Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
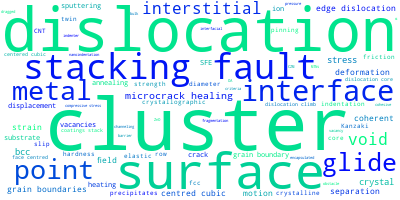
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
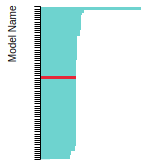
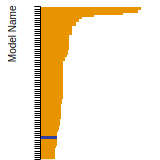
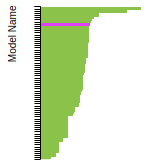
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
483 Citations (297 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) F. Muller, P. Nielaba, J. Cuevas, and F. Pauly, “Phononic heat conductance of gold atomic contacts: Coherent versus incoherent transport,” Physical Review B. 2022. link Times cited: 0 Abstract: We present here a theoretical method to determine the phonon… read moreAbstract: We present here a theoretical method to determine the phononic contribution to the thermal conductance of nanoscale systems in the phase-coherent regime. Our approach makes use of classical molecular dynamics (MD) simulations to calculate the temperature-dependent dynamical matrix, and the phononic heat conductance is subsequently computed within the Landauer-B\"uttiker formalism with the help of nonequilibrium Green's function techniques. Tailored to nanostructures, crucial steps of force constant and heat transport calculations are performed directly in real space. As compared to conventional density functional theory (DFT) approaches, the advantage of our method is two-fold. First, interatomic interactions can be described with the method of choice. Semiempirical potentials may lead to large computational speedups, enabling the study of much larger systems. Second, the method naturally takes into account the temperature dependence of atomic force constants, an aspect that is ignored in typical static DFT-based calculations. We illustrate our method by analyzing the temperature dependence of the phononic thermal conductance of gold (Au) chains with lengths ranging from 1 to 12 atoms. Moreover, in order to evaluate the importance of anharmonic effects in these atomic-scale wires, we compare the phase-coherent approach with nonequilibrium MD (NEMD) simulations. We find that the predictions of the phase-coherent method and the classical NEMD approach largely agree above the Debye temperature for all studied chain lengths, which shows that heat transport is coherent and that our phase-coherent approach is well suited for such nanostructures. read less USED (high confidence) L. Skopinski et al., “Velocity distributions of particles sputtered from supported two-dimensional
MoS2
during highly charged ion irradiation,” Physical Review B. 2022. link Times cited: 2 Abstract: The interaction of highly charged ions (HCI) with solids lea… read moreAbstract: The interaction of highly charged ions (HCI) with solids leads to particle sputtering, which can be used for defect-mediated engineering of the properties of the material. Ions can store energy in the form of kinetic and potential energy (sum of the ionization energies) and transfer it to the solid upon impact. The interaction and sputtering mechanisms depend significantly on the projectile energies. However, the relevance of various interaction mechanisms is unknown. Here we show that for slow HCI (5 keV) the interaction mechanisms leading to particle emission by electronic excitation and transferred kinetic energy are independent from each other, which is consistent with our atomistic simulations. We have irradiated substrate supported (Au, SiO$_2$) monolayers of MoS$_2$ with highly charged xenon ions (charge state: 17$+$ - 40$+$), extracted the emitted neutral, post-ionized Mo particles into a time-of-flight mass spectrometer and determined their velocity distributions. We find two main contributions, one at high velocities and a second one at lower velocities, and assign them to kinetic and potential effects respectively. Our data suggests that the dominant mechanism for potential sputtering is related to electron-phonon coupling, while non-thermal processes play no significant role. We anticipate that our work will be a starting point for further experiments and simulations to determine whether the different processes resulting from E$_{pot}$ and E$_{kin}$ can be separated or whether synergistic effects play a role. read less USED (high confidence) S. Chu et al., “In situ atomic-scale observation of dislocation climb and grain boundary evolution in nanostructured metal,” Nature Communications. 2022. link Times cited: 14 USED (high confidence) E. Pervolarakis, G. Tritsaris, P. Rosakis, and I. Remediakis, “Machine Learning for the edge energies of high symmetry Au nanoparticles,” Surface Science. 2022. link Times cited: 1 USED (high confidence) S. Chu et al., “In Situ Atomic-Scale Observation of Dislocation Climb and Grain Boundary Transformation in Nanostructured Metal.” 2021. link Times cited: 0 Abstract:
We report atomic-scale observations of grain boundary (GB)… read moreAbstract:
We report atomic-scale observations of grain boundary (GB) dislocation climb in nanostructured Au during in situ straining at room temperature. Climb of a dislocation occurs by stress-induced reconstruction of two atomic columns at the edge of an extra half atomic plane in the dislocation core. Different from the conventional belief of dislocation climb by destruction or construction of a single atomic column at the dislocation core, the new atomic route is demonstrated to be energetically favorable by Monte Carlo simulations. Our in situ observations also reveal GB transformation through dislocation climb, which suggests a means of controlling microstructures and properties of nanostructured metals. read less USED (high confidence) A. Lange, A. Samanta, T. Y. Olson, and S. Elhadj, “Quantized Grain Boundary States Promote Nanoparticle Alignment During Imperfect Oriented Attachment.,” Small. 2020. link Times cited: 8 Abstract: Oriented attachment (OA) has become a well-recognized mechan… read moreAbstract: Oriented attachment (OA) has become a well-recognized mechanism for the growth of metal, ceramic, and biomineral crystals. While many computational and experimental studies of OA have shown that particles can attach with some misorientation then rotate to remove adjoining grain boundaries, the underlying atomistic pathways for this "imperfect OA" process remain the subject of debate. In this study, molecular dynamics and in situ transmission electron microscopy (TEM) are used to probe the crystallographic evolution of up to 30 gold nanoparticles during aggregation. It is found that Imperfect OA occurs because 1) grain boundaries become quantized when their size is comparable to the separation between constituent dislocations and 2) kinetic barriers associated with the glide of grain boundary dislocations are small. In support of these findings, TEM experiments show the formation of a single crystal aggregate after annealing nine initially misoriented, agglomerated particles with evidence of dislocation activity and twin formation during particle/grain alignment. These observations motivate future work on assembled nanocrystals with tailored defects and call for a revision of Read-Shockley models for grain boundary energies in nanocrystalline materials. read less USED (high confidence) F. Ojaghnezhad and H. Shodja, “Second strain gradient theory in orthogonal curvilinear coordinates: Prediction of the relaxation of a solid nanosphere and embedded spherical nanocavity,” Applied Mathematical Modelling. 2019. link Times cited: 5 USED (high confidence) G. Lerma, B. Verschueren, B. Gurrutxaga-Lerma, and J. Verschueren, “Generalized Kanzaki force field of extended defects in crystals with applications to the modeling of edge dislocations,” Physical Review Materials. 2019. link Times cited: 4 Abstract: The Kanzaki forces and their associated multipolar moments a… read moreAbstract: The Kanzaki forces and their associated multipolar moments are standard ways of representing point defects in an atomistically informed way in the continuum. In this article, the Kanzaki force approach is extended to other crystalline defects. The article shows how the resulting Kanzaki force fields are to be computed for any general extended defect by first computing the relaxed defect’s structure and then defining an affine mapping between the said defect structure and the original perfect lattice. This methodology can be employed to compute the Kanzaki force field of any mass-conserving defect, including dislocations, grain and twin boundaries, or cracks. Particular focus is then placed on straight edge dislocation in face-centered cubic (fcc) and body-centered cubic (bcc) pure metals, which are studied along different crystallographic directions. The particular characteristics of these force fields are discussed, drawing a distinction between the slip Kanzaki force field associated with the Volterra disregistry that characterizes the dislocation, and the core Kanzaki force field associated with the specific topology of the dislocation’s core. The resulting force fields can be employed to create elastic models of the dislocation that, unlike other regularization procedures, offer a geometrically true representation of the core and the elastic fields in its environs, capturing all three-dimensional effects associated with the core. DOI: 10.1103/PhysRevMaterials read less USED (high confidence) S. Ghaderzadeh, M. Ghorbani-Asl, S. Kretschmer, G. Hlawacek, and A. Krasheninnikov, “Channeling effects in gold nanoclusters under He ion irradiation: insights from molecular dynamics simulations,” Nanotechnology. 2019. link Times cited: 10 Abstract: The interpretation of helium ion microscopy (HIM) images of … read moreAbstract: The interpretation of helium ion microscopy (HIM) images of crystalline metal clusters requires microscopic understanding of the effects of He ion irradiation on the system, including energy deposition and associated heating, as well as channeling patterns. While channeling in bulk metals has been studied at length, there is no quantitative data for small clusters. We carry out molecular dynamics simulations to investigate the behavior of gold nanoparticles with diameters of 5–15 nm under 30 keV He ion irradiation. We show that impacts of the ions can give rise to substantial heating of the clusters through deposition of energy into electronic degrees of freedom, but it does not affect channeling, as clusters cool down between consecutive impact of the ions under typical imaging conditions. At the same time, high temperatures and small cluster sizes should give rise to fast annealing of defects so that the system remains crystalline. Our results show that ion-channeling occurs not only in the principal low-index, but also in the intermediate directions. The strengths of different channels are specified, and their correlations with sputtering-yield and damage production is discussed, along with size-dependence of these properties. The effects of planar defects, such as stacking faults on channeling were also investigated. Finally, we discuss the implications of our results for the analysis of HIM images of metal clusters. read less USED (high confidence) J. Yu, H. Xie, F. Yin, and T. Yu, “Face-centered-cubic to body-centered-cubic phase transformation of Cu nanoplate under [100] tensile loading,” Philosophical Magazine. 2019. link Times cited: 1 Abstract: ABSTRACT Molecular dynamics simulation was used to stretch C… read moreAbstract: ABSTRACT Molecular dynamics simulation was used to stretch Cu nanoplates along its [100] direction at various strain rates and temperatures. Under high strain rate and beyond the elastic limit, the Cu nanoplates underwent an unusual deformation mechanism with expansion along free surface lateral direction and contraction along the other lateral direction, which leaded to the face-centred-cubic phase transforming into unstressed body-centred-cubic phase. Under low strain rate, the deformation of the nanoplate went back to well-known dislocation mechanism. The face-centred-cubic to body-centred-cubic phase transformation mechanism was further discussed in terms of elastic stability theory and free surface stress effect. read less USED (high confidence) D. Chen and Y. Kulkarni, “Atomistic modeling of grain boundary motion as a random walk,” Physical Review Materials. 2018. link Times cited: 4 Abstract: Mechanical behavior of polycrystalline materials, such as cr… read moreAbstract: Mechanical behavior of polycrystalline materials, such as creep and microstructural evolution, is dramatically impacted by the mobility of grain boundaries. Existing methods for extracting mobility that combine atomistic simulations with conventional Brownian-like random walk model for grain boundary motion miss critical insights and are reliable only above very high (and unrealistic) temperatures. In this paper, we present a computational method based on the classical Green-Kubo relation for computing interface mobility over a wide range of temperatures. Our study makes an intriguing revelation that the severe time limitation of molecular dynamics simulations warrants the use of a generalized diffusion equation for Brownian particles not accounted for in current studies. Moreover, the method furnishes analytical expressions for the interface mobility in terms of the velocity autocorrelation functions. Taken together, our results possibly provide the first reliable estimates for interface mobility in the limit of zero driving forces. This is in sharp contrast to studies based on the widely used interface random walk approach, which extracts mobility by fitting the conventional Brownian motion equation to atomistic simulations. The efficiency of the method and applicability over a range of temperatures should open avenues for integration of computations and experiments to understand and engineer material systems with desired properties. read less USED (high confidence) S. Kumar, S. Das, and S. K. Pattanayek, “Evolution of nanostructure and mechanical properties of silver nano-particle in the confined region between graphene sheets: An atomistic investigation,” Computational Materials Science. 2018. link Times cited: 8 USED (high confidence) E. V. Duda and G. V. Kornich, “Construction of a Changed Potential of Interatomic Interaction in the Case of Temperature-Accelerated Dynamics Simulation,” Journal of Surface Investigation: X-ray, Synchrotron and Neutron Techniques. 2018. link Times cited: 2 USED (high confidence) Y. Yang and N. Yan, “Understanding the cooperative atomic motion and shape change of ultrasmall Au nanoparticles below the premelting temperature,” RSC Advances. 2017. link Times cited: 0 Abstract: Surface melting is widely observed in crystalline materials,… read moreAbstract: Surface melting is widely observed in crystalline materials, which has a significant influence on their interfacial properties. In this computational study using molecular dynamics simulations, we observed that at 50 K below the onset temperature of surface melting, the “out-shell” atoms of ultrasmall Au nanoparticles (NPs) have already undergone remarkable rearrangements. Unlike the observations in Ni ultrasmall NPs, the resulting shape change was often isotropic. Further investigations reveal that such interfacial motions are cooperative and string-like. The gold “atom strings” do not migrate through the center of the particle, behaving similarly as those in much larger particles. Therefore, the “spherical” shape was sustained during the atomic motions. This result reveals the dynamic nature of the atomic motions of Au before the commencement of premelting and sheds light on the understanding of the origin of surface melting. read less USED (high confidence) S. Kumar, V. Srivastava, G. K. Mandal, S. K. Pattanayek, and K. L. Sahoo, “Single-Walled Carbon Nanotube Engendered Pseudo-1D Morphologies of Silver Nanowire,” Journal of Physical Chemistry C. 2017. link Times cited: 9 Abstract: Silver nanowires show enhancement in desired properties, due… read moreAbstract: Silver nanowires show enhancement in desired properties, due to high surface area and high aspect ratio, which increases the possibility in design and development of many advanced optoelectronic devices. The organization of silver atoms and the morphology of cylindrical nanowire highly influence the desired properties at various physical conditions during applications. Therefore, the synthesis of nanowires with desired atomic organization becomes essential. In the present study, a pseudo-1D morphology of silver nanowires, encapsulated into a Single Wall Carbon Nanotubes (SWCNTs), has been investigated by employing Molecular Dynamics (MD) simulation. At high temperature (T = 1500 K), molten silver encapsulated into a SWCNT attaining a low energy state followed by restricted thermal motion or vibration. With an increase in SWCNT diameter, various unique pseudo-1D morphologies of encapsulated silver atoms were observed during cooling from 1500 to 10 K. Silver atoms, encapsulated into a SWCNT having a diamete... read less USED (high confidence) A. Lange et al., “Dislocation mediated alignment during metal nanoparticle coalescence,” Acta Materialia. 2016. link Times cited: 29 USED (high confidence) J. Bean and K. McKenna, “Origin of differences in the excess volume of copper and nickel grain boundaries,” Acta Materialia. 2016. link Times cited: 61 USED (high confidence) T. Okita, Y.-Y. Yang, J. Hirabayashi, M. Itakura, and K. Suzuki, “Effects of stacking fault energy on defect formation process in face-centered cubic metals,” Philosophical Magazine. 2016. link Times cited: 14 Abstract: To elucidate the effect of stacking fault energies (SFEs) on… read moreAbstract: To elucidate the effect of stacking fault energies (SFEs) on defect formation by the collision cascade process for face-centred cubic metals, we used six sets of interatomic potentials with different SFEs while keeping the other properties almost identical. Molecular dynamic simulations of the collision cascade were carried out using these potentials with primary knock-on atom energies (EPKA) of 10 and 20 keV at 100 K. Neither the number of residual defects nor the size distributions for both self-interstitial atom (SIA) type and vacancy type clusters were affected by the difference in the SFE. In the case of EPKA = 20 keV, the ratio of glissile SIA clusters increased as the SFE decreased, which was not expected by a prediction based on the classical dislocation theory. The trend did not change after annealing at 1100 K for 100 ps. For vacancy clusters, few stacking fault tetrahedrons (SFTs) formed before the annealing. However, lower SFEs tended to increase the SFT fraction after the annealing, where large vacancy clusters formed at considerable densities. The findings of this study can be used to characterise the defect formation process in low SFE metals such as austenitic stainless steels. read less USED (high confidence) L. Niu et al., “Shear-coupled grain boundary migration assisted by unusual atomic shuffling,” Scientific Reports. 2016. link Times cited: 18 USED (high confidence) I. E. Awad and L. Ladani, “Mechanical integrity of a carbon nanotube/copper-based through-silicon via for 3D integrated circuits: a multi-scale modeling approach,” Nanotechnology. 2015. link Times cited: 10 Abstract: Carbon nanotube (CNT)/copper (Cu) composite material is prop… read moreAbstract: Carbon nanotube (CNT)/copper (Cu) composite material is proposed to replace Cu-based through-silicon vias (TSVs) in micro-electronic packages. The proposed material is believed to offer extraordinary mechanical and electrical properties and the presence of CNTs in Cu is believed to overcome issues associated with miniaturization of Cu interconnects, such as electromigration. This study introduces a multi-scale modeling of the proposed TSV in order to evaluate its mechanical integrity under mechanical and thermo-mechanical loading conditions. Molecular dynamics (MD) simulation was used to determine CNT/Cu interface adhesion properties. A cohesive zone model (CZM) was found to be most appropriate to model the interface adhesion, and CZM parameters at the nanoscale were determined using MD simulation. CZM parameters were then used in the finite element analysis in order to understand the mechanical and thermo-mechanical behavior of composite TSV at micro-scale. From the results, CNT/Cu separation does not take place prior to plastic deformation of Cu in bending, and separation does not take place when standard thermal cycling is applied. Further investigation is recommended in order to alleviate the increased plastic deformation in Cu at the CNT/Cu interface in both loading conditions. read less USED (high confidence) L. Niu et al., “Interplay between intrinsic point defects and low-angle grain boundary in bcc tungsten: effects of local stress field,” Journal of Physics: Condensed Matter. 2015. link Times cited: 19 Abstract: We have used molecular statics in conjunction with an embedd… read moreAbstract: We have used molecular statics in conjunction with an embedded atom method to explore the interplay between native point defects (vacancies and self-interstitials (SIAs)) and a low-angle grain boundary (GB) in bcc tungsten. The low-angle GB has biased absorption of SIAs over vacancies. We emphasize the significance of phenomena such as vacancy delocalization and SIA instant absorption around the GB dislocation cores in stabilizing the defect structures. Interstitial loading into the GB can dramatically enhance the interaction strength between the point defects and the GB due to SIA clustering (SIA cloud formation) or SIA vacancy recombination. We propose that the ‘maximum atom displacement’ can complement the ‘vacancy formation energy’ in evaluating unstable vacancy sites. Calculations of point defect migration barriers in the vicinity of GB dislocation cores show that vacancies and SIAs preferentially migrate along the pathways in the planes immediately above and below the core, respectively. read less USED (high confidence) F. Z. Dai and W.-Z. Zhang, “A simple method for constructing a reliable initial atomic configuration of a general interface for energy calculation,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 9 Abstract: Interfacial energy calculated with an atomistic model is ver… read moreAbstract: Interfacial energy calculated with an atomistic model is very sensitive to adding atoms to or removing atoms from the interface, especially when the interface has a general orientation. Therefore, it is crucial to construct an appropriate initial atomic configuration in order to obtain a reliable value for the interfacial energy from atomistic simulations. In this work, a simple method is proposed for constructing the atomic configuration of a general interface under the condition that the interface is virtually free of interstitial and vacancy. The validity of the method is demonstrated by using it to calculate the equilibrium morphology of precipitates with interfaces in irrational orientations, which shows good agreement with experimental observations. read less USED (high confidence) T. Schablitzki, J. Rogal, and R. Drautz, “Topological fingerprints for intermetallic compounds for the automated classification of atomistic simulation data,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 13 Abstract: We introduce a method to determine intermetallic crystal pha… read moreAbstract: We introduce a method to determine intermetallic crystal phases by creating topological fingerprints using coordination polyhedra. Many intermetallic crystal phases have complex structures that cannot be determined from the information of their nearest neighbour environment alone, but need information from a further reaching local environment. We obtain the coordination polyhedra of each atom in the structure and use this information in a topological fingerprint to determine the crystal phases in the structure as locally as possible. This allows us to analyse complex crystal phases like the topologically close-packed phases and multi-phase structures. With the information extracted from the coordination polyhedra and topological fingerprint, it is also possible to find and identify point and extended defects. Therefore, our method is able to track interface regions in multi-phase structures, and follow structural changes during phase transformations. read less USED (high confidence) F. Dai and W.-Z. Zhang, “A systematic study on the interfacial energy of O-line interfaces in fcc/bcc systems,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 11 Abstract: Habit planes between face-centered cubic (fcc)/body-centered… read moreAbstract: Habit planes between face-centered cubic (fcc)/body-centered cubic (bcc) phases usually exhibit irrational orientations, which often agree with the O-line criterion. Previously, energy calculation was made to test whether the habit planes were energetically favorable, but the values of the energy were found very sensitive to the initial atomic configuration in an irrationally orientated interface. In this paper, under the O-line condition, simple selection criteria are proposed to define and remove interfacial interstitials and vacancies in the initial atomic configuration. The criteria are proved to be effective in obtaining robust energy results. Interfacial energies of two types of O-line interfaces in fcc/bcc systems are calculated following the criteria. The observed transformation crystallography of precipitates in Ni–Cr and Cu–Cr systems can be explained consistently as the irrational habit plane in each system is associated with the lowest energy O-line interface. read less USED (high confidence) W. Gräfe, “Supplement to: a simple quantum mechanical model for the contribution of electronic surface states to surface stress, strength and electrocapillarity of solids,” Journal of Materials Science. 2013. link Times cited: 6 USED (high confidence) C. Scott and R. Smith, “Modelling the sputtering of Au surfaces using a multi time-scale technique,” Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2013. link Times cited: 7 Abstract: We present results from an atomistic computer simulation mod… read moreAbstract: We present results from an atomistic computer simulation model of the sputtering of gold crystal surfaces under 500 eV ion bombardment by Au and Ar ions for doses up to 1014 ions cm−2. The multi time-scale technique uses molecular dynamics to calculate the fast ballistic collision processes in the early stages of the cascade, whereas an on-the fly kinetic Monte Carlo technique is used to model the relaxation and diffusion processes between successive ion impacts when the defect motion has begun to be dominated by rare events. The results indicate a large amount of crystalline recovery between impacts, some facetting of the crystal surfaces but no large sub-surface defect accumulation. Because of this recovery process, sputtering yields and energy distributions are in good agreement with those obtained assuming a perfect crystal surface and also with those experimentally measured. read less USED (high confidence) C. Becker, F. Tavazza, and L. Levine, “Implications of the choice of interatomic potential on calculated planar faults and surface properties in nickel,” Philosophical Magazine. 2011. link Times cited: 15 Abstract: With the increasing use of molecular simulation to understan… read moreAbstract: With the increasing use of molecular simulation to understand deformation mechanisms in transition metals, it is important to assess how well the simulations reproduce physical behavior both near equilibrium and under more extreme conditions. In particular, it is important to examine whether simulations predict unusual deformation paths that are competitive with those experimentally observed. In this work we compare generalized planar fault energy landscapes and surface energies for various interatomic potentials with those from density functional theory calculations to examine how well these more complicated planar faults and surface energies are captured and whether any deformations are energetically competitive with the {111}⟨112⟩ slip observed in FCC crystals. To do this we examine not just the (111) fault orientation, but also the (100), (110), (210), (211), (311), and (331) orientations to test behavior outside of the fitting range of the interatomic potentials. We find that the shape of the (111)[11 ] stacking fault energy curve varies significantly with potential, with the ratio of unstable to stable stacking fault energies ranging from 1.22 to 14.07, and some deformation paths for non-(111) orientations give activation barriers less than 50% higher than the unstable stacking fault energies. These are important considerations when choosing an interatomic potential for deformation simulations. read less USED (high confidence) J.-H. Yoo, S.-I. Oh, and M. Jeong, “The enhanced elastic modulus of nanowires associated with multitwins,” Journal of Applied Physics. 2010. link Times cited: 31 Abstract: In this study, we performed molecular static simulations to … read moreAbstract: In this study, we performed molecular static simulations to investigate the enhanced elastic modulus of multiply twinned nanowires (MTNs) that had fivefold twin planes. MTNs have the same size-dependent elastic behavior as twin-free nanowires (TFNs), however, the simulation results reveal stiffer elastic behavior of MTNs when compared with that of TFNs of the same size. This is because atomic rearrangement occurs inside MTNs through the intrinsic elastic deformation as a result of their unique geometry. These results are supported by the elastic-modulus distribution on the cross-sectional area—the elastic modulus increases close to the central part of MTNs—and the intrinsically strained structure that has a highly compressed central portion in the radial direction. read less USED (high confidence) Y. Osetsky and D. Bacon, “Atomic-scale mechanisms of void hardening in bcc and fcc metals,” Philosophical Magazine. 2010. link Times cited: 98 Abstract: Strengthening due to voids can be a significant effect of rad… read moreAbstract: Strengthening due to voids can be a significant effect of radiation damage in metals, but treatment of this by elasticity theory of dislocations is difficult when the mechanisms controlling the obstacle strength are atomic in nature. Results are reported of atomic-scale modelling to compare edge dislocation–void interaction in fcc copper and bcc iron. Voids of up to 6 nm diameter in iron and 8 nm diameter in copper were studied over the temperature range 0 to 600 K at different applied strain rates. Voids in iron are strong obstacles, for the dislocation has to adopt a dipole-like configuration at the void before breaking away. The dipole unzips at the critical stress when the dislocation is able to climb by absorbing vacancies and leave the void surface. Dislocation dissociation into Shockley partials in copper prevents dislocation climb and affects the strength of small and large voids differently. Small voids are much weaker obstacles than those in iron because the partials break from a void individually. Large voids are at least as strong as those in iron, but the controlling mechanism depends on temperature. read less USED (high confidence) I. Gheewala, S. Kenny, and R. Smith, “Atomistic-scale modelling of nanoindentation into optical coatings,” Philosophical Magazine. 2009. link Times cited: 4 Abstract: Simulations of nanoindentation into a typical optical-coatin… read moreAbstract: Simulations of nanoindentation into a typical optical-coatings stack employed in energy efficient glazing have been performed using classical molecular dynamics (MD) and a coupled finite element/MD methodology. The coatings stack consists of a low-emissivity material, Ag, sandwiched between two layers of a transparent conducting oxide (TCO), ZnO. Simulations into both the ZnO and the coatings stack show a strong interaction between the tip symmetry and crystal symmetry in the observed displacement field. A large amount of elastic recovery is observed for both the ZnO system and the coatings stack, but with an impression left on the surface that looks like a crack but extends no further than the tip imprint at maximum depth. The full stack is observed to have a lower hardness once there is a significant penetration of the displacement field into the Ag, when compared to the pure ZnO system. A comparison between the coupled finite element/MD methodology and the fixed boundary MD-only model shows that the boundary conditions have little influence on the calculated results. read less USED (high confidence) H. Ogawa, “Molecular Dynamics Simulation on the Modification of Crystallographic Orientation in Fragmented Particles in the Aerosol-Deposition Process,” Materials Transactions. 2007. link Times cited: 16 Abstract: Particle fragmentation and modification of crystallographic … read moreAbstract: Particle fragmentation and modification of crystallographic orientation in the aerosol-deposition process were investigated by molecular dynamics simulation. Atomistic models of bcc Fe and fcc Ni particles of 10 nm in diameter were subjected to collision to at substrates, and analyzed for their structural modification due to the impact. It was found that particle fragmentation takes place depending on both crystal structure and initial orientation of incident particles. Modification of crystallographic orientation were observed in both bcc and fcc particles and in bcc substrates. Microscopic mechanisms of particle fragmentation and orientation modification were found to be classified into three types, sliding, compression, and third phase generation, which are related to slip systems and relaxation of interface structures. read less USED (high confidence) M. A. Puigvi, N. de Diego, A. Serra, Y. Osetsky, and D. Bacon, “On the interaction between a vacancy and self-interstitial atom clusters in metals,” Philosophical Magazine. 2007. link Times cited: 14 Abstract: Atomic-scale computer simulation is used to study the intera… read moreAbstract: Atomic-scale computer simulation is used to study the interaction between a vacancy and a cluster of self-interstitial atoms in metals with hcp, fcc and bcc crystal structure: α-zirconium, copper and α-iron. Effects of cluster size, atomic structure, dislocation nature of the cluster side and temperature are investigated. A vacancy can recombine with any interstitial in small clusters and this does not affect cluster mobility. With increasing sizes clusters develop dislocation character and their interaction with vacancies depends on whether the cluster sides dissociate into partial dislocations. A vacancy recombines only on undissociated sides and corners created with undissociated segments. Vacancies inside the cluster perimeter do not recombine but restrict cluster mobility. Temperature enhances recombination by either increasing the number of recombination sites or assisting vacancy diffusion towards such sites. The results are discussed with relevance to differences in irradiation microstructure evolution of bcc, fcc and hcp metals and higher level theoretical modelling techniques. read less USED (high confidence) Y. Osetsky, Y. Matsukawa, R. Stoller, and S. Zinkle, “On the features of dislocation–obstacle interaction in thin films: large-scale atomistic simulation,” Philosophical Magazine Letters. 2006. link Times cited: 30 Abstract: Large-scale atomistic modelling has demonstrated that the dy… read moreAbstract: Large-scale atomistic modelling has demonstrated that the dynamic interactions of dislocations in thin films have a number of remarkable features. A particular example is the interaction between a screw dislocation and a stacking fault tetrahedron (SFT) in Cu, which can be directly compared with in situ observations of quenched or irradiated fcc metals. If the specimen is thin, the dislocation velocity is slow, and the temperature is high enough, a segment of the original SFT can be transported towards the surface via a double cross-slip mechanism and fast glide of an edge dislocation segment formed during the interaction. The mechanisms observed in the simulations provide an explanation for the results of in situ straining experiments and the differences between bulk and thin film experiments. read less USED (high confidence) D. Mulliah, S. Kenny, E. Mcgee, R. Smith, A. Richter, and B. Wolf, “Atomistic modelling of ploughing friction in silver, iron and silicon,” Nanotechnology. 2006. link Times cited: 44 Abstract: Molecular dynamics (MD) simulations of atomic-scale stick–sl… read moreAbstract: Molecular dynamics (MD) simulations of atomic-scale stick–slip have been performed for a diamond tip in contact with the (100) surface of fcc Ag, bcc Fe, Si and H-terminated Si, at a temperature of 300 K. Simulations were carried out at different support displacements between 5 and 15 Å. The simulations illustrate the important mechanisms that take place during stick–slip. In particular, for the case of the metals they show a direct link between tip slip events and the emission of dislocations from the point of contact of the tip with the substrate. This occurs both during indentation and scratching. For the case of silicon, no slip events were observed and no subsurface dislocations were generated underneath the scratch groove. At the deeper support displacement of 15 Å the silicon atoms undergo some local phase transformations and the atom coordination number varies between 5 and 8, with the majority being five-fold or six-fold coordinated. Both the dynamic and the static friction coefficients were found to be higher for Si compared to the corresponding values for H-terminated Si. Comparisons were made between the MD simulations and experimental measurements for indentation on the (100) surface of Si and Al. A good qualitative agreement was observed between the experimental and theoretical results. However, in both the cases of Si and metals the MD simulations give a contact pressure under load that is depth dependent and values that are higher than experimental nanohardness values. read less USED (high confidence) R. Smith et al., “Modeling the pinning of Au and Ni clusters on graphite,” Physical Review B. 2006. link Times cited: 63 Abstract: The pinning of size-selected AuN and NiN clusters on graphit… read moreAbstract: The pinning of size-selected AuN and NiN clusters on graphite, for N=7–100, is investigated by means of molecular dynamics simulations and the results are compared to experiment and previous work with Ag clusters. Ab initio calculations of the binding of the metal adatom and dimers on a graphite surface are used to parametrize the potentials used in the simulations. The clusters are projected normally towards a graphite surface and the value of the energy at which pinning first occurs, EP, is determined. Pinning is shown to occur when a surface defect, made by the cluster interaction, is first produced. The simulations give a good agreement with the experimentally determined pinning energy thresholds and the heights of the clusters on the surface. The gold clusters are shown to be flatter and more spread out than the nickel clusters which are more compact. read less USED (high confidence) Z. Rong, Y. Osetsky, and D. Bacon, “A model for the dynamics of loop drag by a gliding dislocation,” Philosophical Magazine. 2005. link Times cited: 54 Abstract: Clusters of self-interstitial atoms are formed in metals by … read moreAbstract: Clusters of self-interstitial atoms are formed in metals by high-energy displacement cascades, often in the form of small dislocation loops with a perfect Burgers vector. In isolation, they are able to undergo fast, thermally activated glide in the direction of their Burgers vector, but do not move in response to a uniform stress field. The present work considers their ability to glide under the influence of the stress of a gliding dislocation. If loops can be dragged by a dislocation, it would have consequences for the effective cross-section for dislocation interaction with other defects near its glide plane. The lattice resistance to loop drag cannot be simulated accurately by the elasticity theory of dislocations, so here it is investigated in iron and copper by atomic-scale computer simulation. It is shown that a row of loops lying within a few nanometres of the dislocation slip plane can be dragged at very high speed. The drag coefficient associated with this process has been determined as a function of metal, temperature and loop size and spacing. A model for loop drag, based on the diffusivity of interstitial loops, is presented. It is tested against data obtained for the effects of drag on the stress to move a dislocation and the conditions under which a dislocation breaks away from a row of loops. read less USED (high confidence) Y. Osetsky, D. Bacon, Z. Rong, and B. N. Singh, “Dynamic properties of edge dislocations decorated by interstitial loops in α-iron and copper,” Philosophical Magazine Letters. 2004. link Times cited: 27 Abstract: Clusters of self-interstitial atoms (SIAs) in the form of pa… read moreAbstract: Clusters of self-interstitial atoms (SIAs) in the form of parallel crowdions are created directly in high-energy displacement cascades produced in metals by neutron irradiation. They are equivalent to small perfect dislocation loops and, in isolation in pure metals, undergo fast thermally-activated glide in the direction of their Burgers vector. Their strain field and ability to glide allows long-range interaction with other extended defects. Indeed, dislocations decorated by dislocation loops are commonly observed after neutron irradiation. Dislocations gliding under applied stress also encounter these mobile defects. These effects influence mechanical properties and require further investigation. This paper presents results from an atomic-scale study of copper and α-iron at either 0 K or 300 K. Loop drag and breakaway effects are investigated for an edge dislocation under applied stress interacting with a row of SIA loops below its glide plane. The maximum speed at which a loop is dragged is lower in copper than iron, and the applied stress at which this occurs is also lower. These differences in the dynamics of cluster-dislocation interaction are determined by the atomic structure of the defects and cannot be investigated by continuum treatment. read less USED (high confidence) D. Mulliah, S. Kenny, R. Smith, and C. Sanz-Navarro, “Molecular dynamic simulations of nanoscratching of silver (100),” Nanotechnology. 2004. link Times cited: 68 Abstract: The nanometric scale ploughing friction and wear behaviour o… read moreAbstract: The nanometric scale ploughing friction and wear behaviour of a pyramidal diamond indenter sliding against a face-centred cubic silver (100) surface is investigated by means of parallel molecular dynamic (MD) simulations of nanoindentation followed by nanoscratching. The relationship between the friction coefficient, the hardness and the indenter orientation is studied. The simulations were performed using three different indenter orientations. For each orientation, simulations were performed at an indentation depth of 5 and 10??, and a scratching length of 210??. In order to study the behaviour of the friction coefficient and the hardness as a function of depth we performed the simulations for one of the orientations at depths of 5, 10 and 30??. The simulations show that the friction coefficient is dependent on both the orientation of the indenter and the indentation depth. The results also show that the friction coefficient increases as the depth increases, whereas the contact pressure decreases and the scratch hardness decreases slightly. With a shallow indent of 5??, no sub-surface defects were observed beneath the scratch groove, but with the deeper indents of 10 and 30???dislocations in the {111} planes are observed beneath the scratch groove. These dislocations propagate in the direction; each dislocation consists of the intersection of stacking faults on two {111} planes and each stacking fault is bounded by two Shockley partial dislocations. read less USED (high confidence) P. Liu, Y. Zhang, and C. Lu, “A three-dimensional concurrent atomistic/continuum analysis of an epitaxially strained island,” Journal of Applied Physics. 2003. link Times cited: 8 Abstract: A three-dimensional concurrent atomistic/continuum analysis … read moreAbstract: A three-dimensional concurrent atomistic/continuum analysis is performed to study the stress distributions in an epitaxial island. The island and the adjacent substrate are modeled by the molecular dynamics method, and the surrounding zone is modeled by the finite element method in which the nonlinear elasticity is governed by the atomic potential. An overlapping zone is defined to be the location where the atomistic and continuum models exchange necessary material information. It is found that the nonlinear elasticity based on the atomic potential is required for a smooth stress transition in the overlapping zone, and that the linear elasticity assumption significantly overestimates the stress levels at moderate and high loading levels. The implications of the present results on surface roughening driven by strain energy are discussed. read less USED (high confidence) Y. Osetsky, D. Bacon, and V. Mohles, “Atomic modelling of strengthening mechanisms due to voids and copper precipitates in α-iron,” Philosophical Magazine. 2003. link Times cited: 140 Abstract: Recently a model has been developed by Osetsky and Bacon to … read moreAbstract: Recently a model has been developed by Osetsky and Bacon to study edge dislocations moving over large distances on the atomic scale. It permits investigation of motion of a dislocation under different conditions of applied shear stress with constant or variable strain rate and temperature, and in the presence of obstacles. In this paper we apply the model to study the motion of an infinite straight but flexible edge dislocation through a row of either voids or coherent copper precipitates in bcc iron. Stress–strain curves, energy barrier profile and strength characteristics of obstacles and other dislocation configuration information have been obtained from the modelling and compared with continuum treatments. Some specific atomic-scale mechanisms associated with strengthening due to voids and precipitates over a range of size have been observed and discussed. read less USED (high confidence) D. Christopher, R. Smith, and A. Richter, “Atomistic modelling of nanoindentation in iron and silver,” Nanotechnology. 2001. link Times cited: 81 Abstract: Experimental and theoretical investigations of nanoindentati… read moreAbstract: Experimental and theoretical investigations of nanoindentation into fcc silver and bcc iron were performed, including an investigation of the effect near the grain boundaries. Experimentally, micrograph images of the surfaces and force-depth curves were obtained which were used to determine the hardness and Young's modulus of the materials. Molecular dynamics simulations, on smaller systems than those investigated experimentally, exhibit the main experimental attributes, showing the plastic deformation of the substrates with piling-up of the work material along the indenter sides. The simulations also show how defects in the substrates form and these are contrasted with the various materials under investigation. read less USED (high confidence) S.-Q. Li, K. Gao, L. Qiao, F. Zhou, and W. Chu, “Molecular Dynamics Simulation of Microcrack Healing in Copper,” Computational Materials Science. 2001. link Times cited: 17 USED (high confidence) L. Shen, G. Kewei, Q. Lijie, W. Chu, and Z. Fuxin, “Molecular dynamics simulation of the role of dislocations in microcrack healing,” Acta Mechanica Sinica. 2000. link Times cited: 5 USED (high confidence) G. Zhou, K. Gao, L. Qiao, Y. Wang, and W. Chu, “Atomistic simulation of microcrack healing in aluminium,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 12 Abstract: A molecular dynamics method is used to simulate microcrack h… read moreAbstract: A molecular dynamics method is used to simulate microcrack healing during heating or under compressive stress. A centre microcrack in Al crystal could be sealed by a critical compressive stress or by heating over a critical temperature. During microcrack healing, dislocation generation and motion occurred. When there were pre-existing dislocations around the microcrack, the critical temperature necessary for microcrack healing would decrease from 850 to 650 K. The critical temperature necessary for microcrack healing depended upon the orientation of the crack plane. For example, the critical temperature of the crack along the (111) plane was the lowest. read less USED (high confidence) J. Hosson, H. B. Groen, B. Kooi, and V. Vítek, “Metal-ceramic interfaces studied with high-resolution transmission electron microscopy,” Acta Materialia. 1999. link Times cited: 33 USED (high confidence) V. Shenoy, R. E. Miller, E. Tadmor, D. Rodney, R. Phillips, and M. Ortiz, “An adaptive finite element approach to atomic-scale mechanics—the quasicontinuum method,” Journal of The Mechanics and Physics of Solids. 1997. link Times cited: 660 USED (high confidence) M. Yan and V. Vítek, “Atomistic studies of the structure and composition of grain boundaries in Cu3Au and Ni3Al,” Interface Science. 1995. link Times cited: 13 USED (high confidence) B. J. Pestman, J. Hosson, V. Vitek, and F. Schapink, “Interaction between Lattice Dislocations and Grain Boundaries in f.c.c. Materials,” Scripta Metallurgica. 1989. link Times cited: 7 USED (high confidence) E. Bitzek, “Atomistic Study of Twinning in Gold Nanowhiskers,” Journal of Solid Mechanics and Materials Engineering. 2012. link Times cited: 14 Abstract: Twinning can become an important deformation mechanism in fc… read moreAbstract: Twinning can become an important deformation mechanism in fcc metals once the crystal size is reduced to the sub-micron scale, e.g., in nanocrystals or nanowhiskers. The study of the twinning process, the interactions between propagating twins and between dislocations and twins is therefore important for a better understanding of the mechanical properties of metallic nanostructures. Here we show the results of atomistic simulations of defect-free nanowhiskers under tensile load using different EAM potentials for Au. The mechanisms of twin propagation and twin-stacking fault interaction are described and a modification to the criterion by Chen et al. [M. Chen et al., Science 300, 1275, 2003] for predicting twinning and its size dependence is presented. read less USED (high confidence) X. M. Jiang, Y. Wang, and Z. Pan, “Atomistic simulation of brittle fracture in nickel induced by He-doping,” Modelling and Simulation in Materials Science and Engineering. 2007. link Times cited: 1 Abstract: Molecular dynamics was used to study the effects of He-dopin… read moreAbstract: Molecular dynamics was used to study the effects of He-doping on brittle fracture in Ni. It was found that the released strain energy during crack propagation with He-doping (Er) is higher than that without He-doping (Er0). Enhancement of the strain energy due to He-doping contributes to a driving force to promote crack growth. This indicates that doping of He clusters in Ni favors crack growth, particularly for He clusters with high He-to-vacancy ratios, in which strong He–Ni interaction plays an important role. Furthermore, Er decreases non-linearly with increasing distance of He clusters away from the crack tip. Our results show that embrittlement of the materials will be enhanced when either the He concentration or the fracture density is above a critical value. The effect of critical He concentration on embrittlement of the materials has been reported in experimental studies. read less USED (high confidence) G. Zhou, K. Gao, L. Qiao, Y. Wang, and W. Chu, “Atomistic simulation of microcrack healing in aluminium,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 0 Abstract: A molecular dynamics method is used to simulate microcrack h… read moreAbstract: A molecular dynamics method is used to simulate microcrack healing during heating or under compressive stress. A centre microcrack in Al crystal could be sealed by a critical compressive stress or by heating over a critical temperature. During microcrack healing, dislocation generation and motion occurred. When there were pre-existing dislocations around the microcrack, the critical temperature necessary for microcrack healing would decrease from 850 to 650 K. The critical temperature necessary for microcrack healing depended upon the orientation of the crack plane. For example, the critical temperature of the crack along the (111) plane was the lowest. read less USED (low confidence) S. M. Handrigan and S. Nakhla, “Generation of viable nanocrystalline structures using the melt-cool method: the influence of force field selection,” Philosophical Magazine. 2023. link Times cited: 0 USED (low confidence) F. Grigoriev and V. Sulimov, “Atomistic Simulation of Physical Vapor Deposition of Optical Thin Films,” Nanomaterials. 2023. link Times cited: 0 Abstract: A review of the methods and results of atomistic modeling of… read moreAbstract: A review of the methods and results of atomistic modeling of the deposition of thin optical films and a calculation of their characteristics is presented. The simulation of various processes in a vacuum chamber, including target sputtering and the formation of film layers, is considered. Methods for calculating the structural, mechanical, optical, and electronic properties of thin optical films and film-forming materials are discussed. The application of these methods to studying the dependences of the characteristics of thin optical films on the main deposition parameters is considered. The simulation results are compared with experimental data. read less USED (low confidence) S. Borisova and G. Rusina, “The Submonolayer Structure of Ni(111)–(√3 × √3)R30°–Pb: Atomic Relaxation and Vibrational Properties,” Physics of Metals and Metallography. 2023. link Times cited: 0 USED (low confidence) X. Qin, Y.-S. Liang, J. Gu, and G. Peng, “The Effect of Interatomic Potentials on the Nature of Nanohole Propagation in Single-Crystal Nickel: A Molecular Dynamics Simulation Study,” Crystals. 2023. link Times cited: 1 Abstract: Based on a molecular dynamics (MD) simulation, we investigat… read moreAbstract: Based on a molecular dynamics (MD) simulation, we investigated the nanohole propagation behaviors of single-crystal nickel (Ni) under different styles of Ni–Ni interatomic potentials. The results show that the MEAM (the modified embedded atom method potential) potential is best suited to describe the brittle propagation behavior of nanoholes in single-crystal Ni. The EAM/FS (embedded atom method potential developed by Finnis and Sinclair) potential, meanwhile, is effective at characterizing the plastic growth behavior of nanoholes in single-crystal Ni. Furthermore, the results show the difference between the different styles of interatomic potentials in characterizing nanohole propagation in single-crystal Ni and provide a theoretical basis for the selection of interatomic potentials in the MD simulation of Ni crystals. read less USED (low confidence) S. Zinkle and R. Stoller, “Quantifying defect production in solids at finite temperatures: Thermally-activated correlated defect recombination corrections to DPA (CRC-DPA),” Journal of Nuclear Materials. 2023. link Times cited: 3 USED (low confidence) A. B. Sivak, D. N. Demidov, and P. A. Sivak, “Diffusion Characteristics of Self-Point Defects in Copper: Molecular Dynamic Study,” Physics of Atomic Nuclei. 2022. link Times cited: 1 USED (low confidence) L. Wei, F. Zhou, S. Wang, W. Hao, Y. Liu, and J. Zhu, “Description of crystal defect properties in BCC Cr with extended Finnis–Sinclair potential,” Multidiscipline Modeling in Materials and Structures. 2022. link Times cited: 0 Abstract: PurposeThe purpose of this study is to propose extended pote… read moreAbstract: PurposeThe purpose of this study is to propose extended potentials and investigate the applicability of extended Finnis–Sinclair (FS) potential to Cr with the unit cell structure of body-centered cubic (BCC Cr).Design/methodology/approachThe parameters of each potential are determined by fitting the elastic constants, cohesive energy and mono-vacancy formation energy. Furthermore, the ability of the extended FS potential to describe the crystal defect properties is tested. Finally, the applicability of reproducing the thermal properties of Cr is discussed.FindingsThe internal relationship between physical properties and potential function is revealed. The mathematical relationship between physical properties and potential function is derived in detail. The extended FS potential performs well in reproducing physical properties of BCC Cr, such as elastic constants, cohesive energy, surface energy and the properties of vacancy et al. Moreover, good agreement is obtained with the experimental data for predicting the melting point, specific heat and coefficient of thermal expansion.Originality/valueIn this study, new extended potentials are proposed. The extended FS potential is able to reproduce the physical and thermal properties of BCC Cr. Therefore, the new extended potential can be used to describe the crystal defect properties of BCC Cr. read less USED (low confidence) M. N. Magomedov, “Dependencies of the parameters of vacancy formation and self-diffusion in a single-component crystal on temperature and pressure,” Journal of Physics and Chemistry of Solids. 2022. link Times cited: 7 USED (low confidence) X. Jiang et al., “First-principles studies on optical absorption of [010] screw dislocation in KDP crystals,” CrystEngComm. 2021. link Times cited: 3 Abstract: The stress caused by the [010] dislocation in KDP deforms th… read moreAbstract: The stress caused by the [010] dislocation in KDP deforms the crystal structure, introduces extra optical absorption and narrows the band gap. read less USED (low confidence) H. Qadr, “A Molecular Dynamics Study of Temperature Dependence of the Primary State of Cascade Damage Processes,” Russian Journal of Non-Ferrous Metals. 2021. link Times cited: 4 USED (low confidence) D. Vizoso, M. Kosmidou, T. Balk, K. Hattar, C. Deo, and R. Dingreville, “Size-dependent radiation damage mechanisms in nanowires and nanoporous structures,” Acta Materialia. 2021. link Times cited: 8 USED (low confidence) M. N. Khatun and R. Gosh, “Verification of the Stokes-Einstein relation in liquid noble metals over a wide range of temperatures,” Physics Letters A. 2021. link Times cited: 1 USED (low confidence) S. Kim and S. Y. Kim, “Sudden transition of dislocation dynamics in FCC crystals at ultralow temperatures,” International Journal of Plasticity. 2021. link Times cited: 3 USED (low confidence) H. Kim, S. Kim, and S. Y. Kim, “Lattice-based J integral for a steadily moving dislocation,” International Journal of Plasticity. 2021. link Times cited: 3 USED (low confidence) H. Qadr, “A molecular dynamics calculation to cascade damage processes,” The Annals of “Dunarea de Jos” University of Galati. Fascicle IX, Metallurgy and Materials Science. 2020. link Times cited: 6 Abstract: In this work, Molecular dynamics simulation was performed to… read moreAbstract: In this work, Molecular dynamics simulation was performed to study the cascade damage evaluation initial from a 250 eV Primary Knock-on Atoms (PKAs) in gold. For this purpose, the simulations were carried out using the molecular dynamics code GRAPE to study the cooling phase of a cascade from the thermodynamic point view. Interatomic interaction of the cascade was investigated by the Morse potential and it is found that during the cooling phase of the cascade local equilibrium was realized. read less USED (low confidence) A. A. Schratt and V. Mohles, “Efficient calculation of the ECO driving force for atomistic simulations of grain boundary motion,” Computational Materials Science. 2020. link Times cited: 16 USED (low confidence) M. Mazalová, M. Všianská, J. Pavlů, and M. Šob, “The Effect of Vacancies on Grain Boundary Segregation in Ferromagnetic fcc Ni,” Nanomaterials. 2020. link Times cited: 6 Abstract: This work presents a comprehensive and detailed ab initio st… read moreAbstract: This work presents a comprehensive and detailed ab initio study of interactions between the tilt Σ5(210) grain boundary (GB), impurities X (X = Al, Si) and vacancies (Va) in ferromagnetic fcc nickel. To obtain reliable results, two methods of structure relaxation were employed: the automatic full relaxation and the finding of the minimum energy with respect to the lattice dimensions perpendicular to the GB plane and positions of atoms. Both methods provide comparable results. The analyses of the following phenomena are provided: the influence of the lattice defects on structural properties of material such as lattice parameters, the volume per atom, interlayer distances and atomic positions; the energies of formation of particular structures with respect to the standard element reference states; the stabilization/destabilization effects of impurities (in substitutional (s) as well as in tetragonal (iT) and octahedral (iO) interstitial positions) and of vacancies in both the bulk material and material with GBs; a possibility of recombination of Si(i)+Va defect to Si(s) one with respect to the Va position; the total energy of formation of GB and Va; the binding energies between the lattice defects and their combinations; impurity segregation energies and the effect of Va on them; magnetic characteristics in the presence of impurities, vacancies and GBs. As there is very little experimental information on the interaction between impurities, vacancies and GBs in fcc nickel, most of the present results are theoretical predictions, which may motivate future experimental work. read less USED (low confidence) S. Kim, K. Kang, and S. Y. Kim, “Dynamic drags acting on moving defects in discrete dispersive media: From dislocation to low-angle grain boundary,” arXiv: Materials Science. 2019. link Times cited: 6 USED (low confidence) M. Song et al., “Oriented attachment induces fivefold twins by forming and decomposing high-energy grain boundaries,” Science. 2019. link Times cited: 109 Abstract: Finding the fivefold path Crystals formed in nature or synth… read moreAbstract: Finding the fivefold path Crystals formed in nature or synthesized in a lab can have domains with different orientations that are called twins. Having five of these domains is relatively common, but how these fivefold twins form is still a mystery. Song et al. combined in situ transmission electron microscope observations with simulations to find two different pathways for forming fivefold twins in gold, platinum, and palladium nanoparticles. This combination provides a key insight into the twin-forming process, which might be exploited for developing nanoparticles suitable for a variety of applications. Science, this issue p. 40 Step-by-step mechanisms for forming fivefold twinned nanoparticles are demonstrated in gold, platinum, and palladium. Natural and synthetic nanoparticles composed of fivefold twinned crystal domains have distinct properties. The formation mechanism of these fivefold twinned nanoparticles is poorly understood. We used in situ high-resolution transmission electron microscopy combined with molecular dynamics simulations to demonstrate that fivefold twinning occurs through repeated oriented attachment of ~3-nanometer gold, platinum, and palladium nanoparticles. We discovered two different mechanisms for forming fivefold twinned nanoparticles that are driven by the accumulation and elimination of strain. This was accompanied by decomposition of grain boundaries and the formation of a special class of twins with a net strain of zero. These observations allowed us to develop a quantitative picture of the twinning process. The mechanisms provide guidance for controlling twin structures and morphologies across a wide range of materials. read less USED (low confidence) L. Hao et al., “Mechanical behavior of metallic nanowires with twin boundaries parallel to loading axis,” Computational Materials Science. 2019. link Times cited: 5 USED (low confidence) S. Huang, I. Beyerlein, and C. Zhou, “Unusual size effects from tilted twin boundaries in nano-twinned metals,” Extreme Mechanics Letters. 2019. link Times cited: 4 USED (low confidence) Z. Wang et al., “Effects of finite temperature on the surface energy in Al alloys from first-principles calculations,” Applied Surface Science. 2019. link Times cited: 19 USED (low confidence) J. Liu et al., “Investigations on fatigue behavior and surface damage of Cu film by nano impact and molecular dynamics simulation,” Surface and Coatings Technology. 2019. link Times cited: 4 USED (low confidence) S. Kumar, “Spreading and orientation of silver nano-drops over a flat graphene substrate: An atomistic investigation,” Carbon. 2018. link Times cited: 10 USED (low confidence) H. N. Pishkenari, F. S. Yousefi, and A. Taghibakhshi, “Determination of surface properties and elastic constants of FCC metals: a comparison among different EAM potentials in thin film and bulk scale,” Materials Research Express. 2018. link Times cited: 22 Abstract: Three independent elastic constants C11, C12, and C44 were c… read moreAbstract: Three independent elastic constants C11, C12, and C44 were calculated and compared using available potentials of eight different metals with FCC crystal structure; Gold, Silver, Copper, Nickel, Platinum, Palladium, Aluminum and Lead. In order to calculate the elastic constants, the second derivative of the energy density of each system was calculated with respect to different directions of strains. Each set of the elastic constants of the metals in bulk scale was compared with experimental results, and the average relative error was for each was calculated and compared with other available potentials. Then, using the Voigt-Reuss-Hill method, approximated values for Young and shear moduli and Poisson’s ratio of the FCC metals in the bulk scale were found for each potential. Furthermore, to observe the surface effects on the metals in nanoscale, surface elastic constants of the thin films of the metals have been calculated. In the study of the thin films of materials in nanoscale, the number of surface atoms is considerable compared to all atoms of the object. This leads to an increase in the surface effects, which influence the elastic properties. By considering this fact and employing related definitions and equations, the properties of the thin films of the metals were calculated, and the surface effects for different crystallographic directions were compared. Subsequently, in some cases, comparisons among characteristics of the metals in the thin film and bulk material were made. read less USED (low confidence) Z. Pan, V. Borovikov, M. Mendelev, and F. Sansoz, “Development of a semi-empirical potential for simulation of Ni solute segregation into grain boundaries in Ag,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 19 Abstract: An Ag–Ni semi-empirical potential was developed to simulate … read moreAbstract: An Ag–Ni semi-empirical potential was developed to simulate the segregation of Ni solutes at Ag grain boundaries (GBs). The potential combines a new Ag potential fitted to correctly reproduce the stable and unstable stacking fault energies in this metal and the existing Ni potential from Mendelev et al (2012 Phil. Mag. 92 4454–69). The Ag–Ni cross potential functions were fitted to ab initio data on the liquid structure of the Ag80Ni20 alloy to properly incorporate the Ag–Ni interaction at small atomic separations, and to the Ni segregation energies at different sites within a high-energy Σ9 (221) symmetric tilt GB. By deploying this potential with hybrid Monte Carlo/molecular dynamics simulations, it was found that heterogeneous segregation and clustering of Ni atoms at GBs and twin boundary defects occur at low Ni concentrations, 1 and 2 at%. This behavior is profoundly different from the homogeneous interfacial dispersion generally observed for the Cu segregation in Ag. A GB transformation to amorphous intergranular films was found to prevail at higher Ni concentrations (10 at%). The developed potential opens new opportunities for studying the selective segregation behavior of Ni solutes in interface-hardened Ag metals and its effect on plasticity. read less USED (low confidence) Y.-yong Lin, J. Pan, H. Zhou, H. Gao, and Y. Li, “Mechanical properties and optimal grain size distribution profile of gradient grained nickel,” Acta Materialia. 2018. link Times cited: 151 USED (low confidence) S. Kumar, “Wettability and Structural Evolution of Gold over a Single-Walled Carbon Nanotube: An Atomistic Investigation,” The Journal of Physical Chemistry C. 2018. link Times cited: 2 Abstract: Gold nanostructures with high surface area-to-volume ratio d… read moreAbstract: Gold nanostructures with high surface area-to-volume ratio depict many applications for the design and development of advanced materials for the nanoelectronic, catalyst, optoelectronic, antibacterial properties, and so forth. For extensive applications, gold nanostructures can be synthesized by the deposition over various substrates, such as carbon nanotubes, graphite, silica, and so forth. In the present study, a thin molten gold film has been deposited over a single-walled carbon nanotube (SWCNT) to observe the wettability and phase transitions using molecular dynamics simulation. At high temperature (T = 2000 K), the gold film over a SWCNT depicts poor wettability and evolves into a globule. However, during cooling from 2000 to 10 K, the gold globule depicts phase transition from liquid to face-centered cubic crystalline structure. At the interface between gold and SWCNT, gold atoms organized both on- and off-positions over the hexagonal arrangement of carbon atoms of SWCNT. In the case of on-position... read less USED (low confidence) K. Zhao, J. He, A. Mayer, and Z. Zhang, “Effect of Hydrogen on the Collective Behavior of Dislocations in the Case of Nanoindentation,” Acta Materialia. 2018. link Times cited: 26 USED (low confidence) S. M. Rassoulinejad-Mousavi and Y. Zhang, “Interatomic Potentials Transferability for Molecular Simulations: A Comparative Study for Platinum, Gold and Silver,” Scientific Reports. 2018. link Times cited: 33 USED (low confidence) D. Chen, T. Ghoneim, and Y. Kulkarni, “Effect of pinning particles on grain boundary motion from interface random walk,” Applied Physics Letters. 2017. link Times cited: 14 Abstract: Impurities can dramatically influence grain boundary migrati… read moreAbstract: Impurities can dramatically influence grain boundary migration, thereby impacting material properties. In this letter, we present a theoretical model for grain boundary motion in the presence of embedded particles using the interface random walk approach. Based on the fluctuation-dissipation relation, we derive an analytical expression relating the grain boundary fluctuations to the boundary mobility and key parameters governing the drag effect of the particles. In addition to predicting the modified boundary mobility due to pinning particles, the model provides a way to estimate the force acting on the particle-boundary interface from atomistic simulations. The theory facilitates an enriched analysis of atomistic simulations of a grain boundary with embedded particles, revealing that a pinned grain boundary exhibits a response akin to tethered Brownian motion. read less USED (low confidence) J. Liu, J. Li, and B. Liu, “Atomistic Modeling to Design Favoured Compositions for the Metallic Glass Formation.” 2017. link Times cited: 0 USED (low confidence) T. Yang et al., “Ab initio study of stability and migration of point defects in copper-graphene layered composite,” Journal of Alloys and Compounds. 2017. link Times cited: 19 USED (low confidence) K. Li, Z. Zhang, L. Li, P. Zhang, J. Yang, and Z. F. Zhang, “The dissociation behavior of dislocation arrays in face centered cubic metals,” Computational Materials Science. 2016. link Times cited: 2 USED (low confidence) S. M. Rassoulinejad-Mousavi, Y. Mao, and Y. Zhang, “Evaluation of Copper, Aluminum and Nickel Interatomic Potentials on Predicting the Elastic Properties,” arXiv: Computational Physics. 2016. link Times cited: 63 Abstract: Choice of appropriate force field is one of the main concern… read moreAbstract: Choice of appropriate force field is one of the main concerns of any atomistic simulation that needs to be seriously considered in order to yield reliable results. Since, investigations on mechanical behavior of materials at micro/nanoscale has been becoming much more widespread, it is necessary to determine an adequate potential which accurately models the interaction of the atoms for desired applications. In this framework, reliability of multiple embedded atom method based interatomic potentials for predicting the elastic properties was investigated. Assessments were carried out for different copper, aluminum and nickel interatomic potentials at room temperature which is considered as the most applicable case. Examined force fields for the three species were taken from online repositories of National Institute of Standards and Technology (NIST), as well as the Sandia National Laboratories, the LAMMPS database. Using molecular dynamic simulations, the three independent elastic constants, C11, C12 and C44 were found for Cu, Al and Ni cubic single crystals. Voigt-Reuss-Hill approximation was then implemented to convert elastic constants of the single crystals into isotropic polycrystalline elastic moduli including Bulk, Shear and Young's modulus as well as Poisson's ratio. Simulation results from massive molecular dynamic were compared with available experimental data in the literature to justify the robustness of each potential for each species. Eventually, accurate interatomic potentials have been recommended for finding each of the elastic properties of the pure species. Exactitude of the elastic properties was found to be sensitive to the choice of the force fields. Those potentials were fitted for a specific compound may not necessarily work accurately for all the existing pure species. read less USED (low confidence) E. Hahn and M. Meyers, “Grain-size dependent mechanical behavior of nanocrystalline metals,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 162 USED (low confidence) E. S. Wise, M. Liu, and T. Miller, “Sputtering of cubic metal crystals by low-energy xenon-ions,” Computational Materials Science. 2015. link Times cited: 5 USED (low confidence) S. Zhang, G. Li, H. Gong, N. Gao, X. Chen, and Z. Wang, “Molecular dynamics simulation of energetic Cu-55 clusters deposition on a Fe (001) surface,” Computational Materials Science. 2015. link Times cited: 7 USED (low confidence) S. Zhang, H. Gong, X. Chen, G. Li, and Z. Wang, “Low energy Cu clusters slow deposition on a Fe (001) surface investigated by molecular dynamics simulation,” Applied Surface Science. 2014. link Times cited: 10 USED (low confidence) I. E. Awad and L. Ladani, “Cohesive Zone Model for the Interface of Multiwalled Carbon Nanotubes and Copper: Molecular Dynamics Simulation,” Journal of Nanotechnology in Engineering and Medicine. 2014. link Times cited: 12 Abstract: Due to their superior mechanical and electrical properties, … read moreAbstract: Due to their superior mechanical and electrical properties, multiwalled carbon nano- tubes (MWCNTs) have the potential to be used in many nano-/micro-electronic applications, e.g., through silicon vias (TSVs), interconnects, transistors, etc. In particular, use of MWCNT bundles inside annular cylinders of copper (Cu) as TSV is proposed in this study. However, the significant difference in scale makes it difficult to evaluate the interfacial mechanical integrity. Cohesive zone models (CZM) are typically used at large scale to determine the mechanical adherence at the interface. However, at molecular level, no routine technique is available. Molecular dynamic (MD) simulations is used to determine the stresses that are required to separate MWCNTs from a copper slab and generate normal stress–displacement curves for CZM. Only van der Waals (vdW) interaction is considered for MWCNT/Cu interface. A displacement controlled loading was applied in a direction perpendicular to MWCNT’s axis in different cases with different number of walls and at different temperatures and CZM is obtained for each case. Fur- thermore, their effect on the CZM key parameters (normal cohesive strength ð r max Þ and the corresponding displacement ð d n Þ has been studied. By increasing the number of the walls of the MWCNT, r max was found to nonlinearly decrease. Displacement at maximum stress, d n , showed a nonlinear decrease as well with increasing the number of walls. Tem- perature effect on the stress–displacement curves was studied. When temperature was increased beyond 1 K, no relationship was found between the maximum normal stress and temperature. Likewise, the displacement at maximum load did not read less USED (low confidence) H. Lee and V. Tomar, “Understanding the influence of grain boundary thickness variation on the mechanical strength of a nickel-doped tungsten grain boundary,” International Journal of Plasticity. 2014. link Times cited: 14 USED (low confidence) I. E. Awad and L. Ladani, “Interfacial Strength Between Single Wall Carbon Nanotubes and Copper Material: Molecular Dynamics Simulation,” Journal of Nanotechnology in Engineering and Medicine. 2013. link Times cited: 18 Abstract: Due to their promising mechanical and electrical properties,… read moreAbstract: Due to their promising mechanical and electrical properties, carbon nanotubes (CNTs) have the potential to be employed in many nano/microelectronic applications e.g., through silicon vias (TSVs), interconnects, transistors, etc. In particular, use of CNT bundles inside annular cylinders of copper (Cu) as TSV is proposed in this study. To evaluate mechanical integrity of CNT-Cu composite material, a molecular dynamics (MD) simulation of the interface between CNT and Cu is conducted. Different arrangements of single wall carbon nanotubes (SWCNTs) have been studied at interface of a Cu slab. Pullout forces have been applied to a SWCNT while Cu is spatially fixed. This study is repeated for several different cases where multiple CNT strands are interfaced with Cu slab. The results show similar behavior of the pull-out-displacement curves. After pull-out force reaches a maximum value, it oscillates around an average force with descending amplitude until the strand/s is/are completely pulled-out. A linear relationship between pull-out forces and the number of CNT strands was observed. Second order interaction effect was found to be negligible when multiple layers of CNTs were studied at the interface of Cu. C–Cu van der Waals (vdW) interaction was found to be much stronger than C–C vdW's interactions. Embedded length has no significance on the average pull-out force. However, the amplitude of oscillations increases as the length of CNTs increases. As expected when one end of CNT strand was fixed, owing to its extraordinary strength, large amount of force was required to pull it out. Finally, an analytical relationship is proposed to determine the interfacial shear strength between Cu and CNT bundle. read less USED (low confidence) J. Schäfer, A. Stukowski, and K. Albe, “On the hierarchy of deformation processes in nanocrystalline alloys: Grain boundary mediated plasticity vs. dislocation slip,” Journal of Applied Physics. 2013. link Times cited: 10 Abstract: Hybrid molecular dynamics and Monte-Carlo simulations on the… read moreAbstract: Hybrid molecular dynamics and Monte-Carlo simulations on the deformation behavior of nanocrystalline Pd–Au are presented. A semi-grandcanonical Monte-Carlo scheme is employed during straining to allow for local relaxation by chemical equilibration and to effectively shortcut diffusional processes. Altering the balance between an imposed straining and local relaxation reveals a strong correlation of the irreversible plastic deformation and the frequency of local relaxation. Using a novel method to quantify the amount of crystal slip strain from atomistic data, it is demonstrated how plastic deformation carried by dislocations changes as a function of the local relaxation. The results indicate that conventional molecular dynamics simulations overestimate the contributions of dislocation slip to the overall plastic deformation of nanocrystalline samples. read less USED (low confidence) C. Scott and R. Smith, “Sputtering of Au surfaces at realistic dose rates using molecular dynamics and on-the-fly kinetic Monte Carlo,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2013. link Times cited: 2 USED (low confidence) Y. Wang, Y. Liu, and X. Wang, “The Divacancy Formation Energy for B2 FeAl Alloy Simulated with Finnis-Sinclair many-Body Interatomic Potential,” Advanced Materials Research. 2013. link Times cited: 0 Abstract: The Finnis-Sinclair many-body potential was fitted for binar… read moreAbstract: The Finnis-Sinclair many-body potential was fitted for binary FeAl alloy with B2 structure. As the examination to the acquired potential function, some properties were calculated, and the results agree with the experiments well. Further, properties of point defects, such as divacancies were studied as an application. read less USED (low confidence) J. Li, Y. Dai, and X. Dai, “Long-range n-body potential and applied to atomistic modeling the formation of ternary metallic glasses,” Intermetallics. 2012. link Times cited: 20 USED (low confidence) S. Miyashiro, S. Fujita, T. Okita, and H. Okuda, “MD simulations to evaluate effects of applied tensile strain on irradiation-induced defect production at various PKA energies,” Fusion Engineering and Design. 2012. link Times cited: 27 USED (low confidence) C. Scott, S. Blackwell, L. Vernon, S. Kenny, M. Walls, and R. Smith, “Atomistic surface erosion and thin film growth modelled over realistic time scales.,” The Journal of chemical physics. 2011. link Times cited: 28 Abstract: We present results of atomistic modelling of surface growth … read moreAbstract: We present results of atomistic modelling of surface growth and sputtering using a multi-time scale molecular dynamics-on-the-fly kinetic Monte Carlo scheme which allows simulations to be carried out over realistic experimental times. The method uses molecular dynamics to model the fast processes and then calculates the diffusion barriers for the slow processes on-the-fly, without any preconceptions about what transitions might occur. The method is applied to the growth of metal and oxide materials at impact energies typical for both vapour deposition and magnetron sputtering. The method can be used to explain growth processes, such as the filling of vacancies and the formation of stacking faults. By tuning the variable experimental parameters on the computer, a parameter set for optimum crystalline growth can be determined. The method can also be used to model sputtering where the particle interactions with the surface occur at a higher energy. It is shown how a steady state can arise in which interstitial clusters are continuously being formed below the surface during an atom impact event which also recombine or diffuse to the surface between impact events. For fcc metals the near surface region remains basically crystalline during the erosion process with a pitted topography which soon attains a steady state roughness. read less USED (low confidence) H. Ogawa, “Simulation study of structural variation of metallic nanoparticles caused by hydrogenation,” Journal of Alloys and Compounds. 2011. link Times cited: 2 USED (low confidence) J. Schäfer, A. Stukowski, and K. Albe, “Plastic deformation of nanocrystalline Pd–Au alloys: On the interplay of grain boundary solute segregation, fault energies and grain size,” Acta Materialia. 2011. link Times cited: 49 USED (low confidence) G. Was et al., “Assessment of radiation-induced segregation mechanisms in austenitic and ferritic–martensitic alloys,” Journal of Nuclear Materials. 2011. link Times cited: 118 USED (low confidence) J. Li, Y. Dai, Y. Cui, and B. Liu, “Atomistic theory for predicting the binary metallic glass formation,” Materials Science & Engineering R-reports. 2011. link Times cited: 80 USED (low confidence) D. Belashchenko, N. Kravchunovskaya, and O. Ostrovski, “Molecular dynamics calculation of surface tension of liquid metals using the embedded atom model,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2010. link Times cited: 9 USED (low confidence) K. Sato, T. Ihara, H. Sakurai, T. Yoshiie, and Q. Xu, “Comparison of interaction between Cu precipitate and vacancy in Fe using first-principle calculations and empirical N-body potential calculations,” Computational Materials Science. 2009. link Times cited: 6 USED (low confidence) D. Bacon and Y. Osetsky, “Dislocation—Obstacle Interactions at Atomic Level in Irradiated Metals,” Mathematics and Mechanics of Solids. 2009. link Times cited: 29 Abstract: Nano-scale defect clusters, such as voids, dislocation loops… read moreAbstract: Nano-scale defect clusters, such as voids, dislocation loops, stacking-fault tetrahedra and irradiation-induced precipitates, are produced in metals by irradiation with high-energy atomic particles. They are obstacles to dislocation glide and can give rise to substantial changes in the yield and flow stresses and ductility. Atomic-scale computer simulation is able to provide detail of how these effects are influenced by obstacle structure, applied stress, strain rate and temperature. Some recent results from modelling dislocations interacting with obstacles are described. Emphasis is placed on dislocation interaction with voids, copper precipitates and dislocation loops in the BCC metal iron and stacking fault tetrahedra in FCC copper. In the latter case, the importance of surfaces in reactions in TEM foils is highlighted. It is shown that while some atomic processes can be represented adequately by the continuum theory of crystal defects, others cannot. read less USED (low confidence) R. Voskoboinikov, Y. Osetsky, and D. Bacon, “Computer simulation of primary damage creation in displacement cascades in copper. I. Defect creation and cluster statistics,” Journal of Nuclear Materials. 2008. link Times cited: 80 USED (low confidence) R. Rentsch, “Atomistic analysis of discontinuous deformation during cutting processes,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2008. link Times cited: 0 USED (low confidence) S. Ahmad and R. Ramzan, “Interaction of Point Defects with Twin Boundaries in Copper,” Chinese Physics Letters. 2007. link Times cited: 5 Abstract: The interaction between small vacancy clusters and twin boun… read moreAbstract: The interaction between small vacancy clusters and twin boundaries in copper is studied by using many-body potential developed by Ackland et al. for fcc metals. The interaction energies of single-, di- and tri-vacancy clusters with (111) and (112) twin boundaries are computed using well established simulation techniques. For (111) twins the vacancy clusters are highly repelled when they are on the adjacent planes, and are attracted when they are away from the boundary. In the case of (112) twins, vacancy clusters are more attracted to the boundary when they are near the boundary as compared to away from it. Vacancy clusters on both the sides of the boundary are also investigated, and it is observed that the clusters energetically prefer to lie on the off-mirror sites as compared to the mirror position across the twin. read less USED (low confidence) P. Monasterio, B. Wirth, and G. Odette, “Kinetic Monte Carlo modeling of cascade aging and damage accumulation in Fe–Cu alloys,” Journal of Nuclear Materials. 2007. link Times cited: 30 USED (low confidence) R. Rentsch and E. Brinksmeier, “Numerical simulation of residual stresses at the grain and sub-grain length scale using atomistic modeling∗,” HTM Journal of Heat Treatment and Materials. 2006. link Times cited: 2 Abstract: For modeling the deformation and the heat treatment related … read moreAbstract: For modeling the deformation and the heat treatment related change of micro structural material properties, a crystal structure with several grains is analyzed using the molecular dynamics simulation. The generated atom arrangement has been equilibrated, sheared and tempered, and the resulting microstructures and stresses as well as their changes are presented. The shearing of the multiple grain model into the region of plastic deformation caused a significant change in its microstructure and introduced additional stress. On applying the heat treatment simulation, it was possible to show thermally induced relaxation processes in a microstructure using molecular dynamics. read less USED (low confidence) E. H. Megchiche, S. Pérusin, J. Barthelat, and C. Mijoule, “Density functional calculations of the formation and migration enthalpies of monovacancies in Ni: Comparison of local and nonlocal approaches,” Physical Review B. 2006. link Times cited: 45 Abstract: We examine in this work the potential and the functional to … read moreAbstract: We examine in this work the potential and the functional to be used in a density functional theory approach in order to describe correctly the formation and migration energies of monovacancies in nickel. As the formation enthalpy is not well-known experimentally at $0\phantom{\rule{0.3em}{0ex}}\mathrm{K}$, we choose in a first step to determine some structural, magnetic, and elastic properties of the bulk, which are well-established experimentally. The comparison between both approaches, i.e., the local spin density approximation (LSDA) and the generalized gradient approximation (GGA) exchange-correlation functionals is analyzed. We conclude that the contribution of nonlocal GGA terms in order to describe correctly the electronic density is necessary to determine the formation and migration enthalpies and activation energy of monovacancy. The calculated formation ${H}_{v}^{f}$ and migration ${H}_{v}^{m}$ enthalpies differ significantly between both approaches. The overestimation of the LSDA approximation is of $0.25\phantom{\rule{0.3em}{0ex}}\mathrm{eV}$ for ${H}_{v}^{f}$ and of $0.23\phantom{\rule{0.3em}{0ex}}\mathrm{eV}$ for ${H}_{v}^{m}$ with respect to the GGA one, leading to a gap of $0.48\phantom{\rule{0.3em}{0ex}}\mathrm{eV}$ between both methods for the activation energy ${Q}_{1}$. We show that the GGA results are comparable with experimental data if the thermal expansion contribution is taken into account through the lattice parameter variation. Finally, it is shown that the activation energy is nearly independent of the thermal expansion effects; thus we can expect that the curvature of the Arrhenius plot of the diffusion factor near the melting point is essentially due to the contribution of divacancies. read less USED (low confidence) L. Ti-Jiang, W. Yue-xia, P. Zheng-Ying, J. Xiao-mei, Z. Liang, and Z. Jing, “Atomistic Simulation of He Clustering and Defects Produced in Ni,” Chinese Physics Letters. 2006. link Times cited: 3 Abstract: Using the molecular dynamics method, the stability of small … read moreAbstract: Using the molecular dynamics method, the stability of small He-vacancy clusters is studied under the condition of the high He and low vacancy densities. The result shows that there is a competition between He atoms detrapped and self-interstitial atoms (SIAs) emitted during the clustering of He atoms. When the He number is above a critical value of 9, the SIA emission is predominant. The SIA emission can result in deep capture of He atoms since the binding energy of He to a He-vacancy cluster is increased with the number of SIAs created. The cluster thus grows up. In addition, more SIAs are created when the temperature is elevated. The average volume of a He atom is increased. The cluster expansion takes place at high temperature. read less USED (low confidence) Y. Osetsky and D. Bacon, “Comparison of void strengthening in fcc and bcc metals: Large-scale atomic-level modelling,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2005. link Times cited: 67 USED (low confidence) M. Hackett, G. Was, and E. Simonen, “Modeling the Effects of Oversize Solute Additions on Radiation-Induced Segregation in Austenitic Stainless Steels,” Journal of Astm International. 2005. link Times cited: 12 Abstract: Oversize solute additions to stainless steels hold promise f… read moreAbstract: Oversize solute additions to stainless steels hold promise for reducing radiation-induced segregation (RIS), possibly delaying the onset and severity of irradiation-assisted stress-corrosion cracking (IASCC). The Modified Inverse Kirkendall (MIK) model for RIS in austenitic stainless steels was adapted to include the effects of defect trapping, which is expected to be caused by oversized solutes, on RIS. The model accounts for the sensitivity of RIS to both dose rate and temperature. Model results show that the primary contribution to the reduction in RIS occurs through vacancy trapping and recombination with migrating interstitials, requiring strong solute binding energies on the order of 1.0 eV. The maximum reduction in RIS due to defect trapping increases with dose rate and temperature. The general trends of the model are consistent with experimental data from proton and neutron irradiations. read less USED (low confidence) R. Voskoboinikov, Y. Osetsky, and D. Bacon, “Correlated Formation and Stability of SIA Loops and Stacking Fault Tetrahedra in High Energy Displacement Cascades in Copper,” Journal of Astm International. 2005. link Times cited: 4 Abstract: Atomistic modeling was conducted for an investigation of pri… read moreAbstract: Atomistic modeling was conducted for an investigation of primary damage creation, self-interstitial and vacancy clusters formation, and their stability in high energy displacement cascades in copper. The simulations were carried out for a wide range of temperatures (100 K {le} T {le} 900 K) and primary knock-on atom (PKA) energies 5 keV {le} Epka {le} 25 keV. This study of over 400 cascades is the largest yet reported for this metal. At least 20 cascades for each (Epka, T) pair were simulated in order to ensure statistical reliability of the results. The number of surviving point defects for each cascade and the mean value for cascades at the same temperature and PKA energy were found. The corresponding fraction of self-interstitial atoms (SIA) in dislocation loops and vacancies in stacking fault tetrahedron (SFT)-like clusters was calculated. Strong spatial and size correlation of SFTs and SIA clusters at low temperatures were established. In the context of high dose irradiation and the spatial overlap of displacement cascades, the stability of SFTs and dislocation loops inside an overlapping cascade region was investigated. It was observed that an SFT destroyed in the collision phase by a cascade is always recreated. On being completely enveloped bymore » the region of displaced atoms, both SFT and SIA dislocation loops are destroyed with corresponding decrease of the number of residual point defects, whereas partial overlapping leads to increase in size of both types of cluster.« less read less USED (low confidence) O. H. Duparc, A. Larere, B. Lezzar, O. Khalfallah, and V. Paidar, “Comparison of the intergranular segregation for eight dilute binary metallic systems in the Σ 11′ 332 tilt grain boundary,” Journal of Materials Science. 2005. link Times cited: 20 USED (low confidence) B.-J. Lee, B. Wirth, J. Shim, J. Kwon, S. Kwon, and J.-H. Hong, “Modified embedded-atom method interatomic potential for the Fe-Cu alloy system and cascade simulations on pure Fe and Fe-Cu alloys,” Physical Review B. 2005. link Times cited: 55 Abstract: A modified embedded-atom method (MEAM) interatomic potential… read moreAbstract: A modified embedded-atom method (MEAM) interatomic potential for the Fe-Cu binary system has been developed using previously developed MEAM potentials of Fe and Cu. The Fe-Cu potential was determined by fitting to data on the mixing enthalpy and the composition dependencies of the lattice parameters in terminal solid solutions. The potential gives a value of 0.65 eV for the dilute heat of solution and reproduces the increase of lattice parameter of Fe with addition of Cu in good agreement with experiments. The potential was used to investigate the primary irradiation defect formation in pure Fe and Fe-0.5 at. % Cu alloy by a molecular dynamics cascade simulation study with a PKA energy of 2 keV at 573 K. A tendency for self-interstitial atom-Cu binding, the formation of mixed (Fe-Cu) dumbbells and even Cu-Cu dumbbells was observed. Given a positive binding energy between Cu atoms and self-interstitials, Cu transport by an interstitial diffusion mechanism could be proposed to contribute to the formation of Cu-rich precipitates and irradiation-induced embrittlement in nuclear structural steels. read less USED (low confidence) M. Said-Ettaoussi, J. Jiménez‐Sáez, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “Deformation behaviour induced by point defects near a Cu(0 0 1) surface,” Applied Surface Science. 2004. link Times cited: 0 USED (low confidence) Y. Quan-wen, Z. Ru-zeng, W. Jiu-An, and W. Yuhua, “Surface-Induced Melting of Metal Nanoclusters,” Chinese Physics Letters. 2004. link Times cited: 6 Abstract: We investigate the size effect on melting of metal nanoclust… read moreAbstract: We investigate the size effect on melting of metal nanoclusters by molecular dynamics simulation and thermo dynamic theory based on Kofman's melt model. By the minimization of the free energy of metal nanoclusters with respect to the thickness of the surface liquid layer, it has been found that the nanoclusters of the same metal have the same premelting temperature T-pre = T-0 - T-0(gamma(su) - gamma(lv) - gamma(sl))/(rhoLxi) (T-0 is the melting point of bulk metal, gamma(sv) the solid-vapour interfacial free energy, gamma(sl) the liquid-vapour interfacial free energy, gamma(sl),l the solid-liquid interfacial free energy, p the density of metal, L the latent heat of bulk metal, and xi the characteristic length of surface-interface interaction) to be independent of the size of nanoclusters, so that the characteristic length of a metal can be obtained easily by T-pre, which can be obtained by experiments or molecular dynamics (MD) simulations. The premelting temperature T-pre of Cu is obtained by AID simulations, then xi is obtained. The melting point T-cm is further predicted by free energy analysis and is in good agreement with the result of our MD simulations. We also predict the maximum premelting-liquid width of Cu nanoclusters with various sizes and the critical size, below which there is no premelting. read less USED (low confidence) E. Ahmed, J. Akhter, and M. Ahmad, “Molecular dynamics study of thermal properties of noble metals,” Computational Materials Science. 2004. link Times cited: 25 USED (low confidence) H. Gong and B. Liu, “Influence of interfacial texture on solid-state amorphization and associated asymmetric growth in immiscible Cu-Ta multilayers,” Physical Review B. 2004. link Times cited: 8 Abstract: For the immiscible Cu-Ta system, a Finnis-Sinclair potential… read moreAbstract: For the immiscible Cu-Ta system, a Finnis-Sinclair potential is constructed and proven to be realistic in reproducing some static properties of the system. Applying the potential, molecular dynamics simulations reveal that among the nine $\mathrm{Cu}∕\mathrm{Ta}$ interfaces stacked by possible combinations of the (100), (110), and (111) atomic planes, the Ta (110) plane could remain stable up to a temperature of $600\phantom{\rule{0.2em}{0ex}}\ifmmode^\circ\else\textdegree\fi{}\mathrm{C}$, while the Cu (111) plane could remain unchanged only if combined with the Ta (100) and (110) planes. Simulations also show that for the other $\mathrm{Cu}∕\mathrm{Ta}$ interfaces, the interface energy serves as the driving force for interdiffusion of the Cu and Ta atoms across the interface, resulting in solid-state amorphization. Interestingly, it is calculated that the amorphization energy of Cu is smaller than that of Ta, thus resulting in an asymmetric growth behavior of the amorphous interlayer, i.e., amorphization of the Cu lattice is easier and faster than that of the Ta lattice. In general, the agreement between the simulation results and experimental observations is fairly good. read less USED (low confidence) D. Wei, J. Han, A. K. Tieu, and Z. Jiang, “Simulation of crack healing in BCC Fe,” Scripta Materialia. 2004. link Times cited: 27 USED (low confidence) K. Sato, T. Yoshiie, T. Ishizaki, and Q. Xu, “Anisotropic motion of point defects near edge dislocations,” Journal of Nuclear Materials. 2004. link Times cited: 4 USED (low confidence) D. Bacon and Y. Osetsky, “Hardening due to copper precipitates in α-iron studied by atomic-scale modelling,” Journal of Nuclear Materials. 2004. link Times cited: 51 USED (low confidence) L. Zhou, X. Wei, and N. Zhou, “Lattice distortion and thermal stability of nano-crystalline copper,” Computational Materials Science. 2004. link Times cited: 11 USED (low confidence) Y. Osetsky, R. Stoller, and Y. Matsukawa, “Dislocation–stacking fault tetrahedron interaction: what can we learn from atomic-scale modelling ☆,” Journal of Nuclear Materials. 2004. link Times cited: 47 USED (low confidence) X. Dai, H. Gong, and B.-xin Liu, “Structural Stability of the Metastable Solid Solution in the Equilibrium Immiscible Ag-Mo System Predicted by an ab Initio Derived Potential,” Journal of the Physical Society of Japan. 2004. link Times cited: 5 Abstract: Based on the cohesive energies and lattice constants of a fe… read moreAbstract: Based on the cohesive energies and lattice constants of a few possible non-equilibrium Ag–Mo compounds obtained by ab initio calculation, a Finnis–Sinclair (FS) potential of the equilibrium immiscible Ag–Mo system is derived. Applying the proven realistic potential, molecular dynamics simulation is carried out to study, at an atomic scale, the structural stability of the Ag-based solid solutions. The simulation results predict that the fcc crystalline structure can be preserved until the Mo concentration reaches 13 at.% and the correlation of the lattice constant of the Ag-based solid solutions vs the Mo concentration is in good agreement with Vegard’s Law. In addition, the heats of formation of the fcc Ag–Mo solid solutions calculated by the derived potential are quite agreeable with that calculated by Miedema’s theory. read less USED (low confidence) S. Dmitriev, N. Yoshikawa, M. Kohyama, S. Tanaka, R. Yang, and Y. Kagawa, “Atomistic structure of the Cu(111)/α-Al2O3(0001) interface in terms of interatomic potentials fitted to ab initio results,” Acta Materialia. 2004. link Times cited: 54 USED (low confidence) S. Chen and F. Ke, “MD simulation of the effect of contact area and tip radius on nanoindentation,” Science in China Series G: Physics, Mechanics and Astronomy. 2004. link Times cited: 19 Abstract: Molecular dynamics simulations of nanoindentation are perfor… read moreAbstract: Molecular dynamics simulations of nanoindentation are performed on monocrystal copper. A new “contact atoms” method is presented for calculating the contact area. Compared with conventional methods, this method can provide the contact area more accurately not only for sink-in but also for pile-up situation. The effect of tip radius on indentation is investigated too. The results indicate that the measured hardness of the material will become higher as the tip radius increases. read less USED (low confidence) R. Smith, S. Kenny, and D. Ramasawmy, “Molecular-dynamics simulations of sputtering,” Philosophical Transactions of the Royal Society of London. Series A: Mathematical, Physical and Engineering Sciences. 2004. link Times cited: 15 Abstract: The use of molecular–dynamics simulations to understand the … read moreAbstract: The use of molecular–dynamics simulations to understand the ejection processes of particles from surfaces after energetic ion bombardment is discussed. Substrates considered include metals, covalent and ionic materials, polymers and molecular solids. It is shown how the simulations can be used to aid interpretation of experimental results by providing the underlying mechanisms behind the ejection processes. read less USED (low confidence) Y. Osetsky and D. Bacon, “Void and precipitate strengthening in α-iron: what can we learn from atomic-level modelling?,” Journal of Nuclear Materials. 2003. link Times cited: 131 USED (low confidence) D. Bacon, Y. Osetsky, R. Stoller, and R. Voskoboinikov, “MD description of damage production in displacement cascades in copper and α-iron,” Journal of Nuclear Materials. 2003. link Times cited: 127 USED (low confidence) Y. Satoh, T. Yoshiie, Y. Matsukawa, and M. Kiritani, “Simulation of transmission electron microscopy images during tensile fracture of metal foils,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2003. link Times cited: 4 USED (low confidence) K. Sato, T. Yoshiie, Y. Satoh, Q. Xu, and M. Kiritani, “Simulation of vacancy migration energy in Cu under high strain,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2003. link Times cited: 29 USED (low confidence) L. Zepeda-Ruiz, S. Han, D. Srolovitz, R. Car, and B. Wirth, “Molecular dynamics study of the threshold displacement energy in vanadium,” Physical Review B. 2003. link Times cited: 27 Abstract: The threshold displacement energy (TDE) is calculated for va… read moreAbstract: The threshold displacement energy (TDE) is calculated for vanadium as a function of temperature and orientation by molecular dynamics simulations. The TDE varies from 13 to 51 eV, depending on orientation and is nearly temperature independent between 100 and 900 K. The lowest TDE is in the 〈100〉 direction. We characterize the defects associated with the displacement simulations and found that they consist of vacancies and 〈111〉-split dumbbells. read less USED (low confidence) F. Fang and H. Deng, “Surface segregation of Al–Pb immiscible alloy system with Monte Carlo simulation,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 2003. link Times cited: 7 USED (low confidence) D. Mulliah, D. Christopher, S. Kenny, and R. Smith, “Nanoscratching of silver (100) with a diamond tip,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 32 USED (low confidence) S. Han, L. Zepeda-Ruiz, G. Ackland, R. Car, and D. Srolovitz, “Interatomic potential for vanadium suitable for radiation damage simulations,” Journal of Applied Physics. 2003. link Times cited: 69 Abstract: The ability to predict the behavior of point defects in meta… read moreAbstract: The ability to predict the behavior of point defects in metals, particularly interstitial defects, is central to accurate modeling of the microstructural evolution in environments with high radiation fluxes. Existing interatomic potentials of embedded atom method type predict disparate stable interstitial defect configurations in vanadium. This is not surprising since accurate first-principles interstitial data were not available when these potentials were fitted. In order to provide the input information required to fit a vanadium potential appropriate for radiation damage studies, we perform a series of first-principles calculations on six different interstitial geometries and vacancies. These calculations identify the 〈111〉 dumbbell as the most stable interstitial with a formation energy of approximately 3.1 eV, at variance with predictions based upon existing potentials. Our potential is of Finnis–Sinclair type and is fitted exactly to the experimental equilibrium lattice parameter, cohesive energy, e... read less USED (low confidence) N. Nozaki, M. Doyama, and Y. Kogure, “Plastic deformation of copper thin foils,” Thin Solid Films. 2003. link Times cited: 7 USED (low confidence) J. Takano, M. Doyama, and Y. Kogure, “Motion and conversion energies of adatom and adatom clusters on gold (0 0 1) surface,” Thin Solid Films. 2003. link Times cited: 2 USED (low confidence) Y. Osetsky, D. Bacon, and B. N. Singh, “Statistical analysis of cluster production efficiency in MD simulations of cascades in copper,” Journal of Nuclear Materials. 2002. link Times cited: 27 USED (low confidence) P. Szelestey, M. Patriarca, L. Perondi, and K. Kaski, “MODIFIED EAM POTENTIALS FOR MODELLING STACKING–FAULT BEHAVIOR IN Cu, Al, Au, AND Ni,” International Journal of Modern Physics B. 2002. link Times cited: 10 Abstract: In this paper we have developed empirical Embedded Atom Mode… read moreAbstract: In this paper we have developed empirical Embedded Atom Model potentials, following the fitting scheme proposed by Chantasiriwan and Milstein, in order to describe the stacking fault behaviour of copper, gold, nickel and aluminium. We show that the potentials based on this scheme can be modified to provide reasonable stacking-fault energy values and consequently a better description of the plastic properties. Modifications were done by changing the cut-off distance in case of aluminium and nickel, and in case of gold and copper by also modifying the functional form of the pair-potential. In order to validate these modified potentials we have tested them by studying various properties, such as structural, defect, and surface energies, and phonon spectra and comparing results with those from experiments and other model potentials. read less USED (low confidence) T. Harry and D. Bacon, “Computer simulation of the core structure of the screw dislocation in α-iron containing copper precipitates: I. structure in the matrix and a precipitate,” Acta Materialia. 2002. link Times cited: 83 USED (low confidence) K. Sato, T. Yoshiie, Y. Satoh, Q. Xu, E. Kuramoto, and M. Kiritani, “Point Defect Production Under High Internal Stress Without Dislocations in Ni and Cu,” Radiation Effects and Defects in Solids. 2002. link Times cited: 28 Abstract: Kiritani et al. have observed a large number of small vacanc… read moreAbstract: Kiritani et al. have observed a large number of small vacancy clusters without dislocations at the tip of torn portions of fcc metals such as Au, Ag, Cu and Ni. Small vacancy clusters, rather than dislocation cell structures, have also been observed after high-speed compressive deformation, suggesting the possibility of plastic deformation without dislocations. In this paper, in order to investigate the mechanism of deformation without dislocations, change in formation energy of point defects under high internal stress was estimated by computer simulation. Elastic deformation up to - 20% strain was found to provide a remarkable lowering of formation energy of point defects. For example, when Ni is subjected to elastic strain, the formation energy of an interstitial atom decreases to 40% that without strain and the formation energy of a vacancy decreases to 51% that without strain. The number of point defects formed under thermal equilibrium during deformation was evaluated. The number was judged to be insufficient for explaining the formation of vacancy clusters as observed in experiments. read less USED (low confidence) T. Nozaki, M. Doyama, and Y. Kogure, “Computer Simulation of High-speed Bending Deformation in Copper,” Radiation Effects and Defects in Solids. 2002. link Times cited: 1 Abstract: According to one suggested model, bending of a single crysta… read moreAbstract: According to one suggested model, bending of a single crystal introduces edge dislocations of the same sign. In the present study, this model is examined by computer simulation using molecular dynamics. When a notch is present on the tension surface, Heidenreich-Shockley partial dislocations are created near the tip of the notch. In the compression surface, partial dislocations are created due to wrinkling of the crystal plane. The results of simulation shows that dislocations are more easily created in a compressive bending region than in a tension bending region or simple tension region. read less USED (low confidence) Y. Nakamura, Y. Kondo, J. Nakamura, and S. Watanabe, “Theoretical study on the structural phase transition of Si(111)√3 ×√3-Ag surface,” Surface Science. 2001. link Times cited: 26 USED (low confidence) I. Galanakis, N. Papanikolaou, and P. Dederichs, “Applicability of the broken-bond rule to the surface energy of the fcc metals,” Surface Science. 2001. link Times cited: 147 USED (low confidence) M. Doyama and Y. Kogure, “Computer simulation of creation and motion of dislocations during plastic deformation in copper,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2001. link Times cited: 2 USED (low confidence) Y. Osetsky and D. Bacon, “Defect cluster formation in displacement cascades in copper,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 61 USED (low confidence) M. Doyama, T. Ohmae, and Y. Kogure, “Motion and conversion energies of ad-atom, di-adatom and tri-adatoms on copper (001) surface,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 0 USED (low confidence) C. Sanz-Navarro and R. Smith, “Numerical calculations using the hyper-molecular dynamics simulation method,” Computer Physics Communications. 2001. link Times cited: 11 USED (low confidence) B. Liu, W. Lai, and Z. J. Zhang, “Solid-state crystal-to-amorphous transition in metal‐metal multilayers and its thermodynamic and atomistic modelling,” Advances in Physics. 2001. link Times cited: 111 Abstract: In this review article, first a brief summary is presented c… read moreAbstract: In this review article, first a brief summary is presented concerning the formation of amorphous alloys (or metallic glasses) in binary metal systems by solid-state reaction of metallic multilayers. Secondly, under the framework of Miedema's model, thermodynamic modelling of crystal-to-amorphous transition is developed with special consideration of the excess interfacial free energy in metallic multilayers. Thirdly, the results of molecular dynamics simulations in some representative systems are presented, revealing the detailed kinetics of the crystal-to-amorphous transition on the atomic scale, such as the temperature/time dependence of interfacial reactions, the asymmetric growth of amorphous interlayers, and the nucleation and/or presence of growth barriers resulting from the interfacial texture. Fourthly, the critical solid solubilities of some representative systems are directly determined from the inter-atomic potentials through molecular dynamics simulations and then correlated with the metallic-glass-forming ability of the systems as well as their asymmetric growth during solid-state amorphization observed in experiments and/or simulations. read less USED (low confidence) P. Beauchamp and J. Lépinoux, “Image force on a dislocation in a bcc bicrystal: Computer investigation of core effects,” Philosophical Magazine A. 2001. link Times cited: 3 Abstract: The ‘image force’ on a dislocation situated a few interatomi… read moreAbstract: The ‘image force’ on a dislocation situated a few interatomic distances from the interface separating two welded half-crystals is investigated by computer simulation. The two half-crystals have bcc structure. with the same lattice parameter and orientation, but have different elastic moduli. The dislocation has the ½⟨111⟩ Burgers vector and a screw character: the interface is along a {112}- or {110}-type plane, parallel to the dislocation. Using a Finnis-Sinclair type of potential for α-Fe, the total strain energy stored in the bicrystal is calculated as a function of the dislocation position. giving the image force by derivation. The modifications in lattice friction (Peierls stress) and the changes in the dislocation core are investigated in the neighbourhood of the interface. Results are compared with those previously obtained for the planar core <100> screw dislocation. All these simulation results are finally compared with the predictions of the Peierls-Nabarro-based model of Pacheco and Mura. Despite some limitations, it is shown that this model can be extended for deriving a general law for the image force in the interface region, provided that some measure of the core width is explicitly introduced. read less USED (low confidence) K. Ozaki, S. Fukutani, and K. Honda, “Effect of interatomic potential on melting point and thermal expansion of a transition metal,” Jsme International Journal Series A-solid Mechanics and Material Engineering. 2001. link Times cited: 4 Abstract: The present study investigated the relation between the inte… read moreAbstract: The present study investigated the relation between the interatomic potentials and thermal properties. Nineteen potential functions for tantalum, which was a high melting point material, were proposed based on the concept of FS potential with some modification on the functional forms, and their melting points and thermal expansion characteristics were investigated by using molecular dynamics simulations. The melting point was determined by starting NPT ensemble simulations with the initial configurations where the solid and liquid phases coexisted in a basic cell. The thermal expansion characteristic could be estimated without MD simulations by calculating the change in potential energy with isotropic expansions and compressions. The melting point was influenced by amplitude of thermal vibrations of atoms, and was decreased with decreasing of the modified elastic moduli. An empirical potential function for tantalum was derived, which satisfied some properties near 0 K, the lattice constant at 2500 K and the melting point. read less USED (low confidence) H. Li, G. Wang, F. Ding, J. Wang, and W. Shen, “Molecular dynamics computation of clusters in liquid Fe–Al alloy,” Physics Letters A. 2001. link Times cited: 15 USED (low confidence) G. V. Kornich, G. Betz, and A. Bazhin, “Molecular dynamics simulation of mass transport processes in a Ni crystal with Al atoms as impurity under low energy ion bombardment,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 2 USED (low confidence) Y. Osetsky, A. Serra, B. N. Singh, and S. Golubov, “Structure and properties of clusters of self-interstitial atoms in fcc copper and bcc iron,” Philosophical Magazine A. 2000. link Times cited: 72 Abstract: Static and molecular dynamics simulations have been used wit… read moreAbstract: Static and molecular dynamics simulations have been used with different types of interatomic potentials to investigate the structure, properties and stability of self-interstitial atom (SIA) clusters produced during irradiation. In α-iron (Fe), faulted clusters of <110> dumbbells are unstable for all the potentials. The most stable SIA clusters are sets of parallel <111> crowdions. Large clusters of this type form perfect dislocation loops with Burgers vector b = ½⟨111⟩. Small clusters (less than 9 SIAs) of ⟨100⟩ crowdions are stable at 0K, but transform into a set of ⟨111⟩ crowdions on annealing. Larger ⟨100⟩ clusters are stable and form perfect dislocation loops with b = ⟨100⟩. Both types of loops are glissile. In copper (Cu), clusters of parallel ⟨100⟩ dumbbells and ⟨110⟩ crowdions are stable. Large clusters of these types form faulted and perfect dislocation loops with b = ⅓ ⟨111⟩ and ½ ⟨110⟩ respectively. Small faulted clusters (less than 7 SIAs) of irregular shape can transform into a set of ⟨110⟩ crowdions during annealing. Larger faulted clusters are stable as hexagonal ⅓ ⟨111⟩ Frank loops at temperatures of about up to 1050K for a period of several hundred picoseconds. All faulted clusters are sessile. Clusters of ⟨110⟩ crowdions and ½ ⟨110⟩ perfect loops are glissile and stable at all temperatures. When large enough (more than 49–64 SIAs) they can dissociate on their glide prism. Symmetric three-dimensional clusters of ⟨100⟩ dumbbells are stable at 0K but during annealing they transform into sets of ⟨110⟩ crowdions. The results for both iron and copper are discussed and compared with experimental data and provide a basis for investigating and explaining the observed differences in radiation damage accumulation behaviour between fcc and bcc metals. read less USED (low confidence) S. G. Wang, E. Tian, and C. Lung, “Surface energy of arbitrary crystal plane of bcc and fcc metals,” Journal of Physics and Chemistry of Solids. 2000. link Times cited: 113 USED (low confidence) B. Liu, W. Lai, and Q. Zhang, “Irradiation induced amorphization in metallic multilayers and calculation of glass-forming ability from atomistic potential in the binary metal systems,” Materials Science & Engineering R-reports. 2000. link Times cited: 199 USED (low confidence) V. Shenoy, R. Phillips, and E. Tadmor, “Nucleation of dislocations beneath a plane strain indenter,” Journal of The Mechanics and Physics of Solids. 2000. link Times cited: 99 USED (low confidence) H. Rafii-Tabar, “Modelling the nano-scale phenomena in condensed matter physics via computer-based numerical simulations,” Physics Reports. 2000. link Times cited: 163 USED (low confidence) G. Bhuiyan, A. Rahman, M. Khaleque, R. Rashid, and S. M. M. Rahman, “Structural, Thermodynamic and Transport Troperties of Liquid Noble and Transition Metals,” Physics and Chemistry of Liquids. 2000. link Times cited: 7 Abstract: We have investigated a number of structural, thermodynamic a… read moreAbstract: We have investigated a number of structural, thermodynamic and atomic transport properties of various liquid noble and transition metals. The underlying theory combines a simple form of the N-body potential and the thermodynamically self-consistent variational modified hypernetted chain (VMHNC) theory of liquid. The static structure factors calculated by using the VMHNC resemble, as expected, the hard sphere (HS) values. Consequently the HS model is used to calculate thermodynamic properties, viz. the specific heat, entropy, isothermal compressibility and the shear viscosity of liquid Ni, Cu, Ag, and Au. The results are in reasonable accord with the experimental values. read less USED (low confidence) T. Makino, Y. Sato, K. Itoigawa, and S. Matsuda, “Molecular dynamics analysis of nucleation and growth in a structural transformation,” Materials Transactions Jim. 1999. link Times cited: 0 Abstract: Nucleation and growth of fcc/bcc structural transformation a… read moreAbstract: Nucleation and growth of fcc/bcc structural transformation are investigated in modeled crystals. To model the transformation, inter-atomic potentials are changed to initiate the transformation. During the relaxation of the fcc structure, an initial bcc structure is created in the crystals containing two dislocations. After applying bcc potential, the nucleation starts at the region where the structure is already changed before the transformation. This result suggests that the necessary condition of the nucleation site is the displacement of about two dozen atoms relevant to the bcc structure. The interaction between atoms, which reflects the fluctuation of the environmental configuration of atoms, is also necessary for the structural transformation. read less USED (low confidence) M. A. Karolewski, “Classical dynamics simulations of 4 keV Ar sputtering of Ca(100),” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 1 USED (low confidence) T. Makino, K. Itoigawa, and S. Matsuda, “Molecular Dynamics Analysis of Elementary Nucleation Events in Structural Transformation,” Materials Transactions Jim. 1999. link Times cited: 0 Abstract: The structural transformation from fcc to bcc was modeled us… read moreAbstract: The structural transformation from fcc to bcc was modeled using a potential-change procedure and a constraint method in the molecular dynamics calculation. To model the transformation, inter-atomic potentials were changed before and after the transformation. To obtain a definite image of the elementary event of nucleation, the following was investigated: (a) how individual atoms move and (b) how an orientation relationship forms during the formation of a crystal. The elementary event of the structural change consists of a contraction, an expansion and a rotation. The contraction and the expansion relate to the structural change and are described in terms of a Bain deformation. The rotation relates to the development of an orientation relationship. The structure changes at an early stage of the nucleation process. On the other hand, the orientation relationship develops gradually during the process. read less USED (low confidence) H. Ichikawa, R. Sahara, H. Mizuseki, K. Ohno, and Y. Kawazoe, “Monte Carlo Simulation of Cu–Au Alloys on FCC Lattice with a Renormalized Potential,” Materials Transactions Jim. 1999. link Times cited: 4 Abstract: Monte Carlo simulation of an FCC lattice-gas model is carrie… read moreAbstract: Monte Carlo simulation of an FCC lattice-gas model is carried out to study order-disorder phase transitions. To study an actual Cu-Au alloys as quantitatively as possible, a Finnis-Sinclair-type potential, which has been used widely for molecular dynamics (MD) simulations, is mapped onto the FCC model by using the potential renormalization technique proposed by one of us. Using this renormalized potential, we find that the linear expansion coefficient of Cu and Au crystals and the transition temperatures are greatly improved when compared with the case of using the MD potential directly on the lattice. read less USED (low confidence) H. Alper and P. Politzer, “Molecular dynamics simulation of the temperature-dependent behavior of solid copper,” Journal of Molecular Structure-theochem. 1999. link Times cited: 8 USED (low confidence) Y. Osetsky, A. Serra, M. Victoria, S. Golubov, and V. Priego, “Vacancy loops and stacking-fault tetrahedra in copper - I. Structure and properties studied by pair and many-body potentials,” Philosophical Magazine. 1999. link Times cited: 38 Abstract: The structure and properties of vacancy loops (VIs) and stac… read moreAbstract: The structure and properties of vacancy loops (VIs) and stacking-fault tetrahedra (SFTs) in copper have been studied by computer simulation using a long-range pair interatomic potential (LRPP), obtained from the generalized pseudopotential theory, and a many-body potential (MBP) of Finnis-Sinclair type. The results obtained for these different potentials are qualitatively different. Thus, for the LRPP, significant atomic relaxation is observed for all defects. Triangular vacancy platelets relax into regular SFTs, and small hexagonal clusters form Frank loops, whereas large hexagonal clusters (containing more than 37 vacancies) can dissociate into six truncated SFTs with the side equal to the [110] side of the hexagon. Similar features are observed after the relaxation of circular loops. For the MBP, on the other hand, none of the hexagonal, circular and triangular planar vacancy platelets relax into a VL or SFT but remain almost unrelaxed 'holes', with a relative stability which is weakly dependent on the shape. The results obtained are compared with experiment and the results of other computer simulations, and the differences stemming from the use of different interatomic potentials and different simulation methods are discussed. read less USED (low confidence) M. Doyama, “Crystal growth study using combination of molecular dynamics and Monte Carlo methods,” Bulletin of Materials Science. 1999. link Times cited: 2 USED (low confidence) P. Wynblatt and A. Landa, “Computer simulation of surface segregation in ternary alloys,” Computational Materials Science. 1999. link Times cited: 38 USED (low confidence) G. V. Kornich, G. Betz, and A. Bazhin, “MD SIMULATION OF ATOMIC DISPLACEMENTS IN METALS AND METALLIC BILAYERS UNDER LOW ENERGY ION BOMBARDMENT AT 300 K,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 7 USED (low confidence) T. Makino, K. Okouchi, and S. Matsuda, “Molecular Dynamics Analysis of Nucleation in Structural Transformation,” Materials Transactions. 1999. link Times cited: 2 Abstract: The characteristics of the microstructure produced by a soli… read moreAbstract: The characteristics of the microstructure produced by a solid/solid transformation greatly depend on the nucleation process of the product phase. The objective of this study is to obtain a definite image of the atomistic nucleation process of structural transformation, which accompanies a change in atomic density, by clarifying (a) the spatial change with time, and (b) the role of dislocations in the nucleation process in computer-modeled metallic crystals. We employed a NPT ensemble attained by the constraint method proposed by Evans and Morriss. To model a fcc/bcc transformation: (a) relaxation of the initial configuration of perfect and defected fcc lattice is done by using the Finnis-Sinclare potential that stabilizes the fcc structure, and then (b) the system is set to be transformed by applying a bcc potential to all atoms at once. Using Voronoi polyhedra analysis, the crystal structure is distinguished on an atomic scale. Changes in crystal structure are found to occur in the system modeled by the NTP ensemble and the potential-change procedure employed in the present study. In a perfect crystal, the change in the number of bcc constituent atoms saturates at about 40% in all atoms in the system and consequently the structural transformation does not proceed further. It is thought that this is the state of fluctuation. In a crystal containing a dislocation, structural transformation starts around the dislocation and it is observed that the crystallographic orientation of formed crystals is restricted. The number of bcc constituent atoms increases steadily. Whether nucleation of structural transformation occurs or not is clearly distinguished by the initial state of the crystal. read less USED (low confidence) M. A. Karolewski, “Classical dynamics simulation of projectile-surface interactions,” Surface and Interface Analysis. 1999. link Times cited: 16 Abstract: A public-domain package of programs for the personal compute… read moreAbstract: A public-domain package of programs for the personal computer, the Simulation Kit (SK), has been developed for the simulation and visualization of collisions of low-energy (<10 keV) atomic projectiles with solid target lattices. Possible applications of the SK include the simulation of ion scattering spectra, sputtering coefficients, reflection coefficients and projectile ranges. The simulation model used by the SK is based on classical dynamics, and uses a composite screened-Coulomb/Morse pair potential to model interactions between particles in the target. The simulation model also incorporates inelastic scattering effects based on the Lindhard–Schiott–Scharff, Oen–Robinson and Shapiro–Tombrello models, respectively. The physical basis of the simulation model is described, and examples are provided of applications in ion beam analysis (ion scattering spectrometry, sputter yields). Copyright © 1998 John Wiley & Sons, Ltd. read less USED (low confidence) B. Kooi, H. B. Groen, and J. Hosson, “Misfit Dislocations at Ag/Mn3O4, Cu/MnO and Cu/Mn3O4 Interfaces,” Acta Materialia. 1998. link Times cited: 18 USED (low confidence) Y. Shibutani, “Nonlocal Elastic Constants of Centrosymmetric Homogeneous Lattice Structure and Inhomogenous One,” Jsme International Journal Series A-solid Mechanics and Material Engineering. 1998. link Times cited: 6 Abstract: Nonlocal elastic constants have been related to atomistic pr… read moreAbstract: Nonlocal elastic constants have been related to atomistic properties through a macro-micro linking approach proposed in a previous paper(Trans.of JSME, Vol.62, No.601, A(1996), p.2054-2059). Using the derived relations, these material constatns can be estimated only by atomistic quantities of force constants and neighboring lattice configuration. In the present study, centrosymmetric homogeneous bulk is first taken up. Agreement between the number of independent components of these elastic constants and the results from analytical estimation by invariance of transformation on an isotropic tensor is demonstrated. Then, the less symmetric surface and the high Σ-value grain boundary are computationally constructed by molecular dynamics simulations in order to examine inhomogeneous nonlocal effects. It is found that atomistic inhomogeneity increases nonlocal material properties, while the characteristic length derived from the ratio of the 6th-order to the 4th-order elastic constants is smaller than a lattice parameter. read less USED (low confidence) G. V. Kornich and G. Betz, “MD simulation of atomic displacements in pure metals and metallic bilayers during low energy ion bombardment at 0 K,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1998. link Times cited: 13 USED (low confidence) D. Timpel and K. Scheerschmidt, “Molecular dynamics investigations of silver diffusion in glass,” Journal of Non-crystalline Solids. 1998. link Times cited: 6 USED (low confidence) A. Landa, A. Ruban, P. Wynblatt, H. Skriver, A. Girshick, and V. Vítek, “Co-segregation at the surface of Pb-Bi-Ni alloys: combined ab initio and Monte Carlo study,” Journal of Physics: Condensed Matter. 1998. link Times cited: 6 Abstract: A recent study of a Pb-Bi-Ni alloy containing 5 at.% Bi and … read moreAbstract: A recent study of a Pb-Bi-Ni alloy containing 5 at.% Bi and 0.04 at.% Ni reported a strong co-segregation of Bi and Ni at the alloy surface. We have performed ab initio calculations of the segregation profiles at the (111), (100) and (110) surfaces of random alloys by means of the coherent potential approximation and the tight-binding linear muffin-tin-orbitals method. We have found the segregation profiles to be oscillatory (this effect is most pronounced for the (111) surface) with a strong preference for Bi to segregate to the first atom layer and depletion of Bi in the subsurface atom layer. The energetic origin of the oscillatory segregation is discussed in terms of the Connolly-Williams effective cluster interactions. In the ternary alloy we have also found a tendency for Ni to segregate to the subsurface atom layer due its strong interaction with Bi, which is present at high concentrations relative to the bulk in both the first and third atom layers of the (111) surface. In order to include relaxation effects, we have performed Monte Carlo simulations, employing Finnis-Sinclair-type empirical many-body potentials, and computed the segregation profiles at the (111) surface of and alloys. For Pb-Bi alloys, the concentration profiles have also been found to be oscillatory, in fair agreement with results of the ab initio calculations. The calculations on Pb-Bi-Ni show strong segregation of Ni to the subsurface atom layer, accompanied by co-segregation of Bi to several of the outermost atom layers. read less USED (low confidence) M. Duesbery and W. Xu, “The motion of edge dislocations in body-centered cubic metals,” Scripta Materialia. 1998. link Times cited: 47 USED (low confidence) A. Landa, P. Wynblatt, A. Girshick, V. Vítek, A. Ruban, and H. Skriver, “Development of Finnis–Sinclair type potentials for Pb, Pb–Bi, and Pb–Ni systems: application to surface segregation,” Acta Materialia. 1998. link Times cited: 27 USED (low confidence) J. Takano, O. Takai, Y. Kogure, and M. Doyama, “Simulation of atomic-scale surface migration in homoepitaxial growth using embedded-atom method potentials for gold,” Thin Solid Films. 1998. link Times cited: 6 USED (low confidence) A. Girshick, A. Bratkovsky, D. Pettifor, and V. Vítek, “Atomistic simulation of titanium. I. A bond-order potential,” Philosophical Magazine. 1998. link Times cited: 53 Abstract: The bond-order potential for hcp Ti has been constructed in … read moreAbstract: The bond-order potential for hcp Ti has been constructed in the framework of a tight-binding description of the binding energy. In this scheme the energy consists of two parts: the bond part that comprises the d-electron contribution to bonding, and a pairwise part. Both parts contain fitting parameters but are treated independently. The bond part reproduces the most important characteristics of the d-band and Cauchy pressures, and the pairwise part complements the bond part so as to reproduce exactly the equilibrium lattice parameters and to a good approximation the elastic moduli. The potential is tested by examining the mechanical stability of the hcp lattice with respect to a variety of large deformations. It is applied to a study of dislocation behaviour in Ti in the accompanying paper, part II. read less USED (low confidence) A. Marinopoulos, V. Vítek, and J. Bassani, “Local and Effective Elastic Properties of Grain Boundaries in Silicon,” Physica Status Solidi (a). 1998. link Times cited: 13 Abstract: When considering the mechanical behaviour of materials an im… read moreAbstract: When considering the mechanical behaviour of materials an important property is the tensor of elastic moduli. Recently, local elastic moduli of interfaces have been defined and studied for metallic materials [1 to 3]. In these works grain boundaries are regarded as heterogeneous continua composed of ‘phases’ associated with individual atoms which possess elastic moduli identified with the atomic-level moduli evaluated at corresponding atomic positions. From this representation it is possible to define the ‘effective’ moduli of the grain boundary region. In this paper this concept is developed for materials with covalent character of bonding, specifically silicon. Using the Tersoff's potential [4, 5], the atomic-level and effective elastic moduli of the interfacial region have been evaluated for three alternate structures of the Σ = 3 (112-)/[11-0] tilt boundary. These calculations are then compared with the continuum bounds on the effective moduli evaluated using the classical minimum-energy principles of elasticity. The effective moduli calculated in the atomistic framework are generally within the continuum bounds and thus the present study demonstrates that the heterogeneous continuum model of the interfaces is appropriate for the description of the elastic properties of grain boundaries in silicon. An important aspect addressed in this study is the uniqueness of interfacial elastic moduli since their evaluation involves the energy associated with an atom which cannot be defined uniquely. The calculations have been made for two different partitions of the total energy into energies associated with individual atoms. These two partitions lead to almost identical results for the effective moduli and continuum bounds when the tensor of the atomic-level moduli is positive definite. When some atomic-level moduli are not positive definite the results may depend on the chosen energy partition. read less USED (low confidence) D. Timpel, K. Scheerschmidt, and S. Garofalini, “Silver clustering in sodium silicate glasses: a molecular dynamics study,” Journal of Non-crystalline Solids. 1997. link Times cited: 25 USED (low confidence) Y. Osetsky, M. Victoria, A. Serra, S. Golubov, and V. Priego, “Computer simulation of vacancy and interstitial clusters in bcc and fcc metals,” Journal of Nuclear Materials. 1997. link Times cited: 62 USED (low confidence) G. Odette and B. Wirth, “A computational microscopy study of nanostructural evolution in irradiated pressure vessel steels,” Journal of Nuclear Materials. 1997. link Times cited: 133 USED (low confidence) B. Kooi, H. B. Groen, and J. Hosson, “Atomic structure of interfaces between Mn3O4 precipitates and Ag studied with HRTEM,” Acta Materialia. 1997. link Times cited: 18 USED (low confidence) J. Cayphas, M. Hou, and L. Coheur, “The behaviour of helium in neutron irradiated beryllium: a molecular dynamics study,” Journal of Nuclear Materials. 1997. link Times cited: 11 USED (low confidence) R. Smith, B. King, and K. Beardmore, “Molecular dynamics simulation of 0.1–2 keV ion bombardment of Ni100,” Radiation Effects and Defects in Solids. 1997. link Times cited: 12 Abstract: Molecular dynamics simulations are performed of the interact… read moreAbstract: Molecular dynamics simulations are performed of the interaction of energetic 0.1–2 keV Ar ions at normal incidence with Ni{100}. The development of some typical collision cascades are examined in detail and the mechanisms by which the radiation damage occurs is described for both elastic and inelastic collisions cascades. Results are presented on the vacancy, interstitial and adatom formation, the sputtering of atoms and atomic mixing efficiency. These parameters are compared with experiment. read less USED (low confidence) W. Vellinga, J. Hosson, and V. Vítek, “Misfit dislocations: An atomistic and elastic continuum approach,” Acta Materialia. 1997. link Times cited: 47 USED (low confidence) J. R. Fernández, A. M. Monti, and R. Pasianot, “Dynamics of free surfaces in model fcc, hcp and L12 structures,” Philosophical Magazine Part B. 1997. link Times cited: 1 Abstract: Dynamic properties of free surfaces in their relaxed configu… read moreAbstract: Dynamic properties of free surfaces in their relaxed configuration obtained by the static relaxation method, are studied in fcc Ni and Al, hcp Ti and the L12 alloy Ni3Al. The (001), (110) and (111) surfaces are analysed for the fcc and Li2 structures and the (0001), (1210) and (1010) surfaces for the hcp structure. Interatomic potentials of the embedded-atom method type are used to calculate vibrational eigenfrequencies obtained through the Einstein and ‘cluster’ approaches for the atoms on the first few layers. read less USED (low confidence) Y. Mishin and D. Farkas, “Atomistic Simulation of Point Defects and Diffusion in B2 NiAl,” Defect and Diffusion Forum. 1997. link Times cited: 122 Abstract: In part I of this work we studied point defect energetics in… read moreAbstract: In part I of this work we studied point defect energetics in the ordered B2 compound NiAI by means of computer simulations using 'molecular statics' and the embedded atom method. In the present paper we apply the computation technique and results of part I to study atomic mechanisms of tracer self-diffusion in NiAl. We calculate the activation energy of Ni and Al self-diffusion in perfectly stoichiometric NiAl for three atomic mechanisms: the mechanism of next-nearest-neighbour (NNN) vacancy jumps, the 6-jump vacancy mechanism and the 4-ring mechanism. The results of our simulations indicate that self-diffusion in stoichiometric NiAI is dominated by the mechanism of next-nearest-neighbour vacancy jumps. Diffusion of Al by this mechanism is likely to occur more slowly and with a higher activation energy than diffusion of Ni. The mechanism of 6-jump cycles is less favourable but still highly competitive to the NNN vacancy mechanism. The 4-ring mechanism is the least effective for both Ni and Al diffusion. The effect of off-stoichiometry on diffusion in NiAI is briefly discussed. read less USED (low confidence) F. Hori, Y. Kamimura, T. Tsutsumi, and E. Kuramoto, “Positron lifetime calculation in FeCu binary alloy with lattice relaxation,” Journal of Nuclear Materials. 1996. link Times cited: 8 USED (low confidence) D. Timpel, K. Scheerschmidt, and S. Ruvimov, “HREM simulations of particles and interfaces refined by molecular dynamics relaxations,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 1996. link Times cited: 2 USED (low confidence) 陈致英, “Molecular dynamics studies on the dislocation gliding near a tilt boundary,” Acta Mechanica Sinica. 1996. link Times cited: 1 USED (low confidence) M. Breeman, G. Barkema, M. Langelaar, and D. Boerma, “Computer simulation of metal-on-metal epitaxy,” Thin Solid Films. 1996. link Times cited: 22 USED (low confidence) Y. Osetsky, A. Mikhin, and A. Serra, “Study of copper precipitates in α‐iron by computer simulation I. Interatomic potentials and properties of Fe and Cu,” Philosophical Magazine. 1995. link Times cited: 61 Abstract: Pair potentials describing Fe-Fe and Cu-Cu interatomic inter… read moreAbstract: Pair potentials describing Fe-Fe and Cu-Cu interatomic interactions based on the generalized pseudopotential theory are presented. Different properties of pure Fe and Cu such as elastic constants, phonon dispersion curves, vacancy formation and migration energy, interstitial formation energy, thermal expansion, stacking fault energy, pressure-volume dependence and relative stability of the bcc, fcc and hcp structures were studied using these potentials. They are in good agreement with the experimental results. The phase transformations fcc → bcc in iron and bcc → fcc in copper have been simulated by molecular dynamics. It was found that the transformation in iron mainly follows the Bain distortion, while that in copper follows the Burgers type mechanisms. read less USED (low confidence) D. Timpel and K. Scheerschmidt, “HREM simulations of Ag particles in sodium silicate glasses refined by molecular dynamic relaxations,” Physica Status Solidi (a). 1995. link Times cited: 6 Abstract: Providing local information at an atomic level high resoluti… read moreAbstract: Providing local information at an atomic level high resolution electron microscopy (HREM) is applied to investigate Ag particles in sodium silicate glasses. Here, the structure of the embedded metallic particles, which influence the properties of glasses, is described by molecular dynamics relaxation calculations. The possibility of HREM to vizualize the structural modifications owing to relaxations is discussed on the basis of simulated HREM micrographs.
Hochauflosende Elektronenmikroskopie (HREM) wird wegen der direkten atomaren Strukturabbildung zur Untersuchung von Ag-Teilchen in Natriumsilikat-Glasern verwendet. Die Struktur der eingelagerten Metallteilchen, die wesentlichen Einflus auf die Glaseigenschaften hat, wird dabei durch molekulardynamische Relaxationsrechnungen beschrieben. Die Moglichkeiten der Sichtbarmachung struktureller Veranderungen im HREM infolge der Relaxationseffekte werden anhand von computersimulierten Abbildungen diskutiert. read less USED (low confidence) M. Yan, S. P. Chen, T. Mitchell, S. Vyas, and R. Grimes, “Atomistic studies of energies and structures of (hk0) surfaces in NiO,” Philosophical Magazine. 1995. link Times cited: 17 Abstract: In this work we study the energies and structures of (hk0) s… read moreAbstract: In this work we study the energies and structures of (hk0) surfaces in NiO by computer simulation. The short-range interactions are described by Buckingham potentials. The O ion is treated as polarizable by employing the shell model. The coefficients of the potentials and the shell parameters are fitted empirically to properties of the NiO perfect lattice assuming full ionicity. It is shown that discrete atomic structures of (hk0) surfaces can be treated as monatomic steps distributed uniformly or non-uniformly on {100} terraces. The energy of a surface is determined by the density of steps. The energy of the monatomic step is found to be 7·58 × 10 −10Jm−1 before relaxation and 3·32 × 10−10 Jm−1 after relaxation. The elastic interaction between steps is repulsive and increases as the square of the step density. Surface rumpling is found to be a general property, with the anion cores moving outwards from the cations and also outwards from the anion shells, so that the anions are polarized. The pol... read less USED (low confidence) W. Phythian, R. Stoller, A. Foreman, A. Calder, and D. Bacon, “A comparison of displacement cascades in copper and iron by molecular dynamics and its application to microstructural evolution,” Journal of Nuclear Materials. 1995. link Times cited: 266 USED (low confidence) Y. W. Zhang, T. Wang, and Q. Tang, “The effect of thermal activation on dislocation processes at an atomistic crack tip,” Journal of Physics D. 1995. link Times cited: 17 Abstract: The effects of thermal activation on the dislocation emissio… read moreAbstract: The effects of thermal activation on the dislocation emission from an atomistic crack tip are discussed. Molecular dynamics simulations at different constant temperatures are carried out to investigate the thermal effects. The simulated results show that the processes of the partial dislocation generation and emission are temperature dependent. As the temperature increases, the incipient duration of the partial dislocation nucleation becomes longer, the critical stress intensity factor for partial dislocation emission is reduced and, at the same loading level, more dislocations are emitted. The dislocation velocity moving away from the crack tip and the separations of partial dislocations are apparently not temperature dependent. The simulated results also show that, as the temperature increases, the stress distribution along the crack increases slightly. Therefore stress softening at the crack tip induced by thermal activation does not exist in the present simulation. A simple model is proposed to evaluate the relation of the critical stress intensity factor versus temperature. The obtained relation is in good agreement with our molecular dynamics results. read less USED (low confidence) R. Fonda, M. Yan, and D. Luzzi, “Atomic structure of the σ = 5, (310)[001] grain boundary in NiAl: A combined electron microscopy and theoretical study,” Philosophical Magazine Letters. 1995. link Times cited: 17 Abstract: The atomic structure of the σ= 5, (310)[001] grain boundary … read moreAbstract: The atomic structure of the σ= 5, (310)[001] grain boundary in NiAl has been determined by a synergistic approach using high-resolution electron microscopy and atomistic structure calculations. These calculations employ N-body empirical potentials which were developed for the NiAl phase. A minimum-energy structure was determined which produced calculated images which matched experimental images of the boundary. The change in interfacial energy due to various defects at the grain boundary was also examined. This is the, first concurrent application of experimental and theoretical structure determinations to a grain boundary in an intermetallic compound. read less USED (low confidence) Y. W. Zhang, T. Wang, and Q. Tang, “SIMULATION OF NUCLEATION AND EMISSION OF DISLOCATIONS BY MOLECULAR-DYNAMICS METHOD,” Journal of Applied Physics. 1995. link Times cited: 17 Abstract: The nucleation and emission of dislocations from the crack t… read moreAbstract: The nucleation and emission of dislocations from the crack tip under mode II loading are analyzed by the molecular‐dynamics method in which the Finnis–Sinclair potential has been used. A suitable atom lattice configuration is employed to allow one to fully analyze the nucleation, emission, dissociation, and pileup of the dislocations. The calculated results show that although the pure mode II loading is applied, the crack tip generally exhibits a combined mode. The stress distributions before the dislocation emission are in agreement with the elasticity solution, but are not after the emission. The critical stress intensity factor corresponding to the dislocation nucleation KIIe is dependent on the loading rate KII. The separations of a pair of partial dislocations and the full dislocations are also dependent on the loading rate. When the first partial dislocation is blocked, a pileup of dislocations can be set up. It is also found that the dislocation can move at subsonic wave speed (less than the shear... read less USED (low confidence) G. Ackland, S. J. Wooding, and D. Bacon, “Defect, surface and displacement-threshold properties of α-zirconium simulated with a many-body potential,” Philosophical Magazine. 1995. link Times cited: 134 Abstract: A many-body interatomic potential has been developed for the… read moreAbstract: A many-body interatomic potential has been developed for the h.c.p. metal α-zirconium using the same methodology as that used by Ackland for α-titanium. The repulsive pair part of the potential has been constructed so that the model can be employed for simulating atomic collisions. The favoured self-interstitial configurations are the 〈1120〉 crowdion and split defects, and they are highly mobile in the basal plane. The energy of surfaces is not strongly dependent on the crystallographic orientation, and the I2 stacking fault on the basal plane is not stable. The displacement threshold energy in a crystal at 0 K exhibits a similar orientation dependence to that computed recently for α-titanium by Bacon et al. and has the same minimum of 27·5 eV along the 〈1120〉 directions, but the mean value of 55 eV averaged over all orientations is higher than that of 30 eV in titanium. read less USED (low confidence) 张永伟, 王自强, and 汤奇恒, “Molecular dynamics simulation of crack-tip processes in copper,” Acta Mechanica Sinica. 1995. link Times cited: 7 USED (low confidence) M. Breeman, G. Barkema, and D. Boerma, “BINDING-ENERGIES AND STABILITY OF CU-ADATOM CLUSTERS ON CU(100) AND CU(111),” Surface Science. 1995. link Times cited: 37 USED (low confidence) D. Hofmann and M. Finnis, “Theoretical and experimental analysis of near Σ3 (211) boundaries in silver,” Acta Metallurgica Et Materialia. 1994. link Times cited: 44 USED (low confidence) V. Vítek, G. J. Wang, E. S. Alber, and J. Bassani, “Relationship between modeling of the atomic structure of grain boundaries and studies of mechanical properties,” Journal of Physics and Chemistry of Solids. 1994. link Times cited: 9 USED (low confidence) H. Polatoglou, “Wetting at the Σ = 5[001] twist boundary of Cu3Au,” Computational Materials Science. 1994. link Times cited: 8 USED (low confidence) M. Menyhárd, M. Yan, and V. Vítek, “Atomistic vs phenomenological approaches to grain boundary segregation: Computer modeling of CuAg alloys,” Acta Metallurgica Et Materialia. 1994. link Times cited: 46 USED (low confidence) H. Polatoglou, “A comparative study of the Sigma =5 and 13 (001) twist boundaries in Cu3Au,” Journal of Physics: Condensed Matter. 1994. link Times cited: 6 Abstract: The Sigma =5 and 13 (001) twist boundaries in Cu3Au are stud… read moreAbstract: The Sigma =5 and 13 (001) twist boundaries in Cu3Au are studied using the constant-temperature, pressure and chemical potential difference Monte Carlo method. An N-body potential and bulk-like boundary conditions are utilized. The properties studied include the stoichiometry profile, the structure factor and the average strain of the atoms along (001). The two boundaries exhibit similar behaviour at different temperatures below the order-disorder transition. The average strain at the boundary is about 7% and diminishes rapidly, within three atomic layers. No noticeable segregation effects are observed, only a tendency of Au atoms to be depleted from the Sigma =13 boundary. Around the boundaries we find a region with lower order than the rest of the material. This region broadens as the transition temperature is approached, thus establishing the presence of wetting. read less USED (low confidence) H. Deng and D. Bacon, “Simulation of low energy displacement processes in a dilute cu-au alloy,” Radiation Effects and Defects in Solids. 1994. link Times cited: 0 Abstract: Research into displacement cascade processes in alloy system… read moreAbstract: Research into displacement cascade processes in alloy systems has received little attention, yet is potentially of interest because issues such as the effect of solutes on the displacement threshold and the defect distribution and movement in cascades are important. As part of a wider study, we have initially considered the minor substitutional solute Au in a Cu matrix, and have used molecular dynamics to investigate the properties of point defects, the threshold displacement energy Ed, and temporal and spatial distribution of defects in low-energy (≤500 eV) displacement cascades. The results show that the influence of the solute on the properties of defects is important and that Ed is dramatically different from its form in pure copper. In comparison with pure copper, the recoil of the Au solute gives rise to a higher peak at longer times in the number of displaced atoms in the generation of a displacement cascade. The influence of this on defect density in the cascade and the final number and a... read less USED (low confidence) G. Bozzolo, J. Ferrante, and A. M. Rodríguez, “Modelling of surfaces. I. Monatomic metallic surfaces using equivalent crystal theory,” Journal of Computer-Aided Materials Design. 1994. link Times cited: 15 USED (low confidence) B. Peng, M. Cai, G.-hai Li, X. Wu, and F. Zhou, “Molecular dynamics study of the microstructure and properties of nanocrystalline copper,” Nanostructured Materials. 1994. link Times cited: 4 USED (low confidence) A. Foreman, W. Phythian, and C. English, “Molecular dynamics simulation of irradiation damage cascades in copper using a many-body potential,” Radiation Effects and Defects in Solids. 1994. link Times cited: 16 Abstract: The evolution of irradiation damage cascades in copper has b… read moreAbstract: The evolution of irradiation damage cascades in copper has been simulated by molecular dynamics, using a many-body potential. Nearly 200 cascades have been produced with random knock-on directions and primary knock-on atom (PKA) energies in the range from 60 eV to 10 keV. The starting temperature for these simulations was 100 K and 600 K, this report will confine itself to the data obtained at 100 K. The cascade evolution has been followed for times typically up to ∼10 psec and in some cases up to ∼30 psec. The cascades are characterised by the sudden emission of replacement collision sequences and with shape variations due to local channelling events. At the higher energies the core has been shown to be liquid-like structure with cavitation. The annealing phase leaves loosely clustered vacancies at the cascade centre but collapse to a vacancy loop is not generally observed. A feature of the more energetic cascades is the production by a ballistic mechanism of interstitial atom clusters at the pe... read less USED (low confidence) H. Polatoglou and G. Bleris, “Comparison of the constrained and unconstrained Monte-Carlo method: The case of Cu3Au,” Solid State Communications. 1994. link Times cited: 13 USED (low confidence) E. S. Alber, J. Bassani, V. Vítek, and G. J. Wang, “Stoneley waves and grain-boundary phonons,” Modelling and Simulation in Materials Science and Engineering. 1994. link Times cited: 9 Abstract: The lattice dynamics (phonons) of bicrystals with stable gra… read moreAbstract: The lattice dynamics (phonons) of bicrystals with stable grain-boundary structures are computed and the results of these calculations are linked with continuum elasticity solutions of interface waves. Through comparisons of the lattice-dynamics calculations for ideal crystals with those for the corresponding bicrystals, the low-frequency acoustic branches of the dispersion curves associated with the interface vibrations are identified. These vibrational modes, in the limit of long wave length and low frequency, are the ones for which we seek to establish connections with continuum solutions for localized interface waves that decay exponentially with distance from the interface. We find that the perfect-bonding assumption over-restricts the nature of these latter waves, that is, these solutions do not reproduce the atomistic results for continuum-like waves. The reason lies in the fact that these localized waves are significantly influenced by the local properties of the interfacial region associated with its distinct structure. read less USED (low confidence) H. Kitagawa, A. Nakatani, and Y. Shibutani, “Molecular dynamics study of crack processes associated with dislocation nucleated at the tip,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1994. link Times cited: 28 USED (low confidence) H. Polatoglou and G. Bleris, “Constant temperature and pressure monte carlo study of the order-disorder transition of Cu3Au,” Interface Science. 1994. link Times cited: 6 USED (low confidence) M. Azzaoui, J. Pennisson, and V. Pontikis, “Combined high-resolution electron microscopy and computer modeling of a Σ=5 (210) [001] tilt boundary in AuCu3,” Interface Science. 1994. link Times cited: 3 USED (low confidence) M. Breeman, G. Barkema, and D. Boerma, “Annealing of Cu-adatom configurations on Cu(100),” Surface Science. 1994. link Times cited: 16 USED (low confidence) W. Phythian and C. English, “Microstructural evolution in reactor pressure vessel steels,” Journal of Nuclear Materials. 1993. link Times cited: 217 USED (low confidence) D. Bacon, H. Deng, and F. Gao, “Computer simulation of threshold displacement events in alloys,” Journal of Nuclear Materials. 1993. link Times cited: 16 USED (low confidence) A. M. Rodríguez, G. Bozzolo, and J. Ferrante, “Multilayer relaxation and surface energies of fcc and bcc metals using equivalent crystal theory,” Surface Science. 1993. link Times cited: 129 USED (low confidence) D. Kulp, T. Egami, D. Luzzi, and V. Vítek, “A molecular dynamics study of atomic level stress distributions in defective intermetallics,” Journal of Alloys and Compounds. 1993. link Times cited: 5 USED (low confidence) D. Kulp, G. Ackland, M. Šob, V. Vítek, and T. Egami, “MANY-BODY POTENTIALS FOR CU-TI INTERMETALLIC ALLOYS AND A MOLECULAR-DYNAMICS STUDY OF VITRIFICATION AND AMORPHIZATION,” Modelling and Simulation in Materials Science and Engineering. 1993. link Times cited: 17 Abstract: The authors present central force many-body potentials for t… read moreAbstract: The authors present central force many-body potentials for the Cu-Ti system which were constructed so as to reproduce a number of properties of the CuTi2 compound (tetragonal structure). The authors fitted the potentials not only to the available experimental data (equilibrium lattice parameters and enthalpy of mixing) but employed an ab initio method to determine additional data, in particular the bulk modulus. It is shown that these potentials ensure the stability of the CuTi2 crystal structure against alternate structures and changes in the c/a ratio and closely reproduce the melting temperature of CuTi2. The authors then employ these potentials in simulations of the glass transition and amorphization by irradiation and use the concept of atomic level stresses to interpret the results. read less USED (low confidence) U. Wolf, F. Ernst, T. Muschik, M. Finnis, and H. Fischmeister, “The influence of grain boundary inclination on the structure and energy of Σ=3 grain boundaries in copper,” Philosophical Magazine. 1992. link Times cited: 171 Abstract: In a combined theoretical and experimental study, the energi… read moreAbstract: In a combined theoretical and experimental study, the energies and structures of σ = 3, [011] tilt boundaries in Cu were investigated. Equilibrium atomistic structures and grain-boundary energies were calculated by static energy minimization using an embedded-atom potential. Cu bicrystals of the same boundary orientations were fabricated by welding of Cu single crystals. Grain-boundary energies were measured by the thermal grooving technique. The atomistic structure of the {211} twin boundary was investigated by high-resolution transmission electron microscopy (HRTEM). The calculated grain-boundary energies γb plotted against the inclination of the boundary plane show a minimum for the {111} twin boundary and a second minimum at an inclination of about 82° to the {111} boundary. The calculated dependence of γb on inclination is confirmed by the measured energies over the entire range. Common to all calculated boundary structures is a microfaceting into {111} and {211} twin facets. The structures ... read less USED (low confidence) A. Foreman, C. English, and W. Phythian, “Molecular dynamics calculations of displacement threshold energies and replacement collision sequences in copper using a many-body potential,” Philosophical Magazine. 1992. link Times cited: 108 Abstract: The many-body potential of Ackland et al. is currently being… read moreAbstract: The many-body potential of Ackland et al. is currently being used to investigate the production of displacement damage cascades in irradiated copper, using the molecular dynamics computer program MOLDY. The effect of adopting this potential on both the static and the dynamic properties of irradiation-induced defects is evaluated here. It is shown that this potential provides a good representation of the well known properties of point defects. However, for dynamic events involving the close approach of atoms, a short-range ion core repulsive term needs to be incorporated. The modified core term has been optimized to give an excellent fit to the measured threshold displacement energies without significantly affecting the fit to other physical properties. read less USED (low confidence) B. Rice, C. Murthy, and B. Garrett, “Effects of surface structure and of embedded-atom pair functionals on adatom diffusion on fcc metallic surfaces,” Surface Science. 1992. link Times cited: 4 USED (low confidence) T. D. Rubia and W. Phythian, “Molecular dynamics studies of defect production and clustering in energetic displacement cascades in copper,” Journal of Nuclear Materials. 1992. link Times cited: 46 USED (low confidence) W. Wallace and G. Ackland, “Calculated energies and relaxations of the low-index planes of ordered Cu3Au,” Applied Surface Science. 1992. link Times cited: 29 USED (low confidence) I. Alber, J. Bassani, M. Khantha, V. Vítek, and G. J. Wang, “Grain boundaries as heterogeneous systems: atomic and continuum elastic properties,” Philosophical Transactions of the Royal Society of London. Series A: Physical and Engineering Sciences. 1992. link Times cited: 83 Abstract: The relation between atomic structure and elastic properties… read moreAbstract: The relation between atomic structure and elastic properties of grain boundaries is investigated theoretically from both atomistic and continuum points of view. A heterogeneous continuum model of the boundary is introduced where distinct phases are associated with individual atoms and possess their atomic level elastic moduli determined from the discrete model. The effective elastic moduli for sub-blocks from an infinite bicrystal are then calculated for a relatively small number of atom layers above and below the grain boundary. These effective moduli can be determined exactly for the discrete atomistic model, while estimates from upper and lower bounds are evaluated in the framework of the continuum model. The complete fourth-order elastic modulus tensor is calculated for both the local and the effective properties. Comparison between the discrete atomistic results and those for the continuum model establishes the validity of this model and leads to criteria to assess the stability of a given grain boundary structure. For stable structures the continuum estimates of the effective moduli agree well with the exact effective moduli for the discrete model. Metastable and unstable structures are associated with a significant fraction of atoms (phases) for which the atomic-level moduli lose positive definiteness or even strong ellipticity. In those cases, the agreement between the effective moduli of the discrete and continuum systems breaks down. read less USED (low confidence) J. Cserti, M. Khantha, V. Vítek, and D. Pope, “An atomistic study of the dislocation core structures and mechanical behavior of a model D019 alloy,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1992. link Times cited: 31 USED (low confidence) B. Peng, X. Wu, F. Zhou, and Q.-heng Tang, “Molecular dynamic study of grain boundary embrittlement for [101] tilt copper bicrystals induced by bismuth segregation,” Journal of Applied Physics. 1992. link Times cited: 7 Abstract: The selective bismuth segregation and the microprocess of fr… read moreAbstract: The selective bismuth segregation and the microprocess of fracture for the three [101] tilt copper bicrystals Σ9(212) 38.94°, Σ11(323) 50.48°, and Σ33(545) 58.99° have been studied by a molecular dynamics technique. The results show that the Bi segregation and the fracture behavior of the Cu‐Bi bicrystals are strongly dependent on the grain boundary (GB) structure. The Bi segregation is strongly related to the polyhedra constructing the GB cores and the stress fields of the GB dislocations (GBDs), and the GB embrittlement of copper induced by the Bi segregation is determined by the segregated concentration and the distribution of Bi atoms. With the increase of the relative number of pentagonal bipyramids and the localization of the stress fields of the GBDs in the GBs, the bicrystals Σ9, Σ11, and Σ33 show a decreasing propensity for the Bi segregation and subsequent different fracture behaviors. The severe intergranular brittle fracture that happens in the Σ9 bicrystal is mainly caused by the breaki... read less USED (low confidence) 周富信, 彭八一, 吴希俊, and 汤奇恒, “Molecular dynamics study of deformation and fracture of Bi-segregated copper bicrystals,” Acta Mechanica Sinica. 1992. link Times cited: 0 USED (low confidence) 周富信, 彭八一, 吴希俊, and 汤奇恒, “PSEUDO-MOLECULAR DYNAMICS STUDY OF GRAIN BOUNDARY SEGREGATION *,” Acta Mechanica Sinica. 1992. link Times cited: 0 USED (low confidence) S. M. Murphy, “Contribution of interstitial migration to segregation in concentrated alloys,” Journal of Nuclear Materials. 1991. link Times cited: 20 USED (low confidence) F. Zhou, B. Peng, and X. Wu, “Molecular dynamics study of deformation and fracture for pure and bismuth‐segregated tilt copper bicrystals,” Journal of Applied Physics. 1990. link Times cited: 8 Abstract: The microprocess of deformation and fracture for pure and Bi… read moreAbstract: The microprocess of deformation and fracture for pure and Bi‐segregated Σ3(111)/[101] 70.53° and Σ33(545)/[101] 58.99° tilt bicrystals of metal copper has been studied by the molecular dynamics method. It has been found that deformation and fracture are dependent on the grain boundary (GB) structure and bismuth segregation. For pure Σ33 bicrystal, the deformation is mainly due to the glide of partial dislocations generated from the GB structural units where the GB dislocations exist. The ductile fracture is attributed to the dislocation emission, which leads to vacancy generation and void coalescence. The bismuth segregation weakens the atomic bonds between copper atoms in the vicinity of GB. Under the action of the external load, the weakened bonds break and lead to formation of microcracks. Finally, the brittle fracture takes place along the binding weakening region. For Σ3 bicrystal, the ductile fracture is related to the void coalescence generated not by dislocation emission but by lattice distort... read less USED (low confidence) J. Holender, “Molecular dynamics studies of solid and liquid copper using the Finnis-Sinclair many-body potential,” Journal of Physics: Condensed Matter. 1990. link Times cited: 15 Abstract: Molecular dynamics studies of the temperature dependent prop… read moreAbstract: Molecular dynamics studies of the temperature dependent properties of copper have been carried out. The constant pressure and constant temperature (NPT) MD method has been used together with the Finnis-Sinclair many-body potential. On the basis only of the room temperature properties the behaviour of solid and liquid copper over wide temperature range have been simulated and compared with experiment. read less USED (low confidence) E. Brandt, “Molecular-dynamics simulation of amorphous alloys. I. Atomic structure of fully relaxed systems,” Journal of Physics: Condensed Matter. 1989. link Times cited: 7 Abstract: Amorphous structures of one or two types of atoms of various… read moreAbstract: Amorphous structures of one or two types of atoms of various radii and masses and interacting by various central potentials are simulated by molecular dynamics on a computer. Fully relaxed atomic arrangements resulting from simulated annealing are investigated with the aim of getting an indication of possible mechanisms of self-diffusion, which is quantitatively studied in the following paper. Relaxation of random positions yields dense and homogeneous amorphous structures with neither microcrystals nor holes. Defective crystals, depending on the annealing rate and potentials, yield either defective crystals again or completely amorphous structures, which in some cases contain holes or channels. read less USED (low confidence) B. Loisel, D. Gorse, V. Pontikis, and J. Lapujoulade, “Quasidynamic computation of multilayer relaxations, repulsion between steps and kink formation energy on copper vicinal surfaces,” Surface Science. 1989. link Times cited: 48 USED (low confidence) G. Tichy and U. Essmann, “Modelling of edge dislocation dipoles in face-centred-cubic lattices,” Philosophical Magazine Part B. 1989. link Times cited: 19 Abstract: Atomistic computer simulations have been carried out to inve… read moreAbstract: Atomistic computer simulations have been carried out to investigate edge dislocation dipole configurations in metals. A non-pair interatomic potential was used and the stress was kept constant as a boundary condition. Narrow edge dislocation dipoles of vacancy and interstitial type show significant differences. Several metastable configurations of vacancy-type dipoles were found. Our results are discussed with respect to the suggested annihilation of narrow edge dipoles in persistent slip bands of fatigued metals. read less USED (low confidence) C. Matthai, “Molecular Dynamics Simulation of Transition Metals as Silicon Substrates,” Molecular Simulation. 1989. link Times cited: 0 Abstract: In recent years the growth of epitaxial layers and strained … read moreAbstract: In recent years the growth of epitaxial layers and strained layer superlattices have been simulated by using the method of molecular dynamics. We have used this method to study the formation of transition metal silicides as the metal is deposited on a silicon substrate. We propose effective empirical potentials to describe the interaction between the metal and silicon and use the Dodson and Stillinger-Weber potentials to model the silicon-silicon interaction. We report on the evolution of nickel layers on the substrate as a function of temperature. read less USED (low confidence) S. Sinnott, S. Heo, D. Brenner, J. Harrison, and D. Irving, “Computer Simulations of Nanometer-Scale Indentation and Friction.” 2010. link Times cited: 18 USED (low confidence) R. Smith, S. Kenny, J. BelBruno, and R. Palmer, “Chapter 15 Modelling the structure and dynamics of metal nanoclusters deposited on graphite,” The Chemical Physics of Solid Surfaces. 2007. link Times cited: 7 USED (low confidence) R. Voskoboinikov, Y. Osetsky, and D. Bacon, “Statistics of primary damage creation in high-energy displacement cascades in copper and zirconium,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2006. link Times cited: 21 USED (low confidence) T. Fukuta, Y. Akahoshi, and S. Harada, “Interaction Between Copper Nano Cluster and Edge Dislocation Under Shear Strain,” Journal of Solid Mechanics and Materials Engineering. 2005. link Times cited: 0 Abstract: It is well known that nuclear pressure vessel steel shows em… read moreAbstract: It is well known that nuclear pressure vessel steel shows embrittlement under thermal aging and strong neutron irradiation. We focus on nanoscale copper-rich precipitates and try to clarify the effect of the nanoscale copper-rich precipitates on embrittlement of reactor pressure vessel steels. Our final goal is to evaluate such embrittlement from microscopic viewpoint based on atomistic simulation. In this study, we simulate interaction between motion of an edge dislocation and copper clusters using Molecular Dynamics method with a PBC, where uniform shear strain is applied to the boundaries parallel to the slip plane (1 1 2) in the system. As the results, we clarify the effects of size, distance and pinning of the copper clusters on dislocation motion. read less USED (low confidence) D. Rodney, “Atomic-scale modeling of clear band formation in FCC metals,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 30 USED (low confidence) T. Tsuru and Y. Shibutani, “Formation of Prismatic Dislocation Loop of Single Crystalline Aluminum under Nanoindentation.” 2004. link Times cited: 1 USED (low confidence) I. Marinescu, W. Rowe, B. Dimitrov, and I. Inasaki, “7 – Molecular Dynamics for Abrasive Process Simulation.” 2004. link Times cited: 0 USED (low confidence) D. Bacon and Y. Osetsky, “Atomic-scale modelling of dislocation behaviour under stress.” 2004. link Times cited: 0 USED (low confidence) T. Nozaki, Y. Kogure, and M. Doyama, “Plastic Deformation of Thin Films – Bending,” MRS Proceedings. 2001. link Times cited: 0 USED (low confidence) Y. Nakamura, Y. Kondo, J. Nakamura, and S. Watanabe, “Structural ordering on Si(111) $ \sqrt 3 \times \sqrt 3 $ -Ag surface: Monte Carlo simulation based on first-principles calculations.” 2001. link Times cited: 0 USED (low confidence) Y. Osetsky and D. Bacon, “Defect Cluster Formation in High Energy Displacement Cascades in Copper,” MRS Proceedings. 2000. link Times cited: 4 USED (low confidence) Y. Osetsky, D. Bacon, A. Serra, B. N. Singh, and S. Golubov, “Stability and mobility of defect clusters and dislocation loops in metals,” Journal of Nuclear Materials. 2000. link Times cited: 242 USED (low confidence) H. Alper and P. Politzer, “Molecular dynamics simulations of the temperature‐dependent behavior of aluminum, copper, and platinum,” International Journal of Quantum Chemistry. 2000. link Times cited: 17 Abstract: The authors have investigated a molecular dynamics procedure… read moreAbstract: The authors have investigated a molecular dynamics procedure for simulating the temperature-dependent behavior of three face-centered-cubic metals: aluminum, copper, and platinum. A potential due to Cai and Ye, which includes both pairwise additive and multibody terms, was used in conjunction with the CHARMM code. The properties calculated where the interaction energy per atom, the radial distribution function, the isothermal compressibility, and the heat capacities at constant pressure and volume. These properties were evaluated at 100{degree} intervals from 300 to 1,100 K for copper and platinum and from 300 to 800 K for aluminum, at pressures of 1 and 10,000 atm. Overall, the results were quite satisfactory. Except in one instance, the qualitative variations with temperature were reproduced reasonably well, and there was frequently also good quantitative agreement, especially at the lower temperatures. In general, the poorest results were obtained for aluminum. Some possible reasons for this are discussed. read less USED (low confidence) Y. Osetsky, D. Bacon, A. Serra, and B. N. Singh, “Interactions between Edge Dislocations and Interstitial Clusters in Iron and Copper,” MRS Proceedings. 2000. link Times cited: 4 Abstract: Dislocations decorated by both clusters of self-interstitial… read moreAbstract: Dislocations decorated by both clusters of self-interstitial atoms (SIAs) and small dislocation loops, are one of the microstructure features which can play an important role in post-irradiated deformation processes. The interactions between dislocations and clusters are important and are usually treated within the framework of isotropic elasticity theory. However, it is still not clear whether or not these interactions, especially for small clusters at short distances, can be treated accurately by elasticity theory. Comparative studies by atomistic simulation and elasticity theory can clarify this. Here we present a simple example of such a study where interactions between a glissile SIA cluster and an edge dislocation are studied in bcc-Fe and fcc-Cu using both techniques. In Fe we have studied the interaction of a dislocation with Burgers vector b = 1/2 lying along direction with a SIA cluster with the same b situated at different distances below the extra half-plane. In Cu, the dislocation and cluster had b = 1/2 and the dislocation line was along the direction. Interactions with clusters of diameter about 1 nm were simulated. Elastic calculations were made within the isotropic theory with parameters estimated from atomistic simulation. The results obtained by both techniques are discussed and some preliminary conclusions for different cases are drawn. read less USED (low confidence) R. Rentsch, “Atomistic Simulation and Experimental Investigation of Ultra Precision Cutting Processes,” MRS Proceedings. 1999. link Times cited: 6 Abstract: Typical applications for components and equipment with extre… read moreAbstract: Typical applications for components and equipment with extreme quality requirements regarding surface roughness, shape accuracy and integrity of the generated surface structure can be found in optical and semiconductor industry. Ultra precision machine tools equipped with sharp, single crystalline diamond provide the necessary machining accuracy. Here the actual cutting process can take place at atomic level, which makes the acquisition of typical cutting process data difficult or impossible. However a detailed characterization and understanding of the process is vital for its effective control as well as for further tool and process development. Therefore an approach is made that focuses on linking results from atomistic simulations with results and observations from cutting experiments. In this work the potential of molecular dynamics (MD) modeling for studying phenomena related to ultra precision cutting processes will be demonstrated. Observations and first results for machining copper will be presented. read less USED (low confidence) R. Rentsch and V. Vítek, “Modeling of the Dislocation Formation at Pores and Inclusions under Thermo-Mechanical Shear Loads,” MRS Proceedings. 1999. link Times cited: 3 USED (low confidence) Y. Osetsky, A. Serra, and V. Priego, “Mobility of Self-Interstitial Clusters in FE and CU,” MRS Proceedings. 1998. link Times cited: 21 USED (low confidence) B. Wirth and G. Odette, “Kinetic Lattice Monte Carlo Simulations of Cascade Aging in Iron and Dilute Iron-Copper Alloys,” MRS Proceedings. 1998. link Times cited: 13 USED (low confidence) Y. Osetsky, D. Bacon, and A. Serra, “Atomistic Simulation of Mobile Defect Clusters in Metals,” MRS Proceedings. 1998. link Times cited: 3 Abstract: The structure, stability and thermally-activated motion of i… read moreAbstract: The structure, stability and thermally-activated motion of interstitial and vacancy clusters in Fe and Cu have been studied using atomic scale computer simulation. All studied interstitial clusters and perfect interstitial loops (PILs) in Fe are mobile whereas their mobility in Cu can be suppressed at large sizes (bigger than 49--61 self-interstitials depending on the temperature) due to dissociation. A comparative study of relaxed configurations has shown that the structure of small perfect dislocation loops of vacancy and self-interstitial nature is very similar. Molecular dynamics simulation has demonstrated that small perfect vacancy loops (PVLs) in Fe consisting of more than 37 vacancies are stable over a wide temperature range and produce atomic displacements by a thermally-activated movement in the direction of the Burgers vector. The mechanism is qualitatively similar to that of SIA clusters studied earlier. Motion of vacancy loops in Cu does not occur because they transform into sessile configurations similar to stacking fault tetrahedra. These results point to the possibly important contribution of vacancy loop mobility to the difference in radiation damage between bcc and fcc metals, and between fcc metals with different stacking fault energy. read less USED (low confidence) Y. Shibutani, “Instability Analyses of Stress-Induced Phase Transformation.” 1998. link Times cited: 0 USED (low confidence) Y. Osetsky, A. Serra, V. Priego, F. Gao, and D. Bacon, “Mobility of Self-Interstitials in FCC and BCC Metals,” MRS Proceedings. 1998. link Times cited: 7 USED (low confidence) Y. Kogure and M. Doyama, “Characterization of Interatomic Potentials by a Calculation of Defect Energy,” MRS Proceedings. 1997. link Times cited: 0 USED (low confidence) B. Wirth and G. Odette, “Kinetic Lattice Monte Carlo Simulations of Diffusion and Decomposition Kinetics In Fe-Cu Alloys: Embedded Atom and Nearest Neighbor Potentials,” MRS Proceedings. 1997. link Times cited: 9 USED (low confidence) A. Aslanides, M. Hayoun, and V. Pontikis, “Atomic structure of low-index and (11n) surfaces in ordered Cu3Au,” Surface Science. 1997. link Times cited: 12 USED (low confidence) V. Vítek, A. Girshick, R. Siegl, H. Inui, and M. Yamaguchi, “Atomic Structure and Properties of Dislocations and Interfaces in Two-Phase TiAl Compounds.” 1997. link Times cited: 6 USED (low confidence) G. Ackland and U. Pinsook, “Evolution of Microstructure in the BCC-HCP Martensitic Phase Transition in Zirconium,” MRS Proceedings. 1997. link Times cited: 2 USED (low confidence) C. Fell, B. King, M. Petravić, and L. Wielunski, “Ion Beam Mixing of Isotopic Nickel Bilayers,” Ion Beam Modification of Materials. 1996. link Times cited: 0 USED (low confidence) V. Vítek, “Atomistic Studies Of The Structure Of Grain Boundaries and Dislocations.” 1996. link Times cited: 12 USED (low confidence) A. Calder and D. Bacon, “Computer Simulation Study of the Effects of Copper Solutes on Cascade Damage in Fe-Cu Alloys,” MRS Proceedings. 1996. link Times cited: 14 Abstract: The production of small, coherent BCC precipitates of copper… read moreAbstract: The production of small, coherent BCC precipitates of copper during fast neutron irradiation of ferritic steels and their apparent stability at peak hardening size are important phenomena governing in-service irradiation embrittlement. An MD model employing manybody interatomic potentials for the Fe-Cu alloy system has been developed and used to simulate the effect of displacement cascades on both dilute random solid solutions of copper and small coherent BCC copper precipitates in an oa-iron matrix. Within the timescales that can be modelled, cascades caused only small changes to the distribution of copper atoms in both the solid solution and the precipitate studies. Vacancies are observed to bind to copper solute atoms and also to accumulate in precipitates near the interface, where they are mobile. Interstitials are mobile in the solid solution matrix and two mechanisms of interstitial entrapment by precipitates are indicated. read less USED (low confidence) T. Harry and D. Bacon, “Computer Simulation Study of the Effects of Copper Precipitates on Dislocation Core Structure in Ferritic Steels,” MRS Proceedings. 1996. link Times cited: 1 USED (low confidence) Y. Shibutani, V. Vítek, and J. Bassani, “Local Atomic Level Elastic Moduli of Surface Regions.” 1995. link Times cited: 1 USED (low confidence) M. Yan, S. P. Chen, and V. Vítek, “Atomistic Studies of Grain Boundaries in NiAl,” MRS Proceedings. 1994. link Times cited: 0 Abstract: This paper presents the results of atomistic studies of grai… read moreAbstract: This paper presents the results of atomistic studies of grain boundaries in NiAl B2 alloy. The interatomic forces are described by Finnis-Sinclair type N-body potentials, and are fitted to properties of NiAl. The results show that the structure, energy and cohesive strength of a grain boundary depend strongly on its chemical configurations, and a grain boundary with surplus of Al is the weakest. Energies of antisite defects at the grain boundary {Sigma}5 (210) are also calculated, and the results suggest that Al has much larger tendency to segregate at a grain boundary than Ni does. read less USED (low confidence) G. Minchev, L. Pramatarova, B. Pödör, and J. Szabó, “Experimental confirmation of the peculiar behaviour of the coherent‐type twin boundaries in sphalerite crystals,” Crystal Research and Technology. 1994. link Times cited: 6 Abstract: This paper presents an experimental confirmation of the pecu… read moreAbstract: This paper presents an experimental confirmation of the peculiar behaviour of coherent twin boundaries in sphalerite crystals through a “two step” experimental procedure involving free abrasive polishing for the “developing” of the stress pattern and infrared whole-sample optical reflection (“magic mirror”) for the registration. The results are in accordance with the novel static atomistic simulations of the grain boundaries. read less USED (low confidence) R. Fonda, M. Yan, and D. Luzzi, “Solution of the Atomic Structure of the Σ=5 (310) [001] Grain Boundary in Nial by Hrem and Atomistic Simulations,” MRS Proceedings. 1994. link Times cited: 0 USED (low confidence) Y. Kogure, M. Doyama, and T. Kosugi, “Molecular dynamics simulation of dislocations in fee metals.” 1994. link Times cited: 2 USED (low confidence) Y. Zhu, J. Zuo, A. Moodenbaugh, and M. Suenaga, “Grain-boundary constraint and oxygen deficiency in YBa2Cu3O7-δ: Application of the coincidence site lattice model to a non-cubic system,” Philosophical Magazine. 1994. link Times cited: 33 Abstract: Local lattice parameters and oxygen (hole) densities in the … read moreAbstract: Local lattice parameters and oxygen (hole) densities in the vicinity of large-angle grain boundaries in bulk YBa2Cu3O7−δ were measured simultaneously using convergent-beay electron diffraction and electron-energy-loss spectroscopy with a less than 20 A field emission probe respectively. The oxygen (hole) contents were quantified by Gaussian fitting to the observed oxygen K-edge absorption spectra and comparing them with results for a set of standard samples with different oxygen contents. The local lattice parameters were determined by matching the simulated high-order Laue zone patterns with experimental patterns by a least-squares algorithm. We found that large-angle grain boundaries in YBa2Cu3O7−δ often exhibit volume expansion. The increase of the c-lattice parameter near the boundary core can be attributed to lattice distortion and/or to an oxygen deficiency. The lattice distortion extends less than 20 A, while the deficiency can extend more than 100 A. Comparison of the c/a ratio of the lat... read less USED (low confidence) F. Gao, D. Bacon, and G. Ackland, “Point-defect and threshold displacement energies in Ni3Al I. Point-defect properties,” Philosophical Magazine. 1993. link Times cited: 128 Abstract: The energy and volume change associated with the creation of… read moreAbstract: The energy and volume change associated with the creation of point defects in Ni3Al have been calculated by computer simulation using a modified version of the many-body potentials of Vitek, Ackland and Cserti. The potentials have been adjusted to provide a better description of the interaction between atoms at separations inside the normal nearest-neighbour spacing. This region is important for interstitial properties and the interaction of atoms in displacement events associated with radiation damage. The properties of point defects in the pure metals Al and Ni are in good agreement with known values. For Ni3Al, the properties of the Ni defects are very close to those found by Caro, Victoria and Averback with the embedded-atom potentials for Ni3Al of Foiles and Daw, and the most stable interstitial, in particular, is the Ni-Ni⟨100⟩ dumbbell centred on a Ni site in the {100} planes which contain only Ni atoms. For the Al and antisite defects, however, the model used here gives different results.... read less USED (low confidence) M. Breeman, G. Barkema, and D. Boerma, “ATOMISTIC CALCULATIONS ON THE SURFACTANT ROLE OF INDIUM IN THE HOMOEPITAXIAL GROWTH OF CU ON CU(100),” MRS Proceedings. 1993. link Times cited: 0 USED (low confidence) G. J. Wang, V. Vítek, I. Alber, and J. Bassani, “Phonons and Local Elastic Moduli in Symmetrical Tilt Boundaries,” MRS Proceedings. 1992. link Times cited: 1 USED (low confidence) J. Bassani, V. Vítek, and I. Alber, “Atomic-level elastic properties of interfaces and their relation to continua,” Acta Metallurgica Et Materialia. 1992. link Times cited: 30 USED (low confidence) H. Kitagawa and A. Nakatani, “MICROSTRUCTURAL ASPECTS OF CRACK EXTENSION IN A CRYSTALLINE MATERIAL: A MOLECULAR DYNAMIC STUDY.” 1992. link Times cited: 0 USED (low confidence) M. Yan, V. Vítek, and G. Ackland, “Atomic Structure and Chemical Composition of Grain Boundaries in L1 2 Intermetallic Compounds: Relation to Intergranular Brittleness.” 1992. link Times cited: 3 USED (low confidence) G. J. Wang and V. Vítek, “A Study of the Dependence of the Structure of Σ = 3 Tilt Boundaries in Gold on the Inclination of the Boundary Plane,” MRS Proceedings. 1992. link Times cited: 2 Abstract: The atomic structure of Σ = 3 tilt boundaries with variously… read moreAbstract: The atomic structure of Σ = 3 tilt boundaries with variously inclined boundary planes has been studied using the many body potentials for gold. A chain unit model, analogous to the structural unit model, describes the relationship between different boundaries. The basic units in this model are units of the (111) and (112) twin boundaries and the model corresponds to atomic level faceting into these boundaries. read less USED (low confidence) V. Vítek, “Structure of dislocation cores in metallic materials and its impact on their plastic behaviour,” Progress in Materials Science. 1992. link Times cited: 194 USED (low confidence) D. Bacon and A. Serra, “The Properties of Twinning Dislocations in Alpha-Titanium Simulated With A Many-Body Interatomic Potential,” MRS Proceedings. 1991. link Times cited: 3 USED (low confidence) B. J. Pestman, J. Hosson, V. Vítek, and F. Schapink, “Atomic Structure Calculations of the Interaction between Lattice Dislocations and Grain Boundaries,” MRS Proceedings. 1990. link Times cited: 0 Abstract: The interaction between screw dislocations and [1 1 0] symme… read moreAbstract: The interaction between screw dislocations and [1 1 0] symmetric tilt boundaries is investigated by atomistic calculations. In order to study the differences between fcc and ordered alloys and to study the effect of increasing ordering tendency, many-body potentials representing Cu, Cu 3 Au and Ni 3 Al were used. For the ordered alloys, the different possible ordering configurations of the boundaries that were studied are discussed. read less USED (low confidence) J. Boehm and R. Nieminen, “Dynamic Simulations of Partial Dislocation Core Structures in Gold Using Many-Body Interactions,” Physica Scripta. 1990. link Times cited: 0 Abstract: The Schockley partial dislocation core structures in f.c.c. … read moreAbstract: The Schockley partial dislocation core structures in f.c.c. gold are obtained by using recently suggested many-atom force laws in molecular dynamics simulations. read less USED (low confidence) M. Khantha, V. Vítek, and M. Goldman, “Favored Boundaries in the Structural Unit Model of Grain Boundaries,” MRS Proceedings. 1990. link Times cited: 1 USED (low confidence) B. J. Pestman, J. Hosson, V. Vítek, F. Tichelaar, and F. Schapink, “Interaction between Lattice Dislocations and Grain Boundaries in Ordered Compounds: Theory and Experiment,” MRS Proceedings. 1990. link Times cited: 1 Abstract: The interaction between a screw dislocation and symmetric [1… read moreAbstract: The interaction between a screw dislocation and symmetric [1 1 0] tilt boundaries under the influence of an applied shear stress was investigated by atomistic simulation. Many-body potentials representing Cu and Ni 3 Al were used for the description of the interatomic forces. A comparison will be made with in-situ observations in a transmission electron microscope of the interaction of screw dislocations with a coherent twin boundary. read less USED (low confidence) J. Boehm and R. Nieminen, “Dynamic Simulations of Dislocation Core Structures in Gold Using Many-Atom Interactions.” 1990. link Times cited: 0 USED (low confidence) V. Vítek, G. Ackland, and J. Cserti, “Atomistic Modeling of Extended Defects in Metalic Alloys: Dislocations and Grain Boundaries in Ll2 Compounds,” MRS Proceedings. 1990. link Times cited: 32 Abstract: Extended defects, such as dislocations and grain boundaries,… read moreAbstract: Extended defects, such as dislocations and grain boundaries, control a wide variety of material properties and their atomic structure is often a governing factor. A necessary precursor for modeling of these structures is a suitable description of atomic interactions. We present here empirical many-body potentials for alloys which represent a very simple scheme for the evaluation of total energies and yet reflect correctly the basic physical features of the alloy systems modeled. As examples of atomistic studies we show results of calculations of the core structures of screw dislocations in Li2 compounds. The same potentials have also been used to calculate structures of grain boundaries in these compounds. The deformation and fracture behavior of L12 alloys is then discussed in the light of grain boundary and dislocation core studies. read less USED (low confidence) J. Todd and J. Pethica, “Imaging by Sliding Planes in Scanning Tunnelling Microscopy,” MRS Proceedings. 1989. link Times cited: 0 USED (low confidence) G. J. Ackland and V. Vítek, “Effect of Ordering Energy on Grain Boundary Structure in L1 2 Alloys,” MRS Proceedings. 1988. link Times cited: 0 Abstract: Atomic structures of grain boundaries in two L1 2 ordered al… read moreAbstract: Atomic structures of grain boundaries in two L1 2 ordered alloys with very different ordering energies have been calculated. The interatomic forces are represented by many-body empirical potentials based on the second moment approximation to the tight-binding density of states. Grain boundaries in strongly ordered alloys show very little relaxation. This results in the presence of columns of cavities in the boundaries. On the other hand boundaries in weakly ordered alloys, and also in pure f.c.c. materials, show extensive relaxations leading to much more homogeneous structures. We suggest that these structural features may be responsible for the intrinsic brittleness of grain boundaries in compounds such as Ni 3 Al in contrast with boundaries in pure f.c.c. metals or ordered alloys such as Cu 3 Au which are only embrittled by segregation. read less NOT USED (low confidence) W. Yim et al., “Goldilocks Energy Minimum: Peptide-Based Reversible Aggregation and Biosensing.,” ACS applied materials & interfaces. 2023. link Times cited: 0 Abstract: Colorimetric biosensors based on gold nanoparticle (AuNP) ag… read moreAbstract: Colorimetric biosensors based on gold nanoparticle (AuNP) aggregation are often challenged by matrix interference in biofluids, poor specificity, and limited utility with clinical samples. Here, we propose a peptide-driven nanoscale disassembly approach, where AuNP aggregates induced by electrostatic attractions are dissociated in response to proteolytic cleavage. Initially, citrate-coated AuNPs were assembled via a short cationic peptide (RRK) and characterized by experiments and simulations. The dissociation peptides were then used to reversibly dissociate the AuNP aggregates as a function of target protease detection, i.e., main protease (Mpro), a biomarker for severe acute respiratory syndrome coronavirus 2. The dissociation propensity depends on peptide length, hydrophilicity, charge, and ligand architecture. Finally, our dissociation strategy provides a rapid and distinct optical signal through Mpro cleavage with a detection limit of 12.3 nM in saliva. Our dissociation peptide effectively dissociates plasmonic assemblies in diverse matrices including 100% human saliva, urine, plasma, and seawater, as well as other types of plasmonic nanoparticles such as silver. Our peptide-enabled dissociation platform provides a simple, matrix-insensitive, and versatile method for protease sensing. read less NOT USED (low confidence) A. Seko, “Tutorial: Systematic development of polynomial machine learning potentials for elemental and alloy systems,” Journal of Applied Physics. 2023. link Times cited: 0 Abstract: Machine learning potentials (MLPs) developed from extensive … read moreAbstract: Machine learning potentials (MLPs) developed from extensive datasets constructed from density functional theory calculations have become increasingly appealing to many researchers. This paper presents a framework of polynomial-based MLPs, called polynomial MLPs. The systematic development of accurate and computationally efficient polynomial MLPs for many elemental and binary alloy systems and their predictive powers for various properties are also demonstrated. Consequently, many polynomial MLPs are available in a repository website [A. Seko, Polynomial Machine Learning Potential Repository at Kyoto University, https://sekocha.github.io ]. The repository will help many scientists perform accurate and efficient large-scale atomistic simulations and crystal structure searches. read less NOT USED (low confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 NOT USED (low confidence) A. Hernandez and T. Mueller, “Generalizability of Functional Forms for Interatomic Potential Models Discovered by Symbolic Regression,” ArXiv. 2022. link Times cited: 0 Abstract: In recent years there has been great progress in the use of … read moreAbstract: In recent years there has been great progress in the use of machine learning algorithms to develop interatomic potential models. Machine-learned potential models are typically orders of magnitude faster than density functional theory but also orders of magnitude slower than physics-derived models such as the embedded atom method. In our previous work, we used symbolic regression to develop fast, accurate and transferrable interatomic potential models for copper with novel functional forms that resemble those of the embedded atom method. To determine the extent to which the success of these forms was specific to copper, here we explore the generalizability of these models to other face-centered cubic transition metals and analyze their out-of-sample performance on several material properties. We found that these forms work particularly well on elements that are chemically similar to copper. When compared to optimized Sutton-Chen models, which have similar complexity, the functional forms discovered using symbolic regression perform better across all elements considered except gold where they have a similar performance. They perform similarly to a moderately more complex embedded atom form on properties on which they were trained, and they are more accurate on average on other properties. We attribute this improved generalized accuracy to the relative simplicity of the models discovered using symbolic regression. The genetic programming models are found to outperform other models from the literature about 50% of the time in a variety of property predictions, with about 1/10th the model complexity on average. We discuss the implications of these results to the broader application of symbolic regression to the development of new potentials and highlight how models discovered for one element can be used to seed new searches for different elements. read less NOT USED (low confidence) Y. Wang et al., “Curvature-Selective Nanocrystal Surface Ligation Using Sterically-Encumbered Metal-Coordinating Ligands.,” ACS nano. 2022. link Times cited: 4 Abstract: Organic ligands are critical in determining the physiochemic… read moreAbstract: Organic ligands are critical in determining the physiochemical properties of inorganic nanocrystals. However, precise nanocrystal surface modification is extremely difficult to achieve. Most research focuses on finding ligands that fully passivate the nanocrystal surface, with an emphasis on the supramolecular structure generated by the ligand shell. Inspired by molecular metal-coordination complexes, we devised an approach based on ligand anchoring groups that are flanked by encumbering organic substituents and are chemoselective for binding to nanocrystal corner, edge, and facet sites. Through experiment and theory, we affirmed that the surface-ligand steric pressures generated by these organic substituents are significant enough to impede binding to regions of low nanocurvature, such as nanocrystal facets, and to promote binding to regions of high curvature such as nanocrystal edges. read less NOT USED (low confidence) T. Zeng, F. Li, and Y. Huang, “Construction of an n-Body Potential for Revealing the Atomic Mechanism for Direct Alloying of Immiscible Tungsten and Copper,” Materials. 2021. link Times cited: 7 Abstract: W-Cu laminated composites are critical materials used to con… read moreAbstract: W-Cu laminated composites are critical materials used to construct nuclear fusion reactors, and it is very important to obtain direct alloying between W and Cu at the W/Cu interfaces of the composites. Our previous experimental studies showed that it is possible to overcome the immiscibility between W and Cu and obtain direct alloying when the alloying temperature is close to the melting point of Cu. Because the W-Cu interatomic potentials published thus far cannot accurately reproduce the alloying behaviors of immiscible W and Cu, an interatomic potential suitable for the W-Cu system has been constructed in the present study. Based on this potential, direct alloying between W and Cu at high temperature has been verified, and the corresponding diffusion mechanism has been studied, through molecular dynamics (MD) simulations. The results indicate that the formation of an amorphous Cu layer at the W/Cu interface plays a critical role in alloying because it allows Cu atoms to diffuse into W. The simulation results for direct alloying between W and Cu can be verified by experimental results and transmission electron microscopy observations. This indicates that the constructed W-Cu potential can correctly model the high-temperature performance of the W-Cu system and the diffusion mechanism of direct alloying between W and Cu. read less NOT USED (low confidence) Z. Wu, R. Wang, L. Zhu, S. Pattamatta, and D. Srolov, “Revealing and Controlling the Core of Screw Dislocations in BCC Metals.” 2021. link Times cited: 0 Abstract:
Body-centred-cubic (BCC) transition metals (TMs) tend to b… read moreAbstract:
Body-centred-cubic (BCC) transition metals (TMs) tend to be brittle at low temperatures, posing significant challenges in their processing and major concerns for damage tolerance in critical load-carrying applications. The brittleness is largely dictated by the screw dislocation core structure; the nature and control of which has remained a puzzle for nearly a century. Here, we introduce a universal model and a physics-based material index χ that guides the manipulation of dislocation core structure in all pure BCC metals and alloys. We show that the core structure, commonly classified as degenerate (D) or non-degenerate (ND), is governed by the energy difference between BCC and face-centred cubic (FCC) structures and χ robustly captures this key quantity. For BCC TMs alloys, the core structure transition from ND to D occurs when χ drops below a threshold, as seen in atomistic simulations based on nearly all extant interatomic potentials and density functional theory (DFT) calculations of W-Re/Ta alloys. In binary W-TMs alloys, DFT calculations show that χ is related to the valence electron concentration at low to moderate solute concentrations, and can be controlled via alloying. χ can be quantitatively and efficiently predicted via rapid, low-cost DFT calculations for any BCC metal alloys, providing a robust, easily applied tool for the design of ductile and tough BCC alloys. read less NOT USED (low confidence) K. Hyodo, S. Munetoh, T. Tsuchiyama, and S. Takaki, “Empirical interatomic potential for Fe-N binary system based on Finnis–Sinclair potential,” Computational Materials Science. 2020. link Times cited: 7 NOT USED (low confidence) B. Yao and R. F. Zhang, “AADIS: An atomistic analyzer for dislocation character and distribution,” Comput. Phys. Commun. 2020. link Times cited: 19 NOT USED (low confidence) C. Yang and L. Qi, “Modified embedded-atom method potential of niobium for studies on mechanical properties,” Computational Materials Science. 2019. link Times cited: 17 NOT USED (low confidence) A. Fang and A. Chien, “ABFR,” Proceedings of the 27th International Symposium on High-Performance Parallel and Distributed Computing. 2018. link Times cited: 0 NOT USED (low confidence) J. Wang, S. Chen, K. Cui, D. Li, and D. Chen, “Approach and Coalescence of Gold Nanoparticles Driven by Surface Thermodynamic Fluctuations and Atomic Interaction Forces.,” ACS nano. 2016. link Times cited: 57 Abstract: The approach and coalescence behavior of gold nanoparticles … read moreAbstract: The approach and coalescence behavior of gold nanoparticles on a silicon surface were investigated by experiments and molecular dynamics simulations. By analyzing the behavior of the atoms in the nanoparticles in the simulations, it was found that the atoms in a single isolated nanoparticle randomly fluctuated and that the surface atoms showed greater fluctuation. The fluctuation increased as the temperature increased. When there were two or more neighboring nanoparticles, the fluctuating surface atoms of the nanoparticles "flowed" toward the neighboring nanoparticle because of atomic interaction forces between the nanoparticles. With the surface atoms "flowing", the gold nanoparticles approached and finally coalesced. The simulation results were in good agreement with the experimental results. It can be concluded that surface thermodynamic fluctuations and atomic interaction forces are the causes of the approach and coalescence behavior of the gold nanoparticles. read less NOT USED (low confidence) Z. Trautt, F. Tavazza, and C. Becker, “Facilitating the selection and creation of accurate interatomic potentials with robust tools and characterization,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 14 Abstract: The Materials Genome Initiative seeks to significantly decre… read moreAbstract: The Materials Genome Initiative seeks to significantly decrease the cost and time of development and integration of new materials. Within the domain of atomistic simulations, several roadblocks stand in the way of reaching this goal. While the NIST Interatomic Potentials Repository hosts numerous interatomic potentials (force fields), researchers cannot immediately determine the best choice(s) for their use case. Researchers developing new potentials, specifically those in restricted environments, lack a comprehensive portfolio of efficient tools capable of calculating and archiving the properties of their potentials. This paper elucidates one solution to these problems, which uses Python-based scripts that are suitable for rapid property evaluation and human knowledge transfer. Calculation results are visible on the repository website, which reduces the time required to select an interatomic potential for a specific use case. Furthermore, property evaluation scripts are being integrated with modern platforms to improve discoverability and access of materials property data. To demonstrate these scripts and features, we will discuss the automation of stacking fault energy calculations and their application to additional elements. While the calculation methodology was developed previously, we are using it here as a case study in simulation automation and property calculations. We demonstrate how the use of Python scripts allows for rapid calculation in a more easily managed way where the calculations can be modified, and the results presented in user-friendly and concise ways. Additionally, the methods can be incorporated into other efforts, such as openKIM. read less NOT USED (low confidence) Y.-xia Liu, H. Wang, H. Wu, D. Xu, and R. Yang, “A mean-field interatomic potential for a multi-component β-type titanium alloy,” Computational Materials Science. 2014. link Times cited: 2 NOT USED (low confidence) Q. Hou, M. Li, Y. Zhou, J. Cui, Z. Cui, and J.-ling Wang, “Molecular dynamics simulations with many-body potentials on multiple GPUs - The implementation, package and performance,” Comput. Phys. Commun. 2012. link Times cited: 48 NOT USED (low confidence) Y. Wang, Y. Liu, X. Wang, Y. Shan, Z. Zhai, and Z. Wang, “The Construction and Application of Interatomic Potential in FeAl Alloy by the Finnis-Sinclair Many-Body Potential Model,” Advanced Materials Research. 2012. link Times cited: 0 Abstract: The Finnis-Sinclair many-body potential model was utilized t… read moreAbstract: The Finnis-Sinclair many-body potential model was utilized to construct the interatomic potential function in B2 type FeAl alloy at equilibrium state. According to the model, the binding energy, the elastic constants C11, C12 and C44 were calculated, and the results agree well with the experimental results. The formation energy of different defects, such as monovacancy and anti-site, in the B2 FeAl alloy were also studied. read less NOT USED (low confidence) Z. Jian, N. Li, M. Zhu, J. Chen, F. Chang, and W. Jie, “Temperature dependence of the crystal–melt interfacial energy of metals,” Acta Materialia. 2012. link Times cited: 23 NOT USED (low confidence) B. Fu, W. Liu, and Z. Li, “Calculation of the surface energy of fcc-metals with the empirical electron surface model,” Applied Surface Science. 2010. link Times cited: 41 NOT USED (low confidence) O. H. Duparc, “On the origins of the Finnis–Sinclair potentials,” Philosophical Magazine. 2009. link Times cited: 10 Abstract: I trace back the origins of the famous Finnis-Sinclair poten… read moreAbstract: I trace back the origins of the famous Finnis-Sinclair potentials. These potentials mimic the results of tight binding theory through their use of the square root embedding function. From the tentative beginnings of tight binding in the 1930s up to 1984 or so, some of the famous names involved are Bloch, Seitz, Montroll, Friedel, Cyrot-Lackmann, Ducastelle, to name just a few. The application of the method of moments to the description of densities of states and its connexion to the physics of closed paths linking nearest neighbours interacting atoms helped to formalize Friedel's rectangular band model for the d electrons in transition metals. Extension from perfectly periodic structures to defective ones could not be but a slow process due to the change of paradigm for solid state scientists and to the necessary caution to be paid to self-consistency. The British scientists school also contributed significantly in the 80s. Computer progress and pragmatism helped to go from mainly analytical developments to numerical experiments (another change of paradigm). I also digress on various not so well known historical points of interest to this story. read less NOT USED (low confidence) B. Fu, W. Liu, and Z. Li, “Calculation of the surface energy of bcc-metals with the empirical electron theory,” Applied Surface Science. 2009. link Times cited: 140 NOT USED (low confidence) Y.-xia Liu, X. Wang, Y. Ma, C. Zhang, J.-xing Sun, and J.-xing Sun, “Austenite composition design at the atomic scale for the iron-based multi-component alloy as cast,” Computational Materials Science. 2008. link Times cited: 1 NOT USED (low confidence) R. D. Nyilas and R. Spolenak, “Orientation-dependent ductile-to-brittle transitions in nanostructured materials,” Acta Materialia. 2008. link Times cited: 4 NOT USED (low confidence) D. Belashchenko, O. Kuskov, and O. Ostrovski, “Application of the embedded-atom method to liquid Fe-S solutions,” Inorganic Materials. 2007. link Times cited: 7 NOT USED (low confidence) M. Aoki, D. Nguyen-Manh, D. Pettifor, and V. Vítek, “Atom-based bond-order potentials for modelling mechanical properties of metals,” Progress in Materials Science. 2007. link Times cited: 52 NOT USED (low confidence) J.-R. Hill, C. Freeman, and L. Subramanian, “Use of Force Fields in Materials Modeling.” 2007. link Times cited: 26 NOT USED (low confidence) M. Finnis and M. Rühle, “Structures of Interfaces in Crystalline Solids,” Materials Science and Technology. 2006. link Times cited: 3 Abstract: Interfaces in materials may be grain boundaries between like… read moreAbstract: Interfaces in materials may be grain boundaries between like crystals or phase boundaries between unlike crystals. Experimental approaches for the determination of the atomic structures of the interfaces are reviewed with emphasis on high-resolution electron microscopy (HREM). It will be shown that information on orientation relationship between the adjacent grains, the translation state and atomic relaxations can be elaborated with high precision. In a case study, the structures of one specific grain boundary in Al2O3 will be discussed in detail. Such experimental studies have provided a mass of structural information in recent years. read less NOT USED (low confidence) A. Caro, M. Caro, E. M. Lopasso, P. Turchi, and D. Farkas, “Thermodynamics of Fe–Cu alloys as described by a classic potential,” Journal of Nuclear Materials. 2006. link Times cited: 26 NOT USED (low confidence) J. Akhter, E. Ahmed, and M. Ahmad, “Study of diffusion coefficients in liquid noble metals,” Materials Chemistry and Physics. 2005. link Times cited: 22 NOT USED (low confidence) R. Smith, D. Mulliah, S. Kenny, E. Mcgee, A. Richter, and M. Gruner, “Stick slip and wear on metal surfaces,” Wear. 2005. link Times cited: 20 NOT USED (low confidence) A. Caro, P. Turchi, M. Caro, and E. M. Lopasso, “Thermodynamics of an empirical potential description of Fe-Cu alloys,” Journal of Nuclear Materials. 2005. link Times cited: 30 NOT USED (low confidence) H. Bonzel and M. Nowicki, “Absolute surface free energies of perfect low-index orientations of metals and semiconductors,” Physical Review B. 2004. link Times cited: 33 NOT USED (low confidence) W. Rowe et al., “Tribology of Abrasive Machining Processes.” 2004. link Times cited: 408 Abstract: This book draws upon the science of tribology to understand,… read moreAbstract: This book draws upon the science of tribology to understand, predict and improve abrasive machining processes. Pulling together information on how abrasives work, the authors, who are renowned experts in abrasive technology, demonstrate how tribology can be applied as a tool to improve abrasive machining processes. Each of the main elements of the abrasive machining system are looked at, and the tribological factors that control the efficiency and quality of the processes are described. Since grinding is by far the most commonly employed abrasive machining process, it is dealt with in particular detail. Solutions are posed to many of the most commonly experienced industrial problems, such as poor accuracy, poor surface quality, rapid wheel wear, vibrations, work-piece burn and high process costs. This practical approach makes this book an essential tool for practicing engineers. Uses the science of tribology to improve understanding and of abrasive machining processes in order to increase performance, productivity and surface quality of final products. A comprehensive reference on how abrasives work, covering kinematics, heat transfer, thermal stresses, molecular dynamics, fluids and the tribology of lubricants. Authoritative and ground-breaking in its first edition, the 2nd edition includes 30 per cent new and updated material, including new topics such as CMP (Chemical Mechanical Polishing) and precision machining for micro-and nano-scale applications. read less NOT USED (low confidence) M. A. Puigvi, Y. Osetsky, and A. Serra, “Interactions between Vacancy and Glissile Interstitial Clusters in Iron and Copper.,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2004. link Times cited: 14 NOT USED (low confidence) E. M. Lopasso, M. Caro, A. Caro, and P. Turchi, “Phase diagram of an empirical potential: The case of Fe-Cu,” Physical Review B. 2003. link Times cited: 55 Abstract: Molecular dynamics simulations are used to calculate the Gib… read moreAbstract: Molecular dynamics simulations are used to calculate the Gibbs free energy in the entire compositional range of Fe-Cu alloys described with a set of embedded atom potentials available in the literature. Thermodynamic integration and switching Hamiltonian techniques are used to obtain the phase diagram at high temperatures (neglecting phonon quantum effects and electronic contributions) with no further approximations. Limitations of the model were confirmed, such as the absence of the y and δ phases, a bcc to fcc transformation before melting for pure Fe, the unexpected existence of a stable bcc phase in pure Cu at high T, and consequently complete solid solubility of Fe in Cu in the bcc phase in some temperature range. This work seeds light on the power and limitations of the empirical description of complex systems. read less NOT USED (low confidence) H. Rafii-Tabar and A. Chirazi, “Multi-scale computational modelling of solidification phenomena,” Physics Reports. 2002. link Times cited: 39 NOT USED (low confidence) H. Feraoun, H. Aourag, T. Grosdidier, D. Klein, and C. Coddet, “Development of modified embedded atom potentials for the Cu–Ag system,” Superlattices and Microstructures. 2001. link Times cited: 10 Abstract: The modified embedded atom method is tested in the atomistic… read moreAbstract: The modified embedded atom method is tested in the atomistic simulations of binary fcc metallic alloys. As an example the alloying behaviour of Cu–Ag is studied using the molecular dynamics (MD) method. The MD algorithms that we use are based on the extended Hamiltonian formalism and the ordinary experimental conditions are simulated using the constant-pressure, constant temperature (NPT) (MD) method. The enthalpy of mixing values of the random Ag–Cu binary alloys are obtained as functions of concentration after 20 000 steps. read less NOT USED (low confidence) J. Hosson and B. Kooi, “Metal/ceramic interfaces: a microscopic analysis,” Surface and Interface Analysis. 2001. link Times cited: 102 Abstract: This contribution treats the fundamental and practical aspec… read moreAbstract: This contribution treats the fundamental and practical aspects of the analysis of interfaces between metals and ceramic materials. It is shown that, in principle at least, it is possible to assess the bond strength by investigating the atomic structure of the dislocation cores. We have chosen to exemplify our approach with the misfit‐dislocation structures at two, crystallographically different, systems, i.e. cube‐on‐cube and cube‐on‐non cube interfaces. It turns out that from high‐resolution transmission electron microscopy observations in combination with computer modelling studies a qualitative insight can be obtained about the bonding behaviour between these dissimilar materials. In general we may conclude that there is a fair correlation between the atomistic and linear elastic continuum description of interface dislocations. However, it is shown that the linear elastic continuum approach cannot account for the possible configurations at an interface with misfit, because it does not include the effects of different bonding strengths on the interface structure. This contribution directs also to some future experimental work. Segregation of other elements might affect the local bond strength which may become manifest in the experimental observations of atomic structure. Copyright © 2001 John Wiley & Sons, Ltd. read less NOT USED (low confidence) C. Sanz-Navarro and R. Smith, “Numerical implementation of the hyper-molecular dynamics method with examples applied to diffusion,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 2 NOT USED (low confidence) Y. Osetsky, D. Bacon, C. Matthai, and N. H. March, “Cleavage force, tribology and bond breaking in some transition metals,” Journal of Physics and Chemistry of Solids. 2000. link Times cited: 1 NOT USED (low confidence) R. Laskowski, “Double Lattice Inversion Technique — Application to the EAM Potential Construction,” Physica Status Solidi B-basic Solid State Physics. 2000. link Times cited: 3 Abstract: The embedded atom model (EAM) has been proved to be a very e… read moreAbstract: The embedded atom model (EAM) has been proved to be a very effective and powerful approach to energy calculation in metals. In the current work, we present a new approach to the construction of the EAM potential. The EAM functions are determined in a way to reproduce exactly two state equations defined for unit cells of two different metal structures. Our method is in some sense an extension of the lattice inversion algorithm proposed by Carlsson et al. (Phil Mag. A 41, 241 (1980)), where the resulting pair potential reconstructs only one given state equation. The inversion technique has been applied before to the construction of a pair potential and embedded atom potential from a single state equation. Using two input state equations we can determine only two functions, thus, we construct the pair potential and embedding function. The atomic charge density is taken from first-principle calculation. As an example, we apply our method to the determination of the EAM potential for solid lead. In order to estimate the quality of this potential, some bulk and defect properties are also calculated. read less NOT USED (low confidence) G. Bhuiyan, A. Rahman, M. Khaleque, R. Rashid, S. Rahman, and S. Rahman, “Liquid state structure of some noble and transition metals,” Journal of Non-crystalline Solids. 1999. link Times cited: 10 NOT USED (low confidence) A. Landa, P. Wynblatt, A. Girshick, V. Vítek, A. Ruban, and H. Skriver, “Development of finnis-sinclair type potentials for the Pb-Bi-Ni system-II. Application to surface Co-segregation,” Acta Materialia. 1999. link Times cited: 8 NOT USED (low confidence) M. Doyama and Y. Kogure, “Embedded atom potentials in fcc and bcc metals,” Computational Materials Science. 1999. link Times cited: 73 NOT USED (low confidence) Y. Kraftmakher, “EQUILIBRIUM VACANCIES AND THERMOPHYSICAL PROPERTIES OF METALS,” Physics Reports. 1998. link Times cited: 197 NOT USED (low confidence) M. Duesbery and V. Vítek, “Plastic anisotropy in b.c.c. transition metals,” Acta Materialia. 1998. link Times cited: 398 NOT USED (low confidence) Y. Shibutani, V. Vítek, and J. Bassani, “Nonlocal properties of inhomogeneous structures by linking approach of generalized continuum to atomistic model,” International Journal of Mechanical Sciences. 1998. link Times cited: 17 NOT USED (low confidence) M. Doyama and Y. Kogure, “Embedded atom potentials in fcc metals,” Radiation Effects and Defects in Solids. 1997. link Times cited: 24 Abstract: A new embedded atom potential has been proposed in this pape… read moreAbstract: A new embedded atom potential has been proposed in this paper. The potential is expressed by simple functions and is applicable to the molecular dynamics simulations of large atomic systems. The potential parameters are determined from the experimental data using the cohesive energy, Born stability, elastic constants, C 11 C 12 and C 44, the formation energy of a vacancy and the stacking fault energy. The potential functions for copper, silver and gold are presented. read less NOT USED (low confidence) M. Yan, V. Vítek, and S. P. Chen, “Many-body central force potentials and properties of grain boundaries in NiAl,” Acta Materialia. 1996. link Times cited: 41 NOT USED (low confidence) Y. Zhang, T.-C. Wang, and Q. Tang, “On the unstable stacking criterion for ideal and cracked copper-crystals,” Philosophical Magazine. 1995. link Times cited: 2 Abstract: The unstable stacking criteria for an ideal copper crystal u… read moreAbstract: The unstable stacking criteria for an ideal copper crystal under homogeneous shearing and for a cracked copper crystal under pure mode II loading are analysed. For the ideal crystal under homogeneous shearing, the unstable stacking energy γ us defined by Rice in 1992 results from shear with no relaxation in the direction normal to the slip plane. For the relaxed shear configuration, the critical condition for unstable stacking does not correspond to the relative displacement Δ = bp /2 where bp is the Burgers vector magnitude of the Shockley partial dislocation, but to the maximum shear stress. Based on this result, the unstable stacking energy y us is defined for the relaxed lattice. For the cracked crystal under pure mode Ii loading, the dislocation configuration corresponding to Δ = bp /2 is a stable state and no instability occurs during the process of dislocation nucleation. The instability takes place at approximately Δ = 3bp /4. An unstable stacking energy y us is defined which corresponds ... read less NOT USED (low confidence) R. Najafabadi and D. Srolovitz, “Interatomic potentials for elastically isotropic crystals,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 0 Abstract: We introduce a simple approach to fitting interatomic potent… read moreAbstract: We introduce a simple approach to fitting interatomic potentials to represent elastically isotropic solids, in order to make direct comparisons with the predictions of isotropic linear elasticity. The potentials are of the Finnis-Sinclair type and are parametrized to fit Au and Ag. Under uniaxial loading, it was shown that the solids modelled employing the 'elastically isotropic' potentials remain elastically isotropic for strains of 0.5% or less. The properties of the (001), (011) and (111) surfaces were determined and compared with those of elastically anisotropic Au and Ag. It was also found that the surface energy anisotropy is lower in solids modelled employing 'elastically isotropic' potentials than in those represented by elastically anisotropic potentials. read less NOT USED (low confidence) A. Goldstein and H. Jónsson, “An embedded atom method potential for the h.c.p. metal Zr,” Philosophical Magazine Part B. 1995. link Times cited: 10 Abstract: The embedded atom method is extended to the h.c.p. metal Zr.… read moreAbstract: The embedded atom method is extended to the h.c.p. metal Zr. The non-ideal c: α ratio and the elastic responses, including contributions from internal degrees of freedom, are incorporated in the fitting procedure: Simple functional forms are assumed for the pair interaction, atomic electron density and embedding function. The functions are parametrized by fitting to experimental data: cohesive energy, equilibrium lattice constants, single crystal elastic constants and vacancy formation energy. An equation of state of the form proposed by Rose, Smith, Guinea and Ferrante is used to reproduce the pressure dependence of the cohesive energy, taking into account the anisotropic elastic response of the crystal. Dimer data and a high energy sputtering potential are also reproduced to extend the range of validity of the potential into regions of very high and low electron density. Good agreement is obtained between the experimental and calculated properties. The potential is applied to the calculation of... read less NOT USED (low confidence) E. Kuramoto and T. Tsutsumi, “Computer simulation of the bias factor in void swelling in metals,” Journal of Nuclear Materials. 1994. link Times cited: 14 NOT USED (low confidence) S. Romanowski and J. Gomes, “Monte Carlo studies of the chemisorption and work function temperature effects on noble metals,” Journal of Electroanalytical Chemistry. 1994. link Times cited: 7 NOT USED (low confidence) H. Gades and H. Urbassek, “Surface binding energies of alloys: a many-body approach,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1994. link Times cited: 27 NOT USED (low confidence) C. L. Kelchner, D. Halstead, L. S. Perkins, N. M. Wallace, and A. Depristo, “Construction and evaluation of embedding functions,” Surface Science. 1994. link Times cited: 88 NOT USED (low confidence) M. Breeman and D. Boerma, “ATOMIC MOBILITIES ON A STEPPED CU(100) SURFACE,” Surface Science. 1993. link Times cited: 9 NOT USED (low confidence) G. Ackland, “Theoretical study of titanium surfaces and defects with a new many-body potential,” Philosophical Magazine. 1992. link Times cited: 150 Abstract: It is shown that any force model using short-range pair-func… read moreAbstract: It is shown that any force model using short-range pair-functional interactions can only have three independent h.c.p. elastic constants. Empirical data show that these elastic properties are nearly realized in a number of materials. A new parametrization of a Finnis-Sinclair-type many-body potential for titanium is presented using these relations. Particular care is taken to describe the anisotropy of the shear constants and the deviation of the c/a lattice parameter ratio from ideal, while maintaining smooth monotonic functions. Energies, stresses and reconstruction modes of various low-index surfaces are calculated and general rules for surface stability are proposed. Various stacking faults on the basal and pyramidal plane are investigated. read less NOT USED (low confidence) E. Kuramoto, “Computer simulation of fundamental features of a bias factor,” Journal of Nuclear Materials. 1992. link Times cited: 11 NOT USED (low confidence) H. Gades and H. Urbassek, “Pair versus many-body potentials in atomic emission processes from a Cu surface,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1992. link Times cited: 57 NOT USED (low confidence) V. Vítek and S. P. Chen, “Modeling of grain boundary structures and properties in intermetallic compounds,” Scripta Metallurgica Et Materialia. 1991. link Times cited: 43 NOT USED (low confidence) H. Häkkinen, J. Merikoski, and M. Manninen, “Surface reconstruction and many-atom models,” Journal of Physics: Condensed Matter. 1991. link Times cited: 18 Abstract: The (110)(1*2) missing-row reconstruction of the seven FCC m… read moreAbstract: The (110)(1*2) missing-row reconstruction of the seven FCC metals Ni, Pd, Pt, Cu, Ag, Au and Al has been studied using the effective medium theory (EMT). A clear trend in the tendency to reconstruct has been observed when going from the 3d metals Ni and Cu to 5d metals Pt and Au. The results are discussed together with some previous calculations using other many-atom models for total energy calculation in metals. The tendency to undergo reconstruction is found to be related to the anisotropy of surface energies on (111) and (110) surfaces. By investigating the effective two-body and three-body interactions on the surface it is shown that the missing-row reconstruction is related to the effective repulsion between adjacent nearest neighbour rows on the unreconstructed (110) surface. Restriction of the atomic interactions to the nearest neighbours only makes all the recent many-atom models favour the missing-row structure. read less NOT USED (low confidence) H. Rafii-Tabar and A. Sulton, “Long-range Finnis-Sinclair potentials for f.c.c. metallic alloys,” Philosophical Magazine Letters. 1991. link Times cited: 256 Abstract: Many-body, long-range potentials of a Finnis-Sinclair type a… read moreAbstract: Many-body, long-range potentials of a Finnis-Sinclair type are formulated for the atomistic description of binary f.c.c. metallic alloys. These potentials are generalizations of the scheme constructed by Sutton and Chen (1990), for the atomistic modelling of f.c.c. elemental metals. The parameters for the alloy potentials are obtained directly from the parameters for the elemental metals, without any further adjustable fitting. Lattice parameters, elastic constants and enthalpy of formation of 45 binary, random f.c.c. alloys are calculated. read less NOT USED (low confidence) M. Igarashi, M. Khantha, and V. Vítek, “N-body interatomic potentials for hexagonal close-packed metals,” Philosophical Magazine Part B. 1991. link Times cited: 123 Abstract: Finnis-Sinclair (F-S) type many-body potentials have been co… read moreAbstract: Finnis-Sinclair (F-S) type many-body potentials have been constructed for eight hexagonal metals: Co, Zr, Ti, Ru, Hf, Zn, Mg and Be. The potentials are parameterized using cubic splines and fitted to the cohesive energy, unrelaxed vacancy formation energy, five independent second-order elastic constants and two eauilibrium conditions. Hence, each of the constructed potentials represents a stable hexagonal close-packed lattice with a particular non-ideal c/a ratio. In the F-S scheme the many-body part is represented by a sauare root function and this form implies that C12 – C66>0. However, C 12 –C66 is negative for Zn, Be and Ru at Iow temperatures. For this reason a modified many-body function has been employed for these metals. To ensure the applicability of the potentials in modelling of extended lattice defects, the mechanical stability of the corresponding hexagonal close-packed lattice with respect to large homogeneous deformations has been tested. For all the metals considered, the h.c.p. l... read less NOT USED (low confidence) A. Sutton and J. Chen, “Long-range Finnis–Sinclair potentials,” Philosophical Magazine Letters. 1990. link Times cited: 1044 Abstract: Finnis–Sinclair potentials are developed for computer simula… read moreAbstract: Finnis–Sinclair potentials are developed for computer simulations in which van der Waals type interactions between well separated atomic clusters are as important as the description of metallic bonding at short range. The potentials always favour f.c.c. and h.c.p. structures over the b.c.c. structure. They display convenient scaling properties for both length and energy, and a number of properties of the perfect crystal may be derived analytically. read less NOT USED (low confidence) H. Häkkinen and M. Manninen, “The effective-medium theory beyond the nearest-neighbour interaction,” Journal of Physics: Condensed Matter. 1989. link Times cited: 19 Abstract: The effective-medium theory has been extended to include mor… read moreAbstract: The effective-medium theory has been extended to include more than the nearest neighbours in the total energy calculation of a metallic system. This extension has been tested by molecular dynamics simulations of thermal expansion and melting of copper. The calculated thermal expansion coefficient, melting point, latent heat of fusion and diffusion constant of the liquid phase are in good agreement with experimental data. In the liquid phase it is necessary to go beyond the nearest-neighbour interactions. read less NOT USED (low confidence) R. Johnson and D. J. Oh, “Analytic embedded atom method model for bcc metals,” Journal of Materials Research. 1989. link Times cited: 448 Abstract: The requirements for fitting bcc metals within the EAM forma… read moreAbstract: The requirements for fitting bcc metals within the EAM format are discussed and, for comparative purposes, the EAM format is cast in a normalized form. A general embedding function is defined and an analytic first- and second-neighbor model is presented. The parameters in the model are determined from the cohesive energy, the equilibrium lattice constant, the three elastic constants, and the unrelaxed vacancy formation energy. Increasing the elastic constants, increasing the elastic anisotropy ratio, and decreasing the unrelaxed vacancy formation energy favor stability of a close-packed lattice over bcc. A stable bcc lattice relative to close packing is found for nine bcc metals, but this scheme cannot generate a model for Cr because the elastic constants of Cr require a negative curvature of the embedding function. read less NOT USED (low confidence) V. Vítek, “Interatomic forces in relation to the theory of dislocations,” Philosophical Magazine. 1988. link Times cited: 23 Abstract: The early developments of the dislocation theory were mainly… read moreAbstract: The early developments of the dislocation theory were mainly in the framework of the continuum theory of elasticity. However, the importance of understanding the dislocation cores became apparent as investigations turned away from materials with close-packed structures to materials with more complex crystal structures. A necessary precursor for theoretical studies of the atomic structure of dislocations and other lattice defects is an understanding of atomic interactions. Until recently pair potentials were used almost exclusively in such studies. In this paper we first discuss the origin, merits and limitations of pair potentials. In particular, we analyse which features of pair potentials, as well as of the problems studied, are most important for the success of such atomistic studies. This is then demonstrated on two examples: the core structures of screw dislocations in intermetallic compounds with the Ll2 crystal structure and the cores of intrinsic grain-boundary dislocations. Finally we di... read less NOT USED (low confidence) M. Finnis, A. Paxton, D. Pettifor, A. Sutton, and Y. Ohta, “Interatomic forces in transition metals,” Philosophical Magazine. 1988. link Times cited: 45 Abstract: This article reviews some recent progress in the derivation … read moreAbstract: This article reviews some recent progress in the derivation of physical models for calculating the behaviour of defects in transition metals by atomistic simulation. It has long been recognised that it is necessary to go beyond the assumption of pair-wise interactions, but this has only recently been achieved with the advent of simplified models which take account of the electron gas. For d-band metals, the tight-binding model is a useful picture and it can be applied at various levels of approximation. The simplest of these is the second-moment approximation, which is very rapid to compute. Other schemes, such as the embedded-atom method, are also very practical for large scale simulations. These schemes will be described and compared in terms of their physical basis. read less NOT USED (low confidence) D. J. Oh and R. Johnson, “Simple embedded atom method model for fcc and hcp metals,” Journal of Materials Research. 1988. link Times cited: 349 Abstract: A procedure based on the embedded atom method (EAM) is prese… read moreAbstract: A procedure based on the embedded atom method (EAM) is presented for developing atomistic models for use in computer simulation calculations, with an emphasis on simple but general schemes for matching experimental data with fitting parameters. Both the electron density function and the two-body potential are taken as exponentially decreasing functions and the model is derived for any choice of cutoff distance. The model has been applied successfully to seven fcc and three hcp metals, but the extension to bcc metals was unsuccessful because of difficulty in matching the shear anisotropy ratio. read less NOT USED (low confidence) W. Gräfe, “Miscellaneous and Open Questions.” 2015. link Times cited: 0 NOT USED (low confidence) G. Ackland, “1.10 – Interatomic Potential Development.” 2012. link Times cited: 10 NOT USED (low confidence) H. Ogawa and P. Bac, “Classical MD Simulation of Hydrogen Absorption in F.C.C. and B.C.C. Nanoparticles,” MRS Proceedings. 2011. link Times cited: 0 Abstract: Hydrogen absorption in metallic nanoparticles was investigat… read moreAbstract: Hydrogen absorption in metallic nanoparticles was investigated by classical molecular dynamics (MD) simulation. We used a simple model composed of an isolated f.c.c. or b.c.c. nanoparticle of 1, 1.4, 2, 4, 6, 8 and 10 nm in diameter and surrounding hydrogen atoms. The simulated particle sizes are which correspond to about 50 to 44000 atoms. In the case of f.c.c. nanoparticles, atomic configuration with five-fold symmetries was observed in both hydrogenfree and hydrogenated particles smaller than 2 nm. The f.c.c. structure was maintained in larger particles than 4 nm with lattice deformation which varies with M-H interaction. The b.c.t. structure was observed in hydrogenated b.c.c. nanoparticles. Number of H atoms absorbed in a nanoparticle varies depending on particle size and M-H interaction: it increases with increasing particle size and M-H bond strength. read less NOT USED (low confidence) P. Davim and M. Jackson, “Nano and Micromachining.” 2009. link Times cited: 46 Abstract: Preface. Chapter 1. Nanoscale Cutting ( Rudiger RENTSCH ). 1… read moreAbstract: Preface. Chapter 1. Nanoscale Cutting ( Rudiger RENTSCH ). 1.1. Introduction. 1.2. Basic elements of molecular dynamics modeling. 1.3. Design and requirements for state-of-the-art MD cutting process simulations. 1.4. Capabilities of MD for nanoscale material removal process analysis. 1.5. Advances and recent developments in material removal process simulation. 1.6. Summary and outlook. 1.7. References. Chapter 2. Ductile Mode Cutting of Brittle Materials: Mechanism, Chip Formation and Machined Surfaces ( Xiaoping LI ). 2.1. Introduction. 2.2. The mechanism of ductile mode cutting of brittle materials. 2.3. The chip formation in cutting of brittle materials. 2.4. Machined surfaces in relation to chip formation mode. 2.5. References. Chapter 3. Diamond Tools in Micromachining ( Waqar AHMED, Mark J. JACKSON and Michael D. WHITFIELD ). 3.1. Introduction. 3.2. Diamond technology. 3.3. Preparation of substrate. 3.4. Modified HFCVD process. 3.5. Nucleation and diamond growth. 3.6. Deposition on complex substrates. 3.7. Diamond micromachining. 3.8. Conclusions. 3.9. References. Chapter 4. Conventional Processes: Microturning, Microdrilling and Micromilling ( Wit GRZESIK ). 4.1. Introduction. 4.2. Microturning. 4.3. Microdrilling. 4.4. Micromilling. 4.5. Product quality in micromachining. 4.6. References. Chapter 5. Microgrinding and Ultra-precision Processes ( Mark J. JACKSON and Michael D. WHITFIELD ). 5.1. Introduction. 5.2. Micro and nanogrinding. 5.3. Nanogrinding tools. 5.4. Conclusions. 5.5. References. Chapter 6. Non-Conventional Processes: Laser Micromachining ( Grant M. ROBINSON and Mark J. JACKSON ). 6.1. Introduction. 6.2. Fundamentals of lasers. 6.3. Laser microfabrication. 6.4. Laser nanofabrication. 6.5. Conclusions. 6.6. References. Chapter 7.Evaluation of Subsurface Damage in Nano and Micromachining ( Jianmei ZHANG, Jiangang SUN and Zhijian PEI ). 7.1. Introduction. 7.2. Destructive evaluation technologies. 7.3. Non-destructive evaluation technologies. 7.4. Acknowledgements. 7.5. References. Chapter 8. Applications of Nano and Micromachining in Industry ( Jiwang YAN ). 8.1. Introduction. 8.2. Typical machining methods. 8.3. Applications in optical manufacturing. 8.4. Semiconductor and electronics related applications. 8.5. Summary. 8.6. Acknowledgements. 8.7. References. List of Authors. Index. read less NOT USED (low confidence) D. Bacon, Y. Osetsky, and D. Rodney, “Chapter 88 Dislocation–Obstacle Interactions at the Atomic Level.” 2009. link Times cited: 114 NOT USED (low confidence) J. Douin, P. Castany, F. Pettinari-Sturmel, and A. Coujou, “Direct measurement of the variation in the energy of a dislocation locked in specific orientations,” Acta Materialia. 2009. link Times cited: 4 NOT USED (low confidence) J. Li, X. Dai, S. Liang, K. Tai, Y. Kong, and B. Liu, “Interatomic potentials of the binary transition metal systems and some applications in materials physics,” Physics Reports. 2008. link Times cited: 110 NOT USED (low confidence) R. Sahara, H. Mizuseki, K. Ohno, and Y. Kawazoe, “Thermodynamic Properties of Materials Using Lattice-Gas Models with Renormalized Potentials.” 2008. link Times cited: 2 NOT USED (low confidence) R. Dingreville and J. Qu, “A semi-analytical method to compute surface elastic properties,” Acta Materialia. 2007. link Times cited: 97 NOT USED (low confidence) W. Mook et al., “Mesoscopic Length Scales for Deformed Nanostructures.” 2006. link Times cited: 0 NOT USED (low confidence) D. Sander and H. Ibach, “References for 4.4.” 2002. link Times cited: 3 NOT USED (low confidence) L. Pan, D. Metzger, and M. Niewczas, “The Meshless Dynamic Relaxation Techniques for Simulating Atomic Structures of Materials.” 2002. link Times cited: 9 Abstract: Traditionally, Molecular Dynamics combined with pair potenti… read moreAbstract: Traditionally, Molecular Dynamics combined with pair potential functions or the Embedded Atom Method (EAM) is applied to simulate the motion of atoms. When a defect is generated in the crystalline lattice, the equilibrium of atoms around it is destroyed. The atoms move to find a new place where the potential energy in the system is minimum, which could result in a change of the local atomic structure. The present paper introduces new Dynamic Relaxation algorithm, which is based on explicit Finite Element Analysis, and pair or EAM potential function, to find equilibrium positions of the block of atoms containing different structural defects. The internal force and stiffness at the atoms (nodes) are obtained by the first and second derivatives of the potential energy functions. The convergence criterion is based on the Euclidean norm of internal force being close to zero when the potential energy is minimum. The damping ratio affects the solution path so that different damping ratios could lead to different minimum potential energy and equilibrium shapes. The numerical responses and results by applying free boundary conditions and certain periodic boundary conditions are presented. The choice of scaled mass of atoms, proper time step and damping appropriate for the efficient and stable simulation is studied.Copyright © 2002 by ASME read less NOT USED (low confidence) D. Sander and H. Ibach, “4.4.7 Clean surfaces - 4.4.8 Adsorbate-induced surface stress on Pt(111).” 2002. link Times cited: 0 NOT USED (low confidence) D. Sander and H. Ibach, “4.4.4 Experimental determination of changes of surface stress due to adsorption - 4.4.5 Calculations of surface free energy and surface stress.” 2002. link Times cited: 5 NOT USED (low confidence) J. Hosson and B. Kooi, “Microstructure and properties of interfaces between dissimilar materials.” 2001. link Times cited: 10 NOT USED (low confidence) S. Znám, D. Nguyen-Manh, D. Pettifor, and V. Vítek, “Bond-Order Potentials for Transition Metals Based Binary Alloys: Ti-Al and Mo-Si Alloys.” 2000. link Times cited: 3 NOT USED (low confidence) M. Ortiz and R. Phillips, “Nanomechanics of Defects in Solids,” Advances in Applied Mechanics. 1998. link Times cited: 143 NOT USED (low confidence) A. Stoneham, C. English, and W. Phythian, “Challenges in radiation damage : From dynamics of atoms to nuclear plant life management,” Radiation Effects and Defects in Solids. 1998. link Times cited: 8 Abstract: The major challenges in nuclear technology relate to the saf… read moreAbstract: The major challenges in nuclear technology relate to the safe, efficient and economical operation over period of the order of 109 seconds (30 years). Associated with these aims are a need for minimal interruptions for maintenance and, if possible, a sound scientific basis for extending the plant life. The scientific basis requires understanding on very different timescales: certain aspects of the collision cascades (10−15 seconds) and, between these limits of atomic dynamics and of engineering, the diffusion-controlled processes which determine microstructure. We show how studies over a wide range of length and timescales can be linked to provide a basis for successful technology. In particular, we discuss the mechanisms underlying radiation induced embrittlement in reactor pressure vessels. We discuss the characteristic times of the various processes (from femtoseconds to years) and the theoretical and experimental tools available for their study. We show how the scientist can reach out to the e... read less NOT USED (low confidence) J. D. Hosson, W. Vellinga, X. Zhou, and V. Vítek, “Structure-property relationship of metal-ceramic interfaces.” 1996. link Times cited: 9 NOT USED (low confidence) Y. Shimomura, K. Sugio, T. D. Rubia, and M. Guinan, “Small point defect clusters in copper.” 1994. link Times cited: 0 NOT USED (low confidence) Y. Kogure, Y. Abe, O. Kouchi, and M. Doyama, “Determination of parameters in n-body interaction potentials by embedded function.” 1994. link Times cited: 0 NOT USED (low confidence) F. Gao and D. Bacon, “On the structure and energy of dissociated dislocations in F.C.C. metals,” Philosophical Magazine. 1992. link Times cited: 13 Abstract: Atomic-scale computer simulations using many-body interatomi… read moreAbstract: Atomic-scale computer simulations using many-body interatomic potentials have been carried out to investigate the energy changes and equilibrium configurations associated with the dissociation of perfect and Lomer dislocations in copper and silver. The state of minimum energy is close in both cases to that given by the use of linear elasticity theory, and the Lomer dislocation adopts the asymmetric Lomer-Cottrell arrangement predicted by the same approach. The absolute energy values obtained from elasticity for the equilibrium dissociated configurations agree well with those found by computer simulation, but the elastic model does not accurately describe the energy changes associated with dissociation of the Lomer dislocation, thus confirming the conclusions drawn recently by Saada and Douin. read less NOT USED (low confidence) G. Simonelli, R. Pasianot, and E. Savino, “Embedded - Atom - Method Interatomic Potentials for BCC - Iron,” MRS Proceedings. 1992. link Times cited: 87 Abstract: An embedded-atom-method (EAM) interatomic potential [1] for … read moreAbstract: An embedded-atom-method (EAM) interatomic potential [1] for bcc-iron is derived. It is fitted exactly to the lattice parameter, elastic constants, an approximation to the unrelaxed vacancy formation energy, and Rose's expression for the cohesive energy [2]. Formation energies and relaxation volumes of point defects are calculated. We find that the relative energies of the defect configurations depend on the functional fitting details of the potential considered, mainly its range: the experimental interstitial configuration of lowest energy can be reproduced by changing this parameter. This result is confirmed by calculating the same defect energies using other EAM potentials, based on the ones developed by Harrison et al. [3]. read less NOT USED (low confidence) F. Ducastelle, “Tight-Binding Potentials.” 1991. link Times cited: 11 NOT USED (low confidence) H. Häkkinen and M. Manninen, “Molecular Dynamics of Metals with Many-Atom Interactions,” Physica Scripta. 1990. link Times cited: 3 Abstract: The thermal properties of simple metals, Al and Cu, have bee… read moreAbstract: The thermal properties of simple metals, Al and Cu, have been studied using the molecular dynamics simulation method and the effective medium theory (EMT). The results obtained from EMT have a strongly predictive nature, because the model includes many-atom interactions and is based on ab initio-calculations within the framework of the density-functional theory. We have calculated the thermal expansion coefficient of solid and melt phase, melting point and latent heat of fusion for both metals. All the results are in good agreement with the experimental data. The long-range interactions (beyond the nearest neighbours) have been found to be important in the simulations of high-temperature processes (melting, disordered structure) and also in the case of extended crystal defects. The theory gives a good description of the vacancy formation energy and the stacking fault energy. The presence of vacancies lowers the simulated melting point of Al indicating the importance of crystal defects in the melting mechanism. No significant temperature dependence of the vacancy formation energy in Al was observed. read less NOT USED (low confidence) C. Windsor, “Defects in Materials: Their Characterization and Simulation.,” MRS Proceedings. 1990. link Times cited: 0 NOT USED (low confidence) A. Carlsson, “Beyond Pair Potentials in Elemental Transition Metals and Semiconductors,” Journal of Physics C: Solid State Physics. 1990. link Times cited: 169 NOT USED (low confidence) R. Johnson, “Implications of the Embedded-Atom Method Format.” 1990. link Times cited: 5 NOT USED (low confidence) M. Manninen, R. Nieminen, and M. Puska, “Introduction to Many-Atom Interactions in Solids.” 1990. link Times cited: 0 NOT USED (low confidence) M. Igarashi, M. Khantha, and V. Vítek, “Many-Body Potentials for Hexagonal Close-Packed Metals.” 1989. link Times cited: 4 NOT USED (low confidence) A. Carlsson, “Angular Forces in Transition Metals and Diamond Structure Semiconductors.” 1989. link Times cited: 1 NOT USED (low confidence) G. Ackland and V. Vítek, “Application of Many-Body Potentials to Noble Metal Alloys.” 1989. link Times cited: 2 NOT USED (low confidence) A. Sutton, “Modelling of the Atomic and Electronic Structures of Interfaces,” MRS Proceedings. 1988. link Times cited: 6 NOT USED (low confidence) Y. Oh and V. Vítek, “Study of the Structure and Energy of Grain Boundaries Using an Lmto Based Tight-Binding Method,” MRS Proceedings. 1988. link Times cited: 1 NOT USED (high confidence) V. Korolev, J. J. Bean, Y. M. Nevolin, Y. Kucherinenko, K. McKenna, and P. Protsenko, “Comparing Five and Lower-Dimensional Grain Boundary Character and Energy Distributions in Copper: Experiment and Molecular Statics Simulation,” Metallurgical and Materials Transactions A. 2022. link Times cited: 2 NOT USED (high confidence) L. A. Mistryukova, N. P. Kryuchkov, V. Mantsevich, A. Sapelkin, and S. Yurchenko, “Interpolation method for crystals with many-body interactions,” Physical Review B. 2021. link Times cited: 0 Abstract: We propose an interpolation scheme to describe pair correlat… read moreAbstract: We propose an interpolation scheme to describe pair correlations in crystals with many-body interactions that requires only information on relative displacements for the nearest neighbours and in the long range. Using crystalline Ni as a test case, the scheme is shown to deliver the functional form for the radial distribution function at least as well as molecular dynamics simulations. The results provide a fast route for verification of interatomic potentials and study of many-body interactions using a combination of x-ray scattering and x-ray absorption spectroscopy. read less NOT USED (high confidence) Z. Wu, K. Tan, R. Zhang, Q. Wei, and Y. Lin, “Atomistic kinetic Monte Carlo—Embedded atom method simulation on growth and morphology of Cu–Zn–Sn precursor of Cu_2ZnSnS_4 solar cells,” Journal of Materials Research. 2020. link Times cited: 2 Abstract: An atomistic kinetic Monte Carlo coupled with the embedded-a… read moreAbstract: An atomistic kinetic Monte Carlo coupled with the embedded-atom method is used to simulate film growth and morphology evolution of a Cu–Zn–Sn precursor of Cu_2ZnSnS_4 solar cells by single-step electrodeposition. The deposition and diffusion events of three different metallic atoms are described by the simulation. Moreover, the multibody Cu–Zn–Sn potential is used to calculate diffusion barrier energy. The effects of process factors, including temperature and electrode potential, on the cross-section morphology and surface roughness are explored, while keeping the elemental composition ratios constant. The lowest roughness with the smoothest morphology is obtained at the optimal parameters. The distribution and transformation behaviors of cluster sizes are investigated to describe the alloy film growth process. Furthermore, the comparison between deposition events and diffusion events reveals that deposition events depend primarily on individual deposition rates of different metallic atoms, but diffusion events are mainly dependent on the interaction of metallic atoms. The film morphology evolution is visualized by three-dimensional configuration with increasing numbers of atoms, which suggests a competing mechanism between nucleation and growth of the thin film alloy. read less NOT USED (high confidence) N. Chen, Q. Peng, Z. Jiao, I. van Rooyen, W. Skerjanc, and F. Gao, “Analytical bond-order potential for silver, palladium, ruthenium and iodine bulk diffusion in silicon carbide,” Journal of Physics: Condensed Matter. 2019. link Times cited: 6 Abstract: The analytical bond-order potential has been developed for s… read moreAbstract: The analytical bond-order potential has been developed for simulating fission product (Ag, Pd, Ru, and I) behavior in SiC, especially for their diffusion. We have proposed adding experimentally available elastic constants and physical properties of the elements as well as important defect formation energies calculated from density functional theory simulation to the list of typical properties as the extensive fitting database. The results from molecular dynamics simulations are in a reasonable agreement with defect properties and energy barriers of their experimental/computational counterparts. The successful validation of the new potential has established a good reliability and transferability of the potentials, which enables the ability of simulation in extended scale. The kinetic behavior such as diffusion of different interstitials is then realized by applying the new interatomic potentials. The bulk diffusion is less likely to dominate the transport of the four fission products under pure thermal condition, when we refer to their extremely small values of the effective diffusion coefficients. The interstitial mechanism is hard for Pd, Ru, and I to access due to the high formation energy and high migration energy. However, it is found that the migration energy of silver is relatively low, which indicates Ag diffusion via an interstitial mechanism being feasible, especially under irradiation condition, where massive interstitials can be formed in high-temperature nuclear reactors. read less NOT USED (high confidence) J. Spitaler and S. Estreicher, “Perspectives on the Theory of Defects,” Frontiers in Materials. 2018. link Times cited: 19 Abstract: Our understanding of defects in materials science has change… read moreAbstract: Our understanding of defects in materials science has changed tremendously over the last century. While one hundred years ago they were often ignored by scientists, nowadays they are in the spotlight of scientific interest and whole branches of technology have emerged from their skillful handling. The first part of this article gives a historical overview and discusses why defects are so important for modern material science. In the second part, we review the treatment of defects in semiconductors. We start by explaining the assumptions and approximations involved in ab-initio calculations and then discuss the treatment of defects in materials. In the third part, we focus on defects in metals. We discuss the theoretical treatment of vacancies in metals starting from experimental findings. The impact of improved theoretical techniques on the predictive power is discussed. This is illustrated with the role of vacancies in intermetallic compounds and random alloys. The last section deals with dislocations. read less NOT USED (high confidence) S. Longbottom and P. Brommer, “Uncertainty quantification for classical effective potentials: an extension to potfit,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 12 Abstract: Effective potentials are an essential ingredient of classica… read moreAbstract: Effective potentials are an essential ingredient of classical molecular dynamics (MD) simulations. Little is understood of the consequences of representing the complex energy landscape of an atomic configuration by an effective potential or force field containing considerably fewer parameters. The probabilistic potential ensemble method has been implemented in the potfit force matching code. This introduces uncertainty quantification into the interatomic potential generation process. Uncertainties in the effective potential are propagated through MD to obtain uncertainties in quantities of interest (QoI), which are a measure of the confidence in the model predictions. We demonstrate the technique using three potentials for nickel: two simple pair potentials, Lennard-Jones and Morse, and a local density dependent embedded atom method potential. A potential ensemble fit to density functional theory (DFT) reference data is constructed for each potential to calculate the uncertainties in lattice constants, elastic constants and thermal expansion. We quantitatively illustrate the cases of poor model selection and fit, highlighted by the uncertainties in the quantities calculated. This shows that our method can capture the effects of the error incurred in QoI resulting from the potential generation process without resorting to comparison with experiment or DFT, which is an essential part to assess the predictive power of MD simulations. read less NOT USED (high confidence) J. Cuny, N. Tarrat, F. Spiegelman, A. Huguenot, and M. Rapacioli, “Density-functional tight-binding approach for metal clusters, nanoparticles, surfaces and bulk: application to silver and gold,” Journal of Physics: Condensed Matter. 2018. link Times cited: 17 Abstract: Density-functional based tight-binding (DFTB) is an efficien… read moreAbstract: Density-functional based tight-binding (DFTB) is an efficient quantum mechanical method that can describe a variety of systems, going from organic and inorganic compounds to metallic and hybrid materials. The present topical review addresses the ability and performance of DFTB to investigate energetic, structural, spectroscopic and dynamical properties of gold and silver materials. After a brief overview of the theoretical basis of DFTB, its parametrization and its transferability, we report its past and recent applications to gold and silver systems, including small clusters, nanoparticles, bulk and surfaces, bare and interacting with various organic and inorganic compounds. The range of applications covered by those studies goes from plasmonics and molecular electronics, to energy conversion and surface chemistry. Finally, perspectives of DFTB in the field of gold and silver surfaces and NPs are outlined. read less NOT USED (high confidence) A. Fang and A. Chien, “ABFR: convenient management of latent error resilience using application knowledge,” Proceedings of the 27th International Symposium on High-Performance Parallel and Distributed Computing. 2018. link Times cited: 2 Abstract: Exascale systems face high error-rates due to increasing sca… read moreAbstract: Exascale systems face high error-rates due to increasing scale (109 cores), software complexity and rising memory error rates. Increasingly, errors escape immediate hardware-level detection, silently corrupting application states. Such latent errors can often be detected by application-level tests but typically at long latencies. We propose a new approach called application-based focused recovery (ABFR), that defines the application knowledge needed for efficient latent error recovery. This allows the application to pursue strategies exploiting a range of application semantics within a well-defined resilience framework. The ABFR runtime then exploits this knowledge to achieve efficient latent error tolerance. ABFR enables application designers to express resilience without concern for the underlying architectures and systems. Together, these ABFR properties support flexible application-based resilience. To demonstrate its generality, we apply ABFR to three varied scientific computations (stencil, N-Body tree, and Monte Carlo). We measure latent error resilience performance for varied error rates; results indicate significant reductions in error recovery cost (up to 367x) and recovery latency (up to 24x). And ABFR achieves efficient and scalable recovery at scale with high latent error rates for these computations. read less NOT USED (high confidence) D. Chen and Y. Kulkarni, “Thermal Fluctuations as a Computational Microscope for Studying Crystalline Interfaces: A Mechanistic Perspective,” Journal of Applied Mechanics. 2017. link Times cited: 7 Abstract: Interfaces such as grain boundaries are ubiquitous in crysta… read moreAbstract: Interfaces such as grain boundaries are ubiquitous in crystalline materials and have provided a fertile area of research over decades. Their importance stems from the numerous critical phenomena associated with them, such as grain boundary sliding, migration, and interaction with other defects, that govern the mechanical properties of materials. Although these crystalline interfaces exhibit small out-of-plane fluctuations, statistical thermodynamics of membranes has been effectively used to extract relevant physical quantities such as the interface free energy, grain boundary stiffness, and interfacial mobility. In this perspective, we advance the viewpoint that thermal fluctuations of crystalline interfaces can serve as a computational microscope for gaining insights into the thermodynamic and kinetic properties of grain boundaries and present a rich source of future study. [DOI: 10.1115/1.4037885] read less NOT USED (high confidence) L. Cui et al., “Quantized thermal transport in single-atom junctions,” Science. 2017. link Times cited: 155 Abstract: Calorimetry reaches an atomic junction Electrical and therma… read moreAbstract: Calorimetry reaches an atomic junction Electrical and thermal conductivity in metals are linked at the macroscopic length scale because electrons carry both heat and current. Cui et al. found that this relationship, the Wiedemann-Franz law, holds down to the atomic scale in gold and platinum (see the Perspective by Segal). They made thermal and electrical conductance measurements through a point contact only one atom thick. In gold, the thermal and electrical conductance was quantized, owing to the electronic band structure of the metal. The experiments pave the way for high-resolution calorimetry and other thermal measurements at the nanoscale. Science, this issue p. 1192; see also p. 1125 Thermal and electrical conductance remain tightly connected across gold and platinum single-atom junctions. Thermal transport in individual atomic junctions and chains is of great fundamental interest because of the distinctive quantum effects expected to arise in them. By using novel, custom-fabricated, picowatt-resolution calorimetric scanning probes, we measured the thermal conductance of gold and platinum metallic wires down to single-atom junctions. Our work reveals that the thermal conductance of gold single-atom junctions is quantized at room temperature and shows that the Wiedemann-Franz law relating thermal and electrical conductance is satisfied even in single-atom contacts. Furthermore, we quantitatively explain our experimental results within the Landauer framework for quantum thermal transport. The experimental techniques reported here will enable thermal transport studies in atomic and molecular chains, which will be key to investigating numerous fundamental issues that thus far have remained experimentally inaccessible. read less NOT USED (high confidence) M. Bhatnagar, M. Ranjan, K. Jolley, A. L. Lloyd, R. Smith, and S. Mukherjee, “Thermal dynamics of silver clusters grown on rippled silica surfaces,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 4 NOT USED (high confidence) Y.-hua Zhou, R. Smith, S. Kenny, and A. L. Lloyd, “Development of an empirical interatomic potential for the Ag–Ti system,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 5 NOT USED (high confidence) N. Admal, J. Marian, and G. Po, “The atomistic representation of first strain-gradient elastic tensors,” Journal of The Mechanics and Physics of Solids. 2016. link Times cited: 36 NOT USED (high confidence) W. Gräfe, “Quantum Mechanical Models of Metal Surfaces and Nanoparticles,” Quantum Mechanical Models of Metal Surfaces and Nanoparticles. 2015. link Times cited: 4 NOT USED (high confidence) F. Taherkhani and P. F. Seresht, “Doping effect on the Janus-like structure of a copper–iron bimetallic nanocluster and its solid–liquid phase transition,” Progress of Theoretical and Experimental Physics. 2015. link Times cited: 2 NOT USED (high confidence) T. Y. Hu, B. Zheng, M. Hu, P. He, and Z. Yue, “Molecular dynamics simulation of incipient plasticity of nickel substrates of different surface orientations during nanoindentation,” Materials Science and Technology. 2015. link Times cited: 14 Abstract: The present study aims to elucidate the anisotropic characte… read moreAbstract: The present study aims to elucidate the anisotropic characteristics in material responses for crystallographic nickel substrates with (001), (011) and (111) surface orientations during nanoindentation. Molecular dynamic simulation is applied to compensate for the experimental limitation of nanoindentation, particularly for pure nickel substrates. Defect nucleation and evolution in Ni single crystal of these three crystal orientations was examined. Hardness and Young’s modulus are also extracted in different orientations. The Young’s modulus of (111) crystallographic orientation is the largest, while that of (001) surface is the smallest. The sensitivity of the yield point for face centred cubic crystals depends on the crystallographic orientation. The (001) crystallographic orientation reaches the yield point first, while the (111) crystallographic orientation is the most difficult in which to achieve yield. Using a visualisation method of centrosymmetry parameter, the homogeneous nucleation and early evolution of dislocations were investigated, deepening understanding of incipient plasticity at the atomic scale. The present results suggest that defect nucleation and evolution are the root of curve jitter. The indentation depth of the elastic–plastic transition point varies in the different crystallographic orientation models, and appears latest in the (111) model. The strain energy of the substrate exerted by the tip is stored by the formation of homogeneous nucleation and is dissipated by the dislocation slide in the {111} glide plane. The three nickel substrates with different crystallographic orientations exhibit different forms of dislocation propagation. read less NOT USED (high confidence) S. Valone, S. Atlas, and M. Baskes, “Fragment Hamiltonian model potential for nickel: metallic character and defects in crystalline lattices,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 5 Abstract: The Fragment Hamiltonian (FH) model is introduced as the bas… read moreAbstract: The Fragment Hamiltonian (FH) model is introduced as the basis for a new class of atomistic potentials that may be viewed as generalizations of the embedded atom method (EAM) and related atomistic potentials. Many metals and alloys have been successfully modeled by this method and other related methods, but the nature of the metallic character in the models has been lost. Here we attempt to recover this character, at a qualitative level, by defining an embedding energy as a function of two variables through the FH model. One of these variables, called the ionicity, is associated with the established concept of background density in EAM models. The FH embedding energy is composed of two types of energies, one for energies of different states of an atom and the other for hopping energies that transform an atom from one state to another. A combination of the energies for the states of an atom yield a local gap energy that conforms to a generalized definition of the ‘Hubbard-U’ energy. The hopping energies compete with the gap energy to provide a notion of metallic behavior in an atomic-scale model. Lattices of nickel with different coordinations and spatial dimensions, elastic constants, energies for several types of defects in three-dimensional lattices and two surface energies are calculated to show the strengths and limitations of the current implementation and to explore their metallic character. read less NOT USED (high confidence) C. Becker et al., “Thermodynamic modelling of liquids: CALPHAD approaches and contributions from statistical physics,” physica status solidi (b). 2014. link Times cited: 32 Abstract: We describe current approaches to thermodynamic modelling of… read moreAbstract: We describe current approaches to thermodynamic modelling of liquids for the CALPHAD method, the use of available experimental methods and results in this type of modelling, and considerations in the use of atomic‐scale simulation methods to inform a CALPHAD approach. We begin with an overview of the formalism currently used in CALPHAD to describe the temperature dependence of the liquid Gibbs free energy and outline opportunities for improvement by reviewing the current physical understanding of the liquid. Brief descriptions of experimental methods for extracting high‐temperature data on liquids and the preparation of undercooled liquid samples are presented. Properties of a well‐determined substance, B2 O3, including the glass transition, are then discussed in detail to emphasize specific modelling requirements for the liquid. We then examine the two‐state model proposed for CALPHAD in detail and compare results with experiment and theory, where available. We further examine the contributions of atomic‐scale methods to the understanding of liquids and their potential for supplementing available data. We discuss molecular dynamics (MD) and Monte Carlo methods that employ atomic interactions from classical interatomic potentials, as well as contributions from ab initio MD. We conclude with a summary of our findings. read less NOT USED (high confidence) I.-H. Chen, C. I. Hsiao, R. Behera, and W.-D. Hsu, “Defect formation by pristine indenter at the initial stage of nanoindentation,” Journal of Applied Physics. 2013. link Times cited: 2 Abstract: Nano-indentation is a sophisticated method to characterize m… read moreAbstract: Nano-indentation is a sophisticated method to characterize mechanical properties of materials. This method samples a very small amount of material during each indentation. Therefore, this method is extremely useful to measure mechanical properties of nano-materials. The measurements using nanoindentation is very sensitive to the surface topology of the indenter and the indenting surfaces. The mechanisms involved in the entire process of nanoindentation require an atomic level understanding of the interplay between the indenter and the substrate. In this paper, we have used atomistic simulation methods with empirical potentials to investigate the effect of various types of pristine indenter on the defect nucleation and growth. Using molecular dynamics simulations, we have predicted the load-depth curve for conical, vickers, and sperical tip. The results are analyzed based on the coherency between the indenter tip and substrate surface for a fixed depth of 20 A. The depth of defect nucleation and growth is ... read less NOT USED (high confidence) D. Belashchenko, “Computer simulation of liquid metals,” Physics—Uspekhi. 2013. link Times cited: 84 Abstract: Methods for and the results of the computer simulation of li… read moreAbstract: Methods for and the results of the computer simulation of liquid metals are reviewed. Two basic methods, classical molecular dynamics with known interparticle potentials and the ab initio method, are considered. Most attention is given to the simulated results obtained using the embedded atom model (EAM). The thermodynamic, structural, and diffusion properties of liquid metal models under normal and extreme (shock) pressure conditions are considered. Liquid-metal simulated results for the Groups I–IV elements, a number of transition metals, and some binary systems (Fe–C, Fe–S) are examined. Possibilities for the simulation to account for the thermal contribution of delocalized electrons to energy and pressure are considered. Solidification features of supercooled metals are also discussed. read less NOT USED (high confidence) M. Mendelev, C. Deng, C. A. Schuh, and D. Srolovitz, “Comparison of molecular dynamics simulation methods for the study of grain boundary migration,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 51 Abstract: In the present study, grain boundary (GB) mobility was deter… read moreAbstract: In the present study, grain boundary (GB) mobility was determined by molecular dynamics (MD) simulations using two different techniques: the applied strain method and the adapted interface random walk method. The first method involves a driving force while the second method does not. Nevertheless, both methods led to essentially the same values of the GB mobility. This shows that the GB mobility is independent of the nature of the driving force, provided that it is low enough that the linear velocity–driving force relationship is properly sampled. The case studied here can be viewed as a validated reference case that can be used in future studies to test new techniques to determine the GB mobility. For this purpose we provide the full information about the interatomic potential we employed and the initial atomic configurations. Finally, we use the obtained results to discuss whether any existing MD simulation data agree with experimental data on pure metals. read less NOT USED (high confidence) D. Wei, Z. Jiang, and J. Han, “Modelling of the evolution of crack of nanoscale in iron,” Computational Materials Science. 2013. link Times cited: 13 NOT USED (high confidence) D. Wei, Z. Jiang, and J. Han, “Evolution of Internal Crack in BCC Fe under Compressive Loading,” Journal of Modern Physics. 2012. link Times cited: 0 Abstract: A molecular dynamics model has been developed to investigate… read moreAbstract: A molecular dynamics model has been developed to investigate the evolution of the internal crack of nano scale during heating or compressive loading in BCC Fe. The initial configuration does not contain any pre-existing dislocations. In the case of heating, temperature shows a significant effect on crack evolution and the critical temperature at which the crack healing becomes possible is 673 K. In the case of compressive loading, the crack can be healed at 40 K at a loading rate 0.025 × 1018 Pa·m1/2/s in 6 × 10-12 s. The diffusion of Fe atoms into the crack area results in the healing process. However, dislocations and voids appear during healing and their positions change continuously. read less NOT USED (high confidence) D. Belashchenko, “Computer simulation of the properties of liquid metals: Gallium, lead, and bismuth,” Russian Journal of Physical Chemistry A. 2012. link Times cited: 22 NOT USED (high confidence) B. Fu, Z. Li, and W. Liu, “Covalent electron density analysis and surface energy calculation of gold with the empirical electron surface model,” International Journal of Minerals, Metallurgy, and Materials. 2011. link Times cited: 5 NOT USED (high confidence) G. J. Ackland, K. D’Mellow, S. L. Daraszewicz, D. J. Hepburn, M. Uhrin, and K.Stratford, “The MOLDY short-range molecular dynamics package,” Comput. Phys. Commun. 2011. link Times cited: 37 NOT USED (high confidence) T. Klaver, D. Hepburn, and G. Ackland, “Defect and solute properties in dilute Fe-Cr-Ni austenitic alloys from first principles,” Performance Evaluation. 2011. link Times cited: 54 Abstract: We present results of an extensive set of first-principles d… read moreAbstract: We present results of an extensive set of first-principles density functional theory calculations of point defect formation, binding, and clustering energies in austenitic Fe with dilute concentrations of Cr and Ni solutes. A large number of possible collinear magnetic structures were investigated as appropriate reference states for austenite. We found that the antiferromagnetic single- and double-layer structures with tetragonal relaxation of the unit cell were the most suitable reference states and highlighted the inherent instabilities in the ferromagnetic states. Test calculations for the presence and influence of noncollinear magnetism were performed but proved mostly negative. We calculate the vacancy formation energy to be between 1.8 and 1.95 eV. Vacancy cluster binding was initially weak at 0.1 eV for divacancies but rapidly increased with additional vacancies. Clusters of up to six vacancies were studied and a highly stable octahedral cluster and stacking fault tetrahedron were found with total binding energies of 2.5 and 2.3 eV, respectively. The ?100? dumbbell was found to be the most stable self-interstitial with a formation energy of between 3.2 and 3.6 eV and was found to form strongly bound clusters, consistent with other fcc metals. Pair interaction models were found to be capable of capturing the trends in the defect cluster binding energy data. Solute-solute interactions were found to be weak in general, with a maximal positive binding of 0.1 eV found for Ni-Ni pairs and maximum repulsion found for Cr-Cr pairs of ?0.1 eV. Solute cluster binding was found to be consistent with a pair interaction model, with Ni-rich clusters being the most stable. Solute-defect interactions were consistent with Ni and Cr being modestly oversized and undersized solutes, respectively, which is exactly opposite to the experimentally derived size factors for Ni and Cr solutes in type 316 stainless steel and in the pure materials. Ni was found to bind to the vacancy and to the ?100? dumbbell in the tensile site by 0.1 eV and was repelled from mixed and compressive sites. In contrast, Cr showed a preferential binding to interstitials. Calculation of tracer diffusion coefficients found that Ni diffuses significantly more slowly than both Cr and Fe, which is consistent with the standard mechanism used to explain radiation-induced segregation effects in Fe-Cr-Ni austenitic alloys by vacancy-mediated diffusion. Comparison of our results with those for bcc Fe showed strong similarity for pure Fe and no correlation with dilute Ni and Cr. read less NOT USED (high confidence) H. Ogawa, “Molecular Dynamics Study of the Particle Size Dependency of Structural Change in Hydrogenated Model f.c.c. Nanoparticles,” Materials Transactions. 2011. link Times cited: 1 Abstract: Hydrogen absorption in f.c.c. nanoparticles of 1–8 nm diamet… read moreAbstract: Hydrogen absorption in f.c.c. nanoparticles of 1–8 nm diameter was investigated using molecular dynamics simulation with model interatomic potentials. Atomic configuration with five-fold symmetries was observed in both hydrogen-free and hydrogenated particles smaller than 2 nm. The f.c.c. structure was maintained in larger particles after hydrogenation in cases where the M–H interaction is weak. Lattice deformation was induced in cases of strong M–H interaction. A shift of critical size for icosahedral–cubic transition by hydrogenation was also inferred. The number of absorbed H atoms increased concomitantly with increasing particle size and M–H interaction. Most absorbed H atoms located at O-sites when M–H interaction was weak. The T-site occupancy increased with M–H interaction. Analysis using local atomic configuration revealed that structural variation in nanoparticles results from three factors: surface effects, icosahedral transformation, and lattice deformation attributable to M–H interaction. [doi:10.2320/matertrans.MA201017] read less NOT USED (high confidence) M. Chassagne, M. Legros, and D. Rodney, “Atomic-scale simulation of screw dislocation/coherent twin boundary interaction in Al, Au, Cu and Ni,” Acta Materialia. 2011. link Times cited: 124 NOT USED (high confidence) E. H. Megchiche, C. Mijoule, and M. Amarouche, “First principles calculations of vacancy–vacancy interactions in nickel: thermal expansion effects,” Journal of Physics: Condensed Matter. 2010. link Times cited: 21 Abstract: The energetic properties of the divacancy defect in fcc nick… read moreAbstract: The energetic properties of the divacancy defect in fcc nickel are studied by ab initio calculations based on density functional theory. The formation and binding enthalpies of the divacancy in the first (1nn), second (2nn) and third (3nn) nearest-neighbor configurations are presented. Results show that the 1nn divacancy configuration is the most stable with a formation enthalpy H2vf of 2.71 eV and a small binding energy H2vb of 0.03 eV. In the 2nn configuration, the monovacancy–monovacancy interaction is repulsive, and it vanishes in the 3nn configuration. The migration process of the divacancy in its stable configuration is studied. We find that the divacancy migrates in the (111) plane by successive rotational steps of 60°. The corresponding migration enthalpy H2vm is predicted to be 0.59 eV, about half of that found for the monovacancy. For a better comparison of our results with high temperature experimental data, we have analyzed the effects of thermal expansion. Our results show that the inclusion of thermal expansion allows us to reproduce satisfactorily the experimental predictions. read less NOT USED (high confidence) R. Smith, “Surface topography after single particle and cluster impacts on gold surfaces,” Philosophical Magazine. 2009. link Times cited: 0 Abstract: The surface topography arising from energetic Au particle im… read moreAbstract: The surface topography arising from energetic Au particle impacts on Au is investigated. Both individual particle and cluster impacts are considered. For individual Au atoms, 20 successive impacts at 5 keV are considered on a low index face (0 1 0) and a higher index face (3 11 0) with a similar orientation. In both cases, all ejected particles occur in the first 10 ps after impact. It is shown that on the (0 1 0) surface, fairly steep sided flat-bottomed holes surrounded by flat topped adatom structures are formed whereas, on the (3 11 0) surface, facetted structures begin to form. For the cluster impact which deposits the same amount of energy in the surface as the 20 single particle impacts, the ejection yield is much higher as atoms boil off from the surface for times as long as 50 ps after impact. The resulting topography formed on the surface is independent of the cluster shape but is characteristic of the structure of the original crystal surface and substrate. read less NOT USED (high confidence) D. Bacon and Y. Osetsky, “Mechanisms of hardening due to copper precipitates in α-iron,” Philosophical Magazine. 2009. link Times cited: 44 Abstract: A comprehensive atomic-level simulation study has been made … read moreAbstract: A comprehensive atomic-level simulation study has been made of interactions between a moving edge dislocation and copper precipitates that are initially coherent with the body-centred-cubic matrix of alpha-iron. Precipitates with diameter, D, in the range 0.7–6 nm have been considered over the temperature range 0–600 K. For some combinations of temperature and D, the critical applied resolved shear stress, τ c, at which the dislocation overcomes a row of precipitates with centre-to-centre spacing, L, is consistent with an elasticity treatment for strong obstacles, e.g. τ c is proportional to L −1 and ln(D). This has a specific atomic-level origin, for the proportionality holds when the dislocation induces a partial transformation of the copper towards the more stable face-centred-cubic phase. The driving force for the transformation increases with decreasing temperature and increasing D, and so τ c has a strong temperature-dependence for large D. The results of these simulations, which employ a set of interatomic potentials of Finnis–Sinclair type, are seen to correspond well with experiments carried out elsewhere. read less NOT USED (high confidence) T. Yi-zhe, Z. Zhijun, X. Meng-Fen, and B. Yilong, “A Unified Guide to Two Opposite Size Effects in Nano Elastic Materials,” Chinese Physics Letters. 2009. link Times cited: 3 Abstract: The microstructural variation near surface of nano elastic m… read moreAbstract: The microstructural variation near surface of nano elastic materials is analyzed based on different potentials. The atomic/molecular mechanism underlying the variation and its effect on elastic modulus are such that the nature of long-range interactions (attractive or repulsive) in the atomic/molecular potentials essentially governs the variation near surface (looser or tighter) and results in two opposite size effects (decreasing or increasing modulus) with decreasing size. read less NOT USED (high confidence) N. Combe and L. Saviot, “Acoustic modes in metallic nanoparticles: atomistic versus elasticity modeling,” Physical Review B. 2009. link Times cited: 23 Abstract: The validity of the linear elasticity theory is examined at … read moreAbstract: The validity of the linear elasticity theory is examined at the nanometer scale by investigating the vibrational properties of silver and gold nanoparticles whose diameters range from about 1.5 to 4~nm. Comparing the vibration modes calculated by elasticity theory and atomistic simulation based on the Embedded Atom Method, we first show that the anisotropy of the stiffness tensor in elastic calculation is essential to ensure a good agreement between elastic and atomistic models. Second, we illustrate the reduction of the number of vibration modes due to the diminution of the number of atoms when reducing the nanoparticles size. Finally, we exhibit a breakdown of the frequency-spectra scaling of the vibration modes and attribute it to surface effects. Some critical sizes under which such effects are expected, depending on the material and the considered vibration modes are given. read less NOT USED (high confidence) D. Chrzan, J. Morris, Y. Osetsky, R. Stoller, and S. Zinkle, “What is the Limit of Nanoparticle Strengthening?,” MRS Bulletin. 2009. link Times cited: 11 Abstract: The stress required to deform a perfect crystal to its elast… read moreAbstract: The stress required to deform a perfect crystal to its elastic limit while maintaining perfect periodicity, the so-called ideal strength, sets the gold standard for the strength of a given material. Materials this strong would be of obvious engineering importance, potentially enabling more efficient turbines for energy production, lighter materials for transportation applications, and more reliable materials for nuclear reactor applications. In practice, the strength of engineering materials is often more than two orders of magnitude less than the ideal strength due to easily activated deformation processes involving dislocations. For many materials, precipitate strengthening is a promising approach to impede dislocation motion and thereby improves strength and creep resistance. This observation begs the question: What are the limits of nanoparticle strengthening? Can the ideal strength of a matrix material be reached? To answer these questions, we need a detailed, atomic scale understanding of the interactions between dislocations and obstacles. Fortunately, simulations are beginning to explore this interaction. read less NOT USED (high confidence) W. Luo, W. Hu, and S. Xiao, “Size Effect on the Thermodynamic Properties of Silver Nanoparticles,” Journal of Physical Chemistry C. 2008. link Times cited: 186 Abstract: The Gibbs free energy of silver nanoparticles has been obtai… read moreAbstract: The Gibbs free energy of silver nanoparticles has been obtained from the calculations of bulk free energy and surface free energy for both the solid and liquid phase. On the basis of the obtained Gibbs free energy of nanoparticles, thermodynamic properties of silver nanoparticles, such as melting temperature, molar heat of fusion, molar entropy of fusion, and temperature dependences of entropy and specific heat capacity have been investigated. Calculation results indicate that these thermodynamic properties can be divided into two parts: bulk quantity and surface quantity, and surface atoms are dominant for the size effect on the thermodynamic properties of nanoparticles. The method that the intersection of the free-energy curves for solid and liquid nanoparticles decide the melting point of nanoparticles demonstrates that the surface free-energy difference between the solid and liquid phase is a decisive factor for the size-dependent melting of nanostructural materials. read less NOT USED (high confidence) L. Nemirovich-Danchenko, A. Lipnitskii, and S. E. Kul’kova, “Vacancies and their complexes in FCC metals,” Physics of the Solid State. 2007. link Times cited: 14 NOT USED (high confidence) E. Mcgee, R. Smith, and S. Kenny, “Multiscale modelling of nanoindentation,” International Journal of Materials Research. 2007. link Times cited: 14 Abstract: The process of nanoindentation causes physical phenomena not… read moreAbstract: The process of nanoindentation causes physical phenomena not only at the nano-scale, but at multiple length scales up to the macroscopic. We present a model that links atomistic scale molecular dynamics to a finite element model in order to extend the length scales that can be modelled. We illustrate how this has been used in some studies of nanoindentation, to test the coupled model, and secondly to compare the results to atomistic only models. These studies show that by extending the length scales, the results from the model are in closer agreement with the experiment. read less NOT USED (high confidence) M. Mrovec, R. Gröger, A. G. Bailey, D. Nguyen-Manh, C. Elsässer, and V. Vítek, “Bond-Order Potential for Simulations of Extended Defects in Tungsten,” Physical Review B. 2007. link Times cited: 109 Abstract: We present a bond-order potential BOP for the bcc transition… read moreAbstract: We present a bond-order potential BOP for the bcc transition metal tungsten. The bond-order potentials are a real-space semiempirical scheme for the description of interatomic interactions based on the tight-binding approximation. In the hierarchy of atomic-scale-modeling methods the BOPs thus provide a direct bridge between electronic-structure and atomistic techniques. Two variants of the BOP were constructed and extensively tested against accurate first-principles methods in order to assess the potentials’ reliability and applicability. A comparison of the BOP with a central-force potential is used to demonstrate that a correct description of directional mixed covalent and metallic bonds is crucial for a successful and fully transferable model. The potentials are applied in studies of low-index surfaces, symmetrical tilt grain boundaries, and dislocations. read less NOT USED (high confidence) M. Ali, R. Smith, and S. Hobday, “The structure of atomic and molecular clusters, optimised using classical potentials,” Comput. Phys. Commun. 2006. link Times cited: 17 NOT USED (high confidence) Y. Osetsky, D. Rodney, and D. Bacon, “Atomic-scale study of dislocation–stacking fault tetrahedron interactions. Part I: mechanisms,” Philosophical Magazine. 2006. link Times cited: 95 Abstract: Stacking fault tetrahedra (SFTs) are formed under irradiatio… read moreAbstract: Stacking fault tetrahedra (SFTs) are formed under irradiation in fcc metals and alloys. The high number density of SFTs observed suggests that they should contribute to radiation-induced hardening and, therefore, be taken into account when estimating mechanical property changes of irradiated materials. The key issue in this is to describe the interaction between a moving dislocation and an individual SFT, which is distinguished by a small physical size of the order of ∼1–10 nm. We have performed atomistic simulations of edge and screw dislocations interacting with SFTs of different sizes at different temperatures and strain rates. Five possible interaction outcomes have been identified, involving either partial absorption, or shearing or restoration of SFTs. The mechanisms that give rise to these processes are described and their dependence on interaction parameters, such as SFT size, dislocation–SFT geometry, temperature and stress/strain rate are determined. Mechanisms that help to explain the formation of defect-free channels cleared by gliding dislocations, as observed experimentally, are also discussed. Hardening due to the various mechanisms and their dependence on loading conditions will be presented in a following paper (Part II). read less NOT USED (high confidence) X. Dai, Y. Kong, J. H. Li, and B. X. Liu, “Extended Finnis–Sinclair potential for bcc and fcc metals and alloys,” Journal of Physics: Condensed Matter. 2006. link Times cited: 138 Abstract: We propose an extended Finnis–Sinclair (FS) potential by ext… read moreAbstract: We propose an extended Finnis–Sinclair (FS) potential by extending the repulsive term into a sextic polynomial for enhancing the repulsive interaction and adding a quartic term to describe the electronic density function. It turns out that for bcc metals the proposed potential not only overcomes the ‘soft’ behaviour of the original FS potential, but also performs better than the modified FS one by Ackland et al, and that for fcc metals the proposed potential is able to reproduce the lattice constants, cohesive energies, elastic constant, vacancy formation energies, equations of state, pressure–volume relationships, melting points and melting heats. Moreover, for some fcc–bcc systems, e.g. the Ag–refractory metal systems, the lattice constants, cohesive energies and elastic constants of some alloys are reproduced by the proposed potential and are quite compatible with those directly determined by ab initio calculations. read less NOT USED (high confidence) D. Belashchenko, “Embedded atom model for liquid metals: Liquid iron,” Russian Journal of Physical Chemistry. 2006. link Times cited: 21 NOT USED (high confidence) D. Belashchenko and O. I. Ostrovskii, “The embedded atom model for liquid metals: Liquid gallium and bismuth,” Russian Journal of Physical Chemistry. 2006. link Times cited: 23 NOT USED (high confidence) S. Debiaggi, M. Koning, and A. M. Monti, “Theoretical study of the thermodynamic and kinetic properties of self-interstitials in aluminum and nickel,” Physical Review B. 2006. link Times cited: 10 Abstract: The formation thermodynamics and migration properties of sel… read moreAbstract: The formation thermodynamics and migration properties of self-interstitials in aluminum and nickel are investigated as a function of temperature using atomistic simulation techniques and embedded-atom-type interatomic potentials. Molecular dynamics and nonequilibrium free-energy techniques are employed to investigate anharmonic effects on the ${H}_{O}$ $⟨100⟩$ dumbbell formation properties. The equilibrium concentration of this defect is compared to those of vacancies and divacancies. The results are then analyzed in the framework of the interstitialcy model, according to which very high vibrational formation entropies should be expected for self-interstitials at high temperatures. The kinetics of self-interstitial migration is also investigated using different atomistic techniques, revealing the simultaneous activity of more than one distinct interstitial configuration as the temperature increases. read less NOT USED (high confidence) D. Kulikov, L. Malerba, and M. Hou, “On the binding energies and configurations of vacancy and copper–vacancy clusters in bcc Fe–Cu:a computational study,” Philosophical Magazine. 2006. link Times cited: 21 Abstract: Vacancy and copper–vacancy clusters in bcc Fe–Cu alloys have… read moreAbstract: Vacancy and copper–vacancy clusters in bcc Fe–Cu alloys have been studied using a combination of metropolis Monte Carlo (MMC) and molecular dynamics (MD) techniques, to investigate their lowest energy configurations and corresponding binding energies, for sizes up to a few hundreds of elements (∼2 nm). Two different many-body interatomic potentials were used to perform the calculations, in order to assess the robustness of the results 1, 2. Empirical expressions for the binding energies, of immediate use in kinetic Monte Carlo (KMC) or rate theory (RT) models, have been obtained. It is observed that vacancy clusters are three-dimensional cavities whose shape is primarily determined by a criterion of maximisation of the number of first and second nearest neighbour pairs. Copper atoms, when present, tend to coat an inner vacancy cluster, while remaining first nearest neighbours to each other. The inner vacancy cluster, when completely coated, tends to be as close as possible to the surface of the hollow precipitate. These findings are consistent with previous experimental and computational work. The binding energy of these complexes is a monotonously growing function of the ratio number of vacancies to number of copper atoms. Pure copper precipitates appear to follow a loose criterion of maximisation of first nearest neighbour pairs. While the two interatomic potentials used in this work provide largely similar values for the binding energies and comparable configurations, some differences are found and discussed. Subtle differences observed in comparison with ab initio calculations are also discussed. read less NOT USED (high confidence) J. Z. Liu, A. Walle, G. Ghosh, and M. Asta, “Structure, energetics, and mechanical stability of Fe-Cu bcc alloys from first-principles calculations,” Physical Review B. 2005. link Times cited: 119 Abstract: Atomic volumes, magnetic moments, mixing energies, and the e… read moreAbstract: Atomic volumes, magnetic moments, mixing energies, and the elastic properties of bcc Fe1–xCux solid solutions are studied by ab initio calculations based on the cluster expansion framework. For the calculation of concentration-dependent elastic moduli in disordered solid solutions, we introduce a generalization of the cluster expansion technique that is designed to handle tensorial quantities in high-symmetry phases. Calculated mixing energies, atomic volumes, and magnetic moments are found to be in good agreement with available measurements for metastable alloys prepared through nonequilibrium processing techniques. Additionally, the predicted variations of the bulk modulus and shear moduli C44 and C[prime] with respect to copper concentration are calculated for the disordered bcc phase. While the bulk modulus and C44 are positive for all concentrations, C[prime] is predicted to be positive only for Cu concentration less than 50 atomic %, and negative otherwise. Our results thus indicate that the mechanical instability of bcc Cu persists over a wide range of compositions. The implications of the present results are discussed in relation to the observed metastability of bcc Fe-Cu alloys, and the strengthening mechanism of nanoscale bcc precipitates in an alpha-Fe matrix. read less NOT USED (high confidence) S. Kenny, D. Mulliah, C. Sanz-Navarro, and R. Smith, “Molecular dynamics simulations of nanoindentation and nanotribology,” Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2005. link Times cited: 20 Abstract: We present results of parallel molecular dynamics simulation… read moreAbstract: We present results of parallel molecular dynamics simulations of nanoindentation and nanotribology experiments. The models we have developed describe both the sample and the indenter atomistically and model the effect of the cantilevers in an atomic force microscope through the use of springs. We show that the simulations are in good qualitative agreement with experiment and help to elucidate many of the mechanisms that take place during these processes. In particular, we illustrate the role that dislocations play both in nanoindentation and also in stick–slip. Further to this we show how real-time visualization and computational steering have been employed in these simulations to capture the dynamical events that take place. read less NOT USED (high confidence) R. Sahara, H. Mizuseki, K. Ohno, and Y. Kawazoe, “Thermodynamic Properties of Transition Metals Using Face-Centered-Cubic Lattice Model with Renormalized Potentials,” Materials Transactions. 2005. link Times cited: 3 Abstract: The thermodynamic properties of transition metals are studie… read moreAbstract: The thermodynamic properties of transition metals are studied by introducing face-centered cubic (FCC) lattice model. In order to treatactualsystemsasquantitativelyaspossible,empiricalsecondmomentapproximation(SMA)potentialsproposedbyRosatoetal.andbyClerietal., which have been used widely for molecular dynamics (MD) simulations, are employed. To overcome shortcomings of lattice-gas modelssuch as neglecting internal entropy of the system, the potential is mapped onto FCC lattice using the renormalization technique. It is found thatthe computed linear thermal expansion coefficients agree well with the results of MD simulations.(Received January 14, 2005; Accepted March 8, 2005; Published June 15, 2005)Keywords: transition metal, lattice-gas model, thermal expansion, renormalization, potential renormalization, molecular dynamics read less NOT USED (high confidence) G. Ackland, M. Mendelev, D. Srolovitz, S. Han, and A. Barashev, “Development of an interatomic potential for phosphorus impurities in α-iron,” Journal of Physics: Condensed Matter. 2004. link Times cited: 542 Abstract: We present the derivation of an interatomic potential for th… read moreAbstract: We present the derivation of an interatomic potential for the iron–phosphorus system based primarily on ab initio data. Transferability in this system is extremely problematic, and the potential is intended specifically to address the problem of radiation damage and point defects in iron containing low concentrations of phosphorus atoms. Some preliminary molecular dynamics calculations show that P strongly affects point defect migration. read less NOT USED (high confidence) R. Sahara, H. Ichikawa, H. Mizuseki, K. Ohno, H. Kubo, and Y. Kawazoe, “Thermodynamic properties of the Cu-Au system using a face-centered-cubic lattice model with a renormalized potential.,” The Journal of chemical physics. 2004. link Times cited: 9 Abstract: A Monte Carlo simulation is carried out to study thermodynam… read moreAbstract: A Monte Carlo simulation is carried out to study thermodynamic properties of Cu-Au alloys using a face-centered-cubic (fcc) lattice-gas model. To obtain quantitatively accurate results, a Finnis-Sinclair-type potential, which has been widely used for molecular dynamics (MD) simulations, is employed. To overcome some shortcomings of lattice-gas models such as neglecting vibrational entropy, the potential is mapped onto the fcc lattice using the renormalization technique. The renormalized potential gives an improved Cu-Au phase diagram compared to the original MD potential applied directly on the lattice. read less NOT USED (high confidence) M. A. Puigvi, A. Serra, N. de Diego, Y. Osetsky, and D. Bacon, “Features of the interactions between a vacancy and interstitial loops in metals,” Philosophical Magazine Letters. 2004. link Times cited: 11 Abstract: Point defects and defect clusters have been observed in meta… read moreAbstract: Point defects and defect clusters have been observed in metals irradiated by high-energy particles. Interactions of these defects between themselves and with existing microstructure features cause microstructure evolution and lead to changes in mechanical and physical properties of the irradiated materials. Models for prediction of radiation-induced changes should include details of reactions involving defects, and so in this paper we present the results of atomic-scale computer modelling of interactions between a cluster of self-interstitial atoms (SIAs) and a single vacancy in models of bcc, fcc and hcp metals. The vacancy is taken to lie on or within the glide prism of the cluster. This type of reaction is considered to be one of the most frequent because formation of SIA clusters, particularly glissile clusters, is commonly observed in high-energy displacement cascades in all metals. The interaction depends strongly on the dislocation nature of the cluster and therefore these interactions are different in the three crystal structures. Vacancy–SIA recombination, in particular, is inhibited by dissociation of the SIA loop on its glide prism. read less NOT USED (high confidence) M. Mrovec, D. Nguyen-Manh, D. Pettifor, and V. Vítek, “Bond-order potential for molybdenum: Application to dislocation behavior,” Physical Review B. 2004. link Times cited: 116 Abstract: The bond-order potential ~BOP! for transition metals is a re… read moreAbstract: The bond-order potential ~BOP! for transition metals is a real-space semiempirical description of interactions between the atoms, which is based on the tight-binding approximation and the d-band model. This scheme provides a direct bridge between the electronic level modeling and the atomistic modeling, where the electronic degrees of freedom have been coarse grained into many-body interatomic potentials. In this paper we construct BOP in which both the attractive and the repulsive contributions to the binding energy are environmentally dependent due to both the nonorthogonality of the orbitals and the breathing of the screening charges. The construction of the BOP is described and tested in detail. First, the energies of alternative crystal structures ~A15, fcc, hcp, simple cubic! are calculated and compared with those evaluated ab initio. The transferability of the BOP to atomic configurations that deviate significantly from the bcc lattice is studied by computing the energies along tetragonal, trigonal, and hexagonal transformation paths. Next, the phonon spectra are evaluated for several symmetrical crystallographic directions and compared with available experiments. All these calculations highlight the importance of directional bonding and the investigation of phonons demonstrates that the environmental dependence of the bond integrals is crucial for the phonons of the N branch not to be unphysically soft. Finally, the constructed BOP was applied in the modeling of the core structure and glide of the 1/2^111& screw dislocation. The calculated structure of the core agrees excellently with that found in the recent ab initio calculations and the observed glide behavior not only agrees with available ab initio data but is in agreement with many experimental observations and explains the primary reason for the breakdown of the Schmid law in bcc metals. read less NOT USED (high confidence) K. Sato, T. Yoshiie, Y. Satoh, and Q. Xu, “Computer simulation of formation energy and migration energy of vacancies under high strain in Cu,” Materials Transactions. 2004. link Times cited: 9 Abstract: Recently it has been reported that many vacancy clusters are… read moreAbstract: Recently it has been reported that many vacancy clusters are generated at sawtooth-like fractured tips of thin foils. The elastic strain of the torn portion is more than 10% before the fracture, which is expected to cause the generation of many vacancy clusters. In this paper, the formation and migration behaviors of vacancies in Cu under high elastic strain from 10% compression to 20% elongation were studied by computer simulation using the effective medium theory (EMT) potential. The model lattice was elastically deformed along the ‹110› and ‹100› directions. Poisson's ratio was determined to minimize the total energy. After full relaxation of the lattice by the static method (Newton-Raphson method) under fixed boundary conditions, a vacancy was introduced and the change of the total energy (formation energy) was calculated. The migration energy of vacancies was obtained as the total energy difference between the model lattice with an atom at the lattice point and the atom at the saddle point. High strain dependence of these energies is obtained. For example, the formation and the migration energies of vacancies are 1.21 eV and 0.79 eV in the absence of deformation, respectively. The formation energy in the ‹100› deformation is 1.09 eV and 1.13 eV by 10% compression and 10% elongation, respectively. The migration energies and the migration distances vary with the migration direction. For example, the migration energy for the shortest migration distance in the ‹100› deformation is reduced to 0.45 eV and 0.26 eV by 10% compression and 10% elongation, respectively. While that for the longest migration distance increases to 1.29 eV by 10% compression. These results are explained by the configuration of neighboring atoms nearest to the vacancy. read less NOT USED (high confidence) J. Hoyt, J. W. Garvin, E. B. Webb, and M. Asta, “An embedded atom method interatomic potential for the Cu–Pb system,” Modelling and Simulation in Materials Science and Engineering. 2003. link Times cited: 57 Abstract: A simple procedure is used to formulate a Cu–Pb pair interac… read moreAbstract: A simple procedure is used to formulate a Cu–Pb pair interaction function within the embedded atom (EAM) method framework. Embedding, density and pair functions for pure Cu and pure Pb are taken from previously published EAM studies. Optimization of the Cu–Pb potential was achieved by comparing with experiment the computed heats of mixing for Cu–Pb liquid alloys and the equilibrium phase diagram, the latter being determined via a thermodynamic integration technique. The topology of the temperature-composition phase diagram computed with this EAM potential is consistent with experiment and features a liquid–liquid miscibility gap, low solubility of Pb in solid Cu and a monotectic reaction at approximately 1012 K. read less NOT USED (high confidence) Y. Osetsky, D. Bacon, A. Serra, B. Singh, and S. Golubov, “One-dimensional atomic transport by clusters of self-interstitial atoms in iron and copper,” Philosophical Magazine. 2003. link Times cited: 200 Abstract: Atomic-scale computer simulation has been used to study the … read moreAbstract: Atomic-scale computer simulation has been used to study the thermally activated atomic transport of self-interstitial atoms (SIAs) in the form of planar clusters in pure Cu and f-Fe. There is strong evidence that such clusters are commonly formed in metals during irradiation with high-energy particles and play an important role in accumulation and spatial distribution of surviving defects. An extensive study of the mobility of SIA clusters containing two to 331 interstitials has been carried out using the molecular dynamics simulation technique for the temperature range from 180 to 1200 K. The results obtained show that clusters larger than three to four SIAs are one-dimensionally mobile in both Cu and Fe. Large clusters of more than 100 SIAs in Cu and 300 SIAs in Fe have significantly reduced mobility. The problem of describing one-dimensional (1D) motion in three-dimensional space is discussed. An attempt is made to describe the mobility of SIA clusters within the approximation of 1D diffusion. For clusters in both metals the effective migration energy of 1D diffusion as estimated via the jump frequency of the cluster centre of mass is found to be independent of the number of SIAs in the clusters, although the cluster jump frequency decreases with increasing cluster size. Mechanisms of 1D mobility of interstitial clusters are discussed. read less NOT USED (high confidence) H. Koguchi, “Surface deformation induced by a variation in surface stresses in anisotropic half-regions,” Philosophical Magazine. 2003. link Times cited: 6 Abstract: The deformations and the stresses in anisotropic half-region… read moreAbstract: The deformations and the stresses in anisotropic half-regions taking into account surface stresses originating from surface energy, which exists originally at surfaces and interfaces dividing phases, are analysed theoretically. In the present paper, the equilibrium equation of force considering surface stresses is used to calculate the inelastic deformation induced by a variation in surface stresses. The problem of varying surface stresses in a half-surface of a half-infinite anisotropic domain is analysed using the theory of elasticity. This problem is related to the occurrence of cracks in contaminated, oxidized or chemisorbed surfaces. Stress analysis on the basis of continuum mechanics is performed precisely under the boundary condition taking into account surface stresses. The Fourier transform technique is applied to perform the analysis, and the components of stress and displacement are expressed in an explicit form. The shear component of bulk stress attains infinity at the edge of discontinuity of the surface stresses, and the free surface deforms like an edge dislocation. This result suggests that cracking in a chemically contaminant surface is easier than in a clean surface. read less NOT USED (high confidence) S. Znám, D. Nguyen-Manh, D. Pettifor, and V. Vítek, “Atomistic modelling of TiAl I. Bond-order potentials with environmental dependence,” Philosophical Magazine. 2003. link Times cited: 55 Abstract: Bond-order potentials (BOPs) for L10 TiAl have been develope… read moreAbstract: Bond-order potentials (BOPs) for L10 TiAl have been developed and constructed within a tight-binding framework. In addition to the usual attractive bond-energy contribution arising from the formation of covalent bonds and pairwise contribution describing the overlap repulsion and electrostatic interaction, we have included an environmentally dependent term to represent the strong repulsion experienced by the valence sp electrons in transition metals and their alloys. The latter contribution is crucial for reproducing the negative Cauchy pressures of TiAl and other transition-metal-based intermetallic compounds. The constructed BOPs have been tested in the following ways: firstly, examination of the mechanical stability of the tetragonal L10 lattice with respect to large deformations and other crystal structures with the same stoichiometry; secondly, calculation of the γ surface for {111} and related evaluation of the energies of stacking-fault-type defects; thirdly, calculation of energies of the γ-γ interfaces that are present in the lamellar TiAl and energies associated with the formations of point defects in TiAl. The results of all these calculations show very good agreement with various ab-initio calculations. Importantly, we find that this potential is transferable to the different bonding environment in the hexagonal D019 Ti3Al. Hence these BOPs are suitable for atomistic study of dislocations and other extended defects not only in L10 TiAl but also in Ti3Al and possibly structures with other titanium-rich stoichiometries. read less NOT USED (high confidence) G. Ackland and S. Reed, “Two-band second moment model and an interatomic potential for caesium,” Physical Review B. 2002. link Times cited: 39 Abstract: A semiempirical formalism is presented for deriving interato… read moreAbstract: A semiempirical formalism is presented for deriving interatomic potentials for materials such as caesium or cerium which exhibit volume collapse phase transitions. It is based on the Finnis-Sinclair second-moment tight-binding approach, but incorporates two independent bands on each atom. The potential is cast in a form suitable for large-scale molecular dynamics, the computational cost being the evaluation of short-ranged pair potentials. Parameters for a model potential for caesium are derived and tested. read less NOT USED (high confidence) R. Cammarata, T. Trimble, and D. Srolovitz, “Surface stress model for intrinsic stresses in thin films,” Journal of Materials Research. 2000. link Times cited: 193 Abstract: A simple model was presented for intrinsic stress generation… read moreAbstract: A simple model was presented for intrinsic stress generation in thin films resulting from surface stress effects. This mechanism can explain the origin of compressive stresses often observed during island growth prior to coalescence, as well as intrinsic compressive stresses reported for certain continuous, fully grown films. In some cases, surface stress effects may contribute to a sudden change in the intrinsic stress during island coalescence. read less NOT USED (high confidence) T. Ochs, C. Elsässer, M. Mrovec, V. Vítek, J. Belak, and J. Moriarty, “Symmetrical tilt grain boundaries in bcc transition metals: Comparison of semiempirical with ab-initio total-energy calculations,” Philosophical Magazine A. 2000. link Times cited: 34 Abstract: Five different semiempirical total-energy methods, provided … read moreAbstract: Five different semiempirical total-energy methods, provided in the literature and applicable for atomistic simulations of extended defects in bcc transition metals, are investigated in a comparative study. The comparison is made with recent theoretical ab-initio (local-density-functional theory) and experimental (high-resolution transmission electron microscopy) studies for the specific case of the Σ = 5, (310)[001] symmetrical tilt grain boundaries (Σ = 5 STGBs) in Nb and Mo. The considered semiempirical real-space approaches based on different approximations of the tight-binding and related methods are the Finnis–Sinclair central-force potentials, non-central-force bond-order potentials recently advanced by Pettifor and co-workers, and non-central-force potentials based on the model-generalized pseudopotential theory of Moriarty. As semiempirical reciprocal-space methods, a very simple d-basis tight-binding model by Paxton and an elaborate environment dependent spd-basis orthogonal tight-binding model by Haas et al. are included in the analysis. The virtues and deficiencies of these models in their ability to predict the translation states and interfacial energies of the ∑ = 5 STGB are discussed. read less NOT USED (high confidence) O. H. Duparc, S. Poulat, A. Larere, J. Thibault, and L. Priester, “High-resolution transmission electron microscopy observations and atomic simulations of the structures of exact and near Σ = 11, 332 tilt grain boundaries in nickel,” Philosophical Magazine A. 2000. link Times cited: 43 Abstract: A Σ = 11, {332} nickel bicrystal was grown by solidification… read moreAbstract: A Σ = 11, {332} nickel bicrystal was grown by solidification. The aim of this paper is to show the evolution of the grain-boundary (GB) structure depending upon the position in the bicrystal of the extracted sample. Conventional transmission electron microscopy and high-resolution transmission electron microscopy (HRTEM) observations were used to characterize the GBs on microscopic and nanoscopic scales respectively. The detailed atomic structures of the exact {332} and the asymmetrical {111}‖{331} GBs were investigated by numerical calculations and compared with the HRTEM images. There is a perfect agreement between the calculated and the experimental image for the symmetrical {332} GB at the head of the bicrystal. The high GB defect density in the asymmetrical GBs at the end of the bicrystal makes the comparison less straightforward. It is, however, noteworthy that two complete periods of the calculated structure are actually observed in the experimental image. In order to approach the atomic description of the {111}1 ‖{hkl}2 asymmetrical tilt GBs, it is proposed to use for each side the structural units which appear in the corresponding symmetrical {111} and hkl tilt GBs respectively. read less NOT USED (high confidence) H. B. Groen, B. Kooi, W. Vellinga, and D. Hosson, “High-resolution transmission electron microscopy imaging of misfit-dislocation networks at Cu-MgO and Cu-MnO interfaces,” Philosophical Magazine. 1999. link Times cited: 28 Abstract: Misfit dislocation networks at Cu-MgO and Cu-MnO {111}metal/… read moreAbstract: Misfit dislocation networks at Cu-MgO and Cu-MnO {111}metal//{111}oxide interfaces were studied with high-resolution transmission electron microscopy. Experimental results were compared with image simulations of tentative atomic structures of the interface region derived from lattice statics calculations. The calculations take into account the two-dimensional misfit at the interface, which is necessary given the high misfit and short repeat distances at the interfaces. The lattice statics calculations use simplified potentials across the interface which capture essential characteristics that have emerged from recent experimental results and ab-initio calculations. Trigonal networks of edge misfit locations with Burgers vectors ⅙〈112〉 and line direction 〈110〉 follow from these calculations. These misfit-dislocation networks have associated strain fields in the metal, stretching out from the interface with approximately the repeat distance along the interface. These strain fields show up in image s... read less NOT USED (high confidence) S. Psakh’e, K. P. Zol’nikov, R. I. Kadryov, G. E. Rudenskii, and D. Saraev, “Interaction of solitary pulses produced by high-rate loading with a free surface,” Combustion, Explosion and Shock Waves. 1999. link Times cited: 0 NOT USED (high confidence) L. Wang, H. Liu, K. Chen, and Z. Hu, “The development of microstructure of Ni_3Al during rapid cooling and heating,” Journal of Materials Research. 1998. link Times cited: 4 Abstract: The processes of rapid solidification from liquid to solid a… read moreAbstract: The processes of rapid solidification from liquid to solid and heating from glass to crystalline for Ni_3Al are simulated using molecular dynamics method. An amorphous state can be obtained by rapid solidification as long as the cooling rate is sufficiently large, which is very difficult to get in experiment. An fcc-type crystalline is obtained by heating the amorphous with a small heating rate. Based on the pair analysis technique, the microstructures of liquid, supercooled liquid, amorphous, and crystalline states of Ni_3Al have been analyzed. Furthermore, the effects of cooling rate and heating rate on microstructures of Ni_3Al during rapid solidification and heating processes have been discussed. read less NOT USED (high confidence) C. Schmidt, M. Finnis, F. Ernst, and V. Vítek, “Theoretical and experimental investigations of structures and energies of Σ = 3, [112] tilt grain boundaries in copper,” Philosophical Magazine. 1998. link Times cited: 51 Abstract: Using atomistic computer simulations, symmetric and asymmetr… read moreAbstract: Using atomistic computer simulations, symmetric and asymmetric Σ = 3 tilt grain boundaries in Cu were investigated. Equilibrium energies and structures were calculated by static and dynamic energy minimization. A semi-empirical N-body potential served as a model of the interatomic forces in Cu. The atomistic structure of the grain boundary inclined at about 84° to the {111} twin boundary was investigated by high-resolution transmission electron microscopy (HRTEM). Plotted against the inclination angle Φ112 of the boundary plane, the calculated grain boundary energies increase monotonically up to Φ112 ≈ 73°. At larger inclination angles the data indicate an energy minimum at about 80°. The computer simulations predic that boundaries equilibrated at temperatures near T = 0K are planar for inclination angles σ112 < 73°, but consist of a three-dimensional layer of predominantly body-centred-cubic (bcc) Cu for inclination angles greater than 73°. In all three-dimensional boundaries by bcc layer ... read less NOT USED (high confidence) P. Beauchamp and J. Lépinoux, “Atomistic computation of the image force on a dislocation in a bicrystal II. Case of a large difference between the elastic moduli of the two half-crystals,” Philosophical Magazine. 1998. link Times cited: 4 Abstract: The image force on a dislocation near an interface separatin… read moreAbstract: The image force on a dislocation near an interface separating two half-crystals of different elastic moduli is investigated by computer simulation. The 〈001〉 screw dislocation and the (100) interface between two bcc crystals have been chosen because the dislocation is undissociated and the anisotropic boundary conditions have an analytic form allowing accurate calculations. In the homogeneous medium, the dislocation core is found to be planar and to extend along a {110} dense plane but the core widths depend on the type of atomic potential utilized. To construct the bicrystal, the same potential is used in medium 2 as in medium 1, but multiplied by a factor α, equal to the desired ratio μ2/μ1 of shear moduli. The study is restricted here to values of α slightly larger than unity and sufficiently small for the lattice friction to remain everywhere larger than the image force. The dislocation is placed at distances x from the interface, ranging from 0 to 6a0, and for each stable position, the confi... read less NOT USED (high confidence) Z. Guohui, L. Hong, W. Fa-rong, C. Wuyang, and Z. Fuxin, “Molecular dynamics simulations of DFZ,” Acta Mechanica Sinica. 1997. link Times cited: 0 NOT USED (high confidence) J. Purton, S. C. Parker, and D. Bullett, “Computer modelling of metal - oxide interfaces,” Journal of Physics: Condensed Matter. 1997. link Times cited: 34 Abstract: We have used atomistic simulations to model oxide - metal in… read moreAbstract: We have used atomistic simulations to model oxide - metal interfaces. We have, for the first time, allowed the atoms on both sides of the interface to relax. The efficiency of the computational method means that calculations can be performed on complex interfaces containing several thousand atoms and do not require an arbitrary definition of the image plane to model the electrostatics across the dielectric discontinuity. We demonstrate the viability of the approach and the effect of relaxation on a range of MgO - Ag interfaces. Defective and faceted interfaces, as well as the ideal case, have been studied. The latter was chosen for comparison with previous theoretical calculations and experimental results. The wetting angle and work of adhesion for MgO{100} - Ag{100} are in reasonable agreement with experiment. As with ab initio electronic structure calculations the silver atoms have been shown to favour the position above the oxygen site. read less NOT USED (high confidence) V. Paidar, A. Larere, and L. Priester, “Exponential many-body potentials and elastic constants of fcc transition metals,” Modelling and Simulation in Materials Science and Engineering. 1997. link Times cited: 6 Abstract: The elastic constants of fcc transition metals are studied a… read moreAbstract: The elastic constants of fcc transition metals are studied as functions of the exponential parameters of a many-body potential based on the second moment approximation of the d-electron density of states. It is shown that this model gives a reasonable agreement with the experimental values not only for all three independent elastic constants but also for the vacancy formation energy for Cu, Ag, Au and Ni. Less satisfactory results are obtained for Ir, Rh, Pt and Pd. read less NOT USED (high confidence) R. Fonda, M. Yan, and D. Luzzi, “A study of grain-boundary structure in non-stoichiometric NiAl by atomistic simulation and electron microscopy,” Philosophical Magazine. 1997. link Times cited: 3 Abstract: The atomic structure of a Σ =5,(310)[001] grain boundary in … read moreAbstract: The atomic structure of a Σ =5,(310)[001] grain boundary in NiAl has been determined by a synergistic approach using high-resolution electron microscopy (HREM) and atomistic structure calculations. The effect and distribution of point defects at the grain boundary were determined using molecular statics calculations employing N-body empirical potentials to calculate the relaxed grain-boundary structures and energies resulting from various initial structures and stoichiometries. Monte Carlo calculations confirm the stability of the lowest-energy structure, which contains Ni antisite defects adjacent to the grain-boundary plane. Multislice image simulations of this structure are in good agreement with the experimental HREM image. This is the first combined application of experimental and theoretical structure determinations to an intermetallic grain boundary. read less NOT USED (high confidence) G. Ackland, D. Bacon, A. Calder, and T. Harry, “Computer simulation of point defect properties in dilute Fe—Cu alloy using a many-body interatomic potential,” Philosophical Magazine. 1997. link Times cited: 594 Abstract: The behaviour of copper atoms in dilute solution in α-iron i… read moreAbstract: The behaviour of copper atoms in dilute solution in α-iron is important for the microstructural changes that occur in ferritic pressure vessel steels under fastneutron irradiation. To investigate the properties of atomic defects that control this behaviour, a set of many-body interatomic potentials has been developed for the Fe—Cu system. The procedures employed, including modifications to ensure suitability for simulating atomic collisions at high energy, are described. The effect of copper on the lattice parameter of iron in the new model is in good agreement with experiment. The phonon properties of the pure crystals and, in particular, the influence of the instability of the metastable, bcc phase of copper that precipitates during irradiation are discussed. The properties of point defects have been investigated. It is found that the vacancy has lower formation and migration energy in bcc copper than in α-iron, and the self-interstitial atom has very low formation energy in this phase of coppe... read less NOT USED (high confidence) M. Azzaoui and M. Hou, “Temperature dependencies of the relaxation, order and segregation at a tilt grain boundary in,” Journal of Physics: Condensed Matter. 1996. link Times cited: 2 Abstract: The thermal dependence of the relaxation, order and segregat… read moreAbstract: The thermal dependence of the relaxation, order and segregation in the vicinity of a tilt grain boundary in the binary alloy is investigated by means of computer simulation with an empirical N-body potential. Energy minimization is performed in order to estimate the particularly strong relaxation effects in the vicinity of the boundary plane at 0 K. Monte Carlo simulations are carried out for constant chemical potential, number of particles, volume and temperature in order to study the thermal properties of the system. Detail is provided plane by plane, parallel to the boundary, which characterizes the temperature dependencies of the order, segregation, sublattice occupancy and relaxation. The vicinity of the boundary remains strongly affected by atomic relaxation and segregation at all temperatures, although no simple relation between relaxation and segregation is found. The evolution of long-range order and sublattice occupancy are strikingly different in the close vicinity of the boundary plane to those in the bulk. The boundary plane is fully disordered at all temperatures between and , where is the bulk temperature for the order - disorder phase transition. The transition to bulk properties with distance from the boundary is characterized quantitatively. The influence of the potential model is emphasized by means of a comparison between the results obtained with two somewhat different N-body potentials of similar nature. read less NOT USED (high confidence) Y. Zhang and T. Wang, “Molecular dynamics simulation of interaction of a dislocation array from a crack tip with grain boundaries,” Modelling and Simulation in Materials Science and Engineering. 1996. link Times cited: 18 Abstract: The interaction of a dislocation array emitted from a crack … read moreAbstract: The interaction of a dislocation array emitted from a crack tip under mode II loading with asymmetric tilt grain boundaries (GBs) is analysed by the molecular dynamics method. The GBs can generally be described by planar and linear matching zones and unmatching zones. All GBs are observed to emit dislocations. The GBs migrated easily due to their planar and linear matching structure and asymmetrical type. The diffusion induced by stress concentration is found to promote the GB migration. The transmissions of dislocations are either along the matched plane or along another plane depending on tilt angle . Alternate processes of stress concentration and stress relaxation take place ahead of the pileup. The stress concentration can be released either by transmission of dislocations, by atom diffusion along GBs, or by migration of GBs by formation of twinning bands. The simulated results also unequivocally demonstrate two processes, i.e. asymmetrical GBs evolving into symmetrical ones and unmatching zones evolving into matching ones during the loading process. read less NOT USED (high confidence) T.-M. Chang and E. Carter, “Mean-field theory of heteroepitaxial thin metal film morphologies,” Surface Science. 1994. link Times cited: 8 NOT USED (high confidence) R. Cammarata, “SURFACE AND INTERFACE STRESS EFFECTS IN THIN FILMS,” Progress in Surface Science. 1994. link Times cited: 866 NOT USED (high confidence) D. Fuks, J. Pelleg, S. Rashkeev, and S. Dorfman, “Calculations of vacancy formation and migration energies offcc Cs by the LMTO-method: Application to self diffusion,” Zeitschrift für Physik B Condensed Matter. 1993. link Times cited: 0 NOT USED (high confidence) Z. Bangwei and O. Yijang, “Calculations of the thermodynamic properties for binary hcp alloys with simple embedded atom method model,” Zeitschrift für Physik B Condensed Matter. 1993. link Times cited: 3 NOT USED (high confidence) B. J. Pestman, J. Hosson, V. Vítek, and F. Schapink, “Interaction between lattice dislocations and grain boundaries in f.c.c. and ordered compounds: A computer simulation,” Philosophical Magazine. 1991. link Times cited: 65 Abstract: The interaction of ½ screw and 60° dislocations with symmetr… read moreAbstract: The interaction of ½ screw and 60° dislocations with symmetric [110] tilt boundaries was investigated by atomistic simulations using many-body potentials representing a pure f.c.c. metal and ordered intermetallic compounds. The calculations were performed with and without an applied shear stress. The observations were: absorption into the grain boundary, attraction of a lattice Shockley partial dislocation towards the grain boundary and transmission through the grain boundary under the influence of a shear stress. It was found that the structural unit model may help to predict the interaction mechanism for long period boundaries and that the interaction in ordered compounds shows similarities to the interaction in f.c.c. metals. Some comparisons with experimental observations have been made. read less NOT USED (high confidence) J. Hosson and V. Vítek, “Atomic structure of (111) twist grain boundaries in f.c.c. metals,” Philosophical Magazine. 1990. link Times cited: 26 Abstract: In this paper we have studied the atomic structures of (111)… read moreAbstract: In this paper we have studied the atomic structures of (111) twist boundaries and investigated the applicability of the structural unit model which has previously been established for tilt boundaries and (001) twist boundaries by Sutton and Vitek. The calculations were carried out using two different descriptions of interatomic forces. A pair potential for aluminium, for which the calculations were made at constant volume, and a many-body potential for gold, for which the calculations were performed at constant pressure. The atomic structures of all the boundaries studied were found to be very similar for both the descriptions of atomic interactions. This suggests that the principal features of the structure of (111) twist boundaries found in this study are common to all f.c.c. metals. At the same time it supports the conclusion that calculations employing pair potentials are fully capable of revealing the generic features of the structure of grain boundaries in metals. The results obtained here,... read less NOT USED (high confidence) C. Becquart, N. Mousseau, and C. Domain, “Kinetic Monte Carlo Simulations of Irradiation Effects,” Comprehensive Nuclear Materials. 2020. link Times cited: 16 NOT USED (high confidence) M. Jia, Y. Lai, Z. Tian, and Y.-xiang Liu, “Calculation of the surface free energy of fcc copper nanoparticles,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 31 Abstract: Using molecular dynamics simulations with the modified analy… read moreAbstract: Using molecular dynamics simulations with the modified analytic embedded-atom method we calculate the Gibbs free energy and surface free energy for fcc Cu bulk, and further obtain the Gibbs free energy of nanoparticles. Based on the Gibbs free energy of nanoparticles, we have investigated the heat capacity of copper nanoparticles. Calculation results indicate that the Gibbs free energy and the heat capacity of nanoparticles can be divided into two parts: bulk quantity and surface quantity. The molar heat capacity of the bulk sample is lower compared with the molar heat capacity of nanoparticles, and this difference increases with the decrease in the particle size. It is also observed that the size effect on the thermodynamic properties of Cu nanoparticles is not really significant until the particle is less than about 20 nm. It is the surface atoms that decide the size effect on the thermodynamic properties of nanoparticles. read less NOT USED (high confidence) F. Berthier, B. Legrand, and G. Tréglia, “New Structures and Atomistic Analysis of the Polymorphism for the ∑ = 5 (210) [001] Tilt Boundary,” Interface Science. 2000. link Times cited: 17 |
 EAM_Dynamo_AcklandTichyVitek_1987_Cu__MO_179025990738_005
EAM_Dynamo_AcklandTichyVitek_1987_Cu__MO_179025990738_005