2018 Survey Results
OpenKIM Survey on the Future of Molecular Simulation
Ellad B. Tadmor, Daniel S. Karls and Ryan S. Elliott
Recommended Citation: E. B. Tadmor, D. S. Karls and R. S. Elliott, "OpenKIM Survey on the Future of Molecular Simulation", https://openkim.org/survey/2018-future, January 2018.
OpenKIM (openkim.org) is an open source project that aims to make molecular simulations more reliable, reproducible, and portable. We archive interatomic potentials, test them, and make them available in plug-and-play fashion to major simulation codes that support the KIM API standard (including LAMMPS, IMD, DL_POLY, GULP and ASE).
To gain an understanding of the needs of the molecular simulation community, and to help guide future OpenKIM development, the "OpenKIM Survey on the Future of Molecular Simulation" was conducted during Dec 20, 2017 and Jan 8, 2018. The survey was anonymous and included 20 questions on various aspects of molecular modeling in materials science. A total of 449 researchers responded to the survey, which constitutes a response rate of 25%. This is considered quite high for a survey of this length, indicating support for the OpenKIM project and the interest in the topic.
The results of the survey are discussed below. Note that in what follows, “molecular simulations” refer to computer simulations of systems at the level of their constituent atoms or molecules. This includes molecular statics, molecular dynamics, lattice dynamics, Monte Carlo, as well as multiscale methods that resolve structure down to atomic resolution.
Profile of Molecular Simulations in Materials Science
About 79% of respondents to the survey were from academia (university and research institutes), 12.4% from government organizations (national labs and institutes, military labs, and funding agencies), 7.2% from industry (commercial companies and self-employed consultants), and the remaining from non-governmental or unspecified organizations.
The respondents were evenly divided between individuals who regularly perform molecular simulations themselves and those who primarily manage or advise the work of others. The respondents were also equally divided between researchers who have developed their own interatomic potentials or force fields and those who have not.
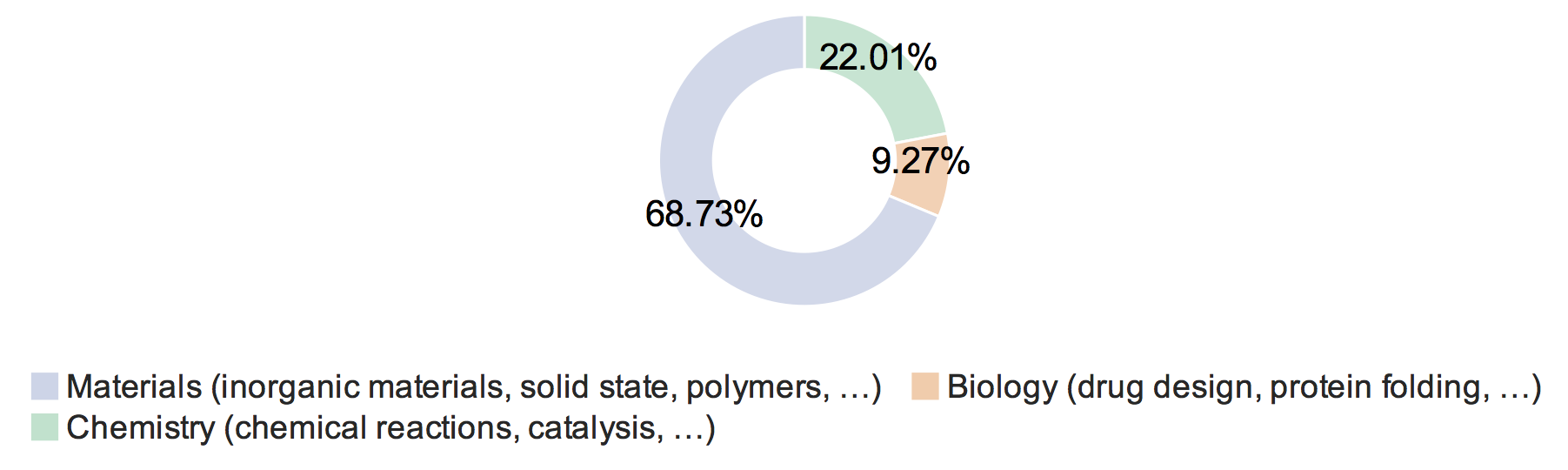
As the ring chart below shows, most responses are from researchers engaged in materials modeling (which was the primary audience to which the survey was sent), but there were also responses from researchers engaged in chemistry and biology.
It is of interest to know which computer systems are primarily used in molecular simulations. The response to "On which of the the following computer operating systems do you typically perform molecular simulations? (check all that apply)" are shown in the bar chart below. We see that Linux/Unix systems are most commonly used (66%), followed by Mac OS (20%) and Windows (14%).
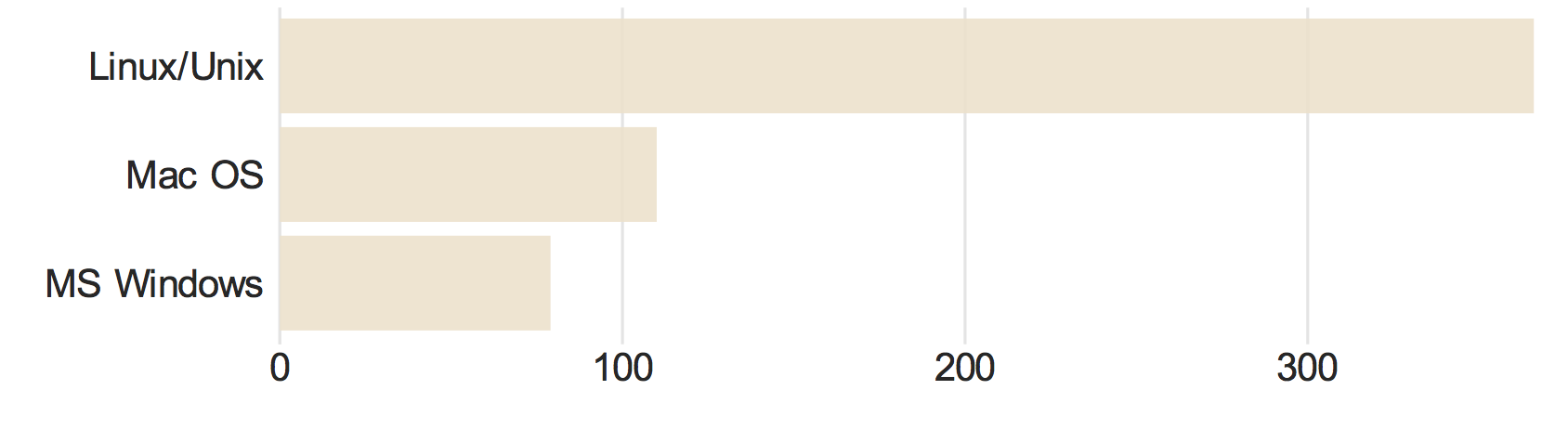
The responses to "Which of the following methodologies do you use in your molecular simulations? (check all that apply)" are below. Most respondents are engaged in classical molecular simulations (which was the target audience of the survey), but density functional theory is also widely employed by these researchers, followed by tight binding and quantum chemistry.
| Field | Count |
|---|---|
| Classical molecular simulations (empirical interatomic potentials and force fields) | 363 |
| Tight binding (semi-empirical quantum calculations) | 73 |
| Density functional theory (quantum simulations) | 213 |
| Quantum chemistry (highly accurate quantum calculations of chemical reactions) | 54 |
| Other | 15 |
Of the respondents who perform classical molecular simulations, the responses to "Which of the following classical interatomic potentials/force fields do you use? (check all that apply)" are shown in the ring chart below in percentages, followed by the count data. (Note that in the ring chart, electrostatic potentials which are included in other classes, and bonded force fields (like CHARMM and AMBER) are not included.)
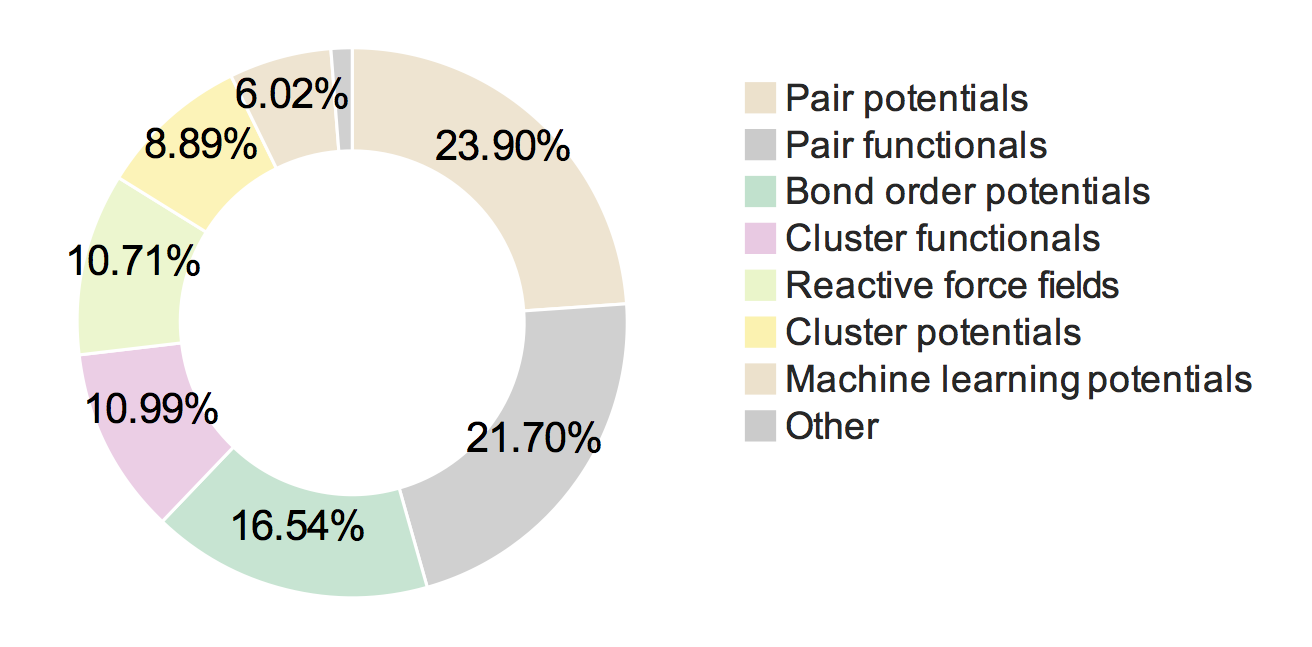
We see that pair potentials and pair functionals are most popular followed by bond-order potentials, cluster functionals, reactive force fields, cluster potentials and machine learning models in that order. See the table below for the exact count number and examples of each of these potential classes.
| Field | Count |
|---|---|
| Bond order potentials (AIREBO, EDIP, REBO, Tersoff, BOP, ...) | 173 |
| Bonded force fields (AMBER, CHARMM, OPLS, TraPPE, ...) | 138 |
| Cluster functionals (ADP, MEAM, ...) | 115 |
| Cluster potentials (Three body (SW, ...), Four body (MGPT, ...)) | 93 |
| Long-range Electrostatics (Born-Mayer, Buckingham, Coulomb, ...) | 151 |
| Machine learning potentials (neural network, GAP, SNAP, ...) | 63 |
| Pair functionals (EAM, EMT, FS, glue potential, ...) | 227 |
| Pair potentials (LJ, Morse, ...) | 250 |
| Reactive force fields (COMB, eFF, ReaxFF, ...) | 112 |
We are also interested to know how frequently molecular simulations are performed to obtained quantitative information versus qualitative insight. The answers to "Typically, what is your objective when performing molecular simulations with classical interatomic potentials/force fields? (select one)" are shown below.
| Field | Count |
|---|---|
| Mostly qualitative understanding of material behavior (e.g. identifying mechanisms or trends). Usually, I don’t expect the calculations to be accurate enough to yield quantitative results. | 126 |
| Mostly quantitative results. Usually, the potential I use is sufficiently accurate to obtain numbers for material properties to be used in analysis or design. | 78 |
| About half the time qualitative, and half the time quantitative. | 151 |
These results show that classical molecular simulations are used qualitatively about 57% of the time, and quantitatively about 43%.
Future Needs in Molecular Simulation
The key motivation for the survey was to identify trends in molecular simulations according to the practitioners in the field. The respondents were asked: "Rank the importance that each of the following currently has to your work, or is likely to have in the future if available." The results on a scale of 1 (not important) to 8 (critical) are displayed below with the mean result and standard deviation (SD).
| Field | Mean | SD |
|---|---|---|
| Portable implementations of interatomic potentials that can be used in "plug-and-play" fashion with different molecular simulation codes. | 7.55 | 2.32 |
| Access to archived interatomic potentials that can be cited in publications (like a DOI), so that simulations can be reproduced. | 7.91 | 2.22 |
| Integration of archived interatomic potentials and codes with workflow management tools (Jupyter notebooks, ExTASY, AiiDA, etc.) | 5.14 | 3.09 |
| Tools that assist researchers in selecting the most accurate interatomic potential for a specific application. | 7.40 | 2.41 |
| Tools that estimate the uncertainty (error bars) associated with the predictions of a given potential (for example through sensitivity analysis). | 7.37 | 2.33 |
| Interatomic potentials that rely on machine learning methods to directly interpolate first-principles (quantum) data rather than using a physically motivated functional form (e.g. neural network potentials, GAP, SNAP, etc). | 6.04 | 2.88 |
| Having access to interatomic potentials that are published together with the complete training set used in their development. (This makes it possible to continue/modify the training of an interatomic potential for new applications.) | 7.37 | 2.43 |
The ability to access archived interatomic potentials that can be cited in publications was identified as the most pressing need. This was closely followed by the ability to use interatomic potentials in plug-and-play fashion across different codes and availability of methods to help researchers select suitable potentials for their applications. These are the top three objectives of the OpenKIM project, with the first two already possible using OpenKIM archiving, permanent KIM IDs and the KIM API.
Other capabilities selected by the community in order of importance are tools for estimating uncertainty in molecular simulations, archiving interatomic potentials along with their training sets, interatomic potentials that are based on machine learning methods, and integration with workflow management tools. These have been identified as important ongoing efforts for OpenKIM in its next development cycle.
Respondents familiar with OpenKIM were asked to rank the importance of specific development goals within the OpenKIM system. The results on a scale of 1 (not important) to 8 (critical) are displayed below.
| Field | Mean | SD |
|---|---|---|
| Extending the KIM framework (KIM API) to support long-range electrostatics and charge equilibration. | 7.32 | 2.34 |
| Extending the KIM framework (KIM API) to support bonded force fields such as CHARMM and AMBER. | 6.20 | 3.02 |
| Support for high-performance computing (HPC) for computationally intensive material properties performed by KIM Tests. | 6.96 | 2.52 |
| Providing KIM-integrated tools to assist developers in fitting traditional physically-motivated interatomic potentials (such as Lennard-Jones, EAM, etc.). | 6.88 | 2.62 |
| Providing KIM-integrated tools to automatically generate machine learning-based interatomic potentials (such as neural network potentials, GAP, SNAP, etc) for specific material systems. | 6.30 | 2.89 |
| In addition to distributing KIM-compliant potentials (which can be used interchangeably with a wide range of simulation codes), OpenKIM should also serve as a distribution hub for potentials specific to different simulation codes (e.g. LAMMPS, IMD, DL_POLY, ...) so that they can be cited in publications and tested within OpenKIM. | 7.38 | 2.54 |
| Developing resources and tools for educators to use in the classroom. | 6.20 | 2.98 |
The items in order of importance are:
-
In addition to distributing KIM Models (i.e. potentials that conform to the KIM API), OpenKIM should also distribute potentials that are native to specific simulators like LAMMPS.
This capability is planned. Potentials that only work with a specific simulator will be called "Simulator Models" (SMs). The OpenKIM framework and potential distribution system will be adapted to work with SMs.
-
Extending the KIM API to support long-range electrostatics and charge equilibration.
This capability is planned. This is not trivial to do because no standard currently exists for electrostatic and charge equilibration methods. We are in process of developing a preliminary standard that will be distributed for public comment.
-
Support of high-performance computing (HPC) tests to enable computation of expensive properties like thermal conductivity.
This capability is planned. OpenKIM is planning to work with the Minnesota Supercomputer Institute to extend the KIM processing pipeline to support KIM Tests running on HPC resources.
-
Providing KIM-integrated tools for fitting traditional physically-motivated (parametric) interatomic potentials.
Preliminary work in this area has been done by extending the Potfit fitting program to support the KIM API. (See here for details.)
-
Providing KIM-integrated tools for fitting machine learning based interatomic potentials.
This is an area for future research for OpenKIM. A first step is to develop a framework for archiving training sets along with interatomic potentials. This capability is planned.
-
Extending the KIM API to support bonded force fields such as CHARMM and AMBER.
We have already invested significant effort in identifying the extensions needed to the KIM API to support bonded force fields. This extension is planned in the future, but extension to electrostatics and charge equilibration takes precedence.
-
Developing resources and tools for educators to use in the classroom.
Although this item received the lowest ranking by the researchers who completed the survey, it is a high priority for the OpenKIM project. To improve the quality of molecular simulation it is vital to improve the training of students in this area. A variety of educational tools are planned for the future.
Conclusion
The insight gained from this survey was invaluable for planning the next steps in the OpenKIM project. The KIM team would like to thank all of the respondents who filled in the survey. A special thanks to those who left us comments with helpful advice and constructive criticism. We do not list the comments here, but they were all taken seriously and many were integrated into our development plan.
We look forward to continued interaction with the molecular simulation community in the coming years.
If you have any thoughts on this survey or have any feedback, please contact us.