Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
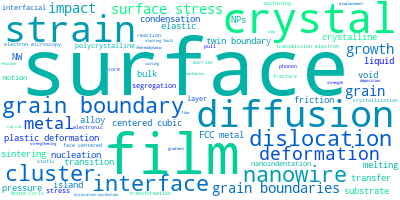
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
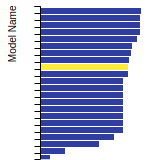
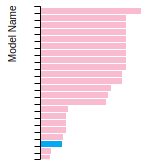
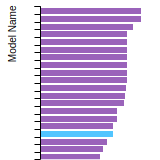
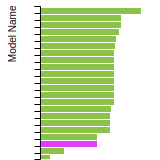
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
3345 Citations (943 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) M. Seiz, H. Hierl, and B. Nestler, “Unravelling densification during sintering by multiscale modelling of grain motion,” Journal of Materials Science. 2023. link Times cited: 1 USED (high confidence) T. Brink, L. Langenohl, H. Bishara, and G. Dehm, “Universality of grain boundary phases in fcc metals: Case study on high-angle [111] symmetric tilt grain boundaries,” Physical Review B. 2022. link Times cited: 6 Abstract: Grain boundaries often exhibit ordered atomic structures. In… read moreAbstract: Grain boundaries often exhibit ordered atomic structures. Increasing amounts of evidence have been provided by transmission electron microscopy and atomistic computer simulations that different stable and metastable grain boundary structures can occur. Meanwhile, theories to treat them thermodynamically as grain boundary phases have been developed. Whereas atomic structures were identified at particular grain boundaries for particular materials, it remains an open question if these structures and their thermodynamic excess properties are material specific or generalizable to, e.g., all fcc metals. In order to elucidate that question, we use atomistic simulations with classical interatomic potentials to investigate a range of high-angle [111] symmetric tilt grain boundaries in Ni, Cu, Pd, Ag, Au, Al, and Pb. We could indeed find two families of grain boundary phases in all of the investigated grain boundaries, which cover most of the standard fcc materials. Where possible, we compared the atomic structures to atomic-resolution electron microscopy images and found that the structures match. This poses the question if the grain boundary phases are simply the result of sphere-packing geometry or if material-specific bonding physics play a role. We tested this using simple model pair potentials and found that medium-ranged interactions are required to reproduce the atomic structures, while the more realistic material models mostly affect the grain boundary (free) energy. In addition to the structural investigation, we also report the thermodynamic excess properties of the grain boundaries, explore how they influence the thermodynamic stability of the grain boundary phases, and detail the commonalities and differences between the materials. read less USED (high confidence) H. Jo et al., “Direct strain correlations at the single-atom level in three-dimensional core-shell interface structures,” Nature Communications. 2022. link Times cited: 8 USED (high confidence) C. Melis et al., “Stiffening of nanoporous gold: experiment, simulation and theory,” The European Physical Journal Plus. 2022. link Times cited: 1 USED (high confidence) E. Pervolarakis, G. Tritsaris, P. Rosakis, and I. Remediakis, “Machine Learning for the edge energies of high symmetry Au nanoparticles,” Surface Science. 2022. link Times cited: 1 USED (high confidence) M. Dupraz et al., “Imaging the facet surface strain state of supported multi-faceted Pt nanoparticles during reaction,” Nature Communications. 2022. link Times cited: 8 USED (high confidence) J. Hu, H. Song, S. Sandfeld, X. Liu, and Y. Wei, “Breakdown of Archard law due to transition of wear mechanism from plasticity to fracture,” Tribology International. 2022. link Times cited: 10 USED (high confidence) Z. Zhu et al., “Molecular Dynamics Study on Nano-Friction and Wear Mechanism of Nickel-Based Polycrystalline Superalloy Coating,” Coatings. 2021. link Times cited: 2 Abstract: In this work, molecular dynamics simulations are employed to… read moreAbstract: In this work, molecular dynamics simulations are employed to study the nanotribological process of nickel-based polycrystalline superalloy coating. A series of simulations were carried out using the method of repeated friction to explore the influence of frictional force, friction coefficient, grinding groove morphology, wear scar depth, debris flow direction, subsurface damage degree and evolution of defects during the nano-friction process. In addition, the change mechanism of different grain sizes on wear scar depth, frictional force, friction coefficient, and internal damage in the repeated friction process is also explored. The results show that the frictional force is related to the direction of the dislocation slip, and that the friction coefficient change is related to the number of repeated frictions. Moreover, it is observed that the grinding ball has a shunting effect on the formed wear debris atoms, and the shunt point is located at the maximum horizontal radius. We reveal that the grain boundary structure has a strengthening effect. When the grinding ball rubs to the grain boundary, the nucleation of dislocation defects inside the workpiece is obviously hindered by it. Simultaneously, we also find that the closer the subsurface is to the bottom of the grinding ball, the greater the degree of damage to the workpiece by friction. Furthermore, with the grain size decreases that the material begins to soften, resulting in a decrease of frictional force, friction coefficient, and smaller defects are formed inside the workpiece. The research of this work can better clarify the microscopic mechanism of the polycrystalline friction process. read less USED (high confidence) C. Zaum, N. Osterloh, R. Darkins, D. Duffy, and K. Morgenstern, “Real-space observation of surface structuring induced by ultra-fast-laser illumination far below the melting threshold,” Scientific Reports. 2021. link Times cited: 3 USED (high confidence) S. Tauchert et al., “Polarized phonons carry angular momentum in ultrafast demagnetization,” Nature. 2021. link Times cited: 72 USED (high confidence) G. D. Leines and J. Rogal, “Template-Induced Precursor Formation in Heterogeneous Nucleation: Controlling Polymorph Selection and Nucleation Efficiency.,” Physical review letters. 2021. link Times cited: 3 Abstract: We present an atomistic study of heterogeneous nucleation in… read moreAbstract: We present an atomistic study of heterogeneous nucleation in Ni employing transition path sampling, which reveals a template precursor-mediated mechanism of crystallization. Most notably, we find that the ability of tiny templates to modify the structural features of the liquid and promote the formation of precursor regions with enhanced bond-orientational order is key to determining their nucleation efficiency and the polymorphs that crystallize. Our results reveal an intrinsic link between structural liquid heterogeneity and the nucleating ability of templates, which significantly advances our understanding toward the control of nucleation efficiency and polymorph selection. read less USED (high confidence) X. Song, S. Wu, and R. Zhang, “Computational Study on Surface Bonding Based on Nanocone Arrays,” Nanomaterials. 2021. link Times cited: 3 Abstract: Surface bonding is an essential step in device manufacturing… read moreAbstract: Surface bonding is an essential step in device manufacturing and assembly, providing mechanical support, heat transfer, and electrical integration. Molecular dynamics simulations of surface bonding and debonding failure of copper nanocones are conducted to investigate the underlying adhesive mechanism of nanocones and the effects of separation distance, contact length, temperature, and size of the cones. It is found that van der Waals interactions and surface atom diffusion simultaneously contribute to bonding strength, and different adhesive mechanisms play a main role in different regimes. The results reveal that increasing contact length and decreasing separation distance can simultaneously contribute to increasing bonding strength. Furthermore, our simulations indicate that a higher temperature promotes diffusion across the interface so that subsequent cooling results in better adhesion when compared with cold bonding at the same lower temperature. It also reveals that maximum bonding strength was obtained when the cone angle was around 53°. These findings are useful in designing advanced metallic bonding processes at low temperatures and pressure with tenable performance. read less USED (high confidence) S. Rawat and S. Chaturvedi, “Evolution dynamics of voids in single crystal copper under triaxial loading condition,” Philosophical Magazine. 2021. link Times cited: 1 Abstract: ABSTRACT An understanding of nucleation, growth and coalesce… read moreAbstract: ABSTRACT An understanding of nucleation, growth and coalescence of voids is required to predict the spall fracture. We employ molecular dynamics simulations to investigate the effect of temperature on the nucleation and growth of voids in single crystal copper under triaxial loading condition. We find that the void density decreases with an increase in temperature. The nucleation rate of the voids diminishes as temperature increases for the range of 300–1250 K. The individual void volume fraction evolves with discrete jumps due to coalescence events. The overall void volume fraction at 1250 K appears earlier than that at 300 K indicating the failure of the material earlier at higher temperatures. The void dimensions evolve with discrete jumps due to coalescence of the voids. The void dimensions change with the applied temperature. The void size distribution at each applied temperature follows a polynomial function. read less USED (high confidence) R. Fahdiran, E. Handoko, I. Sugihartono, and S. Budi, “Melting dynamics of Gold thin film: A Molecular Dynamics study,” IOP Conference Series: Materials Science and Engineering. 2021. link Times cited: 0 Abstract: We studied the melting dynamics of Gold in the form of thin … read moreAbstract: We studied the melting dynamics of Gold in the form of thin film. The thin film thickness is 9.756 nm. The systems are heated from room temperature up to slightly above melting point, Tm = 1338 K. The evolution of the atoms in each system is followed using Molecular Dynamics (MD) scheme up to 20 ps. Thermodynamics analysis indicated that thin film is suppressed by the uniform heat while the temperature is increasing. Structural analysis compared with Common Neighbor Analysis (CNA) expressed the condition that melting conduction occurred at the end of simulation. read less USED (high confidence) R. Fahdiran et al., “Molecular Dynamics study on size dependencies of melting dynamics in Gold thin film,” IOP Conference Series: Materials Science and Engineering. 2021. link Times cited: 0 Abstract: We explore the size dependencies of melting dynamics of Gold… read moreAbstract: We explore the size dependencies of melting dynamics of Gold in the form of thin film. The sizes are 4.896 nm, 7.344 nm and 9.792 nm which is comparable between nanoparticle diameter and thin film thickness. The systems are treated by increasing temperature from 300 K to 1400 K. Molecular Dynamics (MD) scheme is employed to follow the trajectories of the systems up to 20 ps. Structure factor analysis indicated that the melting is suppressed by the increasing size. read less USED (high confidence) H. Kim, D. T. Ho, and S. Y. Kim, “Fracture Behavior Transition in (001) Cracked Metal Nanoplates Induced by the Surface Effect,” Journal of Physical Chemistry C. 2021. link Times cited: 0 Abstract: We gratefully acknowledge the support from the Mid-Career Re… read moreAbstract: We gratefully acknowledge the support from the Mid-Career Researcher Support Program (Grant No. 2019R1A2C2011312) of the National Research Foundation (NRF) of Korea and from the Meta-Structure Based Seismic Shielding Research Fund (Project No. 1.200043.01) of UNIST. We also acknowledge with gratitude the supercomputing resources of the UNIST supercomputing Center. read less USED (high confidence) S. Zhang, Q. Zhang, X. Liu, and S. Luo, “Tunable Poisson’s ratio and tension-compression asymmetry of graphene-copper nanolayered composites,” Journal of Physics D: Applied Physics. 2021. link Times cited: 2 Abstract: The Poisson’s ratios of graphene-copper nanolayered (GrCuNL)… read moreAbstract: The Poisson’s ratios of graphene-copper nanolayered (GrCuNL) composites under tension and compression are investigated by molecular dynamics and theoretical analysis. The Poisson’s ratio of a GrCuNL composite can be tuned by tailoring its repeat layer spacing without changing the topological structures. The effect of constituent nanocrystalline Cu grain size on the Poisson’s ratio is negligible. There are remarkable in-plane anisotropy and tension-compression asymmetry in the Poisson’s ratio due to the chiral difference in compressive stress in graphene layers. A mechanical model considering the chirality and repeat layer spacing is proposed, which can accurately predict the Poisson’s ratio of a GrCuNL composite. For stable GrCuNL composites, the repeat layer spacing should be larger than 2 nm, and their tunable range of Poisson’s ratio is 0.1–0.35. read less USED (high confidence) C. Zhang, R. Fuller, and I. Hijazi, “Quaternary Hydrides Pd1-y-zAgyCuzHx Embedded Atom Method Potentials for Hydrogen Energy Applications,” Journal of Energy and Power Technology. 2021. link Times cited: 0 Abstract: The Pd-H system has attracted extensive attention. Pd can ab… read moreAbstract: The Pd-H system has attracted extensive attention. Pd can absorb considerable amount of H at room temperature, this ability is reversible, so it is suitable for multiple energy applications. Pd-Ag alloys possess higher H permeability, solubility and narrower miscibility gap with better mechanical properties than pure Pd, but sulfur poisoning remains an issue. Pd-Cu alloys have excellent resistance to sulfur and carbon monoxide poisoning and hydrogen embrittlement, good mechanical properties, and broader temperature working environments over pure Pd, but relatively lower hydrogen permeability and solubility than pure Pd and Pd-Ag alloys. This suggests that alloying Pd with Ag and Cu to create Pd-Ag-Cu ternary alloys can optimize the overall performance and substantially lowers the cost. Thus, in this paper, we provide the first embedded atom method potentials for the quaternary hydrides Pd1-y-zAgyCuzHx. The fully analytical potentials are fitted utilizing the central atom method without performing time-consuming molecular dynamics simulations. read less USED (high confidence) M. Liao and L. Duan, “Investigation of Coalescence-Induced Droplet Jumping on Mixed-Wettability Superhydrophobic Surfaces,” Processes. 2021. link Times cited: 2 Abstract: Coalescence-induced droplet jumping has received more attent… read moreAbstract: Coalescence-induced droplet jumping has received more attention recently, because of its potential applications in condensation heat transfer enhancement, anti-icing and self-cleaning, etc. In this paper, the molecular dynamics simulation method is applied to study the coalescence-induced jumping of two nanodroplets with equal size on the surfaces of periodic strip-like wettability patterns. The results show that the strip width, contact angle and relative position of the center of two droplets are all related to the jumping velocity, and the jumping velocity on the mixed-wettability superhydrophobic surfaces can exceed the one on the perfect surface with a 180° contact angle on appropriately designed surfaces. Moreover, the larger both the strip width and the difference of wettability are, the higher the jumping velocity is, and when the width of the hydrophilic strip is fixed, the jumping velocity becomes larger with the increase of the width of the hydrophobic strip, which is contrary to the trend of fixing the width of the hydrophobic strip and altering the other strip width. read less USED (high confidence) G. Martins, B. de P Cardoso, N. Galamba, and B. Cabral, “Exploring a near-Hartree-Fock-Kohn-Sham approach to study electronic properties of azobenzene in interaction with gold: From clusters to the Au(111) surface.,” The Journal of chemical physics. 2020. link Times cited: 3 Abstract: The electronic properties of azobenzene (AB) in interaction … read moreAbstract: The electronic properties of azobenzene (AB) in interaction with gold clusters and adsorbed on the Au(111) surface are investigated by adopting a near-Hartree-Fock-Kohn-Sham (HFKS) scheme. This scheme relies on a hybrid Perdew-Burke-Ernzerhof functional, in which the exact non-local HF exchange contribution to the energy is taken as 3/4. Ionization energies and electron affinities for gas phase AB are in very good agreement with experimental data and outer valence Green's function) calculations. The presence of C-H⋯Au interactions in AB-Aun complexes illustrates the role played by weak interactions between molecular systems and Au nanoparticles, which is in line with recent works on Au-H bonding. In AB-Aun complexes, the frontier orbitals are mainly localized on the gold platform when n ≥ 10, which indicates the transition from a molecular to a semiconducting regime. In the latter regime, the electronic density reorganization in AB-Aun clusters is characterized by significant polarization effects on the Au platform. The accuracy of the near-HFKS scheme for predicting adsorption energies of AB on Au(111) and the interest of combining exact non-local HF exchange with a non-local representation of the dispersion energy are discussed. Taking into account the significant computational cost of the exact non-local HF exchange contribution, calculations for the adsorption energies and density of states for AB adsorbed on Au(111) were carried out by using a quantum mechanics/molecular mechanics approach. The results strongly support near-HFKS as a promising methodology for predicting the electronic properties of hybrid organic-metal systems. read less USED (high confidence) A. Nemati, H. N. Pishkenari, A. Meghdari, and S. Ge, “Directional control of surface rolling molecules exploiting non-uniform heat-induced substrates.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 5 Abstract: Molecular machines, such as nanocars, have shown promising p… read moreAbstract: Molecular machines, such as nanocars, have shown promising potential for various tasks, including manipulation at the nanoscale. In this paper, we examined the influence of temperature gradients on nanocar and nanotruck motion as well as C60 - as their wheel - on a flat gold surface under various conditions. We also compared the accuracy and computational cost of two different approaches for generating the temperature gradient. The results show that severe vibrations and frequent impacts of gold atoms at high temperatures increase the average distance of C60 from the substrate, reducing its binding energy. Moreover, the temperature field drives C60 to move along the temperature variation; still, the diffusive motion of C60 remained unchanged in the direction perpendicular to the temperature gradient. Increasing the magnitude of the temperature gradient speeds up its motion parallel to the gradient, while raising the average temperature of the substrate increases the diffusion coefficient in all directions. The temperature field influences the nanocar motion in the same manner as C60. However, the nanocars have a substantially shorter motion range compared to C60. The relatively larger, heavier, and more flexible chassis of the nanocar makes it more sluggish than the nanotruck. In general, the motion of large and heavy surface rolling molecules is less affected by the temperature field compared to small and light molecules. The results of the study show that concentrated heat sources can be employed to push surface rolling molecules or break down their large clusters. We can exploit a temperature field as a driving force to push nanocars in a desired direction on prebuilt pathways. read less USED (high confidence) M. Mikelani, M. Panjepour, and A. Taherizadeh, “Investigation on mechanical properties of nanofoam aluminum single crystal: using the method of molecular dynamics simulation,” Applied Physics A. 2020. link Times cited: 3 USED (high confidence) M. Bagheripoor and R. Klassen, “Effect of crystal orientation on the size effects of nano-scale fcc metals,” Materials Science and Technology. 2020. link Times cited: 3 Abstract: The present work investigates the dominant mechanisms in the… read moreAbstract: The present work investigates the dominant mechanisms in the plasticity of nano-sized fcc metallic samples. Molecular dynamics simulations of nanopillar compression show that plasticity always starts with the nucleation of dislocations at the free surface, and the crystal orientation affects the subsequent microstructural evolution. The Schmid factor of leading and trailing partials plays a decisive role in leading to the twinning, or slip deformation. A significant difference is observed in the strength of pillars of the same size with different orientations. The power-law equation exponent is completely dependent on the crystal orientations, and a weak or no size effect is observed in the compression of [100]- and [110]-oriented nanopillars. The observed orientation based behaviour decreases by confining the free surface. read less USED (high confidence) J. Liu, X. Fan, Y. Shi, D. J. Singh, and W. Zheng, “The Effect of Strain Rate on the Deformation Processes of NC Gold with Small Grain Size,” Crystals. 2020. link Times cited: 3 Abstract: The strength of nanocrystalline (NC) metal has been found to… read moreAbstract: The strength of nanocrystalline (NC) metal has been found to be sensitive to strain rate. Here, by molecular dynamics simulation, we explore the strain rate effects on apparent Young’s modulus, flow stress and grain growth of NC gold with small size. The simulation results indicate that the apparent Young’s modulus of NC gold decreases with the decrease of strain rate, especially for strain rates above 1 ns−1. The rearrangement of atoms near grain boundaries is a response to the decrease of apparent Young’s modulus. Indeed, the flow stress is also sensitive to the strain rate and decreases following the strain rate’s decrease. This can be found from the change of strain rate sensitivity and activation volume with the strain rate. Temperature has little effect on the activation volume of NC gold with small grain size, but has an obvious effect on that of relatively large grain size (such as 18 nm) under low strain rate (0.01 ns−1). Finally, grain growth in the deformation process is found to be sensitive to strain rate and the critical size for grain growth increases following the decrease of strain rate. read less USED (high confidence) M. Papanikolaou, F. Hernández, and K. Salonitis, “Investigation of the Subsurface Temperature Effects on Nanocutting Processes via Molecular Dynamics Simulations,” Metals. 2020. link Times cited: 5 Abstract: In this investigation, three-dimensional molecular dynamics … read moreAbstract: In this investigation, three-dimensional molecular dynamics simulations have been performed in order to investigate the effects of the workpiece subsurface temperature on various nanocutting process parameters including cutting forces, friction coefficient, as well as the distribution of temperature and equivalent Von Mises stress at the subsurface. The simulation domain consists of a tool with a negative rake angle made of diamond and a workpiece made of copper. The grinding speed was considered equal to 100 m/s, while the depth of cut was set to 2 nm. The obtained results suggest that the subsurface temperature significantly affects all of the aforementioned nanocutting process parameters. More specifically, it has been numerically validated that, for high subsurface temperature values, thermal softening becomes dominant and this results in the reduction of the cutting forces. Finally, the dependency of local properties of the workpiece material, such as thermal conductivity and residual stresses on the subsurface temperature has been captured using numerical simulations for the first time to the authors’ best knowledge. read less USED (high confidence) D. Chatain et al., “Influence of step structure on preferred orientation relationships of Ag deposited on Ni(111),” Acta Materialia. 2020. link Times cited: 1 USED (high confidence) L. Zou et al., “Atomic-scale phase separation induced clustering of solute atoms,” Nature Communications. 2020. link Times cited: 8 USED (high confidence) A. E. Korenchenko, B. R. Gel’chinskii, A. Vorontsov, and A. Zhukova, “Statistical Model for the Energy Exchange during Copper Vapor Condensation in an Inert Gas Atmosphere,” Russian Metallurgy (Metally). 2020. link Times cited: 0 USED (high confidence) J. Zhang, L. Su, and Z. Wang, “Concurrent Multiscale Simulations of Rough Lubricated Contact of Aluminum Single Crystal,” Metals. 2020. link Times cited: 2 Abstract: In this paper, a concurrent multiscale simulation strategy c… read moreAbstract: In this paper, a concurrent multiscale simulation strategy coupling atomistic and continuum models was proposed to investigate the three-dimensional contact responses of aluminum single crystal under both dry and lubricated conditions. The Hertz contact is performed by using both the multiscale and full molecular dynamics (MD) simulations for validation. From the contact area, kinetic energy and stress continuity aspects, the multiscale model shows good accuracy. It can also save at least five times the computational time compared with the full MD simulations for the same domain size. Furthermore, the results of lubricated contact show that the lubricant molecules could effectively cover the contact surfaces; thereby separating the aluminum surfaces and bearing the support loads. Moreover, the surface topography could be protected by the thin film formed by the lubricant molecules. It has been found that the contact area decreases obviously with increasing the magnitude of load under both dry and lubricated contacts. Besides, a decrease in contact area is also seen when the number of lubricant molecules increases. The present study has confirmed that the dimension of lubricated contacts could be greatly expanded during the simulation using the proposed multiscale method without sacrificing too much computational time and accuracy. read less USED (high confidence) P. Brault, “Multiscale Molecular Dynamics Simulations of Fuel Cell Nanocatalyst Plasma Sputtering Growth and Deposition,” Energies. 2020. link Times cited: 2 Abstract: Molecular dynamics simulations (MDs) are carried out for pre… read moreAbstract: Molecular dynamics simulations (MDs) are carried out for predicting platinum Proton Exchange Membrane (PEM) fuel cell nanocatalyst growth on a model carbon electrode. The aim is to provide a one-shot simulation of the entire multistep process of deposition in the context of plasma sputtering, from sputtering of the target catalyst/transport to the electrode substrate/deposition on the porous electrode. The plasma processing reactor is reduced to nanoscale dimensions for tractable MDs using scale reduction of the plasma phase and requesting identical collision numbers in experiments and the simulation box. The present simulations reproduce the role of plasma pressure for the plasma phase growth of nanocatalysts (here, platinum). read less USED (high confidence) S. Ajori, H. Parsapour, and R. Ansari, “A molecular dynamics study on the buckling behavior of single-walled carbon nanotubes filled with gold nanowires,” Journal of Molecular Modeling. 2020. link Times cited: 7 USED (high confidence) D. Xu, Z. Wang, T.-Y. Chang, and F. Chen, “Inverted core–shell potential energy landscape of icosahedral clusters in deeply undercooled metallic liquids and glasses and its effect on the glass forming ability of bcc and fcc metals,” Journal of Physics: Condensed Matter. 2020. link Times cited: 6 Abstract: Current understanding of the origin of icosahedral clusters … read moreAbstract: Current understanding of the origin of icosahedral clusters or icosahedral short-range ordering in undercooled metallic liquids or glasses is based on Frank’s consideration of an isolated icosahedron whose core has lower potential energy than the shell. Using large scale atomistic simulations and statistical analysis of several bcc (body-centered-cubic) and fcc (face-centered-cubic) metals, here we show that the shells of icosahedrons spontaneously formed inside deeply undercooled metallic liquids or glasses in fact have lower (averaged) potential energy than the cores. The shell potential energy deficiency occurs only to the icosahedral clusters but not to the equilibrium-crystal clusters, and, for icosahedral clusters, this deficiency grows with decreasing temperature. Compared with fcc metals, bcc metals exhibit greater potential energy deficiency on the icosahedral shells and produce significantly more icosahedral clusters upon liquid quenching, which explains the higher tendency of bcc metals to be vitrified observed in ultrafast cooling experiments. Inspecting the potential energy deficiency on the icosahedral shells through computation provides a new avenue to the search for amorphous metals (i.e. metallic glasses) with high glass forming ability and processability. read less USED (high confidence) R. P. Patil et al., “Hardening in Au-Ag nanoboxes from stacking fault-dislocation interactions,” Nature Communications. 2020. link Times cited: 24 USED (high confidence) X. Zhang et al., “A powder-metallurgy-based strategy toward three-dimensional graphene-like network for reinforcing copper matrix composites,” Nature Communications. 2020. link Times cited: 130 USED (high confidence) Y. Zhao, A. Gosai, K. Kang, and P. Shrotriya, “Multiscale Modeling Reveals the Cause of Surface Stress Change on Microcantilevers Due to Alkanethiol SAM Adsorption,” Journal of chemical information and modeling. 2020. link Times cited: 4 Abstract: Experimental results show that the adsorption of the self as… read moreAbstract: Experimental results show that the adsorption of the self assembled monolayers (SAMs) on a gold surface induces surface stress change that cause a deformation of the underlying substrate. However, the exact mechanism of stress development is yet to be elucidated. In the present study, multiscale computational models based on molecular dynamics (MD) simulations are applied to study the mechanism governing surface stress change. Distinct mechanisms for adsorption induced surface deformation, namely inter chain repulsion and thiol-gold interaction driven gold surface reconstruction, are investigated. Two different inter-atomic potentials, embedded atom method (EAM) and surface embedded atom method (SEAM), are used in the MD simulations to study the reconstruction induced surface stresses. Comparison of the predicted surface stress changes, resulting from MD and continuum mechanics based models, with observed experimental response, indicate that a modified SEAM based multiscale model can better capture the surface stress changes observed during alkanethiol SAM formation and gold surface reconstruction is the primary factor behind the surface stress change. Inter chain repulsions of SAM are found to have minimal contribution. Also, both the simulations and experiments show that surface stress change increases with surface coverage density and larger grain size. read less USED (high confidence) J. Liu, X. Fan, W. Zheng, D. J. Singh, and Y. Shi, “Nanocrystalline gold with small size: inverse Hall–Petch between mixed regime and super-soft regime,” Philosophical Magazine. 2020. link Times cited: 19 Abstract: ABSTRACT Molecular dynamics simulations were used to study t… read moreAbstract: ABSTRACT Molecular dynamics simulations were used to study the atomic mechanisms of deformation of nanocrystalline gold with 2.65–18 nm in grain size to explore the inverse Hall–Petch effect. Based on the mechanical responses, particularly the flow stress and the elastic-to-plastic transition, one can delineate three regimes: mixed (10–18 nm, dislocation activities and grain boundary sliding), inverse Hall-Petch (5–10 nm, grain boundary sliding), and super-soft (below 5 nm). As the grain size decreases, more grain boundaries present in the nanocrystalline solids, which block dislocation activities and facilitate grain boundary sliding. The transition from dislocation activities to grain boundary sliding leads to strengthening-then-softening due to grain size reduction, shown by the flow stress. It was further found that, samples with large grain exhibit pronounced yield, with the stress overshoot decrease as the grain size decreases. Samples with grain sizes smaller than 5 nm exhibit elastic-perfect plastic deformation without any stress overshoot, leading to the super-soft regime. Our simulations show that, during deformation, smaller grains rotate more and grow in size, while larger grains rotate less and shrink in size. read less USED (high confidence) E. Bird, J. G. Plascencia, and Z. Liang, “Thermal transport across the interface between liquid n-dodecane and its own vapor: A molecular dynamics study.,” The Journal of chemical physics. 2020. link Times cited: 9 Abstract: There are two possible thermal transport mechanisms at liqui… read moreAbstract: There are two possible thermal transport mechanisms at liquid-gas interfaces, namely, evaporation/condensation (i.e., heat transfer by liquid-vapor phase change at liquid surfaces) and heat conduction (i.e., heat exchange by collisions between gas molecules and liquid surfaces). Using molecular dynamics (MD) simulations, we study thermal transport across the liquid-vapor interface of a model n-dodecane (C12H26) under various driving force conditions. In each MD simulation, we restrict the thermal energy to be transferred across the liquid-vapor interface by only one mechanism. In spite of the complex intramolecular interactions in n-dodecane molecules, our modeling results indicate that the Schrage relationships, which were shown to give accurate predictions of evaporation and condensation rates of monatomic fluids, are also valid in the prediction of evaporation and condensation rates of n-dodecane. In the case of heat conduction at the liquid-vapor interface of n-dodecane, the interfacial thermal conductance obtained from MD simulations is consistent with the prediction from the kinetic theory of gases. The fundamental understanding of thermal transport mechanisms at liquid-gas interfaces will allow us to formulate appropriate boundary conditions for continuum modeling of heating and evaporation of small fuel droplets. read less USED (high confidence) J.-H. Kim, S.-H. Cha, S.-H. Kang, Y. Park, and S. Cho, “Atomistic simulation of agglomeration of metal nanoparticles considering the induced charge density of surface atoms,” International Journal of Mechanics and Materials in Design. 2020. link Times cited: 3 USED (high confidence) J.-H. Kim, S.-H. Cha, S.-H. Kang, Y. Park, and S. Cho, “Atomistic simulation of agglomeration of metal nanoparticles considering the induced charge density of surface atoms,” International Journal of Mechanics and Materials in Design. 2020. link Times cited: 0 USED (high confidence) N. Zhou, K. Elkhodary, X. Huang, S. Tang, and Y. Li, “Dislocation structure and dynamics govern pop-in modes of nanoindentation on single-crystal metals,” Philosophical Magazine. 2020. link Times cited: 14 Abstract: ABSTRACT There are two types of pop-in mode that have been w… read moreAbstract: ABSTRACT There are two types of pop-in mode that have been widely observed in nanoindentation experiments: the single pop-in, and the successive pop-in modes. Here we employ the molecular dynamics (MD) modelling to simulate nanoindentation for three face-centred cubic (FCC) metals, including Al, Cu and Ni, and two body-centred cubic (BCC) metals, such as Fe and Ta. We aim to examine the deformation mechanisms underlying these pop-in modes. Our simulation results indicate that the dislocation structures formed in single crystals during nanoindentation are mainly composed of half prismatic dislocation loops. These half prismatic dislocation loops in FCC metals are primarily constituted of extended dislocations. Lomer–Cottrell locks that result from the interactions between these extended dislocations can resist the slipping of half dislocation loops. These locks can build up the elastic energy that is needed to activate the nucleation of new half dislocation loops. A repetition of this sequence results in successive pop-in events in Al and other FCC metals. Conversely, the half prismatic dislocation loops that form in BCC metals after first pop-in are prone to slip into the bulk, which sustains plastic indentation process after first pop-in and prevents subsequent pop-ins. We thus conclude that pop-in modes are correlated with lattice structures during nanoindentation, regardless of their crystal orientations. read less USED (high confidence) A. E. Korenchenko, B. Gelchinski, and A. Vorontsov, “Statistical analysis of homogeneous nucleation of metallic nanoparticles during gas-phase synthesis,” Journal of Physics: Condensed Matter. 2020. link Times cited: 1 Abstract: In this work, we set out to develop a model of gas-phase nuc… read moreAbstract: In this work, we set out to develop a model of gas-phase nucleation in a mixture of copper and argon atoms, which can be further used for analysing macro-systems. Processes occurring at the atomic level are described using coefficients obtained by statistical analysis of molecular dynamic (MD) data on interactions of metal clusters with metal and argon atoms. The MD simulation results are compared with those obtained using the proposed macroscopic model. It is found that the coefficients obtained by averaging the interaction data suitably represent the integral value of the heat of condensation, although result in the smoothing of the energy distribution functions of the clusters. Analysis of the evolution of the number of clusters has shown that the values of their increase rate were lower than those obtained by MD simulation. The conclusion is made, that in order to improve the precision of the developed gas-phase condensation model, it should be supplemented by cluster coagulation. read less USED (high confidence) P. Zhao, Q. Zhang, Y.-bo Guo, H. Liu, and Z. Deng, “Atomistic Simulation Study of Nanoparticle Effect on Nano-Cutting Mechanisms of Single-Crystalline Materials,” Micromachines. 2020. link Times cited: 11 Abstract: Nanoparticle (NP), as a kind of hard-to-machine component in… read moreAbstract: Nanoparticle (NP), as a kind of hard-to-machine component in nanofabrication processes, dramatically affects the machined surface quality in nano-cutting. However, the surface/subsurface generation and the plastic deformation mechanisms of the workpiece still remain elusive. Here, the nano-cutting of a single-crystalline copper workpiece with a single spherical embedded nanoparticle is explored using molecular dynamics (MD) simulations. Four kinds of surface/subsurface cases of nanoparticle configuration are revealed, including being removed from the workpiece surface, moving as a part of the cutting tool, being pressed into the workpiece surface, and not interacting with the cutting tool, corresponding to four kinds of relative depth ranges between the center of the nanoparticle and the cutting tool. Significantly different plastic deformation mechanisms and machined surface qualities of the machined workpiece are also observed, suggesting that the machined surface quality could be improved by adjusting the cutting depth, which results in a change of the relative depth. In addition, the nanoparticle also significantly affects the processing forces in nano-cutting, especially when the cutting tool strongly interacts with the nanoparticle edge. read less USED (high confidence) Y. Ma, S. Zhang, Y. Xu, X. Liu, and S. Luo, “Effects of temperature and grain size on deformation of polycrystalline copper-graphene nanolayered composites.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 19 Abstract: The effects of temperature and grain size on mechanical prop… read moreAbstract: The effects of temperature and grain size on mechanical properties of polycrystalline copper-graphene nanolayered (PCuGNL) composites are investigated by analytical mechanical models and molecular dynamics simulations. The yield of PCuGNL composites under tension depends on temperature, copper grain size, and repeat layer spacing. Graphene-copper interfaces play the dominant role in the ultimate tensile strength of PCuGNL composites. The optimal range for strengthening of repeat layer spacing is 2-10 nm, and the failure stress of PCuGNL composites is weakly dependent on temperature. An analytical model is proposed to accurately characterize the mechanical behaviors of PCuGNL composites. read less USED (high confidence) X. Hu, Y. Ni, and Z. Zhang, “Atomistic Study of Interactions between Intrinsic Kink Defects and Dislocations in Twin Boundaries of Nanotwinned Copper during Nanoindentation,” Nanomaterials. 2020. link Times cited: 8 Abstract: In order to study the effects of kink-like defects in twin b… read moreAbstract: In order to study the effects of kink-like defects in twin boundaries on deformation mechanisms and interaction between dislocations and defects in twin boundaries under localized load, nanotwinned Cu with two defective twin (TDT) boundaries is compared with the nanotwinned Cu with two perfect twin (TPT) boundaries, and nanotwinned Cu with single defective twin (SDT) boundary and single perfect twin boundary by simulating spherical nanoindentations using molecular mechanics. The indenter force-depth and hardness-contact strain responses were analyzed. Results show that the existence of intrinsic defects in twin boundary could reduce the critical load and critical hardness of nanotwinned material. A quantitative parameter was first proposed to evaluate the degree of surface atom accumulation around the indenter during nanoindentation, and it can be inferred that the surface morphology in TDT changes more frequently than the surface morphologies in TPT and SDT. The atomistic configurations of incipient plastic structures of three different models were also analyzed. We found that the intrinsic defects in twin boundary will affect the incipient plastic structures. The formation of twinning partial slip on the defective twin boundary happens before the contact of the dislocation and twin boundary. The kink-like defects could introduce Frank partial dislocation to the twin boundary during interaction between dislocation and twin boundary, which was not detected on the perfect twin boundary. In addition, the area of twinning partial slips on the upper twin boundary in the incipient plastic structures in SDT and TDT are larger than the twinning partial slip area in TPT, which results in the reduction of the critical hardness in SDT and TDT. The kink-like defects could also block the expansion of twinning partial slip on the twin boundary. Furthermore, we investigated the dislocation transmission processes in three different models. It is found that the dislocation transmission event could be delayed in model containing single defective twin boundary, while the transmission process could be advanced in model containing two consecutive defective twin boundaries. The quantitative analysis of dislocation length was also implemented. Result shows that the main emitted dislocation during nanoindentation is Shockley partial, and the dislocation nucleation in SDT and TDT is earlier than the dislocation nucleation in TPT due to the existence of defects. It is inferred that the intrinsic defects on twin boundaries could enhance the interaction between dislocations and twin boundaries, and could strongly change the structure evolution and promote the dislocation nucleation and emission. These findings about kink-like defects in twin boundaries show that the inherent kink-like defects play a crucial role in the deformation mechanisms and it should be taken into consideration in future investigations. Single defective twin boundary structure is recommended to delay the transmission and block the expansion of twin boundary migration. Some of the results are in good agreement with experiments. read less USED (high confidence) G. Sun, A. Hawken, and P. Harrowell, “The displacement field associated with the freezing of a melt and its role in determining crystal growth kinetics,” Proceedings of the National Academy of Sciences. 2020. link Times cited: 11 Abstract: Significance We demonstrate that an accurate estimation of t… read moreAbstract: Significance We demonstrate that an accurate estimation of the displacements associated with the transformation of liquid into crystal is necessary to explain the striking variations in the temperature dependence of the addition rate of liquid atoms to the growing crystal interface during freezing. An assignment algorithm, adapted from operations theory, is shown to provide a good estimate of these atomic displacements. As the assignment algorithm requires only initial and target locations, it is applicable to all forms of structural transformations. In resolving a fundamental feature of the kinetics of freezing, a phenomenon of central importance to material fabrication, this paper also provides the tools to open lines of research into the kinetics of structural transformation. The atomic displacements associated with the freezing of metals and salts are calculated by treating crystal growth as an assignment problem through the use of an optimal transport algorithm. Converting these displacements into timescales based on the dynamics of the bulk liquid, we show that we can predict the activation energy for crystal growth rates, including activation energies significantly smaller than those for atomic diffusion in the liquid. The exception to this success, pure metals that freeze into face-centered cubic crystals with little to no activation energy, are discussed. The atomic displacements generated by the assignment algorithm allows us to quantify the key roles of crystal structure and liquid caging length in determining the temperature dependence of crystal growth kinetics. read less USED (high confidence) P. Cao, “The Strongest Size in Gradient Nanograined Metals.,” Nano letters. 2020. link Times cited: 53 Abstract: Conventional polycrystalline metals become stronger with dec… read moreAbstract: Conventional polycrystalline metals become stronger with decreasing grain size, yet softening starts to take over at nanometer regime, giving rise to the strongest size at which the predominate strengthening mechanism switches to softening. We show that this critical size for the onset of softening shifts from ~12 nm for homogeneous nanograined copper to ~7 nm for its gradient nanograined counterpart, and this strongest size decreases with increasing the grain size gradient. The decrease in the strongest size is prompted by mitigation of grain boundary-mediated softening processes accompanying by enhanced intragranular plastic deformations. We found the nanograins as small as 6 nm, mainly involving intergranular sliding in homogeneous structures, reveal anomalous plastic deformation in gradient systems, which is mediated by partial dislocations nucleation and motion. The results on extended dislocation slip and gradient stress and plasticity, stemming from the structure heterogeneity, shed light on an emerging class of heterogeneous nanostructured materials of improved strength-ductility synergy. read less USED (high confidence) Z. Wei, Z. Duan, Y. Kan, Y. Zhang, and Y. Chen, “Phonon energy dissipation in friction between graphene/graphene interface,” Journal of Applied Physics. 2020. link Times cited: 23 Abstract: The theory of phononic friction attributes that the multipho… read moreAbstract: The theory of phononic friction attributes that the multiphonon processes are the main cause of the mechanical energy dissipation in a wear-free friction process. Unfortunately, it is still impossible to set up a direct relationship between the phonons and the frictional force. In this study, a classical molecular dynamics simulation model is used to mimic a piece of graphene sliding over a supported graphene substrate. It is found that the lifetime of some phonons, especially the modes around the Γ point of the first Brillouin zone, gradually decreases with the increase of the sliding velocity. A phonon lifetime-based model is proposed to explain the variation of the frictional force as a function of the sliding velocity, i.e., the shorter phonon lifetime corresponding to a higher friction force under the same temperature. This model is consistent with the traditional Prandtl-Tomlinson model at a low sliding velocity range, which predicts that the friction force increases logarithmically with the sliding velocity. Once the sliding velocity exceeds a critical value, the lifetime of the excited phonons is far longer than the time for the tip sweeping a lattice constant. In this case, the excited phonons do not have enough time to dissipate the mechanical energy, which leads to the reduced friction force with the increase of the sliding velocity.The theory of phononic friction attributes that the multiphonon processes are the main cause of the mechanical energy dissipation in a wear-free friction process. Unfortunately, it is still impossible to set up a direct relationship between the phonons and the frictional force. In this study, a classical molecular dynamics simulation model is used to mimic a piece of graphene sliding over a supported graphene substrate. It is found that the lifetime of some phonons, especially the modes around the Γ point of the first Brillouin zone, gradually decreases with the increase of the sliding velocity. A phonon lifetime-based model is proposed to explain the variation of the frictional force as a function of the sliding velocity, i.e., the shorter phonon lifetime corresponding to a higher friction force under the same temperature. This model is consistent with the traditional Prandtl-Tomlinson model at a low sliding velocity range, which predicts that the friction force increases logarithmically with the sliding... read less USED (high confidence) F. Ojaghnezhad and H. Shodja, “Second strain gradient theory in orthogonal curvilinear coordinates: Prediction of the relaxation of a solid nanosphere and embedded spherical nanocavity,” Applied Mathematical Modelling. 2019. link Times cited: 5 USED (high confidence) Y. Cui, Y. Ju, and S. Meguid, “Atomistic treatment of periodic gold nanowire array nanofasteners under shear loading,” Nanotechnology. 2019. link Times cited: 5 Abstract: Comprehensive molecular dynamics simulations are conducted t… read moreAbstract: Comprehensive molecular dynamics simulations are conducted to unravel the mechanics and mechanisms associated with the strength and fracture behavior of a highly ordered gold nanowire (Au-NW) array of a pair of nanofasteners (nanoconnectors) under externally applied shear strain. Large-scale atomic/molecular massively parallel simulator (LAMMPS and embedded atom method were adopted to model the atomic interactions of a number of neighboring nanofasteners. This was affected via the use of a periodic simulation box around a pair of highly ordered nanotube arrays to minimize the cost of the computations. Energy minimization using a conjugate gradient algorithm was first performed and followed by atomic relaxation to achieve an equilibrated configuration under the canonical ensemble of constant temperature and volume. The relaxed equilibrated configuration of the nanofastener was then subjected to an externally applied shear strain γ x y at a rate of γ ̇ x y = 0.1 per nanosecond under the canonical ensemble. Our results reveal the importance of the morphology and the overlap depth of the mating nanowire arrays upon the mechanical and fracture behavior of the nanofastener under shear loading. Our work also disclosed the phenomenon of multiple contacts of some displaced nanowires with their neighbors even after their fracture leading to multiple cold-welds with added redundancy to the nanofastener. Finally, in this research, we identified the locations of dislocation emissions and the resulting fracture processes that govern the mechanical integrity and ultimately the functionality of the Au-NW connector. The proposed highly ordered alignment, as conceived numerically herein, can yield a peak stress two to three times higher than that corresponding to a random alignment reported in a previous study. The Au-NW connector also exhibited resistance to fracture, even in cases where small overlap depth is considered in joint bonding. The nanoconnector was also tested at high temperatures (up to 450 K). Our results show that the rising temperature only leads to a minor reduction in the load transmitted by the nanoconnector. read less USED (high confidence) D. Zeng, C. Liu, J. Jiang, and J. Fan, “Langevin dynamics with energy dissipation on periodic lattice structures modeling as an equivalent two-body method for atom-surface interactions,” Materials Research Express. 2019. link Times cited: 0 Abstract: In this work, we propose a classical equivalent two-body (ET… read moreAbstract: In this work, we propose a classical equivalent two-body (ETB) model that can capture more detailed dynamic features arising from energy dissipation and atom oscillations, by introducing the Langevin equation of a harmonic oscillator. The trapping probability, scattering angle and the residence time of Ar interacting with Pt (111) and W (110) surfaces predicted by the ETB model agree well with the measured experimental data or molecular dynamics simulations. Moreover, the ETB model is also used to study the influence of oscillating and dissipating properties on the thermal accommodation coefficients and rainbow scattering of gas atoms colliding with the surface. It is found that the dependence of energy accommodation coefficients and rainbow scattering on the oscillating and dissipating parameters shows nonlinear behaviors, and the associated mechanisms are disclosed. ETB model further provides the possibility to explore the physics beyond the existing two-body models for a better description of energy transferring during atom surface interactions. read less USED (high confidence) M. Islam, M. S. H. Thakur, S. Mojumder, A. A. Amin, and M. M. Islam, “Mechanical and vibrational characteristics of functionally graded Cu–Ni nanowire: A molecular dynamics study,” Composites Part B-engineering. 2019. link Times cited: 27 USED (high confidence) S. Jekal, “Links Between Mechanical Properties and Local Atomic Structures of Cu–Zr Bulk Metallic Glasses,” Research & Development in Material Science. 2019. link Times cited: 0 Abstract: Bulk metallic glasses (BMGs) have drawn much attention due t… read moreAbstract: Bulk metallic glasses (BMGs) have drawn much attention due to their interesting mechanical properties such as extraordinary elastic strain limits and a high tensile yield stress [1-6]. For example, their yield strengths can be up to 1 to 5GPa and elastic strain limits up to ~2% [7-10]. However, their use for engineering applications has been challenging since BMGs exhibit localized strain softening leading to failure and brittleness. The underlying atomicscale plastic mechanisms are believed to be mediated by a local microscopic mechanism [11-14]. Such localized processes have been observed during high-strain deformation atomistuc simulations [15-22] inspiring the development of the shear transformation zone (STZ) concept and the effective temperature theory of athermal glass plasticity [23-25]. read less USED (high confidence) D. Sandbeck, O. Brummel, K. Mayrhofer, J. Libuda, I. Katsounaros, and S. Cherevko, “Dissolution of Platinum Single Crystals in Acidic Medium,” Chemphyschem. 2019. link Times cited: 28 Abstract: Platinum single crystal basal planes consisting of Pt(111), … read moreAbstract: Platinum single crystal basal planes consisting of Pt(111), Pt(100), Pt(110) and reference polycrystalline platinum Pt(poly) were subjected to various potentiodynamic and potentiostatic electrochemical treatments in 0.1 M HClO4. Using the scanning flow cell coupled to an inductively coupled plasma mass spectrometer (SFC‐ICP‐MS) the transient dissolution was detected on‐line. Clear trends in dissolution onset potentials and quantities emerged which can be related to the differences in the crystal plane surface structure energies and coordination. Pt(111) is observed to have a higher dissolution onset potential while the generalized trend in dissolution rates and quantities was found to be Pt(110)>P(100)≈Pt(poly)>Pt(111). read less USED (high confidence) Z. Zhang et al., “Dislocations in Grain Boundary Regions: The Origin of Heterogeneous Microstrains in Nanocrystalline Materials,” Metallurgical and Materials Transactions A. 2019. link Times cited: 27 USED (high confidence) Z. Zhu, B. Peng, R. Feng, L. Wang, S. Jiao, and Y. Dong, “Molecular dynamics simulation of chip formation mechanism in single-crystal nickel nanomachining,” Science China Technological Sciences. 2019. link Times cited: 10 USED (high confidence) L. Y. Zhao and Y. Liu, “The influence mechanism of the strain rate on the tensile behavior of copper nanowire,” Science China Technological Sciences. 2019. link Times cited: 5 USED (high confidence) R. Freitas and E. Reed, “Uncovering the effects of interface-induced ordering of liquid on crystal growth using machine learning,” Nature Communications. 2019. link Times cited: 28 USED (high confidence) Z. Zhao, J. Liu, A. Soh, and C. Tang, “Temperature-mediated fabrication, stress-induced crystallization and transformation: atomistic simulations of additively manufactured amorphous Cu pillars,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 6 Abstract: Additive manufacturing (AM) is an emerging and promising tec… read moreAbstract: Additive manufacturing (AM) is an emerging and promising technology. In this manuscript, an attempt is made to simulate an AM process via molecular dynamics simulations. Amorphous Cu pillars are built by bundling melting Cu wires parallelly one by one, with a particular temperature-controlling procedure through the use of ‘mediate temperature’. Thus, the volume fraction of the amorphous phase becomes adjustable. Uniaxial tests are conducted on the pillars after free relaxation. The mediate temperature is found to play a profound role in atomic arrangement, which governs the volume fractions of amorphous and crystalline phases. The results obtained also show that crystallization prevails when the pillar is subjected to an external tension. Furthermore, such a stress-induced crystallization serves as the dominant plastic mechanism instead of dislocation, and a vibrating uniaxial loading is found to accelerate the transformation from an amorphous to a crystalline phase, compared with a monotonic tension. read less USED (high confidence) A. E. Korenchenko, A. Vorontsov, B. R. Gel’chinskii, and A. Zhukova, “Determination of Small Copper Clusters Based on Simulation of the Process of Gas Phase Condensation,” High Temperature. 2019. link Times cited: 4 USED (high confidence) J. Palomares-Báez, J. M. Montejano-Carrizales, G. Guisbiers, M. José-Yacamán, and J. Rodríguez-López, “The Decmon: a new nanoparticle shape along the truncation path from the icosahedron to the decahedron,” Nanotechnology. 2019. link Times cited: 2 Abstract: The idea that shape and structure determines functionality i… read moreAbstract: The idea that shape and structure determines functionality is one of the leiv-motifs that drives research and applications on fields such as catalysis and plasmonics. The growth and stability of metallic clusters is extensively discussed through faceting and energy minimization mechanisms, respectively. Facet truncations on the regular Mackay-icosahedron (m-Ih) give rise to two sub-families exhibiting five-fold symmetry and external decahedral shape. Such successive truncations made to the regular m-Ih, led to a decahedral motif called ‘Decmon’ (Montejano’s decahedron). This structure expose facets (111) and (100), that after a total energy minimization through molecular dynamics simulations using the embedded atom model, proved to be thermally stable. This result has been confirmed by using nano-thermodynamics. The surface energy competition between the (111) and (100) facets explains its stability at some given cluster sizes, and this truncation path permits to glimpse the potential energy surface in the growth path of nanoparticles from the decahedral (s-Dh) to icosahedral (m-Ih) structures. read less USED (high confidence) X. Zhang, S. Sun, T. Xu, and T.-Y. Zhang, “Temperature dependent Grüneisen parameter,” Science China Technological Sciences. 2019. link Times cited: 12 USED (high confidence) Y. Zhao, X. Liu, J. Zhu, and S. Luo, “Unusually high flexibility of graphene-Cu nanolayered composites under bending.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 7 Abstract: The mechanical properties of graphene-Cu nanolayered (GCuNL)… read moreAbstract: The mechanical properties of graphene-Cu nanolayered (GCuNL) composites under bend loading are investigated via an energy-based analytical model and molecular dynamics (MD) simulations. For an anisotropic material, if it has a weak strength in a certain direction, improving the mechanical properties along this direction is normally difficult for its composites. Here, we find that the flexibility of GCuNL composites can be improved considerably by graphene interfaces, despite graphene's small bending stiffness. The graphene interfaces can delocalize slip bands in the inner Cu layers of GCuNL composites, and impede local nucleation of dislocations, thus greatly increasing the yield and failure bend angles. As the thickness decreases, the flexibility of GCuNL nanofilms increases. However, the GCuNL nanofilms are thermodynamically unstable due to interface instability when the repeat layer spacing is less than 2 nm. The energy-based analytical model for large deformation can accurately characterize the bending response of GCuNL nanofilms. read less USED (high confidence) L. Xiao, J. Zhang, Y. Zhu, T. Shi, and G. Liao, “Molecular dynamics simulation of the tensile mechanical behaviors of axial torsional copper nanorod,” Journal of Nanoparticle Research. 2019. link Times cited: 2 USED (high confidence) H. Yang, Y. Tang, and P. Yang, “Factors influencing thermal transport across graphene/metal interfaces with van der Waals interactions.,” Nanoscale. 2019. link Times cited: 21 Abstract: We implement non-equilibrium Green's function (NEGF) ca… read moreAbstract: We implement non-equilibrium Green's function (NEGF) calculations to investigate thermal transport across graphene/metal interfaces with interlayer van der Waals interactions to understand the factors influencing thermal conductance across the interface. It is found that interfaces with a smaller interfacial lattice mismatch, lighter metal substrate and stronger interfacial bonding strength will show better interfacial thermal transport abilities. Strain induced by the interfacial lattice mismatch in graphene is the key factor for the decrease of interfacial phonon transmission in the main frequency range of metals, which finally results in a decrease of interfacial thermal conductance. A comprehensive interfacial influencing factor is proposed combining the factors of graphene density, metal density and interfacial binding energy to realize the prediction of interfacial thermal conductance across the graphene/metal interface. The results are hoped to promote the understanding of the thermal transport mechanism and design of graphene based 2D/3D materials interfaces. read less USED (high confidence) M. Dou, F. C. Maier, and M. Fyta, “The influence of a solvent on the electronic transport across diamondoid-functionalized biosensing electrodes.,” Nanoscale. 2019. link Times cited: 5 Abstract: Electrodes embedded in nanopores have the potential to detec… read moreAbstract: Electrodes embedded in nanopores have the potential to detect the identity of biomolecules, such as DNA. This identification is typically being done through electronic current measurements across the electrodes in a solvent. In this work, using quantum-mechanical calculations, we qualitatively present the influence of this solvent on the current signals. For this, we model electrodes functionalized with a small diamond-like molecule known as diamondoid and place a DNA nucleotide within the electrode gap. The influence of an aqueous solvent is taken explicitly into account through Quantum-Mechanics/Molecular Mechanics (QM/MM) simulations. From these, we could clearly reveal that at the (111) surface of the Au electrode, water molecules form an adlayer-like structure through hydrogen bond networks. From the temporal evolution of the hydrogen bond between a nucleotide and the functionalizing diamondoid, we could extract information on the conductance across the device. In order to evaluate the influence of the solvent, we compare these results with ground-state electronic structure calculations in combination with the non-equilibrium Green's function (NEGF) approach. These allow access to the electronic transport across the electrodes and show a difference in the detection signals with and without the aqueous solution. We analyze the results with respect to the density of states in the device. In the end, we demonstrate that the presence of water does not hinder the detection of a mutation over a healthy DNA nucleotide. We discuss these results in view of sequencing DNA with nanopores. read less USED (high confidence) T. Vo and B. H. Kim, “A molecular dynamics study on cooling rate effect on atomic structure of solidified silver nanoparticles,” The European Physical Journal D. 2019. link Times cited: 2 USED (high confidence) H. Li, S. Guo, K. Shin, M. Wong, and G. Henkelman, “Design of a Pd–Au Nitrite Reduction Catalyst by Identifying and Optimizing Active Ensembles,” ACS Catalysis. 2019. link Times cited: 134 Abstract: Nitrate (NO3–) is a ubiquitous contaminant in groundwater th… read moreAbstract: Nitrate (NO3–) is a ubiquitous contaminant in groundwater that causes serious public health issues around the world. Though various strategies are able to reduce NO3– to nitrite (NO2–), a rational catalyst design strategy for NO2– removal has not been found, in part because of the complicated reaction network of nitrate chemistry. In this study, we show, through catalytic modeling with density functional theory (DFT) calculations, that the performance of mono- and bimetallic surfaces for nitrite reduction can be rapidly screened using N, N2, and NH3 binding energies as reactivity descriptors. With a number of active surface atomic ensembles identified for nitrite reduction, we have designed a series of “metal-on-metal” bimetallics with optimized surface reactivity and a maximum number of active sites. Choosing Pd-on-Au nanoparticles (NPs) as candidate catalysts, both theory and experiment find that a thin monolayer of Pd-on-Au NPs (size: ∼4 nm) leads to high nitrite reduction performance, outperforming pu... read less USED (high confidence) G. Rusina, S. Borisova, and E. Chulkov, “Structure and Dynamic Stability of a Multilayer Na Film on the Cu(001) Surface,” JETP Letters. 2019. link Times cited: 1 USED (high confidence) D. Datta, R. Balasubramaniam, P. Ranjan, A. Sharma, and T. K. Roy, “Investigation of tool-workpiece interaction in nanoscale cutting: a molecular dynamics study,” International Journal of Precision Technology. 2019. link Times cited: 1 Abstract: Ductile and brittle materials differ in their physical and m… read moreAbstract: Ductile and brittle materials differ in their physical and mechanical properties and pose distinct interaction with the cutting tool while nano-machining. It is thus imperative to analyse the mechanism of material removal and tool-workpiece interaction. Towards this, molecular dynamics simulation (MDS) is carried out to study the diamond tool and workpiece interaction in the nanoscale cutting of Cu (ductile material) and Si (brittle material). Results show that material removal in Cu takes place through shear deformation by dislocations formation and their propagation while in case of Si, it takes place through phase transformation of the material in cutting zone. Force analysis of both the materials shows that machinability of Cu in nanoscale cutting is better compared to Si. Furthermore, tool wear while machining of Si with sharp edge tool is due to chipping whereas radial distribution function reveals that graphitisation of the round edge tool occurs during machining of Si. read less USED (high confidence) P. Peng et al., “Near-ideal compressive strength of nanoporous silver composed of nanowires,” Acta Materialia. 2019. link Times cited: 12 USED (high confidence) I. Talyzin and V. Samsonov, “Molecular Dynamics of Solid State Spreading in a Pb (Nanoparticle)/Cu (Substrate) System,” Bulletin of the Russian Academy of Sciences: Physics. 2019. link Times cited: 0 USED (high confidence) S. A. Peddakotla, K. K. Kammara, and R. Kumar, “Molecular dynamics simulation of particle trajectory for the evaluation of surface accommodation coefficients,” Microfluidics and Nanofluidics. 2019. link Times cited: 10 USED (high confidence) L. Li et al., “Adaptive kinetic Monte Carlo simulations of surface segregation in PdAu nanoparticles.,” Nanoscale. 2019. link Times cited: 16 Abstract: Surface segregation in bimetallic nanoparticles (NPs) is cri… read moreAbstract: Surface segregation in bimetallic nanoparticles (NPs) is critically important for their catalytic activity because the activity is largely determined by the surface composition. Little, however, is known about the atomic scale mechanisms and kinetics of surface segregation. One reason is that it is hard to resolve atomic rearrangements experimentally. It is also difficult to model surface segregation at the atomic scale because the atomic rearrangements can take place on time scales of seconds or minutes - much longer than can be modeled with molecular dynamics. Here we use the adaptive kinetic Monte Carlo (AKMC) method to model the segregation dynamics in PdAu NPs over experimentally relevant time scales, and reveal the origin of kinetic stability of the core@shell and random alloy NPs at the atomic level. Our focus on PdAu NPs is motivated by experimental work showing that both core@shell and random alloy PdAu NPs with diameters of less than 2 nm are stable, indicating that one of these structures must be metastable and kinetically trapped. Our simulations show that both the Au@Pd and the PdAu random alloy NPs are metastable and kinetically trapped below 400 K over time scales of hours. These AKMC simulations provide insight into the energy landscape of the two NP structures, and the diffusion mechanisms that lead to segregation. In the core-shell NP, surface segregation occurs primarily on the (100) facet through both a vacancy-mediated and a concerted mechanism. The system becomes kinetically trapped when all corner sites in the core of the NP are occupied by Pd atoms. Higher energy barriers are required for further segregation, so that the metastable NP has a partially alloyed shell. In contrast, surface segregation in the random alloy PdAu NP is suppressed because the random alloy NP has reduced strain as compared to the Au@Pd NP, and the segregation mechanisms in the alloy require more elastic energy for exchange of Pd and Au and between the surface and subsurface. read less USED (high confidence) S. A. Peddakotla, K. K. Kammara, and R. Kumar, “Molecular dynamics simulation of particle trajectory for the evaluation of surface accommodation coefficients,” Microfluidics and Nanofluidics. 2019. link Times cited: 0 USED (high confidence) K. Zolnikov, D. Kryzhevich, and A. Korchuganov, “Atomic mechanisms of high-speed migration of symmetric tilt grain boundaries in nanocrystalline Ni,” Letters on Materials. 2019. link Times cited: 27 Abstract: Molecular dynamics simulations of structural rearrangements … read moreAbstract: Molecular dynamics simulations of structural rearrangements in nanocrystalline Ni with the symmetric tilt grain boundary (GB) ∑5 (310) [001] under shear loading were conducted. It was found that GB can be displaced in the direction perpendicular to the shear loading direction. To activate the displacement, it is necessary to reach the threshold value of the shear stress. The GB displacement is abrupt and is due to a certain sequence of displacements of the atomic planes adjacent to the GB. These planes are successively rebuilt from the structure of one grain to the structure of another grain in the process of GB migration. The velocity of GB migration can reach several hundred meters per second and depends on the rate of shear loading. The use of periodic boundary conditions prevents the rotations of the grains. As the simulated tilt GB is symmetric, both of the crystallite grains will have the same shear moduli in the direction of the applied loading. The shear loading of the crystallite with such a structure does not lead to any volume driving forces. The GB displacement was entirely due to the coupling effect. The shear stress curve as a function of time has a sawtooth shape. The GB experiences displacement upon reaching the maximum value of the applied shear stresses. Despite the high stress values, the GB displacement did not cause the nucleation of the defect structure in the crystallite. The GB migration is accompanied by a change in the volume of atoms involved in structural rearrangements. read less USED (high confidence) A. Rida, M. Micoulaut, E. Rouhaud, and A. Makke, “Crystals at High Deformation Rates Displaying Glassy Behavior,” physica status solidi (b). 2019. link Times cited: 2 Abstract: When investigated as a function of deformation rate and time… read moreAbstract: When investigated as a function of deformation rate and time, model nano‐crystals exhibit a clear threshold in mechanical and relaxation behavior, this observation being valid in both the Hooke (elastic) and plastic regime. At low deformation rate (ϵ˙ read less USED (high confidence) Y. Cui and H. Chew, “A simple numerical approach for reconstructing the atomic stresses at grain boundaries from quantum-mechanical calculations.,” The Journal of chemical physics. 2019. link Times cited: 4 Abstract: The atomistic stress state at a metal grain boundary is an i… read moreAbstract: The atomistic stress state at a metal grain boundary is an intrinsic attribute which affects many physical and mechanical properties of the metal. While the virial stress is an accepted measure of the atomistic stress in molecular dynamics simulations, an equivalent definition is not well-established for quantum-mechanical density functional theory (DFT) calculations. Here, we introduce a numerical technique, termed the sequential atom removal (SAR) approach, to reconstruct the atomic stresses near a symmetrical-tilt Σ5(310)[001] Cu grain boundary. In the SAR approach, individual atoms near the boundary are sequentially removed to compute the pair (reaction) force between atoms, while correcting for changes to the local electron density caused by atom removal. We show that this SAR approach accurately reproduces the spatially-varying virial stresses at a grain boundary governed by an embedded atom method potential. The SAR approach is subsequently used to extract the atomistic stresses of the grain boundary from DFT calculations, from which we reconstruct a continuum-equivalent grain boundary traction distribution as a quantitative descriptor of the grain boundary atomic structure. read less USED (high confidence) X.-fei Zhu et al., “Graphene coating makes copper more resistant to plastic deformation,” Composites Communications. 2019. link Times cited: 17 USED (high confidence) S.-H. Cha et al., “Fabrication of nanoribbons by dielectrophoresis assisted cold welding of gold nanoparticles on mica substrate,” Scientific Reports. 2019. link Times cited: 17 USED (high confidence) H. Liu, M. Hao, M. Tao, Y. Sun, and W. Xie, “Molecular dynamics simulation of dislocation evolution and surface mechanical properties on polycrystalline copper,” Applied Physics A. 2019. link Times cited: 13 USED (high confidence) Y. Jin, R. Tao, and Z. Li, “Understanding flow enhancement in graphene‐coated nanochannels,” ELECTROPHORESIS. 2019. link Times cited: 18 Abstract: In this work, we investigate pressure‐driven water flows in … read moreAbstract: In this work, we investigate pressure‐driven water flows in graphene‐coated copper nanochannels through molecular dynamics simulations. It is found that the flow rate in bare copper nanochannel can be significantly enhanced by a factor of 45 when the nanochannel is coated with monolayer graphene. The enhancement factor for the flow rate reaches about 90 when the nanochannel is modified with 3 or more graphene layers. The dipole relaxation time and the hydrogen bond lifetime of interfacial water molecules show that the graphene coating promotes the mobility of water molecules at the interface. The distribution of the potential of mean force and the free energy barriers also confirm that graphene coating reduces the flow resistance and 3 layers of graphene can fully screen the surface effects. The results in this work provide important information for the design of graphene‐based nanofluidic systems for flow enhancement. read less USED (high confidence) Z. Pang, B. Deng, Z. Liu, H. Peng, and Y. Wei, “Defects guided wrinkling in graphene on copper substrate,” Carbon. 2019. link Times cited: 22 USED (high confidence) S. Shirsath et al., “Au quantum dots engineered room temperature crystallization and magnetic anisotropy in CoFe2O4 thin films.,” Nanoscale horizons. 2019. link Times cited: 58 Abstract: For the first time, this work presents a novel room temperat… read moreAbstract: For the first time, this work presents a novel room temperature time-effective concept to manipulate the crystallization kinetics and magnetic responses of thin films grown on amorphous substrates. Conventionally, metal-induced crystallization is adopted to minimize the crystallization temperature of the upper-layer thin film. However, due to the limited surface area of the continuous metal under-layer, the degree of crystallization is insufficient and post-annealing is required. To expose a large surface area of the metal under-layer, we propose a simple and novel approach of using an Au nanodots array instead of a continuous metallic under-layer to obtain crystallization of upper-layer thin films. Spinel cobalt ferrite (CFO) thin film as a 'model' was deposited on an Au nano-dots array to realize this methodology. Our findings revealed that the addition of quantum-sized Au nano-dots as a metal under-layer dramatically enhanced the crystallization of the cobalt ferrite upper layer at room temperature. The appearance of major X-ray diffraction peaks with high intensity and well-defined crystallized lattice planes observed via transmission electron microscopy confirmed the crystallization of the CFO thin film deposited at room temperature on 4 nm-sized Au nano-dots. This crystallized CFO thin film exhibits 18-fold higher coercivity (Hc = 4150 Oe) and 4-fold higher saturation magnetization (Ms = 262 emu cm-3) compared to CFO deposited without the Au under-layer. The development of this novel concept of room-temperature crystallization without the aid of additives and solvents represents a crucial breakthrough that is highly significant for exploring the green and energy-efficient synthesis of a variety of oxide and metal thin films. read less USED (high confidence) Z. Zheng, H. Zhan, Y. Nie, A. Bo, X. Xu, and Y. T. Gu, “General existence of flexural mode doublets in nanowires targeting vectorial sensing applications.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 1 Abstract: Nanowires (NWs) are one of the fundamental building blocks f… read moreAbstract: Nanowires (NWs) are one of the fundamental building blocks for nanoscale devices, and have been frequently utilized as mechanical resonators. Earlier studies show that ultra-sensitive vectorial sensing toolkits can be fabricated by changing the flexural mode of NWs to oscillation doublets along two orthogonal directions. Based on in silico studies and the Timoshenko beam theory, this work finds that the dual orthogonal flexural mode of NWs can be effectively controlled through the proper selection of their growth direction. It is found that metallic NWs with a directional-independent shear modulus possess a single flexural mode. However, NWs with a directional-dependent shear modulus naturally exhibit flexural mode doublets, which do not disappear even with increasing slenderness ratio. Further studies show that such a feature generally exists in other NWs, such as Si NWs. Mimicking a pendulum configuration as used in NW-based scanning force microscopy, the cantilevered 110 Si NW demonstrates zeptogram mass resolution and a force sensitivity down to the order of 10-24 N Hz-1/2 (yN Hz-1/2) in both transverse directions. The findings in this work open up a new and facile avenue to fabricate 2D vectorial force sensors, which could enable ultra-sensitive and novel detection devices/systems for 2D effects, such as the anisotropy strength of atomic bonds. read less USED (high confidence) H. Moslemzadeh, O. Alizadeh, and S. Mohammadi, “Quasicontinuum multiscale modeling of the effect of rough surface on nanoindentation behavior,” Meccanica. 2019. link Times cited: 4 USED (high confidence) S. Yang, Y. Zhang, and L. Chen, “Molecular dynamics study on the effect of surface wettability on the performance of water vapor condensation,” AIP Advances. 2019. link Times cited: 11 Abstract: Many studies have focused on the effect of surface wettabili… read moreAbstract: Many studies have focused on the effect of surface wettability on condensation at the nanoscale, while few studies investigated the condensation process of water vapor below 450K. However, water vapor condensation below 450K is common and important in industrial fields. In this paper, molecular dynamics method is used to study the effect of surface wettability on the performance of water vapor condensation below 450K on a copper surface, and a comparison with the performance of water vapor condensation at 450K was performed. The results show that the heat transfer performance of vapor is not the same when condensing on a hydrophilic surface and on a hydrophobic surface. It’s found that lower temperature vapor requires more time in starting to condense on a hydrophobic surface, whose heat transfer efficiency first increases gradually and finally becomes constant. For the first time the process of vapor condensation on a hydrophobic surface was divided into three stages based on the changes in heat transfer efficiency, and the heat transfer performance of each stage was analyzed. The results show that a stronger surface wettability and higher vapor temperature improve the heat transfer performance during the condensation process. Moreover, the lower the vapor temperature is, the greater the impact of the wettability is on the heat transfer efficiency, and the vapor less easily condenses on a hydrophobic surface.Many studies have focused on the effect of surface wettability on condensation at the nanoscale, while few studies investigated the condensation process of water vapor below 450K. However, water vapor condensation below 450K is common and important in industrial fields. In this paper, molecular dynamics method is used to study the effect of surface wettability on the performance of water vapor condensation below 450K on a copper surface, and a comparison with the performance of water vapor condensation at 450K was performed. The results show that the heat transfer performance of vapor is not the same when condensing on a hydrophilic surface and on a hydrophobic surface. It’s found that lower temperature vapor requires more time in starting to condense on a hydrophobic surface, whose heat transfer efficiency first increases gradually and finally becomes constant. For the first time the process of vapor condensation on a hydrophobic surface was divided into three stages based on the changes in heat transfer... read less USED (high confidence) Y. Zhou, W. Jiang, D. Li, and Q. Qin, “Study on Lightweight and Strengthening Effect of Carbon Nanotube in Highly Ordered Nanoporous Nickel: A Molecular Dynamics Study,” Applied Sciences. 2019. link Times cited: 3 Abstract: The mechanical behavior of nanocomposites consisting of high… read moreAbstract: The mechanical behavior of nanocomposites consisting of highly ordered nanoporous nickel (HONN) and its carbon nanotube (CNT)-reinforced composites (CNHONNs) subjected to a high temperature of 900 K is investigated via molecular dynamics (MD) simulations. The study indicates that, out-of-plane mechanical properties of the HONNs are generally superior to its in-plane mechanical properties. Whereas the CNT shows a significant strengthening effect on the out-of-plane mechanical properties of the CNHONN composites. Compared to pure HONNs, through the addition of CNTs from 1.28 wt‰ to 5.22 wt‰, the weight of the composite can be reduced by 5.83‰ to 2.33% while the tensile modulus, tensile strength, compressive modulus and compressive strength can be increased by 2.2% to 8.8%, 1% to 5.1%, 3.6% to 10.2% and 4.9% to 10.7%, respectively. The energy absorption capacity can also be improved due to the existence of CNTs. Furthermore, the MD simulations provide further insights into the deformation mechanism at the atomic scale, including fracture in tension, pore collapse in compression and local changes in lattice structures due to stacking faults. read less USED (high confidence) Q. Fang, Y. Tian, J. Li, Q. Wang, and H. Wu, “Interface-governed nanometric machining behaviour of Cu/Ag bilayers using molecular dynamics simulation,” RSC Advances. 2019. link Times cited: 15 Abstract: The nanometric machining of Cu/Ag bilayers and pure Cu film … read moreAbstract: The nanometric machining of Cu/Ag bilayers and pure Cu film is performed using molecular dynamics (MD) simulations. The mechanical and tribological properties of Cu/Ag bilayers are investigated by comparing with those of pure Cu film. The effects of machining parameters (indenter radius, tool speed and machining depth) on the subsurface damage and material removal are studied by analyzing the dislocation movement, chipping volume, machining force and average temperature of the workpiece. The results show that the hardness of Cu/Ag bilayers is smaller than that of pure Cu film, due to the dislocation nucleation and emission from the Cu/Ag interface. Meanwhile, the friction coefficient of Cu/Ag bilayers is larger than that of pure Cu film. Furthermore, the metal bonding energy at the Cu/Ag interface is weaker than that in pure Cu film, which causes the low hardness in the Cu/Ag bilayers. The Young's moduli in the Cu/Ag bilayers and pure Cu film are calculated by the Hertz contact mechanism and are close to the experimental result. During nanometric machining of Cu/Ag bilayers, the larger indenter radius or higher tool speed would cause a larger indentation force. The chipping volume, machining force and average temperature would increase with the increment of indenter radius, tool speed and machining depth. The subsurface damage can be reduced by selecting the smaller indenter radius, lower tool speed, and smaller machining depth, where fewer lattice defects are produced. In addition, the selection of lower tool speed also plays a crucial role in improving the smoothness of the ground surface. read less USED (high confidence) Q. Wang, H. Xie, Z. Hu, and C. Liu, “The Impact of the Electric Field on Surface Condensation of Water Vapor: Insight from Molecular Dynamics Simulation,” Nanomaterials. 2019. link Times cited: 18 Abstract: In this study, molecular dynamics simulations were carried o… read moreAbstract: In this study, molecular dynamics simulations were carried out to study the coupling effect of electric field strength and surface wettability on the condensation process of water vapor. Our results show that an electric field can rotate water molecules upward and restrict condensation. Formed clusters are stretched to become columns above the threshold strength of the field, causing the condensation rate to drop quickly. The enhancement of surface attraction force boosts the rearrangement of water molecules adjacent to the surface and exaggerates the threshold value for shape transformation. In addition, the contact area between clusters and the surface increases with increasing amounts of surface attraction force, which raises the condensation efficiency. Thus, the condensation rate of water vapor on a surface under an electric field is determined by competition between intermolecular forces from the electric field and the surface. read less USED (high confidence) M. Amini, A. Lohrasebi, and A. Vaez, “Molecular dynamics study of gold nano-clusters aggregation on a model defected graphene,” Indian Journal of Physics. 2018. link Times cited: 0 USED (high confidence) E. Ilker, M. Madran, M. Konuk, and S. Durukanoğlu, “Growth and shape stability of Cu-Ni core-shell nanoparticles: an atomistic perspective.,” Chemical communications. 2018. link Times cited: 5 Abstract: The growth and shape stability of bi-metallic cubic Cu-Ni na… read moreAbstract: The growth and shape stability of bi-metallic cubic Cu-Ni nanoparticles are studied using atomic-level simulations. Cubic nano-crystals coated with an ultra-thin Cu layer can be readily obtained when Ni cubic nanoparticles are used as the seeds. At elevated temperatures, the Cu seed with extending Ni branches preserves its shape compared to the Ni seed with extending Cu branches. read less USED (high confidence) S. Vishnubhotla et al., “Quantitative measurement of contact area and electron transport across platinum nanocontacts for scanning probe microscopy and electrical nanodevices,” Nanotechnology. 2018. link Times cited: 11 Abstract: Conductive modes of atomic force microscopy are widely used … read moreAbstract: Conductive modes of atomic force microscopy are widely used to characterize the electronic properties of materials, and in such measurements, contact size is typically determined from current flow. Conversely, in nanodevice applications, the current flow is predicted from the estimated contact size. In both cases, it is very common to relate the contact size and current flow using well-established ballistic electron transport theory. Here we performed 19 electromechanical tests of platinum nanocontacts with in situ transmission electron microscopy to measure contact size and conductance. We also used molecular dynamics simulations of matched nanocontacts to investigate the nature of contact on the atomic scale. Together, these tests show that the ballistic transport equations under-predict the contact size by more than an order of magnitude. The measurements suggest that the low conductance of the contact cannot be explained by the scattering of electrons at defects nor by patchy contact due to surface roughness; instead, the lower-than-expected contact conductance is attributed to approximately a monolayer of insulating surface species on the platinum. Surprisingly, the low conductance persists throughout loading and even after significant sliding of the contact in vacuum. We apply tunneling theory and extract best-fit barrier parameters that describe the properties of this surface layer. The implications of this investigation are that electron transport in device-relevant platinum nanocontacts can be significantly limited by the presence and persistence of surface species, resulting in current flow that is better described by tunneling theory than ballistic electron transport, even for cleaned pure-platinum surfaces and even after loading and sliding in vacuum. read less USED (high confidence) S. Vishnubhotla, R. Chen, S. Khanal, A. Martini, and T. Jacobs, “Understanding contact between platinum nanocontacts at low loads: The effect of reversible plasticity,” Nanotechnology. 2018. link Times cited: 8 Abstract: Metal nanocontacts play a critical role in atomic force micr… read moreAbstract: Metal nanocontacts play a critical role in atomic force microscopy, functional nanostructures, metallic nanoparticles, and nanoscale electromechanical devices. In all cases, knowledge of the area of contact, and its variation with load, is critical for the quantitative prediction of behavior. Often, the contact area is predicted using continuum mechanics models which relate contact size to geometry, material properties, and load. Here we show for platinum nanoprobes that the contact size deviates significantly from these continuum predictions, even at low applied loads and in the absence of irreversible shape change. We use in situ transmission electron microscopy (TEM) with matched molecular dynamics (MD) simulations to investigate the load-dependent size of the contact. Direct measurements of contact radius from MD and TEM exceed the predictions of continuum mechanics by 24%–164%, depending on the model applied. The physical mechanism for this deviation is found to be dislocation activity in the near-surface material, which is fully reversed upon unloading. These findings demonstrate that contact mechanics models are insufficient for predicting contact area in real-world platinum nanostructures, even at ultra-low applied loads. read less USED (high confidence) D. T. Ho, C. T. Nguyen, S. Y. Kwon, and S. Y. Kim, “Auxeticity in Metals and Periodic Metallic Porous Structures Induced by Elastic Instabilities,” physica status solidi (b). 2018. link Times cited: 24 Abstract: Materials with a negative Poisson's ratio (auxetics) ar… read moreAbstract: Materials with a negative Poisson's ratio (auxetics) are counter intuitive because their mechanical response is unusual. On the other hand, instabilities are usually regarded as deleterious phenomena and thus their prevention is needed. Here, numerical and theoretical evidences have been provided to show that two different elastic instabilities are, rather than deleterious, useful phenomena that cause auxeticity. It has been shown that a negative Poisson's ratio can be found in some face‐centered cubic (FCC) single crystals at a finite strain as they are under uniaxial stress along the [100]‐direction. The auxeticity is associated with a phase transformation induced by the Born–Hill's elastic instability, i.e., an elastic material instability. In addition, it has been found that periodic metallic porous structures can also show a negative Poisson's ratio at finite compressive strain. In this case, buckling of the micro‐structure of the porous structures, which is an elastic and geometric instability, is respondent for the auxeticity. read less USED (high confidence) X. Wang, C. Hou, C. Li, and Y. Han, “Shape-Dependent Aggregation of Silver Particles by Molecular Dynamics Simulation,” Crystals. 2018. link Times cited: 4 Abstract: In crystallization, nanoparticle aggregation often leads to … read moreAbstract: In crystallization, nanoparticle aggregation often leads to the formation of orderly structures, even single crystals. Why can nanoparticles form orderly structures and what is the mechanism dominating their orderly aggregation? These questions raise interesting research problems, but the occurrences that could answer them often fail to be directly observed, since the interaction among particles is invisible. Here, we report an attempt to discover the interaction and aggregation of building blocks through a computer simulation, focusing on the shape effect of building blocks on the aggregation. Four types of silver building blocks were selected, each consisting of (100) and (111) facets, but the ratio of these two facets was different. It was found that the area of facets played an important role in selecting the aggregation mode. The facets with a large area and high energy had a high possibility of aggregation. In addition, the effects of solvent viscosity and temperature were also investigated. High viscosity and low temperature enhanced the orderliness of aggregation. This paper reports a detailed view of the aggregation process of silver nanoparticles, which is expected to be helpful in understanding the structure evolution of materials in nonclassical crystallization. read less USED (high confidence) G. D. Leines and J. Rogal, “Maximum Likelihood Analysis of Reaction Coordinates during Solidification in Ni.,” The journal of physical chemistry. B. 2018. link Times cited: 11 Abstract: Understanding the underlying mechanism of crystal nucleation… read moreAbstract: Understanding the underlying mechanism of crystal nucleation is a fundamental aspect in the prediction and control of materials properties. Classical nucleation theory (CNT) assumes that homogeneous nucleation occurs via random fluctuations within the supercooled liquid, that the structure of the growing clusters resembles the most stable bulk phase, and that the nucleus size is the sole reaction coordinate (RC) of the process. Many materials are, however, known to exhibit multiple steps during crystallization, forming different polymorphs. As a consequence, more complex RCs are often required to capture all relevant information about the process. Here, we employ transition path sampling together with a maximum likelihood analysis of candidate order parameters to identify suitable RCs for the nucleation mechanism during solidification in Ni. In contrast to CNT, the analysis of the reweighted path ensemble shows that a prestructured liquid region that surrounds the crystal cluster is a relevant order parameter that enhances the RC and therefore plays a key role in the description of the nucleus and the interfacial free energy. We demonstrate that prestructured liquid clusters that emerge within the liquid act as precursors of the crystallization in a nonclassical two-step mechanism, which predetermines the coordination of the selected polymorphs. read less USED (high confidence) S. Ma, J. Zhang, P. Li, Y. Zhang, H. Jin, and W. Zhang, “Elastic deformation behavior and microstructure evolution of single crystal nickel nanowire in tensile test along [0 0 1] orientation,” Materialwissenschaft und Werkstofftechnik. 2018. link Times cited: 0 Abstract: Elastic deformation behavior and microstructure evolution of… read moreAbstract: Elastic deformation behavior and microstructure evolution of single crystal nickel nanowire in tensile test have been investigated using molecular dynamics simulations. When the temperature is above 300 K, the deformation of nanowire is inhomogeneous in the elastic stage. It can generate a kind of (body‐centered cubic)‐like structure within the material to guide the elastic deformation, rather than the uniform deformation of all the atoms. The (body‐centered cubic)‐like structure will transform to be a body‐centered cubic structure gradually with the increasing of strain and will revert to be a face‐centered cubic structure when the twin crystal appears. The formation of (body‐centered cubic)‐like structure could reduce the density of the material to resist deformation. Besides, the (body‐centered cubic)‐like structure increase with increasing of strain or temperature, and they will gather together to reduce the interface energy. Our conclusion also can be proved by the radial distribution functions g(r). read less USED (high confidence) J. Jin, P. Yang, J. Cao, S. Li, and Q. Peng, “Quasicontinuum Simulation of the Effect of Lotus-Type Nanocavity on the Onset Plasticity of Single Crystal Al during Nanoindentation,” Nanomaterials. 2018. link Times cited: 4 Abstract: Stress concentration around nanosized defects such as caviti… read moreAbstract: Stress concentration around nanosized defects such as cavities always leads to plastic deformation and failure of solids. We investigate the effects of depth, size, and shape of a lotus-type nanocavity on onset plasticity of single crystal Al during nanoindentation on a (001) surface using a quasicontinuum method. The results show that the presence of a nanocavity can greatly affect the contact stiffness (Sc) and yield stress (σy) of the matrix during nanoindentation. For a circular cavity, the Sc and σy gradually increase with the cavity depth. A critical depth can be identified, over which the Sc and σy are insensitive to the cavity depth and it is firstly observed that the nucleated dislocations extend into the matrix and form a y-shaped structure. Moreover, the critical depth varies approximately linearly with the indenter size, regarding the same cavity. The Sc almost linearly decreases with the cavity diameter, while the σy is slightly affected. For an ellipsoidal cavity, the Sc and σy increase with the aspect ratio (AR), while they are less affected when the AR is over 1. Our results shed light in the mechanical behavior of metals with cavities and could also be helpful in designing porous materials and structures. read less USED (high confidence) S. Zhang, Y. Xu, X. Liu, and S. Luo, “Competing roles of interfaces and matrix grain size in the deformation and failure of polycrystalline Cu-graphene nanolayered composites under shear loading.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 15 Abstract: The roles of interfaces and matrix grain size in the deforma… read moreAbstract: The roles of interfaces and matrix grain size in the deformation and failure of polycrystalline Cu-graphene nanolayered (PCuGNL) composites under shear loading are explored with molecular dynamics simulations for different repeat layer spacings (λ), Cu grain sizes (D) and graphene chiralities, and an analytical model is proposed to describe the shear behavior. At the yield stage, the yield stress of the PCuGNL composites is mainly controlled by λ for λ ≤ 15 nm but mainly by D for λ > 15 nm; the yield strain of the composites is approximately a constant value of 0.056, weakly dependent on λ, D and graphene chirality. The shear failure strain and failure stress are determined only by the Cu-graphene interfaces. Small λ reduces the stability of the composites, while large λ decreases their shear failure strength. Considering the yield, failure and interface stability, the optimum λ value for the PCuGNL composites is 2-15 nm. In this optimum λ range, PCuGNL composites can be designed by tailoring Cu-graphene interfaces, regardless of the microstructures of polycrystalline Cu. read less USED (high confidence) J. Zhang, J. Cui, Z. Yang, and Y. Yu, “Heat capacity and thermal expansion of metal crystalline materials based on dynamic thermal vibration,” Computational Mechanics. 2018. link Times cited: 0 USED (high confidence) G. Arora, K. Rawat, and D. Aidhy, “Effect of atomic order/disorder on Cr segregation in Ni-Fe alloys,” Journal of Applied Physics. 2018. link Times cited: 4 Abstract: Recent irradiation experiments on concentrated random solid … read moreAbstract: Recent irradiation experiments on concentrated random solid solution alloys (CSAs) show that some CSAs can undergo disorder-to-order transition, i.e., the atoms that are initially randomly distributed on a face centered cubic crystal lattice undergo ordering (e.g., L10 or L12) due to irradiation. In this work, we elucidate that the atomic structure could affect the segregation properties of grain boundaries. While working on Ni and Ni-Fe alloys, from static atomistic simulations on 138 grain boundaries, we show that despite identical alloy composition, Cr segregation is higher in the disordered structures compared to ordered structures in both Ni0.50Fe0.50 and Ni0.75Fe0.25 systems. We also show that grain boundary (GB) energy could act as a descriptor for impurity segregation. We illustrate that there is a direct correlation between Cr segregation and grain boundary energy, i.e., segregation increases with the increase in the GB energy. Such correlation is observed in pure Ni and in the Ni-Fe alloys studied in this work.Recent irradiation experiments on concentrated random solid solution alloys (CSAs) show that some CSAs can undergo disorder-to-order transition, i.e., the atoms that are initially randomly distributed on a face centered cubic crystal lattice undergo ordering (e.g., L10 or L12) due to irradiation. In this work, we elucidate that the atomic structure could affect the segregation properties of grain boundaries. While working on Ni and Ni-Fe alloys, from static atomistic simulations on 138 grain boundaries, we show that despite identical alloy composition, Cr segregation is higher in the disordered structures compared to ordered structures in both Ni0.50Fe0.50 and Ni0.75Fe0.25 systems. We also show that grain boundary (GB) energy could act as a descriptor for impurity segregation. We illustrate that there is a direct correlation between Cr segregation and grain boundary energy, i.e., segregation increases with the increase in the GB energy. Such correlation is observed in pure Ni and in the Ni-Fe alloys studi... read less USED (high confidence) A. Herron, S. Coleman, K. Dang, D. Spearot, and E. Homer, “Simulation of kinematic Kikuchi diffraction patterns from atomistic structures,” MethodsX. 2018. link Times cited: 3 USED (high confidence) H. Amekura et al., “Vaporlike phase of amorphous
SiO2
is not a prerequisite for the core/shell ion tracks or ion shaping,” Physical Review Materials. 2018. link Times cited: 9 Abstract: The SHI irradiations were performed under the CommonUse Faci… read moreAbstract: The SHI irradiations were performed under the CommonUse Facility Program of JAEA. H.A. was supported by JSPSKAKENHI Grant No. 18K04898. I.S., V.J., F.D., and K.N.
gratefully acknowledge financial support from the Academy
of Finland MESIOS and NANOIS projects, and CPU capacity
grants from the IT Centre for Science CSC in Espoo, Finland. Part of this work was performed at the SAXS/WAXS
beamline at the Australian Synchrotron, part of ANSTO. P.K.
acknowledges the Australian Research Council for financial
support. read less USED (high confidence) K. Yun, J. Lee, and H. Nam, “Effect of Temperature on Coalescence Behavior of Unsupported Gold Nanoparticles,” Electronic Materials Letters. 2018. link Times cited: 10 USED (high confidence) J. Zhang, “Phase transformation in two-dimensional covalent organic frameworks under compressive loading.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 9 Abstract: As a new class of two-dimensional (2D) materials, 2D covalen… read moreAbstract: As a new class of two-dimensional (2D) materials, 2D covalent organic frameworks (COFs) are proven to possess remarkable electronic and magnetic properties. However, their mechanical behaviours remain almost unexplored. In this work, taking the recently synthesised dimethylmethylene-bridged triphenylamine (DTPA) sheet as an example, we investigate the mechanical behaviours of 2D COFs based on molecular dynamics simulations together with density functional theory calculations. A novel phase transformation is observed in DTPA sheets when a relatively large in-plane compressive strain is applied to them. Specifically, the crystal structures of the transformed phases are topographically different when the compressive loading is applied in different directions. The compression-induced phase transformation in DTPA sheets is attributed to the buckling of their kagome lattice structures and is found to have significant impacts on their material properties. After the phase transformation, Young's modulus, band gap and thermal conductivity of DTPA sheets are greatly reduced and become strongly anisotropic. Moreover, a large in-plane negative Poisson's ratio is found in the transformed phases of DTPA sheets. It is expected that the results of the compression-induced phase transformation and its influence on the material properties observed in the present DTPA sheets can be further extended to other 2D COFs, since most 2D COFs are found to possess a similar kagome lattice structure. read less USED (high confidence) S. Guo, M. Wang, Y. Zhang, X. Lin, and W. Huang, “Region selectivity of nanometer scale crystallization behavior in metallic glass,” Journal of Materials Science. 2018. link Times cited: 0 USED (high confidence) V. Menon and S. James, “Molecular Dynamics Simulation Study of Liquid-Assisted Laser Beam Micromachining Process,” Journal of Manufacturing and Materials Processing. 2018. link Times cited: 7 Abstract: Liquid Assisted Laser Beam Micromachining (LA-LBMM) process … read moreAbstract: Liquid Assisted Laser Beam Micromachining (LA-LBMM) process is an advanced machining process that can overcome the limitations of traditional laser beam machining processes. This research involves the use of a Molecular Dynamics (MD) simulation technique to investigate the complex and dynamic mechanisms involved in the LA-LBMM process both in static and dynamic mode. The results of the MD simulation are compared with those of Laser Beam Micromachining (LBMM) performed in air. The study revealed that machining during LA-LBMM process showed higher removal compared with LBMM process. The LA-LBMM process in dynamic mode showed lesser material removal compared with the static mode as the flowing water carrying the heat away from the machining zone. Investigation of the material removal mechanism revealed the presence of a thermal blanket and a bubble formation in the LA-LBMM process, aiding in higher material removal. The findings of this study provide further insights to strengthen the knowledge base of laser beam micromachining technology. read less USED (high confidence) C. Schöner and T. Pöschel, “Orientation-dependent properties of nanoparticle impact.,” Physical review. E. 2018. link Times cited: 9 Abstract: The mechanical properties of nanoparticles cannot be reliabl… read moreAbstract: The mechanical properties of nanoparticles cannot be reliably described by bulk material characteristics due to their atomic structure, leading to pronounced anisotropic behavior. By means of molecular dynamics simulations, we study the impact of 5-nm Ag particles on an adhesive rigid wall. We show that the main characteristics of the impact such as the coefficient of normal restitution, the sticking probability, the maximal contact force, and the degree of plastic deformation of the particle depend sensitively on the angular orientation of the nanoparticle prior to the impact. We introduce the scalar parameter Ω describing the orientation and show that the impact characteristics can be described as functions of Ω. read less USED (high confidence) K. Chen, L. Zhao, M. Fu, and P. Patnaik, “The Effect of Platinum on β-NiAl/α-Al2O3 Interfacial Tensile Stress: A Combined Ab Initio DFT and Mechanics-Based Model Study,” Oxidation of Metals. 2018. link Times cited: 3 USED (high confidence) N. Beets and D. Farkas, “Mechanical Response of Au Foams of Varying Porosity from Atomistic Simulations,” JOM. 2018. link Times cited: 16 USED (high confidence) J. Fatermans et al., “Single Atom Detection from Low Contrast-to-Noise Ratio Electron Microscopy Images.,” Physical review letters. 2018. link Times cited: 28 Abstract: Single atom detection is of key importance to solving a wide… read moreAbstract: Single atom detection is of key importance to solving a wide range of scientific and technological problems. The strong interaction of electrons with matter makes transmission electron microscopy one of the most promising techniques. In particular, aberration correction using scanning transmission electron microscopy has made a significant step forward toward detecting single atoms. However, to overcome radiation damage, related to the use of high-energy electrons, the incoming electron dose should be kept low enough. This results in images exhibiting a low signal-to-noise ratio and extremely weak contrast, especially for light-element nanomaterials. To overcome this problem, a combination of physics-based model fitting and the use of a model-order selection method is proposed, enabling one to detect single atoms with high reliability. read less USED (high confidence) Y. Akkus, C. Nguyen, A. T. Celebi, and A. Beskok, “A first look at the performance of nano-grooved heat pipes,” International Journal of Heat and Mass Transfer. 2018. link Times cited: 21 USED (high confidence) Y. Yang, J. Cui, Y. Yu, and M. Xiang, “Macroscopic damping model for structural dynamics with random polycrystalline configurations,” Acta Mechanica Sinica. 2018. link Times cited: 3 USED (high confidence) X. Zhang 张, Yun-Xia 运侠 Han 韩, H. Jia 贾, Nuo 诺 Qu 曲, and Z. Liu 刘, “Molecular Dynamics Simulations of the Elastic Anisotropy of Pd at Extreme Conditions,” Communications in Theoretical Physics. 2018. link Times cited: 0 Abstract: It is very interesting to discover the elastic properties of… read moreAbstract: It is very interesting to discover the elastic properties of engineering material palladium, especially its elastic anisotropy along Hugoniot states. We here investigate the evolution of its high pressure and temperature (PT) elastic ansotropy along Hugoniot using molecular dynamics simulations based on accurate classical interatomic potential. In order to testify the validity of the interatomic potential of Pd in describing the high PT elastic properties, we calculate its isothermal and adiabatic elastic moduli using molecular dynamics method. The obtained data are in good agreement with experimental data. From the isothermal elastic constants, we deduce the Hugoniot acoustic velocities and find that the resulting data are in good agreement with experimental acoustic velocity data. Based on the reliable elastic constants, we further investigate the spacial elastic ansotropy along Hugoniot PT states. It is found that the spacial elastic anisotropy of Pd increases along Hugoniot states. read less USED (high confidence) J. Abraham, A. Hinz, T. Strunskus, F. Faupel, and M. Bonitz, “Formation of polymer-based nanoparticles and nanocomposites by plasma-assisted deposition methods,” The European Physical Journal D. 2018. link Times cited: 8 USED (high confidence) E. Dolgusheva and V. Trubitsin, “Lattice Heat Capacity of Nanostructured Materials Based on Titanium/Zirconium and Aluminum,” Physics of the Solid State. 2018. link Times cited: 2 USED (high confidence) J. Zhang, J. Cui, Z. Yang, and Y. Yu, “Heat capacity and thermal expansion of metal crystalline materials based on dynamic thermal vibration,” Computational Mechanics. 2018. link Times cited: 5 USED (high confidence) A. Gola and L. Pastewka, “Embedded atom method potential for studying mechanical properties of binary Cu–Au alloys,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 13 Abstract: We present an embedded atom method (EAM) potential for the b… read moreAbstract: We present an embedded atom method (EAM) potential for the binary Cu–Au system. The unary phases are described by two well-tested unary EAM potentials for Cu and Au. We fitted the interaction between Cu and Au to experimental properties of the binary intermetallic phases Cu3Au, CuAu and CuAu3. Particular attention has been paid to reproducing stacking fault energies in order to obtain a potential suitable for studying deformation in this binary system. The resulting energies, lattice constant, elastic properties and melting points are in good agreement with available experimental data. We use nested sampling to show that our potential reproduces the phase boundaries between intermetallic phases and the disordered face-centered cubic solid solution. We benchmark our potential against four popular Cu–Au EAM parameterizations and density-functional theory calculations. read less USED (high confidence) J. Andrés et al., “Formation of Ag nanoparticles under electron beam irradiation: Atomistic origins from first-principles calculations,” International Journal of Quantum Chemistry. 2018. link Times cited: 20 Abstract: Department of Chemistry, CDMF, Federal University of S~ao Ca… read moreAbstract: Department of Chemistry, CDMF, Federal University of S~ao Carlos, P.O. Box 676, S~ao Carlos 13565-905, Brazil Department of Physical Chemistry, University of Valencia, Burjassot 46100, Spain Faculdade de Ciências Exatas e Tecnol ogicas, Universidade Federal da Grande Dourados, Unidade II, CP 533, 79804-970, Dourados, Mato Grosso do Sul, Brazil Department of Condensed Matter Physics, Institute of Physics ‘Gleb Wataghin’, State University of Campinas, 13083-970, Campinas, S~ao Paulo, Brazil Department of Physical Chemistry, Institute of Chemistry, State University of Campinas, 13083-970, Campinas, S~ao Paulo, Brazil Department of Biotechnology and Exact Sciences, Federal University of Tocantins, 77410-530, Gurupi, Tocantins, Brazil read less USED (high confidence) H. Park, K. Kim, H.-J. Kim, D.-K. Kim, Y. Won, and S. K. Kim, “Electrodeposition-fabricated PtCu-alloy cathode catalysts for high-temperature proton exchange membrane fuel cells,” Korean Journal of Chemical Engineering. 2018. link Times cited: 24 USED (high confidence) M. H. Nazir, Z. Khan, A. Saeed, A. Siddaiah, and P. Menezes, “Synergistic wear-corrosion analysis and modelling of nanocomposite coatings,” Tribology International. 2018. link Times cited: 31 USED (high confidence) D. Farkas, J. Stuckner, R. Umbel, B. Kuhr, and M. Demkowicz, “Indentation response of nanoporous gold from atomistic simulations,” Journal of Materials Research. 2018. link Times cited: 12 Abstract: We present classical potential molecular dynamics simulation… read moreAbstract: We present classical potential molecular dynamics simulations of nanoporous gold (np-Au) impacted by a spherical indenter. The atomic structure was generated using a phase field model as a template. In agreement with previous experiments, we observe densification in the region under the indenter. The hardness values obtained from our simulations exhibit a transition from an initially perfect-plastic plateau to hardening behavior in the later stages of indentation. This transition occurs when the relative density beneath the indenter exceeds ∼0.9. Hardness values obtained from the nanoindentation simulations reach 0.6 GPa, due to the densification of the material under the indenter. Elevated dislocation densities are observed in the densified region. The mechanism of pore collapse in the densified layer under the indenter is seen to switch from uniaxial to triaxial, consistent with a change in deformation mechanism from one based on shearing of individual ligaments in np-Au to one involving dislocation-mediated plasticity around voids in a Au single crystal undergoing uniaxial compression. read less USED (high confidence) L. Lin et al., “Significant enhancement of the performance of hydrogen evolution reaction through shape-controlled synthesis of hierarchical dendrite-like platinum,” Journal of Materials Chemistry. 2018. link Times cited: 25 Abstract: Herein, hierarchical dendrite-like Pt crystals with a distin… read moreAbstract: Herein, hierarchical dendrite-like Pt crystals with a distinct morphology were synthesized via a facile one-pot method without any templates. Formation of this hierarchical structure is dependent on the reaction duration. Interestingly, different hierarchical structures show different catalytic activities. After a 12 hour reaction, tertiary structures of Pt are formed, which can act as outstanding catalysts in the hydrogen evolution reaction (HER). The onset potential of this dendrite-like Pt catalyst for the HER in a 0.5 M H2SO4 solution is 15 mV, which outperforms that of commercial Pt/C (30 mV). Moreover, it shows significantly improved stability for HER as the polarization curve after 10 000 cycles retains a similar performance as in the initial test; this results in a loss of only 2.6% of its initial current density at an overpotential of 0.05 V. The distinct hierarchical dendrite-like structures are maintained after cycling and current–time tests, which can be responsible for the excellent performance of this catalyst. read less USED (high confidence) H. Yang, E. Goudeli, and C. J. Hogan, “Condensation and dissociation rates for gas phase metal clusters from molecular dynamics trajectory calculations.,” The Journal of chemical physics. 2018. link Times cited: 23 Abstract: In gas phase synthesis systems, clusters form and grow via c… read moreAbstract: In gas phase synthesis systems, clusters form and grow via condensation, in which a monomer binds to an existing cluster. While a hard-sphere equation is frequently used to predict the condensation rate coefficient, this equation neglects the influences of potential interactions and cluster internal energy on the condensation process. Here, we present a collision rate theory-molecular dynamics simulation approach to calculate condensation probabilities and condensation rate coefficients. We use this approach to examine atomic condensation onto 6-56-atom Au and Mg clusters. The probability of condensation depends upon the initial relative velocity (v) between atom and cluster and the initial impact parameter (b). In all cases, there is a well-defined region of b-v space where condensation is highly probable, and outside of which the condensation probability drops to zero. For Au clusters with more than 10 atoms, we find that at gas temperatures in the 300-1200 K range, the condensation rate coefficient exceeds the hard-sphere rate coefficient by a factor of 1.5-2.0. Conversely, for Au clusters with 10 or fewer atoms and for 14- and 28-atom Mg clusters, as cluster equilibration temperature increases, the condensation rate coefficient drops to values below the hard-sphere rate coefficient. Calculations also yield the self-dissociation rate coefficient, which is found to vary considerably with gas temperature. Finally, calculations results reveal that grazing (high b) atom-cluster collisions at elevated velocity (>1000 m s-1) can result in the colliding atom rebounding (bounce) from the cluster surface or binding while another atom dissociates (replacement). The presented method can be applied in developing rate equations to predict material formation and growth rates in vapor phase systems. read less USED (high confidence) F. Wang, Z. Tang, and H. He, “Stress-dislocation interaction mechanism in low-temperature thermo-compression sintering of Ag NPs,” AIP Advances. 2018. link Times cited: 10 Abstract: The sintering of metal nanoparticles (NPs) has been widely s… read moreAbstract: The sintering of metal nanoparticles (NPs) has been widely studied in the field of nanotechnology, and low-temperature sintering has become the industry standard. In this study, a molecular dynamics (MD) model was established to study the sintering behaviour of silver NPs during low-temperature thermo-compression. Primarily, we studied the sintering process, in which the ratio of neck radius to particle radius (x/r) changes. Under a uniaxial pressure, the maximum ratio in the temperature range 420–425 K was 1. According to the change of x/r, the process can be broken down into three stages: the neck-formation stage, neck-growth stage, and neck-stability stage. In addition, the relationship between potential energy, internal stress, and dislocation density during sintering is discussed. The results showed that cycling internal stress played an important role in sintering. Under the uniaxial pressure, the stress-dislocation interaction was found to be the major mechanism for thermo-compression sintering bec... read less USED (high confidence) H. Xie, J. Yu, T. Yu, and F. Yin, “Face-centred cubic to body-centred cubic phase transformation under [1 0 0] tensile loading,” Philosophical Magazine. 2018. link Times cited: 2 Abstract: ABSTRACT Molecular dynamics simulation was used to verify a … read moreAbstract: ABSTRACT Molecular dynamics simulation was used to verify a speculation of the existence of a certain face-centred cubic (FCC) to body-centred cubic (BCC) phase transformation pathway. Four FCC metals, Ni, Cu, Au and Ag, were stretched along the [1 0 0] direction at various strain rates and temperatures. Under high strain rate and low temperature, and beyond the elastic limit, the bifurcation of the FCC phase occurred with sudden contraction along one lateral direction and expansion along the other lateral direction. When the lattice constant along the expansion direction converged with that of the stretched direction, the FCC phase transformed into an unstressed BCC phase. By reducing the strain rate or increasing the temperature, dislocation or ‘momentum-induced melting’ mechanisms began to control the plastic deformation of the FCC metals, respectively. read less USED (high confidence) H. Sun, L. Chen, S. Sun, and T.-Y. Zhang, “Size- and temperature-dependent Young’s modulus and size-dependent thermal expansion coefficient of nanowires,” Science China Technological Sciences. 2018. link Times cited: 17 USED (high confidence) H. Sun, L. Chen, S. Sun, and T.-Y. Zhang, “Size- and temperature-dependent Young’s modulus and size-dependent thermal expansion coefficient of nanowires,” Science China Technological Sciences. 2018. link Times cited: 0 USED (high confidence) X. Liu, J. Cai, and S. Luo, “Interfacial anti-fatigue effect in graphene-copper nanolayered composites under cyclic shear loading.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 13 Abstract: Low-cycle fatigue behaviors of graphene-copper nanolayered (… read moreAbstract: Low-cycle fatigue behaviors of graphene-copper nanolayered (GCuNL) composites are explored at different interface configurations and repeat layer spacings. The graphene interfaces can trap dislocations through impeding the propagation of dislocations in copper layers, giving rise to the absence of softening, and an increase in the fatigue strength of GCuNL composites (up to 400% that of pure copper). This anti-fatigue effect is independent of the crystal orientation of copper or the chirality of graphene due to interfacial constraints and can be controlled by tailoring the repeat layer spacing. Low repeat layer spacing increases the instability and nonlinearity of the composites, while high repeat layer spacing decreases the anti-fatigue effect. The optimum value of the repeat layer spacing for the GCuNL composites is 3-7 nm, in order to achieve a balanced anti-fatigue capability and interface stability. read less USED (high confidence) S. Ebrahimi, “The effect of Stone-Wales defects and roughness degree on the lubricity of graphene on gold surfaces,” Journal of Molecular Modeling. 2018. link Times cited: 3 USED (high confidence) X. Qiu and R. Joshi, “Dynamic analysis of material ejection from cathodic metal nano-tips due to local heating and field generated stress,” Physics of Plasmas. 2018. link Times cited: 6 Abstract: The potential for explosive cathode emission due to nanoprot… read moreAbstract: The potential for explosive cathode emission due to nanoprotrusions subjected to Maxwell stress and heating from strong electric fields is probed self-consistently based on non-equilibrium molecular-dynamics. The focus is on determining the electric field magnitudes that could lead to material ejection, assessing dependencies of the instability on the nanoprotrusion height and cross-sectional area, and the role of time-dependent thermal conductivity and local temperature changes. Our results indicate that large aspect ratios would facilitate mass ejection, with protrusion break up occurring over times in the 25 ns range, in agreement with experimental reports on explosive emission. read less USED (high confidence) S. M. Rassoulinejad-Mousavi and Y. Zhang, “Interatomic Potentials Transferability for Molecular Simulations: A Comparative Study for Platinum, Gold and Silver,” Scientific Reports. 2018. link Times cited: 33 USED (high confidence) S. Ajori, S. Haghighi, and R. Ansari, “A molecular dynamics study on the thermal conductivity of endohedrally functionalized single-walled carbon nanotubes with gold nanowires,” The European Physical Journal D. 2018. link Times cited: 23 USED (high confidence) Y. Rosandi, J. Grossi, E. Bringa, and H. Urbassek, “The Laser ablation of a metal foam: The role of electron–phonon coupling and electronic heat diffusivity,” Journal of Applied Physics. 2018. link Times cited: 4 Abstract: The incidence of energetic laser pulses on a metal foam may … read moreAbstract: The incidence of energetic laser pulses on a metal foam may lead to foam ablation. The processes occurring in the foam may differ strongly from those in a bulk metal: The absorption of laser light, energy transfer to the atomic system, heat conduction, and finally, the atomistic processes—such as melting or evaporation—may be different. In addition, novel phenomena take place, such as a reorganization of the ligament network in the foam. We study all these processes in an Au foam of average porosity 79% and an average ligament diameter of 2.5 nm, using molecular dynamics simulation. The coupling of the electronic system to the atomic system is modeled by using the electron–phonon coupling, g, and the electronic heat diffusivity, κe, as model parameters, since their actual values for foams are unknown. We show that the foam coarsens under laser irradiation. While κe governs the homogeneity of the processes, g mainly determines their time scale. The final porosity reached is independent of the value of g. read less USED (high confidence) Y. Yang, J. Cui, Y. Yu, and M. Xiang, “Macroscopic damping model for structural dynamics with random polycrystalline configurations,” Acta Mechanica Sinica. 2017. link Times cited: 0 USED (high confidence) F. Wang, N. Nie, H. He, Z. Tang, Z. Chen, and W. Zhu, “Ultrasonic-Assisted Sintering of Silver Nanoparticles for Flexible Electronics,” Journal of Physical Chemistry C. 2017. link Times cited: 21 Abstract: A silver nanoparticle sintering method is promising for the … read moreAbstract: A silver nanoparticle sintering method is promising for the fabrication of flexible electronics. An ultrasonic-assisted sintering process is presented to obtain conductive patterns with low resistivity on a paper-based substrate. Compared to the conventional hot-pressing sintering process, the proposed method can efficiently fabricate densified microstructures with better electrical properties at low temperature and pressure. In particular, the effects of ultrasonic effective time, entry time, and amplitude were systematically analyzed. It was found that lower resistivity preferred larger ultrasonic amplitude. The ultrasonic effective time happened during the first 3 min of the entire sintering process, which implied that the ultrasonic synergetic effect occurred at the very beginning stage of the sintering process. Additionally, better conductivity of the sintered patterns was obtained when the ultrasonic entry time was shorter. The mechanism of the synergetic effect of ultrasonic sintering was also disc... read less USED (high confidence) Yuan-Yuan 圆圆 Tian 田, Jia 甲 Li 李, Ze-Ying 泽英 Hu 胡, Zhi-Peng 志鹏 Wang 王, and Q. Fang 方, “Molecular dynamics study of plastic deformation mechanism in Cu/Ag multilayers,” Chinese Physics B. 2017. link Times cited: 4 Abstract: The plastic deformation mechanism of Cu/Ag multilayers is in… read moreAbstract: The plastic deformation mechanism of Cu/Ag multilayers is investigated by molecular dynamics (MD) simulation in a nanoindentation process. The result shows that due to the interface barrier, the dislocations pile-up at the interface and then the plastic deformation of the Ag matrix occurs due to the nucleation and emission of dislocations from the interface and the dislocation propagation through the interface. In addition, it is found that the incipient plastic deformation of Cu/Ag multilayers is postponed, compared with that of bulk single-crystal Cu. The plastic deformation of Cu/Ag multilayers is affected by the lattice mismatch more than by the difference in stacking fault energy (SFE) between Cu and Ag. The dislocation pile-up at the interface is determined by the obstruction of the mismatch dislocation network and the attraction of the image force. Furthermore, this work provides a basis for further understanding and tailoring metal multilayers with good mechanical properties, which may facilitate the design and development of multilayer materials with low cost production strategies. read less USED (high confidence) M. H. Nazir et al., “Analyzing and Modelling the Corrosion Behavior of Ni/Al2O3, Ni/SiC, Ni/ZrO2 and Ni/Graphene Nanocomposite Coatings,” Materials. 2017. link Times cited: 22 Abstract: A study has been presented on the effects of intrinsic mecha… read moreAbstract: A study has been presented on the effects of intrinsic mechanical parameters, such as surface stress, surface elastic modulus, surface porosity, permeability and grain size on the corrosion failure of nanocomposite coatings. A set of mechano-electrochemical equations was developed by combining the popular Butler–Volmer and Duhem expressions to analyze the direct influence of mechanical parameters on the electrochemical reactions in nanocomposite coatings. Nanocomposite coatings of Ni with Al2O3, SiC, ZrO2 and Graphene nanoparticles were studied as examples. The predictions showed that the corrosion rate of the nanocoatings increased with increasing grain size due to increase in surface stress, surface porosity and permeability of nanocoatings. A detailed experimental study was performed in which the nanocomposite coatings were subjected to an accelerated corrosion testing. The experimental results helped to develop and validate the equations by qualitative comparison between the experimental and predicted results showing good agreement between the two. read less USED (high confidence) P. Quaino et al., “Understanding the structure and reactivity of NiCu nanoparticles: an atomistic model.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 7 Abstract: The structure of bimetallic NiCu nanoparticles (NP) is inves… read moreAbstract: The structure of bimetallic NiCu nanoparticles (NP) is investigated as a function of their composition and size by means of Lattice MonteCarlo (LMC) and molecular dynamics (MD) simulations. According to our results, copper segregation takes place at any composition of the particles. We found that this feature is not size-dependent. In contrast, nickel segregation depends on the NP size. When the size increases, Ni atoms tend to remain in the vicinity of the surface and deeper. For smaller NPs, Ni atoms are located at the surface as well. Our results also showed that most of the metal atoms segregated at the surface area were found to decorate edges and/or form islands. Our findings agree qualitatively with the experimental data found in the literature. In addition, we comment on the reactivity of these nanoparticles. read less USED (high confidence) S. Borisova, G. Rusina, S. Eremeev, and E. Chulkov, “Submonolayer Adsorption of Potassium on Reconstructed and Unreconstructed Cu(110): Structure and Phonons,” Journal of Physical Chemistry C. 2017. link Times cited: 6 Abstract: We present a theoretical investigation of the structural and… read moreAbstract: We present a theoretical investigation of the structural and vibrational properties of the K/Cu(110) adsorbed systems using the embedded atom method. The surface relaxation, phonon dispersions, and polarization of vibrational modes as well as the local density of states have been calculated as a function of potassium coverage on reconstructed and unreconstructed Cu(110) surface. On the reconstructed surface, two K-substrate stretching modes have been found, which do not show any coverage dependence. The obtained vibrational frequencies of surface states are in close agreement with available experimental data. read less USED (high confidence) K. Chen, L. Wang, Y. Yin, and Q. Wang, “Molecular dynamics simulation of polycrystalline metal under high velocity nanoscale sliding,” Chemical engineering transactions. 2017. link Times cited: 1 Abstract: Three-dimensional non-equilibrium molecular dynamics simulat… read moreAbstract: Three-dimensional non-equilibrium molecular dynamics simulations are performed to investigate the tribological characteristics of polycrystalline Ni-Ni and Ni-Cu high-speed sliding systems. The grain size distribution, temperature profiles and heat density changes are obtained. Founded on simulation results a correlation between surface grain size and heat conduction is proposed which can help us to fully understand the mechanism of polycrystalline friction behavior and design better friction material. This interaction shows that the sizes of the grains located in the interface decrease and it increases the resistance of friction heat conduction during sliding. read less USED (high confidence) M. Shugaev, C.-Y. Shih, E. T. Karim, C. Wu, and L. Zhigilei, “Generation of nanocrystalline surface layer in short pulse laser processing of metal targets under conditions of spatial confinement by solid or liquid overlayer,” Applied Surface Science. 2017. link Times cited: 27 USED (high confidence) G. Rusina, S. Borisova, and E. Chulkov, “Submonolayer adsorption of Na onto the Cu(110) surface: Structure and vibrational properties,” Journal of Experimental and Theoretical Physics. 2017. link Times cited: 2 USED (high confidence) Y. Wei and S. Peng, “The stress-velocity relationship of twinning partial dislocations and the phonon-based physical interpretation,” Science China Physics, Mechanics & Astronomy. 2017. link Times cited: 14 USED (high confidence) Y. Cui and Z. Chen, “Void growth via atomistic simulation: will the formation of shear loops still grow a void under different thermo-mechanical constraints?,” Philosophical Magazine. 2017. link Times cited: 9 Abstract: Molecular dynamics (MD) simulations under different mechanic… read moreAbstract: Molecular dynamics (MD) simulations under different mechanical and thermal constraints are carried out with a nanovoid embedded inside a single-crystal, face-centred-cubic copper. The dislocation emission angles measured from MD plots under 0.1 K, uniaxial-strain simulation are in line with the theoretical model. The dislocation density calculated from simulation is qualitatively consistent with the experimental measurement in terms of a saturation feature. The ‘relatively farthest-travelled’ atoms are employed to reflect the correlation between the dislocation structure and the void growth. At a smaller scale, the incomplete shear dislocation loops on the slip plane contribute to the local material transport. At a larger scale, the dislocation structures formed by those incomplete shear loops further facilitate the growth of nanovoid. Compared to the uniaxial-strain case, the void growth under the uniaxial-stress is very limited. The uniaxial-strain loading results in an octahedron void shape. The uniaxial-stress loading turns the nanovoid into a prolate ellipsoid along the loading direction. In the simulation, the largest specimen contains 12 million atoms and the lowest strain rate applied is 2 × 106 s−1. Under all the different thermomechanical constraints concerned, the formation of incomplete shear dislocation loops are found capable of growing the void. read less USED (high confidence) B. Revard, “Development of an Evolutionary Algorithm for the ab Initio Discovery of Two-Dimensional Materials.” 2017. link Times cited: 0 USED (high confidence) Q. Fang, Q. Wang, J. Li, X. Zeng, and Y.-wen Liu, “Mechanisms of subsurface damage and material removal during high speed grinding processes in Ni/Cu multilayers using a molecular dynamics study,” RSC Advances. 2017. link Times cited: 25 Abstract: The molecular dynamics (MD) simulation of Ni/Cu multilayers … read moreAbstract: The molecular dynamics (MD) simulation of Ni/Cu multilayers under a high speed grinding process with a diamond tip is performed, with the aim of investigating the effects of varying machining parameters on subsurface damage and material removal in the Ni/Cu multilayers. A series of key factors, consisting of grinding speed, tool radius and depth of cut, that influence the deformation of the workpiece are systemically studied in terms of surface morphology, dislocation movement, grinding temperature and average grinding force. Both the grinding temperature and force increase with increasing grinding speed, tool radius and depth of cut. In addition, a relatively small grinding velocity results in more stacking faults (SF) and a greater volume of material pileups on the sides of the groove. A good surface integrity of the Ni/Cu multilayers, with relatively fewer lattice defects, is more easily obtained by a machining process with a smaller tool radius or cutting depth. The results also show that the grinding temperature of Ni/Cu multilayers with varying grinding speeds, tool radii and cutting depths is higher than that of pure Ni thin film. read less USED (high confidence) T. Cheng, H. Xiao, and W. Goddard, “Nature of the Active Sites for CO Reduction on Copper Nanoparticles; Suggestions for Optimizing Performance.,” Journal of the American Chemical Society. 2017. link Times cited: 119 Abstract: Recent experiments show that the grain boundaries (GBs) of c… read moreAbstract: Recent experiments show that the grain boundaries (GBs) of copper nanoparticles (NPs) lead to an outstanding performance in reducing CO2 and CO to alcohol products. We report here multiscale simulations that simulate experimental synthesis conditions to predict the structure of a 10 nm Cu NP (158 555 atoms). To identify active sites, we first predict the CO binding at a large number of sites and select four exhibiting CO binding stronger than the (211) step surface. Then, we predict the formation energy of the *OCCOH intermediate as a descriptor for C-C coupling, identifying two active sites, both of which have an under-coordinated surface square site adjacent to a subsurface stacking fault. We then propose a periodic Cu surface (4 by 4 supercell) with a similar site that substantially decreases the formation energy of *OCCOH, by 0.14 eV. read less USED (high confidence) V. Mazhukin, A. V. Shapranov, V. E. Perezhigin, O. Koroleva, and A. Mazhukin, “Kinetic melting and crystallization stages of strongly superheated and supercooled metals,” Mathematical Models and Computer Simulations. 2017. link Times cited: 17 USED (high confidence) D. T. Ho, S. Y. Kwon, H. S. Park, and S. Y. Kim, “Negative Thermal Expansion of Ultrathin Metal Nanowires: A Computational Study.,” Nano letters. 2017. link Times cited: 24 Abstract: Most materials expand upon heating because the coefficient o… read moreAbstract: Most materials expand upon heating because the coefficient of thermal expansion (CTE), the fundamental property of materials characterizing the mechanical response of the materials to heating, is positive. There have been some reports of materials that exhibit negative thermal expansion (NTE), but most of these have been in complex alloys, where NTE originates from the transverse vibrations of the materials. Here, we show using molecular dynamics simulations that some single crystal monatomic FCC metal nanowires can exhibit NTE along the length direction due to a novel thermomechanical coupling. We develop an analytic model for the CTE in nanowires that is a function of the surface stress, elastic modulus, and nanowire size. The model suggests that the CTE of nanowires can be reduced due to elastic softening of the materials and also due to surface stress. For the nanowires, the model predicts that the CTE reduction can lead to NTE if the nanowire Young's modulus is sufficiently reduced while the nanowire surface stress remains sufficiently large, which is in excellent agreement with the molecular dynamics simulation results. Overall, we find a "smaller is smaller" trend for the CTE of nanowires, leading to this unexpected, surface-stress-driven mechanism for NTE in nanoscale materials. read less USED (high confidence) O. Kovalenko, C. Brandl, L. Klinger, and E. Rabkin, “Self‐Healing and Shape Memory Effects in Gold Microparticles through the Defects‐Mediated Diffusion,” Advanced Science. 2017. link Times cited: 15 Abstract: Some metal alloys subjected to irreversible plastic deformat… read moreAbstract: Some metal alloys subjected to irreversible plastic deformation can repair the inflicted damage and/or recover their original shape upon heating. The conventional shape memory effect in metallic alloys relies on collective, or “military” phase transformations. This work demonstrates a new and fundamentally different type of self‐healing and shape memory in single crystalline faceted nano and microparticles of pure gold, which are plastically deformed with an atomic force microscope tip. It is shown that annealing of the deformed particles at elevated temperatures leads to nearly full recovery of their initial asymmetric polyhedral shape, which does not correspond to global energy minimum shape. The atomistic molecular dynamic simulations demonstrate that the shape recovery of the particles is controlled by the self‐diffusion of gold atoms along the terrace ledges formed during the particles indentation. This ledge‐guided diffusion leads to shape recovery by the irreversible diffusion process. A semiquantitative model of healing developed in this work demonstrates a good agreement with the experimental data. read less USED (high confidence) A. Rida, E. Rouhaud, A. Makke, M. Micoulaut, and B. Mantisi, “Study of the effects of grain size on the mechanical properties of nanocrystalline copper using molecular dynamics simulation with initial realistic samples,” Philosophical Magazine. 2017. link Times cited: 29 Abstract: Molecular dynamics simulations have been performed to study … read moreAbstract: Molecular dynamics simulations have been performed to study the mechanical properties of a columnar nanocrystalline copper with a mean grain size between 9.0 and 24 nm. A melting–cooling method has been used to generate the initial samples: this method produces realistic samples that contain defects inside the grains such as dislocations and vacancies. The results of uniaxial tensile tests applied to these samples reveal the presence of a critical mean grain size between 16 and 20 nm, for which there is an inversion of the conventional Hall–Petch relation. The principal mechanisms of deformation present in the samples correspond to a combination of dislocations and grain boundary sliding. In addition, this analysis shows the presence of sliding planes generated by the motion of perfect edge dislocations that are absorbed by grain boundaries. It is the initial defects present inside the grains that lead to this mechanism of deformation. An analysis of the atomic configurations further shows that nucleation and propagation of cracks are localised on the grain boundaries especially on the triple grains junctions. read less USED (high confidence) C.-Y. Shih, M. Shugaev, C. Wu, and L. Zhigilei, “Generation of Subsurface Voids, Incubation Effect, and Formation of Nanoparticles in Short Pulse Laser Interactions with Bulk Metal Targets in Liquid: Molecular Dynamics Study,” The Journal of Physical Chemistry. C, Nanomaterials and Interfaces. 2017. link Times cited: 75 Abstract: The ability of short pulse laser ablation in liquids to prod… read moreAbstract: The ability of short pulse laser ablation in liquids to produce clean colloidal nanoparticles and unusual surface morphology has been employed in a broad range of practical applications. In this paper, we report the results of large-scale molecular dynamics simulations aimed at revealing the key processes that control the surface morphology and nanoparticle size distributions by pulsed laser ablation in liquids. The simulations of bulk Ag targets irradiated in water are performed with an advanced computational model combining a coarse-grained representation of liquid environment and an atomistic description of laser interaction with metal targets. For the irradiation conditions that correspond to the spallation regime in vacuum, the simulations predict that the water environment can prevent the complete separation of the spalled layer from the target, leading to the formation of large subsurface voids stabilized by rapid cooling and solidification. The subsequent irradiation of the laser-modified surface is found to result in a more efficient ablation and nanoparticle generation, thus suggesting the possibility of the incubation effect in multipulse laser ablation in liquids. The simulations performed at higher laser fluences that correspond to the phase explosion regime in vacuum reveal the accumulation of the ablation plume at the interface with the water environment and the formation of a hot metal layer. The water in contact with the metal layer is brought to the supercritical state and provides an environment suitable for nucleation and growth of small metal nanoparticles from metal atoms emitted from the hot metal layer. The metal layer itself has limited stability and can readily disintegrate into large (tens of nanometers) nanoparticles. The layer disintegration is facilitated by the Rayleigh–Taylor instability of the interface between the higher density metal layer decelerated by the pressure from the lighter supercritical water. The nanoparticles emerging from the layer disintegration are rapidly cooled and solidified due to the interaction with water environment, with a cooling rate of ∼2 × 1012 K/s observed in the simulations. The computational prediction of two distinct mechanisms of nanoparticle formation yielding nanoparticles with different characteristic sizes provides a plausible explanation for the experimental observations of bimodal nanoparticle size distributions in laser ablation in liquids. The ultrahigh cooling and solidification rates suggest the possibility for generation of nanoparticles featuring metastable phases and highly nonequilibrium structures. read less USED (high confidence) P. Wynblatt and D. Chatain, “Two-dimensional versus three-dimensional constraints in hetero-epitaxy/orientation relationships,” Journal of Materials Science. 2017. link Times cited: 4 USED (high confidence) Y. Zheng, L. Ding, H. Ye, and Z. Chen, “Vibration-Induced Property Change in the Melting and Solidifying Process of Metallic Nanoparticles,” Nanoscale Research Letters. 2017. link Times cited: 5 USED (high confidence) G. D. Leines, R. Drautz, and J. Rogal, “Atomistic insight into the non-classical nucleation mechanism during solidification in Ni.,” The Journal of chemical physics. 2017. link Times cited: 25 Abstract: Nucleation is a key step during crystallization, but a compl… read moreAbstract: Nucleation is a key step during crystallization, but a complete understanding of the fundamental atomistic processes remains elusive. We investigate the mechanism of nucleation during solidification in nickel for various undercoolings using transition path sampling simulations. The temperature dependence of the free energy barriers and rate constants that we obtain is consistent with the predictions of classical nucleation theory and experiments. However, our analysis of the transition path ensemble reveals a mechanism that deviates from the classical picture of nucleation: the growing solid clusters have predominantly non-spherical shapes and consist of face-centered-cubic and random hexagonal-close-packed coordinated atoms surrounded by a cloud of prestructured liquid. The nucleation initiates in regions of supercooled liquid that are characterized by a high orientational order with structural features that predetermine the polymorph selection. These results provide atomistic insight not only into the nucleation mechanism of nickel but also into the role of the preordered liquid regions as precursors for crystallization. read less USED (high confidence) Z. Zhang, M. Giesselmann, J. Mankowski, J. Dickens, A. Neuber, and R. Joshi, “Evaluation of high field and/or local heating based material degradation of nanoscale metal emitter tips: a molecular dynamics analysis,” Journal of Physics D: Applied Physics. 2017. link Times cited: 7 Abstract: A molecular dynamics (MD) model is used to study the potenti… read moreAbstract: A molecular dynamics (MD) model is used to study the potential for mass ejection from a metal nanoprotrusion, driven by high fields and temperature increases. Three-dimensional calculations of the electric fields surrounding the metal emitter are used to obtain the Maxwell stress on the metal. This surface loading is coupled into MD simulations. Our results show that mass ejection from the nanotip is possible and indicate that both larger aspect ratios and higher local temperatures will drive the instability. Hence it is predicted that in a nonuniform distribution of emitters, the longer and thinner sites will suffer the most damage, which is generally in keeping with the trends of a recent experimental report (Parson et al 2014 IEEE Trans. Plasma Sci. 42 3982). A possible hypothesis for mass ejection in the absence of a distinct nanoprotrusion is also discussed. read less USED (high confidence) J. Veerababu, S. Goyal, R. Sandhya, and K. Laha, “Understanding the Low Cycle Fatigue Behavior of Single Crystal Cu at the Nano-scale: A Molecular Dynamics Study,” Transactions of the Indian Institute of Metals. 2017. link Times cited: 2 USED (high confidence) T. Xie, S. Sarupria, and R. B. Getman, “A DFT and MD study of aqueous-phase dehydrogenation of glycerol on Pt(1 1 1): comparing chemical accuracy versus computational expense in different methods for calculating aqueous-phase system energies,” Molecular Simulation. 2017. link Times cited: 15 Abstract: Glycerol, which is one of the most abundant by-products in b… read moreAbstract: Glycerol, which is one of the most abundant by-products in biodiesel production, can be converted into H2 through aqueous-phase reforming (APR). Dehydrogenation is one of the main processes in glycerol APR. In this work, we use computational methods to study Pt(1 1 1)-catalysed glycerol dehydrogenation under aqueous conditions. There are 84 intermediates and 250 possible reactions in the dehydrogenation network. Inclusion of the liquid environment adds computational expense, especially if we are to study all the reaction intermediates and reactions under explicit water solvation using quantum methods. In this work, we present a method that can be used to efficiently estimate reaction energies under explicit solvation with reasonable accuracy and computational expense. The method couples a linear scaling relationship for obtaining adsorbate binding energies with Lennard-Jones + Coulomb potentials for obtaining water–adsorbate interaction energies. Comparing reaction energies calculated with this approach to reaction energies obtained from a more extensive approach that attains quantum-level accuracy (published previously by our group), we find good correlation (R2 = 0.84) and reasonable accuracy (the mean absolute error, MAE = 0.28 eV). read less USED (high confidence) J. Wang, J. Bian, X. Niu, and G. Wang, “A universal method to calculate the surface energy density of spherical surfaces in crystals,” Acta Mechanica Sinica. 2017. link Times cited: 6 USED (high confidence) K. Nie, W. Wu, X. Zhang, and S. Yang, “Molecular dynamics study on the grain size, temperature, and stress dependence of creep behavior in nanocrystalline nickel,” Journal of Materials Science. 2017. link Times cited: 47 USED (high confidence) M. Imran et al., “Molecular Dynamics Study of Surface Anisotropy in $\hbox Ag_60; \hbox Cu_40$Ag60Cu40 Alloy at Nanoscale,” International Journal of Thermophysics. 2017. link Times cited: 2 USED (high confidence) A. Hakonen et al., “Hand-Held Femtogram Detection of Hazardous Picric Acid with Hydrophobic Ag Nanopillar SERS Substrates and Mechanism of Elasto-Capillarity.,” ACS sensors. 2017. link Times cited: 75 Abstract: Picric acid (PA) is a severe environmental and security risk… read moreAbstract: Picric acid (PA) is a severe environmental and security risk due to its unstable, toxic, and explosive properties. It is also challenging to detect in trace amounts and in situ because of its highly acidic and anionic character. Here, we assess sensing of PA under nonlaboratory conditions using surface-enhanced Raman scattering (SERS) silver nanopillar substrates and hand-held Raman spectroscopy equipment. The advancing elasto-capillarity effects are explained by molecular dynamics simulations. We obtain a SERS PA detection limit on the order of 20 ppt, corresponding attomole amounts, which together with the simple analysis methodology demonstrates that the presented approach is highly competitive for ultrasensitive analysis in the field. read less USED (high confidence) A. Leonardi and P. Scardi, “Dislocation Effects on the Diffraction Line Profiles from Nanocrystalline Domains,” Metallurgical and Materials Transactions A. 2016. link Times cited: 19 USED (high confidence) J. Wang, J. Bian, X. Niu, and G. Wang, “A universal method to calculate the surface energy density of spherical surfaces in crystals,” Acta Mechanica Sinica. 2016. link Times cited: 0 USED (high confidence) N. He, Y. Liu, and X. Zhang, “Molecular dynamics‐smoothed molecular dynamics (MD‐SMD) adaptive coupling method with seamless transition,” International Journal for Numerical Methods in Engineering. 2016. link Times cited: 8 Abstract: Smoothed molecular dynamics (SMD) method is a recently propo… read moreAbstract: Smoothed molecular dynamics (SMD) method is a recently proposed efficient molecular simulation method by introducing one set of background mesh and mapping process into molecular dynamics (MD) flow chart. SMD can sharply enlarge MD time step size while maintaining global accuracy. MD‐SMD coupling method was proposed to improve the capability to describe local atom disorders. The coupling method is greatly improved in this paper in two essential aspects. Firstly, a transition scheme is proposed to avoid artificial wave reflection at the interface of MD and SMD regions. The new transition scheme has simple formulation and high efficiency, and the wave reflection can be well suppressed. Secondly, an adaptive scheme is proposed to automatically identify the regions requiring MD simulation. Two adaptive criteria, the centro‐symmetry parameter criterion and the displacement criterion, are also proposed. It is found that both the two criteria can achieve good accuracy but the efficiency of the displacement criterion is much better. The coupling method does not demand reduction in mesh size near the interface, and a multiple time stepping scheme is adopted to ensure high efficiency. Numerical results including wave propagation, nano‐indentation, and crack propagation validate the method and show nice accuracy. Copyright © 2016 John Wiley & Sons, Ltd. read less USED (high confidence) G. Rusina et al., “Surface Dynamics of the Wetting Layers and Ultrathin Films on a Dynamic Substrate: (0.5–4) ML Pb/Cu(111),” Journal of Physical Chemistry C. 2016. link Times cited: 13 Abstract: The growth of Pb ultrathin films on Cu(111) has been long st… read moreAbstract: The growth of Pb ultrathin films on Cu(111) has been long studied in connection with electronic quantum-size effects and for the different temperature-dependent growth kinetics. At low temperature the formation of a wetting layer (1 monolayer (ML)), is followed by an instability of the 2 ML film and a regular layer-by-layer growth is then only observed for more than two monolayers. The 2 ML film was, however, shown to be stabilized by alloying Pb with 20% Tl. This work presents a theoretical study of the dynamics of the wetting layer as well as for 2 ML Pb0.8Tl0.2, 3 and 4 ML Pb on Cu(111) in the 4 × 4 commensurate phase, for which detailed inelastic Helium atom scattering (HAS) spectra have been measured. The present calculations based on the embedded atom method (EAM) include the dynamics of the substrate. Besides leading to a detailed interpretation of the HAS experimental data, the present results are compared with a previous density-functional perturbation theory (DFPT) study for 3 to 7 ML Pb on a ri... read less USED (high confidence) T. Otto, J. Ramallo-López, L. Giovanetti, F. Requejo, S. Zones, and E. Iglesia, “Synthesis of stable monodisperse AuPd, AuPt, and PdPt bimetallic clusters encapsulated within LTA-zeolites,” Journal of Catalysis. 2016. link Times cited: 49 USED (high confidence) M. Kroonblawd, N. Mathew, S. Jiang, and T. Sewell, “A generalized crystal-cutting method for modeling arbitrarily oriented crystals in 3D periodic simulation cells with applications to crystal-crystal interfaces,” Comput. Phys. Commun. 2016. link Times cited: 51 USED (high confidence) B. Zhao 赵, Y. Wang 王, C. Liu 刘, and X. Wang 王, “Molecular dynamics simulation of structural change at metal/semiconductor interface induced by nanoindenter,” Chinese Physics B. 2016. link Times cited: 0 Abstract: The structures of the Si/Cu heterogenous interface impacted … read moreAbstract: The structures of the Si/Cu heterogenous interface impacted by a nanoindenter with different incident angles and depths are investigated in detail using molecular dynamics simulation. The simulation results suggest that for certain incident angles, the nanoindenter with increasing depth can firstly increase the stress of each atom at the interface and it then introduces more serious structural deformation of the Si/Cu heterogenous interface. A nanoindenter with increasing incident angle (absolute value) can increase the length of the Si or Cu extended atom layer. It is worth mentioning that when the incident angle of the nanoindenter is between −45° and 45°, these Si or Cu atoms near the nanoindenter reach a stable state, which has a lower stress and a shorter length of the Si or Cu extended atom layer than those of the other incident angles. This may give a direction to the planarizing process of very large scale integration circuits manufacture. read less USED (high confidence) X. W. Zhou, D. Ward, and M. E. Foster, “An analytical bond-order potential for the aluminum copper binary system,” Journal of Alloys and Compounds. 2016. link Times cited: 38 USED (high confidence) S.-H. Cha et al., “Cold welding of gold nanoparticles on mica substrate: Self-adjustment and enhanced diffusion,” Scientific Reports. 2016. link Times cited: 23 USED (high confidence) L. Yang, J. Feng, Y. Ding, J. Bian, and G. Wang, “An analytical description for the elastic compression of metallic polyhedral nanoparticles,” AIP Advances. 2016. link Times cited: 5 Abstract: Metallic nanoparticles are usually polyhedrons instead of pe… read moreAbstract: Metallic nanoparticles are usually polyhedrons instead of perfect spheres, which presents a challenge to characterize their elastic response. In the present paper, the elastic compression of truncated octahedral nanoparticles is investigated through finite element calculations and atomic simulations. An analytical expression of load is obtained for octahedral particles, which is linearly proportional to indent depth, instead of the 3/2 power law relation predicted by Hertzian model for elastic sphere. Comparisons with molecular dynamics simulations demonstrate that the obtained relation can predict the elastic response of polyhedral nanoparticles. This study is helpful to measure the elastic properties of polyhedral nanoparticles, and characterize their elastic response. read less USED (high confidence) P. Gao et al., “Size-dependent concentrations of thermal vacancies in solid films.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 8 Abstract: Solid films are considered as typical model systems to study… read moreAbstract: Solid films are considered as typical model systems to study size effects on thermal vacancy concentration in nanomaterials. By combining the generalized Young-Laplace equation with the chemical potential of vacancies, a strict size-dependent thermodynamic model of vacancies, which includes the surface intrinsic elastic parameters of the eigenstress, Young's modulus and the geometric size of the solid films, was established. The vacancy concentration changes in the film with respect to the bulk value, depending on the geometric size and surface stress sign of the solid films. Atomistic simulations of Au and Pt films verified the developed thermodynamic model. These results provide physical insights into the size-dependent thermal vacancy concentration in nanomaterials. read less USED (high confidence) J. Ling, R. Jones, and J. Templeton, “Machine learning strategies for systems with invariance properties,” J. Comput. Phys. 2016. link Times cited: 288 USED (high confidence) L. Bao, H.-bao Hu, J. Wen, P. Sepri, and K. Luo, “Three-Dimensional Structure of a Simple Liquid at a Face-Centered-Cubic (001) Solid Surface Interface,” Scientific Reports. 2016. link Times cited: 9 USED (high confidence) D. Watvisave, B. Puranik, and U. Bhandarkar, “Modeling wall effects in a micro-scale shock tube using hybrid MD–DSMC algorithm,” Shock Waves. 2016. link Times cited: 3 USED (high confidence) D. T. Ho, S. D. Park, S. Y. Kwon, T. Han, and S. Y. Kim, “Negative Poisson’s ratio in cubic materials along principal directions,” physica status solidi (b). 2016. link Times cited: 33 Abstract: This report employed molecular statics simulation and densit… read moreAbstract: This report employed molecular statics simulation and density‐functional‐theory calculation to study the Poisson's ratios of face‐centered‐cubic materials. We provide numerical and theoretical evidences to show that cubic materials can exhibit auxetic behavior in a principal direction under proper loading conditions. When a stress perpendicular to the loading direction is applied, cubic materials can exhibit a negative Poisson's ratio at finite strain. The negative Poisson's ratio behavior, including its direction and value, is highly dependent on the direction and magnitude of the transversely applied stresses. As a result, we show that it is possible to tune the direction and magnitude of the negative Poisson's ratio behavior of cubic materials by controlling the transverse loadings. read less USED (high confidence) J. Zhang, W. Zhideng, Y. Yan, and T. Sun, “Interface-dependent nanoscale friction of copper bicrystals: tilt versus twist,” RSC Advances. 2016. link Times cited: 13 Abstract: Interfaces with different structural units have a strong imp… read moreAbstract: Interfaces with different structural units have a strong impact on their microscopic deformation behavior and correlated macroscopic mechanical response. In current study, we elucidate the underlying nanoscale friction mechanisms of Cu bicrystals by means of molecular dynamics simulations. Four grain boundaries, i.e., pure tilt and twist with two misorientation angles of 7.63° and 67.38°, are considered to address the grain boundary structure dependence of the friction. While the small- and high-angle tilt grain boundaries are respectively composed of parallel edge dislocation dipoles and edge dislocations of like sign, the small- and high-angle twist ones respectively incorporate two sets of intersecting screw dislocations and a planar defect. Simulation results demonstrate that the grain boundary resistance to dislocation motion and absorption, as well as the grain boundary evolution, are significantly varied with grain boundary structural units. It is found that splitting, annihilation and generation of grain boundary dislocations are the three competing decomposition mechanisms of the well-defined grain boundaries. The anisotropic dislocation-grain boundary interactions in turn results in a strong grain boundary structure dependence of the frictional response for scratching in the vicinity of grain boundaries. These findings will not only advance our understanding of the interface-dependent nanoscale friction behavior of metals, but also provide rational design guidelines for the synthesis of advanced functional nanostructured materials with unique internal interface textures. read less USED (high confidence) X. Liu, G. Zhang, and Y.-W. Zhang, “Thermal conduction across the one-dimensional interface between a MoS2 monolayer and metal electrode,” Nano Research. 2016. link Times cited: 27 USED (high confidence) J. Douglas, B. A. P. Betancourt, X. Tong, and H. Zhang, “Localization model description of diffusion and structural relaxation in glass-forming Cu–Zr alloys,” Journal of Statistical Mechanics: Theory and Experiment. 2016. link Times cited: 56 Abstract: We test the localization model (LM) prediction of a paramete… read moreAbstract: We test the localization model (LM) prediction of a parameter-free relationship between the α-structural relaxation time τ α and the Debye–Waller factor 〈u 2 〉 for a series of simulated glass-forming Cu–Zr metallic liquids having a range of alloy compositions. After validating this relationship between the picosecond (‘fast’) and long-time relaxation dynamics over the full range of temperatures and alloy compositions investigated in our simulations, we show that it is also possible to estimate the self-diffusion coefficients of the individual atomic species (D Cu, D Zr) and the average diffusion coefficient D using the LM, in conjunction with the empirical fractional Stokes–Einstein (FSE) relation linking these diffusion coefficients to τ α . We further observe that the fragility and extent of decoupling between D and τ α strongly correlate with 〈u 2 〉 at the onset temperature of glass-formation T A where particle caging and the breakdown of Arrhenius relaxation first emerge. read less USED (high confidence) A. E. Korenchenko, A. Vorontsov, and B. R. Gel’chinskii, “Statistical analysis of formation and relaxation of atomic clusters based on data of molecular-dynamic modeling of gas-phase nucleation of metallic nanoparticles,” High Temperature. 2016. link Times cited: 9 USED (high confidence) J. Shim, H.-J. L. Voigt, and B. Wirth, “Temperature dependent dislocation bypass mechanism for coherent precipitates in Cu–Co alloys,” Acta Materialia. 2016. link Times cited: 17 USED (high confidence) H. S. Kim, Y. S. Seo, K. Kim, J. Han, Y. Park, and S. Cho, “Concentration Effect of Reducing Agents on Green Synthesis of Gold Nanoparticles: Size, Morphology, and Growth Mechanism,” Nanoscale Research Letters. 2016. link Times cited: 67 USED (high confidence) G. Rusina, S. Borisova, and E. Chulkov, “Structure and phonon spectrum of a submonolayer Ni film on the surface of Cu(100),” Journal of Experimental and Theoretical Physics. 2016. link Times cited: 0 USED (high confidence) E. T. Karim et al., “Experimental characterization and atomistic modeling of interfacial void formation and detachment in short pulse laser processing of metal surfaces covered by solid transparent overlayers,” Applied Physics A. 2016. link Times cited: 16 USED (high confidence) J. Abraham, T. Strunskus, F. Faupel, and M. Bonitz, “Molecular dynamics simulation of gold cluster growth during sputter deposition,” Journal of Applied Physics. 2016. link Times cited: 25 Abstract: We present a molecular dynamics simulation scheme that we ap… read moreAbstract: We present a molecular dynamics simulation scheme that we apply to study the time evolution of the self-organized growth process of metalcluster assemblies formed by sputter-deposited gold atoms on a planar surface. The simulation model incorporates the characteristics of the plasma-assisted deposition process and allows for an investigation over a wide range of deposition parameters. It is used to obtain data for the cluster properties which can directly be compared with recently published experimental data for gold on polystyrene [M. Schwartzkopf et al., ACS Appl. Mater. Interfaces 7, 13547 (2015)]. While good agreement is found between the two, the simulations additionally provide valuable time-dependent real-space data of the surface morphology, some of whose details are hidden in the reciprocal-space scattering images that were used for the experimental analysis. read less USED (high confidence) E. Goudeli and S. Pratsinis, “Crystallinity dynamics of gold nanoparticles during sintering or coalescence,” Aiche Journal. 2016. link Times cited: 55 Abstract: The crystallinity of gold nanoparticles during coalescence o… read moreAbstract: The crystallinity of gold nanoparticles during coalescence or sintering is investigated by molecular dynamics. The method is validated by the attainment of the Au melting temperature that increases with increasing particle size approaching the Au melting point. The morphology and crystal dynamics of nanoparticles of (un)equal size during sintering are elucidated. The characteristic sintering time of particle pairs is determined by tracing their surface area evolution during coalescence. The crystallinity is quantified by the disorder variable indicating the system's degree of disorder. The atoms at the grain boundaries are amorphous, especially during particle adhesion and during sintering when grains of different orientation are formed. Initial grain orientation affects final particle morphology leading to exposure of different crystal surfaces that can affect the performance of Au nanoparticles (e.g., catalytic efficiency). Coalescence between crystalline and amorphous nanoparticles of different size results in polycrystalline particles of increasing crystallinity with time and temperature. Crystallinity affects the sintering rate and mechanism. Such simulations of free-standing Au nanoparticle coalescence are relevant also to Au nanoparticles on supports that do not exhibit strong affinity or strong metal support interactions. © 2015 American Institute of Chemical Engineers AIChE J, 62: 589–598, 2016 read less USED (high confidence) A. Vattré, T. Jourdan, H. Ding, M. Marinica, M. Demkowicz, and M. Demkowicz, “Non-random walk diffusion enhances the sink strength of semicoherent interfaces,” Nature Communications. 2016. link Times cited: 82 USED (high confidence) V. Iacobellis and K. Behdinan, “Bridging cell multiscale modeling of fatigue crack growth in fcc crystals,” International Journal for Numerical Methods in Engineering. 2015. link Times cited: 10 Abstract: The previously developed bridging cell method for modeling c… read moreAbstract: The previously developed bridging cell method for modeling coupled continuum/atomistic systems at finite temperature is used to model fatigue crack growth in single crystal nickel under two crystal orientations at different temperatures. The method is expanded to implement a temperature‐dependent embedded atom method potential for finite temperature simulations avoiding time‐scale restrictions associated with small timesteps. Results for the fatigue simulation were compared with respect to deformation behavior, stress distribution, and crack length. Results showed very different crack growth mechanisms between the two crystal orientations as well as reduced resistance to crack growth with increased temperature. Copyright © 2015 John Wiley & Sons, Ltd. read less USED (high confidence) M. Warrier, U. Bhardwaj, H. Hemani, R. Schneider, A. Mutzke, and M. C. Valsakumar, “Statistical study of defects caused by primary knock-on atoms in fcc Cu and bcc W using molecular dynamics,” Journal of Nuclear Materials. 2015. link Times cited: 20 USED (high confidence) E. Figueroa, D. Tramontina, D. Tramontina, G. Gutiérrez, and E. Bringa, “Mechanical properties of irradiated nanowires – A molecular dynamics study,” Journal of Nuclear Materials. 2015. link Times cited: 11 USED (high confidence) C. Qiao et al., “Inhibition effect on the evolution of a twist grain boundary for an Al/Ni bimetal interface under torsion,” RSC Advances. 2015. link Times cited: 4 Abstract: By using a molecular dynamics method with EAM potential, we … read moreAbstract: By using a molecular dynamics method with EAM potential, we study the evolution phenomena of metal twist grain boundaries (GBs) in the [100], [111] and [110] orientations, together with their bimetal interface, under anticlockwise and clockwise torsions. Our results show that there are different evolution behaviors of the GB screw dislocations for single metals (Al and Ni) and their bimetal interface (Al/Ni) under torsion. Specifically, for the single metals in the [100] and [111] orientations, their GBs evolve toward lower or higher angle twist GBs depending on the twist direction. For Ni in the [110] orientation, the dislocations spread not only in the GB region but also in the grain interior. However, for the bimetal interface, the propagation of dislocations is not only reduced dramatically but also limited to the interface region, showing that there is an inhibition effect. Therefore, such an inhibition effect can enhance the stability of nanomaterials which is very useful for the further design of nanodevices. read less USED (high confidence) O. Trushin, A. N. Kupryanov, S. Ying, E. Granato, and T. Ala‐Nissila, “Atomic mechanisms of strain relaxation in heteroepitaxial Cu/Ni(001) system,” Russian Microelectronics. 2015. link Times cited: 0 USED (high confidence) L. Hu and L. Liu, “From atomistics to continuum: Effects of a free surface and determination of surface elasticity properties,” Mechanics of Materials. 2015. link Times cited: 6 USED (high confidence) C. G. Zhang, Y. Li, W. Zhou, L. Hu, and Z. Zeng, “Anti-radiation mechanisms in nanoporous gold studied via molecular dynamics simulations,” Journal of Nuclear Materials. 2015. link Times cited: 20 USED (high confidence) O. Trushin, A. N. Kupryanov, S. Ying, E. Granato, and T. Ala‐Nissila, “Atomic mechanisms of strain relaxation in heteroepitaxial Cu/Ni(001) system,” Russian Microelectronics. 2015. link Times cited: 0 USED (high confidence) K. Ng et al., “Ion-Desorption Efficiency and Internal-Energy Transfer in Surface-Assisted Laser Desorption/Ionization: More Implication(s) for the Thermal-Driven and Phase-Transition-Driven Desorption Process,” Journal of Physical Chemistry C. 2015. link Times cited: 52 Abstract: Fundamental factors governing the ion-desorption efficiency … read moreAbstract: Fundamental factors governing the ion-desorption efficiency and extent of internal-energy transfer to a chemical thermometer, benzylpyridinium ion ([BP]+), generated in the surface-assisted laser desorption/ionization (SALDI) process, were systematically investigated using noble metal nanoparticles (NPs), including AuNPs, AgNPs, PdNPs, and PtNPs, as substrates, with an average particle size of 1.7–3.1 nm in diameter. In the correlation of ion-desorption efficiency and internal-energy transfer with physicochemical properties of the NPs, laser-induced heating of the NPs, which are dependent on their photoabsorption efficiencies, was found to be a key factor in governing the ion-desorption efficiency and the extent of internal-energy transfer. This suggested that the thermal-driven desorption played a significant role in the ion-desorption process. In addition, a stronger binding affinity of [BP]+ to the surface of the NPs could hinder its desorption from the NPs, and this could be another factor in determin... read less USED (high confidence) W. Pang, G.-cai Zhang, X.-geng Zhao, and P. Zhang, “Dependence of dislocation creation on tensile orientation in face-centered-cubic ductile metals under high strain rate loading,” Journal of Applied Physics. 2015. link Times cited: 4 Abstract: We investigate through molecular dynamic simulations the dep… read moreAbstract: We investigate through molecular dynamic simulations the dependence of dislocation creation on tensile orientation in face-centered-cubic ductile metals under high strain rate loading. It is found that while dislocations generally originate from the double-layer defect clusters consisting of flatted octahedral structures (FOSs), the formation mechanism and the types of FOSs, as well as the types of nucleated dislocations, depend on the applied loading directions. For the loading along the [1¯10], [1¯1¯2], and [111] crystal directions, it is shown that a pair of the nearest-neighboring atoms move away to form the elongated FOS. However, for the loading along the [100] crystal direction, a pair of the next-nearest-neighboring atoms move close to form the compressed FOS. According to the uniform deformation amount of the spacing vector for a pair of neighboring atoms and the stress component along the Burgers vector on the stacking fault plane, we analytically predict the activated types of FOSs and dislocat... read less USED (high confidence) H. Kim and A. Strachan, “Mechanical response of nanocrystalline platinum via molecular dynamics: size effects in bulk versus thin-film samples,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 6 Abstract: We use large-scale molecular dynamics simulations to charact… read moreAbstract: We use large-scale molecular dynamics simulations to characterize the mechanical responses of nanocrystalline bulk and thin-film samples with average grain size ranging from 5 to 40 nm and at two strain rates. Our simulations show Hall–Petch maxima for both yield and flow stresses and for both sets of specimens. We find that the presence of free surface decreases both the yield and flow stresses and, interestingly, the Hall–Petch maximum for slabs occur at a larger grain size than for the bulk samples. A quantitative analysis of plastic slip on grain interiors and boundaries reveals that the shift in the maximum results from a combination of higher intergranular slip and weaker size dependence of dislocation activity in the slabs as compared with the bulk. Finally, increasing strain rate increases both yield and flow stresses and this rate effect is dominated by the plasticity involving full dislocations; plastic slip by partial dislocations and grain boundary processes exhibit weaker size effects. read less USED (high confidence) D. Watvisave, B. Puranik, and U. Bhandarkar, “Modeling wall effects in a micro-scale shock tube using hybrid MD–DSMC algorithm,” Shock Waves. 2015. link Times cited: 0 USED (high confidence) Y. Yuan, T. Sun, J. Zhang, and C. Liu, “Molecular Dynamics Study of Nanoimprint of Single Crystal Aluminium Thin Film.” 2015. link Times cited: 1 Abstract: In current study, molecular dynamics simulations are perform… read moreAbstract: In current study, molecular dynamics simulations are performed to investigate nanoimprint processes on single crystal aluminium thin films with silicon stamp. An EAM potential developed for aluminium is adopted to describe the Al-Al interaction in the aluminium thin film, and the Lennard-Jones potential is used to describe the Al-Si atomic interaction. Simulations show that dislocation events are the main plastic deformation mode of Al thin film in nanoimprint. It is found that stamp size and stamp shape have significant influence on the dislocation events and imprint forces during the nanoimprint processes. Keywords-nanoimprint; molecular dynamics; single crystal aluminium; thin film; stamp read less USED (high confidence) M. Warrier, P. Pahari, and S. Chaturvedi, “Molecular dynamics analysis of the transient temperature increase at void locations in shocked materials: RDX and Cu,” Journal of Molecular Modeling. 2015. link Times cited: 5 USED (high confidence) Y. Geng, J. Zhang, Y. Yan, B. Yu, L. Geng, and T. Sun, “Experimental and Theoretical Investigation of Crystallographic Orientation Dependence of Nanoscratching of Single Crystalline Copper,” PLoS ONE. 2015. link Times cited: 24 Abstract: In the present work, we perform experiments and molecular dy… read moreAbstract: In the present work, we perform experiments and molecular dynamics simulations to elucidate the underlying deformation mechanisms of single crystalline copper under the load-controlled multi-passes nanoscratching using a triangular pyramidal probe. The correlation of microscopic deformation behavior of the material with macroscopically-observed machining results is revealed. Moreover, the influence of crystallographic orientation on the nanoscratching of single crystalline copper is examined. Our simulation results indicate that the plastic deformation of single crystalline Cu under the nanoscratching is exclusively governed by dislocation mechanisms. However, there is no glissile dislocation structure formed due to the probe oscillation under the load-controlled mode. Both experiments and MD simulations demonstrate that the machined surface morphologies in terms of groove depth and surface pile-up exhibit strong crystallographic orientation dependence, because of different geometries of activated slip planes cutting with free surfaces and strain hardening abilities associated with different crystallographic orientations. read less USED (high confidence) D. T. Ho, Y.-H. Im, S. Y. Kwon, Y. Earmme, and S. Y. Kim, “Mechanical Failure Mode of Metal Nanowires: Global Deformation versus Local Deformation,” Scientific Reports. 2015. link Times cited: 15 USED (high confidence) G. Rusina, S. Borisova, and E. Chulkov, “Structure and atomic vibrations in bimetallic Ni13 − nAln clusters,” JETP Letters. 2015. link Times cited: 3 USED (high confidence) S. Ding, Y. Tian, Z. Jiang, and X. He, “Molecular dynamics simulation of joining process of Ag-Au nanowires and mechanical properties of the hybrid nanojoint,” AIP Advances. 2015. link Times cited: 16 Abstract: The nanojoining process of Ag-Au hybrid nanowires at 800K wa… read moreAbstract: The nanojoining process of Ag-Au hybrid nanowires at 800K was comprehensively studied by virtue of molecular dynamics (MD) simulation. Three kinds of configurations including end-to-end, T-like and X-like were built in the simulation aiming to understand the nanojoining mechanism. The detailed dynamic evolution of atoms, crystal structure transformation and defects development during the nanojoining processes were performed. The results indicate that there are two stages in the nanojoining process of Ag-Au nanowires which are atom diffusion and new bonds formation. Temperature is a key parameter affecting both stages ascribed to the energy supply and the optimum temperature for Ag-Au nanojoint with diameter of 4.08 nm has been discussed. The mechanical properties of the nanojoint were examined with simulation of tensile test on the end-to-end joint. It was revealed that the nanojoint was strong enough to resist fracture at the joining area. read less USED (high confidence) K. Krupski, M. Moors, P. Jóźwik, T. Kobiela, and A. Krupski, “Structure Determination of Au on Pt(111) Surface: LEED, STM and DFT Study,” Materials. 2015. link Times cited: 44 Abstract: Low-energy electron diffraction (LEED), scanning tunneling m… read moreAbstract: Low-energy electron diffraction (LEED), scanning tunneling microscopy (STM) and density functional theory (DFT) calculations have been used to investigate the atomic and electronic structure of gold deposited (between 0.8 and 1.0 monolayer) on the Pt(111) face in ultrahigh vacuum at room temperature. The analysis of LEED and STM measurements indicates two-dimensional growth of the first Au monolayer. Change of the measured surface lattice constant equal to 2.80 Å after Au adsorption was not observed. Based on DFT, the distance between the nearest atoms in the case of bare Pt(111) and Au/Pt(111) surface is equal to 2.83 Å, which gives 1% difference in comparison with STM values. The first and second interlayer spacing of the clean Pt(111) surface are expanded by +0.87% and contracted by −0.43%, respectively. The adsorption energy of the Au atom on the Pt(111) surface is dependent on the adsorption position, and there is a preference for a hollow fcc site. For the Au/Pt(111) surface, the top interlayer spacing is expanded by +2.16% with respect to the ideal bulk value. Changes in the electronic properties of the Au/Pt(111) system below the Fermi level connected to the interaction of Au atoms with Pt(111) surface are observed. read less USED (high confidence) B. D. Ngô, A. Stukowski, N. Mameka, J. Markmann, K. Albe, and J. Weissmuller, “Anomalous Compliance and Early Yielding of Nanoporous Gold,” Acta Materialia. 2015. link Times cited: 115 USED (high confidence) J. Ye, “Fabrication of ordered arrays of micro- and nanoscale features with control over their shape and size via templated solid-state dewetting,” Scientific Reports. 2015. link Times cited: 25 USED (high confidence) A. Leonardi, S. Ryu, N. Pugno, and P. Scardi, “Eshelby twist and correlation effects in diffraction from nanocrystals,” Journal of Applied Physics. 2015. link Times cited: 8 Abstract: Molecular dynamics simulations were used to model the Eshelb… read moreAbstract: Molecular dynamics simulations were used to model the Eshelby dislocation inside Pd and Ir nanowires and to predict the powder diffraction pattern using the Debye scattering equation. We find that the ideal dislocation solution by Eshelby is in good agreement with the observed twist angle and deviatoric strain, even though it ignores both the splitting of the Eshelby dislocation into two partials and surface stress. Surface stress plays a significant role only for nanorods with small aspect ratio (∼1:1). We also find that Wilson's prediction on the diffraction peak broadening for the Eshelby dislocation is overestimated because it ignores the fact that the Eshelby twist relaxes the deviatoric strain. Moreover, the twist loosens the correlation along the nanorod, causing additional line profile broadening, which is read by diffraction as a decrease of coherent domain size when the total twist angle is bigger than 1.5°. Overall, our findings suggest a novel way to predict and analyze the dislocations as well as the resulting strain fields in the twisted nanocrystalline rods. read less USED (high confidence) Z. Liang and P. Keblinski, “Slip length crossover on a graphene surface.,” The Journal of chemical physics. 2015. link Times cited: 20 Abstract: Using equilibrium and non-equilibrium molecular dynamics sim… read moreAbstract: Using equilibrium and non-equilibrium molecular dynamics simulations, we study the flow of argon fluid above the critical temperature in a planar nanochannel delimited by graphene walls. We observe that, as a function of pressure, the slip length first decreases due to the decreasing mean free path of gas molecules, reaches the minimum value when the pressure is close to the critical pressure, and then increases with further increase in pressure. We demonstrate that the slip length increase at high pressures is due to the fact that the viscosity of fluid increases much faster with pressure than the friction coefficient between the fluid and the graphene. This behavior is clearly exhibited in the case of graphene due to a very smooth potential landscape originating from a very high atomic density of graphene planes. By contrast, on surfaces with lower atomic density, such as an (100) Au surface, the slip length for high fluid pressures is essentially zero, regardless of the nature of interaction between fluid and the solid wall. read less USED (high confidence) Z. Nourmohammadi, S. Mukherjee, S. Joshi, J. Song, and S. Vengallatore, “Methods for Atomistic Simulations of Linear and Nonlinear Damping in Nanomechanical Resonators,” Journal of Microelectromechanical Systems. 2015. link Times cited: 6 Abstract: Atomistic simulations can be used to compute damping from fi… read moreAbstract: Atomistic simulations can be used to compute damping from first principles and gain unprecedented insights into the mechanisms of dissipation. However, the technique is still in its infancy and many foundational aspects remain unexplored. As a step toward addressing these issues, we present here a comparative study of five different methods for estimating damping under isothermal conditions. Classical molecular dynamics was used to simulate the fundamental longitudinal-mode oscillations of nanowires and nanofilms of silicon and nickel at room temperature (300 K) in the canonical ensemble using the Nosé-Hoover thermostat. In the subresonant regime, damping was quantified using the loss tangent and loss factor during steady-state harmonic vibration. The quality factor was obtained by analyzing the spectrum of thermomechanical noise and also from the Duffing-like nonlinearity in the frequency response under harmonic excitation. In addition, the nonlinear logarithmic decrement was obtained from the Hilbert transform of freely decaying oscillations. We discuss the factors that must be considered while selecting simulation parameters, establish criteria for convergence and linearity, and highlight the relative merits and limitations of each method. read less USED (high confidence) L. Li and M. Han, “Shear behaviors of single crystal nickel at different temperatures: molecular dynamics simulations,” Applied Physics A. 2015. link Times cited: 8 USED (high confidence) L. Li and M. Han, “Shear behaviors of single crystal nickel at different temperatures: molecular dynamics simulations,” Applied Physics A. 2015. link Times cited: 0 USED (high confidence) J. Li, Q. Fang, Y.-wen Liu, and L. Zhang, “Scratching of copper with rough surfaces conducted by diamond tip simulated using molecular dynamics,” The International Journal of Advanced Manufacturing Technology. 2015. link Times cited: 38 USED (high confidence) H. Zhang, Y. Yang, and J. Douglas, “Influence of string-like cooperative atomic motion on surface diffusion in the (110) interfacial region of crystalline Ni.,” The Journal of chemical physics. 2015. link Times cited: 21 Abstract: Although we often think about crystalline materials in terms… read moreAbstract: Although we often think about crystalline materials in terms of highly organized arrays of atoms, molecules, or even colloidal particles, many of the important properties of this diverse class of materials relating to their catalytic behavior, thermodynamic stability, and mechanical properties derive from the dynamics and thermodynamics of their interfacial regions, which we find they have a dynamics more like glass-forming (GF) liquids than crystals at elevated temperatures. This is a general problem arising in any attempt to model the properties of naturally occurring crystalline materials since many aspects of the dynamics of glass-forming liquids remain mysterious. We examine the nature of this phenomenon in the "simple" case of the (110) interface of crystalline Ni, based on a standard embedded-atom model potential, and we then quantify the collective dynamics in this interfacial region using newly developed methods for characterizing the cooperative dynamics of glass-forming liquids. As in our former studies of the interfacial dynamics of grain-boundaries and the interfacial dynamics of crystalline Ni nanoparticles (NPs), we find that the interface of bulk crystalline Ni exhibits all the characteristics of glass-forming materials, even at temperatures well below the equilibrium crystal melting temperature, Tm. This perspective offers a new approach to modeling and engineering the properties of crystalline materials. read less USED (high confidence) A. Leonardi and P. Scardi, “Atomistic Model of Metal Nanocrystals with Line Defects: Contribution to Diffraction Line Profile,” Frontiers in Materials. 2015. link Times cited: 7 Abstract: Molecular Dynamics (MD) was used to simulate cylindrical Pd … read moreAbstract: Molecular Dynamics (MD) was used to simulate cylindrical Pd and Ir domains with ideal dislocations parallel to the axis. Results show significant discrepancies with respect to predictions of traditional continuum mechanics. When MD atomistic models are used to generate powder diffraction patterns, strong deviations are observed from the usual paradigm of a small crystal perturbed by the strain field of lattice defects. The Krivoglaz-Wilkens model for dislocation effects of diffraction line profiles seems correct for the screw dislocation case if most parameters are known or strongly constrained. Nevertheless the practical implementation of the model, i.e., a free refinement of all microstructural parameters, leads to instability. Possible effects of the experimental practice based on Line Profile Analysis are discussed. read less USED (high confidence) A. Ilinov et al., “Sputtering yields exceeding 1000 by 80 keV Xe irradiation of Au nanorods,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2014. link Times cited: 18 USED (high confidence) S. Rawat et al., “Integrated experimental and computational studies of deformation of single crystal copper at high strain rates,” Journal of Applied Physics. 2014. link Times cited: 19 Abstract: Quasi-static (0.0033 s−1) and dynamic (103 s−1) compression … read moreAbstract: Quasi-static (0.0033 s−1) and dynamic (103 s−1) compression experiments were performed on single crystal copper along ⟨100⟩ and ⟨110⟩ directions and best-fit parameters for the Johnson-Cook (JC) material model, which is an important input to hydrodynamic simulations for shock induced fracture, have been obtained. The deformation of single crystal copper along the ⟨110⟩ direction showed high yield strength, more strain hardening, and less strain rate sensitivity as compared to the ⟨100⟩ direction. Although the JC model at the macro-scale is easy to apply and describes a general response of material deformation, it lacks physical mechanisms that describe the influence of texture and initial orientation on the material response. Hence, a crystal plasticity model based on the theory of thermally activated motion of dislocations was used at the meso-scale, in which the evolution equations permit one to study and quantify the influence of initial orientation on the material response. Hardening parameters of the... read less USED (high confidence) E. Guevara-Chapa and S. Mejía-Rosales, “Molecular dynamics of coalescence and collisions of silver nanoparticles,” Journal of Nanoparticle Research. 2014. link Times cited: 9 USED (high confidence) D. Shyrokorad and G. V. Kornich, “Evolution of isolated copper clusters under low-energy argon ion bombardment,” Physics of the Solid State. 2014. link Times cited: 6 USED (high confidence) A. N. Kupriyanov, O. Trushin, and I. Amirov, “Modeling the oscillations of a copper nanorod using the molecular dynamics method,” Technical Physics Letters. 2014. link Times cited: 0 USED (high confidence) Y. Huang, S. Zhu, and T. Li, “Directional transport of molecular mass on graphene by straining,” Extreme Mechanics Letters. 2014. link Times cited: 22 USED (high confidence) K. M. Bal and E. Neyts, “On the time scale associated with Monte Carlo simulations.,” The Journal of chemical physics. 2014. link Times cited: 44 Abstract: Uniform-acceptance force-bias Monte Carlo (fbMC) methods hav… read moreAbstract: Uniform-acceptance force-bias Monte Carlo (fbMC) methods have been shown to be a powerful technique to access longer timescales in atomistic simulations allowing, for example, phase transitions and growth. Recently, a new fbMC method, the time-stamped force-bias Monte Carlo (tfMC) method, was derived with inclusion of an estimated effective timescale; this timescale, however, does not seem able to explain some of the successes the method. In this contribution, we therefore explicitly quantify the effective timescale tfMC is able to access for a variety of systems, namely a simple single-particle, one-dimensional model system, the Lennard-Jones liquid, an adatom on the Cu(100) surface, a silicon crystal with point defects and a highly defected graphene sheet, in order to gain new insights into the mechanisms by which tfMC operates. It is found that considerable boosts, up to three orders of magnitude compared to molecular dynamics, can be achieved for solid state systems by lowering of the apparent activation barrier of occurring processes, while not requiring any system-specific input or modifications of the method. We furthermore address the pitfalls of using the method as a replacement or complement of molecular dynamics simulations, its ability to explicitly describe correct dynamics and reaction mechanisms, and the association of timescales to MC simulations in general. read less USED (high confidence) W. Pang, P. Zhang, G.-cai Zhang, A. Xu, and X.-geng Zhao, “Dislocation creation and void nucleation in FCC ductile metals under tensile loading: A general microscopic picture,” Scientific Reports. 2014. link Times cited: 28 USED (high confidence) R. K. Siripurapu, B. Szpunar, and J. Szpunar, “Molecular Dynamics Study of Hydrogen in α-Zirconium.” 2014. link Times cited: 9 Abstract: Molecular dynamics approach is used to simulate hydrogen (H)… read moreAbstract: Molecular dynamics approach is used to simulate hydrogen (H) diffusion in zirconium. Zirconium alloys are used in fuel channels of many nuclear reactors. Previously developed embedded atom method (EAM) and modified embedded atom method (MEAM) are tested and a good agreement with experimental data for lattice parameters, cohesive energy, and mechanical properties is obtained. Both EAM and MEAM are used to calculate hydrogen diffusion in zirconium. At higher temperatures and in the presence of hydrogen, MEAM calculation predicts an unstable zirconium structure and low diffusion coefficients. Mean square displacement (MSD) of hydrogen in bulk zirconium is calculated at a temperature range of 500–1200 K with diffusion coefficient at 500 K equals 1.92 10−7 cm2/sec and at 1200 K has a value 1.47 10−4 cm2/sec. Activation energy of hydrogen diffusion calculated using Arrhenius plot was found to be 11.3 kcal/mol which is in agreement with published experimental results. Hydrogen diffusion is the highest along basal planes of hexagonal close packed zirconium. read less USED (high confidence) G. Rusina, S. Borisova, and E. Chulkov, “Atomic structure and phonons of a Pb ultrathin film on the Al(100) surface,” JETP Letters. 2014. link Times cited: 6 USED (high confidence) J. Li, Q. Fang, Y.-wen Liu, and L. Zhang, “Scratching of copper with rough surfaces conducted by diamond tip simulated using molecular dynamics,” The International Journal of Advanced Manufacturing Technology. 2014. link Times cited: 0 USED (high confidence) V. Bochenkov, N. Suetin, and S. Shankar, “Extended temperature-accelerated dynamics: enabling long-time full-scale modeling of large rare-event systems.,” The Journal of chemical physics. 2014. link Times cited: 9 Abstract: A new method, the Extended Temperature-Accelerated Dynamics … read moreAbstract: A new method, the Extended Temperature-Accelerated Dynamics (XTAD), is introduced for modeling long-timescale evolution of large rare-event systems. The method is based on the Temperature-Accelerated Dynamics approach [M. Sørensen and A. Voter, J. Chem. Phys. 112, 9599 (2000)], but uses full-scale parallel molecular dynamics simulations to probe a potential energy surface of an entire system, combined with the adaptive on-the-fly system decomposition for analyzing the energetics of rare events. The method removes limitations on a feasible system size and enables to handle simultaneous diffusion events, including both large-scale concerted and local transitions. Due to the intrinsically parallel algorithm, XTAD not only allows studies of various diffusion mechanisms in solid state physics, but also opens the avenue for atomistic simulations of a range of technologically relevant processes in material science, such as thin film growth on nano- and microstructured surfaces. read less USED (high confidence) K. Kunal and N. Aluru, “Intrinsic dissipation in a nano-mechanical resonator,” Journal of Applied Physics. 2014. link Times cited: 16 Abstract: We investigate the effect of size on intrinsic dissipation i… read moreAbstract: We investigate the effect of size on intrinsic dissipation in nano-structures. We use molecular dynamics simulation and study dissipation under two different modes of deformation: stretching and bending mode. In the case of stretching deformation (with uniform strain field), dissipation takes place due to Akhiezer mechanism. For bending deformation, in addition to the Akhiezer mechanism, the spatial temperature gradient also plays a role in the process of entropy generation. Interestingly, we find that the bending modes have a higher Q factor in comparison with the stretching deformation (under the same frequency of operation). Furthermore, with the decrease in size, the difference in Q factor between the bending and stretching deformation becomes more pronounced. The lower dissipation for the case of bending deformation is explained to be due to the surface scattering of phonons. A simple model, for phonon dynamics under an oscillating strain field, is considered to explain the observed variation in diss... read less USED (high confidence) L. Chen, Z. Huang, and S. Kumar, “Impact of bonding at multi-layer graphene/metal Interfaces on thermal boundary conductance,” RSC Advances. 2014. link Times cited: 47 Abstract: We use density functional theory and the atomistic Green&apo… read moreAbstract: We use density functional theory and the atomistic Green's function method (AGF) to study the effect of bonding on phonon transmission and thermal boundary conductance (TBC) at the interface of metals (Au, Cu, and Ti) and single layer graphene (SLG)/multi-layer graphene (MLG). Our analysis shows that the TBC across Ti/SLG/Ti interfaces (∼500 MW m−2 K−1) is significantly larger than the TBC across Cu/SLG/Cu (∼10 MW m−2 K−1) and Au/SLG/Au (∼7 MW m−2 K−1) interfaces. However, the TBC across Ti/MLG/Ti (∼40 MW m−2 K−1) is an order of magnitude lower compared to TBC at the Ti/SLG/Ti interface, whereas the TBC at Cu/MLG/Cu and Au/MLG/Au interfaces are similar to those of Cu/SLG/Cu and Au/SLG/Au, respectively. We find that this substantial decrease in TBC at the Ti/MLG/Ti interface is a result of phonon mismatch between the graphene layer bonded to Ti and the non-bonded graphene layers. The effect of number of graphene layers on TBC at Cu/MLG/Cu and Au/MLG/Au interfaces is relatively insignificant because of the weak interactions at these metal/graphene interfaces. It was observed that the moderate attenuation of Ti/C bonding strength can enhance the phonon coupling between the graphene layers bonded to Ti and non-bonded graphene layers, and can increase the TBC across Ti/MLG/Ti by ∼100%. This impact of interfacial bonding strength on TBC at metal/MLG interfaces, predicted by AGF calculations, is further confirmed by non-equilibrium molecular dynamics simulations which show the transition of thermal transport mechanism from metal/graphene dominated resistance to graphene/graphene dominated resistance as the metal/graphene bonding strength increases in the metal/MLG/metal structure. read less USED (high confidence) J. Niu, M. Li, and Z. Xia, “Growth mechanisms and mechanical properties of 3D carbon nanotube–graphene junctions: molecular dynamic simulations,” RSC Advances. 2014. link Times cited: 15 Abstract: The growth process of carbon nanotube (CNT)–graphene 3D junc… read moreAbstract: The growth process of carbon nanotube (CNT)–graphene 3D junctions on copper templates with nano-holes was simulated with classical molecular dynamic (MD) simulation. The CNT, graphene and their seamlessly C–C bonded junction can form simultaneously on the templates without catalysts. There are two mechanisms of junction formation: (i) CNT growth over the holes that are smaller than 3 nm, and (ii) CNT growth inside the holes that are larger than 3 nm. The tensile strengths of the as-grown C–C junctions, as well as the junctions embedded with metal nanoparticles (catalysts), were determined by a quantum mechanics MD simulation method. Metal nanoparticles as catalysts remaining in the junctions significantly reduce the fracture strength and fracture energy, making them brittle and weak. Among the junctions, the seamlessly C–C bonded junctions show the highest tensile strength and fracture energy due to their unique structure. This work provides a theoretical basis and route for synthesizing high-quality single-layer CNT–graphene nanostructures. read less USED (high confidence) T. Cheng, W. Li, and D. Fang, “Modeling of the temperature-dependent ideal tensile strength of solids,” Physica Scripta. 2014. link Times cited: 11 Abstract: To reveal the fracture failure mechanisms of single crystals… read moreAbstract: To reveal the fracture failure mechanisms of single crystals at elevated temperatures, a new temperature-dependent ideal tensile strength model for solids has been developed, based on the critical strain principle. At the same time, the uniaxial tensile strength model, based on the critical failure energy density principle for isotropic materials that was presented in the previous study, is generalized to multi-axial loading and to cubic single crystals. The relationship between the two models is discussed, and how to obtain the material properties needed in the calculations is summarized. The two well-established models are used to predict the temperature-dependent ideal tensile strength of W, Fe and Al single crystals. The predictions from the critical strain principle agree well with the predictions from the critical failure energy density principle. The theoretical values from the critical strain principle at 0 K is in reasonable agreement with the ab initio results. The study shows that the temperature dependence of the ideal tensile strength is similar to that of Young’s modulus; that is, the ideal tensile strength firstly remains approximately constant and then decreases linearly with the temperature. The fracture failure for single crystals at elevated temperatures has been identified, for the first time, as a strain-controlled criterion. read less USED (high confidence) S. Rawat, M. Warrier, S. Chaturvedi, and V. R. Ikkurthi, “Multiscale simulations of damage of perfect crystal Cu at high strain rates,” Pramana. 2014. link Times cited: 10 USED (high confidence) J. Bian, X. Niu, H. Zhang, and G. Wang, “Atomistic deformation mechanisms in twinned copper nanospheres,” Nanoscale Research Letters. 2014. link Times cited: 8 USED (high confidence) X. Liu, F. Wang, H. Wu, and W. Wang, “Strengthening metal nanolaminates under shock compression through dual effect of strong and weak graphene interface,” Applied Physics Letters. 2014. link Times cited: 78 Abstract: We use molecular dynamics method to study the strengthening … read moreAbstract: We use molecular dynamics method to study the strengthening effect of graphene-metal nanolayered composites under shock loading. The graphene interfaces have the advantages of both strong and weak interfacial features simultaneously, which solves a strengthening paradox of interfacial structures. On one hand, the weak bending stiffness of graphene leads to interlayer reflections and weakening the shock wave. On the other hand, the strong in-plane sp2-bonded structures constrain the dislocations and heal the material. The elastic recovery due to graphene interfacial constraints plays an important role in the strengthening effect, and the shock strength can be enhanced by decreasing the interlayer distance. This interface with strong/weak duality should lead to an improved fundamental understanding on the dynamic mechanism of composites with interfacial structures. read less USED (high confidence) D. Kang et al., “Interfacial Free Energy Controlling Glass-Forming Ability of Cu-Zr Alloys,” Scientific Reports. 2014. link Times cited: 34 USED (high confidence) S.-H. Cheng and C. Sun, “Convergence of local atomistic stress based on periodic lattice,” International Journal of Solids and Structures. 2014. link Times cited: 5 USED (high confidence) E. T. Karim, M. Shugaev, C. Wu, Z. Lin, R. Hainsey, and L. Zhigilei, “Atomistic simulation study of short pulse laser interactions with a metal target under conditions of spatial confinement by a transparent overlayer,” Journal of Applied Physics. 2014. link Times cited: 38 Abstract: The distinct characteristics of short pulse laser interactio… read moreAbstract: The distinct characteristics of short pulse laser interactions with a metal target under conditions of spatial confinement by a solid transparent overlayer are investigated in a series of atomistic simulations. The simulations are performed with a computational model combining classical molecular dynamics (MD) technique with a continuum description of the laser excitation, electron-phonon equilibration, and electronic heat transfer based on two-temperature model (TTM). Two methods for incorporation of the description of a transparent overlayer into the TTM-MD model are designed and parameterized for Ag-silica system. The material response to the laser energy deposition is studied for a range of laser fluences that, in the absence of the transparent overlayer, covers the regimes of melting and resolidification, photomechanical spallation, and phase explosion of the overheated surface region. In contrast to the irradiation in vacuum, the spatial confinement by the overlayer facilitates generation of sustain... read less USED (high confidence) M. Schmidt, R. Sauer, and A. Ismail, “Multiscale treatment of mechanical contact problems involving thin polymeric layers,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 5 Abstract: We propose a strategy to obtain a hyperelastic constitutive … read moreAbstract: We propose a strategy to obtain a hyperelastic constitutive law for film-like systems from molecular dynamics (MD) simulations. The aim is to furnish a computationally efficient continuum model with this description of the material. In particular, two different methods are suggested, both of which consist of virtual experiments that are performed on the material to sample systematically the stress–strain relation. The latter is then fitted to a suitable functional form. We use a polymeric self-assembled monolayer, which spans a height of only a few nanometers, as a test case. Having determined the response function, we then apply it within a finite-element simulation of a continuum mechanical nanoindentation problem. Several contact quantities such as normal reaction forces and the contact geometry are extracted from these calculations and are compared to those from an analogous, fully atomistic nanoindentation simulation. We find that the considered benchmark quantities as obtained from the continuum surrogate model reproduce well the corresponding values of the MD simulation. read less USED (high confidence) J. Zhang, Y. D. Yan, X. Liu, T. Sun, and Y. Liang, “Influence of coherent twin boundaries on three-point bending of gold nanowires,” Journal of Physics D: Applied Physics. 2014. link Times cited: 13 Abstract: In the present work we elucidate the deformation mechanisms … read moreAbstract: In the present work we elucidate the deformation mechanisms of twinned Au nanowires (NWs) under three-point bending by means of molecular dynamics simulations. We further investigate the effects of twin boundary orientation, NW diameter and twin boundary spacing on the mechanical properties and deformation behaviors of NWs. Our simulation results reveal that dislocation slip, twin boundary associated mechanisms and deformation twinning work in parallel in the heterogeneous localized plastic deformation of twinned Au NWs under bending. The dislocation–twin boundary interactions can be significantly altered by changing the twin boundary orientation as well as the bending direction. Furthermore, the NW diameter and twin boundary spacing strongly influence the competition between individual deformation mechanisms, which in turn leads to extrinsic and intrinsic size effects on the strength and the bending ductility of the NWs. read less USED (high confidence) J. Song and W. Curtin, “Mechanisms of hydrogen-enhanced localized plasticity: An atomistic study using α-Fe as a model system,” Acta Materialia. 2014. link Times cited: 172 USED (high confidence) S. Hartmann et al., “Molecular dynamic simulations of maximum pull-out forces of embedded CNTs for sensor applications and validating nano scale experiments,” 2014 15th International Conference on Thermal, Mechanical and Mulit-Physics Simulation and Experiments in Microelectronics and Microsystems (EuroSimE). 2014. link Times cited: 2 Abstract: We present investigations of pull-out tests on CNTs embedded… read moreAbstract: We present investigations of pull-out tests on CNTs embedded in palladium by means of molecular dynamics (MD) and compare our results of maximum pull-out forces with values of nano scale in situ pull-out tests inside a scanning electron microscope (SEM). Our MD model allows the investigation of crucial influencing parameters on the interface behaviour, like CNT diameter, intrinsic CNT defects and functional groups. For the experiments we prepared simple specimens using silicon substrates and wafer level compliant technologies. We realised the nano scale experiment with a nanomanipulation system supporting an AFM cantilever with known stiffness as a force sensing element inside a SEM. Greyscale correlation has been used to evaluate the cantilever deflection. From simulations derived maximum pull-out forces are approximately 17 nN and depend on the existence of intrinsic defects or functional groups and weakly on temperature. Experimentally obtained maximum pull-out forces with values between 16-29 nN are in good agreement with the computational predictions. Our results are of significant interest for the design and a failure-mechanistic treatment of future mechanical sensors with integrated single-walled CNTs showing high piezoresistive gauge factor or other nano scale systems incorporating CNT-metal interfaces. read less USED (high confidence) S. Huang, R.-Z. Li, S.-T. Qi, B. Chen, and J. Shen, “Order–disorder effects on the elastic properties of CuMPt6 (M=Cr and Co) compounds,” Solid State Communications. 2014. link Times cited: 0 USED (high confidence) K. Saini and N. Kumar, “Torsional deformation behavior of cracked gold nano-wires,” Acta Mechanica. 2014. link Times cited: 9 USED (high confidence) S. Goel, N. Faisal, V. Ratia, A. Agrawal, and A. Stukowski, “Atomistic investigation on the structure–property relationship during thermal spray nanoparticle impact,” Computational Materials Science. 2014. link Times cited: 32 USED (high confidence) M. Baskes and S. G. Srinivasan, “The embedded atom method ansatz: validation and violation,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 3 Abstract: The addition of the embedding energy term to pair interactio… read moreAbstract: The addition of the embedding energy term to pair interaction contribution has made the embedded atom method (EAM) potentials a simple and vastly superior alternative to popular classical pair potentials. EAM relies on the ansatz that the embedding energy is a function of a linear superposition of spherically averaged atomic electron densities. This ansatz is taken to be self-evident and inviolate. Using density functional theory (DFT) calculations of a model face-centered cubic (fcc) Cu system, we systematically investigate the validity of this foundational ansatz of EAM. We conclude that it (1) agrees well with DFT calculations along a path with changing coordination and symmetry, (2) captures the exponential decrease of the background electron density with respect to distance, (3) demonstrates transferability as seen by agreement of electron densities for other non-fcc structures with first nearest neighbor (NN) coordination ranging from 4 to 12 and (4) fails to explain the behavior of background electron density with respect to second NN distance and arrangements. This failure may be remedied by including a fraction of the second NN atomic electron density in the background electron density, including angular contributions to the density, or including electron density rearrangement. These insights likely make EAM approaches more broadly applicable, more predictive and perhaps unique, and in the process broadly impact atomistic modeling. A new EAM potential is presented that for the first time reproduces electron densities from DFT calculations as well as experimental properties of Cu in the potential fitting. read less USED (high confidence) V. Péron-Lührs, F. Sansoz, and L. Noels, “Quasicontinuum study of the shear behavior of defective tilt grain boundaries in Cu,” Acta Materialia. 2014. link Times cited: 11 USED (high confidence) S. Meguid and F. A. Jahwari, “Modeling the pullout test of nanoreinforced metallic matrices using molecular dynamics,” Acta Mechanica. 2014. link Times cited: 17 USED (high confidence) S. Meguid and F. A. Jahwari, “Modeling the pullout test of nanoreinforced metallic matrices using molecular dynamics,” Acta Mechanica. 2014. link Times cited: 0 USED (high confidence) Y. Yang and D. Cheng, “Role of Composition and Geometric Relaxation in CO2 Binding to Cu–Ni Bimetallic Clusters,” Journal of Physical Chemistry C. 2014. link Times cited: 24 Abstract: Adsorption and activation of CO2 can be considered as the fi… read moreAbstract: Adsorption and activation of CO2 can be considered as the first steps of the reaction mechanism of methanol synthesis via CO2 hydrogenation on Cu-based bimetallic clusters. In this work, the adsorption properties of CO2 on Cu55, Cu54Ni1, and Cu42Ni13 clusters with highly symmetric cuboctaheral (Cubo), decahedral (Dec), and icosahedral (Ico) structures are studied by density functional theory calculations. It is found that icosahedral Cu42Ni13 cluster exhibits the strongest CO2 adsorption ability with all the −Coo, −Oco, and −OcO– adsorption models, compared to the icosahedral Cu55 and Cu54Ni1 clusters. In addition, the structural transformation from cuboctaheral and decahedral to icosahedral clusters upon CO2 adsorption is found, which is attributed to the fact that the stability of these clusters follows the order Cubo < Dec < Ico. Our results show that composition and geometric relaxation can modify the adsorption properties of CO2 on Cu–Ni bimetallic clusters, which can provide useful insights for the ... read less USED (high confidence) M. Grouchko et al., “Merging of metal nanoparticles driven by selective wettability of silver nanostructures,” Nature Communications. 2014. link Times cited: 10 USED (high confidence) A. Leino, O. Pakarinen, F. Djurabekova, K. Nordlund, P. Kluth, and M. Ridgway, “Swift Heavy Ion Shape Transformation of Au Nanocrystals Mediated by Molten Material Flow and Recrystallization,” Materials Research Letters. 2014. link Times cited: 39 Abstract: Swift heavy ion (SHI) irradiation of amorphous SiO2 that con… read moreAbstract: Swift heavy ion (SHI) irradiation of amorphous SiO2 that contains metal nanocrystals can be used to transform the shape of the particles into peculiar asymmetric ones not easily achievable by other means. Using a molecular dynamics simulation framework augmented to include the electronic excitations of the SHIs, we predict that the reshaping of spherical particles into nanorods occurs continuously during consecutive ion impacts by a dynamic crystal–liquid–crystal phase transition of metal particle with the flow of liquid phase into an underdense track core in silica. The simulated nanocrystals are shown to have a saturation width that agrees with experiments. read less USED (high confidence) S. Khanal et al., “Trimetallic nanostructures: the case of AgPd-Pt multiply twinned nanoparticles.,” Nanoscale. 2013. link Times cited: 40 Abstract: We report the synthesis, structural characterization, and at… read moreAbstract: We report the synthesis, structural characterization, and atomistic simulations of AgPd-Pt trimetallic (TM) nanoparticles. Two types of structure were synthesized using a relatively facile chemical method: multiply twinned core-shell, and hollow particles. The nanoparticles were small in size, with an average diameter of 11 nm and a narrow distribution, and their characterization by aberration corrected scanning transmission electron microscopy allowed us to probe the structure of the particles at an atomistic level. In some nanoparticles, the formation of a hollow structure was also observed, that facilitates the alloying of Ag and Pt in the shell region and the segregation of Ag atoms on the surface, affecting the catalytic activity and stability. We also investigated the growth mechanism of the nanoparticles using grand canonical Monte Carlo simulations, and we have found that Pt regions grow at overpotentials on the AgPd nanoalloys, forming 3D islands at the early stages of the deposition process. We found very good agreement between the simulated structures and those observed experimentally. read less USED (high confidence) K. Suzuki, H. Miura, O. Asai, R. Furuya, J. Sung, and N. Murata, “Micro-texture dependence of stress-induced migration of electroplated copper thin film interconnections used for 3D integration,” 2013 International Conference on Simulation of Semiconductor Processes and Devices (SISPAD). 2013. link Times cited: 0 Abstract: Effect of the micro texture of electroplated copper thin fil… read moreAbstract: Effect of the micro texture of electroplated copper thin film interconnections on stress-induced migration was investigated experimentally and theoretically. The micro texture of electroplated copper thin films changed drastically as a function of the annealing temperature after the electroplating. However, stress-induced migration was activated even though the thin film interconnection was kept at room temperature after annealing. As a result, voids and hillocks appeared on the thin film interconnection. This is because high residual stress was caused by shrinkage of the thin film interconnection due to the densification caused by recrystallization. Molecular dynamics simulations showed that the diffusivity of copper atoms along grain boundaries with low crystallinity was enhanced significantly by high tensile residual stress. Therefore, the grain boundary diffusion accelerated by tensile residual stress is the main reason for the formation of hillocks and voids in the thin film interconnection after annealing. read less USED (high confidence) C. Cassidy, D. V. Singh, P. Grammatikopoulos, F. Djurabekova, K. Nordlund, and M. Sowwan, “Inoculation of silicon nanoparticles with silver atoms,” Scientific Reports. 2013. link Times cited: 37 USED (high confidence) H. Chen and S. Chen, “The peeling behaviour of a graphene sheet on a nano-scale corrugated surface,” Journal of Physics D: Applied Physics. 2013. link Times cited: 13 Abstract: The peeling process and average peeling force of a graphene … read moreAbstract: The peeling process and average peeling force of a graphene (GE) sheet on a corrugated surface are investigated using molecular dynamics simulation. It is found that the peeling behaviour varies with the substrate surface roughness and the peeling angle. Three kinds of typically peeling behaviours include (a) GE sheet directly passing the valley of the substrate roughness; (b) bouncing off from the substrate; and (c) continuously peeling off similarly to that on a flat substrate. As a result, the average peeling force is strongly dependent of the peeling behaviours. Furthermore, some interesting phenomena are caught, such as partial detaching and partial sliding of GE sheet in the valley of the substrate roughness, which are mainly due to the effects of pre-tension in GE sheet and the reduction of friction resistance. The results in this paper should be useful for the design of nano-film/substrate systems. read less USED (high confidence) M. Pozuelo, S. Mathaudhu, S. Kim, B. Li, W. Kao, and J. Yang, “Nanotwins in nanocrystalline Mg–Al alloys: an insight from high-resolution TEM and molecular dynamics simulation,” Philosophical Magazine Letters. 2013. link Times cited: 16 Abstract: Twinning in hexagonal close-packed Mg alloys has been report… read moreAbstract: Twinning in hexagonal close-packed Mg alloys has been reported to be unfavorable when the grain size is reduced below a couple of microns and suppressed at the nanoscale. Using high-resolution transmission electron microscopy, we present evidence of nanotwins (<1 nm) in nanocrystalline Mg–Al alloys processed by cryomilling. The commonly observed twinning modes for coarse-grained Mg are identified and supported with atomistic molecular dynamics simulations. The specific thermomechanical conditions offered by cryomilling facilitate the generation of deformation twins that are not observed with conventional deformation processing methods. read less USED (high confidence) K. Saini and N. Kumar, “Torsional deformation behavior of cracked gold nano-wires,” Acta Mechanica. 2013. link Times cited: 0 USED (high confidence) V. Péron-Lührs, A. Jérusalem, F. Sansoz, L. Stainier, and L. Noels, “A two-scale model predicting the mechanical behavior of nanocrystalline solids,” Journal of The Mechanics and Physics of Solids. 2013. link Times cited: 14 USED (high confidence) E. Marzbanrad, A. Hu, B. Zhao, and Y. Zhou, “Room Temperature Nanojoining of Triangular and Hexagonal Silver Nanodisks,” Journal of Physical Chemistry C. 2013. link Times cited: 43 Abstract: Room temperature nanojoining is an important phenomenon that… read moreAbstract: Room temperature nanojoining is an important phenomenon that has to be understood well for use in different applications, for example, for assembly of nanoscale building blocks into nanoscale and microscale structures and devices. However, the mechanism for nanoparticle joining at room temperature is not well established. In this research, we employed molecular dynamics simulation to explain how and why silver nanodisks are joined/assembled but with their original shape unchanged. To support our theoretical observations, we compared our simulation results to SEM and HRTEM observations of joined silver nanodisks. It was found that joining at a wide temperature range (1–500 K) can be done through short movement and rearrangement of surface atoms and subsequent elastic or plastic deformation of the particles, resulting in perfect crystal alignment at the joint interface. Our simulation shows the crystal defects such as dislocations due to initial lattice mismatch of the crystals can be sintered out to yield ... read less USED (high confidence) X. Ben, P. Cao, and H. S. Park, “The Effect of Planar Defects on the Optical Properties of Silver Nanostructures,” Journal of Physical Chemistry C. 2013. link Times cited: 5 Abstract: We present a computational, atomistic electrodynamics invest… read moreAbstract: We present a computational, atomistic electrodynamics investigation of the effects of planar defects on the optical properties of silver nanocubes, where the planar defects we considered are different surface orientations, twins, partial dislocations, and full dislocations. We find that for nanocubes smaller than about 3 nm, the optical response is very sensitive to the specific surface structure resulting from the defects. However, the sensitivity, as measured by shifts in the plasmon resonance wavelength, is strongly reduced at larger sizes because of the decreasing importance of surface effects even when the majority of the atomic deformation due to the crystal defects is contained within the interior of the nanocube. Overall, this study suggests that the effects of individual crystalline defects on the optical properties of nanostructures can be safely ignored for nanostructure sizes larger than about 5 nm. read less USED (high confidence) Y.-quan Yuan, X.-guo Zeng, H. Chen, A. Yao, and Y. Hu, “Molecular dynamics simulation on microstructure evolution during solidification of copper nanoparticles,” Journal of the Korean Physical Society. 2013. link Times cited: 9 Abstract: The effect of cooling rate on the microstructure evolution o… read moreAbstract: The effect of cooling rate on the microstructure evolution of liquid Cu nanoparticles during their solidification process is investigated by using a molecular dynamics simulation based on the embedded atom method (EAM) potential developed by Foiles et al.. The potential energy analysis, the pair distribution function and the common neighbor analysis have been used. The results show that the solidification point increases with decreasing cooling rate and that the solidification of the microstructure of Cu nanoparticles varies with the cooling rate. The microstructure consists of fcc, hcp and bcc crystals or mixtures, though the fcc structure dominates, except in the amorphous state. An amorphous structure was obtained when the cooling rate reached 1.0 × 1013 K/s or higher while crystallization degree increased with decreasing cooling rate, and the total content of crystal structures reached to 95% when the cooling rate dropped to 4.0 × 1011 K/s, which was nearly a perfect crystal structure. The results also indicate that a single-crystal nanoparticle will not be obtained by quenching the liquid metal under various cooling rates. read less USED (high confidence) Y. Fu and A. To, “On the evaluation of Hardy’s thermomechanical quantities using ensemble and time averaging,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 8 Abstract: An ensemble averaging approach was investigated for its accu… read moreAbstract: An ensemble averaging approach was investigated for its accuracy and convergence against time averaging in computing continuum quantities such as stress, heat flux and temperature from atomistic scale quantities. For this purpose, ensemble averaging and time averaging were applied to evaluate Hardy's thermomechanical expressions (Hardy 1982 J. Chem. Phys. 76 622–8) in equilibrium conditions at two different temperatures as well as a nonequilibrium process due to shock impact on a Ni crystal modeled using molecular dynamics simulations. It was found that under equilibrium conditions, time averaging requires selection of a time interval larger than the critical time interval to obtain convergence, where the critical time interval can be estimated using the elastic properties of the material. The reason for this is because of the significant correlations among the computed thermomechanical quantities at different time instants employed in computing their time average. On the other hand, the computed thermomechanical quantities from different realizations in ensemble averaging are statistically independent, and thus convergence is always guaranteed. The computed stress, heat flux and temperature show noticeable difference in their convergence behavior while their confidence intervals increase with temperature. Contrary to equilibrium settings, time averaging is not equivalent to ensemble averaging in the case of shock wave propagation. Time averaging was shown to have poor performance in computing various thermomechanical fields by either oversmoothing the fields or failing to remove noise. read less USED (high confidence) A. Nazarov, “Disclinations in bulk nanostructured materials: their origin, relaxation and role in material properties,” Advances in Natural Sciences: Nanoscience and Nanotechnology. 2013. link Times cited: 15 Abstract: The role of disclinations in the processing, microstructure … read moreAbstract: The role of disclinations in the processing, microstructure and properties of bulk nanostructured materials is reviewed. Models of grain subdivision during severe plastic deformation (SPD) based on the disclination concept, a structural model of the bulk nanostructured materials processed by SPD are presented. The critical strength of triple junction disclinations is estimated. Kinetics of relaxation of triple junction disclinations and their role in the grain boundary diffusion are studied. read less USED (high confidence) H. Alarifi et al., “Molecular Dynamics Simulation of Sintering and Surface Premelting of Silver Nanoparticles,” Materials Transactions. 2013. link Times cited: 51 Abstract: Sintering of Ag nanoparticles (NPs) is increasingly being us… read moreAbstract: Sintering of Ag nanoparticles (NPs) is increasingly being used as a driving mechanism for joining in the microelectronics industry. We therefore performed molecular dynamics simulations based on the embedded atom method (EAM) to study pressureless sintering kinetics of two Ag NPs in the size range of (4 to 20 nm), and sintering of three and four Ag NPs of 4 nm diameter. We found that the sintering process passed through three main stages. The first was the neck formation followed by a rapid increase of the neck radius at 50K for 20 nm particles and at 10K for smaller NPs. The second was characterized by a gradual linear increase of the neck radius to particle radius ratio as the temperature of the sintered structure was increased to the surface premelting point. Different than previous sintering studies, a twin boundary was formed during the second stage that relaxed the sintered structure and decreased the average potential energy (PE). The third stage of sintering was a rapid shrinkage during surface premelting of the sintered structure. Based on pore geometry, densification occurred during the first stage for three 4 nm particles and during the second stage for four 4 nm particles. Sintering rates obtained by our simulation were higher than those obtained by theoretical models generally used for predicting sintering rates of microparticles. [doi:10.2320/matertrans.MD201225] read less USED (high confidence) D. Zhu, H. Zhang, and D. Li, “Influence of Nanotwin Boundary on the Bauschinger’s Effect in Cu: A Molecular Dynamics Simulation Study,” Metallurgical and Materials Transactions A. 2013. link Times cited: 14 USED (high confidence) S. Hartmann, O. Holck, and B. Wunderle, “Molecular dynamics simulations for mechanical characterization of CNT/gold interface and its bonding strength,” 2013 14th International Conference on Thermal, Mechanical and Multi-Physics Simulation and Experiments in Microelectronics and Microsystems (EuroSimE). 2013. link Times cited: 5 Abstract: CNT/metal interfaces under mechanical loads are investigated… read moreAbstract: CNT/metal interfaces under mechanical loads are investigated using molecular dynamics by simulating pull-out tests of single walled carbon nanotubes (CNTs) emdedded in single crystal gold lattices. As a result of our simulations we present obtained force-displacement data. We investigated the influence of two different Lennard Jones (LJ) coefficients pairs, two CNT types and three lattice directions of the gold matrix with respect to the embedding direction. Additionally we incorporated structural defects into our model and report on their influence. The change of the CNT type leads to a change in the maximum pull-out force. Here we attribute this to the change in CNT diameter, where a bigger diameter entails an increased maximum pull-out force. Changing the LJ coefficient pair has a strong impact on the maximum pull-out forces, where a higher bonding energy results in a higher maximum pull-out force. Defects also show a positive effect on the maximum pull-out force. The presented results have impact on bonding strength of CNT/metal interfaces. read less USED (high confidence) H. Zhang, M. Khalkhali, Q. Liu, and J. Douglas, “String-like cooperative motion in homogeneous melting.,” The Journal of chemical physics. 2013. link Times cited: 68 Abstract: Despite the fundamental nature and practical importance of m… read moreAbstract: Despite the fundamental nature and practical importance of melting, there is still no generally accepted theory of this ubiquitous phenomenon. Even the earliest simulations of melting of hard discs by Alder and Wainwright indicated the active role of collective atomic motion in melting and here we utilize molecular dynamics simulation to determine whether these correlated motions are similar to those found in recent studies of glass-forming (GF) liquids and other condensed, strongly interacting, particle systems. We indeed find string-like collective atomic motion in our simulations of "superheated" Ni crystals, but other observations indicate significant differences from GF liquids. For example, we observe neither stretched exponential structural relaxation, nor any decoupling phenomenon, while we do find a boson peak, findings that have strong implications for understanding the physical origin of these universal properties of GF liquids. Our simulations also provide a novel view of "homogeneous" melting in which a small concentration of interstitial defects exerts a powerful effect on the crystal stability through their initiation and propagation of collective atomic motion. These relatively rare point defects are found to propagate down the strings like solitons, driving the collective motion. Crystal integrity remains preserved when the permutational atomic motions take the form of ring-like atomic exchanges, but a topological transition occurs at higher temperatures where the rings open to form linear chains similar in geometrical form and length distribution to the strings of GF liquids. The local symmetry breaking effect of the open strings apparently destabilizes the local lattice structure and precipitates crystal melting. The crystal defects are thus not static entities under dynamic conditions, such as elevated temperatures or material loading, but rather are active agents exhibiting a rich nonlinear dynamics that is not addressed in conventional "static" defect melting models. read less USED (high confidence) J. Zimmerman and R. Jones, “The application of an atomistic J-integral to a ductile crack,” Journal of Physics: Condensed Matter. 2013. link Times cited: 22 Abstract: In this work we apply a Lagrangian kernel-based estimator of… read moreAbstract: In this work we apply a Lagrangian kernel-based estimator of continuum fields to atomic data to estimate the J-integral for the emission dislocations from a crack tip. Face-centered cubic (fcc) gold and body-centered cubic (bcc) iron modeled with embedded atom method (EAM) potentials are used as example systems. The results of a single crack with a K-loading compare well to an analytical solution from anisotropic linear elastic fracture mechanics. We also discovered that in the post-emission of dislocations from the crack tip there is a loop size-dependent contribution to the J-integral. For a system with a finite width crack loaded in simple tension, the finite size effects for the systems that were feasible to compute prevented precise agreement with theory. However, our results indicate that there is a trend towards convergence. read less USED (high confidence) A. V. Myshlyavtsev and P. Stishenko, “Relative stability of icosahedral and cuboctahedral metallic nanoparticles,” Adsorption. 2013. link Times cited: 10 USED (high confidence) L. Momenzadeh, A. Evteev, E. Levchenko, I. Belova, G. Murch, and Y. Sohn, “Phonon Thermal Conductivity of F.C.C. Cu by Molecular Dynamics Simulation,” Defect and Diffusion Forum. 2013. link Times cited: 7 Abstract: Phonon dynamics and phonon thermal conductivity of f.c.c. Cu… read moreAbstract: Phonon dynamics and phonon thermal conductivity of f.c.c. Cu are investigated in detail in the temperature range 200 1300 K within the framework of equilibrium molecular dynamics simulations making use of the Green-Kubo formalism and one of the most reliable embedded-atom method potentials. It is found that the temporal decay of the heat current autocorrelation function of the f.c.c. Cu model at low and intermediate temperatures demonstrates a more complex behaviour than the two-stage decay observed previously for the f.c.c. Ar model. After the first stage of decay, it demonstrates a peak in the temperature range 200 800 K. The intensity of the peak decreases as the temperature increases. At 900 K, it transforms to a shoulder which diminishes almost entirely at 1200 K. It is suggested that the peak may be activated by the influence of the Cauchy pressure in f.c.c. Cu on the phonon dynamics. A decomposition model of the heat current autocorrelation function of a monatomic f.c.c. lattice is introduced. This model can capture all contributions to the function discussed in the literature. It is found that the temperature dependence of the phonon thermal conductivity of the f.c.c. Cu model is in good agreement with previous calculations on the f.c.c. Ar model which follows an exponent close to-1.4, i.e. varies more rapidly than the T-1 law predicted by the theory. The calculated phonon thermal conductivity of the f.c.c. Cu is found to be about one order of magnitude higher than the f.c.c. Ar. This is explained by the inclusion of the electronic contribution to the bulk lattice properties during the fitting of the embedded-atom method potential functions to the experimental or ab initio data. It is demonstrated that the electronic contribution to the total thermal conductivity of f.c.c. Cu dominates over the whole studied temperature range. Nevertheless, the phonon contribution increases as the temperature decreases. The contribution can be estimated to be about 0.5 % at 1300 K and about 5 % at 200 K. read less USED (high confidence) D. Shan, L. Wang, and L. Yuan, “Effect of the ∑5(310)/[001]θ = 53.1° grain boundary on the incipient yield of bicrystal copper: A quasicontinuum simulation and nanoindentation experiment,” Journal of Materials Research. 2013. link Times cited: 8 Abstract: To study the initial plasticity during the nanoindentation o… read moreAbstract: To study the initial plasticity during the nanoindentation of face-centered cubic bicrystal materials at the micro- and nanoscales, a $$\sum {5(310)/[001]} {{\theta }} = 53.1\circ $$ symmetrical tilt grain boundary (GB) model of bicrystal copper was constructed using the quasicontinuum method. The nanoindentation process of the model was then simulated, and a group of indents across the GB during a bicrystal copper nanoindentation experiment were performed. The effect of the GB on the incipient yield was studied when the transition from elastic to plastic deformation and the first dislocation emission occur. The results show that the maximum incipient load appears in the center of the boundary; the load first increases and then gradually decreases until it presents no further significant changes when the indenter is far from the GB. It is observed that theoretical simulation results are in good agreement with those of the experimental measurement. The incipient yield force was affected by the size and the position of the indenter, the structure of the boundary, and the first dislocation emission. read less USED (high confidence) N. Abdolrahim et al., “The mechanical response of core-shell structures for nanoporous metallic materials,” Philosophical Magazine. 2013. link Times cited: 32 Abstract: Nanoporous gold (NP-Au) exhibits microscale plasticity, but … read moreAbstract: Nanoporous gold (NP-Au) exhibits microscale plasticity, but macroscopically fails in a relatively brittle manner. This current study suggests that a core-shell structure can increase both ductility and strength of NP-Au. A core Au foam structure was created using conventional dealloying methods with average ligament size of 60 nm. Nickel was then electroplated on to the NP-Au with layer thicknesses ranging from 2.5 nm to 25 nm. Nanoindentation demonstrated a significant increase in the hardness of the coated Np-Au, to about five times of that of the pure Np-Au, and a decrease in creep by increasing the thickness of the coated Ni layer. Molecular dynamics simulations of Au–Ni ligaments show the same trend of strengthening behavior with increasing Ni thickness suggesting that the strengthening mechanisms of the Np-Au are comparable to those for fcc nano ligaments. The simulations demonstrate two different strengthening mechanisms with the increased activity of the twins in plated Au–Ni ligaments, which leads to more ductile behavior, as opposing to the monolithic Au ligaments where nucleation of dislocations govern the plasticity during loading. read less USED (high confidence) Y. Gan and S. Jiang, “Ultrafast laser-induced premelting and structural transformation of gold nanorod,” Journal of Applied Physics. 2013. link Times cited: 32 Abstract: Femtosecond laser irradiation of a gold nanorod has been sim… read moreAbstract: Femtosecond laser irradiation of a gold nanorod has been simulated by a method that couples two-temperature model into molecular dynamics. Numerical results show that the surface premelting occurs prior to the initiation of planar defect and propagates from the surface layer into the inside of nanorod. Pressure relaxation leads to high-frequency temperature oscillation and two-way transformation between fcc and disordered atoms produced by the dynamic stresses. Partial dislocation cores are initiated on the crystal surfaces due to high stresses, and then noticeable planar defects including stacking faults and twin boundaries on {111} close-packed planes are developed. Finally, only parallel twin boundaries are present in the nanorod, showing favorable agreement with the experimental observation. read less USED (high confidence) D. Schebarchov, D. Schebarchov, B. Lefevre, W. Somerville, S. C. Hendy, and S. C. Hendy, “Filling a nanoporous substrate by dewetting of thin films.,” Nanoscale. 2013. link Times cited: 10 Abstract: Following a simple thermodynamic model, which predicts that … read moreAbstract: Following a simple thermodynamic model, which predicts that an array of non-wettable pores can be filled by dewetting of sufficiently thin films, we use molecular dynamics to simulate the rupture of nanometre-thick liquid Au films on nanoporous substrates. Our simulations clearly exhibit spinodal dewetting and hole nucleation, and some of the metal is indeed absorbed by non-wettable pores solely as a virtue of the Laplace pressure acting on dewetted droplets and rivulet-like structures. Finally, we show that the fraction of absorbed Au can be increased through patterning of the initial film. read less USED (high confidence) Z. Liang, W. Evans, T. Desai, and P. Keblinski, “Improvement of heat transfer efficiency at solid-gas interfaces by self-assembled monolayers,” Applied Physics Letters. 2013. link Times cited: 36 Abstract: Using molecular dynamics simulations, we demonstrate that th… read moreAbstract: Using molecular dynamics simulations, we demonstrate that the efficiency of heat exchange between a solid and a gas can be maximized by functionalizing solid surface with organic self-assembled monolayers (SAMs). We observe that for bare metal surfaces, the thermal accommodation coefficient (TAC) strongly depends on the solid-gas interaction strength. For metal surfaces modified with organic SAMs, the TAC is close to its theoretical maximum and is essentially independent from the SAM-gas interaction strength. The analysis of the simulation results indicates that softer and lighter SAMs, compared to the bare metal surfaces, are responsible for the greatly enhanced TAC. read less USED (high confidence) J. Zhang, F. Xu, Y. Yan, and T. Sun, “Detwinning-induced reduction in ductility of twinned copper nanowires,” Chinese Science Bulletin. 2013. link Times cited: 15 USED (high confidence) A. Leonardi, M. Leoni, and P. Scardi, “Directional pair distribution function for diffraction line profile analysis of atomistic models,” Journal of Applied Crystallography. 2013. link Times cited: 12 Abstract: The concept of the directional pair distribution function is… read moreAbstract: The concept of the directional pair distribution function is proposed for an atomistic level interpretation of the line profile broadening in powder diffraction patterns of nanocrystalline materials. read less USED (high confidence) X.-Y. Sun, G. Xu, X. Li, X.-Q. Feng, and H. Gao, “Mechanical properties and scaling laws of nanoporous gold,” Journal of Applied Physics. 2013. link Times cited: 170 Abstract: Nanoporous metals are a class of novel nanomaterials with po… read moreAbstract: Nanoporous metals are a class of novel nanomaterials with potential applications in many fields such as sensing, catalysis, and fuel cells. The present paper is aimed to investigate atomic mechanisms associated with the uniaxial tensile deformation behavior of nanoporous gold. A phase field method is adopted to generate the bicontinuous open-cell porous microstructure of the material. Molecular dynamics simulations then reveal that the uniaxial tensile deformation in such porous materials is accompanied by an accumulation of stacking faults in ligaments along the loading direction and their junctions with neighboring ligaments, as well as the formation of Lomer–Cottrell locks at such junctions. The tensile strain leads to progressive necking and rupture of some ligaments, ultimately resulting in failure of the material. The simulation results also suggest scaling laws for the effective Young's modulus, yield stress, and ultimate strength as functions of the relative mass density and average ligament size ... read less USED (high confidence) W. Kim, S. Y. Rhee, and M. Cho, “Molecular dynamics-based continuum models for the linear elasticity of nanofilms and nanowires with anisotropic surface effects,” Journal of Mechanics of Materials and Structures. 2012. link Times cited: 7 USED (high confidence) Y. Hakobyan, E. Tadmor, and R. James, “Objective quasicontinuum approach for rod problems,” Physical Review B. 2012. link Times cited: 20 Abstract: An objective quasicontinuum (OQC) method is developed for si… read moreAbstract: An objective quasicontinuum (OQC) method is developed for simulating rodlike systems that can be represented as a combination of locally objective structures. An objective structure (OS) is one for which a group of atoms, called a “fundamental domain” (FD), is repeated using specific rules of translation and rotation to build a more complex structure. An objective Cauchy-Born rule defines the kinematics of the OS atoms in terms of a set of symmetry parameters and the positions of the FD atoms. The computational advantage lies in the capability of representing a large system of atoms through a small set of symmetry parameters and FD atom positions. As an illustrative example, we consider the deformation of a copper single-crystal nanobeam which can be described as an OS. OQC simulations are performed for uniform and nonuniform bending for two different orientations (nanobeam axis oriented along [111] and [100]) and compared with elastica results. In the uniform bending case, the [111]-oriented single-crystal nanobeam experiences elongation, while the [100]-oriented nanobeam experiences contraction in total length. The nonuniform bending allows for stretching, contraction, and bending as deformation. Under certain loading conditions, dislocation nucleation is observed within the FD. read less USED (high confidence) A. Vorontsov, B. R. Gel’chinskii, and A. E. Korenchenko, “Kinetics and energy states of nanoclusters in the initial stage of homogeneous condensation at high supersaturation degrees,” Journal of Experimental and Theoretical Physics. 2012. link Times cited: 20 USED (high confidence) A. Bisht, K. Albe, and R. Jayaganthan, “Effect of porosity on mechanical behaviour of nanocrystalline metals,” 2012 International Conference on Emerging Electronics. 2012. link Times cited: 0 Abstract: The understanding of deformation mechanisms in nanocrystalli… read moreAbstract: The understanding of deformation mechanisms in nanocrystalline metals is still not clear. At present, the role of grain boundary is gaining importance. Most of the studies are carried out through simulation methods. Nanocrystalline materials are normally porous in nature. In this work, the effect of porosity on the deformation behaviour (under compression) of nanocrystalline FCC metal (Pd) is investigated by molecular dynamics simulation. The results show that modulus of elasticity and flow stress of the nanocrystalline FCC metals decreases with increase in porosity. The atoms in grains adjacent to the pores are observed to move towards the void space. For a relaxed nanocrystalline metal, the grain boundary atoms are under compressive stress. read less USED (high confidence) H. Zhan and Y. Gu, “Surface effects on the dual-mode vibration of 〈1 1 0〉 silver nanowires with different cross-sections,” Journal of Physics D: Applied Physics. 2012. link Times cited: 20 Abstract: Dual-mode vibration of nanowires (NWs) has been reported exp… read moreAbstract: Dual-mode vibration of nanowires (NWs) has been reported experimentally through actuation of the NW at its resonance frequency, which is expected to open up a variety of new modalities for nanoelectromechanical systems that could operate in the nonlinear regime. In this work, we utilize large-scale molecular dynamics simulations to investigate the dual-mode vibration of 〈1 1 0〉 Ag NWs with triangular, rhombic and truncated rhombic cross-sections. By incorporating the generalized Young–Laplace equation into the Euler–Bernoulli beam theory, the influence of surface effects on the dual-mode vibration is studied. Due to the different lattice spacings in the principal axes of inertia of the {1 1 0} atomic layers, the NW is also modelled as a discrete system to reveal the influence from such a specific atomic arrangement. It is found that the 〈1 1 0〉 Ag NW will be under a dual-mode vibration if the actuation direction deviates from the two principal axes of inertia. The predictions of the two first mode natural frequencies by the classical beam model appear underestimated compared with the MD results, which are found to be enhanced by the discrete model. Particularly, the predictions by the beam theory with the contribution of surface effects are uniformly larger than the classical beam model, which exhibit better agreement with MD results for a larger cross-sectional size. However, for ultrathin NWs, current consideration of surface effects still experiences certain inaccuracy. In all, for all different cross-sections, the inclusion of surface effects is found to reduce the difference between the two first mode natural frequencies. This trend is observed to be consistent with MD results. This study provides a first comprehensive investigation on the dual-mode vibration of 〈1 1 0〉 oriented Ag NWs, which is supposed to benefit the applications of NWs that act as a resonating beam. read less USED (high confidence) T. Frolov, D. Olmsted, M. Asta, and Y. Mishin, “Structural phase transformations in metallic grain boundaries,” Nature Communications. 2012. link Times cited: 314 USED (high confidence) H. Zhan, Y. T. Gu, P. Yarlagadda, and C. Yan, “Influence of pre-exsiting surface defects on the vibrational properties of Ag nanowires,” 2012 12th IEEE International Conference on Nanotechnology (IEEE-NANO). 2012. link Times cited: 1 Abstract: Large-scale molecular dynamics simulations are performed to … read moreAbstract: Large-scale molecular dynamics simulations are performed to characterize the effects of pre-existing surface defects on the vibrational properties of Ag nanowires. It is found that the first order natural frequency of the nanowire appears insensitive to different surface defects, indicating a defect insensitivity property of the nanowire's Young's modulus. In the meanwhile, an increase of the quality (Q)-factor is observed due to the presence of defects. Particular, a beat phenomenon is observed for the nanowire with the presence of a surface edge defect, which is driven by a single actuation. It is concluded that different surface defects could act as an effective mean to tune the vibrational properties of nanowires. This study sheds lights on the better understanding of nanowire's mechanical performance when surface defects are presented, which would benefit the development of nanowire-based devices. read less USED (high confidence) R. Durairaj et al., “Pressure free sintering of silver nanoparticles to silver substrate using weakly binding ligands,” 2012 12th IEEE International Conference on Nanotechnology (IEEE-NANO). 2012. link Times cited: 7 Abstract: The use of a weakly binding ligand to facilitate sintering b… read moreAbstract: The use of a weakly binding ligand to facilitate sintering between particles and a planar substrate in the absence of pressure and at low homologous temperature has been explored. Ag nanoparticles in the 5-15 nm range suspended in water and stabilized by a BH4 complex were dropped onto a polished Ag substrate heated to 333 K. The Ag particles sintered to each other and to the substrate to form a largely pore free system. A molecular dynamics simulation is used to understand the theoretical limits of pressure free sintering and practical implications for adhesive systems based on Ag nanoparticle suspensions are discussed. read less USED (high confidence) M. Barisik and A. Beskok, “Boundary treatment effects on molecular dynamics simulations of interface thermal resistance,” J. Comput. Phys. 2012. link Times cited: 51 USED (high confidence) N. Abdolrahim, I. Mastorakos, and H. Zbib, “Precipitate strengthening in nanostructured metallic material composites,” Philosophical Magazine Letters. 2012. link Times cited: 17 Abstract: Nanostructured metallic material (NMM) composites are a new … read moreAbstract: Nanostructured metallic material (NMM) composites are a new class of materials that exhibit high structural stability, mechanical strength, high ductility, toughness and resistance to fracture and fatigue; these properties suggest that these materials can play a leading role in the future micromechanical devices. However, before those materials are put into service in any significant applications, many important fundamental issues remain to be understood. Among them, is the question of the strengthening of NMM using second phase particles and if the addition of precipitates will strengthen the structures in the same manner as in bulk crystalline solids. This issue is addressed in this work by performing molecular dynamics simulations on NMM with precipitates of various sizes and comparing the results with the same structure without precipitates. In this view, Cu/Nb bilayer thin films with spherical Nb particles inside the Cu layer were examined using molecular dynamics simulations and show a significant improvement on their mechanical behavior, compared to similar structures without particles. Furthermore, an analytical model is developed that explains the strengthening behavior of an NMM that has precipitates inside one layer. The theoretical results show a qualitative agreement with the finding of the atomistic simulations. read less USED (high confidence) S. Kim, H. Chew, E. Chason, V. Shenoy, and K.-S. Kim, “Nanoscale mechanisms of surface stress and morphology evolution in FCC metals under noble-gas ion bombardments,” Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2012. link Times cited: 16 Abstract: Here, we uncover three new nanoplasticity mechanisms, operat… read moreAbstract: Here, we uncover three new nanoplasticity mechanisms, operating in highly stressed interstitial-rich regions in face-centred-cubic (FCC) metals, which are particularly important in understanding evolution of surface stress and morphology of a FCC metal under low-energy noble-gas ion bombardments. The first mechanism is the configurational motion of self-interstitials in subsonic scattering during ion bombardments. We have derived a stability criterion of self-interstitial scattering during ion embedding, which consistently predicts the possibility of vacancy- and interstitial-rich double-layer formation for various ion bombardments. The second mechanism is the growth by gliding of prismatic dislocation loops (PDLs) in a highly stressed interstitial-rich zone. This mechanism allows certain prismatic dislocations with their Burgers vectors parallel to the surface to grow in subway-glide mode (SGM) during ion bombardment. The SGM growth creates a large population of nanometre-sized prismatic dislocations beneath the surface. The third mechanism is the Burgers vector switching of a PDL that leads to unstable eruption of adatom islands during certain ion bombardments of FCC metals. We have also derived the driving force and kinetics for the growth by gliding of prismatic dislocations in an interstitial-rich environment as well as the criterion for Burgers vector switching, which consistently clarifies previously unexplainable experimental observations. read less USED (high confidence) Z. Chen, S. Jiang, and Y. Gan, “The ‘Inverse Hall-Petch’ effect on the impact response of single crystal copper,” Acta Mechanica Sinica. 2012. link Times cited: 5 USED (high confidence) L. Karkina and L. Yakovenkova, “Dislocation core structure and deformation behavior of Ti3Al,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 9 Abstract: Using molecular dynamics simulations with the embedded atom … read moreAbstract: Using molecular dynamics simulations with the embedded atom method we calculate the core structure of a superdislocations in prism and basal planes and 2c + a superdislocations in pyramid planes. An analysis of the structure of the cores showed that the core is planar in the prismatic plane and nonplanar for screw superpartial dislocations in the basal plane. It is shown that the glissile 2c + a superdislocations have higher energy than the configurations of dislocation barriers. The influence of the core structure of superdislocations on the orientation dependence of the deformation behavior of Ti3Al is discussed. read less USED (high confidence) Y.-bo Guo, T. Xu, and M. Li, “Atomistic calculation of internal stress in nanoscale polycrystalline materials,” Philosophical Magazine. 2012. link Times cited: 18 Abstract: Internal stress in polycrystalline materials is an intrinsic… read moreAbstract: Internal stress in polycrystalline materials is an intrinsic attribute of the microstructure that affects a broad range of material properties. It is usually acquired through experiment in conjunction with continuum mechanics modelling, but its determination at nanometre and submicron scales is extremely difficult. Here, we report a bottom-up approach using atomistic calculation. We obtain the internal stress in polycrystalline copper with nanosized grains by first computing the stress associated with each atom and then sorting the stress into those associated with different self-equilibrating length scales, i.e. sample scale and grain cell, which gives type I, II and III residual stresses, respectively. The result shows highly non-uniform internal stress distribution; the internal stress depends sensitively on grain size and the grain shape anisotropy. Statistical distributions of the internal stresses, along with the means and variance, are calculated as a function of the mean grain size and temperature. The implementation of this work in assisting the interpretation of experimental results and predicting material properties is discussed. read less USED (high confidence) H. Shang and W. Wang, “Hypervelocity impact properties of graphene armor via molecular dynamics simulations.” 2012. link Times cited: 4 Abstract: Hypervelocity impact properties of two different graphene ar… read moreAbstract: Hypervelocity impact properties of two different graphene armor systems are investigated using molecular dynamics simulations. One system is the so-called spaced armor which consists of a number of graphene plates spaced certain distance apart. Its response under normal impact of a spherical projectile is studied, focusing on the effect of the number of graphene monolayers per plate (denoted by n ) on the penetration resistance of the armor. We find that under normal impact by a spherical projectile the penetration resistance increases with decreasing number of monolayers per plate ( n ), and the best penetration resistance is achieved in the system with one graphene layer for each plate. Note that the monolayers in all the simulated multilayer graphene plates were AB-stacked. The second system being studied is the laminated copper/graphene composites with the graphene layers inside copper, on impact or back surface, or on both the impact and back surfaces. The simulation results show that under normal impact by a spherical projectile the laminated copper/graphene composite has much higher penetration resistance than the monolithic copper plate. The best efficiency is achieved when the graphene layers are on both the impact and back surfaces. read less USED (high confidence) Z. Chen, S. Jiang, and Y. Gan, “The ‘Inverse Hall-Petch’ effect on the impact response of single crystal copper,” Acta Mechanica Sinica. 2012. link Times cited: 0 USED (high confidence) C. Yan and Q. Zhang, “Study on low-energy sputtering near the threshold energy by molecular dynamics simulations,” AIP Advances. 2012. link Times cited: 11 Abstract: Using molecular dynamics simulation, we have studied the low… read moreAbstract: Using molecular dynamics simulation, we have studied the low-energy sputtering at the energies near the sputtering threshold. Different projectile-target combinations of noble metal atoms (Cu, Ag, Au, Ni, Pd, and Pt) are simulated in the range of incident energy from 0.1 to 200 eV. It is found that the threshold energies for sputtering are different for the cases of M1 < M2 and M1 ≥ M2, where M1 and M2 are atomic mass of projectile and target atoms, respectively. The sputtering yields are found to have a linear dependence on the reduced incident energy, but the dependence behaviors are different for the both cases. The two new formulas are suggested to describe the energy dependences of the both cases by fitting the simulation results with the determined threshold energies. With the study on the energy dependences of sticking probabilities and traces of the projectiles and recoils, we propose two different mechanisms to describe the sputtering behavior of low-energy atoms near the threshold energy for the cases of M1 < M2 and M1 ≥ M2, respectively.Using molecular dynamics simulation, we have studied the low-energy sputtering at the energies near the sputtering threshold. Different projectile-target combinations of noble metal atoms (Cu, Ag, Au, Ni, Pd, and Pt) are simulated in the range of incident energy from 0.1 to 200 eV. It is found that the threshold energies for sputtering are different for the cases of M1 < M2 and M1 ≥ M2, where M1 and M2 are atomic mass of projectile and target atoms, respectively. The sputtering yields are found to have a linear dependence on the reduced incident energy, but the dependence behaviors are different for the both cases. The two new formulas are suggested to describe the energy dependences of the both cases by fitting the simulation results with the determined threshold energies. With the study on the energy dependences of sticking probabilities and traces of the projectiles and recoils, we propose two different mechanisms to describe the sputtering behavior of low-energy atoms near the threshold energy for the... read less USED (high confidence) M. Mariscal, O. A. Oviedo, and E. Leiva, “On the selective decoration of facets in metallic nanoparticles,” Journal of Materials Research. 2012. link Times cited: 13 Abstract: This work presents key modeling aspects that are central to … read moreAbstract: This work presents key modeling aspects that are central to the manipulation of the decoration of metallic nanoparticles by a thin shell of a metal of different chemical nature. The concept of underpotential deposition is generalized to nanoparticles. An all-atom model, taking into account many-body interactions by means of the embedded atom potential, was used to represent nanoparticles of different sizes and atomic adsorbates on them. A full set of state-of-the-art computer simulations are performed for a model system, showing that selective decoration of facets is possible. The trends observed in the present work are in good qualitative agreement with experimental data reported very recently. read less USED (high confidence) Y. Gan and J. K. Chen, “Nonequilibrium phase change in gold films induced by ultrafast laser heating.,” Optics letters. 2012. link Times cited: 8 Abstract: Ultrafast laser-induced melting in a gold thin film is simul… read moreAbstract: Ultrafast laser-induced melting in a gold thin film is simulated by an integrated continuum-atomistic method with the extended Drude model for dynamic optical properties. The local order parameter of atoms is used to identify solid and liquid regions. It is shown that the film is superheated in the early nonequilibrium stage and the melted region grows very quickly with a very high rate of melting up to ∼13,300 m/s. It is also found that the continuum approach could significantly underestimate the ultrafast phase-change response, and temperature-dependent optical properties should be considered in atomic-level modeling for ultrafast laser heating. read less USED (high confidence) X. Shi, Q. Yin, and Y. Wei, “A theoretical analysis of the surface dependent binding, peeling and folding of graphene on single crystal copper,” Carbon. 2012. link Times cited: 47 USED (high confidence) C. C. Asuquo and R. Bowles, “Molecular Dynamics Simulations of Competitive Freezing in Gold Nanoclusters,” Journal of Physical Chemistry C. 2012. link Times cited: 14 Abstract: Molecular dynamics simulations are used to study the competi… read moreAbstract: Molecular dynamics simulations are used to study the competitive freezing of icosahedral, decahedral, polydecahedral, and face-centered cubic structures in gold nanoclusters for a range of cluster sizes and temperatures. Measuring the probability of observing each cluster type in an ensemble of freezing events, along with the overall rate at which liquid drops freeze to any structure, allows us to calculate the rate of formation for each structure. An analysis of the freezing trajectories of the clusters reveals that both the icosahedron and the decahedron structures nucleate through the initial formation of a 5-fold symmetric cap. The icosahedron then continues to grow through the sequential growth of neighboring tetrahedral subunits, while the decahedral cluster grows out from the cap along the central 5-fold axis of symmetry. read less USED (high confidence) Z. Chen and S. Jiang, “Recent Findings About Combined Size And RateEffects On Material Properties,” WIT Transactions on the Built Environment. 2012. link Times cited: 0 Abstract: The need for modeling and simulating multiscale structural r… read moreAbstract: The need for modeling and simulating multiscale structural responses to extreme loading conditions has brought about the challenging tasks of bridging different spatial and temporal scales within a unified framework. Based on the available experimental and computational capabilities, a simple approach has been proposed to formulate a hyper-surface in both spatial and temporal domains to predict combined specimen size and loading rate effects on the material properties. A systematic investigation has been performed over the last several years to understand the combined size, rate and thermal effects on the properties and deformation patterns of representative materials with different nanostructures and under various types of loading conditions. In this presentation, recent findings are presented about combined size and rate effects on the material properties of single crystal copper nanobeams under impact loading, with a focus on the link between the inverse Hall-Petch phenomenon and classical Hall-Petch phenomenon. It appears from the preliminary results that the inverse Hall-Petch behavior in single crystal materials is mainly due to the formation and evolution of disordered atoms as compared with the physics behind the inverse Hall-Petch behavior in nanocrystalline materials. read less USED (high confidence) H. Zhan and Y. T. Gu, “A fundamental numerical and theoretical study for the vibrational properties of nanowires,” Journal of Applied Physics. 2012. link Times cited: 36 Abstract: Based on the molecular dynamics (MD) simulation and the clas… read moreAbstract: Based on the molecular dynamics (MD) simulation and the classical Euler-Bernoulli beam theory, a fundamental study of the vibrational performance of the Ag nanowire (NW) is carried out. A comprehensive analysis of the quality (Q)-factor, natural frequency, beat vibration, as well as high vibration mode is presented. Two excitation approaches, i.e., velocity excitation and displacement excitation, have been successfully implemented to achieve the vibration of NWs. Upon these two kinds of excitations, consistent results are obtained, i.e., the increase of the initial excitation amplitude will lead to a decrease to the Q-factor, and moderate plastic deformation could increase the first natural frequency. Meanwhile, the beat vibration driven by a single relatively large excitation or two uniform excitations in both two lateral directions is observed. It is concluded that the nonlinear changing trend of external energy magnitude does not necessarily mean a non-constant Q-factor. In particular, the first order ... read less USED (high confidence) Z. Chen, S. Jiang, Y. Gan, Y. S. Oloriegbe, T. Sewell, and D. Thompson, “Size effects on the impact response of copper nanobeams,” Journal of Applied Physics. 2012. link Times cited: 19 Abstract: Molecular dynamics simulations are performed to study size e… read moreAbstract: Molecular dynamics simulations are performed to study size effects on the impact response of copper nanobeam targets subjected to impacts by copper nanobeam flyers with different impact velocities. It is found that the Hugoniot response is size-dependent, while the aspect ratio – that is, the ratio of flyer and target nanobeam heights – has a small effect. It is also observed that the propagation speed of a disordering front generated at the impact surface is close to the shock wave speed initially, but decreases as dislocations form. The thermal gradient in the target is mainly due to the quasi-temperature difference (transient spatial localization of kinetic energy) between hexagonal-close-packed atoms and face-centered-cubic atoms. The findings for the impact stress, defect evolution, and quasi-temperature could be useful for better understanding the responses of nanosystems to extreme loading conditions. read less USED (high confidence) M. Mees, G. Pourtois, E. Neyts, B. Thijsse, and A. Stesmans, “Uniform-acceptance force-bias Monte Carlo method with time scale to study solid-state diffusion,” Physical Review B. 2012. link Times cited: 52 Abstract: Monte Carlo (MC) methods have a long-standing history as par… read moreAbstract: Monte Carlo (MC) methods have a long-standing history as partners of molecular dynamics (MD) to simulate the evolution of materials at the atomic scale. Among these techniques, the uniform-acceptance force-bias Monte Carlo (UFMC) method [ G. Dereli Mol. Simul. 8 351 (1992)] has recently attracted attention [ M. Timonova et al. Phys. Rev. B 81 144107 (2010)] thanks to its apparent capacity of being able to simulate physical processes in a reduced number of iterations compared to classical MD methods. The origin of this efficiency remains, however, unclear. In this work we derive a UFMC method starting from basic thermodynamic principles, which leads to an intuitive and unambiguous formalism. The approach includes a statistically relevant time step per Monte Carlo iteration, showing a significant speed-up compared to MD simulations. This time-stamped force-bias Monte Carlo (tfMC) formalism is tested on both simple one-dimensional and three-dimensional systems. Both test-cases give excellent results in agreement with analytical solutions and literature reports. The inclusion of a time scale, the simplicity of the method, and the enhancement of the time step compared to classical MD methods make this method very appealing for studying the dynamics of many-particle systems. read less USED (high confidence) X. Liu, Z. Liu, and Y. Wei, “Nanoscale Friction Behavior of the Ni-Film/Substrate System Under Scratching Using MD Simulation,” Tribology Letters. 2012. link Times cited: 25 USED (high confidence) M. Toimil-Molares, L. Röntzsch, W. Sigle, K. Heinig, C. Trautmann, and R. Neumann, “Pipetting Nanowires: In Situ Visualization of Solid‐State Nanowire‐to‐Nanoparticle Transformation Driven by Surface Diffusion‐Mediated Capillarity,” Advanced Functional Materials. 2012. link Times cited: 12 Abstract: The most interesting applications of nanotubes include their… read moreAbstract: The most interesting applications of nanotubes include their use as storage media for atoms and small molecules, as nanoscale capsules for chemical reactions, and as nanopipettes for material delivery. The geometrical transformation of metallic copper nanowires, confined in graphitic coating, into crystalline nanoparticles of up to tenfold increased diameter is reported. In situ transmission electron microscopy images at 500 °C, recorded as movies, provide an exceptional real‐time visualization of Cu draining out of the carbon coating. The solid content of the carbon tube is effectively evacuated over micrometer distances towards the open end, transforming each nanowire into a single monocrystalline, facetted Cu particle. Kinetic Monte Carlo simulations propose that this dramatic morphological transformation is driven by surface diffusion of Cu atoms along the wire/tube interface, thus minimizing the total free energy of the system. read less USED (high confidence) Y. Shim and J. Amar, “Shape transitions in strained Cu islands on Ni(100): kinetics versus energetics.,” Physical review letters. 2012. link Times cited: 7 Abstract: We examine the ramified islands observed in submonolayer Cu/… read moreAbstract: We examine the ramified islands observed in submonolayer Cu/Ni(100) growth. Our results indicate that the strain-energy contribution to the dependence of island energy on shape is surprisingly weak. In contrast, our accelerated dynamics simulations indicate that unexpected concerted popout processes occurring at step edges may be responsible. Kinetic Monte Carlo (KMC) simulations which include these processes produce island shapes which are very similar to those observed in experiment. These results suggest that the shape transition is of kinetic origin but is strongly mediated by strain. read less USED (high confidence) D. Zhu, H. Zhang, and D. Li, “Molecular dynamics simulation of Bauschinger’s effect in deformed copper single crystal in different strain ranges,” Journal of Applied Physics. 2011. link Times cited: 12 Abstract: Plastic pre-strain may decrease the yield strength of metall… read moreAbstract: Plastic pre-strain may decrease the yield strength of metallic materials when stressed in the opposite direction, known as Bauschinger’s effect, which could considerably influence the performance of the materials. However, various processes on microscopic level associated with the Bauschinger’s effect are still not clear. In this study, defect generation, movement and annihilation in single crystal copper during cyclic tension-compression loading processes were simulated using the molecular dynamics method. It was observed that Bauschinger’s effect was asymmetrical and the strain hardening was more profound during compression. After plastic compression, the tensile fracture strain was increased. The absorbed energy was self-adjusted during the tension-compression cycles and kept relatively stable during the loading cycles. Efforts were made to understand the mechanisms responsible for these phenomena. read less USED (high confidence) R. González, G. García, R. Ramírez, and M. Kiwi, “Role of the substrate dynamics: Iron clusters deposited on an iron slab,” Surface Science. 2011. link Times cited: 3 USED (high confidence) Z. Pereira and E. Silva, “Cold Welding of Gold and Silver Nanowires: A Molecular Dynamics Study,” Journal of Physical Chemistry C. 2011. link Times cited: 92 Abstract: Recently a new possibility of welding was experimentally sho… read moreAbstract: Recently a new possibility of welding was experimentally shown in the case of gold nanowires (NWs) at ambient temperatures, without need of additional heat and with low pressures, called cold weldi... read less USED (high confidence) S.-P. Kim et al., “Anisotropic rearrangement of the substrate atoms during Ar bombardment on Pd(0 0 1) surface,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2011. link Times cited: 3 USED (high confidence) Y. Lu, J. Song, J. Huang, and J. Lou, “Fracture of Sub‐20nm Ultrathin Gold Nanowires,” Advanced Functional Materials. 2011. link Times cited: 113 Abstract: Fracture of metals at the nanoscale and corresponding failur… read moreAbstract: Fracture of metals at the nanoscale and corresponding failure mechanisms have recently attracted considerable interest. However, quantitative in situ fracture experiments of nanoscale metals are rarely reported. Here it is shown that, under uni‐axial tensile loading, single crystalline ultrathin gold nanowires may fracture in two modes, displaying distinctively different fracture morphologies and ductility. In situ high resolution transmission electron microscopy (HRTEM) studies suggest that the unexpected brittle‐like fracture was closely related to the observed twin structures, which is very different from surface dislocation nucleation/propagation mediated mechanism in ductile fracture mode. Molecular dynamics (MD) simulations further reveal the processes of shear‐induced twin formation and damage initiation at the twin structure/free surface interface, confirming the experimentally observed differences in fracture morphology and ductility. Finally, a fracture criterion based on competition between twin formation and surface dislocation nucleation/propagation as a function of misalignment angle is discussed. read less USED (high confidence) L. Kong, “Phonon dispersion measured directly from molecular dynamics simulations,” Comput. Phys. Commun. 2011. link Times cited: 215 USED (high confidence) W. Zhu and W. Yang, “Molecular dynamics study of configuration and stability of vacancy clusters in fcc Ag,” Philosophical Magazine. 2011. link Times cited: 2 Abstract: Vacancies may agglomerate to form vacancy Frank loops of dif… read moreAbstract: Vacancies may agglomerate to form vacancy Frank loops of different shapes, as observed by transmission electron microscopy in quenched and irradiated fcc metals. The dynamics for the dissociation of vacancy Frank loops and the subsequent evolution of defect nanostructures were explored by means of the molecular dynamics method and displayed by the local crystalline order method. Frank loops of different initial shapes were found to transform to a variety of defect nanostructures: triangle to stacking fault tetrahedra, equilateral hexagon to quasi-heptahedron, and scalene hexagon to various intermediate structures depending on the length of the short side. The formation energies for vacancy Frank loops of different geometries are introduced to categorize various final configurations. Crystallographic analysis and elasticity calculations were performed to elucidate the transform mechanisms in fcc Ag. read less USED (high confidence) G. Nandipati, A. Kara, S. I. Shah, and T. Rahman, “Off-lattice pattern recognition scheme for kinetic Monte Carlo simulations,” J. Comput. Phys. 2011. link Times cited: 19 USED (high confidence) C. Mi, D. Buttry, P. Sharma, and D. Kouris, “Atomistic insights into dislocation-based mechanisms of void growth and coalescence,” Journal of The Mechanics and Physics of Solids. 2011. link Times cited: 62 USED (high confidence) J. Deng et al., “Accelerated molecular dynamics and equation-free methods for simulating diffusion in solids.” 2011. link Times cited: 1 Abstract: Many of the most important and hardest-to-solve problems rel… read moreAbstract: Many of the most important and hardest-to-solve problems related to the synthesis, performance, and aging of materials involve diffusion through the material or along surfaces and interfaces. These diffusion processes are driven by motions at the atomic scale, but traditional atomistic simulation methods such as molecular dynamics are limited to very short timescales on the order of the atomic vibration period (less than a picosecond), while macroscale diffusion takes place over timescales many orders of magnitude larger. We have completed an LDRD project with the goal of developing and implementing new simulation tools to overcome this timescale problem. In particular, we have focused on two main classes of methods: accelerated molecular dynamics methods that seek to extend the timescale attainable in atomistic simulations, and so-called 'equation-free' methods that combine a fine scale atomistic description of a system with a slower, coarse scale description in order to project the system forward over long times. read less USED (high confidence) L. Fan et al., “Step driven competitive epitaxial and self-limited growth of graphene on copper surface,” AIP Advances. 2011. link Times cited: 20 Abstract: The existence of surface steps was found to have significant… read moreAbstract: The existence of surface steps was found to have significant function and influence on the growth of graphene on copper via chemical vapor deposition. The two typical growth modes involved were found to be influenced by the step morphologies on copper surface, which led to our proposed step driven competitive growth mechanism. We also discovered a protective role of graphene in preserving steps on copper surface. Our results showed that wide and high steps promoted epitaxial growth and yielded multilayer graphene domains with regular shape, while dense and low steps favored self-limited growth and led to large-area monolayer graphene films. We have demonstrated that controllable growth of graphene domains of specific shape and large-area continuous graphene films are feasible. read less USED (high confidence) G. Schröder-Turk et al., “Minkowski Tensor Shape Analysis of Cellular, Granular and Porous Structures,” Advanced Materials. 2011. link Times cited: 143 Abstract: Predicting physical properties of materials with spatially c… read moreAbstract: Predicting physical properties of materials with spatially complex structures is one of the most challenging problems in material science. One key to a better understanding of such materials is the geometric characterization of their spatial structure. Minkowski tensors are tensorial shape indices that allow quantitative characterization of the anisotropy of complex materials and are particularly well suited for developing structure‐property relationships for tensor‐valued or orientation‐dependent physical properties. They are fundamental shape indices, in some sense being the simplest generalization of the concepts of volume, surface and integral curvatures to tensor‐valued quantities. Minkowski tensors are based on a solid mathematical foundation provided by integral and stochastic geometry, and are endowed with strong robustness and completeness theorems. The versatile definition of Minkowski tensors applies widely to different types of morphologies, including ordered and disordered structures. Fast linear‐time algorithms are available for their computation. This article provides a practical overview of the different uses of Minkowski tensors to extract quantitative physically‐relevant spatial structure information from experimental and simulated data, both in 2D and 3D. Applications are presented that quantify (a) alignment of co‐polymer films by an electric field imaged by surface force microscopy; (b) local cell anisotropy of spherical bead pack models for granular matter and of closed‐cell liquid foam models; (c) surface orientation in open‐cell solid foams studied by X‐ray tomography; and (d) defect densities and locations in molecular dynamics simulations of crystalline copper. read less USED (high confidence) C. Wu, D. A. Thomas, Z. Lin, and L. Zhigilei, “Runaway lattice-mismatched interface in an atomistic simulation of femtosecond laser irradiation of Ag film–Cu substrate system,” Applied Physics A. 2011. link Times cited: 39 USED (high confidence) V. K. Sutrakar and D. Mahapatra, “Universal Stability and Temperature Dependent Phase Transformation in Group VIIIB–IB Transition Metal FCC Nanowires,” Journal of Physical Chemistry C. 2011. link Times cited: 18 Abstract: Atomistic simulation of Ag, Al, Au, Cu, Ni, Pd, and Pt FCC m… read moreAbstract: Atomistic simulation of Ag, Al, Au, Cu, Ni, Pd, and Pt FCC metallic nanowires show a universal FCC → HCP phase transformation below a critical cross-sectional size, which is reported for the first time in this paper. The newly observed HCP structure is also confirmed from previous experimental results. Above the critical cross-sectional size, initial ⟨100⟩/{100} FCC metallic nanowires are found to be metastable. External thermal heating shows the transformation of metastable ⟨100⟩/{100} FCC nanowires into ⟨110⟩/{111} stable configuration. Size dependent metastability/instability is also correlated with initial residual stresses of the nanowire by use of molecular static simulation using the conjugant gradient method at a temperature of 0 K. It is found that a smaller cross-sectional dimension of an initial FCC nanowire shows instability due to higher initial residual stresses, and the nanowire is transformed into the novel HCP structure. The initial residual stress shows reduction with an increase in the ... read less USED (high confidence) L. Yang, Y. Zhang, and J. Chen, “Molecular dynamics simulation of deposition of nickel nanocluster on copper surface,” Journal of Nanoparticle Research. 2011. link Times cited: 2 USED (high confidence) S. S. Hayat, I. Ahmad, and M. A. Choudhry, “Diffusion of Six-Atom Cu Islands on Cu(111) and Ag(111),” Chinese Physics Letters. 2011. link Times cited: 3 Abstract: Diffusion of Cu hexamer islands on Cu(111) and Ag(111) is st… read moreAbstract: Diffusion of Cu hexamer islands on Cu(111) and Ag(111) is studied using a molecular dynamics simulation technique with many-body potentials obtained from the embedded atom method. Simulations are carried out at temperatures 300, 500 and 700 K, showing that shape-changing multiple-atom processes are more helpful for the diffusion rather than concerted motion of islands. Arrhenius plots of the diffusion coefficients provide effective energy barrier values of 161.29 ± 5 meV for Cu(111) and 179.34 ± 5 meV for Ag(111) surfaces. At 700K, one popup atom among island atoms is observed with correlative changes in the position and shape of the lower-layer adatoms. read less USED (high confidence) K. Edgar, S. Hendy, D. Schebarchov, and R. Tilley, “Reverse capillary action in carbon nanotubes: sucking metal nanoparticles out of nanotubes.,” Small. 2011. link Times cited: 13 USED (high confidence) B. R. Gel’chinskii, A. Vorontsov, A. E. Korenchenko, and L. Leont’ev, “Multiscale computer modeling of gas-phase synthesis of metal nanoparticles,” Doklady Physical Chemistry. 2011. link Times cited: 4 USED (high confidence) S. Rawat, M. Warrier, S. Chaturvedi, and V. Chavan, “Temperature sensitivity of void nucleation and growth parameters for single crystal copper: a molecular dynamics study,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 26 Abstract: The effect of temperature on the void nucleation and growth … read moreAbstract: The effect of temperature on the void nucleation and growth is studied using the molecular dynamics (MD) code LAMMPS (Large-Scale Atomic/Molecular Massively Parallel Simulator). Single crystal copper is triaxially expanded at 5 × 109 s−1 strain rate keeping the temperature constant. It is shown that the nucleation and growth of voids at these atomistic scales follows a macroscopic nucleation and growth (NAG) model. As the temperature increases there is a steady decrease in the nucleation and growth thresholds. As the melting point of copper is approached, a double-dip in the pressure–time profile is observed. Analysis of this double-dip shows that the first minimum corresponds to the disappearance of the long-range order due to the creation of stacking faults and the system no longer has a FCC structure. There is no nucleation of voids at this juncture. The second minimum corresponds to the nucleation and incipient growth of voids. We present the sensitivity of NAG parameters to temperature and the analysis of double-dip in the pressure–time profile for single crystal copper at 1250 K. read less USED (high confidence) S. A. Paz, E. Leiva, J. Jellinek, and M. Mariscal, “Properties of rotating nanoalloys formed by cluster collision: a computer simulation study.,” The Journal of chemical physics. 2011. link Times cited: 20 Abstract: Results of dynamical simulations of collision-induced format… read moreAbstract: Results of dynamical simulations of collision-induced formation and properties of bimetallic nanoparticles are presented and analyzed. The analysis includes the effects of the collision energy and the impact parameter. For nonzero impact parameters, the formed (in many cases Janus-type) nanoparticles are rotating. The energy of the rotating nanoparticles is decomposed into the rotational and vibrational components, and the structural effects of these components are analyzed. Comparison is made with the case of the corresponding homoatomic systems, formed by collision of nanoparticles with the same elemental composition. read less USED (high confidence) Y. Liu et al., “Surfactant-induced postsynthetic modulation of Pd nanoparticle crystallinity.,” Nano letters. 2011. link Times cited: 92 Abstract: Modulation of Pd nanoparticle (NP) crystallinity is achieved… read moreAbstract: Modulation of Pd nanoparticle (NP) crystallinity is achieved by switching the surfactants of different binding strengths. Pd NPs synthesized in the presence of weak binding surfactants such as oleylamine possess polyhedral shapes and a polycrystalline nature. When oleylamine is substituted by trioctylphosphine, a much stronger binding surfactant, the particles become spherical and their crystallinity decreases significantly. Moreover, the Pd NPs reconvert their polycrystalline structure when the surfactant is switched back to oleylamine. Through control experiments and molecular dynamics simulation, we propose that this unusual nanocrystallinity transition induced by surfactant exchange was resulted from a counterbalance between the surfactant binding energy and the nanocrystal adhesive energy. The findings represent a novel postsynthetic approach to tailoring the structure and corresponding functional performance of nanomaterials. read less USED (high confidence) D. Schebarchov and S. Hendy, “Uptake and withdrawal of droplets from carbon nanotubes.,” Nanoscale. 2011. link Times cited: 24 Abstract: We give an account of recent studies of droplet uptake and w… read moreAbstract: We give an account of recent studies of droplet uptake and withdrawal from carbon nanotubes using simple theoretical arguments and molecular dynamics simulations. Firstly, the thermodynamics of droplet uptake and release is considered and tested via simulation. We show that the Laplace pressure acting on a droplet assists capillary uptake, allowing sufficiently small non-wetting droplets to be absorbed. We then demonstrate how the uptake and release of droplets of non-wetting fluids can be exploited for the use of carbon nanotubes as nanopipettes. Finally, we extend the Lucas-Washburn model to deal with the dynamics of droplet capillary uptake, and again test this by comparison with molecular dynamics simulations. read less USED (high confidence) W. Kim and M. Cho, “Surface effect on the self-equilibrium state and size-dependent elasticity of FCC thin films,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 19 Abstract: On the {1 1 0} solid surface, surface stresses of two perpen… read moreAbstract: On the {1 1 0} solid surface, surface stresses of two perpendicular directions, such as ⟨1 0 0⟩ and ⟨1 1 0⟩, will differ. These different surface stresses cause different equilibrium strains in the ⟨1 0 0⟩ and ⟨1 1 0⟩ directions on a {1 1 0} thin film. In this paper, an analytical formulation is presented to predict the equilibrium strains of orthotropic thin films with {1 0 0}, {1 1 1} or {1 1 0} surfaces. This formulation is composed of the elastic constants of bulk materials and surface parameters such as surface stresses and surface elastic tensors. Transition metals with FCC structures (Cu, Ag, Au, Ni, Pd and Pt) are used in this study, along with a surface relaxation model that calculates surface parameters using atomistic calculations. Molecular dynamics simulations are performed and compared with the results of this suggested method, and both results show good agreement. In addition, size-dependent elastic constants are calculated analytically from the predicted equilibrium state. read less USED (high confidence) Shang-Da 尚达 Chen 陈, T. Wang 王, De-Li 德立 Zheng 郑, and Yi-Chun 益春 Zhou 周, “The effect of deposition temperature on the intermixing and microstructure of Fe/Ni thin film,” Chinese Physics B. 2010. link Times cited: 1 Abstract: The physical vapour deposition of Ni atoms on α-Fe(001) surf… read moreAbstract: The physical vapour deposition of Ni atoms on α-Fe(001) surface under different deposition temperatures were simulated by molecular dynamics to study the intermixing and microstructure of the interfacial region. The results indicate that Ni atoms hardly penetrate into Fe substrate while Fe atoms easily diffuse into Ni deposition layers. The thickness of the intermixing region is temperature-dependent, with high temperatures yielding larger thicknesses. The deposited layers are mainly composed of amorphous phase due to the abnormal deposition behaviour of Ni and Fe. In the deposited Ni-rich phase, the relatively stable metallic compound B2 structured FeNi is found under high deposition temperature conditions. read less USED (high confidence) B. An et al., “Atomic structure of interface between monolayer Pd film and Ni(111) determined by low-energy electron diffraction and scanning tunneling microscopy,” Journal of Applied Physics. 2010. link Times cited: 3 Abstract: The atomic structure of Pd ultrathin films grown on Ni(111) … read moreAbstract: The atomic structure of Pd ultrathin films grown on Ni(111) at 300 K is investigated by low-energy electron diffraction and scanning tunneling microscopy. It is determined atomically that the growth of monolayer Pd films leads to a periodic arrangement of triangular misfit dislocation loops in the underlying Ni(111) surface, resulting in a triangular superstructure on the monolayer Pd surface. The triangular dislocation loops tend to align at an angle of about 5° from the Ni atom row, owing to a slight rotation of the Pd films with respect to the Ni substrate, and appear as a moirelike superstructure on the multilayer Pd surfaces. Atomistic simulations indicate that the slight rotation of monolayer Pd films and the formation of misfit dislocation loops in the Ni surface minimize the Pd–Ni interface energy. read less USED (high confidence) Y.-S. Geng and X.-wu Du, “The Research of Data Mining Based Sales Forecast,” 2010 International Conference on Multimedia Technology. 2010. link Times cited: 5 Abstract: The accuracy of sales forecast has great impact on manufactu… read moreAbstract: The accuracy of sales forecast has great impact on manufacturing and sales. With the quick development of the society, it is really hard for traditional forecast system to meet new demand in dealing very large amount of data and sales forecasting with high accuracy. Obviously, data mining technology is the key to solve those problems. In this article, the first two parts are a brief introduction about the concept of Sales Forecast, then an introduction of two popular Extract-Transform-Load (ETL) tools and a comprehensive analysis of four most often used forecast method are given in the following parts. Finally, based on analysis and combined with a variety of technical advantages, we propose a new sales forecast system model based on data mining and Grey-Markov prediction model is used as an example to illustrate its working principle and to verify its feasibility theoretically. read less USED (high confidence) Z.-J. Wang, C. Liu, Z. Li, and T.-Y. Zhang, “Size-dependent elastic properties of Au nanowires under bending and tension—Surfaces versus core nonlinearity,” Journal of Applied Physics. 2010. link Times cited: 37 Abstract: The present work investigates contributions from surfaces an… read moreAbstract: The present work investigates contributions from surfaces and core nonlinearity to the size-dependent elastic properties of nanowires under bending and tension-compression. When a nanowire is formed by removing it from its parent bulk material, relaxation occurs inevitably because of high energy of newly created surfaces or born high surface eigenstress. Relaxation-induced initial strain could be large and nonlinear, which causes the size-dependent elastic properties of nanowires. If relaxation-induced initial strain is small and linear, the size-dependent elastic properties of nanowires are caused by surface Young’s modulus. The eigenstress model for surface stress of solids {Zhang et al. [Phys. Rev. B 81, 195427 (2010)]} is further developed here for nanowires under bending and tension-compression. The developed eigenstress model leads to general scaling laws for nanowires under bending and tension-compression. In the scaling laws, there are the surface and nonlinearity factors, which measure quantitati... read less USED (high confidence) M. Mendelev, M. J. Rahman, J. Hoyt, and M. Asta, “Molecular-dynamics study of solid–liquid interface migration in fcc metals,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 85 Abstract: In order to establish a link between various structural and … read moreAbstract: In order to establish a link between various structural and kinetic properties of metals and the crystal–melt interfacial mobility, free-solidification molecular-dynamics simulations have been performed for a total of nine embedded atom method interatomic potentials describing pure Al, Cu and Ni. To fully explore the space of materials properties three new potentials have been developed. The new potentials are based on a previous description of Al, but in each case the liquid structure, the melting point and/or the latent heat are varied considerably. The kinetic coefficient, μ, for all systems has been compared with several theoretical predictions. It is found that at temperatures close to the melting point the magnitude of μ correlates well with the value of the diffusion coefficient in the liquid. read less USED (high confidence) H. S. Park, “A multiscale finite element method for the dynamic analysis of surface‐dominated nanomaterials,” International Journal for Numerical Methods in Engineering. 2010. link Times cited: 5 Abstract: The purpose of this article is to present a multiscale finit… read moreAbstract: The purpose of this article is to present a multiscale finite element method that captures nanoscale surface stress effects on the dynamic mechanical behavior of nanomaterials. The method is based upon arguments from crystal elasticity, i.e. the Cauchy–Born rule, but significantly extends the capability of the standard Cauchy–Born rule by accounting for critical nanoscale surface stress effects, which are well known to have a significant effect on the mechanics of crystalline nanostructures. We present the governing equations of motion including surface stress effects, and demonstrate that the methodology is general and thus enables simulations of both metallic and semiconducting nanostructures. The numerical examples on elastic wave propagation and dynamic tensile and compressive loading show the ability of the proposed approach to capture surface stress effects on the dynamic behavior of both metallic and semiconducting nanowires, and demonstrate the advantages of the proposed approach in studying the deformation of nanostructures at strain rates and time scales that are inaccessible to classical molecular dynamics simulations. Copyright © 2010 John Wiley & Sons, Ltd. read less USED (high confidence) R. Jones, J. Templeton, G. Wagner, D. Olmsted, and N. A. Modine, “Electron transport enhanced molecular dynamics for metals and semi‐metals,” International Journal for Numerical Methods in Engineering. 2010. link Times cited: 21 Abstract: In this work we extend classical molecular dynamics by coupl… read moreAbstract: In this work we extend classical molecular dynamics by coupling it with an electron transport model known as the two temperature model. This energy balance between free electrons and phonons was first proposed in 1956 by Kaganov et al. but has recently been utilized as a framework for coupling molecular dynamics to a continuum description of electron transport. Using finite element domain decomposition techniques from our previous work as a basis, we develop a coupling scheme that preserves energy and has local control of temperature and energy flux via a Gaussian isokinetic thermostat. Unlike the previous work on this subject, we employ an efficient, implicit time integrator for the fast electron transport which enables larger stable time steps than the explicit schemes commonly used. A number of example simulations are given that validate the method, including Joule heating of a copper nanowire and laser excitation of a suspended carbon nanotube with its ends embedded in a conducting substrate. Published in 2010 by John Wiley & Sons, Ltd. read less USED (high confidence) X. Tian, J. Cui, B. Li, and M. Xiang, “Investigations on the deformation behavior of polycrystalline Cu nanowires and some factors affecting the modulus and yield strength,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 6 Abstract: This paper consists of two parts. In the first part, the def… read moreAbstract: This paper consists of two parts. In the first part, the deformation behavior of polycrystalline Cu nanowires under tension, bending and torsion is studied by using molecular dynamics simulations. The results show that both free surfaces and grain boundaries can control the plastic deformation. In detail, for polycrystalline Cu nanowires under tension, the stress-assisted grain growth caused by atomic diffusion and grain boundary migration is found, which is thought to be the reason for necking. Then under the bending effect, full dislocations and deformation twins of polycrystalline Cu nanowires are found in the grains. Moreover, the twin boundaries act as obstacles to dislocation motion. Finally, full dislocations and fivefold deformation twins are detected in polycrystalline Cu nanowires in the torsional state. These phenomena are in good agreement with the experimental observations of Liao et al (2005 Appl. Phys. Lett. 86 103112). The second part investigates the effects of sample shape and crystallographic structure on the modulus and yield strength under tension, bending and torsion. The results demonstrate that the modulus (in particular the bending and torsion modulus) can be significantly influenced by both the effects; however, remarkable difference in yield strength can merely be caused by different crystallographic structures (here, different crystallographic structures refer to polycrystalline and single-crystalline structures). read less USED (high confidence) G. Nandipati, A. Kara, S. I. Shah, and T. Rahman, “Island-size selectivity during 2D Ag island coarsening on Ag(111),” Journal of Physics: Condensed Matter. 2010. link Times cited: 5 Abstract: We report on the early stages of submonolayer Ag island coar… read moreAbstract: We report on the early stages of submonolayer Ag island coarsening on the Ag(111) surface carried out using kinetic Monte Carlo simulations for several temperatures. Our simulations were performed using a very large database of processes identified by their local environment and whose activation barriers were calculated using the semi-empirical interaction potentials based on the embedded-atom method. We find that during the early stages, coarsening proceeds as a sequence of selected island sizes, creating peaks and valleys in the island-size distribution. This island-size selectivity is independent of initial conditions and results from the formation of kinetically stable islands for certain sizes as dictated by the relative energetics of edge atom detachment/attachment processes together with the large activation barrier for kink detachment. Our results indicate that by tuning the growth temperature it is possible to enhance the island-size selectivity read less USED (high confidence) T.-Y. Zhang, Z.-J. Wang, and W. Chan, “Eigenstress model for surface stress of solids,” Physical Review B. 2010. link Times cited: 64 Abstract: Solid films are taken here as a typical example to study sur… read moreAbstract: Solid films are taken here as a typical example to study surface stress of solids. When a thin film is created by removing it from a bulk material, relaxation occurs inevitably because of high energy of newly created surfaces. We separate the relaxation process into normal and parallel relaxations and propose an eigenstress model to calculate the strain energy released during parallel relaxation. After parallel relaxation, a tensile (or compressive) surface eigenstress causes a compressive (or tensile) initial strain in the thin film with respect to its bulk lattice. Due to initial deformation, surface energy density and surface stress are both dependent on the film thickness, whereas surface elastic constants are independent of the film thickness. The nominal modulus of a thin film is determined by nonlinear elastic properties of its core and surfaces with initial strain. A tensile (or compressive) eigenstress makes the nominal modulus of a thin film larger (or smaller), resulting in the thinner, the harder (or softer) elastic behavior in thin films. Atomistic simulations on Au (001), Cu (001), Si (001), and diamond (001) thin films verify the developed eigenstress model. read less USED (high confidence) F. Liu et al., “Dynamics diffusion behaviors of Pd small clusters on a Pd(1 1 1) surface,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 10 Abstract: Using molecular dynamics, nudged elastic band and modified a… read moreAbstract: Using molecular dynamics, nudged elastic band and modified analytic embedded atom methods, the self-diffusion dynamics properties of palladium atomic clusters up to seven atoms on the Pd (1 1 1) surface have been studied at temperatures ranging from 300 to 1000 K. The simulation time varies from 20 to 75 ns according to the cluster sizes and the temperature ranges. The heptamer and trimer are more stable than the other neighboring clusters. The diffusion coefficients of the clusters are derived from the mean square displacement of the cluster's mass-center, and the diffusion prefactors D0 and activation energies Ea are derived from the Arrhenius relation. The activation energy of the clusters increases with the increasing atom number in the clusters, especially for Pd6 to Pd7. The analysis of trajectories shows the noncompact clusters diffuse by the local diffusion mechanism but the compact clusters diffuse mainly by the whole gliding mechanism, and some static energy barriers of the diffusion modes are calculated. From Pd2 to Pd6, the prefactors are in the range of the standard value 10−3 cm2 s−1, and the prefactor of Pd7 cluster is 2 orders of magnitude greater than that of the single Pd adatom because of a large number of nonequivalent diffusion processes. The heptamer can be the nucleus in the room temperature range according to nucleation theory. read less USED (high confidence) A. Gerlich, L. Yue, P. Mendez, and H. Zhang, “Plastic deformation of nanocrystalline aluminum at high temperatures and strain rate,” Acta Materialia. 2010. link Times cited: 29 USED (high confidence) J. Crill, X. Ji, D. Irving, D. Brenner, and C. Padgett, “Atomic and multi-scale modeling of non-equilibrium dynamics at metal–metal contacts,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 12 Abstract: A coarse graining method that introduces Joule heating and i… read moreAbstract: A coarse graining method that introduces Joule heating and improves heat transport in a classical molecular dynamics simulation is reviewed, and two example sets of simulations, opening of gold–gold nano-asperity contacts and nano-asperity sliding at loaded copper–aluminum interfaces are discussed. For the gold contact, dislocations nucleate from the edges of where the asperity contacts the substrates and move along the close-packed planes, resulting in stacking faults that form two subsurface Thompson tetrahedra. For a null voltage, a nanowire with a diameter much smaller than the initial contact area is created when the two tetrahedra are completed, and as the wire yields the partial dislocations retreat to the surface. Opening with Joule heating enhances dislocation mobility and intransient subsurface plasticity. Constant current simulations show melting and boiling of the nanowires depending on the voltage cap. Sliding of an aluminum asperity on copper with a null voltage shows dislocation formation in the copper and aluminum, while heating from an applied voltage eliminates damage in the copper. Sliding with a copper asperity enhances plastic damage in the copper substrate compared with the aluminum asperity, while Joule heating enhances aluminum pile-up in front of the copper asperity due to plowing. read less USED (high confidence) M. Aminpour, O. Trushin, and T. Rahman, “Effect of misfit dislocation on surface diffusion,” Bulletin of the American Physical Society. 2010. link Times cited: 8 Abstract: We apply molecular dynamics and molecular static methods to … read moreAbstract: We apply molecular dynamics and molecular static methods to study the effect of misfit dislocations on adatom diffusion in close proximity to the dislocation core in heteroepitaxial systems, using many-body interaction potentials. Our system consists of several layers (three–seven) of Cu on top of a Ni(111) substrate. The misfit dislocations are created with the core located at the interface between the Cu film and the Ni substrate, using the repulsive biased potential method described earlier. We find that presence of the defect under the surface strongly affects the adatom trajectory, creating anisotropy in atomic diffusion, independent of the thickness of the Cu film. We also calculate the potential energy surface available to the adatom and compare the energy barriers for adatom diffusion in the proximity of the core region and on the defect-free surface. read less USED (high confidence) J. Song, M. Soare, and W. A. Curtin, “Testing continuum concepts for hydrogen embrittlement in metals using atomistics,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 44 Abstract: Hydrogen embrittlement is a pervasive mode of degradation in… read moreAbstract: Hydrogen embrittlement is a pervasive mode of degradation in many metallic systems that can occur via several mechanisms. Here, the competition between dislocation emission and cleavage at a crack tip is evaluated in the presence of H. At this level, embrittlement is predicted when the critical stress intensity required for emission rises above that needed for cleavage, eliminating crack tip plasticity and blunting as toughening mechanisms. Continuum predictions for emission and cleavage are made using computed generalized stacking fault energies and surface energies in a model Ni–H system, and embrittlement is predicted at a critical H concentration. An atomistic model is then used to investigate actual crack tip behavior in the presence of controlled arrays of H atoms around the crack tip. The continuum models are accurate at low H concentrations, below the embrittlement point, but at higher H concentrations the models deviate from the atomistic behavior due to alternative dislocation emission modes. Additional H configurations are investigated to understand controlling features of the emission process. In no cases does crack propagation occur in preference to dislocation emission in geometries where emission is possible, indicating that embrittlement can be more complicated than envisioned by the basic brittle–ductile transition. read less USED (high confidence) C. Pignedoli, T. Laino, M. Treier, R. Fasel, and D. Passerone, “A simple approach for describing metal-supported cyclohexaphenylene dehydrogenation,” The European Physical Journal B. 2010. link Times cited: 10 USED (high confidence) K. Zhao, L. Fan, and C. Chen, “Multiaxial behavior of nanoporous single crystal copper: a molecular dynamics study,” Acta Mechanica Solida Sinica. 2009. link Times cited: 24 USED (high confidence) K. Kolluri, M. Gungor, and D. Maroudas, “Atomistic analysis of strain relaxation in [11¯0]-oriented biaxially strained ultrathin copper films,” Journal of Applied Physics. 2009. link Times cited: 2 Abstract: Results are reported of a systematic atomic-scale computatio… read moreAbstract: Results are reported of a systematic atomic-scale computational analysis of strain relaxation mechanisms and the associated defect dynamics in nanometer-scale thin or ultrathin Cu films that are subjected to a broad range of biaxial tensile strains. The films contain pre-existing voids and the film planes are oriented normal to the [11¯0] crystallographic direction. The analysis is based on isothermal-isostrain molecular-dynamics simulations according to an embedded-atom-method parameterization for Cu and employing multimillion-atom slab supercells. In addition to an initial elastic response for an applied biaxial strain level e<2%, our analysis reveals three regimes in the thin-film mechanical response as e increases. For 2%≤e≤6%, biaxial strain relaxation is dominated by emission and propagation of dislocations (plastic flow) from the surface of the void accompanied by ductile void growth. For 6% read less USED (high confidence) Y. Sun and E. Webb, “The atomistic mechanism of high temperature contact line advancement: results from molecular dynamics simulations,” Journal of Physics: Condensed Matter. 2009. link Times cited: 13 Abstract: Atomic scale phenomena driving contact line advancement duri… read moreAbstract: Atomic scale phenomena driving contact line advancement during the wetting of a solid by a liquid are investigated via molecular dynamics simulations of Ag(l) drops spreading on Ni substrates. For the homologous temperature ∼5% above melting for Ag, essentially non-reactive wetting is observed with relatively high spreading velocity. Analyzing atomic positions with time, including computing flow fields, permits investigation of atomic scale transport mechanisms associated with advancement of the contact line. Delivery of material to the contact line occurs preferentially along the liquid/vapor interface. Ag(l) atoms transported along the liquid/vapor interface become new droplet edge material, effectively displacing existing edge material. Evidence is also shown of a prominent transport and flow mechanism more typically associated with the molecular kinetic theory of spreading: some portion of Ag(l) atoms move along the solid/liquid interface to eventually occupy the contact line region. Selected atomic trajectories are shown to illustrate atoms moving with the contact line, detaching and re-attaching at sites along the solid/liquid interface. However, this latter solid/liquid interface transport mechanism contributed a lower percentage of new material to the advancing contact line compared to the liquid/vapor interface transport mechanism. Features of the AgNi system that may contribute to the dominance of a liquid/vapor interface transport mechanism are highlighted, including a relatively low liquid/vapor surface tension. read less USED (high confidence) Y. Kulkarni and R. Asaro, “Are some nanotwinned fcc metals optimal for strength, ductility and grain stability?,” Acta Materialia. 2009. link Times cited: 81 USED (high confidence) H. H. Wu, A. Signor, and D. Trinkle, “Island Shape Controls Magic-Size Effect for Heteroepitaxial Diffusion,” arXiv: Materials Science. 2009. link Times cited: 11 Abstract: Lattice mismatch of Cu on Ag(111) produces fast diffusion fo… read moreAbstract: Lattice mismatch of Cu on Ag(111) produces fast diffusion for special "magic sizes" of islands. A size- and shape-dependent reptation mechanism is responsible for low diffusion barriers. Initiating the reptation mechanism requires a suitable island shape, a property not considered in previous studies of 1D island chains and 2D closed-shell islands. Shape determines the dominant diffusion mechanism and leads to multiple clearly identifiable magic-size trends for diffusion depending on the number of atoms whose bonds are shortened during diffusion. read less USED (high confidence) B. Morrow and A. Striolo, “Assessing how metal–carbon interactions affect the structure of supported platinum nanoparticles,” Molecular Simulation. 2009. link Times cited: 17 Abstract: Towards understanding the effect of solid supports on the ca… read moreAbstract: Towards understanding the effect of solid supports on the catalytic activity of supported metal nanoparticles, all-atom molecular dynamics (MD) simulations are often conducted. However, these calculations are hampered by the uncertainty related to describing metal–support interactions (typically described as Lennard-Jones (LJ) potentials) at the atomic length scale. Ab initio electron-structure calculations are expected to refine such calculations by providing better estimates for the metal–support pair interaction potential. In the case of platinum nanoparticles supported on graphite, recent ab initio results suggest the correct energetic LJ parameter should be about four times that used in previous simulation studies from our group, as well as from others. Stimulated by these findings, MD simulations have been used here to investigate the effect of the magnitude of the metal–carbon interaction on the structure of supported metal nanoparticles. The LJ potential was used to model the metal–carbon interactions, and the embedded-atom method was used to model the metal–metal interactions. The morphology of platinum nanoparticles of 130, 249 and 498 atoms supported on graphite and various bundles of carbon nanotubes (CNTs) was studied. For the larger nanoparticle it was found that, although the details of platinum–carbon interactions are important for correctly capturing the morphological details, the morphology of the support is the primary factor that determines such features. Platinum–carbon interactions affect more significantly the results obtained for metal nanoparticles supported by CNT bundles. In this case, we found that the deviations become significant for small supported nanoparticles, as well as for nanoparticles of any size supported on CNTs of small diameter. read less USED (high confidence) I. Shabib and R. Miller, “A molecular dynamics study of twin width, grain size and temperature effects on the toughness of 2D-columnar nanotwinned copper,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 33 Abstract: The introduction of twin boundaries (TBs) within nanocrystal… read moreAbstract: The introduction of twin boundaries (TBs) within nanocrystalline grains has given scientists an opportunity to enhance mechanical properties that are usually mutually exclusive: strength and ductility. This research is focused on developing a complete understanding of the influences of twin width, grain size and temperature on the deformation characteristics and properties of nanotwinned Cu by large-scale molecular dynamics simulations. Simulation results have shown that a material's toughness can be enhanced by introducing nanotwins, and the enhancement is more pronounced for the higher twin density structures and at lower temperatures. Nanotwinned grains are found to be highly anisotropic in their plastic response; ductile along TBs but strong across them. A random polycrystalline sample gains toughness through the combined response of variously oriented grains. At extremely low temperature, toughness values are elevated further due to depressed dislocation activities inside the grains. The study has also revealed that, unlike twin width refinement, grain size refinement may not always yield superior properties, and may deteriorate material toughness. read less USED (high confidence) S. Borisova, G. Rusina, S. Eremeev, and E. Chulkov, “Vibrational properties of small cobalt clusters on the Cu(111) surface,” Physics of the Solid State. 2009. link Times cited: 4 USED (high confidence) J. Ferrón, R. Miranda, and J. Miguel, “Atomic jumps during surface diffusion,” Physical Review B. 2009. link Times cited: 14 Abstract: Fil: Ferron, Julio. Consejo Nacional de Investigaciones Cien… read moreAbstract: Fil: Ferron, Julio. Consejo Nacional de Investigaciones Cientificas y Tecnicas. Centro Cientifico Tecnologico Conicet - Santa Fe. Instituto de Desarrollo Tecnologico para la Industria Quimica. Universidad Nacional del Litoral. Instituto de Desarrollo Tecnologico para la Industria Quimica; Argentina read less USED (high confidence) L. Kong, G. Bartels, C. Campañá, C. Denniston, and M. Müser, “Implementation of Green’s function molecular dynamics: An extension to LAMMPS,” Comput. Phys. Commun. 2009. link Times cited: 56 USED (high confidence) K. Mccarty et al., “How metal films de-wet substrates—identifying the kinetic pathways and energetic driving forces,” New Journal of Physics. 2009. link Times cited: 25 Abstract: We study how single-crystal chromium films of uniform thickn… read moreAbstract: We study how single-crystal chromium films of uniform thickness on W(110) substrates are converted to arrays of three-dimensional (3D) Cr islands during annealing. We use low-energy electron microscopy (LEEM) to directly observe a kinetic pathway that produces trenches that expose the wetting layer. Adjacent film steps move simultaneously uphill and downhill relative to the staircase of atomic steps on the substrate. This step motion thickens the film regions where steps advance. Where film steps retract, the film thins, eventually exposing the stable wetting layer. Since our analysis shows that thick Cr films have a lattice constant close to bulk Cr, we propose that surface and interface stress provide a possible driving force for the observed morphological instability. Atomistic simulations and analytic elastic models show that surface and interface stress can cause a dependence of film energy on thickness that leads to an instability to simultaneous thinning and thickening. We observe that de-wetting is also initiated at bunches of substrate steps in two other systems, Ag/W(110) and Ag/Ru(0001). We additionally describe how Cr films are converted into patterns of unidirectional stripes as the trenches that expose the wetting layer lengthen along the W[001] direction. Finally, we observe how 3D Cr islands form directly during film growth at elevated temperature. The Cr mesas (wedges) form as Cr film steps advance down the staircase of substrate steps, another example of the critical role that substrate steps play in 3D island formation. read less USED (high confidence) J. Wang, R. Hoagland, and A. Misra, “Room-temperature dislocation climb in metallic interfaces,” Applied Physics Letters. 2009. link Times cited: 69 Abstract: Using atomistic simulations, we show that dislocations effic… read moreAbstract: Using atomistic simulations, we show that dislocations efficiently climb in metallic interfaces, such as Cu–Nb, through absorption and emission of vacancies and a counter diffusion of Cu atoms in the interfacial plane. The efficiency of dislocation climb is ascribed to the high vacancy concentration of 0.05 in the interfacial plane, the low formation energy of 0.12 eV with respect to removal or insertion of Cu atoms, and the low kinetic barrier of 0.10 eV for vacancy migration. Dislocation climb facilitates reactions of interfacial dislocations and enables interfaces to be in the equilibrium state with respect to concentrations of point defects. read less USED (high confidence) O. Trushin, J. Jalkanen, E. Granato, S. Ying, and T. Ala‐Nissila, “Atomistic studies of strain relaxation in heteroepitaxial systems,” Journal of Physics: Condensed Matter. 2009. link Times cited: 9 Abstract: We present a review of recent theoretical studies of differe… read moreAbstract: We present a review of recent theoretical studies of different atomistic mechanisms of strain relaxation in heteroepitaxial systems. We explore these systems in two and three dimensions using different semi-empirical interatomic potentials of Lennard-Jones and many-body embedded atom model type. In all cases we use a universal molecular static method for generating minimum energy paths for transitions from the coherent epitaxial (defect free) state to the state containing an isolated defect (localized or extended). This is followed by a systematic search for the minimum energy configuration as well as self-organization in the case of a periodic array of islands. In this way we are able to understand many general features of the atomic mechanisms and energetics of strain relaxation in these systems. Finally, for the special case of Pd/Cu(100) and Cu/Pd(100) heteroepitaxy we also use conventional molecular dynamics simulation techniques to compare the compressively and tensilely strained cases. The results for this case are in good agreement with the existing experimental data. read less USED (high confidence) K. Fichthorn, R. Miron, Y. Wang, and Y. Tiwary, “Accelerated molecular dynamics simulation of thin-film growth with the bond-boost method,” Journal of Physics: Condensed Matter. 2009. link Times cited: 29 Abstract: We review the bond-boost method for accelerated molecular dy… read moreAbstract: We review the bond-boost method for accelerated molecular dynamics (MD) simulation and we demonstrate its application to kinetic phenomena relevant to thin-film growth. To illustrate various aspects of the method, three case studies are presented. We first illustrate aspects of the bond-boost method in studies of the diffusion of Cu atoms on Cu(001). In these studies, Cu interactions are described using a semi-empirical embedded-atom method potential. We recently extended the bond-boost method to perform accelerated ab initio MD (AIMD) simulations and we present results from preliminary studies in which we applied the bond-boost method in AIMD to uncover diffusion mechanisms of Al adatoms on Al(110). Finally, a problem inherent to many rare-event simulation methods is the ‘small-barrier problem’, in which the system resides in a group of states connected by small energy barriers and separated from the rest of phase space by large barriers. We developed the state-bridging bond-boost method to address this problem and we discuss its application for studying the diffusion of Co clusters on Cu(001). We discuss the outlook for future applications of the bond-boost method in materials simulation. read less USED (high confidence) F. Ma, S. L. Ma, K. Xu, and P. Chu, “Asymmetrical reorientation of bimetallic core–shell nanowires,” Nanotechnology. 2009. link Times cited: 3 Abstract: The reorientation mechanism of core–shell nanowires is inves… read moreAbstract: The reorientation mechanism of core–shell nanowires is investigated and our theoretical studies reveal the significance of the structural configuration. In nanowires which have a larger lattice in the core region than in the shell, for example, Au-core and Pd-shell, the surface stress and interfacial stress may synergistically cause them to reorient spontaneously, but they can revert back to the original state upon an appropriate tensile loading. In contrast, the misfit interface is detrimental to spontaneous reorientation in nanowires which have a smaller lattice in the core than in the shell such as the Pd-core and Au-shell structure, but uniaxial tensile loading causes the nanowires to transform in another way. This asymmetrical reorientation is caused by the different intrinsic stress as well as distinctive slipping characteristics, namely partial slipping and perfect slipping in the compressive and tensile processes. read less USED (high confidence) Y. Yan, J. Zhang, T. Sun, W. Fei, Y. Liang, and S. Dong, “Nanobending of nanowires: A molecular dynamics study,” Applied Physics Letters. 2008. link Times cited: 12 Abstract: Three-dimensional molecular dynamics simulations of the nano… read moreAbstract: Three-dimensional molecular dynamics simulations of the nanobending of copper nanowires are carried out. Simulation results show that the loading and unloading cycles of the nanobending test can reveal the full spectrum of the nanowires’ mechanical properties. Up-tensile and bottom-compressive features have been observed along with the neck zone formation. Amorphous region formation is the mechanism of fracture and final breakage. The measured elastic modulus and yield stress are 49 and 7.6 GPa, respectively. Moreover, the effect of the adhesion on the nanobending process is revealed. read less USED (high confidence) A. Kara, O. Trushin, H. Yildirim, and T. Rahman, “Off-lattice self-learning kinetic Monte Carlo: application to 2D cluster diffusion on the fcc(111) surface,” Journal of Physics: Condensed Matter. 2008. link Times cited: 47 Abstract: We report developments of the kinetic Monte Carlo (KMC) meth… read moreAbstract: We report developments of the kinetic Monte Carlo (KMC) method with improved accuracy and increased versatility for the description of atomic diffusivity on metal surfaces. The on-lattice constraint built into our recently proposed self-learning KMC (SLKMC) (Trushin et al 2005 Phys. Rev. B 72 115401) is released, leaving atoms free to occupy ‘off-lattice’ positions to accommodate several processes responsible for small-cluster diffusion, periphery atom motion and heteroepitaxial growth. This technique combines the ideas embedded in the SLKMC method with a new pattern-recognition scheme fitted to an off-lattice model in which relative atomic positions are used to characterize and store configurations. Application of a combination of the ‘drag’ and the repulsive bias potential (RBP) methods for saddle point searches allows the treatment of concerted cluster, and multiple- and single-atom, motions on an equal footing. This tandem approach has helped reveal several new atomic mechanisms which contribute to cluster migration. We present applications of this off-lattice SLKMC to the diffusion of 2D islands of Cu (containing 2–30 atoms) on Cu and Ag(111), using the interatomic potential from the embedded-atom method. For the hetero-system Cu/Ag(111), this technique has uncovered mechanisms involving concerted motions such as shear, breathing and commensurate–incommensurate occupancies. Although the technique introduces complexities in storage and retrieval, it does not introduce noticeable extra computational cost. read less USED (high confidence) H. S. Park and P. Klein, “Surface stress effects on the resonant properties of metal nanowires: The importance of finite deformation kinematics and the impact of the residual surface stress,” Journal of The Mechanics and Physics of Solids. 2008. link Times cited: 150 USED (high confidence) E. Bleda, X. Gao, and M. Daw, “Calculations of diffusion in FCC binary alloys using on-the-fly kinetic Monte Carlo,” Computational Materials Science. 2008. link Times cited: 8 USED (high confidence) H. S. Park, “Strain sensing through the resonant properties of deformed metal nanowires,” Journal of Applied Physics. 2008. link Times cited: 13 Abstract: In this article, we study the potential of gold nanowires as… read moreAbstract: In this article, we study the potential of gold nanowires as resonant nanoscale strain sensors. The sensing ability of the nanowires is determined by calculating the variations in resonant frequency that occur due to applied uniaxial tensile and compressive strain. The resonant frequencies are obtained using the surface Cauchy–Born model, which captures surface stress effects on the nanowires through a nonlinear continuum mechanics framework; due to the continuum formulation, the strain-dependent nanowire resonant frequencies are calculated through the solution of a standard finite element eigenvalue problem, where the coupled effects of the applied uniaxial strain and surface stress are naturally included through the finite element stiffness matrix. The nanowires are found to be more sensitive to compressive than tensile strain, with resonant frequency shifts around 200–400 MHz with the application of 1% tensile and compressive strain. In general, the strain sensitivity of the nanowires is found to incre... read less USED (high confidence) L. Wang, H. Zhang, and X. Deng, “Influence of defects on mechanical properties of bicrystal copper grain boundary interfaces,” Journal of Physics D: Applied Physics. 2008. link Times cited: 10 Abstract: Defects play a key role in the determination of material pro… read moreAbstract: Defects play a key role in the determination of material properties, especially at small scales. The influence of several kinds of defects (point vacancies, line vacancies and cracks) on the deformation and fracture characteristics of a planar copper grain boundary interface with a 45° lattice misorientation is explored using molecular dynamics simulations and the embedded-atom method. Both tensile and shear modes of interfacial separation are considered. The results show that the crystalline defects can have a strong influence on the interfacial behaviours. The sensitivity of the mechanical properties of the interface to a defect type may be different under tension than under shear. It is found that some defect topologies can improve certain properties (e.g. strength and fracture strain) of the bicrystal interface system. read less USED (high confidence) A. K. Subramaniyan and C. Sun, “Continuum interpretation of virial stress in molecular simulations,” International Journal of Solids and Structures. 2008. link Times cited: 388 USED (high confidence) K. Kolluri, M. Gungor, and D. Maroudas, “Atomic-scale analysis of defect dynamics and strain relaxation mechanisms in biaxially strained ultrathin films of face-centered cubic metals,” Journal of Applied Physics. 2008. link Times cited: 20 Abstract: We report results of a detailed systematic computational ana… read moreAbstract: We report results of a detailed systematic computational analysis of strain relaxation mechanisms and the associated defect dynamics in ultrathin, i.e., a few nanometers thick, Cu films subjected to a broad range of biaxial tensile strains. The analysis is based on isothermal-isostrain molecular-dynamics simulations of the response of Cu films that are oriented normal to the [111] crystallographic direction using an embedded-atom-method parametrization for Cu and multimillion-atom slab supercells. Our analysis reveals five regimes in the thin film’s mechanical response with increasing strain. Within the considered strain range, after an elastic response up to a biaxial strain level e=5.5%, the strain in the metallic thin film is relaxed by plastic deformation. At low levels of the applied biaxial strain above the yield strain (e∼6%), threading dislocation nucleation at the surface of the thin film in conjunction with vacancy cluster formation in the film leads eventually to the formation of voids that ext... read less USED (high confidence) S. S. Hayat, M. A. Choudhry, and S. A. Ahmad, “Effect of twin-boundaries on melting of aluminum,” Journal of Materials Science. 2008. link Times cited: 10 USED (high confidence) B. Morrow and A. Striolo, “Platinum nanoparticles on carbonaceous materials: the effect of support geometry on nanoparticle mobility, morphology, and melting,” Nanotechnology. 2008. link Times cited: 35 Abstract: Molecular dynamics simulations have been used to investigate… read moreAbstract: Molecular dynamics simulations have been used to investigate the morphology and mobility of platinum nanoparticles of various sizes supported by carbon materials. The embedded-atom method was used to model Pt–Pt interactions, and the Lennard-Jones potential was used to model the Pt–C interactions. The C atoms in the supports were held fixed during the simulations. The supports considered were a single graphite sheet and three bundles of carbon nanotubes. Three sizes of Pt nanoparticles were considered: 130 atoms, 249 atoms, and 498 atoms (Pt130, Pt249, and Pt498 respectively). It was found that for all three sizes, diffusion coefficients were approximately one order of magnitude higher for graphite-supported nanoparticles than for carbon nanotube-supported nanoparticles. In addition, increasing the size of the nanoparticle decreased its diffusion coefficient, with Pt130 having the highest and Pt498 the lowest diffusion coefficients. More interestingly, we found that for the Pt nanoparticles of all three sizes the diffusion coefficient increases as temperature increases, reaches a maximum at the melting temperature of the nanoparticle, and then decreases. The melting temperature was found to be strongly dependent on the particle size, but only slightly dependent on the features of the supports. While the size of the nanoparticle was seen to affect the particles’ mobility, it did not significantly affect their structure. The nanoparticles supported by graphite have density profiles that indicate a highly ordered, fcc-like structure, while the particles supported by carbon nanotubes have a more disordered structure. An order parameter confirms that the nanoparticles’ structure depends on the support morphology. read less USED (high confidence) Y. Wang, D. Ping, and D. X. Li, “The potential for ordered grain boundary sliding in nanocrystalline palladium,” Philosophical Magazine Letters. 2008. link Times cited: 2 Abstract: In nanocrystalline (nc) materials, the misfit of lattice str… read moreAbstract: In nanocrystalline (nc) materials, the misfit of lattice structures at the junctions of grain boundaries (GBs) generally increases local stresses that can be released by GB sliding. In this article, the sliding behaviors of the ordered Σ = 3 (111) GB (twin) and Σ = 11(113) GB in nc palladium have been investigated by means of high-resolution transmission electron microscopy observations and molecular statics calculations. It is found that the twin boundary (TB) can release either large stresses by sliding with a DSC-lattice vector of 1/6 or small stresses by sliding over an imperfect vector. The observed Σ = 11 GB image is exactly symmetrical from which we can clearly identify no GB sliding along the [332] direction, but we cannot determine a sliding component along the direction. The image simulations indicate that the latter is experimentally unidentifiable. read less USED (high confidence) O. Melikhova et al., “Simulation of Positron Annihilation Response to Mechanical Deformation of Nanostructured AgCo,” Acta Physica Polonica A. 2008. link Times cited: 1 Abstract: Department of Low Temperature Physics, Faculty of Mathematic… read moreAbstract: Department of Low Temperature Physics, Faculty of Mathematics and Physics Charles University in Prague V Holesovickach 2, CZ-180 00 Prague 8, Czech Republic Physique des Solides Irradiés et des Nanostructures CP234 Université Libre de Bruxelles, Bd du Triomphe, B-1050 Brussels, Belgium Faculty of Physics, University of Sofia 5 James Bourchier str., 1164 Sofia, Bulgaria Department of Experimental Nuclear Physics, Physical & Mechanical Faculty K-89, St. Petersburg State Polytechnical University 29 Polytekhnicheskaya str., 195251 St. Petersburg, Russia read less USED (high confidence) X. Liu, J. Wang, and S. Biner, “Hydrogen and self-interstitial interactions with edge dislocations in Ni: atomistic and elasticity comparisons,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 8 Abstract: The interactions of hydrogen interstitial and self-interstit… read moreAbstract: The interactions of hydrogen interstitial and self-interstitial with dissociated Shockley partial dislocations in fcc Ni were studied with embedded atom method calculations and the results were compared with those obtained from elasticity solutions. Such cross-correlations are important for efficient and accurate inclusion of the point defects into the dislocation dynamics simulations that are usually based on elasticity theories. The simulations were carried out using a dipole dislocation cell having periodic boundaries. The size effect, tetragonal distortions and the modulus effect were considered in the elasticity analysis. The results indicate that the elasticity solutions compare well with the atomistic results for the regions outside the Shockley partial cores, even though the interaction energies differed by approximately one order of magnitude for these two types of point defects. The range where the elasticity description of the interstitial–dislocation interaction breaks down was identified. In the self-interstitial case, the core reaction with the interstitial was observed, resulting in a larger core interaction range. read less USED (high confidence) C. Özdogan, M. Atis, and Z. B. Güvenç, “Surface modification by 1 keV ion impact: molecular dynamics study of an Ar+–Ni(1 0 0) collision system,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 1 Abstract: An Ar+–Ni(1 0 0) collision system at 1 keV impact energy was… read moreAbstract: An Ar+–Ni(1 0 0) collision system at 1 keV impact energy was investigated by using realistic isoenergetic molecular dynamics (MD) simulations. The sputtering process upon Ar+ ion impact and damage to the Ni(1 0 0) surface are examined in detail. Studying of high bombarding energy regions leads to the necessity of larger and thick enough slabs, otherwise incoming ions can easily pass through the slab; as a result, investigated physical properties may not be revealed. In addition the simulation time should be long enough to observe and to calculate a reliable macroscopic property such as sputtering yield that is addressed in this study. In order to preserve the total energy in the simulation at this collision energy a small time-step (0.1 fs) is used. We have made use of our developed linear scaling parallel MD program to overcome these demands. The Ni(1 0 0) slab is formed by 63700 atoms (122 Å × 122 Å × 44 Å) and the total observation time for each collision event is about 2.25 ps. Several properties such as penetration depths, angular and energy distributions of the reflected Ar and sputtered Ni atoms as well as dissociation time, embedded, scattering, sputtering patterns and geometries of the sputtered clusters are also reported, and the calculated sputtering yield is found to be in good agreement with the available experimental results. read less USED (high confidence) J. A. Sanchez and M. P. Mengüç, “Melting and vaporization of Cu and Ni films during electron-beam heating,” Journal of Applied Physics. 2008. link Times cited: 12 Abstract: In this paper, we present a study of the phase change proces… read moreAbstract: In this paper, we present a study of the phase change processes that take place in Cu and Ni films when they are heated with an electron-beam produced by field emission from an array of carbon nanotubes. A Monte Carlo method is adapted to solve the electron-beam Boltzmann transport equation to determine the electron distribution inside these materials. A hybrid approach is implemented to couple the two-temperature model with molecular dynamics simulations. We consider an analysis based on an order parameter and a radial distribution function to characterize the transition point at which the materials change phase. Slower electron diffusion in Ni produces more pronounced temperature gradients in both the electron system and the lattice, whereas the temperature rise throughout the Cu film is more uniform due to the faster electronic diffusion. We found that the phase change process is a combination of speed of the energy diffusion into the materials accompanied by a concentration of tensile stresses that co... read less USED (high confidence) H. Zhang, D. Du, and D. Srolovitz, “Effects of boundary inclination and boundary type on shear-driven grain boundary migration,” Philosophical Magazine. 2008. link Times cited: 54 Abstract: A series of molecular dynamics simulations was performed on … read moreAbstract: A series of molecular dynamics simulations was performed on a bicrystal to which a fixed shear rate was applied parallel to the boundary plane. Under some conditions, grain boundary motion is coupled to the relative tangential motion of the two grains. In order to investigate the generality of this type of coupled shear/boundary motion, simulations were performed for both special (low Σ) and general (non-Σ) [010] tilt boundaries over a wide range of grain boundary inclinations. The data point to the existence of two critical stresses: one for coupled shear/boundary motion and the other for grain boundary sliding. For the non-Σ boundaries, the critical stress for coupled shear/boundary motion is typically smaller than that for sliding; coupled shear/boundary motion occurs for all inclinations. For Σ5 boundaries, for which the critical stress is smaller and depends on boundary inclination, coupled shear/boundary motion occurs for some, but not all inclinations. read less USED (high confidence) F. Römer and T. Kraska, “Homogeneous nucleation and growth in supersaturated zinc vapor investigated by molecular dynamics simulation.,” The Journal of chemical physics. 2007. link Times cited: 37 Abstract: Homogeneous nucleation and growth of zinc from supersaturate… read moreAbstract: Homogeneous nucleation and growth of zinc from supersaturated vapor are investigated by nonequilibrium molecular dynamics simulations in the temperature range from 400 to 800 K and for a supersaturation ranging from log S=2 to 11. Argon is added to the vapor phase as carrier gas to remove the latent heat from the forming zinc clusters. A new parametrization of the embedded atom method for zinc is employed for the interaction potential model. The simulation data are analyzed with respect to the nucleation rates and the critical cluster sizes by two different methods, namely, the threshold method of Yasuoka and Matsumoto [J. Chem. Phys. 109, 8451 (1998)] and the mean first passage time method for nucleation by Wedekind et al. [J. Chem. Phys. 126, 134103 (2007)]. The nucleation rates obtained by these methods differ approximately by one order of magnitude. Classical nucleation theory fails to describe the simulation data as well as the experimental data. The size of the critical cluster obtained by the mean first passage time method is significantly larger than that obtained from the nucleation theorem. read less USED (high confidence) N. Pradeep, D.-in Kim, J. Grobelny, T. Hawa, B. Henz, and M. Zachariah, “Ductility at the nanoscale: Deformation and fracture of adhesive contacts using atomic force microscopy,” Applied Physics Letters. 2007. link Times cited: 16 Abstract: Fracture of nanosize contacts formed between spherical probe… read moreAbstract: Fracture of nanosize contacts formed between spherical probes and flat surfaces is studied using an atomic force microscope in an ultrahigh vacuum environment. Analysis of the observed deformation during the fracture process indicates significant material extensions for both gold and silica contacts. The separation process begins with an elastic deformation followed by plastic flow of material with atomic rearrangements close to the separation. Classical molecular dynamics studies show similarity between gold and silicon, materials that exhibit entirely different fracture behavior at macroscopic scale. This direct experimental evidence suggests that fracture at nanoscale occurs through a ductile process. read less USED (high confidence) B. H. Morrow and and A. Striolo, “Morphology and Diffusion Mechanism of Platinum Nanoparticles on Carbon Nanotube Bundles,” Journal of Physical Chemistry C. 2007. link Times cited: 42 Abstract: Molecular dynamics simulations have been used to investigate… read moreAbstract: Molecular dynamics simulations have been used to investigate the mobility and morphology of platinum nanoparticles supported on carbonaceous materials. The embedded-atom method was used to model Pt−Pt interactions. The Pt−C interactions were modeled using the Lennard−Jones potential. Carbon atoms were treated as rigid. The supports considered include a single graphite layer as well as carbon nanotubes, regarded as bundles. The goal of our work is to assess the effect of the substrate morphology on the properties of the metal nanoparticles. The properties of interest include the mobility and morphology of the supported nanoparticles. Our results show that the diffusion coefficients of Pt nanoparticles on carbon nanotube bundles are 1 order of magnitude lower than those of Pt nanoparticles supported by graphite. Density profiles, radial distribution functions, and average coordination numbers were calculated to study the structure of the supported nanoparticles. Platinum nanoparticles deposited on carbon na... read less USED (high confidence) K. E. Ryan, I. A. Wojciechowski, and B. Garrison, “Reaction dynamics following keV cluster bombardment,” Journal of Physical Chemistry C. 2007. link Times cited: 25 Abstract: The nature of the regions that are favorable for chemical re… read moreAbstract: The nature of the regions that are favorable for chemical reactions has been investigated for a pure amorphous water ice substrate following energetic bombardment by C60 and Au3 cluster projectiles using molecular dynamics (MD) simulations. The simulations show that both projectiles, especially C60, produce regions where a plethora of reactions occur at elevated densities indicating that multiple atoms or molecules are involved simultaneously in the reactions initiated by cluster bombardment. The total number of reacted water molecules is significantly less than the total sputtering yield, which confirms that both cluster projectiles are useful for molecular depth profiling experiments. read less USED (high confidence) M. Tschopp and D. McDowell, “Structures and energies of Σ 3 asymmetric tilt grain boundaries in copper and aluminium,” Philosophical Magazine. 2007. link Times cited: 224 Abstract: The objective of this research is to use atomistic simulatio… read moreAbstract: The objective of this research is to use atomistic simulations to investigate the energy and structure of symmetric and asymmetric Σ3 ⟨110⟩ tilt grain boundaries. A nonlinear conjugate gradient algorithm was employed along with an embedded atom method potential for Cu and Al to generate the equilibrium 0 K grain boundary structures. A total of 25 ⟨110⟩ grain boundary structures were explored to identify the various equilibrium and metastable structures. Simulation results show that the Σ3 asymmetric tilt grain boundaries in the ⟨110⟩ system are composed of only structural units of the two Σ3 symmetric tilt grain boundaries. The energies for the Σ3 grain boundaries are similar to previous experimental and calculated grain boundary energies. A structural unit and faceting model for Σ3 asymmetric tilt grain boundaries fits all of the calculated asymmetric grain boundary structures. The significance of these results is that the structural unit and facet description of all Σ3 asymmetric tilt grain boundaries may be predicted from the structural units of the Σ3 coherent twin and incoherent twin boundaries for both Cu and Al. read less USED (high confidence) C. M. Doelling, T. Vanderlick, J. Song, and D. Srolovitz, “Nanospot welding and contact evolution during cycling of a model microswitch,” Journal of Applied Physics. 2007. link Times cited: 11 Abstract: The useful lifetime of microelectromechanical system switche… read moreAbstract: The useful lifetime of microelectromechanical system switches is shortened during repetitive contact when the continual making and breaking of an electrical circuit accelerates damage done to the metallic contact points in the switch. In this study the interfacial force microscope is used as a model switch, and we explore the fundamental processes involved in switch failure. We find that repeated indentation (cyclic contact) causes protective coatings (in the form of self-assembled monolayers) to fail allowing metal-metal intimacy and formation of a malleable “nanospot weld.” The weld is stretched during separation of the contacting surfaces, leading to the development of nanoasperities. With the help of atomistic simulations, which provide insight into material transfer and consequential roughening of the surfaces, we show that asperity length grows with continued repetition, drastically changing the resistance of the contact over the lifetime of the switch. Controlling the amount of current passed throu... read less USED (high confidence) G. Rusina, S. Eremeev, S. Borisova, I. Sklyadneva, P. Echenique, and E. Chulkov, “Phonons in the ordered c(2 × 2) phases of Na and Li on Al(001),” Journal of Physics: Condensed Matter. 2007. link Times cited: 5 Abstract: The vibrational properties of the Al(001)-c(2 × 2)–Na (Li) o… read moreAbstract: The vibrational properties of the Al(001)-c(2 × 2)–Na (Li) ordered phases formed by alkali atoms (Na and Li) on the Al(001) surface at low and room temperatures are presented. The equilibrium structural characteristics, phonon dispersions and polarization of vibrational modes as well as the local density of phonon states are calculated using the embedded-atom method. The obtained structural parameters are in close agreement with experimental data. read less USED (high confidence) E. Kesälä, A. Kuronen, and K. Nordlund, “Molecular dynamics simulation of pressure dependence of cluster growth in inert gas condensation,” Physical Review B. 2007. link Times cited: 44 Abstract: The growth speed of nanoclusters during inert gas condensati… read moreAbstract: The growth speed of nanoclusters during inert gas condensation has been studied for copper, silver, aluminum, and platinum by using molecular dynamics simulations. We determine the condensation time for vapor atoms in a particular volume to be inversely proportional to the initial partial vapor pressure. We further find that the condensation time depends on the molecular mass and lattice constant, but not on other material properties. An analytical model for the condensation time is derived from kinetic gas theory by using the basic approximations of classical nucleation theory for a homogeneous vapor. read less USED (high confidence) E. Mendez-Villuendas, I. Saika-Voivod, and R. Bowles, “A limit of stability in supercooled liquid clusters.,” The Journal of chemical physics. 2007. link Times cited: 13 Abstract: We examine the metastable liquid phase of a supercooled gold… read moreAbstract: We examine the metastable liquid phase of a supercooled gold nanocluster by studying the free energy landscape using the largest solidlike embryo as an order parameter. Just below freezing, the free energy exhibits a local minimum at small embryo sizes and a maximum at a larger critical embryo size. At T=660 K the free energy becomes a monotonically decreasing function of the order parameter as the liquid phase becomes unstable, indicating that we have reached a limit of stability. In contrast to the mean-field theory predictions for a spinodal, the size of the critical embryo remains finite as the limit of stability is approached. We also calculate the rate of nucleation, independently from our free energy calculations, and observe a rapid increase in its temperature dependence when the free energy barrier is on the order of kT. We suggest that this supports the idea that freezing becomes a barrierless process at low temperatures. read less USED (high confidence) S. T. Choi and K. Kim, “Nanoscale planar field projections of atomic decohesion and slip in crystalline solids. Part I. A crack-tip cohesive zone,” Philosophical Magazine. 2007. link Times cited: 22 Abstract: The field projection method of Hong and Kim (2003) to identi… read moreAbstract: The field projection method of Hong and Kim (2003) to identify the crack-tip cohesive zone constitutive relations in an isotropic elastic solid is extended to a nanoscale planar field projection of a cohesive crack tip on an interface between two anisotropic solids. This formulation is applicable to the elastic field of a cohesive crack tip on an interface or in a homogeneous material for any combination of anisotropies. This method is based on a new orthogonal eigenfunction expansion of the elastic field around an interfacial cohesive crack, as well as on the effective use of interaction J integrals. The nanoscale planar field projection is applied to characterizing a crack-tip cohesive zone naturally arising in the fields of atomistics. The atomistic fields analyzed are obtained from molecular statics simulations of decohesion in a gold single crystal along a direction in a (111) plane, for which the interatomic interactions are described by an embedded atom method potential. The field projection provides cohesive traction, interface separation, and the surface-stress gradient caused by the gradual variation of surface formation within the cohesive zone. Therefore, the cohesive traction and surface energy gradient can be measured as functions of the cohesive zone displacements. The introduction of an atomistic hybrid reference configuration for the deformation analysis has made it possible to complete the field projection and to evaluate the energy release rate of decohesion with high precision. The results of the hybrid analyses of the atomistics and continuum show that there is a nanoscale mechanism of decohesion lattice trapping or hardening caused by the characteristics of non-local atomistic deformations near the crack tip. These characteristics are represented by surface relaxation and the development of surface stresses in the cohesive zone. read less USED (high confidence) M. Mariscal, E. Leiva, K. Pötting, and W. Schmickler, “The structure of electrodeposits – a computer simulation study,” Applied Physics A. 2007. link Times cited: 12 USED (high confidence) H. Neubauer and S. G. Mayr, “Dealloying of liquid CuAu nanoclusters during rotary motion: A molecular dynamics study,” Journal of Applied Physics. 2007. link Times cited: 0 Abstract: Dealloying effects caused by centrifugal forces in rotating … read moreAbstract: Dealloying effects caused by centrifugal forces in rotating multicomponent liquid clusters are investigated with the help of classical molecular dynamics computer simulations. The model system, CuAu, is chosen to investigate the phenomenon as a function of composition and cluster size. In addition to well-established surface energy related effects, we find an enrichment of Au on the surface due to centrifugal forces, which forms a bulge surrounding the rotating cluster. Fluctuations occurring during rotation are found to counteract the dealloying effects by mixing the constituents, leading to a stationary state in the system. read less USED (high confidence) T. Nogaret, C. Robertson, and D. Rodney, “Atomic-scale plasticity in the presence of Frank loops,” Philosophical Magazine. 2007. link Times cited: 59 Abstract: The different reactions between edge or screw dislocations a… read moreAbstract: The different reactions between edge or screw dislocations and interstitial Frank loops were studied by means of molecular dynamics simulations. The calculations were performed at 600 K using an embedded atom method (EAM) potential describing a model FCC material with a low stacking fault energy. An interaction matrix that provides the corresponding interaction strength was determined. In an attempt to investigate the role of pile-ups, simulations with either one or two dislocations in the cell were performed. We find that screw and edge dislocations behave very differently. Edge dislocations shear Frank loops in two out of three cases, while screw dislocations systematically unfault Frank loops by mechanisms that involve cross-slip. After unfaulting, they are strongly pinned by the formation of extended helical turns. The simulations show an original unpinning effect that leads to clear band broadening. This process involves the junction of two screw dislocations around a helical turn (arm-exchange) and the transfer of a dislocation from its initial glide plane to an upper glide plane (elevator effect). read less USED (high confidence) F. Sansoz and J. Molinari, “Size and microstructure effects on the mechanical behavior of FCC bicrystals by quasicontinuum method,” Thin Solid Films. 2007. link Times cited: 15 USED (high confidence) H. S. Park and P. Klein, “Surface Cauchy-Born analysis of surface stress effects on metallic nanowires,” Physical Review B. 2007. link Times cited: 138 Abstract: We present a surface Cauchy-Born approach to modeling FCC me… read moreAbstract: We present a surface Cauchy-Born approach to modeling FCC metals with nanometer scale dimensions for which surface stresses contribute significantly to the overall mechanical response. The model is based on an extension of the traditional Cauchy-Born theory in which a surface energy term that is obtained from the underlying crystal structure and governing interatomic potential is used to augment the bulk energy. By doing so, solutions to three-dimensional nanomechanical boundary value problems can be found within the framework of traditional nonlinear finite element methods. The major purpose of this work is to utilize the surface Cauchy-Born model to determine surface stress effects on the minimum energy configurations of single crystal gold nanowires using embedded atom potentials on wire sizes ranging in length from 6 to 280 nm with square cross sectional lengths ranging from 6 to 35 nm. The numerical examples clearly demonstrate that other factors beside surface area to volume ratio and total surface energy minimization, such as geometry and the percentage of transverse surface area, are critical in determining the minimum energy configurations of nanowires under the influence of surface stresses. read less USED (high confidence) P. Wynblatt, D. Chatain, and Y. Pang, “Some aspects of the anisotropy of grain boundary segregation and wetting,” Journal of Materials Science. 2006. link Times cited: 14 USED (high confidence) S. Borisova et al., “Vibrations in submonolayer structures of Na on Cu(111),” Physical Review B. 2006. link Times cited: 39 Abstract: We present the results of a comparative study of the equilib… read moreAbstract: We present the results of a comparative study of the equilibrium crystal structure and vibrational properties of the Na/Cu (cid:1) 111 (cid:2) system at coverages up to monolayer saturation. The calculations are performed with interaction potentials from the embedded-atom method. The following ordered structures are considered: p (cid:1) 3 (cid:1) 3 (cid:2) , p (cid:1) 2 (cid:1) 2 (cid:2) , (cid:1) (cid:3) 3 (cid:1) (cid:3) 3 (cid:2) 30°, and (cid:1) 3/2 (cid:1) 3/2 (cid:2) . The surface relaxation, phonon dispersion, and polarization of vibrational modes for the adsorbate and substrate atoms as well as the local density of states are discussed. It is found that the bond length between an adsorbate and the nearest-neighbor substrate atom slightly increases with increasing coverage. Adsorption of sodium also results in a small rumpling in two upper substrate layers. The mode associated with adatom-substrate stretch vibrations was obtained in our calculation at about 22 meV for all the structures considered. The strength of this mode decreases with increasing coverage in accordance with the experiment. On the other hand, we find that the frustrated translation mode frequency of sodium on Cu (cid:1) 111 (cid:2) is strongly coverage dependent. read less USED (high confidence) P. Wynblatt and D. Chatain, “Anisotropy of segregation at grain boundaries and surfaces,” Metallurgical and Materials Transactions A. 2006. link Times cited: 136 USED (high confidence) D. Schebarchov and S. Hendy, “Thermal instability of decahedral structures in platinum nanoparticles,” The European Physical Journal D. 2006. link Times cited: 13 USED (high confidence) M. Walker, C. Parkinson, M. Draxler, M. Brown, and C. McConville, “Initial growth of platinum on oxygen-covered Ni(110) surfaces,” Surface Science. 2006. link Times cited: 4 USED (high confidence) R. Murzaev and A. A. Nazarov, “Energies of formation and activation for migration of grain-boundary vacancies in a nickel bicrystal containing a disclination,” The Physics of Metals and Metallography. 2006. link Times cited: 0 USED (high confidence) P. L. Williams, Y. Mishin, and J. C. Hamilton, “An embedded-atom potential for the Cu–Ag system,” Modelling and Simulation in Materials Science and Engineering. 2006. link Times cited: 430 Abstract: A new embedded-atom method (EAM) potential has been construc… read moreAbstract: A new embedded-atom method (EAM) potential has been constructed for Ag by fitting to experimental and first-principles data. The potential accurately reproduces the lattice parameter, cohesive energy, elastic constants, phonon frequencies, thermal expansion, lattice-defect energies, as well as energies of alternate structures of Ag. Combining this potential with an existing EAM potential for Cu, a binary potential set for the Cu–Ag system has been constructed by fitting the cross-interaction function to first-principles energies of imaginary Cu–Ag compounds. Although properties used in the fit refer to the 0 K temperature (except for thermal expansion factors of pure Cu and Ag) and do not include liquid configurations, the potentials demonstrate good transferability to high-temperature properties. In particular, the entire Cu–Ag phase diagram calculated with the new potentials in conjunction with Monte Carlo simulations is in satisfactory agreement with experiment. This agreement suggests that EAM potentials accurately fit to 0 K properties can be capable of correctly predicting simple phase diagrams. Possible applications of the new potential set are outlined. read less USED (high confidence) D. Warner, F. Sansoz, and J. Molinari, “Atomistic based continuum investigation of plastic deformation in nanocrystalline copper,” International Journal of Plasticity. 2006. link Times cited: 132 USED (high confidence) J. M. Montejano-Carrizales, J. Rodríguez-López, U. Pal, M. Miki-Yoshida, and M. José-Yacamán, “The completion of the platonic atomic polyhedra: the dodecahedron.,” Small. 2006. link Times cited: 23 Abstract: The shape of metallic nanoparticles with diameters in the re… read moreAbstract: The shape of metallic nanoparticles with diameters in the region of few nanometers is a topic of extreme importantance in nanotechnology and catalysis. The atomic packing of a particle will define its surface orientation and, as a consequence, its catalytic activity, as well as many other properties. In addition, the shape of nanoparticles is strongly dependent on the preparation method and can be in a metastable or even in non-equilibrium state. 9] Dramatic variations in their structure can be understood in terms of the “energy landscape” concept, whereby many structures, or isomers, are represented in a free-energy landscape by minima of similar energies and separated by relatively small energy barriers. This fact is clearly observed in the socalled quasi-melting states, where a nanoparticle changes its shape in a very dramatic way under an electron beam, an effect that has been extensively studied by several authors and is a direct proof of the energy landscape concept. 9] When metallic nanoparticles are synthesized, their three-dimensional (3D) shape or structure results in 2D projections in the transmission electron microscopy (TEM) image corresponding to different shapes such as squares, triangles, hexagons, quasi-circles, and pentagons. These shapes can be associated in three dimensions to the cube, tetrahedron, cubo-octahedron, icosahedron, and decahedron, respectively, depending on the orientation of the particles on the substrate surface. When the particles are of 1–2 nm size, it is possible to observe the fivefold orientation more frequently, and this is probably due to truncations on the surface in contact with the substrate. Also, when bimetallic nanoparticles are synthesized, the frequency of the icosahedral particle increases, particularly for the smallest sizes. Platonic solids are polyhedra formed by the faces made of a same regular polygon. In total there exist five Platonic solids, namely, the tetrahedron, cube, octahedron, dodecahedron, and icosahedron. Composed of regular triangles, there exists the tetrahedron, octahedron, and icosahedron with 4, 8, and 20 faces, respectively. The cube, with 6 faces, is the only Platonic solid composed of squares, and the dodecahedron, with 12 faces, is the only one composed of pentagons. No other solid can be constructed from regular polygons. This is the reason that ancient Greeks attributed five shapes of special significance to the five elements (fire, earth, water, air, and cosmos), believing they must constitute the basic building blocks of the universe. Interestingly, today:s modern technology has confirmed that many nanoparticles take on some Platonic-solid-related shapes. Each Platonic solid looks the same from any vertex, and intuitively they appear as good candidates for shapes found at atomic equilibrium. A very clear example is the icosahedral (Ih) particle, which only exhibits (111) facets that contribute to produce a more rounded structure. Indeed, many studies report the Ih form as the most stable particle at the size range r 20 ; for noble gases and for some metals. However, in Platonic-solid clusters of a particular shape, an internal strain builds up with size; at some point the surface tension is suppressed in expense of strain energy and a new shape is favored. In that limit two different faces appear and an Archimedean solid is generated. A very elegant analytical treatment of the transition between an icosahedron (Platonic) to a Wulff polyhedron (Archimedean) has been presented by Nagaev, and we refer the reader to that article for more details. This transition is size-dependent and for noble metals, r 20 ; corresponds to an optimum value for the transition. The other Platonic solid structures such as tetrahedra, cubes, and octahedra have also been reported, but surprisingly dodecahedral structures have not been reported in this small size range. In this Communication we report the structure of small bimetallic nanoparticles of size r 2 nm. The profiles that these binary particles show in the microscope do not match with any of the known profiles in this size range. Therefore, we propose some very likely structures, based on the dodecahedron, with a close resemblance to the experimental profiles. We present some detailed models based on dodecahedral atomic growth packing, their corresponding HRTEM simulations, as well as their energetic stability through ab initio and embedded-atom method (EAM) calculations in order to discern/assign the structure for these binary AuPd nanoparticles. [*] Dr. J. M. Montejano-Carrizales Instituto de F sica, Universidad Aut noma de San Luis Potos 78000 San Luis Potos (Mexico) read less USED (high confidence) S. Psakhie, K. Zolnikov, D. S. Kryzhevich, and A. Lipnitskii, “Molecular-dynamics study of crystal structure defect formation by the thermal fluctuation mechanism during high-rate deformation,” Technical Physics Letters. 2006. link Times cited: 1 USED (high confidence) N. Sahoo, S. Thakur, and M. Senthilkumar, “Optical multilayer post growth instabilities: Analyses of Gd2O3/SiO2 system in combination with scanning probe force spectroscopy,” Applied Surface Science. 2005. link Times cited: 3 USED (high confidence) M. Mariscal, S. A. Dassie, and E. Leiva, “Collision as a way of forming bimetallic nanoclusters of various structures and chemical compositions.,” The Journal of chemical physics. 2005. link Times cited: 81 Abstract: In the present work, a new way to obtain bimetallic nanoclus… read moreAbstract: In the present work, a new way to obtain bimetallic nanoclusters of different structures and chemical compositions is proposed, which is based on computer simulations. Collision processes between two metal clusters of different natures are simulated through molecular-dynamics simulations using many-body potentials. Diverse diffusion mechanisms and structures can be observed, depending on the metals combined and the initial kinetic energies. The nanostructures we have found are core-shell (Pt-Au), alloyed (Pd-Au), and three-shell onionlike (Cu-Ag). read less USED (high confidence) D. Saraev and R. E. Miller, “Atomistic simulation of nanoindentation into copper multilayers,” Modelling and Simulation in Materials Science and Engineering. 2005. link Times cited: 48 Abstract: Atomic-scale simulations are used to examine the plastic beh… read moreAbstract: Atomic-scale simulations are used to examine the plastic behaviour of copper multi-layered thin films during nanoindentation tests. It is found that glide of nucleated dislocation loops and slip in the grain boundaries are the main operating deformation mechanisms in such multi-layered polycrystals. Furthermore, for a very small layer thickness, slip in the grain boundary dominates over the dislocation mediated plasticity. In order to survey the resistance of a multi-layered metal thin film to plastic deformation during nanoindentation, hardness versus penetration depth curves are plotted. Hardness curves reveal softening of multi-layer films, i.e. a reverse Hall–Petch effect is found for layer thicknesses in the nanometre range. read less USED (high confidence) M. Meyers, J. Rickman, and T. Delph, “The calculation of elastic constants from displacement fluctuations,” Journal of Applied Physics. 2005. link Times cited: 14 Abstract: We present a methodology for the accurate and efficient extr… read moreAbstract: We present a methodology for the accurate and efficient extraction of elastic constants in homogeneous solids via the calculation of the atomic displacement correlation function. This approach is validated for cubic solids parametrized by both Lennard-Jones and embedded-atom method potentials. Finally, we also discuss the extension of this method to obtain the elastic properties of inhomogeneous solids. read less USED (high confidence) D. Schebarchov and S. Hendy, “Static, transient, and dynamic phase coexistence in metal nanoclusters.,” The Journal of chemical physics. 2005. link Times cited: 32 Abstract: Molecular dynamics simulations are used to examine static an… read moreAbstract: Molecular dynamics simulations are used to examine static and dynamic coexistence between solid and liquid phases in nanoscale silver, copper, and nickel clusters. We find static coexistence in the 561-atom copper icosahedron, the 561-atom silver icosahedron, and the 923-atom nickel icosahedron, and in cluster sizes above these thresholds, but not in smaller clusters. Nonetheless, in smaller clusters we typically observe either dynamic coexistence between fully solid and liquid states or transient coexistence which is essentially dynamic coexistence between a fully solid state and a solid-liquid state. read less USED (high confidence) M. Gungor and D. Maroudas, “Atomistic mechanisms of strain relaxation due to ductile void growth in ultrathin films of face-centered-cubic metals,” Journal of Applied Physics. 2005. link Times cited: 18 Abstract: A comprehensive computational analysis is reported of the at… read moreAbstract: A comprehensive computational analysis is reported of the atomistic mechanisms of strain relaxation and failure in free-standing Cu thin films under applied biaxial tensile strain for strain levels up to 6%. The analysis focuses on nanometer-scale-thick films with a preexisting void extending across the film thickness and the film plane oriented normal to the [111] crystallographic direction. Our computational study is based on isothermal-isostrain large-scale molecular-dynamics simulations within an embedded-atom-method parametrization for Cu. Our analysis has revealed various regimes in the film’s mechanical response as the applied strain level increases. Within the considered strain range, after an elastic response at a low strain (<2%), void growth is the major strain relaxation mechanism mediated by the emission of perfect screw dislocation pairs from the void surface and subsequent dislocation propagation; as a result, a plastic zone forms around the void. Plastic deformation is accompanied by the g... read less USED (high confidence) M. Mariscal, C. Narambuena, M. G. D. Pópolo, and E. Leiva, “Effects of tip structure on the generation of metal clusters by an STM tip: a way to control the orientation of nanocrystallites?,” Nanotechnology. 2005. link Times cited: 11 Abstract: The role of the crystalline orientation of the STM tip in th… read moreAbstract: The role of the crystalline orientation of the STM tip in the generation of metal clusters is studied by atom dynamics simulations. When a (111) facet is facing the surface, the process is accompanied by a perturbation of the surface stronger than that observed for more open tip structures. This implies a technological application: the possibility of orienting a nanocrystallite deposited on a tip according to the changes observed in the force on the tip. read less USED (high confidence) G. Rusina, S. Eremeev, S. Borisova, I. Sklyadneva, and E. Chulkov, “Surface phonons on Al(111) surface covered by alkali metals,” Physical Review B. 2005. link Times cited: 18 Abstract: We investigated the vibrational and structural properties of… read moreAbstract: We investigated the vibrational and structural properties of the Al s 111 d - s ˛ read less USED (high confidence) F. Sansoz and J. Molinari, “Mechanical behavior of Σ tilt grain boundaries in nanoscale Cu and Al: A quasicontinuum study,” Acta Materialia. 2005. link Times cited: 220 USED (high confidence) S. Eremeev, G. Rusina, I. Sklyadneva, S. Borisova, and E. Chulkov, “Diffusional and vibrational properties of Cu(001)-c(2×2)-Pd surface alloys,” Physics of the Solid State. 2005. link Times cited: 2 USED (high confidence) M. Prasad and T. Sinno, “Feature activated molecular dynamics: an efficient approach for atomistic simulation of solid-state aggregation phenomena.,” The Journal of chemical physics. 2004. link Times cited: 3 Abstract: An efficient approach is presented for performing efficient … read moreAbstract: An efficient approach is presented for performing efficient molecular dynamics simulations of solute aggregation in crystalline solids. The method dynamically divides the total simulation space into "active" regions centered about each minority species, in which regular molecular dynamics is performed. The number, size, and shape of these regions is updated periodically based on the distribution of solute atoms within the overall simulation cell. The remainder of the system is essentially static except for periodic rescaling of the entire simulation cell in order to balance the pressure between the isolated molecular dynamics regions. The method is shown to be accurate and robust for the Environment-Dependant Interatomic Potential (EDIP) for silicon and an Embedded Atom Method potential (EAM) for copper. Several tests are performed beginning with the diffusion of a single vacancy all the way to large-scale simulations of vacancy clustering. In both material systems, the predicted evolutions agree closely with the results of standard molecular dynamics simulations. Computationally, the method is demonstrated to scale almost linearly with the concentration of solute atoms, but is essentially independent of the total system size. This scaling behavior allows for the full dynamical simulation of aggregation under conditions that are more experimentally realizable than would be possible with standard molecular dynamics. read less USED (high confidence) P. Cha, J. Song, T. Vanderlick, and D. Srolovitz, “Molecular dynamics simulation of single asperity contact,” Acta Materialia. 2004. link Times cited: 67 USED (high confidence) F. E. Gabaly, R. Miranda, and J. Figuera, “Properties of dislocation half loops inAu(100): Structure, formation energy, and diffusion barrier,” Physical Review B. 2004. link Times cited: 2 Abstract: Dislocation half-loops intersecting the surface have been de… read moreAbstract: Dislocation half-loops intersecting the surface have been detected by Scanning Tunneling Microscopy after mild sputtering on Aus100d. We determine their structure and formation energy by means of atomistic simulations. The Peierls barrier is also estimated from the simulations and found to be a few meV. This represents a very efficient way of moving mass parallel to the surface under applied stress, where a net movement of hundreds of atoms can take place with barriers smaller than that for the diffusion of a single adatom. Thermal diffusion is not observed. read less USED (high confidence) Y. Zhukovskii, E. Kotomin, D. Fuks, S. Dorfman, A. Stoneham, and G. Borstel, “Adhesion trends and growth mode of ultra-thin copper films on MgO,” Journal of Physics: Condensed Matter. 2004. link Times cited: 18 Abstract: Ab initio simulations are performed for Cu atoms adsorbed on… read moreAbstract: Ab initio simulations are performed for Cu atoms adsorbed on the perfect MgO(001) substrate, with an ordered metal coverage varied from 1 monolayer (ML), i.e. almost single atoms, up t o1M L. As trong dependence of the adhesion energy and the sub-monolayer film distance from the substrate on the surface coverage and adsorbate positions (Mg 2+ or O 2− )i s discussed. The nature of interfacial bonding at all coverages is physisorption .W hen increasing Cu atomic fraction, a decrease of the substrate-induced polarization of adatoms accompanied by an increase of both in-plane metallic bonding and the interfacial distance has been found. Combining results of ab initio calculations with thermodynamic theory (taking into account the lattice mismatch), we show that the metal cluster formation becomes the predominant growth mode even at low Cu coverages, in agreement with experiment. (Some figures in this article are in colour only in the electronic version) read less USED (high confidence) F. Sansoz and J. Molinari, “Incidence of atom shuffling on the shear and decohesion behavior of a symmetric tilt grain boundary in copper,” Scripta Materialia. 2004. link Times cited: 82 USED (high confidence) J. Hoyt, M. Asta, and A. Karma, “Atomistic and continuum modeling of dendritic solidification,” Materials Science & Engineering R-reports. 2003. link Times cited: 332 USED (high confidence) M. G. D. Pópolo, E. Leiva, M. Mariscal, and W. Schmickler, “The basis for the formation of stable metal clusters on an electrode surface,” Nanotechnology. 2003. link Times cited: 18 Abstract: Metal nanoclusters can be produced cheaply and precisely in … read moreAbstract: Metal nanoclusters can be produced cheaply and precisely in an electrochemical environment. Experimentally this method works in some systems, but not in others, and the unusual stability of the clusters has remained a mystery. We have simulated the deposition of the clusters using classical molecular dynamics and studied their stability by grand-canonical Monte Carlo simulations. We find that electrochemically stable clusters occur only in those cases where the two metals involved form stable alloys. read less USED (high confidence) R. Enrique, K. Nordlund, R. Averback, and P. Bellon, “Simulations of dynamical stabilization of Ag-Cu nanocomposites by ion-beam processing,” Journal of Applied Physics. 2003. link Times cited: 57 Abstract: Recent theoretical results indicate that ion-beam mixing can… read moreAbstract: Recent theoretical results indicate that ion-beam mixing can be used to synthesize nanocomposite structures from immiscible elements, relying on a self-organization phenomenon at steady state under irradiation. According to this modeling, self organization requires that the range of the forced atomic relocations in displacement cascades exceeds a critical value. Experimental evidence supporting the formation of nanocomposites by this mechanism has been found in the immiscible system Ag‐Cu irradiated with 1 MeV Kr ions. To address this experimentally relevant model system, and to test the theoretical predictions, we study, by molecular dynamics ~MD!, the characteristics of irradiation mixing in a Ag‐Cu alloy subjected to bombardment with 62 keV He, 270 keV Ne, 500 keV Ar, and 1 MeV Kr ions. The distribution of atomic relocations measured by MD is then used to perform lattice kinetic Monte Carlo ~KMC! simulations of phase evolution, during which both thermal decomposition and irradiation mixing operate simultaneously. The KMC results show that, in the framework of this self-organization mechanism, a nanocomposite structure can be stabilized at steady state by irradiation with heavy ions ~Ne, Ar, and Kr!, but not with He ions. As the characteristic relocation range for He ions is half of that measured for the heavy ions, these results support the theoretical prediction of the existence of a critical relocation range for compositional patterning to take place under irradiation. © 2003 American Institute of Physics. @DOI: 10.1063/1.1540743# read less USED (high confidence) J. Miguel and R. Miranda, “TOPICAL REVIEW: Atomic aspects in the epitaxial growth of metallic superlattices and nanostructures,” Journal of Physics: Condensed Matter. 2002. link Times cited: 38 Abstract: The properties of materials (mechanical, electronic, magneti… read moreAbstract: The properties of materials (mechanical, electronic, magnetic, etc) derive ultimately from the identity and spatial arrangement of their constituents. Nowadays, with the dimensions of technological devices and nanostructures reaching a few atomic constants, descriptions in terms of macroscopic concepts appear to be frequently inadequate and must give way to atomistic formulations based on elementary processes. Focusing on metallic materials, and more specifically on low-dimensional systems such as ultrathin films, superlattices or nanostructures, this paper reviews the atomic scale phenomena responsible for the most common types of defects (interfacial alloying, etching and roughness, formation of dislocations and pinholes, film discontinuities and twinning). It is shown that many of these features are related to the different mechanisms of strain relaxation in heteroepitaxial systems as well as to specific characteristics of atomic diffusion, such as the presence of Ehrlich–Schwoebel barriers hindering step crossings. Some special growth techniques (use of surfactants and codeposition) are also presented together with experimental examples demonstrating their usefulness to overcome the elements' natural limitations and produce accurately controlled, custom-designed epitaxial samples. Finally, a brief overview is given of different phenomena that can be exploited to produce self-assembled or self-organized structures. read less USED (high confidence) M. D. Pópolo et al., “Generation of palladium clusters on Au(111) electrodes: Experiments and simulations,” Applied Physics Letters. 2002. link Times cited: 37 Abstract: The properties of palladium clusters, generated with the ele… read moreAbstract: The properties of palladium clusters, generated with the electrochemical scanning tunneling microscope, have been investigated both by experiments and by computer simulations. The clusters are found to be larger and more stable if the tip is moved further towards the electrode surface in the generation process. The simulations suggest that the larger clusters consist of a palladium–gold mixture, which is more stable than pure palladium. Dissolution of the clusters occurs from the edges rather than layer by layer. read less USED (high confidence) V. Rodrigues and D. Ugarte, “Metal nanowires: atomic arrangement and electrical transport properties,” Nanotechnology. 2002. link Times cited: 34 Abstract: We have studied the atomic arrangement and defect formation … read moreAbstract: We have studied the atomic arrangement and defect formation in metal nanowires (NWs) generated by mechanical elongation using in situ high resolution transmission electron microscopy. It has been observed that the narrowest constriction of gold and platinum NWs is crystalline and defect-free; in particular, gold NWs adopt only three kinds of atomic arrangement. A model correlating these gold structures and the quantum conductance behaviour is proposed, which showed a remarkable agreement with ultrahigh-vacuum mechanically controllable break junction electrical transport measurements. read less USED (high confidence) R. Miron and K. Fichthorn, “The Step and Slide method for finding saddle points on multidimensional potential surfaces,” Journal of Chemical Physics. 2001. link Times cited: 50 Abstract: We present the Step and Slide method for finding saddle poin… read moreAbstract: We present the Step and Slide method for finding saddle points between two potential-energy minima. The method is applicable when both initial and final states are known. The potential-energy surface is probed by two replicas of the system that converge to the saddle point by following isoenergetic surfaces. The value of the transition-state potential is bracketed in the process, such that a convergence criterion based on the potential can be used. We applied the method to study diffusion mechanisms of a small Ag cluster on a Ag(111) surface using an embedded-atom method potential. Our approach is comparable in efficiency to other commonly used methods. read less USED (high confidence) M. I. Rojas, G. Amilibia, M. D. Pópolo, and E. Leiva, “2D-drop model applied to the calculation of step formation energies on a (111) substrate,” Surface Science. 2001. link Times cited: 4 USED (high confidence) M. Gungor and D. Maroudas, “Modeling of electromechanically-induced failure of passivated metallic thin films used in device interconnections,” International Journal of Fracture. 2001. link Times cited: 52 USED (high confidence) K. P. Zol’nikov, T. Uvarov, and S. Psakh’e, “Anisotropy of the plastic deformation and fracture processes in a dynamically loaded crystallite,” Technical Physics Letters. 2001. link Times cited: 25 USED (high confidence) T. Yokoyama, “Path-integral approach to anharmonic vibration of solids and solid interfaces.,” Journal of synchrotron radiation. 2001. link Times cited: 1 Abstract: The temperature dependence of EXAFS (extended X-ray absorpti… read moreAbstract: The temperature dependence of EXAFS (extended X-ray absorption fine structure) cumulants was investigated for bulk Cu and a thin film of Cu by means of the path-integral effective classical potential method. By using the semi-empirical embedded-atom method as a potential, agreement between the experiments and calculations is found to be excellent for bulk Cu. In the thin Cu(111) film, anisotropic anharmonic vibration was clearly observed; the out-of-plane vibration is much more enhanced and more anharmonic than the lateral vibration. The results are semiquantitatively consistent with the previous experimental data on Cu(111)/graphite. Such a vibrational enhancement should be the driving force for the roughening transition and/or the surface pre-melting at higher temperature. The practical usefulness of the path-integral effective classical potential method combined with the embedded-atom method is demonstrated. read less USED (high confidence) E. Kentzinger and H. Schober, “Migration energies in L12 intermetallic compounds,” Journal of Physics: Condensed Matter. 2000. link Times cited: 11 Abstract: The migration energies EM for the jumps into nearest-neighbo… read moreAbstract: The migration energies EM for the jumps into nearest-neighbour vacancies in L12 intermetallic compounds are related to the static lattice Green functions that can be calculated from the measured phonon dispersions. The present approach is an extension of a similar approach used earlier for BCC and FCC pure metals (Schober H R, Petry W and Trampenau J 1992 J. Phys.: Condens. Matter 4 9321). In A3B compounds with the L12 structure, three first-nearest-neighbour jumps into vacancies have to be distinguished and in some cases there is a bias, ΔE, between final and initial configurations. As was done earlier for monatomic lattices by Schober et al, the migration energy is split into two terms, one depending only on structure and a material-dependent term, given by the Green function elements. The difference in size between A and B atoms has to be taken into account and the compounds have to be separated into two groups depending on the size of the majority atoms relative to the minority ones. The formulae have been checked by computer simulations. The values of EM or EM-ΔE/2 are calculated for those L12 compounds where the phonon dispersions have been measured. In the case of the Ni3Al system, other theoretical and experimental determinations compare well with our model. Comparing these energies with the critical temperatures of stability of the L12 structure, we note a significant contribution of the ordering energy to the migration energy for all three jump types. read less USED (high confidence) M. Gungor, D. Maroudas, and S. Zhou, “Molecular-dynamics study of the mechanism and kinetics of void growth in ductile metallic thin films,” Applied Physics Letters. 2000. link Times cited: 25 Abstract: A molecular-dynamics study is presented of the mechanism and… read moreAbstract: A molecular-dynamics study is presented of the mechanism and kinetics of void growth and morphological evolution in ductile metallic thin films subject to biaxial tensile strains. The void becomes faceted, grows, and relieves strain by emission from its surface of pairs of screw dislocations with opposite Burgers vectors. Repeated dislocation generation and propagation leads to formation of a step pattern on the film’s surfaces. A simple phenomenological kinetic model of void growth is derived. Such kinetic equations can be used to formulate constitutive theories of plastic deformation for continuum-scale modeling of void evolution. read less USED (high confidence) O. Trushin, V. F. Bochkarev, and V. Naumov, “Simulation of epitaxial growth under ion-beam sputtering,” Russian Microelectronics. 2000. link Times cited: 0 USED (high confidence) X. W. Zhou and H. Wadley, “The low energy ion assisted control of interfacial structure: Ion incident energy effects,” Journal of Applied Physics. 2000. link Times cited: 25 Abstract: The properties of multilayered materials are often dependent… read moreAbstract: The properties of multilayered materials are often dependent upon their interfacial structure. For low temperature deposition processes where the structure is kinetically controlled, the interfacial roughness and the extent of interlayer mixing are primarily controlled by the adatom energy used in the deposition. Inert gas ion assistance during the growth process also enables manipulation of the interfacial roughness and intermixing. To explore inert gas ion assistance, a molecular dynamics approach has been used to investigate the role of ion energy and ion species upon the flattening of various surfaces formed during the growth of the Ni/Cu/Ni multilayers. The results indicated that ion energies in the 1–4 eV range could flatten the “rough” copper islands on either copper or nickel crystals. To flatten the rough nickel islands on copper or nickel crystals, higher ion energies in the 9–15 eV range would have to be used. Significant mixing between nickel island atoms and the underlying copper crystal atom... read less USED (high confidence) K. P. Zol’nikov, T. Uvarov, V. Skripnyak, A. Lipnitskii, D. Saraev, and S. Psakh’e, “Effect of grain boundary on the character of pulse-train-induced cleavage fracture in copper crystal,” Technical Physics Letters. 2000. link Times cited: 0 USED (high confidence) O. Trushin, K. Kokko, and P. Salo, “Film–substrate interface mixing in the energetic deposition of Ag on Cu(001),” Surface Science. 1999. link Times cited: 17 USED (high confidence) P. Keblinski, D. Wolf, S. Phillpot, and H. Gleiter, “Self-diffusion in high-angle fcc metal grain boundaries by molecular dynamics simulation,” Philosophical Magazine. 1999. link Times cited: 98 Abstract: Recent molecular dynamics simulations of high-energy high-an… read moreAbstract: Recent molecular dynamics simulations of high-energy high-angle twist grain boundaries (GBs) in Si revealed a universal liquid-like high-temperature structure which, at lower temperatures, undergoes a reversible structural and dynamical transition from a confined liquid to a solid; low-energy boundaries, by contrast, were found to remain solid all the way up to the melting point. Here we demonstrate for the case of palladium that fcc metal GBs behave in much the same manner. Remarkably, at high temperatures the few representative high-energy high-angle (tilt or twist) boundaries examined here exhibit the same, rather low self-diffusion activation energy and an isotropic liquid-like diffusion mechanism that is independent of the boundary misorientation. These observations are in qualitative agreement with recent GB self- and impurity-diffusion experiments by Budke et al. on Cu. Our simulations demonstrate that the decrease in the activation energy at elevated temperatures is caused by a structural... read less USED (high confidence) N. A. Levanov, A. A. Katsnel’son, A. Moroz, V. Stepanyuk, W. Hergert, and K. Kokko, “Structure and stability of clusters on metal surfaces,” Physics of the Solid State. 1999. link Times cited: 5 USED (high confidence) Z. Qing-yu, P. Zheng-Ying, and T. Jia-yong, “Molecular dynamics simulation of energetic atom depositions of Au/Au(100) film,” Acta Physica Sinica (overseas Edition). 1999. link Times cited: 5 Abstract: The energetic atom deposition of thin Au/Au(100) film has be… read moreAbstract: The energetic atom deposition of thin Au/Au(100) film has been studied by molecular dynamics simulation using the Au-Au interatomic interaction potential with embedded atom method. By investigating the variation of coverage curves and Bragg diffraction intensities during the film growth, the transition of Stranski-Kranstanov growth mode to Frank-van der Merwe growth mode was observed with the increase of the incident energy of deposition atoms. The role of energetic atoms in the film growth is discussed by analyzing the transport properties of deposited atoms and the evolution of incident energy and substrate temperatures. read less USED (high confidence) J. Wan, Y. L. Fan, D. W. Gong, S. G. Shen, and X. Q. Fan, “Surface relaxation and stress of fcc metals: Cu, Ag, Au, Ni, Pd, Pt, Al and Pb,” Modelling and Simulation in Materials Science and Engineering. 1999. link Times cited: 135 Abstract: The multilayer relaxation at (100), (110), (111), (210), (21… read moreAbstract: The multilayer relaxation at (100), (110), (111), (210), (211), (310), (311) and (331) surfaces of the fcc metals Cu, Ag, Au, Ni, Pd, Pt, Al and Pb are calculated using the modified embedded atom method. The `anomalous' outward relaxation at Al (100), (111), Pt (111), and Cu (111) surfaces is described correctly. The relief of surface stress and tension on the relaxation is studied on (111), (100) and (110) surfaces. In general, the surface stress in the direction of the surface normal determines the relaxation direction except for the Al (110) surface. When the surface stress is negative, the surface relaxation is inward; otherwise, the relaxation is outward. An interesting result is that the surface tension does not always decrease after relaxation. The outward relaxation will induce the increase in surface tension while the inward relaxation induces the decrease in surface tension. read less USED (high confidence) X. W. Zhou and H. Wadley, “Twin formation during the atomic deposition of copper,” Acta Materialia. 1999. link Times cited: 52 USED (high confidence) R. Najafabadi and D. Srolovitz, “Elastic step interactions on vicinal surfaces of fcc metals,” Surface Science. 1994. link Times cited: 38 USED (high confidence) R. Najafabadi and D. Srolovitz, “The effect of surface relaxation and atomic vibration on the equilibrium shape of gold and copper crystallites,” Journal of Computer-Aided Materials Design. 1994. link Times cited: 4 USED (high confidence) R. Najafabadi and D. Srolovitz, “Order-disorder transitions at and segregation to (001) Ni-Pt surfaces,” Surface Science. 1993. link Times cited: 19 USED (high confidence) H. Y. Wang, R. Najafabadi, D. Srolovitz, and R. LeSar, “Interfacial segregation in Ag-Au, Au-Pd, and Cu-Ni alloys: II. [001] Σ5 twist grain boundaries,” Interface Science. 1993. link Times cited: 15 USED (high confidence) A. Patil, D. Paithankar, N. Otsuka, and R. P. Andres, “The minimum-energy structure of nanometer-scale gold clusters,” Zeitschrift für Physik D Atoms, Molecules and Clusters. 1993. link Times cited: 30 USED (high confidence) J. Hirth and B. Carnahan, “Anisotropic elastic fields of twist boundaries,” Acta Metallurgica Et Materialia. 1992. link Times cited: 10 USED (high confidence) H. P. Ho, D. Yao, R. F. Ilmasani, M. A. Salam, D. Creaser, and L. Olsson, “The Effect of Pt/Pd Ratio on the Oxidation Activity and Resistance to Sulfur Poisoning for Pt-Pd/Bea Diesel Oxidation Catalysts with High Siliceous Content,” SSRN Electronic Journal. 2022. link Times cited: 8 Abstract: This study investigates the effect of the Pt/Pd ratio on the… read moreAbstract: This study investigates the effect of the Pt/Pd ratio on the oxidation activity and sulfur poisoning/regeneration of diesel oxidation catalysts (DOC) using beta zeolites with high siliceous content as support. Formation of Pt-Pd alloy leads to contraction of the cell lattice of Pt in the bimetallic catalysts, improving not only the sintering resistance of Pt but also retaining a high fraction of Pd in Pd 2 + form. Moreover, the Pt-Pd alloy also improves the oxidation resistance of the particles, which enhances the activity of the catalysts for CO and C 3 H 6 oxidation. Bimetallic catalysts also favor NO reduction at a lower temperature than the monometallic Pt although they showed lower values for the absolute conversion of NO due to a decrease in the total number of the Pt active sites. In addition, the bimetallic catalysts significantly improved the sulfur resistance as compared to the monometallic Pd catalyst. Moreover, the bimetallic catalysts could easily recover their activity for NO and C 3 H 6 oxidation by thermal treatment either in lean conditions or in H 2 . The reduction with H 2 was necessary to recover completely the activity of the CO and C 3 H 8 oxidation. read less USED (high confidence) V. Mazhukin, A. V. Shapranov, and O. Koroleva, “Atomistic modeling of crystal-melt interface mobility of fcc (Al, Cu) and bcc (Fe) metals in STRONG superheating/undercooling states.” 2020. link Times cited: 5 Abstract: A detailed study of the mobility and kinetic properties of s… read moreAbstract: A detailed study of the mobility and kinetic properties of solid liquid interfaces (SLI) with different types of crystal lattices (fcc Al, Cu) and (bcc Fe) metals in a wide range of temperatures and pressures was carried out using atomistic modeling. The ranges of maximum permissible values of superheated/undercooled states for each metal have been determined. The ultimate goal of the study was to determine the temperature dependences of the stationary front velocity υsl(ΔT) describing the SLI mobility in each of the metals in an analytical form. The analytical dependence υsl(ΔT) was constructed by comparing the results of atomistic modeling in the area of maximum permissible superheating/undercooling values with the data of the main kinetic models of Wilson Frenkel (WF) and Broughton, Gilmer and Jackson (BGJ). An acceptable agreement was achieved by introducing appropriate correction parameters into the kinetic models using the least squares method. The influence of the crystallographic orientation of metals and external pressure on the SLI mobility is investigated. read less USED (high confidence) A. Tal, “Electronic and structural properties of nanoclusters.” 2018. link Times cited: 1 Abstract: Nanoclusters have gained a huge interest due to their unique… read moreAbstract: Nanoclusters have gained a huge interest due to their unique properties. They represent an intermediate state between an atom and a solid, which manifests itself in their atomic configurations and ... read less USED (high confidence) M. Kroonblawd, “Anisotropic energy transport properties of 1,3,5-Triamino-2,4,6-Trinitrobenzene (TATB).” 2016. link Times cited: 1 Abstract: Anisotropic energy transport properties were determined theo… read moreAbstract: Anisotropic energy transport properties were determined theoretically for crystals of the insensitive explosive 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) using molecular dynamics simulations. Determination of these properties is necessary to predict and understand processes such as shock response and hot spot formation/relaxation and is also important for accurate parameterization of engineering models. Many properties of TATB exhibit significant anisotropy, which is thought to be due to the triclinic, graphitic-like layered packing structure. The thermal conductivity was determined as a function of temperature, pressure, and defect density; conduction within the layers is approximately 70% greater than conduction between the layers. Hot spot relaxation simulations were compared with and fit to solutions for the 1D heat equation to assess the validity of using continuum models to describe heat transport in TATB on nanometer length scales. A dissipative particle dynamics at constant energy coarse-grained model was developed for TATB and applied to micron-scale shock simulations. The predicted shock response is shown to be highly sensitive to a model parameter controlling energy transport kinetics, underscoring the need for a physics-based upscaling approach. A generalized crystal-cutting method was developed to construct 3D periodic simulation cells containing arbitrarily oriented single crystals and crystalcrystal interfaces for materials of arbitrary symmetry class. Strategies for non-uniform sampling of simulated transient phenomena were proposed that drastically reduce data storage costs. read less USED (high confidence) Y. Gao, G. Raos, C. Cavallotti, and M. Takahashi, “Molecular Dynamics Simulation on Physical Properties of Liquid Lead, Bismuth and Lead-bismuth Eutectic (LBE),” Procedia Engineering. 2016. link Times cited: 7 USED (high confidence) X.-J. Zhang and C.-L. Chen, “Phonon dispersion on Ag (100) surface: A modified analytic embedded atom method study*,” Chinese Physics B. 2016. link Times cited: 0 Abstract: Within the harmonic approximation, the analytic expression o… read moreAbstract: Within the harmonic approximation, the analytic expression of the dynamical matrix is derived based on the modified analytic embedded atom method (MAEAM) and the dynamics theory of surface lattice. The surface phonon dispersions along three major symmetry directions , and are calculated for the clean Ag (100) surface by using our derived formulas. We then discuss the polarization and localization of surface modes at points and by plotting the squared polarization vectors as a function of the layer index. The phonon frequencies of the surface modes calculated by MAEAM are compared with the available experimental and other theoretical data. It is found that the present results are generally in agreement with the referenced experimental or theoretical results, with a maximum deviation of 10.4%. The agreement shows that the modified analytic embedded atom method is a reasonable many-body potential model to quickly describe the surface lattice vibration. It also lays a significant foundation for studying the surface lattice vibration in other metals. read less USED (high confidence) L. Hu, “Continuum and atomistic models of surface elasticity and applications.” 2015. link Times cited: 0 Abstract: OF THE DISSERTATION Continuum and atomistic models of surfac… read moreAbstract: OF THE DISSERTATION Continuum and atomistic models of surface elasticity and applications by Lixin Hu Dissertation Director: Professor Liping Liu We present an analysis of surface elasticity from the Born-Oppenheimer approximation for monatomic crystals. The analysis shows that the relaxations of crystal planes parallel to a free surface can be sufficiently determined by a low-rank algebraic Riccati equation instead of a full-scale molecular dynamic (MD) simulation, and gives new restrictions on physically reasonable atomistic models and simple criteria for surface reconstructions. In the case of surface relaxations, we calculate surface elasticity properties from atomistic models, which are compared with experimental data and prior simulation results. This fundamental research is useful in a variety of applications. First, with the help of the proposed algorithm we quickly calculate the surface tension and determine the equilibrium shape of crystals. Secondly, in previous studies of wave propagation the impact of surface elasticity was not noticed. We find that when the surface/interface gains its own elasticity, the inhomogeneities between the bulk and the surface/interface result in nonlinearity for both interfacial and bulk wave propagation aspects. We study the interfacial wave between two half-spaces with surface elasticity taken into account. read less USED (high confidence) E. T. Karim, C. Wu, and L. Zhigilei, “Molecular Dynamics Simulations of Laser-Materials Interactions: General and Material-Specific Mechanisms of Material Removal and Generation of Crystal Defects.” 2014. link Times cited: 11 USED (high confidence) H. Zhang and J. Douglas, “Glassy Interfacial Dynamics of Ni Nanoparticles: Part II Discrete Breathers as an Explanation of Two-Level Energy Fluctuations.,” Soft matter. 2013. link Times cited: 26 Abstract: Recent studies of the dynamics of diverse condensed amorphou… read moreAbstract: Recent studies of the dynamics of diverse condensed amorphous materials have indicated significant heterogeneity in the local mobility and a progressive increase in collective particle motion upon cooling that takes the form of string-like particle rearrangements. In a previous paper (Part I), we examined the possibility that fluctuations in potential energy E and particle mobility μ associated with this 'dynamic heterogeneity' might offer information about the scale of collective motion in glassy materials based on molecular dynamics simulations of the glassy interfacial region of Ni nanoparticles (NPs) at elevated temperatures. We found that the noise exponent associated with fluctuations in the Debye-Waller factor, a mobility related quantity, was directly proportional to the scale of collective motion L under a broad range of conditions, but the noise exponent associated with E(t) fluctuations was seemingly unrelated to L. In the present work, we focus on this unanticipated difference between potential energy and mobility fluctuations by examining these quantities at an atomic scale. We find that the string atoms exhibit a jump-like motion between two well-separated bands of energy states and the rate at which these jumps occur seems to be consistent with the phenomenology of the 'slow-beta' relaxation process of glass-forming liquids. Concurrently with these local E(t) jumps, we also find 'quake-like' particle displacements having a power-law distribution in magnitude so that particle displacement fluctuations within the strings are strikingly different from local E(t) fluctuations. An analysis of these E(t) fluctuations suggests that we are dealing with 'discrete breather' excitations in which large energy fluctuations develop in arrays of non-linear oscillators by virtue of large anharmonicity in the interparticle interactions and discreteness effects associated with particle packing. We quantify string collective motions on a fast caging times scale (picoseconds) and explore the significance of these collective motions for understanding the Boson peak of glass-forming materials. read less USED (high confidence) S. Hartmann, B. Wunderle, and O. Holck, “Pull-Out Testing of SWCNTs Simulated by Molecular Dynamics.” 2012. link Times cited: 12 Abstract: In this paper we present our results of simulating a pull-ou… read moreAbstract: In this paper we present our results of simulating a pull-out test of single walled carbon nanotubes (SWCNT) out of a single crystal gold lattice by means of molecular dynamics. We compare the obtained force-displacement data of the pullout test to results of simulated uniaxial tensile strain tests of SWCNTs. In doing so, we make a theoretical estimation about the quality of the clamping of SWCNTs in a gold crystal. We investigated the influence of chirality of SWCNTs and of the system temperature. Dependent on SWCNT chirality two different pull-out behaviours can be described. Zigzag nanotubes show stronger pull-out resistance than chiral or armchair nanotubes. Our results indicate a minor influence of embedding length of the SWCNT in the gold matrix on pull-out forces. The system temperature has only little effect on the maximum pull-out forces. The presented results have impact on design criteria of SWCNT-metal interfaces. read less USED (high confidence) A. Leonardi, M. Leoni, and P. Scardi, “Interference Effects in Nanocrystalline Systems,” Metallurgical and Materials Transactions A. 2012. link Times cited: 4 USED (high confidence) S. Rawat, M. Warrier, S. Chaturvedi, and V. Chavan, “Effect of material damage on the spallation threshold of single crystal copper: a molecular dynamics study,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 17 Abstract: High velocity impact of copper plates using molecular dynami… read moreAbstract: High velocity impact of copper plates using molecular dynamics has been performed to study the spallation of single crystal copper at impact velocities of 1100 and 1000 m s−1. The molecular dynamics code LAMMPS (Large-Scale Atomic/Molecular Massively Parallel Simulator) with the embedded atom method potential is used for this study. It is found that for an impact velocity of 1100 m s−1, nucleation and growth of multiple voids take place which lead to the spallation of the material. For the impact at 1000 m s−1 in the ⟨1 0 0⟩ impact direction, the material does not undergo spallation but gives a spall-like signal in the free surface velocity of the target. We show that the tension developed by first traversal of the shock wave creates various kinds of defects in the target. These become void nucleation sites during the subsequent traversal of the shock wave. The presence of void nucleation sites due to the first traversal of the shock leads to the nucleation of the voids at a lower tensile pressure. We also show that the spall-like signal in the free surface velocity of the target at 1000 m s−1 impact along the ⟨1 0 0⟩ direction occurs due to stress relaxation resulting from the nucleation and growth of the voids without physical separation of scab from the target. read less USED (high confidence) Z. Postawa, L. Rzeznik, R. Paruch, M. F. Russo, N. Winograd, and B. Garrison, “Depth profiling by cluster projectiles as seen by computer simulations,” Surface and Interface Analysis. 2011. link Times cited: 25 Abstract: Molecular dynamics computer simulations are used to probe th… read moreAbstract: Molecular dynamics computer simulations are used to probe the development of the surface morphology and the processes that determine the depth resolution in depth profiling experiments performed by secondary ion and neutral mass spectrometry (SIMS/SNMS). The Ag(111) surface is irradiated by an impact of 20‐keV Au3, C60 and Ar872 clusters that represent a broad range of cluster projectiles used in SIMS/SNMS experiments. Improvements in the simulation protocol including automation and optimal sample shape allow for at least 1000 consecutive impacts for each set of initial conditions. This novel approach allows to shrink the gap between single‐impact simulations and real experiments in which numerous impacts are used. Copyright © 2010 John Wiley & Sons, Ltd. read less USED (high confidence) F. Grillo, H. A. Früchtl, S. M. Francis, and N. V. Richardson, “Site selectivity in the growth of copper islands on Au (111),” New Journal of Physics. 2011. link Times cited: 29 Abstract: The room temperature deposition of copper onto a Au(111)-(22… read moreAbstract: The room temperature deposition of copper onto a Au(111)-(22×√3) reconstructed surface has been investigated using scanning tunnelling microscopy (STM), up to a copper coverage of approximately 0.7 monolayer (ML). At extremely low coverage (∼0.02 ML), preferential adsorption is observed to occur by displacement of gold atoms and incorporation of copper into the top gold layer at alternate herringbone elbows along the ⟨112⟩ directions. Both fcc regions and hcp regions are occupied. With increasing coverage, incorporation of copper continues but copper is also deposited on top of the incorporated copper islands. When full coverage of these islands to monolayer thickness is reached, further deposition leads to preferential growth of those islands located in hcp regions through both the deposition process and migration of copper from other elbows, predominantly those in fcc regions. Eventually, a critical island size is reached above which atomically thick copper islands exhibit a reconstructed surface similar, in essence, to that of the clean gold surface. Models for the initial adsorption mechanism, island formation and the eventual reconstruction of the copper islands are discussed qualitatively in terms of surface strain within the gold and copper surfaces. read less USED (high confidence) T. Ito, “Molecular Dynamics Study on Melting Phenomena in Cu-Ag Eutectic System,” Journal of Power and Energy Systems. 2009. link Times cited: 7 Abstract: This study is made to elucidate the primary mechanism domina… read moreAbstract: This study is made to elucidate the primary mechanism dominating the inception and progress of eutectic melting between two solid metals contacting with each other. In this study, Cu-Ag eutectic system is simulated with classical molecular dynamics using Embedded Atom Method (1) . First, melting temperature of solid solution of Cu-Ag binary system is investigated. The melting temperature depends on the atomic concentration of Cu and follows the liquidus line obtained in the experiments. The minimum melting temperature was obtained at the eutectic concentration. The melting behavior on the interface between two pure Cu and Ag slabs are then simulated. The mutual diffusion at the interface was considerably enhanced by the surface melting of both the metals. It is shown that the melting temperature at the interface is lowered depending on the local value of the atomic fraction and is almost identical to that of the solid solution with the corresponding atomic fraction. read less USED (high confidence) J. Monk, Y. Yang, M. Mendelev, M. Asta, J. Hoyt, and D. Sun, “Determination of the crystal-melt interface kinetic coefficient from molecular dynamics simulations,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 92 Abstract: The generation and dissipation of latent heat at the moving … read moreAbstract: The generation and dissipation of latent heat at the moving solid–liquid boundary during non-equilibrium molecular dynamics (MD) simulations of crystallization can lead to significant underestimations of the interface mobility. In this work we examine the heat flow problem in detail for an embedded atom description of pure Ni and offer strategies to obtain an accurate value of the kinetic coefficient, μ. For free-solidification simulations in which the entire system is thermostated using a Nose–Hoover or velocity rescaling algorithm a non-uniform temperature profile is observed and a peak in the temperature is found at the interface position. It is shown that if the actual interface temperature, rather than the thermostat set point temperature, is used to compute the kinetic coefficient then μ is approximately a factor of 2 larger than previous estimates. In addition, we introduce a layered thermostat method in which several sub-regions, aligned normal to the crystallization direction, are indepently thermostated to a desired undercooling. We show that as the number of thermostats increases (i.e., as the width of each independently thermostated layer decreases) the kinetic coefficient converges to a value consistent with that obtained using a single thermostat and the calculated interface temperature. Also, the kinetic coefficient was determined from an analysis of the equilibrium fluctuations of the solid–liquid interface position. We demonstrate that the kinetic coefficient obtained from the relaxation times of the fluctuation spectrum is equivalent to the two values obtained from free-solidification simulations provided a simple correction is made for the contribution of heat flow controlled interface motion. Finally, a one-dimensional phase field model that captures the effect of thermostats has been developed. The mesoscale model reproduces qualitatively the results from MD simulations and thus allows for an a priori estimate of the accuracy of a kinetic coefficient determination for any given classical MD system. The model also elucidates that the magnitude of the temperature gradients obtained in simulations with a single thermostat depends on the length of the simulation system normal to the interface; the need for the corrections discussed in this paper can thus be gauged from a study of the dependence of the calculated kinetic coefficient on system size. read less USED (high confidence) W. Lee, K. Cho, and H. Jang, “Molecular Dynamics Simulation of Rolling Friction Using Nanosize Spheres,” Tribology Letters. 2008. link Times cited: 20 USED (high confidence) R. B. Getman and W. Schneider, “DFT-Based Characterization of the Multiple Adsorption Modes of Nitrogen Oxides on Pt(111),” Journal of Physical Chemistry C. 2007. link Times cited: 84 Abstract: Pt is the most common catalyst for NO oxidation to NO2, a ke… read moreAbstract: Pt is the most common catalyst for NO oxidation to NO2, a key reaction in NOx remediation chemistry. In this work, density functional theory calculations and plane-wave supercell models are used to calculate the energies, charge distributions, and vibrational spectra of the stable and metastable states of adsorbed NO, NO2, and NO3 on Pt(111), the most likely active metal face for this catalytic oxidation. NO, NO2, and NO3 are all strong electron acceptors and bind to the Pt(111) surface via charge donation from the surface. NO and NO2, in particular, exhibit a variety of adsorption geometries, the most favorable at low coverage being those that maximize surface−adsorbate charge transfer through binding to multiple surface Pt. At low coverage, the order of binding energies is NO > NO3> NO2, and the oxidation of adsorbed NO to NO2 is endothermic by 0.78 eV. Higher surface coverages favor migration of NO and NO2 to lower-coordination surface sites due to competition for metal d charge density. These changes ... read less USED (high confidence) R. Murzaev and A. A. Nazarov, “Activation energy for vacancy migration in [001] tilt boundaries in nickel,” The Physics of Metals and Metallography. 2006. link Times cited: 5 USED (high confidence) H. Sharma and S. Prakash, “Strain field due to self-interstitial impurity in Ni,” Pramana. 2003. link Times cited: 2 USED (high confidence) H. Yu, P. Shrotriya, J. Wang, and K.-S. Kim, “Dislocation nucleation and segregation in nano-scale contact of stepped surfaces,” MRS Proceedings. 2003. link Times cited: 8 Abstract: A myriad of engineering applications involve contact between… read moreAbstract: A myriad of engineering applications involve contact between two surfaces, which induces localized plastic deformation near the surface asperities. As a generic problem in studying nanometer scale plastic deformation of solid surfaces, a unit process model of dislocation formation near a surface step under contact loading of a flat rigid surface is considered. The driving force on the dislocation is calculated using conservation integrals. The effect of surface adhesion, step size and lattice resistance on the dislocation driving force are analyzed in a continuum dislocation model, while the nucleation process is simulated atomistically. The driving force formula is used for a dislocation nucleation criterion and to get the equilibrium distance traveled by the dislocation away from the surface step. Results of the unit process model show that under a normal contact load dislocations nucleated in certain slip planes can only stay in a thin layer near the surface, while dislocations nucleated along other slip planes easily move away from the surface into the bulk material. The former dislocation is named anti-load dislocation and the latter dislocation is called pro-load dislocation. Embedded atom method (EAM) is utilized to perform the atomistic simulation of the unit-process model. As predicted by the continuum dislocation model, the atomistic simulations also indicate that surface adhesion plays significant role in dislocation nucleation process. Varying the surface adhesion leads to three different regimes of load-deflection instabilities, namely, just dislocation nucleation instability for no adhesive interaction, two distinct surface adhesion and dislocation nucleation instabilities for weak adhesive interaction and a simultaneous surface adhesion and dislocation nucleation instability for strong adhesive interaction. The atomistic simulations provide additional information on dislocation nucleation and growth near the surface steps. The results of dislocation segregation predict existence of a thin tensile-stress layer near the deformed surface and the results on the adhesion effect provides a cold-welding criterion. read less USED (high confidence) D. Warner, F. Sansoz, and J. Molinari, “Modeling of Deformation in Nanocrystalline Copper Using An Atomistic-Based Continuum Approach,” MRS Proceedings. 2003. link Times cited: 4 Abstract: The deformation of copper with grain size less than 10 nm is… read moreAbstract: The deformation of copper with grain size less than 10 nm is investigated using a 2D continuum model incorporating atomistically-based constitutive relations. The local constitutive response of a series of symmetric and asymmetric tilt grain boundaries is obtained using an atomistic quasicontinuum method under tension and shear. The atomistic results show that it is possible to associate a constant maximum stress with each deformation mechanism triggered in the GB vicinity, i.e. GB sliding and decohesion, atom shuffling and partial dislocation emission. The GB strength is always found weaker in shear than in tension. This information is incorporated into a continuum polycrystalline model tested under compression. This model provides useful insights, in the absence of intragranular plasticity, into the onset of macroscopic quasi-plasticity, which results from GB sliding and collective grain rotation mechanisms. read less USED (high confidence) E. M. Lopasso, A. Caro, E. Arregui, and M. Caro, “Nanoscale compositional changes along fast ion tracks in equilibrium solid solutions: A computer simulation of ultra-fast solidification and thermomigration,” MRS Proceedings. 2001. link Times cited: 0 Abstract: : Starting from two equilibrium solid solutions in the Au-Ni… read moreAbstract: : Starting from two equilibrium solid solutions in the Au-Ni system, we analyze the change in composition due to a 400 eV/A fast ion track simulated by molecular dynamics in the Embedded Atom approximation. We aim at determining the influence of the thermodynamic forces derived from the large thermal gradients and the rapid solidification across the solidus and liquidus on the motion of solute atoms. One dimensional gradients as well as analytic models are used to quantitatively determine the domains of influence of these forces. Evidence shows that the liquidus and solidus equilibrium solidification predicted by the phase diagram is not reached during the track. The solute concentration is mainly determined by the combined diffusion and thermomigration mechanisms in the liquid stage. read less USED (high confidence) G. D. Billing, “Electron–hole pair excitation in molecule–surface collisions,” Journal of Chemical Physics. 2000. link Times cited: 17 Abstract: We investigate the role of electron–hole pair excitation in … read moreAbstract: We investigate the role of electron–hole pair excitation in molecule–surface collisions by using a semiclassical model which incorporates coupling to phonons and electrons in the substrate. The model treats the dynamics of the incoming molecule by classical mechanics but quantizes the phonons and electrons using second quantization techniques. We find that neither phonons nor electron–hole pair excitation can be neglected for an accurate description of molecule–surface collisions. read less USED (high confidence) B. Cappella and G. Dietler, “Force-distance curves by atomic force microscopy,” Surface Science Reports. 1999. link Times cited: 1467 USED (high confidence) G. D. Billing, “Second quantization formulation of molecular dynamics,” Physical Chemistry Chemical Physics. 1999. link Times cited: 2 Abstract: By introducing an expansion of the wavefunction for an N-ato… read moreAbstract: By introducing an expansion of the wavefunction for an N-atomic system in a Gauss–Hermite basis set is it possible to derive a trajectory based second quantization formulation of molecular dynamics. In the present paper we discuss some of the computational details of the theory and use it to treat an M=4 dimensional quantum mechanical system, namely that of hydrogen scattered from a solid surface. The full coupling to the surface phonons is also included in the calculations. read less USED (high confidence) H. Mehrez and S. Ciraci, “Conductance in nanowires.” 1997. link Times cited: 0 USED (high confidence) C. Massobrio and P. Fernandez, “Cluster adsorption on metallic surfaces: Structure and diffusion in the Cu/Pd(110) and Pd/Pd(110) systems,” Journal of Chemical Physics. 1995. link Times cited: 10 Abstract: We investigate structural and diffusion properties of CuN an… read moreAbstract: We investigate structural and diffusion properties of CuN and PdN (with N going from 1 to 30) clusters adsorbed on the (110) surface of Pd via atomistic simulations performed by employing embedded atom method potentials. For both systems, one dimensional linear chains are lower in energy than two dimensional structures, although the linear chain stability is more enhanced in the case of Cu/Pd(110). Our results on cluster stability are analyzed in terms of effective interactions and adsorbate arrangement upon relaxation. In close connection with STM experiments performed recently on Cu/Pd(110) [Roeder et al., Nature 366, 141 (1993)] we evaluate the diffusion barrier for atomic movement along and across the [110] direction. A cross exchange mechanism is found to lower significantly the diffusion barrier across the [110] direction, consistent with the value of the diffusion anisotropy found experimentally and the phase separation observed at the uppermost layer level. read less USED (high confidence) A. Biedermann, M. Schmid, and P. Varga, “Chemical analysis of PtxNi1−x alloy single crystal surfaces by scanning tunnelling microscopy,” Fresenius’ Journal of Analytical Chemistry. 1994. link Times cited: 2 USED (low confidence) Y. Jin et al., “Design and catalytic performance of Cu/?-Al2O3?for electrocatalytic methanol oxidation reaction by using density functional theory and molecular dynamics,” Applied Surface Science. 2023. link Times cited: 0 USED (low confidence) Y. Kaddar, V. Chaudhary, H. Bouhani, P. Neugebauer, A. Belhboub, and A. Fatimy, “Growth and stability of blue phosphorene on copper substrates: a molecular dynamics study,” Applied Physics A. 2023. link Times cited: 0 USED (low confidence) S. Meguid, S. I. Kundalwal, and A. Alian, “Role played by phonon drag on accuracy of MD simulations of nanowires due to deficiently selected strain rates,” International Journal of Mechanics and Materials in Design. 2023. link Times cited: 0 USED (low confidence) Z. Zhen-yu et al., “Influence of surface burnishing process with single strain path and reciprocating strain path on copper wear behavior,” Wear. 2023. link Times cited: 1 USED (low confidence) Z. Zhu et al., “Nano-cutting deformation characteristics and atomic-scale behavior of two-phase γ/γ′ nickel-based single crystal superalloy,” Intermetallics. 2023. link Times cited: 1 USED (low confidence) J. C. Jiménez-García, D. F. F. R. Flores, R. H. Acosta, M. I. Velasco, E. Franceschini, and M. M. Mariscal, “Water behavior at PEMFC triple phase boundary: Exploring ionomer and catalytic layer effects via molecular dynamic simulations and NMR experiments,” International Journal of Hydrogen Energy. 2023. link Times cited: 1 USED (low confidence) O. Politano and F. Baras, “Thermocapillary convection in a laser-heated Ni melt pool: A molecular dynamics study,” Journal of Applied Physics. 2023. link Times cited: 0 Abstract: Thermocapillary convection was investigated in a metallic sy… read moreAbstract: Thermocapillary convection was investigated in a metallic system of pure Ni, at the nanoscale, by molecular dynamics. The system interface was irradiated by a heat flux, mimicking a focused laser source. The melt pool was submitted to a large temperature gradient that modified the surface tension along the interface. In liquid metal, because surface tension typically decreases with increasing temperature, the result is a gradient of surface tension along the free surface. The liquid metal, therefore, started to flow in the direction of high surface tension. Two counter-rotating convection cells developed, characteristic of those observed in welding and other material processing. A systematic estimation of relevant parameters in hydrodynamics allowed us to interpret the results in terms of Prandtl, Marangoni, and Péclet numbers. This study demonstrates the influence of laser power and system size on pool shape and flow characteristics. read less USED (low confidence) Y.-C. Wu, J.-L. Shao, Y. Mei, X. Mu, and P. Chen, “Spall characteristics of three-dimensional graphene networks with embedded copper: A molecular dynamics study,” Mechanics of Materials. 2023. link Times cited: 0 USED (low confidence) Z. Zhang, Q. Huang, and H. Zhou, “High-entropy alloy nanocrystals with low-angle grain boundary for superb plastic deformability and recoverability,” International Journal of Plasticity. 2023. link Times cited: 3 USED (low confidence) H. Akbarzadeh, N. Abareshi, and M. Kamrani, “Role of the Middle-Shell in the Stability of Three-Shell Nanoparticles: A Molecular Dynamics Study,” Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2023. link Times cited: 0 USED (low confidence) Y. Chen, Y. Sun, W. Cheng, A. Meng, S. Zhang, and P. Wang, “Tissue evolution of Al0.67Cu0.33 alloy during melting and solidification by molecular dynamics simulation,” Chemical Physics. 2023. link Times cited: 0 USED (low confidence) G. Guo et al., “Investigating the effect of external force on the collision of an iron bullet with shear-thickening fluid nanocomposites using molecular dynamics simulation,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) A. Kubo, E. Kawai, T. Sumigawa, H. Shima, and Y. Umeno, “Defect formation mechanisms in metal nanowire under cyclic loading: a molecular dynamics study,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 0 Abstract: A series of molecular dynamics simulations were conducted to… read moreAbstract: A series of molecular dynamics simulations were conducted to reveal the fatigue mechanisms in metal nanowires. We applied axial cyclic loading deformation on a copper single-crystal nanowire model and observed the deformation process during cycle evolution. The detailed observation revealed that the deformation mechanisms in the nanowire is essentially different from the case of the macro- and micro-scaled materials because of the lack of dislocation sources. We also found that atomic vacancies were formed continually by dislocation motion even under a simple single-slip condition. The accumulation of vacancies is expected to be a probable mechanism of fatigue in nanomaterials. read less USED (low confidence) W. Zhang et al., “Effect of Loading Method, Temperature, and Twin Defects on the Mechanical Behavior of Nanocrystalline Ni with Gradient Spacing Twin Structure,” Crystal Growth & Design. 2023. link Times cited: 0 USED (low confidence) C. Mieszczyński et al., “Combining MD-LAMMPS and MC-McChasy2 codes for dislocation simulations of Ni single crystal structure,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2023. link Times cited: 0 USED (low confidence) H. Han, W. Ye, F. Zhang, D.-sheng Zhu, Y. Shen, and X. Xiong, “Molecular dynamics simulation study of nano-cutting interaction mechanisms in grain boundary affect zone segregated Cu alloys,” Journal of Nanoparticle Research. 2023. link Times cited: 0 USED (low confidence) E. Kawai, A. Kubo, and Y. Umeno, “Atomistic simulation for initiation of crystal slip deformation from surface of nanoscale copper single-crystal nanowires,” Computational Materials Science. 2023. link Times cited: 2 USED (low confidence) G. Zhu, M. Han, B. Xiao, and Z. Gan, “On the Microcrystal Structure of Sputtered Cu Films Deposited on Si(100) Surfaces: Experiment and Integrated Multiscale Simulation,” Molecules. 2023. link Times cited: 0 Abstract: Sputtered Cu/Si thin films were experimentally prepared at d… read moreAbstract: Sputtered Cu/Si thin films were experimentally prepared at different sputtering pressures and characterized using X-ray diffraction (XRD) and an atomic force microscope (AFM). Simultaneously, an application-oriented simulation approach for magnetron sputtering deposition was proposed in this work. In this integrated multiscale simulation, the sputtered atom transport was modeled using the Monte Carlo (MC) and molecular dynamics (MD) coupling method, and the deposition of sputtered atoms was simulated using the MD method. This application-oriented simulation approach was used to simulate the growth of Cu/Si(100) thin films at different sputtering pressures. The experimental results unveiled that, as the sputtering pressure decreased from 2 to 0.15 Pa, the surface roughness of Cu thin films gradually decreased; (111)-oriented grains were dominant in Cu thin films and the crystal quality of the Cu thin film was gradually improved. The simulation results were consistent with the experimental characterization results. The simulation results revealed that the transformation of the film growth mode from the Volmer–Weber growth mode to the two-dimensional layered growth mode resulted in a decrease in the surface roughness of Cu thin films; the increase in the amorphous compound CuSix and the hcp copper silicide with the decrease in the sputtering pressure was responsible for the improvement of the crystal quality of the Cu thin film. This work proposed a more realistic, integrated simulation scheme for magnetron sputtering deposition, providing theoretical guidance for the efficient preparation of high-quality sputtered films. read less USED (low confidence) V. Samsonov, I. Talyzin, S. Vasilyev, V. Puytov, and A. A. Romanov, “On surface pre-melting of metallic nanoparticles: molecular dynamics study,” Journal of Nanoparticle Research. 2023. link Times cited: 0 USED (low confidence) Z. Wang, S. Wang, D.-Q. Wang, Y.-R. Yang, X. Wang, and D.-J. Lee, “Water vapor condensation on substrates with nanoscale hydrophilic spots: A molecular dynamics study,” International Journal of Heat and Mass Transfer. 2023. link Times cited: 3 USED (low confidence) S. González-Tortuero, M. A. Garrido, and J. Rodríguez, “An adhesion study in Ni and Cu nanocontacts from a molecular dynamics perspective,” European Journal of Mechanics - A/Solids. 2023. link Times cited: 3 USED (low confidence) “Asymmetric and symmetric spreading for a nanodroplet on an isothermally heated surface in the presence of a parallel electric field,” Physics of Fluids. 2023. link Times cited: 0 Abstract: Under parallel electric fields and free evaporation conditio… read moreAbstract: Under parallel electric fields and free evaporation conditions, the statics and dynamics of spreading–evaporating nanodroplets are investigated on an isothermally heated surface via molecular dynamics (MD) simulations. The simulation results show that at the substrate temperature of Ts = 320 K, the static and dynamic contact angles on the left and right edges are initially asymmetric and then symmetric with increasing field strengths of E = 0.00–0.06 V Å−1, resulting in the asymmetric-to-symmetric spreading transition of spreading–evaporating nanodroplets. Under weak evaporation condition, that is, at Ts = 320 K, the asymmetric-to-symmetric spreading transition is triggered by enhancing the intrinsic surface wettability θ0 = 49°–80° at a constant field strength of E = 0.03 V Å−1. However, at the substrate temperature of Ts = 350 K, the symmetric-to-asymmetric spreading transition first appears for the static and dynamic contact angles on the left and right edges, and then the asymmetric-to-symmetric spreading transition appears with increasing field strength. Under strong evaporation condition, that is, at Ts = 350 K, as the field strength is constant at E = 0.03 V Å−1, the asymmetric-to-symmetric spreading transition also appears with increasing surface wettability. read less USED (low confidence) D.-W. Zheng, M.-bo Zhou, S. Liu, C. Ke, and X.-P. Zhang, “An Intensive Study of Effects of Orientations of Cu Bumps on Cu-Cu Direct Bonding for 3D Integration by Molecular Dynamics Simulation,” 2023 IEEE 73rd Electronic Components and Technology Conference (ECTC). 2023. link Times cited: 0 Abstract: In this paper, the bonding process of (100)- and (111)-orien… read moreAbstract: In this paper, the bonding process of (100)- and (111)-oriented single crystal Cu bumps as well as (111)-oriented nanotwinned Cu (nt-Cu) bumps is studied intensively by molecular dynamics simulation. The influence of bonding temperature (473, 523 and 573 K) and bonding stress (0.5, 1 and 1.5 MPa) on the bonding process of the above three Cu bumps are characterized. It is found that the bonding degree of (111)-oriented single crystal Cu bumps is better than that of (100)-oriented ones, the shear strength of (111)-oriented Cu bump interconnects obtained under the condition of bonding temperature 473 K without bonding stress is about 22% higher than that of (100)-oriented ones. The bonding degree of (111)-oriented nt-Cu bump interconnects is slightly better than that of (111)-oriented ones, at bonding temperature 473 K and without bonding stress whose shear strength is about 18% higher than that of the latter. The use of (111)-oriented nt-Cu bumps can effectively improve the plasticity of Cu-Cu direct bonding interconnects. Moreover, the increase in bonding temperature can significantly improve the bonding degree of Cu bump interconnects, but the influence of bonding stress on the bonding degree of Cu bump interconnects is not completely positive. read less USED (low confidence) S. Garg, N. Kaur, N. Goel, M. Molayem, V. Grigoryan, and M. Springborg, “Properties of Naked Silver Clusters with Up to 100 Atoms as Found with Embedded-Atom and Density-Functional Calculations,” Molecules. 2023. link Times cited: 1 Abstract: The structural and energetic properties of small silver clus… read moreAbstract: The structural and energetic properties of small silver clusters Agn with n = 2–100 atoms are reported. For n = 2–100 the embedded atom model for the calculation of the total energy of a given structure in combination with the basin-hopping search strategy for an unbiased structure optimization has been used to identify the energies and structures of the three energetically lowest-lying isomers. These optimized structures for n = 2–11 were subsequently studied further through density-functional-theory calculations. These calculations provide additional information on the electronic properties of the clusters that is lacking in the embedded-atom calculations. Thereby, also quantities related to the catalytic performance of the clusters are studied. The calculated properties in comparison to other available theoretical and experimental data show a good agreement. Previously unidentified magic (i.e., particularly stable) clusters have been found for n>80. In order to obtain a more detailed understanding of the structural properties of the clusters, various descriptors are used. Thereby, the silver clusters are compared to other noble metals and show some similarities to both copper and nickel systems, and also growth patterns have been identified. All vibrational frequencies of all the clusters have been calculated for the first time, and here we focus on the highest and lowest frequencies. Structural effects on the calculated frequencies were considered. read less USED (low confidence) Y. Q. Tang, A. Kumar, D. L. Chen, D. Y. Li, Q. Y. Li, and W. Li, “Bauschinger effect on wear of cold-worked Cu and Mg – A study combining molecular dynamics modeling and experimental investigation,” Wear. 2023. link Times cited: 0 USED (low confidence) P. Simonnin, D. Schreiber, B. Uberuaga, and K. Rosso, “Atomic Diffusion, Segregation, and Grain Boundary Migration in Nickel-Based Alloys from Molecular Dynamics Simulations,” SSRN Electronic Journal. 2023. link Times cited: 4 USED (low confidence) X. Zhang and J. Boland, “Core shift controls grain boundary energy scaling in Cu and Al,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) Z. Wang, L. Lin, Y. Feng, and F. Sun, “Enhancing the thermal boundary conductance of Cu/Diamond interface via diamond surface amorphization by molecular dynamics simulation,” Advances in Engineering Technology Research. 2023. link Times cited: 0 Abstract: Based on the non-equilibrium molecular dynamics simulation, … read moreAbstract: Based on the non-equilibrium molecular dynamics simulation, a Cu/amorphous diamond/crystalline diamond sandwich model was established to investigate the effects of the amorphous degree diamond surface and the thickness of the amorphous layer on the thermal boundary conductance of Cu/crystalline diamond. The simulation results show that the thermal boundary conductance can be enhanced by diamond surface amorphization, and increases with the increase of the amorphous degree. For the fully amorphous layer, the thermal boundary conductance increases gradually with the increase of the thickness of the amorphous layer and can be enhanced up to 4 times. The analysis of the vibrational density of states, overlap energy and phonon participation ratio shows that the diamond surface amorphization promotes the vibrational coupling between diamond and Cu atoms at low frequencies, as well as the occurrence of phonon inelastic scattering, and thus improves the thermal transport capacity of interface. read less USED (low confidence) P. Gao, C. Zhang, R. Wang, G. Deng, J. Li, and L. Su, “Tamping effect during additive manufacturing of copper coating by cold spray: A comprehensive molecular dynamics study,” Additive Manufacturing. 2023. link Times cited: 4 USED (low confidence) N. Dhariwal, A. S. M. Miraz, W. Meng, and C. Wick, “Strengthening the Ti/Tin Interface Against Shear Failure with Al Dopants:A Molecular Dynamics Study,” SSRN Electronic Journal. 2022. link Times cited: 1 USED (low confidence) B. Wu, Y. Wu, Y. Pan, and Z. Liu, “Nanoscale deformation of crystalline metals: Experiments and simulations,” International Journal of Plasticity. 2022. link Times cited: 2 USED (low confidence) Y. Tian and F. Chen, “Strength of nano-twinned gradient nano-grained copper: Molecular dynamic simulation,” IOP Conference Series: Materials Science and Engineering. 2022. link Times cited: 0 Abstract: Molecular dynamics simulations are performed to reveal the u… read moreAbstract: Molecular dynamics simulations are performed to reveal the underlying deformation mechanisms of gradient nano-grained materials with different-sized twins. The results indicate that the critical twin boundary spacing where the strength begins to soften decreases and the maximum strength of the material increases with the declining of the gradient. Below the critical value, the plastic deformation mechanism is dominated by the partial dislocations paralleling to the twin boundary, but when the twin boundary spacing exceed the critical value, the dislocation moved by the way of intersecting to the twin boundary. read less USED (low confidence) J. Li, J. Li, Y. Chen, and J. Chen, “Strengthening Modulus and Softening Strength of Nanoporous Gold in Multiaxial Tension: Insights from Molecular Dynamics,” Nanomaterials. 2022. link Times cited: 1 Abstract: The functionalized applications of nanoporous metals place c… read moreAbstract: The functionalized applications of nanoporous metals place clear requirements on their basic mechanical properties, yet there is a lack of research on the mechanical response under multiaxial loading conditions. In this work, the mechanical behaviors of nanoporous gold under multiaxial tension are investigated via molecular dynamics simulations. The mechanical properties under different loading conditions are compared and the microstructure evolution is analyzed to clarify the deformation mechanisms of nanoporous gold in biaxial and triaxial tension. It is found that the modulus of nanoporous gold in multiaxial tension is strengthened and the strength is softened compared to uniaxial tension. The failure of nanoporous gold in multiaxial tension is dominated by the progressive yielding, necking, and rupture of ligaments along the multiple uniaxial loading directions. The dislocation activity under multiaxial loads is more intense and more prone to plastic deformation, ultimately resulting in lower strength and smaller failure strain. The findings provide more insight into the understanding of the deformation mechanisms of nanoporous metals under complex stress states. read less USED (low confidence) S. Madhavan, V. Mishra, P. L. Narayana, and M. Warrier, “Dynamic Response of Single Crystal Al, Cu & Ni Upon Impact : MD and Ab-Initio Calculations,” Journal of Dynamic Behavior of Materials. 2022. link Times cited: 5 USED (low confidence) L. Zepeda-Ruiz, “Melting temperature, critical nucleus size, and interfacial free energy in single FCC metals — A Molecular Dynamics study of liquid–solid phase equilibria,” Journal of Crystal Growth. 2022. link Times cited: 0 USED (low confidence) Z. Fang, Y. Feng, Y. Yan, and Y. Geng, “Molecular dynamics simulation study on the effect of crystal orientation on bi-crystal gold nanocrystals in nanoskiving process,” Journal of Manufacturing Processes. 2022. link Times cited: 5 USED (low confidence) M. Eidani, H. Akbarzadeh, E. Mehrjouei, M. Abbaspour, S. Salemi, and H. Yaghoubi, “Thermal Stability and Melting Mechanism of Diamond Nanothreads: Insight from Molecular Dynamics Simulation,” Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2022. link Times cited: 0 USED (low confidence) D.-W. Zheng, M.-bo Zhou, S. Liu, C. Ke, and X.-P. Zhang, “Atomic-scale study of Cu–Cu direct bonding with assistance of Cu nanoparticles as joining medium by molecular dynamics simulation,” 2022 23rd International Conference on Electronic Packaging Technology (ICEPT). 2022. link Times cited: 0 Abstract: The application of Cu–Cu direct bonding in stacked chips is … read moreAbstract: The application of Cu–Cu direct bonding in stacked chips is becoming an alternative to the traditional solder-bump joint during the development of three-dimensional integrated circuits (3D IC). In this paper, the self-sintering between copper nanoparticles (Cu NPs) and the sintering between copper nanoparticle (Cu NP) and copper substrate (metallization) are simulated by molecular dynamics method. The results show that Cu atoms move mainly by interdiffusion between adjacent Cu NPs during the early and middle stages of sintering process, and the self-sintering of Cu NPs occurs first. In the late stage of sintering process, Cu atoms begin to diffuse at the contact interface between Cu NP and Cu metallization, and Cu NPs are sintered with Cu metallization. In addition, the simulation results of the effects of sintering temperature and particle size of Cu NPs on the Cu–Cu sintering behavior manifest that a relatively higher sintering temperature and smaller particle size of Cu NPs can better promote the self-sintering between Cu NPs and the sintering between Cu NP and Cu metallization. It is also found that the suitable sintering temperature of Cu NP and Cu metallization is between 450 K and 600 K, and the particle size has little effect on the diffusion distribution of Cu atoms in Cu NPs and Cu metallization during the sintering process. read less USED (low confidence) L. Xu, Z. Huang, Q. Shen, and F. Chen, “Atomistic Simulations of Plasticity Heterogeneity in Gradient Nano-grained FCC Metals,” Materials & Design. 2022. link Times cited: 10 USED (low confidence) D. P. Ranjan, M. A. Owhal, D. Chakrabarti, D. S. Belgamwar, T. Roy, and D. R. Balasubramaniam, “Fundamental Insights of Mechanical Polishing on Polycrystalline Cu Through Molecular Dynamics Simulations,” SSRN Electronic Journal. 2022. link Times cited: 9 USED (low confidence) Y.-C. Wu, J. Shao, and H. Zhan, “Deformation and Damage Characteristics of Copper/Honeycomb-Graphene under Shock Loading,” International Journal of Mechanical Sciences. 2022. link Times cited: 7 USED (low confidence) Y. Kashyrina, A. S. Muratov, V. Kazimirov, and O. S. Roik, “X-ray diffraction study and molecular dynamic simulation of liquid Al-Cu alloys: a new data and interatomic potentials comparison,” Journal of Molecular Modeling. 2022. link Times cited: 0 USED (low confidence) A. A. Joneidi, M. Shamshirsaz, and A. Taghvaeipour, “Investigation of the adhesive and abrasive wear mechanisms at the atomic scale using molecular dynamic simulations,” Proceedings of the Institution of Mechanical Engineers, Part J: Journal of Engineering Tribology. 2022. link Times cited: 2 Abstract: In this study, the effect of adhesion on material removal is… read moreAbstract: In this study, the effect of adhesion on material removal is numerically investigated at the nanoscale level. In this regard, Molecular Dynamics (MD) simulations are conducted to distinguish the contribution of adhesive and abrasive wear mechanisms during a scratching process in terms of the degree of interfacial adhesion and scratching depth. The numerical model simulates the scratching of a flat workpiece made of a single crystalline aluminum using a rigid conical indenter with a blunted spherical tip at four different indentation depths (i.e. 0, 3, 7, and 10 Å). The classical Leonard-Jones interatomic potential is used to mimic the degree of adhesion by varying the adhesion parameter between 5–70% of the aluminum bonding energy. It is shown that, at shallow scratching depths, the contribution of adhesive work to the frictional work is much larger than the ploughing work. On the contrary, the ratio of adhesive to ploughing work reduces by increasing the scratching depth. read less USED (low confidence) S. Wang, Z. Wang, S. Wang, Y. Yang, C. Huang, and X. Wang, “Non-Isothermal Dissolutive Wetting of Al-Ni and Cu-Ni Alloy Nanodroplets on a Cu(100) Substrate,” Journal of Thermal Science. 2022. link Times cited: 0 USED (low confidence) Y. Zhang et al., “Mechanical behavior of Pt-graphene porous biocompatible nanocomposites prepared by powder metallurgy using molecular dynamics simulation,” Journal of Molecular Liquids. 2022. link Times cited: 3 USED (low confidence) Z. Qiu-yang et al., “Mechanical response of single-crystal copper under vibration excitation based on molecular dynamics simulation,” Journal of Manufacturing Processes. 2022. link Times cited: 10 USED (low confidence) G. Zhang, J. Han, Y. Chen, J. Xiong, J. Wang, and J. Ran, “Generation mechanism and dual-dynamics simulation of surface patterns in single-point diamond turning of single-crystal copper,” Journal of Manufacturing Processes. 2022. link Times cited: 11 USED (low confidence) J. Wen et al., “Morphological evolution of Pt-films on sapphire and quartz substrates at various temperatures: an experimental and molecular dynamics study,” Applied Surface Science. 2022. link Times cited: 2 USED (low confidence) S. Zhou and H. Shen, “Experimental and numerical studies on micro-bumps without melting of gold films with different thicknesses induced by ultrafast laser,” Optics Communications. 2022. link Times cited: 2 USED (low confidence) M. Gu, T. Liu, X. Xiao, G. Li, and W. Liao, “Simulation and Experimental Study of the Multisized Silver Nanoparticles Sintering Process Based on Molecular Dynamics,” Nanomaterials. 2022. link Times cited: 5 Abstract: Multisized nanoparticles (MPs) are widely employed as electr… read moreAbstract: Multisized nanoparticles (MPs) are widely employed as electronic materials to form conductive patterns, benefitting from their excellent sintering properties and mechanical reliability. However, due to the lack of effective detection methods for the real-time sintering process, it is difficult to reveal the sintering behavior during the MPs sintering process. In this work, a molecular dynamics method is used to track the trajectory of silver atoms. The melting behavior of a single nanoparticle (SP) is first discussed. The structural evolution of equally sized nanoparticles (EPs) and unequally sized nanoparticles (UPs) during the sintering process is analyzed alongside morphology changes. It is proposed that the UPs sintering process benefits from the wetting behavior of small-sized nanoparticles on the surface of large-sized nanoparticles, and the sintering angle (θ) is proposed as an index to estimate the sintering result of UPs. Based on the works above, three basic sintering modes and one advanced sintering mode in the MP sintering process are analyzed emphatically in this paper, and the roles of different-sized nanoparticles in MPs are concluded from simulation and experimental results. This work provides theoretical support for conductive ink composition design and sintering process optimization. read less USED (low confidence) W. Li et al., “Deformation Mechanism of Depositing Amorphous Cu-Ta Alloy Film via Nanoindentation Test,” Nanomaterials. 2022. link Times cited: 2 Abstract: As a representative of immiscible alloy systems, the Cu-Ta s… read moreAbstract: As a representative of immiscible alloy systems, the Cu-Ta system was the research topic because of its potential application in industry, military and defense fields. In this study, an amorphous Cu-Ta alloy film was manufactured through magnetron sputter deposition, which was characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Mechanical properties of Cu-Ta film were detected by the nanoindentation method, which show that the elastic modulus of Cu3.5Ta96.5 is 156.7 GPa, and the hardness is 14.4 GPa. The nanoindentation process was also simulated by molecular dynamic simulation to indicate the deformation mechanism during the load-unload stage. The simulation results show that the structure <0,2,8,4> and <0,2,8,5> Voronoi cells decreased by 0.1% at 50 Ps and then remained at this value during the nanoindentation process. In addition, the number of dislocations vary rapidly with the depth between indenter and surface. Based on the experimental and simulation results, the Voronoi structural changes and dislocation motions are the key reasons for the crystallization of amorphous alloys when loads are applied. read less USED (low confidence) A. Shinde, A. Owhal, A. Sharma, P. Ranjan, T. Roy, and R. Balasubramaniam, “Comparative analysis of mechanical properties for mono and poly-crystalline copper under nanoindentation – Insights from molecular dynamics simulations,” Materials Chemistry and Physics. 2022. link Times cited: 10 USED (low confidence) P. Hansson, A. Ahadi, and S. Melin, “Molecular dynamic modelling of the combined influence from strain rate and temperature at tensile loading of nanosized single crystal Cu beams,” Materials Today Communications. 2022. link Times cited: 3 USED (low confidence) A. Markidonov, M. Starostenkov, D. Lubyanoi, P. Zakharov, and V. N. Lipunov, “Modeling of Healing Cylindrical Cavities Exposed to Shock Waves in Crystal Subjected to Shear Deformation,” Steel in Translation. 2022. link Times cited: 1 USED (low confidence) O. A. Sanders-Gutierrez, A. Luna-Valenzuela, A. Posada-Borbón, J. C. Schön, and A. Posada-Amarillas, “Molecular dynamics and DFT study of 38-atom coinage metal clusters,” Computational Materials Science. 2022. link Times cited: 6 USED (low confidence) J. Li et al., “Deformation mechanism of copper reinforced by three-dimensional graphene under torsion and tension,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 3 Abstract: The mechanical behaviors of uniaxial torsional and tensional… read moreAbstract: The mechanical behaviors of uniaxial torsional and tensional copper nanorod embedded with sp2-type hybrid graphene nanosheets (3DG/Cu) were investigated systematically using molecular dynamics methods. During the torsion process, graphene expanded the plastic deformation region of copper, while the plastic deformation in monocrystalline Cu cases was limited to a smaller area. 3DG/Cu responded to the torsion by one more plastic stage when plastic deformation spread along the length after the elastic response. Graphene improved the torsional loading capacity of the composite material, greatly extending the effective response range of the material by distributing the deformation of copper along with the graphene rather than being concentrated at a certain position like monocrystalline Cu. Generally, as the length of the model increased, this enhancement decreased. The copper portion of 3DG/Cu was divided into three areas during uniaxial tensile, a static region, a quasi-static region of the middle portion where the shear and necking occurred, and a dynamic area near the loading end. However, the inside graphene kept continuous until fracture. Furthermore, graphene improved the yield strain of copper by maintaining intact after copper failure. The greater the pre-loaded torsion angle, the smaller the yield strength and Young’s modulus of 3DG/Cu. read less USED (low confidence) M. Saffarini, G. Voyiadjis, and C. Ruestes, “Temperature effect on nanoporous gold under uniaxial tension and compression,” Computational Materials Science. 2021. link Times cited: 11 USED (low confidence) W. Lu et al., “Atomistic Simulation Study of the FCC and BCC Crystal-Melt Interface Stresses,” Surfaces and Interfaces. 2021. link Times cited: 4 USED (low confidence) G. Zhu, W. Liu, Z. Gan, and B. Xiao, “Mechanism for anisotropic ejection of atoms from fcc (100) metal surface by low-energy argon ion bombardment: Molecular dynamics simulation,” Vacuum. 2021. link Times cited: 6 USED (low confidence) Z. Wang, S. Wang, D.-Q. Wang, Y.-R. Yang, X. Wang, and D.-J. Lee, “Water vapor condensation on binary mixed substrates: A molecular dynamics study,” International Journal of Heat and Mass Transfer. 2021. link Times cited: 9 USED (low confidence) S. Maqbool, Y. Li, S. Muhammad, and Z. Yan, “Phase-field simulation of Cu enriched nanoparticles with variation of defects migration energy under neutron irradiation,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 3 Abstract: Neutron radiation induces point defects and affects the diff… read moreAbstract: Neutron radiation induces point defects and affects the diffusivity of atoms and the kinetics of precipitation. The phase-field simulation reveals the influence of migration energy of vacancy on the radiation-enhanced precipitation in Fe–Cu alloy. The study shows that radiation-enhanced diffusion (RED) also depends on the diffusivity of vacancy-associated migration energy and not only on the dose rate; the low migration energy of vacancy results in accelerated precipitation and a higher volume fraction of Cu precipitates. Interestingly, decreasing migration energy from 1.0 eV to 0.9 eV results in a 30% increase in the precipitates’ volume fraction. Also, the combination of the lowest dose rate 5.0 × 10−3 dpa s−1 and highest migration energy 1.0 eV delays the precipitation. The study also examines the influence of migration energy of vacancy on the radius of Cu precipitates. The lowest migration energy, 0.9 eV, increases the radius up to one-third. Finally, the work presents the drawbacks of the analytical digital image processing technique in the quantitative comparison with the script. read less USED (low confidence) Z. Zhu et al., “Study on Nanoscale Friction and Wear Mechanism of Nickel-based Single Crystal Superalloy by Molecular Dynamics Simulations,” Tribology International. 2021. link Times cited: 25 USED (low confidence) Y. Zhou, G. Luo, Y. Hu, D. Wu, and Z. Yao, “Interaction properties between molten metal and quartz by molecular dynamics simulation,” Journal of Molecular Liquids. 2021. link Times cited: 2 USED (low confidence) Y. Qin, J. Zhao, Z. Liu, C. Wang, and H. Zhang, “Study on effect of different surface roughness on nanofluid flow in nanochannel by using molecular dynamics simulation,” Journal of Molecular Liquids. 2021. link Times cited: 8 USED (low confidence) C. Jiang, Y. Mo, H. Wang, R. Li, M. Huang, and S. Jiang, “Molecular dynamics simulation of the production of hollow silver nanoparticles under ultrafast laser irradiation,” Computational Materials Science. 2021. link Times cited: 10 USED (low confidence) A. Markidonov, M. Starostenkov, D. Lubyanoi, P. Zakharov, and V. N. Lipunov, “Modeling of healing pores of cylindrical form under the action of shock waves in a crystal subjected to shear deformation,” Izvestiya. Ferrous Metallurgy. 2021. link Times cited: 0 Abstract: Volumetric defects in crystals worsen operational properties… read moreAbstract: Volumetric defects in crystals worsen operational properties of structural materials; therefore, the problem of reducing discontinuities in solid is one of the most important in modern materials science. In the present work, the results of computer simulation are presented that demonstrate possibility of collapse of pores in a crystal in state of shear deformation under the influence of shock waves. Similar waves can occur in a solid under external high-intensity exposure. For example, in the zone of propagation of displacement cascade, there are regions in which occurs a mismatch between the thermalization times of atomic vibrations and the removal of heat from them. As a result of the expansion of such a region, a shock after cascade wave arises. The simulation was carried out based on molecular dynamics method using the potential calculated by means of mmersed atom method. As a bulk defect, we considered extended pores of cylindrical shape, which can be formed after passing of high-energy ions through a crystal, or, for example, when superheated closed fluid inclusions (mother liquor) reach the surface. The study has shown that such defects are the source of heterogeneous nucleation of dislocation loops, contributing to a decrease in the shear stresses in simulated structure. Dependences of the average dislocation density on the shear angle and temperature of the designed cell were established, and the loop growth rate was estimated. Generated shock waves create additional tangential stresses that contribute to the formation of dislocation loops; therefore, in this case, dislocations are observed even with a small shear strain. If during simulation the thermal effect increases, the pore collapses. read less USED (low confidence) C. Zhang, C. Lu, G. Michal, J. Li, and R. Wang, “Strong strain hardening in graphene/nanotwinned metal composites revealed by molecular dynamics simulations,” International Journal of Mechanical Sciences. 2021. link Times cited: 15 USED (low confidence) P. Zhao, J. Wu, H. Chen, H. Liu, D. Li, and J. Tan, “Molecular dynamics simulation study of interaction mechanism between grain boundaries and subgrain boundaries in nano-cutting,” Journal of Manufacturing Processes. 2021. link Times cited: 30 USED (low confidence) J. Xiong, Y. Chen, Y. Dai, G. Zhang, J. Ran, and S. To, “Generation Mechanism and Dual Dynamic Simulations of Surface Patterns in Single-Point Diamond Turning of Single-Crystal Copper.” 2021. link Times cited: 0 Abstract:
Single-crystal copper (Cu), whose atom arrangement is in t… read moreAbstract:
Single-crystal copper (Cu), whose atom arrangement is in the same direction and has no grain boundary, is widely used in defense technology, civil electronics and network communication. As a diamond turnable material, fan-shaped patterns appear on the machined surface, which affects the machined surface quality and the optical function it carries. Previous studies on the surface generation mechanism in single-point diamond turning (SPDT) of Cu were limited to experimental analysis, while there is a lack of fundamental understanding of the fan-shaped pattern generation mechanism and suppression method. In the present study, the different fan-shaped patterns, surface quality, cutting force and chip morphology of the typical crystal planes (100), (110) and (111) planes of Cu were studied by both theoretical and experimental analyses. A molecular dynamics (MD) simulation was conducted to present the fundamental generation mechanism of the fan-shaped patterns from atom arrangement directions and its angle change with the main cutting direction, while a cutting dynamics model was established to simulate the generation of fan-shaped patterns on the machined surface. Based on theoretical and experimental analysis, it was found that the atom density arrangement directions of Cu and its angle change with the main cutting direction of SPDT caused fluctuations in the friction coefficient, which further caused the vibration of the cutting system and generated the fan-shaped patterns. The SPDT of crystal planes (100) can achieve the best surface quality. The present research provides a fundamental understanding of fan-shaped pattern formation on the machined surface, and provides an instruction for machining Cu to obtain better surface quality. read less USED (low confidence) Y.-C. Wu, J. Shao, and H. Zhan, “Damage and self-healing characteristics of monolayer graphene enhanced Cu under ballistic impact,” Mechanics of Materials. 2021. link Times cited: 13 USED (low confidence) Y. J. Shen, S. Mi, L. Sun, L. Yang, and H. Gong, “Mechanical properties and dislocation evolution of Cu–Fe interfaces from molecular dynamics simulation,” Materials Chemistry and Physics. 2021. link Times cited: 12 USED (low confidence) B.-X. Zhang, S. Wang, X.-lei He, Y.-R. Yang, and X. Wang, “Dynamic spreading of a water nanodroplet on a nanostructured surface in the presence of an electric field,” Journal of Molecular Liquids. 2021. link Times cited: 8 USED (low confidence) L. Wang et al., “Effect of sink strength on coherency loss of precipitates in dilute Cu-base alloys during in situ ion irradiation,” Acta Materialia. 2021. link Times cited: 7 USED (low confidence) Q. Han et al., “In-situ TEM observation of the evolution of helium bubbles & dislocation loops and their interaction in Pd during He+ irradiation,” Journal of Materials Science & Technology. 2021. link Times cited: 26 USED (low confidence) H. Mes-adi, K. Saadouni, and M. Mazroui, “Effect of incident angle on the microstructure proprieties of Cu thin film deposited on Si (001) substrate,” Thin Solid Films. 2021. link Times cited: 8 USED (low confidence) Y. Cui, Y. Toku, Y. Kimura, and Y. Ju, “The deformation mechanism in cold-welded gold nanowires due to dislocation emission,” Computational Materials Science. 2021. link Times cited: 4 USED (low confidence) L. Zhang, L. Tian, A. Zhang, Y. Jing, and P. Qu, “Molecular Dynamics Simulations of the Effects of a Nanoparticle Surface Adsorption Layer on the Thermal Conductivity of a Cu–Ar Nanofluid,” International Journal of Thermophysics. 2021. link Times cited: 6 USED (low confidence) F. Shuang and K. Aifantis, “Dislocation-graphene interactions in Cu/graphene composites and the effect of boundary conditions: a molecular dynamics study,” Carbon. 2021. link Times cited: 43 USED (low confidence) S. Luan et al., “Length effects on tensile behavior of Au-Ag heterostructured nanowires with the load on different ends: A molecular dynamics study,” Physics Letters A. 2020. link Times cited: 2 USED (low confidence) J. Cao, L. Li, and C. Zhang, “The coalescence of Cu nanoparticles with different interfacial lattice structures: A molecular dynamics study,” Modern Physics Letters B. 2020. link Times cited: 1 Abstract: With the popularization of 3D printing technology, micro/nan… read moreAbstract: With the popularization of 3D printing technology, micro/nanoparticles sintering technology has drawn lots of attentions all over the world. Here, molecular dynamic simulation is employed to discuss the effects of different interfacial lattice structures, different diameter of nanoparticles, and different heating rates on the coalescence of metallic Cu nanoparticles. The results showed that the diameter of nanoparticles determine the melting point of the system. Besides, the interfacial lattice structure, diameter of nanoparticles, and heating rate have an influence on the initial sintering temperature. This is because the melting point is the inherent property of material which relies on the mass of substance. However, the initial sintering temperature is sensitive to many factors, including the temperature, interfacial, and intermolecular interactions. read less USED (low confidence) C. Zhao, W. An, and N. Gao, “Light-induced latent heat reduction of silver nanofluids: A molecular dynamics simulation,” International Journal of Heat and Mass Transfer. 2020. link Times cited: 10 USED (low confidence) L. Y. Zhao and Y. Liu, “Investigation on void growth and coalescence in single crystal copper under high-strain-rate tensile loading by atomistic simulation,” Mechanics of Materials. 2020. link Times cited: 11 USED (low confidence) F. Eddiai, M. Dardouri, H. E. Azrak, A. Hassani, K. Sbiaai, and A. Hassnaoui, “Molecular-Dynamics Study Of Self-Diffusion: Of The Au4;Au4/Ag(110) System,” IOP Conference Series: Materials Science and Engineering. 2020. link Times cited: 1 Abstract: In this work, we will shed light on the results obtained fro… read moreAbstract: In this work, we will shed light on the results obtained from the molecular dynamics method in the temperature ranging of 300-700K. This investigation concerning the coalescence for the two tetramers islands of system having different forms (SS, TT and NN) deposited at different spacing sites in-channel and cross-channel. The stability of the systems, the diffusion phenomena produced during the dynamics, as well as the diffusion/coalescence lifetime during the evolution of temperature are dependent. The dynamic study shows that between 450K and 650K the homogeneous partial coalescence occurs through the mechanism of the jump. As time goes by increasing the temperature which favors the process of exchange the heterogeneous total coalescence is obtained. read less USED (low confidence) S. Sarangi, “Study on Young’s modulus of metallic nanowires using classical molecular dynamics simulations,” Materials Today: Proceedings. 2020. link Times cited: 4 USED (low confidence) A. Samiri, A. Khmich, H. Haouas, A. Hassani, and A. Hasnaoui, “Structural and mechanical behaviors of Mg-Al metallic glasses investigated by molecular dynamics simulations,” Computational Materials Science. 2020. link Times cited: 15 USED (low confidence) A. Sharma, P. Ranjan, and R. Balasubramaniam, “Investigation of effect of uncut chip thickness to edge radius ratio on nanoscale cutting behavior of single crystal copper: MD simulation approach,” Journal of Micromanufacturing. 2020. link Times cited: 5 Abstract: Extremely small cutting depths in nanoscale cutting makes it… read moreAbstract: Extremely small cutting depths in nanoscale cutting makes it very difficult to measure the thermodynamic properties and understand the underlying mechanism and behavior of workpiece material. Highly precise single-crystal Cu is popularly employed in optical and electronics industries. This study, therefore, implements the molecular dynamics technique to analyze the cutting behavior and surface and subsurface phenomenon in the nanoscale cutting of copper workpieces with a diamond tool. Molecular dynamics simulation is carried out for different ratios of uncut chip thickness (a) to cutting edge radius (r) to investigate material removal mechanism, cutting forces, surface and subsurface defects, material removal rate (MRR), and stresses involved during the nanoscale cutting process. Calculation of forces and amount of plowing indicate that a/r = 0.5 is the critical ratio for which the average values of both increase to maximum. Material deformation mechanism changes from shear slip to shear zone deformation and then to plowing and elastic rubbing as the cutting depth/uncut chip thickness is reduced. The deformation during nano-cutting in terms of dislocation density changes with respect to cutting time. During the cutting process, it is observed that various subsurface defects like point defects, dislocations and dislocation loops, stacking faults, and stair-rod dislocation take place. read less USED (low confidence) F. Eddiai, M. Dardouri, A. Hassani, M. Badawi, K. Sbiaai, and A. Hassnaoui, “Structure, stability, and surface diffusion of clusters: Pt4/Cu (110) AND Au4/Ag (110) surface by molecular dynamics,” European Physical Journal-applied Physics. 2020. link Times cited: 2 Abstract: In this work, molecular dynamics simulations have been used … read moreAbstract: In this work, molecular dynamics simulations have been used to simulate the behavior of tetramer clusters behavior in Pt4 /Cu (110) and Au4 /Ag (110) systems, in the temperature range 300-600 K. All activation barriers and formation energies related to different tetramer shapes (4S, 4L, 4T, 4N and 4l) have been calculated by embedded atom method (EAM) at static regime (0 K). From an energetical point of view, the adatoms tend to diffuse via simple jumps and exchange mechanisms leading to a transition between all forms during tetramer diffusion. Statistical analysis after molecular dynamics simulations confirms that the linear 4l shape is more stable and needs high energy to be disintegrated in both systems. The lifetime study of each shape for different temperatures (from 300 K to 600 K) proves that the 4 l form is more stiff, which is in a good agreement with the formation energy predictions. read less USED (low confidence) X. Ren, X. Li, C. Huang, H. Yin, and F. Wei, “Molecular dynamics simulation of thermal welding morphology of Ag/Au/Cu nanoparticles distributed on Si substrates,” Ferroelectrics. 2020. link Times cited: 3 Abstract: The behavior of Ag/Au/Cu nanoparticles dispersed on Si subst… read moreAbstract: The behavior of Ag/Au/Cu nanoparticles dispersed on Si substrates during heat treatment was studied by molecular dynamics in this paper. Whether for the thermal welding of homogeneous metal nanoparticles or heterogeneous metal nanoparticles, it was found that the fundamental reason for the contact between the surface of metal nanoparticles is the electron exchange, which reduces the energy of the system and leads to welding contact, then the atoms migrate toward the interior of nanoparticles through nucleation and growth process, resulting in the decrease of the porosity and the shrinkage of the sample. At the same time, the simulation results show that when the applied temperature rises to a certain extent, the adjacent nanoparticles may condense into isolated structures under their own surface tension and stress, as well as the adhesion of the substrate, which will make the conductive structure discontinuous. However, by adjusting the size and proportion of Ag/Au/Cu metal nanoparticles, besides saving the cost of printing circuits, the conductive structure can be more continuous and the conductivity can be enhanced. read less USED (low confidence) Y. Long, B. He, W. Cui, Y. Ji, X. Zhuang, and J. Twiefel, “Investigations on the mechanism of microweld changes during ultrasonic wire bonding by molecular dynamics simulation,” Materials & Design. 2020. link Times cited: 19 USED (low confidence) Y. Jiang, S. Dehghan, A. Karimipour, D. Toghraie, Z. Li, and I. Tlili, “Effect of copper nanoparticles on thermal behavior of water flow in a zig-zag nanochannel using molecular dynamics simulation,” International Communications in Heat and Mass Transfer. 2020. link Times cited: 16 USED (low confidence) P. Hansson, A. Ahadi, and S. Melin, “Molecular dynamics modelling of metric scaling effects in nanosized Cu beams holding a grain boundary,” Theoretical and Applied Fracture Mechanics. 2020. link Times cited: 2 USED (low confidence) S. Zhang, P. Huang, and F. Wang, “Graphene-boundary strengthening mechanism in Cu/graphene nanocomposites: A molecular dynamics simulation,” Materials & Design. 2020. link Times cited: 41 USED (low confidence) Y. Lachtioui, M. Kbirou, K. Saadouni, M. Sajieddine, and M. Mazroui, “Glass formation and structure evolution in the rapidly solidified monatomic metallic liquid Pt under high pressure,” Chemical Physics. 2020. link Times cited: 11 USED (low confidence) C.-Y. Shih, M. Shugaev, C. Wu, and L. Zhigilei, “Correction: The effect of pulse duration on nanoparticle generation in pulsed laser ablation in liquids: insights from large-scale atomistic simulations.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 9 Abstract: Correction for 'The effect of pulse duration on nanopar… read moreAbstract: Correction for 'The effect of pulse duration on nanoparticle generation in pulsed laser ablation in liquids: insights from large-scale atomistic simulations' by Cheng-Yu Shih et al., Phys. Chem. Chem. Phys., 2020, 22, 7077-7099, DOI: 10.1039/d0cp00608d. read less USED (low confidence) Y. Yildiz, A. Ahadi, and M. Kırca, “Strain rate effects on tensile and compression behavior of nano-crystalline nanoporous gold: A molecular dynamic study,” Mechanics of Materials. 2020. link Times cited: 11 USED (low confidence) D. Utt, A. Stukowski, and K. Albe, “Grain boundary structure and mobility in high-entropy alloys: A comparative molecular dynamics study on a Σ11 symmetrical tilt grain boundary in face-centered cubic CuNiCoFe,” Acta Materialia. 2020. link Times cited: 55 USED (low confidence) A. Rida, M. Micoulaut, E. Rouhaud, and A. Makke, “Understanding the strain rate sensitivity of nanocrystalline copper using molecular dynamics simulations,” Computational Materials Science. 2020. link Times cited: 25 USED (low confidence) J. Li, B. Lu, Y. Zhang, H. Zhou, G. Hu, and R. Xia, “Nanoindentation response of nanocrystalline copper via molecular dynamics: Grain-size effect,” Materials Chemistry and Physics. 2020. link Times cited: 35 USED (low confidence) Y. Su et al., “The relationship between viscosity and local structure in liquid zirconium via electromagnetic levitation and molecular dynamics simulations,” Journal of Molecular Liquids. 2020. link Times cited: 15 USED (low confidence) H. Chabba and D. Dafir, “Compression Behavior of Al-Mg Phases, Molecular Dynamics Simulation,” International Journal of Engineering Research in Africa. 2020. link Times cited: 2 Abstract: Aluminum alloys development always exit in the manufacturing… read moreAbstract: Aluminum alloys development always exit in the manufacturing process. Al/Mg alloys have been attracted significant attention because of their excellent mechanical properties. The microstructural evolution and deformation mechanisms are still challenging issues, and it is hard to observe directly by experimental methods. Accordingly, in this paper atomic simulations are performed to investigate the uniaxial compressive behavior of Al/Mg phases; with different ratio of Mg ranging from 31% to 56%. The compression is at the same strain rate (3.1010 s⁻¹), at the same temperature (300K) and pressure, using embedded atom method (EAM) potential to model the interactions and the deformation behavior between Al and Mg.From these simulations, we get the radial distribution function; the stress–strain responses to describe the elastic and plastic behaviors of β-Al3Mg2, ε-Al30Mg23, Al1Mg1 and γ-Al12Mg17 phases with 31, 41, 50 and 56% of Mg added to pure aluminum, respectively. The mechanical properties, such as Young’s modulus, elasticity limit and rupture pressure, are determined and presented. The engineering equation was used to plot the stress-strain curve for each phase.From the results obtained, the chemical composition has a significant effect on the properties of these phases. The stress-strain behavior comprised elastic, yield, strain softening and strain hardening regions that were qualitatively in agreement with previous simulations and experimental results. These stress-strain diagrams obtained show a rapid increase in stress up to a maximum followed by a gradual drop when the specimen fails by ductile fracture. Under compression, the deformation behavior of β-Al3Mg2 and γ-Al12Mg17 phases is slightly similar. From the results, it was found that ε-Al30Mg23 phase are brittle under uniaxial compressive loading and γ-Al12Mg17 phase is very ductile under the same compressive loading.The engineering stress-strain relationship suggests that β-Al3Mg2 and γ-Al12Mg17 phases have high elasticity limit, ability to resist deformation and also have the advantage of being highly malleable. From this simulation, we also find that the mechanical properties under compressive load of ε-Al30Mg23 phase are evidently less than other phases, which makes it the weakest phase. The obtained results were compared with the previous experimental studies, and generally, there is a good correlation.The Al-Mg system was built and simulated using molecular dynamics (MD) software LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator). read less USED (low confidence) R. Vidal, J. Ferrón, C. I. Meyer, V. Q. Riascos, and F. Bonetto, “The influence of surface defects on the low energy scattering of Ar ions from a Cu(111) surface,” Surface Science. 2019. link Times cited: 4 USED (low confidence) H.-H. Zhang, B. Wang, Z. Xu, X. Li, and W. M. Yan, “Molecular dynamics simulation on evaporation enhancement of water and aqueous nano-films by the application of alternating electric field,” International Journal of Heat and Mass Transfer. 2019. link Times cited: 12 USED (low confidence) R. Fahdiran, E. Handoko, and I. Sugihartono, “Size dependencies on melting of Gold nanoparticle: A Molecular Dynamics study,” Journal of Physics: Conference Series. 2019. link Times cited: 4 Abstract: We study the size dependencies on melting of Gold nanopartic… read moreAbstract: We study the size dependencies on melting of Gold nanoparticle. The nanoparticles are built with different sizes and heated up with same temperature gradient from room temperature up to 1400 K. The trajectories of the atoms are investigated based on Molecular Dynamics (MD) simulation. Pressure evolution as a function of time shows the oscillation pattern as in the case of thermal induced melting. Analysis based on structure factor combined with Common Neighbour Analysis (CNA) indicated the properties of melting depends on nanoparticle sizes. read less USED (low confidence) H. Wu, P. Torkian, A. Zarei, I. Moradi, A. Karimipour, and M. Afrand, “Hydrodynamic and thermal flow in nanochannel to study effects of roughness by estimation the atoms positions via MD method,” International Journal of Numerical Methods for Heat & Fluid Flow. 2019. link Times cited: 11 Abstract: This paper aims to investigate atoms type and channel roughn… read moreAbstract: This paper aims to investigate atoms type and channel roughness effects on fluid behavior in nanochannel.,The results of mechanical properties of these structures are reported in this work by using molecular dynamics method.,The results show that nanochannel roughness is a limiting factor in flowing fluid in nanochannel. Moreover, fluids with less atomic weight have more free movement in ideal and non-ideal nanochannels.,For the study of mechanical properties of fluid/nanochannel system, the authors calculated parameters such as potential energy, density, temperature and velocity profiles of simulated fluids. read less USED (low confidence) M. Dardouri et al., “Anisotropy diffusion in monolayer growth of Au on Cu (110) by kinetic Monte Carlo method,” Molecular Crystals and Liquid Crystals. 2019. link Times cited: 2 Abstract: Morphological and Structural transitions during epitaxial gr… read moreAbstract: Morphological and Structural transitions during epitaxial growth of surface and interface are an important nanoscal phenomena. Understanding the diffusion of adatom in anisotropic surface on the fc... read less USED (low confidence) M. Zarringhalam, H. Ahmadi‐Danesh‐Ashtiani, D. Toghraie, and R. Fazaeli, “The effects of suspending Copper nanoparticles into Argon base fluid inside a microchannel under boiling flow condition by using of molecular dynamic simulation,” Journal of Molecular Liquids. 2019. link Times cited: 41 USED (low confidence) Z. Wang et al., “The interaction between grain boundary and tool geometry in nanocutting of a bi-crystal copper,” International Journal of Extreme Manufacturing. 2019. link Times cited: 17 Abstract: Anisotropy is one central influencing factor on achievable u… read moreAbstract: Anisotropy is one central influencing factor on achievable ultimate machined surface integrity of metallic materials. Specifically, grain boundary has a strong impact on the deformation behaviour of polycrystalline materials and correlated material removal at the microscale. In the present work, we perform molecular dynamics simulations and experiments to elucidate the underlying grain boundary-associated mechanisms and their correlations with machining results of a bi-crystal Cu under nanocutting using a Berkovich tool. Specifically, crystallographic orientations of simulated bi-crystal Cu with a misorientation angle of 44.1° are derived from electron backscatter diffraction characterization of utilized polycrystalline copper specimen. Simulation results reveal that blocking of dislocation motion at grain boundaries, absorption of dislocations by grain boundaries and dislocation nucleation from grain boundaries are operating deformation modes in nanocutting of the bi-crystal Cu. Furthermore, heterogeneous grain boundary-associated mechanisms in neighbouring grains lead to strong anisotropic machining behaviour in the vicinity of the grain boundary. Simulated machined surface morphology and machining force evolution in the vicinity of grain boundary qualitatively agree well with experimental results. It is also found that the geometry of Berkovich tool has a strong impact on grain boundary-associated mechanisms and resultant ploughing-induced surface pile-up phenomenon. read less USED (low confidence) Y. Peng, M. Zarringhalam, M. Hajian, D. Toghraie, S. J. Tadi, and M. Afrand, “Empowering the boiling condition of Argon flow inside a rectangular microchannel with suspending Silver nanoparticles by using of molecular dynamics simulation,” Journal of Molecular Liquids. 2019. link Times cited: 41 USED (low confidence) Q. Xiong, T. Kitamura, and Z. Li, “Thermomechanical responses in metal films under mechanical shock: A molecular dynamics study,” Journal of Thermal Stresses. 2019. link Times cited: 2 Abstract: A series of dynamic shock compressions of metal films are si… read moreAbstract: A series of dynamic shock compressions of metal films are simulated using molecular dynamics method. Characteristics of mechanical shock load are analyzed theoretically. Thermomechanical responses induced by mechanical shock, including pressure, temperature, volumetric strain, and shear strain, are presented graphically and analyzed from viewpoint of thermomechanical coupling. The results show that the temperature rise caused by volumetric strain (elastic) is reversible, while that caused by shear strain is irreversible. Under the same mechanical shock load, several metal films exhibit significant differences in thermomechanical responses and the potential reasons are analyzed from the differences of thermophysical parameters. read less USED (low confidence) J. Ren, G. Liang, and M. Lv, “Effect of different crystal orientations on the surface integrity during nanogrinding of monocrystalline nickel,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 7 Abstract: During nanofabrication, the processing of different crystal … read moreAbstract: During nanofabrication, the processing of different crystal faces along the surface of a workpiece varies because of size effects. The burr height and subsurface damage during nanogrinding of different crystal orientations in monocrystalline nickel are simulated by molecular dynamics in this study. The burr height and depth of the subsurface deformation layer are calculated quantitatively by atomic displacement, common neighbor analysis and dislocation analysis techniques. Among the directions considered in this paper, the burr height is the smallest when grinding in the (110)[110] direction, and the depth of the subsurface deformation layer is small when grinding along the (111) plane. This study demonstrates the effect of different slip paths on burr height and provides a reference for the actual processing of face-centered cubic crystals. read less USED (low confidence) B. Wang, H.-H. Zhang, Z. Xu, X.-dong Wang, Q. Zhao, and W. M. Yan, “Acceleration of aqueous nano-film evaporation by applying parallel electric field: A molecular dynamics simulation,” International Journal of Heat and Mass Transfer. 2019. link Times cited: 15 USED (low confidence) J. Wang, J. Bian, and G. Wang, “Calculation of surface energy density of rough surface by atomic simulations,” Applied Surface Science. 2019. link Times cited: 11 USED (low confidence) S. Ajori, H. Parsapour, R. Ansari, and A. Ameri, “Buckling behavior of various metallic glass nanocomposites reinforced by carbon nanotube and Cu nanowire: A molecular dynamics simulation study,” Materials Research Express. 2019. link Times cited: 19 Abstract: The reinforcement of various materials by nanofillers as nan… read moreAbstract: The reinforcement of various materials by nanofillers as nanocomposites has recently received the attention of many researchers. In the present research, molecular dynamics simulations are used to investigate the influence of nanowire (NW)/carbon nanotube (CNT) reinforcement on the buckling behavior of metallic glass matrix nanocomposites (MGMNCs). The buckling characteristics of nanocomposites made by adding Cu NWs, CNTs and Cu NW-encapsulated CNTs to metallic glass matrices are studied. The results demonstrate that MG alloys comprising just two elements (Cu and Zr) with higher Cu percentage have higher mechanical stability. Also, it is observed that adding NW leads to a negative effect on the buckling behavior, while adding CNT and NW-encapsulated CNT considerably increases the buckling force and strain of the metallic glass models. Moreover, it is found that the filled CNT is the most effective nanofiller for amending the buckling behavior of metallic glasses. Furthermore, as the size of nanofillers gets larger, the critical force increases and the critical strain decreases. read less USED (low confidence) S. Tiwari, G. Tucker, and D. McDowell, “The effect of hydrostatic pressure on the shear deformation of Cu symmetric tilt interfaces,” International Journal of Plasticity. 2019. link Times cited: 7 USED (low confidence) M. Dardouri, A. Hassani, A. Hasnaoui, A. Arbaoui, Y. Boughaleb, and K. Sbiaai, “Kinetic Monte Carlo simulations of coverage effect on Ag and Au monolayers growth on Cu (1 1 0),” Journal of Crystal Growth. 2019. link Times cited: 5 USED (low confidence) Z. Wang et al., “Effects of finite temperature on the surface energy in Al alloys from first-principles calculations,” Applied Surface Science. 2019. link Times cited: 19 USED (low confidence) J. Li et al., “Molecular dynamics simulation of mechanical properties of nanocrystalline platinum: Grain-size and temperature effects,” Physics Letters A. 2019. link Times cited: 41 USED (low confidence) W. Wei, L. C. Liu, H. Gong, M. Song, M. Chang, and D. C. Chen, “Fundamental mechanism of BCC-FCC phase transition from a constructed PdCu potential through molecular dynamics simulation,” Computational Materials Science. 2019. link Times cited: 20 USED (low confidence) H. Parsapour, S. Ajori, R. Ansari, and S. Haghighi, “Tensile characteristics of single-walled carbon nanotubes endohedrally decorated with gold nanowires: A molecular dynamics study,” Diamond and Related Materials. 2019. link Times cited: 12 USED (low confidence) P. Zhao and Y.-bo Guo, “Effect of Initial Indentation Position on Plastic Deformation Behaviors of Polycrystalline Materials via Molecular Dynamics Simulation,” Nano. 2019. link Times cited: 4 Abstract: Polycrystalline materials can be divided into four types of … read moreAbstract: Polycrystalline materials can be divided into four types of microstructural components, including grain cell (GC), grain boundary (GB), triple junction (TJ) and vertex points (VP). Nanoindentation at different microstructural components on the polycrystalline materials surface can lead to different plastic deformation behaviors of the polycrystalline materials. Due to experimental limitations, the indentation-induced internal stress and defect evolution process are difficult to investigate directly, especially for the polycrystalline materials with grain size less than 100[Formula: see text]nm. The molecular dynamics (MD) simulations were performed to unravel the initial indentation position effect on the elasticity/plastic deformation mechanism of polycrystalline copper. The results reveal that the initial indentation position governs the indentation force variation and defect distribution range due to the different dimensionalities of the microstructural components. The defect propagation as well as the internal stress transmission in the GC regions tend to transfer to the low-dimensional microstructural components of the interfaces. In addition, the atomic internal stress and potential energy accumulation/release of the microstructural component atoms during the nanoindentation process are also investigated, revealing that the atomic internal stress and potential energy in the VPs vary earliest, followed by the TJs, GBs and GCs. read less USED (low confidence) S. Ajori, H. Parsapour, and R. Ansari, “Stability analysis of endohedrally functionalized carbon nanotubes with pentagonal metallic nanowires: a molecular dynamics simulation approach,” Materials Research Express. 2019. link Times cited: 8 Abstract: The endohedral functionalization of single-walled carbon nan… read moreAbstract: The endohedral functionalization of single-walled carbon nanotubes with molecular species, nanowires (NWs) and nanoparticles is of great importance for fabrication and development of nanoelecronic devices, drug delivery and energy storage applications. This research intends to explore the axial buckling behavior of the endohedrally functionalized single-walled carbon nanotubes (SWCNTs) by various metallic NWs (mNW@SWCNT), i.e. aluminum, copper, iron, sodium, nickel (AlNW, CuNW, FeNW, NaNW, NiNW), considering all possible pentagonal configurations. Employing the molecular dynamics (MD) simulations, the results demonstrate that the structurally stable radius of SWCNTs for successful endohedral functionalization of SWCNTs with pentagonal NWs are different. Considering buckling analysis of models, it is observed that NWs, solely, cannot tolerate any axial compressive load and their structure becomes dramatically unstable under mechanical force. By inserting NWs inside SWCNTs, their pentagonal structures during simulation are preserved due to Vdw interaction of NW and SWCNT until buckling occurs. Moreover, the buckling simulation results indicate that by increasing the length, the critical force of mNW@SWCNT decreases and approximately tends to that of pure SWCNTs which is more considerable for AlNWs. Also, in the particular length, the encapsulation of NWs inside the SWCNTs causes a considerable increase in the critical buckling forces particularly in smaller lengths. According to the attained results, functionalization of SWCNTs with E and S configuration of AlNWs improves the structural stability of SWCNTs more pronounced than other pentagonal NWs. read less USED (low confidence) Z. Hao, R. Cui, Y. Fan, and J. Lin, “Diffusion mechanism of tools and simulation in nanoscale cutting the Ni–Fe–Cr series of Nickel-based superalloy,” International Journal of Mechanical Sciences. 2019. link Times cited: 43 USED (low confidence) L. Lijia et al., “Nanoindentation response of monocrystalline copper under various tensile pre-deformations via molecular dynamic simulations:” Advances in Mechanical Engineering. 2018. link Times cited: 1 Abstract: The mechanical properties of a material can be positively or… read moreAbstract: The mechanical properties of a material can be positively or negatively affected by its applied or residual stress. In this article, a series of molecular dynamic simulations were adopted to invest... read less USED (low confidence) A. A. Nazarov, “Molecular dynamics simulation of the effect of cyclic stresses on nanocrystals with nonequilibrium grain boundaries: the role of the grain size,” IOP Conference Series: Materials Science and Engineering. 2018. link Times cited: 1 Abstract: Grain boundaries (GBs) in bulk nanostructured materials proc… read moreAbstract: Grain boundaries (GBs) in bulk nanostructured materials processed by severe plastic deformation (SPD) have a nonequilibrium structure caused by extrinsic grain boundary dislocations (EGBDs) absorbed during deformation. Under external influences (annealing or cyclic straining) these GBs relax towards equilibrium resulting in a release of the excess energy of nanomaterials. Using our earlier developed method, atomic models of nanocrystals with nonequilibrium GBs having grain sizes of 10, 15, and 20 nm are constructed and relaxed by molecular dynamics. Then, the effect of oscillating tension-compression stresses with amplitudes of 2, 3 and 4 GPa on the systems thus obtained is simulated. Relaxation of nonequilibrium GBs by dislocation emission is found and the effect is shown to be independent of the grain size. read less USED (low confidence) L. Wang, J. Hou, H. Lu, W.-jun Lu, Y. Dai, and C.-L. Luo, “The liquid-solid phase transition characteristics of AgxCu(500−x) alloy particles: a molecular dynamics study,” Materials Research Express. 2018. link Times cited: 2 Abstract: The phase transition characteristics of AgxCu(500−x) (x = 0,… read moreAbstract: The phase transition characteristics of AgxCu(500−x) (x = 0, 100, 125, 200, 250, 300, 375, 400, 500) alloy particles are studied by molecular dynamics using the embedded atom potentials. We find that the Ag-Cu alloy particles tend to be Cu-core/Ag-shell structure. Ag300Cu200 alloy particle forms a glass phase at a quenching rate of more than 2 × 1011 K s−1. It is an atom-level amorphous alloying particle. The binary Ag-Cu alloy particle with stoichiometry Ag3Cu2 is favorable for producing amorphous phase and the phase transition temperature of Ag-Cu alloy particles is typically less than or close to the phase transition temperature of the Ag particle at the same size. read less USED (low confidence) T. D. Gupta, D. Dutta, and M. R. B. Shahadat, “Temperature and Strain Rate Dependent Mechanical Properties of a Square Nickel Plate with Different Shaped Central Cracks: A Molecular Dynamics Study,” Journal of Nano Research. 2018. link Times cited: 2 Abstract: In our present study, under uniaxial tension, atomistic simu… read moreAbstract: In our present study, under uniaxial tension, atomistic simulations were conducted to explore the crack propagation mechanism of Square Nickel Plate (SNP) for two distinct shaped cracks (Rectangular and Circular) at center separately. Here, for modeling the inter-atomic potential between atoms, Embedded Atom Model (EAM) was used. In case of both types, the crack size was varied keeping a constant strain rate of 2×109 s-1 and temperature of 300 k for investigation of the effects of crack geometry and size on the behavior of crack propagation. Along with the size and geometry of crack, the effects of different strain rates (1×109, 2×109 and 4×109 s-1) and temperatures (300 K, 600 K and 900 k) were also studied. From the simulations, the declination nature of peak stress can be deduced for both of the geometries by increasing the crack size. It can also be concluded that when crack area was same, the peak stresses were higher in SNP with Circular crack than with the SNP with Rectangular one. Besides, increasing and decreasing nature of peak stress were found for two genres with the increment of strain rate and temperature separately. read less USED (low confidence) J. González, J. Ortega, and Z. Liang, “Prediction of thermal conductance at liquid-gas interfaces using molecular dynamics simulations,” International Journal of Heat and Mass Transfer. 2018. link Times cited: 23 USED (low confidence) I. Matrane, M. Mazroui, and Y. Boughaleb, “Diffusion and adsorption of Au and Pt adatoms on ideal and missing row reconstructed surfaces of Au(110): DFT and EAM calculations,” Surface Science. 2018. link Times cited: 3 USED (low confidence) S. Ajori, H. Parsapour, and R. Ansari, “Vibrational analysis of single-walled carbon nanotubes filled with gold nanowires using MD simulations,” Physica E: Low-dimensional Systems and Nanostructures. 2018. link Times cited: 10 USED (low confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “Molecular dynamics simulation to investigate the orientation effects on nanoscale cutting of single crystal copper,” Computational Materials Science. 2018. link Times cited: 66 USED (low confidence) A. Nazarov and R. Murzaev, “Nonequilibrium grain boundaries and their relaxation under oscillating stresses in columnar nickel nanocrystals studied by molecular dynamics,” Computational Materials Science. 2018. link Times cited: 15 USED (low confidence) Z. Kang and B. Wu, “Coalescence of gold nanoparticles around the end of a carbon nanotube: A molecular-dynamics study,” Journal of Manufacturing Processes. 2018. link Times cited: 5 USED (low confidence) B. Mobedpour, S. Rajabdoust, and R. Roumina, “Melting of graphene supported Pd-Pt core-shell nanoparticles: A molecular dynamics study,” Computational Materials Science. 2018. link Times cited: 4 USED (low confidence) A. Nazarov and R. Murzaev, “Molecular Dynamics Simulation of Nonequilibrium Grain Boundaries in Ultrafine-Grained Nickel and their Relaxation under Cyclic Loading,” Defect and Diffusion Forum. 2018. link Times cited: 0 Abstract: Atomistic simulations of the structure, energy and relaxatio… read moreAbstract: Atomistic simulations of the structure, energy and relaxation under the action of high frequency cyclic straining are carried out for columnar nickel nanocrystals with [112] column axis, the grain boundaries (GBs) of which are in a nonequilibrium state caused by the presence of extrinsic grain boundary dislocations (EGBDs). A special method of introducing EGBDs is used to create initial structures with nonequilibrium GBs. Energy of GBs as a function of the degree of nonequilibrium is evaluated and qualitatively compared to the results of dislocation and disclination modeling. It is shown that under loading by symmetrically oscillating stresses the nonequilibrium GBs generate lattice dislocations, which travel across the grains and are absorbed by opposite GBs thus resulting in a relaxation of the structure, long-range stress fields and the energy of GBs. read less USED (low confidence) T. T. Li, C. He, W. Zhang, and M. Cheng, “Structural and melting properties of Cu-Ni clusters: A simulation study,” Journal of Alloys and Compounds. 2018. link Times cited: 20 USED (low confidence) X. Yuan and Y. Wang, “Radial deformation of single-walled carbon nanotubes adhered to solid substrates and variations of energy: Atomistic simulations and continuum analysis,” International Journal of Solids and Structures. 2018. link Times cited: 8 USED (low confidence) G. Zhu, J. Sun, L. Zhang, and Z. Gan, “Molecular dynamics simulation of temperature effects on deposition of Cu film on Si by magnetron sputtering,” Journal of Crystal Growth. 2018. link Times cited: 20 USED (low confidence) M. Al and E. Webb, “A case study of thin film stress evolution at a dissimilar material interface via molecular dynamics simulations,” Nanomaterials and Nanotechnology. 2018. link Times cited: 6 Abstract: Evolution of deformation and stress in growing thin films ha… read moreAbstract: Evolution of deformation and stress in growing thin films has been studied in this work using computational simulations that resolve matter at atomic length and time scales. For the surface layers of films laying on the substrate of a dissimilar material, the stress distribution analysis around defects becomes more challenging. Herein, spatial and temporal distribution of deformation and associated stress evolution are presented for different thin film formation events including (1) sub-monolayer growth during an early film nucleation stage and (2) coalescence of adjacent monolayer “islands.” Validity of the stress computed via local computations of the virial expression for stress in a system of interacting particles was checked by comparing to results obtained from considerations of local atomic deformation in conjunction with existing expressions for epitaxial thin film growth stress. For the geometries studied here, where a monolayer of film with a highly characterized linear defect, as in the case of a stacking fault, was simulated for coalescence, fairly good agreement was found. This result demonstrates that, for similar defects at the surface layer, with sufficient sub-ensemble averaging of the standard virial expression for stress, semiquantitative spatial stress distribution information can be obtained from atomic scale simulations. Using our validated stress computation method, we reveal significant stress localization during thin film growth processes, leading to pronounced differences in maximum and minimum stress observed over very small spatial extent (of order multiple GPa over 3–6 nm distances). One prominent mechanism of stress localization revealed here is coalescence between adjacent growing islands. For geometries explored here, stress manifesting during coalescence is highly localized. read less USED (low confidence) P. Reyes et al., “The stability of hollow nanoparticles and the simulation temperature ramp,” Inorganic chemistry frontiers. 2018. link Times cited: 6 Abstract: Hollow nanoparticles (hNPs) are of interest because their la… read moreAbstract: Hollow nanoparticles (hNPs) are of interest because their large cavities and small thickness give rise to a large surface to volume ratio. However, in general they are not in equilibrium and far from their global energy minimum, which often makes them unstable against perturbations. In fact, a temperature increase can induce a structural collapse into a nanoparticle, and consequently a loss of their unique properties. This problem has been studied by means of molecular dynamics (MD) simulations, but without emphasis on the speed of the temperature increase. Here we explore how the temperature variation, and the rate at which it is varied in MD simulations, determines the final conformation of the hNPs. In particular, we show how different temperature ramps determine the final shape of Pt hNPs that initially have an external radius between 0.7 and 24 nm, and an internal radius between 0.19 and 2.4 nm. In addition, we also perform the simulations of other similar metals like Ag and Au. Our results indicate that the temperature ramp strongly modifies the final hNP shape, even at ambient temperature. In fact, a rapid temperature increase leads to the formation of stacking faults and twin boundaries which are not generated by a slower temperature increase. Quantitative criteria are established and they indicate that the stacking fault energy is the dominant parameter. read less USED (low confidence) Y. Zhang, W. Xiao, and P. Peng, “Study of Processability of Cu/Ni Bilayers Using Molecular Dynamics Simulations,” Journal of Nano Research. 2018. link Times cited: 2 Abstract: Nanoscratching and nanoindentation simulations are performed… read moreAbstract: Nanoscratching and nanoindentation simulations are performed to study the processability of Cu/Ni bilayers with interfaces using molecular dynamics (MD) method. Single crystals Cu and Ni are served as comparisons. In the nanoscratching processes, the interfaces of Cu/Ni bilayers appear as a barrier of dislocations gliding, and lead to larger friction forces and normal forces. For single crystals and bilayers, both their friction forces and normal forces increase with the increasement of scratch velocity at 100-300 m/s. Friction coefficients under scratching processes are calculated, and they are smaller than macrosacle scratching process because of coating effects of nano-chips on the tool. The effects are analyzed by conducting both molecular dynamics simulations in nanoscale and finite element simulations (FES) in macroscale. In the indentation process, the processing properties of Cu-Ni and Ni-Cu bilayers are different from each other, and their indentation forces are both larger than their single crystals. Recovery deformation takes place during the relaxation stage. When the tool is unloading, some workpiece atoms adhere to the tool. The simulation results of the two nanoscale machining processes reveal the strengthening mechanism of interface, and show comprehensive processability of metal bilayers. read less USED (low confidence) Y. Gao, M. Takahashi, C. Cavallotti, and G. Raos, “Molecular dynamics simulation of metallic impurity diffusion in liquid lead-bismuth eutectic (LBE),” Journal of Nuclear Materials. 2018. link Times cited: 13 USED (low confidence) Y. Xian, J. Li, R. Wu, and R. Xia, “Softening of nanocrystalline nanoporous platinum: A molecular dynamics simulation,” Computational Materials Science. 2018. link Times cited: 29 USED (low confidence) M. Jamshidian, A. Dehghani, M. S. Talaei, and T. Rabczuk, “Size dependent surface energy of nanoplates: Molecular dynamics and nanoscale continuum theory correlations,” Physics Letters A. 2018. link Times cited: 7 USED (low confidence) A. Nazarov and R. Murzaev, “Molecular Dynamics Study of Nonequilibrium [112] Tilt Grain Boundaries in Ni and their Relaxation under Cyclic Deformation,” Journal of Metastable and Nanocrystalline Materials. 2018. link Times cited: 3 Abstract: Atomic structure of nonequilibrium [112] tilt grain boundari… read moreAbstract: Atomic structure of nonequilibrium [112] tilt grain boundaries in nickel containing disclination dipoles is studied by means of molecular dynamics simulations. Initial systems for simulations are constructed by joining together pieces of two bicrystals one of which contains a symmetric tilt GB S=11 / 62.96° and the other a GB S=105 / 57.12°, or S=125 / 55.39°, or S=31 / 52.20°, so disclination dipoles with strengths w = 5.84°, 7.58° and 10.76° are created. Stress maps plotted after relaxation at zero temperature indicate the presence of high long-range stresses induced by disclination dipoles. Excess energy of GBs due to the nonequilibrium structure is calculated. Effect of oscillating tension-compression stresses on the nonequilibrium GB structure is studied at temperature T = 300 K. The simulations show that the oscillating stress results in a generation of partial lattice dislocations by the GB, their glide across grains and sink at appropriate surfaces that results in a compensation of the disclination stress fields and recovery of an equilibrium GB structure and energy. read less USED (low confidence) C.-Y. Shih, C. Wu, H.-ming Wu, M. Shugaev, and L. Zhigilei, “Atomistic Simulations of the Generation of Nanoparticles in Short-Pulse Laser Ablation of Metals: Effect of Background Gas and Liquid Environments.” 2018. link Times cited: 1 Abstract: in the investigation of the fundamental mechanisms of laserm… read moreAbstract: in the investigation of the fundamental mechanisms of lasermaterial interactions. The advancements in the computational methodology and fast growth of available computing resources are rapidly expanding the range of problems amenable to atomistic modeling. This chapter provides an overview of the results obtained in recent simulations of laser ablation of metal targets in vacuum, a background gas, and a liquid environment. A comparison of the Chapter 12 read less USED (low confidence) F. Faraji and A. Rajabpour, “Fluid heating in a nano-scale Poiseuille flow: A non-equilibrium molecular dynamics study,” Current Applied Physics. 2017. link Times cited: 6 USED (low confidence) A. Rida, A. Makke, E. Rouhaud, and M. Micoulaut, “Influence of grain size on the mechanical properties of nano-crystalline copper; insights from molecular dynamics simulation.” 2017. link Times cited: 0 Abstract: We use molecular dynamics simulations to study the mechanica… read moreAbstract: We use molecular dynamics simulations to study the mechanical properties of a columnar nanocrystalline copper with a mean grain size between 8.91 nm and 24 nm. The used samples were generated by using a melting cooling method. These samples were submitted to uniaxial tensile test. The results reveal the presence of a critical mean grain size between 16 and 20 nm, where there is an inversion in the conventional Hall-Petch tendency. This inversion is illustrated by the increase of flow stress with the increase of the mean grain size. This transition is caused by shifting of the deformation mechanism from dislocations to a combination of grain boundaries sliding and dislocations. Moreover, the effect of temperature on the mechanical properties of nanocrystalline copper has been investigated. The results show a decrease of the flow stress and Young’s modulus when the temperature increases. read less USED (low confidence) M. Kozłowski, D. Scopece, J. Janczak-Rusch, L. Jeurgens, R. Abdank-Kozubski, and D. Passerone, “Validation of an Embedded-Atom Copper Classical Potential via Bulk and Nanostructure Simulations,” Diffusion Foundations. 2017. link Times cited: 0 Abstract: The validation of classical potentials for describing multic… read moreAbstract: The validation of classical potentials for describing multicomponent materials in complex geometries and their high temperature structural modifications (disordering and melting) requires to verify both a faithful description of the individual phases and a convincing scheme for the mixed interactions, like it is the case of the embedded atom scheme. The present paper addresses the former task for an embedded atom potential for copper, namely the widely adopted parametrization by Zhou, through application to bulk, surface and nanocluster systems. It is found that the melting point is underestimated by 200 degrees with respect to experiment, but structural and temperature-dependent properties are otherwise faithfully reproduced. read less USED (low confidence) W. Yu, Z. Wang, and S. Shen, “Edge dislocations interacting with a Σ11 symmetrical grain boundary in copper upon mixed loading: A quasicontinuum method study,” Computational Materials Science. 2017. link Times cited: 11 USED (low confidence) N. Cherbal, E. H. Megchiche, H. Zenia, K. Lounis, and M. Amarouche, “Enhanced atomic oxygen adsorption on defective nickel surfaces: An ab initio study,” Surface Science. 2017. link Times cited: 5 USED (low confidence) S. Acharya, S. I. Shah, and T. Rahman, “Diffusion of small Cu islands on the Ni(111) surface: A self-learning kinetic Monte Carlo study,” Surface Science. 2017. link Times cited: 15 USED (low confidence) G. Zhu, J. Sun, X. Guo, X. Zou, L. Zhang, and Z. Gan, “Molecular dynamics simulation of temperature effects on low energy near-surface cascades and surface damage in Cu,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 6 USED (low confidence) E. Symianakis and A. Kucernak, “Embedded atom method interatomic potentials fitted upon density functional theory calculations for the simulation of binary Pt Ni nanoparticles,” Computational Materials Science. 2017. link Times cited: 4 USED (low confidence) Y. Zhang, S. Jiang, X.-ming Zhu, and Y. Zhao, “Influence of void density on dislocation mechanisms of void shrinkage in nickel single crystal based on molecular dynamics simulation,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 29 USED (low confidence) L. Lin, S. Hui, G. Lu, S. Wang, X.-dong Wang, and D.-J. Lee, “Molecular dynamics simulations on dissolutive wetting of Al–Ni alloy droplets on NiAl substrate,” Journal of The Taiwan Institute of Chemical Engineers. 2017. link Times cited: 13 USED (low confidence) Y. Jia, S. Li, W. Qi, M. Wang, Z. Li, and Z. Wang, “Thermal stability of marks gold nanoparticles: A molecular dynamics simulation,” International Journal of Modern Physics B. 2017. link Times cited: 0 Abstract: Molecular dynamics (MDs) simulations were used to explore th… read moreAbstract: Molecular dynamics (MDs) simulations were used to explore the thermal stability of Au nanoparticles (NPs) with decahedral, cuboctahedral, icosahedral and Marks NPs. According to the calculated cohesive energy and melting temperature, the Marks NPs have a higher cohesive energy and melting temperature compared to these other shapes. The Lindemann index, radial distribution function, deformation parameters, mean square displacement and self-diffusivity have been used to characterize the structure variation during heating. This work may inspire researchers to prepare Marks NPs and apply them in different fields. read less USED (low confidence) Y. Zhang, S. Jiang, X.-ming Zhu, and Y. Zhao, “Influence of twist angle on crack propagation of nanoscale bicrystal nickel film based on molecular dynamics simulation,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 10 USED (low confidence) A. Ahadi, P. Hansson, and S. Melin, “Simulating Nanoindentation of Thin Cu Films Using Molecular Dynamics and Peridynamics,” Solid State Phenomena. 2016. link Times cited: 1 Abstract: Nanoindentation is a useful experimental method to character… read moreAbstract: Nanoindentation is a useful experimental method to characterize the micromechanical properties of materials. In this study molecular dynamics and peridynamics are used to simulate nanoindentation, with a spherical indenter targeting a thin single crystal Cu film, resting on an infinitely stiff substrate. The objective is to compare the results obtained from molecular dynamic simulations to those obtained using a peridynamic approach as regards the force-displacement curves and the deformation patterns after that the material parameters in the peridynamic model have been fitted to the force displacement curve from the molecular dynamic simulation. read less USED (low confidence) Y. Zhang, S. Jiang, X.-ming Zhu, and Y. Zhao, “A molecular dynamics study of intercrystalline crack propagation in nano-nickel bicrystal films with (0 1 0) twist boundary,” Engineering Fracture Mechanics. 2016. link Times cited: 18 USED (low confidence) Y. Zhang, S. Jiang, X.-ming Zhu, and D. Sun, “Orientation dependence of void growth at triple junction of grain boundaries in nanoscale tricrystal nickel film subjected to uniaxial tensile loading,” Journal of Physics and Chemistry of Solids. 2016. link Times cited: 12 USED (low confidence) T. Fu et al., “Molecular dynamics simulation of nanoindentation on Cu/Ni nanotwinned multilayer films using a spherical indenter,” Scientific Reports. 2016. link Times cited: 118 USED (low confidence) M. Trybula, “Structure and transport properties of the liquid Al80Cu20 alloy – A molecular dynamics study,” Computational Materials Science. 2016. link Times cited: 22 USED (low confidence) W. Shi, X. Luo, Z. Zhang, Y. Liu, and W. Lu, “Influence of external load on the frictional characteristics of rotary model using a molecular dynamics approach,” Computational Materials Science. 2016. link Times cited: 12 USED (low confidence) D. Bolmatov, M. Zhernenkov, D. Zav’yalov, Y. Cai, and A. Cunsolo, “Terasonic Excitations in 2D Gold Nanoparticle Arrays in a Water Matrix as Revealed by Atomistic Simulations,” Journal of Physical Chemistry C. 2016. link Times cited: 13 Abstract: In this work we report on terahertz phononic excitations in … read moreAbstract: In this work we report on terahertz phononic excitations in two-dimensional (2D) gold nanoparticle arrays in a water matrix through a series of large-scale molecular dynamics simulations. For the first time, we observe acoustic Dirac-like crossings in H (H2O) atomic (molecular) networks that emerge due to an intraband phononic scattering. These crossings are the phononic fingerprints of ice-like arrangements of H (H2O) atomic (molecular) networks at nanometer scale. We reveal how phononic excitations in metallic nanoparticles and the water matrix reciprocally impact on one another providing the mechanism for the THz phononics manipulation via structural engineering. Furthermore, we show that by tuning the arrangement of 2D gold nanoparticle assemblies, the Au phononic polarizations experience subterahertz hybridization (Kohn anomaly) due to surface electron–phonon relaxation processes. This opens the way for the sound control and manipulation in soft matter metamaterials at nanoscale. read less USED (low confidence) A. Nassour, “Embedded atom approach for gold–silicon system from ab initio molecular dynamics simulations using the force matching method,” Bulletin of Materials Science. 2016. link Times cited: 2 USED (low confidence) Y. Zhang, S. Jiang, X.-ming Zhu, and Y. Zhao, “Dislocation mechanism of void growth at twin boundary of nanotwinned nickel based on molecular dynamics simulation,” Physics Letters A. 2016. link Times cited: 26 USED (low confidence) K. Sebeck, C. Shao, and J. Kieffer, “Alkane-Metal Interfacial Structure and Elastic Properties by Molecular Dynamics Simulation.,” ACS applied materials & interfaces. 2016. link Times cited: 9 Abstract: The structure of amorphous materials near the interface with… read moreAbstract: The structure of amorphous materials near the interface with an ordered substrate can be affected by various characteristics of the adjoining phases, such as the lattice spacing of the adherent surface, polymer chain length, and adhesive strength. To discern the influence of each of these factors, four FCC metal lattices are examined for three chain lengths of n-alkane and van der Waals interfacial interactions are controlled by adjusting the Lennard-Jones 12-6 potential parameters. The role of interaction strength is investigated for a single chain length and substrate combination. Four nanoconfined systems are also analyzed in terms of their mechanical strength. A strong layering effect is observed near the interface for all systems. The distinctiveness of polymer layering, i.e., the maximum density and spatial extent, exhibits a logarithmic dependence on the interaction strength between polymer and substrate. Congruency with the substrate lattice parameter further enhances this effect. Moreover, the elastic modulus of the alkane phase as a function of layer thickness indicates that the effects of ordering within the structure extend beyond the immediately obvious interfacial region. read less USED (low confidence) S. Chandra, M. K. Samal, V. Chavan, and R. Patel, “Edge cracks in nickel and aluminium single crystals: A molecular dynamics study.” 2016. link Times cited: 1 Abstract: A molecular dynamics study of edge cracks in Ni and Al singl… read moreAbstract: A molecular dynamics study of edge cracks in Ni and Al single crystals under mode-I loading conditions is presented. Simulations are performed using embedded-atom method potentials for Ni and Al at a temperature of 0.5K. The results reveal that Ni and Al show different fracture mechanisms. Overall failure behavior of Ni is brittle, while fracture in Al proceeds through void nucleation and coalescence with a zig-zag pattern of crack growth. The qualitative nature of results is discussed in the context of vacancy-formation energies and surface energies of the two FCC metals. read less USED (low confidence) W. Xiao et al., “MD and OKMC simulations of the displacement cascades in nickel,” Nuclear Science and Techniques. 2016. link Times cited: 3 USED (low confidence) W. Xiao et al., “MD and OKMC simulations of the displacement cascades in nickel,” Nuclear Science and Techniques. 2016. link Times cited: 0 USED (low confidence) P. Hansson, “Influence of surface roughening on indentation behavior of thin copper coatings using a molecular dynamics approach,” Computational Materials Science. 2016. link Times cited: 8 USED (low confidence) J. Wu, S. Nagao, Z. Zhang, and J. He, “Deformation and fracture of nano-sized metal-coated polymer particles: A molecular dynamics study,” Engineering Fracture Mechanics. 2015. link Times cited: 9 USED (low confidence) A. Prokhoda, “Formation of multiply twinned nanoparticles of pure (Al, Cu, Ni) metals during crystallization: Results of molecular dynamics simulation,” 2015 International Young Scientists Forum on Applied Physics (YSF). 2015. link Times cited: 0 Abstract: Structures of simulated metals (Al, Cu, Ni) obtained in resu… read moreAbstract: Structures of simulated metals (Al, Cu, Ni) obtained in result of isothermal annealing after quick cooling to certain temperatures are studied in detail. Obtained large enough structures heave a core skeleton from hcp-planes in the form of icosahedron that already are named “Ih-fractal” (tetrahedra with internal fcc structure are divided one from another by twinning hcp-planes). Rows from elementary decahedra in structures of hcp-planes give the basis of new 60 twinning planes for forming new sectors at growing of such formation. New rows from decahedra are appearing in the places of intersections of the secondary twinning planes, and a new family of twinning planes is forming. read less USED (low confidence) F. T. Latypov and A. Mayer, “Shear strength of metals under uniaxial deformation and pure shear,” Journal of Physics: Conference Series. 2015. link Times cited: 5 Abstract: In this paper, we investigate the dynamic shear strength of … read moreAbstract: In this paper, we investigate the dynamic shear strength of perfect monocrystalline metals using the molecular dynamics simulation. Three types of deformation (single shear, uniaxial compression and tension) are investigated for five metals of different crystallographic systems (fcc, bcc and hcp). A strong dependence of the calculated shear strength on the deformation type is observed. In the case of bcc (iron) and hcp (titanium) metals, the maximal shear strength is achieved at the uniaxial compression, while the minimal shear strength is observed at the uniaxial tension. In the case of fcc metals (aluminum, copper, nickel) the largest strength is achieved at the pure shear, the lowest strength is obtained at the uniaxial compression. read less USED (low confidence) M. M. Sichani and D. Spearot, “A molecular dynamics study of the role of grain size and orientation on compression of nanocrystalline Cu during shock,” Computational Materials Science. 2015. link Times cited: 25 USED (low confidence) J. Zhang, L. Geng, Y. Yan, and T. Sun, “Effect of tool geometry in nanometric cutting of nanotwinned Cu: a molecular dynamics study,” International Journal of Nanomanufacturing. 2015. link Times cited: 5 Abstract: In the present work we investigate the nanometric cutting of… read moreAbstract: In the present work we investigate the nanometric cutting of a nanotwinned Cu containing 26° inclined twin boundaries using a diamond cutting tool by means of molecular dynamics simulations, with a focus on examining the influence of rake angle of cutting tools on the cutting processes. The underlying deformation mechanisms of the material are elucidated and are further correlated with the evolution of machining forces and the formation of machined surface and chips. Our simulation results indicate that dislocation slip, interaction of dislocation with twin boundaries and twin boundaries-associated mechanisms work in parallel in the plastic deformation of the nanotwinned Cu. It is found that the rake angle has a significant influence on the deformation behaviour of the material, chip formation and machined surface quality. A rake angle of 45° results in smaller energy dissipation and better machined surface quality than the other two rake angles of 0° and −45°. read less USED (low confidence) A. Pavlova et al., “Local anodic oxidation of Ni films with (2 0 0) and (1 1 1) texture,” Applied Surface Science. 2015. link Times cited: 4 USED (low confidence) A. Vorontsov, “Structure of overheated metal clusters: MD simulation study.” 2015. link Times cited: 0 Abstract: The structure of overheated metal clusters appeared in conde… read moreAbstract: The structure of overheated metal clusters appeared in condensation process was studied by computer simulation techniques. It was found that clusters with size larger than several tens of atoms have three layers: core part, intermediate dense packing layer and a gas- like shell with low density. The change of the size and structure of these layers with the variation of internal energy and the size of cluster is discussed. read less USED (low confidence) J. Li, Q. Fang, B. Liu, Y.-xiang Liu, Y. W. Liu, and P. Wen, “Mechanism of crack healing at room temperature revealed by atomistic simulations,” Acta Materialia. 2015. link Times cited: 23 USED (low confidence) S. Mandal, T. Sahoo, S. Ghosh, and S. Adhikari, “The effect of surface temperature on H2/D2(v = 0, j = 0)–Ni(100) scattering processes,” Molecular Physics. 2015. link Times cited: 5 Abstract: We carry out both four-dimensional (4D×2D) and six-dimension… read moreAbstract: We carry out both four-dimensional (4D×2D) and six-dimensional (6D) quantum dynamics on a parametrically time- and temperature-dependent effective Hamiltonian for H2/D2(v = 0,j = 0)–Ni(100) collision process. Such an effective potential was derived within a theoretical framework of mean-field approximation by considering weakly correlated interaction between molecular degrees of freedom, phonon modes and electron– hole pair (elhp) coupling through a Hartree-product-type wave function, where the initial state distribution of the surface modes and elhp coupling were introduced through Bose– Einstein and Fermi– Dirac probability factor, respectively. The temperature-dependent dissociation and state-to-state transition probabilities for H2/D2(v = 0,j = 0)–Ni(100) system are depicted as a function of initial kinetic energ of the incoming diatom. Though such effect appears negligibly small for H2(v = 0,j = 0)–Ni(100) system, it is prominent in the case of D2(v = 0,j = 0)–Ni(100) collision. It appears that the change of dissociation and transition probabilities of D2 with the increase of surface temperature is exclusively dictated by the phonon modes directed along Z-axis, but the effect of elhp coupling particularly for transition probabilities is insignificant. read less USED (low confidence) S. Fensin, S. Valone, E. Cerreta, P. Rigg, and G. Gray, “Nucleation and Evolution of Dynamic Damage at Cu/Pb Interfaces using Molecular Dynamics,” Bulletin of the American Physical Society. 2015. link Times cited: 4 Abstract: For ductile metals, the process of dynamic fracture occurs t… read moreAbstract: For ductile metals, the process of dynamic fracture occurs through nucleation, growth and coalescence of voids. For high purity single-phase metals, it has been observed by numerous investigators that voids tend to heterogeneously nucleate at grain boundaries and all grain boundaries are not equally susceptible to void nucleation. However, for materials of engineering significance, especially those with second phase particles, it is less clear if the type of bi-metal interface between the two phases will affect void nucleation and growth. To approach this problem in a systematic manner two bi-metal interfaces between Cu and Pb have been investigated: {111} and {100}. Qualitative and quantitative analysis of the collected data from molecular dynamics shock and spall simulations suggests that Pb becomes disordered during shock compression and is the preferred location for void nucleation under tension. Despite the interfaces being aligned with the spall plane (by design), they are not the preferred location... read less USED (low confidence) S. Zhang and Y. Wang, “Molecular dynamics simulation of tension–compression asymmetry in plasticity of fivefold twinned Ag nanopillars,” Physics Letters A. 2015. link Times cited: 6 USED (low confidence) J. Li, Q. Fang, L. Zhang, and Y.-wen Liu, “The effect of rough surface on nanoscale high speed grinding by a molecular dynamics simulation,” Computational Materials Science. 2015. link Times cited: 61 USED (low confidence) S. Zhang, “Mechanical behaviors of single crystalline and fivefold twinned Ag nanowires under compression,” Computational Materials Science. 2015. link Times cited: 4 USED (low confidence) H. Su and Q. Tang, “Chip formation dependence of machining velocities in nano-scale by molecular dynamics simulations,” Science China Technological Sciences. 2014. link Times cited: 9 USED (low confidence) A. V. Sales, “Collective dynamics of bulk metallic glasses studied by molecular dynamics simulations.” 2014. link Times cited: 0 Abstract: The development of new materials impacts on all branches of … read moreAbstract: The development of new materials impacts on all branches of engineering and, in particular, in aerospace engineering. Metallic glasses (MG) are relatively newcomers to the world of materials science and have excellent mechanical properties; its study is mandatory to allow its technological implantation.
The macroscopic mechanical properties of a material are linked to its atomic structure. In particular, the fracture behaviour of brittle materials is initiated by the generation of vibrational modes. In metallic glasses, with amorphous structure, the vibrational spectrum has specific features.
In this work, the vibrational properties of metallic glasses are examined by Molecular Dynamics simulations. The study was focused in binary systems, which were simulated using different interatomic potentials: Lennard-Jones (LJ), Morse and the semiempirical Embedded atom (EAM). As in Pd-based metallic glasses, the ratio of masses of both species was high, namely 2 in Lennard-Jones potentials and 1.67 in Morse and EAM potentials. The large scale simulations allowed us to simulate systems with nm-scale heterogeneities frozen-in during the quenching process. Different relaxation states were obtained by changing the quenching rates of the simulated MGs.
The collective vibrational atomic dynamics of metallic glasses is a longstanding subject of debate. The origin of the excess of vibrational modes known as Boson Peak (BP) is not clear. In the systems analysed the dependence of the BP position and intensity on the system size is found to be weak. On the contrary, the BP intensity increases with the quenching rate, while its position shifts slightly to smaller frequencies. The results obtained by using realistic, semiempirical EAM potentials compare well with the experimental data available in glasses of similar compositions.
The dynamic structure factor, S(q, ¿) is also computed in large systems to get information on the behaviour of acoustic excitations at low wavenumbers. The dominant frequencies O L,T (q) are determined for each considered wavevector, in order to compute the relation of dispersion of longitudinal and transverse phonons. In all studied cases the width of the peak, GL,T(q), increases as the frequency increases. A linear region at low wavenumbers and a bending when approaching the limit of the first pseudo-Brillouin zone are found. This behaviour is the same than that observed experimentally by Inelastic X-Ray scattering.
The macroscopic sound speed is obtained for wavenumbers tending to zero. The values obtained with EAM and Morse potentials are in qualitative agreement with those obtained experimentally in systems of similar composition.
The Ioffer-Regel limit (IR), where the coherence length of the phonon is similar to the phonon wavelenght, was computed. It is found that the Ioffe-Regel frequency decreases slightly when applying faster quenching rates. The longitudinal Ioffe-Regel limit was found at frequencies higher than the Boson peak frequency for all the cases, although the diference in EAM systems is much reduced.
Contrary to the typical results obtained in the LJ systems, using EAM potentials in both Cu20Pd80 and Cu50Pd50 the longitudinal IR limit is very close to the position of the BP while the transversal IR limit is found well below. This behaviour is coherent with that found by IXS measurements. It is inferred that the EAM potential increases the interaction between the longitudinal modes and the BP excess states.
Finally the fragility of the studied systems was obtained by calculating the viscosity at different temperatures. Lennard-Jones systems showed a much larger fragility than EAM and Morse systems. However, even systems simulated with more realistic potentials showed fragility values much higher than those obtained experimentally. This is attributed to the extremely high quenching rates used in simulations, as it is known experimentally that fragility increases with the quenching rate.
El desenvolupament de nous materials té un gran impacte en totes les àrees de l'enginyeria, i en particular a l'enginyeria aeronàutica. Els vidres metàl·lics son materials relativament nous, amb excel·lents propietats mecàniques; el seu estudi és imprescindible per la seva implantació tecnològica. Les propietats mecàniques macroscòpics d'un material estan determinades per la seva estructura atòmica. En particular, la fractura de materials fràgils s'inicia per la generació de modes de vibració. Als vidres metàl·lics, d'estructura amorfa, l'espectre vibracional té característiques específiques. En aquest treball s'estudien les propietats vibracionals dels vidres metàl·lics emprant simulacions de dinàmica Molecular. L'estudi es centra en sistemes binaris simulats emprant potencials atòmics de Lennard-Jones (LJ), Morse i Embedded Atom method (EAM), de tipus semiempíric. Com als vidres metàl.lics basats en Pd, el quocient de les masses d'ambdues espècies en les simulacions és alt, essent 2 en potencials LJ i 1.67 en potencials Morse i EAM. Simulacions a gran escala permeten simular sistemes amb heterogeneities a escala nm, i la simulació a velocitats de refredament diferents permet obtenir configuracions en diferents estats de relaxació. La dinàmica de les vibracions atòmiques dels vidres metàl·lics és un tema de debat. L'origen de l'excés dels modes de vibració conegut com Boson Peak (BP) no és clar. Als sistemes analitzats s'observa que la dependència de la posició i la intensitat del BP amb la mida del sistema és feble. Al contrari, la intensitat de la BP augmenta amb la velocitat de refredament, mentre que la seva posició es desplaça a freqüències menors. Els resultats obtinguts emprant potencials de realista tipus EAM coincideixen amb les dades experimentals disponibles en vidres de composicions similars. El factor d'estructura dinàmica, S (q, ¿) es calculà també en sistemes grans per obtenir informació sobre el comportament de les excitacions acústiques en vectors d'ona baixos. També es va obtenir la relació de dispersió de fonons longitudinals i transversals. En tots els casos estudiats, l'amplada del pic, GL,T(q), augmenta amb la freqüència. S'observa una regió lineal a baixos nombres d'ona i una flexió quan s'aproxima el límit de la pseudo-zona de Brillouin. Aquest comportament és el mateix que l'observat experimentalment emprant Inelàstic X-Ray scattering (IXS). La velocitat del so macroscòpica s'obté quan el nombre d'ona tendeix a zero. Els valors obtinguts amb potencials EAM i Morse estan d'acord amb els valors mesurats experimentalment en sistemes de composició similar. El límit Ioffe-Regel (IR) es defineix com la freqüència a la que la longitud de coherència del fonó és similar a la longitud d'ona de fonó. S'observa que la freqüència IR augmenta lleugerament als sistemes més relaxats. El límit IR longitudinal és troba en tots els casos a freqüències superiors a la freqüència del BP. Contràriament als resultats obtinguts en els sistemes LJ, el límit longitudinal IR en Cu20Pd80 y Cu50Pd50 és molt a prop de la posició del BP, mentre que el límit transversal IR és molt per sota d'aquest valor; aquests resultats estan d'acord amb els obtinguts experimentalment per IXS. S'infereix que el potencial EAM augmenta la interacció entre modes longitudinals i l'excés de modes del BP. Finalment s'obtingué la fragilitat en els sistemes estudiats mitjançant el càlcul de la viscositat a temperatures diferents. És conegut experimentalment que la fragilitat augmenta amb la velocitat de refredament. Els sistemes LJ mostraren una fragilitat molt més alta que els Morse i EAM. No obstant això, fins i tot els sistemes simulats amb potencials realistes van mostrar valors de la fragilitat molt superiors als obtinguts experimentalment a causa de l'extrema velocitat extrema de refredament de les simulacions
El desarrollo de nuevos materiales tiene impacto en todas las áreas de la ingeniería, y en particular en la ingeniería aeronáutica. Los vidrios metálicos son materiales relativamente nuevos, con excelentes propiedades mecánicas; su estudio es imprescindible para su implantación tecnológica. Las propiedades mecánicas macroscópicas de un material están determinadas por su estructura atómica. En particular, la fractura
de materiales frágiles se inicia mediante la generación de modos de vibración. En los vidrios metálicos, con estructura amorfa, el espectro vibracional tiene características específicas. En este trabajo es estudian las propiedades vibracionales de los vidrios metálicos usando simulaciones de Dinámica Molecular. Se estudian sistemas binarios utilizando potenciales atómicos de Lennard-Jones (LJ), Morse y Embedded atom method (EAM), de tipo semiempírico. Al igual que en los vidrios metálicos a base de Pd, la relación de las masas de ambas especies en las simulaciones es alta, siendo 2 en potenciales LJ y 1,67 en potenciales Morse y EAM. Las simulaciones a gran escala permiten simular sistemas
con heterogeneidades a escala nm, y la simulación a diferente velocidad de enfriamiento permite obtener configuraciones en diferentes estados de relajación.
La dinámica de las vibraciones atómicas de los vidrios metálicos es un tema de debate. El origen del exceso de los modos de vibración conocidos como Boson Peak (BP) no es claro. En los sistemas analizados se observa que la dependencia de la posición y la intensitat del BP con el tamaño del sistema es débil. Por el contrario, la intensidad del BP aumenta con la velocidad de enfriamiento, mientras que su posición se desplaza a frecuencias menores. Los resultados obtenidos mediante el uso de potenciales realistas tipo EAM coinciden con los datos experimentales disponibles en vidres de composiciones similares.
El factor de estructura dinámica, S (q, ω) se calculó también en sistemas grandes para obtener información sobre el comportamiento de las excitaciones acústicas en vectores de onda bajos. También se obtuvo la relación de dispersión de fonones longitudinal read less USED (low confidence) W.-jin Zhang, Z.-L. Liu, and Y. Peng, “Molecular dynamics simulations of the melting curves and nucleation of nickel under pressure,” Physica B-condensed Matter. 2014. link Times cited: 11 USED (low confidence) Y. Wen, X. Fang, and X. Jia, “Atomistic Simulation of the Self-Diffusion in Very Thin Cu (001) Film by Using MAEAM,” Advanced Materials Research. 2014. link Times cited: 0 Abstract: The self-diffusion in very thin Cu (001) film that formed by… read moreAbstract: The self-diffusion in very thin Cu (001) film that formed by 2~11 atomic layers have been studied by using modified analytic embedded atom method (MAEAM) and a molecular dynamic (MD) simulation. The vacancy formation is the most easily in of Cu (001) thin film formed by any layers. The vacancy formation energy 0.5054eV in of the Cu (001) thin film formed by layers is the highest in all the values in the ones that formed by layers. The vacancy in and 3 is easily migrated to layer, and the vacancy in is easily migrated in intra-layer, and the vacancy in is easily migrated to when the corresponding atomic layer is existed. The vacancy formation and diffusion will not be affected by the atomic layer when the Cu (001) thin film is formed by more than ten layers (). read less USED (low confidence) T. Zientarski and D. Chocyk, “Strain and structure in nano Ag films deposited on Au: Molecular dynamics simulation,” Applied Surface Science. 2014. link Times cited: 9 USED (low confidence) T. Zientarski and D. Chocyk, “Structure and stress in Cu/Au and Fe/Au systems: A molecular dynamics study,” Thin Solid Films. 2014. link Times cited: 12 USED (low confidence) F. Granberg, S. Parviainen, F. Djurabekova, and K. Nordlund, “Investigation of the thermal stability of Cu nanowires using atomistic simulations,” Journal of Applied Physics. 2014. link Times cited: 14 Abstract: We present a method for determining the melting point of cop… read moreAbstract: We present a method for determining the melting point of copper nanowires based on classical molecular dynamics simulations and use it to investigate the dependence of the melting point on wire diameter. The melting point is determined as the temperature at which there is a significant change in the fraction of liquid atoms in the wire, according to atomic bond angle analysis. The results for the wires with diameters in the range 1.5 nm to 20 nm show that the melting point is inversely proportional to the diameter while the cross-sectional shape of the wire does not have a significant impact. Comparison of results obtained using different potentials show that while the absolute values of the melting points may differ substantially, the melting point depression is similar for all potentials. The obtained results are consistent with predictions based on the semi-empirical liquid drop model. read less USED (low confidence) L. Xie, P. Brault, J. Bauchire, A. Thomann, and L. Bedra, “Molecular dynamics simulations of clusters and thin film growth in the context of plasma sputtering deposition,” Journal of Physics D: Applied Physics. 2014. link Times cited: 40 Abstract: Carrying out molecular dynamics (MD) simulations is a releva… read moreAbstract: Carrying out molecular dynamics (MD) simulations is a relevant way to understand growth phenomena at the atomic scale. Initial conditions are defined for reproducing deposition conditions of plasma sputtering experiments. Two case studies are developed to highlight the implementation of MD simulations in the context of plasma sputtering deposition: ZrxCu1−x metallic glass and AlCoCrCuFeNi high entropy alloy thin films deposited onto silicon. Effects of depositing atom kinetic energies and atomic composition are studied in order to predict the evolution of morphologies and atomic structure of MD grown thin films. Experimental and simulated x-ray diffraction patterns are compared. read less USED (low confidence) C. Rouleau, C.-Y. Shih, C. Wu, L. Zhigilei, A. Puretzky, and D. Geohegan, “Nanoparticle generation and transport resulting from femtosecond laser ablation of ultrathin metal films: Time-resolved measurements and molecular dynamics simulations,” Applied Physics Letters. 2014. link Times cited: 46 Abstract: The synthesis of metal nanoparticles by ultrafast laser abla… read moreAbstract: The synthesis of metal nanoparticles by ultrafast laser ablation of nanometers-thick metal films has been studied experimentally and computationally. Near-threshold backside laser ablation of 2–20 nm-thick Pt films deposited on fused silica substrates was found to produce nanoparticles with size distributions that were bimodal for the thicker films, but collapsed into a single mode distribution for the thinnest film. Time-resolved imaging of blackbody emission from the Pt nanoparticles was used to reveal the nanoparticle propagation dynamics and estimate their temperatures. The observed nanoparticle plume was compact and highly forward-directed with a well-defined collective velocity that permitted multiple rebounds with substrates to be revealed. Large-scale molecular dynamics simulations were used to understand the evolution of compressive and tensile stresses in the thicker melted liquid films that lead to their breakup and ejection of two groups of nanoparticles with different velocity and size distributions. Ultrafast laser irradiation of ultrathin (few nm) metal films avoids the splitting of the film and appears to be a method well-suited to cleanly synthesize and deposit nanoparticles from semitransparent thin film targets in highly directed beams. read less USED (low confidence) B. Peng, W. He, X. Hao, Y. Chen, and Y. Liu, “Interfacial thermal conductance and thermal accommodation coefficient of evaporating thin liquid films: A molecular dynamics study,” Computational Materials Science. 2014. link Times cited: 8 USED (low confidence) W. Yu and Z. Wang, “‘Positive’ and ‘negative’ edge dislocations simultaneously interacting with Σ11 GB during nanoindentation,” Computational Materials Science. 2014. link Times cited: 7 USED (low confidence) C. V. D. Walt, J. J. Terblans, and H. Swart, “Molecular dynamics study of the temperature dependence and surface orientation dependence of the calculated vacancy formation energies of Al, Ni, Cu, Pd, Ag, and Pt,” Computational Materials Science. 2014. link Times cited: 17 USED (low confidence) Y. Han, J. Zhou, J. Dong, and K. Yoshiyuki, “Formation of single-walled bimetallic coinage alloy nanotubes in confined carbon nanotubes: molecular dynamics simulations.,” Physical chemistry chemical physics : PCCP. 2013. link Times cited: 8 Abstract: The growth of single-walled bimetallic Au-Ag, Au-Cu and Ag-C… read moreAbstract: The growth of single-walled bimetallic Au-Ag, Au-Cu and Ag-Cu alloy nanotubes (NTs) and nanowires (NWs) in confined carbon nanotubes (CNTs) has been investigated by using the classical molecular dynamics (MD) method. It is found that three kinds of single-walled gold-silver, gold-copper and silver-copper alloy NTs could indeed be formed in confined CNTs at any alloy concentration, whose geometric structures are less sensitive to the alloy concentration. And an extra nearly pure Au (Cu) chain will exist at the center of Au-Ag (Au-Cu and Ag-Cu) NTs when the diameters of the outside CNTs are big enough, thus producing a new type of tube-like alloy NWs. The bonding energy differences between the mono- and hetero-elements of the coinage metal atoms and the quasi-one-dimensional confinement from the CNT play important roles in suppressing effectively the "self-purification" effects, leading to formation of these coinage alloy NTs. In addition, the fluid-solid phase transition temperatures of the bimetallic alloy NTs are found to locate between those of the corresponding pure metal tubes. Finally, the dependences of the radial breathing mode (RBM) frequencies and the tube diameters of the alloy NTs on the alloying concentration were obtained, which will be very helpful for identifying both the alloying concentration and the alloy tube diameters in future experiments. read less USED (low confidence) P. Zhang, H. Zhao, C. Shi, L. Zhang, H. Huang, and L. Ren, “Influence of double-tip scratch and single-tip scratch on nano-scratching process via molecular dynamics simulation,” Applied Surface Science. 2013. link Times cited: 59 USED (low confidence) S. Yu, B. Bahrim, B. Makarenko, and J. Rabalais, “Dynamics conditions for channeling during H− scattering on Cu(111),” Surface Science. 2013. link Times cited: 3 USED (low confidence) H. Shang and W. Wang, “Shock responses of graphene reinforced composites via molecular dynamics simulations,” Journal of Physics: Conference Series. 2013. link Times cited: 10 Abstract: Shock responses of graphene reinforced composites are invest… read moreAbstract: Shock responses of graphene reinforced composites are investigated using molecular dynamics simulations. The first case studied is the response of spaced multilayer graphene plates under normal impact of a spherical projectile, focusing on the effect of the number of graphene monolayers per plate on the penetration resistance of the armor. The simulation results indicate that the penetration resistance increases with decreasing number of graphene monolayers per plate. The second case studied is the penetration resistance of laminated copper/graphene composites. The simulation results demonstrate that under normal impact by a spherical projectile the penetration resistance of copper can be improved significantly by laminating the copper plates with graphene. The results of this research have revealed the possibility that graphene might be used in hyper velocity-relevant armor systems to enhance their penetration resistance. read less USED (low confidence) D. Chocyk and T. Zientarski, “Study of Structure and Strain in Au/Cu Systems Using Molecular Dynamics Simulation: X-Ray Scattering Analysis,” Solid State Phenomena. 2013. link Times cited: 1 Abstract: The aim of this work is to investigate structure and stress … read moreAbstract: The aim of this work is to investigate structure and stress evolution in Au/Cu bilayer systems during deposition. The approach used here is based on an embedded atom method (EAM). interatomic potential database for different metal elements, their alloys and multilayers. We applied the kinematical scattering theory to calculate the X-ray scattering profiles. In this case the X-ray scattering techniques are used for the structural characterization of crystal structures obtained from simulation data. This method was applied to determine the lattice parameters in any directions. The lattice parameters in deposited layers were directly determined by the analysis of X-ray diffraction profiles. Results shows that on the interface of Au/Cu system, the crystalline lattice of Au layer is fitted to crystalline lattice of Cu layer. We found that deformation of the crystal lattice near the interface has a major influence on the stress. read less USED (low confidence) H. Alarifi, M. Atis, C. Özdogan, A. Hu, M. Yavuz, and Y. Zhou, “Determination of Complete Melting and Surface Premelting Points of Silver Nanoparticles by Molecular Dynamics Simulation,” Journal of Physical Chemistry C. 2013. link Times cited: 105 Abstract: A molecular dynamics simulation based on the embedded-atom m… read moreAbstract: A molecular dynamics simulation based on the embedded-atom method was conducted at different sizes of single-crystal Ag nanoparticles (NPs) with diameters of 4 to 20 nm to find complete melting and surface premelting points. Unlike the previous theoretical models, our model can predict both complete melting and surface premelting points for a wider size range of NPs. Programmed heating at an equal rate was applied to all sizes of NPs. Melting kinetics showed three different trends that are, respectively, associated with NPs in the size ranges of 4 to 7 nm, 8 to 10 nm, and 12 to 20 nm. NPs in the first range melted at a single temperature without passing through a surface premelting stage. Melting of the second range started by forming a quasi-liquid layer that expanded to the core, followed by the formation of a liquid layer of 1.8 nm thickness that also subsequently expanded to the core with increasing temperature and completed the melting process. For particles in the third range, the 1.8 nm liquid laye... read less USED (low confidence) S. Sun, X.-min Chen, F. Zhang, and B. Yang, “Ab Initio Molecular Dynamics Simulations of Cu under Vacuum and 473 1573K,” Advanced Materials Research. 2013. link Times cited: 0 Abstract: The structure and properties simulation of Cu under vacuum w… read moreAbstract: The structure and properties simulation of Cu under vacuum were studied by ab initio molecular dynamics simulation. The calculation results were characterized in terms of radial distribution function (RDF), coordination number (CN) and partial density of states (PDOS). The results show that the average distance between atoms increased with the temperature, while CN decreased, which indicated an obvious improvement of the thermal motion between atoms. The simulation datas showed that the liquid phase appeared in the system when the temperature arrived 1373K,which close to the melting point(1357K) of copper. read less USED (low confidence) S. Sun, X.-min Chen, F. Zhang, and B. Yang, “Molecular Dynamics Simulations of the Structure Transformation during the Cu Heating Process Under Vacuum.” 2013. link Times cited: 0 USED (low confidence) H. Gong, W. Lu, L. Wang, G. Li, and S. Zhang, “The effect of deposition velocity and cluster size on thin film growth by Cu cluster deposition,” Computational Materials Science. 2012. link Times cited: 23 USED (low confidence) W. Zhu, H. Wang, and W. Yang, “Orientation- and microstructure-dependent deformation in metal nanowires under bending,” Acta Materialia. 2012. link Times cited: 21 USED (low confidence) Y. Cheng, M. X. Shi, and Y.-W. Zhang, “Atomistic simulation study on key factors dominating dislocation nucleation from a crack tip in two FCC materials: Cu and Al,” International Journal of Solids and Structures. 2012. link Times cited: 27 USED (low confidence) W. Yu and Z. Wang, “Interactions between edge lattice dislocations and Σ11 symmetrical tilt grain boundaries in copper: A quasi-continuum method study,” Acta Materialia. 2012. link Times cited: 29 USED (low confidence) S. Rawat, M. Warrier, S. Chaturvedi, and V. Chavan, “Fracture during high-velocity impact of copper plates: a molecular dynamics study,” Journal of Physics: Conference Series. 2012. link Times cited: 0 Abstract: High velocity impact of copper plates has been studied using… read moreAbstract: High velocity impact of copper plates has been studied using the molecular dynamics code LAMMPS. The impact of copper plates at 1100 m/s impact velocity shows that spall of the material takes place at 160 kbar tensile pressure. Void nucleation is not observed at lower impact velocities where the peak tensile pressure stays below 160 kbar. No significant void nucleation occurs in the neighbouring regions where the peak tensile pressure stays below 160 kbar. For impact velocities close to the threshold, we observe stochastic behaviour of the spall process with respect to small changes in the initial atomic coordinates. read less USED (low confidence) C. Yan, D. Junhong, and X. D. He, “Molecular dynamics simulation of low-energy bombardment of Pt (1 1 1) surface by Cu atoms with various incident angles,” Physica B-condensed Matter. 2012. link Times cited: 1 USED (low confidence) A. Leino, O. Pakarinen, F. Djurabekova, and K. Nordlund, “A study on the elongation of embedded Au nanoclusters in SiO2 by swift heavy ion irradiation using MD simulations,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2012. link Times cited: 19 USED (low confidence) S. Eich, M. Kasprzak, A. Gusak, and G. Schmitz, “On the mechanism of diffusion-induced recrystallization: Comparison between experiment and molecular dynamics simulations,” Acta Materialia. 2012. link Times cited: 6 USED (low confidence) P. Zhu, Y.-zhong Hu, F. Fang, and H. Wang, “Multiscale simulations of nanoindentation and nanoscratch of single crystal copper,” Applied Surface Science. 2012. link Times cited: 39 USED (low confidence) V. Timoshenko, V. Bochenkov, V. Traskine, and P. Protsenko, “Anisotropy of Wetting and Spreading in Binary Cu-Pb Metallic System: Experimental Facts and MD Modeling,” Journal of Materials Engineering and Performance. 2012. link Times cited: 16 USED (low confidence) X. Jing, Z. Liu, and K. Yao, “Molecular dynamics investigation of deposition and annealing behaviors of Cu atoms onto Cu(0 0 1) substrate,” Applied Surface Science. 2012. link Times cited: 18 USED (low confidence) C. Mottet, “Relaxations on the Nanoscale: An Atomistic View by Numerical Simulations.” 2011. link Times cited: 1 USED (low confidence) O. Yermolenko, G. V. Kornich, and G. Betz, “Molecular dynamics simulations of low-energy argon ion sputtering of copper clusters on polyethylene surfaces,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2011. link Times cited: 1 USED (low confidence) X. W. Zhou, F. Doty, and P. Yang, “Atomistic simulation study of atomic size effects on B1 (NaCl), B2 (CsCl), and B3 (zinc-blende) crystal stability of binary ionic compounds,” Computational Materials Science. 2011. link Times cited: 15 USED (low confidence) P. Zhu, Y.-zhong Hu, H. Wang, and T. Ma, “Study of effect of indenter shape in nanometric scratching process using molecular dynamics,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2011. link Times cited: 52 USED (low confidence) S. V. Hosseini, M. Vahdati, and A. Shokuhfar, “Effect of Tool Nose Radius on Nano-Machining Process by Molecular Dynamics Simulation,” Defect and Diffusion Forum. 2011. link Times cited: 5 Abstract: Today, there is a need to understand the micro mechanism of … read moreAbstract: Today, there is a need to understand the micro mechanism of material removal to achieve a better roughness in ultra precision machining (UPM). The conventional finite element method becomes impossible to use because the target region and grids are very tiny. In addition, FEM cannot consider the micro property of the material such as atomic defect and dislocation. As an alternative, molecular dynamics (MD) simulation is significantly implemented in the field of nano-machining and nano-tribological problems to investigate deformation mechanism like work hardening, stick-slip phenomenon, frictional resistance and surface roughness [1]. One of the machining parameters than can affect nano-cutting deformation and the machined surface quality is tool nose radius [2]. In this paper molecular dynamics simulations of the nano-metric cutting on single-crystal copper were performed with the embedded atom method (EAM). To investigate the effect of tool nose radius, a comparison was done between a sharp tool with no edge radius and tools with a variety of edge radii. Tool forces, coefficient of friction, specific energy and nature of material removal with distribution of dislocations were simulated. Results show that in the nano-machining process, the tool nose radius cannot be ignored compared with the depth of cut and the edge of tool can change micro mechanism of chip formation. It appears that a large edge radius (relative to the depth of cut) of the tool used in nano-metric cutting, provides a high hydrostatic pressure. Thus, the trust force and frictional force of the tool is raised. In addition, increasing the tool edge radius and the density of generated dislocation in work-piece is scaled up that is comparable with TEM photographs [6]. read less USED (low confidence) S. V. Hosseini, M. Vahdati, and A. Shokuhfar, “Investigation of Interatomic Potential on Chip Formation Mechanism in Nanometric Cutting Using MD Simulation,” Defect and Diffusion Forum. 2011. link Times cited: 2 Abstract: Nowadays, the nano-machining process is used to produce high… read moreAbstract: Nowadays, the nano-machining process is used to produce high quality finished surfaces with precise form accuracy. To understand and analyze the chip formation mechanism of nano-machining process on an atomistic scale, since the experimentation is not an easy task, numerical simulation such as molecular dynamic (MD) simulation is a very useful method. In this paper, MD simulation of the nano-metric cutting of single-crystal copper was performed with a single crystal diamond tool. The model was solved with both pair wise Morse potential function and embedded atom method (EAM) potential to simulate the inter-atomic force between the work-piece and a rigid tool. The chip formation mechanism, dislocation generation, tool forces and generated temperature were investigated. Results show that the Morse potential cannot perform an appropriate defect formation and plastic deformation in nano-metric cutting of metals. Also, tool forces in Morse potential are more than the forces in EAM potential. Furthermore, the fluctuations of resultant forces in Morse potential are greater than that of EAM. In addition, using many-body interaction potentials like EAM can lead to substantial changes in surface energies, elastic-plastic properties and atomic displacement, compared with the pair-wise potentials like Morse. Finally, the atomic displacement investigation shows that in EAM potential study, only the atoms in a local region near the cutting process are displaced, but in Morse potential a large portion of atoms has affected during cutting process. Subsequently, the chip temperature in EAM potential is more than that of Morse potential. read less USED (low confidence) M. Benhassine, E. Saiz, A. Tomsia, and J. Coninck, “Nonreactive wetting kinetics of binary alloys: A molecular dynamics study,” Acta Materialia. 2011. link Times cited: 21 USED (low confidence) X. Ma, J. Yin, and J. J. Zhang, “Molecular Dynamics Study of Size Effect on Uniaxial Tension of Single Crystal Cu Nanowires,” Advanced Materials Research. 2010. link Times cited: 1 Abstract: The uniaxial tensions of single crystal Cu nanowires (NWs) w… read moreAbstract: The uniaxial tensions of single crystal Cu nanowires (NWs) with different circular cross-section radiuses are simulated by molecular dynamics (MD) method. Atomic interactions in Cu NWs are described by EAM potential. The results show that the plastic deformation of NWs under uniaxial tension is dominantly controlled by dislocation nucleation and gliding. The mechanical properties of NWs are size-dependent. The NWs with larger radius of cross-section possess higher strength and ductility. Furthermore, the site of neck formation and following break of NWs has strong dependence on size scale of NWs. read less USED (low confidence) L. Yue, H. Zhang, and D. Li, “Defect generation in nano-twinned, nano-grained and single crystal Cu systems caused by wear: A molecular dynamics study,” Scripta Materialia. 2010. link Times cited: 22 USED (low confidence) W. Yu and S. Shen, “Initial dislocation topologies of nanoindentation into copper (001) film with a nanocavity,” Engineering Fracture Mechanics. 2010. link Times cited: 8 USED (low confidence) D. Terentyev, X. He, A. Serra, and J. Kuriplach, “Structure and strength of 〈1 1 0〉 tilt grain boundaries in bcc Fe: An atomistic study,” Computational Materials Science. 2010. link Times cited: 52 USED (low confidence) H. Song, S. Fensin, M. Asta, and J. Hoyt, “A molecular dynamics simulation of (1 1 0) surface premelting in Ni,” Scripta Materialia. 2010. link Times cited: 17 USED (low confidence) X. Yan and H. Zhang, “On the atomistic mechanisms of grain boundary migration in [0 0 1] twist boundaries: Molecular dynamics simulations,” Computational Materials Science. 2010. link Times cited: 14 USED (low confidence) Z.-L. Liu, J. Yang, Z. Zhao, L. Cai, and F. Jing, “The anisotropy of shock-induced melting of Pt observed in molecular dynamics simulations,” Physics Letters A. 2010. link Times cited: 21 USED (low confidence) X. Liu and P. Feng, “Picosecond Pulsed Laser Induced Melting of Monocrystalline Copper: A Hybrid Simulation,” Advanced Materials Research. 2010. link Times cited: 0 Abstract: A hybrid method combing molecular dynamics and two-step radi… read moreAbstract: A hybrid method combing molecular dynamics and two-step radiation heating model is used to study the kinetics and microscopic mechanisms of picosecond laser melting of monocrystalline copper in stress confinement regime. The nonequilibrium processes of laser melting are simulated by classical MD method, and laser excitation as well as subsequent relaxation of the conduction band electrons are described continually by two-step radiation heating model. The mechanism responsible for melting of copper under picosecond laser pulse irradiation can be attributed to homogeneous nucleation of the liquid phase inside the solid region. The speed of stress wave is predicted to be 4400m/s equal to that of sound. The liquid and crystal regions are identified definitely in the atomic configurations by means of Local Order Parameter, in-plane structure and number density of atoms. Velocity-reducing technique is proved efficient in avoiding the influence of the reflected stress wave on melting process by comparing two models with velocity-reducing technique and free boundary condition at the bottom respectively. read less USED (low confidence) H. Yildirim, A. Kara, T. Rahman, R. Heid, and K. Bohnen, “Surface vibrational thermodynamics from ab initio calculations for fcc(1 0 0),” Surface Science. 2010. link Times cited: 5 USED (low confidence) H. Yildirim and T. Rahman, “Diffusion barriers for Ag and Cu adatoms on the terraces and step edges on Cu(100) and Ag(100): An ab initio study,” Physical Review B. 2009. link Times cited: 43 Abstract: We present the results of density-functional-theory-based ca… read moreAbstract: We present the results of density-functional-theory-based calculations for the activation energies for the diffusion of adatoms (Cu or Ag) on Cu(100) and Ag(100) with and without steps. We find that only for Cu on Ag(100), exchange is the dominant mechanism for the diffusion on terraces. On the other hand, for diffusion at step edges, exchange is the dominant mechanism except for Ag on Cu(100). This result also indicates that incorporation of Cu atoms into the step edges of Ag(100) costs only 330 meV, while the energy cost for Ag incorporation into Cu(100) step edge is much higher (about 700 meV). We find the hierarchy of Ehrlich-Schwoebel barriers to be: 170 meV for Ag on Cu(100); 60 meV for Cu on Cu(100); 20 meV for Ag on Ag(100), and $\ensuremath{-}30\text{ }\text{meV}$ $(\ensuremath{-}270\text{ }\text{meV})$ for Cu on Ag(100). These barriers point to a striking difference in the growth modes for Ag layers on Cu(100) and Cu layers on Ag(100). read less USED (low confidence) T. Kogita, M. Kohyama, and Y. Kido, “Structure and dynamics of NiAl(110) studied by high-resolution ion scattering combined with density functional calculations,” Physical Review B. 2009. link Times cited: 3 Abstract: The surface relaxation and the rumpling of the top and the 2… read moreAbstract: The surface relaxation and the rumpling of the top and the 2nd layer together with the mean thermal vibration amplitudes of NiAl(110) are determined by high-resolution medium energy ion scattering (MEIS) with an excellent depth resolution of typically ±0.01 A. We also perform classical molecular dynamics (MD) simulations employing the embedded atom method and the first principles calculations using the VASP (Vienna Ab-initio Simulation Package) code. The results obtained by MEIS are compared with the theoretical predictions and experimental analysis reported so far. Interestingly, the present MEIS analysis observes slightly expanded relaxation 12 e Δ , which is supported by the present MD and VASP calculations and by X-ray diffraction analysis, whereas other experimental and theoretical analyses give contracted relaxation. The root mean square thermal vibration amplitude of the bulk Ni atoms is determined to be 0.10±0.005 A, which agrees well with the value of 0.097 A derived from the phonon dispersion relation calculated from VASP. A slightly enhanced thermal vibration amplitude of the top layer Ni in the surface normal direction observed is consistent with the MD simulation. 1 Department of Physics, Ritsumeikan University, Kusatsu, Shiga-ken 525-8577, Japan 2 Advanced Industrial Science and Technology, AIST, Ikeda, Osaka 563-8577, Japan read less USED (low confidence) E. Hristova, V. Grigoryan, and M. Springborg, “Structures and stability of Ag clusters on Ag(111) and Ni(111) surfaces,” Surface Science. 2009. link Times cited: 5 USED (low confidence) W. Yu and S. Shen, “Effects of small indenter size and its position on incipient yield loading during nanoindentation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2009. link Times cited: 12 USED (low confidence) L. Guan et al., “Relaxation and electronic states of Au(100), (110) and (111) surfaces,” Solid State Communications. 2009. link Times cited: 13 USED (low confidence) I. Shabib and R. E. Miller, “Deformation characteristics and stress–strain response of nanotwinned copper via molecular dynamics simulation,” Acta Materialia. 2009. link Times cited: 82 USED (low confidence) K. Pötting, N. B. Luque, P. Quaino, H. Ibach, and W. Schmickler, “Island dynamics on charged silver electrodes: Kinetic Monte-Carlo simulations,” Electrochimica Acta. 2009. link Times cited: 18 USED (low confidence) N. B. Luque, L. Reinaudi, P. Serra, and E. Leiva, “Electrochemical deposition on surface nanometric defects: Thermodynamics and grand canonical Monte Carlo simulations,” Electrochimica Acta. 2009. link Times cited: 12 USED (low confidence) T. Liu, G. Liu, P. Wriggers, and S. Zhu, “Study on Contact Characteristic of Nanoscale Asperities by Using Molecular Dynamics Simulations,” Journal of Tribology-transactions of The Asme. 2009. link Times cited: 17 Abstract: The nanoscale contacts, which play a key role in nanotechnol… read moreAbstract: The nanoscale contacts, which play a key role in nanotechnology and micro-/ nanoelectromechanical systems, are fundamentally important for a wide range of problems including adhesion, contact formation, friction and wear, etc. Because continuum contact mechanics has limitations when it is applied at length of nanoscale, molecular dynamics (MD) simulations, which can investigate internal physical mechanisms of nanostructures by atomic motions in detail, become one of the most promising approaches for investigating mechanical behaviors of contacts in nanoscale. First, contacts between rigid cylindrical probes with different radii and an elastic half-space substrate are studied by using MD simulations with the assistance of the classical Lennard-Jones potential. For contacts without adhesion, the relationship between the applied force and the contact half-width is analyzed. The von Mises stress distributions are then discussed. For contacts with adhesion, the phenomena of the jump-to-contact, the break-off contact, and the hysteresis are observed. The pressure distributions and the von Mises stress contours in the contact region agree with the existing solutions. Second, the effects of the surface topography on adhesive contacts are studied by using MD simulations with the embedded atom method potential. The adhesive contact mechanical characteristic of a series of asperities with different shapes, different sizes, and different numbers on contacting surfaces are discovered and compared. The results show that the surface topography is one of the major factors, which may influence the contact behaviors between the interfaces of nanoscale components. read less USED (low confidence) J. Zhong, L. Hector, and J. B. Adams, “Dynamic decomposition of aliphatic molecules on Al(111) from ab initio molecular dynamics,” Physical Review B. 2009. link Times cited: 8 Abstract: Ab initio molecular dynamics based on density functional the… read moreAbstract: Ab initio molecular dynamics based on density functional theory within the generalized gradient approximation was used to explore decomposition on Al(111) of butanol-alcohol and butanoic-acid, two important boundary additives in Al processing. Each molecule was oriented with its functional group closest to the surface and then given an initial velocity toward the surface. Decomposition occurred upon collision with Al(111) resulting in the formation of adhered fragments that represent the very initial stages in additive film formation during plastic deformation where nascent Al is liberated. Bonding interactions over the simulation time frames were explored with contours of the electron localization function. Results of the simulations were compared with existing experimental studies of chemical decomposition on clean Al surfaces and found to be in qualitative accord. The effects of other initial molecular orientations on decomposition were explored in ancillary calculations where the molecules were rotated through 90 deg. and 180 deg. prior to collision with Al(111) read less USED (low confidence) R. K. Rajgarhia, D. Spearot, and A. Saxena, “Interatomic potential for copper–antimony in dilute solid–solution alloys and application to single crystal dislocation nucleation,” Computational Materials Science. 2009. link Times cited: 10 USED (low confidence) Y. Shibuta, S. Takamoto, and T. Suzuki, “Dependence of the grain boundary energy on the alloy composition in the bcc iron–chromium alloy: A molecular dynamics study,” Computational Materials Science. 2009. link Times cited: 33 USED (low confidence) Y. Zheng, H. W. Zhang, Z. Chen, C. Lu, and Y. Mai, “Roles of grain boundary and dislocations at different deformation stages of nanocrystalline copper under tension,” Physics Letters A. 2009. link Times cited: 37 USED (low confidence) A. Setoodeh, H. Attariani, and M. Khosrownejad, “Nickel Nanowires Under Uniaxial Loads: A Molecular Dynamics Simulation Study,” Computational Materials Science. 2008. link Times cited: 70 USED (low confidence) P. Xiao-dong, G. Zhi-gang, and L. Ping, “Energy and structure of copper clusters (n=70-150) studied by the Monte Carlo computer simulation,” Chinese Physics B. 2008. link Times cited: 10 Abstract: The structure and binding energy of copper clusters of the s… read moreAbstract: The structure and binding energy of copper clusters of the size range 70 to 150 were studied by using the embedded-atom method. The stability of the structure of the clusters was studied by calculating the average binding energy per atom, first difference energy and second difference energy of copper cluster. Most of the copper clusters of the size n = 70–150 adopt an icosahedral structure. The results show that the trends are in agreement with theoretic prediction for copper clusters. The most stable structures for copper clusters are found at n = 77, 90, 95, 131, 139. read less USED (low confidence) P. Hirel, J. Godet, S. Brochard, L. Pizzagalli, and P. Beauchamp, “Determination of activation parameters for dislocation formation from a surface in fcc metals by atomistic simulations,” Physical Review B. 2008. link Times cited: 31 Abstract: Defects in free surfaces are expected to be seeds for the nu… read moreAbstract: Defects in free surfaces are expected to be seeds for the nucleation of dislocations, which is the likely way nanoscale materials suffer plastic deformation. The nucleation results in the competition between the image force attracting the dislocation to the surface and the applied strain. In this work, two methods based on molecular dynamics simulations using an embedded atom method (EAM) potential are used to determine the activation energy and the critical radius for the formation of dislocations from a surface defect in a typical fcc metal. read less USED (low confidence) J. Monk, J. Hoyt, and D. Farkas, “Metastability of multitwinned Ag nanorods: Molecular dynamics study,” Physical Review B. 2008. link Times cited: 20 Abstract: Nanoscale rods have been shown to exhibit a multiple twinned… read moreAbstract: Nanoscale rods have been shown to exhibit a multiple twinned structure. The rods grow along a [110]-type crystallographic direction and have a pentagonal cross section with five (111) twins connecting the wire center to the corners of the pentagon. Here, we use molecular dynamics simulations with an embedded atom method interatomic potential for Ag to compute the ground-state energies of the multitwinned rods and compare with the bulk equilibrium crystal shape, as estimated from a Wulff construction. The excess energy of the nontwinned equilibrium nanorods and the multitwinned nanorods was obtained as a function of the wire length $(L)$ as well as the cross sectional area $({A}_{\text{cs}})$. Various contributions to the total energy, such as twin boundary energy and surface energies, are discussed and included in an analytical model that compares favorably with the simulation results. Our results show that for infinitely long nanowires with ${A}_{\text{cs}}l1500\text{ }{\text{nm}}^{2}$, the nontwinned structure is always energetically favorable. However, if the energy of the dipyramidal atomic structure at the nanorod ends is included in the model then the twinned nanorods are stable with respect to the nontwinned rods below a critical aspect ratio $(L/\sqrt{{A}_{\text{cs}}})$. read less USED (low confidence) J.-min Zhang, H.-yan Li, K. Xu, and V. Ji, “Calculation of surface energy and simulation of reconstruction for diamond cubic crystals (001) surface,” Applied Surface Science. 2008. link Times cited: 11 USED (low confidence) S. Kim, S. Kim, D. Kim, Y. Chung, and K. Lee, “Molecular dynamics study of redeposition effect by Ar ion bombardments on Au, Pd(001).” 2008. link Times cited: 2 Abstract: Atomic behavior during ion beam sputtering was investigated … read moreAbstract: Atomic behavior during ion beam sputtering was investigated by using classical molecular dynamics simulation. When Ar ion bombards on Au and Pd(001) surface with various incidence energies and angles, some atoms which gained substantial energy by impacting Ar ion were sputtered out and, simultaneously, others were landed on the surface as if surface atoms were redeposited. It was observed that the redeposited atoms are five times for Au and three times for Pd as many as sputtered atoms irrespective of both incidence energy and angle. From sequential ion bombarding calculations, contrary to the conventional concepts which have described the mechanism of surface pattern formation based only on the erosion theory, the redeposition atoms were turned out to play a significant role in forming the surface patterns. read less USED (low confidence) Y. Zheng, H. W. Zhang, Z. Chen, L. Wang, Z. Zhang, and J. B. Wang, “Formation of two conjoint fivefold deformation twins in copper nanowires with molecular dynamics simulation,” Applied Physics Letters. 2008. link Times cited: 30 Abstract: The formation of two conjoint fivefold deformation twins (DT… read moreAbstract: The formation of two conjoint fivefold deformation twins (DTs) in copper nanowires under bending is reported based on molecular dynamics simulations. It is found that an intermediate icosahedral phase is formed to facilitate the transformation from a low dense (010) plane in a face-centered-cubic lattice to a {111} close-packed fashion, forming trijunctions composed of three DTs. These trijunctions can easily interact with other DTs, forming two conjoint fivefold DTs. This formation process differs from the one observed by Cao and Wei [Appl. Phys. Lett. 89, 041919 (2006)], that is, fivefold DTs could be formed without introducing initial imperfections in simulations. read less USED (low confidence) M. Demkowicz and R. Hoagland, “Structure of Kurdjumov-Sachs interfaces in simulations of a copper-niobium bilayer,” Journal of Nuclear Materials. 2008. link Times cited: 94 USED (low confidence) J. A. Sanchez and M. P. Mengüç, “Phase-change phenomena during electron-beam heating : Molecular dynamics simulations,” Physical Review B. 2007. link Times cited: 10 USED (low confidence) S. Sankaranarayanan, V. Bhethanabotla, and B. Joseph, “Molecular dynamics simulation of temperature and strain rate effects on the elastic properties of bimetallic Pd-Pt nanowires,” Physical Review B. 2007. link Times cited: 63 Abstract: Molecular dynamics simulation is used to investigate the mec… read moreAbstract: Molecular dynamics simulation is used to investigate the mechanical properties of infinitely long, cylindrical bimetallic Pd-Pt nanowires, with an approximate diameter of $1.4\phantom{\rule{0.3em}{0ex}}\mathrm{nm}$ and two different compositions (25% and 50% Pt). The nanowires are subjected to uniaxial tensile strain along the [001] axis with varying strain rates of $0.05%\phantom{\rule{0.2em}{0ex}}{\mathrm{ps}}^{\ensuremath{-}1}$, and $5.0%\phantom{\rule{0.2em}{0ex}}{\mathrm{ps}}^{\ensuremath{-}1}$, at simulation temperatures of 50 and $300\phantom{\rule{0.3em}{0ex}}\mathrm{K}$, to study the effects of strain rates and thermal conditions on the deformation characteristics and mechanical properties of the nanowire. The deformation and rupture mechanism of these nanowires is explored in detail. Comparisons to the behavior exhibited by pure Pd and Pt nanowires of similar diameter are also made. The effect of lattice mismatch on the observed deformation modes in bimetallic nanowires is also discussed. Our simulations indicate that Pd-Pt alloy nanowires of various compositions, with little lattice mismatch between Pd and Pt atoms, undergo similar deformation and rupture upon uniaxial stretching. It is found that yielding and fracture mechanisms depend on the applied strain rate as well as atomic arrangement and temperature. At low temperature and strain rate, where crystal order and stability are highly preserved, the calculated stress-strain response of pure Pt and Pd as well as Pd-Pt alloy nanowires showed clear periodic, stepwise dislocation-relaxation behavior. Crystalline to amorphous transformation takes place at high strain rates $(5%\phantom{\rule{0.2em}{0ex}}{\mathrm{ps}}^{\ensuremath{-}1})$, with amorphous melting detected at $300\phantom{\rule{0.3em}{0ex}}\mathrm{K}$. Deformation of nanowires at higher strain rates and low temperature, where the superplasticity characteristic is significantly enhanced, results in the development of a multishell helical structure. Mechanical properties of the alloy nanowires are significantly different from those of bulk phase and are dictated by the applied strain rate, temperature, alloy composition, as well as the structural rearrangement associated with nanowire elongation. We find that Young's modulus of both the single component as well as alloy nanowires depends on the applied strain rate and is about 70%--75% of the bulk value. Ductility of the studied nanowires showed a nonmonotonic variation with Pd composition at low strain rates and was significantly enhanced for wires developing and rearranging into a multishell helical structure which occurred at higher strain rates. The Poisson ratio of Pd rich alloys is 60%--70% of its bulk value, whereas that of Pt rich alloys is not significantly changed at the nanoscale. The calculated differences in the nanowire mechanical properties are shown to have significant effect on their applicability in areas such as sensing and catalysis. read less USED (low confidence) A. J. Francis, C. G. Roberts, Y. Cao, A. Rollett, and P. Salvador, “Monte Carlo simulations and experimental observations of templated grain growth in thin platinum films,” Acta Materialia. 2007. link Times cited: 10 USED (low confidence) C. Zhang, C.-Y. Yan, X. Tang, Y.-liang Wang, and Q. Zhang, “Study of low-energy impact of Pt atoms on Pt (111) doped with noble metal atoms by molecular dynamics simulation,” Surface & Coatings Technology. 2007. link Times cited: 3 USED (low confidence) H. Zhang, D. Srolovitz, J. Douglas, and J. Warren, “Atomic Motion During the Migration of General [001] Tilt Grain Boundaries in Ni,” Acta Materialia. 2007. link Times cited: 41 USED (low confidence) C. Ghosh, A. Kara, and T. Rahman, “Usage of Pattern Recognition Scheme in Kinetic Monte Carlo Simulations: Application to Cluster Diffusion on Cu(111),” Surface Science. 2007. link Times cited: 5 USED (low confidence) S. G. Mayr and D. Bedorf, “Stabilization of Cu nanostructures by grain boundary doping with Bi : Experiment versus molecular dynamics simulation,” Physical Review B. 2007. link Times cited: 16 USED (low confidence) C. Becker, J. Hoyt, D. Buta, and M. Asta, “Crystal-melt interface stresses: atomistic simulation calculations for a Lennard-Jones binary alloy, Stillinger-Weber Si, and embedded atom method Ni.,” Physical review. E, Statistical, nonlinear, and soft matter physics. 2007. link Times cited: 19 Abstract: Molecular-dynamics and Monte Carlo simulations have been use… read moreAbstract: Molecular-dynamics and Monte Carlo simulations have been used to compute the crystal-melt interface stress (f) in a model Lennard-Jones (LJ) binary alloy system, as well as for elemental Si and Ni modeled by many-body Stillinger-Weber and embedded-atom-method (EAM) potentials, respectively. For the LJ alloys the interface stress in the (100) orientation was found to be negative and the f vs composition behavior exhibits a slight negative deviation from linearity. For Stillinger-Weber Si, a positive interface stress was found for both (100) and (111) interfaces: f{100}=(380+/-30)mJ/m{2} and f{111}=(300+/-10)mJ/m{2}. The Si (100) and (111) interface stresses are roughly 80 and 65% of the value of the interfacial free energy (gamma) , respectively. In EAM Ni we obtained f{100}=(22+/-74)mJ/m{2}, which is an order of magnitude lower than gamma. A qualitative explanation for the trends in f is discussed. read less USED (low confidence) C. E. Lekka and G. Evangelakis, “Dynamical properties of the Ni3Al low index surfaces with and without Ni or Al adatoms from molecular dynamics simulations,” Materials Chemistry and Physics. 2007. link Times cited: 0 USED (low confidence) J.-min Zhang, Y. Wen, and K. Xu, “Atomic simulation of the point defects in three low‐index surfaces of BCC transition metals with the MAEAM,” Surface and Interface Analysis. 2007. link Times cited: 0 Abstract: The favorable position of an adatom and the formation energi… read moreAbstract: The favorable position of an adatom and the formation energies of a single vacancy and an adatom‐vacancy pair in three low‐index surfaces of body‐centered cubic (BCC) transition metals have been calculated by using the modified analytical embedded atom method (MAEAM). The favorable position of an adatom is at the fourfold and twofold positions above the (100) and (110) surfaces respectively, but it is deviated $(3 - \sqrt{6})a/3$ from the threefold position of the (111) surface. Either the heights of the adatom from the top atomic layer, or the formation energies of a single vacancy, or an adatom‐vacancy pair decrease in sequence of the (110), (100) and (111) surfaces for each metal. Furthermore, the formation energy of an adatom‐vacancy pair is always lower than that of a single vacancy for each low‐index surface of each metal, which shown the formation of adatom‐vacancy pair is more energetically favorable than the vacancy for the BCC transition metals. Copyright © 2007 John Wiley & Sons, Ltd. read less USED (low confidence) M. Tschopp, G. Tucker, and D. McDowell, “Structure and free volume of 〈1 1 0〉 symmetric tilt grain boundaries with the E structural unit,” Acta Materialia. 2007. link Times cited: 106 USED (low confidence) V. Pierron-Bohnes et al., “Atomic Migration in Bulk and Thin Film L10 Alloys: Experiments and Molecular Dynamics Simulations,” Defect and Diffusion Forum. 2007. link Times cited: 4 Abstract: Ferromagnetic L10 ordered alloys are extensively studied now… read moreAbstract: Ferromagnetic L10 ordered alloys are extensively studied nowadays as good candidates for high density magnetic storage media due to their high magnetic anisotropy, related to their chemical order anisotropy. Epitaxial thin bilayers NiPt/FePt/MgO(001) have been grown at 700 K and annealed at 800 K and 900 K. At 800 K, the L10 long-range order increases without measurable interdiffusion. At 900 K, the interdiffusion takes place without destroying the L10 long-range order. This surprising observation can be explained by different diffusion mechanisms that are energetically compared using molecular dynamics simulations in CoPt in the second moment tight binding approximation. In addition, the frequencies of the normal modes of vibration have been measured in FePd, CoPt and FePt single crystals using inelastic neutron scattering. The measurements were performed in the L10 ordered structure at 300 K. From a Born-von Karman fit, we have calculated the phonon densities of states. The migration energies in the 3 systems have been estimated using the model developed by Schober et al. (1981). The phonon densities of states have also been used to calculate several thermodynamic quantities as the vibration entropy and the Debye temperature. read less USED (low confidence) H. Yu, P. Shrotriya, Y. Gao, and K.-S. Kim, “Micro-plasticity of surface steps under adhesive contact Part I-Surface yielding controlled by single-dislocation nucleation,” Journal of The Mechanics and Physics of Solids. 2007. link Times cited: 31 USED (low confidence) J.-min Zhang, X.-L. Song, and K. Xu, “Atomistic simulation of the vacancy in Ni (1 1 0) surface,” Applied Surface Science. 2007. link Times cited: 7 USED (low confidence) S. Sankaranarayanan, V. Bhethanabotla, and B. Joseph, “Molecular dynamics simulation study of the melting and structural evolution of bimetallic Pd-Pt nanowires,” Physical Review B. 2006. link Times cited: 43 Abstract: Thermal characteristics of $\mathrm{Pd}\text{\ensuremath{-}}… read moreAbstract: Thermal characteristics of $\mathrm{Pd}\text{\ensuremath{-}}\mathrm{Pt}$ metal nanowires with diameters ranging from 2.3 to $3.5\phantom{\rule{0.3em}{0ex}}\mathrm{nm}$ and of several compositions were studied by molecular dynamics simulations utilizing the quantum Sutton-Chen potential function. Monte Carlo simulations employing bond order simulation model were used to generate the initial wire configurations that consisted of surface segregated structures. Melting temperatures were estimated based on variations in thermodynamic properties such as potential energy and specific heat capacity. We find that the melting transition temperatures for the nanowires are much lower than those of bulk alloys of the same composition and at least $100--200\phantom{\rule{0.3em}{0ex}}\mathrm{K}$ higher than those of nanoclusters of the same diameter. Density distributions along the nanowire cross section and axis as well as components of shell-based diffusion coefficients and velocity autocorrelation functions were used to investigate the melting mechanism in these nanowires. Our findings indicate a surface-initiated melting process characterized by predominantly larger cross-sectional movement. This two-dimensional surface melting mechanism in nanowires differs from that in nanoclusters in which atomic movement is more isotropic in all three dimensions. Differences in the surface melting mechanism result in structural transformations from fcc-hcp type and lead to simulated phase boundaries for nanowires that are different from bulk alloys as well as from same-diameter nanoclusters. A composition and temperature dependent fcc-hcp transformation occurs prior to the melting transition in both nanowires and nanoclusters. Hcp phase occurs over a wider temperature range at Pd-rich compositions and a narrower range at low Pd compositions with the fcc-hcp and hcp-liquid transition temperatures showing a minimum at 25% Pt composition. In contrast, the nanoclusters exhibit a near-linear dependence of melting temperature on Pd composition with the hcp phase existing over a much narrower range of temperatures, closer to the melting transition. Thermal stability of the solid phases of these nanowires was investigated by simulating two alternative starting configurations such as a hypothetical hcp and an annealed-solid structure for two compositions. The size and composition dependence of nanowire melting temperatures are consistent with those predicted by available melting theories. read less USED (low confidence) V. Yamakov, D. Moldovan, K. Rastogi, and D. Wolf, “Relation between grain growth and grain-boundary diffusion in a pure material by molecular dynamics simulations,” Acta Materialia. 2006. link Times cited: 28 USED (low confidence) J. Hoyt, M. Asta, and D. Sun, “Molecular dynamics simulations of the crystal–melt interfacial free energy and mobility in Mo and V,” Philosophical Magazine. 2006. link Times cited: 40 Abstract: Molecular dynamics simulations, based on embedded-atom metho… read moreAbstract: Molecular dynamics simulations, based on embedded-atom method potentials, have been used to compute thermodynamic and kinetic properties of crystal–melt interfaces in the bcc metals Mo and V. The interfacial free energy and its associated crystalline anisotropy have been obtained with the capillary fluctuation method and for both metals the anisotropy and the value of the Turnbull coefficient are found to be significantly lower than for the case of fcc materials. The interface mobility, or kinetic coefficient, which relates the isothermal crystallization rate to interface undercooling, was computed by non-equilibrium molecular dynamics simulations. Mobilities in the range 9-16 cm s−1K−1 are obtained. For Mo the mobility in the (110) crystallographic growth direction is larger than in the (100) and (111) directions, whereas for V the growth is found to be isotropic within numerical uncertainty. The kinetic-coefficient results are discussed within the framework of a density-functional-based theory of crystal growth. read less USED (low confidence) A. Cao and Y. Wei, “Formation of fivefold deformation twins in nanocrystalline face-centered-cubic copper based on molecular dynamics simulations,” Applied Physics Letters. 2006. link Times cited: 72 Abstract: Fivefold deformation twins were reported recently to be obse… read moreAbstract: Fivefold deformation twins were reported recently to be observed in the experiment of the nanocrystalline face-centered-cubic metals and alloys. However, they were not predicted previously based on the molecular dynamics (MD) simulations and the reason was thought to be a uniaxial tension considered in the simulations. In the present investigation, through introducing pretwins in grain regions, using the MD simulations, the authors predict out the fivefold deformation twins in the grain regions of the nanocrystal grain cell, which undergoes a uniaxial tension. It is shown in their simulation results that series of Shockley partial dislocations emitted from grain boundaries provide sequential twining mechanism, which results in fivefold deformation twins. read less USED (low confidence) J. Marian and A. Caro, “Moving dislocations in disordered alloys: Connecting continuum and discrete models with atomistic simulations,” Physical Review B. 2006. link Times cited: 89 Abstract: Using atomistic simulations of dislocation motion in Ni and … read moreAbstract: Using atomistic simulations of dislocation motion in Ni and $\mathrm{Ni}\text{\ensuremath{-}}\mathrm{Au}$ alloys we report a detailed study of the mobility function as a function of stress, temperature, and alloy composition. We analyze the results in terms of analytic models of phonon radiation and their selection rules for phonon excitation. We find a remarkable agreement between the location of the cusps in the $\ensuremath{\sigma}$-$v$ relation and the velocity of waves propagating in the direction of dislocation motion. We identify and characterize three regimes of dissipation whose boundaries are essentially determined by the direction of motion of the dislocation, rather than by its screw or edge character. read less USED (low confidence) G. F. Cabeza, N. Castellani, and P. Légaré, “Adhesion and bonding of Pt/Ni and Pt/Co overlayers : Density functional calculations,” Journal of Physics and Chemistry of Solids. 2006. link Times cited: 8 USED (low confidence) Z. Sun, X. Wang, A. K. Soh, and H. Wu, “On stress calculations in atomistic simulations,” Modelling and Simulation in Materials Science and Engineering. 2006. link Times cited: 32 Abstract: A systematic comparison between the decomposed virial formul… read moreAbstract: A systematic comparison between the decomposed virial formula and the Tsai formula shows that they are mathematically equivalent in calculating the overall average stress of an atomistic system. But in the case of calculating local stress distribution, the former gives ambiguous results, e.g. it gives nonzero normal stress at free surfaces and it typically ‘underestimates’ the inhomogeneity of microstructures and deformations in material. With a highly degenerate atomic chain model, we show mathematically that the results obtained by the decomposed virial formula are accurate only if the deformation is homogeneous within the neighbourhood of an interaction-cutoff radius, centred at the atomic site considered. Thus it is worth noting that the Tsai formula is more adequate for calculating both the overall average stress and local stress distribution. read less USED (low confidence) N. Lümmen and T. Kraska, “Influence of the carrier gas on the formation of iron nano-particles from the gas phase: A molecular dynamics simulation study,” Computational Materials Science. 2006. link Times cited: 18 USED (low confidence) H. Zhang and D. Srolovitz, “Simulation and analysis of the migration mechanism of Σ5 tilt grain boundaries in an fcc metal,” Acta Materialia. 2006. link Times cited: 44 USED (low confidence) N. Lümmen and T. Kraska, “Homogeneous nucleation of iron from supersaturated vapor investigated by molecular dynamics simulation,” Journal of Aerosol Science. 2005. link Times cited: 33 USED (low confidence) S. Aubry and D. Hughes, “Reductions in stacking fault widths in fcc crystals: Semiempirical calculations,” Physical Review B. 2005. link Times cited: 22 Abstract: Recent molecular dynamics simulations of nanocrystals have i… read moreAbstract: Recent molecular dynamics simulations of nanocrystals have illustrated the importance of stacking fault width for mechanical behavior and microstructure. Stacking fault SF width is a balance between elastic strain energy and the SF energy. While the latter is a material constant, the former strain energy may vary due to local internal stresses thereby effecting SF width. The presence and intensity of these stresses are functions of dislocation interactions in the crystal. Recent high resolution electron microscopy observations reveal a range of narrow SF widths within grains of different sizes including those less than 10 nm. To understand the reason for the presence of dislocations with small SF widths in metals, we first demonstrate theoretically and numerically with molecular statics simulation, the reductions in SF width due to dislocation stress screening. Different dislocation arrangements are examined including a dislocation wall. Second, the reduction in SF width in a thin film is examined via the introduction of free surfaces. These results reveal a wide variation of SF widths depending on structure and indicate that stress screening is an important mechanism for creating the narrow widths observed experimentally. read less USED (low confidence) T. Tsuji, “The Void Growth Simulations in the Hyper-Elastic Material with Multiple Seeds,” Materials Science Forum. 2005. link Times cited: 0 Abstract: The behaviors of a material are nonlinear in the large defor… read moreAbstract: The behaviors of a material are nonlinear in the large deformed region. The hyper elastic models can describe such non linear materials. If the hyper elastic material is applied to the hydrostatic tensile load, the void begins to grow when the load exceed the critical value. It is important to study the coalescence of the void growth in order to consider the destruction of the material. In this paper, the void growth simulations in the hyper-elastic material with multiple seeds are studied. The unit rectangular cell with small voids is subjected to the hydrostatic tensile load. This problem can be analyzed by FEM. However, the simulation with the larger number of the voids is not possible. Thus, the CA (Cellular Automaton) is used to describe the behaviors of the void coalescence and the possibility of CA is discussed. read less USED (low confidence) G. Zhou and J. C. Yang, “Initial oxidation kinetics of Cu(100), (110), and (111) thin films investigated by in situ ultra-high-vacuum transmission electron microscopy,” Journal of Materials Research. 2005. link Times cited: 82 Abstract: The initial oxidation stages of Cu(100), (110), and (111) su… read moreAbstract: The initial oxidation stages of Cu(100), (110), and (111) surfaces have been investigated by using in situ ultra-high-vacuum transmission electron microscopy (TEM) techniques to visualize the nucleation and growth of oxide islands. The kinetic data on the nucleation and growth of oxide islands shows a highly enhanced initial oxidation rate on the Cu(110) surface as compared with Cu(100), and it is found that the dominant mechanism for the nucleation and growth is oxygen surface diffusion in the oxidation of Cu(100) and (110). The oxidation of Cu(111) shows a dramatically different behavior from that of the other two orientations, and the in situ TEM observation reveals that the initial stages of Cu(111) oxidation are dominated by the nucleation of oxide islands at temperatures lower than 550 °C, and are dominated by two-dimensional oxide growth at temperatures higher than 550 °C. This dependence of the oxidation behavior on the crystal orientation and temperature is attributed to the structures of the oxygen-chemisorbed layer, oxygen surface diffusion, surface energy, and the interfacial strain energy. read less USED (low confidence) S. Sankaranarayanan, V. Bhethanabotla, and B. Joseph, “Molecular dynamics simulation study of the melting of Pd-Pt nanoclusters,” Physical Review B. 2005. link Times cited: 0 Abstract: Bimetallic nanoclusters are of interest because of their uti… read moreAbstract: Bimetallic nanoclusters are of interest because of their utility in catalysis and sensors. The thermal characteristics of bimetallic Pt-Pd nanoclusters of different sizes and compositions were investigated through molecular dynamics simulations using quantum Sutton-Chen (QSC) many-body potentials. Monte Carlo simulations employing the bond order simulation model were used to generate minimum energy configurations, which were utilized as the starting point for molecular dynamics simulations. The calculated initial configurations of the Pt-Pd system consisted of surface segregated Pd atoms and a Pt-rich core. Melting characteristics were studied by following the changes in potential energy and heat capacity as functions of temperature. Structural changes accompanying the thermal evolution were studied by the bond order parameter method. The Pt-Pd clusters exhibited a two-stage melting: surface melting of the external Pd atoms followed by homogeneous melting of the Pt core. These transitions were found to depend on the composition and size of the nanocluster. Melting temperatures of the nanoclusters were found to be much lower than those of bulk Pt and Pd. Bulk melting temperatures of Pd and Pt simulated using periodic boundary conditions compare well with experimental values, thus providing justification for the use of QSC potentials in these simulations. Deformation parameters were calculated to characterize the structural evolution resulting from diffusion of Pd and Pt atoms. The results indicate that in Pd-Pt clusters, Pd atoms prefer to remain at the surface even after melting. In addition, Pt also tends to diffuse to the surface after melting due to reduction of its surface energy with temperature. This mixing pattern is different from those reported in some of the earlier studies on melting of bimetallics. read less USED (low confidence) O. A. Oviedo, M. I. Rojas, and E. Leiva, “Off lattice Monte-Carlo simulations of low-dimensional surface defects and metal deposits on Pt(111),” Electrochemistry Communications. 2005. link Times cited: 2 USED (low confidence) Z. B. Güvenç, R. Hippler, and B. Jackson, “Bombardment of Ni(100) surface with low-energy argons: molecular dynamics simulations,” Thin Solid Films. 2005. link Times cited: 6 USED (low confidence) G. Adebayo, O. Akinlade, and L. Hussain, “Structures and autocorrelation functions of liquid Al and Mg modelled via Lennard-Jones potential from molecular dynamics simulation,” Pramana. 2005. link Times cited: 5 USED (low confidence) M. Alatalo, S. Jaatinen, P. Salo, and K. Laasonen, “Oxygen adsorption on Cu(100): First-principles pseudopotential calculations,” Physical Review B. 2004. link Times cited: 51 Abstract: We have studied the adsorption characteristics of atomic and… read moreAbstract: We have studied the adsorption characteristics of atomic and molecular oxygen, incident on the Cu(100) surface. Our pseudopotential first-principles calculations yield trajectories for the ${\mathrm{O}}_{2}$ molecule without dissociation barriers at the entrance channel. We discuss the energetics of the ${\mathrm{O}}_{2}$ adsorption and dissociation in terms of the elbow plots which are two-dimensional cuts of the full six-dimensional potential-energy surface. The top site is found to be the most reactive one while at the fcc site molecular adsorption takes place. The adsorption energies at horizontal configurations of the ${\mathrm{O}}_{2}$ molecule are found to be larger than those at the vertical configurations. The local densities of states reveal differences between the sites with direct dissociative adsorption and the ones with molecular precursor states. We also discuss the interactions between O and Cu atoms adsorbed at the hollow sites on Cu(100), and the corresponding diffusion barriers. read less USED (low confidence) M. Said-Ettaoussi, J. Jiménez‐Sáez, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “Deformation behaviour induced by point defects near a Cu(0 0 1) surface,” Applied Surface Science. 2004. link Times cited: 0 USED (low confidence) N. B. Luque, M. D. Pópolo, and E. Leiva, “Monte Carlo simulation of cluster growth in surface defects induced by the tip of a scanning tunnelling microscope,” Surface Science. 2004. link Times cited: 6 USED (low confidence) M. I. Rojas, “Off lattice Monte Carlo simulation study for different metal adlayers onto (1 1 1) substrates,” Surface Science. 2004. link Times cited: 14 USED (low confidence) L. Qi, H. Zhang, and Z. Hu, “Molecular dynamic simulation of glass formation in binary liquid metal: Cu-Ag using EAM,” Intermetallics. 2004. link Times cited: 46 USED (low confidence) M. Basham, P. Mulheran, and F. Montalenti, “Diffusion and stability of small vacancy clusters on Cu(1 0 0)––a simulation study,” Surface Science. 2004. link Times cited: 12 USED (low confidence) M. Upmanyu, Z. Trautt, and B. Kappes, “Anisotropy in Grain Boundary Thermo-Kinetics: Atomic-Scale Computer Simulations,” Materials Science Forum. 2004. link Times cited: 2 Abstract: Anisotropy in grain boundary “thermo-kinetics” is central to… read moreAbstract: Anisotropy in grain boundary “thermo-kinetics” is central to our understanding of microstructural evolution during grain growth and recrystallization. This paper focusses on role of atomic-scale computer simulation techniques, in particular molecular dynamics (MD), in extracting fundamental grain boundary properties and elucidating the atomic-scale mechanisms that determine these properties. A brief overview of recent strides made in extraction of grain boundary mobility and energy is presented, with emphasis on plastic strain induced boundary motion (p-SIBM) during recrystallization and curvature driven boundary motion (CDBM) during grain growth. Simulations aimed at misorientation dependence of the grain boundary properties during p-SIBM and CDBM show that boundary mobility and energy exhibit extrema at high symmetry misorientations and boundary mobility is comparatively more anisotropic during CDBM. This suggests that boundary mobility is dependent on the driving force. Qualitative observations of the atomic-scale mechanisms in play during boundary motion corroborate the simulation data. p-SIBM is dominated by motion of dislocation-interaction induced stepped structure of the grain boundaries, while correlated shuffling of group of atoms preceded by rearrangement of grain boundary free volume due to single atomic-hops across the grain boundary is frequently observed during CDBM. Comparison of the simulation results with high-purity experimental data extracted in Al indicates that while there is excellent agreement in misorientation dependent anisotropic properties, there are significant differences in values of boundary mobility and migration activation enthalpy. This strongly suggests that minute concentration of impurities retard grain boundary kinetics via impurity drag. Finally, the paper briefly discusses current and future challenges facing the computer simulation community in studying grain boundary systems in real materials where extrinsic effects (vacancy, impurity, segregation and particle effects) significantly alter the microscopic structure-mechanism relations and play a decisive role in determining the boundary properties. read less USED (low confidence) J. Diao, K. Gall, M. Dunn, and J. Zimmerman, “Atomistic simulations of the yielding of gold nanowires,” Acta Materialia. 2004. link Times cited: 248 USED (low confidence) L. Qi, H. Zhang, Z.-ning Hu, and P. Liaw, “Molecular dynamic simulation studies of glass formation and atomic-level structures in Pd–Ni alloy,” Physics Letters A. 2004. link Times cited: 33 USED (low confidence) E. Webb, G. Grest, D. Heine, and J. Hoyt, “Dissolutive wetting of Ag on Cu: A molecular dynamics simulation study,” Acta Materialia. 2004. link Times cited: 60 USED (low confidence) A. J. Haslam, V. Yamakov, D. Moldovan, D. Wolf, S. Phillpot, and H. Gleiter, “Effects of grain growth on grain-boundary diffusion creep by molecular-dynamics simulation,” Acta Materialia. 2004. link Times cited: 65 USED (low confidence) Y. Shen, H. Gong, L. Kong, and B. Liu, “Structural phase transitions in the Cu-based Cu–V solid solutions studied by molecular dynamics simulation,” Journal of Alloys and Compounds. 2004. link Times cited: 6 USED (low confidence) N. H. Leeuw, C. J. Nelson, C. Catlow, P. Sautet, and W. Dong, “Density-functional theory calculations of the adsorption of Cl at perfect and defective Ag(111) surfaces,” Physical Review B. 2004. link Times cited: 22 Abstract: Density functional theory calculations of adsorption of chlo… read moreAbstract: Density functional theory calculations of adsorption of chlorine at the perfect and defective silver (111) surface have shown that the energies of adsorption of chlorine atoms show little variation (less than 30 kJ mol - 1 ) between the different sites, from -136 kJ mol - 1 next to a silver adatom, through -159 kJ mol - 1 at the perfect surface to - 166 kJ mol - 1 next to a silver vacancy at the surface. Molecular chlorine adsorbs in a series of energetically similar overlayers, which are in good agreement with experimentally found structures. The lowest energy configuration is a planar hexagonal honeycomb structure of chlorine atoms adsorbed in fcc and hep hollow sites on the silver surface. An energetically similar structure is geometrically nonplanar, but has a planar electronic structure. Although the chlorine molecules are virtually dissociated (Cl-Cl distance = 3.40 A), significant electron density is distributed along the Cl-Cl axes, leading to a network of electronic interactions between the adsorbed chlorine atoms. The adsorption energy for Cl 2 is calculated at -231 kJ mol - 1 , in good agreement with experiment. Calculated Ag-Cl bond lengths of 2.69, 2.47, and 2.33 A agree with several experimental studies and show that the different bond lengths found experimentally are not anomalous, but due to the formation of geometrically different but energetically almost identical chlorine overlayer structures. read less USED (low confidence) B.-J. Lee, C. Lee, and J.-C. Lee, “Stress induced crystallization of amorphous materials and mechanical properties of nanocrystalline materials: a molecular dynamics simulation study,” Acta Materialia. 2003. link Times cited: 41 USED (low confidence) M. Shiga, M. Yamaguchi, and H. Kaburaki, “Structure and energetics of clean and hydrogenated Ni surfaces and symmetrical tilt grain boundaries using the embedded-atom method,” Physical Review B. 2003. link Times cited: 29 Abstract: Multilayer relaxations, surface/interface energies, and hydr… read moreAbstract: Multilayer relaxations, surface/interface energies, and hydrogen binding sites and energies were systematically studied using the embedded-atom method, for a series of the clean and hydrogenated Ni surfaces and symmetrical tilt Ni grain boundaries. The hydrogen binding energies were in the range of 2.7-2.9 eV at the surface sites while 2.1-2.6 eV at the grain-boundary sites, both being larger than at the fcc crystal interior site, 2.1 eV. These data are consistent with the conclusions from ab initio calculations that intergranular embrittlement is seen by hydrogen segregation at the Ni grain boundaries. It was found that multilayer relaxation of Ni plays a key role in trapping hydrogen at the grain boundaries. read less USED (low confidence) P. Torelli, F. Sirotti, and P. Ballone, “Surface alloying and mixing at the Mn/Fe(001) interface: Real-time photoelectron spectroscopy and modified embedded atom simulations,” Physical Review B. 2003. link Times cited: 20 Abstract: Structural and magnetic properties of thin Mn films on the F… read moreAbstract: Structural and magnetic properties of thin Mn films on the Fe(001) surface have been investigated by a combination of photoelectron spectroscopy and computer simulation in the temperature range 300 K≤T ≤750 K. Room-temperature as deposited Mn overlayers are found to be ferromagnetic up to 2.5-monolayer (ML) coverage, with a magnetic moment parallel to that of the iron substrate. The Mn atomic moment decreases with increasing coverage, and thicker samples (4-ML and 4.5-ML coverage) are antiferromagnetic. Photoemission measurements performed while the system temperature is rising at constant rate (dT/dt ∼0.5 K/s) detect the first signs of Mn-Fe interdiffusion at T=450 K, and reveal a broad temperature range (610 K≤T≤680 K) in which the interface appears to be stable. Interdiffusion resumes at T≥680 K. Molecular dynamics and Monte Carlo simulations allow us to attribute the stability plateau at 610 K≤T≤680 K to the formation of a single-layer MnFe surface alloy with a 2 ×2 unit cell and a checkerboard distribution of Mn and Fe atoms. X-ray-absorption spectroscopy and analysis of the dichroic signal show that the alloy has a ferromagnetic spin structure, collinear with that of the substrate. The magnetic moments of Mn and Fe atoms in the alloy are estimated to be 0.8μ B and 1.1μ B , respectively. read less USED (low confidence) C. Creemers, P. Deurinck, S. Helfensteyn, and J. Luyten, “Segregation and ordering at alloys surfaces: modelling and experiment confronted,” Applied Surface Science. 2003. link Times cited: 30 USED (low confidence) J. Zhong, J. B. Adams, and L. Hector, “Molecular dynamics simulations of asperity shear in aluminum,” Journal of Applied Physics. 2003. link Times cited: 26 Abstract: One important wear mechanism involves the shear of asperitie… read moreAbstract: One important wear mechanism involves the shear of asperities by other asperities. Molecular dynamics is used to simulate the shearing of aluminum asperities by a “hard” (Lennard-Jones) asperity. These simulations involve the use of a reliable interatomic potential based on the embedded atom method for aluminum that was developed by fitting a large database of density functional calculated forces and experimental data. The simulations are repeated for a wide range of conditions, including velocities, temperatures, asperity shapes, degree of intersection, crystal orientations and adhesive strengths, to determine their effects on the wear process. The design-of-experiment approach is used to analyze the relative importance of each factor and its interactions. Thermal distributions and mechanical deformation in the residual aluminum substrate during asperity shear are analyzed. The final results show that the most significant factor in determining the wear process is the interasperity bonding. The degree of overlap between two asperities is also important. The temperature, the translational velocity, and the crystal orientation play smaller roles. read less USED (low confidence) H. F. Lu, C. Zhang, and Q. Zhang, “Adatom, vacancy and sputtering yields of low energy Pt atoms impacts on Pt(111) by molecular dynamics simulation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 7 USED (low confidence) E. Lilleodden, J. Zimmerman, S. Foiles, and W. Nix, “Atomistic simulations of elastic deformation and dislocation nucleation during nanoindentation,” Journal of The Mechanics and Physics of Solids. 2003. link Times cited: 269 USED (low confidence) A. J. Haslam, D. Moldovan, V. Yamakov, D. Wolf, S. Phillpot, and H. Gleiter, “Stress-enhanced grain growth in a nanocrystalline material by molecular-dynamics simulation,” Acta Materialia. 2003. link Times cited: 217 USED (low confidence) L. Wang, Y. Zhang, X. Bian, and Y. Chen, “Melting of Cu nanoclusters by molecular dynamics simulation,” Physics Letters A. 2003. link Times cited: 69 USED (low confidence) D. Spearot, K. Jacob, and D. McDowell, “MD SIMULATION OF GRAIN BOUNDARY FRACTURE IN COPPER AND CONNECTIONS TO INTERFACE SEPARATION POTENTIALS.” 2003. link Times cited: 0 Abstract: Molecular dynamics (MD) simulations of metallic interface se… read moreAbstract: Molecular dynamics (MD) simulations of metallic interface separation are presented. A copper grain boundary interface model with 45-degree tilt lattice misorientation is subjected to normal and tangential separations. Nanoscale porosity and dislocation development within the interface region are monitored during the deformation process. In addition, the elastic relaxation response of the interface model is examined after tensile and shear separations. Atomistic calculations of interface separation are motivated by a proposed framework to characterize fracture through continuum interface separation potentials. These potentials are distinguished from previous continuum models in that discrete atomistics are used to determine a set of nanoscale effects, accounting for the influence of atomic structure and imperfections on interface separation or fracture. Atomistic calculations presented in this work may be used to develop functional forms for the advanced interface separation potentials. Identification of the elastic unloading response after normal and tangential separation shows that a distinction must be made regarding elastic relaxation after normal and tangential separation modes. This is contrary to assumptions made in previous damagedependent interface separation potentials. In general, continuum calculations motivated by discrete nanoscale computations facilitate a potentially more complete description of real material separation processes. read less USED (low confidence) R. Schaeublin et al., “Correlating TEM images of damage in irradiated materials to molecular dynamics simulations,” Journal of Nuclear Materials. 2002. link Times cited: 6 USED (low confidence) J. Sprague, F. Montalenti, B. Uberuaga, J. Kress, and A. Voter, “Simulation of growth of Cu on Ag(001) at experimental deposition rates,” Physical Review B. 2002. link Times cited: 53 Abstract: The initial stages of growth of (001)Cu films on (001)Ag sub… read moreAbstract: The initial stages of growth of (001)Cu films on (001)Ag substrates have been investigated using the temperature-accelerated dynamics (TAD) simulation method. The acceleration provided by TAD made it possible to simulate the deposition of Cu on (001)Ag at 77 K using a deposition rate of 0.04 ML/s, which matched previously reported experiments. This simulation was achieved without a priori knowledge of the significant atomic processes. The results showed that the increased in-plane lattice parameter of the pseudomorphic Cu reduces the activation energy for the exchange mode of surface diffusion, allowing short-range terrace diffusion and the formation of compact Cu islands on the second film layer at 77 K. Some unexpected complex surface diffusion processes and off-lattice atomic configurations were also observed. read less USED (low confidence) C. M. Gilmore and J. Sprague, “Molecular dynamics simulation of defect formation during energetic Cu deposition,” Thin Solid Films. 2002. link Times cited: 16 USED (low confidence) L.-P. Zheng, D.-xing Li, Z. G. Zhu, W.-Z. Jiang, B. Jiang, and X.-huai Liu, “Monte Carlo simulation study of the bulk effects of boron on the Ni3Al-x at.% B grain boundary,” Materials Letters. 2002. link Times cited: 2 USED (low confidence) Y. Ye, R. Biswas, A. Bastawros, and A. Chandra, “Simulation of chemical mechanical planarization of copper with molecular dynamics,” Applied Physics Letters. 2002. link Times cited: 25 Abstract: With an aim to understanding the fundamental mechanisms unde… read moreAbstract: With an aim to understanding the fundamental mechanisms underlying chemical mechanical planarization (CMP) of copper, we simulate the nanoscale polishing of a copper surface with molecular dynamics utilizing the embedded atom method. Mechanical abrasion produces rough planarized surfaces with a large chip in front of the abrasive particle, and dislocations in the bulk of the crystal. The addition of chemical dissolution leads to very smooth planarized copper surfaces and considerably smaller frictional forces that prevent the formation of bulk dislocations. This is a first step towards understanding the interplay between mechanistic material abrasion and chemical dissolution in chemical mechanical planarization of copper interconnects. read less USED (low confidence) J. Shim, B.-J. Lee, and Y. Cho, “Thermal stability of unsupported gold nanoparticle: a molecular dynamics study,” Surface Science. 2002. link Times cited: 160 USED (low confidence) J. Cai and J.-S. Wang, “ADSORPTION AND DIFFUSION OF Si ON THE Si(001): AN EMPIRICAL POTENTIAL CALCULATION,” International Journal of Modern Physics B. 2002. link Times cited: 1 Abstract: A recently developed potential function for covalent materia… read moreAbstract: A recently developed potential function for covalent materials (Phys. Stat. Sol.B212, 9 (1999)) is used to simulate the surface adsorption, and diffusion of Si adtom and ad-dimer on the Si(001) surface. We calculate the formation energies and diffusion activation energies of several possible binding sites. The predicted stable and metastable configurations and diffusion paths of Si ad-atom and Si ad-dimer on Si(001)-(2×1) surface are in agreement with that from the first principle calculations or experiments. read less USED (low confidence) M. Jiang, K. Oikawa, C. Liew, and T. Ikeshoji, “Molecular Dynamics Simulations of Nucleation Process from Supercooled Liquid Pt with EAM Potentials,” Materials Transactions. 2001. link Times cited: 2 Abstract: The homogeneous nucleation process induced by supercooling l… read moreAbstract: The homogeneous nucleation process induced by supercooling liquid Pt has been studied by means of molecular dynamics employing the embedded-atom method for the potential energy. The process was simulated by cooling an equilibrated liquid to a low temperature, while structure analysis was performed during the subsequent time evolution of the system under constant temperature and pressure conditions. In order to investigate the effects of degree of supercooling and cooling rate on crystallization, cooling temperature was varied from 900 to 1300 K and three processes at different cooling rates were studied. The results proved that certain degree of supercooling is necessary for homogeneous nucleation of crystals due to the existence of a free energy barrier in forming critical nuclei from supercooled liquid, as described by the classical theory of homogeneous nucleation. The scale of the degree of supercooling has much effect on the incubation period of homogeneous nucleation and nucleation rate. The crystallization towards fcc and hcp phases takes place in all homogeneous nucleation processes from liquid in our simulations. The progression of crystallization is sensitive to cooling rate. A very high cooling rate has been found to prolong incubation period, decrease nucleation rate, thus suppress crystallization. This may be associated with the phenomenon that high cooling rate prohibits the rearrangement of atoms towards forming a crystal lattice at high temperatures during the early stage of cooling. read less USED (low confidence) L.-P. Zheng, D.-xing Li, S. Qiu, W. Zhou, and B. Jiang, “Dependence of Ni, Al and B boundary concentrations on the B bulk concentration for the Ni3Al-x at.% B grain boundary,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 9 USED (low confidence) J.-C. Zheng, “RELAXATION OF Cu(100), (110) AND (111) SURFACES USING AB INITIO PSEUDOPOTENTIALS,” Surface Review and Letters. 2001. link Times cited: 12 USED (low confidence) J. R. Hu, S. Chang, F.-R. Chen, and J. Kai, “HRTEM investigation of a Σ = 9 [011]/(122) symmetric tilt grain boundary in Cu,” Scripta Materialia. 2001. link Times cited: 10 USED (low confidence) A. Ghazali and J. Lévy, “Low temperature Pb deposits on Cu(001): Monte Carlo structural studies,” Surface Science. 2001. link Times cited: 3 USED (low confidence) C. E. Lekka, N. Papanicolaou, and G. Evangelakis, “Molecular dynamics study of the ordered Cu3Au. I. Vibrational and structural properties of the low indexed surfaces,” Surface Science. 2001. link Times cited: 18 USED (low confidence) T. Iizuka, A. Onoda, and T. Hoshide, “Molecular Dynamics Simulation on Microstructure and Deformation Properties Related to Porosity in Al Thin Film Sputtered on Si Substrate,” Jsme International Journal Series A-solid Mechanics and Material Engineering. 2001. link Times cited: 0 Abstract: A material system of Al sputtered on crystalline Si was deal… read moreAbstract: A material system of Al sputtered on crystalline Si was dealt with as one of the simple material models for semiconductor material systems. To investigate qualitative properties of sputtered films on an atomic scale, simulations were conducted by a molecular dynamics (MD) method using two film models; i.e. a deposition model based on MD simulations of sputtering process and a crystal model using a crystalline Al film instead of a deposited one. The surface roughness and porosity, which are defined in this work, were found to decrease with an increase in the incident energy of atoms. Relationships between tensile deformation properties and porosities in simulated thin films were also investigated. Although the porosity was found to affect the tensile strength in the direction parallel to the substrate surface, it was revealed that the tensile strength in the direction perpendicular to the substrate surface was hardly influenced by the difference in the porosity. read less USED (low confidence) L.-P. Zheng, D.-zhang Zhu, B. Jiang, X. Liu, and D.-xing Li, “Monte Carlo simulation of concentration distribution at Ni3Al-5 at.% Mg grain boundary,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 4 USED (low confidence) O. Shenderova, J. Mewkill, and D. Brenner, “Nanoindentation as a Probe of Nanoscale Residual Stresses: Atomistic Simulation Results,” Molecular Simulation. 2000. link Times cited: 3 Abstract: Results from two sets of molecular dynamics simulations are … read moreAbstract: Results from two sets of molecular dynamics simulations are reported. In the first set of simulations a nanoscale tip was used to indent single-crystal gold lattices subjected to external strains. These were carried out to explore possible relationships between nanoindentation curves and elastic properties of uniformly strained films. The changes in the slope of the loading curves reflect the stress state of the sample. In the second set of simulations the use of shallow nanoindentation for mapping nonuniform residual surface stress near a dislocation intersecting a surface was tested. Correlation between the maximum force on the tip and the initial local stresses at the point of indentation were observed. Preliminary atomistic simulations indicate that atomic-force microscopy can be used as a nondestructive, nanoscale probe of the surface stress distributions. read less USED (low confidence) R. Longo, C. Rey, and L. J. Gallego, “Molecular dynamics study of the melting behaviour of seven-atom clusters of fcc transition and noble metals on the (111) surface of the same metal using the embedded atom model,” Surface Science. 2000. link Times cited: 10 USED (low confidence) B. Jiang, X.-huai Liu, L.-P. Zheng, and D.-xing Li, “Monte Carlo simulation of Mg segregation to Ni3Al grain boundary,” Materials Letters. 2000. link Times cited: 3 USED (low confidence) Q. Zhang, T. Ma, Z. Pan, and J. Tang, “The role of energetic atoms in the deposition of Au/Au (001) thin films — a computer simulation study,” Surface & Coatings Technology. 2000. link Times cited: 7 USED (low confidence) K. Takahashi, C. Nara, T. Yamagishi, and T. Onzawa, “Calculation of surface energy and simulation of reconstruction for Si(111) 3 × 3, 5 × 5, 7 × 7, and 9 × 9 DAS structure,” Applied Surface Science. 1999. link Times cited: 18 USED (low confidence) M. Katagiri, D. Patrick, and R. Lynden-Bell, “Molecular dynamics simulation of atomic force microscopy: imaging single-atom vacancies on Ag(001) and Pt(001),” Surface Science. 1999. link Times cited: 6 USED (low confidence) S. Todorov, H. Bu, K. Boyd, J. Rabalais, C. M. Gilmore, and J. Sprague, “Ion beam deposition of 107Ag(111) films on Ni(100),” Surface Science. 1999. link Times cited: 5 USED (low confidence) A. Almazouzi, M. Caturla, M. Alurralde, T. D. Rubia, and M. Victoria, “Defect production and damage evolution in Al: a molecular dynamics and Monte Carlo computer simulation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 18 USED (low confidence) U. Kürpick and T. Rahman, “The role of vibrational entropy in surface diffusion: adatoms and vacancies on Ag(100), Cu(100), and Ni(100),” Surface Science. 1999. link Times cited: 9 USED (low confidence) A. Tsukamoto, T. Fujita, K. Nakagawa, and A. Itoh, “The structure at the interface of Ni/Pd bilayer films with different deposition sequences estimated from molecular dynamics simulation,” IEEE International Magnetics Conference. 1999. link Times cited: 1 Abstract: Molecular dynamics simulations of the formation process of N… read moreAbstract: Molecular dynamics simulations of the formation process of Ni/Pd bilayer films were performed by employing the embedded atom method with high (10 eV) and low (0.1 eV) kinetic energies of incident atoms for two different crystal orientations: fcc [111] and fcc [100] of Ni or Pd substrate. The tendencies of the film formation process and the interdiffusion at the interface were similar in both the [111] and [100] cases. However, the strain at the interface of films on the [100] substrate was larger than that on the [111] substrate. At the case of Pd deposited on Ni [100], the Pd film grew with the [111] symmetry and partially observed Ni[100]-c(16/spl times/2) structure. The crystal orientation of Pd layer varied from the value of the strain in Ni [100] under layer. In the crystal orientation dependencies, the value of strain in the magnetic layer at interfaces derived from simulations corresponds well to the experimental value of perpendicular magnetic anisotropy. read less USED (low confidence) Y. Isono and T. Tanaka, “Molecular Dynamics Simulations of Atomic Scale Indentation and Cutting Process with Atomic Force Microscope,” Jsme International Journal Series A-solid Mechanics and Material Engineering. 1999. link Times cited: 23 Abstract: This paper describes the effect of the material used for a t… read moreAbstract: This paper describes the effect of the material used for a tool on atomic scale indentation and cutting mechanisms of metal workpieces, by means of molecular dynamics simulations. The interatomic force between the tool and workpiece is assumed to be a two-body interatomic potential using parameters based on the ab-initio molecular orbital calculation for a(Cr, Ni)-(C, Si)6H9 atom cluster. Molecular dynamics simulated the atomic scale indentation and cutting process of the chromium and nickel workpieces using the diamond, silicon and diamond-like carbon(DLC) tools. The diamond and DLC tools formed the indentation mark. Young's modulus of the chromium and nickel in indentation simulations was larger than that in experiments. This was qualitatively explained by the effect of the surface energy for the workpiece on the elastic modulus. The machinability of the chromium and nickel with the diamond tool was better than that of the silicon tool in atomic scale cutting simulations. The depth of the cut for the workpieces in nano scale cutting experiments with AFM, was similar to that in atomic scale cutting by molecular dynamics simulations. read less USED (low confidence) P. Deurinck and C. Creemers, “Monte Carlo simulation of Cu segregation and ordering at the (110) surface of Cu75Pd25,” Surface Science. 1998. link Times cited: 24 USED (low confidence) C. L. Kelchner, S. Plimpton, and J. C. Hamilton, “Dislocation nucleation and defect structure during surface indentation,” Physical Review B. 1998. link Times cited: 1801 Abstract: We model indentation of a metal surface by combining an atom… read moreAbstract: We model indentation of a metal surface by combining an atomistic metal with a hard-sphere indenter. This work provides atomistic imaging of dislocation nucleation during displacement controlled indentation on a passivated surface. Dislocations and defects are located and imaged by local deviations from centrosymmetry. For a Au(111) surface, nucleation of partial dislocation loops occurs below the surface inside the indenter contact area. We compare and contrast these observations with empirical criteria for dislocation nucleation and corresponding continuum elasticity solutions. read less USED (low confidence) M. Çivi, H. Avcı, A. Günen, and Z. B. Güvenç, “Collisionless fragmentation of small super-heated Nin, n = 4−6, clusters: molecular dynamics computer-simulation study,” ARI - An International Journal for Physical and Engineering Sciences. 1998. link Times cited: 0 USED (low confidence) D. Medlin, G. Campbell, and C. B. Carter, “Stacking defects in the 9R phase at an incoherent twin boundary in copper,” Acta Materialia. 1998. link Times cited: 65 USED (low confidence) T. D. de la Rubia, M. Caturla, E. Alonso, M. Fluss, and J. Perlado, “Self-decay-induced damage production and micro-structure evolution in fcc metals: An atomic-scale computer simulation approach,” Journal of Computer-Aided Materials Design. 1998. link Times cited: 9 USED (low confidence) Q. Zhang, J. Tang, and G.-qing Zhao, “Study on ion beam-assisted deposition of thin Au films by molecular dynamics simulation,” Surface & Coatings Technology. 1998. link Times cited: 1 USED (low confidence) M. H. Shapiro, “Using molecular dynamics simulations to investigate surface modification processes,” Surface & Coatings Technology. 1998. link Times cited: 3 USED (low confidence) C. Höfner and J. Rabalais, “Surface and subsurface distortions of the Au110-(1×2) structure,” Surface Science. 1998. link Times cited: 7 USED (low confidence) R. Zarić, B. Pearson, K. D. Krantzman, and B. Garrison, “Molecular dynamics simulations to explore the effect of projectile size on the ejection of organic targets from metal surfaces,” International Journal of Mass Spectrometry and Ion Processes. 1998. link Times cited: 20 USED (low confidence) D. Udler and D. Seidman, “Solute segregation at [001] tilt boundaries in dilute f.c.c. alloys,” Acta Materialia. 1998. link Times cited: 21 USED (low confidence) Q. Zhang, J. Tang, and G.-qing Zhao, “Investigation of the energetic deposition of Au (0 0 1) thin films by molecular-dynamics simulation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1998. link Times cited: 8 USED (low confidence) S. Tan, A. Ghazali, and J. C. S. Le´vy, “Pb/Cu (100) surface superstructures: Monte Carlo and molecular dynamics simulations,” Surface Science. 1997. link Times cited: 6 USED (low confidence) D. Timpel, K. Scheerschmidt, and S. Garofalini, “Silver clustering in sodium silicate glasses: a molecular dynamics study,” Journal of Non-crystalline Solids. 1997. link Times cited: 25 USED (low confidence) Y. Shimomura, “Point defects and their clusters in f.c.c. metals studied by computer simulations,” Materials Chemistry and Physics. 1997. link Times cited: 13 USED (low confidence) L. Wang, H. Liu, K. Chen, and Z. Hu, “The local orientational orders and structures of liquid and amorphous metals Au and Ni during rapid solidification,” Physica B-condensed Matter. 1997. link Times cited: 23 USED (low confidence) C. Massobrio, B. Nacer, T. Bekkay, G. Vandoni, and C. Félix, “Low energy impact of silver atoms on Pd(100): comparison between helium scattering and microscopic scale simulation results,” Surface Science. 1997. link Times cited: 2 USED (low confidence) M. Shapiro, “Using molecular dynamics simulations to investigate sputtering processes,” Radiation Effects and Defects in Solids. 1997. link Times cited: 8 Abstract: The popularity of molecular dynamics (MD) techniques for the… read moreAbstract: The popularity of molecular dynamics (MD) techniques for the investigation of sputtering processes continues to grow, driven in large measure by the rapid increase in the performance-to-price ratio for modern workstations and high-end personal computers. The ready availability of these inexpensive, powerful computing platforms has encouraged researchers to use MD methods to better understand a variety of problems in sputtering. These include studies of: (1) sputtering induced by complex projectiles: (2) the ejection of small clusters during sputtering: (3) the role of inelastic effects during sputtering: (4) sputtering from complex target materials; and (5) chemical effects during sputtering. Increases in computing power also have made it possible to use more realistic many-body potentials in these simulations. This paper reviews some of the recent literature in these areas, and provides an overview of the progress made in the past few years. read less USED (low confidence) J. R. Fernández, A. M. Monti, and R. Pasianot, “Dynamics of free surfaces in model fcc, hcp and L12 structures,” Philosophical Magazine Part B. 1997. link Times cited: 1 Abstract: Dynamic properties of free surfaces in their relaxed configu… read moreAbstract: Dynamic properties of free surfaces in their relaxed configuration obtained by the static relaxation method, are studied in fcc Ni and Al, hcp Ti and the L12 alloy Ni3Al. The (001), (110) and (111) surfaces are analysed for the fcc and Li2 structures and the (0001), (1210) and (1010) surfaces for the hcp structure. Interatomic potentials of the embedded-atom method type are used to calculate vibrational eigenfrequencies obtained through the Einstein and ‘cluster’ approaches for the atoms on the first few layers. read less USED (low confidence) C. Jun and Y. Yi-ying, “Theoretical strength and phase stability of Cu and Ni under [100] uniaxial loading,” Acta Physica Sinica (overseas Edition). 1996. link Times cited: 0 Abstract: Based on Born's criteria we studied phase stability and… read moreAbstract: Based on Born's criteria we studied phase stability and theoretical strength of fcc crystals of copper and nickel under [100] uniaxial loading. The calculation was carried out using a simple and completely analytical embedded atom method (EAM) potential proposed by the present authors. For Cu, the calculated value of its theoretical strength (0.33 ? 1011 dyn?cm-2) agrees well with the experimental value (0.30 ? 1011 dyn?cm-2), while the calculated strain (9.76%) is somewhat larger than the experimental one (2.8%). For Ni, its theoretical strength and strain predicted using the EAM potential are found smaller than those predicted using a pair potential. It is worthy to note that unlike previous calculations, in which pair potentials were used and three unstressed fcc, bcc, and fct structures included (for Ni only fcc state is found stable, while for Cu both fcc and bcc states are predicted stable), in present calculations using EAM potential the [100] primary loading path passes through only two zeroes (a stable unstressed fcc structure and an unstable stress-free bcc structure) either for Cu or for Ni. read less USED (low confidence) C. Kui-ying, S. Xianwei, Z. Xiumu, and L. Yiyi, “Rapid solidification of Cu25at.% Ni alloy: molecular dynamics simulations using embedded atom method,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1996. link Times cited: 19 USED (low confidence) M. Katagiri et al., “Forces of a Pt adatom on a Pt(100) surface by the embedded-atom method,” Surface Science. 1996. link Times cited: 1 USED (low confidence) A. Seki, O. Hellman, and S. Tanaka, “Calculation of grain boundary energies and structures in copper [001] twist boundaries using the modified embedded atom method,” Scripta Materialia. 1996. link Times cited: 11 USED (low confidence) S. Allameh, S. A. Dregia, and P. Shewmon, “Energy of (110) twist boundaries in AgNi and its variation with induced strain,” Acta Materialia. 1996. link Times cited: 8 USED (low confidence) L. Pizzagalli, D. Stoeffler, and F. Gautier, “Magnetism of Fe atoms deposited on W(001) and W(110): A theoretical study of the role of the interplanar relaxation,” Journal of Magnetism and Magnetic Materials. 1996. link Times cited: 2 USED (low confidence) M. Katagiri, A. Miyamoto, T. Coley, Y. S. Li, and J. Newsam, “Deposition and Surface Dynamic of Metals Studied by the Embedded-Atom Molecular Dynamics Method,” Molecular Simulation. 1996. link Times cited: 8 Abstract: The embedded atom method (EAM) was used to simulate the dyna… read moreAbstract: The embedded atom method (EAM) was used to simulate the dynamics of deposition of supported metal clusters on fcc transition metals and the dynamic behavior of surfaces. The formation of metal clusters was observed before deposition and the geometries, stabilities and dynamics of a series of metal clusters were discussed as obtained by EAM and compared with first-principle calculations. The calculations indicate that the structure of a cluster in the vapor phase is governed by its zero kelvin stability. We find that if the cluster forms in the vapor phase, it is difficult to get epitaxial growth since the metal cohesive energy is large and the chester can not be broken by thermal motions. The effect of impurities on the deposition is also discussed. The anharmonicity, roughening and reconstruction of surface were also investigated. Three-body interactions were found to he important to understand these phenomena read less USED (low confidence) W. Xu and J. Moriarty, “Atomistic simulation of point defects and dislocations in bcc transition metals from first principles,” Journal of Computer-Aided Materials Design. 1996. link Times cited: 3 USED (low confidence) M. Ghaly, R. Averback, and T. D. Rubia, “Surface effects on damage production during ion bombardment: A molecular dynamics study,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1995. link Times cited: 28 USED (low confidence) H. Gades and H. Urbassek, “Preferential sputtering of alloys: a molecular-dynamics study,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1995. link Times cited: 22 USED (low confidence) D. Udler and D. Seidman, “Solute-atom segregation is high-angle (002) twist boundaries in dilute Au–Pt alloys,” Journal of Materials Research. 1995. link Times cited: 9 Abstract: Solute-atom segregation is studied by Monte Carlo simulation… read moreAbstract: Solute-atom segregation is studied by Monte Carlo simulations for three high-angle symmetrical (002) twist boundaries in Au-1 at. % Pt and Pt-1 at. % Au alloys at T = 850 K. It complements our previous study, that focused mainly on low-angle boundaries in the same alloys. Solute enhancement occurs on the Pt-rich side of the phase diagram, while on the Au-rich side net depletion in solute is observed. Following the trend observed for low-angle boundaries, Au as a solute prefers the structural units of the perfect crystal type, while Pt as a solute is depleted at those sites. The solutc concentration at structural units depends on the planar fraction of those units in the boundary. read less USED (low confidence) C.-L. Liu and S. Plimpton, “Molecular dynamics simulations of grain boundary diffusion in Al using embedded atom method potentials,” Journal of Materials Research. 1995. link Times cited: 9 Abstract: Molecular dynamics (MD) simulations of diffusion in a Σ5(310… read moreAbstract: Molecular dynamics (MD) simulations of diffusion in a Σ5(310) [001] Al tilt grain boundary were performed using for the first time three different potentials based on the embedded atom method (EAM). The EAM potentials that produce more accurate melting temperatures also yield activation energies in better agreement with experimental data. Compared to pair potentials, the EAM potentials also give more accurate results. read less USED (low confidence) E. Chulkov and I. Sklyadneva, “Phonon states on the (100), (110) and (111) aluminium surfaces,” Surface Science. 1995. link Times cited: 10 USED (low confidence) H. Zhu, R. Averback, and M. Nastasi, “Molecular dynamics simulations of a 10keV cascade in β-NiAl,” Philosophical Magazine. 1995. link Times cited: 61 Abstract: Molecular dynamics computer simulations were employed to inv… read moreAbstract: Molecular dynamics computer simulations were employed to investigate defect production, atomic mixing and chemical disordering in β-NiA1 owing to 10keV cascades. The embedded atom potentials of Voter and Chen (1989) were employed. Point defect energies and threshold displacement energies were also obtained. The defect production efficiency for the 10 keV cascade was 0.27, which is somewhat larger than that found in simulations of metals with a f.c.c. structure. No interstitial clustering and very little vacancy clustering were observed. The threshold displacement energy is lowest for Ni atoms recoiling along 〈100〉, 34 eV, followed by the 〈110〉 ≊54eV, and 〈111〉, ≊130eV. The 〈111〉 crowd-ion is the stable interstitial defect. The mixing parameter was small, 6.8 A5/eV. Efficient chemical disordering was nevertheless observed near the centre of the cascade; there the short range order parameter was reduced to 0.24. Statistical methods were developed to follow the atomic structure during the evolution ... read less USED (low confidence) C. Kui-ying, L. Hongbo, L. Xiaoping, H. Qiyong, and H. Zhuangqi, “Molecular dynamics simulation of local structure of aluminium and copper in supercooled liquid and solid state by using EAM,” Journal of Physics: Condensed Matter. 1995. link Times cited: 38 Abstract: Based on the embedded-atom method, molecular dynamics simula… read moreAbstract: Based on the embedded-atom method, molecular dynamics simulations have been performed to study the structural features of Al and Cu in the liquid and solid states during rapid solidification. The calculated pair correlation functions above the melting points of Al and Cu are found to be in good agreement with experiment, especially for Cu. The results show that the EAM can correctly and efficiently predict the glass transition and crystallization during rapid solidification from liquid metals, and can also describe microstructures of liquids, supercooled liquids, glasses and crystalline phases. In addition, structural analysis using bond orientational order and pair analysis techniques have been made in detail, and the effect of cooling rate on microstructures during rapid solidification has been analysed. read less USED (low confidence) G. Derry, C. B. McVey, and P. Rous, “The surface structure and segregation profile of Ni50Pd50(100) : a dynamical LEED study,” Surface Science. 1995. link Times cited: 44 USED (low confidence) R. Taylor and B. Garrison, “Molecular dynamics simulations of keV particle bombardment. Correlation of intact molecular ejection with adsorbate size,” Chemical Physics Letters. 1994. link Times cited: 13 USED (low confidence) E. Webb and S. Garofalini, “Effect of adsorption of EAM metal atoms on the structure of a sodium-alumino silicate glass surface: a molecular dynamics simulation,” Surface Science. 1994. link Times cited: 11 USED (low confidence) I. Majid, C. Counterman, P. Bristowe, and R. Balluffi, “X-ray diffraction and computer simulation studies of the structure of [001] twist boundaries in Au-Ag alloys,” Acta Metallurgica Et Materialia. 1994. link Times cited: 0 USED (low confidence) S. Allameh, S. A. Dregia, and P. Shewmon, “Structure and energy of (110) twist boundaries in the Ag/Ni system,” Acta Metallurgica Et Materialia. 1994. link Times cited: 8 USED (low confidence) S. Khare and T. Einstein, “Energetics of steps and kinks on Ag and Pt using equivalent crystal theory (ECT),” Surface Science. 1994. link Times cited: 23 USED (low confidence) Y. Shimomura, R. Nishiguchi, T. D. Rubia, and M. Guinan, “Relaxation and movement of point defect clusters in copper by molecular dynamics simulation,” Radiation Effects and Defects in Solids. 1994. link Times cited: 5 Abstract: The structures of point defect clusters of both interstitial… read moreAbstract: The structures of point defect clusters of both interstitial and vacancy type were examined by computer simulation using molecular dynamics and molecular statics with the DYNAMO code (Daw, Foiles and Baskes [6]). The code implements an isotropic potential of embedded atom method (EAM) developed by Daw and Baskes [5]. Interstitial clusters relax to either the immobile mixture of dumbbell and bcc interstitials or a mobile platelet of parallel interstitials. The latter cluster moves along directions. A tri-vacancy relaxes to an un-collapsed stacking fault tetrahedron (sft) of Damask-Dienes type (3v-sft) containing a central atom that vibrates with a large amplitude. A hexa-vacancy relaxes to a stacking fault tetrahedron the structure of which fluctuates between a sft and void. Larger vacancy clusters are stable as a combination of sft and 3v-sft. In these vacancy clusters, atoms show significant vibration with large amplitude. Voids form only with the inclusion of gas-atoms into va... read less USED (low confidence) T. Honglai and Y. Wei, “Atomistic/continuum simulation of interfacial fracture part I: Atomistic simulation,” Acta Mechanica Sinica. 1994. link Times cited: 28 USED (low confidence) P. Fernandez, C. Massobrio, P. Blandin, and J. Buttet, “Embedded atom method computations of structural and dynamical properties of Cu and Ag clusters adsorbed on Pd(110) and Pd(100) : evolution of the most stable geometries versus cluster size,” Surface Science. 1994. link Times cited: 6 USED (low confidence) S. Phillpot, “Reconstruction of grain boundaries in copper and gold by simulation,” Journal of Materials Research. 1994. link Times cited: 3 Abstract: The reconstructions of high-angle twist grain boundaries on … read moreAbstract: The reconstructions of high-angle twist grain boundaries on the four densest atomic planes in fcc copper, as described by a Lennard-Jones potential, and gold, as described by an embedded-atom-method potential, are investigated using the recently developed method of grand-canonical simulated quenching. It is found that the grain boundaries on the widely spaced (111) and (100) planes do not reconstruct, while those on the less widely spaced (110) and (113) planes do reconstruct. The effect that reconstruction can have on the physical properties of an interfacial system is illustrated by comparing the elastic properties and ideal cleavage energies of reconstructed grain boundaries with those of corresponding unreconstructed grain boundaries. read less USED (low confidence) D. N. Bernardo, R. Bhatia, and B. Garrison, “keV particle bombardment of solids: molecular dynamics simulations and beyond,” Computer Physics Communications. 1994. link Times cited: 16 USED (low confidence) P. Fay, J. R. Ray, and R. J. Wolf, “Detailed balance method for chemical potential determination in Monte Carlo and molecular dynamics simulations,” Journal of Chemical Physics. 1994. link Times cited: 7 Abstract: We present a new, nondestructive, method for determining che… read moreAbstract: We present a new, nondestructive, method for determining chemical potentials in Monte Carlo and molecular dynamics simulations. The method estimates a value for the chemical potential such that one has a balance between fictitious successful creation and destruction trials in which the Monte Carlo method is used to determine success or failure of the creation/destruction attempts; we thus call the method a detailed balance method. The method allows one to obtain estimates of the chemical potential for a given species in any closed ensemble simulation; the closed ensemble is paired with a ‘‘natural’’ open ensemble for the purpose of obtaining creation and destruction probabilities. We present results for the Lennard‐Jones system and also for an embedded atom model of liquid palladium, and compare to previous results in the literature for these two systems. We are able to obtain an accurate estimate of the chemical potential for the Lennard‐Jones system at higher densities than reported in the literature. read less USED (low confidence) M. Schmid, A. Biedermann, C. Slama, H. Stadier, P. Weigand, and P. Varga, “Preferential sputtering of Pt-Ni alloy single crystals studied by scanning tunneling microscopy,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1993. link Times cited: 22 USED (low confidence) J. Black, Z. Tian, and T. Rahman, “Structure and dynamics of an Ag overlayer on Cu(100): a study using the embedded atom method,” Surface Science. 1993. link Times cited: 14 USED (low confidence) Z. C. Li and S. Whang, “Planar defects in 113 planes of L10 type TiAl their structures and energies,” Philosophical Magazine. 1993. link Times cited: 0 Abstract: Stacking faults, complex stacking faults and antiphase bound… read moreAbstract: Stacking faults, complex stacking faults and antiphase boundary in a {113} plane of L10 type TiAl were introduced into the perfect crystal by a shear model. The structure and the stacking sequences of these defects were studied. The energies of these defects were calculated by an embedded atom method. The results show that metastable stacking faults and antiphase boundary exist in the (113) plane while no metastable complex stacking faults can be found. The determined energies of the intrinsic stacking faults, the extrinsic stacking faults and the antiphase boundary by the embedded atom method were 1195, 1217 and 976 (mJm−2), respectively. read less USED (low confidence) D. Koleske and S. Sibener, “Molecular dynamics simulations of the basal planes of Ni and Cu using Finnis-Sinclair potentials,” Surface Science. 1993. link Times cited: 15 USED (low confidence) T. D. Rubia, A. Caro, M. Spaczér, G. Janaway, M. Guinan, and M. Victoria, “Radiation-induced disordering and defect production in Cu3Au and Ni3Al studied by molecular dynamics simulation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1993. link Times cited: 23 USED (low confidence) J. Hayoz, D. Naumović, R. Fasel, P. Aebi, and L. Schlapbach, “Growth of Ag on Cu(001) studied by full-hemispherical X-ray photoelectron diffraction,” Surface Science. 1993. link Times cited: 19 USED (low confidence) C. M. Gilmore and J. A. Spraque, “Molecular dynamics simulations of thin film growth on Ag(100) and (111) with energetic Ag atoms,” Nanostructured Materials. 1993. link Times cited: 6 USED (low confidence) H. Y. Wang, R. Najafabadi, D. Srolovitz, and R. LeSar, “Interfacial segregation in Ag-Au, Au-Pd, and Cu-Ni alloys: I. (100) surfaces,” Interface Science. 1993. link Times cited: 15 USED (low confidence) U. Wolf, F. Ernst, T. Muschik, M. Finnis, and H. Fischmeister, “The influence of grain boundary inclination on the structure and energy of Σ=3 grain boundaries in copper,” Philosophical Magazine. 1992. link Times cited: 171 Abstract: In a combined theoretical and experimental study, the energi… read moreAbstract: In a combined theoretical and experimental study, the energies and structures of σ = 3, [011] tilt boundaries in Cu were investigated. Equilibrium atomistic structures and grain-boundary energies were calculated by static energy minimization using an embedded-atom potential. Cu bicrystals of the same boundary orientations were fabricated by welding of Cu single crystals. Grain-boundary energies were measured by the thermal grooving technique. The atomistic structure of the {211} twin boundary was investigated by high-resolution transmission electron microscopy (HRTEM). The calculated grain-boundary energies γb plotted against the inclination of the boundary plane show a minimum for the {111} twin boundary and a second minimum at an inclination of about 82° to the {111} boundary. The calculated dependence of γb on inclination is confirmed by the measured energies over the entire range. Common to all calculated boundary structures is a microfaceting into {111} and {211} twin facets. The structures ... read less USED (low confidence) T. D. Rubia, M. Guinan, A. Caro, and P. Scherrer, “Radiation effects in FCC metals and intermetallic compounds: A molecular dynamics computer simulation study,” Radiation Effects and Defects in Solids. 1992. link Times cited: 21 Abstract: We review recent results on atomic mixing, radiation-induced… read moreAbstract: We review recent results on atomic mixing, radiation-induced disordering, and defect production and clustering induced by displacement cascades in Cu, Ag, Cu{sub 3}Au and Ni{sub 3}Al. We employ molecular dynamics computer simulation methods with isotropic many body potentials and recoil energies near subcascade formation regime up the locally molten cascade core in the pure metals and intermetallics. Disordering of intermetallics takes place in the cascade core, but because of the short lifetime of the displacement cascade, chemical short range order is preserved in molten zone. Results reveal very large vacancy and interstitial type defect clusters at high recoil energy and cascade energy density. Vacancies agglomerate and collapse into Frank dislocation loops in quenching of the cascade molten core. Large interstitial clusters are directly produced in cascades and form prismatic dislocation loops. Fraction of defects in clusters for low temperature cascades increases with recoil energy and approaches {approx} 70 and 60% for interstitials and vacancies, at recoil energies near threshold for subcascades. In the case of intermetallics the large energy required to produce and transport a superdislocation appears to inhibit interstitial prismatic loop punching and interstitials appear as isolated (100) dumbbells. read less USED (low confidence) D. Udler and D. Seidman, “Solute‐Atom Segregation at Symmetrical Twist Boundaries Studied by Monte Carlo Simulation,” Physica Status Solidi B-basic Solid State Physics. 1992. link Times cited: 26 Abstract: Detailed Monte Carlo simulations are performed of solute-ato… read moreAbstract: Detailed Monte Carlo simulations are performed of solute-atom segregation at (002) twist boundaries in the Au–Pt system at 850 K; the particular single-phase bicrystal alloys studied are Pt–1 at% Au and Au–1 at% Pt. The emphasis in this paper is on studying the distribution of solute atoms at low-angle boundaries. For the Pt–1 at% Au alloy the distribution of sites enhanced in the solute species Au is found to form a bipyramid based on the square cells of the orthogonal primary grain boundary screw dislocations. In the case of the Au–1 at% Pt alloy the solute species Pt is found to be depleted and it also forms a similar bipyramidal pattern. The Gibbsian interfacial excesses of Au and Pt are found to be positive and negative, respectively, for the Pt–1 at% Au and Au–1 at% Pt bicrystal alloys. The absolute values of these Gibbsian interfacial excesses both increase with increasing twist angle.
An (002)-Dreh-Korngrenzen im Au–Pt System werden ausfuhrliche Monte-Carlo-Simulationen zur Segregation geloster Atome bei 850 K durchgefuhrt; speziell untersuchte einphasige Bikristalle sind Pt–1 At% Au und Au–1 At% Pt. Schwerpunkt der Arbeit ist das Studium der Verteilung der gelosten Atome an Klein-Winkelkorngrenzen. Fur Pt–1 At% Au bilden die Au-angereicherten Platze eine Bipyramide mit den quadratischen Zellen der orthogonalen primaren Korngrenzen-Schraubenversetzungen als Basis. Im Falle der Au–1 At% Pt Legierung findet man auf einem ahnlichen Bipyramiden-Muster eine Abnahme an Pt Atomen. Der Gibbssche Grenzflachen-Uberschus an Au und Pt is positiv bzw. negativ fur Pt–1 At% Au bzw. Au–1 At% Pt Bikristalle. Die Absolutwerte des Gibbsschen Grenzflachen-Uberschusses wachsen mit zunehmendem Drehwinkel jeweils an. read less USED (low confidence) I. Majid and P. Bristowe, “An x-ray diffraction and computer simulation study of [111] twist boundaries in gold,” Philosophical Magazine. 1992. link Times cited: 6 Abstract: Quantitative X-ray diffraction measurements have been made f… read moreAbstract: Quantitative X-ray diffraction measurements have been made from a series of (111) gold bicrystals containing twist grain boundaries with controlled geometry. Each bicrystal was prepared at a specific angle of misorientation Θ° but because of double positioning both Θ° and 60-Θ° twist boundaries were present in the specimens. Since these pairs of boundaries have distinct structures, the intensity measurements from each bicrystal contained superimposed information representing an average from both boundary types. In order to understand these measurements in terms of the boundary structures involved, computer calculations were performed to characterize the theoretical scattering effects expected from [111] twist boundaries related by double positioning. By comparing the measurements with the calculations, it was possible to determine the conditions under which reliable structural information concerning doubly positioned twist boundaries could be obtained. From an appropriate analysis of the data, it... read less USED (low confidence) A. Wucher and B. Garrison, “Internal and translational energy of sputtered silver dimers: a molecular dynamics study,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1992. link Times cited: 15 USED (low confidence) M. Mills, M. Daw, G. Thomas, and F. Cosandey, “High-resolution transmission electron microscopy of grain boundaries in aluminum and correlation with atomistic calculations,” Ultramicroscopy. 1992. link Times cited: 48 USED (low confidence) V. Chirita and B. Pailthorpe, “Molecular dynamics simulation of the Ni(001) surface,” Thin Solid Films. 1992. link Times cited: 5 USED (low confidence) 周富信, 彭八一, 吴希俊, and 汤奇恒, “PSEUDO-MOLECULAR DYNAMICS STUDY OF GRAIN BOUNDARY SEGREGATION *,” Acta Mechanica Sinica. 1992. link Times cited: 0 USED (low confidence) A. Seki, D. Seidman, Y. Oh, and S. Foiles, “Monte Carlo simulations of segregation at [001] twist boundaries in a Pt(Au) alloy—I. Results,” Acta Metallurgica Et Materialia. 1991. link Times cited: 41 USED (low confidence) P. Bacher, P. Wynblatt, and S. Foiles, “A Monte Carlo study of the structur and composition of (001) semicoherent interphase boundaries in CuAgAu alloys,” Acta Metallurgica Et Materialia. 1991. link Times cited: 32 USED (low confidence) D. Seidman, “Solute-atom segregation at internal interfaces on an atomic scale: atom-probe experiments and computer simulations,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1991. link Times cited: 30 USED (low confidence) D. Wolf, “Correlation between the energy and structure of grain boundaries in b.c.c. metals. II. Symmetrical tilt boundaries,” Philosophical Magazine. 1990. link Times cited: 117 Abstract: The correlation between the structure and zero-temperature e… read moreAbstract: The correlation between the structure and zero-temperature energy of symmetrical tilt grain boundaries (STGBs) in b.c.c. metals is investigated using a many-body potential of the Finnis-Sinclair type for Mo and Johnson's pair potential spline-fitted for α-Fe. As in free surfaces, the misorientation phase space associated with these simple planar defects consists of only two macroscopic degrees of freedom, namely those defining the grain-boundary (GB) plane normal. In a novel approach, the two-dimensional phase space is investigated in terms of a stereographic triangle and not in terms of the usual three parameters associated with the boundary misorientation. A comparison with similar calculations for free surfaces demonstrates the importance of the translational (microscopic) degrees of freedom in grain boundaries, while the comparison with STGBs in f.c.c. metals elucidates the important role played by the GB plane. read less USED (low confidence) R.-J. Jhan and P. Bristowe, “A molecular dynamics study of grain boundary migration without the participation of secondary grain boundary dislocations,” Scripta Metallurgica Et Materialia. 1990. link Times cited: 33 USED (low confidence) S. P. Chen, A. Voter, R. Albers, A. M. Boring, and P. Hay, “Investigation of the effects of boron on Ni_3Al grain boundaries by atomistic simulations,” Journal of Materials Research. 1990. link Times cited: 126 Abstract: A series of simulations has been performed on grain boundari… read moreAbstract: A series of simulations has been performed on grain boundaries in Ni and Ni_3Al with and without boron doping using embedded atom-style potentials. A new procedure of obtaining “reference” data for boron related properties from electronic band structure calculations has been employed. Good agreement with existing experimental structural and energetic determinations was obtained. Boron is found to segregate more strongly to grain boundaries than to free surfaces. Adding boron to grain boundaries in Ni and Ni_3Al increases their cohesive strength and the work required to pull apart the boundary. This effect is much more dramatic for Ni-rich boundaries than for stoichiometric or Al-rich boundaries. In some Ni-rich cases, adding boron increases the cohesive strength of the boundary to such an extent that the boundaries become stronger than the bulk. Bulk Ni_3Al samples that are Ni-rich produce Ni-rich grain boundaries. The best cohesive properties of Ni_3Al grain boundaries are obtained when the boundary is Ni saturated and also with boron present. Boron and nickel are found to cosegregate to the grain boundaries. read less USED (low confidence) M. Kluge, D. Wolf, J. Lutsko, and S. Phillpot, “Formalism for the calculation of local elastic constants at grain boundaries by means of atomistic simulation,” Journal of Applied Physics. 1990. link Times cited: 108 Abstract: A new formalism for use in atomistic simulations to calculat… read moreAbstract: A new formalism for use in atomistic simulations to calculate the full local elastic‐constant tensor in terms of local stresses and strains is presented. Results of simulations on a high‐angle (001) twist grain boundary are illustrated, using both a Lennard–Jones potential for Cu and an embedded‐atom potential for Au. The two conceptionally rather different potentials show similar anomalies in all elastic constants, confined to within a few lattice planes of the grain boundary, with an especially dramatic reduction in the resistance to shear parallel to the grain‐boundary plane. It is found that the primary cause of the anomalies is the atomic disorder near the grain boundary, as evidenced by the slice‐by‐slice radial distribution functions for the inhomogeneous interface system. read less USED (low confidence) R. Najafabadi, D. Srolovitz, and R. LeSar, “Influence of anisotropic vibrational motion on diffraction from grain boundaries,” Scripta Metallurgica Et Materialia. 1990. link Times cited: 4 USED (low confidence) J. P. Rogers, P. Wynblatt, S. Foiles, and M. Baskes, “Monte Carlo simulation of the CuAg (001) semicoherent interphase boundary,” Acta Metallurgica Et Materialia. 1990. link Times cited: 31 USED (low confidence) Y. Gao, P. Shewmon, and S. A. Dregia, “Investigation of low energy interphase boundaries in agni by computer simulation and crystallite rotation,” Acta Metallurgica. 1989. link Times cited: 39 USED (low confidence) D. Wolf, “Structure-energy correlation for grain boundaries in F.C.C. metals—I. Boundaries on the (111) and (100) planes,” Acta Metallurgica. 1989. link Times cited: 168 USED (low confidence) S. P. Chen, D. Srolovitz, and A. Voter, “Computer simulation on surfaces and [001] symmetric tilt grain boundaries in Ni, Al, and Ni_3Al,” Journal of Materials Research. 1989. link Times cited: 141 Abstract: We have used “local volume” (embedded atom) type potentials … read moreAbstract: We have used “local volume” (embedded atom) type potentials to study the surfaces and grain boundaries of Ni, Al, and Ni_3Al. The simulations show that with appropriately fit potentials, the surface and grain boundary structure can be realistically calculated. The surface rippling and relaxation show good agreement with experiments. The energies of most surfaces and grain boundaries also agree with existing data. The structural unit model for grain boundaries in Ni_3Al shows the same generic units as in pure metals, but with large variations due to distortions and multiplicity. The utility of the structural unit model is thus more limited for alloys. The grain boundary energies were found to be the highest for Al-rich Ni_3Al grain boundaries, and depend significantly on the local composition of the grain boundary. The cusps in the grain boundary energy as a function of misorientation angle are different for different grain boundary stoichiometries. The Ni_3Al grain boundaries have approximately the same grain boundary energy and cohesive energy as that of Ni. read less USED (low confidence) J. Lutsko, D. Wolf, and S. Phillpot, “High-temperature behavior of grain boundaries from embedded atom method molecular dynamics simulation,” MRS Proceedings. 1988. link Times cited: 0 Abstract: The behavior of a metallic grain boundary at high temperatur… read moreAbstract: The behavior of a metallic grain boundary at high temperatures is studied using an embedded atom potential. A recently developed molecular dynamics code is used which allows the simulation of an isolated grain boundary at temperatures as high as the bulk melting point. The stability of the boundary below the melting point is studied and compared with earlier investigations which have suggested the existence of a ''premelting'' transition. It is found that the boundary migrates at high temperature but remains well defined up to the bulk melting point. In contrast to simulations of ideal crystals, it was not possible to superheat the grain boundary due to the nucleation of bulk melting at the boundary. 14 refs., 4 figs. read less USED (low confidence) K. Blixt, L. Christierson, A. Ahadi, P. Hansson, and S. Melin, “Molecular dynamics simulations of nanometric cutting of single crystal copper sheets using a diamond tool,” Procedia Structural Integrity. 2023. link Times cited: 1 USED (low confidence) J. Li, Y. Tian, and Q. Fang, “Molecular dynamics simulation of friction, lubrication, and tool wear during nanometric machining,” Machining and Tribology. 2022. link Times cited: 5 USED (low confidence) J. G. Plascencia, E. Bird, and Z. Liang, “Thermal and mass transfer resistance at a liquid-gas interface of an evaporating droplet: A molecular dynamics study,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 5 USED (low confidence) B.-X. Zhang, S. Wang, Y.-B. Wang, S.-R. Gao, Y.-R. Yang, and X.-dong Wang, “Spreading of a nanodroplet over isothermally heated smooth and nanostructured surfaces: A molecular dynamics study,” International Journal of Thermal Sciences. 2021. link Times cited: 7 USED (low confidence) X.-feng Yang et al., “The effect of grain boundary structures on crack nucleation in nickel nanolaminated structure: A molecular dynamics study,” Computational Materials Science. 2021. link Times cited: 2 USED (low confidence) A. K. Balerba et al., “Graphene nano-flakes on Cu low-index surfaces by density functional theory and molecular dynamics simulations.” 2020. link Times cited: 2 USED (low confidence) L. Wang et al., “Effect of Sink Strength on Coherency Loss of Precipitates in Dilute Cu Base Alloys During in Situ Ion Irradiation,” Mechanical Engineering eJournal. 2020. link Times cited: 0 Abstract: In situ irradiations with 1 MeV Kr ions at 50~613 K up to a … read moreAbstract: In situ irradiations with 1 MeV Kr ions at 50~613 K up to a fluence of 6.25 × 10 14 ions/cm 2 (~1.25 dpa) have been performed on pre-aged dilute Cu-0.9%Co, Cu-0.9%Fe and Cu-0.8%Cr alloys containing uniform matrix dispersions of coherent precipitates in order to study the effects of initial precipitate sink strength, damage dose and irradiation temperature on radiation-induced coherency loss of precipitates. Coherent precipitates with different sink strengths (2πNd , where N and d are the precipitate density and diameter) were used in this work to examine potential differences in atomic relaxation during absorption of point defects. At low sink strengths (~10 13 m -2 ), loss of precipitate coherency could be induced for doses below ~0.1 dpa. There might be a preferential medium-range strain-induced bias for absorption of interstitial defects due to tensile strains at the undersized precipitates, which induces relatively rapid loss of coherency at low precipitate sink strengths. High sink strength (~10 14 m -2 ) conditions were relatively resistant to the radiation-induced loss of coherency and this might be due to nearly equal numbers of interstitial and vacancy defects arriving at the precipitate interface for such high sink strength conditions. The precipitate coherency loss was observed to have a weak dependence on irradiation temperature. Molecular dynamics simulations confirm a strong effect of precipitate sink strength on the probability of interstitial absorption at precipitates. read less USED (low confidence) K. Liu, H. Wang, and X. Zhang, “Molecular Dynamics Simulation of Ductile Mode Cutting,” Springer Series in Advanced Manufacturing. 2019. link Times cited: 1 USED (low confidence) S. Melin, P. Hansson, and A. Ahadi, “Grain boundary influence on the mechanical response to tensile loading for nanosized Cu beams modelled by MD simulations,” Procedia Structural Integrity. 2019. link Times cited: 3 USED (low confidence) C.-H. Yu, K. Lin, and C.-S. Chen, “Nanoindentation and Indentation Size Effects: Continuum Model and Atomistic Simulation,” Handbook of Mechanics of Materials. 2019. link Times cited: 1 USED (low confidence) S. Paul, Nagahanumaiah, S. Mitra, and D. Roy, “Molecular Dynamics Simulation Study of Neck Growth in Micro-selective Laser Sintering of Copper Nanoparticles.” 2018. link Times cited: 6 USED (low confidence) A. Joshi and S. James, “Molecular Dynamics Simulation Study on Effect of Process Parameters on Coatings during Cold Spray Process,” Procedia Manufacturing. 2018. link Times cited: 20 USED (low confidence) H. Mori and N. Matubayasi, “MD simulation analysis of resin filling into nano-sized pore formed on metal surface,” Applied Surface Science. 2018. link Times cited: 13 USED (low confidence) G. P. Sannikov and A. E. Korenchenko, “ESTIMATED PROBABILITY OF COPPER LONG-LIVED DIMER FORMATION IN TWO PARTICLE COLLISIONS BASED ON THE MOLECULAR DYNAMICS SIMULATION.” 2017. link Times cited: 0 USED (low confidence) Y. Zhang, S. Jiang, X.-ming Zhu, and Y. Zhao, “Mechanisms of crack propagation in nanoscale single crystal, bicrystal and tricrystal nickels based on molecular dynamics simulation,” Results in physics. 2017. link Times cited: 30 USED (low confidence) A. Ahadi, P. Hansson, and S. Melin, “Indentation of thin copper film using molecular dynamics and peridynamics,” Procedia structural integrity. 2016. link Times cited: 12 USED (low confidence) Z. Fan, O. H. Duparc, and M. Sauzay, “Molecular dynamics simulation of surface step reconstruction and irreversibility under cyclic loading,” Acta Materialia. 2016. link Times cited: 14 USED (low confidence) L. Cui, Y. Feng, J.-kun Tang, P. Tan, and X. Zhang, “Heat conduction in coaxial nanocables of Au nanowire core and carbon nanotube shell: A molecular dynamics simulation,” International Journal of Thermal Sciences. 2016. link Times cited: 19 USED (low confidence) A. Evteev, L. Momenzadeh, E. Levchenko, I. Belova, and G. Murch, “Vibrational contribution to thermal transport in liquid cooper: Equilibrium molecular dynamics study,” Computational Materials Science. 2015. link Times cited: 5 USED (low confidence) S. Hosseini, M. Vahdati, and A. Shokuhfar, “Molecular Dynamics Simulation on Nano-Machining of Single Crystal Copper with a Void.” 2012. link Times cited: 2 USED (low confidence) E. Hristova, R. Janisch, R. Drautz, and A. Hartmaier, “Solubility of carbon in α-iron under volumetric strain and close to the Σ5(3 1 0)[0 0 1] grain boundary: Comparison of DFT and empirical potential methods,” Computational Materials Science. 2011. link Times cited: 46 USED (low confidence) F. V. Mackenzie and B. Thijsse, “Atomistic Simulations of the Mechanical Response of Copper/Polybutadiene Joints under Stress,” MRS Proceedings. 2009. link Times cited: 1 Abstract: Metal/polymer system joints are widely encountered nowadays … read moreAbstract: Metal/polymer system joints are widely encountered nowadays in microscopic structures such as displays and microchips. In several critical cases they undergo thermal and mechanical loading, with contact failure due to fracture as a possible consequence. Because of their variety in nature and composition metal/polymer joints have become major challenges for experimental, theoretical, and numerical studies. Here we report on results of molecular dynamics simulations carried out to study the mechanical response of a metal/polymer joint, in this case the Cu/polybutadiene model system. The behavior of Cu and the cross-linked polybutadiene are modeled, respectively, by the Embedded Atom Method (EAM) and the Universal Force Field (UFF). Loading is applied under compression. Different potentials are used to describe the interactions in the metal/polymer interface, which allows us to qualitatively analyze possible mechanisms of failure in these joints, below the metal melting point and above the polymer glass transition temperatures. read less USED (low confidence) M. Hou and O. Melikhova, “Internal stress and mechanical deformation of Al and Al/Ni multilayered nanowires,” Acta Materialia. 2009. link Times cited: 9 USED (low confidence) Y. Ma and P. Balbuena, “Pt surface segregation in bimetallic Pt3M alloys: A density functional theory study,” Surface Science. 2008. link Times cited: 197 USED (low confidence) J. Rodríguez-López, J. M. Montejano-Carrizales, and M. José-Yacamán, “How does a crystal grow? Experiments, models and simulations from the nano- to the micro-scale regime.” 2007. link Times cited: 2 USED (low confidence) N. Lümmen, B. Fischer, and T. Kraska, “Homogeneous nucleation and growth from highly supersaturated vapor by molecular dynamics simulation.” 2007. link Times cited: 2 USED (low confidence) T. Muramoto, K. Itabasi, and Y. Yamamura, “MD simulation of surface smoothing due to cluster impact: estimation of radiation damage,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 4 USED (low confidence) J. Jiménez‐Sáez, Pérez-Martı́n A., M. Said-Ettaoussi, and Jiménez-Rodrı́guez J. J., “Molecular dynamics simulation of Ni cluster deposition on Cu(0 0 1) surfaces,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 6 USED (low confidence) A. Dalis and S. Friedlander, “A Molecular Dynamics Study of Short Nanoparticle Chains under Mechanical Strain: Linear and Kinked Configurations,” MRS Proceedings. 2004. link Times cited: 0 Abstract: Previous studies in our laboratory have shown that individua… read moreAbstract: Previous studies in our laboratory have shown that individual nanoparticle chain aggregates (NCA) exhibit remarkable mechanical behavior when under strain inside the transmission electron microscope. NCA made of various materials (e.g. carbon, metal oxides, metals, etc.) were strained by as much as 100% when tension was applied to them. After breaking, the NCA rapidly contracted to form more compact structures. In this study, molecular dynamics (MD) computer simulations are employed to investigate, at the atomic scale, the behavior of short nanoparticle chains under strain and to obtain quantitative information of the forces involved in chain straining and fracturing. The interaction potential used is that of copper obtained with the embedded atom method (EAM). Although the methodology is generally applicable, copper was selected as a test material because reliable interatomic potentials are available. Seven single- crystal nanoparticles, each 2.452 nm in diameter, are placed in contact in two chain configurations, linear and kinked. The structures are initially relaxed adiabatically with MD steps for 225 ps, at a starting temperature of 300 K. The bonding energy between any two nanoparticles in contact ranges from about 20 eV to 30 eV at 0 K. The two relaxed chain configurations are strained along their longest dimension, to the breaking point, at strain rates spanning from 0.3 m/s to 10 m/s. We identify mechanisms of stress accommodation that lead to plastic deformation and eventually fracture for both chain configurations, linear and kinked, and we construct the corresponding stress-strain curves. The two chain configurations exhibit different mechanical behavior. Applications of our experimental and simulation studies on NCA are to the behavior of nanocomposite materials, including carbon black reinforced rubber, sampling of aggregates by high speed impactors and the formation of flexible coatings of nanoparticles. read less USED (low confidence) C. Massobrio, M. Celino, Y. Pouillon, and I. Billas, “7. From the Cluster to the Liquid: Ab-Initio Calculations on Realistic Systems Based on First-Principles Molecular Dynamics.” 2004. link Times cited: 0 USED (low confidence) C. M. Gilmore and J. Sprague, “Molecular Dynamics Simulation of Thin Film Growth with Energetic Atoms.” 2002. link Times cited: 2 USED (low confidence) T. Ikeshoji, “Formation of Free Clusters and Their Structures: Molecular Dynamics Simulations.” 2002. link Times cited: 0 USED (low confidence) D. Seidman, “Subnanoscale Studies of Segregation at Grain Boundaries: Simulations and Experiments,” Annual Review of Materials Research. 2002. link Times cited: 56 Abstract: ▪ Abstract Lattice statics (0 K) and Monte Carlo (Metropolis… read moreAbstract: ▪ Abstract Lattice statics (0 K) and Monte Carlo (Metropolis algorithm) simulation are utilized to determine equilibrium and metastable structures of 21 [110] symmetric tilt boundaries between 0° and 180° at 800 K, employing a Ni embedded-atom method potential; attention is paid to the effects of the macroscopic and microscopic degrees of freedom (DOFs) on grain boundary (GB) structure. Segregation of Pd is studied at all GB structures at 800 K, employing Monte Carlo and overlapping distributions Monte Carlo simulation, which yield the Gibbsian interfacial excess of Pd (ΓPd) as a function of tilt angle for both stable and metastable structures, thereby demonstrating that ΓPd is an anisotropic function of a GB's five macroscopic DOFs. In addition, atom-probe experiments on GBs on an Fe-3 at.% Si alloy, whose five macroscopic DOFs are measured by transmission electron microscopy, directly yield ΓSi and thereby demonstrate experimentally that this quantity is an anisotropic function of these DOFs. read less USED (low confidence) T. Nozaki, Y. Kogure, and M. Doyama, “Plastic Deformation of Thin Films – Bending,” MRS Proceedings. 2001. link Times cited: 0 USED (low confidence) A. C. Pillai and R. E. Miller, “Crack Behaviour at Bi-Crystal Interfaces: A Mixed Atomistic and Continuum Approach,” MRS Proceedings. 2000. link Times cited: 10 Abstract: Interfacial defects like grain boundaries and phase boundari… read moreAbstract: Interfacial defects like grain boundaries and phase boundaries play an important role in the mechanical behaviour of engineering alloys. In this work the problem of a crack on a bi-crystal interface is studied at the atomic scale, with the goal of elucidating the effects of varrying interatomic interaction on crack behaviour and to assess the suitability of existing fracture criteria to the anisotropic bi-crystal case. Calculations are performed using the Quasicontinuum (QC) method [1]. Using suitable approximations, some of the existing fracture criteria were used to predict ductile or brittle fracture and compared to the QC results. read less USED (low confidence) J. Zimmerman, P. Klein, and S. Foiles, “Effect of Surface Steps on Dislocation Structure During Nanoindentation,” MRS Proceedings. 2000. link Times cited: 5 USED (low confidence) P. Ballo, N. Kioussis, and G. Lu, “Monte Carlo Simulations of Grain Boundary Sliding and Migration: Effect of Temperature and Vacancy,” MRS Proceedings. 2000. link Times cited: 2 USED (low confidence) J. Mortensen, B. Hammer, O. H. Nielsen, K. Jacobsen, and J. Nørskov, “Density Functional Theory Study of Self-Diffusion on the (111) Surfaces of Ni, Pd, Pt, Cu, Ag and Au.” 1996. link Times cited: 5 USED (low confidence) R. Averback, M. Ghaly, and H. Zhu, “Surface Damage During Kev Ion Irradiation: Results of Computer Simulations,” MRS Proceedings. 1995. link Times cited: 3 USED (low confidence) C. Battaile, R. NajafBdi, and D. Srolovitz, “Atomistic Monte Carlo Simulations of Surface Segregation in (Fe x Mn 1-x )O and (Ni x Co 1-x )O,” MRS Proceedings. 1995. link Times cited: 0 Abstract: An atomistic Monte Carlo (MC) method has been used to predic… read moreAbstract: An atomistic Monte Carlo (MC) method has been used to predict equilibrium segregation of isovalent cations to (001) surfaces in (Fe x .Mn-x)O and (Ni x Col-x)O. The surface is found to be enriched with solvent in both systems. Long-range electrostatic interactions and atomic motions that occur on small time scales make the MC approach very computationally demanding. The Free Energy Minimization (FEM) method is a more efficient alternative for performing such segregation simulations, but involves several approximations. Comparison of the surface segregation profiles determined using the MC and FEM simulation methods show that the two are essentially indistinguishable. The FEM results can be obtained about 1,000 times faster than the MC predictions. Therefore, the FEM method is a practical and accurate alternative to the more cumbersome MC approach. read less USED (low confidence) A. Landa, H. Häkkinen, R. Barnett, P. Wynblatt, and U. Landman, “Molecular Dynamics Study of Disordering and Premelting of the Pb(110) Surface,” MRS Proceedings. 1994. link Times cited: 1 USED (low confidence) R. Averback, M. Ghaly, and H. Zhu, “Defect production mechanisms during keV ion irradiation: results of computer simulations,” MRS Proceedings. 1994. link Times cited: 3 Abstract: Production of Defects in Metals by Collision Cascades: TEM E… read moreAbstract: Production of Defects in Metals by Collision Cascades: TEM Experiments p. 47 Effect of Copper and Nickel on the Neutron Irradiation Damage in Iron Alloys p. 57 Cascade Damage in the Ordered Alloy Ni[subscript 3]Al p. 63 Void Swelling Resistance in Fe-Cr Alloys at 200 DPA p. 69 Nucleation with Radiation Dissolution and Resolution p. 75 Dissolution of Ordered Precipitates Under Ion Irradiation p. 83 Monte Carlo Simulations for BCC Alloys Under Irradiation: Phase Stability and Microstructural Evolutions p. 89 read less USED (low confidence) Q. Ma, C. L. Liu, J. B. Adams, and R. Balluffi, “Diffusion along [001] tilt boundaries in the Au/Ag system—II. atomistic modeling and interpretation,” Acta Metallurgica Et Materialia. 1993. link Times cited: 45 USED (low confidence) F. Ercolessi, O. Tomagnini, S. Iarlori, and E. Tosatti, “Molecular Dynamics Simulations of Metal Surfaces: Surface Melting and Non-Melting, and Tip-Surface Interactions.” 1993. link Times cited: 7 USED (low confidence) R. Berry, H.-P. Cheng, and J. Rose, “Cluster Simulations: Melting and Sintering.” 1993. link Times cited: 0 USED (low confidence) A. Wucher and B. Garrison, “Sputtering of silver dimers: a molecular dynamics calculation using a many-body embedded-atom potential,” Surface Science. 1992. link Times cited: 44 USED (low confidence) U. Landman, W. Luedtke, and E. Ringer, “Molecular Dynamics Simulations of Adhesive Contact Formation and Friction.” 1992. link Times cited: 32 USED (low confidence) Y. Li and G. Wahnström, “H Motion in Pd and Nb: A Molecular-Dynamics Study,” MRS Proceedings. 1992. link Times cited: 0 USED (low confidence) B. Bolding and E. Carter, “Effect of strain on thin film growth: deposition of Ni on Ag(100),” Surface Science. 1992. link Times cited: 15 USED (low confidence) D. Udler and D. Seidman, “Monte Carlo Simulations of Solute-Atom Segregation at [001] Symmetrical Twist Boundaries in the Ni-Pi System,” MRS Proceedings. 1992. link Times cited: 0 USED (low confidence) C. Counterman, I. Majid, P. Bristowe, and R. Balluffi, “On The Study Of Grain Boundary Segregation Using X-Ray Diffraction And Computer Simulation,” MRS Proceedings. 1991. link Times cited: 4 USED (low confidence) P. C. Clapp, Y. Shao, and J. Rifkin, “Coherent Nucleation and Growth of Marte1Nite in B2 Nial Oberved in Computer Simulations,” MRS Proceedings. 1991. link Times cited: 7 USED (low confidence) A. F. Wright, M. Daw, and C. Y. Fong, “Energetics and Structures of Pt, Pd, and Ni Adatom Clusters on the Pt (001) Surface,” MRS Proceedings. 1990. link Times cited: 0 USED (low confidence) B. Voigtländer, S. Lehwald, and H. Ibach, “Phonon dispersion on Au(110)-EELS results compared to glue-model and first principles calculations,” Vacuum. 1990. link Times cited: 0 USED (low confidence) Y. Sun, J. Rice, and L. Truskinovsky, “Dislocation Nucleation Versus Cleavage in Ni 3 AI and Ni,” MRS Proceedings. 1990. link Times cited: 10 USED (low confidence) J. S. Nelson, M. Daw, and E. C. Sowa, “Embedded atom calculations of the Cu (001), (111), and (110) surface phonon spectra,” Superlattices and Microstructures. 1990. link Times cited: 4 USED (low confidence) M. Mills and M. Daw, “The Study of Defects in Metals Using High Resolution Transmission Electron Microscopy and Atomistic Calculations,” MRS Proceedings. 1990. link Times cited: 17 USED (low confidence) H. Hsieh and R. Averback, “Molecular Dynamics Simulations of the Impact of Energetic Cu Clusters on Cu and Ni Substrates,” MRS Proceedings. 1990. link Times cited: 0 USED (low confidence) W. Hoover, C. G. Hoover, I. Stowers, A. J. D. Groot, and B. Moran, “Simulation of Mechanical Deformation via Nonequilibrium Molecular Dynamics.” 1990. link Times cited: 1 USED (low confidence) B. Legrand and M. Guillopé, “Stability of the (110) Face in Noble Metals Analyzed within a Tight-Binding Scheme.” 1989. link Times cited: 2 USED (low confidence) D. Price and B. Cooper, “Full Potential, Total Energy Lmto Calculation of Interface Structure in Ti-C and W-C Superlattices,” MRS Proceedings. 1988. link Times cited: 2 Abstract: We discuss calculations of the electronic and crystallograph… read moreAbstract: We discuss calculations of the electronic and crystallographic structure at the interfaces of titanium-carbon and tungsten-carbon superlattices. Specifically, we present total energy calculations for an arrangement of atoms designed to allow direct investigation of the competition between the formation of M-C bonds and C-C bonds. We conclude that the equilibrium structure is dominated by C-C bonding and so find that the interface has a graphite-like atomic arrangement rather than a carbide-like arrangement. These total energy calculations have been performed using a recently developed self-consistent linear combination of muffin-tin orbitals electronic structure method. This is a full-potential, all-electron, variation on standard LMTO electronic structure methods and, along with careful self-consistent determination of the parameters involved, allows accurate total energy calculations of the type of low symmetry systems involved in this study. read less NOT USED (low confidence) B. Liu, Y. Wang, and H. Yang, “Material Removal Mechanism and Evolution of Subsurface Defects during Nanocutting of Monocrystalline Cu,” Nanomaterials and Nanotechnology. 2023. link Times cited: 0 Abstract: Multigroup large-scalenanocutting models of monocrystalline … read moreAbstract: Multigroup large-scalenanocutting models of monocrystalline Cu were established by molecular dynamics simulations to investigate the influence of cutting parameters on the material removal mechanism. The formation and distribution of subsurface defect structures were revealed, and the evolution behavior of the complete prismatic dislocation loop was analyzed in depth. It was demonstrated that the chips and machined surface of monocrystalline Cu were mainly formed under the coupling effect of shearing and extrusion forces. A diamond tool with a larger edge radius or a negative rake angle could produce a noticeable suppression on the chip formation. The corresponding relationship between the location of defect atoms and the distribution of von Mises stress was studied, which indicated that the shear stress would become larger at the subgrain boundaries, dislocation lines, and the amorphous atoms than that in their nearby regions. The complete prismatic dislocation loop was formed by cross-slip between two sets of stacking faults; meanwhile, the generated multiple Lomer–Cottrell locks hindered its movement and promoted the work-hardening phenomenon. These research results are of great theoretical value to enrich the nanocutting mechanism and technology of plastic materials. read less NOT USED (low confidence) M. E. Ayoubi, A. Khmich, A. Samiri, and A. Hasnaoui, “Investigating medium range order in Mg-Al binary metallic glasses: Molecular dynamics approach,” Journal of Non-Crystalline Solids. 2023. link Times cited: 0 NOT USED (low confidence) Z. Lv, Y. Mao, Q. Zhang, Y. Liu, and R. Li, “Molecular dynamics analysis on the indentation hardness of nano-twinned nickel,” Materials Today Communications. 2023. link Times cited: 0 NOT USED (low confidence) Z. Wang, Y. Li, B. Xu, and H. Yang, “Calculation of formation enthalpies for Alfcc-Xbcc (X=Cr, Fe, Mo, Ta, V and W) binary alloys with MAEAM,” Physica B: Condensed Matter. 2023. link Times cited: 0 NOT USED (low confidence) Q.-yang Zheng, Z.-yu Zhou, L. Yu, J. Chen, S.-bin Ye, and P. Zhong-yu, “Molecular dynamic simulation of the influence of vibration effects on scratching processes in Varied crystal orientations,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 0 Abstract: The research delves into the uncharted terrain of crystal or… read moreAbstract: The research delves into the uncharted terrain of crystal orientation’s effect on high-frequency vibration-assisted processing of single-crystal copper, employing molecular dynamics to devise non-vibration, one-dimensional (1D), and two-dimensional (2D) vibration-assisted scratching models. The innovative discovery is the ‘peak-shaving’ effect, invoked by high-frequency vibration, which significantly mitigates surface irregularities on single-crystal copper, enhancing surface quality and material plasticity, thereby facilitating machinability. A key revelation is the superior efficacy of 2D vibration in material fortification relative to 1D vibration. Another novel finding is the amplified plasticity of single-crystal copper with a (111) crystal orientation under vibration-assisted excitation, linked to the varying directions of dislocation slip contingent upon crystal orientations. The pioneering observation that the induction of vibration during scratching dynamically propels dislocation defect structures, leading to the generation of a significant volume of vacant and interstitial atomic sites, underscores the pronounced influence of 2D vibration. This research contributes invaluable microscopic perspectives into the operative mechanism of crystal orientation’s impact on high-frequency vibration-assisted processing. read less NOT USED (low confidence) В. И. Мажукин et al., “Модификация кинетической модели Вильсона-Френкеля и атомистическое моделирование скорости плавления/кристаллизации металлов,” Математическое моделирование. 2023. link Times cited: 0 Abstract: В рамках кинетико-атомистического подхода предложен новый по… read moreAbstract: В рамках кинетико-атомистического подхода предложен новый подход к построению температурной зависимости стационарной скорости распространения межфазной границы «твeрдое тело-жидкость» в металлах (алюминий, медь и железо) с различной кристаллографической ориентацией. Рассматриваемый температурный диапазон включает область предельно допустимых значений перегрева/переохлаждения для каждого из металлов. Выполнена существенная модификация известной кинетической модели с диффузионным ограничением Вильсона-Френкеля, которая использовалась для построения функции отклика. Проведено атомистическое моделирование процессов плавления/кристаллизации алюминия, меди и железа в рассматриваемом температурном диапазоне с использованием трех потенциалов взаимодействия семейства потенциалов «погруженного атома». Из сопоставления результатов моделирования с данными модифицированной кинетической модели построена функция отклика скорости интерфейса в области предельно допустимых значений перегрева/переохлаждения в металлах с использованием критерия наименьших квадратов. Использование в расчeтах модифицированной кинетической модели Вильсона-Френкеля существенно увеличивает точность функции отклика в рассматриваемом температурном диапазоне. Полученная температурная зависимость скорости движения межфазной границы является диффузионно-ограниченной и описывается одним и тем же уравнением для каждого металла в рассматриваемом температурном диапазоне. read less NOT USED (low confidence) Y. Zhang, Y. Sun, and S. Wu, “Nanoscale friction analysis using asperity cross-section and longitudinal section area,” Materials Today Communications. 2023. link Times cited: 0 NOT USED (low confidence) A. Khoei, M. R. Seddighian, and A. R. Sameti, “Machine learning-based multiscale framework for mechanical behavior of nano-crystalline structures,” International Journal of Mechanical Sciences. 2023. link Times cited: 0 NOT USED (low confidence) R. Nilsson, S. Choupanian, C. Ronning, K. Nordlund, and F. Granberg, “Investigation of surface orientation dependent sputtering of Ag,” Journal of Physics: Condensed Matter. 2023. link Times cited: 0 Abstract: Sputtering of metal surfaces can be both a beneficial phenom… read moreAbstract: Sputtering of metal surfaces can be both a beneficial phenomenon, for instance in the coating industry, or an undesired side-effect, for instant materials subjected to irradiation. While the average sputtering yields are well known in common metals, recent studies have shown that the yields can depend on the crystallographic orientation of the surface much stronger than commonly appreciated. In this study, we investigate by computational means, molecular dynamics, the sputtering of single crystalline Ag surfaces under various incoming energies. The results at low and high energy are compared to experimental results for single crystalline Ag nanocubes of different orientations. We observe strong differences between the sputtering yields of different surface directions and ion energies. We analyze the results in terms of the atom cluster size of the sputtered materials, and show that the cluster size distribution is a key factor to understand the correspondence between simulations and experiments. At low energies mainly single atoms are sputtered, whereas at higher energies the sputtered material is mainly in atom clusters. read less NOT USED (low confidence) D. G. Kizzire et al., “Modified embedded atom method interatomic potential for FCC γ-cerium,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) T. Fedyaeva, S. Mathesan, A. Bisht, Z. Liang, D. Mordehai, and E. Rabkin, “The effects of composition and microstructure on compressive strength of Ag-Au nanoparticles,” Acta Materialia. 2023. link Times cited: 0 NOT USED (low confidence) J. Song et al., “Achieving atomically flat copper surface: formation of mono-atomic steps and associated strain energy mechanisms,” Acta Materialia. 2023. link Times cited: 0 NOT USED (low confidence) J. Liu, W. Lv, Y. Mou, C. Chen, and Y. Kang, “Coalescence Behavior of Cu Nanoparticles during Sintering: Based on Atomic Scale to Macro Scale,” Journal of Materials Research and Technology. 2023. link Times cited: 1 NOT USED (low confidence) S. Wang, W. Zhao, L. Zhou, and X. Du, “Quantum corrections to molecular dynamics simulations of specific heat capacities of thin ices: Role of adsorption and quasi-liquid layers at interfaces,” Journal of Molecular Liquids. 2023. link Times cited: 0 NOT USED (low confidence) M. Bakhtiari, S. N. Tavoosi, H. Shaygani, M. Tohidloo, S. Seifi, and A. Shamloo, “Effects of different wheels on the mobility of thermally driven fullerenes-based nanotrucks,” Sensors and Actuators A: Physical. 2023. link Times cited: 0 NOT USED (low confidence) J. Zhou et al., “The effect of binding energy on optimizing the interfacial thermal transport in metal-MoS2-dielectric nanostructures,” Materials Today Physics. 2023. link Times cited: 0 NOT USED (low confidence) J. Pang et al., “Defects induced the bilayer graphene-copper hybrid and its effect on mechanical properties of graphene reinforced copper matrix composites,” Applied Surface Science. 2023. link Times cited: 0 NOT USED (low confidence) W. Chen et al., “Effect of pores on microscopic wear properties and deformation behavior of Ni-Cr alloy coating,” Journal of Molecular Modeling. 2023. link Times cited: 0 NOT USED (low confidence) Y. F. Woguem, P. Godard, J. Durinck, and S. Brochard, “Elastic energy and interactions between twin boundaries in nanotwinned gold,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) M. H. Saffarini, T. Sewell, Y.-C. Su, and Z. Chen, “Atomistic study of the impact response of bicontinuous nanoporous gold as a protection medium: Effect of porous-nonporous interface on failure evolution,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) Y.-G. Lee, D. Y. Shin, C. W. Yoon, and D.-H. Lim, “BN-bicyclohexyl material for enhanced reversible dehydrogenation reaction for hydrogen storage: Density functional theory approach,” Applied Surface Science. 2023. link Times cited: 0 NOT USED (low confidence) M.-C. Dragassi, S. Haj-Khlifa, N. Menguy, M. Redolfi, and S. Ammar, “Can be metal layered hydroxide an alternative for hydride production by cold H2/Ar plasma treatment?,” International Journal of Hydrogen Energy. 2023. link Times cited: 1 NOT USED (low confidence) R. Fahdiran, A. Susila, I. Sugihartono, and E. Handoko, “Structural and Small-Angle Scattering Analysis on Melting of Gold Nanoparticle,” Journal of Physics: Conference Series. 2023. link Times cited: 0 Abstract: Molecular Dynamics (MD) simulation was used for time evoluti… read moreAbstract: Molecular Dynamics (MD) simulation was used for time evolution of melting dynamics on gold nanoparticle with thickness 8 nm. The systems are heated up from room temperature up to three times melting point in 10 ps to ensure that the system is melted and expanding. After t = 7 ps, the system is collapsed, hence pressure oscillation is vanished. Common Neighbor Analysis (CNA) along with wide angle scattering confirmed the melting state at the end of simulation. While small-angle scattering indicated the expansion of the system, it cannot precisely calculate system radius due to its small size. read less NOT USED (low confidence) H. Sun and L. Béland, “Calculation of the recombination radii between point defects and defect clusters in nickel via kinetic Activation Relaxation Technique,” Journal of Nuclear Materials. 2023. link Times cited: 0 NOT USED (low confidence) M. R. E. Tanjil et al., “Nanoscale goldbeating: Solid-state transformation of 0D and 1D gold nanoparticles to anisotropic 2D morphologies,” PNAS Nexus. 2023. link Times cited: 0 Abstract: Goldbeating is the ancient craft of thinning bulk gold (Au) … read moreAbstract: Goldbeating is the ancient craft of thinning bulk gold (Au) into gossamer leaves. Pioneered by ancient Egyptian craftsmen, modern mechanized iterations of this technique can fabricate sheets as thin as ∼100 nm. We take inspiration from this millennia-old craft and adapt it to the nanoscale regime, using colloidally synthesized 0D/1D Au nanoparticles (AuNPs) as highly ductile and malleable nanoscopic Au ingots and subjecting them to solid-state, uniaxial compression. The applied stress induces anisotropic morphological transformation of AuNPs into 2D leaf form and elucidates insights into metal nanocrystal deformation at the extreme length scales. The induced 2D morphology is found to be dependent on the precursor 0D/1D NP morphology, size (0D nanosphere diameter and 1D nanorod diameter and length), and their on-substrate arrangement (e.g., interparticle separation and packing order) prior to compression. Overall, this versatile and generalizable solid-state compression technique enables new pathways to synthesize and investigate the anisotropic morphological transformation of arbitrary NPs and their resultant emergent phenomena. read less NOT USED (low confidence) A. Sarmah, M. Jain, S. Asqardoust, and P. Mohammadpour, “Multiscale modelling of particle-induced damage in AA7075 aluminum sheet at large plastic strains,” International Journal of Plasticity. 2023. link Times cited: 0 NOT USED (low confidence) U. Nwankwo, Y.-D. Wang, C. Lam, and N. Onofrio, “Charge equilibration model with shielded long-range Coulomb for reactive molecular dynamics simulations.,” The Journal of chemical physics. 2023. link Times cited: 1 Abstract: Atomic description of electrochemical systems requires react… read moreAbstract: Atomic description of electrochemical systems requires reactive interaction potential to explicitly describe the chemistry between atoms and molecules and the evolving charge distribution and polarization effects. Calculating Coulomb electrostatic interactions and polarization effects requires a better estimate of the partial charge distribution in molecular systems. However, models such as reactive force fields and charge equilibration (QEq) include Coulomb interactions up to a short-distance cutoff for better computational speeds. Ignoring long-distance electrostatic interaction affects the ability to describe electrochemistry in large systems. We studied the long-range Coulomb effects among charged particles and extended the QEq method to include long-range effects. By this extension, we anticipate a proper account of Coulomb interactions in reactive molecular dynamics simulations. We validate the approach by computing charges on a series of metal-organic frameworks and some simple systems. Results are compared to regular QEq and quantum mechanics calculations. The study shows slightly overestimated charge values in the regular QEq approach. Moreover, our method was combined with Ewald summation to compute forces and evaluate the long-range effects of simple capacitor configurations. There were noticeable differences between the calculated charges with/without long-range Coulomb interactions. The difference, which may have originated from the long-range influence on the capacitor ions, makes the Ewald method a better descriptor of Coulomb electrostatics for charged electrodes. The approach explored in this study enabled the atomic description of electrochemical systems with realistic electrolyte thickness while accounting for the electrostatic effects of charged electrodes throughout the dielectric layer in devices like batteries and emerging solid-state memory. read less NOT USED (low confidence) L. Guo et al., “Structural transformations in single-crystalline AgPd nanoalloys from multiscale deep potential molecular dynamics.,” The Journal of chemical physics. 2023. link Times cited: 1 Abstract: AgPd nanoalloys often undergo structural evolution during ca… read moreAbstract: AgPd nanoalloys often undergo structural evolution during catalytic reactions; the mechanism underlying such restructuring remains largely unknown due to the use of oversimplified interatomic potentials in simulations. Herein, a deep-learning potential is developed for AgPd nanoalloys based on a multiscale dataset spanning from nanoclusters to bulk configurations, exhibits precise predictions of mechanical properties and formation energies with near-density functional theory accuracy, calculates the surface energies closer to experimental values compared to those obtained by Gupta potentials, and is applied to investigate the shape reconstruction of single-crystalline AgPd nanoalloys from cuboctahedron (Oh) to icosahedron (Ih) geometries. The Oh to Ih shape restructuring is thermodynamically favorable and occurs at 11 and 92 ps for Pd55@Ag254 and Ag147@Pd162 nanoalloys, respectively. During the shape reconstruction of Pd@Ag nanoalloys, concurrent surface restructuring of the (100) facet and internal multi-twinned phase change are observed with collaborative displacive characters. The presence of vacancies can influence the final product and reconstructing rate of Pd@Ag core-shell nanoalloys. The Ag outward diffusion on Ag@Pd nanoalloys is more pronounced in Ih geometry compared to Oh geometry and can be further accelerated by the Oh to Ih deformation. The deformation of single-crystalline Pd@Ag nanoalloys is characterized by a displacive transformation involving the collaborative displacement of a large number of atoms, distinguishing it from the diffusion-coupled transformation of Ag@Pd nanoalloys. read less NOT USED (low confidence) Y. Fang et al., “Structural prediction of Fe-Mg-O compounds at super-Earth’s pressures,” Physical Review Materials. 2023. link Times cited: 0 Abstract: Terrestrial exoplanets are of great interest for being simul… read moreAbstract: Terrestrial exoplanets are of great interest for being simultaneously similar to and different from Earth. Their compositions are likely comparable to those of solar-terrestrial objects, but their internal pressures and temperatures can vary significantly with their masses/sizes. The most abundant non-volatile elements are O, Mg, Si, Fe, Al, and Ca, and there has been much recent progress in understanding the nature of magnesium silicates up to and beyond ~3 TPa. However, a critical element, Fe, has yet to be systematically included in materials discovery studies of potential terrestrial planet-forming phases at ultra-high pressures. Here, using the adaptive genetic algorithm (AGA) crystal structure prediction method, we predict several unreported stable crystalline phases in the binary Fe-Mg and ternary Fe-Mg-O systems up to pressures of 3 TPa. The analysis of the local packing motifs of the low-enthalpy Fe-Mg-O phases reveals that the Fe-Mg-O system favors a BCC motif under ultra-high pressures regardless of chemical composition. Besides, oxygen enrichment is conducive to lowering the enthalpies of the Fe-Mg-O phases. Our results extend the current knowledge of structural information of the Fe-Mg-O system to exoplanet pressures. read less NOT USED (low confidence) Y. Zhang et al., “Amorphous Carbon Interlayer Modulated Interfacial Thermal Conductance between Cu and Diamond,” Applied Surface Science. 2023. link Times cited: 0 NOT USED (low confidence) M. Kang, H. Lee, S. Hong, and J. Choi, “Nanoscale contact state design for nodal energy transfer at crossed Ag nanowires,” Extreme Mechanics Letters. 2023. link Times cited: 0 NOT USED (low confidence) A. Abu-Odeh, B. Uberuaga, and M. Asta, “Barrier-free predictions of short-range ordering/clustering kinetics in binary FCC solid solutions,” Acta Materialia. 2023. link Times cited: 1 NOT USED (low confidence) K. C. Lai, S. Matera, C. Scheurer, and K. Reuter, “A fuzzy classification framework to identify equivalent atoms in complex materials and molecules.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: The nature of an atom in a bonded structure-such as in molec… read moreAbstract: The nature of an atom in a bonded structure-such as in molecules, in nanoparticles, or in solids, at surfaces or interfaces-depends on its local atomic environment. In atomic-scale modeling and simulation, identifying groups of atoms with equivalent environments is a frequent task, to gain an understanding of the material function, to interpret experimental results, or to simply restrict demanding first-principles calculations. However, while routine, this task can often be challenging for complex molecules or non-ideal materials with breaks in symmetries or long-range order. To automatize this task, we here present a general machine-learning framework to identify groups of (nearly) equivalent atoms. The initial classification rests on the representation of the local atomic environment through a high-dimensional smooth overlap of atomic positions (SOAP) vector. Recognizing that not least thermal vibrations may lead to deviations from ideal positions, we then achieve a fuzzy classification by mean-shift clustering within a low-dimensional embedded representation of the SOAP points as obtained through multidimensional scaling. The performance of this classification framework is demonstrated for simple aromatic molecules and crystalline Pd surface examples. read less NOT USED (low confidence) M. Liao, X. Ren, Z. Liu, W. Hong, and F.-F. Xie, “Study on the Coalescence-Induced Jumping of Droplets with Different Radii on Superhydrophobic Surface,” Processes. 2023. link Times cited: 0 Abstract: The phenomenon of droplet coalescence and jumping has receiv… read moreAbstract: The phenomenon of droplet coalescence and jumping has received increasing attention due to its potential applications in the fields of condensation heat transfer and surface self-cleaning. Basic research on the process and mechanism of coalescence-induced droplet jumping has been carried out, and some universal laws have been established. However, it is found that the focus of these studies is based on two identical droplets, and the coalescence-induced jumping with different radii is rarely investigated, which is commonly encountered in nature. Therefore, it is essential to proceed with the research of coalescence and jumping of droplets with unequal radii. In this paper, molecular dynamics (MD) simulations are performed to reveal the effects of radius ratio and radius of small droplets on jumping velocity. The results show that as the increasing of radius ratio with an unchanged small droplet radius of 8.1 nm, the jumping velocity increases then decreases, which indicates there is an optimal radius ratio to maximize the jumping velocity. Additionally, it is found that if the small droplet radius is changed, the critical radius ratio for characterizing whether the coalesced droplet jumping increases with increasing the small droplet radius. Furthermore, according to energy conservation, the conversion efficiency of energy is discussed. The results show that when the radius ratio is greater than 1.3 with three different small droplet radii, the energy conversion efficiency rapidly decreases to below 1.0%; and the critical radius ratios are consistent with the result obtained from the velocity analysis. This work broadens the understanding of the more general phenomenon of coalescence-induced droplet jumping and can better guide industrial applications. read less NOT USED (low confidence) H. Lan and X. Wei, “Simplicial Message Passing for Chemical Property Prediction,” ArXiv. 2023. link Times cited: 0 Abstract: Recently, message-passing Neural networks (MPNN) provide a p… read moreAbstract: Recently, message-passing Neural networks (MPNN) provide a promising tool for dealing with molecular graphs and have achieved remarkable success in facilitating the discovery and materials design with desired properties. However, the classical MPNN methods also suffer from a limitation in capturing the strong topological information hidden in molecular structures, such as nonisomorphic graphs. To address this problem, this work proposes a Simplicial Message Passing (SMP) framework to better capture the topological information from molecules, which can break through the limitation within the vanilla message-passing paradigm. In SMP, a generalized message-passing framework is established for aggregating the information from arbitrary-order simplicial complex, and a hierarchical structure is elaborated to allow information exchange between different order simplices. We apply the SMP framework within deep learning architectures for quantum-chemical properties prediction and achieve state-of-the-art results. The results show that compared to traditional MPNN, involving higher-order simplex can better capture the complex structure of molecules and substantially enhance the performance of tasks. The SMP-based model can provide a generalized framework for GNNs and aid in the discovery and design of materials with tailored properties for various applications. read less NOT USED (low confidence) K. Chen, Y. Song, P. Ding, Z. Ke, and Z. Zhao, “The MD simulation of friction heat dissipation and generation in high-speed dry sliding system,” Energy Reports. 2023. link Times cited: 0 NOT USED (low confidence) Y. Peng, Z. Tian, L. Liu, and Q. Zheng, “Autonomous identification of Lindemann atoms based on deep learning,” Materials Today Communications. 2023. link Times cited: 0 NOT USED (low confidence) H.-N. Zhang, Y. Fan, and H. S. Shen, “Prediction of Temperature-Dependent Mechanical Properties for SWCNT/Cu Nanocomposite Metamaterials: A Molecular Dynamics Study,” Nanomaterials. 2023. link Times cited: 0 Abstract: Single-walled carbon nanotube (SWCNT) is a promising candida… read moreAbstract: Single-walled carbon nanotube (SWCNT) is a promising candidate for strengthening nanocomposite. As the matrix of nanocomposite, a single crystal of copper is designed to be in-plane auxetic along the crystal orientation [1 1 0]. In that way, the nanocomposite could also be auxetic when enhanced by (7, 2) a single-walled carbon nanotube with relatively small in-plane Poisson’s ratio. A series of molecular dynamics (MD) models of the nanocomposite metamaterial are then established to study mechanical behaviors of the nanocomposite. In the modelling, the gap between copper and SWCNT is determined following the principle of crystal stability. The enhanced effect for different content and temperature in different directions is discussed in detail. This study provides a complete set of mechanical parameters of nanocomposite including thermal expansion coefficients (TECs) from 300 K to 800 K for five weight fractions, which is essential for a wide range of applications of auxetic nanocomposites in the future. read less NOT USED (low confidence) X. Du et al., “Machine-learning-accelerated simulations to enable automatic surface reconstruction,” Nature Computational Science. 2023. link Times cited: 2 NOT USED (low confidence) C. Wu and G.-W. Hong, “Atomistic simulation of the effect of porosity on shock response of nanoporous gold,” Journal of Molecular Modeling. 2023. link Times cited: 0 NOT USED (low confidence) J. Song et al., “Reorientation Mechanisms of Graphene Coated Copper 001 Surfaces,” Metals. 2023. link Times cited: 3 Abstract: Engineering the surface orientation of face-centered cubic (… read moreAbstract: Engineering the surface orientation of face-centered cubic (fcc) metals to the close-packed {111} plane can significantly enhance their oxidation resistance. However, owing to the synergetic effect of surface energy density (γ˙) and strain energy density (ω), such close-packed surface orientation can currently only be achieved by atomic-level thin film epitaxy or monocrystallization of polycrystalline metals. In this study, we characterized the microstructures of pure copper (Cu) foil and two types of graphene-coated Cu (Gr/Cu) foils and observed a 12~14 nm thick reconstructed surface layer with the {111} orientation in the high-temperature deposited Gr/{001} Cu surface. Combining the statistical results with thermodynamic analysis, we proposed a surface melting-solidification mechanism for the reconstruction of the Cu surface from {001} orientation to {111} orientation. This process is dominated by Gr/Cu interfacial energy and is particularly promoted by high-temperature surface melting. We also validated such a mechanism by examining Cu surfaces coated by h-BN (hexagonal boron nitride) and amorphous carbon. Our findings suggest a possible strategy to enhance the surface properties of fcc metals via engineering surface crystallography. read less NOT USED (low confidence) W. Y. Wu et al., “Revealing the adhesion, stability, and electronic structure of SiC/M (M=Au, Pt) interface: A first-principles study,” Vacuum. 2023. link Times cited: 4 NOT USED (low confidence) A. Khoei, H. Mofatteh, and A. R. Sameti, “A multiscale framework for atomistic–continuum transition in nano-powder compaction process using a cap plasticity model,” International Journal of Mechanical Sciences. 2023. link Times cited: 2 NOT USED (low confidence) S. Rezaee, H. Araghi, H. Noshad, and Z. Zabihi, “Physical characteristics of nickel thin-films and nickel thin-film foams as Li-air batteries anode and cathode current collectors,” Journal of Molecular Liquids. 2023. link Times cited: 0 NOT USED (low confidence) M. Buehler, “A computational building block approach towards multiscale architected materials analysis and design with application to hierarchical metal metamaterials,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 3 Abstract: In this study we report a computational approach towards mul… read moreAbstract: In this study we report a computational approach towards multiscale architected materials analysis and design. A particular challenge in modeling and simulation of materials, and especially the development of hierarchical design approaches, has been to identify ways by which complex multi-level material structures can be effectively modeled. One way to achieve this is to use coarse-graining approaches, where physical relationships can be effectively described with reduced dimensionality. In this paper we report an integrated deep neural network architecture that first learns coarse-grained representations of complex hierarchical microstructure data via a discrete variational autoencoder and then utilizes an attention-based diffusion model solve both forward and inverse problems, including a capacity to solve degenerate design problems. As an application, we demonstrate the method in the analysis and design of hierarchical highly porous metamaterials within the context of nonlinear stress–strain responses to compressive deformation. We validate the mechanical behavior and mechanisms of deformation using embedded-atom molecular dynamics simulations carried out for copper and nickel, showing good agreement with the design objectives. read less NOT USED (low confidence) S. Deng, Y. Huang, C. Mao, and J. Wang, “Size-dependent interfacial thermal transport in supported platinum nanocatalysts,” Chemical Engineering Science. 2023. link Times cited: 0 NOT USED (low confidence) S. Zhou et al., “Optimal grain size distribution in gradient nano-grained nickel,” Vacuum. 2023. link Times cited: 6 NOT USED (low confidence) H. Mes-adi, R. Herbazi, M. Lablali, K. Saadouni, and M. Mazroui, “NiAl (0 0 1) terminated surface effect on the growth of the Al thin film,” Computational Materials Science. 2023. link Times cited: 3 NOT USED (low confidence) A. Abdelrazik et al., “Potential of Molecular Dynamics in the simulation of nanofluids properties and stability,” Journal of Molecular Liquids. 2023. link Times cited: 1 NOT USED (low confidence) X. Feng, Z. Zhu, Z. Wu, M. Zheng, W. Chen, and X. Wei, “Atomic-scale study of the repeated friction processes of γ/γ’ phase nickel-based single crystal alloys,” Tribology International. 2023. link Times cited: 0 NOT USED (low confidence) T. Hu, J. Z. Yang, and C. Yuan, “DPK: Deep Neural Network Approximation of the First Piola-Kirchhoff Stress,” Advances in Applied Mathematics and Mechanics. 2023. link Times cited: 0 Abstract: . This paper presents a specific network architecture for app… read moreAbstract: . This paper presents a specific network architecture for approximation of the first Piola-Kirchhoff stress. The neural network enables us to construct the constitutive relation based on both macroscopic observations and atomistic simulation data. In contrast to traditional deep learning models, this architecture is intrinsic symmetric, guarantees the frame-indifference and material-symmetry of stress. Specifically, we build the approximation network inspired by the Cauchy-Born rule and virial stress formula. Several numerical results and theory analyses are presented to illustrate the learnability and effectiveness of our network read less NOT USED (low confidence) Z. Zhu et al., “Atomic-scale study of the nano-cutting deformation mechanism of nickel-based single crystal superalloy containing Cr, Co, and γ/γ´,” Applied Physics A. 2023. link Times cited: 0 NOT USED (low confidence) X. Qin, Y.-S. Liang, J. Gu, and G. Peng, “The Effect of Interatomic Potentials on the Nature of Nanohole Propagation in Single-Crystal Nickel: A Molecular Dynamics Simulation Study,” Crystals. 2023. link Times cited: 1 Abstract: Based on a molecular dynamics (MD) simulation, we investigat… read moreAbstract: Based on a molecular dynamics (MD) simulation, we investigated the nanohole propagation behaviors of single-crystal nickel (Ni) under different styles of Ni–Ni interatomic potentials. The results show that the MEAM (the modified embedded atom method potential) potential is best suited to describe the brittle propagation behavior of nanoholes in single-crystal Ni. The EAM/FS (embedded atom method potential developed by Finnis and Sinclair) potential, meanwhile, is effective at characterizing the plastic growth behavior of nanoholes in single-crystal Ni. Furthermore, the results show the difference between the different styles of interatomic potentials in characterizing nanohole propagation in single-crystal Ni and provide a theoretical basis for the selection of interatomic potentials in the MD simulation of Ni crystals. read less NOT USED (low confidence) Y. Yildiz, “Morphological evolution of irregularly shaped Au nanoparticles during the sintering process and their mechanical performance,” Computational Particle Mechanics. 2023. link Times cited: 0 NOT USED (low confidence) “Atomistic-Continuum Constitutive Modeling Connection for Gold Foams under Compression at High Strain Rates: The Dislocation Density Effect,” Metals. 2023. link Times cited: 2 Abstract: Constitutive description of the plastic flow in metallic foa… read moreAbstract: Constitutive description of the plastic flow in metallic foams has been rarely explored in the literature. Even though the material is of great interest to researchers, its plasticity remains a topic that has a much room for exploration. With the help of the rich literature that explored the material deformation mechanism, it is possible to introduce a connection between the results of the atomistic simulations and the well-established continuum constitutive models that were developed for various loading scenarios. In this work, we perform large-scale atomistic simulations of metallic gold foams of two different sizes at a wide range of strain rates (107−109 s−1) under uniaxial compression. By utilizing the results of those simulations, as well as the results we reported in our previous works, a physical atomistic-continuum dislocations-based constitutive modeling connection is proposed to capture the compressive plastic flow in gold foams for a wide range of sizes, strain rates, temperatures, and porosities. The results reported in this work present curated datasets that can be of extreme usefulness for the data-driven AI design of metallic foams with tunable nanoscale properties. Eventually, we aim to produce an optimal physical description to improve integrated physics-based and AI-enabled design, manufacture, and validation of hierarchical architected metallic foams that deliver tailored mechanical responses and precision failure patterns at different scales. read less NOT USED (low confidence) J. Laakso et al., “Updates to the DScribe library: New descriptors and derivatives.,” The Journal of chemical physics. 2023. link Times cited: 2 Abstract: We present an update of the DScribe package, a Python librar… read moreAbstract: We present an update of the DScribe package, a Python library for atomistic descriptors. The update extends DScribe's descriptor selection with the Valle-Oganov materials fingerprint and provides descriptor derivatives to enable more advanced machine learning tasks, such as force prediction and structure optimization. For all descriptors, numeric derivatives are now available in DScribe. For the many-body tensor representation (MBTR) and the Smooth Overlap of Atomic Positions (SOAP), we have also implemented analytic derivatives. We demonstrate the effectiveness of the descriptor derivatives for machine learning models of Cu clusters and perovskite alloys. read less NOT USED (low confidence) D. Li, Y. Liu, and Q. Liu, “Study on Interface Mechanical Properties of Graphene/Copper Matrix Composites,” Applied Sciences. 2023. link Times cited: 0 Abstract: Graphene/copper matrix composites have a wide range of appli… read moreAbstract: Graphene/copper matrix composites have a wide range of application prospects, but the mechanical properties of the interface have been one of the key problems restricting their wide application. In this paper, the mechanical behaviors at the interface of graphene/copper matrix composites, such as pulling up, pulling out, and cohesion, and the effects of temperature and graphene content on them were studied by the molecular dynamics method. The results show that the pull-up force and cohesiveness show two stages in the whole process. The pulling force increases rapidly and then decreases to 0 slowly. The pull-out force shows three stages: it rises rapidly at first, then fluctuates continuously, and finally drops to 0. The mechanical properties of the interface deteriorated with the increase in temperature. When the temperature increased from 0 K to 1100 K, the interface normal strength, shear strength, and cohesion strength of the interface decreased by 26.3%, 32.9%, and 24.8%, respectively. In addition, with the increase in graphene content, the normal strength of the interface increases, the shear strength decreases, and the cohesion strength almost stays the same. When the graphene content increases from 6.71 at% to 11.75 at%, the normal strength increases by 6.8%, while the shear strength decreases by 37.4%. The influence mechanism of temperature and content is explained from the aspects of the influence of atomic thermal motion and the hindering effect of graphene on the dislocation motion of the copper matrix. The relevant results have certain reference values for the engineering application and theoretical research of graphene/copper composites. read less NOT USED (low confidence) W. Siswanto et al., “The characterization of plastic behavior and mechanical properties in the gradient nanostructured copper,” Proceedings of the Institution of Mechanical Engineers, Part L: Journal of Materials: Design and Applications. 2023. link Times cited: 8 Abstract: Due to their individual plastic deformation behavior and mec… read moreAbstract: Due to their individual plastic deformation behavior and mechanistic performances, gradient nano-grained (GNG) metals have been identified as promising materials for application in advanced engineering applications. However, their plasticity mechanism has not been thoroughly investigated. Hence, in this work, molecular dynamics simulation was employed to characterize the atomistic behavior of copper GNG structures under tensile loading with different grain sizes (2–9 nm) at varied temperatures (100, 300, and 600 K). The results indicated that there existed a critical grain size value (6 nm) for the Hall–Petch and inverse Hall–Petch relationship. Besides, it was found that the high-temperature loading (600 K) negatively affected the plasticity-induced strengthening, while low-temperature loading induced outstanding plasticity-induced strengthening owing to the excessive strain hardening events. The details of plastic deformation from the perspective of grain boundary, deformation twining, stacking faults, and dislocations were analyzed and a thorough discussion was presented to elucidate the grain size and temperature effects on the plasticity mechanism. read less NOT USED (low confidence) B.-X. Zhang et al., “Explosive boiling of suspended argon films in the cassie-baxter state on nanopillar-arrayed surfaces,” International Journal of Thermal Sciences. 2023. link Times cited: 2 NOT USED (low confidence) Z. Zhang et al., “Atomistic understanding towards twin boundary on the effect of crack propagation in FeNiCrCoCu high-entropy alloy and Ni,” Materials Today Communications. 2023. link Times cited: 1 NOT USED (low confidence) M. H. Nazir, Z. Khan, M. M. Hussain, A. Rahil, and S. J. Zaidi, “A comprehensive experimental study and numerical analysis of coefficient of friction of nanocomposite coatings,” Materials Chemistry and Physics. 2023. link Times cited: 1 NOT USED (low confidence) A. Khoei, A. R. Sameti, and H. Mofatteh, “Multiscale analysis of nano-powder compaction process using the FEM–MD technique,” Powder Technology. 2023. link Times cited: 2 NOT USED (low confidence) Y. Nikravesh, M. Latypov, G. Frantziskonis, and K. Muralidharan, “Atomistic Characterization of Impact Bonding in Cold Spray Deposition of Copper,” SSRN Electronic Journal. 2023. link Times cited: 1 NOT USED (low confidence) W. Chen et al., “Study on the Nano-Friction Behavior of Nickel-Based Ag Film Composites Based on Molecular Dynamics,” Lubricants. 2023. link Times cited: 0 Abstract: The nano-friction behavior of nickel-based Ag film composite… read moreAbstract: The nano-friction behavior of nickel-based Ag film composites was evaluated using molecular dynamics simulations. The mechanical properties, the surface morphology, the migration behavior of Ag atoms and the defect evolution during repeated friction were investigated. Our results show that the poor mechanical properties of the Ag film surface at the first stage of friction are related to a large amount of abrasive chip pileup. The slip channel with low shear strength formed by secondary friction significantly reduces the friction coefficient of the Ag film surface. Meanwhile, the migration of Ag atoms at the two-phase interface relies mainly on the repeated friction of the grinding ball, and the friction coefficient of the nickel surface decreases as the number of migrating atoms increases. In addition, the extension of defects inside the Ag film and atomic displacement is hindered by the two-phase interface. The defects inside the Ag film near the friction zone gradually evolve from an intrinsic stacking fault to a horizontal stacking fault as the friction proceeds. This is attributed to the horizontal layer-by-layer motion of Ag atoms, promoting the formation of horizontal stacking faults. read less NOT USED (low confidence) H. Dorrani and A. Mohebbi, “Molecular dynamics insight into the best governing mechanism for thermophysical properties changes in nanofluids,” Journal of Thermal Analysis and Calorimetry. 2023. link Times cited: 0 NOT USED (low confidence) N. Amadou, A. R. A. Abdoulaye, T. de Rességuier, and A. Dragon, “Strain-Rate Dependence of Plasticity and Phase Transition in [001]-Oriented Single-Crystal Iron,” Crystals. 2023. link Times cited: 1 Abstract: Non-equilibrium molecular dynamics simulations have been use… read moreAbstract: Non-equilibrium molecular dynamics simulations have been used to investigate strain-rate dependence of plasticity and phase transition in [001]-oriented single-crystal iron under ramp compression. Here, plasticity is governed by deformation twinning, in which kinetics is tightly correlated with the loading rate. Over the investigated range of strain rates, a hardening-like effect is found to shift the onset of the structural bcc-to-hcp phase transformation to a high, almost constant stress during the ramp compression regime. However, when the ramp evolves into a shock wave, the bcc–hcp transition is triggered whenever the strain rate associated with the plastic deformation reaches some critical value, which depends on the loading rate, leading to a constitutive functional dependence of the transition onset stress on the plastic deformation rate, which is in overall consistence with the experimental data under laser compression. read less NOT USED (low confidence) A. D. Backer, S. Bals, and S. V. Aert, “A decade of atom-counting in STEM: From the first results toward reliable 3D atomic models from a single projection.,” Ultramicroscopy. 2023. link Times cited: 2 NOT USED (low confidence) M. Daw and M. Chandross, “Simple Parameterization of Embedded Atom Method Potentials for FCC Alloys,” Acta Materialia. 2023. link Times cited: 1 NOT USED (low confidence) M. Daw and M. Chandross, “Simple Parameterization of Embedded Atom Method Potentials for FCC Metals,” Acta Materialia. 2023. link Times cited: 1 NOT USED (low confidence) K. Wakamoto, T. Otsuka, K. Nakahara, V. Mugilgeethan, R. Matsumoto, and T. Namazu, “Degradation Mechanism of Silver Sintering Die Attach Based on Thermal and Mechanical Reliability Testing,” IEEE Transactions on Components, Packaging and Manufacturing Technology. 2023. link Times cited: 0 Abstract: This article investigates the degradation mechanism of sinte… read moreAbstract: This article investigates the degradation mechanism of sintered silver (s-Ag) layer in silicon carbide (SiC) die attach by means of thermal shocked test (TST) and nine-point bending test (NBT). TST can thermally provide out-of-plane deformation with s-Ag die layer, whereas NBT developed by the authors can mechanically provide the out-of-plane deformation. Two types of Ag paste (NP: nanopaste and NMP: nano- and micropaste) are employed, which are sintered with 300 °C for 10 min under 60-MPa pressure for SiC die attach. TSTs are conducted under thermal cyclic loading between −40 °C and 150 °C with trapezoidal waveform for 60 min per a cycle. NBTs are conducted under cyclic mechanical loading between 0 and 300 N with triangle waveform for 3 min per a cycle at 150 °C. Scanning acoustic tomography (SAT) evaluates the die-attach delamination during TST and NBT, which starts from the edge of the die. Cross-sectional scanning electron microscopy (SEM) observation demonstrates that mechanical cracking and material aging in s-Ag layer coexist after TST under the −40 °C to 150 °C conditions only. A damage parameter (DP) proposed in this study works well to fit the delamination area ratio in the cracking and cracking-aging specimens separately after 1000 cycles. Molecular dynamics (MD) simulation and classical pore growth discussion suggest that pores can grow under only tensile stress state at high temperature, which would have given rise to the difference between the two degradation mechanisms. read less NOT USED (low confidence) Y. Sun, H. Zheng, Y. Geng, G. Li, and Y. Xiao, “Molecular Dynamics Simulations of Warm Laser Shock Peening for Monocrystalline Nickel,” SSRN Electronic Journal. 2023. link Times cited: 0 NOT USED (low confidence) S. Wang et al., “Nucleation of water vapor on nanodimpled surfaces: Effects of curvature radius and surface wettability,” Applied Thermal Engineering. 2023. link Times cited: 7 NOT USED (low confidence) Y. Tian, F.-cai Ren, and F. Chen, “Twin-boundary-spacing-dependent strength in gradient nano-grained copper,” Materials Today Communications. 2022. link Times cited: 2 NOT USED (low confidence) G. Tang, F. Su, X. Liu, Z. Liang, T. Zou, and P. Chu, “Origin of superlubricity promoted by black phosphorus dotted with gold nanoparticles,” Applied Surface Science. 2022. link Times cited: 2 NOT USED (low confidence) Z. Fang, Y. Yan, Z. Li, A. Zhang, and Y. Geng, “Tool Geometry Effect on Nanowire Formation Behavior During Nanoskiving,” SSRN Electronic Journal. 2022. link Times cited: 13 NOT USED (low confidence) H.-L. Wang, L. Yang, D. Zhai, L. Sun, and W. Deng, “Global optimization of gold nanocrystals based on an iterative QM/MM method,” Chemical Physics Letters. 2022. link Times cited: 0 NOT USED (low confidence) N. Tuchinda and C. Schuh, “The Vibrational Entropy Spectra of Grain Boundary Segregation in Polycrystals,” Acta Materialia. 2022. link Times cited: 7 NOT USED (low confidence) A. A. Nazarov, D. Bachurin, and Z. Ni, “Atomistic Simulation of Ultrasonic Welding of Copper,” Metals. 2022. link Times cited: 1 Abstract: Molecular dynamics simulations of ultrasonic welding of two … read moreAbstract: Molecular dynamics simulations of ultrasonic welding of two blocks of fcc copper containing asperities under the conditions of a constant clamping pressure and sinusoidal shear displacements were performed. Two different atomistic models of blocks were simulated: Model I with no misorientation between the lattices, and Model II with a special misorientation of 78.46°. Alternating shearing results in a plastic deformation of the interface layers and is accompanied by the emission of partial dislocations. Misorientation between the joined blocks contributes significantly to an interface sliding, interface migration, and pores healing during ultrasonic processing. A significantly larger increase in temperature occurs during shearing in Model II than in Model I. The applied pressure has almost no effect on the interface temperature in both studied models. The temperature increases almost up to maximum values after the first shear cycle, and then practically does not undergo changes in the next four cycles. The temperature at the interface in Model II is significantly higher than that in Model I. The change in the porosity of the interface and its structure are analyzed. The results obtained in the present work contribute to a deeper understanding of the processes occurring at the atomic level during ultrasonic welding of metals. read less NOT USED (low confidence) A. Clément and T. Auger, “An EAM potential for α-brass copper–zinc alloys: application to plasticity and fracture,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 1 Abstract: An embedded atom method potential has been developed for cop… read moreAbstract: An embedded atom method potential has been developed for copper–zinc alloys valid from 0% to 37% zinc content (dedicated to describe the α fcc phase). It has been fit to a set of first-principles data for the fcc copper, the fcc Cu3Zn DO 23 phase and Zn on a fcc lattice. Elastic anisotropies, the lattice parameter, cohesive energy are used as input. Ponctual defects, surface energies, intrinsic stacking fault and phonon spectrum have been computed and compare well with experimental trends. This potential has been used to study dislocation dissociation and dislocation emission at a crack tip up to 30% Zn. Dislocation emission at the crack tip is correctly described compared with recent parametrization including the surface energy. It is found that with alloying, dislocation emission becomes easier following the decrease of the unstable stacking fault energy with Zn concentration, a non-trivial finding. This potential is therefore well suited to carry out basic studies of plasticity and fracture in α-brass alloys. read less NOT USED (low confidence) M. Bakhtiari, S. Seifi, M. Tohidloo, and A. Shamloo, “Investigation of the motion of fullerene-wheeled nano-machines on thermally activated curved gold substrates,” Scientific Reports. 2022. link Times cited: 3 NOT USED (low confidence) A. Hernandez and T. Mueller, “Generalizability of Functional Forms for Interatomic Potential Models Discovered by Symbolic Regression,” ArXiv. 2022. link Times cited: 0 Abstract: In recent years there has been great progress in the use of … read moreAbstract: In recent years there has been great progress in the use of machine learning algorithms to develop interatomic potential models. Machine-learned potential models are typically orders of magnitude faster than density functional theory but also orders of magnitude slower than physics-derived models such as the embedded atom method. In our previous work, we used symbolic regression to develop fast, accurate and transferrable interatomic potential models for copper with novel functional forms that resemble those of the embedded atom method. To determine the extent to which the success of these forms was specific to copper, here we explore the generalizability of these models to other face-centered cubic transition metals and analyze their out-of-sample performance on several material properties. We found that these forms work particularly well on elements that are chemically similar to copper. When compared to optimized Sutton-Chen models, which have similar complexity, the functional forms discovered using symbolic regression perform better across all elements considered except gold where they have a similar performance. They perform similarly to a moderately more complex embedded atom form on properties on which they were trained, and they are more accurate on average on other properties. We attribute this improved generalized accuracy to the relative simplicity of the models discovered using symbolic regression. The genetic programming models are found to outperform other models from the literature about 50% of the time in a variety of property predictions, with about 1/10th the model complexity on average. We discuss the implications of these results to the broader application of symbolic regression to the development of new potentials and highlight how models discovered for one element can be used to seed new searches for different elements. read less NOT USED (low confidence) L. Y. Zhao, S. Wang, and Y. Liu, “Parallel algorithm for particle-grid dual discretization,” Computational Mechanics. 2022. link Times cited: 1 NOT USED (low confidence) L. Yu, Z. Qiu-yang, Z. Zhen-yu, D. Cong, Y. Sen-bin, and P. Zhong-yu, “Molecular dynamics study on the effect of electric current on electrically-assisted scratching for crystal copper,” Physica Scripta. 2022. link Times cited: 1 Abstract: Investigation of the effect of electric current on the plast… read moreAbstract: Investigation of the effect of electric current on the plastic deformation mechanism of metals during the electrically-assisted machining process is significant in further improving surface properties. In this paper, the molecular dynamics (MD) method is adopted to simulate the electrically-assisted scratching process of crystal copper, obtaining and analyzing the surface morphology, potential energy change, von Mises stress distribution, and crystal defect structure evolution. The MD simulation results show that the electric current effectively expands the dislocation slip range, resulting in a larger plastic deformation zone. Meanwhile, the combined action of the electron wind forces and Joule heating causes more dislocations to proliferate and increases the dislocation density limit, enhancing the plastic deformation ability of the single-crystal copper. Furthermore, the electric current strengthens the dislocation-grain boundary interactions and reduces the hindering effect of the grain boundaries on dislocations, promoting more dislocations to cross the grain boundaries. This work will be helpful for guiding the optimization of surface strengthening techniques to get better surface properties of metals. read less NOT USED (low confidence) C. Mieszczyński, L. Nowicki, K. Skrobas, P. Jóźwik, and J. Jagielski, “Edge dislocations in Ni monocrystalline structure studied by McChasy 2.0 Monte Carlo code,” Journal of Physics: Conference Series. 2022. link Times cited: 3 Abstract: The main goal of the McChasy code, in general, is to reprodu… read moreAbstract: The main goal of the McChasy code, in general, is to reproduce Rutherford Backscattering Spectrometry experimental spectra recorded in channeling direction (RBS/C) by simulating He-ions travelling inside crystalline structures and to calculate the probability of the backscattering process. The 2.0 version of the code provides the possibility to simulate channeling spectra in large (ca. 108 atoms) arbitrary structures that are created based on crystallographic data or Molecular Dynamic (MD) calculations. In this work, we present the current status of the code and the results of recent investigations of extended structural defects (edge dislocations and dislocation loops) formed inside nickel (Ni) single crystals. Two ways of modelling extended defects are described: one developed using the McChasy code (Peierls-Nabarro approach) and the other one obtained by modification and thermalization of Ni structures by MD (LAMMPS code). The atomic local environment was studied qualitatively and quantitatively by the local projectile-flux density distributions around the defects. read less NOT USED (low confidence) K. Sikdar, A. Mahata, B. Roy, and D. Roy, “Thermokinetic stabilisation of nanocrystalline Cu by ternary approach,” Philosophical Magazine. 2022. link Times cited: 0 Abstract: ABSTRACT Nanocrystalline alloy design with the synergistic c… read moreAbstract: ABSTRACT Nanocrystalline alloy design with the synergistic contribution of ‘thermodynamic’ and ‘kinetic’ stabilisation mechanism leads to much superior microstructural stabilisation at elevated temperatures. Ternary Cu98.5W1Zr0.5 (at. %) alloy, synthesised by mechanical milling under cryogenic temperature followed by consolidation through hot pressing at 550°C, has been examined to access the potential of their concurrence. A meager drop in hardness (∼0.5 GPa) confirms the stability of the alloy up to 800°C. The effect of alloy addition has been studied in terms of microstructure alteration, measured by X-ray diffraction, transmission electron microscopy, and Molecular dynamics (MD) simulation. In addition, the shear punch test (SPT) has been employed to assess the mechanical property of the consolidated alloy. Results suggest that the current approach provides a framework en route to designing bulk nanostructured alloys adapting the bottom-up method. read less NOT USED (low confidence) Y. Zhu et al., “Molecular dynamic simulation of Cs corrosion in Cs oven for negative ion source applications,” AIP Advances. 2022. link Times cited: 0 Abstract: Molecular dynamic simulation is used to simulate the corrosi… read moreAbstract: Molecular dynamic simulation is used to simulate the corrosion process of Fe or Ni in liquid Cs by Large-scale Atomic/Molecular Massively Parallel Simulator. The embedded-atom method potential is used to describe the interaction of Fe–Fe, Ni–Ni, and Cs–Cs, and Morse two-body potential is used to describe the Fe–Cs and Ni–Cs atomic interaction. Temperature is considered as a critical condition in this work. Results indicate that corrosion is easy to occur in the systems. The increase in temperature can help the process of Cs corrosion. Compared to the Ni–Cs system, the Fe–Cs system has a higher atomic concentration function. The radial distribution function shows that Cs atoms are dissolved into the substrates, but the Fe and Ni substrates are still crystalline structures. Moreover, Cs in Fe or Ni is still a liquid phase. read less NOT USED (low confidence) A. Shamloo, M. Bakhtiari, M. Tohidloo, and S. Seifi, “Investigation of fullerene motion on thermally activated gold substrates with different shapes,” Scientific Reports. 2022. link Times cited: 3 NOT USED (low confidence) Y.-C. Hu and H. Tanaka, “Revealing the role of liquid preordering in crystallisation of supercooled liquids,” Nature Communications. 2022. link Times cited: 19 NOT USED (low confidence) B. Yao, Z. R. Liu, and R. F. Zhang, “EAPOTc: An integrated empirical interatomic potential optimization platform for compound solids,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) S. Doronin, N. Dokhlikova, and M. Grishin, “Descriptor of catalytic activity nanoparticles surface: Atomic and molecular hydrogen on gold,” Molecular Catalysis. 2022. link Times cited: 2 NOT USED (low confidence) M. Cioni, D. Polino, D. Rapetti, L. Pesce, M. D. Piane, and G. Pavan, “Innate dynamics and identity crisis of a metal surface unveiled by machine learning of atomic environments.,” The Journal of chemical physics. 2022. link Times cited: 5 Abstract: Metals are traditionally considered hard matter. However, it… read moreAbstract: Metals are traditionally considered hard matter. However, it is well known that their atomic lattices may become dynamic and undergo reconfigurations even well below the melting temperature. The innate atomic dynamics of metals is directly related to their bulk and surface properties. Understanding their complex structural dynamics is, thus, important for many applications but is not easy. Here, we report deep-potential molecular dynamics simulations allowing to resolve at an atomic resolution the complex dynamics of various types of copper (Cu) surfaces, used as an example, near the Hüttig (∼1/3 of melting) temperature. The development of deep neural network potential trained on density functional theory calculations provides a dynamically accurate force field that we use to simulate large atomistic models of different Cu surface types. A combination of high-dimensional structural descriptors and unsupervized machine learning allows identifying and tracking all the atomic environments (AEs) emerging in the surfaces at finite temperatures. We can directly observe how AEs that are non-native in a specific (ideal) surface, but that are, instead, typical of other surface types, continuously emerge/disappear in that surface in relevant regimes in dynamic equilibrium with the native ones. Our analyses allow estimating the lifetime of all the AEs populating these Cu surfaces and to reconstruct their dynamic interconversions networks. This reveals the elusive identity of these metal surfaces, which preserve their identity only in part and in part transform into something else under relevant conditions. This also proposes a concept of "statistical identity" for metal surfaces, which is key to understanding their behaviors and properties. read less NOT USED (low confidence) J. Zhou et al., “Graphene Layer Number-Dependent Heat Transport across Nickel/Graphene/Nickel Interfaces.,” ACS applied materials & interfaces. 2022. link Times cited: 4 Abstract: As a typical two-dimensional material, graphene (Gr) has sho… read moreAbstract: As a typical two-dimensional material, graphene (Gr) has shown great potential to be used in thermal management applications due to its ultrahigh in-plane thermal conductivity (k). However, low interface thermal conductance (ITC) between Gr and metals to a large extent limits the effective heat dissipation in Gr-based devices. Therefore, having a deep understanding on heat transport at Gr-metal interfaces is essential. Because of the semimetallic nature of Gr, electrons would possibly play a role in the heat transport across Gr-metal interfaces as heat carriers, whereas, However, how much the electron can participate in this process and how to optimize the total ITC considering both electron and phonon transportations have not yet been revealed yet. Therefore, in this work, hydrogenation-treated Gr (H-Gr) was sandwiched by nickel (Ni) nanofilms to compare with the samples containing pure Gr for investigating the interfacial electron behaviors. Moreover, both Gr and H-Gr sets of the samples were prepared with different layer numbers (N) ranging from 1 to 7, and the corresponding ITC was systematically studied based on both time-domain thermoreflectance measurements and theoretical calculations. We found that a larger ITC can be obtained when N is low, and the ITC may reach a peak value when N is 2 in certain circumstances. The present findings not only provide a comprehensive understanding on heat transport across Gr-metal interfaces byconsidering a combined effect of the interfacial interaction strength, phonon mode mismatch, and electron contributions, but also shed new lights on interface strucure optimiazations of Gr-based devices. read less NOT USED (low confidence) Y. Lei, R. Zhou, and B. Zhang, “The influence of atomic delocalization on dynamic behavior in Ce-Ni metallic melts,” Computational Materials Science. 2022. link Times cited: 0 NOT USED (low confidence) L. Wu et al., “A new method for computing the anisotropic free energy of the crystal-melt interface,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) Y. Fan and H. S. Shen, “Non-symmetric stiffness of origami-graphene metamaterial plates,” Composite Structures. 2022. link Times cited: 10 NOT USED (low confidence) Y. Zhang et al., “Atomistic modeling of surface and grain boundary dislocation nucleation in FCC metals,” Acta Materialia. 2022. link Times cited: 11 NOT USED (low confidence) Z. Fang, Y. Yan, and Y. Geng, “Uncovering the machining mechanism of polycrystalline gold nanowires by nanoskiving,” International Journal of Mechanical Sciences. 2022. link Times cited: 6 NOT USED (low confidence) S. Wang, D. Zhao, Y.-hong Niu, Z. Wang, H. Yang, and H. Zhao, “Investigations of Micro-Deformation in Monocrystalline Copper at Low Temperatures via Indentation,” Micromachines. 2022. link Times cited: 1 Abstract: Indentation experiments on differently oriented faces of mon… read moreAbstract: Indentation experiments on differently oriented faces of monocrystalline copper were conducted to investigate the micro-deformation process at temperatures ranging from room temperature to 150 K. The morphologies and textures of the residual imprints were observed using electron microscopy. Distinct slip bands were observed inside the imprints at 150 K compared to smooth surfaces at room temperature. Molecular dynamics simulations were performed to identify the deformation process beneath the indentation region. The results showed that plastic deformation was inhibited with decreasing temperature, but elastic recovery during the unloading process was enhanced, resulting in inner slip bands (ISBs) being observable in the residual imprints. The performances of these ISBs were strongly associated with the angles between the indentation direction and major slip surfaces and could be considered microscopic forms on the surfaces of aggregated geometrically necessary dislocations (GNDs). This work helped reveal the micro-deformation mechanism of indentations inside imprints. read less NOT USED (low confidence) Y. Zhao et al., “Molecular dynamics study of acoustic softening effect in ultrasonic vibration assisted tension of monocrystalline/polycrystalline coppers,” Journal of Materials Processing Technology. 2022. link Times cited: 9 NOT USED (low confidence) D. F. Rojas, M. Isiet, and M. Ponga, “Dynamic recrystallization in face-centered cubic particles during high-velocity impacts,” Mechanics of Materials. 2022. link Times cited: 6 NOT USED (low confidence) Y. Zhou, D. Wu, G. Luo, Y. Hu, and Y. Qin, “Efficient modeling of metal ablation irradiated by femtosecond laser via simplified two-temperature model coupling molecular dynamics,” Journal of Manufacturing Processes. 2022. link Times cited: 5 NOT USED (low confidence) D. Trong, V. C. Long, and Ș. Ţălu, “Molecular Dynamics Simulation of Bulk Cu Material under Various Factors,” Applied Sciences. 2022. link Times cited: 4 Abstract: In this paper, the molecular dynamics (MD) method was used t… read moreAbstract: In this paper, the molecular dynamics (MD) method was used to study the influence of factors of bulk Cu material, such as the effect of the number of atoms (N) at temperature (T), T = 300 K, temperature T, and annealing time (t) with Cu5324 on the structure properties, phase transition, and glass temperature Tg of the bulk Cu material. The obtained results showed that the glass transition temperature (Tg) of the bulk Cu material was Tg = 652 K; the length of the link for Cu-Cu had a negligible change; r = 2.475 Å; and four types of structures, FCC, HCP, BCC, Amor, always existed. With increasing the temperature the FCC, HCP, and BCC decrease, and Amorphous (Amor) increases. With an increasing number of atoms and annealing time, the FCC, HCP, and BCC increased, and Amor decreased. The simulated results showed that there was a great influence of factors on the structure found the gradient change, phase transition, and successful determination of the glass temperature point above Tg of the bulk Cu material. On the basis of these results, essential support will be provided for future studies on mechanical, optical, and electronic properties. read less NOT USED (low confidence) D. Jacobson and G. Thompson, “Revisting Lennard Jones, Morse, and N-M potentials for metals,” Computational Materials Science. 2022. link Times cited: 9 NOT USED (low confidence) J. Byggmästar, K. Nordlund, and F. Djurabekova, “Simple machine-learned interatomic potentials for complex alloys,” Physical Review Materials. 2022. link Times cited: 5 Abstract: Developing data-driven machine-learning interatomic potentia… read moreAbstract: Developing data-driven machine-learning interatomic potentials for materials containing many elements becomes increasingly challenging due to the vast configuration space that must be sampled by the training data. We study the learning rates and achievable accuracy of machine-learning interatomic potentials for many-element alloys with different combinations of descriptors for the local atomic environments. We show that for a five-element alloy system, potentials using simple low-dimensional descriptors can reach meV/atom-accuracy with modestly sized training datasets, significantly outperforming the high-dimensional SOAP descriptor in data efficiency, accuracy, and speed. In particular, we develop a computationally fast machine-learned and tabulated Gaussian approximation potential (tabGAP) for Mo–Nb–Ta–V–W alloys with a combination of two-body, three-body, and a new simple scalar many-body density descriptor based on the embedded atom method. read less NOT USED (low confidence) C. Atlan et al., “Imaging the strain evolution of a platinum nanoparticle under electrochemical control,” Nature Materials. 2022. link Times cited: 4 NOT USED (low confidence) M. Zheng et al., “Molecular dynamics study on the nanoscale repeated friction and wear mechanisms of TiC/Ni composites,” Applied Physics A. 2022. link Times cited: 7 NOT USED (low confidence) M. Dinpajooh and A. Nitzan, “Heat conduction in polymer chains: Effect of substrate on the thermal conductance.,” The Journal of chemical physics. 2022. link Times cited: 7 Abstract: In standard molecular junctions, a molecular structure is pl… read moreAbstract: In standard molecular junctions, a molecular structure is placed between and connected to metal leads. Understanding how mechanical tuning in such molecular junctions can change heat conductance has interesting applications in nanoscale energy transport. In this work, we use nonequilibrium molecular dynamics simulations to address the effect of stretching on the phononic contribution to the heat conduction of molecular junctions consisting of single long-chain alkanes and various metal leads, such as Ag, Au, Cu, Ni, and Pt. The thermal conductance of such junctions is found to be much smaller than the intrinsic thermal conductance of the polymer and significantly depends on the nature of metal leads as expressed by the metal-molecule coupling and metal vibrational density of states. This behavior is expected and reflects the mismatch of phonon spectra at the metal molecule interfaces. As a function of stretching, we find a behavior similar to what was observed earlier [M. Dinpajooh and A. Nitzan, J. Chem. Phys. 153, 164903 (2020)] for pure polymeric structures. At relatively short electrode distances, where the polyethylene chains are compressed, it is found that the thermal conductances of the molecular junctions remain almost constant as one stretches the polymer chains. At critical electrode distances, the thermal conductances start to increase, reaching the values of the fully extended molecular junctions. Similar behaviors are observed for junctions in which several long-chain alkanes are sandwiched between various metal leads. These findings indicate that this behavior under stretching is an intrinsic property of the polymer chain and not significantly associated with the interfacial structures. read less NOT USED (low confidence) M. Zheng et al., “Molecular dynamics study on the nanoscale repeated friction and wear mechanisms of TiC/Ni composites,” Applied Physics A. 2022. link Times cited: 0 NOT USED (low confidence) Y. Nikravesh, A. R. Sameti, and A. Khoei, “An atomistic–continuum multiscale analysis for heterogeneous nanomaterials and its application in nanoporous gold foams,” Applied Mathematical Modelling. 2022. link Times cited: 9 NOT USED (low confidence) H. Sun and L. Béland, “Statistical distribution of spontaneous recombination radii of Frenkel pairs in FCC and BCC metals,” Acta Materialia. 2022. link Times cited: 6 NOT USED (low confidence) X. Chen, R. Dingreville, T. Richeton, and S. Berbenni, “Invariant surface elastic properties in FCC metals and their correlation to bulk properties revealed by machine learning methods,” Journal of the Mechanics and Physics of Solids. 2022. link Times cited: 2 NOT USED (low confidence) Z. Wang, J. Zhang, and J. Lu, “Atomic-scale study of dislocation-grain boundary interactions in Cu bicrystal by Berkovich nanoindentation,” Materials Science and Engineering: A. 2022. link Times cited: 4 NOT USED (low confidence) Y. Xu, G. Wang, P. Qian, and Y. Su, “Element segregation and thermal stability of Ni–Rh nanoparticles,” Journal of Solid State Chemistry. 2022. link Times cited: 6 NOT USED (low confidence) Z. Zhao, J. Liu, A. Soh, and C. Tang, “On the snap-through time of a nanoscale elastic strip,” Acta Mechanica Sinica. 2022. link Times cited: 1 NOT USED (low confidence) B.-X. Zhang et al., “Explosive boiling of argon nanofilms in the Wenzel or Cassie state on high-temperature nanopillar-arrayed surfaces,” International Journal of Thermal Sciences. 2022. link Times cited: 8 NOT USED (low confidence) D.-Q. Doan, T. Fang, and T.-H. Chen, “Nanomachining characteristics of textured polycrystalline NiFeCo alloy using molecular dynamics,” Journal of Manufacturing Processes. 2022. link Times cited: 15 NOT USED (low confidence) C.-H. Tsai, W.-C. Huang, and C. Kao, “Development of Ag–In Alloy Pastes by Mechanical Alloying for Die Attachment of High-Power Semiconductor Devices,” Materials. 2022. link Times cited: 5 Abstract: Sintered silver paste is widely used as the die-attachment m… read moreAbstract: Sintered silver paste is widely used as the die-attachment material for power semiconductors. However, sintered silver joints encounter problems, such as severe coarsening of sintered pores and oxidation issues, in harsh high-temperature environments. These lead to the deterioration of the die-attachment joints. In this paper, a novel method of sintering silver joints is demonstrated, where silver–indium alloy paste is used to improve the reliability of sintered Ag joints. The silver–indium (Ag–In) alloy paste was fabricated through mechanical alloying using the ball-milling technique. The well-bonded sintered Ag–In alloy joints inhibited pore coarsening better than pure sintered Ag joints and significantly enhanced the mechanical properties at high operating temperatures. Lastly, an oxidation mechanism for the sintered joint was proposed, and strategies to prevent such high-temperature oxidation were discussed. read less NOT USED (low confidence) S. Dutta, S. Corni, and G. Brancolini, “Atomistic Simulations of Functionalized Nano-Materials for Biosensors Applications,” International Journal of Molecular Sciences. 2022. link Times cited: 7 Abstract: Nanoscale biosensors, a highly promising technique in clinic… read moreAbstract: Nanoscale biosensors, a highly promising technique in clinical analysis, can provide sensitive yet label-free detection of biomolecules. The spatial and chemical specificity of the surface coverage, the proper immobilization of the bioreceptor as well as the underlying interfacial phenomena are crucial elements for optimizing the performance of a biosensor. Due to experimental limitations at the microscopic level, integrated cross-disciplinary approaches that combine in silico design with experimental measurements have the potential to present a powerful new paradigm that tackles the issue of developing novel biosensors. In some cases, computational studies can be seen as alternative approaches to assess the microscopic working mechanisms of biosensors. Nonetheless, the complex architecture of a biosensor, associated with the collective contribution from “substrate–receptor–analyte” conjugate in a solvent, often requires extensive atomistic simulations and systems of prohibitive size which need to be addressed. In silico studies of functionalized surfaces also require ad hoc force field parameterization, as existing force fields for biomolecules are usually unable to correctly describe the biomolecule/surface interface. Thus, the computational studies in this field are limited to date. In this review, we aim to introduce fundamental principles that govern the absorption of biomolecules onto functionalized nanomaterials and to report state-of-the-art computational strategies to rationally design nanoscale biosensors. A detailed account of available in silico strategies used to drive and/or optimize the synthesis of functionalized nanomaterials for biosensing will be presented. The insights will not only stimulate the field to rationally design functionalized nanomaterials with improved biosensing performance but also foster research on the required functionalization to improve biomolecule–surface complex formation as a whole. read less NOT USED (low confidence) J. Ren, H. Yue, G. Liang, and M. Lv, “Influence of Tool Shape on Surface Quality of Monocrystalline Nickel Nanofabrication,” Molecules. 2022. link Times cited: 1 Abstract: In this paper, the influence of tool shape on the surface qu… read moreAbstract: In this paper, the influence of tool shape on the surface quality of monocrystalline nickel nanofabrication is studied. The research mainly adopts the method of molecular dynamics simulation, through the statistics of the atomic coordinates of the machined surface, then calculates the influence of different tool rake angles on the surface roughness of monocrystalline nickel. It is concluded that the surface roughness distribution is ‘W’ when the rake angle of the diamond tool changes from −45° to +45°. When analyzing the relationship between the tool shape and the processing temperature, it is found that when the clearance angle of the tool reaches a certain range, the clearance angle is further increased, and the temperature of the workpiece does not change during machining. Therefore, a large number of simulations were carried out, and it was concluded that there is a critical clearance angle, and the critical clearance angle of the tool in the research conditions is 8–10°. read less NOT USED (low confidence) S. Kazanç, “Kristal Yöneliminin ve Sıcaklığın Cu Nano Telinin Mekanik Özelliklerine Etkisinin Moleküler Dinamik Benzetimi ile İncelenmesi,” Deu Muhendislik Fakultesi Fen ve Muhendislik. 2022. link Times cited: 0 Abstract: In this study, the effect of uniaxial tensile strain applied… read moreAbstract: In this study, the effect of uniaxial tensile strain applied to Cu nanowire along the <100>, <110> and <111> highly symmetric crystallographic orientations on the mechanical properties was investigated by Molecular Dynamics (MD) simulation method. The forces acting on atoms were obtained from the derivative of the Embedded Atom Method (EAM) potential function, which includes many-body interactions. The Stress-strain curves, Young's modulus (E), yielding strength, and values of Cu model nanowires with different crystal orientations were determined under different temperatures. By using the obtained atomic images and the Common Neighbor Analysis method (CNA), it was determined that the plastic deformation occurred in the nanowires as a result of the stress applied as a result of the activation of Shockley partial dislocations and stacking faults defects for all orientations. In addition, for the <100> and <110> orientations, the formation of twin Kristal Yöneliminin ve Sıcaklığın Cu Nano Telinin Mekanik Özelliklerine Etkisinin Moleküler Dinamik Benzetimi ile İncelenmesi Investigation of the Effect of Crystal Orientation and Temperature on Mechanical Properties of Cu Nanowire by Molecular Dynamics Simulation read less NOT USED (low confidence) Z. Yan, B. Xu, J. Li, and L. Kong, “Defect-mediated crystal growth from deeply undercooled melts,” Computational Materials Science. 2022. link Times cited: 2 NOT USED (low confidence) A. Samiri, A. Khmich, A. Hassani, and A. Hasnaoui, “Elastic and structural properties of Mg25Al75 binary metallic glass under different cooling conditions,” Journal of Alloys and Compounds. 2022. link Times cited: 8 NOT USED (low confidence) F. Taherkhani and F. Taherkhani, “Ir nanocluster shape effects on melting, surface energy and scaling behavior of self-diffusion coefficient near melting temperature,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) A. Mahata, T. Mukhopadhyay, and M. A. Zaeem, “Modified embedded-atom method interatomic potentials for Al-Cu, Al-Fe and Al-Ni binary alloys: From room temperature to melting point,” Computational Materials Science. 2022. link Times cited: 27 NOT USED (low confidence) J. Dutta, S. Mandal, and S. Adhikari, “Formulation of temperature dependent effective Hartree potential incorporating quadratic over linear molecular DOFs-surface modes couplings and its effect on quantum dynamics of D2 (v = 0, j = 0)/D2 (v = 0, j = 2) on Cu(111) metal surface,” Chemical Physics. 2022. link Times cited: 1 NOT USED (low confidence) R. Yasbolaghi and A. Khoei, “A continuum–atomistic multi-scale analysis of temperature field problems and its application in phononic nano-structures,” Finite Elements in Analysis and Design. 2022. link Times cited: 1 NOT USED (low confidence) J. Li, Y. Hu, Y. Zhang, and R. Xia, “Optimum atomic concentration in structurally disordered nanoporous Pt–Co alloys with the strongest mechanical properties,” Microporous and Mesoporous Materials. 2022. link Times cited: 4 NOT USED (low confidence) T. Akande, F. Matthew-Ojelabi, G. Agunbiade, G. Adesakin, T. A. Ojuola, and A. Fasiku, “Modeling heats of solution for CuNixM(1−x): M = Rh, Sr, and Ir alloys,” AIP Advances. 2022. link Times cited: 0 NOT USED (low confidence) C. D. Reddy, Z. Zhang, S. Msolli, J. Guo, and N. Sridhar, “Impact velocity-dependent bonding mechanisms in metal cold spray,” Surface and Coatings Technology. 2022. link Times cited: 8 NOT USED (low confidence) A. Galashev, K. A. Ivanichkina, and O. Rakhmanova, “Advanced hybrid-structured anodes for lithium-ion batteries,” Computational Materials Science. 2021. link Times cited: 5 NOT USED (low confidence) E. Bird, J. G. Plascencia, P. Keblinski, and Z. Liang, “Molecular simulation of steady-state evaporation and condensation of water in air,” International Journal of Heat and Mass Transfer. 2021. link Times cited: 10 NOT USED (low confidence) P. Wynblatt, D. Chatain, and U. Dahmen, “Heteroepitaxy of FCC-on-FCC Systems of Large Misfit,” Acta Materialia. 2021. link Times cited: 5 NOT USED (low confidence) J. A. Hossain and B. H. Kim, “Scale effects in the nanoscale heat transfer of molecular interfaces with different lattice orientations,” AIP Advances. 2021. link Times cited: 3 NOT USED (low confidence) H. Kawano, “Effective Work Functions of the Elements,” Progress in Surface Science. 2021. link Times cited: 24 NOT USED (low confidence) S. Starikov, A. R. Kuznetsov, and V. Sagaradze, “Crowdion in Deformed FCC Metal. Atomistic Modeling,” Physics of Metals and Metallography. 2021. link Times cited: 3 NOT USED (low confidence) M. Kianezhad, M. Youzi, M. Vaezi, and H. N. Pishkenari, “Rectilinear Motion of Carbon Nanotube on Gold Surface,” International Journal of Mechanical Sciences. 2021. link Times cited: 10 NOT USED (low confidence) Y. Wang, K. Niu, and Y. Wu, “Multiscale modelling of graphene sheet and its application in laminated composites,” Composite Structures. 2021. link Times cited: 8 NOT USED (low confidence) L. G. Gonçalves, J. P. D. Souza, and E. D. Zanotto, “Assessment of the classical nucleation theory in supercooled nickel by molecular dynamics,” Materials Chemistry and Physics. 2021. link Times cited: 5 NOT USED (low confidence) B.-X. Zhang, S. Wang, X.-lei He, Y.-R. Yang, X. Wang, and D.-J. Lee, “Statics and dynamics of nanodroplet electrowetting on an isothermally heated nanostructured surface,” Journal of Molecular Liquids. 2021. link Times cited: 2 NOT USED (low confidence) N. Dhariwal, A. S. M. Miraz, W. Meng, B. Ramachandran, and C. Wick, “Impact of metal/ceramic interactions on interfacial shear strength: Study of Cr/TiN using a new modified embedded-atom potential,” Materials & Design. 2021. link Times cited: 4 NOT USED (low confidence) L. Gao, X. Fan, S. Zhang, D. Che, and B. Sun, “Palmitic acid graphene composite phase change materials: a molecular dynamics simulation,” Thermochimica Acta. 2021. link Times cited: 8 NOT USED (low confidence) V. Guder, S. Sengul, M. Celtek, and U. Domekeli, “Pressure dependent evolution of microstructures in Pd80Si20 bulk metallic glass,” Journal of Non-Crystalline Solids. 2021. link Times cited: 6 NOT USED (low confidence) L. Shi et al., “Achieving high strength and ductility in copper matrix composites with graphene network,” Materials Science and Engineering: A. 2021. link Times cited: 18 NOT USED (low confidence) O. Bachurina and A. A. Kudreyko, “Two-component localized vibrational modes in fcc metals,” The European Physical Journal B. 2021. link Times cited: 4 NOT USED (low confidence) D. Zhao, J. Wang, and Z. Xu, “Surface Effect on Vibration of Timoshenko Nanobeam Based on Generalized Differential Quadrature Method and Molecular Dynamics Simulation,” Nanomanufacturing and Metrology. 2021. link Times cited: 2 NOT USED (low confidence) D. Zhao, J. Wang, and Z. Xu, “Surface Effect on Vibration of Timoshenko Nanobeam Based on Generalized Differential Quadrature Method and Molecular Dynamics Simulation,” Nanomanufacturing and Metrology. 2021. link Times cited: 0 NOT USED (low confidence) X. Yue, J. Fan, Q. Li, X. Yang, Z.-J. Xu, and Z. Chen, “Influence of discharge gap on material removal and melt pool movement in EDM discharge process,” The International Journal of Advanced Manufacturing Technology. 2021. link Times cited: 3 NOT USED (low confidence) S. Ajori, S. Haghighi, H. Parsapour, and R. Ansari, “Fundamental frequency analysis of endohedrally functionalized carbon nanotubes with metallic nanowires: a molecular dynamics study,” Journal of Molecular Modeling. 2021. link Times cited: 2 NOT USED (low confidence) A. Kedharnath, R. Kapoor, and A. Sarkar, “Classical molecular dynamics simulations of the deformation of metals under uniaxial monotonic loading: A review,” Computers & Structures. 2021. link Times cited: 16 NOT USED (low confidence) A. Kotri, Y. Belkassmi, E. Elkoraychy, M. Mazroui, and L. Elmaimouni, “Static investigation of the small clusters on the Cu(111) and Au(111) surfaces,” Chinese Journal of Physics. 2021. link Times cited: 4 NOT USED (low confidence) W. Wanmolee et al., “Phase speciation and surface analysis of copper phosphate on high surface area silica support by in situ XAS/XRD and DFT: Assessment for guaiacol hydrodeoxygenation,” Applied Surface Science. 2021. link Times cited: 6 NOT USED (low confidence) Y. Cui and H. Chew, “Machine-Learning Prediction of Atomistic Stress along Grain Boundaries,” Acta Materialia. 2021. link Times cited: 8 NOT USED (low confidence) A. J. Klomp, A. Stukowski, R. Müller, K. Albe, and F. Diewald, “Influence of surface stress on the mechanical response of nanoporous metals studied by an atomistically informed continuum model,” Acta Materialia. 2021. link Times cited: 1 NOT USED (low confidence) H. Yang, G. Song, and C. J. Hogan, “A molecular dynamics study of collisional heat transfer to nanoclusters in the gas phase,” Journal of Aerosol Science. 2021. link Times cited: 8 NOT USED (low confidence) K. Ivanichkina, A. Galashev, and A. Isakov, “Computational modeling of electrolytic deposition of a single-layer silicon film on silver and graphite substrates,” Applied Surface Science. 2021. link Times cited: 3 NOT USED (low confidence) X. Zhang, H. Zhang, Z. Zong, Z. Li, and X. Chen, “From regular arrays of liquid metal nano-islands to single crystalline biatomic-layer gallium film: Molecular dynamics and first principle study,” Journal of Applied Physics. 2021. link Times cited: 0 Abstract: The two-dimensional (2D) materials provide an excellent plat… read moreAbstract: The two-dimensional (2D) materials provide an excellent platform for the study of the dimensional effect. The richer the types of 2D materials, the broader the unknown field we can explore. However, among the large number of 2D materials manufactured by humans, true single-crystalline (SC) atomically thin 2D metals are rare. The instability of SC 2D metal materials puts high demands on its fabrication process. By implementing molecular dynamics (MD) simulations, we proved that the SC biatomic-layer (BL) gallium film can be formed at the interface between two graphene layers. The Ga atoms deposited on the surface of the graphene on the copper substrate will spontaneously evolve into independent liquid nano-islands, and then cover the nano-island with a monolayer graphene. When the Ga nano-islands confined under the graphene layer are heated to 500 °C, they will expand into a BL Ga film, and finally, the entire system is cooled to room temperature to obtain the SCBL Ga film. It is found that these nano-islands are in the liquid state at ∼400 °C, but they undergo a phase transition and evolve into the solid state at ∼500°C. At the same time, the nano-islands also drop from 3D to 2D. In addition, the vertical heterostructure with moire superstructure is formed between the SCBL Ga and the top layer graphene. The calculations of the electronic properties show that the Dirac conical point of the graphene in the heterostructure is shifted below the Fermi level, which proves that SCBL Ga is able to induce semimetallic to metallic conversion in graphene, indicating SCBL Ga can be used for metal contacts in 2D devices. read less NOT USED (low confidence) B. Fu, Z. Zhang, L. Li, X. Qin, and X. Ye, “Compressive properties and behavior of copper nanowires wrapped by carbon nanotube,” Applied Physics A. 2021. link Times cited: 0 NOT USED (low confidence) G.-M. Lin, J. Guo, and P. Ji, “Molecular dynamics study on the diffusion process of AuAgCuNiPd high-entropy alloy metallurgy induced by pulsed laser heating.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 3 Abstract: As novel alloy materials with outstanding mechanical propert… read moreAbstract: As novel alloy materials with outstanding mechanical properties, high-entropy alloys have a wide range of promising applications. By establishing individual Au, Ag, Cu, Ni, and Pd nanolaminates with face-centered-cubic lattice structure arrangements, molecular dynamics simulation is carried out to track the diffusion process of AuAgCuNiPd high-entropy alloy metallurgy, which is induced by pulsed laser heating. The temperature, potential energy, and kinetic energy are analyzed to evaluate the metallurgy. The snapshots and atomic fractions are presented to show the mass transfer between metallic nanolaminates. The diffusion process is firstly observed 0.3 ns after the central point for pulsed laser heating (absorbed laser energy density at 7 kJ cm-3 and pulse duration of 0.5 ns). Meanwhile, the degrees of atomic activity for Au, Ag, Cu, Ni, and Pd are assessed by calculating the mean square displacement and diffusion coefficient. Ni has a slightly larger diffusion coefficient than the other four metallic elements. Moreover, after the central point of laser irradiation, the kinetic energy of the system reduces, while the potential energy increases, which relates to the transition from nanolaminates to high-entropy alloys. A critical absorbed laser energy density of 6 kJ cm-3 with a relative error of 8.3% for the generation of AuAgCuNiPd high-entropy alloys is found. The order of constituent nanolaminates configured with the earlier initiation of diffusion between atoms in the neighboring nanolaminates speeds up the metallurgy. read less NOT USED (low confidence) X. Shen, B. Yao, Z. R. Liu, D. Legut, H. J. Zhang, and R. Zhang, “Mechanistic insights into interface-facilitated dislocation nucleation and phase transformation at semicoherent bimetal interfaces,” International Journal of Plasticity. 2021. link Times cited: 11 NOT USED (low confidence) B. Yao, Z. Liu, and R. Zhang, “EAPOTs: An integrated empirical interatomic potential optimization platform for single elemental solids,” Computational Materials Science. 2021. link Times cited: 3 NOT USED (low confidence) Y. Yang, M. Liu, S. Zhou, W. Ren, Q. Zhou, and W. Zhang, “Strengthening behaviour of continuous graphene network in metal matrix composites,” Carbon. 2021. link Times cited: 18 NOT USED (low confidence) X. Yue, J. Liu, and Y. Liu, “Investigation of electric field distribution and material removal process in nano electro machining,” Materials today communications. 2021. link Times cited: 1 NOT USED (low confidence) I. I. Fairushin, A. Shemakhin, and A. A. Khabir’yanova, “Molecular Dynamics Simulation of Copper Nanofilm Self-Assembly on Silicon Substrate under Gas-Discharge Plasma Conditions,” High Energy Chemistry. 2021. link Times cited: 0 NOT USED (low confidence) H. Qadr, “A Molecular Dynamics Study of Temperature Dependence of the Primary State of Cascade Damage Processes,” Russian Journal of Non-Ferrous Metals. 2021. link Times cited: 4 NOT USED (low confidence) A. J. Shih, N. Arulmozhi, and M. Koper, “Electrocatalysis under Cover: Enhanced Hydrogen Evolution via Defective Graphene-Covered Pt(111),” ACS Catalysis. 2021. link Times cited: 15 Abstract: : The production of hydrogen via water electrolysis using re… read moreAbstract: : The production of hydrogen via water electrolysis using renewable electricity is a promising carbon-neutral technology. In this contribution, we report insights into the hydrogen evolution reaction (HER) in H 2 SO 4 on Pt(111) and graphene-covered Pt(111), in addition to the electrochemical properties of graphene overlayers. As-prepared graphene overlayers are selectively permeable to H + ions in the electrolyte, allowing H + ions into the con fi ned layer between graphene and Pt(111) while excluding SO 42 − and other anions. We demonstrate that defects in these as-prepared graphene overlayers can be generated from oxidation at high overpotentials or reduction from the production of H 2 bubbles and postulate that HER occurs locally at only Pt(111) in the proximity of defects in graphene overlayers on as-prepared G/Pt(111) electrodes, and as defects in graphene increases, more of the Pt(111) surface becomes utilized for HER. Kinetically, the addition of defective graphene overlayers can increase the geometric HER rate by up to 200%, while Tafel slopes and [H + ] reaction orders remain unchanged. These results shed kinetic insight into the nature of graphene overlayers and their e ff ect on HER catalysis and also demonstrate the promise of con fi nement modi fi cations in designing catalysts with properties closer to achieving optimum rates. read less NOT USED (low confidence) B. Zandersons, L. Lührs, Y. Li, and J. Weissmüller, “On factors defining the mechanical behavior of nanoporous gold,” Acta Materialia. 2021. link Times cited: 21 NOT USED (low confidence) D. Vizoso, M. Kosmidou, T. Balk, K. Hattar, C. Deo, and R. Dingreville, “Size-dependent radiation damage mechanisms in nanowires and nanoporous structures,” Acta Materialia. 2021. link Times cited: 8 NOT USED (low confidence) C. M. Andolina, M. Bon, D. Passerone, and W. Saidi, “Robust, Multi-Length-Scale, Machine Learning Potential for Ag–Au Bimetallic Alloys from Clusters to Bulk Materials,” The Journal of Physical Chemistry C. 2021. link Times cited: 22 NOT USED (low confidence) T. Wang, X. Liu, H. Liu, and M. He, “Synergistic effect of supercritical water and nano-catalyst on lignin gasification,” International Journal of Hydrogen Energy. 2021. link Times cited: 12 NOT USED (low confidence) X.-lei He et al., “Electrowetting-based control of wetting transition of a nanodroplet on pillar-arrayed surfaces,” Journal of Molecular Liquids. 2021. link Times cited: 15 NOT USED (low confidence) G. Rusina, S. Borisova, and E. Chulkov, “Structural Relaxation and Vibrational Properties of a Surface with Point Defects,” JETP Letters. 2021. link Times cited: 0 NOT USED (low confidence) S. Gao et al., “Core-shell PdAu nanocluster catalysts to suppress sulfur poisoning.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 2 Abstract: Reducing sulfur poisoning is significant for maintaining the… read moreAbstract: Reducing sulfur poisoning is significant for maintaining the catalytic efficiency and durability of heterogeneous catalysts. We screened PdAu nanoclusters with specific Pd : Au ratios based on Monte Carlo simulations and then carried out density functional calculations to reveal how to reduce sulfur poisoning via alloying. Among various nanoclusters, the core-shell structure Pd13Au42 (Pd@Au) exhibits a low adsorption energy of SO2 (-0.67 eV), comparable with O2 (-0.45 eV) and lower than CO (-1.25 eV), thus avoiding sulfur poisoning during the CO catalytic oxidation. Fundamentally, the weak adsorption of SO2 originates from the negative d-band center of the shell and delocalized charge distribution near the Fermi level, due to the appropriate charge transfer from the core to shell. Core-shell nanoclusters with a different core (Ni, Cu, Ag, Pt) and a Pd@Au slab model were further constructed to validate and extend the results. These findings provide insights into designing core-shell catalysts to suppress sulfur poisoning while optimizing catalytic behaviors. read less NOT USED (low confidence) A. Galashev and A. Vorob’ev, “DFT study of silicene on metal (Al, Ag, Au) substrates of various thicknesses,” Physics Letters A. 2021. link Times cited: 7 NOT USED (low confidence) C.-M. Wu, X. Wei, and Y.-R. Li, “Investigation on the mechanisms of cluster formation and transition from adsorption to condensation,” International Journal of Heat and Mass Transfer. 2021. link Times cited: 5 NOT USED (low confidence) G. Voyiadjis, M. Saffarini, and C. Ruestes, “Characterization of the strain rate effect under uniaxial loading for nanoporous gold,” Computational Materials Science. 2021. link Times cited: 12 NOT USED (low confidence) W. Lv et al., “Development of modified embedded-atom model and molecular dynamics simulation of cesium,” Computational Materials Science. 2021. link Times cited: 1 NOT USED (low confidence) S. Wang, E. Zhu, Y. Huang, and H. Heinz, “Direct correlation of oxygen adsorption on platinum-electrolyte interfaces with the activity in the oxygen reduction reaction,” Science Advances. 2021. link Times cited: 31 Abstract: The adsorption of oxygen molecules to Pt nanostructures in s… read moreAbstract: The adsorption of oxygen molecules to Pt nanostructures in solution is shown to predict the relative ORR activity in fuel cells. The oxygen reduction reaction (ORR) on platinum catalysts is essential in fuel cells. Quantitative predictions of the relative ORR activity in experiments, in the range of 1 to 50 times, have remained challenging because of incomplete mechanistic understanding and lack of computational tools to account for the associated small differences in activation energies (<2.3 kilocalories per mole). Using highly accurate molecular dynamics (MD) simulation with the Interface force field (0.1 kilocalories per mole), we elucidated the mechanism of adsorption of molecular oxygen on regular and irregular platinum surfaces and nanostructures, followed by local density functional theory (DFT) calculations. The relative ORR activity is determined by oxygen access to platinum surfaces, which greatly depends on specific water adlayers, while electron transfer occurs at a similar slow rate. The MD methods facilitate quantitative predictions of relative ORR activities of any platinum nanostructures, are applicable to other catalysts, and enable effective MD/DFT approaches. read less NOT USED (low confidence) Y. Jiang, S. Sun, and T.-Y. Zhang, “Thickness- and temperature-dependent Grüneisen parameter in thin films.,” Nanoscale. 2021. link Times cited: 2 Abstract: The Grüneisen formula is one of the most important equations… read moreAbstract: The Grüneisen formula is one of the most important equations of state, in which the Grüneisen parameter plays a key role in the linkage of mechanical and thermal properties of materials. In the present work, for the first time, we investigate the dependence of the Grüneisen parameter on film-thickness and temperature via theoretical modeling and molecular dynamics (MD) simulations. The theoretical analysis gives two analytic expressions of a thickness- and temperature-dependent Grüneisen parameter, and the difference between the two analytic expressions lies in the quadratic or linear dependence on temperature. MD simulations are conducted on face-centered cubic (FCC) Ni, Cu, and Au (001) thin films and their bulk counterparts. The simulation results completely verify the theoretical results and determine the values of parameters involved in the theoretical modeling. The thickness- and temperature-dependent film heat capacity density is also investigated during the course of the Grüneisen parameter study. read less NOT USED (low confidence) P. Schultz et al., “First-principles calculations of metal surfaces. II. Properties of low-index platinum surfaces toward understanding electron emission,” Physical Review B. 2021. link Times cited: 4 Abstract: The stability of low-index platinum surfaces and their elect… read moreAbstract: The stability of low-index platinum surfaces and their electronic properties is investigated with density functional theory, toward the goal of understanding the surface structure and electron emission, and identifying precursors to electrical breakdown, on nonideal platinum surfaces. Propensity for electron emission can be related to a local work function, which, in turn, is intimately dependent on the local surface structure. The $(1\ifmmode\times\else\texttimes\fi{}N)$ missing row reconstruction of the Pt(110) surface is systematically examined. The $(1\ifmmode\times\else\texttimes\fi{}3)$ missing row reconstruction is found to be the lowest in energy, with the $(1\ifmmode\times\else\texttimes\fi{}2)$ and $(1\ifmmode\times\else\texttimes\fi{}4)$ slightly less stable. In the limit of large $(1\ifmmode\times\else\texttimes\fi{}N)$ with wider (111) nanoterraces, the energy accurately approaches the asymptotic limit of the infinite Pt(111) surface. This suggests a local energetic stability of narrow (111) nanoterraces on free Pt surfaces that could be a common structural feature in the complex surface morphologies, leading to work functions consistent with those on thermally grown Pt substrates. read less NOT USED (low confidence) C. D. Reddy, Z.-qian Zhang, S. Msolli, J. Guo, and N. Sridhar, “Impact induced metallurgical and mechanical interlocking in metals,” Computational Materials Science. 2021. link Times cited: 8 NOT USED (low confidence) Z. Cui, X. Fan, and G. Zhang, “Molecular dynamic study for concentration-dependent volume relaxation of vacancy,” Microelectronics Reliability. 2021. link Times cited: 10 NOT USED (low confidence) N. Amadou, T. de Rességuier, and A. Dragon, “Influence of point defects and grain boundaries on plasticity and phase transition in uniaxially-compressed iron,” Computational Condensed Matter. 2021. link Times cited: 3 NOT USED (low confidence) M. N. Khatun and R. Gosh, “Verification of the Stokes-Einstein relation in liquid noble metals over a wide range of temperatures,” Physics Letters A. 2021. link Times cited: 1 NOT USED (low confidence) Y. Zhu, X. Hu, and Y. Ni, “Molecular dynamics simulation of microstructure evolution during the fracture process of nano-twinned Ag,” Engineering Fracture Mechanics. 2021. link Times cited: 7 NOT USED (low confidence) J. Liu, X. Fan, Y. Shi, D. J. Singh, and W. Zheng, “Deformation and ductile fracture of nanocrystalline gold ultrathin nanoribbon: Width effect,” Fatigue & Fracture of Engineering Materials & Structures. 2021. link Times cited: 3 NOT USED (low confidence) M. Daw and M. Chandross, “Sluggish diffusion in random equimolar FCC alloys,” Physical Review Materials. 2021. link Times cited: 16 Abstract: We examine vacancy-assisted diffusion in the 57 random, equi… read moreAbstract: We examine vacancy-assisted diffusion in the 57 random, equimolar alloys that can be formed from Cu, Ag, Au, Ni, Pd, and Pt based on the well-tested embedded atom method functions of Foiles, Baskes, and Daw [Phys. Rev. B 33 , 7983 (1986)]. We address the suggestion [Yeh et al. , Adv. Eng. Mater. 6 , 299 (2004)] that increasing the number of constituents causes diffusion to be “sluggish” in random, equimolar alloys. Using molecular dynamics (MD) simulations of random alloys with a single vacancy, combined with calculations of vacancy formation, we extract vacancy-assisted diffusivities in each alloy. After developing and applying several possible criteria for evaluating “sluggishness,” we find that only a small minority (from 1 to 8, depending on how sluggishness is defined) of the alloys exhibit sluggish diffusion whereas in the large majority of alloys diffusion is faster and in quite a few cases ought to be considered vigorous (that is, faster than in any of the constituents). We correlate diffusivity with a combination of the mean of the constituent diffusivity and a simple function of lattice mismatch. We conclude that simply increasing the number of constituents in such alloys does not systematically alter the diffusion, but that instead lattice mismatch plays a primary factor; sluggish diffusion is more likely to occur in a window of small lattice mismatch (1–3%) even in binary alloys. Quantitatively, our calculated diffusivities correlate with a combination of (1) rule of mixtures of the diffusivities of the constituents, and (2) a simple function of the lattice mismatch; this accounts for the large majority of our calculated diffusivities to within a factor of 2 (over a range of three orders of magnitude). We also find that while lattice mismatch on the order of 1–3% is necessary for sluggish diffusion, it is not sufficient. read less NOT USED (low confidence) P. P. Shinde et al., “Graphene nanoribbons with mixed cove-cape-zigzag edge structure,” Carbon. 2021. link Times cited: 9 NOT USED (low confidence) B. Xu et al., “Weaker bonding can give larger thermal conductance at highly mismatched interfaces,” Science Advances. 2021. link Times cited: 28 Abstract: Thermal conductance at interface with weak van der Waals bon… read moreAbstract: Thermal conductance at interface with weak van der Waals bonding can become larger than that with strong covalent bonding. Thermal boundary conductance is typically positively correlated with interfacial adhesion at the interface. Here, we demonstrate a counterintuitive experimental result in which a weak van der Waals interface can give a higher thermal boundary conductance than a strong covalently bonded interface. This occurs in a system with highly mismatched vibrational frequencies (copper/diamond) modified by a self-assembled monolayer. Using finely controlled fabrication and detailed characterization, complemented by molecular simulation, the effects of bridging the vibrational spectrum mismatch and bonding at the interface are systematically varied and understood from a molecular dynamics viewpoint. The results reveal that the bridging and binding effects have a trade-off relationship and, consequently, that the bridging can overwhelm the binding effect at a highly mismatched interface. This study provides a comprehensive understanding of phonon transport at interfaces, unifying physical and chemical understandings, and allowing interfacial tailoring of the thermal transport in various material systems. read less NOT USED (low confidence) G. Park, B. Beeler, and M. Okuniewski, “An atomistic study of defect energetics and diffusion with respect to composition and temperature in γU and γU-Mo alloys,” Journal of Nuclear Materials. 2021. link Times cited: 10 NOT USED (low confidence) Z. Aitken, V. Sorkin, Z. Yu, S. Chen, Z. Wu, and Y.-W. Zhang, “Modified embedded-atom method potentials for the plasticity and fracture behaviors of unary fcc metals,” Physical Review B. 2021. link Times cited: 5 NOT USED (low confidence) D. J. Kim et al., “Ultrahigh-strength multi-layer graphene-coated Ni film with interface-induced hardening,” Carbon. 2021. link Times cited: 15 NOT USED (low confidence) M. Khodabakhshi, J. Wen, and Z. Tan, “Coefficient of restitution for silver nanoparticles colliding on a wet silver substrate,” Applied Surface Science. 2021. link Times cited: 2 NOT USED (low confidence) S. Zhang, F. Wang, and P. Huang, “Enhanced Hall-Petch strengthening in graphene/Cu nanocomposites,” Journal of Materials Science & Technology. 2021. link Times cited: 24 NOT USED (low confidence) Q. Huang, Z. Zhang, Z. Liu, Z. Fujian, G. Cheng, and J. Ding, “Pinning effect in droplet self-driving and its reduction mechanism by monolayer graphene,” Applied Surface Science. 2021. link Times cited: 6 NOT USED (low confidence) C. Balbuena, M. M. Gianetti, and E. Soulé, “Molecular dynamics simulations of the formation of Ag nanoparticles assisted by PVP.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 6 Abstract: Understanding the formation mechanisms of nanoparticles is e… read moreAbstract: Understanding the formation mechanisms of nanoparticles is essential for the synthesis of nanomaterials with controlled properties. In solution synthesis, capping agents are used to mediate this process and control the final size and shape of the particles. In this work, the synthesis of silver nanoparticles, with polyvinylpyrrolidone (PVP) as the capping agent, is studied through molecular dynamics simulations. Nucleation of clusters of atoms and subsequent growth to form nanoparticles are analyzed, with focus on the role of PVP. No finite critical nucleus is detected, and amorphous particles seem to form by spinodal growth. In this timescale, PVP seems to have no effect on particle growth, which is ascribed to the competition between the protective effect and "bridging" (where a molecule of PVP is adsorbed to two different clusters, bringing them together). As the process evolves, a sequence of ordered structures appears within the particles: icosahedral, BCC, and FCC, the last one being the equilibrium configuration of bulk silver. In addition, for a low PVP content an apparent acceleration is observed in particle growth after these ordered phases appear, indicating that the growth of ordered particles from the solution is faster than the growth of amorphous particles. For a high PVP content, this acceleration is not observed, indicating that the protective effect prevails on particle growth in this regime. In addition, due to the bridging effect, the final overall configuration is strongly dependent on the PVP content. In the absence of PVP, large but dispersed particles are observed. When the PVP content is low, due to strong bridging, particles form agglomerates (with no strong coalescence in the timescale of simulations). When the PVP content is large enough, particles are smaller in size and do not show a strong tendency to agglomerate. read less NOT USED (low confidence) G. M. Faccin, Z. Pereira, and E. D. da Silva, “How Crystallization Affects the Oriented Attachment of Silver Nanocrystals,” Journal of Physical Chemistry C. 2021. link Times cited: 4 Abstract: Oriented attachment processes between nanocrystals provide a… read moreAbstract: Oriented attachment processes between nanocrystals provide a promising route for the synthesis of mesocrystals that, although made of the same elements as their usual crystal counterparts, neverthe... read less NOT USED (low confidence) J. V. D. van der Hoeven et al., “Structural Control over Bimetallic Core–Shell Nanorods for Surface-Enhanced Raman Spectroscopy,” ACS Omega. 2021. link Times cited: 20 Abstract: Bimetallic nanorods are important colloidal nanoparticles fo… read moreAbstract: Bimetallic nanorods are important colloidal nanoparticles for optical applications, sensing, and light-enhanced catalysis due to their versatile plasmonic properties. However, tuning the plasmonic resonances is challenging as it requires a simultaneous control over the particle shape, shell thickness, and morphology. Here, we show that we have full control over these parameters by performing metal overgrowth on gold nanorods within a mesoporous silica shell, resulting in Au–Ag, Au–Pd, and Au–Pt core–shell nanorods with precisely tunable plasmonic properties. The metal shell thickness was regulated via the precursor concentration and reaction time in the metal overgrowth. Control over the shell morphology was achieved via a thermal annealing, enabling a transition from rough nonepitaxial to smooth epitaxial Pd shells while retaining the anisotropic rod shape. The core–shell synthesis was successfully scaled up from micro- to milligrams, by controlling the kinetics of the metal overgrowth via the pH. By carefully tuning the structure, we optimized the plasmonic properties of the bimetallic core–shell nanorods for surface-enhanced Raman spectroscopy. The Raman signal was the most strongly enhanced by the Au core–Ag shell nanorods, which we explain using finite-difference time-domain calculations. read less NOT USED (low confidence) S. Zhao, G. Ran, P. Chen, Q. Han, H. Deng, and X. Ye, “DFT study on the nucleation of He bubbles in Pd: Effect of H and self-interstitial atoms,” Journal of Nuclear Materials. 2021. link Times cited: 2 NOT USED (low confidence) J. Liu, X. Fan, C. Gu, Y. Shi, D. J. Singh, and W. Zheng, “Effect of voids on nanocrystalline gold ultrathin film,” Computational Materials Science. 2021. link Times cited: 3 NOT USED (low confidence) P. Simonnin, D. Schreiber, and K. Rosso, “Predicting the temperature dependence of self-diffusion behavior in Ni-Cr alloys via molecular dynamics,” Materials today communications. 2021. link Times cited: 6 NOT USED (low confidence) S. Yang and L. Zhang, “Characterization of mechanical properties and failure of potassium dihydrogen phosphate under mechanical stressing,” Ceramics International. 2021. link Times cited: 10 NOT USED (low confidence) P. D. Hengfei Gu et al., “Deformation Twinning in Octahedron-Based Face-Centered Cubic Metallic Structures: Localized Shear-Force Dipoles Drive Atomic Displacements,” Chemical Engineering (Engineering) eJournal. 2021. link Times cited: 6 Abstract: Twinning is found to impart favorable mechanical, physical a… read moreAbstract: Twinning is found to impart favorable mechanical, physical and chemical properties to nanostructured materials. One important twinning mode, deformation twinning, prevails in coarse-grained hexagonal close-packed (HCP) crystalline materials and body-centered cubic (BCC) and face-centered cubic (FCC) nanomaterials under high-stress conditions. In FCC structures, the {111} deformation twinning is traditionally believed to nucleate and grow through layer-by-layer emission of 1/6 Shockley partial dislocations on consecutive {111} planes. Here, we report that by conducting high-resolution transmission electron microscopy (HRTEM) observation, deformation twinning is, for the first time, found to occur in nanocrystalline (Fe, Nb)23Zr6 particles with a Mn23Th6-type FCC structure that is composed of a Zr-octahedron-based FCC network connected by alloying elements Fe and Nb like the large FCC structure such as metal-organic-framework (MOF). Based on direct atomic-scale observations, we discover a new mechanism for the {111} deformation twinning in FCC structures. To form a [112]/(111) twin, for example, short ( (‾1‾11) planes within two adjacent (111) plane layers in the repeated three-layer sequence of (111) planes are shear deformed continuously by a shear-force dipole along the [112] direction like a domino effect, whereas the other (111) plane in the repeated sequence remains intact. Through this route, a small energy for twinning is expected because only 2/3 (111) planes need to be transformed to form a twin. In addition, a loading criterion for deformation twinning of a FCC NP under uniaxial compression is proposed based on our results. Our work here not only provides a fundamental understanding on deformation twinning in FCC structures, but also opens up studies of deformation behaviors in a class of Mn23Th6-type FCC materials. read less NOT USED (low confidence) D. Scheiber, “Segregation and embrittlement of gold grain boundaries,” Computational Materials Science. 2021. link Times cited: 12 NOT USED (low confidence) Y. Nagaoka et al., “Bulk Grain-Boundary Materials from Nanocrystals,” Chem. 2021. link Times cited: 10 NOT USED (low confidence) M. He, E. T. Karim, M. Shugaev, and L. Zhigilei, “Atomistic simulation of the generation of vacancies in rapid crystallization of metals,” Acta Materialia. 2021. link Times cited: 7 NOT USED (low confidence) A. Galashev, K. Ivanichkina, A. Vorob’ev, O. Rakhmanova, K. Katin, and M. Maslov, “Improved lithium-ion batteries and their communication with hydrogen power,” International Journal of Hydrogen Energy. 2021. link Times cited: 9 NOT USED (low confidence) D. S. Bertoldi, E. N. Millán, and A. F. Guillermet, “Phenomenology of the heating, melting and diffusion processes in Au nanoparticles.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 4 Abstract: The paper reports the results of a molecular dynamics study … read moreAbstract: The paper reports the results of a molecular dynamics study of the heating and melting process of nanoparticles with 1985 to 84 703 atoms. Building on a previous study by the present authors [Bertoldi, et al., J. Phys. Chem. Solids, 2017, 111, 286-293] involving the energy versus temperature, the Lindemann index and the radial distribution function, the current work relies on the mean-square displacement, the Lindemann ratio and the simulated snapshots to characterize four regions in the process of heating-to-melting. A general pattern of the atomic configuration evolution upon heating and a systematics of the transition temperatures between the various identified steps, is proposed. In addition, the most significant, so-called "melting step" in this process is analyzed in terms of the quasi-chemical approach proposed by Bertoldi et al., which treats this step by invoking a dynamic equilibrium of the type Au (LEA-SPL) ⇌ Au (HEA-LPL) involving low-energy atoms (LEA) and high-energy atoms (HEA) forming the solid phase-like (SPL) and the liquid phase-like (LPL) states of the system, respectively. The "melting step" is characterized by evaluating the equal-Gibbs energy temperature, i.e., the "T0 temperature", previously introduced by the current authors, which is the thermodynamic counterpart of the temperature of fusion of macroscopic elemental solids. The diffusion coefficients at T0 are determined, and their spatial and temperature dependence is discussed. In particular, the activation energy for the atom movements in the HEA-LPL/LEA-SPL mixture at T0 is reported. The consistency between the current phenomenological picture and microscopic interpretation of the thermodynamic, kinetic and atomic configuration information obtained is highlighted. read less NOT USED (low confidence) H. Liu, Y.-bo Guo, and P. Zhao, “Surface generation mechanism of monocrystalline materials under arbitrary crystal orientations in nanoscale cutting,” Materials today communications. 2020. link Times cited: 20 NOT USED (low confidence) X. Li and A. Minor, “Precise Measurement of Activation Parameters for Individual Dislocation Nucleation During in Situ Tem Tensile Testing of Single Crystal Nickel,” Materials Engineering eJournal. 2020. link Times cited: 6 Abstract: Nucleation of crystalline defects such as dislocations lies … read moreAbstract: Nucleation of crystalline defects such as dislocations lies at the heart of mechanical deformation. Here, we demonstrate a technique for observing the nucleation of individual dislocations during in situ transmission electron microscopy (TEM) tensile testing and measuring fundamental parameters relevant for plasticity from the individual events. Our method relies on systematic detection of dislocation slip traces with automated image analysis in an oriented single crystal Ni sample. Using the identification of individual defect traces from in situ testing, a cumulative probabilistic function is applied to correlate the relationship between a dislocation nucleation event and the corresponding stress level. Our analysis allows for the extrapolation of the activation parameters for individual dislocation nucleation events using the data on one sample in one tensile test. Precise and quantitative correlation of activation parameters for dislocation nucleation from in situ TEM nanomechanical testing can provide direct quantitative measurements useful for computational models of plasticity. read less NOT USED (low confidence) K. Sbiaai et al., “First Au monolayer formation on Cu(110) surface,” IOP Conference Series: Materials Science and Engineering. 2020. link Times cited: 0 Abstract: We study in this work the growth of a gold monolayer on a Cu… read moreAbstract: We study in this work the growth of a gold monolayer on a Cu (110) surface thanks to the kinetic Monte Carlo method. We considered a coverage of 0.1% of the monolayer. Indeed, the growth of a monolayer is done through the intermediary of several diffusion processes. All activation energies for each process were calculated using the static method coupled with the KMC method. This coupling is established by the ABBM anisotropic bond breaking model. The study of the results obtained in the temperature range (100-300K) at different deposition rates shows the formation of small 1D islands and certain 2D islands at high temperature (280 k-300 K). read less NOT USED (low confidence) S. Zamani and K. Behdinan, “A molecular dynamics study of the mechanical and electrical properties of Polydimethylsiloxane-Ni conductive nanocomposites,” Composites Science and Technology. 2020. link Times cited: 7 NOT USED (low confidence) W. Xie and F. Fang, “Crystallographic orientation effect on cutting-based single atomic layer removal,” Frontiers of Mechanical Engineering. 2020. link Times cited: 2 NOT USED (low confidence) A. S. Davis and V. Agrawal, “One-dimensional moving window atomistic framework to model long-time shock wave propagation,” Computer Methods in Applied Mechanics and Engineering. 2020. link Times cited: 3 NOT USED (low confidence) P. Zhao, Q. Zhang, Y.-bo Guo, H. Liu, and Z. Deng, “Atomic simulation of crystal orientation effect on coating surface generation mechanisms in cold spray,” Computational Materials Science. 2020. link Times cited: 15 NOT USED (low confidence) X. Yue and X. Yang, “Molecular dynamics simulation of material removal process and mechanism of EDM using a two-temperature model,” Applied Surface Science. 2020. link Times cited: 24 NOT USED (low confidence) J. Harrison, S. Stuart, and D. Brenner, “Atomic-Scale Simulation of Tribological and Related Phenomena,” Handbook of Micro/Nano Tribology. 2020. link Times cited: 3 NOT USED (low confidence) K. Yeh, Y. Chiang, and S. W. Chang, “Full Atomistic Simulation of Cross-Linked Gold Nanoparticle Assemblies,” Multiscale Science and Engineering. 2020. link Times cited: 4 NOT USED (low confidence) J. Zhao, Á. Mayoral, L. Martínez, M. P. Johansson, F. Djurabekova, and Y. Huttel, “Core–Satellite Gold Nanoparticle Complexes Grown by Inert Gas-Phase Condensation,” The Journal of Physical Chemistry. C, Nanomaterials and Interfaces. 2020. link Times cited: 8 Abstract: Spontaneous growth of complexes consisted of a number of ind… read moreAbstract: Spontaneous growth of complexes consisted of a number of individual nanoparticles in a controlled manner, particularly in demanding environments of gas-phase synthesis, is a fascinating opportunity for numerous potential applications. Here, we report the formation of such core–satellite gold nanoparticle structures grown by magnetron sputtering inert gas condensation. Combining high-resolution scanning transmission electron microscopy and computational simulations, we reveal the adhesive and screening role of H2O molecules in formation of stable complexes consisted of one nanoparticle surrounded by smaller satellites. A single layer of H2O molecules, condensed between large and small gold nanoparticles, stabilizes positioning of nanoparticles with respect to one another during milliseconds of the synthesis time. The lack of isolated small gold nanoparticles on the substrate is explained by Brownian motion that is significantly broader for small-size particles. It is inferred that H2O as an admixture in the inert gas condensation opens up possibilities of controlling the final configuration of the different noble metal nanoparticles. read less NOT USED (low confidence) E. Cihan, “Structure evolution in tribological interfaces studied by multilayer model alloys.” 2020. link Times cited: 0 Abstract: Das Verstandnis von Reibung und Verschleis metallischer Ober… read moreAbstract: Das Verstandnis von Reibung und Verschleis metallischer Oberflachen ist eine kontinuierliche Herausforderung fur Ingenieure und Wissenschaftler – wurde man die industriellen Prozesse des 21. Jahrhunderts insgesamt betrachten, ergabe sich eine beachtlicher Energieaufwand durch Reibungsprozesse mit entsprechend grosen wirtschaftlichen Auswirkungen. Es ist daruber hinaus sehr schwierig, Reibung und Verschleis metallischer Oberflachen vorherzusagen, wenn der Einfluss der Randzonenverformung berucksichtigt werden soll. Studien zu den Verformungsmechanismen von Metallen und Legierungen ermoglichen ein besseres Verstandnis der Reibungs- und Verschleismechanismen von Werkstoffen mit genau definierten Ausgangsgefugen. In diesem Rahmen wurde in der vorliegenden Arbeit der Einfluss der Randzonenverformung auf das gemessene Reibungs- und Verschleisverhalten uber eine systematische experimentelle Studie an einem Au-Ni-Multilagenmodellsystem untersucht.
Die Reibexperimente, die an Au-Ni-Multilagenproben unter Ultrahochvakuumbedingungen (UHV) durchgefuhrt wurden, zeigten, dass die Dicke der einzelnen Schichten im Multilagensystem einen starken Einfluss auf die Reibung hat. Grund dafur ist der Ubergang der vorherrschenden Verformungsmechanismen nahe der Oberflache. Die Versuche geben auch einen neuen Ansatz vor fur metallische Systeme in trockener und sauberer Umgebung vor: Die Verringerung der Reibkraft kann durch stabile Gefuge, z.B. uber Legierungen, erreicht werden. Durch die ultrafeinen Korner, die durch mechanische Vermischung gebildet wurden, erhoht sich die Zahl der Korngrenzen. Die Verformung uber die Korngrenzen ergibt einen niedrigen Reibungskoeffizienten. Mit zunehmender Zyklenzahl bildete sich ein metastabiles Gefuge aus Au und Ni, das bis zum Bruch der Triboschicht erhalten blieb.
Um den Einfluss der Umgebung auf Reibung und Verschleis metallischer Werkstoffe zu betrachten, wurde der zweite Teil der Versuche unter definierter Stickstoffatmosphare durchgefuhrt. Wie bereits unter UHV-Bedingungen wurde ein deutlicher Anstieg des Reibungskoeffizienten mit zunehmender Schichtdicke gefunden. Die Reibungskrafte waren jedoch insgesamt hoher als unter UHV-Bedingungen. Aufgrund der anderen Grenzflacheneigenschaften wurde verglichen zu den UHV-Bedingungen ein unterschiedliches Verschleisverhalten als auch eine andere Randzonengefugeentwicklung gefunden. read less NOT USED (low confidence) L. Reinaudi, C. Negre, and M. C. Giménez, “Monte Carlo simulations for understanding the transport properties of metallic nanowires,” Physica E-low-dimensional Systems & Nanostructures. 2020. link Times cited: 1 NOT USED (low confidence) J. Li, H.-W. Chen, Q. Fang, C. Jiang, Y. Liu, and P. Liaw, “Unraveling the dislocation–precipitate interactions in high-entropy alloys,” International Journal of Plasticity. 2020. link Times cited: 68 NOT USED (low confidence) E. Bird and Z. Liang, “Maximum evaporating flux of molecular fluids from a planar liquid surface.,” Physical review. E. 2020. link Times cited: 6 Abstract: In this work, we use the kinetic theory of gases (KTG) to de… read moreAbstract: In this work, we use the kinetic theory of gases (KTG) to develop a theoretical model to understand the role of internal motions of molecules on the maximum evaporation flux from a planar liquid surface. The kinetic theory is applied to study the evaporation of molecular fluids into a vacuum and predict the dimensionless maximum evaporation flux (J_{R,max}, i.e., the ratio of the maximum evaporation flux to the molar flux emitted from a liquid surface). The key assumptions regarding the velocity distribution function (VDF) of polyatomic molecules in the highly nonequilibrium vapor near the evaporating surface are validated by the VDF obtained directly from molecular dynamics (MD) simulations. Our KTG-based analysis shows that J_{R,max} is affected by the specific heat (c_{V,int}) associated with internal degrees of freedom of fluid molecules. When the maximum evaporation flux is reached, the isotropic evaporating vapor far from the liquid surface moves at its speed of sound regardless of whether it is a monatomic vapor or polyatomic vapor. To fundamentally understand the evaporation of a molecular fluid into a vacuum, we solve the Boltzmann transport equation (BTE) to obtain the temperature, density, and flow speed distributions in the highly nonequilibrium evaporating vapor flow. Our BTE solutions indicate that there are several universal features of the evaporating vapor when the maximum evaporation flux occurs. In particular, we find that the evaporating vapor flow speed reaches the maximum value of sqrt[1.5] times the most probable thermal speed in the vapor flow direction at the vacuum boundary, and this maximum value is independent of fluid properties. All theoretical predictions in this work are verified by the MD simulation results of the evaporation of the model liquid Ar and the model liquid n-dodecane into a vacuum, and existing experimental data. read less NOT USED (low confidence) L. Stepanova and S. Bronnikov, “A computational study of the mixed–mode crack behavior by molecular dynamics method and the multi – Parameter crack field description of classical fracture mechanics,” Theoretical and Applied Fracture Mechanics. 2020. link Times cited: 10 NOT USED (low confidence) A. V. Yakutovich et al., “AiiDAlab – an ecosystem for developing, executing, and sharing scientific workflows,” Computational Materials Science. 2020. link Times cited: 33 NOT USED (low confidence) M. S. Nitol, S. Adibi, C. Barrett, and J. Wilkerson, “Solid solution softening in dislocation-starved Mg–Al alloys,” Mechanics of Materials. 2020. link Times cited: 15 NOT USED (low confidence) J. Chapman and R. Ramprasad, “Nanoscale Modeling of Surface Phenomena in Aluminum Using Machine Learning Force Fields,” The Journal of Physical Chemistry C. 2020. link Times cited: 7 Abstract: The study of nano-scale surface phenomena is essential in un… read moreAbstract: The study of nano-scale surface phenomena is essential in understanding the physical processes that aid in technologically relevant applications, such as catalysis, material growth, and failure nuc... read less NOT USED (low confidence) D. Benz, “Modifying TiO2 Nanoparticles by Atomic Layer Deposition for Enhanced Photocatalytic Water Purification.” 2020. link Times cited: 0 Abstract: Photocatalysts, contrary to conventional catalysts, utilize … read moreAbstract: Photocatalysts, contrary to conventional catalysts, utilize light to drive a reaction. By absorption of photons, electrons are excited and reach higher states to initiate a redox reaction. This principle can be broadly used to produce chemicals such as hydrogen via the reduction of water or transform carbon dioxide into solar fuels, i.e., methane, methanol, and formaldehyde. Besides chemical production, the reductive/oxidative potential of the excited electrons/holes can be utilized to degrade molecules. This is especially useful for self-cleaning materials or for cleaning waste water streams from molecules such as dyes, pesticides, or pharmaceuticals, as water pollution levels increasingly harm human health, especially in developing countries. Despite the great potential and vast research activities over the last decades, the development of cheap and efficient photocatalysts and especially the evaluation of the mechanism of photocatalytic degradation pathways are still lacking, which hampers the translation from lab to application. TiO2 (P25) is a cheap and non-toxic photoactive material, yet too inefficient for water cleaning. This thesis focuses on the surface modification of TiO2 (P25), minimizing various drawbacks of TiO2 (P25) and on the evaluation of the photocatalytic mechanisms. Atomic layer deposition (ALD) allows us to precisely control the deposition of many different materials at the atomic level, an advantage that could improve the photocatalyst design and optimize the activity. read less NOT USED (low confidence) O. Bachurina and A. Kudreyko, “Two-dimensional discrete breathers in fcc metals,” Computational Materials Science. 2020. link Times cited: 15 NOT USED (low confidence) Y. Yao, Q. Huang, and S. Wang, “Effects of porosity and pore microstructure on the mechanical behavior of nanoporous silver,” Materials today communications. 2020. link Times cited: 11 NOT USED (low confidence) K. K. Kammara and R. Kumar, “Development of empirical relationships for surface accommodation coefficients through investigation of nano-poiseuille flows using molecular dynamics method,” Microfluidics and Nanofluidics. 2020. link Times cited: 8 NOT USED (low confidence) L. Cao, Z. Zeng, and F. Fan, “Effect of lattice defects on the plastic Poisson’s ratio of nanoporous gold,” Scripta Materialia. 2020. link Times cited: 2 NOT USED (low confidence) M. Kappeler, A. Marusczyk, and B. Ziebarth, “Simulation of nickel surfaces using ab-initio and empirical methods,” Materialia. 2020. link Times cited: 2 NOT USED (low confidence) W. Xie and F. Fang, “Rake angle effect in cutting-based single atomic layer removal,” Journal of Manufacturing Processes. 2020. link Times cited: 13 NOT USED (low confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “Prediction of tool wear constants for diamond turn machining of CuBe,” Journal of Micromanufacturing. 2020. link Times cited: 3 Abstract: While several studies in diamond turning of homogeneous mate… read moreAbstract: While several studies in diamond turning of homogeneous materials like Cu, Al, and Si are well published, there is a lack of understanding about tool wear in case of heterogeneous materials like CuBe. Severity of the tool wear can be understood from the magnitude of the wear coefficients, and the magnitude of these coefficients is influenced by the wear mechanism. Hence, this study is aimed to calculate the wear coefficients from a known tool wear model in diamond turn machining of CuBe. Molecular dynamics simulation (MDS) results show that stress and temperature are responsible for increasing rate of tool wear. From the experimental results, change in the tool cutting edge radius due to wear was obtained and the temperature and stress for various a/r were found out using MDS. With these data, the wear coefficients, A & B, from a wear model for diamond turning were calculated. This methodology of using MDS to obtain stress and temperature for various a/r wherein, values of r are obtained from a single machining trial on actual material, will be useful for calculating the wear coefficients for the combination of single crystal diamond tool with various work piece materials and their activation energies. read less NOT USED (low confidence) M. Porterfield and D. Borca-Tasciuc, “Molecular Dynamics Simulation of Ultra-Fast Phase Transition in Water Nanofilms,” Journal of Heat Transfer. 2020. link Times cited: 1 Abstract:
Molecular dynamics simulations are used to explore explosi… read moreAbstract:
Molecular dynamics simulations are used to explore explosive boiling of thin water films on a gold substrate. In particular, water films of 0.7, 1.6, and 2.5 nanometer thickness were examined. Three different surface wettabilities with contact angles of 11 deg, 47 deg, and 110 deg were simulated along with substrate temperatures of 400 K, 600 K, 800 K, and 1000 K. The 11 and 47 deg contact angles were obtained using a Morse interaction potential between the water film and gold substrate while the 47 and 110 deg contact angles were obtained via a Lennard-Jones potential. Evaporation was the first mode of phase change observed in all cases and explosive boiling did not occur until the substrate reached a temperature of 800 K. When explosive boiling was present for all three contact angles, it was consistently shown to occur first for the surface with a 47 deg contact angle and Lennard-Jones potential. These results suggest that explosive boiling onset is strongly dependent on the particularities of the interaction potential. For instance, the Morse potential is smoother when compared to the Lennard-Jones potential, but has more interaction sites per molecule—two hydrogen atoms and one oxygen atom versus one oxygen atom. Thus, even when the water film reaches a higher temperature with the Morse potential, explosive boiling onset is delayed as more interaction sites have to be disrupted. These results suggest that contact angle alone is insufficient and both the interaction strength and the number of atoms interacting at the interface must be considered when investigating trends of explosive boiling with surface wettability. read less NOT USED (low confidence) S. Lee et al., “Dynamic metal-polymer interaction for the design of chemoselective and long-lived hydrogenation catalysts,” Science Advances. 2020. link Times cited: 24 Abstract: Dynamic interaction between Pd and polyphenylene sulfide ena… read moreAbstract: Dynamic interaction between Pd and polyphenylene sulfide enables the design of selective and long-lived hydrogenation catalyst. Metal catalysts are generally supported on hard inorganic materials because of their high thermochemical stabilities. Here, we support Pd catalysts on a thermochemically stable but “soft” engineering plastic, polyphenylene sulfide (PPS), for acetylene partial hydrogenation. Near the glass transition temperature (~353 K), the mobile PPS chains cover the entire surface of Pd particles via strong metal-polymer interactions. The Pd-PPS interface enables H2 activation only in the presence of acetylene that has a strong binding affinity to Pd and thus can disturb the Pd-PPS interface. Once acetylene is hydrogenated to weakly binding ethylene, re-adsorption of PPS on the Pd surface repels ethylene before it is further hydrogenated to ethane. The Pd-PPS interaction enables selective partial hydrogenation of acetylene to ethylene even in an ethylene-rich stream and suppresses catalyst deactivation due to coke formation. The results manifest the unique possibility of harnessing dynamic metal-polymer interaction for designing chemoselective and long-lived catalysts. read less NOT USED (low confidence) S. Ghahremanian, A. Abbassi, Z. Mansoori, and D. Toghraie, “Molecular dynamics simulation of annular condensation of vapor argon through a nanochannel for different saturation conditions with focusing on the flow and heat transfer,” International Communications in Heat and Mass Transfer. 2020. link Times cited: 10 NOT USED (low confidence) M. Settem, “Novel structural motifs: Chiral AgCu nanoalloys with chiral Cu core,” Journal of Alloys and Compounds. 2020. link Times cited: 5 NOT USED (low confidence) J. Yang and K. Komvopoulos, “A stress analysis method for molecular dynamics systems,” International Journal of Solids and Structures. 2020. link Times cited: 16 NOT USED (low confidence) Z. Yang, H. Gong, and F. Li, “Formation of Pt-enriched area in acidic media at the surface of Pt-Pd-Cu nanoparticles for electro-catalysis,” Catalysis Today. 2020. link Times cited: 2 NOT USED (low confidence) F. Shuang, “Accelerated Molecular Statics Based on Atomic Inertia Effect,” Communications in Computational Physics. 2020. link Times cited: 1 Abstract: Molecular statics (MS) based on energy minimization serves a… read moreAbstract: Molecular statics (MS) based on energy minimization serves as a useful simulation technique to study mechanical behaviors and structures at atomic level. The efficiency of MS, however, still remains a challenge due to the complexity of mathematical optimization in large dimensions. In this paper, the Inertia Accelerated Molecular Statics (IAMS) method is proposed to improve computational efficiency in MS simulations. The core idea of IAMS is to let atoms move to meta positions very close to their final equilibrium positions before minimization starts at a specific loading step. It is done by self-learning from historical movements (atomic inertia effect) without knowledge of external loadings. Examples with various configurations and loading conditions indicate that IAMS can effectively improve efficiency without loss of fidelity. In the simulation of three-point bending of nanopillar, IAMS shows efficiency improvement of up to 23 times in comparison with original MS. Particularly, the sizeindependent efficiency improvement makes IAMS more attractive for large-scale simulations. As a simple yet efficient method, IAMS also sheds light on improving the efficiency of other energy minimization-based methods. AMS subject classifications: 74G65, 74A25, 74S30 read less NOT USED (low confidence) M. F.-F. Xie, M. D.-Q. Wang, Y.-B. Wang, Y.-R. Yang, and X.-dong Wang, “Coalescence-induced jumping of nanodroplets on mixed-wettability superhydrophobic surfaces,” Canadian Journal of Physics. 2020. link Times cited: 4 Abstract: Coalescence-induced droplet jumping on superhydrophobic surf… read moreAbstract: Coalescence-induced droplet jumping on superhydrophobic surfaces has been observed at microscale and even nanoscale. The enhancement in jumping velocity of coalescing droplets is crucial for conden... read less NOT USED (low confidence) R. Otani, S. Kiyohara, K. Shibata, and T. Mizoguchi, “Prediction of interface and vacancy segregation energies at silver interfaces without determining interface structures,” Applied Physics Express. 2020. link Times cited: 3 Abstract: Interfaces play a crucial role in determining the functional… read moreAbstract: Interfaces play a crucial role in determining the functional and mechanical properties of materials. However, predicting interface properties is not straightforward because the atomic arrangements at the interface are different from those in the bulk. Hence, in this study, we discovered a descriptor from the bulk that helps predict the interface properties without the need to determine the interface structure. The descriptors related to the angle of elevation effectively described the structure units on both the bulk surface and optimized interface. Our model successfully predicted the interface and vacancy segregation energies at silver interfaces without using the interface structure. read less NOT USED (low confidence) X. Zhang et al., “Temperature dependent vacancy formation energy of metallic materials,” Physica B-condensed Matter. 2020. link Times cited: 4 NOT USED (low confidence) S. Hernandez, F. Freibert, B. Uberuaga, and J. Wills, “Role of electronic and magnetic interactions in defect formation and anomalous diffusion in δ-Pu,” Journal of Nuclear Materials. 2020. link Times cited: 6 NOT USED (low confidence) S. Ajori, H. Parsapour, and R. Ansari, “A comprehensive analysis of the mechanical properties and fracture analysis of metallic glass nanocomposites reinforced by carbon nanotubes and Cu nanowires: A molecular dynamics study,” Mechanics of Advanced Materials and Structures. 2020. link Times cited: 9 Abstract: Reinforced by nanowires (NWs), carbon nanotubes (CNTs) and N… read moreAbstract: Reinforced by nanowires (NWs), carbon nanotubes (CNTs) and NW encapsulated CNT (NW@CNT), tensile behavior of various types of Cu-Zr based metallic glass (MG) nanocomposites are studied using molecular dynamics (MD) simulations. It is observed that pure two-toms alloy MG and the one reinforced with bigger CNT demonstrates higher tensile properties than other types of MGs. Further, it is observed that the ultimate strength of reinforced MGs with individual CNTs is slightly higher than that of NW@CNT reinforced analogous. In this case, it is noticed that reinforced three-atoms Cu-Zr MG nanocomposites including Ti atoms demonstrate the highest ultimate strength and strain. read less NOT USED (low confidence) M. Bagheripoor and R. Klassen, “The effect of crystal anisotropy and pre-existing defects on the incipient plasticity of FCC single crystals during nanoindentation,” Mechanics of Materials. 2020. link Times cited: 22 NOT USED (low confidence) Z. Lin et al., “Ternary heterogeneous Pt-Ni-Au nanowires with enhanced activity and stability for PEMFCs.,” Chemical communications. 2020. link Times cited: 12 Abstract: Heterogeneous ternary Pt-Ni-Au nanowires (NWs) with randomly… read moreAbstract: Heterogeneous ternary Pt-Ni-Au nanowires (NWs) with randomly distributed Pt-Ni and Pt-Au micro phases were successfully synthesized following an oriented attachment mechanism. The as-prepared NWs exhibit enhanced activity and durability in both a rotating disk electrode (RDE) and single-cell, originating from the one dimensional (1D) heterogeneous structure. read less NOT USED (low confidence) H. Mes-adi, Y. Lachtioui, K. Saadouni, and M. Mazroui, “Morphology and surface properties of Cu thin film on Si (001),” Thin Solid Films. 2020. link Times cited: 12 NOT USED (low confidence) J. Li, Y. Zhang, C. Tian, H. Zhou, G. Hu, and R. Xia, “Structurally ordered nanoporous Pt–Co alloys with enhanced mechanical behaviors in tension,” Microporous and Mesoporous Materials. 2020. link Times cited: 18 NOT USED (low confidence) J. Chen et al., “Heating-Rate and Particle-Size Effects on Melting Process of Au Nanoparticles,” The Journal of Physical Chemistry C. 2020. link Times cited: 11 Abstract: The atomic mechanisms of the melting process of gold nanopar… read moreAbstract: The atomic mechanisms of the melting process of gold nanoparticles were investigated by molecular dynamics simulations. The melting under high heating rate is found to be much different from the ne... read less NOT USED (low confidence) G. Pilania, P. Balachandran, J. Gubernatis, and T. Lookman, “Data-Based Methods for Materials Design and Discovery: Basic Ideas and General Methods.” 2020. link Times cited: 11 Abstract: Machine learning methods are changing the way we design and … read moreAbstract: Machine learning methods are changing the way we design and discover new materials. This book provides an overview of approaches successfully used in addressing materials problems (alloys,... read less NOT USED (low confidence) F. Ning and W. Cong, “Ultrasonic vibration-assisted (UV-A) manufacturing processes: State of the art and future perspectives,” Journal of Manufacturing Processes. 2020. link Times cited: 93 NOT USED (low confidence) A.-V. Pham, T. Fang, A.-S. Tran, and T.-H. Chen, “Effect of annealing and deposition of Cu atoms on Ni trench to interface formation and growth mechanisms of Cu coating,” Superlattices and Microstructures. 2020. link Times cited: 8 NOT USED (low confidence) Z. Liang, A. Chandra, E. Bird, and P. Keblinski, “A molecular dynamics study of transient evaporation and condensation,” International Journal of Heat and Mass Transfer. 2020. link Times cited: 22 NOT USED (low confidence) J. Chapman, R. Batra, and R. Ramprasad, “Machine learning models for the prediction of energy, forces, and stresses for Platinum,” Computational Materials Science. 2020. link Times cited: 18 NOT USED (low confidence) N. Beets, J. Stuckner, M. Murayama, and D. Farkas, “Fracture in nanoporous gold: An integrated computational and experimental study,” Acta Materialia. 2020. link Times cited: 21 NOT USED (low confidence) W. Xie and F. Fang, “Mechanism of atomic and close-to-atomic scale cutting of monocrystalline copper,” Applied Surface Science. 2020. link Times cited: 21 NOT USED (low confidence) Q. Ye et al., “Theoretical development and experimental validation on the measurement of temperature by extended X-ray absorption fine structure.,” Journal of synchrotron radiation. 2020. link Times cited: 1 Abstract: A systematic investigation on the theoretical framework of t… read moreAbstract: A systematic investigation on the theoretical framework of the ultra-fast measurement of temperature by extended X-ray absorption fine structure (EXAFS) applied in laser-driven-compression experiments has been carried out and a new temperature measurement scheme based on the EXAFS cumulant expansion analysis and anharmonic correlated Debye model has been advanced. By considering the anharmonic effect of thermal vibration and avoiding the employment of the empirical model as well as parameters which have large inherent uncertainties in the temperature determination, this new scheme is theoretically more accurate than traditional ones. Then the performance of the new measurement scheme and traditional methods were validated on a synchrotron radiation platform by temperature-dependent EXAFS (TDEXAFS) experiments on Au, Fe, V and Ti; the results showed that the new scheme could provide the most accurate measured temperatures with much lower uncertainties. This accurate scheme gives a firmer physical ground to the EXAFS temperature measurement technique and can expect to be applied in laser-driven compression experiments and promote the development of matter state research at extreme conditions. read less NOT USED (low confidence) Y. Fan, Y. Xiang, and H. S. Shen, “Temperature-Dependent Mechanical Properties of Graphene/Cu Nanocomposites with In-Plane Negative Poisson’s Ratios,” Research. 2020. link Times cited: 35 Abstract: Negative Poisson's ratio (NPR), also known as “auxetic”… read moreAbstract: Negative Poisson's ratio (NPR), also known as “auxetic”, is a highly desired property in a wide range of future industry applications. By employing molecular dynamics (MD) simulation, metal matrix nanocomposites reinforced by graphene sheets are studied in this paper. In the simulation, single crystal copper with crystal orientation [1 1 0] is selected as the matrix and an embedded-atom method (EAM) potential is used to describe the interaction of copper atoms. An aligned graphene sheet is selected as reinforcement, and a hybrid potential, namely, the Erhart-Albe potential, is used for the interaction between a pair of carbon atoms. The interaction between the carbon atom and copper atom is approximated by the Lennard-Jones (L-J) potential. The simulation results showed that both graphene and copper matrix possess in-plane NPRs. The temperature-dependent mechanical properties of graphene/copper nanocomposites with in-plane NPRs are obtained for the first time. read less NOT USED (low confidence) A. E. Korenchenko, A. Vorontsov, and A. Zhukova, “Macroscopic Model of Nucleation during the Condensation of Copper Vapor in an Inert Gas,” Russian Metallurgy (Metally). 2020. link Times cited: 2 NOT USED (low confidence) A. Sharma, S. Joshi, D. Datta, and R. Balasubramaniam, “Investigation of Tool and Workpiece Interaction on Surface Quality While Diamond Turning of Copper Beryllium Alloy,” Journal of Manufacturing Science and Engineering-transactions of The Asme. 2020. link Times cited: 9 Abstract:
Among all the materials, diamond turning of heterogeneous … read moreAbstract:
Among all the materials, diamond turning of heterogeneous materials like copper beryllium (CuBe) poses serious machining challenges as the heterogeneity in the workpiece affects the quality of generated surface. Therefore, the present study is aimed to understand the effect of tool–workpiece interactions on the surface characteristics of heterogeneous CuBe workpiece material. Experiments and molecular dynamics simulation (MDS) were carried out to analyze the various surface and subsurface interactions during cutting. Results from the experiments on both the materials for whole cutting length show that the average roughness values on CuBe-machined surface are found to be ∼48% higher than those of copper (Cu). Scanning electron microscopy (SEM) results show that while deterministic lay pattern is obtained in the case of Cu, the CuBe-machined surface possesses near-random lay pattern, which is also reflected by the fast Fourier transform (FFT) spectrum of surface roughness profiles. Experimental and MDS results reveal that the hard precipitate suffers cracks which propagate vertically as well as radially and as the tool travels from Cu-rich phase to Be-rich phase, ductile to brittle transition in cutting mechanism is observed. Furthermore, it is observed that diamond-turned Cu and CuBe surfaces are contaminated by the oxides of C and Cu. MDS results verify the mechanisms involved in the surface and subsurface interactions during diamond turning. read less NOT USED (low confidence) Z. Weng, F. Zhang, C. Xu, and J. Zhou, “The effect of incident energy, incident angle and substrate temperature on surface morphology and atomic distribution of NiTi films,” Materials & Design. 2020. link Times cited: 11 NOT USED (low confidence) T. Krauss and S. Eich, “Development of a segregation model beyond McLean based on atomistic simulations,” Acta Materialia. 2020. link Times cited: 6 NOT USED (low confidence) P. Valerius et al., “Reversible crystalline-to-amorphous phase transformation in monolayer MoS2 under grazing ion irradiation,” 2D Materials. 2020. link Times cited: 13 Abstract: By combining scanning tunneling microscopy, low-energy elect… read moreAbstract: By combining scanning tunneling microscopy, low-energy electron diffraction, photoluminescence and Raman spectroscopy experiments with molecular dynamics simulations, a comprehensive picture of the structural and electronic response of a monolayer of MoS2 to 500 eV Xe+ irradiation is obtained. The MoS2 layer is epitaxially grown on graphene/Ir(1 1 1) and analyzed before and after irradiation in situ under ultra-high vacuum conditions. Through optimized irradiation conditions using low-energy ions with grazing trajectories, amorphization of the monolayer is induced already at low ion fluences of ions cm−2 and without inducing damage underneath the MoS2 layer. The crystalline-to-amorphous transformation is accompanied by changes in the electronic properties from semiconductor-to-metal and an extinction of photoluminescence. Upon thermal annealing, the re-crystallization occurs with restoration of the semiconducting properties, but residual defects prevent the recovery of photoluminescence. read less NOT USED (low confidence) D. Nguyen-Trong and P. Nguyen-Tri, “Factors affecting the structure, phase transition and crystallization process of AlNi nanoparticles,” Journal of Alloys and Compounds. 2020. link Times cited: 27 NOT USED (low confidence) B. R. S. Kouamé et al., “Insights on the unique electro-catalytic behavior of PtBi/C materials,” Electrochimica Acta. 2020. link Times cited: 15 NOT USED (low confidence) S. Wang, S. Wang, Y.-R. Yang, X.-dong Wang, and D.-J. Lee, “High-temperature reactive wetting systems: Role of lattice constant,” Chemical Engineering Science. 2019. link Times cited: 12 NOT USED (low confidence) W. Liu et al., “Molecular dynamics simulation studies of displacement cascade induced defects in gold nanotubes,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2019. link Times cited: 0 NOT USED (low confidence) M. Mirakhory, M. M. Khatibi, and T. Rabczuk, “Attenuation of nanoparticle mass detection by single layer graphene sheets via mono-vacancy and plate geometry,” Diamond and Related Materials. 2019. link Times cited: 2 NOT USED (low confidence) N. Sdobnyakov et al., “Simulation of phase transformations in titanium nanoalloy at different cooling rates,” Materials Chemistry and Physics. 2019. link Times cited: 11 NOT USED (low confidence) R. Fahdiran et al., “Melting of gold nanoparticle: study on structural evolution,” Journal of Physics: Conference Series. 2019. link Times cited: 3 Abstract: We investigate the structural evolution on melting of Gold n… read moreAbstract: We investigate the structural evolution on melting of Gold nanoparticle due to heat treatment from room temperature up to slightly above melting point. The evolution is followed using Molecular Dynamics (MD) simulation. Structure factor and pair distribution function calculation indicated that the system is melted at the end of simulation. Common Neighbour Analysis (CNA) method shows the local lattice structure transformation confirming phase transition. read less NOT USED (low confidence) B. Sharma and J. Myung, “Pd-based ternary alloys used for gas sensing applications: A review,” International Journal of Hydrogen Energy. 2019. link Times cited: 26 NOT USED (low confidence) A. Sobolev, O. Golovnia, and A. Popov, “Embedded atom potential for Sm–Co compounds obtained by force-matching,” Journal of Magnetism and Magnetic Materials. 2019. link Times cited: 3 NOT USED (low confidence) H. Haowei, L. Qin, L. Shuang, and T. Fang, “Molecular dynamics study on water vapor condensation and infiltration characteristics in nanopores with tunable wettability,” Applied Surface Science. 2019. link Times cited: 14 NOT USED (low confidence) R. Marimpul, T. Winata, F. A. Noor, I. Syuhada, and A. Rosikhin, “Molecular dynamics simulation of platinum film growth based on thermal evaporation method,” IOP Conference Series: Materials Science and Engineering. 2019. link Times cited: 0 Abstract: Platinum film growth using thermal evaporation method was st… read moreAbstract: Platinum film growth using thermal evaporation method was studied using molecular dynamics simulation. This platinum film was intended as catalyst film for graphene growth. Tersoff, Eam and Lennard-Jones potential were used to describe interaction of Si-Si, Pt-Pt and Pt-Si respectively. Deposition process was performed with low incident energy to represent thermal evaporation method. Our simulation found that heating temperature at 400 K produced platinum film with higher percentage of crystal structure than other heating condition 300K, 500K & 600K. We also found transition phase from fcc to bcc at 600K. read less NOT USED (low confidence) V. Tikare, R. Devanathan, and M. Caturla, “List of Authors,” 2019 Panhellenic Conference on Electronics & Telecommunications (PACET). 2019. link Times cited: 0 NOT USED (low confidence) L. Zhang, Q. Li, S. Tian, and G.-W. Hong, “Molecular Dynamics Simulation of the Cu/Au Nanoparticle Alloying Process,” Journal of Nanomaterials. 2019. link Times cited: 8 Abstract: Sintering is an important approach for the alloying of diffe… read moreAbstract: Sintering is an important approach for the alloying of different metals, which is affected by factors such as temperature, grain size, and material properties. And it represents a complex thermodynamic process. This paper had adopted the molecular dynamics methods to investigate the evolution process of nanostructure during the sintering of Cu and Au nanoparticles. The changes in crystalline during the nanosintering process were observed, and the radial distribution function of atoms, the shrinkage ratio, and the sintering neck of the systems were discussed. The initial sintering temperature and melting temperature of the system were obtained; at the same time, the characteristics of the sintering neck with changes in temperature during the nanosintering process were revealed. read less NOT USED (low confidence) S. Ding and X.-qiang Wang, “A systematic study on the MEAM interatomic potentials of the transition metal nitrides TMNs (TM=Ti, V, Cr, Fe) binary systems,” Journal of Alloys and Compounds. 2019. link Times cited: 10 NOT USED (low confidence) J. Méndez and M. Ponga, “MXE: A package for simulating long-term diffusive mass transport phenomena in nanoscale systems,” Comput. Phys. Commun. 2019. link Times cited: 8 NOT USED (low confidence) E. Bird and Z. Liang, “Transport phenomena in the Knudsen layer near an evaporating surface.,” Physical review. E. 2019. link Times cited: 22 Abstract: Using the combination of the kinetic theory of gases (KTG), … read moreAbstract: Using the combination of the kinetic theory of gases (KTG), Boltzmann transport equation (BTE), and molecular dynamics (MD) simulations, we study the transport phenomena in the Knudsen layer near a planar evaporating surface. The MD simulation is first used to validate the assumption regarding the anisotropic velocity distribution of vapor molecules in the Knudsen layer. Based on this assumption, we use the KTG to formulate the temperature and density of vapor at the evaporating surface as a function of the evaporation rate and the mass accommodation coefficient (MAC), and we use these vapor properties as the boundary conditions to find the solution to the BTE for the anisotropic vapor flow in the Knudsen layer. From the study of the evaporation into a vacuum, we show the ratio of the macroscopic speed of vapor to the most probable thermal speed of vapor molecules in the flow direction will always reach the maximum value of sqrt[1.5] at the vacuum boundary. The BTE solutions predict that the maximum evaporation flux from a liquid surface at a given temperature depends on both the MAC and the distance between the evaporating surface and the vacuum boundary. From the study of the evaporation and condensation between two parallel plates, we show the BTE solutions give good predictions of transport phenomena in both the anisotropic vapor flow within the Knudsen layer and the isotropic flow out of the Knudsen layer. All the predictions from the BTE are verified by the MD simulation results. read less NOT USED (low confidence) I. I. Fairushin, A. Saifutdinov, A. O. Sofronitskiy, B. Timerkaev, and G. Dautov, “Development of plasma reactor design for synthesis of copper nanoparticles using multi-scale simulation,” Journal of Physics: Conference Series. 2019. link Times cited: 11 Abstract: On the basis of the hybrid hydrodynamic model for the gas di… read moreAbstract: On the basis of the hybrid hydrodynamic model for the gas discharge, numerical experiments were performed. The main parameters of the electric discharge of a direct current with copper cathode and anode were obtained. The obtained data were used as the main conditions for molecular-dynamics simulations of the copper vapor nucleation process in the argon. The results of the simulation formed the basis for the development of an experimental setup for the plasma synthesis of copper nanoparticles. read less NOT USED (low confidence) S. Dang, C. Li, and P. Han, “Synergetic effects of impurities and alloying element Cr on oxidation and dissolution corrosion of Ni (111) surfaces: A DFT study,” Chinese Journal of Physics. 2019. link Times cited: 4 NOT USED (low confidence) L. Zhu, X. Zhang, J. Zhou, and Z. Sun, “Interfacial graphene modulated energetic behavior of the point-defect at the Au/HfO2 interface,” Applied Surface Science. 2019. link Times cited: 3 NOT USED (low confidence) F. Yang et al., “Mechanochemical Effects of Adsorbates at Nanoelectromechanical Switch Contacts.,” ACS applied materials & interfaces. 2019. link Times cited: 4 Abstract: Herein, classical molecular dynamics simulations are used to… read moreAbstract: Herein, classical molecular dynamics simulations are used to examine nanoscale adsorbate reactions during the cyclic opening and closing of nanoelectromechanical systems (NEMS) switches. We focus upon how reactions change metal/metal conductive contact area, asperity morphology, and plastic deformation. We specifically consider Pt, which is often used as an electrode material for NEMS switches. The structural evolution of asperity contacts in gaseous environments with molecules which can potentially form tribopolymers is determined by various factors, e.g., contact forces, partial pressure and molecular weight of gas, and the fundamental reaction rates of surface adsorption and adsorbate linkage. The modeled systems exhibit significant changes during the first few cycles, but as the number of contact cycles increases, the system finds a steady-state where the morphologies, Pt/Pt contact area, oligomer chain lengths, amount of Pt transfer between opposing surfaces, and deformation rate stabilize. The stress generated during asperity contact increases the rate of reactions amongst the adsorbates in the contact region. This makes the size of the adsorbate molecules increase, and thus more exposed metal, which implies higher electrical conductance in the closed contact, but more plastic deformation, metal-metal transfer, and mechanical work expended in each contact cycle. read less NOT USED (low confidence) H. Yang, Y. Drossinos, and C. J. Hogan, “Excess thermal energy and latent heat in nanocluster collisional growth.,” The Journal of chemical physics. 2019. link Times cited: 19 Abstract: Nanoclusters can form and grow by nanocluster-monomer collis… read moreAbstract: Nanoclusters can form and grow by nanocluster-monomer collisions (condensation) and nanocluster-nanocluster collisions (coagulation). During growth, product nanoclusters have elevated thermal energies due to potential and thermal energy exchange following a collision. Even though nanocluster collisional heating may be significant and strongly size dependent, no prior theory describes this phenomenon for collisions of finite-size clusters. We derive a model to describe the excess thermal energy of collisional growth, defined as the kinetic energy increase in the product cluster, and latent heat of collisional growth, defined as the heat released to the background upon thermalization of the nonequilibrium cluster. Both quantities are composed of a temperature-independent term related to potential energy minimum differences and a size- and temperature-dependent term, which hinges upon heat capacity and energy partitioning. Example calculations using gold nanoclusters demonstrate that collisional heating can be important and strongly size dependent, particularly for reactive collisions involving nanoclusters composed of 14-20 atoms. Excessive latent heat release may have considerable implications in cluster formation and growth. read less NOT USED (low confidence) K. Lee, D. Yoo, W. Jeong, and S. Han, “SIMPLE-NN: An efficient package for training and executing neural-network interatomic potentials,” Comput. Phys. Commun. 2019. link Times cited: 75 NOT USED (low confidence) A. Alian, Y. Ju, and S. Meguid, “Comprehensive atomistic modeling of copper nanowires-based surface connectors,” Materials & Design. 2019. link Times cited: 12 NOT USED (low confidence) O. Bachurina, R. Murzaev, and D. Bachurin, “Molecular dynamics study of two-dimensional discrete breather in nickel,” Journal of Micromechanics and Molecular Physics. 2019. link Times cited: 9 Abstract: A discrete breather (DB) is a spatially localized vibrationa… read moreAbstract: A discrete breather (DB) is a spatially localized vibrational mode of large amplitude in a defect-free anharmonic lattice. Generally, zero-dimensional DB is considered to be localized in all [Formula: see text] directions of the [Formula: see text]-dimensional lattice. However, the question of existence of DBs localized in [Formula: see text]–[Formula: see text] directions and delocalized in other [Formula: see text] directions remains open. In the present paper, for the first time, the case of [Formula: see text] and [Formula: see text] is considered by constructing a two-dimensional (2D) DB in the fcc nickel lattice using molecular dynamics methods. In order to excite such DB, one of the delocalized vibrational modes of the triangular lattice was used (the (111) plane in fcc crystal is a triangular lattice). All simulations were carried out at zero temperature. The investigated 2D DB demonstrates hard-type nonlinearity, when its oscillation frequency increases with increasing amplitude. The oscillation frequencies of the DB are above the upper edge of the phonon spectrum for nickel, which is 10.3[Formula: see text]THz. The maximum DB lifetime is found to be 9.5[Formula: see text]ps. The obtained results expand our understanding of diversity of nonlinear spatially localized vibrational modes in nonlinear lattices. read less NOT USED (low confidence) Z. Hu and X. Yu, “Controlling the chemical reactivity of nanostructured electrode materials by surface reactive sites,” Materials Research Express. 2019. link Times cited: 2 Abstract: To better control the chemical reactivity of nanostructured … read moreAbstract: To better control the chemical reactivity of nanostructured electrode materials, a surface reactive site model is proposed based on thermodynamic theory. Impressively, due to the quantum size effect, the close existence of the photoformed electron and hole pairs in nanocrystalline materials and their efficient contribution to the reaction, resulting in chemical activity much enhanced over that of bulk materials. The ratios of the surface vacancy formation energies of Au, Ag, and Cu (111) nanocrystal planes to the volume vacancy formation energies are all 0.25, much lower than the corresponding bulk values. Remarkably, the chemical reactivity of carbon nanotubes, TiO2 nanotubes, MgO nanoparticle, and ZnO nanoparticle can be attributed to the number of surface reactive sites. In addition, we have also found that stress and vacancies have similar effects, indicating that mechanical activation can effectively improve chemical reactivity. Our model is applicable to nanocrystalline materials of different shapes, dimensions, and crystal structures. read less NOT USED (low confidence) Y. Cui, Z. Chen, and Y. Ju, “Mass transfer and morphology change via dislocation emission in a macroporous FCC metal,” Materials Letters. 2019. link Times cited: 13 NOT USED (low confidence) Z.-qiang Zhang, X. Guo, H. Tang, J. Ding, Y.-G. Zheng, and S. Li, “Unidirectional self-driving liquid droplet transport on a monolayer graphene-covered textured substrate.,” ACS applied materials & interfaces. 2019. link Times cited: 29 Abstract: Controllable directional transport of liquid droplets on a f… read moreAbstract: Controllable directional transport of liquid droplets on a functionalized surface has been a challenge in the field of microfluidics since it does not require energy supply, and the physical mechanism of such self-driving transport exhibits extraordinary contribution to fundamental understanding of some biological processes and the design of microfluidic apparatus. In this paper, we report a novel design of surface microstructure that can realize unidirectional self-driving liquid mercury (Hg) droplet transport on a graphene-covered copper (Cu) substrate with three-dimensional surface microstructure. We have demonstrated that a liquid Hg droplet spontaneously propagates on a gradient Cu groove covered by a monolayer graphene without any external force fields. Classical molecular dynamics (MD) results provide a profound insight on self-driving process of Hg droplets. It shows that Hg droplets undergo acceleration, deceleration and return stages successively from the narrow to wide ends of the gradient groove. Intriguingly, Hg droplets can be transferred continuously and unidirectionally on the three-dimensional graphene-covered surface microstructure when they capture enough kinetic energy from the gradient groove to break the energy barrier at the step junctions between the two neighboring unit cells. The design of the zigzag textured surface covered by a monolayer graphene artfully uses the fact that the monolayer graphene can effectively reduce the droplet pinning on the textured surface, and meanwhile the textured surface can permeably interact with the droplets through the monolayer graphene. The findings reported here may open a door to explore the graphene-covered functional surface to directional transport of liquid droplets and provide an in-depth understanding of the self-driving mechanism for liquid droplets on graphene-covered textured substrates. read less NOT USED (low confidence) E. Cihan, H. Störmer, H. Leiste, M. Stüber, and M. Dienwiebel, “Low friction of metallic multilayers by formation of a shear-induced alloy,” Scientific Reports. 2019. link Times cited: 9 NOT USED (low confidence) C. Zhao et al., “2D-3D transformation of palladium and gold nanoparticles on functionalized Mo2C by multiscale simulation,” Applied Surface Science. 2019. link Times cited: 9 NOT USED (low confidence) Y. Han, K. C. Lai, A. Lii-Rosales, M. Tringides, J. Evans, and P. Thiel, “Surface energies, adhesion energies, and exfoliation energies relevant to copper-graphene and copper-graphite systems,” Surface Science. 2019. link Times cited: 58 NOT USED (low confidence) V. Cruz and J. Enrique., “Estudio de la inhomogeneidad elástica en vidrios metalicos en la mesoescala.” 2019. link Times cited: 0 Abstract: Metallic glasses are amorphous solids produced by rapid cool… read moreAbstract: Metallic glasses are amorphous solids produced by rapid cooling, with disordered atomic structure and lacking long-range order. This structural disorder makes them to show mechanical properties different from those observed in a crystalline solid.
Metallic glasses are ideally isotropic, but they can become anisotropic during the manufacturing process or as a result of non-homogeneous plastic deformation, creep, etc. Experimental studies showed remnant anisotropy in amorphous PdSiCuP under a homogeneous deformation regime at temperatures close to the glass transition (Tg). In this thesis we studied the remnant anisotropy induced by shear in two amorphous systems by using molecular dynamics simulation. The Cu13Ni34Pd53 system was chosen to approach the PdCuSiP compositions, while the Zr46Cu46Al8 system is an excellent metallic glass former.
Amorphous systems were obtained by fast cooling from the liquid and subsequent thermal annealing. Both systems were then sheared in the [100] direction at a deformation speed of 10^10 s^(-1), and returned to its original form at the same speed. Shearing simulations were performed in isothermal-isobaric conditions at different temperatures. The resulting states were examined using the directional pair distribution function (d-PDF), calculated from the distribution function of interatomic distances in planes perpendicular to the selected axis.
Remnant anisotropy was detected in both systems after a deformation-recovery cycle. Cu13Ni34Pd53 displays remnant anisotropy below the glass transition at 0 and 300 K after the shear process, and again at 700 K after the full deformation-recovery cycle.
The intensity of anisotropy in the d-PDF of Zr46Cu46Al8 is lower than in Cu13Ni34Pd53, possibly due to the different atomic radii. However, it is found in all the studied temperature range, with decreasing intensity with temperature.
Anisotropy is concentrated in the [110] planes and [1(-1)0], corresponding to the planes of maximum and minimum shear respectively. This result indicates that remnant anisotropy is highly directional and can go unnoticed if not searched for. It also states that the shear process is essentially symmetrical with respect to the plane [110].
The shear and recovery process induces the creation of free volume, falling into the category of rejuvenation processes discussed in the literature. Counterintuitively, the process of free volume creation is accompanied by a reduction of the distance of the first maximum of the pair distribution function g (r), indicating a decrease of the most likely distance (mode) between first neighbors. However, this decrease in mode is accompanied by an increase of the width of the first peak, increasing the standard deviation of the distribution of distances between first neighbors. This explains the apparent contradiction between the increase in the volume and reduction of the most probable distance between first neighbors. Simultaneously, the second coordination sphere is more homogenous after the deformation and recovery process. As a result, medium range order is increased.
The discrepancy between the results obtained between Cu13Ni34Pd53 and Zr46Cu46Al8 may be due to the fact that the potential used in the simulations of Zr46Cu46Al8 is more representative of the behavior of metallic glasses. As a result, we hypothesize that the presence of remnant anisotropy after mechanical deformation could be a general feature in metallic glasses.
The computational cost of this study was very high, since it was necessary to simulate million-atom systems to avoid that results were dependent on the size of the simulation box.
Los vidrios metálicos son sólidos amorfos producidos por enfriamiento rápido, con estructura atómica desordenada y sin orden de largo alcance. Este desorden estructural les proporciona propiedades mecánicas diferentes de las observadas en un sólido cristalino. Idealmente los vidrios metálicos son isotrópicos pero pueden convertirse en anisotrópicos durante el proceso de fabricación o como consecuencia de una deformación plástica no homogénea, fluencia, etc. Estudios experimentales mostraron anisotropía remanente en PdSiCuP amorfo bajo un régimen de deformación homogéneo a temperaturas cercanas a la transición vítrea (Tg). En esta tesis estudiamos la anisotropía remanente inducida por cizalla en dos sistemas amorfos mediante simulación por dinámica molecular. El sistema Cu13Ni34Pd53 se eligió intentando aproximarse a las composiciones PdCuSiP. Por otra parte, el sistema Zr46Cu46Al8 es un excelente formador de vidrios metálicos. Tras la obtención del sistema amorfo por enfriamiento rápido y equilibrado térmico ambos modelos fueron sometidos a deformación por cizalladura en la dirección [100], a una velocidad de deformación de 10^10 s^(-1), y retornados a su forma original con la misma velocidad, en condiciones isotérmicas-isobáricas a diferentes temperaturas. Los estados obtenidos fueron examinados utilizando la función de distribución par direccional (dPDF), calculada a partir de la distribución de distancias interatómicas en los planos perpendiculares al eje escogido. En ambos sistemas se detectó anisotropía remanente después de un ciclo deformación- recuperación. En Cu13Ni34Pd53 se observa anisotropía remanente por debajo de la transición vítrea en 0 y 300 K al final del proceso de cizalla, y nuevamente en el estado deformado a 700 K. En Zr46Cu46Al8, la intensidad de la anisotropía determinada en las d-PDF es menor que en Cu13Ni34Pd53, posiblemente debido a los radios atómicos, pero se observa en todo el rango de temperaturas estudiadas con intensidad decreciente con la temperatura. La anisotropía se concentra en los planos [110] y [1(-1)0], correspondientes a los planos de máxima y mínima cizalla respectivamente. Este resultado indica que la anisotropía remanente es altamente direccional y puede pasar desapercibida si no se busca específicamente. Asimismo, se observa que el proceso de cizalla es esencialmente simétrico con respecto al plano [1(-1)0]. El proceso de cizalla y recuperación induce la creación de volumen libre, entrando dentro de la categoría de los procesos de rejuvenecimiento analizados en la literatura. Contraintuitivamente, el proceso de creación de volumen libre va acompañando de una reducción de la distancia del primer máximo de la función de distribución de pares g(r), indicando una disminución de la distancia más probable (moda) entre primeros vecinos. Sin embargo, esta disminución de la moda va acompañada de un aumento de la anchura del primer pico, aumentando la desviación estándard de la distribución de distancias entre primeros vecinos. Esto explica la aparente contradicción entre el aumento del volumen y la reducción de la distancia más probable entre primeros vecinos. Simultáneamente, la segunda esfera de coordinación es más homogénea después del proceso de deformación y recuperación. En consecuencia, se produce un aumento del orden a media distancia. La discrepancia entre los resultados obtenidos entre Cu13Ni34Pd53 y Zr46Cu46Al8 puede deberse a que el potencial en Zr46Cu46Al8 es más representativo del comportamiento de los vidrios metálicos. Consecuentemente, la presencia de anisotropía remanente después de deformación mecánica podría ser una característica general en los vidrios metálicos. El coste computacional de este estudio ha sido muy alto, puesto que ha sido necesario simular sistemas con muchos átomos para evitar que los resultados obtenidos fuesen dependientes del tamaño de la caja de simulación utilizadas read less NOT USED (low confidence) I. Napier, V. Chang, T. Noakes, and N. Harrison, “From Electronic Structure to Design Principles for Photocathodes:
Cu
-
Ba
Alloys,” Physical Review Applied. 2019. link Times cited: 1 Abstract: Producing a metal photocathode with a low work function (WF)… read moreAbstract: Producing a metal photocathode with a low work function (WF), low emissivity, and high quantum efficiency is a matter of importance in the design of the next generation of free-electron laser facilities. General rules for the design of appropriate materials are currently unclear and difficult to elucidate from observations of structure-composition relationships of known photocathodes. In this work, high-quality density-functional-theory electronic structure calculations and a simple physical model are employed to develop design rules for photocathodes based on metallic alloys. A theoretical study of metal alloys for photocathode applications is presented, in which high WF, stable copper is paired with low WF, unstable barium in two alloys, Cu13Ba and CuBa. Surfaces terminating in a plane of Ba atoms have a lower computed surface energy than those terminating in Cu atoms due to surface segregation of the larger Ba atoms. This results in a significant surface dipole due to the interatomic charge transfer from the differences in electronegativity of the species. The details of the surface structure determine the direction of the dipole and thus have a strong influence on the computed WF. The computed WF of the Cu13Ba Ba-terminated (100) surface is even lower than that of pure Ba, at 1.95 eV. The computed quantum efficiency (QE) of the best-performing pure Cu surface is 5.86 × 10−6, whereas the best-performing Cu13Ba surface terminates in a plane of Ba atoms and has a significantly increased QE of 5.09 × 10−3. A surface terminating in two planes of Ba atoms, the (001) surface of CuBa, has an even higher computed QE of 1.38 × 10−2. read less NOT USED (low confidence) Y. Zuo et al., “A Performance and Cost Assessment of Machine Learning Interatomic Potentials.,” The journal of physical chemistry. A. 2019. link Times cited: 413 Abstract: Machine learning of the quantitative relationship between lo… read moreAbstract: Machine learning of the quantitative relationship between local environment descriptors and the potential energy surface of a system of atoms has emerged as a new frontier in the development of interatomic potentials (IAPs). Here, we present a comprehensive evaluation of ML-IAPs based on four local environment descriptors --- atom-centered symmetry functions (ACSF), smooth overlap of atomic positions (SOAP), the Spectral Neighbor Analysis Potential (SNAP) bispectrum components, and moment tensors --- using a diverse data set generated using high-throughput density functional theory (DFT) calculations. The data set comprising bcc (Li, Mo) and fcc (Cu, Ni) metals and diamond group IV semiconductors (Si, Ge) is chosen to span a range of crystal structures and bonding. All descriptors studied show excellent performance in predicting energies and forces far surpassing that of classical IAPs, as well as predicting properties such as elastic constants and phonon dispersion curves. We observe a general trade-off between accuracy and the degrees of freedom of each model, and consequently computational cost. We will discuss these trade-offs in the context of model selection for molecular dynamics and other applications. read less NOT USED (low confidence) Y. Chen, Y. Huang, T. Cheng, and W. Goddard, “Identifying Active Sites for CO2 Reduction on Dealloyed Gold Surfaces by Combining Machine Learning with Multiscale Simulations.,” Journal of the American Chemical Society. 2019. link Times cited: 85 Abstract: Gold nanoparticles (AuNPs) and dealloyed Au3Fe core-shell NP… read moreAbstract: Gold nanoparticles (AuNPs) and dealloyed Au3Fe core-shell NP surfaces have been shown to have dramatically improved performance in reducing CO2 to CO (CO2RR), but the surface features responsible for these improvements are not known. The active sites cannot be identified with surface science experiments, and quantum mechanics (QM) is not practical for the 10 000 surface sites of a 10 nm NP (200 000 bulk atoms). Here, we combine machine learning, multiscale simulations, and QM to predict the performance (a-value) of all 5000-10 000 surface sites on AuNPs and dealloyed Au surfaces. We then identify the optimal active sites for CO2RR on dealloyed gold surfaces with dramatically reduced computational effort. This approach provides a powerful tool to visualize the catalytic activity of the whole surface. Comparing the a-value with descriptors from experiment, computation, or theory should provide new ways to guide the design of high-performance electrocatalysts for applications in clean energy conversion. read less NOT USED (low confidence) A. Galashev and K. Ivanichkina, “Computational investigation of a promising Si-Cu anode material.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 21 Abstract: The lack of suitable anode materials is a limiting factor in… read moreAbstract: The lack of suitable anode materials is a limiting factor in the creation of a new generation of lithium-ion batteries. We use the molecular dynamics method to explore the processes of intercalation and deintercalation of lithium in the anode element, represented by two sheets of silicene, on a copper substrate. It is shown that the presence of vacancy-type defects in silicene increases the electrode capacitance, which becomes especially significant with bivacancies. However, the enlargement of defect sizes reduces the strength of the silicene channel during cycling and in the presence of hexavacancies it suffers a strong deformation and becomes impassable for Li+ ions during intercalation. The presence of a copper substrate greatly changes the electronic properties of silicene. The calculated DOS spectrum shows that silicon on a copper substrate acquires metallic properties. To analyze the structure we used the statistical geometry method. Lithium atoms in the channel are predominantly irregularly packed. However, part of the Li atoms are located above the hexagonal Si cells. The average stresses in silicene, calculated with limiting filling of the channel with lithium, are usually small. However, in the case of silicene with monovacancies, the tensile stress reaches 12.5% of the ultimate tensile stress. Evaluation of the dynamic stress observed in silicene during cycling shows that its value is less than 5% of the ultimate tensile stress. read less NOT USED (low confidence) A. Naghilou, M. He, J. S. Schubert, L. Zhigilei, and W. Kautek, “Femtosecond laser generation of microbumps and nanojets on single and bilayer Cu/Ag thin films.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 11 Abstract: The formation mechanisms of microbumps and nanojets on films… read moreAbstract: The formation mechanisms of microbumps and nanojets on films composed of single and double Cu/Ag layers deposited on a glass substrate and irradiated by a single 60 fs laser pulse are investigated experimentally and in atomistic simulations. The composition of the laser-modified bilayers is probed with the energy dispersive X-ray spectroscopy and used as a marker for processes responsible for the modification of the film morphology. For the bilayer with the top Ag layer facing the laser, the increase in fluence is found to result in a sequential appearance of a Ag microbump, the exposure of the Cu underlayer by removal of the Ag layer, a Cu microbump, and a frozen nanojet. The Cu on Ag bilayer exhibits a partial spallation of the top Cu film, followed by the generation of surface structures that mainly consist of Ag at higher fluences. The experimental observations are explained with atomistic simulations, which reveal that the stronger electron-phonon coupling of Cu results in the confinement of the deposited laser energy in the top Cu layer in the Cu on Ag case and channelling of the energy from the top Ag layer to the underlying Cu layer in the Ag on Cu case. This difference in the energy (re)distribution directly translates into differences in the morphology of the laser-modified bilayers. In all systems, the generation of microbumps and nanojets occurs in the molten state. It is driven by the dynamic relaxation of the laser-induced stresses and, at higher fluences, the release of vapor at the interface with the substrate. The resistance of the colder periphery of the laser spot to the ejection of spalled layers as well as the rapid solidification of the transient molten structures are largely defining the final shapes of the surface structures. read less NOT USED (low confidence) W. Zhou, Y. Li, M. Li, J. Wei, and W. Tao, “Bubble nucleation over patterned surfaces with different wettabilities: Molecular dynamics investigation,” International Journal of Heat and Mass Transfer. 2019. link Times cited: 49 NOT USED (low confidence) R. Babicheva, I. Evazzade, E. Korznikova, I. Shepelev, K. Zhou, and S. Dmitriev, “Low-energy channel for mass transfer in Pt crystal initiated by molecule impact,” Computational Materials Science. 2019. link Times cited: 25 NOT USED (low confidence) J. Song, Z. Gao, L. Zhang, W.-heng Wu, B. He, and L. Lu, “Prediction on elastic properties of Nb-doped Ni systems,” Molecular Simulation. 2019. link Times cited: 8 Abstract: ABSTRACT On the basis of first-principles simulation, the st… read moreAbstract: ABSTRACT On the basis of first-principles simulation, the structure, formation enthalpy and mechanical properties (elastic constant, bulk and shear modulus and hardness) of five Nb-doped Ni systems are systematically studied. The calculated equilibrium volume increases with the Nb concentration increasing. The computational elastic constants and formation enthalpy indicate that all Nb-doped Ni systems are mechanically and thermodynamically stable in our research. The hardness of these systems was predicted after the bulk modulus and shear modulus had been accurately calculated. The results show that the hardness increases with the Nb concentration increasing when the Nb concentration was below 4.9%, beyond which the hardness will decrease; this is within the scope of our study. read less NOT USED (low confidence) H.-J. Stromberg, N. Gunkelmann, and A. Lohrengel, “A Novel Approach to Multiscale MD/FE Simulations of Frictional Contacts.” 2019. link Times cited: 0 NOT USED (low confidence) C. Zhang et al., “Impact of bonding energy on thermal conductance of metal/graphene/metal interfaces,” Materials Research Express. 2019. link Times cited: 4 Abstract: The thermal conductance at the interface between graphene an… read moreAbstract: The thermal conductance at the interface between graphene and other metals is an important issue for graphene-based applications in the near future. The non-equilibrium molecular dynamics simulation is performed to investigate the effects of bonding energy between graphene and metal films on the metal/graphene/metal interfacial thermal conductance by varying the energy parameters between them, with other parameters unchanged. The calculation results demonstrate that the thermal conductance at the metal/graphene/metal interface increases initially to a peak, and subsequently decreases and saturates at a certain value with the bonding energy of one single graphene-metal side increasing. However, for the case that bonding energies of both graphene-metal sides are increased, the metal/graphene/metal interfacial thermal conductance increases rapidly and keeps at a certain value. The simulation results present a hint to tune effectively the interfacial thermal conductance for graphene-based devices. read less NOT USED (low confidence) O. Bachurina, “Plane and plane-radial discrete breathers in fcc metals,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 16 Abstract: Discrete breather (DB) is a time-periodic, spatially localiz… read moreAbstract: Discrete breather (DB) is a time-periodic, spatially localized vibrational mode in a perfect nonlinear lattice. In this study, two new types of DBs are reported in fcc metals (Al, Cu and Ni), based on the molecular dynamics simulations using standard embedded atom method interatomic potentials. All calculations are performed at a zero temperature in a three-dimensional computational cell with the use of periodic boundary conditions. A plane DB is excited in a single (111) atomic plane by displacing the atoms from their equilibrium lattice sites according to a specific pattern corresponding to a delocalized vibrational mode in a two-dimensional triangular lattice. This plane DB is delocalized in two dimensions and localized in one dimension, normal to the excited (111) plane. It is shown that in all studied metals the plane DBs have maximal lifetimes of 17–22 ps in the range of initial amplitudes of 0.15–0.30 Å. Herewith the studied mode demonstrates a hard type of nonlinearity, i.e. its frequency increases with amplitude. The second new type of DB is the plane-radial DB obtained by imposing a radial localizing function on the plane DB. This disk-type DB is localized in all three dimensions. The time evolution of the atomic displacement amplitudes and the kinetic energy are studied. The DBs of this type can exist for 9 and 4 ps in Cu and Ni, respectively, and then they decay by dissipating their vibrational energy onto neighboring atoms. The long-lived plane-radial DB in Al is not found. read less NOT USED (low confidence) J. Ren, M. Hao, G. Liang, S. Wang, and M. Lv, “Study of subsurface damage of monocrystalline nickel in nanometric grinding with spherical abrasive grain,” Physica B: Condensed Matter. 2019. link Times cited: 11 NOT USED (low confidence) M. Bon, N. Ahmad, R. Erni, and D. Passerone, “Reliability of two embedded atom models for the description of Ag@Au nanoalloys,” The Journal of Chemical Physics. 2019. link Times cited: 7 Abstract: The validation of embedded atom models (EAM) for modelling n… read moreAbstract: The validation of embedded atom models (EAM) for modelling nanoalloys requires to verify both a faithful description of the individual phases and a convincing scheme for the mixed interactions. In this work, we present a systematic benchmarking of two widely adopted EAM parameterizations, i.e. by Foiles [S. M. Foiles et al. Phys. Rev. B 33, 7983 (1986)] and by Zhou [X. W. Zhou et al. Phys. Rev. B, 69, 144113 (2004)] with density functional theory calculations for the description of processes at Ag@Au nanoalloys surfaces and nanoclusters. read less NOT USED (low confidence) M. Guillotte, J. Godet, and L. Pizzagalli, “A fully molecular dynamics-based method for modeling nanoporous gold,” Computational Materials Science. 2019. link Times cited: 12 NOT USED (low confidence) J. Wei et al., “Modified Embedded Atom Method Potential for Modeling the Thermodynamic Properties of High Thermal Conductivity Beryllium Oxide,” ACS Omega. 2019. link Times cited: 8 Abstract: Modified embedded atom method potential parameters of beryll… read moreAbstract: Modified embedded atom method potential parameters of beryllium oxide (BeO) have been developed, which can well reproduce the thermodynamic properties of beryllium oxide. To accurately describe the interactions between the atoms in the BeO structure, the density functional theory is used to calculate the fundamental properties such as the lattice constant, bulk modulus, and elastic constant, which are used for the potential fitting. The properties such as the enthalpy and specific heat are used to test the validity of the potential parameters. The calculated results by the developed potential parameters are compared with the experimental and other theoretical data as a function of temperature. The good agreement between the calculated results by the new potential and the experimental data verifies the potential parameters. The developed potential parameters have also been used to predict the thermal conductivity of BeO as a function of temperature for further applications of beryllium oxide. read less NOT USED (low confidence) A. Hernandez, A. Balasubramanian, F. Yuan, S. Mason, and T. Mueller, “Fast, accurate, and transferable many-body interatomic potentials by symbolic regression,” npj Computational Materials. 2019. link Times cited: 51 NOT USED (low confidence) O. Bachurina, “Linear discrete breather in fcc metals,” Computational Materials Science. 2019. link Times cited: 21 NOT USED (low confidence) Z. Wei, F. Yang, K. Bi, J. Yang, and Y. Chen, “Tuning the interfacial thermal conductance via the anisotropic elastic properties of graphite,” Carbon. 2019. link Times cited: 18 NOT USED (low confidence) Y.-hong Wang, S. Wang, G. Lu, and X.-dong Wang, “Effects of wettability on explosive boiling of nanoscale liquid films: Whether the classical nucleation theory fails or not?,” International Journal of Heat and Mass Transfer. 2019. link Times cited: 51 NOT USED (low confidence) J. Byggmästar, M. J. Nagel, K. Albe, K. Henriksson, and K. Nordlund, “Analytical interatomic bond-order potential for simulations of oxygen defects in iron,” Journal of Physics: Condensed Matter. 2019. link Times cited: 11 Abstract: We present an analytical bond-order potential for the Fe–O s… read moreAbstract: We present an analytical bond-order potential for the Fe–O system, capable of reproducing the basic properties of wüstite as well as the energetics of oxygen impurities in -iron. The potential predicts binding energies of various small oxygen-vacancy clusters in -iron in good agreement with density functional theory results, and is therefore suitable for simulations of oxygen-based defects in iron. We apply the potential in simulations of the stability and structure of Fe/FeO interfaces and FeO precipitates in iron, and observe that the shape of FeO precipitates can change due to formation of well-defined Fe/FeO interfaces. The interface with crystalline Fe also ensures that the precipitates never become fully amorphous, no matter how small they are. read less NOT USED (low confidence) P. Grammatikopoulos, M. Sowwan, and J. Kioseoglou, “Computational Modeling of Nanoparticle Coalescence,” Advanced Theory and Simulations. 2019. link Times cited: 68 Abstract: The coalescence of nanoclusters fabricated in the gas phase … read moreAbstract: The coalescence of nanoclusters fabricated in the gas phase is a fundamental growth mechanism determining cluster shapes, sizes, compositions, and structures, with resultant effects on practically all of their physical and chemical properties. Furthermore, coalescence can affect properties of larger structures that consist of nanoparticles as their elementary building blocks, such as the fractal dimension of cluster aggregates and the porosity and conductance of thin films. Therefore, it comes as no surprise that a great body of research, both experimental and theoretical, has focused on nanoparticle coalescence over the course of the past few decades. This review attempts to summarize the most important recent results from computational studies on nanoparticle coalescence and draw parallels between theoretical and experimental findings. The approach used here aspires to explain nanoparticle coalescence within the framework of a single intuitive narrative by integrating previous results obtained using various methods by the authors and others. Simultaneously, it is discussed where understanding and controlling (i.e., enhancing or inhibiting) nanoparticle coalescence can have great technological interest. read less NOT USED (low confidence) D. Yoo, K. Lee, W. Jeong, D. Lee, S. Watanabe, and S. Han, “Atomic energy mapping of neural network potential,” Physical Review Materials. 2019. link Times cited: 22 Abstract: We show that the intelligence of the machine-learning potent… read moreAbstract: We show that the intelligence of the machine-learning potential arises from its ability to infer the reference atomic-energy function from a given set of total energies. By utilizing invariant points in the feature space at which the atomic energy has a fixed reference value, we examine the atomic energy mapping of neural network potentials. Through a series of examples on Si, we demonstrate that the neural network potential is vulnerable to 'ad hoc' mapping in which the total energy appears to be trained accurately while the atomic energy mapping is incorrect in spite of its capability. We show that the energy mapping can be improved by choosing the training set carefully and monitoring the atomic energy at the invariant points during the training procedure. read less NOT USED (low confidence) J. Mattei et al., “Gas-Phase Synthesis of Trimetallic Nanoparticles,” Chemistry of Materials. 2019. link Times cited: 58 Abstract: To this day, engineering nanoalloys beyond bimetallic compos… read moreAbstract: To this day, engineering nanoalloys beyond bimetallic compositions has scarcely been within the scope of physical deposition methods due to the complex, nonequilibrium processes they entail. Here, ... read less NOT USED (low confidence) C.-Y. Chang, W. Chen, C.-H. Su, P.-C. Chang, Y. T. Huang, and K.-C. Hsu, “Enhanced bioconjugation on sputtered palladium nano-thin-film electrode,” Applied Physics Letters. 2019. link Times cited: 4 Abstract: A highly reactive surface with an enhanced ability for chemi… read moreAbstract: A highly reactive surface with an enhanced ability for chemical bonding relies on the presence of specifically coordinated atoms and step edges at the surface. In this study, an electrode with a unique Stranski-Krastanov-like thin film, with an epitaxial sputtering of a palladium (Pd) nanoparticle double layer on the polyethylene terephthalate substrate, was developed. On the surface of this flexible Pd-nano-thin-film (NTF) electrode with a (1 1 1) containing surface, DNA probes can be quickly immobilized in as short a period as 20 min, which is 24 times faster than that on the gold electrode. A DNA-based anticancer compound (ACC) sensing and screening process that would use the DNA functionalized Pd-NTF electrode as the biosensor was then proposed. Interestingly, the developed biosensor could detect DNA and ACCs, such as doxorubicin, tetra-O-methyl nordihydroguaiaretic acid, and Taxol via interactions with solutions containing 1 μl ACCs within 11 min, and the sensitivity of the ACC solution is ∼0.1 μM (∼36 pg per-test), as detected by electrochemical impedance spectroscopy. Moreover, this highly reactive surface can be used in regular sensors and other interfaces, in scientific applications.A highly reactive surface with an enhanced ability for chemical bonding relies on the presence of specifically coordinated atoms and step edges at the surface. In this study, an electrode with a unique Stranski-Krastanov-like thin film, with an epitaxial sputtering of a palladium (Pd) nanoparticle double layer on the polyethylene terephthalate substrate, was developed. On the surface of this flexible Pd-nano-thin-film (NTF) electrode with a (1 1 1) containing surface, DNA probes can be quickly immobilized in as short a period as 20 min, which is 24 times faster than that on the gold electrode. A DNA-based anticancer compound (ACC) sensing and screening process that would use the DNA functionalized Pd-NTF electrode as the biosensor was then proposed. Interestingly, the developed biosensor could detect DNA and ACCs, such as doxorubicin, tetra-O-methyl nordihydroguaiaretic acid, and Taxol via interactions with solutions containing 1 μl ACCs within 11 min, and the sensitivity of the ACC solution is ∼0.1 μM (∼... read less NOT USED (low confidence) A. Nomoev et al., “Receiving Copper Nanoparticles: Experiment and Modelling,” Solid State Phenomena. 2019. link Times cited: 2 Abstract: The copper nanoparticles were obtained by evaporating the me… read moreAbstract: The copper nanoparticles were obtained by evaporating the metal by the relativistic electron beam. The average size of synthesized particles was about 120 nm. They were characterized by X-ray diffraction, transmission electron microscopy. The results of the X-ray diffraction showed high content of the pure copper for closed setup with an inert gas. Transmission electron microscopy cleared some particles to have an icosahedral structure. These nanoparticles were obtained when the target was exposed by the beam with the highest current strength used in the experiment and the highest cooling of the copper vapor. The process of copper nanoparticle formation by the molecular dynamic method using EAM – potentials (potentials in the Embedded Atom Method form) was studied. read less NOT USED (low confidence) P. Grammatikopoulos, “Atomistic modeling of the nucleation and growth of pure and hybrid nanoparticles by cluster beam deposition,” Current Opinion in Chemical Engineering. 2019. link Times cited: 15 NOT USED (low confidence) H. Loulijat and H. Zerradi, “The Effect of the Liquid Layer Around the Spherical and Cylindrical Nanoparticles in Enhancing Thermal Conductivity of Nanofluids,” Journal of Heat Transfer. 2019. link Times cited: 13 Abstract: In this work, the equilibrium molecular dynamics (MD) simula… read moreAbstract: In this work, the equilibrium molecular dynamics (MD) simulation combined with the Green–Kubo method is employed to calculate the thermal conductivity and investigate the impact of the liquid layer around the solid nanoparticle (NP) in enhancing thermal conductivity of nanofluid (argon–copper), which contains the liquid argon as a base fluid surrounding the spherical or cylindrical NPs of copper. First, the thermal conductivity is calculated at temperatures 85, 85.5, 86, and 86.5 K and for different volume fractions ranging from 4.33% to 11.35%. Second, the number ΔN of argon atoms is counted in the liquid layer formed at the solid–liquid interface with the thickness of Δr = 0.3 nm around the NP. Finally, the number density n of argon atoms in this layer formed is calculated in all cases. Also, the results for spherical and cylindrical NPs are compared with one another. It is observed that the thermal conductivity of the nanofluid increased with the increasing volume fraction and the number ΔN. Likewise, the thermal conductivity of nanofluid containing spherical NPs is higher than that of nanofluid containing cylindrical NPs. Furthermore, the number density n of argon atoms near the surface of spherical NPs is higher than that of argon atoms attached in the curved surface of cylindrical NPs. As a result, the liquid layer around the solid NP has been considered one of the mechanisms responsible contributing to the thermal conductivity enhancement in nanofluids. read less NOT USED (low confidence) A. Brant and M. Sundaram, “Molecular dynamics study of direct localized overpotential deposition for nanoscale electrochemical additive manufacturing process,” Precision Engineering. 2019. link Times cited: 7 NOT USED (low confidence) X. Zhou, X.-xiang Yu, D. Jacobson, and G. Thompson, “A molecular dynamics study on stress generation during thin film growth,” Applied Surface Science. 2019. link Times cited: 25 NOT USED (low confidence) F. C. Maier, C. S. Sarap, M. Dou, G. Sivaraman, and M. Fyta, “Diamondoid-functionalized nanogaps: from small molecules to electronic biosensing,” The European Physical Journal Special Topics. 2019. link Times cited: 4 NOT USED (low confidence) H. S. Kim, J. H. Kim, S.-H. Cha, and S. Cho, “Optimal determination of force field parameters for reduced molecular dynamics model,” Comput. Phys. Commun. 2019. link Times cited: 2 NOT USED (low confidence) J. Liu, X. Fan, Y. Shi, D. J. Singh, and W. Zheng, “Nanopores in nanocrystalline gold,” Materialia. 2019. link Times cited: 7 NOT USED (low confidence) N. Beets, D. Farkas, and S. Corcoran, “Deformation mechanisms and scaling relations in the mechanical response of nano-porous Au,” Acta Materialia. 2019. link Times cited: 23 NOT USED (low confidence) K. Yoshida et al., “Influence of gas environment and heating on atomic structures of platinum nanoparticle catalysts for proton-exchange membrane fuel cells,” Nanotechnology. 2019. link Times cited: 3 Abstract: Atomic-scale relaxations of platinum nanoparticles (Pt NPs) … read moreAbstract: Atomic-scale relaxations of platinum nanoparticles (Pt NPs) for fuel-cell catalysts are evaluated by spherical-aberration corrected environmental transmission electron microscopy (ETEM) under reference high-vacuum and N2 atmospheres, and then under reactive H2, CO and O2 atmospheres, combined with ex situ durability test using an electrochemical half-cell. In high-vacuum, increasing roughness due to continuous relaxation of surface-adsorbed Pt atoms is quantified in real-space. Under H2 and N2 atmospheres at a critical partial pressure of 1 × 10−2 Pa the stability of the surface facets is for the first time found to be improved. The adsorption behaviour of CO molecules is investigated using experimentally measured Pt–Pt bond lengths on the topmost surface layer of Pt NPs. The deactivation of Pt NPs in the anode environment of a proton-exchange-membrane fuel-cell is demonstrated at the atomic-scale in the ETEM, and the transformation of NPs into disordered nanoclusters is systematically quantified using the partial size distribution of Pt atomic clusters under controlled heating experiments at 423, 573 and 723 K. read less NOT USED (low confidence) C. T. Nguyen, D. T. Ho, S. Choi, D. Chun, and S. Y. Kim, “Pattern transformation induced by elastic instability of metallic porous structures,” Computational Materials Science. 2019. link Times cited: 8 NOT USED (low confidence) J. Pang et al., “Photothermal effects induced by surface plasmon resonance at graphene/gold nanointerfaces: A multiscale modeling study.,” Biosensors & bioelectronics. 2019. link Times cited: 15 NOT USED (low confidence) J. Liu, X. Fan, Y. Shi, D. J. Singh, and W. Zheng, “Melting of Nanocrystalline Gold,” The Journal of Physical Chemistry C. 2019. link Times cited: 4 Abstract: We report atomistic simulations of the melting of nanocrysta… read moreAbstract: We report atomistic simulations of the melting of nanocrystalline gold with mean grain sizes from 1.7 to 23 nm. Analysis of the structural changes near melting point at the atomic scale confirms that in the melting process, the solid–liquid interface sweeps rapidly from grain boundary into inner grain as the temperature increases. We find a linear relation between the melting point and the reciprocal grain size for the larger grain size samples, above 7.7 nm, similar to the observations in nanoparticles. However, based on the critical Lindemann ratio, the grain boundaries in these cases should be liquefied at very low temperature (less than 800 K for grain size of 9.7 nm). At a small grain size, this relation between grain size and melting temperature was broken. In particular, at grain sizes below 4 nm, the melting point was found to be approximately constant. It was proposed that the growth and/or merging of grains at low temperature far from melting was contributed to this observation. read less NOT USED (low confidence) B. Zhang, X. Liao, Y.-N. Chen, H. Xiao, Y. Ni, and X. Chen, “Rapid Programmable Nanodroplet Motion on a Strain-Gradient Surface.,” Langmuir : the ACS journal of surfaces and colloids. 2019. link Times cited: 11 Abstract: When a nanodroplet is placed on a lattice surface, an inhomo… read moreAbstract: When a nanodroplet is placed on a lattice surface, an inhomogeneous surface strain field perturbs the balance of van der Waals force between the nanodroplet and surface, thus providing a net driving force for nanodroplet motion. Using molecular dynamics and theoretical analysis, we study the effect of strain gradient on modulating the movement of a nanodroplet. Both modeling and simulation show that the driving force is opposite to the direction of strain gradient, with a magnitude that is proportional to the strain gradient as well as nanodroplet size. Two representative surfaces, graphene and copper (111) plane, are exemplified to demonstrate the controllable motion of the nanodroplet. When the substrate undergoes various types of reversible deformations, multiple motion modes of nanodroplets can be feasibly achieved, including acceleration, deceleration, and turning, becoming a facile strategy to manipulate nanodroplets along a designed two-dimensional pathway. read less NOT USED (low confidence) B.-X. Zhang, S. Wang, and X.-dong Wang, “Wetting Transition from the Cassie-Baxter State to the Wenzel State on Regularly Nanostructured Surfaces Induced by an Electric Field.,” Langmuir : the ACS journal of surfaces and colloids. 2019. link Times cited: 38 Abstract: When droplets are placed on hydrophobic textured surfaces, d… read moreAbstract: When droplets are placed on hydrophobic textured surfaces, different wetting states Cassie-Baxter (CB) state or Wenzel (W) state may occur depending on materials and structures of surfaces, types and sizes of droplets, thermal fluctuations, and external stimuli. The wetting transition from the CB to the W state and the opposite process have attracted a great deal of attention because of their primary importance for designing and fabricating textured surfaces. In this work, molecular dynamics (MD) simulations are employed to understand the mechanism behind the CB-to-W transition for a nanoscale water film placed on a surface decorated with a single nanogroove when an external electric field is applied. The free energy variation during the transition process is computed on the basis of the restrained MD simulations. Water intrusion into the groove is observed by simulation snapshots, which provides direct evidence for the electric field-induced CB-to-W transition. In the previous experiments, however, only a sharp reduction in the apparent contact angle is employed to judge whether the transition takes place. The free energy curves reveal that there are two energy barriers separating the CB and W states (Δ E1) as well as separating the W and CB states (Δ E2). Owing to the presence of Δ E1, although the CB state has a higher free energy than the W state, it cannot spontaneously convert to the W state. When the external energy input exceeds Δ E1, the CB-to-W transition can be triggered, otherwise the transition will stop, and the water film will return to the CB state. Moreover, it is found that the maximum of free energy always occurs after the film touches the groove bottom. Thus, the requirement that the film should touch the groove bottom is responsible for the presence of the energy barrier Δ E1. Finally, the dependence of the two energy barriers on the electric field strength, groove aspect ratio, and intrinsic contact angle of the groove is also discussed. read less NOT USED (low confidence) H. Shodja and C. Enzevaee, “Surface characterization of face-centered cubic crystals,” Mechanics of Materials. 2019. link Times cited: 14 NOT USED (low confidence) C. L. Wu, H.-T. Lin, H.-A. Chen, S.-Y. Lin, M. Shih, and C. Pao, “Defect formation and modulation during patterning supported graphene sheets using focused ion beams,” Materials Today Communications. 2018. link Times cited: 6 NOT USED (low confidence) M. H. Nazir, Z. Khan, A. Saeed, V. Bakolas, W. Braun, and R. Bajwa, “Experimental analysis and modelling for reciprocating wear behaviour of nanocomposite coatings,” Wear. 2018. link Times cited: 19 NOT USED (low confidence) T. R. Dhakal and D. Zhang, “Combining dual domain material point method with molecular dynamics for thermodynamic nonequilibriums,” J. Comput. Phys. 2018. link Times cited: 11 NOT USED (low confidence) X. Fu, L. Liang, and Y. Wei, “Atomistic simulation study on the shear behavior of Ag/MgO interface,” Computational Materials Science. 2018. link Times cited: 10 NOT USED (low confidence) P. Zhao and Y.-bo Guo, “Grain size effects on indentation-induced defect evolution and plastic deformation mechanism of ploycrystalline materials,” Computational Materials Science. 2018. link Times cited: 20 NOT USED (low confidence) W. Ko and J. Jeon, “Atomistic simulations of PdTi high-temperature shape-memory alloys,” Intermetallics. 2018. link Times cited: 2 NOT USED (low confidence) A. Khoei, A. R. Sameti, and Y. Nikravesh, “A continuum-atomistic multi-scale technique for nonlinear behavior of nano-materials,” International Journal of Mechanical Sciences. 2018. link Times cited: 20 NOT USED (low confidence) Z. Xu, D. Ge, and L. Zhang, “Simulation and prediction on phonon thermal conductivity of Al/Cu interface,” Journal of Physics and Chemistry of Solids. 2018. link Times cited: 12 NOT USED (low confidence) F. Trillitzsch, R. Guerra, A. Janas, N. Manini, F. Krok, and E. Gnecco, “Directional and angular locking in the driven motion of Au islands on
MoS2,” Physical Review B. 2018. link Times cited: 17 NOT USED (low confidence) H. N. Pishkenari, F. S. Yousefi, and A. Taghibakhshi, “Determination of surface properties and elastic constants of FCC metals: a comparison among different EAM potentials in thin film and bulk scale,” Materials Research Express. 2018. link Times cited: 22 Abstract: Three independent elastic constants C11, C12, and C44 were c… read moreAbstract: Three independent elastic constants C11, C12, and C44 were calculated and compared using available potentials of eight different metals with FCC crystal structure; Gold, Silver, Copper, Nickel, Platinum, Palladium, Aluminum and Lead. In order to calculate the elastic constants, the second derivative of the energy density of each system was calculated with respect to different directions of strains. Each set of the elastic constants of the metals in bulk scale was compared with experimental results, and the average relative error was for each was calculated and compared with other available potentials. Then, using the Voigt-Reuss-Hill method, approximated values for Young and shear moduli and Poisson’s ratio of the FCC metals in the bulk scale were found for each potential. Furthermore, to observe the surface effects on the metals in nanoscale, surface elastic constants of the thin films of the metals have been calculated. In the study of the thin films of materials in nanoscale, the number of surface atoms is considerable compared to all atoms of the object. This leads to an increase in the surface effects, which influence the elastic properties. By considering this fact and employing related definitions and equations, the properties of the thin films of the metals were calculated, and the surface effects for different crystallographic directions were compared. Subsequently, in some cases, comparisons among characteristics of the metals in the thin film and bulk material were made. read less NOT USED (low confidence) A. Ahadi and S. Melin, “Capturing nanoscale effects by peridynamics,” Mechanics of Advanced Materials and Structures. 2018. link Times cited: 14 Abstract: ABSTRACT Molecular dynamic simulations inevitably demand lar… read moreAbstract: ABSTRACT Molecular dynamic simulations inevitably demand large computational resources for structures of liner measures even as small as a few tens or hundreds of nanometers. Thus, a computationally efficient method to simulate larger structures and, at the same time, retain the properties and the mechanical response at the atomic scale is in demand. One such approach is peridynamics, which is a nonlocal extension of continuum mechanics. In this study, we investigate the possibility to efficiently reproduce results from molecular dynamic (MD) simulations by calibration of two parameters inherent in peridynamics: the length scale parameter and the interparticle bond strength. The free-ware LAMMPS supports both numerical approaches, and thus LAMMPS has been used as the common framework. Beams of single-crystal fcc copper of various sizes and under tension along the crystallographic [100]- and [110]-directions act as the modeling example. The force–displacement curves and the elastic–plastic transitions have been compared between the approaches. The conclusion is that proper calibration of the peridynamic two parameters to MD simulations results in proper reproduction of the molecular dynamic results. This in turn allows for geometrical upscaling or simulation of geometrically more complicated structures, without loss of features derived from the atomic scale but to a much lower computational cost. read less NOT USED (low confidence) D. Gulmez, Y. Yildiz, and M. Kırca, “Nanoporous gold reinforced with carbon based nanomaterials: A molecular dynamics study,” Composites Part B: Engineering. 2018. link Times cited: 7 NOT USED (low confidence) J. Ren, M. Hao, M. Lv, S. Wang, and B. Zhu, “Molecular dynamics research on ultra-high-speed grinding mechanism of monocrystalline nickel,” Applied Surface Science. 2018. link Times cited: 50 NOT USED (low confidence) Z. Meng and S. Keten, “Unraveling the Effect of Material Properties and Geometrical Factors on Ballistic Penetration Energy of Nanoscale Thin Films,” Journal of Applied Mechanics. 2018. link Times cited: 22 Abstract: It is crucial to investigate the dynamic mechanical behavior… read moreAbstract: It is crucial to investigate the dynamic mechanical behavior of materials at the nanoscale to create nanostructured protective systems that have superior ballistic impact resistance. Inspired from recent experimental advances that enable ballistic materials testing at small scales, here we report a comparative analysis of the dynamic behavior of nanoscale thin films made from multilayer graphene (MLG), polymer, gold, and aluminum under high-speed projectile impact. We employ atomistic and coarse-grained (CG) molecular dynamics (MD) simulations to measure the ballistic limit velocity (V50) and penetration energy (Ep) of these nanoscale films and investigate their distinctive failure mechanisms over a wide range of impact velocities (Vi). For the local penetration failure mechanism observed in polymer and metal films, we find that the intrinsic mechanical properties influence Ep at low Vi, while material density tends to govern Ep at high Vi. MLG films uniquely show a large impact propagation zone (IPZ), which transfers the highly localized impact energy into elastic deformation energy in a much larger area through cone wave propagation. We present theoretical analyses that corroborate that the size of IPZ should depend not only on material properties but also on a geometrical factor, specifically, the ratio between the projectile radius and film thickness. This study clearly illustrates how material properties and geometrical factors relate to the ballistic penetration energy, thereby allowing a quantitative comparison of the nanoscale ballistic response of different materials. read less NOT USED (low confidence) Y. Cai, Y.-L. Chen, Y. Shimizu, S. Ito, and W. Gao, “Molecular dynamics simulation of elastic–plastic deformation associated with tool–workpiece contact in force sensor–integrated fast tool servo,” Proceedings of the Institution of Mechanical Engineers, Part B: Journal of Engineering Manufacture. 2018. link Times cited: 6 Abstract: The tool–workpiece interactions when a single-point diamond … read moreAbstract: The tool–workpiece interactions when a single-point diamond cutting tool with specific tool edge geometry is made to contact with a copper workpiece are evaluated by the molecular dynamics simulations under different temperatures, boundary conditions and model sizes for ultra-precision microcutting and in-process surface form measurement based on a force sensor–integrated fast tool servo. It is confirmed that the proposed multi-relaxation time method is effective to stabilize the workpiece molecular dynamics model over a wide temperature range up to the room temperature under which a practical microcutting and on-machine surface form metrology process are conducted. The boundary condition and model size of the molecular dynamics model are then optimized to make reliable and cost-effective simulations for evaluation of the elastic–plastic transition contact depth and the corresponding contact force when a diamond tool with a practical edge sharpness of up to 30 nm is employed for microcutting and on-machine surface form metrology. read less NOT USED (low confidence) S. E. Muller, R. R. Santhapuram, and A. Nair, “Failure mechanisms in pre-cracked Ni-graphene nanocomposites,” Computational Materials Science. 2018. link Times cited: 19 NOT USED (low confidence) L. V. Stepanova and O. Belova, “Estimation of crack propagation direction angles under mixed mode loading in linear elastic isotropic materials by generalized fracture mechanics criteria and by molecular dynamics method,” Journal of Physics: Conference Series. 2018. link Times cited: 7 Abstract: The crack growth directional angles in the isotropic linear … read moreAbstract: The crack growth directional angles in the isotropic linear elastic plane with the central crack under mixed-mode loading conditions for the full range of the mixity parameter are found. The traditional strain energy density, maximum tangential stress and maximum tangential strain criteria are introduced and analyzed. The accuracy of each model is investigated and discussed by comparing its results to the molecular dynamics modelling. Three fracture criteria of traditional linear fracture mechanics (maximum tangential stress, and minimum strain energy density criteria) are generalized by mean the multi-parameter stress expansion in the vicinity of the crack tip. Atomistic simulations of the central crack growth process in an infinite plane medium under mixed-mode loading using Large-scale Molecular Massively Parallel Simulator (LAMMPS), a classical molecular dynamics code, are performed. The inter-atomic potential used in this investigation is Embedded Atom Method (EAM) potential. The plane specimens with initial central crack were subjected to Mixed-Mode loadings. The simulation cell contains 400000 atoms. The crack propagation direction angles under different values of the mixity parameter in a wide range of values from pure tensile loading to pure shear loading in a wide diapason of temperatures (from 0.1 К to 800 К) are obtained and analyzed. It is shown that the crack propagation direction angles obtained by molecular dynamics method coincide with the crack propagation direction angles given by the multi-parameter fracture criteria based on the strain energy density and the multi-parameter description of the crack-tip fields. read less NOT USED (low confidence) R. Shi and A. Luo, “Applications of CALPHAD modeling and databases in advanced lightweight metallic materials,” Calphad. 2018. link Times cited: 67 NOT USED (low confidence) J. Harrison, J. Schall, S. Maskey, P. Mikulski, M. T. Knippenberg, and B. Morrow, “Review of force fields and intermolecular potentials used in atomistic computational materials research,” Applied Physics Reviews. 2018. link Times cited: 99 Abstract: Molecular simulation is a powerful computational tool for a … read moreAbstract: Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferability. When selecting a potential energy function for use in molecular simulations, the accuracy, transferability, and computational speed must all be considered. In this focused review, some of the more commonly used potential energy functions for molecular simulations are reviewed with an eye toward presenting their general forms, strengths, and weaknesses.Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferabilit... read less NOT USED (low confidence) F.-F. Xie, G. Lu, X.-dong Wang, and D.-Q. Wang, “Enhancement of Coalescence-Induced Nanodroplet Jumping on Superhydrophobic Surfaces.,” Langmuir : the ACS journal of surfaces and colloids. 2018. link Times cited: 33 Abstract: Coalescence-induced droplet self-jumping on superhydrophobic… read moreAbstract: Coalescence-induced droplet self-jumping on superhydrophobic surfaces has received extensive attentions over the past decade because of its potential applications ranging from anti-icing materials to self-sustained condensers, in which a higher jumping velocity vj is always expected and favorable. However, the previous studies have shown that there is a velocity limit with vj ≤ 0.23 uic for microscale droplets and vj ≤ 0.127 uic for nanoscale droplets, where uic is referred to as the inertial-capillary velocity. Here, we show that the jumping velocity can be significantly increased by patterning a single groove, ridge, or more hydrophobic strip, whose size is comparable with the radius of coalescing droplets, on a superhydrophobic surface. We implement molecular dynamics simulations to investigate the coalescence of two equally sized nanodroplets (8.0 nm in radius) on these surfaces. We found that a maximum vj = 0.23 uic is achieved on the surface with a 1.6 nm high and 5.9 nm wide ridge, which is 1.81 times higher than the nanoscale velocity limit. We also demonstrate that the presence of groove, ridge, and strip alters coalescence dynamics of droplets, leading to a significantly shortened coalescence time which remarkably reduces viscous dissipation during coalescence; thus, we believe that the present approach is also effective for microscale droplet jumping. read less NOT USED (low confidence) S. Hernandez, F. Freibert, R. Hoagland, B. Uberuaga, and J. Wills, “Spin density stabilization of local distortions induced by a monovacancy in
δ
-Pu,” Physical Review Materials. 2018. link Times cited: 5 NOT USED (low confidence) W. Jeong, K. Lee, D. Yoo, D. Lee, and S. Han, “Toward Reliable and Transferable Machine Learning Potentials: Uniform Training by Overcoming Sampling Bias,” The Journal of Physical Chemistry C. 2018. link Times cited: 29 Abstract: The neural network interatomic potential (NNP) is anticipate… read moreAbstract: The neural network interatomic potential (NNP) is anticipated to be a promising next-generation atomic potential for its self-learning capability and universal mathematical structure. While various examples demonstrate the usefulness of NNPs, we find that the NNP suffers from highly inhomogeneous feature-space sampling in the training set. As a result, underrepresented atomic configurations, often critical for simulations, cause large errors even though they are included in the training set. Using the Gaussian density function (GDF) that quantifies the sparsity of training points, we propose a weighting scheme that can effectively rectify the sampling bias. Various examples confirm that GDF weighting significantly improves the reliability and transferability of NNPs compared to the conventional training method, which is attributed to accurate mapping of atomic energies. By addressing a detrimental problem that is inherent in every machine learning potential, the present work will extend the application ra... read less NOT USED (low confidence) T. Sipkens and K. Daun, “Effect of Surface Interatomic Potential on Thermal Accommodation Coefficients Derived from Molecular Dynamics,” The Journal of Physical Chemistry C. 2018. link Times cited: 14 Abstract: This work investigates how the interatomic surface potential… read moreAbstract: This work investigates how the interatomic surface potential influences molecular dynamics (MD)-derived thermal accommodation coefficients (TACs). Iron, copper, and silicon surfaces are considered over a range of temperatures that include their melting points. Several classes of potentials are reviewed, including two-body, three-body, and bond-order force fields. MD-derived densities and visualization of the surfaces are used to explain the differences in the parameterizations of these potentials within the context of gas–surface scattering. Finally, TACs are predicted for a range of gas–surface combinations, and recommended values of the TAC are selected that take into account the robustness and uncertainties of each of the considered parameterizations. Further, it is observed that there is a significant change in the TAC about phase changes that must be taken into account for applications with a large range of surface temperatures. read less NOT USED (low confidence) G. Sun, J. Xu, and P. Harrowell, “The mechanism of the ultrafast crystal growth of pure metals from their melts,” Nature Materials. 2018. link Times cited: 59 NOT USED (low confidence) M. Widom, “Modeling the structure and thermodynamics of high-entropy alloys,” Journal of Materials Research. 2018. link Times cited: 72 Abstract: High-entropy and multiprincipal element alloys present excit… read moreAbstract: High-entropy and multiprincipal element alloys present exciting opportunities and challenges for computational modeling of their structure and phase stability. Recent interest has catalyzed rapid development of techniques and equally rapid growth of new results. This review surveys the essential concepts of thermodynamics and total energy calculation, and the bridge between them provided by statistical mechanics. Specifically, we review the electronic density functional theory of alloy total energy as applied to supercells and special quasirandom structures. We contrast these with the coherent potential approximation and semi-empirical approximations. Statistical mechanical approaches include cluster expansions, hybrid Monte Carlo/molecular dynamics simulations, and extraction of entropy from correlation functions. We also compare first-principles approaches with Calculation of Phase Diagrams (CALPHAD) and highlight the need to augment experimental databases with first-principles derived data. Numerous example applications are given highlighting recent progress utilizing the concepts and methods that are introduced. read less NOT USED (low confidence) N. Amadou, T. D. Rességuier, A. Dragon, and E. Brambrink, “Coupling between plasticity and phase transition in shock- and ramp-compressed single-crystal iron,” Physical Review B. 2018. link Times cited: 27 Abstract: Molecular dynamics simulations have been used to investigate… read moreAbstract: Molecular dynamics simulations have been used to investigate the coupling process between plasticity and structural phase transformation in single-crystal iron under both shock and ramp compressions. In both cases, iron was found to yield via twinning. Then, the onset of the bcc-hcp phase transformation was shown to be tightly dependent on the plasticity history through a hardening-like effect, which in some conditions may inhibit the nucleation of the hcp phase. read less NOT USED (low confidence) E. Zasimchuk, O. Baskova, O. Gatsenko, and T. Turchak, “Universal Mechanism of Viscoplastic Deformation of Metallic Materials Far from Thermodynamics Equilibrium,” Journal of Materials Engineering and Performance. 2018. link Times cited: 10 NOT USED (low confidence) Z. Xiong and L. Cao, “Red-ultraviolet photoluminescence tuning by Ni nanocrystals in epitaxial SrTiO 3 matrix,” Applied Surface Science. 2018. link Times cited: 23 NOT USED (low confidence) D. R. Gomes, A. Turkin, D. Vainchtein, and J. D. D. Hosson, “On the mechanism of ion-induced bending of nanostructures,” Applied Surface Science. 2018. link Times cited: 16 NOT USED (low confidence) X. Yue, X. Yue, X. Yang, and M. Kunieda, “Influence of metal vapor jets from tool electrode on material removal of workpiece in EDM,” Precision Engineering. 2018. link Times cited: 27 NOT USED (low confidence) H. Liu et al., “Confinement Impact for the Dynamics of Supported Metal Nanocatalyst.,” Small. 2018. link Times cited: 5 Abstract: Supported metal nanoparticles play key roles in nanoelectron… read moreAbstract: Supported metal nanoparticles play key roles in nanoelectronics, sensors, energy storage/conversion, and catalysts for the sustainable production of fuels and chemicals. Direct observation of the dynamic processes of nanocatalysts at high temperatures and the confinement of supports is of great significance to investigate nanoparticle structure and functions for practical utilization. Here, in situ high-resolution transmission electron microscopy photos and videos are combined with dynamics simulations to reveal the real-time dynamic behavior of Pt nanocatalysts at operation temperatures. Amorphous Pt surface on moving and deforming particles is the working structure during the high operation temperature rather than a static crystal surface and immobilization on supports as proposed before. The free rearrangement of the shape of Pt nanoparticles allows them to pass through narrow windows, which is generally considered to immobilize the particles. The Pt particles, no matter what their sizes, prefer to stay inside nanopores even when they are fast moving near an opening at temperatures up to 900 °C. The porous confinement also blocks the sintering of the particles under the confinement size of pores. These contribute to the continuous high activity and stability of Pt nanocatalysts inside nanoporous supports during a long-term evaluation of catalytic reforming reaction. read less NOT USED (low confidence) A. Joshi and S. James, “Molecular dynamics simulation study of cold spray process,” Journal of Manufacturing Processes. 2018. link Times cited: 35 NOT USED (low confidence) Y. Akkus, A. Koklu, and A. Beskok, “Atomic Scale Interfacial Transport at an Extended Evaporating Meniscus.,” Langmuir : the ACS journal of surfaces and colloids. 2018. link Times cited: 21 Abstract: Recent developments in fabrication techniques have enabled t… read moreAbstract: Recent developments in fabrication techniques have enabled the production of nano- and Ångström-scale conduits. While scientists are able to conduct experimental studies to demonstrate extreme evaporation rates from these capillaries, theoretical modeling of evaporation from a few nanometers or sub-nanometer meniscus interfaces, where the adsorbed film, the transition film, and the intrinsic region are intertwined, is absent in the literature. Using the computational setup constructed, we first identified the detailed profile of a nanoscale evaporating interface and then discovered the existence of lateral momentum transport within and associated net evaporation from adsorbed liquid layers, which are long believed to be at the equilibrium established between equal rates of evaporation and condensation. Contribution of evaporation from the adsorbed layer increases the effective evaporation area, reducing the excessively estimated evaporation flux values. This work takes the first step toward a comprehensive understanding of atomic/molecular scale interfacial transport at extended evaporating menisci. The modeling strategy used in this study opens an opportunity for computational experimentation of steady-state evaporation and condensation at liquid-vapor interfaces located in capillary nanoconduits. read less NOT USED (low confidence) A. Gola, P. Gumbsch, and L. Pastewka, “Atomic-scale simulation of structure and mechanical properties of Cu1−xAgx|Ni multilayer systems,” Acta Materialia. 2018. link Times cited: 17 NOT USED (low confidence) M. M. Devi et al., “Morphology controlled graphene-alloy nanoparticle hybrids with tunable carbon monoxide conversion to carbon dioxide.,” Nanoscale. 2018. link Times cited: 5 Abstract: Selective oxidation of CO to CO2 using metallic or alloy nan… read moreAbstract: Selective oxidation of CO to CO2 using metallic or alloy nanoparticles as catalysts can solve two major problems of energy requirements and environmental pollution. Achieving 100% conversion efficiency at a lower temperature is a very important goal. This requires sustained efforts to design and develop novel supported catalysts containing alloy nanoparticles. In this regard, the decoration of nanoalloys with graphene, as a support for the catalyst, can provide a novel structure due to the synergic effect of the nanoalloys and graphene. Here, we demonstrate the effect of nano-PdPt (Palladium-Platinum) alloys having different morphologies on the catalytic efficiency for the selective oxidation of CO. Efforts were made to prepare different morphologies of PdPt alloy nanoparticles with the advantage of tuning the capping agent (PVP - polyvinyl pyrollidone) and decorating them on graphene sheets via the wet-chemical route. The catalytic activity of the G-PdPt hybrids with an urchin-like morphology has been found to be superior (higher % conversion at 135 °C lower) to that with a nanoflower morphology. The above experimental observations are further supported by molecular dynamics (MD) simulations. read less NOT USED (low confidence) S. Trady, M. Mazroui, A. Hasnaoui, and K. Saadouni, “Microstructural evolutions and fractal characteristics in medium range level in AlxNi100-x alloys during rapid solidification process,” Journal of Alloys and Compounds. 2018. link Times cited: 13 NOT USED (low confidence) A. Cherala and S. Sreenivasan, “Molecular dynamics modeling framework for overcoming nanoshape retention limits of imprint lithography,” Microsystems & Nanoengineering. 2018. link Times cited: 4 NOT USED (low confidence) Y. Akkus and A. Beskok, “Molecular diffusion replaces capillary pumping in phase-change-driven nanopumps,” Microfluidics and Nanofluidics. 2018. link Times cited: 12 NOT USED (low confidence) R. Ramachandramoorthy, M. Milan, Z. Lin, S. Trolier-McKinstry, A. Corigliano, and H. Espinosa, “Design of piezoMEMS for high strain rate nanomechanical experiments,” Extreme Mechanics Letters. 2018. link Times cited: 13 NOT USED (low confidence) J.-S. Kim, D. Seol, and B.-J. Lee, “Critical assessment of Pt surface energy – An atomistic study,” Surface Science. 2018. link Times cited: 10 NOT USED (low confidence) S. Eich and G. Schmitz, “Embedded-atom study of grain boundary segregation and grain boundary free energy in nanosized iron–chromium tricrystals,” Acta Materialia. 2018. link Times cited: 22 NOT USED (low confidence) A. E. Korenchenko, A. Vorontsov, B. Gelchinski, and G. P. Sannikov, “Statistical analysis of dimer formation in supersaturated metal vapor based on molecular dynamics simulation,” Physica A-statistical Mechanics and Its Applications. 2018. link Times cited: 8 NOT USED (low confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “An investigation of tool and hard particle interaction in nanoscale cutting of copper beryllium,” Computational Materials Science. 2018. link Times cited: 35 NOT USED (low confidence) T. Roy, A. Sharma, D. Datta, and R. Balasubramaniam, “Molecular dynamics study on the effect of discharge on adjacent craters on micro EDMed surface,” Precision Engineering-journal of The International Societies for Precision Engineering and Nanotechnology. 2018. link Times cited: 5 NOT USED (low confidence) S. Brauer et al., “Multiscale Modeling of Pure Nickel.” 2018. link Times cited: 0 NOT USED (low confidence) T. Stone and Y. Hammi, “Nickel Powder Metal Modeling Illustrating Atomistic-Continuum Friction Laws.” 2018. link Times cited: 0 NOT USED (low confidence) S. Bukkuru, U. Bhardwaj, K. S. Rao, A. Rao, M. Warrier, and M. C. Valsakumar, “Kinetics of self-interstitial migration in bcc and fcc transition metals,” Materials Research Express. 2018. link Times cited: 11 Abstract: Radiation damage is a multi-scale phenomenon. A thorough und… read moreAbstract: Radiation damage is a multi-scale phenomenon. A thorough understanding of diffusivities and the migration energies of defects is a pre-requisite to quantify the after-effects of irradiation. We investigate the thermally activated mobility of self-interstitial atom (SIA) in bcc transition metals Fe, Mo, Nb and fcc transition metals Ag, Cu, Ni, Pt using molecular dynamics (MD) simulations. The self-interstitial diffusion involves various mechanisms such as interstitialcy, dumbbell or crowdion mechanisms. Max-Space Clustering (MSC) method has been employed to identify the interstitial and its configuration over a wide range of temperature. The self-interstitial diffusion is Arrhenius like, however, there is a slight deviation at high temperatures. The migration energies, pre-exponential factors of diffusion and jump-correlation factors, obtained from these simulations can be used as inputs to Monte Carlo simulations of defect transport. The jump-correlation factor shows the degree of preference of rectilinear or rotational jumps. We obtain the average jump-correlation factor of 1.4 for bcc metals and 0.44 for fcc metals. It indicates that rectilinear jumps are preferred in bcc metals and rotational jumps are preferred in fcc metals. read less NOT USED (low confidence) P. S. Cappellari, G. J. Soldano, and M. Mariscal, “A density functional study on the reactivity enhancement induced by gold in IrAu nanoalloys,” RSC Advances. 2018. link Times cited: 1 Abstract: IrAu nanoalloys have been proven to have remarkable reactivi… read moreAbstract: IrAu nanoalloys have been proven to have remarkable reactivity for several reactions. In this work, mixed IrAu nanoalloys of 8, 27, 48 and 64 total atoms were studied in different atomic compositions (IrmAun) using Density Functional Theory (DFT). A notable segregation tendency is observed, where Ir atoms are located in the inner part and Au atoms in the outermost region of the nanostructure. We found that IrAu nanoalloys present a distinctive synergistic effect with respect to reactivity. In addition, the projected density of electronic states (PDOS) energies were analyzed by examining the d-band shift to estimate the reactivity of various IrAu nanoalloys. Furthermore, the adsorption energies for the CO molecule in the domains of the Ir–Au interface were evaluated. In this sense, the addition of Au atoms to Ir clusters increases the reactivity of Ir by generating unoccupied orbitals near the Fermi level as indicated by the PDOS study. read less NOT USED (low confidence) B. Yang and B. Zheng, “The Adhesion Force in Nano-Contact During Approaching and Retrieving Processes.” 2018. link Times cited: 0 NOT USED (low confidence) F. Wang and B. Li, “Surface and Interfacial Energies of Mg 17 Al 12 –Mg System.” 2018. link Times cited: 1 NOT USED (low confidence) X. Liu, X. Wen, and R. Hoffmann, “Surface Activation of Transition Metal Nanoparticles for Heterogeneous Catalysis: What We Can Learn from Molecular Dynamics,” ACS Catalysis. 2018. link Times cited: 43 Abstract: Many heterogeneous reactions catalyzed by nanoparticles occu… read moreAbstract: Many heterogeneous reactions catalyzed by nanoparticles occur at relatively high temperatures, which may modulate the surface morphology of nanoparticles during reaction. Inspired by the discovery of dynamic formation of active sites on gold nanoparticles, we explore theoretically the nature of the highly mobile atoms on the surface of nanoparticles of various sizes for 11 transition metals. Using molecular dynamics simulations, on a 3 nm Fe nanoparticle as an example, the effect of surface premelting and overall melting on the structure and physical properties of the nanoparticles is analyzed. When the nanoparticle is heated up, the atoms in the outer shell appear amorphous already at 900 K. Surface premelting is reached at 1050 K, with more than three liquid atoms, based on the Lindemann criterion. The activated atoms may transfer their extra kinetic energy to the rest of the nanoparticle and activate other atoms. The dynamic studies indicate that the number of highly mobile atoms on the surface increas... read less NOT USED (low confidence) C.-Y. Shih et al., “Two mechanisms of nanoparticle generation in picosecond laser ablation in liquids: the origin of the bimodal size distribution† †Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr08614h,” Nanoscale. 2018. link Times cited: 136 Abstract: Novel mechanisms of nanoparticle generation in laser ablatio… read moreAbstract: Novel mechanisms of nanoparticle generation in laser ablation in liquids are revealed in atomistic simulations and verified in experiments. read less NOT USED (low confidence) H. Zhang et al., “Effects of high pressure on microstructure evolution and crystallization mechanisms during solidification of nickel,” Materials Research Express. 2018. link Times cited: 4 Abstract: To deeply understand the effects of high pressure on microst… read moreAbstract: To deeply understand the effects of high pressure on microstructural evolutions and crystallization mechanisms of liquid metal Ni during solidification process, MD simulation studies have been performed under 7 pressures of 0 ∼ 30 GPa, at cooling rate of 1.0 × 1011 K s−1. Adopting several microstructural analyzing methods, especially the cluster-type index method (CTIM-2) to analyze the local microstructures in the system. It is found that the pressure has important influence on the formation and evolution of microstructures, especially of the main basic clusters in the system. All the simulation systems are directly solidified into crystal structures, and the 1421, 1422, 1441 and 1661 bond-types, as well the FCC (12 0 0 0 12 0), HCP (12 0 0 0 6 6) and BCC (14 6 0 8 0 0) clusters play a key role in the microstructure transitions from liquid to crystal structures. The crystallization temperature Tc is enhanced almost linearly with the increase of pressure. Highly interesting, it is found for the first time that there is an important phase transformation point from FCC to BCC structures between 20 ∼ 22.5 GPa during the solidification processes from the same initial liquid system at the same cooling rate. And the effect of increasing pressure is similar to that of decreasing cooling rate for the phase transformation of microstructures during solidification process of liquid metal Ni system, though they have different concrete effecting mechanisms. read less NOT USED (low confidence) L. Stepanova and S. Bronnikov, “Mathematical modeling of the crack growth in linear elastic isotropic materials by conventional fracture mechanics approaches and by molecular dynamics method: crack propagation direction angle under mixed mode loading,” Journal of Physics: Conference Series. 2018. link Times cited: 9 Abstract: The crack growth directional angles in the isotropic linear … read moreAbstract: The crack growth directional angles in the isotropic linear elastic plane with the central crack under mixed-mode loading conditions for the full range of the mixity parameter are found. Two fracture criteria of traditional linear fracture mechanics (maximum tangential stress and minimum strain energy density criteria) are used. Atomistic simulations of the central crack growth process in an infinite plane medium under mixed-mode loading using Large-scale Molecular Massively Parallel Simulator (LAMMPS), a classical molecular dynamics code, are performed. The inter-atomic potential used in this investigation is Embedded Atom Method (EAM) potential. The plane specimens with initial central crack were subjected to Mixed-Mode loadings. The simulation cell contains 400000 atoms. The crack propagation direction angles under different values of the mixity parameter in a wide range of values from pure tensile loading to pure shear loading in a wide diapason of temperatures (from 0.1 К to 800 К) are obtained and analyzed. It is shown that the crack propagation direction angles obtained by molecular dynamics method coincide with the crack propagation direction angles given by the multi-parameter fracture criteria based on the strain energy density and the multi-parameter description of the crack-tip fields. read less NOT USED (low confidence) A. Erturk, Y. Yildiz, and M. Kırca, “Mechanical behavior of a novel carbon-based nanostructured aluminum material,” Computational Materials Science. 2018. link Times cited: 5 NOT USED (low confidence) M. Kbirou, M. Mazroui, and A. Hasnaoui, “Atomic packing and fractal behavior of Al-Co metallic glasses,” Journal of Alloys and Compounds. 2018. link Times cited: 17 NOT USED (low confidence) L. Deng et al., “Local identification of chemical ordering: Extension, implementation, and application of the common neighbor analysis for binary systems,” Computational Materials Science. 2018. link Times cited: 8 NOT USED (low confidence) J. Zhang et al., “Molecular dynamics simulation of the melting behavior of copper nanorod,” Computational Materials Science. 2018. link Times cited: 30 NOT USED (low confidence) J. Hinks et al., “Effects of crystallographic and geometric orientation on ion beam sputtering of gold nanorods,” Scientific Reports. 2018. link Times cited: 10 NOT USED (low confidence) M. Park, C. Hwang, and K. Jeong, “Nanoplasmonic Alloy of Au/Ag Nanocomposites on Paper Substrate for Biosensing Applications.,” ACS applied materials & interfaces. 2018. link Times cited: 43 Abstract: Plasmonic alloy has attracted much interest in tailoring loc… read moreAbstract: Plasmonic alloy has attracted much interest in tailoring localized surface plasmon resonance (LSPR) for recent biosensing techniques. In particular, paper-based plasmonic substrates allow capillary-driven lateral flow as well as three-dimensional metal nanostructures, and therefore they become actively transferred to LSPR-based biosensing such as surface-enhanced Raman spectroscopy (SERS) or metal-enhanced fluorescence (MEF). However, employing plasmonic alloy nanoislands on heat-sensitive substrate is still challenging, which significantly inhibits broad-range tailoring of the plasmon resonance wavelength (PRW) for superior sensitivity. Here we report paper-based plasmonic substrate with plasmonic alloy of Au/Ag nanocomposites for highly sensitive MEF and SERS biosensing applications. The nanofabrication procedures include concurrent deposition of Au and Ag below 100 °C without any damage on cellulose fibers. The Au/Ag nanocomposites feature nanoplasmonic alloy with single plasmon peak as well as broad-range tunability of PRW by composition control. This paper-based plasmonic alloy substrate enables about twofold enhancement of fluorescence signals and selective MEF after paper chromatography. The experimental results clearly demonstrate extraordinary enhancement in SERS signals for picomolar detection of folic acid as a cancer biomarker. This new method provides huge opportunities for fabricating plasmonic alloy on heat-sensitive substrate and biosensing applications. read less NOT USED (low confidence) C. Mahr et al., “Measurement of local crystal lattice strain variations in dealloyed nanoporous gold,” Materials Research Letters. 2018. link Times cited: 10 Abstract: ABSTRACT Reversible macroscopic length changes in nanoporous… read moreAbstract: ABSTRACT Reversible macroscopic length changes in nanoporous structures can be achieved by applying electric potentials or by exposing them to different gases or liquids. Thus, these materials are interesting candidates for applications as sensors or actuators. Macroscopic length changes originate from microscopic changes of crystal lattice parameters. In this report, we show spatially resolved measurements of crystal lattice strain in dealloyed nanoporous gold. The results confirm theory by indicating a compression of the lattice along the axis of cylindrically shaped ligaments and an expansion in radial direction. Furthermore, we show that curved npAu surfaces show inward relaxation of the surface layer. GRAPHICAL ABSTRACT IMPACT STATEMENT We show spatially resolved measurements of strain in nanoporous gold confirming theory: Crystal lattice is compressed along the axis of cylindrical ligaments and expanded in radial direction, surfaces relax inward. read less NOT USED (low confidence) L. Stepanova, “Modeling of crack growth under mixed-mode loading by a molecular dynamics method and a linear fracture mechanics approach.” 2017. link Times cited: 2 Abstract: Atomistic simulations of the central crack growth process in… read moreAbstract: Atomistic simulations of the central crack growth process in an infinite plane medium under mixed-mode loading using Large-Scale Atomic/Molecular Massively Parallel Simulator (LAMMPS), a classical molecular dynamics code, are performed. The inter-atomic potential used in this investigation is the Embedded Atom Method (EAM) potential. Plane specimens with an initial central crack are subjected to mixed-mode loadings. The simulation cell contains 400,000 atoms. The crack propagation direction angles under different values of the mixity parameter in a wide range of values from pure tensile loading to pure shear loading in a wide range of temperatures (from 0.1 K to 800 K) are obtained and analyzed. It is shown that the crack propagation direction angles obtained by molecular dynamics coincide with the crack propagation direction angles given by the multi-parameter fracture criteria based on the strain energy density and the multi-parameter description of the crack-tip fields. The multi-parameter fracture cri... read less NOT USED (low confidence) B. Deng et al., “Wrinkle-Free Single-Crystal Graphene Wafer Grown on Strain-Engineered Substrates.,” ACS nano. 2017. link Times cited: 154 Abstract: Wrinkles are ubiquitous for graphene films grown on various … read moreAbstract: Wrinkles are ubiquitous for graphene films grown on various substrates by chemical vapor deposition at high temperature due to the strain induced by thermal mismatch between the graphene and substrates, which greatly degrades the extraordinary properties of graphene. Here we show that the wrinkle formation of graphene grown on Cu substrates is strongly dependent on the crystallographic orientations. Wrinkle-free single-crystal graphene was grown on a wafer-scale twin-boundary-free single-crystal Cu(111) thin film fabricated on sapphire substrate through strain engineering. The wrinkle-free feature of graphene originated from the relatively small thermal expansion of the Cu(111) thin film substrate and the relatively strong interfacial coupling between Cu(111) and graphene, based on the strain analyses as well as molecular dynamics simulations. Moreover, we demonstrated the transfer of an ultraflat graphene film onto target substrates from the reusable single-crystal Cu(111)/sapphire growth substrate. The wrinkle-free graphene shows enhanced electrical mobility compared to graphene with wrinkles. read less NOT USED (low confidence) F. Liu, Z. Liu, X. Pei, J. Hu, and Z. Zhuo, “Modeling high temperature anneal hardening in Au submicron pillar by developing coupled dislocation glide-climb model,” International Journal of Plasticity. 2017. link Times cited: 26 NOT USED (low confidence) J. Liu et al., “Lateral force modulation by moiré superlattice structure: Surfing on periodically undulated graphene sheets,” Carbon. 2017. link Times cited: 14 NOT USED (low confidence) S. S. Hayat, Z. Rehman, and Z. Shah, “A study of dynamical evolution of small two-dimensional Copper islands’ diffusion on Ag(111) surface and observed surface effects,” Modern Physics Letters B. 2017. link Times cited: 3 Abstract: We study the diffusion of two-dimensional Cun(1 ≤ n ≤ 9) isl… read moreAbstract: We study the diffusion of two-dimensional Cun(1 ≤ n ≤ 9) islands on Ag(111) surface using molecular dynamics (MD) simulations. The work is the extension of calculations of monomer and dimer Hayat et al. [Phys. Rev. B 82 (2010) 085411] and trimer results Shah et al. [Phys. Lett. A 378 (2014) 1732]. Simulations carried out at three different temperatures — 300, 500, and 700 K — show the concerted motion to be dominant for the smaller islands (2- to 4-atoms), while the shape-changing multiple-atom processes are responsible for the diffusion of larger islands. Arrhenius plots of the diffusion coefficients reveal that the effective energy barrier is less than 260 ± 5 meV for the largest island size of Cu/Ag(111). There is a scaling of the effective energy barrier with size to some extent, but most notably it remains constant for islands with 4- to 6-atoms. The diffusion coefficient increases within a factor of 10 at the three temperatures 300, 500, and 700 K. The observed anharmonic features of the Cun adislands (breakage and pop–up) at Ag(111) surface as well as the surface anharmonicity of the Ag-substrate (fissures, dislocations, vacancy generation, and atomic exchange), are also presented. These findings can serve as an input for kinetic Monte Carlo (KMC) simulations. For the smaller sized islands the variation in the effective energy barrier with the island size is in good agreement with the experimental findings. read less NOT USED (low confidence) H. Liu, X. Zhu, Y. Sun, and W. Xie, “Evolution of stacking fault tetrahedral and work hardening effect in copper single crystals,” Applied Surface Science. 2017. link Times cited: 33 NOT USED (low confidence) H. Yang et al., “Substrate Effects in Graphene‐Based Electric Double‐Layer Capacitors: The Pivotal Interplays between Ions and Solvents.” 2017. link Times cited: 9 Abstract: Graphene has been considered as a promising active material … read moreAbstract: Graphene has been considered as a promising active material for electric double-layer capacitors (EDLCs) primarily owing to its extraordinary monolayer properties, whereas the interfacial behaviors are conspicuously impacted by underlying substrates. In this work, substrate effects on the interfacial wettability, EDL structure, and capacitive behavior of graphene-based EDLCs are delineated with numerical simulation. Unlike previous studies, a partially wetting transparency of topmost graphene is recognized for hydrophilic supports. Especially, a virtually identical capacitance is demonstrated for graphene with various supports, albeit the substantially different EDL structures stemmed from substrate effect. The achieved invariant capacitance is prominently attributed to the counterbalancing correlations between ions and proximal solvents, going beyond traditional views of modulating capacitance preferentially via ion structural evolutions. Specifically, the suppressed permittivity of apparently-ordered water dipoles (i.e., detrimental solvent effects) attenuates the beneficial ionic influences (i.e., reinforced population and closer approach) on shielding the external electric fields. The as-obtained findings illuminate the paramount importance of substrate in mediating interfacial behaviors within electrified EDLC system, and highlight that exploiting the pivotal interplay between ions and solvents could be a novel avenue to further manipulate electrochemical performances. read less NOT USED (low confidence) J. Che et al., “Selective suppression of toluene formation in solvent-free benzyl alcohol oxidation using supported Pd-Ni bimetallic nanoparticles,” Chinese Journal of Catalysis. 2017. link Times cited: 21 NOT USED (low confidence) Z. Liang, T. Biben, and P. Keblinski, “Molecular simulation of steady-state evaporation and condensation: Validity of the Schrage relationships,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 71 NOT USED (low confidence) G. Lu, L. Lin, S. Hui, S. Wang, X.-dong Wang, and D.-J. Lee, “Dewetting kinetics of metallic liquid films: Competition between unbalanced Young’s force and dissolutive reaction,” Chemical Physics Letters. 2017. link Times cited: 7 NOT USED (low confidence) H. Yang, Z. Bo, J. Yang, J.-hua Yan, and K. Cen, “Towards understanding the effects of van der Waals strengths on the electric double-layer structures and capacitive behaviors,” Journal of Power Sources. 2017. link Times cited: 13 NOT USED (low confidence) B. R. H. de Aquino, M. Neek‐Amal, and M. Milošević, “Unconventional two-dimensional vibrations of a decorated carbon nanotube under electric field: linking actuation to advanced sensing ability,” Scientific Reports. 2017. link Times cited: 1 NOT USED (low confidence) H. Tran, H. Tummala, L. Duchêne, T. Pardoen, M. Fivel, and A. Habraken, “Quasicontinuum analysis of dislocation-coherent twin boundary interaction to provide local rules to discrete dislocation dynamics.” 2017. link Times cited: 0 Abstract: The interaction of a pure screw dislocation with a Coherent … read moreAbstract: The interaction of a pure screw dislocation with a Coherent Twin Boundary Σ3 in copper was studied using the Quasicontinuum method. Coherent Twin Boundary behaves as a strong barrier to dislocation glide and prohibits slip transmission across the boundary. Dislocation pileup modifies the stress field at its intersection with the Grain Boundary (GB). A methodology to estimate the strength of the barrier for a dislocation to slip across CTB is proposed. A screw dislocation approaching the boundary from one side either propagates into the adjacent twin grain by cutting through the twin boundary or is stopped and increases the dislocation pileup amplitude at the GB. Quantitative estimation of the critical stress for transmission was performed using the virial stress computed by Quasicontinuum method. The transmission mechanism and critical stress are in line with the literature. Such information can be used as input for dislocation dynamic simulations for a better modeling of grain boundaries. read less NOT USED (low confidence) Y. Zhang, Q. Liu, and B. Xu, “Liquid-assisted, etching-free, mechanical peeling of 2D materials,” Extreme Mechanics Letters. 2017. link Times cited: 22 NOT USED (low confidence) S. Zimnik, M. Dickmann, and C. Hugenschmidt, “In-situ observation of the temperature and orientation dependence of the surface concentration of Ni adatoms deposited on Pd,” Surface Science. 2017. link Times cited: 0 NOT USED (low confidence) D. Niu, L. Guo, H. Hu, and G. Tang, “Dropwise condensation heat transfer model considering the liquid-solid interfacial thermal resistance,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 42 NOT USED (low confidence) H. Zhang, H. Zhang, F. Liu, Y. Yang, and D. Sun, “The Molecular Dynamics Study of Vacancy Formation During Solidification of Pure Metals,” Scientific Reports. 2017. link Times cited: 24 NOT USED (low confidence) 高 廷红, “Influence of Ag/Cu Micro-Doping on the Fusing Time and Fusing Position of Au Nanowires,” Modern Physics. 2017. link Times cited: 0 Abstract: 随着高精密仪器设备对熔断器要求的不断增加,设计高精度熔断器成为研究重点。本文基于分子动力学模拟方法,研究Ag、Cu原子掺… read moreAbstract: 随着高精密仪器设备对熔断器要求的不断增加,设计高精度熔断器成为研究重点。本文基于分子动力学模拟方法,研究Ag、Cu原子掺杂对Au纳米线熔断时间的影响和熔断位置的调控机制。统计分析了不同掺杂形式对Au纳米线熔断时间的影响,结合可视化技术研究熔断过程的微观结构变化情况。研究结果表明:单原子掺杂会一定程度缩短熔断时间,但不会对熔断位置产生明显影响。三层Ag或Cu原子掺杂对纳米线的熔断时间具有较大影响,明显降低了熔断时间。三层Ag、Cu原子掺杂对纳米线熔断位置的影响截然不同,Ag掺杂纳米线的熔断位置出现在掺Ag区域,而Cu掺杂纳米线的熔断位置远离掺Cu区域。Ag和Cu掺杂能够向相反方向调控纳米线的熔断位置,这对设计高精度熔断器意义重大。 With the increasing requirement on the high precision fuses of the instruments and equipments, designing high precision fuses becomes the focus of research. In this paper, the effects of Ag and Cu doping on fusing times and fusing position of the Au nanowires during the fusion processes are investigated based on molecular dynamics simulations. The effects of different doping on the fusing time of Au nanowires are analyzed by the statistical analysis method. The visualization technology has been used to trace the structural evolution of nanowires during the fusion processes. The results reveal that the fusing times are abbreviated by the single atom doping, but have no obvious influence on the fusing position to some extent. Three atomic layers doping with Ag and Cu atoms in nanowires both significantly reduce the fusing time. However, the fusing positions are quite different by doping three atomic layers of Ag or Cu atoms. The fusing position appears in the place where dopes many Ag atoms, while the situation is quite different by doping three atomic layers of Cu atoms. Ag and Cu doping can regulate the fusing time and position of nanowires, which is of great significance to design high precision fuses. read less NOT USED (low confidence) W. Liu et al., “A molecular dynamics simulation study of irradiation induced defects in gold nanowire,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 13 NOT USED (low confidence) A. Kotri, E. E. koraychy, M. Mazroui, and Y. Boughaleb, “Static investigation of adsorption and hetero‐diffusion of copper, silver, and gold adatoms on the (111) surface,” Surface and Interface Analysis. 2017. link Times cited: 16 Abstract: In this work, we have used the static molecular simulations … read moreAbstract: In this work, we have used the static molecular simulations combined with an interatomic potential derived from the embedded‐atom method to study the adsorption and hetero‐diffusion on the (111) surface of Cu, Ag, and Au adatoms by using LAMMPS code. The investigation is performed for six heterogeneous systems such as Ag/Au(111), Ag/Cu(111), Au/Ag(111), Au/Cu(111), Cu/Ag(111), and Cu/Au(111). First, we have investigated the relaxation trends and the bond lengths of the atoms in the systems. The calculation results show that, the top layer spacing between the first and second layers of the Au(111), Ag(111), and Cu(111) substrates is contracted. This contraction is found to be more important in the Au(111) substrate. On the other hand, the strong reduction of the binding length is found in Au/Cu(111) for the different adsorption sites. In addition, the binding, adsorption, and static activation energies for all studied systems were examined. The results indicated that the binding and adsorption energies reached their maximum values in the Au/Cu(111) and Au/Ag(111) systems, respectively. Moreover, the static activation barriers for hopping diffusion on the (111) surfaces are found to be low compared with those found in the (100) and (110) surfaces. Therefore, our calculations showed that the difference in energy between the hcp and fcc sites on the (111) surfaces is very small. Copyright © 2017 John Wiley & Sons, Ltd. read less NOT USED (low confidence) A. Ito, S. Kato, A. Takayama, and H. Nakamura, “Automatic kinetic Monte-Carlo modeling for impurity atom diffusion in grain boundary structure of tungsten material,” Nuclear materials and energy. 2017. link Times cited: 6 NOT USED (low confidence) J. Han, L. Zhang, R. Car, and E. Weinan, “Deep Potential: a general representation of a many-body potential energy surface,” arXiv: Computational Physics. 2017. link Times cited: 153 Abstract: We present a simple, yet general, end-to-end deep neural net… read moreAbstract: We present a simple, yet general, end-to-end deep neural network representation of the potential energy surface for atomic and molecular systems. This methodology, which we call Deep Potential, is "first-principle" based, in the sense that no ad hoc approximations or empirical fitting functions are required. The neural network structure naturally respects the underlying symmetries of the systems. When tested on a wide variety of examples, Deep Potential is able to reproduce the original model, whether empirical or quantum mechanics based, within chemical accuracy. The computational cost of this new model is not substantially larger than that of empirical force fields. In addition, the method has promising scalability properties. This brings us one step closer to being able to carry out molecular simulations with accuracy comparable to that of quantum mechanics models and computational cost comparable to that of empirical potentials. read less NOT USED (low confidence) A. Ahadi, P. Hansson, and S. Melin, “Tensile behavior of single-crystal nano-sized Cu beams – Geometric scaling effects,” Computational Materials Science. 2017. link Times cited: 12 NOT USED (low confidence) B. Wang, X.-dong Wang, T.-H. Wang, G. Lu, and W. M. Yan, “Enhancement of boiling heat transfer of thin water film on an electrified solid surface,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 34 NOT USED (low confidence) K. Chen, L. Wang, Y. Chen, and Q. Wang, “Molecular dynamics simulation of microstructure evolution and heat dissipation of nanoscale friction,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 21 NOT USED (low confidence) C. Zhang et al., “Electron contributions to the heat conduction across Au/graphene/Au interfaces,” Carbon. 2017. link Times cited: 22 NOT USED (low confidence) Z. Zhao, J.-S. Sun, X.-L. Zhang, H. Yang, and Z.-L. Liu, “Anharmonic Properties of Aluminum from Direct Free Energy Interpolation Method,” Communications in Theoretical Physics. 2017. link Times cited: 2 Abstract: We compare the direct free energy interpolation (DFEI) metho… read moreAbstract: We compare the direct free energy interpolation (DFEI) method and the quasi-harmonic approximation (QHA) in calculating of the equation of states and thermodynamic properties of prototype Al. The Gibbs free energy of Al is calculated using the DFEI method based on the high-temperature phonon density of states reduced from classical molecular dynamics simulations. Then, we reproduce the thermal expansion coefficients, the specific heat, the isothermal bulk modulus of Al accurately. By comparing the results from the DFEI method and the QHA, we find that the DFEI method is indeed more accurate in calculating anharmonic properties than the QHA. read less NOT USED (low confidence) O. Peña-Rodríguez et al., “Understanding the ion-induced elongation of silver nanoparticles embedded in silica,” Scientific Reports. 2017. link Times cited: 62 NOT USED (low confidence) A. Subashiev and H. Nee, “Hydrogen trapping at divacancies and impurity-vacancy complexes in nickel: First principles study,” Journal of Nuclear Materials. 2017. link Times cited: 21 NOT USED (low confidence) L. Koch et al., “Local segregation versus irradiation effects in high-entropy alloys : Steady-state conditions in a driven system,” Journal of Applied Physics. 2017. link Times cited: 61 Abstract: We study order transitions and defect formation in a model h… read moreAbstract: We study order transitions and defect formation in a model high-entropy alloy (CuNiCoFe) under ion irradiation by means of molecular dynamics simulations. Using a hybrid Monte-Carlo/molecular dynamics scheme, a model alloy is generated which is thermodynamically stabilized by configurational entropy at elevated temperatures, but partly decomposes at lower temperatures by copper precipitation. Both the high-entropy and the multiphase sample are then subjected to simulated particle irradiation. The damage accumulation is analyzed and compared to an elemental Ni reference system. The results reveal that the high-entropy alloy—independent of the initial configuration—installs a certain fraction of short-range order even under particle irradiation. Moreover, the results provide evidence that defect accumulation is reduced in the high-entropy alloy. This is because the reduced mobility of point defects leads to a steady state of defect creation and annihilation. The lattice defects generated by irradiation are ... read less NOT USED (low confidence) L. Luo, Z. Duan, H. Li, J. Kim, G. Henkelman, and R. Crooks, “Tunability of the Adsorbate Binding on Bimetallic Alloy Nanoparticles for the Optimization of Catalytic Hydrogenation.,” Journal of the American Chemical Society. 2017. link Times cited: 85 Abstract: In this paper, we show that PtAu and PdAu random alloy dendr… read moreAbstract: In this paper, we show that PtAu and PdAu random alloy dendrimer-encapsulated nanoparticles with an average size of ∼1.6 nm have different catalytic activity trends for allyl alcohol hydrogenation. Specifically, PtAu nanoparticles exhibit a linear increase in activity with increasing Pt content, whereas PdAu dendrimer-encapsulated nanoparticles show a maximum activity at a Pd content of ∼60%. Both experimental and theoretical results suggest that this contrasting behavior is caused by differences in the strength of H binding on the PtAu and PdAu alloy surfaces. The results have significant implications for predicting the catalytic performance of bimetallic nanoparticles on the basis of density functional theory calculations. read less NOT USED (low confidence) R. Ramakrishnan and R. Sankarasubramanian, “Crystal-melt kinetic coefficients of Ni3Al,” Acta Materialia. 2017. link Times cited: 18 NOT USED (low confidence) Z.-L. Liu, R. Li, X.-L. Zhang, N. Qu, and L. Cai, “Direct anharmonic correction method by molecular dynamics,” Comput. Phys. Commun. 2017. link Times cited: 3 NOT USED (low confidence) M. N. Esfahani, M. Yilmaz, M. R. Sonne, J. Hattel, and B. E. Alaca, “Selecting the optimum engineering model for the frequency response of fcc nanowire resonators,” Applied Mathematical Modelling. 2017. link Times cited: 7 NOT USED (low confidence) N. T. Brown, J. Qu, and E. Martínez, “Modeling material interfaces with hybrid adhesion method,” Computational Materials Science. 2017. link Times cited: 1 NOT USED (low confidence) G. Demange, L. Lunéville, V. Pontikis, and D. Simeone, “Prediction of irradiation induced microstructures using a multiscale method coupling atomistic and phase field modeling: Application to the AgCu model alloy,” Journal of Applied Physics. 2017. link Times cited: 18 Abstract: Microstructure patterning using the ion beam mixing process … read moreAbstract: Microstructure patterning using the ion beam mixing process results from the competition between thermal diffusion and ballistic disordering induced by impinging ions. Although microstructure patterning under irradiation is now qualitatively understood, so far, no study could quantitatively estimate irradiation conditions leading to patterning. In this work, a new multiscale approach based on phase field was developed to simulate the microstructure evolution, and the occurrence of patterning due to ion irradiation in a silver-copper alloy, from atomic to microstructural scale. For that purpose, an efficient numerical scheme was developed to simulate the microstructure dynamics, within the framework of phase field. Equilibrium parameters of AgCu were computed using a mixed Monte Carlo-molecular dynamics approach. Ballistic effects induced by krypton ion irradiation, and point defect recreation leading to irradiation enhanced diffusion, were estimated using the binary collision approximation framework. As a... read less NOT USED (low confidence) B. Li et al., “Superfast assembly and synthesis of gold nanostructures using nanosecond low-temperature compression via magnetic pulsed power,” Nature Communications. 2017. link Times cited: 28 NOT USED (low confidence) S. Zimnik, C. Piochacz, S. Vohburger, and C. Hugenschmidt, “Comparative Study of Time-Dependent PAES and XPS on a Ni/Pd Surface,” Defect and Diffusion Forum. 2017. link Times cited: 1 Abstract: We report on time-dependent Positron annihilation induced Au… read moreAbstract: We report on time-dependent Positron annihilation induced Auger Electron Spectroscopy (PAES) study on 0.5 monolayers (ML) Ni on polycrystalline Pd accompanied by complementary X-ray induced Photoelectron Spectroscopy (XPS). The normalized PAES spectra showed a significant decrease in the Ni intensity and an increase in the Pd intensity as a function of time. To rule out varying influence on the elements e.g. from surface contaminates due to the residual gas, a time-dependent XPS analysis was performed on pure Ni and Pd as well as to analyze the main contaminants C and O. The O fraction was found to be constant within the measurement time and the time constants for C significantly differ from those of Ni and Pd in the PAES data. Consequently, it was concluded that the PAES data show a superposition of C contamination and structural changes at the surface of Ni/Pd. read less NOT USED (low confidence) Y. Moon, H. D. Mai, and H. Yoo, “Platinum Overgrowth on Gold Multipod Nanoparticles: Investigation of Synergistic Catalytic Effects in a Bimetallic Nanosystem.” 2017. link Times cited: 17 Abstract: Highly-ordered heteronanostructures can be synthesized by ov… read moreAbstract: Highly-ordered heteronanostructures can be synthesized by overgrowth of a secondary material on the as-prepared seed nanocrystal. The unique morphology of seed nanocrystals directs the shape of the secondary structure, which is exploited to develop structure-controlled composite nanomaterials. Herein, we report a facile and controllable synthetic strategy toward noble star-shaped bimetallic nanostructures. Gold multipod nanoparticle (GMN) core-platinum shell nanoparticles (GMN@Pt NPs) are successfully synthesized through the overgrowth of Pt on the surface of GMN seeds. Due to the structural uniqueness of GMNs, GMN@Pt NPs show bimetallic core-shell geometries, which have surface roughness and star-shaped multi-branches with sharp edges and tips. The island nucleation and growth of individual Pt particles to form a shell provide larger surface areas and plenty of defects on the bimetallic nanocrystals. GMN@Pt NPs with optimized Pt shells show much higher catalytic activities in 4-nitrophenol reduction and ethanol electrooxidation than those of GMNs or Pt nanoparticles, mainly due to the synergistic effects of the bicomponent nanosystem. read less NOT USED (low confidence) V. Zhdanov and C. Langhammer, “Charge transfer between sensing and targeted metal nanoparticles in indirect nanoplasmonic sensors,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 3 NOT USED (low confidence) J. Wang, L. Niu, and Y. Zhang, “Ab initio study of He-He interactions in homogeneous electron gas,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 2 NOT USED (low confidence) H. Huan, B. Fu, and X. Ye, “The torsional mechanical properties of copper nanowires supported by carbon nanotubes,” Physics Letters A. 2017. link Times cited: 8 NOT USED (low confidence) E. E. koraychy, K. Sbiaai, M. Mazroui, R. Ferrando, and Y. Boughaleb, “Heterodiffusion of Ag adatoms on imperfect Au(110) surfaces,” Chemical Physics Letters. 2017. link Times cited: 7 NOT USED (low confidence) S. Bukkuru, U. Bhardwaj, M. Warrier, A. Rao, and M. C. Valsakumar, “Identifying self-interstitials of bcc and fcc crystals in molecular dynamics,” Journal of Nuclear Materials. 2017. link Times cited: 6 NOT USED (low confidence) F. Yang, R. Carpick, and D. Srolovitz, “Mechanisms of Contact, Adhesion, and Failure of Metallic Nanoasperities in the Presence of Adsorbates: Toward Conductive Contact Design.,” ACS nano. 2017. link Times cited: 17 Abstract: The properties of contacting interfaces are strongly affecte… read moreAbstract: The properties of contacting interfaces are strongly affected not only by the bulk and surface properties of contacting materials but also by the ubiquitous presence of adsorbed contaminants. Here, we focus on the properties of single asperity contacts in the presence of adsorbates within a molecular dynamics description of metallic asperity normal contact and a parametric description of adsorbate properties. A platinum-platinum asperity contact is modeled with adsorbed oligomers with variable properties. This system is particularly tailored to the context of nanoelectromechanical system (NEMS) contact switches, but the results are generally relevant to metal-metal asperity contacts in nonpristine conditions. Even though mechanical forces can displace adsorbate out of the contact region, increasing the adsorbate layer thickness and/or adsorbate/metal adhesion makes it more difficult for metal asperity/metal surface contact to occur, thereby lowering the electrical contact conductance. Contact separation is a competition between plastic necking in the asperity or decohesion at the asperity/substrate interface. The mechanism which operates at a lower tensile stress dominates. Necking dominates when the adsorbate/metal adhesion is strong and/or the adsorbate layer thickness is small. In broad terms, necking implies larger asperity deformation and mechanical work, as compared with decohesion. Optimal NEMS switch performance requires substantial contact conductance and minimal asperity deformation; these results indicate that these goals can be achieved by balancing the quantity of adsorbates and their adhesion to the metal surface. read less NOT USED (low confidence) P. Brault et al., “Molecular dynamics simulations of ternary PtxPdyAuz fuel cell nanocatalyst growth,” International Journal of Hydrogen Energy. 2016. link Times cited: 8 NOT USED (low confidence) J. Zhang, Z. Wang, T. Sun, and Y. Yan, “Influence of stacking fault energy on friction of nanotwinned metals,” Materials Research Express. 2016. link Times cited: 1 Abstract: The unique dislocation–twin boundary (TB) interactions that … read moreAbstract: The unique dislocation–twin boundary (TB) interactions that govern the extraordinary mechanical properties of nanotwinned (NT) metals have the strong intrinsic effect of material energy and the extrinsic effect of feature size. In this work, we perform molecular dynamics (MD) simulations to elucidate fundamental deformation mechanisms of two NT face-centered cubic (FCC) metals (Cu and Pd) under probe-based friction, with an emphasis on evaluating the influence of both material’s intrinsic energy barrier and extrinsic grain size on the microscopic deformation behavior and correlated macroscopic frictional results of the materials. Simulation results reveal that individual deformation modes of dislocation mechanisms, dislocation–TB interactions, TB-associated mechanisms, deformation twinning and grain boundary (GB) accommodation work in parallel in the plastic deformation of the materials, and their competition is strongly influenced by both the intrinsic energy barriers for the nucleation of stacking faults and twin faults, and the extrinsic grain size. Consequently, both the frictional response and worn surface morphology present strong anisotropic characteristics. It is also found that the deformation behavior of NT Pd under a localized multi-axis stress state is significantly different from that which occurs under a uniaxial stress state. These findings will advance the rational design and synthesis of nanostructured materials with advanced frictional properties. read less NOT USED (low confidence) S. Melin, A. Ahadi, and P. Hansson, “Tensile Behavior of Single-Crystal Nanosized Copper Beams with Voids,” Solid State Phenomena. 2016. link Times cited: 0 Abstract: The tensile response under displacement controlled loading o… read moreAbstract: The tensile response under displacement controlled loading of nanosized single crystal Cu beams, solid or holding square shaped through-the thickness voids, have been investigated through 3D molecular dynamics simulations using free-ware LAMMPS [1]. For the same beam size and void height, the void width along the beam length axis was varied. Two different crystallographic orientations were considered. It was found that, under some circumstances, voids were able to close and heal the beam cross section, causing final failure through necking in the region of the initial void. For other cases instead the void split in two, smaller voids that both eventually healed. A third scenario was that the void widened, splitting the beam in two ligaments that each necked individually. As expected, both defect geometry and crystal orientation influences the mechanical behavior. read less NOT USED (low confidence) U. Jameel, M.-qiao Zhu, W. Tikkanen, X. Chen, and Z. Tong, “Recent fuel cell progress in nano gold hybrid materials for oxygen reduction reaction in alkaline media,” Materials Research Bulletin. 2016. link Times cited: 11 NOT USED (low confidence) Z. Yang, D. Wang, Z.-X. Lu, and W. Hu, “Atomistic simulation on the plastic deformation and fracture of bio-inspired graphene/Ni nanocomposites,” Applied Physics Letters. 2016. link Times cited: 37 Abstract: Molecular dynamics simulations were performed to investigate… read moreAbstract: Molecular dynamics simulations were performed to investigate the plastic deformation and fracture behaviors of bio-inspired graphene/metal nanocomposites, which have a “brick-and-mortar” nanostructure, consisting of hard graphene single-layers embedded in a soft Ni matrix. The plastic deformation mechanisms of the nanocomposites were analyzed as well as their effects on the mechanical properties with various geometrical variations. It was found that the strength and ductility of the metal matrix can be highly enhanced with the addition of the staggered graphene layers, and the plastic deformation can be attributed to the interfacial sliding, dislocation nucleation, and cracks' combination. The strength of the nanocomposites strongly depends on the length scale of the nanostructure and the interlayer distance as well. In addition, slip at the interface releases the stress in graphene layers, leading to the stress distribution on the graphene more uniform. The present results are expected to contribute to the design of the nanolayered graphene/metal composites with high performance. read less NOT USED (low confidence) Y. Fu, J. Michopoulos, and J. H. Song, “Bridging the multi phase-field and molecular dynamics models for the solidification of nano-crystals,” J. Comput. Sci. 2016. link Times cited: 25 NOT USED (low confidence) S. Sadeghzadeh, “Computational design of graphene sheets for withstanding the impact of ultrafast projectiles.,” Journal of molecular graphics & modelling. 2016. link Times cited: 17 NOT USED (low confidence) M. S. Talaei, N. Nouri, and S. Ziaei-Rad, “Grain boundary effects on nanoindentation of Fe bicrystal using molecular dynamic,” Mechanics of Materials. 2016. link Times cited: 19 NOT USED (low confidence) X. Liu, F. Wang, W. Wang, and H. Wu, “Interfacial strengthening and self-healing effect in graphene-copper nanolayered composites under shear deformation,” Carbon. 2016. link Times cited: 76 NOT USED (low confidence) S. Bukkuru, U. Bhardwaj, A. Rao, and M. Warrier, “Identifying Interstitials in MD Simulations — Max Space Clustering Method,” Journal of Physics: Conference Series. 2016. link Times cited: 1 Abstract: Molecular Dynamics (MD) simulations are suitable to study th… read moreAbstract: Molecular Dynamics (MD) simulations are suitable to study the details of atomistic processes. Identification of interstitials is one of the main difficulties in the analysis of defect dynamics. To identify the interstitials, different methods use different criteria. All the methods, except the Wigner-Seitz method, assume some input parameters which may lead to an erroneous number of interstitials. We describe an unsupervised clustering algorithm, called Max Space clustering method, to identify interstitials without assuming any input parameters. We show that the algorithm is suited for identifying interstitials at high temperatures when amplitude of atomic oscillations become larger, a fact ignored by other models. read less NOT USED (low confidence) Y. D. Wei et al., “A comparative study on local atomic configurations characterized by cluster-type-index method and Voronoi polyhedron method,” Computational Materials Science. 2016. link Times cited: 30 NOT USED (low confidence) L. Sun, H. Hu, A. Rokoni, and Y. Sun, “Intrinsic instability of thin liquid films on nanostructured surfaces,” Applied Physics Letters. 2016. link Times cited: 3 Abstract: The instability of a thin liquid film on nanostructures is n… read moreAbstract: The instability of a thin liquid film on nanostructures is not well understood but is important in liquid-vapor two-phase heat transfer (e.g., thin film evaporation and boiling), lubrication, and nanomanufacturing. In thin film evaporation, the comparison between the non-evaporating film thickness and the critical film breakup thickness determines the stability of the film: the film becomes unstable when the critical film breakup thickness is larger than the non-evaporating film thickness. In this study, a closed-form model is developed to predict the critical breakup thickness of a thin liquid film on 2D periodic nanostructures based on the minimization of system free energy in the limit of a liquid monolayer. Molecular dynamics simulations are performed for water thin films on square nanostructures of varying depth and wettability, and the simulations agree with the model predictions. The results show that the critical film breakup thickness increases with the nanostructure depth and the surface wettabi... read less NOT USED (low confidence) J. Davoodi, M. Safaralizade, and M. Yarifard, “Molecular dynamics simulation of a Gold nanodroplet in contact with graphene,” Materials Letters. 2016. link Times cited: 11 NOT USED (low confidence) D. Kochmann and J. S. Amelang, “The Quasicontinuum Method: Theory and Applications.” 2016. link Times cited: 18 NOT USED (low confidence) M. Lodge et al., “Lubricity of gold nanocrystals on graphene measured using quartz crystal microbalance,” Scientific Reports. 2016. link Times cited: 17 NOT USED (low confidence) X.-Y. Zhou, B. Huang, and T.-Y. Zhang, “Size- and temperature-dependent Young’s modulus and size-dependent thermal expansion coefficient of thin films.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 16 Abstract: Nanomaterials possess a high surface/volume ratio and surfac… read moreAbstract: Nanomaterials possess a high surface/volume ratio and surfaces play an essential role in size-dependent material properties. In the present study, nanometer-thick thin films were taken as an ideal system to investigate the surface-induced size- and temperature-dependent Young's modulus and size-dependent thermal expansion coefficient. The surface eigenstress model was further developed with the consideration of thermal expansion, leading to analytic formulas of size- and temperature-dependent Young's modulus, and size-dependent thermal expansion coefficient of thin films. Molecular dynamics (MD) simulations on face-centered cubic (fcc) Ag, Cu, and Ni(001) thin films were conducted at temperatures ranging from 300 K to 600 K. The MD simulation results are perfectly consistent with the theoretical predictions, thereby verifying the theoretical approach. The newly developed surface eigenstress model will be able to attack similar problems in other types of nanomaterials. read less NOT USED (low confidence) A. Lipnitskii and V. Saveliev, “Development of n-body expansion interatomic potentials and its application for V,” Computational Materials Science. 2016. link Times cited: 20 NOT USED (low confidence) M. L. Jackson, P. Fossati, and R. Grimes, “Simulations of threshold displacement in beryllium,” Journal of Applied Physics. 2016. link Times cited: 9 Abstract: Atomic scale molecular dynamics simulations of radiation dam… read moreAbstract: Atomic scale molecular dynamics simulations of radiation damage have been performed on beryllium. Direct threshold displacement simulations along a geodesic projection of directions were used to investigate the directional dependence with a high spatial resolution. It was found that the directionally averaged probability of displacement increases from 0 at 35 eV, with the energy at which there is a 50% chance of a displacement occurring is 70 eV and asymptotically approaching 1 for higher energies. This is, however, strongly directionally dependent with a 50% probability of displacement varying from 35 to 120 eV, with low energy directions corresponding to the nearest neighbour directions. A new kinetic energy dependent expression for the average maximum displacement of an atom as a function of energy is derived which closely matches the simulated data. read less NOT USED (low confidence) S. Trady, M. Mazroui, A. Hasnaoui, and K. Saadouni, “Molecular dynamics study of atomic-level structure in monatomic metallic glass,” Journal of Non-crystalline Solids. 2016. link Times cited: 46 NOT USED (low confidence) R. Zhang, I. Beyerlein, S. Zheng, S. H. Zhang, A. Stukowski, and T. Germann, “Manipulating dislocation nucleation and shear resistance of bimetal interfaces by atomic steps,” Acta Materialia. 2016. link Times cited: 39 NOT USED (low confidence) P. N. Ram, V. Gairola, and P. D. Semalty, “Vibrational properties of vacancy in Au using modified embedded atom method potentials,” Journal of Physics and Chemistry of Solids. 2016. link Times cited: 2 NOT USED (low confidence) X. Long, B. Li, L. Wang, J. Huang, J. Zhu, and S. Luo, “Shock response of Cu/graphene nanolayered composites,” Carbon. 2016. link Times cited: 78 NOT USED (low confidence) D. T. Ho, S. Y. Kwon, and S. Y. Kim, “Metal [100] Nanowires with Negative Poisson’s Ratio,” Scientific Reports. 2016. link Times cited: 20 NOT USED (low confidence) Z. Fu et al., “First Principles Study of Structural and Electronic Properties of Pentagonal and Hexagonal Noble Metal Nanowires,” NANO. 2016. link Times cited: 0 Abstract: The equilibrium structure and electronic properties of four … read moreAbstract: The equilibrium structure and electronic properties of four ultrathin free-standing pentagonal and hexagonal noble metal nanowires, that is, copper nanowires (CuNWs), silver nanowires (AgNWs), gold nanowires (AuNWs) and platinum nanowires (PtNWs), have been studied comprehensively by adopting a first-principles simulation based on the density-functional theory. The staggered topologies are more stable than the eclipsed ones by analyzing the bonding energy. The staggered ones with a linear atom chain in the center of the pentagonal or hexagons topologies are the preferred structures for CuNWs and AgNWs, but the staggered ones without a linear atom chain in the center of the pentagon or hexagon are the preferred structures for AuNWs and PtNWs due to the increasing core–core repulsions. The calculated electronic band structures and density of states present that all the noble metal nanowires are metallic. The projected densities of states (PDOS) of dominant d-states and the charge density show that the narro... read less NOT USED (low confidence) H. Ma et al., “Eigenstress model for electrochemistry of solid surfaces,” Scientific Reports. 2016. link Times cited: 18 NOT USED (low confidence) L. Chang, A. Fisher, Z. Liu, and D. Cheng, “A density functional theory study of sulfur adsorption on Ag–Au nanoalloys,” Computational and Theoretical Chemistry. 2016. link Times cited: 3 NOT USED (low confidence) K. Lounis, H. Zenia, E. H. Megchiche, and C. Mijoule, “Stability of vacancy clusters in nickel: A molecular statics study,” Computational Materials Science. 2016. link Times cited: 8 NOT USED (low confidence) S. M. Rassoulinejad-Mousavi, Y. Mao, and Y. Zhang, “Evaluation of Copper, Aluminum and Nickel Interatomic Potentials on Predicting the Elastic Properties,” arXiv: Computational Physics. 2016. link Times cited: 63 Abstract: Choice of appropriate force field is one of the main concern… read moreAbstract: Choice of appropriate force field is one of the main concerns of any atomistic simulation that needs to be seriously considered in order to yield reliable results. Since, investigations on mechanical behavior of materials at micro/nanoscale has been becoming much more widespread, it is necessary to determine an adequate potential which accurately models the interaction of the atoms for desired applications. In this framework, reliability of multiple embedded atom method based interatomic potentials for predicting the elastic properties was investigated. Assessments were carried out for different copper, aluminum and nickel interatomic potentials at room temperature which is considered as the most applicable case. Examined force fields for the three species were taken from online repositories of National Institute of Standards and Technology (NIST), as well as the Sandia National Laboratories, the LAMMPS database. Using molecular dynamic simulations, the three independent elastic constants, C11, C12 and C44 were found for Cu, Al and Ni cubic single crystals. Voigt-Reuss-Hill approximation was then implemented to convert elastic constants of the single crystals into isotropic polycrystalline elastic moduli including Bulk, Shear and Young's modulus as well as Poisson's ratio. Simulation results from massive molecular dynamic were compared with available experimental data in the literature to justify the robustness of each potential for each species. Eventually, accurate interatomic potentials have been recommended for finding each of the elastic properties of the pure species. Exactitude of the elastic properties was found to be sensitive to the choice of the force fields. Those potentials were fitted for a specific compound may not necessarily work accurately for all the existing pure species. read less NOT USED (low confidence) A. Ulvestad et al., “In Situ 3D Imaging of Catalysis Induced Strain in Gold Nanoparticles.,” The journal of physical chemistry letters. 2016. link Times cited: 30 Abstract: Multielectron transfer processes are crucially important in … read moreAbstract: Multielectron transfer processes are crucially important in energy and biological science but require favorable catalysts to achieve fast kinetics. Nanostructuring catalysts can dramatically improve their properties, which can be difficult to understand due to strain- and size-dependent thermodynamics, the influence of defects, and substrate-dependent activities. Here, we report three-dimensional (3D) imaging of single gold nanoparticles during catalysis of ascorbic acid decomposition using Bragg coherent diffractive imaging (BCDI). Local strains were measured in single nanoparticles and modeled using reactive molecular dynamics (RMD) simulations and finite element analysis (FEA) simulations. RMD reveals the pathway for local strain generation in the gold lattice: chemisorption of hydroxyl ions. FEA reveals that the RMD results are transferable to the nanocrystal sizes studied in the experiment. Our study probes the strain-activity connection and opens a powerful avenue for theoretical and experimental studies of nanocrystal catalysis. read less NOT USED (low confidence) R. Wilhelm and W. Möller, “Charge-state-dependent energy loss of slow ions. II. Statistical atom model,” Physical Review A. 2016. link Times cited: 13 Abstract: A model for charge-dependent energy loss of slow ions is dev… read moreAbstract: A model for charge-dependent energy loss of slow ions is developed based on the Thomas-Fermi statistical model of atoms. Using a modified electrostatic potential which takes the ionic charge into account, nuclear and electronic energy transfers are calculated, the latter by an extension of the Firsov model. To evaluate the importance of multiple collisions even in nanometer-thick target materials we use the charge-state-dependent potentials in a Monte Carlo simulation in the binary collision approximation and compare the results to experiment. The Monte Carlo results reproduce the incident charge-state dependence of measured data well [see R. A. Wilhelm et al., Phys. Rev. A 93, 052708 (2016)], even though the experimentally observed charge exchange dependence is not included in the model. read less NOT USED (low confidence) S. Uniyal, M. Chand, S. Joshi, and P. D. Semalty, “Divacancy binding energy, formation energy and surface energy of BCC transition metals using MEAM potentials.” 2016. link Times cited: 2 Abstract: The modified embedded atom method (MEAM) potential parameter… read moreAbstract: The modified embedded atom method (MEAM) potential parameters have been employed to calculate the unrelaxed divacancy formation energy, binding energy and surface energies for low index planes in bcc transition metals. The calculated results of divacancy binding energy and vacancy formation energy compare well with experimental and other available calculated results. read less NOT USED (low confidence) S. Joshi, M. Chand, K. Dabral, and P. D. Semalty, “Phonon dispersion and local density of states in NiPd alloy using modified embedded atom method potential.” 2016. link Times cited: 0 Abstract: A modified embedded atom method (MEAM) potential model up to… read moreAbstract: A modified embedded atom method (MEAM) potential model up to second neighbours has been used to calculate the phonon dispersions for Ni0.55Pd0.45 alloy in which Pd is introduced as substitutional impurity. Using the force-constants obtained from MEAM potential, the local vibrational density of states in host Ni and substitutional Pd atoms using Green’s function method has been calculated. The calculation of phonon dispersions of NiPd alloy shows a good agreement with the experimental results. Condition of resonance mode has also been investigated and resonance mode in the frequency spectrum of impurity atom at low frequency is observed. read less NOT USED (low confidence) J. Ren, Z. G. Dong, J. Zhao, and P. Liu, “A novel approach for determining the minimum feed in nanochannels processing via molecular dynamics simulation,” Applied Surface Science. 2016. link Times cited: 8 NOT USED (low confidence) Y. Cai, Y.-L. Chen, Y. Shimizu, S. Ito, W. Gao, and L. Zhang, “Molecular dynamics simulation of subnanometric tool-workpiece contact on a force sensor-integrated fast tool servo for ultra-precision microcutting,” Applied Surface Science. 2016. link Times cited: 12 NOT USED (low confidence) C. Denton, J. Moreno-Marín, and S. Heredia‐Avalos, “Multiwalled carbon nanotubes as masks against carbon and argon irradiation. A molecular dynamics study,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2016. link Times cited: 1 NOT USED (low confidence) K. Lee and J. Ye, “Significantly improved thickness uniformity of graphene monolayers grown by chemical vapor deposition by texture and morphology control of the copper foil substrate,” Carbon. 2016. link Times cited: 27 NOT USED (low confidence) Z. Wu, A. Hu, Y.-ting Cui, and P. Yu, “Melting and crystallization in large sized copper cluster,” Integrated Ferroelectrics. 2016. link Times cited: 0 Abstract: ABSTRACT The melting and crystallization with two different … read moreAbstract: ABSTRACT The melting and crystallization with two different cooling rates of large sized CuN (N = 1956, 2112, 2208, and 2340) nanoclusters are simulated by using molecular dynamics technique with the frame work of embedded atom method. The potential energy as a function of temperatures is obtained and the structural details are analyzed. The results reveal that the melting and freezing temperatures increase almost linearly with the atom number of the clusters slowly. All the copper nanoclusters have negative heat capacity around the phase transition temperature, and the clusters with slow cooling rates have icosahedral structure at 300 K. read less NOT USED (low confidence) T. Fu et al., “Molecular dynamics simulation of effects of twin interfaces on Cu/Ni multilayers,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2016. link Times cited: 54 NOT USED (low confidence) E. T. Karim et al., “Experimental characterization and atomistic modeling of interfacial void formation and detachment in short pulse laser processing of metal surfaces covered by solid transparent overlayers,” Applied Physics A. 2016. link Times cited: 0 NOT USED (low confidence) X. Wu et al., “Thermal Transport across Surfactant Layers on Gold Nanorods in Aqueous Solution.,” ACS applied materials & interfaces. 2016. link Times cited: 38 Abstract: Ultrafast transient absorption experiments and molecular dyn… read moreAbstract: Ultrafast transient absorption experiments and molecular dynamics simulations are utilized to investigate the thermal transport between aqueous solutions and cetyltrimethylammonium bromide (CTAB)- or polyethylene glycol (PEG)-functionalized gold nanorods (GNRs). The transient absorption measurement data are interpreted with a multiscale heat diffusion model, which incorporates the interfacial thermal conductances predicted by molecular dynamics. According to our observations, the effective thermal conductance of the GNR/PEG/water system is higher than that of the GNR/CTAB/water system with a surfactant layer of the same length. We attribute the enhancement of thermal transport to the larger thermal conductance at the GNR/PEG interface as compared with that at the GNR/CTAB interface, in addition to the water penetration into the hydrophilic PEG layer. Our results highlight the role of the GNR/polymer thermal interfaces in designing biological and composite-based heat transfer applications of GNRs, and the importance of multiscale analysis in interpreting transient absorption data in systems consisting of low interfacial thermal conductances. read less NOT USED (low confidence) M. Cesaria, A. Taurino, M. Catalano, A. Caricato, and M. Martino, “Edge-melting: nanoscale key-mechanism to explain nanoparticle formation from heated TEM grids,” Applied Surface Science. 2016. link Times cited: 0 NOT USED (low confidence) S. Kawai et al., “Superlubricity of graphene nanoribbons on gold surfaces,” Science. 2016. link Times cited: 284 Abstract: A golden opportunity for graphene Reducing friction can limi… read moreAbstract: A golden opportunity for graphene Reducing friction can limit wear and improve the energy efficiency of mechanical devices. Graphene is a promising lubricant because the friction between sheets is minuscule under certain circumstances. Kawai et al. show that the same ultra-low frictional properties extend to other surfaces. They find ultralow friction when dragging graphene nanoribbons across a gold surface using an atomic force microscope. This discovery sets up the potential for developing nanographene frictionless coatings. Science, this issue p. 957 Experiments reveal ultralow friction when graphene nanoribbons slide across an oriented gold surface. The state of vanishing friction known as superlubricity has important applications for energy saving and increasing the lifetime of devices. Superlubricity, as detected with atomic force microscopy, appears when sliding large graphite flakes or gold nanoclusters across surfaces, for example. However, the origin of the behavior is poorly understood because of the lack of a controllable nanocontact. We demonstrated the superlubricity of graphene nanoribbons when sliding on gold with a joint experimental and computational approach. The atomically well-defined contact allows us to trace the origin of superlubricity, unraveling the role played by ribbon size and elasticity, as well as by surface reconstruction. Our results pave the way to the scale-up of superlubricity and thus to the realization of frictionless coatings. read less NOT USED (low confidence) F. Kong et al., “A large-scale simulation method on complex ternary Li–Mn–O compounds for Li-ion battery cathode materials,” Computational Materials Science. 2016. link Times cited: 12 NOT USED (low confidence) A. Zaretski et al., “Metallic Nanoislands on Graphene as Highly Sensitive Transducers of Mechanical, Biological, and Optical Signals,” Nano Letters. 2016. link Times cited: 61 Abstract: This article describes an effect based on the wetting transp… read moreAbstract: This article describes an effect based on the wetting transparency of graphene; the morphology of a metallic film (≤20 nm) when deposited on graphene by evaporation depends strongly on the identity of the substrate supporting the graphene. This control permits the formation of a range of geometries, such as tightly packed nanospheres, nanocrystals, and island-like formations with controllable gaps down to 3 nm. These graphene-supported structures can be transferred to any surface and function as ultrasensitive mechanical signal transducers with high sensitivity and range (at least 4 orders of magnitude of strain) for applications in structural health monitoring, electronic skin, measurement of the contractions of cardiomyocytes, and substrates for surface-enhanced Raman scattering (SERS, including on the tips of optical fibers). These composite films can thus be treated as a platform technology for multimodal sensing. Moreover, they are low profile, mechanically robust, semitransparent and have the potential for reproducible manufacturing over large areas. read less NOT USED (low confidence) D. Niu and G. Tang, “The effect of surface wettability on water vapor condensation in nanoscale,” Scientific Reports. 2016. link Times cited: 65 NOT USED (low confidence) M. Konuk and S. Durukanoğlu, “Shape-controlled growth of metal nanoparticles: an atomistic view.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 11 Abstract: Recent developments in shape-controlled synthesis of metalli… read moreAbstract: Recent developments in shape-controlled synthesis of metallic nano-particles present a promising path for precisely tuning chemical activity, selectivity, and stability of nano-materials. While previous studies have highlighted the macroscopic description of synthesis processes, there is less understanding as to whether individual atomic-scale processes possess any significant role in controlling the growth of nano-products. The presented molecular static and dynamic simulations are the first simulations to understand the underlying atomistic mechanisms of the experimentally determined growth modes of metal nano-clusters. Our simulations on Ag nano-cubes confirm that metal nano-seeds enclosed by {100} facets can be directed to grow into octopods, concave, truncated cubes, and cuboctahedra when the relative surface diffusion and deposition rates are finely tuned. Here we further showed that atomic level processes play a significant role in controllably fine tuning the two competing rates: surface diffusion and deposition. We also found that regardless of temperature and the initial shape of the nano-seeds, the exchange of the deposited atom with an edge atom of the seed is by far the governing diffusion mechanism between the neighboring facets, and thus is the leading atomistic process determining the conditions for fine tuning of macroscopic processes. read less NOT USED (low confidence) E. Antolini, “Iron-containing platinum-based catalysts as cathode and anode materials for low-temperature acidic fuel cells: a review,” RSC Advances. 2016. link Times cited: 38 Abstract: The high availability and low cost of Fe make it an interest… read moreAbstract: The high availability and low cost of Fe make it an interesting element for use in non-precious Pt-free catalysts and Pt-based catalysts for low-temperature fuel cells. Pt–Fe compounds can present three crystal structures, these are a disordered fcc PtxFe alloy and two ordered intermetallic alloys (fcc Pt3Fe and fct PtFe types). Fe-containing Pt-based binary and ternary catalysts in the different crystal structures have been tested both as anode and cathode materials in low-temperature acid fuel cells. In this work an overview of the application of Fe-containing catalysts as cathode materials for oxygen reduction and as anode materials for methanol and ethanol oxidation in low-temperature polymer electrolyte fuel cells fuelled with hydrogen or low molecular weight alcohols, is presented. Moreover, the stability of iron in Pt-based binary and ternary catalysts towards dissolution in acid medium is discussed. read less NOT USED (low confidence) X. Zhou, S. Song, L. Li, and R.-jie Zhang, “Molecular dynamics simulation for mechanical properties of magnesium matrix composites reinforced with nickel-coated single-walled carbon nanotubes,” Journal of Composite Materials. 2016. link Times cited: 17 Abstract: As the interfacial structure and bonding strength play an im… read moreAbstract: As the interfacial structure and bonding strength play an important role in determining the mechanical performance of carbon nanotube reinforced metal matrix composite, investigating the interfacial mechanical properties of surface modified carbon nanotube reinforced metal matrix composite becomes one of the key factors for the improvement. The mechanical behaviors of nickel-coated single-walled carbon nanotube reinforced magnesium matrix composites were investigated using molecular dynamics simulation method. The results show that the Young's modulus of the nickel-coated single-walled carbon nanotube/Mg composite is obviously larger than that of the uncoated single-walled carbon nanotube/Mg composite. The results also show that the interfacial bonding of single-walled carbon nanotube/Mg composite can be drastically increased by addition of nickel coating to improve the wettability of the nanotube surface and Mg matrix. Furthermore, the influences of nickel coating number on the interfacial bonding characteristics of single-walled carbon nanotube/Mg composites also were studied. For three types of nickel coating number, i.e. without nickel coating, with one layer of nickel and two layers of nickel, the final pullout interfacial bonding strength of the nickel-coated single-walled carbon nanotube from Mg matrix about are 3.9 and 11.9 times larger, respectively, than that of the uncoated single-walled carbon nanotube. The simulation results have proved that such interfaces can effectively transfer load between the nanotube and magnesium matrix in the carbon nanotube/Mg composite, and this will provide the theoretical and experimental basis for the interface mechanics design of the carbon nanotube reinforced composites. read less NOT USED (low confidence) X. Wang, J. Li, J. Wang, X. He, and N. Nie, “Kernel Optimization on Short-Range Potentials Computations in Molecular Dynamics Simulations.” 2015. link Times cited: 5 NOT USED (low confidence) J. Zhang, Y. Feng, H. Yuan, D. Feng, X. Zhang, and G. Wang, “Thermal properties of C17H36/MCM-41 composite phase change materials,” Computational Materials Science. 2015. link Times cited: 23 NOT USED (low confidence) N. He, Y. Liu, and X. Zhang, “An improved smoothed molecular dynamics method by alternating with molecular dynamics,” Computer Methods in Applied Mechanics and Engineering. 2015. link Times cited: 6 NOT USED (low confidence) C. Hu, M.-li Bai, J. Lv, and X.-jie Li, “Molecular dynamics simulation of mechanism of nanoparticle in improving load-carrying capacity of lubricant film,” Computational Materials Science. 2015. link Times cited: 20 NOT USED (low confidence) H. Loulijat, H. Zerradi, S. Mizani, E. Achhal, A. Dezairi, and S. Ouaskit, “The behavior of the thermal conductivity near the melting temperature of copper nanoparticle,” Journal of Molecular Liquids. 2015. link Times cited: 38 NOT USED (low confidence) H. N. Pishkenari, A. Nemati, A. Meghdari, and S. Sohrabpour, “A close look at the motion of C60 on gold,” Current Applied Physics. 2015. link Times cited: 30 NOT USED (low confidence) L. Peng, E. Ringe, R. V. V. Duyne, and L. Marks, “Segregation in bimetallic nanoparticles.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 60 Abstract: Bimetallic nanoparticles are of interest due to their physic… read moreAbstract: Bimetallic nanoparticles are of interest due to their physical and chemical properties, which differ from their monometallic counterparts, and are dependent on size, composition and structure. Their unique chemical and physical properties make them useful in many optical, electronic and catalytic applications. In this perspective article we discuss segregation in bimetallic nanoparticles and highlight a recent analytical model based on minimization of energy. Computational approaches are discussed, along with a few examples and a comparison with the analytical approach. Experimental evidence for surface segregation is described, and finally, future directions are suggested. From this review of theoretical and experimental information it appears that a general consensus is starting to emerge that there are size-dependent variations in segregation in nanoparticles with the experimental data reasonably consistent with the theoretical models. read less NOT USED (low confidence) A. Afzali, S. Maghsoodlou, and A. K. Haghi, “The ‘How-to’ Guide to Atomistic Simulations of Nanomaterials.” 2015. link Times cited: 0 NOT USED (low confidence) E. Hahn and M. Meyers, “Grain-size dependent mechanical behavior of nanocrystalline metals,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 162 NOT USED (low confidence) Z. Liang and P. Keblinski, “Coalescence-induced jumping of nanoscale droplets on super-hydrophobic surfaces,” Applied Physics Letters. 2015. link Times cited: 55 Abstract: The coalescence-induced jumping of tens of microns size drop… read moreAbstract: The coalescence-induced jumping of tens of microns size droplets on super-hydrophobic surfaces has been observed in both experiments and simulations. However, whether the coalescence-induced jumping would occur for smaller, particularly nanoscale droplets, is an open question. Using molecular dynamics simulations, we demonstrate that in spite of the large internal viscous dissipation, coalescence of two nanoscale droplets on a super-hydrophobic surface can result in a jumping of the coalesced droplet from the surface with a speed of a few m/s. Similar to the coalescence-induced jumping of microscale droplets, we observe that the bridge between the coalescing nano-droplets expands and impacts the solid surface, which leads to an acceleration of the coalesced droplet by the pressure force from the solid surface. We observe that the jumping velocity decreases with the droplet size and its ratio to the inertial-capillary velocity is a constant of about 0.126, which is close to the minimum value of 0.111 predi... read less NOT USED (low confidence) Y. Cui and Z. Chen, “Molecular dynamics simulation of the influence of elliptical void interaction on the tensile behavior of aluminum,” Computational Materials Science. 2015. link Times cited: 29 NOT USED (low confidence) K. M. Bal and E. Neyts, “Merging metadynamics into hyperdynamics: accelerated molecular simulations reaching time scales from microseconds to seconds.,” Journal of chemical theory and computation. 2015. link Times cited: 7 Abstract: The hyperdynamics method is a powerful tool to simulate slow… read moreAbstract: The hyperdynamics method is a powerful tool to simulate slow processes at the atomic level. However, the construction of an optimal hyperdynamics potential is a task that is far from trivial. Here, we propose a generally applicable implementation of the hyperdynamics algorithm, borrowing two concepts from metadynamics. First, the use of a collective variable (CV) to represent the accelerated dynamics gives the method a very large flexibility and simplicity. Second, a metadynamics procedure can be used to construct a suitable history-dependent bias potential on-the-fly, effectively turning the algorithm into a self-learning accelerated molecular dynamics method. This collective variable-driven hyperdynamics (CVHD) method has a modular design: both the local system properties on which the bias is based, as well as the characteristics of the biasing method itself, can be chosen to match the needs of the considered system. As a result, system-specific details are abstracted from the biasing algorithm itself, making it extremely versatile and transparent. The method is tested on three model systems: diffusion on the Cu(001) surface and nickel-catalyzed methane decomposition, as examples of “reactive” processes with a bond-length-based CV, and the folding of a long polymer-like chain, using a set of dihedral angles as a CV. Boost factors up to 109, corresponding to a time scale of seconds, could be obtained while still accurately reproducing correct dynamics. read less NOT USED (low confidence) H. T. Chen and C.-W. Chan, “Promoting ethylene epoxidation on gold nanoclusters: self and CO induced O2 activation.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 12 Abstract: We have investigated the epoxidation of ethylene heterogeneo… read moreAbstract: We have investigated the epoxidation of ethylene heterogeneously catalyzed by small gold nanoclusters based on density functional theory calculations. A promising trimolecular Langmuir-Hinshelwood mechanism via co-adsorbed ethylene- and CO-assisted reaction is addressed which provides significant insights into the fundamental catalytic mechanism for ethylene oxidation on small Au nanoclusters. O2 activation is found to be a key step for accelerating ethylene oxidation. Especially, the coadsorbed neighboring CO is found to be more robust for promoting the activation of the O-O bond, resulting in the formation of epoxide and CO2 due to the barrierless process. The new CO-promoted oxidation mechanism has also been clarified by the ab initio MD simulations. read less NOT USED (low confidence) T. Owolabi, K. O. Akande, and O. O. Sunday, “Modeling of average surface energy estimator using computational intelligence technique,” Multidiscipline Modeling in Materials and Structures. 2015. link Times cited: 15 Abstract: Purpose – The surface energy per unit area of material is kn… read moreAbstract: Purpose – The surface energy per unit area of material is known to be proportional to the thermal energy at the melting point of the material. The purpose of this paper is to employ the values of the melting points of metals to develop a model that estimates the average surface energies of metals. Average surface energy estimator (ASEE) was developed with the aid of computational intelligence technique on the platform of support vector regression (SVR) using the values of the melting point of the materials as the descriptor. Design/methodology/approach – The development of ASEE which involves 12 data set was conducted by training and testing SVR model using test-set-cross-validation technique. The developed model (ASEE) was used to estimate average surface energies of 3d, 4d, 5d and other selected metals in the periodic table. The average surface energies obtained from ASEE are in good agreement with the experimental values and with the values from other theoretical models. Findings – The accuracy of this... read less NOT USED (low confidence) A. Afzali and S. Maghsoodlou, “Understanding an Atomistic Description of Diffusion Dynamics in Nanomaterials.” 2015. link Times cited: 0 NOT USED (low confidence) M. Mesgar, “Multi-scale modeling of island formation and surface dynamics on the Au(100) surface.” 2015. link Times cited: 3 NOT USED (low confidence) L. Y. Chen, M. He, J. Shin, G. Richter, and D. Gianola, “Measuring surface dislocation nucleation in defect-scarce nanostructures.,” Nature materials. 2015. link Times cited: 150 NOT USED (low confidence) W. Kim, H. Chung, and M. Cho, “Anisotropic hyperelastic modeling for face-centered cubic and diamond cubic structures,” Computer Methods in Applied Mechanics and Engineering. 2015. link Times cited: 10 NOT USED (low confidence) A. Januszko and S. K. Bose, “Phonon spectra and temperature variation of bulk properties of Cu, Ag, Au and Pt using Sutton-Chen and modified Sutton-Chen potentials,” Journal of Physics and Chemistry of Solids. 2015. link Times cited: 14 NOT USED (low confidence) V. Zhdanov, “Ostwald ripening of charged supported metal nanoparticles: Schottky model,” Physica E-low-dimensional Systems & Nanostructures. 2015. link Times cited: 5 NOT USED (low confidence) V. Zhdanov, F. Schweinberger, U. Heiz, and C. Langhammer, “Ostwald ripening of supported Pt nanoclusters with initial size-selected distributions,” Chemical Physics Letters. 2015. link Times cited: 16 NOT USED (low confidence) A. Afzali and S. Maghsoodlou, “Atomistic Simulations Investigation in Nanoscience: A Detailed Review.” 2015. link Times cited: 0 NOT USED (low confidence) S. Eich, D. Beinke, and G. Schmitz, “Embedded-atom potential for an accurate thermodynamic description of the iron–chromium system,” Computational Materials Science. 2015. link Times cited: 24 NOT USED (low confidence) T. Owolabi, K. O. Akande, and S. Olatunji, “Estimation of surface energies of hexagonal close packed metals using computational intelligence technique,” Appl. Soft Comput. 2015. link Times cited: 50 NOT USED (low confidence) X. Zhou et al., “3He retention and structural evolution in erbium tritides: Phase and aging effects,” Journal of Nuclear Materials. 2015. link Times cited: 5 NOT USED (low confidence) C. Taylor, J. Gale, H. Strehblow, and P. Marcus, “An Introduction to Corrosion Mechanisms and Models.” 2015. link Times cited: 4 NOT USED (low confidence) C. Taylor, “Molecular Modeling of Structure and Reactivity at the Metal/Environment Interface.” 2015. link Times cited: 1 NOT USED (low confidence) N. B. Luque, W. Schmickler, E. Santos, and P. Quaino, “Processes at Metal–Solution Interfaces.” 2015. link Times cited: 0 NOT USED (low confidence) H. Zhang et al., “Role of string-like collective atomic motion on diffusion and structural relaxation in glass forming Cu-Zr alloys.,” The Journal of chemical physics. 2015. link Times cited: 84 Abstract: We investigate Cu-Zr liquid alloys using molecular dynamics … read moreAbstract: We investigate Cu-Zr liquid alloys using molecular dynamics simulation and well-accepted embedded atom method potentials over a wide range of chemical composition and temperature as model metallic glass-forming (GF) liquids. As with other types of GF materials, the dynamics of these complex liquids are characterized by "dynamic heterogeneity" in the form of transient polymeric clusters of highly mobile atoms that are composed in turn of atomic clusters exhibiting string-like cooperative motion. In accordance with the string model of relaxation, an extension of the Adam-Gibbs (AG) model, changes in the activation free energy ΔGa with temperature of both the Cu and Zr diffusion coefficients D, and the alpha structural relaxation time τα can be described to a good approximation by changes in the average string length, L. In particular, we confirm that the strings are a concrete realization of the abstract "cooperatively rearranging regions" of AG. We also find coexisting clusters of relatively "immobile" atoms that exhibit predominantly icosahedral local packing rather than the low symmetry packing of "mobile" atoms. These two distinct types of dynamic heterogeneity are then associated with different fluid structural states. Glass-forming liquids are thus analogous to polycrystalline materials where the icosahedrally packed regions correspond to crystal grains, and the strings reside in the relatively disordered grain boundary-like regions exterior to these locally well-ordered regions. A dynamic equilibrium between localized ("immobile") and wandering ("mobile") particles exists in the liquid so that the dynamic heterogeneity can be considered to be type of self-assembly process. We also characterize changes in the local atomic free volume in the course of string-like atomic motion to better understand the initiation and propagation of these fluid excitations. read less NOT USED (low confidence) A. Akimov and O. Prezhdo, “Large-Scale Computations in Chemistry: A Bird’s Eye View of a Vibrant Field.,” Chemical reviews. 2015. link Times cited: 171 NOT USED (low confidence) L. H. Li, W.-li Wang, and B. Wei, “First-principle and molecular dynamics calculations for physical properties of Ni–Sn alloy system,” Computational Materials Science. 2015. link Times cited: 22 NOT USED (low confidence) S. Li, W. Qi, H. Peng, and J. Wu, “A comparative study on melting of core–shell and Janus Cu–Ag bimetallic nanoparticles,” Computational Materials Science. 2015. link Times cited: 32 NOT USED (low confidence) X.-Y. Zhou, H. Ren, B. Huang, and T.-Y. Zhang, “Surface-induced size-dependent ultimate tensile strength of thin films,” Physics Letters A. 2015. link Times cited: 8 NOT USED (low confidence) C. Zou, Y. Shin, A. V. van Duin, H. Fang, and Z.-kui Liu, “Molecular dynamics simulations of the effects of vacancies on nickel self-diffusion, oxygen diffusion and oxidation initiation in nickel, using the ReaxFF reactive force field,” Acta Materialia. 2015. link Times cited: 76 NOT USED (low confidence) Z. W. Wu, M. Li, W. Wang, and K. Liu, “Hidden topological order and its correlation with glass-forming ability in metallic glasses,” Nature Communications. 2015. link Times cited: 111 NOT USED (low confidence) E. Marzbanrad, G. Rivers, P. Peng, B. Zhao, and N. Zhou, “How morphology and surface crystal texture affect thermal stability of a metallic nanoparticle: the case of silver nanobelts and pentagonal silver nanowires.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 40 Abstract: Thermal instability of metallic nanoparticles is typically a… read moreAbstract: Thermal instability of metallic nanoparticles is typically attributed to chemical attack by contaminants. However, thermodynamic stability is independent of other affecting parameters. The importance of this will be clarified when the structural change toward a more stable thermodynamic condition may be followed by a chemical reaction with the surroundings, which may cause a wrong diagnosis. In this research, molecular dynamics simulations and experimental observations were performed to investigate the effect of crystallography and surface texture on stability at high temperature using two closely related model nanoparticles: silver nanobelts and pentagonal nanowires. Previously, the instability of silver nanowires was associated with sulfidation of the wire at high temperature. However, we found that the silver nanowires are inherently unstable at high temperature, degrading due to the high-energy nature of the nanowire's predominately (100) crystallographic surface and pentagonal geometry. In contrast, the silver nanobelts resist thermal degradation up to 500 °C because of their predominately low-energy (111) crystallographic surfaces. In this case study, we successfully demonstrate that inherent thermodynamic stability driven by morphology is significant in metallic nanoparticles, and should be investigated when selecting a nanoparticle for high temperature applications. Moreover, we identify a new one-dimensional nanoparticle, the silver nanobelt, with inherent high-temperature stability. read less NOT USED (low confidence) K. Avchaciov, Y. Ritter, F. Djurabekova, K. Nordlund, and K. Albe, “Effect of ion irradiation on structural properties of Cu64Zr36 metallic glass,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2014. link Times cited: 10 NOT USED (low confidence) S. Zhan, X.-jing Yang, and Y. Chi, “Molecular dynamics study of nanoscale contact process for defective monocrystalline copper,” Proceedings of the Institution of Mechanical Engineers, Part N: Journal of Nanoengineering and Nanosystems. 2014. link Times cited: 0 Abstract: The research of nanoscale contact problem has important sign… read moreAbstract: The research of nanoscale contact problem has important significance for the micro-/nano-machinery, especially for micro-/nano-contact problem of material defects, from which deformation and failure mechanism of materials could be revealed. Molecular dynamics method is a valid approach that describes microscopic phenomenon. Taking the rigid hemispherical surface in contact and indentation process with the perfect and defective monocrystalline copper as the research object, the molecular dynamics model of nanoscale contact was established, after solving and simulating analysis. Results showed that the substrates above the square void collapse at contact depths of 0.20 and 0.37 nm when the depths of the square void (d) are 0.8 and 1.6 nm, respectively. The dislocations and glide band increase as the d increases; at the same time, a larger depth of square void results in a bigger contact force between the hemispherical surface and the substrate surface. In the process of disengagement, there is a sudden drop in the contact force, and unrecoverable plastic deformation of the copper material occurs. Moreover, with the increase in void depth, the number of high-stress atoms increases between the hemisphere and the surface of substrate. The stress is mainly distributed in the diagonal of the square void, and the contact area generates stress concentration. read less NOT USED (low confidence) Y.-xia Liu, H. Wang, H. Wu, D. Xu, and R. Yang, “A mean-field interatomic potential for a multi-component β-type titanium alloy,” Computational Materials Science. 2014. link Times cited: 2 NOT USED (low confidence) J.-feng Tang, L. Deng, H. Deng, S. Xiao, X. Zhang, and W. Hu, “Surface Segregation and Chemical Ordering Patterns of Ag-Pd Nanoalloys: Energetic Factors, Nanoscale Effects, and Catalytic Implication,” Journal of Physical Chemistry C. 2014. link Times cited: 31 Abstract: We performed Monte Carlo simulations to determine the roles … read moreAbstract: We performed Monte Carlo simulations to determine the roles of energetic factors and nanoscale effects in the surface segregation and chemical ordering patterns of Ag–Pd nanoalloy particles. Ag atoms significantly segregate onto the surface and preferentially occupy the low-coordinated sites, which significantly reduces the surface and strain energies of the nanoalloys. The segregation isotherms reveal that surface Ag composition is enhanced with increasing particle size or Ag concentration to circumvent the finite matter effects. Chemical ordering favored by attractive heterobonds can coexist and compete with surface segregation. Accordingly, small and Pd-rich nanoalloys display a continuous transition from Pd-core/mixing-shell to mixing-core/Ag-shell, where an ordered core is absent as a result of surface segregation and limited Ag supply. By contrast, large nanoalloys with equimolar or Ag-rich concentration exhibit the strong core ordering characteristics of bulk alloys. Particularly, surface patterns ... read less NOT USED (low confidence) B. Li, J. Cui, X. Tian, X. Yu, and M. Xiang, “The calculation of mechanical behavior for metallic devices at nano-scale based on Atomic–Continuum Coupled model,” Computational Materials Science. 2014. link Times cited: 8 NOT USED (low confidence) Z. Liang and P. Keblinski, “Parametric studies of the thermal and momentum accommodation of monoatomic and diatomic gases on solid surfaces,” International Journal of Heat and Mass Transfer. 2014. link Times cited: 35 NOT USED (low confidence) J. Rodriguez-Nieva, C. Ruestes, Y. Tang, and E. Bringa, “Atomistic simulation of the mechanical properties of nanoporous gold,” Acta Materialia. 2014. link Times cited: 41 NOT USED (low confidence) K. Volgmann et al., “Local determination of the amount of integration of an atom into a crystal surface,” Nature Communications. 2014. link Times cited: 11 NOT USED (low confidence) C. Hu, M.-li Bai, J. Lv, P. Wang, and X.-jie Li, “Molecular dynamics simulation on the friction properties of nanofluids confined by idealized surfaces,” Tribology International. 2014. link Times cited: 30 NOT USED (low confidence) L. Chang, H. Xu, and D. Cheng, “Role of ligand type on the geometric and electronic properties of Ag–Au bimetallic clusters,” Computational and Theoretical Chemistry. 2014. link Times cited: 9 NOT USED (low confidence) Z. Shah, S. S. Hayat, Z. Rehman, S. S. U. Rahman, and F. Bouafia, “VACANCY GENERATION AND ADSORPTION OF Cu ATOM AT Ag(1 1 1) SURFACE DURING DIFFUSION OF Cu-TRIMER,” Surface Review and Letters. 2014. link Times cited: 2 Abstract: In this paper, the vacancy generation at (1 1 1) surface of … read moreAbstract: In this paper, the vacancy generation at (1 1 1) surface of silver (Ag) and the adsorption of copper (Cu) atom in the vacant site at Ag(1 1 1) surface has been studied using molecular dynamics (MD) simulation technique. The run of micro-canonical constant energy (NVE) ensemble results the generation of vacancy at 700 K in the presence of Cu-trimer, and its migration is observed toward the island as a result of self-diffusion of the surface layer atom. The energy of vacancy generation at Ag surface is found to be Ev = 1.078 eV, in the presence of Cu-trimer. The popped-up Ag adatom combines with Cu-trimer, making a mixed-tetramer (Cu3–Ag island). The adsorption energy of the Cu-atom among the mixed-tetramer island into the substitutional site is Es = ~ -0.813 eV, leaving a mixed-trimer (Cu2–Ag island) at Ag(1 1 1). Moreover, the presence of the island creates some anharmonic effects like dislocations and fissures at the surface. These dislocations and fissures show a trend of attraction to each other and also migrate toward the island, during the time evolution of the system. read less NOT USED (low confidence) J. Hu, Z. Liu, Y. Cui, Z. Wang, Z. Shan, and Z. Zhuang, “Sensitive Material Behavior: Theoretical Model and Experiment for Compression Collapse of Gold Particles at Submicron Scale,” Journal of Applied Mechanics. 2014. link Times cited: 6 NOT USED (low confidence) J. Mei and Y. Ni, “The study of anisotropic behavior of nano-adhesive contact by multiscale simulation,” Thin Solid Films. 2014. link Times cited: 6 NOT USED (low confidence) W. Li, H. Fan, and J. Li, “Deviatoric stress-driven fusion of nanoparticle superlattices.,” Nano letters. 2014. link Times cited: 35 Abstract: We model the mechanical response of alkanethiol-passivated g… read moreAbstract: We model the mechanical response of alkanethiol-passivated gold nanoparticle superlattice (supercrystal) at ambient and elevated pressures using large-scale molecular dynamics simulation. Because of the important roles of soft organic ligands in mechanical response, the supercrystals exhibit entropic viscoelasticity during compression at ambient pressure. Applying a hydrostatic pressure of several hundred megapascals on the superlattice, combined with a critical deviatoric stress of the same order along the [110] direction of the face-centered-cubic supercrystal, can drive the room-temperature sintering ("fusion") of gold nanoparticles into ordered gold nanowire arrays. We discuss the molecular-level mechanism of such phenomena and map out a nonequilibrium stress-driven processing diagram, which reveals a region in stress space where fusion of nanoparticles can occur, instead of other competing plasticity or phase transformation processes in the supercrystal. We further demonstrate that, for silver-gold (Ag-Au) binary nanoparticle superlattices in sodium chloride-type superstructure, stress-driven fusion along the [100] direction leads to the ordered formation of Ag-Au multijunction nanowire arrays. read less NOT USED (low confidence) T. Milek and D. Zahn, “Molecular simulation of AG nanoparticle nucleation from solution: redox-reactions direct the evolution of shape and structure.,” Nano letters. 2014. link Times cited: 30 Abstract: The association of Ag(+) ions and the early stage of Ag nano… read moreAbstract: The association of Ag(+) ions and the early stage of Ag nanoparticle nucleation are investigated from molecular dynamics simulations. Combining special techniques for tackling crystal nucleation from solution with efficient approaches to model redox-reactions, we unravel the structural evolution of forming silver nanoparticles as a function of the redox-potential in the solution. Within a range of only 1 eV, the redox-potential is demonstrated to have a drastic effect on both the inner structure and the overall shape of the forming particles. On the basis of our simulations we identify surface charge and its distribution as an atomic scale mechanism that accounts for creating/avoiding 5-fold coordination polyhedra and thus the degree of (multiple)-twinning in silver nanoparticles. read less NOT USED (low confidence) E. Webb and B. Shi, “Early stage spreading: Mechanisms of rapid contact line advance,” Current Opinion in Colloid and Interface Science. 2014. link Times cited: 7 NOT USED (low confidence) Y. Wu, T. Hang, J. Komadina, H. Ling, and M. Li, “High-adhesive superhydrophobic 3D nanostructured silver films applied as sensitive, long-lived, reproducible and recyclable SERS substrates.,” Nanoscale. 2014. link Times cited: 40 Abstract: Silver films with different morphologies were chemically dep… read moreAbstract: Silver films with different morphologies were chemically deposited by controlling the bath composition. It is found that the wettability and surface enhanced Raman scattering (SERS) properties were closely connected with the surface morphology. Due to the perfect 3D morphology and the 3D electromagnetic field enhanced by three types of nanogaps distributed uniformly, the 3D microball/nanosheet (MN) silver film shows better SERS properties than those of 2D nanosheets (NSs) and nanoparticles (NPs). The MN silver film showed high adhesive superhydrophobic properties after an oxidation process without any functionalization. It can hold the liquid droplet and trace the target molecules in a rather small volume. The SERS properties of the oxidized MN substrate were enhanced remarkably compared to those of the freshly prepared substrate because of the concentrating effect of the superhydrophobicity. The as-prepared 3D MN silver substrate has also exhibited good performances in reproducibility and reutilization which makes it a promising substrate for molecule tracing. read less NOT USED (low confidence) N. Saendig and F. Zerbetto, “Molecules on Gold Surfaces: What They Do and How They Go Around to Do It.” 2014. link Times cited: 0 NOT USED (low confidence) Y. Wu, T. Hang, Z. Yu, L. Xu, and M. Li, “Lotus leaf-like dual-scale silver film applied as a superhydrophobic and self-cleaning substrate.,” Chemical communications. 2014. link Times cited: 46 Abstract: Lotus leaf-like and petal-like substrates were fabricated by… read moreAbstract: Lotus leaf-like and petal-like substrates were fabricated by chemical deposition, which have quite different superhydrophobic properties. Excellent, non-sticky, self-cleaning and durable properties were obtained based on the lotus leaf-like substrate. read less NOT USED (low confidence) D. V. Singh, C. Cassidy, P. Grammatikopoulos, F. Djurabekova, K. Nordlund, and M. Sowwan, “Heterogeneous Gas-Phase Synthesis and Molecular Dynamics Modeling of Janus and Core–Satellite Si–Ag Nanoparticles,” Journal of Physical Chemistry C. 2014. link Times cited: 73 Abstract: Heterogeneous gas-phase condensation is a promising method o… read moreAbstract: Heterogeneous gas-phase condensation is a promising method of producing hybrid multifunctional nanoparticles with tailored composition and microstructure but also intrinsically introduces greater complexity to the nucleation process and growth kinetics. Herein, we report on the synthesis and growth modeling of silicon–silver (Si–Ag) hybrid nanoparticles using gas-aggregated cosputtering from elemental Si and Ag source targets. The final Si–Ag ensemble size was manipulated in the range 5–15 nm by appropriate tuning of the deposition parameters, while variations in the Si–Ag sputtering power ratio, from 1.8 to 2.25, allowed distinctive Janus and core–satellite structures, respectively, to be produced. Molecular dynamics simulations indicate that the individual species first form independent clusters of Si and Ag without significant intermixing. Collisions between unlike species are unstable in the early stages of growth (<100 ns), with large temperature differences resulting in rapid energy exchange and sep... read less NOT USED (low confidence) F. Liu et al., “Atomic self-diffusion behaviors relevant to 2D homoepitaxy growth on stepped Pd(001) surface,” Surface Science. 2014. link Times cited: 10 NOT USED (low confidence) J. Li, Q. Fang, Y.-wen Liu, and L. Zhang, “A molecular dynamics investigation into the mechanisms of subsurface damage and material removal of monocrystalline copper subjected to nanoscale high speed grinding,” Applied Surface Science. 2014. link Times cited: 120 NOT USED (low confidence) S. Debiaggi, E. Crespo, F. U. Braschi, E. Bringa, M. Alí, and M. Ruda, “Hydrogen absorption in Pd thin-films,” International Journal of Hydrogen Energy. 2014. link Times cited: 18 NOT USED (low confidence) M. A. Ortigoza, R. Heid, K. Bohnen, and T. Rahman, “Anomalously Soft and Stiff Modes of Transition-Metal Nanoparticles,” Journal of Physical Chemistry C. 2014. link Times cited: 11 Abstract: We propose an explanation for the enhanced low- and high-ene… read moreAbstract: We propose an explanation for the enhanced low- and high-energy tails of the vibrational density of states (VDOS) of nanoparticles (NPs) with respect to their bulk counterparts. Density functional theory calculations of the frequency and eigenvector of each mode allow us to identify radial breathing/multipolar and nonraclial tidal/shear/torsional vibrations as the modes that populate such tails. These modes have long been obtained from elasticity theory and are thus analogous to the widely studied and observed pulsations in variable stars. The features particular to the VDOS of NPs are rationalized in terms of the charge density distribution around low-coordinated atoms, the quasi-radial geometric distribution of NPs, force constant variations, degree of symmetry of the nanoparticle, discreteness of the spectrum, and the confinement of the eigenmodes. Our results indicate that the high- and low-energy tails of the VDOS may be a powerful tool to reveal information about the chemical composition and geometric structure of small NPs. In particular, the size of the confinement gap at the low-frequency end of the VDOS and the extent by which the high-frequency end surpasses the bulk limit signal whether a NP is bulk-like or non-bulk-like and the extent to which it is disordered. read less NOT USED (low confidence) Z. Shah, S. S. Hayat, Z. Rehman, and F. Bouafia, “The molecular dynamic study of anharmonic effects at Cu(111) and Ag(111) surfaces in the presence of Cu- and Ag-trimer island,” Physics Letters A. 2014. link Times cited: 5 NOT USED (low confidence) S. Karewar, N. Gupta, A. Caro, and S. G. Srinivasan, “A concentration dependent embedded atom method potential for the Mg–Li system,” Computational Materials Science. 2014. link Times cited: 10 NOT USED (low confidence) R. Xiong, H. Peng, H. Si, W. Zhang, and Y. Wen, “Thermodynamic calculation of stacking fault energy of the Fe–Mn–Si–C high manganese steels,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2014. link Times cited: 61 NOT USED (low confidence) V. Vasumathi and M. Cordeiro, “How reliable is the ReaxFF Potential for Describing the Structure of Alkanethiols on Gold? A Molecular Dynamics Study,” Journal of Physics: Conference Series. 2014. link Times cited: 2 Abstract: The structures of self-assembled monolayers (SAMs) of short … read moreAbstract: The structures of self-assembled monolayers (SAMs) of short (methyl) and long (hexyl) chain alkyl thiols on the clean gold (111) surface were modelled using for the Au-S interactions either the reactive ReaxFF potential or the well known non-reactive Morse potential, while for the Au-Au interactions either the ReaxFF potential or an embedded-atom method (EAM). Analysis of the MD trajectories of possible SAM structures suggests that disordering of interfacial Au atoms is definitely driven by the gold-sulphur interactions. Our MD results reveal a novel structure where two methanethiol molecules are bound to a gold adatom that has been lifted from the surface at 300 K, and the same kind of RS-Au-SR motif was also observed for hexanethiol at 600 K but not at 300 K. What is more, the above motif is only observed for the reactive ReaxFF potential. Moreover, these results are in clear agreement with recent experiments and more costly first principles-based MD simulations. These findings strongly support the use of reactive potentials such as ReaxFF for gathering an accurate description of Au-S interactions in inexpensive classical MD simulations. read less NOT USED (low confidence) X. Yang and S. Zhan, “Molecular dynamics simulation of nanoscale contact and sliding processes for probes with different tip radius of curvature,” Chinese Science Bulletin. 2014. link Times cited: 1 NOT USED (low confidence) P. Chen et al., “Spatiotemporal catalytic dynamics within single nanocatalysts revealed by single-molecule microscopy.,” Chemical Society reviews. 2014. link Times cited: 93 Abstract: This review discusses the latest advances in using single-mo… read moreAbstract: This review discusses the latest advances in using single-molecule microscopy of fluorogenic reactions to examine and understand the spatiotemporal catalytic behaviors of single metal nanoparticles of various shapes including pseudospheres, nanorods, and nanoplates. Real-time single-turnover kinetics reveal size-, catalysis-, and metal-dependent temporal activity fluctuations of single pseudospherical nanoparticles (<20 nm in diameter). These temporal catalytic dynamics can be related to nanoparticles' dynamic surface restructuring whose timescales and energetics can be quantified. Single-molecule super-resolution catalysis imaging further enables the direct quantification of catalytic activities at different surface sites (i.e., ends vs. sides, or corner, edge vs. facet regions) on single pseudo 1-D and 2-D nanocrystals, and uncovers linear and radial activity gradients within the same surface facets. These spatial activity patterns within single nanocrystals can be attributed to the inhomogeneous distributions of low-coordination surface sites, including corner, edge, and defect sites, among which the distribution of defect sites is correlated with the nanocrystals' morphology and growth mechanisms. A brief discussion is given on the extension of the single-molecule imaging approach to catalysis that does not involve fluorescent molecules. read less NOT USED (low confidence) M. Bhattacharya, A. Dutta, and P. Barat, “Stick Slip Response of Dislocation Core.” 2014. link Times cited: 0 NOT USED (low confidence) J. Tsai and C. Hong, “Investigate Mechanical Behavior of Gold Nanowire with Defect,” Applied Mechanics and Materials. 2013. link Times cited: 0 Abstract: This study aims to investigate the mechanical properties of … read moreAbstract: This study aims to investigate the mechanical properties of gold nanowires using molecular dynamics (MD) simulation. The effects of the cross section size and the defects on the stress strain curves of the nanowires are examined. Moreover, the inception as well as the processing of dislocationin the nanowire is accounted by means of the centro-symmetry parameter and meanwhile, the energy variation during the dislocation is calculated. Results indicated for the pristine gold nanowire, as the cross section size increases, Youngs modulus increases, but the yielding stress decreases accordingly. Once the ultimate linear point is attained, the dislocation takes place abruptly from the nanowire surfaceand extended along the {111} planes. On the other hand, for the nanowire with defect, it was found that the dislocation is initiated from the defect which can significantlyreduce the yielding stress of the nanowires. read less NOT USED (low confidence) C. Taylor, “Oxygen induced transformations of the δ-Pu(111) surface,” Surface Science. 2013. link Times cited: 7 NOT USED (low confidence) Y.-J. Lee et al., “Ultrasmooth, highly spherical monocrystalline gold particles for precision plasmonics.,” ACS nano. 2013. link Times cited: 99 Abstract: Ultrasmooth, highly spherical monocrystalline gold particles… read moreAbstract: Ultrasmooth, highly spherical monocrystalline gold particles were prepared by a cyclic process of slow growth followed by slow chemical etching, which selectively removes edges and vertices. The etching process effectively makes the surface tension isotropic, so that spheres are favored under quasi-static conditions. It is scalable up to particle sizes of 200 nm or more. The resulting spherical crystals display uniform scattering spectra and consistent optical coupling at small separations, even showing Fano-like resonances in small clusters. The high monodispersity of the particles we demonstrate should facilitate the self-assembly of nanoparticle clusters with uniform optical resonances, which could in turn be used to fabricate optical metafluids. Narrow size distributions are required to control not only the spectral features but also the morphology and yield of clusters in certain assembly schemes. read less NOT USED (low confidence) D. Zhu, H. Zhang, and D. Li, “Effects of nano-scale grain boundaries in Cu on its Bauschinger’s effect and response to cyclic deformation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2013. link Times cited: 11 NOT USED (low confidence) S. Huang, C. Zhang, J. Sun, and J. Shen, “ELASTIC AND VIBRATIONAL PROPERTIES OF ORDERED AND DISORDERED CuMnPt6,” Modern Physics Letters B. 2013. link Times cited: 1 Abstract: In this paper, we have measured the elastic and vibrational … read moreAbstract: In this paper, we have measured the elastic and vibrational properties of CuMnPt6 with embedded-atom method in three states of chemical order: as a disordered FCC (face-centered cubic) solid solution and as the equilibrium L12 and ABC6 ordered structures. Our results agree quite well with the comparable experimental values. The present calculations indicate that the ABC6 and L12 phases are energetically more stable than FCC phase. Numerical estimates of a set of elastic parameters, including aggregate elastic modulus, Poisson's ratio, and elastic anisotropy are performed, and the results demonstrate that the FCC phase is much softer than other two phases. Further analysis from the point of view of vibrational properties such as phonon density of states and vibrational entropy difference are also presented in this study. read less NOT USED (low confidence) D. Golze, M. Iannuzzi, M. T. Nguyen, D. Passerone, and J. Hutter, “Simulation of Adsorption Processes at Metallic Interfaces: An Image Charge Augmented QM/MM Approach.,” Journal of chemical theory and computation. 2013. link Times cited: 60 Abstract: A novel method for including polarization effects within hyb… read moreAbstract: A novel method for including polarization effects within hybrid quantum mechanics/molecular mechanics (QM/MM) simulations of adsorbate-metal systems is presented. The interactions between adsorbate (QM) and metallic substrate (MM) are described at the MM level of theory. Induction effects are additionally accounted for by applying the image charge formulation. The charge distribution induced within the metallic substrate is modeled by a set of Gaussian charges (image charges) centered at the metal atoms. The image charges and the electrostatic response of the QM potential are determined self-consistently by imposing the constant-potential condition within the metal. The implementation is embedded in a highly efficient Gaussian and plane wave framework and is naturally suited for periodic systems. Even though the electronic properties of the metallic substrate are not taken into account explicitly, the augmented QM/MM scheme can reproduce characteristic polarization effects of the adsorbate. The method is assessed through the investigation of structural and electronic properties of benzene, nitrobenzene, thymine, and guanine on Au(111). The study of small water clusters adsorbed on Pt(111) is also reported in order to demonstrate that the approach provides a sizable correction of the MM-based interactions between adsorbate and substrate. Large-scale molecular dynamics (MD) simulations of a water film in contact with a Pt(111) surface show that the method is suitable for simulations of liquid/metal interfaces at reduced computational cost. read less NOT USED (low confidence) J. Los, C. Mottet, and G. Tréglia, “Surface segregation trends in transition metal alloys,” Physical Review B. 2013. link Times cited: 9 NOT USED (low confidence) K. Davami et al., “Thermal conductivity of ZnTe nanowires,” Journal of Applied Physics. 2013. link Times cited: 18 Abstract: The thermal conductivity of individual ZnTe nanowires (NWs) … read moreAbstract: The thermal conductivity of individual ZnTe nanowires (NWs) was measured using a suspended micro-bridge device with built-in resistance thermometers. A collection of NWs with different diameters were measured, and strong size-dependent thermal conductivity was observed in these NWs. Compared to bulk ZnTe, NWs with diameters of 280 and 107 nm showed approximately three and ten times reduction in thermal conductivity, respectively. Such a reduction can be attributed to phonon-surface scattering. The contact thermal resistance and the intrinsic thermal conductivities of the nanowires were obtained through a combination of experiments and molecular dynamic simulations. The obtained thermal conductivities agree well with theoretical predictions. read less NOT USED (low confidence) V. Grigoryan, M. Springborg, H. Minassian, and A. Melikyan, “Optical properties of silver and copper clusters with up to 150 atoms,” Computational and Theoretical Chemistry. 2013. link Times cited: 8 NOT USED (low confidence) M. Burgess, A. Leonardi, M. Leoni, and P. Scardi, “Diffraction line broadening from nanocrystals under large hydrostatic pressures,” Powder Diffraction. 2013. link Times cited: 1 Abstract: Atomistic copper nanocrystals were investigated via Molecula… read moreAbstract: Atomistic copper nanocrystals were investigated via Molecular Dynamics (MD) under hydrostatic pressure to probe the relationship between applied load and structure deformation. The corresponding X-ray powder diffraction patterns were generated from the atomic coordinates. The analysis followed both the traditional Williamson-Hall approach based on pseudo-Voigt fitting and an alternative, more accurate method able to derive the integral breadths without applying a fitting. The Williamson-Hall results show discrepancies not fully associated with an issue of fitting. read less NOT USED (low confidence) A. Fraile, S. Cuesta-López, R. Iglesias, A. Caro, and J. Perlado, “Atomistic molecular point of view for liquid lead and lithium in Nuclear Fusion technology,” Journal of Nuclear Materials. 2013. link Times cited: 16 NOT USED (low confidence) K. Yu et al., “Comparisons of radiation damage in He ion and proton irradiated immiscible Ag/Ni nanolayers,” Journal of Nuclear Materials. 2013. link Times cited: 66 NOT USED (low confidence) K. Elder et al., “Modeling Self-Organization of Thin Strained Metallic Overlayers from Atomic to Micron Scales,” Physical Review B. 2013. link Times cited: 12 Abstract: K. R. Elder,1 G. Rossi,2 P. Kanerva,2 F. Sanches,1 S-C. Ying… read moreAbstract: K. R. Elder,1 G. Rossi,2 P. Kanerva,2 F. Sanches,1 S-C. Ying,3 E. Granato,3,4 C. V. Achim,2 and T. Ala-Nissila2,3 1Department of Physics, Oakland University, Rochester, Michigan 48309-4487, USA 2Department of Applied Physics and COMP Centre of Excellence, Aalto University School of Science, P.O. Box 11000, FI-00076 AALTO, Finland 3Department of Physics, Brown University, P.O. Box 1843, Providence, Rhode Island 02912-1843, USA 4Laboratório Associado de Sensores e Materiais, Instituto Nacional de Pesquisas Espaciais, 12227-010 São José dos Campos, SP, Brazil (Received 7 May 2013; published 16 August 2013) read less NOT USED (low confidence) X. Liu, F. Yuan, and Y. Wei, “Grain size effect on the hardness of nanocrystal measured by the nanosize indenter,” Applied Surface Science. 2013. link Times cited: 80 NOT USED (low confidence) G. Bozzolo, J. Garcés, R. Noebe, P. Abel, and H. O. Mosca, “Atomistic Modeling of Surface and Bulk Properties of Cu, Pd and the Cu-Pd System.” 2013. link Times cited: 17 NOT USED (low confidence) T. Matsuda, Y. Aiba, J. Morimoto, K. Mitsuhara, and Y. Kido, “Enhanced and correlated lattice vibrations of relaxed Cu(0 0 1) surface studied by high-resolution medium energy ion scattering,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2013. link Times cited: 1 NOT USED (low confidence) Y.-bo Guo, T. Xu, and M. Li, “Generalized type III internal stress from interfaces, triple junctions and other microstructural components in nanocrystalline materials,” Acta Materialia. 2013. link Times cited: 15 NOT USED (low confidence) S. Jiang, Y. Shen, Y. Zheng, and Z. Chen, “Formation of quasi-icosahedral structures with multi-conjoint fivefold deformation twins in fivefold twinned metallic nanowires,” Applied Physics Letters. 2013. link Times cited: 15 Abstract: We show by molecular dynamics simulations that symmetrical q… read moreAbstract: We show by molecular dynamics simulations that symmetrical quasi-icosahedral structures can be formed in fivefold twinned metallic nanowires (Cu, Au, and Ag) under dynamic tensile loading. The quasi-icosahedral structure, different from the icosahedral nanoclusters found in the past, consists of a twisted original fivefold twinned axis and ten secondary fivefold deformation twins, with five preexisting prismatic and fifteen tetrahedral subunits joined adjacently. Formation of these structures is observed in the necking region during the plastic deformation with successive twinning processes and is found to be independent on the cross-sectional shape as well as the tensile strain rate of the nanowires. read less NOT USED (low confidence) L. Zepeda-Ruiz, E. Martínez, M. Caro, E. Fu, and A. Caro, “Deformation mechanisms of irradiated metallic nanofoams,” Applied Physics Letters. 2013. link Times cited: 25 Abstract: It was recently proposed that within a particular window in … read moreAbstract: It was recently proposed that within a particular window in the parameter space of temperature, ion energy, dose rate, and filament diameter, nanoscale metallic foams could show radiation tolerance [Bringa et al., Nano Lett. 12, 3351 (2012)]. Outside this window, damage appears in the form of vacancy-related stacking fault tetrahedra (SFT), with no effects due to interstitials [Fu et al., Appl. Phys. Lett. 101, 191607 (2012)]. These SFT could be natural sources of dislocations within the ligaments composing the foam and determine their mechanical response. We employ molecular dynamics simulations of cylindrical ligaments containing an SFT to obtain an atomic-level picture of their deformation behavior under compression. We find that plastic deformation originates at the edges of the SFT, at lower stress than needed to create dislocations at the surface. Our results predict that nanoscale foams soften under irradiation, a prediction not yet tested experimentally. read less NOT USED (low confidence) S. Wang et al., “Porous Pt-M (M = Cu, Zn, Ni) nanoparticles as robust nanocatalysts.,” Chemical communications. 2013. link Times cited: 25 Abstract: Porous Pt-M (M = Cu, Zn, Ni) nanoparticles (NPs) were obtain… read moreAbstract: Porous Pt-M (M = Cu, Zn, Ni) nanoparticles (NPs) were obtained by reduction of [Pt(CH3NH2)4][PtCl4] and M(Ac)2 (or MCl2) with oleylamine under mild conditions. The porous Pt-Cu NPs exhibited superior catalytic activities over a Pt/Cu NP mixture and Cu NPs as references for CO oxidation processes. read less NOT USED (low confidence) T. Liang et al., “Reactive Potentials for Advanced Atomistic Simulations,” Materials Research-ibero-american Journal of Materials. 2013. link Times cited: 180 Abstract: This article reviews recent advances in the development of r… read moreAbstract: This article reviews recent advances in the development of reactive empirical force fields or potentials. In particular, we compare two widely used reactive potentials with variable-charge schemes that are desirable for multicomponent or multifunctional systems: the ReaxFF (reactive force field) and charge-optimized many-body (COMB) potentials. Several applications of these approaches in atomistic simulations that involve metal-based heterogeneous systems are also discussed. read less NOT USED (low confidence) S. Dudarev, “Density Functional Theory Models for Radiation Damage,” Annual Review of Materials Research. 2013. link Times cited: 79 Abstract: Density functional theory models developed over the past dec… read moreAbstract: Density functional theory models developed over the past decade provide unique information about the structure of nanoscale defects produced by irradiation and about the nature of short-range interaction between radia- tion defects, clustering of defects, and their migration pathways. These ab initio models, involving no experimental input parameters, appear to be as quantitatively accurate and informative as the most advanced experimental techniques developed for the observation of radiation damage phenomena. Density functional theory models have effectively created a new paradigm for the scientific investigation and assessment of radiation damage effects, offering new insight into the origin of temperature- and dose-dependent response of materials to irradiation, a problem of pivotal significance for applications. read less NOT USED (low confidence) C. Dong, W. Zhu, S. Zhao, P. Wang, H. Wang, and W. Yang, “Evolution of Pt Clusters on Graphene Induced by Electron Irradiation,” Journal of Applied Mechanics. 2013. link Times cited: 8 Abstract: In situ low-voltage transmission electron microscopy (TEM) w… read moreAbstract: In situ low-voltage transmission electron microscopy (TEM) was performed to study the evolution of small Pt clusters on suspended graphene. Pt clusters, trapped by the edge of holes, generally take a stable shape of truncated octahedron for sizes ranging from sub-1 to � 5nm. The interaction to the graphene dots takes in charge when they form composite nanostructures embedded in graphene. The Pt clusters are slowly flattened due to hole enlargement under electron irradiation. The planar structure is maintained by the peripheral Pt-C bonds and instantly collapses into a three-dimensional (3D) cluster if one side is detached from the edge. Based on the heat transfer model, the thermal effect can be excluded under the experimental condition. Atomistic evolution can be attributed to the electron irradiation. Molecular dynamics simulations revealed that the evolution kinetics was found to be dominated by the surface diffusion (characterized by the migration barrier Em), the temperature (the thermal activation energy � 5kBT), and the scattering from electrons (the maximum transferred energy Emax). The corresponding energies are comparable for the Pt cluster system, leading to similar evolution behaviors. A different scenario in graphene systems is due to the large difference in agitations, i.e., Emax � Em � 5kBT at 3000K. This unique behavior comes from TEM observation, implying that electron beam irradiation can be utilized as a unique tool in shaping carbon nanostructures. [DOI: 10.1115/1.4024168] read less NOT USED (low confidence) S. Verma, T. Rehman, and A. Chatterjee, “A cluster expansion model for rate constants of surface diffusion processes on Ag, Al, Cu, Ni, Pd and Pt(100) surfaces,” Surface Science. 2013. link Times cited: 30 NOT USED (low confidence) Y. Fu and A. To, “A modification to Hardy’s thermomechanical theory that conserves fundamental properties more accurately,” Journal of Applied Physics. 2013. link Times cited: 8 Abstract: This work proposes a modification to Hardy's atomistic-… read moreAbstract: This work proposes a modification to Hardy's atomistic-to-continuum thermomechanical theory, so that it can more accurately conserve mass, momentum, and energy for non-equilibrium thermomechanical processes. The modification proposed is a new normalization rule for the localization function employed in the theory. The improved accuracy of the modified theory is demonstrated based on several molecular dynamics (MD) simulation examples of elastic and shock wave propagation in metals. Through the simulation results, it is also found that Hardy's theory remains valid to a large extent, regardless of the width of the localization function, the interatomic potential, and crystal structure, with and without ensemble averaging. The results from this work will help inject confidence in employing the modified Hardy's theory with the proposed modifications to analyze MD simulation results for non-equilibrium thermomechanical processes and pave the way for concurrent atomistic/continuum coupled simulations. read less NOT USED (low confidence) H. Lou et al., “Negative expansions of interatomic distances in metallic melts,” Proceedings of the National Academy of Sciences. 2013. link Times cited: 105 Abstract: When a material is heated, generally, it dilates. Here, we f… read moreAbstract: When a material is heated, generally, it dilates. Here, we find a general trend that the average distance between a center atom and atoms in the first nearest-neighbor shell contracts for several metallic melts upon heating. Using synchrotron X-ray diffraction technique and molecular dynamics simulations, we elucidate that this anomaly is caused by the redistribution of polyhedral clusters affected by temperature. In metallic melts, the high-coordinated polyhedra are inclined to evolve into low-coordinated ones with increasing temperature. As the coordination number decreases, the average atomic distance between a center atom and atoms in the first shell of polyhedral clusters is reduced. This phenomenon is a ubiquitous feature for metallic melts consisting of various-sized polyhedra. This finding sheds light on the understanding of atomic structures and thermal behavior of disordered materials and will trigger more experimental and theoretical studies of liquids, amorphous alloys, glasses, and casting temperature effect on solidification process of crystalline materials. read less NOT USED (low confidence) F. Ma, K. Xu, and P. Chu, “Surface-induced structural transformation in nanowires,” Materials Science & Engineering R-reports. 2013. link Times cited: 24 NOT USED (low confidence) K. Avchaciov, Y. Ritter, F. Djurabekova, K. Nordlund, and K. Albe, “Controlled softening of Cu64Zr36 metallic glass by ion irradiation,” Applied Physics Letters. 2013. link Times cited: 29 Abstract: We study the effect of irradiation with 5–20 keV recoils on … read moreAbstract: We study the effect of irradiation with 5–20 keV recoils on the topological and chemical short-range order of Cu 64Zr36 metallic glass using molecular dynamics simulations. We show that within the cascade region, the structural backbone of stiff Cu-centered icosahedral units is destroyed, leading to locally softened areas. Under mechanical load, the formation of shear transformation zones is thus promoted in the damaged area. Our results suggest that irradiation is a means to introduce nucleation sites for multiple shear bands and thus prevents catastrophic failure due to the presence of a single critical shear band. read less NOT USED (low confidence) A. Logsdail, A. Logsdail, Z. Li, and R. Johnston, “Faceting preferences for Au(N) and Pd(N) nanoclusters with high-symmetry motifs.,” Physical chemistry chemical physics : PCCP. 2013. link Times cited: 12 Abstract: The structural preferences of nanoparticles are important fo… read moreAbstract: The structural preferences of nanoparticles are important for understanding their chemical properties and potential applications, and remain widely debated. Based on recent experimental observations, we present calculations on the stability of high-symmetry AuN and PdN clusters of various structural motifs, performing a systematic search of faceting preferences using mathematical constructs, a semi-empirical potential with two different parameter sets, and a quasi-Newtonian minimisation technique. We have studied the preferred ratios of (100) and (111) faces for two experimentally observed nanostructures: (a) FCC crystals, comparing octahedra with 8 (111) faces to cuboctahedra where the vertices have been systematically removed (for N < 1500); and (b) Marks-decahedra, with differing "stellation" depths (for N < 6000). For PdN and AuN we see preference towards minimisation of (100) surfaces using the parameter sets of both Cleri and Rosato [Cleri and Rosato, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 22] and Baletto et al. [Baletto et al., J. Chem. Phys., 2002, 116, 3856]. Fully stellated Marks-decahedra are found to be unfavourable at large sizes, with truncated facets identified which are similar to recent experimental observations. We find however that these stellations are deeper in PdN particles than AuN. Truncated-octahedra are found to prefer much reduced (100) surfaces and increased (111) surface areas. read less NOT USED (low confidence) P. He, D. Xu, T. Lin, and Z. Jiao, “Joint properties between carbon nanotube and gold at different energy levels from molecular dynamics,” Computational Materials Science. 2013. link Times cited: 8 NOT USED (low confidence) W. Yao, N. Wang, and J. Lee, “Influence of Rear Earth Yb on the Specific Heat of Liquid Fe-Co-Yb Alloy,” Advanced Materials Research. 2013. link Times cited: 1 Abstract: By Monte Carlo method with EAM potentials, the specific heat… read moreAbstract: By Monte Carlo method with EAM potentials, the specific heat of liquid Co50Fe50 and Co48Fe48Th4 alloys at different temperatures are obtained. The effect of Th on the thermophysical parameters is examined. Over the temperature range from 1400 to 2000 K, the specific heat decreases from to Jmol-1K-1 after Th was added. It is the addition of Th element which has bigger atomic radius, larger atomic mass, and complex arrangement of extranuclear electron, leads to the reduced specific heat. read less NOT USED (low confidence) D. Farkas, A. Caro, E. Bringa, and D. Crowson, “Mechanical response of nanoporous gold,” Acta Materialia. 2013. link Times cited: 133 NOT USED (low confidence) J. Fennell et al., “A Selective Blocking Method To Control the Overgrowth of Pt on Au Nanorods,” Journal of the American Chemical Society. 2013. link Times cited: 69 Abstract: A method for the preparation of smooth deposits of Pt on Au … read moreAbstract: A method for the preparation of smooth deposits of Pt on Au nanorods is described, involving sequential deposition steps with selective blocking of surface sites that reduces Pt-on-Pt deposition. The Au–Pt nanorods prepared by this method have higher long-term stability than those prepared by standard Pt deposition. Electrochemical data show that the resulting structure has more extended regions of Pt surface and enhanced activity toward the carbon monoxide oxidation and oxygen reduction reactions. read less NOT USED (low confidence) K. Elkhodary, S. Tang, and W. K. Liu, “Inclusion clusters in the archetype-blending continuum theory.” 2013. link Times cited: 13 Abstract: In this chapter, we will present a contemporary review of th… read moreAbstract: In this chapter, we will present a contemporary review of the hitherto numerical characterization of nanowires (NWs). The bulk of the research reported in the literatures concern metallic NWs including Al, Cu, Au, Ag, Ni, and their alloys NWs. Research has also been reported for the investigation of some nonmetallic NWs, such as ZnO, GaN, SiC, SiO2. A plenty of researches have been conducted regarding the numerical investigation of NWs. Issues analyzed include structural changes under different loading situations, the formation and propagation of dislocations, and the effect of the magnitude of applied loading on deformation mechanics. Efforts have also been made to correlate simulation results with experimental measurements. However, direct comparisons are difficult since most simulations are carried out under conditions of extremely high strain/loading rates and small simulation samples due to computational limitations. Despite of the immense numerical studies of NWs, a significant work still lies ahead in terms of problem formulation, interpretation of results, identification and delineation of deformation mechanisms, and constitutive characterization of behavior. In this chapter, we present an introduction of the commonly adopted experimental and numerical approaches in studies of the deformation of NWs in Section 1. An overview of findings concerning perfect NWs under different loading situations, such as tension, compression, torsion, and bending are presented in Section 2. In Section 3, we will detail some recent results from the authors’ own work with an emphasis on the study of influences from different pre-existing defect on NWs. Some thoughts on future directions of the computational mechanics of NWs together with Conclusions will be given in the last section. read less NOT USED (low confidence) H. Zhang and J. Douglas, “Similarities of the Collective Interfacial Dynamics of Grain Boundaries and Nanoparticles to Glass‐Forming Liquids.” 2013. link Times cited: 3 NOT USED (low confidence) X. Dai, J.-yu Yang, and W. Hu, “Surface self-diffusion of Re adatom on the Re cluster with hexahedral structure,” Physica B-condensed Matter. 2013. link Times cited: 3 NOT USED (low confidence) Y. Fu and A. To, “Application of Many‐Realization Molecular Dynamics Method to Understand the Physics of Nonequilibrium Processes in Solids.” 2013. link Times cited: 0 NOT USED (low confidence) E. Aghemenloh, S. Yusuf, and J. Idiodi, “Surface energy calculation of hcp metals using the analytical equivalent crystal theory,” Materials Chemistry and Physics. 2013. link Times cited: 11 NOT USED (low confidence) A. Leonardi, M. Leoni, and P. Scardi, “Atomistic interpretation of microstrain in diffraction line profile analysis,” Thin Solid Films. 2013. link Times cited: 11 NOT USED (low confidence) D. Davydov, A. Javili, and P. Steinmann, “On molecular statics and surface-enhanced continuum modeling of nano-structures,” Computational Materials Science. 2013. link Times cited: 60 NOT USED (low confidence) Z. Xiong et al., “Size dependence of plasmon absorption of Ni nanoparticles embedded in BaTiO3/SrTiO3 superlattices,” Applied Surface Science. 2013. link Times cited: 21 NOT USED (low confidence) Z. Liang, W. Evans, and P. Keblinski, “Equilibrium and nonequilibrium molecular dynamics simulations of thermal conductance at solid-gas interfaces.,” Physical review. E, Statistical, nonlinear, and soft matter physics. 2013. link Times cited: 42 Abstract: The thermal conductance at solid-gas interfaces with differe… read moreAbstract: The thermal conductance at solid-gas interfaces with different interfacial bonding strengths is calculated through Green-Kubo equilibrium molecular dynamics (EMD) simulations. Due to the finite size of the simulation system, the long-time integral of the time correlation function of heat power across the solid-gas interface exhibits an exponential decay, which contains the information on interfacial thermal conductance. If an adsorbed gas layer is formed on the solid surface, it is found that the solid-gas interface needs to be defined at a plane outside the adsorbed layer so as to obtain the correct result from the Green-Kubo formula. The EMD simulation result agrees very well with that obtained from nonequilibrium molecular dynamics simulations. By calculating the average solid-gas interaction time as a function of solid-gas interaction strength, we find the incident gas atoms thermalize with the metal surface much more rapidly when the surface is covered by adsorbed gas molecules. read less NOT USED (low confidence) V. Karthik, S. Sahoo, S. K. Pabi, and S. Ghosh, “On the phononic and electronic contribution to the enhanced thermal conductivity of water-based silver nanofluids,” International Journal of Thermal Sciences. 2013. link Times cited: 26 NOT USED (low confidence) L. Yang and A. Martini, “Nano-scale roughness effects on hysteresis in micro-scale adhesive contact,” Tribology International. 2013. link Times cited: 8 NOT USED (low confidence) 陈国祥 and 李晓莉, “面心立方过渡金属自扩散的改进分析型嵌入原子法分析 Analysis of Self-Diffusion of FCC Transition Metals by Modified Analytical Embedded-Atom Method,” Applied physics. 2013. link Times cited: 6 Abstract: 采用改进分析型嵌入原子法(MAEAM)计算了面心立方过渡金属Ni、Pd、Pt、Cu、Ag、Au和Al自扩散过程中的能量。… read moreAbstract: 采用改进分析型嵌入原子法(MAEAM)计算了面心立方过渡金属Ni、Pd、Pt、Cu、Ag、Au和Al自扩散过程中的能量。对于最近邻(NN)和次近邻(NNN)二种扩散机制,其能量曲线均为对称曲线且能量的最大值均位于各自扩散路径的中点。计算得到的单空位形成能、迁移能和自扩散激活能比用嵌入原子法(EAM)计算的结果更接近NN扩散的实验数据。计算结果表明NN扩散的激活能最低(迁移能也为最低),因此面心立方过渡金属中的最可几扩散为单空位最近邻扩散。 The energy during the process of self-diffusion in FCC transition metals Ni, Pd, Pt, Cu, Ag, Au and Al has been calculated by using modified analytic embedded-atom method (MAEAM). For each kind of two diffusion mechanisms nearest-neighbor (NN) and next-nearest-neighbor (NNN), the energy curve is symmetric and the maximum value of the energy appears at the middle point of the diffusion path. Determined mono-vacancy formation energy , migration energy and activation energy for self-diffusion agree well with available experimental data of NN diffusion and are better than those obtained by the embedded-atom method (EAM). Compared the energies corresponding to two diffusion mechanisms, the NN diffusion needs the lowest activation energy (and thus the lowest migration energy). So that, the NN mono-vacancy diffusion is favorable in FCC transition metals. read less NOT USED (low confidence) H. Zhang and J. Douglas, “Glassy Interfacial Dynamics of Ni Nanoparticles: Part I Colored Noise, Dynamic Heterogeneity and Collective Atomic Motion.,” Soft matter. 2013. link Times cited: 27 Abstract: Most condensed materials exhibit a significant fraction of a… read moreAbstract: Most condensed materials exhibit a significant fraction of atoms, molecules or particles that are strongly interacting with each other, while being configured geometrically at any instant of time in an 'amorphous' state having a relatively uniform density. Recently, both simulations and experiments have revealed that the dynamics of diverse condensed amorphous materials is generally characterized by significant heterogeneity in the local mobility and by progressively increasing collective motion upon cooling that takes the form of string-like collective particle rearrangements. The direct experimental observation of this type of collective motion, which has been directly linked to the growing relaxation times of glass-forming materials, and its quantification under different thermodynamic conditions, has so far been restricted to colloidal and driven granular fluids. The present work addresses the fundamental problem of how to determine the scale of this type of collective motion in materials composed of molecules or atoms. The basic premise of our work is that large scale dynamic particle clustering in amorphous materials must give rise to large fluctuations in particle mobility so that transport properties, especially those related to particle mobility, should naturally exhibit noise related to the cooperative motion scale. In our initial exploratory study seeking a relationship of this kind, we find 1/fα or 'colored noise', in both potential energy and particle displacements fluctuations of the atoms within the glassy interfacial layer of Ni nanoparticles (NPs). A direct relation between the particle displacement (mobility) noise exponent α and the average polymerization index of the string-like collective motion L is observed for a range of NP sizes, temperatures and for surface doping of the NPs with other metal atoms (Ag, Au, Pt) to change of fragility of the glassy interfacial layer at the surface of the Ni NPs. We also introduce a successful analytic model to understand this relationship between α and L. read less NOT USED (low confidence) W. Luo, W. Hu, K. Su, and F. Liu, “The calculation of surface free energy based on embedded atom method for solid nickel,” Applied Surface Science. 2013. link Times cited: 27 NOT USED (low confidence) T.-X. Yang, X. Ye, L. Huang, Y. Xie, and S. Ke, “Assembling three-dimensional nanostructures on metal surfaces with a reversible vertical single-atom manipulation: A theoretical modeling,” Applied Surface Science. 2012. link Times cited: 1 NOT USED (low confidence) W. Kim, H. Chung, and M. Cho, “Length and boundary effects on a nanorod,” AIP Advances. 2012. link Times cited: 3 Abstract: We investigate length and boundary effects on the equilibriu… read moreAbstract: We investigate length and boundary effects on the equilibrium strain of a ⟨100⟩ copper nanorod with {100} or {110} surfaces. Unlike a nanowire, a free-edged nanorod has finite length and has two more surfaces at both tip and root. Although the area of these two edge surfaces is generally much smaller than that of side surfaces, the effect of the edge surfaces should not be ignored in the equilibrium configuration of a nanorod. In this letter, an analytical model to estimate the equilibrium strain of the nanorod is proposed, and molecular statics simulations are performed to prove the proposed model. As the length of a nanorod increases, the equilibrium strain increases and converges to that of a nanowire. As for the boundary effect, we compare the equilibrium strain of a clamped nanorod with that of a free-edged nanorod. read less NOT USED (low confidence) C. Wang, F. Wang, Y. Zhang, Q. Sun, and Y. Jia, “Magic size effects of small Cu clusters diffusion on Ag (1 1 1) surface,” Applied Surface Science. 2012. link Times cited: 5 NOT USED (low confidence) H. Kim, G. Venturini, and A. Strachan, “Molecular dynamics study of dynamical contact between a nanoscale tip and substrate for atomic force microscopy experiments,” Journal of Applied Physics. 2012. link Times cited: 14 Abstract: We propose a molecular dynamics approach to model the dynami… read moreAbstract: We propose a molecular dynamics approach to model the dynamical interaction of a nanoscale tip with a substrate under conditions relevant to dynamic atomic force microscopy (AFM). We apply this approach to a half-sphere platinum tip contacting a flat surface of the same metal and study how the input dynamical variables (loading rate during contact and nominal separation between the tip and the substrate) affect the observed interaction between the tip and the substrate. We predict the energy dissipated per cycle and study the use of force-displacement curves to extract local stiffness. We find that, when using nanoscale probes, accurate values can only be obtained for a narrow range of indentations, large enough for continuum elasticity to apply and small enough to avoid plastic deformation. Simulations using the proposed approach are expected to be useful to explore operating conditions of AFM and interpret their results. read less NOT USED (low confidence) E. Fu et al., “Surface effects on the radiation response of nanoporous Au foams,” Applied Physics Letters. 2012. link Times cited: 77 Abstract: We report on an experimental and simulation campaign aimed a… read moreAbstract: We report on an experimental and simulation campaign aimed at exploring the radiation response of nanoporous Au (np-Au) foams. We find different defect accumulation behavior by varying radiation dose-rate in ion-irradiated np-Au foams. Stacking fault tetrahedra are formed when np-Au foams are irradiated at high dose-rate, but they do not seem to be formed in np-Au at low dose-rate irradiation. A model is proposed to explain the dose-rate dependent defect accumulation based on these results. read less NOT USED (low confidence) P. White, “Molecular dynamic modelling of fatigue crack growth in aluminium using LEFM boundary conditions,” International Journal of Fatigue. 2012. link Times cited: 27 NOT USED (low confidence) T. Rahman, “Molecular‐Dynamics Simulation of Surface Phenomena,” Characterization of Materials. 2012. link Times cited: 2 Abstract: Molecular-dynamics (MD) simulation is a well-developed numer… read moreAbstract: Molecular-dynamics (MD) simulation is a well-developed numerical technique that involves the use of a suitable algorithm to solve the classical equations of motion for atoms interacting with a known interatomic potential. This method has been used for several decades now to illustrate and understand the temperature and pressure dependencies of dynamical phenomena in liquids, solids, and liquid-solid interfaces.
MD simulation techniques are also well suited for studying surface phenomena, as they provide a qualitative understanding of surface structure and dynamics. This article considers the use of MD techniques to better understand surface disorder and premelting. Specifically, it examines the temperature dependence of structure and vibrational dynamics at surfaces of face-centered cubic (fcc) metals—mainly Ag, Cu, and Ni. It also makes contact with results from other theoretical and experimental methods.
While the emphasis in this article is on metal surfaces, the MD technique has been applied over the years to a wide variety of surfaces including those of semiconductors, insulators, alloys, glasses, and simple or binary liquids. A full review of the pros and cons of the method as applied to these very interesting systems is beyond the scope of this article. However, it is worth pointing out that the success of the classical MD simulation depends to a large extent on the exactness with which the forces acting on the ion cores can be determined. On semiconductor and insulator surfaces, the success of ab initio molecular dynamics simulations has made them more suitable for such calulations, rather than classical MD simulations.
Keywords:
molecular-dynamics simulation;
surface behavior;
temperature;
related techniques;
data analysis;
interpretation;
principles;
problems read less NOT USED (low confidence) T. Filleter et al., “Nucleation-controlled distributed plasticity in penta-twinned silver nanowires.,” Small. 2012. link Times cited: 101 Abstract: A unique size-dependent strain hardening mechanism, that ach… read moreAbstract: A unique size-dependent strain hardening mechanism, that achieves both high strength and ductility, is demonstrated for penta-twinned Ag nanowires (NWs) through a combined experimental-computational approach. Thin Ag NWs are found to deform via the surface nucleation of stacking fault decahedrons (SFDs) in multiple plastic zones distributed along the NW. Twin boundaries lead to the formation of SFD chains that locally harden the NW and promote subsequent nucleation of SFDs at other locations. Due to surface undulations, chain reactions of SFD arrays are activated at stress concentrations and terminated as local stress decreases, revealing insensitivity to defects imparted by the twin structures. Thick NWs exhibit lower flow stress and number of distributed plastic zones due to the onset of necking accompanied by more complex dislocation structures. read less NOT USED (low confidence) L. Li and X. F. Zhang, “Molecular Dynamics Simulation of Thermodynamic Properties for Al-Si Alloys in Material Manufacturing Engineering,” Advanced Materials Research. 2012. link Times cited: 0 Abstract: The molecular dynamics method was used to simulate thermodyn… read moreAbstract: The molecular dynamics method was used to simulate thermodynamic properties of two binary alloys: Al75 Si25, Al85 Si15..They were calculated of the energy functions, including cohesive energy , formation energy. Results display, formation energy and excess free energy are all negative values, so Al-Si alloys belong to negative system. The atomic interactions were analyzed in macroscopic and microcosmic views. The calculated formation energy can describe the deviation degree between the actual alloy and the ideal melt quantitatively. read less NOT USED (low confidence) D. He, Y. Han, J. Fennell, S. L. Horswell, and Z. Li, “Growth and stability of Pt on Au nanorods,” Applied Physics Letters. 2012. link Times cited: 12 Abstract: We present a systematic study of Pt overgrowth on Au nanorod… read moreAbstract: We present a systematic study of Pt overgrowth on Au nanorods via a wet chemistry approach. The atomic resolved imaging provides direct evidence of initial epitaxial growth of Pt on the surface of Au nanorods, with a preferential deposition occurring at the rod ends. Over a period of one and half years, the carbon-supported nanorods are shown to have undergone a structural transformation when they are kept at ambient conditions, in contrast to the rods kept in solutions whose structure remains stable. Further controlled experiments show morphological changes of the nanorods upon annealing. We discuss the results in terms of the role that kinetics vs. thermodynamics plays in the observed phenomena. read less NOT USED (low confidence) T. Fujita et al., “Atomic origins of the high catalytic activity of nanoporous gold.,” Nature materials. 2012. link Times cited: 754 NOT USED (low confidence) Y. Gan, J. Shi, and S. Jiang, “Atomic-level study of a thickness-dependent phase change in gold thin films heated by an ultrafast laser.,” Applied optics. 2012. link Times cited: 1 Abstract: An ultrafast laser-induced phase change in gold thin films w… read moreAbstract: An ultrafast laser-induced phase change in gold thin films with different thicknesses has been simulated by the method of coupling the two-temperature model and the molecular dynamics, including transient optical properties. Numerical results show that the decrease of film thickness leads to faster melting in the early nonequilibrium time and a larger melting depth. Moreover, earlier occurrence and a higher rate of resolidification are observed for the thicker film. Further analysis reveals that the mechanism for the thickness-dependent phase change in the films is the fast electron thermal conduction in the nonequilibrium state. read less NOT USED (low confidence) O. A. Oviedo, L. Reinaudi, M. Mariscal, and E. Leiva, “Thermodynamic stability of electrochemically decorated Au–Pd core@shell nanoparticles,” Electrochimica Acta. 2012. link Times cited: 10 NOT USED (low confidence) S. Rawat, M. Warrier, D. Raju, S. Chaturvedi, and V. Chavan, “Excitation of characteristic modes of a crystal during solid fracture at high tensile pressure,” Journal of Physics: Conference Series. 2012. link Times cited: 0 Abstract: We have performed uniform triaxial deformation of single cry… read moreAbstract: We have performed uniform triaxial deformation of single crystal copper at high strain rate using molecular dynamics code LAMMPS. The best-fit void nucleation and growth parameters are obtained using a macroscopic nucleation and growth (NAG) model. The detailed analysis of the data shows that voids nucleate at specific locations in the domain, and subsequently grow and coalesce. We explain the spatial location of first void nucleation in terms of the excitation and interaction of characteristic modes in the crystal domain using singular value decomposition analysis. read less NOT USED (low confidence) E. Bringa et al., “Are nanoporous materials radiation resistant?,” Nano letters. 2012. link Times cited: 202 Abstract: The key to perfect radiation endurance is perfect recovery. … read moreAbstract: The key to perfect radiation endurance is perfect recovery. Since surfaces are perfect sinks for defects, a porous material with a high surface to volume ratio has the potential to be extremely radiation tolerant, provided it is morphologically stable in a radiation environment. Experiments and computer simulations on nanoscale gold foams reported here show the existence of a window in the parameter space where foams are radiation tolerant. We analyze these results in terms of a model for the irradiation response that quantitatively locates such window that appears to be the consequence of the combined effect of two length scales dependent on the irradiation conditions: (i) foams with ligament diameters below a minimum value display ligament melting and breaking, together with compaction increasing with dose (this value is typically ∼5 nm for primary knock on atoms (PKA) of ∼15 keV in Au), while (ii) foams with ligament diameters above a maximum value show bulk behavior, that is, damage accumulation (few hundred nanometers for the PKA's energy and dose rate used in this study). In between these dimensions, (i.e., ∼100 nm in Au), defect migration to the ligament surface happens faster than the time between cascades, ensuring radiation resistance for a given dose-rate. We conclude that foams can be tailored to become radiation tolerant. read less NOT USED (low confidence) H. Zhou, L. Zhang, and S. Qu, “Temperature effect on critical shear stress for twin boundary migration,” Computational Materials Science. 2012. link Times cited: 10 NOT USED (low confidence) Y. T. Gu, H. Zhan, and X. Xu, “Numerical investigation of mechanical properties of nanowires : a review,” Interaction and multiscale mechanics. 2012. link Times cited: 4 Abstract: Nanowires (NWs) have attracted intensive researches owing to… read moreAbstract: Nanowires (NWs) have attracted intensive researches owing to the broad applications that arise from their remarkable properties. Over the last decade, immense numerical studies have been conducted for the numerical investigation of mechanical properties of NWs. Among these numerical simulations, the molecular dynamics (MD) plays a key role. Herein we present a brief review on the current state of the MD investigation of nanowires. Emphasis will be placed on the FCC metal NWs, especially the Cu NWs. MD investigations of perfect NWs’ mechanical properties under different deformation conditions including tension, compression, torsion and bending are firstly revisited. Following in succession, the studies for defected NWs including the defects of twin boundaries (TBs) and pre-existing defects are discussed. The different deformation mechanism incurred by the presentation of defects is explored and discussed. This review reveals that the numerical simulation is an important tool to investigate the properties of NWs. However, the substantial gaps between the experimental measurements and MD results suggest the urgent need of multi-scale simulation technique. read less NOT USED (low confidence) S. M. Amir, M. Gupta, and A. Gupta, “Surfactant controlled interfacial alloying in thermally evaporated Cu/Co multilayers,” Journal of Alloys and Compounds. 2012. link Times cited: 12 NOT USED (low confidence) M. I. Pascuet, G. Bonny, and J. R. Fernández, “Many-body interatomic U and Al–U potentials,” Journal of Nuclear Materials. 2012. link Times cited: 28 NOT USED (low confidence) H. Shi and Z. Ji, “Multiscale simulation of onset plasticity during nanoshearing process of copper film,” Microelectronic Engineering. 2012. link Times cited: 1 NOT USED (low confidence) P. Phillips, M. Graef, L. Kovarik, A. Agrawal, W. Windl, and M. Mills, “Atomic-resolution defect contrast in low angle annular dark-field STEM,” Ultramicroscopy. 2012. link Times cited: 84 NOT USED (low confidence) V. Zhdanov, E. M. Larsson, and C. Langhammer, “Novel aspects of Ostwald ripening of supported metal nanoparticles,” Chemical Physics Letters. 2012. link Times cited: 30 NOT USED (low confidence) H. Zhan and Y. T. Gu, “Theoretical and numerical investigation of bending properties of Cu nanowires,” Computational Materials Science. 2012. link Times cited: 33 NOT USED (low confidence) M. Jakšić, W. Schmickleer, and G. Botton, “Advances in Electrocatalysis,” Advances in Physical Chemistry. 2012. link Times cited: 4 Abstract: 1 Institute of Chemical Engineering and High Temperature Che… read moreAbstract: 1 Institute of Chemical Engineering and High Temperature Chemical Processes-Patras, FORTH, 26500 Patras, Greece 2 Institute of Food Technology, Faculty of Agriculture, University of Belgrade, 11080 Belgrade, Serbia 3 Institute of Theoretical Chemistry, Ulm University, 89081 Ulm, Germany 4Department of Material Science and Engineering and Canadian Centre for Electron Microscopy, McMaster University, Hamilton ON, Canada L8S4M1 read less NOT USED (low confidence) A.-Y. Lu et al., “Decoupling of CVD graphene by controlled oxidation of recrystallized Cu,” RSC Advances. 2012. link Times cited: 83 Abstract: Large-area graphene grown by chemical vapour deposition (CVD… read moreAbstract: Large-area graphene grown by chemical vapour deposition (CVD) is promising for applications; however, the interaction between graphene and the substrate is still not well understood. In this report, we use a combination of two non-destructive characterization techniques, i.e., electron backscatter diffraction (EBSD) and Raman mapping to locally probe the interface between graphene and copper lattices without removing graphene. We conclude that the crystal structure of the Cu grains under graphene layers is governed by two competing processes: (1) graphene induced Cu surface reconstruction favoring the formation of Cu(100) orientation, and (2) recrystallization from bulk Cu favoring Cu(111) formation. The underlying Cu grains, regardless of reconstruction or recrystallization, induce a large hydrostatic compression to the graphene lattice. Interestingly, the strong interaction could be decoupled by allowing the intercalation of a thin cuprous oxide interfacial-layer. The Cu2O layer is mechanically and chemically weak; hence, graphene films can be detached and transferred to arbitrary substrates and the Cu substrates could be re-used for graphene growth. read less NOT USED (low confidence) F. Muñoz, A. Romero, J. Mejía‐López, and J. Morán‐López, “First-principles theoretical investigation of monoatomic and dimer Mn adsorption on noble metal (111) surfaces,” Physical Review B. 2012. link Times cited: 12 Abstract: The authors thanks the support from FONDECYT through
Grants… read moreAbstract: The authors thanks the support from FONDECYT through
Grants No. 1100365 (J.M.-L.) and No. 11110510 (F.M.),
from Grant ICM P10-061-F by Fondo de Innovaci´on para
la Competitividad-MINECON, from Financiamiento Basal
para Centros Cient´ificos y Tecnol´ogicos de Excelencia, under
Project No. FB 0807, and from CONACYT (M´exico) through
Grants No. 61417 and No. J-152153-F. read less NOT USED (low confidence) L. Chen and S. Kumar, “Thermal transport in graphene supported on copper,” Journal of Applied Physics. 2012. link Times cited: 91 Abstract: We investigate the thermal transport in isolated single laye… read moreAbstract: We investigate the thermal transport in isolated single layer graphene (SLG) and SLG supported on Cu substrate using equilibrium molecular dynamics simulations and relaxation time approximation (RTA) method. We observe significant changes in the SLG dispersion curve in low frequency and low wave-vector region due to the interaction with Cu substrate. Several new phonon modes related to out-of-plane vibrations appear at the low frequency and small wave vector regions, but their contribution to graphene thermal conductivity is negligible. The thermal conductivity of graphene decreases by 44% due to the interactions with Cu substrate for high interaction strength parameter in Lennard-Jones potential formulation for graphene-Cu interaction. The phonon mode analysis through the RTA approach shows that the acoustic phonons dominate the thermal transport for both isolated and supported graphenes. The longitudinal acoustic (LA), transverse acoustic (TA), and out-of-plane acoustic (ZA) phonons contribute 654, 330,... read less NOT USED (low confidence) I. Chang and F.-R. Chang, “The atomistic study on the thermal expansion behaviors of nanowires,” Computational Materials Science. 2012. link Times cited: 7 NOT USED (low confidence) O. A. Oviedo, C. Negre, M. Mariscal, C. Sánchez, and E. Leiva, “Underpotential deposition on free nanoparticles: Its meaning and measurement,” Electrochemistry Communications. 2012. link Times cited: 23 NOT USED (low confidence) L. G. Gonçalves, C. DaSilva, and J. Rino, “Glass forming ability and alloying effect of a noble-metal-based glass former.,” The journal of physical chemistry. B. 2012. link Times cited: 6 Abstract: This work addresses the question on how the glass-forming ab… read moreAbstract: This work addresses the question on how the glass-forming ability (GFA) of a binary Pd-Ni metallic glass can be enhanced by the alloying effect of Pt. The structural features and slow dynamics of liquid and glassy states on both alloys are investigated by molecular dynamics simulations. Both alloys show typical features of glassy dynamics, namely, the non-Arrhenian behavior of diffusion and relaxation and the fractional Stokes-Einstein relation validity at low temperatures. On the basis of the analysis of the dynamical susceptibilities, we demonstrate that there is a strong influence of the alloying effect on the collective motion of the species, revealing that the GFA of the binary liquid increases with Pt alloying. read less NOT USED (low confidence) S. Cheng and M. Chandross, “Atomic Origins of Friction Reduction in Metal Alloys,” Tribology Letters. 2012. link Times cited: 0 NOT USED (low confidence) P. Han and P. Weiss, “Electronic substrate-mediated interactions,” Surface Science Reports. 2012. link Times cited: 69 NOT USED (low confidence) C. Goyhenex, “Revised tight-binding second moment potential for transition metal surfaces,” Surface Science. 2012. link Times cited: 14 NOT USED (low confidence) K.-S. Han, G. Liu, X. Zhou, R. E. Medina, and P. Chen, “How does a single Pt nanocatalyst behave in two different reactions? A single-molecule study.,” Nano letters. 2012. link Times cited: 89 Abstract: Using single-molecule microscopy of fluorogenic reactions we… read moreAbstract: Using single-molecule microscopy of fluorogenic reactions we studied Pt nanoparticle catalysis at single-particle, single-turnover resolution for two reactions: one an oxidative N-deacetylation and the other a reductive N-deoxygenation. These Pt nanoparticles show distinct catalytic kinetics in these two reactions: one following noncompetitive reactant adsorption and the other following competitive reactant adsorption. In both reactions, single nanoparticles exhibit temporal activity fluctuations attributable to dominantly spontaneous surface restructuring. Depending on the reaction sequence, single Pt nanoparticles may or may not show activity correlations in catalyzing both reactions, reflecting the structure insensitivity of the N-deacetylation reaction and the structure sensitivity of the N-deoxygenation reaction. read less NOT USED (low confidence) M. Arafin, J. Lu, and J. Szpunar, “A Multi-Scale Modeling Approach to Simulate Intergranular Crack Propagation in Textured Polycrystalline Materials,” Materials Science Forum. 2011. link Times cited: 0 Abstract: In this paper, a multiscale modeling approach has been devel… read moreAbstract: In this paper, a multiscale modeling approach has been developed to simulate the intergranular crack propagation in textured polycrystalline materials. Embedded Atom Method (EAM) and Molecular Dynamics (MD) simulations were carried out to determine the energy and fracture strength of different types of grain boundaries in Ni3Al. Subsequently, the atomistic model has been integrated with the microstructure based model of crack propagation using the Voronoi-Markov Chain-Monte Carlo approach. The model has been utilized to evaluate the crack length for various scenarios and reasonable results are obtained. read less NOT USED (low confidence) Z.-L. Liu, X. Li, X.-L. Zhang, L. Cai, and F. Jing, “Phase transition and thermodynamic properties of Sr under high pressure,” Physica B-condensed Matter. 2011. link Times cited: 4 NOT USED (low confidence) C. Zhang, J. Han, S. Huang, and J. Shen, “Chen’s Lattice Inversion Embedded-Atom Method for Nial and Ni3Al Alloy,” Applied Mechanics and Materials. 2011. link Times cited: 0 Abstract: We explored a new type alloy EAM potential (CLI-EAM) that th… read moreAbstract: We explored a new type alloy EAM potential (CLI-EAM) that the value of atomic electron density and pair potential between distinct atoms are obtained by Chen’s lattice inversion based on first-principles calculations. The alloy CLI-EAM potential acquired from NiAl alloy can also apply in Ni3Al successfully and the results of basic properties agreed with the experiments. The results of formation energy of point defects of NiAl and Ni3Al alloy indicate that the structural defects are anti-site defects of Al when enrichments of Al atoms. read less NOT USED (low confidence) E. Aghemenloh, J. Umukoro, S. Azi, S. Yusuf, and J. Idiodi, “Surface energy calculation of bcc metals using the analytical equivalent crystal theory method,” Computational Materials Science. 2011. link Times cited: 29 NOT USED (low confidence) E. Tadmor and R. E. Miller, “Modeling Materials: Continuum, Atomistic and Multiscale Techniques.” 2011. link Times cited: 395 Abstract: 1. Introduction Part I. Continuum Mechanics and Thermodynami… read moreAbstract: 1. Introduction Part I. Continuum Mechanics and Thermodynamics: 2. Essential continuum mechanics and thermodynamics Part II. Atomistics: 3. Lattices and crystal structures 4. Quantum mechanics of materials 5. Empirical atomistic models of materials 6. Molecular statics Part III. Atomistic Foundations of Continuum Concepts: 7. Classical equilibrium statistical mechanics 8. Microscopic expressions for continuum fields 9. Molecular dynamics Part IV. Multiscale Methods: 10. What is multiscale modeling? 11. Atomistic constitutive relations for multilattice crystals 12. Atomistic/continuum coupling: static methods 13. Atomistic/continuum coupling: finite temperature and dynamics Appendix References Index. read less NOT USED (low confidence) J. Zhang, T. Sun, Y. Yan, Y. Liang, and S. Dong, “Molecular dynamics investigation of incipient plasticity during nanomachining of Cu (111) surface,” International Journal of Nanomanufacturing. 2011. link Times cited: 1 Abstract: Large-scale molecular dynamics (MD) simulation of the probe-… read moreAbstract: Large-scale molecular dynamics (MD) simulation of the probe-based mechanical nanomachining is performed to explore the dislocation activities governed incipient plasticity in single crystalline Cu (111) surface with a spherical probe. The atomic interactions between approximate 6 million Cu atoms are described by EAM potential. Simulation results show that in indentation process emergence of plastic deformation is accompanied with force drop phenomena caused by initial dislocation nucleation, and surface pile up pattern is dominantly determined by geometry of slip planes activated. It is indicated that the dislocation activity is the dominant deformation mechanisms in nanomachining of single crystal Cu. Three serial phases of dislocation motions during the following scratching process are observed. Furthermore, there are more kinds of defects generated in scratching process than in indentation process. read less NOT USED (low confidence) Y. H. Park and I. Hijazi, “Simple analytic embedded atom potential for FCC materials,” International Journal of Microstructure and Materials Properties. 2011. link Times cited: 5 Abstract: A simple empirical embedded-atom potential that includes a l… read moreAbstract: A simple empirical embedded-atom potential that includes a long range force is developed for FCC metals. The potential parameters of this model are determined by fitting lattice constant, three elastic constants, cohesive energy, and vacancy formation energy using an optimisation technique (Arora, 2007). Parameters for Cu, Ag, Au, Al, Ni, Pd and Pt have been obtained. The obtained parameters are used to calculate bulk modulus, divacancy formation energy, and melting point. The predicted values are in good agreement with experimental results. The predicted total energy as a function of lattice parameter is also in good agreement with the equation of state of Rose et al. read less NOT USED (low confidence) M. Chen, X. Song, Q. Lv, Z. Gan, and S. Liu, “Bonding of carbon nanotubes onto microelectrodes by localized induction heating,” Sensors and Actuators A-physical. 2011. link Times cited: 14 NOT USED (low confidence) H. Zhan and Y. T. Gu, “Atomistic Exploration of Deformation Properties of Copper Nanowires with Pre-Existing Defects,” Cmes-computer Modeling in Engineering & Sciences. 2011. link Times cited: 11 Abstract: Free to read full-text Based on the embedded atom method (EA… read moreAbstract: Free to read full-text Based on the embedded atom method (EAM) and molecular dynamics (MD) method, in this paper, the tensile deformation properties of Cu nanowires (NWs) with different pre-existing defects, including single surface defects, surface bi-defects and single internal defects, are systematically studied. In-depth deformation mechanisms of NWs with pre-existing defects are also explored. It is found that Young's modulus is insensitive to different pre-existing defects, but yield strength shows an obvious decrease. Defects are observed influencing greatly on NWs' tensile deformation mechanisms, and playing a role of dislocation sources. Besides of the traditional deformation process dominated by the nucleation and propagation of partial dislocations, the generations of twins, grain boundaries, fivefold deformation twins, hexagonal close-packed (HCP) structure and phase transformation from face-centred cubic (FCC) structure to HCP structure have been triggered by pre-existing defects. It is found that surface defect intends to induce larger influence to yield strength than internal defect. Most importantly, the defect that lies on slip planes exerts larger influence than other defects. As expected, it is also found that the more or longer of the defect, the bigger influence will be induced. read less NOT USED (low confidence) T. Böhme, T. Hammerschmidt, R. Drautz, and T. Pretorius, “Closing the Gap Between Nano- and Macroscale: Atomic Interactions vs. Macroscopic Materials Behavior.” 2011. link Times cited: 1 Abstract: In order to meet the continuously increasing requirements in… read moreAbstract: In order to meet the continuously increasing requirements in nearly all fields of technology, an ongoing development and optimization of new and existing materials, components and manufacturing facilities is necessary. The rapidly growing demand on the application side implies a constant acceleration of the complete development process. In the past, development and optimization were often based on experiments. Indeed, the efforts for this approach are mostly extensive, time consuming and expensive, which significantly restricts the development speed. The development of numerical methods and physical models as well as steadily increasing computer capacities allow for the employment of numerical simulations during materials development and optimization. Thus the experimental efforts can be considerably reduced. Moreover, the application of computational methods allows for the investigations of physical phenomena, which are "inaccessible" from the experimental point-of-view, such as trapping behaviour of hydrogen or carbon at different lattice defects (vacancies, dislocations, grain boundaries, etc.) within an Fe-based matrix, see e.g. (Desai et al., 2010; Hristova et al., 2011; Lee, 2006; Lee & Jang, 2007; Nazarov et.al., 2010). In steel production for example, the goal is pursued to set up a so-called ’digital plant’, in which it is possible to calculate the behavior of material and components up to the application level, see Figure 1. Such a digital production line provides deep insight into the materials response and the involved physical effects at each step of the process chain. Furthermore material parameters can be calculated, which will be used as input data to perform calculations of subsequently following process steps. In fact, if the production process chain can be completely reproduced, a backwards approach will be possible, which allows for the transfer from application requirements to the materials design (computer aided material design). A fully theoretical, sufficiently accurate reproduction of all steps of materials processing is as far as we know still not possible. To achieve reliable simulation results in manageable computational times, (semi-)empirical models are widely used at nearly all production 6 read less NOT USED (low confidence) S. Nieves-Torres, E. Mo, and G. López, “Bimetallic Ni/Pd finite systems: Structure and thermodynamics of bimetallic Ni/Pd nanostructures in two and three dimensions,” Materials Chemistry and Physics. 2011. link Times cited: 12 NOT USED (low confidence) A. Karim, A. Kara, O. Trushin, and T. Rahman, “The crossover from collective motion to periphery diffusion for two-dimensional adatom-islands on Cu(111),” Journal of Physics: Condensed Matter. 2011. link Times cited: 13 Abstract: The diffusion of two-dimensional adatom-islands (up to 100 a… read moreAbstract: The diffusion of two-dimensional adatom-islands (up to 100 atoms) on Cu(111) has been studied, using the self-learning kinetic Monte Carlo method (Trushin et al 2005 Phys. Rev. B 72 115401). A variety of multiple- and single-atom processes are revealed in the simulations, and the size dependences of the diffusion coefficients and effective diffusion barriers are calculated for each. From the tabulated frequencies of events found in the simulation, we show a crossover from diffusion due to the collective motion of the island to a regime in which the island diffuses through periphery-dominated mass transport. This crossover occurs for island sizes between 13 and 19 atoms. For islands containing 19–100 atoms the scaling exponent is 1.5, which is in good agreement with previous work. The diffusion of islands containing 2–13 atoms can be explained primarily on the basis of a linear increase of the barrier for the collective motion with the size of the island. read less NOT USED (low confidence) D. Schebarchov and S. Hendy, “Effects of epitaxial strain on the melting of supported nickel nanoparticles,” Physical Review B. 2011. link Times cited: 7 Abstract: We use molecular dynamics to investigate the effects of subs… read moreAbstract: We use molecular dynamics to investigate the effects of substrate-induced epitaxial strain on the melting temperature and equilibrium structure of supported metal nanoparticles. Our model system comprises Ni clusters supported on strained graphene. The clusters are modeled using an embedded atom potential, and the nickel-carbon interactions are described by a Lennard-Jones field with one parameter varied to control the substrate binding strength. We find that, after adjusting for curvature effects due to the clusters' free surface, the melting temperature of supported Ni clusters can shift by hundreds of degrees depending on the cluster-substrate epitaxial relationship. The order of magnitude of this effect is shown to be consistent with prior predictions based on thermodynamic modelling. We also find that sufficiently strong substrate binding leads to a solid-solid transition from icosahedral to lamellar-twinned fcc particles, which occurs via a melt-freeze process. These results illustrate how substrate-induced epitaxial strain can be used to control the phase of metal nanoparticles. read less NOT USED (low confidence) Z. Kou and M.-li Bai, “Effects of wall slip and temperature jump on heat and mass transfer characteristics of an evaporating thin film,” International Communications in Heat and Mass Transfer. 2011. link Times cited: 16 NOT USED (low confidence) C. Zhang, J. Han, S. Huang, and J. Shen, “Chen’s Lattice Inversion Embedded-Atom Method for FCC Metal,” Advanced Materials Research. 2011. link Times cited: 10 Abstract: We explored a new type EAM potential (CLI-EAM) that the valu… read moreAbstract: We explored a new type EAM potential (CLI-EAM) that the value of atomic electron density and pair potential functions are obtained by Chen’s lattice inversion based on first-principles calculations. This EAM potential is applied to Cu, Ag, Cu and Pt metals successfully and the results of basic properties agreed with the experiments. For the same metal, the cohesive energy of fcc structures are the lower than bcc structures. read less NOT USED (low confidence) Z. Wu, C. Kong, and P. Yu, “Melting and Freezing of Free Silver Nanoclusters,” Advanced Materials Research. 2011. link Times cited: 2 Abstract: The melting and freezing with two different cooling rates of… read moreAbstract: The melting and freezing with two different cooling rates of AgN (N= 140, 360, 532, 784, and 952) nanoclusters are simulated by using molecular dynamics technique with the frame work of embedded atom method. The potential energy as a function of temperature is obtained and the structural details are analyzed. The results reveal that the melting and freezing temperature increases almost linearly with the atom number of the clusters except for Ag360. All the silver nanoclusters have negative heat capacity around the phase transition temperature, and the clusters with slow cooling rate have icosahedral structure at 300 K. read less NOT USED (low confidence) L. Trandinh, Y.-M. Ryu, and S. Cheon, “Nanoindentation on the Layered Ag/Cu for Investigating Slip of Misfit Dislocation.” 2011. link Times cited: 0 Abstract: The EAM simulation of nanoindentation was performed to inves… read moreAbstract: The EAM simulation of nanoindentation was performed to investigate misfit dislocation slip in the Ag/Cu. The film layer, whose thickness in the range of 2-5nm, was indented by a spherical indenter with the Nse-Hoover thermostat condition. The simulation shows that the indentation position relative to misfit dislocation (MFD) has the effect on the dislocation, glide up or cross slip, for Ag film layer thickness less than 4 nm. Elastic energy variation during MFDs slip was revealed to be a key factor for the softening of Ag/Cu. The critical film layer thickness was evaluated for each case of Ag/Cu according to the spline extrapolation technique. read less NOT USED (low confidence) D. Gross, R. Müller, M. Müller, B.-X. Xu, and K. Albe, “On the origin of inhomogeneous stress and strain distributions in single-crystalline metallic nanoparticles,” International Journal of Materials Research. 2011. link Times cited: 22 Abstract: The internal stress state in a facetted nanoparticle of fcc … read moreAbstract: The internal stress state in a facetted nanoparticle of fcc copper is investigated by means of finite element calculations based on a linear elastic continuum description. By comparing with atomistic simulations using the embedded-atom method we can show that the elastic anisotropy, particle geometry and surface stresses determine the internal stress state. The calculated internal stresses are much lower than predictions by the Laplace–Young equation. Even under positive surface stresses a negative hydrostatic pressure may develop within the particle, which can be attributed to the strong elastic anisotropy of copper. read less NOT USED (low confidence) F. Muñoz, A. Romero, J. Mejía‐López, and J. Morán‐López, “Monoatomic and dimer Mn adsorption on the Au(111) surface from first principles,” Physical Review B. 2011. link Times cited: 9 Abstract: Francisco Munoz,1,2 Aldo H. Romero,3 Jose Mejia-Lopez,1,2 an… read moreAbstract: Francisco Munoz,1,2 Aldo H. Romero,3 Jose Mejia-Lopez,1,2 and J. L. Moran-Lopez4 1Departamento de Fisica, Facultad de Ciencias, Universidad Nacional Autonoma de Mexico, Mexico D.F. Mexico 2Centro para el Desarrollo de la Nanociencia y la Nanotecnologia CEDENNA, Avda. Ecuador 3493, Santiago, Chile 3CINVESTAV, Unidad Queretaro, Libramiento Norponiente 2000, Real de Juriquilla, CP 76230, Queretaro, Mexico 4Departamento de Fisica, Facultad de Ciencias, Universidad Nacional Autonoma de Mexico (UNAM), Mexico D. F. Mexico (Received 9 December 2010; revised manuscript received 14 March 2011; published 23 May 2011) read less NOT USED (low confidence) X. Jing, Z. Liu, H. Wei, and K. Yao, “The influences of the local impact site and incident energy on the transport behaviors of single copper atom onto Cu (0 0 1) surface,” Applied Surface Science. 2011. link Times cited: 3 NOT USED (low confidence) W. Li, H. Duan, K. Albe, and J. Weissmüller, “Line stress of step edges at crystal surfaces,” Surface Science. 2011. link Times cited: 13 NOT USED (low confidence) J. Wang, R. Hoagland, X.-Y. Liu, and A. Misra, “The influence of interface shear strength on the glide dislocation–interface interactions,” Acta Materialia. 2011. link Times cited: 123 NOT USED (low confidence) Y. Cheng and E. Ma, “Atomic-level structure and structure–property relationship in metallic glasses,” Progress in Materials Science. 2011. link Times cited: 1296 NOT USED (low confidence) Y. Fu, M. Kırca, and A. To, “On determining the thermal state of individual atoms in molecular dynamics simulations of nonequilibrium processes in solids,” Chemical Physics Letters. 2011. link Times cited: 6 NOT USED (low confidence) J. Ye and C. Thompson, “Templated Solid‐State Dewetting to Controllably Produce Complex Patterns,” Advanced Materials. 2011. link Times cited: 127 NOT USED (low confidence) F. Li, J. Pan, and C. Sinka, “Modelling adhesive contact between fine particles using material point method,” Mechanics of Materials. 2011. link Times cited: 7 NOT USED (low confidence) W. Luo, L. Deng, K. Su, K. Li, G. Liao, and S. Xiao, “Gibbs free energy approach to calculate the thermodynamic properties of copper nanocrystals,” Physica B-condensed Matter. 2011. link Times cited: 15 NOT USED (low confidence) K. Kleiner et al., “Multiscale Modeling of Au-Island Ripening on Au(100),” Advances in Physical Chemistry. 2011. link Times cited: 11 Abstract: We describe a multiscale modeling hierarchy for the particul… read moreAbstract: We describe a multiscale modeling hierarchy for the particular case of Au-island ripening on Au(100). Starting at the microscopic scale, density functional theory was used to investigate a limited number of self-diffusion processes on perfect and imperfect Au(100) surfaces. The obtained structural and energetic information served as basis for optimizing a reactive forcefield (here ReaxFF), which afterwards was used to address the mesoscopic scale. Reactive force field simulations were performed to investigate more diffusion possibilities at a lower computational cost but with similar accuracy. Finally, we reached the macroscale by means of kinetic Monte Carlo (kMC) simulations. The reaction rates for the reaction process database used in the kMC simulations were generated using the reactive force field. Using this strategy, we simulated nucleation, aggregation, and fluctuation processes for monoatomic high islands on Au(100) and modeled their equilibrium shape structures. Finally, by calculating the step line tension at different temperatures, we were able to make a direct comparison with available experimental data. read less NOT USED (low confidence) J. Song and W. Curtin, “A nanoscale mechanism of hydrogen embrittlement in metals,” Acta Materialia. 2011. link Times cited: 193 NOT USED (low confidence) Y. Wei, “The kinetics and energetics of dislocation mediated de-twinning in nano-twinned face-centered cubic metals,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2011. link Times cited: 54 NOT USED (low confidence) M. Sushko, P. Sushko, I. Abarenkov, and A. Shluger, “QM/MM method for metal–organic interfaces,” Journal of Computational Chemistry. 2010. link Times cited: 11 Abstract: We present a QM/MM method for modeling metal/organic interfa… read moreAbstract: We present a QM/MM method for modeling metal/organic interfaces, which incorporates contributions from long‐range electron correlation, characteristic to metals and non‐bonded interactions in organic systems. This method can be used to study structurally irregular systems. We apply the method to model finite size domains of self‐assembled monolayers on the gold (111) surface and discuss the influence of boundary effects on the electrostatic and electronic properties of these systems. © 2010 Wiley Periodicals, Inc. J Comput Chem, 2010 read less NOT USED (low confidence) A. Oluwajobi and X. Chen, “The fundamentals of modelling abrasive machining using molecular dynamics,” International Journal of Abrasive Technology. 2010. link Times cited: 17 Abstract: The development of ultra-precision processes which can achie… read moreAbstract: The development of ultra-precision processes which can achieve excellent surface finish and tolerance at the nanometre level is now a critical requirement for many industrial applications. At present, it is very difficult to observe the diverse microscopic physical phenomena occurring in nanometric machining through experiments. The use of molecular dynamics (MD) simulation has proved to be an effective tool for the prediction and the analysis of these processes at the nanometre scale. The crucial task in a MD simulation is the selection of the potential function. The lack of clear understanding about the scope and the limitations of a given potential function may lead to nonsensical results. This article presents the backgrounds of popular potentials used in the modelling of materials processes and the algorithms for the solution of the equations encountered in the simulation. Current applications of MD in abrasive machining are reviewed. read less NOT USED (low confidence) H. Zhang, P. Kalvapalle, and J. Douglas, “String-like collective atomic motion in the interfacial dynamics of nanoparticles,” Soft Matter. 2010. link Times cited: 51 Abstract: The exploding field of nanotechnology is largely driven by t… read moreAbstract: The exploding field of nanotechnology is largely driven by the significant physical and chemical property changes of nanoparticles (NPs) and other nanostructures in comparison to bulk materials and by an increasing capacity for measuring these property changes and synthetically tuning these property changes by controlling particle and feature size, along with surface chemistry. Given the large size-dependent shifts of NP melting temperatures Tm, we can expect at least some of these NP property changes to be associated with an alteration in the NP atomic mobility as the particle size and surface chemistry are varied. Since recent electron microscopy studies on metal NPs of interest in heterogeneous catalysis (e.g., fuel cells, carbon nanotube growth) have often indicated a high NP interfacial mobility and have suggested the relevance of this phenomenon to catalysis, we performed molecular dynamics (MD) simulations for a range of NP sizes in the catalytically relevant temperature range with a focus on quantifying the NP interfacial dynamics. Our illustrative computations were performed for Ni NPs because these particles have been considered in fundamental studies of both fuel cell catalysis and carbon nanotubes growth. Instead of a simple fluid layer on the NP surface, we find a prevalence of string-like collective atomic motions where the geometrical nature of these collective excitations is found to be quantitatively like the collective atomic motions found in glass-forming liquids and in the grain boundary dynamics of polycrystalline materials. We illustrate our new perspective on NP interfacial dynamics by showing metal atom additives (Ag and Pt) alter the length of the string excitations (decreasing and increasing the average length, respectively), as in previous studies of molecular and NP additives in glass-forming polymer liquids. read less NOT USED (low confidence) J. Tucker, R. Najafabadi, T. Allen, and D. Morgan, “Ab initio-based diffusion theory and tracer diffusion in Ni–Cr and Ni–Fe alloys,” Journal of Nuclear Materials. 2010. link Times cited: 120 NOT USED (low confidence) S. Kuang and J. Gezelter, “A gentler approach to RNEMD: nonisotropic velocity scaling for computing thermal conductivity and shear viscosity.,” The Journal of chemical physics. 2010. link Times cited: 33 Abstract: We present a new method for introducing stable nonequilibriu… read moreAbstract: We present a new method for introducing stable nonequilibrium velocity and temperature gradients in molecular dynamics simulations of heterogeneous systems. This method extends earlier reverse nonequilibrium molecular dynamics (RNEMD) methods which use momentum exchange swapping moves. The standard swapping moves can create nonthermal velocity distributions and are difficult to use for interfacial calculations. By using nonisotropic velocity scaling (NIVS) on the molecules in specific regions of a system, it is possible to impose momentum or thermal flux between regions of a simulation while conserving the linear momentum and total energy of the system. To test the method, we have computed the thermal conductivity of model liquid and solid systems as well as the interfacial thermal conductivity of a metal-water interface. We find that the NIVS-RNEMD improves the problematic velocity distributions that develop in other RNEMD methods. read less NOT USED (low confidence) T. Uehara, “Molecular Dynamics Simulation of Shape-Memory Behavior.” 2010. link Times cited: 2 Abstract: Mechanical properties of shape-memory alloys (SMAs) are typi… read moreAbstract: Mechanical properties of shape-memory alloys (SMAs) are typically represented by the characteristic stress–strain curve, which forms a hysteresis loop in a loading, unloading and shaperecovering process. To represent the deformation behavior of SMAs, various constitutive equations have been developed, and prediction of the macroscopic behavior has been possible using finite-element simulations. The atomistic behavior leading to the deformation and shape-recovery is explained on the basis of the phase transformation between austenite and martensite phases and the characteristics of the crystal structure. One well-known atomistic mechanism is illustrated in Fig. 1. The stable phase depends on the temperature, and phases at high and low temperature are body-centered cubic (bcc or B2) and martensite, respectively. The martensite phase consists of many variants, and each variant has a directional unit cell. In Fig. 1(b), for example, a unit cell of the martensite is illustrated as a box leaning in the positive or negative direction along the x-axis. Cells leaning in the same direction constitute a layer, and the direction of the lean alternates between layers. In this paper, the layer is called a variant, although a realistic variant is defined as a rather larger domain. The martensite phase is generated by cooling the B2 structure shown in Fig. 1(a). Randomly orientated variants are then generated, as shown in Fig. 1(b). When a shear load is imposed on this state, some of the layers change their orientation, as shown in Fig. 1(c). This structural change induces macroscopic deformation. When the external shear load is released, the strain does not return to the original state except for slight elastic recovery. When the specimen is heated to the transformation temperature, the martensite transforms into the B2 structure, and martensite appears again with cooling of the specimen. Since the B2 structure is cubic, the shape of the unit cell is independent of the orientation of the martensite layers. Therefore, the specimen macroscopically regains its original shape. This mechanism is well known but has not been fully verified since direct observation of dynamic behavior in a wide range of temperatures is difficult. Therefore, computer simulation is expected to provide evidence for and further extend the mechanism. The molecular dynamics method has become a powerful and effective tool to investigate material properties and dynamic behavior on an atomistic scale, and it has also been applied in the case of SMAs. The stable structure of Ni3Al, for instance, was investigated by Foiles and Daw (Foiles & Daw, 1987), Chen et al. (Chen et al., 1989) using an interatomic potential based on the embeddedatom method (EAM) with suitable parameters (Daw & Baskes, 1984; Foiles et al., 1986). The phase stability and transformation between B2 and martensite structures in NiAl was also 1 read less NOT USED (low confidence) M. Hu, K. Giapis, J. Goicochea, and D. Poulikakos, “Surface segregation of bimetallic alloys in nanoscale confinement,” Applied Physics Letters. 2010. link Times cited: 9 Abstract: The surface segregation of Pt atoms in liquid bimetallic all… read moreAbstract: The surface segregation of Pt atoms in liquid bimetallic alloys confined in carbon nanotube cavities was studied using molecular dynamics simulations. Considerable enrichment in the Pt-atom surface density was found to occur in Pt alloys, when the complementary metal has surface energy higher than Pt and simultaneously metal-wall interaction strength lower than that of Pt with the confining wall. The results suggest that solidification of liquid binary alloys in nanochannels could produce core-shell nanorods with the shell enriched in one of the components for catalytic and other applications. read less NOT USED (low confidence) V. K. Sutrakar and D. Mahapatra, “Comment on ‘Surface stress induced structural transformations and pseudoelastic effects in palladium nanowires’ †Appl. Phys. Lett. 93, 093108 ‘2008…‡,” Applied Physics Letters. 2010. link Times cited: 6 Abstract: Recently, Lao and Moldovan have reported that 100 Pd nanowir… read moreAbstract: Recently, Lao and Moldovan have reported that 100 Pd nanowire of cross-sectional dimensions 2.18 2.18 nm2 undergoes spontaneous reversible phase transformation from face-centered-cubic fcc to body-centeredtetragonal bct structure, provided that temperature is above a critical value of 22.5 K. The results reported by Lao and Moldovan have been re-examined in the present paper, which is due to inconsistent results reported for Pd nanowires in their study as compared to other FCC metal nanowires. For example, Diao et al. have shown that surface stress alone causes fcc→bct phase transformation in 100 gold nanowire of cross-sectional dimensions 2.0 2.0 nm2 during the energy minimization process i.e., T =0 K . Lao and Moldovan reported that Pd nanowire of cross-sectional dimensions 2.18 2.18 nm2 do not show fcc→bct phase transformation during energy minimization, instead a critical temperature of 22.5 K is reported for such phase transformation. We believe that Pd nanowire of smaller cross-sectional size 2.0 2.0 nm2 approximately should be unstable in nature instead of metastable as reported by Lao and Moldovan due to very high surface stresses and it should get transforms into a stable structure via spontaneous phase transformation during the energy minimization process, as observed in other fcc metal nanowire. Further to confirm this, we have performed extensive molecular dynamics simulations using embedded-atommethod potential of Foils et al. The same potential was considered by Lao and Moldovan. We found that Pd nanowires of cross-sectional size 2.0 2.0 nm2 show phase transformation during energy minimization process itself, as shown in Figs. 1 a and 1 b . The results shown here are contradicting the result reported in Ref. 1, whereas our present results show similar trends as reported by Diao et al. for gold nanowires. Snapshot a in Fig. 1 shows an initial 100 / 100 Pd nanowire, which show phase transformation during the energy minimization process leading to Fig. 1 b . Interestingly, we observed that the newly transformed structure is hexagonally closed packed hcp instead of bct structure that was reported by Lao and Moldovan see Fig. 1 and 2 of Ref. 1 . It is also important to mentioned here that some of the nanowires having cross-sectional dimensions less than 2.0 2.0 nm2 shows 100 / 100 to 110 / 111 lattice reorientation during the energy minimization process. Whereas Lao and Moldovan have mentioned that such lattice reorientation is only possible above cross-sectional dimensions of 2.18 2.18 nm2, which contradict our present results. Such 100 / 100 to 110 / 111 lattice reorientation for an initial cross-sectional dimensions of 0.9875 0.9875 nm2 i.e., 2.5 2.5 atomic lattice units is shown in Fig. 1 c . Lao and Moldovan have also mentioned that Pd nanowire with larger cross-sectional dimensions such as 2.57 2.57 nm2 shows 100 to 110 / 111 reorientation at 100 K. In other studies, i.e., in gold and copper nanowires, 100 to 110 / 111 reorientation is also reported by Diao et al. and Liang et al., respectively. In fact, it has been established now that 100 to 110 / 111 spontaneous reorientation de- read less NOT USED (low confidence) D. Irving, “Understanding Metal/Metal Electrical Contact Conductance from the Atomic to Continuum Scales.” 2010. link Times cited: 2 NOT USED (low confidence) F. Wang, J.-min Zhang, Y. Zhang, and V. Ji, “Structural properties and diffusion processes of the Cu3Au (0 0 1) surface,” Applied Surface Science. 2010. link Times cited: 5 NOT USED (low confidence) P. Zhu, Y.-zhong Hu, T. Ma, and H. Wang, “Study of AFM-based nanometric cutting process using molecular dynamics,” Applied Surface Science. 2010. link Times cited: 100 NOT USED (low confidence) R. Jones and J. Zimmerman, “The construction and application of an atomistic J-integral via Hardy estimates of continuum fields,” Journal of The Mechanics and Physics of Solids. 2010. link Times cited: 47 NOT USED (low confidence) B. Fu, W. Liu, and Z. Li, “Calculation of the surface energy of fcc-metals with the empirical electron surface model,” Applied Surface Science. 2010. link Times cited: 41 NOT USED (low confidence) H. Gong, “Electronic structures and related properties of Ag–Au bulks and surfaces,” Materials Chemistry and Physics. 2010. link Times cited: 13 NOT USED (low confidence) X. Wu, R. Wang, S. Wang, and Q. Wei, “Ab initio calculations of generalized-stacking-fault energy surfaces and surface energies for FCC metals,” Applied Surface Science. 2010. link Times cited: 81 NOT USED (low confidence) Y. Şengün and S. Durukanoğlu, “Vibrational properties of Cu nanowires,” Physica A-statistical Mechanics and Its Applications. 2010. link Times cited: 3 NOT USED (low confidence) N. B. Luque, H. Ibach, K. Pötting, and W. Schmickler, “A simulation of two-dimensional Ostwald ripening on silver electrodes,” Electrochimica Acta. 2010. link Times cited: 15 NOT USED (low confidence) G. Zhou, “Nucleation-induced kinetic hindrance to the oxide formation during the initial oxidation of metals,” Physical Review B. 2010. link Times cited: 19 Abstract: A kinetic model is developed to elucidate the nucleation rat… read moreAbstract: A kinetic model is developed to elucidate the nucleation rate of oxide islands during the initial stages of oxidation of metals. Our theoretical analysis shows that the nucleation of oxide islands requires a critical oxygen pressure below which the nucleation rate is practically equal to zero and increases dramatically beyond it. The kinetic model shows that this critical oxygen pressure is many orders of magnitude larger than the equilibrium oxygen pressure predicted from the bulk thermodynamics. Comparison between the kinetic model and experimental data is made over a wide range of oxidation temperature. read less NOT USED (low confidence) X. Zhao, J. Li, T. A. Yeung, C. Kam, Q.-H. Chen, and C. Sun, “Phonon transport in atomic chains coupled by thermal contacts: The role of buffer layer,” Journal of Applied Physics. 2010. link Times cited: 9 Abstract: In this work, ballistic phonon transport in atomic chain nan… read moreAbstract: In this work, ballistic phonon transport in atomic chain nanostructures is investigated by atomic nonequilibrium Green’s functions and embedded atom method. Bond length and strength modification in atomic chain (low-dimensional structure) was taken into consideration by using bond-order-length-strength correlation premise. We especially focus on the contact interface effects on phonon transmission and thermal conductance. It is found that the contact interfaces between an atomic chain and contact reservoir, i.e., neck region or buffer layers, play an important role in phonon transport. The more buffer layers the less thermal conductance. read less NOT USED (low confidence) S. Psakhie, K. Zolnikov, D. Kryzhevich, A. Abdrashitov, and M. Lerner, “Stage character of cluster formation in metal specimens in electrothermal pulse dispersion,” Physical Mesomechanics. 2010. link Times cited: 26 NOT USED (low confidence) 崇 徳増 and 大吾 伊藤, “原子・分子の運動が白金表面での水素分子の解離確率に与える影響 : 第1報,EAMポテンシャルの改良および妥当性の検証(熱工学,内燃機関,動力など),” Transactions of the Japan Society of Mechanical Engineers. B. 2010. link Times cited: 0 NOT USED (low confidence) H. S. Park and X. Qian, “Surface-Stress-Driven Lattice Contraction Effects on the Extinction Spectra of Ultrasmall Silver Nanowires,” Journal of Physical Chemistry C. 2010. link Times cited: 14 Abstract: We utilize numerical simulations based on the discrete dipol… read moreAbstract: We utilize numerical simulations based on the discrete dipole approximation to study the effects of surface-stress-driven lattice contraction on the extinction spectra of silver nanowires with a square cross section of length 2 nm. The novel finding of the present work is the determination that the blue shift that is induced in the silver nanowires due to surface-stress-driven lattice contraction increases with an increase in the nanowire aspect ratio; the blue shift in the longitudinal plasmon resonance wavelength reaches 20 nm in air and 30 nm in water when the nanowire aspect ratio increases to six. Furthermore, we have delineated the lattice contraction effects on the relative contributions of the free (conduction) electrons and the ionic core (bound) electrons to the observed blue shift; specifically, due to the increasingly free electron optical response of the nanowires with increasing aspect ratio, the blue shift due to the contraction-driven increase in the free electron density is found to domin... read less NOT USED (low confidence) Y. Hundur, Z. B. Güvenç, and R. Hippler, “Molecular dynamics of thermal vibration effects: Ar + Ni(1 0 0) collision system,” Communications in Nonlinear Science and Numerical Simulation. 2010. link Times cited: 3 NOT USED (low confidence) W. Yu and S. Shen, “Multiscale study on the tensile fracture of Al-terminated Cu(1 1 1)/α-Al2O3(0 0 0 1) interfaces,” Computational Materials Science. 2010. link Times cited: 4 NOT USED (low confidence) L. Yue, H. Zhang, and D. Li, “A closer look at the local responses of twin and grain boundaries in Cu to stress at the nanoscale with possible transition from the P–H to the inverse P–H relation,” Acta Materialia. 2010. link Times cited: 14 NOT USED (low confidence) M. Benhassine, E. Saiz, A. Tomsia, and J. Coninck, “Role of substrate commensurability on non-reactive wetting kinetics of liquid metals,” Acta Materialia. 2010. link Times cited: 22 NOT USED (low confidence) H. M. Khan and S.-G. Kim, “Atomistic modeling of scratching process based on Atomic Force Microscope: Effects of temperature,” 2010 3rd International Nanoelectronics Conference (INEC). 2010. link Times cited: 3 Abstract: A three-dimensional molecular dynamics model has been used t… read moreAbstract: A three-dimensional molecular dynamics model has been used to investigate the effects of temperature during Atomic Force Microscopy (AFM) based scratching process. Effects of temperature have been taken into consideration as tribological properties are affected significantly by temperature. Deformation behavior, force components, tribological behavior and dislocations generation have been taken into consideration. It has been found that, low temperature like 200K is better choice considering these aspects of nanometric scratching process. read less NOT USED (low confidence) K. A. Bhatti, M. Khaleeq-ur-Rahman, M. Rafique, K. Chaudhary, and A. Latif, “Electrons emission from laser induced metallic plasmas,” Vacuum. 2010. link Times cited: 17 NOT USED (low confidence) Q. To, C. Bercegeay, G. Lauriat, C. Léonard, and G. Bonnet, “A slip model for micro/nano gas flows induced by body forces,” Microfluidics and Nanofluidics. 2010. link Times cited: 21 NOT USED (low confidence) D. Belashchenko, N. Kravchunovskaya, and O. Ostrovski, “Molecular dynamics calculation of surface tension of liquid metals using the embedded atom model,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2010. link Times cited: 9 NOT USED (low confidence) Y. Y. Huang, Y. Zhou, and Y. Pan, “Effects of hydrogen adsorption on the surface-energy anisotropy of nickel,” Physica B-condensed Matter. 2010. link Times cited: 12 NOT USED (low confidence) F. Ma, Z. Song, Y. Li, and K. Xu, “Plastic deformation in bi-metal multilayer nanowires,” Microelectronic Engineering. 2010. link Times cited: 8 NOT USED (low confidence) Z. Duan and W. Xiao, “Cu dimer diffusion on strained Cu(0 0 1),” Surface Science. 2010. link Times cited: 8 NOT USED (low confidence) N. Saendig and F. Zerbetto, “Molecules on gold.,” Chemical communications. 2010. link Times cited: 25 Abstract: The high stability of gold makes its surface an attractive s… read moreAbstract: The high stability of gold makes its surface an attractive substrate for the deposition of molecular materials and their functioning in devices. There are many substantial and profound experimental and theoretical issues that the use of any metal interacting with molecules requires to address. Here, we examine in some detail a semi-empirical computational model able to describe the metal-molecules interactions and provide an overview of the measure of success that it has reached. A number of specific examples are illustrated for a variety of rather large molecular systems. Challenges and future goals are also discussed. read less NOT USED (low confidence) H. Gong, “The work function and electronic structure of coherent Ag–Au interfaces,” Solid State Communications. 2009. link Times cited: 13 NOT USED (low confidence) A. Iskandarov, N. Medvedev, P. Zakharov, and S. Dmitriev, “Crowdion mobility and self-focusing in 3D and 2D nickel,” Computational Materials Science. 2009. link Times cited: 21 NOT USED (low confidence) F. Cinquini, F. Delbecq, and P. Sautet, “A DFT comparative study of carbon adsorption and diffusion on the surface and subsurface of Ni and Ni3Pd alloy.,” Physical chemistry chemical physics : PCCP. 2009. link Times cited: 37 Abstract: Carbon diffusion in transition metal nanoparticles is assume… read moreAbstract: Carbon diffusion in transition metal nanoparticles is assumed to be a key factor in the catalyzed growth of carbon nanotubes (CNT). Aiming at designing more efficient catalysts, we have compared this carbon diffusion process in the near surface and in the bulk of Ni and Ni(3)Pd by means of density functional theory (DFT) calculations. Ni nanoparticles are indeed the most largely used catalysts and the alloying with Pd could modify and improve their properties. The alloy has the same crystal structure as pure Ni, with a slight lattice expansion due to the presence of palladium. For both systems, the subsurface octahedral site is the most stable adsorption site, but the thermodynamic trend favoring the penetration to the subsurface is larger on the alloy than on the Ni. As a result, in the conditions of temperature and pressure for nanotube growth, the population of the subsurface sites is a more exothermic process on the alloy. In addition, while on pure nickel the diffusion over the (111) surface is easy, on the alloy the vertical process leading the carbon to the subsurface is preferred. Palladium atoms have the double effect to expand the lattice parameter providing more adapted diffusion channels for the carbon and to create new adsorption sites less stable than the all-nickel ones. The results can be related to more selective formation of nanotubes on the alloy at low temperature, where Ni produces fibers. read less NOT USED (low confidence) C. Zhou, J. Wu, L. Chen, Y. Wang, H. Cheng, and R. C. Forrey, “Force field for copper clusters and nanoparticles,” Journal of Computational Chemistry. 2009. link Times cited: 2 Abstract: An atomic force field for simulating copper clusters and nan… read moreAbstract: An atomic force field for simulating copper clusters and nanoparticles is developed. More than 2000 cluster configurations of varying size and shape are used to constrain the parametrization of the copper force field. Binding energies for these training clusters were computed using density functional theory. Extensive testing shows that the copper force field is fast and reliable for near‐equilibrium structures of clusters, ranging from only a few atoms to large nanoparticles that approach bulk structure. Nonequilibrium dissociation and compression structures that are included in the training set are also well described by the force field. Implications for molecular dynamics simulations and extensions to other metallic and covalent systems are discussed. © 2009 Wiley Periodicals, Inc. J Comput Chem, 2009 read less NOT USED (low confidence) R. Bachelet et al., “Enhanced thermal stability of Pt electrodes for flat epitaxial biferroic-YMnO3/Pt heterostructures,” Applied Physics Letters. 2009. link Times cited: 4 Abstract: We have investigated the thermal stability of platinum elect… read moreAbstract: We have investigated the thermal stability of platinum electrodes on oxide substrates for oxide-based devices. We show that flat epitaxial Pt(111) bottom electrodes, deposited on SrTiO3(111) and Al2O3(0001) substrates, can be stable against dewetting up to usual oxide-deposition temperatures (Ts) by increasing Pt film thickness (tPt) and preferably using SrTiO3(111) rather than Al2O3(0001) substrates. Subsequently, high-quality epitaxial biferroic YMnO3/Pt/oxide-substrate heterostructures have been grown. A diagram of morphological and crystalline quality versus tPt and Ts is given for both YMnO3/Pt/SrTiO3(111) and YMnO3/Pt/Al2O3(0001) heterostructures. These results shall guideline the growth of other functional oxide thin films on Pt electrodes. read less NOT USED (low confidence) S. W. Lee et al., “Roles of surface steps on Pt nanoparticles in electro-oxidation of carbon monoxide and methanol.,” Journal of the American Chemical Society. 2009. link Times cited: 173 Abstract: Design of highly active nanoscale catalysts for electro-oxid… read moreAbstract: Design of highly active nanoscale catalysts for electro-oxidation of small organic molecules is of great importance to the development of efficient fuel cells. Increasing steps on single-crystal Pt surfaces is shown to enhance the activity of CO and methanol electro-oxidation up to several orders of magnitude. However, little is known about the surface atomic structure of nanoparticles with sizes of practical relevance, which limits the application of fundamental understanding in the reaction mechanisms established on single-crystal surfaces to the development of active, nanoscale catalysts. In this study, we reveal the surface atomic structure of Pt nanoparticles supported on multiwall carbon nanotubes, from which the amount of high-index surface facets on Pt nanoparticles is quantified. Correlating the surface steps on Pt nanoparticles with the electrochemical activity and stability clearly shows the significant role of surface steps in enhancing intrinsic activity for CO and methanol electro-oxidation. Here, we show that increasing surface steps on Pt nanoparticles of approximately 2 nm can lead to enhanced intrinsic activity up to approximately 200% (current normalized to Pt surface area) for electro-oxidation of methanol. read less NOT USED (low confidence) J. Zimmerman, B. M. Wong, R. Jones, J. Templeton, and J. W. Lee, “Enhanced molecular dynamics for simulating porous interphase layers in batteries.” 2009. link Times cited: 0 Abstract: Understanding charge transport processes at a molecular leve… read moreAbstract: Understanding charge transport processes at a molecular level using computational techniques is currently hindered by a lack of appropriate models for incorporating anistropic electric fields in molecular dynamics (MD) simulations. An important technological example is ion transport through solid-electrolyte interphase (SEI) layers that form in many common types of batteries. These layers regulate the rate at which electro-chemical reactions occur, affecting power, safety, and reliability. In this work, we develop a model for incorporating electric fields in MD using an atomistic-to-continuum framework. This framework provides the mathematical and algorithmic infrastructure to couple finite element (FE) representations of continuous data with atomic data. In this application, the electric potential is represented on a FE mesh and is calculated from a Poisson equation with source terms determined by the distribution of the atomic charges. Boundary conditions can be imposed naturally using the FE description of the potential, which then propagates to each atom through modified forces. The method is verified using simulations where analytical or theoretical solutions are known. Calculations of salt water solutions in complex domains are performed to understand how ions are attracted to charged surfaces in the presence of electric fields and interfering media. read less NOT USED (low confidence) B. Fu, W. Liu, and Z. Li, “Calculation of the surface energy of bcc-metals with the empirical electron theory,” Applied Surface Science. 2009. link Times cited: 140 NOT USED (low confidence) A. Dannenberg, M. Gruner, A. Hucht, and P. Entel, “Surface energies of stoichiometric FePt and CoPt alloys and their implications for nanoparticle morphologies,” Physical Review B. 2009. link Times cited: 116 Abstract: We have calculated surface energies and surface magnetic ord… read moreAbstract: We have calculated surface energies and surface magnetic order of various low-indexed surfaces of monoatomic Fe, Co, and Pt, and binary, ordered FePt, CoPt, and MnPt using density-functional theory. Our results for the binary systems indicate that elemental, Pt-covered surfaces are preferred over Fe and Co covered and mixed surfaces of the same orientation. The lowest energy orientation for mixed surfaces is the highly coordinated (111) surface. We find Pt-covered (111) surfaces, which can be realized in the $\text{L}{1}_{1}$ structure only, to be lower in energy by about 400 meV/atom compared to the mixed $\text{L}{1}_{0}$ (111) surface. We conclude that in small nanoparticles this low surface energy can stabilize the $\text{L}{1}_{1}$ structure, which is suppressed in bulk alloys. From the interplay of surface and bulk energies, equilibrium shapes of single-crystalline ordered nanoparticles and crossover sizes between the different orderings can be estimated. read less NOT USED (low confidence) F. Tavazza, L. Levine, and A. Chaka, “Elongation and breaking mechanisms of gold nanowires under a wide range of tensile conditions,” Journal of Applied Physics. 2009. link Times cited: 39 Abstract: Semistatic density functional theory is used to explore the … read moreAbstract: Semistatic density functional theory is used to explore the evolution of [1 1 0] and [1 1 1] gold nanowires during tensile deformation under a wide range of conditions, including different tensile axes (along high- and low-symmetry directions), nanowire shapes, and effective strain rates. Large structural changes are observed during the elongation. The analysis of such low-energy intermediate configurations provides quantitative information about the underlying energy landscape that cannot be obtained through experiments or more approximate modeling methods, and four stable intermediate atomic structures are identified. A rich diversity of deformation pathways is uncovered that converge to only two final local configurations with reproducible breaking strengths, in agreement with experimental results. Such a high reproducibility in the breaking force makes gold nanowires excellent candidates as intrinsic force standards at the nanolevel. read less NOT USED (low confidence) Z. Xu and M. Buehler, “Nanoengineering heat transfer performance at carbon nanotube interfaces.,” ACS nano. 2009. link Times cited: 203 Abstract: Carbon nanotubes are superb materials for nanoscale thermal … read moreAbstract: Carbon nanotubes are superb materials for nanoscale thermal management and phononic devices applications, due to their extremely high thermal conductivity (3000-6600 W/mK) and quasi-one-dimensional geometry. However, the presence of interfaces between individual carbon nanotubes as found widely in nanocomposites, nanoelectronics, and nanodevices severely limits their performance for larger scale applications. Solving this issue requires a deep understanding of the heat transfer mechanism at this nanoscale interface between low-dimensional structures, where conventional models developed for interfaces in bulk materials do not apply. Here we address this challenge through a bottom-up approach based on atomistic simulations. We demonstrate that the huge thermal resistance of carbon nanotube junctions can be significantly improved through modifying the molecular structure at the interface to enhance both the matching of phonon spectra and phonon mode coupling. Specifically, two approaches based on polymer wrapping and metal coatings are investigated here and have shown to improve both the structural stability and interfacial thermal conductivity of carbon nanotube junctions. By properly designing the interface molecular structure between individual carbon nanotubes, significant performance gains up to a factor of 4 can be achieved. These results pave the way for future designs of thermal management networks and phononic devices with thermally transparent and structurally stable interfaces. read less NOT USED (low confidence) E. Aghemenloh, J. Idiodi, and S. Azi, “Surface energies of hcp metals using equivalent crystal theory,” Computational Materials Science. 2009. link Times cited: 24 NOT USED (low confidence) W. Yu and S. Shen, “Multiscale analysis of the effects of nanocavity on nanoindentation,” Computational Materials Science. 2009. link Times cited: 19 NOT USED (low confidence) R. Dingreville and J. Qu, “A semi-analytical method to estimate interface elastic properties,” Computational Materials Science. 2009. link Times cited: 26 NOT USED (low confidence) H. Akbarzadeh and G. Parsafar, “A molecular-dynamics study of thermal and physical properties of platinum nanoclusters,” Fluid Phase Equilibria. 2009. link Times cited: 24 NOT USED (low confidence) J. C. Hamilton and W. G. Wolfer, “Theories of surface elasticity for nanoscale objects,” Surface Science. 2009. link Times cited: 52 NOT USED (low confidence) S. Giuffrida, G. Barone, and D. Duca, “Adsorbed CO on Group 10 Metal Fragments: A DFT Study,” Journal of chemical information and modeling. 2009. link Times cited: 5 Abstract: DFT calculations on the helicopter and cartwheel rotations o… read moreAbstract: DFT calculations on the helicopter and cartwheel rotations of one CO molecule adsorbed at the bridge site on metal-surface fragments, characterized by two (M(8)) or three (M(14)) metal-atom layers (M = Ni, Pd, Pt) were performed by the B3LYP[LANL2DZ+6-31 g(d,p)] method, to rationalize the adsorption energetics and the steric hindrance characteristics of surface CO molecules. Potential Energy Surfaces were obtained, either fixing the C-O bond-length or allowing it to change. The behavior of the three metals, as obtained from the study of the configurational space characterizing the CO adsorption on the fragments was explained on the basis of the interaction energies involved in the different CO/M systems. The results, obtained by using the M(14) fragments and varying both the C-O and the CO/M distances, point out that the CO adsorption on the Ni fragment is stabilized by surface-configurations in which the O atom is pointing toward a metal center. At variance, C-O bond elongation and stabilization occur on Pd when the O atom is situated between two palladium atoms. The CO adsorption on Pt displays similar characteristics to those observed on the Pd systems, but with the fundamental difference caused by the destabilization of the Pt-O interactions when the O atom is situated exactly between two Pt atoms. The calculations allowed us to estimate the IR spectroscopy frequency and band-broadening of the adsorbed CO stretching by a statistic analysis on a large set of energy / bond-length computed data. Good agreement with the experimental results was obtained for all the metals, in particular concerning the frequencies. Reliable band-broadenings were also obtained for the CO/Ni and CO/Pt systems, while the lower band-broadening value for the CO/Pd system was related to the small extent of the configurational sampling space. read less NOT USED (low confidence) I. Mastorakos, H. Zbib, and D. Bahr, “Deformation mechanisms and strength in nanoscale multilayer metallic composites with coherent and incoherent interfaces,” Applied Physics Letters. 2009. link Times cited: 77 Abstract: We investigate the deformation behavior of bimetallic and tr… read moreAbstract: We investigate the deformation behavior of bimetallic and trimetallic nanoscale multilayer metallic composites under biaxial loading using molecular dynamics. Three types of structures were studied: (a) Cu–Ni fcc/fcc bilayer, (b) Cu–Nb fcc/bcc bilayer, and (c) Ni–Cu–Nb fcc/fcc/bcc trilayer. A configuration with a dislocation structure inside is generated by initially loading a perfect structure to a high strain to nucleate dislocations, then completely unloading it and loading it again. The comparison between the deformation behavior of bilayer and trilayer structures revealed that the Cu–Ni is more ductile, the Cu–Nb is stronger, and the trilayer structure exhibits both high strength and ductility. read less NOT USED (low confidence) J. Zhang, T. Sun, Y. Yan, and Y. Liang, “Molecular dynamics study of scratching velocity dependency in AFM-based nanometric scratching process,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2009. link Times cited: 54 NOT USED (low confidence) J. Song and D. Srolovitz, “Molecular dynamics investigation of patterning via cold welding,” Journal of The Mechanics and Physics of Solids. 2009. link Times cited: 9 NOT USED (low confidence) A. Stukowski, J. Markmann, J. Weissmüller, and K. Albe, “Atomistic origin of microstrain broadening in diffraction data of nanocrystalline solids,” Acta Materialia. 2009. link Times cited: 70 NOT USED (low confidence) J. Zimmerman, D. Bammann, and H. Gao, “Deformation gradients for continuum mechanical analysis of atomistic simulations,” International Journal of Solids and Structures. 2009. link Times cited: 121 NOT USED (low confidence) T. Qin, R. Drautz, and D. Pettifor, “Binding-energy relations and equations of state for the4dand5dtransition metals,” Physical Review B. 2008. link Times cited: 15 NOT USED (low confidence) B. Hamad, “Structural and dynamical properties of Ru(0 0 0 1) surface,” Surface Science. 2008. link Times cited: 7 NOT USED (low confidence) X. W. Zhou and F. Doty, “Embedded-ion method: An analytical energy-conserving charge-transfer interatomic potential and its application to the La-Br system,” Physical Review B. 2008. link Times cited: 30 NOT USED (low confidence) T. Tokumasu, K. Hara, and D. Ito, “Molecular dynamics study for dissociation phenomena of a gas molecule on a metal surface,” Heat Transfer Research. 2008. link Times cited: 1 Abstract: The dissociation phenomena of a gas molecule on a metal surf… read moreAbstract: The dissociation phenomena of a gas molecule on a metal surface were analyzed by the molecular dynamics method. A platinum (111) surface and hydrogen were chosen as the metal surface and the gas molecule, respectively. The embedded atom method was used as the interaction between atoms in order to express the dependence of electron density. The parameters were determined so that the results such as the electron density, adsorption energy of an H atom on a Pt(111) surface, and the interaction between H atoms of an H2 molecule obtained by the EAM method were consistent with those obtained by the density functional theory or empirical function. Collisions between a hydrogen molecule and the platinum surface were simulated by the molecular dynamics method, and the dissociation probability was obtained. Using these results, the effect of the motion of the surface atoms or the hydrogen molecule on the dissociation probability was analyzed. © 2008 Wiley Periodicals, Inc. Heat Trans Asian Res; Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/htj.20222 read less NOT USED (low confidence) K. Zhou, M. S. Wu, and A. A. Nazarov, “Relaxation of a disclinated tricrystalline nanowire,” Acta Materialia. 2008. link Times cited: 63 NOT USED (low confidence) A. Setoodeh and H. Attariani, “Nanoscale simulations of Bauschinger effects on a nickel nanowire,” Materials Letters. 2008. link Times cited: 22 NOT USED (low confidence) O. Melikhova et al., “Simulation of positron annihilation response to mechanical deformation of nanostructured Ni3Al,” Applied Surface Science. 2008. link Times cited: 4 NOT USED (low confidence) X. W. Zhou, J. Zimmerman, E. Reedy, and N. Moody, “Molecular dynamics simulation based cohesive surface representation of mixed mode fracture,” Mechanics of Materials. 2008. link Times cited: 86 NOT USED (low confidence) J. Li, Y. Dai, X. Dai, T. Wang, and B. Liu, “Development of n-body potentials for hcp–bcc and fcc–bcc binary transition metal systems,” Computational Materials Science. 2008. link Times cited: 20 NOT USED (low confidence) A. Bittner, T. Bohnenberger, R. Engel, H. Seidel, and U. Schmid, “Morphological and Electrical Properties of Silver Thin Films Sputter Deposited on LTCC Substrates,” Journal of microelectronics and electronic packaging. 2008. link Times cited: 3 Abstract: Screen printed noble metal thick films are commonly used as … read moreAbstract: Screen printed noble metal thick films are commonly used as metallization on LTCC (low temperature cofired ceramics) substrates. When, however, geometries with a lateral resolution below 20 μm are needed for the realization of devices, alternative techniques are needed, and they are provided by standard thin film technology. To minimize conduction losses, silver (Ag) is favored due to a low bulk resistivity. To evaluate the potential of Ag as metallization, thin films are sputter deposited on glass and LTCC substrates under varying conditions (i.e., plasma power) with different film thicknesses ranging up to 1.75 μm. The microstructure of the Ag films is analyzed applying techniques such as scanning electron microscopy, focused ion beam, and x-ray diffraction. With the latter approach, a mean grain size of about 33 nm is measured independent of plasma power used for Ag deposition. In contrast, the texture strongly varies with deposition parameters resulting in an enhanced generation of (111) planes at hig... read less NOT USED (low confidence) T. Järvi et al., “Development of a ReaxFF description for gold,” The European Physical Journal B. 2008. link Times cited: 61 NOT USED (low confidence) J. Lao and D. Moldovan, “Surface stress induced structural transformations and pseudoelastic effects in palladium nanowires,” Applied Physics Letters. 2008. link Times cited: 40 Abstract: Using molecular dynamics simulations, we investigate the sur… read moreAbstract: Using molecular dynamics simulations, we investigate the surface stress induced structural transformations and pseudoelastic behaviors in palladium nanowires. For wires with a ⟨100⟩ initial orientation, the simulations indicate that when the cross-sectional area is less than 2.18×2.18nm2, the nanowire undergoes spontaneous reversible phase transformation from fcc to body-centered tetragonal structure. In wires with larger cross-sectional areas, the structural transformation is achieved via spontaneous reversible lattice reorientation. In both cases, under tensile loading and unloading, Pd nanowires reverse between the corresponding transformed structure and the original structure, exhibiting pseudoelastic behaviors characterized by fully recoverable strains of up to 50%. read less NOT USED (low confidence) J. Jin, S. Shevlin, and Z. Guo, “Multiscale simulation of onset plasticity during nanoindentation of Al (001) surface,” Acta Materialia. 2008. link Times cited: 59 NOT USED (low confidence) H.-K. Kim, W. Jung, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for the Fe–Ti–C and Fe–Ti–N ternary systems,” Acta Materialia. 2008. link Times cited: 121 NOT USED (low confidence) H. Sa’adi and B. Hamad, “Structural and dynamical properties of iridium surfaces: First principles and molecular dynamics investigations,” Physica B-condensed Matter. 2008. link Times cited: 0 NOT USED (low confidence) Z. Sun, X.-xi Wang, and H. Wu, “Surface relaxation effect on the distributions of energy and bulk stresses in the vicinity of Cu surface: An embedded-atom method study,” Journal of Applied Physics. 2008. link Times cited: 7 Abstract: Molecular statics simulations combined with an embedded-atom… read moreAbstract: Molecular statics simulations combined with an embedded-atom method potential were performed to calculate the distributions of energy and bulk stresses in crystalline and amorphous Cu slabs and to examine the effect of surface relaxation on the distributions of energy and bulk stresses in the surface region. The results reveal that a release of energy and bulk stresses in the surface region occurs upon surface relaxation. The profile of energy release upon surface relaxation of a crystalline Cu slab exhibits oscillatory damping from the topmost layer to the interior layers, while that of an amorphous Cu slab exhibits monotonic decreasing. A great diversity of patterns was observed in the profiles of bulk stresses released upon surface relaxation, which may exhibit anisotropy according to the symmetry of the surface considered. Both the profiles of energy release and bulk stress release exhibit surface-orientation dependence. The present results may provide useful information to analyze different phenomena... read less NOT USED (low confidence) Y. Han, J. Evans, and D. J. Liu, “Quantum stabilities and growth modes of thin metal films: Unsupported and NiAl-supported Ag(1 1 0) and Ag(1 0 0),” Surface Science. 2008. link Times cited: 15 NOT USED (low confidence) J. Zhang, T. Sun, Y. Yan, Y. Liang, and S. Dong, “Molecular dynamics simulation of subsurface deformed layers in AFM-based nanometric cutting process,” Applied Surface Science. 2008. link Times cited: 90 NOT USED (low confidence) J. R. Morris, R. Aga, V. A. Levashov, and T. Egami, “Many-body effects in bcc metals: An embedded atom model extension of the modified Johnson pair potential for iron,” Physical Review B. 2008. link Times cited: 8 Abstract: In this work, we generalize a many-body extension of pairwis… read moreAbstract: In this work, we generalize a many-body extension of pairwise interatomic potentials originally proposed by Baskes, in particular, showing how a pair potential interacting with multiple near neighbor shells may be extended to an embedded atom form without changing the cohesive energy or lattice constant. This is important for parametric studies of interatomic potentials, particularly how elastic constants affect other properties. Specifically, we apply this to the modified Johnson potential, a pair potential for Fe that has been used extensively for understanding liquid and amorphous metals. read less NOT USED (low confidence) W. Chan, K. Zhao, N. Q. Vo, Y. Ashkenazy, D. Cahill, and R. Averback, “Stress evolution in platinum thin films during low-energy ion irradiation,” Physical Review B. 2008. link Times cited: 22 Abstract: Stress evolution of Pt thin films during low-energy ion irra… read moreAbstract: Stress evolution of Pt thin films during low-energy ion irradiation is investigated by using wafer bending measurements and molecular dynamics simulations. Noble gas ions ranging in mass from He to Xe and energy from 0.5 to 5 keV are used. Depending on the type and energy of the ion, the change in stress can either be tensile or compressive. Heavier or higher-energy ions tend to create tensile stress, while lighter ions such as He always induce compressive stress. The stress evolution also depends on the initial state of stress in the thin films. The results are explained by a competition between the tensile stress induced by local melting along the ion track and the compressive stress induced by the accumulation of ion-induced interstitials in defect clusters or grain boundaries, often beyond the calculated ion penetration depth. Anisotropic diffusion of interstitials under an external stress field also plays an important role in the stress evolution. Molecular dynamics simulation is employed to evaluate the importance of each of these microscopic mechanisms. read less NOT USED (low confidence) E. Webb, J. Zimmerman, and S. Seel, “Reconsideration of Continuum Thermomechanical Quantities in Atomic Scale Simulations,” Mathematics and Mechanics of Solids. 2008. link Times cited: 64 Abstract: As motivation builds to consider mechanics of nanometer scal… read moreAbstract: As motivation builds to consider mechanics of nanometer scale objects, it is increasingly advantageous to implement models with finer resolution than standard continuum approaches. For such exercises to prove fruitful, these models must be able to quantify continuum thermomechanical quantities; furthermore, it may be necessary to do so on a sub-system level in order to assess gradients or distributions in a given property. Herein we review the calculation of stress, heat flux, and temperature in atomic scale numerical simulations such as the molecular dynamics method. read less NOT USED (low confidence) Y. Wen and J.-min Zhang, “Surface energy calculation of the bcc metals by using the MAEAM,” Computational Materials Science. 2008. link Times cited: 47 NOT USED (low confidence) D. Alamanova, V. Grigoryan, and M. Springborg, “Deposition of copper clusters on the Cu(1 1 1) surface,” Surface Science. 2008. link Times cited: 21 NOT USED (low confidence) W. Huang et al., “Coordination-dependent surface atomic contraction in nanocrystals revealed by coherent diffraction.,” Nature materials. 2008. link Times cited: 319 NOT USED (low confidence) K. Nordlund and S. Dudarev, “Interatomic potentials for simulating radiation damage effects in metals,” Comptes Rendus Physique. 2008. link Times cited: 29 NOT USED (low confidence) E.-H. Kim, Y.-H. Shin, and B.-J. Lee, “A modified embedded-atom method interatomic potential for Germanium,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2008. link Times cited: 86 NOT USED (low confidence) M. Mariscal, N. Oldani, S. A. Dassie, and E. Leiva, “Atomistic computer simulations on the generation of bimetallic nanoparticles.,” Faraday discussions. 2008. link Times cited: 17 Abstract: Computer simulations on the generation of bimetallic nanopar… read moreAbstract: Computer simulations on the generation of bimetallic nanoparticles are presented in this work. Two different generation mechanisms are simulated: (a) cluster-cluster collision by means of atom dynamics simulations; and (b) nanoparticle growth from a previous seed through grand canonical Monte Carlo (gcMC) calculations. When two metal nanoparticles collide, different structures are found: core/shell, alloyed and three-shell (A-B-A). On the other hand, the growth mechanism at different chemical potentials by means of gcMC reveals the same results as atom dynamics collisions do. read less NOT USED (low confidence) W. Luo, W. Hu, and S. Xiao, “Size Effect on the Thermodynamic Properties of Silver Nanoparticles,” Journal of Physical Chemistry C. 2008. link Times cited: 186 Abstract: The Gibbs free energy of silver nanoparticles has been obtai… read moreAbstract: The Gibbs free energy of silver nanoparticles has been obtained from the calculations of bulk free energy and surface free energy for both the solid and liquid phase. On the basis of the obtained Gibbs free energy of nanoparticles, thermodynamic properties of silver nanoparticles, such as melting temperature, molar heat of fusion, molar entropy of fusion, and temperature dependences of entropy and specific heat capacity have been investigated. Calculation results indicate that these thermodynamic properties can be divided into two parts: bulk quantity and surface quantity, and surface atoms are dominant for the size effect on the thermodynamic properties of nanoparticles. The method that the intersection of the free-energy curves for solid and liquid nanoparticles decide the melting point of nanoparticles demonstrates that the surface free-energy difference between the solid and liquid phase is a decisive factor for the size-dependent melting of nanostructural materials. read less NOT USED (low confidence) R. Zhang, X. Wang, P. Shrotriya, R. Biswas, A. Bastawros, and A. Chandra, “MOLECULAR APPROACH TO MATERIAL DETACHMENT MECHANISM DURING CHEMICAL MECHANICAL PLANARIZATION,” Machining Science and Technology. 2007. link Times cited: 5 Abstract: Mechanistic numerical analysis and molecular dynamics (MD) s… read moreAbstract: Mechanistic numerical analysis and molecular dynamics (MD) simulation are employed to understand the material detachment mechanism associated with chemical mechanical polishing. We investigate the mechanics of scratch intersection mechanism to obtain a characteristic length scale and compare the theoretical predictions with previous experimental observations on ductile copper discs at the micro-scale. First, an analytical model is developed based on mechanics of materials approach. The analytical model includes the effects of strain hardening during material removal as well as the geometry of indenter tip. In the next step, molecular simulations of the scratch intersection are performed at the atomistic scale. The embedded atom method (EAM) is utilized as the force field for workpiece material and a simplified tool-workpiece interaction is assumed to simulate material removal through scratch intersection mechanism. Both models are utilized to predict a characteristic length of material detachment related to material removal during scratch intersection. The predictions from two approaches are compared with experimental observations in order to draw correlations between experiment and simulation. The insights obtained from this work may assist in understanding the mechanism for chemical mechanical planarization (CMP), and even be applied to other different machining and polishing events. read less NOT USED (low confidence) H. Yildirim, A. Kara, and T. Rahman, “Origin of quasi-constant pre-exponential factors for adatom diffusion on Cu and Ag surfaces,” Physical Review B. 2007. link Times cited: 17 NOT USED (low confidence) J. R. Morris, U. Dahlborg, and M. Calvo-Dahlborg, “Recent developments and outstanding challenges in theory and modeling of liquid metals,” Journal of Non-crystalline Solids. 2007. link Times cited: 23 NOT USED (low confidence) M. S. Wu, K. Zhou, and A. A. Nazarov, “Crack nucleation at disclinated triple junctions,” Physical Review B. 2007. link Times cited: 56 NOT USED (low confidence) Y. Wen and J.-min Zhang, “Surface energy calculation of the fcc metals by using the MAEAM,” Solid State Communications. 2007. link Times cited: 199 NOT USED (low confidence) L. Zepeda-Ruiz, B. Sadigh, J. Biener, A. Hodge, and A. Hamza, “Mechanical response of freestanding Au nanopillars under compression,” Applied Physics Letters. 2007. link Times cited: 47 Abstract: We employ molecular dynamics simulations of defect-free nano… read moreAbstract: We employ molecular dynamics simulations of defect-free nanopillars with realistic cylindrical geometries to obtain an atomic-level picture of their deformation behavior under compression. We find that dislocations are nucleated in the two outermost surface layers. Furthermore, plastic yield depends crucially on the particular arrangement of steps and facets at the surface of the nanopillars. We show that different facet orientations can differ dramatically in their response to external stresses. Freestanding nanopillars exhibit a highly nonuniform distribution of stresses along their height. This causes an elastic deformation that leads to a barrel-like shape attained by the nanopillars under compression. The stress concentration at the center of the pillars due to barreling causes dislocations to preferentially nucleate in this region. read less NOT USED (low confidence) X. Zheng, D. Cahill, P. Krasnochtchekov, R. Averback, and J.-C. Zhao, “High-throughput thermal conductivity measurements of nickel solid solutions and the applicability of the Wiedemann–Franz law,” Acta Materialia. 2007. link Times cited: 79 NOT USED (low confidence) D. Belashchenko, O. Kuskov, and O. Ostrovski, “Application of the embedded-atom method to liquid Fe-S solutions,” Inorganic Materials. 2007. link Times cited: 7 NOT USED (low confidence) S. Eremeev, G. Rusina, and E. Chulkov, “Diffusion properties of Cu(0 0 1)-c(2 × 2)–Pd surface alloys,” Surface Science. 2007. link Times cited: 7 NOT USED (low confidence) J. Song and D. Srolovitz, “Atomistic simulation of multicycle asperity contact,” Acta Materialia. 2007. link Times cited: 39 NOT USED (low confidence) S. Psakhie, K. Zolnikov, and D. S. Kryzhevich, “Elementary atomistic mechanism of crystal plasticity,” Physics Letters A. 2007. link Times cited: 36 NOT USED (low confidence) F. Ma and K. Xu, “Using dangling bond density to characterize the surface energy of nanomaterials,” Surface and Interface Analysis. 2007. link Times cited: 30 Abstract: Taking f.c.c Ag, Al, Au, Ir, Pd, Pt, Rh and b.c.c Cr, Fe, Mo… read moreAbstract: Taking f.c.c Ag, Al, Au, Ir, Pd, Pt, Rh and b.c.c Cr, Fe, Mo, Nb, Ta, V, W as examples, the energetic and bonding features of unrelaxed cubic nanoparticles were investigated by the modified embedded atom method. The surface free energy increases almost inversely with the decreasing feature sizes. This is the essential reason for the fantastic microstructures and distinct properties observed at the nanometer scale. According to the analysis on atomic bonding states, we further found that the size‐dependent surface energy is directly associated with the dangling bond density. Summing up these two aspects, the dangling bond density, a microscopic parameter, is believed to be one of the intrinsic physical quantities characterizing the structures and properties of nanomaterials. Copyright © 2007 John Wiley & Sons, Ltd. read less NOT USED (low confidence) B. Henz and M. R. Zachairah, “Molecular Dynamics Study of Alkanethiolate Self-Assembled Monolayer coated Gold Nanoparticle,” 2007 DoD High Performance Computing Modernization Program Users Group Conference. 2007. link Times cited: 5 Abstract: Through molecular simulations we have observed that the surf… read moreAbstract: Through molecular simulations we have observed that the surface of gold nanoparticles become highly corrugated by the adsorption of alkanethiolate self-assembled monolayers (SAMs). Furthermore, as the temperature is increased, the SAMs dissolve into the gold nanoparticle, creating a liquid mixture at temperatures much lower than the melting temperature of the gold nanoparticle. By analyzing the mechanical and chemical properties of gold nanoparticles at temperatures below the melting point of gold, with different SAM chain lengths and surface coverage properties, we have determined that the system is metastable. The model and computational results that provide support for this hypothesis are presented in this paper. read less NOT USED (low confidence) X.-L. Song, J.-min Zhang, and K. Xu, “Atomistic simulation of point defects in L12-type Au3Cu ordered alloy,” Journal of Alloys and Compounds. 2007. link Times cited: 8 NOT USED (low confidence) D. Crowson, D. Farkas, and S. Corcoran, “Geometric relaxation of nanoporous metals: The role of surface relaxation,” Scripta Materialia. 2007. link Times cited: 74 NOT USED (low confidence) Y. Chen and S. Liao, “MONTE CARLO SIMULATION OF THE SURFACE SEGREGATION OF Au75Pd25 AT (110) SURFACE USING AN ANALYTIC EMBEDDED ATOM METHOD,” Surface Review and Letters. 2007. link Times cited: 2 Abstract: The surface concentrations and concentration depth profiles … read moreAbstract: The surface concentrations and concentration depth profiles to the (110) surface of an Au75Pd25 alloy is studied by modified analytical embedded atom method (MAEAM) with the Monte Carlo simulations. The results indicate that Au enriched in the two topmost layers, but depleted in the third layer. The Au concentration in the non-reconstructed surface is less than that in the reconstructed surface. Au concentration in third layer of reconstructed surface, which is more agreement with experimental data in present simulations, is about 63% 61% and 55%, at 800K, 600K and 400K respectively. Thus the present simulations are helpful for a better understanding of surface segregation of AuPd alloys. read less NOT USED (low confidence) K. Kolluri, M. Gungor, and D. Maroudas, “Void nucleation in biaxially strained ultrathin films of face-centered cubic metals,” Applied Physics Letters. 2007. link Times cited: 9 Abstract: We report an analysis of void nucleation as a relaxation mec… read moreAbstract: We report an analysis of void nucleation as a relaxation mechanism in freestanding biaxially strained ultrathin films of face-centered cubic metals based on large-scale molecular-dynamics simulations. Above a critical strain level, multiple threading dislocations are emitted from the film surface. The surface step traces formed by gliding dislocations on intersecting and on adjacent parallel glide planes lead to formation and growth of surface pits and grooves, while vacancies form due to gliding of jogged dislocations and dislocation intersections. Coalescence of the surface pits with vacancy clusters is the precursor to the formation of a larger void extending across the film. read less NOT USED (low confidence) H. Yildirim, A. Kara, and T. Rahman, “Tip-induced adatom extraction and cluster manipulation,” Physical Review B. 2007. link Times cited: 23 Abstract: We present results for tip-induced extraction of a Cu adatom… read moreAbstract: We present results for tip-induced extraction of a Cu adatom from Cu mound on Cu(111), and compare the characteristics to that for a similar Ag system. Molecular-dynamics and molecular static simulations were carried out using interaction potentials from the embedded atom method. Molecular-dynamics simulations revealed differences in the modes of extraction for the cases of Ag and Cu systems and their dependence on tip geometry. For the case of a sharp Ag tip, the extraction of a Ag adatom occurs via the pulling mode, while with a blunt Ag tip, the extraction is more complex involving a two-step motion. On the other hand, the relatively stronger Cu-Cu interaction leads to a sliding and/or dragging mode in which the whole three-dimensional cluster is dragged followed by the extraction of the adatom from the cluster. Molecular static simulations provide a detailed analysis of the changes in the energy landscape in the presence of the tip, resulting in a substantial decrease of the energy barrier for an adatom to descend from the mound. read less NOT USED (low confidence) M. Müller and K. Albe, “Concentration of thermal vacancies in metallic nanoparticles,” Acta Materialia. 2007. link Times cited: 42 NOT USED (low confidence) E. Rabkin, H. Nam, and D. Srolovitz, “Atomistic simulation of the deformation of gold nanopillars,” Acta Materialia. 2007. link Times cited: 110 NOT USED (low confidence) P. Wynblatt, D. Chatain, A. Ranguis, J. Monchoux, J. Moon, and S. Garoff, “Factors Affecting the Coverage Dependence of the Diffusivity of One Metal over the Surface of Another,” International Journal of Thermophysics. 2007. link Times cited: 3 NOT USED (low confidence) T. Sondón, J. Guevara, and A. Saúl, “Study of the structure, segregation, and magnetic properties of Ni-Rh clusters,” Physical Review B. 2007. link Times cited: 10 Abstract: We studied the effects of size reduction and alloying on the… read moreAbstract: We studied the effects of size reduction and alloying on the determination of structural, segregation, and magnetic properties of 55-atom mixed Ni and Rh clusters in the whole range of concentrations. Molecular-dynamics simulations were performed to determine the cluster structures with energies and forces calculated with a semiempirical many-body potential parametrized to the alloy thermodynamic data. Magnetic properties were calculated by solving self-consistently a tight-binding Hamiltonian in the unrestricted Hartree-Fock approximation. We relate segregation behavior to magnetic properties, and we show that for low Rh concentrations there is an enhancement of the cluster magnetic moment with respect to the pure Ni one. For the central range of concentrations, we found that chemical isomers whose structures lie very close in energy present very different magnetic properties. read less NOT USED (low confidence) M. Schurmans, J. Luyten, and C. Creemers, “The BFS Method Combined with Chemical Cluster Interactions for the Study of Order-Disorder Transitions,” Defect and Diffusion Forum. 2007. link Times cited: 1 Abstract: First Principles (FP) methods are invoked to improve the acc… read moreAbstract: First Principles (FP) methods are invoked to improve the accuracy of Bozzolo-Ferrante- Smith (BFS) model, one of the quantum-approximate modeling techniques for the computation of thermodynamic properties that involve a large number of particles. The BFS method calculates the energy of an atom in an alloy in two steps [1]. A first term pertains to the structural contribution. A recent improvement [2] allows to calculate the strain energy depending on the local environment [1,2] and this involves only pure element properties of the different atomic species. In the second step, binary chemical interactions are taken into account. This was originally done by only two interaction parameters for each atom pair in an alloy. In contrast, the adaptable parameterization of Cluster Expansion Methods (CEM) routinely incorporates any number of FP data to describe ordering in alloy systems. But in standard CEM calculations, no explicit information on local atomic displacements is used. In this work, the BFS chemical energy term is successfully replaced by a CEM chemical term to combine the ability of BFS to account for local displacements and the ability of CEM to include as many FP results as needed for the correct evaluation of alloying effects. read less NOT USED (low confidence) Y. Qi and R. Mishra, “Ab Initio Study of the Effect of Solute Atoms on Stacking Fault Energy in Aluminum,” Bulletin of the American Physical Society. 2007. link Times cited: 78 Abstract: Received 11 December 2006; revised manuscript received 16 Fe… read moreAbstract: Received 11 December 2006; revised manuscript received 16 February 2007; published 6 June 2007The stacking fault energy SFE in binary and ternary alloys of Al with common alloying elements wasstudied using density functional theory. Among these alloying elements, Fe further increases the SFE and Gereduces the SFE of Al. The alloying elements increase the SFE by increasing the directional inhomogeneity inthe electronic charge distribution of Al. The maximum value of charge difference on the fault plane, Max ,is used to characterize how many electrons have been redistributed due to the stacking fault formation, and theSFE increases with Max .DOI: 10.1103/PhysRevB.75.224105 PACS number s : 61.72.Nn, 62.20.Fe, 71.15.Nc read less NOT USED (low confidence) P. Ferreira and Y. Shao-horn, “Formation Mechanism of Pt Single-Crystal Nanoparticles in Proton Exchange Membrane Fuel Cells,” Electrochemical and Solid State Letters. 2007. link Times cited: 75 Abstract: In proton exchange membrane fuel cells, hydrogen permeated f… read moreAbstract: In proton exchange membrane fuel cells, hydrogen permeated from the anode to the cathode was found to reduce soluble Pt species and produce faceted and dendritic Pt nanoparticles in the cathode ionomer. Moving away from the carbon support particles, the morphology of Pt nanoparticles changed from dendritic shapes to truncated tetrahedrons, truncated octahedrons, and truncated square cuboids. Transmission electron microscopy results suggest that the homogeneity of the driving force (supersaturation) for reduction of soluble Pt at the growing surface could dictate the transition from dendritic to faceted growth, and the competition between surface energy and interfacial kinetics of Pt reduction could govern the shape of faceted Pt nanoparticles. read less NOT USED (low confidence) G. X. Chen, J.-min Zhang, K. Xu, and V. Ji, “Computer simulation study of self-diffusion in Pd(0 0 1) surface,” Journal of Physics and Chemistry of Solids. 2007. link Times cited: 2 NOT USED (low confidence) G. Antczak and G. Ehrlich, “Jump processes in surface diffusion,” Surface Science Reports. 2007. link Times cited: 158 NOT USED (low confidence) J. C. Flores, B. Aguilar, A. Coronado, and H.-C. Huang, “Double rotation mechanism in small Cu clusters concerted diffusion over Cu1 1 1 surfaces,” Surface Science. 2007. link Times cited: 8 NOT USED (low confidence) J.-min Zhang, Y. Wen, and K. Xu, “Calculation of the formation energies of isolated vacancy and adatom–vacancy pair at low-index surfaces of fcc metals with MAEAM,” Applied Surface Science. 2007. link Times cited: 13 NOT USED (low confidence) K. Narayan, K. Behdinan, and Z. Fawaz, “An engineering-oriented embedded-atom-method potential fitting procedure for pure fcc and bcc metals,” Journal of Materials Processing Technology. 2007. link Times cited: 22 NOT USED (low confidence) R. Pasianot and L. Malerba, “Interatomic potentials consistent with thermodynamics: The Fe–Cu system,” Journal of Nuclear Materials. 2007. link Times cited: 68 NOT USED (low confidence) F. Ma and K. Xu, “Size-dependent multilayer relaxation of nanowires and additional effect of surface stresses,” Solid State Communications. 2007. link Times cited: 2 NOT USED (low confidence) Y. Shu, J.-min Zhang, K. Xu, and V. Ji, “Anisotropy analysis of the surface stress and surface energy in Cu surfaces with the modified embedded atom method,” Solid State Communications. 2007. link Times cited: 7 NOT USED (low confidence) K.-H. Hong, J. Yoon, and P. Cha, “Influence of epitaxial strain on the terrace and inter-layer diffusions in metal epitaxy,” Applied Surface Science. 2006. link Times cited: 2 NOT USED (low confidence) J. Rogan, G. García, C. Loyola, W. Orellana, R. Ramírez, and M. Kiwi, “Alternative search strategy for minimal energy nanocluster structures: the case of rhodium, palladium, and silver.,” The Journal of chemical physics. 2006. link Times cited: 35 Abstract: An alternative strategy to find the minimal energy structure… read moreAbstract: An alternative strategy to find the minimal energy structure of nanoclusters is presented and implemented. We use it to determine the structure of metallic clusters. It consists in an unbiased search, with a global minimum algorithm: conformational space annealing. First, we find the minima of a many-body phenomenological potential to create a data bank of putative minima. This procedure assures us the generation of a set of cluster configurations of large diversity. Next, the clusters in this data bank are relaxed by ab initio techniques to obtain their energies and geometrical structures. The scheme is successfully applied to magic number 13 atom clusters of rhodium, palladium, and silver. We obtained minimal energy cluster structures not previously reported, which are different from the phenomenological minima. Moreover, they are not always highly symmetric, thus casting some doubt on the customary biased search scheme, which consists in relaxing with density functional theory global minima chosen among high symmetry structures obtained by means of phenomenological potentials. read less NOT USED (low confidence) Y. Puzyrev and J. Faulkner, “Atomic displacements in alloys,” Metallurgical and Materials Transactions A. 2006. link Times cited: 0 NOT USED (low confidence) K. Liu and S. Gao, “Adsorbate vibration and resonance lifetime broadening of a cobalt adatom on a Cu(111) surface,” Physical Review B. 2006. link Times cited: 9 Abstract: We present a theoretical calculation of the vibrational spec… read moreAbstract: We present a theoretical calculation of the vibrational spectrum and resonance lifetime of Co/Cu(111) surface based on a well-tested empirical potential model. The phonon dispersions and local vibrational densities are obtained as a function of adsorbate coverage. The frequencies of the adsorbate-induced vibrational modes are in close agreement with those given by first-principles calculations and comparable with available experimental data. Analysis of the polarization distributions shows that both the frustrated translation (FT) and vertical vibration of the adsorbate are strongly coupled with atomic vibrations in the substrate. We also determined the lifetime of the FT mode caused by resonance broadening in the single-adsorbate limit. These results provide the basis for detailed study of nonadiabatic dynamics of vibrational excitation and damping of adatoms at metal surfaces. read less NOT USED (low confidence) J. Wang, Y. Wang, R. Schäublin, C. Abromeit, and R. Gotthardt, “The effect of point defects on the martensitic phase transformation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2006. link Times cited: 10 NOT USED (low confidence) M. Amini and B. Laird, “Kinetic coefficient for hard-sphere crystal growth from the melt.,” Physical review letters. 2006. link Times cited: 62 Abstract: Using molecular-dynamics simulation, we determine the magnit… read moreAbstract: Using molecular-dynamics simulation, we determine the magnitude and anisotropy of the kinetic coefficient (mu) for the crystal growth from the melt for the hard-sphere system through an analysis of equilibrium capillary fluctuations in interfacial height. We find mu100 = 1.44(7), mu110 = 1.10(5), and mu111 = 0.64(3) in units of square root (kB/(mTm)), where kB is Boltzmann's constant, m is the particle mass, and Tm is the melting temperature. These values are shown to be consistent, with some exceptions, with those obtained in recent simulation results a variety of fcc metals, when expressed in hard-sphere units. This suggests that the kinetic coefficient for fcc metals can be roughly estimated from C square root (R/(MTm)), where R is the gas constant, M is the molar mass, and C is a constant that varies with interfacial orientation. read less NOT USED (low confidence) R. V. P. Montsouka et al., “Preparation of anisotropic magneticFeNiPt2films on MgO(001): Atomistic mechanisms for the interdiffusion of twoL10phases,” Physical Review B. 2006. link Times cited: 11 Abstract: $\mathrm{L}{1}_{0}$-ordered $\mathrm{Fe}\mathrm{Ni}{\mathrm{… read moreAbstract: $\mathrm{L}{1}_{0}$-ordered $\mathrm{Fe}\mathrm{Ni}{\mathrm{Pt}}_{2}(001)$ thin films were prepared by the interdiffusion of FePt(001) and NiPt(001) layers codeposited on MgO(001) substrates by molecular beam epitaxy (MBE). A large uniaxial magnetic anisotropy $({K}_{u}={9.10}^{5}\phantom{\rule{0.3em}{0ex}}\mathrm{J}∕{\mathrm{m}}^{3})$ and a reduced magnetic transition temperature $({T}_{c}=400\phantom{\rule{0.3em}{0ex}}\mathrm{K})$ were obtained. Growth at $700\phantom{\rule{0.3em}{0ex}}\mathrm{K}$ and a first annealing at $800\phantom{\rule{0.3em}{0ex}}\mathrm{K}$ result in a large long-range order parameter reflecting the concentration modulation along the growth direction. This high long-range order parameter is conserved in the $\mathrm{Fe}\mathrm{Ni}{\mathrm{Pt}}_{2}$ layers after interdiffusion at $900\phantom{\rule{0.3em}{0ex}}\mathrm{K}$, contrary to what is expected from a simple vacancy migration process. This experimental observation can be explained either by a 6-jump cycle mechanism or by the alternate diffusion of a double vacancy, which are both favored energetically over a second-nearest-neighbor jump mechanism or the simultaneous diffusion of a double vacancy as shown by quenched molecular dynamics simulations. read less NOT USED (low confidence) M. C. Giménez, A. Ramirez-Pastor, and E. Leiva, “Monte Carlo simulation of metal deposition on foreign substrates,” Surface Science. 2006. link Times cited: 12 NOT USED (low confidence) G. X. Chen, J.-min Zhang, K. Xu, and V. Ji, “Computer simulation study of self-diffusion in Pd(1 1 1) surface,” Journal of Molecular Catalysis A-chemical. 2006. link Times cited: 2 NOT USED (low confidence) M. Dunn et al., “Atomistic modeling of nanowires, small-scale fatigue damage in cast magnesium, and materials for MEMS.” 2006. link Times cited: 0 Abstract: Lightweight and miniaturized weapon systems are driving the … read moreAbstract: Lightweight and miniaturized weapon systems are driving the use of new materials in design such as microscale materials and ultra low-density metallic materials. Reliable design of future weapon components and systems demands a thorough understanding of the deformation modes in these materials that comprise the components and a robust methodology to predict their performance during service or storage. Traditional continuum models of material deformation and failure are not easily extended to these new materials unless microstructural characteristics are included in the formulation. For example, in LIGA Ni and Al-Si thin films, the physical size is on the order of microns, a scale approaching key microstructural features. For a new potential structural material, cast Mg offers a high stiffness-to-weight ratio, but the microstructural heterogeneity at various scales requires a structure-property continuum model. Processes occurring at the nanoscale and microscale develop certain structures that drive material behavior. The objective of the work presented in this report was to understand material characteristics in relation to mechanical properties at the nanoscale and microscale in these promising new material systems. Research was conducted primarily at the University of Colorado at Boulder to employ tightly coupled experimentation and simulation to study damage at various material sizemore » scales under monotonic and cyclic loading conditions. Experimental characterization of nano/micro damage will be accomplished by novel techniques such as in-situ environmental scanning electron microscopy (ESEM), 1 MeV transmission electron microscopy (TEM), and atomic force microscopy (AFM). New simulations to support experimental efforts will include modified embedded atom method (MEAM) atomistic simulations at the nanoscale and single crystal micromechanical finite element simulations. This report summarizes the major research and development accomplishments for the LDRD project titled 'Atomistic Modeling of Nanowires, Small-scale Fatigue Damage in Cast Magnesium, and Materials for MEMS'. This project supported a strategic partnership between Sandia National Laboratories and the University of Colorado at Boulder by providing funding for the lead author, Ken Gall, and his students, while he was a member of the University of Colorado faculty.« less read less NOT USED (low confidence) O. A. Oviedo, C. Mayer, G. Staikov, E. Leiva, and W. Lorenz, “Low-dimensional metallic nanostructures and their electrochemical relevance: Energetics and phenomenological approach,” Surface Science. 2006. link Times cited: 6 NOT USED (low confidence) B.-J. Lee, T.-H. Lee, and S.-J. Kim, “A modified embedded-atom method interatomic potential for the Fe–N system: A comparative study with the Fe–C system,” Acta Materialia. 2006. link Times cited: 72 NOT USED (low confidence) J.-min Zhang, D.-D. Wang, and K. Xu, “Calculation of the surface energy of bcc transition metals by using the second nearest neighbor modified embedded atom method,” Applied Surface Science. 2006. link Times cited: 48 NOT USED (low confidence) M. Finnis and M. Rühle, “Structures of Interfaces in Crystalline Solids,” Materials Science and Technology. 2006. link Times cited: 3 Abstract: Interfaces in materials may be grain boundaries between like… read moreAbstract: Interfaces in materials may be grain boundaries between like crystals or phase boundaries between unlike crystals. Experimental approaches for the determination of the atomic structures of the interfaces are reviewed with emphasis on high-resolution electron microscopy (HREM). It will be shown that information on orientation relationship between the adjacent grains, the translation state and atomic relaxations can be elaborated with high precision. In a case study, the structures of one specific grain boundary in Al2O3 will be discussed in detail. Such experimental studies have provided a mass of structural information in recent years. read less NOT USED (low confidence) G. Rusina, S. Eremeev, S. Borisova, I. Sklyadneva, and E. Chulkov, “Vibrations on Al surfaces covered by sodium,” Surface Science. 2006. link Times cited: 4 NOT USED (low confidence) S. Zhao, K. Albe, and H. Hahn, “Grain size dependence of the bulk modulus of nanocrystalline nickel,” Scripta Materialia. 2006. link Times cited: 46 NOT USED (low confidence) F. Streitz et al., “Simulating Solidification in Metals at High Pressure: The Drive to Petascale Computing.” 2006. link Times cited: 36 Abstract: We investigate solidification in metal systems ranging in si… read moreAbstract: We investigate solidification in metal systems ranging in size from 64,000 to 524,288,000 atoms on the IBM BlueGene/L computer at LLNL. Using the newly developed ddcMD code, we achieve performance rates as high as 103 TFlops, with a performance of 101.7 TFlop sustained over a 7 hour run on 131,072 cpus. We demonstrate superb strong and weak scaling. Our calculations are significant as they represent the first atomic-scale model of metal solidification to proceed, without finite size effects, from spontaneous nucleation and growth of solid out of the liquid, through the coalescence phase, and into the onset of coarsening. Thus, our simulations represent the first step towards an atomistic model of nucleation and growth that can directly link atomistic to mesoscopic length scales. read less NOT USED (low confidence) L. Kong and L. J. Lewis, “Transition state theory of the preexponential factors for self-diffusion on Cu, Ag, and Ni surfaces,” Physical Review B. 2006. link Times cited: 32 NOT USED (low confidence) H. Zhang and C. Sun, “A multiscale mechanics approach for modeling textured polycrystalline thin films with nanothickness,” International Journal of Mechanical Sciences. 2006. link Times cited: 9 NOT USED (low confidence) S. Valone, M. Baskes, and R. L. Martin, “Atomistic model of helium bubbles in gallium-stabilized plutonium alloys,” Physical Review B. 2006. link Times cited: 66 Abstract: The varying thermodynamic stability of gallium- (Ga-) stabil… read moreAbstract: The varying thermodynamic stability of gallium- (Ga-) stabilized plutonium (Pu) alloys with temperature affords a unique setting for the development of self-irradiation damage. Here, fundamental characteristics of helium (He) bubbles in these alloys with respect to temperature, gallium concentration, and He-to-vacancy ratio are modeled at the atomistic level with a modified embedded atom potential that takes account of this varying stability. Aside from the bubbles themselves, the surrounding matrix material is single-crystal metal or alloy. As a function of temperature, with a 2:1 He-to-vacancy ratio in a 5-at. % Ga fcc lattice, a 1.25-nm bubble is very stable up to about 1000 K. At 1000 K, the bubble distorts the surrounding lattice and precipitates a liquid zone, as is consistent with the phase diagram for the model material. Between 300 and 500 K, this same bubble relaxes slightly through interstitial emission. At 300 K, with a 2:1 He-to-vacancy ratio in a 2.5-at. % Ga fcc lattice, the Ga stabilization is less effective in the model to the point where the bubble distorts the local lattice and expands significantly. Similarly, at 300 K, if the He-to-vacancy ratio is increased to 3:1, there is significant local lattice distortion, as well as ejectionmore » of some He atoms into the lattice. The formation of new bubbles is not observed, because those events take place on a longer time scale than can be simulated with the present approach.« less read less NOT USED (low confidence) R. Iglesias and E. Leiva, “Two-grain nanoindentation using the quasicontinuum method: Two-dimensional model approach,” Acta Materialia. 2006. link Times cited: 30 NOT USED (low confidence) W. K. Liu, E. Karpov, and H. S. Park, “Classical Molecular Dynamics.” 2006. link Times cited: 0 NOT USED (low confidence) J.-yu Yang and W. Hu, “Anharmonicity in Al vicinal surfaces of (1 0 0) with (1 1 1) step,” Applied Surface Science. 2006. link Times cited: 2 NOT USED (low confidence) S. Eremeev, S. Kulkova, and P. Potapov, “The electronic structure of grain boundaries in metals and alloys,” Computational Materials Science. 2006. link Times cited: 6 NOT USED (low confidence) O. A. Oviedo, E. Leiva, and M. I. Rojas, “Energetic and entropic contributions to the underpotential/overpotential deposition shifts on single crystal surfaces from lattice dynamics,” Electrochimica Acta. 2006. link Times cited: 18 NOT USED (low confidence) J.-min Zhang, X.-L. Song, X. Zhang, and K. Xu, “The properties and structures of the mono- and the di- vacancy in Cu crystal,” Journal of Physics and Chemistry of Solids. 2006. link Times cited: 22 NOT USED (low confidence) S. Durukanoğlu, O. Trushin, and T. Rahman, “Effect of step-step separation on surface diffusion processes,” Physical Review B. 2006. link Times cited: 12 NOT USED (low confidence) P. Klein and J. Zimmerman, “Coupled atomistic-continuum simulations using arbitrary overlapping domains,” J. Comput. Phys. 2006. link Times cited: 81 NOT USED (low confidence) A. Saedi, “A study on mutual interaction between atomistic and macroscopic phenomena during electrochemical processes using coupled finite difference – kinetic Monte Carlo model: Application to potential step test in simple copper sulfate bath,” Journal of Electroanalytical Chemistry. 2006. link Times cited: 14 NOT USED (low confidence) T. Uehara and T. Tamai, “An Atomistic Study on Shape-Memory Effect by Shear Deformation and Phase Transformation,” Mechanics of Advanced Materials and Structures. 2006. link Times cited: 14 Abstract: An atomistic study on the shape-memory effect is carried out… read moreAbstract: An atomistic study on the shape-memory effect is carried out by molecular dynamics simulations with the EAM potential for Ni-Al alloy. As preliminary simulations, the stable structures for several Ni contents at various temperatures are investigated, and reveal that the martensite structure is obtained at low temperature for models with 62% or higher Ni content, while bcc is stable at high temperatures. A 68% Ni model with martensite structure, which consists of several variants in two opposite orientations without macroscopic deformation, is applied for MD simulation under a series of thermo-mechanical conditions of shear loading, unloading, heating and cooling. When shear load is applied, some of the variants with unstable orientation against the load change orientation, and a permanent macroscopic deformation is obtained. When this deformed martensite is heated up, a phase transformation to bcc occurs and the deformation is diminished. The original martensite is regained by cooling. Since the orientation of the variants are random, the macroscopic deformation is not observed, which means that the deformation imposed by the external load recovers the original shape by the heat treatment. read less NOT USED (low confidence) H. Wu, “Molecular dynamics study of the mechanics of metal nanowires at finite temperature,” European Journal of Mechanics A-solids. 2006. link Times cited: 133 NOT USED (low confidence) C.-yu Wang and X. Zhang, “Multiscale modeling and related hybrid approaches,” Current Opinion in Solid State & Materials Science. 2006. link Times cited: 17 NOT USED (low confidence) J.-min Zhang, Y. Shu, and K. Xu, “Multilayer relaxation of fcc metals (001) surface: A modified embedded atom method study,” Solid State Communications. 2006. link Times cited: 14 NOT USED (low confidence) S. Psakhie, K. Zolnikov, D. S. Kryzhevich, and A. Lipnitskii, “On structural defect generation induced by thermal fluctuations in materials with a perfect lattice under dynamic loading,” Physics Letters A. 2006. link Times cited: 28 NOT USED (low confidence) K. Kádas et al., “Surface relaxation and surface stress of 4d transition metals,” Surface Science. 2006. link Times cited: 29 NOT USED (low confidence) K. Zhou, A. A. Nazarov, and M. Wu, “Continuum and atomistic studies of a disclinated crack in a bicrystalline nanowire,” Physical Review B. 2006. link Times cited: 71 NOT USED (low confidence) W. Schmickler, K. Pötting, and M. Mariscal, “A new simulation model for electrochemical metal deposition,” Chemical Physics. 2006. link Times cited: 18 NOT USED (low confidence) M. D. Pópolo, E. Leiva, M. Mariscal, and W. Schmickler, “On the generation of metal clusters with the electrochemical scanning tunneling microscope,” Surface Science. 2005. link Times cited: 28 NOT USED (low confidence) D. Shan, L. Yuan, and B. Guo, “Multiscale simulation of surface step effects on nanoindentation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2005. link Times cited: 36 NOT USED (low confidence) Y. Yin, K. Takahashi, X. Yuan, and T. Onzawa, “Dimer Reference Embedded Atom Method (DR-EAM) and Its Application to Vacancy Formation Energy of FCC Metals,” Quarterly Journal of The Japan Welding Society. 2005. link Times cited: 1 Abstract: Embedded atom method (EAM) has been successfully applied to … read moreAbstract: Embedded atom method (EAM) has been successfully applied to investigate surface properties and simulate diffusion phenomena, which do a great help to study micro-joining process. However, because its theory and parameterization are based on bulk system it fails in solving some problems of non-bulk system. In order to increase the applicability of EAM to non-bulk system, a new scheme of EAM, DR-EAM is proposed. In this scheme, the dimer structure is selected as a common reference structure and the parameters are derived from dimer and some bulk properties. In this work, the DR-EAM parameters of 7 kinds of FCC metals are renewed by revising the modeling system and including some experimental data of dimer. The features of their energy-distance curves are discussed and it shows the need of including angular dependency of electronic density. The vacancy formation energies, which play an important role in the diffusion process, are calculated and compared with experimental data. read less NOT USED (low confidence) S. Sankaranarayanan, V. Bhethanabotla, and B. Joseph, “Molecular dynamics simulations of the structural and dynamic properties of graphite-supported bimetallic transition metal clusters,” Physical Review B. 2005. link Times cited: 57 NOT USED (low confidence) M. Gungor and D. Maroudas, “Relaxation of biaxial tensile strain in ultrathin metallic films: Ductile void growth versus nanocrystalline domain formation,” Applied Physics Letters. 2005. link Times cited: 12 Abstract: We report a computational analysis of the atomistic mechanis… read moreAbstract: We report a computational analysis of the atomistic mechanisms of relaxation of biaxially applied tensile strains over a range of strain levels up to 17% in free-standing ultrathin metallic films with the film plane oriented normal to the [111] crystallographic direction. The analysis is based on molecular-dynamics simulations using slab supercells that contain millions of atoms to model single-crystalline thin films without and with cylindrical voids oriented normal to the film plane and penetrating through the film thickness. At high levels of applied strain (>8%), a strain relaxation regime other than the ductile void growth is revealed that gives rise to a practically uniform distribution of dislocations in the film and subsequent formation of nanometer-scale face-centered-cubic crystalline domains, i.e., a single-to-polycrystalline structural transition. It is demonstrated that in this strain relaxation regime, void growth is inhibited as the dislocations emitted from the void surface are pinned by t... read less NOT USED (low confidence) J. Akhter, E. Ahmed, and M. Ahmad, “Study of diffusion coefficients in liquid noble metals,” Materials Chemistry and Physics. 2005. link Times cited: 22 NOT USED (low confidence) W. Liang and M. Zhou, “Pseudoelasticity of Single Crystalline Cu Nanowires Through Reversible Lattice Reorientations,” Journal of Engineering Materials and Technology-transactions of The Asme. 2005. link Times cited: 85 Abstract: Molecular dynamics simulations are carried out to analyze th… read moreAbstract: Molecular dynamics simulations are carried out to analyze the structure and mechanical behavior of Cu nanowires with lateral dimensions of 1.45-2.89 nm. The calculations simulate the formation of nanowires through a top-down fabrication process by slicing square columns of atoms from single-crystalline bulk Cu along the [001], [010], and [100] directions and by allowing them to undergo controlled relaxation which involves the reorientation of the initial configuration with a (001) axis and {001} surfaces into a new configuration with a axis and {111} lateral surfaces. The propagation of twin planes is primarily responsible for the lattice rotation. The transformed structure is the same as what has been observed experimentally in Cu nanowires. A pseudoelastic behavior driven by the high surface-to-volume ratio and surface stress at the nanoscale is observed for the transformed wires. Specifically, the relaxed wires undergo a reverse transformation to recover the configuration it possessed as part of the bulk crystal prior to relaxation when tensile loading with sufficient magnitude is applied. The reverse transformation progresses with the propagation of a single twin boundary in reverse to that observed during relaxation. This process has the diffusion less nature and the invariant-plane strain of a martensitic transformation and is similar to those in shape memory alloys in phenomenology. The reversibility of the relaxation and loading processes endows the nanowires with the ability for pseudoelastic elongations of up to 41% in reversible axial strain which is well beyond the yield strain of the approximately 0.25% of bulk Cu and the recoverable strains on the order of 8% of most bulk shape memory materials. The existence of the pseudoelasticity observed in the single-crystalline, metallic nanowires here is size and temperature dependent. At 300 K, this effect is observed in wires with lateral dimensions equal to or smaller than 1.81 X 1.81 nm. As temperature increases, the critical wire size for observing this effect increases. This temperature dependence gives rise to a novel shape memory effect to Cu nanowires not seen in bulk Cu. read less NOT USED (low confidence) R. Hong, M. Huang, and J. Yang, “Molecular dynamics study of copper trench filling in damascene process,” Materials Science in Semiconductor Processing. 2005. link Times cited: 7 NOT USED (low confidence) A. Jiang, T. Tyson, and L. Axe, “The structure of small Ta clusters,” Journal of Physics: Condensed Matter. 2005. link Times cited: 13 Abstract: The structure of small tantalum clusters is investigated by … read moreAbstract: The structure of small tantalum clusters is investigated by using molecular dynamics simulations. A structural evolution from polytetrahedral structures to layered Frank–Kasper-type structures is revealed as cluster size increases to N∼100 atoms. The lowest-energy structures have been located for clusters with N≤78. The bulk-like (bcc) structure becomes the most stable structure beyond N∼100. The stabilized structure strongly depends on the cooling rate. A structure similar to β-Ta, a σ-type Frank–Kasper structure, can be obtained by rapid cooling. The structural properties of small Ta clusters presented in the paper provide insight into the formation and origin of β-phase Ta. The growth of β-Ta films in practice may be due to the nucleation of Ta clusters with layered Frank–Kasper-type structure during the initial stage of film growth. read less NOT USED (low confidence) H. S. Park, E. Karpov, and W. K. Liu, “Non‐reflecting boundary conditions for atomistic, continuum and coupled atomistic/continuum simulations,” International Journal for Numerical Methods in Engineering. 2005. link Times cited: 57 Abstract: We present a method to numerically calculate a non‐reflectin… read moreAbstract: We present a method to numerically calculate a non‐reflecting boundary condition which is applicable to atomistic, continuum and coupled multiscale atomistic/continuum simulations. The method is based on the assumption that the forces near the domain boundary can be well represented as a linear function of the displacements, and utilizes standard Laplace and Fourier transform techniques to eliminate the unnecessary degrees of freedom. The eliminated degrees of freedom are accounted for in a time‐history kernel that can be calculated for arbitrary crystal lattices and interatomic potentials, or regular finite element meshes using an automated numerical procedure. The new theoretical developments presented in this work allow the application of the method to non‐nearest neighbour atomic interactions; it is also demonstrated that the identical procedure can be used for finite element and mesh‐free simulations. We illustrate the effectiveness of the method on a one‐dimensional model problem, and calculate the time‐history kernel for FCC gold using the embedded atom method (EAM). Copyright © 2005 John Wiley & Sons, Ltd. read less NOT USED (low confidence) Z. Chen and C.-yu Wang, “First-principles study on the effect of impurities at the front of cracks in α-Fe,” Physical Review B. 2005. link Times cited: 6 Abstract: By using two first-principles methods, discrete variational … read moreAbstract: By using two first-principles methods, discrete variational method (DVM) and DMol, within the framework of density-functional theory, the effects of nonmetal light impurities (C, N, and O) on a fracture in $\ensuremath{\alpha}\text{\ensuremath{-}}\mathrm{Fe}$ are studied. We calculate the curve of binding energy $({E}_{b})$ vs the displacements of the nearest-neighbor Fe atoms of the impurity, and find that C and N tend to hold onto these two Fe atoms. Calculated PDOS and deformation charge density reveal that C and N prefer to form polarized bonds vertical to the propagation direction of the crack with the nearest-neighbor Fe atoms. Furthermore, interaction of the C $(\mathrm{N})\ensuremath{-}\mathrm{Fe}$ pair which is vertical to the crack plane is much larger than that of the C $(\mathrm{N})\ensuremath{-}\mathrm{Fe}$ pair which is parallel to the plane. This anisotropic bonding character indicates that doping C or N impedes the propagation of the crack, thus can improve the mechanical property of $\ensuremath{\alpha}\text{\ensuremath{-}}\mathrm{Fe}$. On the contrary, O tends to push its nearest-neighbor Fe atoms away, and exhibits weak interaction with its nearest-neighbor atoms. O also weakens the interaction of $\mathrm{Fe}\ensuremath{-}\mathrm{Fe}$ pair in bulk. Thus O has detrimental effects on the ductility of $\ensuremath{\alpha}\text{\ensuremath{-}}\mathrm{Fe}$. read less NOT USED (low confidence) Q. Lu and B. Bhattacharya, “The role of atomistic simulations in probing the small-scale aspects of fracture—a case study on a single-walled carbon nanotube,” Engineering Fracture Mechanics. 2005. link Times cited: 71 NOT USED (low confidence) M. Caffio, A. Atrei, U. Bardi, and G. Rovida, “Growth mechanism and structure of nickel deposited on Ag(001),” Surface Science. 2005. link Times cited: 6 NOT USED (low confidence) M. Mariscal and W. Schmickler, “On the surface properties of lead structures on Au(111), an atom dynamic approach,” Journal of Electroanalytical Chemistry. 2005. link Times cited: 2 NOT USED (low confidence) C. Vardeman, P. Conforti, M. M. Sprague, and J. Gezelter, “Breathing mode dynamics and elastic properties of gold nanoparticles.,” The journal of physical chemistry. B. 2005. link Times cited: 8 Abstract: We present calculations of the bulk modulus, heat capacity, … read moreAbstract: We present calculations of the bulk modulus, heat capacity, and the period of the breathing mode for spherical nanoparticles following excitation by ultrafast laser pulses. The bulk modulus and heat capacities both exhibit clear transitions upon bulk melting of the particles. Equilibrium calculations of the heat capacity show that the melting transition is sharper and occurs at a lower temperature than one would observe from an ultrafast experiment. We also observe an intriguing splitting in the low-frequency spectra of the nanoparticles and analyze this splitting in terms of Lamb's classical theory of elastic spheres. We conclude that the particles either (1) melt during the observation period following laser excitation or (2) melt an outer shell while maintaining a crystalline core. Both mechanisms for melting are commensurate with our observations. read less NOT USED (low confidence) S. Durukanoğlu, A. Kara, and T. Rahman, “The role of lattice vibrations in adatom diffusion at metal stepped surfaces,” Surface Science. 2005. link Times cited: 7 NOT USED (low confidence) A. Kubota and W. Wolfer, “Transition pathways in the unfaulting of dislocation loops,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2005. link Times cited: 9 NOT USED (low confidence) F. E. Gabaly, W. Ling, K. Mccarty, and J. de la Figuera, “The Importance of Threading Dislocations on the Motion of Domain Boundaries in Thin Films,” Science. 2005. link Times cited: 14 Abstract: Thin films often present domain structures whose detailed ev… read moreAbstract: Thin films often present domain structures whose detailed evolution is a subject of debate. We analyze the evolution of copper films, which contain both rotational and stacking domains, on ruthenium. Real-time observation by low-energy electron microscopy shows that the stacking domains evolve in a seemingly complex way. Not only do the stacking boundaries move in preferred directions, but their motion is extremely uneven and they become stuck when they reach rotational boundaries. We show that this behavior occurs because the stacking-boundary motion is impeded by threading dislocations. This study underscores how the coarse-scale evolution of thin films can be controlled by defects. read less NOT USED (low confidence) N. B. Luque and E. Leiva, “On the application of computer simulations to the study of electrochemical nanostructuring and surface phase formation,” Electrochimica Acta. 2005. link Times cited: 11 NOT USED (low confidence) N. Lümmen and T. Kraska, “Molecular dynamics investigations of the coalescence of iron clusters embedded in an inert-gas heat bath,” Physical Review B. 2005. link Times cited: 32 Abstract: A detailed analysis of the coalescence of iron clusters over… read moreAbstract: A detailed analysis of the coalescence of iron clusters over the course of their growth in an inert-gas atmosphere is presented. The investigation is performed by molecular dynamics simulations, using a recent version of the embedded atom method for iron. For several coalescence events extracted from realistic particle-growth simulations, the change of temperature, the atomic structure, and the morphology are analyzed. Here, the change in morphology is investigated by the relative number of atoms in the surface related to the driving force of the coalescence, the surface energy. The duration of the coalescence depends on the state of the colliding clusters, which is related to their temperature. At elevated temperatures an exponential decay of the relaxation of the cluster shape is found in case of liquid clusters. Clusters at lower temperatures exhibit a regular atomic structure. The coalescence includes the restructuring of the clusters, leading to deviations from the exponential decay of the cluster properties. Here, a distinct three-step coalescence process has been identified for structured clusters under nonadiabatic conditions. Each of these steps is related to a different extent of heat exchange with the carrier gas. read less NOT USED (low confidence) B.-J. Lee, B. Wirth, J. Shim, J. Kwon, S. Kwon, and J.-H. Hong, “Modified embedded-atom method interatomic potential for the Fe-Cu alloy system and cascade simulations on pure Fe and Fe-Cu alloys,” Physical Review B. 2005. link Times cited: 55 Abstract: A modified embedded-atom method (MEAM) interatomic potential… read moreAbstract: A modified embedded-atom method (MEAM) interatomic potential for the Fe-Cu binary system has been developed using previously developed MEAM potentials of Fe and Cu. The Fe-Cu potential was determined by fitting to data on the mixing enthalpy and the composition dependencies of the lattice parameters in terminal solid solutions. The potential gives a value of 0.65 eV for the dilute heat of solution and reproduces the increase of lattice parameter of Fe with addition of Cu in good agreement with experiments. The potential was used to investigate the primary irradiation defect formation in pure Fe and Fe-0.5 at. % Cu alloy by a molecular dynamics cascade simulation study with a PKA energy of 2 keV at 573 K. A tendency for self-interstitial atom-Cu binding, the formation of mixed (Fe-Cu) dumbbells and even Cu-Cu dumbbells was observed. Given a positive binding energy between Cu atoms and self-interstitials, Cu transport by an interstitial diffusion mechanism could be proposed to contribute to the formation of Cu-rich precipitates and irradiation-induced embrittlement in nuclear structural steels. read less NOT USED (low confidence) J. Braithwaite and P. Rez, “Grain boundary impurities in iron,” Acta Materialia. 2005. link Times cited: 91 NOT USED (low confidence) M. I. Rojas, M. C. Giménez, and E. Leiva, “Kinetic Monte Carlo simulation of Pt discontinuous thin film formation adsorbed on Au,” Surface Science. 2005. link Times cited: 17 NOT USED (low confidence) J. Jiménez‐Sáez, Pérez-Martı́n A., M. Said-Ettaoussi, and Jiménez-Rodrı́guez J. J., “Atomic structure of Ni nanoclusters on Cu(001) surfaces,” Nanotechnology. 2005. link Times cited: 2 Abstract: Depositions of Ni clusters on a Cu(001) surface have been si… read moreAbstract: Depositions of Ni clusters on a Cu(001) surface have been simulated by molecular dynamics in order to produce magnetic nanostructures. Two arrangements of the atoms at the interface between the Ni clusters (a few monolayers) and the Cu substrate, overlapped and non-overlapped, have been analysed. The difference between Ni and Cu lattice parameters (2.6%) gives rise to strain at the interface, which is the cause of magnetoelastic anisotropy. We have focused our interest especially on matching effects. The bombardment energy was varied between 0 and 1 eV/atom. Differences in the nanocluster morphology due to this have been discussed. Lattice defects which develop in the deposited clusters have been analysed. Final atomic distances, especially mean changes in lattice parameters, have been quantified at the interface. A study of atomic mixing and of its influence in spacing between layers has been also accomplished. read less NOT USED (low confidence) F. Ma, J.-min Zhang, and K. Xu, “Surface-energy-driven abnormal grain growth in Cu and Ag films,” Applied Surface Science. 2005. link Times cited: 32 NOT USED (low confidence) F. Aguilera-Granja, J. M. Montejano-Carrizales, and A. Vega, “Twining effects in the magnetism of small Pd clusters,” Solid State Communications. 2005. link Times cited: 11 NOT USED (low confidence) S. Ö. Kart, M. Tomak, and T. Çagin, “Phonon dispersions and elastic constants of disordered Pd–Ni alloys,” Physica B-condensed Matter. 2005. link Times cited: 11 NOT USED (low confidence) A. Kara and T. Rahman, “Structure, dynamics and thermodynamics of a metal chiral surface: Cu(532),” Journal of Physics: Condensed Matter. 2005. link Times cited: 3 Abstract: The structure, vibrational dynamics and thermodynamics of a … read moreAbstract: The structure, vibrational dynamics and thermodynamics of a chiral surface, Cu(532), have been calculated using a local approach and the harmonic approximation, with interatomic potentials based on the embedded atom method. Relaxation of the atomic positions to the optimum configuration results in a complex relaxation pattern with strong contractions in the bond length of atoms near the kink and the step sites and an equivalently large expansion near the least under-coordinated surface atoms. The low coordination of the atoms on the surface substantially affects the vibrational dynamics and thermodynamics of this system. The local vibrational density of states shows a deviation from the bulk behaviour that persists down to the tenth layer, resulting in a substantial contribution of the vibrational entropy to the excess free energy amounting to about 90 meV per unit cell at 300 K. read less NOT USED (low confidence) F. Aguilera-Granja, J. M. Montejano-Carrizales, E. O. Berlanga-Ramírez, and A. Vega, “Magnetic behavior of Pd nanoclusters,” Physica B-condensed Matter. 2004. link Times cited: 4 NOT USED (low confidence) J. Hoyt et al., “Crystal–Melt Interfaces and Solidification Morphologies in Metals and Alloys,” MRS Bulletin. 2004. link Times cited: 100 Abstract: When liquids solidify, the interface between a crystal and i… read moreAbstract: When liquids solidify, the interface between a crystal and its melt often forms branching structures (dendrites), just as frost spreads across a window.The development of a quantitative understanding of dendritic evolution continues to present a major theoretical and experimental challenge within the metallurgical community. This article looks at key parameters that describe the interface—excess free energy and mobility—and discusses how these important properties relate to our understanding of crystal growth and other interfacial phenomena such as wetting and spreading of droplets and nucleation of the solid phase from the melt. In particular, two new simulation methods have emerged for computing the interfacial free energy and its anisotropy: the cleaving technique and the capillary fluctuation method. These are presented, along with methods for extracting the kinetic coefficient and a comparison of the results to several theories of crystal growth rates. read less NOT USED (low confidence) Y.-S. Kim, K. Na, S.-W. Choi, and S. Yang, “Atomic force microscopy-based nano-lithography for nano-patterning: a molecular dynamic study,” Journal of Materials Processing Technology. 2004. link Times cited: 30 NOT USED (low confidence) F. Aguilera-Granja, J. M. Montejano-Carrizales, and A. Vega, “Magnetism in small Pd clusters,” Physics Letters A. 2004. link Times cited: 13 NOT USED (low confidence) E. Ahmed, J. Akhter, and M. Ahmad, “Molecular dynamics study of thermal properties of noble metals,” Computational Materials Science. 2004. link Times cited: 25 NOT USED (low confidence) A. Vega, J. Parlebas, and C. Demangeat, “Electronic Structure Calculations of Low‐Dimensional Transition Metals,” ChemInform. 2004. link Times cited: 9 Abstract: Publisher Summary This chapter discusses the state of the ar… read moreAbstract: Publisher Summary This chapter discusses the state of the art in the electronic and magnetic calculations of low-dimensional transition metals. It discusses several aspects of electronic structure calculations of low-dimensional transition metals, including (1) magnetic multilayered systems, mostly described currently by density functional theory (DFT) calculations with usually a constrained collinear magnetization and (2) a more semiempirical-like aspect related to the fundamental interest in considering not only the electronic and magnetic properties of a given geometrical configuration but also to find the ground state by minimization of the forces on a system where only the number of electrons are given. Most of the calculations to derive magnetic maps of magnetic nanostructures have been performed within a constrained direction of the polarization—that is, within the approximation of collinear magnetism. The chapter also discusses several problems that arise from a realistic description of the electronic and magnetic (scalar relativistic) properties of multilayers and clusters along with a short introduction of thin films and multilayers. read less NOT USED (low confidence) X. Xu, C. Cheng, and I. Chowdhury, “Molecular dynamics study of phase change mechanisms during femtosecond laser ablation,” Journal of Heat Transfer-transactions of The Asme. 2004. link Times cited: 47 Abstract: In this work, Molecular Dynamics (MD) simulation is employed… read moreAbstract: In this work, Molecular Dynamics (MD) simulation is employed to investigate femtosecond laser ablation of copper, with an emphasis on the understanding of the mechanism of phase change during laser ablation. Laser induced heat transfer, melting, surface evaporation, and material ablation are studied. Theoretically, it has been suggested that under intense femtosecond laser irradiation, the material undergoes a volumetric phase change process; its maximum temperature can be close to or even above the thermodynamic critical point. The MD simulations allow us to determine the transient temperature history of the irradiated material and to reveal the exact phase change process, which explains the mechanisms of femtosecond laser ablation. A finite difference calculation is also performed, which is used to compare results of heating and melting prior to a significant amount of material being ablated. read less NOT USED (low confidence) R. Song, W. Dietzel, B. Zhang, W. Liu, M. Tseng, and A. Atrens, “Stress corrosion cracking and hydrogen embrittlement of an Al–Zn–Mg–Cu alloy,” Acta Materialia. 2004. link Times cited: 208 NOT USED (low confidence) F. Aguilera-Granja, J. M. Montejano-Carrizales, E. O. Berlanga-Ramírez, and A. Vega, “Magnetic behaviour of selected geometries of Pd clusters: icosahedral versus fcc structures,” Physics Letters A. 2004. link Times cited: 13 NOT USED (low confidence) W. Hoover and C. G. Hoover, “Searching for auxetics with DYNA3D and ParaDyn,” physica status solidi (b). 2004. link Times cited: 67 Abstract: We sought to simulate auxetic behavior by carrying out dynam… read moreAbstract: We sought to simulate auxetic behavior by carrying out dynamic analyses of mesoscopic model structures. We began by generating nearly periodic cellular structures. Four‐node “Shell” elements and eight‐node “Brick” elements are the basic building blocks for each cell. The shells and bricks obey standard elastic‐plastic continuum mechanics. The dynamical response of the structures was next determined for a three‐stage loading process: (1) homogeneous compression; (2) viscous relaxation; (3) uniaxial compression. The simulations were carried out with both serial and parallel computer codes – DYNA3D and ParaDyn – which describe the deformation of the shells and bricks with a robust contact algorithm. We summarize the results found here. (© 2005 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (low confidence) D. Spearot, K. Jacob, and D. McDowell, “Non-local separation constitutive laws for interfaces and their relation to nanoscale simulations,” Mechanics of Materials. 2004. link Times cited: 97 NOT USED (low confidence) J. Diao, K. Gall, and M. Dunn, “Atomistic simulation of the structure and elastic properties of gold nanowires,” Journal of The Mechanics and Physics of Solids. 2004. link Times cited: 318 NOT USED (low confidence) R. F. Zhang, Y. Shen, H. Gong, L. Kong, and B.-xin Liu, “Atomistic Modeling of Metastable Phase Selection of a Highly Immiscible Ag-W System,” Journal of the Physical Society of Japan. 2004. link Times cited: 6 Abstract: Ab initio calculation is performed for predicting the cohesi… read moreAbstract: Ab initio calculation is performed for predicting the cohesive energies and lattice constants of some possible metastable compounds in the Ag-W system with the largest positive formation enthalpy among the binary transition metal alloys. The calculated results together with the experimental data are then used in deriving an n-body Ag-W potential under the framework of the embedded atom method. Based on the proven realistic Ag-W potential, molecular dynamics simulations are performed to reveal the metastable phase selection over the entire composition of the system and the results predict that the metastable Ag 100-x W x alloy in an fcc structure is more stable than in the bcc structure when 0 ≤ x < 57, whereas the bcc structure becomes energetically favored when 57 < x≤100. Interestingly, the prediction is in good agreement with the experimental results. read less NOT USED (low confidence) W. Xiao, P. Greaney, and D. Chrzan, “Pt adatom diffusion on strained Pt(001),” Physical Review B. 2004. link Times cited: 14 Abstract: The diffusion of Pt on unreconstructed, strained Pt~001! is … read moreAbstract: The diffusion of Pt on unreconstructed, strained Pt~001! is considered. The strain dependence for the energy barrier to adatom diffusion is computed using the embedded atom method. The computations predict that for a broad range of accessible strain states, the dominant diffusion mechanism is mediated by the formation and motion of a surface crowdion. Further, the saddle-point configuration for ‘‘simple’’ hopping is predicted to extend over a number of lattice sites. The diffusion energy barriers for both exchange and hopping mechanisms depend on the size of the cell used in the calculation. read less NOT USED (low confidence) R. E. Miller and A. Acharya, “A stress-gradient based criterion for dislocation nucleation in crystals,” Journal of The Mechanics and Physics of Solids. 2004. link Times cited: 62 NOT USED (low confidence) S. Arcidiacono, N. Bieri, D. Poulikakos, and C. Grigoropoulos, “On the coalescence of gold nanoparticles,” International Journal of Multiphase Flow. 2004. link Times cited: 224 NOT USED (low confidence) X. Liu, F. Ercolessi, and J. B. Adams, “Aluminium interatomic potential from density functional theory calculations with improved stacking fault energy,” Modelling and Simulation in Materials Science and Engineering. 2004. link Times cited: 147 Abstract: A new Al potential with improved stacking fault energy is co… read moreAbstract: A new Al potential with improved stacking fault energy is constructed using the force-matching method. The potential is fitted to an ab initio forces database and various experimental data. By using a slightly larger cut-off, we found that the new potential gives the relaxed stacking fault energy in the experimental range without changing the excellent thermal and surface properties of the original force-matching Al potential given by Ercolessi and Adams (1994 Europhys. Lett. 26 583). read less NOT USED (low confidence) V. I. Zubov and A. A. Caparica, “A STATISTICAL MECHANICAL STUDY OF THERMODYNAMIC PROPERTIES OF SOLID SODIUM UNDER PRESSURE BASED ON AN EFFECTIVE INTERIONIC POTENTIAL,” International Journal of Modern Physics B. 2004. link Times cited: 2 Abstract: Basing on the Schiff effective interionic potential that has… read moreAbstract: Basing on the Schiff effective interionic potential that has an oscillatory character and the correlative method of unsymmetrized self-consistent field (CUSF) that enables one to take into account the strong anharmonicity of the crystal lattice vibrations, we have calculated a complete set of equilibrium thermodynamic properties of solid sodium as functions of pressure and temperature: the lattice parameter, the elastic moduli, the thermal expansion coefficient, the Gruneisen parameter and the isochoric and isobaric heat capacities. Our results are compared with available experimental data. We also discuss the thermodynamic stability of the BCC lattice, the mechanism of its loss and its change under pressure. read less NOT USED (low confidence) Y.-M. Kim and B.-J. Lee, “A modified embedded-atom method interatomic potential for the Cu–Zr system,” Journal of Materials Research. 2004. link Times cited: 65 NOT USED (low confidence) H. Yildirim and S. Durukanoğlu, “Structural relaxations of Cu vicinals,” Surface Science. 2004. link Times cited: 16 NOT USED (low confidence) J.-min Zhang, F. Ma, and K. Xu, “Calculation of the surface energy of fcc metals with modified embedded-atom method,” Chinese Physics. 2004. link Times cited: 311 Abstract: The surface energies for 38 surfaces of fcc metals Cu, Ag, A… read moreAbstract: The surface energies for 38 surfaces of fcc metals Cu, Ag, Au, Ni, Pd, Pt, Al, Pb, Rh and Ir have been calculated by using the modified embedded-atom method. The results show that, for Cu, Ag, Ni, Al, Pb and Ir, the average values of the surface energies are very close to the polycrystalline experimental data. For all fcc metals, as predicted, the close-packed (111) surface has the lowest surface energy. The surface energies for the other surfaces increase linearly with increasing angle between the surfaces (hkl) and (111). This can be used to estimate the relative values of the surface energy. read less NOT USED (low confidence) H. Fan, C. Wong, and M. Yuen, “Molecular simulation of cu-sam adhesion force,” 5th International Conference on Thermal and Mechanical Simulation and Experiments in Microelectronics and Microsystems, 2004. EuroSimE 2004. Proceedings of the. 2004. link Times cited: 2 Abstract: The interface of Copper-EMC (Epoxy Molding Compound) is know… read moreAbstract: The interface of Copper-EMC (Epoxy Molding Compound) is known to be the weakest joint in the electronic package design, which causes delamination during reliability test. A prime reason is the poor adhesion between Cu and epoxy compound. To solve the problem, We have used self-assembly monolayer (SAM) to improve adhesion of copper-epoxy system. This paper focuses on simulation of adhesion in Cu-SAM system. In this study, molecular models of bi-material system, which consists of SAM and Cu, were built to evaluate adhesion force of the Cu-SAM system. In order to dramatically reduce the computational time, only the copper tip and SAM substrate were modeled with a limited number of atoms. After energy minimization of the whole structure, tensile stresses were applied to the whole structure to simulate the debonding process, and the ultimate stress was obtained when the two materials delaminates completely. The adhesion force between the copper tip and the SAM can be evaluated from the ultimate tensile stress. The molecular model results were compared with AFM results in which adhesion force between Cu tip and SAM coated copper substrate were measured. Two different types of SAM material were used in this study. The paper intended to relate closely adhesion by using MD simulation, and the underlying physics for the explanation to the adhesion phenomenon for further understanding of failure mechanism of interfacial delamination. read less NOT USED (low confidence) O. Ajayi, K. Ludema, J. Hammerberg, and B. Holian, “Simulation Methods for Interfacial Friction in Solids.” 2004. link Times cited: 2 NOT USED (low confidence) S. Chen, J. Xu, and H. Zhang, “A new scheme of many-body potentials for hcp metals,” Computational Materials Science. 2004. link Times cited: 12 NOT USED (low confidence) L. Shilkrot, R. E. Miller, and W. Curtin, “Multiscale plasticity modeling: coupled atomistics and discrete dislocation mechanics,” Journal of The Mechanics and Physics of Solids. 2004. link Times cited: 278 NOT USED (low confidence) R. Robles, R. Longo, E. Noya, A. Vega, and L. J. Gallego, “Structural and magnetic properties ofFenclusters at the Al (001) surface: Early transition from paramagnetic to ferromagneticFen,” Physical Review B. 2004. link Times cited: 10 Abstract: Using the (re)modified embedded atom method, an extension of… read moreAbstract: Using the (re)modified embedded atom method, an extension of the embedded atom model that includes angular forces and second-nearest-neighbor interactions, we performed quenched molecular-dynamics simulations to compute the lowest-energy structures of Fe n clusters (n=2-20,25) supported on or embedded in the top few layers of the Al (001) surface. Embedded clusters were always more stable than adsorbed clusters, and formed either linear chains (for n=3 or 5) or warped single-layer close-packed islands (for n=4 or n≥6). Determination of the spin-polarized electronic structure using a self-consistent tight-binding method showed that, due to hybridization between the Al sp and Fe d states, the embedded Fe n clusters are nonmagnetic for n=2-10. However, for larger sizes they can sustain magnetic moments. read less NOT USED (low confidence) X.-C. Wang, Y. Jia, Q. Yao, F. Wang, J.-xin Ma, and X. Hu, “The calculation of the surface energy of high-index surfaces in metals at zero temperature,” Surface Science. 2004. link Times cited: 39 NOT USED (low confidence) J. Peltola and K. Nordlund, “Ion beam induced coherent displacement in Al on Au system,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2004. link Times cited: 4 NOT USED (low confidence) P. Jensen, A. Clement, and L. J. Lewis, “Diffusion of nanoclusters on non-ideal surfaces,” Physica E-low-dimensional Systems & Nanostructures. 2004. link Times cited: 14 NOT USED (low confidence) R. Huang, X. Ma, and Y. Wang, “Computational investigations of the monolayer heteroepitaxial growth of Au on Ni(110),” Surface Science. 2004. link Times cited: 5 NOT USED (low confidence) S. Chen and F. Ke, “MD simulation of the effect of contact area and tip radius on nanoindentation,” Science in China Series G: Physics, Mechanics and Astronomy. 2004. link Times cited: 19 Abstract: Molecular dynamics simulations of nanoindentation are perfor… read moreAbstract: Molecular dynamics simulations of nanoindentation are performed on monocrystal copper. A new “contact atoms” method is presented for calculating the contact area. Compared with conventional methods, this method can provide the contact area more accurately not only for sink-in but also for pile-up situation. The effect of tip radius on indentation is investigated too. The results indicate that the measured hardness of the material will become higher as the tip radius increases. read less NOT USED (low confidence) G. Betz and W. Husinsky, “Modelling of cluster emission from metal surfaces under ion impact,” Philosophical Transactions of the Royal Society of London. Series A: Mathematical, Physical and Engineering Sciences. 2004. link Times cited: 34 Abstract: Using the molecular–dynamics technique, cluster emission for… read moreAbstract: Using the molecular–dynamics technique, cluster emission for 5 keV Ar bombardment of a Cu (111) surface has been investigated using a many–body (tight binding) potential for the Cu–Cu interaction. The calculations allow us to analyse the basic processes underlying cluster emission. It is found that two distinct processes can be distinguished which lead to cluster emission under energetic ion bombardment. The first process causes the emission of small clusters, which are emitted by a collective motion during the development of the collision cascade within the first picosecond after impact. Thus, emission times of such clusters agree with the emission times of atoms in sputtering. Such a process can be envisioned if, for example, a few layers below the surface, an energetic recoil causes the development of a subcascade. Energy transferred by this event to the surface is strongly directional and can lead to the simultaneous emission of a group of neighbouring surface atoms, which in some cases will remain bounded and form a cluster after emission. Typically, clusters emitted by this mechanism consist of atoms, which are neighbouring in the target and are almost exclusively surface atoms, similar to all sputtered atoms. Emission of large clusters (cluster sizes of 10 or more atoms), as observed experimentally, is a puzzling phenomenon. From our calculations we conclude that the emission of such large clusters does not occur during the collisional phase of sputtering, but happens much later (5–10 ps after ion impact). Emission can occur for spike events, where all the energy of the impinging ion is deposited locally in a small volume near to the surface, and the sputtering yield is 3–5 times the average yield. Such events are rare, but we have found a few cases in our calculations where stable clusters consisting of more than 20 atoms were emitted. Melting of the spike volume occurs, and the high temperatures and pressures produced can cause emission of large fragments during the thermal phase. The composition of such large clusters is quite different from that of small clusters. They consist of atoms from different layers and the constituents are also generally not next–neighbour atoms. This change in origin of the cluster atoms reflects the mixing and diffusion processes occurring in the melted zone before emission. The calculations indicate that hydrodynamical phenomena might play a role in the emission of large fragments. Additional calculations, where the energy was distributed ‘thermall’ in a three–dimensional volume under the surface for 500 fs, give very similar results, even in such cases where the kinetic phase of the collision–cascade development was absent. read less NOT USED (low confidence) R. Kojima and M. Susa, “Second moment approximation of tight-binding potential for γFe applicable up to 1700 K,” Science and Technology of Advanced Materials. 2004. link Times cited: 3 NOT USED (low confidence) A. Lozovoi and A. Alavi, “Reconstruction of charged surfaces: General trends and a case study of Pt(110) and Au(110),” Physical Review B. 2003. link Times cited: 94 Abstract: The stability of missing-row reconstructions of (110) surfac… read moreAbstract: The stability of missing-row reconstructions of (110) surfaces with respect to surface charging has been investigated using ab initio theory, taking Pt and Au as representative systems. A thermodynamic formulation is derived to compare the relative stability of charged surfaces either in constant-potential or constant-charge mode. By generalizing Koopmans' theorem to charged metallic surfaces, we obtain an expression for the surface (excess) energy as a function of charge (or potential) in terms of the neutral surface energy, work function, and the position of the image plane. A surface is shown to reconstruct in constant-charge mode if and only if it reconstructs in constant-potential mode. We next address the question of whether a positive (negative) surface charge can lift (induce) the reconstruction, as suggested in the literature. This turns out not to be the case. Instead the following consistent picture arises: at small surface charges, the effect of the charge follows the difference of the work functions; i.e., positive charge favors a surface having a smaller work function and vice versa. Larger charges, either positive or negative, tend to stabilize the reconstructed surface or, more generally, the 1 × r reconstruction with larger r. The latter essentially results in that the 1 × 2 reconstruction in either Pt or Au is never lifted in our study, although the 1 × 3 surface in Au eventually becomes more stable. read less NOT USED (low confidence) E. M. Lopasso, A. Caro, and M. Caro, “How fast are the ultra-fast nano-scale solid–liquid phase transitions induced by energetic particles in solids?,” Journal of Nuclear Materials. 2003. link Times cited: 1 NOT USED (low confidence) E. Salonen, K. Nordlund, and J. Keinonen, “Au irradiation by 25-keV Aun (n = 1-65600) clusters,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 12 NOT USED (low confidence) X. Ma and Y. Wei, “MD simulation for nanocrystals,” Acta Mechanica Sinica. 2003. link Times cited: 5 NOT USED (low confidence) S. Huang, D. S. Mainardi, and P. Balbuena, “Structure and dynamics of graphite-supported bimetallic nanoclusters,” Surface Science. 2003. link Times cited: 154 NOT USED (low confidence) R. Heid and K. Bohnen, “Ab initio lattice dynamics of metal surfaces,” Physics Reports. 2003. link Times cited: 76 NOT USED (low confidence) B.-J. Lee, J. Shim, and M. Baskes, “Semiempirical atomic potentials for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, Al, and Pb based on first and second nearest-neighbor modified embedded atom method,” Physical Review B. 2003. link Times cited: 426 Abstract: Modified embedded atom method (MEAM) potentials for fcc elem… read moreAbstract: Modified embedded atom method (MEAM) potentials for fcc elements Cu, Ag, Au, Ni, Pd, Pt, Al, and Pb have been newly developed using the original first nearest-neighbor MEAM and the recently developed second nearest-neighbor MEAM formalisms. It was found that the original MEAM potentials for fcc elements show some critical shortcomings such as structural instability and incorrect surface reconstructions on (100), (110), and/or (111) surfaces. The newly developed MEAM potentials solve most of the problems and describe the bulk properties (elastic constants, structural energy differences), point defect properties (vacancy and interstitial formation energy and formation volume, activation energy of vacancy diffusion), planar defect properties (stacking fault energy, surface energy, surface relaxation and reconstruction), and thermal properties (thermal expansion coefficients, specific heat, melting point, heat of melting) of the fcc elements considered, in good agreement with relevant experimental information. It has been shown that in the MEAM the degree of many-body screening (C m i n ) is an important material property and that structural stability at finite temperatures should be included as a checkpoint during development of semiempirical potentials. read less NOT USED (low confidence) H. Ishida, S. Motoyama, K. Mae, and Y. Hiwatari, “Molecular Dynamics Simulation of Martensitic Transformations in NiAl Alloy Using the Modified Embedded Atom Method,” Journal of the Physical Society of Japan. 2003. link Times cited: 6 Abstract: The martensitic transformations in NiAl alloys were studied … read moreAbstract: The martensitic transformations in NiAl alloys were studied using molecular dynamics simulations. The modified embedded atom method was used with the pseudo monoatomic potentials which included angular dependence of each atoms. The thermally induced B2 → 3R martensitic and 3R → B2 reverse martensitic transformations have been obtained in the present molecular dynamics simulations for the first time with a bulk (no surface) computational model. The transformation is accompanied by a twin in the 3R phase which leads to a lattice-invariant deformation and minimize the transformation strain energy. The concentration dependence of the transformation temperature for Ni x Al 1- x (0.58 < x < 0.69) alloys have been observed. read less NOT USED (low confidence) J.-min Zhang, F. Ma, K. Xu, and X. Xin, “Anisotropy analysis of the surface energy of diamond cubic crystals,” Surface and Interface Analysis. 2003. link Times cited: 72 Abstract: The surface energies for 24 surfaces of diamond structure cu… read moreAbstract: The surface energies for 24 surfaces of diamond structure cubic crystals of C, Si and Ge have been calculated using the modified embedded‐atom method. The results show that the three lowest surface energies correspond to the (111), (211) and (433) surfaces. Considering surface energy minimization solely, the (111), (211) and (433) textures should be favourable successively in diamond cubic films. The appearance of abnormal grains or textures with (111) and (211) orientations in Si, Ge and C films results from surface energy minimization. Copyright © 2003 John Wiley & Sons, Ltd. read less NOT USED (low confidence) D. Papageorgiou, I. Lagaris, N. Papanicolaou, G. Petsos, and H. Polatoglou, “MERLIN a versatile optimization environment applied to the design of metallic alloys and intermetallic compounds,” Computational Materials Science. 2003. link Times cited: 3 NOT USED (low confidence) E. León, A. Maccabe, and R. Brightwell, “An MPI Tool to Measure Application Sensitivity to Variation in Communication Parameters,” PVM/MPI. 2003. link Times cited: 2 NOT USED (low confidence) Y. Zhong, Y. Ashkenazy, K. Albe, and R. Averback, “Ion beam smoothening of metal surfaces,” Journal of Applied Physics. 2003. link Times cited: 14 Abstract: Ion beam induced smoothening of crystalline and amorphous su… read moreAbstract: Ion beam induced smoothening of crystalline and amorphous substrates were investigated using molecular dynamics simulations. Rough surfaces created by depositing small nanoparticles, 2–3 nm in diameter, onto flat substrates were subjected to repeated impacts with 40 keV Xe atoms. Two smoothening processes are identified: The nanoparticle either burrows into the substrate, with the underlying substrate atoms flowing around it to the surface, or it flows over the substrate surface, wetting it. Generally, these two mechanisms operate simultaneously in both amorphous and crystalline substrates. The burrowing mechanism in amorphous substrates was additionally investigated by creating low energy recoils, 20 or 200 eV in the substrate beneath the nanoparticle. Roughening of initially smooth amorphous substrates during ion impact was also studied for comparison with the smoothening process. read less NOT USED (low confidence) R. Miron and K. Fichthorn, “Accelerated molecular dynamics with the bond-boost method,” Journal of Chemical Physics. 2003. link Times cited: 139 Abstract: We present a new method for accelerating molecular-dynamics … read moreAbstract: We present a new method for accelerating molecular-dynamics simulations of infrequent events. The method targets simulation of systems that spend most of the time in local energy minima, with slow transitions in between, as is the case with low-temperature surface diffusion. The potential-energy surface is modified by adding a boost potential in regions close to the local minima, such that all transition rates are increased while relative rates are preserved. The boost potential is an empirical function determined by the deviation of the bond lengths of a specified set of atoms from equilibrium. The method requires no previous knowledge of the processes involved and it can be applied to a wide variety of interaction potentials. Application to the diffusion of Cu atoms on the Cu(100) surface using an embedded-atom potential yields correct rates for adatom hopping, exchange, as well as vacancy and dimer diffusion with speed-ups up to several orders of magnitude. read less NOT USED (low confidence) S. G. Mayr and R. Averback, “Evolution of morphology in nanocrystalline thin films during ion irradiation,” Physical Review B. 2003. link Times cited: 26 Abstract: Changes in morphology of nanocrystalline AgCo and AgNi films… read moreAbstract: Changes in morphology of nanocrystalline AgCo and AgNi films during ion beam bombardment are investigated using a combination of experiments and molecular dynamics (MD) computer simulations. It is shown as long as the grain size is smaller than the typical dimension of the thermal spike, that surface topography relaxation, stress relaxation, domain and grain growth are governed by a viscous flow mechanism. A detailed analysis of the simulation data reveals that the domain growth proceeds by a mechanism similar to liquid-phase sintering of nanoparticles, whereas grain growth proceeds by grain reorientation. With increasing grain size beyond the thermal spike size, a strong reduction in the rate of change in the surface morphology and film stress is observed. read less NOT USED (low confidence) J.-min Zhang, F. Ma, and K. Xu, “Calculation of the surface energy of bcc metals by using the modified embedded‐atom method,” Surface and Interface Analysis. 2003. link Times cited: 127 Abstract: The surface energies for 24 surfaces of bcc metals Li, Na, K… read moreAbstract: The surface energies for 24 surfaces of bcc metals Li, Na, K, V, Nb, Ta, Cr, Mo, W and Fe have been calculated by using the modified embedded‐atom method. The results show that for most bcc metals the lowest surface energies correspond to the (110) surface, as predicted from the bcc lattice, and the highest surface energies correspond to the (433) surface. From surface energy minimization, the (110) texture should be favourable in the bcc films. This is consistent with experimental results. Copyright © 2003 John Wiley & Sons, Ltd. read less NOT USED (low confidence) G. Mattei et al., “De-alloying behaviour of metal nanoclusters in SiO2 upon irradiation and thermal treatments,” Journal of Non-crystalline Solids. 2003. link Times cited: 15 NOT USED (low confidence) S. Durukanoğlu, A. Kara, and T. Rahman, “Local and excess vibrational free energies of stepped metal surfaces,” Physical Review B. 2003. link Times cited: 19 Abstract: We present a comparative study of the local and excess vibra… read moreAbstract: We present a comparative study of the local and excess vibrational thermodynamic functions for the (511), (211), and (331) surfaces of Ag and Cu, in the harmonic approximation, employing interaction potentials based on the embedded atom method. Related thermodynamic functions of the corresponding (100) and (111) surfaces are also examined. For these surfaces, local contributions to the vibrational free energy for atoms in layers down to the fifth are found to deviate from that of the atoms in the bulk, as a result of complex multilayer relaxations. These contributions show a linear dependence on temperature, beyond 200 K, and a correlation with the local coordination but without a simple relationship. The vibrational contribution to the step excess free energy for vicinals of (111) is found to be significant. read less NOT USED (low confidence) M. Karabacak, S. Özçelik, and Z. B. Güvenç, “Structures and energetics of Pd21–Pd55 clusters,” Surface Science. 2003. link Times cited: 9 NOT USED (low confidence) S. Helfensteyn, J. Luyten, L. Feyaerts, and C. Creemers, “Modelling surface phenomena in PdNi alloys,” Applied Surface Science. 2003. link Times cited: 30 NOT USED (low confidence) H. Sharma and S. Prakash, “Atomic Displacements Due to Point Defects in Ni Dilute Alloys,” International Journal of Modern Physics B. 2003. link Times cited: 2 Abstract: The Embedded atom method has been used to investigate the st… read moreAbstract: The Embedded atom method has been used to investigate the strain field due to substitutional transition metal impurities in Ni. The calculations are carried out in the discrete lattice model of the metal using Kanzaki lattice static method. The results for atomic displacements due to 3d, 4d and 5d impurities (Cu, Pd, Pt and Au) in Ni are given up to 20 NN's of impurity and are compared with the earlier calculations and with the available experimental data. The maximum displacements of 3.6% of 1NN distance are found for NiAu, while the minimum displacements of 0.78% of 1NN distance are found for NiCu alloy respectively. The relaxation energy for Cu are found less than those for Pd, Au and Pt impurities in the Ni host. read less NOT USED (low confidence) S. Ciraci and A. Buldum, “Atomic-scale study of friction and energy dissipation,” Wear. 2003. link Times cited: 18 NOT USED (low confidence) M. D. Pópolo et al., “A combined experimental and theoretical study of the generation of palladium clusters on Au(111) with a scanning tunnelling microscope,” Electrochimica Acta. 2003. link Times cited: 18 NOT USED (low confidence) T. Muramoto, N. Hirotani, K. Itabasi, A. Harada, and Y. Yamamura, “MD simulation of surface smoothing due to cluster impact,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 6 NOT USED (low confidence) W. Voegeli, K. Albe, and H. Hahn, “Simulation of grain growth in nanocrystalline nickel induced by ion irradiation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 55 NOT USED (low confidence) A. Kislov, V. Mazurenko, and K. N. Korzov, “Calculation of the vibrational spectra of copper crystals with vacancies,” Physics of the Solid State. 2003. link Times cited: 1 NOT USED (low confidence) P. Pochet, “Modeling of aging in plutonium by molecular dynamics,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 23 NOT USED (low confidence) F. Cherne, M. Baskes, and B. Holian, “Predicted transport properties of liquid plutonium,” Physical Review B. 2003. link Times cited: 14 Abstract: The fluid-phase transport properties, diffusivity and viscos… read moreAbstract: The fluid-phase transport properties, diffusivity and viscosity, are calculated by equilibrium and nonequilibrium techniques for plutonium, whose interatomic interactions are described by the modified embedded-atom method. The transport coefficients are evaluated at zero pressure, for temperatures between 950 K and 1300 K. We find the calculated viscosity to be noticeably higher than experiment, while the structure of liquid Pu appears to be similar to other liquid metals. read less NOT USED (low confidence) Y. Shen, L. Kong, H. Gong, W. Lai, and B.-xin Liu, “Construction of an Embedded-Atom Potential for an Immiscible Cu–V System,” Journal of the Physical Society of Japan. 2003. link Times cited: 2 Abstract: A realistic many-body potential based on the embedded atom m… read moreAbstract: A realistic many-body potential based on the embedded atom method is derived for an equilibrium immiscible Cu–V system by fitting some physical properties of the nonequilibrium B2 CuV and L1 2 Cu 3... read less NOT USED (low confidence) K. Masuda-Jindo, V. Hùng, and P. D. Tam, “Thermodynamic quantities of metals investigated by an analytic statistical moment method,” Physical Review B. 2003. link Times cited: 55 Abstract: The thermodynamic properties of metals are studied by includ… read moreAbstract: The thermodynamic properties of metals are studied by including explicitly the anharmonic effects of the lattice vibrations going beyond the quasiharmonic approximations. The free energy, thermal lattice expansion coefficients, mean-square atomic displacements, and specific heats at the constant volume and those at the constant pressure, ${C}_{v}$ and ${C}_{p},$ are derived in closed analytic forms in terms of the power moments of the atomic displacements. The analytical formulas give highly accurate values of the thermodynamic quantities, which are comparable to those of the molecular dynamics or Monte Carlo simulations for a wide temperature range. The present formalism is well suited to calculate the thermodynamic quantities of metals and alloys by including the many body electronic effects and by combining it with the first-principles approaches. read less NOT USED (low confidence) D. Papaconstantopoulos and M. Mehl, “TOPICAL REVIEW: The Slater Koster tight-binding method: a computationally efficient and accurate approach,” Journal of Physics: Condensed Matter. 2003. link Times cited: 122 Abstract: In this article we discuss the Slater–Koster (SK) tight-bind… read moreAbstract: In this article we discuss the Slater–Koster (SK) tight-binding (TB) method from the perspective of our own developments and applications to this method. We first present an account of our work in constructing TB Hamiltonians and applying them to a variety of calculations which require an accurate representation of the electronic energy bands and density of states. In the second part of the article we present the Naval Research Laboratory TB method, wherein we demonstrate that this elaborate scheme can accurately account for both the band structure and total energy of a given system. The SK parameters generated by this method are transferable to other structures and provide the means for performing computationally demanding calculations of fairly large systems. These calculations, including molecular dynamics, are of comparable accuracy to first-principles calculations and three orders of magnitude faster. read less NOT USED (low confidence) N. Papanicolaou, H. Chamati, G. Evangelakis, and D. Papaconstantopoulos, “Second-moment interatomic potential for Al, Ni and Ni-Al alloys, and molecular dynamics application,” Computational Materials Science. 2003. link Times cited: 37 NOT USED (low confidence) Y. Ye, R. Biswas, J. R. Morris, A. Bastawros, and A. Chandra, “Molecular dynamics simulation of nanoscale machining of copper,” Nanotechnology. 2003. link Times cited: 138 Abstract: Molecular dynamics simulations of the nanometric cutting of … read moreAbstract: Molecular dynamics simulations of the nanometric cutting of single-crystal copper were performed with the embedded atom method. The nature of material removal, chip formation, material defects and frictional forces were simulated. Nanometric cutting was found to comprise two steps: material removal as the tool machines the top surface, followed by relaxation of the work material to a low defect configuration, after the tool or abrasive particle has passed over the machined region. During nanometric cutting there is a local region of higher temperature and stress below the tool, for large cutting speeds. Relaxation anneals this excess energy and leads to lower dislocation work material. At high cutting speeds (180 m s−1), the machined surface is rough but the work material is dislocation free after the large excess energy has annealed the work material. At lower cutting speeds (1.8– 18 m s−1), the machined surface is smooth, with dislocations remaining in the substrate, and there is only a small excess temperature in the work material after machining. The size of the chip grows with increasing cutting speed. read less NOT USED (low confidence) C. Henry, “Adsorption and Reaction at Supported Model Catalysts.” 2003. link Times cited: 5 NOT USED (low confidence) N. Nozaki, M. Doyama, and Y. Kogure, “Plastic deformation of copper thin foils,” Thin Solid Films. 2003. link Times cited: 7 NOT USED (low confidence) J. Takano, M. Doyama, and Y. Kogure, “Motion and conversion energies of adatom and adatom clusters on gold (0 0 1) surface,” Thin Solid Films. 2003. link Times cited: 2 NOT USED (low confidence) S. Sawada, Y. Shimizu, and K. Ikeda, “Molecular-dynamics simulations of rapid alloying of microclusters using a many-body potential,” Physical Review B. 2003. link Times cited: 5 Abstract: Rapid alloying in $4d$-transition-metal clusters is studied … read moreAbstract: Rapid alloying in $4d$-transition-metal clusters is studied by isoenergetic molecular-dynamics simulations. A many-body potential is proposed by extending Pettifor's theory of alloys. Numerical results of simulations for two-dimensional clusters by using this potential are reported. It is found that clusters always exhibit surface structural fluctuation at temperatures even much below the melting temperature, which causes every atom in clusters to change its equilibrium position. It is a main driving factor of rapid alloying. Moreover, the presence of enough of a proportion of guest atoms is a necessary condition for the onset of the rapid alloying. The temperature dependence of time spent by alloying (alloying time) is found to obey the Arrhenius law. By extrapolating this result to room temperature, we show that the alloying time is of the order of one second at room temperature, which is consistent with experimental observation. read less NOT USED (low confidence) T. Muramoto and Y. Yamamura, “MD simulation of cluster ejection due to sputtering by polyatomic projectiles,” Applied Surface Science. 2003. link Times cited: 4 NOT USED (low confidence) F. M. Poveda, J. Fernández-Sanz, and F. Ruette, “A parametrical embedding method for catalytic modeling,” Journal of Molecular Catalysis A-chemical. 2003. link Times cited: 3 NOT USED (low confidence) M. Hou, V. Kharlamov, and E. Zhurkin, “Atomic-scale modeling of cluster-assembled (formula presented) thin films,” Physical Review B. 2002. link Times cited: 33 Abstract: Thermodynamic and structural properties of Ni-Al cluster ass… read moreAbstract: Thermodynamic and structural properties of Ni-Al cluster assembled materials are investigated at the atomic scale. Model predictions are available for elemental systems but the field of bimetallic nanostructured systems remains close to unexplored. The aim of the present work is to model at the atomic scale the structural and segregation properties in the Ni x Al 1 - x bimetallic cluster assembled materials that are synthesized in two different ways. In the first, isolated clusters are compacted at high pressure. We consider the L1 1 2 and B2 phases of the initial free clusters. Compaction of clusters at thermodynamic equilibrium is modeled by classical molecular dynamics combining isobaric and isothermal schemes. After compaction, interface segregation is computed by Metropolis Monte Carlo importance sampling in the semigrand canonical ensemble. After this model treatment, clusters are found to keep their identity, and their structural and segregation states do not differ much from those in the initial free clusters. The cluster cores keep the stable bulk phases while segregation may take place at the interfaces. The second method is low-energy cluster beam deposition. Cluster impact is found to influence chemical and structural order in the films formed. This is shown and discussed on the example of L1 2 cluster deposition. Molecular dynamics is used therefore, which accounts for electron-phonon coupling in the equations of motion. The slowing down of a single cluster is examined in detail. It is found that the expitaxial accommodation of the cluster with the substrate and chemical order in the cluster depend on the mechanical properties of the substrate material. Competition between chemical order and epitaxy is observed. The harder the material, the higher the epitaxy and the lower the chemical order. The cluster impact induces significant chemical disorder but the clusters forming the cluster assembled film keep their initial identities. Similarly to the sample obtained by compaction, this one displays partial structural and chemical order at its interfaces. The film density is particularly low and the open volumes form a fully interconnected network of pores. read less NOT USED (low confidence) C. Tully and G. B. Billing, “Dissociation of N2 on ruthenium using an embedded diatoms in molecules potential,” Chemical Physics Letters. 2002. link Times cited: 2 NOT USED (low confidence) V. Rodrigues and D. Ugarte, “Quantum conductance properties of metal nanowires,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 2002. link Times cited: 6 NOT USED (low confidence) J. Moon, J. Yoon, P. Wynblatt, S. Garoff, and R. Suter, “Simulation of spreading of precursing Ag films on Ni(100),” Computational Materials Science. 2002. link Times cited: 22 NOT USED (low confidence) L.-P. Zheng, H. F. Zhang, D.-xing Li, and F. Cui, “Calculation of the Griffith Cohesive Energy of the Ni3AlBx Symmetrical Grain Boundary,” Chinese Physics Letters. 2002. link Times cited: 3 Abstract: A Monte Carlo simulation, with the energetics described by t… read moreAbstract: A Monte Carlo simulation, with the energetics described by the embedded atom method, has been employed to study the physical behaviour of boron atoms during relaxation of the Ni3AlBinfinity grain boundary. It has also been used to calculate not only the peak concentrations of Ni and B and the valley concentration of Al at the grain boundary, but also the dependence of the grain boundary cohesion on the B bulk concentration. During relaxation of impure Ni3Al grain boundaries, we suggest that, as the segregating species, the B atoms either insert into interstices in the grain boundary or substitute Ni atoms. Meanwhile, as the inducing species, they induce Ni atoms to substitute for Al atoms. Calculations show that in the equilibrium, when the B bulk concentration x increases from 0.1 to 0.9, the peak concentration of B increases, the peak concentration of Ni maximizes while the valley concentration of Al minimizes at x = 0.5. The calculations also show the best cohesion of the grain boundary at x = 0.5. read less NOT USED (low confidence) H. Deng, W. Hu, X. Shu, L. Zhao, and B. Zhang, “Monte Carlo simulation of the surface segregation of Pt–Pd and Pt–Ir alloys with an analytic embedded-atom method,” Surface Science. 2002. link Times cited: 42 NOT USED (low confidence) W. Fan, X. Gong, and W. Lau, “Instability of an atomic chain arising from lattice misfit,” Physical Review B. 2002. link Times cited: 3 NOT USED (low confidence) N. Agrait, A. Yeyati, and J. M. Ruitenbeek, “Quantum properties of atomic-sized conductors,” Physics Reports. 2002. link Times cited: 1171 NOT USED (low confidence) E. Arregui, M. Caro, and A. Caro, “Numerical evaluation of the exact phase diagram of an empirical Hamiltonian: Embedded atom model for the Au-Ni system,” Physical Review B. 2002. link Times cited: 34 Abstract: Molecular-dynamics simulations were used to calculate the Gi… read moreAbstract: Molecular-dynamics simulations were used to calculate the Gibbs free energy on the entire compositional range of gold-nickel alloys described with a set of embedded atom potentials available in the literature. Thermodynamic integration and switching Hamiltonian techniques were used to obtain the exact phase diagram (with no approximations), and that corresponding to the regular approximation. Remarkable agreement for some properties, such as the solvus critical point, the congruential melting, the melting points of the pure elements, and the formation entropy of the alloy, contrasts with the poor prediction of the location of the solidus-liquidus lines, reflecting errors in the heat of solution in the liquid phase. The results are compared with recent experimental reassessment of the Au-Ni phase diagram and with ab initio calculations. read less NOT USED (low confidence) A. Strachan, T. Çagin, O. Gulseren, S. Mukherjee, R. E. Cohen, and W. A. Goddard, “First principles force field for metallic tantalum,” Modelling and Simulation in Materials Science and Engineering. 2002. link Times cited: 32 Abstract: We develop a many-body force field (FF) for tantalum based o… read moreAbstract: We develop a many-body force field (FF) for tantalum based on extensive ab initio quantum mechanical (QM) calculations and illustrate its application with molecular dynamics (MD). As input data to the FF we use ab initio methods (LAPW-GGA) to calculate: (i) the zero temperature equation of state (EOS) of Ta for bcc, fcc, and hcp crystal structures for pressures up to ∼500 GPa, and (ii) elastic constants. We use a mixed-basis pseudopotential code to calculate: (iii) volume-relaxed vacancy formation energy also as a function of pressure. In developing the Ta FF we also use previous QM calculations of: (iv) the EOS for the A15 structure; (v) the surface energy bcc (100); (vi) energetics for shear twinning of the bcc crystal. We find that, with appropriate parameters, an embedded atom model FF (denoted as qEAM FF) is able to reproduce all this QM data. We illustrate the use of the qEAM FF with MD to calculate such finite temperature properties as the melting curve up to 300 GPa and thermal expansivity in a wide temperature range. Both our predictions agree well with experimental values. read less NOT USED (low confidence) P. Szelestey, M. Patriarca, L. Perondi, and K. Kaski, “MODIFIED EAM POTENTIALS FOR MODELLING STACKING–FAULT BEHAVIOR IN Cu, Al, Au, AND Ni,” International Journal of Modern Physics B. 2002. link Times cited: 10 Abstract: In this paper we have developed empirical Embedded Atom Mode… read moreAbstract: In this paper we have developed empirical Embedded Atom Model potentials, following the fitting scheme proposed by Chantasiriwan and Milstein, in order to describe the stacking fault behaviour of copper, gold, nickel and aluminium. We show that the potentials based on this scheme can be modified to provide reasonable stacking-fault energy values and consequently a better description of the plastic properties. Modifications were done by changing the cut-off distance in case of aluminium and nickel, and in case of gold and copper by also modifying the functional form of the pair-potential. In order to validate these modified potentials we have tested them by studying various properties, such as structural, defect, and surface energies, and phonon spectra and comparing results with those from experiments and other model potentials. read less NOT USED (low confidence) C. Mottet, G. Tréglia, and B. Legrand, “Theoretical investigation of chemical and morphological ordering in Pd c Cu 1 − c clusters,” Physical Review B. 2002. link Times cited: 29 Abstract: We present a theoretical study of ${\mathrm{Pd}}_{c}{\mathrm… read moreAbstract: We present a theoretical study of ${\mathrm{Pd}}_{c}{\mathrm{Cu}}_{1\ensuremath{-}c}$ clusters from a few hundred to a few thousand of atoms, using Monte Carlo simulations and quenched molecular dynamics. This is performed within tight-binding many-body potentials, the tight-binding Ising model and the second moment approximation, which properly account for chemical and structural changes at transition-metal surfaces. The respective stabilities of the fcc, bcc, and icosahedral cluster shape are discussed in terms of competition or synergy between surface segregation and bulk ordering. Besides a finite-size effect on surface segregation, due to the limited quantity of matter, we show that chemical ordering can induce some geometrical frustrations and enhance the stability of ``stoichiometric'' clusters, the composition of which is strongly size dependent. Finally, it is found that chemical ordering leads to morphological transitions at equiconcentration. read less NOT USED (low confidence) F. Palomares, M. E. C. Serrano, A. Ruiz, F. Soria, K. Horn, and M. Alonso, “ARPES study of the surface states from Au/Ag(111): evolution with coverage and photon energy,” Surface Science. 2002. link Times cited: 6 NOT USED (low confidence) H. Rafii-Tabar and A. Chirazi, “Multi-scale computational modelling of solidification phenomena,” Physics Reports. 2002. link Times cited: 39 NOT USED (low confidence) B. Wirth, V. Bulatov, and T. D. Rubia, “Dislocation-Stacking Fault Tetrahedron Interactions in Cu,” Journal of Engineering Materials and Technology-transactions of The Asme. 2002. link Times cited: 111 Abstract: In copper and other face centered cubic metals, high-energy … read moreAbstract: In copper and other face centered cubic metals, high-energy particle irradiation produces hardening and shear localization. Post-irradiation microstructural examination in Cu reveals that irradiation has produced a high number density of nanometer sized stacking fault tetrahedra. The resultant irradiation hardening and shear localization is commonly attributed to the interaction between stacking fault tetrahedra and mobile dislocations, although the mechanism of this interaction is unknown. In this work, we present results from a molecular dynamics simulation study to characterize the motion and velocity of edge dislocations at high strain rate and the interaction and fate of the moving edge dislocation with slacking fault tetrahedra in Cu using an EAM interatomic potential. The results show that a perfect SFT acts as a hard obstacle for dislocation motion and although the SFT is sheared by the dislocation passage, it remains largely intact. How-ever, our simulations show that an overlapping, truncated SFT is absorbed by the passage of an edge dislocation, resulting in dislocation climb and the formation of a pair of less mobile super-jogs on the dislocation. read less NOT USED (low confidence) M. Karabacak, S. Özçelik, and Z. B. Güvenç, “Structures and energetics Of Pdn (n = 2-20) clusters using an embedded-atom model potential,” Surface Science. 2002. link Times cited: 27 NOT USED (low confidence) J. Jiménez‐Sáez, J. Domínguez-Vázquez, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “A molecular dynamics study of Ni/Cu(001) interfaces,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2002. link Times cited: 7 NOT USED (low confidence) T. Rahman, J. Spangler, and A. Al-Rawi, “Temperature variation of surface phonon line width: low Miller index surfaces of Ag and Cu,” Surface Science. 2002. link Times cited: 10 NOT USED (low confidence) J. Shu, W. Zheng, Q. Lu, H.-C. Huang, and W. Wong, “Parallel computing for lattice Monte Carlo simulation of large-scale thin film growth,” Science in China Series : Information Sciences. 2002. link Times cited: 0 Abstract: This paper proposes two viable computing strategies for dist… read moreAbstract: This paper proposes two viable computing strategies for distributed parallel systems: domain division with sub-domain overlapping and asynchronous communication. We have implemented a parallel computing procedure for simulation of Ti thin film growing process of a system with 1000 × 1000 atoms by means of the Monte Carlo (MC) method. This approach greatly reduces the computation time for simulation of large-scale thin film growth under realistic deposition rates. The multi-lattice MC model of deposition comprises two basic events: deposition, and surface diffusion. Since diffusion constitutes more than 90% of the total simulation time of the whole deposition process at high temperature, we concentrated on implementing a new parallel diffusion simulation that reduces communication time during simulation. Asynchronous communication and domain overlapping techniques are used to reduce the waiting time and communication time among parallel processors. The parallel algorithms we propose can simulate the thin film growth of a system with many more particles than before under realistic deposition rates, and can provide a more efficient means for computer simulation of thin film growth. read less NOT USED (low confidence) A. Franchini, C. Magherini, and G. Santoro, “Surface thermal expansion of Ag(110),” Surface Science. 2002. link Times cited: 2 NOT USED (low confidence) M. C. Giménez et al., “Theoretical Considerations of Electrochemical Phase Formation for an Ideal Frank-van der Merwe System Ag on Au(111) and Au(100),” Journal of The Electrochemical Society. 2002. link Times cited: 25 Abstract: Static calculation and preliminary kinetic Monte Carlo simul… read moreAbstract: Static calculation and preliminary kinetic Monte Carlo simulation studies are undertaken for the nucleation and growth on a model system which follows a Frank-van der Merwe mechanism. In the present case, we consider the deposition of Ag on Au(100) and Au(111) surfaces. The interactions were calculated using the embedded atom model. The kinetics of formation and growth of 2D Ag structures on Au(100) and Au(111) is investigated and the influence of surface steps on this phenomenon is studied. Very different time scales are predicted for Ag diffusion on Au(100) and Au(111), thus rendering very different regimes for the nucleation and growth of the related 2D phases. These observations are drawn from the application of a model free of any adjustable parameter. read less NOT USED (low confidence) D. Maroudas and M. Gungor, “Continuum and atomistic modeling of electromechanically-induced failure of ductile metallic thin films,” Computational Materials Science. 2002. link Times cited: 17 NOT USED (low confidence) V. Rodrigues and D. Ugarte, “Atomic Arrangement and Conductance of Metal Nanowires,” Physica Status Solidi B-basic Solid State Physics. 2002. link Times cited: 6 Abstract: We have used high resolution transmission electron microscop… read moreAbstract: We have used high resolution transmission electron microscopy and mechanically controllable break junction technique to study metal NW structure and electrical transport properties. In the last stages just before rupture, gold and platinum nanowires are crystalline and defect-free. In particular, gold NWs assume merely three kinds of atomic arrangements, which were correlated to observed conductance behaviors. read less NOT USED (low confidence) J. R. Hu, S. Chang, F. Chen, and J. Kai, “HRTEM investigation of the multiplicity of Σ=9[01̄1]/(122) symmetric tilt grain boundary in Cu,” Materials Chemistry and Physics. 2002. link Times cited: 17 NOT USED (low confidence) K. Nordlund, “Computational materials science of ion irradiation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2002. link Times cited: 18 NOT USED (low confidence) M. Murty, “Sputtering: the material erosion tool,” Surface Science. 2002. link Times cited: 54 NOT USED (low confidence) R. Benedek, D. Seidman, and C. Woodward, “The effect of misfit on heterophase interface energies,” Journal of Physics: Condensed Matter. 2002. link Times cited: 63 Abstract: Most previous atomistic simulations of heterophase interface… read moreAbstract: Most previous atomistic simulations of heterophase interfaces have neglected misfit, the discrepancy between the interatomic length scales parallel to the interface of the two phases. The obstacles to quantitative calculations of interface energies in the presence of misfit are assessed. The most straightforward approach is to perform simulations for a supercell whose size is of the order of the cube of the smallest common periodic length scale (essentially the coincidence-site-lattice periodicity), which varies inversely with the misfit parameter. Such supercells typically contain at least thousands of atoms. First-principles simulations are highly accurate, but are feasible only for a few selected heterophase interfaces with large misfit. Classical interatomic potentials, on the other hand, are efficient numerically, but their accuracy has not been demonstrated in the context of heterophase interface calculations. An approximate formulation of the interface energy is presented here which can be evaluated numerically by first-principles calculations for supercells of only moderate size. This formulation explores the relationship between the interface energies for coherent and semi-coherent interfaces. A numerical application to an interface between tetragonal TiAl and perovskite Ti3AlC is presented. read less NOT USED (low confidence) E. Webb and G. Grest, “Molecular dynamics simulations of reactive wetting,” Scripta Materialia. 2002. link Times cited: 38 NOT USED (low confidence) X. Yuan, K. Takahashi, and T. Onzawa, “Formalism of Modified Embedded Atom Method in Application to Dimer Systems : Fundamental Study of Applicability of Modified Embedded Atom Method to Joining and Adhesion Problems (Report 1),” Quarterly Journal of The Japan Welding Society. 2002. link Times cited: 2 NOT USED (low confidence) M. D. Pópolo, E. Leiva, and W. Schmickler, “Model calculations for copper clusters on Au(111) surfaces,” Journal of Electroanalytical Chemistry. 2002. link Times cited: 10 NOT USED (low confidence) S. Narasimhan, “Ab initio lattice dynamics of Ag(1 1 0),” Surface Science. 2002. link Times cited: 10 NOT USED (low confidence) T. Nozaki, M. Doyama, and Y. Kogure, “Computer Simulation of High-speed Bending Deformation in Copper,” Radiation Effects and Defects in Solids. 2002. link Times cited: 1 Abstract: According to one suggested model, bending of a single crysta… read moreAbstract: According to one suggested model, bending of a single crystal introduces edge dislocations of the same sign. In the present study, this model is examined by computer simulation using molecular dynamics. When a notch is present on the tension surface, Heidenreich-Shockley partial dislocations are created near the tip of the notch. In the compression surface, partial dislocations are created due to wrinkling of the crystal plane. The results of simulation shows that dislocations are more easily created in a compressive bending region than in a tension bending region or simple tension region. read less NOT USED (low confidence) G. Garda, R. Ferullo, and N. Castellani, “NO ADSORPTION ON Pd(111): THE FORMATION OF THE ISOCYANATE GROUP,” Surface Review and Letters. 2001. link Times cited: 2 NOT USED (low confidence) S. Psakhie et al., “Movable cellular automata method for simulating materials with mesostructure,” Theoretical and Applied Fracture Mechanics. 2001. link Times cited: 127 NOT USED (low confidence) K. Nordlund, K. Henriksson, and J. Keinonen, “Melting temperature effects on the size of ion-induced craters,” Applied Physics Letters. 2001. link Times cited: 24 Abstract: Recent work on the sizes of craters produced by ion impacts … read moreAbstract: Recent work on the sizes of craters produced by ion impacts of solids has shown that the size of the crater scales with the inverse square of the cohesive energy. This observation is in contrast to the size of craters produced in macroscopic impacts, which scale directly with the inverse of the cohesive energy. It has relied on the assumption that the melting temperature is proportional to the cohesive energy. Using computer simulations, we now show that the size scales in fact with the inverse of the product of the melting temperature and cohesive energy. This provides direct proof that the reason to the different behavior of macroscopic and ion-induced cratering is flow of the liquid produced by the ions. read less NOT USED (low confidence) A. Kara and T. Rahman, “Phonons of Metallic Vicinal Surfaces,” Surface Science. 2001. link Times cited: 3 NOT USED (low confidence) C. Barnes, E. AlShamaileh, T. Pitkänen, P. Kaukasoina, and M. Lindroos, “The kinetics of formation and structure of an underlayer alloy: the Cu(1 0 0)-c(2×2)–Pd system,” Surface Science. 2001. link Times cited: 23 NOT USED (low confidence) J. Zimmerman, C. L. Kelchner, P. Klein, J. C. Hamilton, and S. Foiles, “Surface step effects on nanoindentation.,” Physical review letters. 2001. link Times cited: 438 Abstract: Atomistic simulation is used to examine nanoindentation of a… read moreAbstract: Atomistic simulation is used to examine nanoindentation of a Au(111) crystal both near and far from a surface step. While the load needed to nucleate dislocations decreases significantly when indenting close to the step, the extent of the step's influence is not as great as seen experimentally. This behavior is explained by measuring the contact area from the simulation data. A new metric, the slip vector, shows material slip coinciding with the <112> directions of a lowest unstable stacking fault barrier. The slip vector is used to calculate an atomic critical resolved shear stress, which is shown to be a good dislocation nucleation criterion. read less NOT USED (low confidence) Y. Wang, W. Wang, K. Fan, and J. Deng, “Structural and electronic properties of silver surfaces: ab initio pseudopotential density functional study,” Surface Science. 2001. link Times cited: 40 NOT USED (low confidence) Y. Wang, W. Wang, K. Fan, and J. Deng, “The first-principle study of the iodine-modified silver surfaces,” Surface Science. 2001. link Times cited: 17 NOT USED (low confidence) R. Kissel and H. Urbassek, “Sputtering of a Au surface covered with large spherical clusters,” International Journal of Mass Spectrometry. 2001. link Times cited: 27 NOT USED (low confidence) M. Doyama and Y. Kogure, “Computer simulation of creation and motion of dislocations during plastic deformation in copper,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2001. link Times cited: 2 NOT USED (low confidence) M. Caturla, T. D. de la Rubia, M. Victoria, R. Corzine, M. James, and G. Greene, “Multiscale modeling of radiation damage: applications to damage production by GeV proton irradiation of Cu and W, and pulsed irradiation effects in Cu and Fe,” Journal of Nuclear Materials. 2001. link Times cited: 41 NOT USED (low confidence) O. Trushin, P. Salo, M. Alatalo, and T. Ala‐Nissila, “Atomic mechanisms of cluster diffusion on metal fcc(100) surfaces,” Surface Science. 2001. link Times cited: 19 NOT USED (low confidence) M. Doyama, T. Ohmae, and Y. Kogure, “Motion and conversion energies of ad-atom, di-adatom and tri-adatoms on copper (001) surface,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 0 NOT USED (low confidence) R. Kissel and H. Urbassek, “Sputtering from spherical Au clusters by energetic atom bombardment,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 62 NOT USED (low confidence) T. Muramoto, M. Okai, Y. Yamashita, K. Yorizane, and Y. Yamamura, “MD simulation of cluster formation during sputtering,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 7 NOT USED (low confidence) C. R. S. Silva and C. Scherer, “Ion-induced energy propagating front and migration of point defects in metals – II,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 0 NOT USED (low confidence) H. Bonzel, “Equilibrium crystal shapes: towards absolute energies,” Progress in Surface Science. 2001. link Times cited: 29 NOT USED (low confidence) Y. Wang, J. Shen, N. Chen, and J. Wang, “Theoretical investigation on site preference of foreign atoms in rare-earth intermetallics,” Journal of Alloys and Compounds. 2001. link Times cited: 19 NOT USED (low confidence) K. Nordlund, J. Nord, and J. Keinonen, “Chemical effects in collision cascades,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 3 NOT USED (low confidence) S. J. Zhao, D. Cheng, S. Wang, and H. Ye, “Melting of the Symmetrical Tilt Boundaries in Copper,” Journal of the Physical Society of Japan. 2001. link Times cited: 6 Abstract: The melting transition in three high-energy and two low-ener… read moreAbstract: The melting transition in three high-energy and two low-energy fee Cu symmetrical tilt grain boundaries (STGBs) has been investigated by molecular-dynamics (MD) simulations. Low-energy STGBs are found to be crystalline from room temperature all the way up to T-0 (T-0 is the thermodynamic equilibrium melting point). In contrast, all high-energy STGBs undergo premelting which is initiated at a transition temperature T-c (T-c < T-0). Moreover, the higher the zero-temperature GB energy, the lower the T-c. read less NOT USED (low confidence) O. Trushin et al., “Adatom island diffusion on metal fcc(100) surfaces.” 2001. link Times cited: 1 NOT USED (low confidence) T. Swiler, “The role of liquid–substrate interactions on wetting in metallic embedded atom method systems,” Acta Materialia. 2000. link Times cited: 18 NOT USED (low confidence) Y. Osetsky, D. Bacon, C. Matthai, and N. H. March, “Cleavage force, tribology and bond breaking in some transition metals,” Journal of Physics and Chemistry of Solids. 2000. link Times cited: 1 NOT USED (low confidence) B. Wirth, V. Bulatov, and T. D. Rubia, “Atomistic simulation of stacking fault tetrahedra formation in Cu,” Journal of Nuclear Materials. 2000. link Times cited: 80 NOT USED (low confidence) T. Swiler and R. Loehman, “Molecular dynamics simulations of reactive wetting in metal–ceramic systems,” Acta Materialia. 2000. link Times cited: 40 NOT USED (low confidence) R. Schäublin, P. Almeida, A. Almazouzi, and M. Victoria, “Correlation of simulated TEM images with irradiation induced damage,” Journal of Nuclear Materials. 2000. link Times cited: 7 NOT USED (low confidence) R. Laskowski, “Double Lattice Inversion Technique — Application to the EAM Potential Construction,” Physica Status Solidi B-basic Solid State Physics. 2000. link Times cited: 3 Abstract: The embedded atom model (EAM) has been proved to be a very e… read moreAbstract: The embedded atom model (EAM) has been proved to be a very effective and powerful approach to energy calculation in metals. In the current work, we present a new approach to the construction of the EAM potential. The EAM functions are determined in a way to reproduce exactly two state equations defined for unit cells of two different metal structures. Our method is in some sense an extension of the lattice inversion algorithm proposed by Carlsson et al. (Phil Mag. A 41, 241 (1980)), where the resulting pair potential reconstructs only one given state equation. The inversion technique has been applied before to the construction of a pair potential and embedded atom potential from a single state equation. Using two input state equations we can determine only two functions, thus, we construct the pair potential and embedding function. The atomic charge density is taken from first-principle calculation. As an example, we apply our method to the determination of the EAM potential for solid lead. In order to estimate the quality of this potential, some bulk and defect properties are also calculated. read less NOT USED (low confidence) K. P. Zol’nikov, T. Uvarov, A. Lipnitskii, D. Saraev, and S. Psakh’e, “Specific features of the nanoscopic spalling fracture near the grain boundary,” Combustion, Explosion and Shock Waves. 2000. link Times cited: 0 NOT USED (low confidence) S. Erkoç, “General analytic density distribution function for atoms,” International Journal of Modern Physics C. 2000. link Times cited: 0 Abstract: A general analytic function is proposed to represent the rad… read moreAbstract: A general analytic function is proposed to represent the radial distribution of charge density for atoms. The proposed function fits perfectly to the radial charge density generated numerically by local spin density functional method for atoms from He to Kr. read less NOT USED (low confidence) D. Bazin, C. Mottet, G. Tréglia, and J. Lynch, “New trends in heterogeneous catalysis processes on metallic clusters from synchrotron radiation and theoretical studies,” Applied Surface Science. 2000. link Times cited: 13 NOT USED (low confidence) S. G. Wang, E. Tian, and C. Lung, “Surface energy of arbitrary crystal plane of bcc and fcc metals,” Journal of Physics and Chemistry of Solids. 2000. link Times cited: 113 NOT USED (low confidence) L. Yang, “Reverse surface segregation in Cu–Pd bimetallic catalysts at low concentrations of Cu,” Philosophical Magazine A. 2000. link Times cited: 9 Abstract: This work presents atomic-level simulations that study the s… read moreAbstract: This work presents atomic-level simulations that study the surface segregation profiles in Cu–Pd catalysts at various concentrations of Cu. It is found that Cu normally enriches on the surface of 201-atom Cu–Pd cluster but depletes from the surface at very low Cu concentrations. This phenomenon was observed in experiments previously and can be reproduced by the molecular dynamics/Monte Carlo-corrected effective medium theory if the latest modifications, to fit the alloy heats of mixing, are included in the interaction potential. This study also reveals different surface segregation behaviours for clusters having different shapes. It appears that most of the thermally accessible shapes of the cluster exhibit reverse surface segregation. However, these shapes are metastable. The lowest-energy configuration tested, having the cubo-octahedral shape, does not show reverse surface segregation; instead, it shows normal surface segregation with Cu enrichment on the surface at all concentrations. Thus, this work suggests that the experimentally observed clusters that demonstrate reverse surface segregation are metastable. read less NOT USED (low confidence) U. Kürpick and B. Fricke, “Manipulation of Cu adatoms on anisotropic Cu surfaces using scanning tunneling microscopy,” Surface Science. 2000. link Times cited: 4 NOT USED (low confidence) J. Ferrón et al., “Influence of surfactants on atomic diffusion,” Surface Science. 2000. link Times cited: 30 NOT USED (low confidence) L. G. González and J. M. Montejano-Carrizales, “Embedded Atom Method Applied to Ni, Cu, Ag, and Pd,” Physica Status Solidi B-basic Solid State Physics. 2000. link Times cited: 8 Abstract: We analyze the structural stability and symmetry of Ni, Cu, … read moreAbstract: We analyze the structural stability and symmetry of Ni, Cu, Ag, and Pd metal clusters using the Embedded Atom Method (EAM), developed by Foiles. The structures used in the study are based in polyhedra such as: tetrahedron, hexahedron, triangular antiprism, decahedron, trigonal prism and dodecahedron. To get the various cluster sizes for each one of the polyhedron, top sites over the faces of the polyhedron are placed at the same distance from the face sites. The faces in the polyhedra can be triangular or square faces, f.c.c. clusters are also used. Comparison between the cohesive energy of the various clusters with the same number of atoms but different structured is performed to find out the structural stability, the cluster symmetry is also determined. read less NOT USED (low confidence) H. Skriver, A. Ruban, J. Nørskov, L. Vitos, and J. Kollár, “Steps, kinks, and segregation at metallic surfaces,” Progress in Surface Science. 2000. link Times cited: 10 NOT USED (low confidence) M. I. Rojas, C. Sánchez, M. D. Pópolo, and E. Leiva, “Erratum to: ‘An embedded atom approach to underpotential deposition phenomena’: [Surf. Sci. 421 (1999) 59],” Surface Science. 2000. link Times cited: 18 NOT USED (low confidence) M. Asta, V. Ozoliņš, and J. Hoyt, “The energetics of surface-alloy formation: an embedded-atom-method, second-order-expansion study,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 0 Abstract: Chemical ordering and clustering instabilities in alloys are… read moreAbstract: Chemical ordering and clustering instabilities in alloys are governed by the Fourier transform of the effective pair interactions, V(k). We make use of a second-order-expansion formalism, based upon embedded-atom-method interatomic potentials, to calculate chemical and elastic contributions to V(k) for monolayer surface alloys on single-crystal substrates. It is demonstrated that the elastic contribution to V(k) is characterized by a finite slope at the origin, consistent with continuum models which predict that V(k)∝-|k| for small wavevectors. As a consequence, the global minimum in V(k) always occurs at finite k, and therefore compositional instabilities in ultrathin surface-alloy films are generally of an ordering (as opposed to clustering, k = 0) type. read less NOT USED (low confidence) M. Polak and L. Rubinovich, “The interplay of surface segregation and atomic order in alloys,” Surface Science Reports. 2000. link Times cited: 128 NOT USED (low confidence) D. Morgan, A. Walle, G. Ceder, J. Althoff, and D. Fontaine, “Vibrational thermodynamics: coupling of chemical order and size effects,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 19 Abstract: The effects of chemical order on the vibrational entropy hav… read moreAbstract: The effects of chemical order on the vibrational entropy have been studied using first-principles and semi-empirical potential methods. Pseudopotential calculations on the Pd_3V system show that the vibrational entropy decreases by 0.07k_B upon disordering in the high-temperature limit. The decrease in entropy contradicts what would be expected from simple bonding arguments, but can be explained by the influence of size effects on the vibrations. In addition, the embedded-atom method is used to study the effects of local environments on the entropic contributions of individual Ni and Al atoms in Ni_3Al. It is found that increasing numbers of Al nearest neighbours decreases the vibrational entropy of an atom when relaxations are not included. When the system is relaxed, this effect disappears, and the local entropy is approximately uniform with increasing number of Al neighbours. These results are explained in terms of the large size mismatch between Ni and Al. In addition, a local cluster expansion is used to show how the relaxations increase the importance of long-range and multisite interactions. read less NOT USED (low confidence) A. Ray and N. H. March, “Relation between vacancy properties and surface energies in three noble or transition metals,” Journal of Physics and Chemistry of Solids. 2000. link Times cited: 0 NOT USED (low confidence) E. Leiva, M. D. Pópolo, and W. Schmickler, “Changes in surface stress caused by the adsorption of an epitaxial metal monolayer,” Chemical Physics Letters. 2000. link Times cited: 14 NOT USED (low confidence) Z. Wang, Y. Li, and J. B. Adams, “Kinetic lattice Monte Carlo simulation of facet growth rate,” Surface Science. 2000. link Times cited: 52 NOT USED (low confidence) Y. Devyatko and S. Rogozhkin, “Point defects at low-index surfaces of fcc metals and the anomalous behaviour of surface atoms at elevated temperatures,” Vacuum. 2000. link Times cited: 6 NOT USED (low confidence) H. Rafii-Tabar, “Modelling the nano-scale phenomena in condensed matter physics via computer-based numerical simulations,” Physics Reports. 2000. link Times cited: 163 NOT USED (low confidence) T. Yamaghishi, K. Takahashi, and T. Onzawa, “Modified embedded atom method calculations for reconstructed (110) surfaces of face-centered cubic metals,” Surface Science. 2000. link Times cited: 11 NOT USED (low confidence) S. Jiang and J. Belak, “Chapter 15 - Molecular Dynamics of Thin Films under Shear,” Theoretical and Computational Chemistry. 1999. link Times cited: 1 NOT USED (low confidence) A. Bilić, B. King, and D. J. O’connor, “Embedded atom method study of surface-confined Al on Ni(001),” Surface Science. 1999. link Times cited: 7 NOT USED (low confidence) P. Deurinck and C. Creemers, “Face-related segregation reversal at Pt50Ni50 surfaces studied with the embedded atom method,” Surface Science. 1999. link Times cited: 16 NOT USED (low confidence) M. Alemany, C. Rey, and L. J. Gallego, “Embedded atom model calculations of the diffusion coefficient of Ni impurity in liquid Al,” Journal of Chemical Physics. 1999. link Times cited: 7 Abstract: Using the embedded atom model potential for solid Ni–Al syst… read moreAbstract: Using the embedded atom model potential for solid Ni–Al systems proposed by Voter and Chen, we performed molecular dynamics simulations to compute the diffusion coefficient of Ni impurity in liquid Al. Our results are in excellent agreement with available experimental data. read less NOT USED (low confidence) J. Hoyt, B. Sadigh, M. Asta, and S. Foiles, “Kinetic phase field parameters for the Cu–Ni system derived from atomistic computations,” Acta Materialia. 1999. link Times cited: 102 NOT USED (low confidence) X.-Y. Liu, C.-L. Liu, and L. J. Borucki, “A new investigation of copper’s role in enhancing Al-Cu interconnect electromigration resistance from an atomistic view,” Acta Materialia. 1999. link Times cited: 44 NOT USED (low confidence) M. Alemany, M. Calleja, C. Rey, L. J. Gallego, J. Casas, and L. González, “A theoretical and computer simulation study of the static structure and thermodynamic properties of liquid transition metals using the embedded atom model,” Journal of Non-crystalline Solids. 1999. link Times cited: 18 NOT USED (low confidence) I. Sklyadneva, G. Rusina, and E. Chulkov, “Vibrations on Cu surfaces covered with Ni monolayer,” Surface Science. 1999. link Times cited: 8 NOT USED (low confidence) N. Singh, “Structural phase transformation of Cu, Pd and Au using transition metal pair potential,” Physica B-condensed Matter. 1999. link Times cited: 12 NOT USED (low confidence) M. Doyama, “Crystal growth study using combination of molecular dynamics and Monte Carlo methods,” Bulletin of Materials Science. 1999. link Times cited: 2 NOT USED (low confidence) T. D. Rubia, M. Caturla, E. Alonso, N. Soneda, and M. Johnson, “The primary damage state and its evolution over multiple length and time scales,” Radiation Effects and Defects in Solids. 1999. link Times cited: 12 Abstract: During his long and illustrious career, Professor Kiritani m… read moreAbstract: During his long and illustrious career, Professor Kiritani made many of the most significant and revealing observations regarding the nature of the primary damage state and the fate of the produced defects in irradiated metals and semiconductors. We present a review of recent results of molecular dynamics (MD) and kinetic Monte Carlo (KMC) simulations of defect production and annealing in irradiated metals and semiconductors. The MD simulations describe the primary damage state in two prototypical elemental metals and in one-elemental semiconductor, namely Fe, Au, and Si. These materials were all thoroughly investigated by Prof. Kiritani and his colleagues using neutron irradiation followed by TEM observation, and here we attempt to provide some further understanding of the experimental observations by using atomic-scale computer simulation tools. We describe the production of interstitial and vacancy clusters in the cascades and highlight the differences among the various materials. In particula... read less NOT USED (low confidence) Y. Shimomura, I. Mukouda, K. Sugio, and P. Zhao, “Atomistic processes of damage evolution in neutron-irradiated Cu and Ni at high temperature,” Radiation Effects and Defects in Solids. 1999. link Times cited: 1 Abstract: This paper consists of two parts. In part 1, the experimenta… read moreAbstract: This paper consists of two parts. In part 1, the experimental results of damage evolution of neutron-irradiated Cu and Ni are described. In part 2, results of computer simulations are described with linkage of experimental data to explore the atomistic process of damage evolution. To study experimentally the atomistic processes of damage evolution in neutron-irradiated Cu and Ni in part 1, we prepare two types of specimens for both metals. One is as-received specimen from manufacturer. Another is a residual-gas-free specimen which is prepared by melting as-received metals in highly evacuated vacuum at 10−5 Pa. Specimens are irradiated with fission neutrons in the temperature-controlled-irradiation capsule at JMTR (Japan Materials Testing Reactor). TEM (Transmission Electron Microscope) observation shows that the dislocation structure is developed by the aggregation of interstitial clusters in irradiated metals. It is found that the number density of void which are observed in specimens, both as-r... read less NOT USED (low confidence) X. W. Zhou and H. Wadley, “Hyperthermal vapor deposition of copper: athermal and biased diffusion effects,” Surface Science. 1999. link Times cited: 62 NOT USED (low confidence) K. Yorizane, T. Muramoto, and Y. Yamamura, “Computer studies on reflection and sputtering due to low-energy cluster impacts,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 3 NOT USED (low confidence) P. Wynblatt and A. Landa, “Computer simulation of surface segregation in ternary alloys,” Computational Materials Science. 1999. link Times cited: 38 NOT USED (low confidence) L. Zhenyun, H. Bai-yun, and L. Dongliang, “Computer simulation of dislocation cores in B2 NiAl intermetallics,” Journal of Central South University of Technology. 1999. link Times cited: 13 NOT USED (low confidence) G. D. Billing, “Trajectory driven second quantization approach to quantum dynamics.” 1999. link Times cited: 5 NOT USED (low confidence) R. Longo, C. Rey, and L. J. Gallego, “Structure and melting of small Ni clusters on Ni surfaces,” Surface Science. 1999. link Times cited: 15 NOT USED (low confidence) P. Gumbsch and R. Schroll, “Atomistic aspects of the deformation of NiAl,” Intermetallics. 1999. link Times cited: 25 NOT USED (low confidence) C. Sánchez, M. D. Pópolo, and E. Leiva, “An embedded atom approach to underpotential deposition phenomena,” Surface Science. 1999. link Times cited: 36 NOT USED (low confidence) M. A. Karolewski, “Classical dynamics simulation of projectile-surface interactions,” Surface and Interface Analysis. 1999. link Times cited: 16 Abstract: A public-domain package of programs for the personal compute… read moreAbstract: A public-domain package of programs for the personal computer, the Simulation Kit (SK), has been developed for the simulation and visualization of collisions of low-energy (<10 keV) atomic projectiles with solid target lattices. Possible applications of the SK include the simulation of ion scattering spectra, sputtering coefficients, reflection coefficients and projectile ranges. The simulation model used by the SK is based on classical dynamics, and uses a composite screened-Coulomb/Morse pair potential to model interactions between particles in the target. The simulation model also incorporates inelastic scattering effects based on the Lindhard–Schiott–Scharff, Oen–Robinson and Shapiro–Tombrello models, respectively. The physical basis of the simulation model is described, and examples are provided of applications in ion beam analysis (ion scattering spectrometry, sputter yields). Copyright © 1998 John Wiley & Sons, Ltd. read less NOT USED (low confidence) K. Yorizane and Y. Yamamura, “Molecular dynamics studies of thin film growth by ionized cluster beam deposition,” Computational Materials Science. 1999. link Times cited: 7 NOT USED (low confidence) P. Zhao and Y. Shimomura, “Molecular dynamics calculations of properties of the self-interstitials in copper and nickel,” Computational Materials Science. 1999. link Times cited: 30 NOT USED (low confidence) P. Gumbsch, “Atomistic modelling of diffusion-controlled interfacial decohesion,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1999. link Times cited: 12 NOT USED (low confidence) M. Doyama and Y. Kogure, “Embedded atom potentials in fcc and bcc metals,” Computational Materials Science. 1999. link Times cited: 73 NOT USED (low confidence) C.-L. Liu, X.-Y. Liu, and L. Borucki, “Defect generation and diffusion mechanisms in Al and Al–Cu,” Applied Physics Letters. 1999. link Times cited: 36 Abstract: A defect generation mechanism, namely, the grain-boundary Fr… read moreAbstract: A defect generation mechanism, namely, the grain-boundary Frenkel pair model, and corresponding diffusion mechanisms during electromigration are developed using atomic simulation techniques in Al and Al–Cu. We contend that large numbers of interstitials and vacancies exist at grain boundaries and both contribute to mass transport. Cu preferentially segregates to the interstitial sites at grain boundaries via a Frenkel pair generation process and reduces the overall grain-boundary diffusivity due to the stronger Al–Cu binding. Predictions from our models are in excellent agreement with available experimental data and observations. read less NOT USED (low confidence) J. Wang, J. Plessis, J. J. Terblans, and G. V. Wyk, “The discontinuous surface transition in the Cu(111)(Ag) binary segregating system,” Surface Science. 1999. link Times cited: 12 NOT USED (low confidence) N. Papanicolaou, G. C. Kallinteris, G. Evangelakis, D. Papaconstantopoulos, and M. J. Mehl, “Second-moment interatomic potential for Cu-Au alloys based on total-energy calculations and its application to molecular-dynamics simulations,” Journal of Physics: Condensed Matter. 1998. link Times cited: 31 Abstract: We have evaluated interatomic potentials of Cu, Au and Cu-Au… read moreAbstract: We have evaluated interatomic potentials of Cu, Au and Cu-Au ordered alloys in the framework of the second-moment approximation to the tight-binding theory by fitting to the volume dependence of the total energy of these materials computed by first-principles augmented-plane-wave calculations. We have applied this scheme to calculate the bulk modulus and elastic constants of the pure elements and alloys and we have obtained a good agreement with experiment. We also have performed molecular-dynamics simulations at various temperatures, deducing the temperature dependence of the lattice constants and the atomic mean square displacements, as well as the phonon density of states and the phonon-dispersion curves of the ordered alloys. A satisfactory accuracy was obtained, comparable to previous works based on the same approximation, but resulting from fitting to various experimental quantities. read less NOT USED (low confidence) D. Morgan, J. Althoff, and D. Fontaine, “Local environment effects in the vibrational properties of disordered alloys: An embedded-atom method study of Ni3Al and Cu3Au,” Journal of Phase Equilibria. 1998. link Times cited: 17 NOT USED (low confidence) C. Sánchez and E. Leiva, “Underpotential versus overpotential deposition: a first-principles calculation,” Journal of Electroanalytical Chemistry. 1998. link Times cited: 18 NOT USED (low confidence) P. Geysermans, M. Mareschal, and V. Pontikis, “Thermodynamic properties of liquid copper modelled via an n-body potential,” Molecular Physics. 1998. link Times cited: 7 Abstract: Using molecular dynamics simulations the ability of a semi-e… read moreAbstract: Using molecular dynamics simulations the ability of a semi-empirical n-body potential to reproduce thermodynamical properties of liquid copper is tested. Density, pair distribution function, diffusion coefficient and surface tension are computed as functions of the temperature. Calculated values are analysed by resorting to well established empirical relationships in liquid metals and are in good agreement with experimental data. By scaling the temperature in terms of the melting point, excellent agreement is obtained between experiment and computed diffusivity values. However, surface tension is considerably underestimated, a result that is consistent with known shortcomings of n-body potentials of the kind used in present work. read less NOT USED (low confidence) W. Lee, L. P. Ford, P. Blowers, H. Nigg, and R. Masel, “Why do heats of adsorption of simple gases on platinum surfaces vary so little with surface structure,” Surface Science. 1998. link Times cited: 28 NOT USED (low confidence) I. Sklyadneva, G. Rusina, and E. Chulkov, “Vibrational states on vicinal surfaces of Al, Ag, Cu and Pd,” Surface Science. 1998. link Times cited: 49 NOT USED (low confidence) B. Mutasa and D. Farkas, “Atomistic structure of high-index surfaces in metals and alloys,” Surface Science. 1998. link Times cited: 30 NOT USED (low confidence) A. Bilić, Y. Shen, B. King, and D. J. O’connor, “Embedded Atom Method Study of Pd Thin Films on Cu(001),” Surface Review and Letters. 1998. link Times cited: 3 Abstract: We have studied the structures formed by the deposition of 0… read moreAbstract: We have studied the structures formed by the deposition of 0.5 and 1 monolayer (ML) of Pd on a Cu(001) surface using Monte Carlo (MC) simulations and static optimizations. The energetics are given by the semiempirical embedded atom method (EAM). At 0.5 ML Pd coverage we find that a Pd–Cu c(2×2) single layer surface alloy is created, consistent with experimental observations. At 1 ML Pd coverage a double layer c(2×2) Cu–Pd surface alloy is found to be energetically favored over structures with a clock-reconstructed topmost layer. However, metastable configurations of the top layer consisting of a clock-rotated phase with the (2×2)p4g symmetry coexisting with phases with c(2×2) and p(2×2) symmetries can also be obtained, in agreement with the experimental results. read less NOT USED (low confidence) R. E. Miller, M. Ortiz, R. Phillips, V. Shenoy, and E. Tadmor, “Quasicontinuum models of fracture and plasticity,” Engineering Fracture Mechanics. 1998. link Times cited: 167 NOT USED (low confidence) D. Timpel and K. Scheerschmidt, “Molecular dynamics investigations of silver diffusion in glass,” Journal of Non-crystalline Solids. 1998. link Times cited: 6 NOT USED (low confidence) S. Durukanoğlu and T. Rahman, “Atomic relaxations and thermodynamics on Cu(410),” Surface Science. 1998. link Times cited: 19 NOT USED (low confidence) M. Alemany, C. Rey, and L. J. Gallego, “Computer simulation study of the dynamic properties of liquid Ni using the embedded-atom model,” Physical Review B. 1998. link Times cited: 46 NOT USED (low confidence) K. Yorizane, T. Muramoto, and Y. Yamamura, “Computer studies on projected range due to few eV to keV cluster impacts,” 1998 International Conference on Ion Implantation Technology. Proceedings (Cat. No.98EX144). 1998. link Times cited: 0 Abstract: The time-evolutionary Monte Carlo simulation code DYACAT and… read moreAbstract: The time-evolutionary Monte Carlo simulation code DYACAT and the molecular dynamics simulation code have been applied to investigate the cluster effects on the projected range of (Au)/sub 55/ cluster impacts on Cu targets, changing the cluster energy from 10 eV/atom to 50 keV/atom. For high-energy incidence (>10 keV/atom), the projected range of cluster atoms is nearly equal to that of monatomic ions. For low-energy incidence (<10 keV/atom), the projected range of cluster atoms is much larger than that of monatomic ions due to the clear-the-way effect. For very-low-energy incidence (<30 eV/atom), the many-body effect plays an important role in the projected range. For the monatomic ion bombardment, it prevents monatomic ions from penetrating into the solid, where it restrains one of the clear-the-way effects for the cluster impact. read less NOT USED (low confidence) M. Haftel and M. Rosen, “New ballistically and thermally activated exchange processes in the vapor deposition of Au on Ag(111): a molecular dynamics study,” Surface Science. 1998. link Times cited: 7 NOT USED (low confidence) J. Sánchez and P. Besser, “Modelling microstructure development in trench-interconnect structures,” Proceedings of the IEEE 1998 International Interconnect Technology Conference (Cat. No.98EX102). 1998. link Times cited: 3 Abstract: The effects of surface and interfacial energies and trench g… read moreAbstract: The effects of surface and interfacial energies and trench geometry on microstructure evolution in damascene-processed interconnect structures are modelled. Grain growth and texture evolution are shown to depend on the magnitude of surface and interfacial energy variations with crystallographic orientation and with trench aspect ratio. Grain texture evolution in high aspect ratio trenches is driven by the minimization of grain sidewall interfacial energy, whereas the crystallographic evolution of grains within low aspect ratio structures is determined by more typical surface and lower interface energy minimization considerations. Comparisons are made to recent experimental results which characterize the development of crystallographic texture and grain size in damascene-processed interconnects. read less NOT USED (low confidence) G. Bhuiyan and M. Khaleque, “Structure and thermodynamic properties of liquid rare earth metals: an embedded atom method approach,” Journal of Non-crystalline Solids. 1998. link Times cited: 10 NOT USED (low confidence) J. Takano, O. Takai, Y. Kogure, and M. Doyama, “Simulation of atomic-scale surface migration in homoepitaxial growth using embedded-atom method potentials for gold,” Thin Solid Films. 1998. link Times cited: 6 NOT USED (low confidence) K. Sugio, Y. Shimomura, and T. D. Rubia, “Computer simulation of displacement damage cascade formation near sigma 5 twist boundary in silver,” Journal of the Physical Society of Japan. 1998. link Times cited: 25 Abstract: A computer simulation of molecular dynamics is carried out t… read moreAbstract: A computer simulation of molecular dynamics is carried out to study the effects of a sigma 5 twist boundary on the formation of displacement damage cascades in silver. When a displacement damage cascade forms near the boundary, interstitial atoms are attracted and segregate on the boundary. The number of Frenkel defects after the formation of damage cascade near the boundary is more than that in a perfect crystal. The segregation of interstitial atoms is due to the interaction with the boundary through the strain field. The movement of interstitial clusters in a strain field is due to a crowdion migration. The larger interstitial clusters are attracted to the boundary at the larger separation. When a damage cascade forms on the boundary, the crystal structure of sigma 5 boundary is modified during the solidification of its molten core. The segregation of interstitial atoms on stacking fault and twin boundary on the formation of damage cascade was also studied. The segregation of interstitial to thes... read less NOT USED (low confidence) R. Schroll, V. Vítek, and P. Gumbsch, “Core properties and motion of dislocations in NiAl,” Acta Materialia. 1998. link Times cited: 58 NOT USED (low confidence) Y. Shen, A. Bilić, D. J. O’connor, and B. King, “Reinvestigation of the surface reconstruction of Cu(001)-(2 × 2)p4g-Pd,” Surface Science. 1997. link Times cited: 18 NOT USED (low confidence) X. Chen, D. Ellis, and G. Olson, “Effects of alloying elements on iron grain boundary cohesion,” MRS Proceedings. 1997. link Times cited: 1 Abstract: For a long time, understanding the mechanisms of impurity-pr… read moreAbstract: For a long time, understanding the mechanisms of impurity-promoted embrittlement in iron and the consequent cohesion(decohesion) effects has been a challenge for materials scientists. The role alloying elements play in impurity-promoted embrittlement is important due to either their direct intergranular cohesion(decohesion) effects or effects upon embrittling potency of other impurities. Some alloying elements like Pd and Mo are known to be helpful for intergranular cohesion in iron and some other alloying elements like Mn are known to segregate to and weaken iron grain boundaries dramatically[1]. There have been intensive investigations on these mechanisms for a long time and especially, with the progress in computing techniques in recent years, calculations on more realistic models have become possible[2–4]. In this paper we briefly present our studies on some selected alloying-element/iron grain boundaries(GB) and free surface(FS) systems. The effects of Pd, Mo, Mn and Cr on the Fe Σ5 (031) grain boundary and its corresponding (031) free surface are examined, using a combination of molecular dynamics(MD) and first-principles electronic structure calculations. Section 2 gives a brief introduction to the methods used and Section 3 gives the main results. read less NOT USED (low confidence) D. Papaconstantopoulos and M. Mehl, “The tight-binding method for interpolating first-principles total energy results,” Journal of Phase Equilibria. 1997. link Times cited: 4 NOT USED (low confidence) J. Akhter and K. Yaldram, “Self Diffusion and Activation Energy of Liquid Palladium,” International Journal of Modern Physics C. 1997. link Times cited: 5 Abstract: Molecular dynamics studies of the temperature dependence of … read moreAbstract: Molecular dynamics studies of the temperature dependence of self diffusion coefficient of palladium has been carried out using the many body potential generated by the Embedded Atom Method of Daw and Baskes. These values as well as the results for activation energy are compared with similar results for other fcc metals. read less NOT USED (low confidence) T. D. Rubia, N. Soneda, M. Caturla, and E. Alonso, “Defect production and annealing kinetics in elemental metals and semiconductors,” Journal of Nuclear Materials. 1997. link Times cited: 39 NOT USED (low confidence) L. Shilkrot and D. Srolovitz, “Anisotropic elastic analysis and atomistic simulation of adatom-adatom interactions on solid surfaces,” Journal of The Mechanics and Physics of Solids. 1997. link Times cited: 11 NOT USED (low confidence) J. Merikoski, I. Vattulainen, J. Heinonen, and T. Ala‐Nissila, “Effect of kinks and concerted diffusion mechanisms on mass transport and growth on stepped metal surfaces,” Surface Science. 1997. link Times cited: 58 NOT USED (low confidence) G. Barrera and R. Tendler, “Simulation of metals and alloys using quasi-harmonic lattice dynamics,” Computer Physics Communications. 1997. link Times cited: 14 NOT USED (low confidence) J. Rittner and D. Seidman, “SOLUTE-ATOM SEGREGATION TO (1 IO) SYMMETRIC TILT GRAIN BOUNDARIES.” 1997. link Times cited: 87 NOT USED (low confidence) S. Eremeev, A. Lipnitskii, A. Potekaev, and E. Chulkov, “Vacancies at low-index surfaces of transition metals and aluminum,” Physics of the Solid State. 1997. link Times cited: 7 NOT USED (low confidence) J. Koplik and J. Banavar, “Continuum Deductions from Molecular Hydrodynamics,” Annual Review of Fluid Mechanics. 1997. link Times cited: 313 Abstract: Since a fluid is composed of molecules, one always has the o… read moreAbstract: Since a fluid is composed of molecules, one always has the option of calculating its static or dynamic properties by computing the motion of these constituents. For most purposes such a procedure is very inefficient, because it provides detailed information at molecular length scales, which are far beneath the usual realm of interest for continuum fluid mechanics. There are, however, situations where the microscopic details of a fluid flow are interesting if not crucial. For example, fluids in microscopic geometries or under high stress may exhibit deviations from the continuum equations, and one may wish to calculate such effects from first principles. Alternatively, in some problems the boundary conditions to be applied to the Navier-Stokes equations are not fully established or are unsatisfactory, as in the presence of moving contact lines or at the edge of a porous read less NOT USED (low confidence) M. Grujicic, S. Lai, and P. Gumbsch, “Atomistic simulation study of the effect of martensitic transformation volume change on crack-tip material evolution and fracture toughness,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1997. link Times cited: 19 NOT USED (low confidence) U. Kürpick and T. Rahman, “Vibrational free energy contribution to self-diffusion on Ni(100), Cu(100) and Ag(100),” Surface Science. 1997. link Times cited: 17 NOT USED (low confidence) U. Kürpick, P. Kürpick, and T. Rahman, “Atomic processes in vacancy island motion on Ag(111),” Surface Science. 1997. link Times cited: 13 NOT USED (low confidence) M. Doyama and Y. Kogure, “Embedded atom potentials in fcc metals,” Radiation Effects and Defects in Solids. 1997. link Times cited: 24 Abstract: A new embedded atom potential has been proposed in this pape… read moreAbstract: A new embedded atom potential has been proposed in this paper. The potential is expressed by simple functions and is applicable to the molecular dynamics simulations of large atomic systems. The potential parameters are determined from the experimental data using the cohesive energy, Born stability, elastic constants, C 11 C 12 and C 44, the formation energy of a vacancy and the stacking fault energy. The potential functions for copper, silver and gold are presented. read less NOT USED (low confidence) Y. Shimomura and R. Nishiguchi, “Vacancy clustering to faulted loop, stacking fault tetrahedron and void in fcc metals,” Radiation Effects and Defects in Solids. 1997. link Times cited: 16 Abstract: An atomistic step of growth to a faulted loop, a stacking fa… read moreAbstract: An atomistic step of growth to a faulted loop, a stacking fault tetrahedron (sft) and a void by clustering of vacancies in fcc metals was studied by molecular dynamics computer simulation with an isotropic EAM potential due to Daw and Baskes [1]. In aluminum, a tri-vacancy relaxes to the Damask-Dienes-Weizer structure (3v-sft). A penta-vacancy relaxes to an octahedral 6v in which an atom is included. The relaxed 5v is stable so that a 6v grows to doubly linked relaxed 5v. This is a critical step of faulted loop formation. By further absorption of vacancy, a cluster grows to an array of relaxed 5vs on a (111) plane and finally collapses to a faulted loop. In gold, a stable structure of vacancy cluster below 15v is a void. A tri-vacancy in gold does not relax to the Damask-Dienes-Weizer structure. Above the size of 6v, partial relaxation of 3v-sft type was observed. The relaxation of a micro-void to a sft is the thermal activated process. In nickel, a void is the most stable cluster below 20v and a... read less NOT USED (low confidence) S. Eremeev, A. Lipnitskii, A. Potekaev, and E. Chulkov, “Activation energy for diffusion of point defects at the surfaces of F.C.C. metals,” Russian Physics Journal. 1997. link Times cited: 12 NOT USED (low confidence) S. Eremeev, A. Lipnitskii, A. Potekaev, and E. Chulkov, “Divacancy binding energy at metal surfaces,” Russian Physics Journal. 1997. link Times cited: 2 NOT USED (low confidence) J. Chladek and G. Betz, “Deposition of Cu atoms on a Pb single crystal surface,” Radiation Effects and Defects in Solids. 1997. link Times cited: 3 Abstract: Surface alloying and the growth of submonolayer films was st… read moreAbstract: Surface alloying and the growth of submonolayer films was studied for the system Cu–Pb using the Molecular Dynamics (MD) technique. Cu atoms at thermal energies were deposited on Pb(100) and Pb(111) surfaces near room temperature. For the Cu–Cu and Pb–Pb interactions different many-body potentials were used. The Cu–Pb interaction was derived from pure element interactions using different models proposed in the literature, such as weighted and simple-arithmetic mean of the dimer potentials. The results of the simulations were compared against each other as well as experimental results from the literature using Scanning Tunnelling Microscopy (STM). Despite the known bulk immiscibility of Pb in Cu, during early stages of growth the following behaviour was observed: at very low deposition stages single Cu atoms move into the Pb surface layer. At higher coverage Cu atoms agglomerate forming islands. However, these islands are partly submerged into the crystal (up to 3 layers deep) and are covered by P... read less NOT USED (low confidence) J. Wang, J. Li, S. Yip, D. Wolf, and S. Phillpot, “Unifying two criteria of Born: Elastic instability and melting of homogeneous crystals,” Physica A-statistical Mechanics and Its Applications. 1997. link Times cited: 51 NOT USED (low confidence) R. Chatterjee and B. Garrison, “Pushing the limits of classical modeling of bombardment events in solids,” Radiation Effects and Defects in Solids. 1997. link Times cited: 5 Abstract: Bombardment of solids with keV atoms leads to violent collis… read moreAbstract: Bombardment of solids with keV atoms leads to violent collisions with subsequent ejection of target particles. This review discusses how classical molecular dynamics simulations designed to describe the bombardment events can give insight into microscopic processes where not only classical but also quantum effects such as electronic excitation and organic reactions play an important role. By incorporating a simple excitation/de-excitation model into the simulation, we have shown that collisional events are important to describe the distribution of excited state atoms measured experimentally. Molecular dynamics simulations employing a reactive many-body potential of small hydrocarbon molecules adsorbed on a metal surface predict the occurrence of various collision induced organic reactions prior to ejection. Lateral motion of particles in the region right above the surface plays an important role in signal enhancement. The calculations predict several processes such as direct molecular ejection, d... read less NOT USED (low confidence) A. Buldum and S. Ciraci, “Interplay between stick-slip motion and structural phase transitions in dry sliding friction,” Physical Review B. 1997. link Times cited: 10 Abstract: Simulations of dry sliding friction between a metal asperity… read moreAbstract: Simulations of dry sliding friction between a metal asperity and an incommensurate metal surface reveal unusual atomic processes. The lateral force exhibits a quasiperiodic variation with the displacement of an asperity; each period consists of two different stick-slip processes involving structural transitions. While one layer of asperity changes and matches the substrate lattice in the first slip, two asperity layers merge into a new one through a structural transition during the second slip. This leads to wear. The lateral force decreases abruptly during these slip stages, but it increases between two consecutive slips and resists the relative motion. The analysis of the order suggests that each structural transition is associated with a first-order phase transition. Nonadiabatic atomic rearrangements during these phase transitions involve a new kind of mechanism of energy dissipation in the dry sliding friction. (cid:64) S0163-1829 (cid:126) 97 (cid:33) 03720-X (cid:35) read less NOT USED (low confidence) I. Sklyadneva, G. Rusina, and E. Chulkov, “Vibrational states on Pd surfaces,” Surface Science. 1997. link Times cited: 10 NOT USED (low confidence) T. Rahman, Z. Tian, and J. Black, “Surface disordering, roughening and premelting of Ag(110),” Surface Science. 1997. link Times cited: 24 NOT USED (low confidence) S. Eremeev, A. Lipnitskii, A. Potekaev, and E. Chulkov, “Vacancies at the surfaces of F.C.C. metals,” Russian Physics Journal. 1997. link Times cited: 3 NOT USED (low confidence) R. A. McCoy and Y. Deng, “Parallel embedded-atom method simulations with delayed electron density approximations,” Computer Physics Communications. 1997. link Times cited: 4 NOT USED (low confidence) D. Farkas, S. J. Zhou, C. Vailhé, B. Mutasa, and J. Panova, “Embedded atom calculations of unstable stacking fault energies and surface energies in intermetallics,” Journal of Materials Research. 1997. link Times cited: 33 Abstract: We performed embedded atom method calculations of surface en… read moreAbstract: We performed embedded atom method calculations of surface energies and unstable stacking fault energies for a series of intermetallics for which interatomic potentials of the embedded atom type have recently been developed. These results were analyzed and applied to the prediction of relative ductility of these materials using the various current theories. Series of alloys with the B2 ordered structure were studied, and the results were compared to those in pure body-centered cubic (bcc) Fe. Ordered compounds with L 1_2 and L 1_0 structures based on the face-centered cubic (fcc) lattice were also studied. It was found that there is a correlation between the values of the antiphase boundary (APB) energies in B2 alloys and their unstable stacking fault energies. Materials with higher APB energies tend to have higher unstable stacking fault energies, leading to an increased tendency to brittle fracture. read less NOT USED (low confidence) T. D. Rubia, “Defect production mechanisms in metals and covalent semiconductors,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1996. link Times cited: 14 NOT USED (low confidence) K. Jacobsen, P. Stoltze, and J. Nørskov, “A semi-empirical effective medium theory for metals and alloys,” Surface Science. 1996. link Times cited: 179 NOT USED (low confidence) G. H. Campbell, D. K. Chan, D. Medlin, J. E. Angelo, and C. B. Carter, “Dynamic observation of the fcc to 9r shear transformation in a copper ∑ = 3 incoherent twin boundary,” Scripta Materialia. 1996. link Times cited: 40 NOT USED (low confidence) F. Zypman and J. Ferrante, “Impurity induced correction to the embedded atom method embedding function,” Physica A-statistical Mechanics and Its Applications. 1996. link Times cited: 1 NOT USED (low confidence) J. Domínguez-Vázquez, E. P. Andribet, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “Molecular dynamics study of the relaxation processes induced by defects in metals,” Surface & Coatings Technology. 1996. link Times cited: 0 NOT USED (low confidence) X. M. Li and Y. Chou, “High angle grain boundary diffusion of chromium in niobium bicrystals,” Acta Materialia. 1996. link Times cited: 21 NOT USED (low confidence) C. M. Gilmore and J. Sprague, “Interface mixing of energetic metals deposited onto metals,” Surface & Coatings Technology. 1996. link Times cited: 6 NOT USED (low confidence) A. Mazzone, “Molecular dynamics simulations of sequential deposition of metallic superlattices,” Applied Physics A. 1996. link Times cited: 2 NOT USED (low confidence) D. Farkas, R. Politano, and I. C. Oppenheim, “Atomistic structure of stepped surfaces,” Surface Science. 1996. link Times cited: 4 NOT USED (low confidence) I. Sklyadneva, E. Chulkov, and A. Bertsch, “Vibrational states on lithium and sodium surfaces,” Surface Science. 1996. link Times cited: 11 NOT USED (low confidence) C. Jayanthi, S. Wu, and J. Cocks, “Dynamics of random overlayers,” Physica B-condensed Matter. 1996. link Times cited: 0 NOT USED (low confidence) K. Tsai, V. Kuznetsov, P. P. Kaminskii, and T. Turkebaev, “Calculation of the thermodynamic properties of the phases and analysis of their stability for the Ni-Al system in the model functional electron density method,” Russian Physics Journal. 1996. link Times cited: 0 NOT USED (low confidence) K. Tsai, V. Kuznetsov, P. P. Kaminskii, T. Turkebaev, and S. A. Zambarnyi, “Multiparticle interatiomic interaction potentials for alloys using the model electron density functional method,” Russian Physics Journal. 1996. link Times cited: 0 NOT USED (low confidence) C. W. Finley and J. Ferrante, “Spectroscopic parameters of the heteronuclear diatomic molecules of Ni, Pd, Pt, Cu, Ag and Au,” Surface and Interface Analysis. 1996. link Times cited: 2 Abstract: The Embedded Atom Method (EAM) is used to calculate the bind… read moreAbstract: The Embedded Atom Method (EAM) is used to calculate the binding energy curves for heteronuclear diatomic molecules of Ni, Pd, Pt, Cu, Ag and Au. The EAM potentials for the homonuclear metals are modified using two-body potentials for the AB alloys developed by R. A. Johnson. The binding energy curves are fit to the Morse potential and the spectroscopic parameters are determined. For the diatomic alloys for which there are experimental data, EAM gives good agreement in binding energy and the equilibrium harmonic frequency and good agreement for the anharmonicity constant. read less NOT USED (low confidence) R. Averback, M. Ghaly, and P. Bellon, “Interfacial effects during ion beam processing of metals,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 1996. link Times cited: 6 NOT USED (low confidence) J. Florêncio, D. Ren, and T. Tsong, “Absolute composition depth-profiles in surface segregation of PtRh alloys,” Surface Science. 1996. link Times cited: 27 NOT USED (low confidence) W. Husinsky and G. Betz, “Fundamental aspects of SNMS for thin film characterization: experimental studies and computer simulations,” Thin Solid Films. 1996. link Times cited: 17 NOT USED (low confidence) B. Kang and K. Sohn, “Barriers for diffusion and interactions with hydrogen in palladium,” Physica B-condensed Matter. 1996. link Times cited: 7 NOT USED (low confidence) P. Gumbsch, “An atomistic study of brittle fracture: Toward explicit failure criteria from atomistic modeling,” Journal of Materials Research. 1995. link Times cited: 122 Abstract: Atomistic techniques are used to study brittle fracture unde… read moreAbstract: Atomistic techniques are used to study brittle fracture under opening mode and mixed mode loading conditions. The influence of the discreteness of the lattice and of the lattice-trapping effect on crack propagation is studied using an embedded atom potential for nickel to describe the crack tip. The recently developed FEAt (Finite Element-Atomistic) coupling scheme provides the atomistic core region with realistic boundary conditions. Several crystallographically distinct crack-tip configurations are studied and commonly reveal that brittle cracks under general mixed mode loading situations follow an energy criterion ( G -criterion) rather than an opening-stress criterion ( K _l-criterion). However, if there are two competing failure modes, they seem to unload each other, which leads to an increase in lattice trapping. Blunted crack tips are studied in the last part of the paper and are compared to the atomically sharp cracks. Depending on the shape of the blunted crack tip, the observed failure modes differ significantly and can drastically disagree with what one would anticipate from a continuum mechanical analysis. read less NOT USED (low confidence) M. Grujicic and J. Du, “Atomistic simulation of transformation toughening in Fe-Ni austenite,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 9 Abstract: The progress of the FCC to BCC martensitic transformation in… read moreAbstract: The progress of the FCC to BCC martensitic transformation in the region surrounding a crack, the morphology of the transformation product, the parent structure-product structure orientation relationship and the accompanying enhancement in toughness in Fe-(20-40) at.% Ni austenite are studied using molecular dynamic simulations. The interactions between the atoms in the two crystal structures are described using the appropriate embedded-atom method type of potential functions. The amount of nickel in the alloys is varied to examine the effect of the thermodynamic stabilities of austenite on the morphology of the crack-tip FCC to BCC transformation and the resulting toughening effect. The results obtained show that the FCC to BCC martensitic transformation is accompanied by a twinning deformation of the transformation product. This deformation gives rise to the relaxation of lattice mismatch stresses built up during the FCC to BCC transformation. The transformation, together with the emission of dislocations from the crack tip, causes the crack tip to blunt and the crack propagation to stop and hence enhances materials toughness. To quantify the effect of the crack-tip FCC to BCC martensitic transformation on the material's toughness, the Eshelby F conservation integral is evaluated. It is found that the component of the F integral in the cracking direction, which represents the force acting to propagate the crack tip, becomes quite small or even negative when the crystal region surrounding the crack undergoes the FCC to BCC transformation. This, in turn, causes the crack propagation to cease. read less NOT USED (low confidence) N. Sonwalkar, S. Yip, and S. Sunder, “A combined molecular dynamics and Raman spectroscopy approach for designing ice-metal interfaces,” Journal of Computer-Aided Materials Design. 1995. link Times cited: 3 NOT USED (low confidence) M. Nomura and J. B. Adams, “Interstitial diffusion along twist grain boundaries in Cu,” Journal of Materials Research. 1995. link Times cited: 14 Abstract: Interstitial diffusion along twist grain boundaries was stud… read moreAbstract: Interstitial diffusion along twist grain boundaries was studied using the Embedded Atom Method (EAM). Six (100) twist grain boundaries (8.79°–43.6°) in copper were investigated. Interstitial formation energies were found to be much lower (0.26-0.78 eV) than in the bulk, and migration energies were found to be comparable (0.01 eV-0.24 eV) to the bulk values (0.09 eV). The vacancy mechanism is favored for low angle boundaries, and the interstitial mechanism is favored for high angle boundaries. The trends in formation energy versus twist angle were partly explained in terms of the volume expansion of the grain boundary. The total diffusion rate due to both mechanisms was calculated, and agreed reasonably well with experimental data for Cu in polycrystalline Cu. The calculated diffusion rate for specific twist boundaries also agreed well with experimental measurements for Zn/Al. read less NOT USED (low confidence) J. Gao, W. Luedtke, and U. Lman, “Nano-Elastohydrodynamics: Structure, Dynamics, and Flow in Nonuniform Lubricated Junctions,” Science. 1995. link Times cited: 63 Abstract: Structure, flow, and response characteristics of molecularly… read moreAbstract: Structure, flow, and response characteristics of molecularly thin films of hexadecane, sheared by topographically nonuniform solid gold surfaces sliding at a relative velocity of 10 meters per second, were investigated with molecular dynamics simulations. The simulations reveal three characteristics: spatial and temporal variations in the density and pressure of the lubricant in the region confined by the approaching asperities, accompanied by asperity-induced molecular layering transitions that are reflected in oscillatory patterns in the friction force; asperity deformations and microstructural transformations mediated by the lubricant; and an onset of cavitated zones in the lubricant after the asperity-asperity collision process. The simulations extend micrometer-scale elastohydrodynamic investigations into the nanometer-scale regime and provide molecular-scale insights into the fundamental mechanisms of ultrathin film lubrication phenomena under extreme conditions. read less NOT USED (low confidence) M. Sambi, E. Pin, and G. Granozzi, “Photoelectron diffraction study of ultrathin film growth of Ni on Pt(111),” Surface Science. 1995. link Times cited: 18 NOT USED (low confidence) R. Smith, R. Najafabadi, and D. Srolovitz, “Segregation to an (Asub 0/2)[1bar 10] edge dislocation in Cusub 0.1Nisub 0.9,” Acta Metallurgica Et Materialia. 1995. link Times cited: 10 NOT USED (low confidence) Q. Sun, J. Xie, and T. Zhang, “Chemisorption of hydrogen on stepped (410) surfaces of Ni and Cu,” Surface Science. 1995. link Times cited: 16 NOT USED (low confidence) H.-C. Huang, N. Ghoniem, J. Wong, and M. Baskes, “Molecular dynamics determination of defect energetics in beta -SiC using three representative empirical potentials,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 102 Abstract: The determination of formation and migration energies of poi… read moreAbstract: The determination of formation and migration energies of point and clustered defects in SiC is of critical importance to a proper understanding of atomic phenomena in a wide range of applications. We present here calculations of formation and migration energies of a number of point and clustered defect configurations. A newly developed set of parameters for the modified embedded-atom method (MEAM) is presented. Detailed molecular dynamics calculations of defect energetics using three representative potentials, namely the Pearson potential, the Tersoff potential and the MEAM, have been performed. Results of the calculations are compared to first-principles calculations and to available experimental data. The results are analysed in terms of developing a consistent empirical interatomic potential and are used to discuss various atomic migration processes. read less NOT USED (low confidence) P. Gumbsch and G. Beltz, “On the continuum versus atomistic descriptions of dislocation nucleation and cleavage in nickel,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 71 Abstract: A hybrid atomistic-finite-element model is compared with the… read moreAbstract: A hybrid atomistic-finite-element model is compared with the continuum-based Peierls-Nabarro model for several crack orientations in a nickel crystal. Both methods incorporate the same embedded-atom potential for Ni, in order to make the comparison as valid as possible. The agreement (expressed in terms of a stability diagram showing envelopes in loading space where fracture or dislocation nucleation are likely to occur) is excellent in the case of a crack lying on a (111) plane, with a crack front running along a (211)-type direction, subject to mixed-mode I-II loadings. That orientation involves dislocation nucleation on the prolongation of the crack plane, and hence no ledge is formed upon dislocation nucleation. In other geometries considered (involving a crack on a (100)-type plane), the agreement seems to get poorer with increasing size of the ledge the is created when a dislocation nucleates. In all geometries, the atomistic model shows that incipient dislocation-like features are present before dislocation nucleation takes place, which serves as additional validation of the continuum Peierls-Nabarro model. read less NOT USED (low confidence) O. H. Nielsen, “Data-Parallel Molecular Dynamics with Neighbor-Lists,” Workshop on Applied Parallel Computin. 1995. link Times cited: 4 NOT USED (low confidence) M. Grujicic and P. Dang, “Molecular dynamics embedded atom method simulations of crack-tip transformation toughening in FeNi austenite,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1995. link Times cited: 14 NOT USED (low confidence) T. Deutsch, P. Bayle, F. Lançon, and J. Thibault, “Computer simulation of Au(001)/Ni multilayers: comparison with experiments,” Journal of Physics: Condensed Matter. 1995. link Times cited: 22 Abstract: The structure of thin Ni film on Au(001) and Au(001)/Ni mult… read moreAbstract: The structure of thin Ni film on Au(001) and Au(001)/Ni multilayers is studied as a function of the thickness of Ni using a semiempirical potential based on the tight-binding second-moment approximation. It is shown that the stable structure is pseudomorphic for a thickness less than five monolayers and (2 1 1 0) HCP or 4H for a greater thickness with close-packed planes perpendicular to the interfaces. Moreover, we show that a thin Ni film grown on Au(001) substrate is covered by at least one monolayer of Au. We compare our results with high-resolution electronic microscopy (HREM) and X-ray diffraction. read less NOT USED (low confidence) T. D. Pope et al., “A structural study of PdCu(100) surface alloys,” Surface Science. 1995. link Times cited: 29 NOT USED (low confidence) M. Spaczér, A. Caro, M. Victoria, and T. D. de la Rubia, “Computer simulations of disordering and amorphization kinetics in intermetallic compounds,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1995. link Times cited: 9 NOT USED (low confidence) G. Betz and W. Husinsky, “Molecular dynamics studies of cluster emission in sputtering,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1995. link Times cited: 63 NOT USED (low confidence) M. Yan, S. P. Chen, T. Mitchell, S. Vyas, and R. Grimes, “Atomistic studies of energies and structures of (hk0) surfaces in NiO,” Philosophical Magazine. 1995. link Times cited: 17 Abstract: In this work we study the energies and structures of (hk0) s… read moreAbstract: In this work we study the energies and structures of (hk0) surfaces in NiO by computer simulation. The short-range interactions are described by Buckingham potentials. The O ion is treated as polarizable by employing the shell model. The coefficients of the potentials and the shell parameters are fitted empirically to properties of the NiO perfect lattice assuming full ionicity. It is shown that discrete atomic structures of (hk0) surfaces can be treated as monatomic steps distributed uniformly or non-uniformly on {100} terraces. The energy of a surface is determined by the density of steps. The energy of the monatomic step is found to be 7·58 × 10 −10Jm−1 before relaxation and 3·32 × 10−10 Jm−1 after relaxation. The elastic interaction between steps is repulsive and increases as the square of the step density. Surface rumpling is found to be a general property, with the anion cores moving outwards from the cations and also outwards from the anion shells, so that the anions are polarized. The pol... read less NOT USED (low confidence) P. Blandin and P. Ballone, “Diffusion of metal adatom on compact metal surfaces in the presence of defects and impurities,” Surface Science. 1995. link Times cited: 4 NOT USED (low confidence) P. Blandin, C. Massobrio, and P. Ballone, “Surface vibrations of ad-clusters on metal surfaces: AgnPt(111),” Surface Science. 1995. link Times cited: 0 NOT USED (low confidence) B. Szpunar, R. Zugic, U. Erb, and L. J. Lewis, “Modelling the influence of structural and compositional disorder on the magnetic properties of grain boundaries,” Canadian Metallurgical Quarterly. 1995. link Times cited: 2 NOT USED (low confidence) A. Goldstein and H. Jónsson, “An embedded atom method potential for the h.c.p. metal Zr,” Philosophical Magazine Part B. 1995. link Times cited: 10 Abstract: The embedded atom method is extended to the h.c.p. metal Zr.… read moreAbstract: The embedded atom method is extended to the h.c.p. metal Zr. The non-ideal c: α ratio and the elastic responses, including contributions from internal degrees of freedom, are incorporated in the fitting procedure: Simple functional forms are assumed for the pair interaction, atomic electron density and embedding function. The functions are parametrized by fitting to experimental data: cohesive energy, equilibrium lattice constants, single crystal elastic constants and vacancy formation energy. An equation of state of the form proposed by Rose, Smith, Guinea and Ferrante is used to reproduce the pressure dependence of the cohesive energy, taking into account the anisotropic elastic response of the crystal. Dimer data and a high energy sputtering potential are also reproduced to extend the range of validity of the potential into regions of very high and low electron density. Good agreement is obtained between the experimental and calculated properties. The potential is applied to the calculation of... read less NOT USED (low confidence) C. D. Silva and C. Scherer, “Ion-induced energy propagating front and migration of point defects in metals,” Solid State Communications. 1995. link Times cited: 1 NOT USED (low confidence) C. Peng, X. Qian, and H. Mei-chun, “CALCULATION OF THERMODYNAMICAL PROPERTIES OF SILVER USING A LATTICE-INVERTED MANY-BODY POTENTIAL,” Chinese Physics Letters. 1995. link Times cited: 1 Abstract: The thermodynamical properties of silver are calculated by u… read moreAbstract: The thermodynamical properties of silver are calculated by using a recent model of many-body potential from lattice inversion method. The predictions of the phonon dispersion relation, the Gruneisen constant and the linear thermal expansion coefficient are all in coincidence with experiments. Of more importance, the present approach represents an efficient way of building potential functions capable of depicting the thermodynamics of metals for the Finnis-Sinclair model. read less NOT USED (low confidence) R. Taylor and B. Garrison, “A microscopic view of particle bombardment of organic films,” International Journal of Mass Spectrometry and Ion Processes. 1995. link Times cited: 11 NOT USED (low confidence) A. Skinner and J. Broughton, “Neural networks in computational materials science: training algorithms,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 70 Abstract: Neural networks can be used in principle in an unbiased way … read moreAbstract: Neural networks can be used in principle in an unbiased way for a multitude of pattern recognition and interpolation problems within computational material science. Reliably finding the weights of large feed-forward neural networks with both accuracy and speed is crucial to their use. In this paper, the rate of convergence of numerous optimization techniques that can be used to determine the weights is compared for two problems related to the construction of atomistic potentials. Techniques considered were back propagation (steepest descent), conjugate gradient methods, real-string genetic algorithms, simulated annealing and a new swarm search algorithm. For small networks, where only a few optimal solutions exist, we find that conjugate-gradient methods are most successful. However, for larger networks where the parameter space to be searched is more complex, a hybrid scheme is most effective; genetic algorithm or simulated annealing to find a good initial starting set of weights, followed by a conjugate-gradient approach to home in on a final solution. These hybrid approaches are now our method of choice for training large networks. read less NOT USED (low confidence) X. Nie et al., “A new embedded-atom potential for metals and its applications,” Solid State Communications. 1995. link Times cited: 1 NOT USED (low confidence) X. Qian and H. Mei-chun, “INVERSE LATTICE PROBLEM AND FINNIS-SINCLAIR MODEL,” Chinese Physics Letters. 1995. link Times cited: 2 Abstract: The lattice inversion method is used to construct the pair p… read moreAbstract: The lattice inversion method is used to construct the pair potential and the hopping integral in Finnis-Sinclair model. In the approach, the lattice sum of square hopping integral is assumed to be an exponential function versus the nearest-neighbour distance, with two parameters determined from the Cauchy discrepancy and the difference of the unrelaxed vacancy-formation energy with the sublimation energy. The individual hopping integral is inverted from the exponential function and the pair potential is inverted from the remaining part of total cohesive energy. read less NOT USED (low confidence) C. L. Rohrer, “Cluster/dislocation interactions in dilute aluminum-based solid solutions,” Journal of Materials Research. 1995. link Times cited: 6 NOT USED (low confidence) M. Mehl, D. Papaconstantopoulos, and R. Cohen, “A tight-binding method for the evaluation of the total energy of large systems,” International Journal of Thermophysics. 1995. link Times cited: 4 NOT USED (low confidence) M. Grujicic and P. Dang, “Assessment of thermodynamic properties of alloys by combining the embedded-atom and the quasiharmonic methods,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 1995. link Times cited: 2 NOT USED (low confidence) B. Kang and K. Sohn, “Diffusion processes and interactions of hydrogen atoms in Pd,” Physica B-condensed Matter. 1995. link Times cited: 3 NOT USED (low confidence) E. Ma and M. Atzmon, “Phase transformations induced by mechanical alloying in binary systems,” Materials Chemistry and Physics. 1995. link Times cited: 117 NOT USED (low confidence) F. Streitz and J. Mintmire, “Electrostatic-based model for alumina surfaces,” Thin Solid Films. 1994. link Times cited: 12 NOT USED (low confidence) Q. Xie and M.-chun Huang, “A LATTICE INVERSION METHOD TO CONSTRUCT THE ALLOY PAIR POTENTIAL FOR THE EMBEDDED-ATOM METHOD,” Journal of Physics: Condensed Matter. 1994. link Times cited: 10 Abstract: The lattice inversion method is used to construct the pair p… read moreAbstract: The lattice inversion method is used to construct the pair potential between a pair of unlike atoms for Cu-Au and Cu-Pd intermetallic compounds within the framework of Johnson's analytical model of the embedded-atom method. Compared with previous treatises, the alloy potential obtained by the present method is based on the Ll2 superstructures Cu3Au and Cu3Pd as references so that the usual assumption that the pair potential between distinct atoms is a function of monatomic pair potentials is cancelled. The alloy potentials from inversion fall in with those from the average schemes of Foiles el al. and Johnson in the short range but show deviation in the long range. The present method is used to solve the considerable disagreements of Johnson's calculations for the dilute-limit heats of solution and the phase stabilities of the intermetallic compounds of palladium with noble metals. While the overall degree of agreement is substantially improved, it is not good in some cases, nor is the phonon spectrum of gold. read less NOT USED (low confidence) Y. Sun and G. Beltz, “Dislocation nucleation from a crack tip: A formulation based on anisotropic elasticity,” Journal of The Mechanics and Physics of Solids. 1994. link Times cited: 60 NOT USED (low confidence) G. Betz and K. Wien, “Energy and angular distributions of sputtered particles,” International Journal of Mass Spectrometry and Ion Processes. 1994. link Times cited: 198 NOT USED (low confidence) Q. Xie and M.-chun Huang, “APPLICATION OF LATTICE INVERSION METHOD TO EMBEDDED-ATOM METHOD,” Physica Status Solidi B-basic Solid State Physics. 1994. link Times cited: 10 Abstract: A model for the embedded-atom method with the use of the lat… read moreAbstract: A model for the embedded-atom method with the use of the lattice inversion method is presented. The lattice sums of electron density and pair potential are assumed to be exponential functions of the lattice parameter. The individual functions of potential and density are inverted from the corresponding lattice sums by using the lattice inversion method. The model parameters are explicitly written by five physical inputs, i.e. the equilibrium lattice constant, the bulk modulus, the Voigt average shear modulus, the sublimation energy, and the unrelaxed vacancy-formation energy. As applications, the 〈100〉 uniaxial stress strain curves in the absence of lateral contraction and shear-mode failure for Cu, Ag, Au, Ni, Pd, and Pt are calculated. The predictions of tensile strengths and failure strains by the present method are found smaller than those by the pair-potential model. The results are in agreement with first-principles calculation of Esposito et al. for Cu and with empirical calculations of Milstein for Ni. read less NOT USED (low confidence) R. Smith and D. Srolovitz, “Simulation of dynamic fracture of an impact-loaded brittle solid,” Modelling and Simulation in Materials Science and Engineering. 1994. link Times cited: 2 Abstract: A new model for simulating dynamic fracture in impact-loaded… read moreAbstract: A new model for simulating dynamic fracture in impact-loaded solids is presented. This model is based upon the traditional molecular dynamics procedure, but accounts for the irreversible nature of the fracture process by deleting the attractive part of the particle interaction potential when the bond between two particles is stretched beyond a critical length. This critical length is determined by comparison with Griffith theory. In the present paper, the model is applied to a two-dimensional homogeneous solid in the absence of microstructure (microstructural effects are treated in a subsequent publication). When the impact zone is much smaller man the size of the sample, or the impact zone is wide and the impact amplitude is large, the first crack forms a finite distance ahead of the impact zone. Static continuum elasticity theory shows that the position of this first crack occurs at the position of the maximum tensile stress. This crack then propagates back to the edges of the impact zone and forward into the sample, thereby creating an X-shaped crack pattern. The tips of the X-shaped crack propagate more slowly than the stress wave and hence strong deviations from this pattern are observed when the stress wave passes the crack tips. When the predominantly compressive stress wave reflects off the back free surface, a tensile wave propagates back into the sample creating even more damage. This damage occurs in bands parallel to and set back from the back surface. read less NOT USED (low confidence) M. Mills, M. Daw, and S. Foiles, “High-resolution transmission electron microscopy studies of dislocation cores in metals and intermetallic compounds,” Ultramicroscopy. 1994. link Times cited: 34 NOT USED (low confidence) M. Shapiro and T. Tombrello, “Molecular dynamics simulations of inelastic energy loss effects in sputtering II,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1994. link Times cited: 12 NOT USED (low confidence) F. Streitz and J. Mintmire, “Electrostatic potentials for metal-oxide surfaces and interfaces.,” Physical review. B, Condensed matter. 1994. link Times cited: 288 Abstract: As most technologically important metals will form oxides re… read moreAbstract: As most technologically important metals will form oxides readily, any complete study of adhesion at real metal surfaces must include the metal-oxide interface. The role of this ubiquitous oxide layer cannot be overlooked, as the adhesive properties of the oxide or oxide-metal system can be expected to differ profoundly from the adhesive properties of a bare metal surface. We report on the development of a computational method for molecular-dynamics simulations, which explicitly includes variable charge transfer between anions and cations. This method is found to be capable of describing the elastic properties, surface energies, and surface relaxation of crystalline metal oxides accurately. We discuss in detail results using this method for \ensuremath{\alpha}-alumina and several of its low-index faces. read less NOT USED (low confidence) U. Bardi, “The atomic structure of alloy surfaces and surface alloys,” Reports on Progress in Physics. 1994. link Times cited: 217 Abstract: This paper reviews the available data about the structure of… read moreAbstract: This paper reviews the available data about the structure of bimetallic surfaces where the constituent elements are intermixed at atomic level. These systems include the surface of bulk alloys ('alloy surfaces') and systems resulting from diffusion at metal-metal interfaces ('surface alloys'). All cases where a complete surface crystallographic determination exists are listed and discussed, as well as selected cases where the structural data are not complete but can lead to a model of the atomic structure. read less NOT USED (low confidence) I. Majid, C. Counterman, R. Najafabadi, and P. Bristowe, “X-ray diffraction and computer simulation studies on the structure of homophase and heterophase interfaces in metals,” Journal of Physics and Chemistry of Solids. 1994. link Times cited: 1 NOT USED (low confidence) F. Reniers, M. Delplancke, A. Asskali, M. Jardinier-Offergeld, and F. Bouillon, “Surface segregation study of Ib-VIII single-crystal alloys,” Applied Surface Science. 1994. link Times cited: 11 NOT USED (low confidence) J. B. Adams et al., “Atomic-level computer simulation,” Journal of Nuclear Materials. 1994. link Times cited: 17 NOT USED (low confidence) D. Hofmann and M. Finnis, “Theoretical and experimental analysis of near Σ3 (211) boundaries in silver,” Acta Metallurgica Et Materialia. 1994. link Times cited: 44 NOT USED (low confidence) D. Bacon and T. D. Rubia, “Molecular dynamics computer simulations of displacement cascades in metals,” Journal of Nuclear Materials. 1994. link Times cited: 226 NOT USED (low confidence) C.-L. Liu, “Energetics of diffusion processes during nucleation and growth for the Cu/Cu(100) system,” Surface Science. 1994. link Times cited: 22 NOT USED (low confidence) M. Spaczér, A. Caro, M. Victoria, and T. D. de la Rubia, “Computer simulation of disordering kinetics in irradiated A3B intermetallic compounds,” Journal of Nuclear Materials. 1994. link Times cited: 8 NOT USED (low confidence) N. Singh and B. Yadav, “Lattice dynamics of transition metals in real space,” Physica B-condensed Matter. 1994. link Times cited: 1 NOT USED (low confidence) L. Zhao, R. Najafabadi, and D. Srolovitz, “Statistical mechanical-atomistic determination of vacancy formation free energies in Cu-Ni alloys,” Philosophical Magazine. 1994. link Times cited: 5 Abstract: Atomistic and statistical mechanical methods are combined to… read moreAbstract: Atomistic and statistical mechanical methods are combined to determine the vacancy formation free energy in binary solid solutions. The first-shell grey model is based on the assumption that all the atoms are effective or mean-field atoms, but with the concentration of the first shell different from the bulk. Comparison of the vacancy formation free energy and the average local concentration profile around the vacancy obtained from the first-shell grey model and the more accurate but much less computationally efficient first-shell black-white model (where the mean-field approximation is not used) suggest that only the composition of the first atomic shell adjacent to the vacancy must be determined. The temperature dependence of the vacancy formation free energy for a Cu-Ni system can be efficiently determined using the first-shell grey atom approximation. Our results show that the vacancy has a strong tendency to form in Cu-rich regions of the Cu-Ni alloy at low temperatures and low Cu bulk conce... read less NOT USED (low confidence) M. Giesen-Scibert and H. Ibach, “On the time structure of tunneling images of steps,” Surface Science. 1994. link Times cited: 54 NOT USED (low confidence) R. Johnson, “Gold on silver(110): an embedded-atom-method study,” Modelling and Simulation in Materials Science and Engineering. 1994. link Times cited: 7 Abstract: Calculations for trends in the energy of various configurati… read moreAbstract: Calculations for trends in the energy of various configurations of small quantities of gold deposited on a silver(110) surface have been carried out using a simple nearest-neighbor embedded-atom-method model. The primary result is that it is energetically preferable for the outermost plane of atoms to be silver rather than gold. Isolated gold atoms on top of a (110)) surface can readily exchange with silver atoms, but pairs of gold atoms are tightly bound and impede interchange. Gold atoms have little interaction with ledges parallel to tight-packed rows on the (110) surface but bind strongly to ledges parallel to (001) and to kinks. Kinetic effects should play a major role in determining the actual configurations found in experiments, with little initial barrier to the interchange of single adatoms with surface atoms on the (110) surface, but with limited interchange with increasing coverage. Clustering of gold on this surface occurs for the same reason that the gold (110) surface reconstructs: the small ratio of the gold (111)-to-(110) surface energies. The primary unique property of gold that lends to all of these effects is the large positive Cauchy pressure, i.e., the large ratio of the bulk modulus to the average shear modulus. This leads to a large curvature in the embedding function of gold so that the energy penalty is greater in gold than in silver to be a given fraction away from its equilibrium electron density. It is also found that, depending on the range of local volume dependence as given by the range of the electron density function used in the model, there may be a significant repulsion between vacancies and free surfaces one or two atomic layers below the surface. read less NOT USED (low confidence) Y. Ouyang and B. Zhang, “Analytic embedded-atom potentials for bcc metals: application to calculating the thermodynamic data of bcc alloys,” Physics Letters A. 1994. link Times cited: 13 NOT USED (low confidence) P. Rous, T. Einstein, and E. Williams, “Theory of surface electromigration on metals: application to self-electromigration on Cu(111),” Surface Science. 1994. link Times cited: 24 NOT USED (low confidence) G. Bozzolo, A. M. Rodríguez, and J. Ferrante, “Multilayer Relaxation and Surface Energies of Metallic Surfaces,” Surface Science. 1994. link Times cited: 4 NOT USED (low confidence) G. Betz, R. Kirchner, W. Husinsky, F. Rüdenauer, and H. Urbassek, “Molecular dynamics study of sputtering of Cu (111) under Ar ion bombardment,” Radiation Effects and Defects in Solids. 1994. link Times cited: 41 Abstract: We have used the molecular dynamics (MD) technique using man… read moreAbstract: We have used the molecular dynamics (MD) technique using many-body interaction potentials to analyse in detail the processes leading to sputter emission, in order to gain a microscopic understanding of low energy bombardment phenomena. Calculations were performed for a Cu (111) single crystal surface bombarded with Ar atoms in the energy range from 10–1000 eV. The results presented for low bombarding energies are mainly concerned with the near sputtering threshold behaviour, yields and depth of origin of sputtered atoms. Furthermore, it is found, that in addition to sputtered atoms, a large number of ad-atoms at the surface are generated during the evolution of the collision cascade. At higher energies the question of cluster emission and especially their energy distribution and angular distribution are addressed. It was found that the energy distributions for the dimers and monomer atoms exhibit a similar dependence on emission energy as has been observed recently also experimentally. For atoms ... read less NOT USED (low confidence) G. Gilmer and C. Roland, “Applications of molecular dynamics methods to low energy ion beams and film deposition processes,” Radiation Effects and Defects in Solids. 1994. link Times cited: 12 Abstract: Molecular dynamics methods are used to model the impingement… read moreAbstract: Molecular dynamics methods are used to model the impingement of low energy ions onto crystalline targets, and the effects of these beams on thin film deposition. Simulations of the deposition of silicon films show that the structure of deposits can often be improved by the use of low energy ion beams instead of the conventional thermal beam. We examine the influence of beam energy on the formation of amorphous or crystalline deposits. The influence of ion beams on surface diffusion rates and the interdiffusion between atomic layers near the surface are also considered. Cluster deposition is treated, and the results suggest that cluster beams would be effective for depositing smooth films of materials that do not wet the substrate. We discuss the use of special purpose computers and signal processing boards to extend the time scales of molecular dynamics simulations. Rapid advances in computer hardware, algorithms, and the development of accurate interatomic potentials are dramatically increasing ... read less NOT USED (low confidence) P. Feibelman, “Diffusion barrier for a Ag adatom on Pt(111),” Surface Science. 1994. link Times cited: 28 NOT USED (low confidence) M. Nomura and J. B. Adams, “Mechanical properties of twist grain boundaries in Cu,” Interface Science. 1994. link Times cited: 1 NOT USED (low confidence) R. Bhatia and B. Garrison, “Electronic distortion in keV particle bombardment,” Journal of Chemical Physics. 1994. link Times cited: 11 Abstract: The angle resolved velocity distributions of excited (4F7/2)… read moreAbstract: The angle resolved velocity distributions of excited (4F7/2) and ground state (4F9/2) Rh atoms ejected from the Rh {100} surface due to keV Ar+ ion bombardment are described with a model that takes into account the local electronic environment. The lifetime of the excitation probability for each excited Rh atom is assumed to depend on the local embedded‐atom method (EAM) density. It is thus possible to distinguish between ejected atoms that experience very little difference in their electronic environments. Although most excited atoms that survive with significantly high excitation probabilities originate from the surface layer, it is not uncommon for an atom beneath the surface to eject from a disrupted environment and end up with a high excitation probability. This model improves upon a previous one, where the lifetime was assumed to vary with the height above the original surface. read less NOT USED (low confidence) J. Kress and A. Voter, “Model description of transition metals using the rotated second moment approximation,” Radiation Effects and Defects in Solids. 1994. link Times cited: 1 Abstract: An interatomic potential is described, the rotated second mo… read moreAbstract: An interatomic potential is described, the rotated second moment approximation (RSMA), which incorporates directional bonding through energy moments evaluated over directional atomic orbitals. When non- directional orbitals are used, RSMA reduces to the standard SMA, and is thus capable of describing metallic systems. A model RSMA potential is demonstrated for 3d transition metals, with only first neighbor shell interactions, which can correctly predict the experimental trend in the relative stability of the fcc and hcp structures. A generalization of RSMA is proposed that is related in the same way that the embedded atom method (EAM) is related to the SMA. A generalized fcc RSMA potential is constructed for which the Cauchy pressure can be made negative by systematically adjusting one parameter. This behavior results from the angular (directional) forces and not from a physically-unreasonable embedding function. 17 refs., 1 fig., 1 tab. read less NOT USED (low confidence) K. Morishita, N. Sekimura, and S. Ishino, “A molecular dynamics study on the collisional and cooling phases of cascade damage,” Radiation Effects and Defects in Solids. 1994. link Times cited: 0 Abstract: Molecular dynamics calculations were performed to study the … read moreAbstract: Molecular dynamics calculations were performed to study the cascade damage evolution initiated from a primary knock-on atom of 250 eV. The thermodynamic characterization of the cascade was investigated by the Morse potential and it is found that during the cooling phase of the cascade local equilibrium was realized. Cascade evolution by the simple embedded atom method (EAM) potential was also calculated and the effects of initial target temperatures on the cascade processes were discussed read less NOT USED (low confidence) T. D. Rubia and M. Guinan, “MD studies of high energy cascades in Cu,” Radiation Effects and Defects in Solids. 1994. link Times cited: 3 Abstract: We have performed molecular dynamics computer simulation stu… read moreAbstract: We have performed molecular dynamics computer simulation studies of 25 keV displacement cascades in Cu at low temperature. At this energy we observe the initial splitting of a cascade into subcascades and show that at low temperatures in metals displacement cascades can lead to the formation of both vacancy and interstitial dislocations loops. We discuss a new mechanism of defect production based on the observation of interstitial prismatic dislocation loop punching from cascades at 10 K. We also show that below the subcascade threshold, atomic mixing in the cascade is recoil-energy dependent. We obtain a mixing efficiency that is proportional to the square root of the recoil energy. read less NOT USED (low confidence) Y. Shimomura, R. Nishiguchi, T. D. Rubia, and M. Guinan, “Structural relaxation of point defect clusters in pure Cu,” Radiation Effects and Defects in Solids. 1994. link Times cited: 0 Abstract: We employ molecular statics and molecular dynamics computer … read moreAbstract: We employ molecular statics and molecular dynamics computer simulation methods to study structural relaxation of small (〈10) vacancy and interstitial clusters in Cu. Vacancy clusters whose sizes are below four do not relax much, but clusters larger than five were found to relax appreciably, often into a stacking fault tetrahedron and an octahedral void. It was found that stacking fault tetrahedra of hexa-vacancies relax to a void. Interstitials were introduced into bcc positions. A di-interstitial was found to relax to two parallel 〈110〉 split interstitials and tri- and tetra-interstitials relaxed to composite clusters of 〈100〉 split interstitials and a bcc interstitial. Penta- and hexa-interstitials form agglomerates of parallel 〈110〉 crowdions whose central portions are not on a single (111) plane. Such structures allow easy motion of these clusters along 〈110〉 directions under stress. Such movement of interstitial clusters has been observed by electron microscopy in neutron-irradiated Au and Cu. read less NOT USED (low confidence) M. Fallis, A. F. Wright, C. Y. Fong, and M. Daw, “Trapping of a diffusing adatom by a substitutional surface defect,” Surface Science. 1994. link Times cited: 6 NOT USED (low confidence) J. Majerus, N. Castellani, and P. Légaré, “Semi-empirical approaches of surface phenomena: Pt and Ni adatoms on Pt(111) and Ni(111),” Surface Science. 1994. link Times cited: 2 NOT USED (low confidence) H. Gades and H. Urbassek, “Surface binding energies of alloys: a many-body approach,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1994. link Times cited: 27 NOT USED (low confidence) R. Averback and M. Ghaly, “MD studies of the interactions of low energy particles and clusters with surfaces,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1994. link Times cited: 41 NOT USED (low confidence) H. Polatoglou and G. Bleris, “Comparison of the constrained and unconstrained Monte-Carlo method: The case of Cu3Au,” Solid State Communications. 1994. link Times cited: 13 NOT USED (low confidence) C. L. Kelchner, D. Halstead, L. S. Perkins, N. M. Wallace, and A. Depristo, “Construction and evaluation of embedding functions,” Surface Science. 1994. link Times cited: 88 NOT USED (low confidence) C. Liu, J. B. Adams, and R. W. Siegel, “Molecular dynamics simulations of consolidation processes during fabrication of nanophase palladium,” Nanostructured Materials. 1994. link Times cited: 19 NOT USED (low confidence) N. Singh and B. Yadav, “The elastic moduli, the volume contribution and the Cauchy ratio ford andf shell metals,” Pramana. 1994. link Times cited: 7 NOT USED (low confidence) P. Alinaghian, S. R. Nlshltani, and D. Pettifor, “Shear constants using angularly dependent bond order potentials,” Philosophical Magazine Part B. 1994. link Times cited: 19 Abstract: Analytic expressions for the shear constants of sp-valent zi… read moreAbstract: Analytic expressions for the shear constants of sp-valent zincblende and f.c.c. structure types are obtained using a first-nearest-neighbour bond order potential. Novel expressions for the tetragonal and trigonal zincblende shear constant C' and C 44 are derived. ImportantlyC' is found to vary as the cube of the bond order. The angular character of the bond order potential is shown to remove the anisotropy constraint of C 44/C' = 2 for f.c.c. lattices within a nearest-neighbour model. read less NOT USED (low confidence) S. Ouannasser, J. Eugène, H. Dreyssé, C. Wolverton, and D. Fontaine, “Study of surface segregation and order in AgPd alloys,” Surface Science. 1994. link Times cited: 7 NOT USED (low confidence) M. Hugenschmidt and C. Beauvais, “Surface morphology after low coverage Pt deposition on Cu(110),” Surface Science. 1994. link Times cited: 6 NOT USED (low confidence) M. Brejnak, J. Kudrnovský, and P. Modrak, “Electronic theory of surface segregation in transition metal alloys,” Surface Science. 1994. link Times cited: 3 NOT USED (low confidence) W. D. Otter, H. Brongersma, and H. Feil, “Molecular dynamics simulations of ion impact on a supported rhodium cluster,” Surface Science. 1994. link Times cited: 7 NOT USED (low confidence) J. M. Cohen, “Long range adatom diffusion mechanism on fcc (100) EAM modeled materials,” Surface Science. 1994. link Times cited: 11 NOT USED (low confidence) H. Polatoglou and G. Bleris, “Constant temperature and pressure monte carlo study of the order-disorder transition of Cu3Au,” Interface Science. 1994. link Times cited: 6 NOT USED (low confidence) M. Shapiro and T. Tombrello, “A molecular dynamics study of Cu dimer sputtering mechanisms,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1994. link Times cited: 31 NOT USED (low confidence) Z. Tian and J. Black, “Phonon spectra and mean square displacements on Cu(11n) vicinal surfaces,” Surface Science. 1994. link Times cited: 15 NOT USED (low confidence) K. Fang and G.-C. Wang, “The rotational disorder of Pb/W(112),” Surface Science. 1994. link Times cited: 0 NOT USED (low confidence) X. Jian-jun, J. Ping, and Z. Kaiming, “Defect effects on H2 dissociative adsorption on the Ni(100) surface,” Journal of Physics: Condensed Matter. 1994. link Times cited: 5 Abstract: The dissociative adsorption of a hydrogen molecule on the ni… read moreAbstract: The dissociative adsorption of a hydrogen molecule on the nickel(100) surface with point defects is investigated using the embedded-atom method (EAM). The potential-energy surfaces (PES) for H2 dissociation on both perfect and imperfect Ni(100) surfaces are presented, based on total-energy calculations. it is clearly shown that as the H2 approaches the Ni(100) surface along the entrance channel, the H-H bond is progressively weakened while the H-metal bonds begin to form; finally the H2 is adsorbed on the surface in the form of two independent H atoms. This dissociation process is affected by the vacancy and impurity atoms existing in the Ni substrate. The activation barriers (Ea) for the dissociation of H2 through various pathways are calculated. The barriers for the dissociation of H2 on the perfect Ni(100) surface are found to be low (about 0.08-0.09 eV. corresponding to different dissociation pathways). The existence of vacancies enhances the dissociation of H2 by lowering the activation barrier height and providing more adsorption sites. However, the impurity atoms (Cu, Pd) can impede the dissociation of H2 on the Ni(100) surface by increasing the activation barrier height. The adsorption heat of H2 chemisorption on the contaminated Ni(100) surface is also calculated. It is found that the effects of impurities on the dissociation of H2 vary with the dissociation pathways and the impurity sites. read less NOT USED (low confidence) W. Xu and J. B. Adams, “Fourth moment approximation to tight binding: application to bcc transition metals,” Surface Science. 1994. link Times cited: 35 NOT USED (low confidence) Q. Xie and M.-chun Huang, “CHEN-MOBIUS INVERSION THEOREM AND A STRUCTURAL REPRESENTATION OF CRYSTALLOGRAPHIC DIRECTION FAMILIES,” Physics Letters A. 1993. link Times cited: 6 NOT USED (low confidence) S. Rao, D. Dimiduk, and M. Mendiratta, “Anisotropic analysis of dislocation line energies and possible dislocation core dissociations in MoSi2,” Philosophical Magazine. 1993. link Times cited: 15 Abstract: The line energy factors of(110)[001], [331], [11],[11];(100)… read moreAbstract: The line energy factors of(110)[001], [331], [11],[11];(100)[010];(103)[331], [010]; and (011)[100], [111] dislocations as well as the elastic interaction forces between two 1/4<331] and two 1/4<111] partials, in MoSi2, are calculated using anisotropic elasticity theory. The line energy factors are found to be relatively large (170–250 GPa) and isotropic, whereas the non-radial interaction forces are found to be a small fraction of the radial forces. The atomic configurations around planar faults on the {110), {013) and {116) planes are analysed using the embedded atom method technique. Results of such calculations are used to energetically rank the possible core dissociations of 1/2<331], 1/2<111], <110], and <100] dislocations on the {110), {013) amd {116) planes in MoSi2. Collectively, these results suggest that the core structures of 1/2<331], 1/2<111], <110] and <100] dislocations are expected to be complicated and non-planar, similar to dislocation cores in b.c.c derivative B2 structures. T... read less NOT USED (low confidence) K. Bohnen and K. Ho, “Structure and dynamics at metal surfaces,” Vacuum. 1993. link Times cited: 76 NOT USED (low confidence) I. Markov, “Recent theoretical developments in epitaxy,” Materials Chemistry and Physics. 1993. link Times cited: 10 NOT USED (low confidence) Y. Shimomura, M. Guinan, and T. D. Rubia, “Atomistics of void formation in irradiated copper,” Journal of Nuclear Materials. 1993. link Times cited: 20 NOT USED (low confidence) A. Wucher, “The mass distribution of sputtered metal clusters: II. Model calculation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1993. link Times cited: 38 NOT USED (low confidence) A. Wucher, M. Wahl, and H. Oechsner, “The mass distribution of sputtered metal clusters. I. Experiment,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1993. link Times cited: 53 NOT USED (low confidence) S. Allameh, S. A. Dregia, and P. Shewmon, “The role of interfacial energy in crystallite reorientation by twinning,” Acta Metallurgica Et Materialia. 1993. link Times cited: 5 NOT USED (low confidence) D. Zhang, G. Rao, and P. Wynblatt, “Simulation of segregation at interphase boundaries in Ni-Ag-Cu alloys,” Modelling and Simulation in Materials Science and Engineering. 1993. link Times cited: 7 Abstract: Monte Carlo simulation, in conjunction with the embedded ato… read moreAbstract: Monte Carlo simulation, in conjunction with the embedded atom method, has been used to model the composition and structure of semicoherent (111) and (001) interphase boundaries separating Ni-rich from Ag-rich phases in ternary Ni-Ag-Cu alloys. Cu is found to segregate strongly at both interfaces, although the interfacial excess of Cu is larger at the (001) than at the (111) interface. The results show that the (001) interphase boundary is unstable and tends to break down into truncated pyramidal facets bounded by (111) and (100) faces. The periodicity of the facets is related to the period of the interfacial dislocations lying in the interface. In contrast, the (111) interphase boundary is stable. Comparison of the behaviour of the (001) interface in the ternary alloy with the binary Ni-Ag alloy implies that the segregation of Cu at the (001) interface tends to stabilize (001) facets relative to (111) facets. This latter behaviour is different from that of the (001) interface in Cu-Ag-Au alloys, where weak segregation of Au at the Cu-Ag interface tends to stabilize (111) facets. read less NOT USED (low confidence) R. C. Nelson, T. Einstein, S. Khare, and P. Rous, “Energies of steps, kinks, and defects on Ag?100? and Ag?111? using the embedded atom method, and some consequences,” Surface Science. 1993. link Times cited: 71 NOT USED (low confidence) C. Liu and J. B. Adams, “Diffusion behavior of single adatoms near and at steps during growth of metallic thin films on Ni surfaces,” Surface Science. 1993. link Times cited: 25 NOT USED (low confidence) Y. Sasajima, S. Taya, and R. Yamamoto, “A computer simulation study of the origin of the supermodulus effect of metallic superlattices,” Journal of Magnetism and Magnetic Materials. 1993. link Times cited: 1 NOT USED (low confidence) R. Najafabadi, D. Srolovitz, E. Ma, and M. Atzmon, “Thermodynamic properties of metastable Ag‐Cu alloys,” Journal of Applied Physics. 1993. link Times cited: 99 Abstract: The enthalpies of formation of metastable fcc Ag‐Cu solid so… read moreAbstract: The enthalpies of formation of metastable fcc Ag‐Cu solid solutions, produced by ball milling of elemental powders, were determined by differential scanning calorimetry. Experimental thermodynamic data for these metastable alloys and for the equilibrium phases are compared with both calculation of phase diagrams (CALPHAD) and atomistic simulation predictions. The atomistic simulations were performed using the free‐energy minimization method (FEMM). The FEMM determination of the equilibrium Ag‐Cu phase diagram and the enthalpy of formation and lattice parameters of the metastable solid solutions are in good agreement with the experimental measurements. CALPHAD calculations made in the same metastable regime, however, significantly overestimate the enthalpy of formation. Thus, the FEMM is a viable alternative approach for the calculation of thermodynamic properties of equilibrium and metastable phases, provided reliable interatomic potentials are available. The FEMM is also capable of determining such properties as the lattice parameter which are not available from CALPHAD calculations. read less NOT USED (low confidence) K. Merkle and D. Wolf, “Quasiperiodic features in the atomic structure of long-period grain boundaries,” Materials Letters. 1993. link Times cited: 8 NOT USED (low confidence) U. Engberg, Y. Li, and G. Wahnström, “Some tests of basic assumptions in transition state theory for hydrogen diffusion in FCC-metals,” Journal of Physics: Condensed Matter. 1993. link Times cited: 3 Abstract: Transition state theory is frequently used to describe inter… read moreAbstract: Transition state theory is frequently used to describe interstitial diffusion in solids. A basic assumption in this theory is that equilibrium statistical mechanics can be used to characterize the different configurations in the transition state, the region in configuration space which acts as a bottleneck for the motion of the interstitial. The authors have performed a detailed test of this assumption for hydrogen diffusion in palladium by combining the molecular dynamics and the Monte Carlo techniques. The study clearly confirms that equilibrium statistical mechanics can be used to characterize the different transition state configurations even though the presence of the hydrogen atom in the transition state strongly influences the fluctuations in the system and despite the fact that the time-scales for the motion of the H atom and the Pd atoms differ considerably. read less NOT USED (low confidence) C. Ko and R. Mclellan, “The thermodynamics of NiCuH solid solutions,” Acta Metallurgica Et Materialia. 1993. link Times cited: 1 NOT USED (low confidence) S. Foiles, “Unexpected relaxation of a Ag layer on Cu(111),” Surface Science. 1993. link Times cited: 21 NOT USED (low confidence) M. Haftel, “Surface reconstruction of platinum and gold and the embedded-atom model.,” Physical review. B, Condensed matter. 1993. link Times cited: 74 Abstract: We investigate the surface reconstructions of various faces … read moreAbstract: We investigate the surface reconstructions of various faces of gold and platinum theoretically with a number of embedded atom potentials. We find that whereas all potentials examined predict equivalent bulk properties, only those with a relatively steep embedding function at surface electron densities predict the experimentally observed buckled quasihexagonal reconstruction for the (100) faces and surface energies in close agreement with experiment. Almost all embedded atom potentials considered predict the observed 2×1 missing row structure for the (110) faces, and some the observed missing row structure for the (311) face of platinum. read less NOT USED (low confidence) A. Wucher and B. Garrison, “Ro-vibrational population of sputtered metal dimers: the influence of unimolecular decomposition,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1993. link Times cited: 6 NOT USED (low confidence) U. Landman, R. Barnett, C. L. Cleveland, and W. Luedtke, “Materials by numbers,” Physica D: Nonlinear Phenomena. 1993. link Times cited: 2 NOT USED (low confidence) J. Idiodi and G. N. Obodi, “The Separable Potential Method and Its Connection with the Embedded‐Atom Method,” Physica Status Solidi B-basic Solid State Physics. 1993. link Times cited: 4 Abstract: A detailed comparison of the separable potential method (SPM… read moreAbstract: A detailed comparison of the separable potential method (SPM), recently proposed by one of us, with the embedded-atom method (EAM) allows a characterization of the embedding function F through a nonlinear differential equation. The complete solutions of the nonlinear differential equation are given, and as a demonstration of the applicability of the method, one of the solutions is employed to calculate various physical quantities for some f. c. c. metals. The applicability of the unified approach discussed here, goes beyond f. c. c. metals. We illustrate this important point by comparing some of our results obtained for b. c. c. metallic vanadium with similar results obtained recently by Adam and Foiles in 1990.
Une comparaison detaillee de la “separable potential method” (SPM) recement propose par l'un de nous, avec la “embedded-atom method” (EAM) permis une caracterisation de la fonction d'incrustration F au moyen d'une equation differentielle non-lineaire. Etant donnes les solutions de l'equations differentielle non-lineaires, et pour demontrer le mode d'application de la methode, une des solutions sert a calculer les differents quantites physiques pour quelques metaux c. f. c. Le mode d'application de la methode unie, dont il est ici question, est au dela des metaux c. f. c. Nous illustrons cette point important avec des comparaisons de nos resultats pour le b. c. c. vanadium metallique avec les resultats similaire recement obtenus par Adam et Foiles. read less NOT USED (low confidence) A. Turnbull, “Modelling of environment assisted cracking,” Corrosion Science. 1993. link Times cited: 160 NOT USED (low confidence) C. Massobrio, “Comment on ‘Structure and diffusion of clusters on Ni surfaces by C.-L. Liu and J.B. Adams,’” Surface Science. 1993. link Times cited: 3 NOT USED (low confidence) G. Bozzolo, B. Good, and J. Ferrante, “Heat of segregation of single substitutional impurities,” Surface Science. 1993. link Times cited: 27 NOT USED (low confidence) H. Stadler, W. Hofer, M. Schmid, and P. Varga, “Embedded-atom method calculations applied to surface segregation of PtNi single crystals,” Surface Science. 1993. link Times cited: 16 NOT USED (low confidence) D. Halstead and A. Depristo, “Thin film growth and the scattering of atoms from surface island defects,” Surface Science. 1993. link Times cited: 15 NOT USED (low confidence) J. Rickman and D. Srolovitz, “A modified-local-harmonic model for solids,” Philosophical Magazine. 1993. link Times cited: 14 Abstract: We present a modified-local-harmonic model for solids that p… read moreAbstract: We present a modified-local-harmonic model for solids that permits quick and accurate calculations of the finite-temperature properties of perfect crystals and defects. The results obtained with this simple modification are more accurate than those obtained by using the local harmonic approximation, and this approach still retains the simplicity inherent in the local-harmonic approximation. The utility of this new approach is demonstrated by a calculation of the free energy of a perfect solid and the formation free energy of an isolated point defect and a comparison of the results with those obtained by using the local-harmonic and quasiharmonic approximations. Finally, we relate both the local-harmonic and the modified-local-harmonic models to a model based on a local vibrational density of states that has been proposed by Sutton. read less NOT USED (low confidence) W. Zhong, Y. Cai, and D. Tománek, “Computer simulation of hydrogen embrittlement in metals,” Nature. 1993. link Times cited: 51 NOT USED (low confidence) P. Blandin, C. Massobrio, and J. Buttet, “Equilibrium properties of Ag clusters on Pt(111),” Zeitschrift für Physik D Atoms, Molecules and Clusters. 1993. link Times cited: 1 NOT USED (low confidence) R. Kalia, S. W. Leeuw, A. Nakano, and P. Vashishta, “Molecular dynamics simu-lations of Coulombic systems on ditributed-memory MIMD machines,” Computer Physics Communications. 1993. link Times cited: 33 NOT USED (low confidence) J. R. Ray and R. J. Wolf, “Monte Carlo simulations at constant chemical potential and pressure,” Journal of Chemical Physics. 1993. link Times cited: 21 Abstract: The usual way of carrying out Monte Carlo simulations of ope… read moreAbstract: The usual way of carrying out Monte Carlo simulations of open systems is by using the grand canonical ensemble. In the grand canonical (TVμ) ensemble a system of fixed volume V is in contact with a temperature reservoir having temperature T, and a particle reservoir having chemical potential μ. In order to obtain values for thermodynamic functions for a given pressure in TVμ Monte Carlo simulations a series of simulations at fixed T and V and different chemical potentials is carried out to determine P(μ), and then an extrapolation to the desired pressure must be carried out. Here we discuss Monte Carlo simulations of open systems in the RPμ ensemble in which an energy R, the chemical potential μ, and the pressure are held fixed during the simulation. Thus each RPμ ensemble simulation replaces a set of TVμ simulations. Simulations of an embedded atom model of liquid palladium are discussed using the new method. read less NOT USED (low confidence) R. Eibler, H. Erschbaumer, C. Temnitschka, R. Podloucky, and A. Freeman, “Ab-initio calculation of the electronic structure and energetics of the unreconstructed Au(001) surface,” Surface Science. 1993. link Times cited: 8 NOT USED (low confidence) M. Brejnak and P. Modrak, “Electronic theory of surface segregation in the coherent potential approximation: The surface segregation in CoNi, IrPt and RhPt,” Surface Science Letters. 1993. link Times cited: 0 NOT USED (low confidence) B. Holian, “Shock waves and spallation by molecular dynamics.” 1993. link Times cited: 7 NOT USED (low confidence) P. Blandin and C. Massobrio, “Diffusion properties and collisional dynamics of Ag adatoms and dimers on Pt(111),” Surface Science. 1992. link Times cited: 11 NOT USED (low confidence) S. Phillpot, “Thermoelastic behavior of grain‐boundary superlattices,” Journal of Applied Physics. 1992. link Times cited: 5 Abstract: The thermal‐expansion coefficients of superlattices of twist… read moreAbstract: The thermal‐expansion coefficients of superlattices of twist grain boundaries on the (100) plane in fcc Cu and Au are calculated using a generalization of the Gruneisen relation. A strong anisotropy in the in‐plane and out‐of‐plane thermal‐expansion coefficients is found. The thermal expansion of these superlattices is found to be determined from a complex interplay of the structural disorder arising from the interfaces with the associated zero‐temperature lattice‐parameter changes. The relationship between the thermal expansion and the elastic properties is explored. read less NOT USED (low confidence) L. Yang, T. Rahman, and J. Black, “Structure and dynamics of an Ag overlayer on Ni(100) : comparison of embedded atom and pair potential results,” Surface Science. 1992. link Times cited: 5 NOT USED (low confidence) A. Sutton, “Direct free energy minimization methods: application to grain boundaries,” Philosophical Transactions of the Royal Society of London. Series A: Physical and Engineering Sciences. 1992. link Times cited: 29 Abstract: A critical review is given of recently developed methods for… read moreAbstract: A critical review is given of recently developed methods for determining the atomic structures and solute concentration profiles at defects in elemental solids and substitutional alloys as a function of temperature. Exact results are given for the effective force on an atom arising from the vibrational entropy in the quasiharmonic approximation and for the occupancy of a site in the pair potential approximation. An improved, approximate formula is given for the effective force arising from the vibrational entropy. The mean field approximation that is used in the alloy problem is compared with the auto-correlation approximation. It is shown that the better statistical averaging of the auto-correlation approximation leads to effective pair interactions that are temperature and concentration dependent. read less NOT USED (low confidence) H. Y. Wang, R. Najafabadi, D. Srolovitz, and R. LeSar, “Segregation effects on intergranular fracture:” Metallurgical Transactions A. 1992. link Times cited: 7 NOT USED (low confidence) S. Phillpot, “Reconstruction of twist grain boundaries in gold,” MRS Proceedings. 1992. link Times cited: 0 Abstract: The reconstruction of high-angle twist grain boundaries on t… read moreAbstract: The reconstruction of high-angle twist grain boundaries on the four densest atomic planes in gold are investigated using the recently developed method of grand-canonical simulated quenching. It is found that the grain boundaries on the two densest planes, (111) and (100), do not reconstruct, while those on the (110) and (113) planes do. read less NOT USED (low confidence) M. Schmid, A. Biedermann, H. Stadler, C. Slama, and P. Varga, “Mismatch dislocations caused by preferential sputtering of a platinum-nickel alloy surface,” Applied Physics A. 1992. link Times cited: 23 NOT USED (low confidence) D. Wolf and J. Jaszczak, “On the interaction between steps in vicinal fcc surfaces : I. Steps along 〈001〉,” Surface Science. 1992. link Times cited: 15 NOT USED (low confidence) D. Wolf and J. Jaszczak, “On the interaction between steps in vicinal fcc surfaces,” Surface Science Letters. 1992. link Times cited: 0 NOT USED (low confidence) W. Luedtke and U. Landman, “Solid and liquid junctions,” Computational Materials Science. 1992. link Times cited: 33 NOT USED (low confidence) P. Bacher, G. Rao, and P. Wynblatt, “Comparisons of the behavior of (111) and (001) interphase boundaries in CuAg and CuAgAu alloys,” Computational Materials Science. 1992. link Times cited: 5 NOT USED (low confidence) D. Lynch, S. Rick, M. A. Gomez, B. W. Spath, J. Doll, and L. Pratt, “Spectroscopic studies of surface and subsurface hydrogen/metal systems,” Journal of Chemical Physics. 1992. link Times cited: 14 Abstract: Recent experiments on the H/Ni(111) system have demonstrated… read moreAbstract: Recent experiments on the H/Ni(111) system have demonstrated that high‐resolution electron‐energy‐loss spectra of subsurface absorbate species can be observed. We report here molecular‐dynamics simulations for both the H/Ni(111) and H/Pd(111) systems. The necessary atomic forces are obtained from embedded atom method (EAM) potentials. From such calculations we have obtained the power spectra and compare our results to the available experimental data. These calculations reasonably reproduce the observed shifts upon embedding the H subsurface and we comment on the possibility of subsurface absorbates interfering with surface adsorbate assignments. Lastly, we illustrate the sensitivity of our results to the parametrization of the EAM potential. read less NOT USED (low confidence) J. R. Smith and D. Srolovitz, “Developing potentials for atomistic simulations,” Modelling and Simulation in Materials Science and Engineering. 1992. link Times cited: 14 Abstract: A small group of researchers met recently to review the new … read moreAbstract: A small group of researchers met recently to review the new and rapidly growing field of many-atom potentials for solids. The workshop was held on 25-27 September 1991, in Ann Arbor, MI, and was commissioned by the Air Force Office of Scientific Research. Some classes of materials are being treated well by many-atom potentials, while others are only now being considered. Combinations of materials including more than one type of bond seem clearly beyond our present capabilities. The systematics of many-atom potential development is in its infancy, and progress appears to be rapid. read less NOT USED (low confidence) M. Stave and A. Depristo, “The structure of NiN and PdN clusters: 4≤N≤23,” Journal of Chemical Physics. 1992. link Times cited: 110 Abstract: Stable geometrical structures of NiN and PdN clusters (N=4–2… read moreAbstract: Stable geometrical structures of NiN and PdN clusters (N=4–23) are identified using a corrected effective medium (CEM) theory. Structural optimization is accomplished by simulated annealing using analytic derivatives to determine the interatomic forces. Unique structural features of these metal clusters are noted, especially in relation to the bulk and surface phases of these metals and to structures commonly associated with rare gas clusters. Elucidation of the general features of cluster growth leads to the principle that transition metal clusters generally maximize the minimum coordination of any atom. By contrast, rare gas clusters maximize the number of interatomic distances close to the optimal distance for the pairwise interaction between rare gas atoms. The latter can be interpreted as the packing of hard balls. Structural transformations between isomers of similar energy are also examined for selected sizes. read less NOT USED (low confidence) A. Miedema, “Energy effects and charge transfer in metal physics; modelling in real space,” Physica B-condensed Matter. 1992. link Times cited: 80 NOT USED (low confidence) K. Morishita, N. Sekimura, T. Toyonaga, and S. Ishino, “Thermodynamical evaluation of cascade damage evolution by molecular dynamics calculation,” Journal of Nuclear Materials. 1992. link Times cited: 5 NOT USED (low confidence) T. D. Rubia and W. Phythian, “Molecular dynamics studies of defect production and clustering in energetic displacement cascades in copper,” Journal of Nuclear Materials. 1992. link Times cited: 46 NOT USED (low confidence) Z. Xi, B. Chakraborty, K. Jacobsen, and J. Nørskov, “An effective-medium theory approach to ordering in Cu-Au alloys,” Journal of Physics: Condensed Matter. 1992. link Times cited: 18 Abstract: The authors present a new approach, based on the effective-m… read moreAbstract: The authors present a new approach, based on the effective-medium theory of metallic cohesion, to the study of alloy phase stability. The efficiency and applicability of the method is demonstrated by studying ordering in Cu-Au alloys. The ground-state properties of the three stoichiometric Cu-Au structures are found to be in excellent agreement with experiment. Monte Carlo simulations have been carried out in an effort to understand the finite-temperature phase transitions. These simulations give a very good description of the complete temperature-concentration phase diagram, and illustrate the importance of going beyond a fixed-lattice Ising model in describing phase transitions in these alloys. read less NOT USED (low confidence) W. G. Hoover and W. G. Hoover, “Nonequilibrium molecular dynamics,” Nuclear Physics. 1992. link Times cited: 2 NOT USED (low confidence) A. Sachdev, R. Masel, and J. B. Adams, “Embedded atom calculations of the equilibrium shape of small platinum clusters,” Journal of Catalysis. 1992. link Times cited: 14 NOT USED (low confidence) S. P. Chen, “Local volume potentials for actinide metals,” Journal of Alloys and Compounds. 1992. link Times cited: 4 NOT USED (low confidence) J. Eckert, J. C. Holzer, C. Krill, and W. L. Johnson, “Structural and thermodynamic properties of nanocrystalline fcc metals prepared by mechanical attrition,” Journal of Materials Research. 1992. link Times cited: 427 Abstract: Nanocrystalline fcc metals have been synthesized by mechanic… read moreAbstract: Nanocrystalline fcc metals have been synthesized by mechanical attrition. The crystal refinement and the development of the microstructure have been investigated in detail by x-ray diffraction, differential scanning calorimetry, and transmission electron microscopy. The deformation process causes a decrease of the grain size of the fcc metals to 6–22 nm for the different elements. The final grain size scales with the melting point and the bulk modulus of the respective metal: the higher the melting point and the bulk modulus, the smaller the final grain size of the powder. Thus, the ultimate grain size achievable by this technique is determined by the competition between the heavy mechanical deformation introduced during milling and the recovery behavior of the metal. X-ray diffraction and thermal analysis of the nanocrystalline powders reveal that the crystal size refinement is accompanied by an increase in atomic-level strain and in the mechanically stored enthalpy in comparison to the undeformed state. The excess stored enthalpies of 10–40% of the heat of fusion exceed by far the values known for conventional deformation processes. The contributions of the atomic-level strain and the excess enthalpy of the grain boundaries to the stored enthalpies are critically assessed. The kinetics of grain growth in the nanocrystalline fcc metals are investigated by thermal analysis. The activation energy for grain boundary migration is derived from a modified Kissinger analysis, and estimates of the grain boundary enthalpy are given. read less NOT USED (low confidence) D. E. Sanders, M. Stave, L. S. Perkins, and A. Depristo, “SCT89: a computer code for atomic and molecular scattering from clean and adsorbate covered surfaces,” Computer Physics Communications. 1992. link Times cited: 12 NOT USED (low confidence) M. S. Singh, “Effective pair potential and structural phase transitions of Cr, Mo, and W.,” Physical review. B, Condensed matter. 1992. link Times cited: 7 Abstract: A fast-converging transition-metal pair potential is used to… read moreAbstract: A fast-converging transition-metal pair potential is used to interpret the structural phase transitions of Cr, Mo, and W under pressure. The bcc structure at equilibrium is found to be most stable for these three metals. The elastic constants, bulk modulus, binding energy, and phonon frequencies of these metals at the observed volume are also calculated. The calculated values and experimental results are found to agree within 15-20%, with a few exceptions read less NOT USED (low confidence) U. Breuer and H. Bonzel, “Morphology of periodic surface profiles on Au single crystals and the anisotropy of the surface free energy of Au,” Surface Science. 1992. link Times cited: 26 NOT USED (low confidence) H. Gades and H. Urbassek, “Pair versus many-body potentials in atomic emission processes from a Cu surface,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1992. link Times cited: 57 NOT USED (low confidence) M. Yoo and C. Fu, “Cleavage fracture of ordered intermetallic alloys,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1992. link Times cited: 47 NOT USED (low confidence) J. Rickman, R. Najafabadi, L. Zhao, and D. Srolovitz, “Finite-temperature properties of perfect crystals and defects from zero-temperature energy minimization,” Journal of Physics: Condensed Matter. 1992. link Times cited: 9 Abstract: Abslmcl. A method for alculating lhe lhermodynamic propcnis … read moreAbstract: Abslmcl. A method for alculating lhe lhermodynamic propcnis of both dassical perfect ayslals and defecu by performing a smgk zero-temperature energy minimization is described. ?lis melhod is based upon the ralNk4iOn of a loel harmonic fref energy as performed by Mar U ol and by Suuon, and invokes determining the dynamical matrix, Gruneisen parameters, and the elastic properties of the system. The dependence of the free energy and the !atlice parameter of a perfecl Au ayslal on lemperature are accuralely determined with this melhod. 'The validity of lhis method is demonstraled by armraleiy determining the temperature dependence of the vacancy formation energy, the acess free energy of a (100) surface, and the excess free energy of a C13 [Oal] (22.62') hvist grain boundary. read less NOT USED (low confidence) Z. C. Li and S. Whang, “Planar defect energies by the embedded atom method and dissociated superdislocation configurations in the L10-type TiAl compound,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1992. link Times cited: 8 NOT USED (low confidence) G. Bozzolo and J. Ferrante, “Lattice parameters of fcc binary alloys using a new semiempirical method,” Scripta Metallurgica Et Materialia. 1992. link Times cited: 9 NOT USED (low confidence) C. M. Gilmore and J. Sprague, “A molecular dynamics analysis of low energy atom-surface interaction during energetic deposition of silver thin films,” Surface & Coatings Technology. 1992. link Times cited: 14 NOT USED (low confidence) S. Mohney and Y. Chang, “Phase equilibria and ternary phase formation in the In–Ni–P system,” Journal of Materials Research. 1992. link Times cited: 11 Abstract: Isothermal sections of the In–Ni–P phase diagram have been d… read moreAbstract: Isothermal sections of the In–Ni–P phase diagram have been determined at 600 °C and 470 °C. Two new ternary phases not previously identified in bulk samples were found. A phase with the composition Ni_57In_22P_21 is present at both 600 °C and 470 °C and was found by differential thermal analysis to melt at 736 °C. The phase Ni_2InP was found to be in equilibrium with InP at 470 °C, but it does not appear in the 600 °C isotherm since it melts at 526 °C. The phases (In), Ni_2P, Ni_5P_4, and NiP_2 are in equilibrium with InP at both temperatures studied. read less NOT USED (low confidence) C. Liu and J. B. Adams, “Diffusion mechanisms on Ni surfaces,” Surface Science. 1992. link Times cited: 48 NOT USED (low confidence) A. Wucher, M. Watgen, C. Möβner, H. Oechsner, and B. Garrison, “Energy dependent studies of anisotropic atomic sputtering of Ni(111),” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1992. link Times cited: 13 NOT USED (low confidence) R. Johnson, “Stability of tight-packed metals with the embedded-atom method,” Journal of Materials Research. 1992. link Times cited: 7 Abstract: Relationships between embedded-atom method parameters and th… read moreAbstract: Relationships between embedded-atom method parameters and the energies of fcc-hcp stability and intrinsic and extrinsic fcc stacking-faults were studied for Cu, Ag, Au, Ni, Pd, and Pt. It was found that the relative magnitudes of these energies for different metals are determined primarily by the physical input data and are almost independent of the cutoff distance and the functions used in the model. These energies increase with increasing vacancy formation energy, decrease with increasing atomic volume and shear modulus, and are almost independent of variations in the cohesive energy and the bulk modulus. However, the shape of the energy versus cutoff distance curve is almost the same for all six metals and is determined primarily by the cutoff distance and the functions used in the model. The shape for a given model is almost independent of the physical input parameters used for fitting to specific metals, can yield either positive or negative values (determined primarily by the cutoff distance), and is similar for all three energies. read less NOT USED (low confidence) U. Landman, W. Luedtke, and E. Ringer, “Atomistic mechanisms of adhesive contact formation and interfacial processes,” Wear. 1992. link Times cited: 114 NOT USED (low confidence) K. Merkle and D. Wolf, “Low-energy configurations of symmetric and asymmetric tilt grain boundaries,” Philosophical Magazine. 1992. link Times cited: 109 Abstract: High-resolution electron-microscopy experiments are combined… read moreAbstract: High-resolution electron-microscopy experiments are combined with computer simulations of tilt grain boundaries (GBs) in Au to investigate the preferred GB planes in ∑ = 9 and ∑ = 11 bicry... read less NOT USED (low confidence) P. Clancy, “Computer Simulation of Crystal Growth and Dissolution in Metals and Semiconductors,” International Journal of High Performance Computing Applications. 1991. link Times cited: 0 Abstract: understanding of the underlying atomic-scale mechanisms resp… read moreAbstract: understanding of the underlying atomic-scale mechanisms responsible for a variety of phenomena concerned with materials processing (for a nontechnical introduction to the subject, see Phillpot, Yip, and Wolf, 1989). For example, atomic-scale simulations using both Monte Carlo and molecular dynamics techniques have been used to emulate processes such as crystal growth of semiconductors (Landman et al., 1988; Broughton and Abraham, 1986; Grabow, Gilmer, and Bakker, 1990), molecular beam epitaxy (Das Sarma, 1990; Srivastava, Garrison, and Brenner, 1989), amorphization (Hsieh and Yip, 1989), and chemical vapor deposition (Brenner, 1990). Molecular Dynamics simulation techniques are a statistical mechanical formulation of appropriate equations of motion for a system of atoms. A semi-empirical model is used to describe the interatomic or intermolecular potential energy function, allowing the forces between particles to be established. For the microcanonical ensemble (constant number of particles, volume, and total energy), Newton’s second law of motion can then be solved, knowing the forces between particles and hence obtaining the accelerations for a system of particles. From these accelerations, the particle velocities and positions as a function of time can thus be established using one of many algorithms developed for the purpose of projecting forward (or backward) in time (Allen and Tildesley, 1987; Heerman, 1986). The solution of these differential equations is repeated for as long as is necessary to capture the event of interest (or as long as one’s computing budget will allow!). A number of &dquo;tricks&dquo; to speed the computation have been devised, such as the use of so-called neighbor lists, or the optimization of the coding for the subroutine which calculates the forces, as well as vectorization read less NOT USED (low confidence) R. Najafabadi, H. Y. Wang, D. Srolovitz, and R. LeSar, “A new method for the simulation of alloys: Application to interfacial segregation,” Acta Metallurgica Et Materialia. 1991. link Times cited: 35 NOT USED (low confidence) I. Robertson, M. Payne, and V. Heine, “Self-consistency in total energy calculations: implications for empirical and semi-empirical schemes,” Journal of Physics: Condensed Matter. 1991. link Times cited: 7 Abstract: The authors assess the viability of nonself-consistent total… read moreAbstract: The authors assess the viability of nonself-consistent total energy calculations using the Harris-Foulkes energy functional. The self-consistent electron density in 11 aluminium structures is resolved into components that are qualitatively similar to the free pseudo-atomic density. For many of the structures the components display significant anisotropies, whilst between the structures there are also important differences. By studying the sensitivity of the Harris-Foulkes energy functional to perturbations in the input electron density, they are able to relate these differences in atomic-like densities to differences in energy. They conclude that no estimate for the electron density based on the superposition of spherical densities can be expected to give errors in the energy of less than 0.03 eV per atom. Given the significant variation in atomic-like electron densities from structure to structure, any transferable density scheme is also prone to energy errors. By constructing a least-squares fit of electron density data to a given functional form, they conclude that the errors in the absolute energies per atom are typically of the order of 0.05 eV whilst for energy differences they drop to 0.01 eV. read less NOT USED (low confidence) J. Idiodi, E. Garba, and O. Akinlade, “Analytic nearest-neighbour model for fcc metals,” Surface Science. 1991. link Times cited: 5 NOT USED (low confidence) C. F. Richardson and P. Clancy, “Picosecond Laser Processing of Copper and Gold,” Molecular Simulation. 1991. link Times cited: 18 Abstract: Non-equilibrium Molecular Dynamics Simulation methods have b… read moreAbstract: Non-equilibrium Molecular Dynamics Simulation methods have been used to study the ability of Embedded Atom Method models of the metals copper and gold to reproduce the equilibrium and non-equilibrium behavior of metals at a stationary and at a moving solid/liquid interface. The equilibrium solid/vapor interface was shown to display a simple termination of the bulk until the temperature of the solid reaches ≊90% of the bulk melting point. At and above such temperatures the systems exhibit a surface disordering known as surface melting. Non-equilibrium simulations emulating the action of a picosecond laser on the metal were performed to determine the regrowth velocity. For copper, the action of a 20 ps laser with an absorbed energy of 2–5 mJ/cm2 produced a regrowth velocity of 83–100 m/s, in reasonable agreement with the value obtained by experient (> 60 m/s). For gold, similar conditions produced a slower regrowth velocity of 63 m/s at an absorbed energy of 5 mJ/c2. This is almost a factor of two ... read less NOT USED (low confidence) M. A. Hoffmann and P. Wynblatt, “Surface composition of ternary cu-ag-au alloys: part ii. a comparison of experiment with theoretical models,” Metallurgical Transactions A. 1991. link Times cited: 6 NOT USED (low confidence) W. Hetterich, C. Höfner, and W. Heiland, “An ion scattering study of the surface structure and thermal vibrations on Ir(110),” Surface Science. 1991. link Times cited: 12 NOT USED (low confidence) Z. Lu, S. Wei, A. Zunger, S. Frota-pessôa, and L. G. Ferreira, “First-principles statistical mechanics of structural stability of intermetallic compounds.,” Physical review. B, Condensed matter. 1991. link Times cited: 188 Abstract: While as elemental solids, Al, Ni, Cu, Rh, Pd, Pt, and Au cr… read moreAbstract: While as elemental solids, Al, Ni, Cu, Rh, Pd, Pt, and Au crystallize in the face-centered-cubic (fcc) structure, at low temperatures, their 50%-50% compounds exhibit a range of structural symmetries: CuAu has the fcc-based L1o structure, CuPt has the rhombohedral L1& structure, and CuPd and A1Ni have the body-centered-cubic B2 structure, while CuRh does not exist (it phase separates into Cu and Rh). Phenomenological approaches attempt to rationalize this type of structural selectivity in terms of classical constructs such as atomic sizes, electronegativities, and electron/atom ratios. More recently, attempts have been made at explaining this type of selectivity in terms of the (quantum-mechanical) electronic structure, e.g. , by contrasting the self-consistently calculated total electron+ion energy of various ordered structures. Such calculations, however, normally select but a small, O(10) subset of "intuitive structures" out of the 2 possible configurations of two types of atoms on a fixed lattice with X sites, searching for the lowest energy. We use instead first-principles calculations of the total energies of O(10) structures to define a multispin Ising Hamiltonian, whose ground-state structures can be systematically searched by using methods of lattice theories. Extending our previous work on semiconductor alloys [S.-H. Wei, L. G. Ferreira, and A. Zunger, Phys. Rev. B 41, 8240 (1990)], this is illustrated here for the intermetallic compounds A1Ni, CuRh, CuPd, CuPt, and CuAu, for which the correct ground states are identified out of -65000 configurations, through the combined use of the densityfunctional formalism (to extract Ising-type interaction energies) with a simple configurational-search strategy (to find ground states). This establishes a direct and systematic link between the electronic structure and phase stability. read less NOT USED (low confidence) B. Dodson, “Chemical effects in sub-keV ion-solid interactions,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1991. link Times cited: 8 NOT USED (low confidence) P. Godowski, “Analysis of surface segregation in Co-Ru alloy using Auger electron spectroscopy,” Applied Surface Science. 1991. link Times cited: 4 NOT USED (low confidence) F. Reniers, M. Jardinier-Offergeld, and F. Bouillon, “Auger electron spectroscopy study of the surface segregation in silver–palladium alloys,” Surface and Interface Analysis. 1991. link Times cited: 11 Abstract: An AES study of the surface composition of silver–palladium … read moreAbstract: An AES study of the surface composition of silver–palladium alloys is described. The influence of sulphur contamination, matrix effects and attenuation coefficients on the quantification of the first layer composition is discussed. The experimental Auger data are treated following the Pons et al. model. A weak silver segregation is observed and the results are compared with theoretical models and other experimental studies. read less NOT USED (low confidence) C. L. Cleveland and U. Landman, “The energetics and structure of nickel clusters: Size dependence,” Journal of Chemical Physics. 1991. link Times cited: 297 Abstract: The energetics of nickel clusters over a broad size range ar… read moreAbstract: The energetics of nickel clusters over a broad size range are explored within the context of the many‐body potentials obtained via the embedded atom method. Unconstrained local minimum energy configurations are found for single crystal clusters consisting of various truncations of the cube or octahedron, with and without (110) faces, as well as some monotwinnings of these. We also examine multitwinned structures such as icosahedra and various truncations of the decahedron, such as those of Ino and Marks. These clusters range in size from 142 to over 5000 atoms. As in most such previous studies, such as those on Lennard‐Jones systems, we find that icosahedral clusters are favored for the smallest cluster sizes and that Marks’ decahedra are favored for intermediate sizes (all our atomic systems larger than about 2300 atoms). Of course very large clusters will be single crystal face‐centered‐cubic (fcc) polyhedra: the onset of optimally stable single‐crystal nickel clusters is estimated to occur at 17 000 at... read less NOT USED (low confidence) D. Wolf, “Surface-stress-induced structure and elastic behavior of thin films,” Applied Physics Letters. 1991. link Times cited: 46 Abstract: Computer simulations of unsupported (111), (001), and (011) … read moreAbstract: Computer simulations of unsupported (111), (001), and (011) thin films of gold, using an embedded‐atom‐method potential, demonstrate a direct correlation between the bulk‐surface stress and the film dimensions. The considerably more complex elastic behavior, by contrast, appears to be dominated by the atomic structure of the film surfaces, and not by the stress‐induced anisotropic lattice parameter changes. read less NOT USED (low confidence) E. C. Sowa, A. Gonis, and X. G. Zhang, “The real-space multiple-scattering theory and the electronic structure of grain boundaries,” MRS Proceedings. 1991. link Times cited: 1 Abstract: We describe the recently developed real-space multiple-scatt… read moreAbstract: We describe the recently developed real-space multiple-scattering theory (RSMST), which is designed for performing first-principles electronic-structure calculations of extended defects, such as surfaces and interfaces including atomic relaxations and with or without impurities, without using artificial periodic boundary conditions. We present the results of non-charge-self-consistent RSMST calculations of the local electronic densities of states at twist and tilt grain boundaries in fcc Cu and bcc Nb, and report on progress towards the implementation of charge self-consistency and total-energy capabilities. read less NOT USED (low confidence) H. Häkkinen, J. Merikoski, and M. Manninen, “Surface reconstruction and many-atom models,” Journal of Physics: Condensed Matter. 1991. link Times cited: 18 Abstract: The (110)(1*2) missing-row reconstruction of the seven FCC m… read moreAbstract: The (110)(1*2) missing-row reconstruction of the seven FCC metals Ni, Pd, Pt, Cu, Ag, Au and Al has been studied using the effective medium theory (EMT). A clear trend in the tendency to reconstruct has been observed when going from the 3d metals Ni and Cu to 5d metals Pt and Au. The results are discussed together with some previous calculations using other many-atom models for total energy calculation in metals. The tendency to undergo reconstruction is found to be related to the anisotropy of surface energies on (111) and (110) surfaces. By investigating the effective two-body and three-body interactions on the surface it is shown that the missing-row reconstruction is related to the effective repulsion between adjacent nearest neighbour rows on the unreconstructed (110) surface. Restriction of the atomic interactions to the nearest neighbours only makes all the recent many-atom models favour the missing-row structure. read less NOT USED (low confidence) H. Rafii-Tabar and A. Sulton, “Long-range Finnis-Sinclair potentials for f.c.c. metallic alloys,” Philosophical Magazine Letters. 1991. link Times cited: 256 Abstract: Many-body, long-range potentials of a Finnis-Sinclair type a… read moreAbstract: Many-body, long-range potentials of a Finnis-Sinclair type are formulated for the atomistic description of binary f.c.c. metallic alloys. These potentials are generalizations of the scheme constructed by Sutton and Chen (1990), for the atomistic modelling of f.c.c. elemental metals. The parameters for the alloy potentials are obtained directly from the parameters for the elemental metals, without any further adjustable fitting. Lattice parameters, elastic constants and enthalpy of formation of 45 binary, random f.c.c. alloys are calculated. read less NOT USED (low confidence) V. Heine, I. Robertson, and M. Payne, “Many-atom interactions in solids,” Philosophical Transactions of the Royal Society of London. Series A: Physical and Engineering Sciences. 1991. link Times cited: 19 Abstract: Computer simulation of complex processes in condensed matter… read moreAbstract: Computer simulation of complex processes in condensed matter comprises a large and broad research effort. These require good models of the interatomic interactions, valid over a wide range of circumstances. In most processes of interest, the crucial atoms are in positions far from standard bonding patterns, at least temporarily: at surfaces and defects, in clusters and in open structures like silicates, the coordination number varies widely. The difficulty of modelling interatomic interactions in such circumstances arises from the existence of strong many-atom forces, originating from the uncertainty principle and the variational principle of quantum mechanics. Some theory of many-atom interactions, and some evidence for them, will be reviewed briefly. In particular a series based on two-, three-, four-atom, etc., interactions is almost certainly not convergent in some cases. In recent years several empirical and semi-empirical, broadly similar approaches to modelling many-atom interactions have come into use, though there are few hard tests of how good they are. An alternative approach, requiring the largest computations possible, involves the Schrödinger equation for the whole simulation to a high relative accuracy. read less NOT USED (low confidence) U. Wolf, S. Foiles, and H. Fischmeister, “Study of misfit dislocations at the interface of weakly bonded metal/metal systems,” Acta Metallurgica Et Materialia. 1991. link Times cited: 16 NOT USED (low confidence) S. P. Chen and A. Voter, “Reconstruction of the (310), (210) and (110) surfaces in fcc metals,” Surface Science. 1991. link Times cited: 25 NOT USED (low confidence) S. Prönnecke, A. Caro, M. Victoria, T. D. Rubia, and M. Guinan, “The effect of electronic energy loss on the dynamics of thermal spikes in Cu,” Journal of Materials Research. 1991. link Times cited: 66 Abstract: We present results of a molecular dynamics simulation study … read moreAbstract: We present results of a molecular dynamics simulation study of the effect of electron-ion interactions on the dynamics of the thermal spike in Cu. Interatomic forces are described with a modified embedded atom method potential. We show that the electron-ion interaction acts to reduce the lifetime of the thermal spike and therefore the amount of atomic rearrangement that takes place in energetic displacement cascades in Cu. The results point toward the important effect that inelastic energy losses might have on the dynamics of displacement cascades in the subcascade energy regime where the lifetime of the thermal spike is expected to exceed the electron-phonon coupling time. read less NOT USED (low confidence) M. Igarashi, M. Khantha, and V. Vítek, “N-body interatomic potentials for hexagonal close-packed metals,” Philosophical Magazine Part B. 1991. link Times cited: 123 Abstract: Finnis-Sinclair (F-S) type many-body potentials have been co… read moreAbstract: Finnis-Sinclair (F-S) type many-body potentials have been constructed for eight hexagonal metals: Co, Zr, Ti, Ru, Hf, Zn, Mg and Be. The potentials are parameterized using cubic splines and fitted to the cohesive energy, unrelaxed vacancy formation energy, five independent second-order elastic constants and two eauilibrium conditions. Hence, each of the constructed potentials represents a stable hexagonal close-packed lattice with a particular non-ideal c/a ratio. In the F-S scheme the many-body part is represented by a sauare root function and this form implies that C12 – C66>0. However, C 12 –C66 is negative for Zn, Be and Ru at Iow temperatures. For this reason a modified many-body function has been employed for these metals. To ensure the applicability of the potentials in modelling of extended lattice defects, the mechanical stability of the corresponding hexagonal close-packed lattice with respect to large homogeneous deformations has been tested. For all the metals considered, the h.c.p. l... read less NOT USED (low confidence) D. Wolf, “Correlation between structure, energy and surface tension for free surfaces in b.c.c. metals,” Philosophical Magazine. 1991. link Times cited: 14 Abstract: A many-body potential of the Finnis-Sinclair type for Mo and… read moreAbstract: A many-body potential of the Finnis-Sinclair type for Mo and a Johnson-type pair potential for α-Fe are used to determine the zero-temperature structures, energies and surface tensions of a variety of free surfaces in b.c.c. metals. The variation in the surface energy and tension as a function of the two variables characterizing the surface normal A is found to be similar for the two potentials, and our results for the principal surfaces of Mo agree with those presented recently by Ackland and Finnis. However, while the pair potential yields an outward relaxation of the surface plane in some cases and an inward relaxation in others, the many-body potential yields an inward relaxation for all surfaces considered. This difference is shown to arise from the very different magnitudes of the surface tensions obtained for the two types of potential which, in turn, are shown to be closely connected with the volume dependence of the related perfect-crystal cohesive energies. For both potentials a strong ... read less NOT USED (low confidence) L. M. Holzman, J. B. Adams, S. Foiles, and W. Hitchon, “Properties of the liquid-vapor interface of fcc metals calculated using the embedded atom method,” Journal of Materials Research. 1991. link Times cited: 18 Abstract: The Embedded Atom Method (EAM) is used to compute density, i… read moreAbstract: The Embedded Atom Method (EAM) is used to compute density, internal energy, and structure factor for bulk liquids of the fcc metals at several temperatures above and below the melting temperature. The calculated values are found to be in generally good agreement with experiment, although the volume expansion upon melting does differ by up to 50% from the expected result for some of the elements studied. The total energy of a liquid system with surfaces is calculated, and the results are compared with the bulk liquid results to determine the enthalpy and thickness of the liquid-vapor interface. Also, the surface tension is found for Cu near the melting temperature. The EAM values for surface enthalpy and surface tension are found to be smaller than experimental values, which is consistent with results for EAM calculations of the surface energy of crystalline solids. read less NOT USED (low confidence) R. Hyland and R. Stiffler, “Determination of the elastic constants of polycrystalline Al3Sc,” Scripta Metallurgica Et Materialia. 1991. link Times cited: 58 NOT USED (low confidence) M. Nomura, S. Lee, and J. B. Adams, “Vacancy diffusion along twist grain boundaries in copper,” Journal of Materials Research. 1991. link Times cited: 47 Abstract: Vacancy diffusion along two different high-angle twist grain… read moreAbstract: Vacancy diffusion along two different high-angle twist grain boundaries (Σ5 and Σ13) was studied using the Embedded Atom Method (EAM). Vacancy formation energies in all the possible sites were calculated and found to be directly related to the degree of coincidence with the neighboring crystal planes. Optimal migration paths and migration energies were determined and found to be very low. The activation energies for self-diffusion at the boundaries were found to be less than half of the bulk value. read less NOT USED (low confidence) I. Majid, D. Wang, and P. Bristowe, “Diffraction effects from (111) twist boundaries in gold,” MRS Proceedings. 1990. link Times cited: 0 Abstract: The structural characteristics of (111) twist boundaries in … read moreAbstract: The structural characteristics of (111) twist boundaries in gold are investigated using a combination of x-ray diffraction and computer modeling techniques. Comparison of the measured scattering effects with those generated from EAM computer models reveals that the (111) boundary displacement field is weak, rotational in form and centered on O' lattice sites. Furthermore, the measured intensities of the strong O' lattice reflections decrease smoothly with increasing boundary angle up to 30{degree}, as calculated from the model. The effect of double positioning on the diffraction pattern and the structural analysis is discussed. 9 refs., 2 figs. read less NOT USED (low confidence) Y. Liu and P. Wynblatt, “COMPUTER SIMULATION OF PHASE TRANSITIONS ASSOCIATED WITH SURFACE MISCIBILITY GAPS,” Surface Science. 1990. link Times cited: 40 NOT USED (low confidence) T. D. de la Rubia and M. Guinan, “Progress in the development of a molecular dynamics code for high-energy cascade studies,” Journal of Nuclear Materials. 1990. link Times cited: 146 NOT USED (low confidence) R. Najafabadi, D. Srolovitz, and R. LeSar, “Finite temperature structure and thermodynamics of the Au Σ5 (001) twist boundary,” Journal of Materials Research. 1990. link Times cited: 29 Abstract: The structure and thermodynamic properties of a Σ5 (001) twi… read moreAbstract: The structure and thermodynamic properties of a Σ5 (001) twist boundary in gold are studied as a function of temperature. This study was performed within the framework of the Local Harmonic (LH) model and employed an Embedded Atom Method (EAM) potential for gold. We find that for the Σ5 (001) twist boundary in gold, a distorted CSL structure is stable at low temperatures, but undergoes a phase transformation to a DSC related structure near room temperature. This transformation is shown to be first order. The temperature dependences of the excess grain boundary free energy, enthalpy, entropy, specific heat, and excess volume are calculated. Discontinuities are observed in the slope of the grain boundary excess free energy (versus temperature), in the value of the grain boundary excess specific heat and excess volume. The stable high temperature grain boundary structure has a smaller excess volume than does the lower temperature structure, and both structures have a coefficient of thermal expansion which is in excess of that for the perfect crystal. read less NOT USED (low confidence) M. A. Hoffmann and P. Wynblatt, “Surface composition of dilute copper-gold alloys,” Surface Science. 1990. link Times cited: 17 NOT USED (low confidence) M. Zoli, “Many-body anharmonic interactions in Ag and Ni,” Philosophical Magazine. 1990. link Times cited: 2 Abstract: A many-body force approach is adopted to calculate the therm… read moreAbstract: A many-body force approach is adopted to calculate the thermoelastic properties of Ag and Ni. The method of homogeneous deformation permits the anharmonic potential parameters to be related to the experimentally known thirdorder elastic constants. Both second- and third-order Cauchy relations are violated by this many-body potential. We have used a strain-dependent perturbative Helmholtz free energy to evaluate the linear thermal expansion coefficient and the thermodynamic Gruneisen parameter of both metals. Good agreement between theory and experiment is found. Our results suggest that, to fit thermodynamical properties, many-body interactions have to be included in the anharmonic part of the potential. read less NOT USED (low confidence) J. K. Norsko, “Chemisorption on metal surfaces,” Reports on Progress in Physics. 1990. link Times cited: 263 Abstract: Presents a coherent summary of the current understanding of … read moreAbstract: Presents a coherent summary of the current understanding of chemisorption phenomena, stressing theoretical concepts and developments rather than experimental techniques. The rapid experimental development of surface science requires a more systematic approach to the explosion of chemisorption data. To this end, the common characteristics of chemisorption systems are first reviewed before the differences are discussed and trends identified, in a search for physical concepts that can systematise the observational data and perhaps make it possible to predict the behaviour of systems that have not yet been studied. The major topics discussed are the adiabatic potential energy surface, the electronic structure problem, the Newns-Anderson model, atomic and molecular chemisorption, and reactions and heterogeneous catalysis. A comprehensive review of experimental results is not attempted within the concept-oriented approach of this study. It is shown that simple models are able to describe semi-quantitatively the chemisorption bond for simple gas atoms, and that there is some understanding of the surface and adsorbate parameters that determine important experimental observables such as the chemisorption energy, bond lengths, and vibrational frequencies. For molecular chemisorption and the dissociation of molecules on metal surfaces the understanding is less well developed, but there is some qualitative understanding of a number of trends. read less NOT USED (low confidence) K. Merkle and D. Wolf, “Structure and Energy of Grain Boundaries in Metals,” MRS Bulletin. 1990. link Times cited: 23 Abstract: The investigation of structure-property correlations is a ra… read moreAbstract: The investigation of structure-property correlations is a rather complex endeavor not only because interfacial Systems are intrinsically inhomogeneous, with chemical composition and physical properties differing from the surrounding bulk material, but also since three different aspects of the geometrical structure are involved — namely the macroscopic, microscopic, and atomic structures. As outlined in the Guest Editors' introduction, in addition to the choice of the materials which form the interface, five macroscopic and three microscopic degrees of freedom (DOFs) are needed to characterize a single bicrystalline interface. The importance of the atomic structure at the interface as well as the local interfacial chemistry, extrinsic (i.e., impurity segregation) or intrinsic (for example, via interfacial reactions or space-charge phenomena), greatly add to the task's complexity. Grain boundaries (GBs) in pure metals represent ideal model Systems for investigating the strictly geometrical aspects of structure-property correlations for the following three reasons. First, the complexity due to the myriad of possible choices of materials combinations forming the interface is avoided, enabling a focus on the different roles of the three distinct geometrical aspects of the structure. Second, because GBs are bulk interfaces, dimensional interface parameters (such as the modulation wavelength in strained-layer superlattices, or the thickness of epitaxial layers) do not enter into the problem. Finally, the GB energy is thought to play a central role in various GB properties, such as impurity segregation, GB mobility and fracture, GB diffusion and cavitation, to name a few. A better understanding of the correlation between the structure and energy of GBs, therefore, promises to offer insights into more complex structure-property correlations, as well. read less NOT USED (low confidence) Y. Gao and K. Merkle, “High-resolution electron microscopy of metal/metal and metal/metal-oxide interfaces in the Ag/Ni and Au/Ni systems,” Journal of Materials Research. 1990. link Times cited: 39 Abstract: The atomic structures of heterophase interfaces with large m… read moreAbstract: The atomic structures of heterophase interfaces with large misfits (>14% in Ag/Ni and Au/Ni) and with small misfits (∼2% in Ag/NiO and Au/NiO) have been studied by high-resolution electron microscopy (HREM). It is found that all interfaces are strongly faceted on (111) planes. This indicates that (111) interfaces have the lowest interfacial energy in both metal/metal and metal/metal-oxide systems. For the metal interfaces, this also agrees with determinations of interfacial energies by lattice statics calculations. The large misfit of Ag/Ni and Au/Ni interfaces is accommodated by misfit dislocations. Observations of misfit localization by HREM are in good agreement with images derived from computer simulation, based on relaxed structures, obtained in embedded atom calculations. All misfit dislocations at the Ag/Ni and Au/Ni interfaces lie exactly in the plane of the interfaces, while the dislocations at Ag/NiO and Au/NiO interfaces reside at a stand-off distance, 3 to 4 (111)_Ag or (111)_Au interplanar spacings from the interfaces. read less NOT USED (low confidence) T. Raeker and A. Depristo, “Corrected effective medium calculations of the chemisorption of H and N on Fe(100), Fe(110) and W(110),” Surface Science. 1990. link Times cited: 33 NOT USED (low confidence) M. Yoo and A. King, “Intergranular fracture by slip/grain boundary interaction,” Metallurgical Transactions A. 1990. link Times cited: 16 NOT USED (low confidence) K. Merkle, “High resolution electron microscopy of interfaces in fcc materials,” Ultramicroscopy. 1990. link Times cited: 74 NOT USED (low confidence) D. Wolf, “Correlation between structure, energy, and ideal cleavage fracture for symmetrical grain boundaries in fcc metals,” Journal of Materials Research. 1990. link Times cited: 87 Abstract: The misorientation phase space for symmetrical grain boundar… read moreAbstract: The misorientation phase space for symmetrical grain boundaries is explored by means of atomistic computer simulations, and the relationship between the tilt and twist boundaries in this three-parameter phase space is clucidated. The so-called random-boundary model (in which the interactions of atoms across the interface are assumed to be entirely random) is further developed to include relaxation of the interplanar spacings away from the grain boundary. This model is shown to include fully relaxed free surfaces naturally, thus permitting a direct comparison of the physical properties of grain boundaries and free surfaces, and hence the determination of ideal cleavage-fracture energies of grain boundaries. An extensive comparison with computer-simulation results for symmetrical tilt and twist boundaries shows that the random-boundary model also provides a good description of the overall structure-energy correlation for both low- and high-angle tilt and twist boundaries. Finally, the role of the interplanar spacing parallel to the grain boundary in both the grain-boundary and cleavage-fracture energies is elucidated. read less NOT USED (low confidence) L. Roelofs, S. Foiles, M. Daw, and M. Baskes, “The (1 × 2) missing-row phase of Au(110): energetics determined from an extended embedded atom method,” Surface Science. 1990. link Times cited: 57 NOT USED (low confidence) A. Trayanov, A. C. Levi, and E. Tosatti, “Anisotropic roughening theory of the (110) faces of Cu, Ni, Pd, and Ag,” Surface Science. 1990. link Times cited: 28 NOT USED (low confidence) D. Brenner and B. Garrison, “Gas‐Surface Reactions: Molecular Dynamics Simulations of Real Systems,” Advances in Chemical Physics. 1990. link Times cited: 8 NOT USED (low confidence) D. Wolf, “Supermodulus effect in metallic superlattices of grain boundaries,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1990. link Times cited: 15 NOT USED (low confidence) J. Mintmire, “Model studies of composition-modulated CuNi superlattices,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1990. link Times cited: 4 NOT USED (low confidence) D. Wolf, “Structure-energy correlation for grain boundaries in F.C.C. metals—III. Symmetrical tilt boundaries,” Acta Metallurgica Et Materialia. 1990. link Times cited: 221 NOT USED (low confidence) D. Wolf and M. Kluge, “Relationship between shear resistance and local atomic structure at grain boundaries in FCC metals,” Scripta Metallurgica Et Materialia. 1990. link Times cited: 25 NOT USED (low confidence) U. Landman, W. Luedtke, N. Burnham, and R. Colton, “Atomistic Mechanisms and Dynamics of Adhesion, Nanoindentation, and Fracture,” Science. 1990. link Times cited: 824 Abstract: Molecular dynamics simulations and atomic force microscopy a… read moreAbstract: Molecular dynamics simulations and atomic force microscopy are used to investigate the atomistic mechanisms of adhesion, contact formation, nanoindentation, separation, and fracture that occur when a nickel tip interacts with a gold surface. The theoretically predicted and experimentally measured hysteresis in the force versus tip-to-sample distance relationship, found upon approach and subsequent separation of the tip from the sample, is related to inelastic deformation of the sample surface characterized by adhesion of gold atoms to the nickel tip and formation of a connective neck of atoms. At small tipsample distances, mechanical instability causes the tip and surface to jump-to-contact, which in turn leads to adhesion-induced wetting of the nickel tip by gold atoms. Subsequent indentation of the substrate results in the onset of plastic deformation of the gold surface. The atomic-scale mechanisms underlying the formation and elongation of a connective neck, which forms upon separation, consist of structural transformations involving elastic and yielding stages. read less NOT USED (low confidence) J. Moriarty and R. Phillips, “First-Principles Interatomic Potentials for Transition Metals and Their Surfaces,” MRS Proceedings. 1990. link Times cited: 0 Abstract: For bulk transition metals, a first-principles generalized p… read moreAbstract: For bulk transition metals, a first-principles generalized pseudopotential theory (GPT) of interatomic potentials has been developed in which the cohesive-energy functional takes form of a volume term plus sums over widely transferable two-, three-, and four-ion potentials. The GPT has been further extended to surfaces by making an internal transformation of this functional to an embedded-atom-like format in which the embedding function is identified as the bulk volume term and the atomic volume is replaced by an average electron density. Applications of the bulk and surface GPT to the calculation of structural, vacancy-formation, and surface energies in Cu and Mo, and to the investigation of surface relaxation and reconstruction in Mo are discussed. 26 refs., 2 figs., 4 tabs. read less NOT USED (low confidence) Y. Gao and K. Merkle, “Atomic Structure of Ag/Ni Interfaces,” MRS Proceedings. 1990. link Times cited: 7 Abstract: While in heterophase systems of small lattice parameter diff… read moreAbstract: While in heterophase systems of small lattice parameter differences, misfit dislocations are often formed at the interface, it is not known, whether and in which form, misfit localization occurs when the misfit is very large. The atomic structure of Ag/Ni interfaces (misfit 14%) was studied by high-resolution electron microscopy (HREM). A special technique was developed to prepare interface specimens suitable for HREM observations. Lattice statics calculations, using embedded-atom potentials, were performed to determine the structure and energies of Ag/Ni interfaces. The lowest interfacial energy was found for the cube-on-cube orientation and (111) interfaces. This is in agreement with the experimental observation, that all interfaces are strongly faceted with (111)Ag/(111)Ni facets. Misfit localization was found by HREM and computer simulation. The HREM observations will be compared to images derived from image simulations, based on model structures obtained from embedded atom calculations. 8 refs., 3 figs. read less NOT USED (low confidence) T. Einstein, M. Daw, and S. Foiles, “Indirect interactions of H/Ni(111) and H/Pd(100) using embedded atom method,” Surface Science. 1990. link Times cited: 26 NOT USED (low confidence) D. Wolf, “Correlation between energy, surface tension and structure of free surfaces in fcc metals,” Surface Science. 1990. link Times cited: 67 NOT USED (low confidence) C. W. Finley and J. Ferrante, “Energetics of nickel and palladium,” Surface and Interface Analysis. 1990. link Times cited: 4 Abstract: We present several model calculations involving nickel and p… read moreAbstract: We present several model calculations involving nickel and palladium using the embedded atom method. Adhesion in the crystalline form of these substances is studied by calculating surface energy as a function of separation of a solid along a crystalline plane, and slip barrier height as a function of displacement. We also investigate a bulk property, Poisson's ratio, as a function of tensile strain. In addition, we examine the equilibrium configuration of metal clusters by finding the minimum energy configuration of clusters consisting of a small number (N = 2–6) of nickel atoms. read less NOT USED (low confidence) M. Brejnak and P. Modrak, “Surface segregation effect for transition-metal alloys in the coherent-potential approximation: general considerations and calculations for Cu-Ni alloys,” Journal of Physics: Condensed Matter. 1990. link Times cited: 7 Abstract: The surface segregation is calculated in the coherent-potent… read moreAbstract: The surface segregation is calculated in the coherent-potential approximation. The influence of the surface potential, d-band fillings and d-level splitting of alloys components on the segregation is examined for a model density of states. The realistic tight-binding Hamiltonian is used to calculate the segregation for Cu-Ni alloys. The model for all reasonable values of parameters predicts the segregation of copper for all alloy compositions. read less NOT USED (low confidence) D. J. Oh and R. Johnson, “Relationship between ratio and point defect properties in HCP metals,” Journal of Nuclear Materials. 1989. link Times cited: 38 NOT USED (low confidence) J. Ferrante, “Applications of surface analysis and surface theory in tribology,” Surface and Interface Analysis. 1989. link Times cited: 7 Abstract: Presentation des applications de l'analyse de surface a… read moreAbstract: Presentation des applications de l'analyse de surface aux etudes de tribologie et de leurs limitations. On insiste sur les etudes fondamentales comportant les effets d'une monocouche ou de couches epaisses sur le forttement et l'usure. Discussion de techniques theoriques nouvelles permettant des calculs simplifies, quantitatifs de l'adherence, de la rupture et du frottement read less NOT USED (low confidence) B. Loisel, D. Gorse, V. Pontikis, and J. Lapujoulade, “Quasidynamic computation of multilayer relaxations, repulsion between steps and kink formation energy on copper vicinal surfaces,” Surface Science. 1989. link Times cited: 48 NOT USED (low confidence) S. Foiles, “Calculation of the atomic structure of the ∑ = 13 (θ = 22.6°) [001] twist boundary in gold,” Acta Metallurgica. 1989. link Times cited: 25 NOT USED (low confidence) D. Wolf, “A read-shockley model for high-angle grain boundaries,” Scripta Metallurgica. 1989. link Times cited: 100 NOT USED (low confidence) G. Tichy and U. Essmann, “Modelling of edge dislocation dipoles in face-centred-cubic lattices,” Philosophical Magazine Part B. 1989. link Times cited: 19 Abstract: Atomistic computer simulations have been carried out to inve… read moreAbstract: Atomistic computer simulations have been carried out to investigate edge dislocation dipole configurations in metals. A non-pair interatomic potential was used and the stress was kept constant as a boundary condition. Narrow edge dislocation dipoles of vacancy and interstitial type show significant differences. Several metastable configurations of vacancy-type dipoles were found. Our results are discussed with respect to the suggested annihilation of narrow edge dipoles in persistent slip bands of fatigued metals. read less NOT USED (low confidence) R. Johnson and D. J. Oh, “Analytic embedded atom method model for bcc metals,” Journal of Materials Research. 1989. link Times cited: 448 Abstract: The requirements for fitting bcc metals within the EAM forma… read moreAbstract: The requirements for fitting bcc metals within the EAM format are discussed and, for comparative purposes, the EAM format is cast in a normalized form. A general embedding function is defined and an analytic first- and second-neighbor model is presented. The parameters in the model are determined from the cohesive energy, the equilibrium lattice constant, the three elastic constants, and the unrelaxed vacancy formation energy. Increasing the elastic constants, increasing the elastic anisotropy ratio, and decreasing the unrelaxed vacancy formation energy favor stability of a close-packed lattice over bcc. A stable bcc lattice relative to close packing is found for nine bcc metals, but this scheme cannot generate a model for Cr because the elastic constants of Cr require a negative curvature of the embedding function. read less NOT USED (low confidence) H. Metiu and A. Depristo, “Surface damage caused by bombardment with low‐energy (10–30 eV) argon,” Journal of Chemical Physics. 1989. link Times cited: 27 Abstract: We apply a recently developed combined molecular dynamics–lo… read moreAbstract: We apply a recently developed combined molecular dynamics–local Langevin equation method to the simulation of the scattering of Ar by the (100) face of a face‐centered cubic solid. The kinetic energies of the Ar are chosen to be low compared to the typical energies used in sputtering. We find that even at low energies, a significant amount of surface damage is inflicted by the Ar, leading to ejection of metal atoms into the gas phase, the formation of dislocations, and the production of isolated atoms trapped on the surface. We study both the probability that such events occur and individual trajectories which display the dynamic processes through which sputtering takes place or defects are created. read less NOT USED (low confidence) M. Lindroos, C. Barnes, M. Valden, and D. King, “A re-examination of multilayer relaxation of Ag(110) by LEED structural analysis,” Surface Science. 1989. link Times cited: 14 NOT USED (low confidence) C. M. Gilmore, J. Sprague, J. Eridon, and V. Provenzano, “An embedded atom analysis of Au clusters on a Ni surface,” Surface Science. 1989. link Times cited: 8 NOT USED (low confidence) J. B. Adams and W. G. Wolfer, “Formation energies of helium-void complexes in nickel,” Journal of Nuclear Materials. 1989. link Times cited: 59 NOT USED (low confidence) Y. Gao, S. A. Dregia, and P. Shewmon, “Energy and structure of (001) twist interphase boundaries in the Ag/Ni system,” Acta Metallurgica. 1989. link Times cited: 38 NOT USED (low confidence) M. Guillopé and B. Legrand, “110) Surface stability in noble metals,” Surface Science. 1989. link Times cited: 134 NOT USED (low confidence) P. Ruppa and C. M. Gilmore, “An embedded atom analysis of Au and Pt substitutional atoms in Ni,” Journal of Materials Research. 1989. link Times cited: 5 Abstract: The Embedded Atom Model (EAM) was utilized to analyze the en… read moreAbstract: The Embedded Atom Model (EAM) was utilized to analyze the energy and strain surrounding Au and Pt atoms substituted into a Ni crystal. Au substituted into Ni has a large positive size difference and a positive heat of mixing; Pt substituted into Ni has a large positive size difference and a negative heat of mixing. The EAM predicted highly anisotropic strain fields around the substitutional atom with large positive strains in the [110] direction and small or negative strains in the [100] and [111] directions. This is in contrast to the normal assumption that the strain around a substitution atom is spherically symmetric. The cohesive energy of the atoms around the substitutional atom was found to depend upon the crystallographic direction relative to the substitutional atom in addition to the radial distance from the substitutional atom. The EAM predicted an energy increase for Au substituted into Ni and an energy decrease for Pt substituted into Ni. The EAM results were compared with an elastic analysis. read less NOT USED (low confidence) S. Myers, P. M. Richards, W. Wampler, and F. Besenbacher, “Ion-beam studies of hydrogen-metal interactions,” Journal of Nuclear Materials. 1989. link Times cited: 154 NOT USED (low confidence) J. K. Strohl and T. S. King, “Monte Carlo simulations of supported bimetallic catalysts,” Journal of Catalysis. 1989. link Times cited: 83 NOT USED (low confidence) D. Wolf, “Are symmetrical tilt boundaries ‘true’ high-angle grain boundaries?,” Scripta Metallurgica. 1989. link Times cited: 8 NOT USED (low confidence) W. G. Wolfer, C. V. Siclen, S. Foiles, and J. B. Adams, “Helium solubility in solid and liquid nickel,” Acta Metallurgica. 1989. link Times cited: 14 NOT USED (low confidence) P. Sigmund et al., “Round Robin computer simulation of ejection probability in sputtering,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1989. link Times cited: 73 NOT USED (low confidence) J. B. Adams, S. Foiles, and W. G. Wolfer, “Self-diffusion and impurity diffusion of fcc metals using the five-frequency model and the Embedded Atom Method,” Journal of Materials Research. 1989. link Times cited: 228 Abstract: The activation energies for self-diffusion of transition met… read moreAbstract: The activation energies for self-diffusion of transition metals (Au, Ag, Cu, Ni, Pd, Pt) have been calculated with the Embedded Atom Method (EAM); the results agree well with available experimental data for both mono-vacancy and di-vacancy mechanisms. The EAM was also used to calculate activation energies for vacancy migration near dilute impurities. These energies determine the atomic jump frequencies of the classic “five-frequency formula,” which yields the diffusion rates of impurities by a mono-vacancy mechanism. These calculations were found to agree fairly well with experiment and with Neumann and Hirschwald's “T_ m ” model. read less NOT USED (low confidence) S. Reindl, A. Aligia, and K. Bennemann, “Electronic theory for adsorbate (N, O) induced reconstruction at the Ni(100) surface,” Surface Science. 1988. link Times cited: 14 NOT USED (low confidence) T. Ning, Q. X. Yu, and Y. Ye, “Multilayer relaxation at the surface of fcc metals: Cu, Ag, Au, Ni, Pd, Pt, Al,” Surface Science. 1988. link Times cited: 87 NOT USED (low confidence) P. Pershan, “STRUCTURE OF SURFACES.” 1988. link Times cited: 11 NOT USED (low confidence) Š. Pick, “On application of an empirical theory to the Si (011) surface,” Czechoslovak Journal of Physics B. 1988. link Times cited: 0 NOT USED (low confidence) A. J. Adams and W. G. Wolfer, “On the diffusion mechanisms of helium in nickel,” Journal of Nuclear Materials. 1988. link Times cited: 36 NOT USED (low confidence) A. Hamelin, L. Stoicoviciu, L. Doubova, and S. Trasatti, “Influence of the crystallographic orientation of the surface on the potential of zero charge of silver electrodes,” Surface Science. 1988. link Times cited: 25 NOT USED (low confidence) M. Finnis, A. Paxton, D. Pettifor, A. Sutton, and Y. Ohta, “Interatomic forces in transition metals,” Philosophical Magazine. 1988. link Times cited: 45 Abstract: This article reviews some recent progress in the derivation … read moreAbstract: This article reviews some recent progress in the derivation of physical models for calculating the behaviour of defects in transition metals by atomistic simulation. It has long been recognised that it is necessary to go beyond the assumption of pair-wise interactions, but this has only recently been achieved with the advent of simplified models which take account of the electron gas. For d-band metals, the tight-binding model is a useful picture and it can be applied at various levels of approximation. The simplest of these is the second-moment approximation, which is very rapid to compute. Other schemes, such as the embedded-atom method, are also very practical for large scale simulations. These schemes will be described and compared in terms of their physical basis. read less NOT USED (low confidence) D. J. Oh and R. Johnson, “Simple embedded atom method model for fcc and hcp metals,” Journal of Materials Research. 1988. link Times cited: 349 Abstract: A procedure based on the embedded atom method (EAM) is prese… read moreAbstract: A procedure based on the embedded atom method (EAM) is presented for developing atomistic models for use in computer simulation calculations, with an emphasis on simple but general schemes for matching experimental data with fitting parameters. Both the electron density function and the two-body potential are taken as exponentially decreasing functions and the model is derived for any choice of cutoff distance. The model has been applied successfully to seven fcc and three hcp metals, but the extension to bcc metals was unsuccessful because of difficulty in matching the shear anisotropy ratio. read less NOT USED (low confidence) Garrison et al., “Many-body embedded-atom potential for describing the energy and angular distributions of Rh atoms desorbed from ion-bombarded Rh111,” Physical review. B, Condensed matter. 1988. link Times cited: 90 Abstract: In this paper, we show that many-body interactions are impor… read moreAbstract: In this paper, we show that many-body interactions are important for describing the energy- and angle-resolved distributions of neutral Rh atoms ejected from keV-ion-bombarded Rh{111}. We compare separate classical-dynamics simulations of the sputtering process assuming either a many-body potential or a pairwise additive potential. The many-body potential is constructed using the embedded-atom method to describe equilibrium properties of the crystal, parameters from the Moliere potential to describe close encounters between energized atoms, and parameters from a Rh2 potential to aid the description of the desorption event. The most dramatic difference between the many-body potential and the pair potential is in the predicted kinetic energy distributions. The pair-potential kinetic energy distribution peaks at ∼2 eV, whereas the many-body potential predicts a broader peak at ∼4 eV, giving much better agreement with experiment. This difference between the model potentials is due to the predicted nature of the attractive interaction in the surface region through which all ejecting particles pass. Variations of the many-body-potential parameters are examined in order to ascertain their effect on the predicted energy and angular distributions. A specific set of parameters has been found which leads to excellent agreement with recent experimental trajectory measurements of desorbed Rh atoms. read less NOT USED (low confidence) T. Matsumiya, “Current Movements in Molecular Dynamics Study with Regard to Its Application to Materials Science and Engineering,” Tetsu To Hagane-journal of The Iron and Steel Institute of Japan. 1988. link Times cited: 3 NOT USED (low confidence) R. Johnson, “Analytic nearest-neighbor model for fcc metals.,” Physical review. B, Condensed matter. 1988. link Times cited: 559 Abstract: The implications of the mathematical format of the embedded-… read moreAbstract: The implications of the mathematical format of the embedded-atom method of computer modeling of metals have been studied with use of a simple nearest-neighbor analytic model for the fcc lattice. The physical inputs into the model are the atomic volume, the cohesive energy, the bulk modulus, the average shear modulus, the vacancy-formation energy, and the slope at the nearest-neighbor distance of the spherically averaged free-atom electron density calculated with Hartree-Fock theory. The model employs an exponential repulsion between nearest-neighboring atoms, an exponentially decreasing function for the free-atom electron density, and a universal equation relating the crystal energy and the lattice constant. The anisotropy ratio of the cubic shear moduli is constrained to be 2 with this model. The dependence of the energies for unrelaxed configurations for vacancy formation, divacancy binding, and low-index plane surfaces on the model parameters has been analyzed. The average shear modulus plays a dominant role in determining these energies relative to the bulk modulus or the cohesive energy because the slope of the embedding function at the equilibrium electron density is linear in the average shear modulus. Embedding functions are not uniquely determined in specific models, and it is shown that the embedding functions used inmore » several models are essentially equivalent.« less read less NOT USED (low confidence) M. Baskes, M. Daw, B. Dodson, and S. Foiles, “Atomic-Scale Simulation in Materials Science,” MRS Bulletin. 1988. link Times cited: 26 Abstract: The potential of applying atomistic simulations in applied m… read moreAbstract: The potential of applying atomistic simulations in applied material science has been established by recent efforts such as those outlined above. Continued development of interatomic potentials suitable for wider classes of materials should result in a tool capable of strong interaction with experiment and of guiding future technological advances. read less NOT USED (low confidence) G. Ackland, G. Tichy, V. Vítek, and M. Finnis, “Simple N-body potentials for the noble metals and nickel,” Philosophical Magazine. 1987. link Times cited: 510 Abstract: Using the approach of Finnis and Sinclair, N-body potentials… read moreAbstract: Using the approach of Finnis and Sinclair, N-body potentials for copper, silver, gold and nickel have been constructed. The total energy is regarded as consisting of a pair-potential part and a many body cohesive part. Both these parts are functions of the atomic separations only and are represented by cubic splines, fitted to various bulk properties. For the noble metals, the pair-potentials were fitted at short range to pressure-volume relationships calculated by Christensen and Heine so that interactions at separations smaller than that of the first-nearest neighbours can be treated in this scheme. Using these potentials, point defects, surfaces (including the surface reconstructions) and grain boundaries have been studied and satisfactory agreement with available experimental data has been found. read less NOT USED (low confidence) S. Foiles, “Ordered surface phases of Au on Cu,” Surface Science. 1987. link Times cited: 91 NOT USED (low confidence) S. Foiles, “Reconstruction of fcc (110) surfaces,” Surface Science. 1987. link Times cited: 82 NOT USED (low confidence) M. Garofalo, E. Tosatti, and F. Ercolessi, “Structure, energetics, and low-temperature behaviour of the Au(110) reconstructed surface,” Surface Science. 1987. link Times cited: 69 NOT USED (low confidence) B. W. Dodson, “Structural energetics of thin coherently strained metallic overlayers,” Surface Science. 1987. link Times cited: 9 NOT USED (low confidence) R. Rebonato, D. Welch, R. Hatcher, and J. Bilello, “A modification of the Finnis-Sinclair potentials for highly deformed and defective transition metals,” Philosophical Magazine. 1987. link Times cited: 44 Abstract: A modification is introduced to the original Finnis-Sinclair… read moreAbstract: A modification is introduced to the original Finnis-Sinclair potentials for transition metals which substantially improves their pressure versus volume behaviour and overcomes their high-compression instability. With this modification good results are obtained for the energetics instability. With this modification good results are obtained for the energetics and the stable configuration of vacancies and interstitials. Excellent agreement is also found between the lattice parameter dependence of the cohesive energy of Mo as obtained using the modified potentials and via self-consistent augmented-spherical-wave calculations. With the proposed modification, the repulsive component of the modified potentials turns out to be akin to a Wedepohl potential. read less NOT USED (low confidence) S. Foiles, M. Baskes, C. Melius, and M. Daw, “Calculation of hydrogen dissociation pathways on nickel using the embedded atom method,” Journal of The Less Common Metals. 1987. link Times cited: 38 NOT USED (low confidence) B. Garrison, N. Winograd, D. Lo, T. Tombrello, M. Shapiro, and D. E. Harrison, “Energy cost to sputter an atom from a surface in keV ion bombardment processes,” Surface Science. 1987. link Times cited: 39 NOT USED (low confidence) S. Foiles and M. Daw, “Application of the embedded atom method to Ni_3Al,” Journal of Materials Research. 1987. link Times cited: 288 Abstract: The embedded atom method [M. S. Daw and M. I. Baskes, Phys. … read moreAbstract: The embedded atom method [M. S. Daw and M. I. Baskes, Phys. Rev. B 29 , 6443 (1984) used to calculate phase stability, lattice vibrational frequencies, point defect properties, antiphase boundary energies, and surface energies and relaxations for Ni_3Al. The empirical embedding functions and core-core repulsions used by this method are obtained. The equilibrium phases for the Ni-rich half of the composition range of Ni–Al are determined for 1000 K and compared with experiment. The elastic constants and vibrational modes of Ni_3Al are calculated and the elastic constants are compared with experiment. The formation energy, formation volume, and migration energies of vacancies are computed, and it is found that the formation energy of vacancies on the Ni sublattice is less than that on the Al sublattice. The (100) antiphase boundary is shown to be significantly lower in energy than the (111) antiphase boundary. The surface energies and atomic relaxations of the low index faces are computed, and it is shown that for the (100) and (110) faces that the preferred surface geometry corresponds to the bulk lattice with the mixed composition plane exposed. read less NOT USED (low confidence) Y. Xu and Y. Zhou, “Collaborative mechanisms boost the nanoscale boiling heat transfer at functionalized gold surfaces,” International Journal of Heat and Mass Transfer. 2023. link Times cited: 0 NOT USED (low confidence) X. Du et al., “Machine-learning-accelerated simulations enable heuristic-free surface reconstruction,” ArXiv. 2023. link Times cited: 2 Abstract: Understanding material surfaces and interfaces is vital in a… read moreAbstract: Understanding material surfaces and interfaces is vital in applications like catalysis or electronics. Ab initio simulations, combining energies from electronic structure with statistical mechanics, can, in principle, predict the structure of material surfaces as a function of thermodynamic variables. However, accurate energy simulations are prohibitive when coupled to the vast phase space that must be statistically sampled. Here, we present a bi-faceted computational loop to predict surface phase diagrams of multi-component materials that accelerates both the energy scoring and statistical sampling methods. Fast, scalable, and data-efficient machine learning interatomic potentials are trained on high-throughput density-functional theory calculations through closed-loop active learning. Markov-chain Monte Carlo sampling in the semi-grand canonical ensemble is enabled by using virtual surface sites. The predicted surfaces for GaN(0001) and SrTiO 3 (001) are in agreement with past work and suggest that the proposed strategy can model complex material surfaces and discover previously unreported surface terminations. read less NOT USED (low confidence) L. Zhou, J. Zhu, Y. Zhao, and H. Ma, “A molecular dynamics study on thermal conductivity enhancement mechanism of nanofluids – Effect of nanoparticle aggregation,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 26 NOT USED (low confidence) X. Wei, C.-M. Wu, and Y. Li, “Characterizing on the interfacial thermal transport through adsorption clusters and vibrational behaviors,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 6 NOT USED (low confidence) G. Wang, Y. Xu, P. Qian, and Y. Su, “ADP potential for the Au-Rh system and its application in element segregation of nanoparticles,” Computational Materials Science. 2021. link Times cited: 6 NOT USED (low confidence) H. Parsapour, S. Ajori, and R. Ansari, “A molecular dynamics study on the tensile characteristics of various metallic glass nanocomposites reinforced by Weyl semimetals three-dimensional graphene network,” European Journal of Mechanics A-solids. 2021. link Times cited: 7 NOT USED (low confidence) M. Saffarini, G. Voyiadjis, C. Ruestes, and M. Yaghoobi, “Ligament size dependency of strain hardening and ductility in nanoporous gold,” Computational Materials Science. 2021. link Times cited: 14 NOT USED (low confidence) Z. Yan, H. Sheng, E. Ma, B. Xu, J. Li, and L. Kong, “Intermediate structural evolution preceding growing BCC crystal interface in deeply undercooled monatomic metallic liquids,” Acta Materialia. 2021. link Times cited: 6 NOT USED (low confidence) J. Kim, J. W. Lim, J. K. Kim, D. H. Kim, E. Park, and H. Chang, “Suppressed radiation-induced dynamic recrystallization in CrFeCoNiCu high-entropy alloy,” Scripta Materialia. 2021. link Times cited: 5 NOT USED (low confidence) A. Panda et al., “Molecular dynamics studies on formation of stacking fault tetrahedra in FCC metals,” Computational Materials Science. 2021. link Times cited: 11 NOT USED (low confidence) J. Fatermans, A. Backer, A. Dekker, and S. Aert, “Atom column detection.” 2021. link Times cited: 2 NOT USED (low confidence) F. C. Maier, M. Dou, and M. Fyta, “Dynamics and Solvent Effects in Functionalized DNA Sensing Nanogaps.” 2021. link Times cited: 0 NOT USED (low confidence) A. Marusczyk, S. Ramakers, M. Kappeler, P. Haremski, M. Wieler, and P. Lupetin, “Atomistic Simulation of Nickel Surface and Interface Properties.” 2021. link Times cited: 0 NOT USED (low confidence) E. E. koraychy, M. Meddad, M. Badawi, and M. Mazroui, “Sintering and deposition of homo- and heteronanoparticles of aluminum and nickel on aluminum (100) substrate,” Chemical Physics. 2021. link Times cited: 7 NOT USED (low confidence) F. Cassin, “Crystallographic influences on the nanomanipulation of gold nanoclusters on molybdenum disulfide.” 2021. link Times cited: 0 Abstract: This work investigates the manipulation behavior of thermall… read moreAbstract: This work investigates the manipulation behavior of thermally deposited gold nanoclusters with tens of nanometers in size on monocrystalline Molybdenum Disulfide (MoS2) surfaces. Using scan raster patterns in the order of several m, dozens of Au islands can be displaced with a single scan, revealing a directional locking effect caused by the epitaxial nature of the nanoparticle growth on the MoS2 surface. Statistical analysis of tapping mode manipulation scans using pyramidal and conical AFM tips along with MD simulations lead to the conclusion that frictional anitrosopy governs the direction of displacement, with the preference to move along the zigzag- or armchair direction of the hexagonally structured surface. It further investigates the manipulation behavior on CVD grown mono- and bilayer MoS2 with the goal of formation of gold nanowires. For this several nanomanipulation and nanoscratching techniques are deployed to exploit the unique movement behavior of gold islands on a crystalline surface. read less NOT USED (low confidence) P. Chen et al., “Simulation Science: Second International Workshop, SimScience 2019, Clausthal-Zellerfeld, May 8-10, 2019, Revised Selected Papers,” Simulation Science. 2020. link Times cited: 0 NOT USED (low confidence) L. Morrissey, S. M. Handrigan, and S. Nakhla, “Discrepancies in the mechanical properties of gold nanowires: The importance of potential type and equilibration method,” Computational Materials Science. 2020. link Times cited: 6 NOT USED (low confidence) D. T. Ho, S. Kim, S. Y. Kwon, and S. Y. Kim, “Ideal strength of nanoscale materials induced by elastic instability,” Mechanics of Materials. 2020. link Times cited: 2 NOT USED (low confidence) M. Demin, O. Koroleva, A. A. Aleksashkina, and V. Mazhukin, “Atomistic modeling of the characteristics of the phonon subsystem of copper in a wide temperature range,” Keldysh Institute Preprints. 2020. link Times cited: 0 NOT USED (low confidence) K. Nordlund and F. Djurabekova, “Molecular Dynamics Simulations of Non-equilibrium Systems,” Handbook of Materials Modeling. 2020. link Times cited: 3 NOT USED (low confidence) T. Rahman, “Surface Thermodynamics and Vibrational Entropy.” 2020. link Times cited: 2 NOT USED (low confidence) R. Ferrando, “Kinetics of Nanoalloys: Nucleation, Mixing, Coalescence.” 2020. link Times cited: 0 NOT USED (low confidence) W. G. Wolfer, “Fundamental Properties of Defects in Metals,” Comprehensive Nuclear Materials. 2020. link Times cited: 33 NOT USED (low confidence) R. Zhang, “Applications for two aspects of molecular simulations: static and dynamic.” 2020. link Times cited: 0 NOT USED (low confidence) Z. Chen and A. Akbarzadeh, “Basic Problems of Non-Fourier Heat Conduction,” Structural Integrity. 2019. link Times cited: 2 NOT USED (low confidence) X. Liu, “Mechanical Behaviors of Graphene Nanolayered Composites,” Springer Theses. 2019. link Times cited: 0 NOT USED (low confidence) Y. Gan and Z. Sun, “Crystal structure dependence of the breathing vibration of individual gold nanodisks induced by the ultrafast laser.,” Applied optics. 2019. link Times cited: 3 Abstract: The ultrafast laser-excited breathing vibrations of gold nan… read moreAbstract: The ultrafast laser-excited breathing vibrations of gold nanodisks with different crystal structures have been studied via atomistic simulations. The vibrational periods and damping rates of nanodisks are obtained by the analysis of the simulated transient responses of nanodisks. It is shown that the breathing period of nanodisks is considerably dependent on their crystal structure, which is contrary to the cases for the breathing vibration of metal nanospheres and nanorods. Furthermore, single-crystal nanodisks exhibit much lower intrinsic damping as compared with polycrystalline nanodisks, for which the additional energy dissipation by the grain boundaries in the polycrystalline nanodisks could be one major factor. read less NOT USED (low confidence) S. A. Peddakotla, K. K. Kammara, and R. Kumar, “Single particle trajectory analysis for the evaluation of surface accommodation coefficients,” 31ST INTERNATIONAL SYMPOSIUM ON RAREFIED GAS DYNAMICS: RGD31. 2019. link Times cited: 0 NOT USED (low confidence) J. Liu and X. Liu, “Adsorption and Desorption Characteristics of Deuterides,” Deuteride Materials. 2019. link Times cited: 0 NOT USED (low confidence) S. Dong, C. Zhu, Y. Chen, and J. Zhao, “Buckling behaviors of metal nanowires encapsulating carbon nanotubes by considering surface/interface effects from a refined beam model,” Carbon. 2019. link Times cited: 21 NOT USED (low confidence) M. Caturla, “Object kinetic Monte Carlo methods applied to modeling radiation effects in materials,” Computational Materials Science. 2019. link Times cited: 10 NOT USED (low confidence) R. Ferrando, “Stress-driven structural transitions in bimetallic nanoparticles.” 2019. link Times cited: 7 NOT USED (low confidence) P. Zakharov, E. Korznikova, S. Dmitriev, E. Ekomasov, and K. Zhou, “Surface discrete breathers in Pt3Al intermetallic alloy,” Surface Science. 2019. link Times cited: 36 NOT USED (low confidence) “References,” Basic Physics of Nanoscience. 2019. link Times cited: 0 NOT USED (low confidence) “References,” Dislocation Mechanism-Based Crystal Plasticity. 2019. link Times cited: 0 NOT USED (low confidence) С. Волегов, Р. М. Герасимов, and Р. П. Давлятшин, “MODELS OF MOLECULAR DYNAMICS: A REVIEW OF EAM-POTENTIALS. PART 2. POTENTIALS FOR MULTI-COMPONENT SYSTEMS.” 2018. link Times cited: 1 Abstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликова… read moreAbstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликована: 29 июня 2018 г. В статье представлена вторая часть обзора современных подходов и работ, посвященных построению потенциалов межатомного взаимодействия с использованием методологии погруженного атома (EAM-потенциалы). Эта часть обзора посвящена одной из наиболее остро стоящих проблем в молекулярной динамике – вопросам построения потенциалов, которые были бы пригодны для описания структуры и физико-механических свойств многокомпонентных (в первую очередь – бинарных и тернарных) материалов. Отмечены первые работы, в которых предлагались подходы к построению функций перекрестного взаимодействия для сплавов никеля и меди – как с использованием методологии EAM, так и несколько отличающийся по процедуре построения потенциал типа Финисса-Синклера. Рассматриваются работы, в которых производится сопоставление различных подходов к построению потенциалов, а также к процедуре идентификации их параметров на примере одних и тех же многокомпонентных систем (типа Al-Ni или Cu-Au). Кроме того, особый интерес представляют некоторые тернарные системы, например Fe–Ni–Cr, W–H– He или U–Mo–Xe, которые являются ключевыми для материалов атомной энергетики и которые в последние годы активно изучаются как возможные материалы для использования в термоядерных ректорах. Приведены примеры работ, в которых предлагаются и исследуются потенциалы для описания многокомпонентных систем, пригодных для использования в аэрокосмической промышленности и изготовленных прежде всего на основе никеля. Рассмотрены результаты исследований различных интерметаллических соединений, отмечены работы, в которых при помощи построенного EAM потенциала удалось количественно точно описать фазовые диаграммы соединений и вычислить характеристики фазовых переходов. read less NOT USED (low confidence) C. Hugenschmidt, “Surface Segregation in Cu/Pd and Ni/Pd Observed by Positron Annihilation Induced Auger Electron Spectroscopy.” 2018. link Times cited: 0 NOT USED (low confidence) H. Barrón, “Catalytic Efficiency in Metallic Nanoparticles: A Computational Approach.” 2018. link Times cited: 0 NOT USED (low confidence) R. Promyoo, H. El-Mounayri, and M. Agarwal, “An Experimental Study to Guide AFM-Based TBN of Nanochannels.” 2018. link Times cited: 1 NOT USED (low confidence) J. Lane, K. Salerno, G. Grest, and H. Fan, “Modeling pressure-driven assembly of polymer coated nanoparticles,” Bulletin of the American Physical Society. 2018. link Times cited: 4 NOT USED (low confidence) S. Mejía-Rosales, “Simulation of Metal Clusters and Nanostructures.” 2018. link Times cited: 3 NOT USED (low confidence) S. Borisova and G. Rusina, “Lattice dynamic of Cu (110) surface covered by 0.16 ML of the lithium.” 2018. link Times cited: 1 NOT USED (low confidence) X. Yue, X. Yue, X. Yang, and M. Kunieda, “Comparison of Electrical Discharge Machining Speed of Tool Electrodes with Different Thermo-physical Properties Related to Ease of Boiling,” Procedia CIRP. 2018. link Times cited: 2 NOT USED (low confidence) G. Raabe, “Molecular Models (Force Fields).” 2017. link Times cited: 0 NOT USED (low confidence) O. A. Oviedo, L. Reinaudi, S. García, and E. Leiva, “Modelling of Underpotential Deposition on Bulk Electrodes.” 2016. link Times cited: 0 NOT USED (low confidence) R. Ferrando, “Nonequilibrium phenomena in nanoalloys: From nucleation to ageing.” 2016. link Times cited: 1 NOT USED (low confidence) L. Talirz, P. P. Shinde, D. Passerone, and C. Pignedoli, “Synthesis of Atomically Precise Graphene-Based Nanostructures: A Simulation Point of View.” 2016. link Times cited: 5 NOT USED (low confidence) O. Alizadeh, G. T. Eshlaghi, and S. Mohammadi, “Nanoindentation simulation of coated aluminum thin film using quasicontinuum method,” Computational Materials Science. 2016. link Times cited: 12 NOT USED (low confidence) S. Melin, P. Hansson, and A. Ahadi, “Defect sensitivity of single-crystal nano-sized Cu beams,” Procedia structural integrity. 2016. link Times cited: 3 NOT USED (low confidence) A. Ahadi and S. Melin, “Size dependence of the Poisson’s ratio in single-crystal fcc copper nanobeams,” Computational Materials Science. 2016. link Times cited: 25 NOT USED (low confidence) R. Ferrando, “Theoretical and computational methods for nanoalloy structure and thermodynamics.” 2016. link Times cited: 5 NOT USED (low confidence) O. A. Oviedo, L. Reinaudi, S. G. García, and E. P. M. Leiva, “Underpotential Deposition and Related Phenomena at the Nanoscale: Theory and Applications.” 2016. link Times cited: 2 NOT USED (low confidence) R. Jones, J. Templeton, and J. Zimmerman, “Principles of Coarse-Graining and Coupling Using the Atom-to-Continuum Method.” 2016. link Times cited: 6 NOT USED (low confidence) S. Zimnik, C. Piochacz, S. Vohburger, and C. Hugenschmidt, “Time-dependent investigation of sub-monolayers of Ni on Pd using Positron-annihilation induced Auger Electron Spectroscopy and XPS,” Surface Science. 2016. link Times cited: 6 NOT USED (low confidence) P. Brandãoa et al., “Thermo-mechanical modeling of a high pressure turbine blade of an airplane gas turbine engine,” Procedia structural integrity. 2016. link Times cited: 41 NOT USED (low confidence) O. A. Oviedo, P. Vélez, V. Macagno, and E. Leiva, “Underpotential deposition: From planar surfaces to nanoparticles,” Surface Science. 2015. link Times cited: 33 NOT USED (low confidence) S. Chandra, M. K. Samal, V. Chavan, and R. Patel, “Multiscale modeling of plasticity in a copper single crystal deformed at high strain rates.” 2015. link Times cited: 12 NOT USED (low confidence) A. Khammang, “Investigating Mechanical Properties of Metallic Nanowires using Molecular Dynamics.” 2014. link Times cited: 0 NOT USED (low confidence) N. Hiroshi, “Defects in Metals.” 2014. link Times cited: 1 NOT USED (low confidence) S. Hartmann, O. Holck, and B. Wunderle, “Mechanics of CNT-palladium Interfaces for Sensor Applications Simulated with Molecular Dynamics,” Procedia Materials Science. 2014. link Times cited: 8 NOT USED (low confidence) N. Abdolrahim, I. Mastorakos, D. Bahr, and H. Zbib, “Observation of pseudoelastic behavior in large Cu-Ni composite multilayer nanowires,” MRS Proceedings. 2014. link Times cited: 0 Abstract: In recent years, studies have shown that single crystal meta… read moreAbstract: In recent years, studies have shown that single crystal metallic nanowires (NWs) can exhibit unique pseudoelastic behavior when their cross-sectional area is smaller than a certain critical value, which is on the order of a few nms. The mechanism responsible for this behavior is the formation of partial dislocations (twinning). In this paper we demonstrate using molecular dynamics simulations that thicker composite nanowires can exhibit pseudoelastic behavior at large cross-sectional dimensions to 28 nm and higher, as long as the individual layer thickness do not exceed a critical value of 1.8-2 nm, thus making their manufacturing feasible and more attractive. In two previous papers we studied the possibility of increasing the critical dimensions of nanowires for exhibiting pseudoelasticity by adding the coherency stresses found in coppernickel (Cu-Ni) interfaces, using molecular dynamics (MD) simulations. In particular, our simulations showed that trilayer composite nanowires, made of alternating copper and nickel layers, would still exhibit pseudoelastic behavior for total thicknesses that are about 3 times larger than that of single crystalline nanowires. In this paper we expand this work to composite nanowires with several numbers of layers with total dimensions much larger (15 times and more) than single crystalline nanowires. Our results show that even these ”thick” structures exhibit pseudoelasticity. The key parameter to this behavior is the restriction of the individual layer thickness below a critical value that is related to the maximum thickness of a single metal nanowire to exhibit pseudoelasticity. The resulted structures could maintain their pseudoelastic behavior with very few residual dislocations under several loading/unloading processes. It is our belief that these composite wires can be manufactured from nano-metallic multilayers using already established techniques. Metallic nanowires have many potential applications in nanoscale electromechanical systems (NEMS) due to their unique physical, electronic, magnetic and optical properties that are coupled with their mechanical behavior. Hence, it is important to understand the mechanical properties of nanowires to recognize their future applications in nanotechnology. Among metallic nanowires, fcc nanowires exhibit very unique behavior of pseudoelasticity upon applying and removal of tensile loading. They can recover plastic strains of up to 40% which is far beyond the limits for conventional shape memory bulk materials that is about 5-8% . This behavior of pseudoelasticity is very important in the area of self-healing materials used as 4 6 9 read less NOT USED (low confidence) D. Davydov, A. Javili, P. Steinmann, and A. McBride, “A Comparison of Atomistic and Surface Enhanced Continuum Approaches at Finite Temperature.” 2013. link Times cited: 7 NOT USED (low confidence) R. Ferrando, “Kinetic aspects: nucleation, mixing, coalescence.” 2013. link Times cited: 0 NOT USED (low confidence) O. A. Oviedo and E. Leiva, “Thermodynamic Modeling of Metallic Nanoclusters.” 2013. link Times cited: 5 NOT USED (low confidence) N. Bulgakova, “Fundamentals of ultrafast laser processing.” 2013. link Times cited: 5 Abstract: When laser light of visible, near-IR or UV spectral range hi… read moreAbstract: When laser light of visible, near-IR or UV spectral range hits condensed matter, it interacts with the valance and/or conduction electrons of the system under action. Depending on laser intensity and irradiation geometry, the interaction can have different far-reaching consequences such as melting, ablation, changing of optical properties, mechanical and chemical transformations. Among existing laser systems, ultrafast lasers have become an extraordinary tool for processing of any kind of materials. With proper choosing the irradiation conditions, laser action allows either inducing highly-localized gentle modifications or obtaining strongly damaged material sites with desired or deleterious structures such as voids, periodic nanocracks, periodic surface structures or craters of various shapes and dimensions. This chapter presents a review on tremendous efforts of researchers in order to achieve clearer insights into laser-matter interactions in ultrashort irradiation regimes. The review does not pretend to completeness and aims to outline main ideas, achievements, and most intriguing findings still waiting for explanations and theoretical treatments. read less NOT USED (low confidence) D. Seif and N. Ghoniem, “Dislocation Bias Calculations in Metals Using a Combined Finite-Element Rate-Theory Approach.” 2013. link Times cited: 2 NOT USED (low confidence) C. Mottet, “Structure and Chemical Ordering in Nanoalloys: Toward Nanoalloy Phase Diagrams.” 2013. link Times cited: 3 NOT USED (low confidence) F. Baletto, “Modelling Janus Nanoparticles.” 2013. link Times cited: 2 NOT USED (low confidence) A. Peles, “Nanostructured Electrocatalysts for Oxygen Reduction Reaction: First-Principles Computational Insights.” 2013. link Times cited: 0 NOT USED (low confidence) R. Callejas-Tovar and P. Balbuena, “Understanding Activity and Durability of Core/ Shell Nanocatalysts for Fuel Cells.” 2013. link Times cited: 0 NOT USED (low confidence) M. Strangwood, “Fundamentals of ferrite formation in steels.” 2012. link Times cited: 11 Abstract: Abstract: The diffusional formation of ferrite at low underc… read moreAbstract: Abstract: The diffusional formation of ferrite at low undercoolings has, historically, been extensively researched, which has led to well-established mechanisms for nucleation and growth. These have been verified against experimental data to allow modelling with good agreement to experiment, often as transformation diagrams. These mechanisms are summarised here and opportunities for improvement through advances in experimentation and computation are highlighted. read less NOT USED (low confidence) P. A. Autreto, M. Lagos, D. Ugarte, and D. Galvão, “Correlation Between Quantum Conductance and Atomic Arrangement of Silver Atomic-Size Nanocontacts,” MRS Proceedings. 2012. link Times cited: 1 NOT USED (low confidence) E. Lin, L. Niu, H.-ji Shi, and Z. Duan, “Molecular dynamics simulation of nano-scale interfacial friction characteristic for different tribopair systems,” Applied Surface Science. 2012. link Times cited: 26 NOT USED (low confidence) W. Wolfer, “1.01 – Fundamental Properties of Defects in Metals.” 2012. link Times cited: 66 NOT USED (low confidence) D. Watvisave, U. Bhandarkar, and B. Puranik, “A DSMC-MD Investigation of Wall Effects in a Shock Tube Operating at High Knudsen Numbers.” 2012. link Times cited: 2 NOT USED (low confidence) M. Springborg, “Theoretical Studies of Structural and Electronic Properties of Clusters.” 2012. link Times cited: 3 NOT USED (low confidence) S. Koh, A. Saxena, W. Driel, G. Zhang, and R. Tummala, “Semi Emprical Low Cycle Fatigue Crack Growth Analysis of Nanostructure Chip-To-Package Copper Interconnect Using Molecular Simulation.” 2012. link Times cited: 1 NOT USED (low confidence) D. Alloyeau, C. Mottet, and C. Ricolleau, “Nanoalloys: Synthesis, Structure and Properties,” Nanoalloys. 2012. link Times cited: 47 NOT USED (low confidence) J. Lao, “Molecular dynamics simulation studies of surface-stress effects in metallic nanostructures.” 2011. link Times cited: 0 Abstract: ............................................................… read moreAbstract: ......................................................................................ix CHAPTER read less NOT USED (low confidence) H. Haftbaradaran, J. Song, W. Curtin, and H. Gao, “Continuum and atomistic models of strongly coupled diffusion, stress, and solute concentration,” Journal of Power Sources. 2011. link Times cited: 186 NOT USED (low confidence) M. Walker, M. Brown, M. Draxler, L. Fishwick, M. Dowsett, and C. McConville, “Analysis of the interactions between He+ ions and transition metal surfaces using co-axial impact collision ion scattering spectroscopy,” Surface Science. 2011. link Times cited: 9 NOT USED (low confidence) L. Yuan-yuan, J. Guo-bin, Y. Bin, and L. Dachun, “Molecular Dynamics Simulation of Thermodynamic Properties for Pb-Au Alloys,” Rare Metal Materials and Engineering. 2011. link Times cited: 2 NOT USED (low confidence) P. Misra, “Chapter 16 – Metallic Nanoclusters,” European Physical Journal B. 2011. link Times cited: 0 NOT USED (low confidence) T. Koido, D. Ito, T. Tokumasu, K. Tomarikawa, and S. Yonemura, “A Molecular Dynamics Study of the Effect of the Incidence Angle on the Dissociation Probability of H2 on Pt(111),” Journal of Thermal Science and Technology. 2011. link Times cited: 0 Abstract: Dissociative adsorption processes of hydrogen molecules on a… read moreAbstract: Dissociative adsorption processes of hydrogen molecules on a platinum surface were simulated by a molecular dynamics (MD) method. The effect of the incidence angle of the impinging gas molecules on the dissociation probability was analyzed. The embedded atom method (EAM) was used to construct an interaction potential between the gas molecule and the Pt(111) surface. Initially, the effect of the motions of atoms or molecules (dynamic effects) on the dissociation probability was studied. It showed that dynamic effects on the dissociation phenomena are very large for adsorption at the top site. A number of MD simulations of H2 or D2 molecules impinging on a Pt(111) surface were also carried out, with varying initial orientations and rotational energy of the molecule, and differing collision locations on the surface. The results were averaged and the effects of the isotopic nature, incident angle, and translational energy of the adsorbate were analyzed. The results were also compared to those obtained from molecular beam experiments to check the validity of the simulations. This comparison showed that the dependences of the dissociation probability on translational energy and incident angle roughly agree with experiments. read less NOT USED (low confidence) N. Khorshidi, “In-situ X-ray studies of model electrode surfaces for solid oxide fuel cells.” 2010. link Times cited: 2 Abstract: Fuel cells are considered as a promising way to produce clea… read moreAbstract: Fuel cells are considered as a promising way to produce clean energy. These cells convert the chemical energy created by the reaction of hydrogen and oxygen to water into electric energy. Regarding the difficulties connected with the production and more importantly storage of hydrogen, solid oxide fuel cells (SOFCs) play an outstanding role among the diverse fuel cell types. SOFCs are able to use not only pure hydrogen as a fuel, but also hydrocarbons. This ability leads to impressive efficiencies and allows to integrate SOFCs into existing structures. SOFCs are usually operated at temperatures above 800°C which leads to extreme conditions for the used materials and limits the lifetime of the cells. One of the major goals in the future is thus to develop low temperature SOFCs. In order to achieve such a goal, an atomic understanding of the chemicals reactions on the electrodes is essential.
A typical SOFC is made of a cathode consisting of lanthanum strontium manganate (LSM) and yttria-stabilized zirconia (YSZ). Due to its electronic and thermal isolation and the conduction of oxygen ions, YSZ also serves as an electrolyte and at the same time as a part of the anode, which in addition is covered with nickel particles. An atomic understanding of the respective chemical reactions thus requires to have atomic models of the YSZ surface being present at the cathode and the anode surface. Another fundamental component to know are the nickel particles covering the anode. The superb importance of the anode or fuel cathode is to be found by the diverse fuels to be processed here.
The aim of this work is to experimentally determine the atomic surface structure of the two important orientations (111) and (100) of YSZ under relevant conditions. Additionally the growth and shape of Ni nano particles grown on these surfaces as well as their shape changes under related conditions are studied. The gathered knowledge can be assembled to a model anode and a part of a model cathode. The found results are also of importance for the growth of thin films, where YSZ is a frequent substrate.
The main experimental tool is surface X-ray diffraction (SXRD), which allows to derive atomic structures of surfaces regardless of their conductivity properties. The experiments were carried out in a mobile ultra-high vacuum chamber using synchrotron radiation.
Brennstoffzellen gelten als eine vielversprechende Moglichkeit saubere Energie zu erzeugen. Dabei wird die bei der Reaktion von Sauerstoff und Wasserstoff zu Wasser entstandene chemische in elektrische Energie umgewandelt. Betrachtet man die Schwierigkeiten, die mit der Erzeugung und vor allem Speicherung von reinem Wasserstoff verbunden sind, so spielt die Festoxidbrennstoffzelle (SOFC aus dem englischen Solid Oxide Fuel Cell) eine besondere Rolle. Die SOFC ist in der Lage nicht nur reinen Wasserstoff, sondern auch Kohlenwasserstoffe als Brennstoff zu verwenden. Somit erreicht sie beachtliche Wirkungsgrade und kann in bestehende Strukturen integriert werden. SOFCs werden ublicherweise bei Temperaturen uber 800°C betrieben, was eine enorme Anforderung an die verwendeten Materialien stellt und die Lebensdauer der Zellen beschrankt. Eine der grosen Ziele der SOFC-Forschung ist daher die Betriebstemperatur zu senken. Um ein solches Ziel zu erreichen, ist es notwendig die auf den Elektroden ablaufenden chemischen Reaktionen auf atomarer Skala zu verstehen und nachzuvollziehen.
Eine typische SOFC besteht aus einer Kathode aus Lanthan-Strontium-Manganat (LSM) und Yttrium-Stabilisiertes Zirconiumdioxid (YSZ). YSZ dient auf Grund seiner elektronischen und thermischen Isolierung und gleichzeitiger Leitfahigkeit von Sauerstoffionen als Elektrolyt aber auch als Teiloberflache der Anode, die zusatzlich mit Nickelteilchen bedeckt ist. Als grundlegend fur ein atomares Verstandnis der SOFC konnen daher Oberflachenmodelle von YSZ definiert werden, die sowohl auf der Kathodenseite als auch auf der Anodenseite prasent sind. Als weiteres wichtiges Element sind die Nickelteilchen zu betrachten, die die Anode bedecken. Die besondere Rolle der Anode ist mit der Vielzahl von Kohlenwasserstoffen, die hier verarbeitet werden zu begrunden.
Ziel der vorliegenden Arbeit ist es daher, experimentell atomare Oberflachenmodelle von den zwei wichtigen YSZ-Orientierungen (111) und (100) unter relevanten Bedingungen zu finden. Desweiteren werden Ni-Nanoteilchen auf diesen Oberflachen erzeugt und das Wachstum, sowie Form und Formanderungen unter relevanten Bedingungen bestimmt. Somit werden Modell-Anoden und, mit den YSZ-Oberflachenstrukturen, ein Teil der Kathode mit atomarer Auflosung untersucht. Die vorliegenden Ergebnisse sind auch von Bedeutung fur das Dunnschichtwachstum, wo YSZ haufig als Substrat verwendet wird.
Als Hauptuntersuchungsmethode wurde oberflachensensitive Rontgenbeugung (SXRD aus dem englischen Surface X-ray Diffraction) angewandt, die eine Oberflachenanalyse auf atomarer Skala unabhangig von der Leitfahigkeitseigenschaften des untersuchten Materials erlaubt. Die Experimente wurden in einer mobilen Kammer unter Ultrahochvakuum-Bedingungen und an Synchrotronquellen durchgefuhrt. read less NOT USED (low confidence) X. Wei, “Multiscale modeling and simulation of material phase change problems: ice melting and copper crystallization.” 2010. link Times cited: 0 Abstract: Recommended Citation Wei, Xiupeng. "Multiscale modeling… read moreAbstract: Recommended Citation Wei, Xiupeng. "Multiscale modeling and simulation of material phase change problems: ice melting and copper crystallization." MS ABSTRACT The primary objective of this work is to propose a state-of-the-art physics based multiscale modeling framework for simulating material phase change problems. Both ice melting and copper crystallization problems are selected to demonstrate this multiscale modeling and simulation. The computational methods employed in this thesis include: classical molecular dynamics, finite element method, phase-field method, and multiscale (nano/micro coupling) methods. Classical molecular dynamics (MD) is a well-known method to study material behaviors at atomic level. Due to the limit of MD, it is not realistic to provide a complete molecular model for simulations at large length and time scales. Continuum methods, including finite element methods, should be employed in this case. In this thesis, MD is employed to study phase change problems at the nanoscale. In order to study material phase change problems at the microscale, a thermal wave method one-way coupling with the MD and a phase-field method one-way coupling with MD are proposed. The thermal wave method is more accurate than classical thermal diffusion for the study of heat transfer problems especially in crystal based structures. The second model is based on the well-known phase-field method. It is modified to respond to the thermal propagation in the crystal matrix by the thermal wave method, as well as modified to respond to temperature gradients and heat fluxes by employing the Dual-Phase-Lag method. Both methods are coupled with MD to obtain realistic results. It should be noted that MD simulations can be conducted to obtain material/thermal properties for microscopic and/or macroscopic simulations for the purpose of hierarchical/sequential multiscale modeling. These material parameters include thermal conductivity, specific heat, latent heat, and relaxation time. Other type of interfacial parameters that occur during the phase change process, such as nucleus shape, iv interfacial energy, interfacial thickness, etc., are also obtained by MD simulation since these have so far been too difficult to measure experimentally. I consider two common phase change phenomena, ice melting and copper crystallization, in this thesis. For the case of ice melting, MD is first employed to study its phase change process and obtain thermal properties of ice and water. Several potential models are used. I conduct simulations of both bulk ice and ice/water contacting cases. It is found that various potential models result in similar melting phenomena, especially melting speed. … read less NOT USED (low confidence) L. Yang, “Molecular dynamics simulation of nanosintering processes.” 2010. link Times cited: 1 Abstract: In this thesis, two scenarios of nanosintering simulations a… read moreAbstract: In this thesis, two scenarios of nanosintering simulations are studied by molecular dynamics (MD) method. The first one will investigate the neck growth in the laser sintering of different-size gold (100) nanoparticles under different heating rates. The numerical simulations are carried out for four pairs of two spherical nanoparticles where one particle has the same diameter of 4 nm and the other one varied from 4 nm to 20 nm. The results show that the stable neck width increases as the size of the other nanoparticle increases. Once the limit stable neck width is reached, it no longer is affected by the nanoparticle size. For the subsequent laser heating to the same final temperature, a lower heating rate results in a larger stable neck width due to the longer sintering process. The other one will study the deposition of Ni(100) nanocluster with zero initial kinetic energy on Cu(100, 110) surface. It’s found that the burrowing process goes extremely slow as temperature is equal or less than 900K. There is no burrowing below 500 K. The completeness of burrowing Ni on Cu(110) is earlier than Cu(100) due to the lower surface energy of Cu(110). Vacancy migration is found to be the main cause of the site-exchange of atoms between Ni cluster and Cu substrate. read less NOT USED (low confidence) H. Bulou, C. Goyhenex, and C. Massobrio, “Surface Diffusion on Inhomogeneous Surfaces.” 2010. link Times cited: 1 NOT USED (low confidence) H. S. Park and P. Klein, “Multiscale Modeling of Surface Effects on the Mechanical Behavior and Properties of Nanowires.” 2010. link Times cited: 0 NOT USED (low confidence) Y. Wang, Z. Su, and D. Ping, “Microstructure Investigation on the Triple Junction with an Adjoining Twin Boundary in Nanocrystalline Palladium,” Journal of Materials Science & Technology. 2010. link Times cited: 6 NOT USED (low confidence) Y. Shao and S. Wang, “An Examination on Atomic-level Stress Calculations by Nanoindentation Simulation via the Quasicontinuum Method,” Journal of Materials Science & Technology. 2010. link Times cited: 3 NOT USED (low confidence) E. Leiva and W. Schmickler, “Theories and Simulations for Electrochemical Nanostructures.” 2009. link Times cited: 1 NOT USED (low confidence) M. Cherkaoui and L. Capolungo, “Predictive Capabilities and Limitations of Molecular Simulations.” 2009. link Times cited: 3 NOT USED (low confidence) 尚樹 岡本, 祐子 藤井, 宏明 栗原, and 和夫 近藤, “無電解 Sn めっき膜上に形成されるウィスカ発生に対する Sn めっき膜の結晶粒径と基板の結晶配向性の影響,” Journal of The Japan Institute of Metals. 2009. link Times cited: 1 Abstract: In this study, we studied how the orientation indexes of the… read moreAbstract: In this study, we studied how the orientation indexes of the Cu foil, which is used for a substrate, and the crystal grain size of the Sn deposited film affects tin whisker formation and the structure of the intermetallic compound deposits. In particular, we considered the relationship between the crystal grain size of Sn deposited films and the amount of intermetallic compound deposits that is formed at the interface between Sn deposited films with aging and Cu foils. We used two types of Cu foils for a substrate. One is “with gelatin additive” that has granular-shaped grains which are 0.5~1 μm in size and the other is “with Cl- ion additive” that has pillar-shaped grains and their nodules. In addition to this, we used Cu (100), (110), (111) single crystals for a substrate. We used two types of Sn plating bath for plating. One is hydrofluoroboric acid bath and another is organic acid bath. The structure of tin whiskers, tin films and Cu foils were investigated by TEM and SEM. The number of the whiskers that formed on the Sn deposited film increased after aging. And the number of the whiskers that formed on the Sn films, which were made by using hydrofluoroboric acid bath, was larger than that formed on the Sn films, which were made by using organic acid bath. We also investigated the mechanisms of heat treatment for inhibiting whiskers formation. There are no whiskers on the Sn deposited film after an aging with heat treatment. From the analysis of TEM diffraction patterns that were obtained by after-aging with heat treatment sample, there were two kinds of intermetallic compound deposits at the interface between the Sn deposited film and the Cu foils. read less NOT USED (low confidence) A. U. Nilekar, A. Ruban, and M. Mavrikakis, “Surface segregation energies in low-index open surfaces of bimetallic transition metal alloys,” Surface Science. 2009. link Times cited: 89 NOT USED (low confidence) M. C. Giménez, E. P. M. Leiva, and E. Albano, “Monte Carlo Simulations of the Underpotential Deposition of Metal Layers on Metallic Substrates: Phase Transitions and Critical Phenomena.” 2009. link Times cited: 0 NOT USED (low confidence) Y.-M. Kim, Y.-H. Shin, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for pure Mn and the Fe–Mn system,” Acta Materialia. 2009. link Times cited: 64 NOT USED (low confidence) J. R. Morris, R. Aga, T. Egami, and V. A. Levashov, “SIMULATING THE EFFECT OF POISSON RATIO ON METALLIC GLASS PROPERTIES,” International Journal of Modern Physics B. 2009. link Times cited: 4 Abstract: Recent work has shown that many metallic glass properties co… read moreAbstract: Recent work has shown that many metallic glass properties correlate with the Poisson ratio of the glass. We have developed a new model for simulating the atomistic behavior of liquids and glasses that allows us to change the Poisson ratio, while keeping the crystalline phase cohesive energy, lattice constant, and bulk modulus fixed. A number of liquid and glass properties are shown to be directly affected by the Poisson ratio. An increasing Poisson ratio stabilizes the liquid structure relative to the crystal phase, as indicated by a significantly lower melting temperature and by a lower enthalpy of the liquid phase. The liquids clearly exhibit two changes in behavior: one at low temperatures, associated with the conventional glass transition Tg, and a second, higher temperature change associated with the shear properties of the liquids. This second crossover has a characteristic, measurable change in the liquid structure. read less NOT USED (low confidence) M. Luo, “Surface-induced size-dependent Young’s modulus in nanomaterials.” 2008. link Times cited: 0 Abstract: Nanowires and ultra-thin films have wide applications in the… read moreAbstract: Nanowires and ultra-thin films have wide applications in the quickly developed nanotechnology and nanoscience. However, their Young’s modulus varies with the size, which is seemingly contradictory to the conventional continuum elasticity. Investigating and understanding the underlying mechanism of the size-dependent elastic properties in nanomaterials is of both academic and practical significance. In this work, both theoretical modeling and virtual experiments have been made on this issue. A nanoelement, from the traction free bulk lattice, undergoes an initial relaxation, during which its morphology changes and energy reduces, which is an emphasis in this developed methodology and is a distinction from almost other existing models. With different definitions of surfaces and edges, two models for a nanomaterial – a nanowire or an ultra-thin film – are derived based on the same thermodynamics framework. Comparing with most of others’ treatment, Model I has an advantage to mathematically treat a surface phase to be two-dimensional and an edge phase to be one-dimensional. Under external loadings, the initial relaxed state is taken as the reference. Experimentally, relaxation and tension/compression tests in different loading directions have been conducted on SiC, Si and Cu crystalline nanowires with different cross-sectional sizes and ultra-thin films with different thicknesses by Molecular Dynamics (MD) simulations. This systematic study successfully illustrates the intrinsic mechanism of the size-dependent Young’s modulus in nanomaterials and the proposed methodology facilitate characterizing mechanical properties of nanomaterials to some extent when continuum concepts, such as, surface energy and surface elastic constants, are used. read less NOT USED (low confidence) J. Li, X. Dai, S. Liang, K. Tai, Y. Kong, and B. Liu, “Interatomic potentials of the binary transition metal systems and some applications in materials physics,” Physics Reports. 2008. link Times cited: 110 NOT USED (low confidence) J. Blackman, “Chapter 6 Structure of Isolated Clusters,” Handbook of Metal Physics. 2008. link Times cited: 1 NOT USED (low confidence) C. Henry, “Size Effects on Structure and Morphology of Free or Supported Nanoparticles.” 2008. link Times cited: 13 NOT USED (low confidence) C. Creemers, S. Helfensteyn, J. Luyten, and M. Schurmans, “Synergy between material, surface science experiments and simulations.” 2007. link Times cited: 3 NOT USED (low confidence) R. Dingreville and J. Qu, “A semi-analytical method to compute surface elastic properties,” Acta Materialia. 2007. link Times cited: 97 NOT USED (low confidence) W. Liang and M. Zhou, “Shape Memory Effect and Pseudoelasticity in Cu Nanowires.” 2007. link Times cited: 0 NOT USED (low confidence) M. Patriarca, A. Kuronen, M. Robles, and K. Kaski, “Three-dimensional interactive Molecular Dynamics program for the study of defect dynamics in crystals,” Comput. Phys. Commun. 2007. link Times cited: 4 NOT USED (low confidence) J. Schall, P. Mikulski, G. M. Chateauneuf, G. Gao, and J. Harrison, “Molecular Dynamics Simulations of Tribology.” 2007. link Times cited: 8 NOT USED (low confidence) W. Liang and M. Zhou, “Surface-Stress-Driven Pseudoelasticity and Shape Memory Effect at the Nanoscale.” 2006. link Times cited: 1 NOT USED (low confidence) K. Subramanian, R. Zhang, P. Shrotriya, A. Chandra, and S. Sundararajan, “Surface Stress Generation During Formation of Alkanethiol Self-assembled Monolayer (SAM),” MRS Proceedings. 2006. link Times cited: 5 Abstract: A high resolution curvature interferometer [Wang, J., Shrotr… read moreAbstract: A high resolution curvature interferometer [Wang, J., Shrotriya, P., Kim, K.S., 2006. Surface residual stress measurement using curvature interferometry. Experimental Mechanics 46 (1), 39–46] is utilized to measure surface stress development associated with formation of self-assembled monolayers (SAM) of octadecanethiols on macroscale domains (25 mm × 25 mm). Atomistic simulations are performed to investigate surface stress generation associated with SAM formation. The results of the molecular simulations are incorporated into the multiscale framework to understand the surface stress generation and curvature change observed during experiments at continuum scale. read less NOT USED (low confidence) G. Betz, “Interaction of Ions and Electrons with Solid Surfaces.” 2006. link Times cited: 1 NOT USED (low confidence) V. Ignatova, D. Karpuzov, I. Chakarov, and I. Katardjiev, “Computer simulations of surface analysis using ion beams,” Progress in Surface Science. 2006. link Times cited: 15 NOT USED (low confidence) D. Saraev and R. E. Miller, “Atomic-scale simulations of nanoindentation-induced plasticity in copper crystals with nanometer-sized nickel coatings,” Acta Materialia. 2006. link Times cited: 117 NOT USED (low confidence) T. Liu, G. Liu, Q. Xie, and Q. J. Wang, “An EFG-FE Coupling Method for Microscale Adhesive Contacts,” Journal of Tribology-transactions of The Asme. 2006. link Times cited: 10 Abstract: An elastic adhesive contact model based on the element-free … read moreAbstract: An elastic adhesive contact model based on the element-free Galerkin-finite element (EFG-FE) coupling method is presented in this paper. The model is first validated though comparison to theoretical solutions. A numerical simulation of the adhesive contact between a microelastic cylinder and a rigid half-space is then conducted. The adhesive contact characteristics of three metals (Al, Cu, and Fe) are studied at different Tabor parameters. The relationships of the applied load and contact half-width of the adhesive contacts are analyzed. Contact pressures, stress contours and deformed profiles of different cylinder sizes and applied loads are illustrated and discussed. The results are compared to published solutions, and good agreements are observed. read less NOT USED (low confidence) D. Rodney, “Atomic-scale modeling of clear band formation in FCC metals,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 30 NOT USED (low confidence) S. Heo, S. Sinnott, D. Brenner, and J. Harrison, “Computational Modeling of Nanometer-Scale Tribology.” 2005. link Times cited: 19 NOT USED (low confidence) D. Wolf, “Elastic Behavior of Interfaces.” 2005. link Times cited: 0 NOT USED (low confidence) B. Hyde, H. Espinosa, and D. Farkas, “An atomistic investigation of elastic and plastic properties of Au nanowires,” JOM. 2005. link Times cited: 70 NOT USED (low confidence) D. Crowson, D. Farkas, and S. Corcoran, “Surface Stress Effects on the Elastic Behavior of Nanoporous Metals,” MRS Proceedings. 2005. link Times cited: 0 Abstract: Atomic scale computer simulations were used to investigate t… read moreAbstract: Atomic scale computer simulations were used to investigate the surface stress induced deformation in nanoporous metals. A phase field model was used to generate digital nanoporous structures that are quantitatively similar to those created experimentally via dealloying. We analyze the important effects of surface relaxations on the macroscopic deformation in these samples as well as in small spherical clusters. read less NOT USED (low confidence) E. Yildirim, M. Atis, and Z. B. Güvenç, “Structure and Dynamical Properties of AuN, N = 12-14 Clusters:” International Journal of Modern Physics C. 2005. link Times cited: 7 Abstract: Using molecular dynamics and thermal quenching methods on th… read moreAbstract: Using molecular dynamics and thermal quenching methods on the basis of Voter–Chen version of the embedded-atom method, we have studied the melting behavior of AuN (N = 12, 13, 14) clusters. This behavior is described in terms of overall and atom resolved root-mean-square bond-length fluctuations, specific-heat, short- and long-time average coordination numbers of each atom and short-time average temperatures of the clusters. The isomer sampling probabilities are obtained from the thermal quenching of the molten clusters, and their energy-spectrum widths are investigated. Phase change of a cluster takes place with the collective and simultaneous motion of all the atoms. read less NOT USED (low confidence) A. Kara and T. Rahman, “Vibrational dynamics and thermodynamics of surfaces and nanostructures,” Surface Science Reports. 2005. link Times cited: 45 NOT USED (low confidence) K. Nordlund and R. Averback, “Point Defects in Metals.” 2005. link Times cited: 2 NOT USED (low confidence) N. Bieri, “Transport phenomena in the microprinting and laser annealing of gold nanoparticle inks.” 2004. link Times cited: 1 Abstract: In the first part of this chapter the coalescence process of… read moreAbstract: In the first part of this chapter the coalescence process of two gold nanoparticles in vacuum is investigated for a host of initial temperatures and starting radii with a phenomenological macroscopic model. The model is based on an energy balance and supplemented by a model for the surface variation of the nanosystem under consideration (Lehtinen and Zachariah, 2001; 2002). The model is modified to account for the curvature dependence of the melting temperature of the nanoparticles. The results are compared with the Molecular Dynamics (MD) simulations (Arcidiacono et al., 2004) using the glue potential for the gold simulation (Ercolessi et al., 1988). Accounting for the findings of the MD simulations for the neck growth rate, the validity of the analytical model with the initial temperature and radius of the particle is shown. The calculations are extended to particles having radius between 0.95 to 2.5 nm. In the second part of the chapter the effects of the environment and laser heating are investigated with the macroscopic model. read less NOT USED (low confidence) M. Caturla, A. G. Martí, J. Jiménez-Rodríguez, J.-C. J. Saez, and Pérez-Martı́n M., “Molecular Dynamics Simulations of Energy Deposition in Solids,” Advances in Quantum Chemistry. 2004. link Times cited: 4 NOT USED (low confidence) Y. T. Kim, S. Choi, S. Lee, and J. Kim, “An Atomic Simulation of AFM-Based Nano Lithography Process for Nano Patterning.” 2004. link Times cited: 6 NOT USED (low confidence) D. Sun, M. Asta, and J. Hoyt, “Kinetic coefficient of Ni solid-liquid interfaces from molecular-dynamics simulations .,” Physical Review B. 2004. link Times cited: 100 Abstract: The kinetics of isothermal crystallization and melting are s… read moreAbstract: The kinetics of isothermal crystallization and melting are studied for elemental Ni employing non-equilibrium molecular-dynamics simulations based on interatomic potentials of the embedded-atom-method form. These simulations form the basis for calculations of the magnitude and crystalline anisotropy of the kinetic coefficient μ, defined as the constant of proportionality between interface velocity and undercooling. We obtain highly symmetric rates for crystallization and melting, from which we extract the following values of μ for low index {100}, {110}, and {111} interfaces: μ 1 0 0 =35.8′22, μ 1 1 0 =25.5′1.6, and μ 1 1 1 =24.1 ′4.0 in units of cm/s K. The results of the present study are discussed in the context of previous molecular-dynamics simulations for related systems, and kinetic models based upon transition-state and density-functional theories. read less NOT USED (low confidence) Y. Ye, R. Biswas, A. Bastawros, and A. Chandra, “Atomistic Mechanisms Underlying Chemical Mechanical Planarization of Copper.” 2003. link Times cited: 3 NOT USED (low confidence) W. Gerberich and W. Yang, “8.01 - Interfacial and Nanoscale Failure.” 2003. link Times cited: 36 NOT USED (low confidence) M. Asta, D. Sun, and J. Hoyt, “Role of Atomic-Scale Simulation in the Modeling of Solidification Microstructure.” 2003. link Times cited: 0 NOT USED (low confidence) V. Rodrigues and D. Ugarte, “Structural Study of Metal Nanowires.” 2003. link Times cited: 8 NOT USED (low confidence) J. Zimmerman, E. Webb, J. Hoyt, R. Jones, P. Klein, and D. Bammann, “Evaluation of continuum stress in atomistic simulation.” 2003. link Times cited: 2 NOT USED (low confidence) P. Vashishta, “Applications: Physical and Electronic Materials.” 2002. link Times cited: 0 NOT USED (low confidence) S. Li and W. K. Liu, “Meshfree and particle methods and their applications,” Applied Mechanics Reviews. 2002. link Times cited: 866 Abstract: Recent developments of meshfree and particle methods and the… read moreAbstract: Recent developments of meshfree and particle methods and their applications in applied mechanics are surveyed. Three major methodologies have been reviewed. First, smoothed particle hydrodynamics ~SPH! is discussed as a representative of a non-local kernel, strong form collocation approach. Second, mesh-free Galerkin methods, which have been an active research area in recent years, are reviewed. Third, some applications of molecular dynamics ~MD! in applied mechanics are discussed. The emphases of this survey are placed on simulations of finite deformations, fracture, strain localization of solids; incompressible as well as compressible flows; and applications of multiscale methods and nano-scale mechanics. This review article includes 397 references. @DOI: 10.1115/1.1431547# read less NOT USED (low confidence) G. Derry, “Chapter 5 – SURFACE SEGREGATION IN BINARY METAL ALLOYS.” 2001. link Times cited: 4 NOT USED (low confidence) L. G. González and J. M. Montejano-Carrizales, “An Energetical Study of Transition-Metal Nanoclusters within the Embedded Atom Method.” 2001. link Times cited: 0 NOT USED (low confidence) M. Caturla, N. Soneda, E. Alonso, B. Wirth, T. D. Rubia, and J. Perlado, “Comparative study of radiation damage accumulation in Cu and Fe,” Journal of Nuclear Materials. 2000. link Times cited: 208 NOT USED (low confidence) B. Wirth, V. Bulatov, and T. D. Rubia, “Atomistic Simulation of Dislocation-Defect Interactions in Cu,” MRS Proceedings. 2000. link Times cited: 5 Abstract: The mechanisms of dislocation-defect interactions are of pra… read moreAbstract: The mechanisms of dislocation-defect interactions are of practical importance for developing quantitative structure-property relationships, mechanistic understanding of plastic flow localization and predictive models of mechanical behavior in metals under irradiation. In copper and other face centered cubic metals, high-energy particle irradiation produces hardening and shear localization. Post-irradiation microstructural examination in Cu reveals that irradiation has produced a high number density of nanometer sized stacking fault tetrahedra. Thus, the resultant irradiation hardening and shear localization is commonly attributed to the interaction between stacking fault tetrahedra and mobile dislocations, although the mechanism of this interaction is unknown. In this work, we present a comprehensive molecular dynamics simulation study that characterizes the interaction and fate of moving dislocations with stacking fault tetrahedra in Cu using an EAM interatomic potential. This work is intended to produce atomistic input into dislocation dynamics simulations of plastic flow localization in irradiated materials. read less NOT USED (low confidence) S. Phillpot, “AN INTRODUCTION TO THE MOLECULAR-DYNAMICS SIMULATION OF MATERIALS*.” 2000. link Times cited: 1 NOT USED (low confidence) D. Srivastava, F. N. Dzegilenko, S. Barnard, S. Saini, M. Menon, and S. Weeratunga, “Carbon-nanotube-based nanotechnology in an integrated modeling and simulation environment.” 2000. link Times cited: 2 NOT USED (low confidence) A. Rakotomahevitra, L. Wille, and M. Rakotomalala, “Atomistic Modeling of Ultrathin Fe Films on Cu (111),” MRS Proceedings. 2000. link Times cited: 1 NOT USED (low confidence) S. Ciraci, “Atomic Structure, Quantized Electrical and Thermal Conductance of Nanowires.” 2000. link Times cited: 0 NOT USED (low confidence) J. Zimmerman, F. F. Abraham, and H. Gao, “Atomistic Simulation of Transonic Dislocations,” MRS Proceedings. 1999. link Times cited: 0 NOT USED (low confidence) J. C. Schön and M. Jansen, “Predicting structures of compounds in the solid state by the global optimisation approach,” Theoretical and Computational Chemistry. 1999. link Times cited: 2 NOT USED (low confidence) C.-L. Liu, X.-Y. Liu, and L. Borucki, “Defect Generation and Diffusion Mechanisms in Al and Al-Cu,” MRS Proceedings. 1998. link Times cited: 6 Abstract: We describe a newly-developed defect generation mechanism, n… read moreAbstract: We describe a newly-developed defect generation mechanism, namely the grain boundary Frenkel pair (GBFP) model, and corresponding diffusion mechanisms during electromigration developed using atomic molecular statics (MS), Monte Carlo (MC), and molecular dynamics (MD) simulation techniques in Al and Al-Cu. We contend that large numbers of interstitials and vacancies exist at grain boundaries and both contribute to mass transport. Cu preferentially segregates to the interstitial sites at grain boundaries via a Frenkel pair generation process and reduces the overall grain boundary diffusivity due to the strong binding in the Al-Cu dimer. Predictions from our models are in excellent agreement with available experimental data and observations. read less NOT USED (low confidence) J. Howe, “Atomic structure, composition, mechanisms and dynamics of transformation interfaces in diffusional phase transformations,” Materials Transactions Jim. 1998. link Times cited: 15 Abstract: This overview describes progress that has been made in under… read moreAbstract: This overview describes progress that has been made in understanding the atomic structure, composition, mechanisms and dynamics of transformation interfaces in diffusional transformations involving plate-shaped transformation products in the past eight years. Some of the important developments in the area of structure include recognition that diffusional phase transformations obey many of the same crystallographic principles developed for martensite, widespread application of high-resolution transmission electron microscopy (HRTEM) and atomistic modelling to understand interphase boundary structure and energetics, and determination of the atomic structure and energetics of high-index ledged interfaces. In the area of composition, recent development of thermal field-emission and energy-filtering HRTEM allows compositional profiling at precipitate interfaces with subnanometer spatial resolution. In addition, the position sensitive atom-probe (PoSAP), which can determine both the mass and position of individual atoms, makes it possible to map elemental distributions in some materials with atomic resolution. These techniques are expected to provide an abundance of data on composition at transformation interfaces approaching the atomic level in the near future. In terms of the dynamics of transformation interfaces, application of in situ hot-stage HRTEM to precipitate growth and dissolution has provided information on the atomic mechanisms and kinetics of ledge motion and interaction, the mechanisms and kinetics of kink nucleation, and mechanisms of faceting/roughening of precipitate plates at the atomic level. Also, kinetic models for ledge growth which are able to treat multiple-ledge interactions and time-dependent growth have been developed and are available for comparison with experimental data. In addition to discussion of these and other major developments, effort is made to emphasize current strengths and limitations in each area and indicate possible directions for future research. read less NOT USED (low confidence) M. Gungor, L. Gray, S. J. Zhou, and D. Maroudas, “Modeling of Failure in Metallic Thin Films Induced by Stress and Electromigration: A Multiscale Computational Analysis,” MRS Proceedings. 1998. link Times cited: 7 Abstract: A common failure mechanism in metallic thin-film interconnec… read moreAbstract: A common failure mechanism in metallic thin-film interconnects is void propagation driven by electric fields and thermomechanical stresses. In this paper, a multiscale computational analysis is presented for predictive modeling of transgranular void dynamics. The modeling approach is hierarchical and involves atomistic simulations for property database development, molecular-dynamics simulations based on boundary=element methods and techniques for moving boundary propagation. An extremely rich void dynamical behavior is predicted, which includes faceting, facet selection, propagation of slits from the void surface, as well as formation of fine-scale crack-like features on the void surface, in agreement with recent experimental data. read less NOT USED (low confidence) U. Kürpick and T. Rahman, “Diffusion Processes and Pre-Exponential Factors in Homo-Epitaxial Growth on Ag(100),” MRS Proceedings. 1997. link Times cited: 1 NOT USED (low confidence) L. J. Lewis, P. Deltour, P. Jensen, and J. Barrat, “Melting, Freezing, Diffusion, and Colescence of Gold Nanoclusters,” MRS Proceedings. 1997. link Times cited: 0 Abstract: We present a detailed molecular-dynamics study of the coales… read moreAbstract: We present a detailed molecular-dynamics study of the coalescence of gold nanoclusters within the framework of the embedded-atom method. We find macroscopic sintering theories to be unable to describe the coalescing behavior of two small clusters, a failure we attribute to the fact that the nanocrystals are faceted; this has important consequences for the morphology of cluster-assembled materials. We also examine the static and dynamic properties of a 249-atom gold cluster on nickel surfaces. read less NOT USED (low confidence) S. Durukanoğlu, A. Kara, and T. Rahman, “VIBRATIONAL MODES AND RELATIVE STABILITY OF STEPPED SURFACES OF COPPER.” 1997. link Times cited: 1 NOT USED (low confidence) P. Besser, J. Sanchez, D. Field, S. P. Anick, and K. Sahota, “The Microstructure and Electromigration Performance of Damascene-Fabricated Aluminum Interconnects,” MRS Proceedings. 1997. link Times cited: 2 Abstract: Novel metal deposition stack and damascene processing method… read moreAbstract: Novel metal deposition stack and damascene processing methods have been used to fabricate electrically isolated parallel arrays of 1.0 νm deep aluminum-alloy interconnect trenches varying in width from 0.5 μm to 16 μm. The grain size and crystallographic texture of the Al in these trenches has been characterized using transmission electron microscopy (TEM) and local electron backscattered diffraction (EBSD), respectively. Narrow lines (0.5 and 1.0 μm wide) have a bamboo microstructure, intermediate widths (2.0 μm wide) are nearly bamboo, and wide lines (4.0 μm and wider) are polycrystalline. The texture of the lines degrades with decreasing linewidth. A secondary component is demonstrated and its origin proposed. The electromigration reliability of the narrow damascene Al lines was measured, and the observed enhancement of damascene Al interconnects compared to conventionally-fabricated Al interconnects is correlated with the microstructure. read less NOT USED (low confidence) G. Bozzolo, J. Ferrante, and B. Good, “A B.F.S. Method Survey of Surface and Interfacial Properties of Multicomponent Metallic Systems,” MRS Proceedings. 1997. link Times cited: 0 Abstract: Computer modeling of atomic processes on surfaces and bulk m… read moreAbstract: Computer modeling of atomic processes on surfaces and bulk materials has seen noticeable growth due to the availability of new, powerful semiempirical techniques that provide both the numerical simplicity, physical foundation and computational efficiency necessary for the study of complex systems. In an attempt to partially summarize the current status of this work, we apply one of these techniques, the BFS method for alloys, to the study of several basic topics in surface structure and analysis: surface energies of alloy surfaces, multilayer relaxation of ordered alloy surfaces, temperature-dependent segregation profiles, surface alloying and thin film growth of multicompo-nent systems. read less NOT USED (low confidence) U. Kürpick and T. Rahman, “The Influence of Lattice Vibrations on Surface Self Diffusion.” 1997. link Times cited: 0 NOT USED (low confidence) G. Tréglia, “Electronic Structure of Metals and Alloys: from Bulk to Surfaces and Clusters,” ChemInform. 1997. link Times cited: 0 NOT USED (low confidence) D. Ellis, X. Chen, and G. Olson, “Effects of Impurities And Alloying Elements on Iron Grain Boundary Cohesion,” MRS Proceedings. 1997. link Times cited: 1 NOT USED (low confidence) G. Tréglia and B. Legrand, “How Far to Use Tight-Binding Potentials for Bimetallic Surface Modelling?,” MRS Proceedings. 1997. link Times cited: 5 NOT USED (low confidence) T. Einstein, “Chapter 11 – Interactions Between Adsorbate Particles.” 1996. link Times cited: 28 NOT USED (low confidence) C. Massobrio and P. Blandin, “Classical and First Principles Molecular Dynamics Simulations in Material Science: Application to Structural and Dynamical Properties of Free and Supported Clusters.” 1996. link Times cited: 1 NOT USED (low confidence) H. Liu, H. Zhang, H. Ren, S. Ouyang, and R. Yuan, “The influence of the cluster models on the study of electronic structure of MgO/Ag interface,” Ceramics International. 1996. link Times cited: 5 NOT USED (low confidence) A. Carol, M. Alurralde, R. Saliba, and M. Carol, “Heat and Mass Transport Induced by Collision Cascades,” MRS Proceedings. 1996. link Times cited: 0 Abstract: Irradiation of materials with energetic particles produces c… read moreAbstract: Irradiation of materials with energetic particles produces changes in the microstructure that affect mechanical properties. In previous work the authors studied the thermal aspects of the quenching of collision cascades that involve nanoscale phase transitions between the solid and the liquid states of the target. In this work they present a rigorous treatment of these phenomena, including a detailed description of the Stefan problem in three dimensions and diffusion in thermal gradients. This approach is oriented to give a quantitative description of the influence of the primary knock-on spectrum on the microstructure short after the quenching of the heat spike. read less NOT USED (low confidence) C. Massobrio, “The Microscopic World of Atomic Diffusion: Dynamical Simulations as a Tool to Probe the Temporal Evolution of Complex Systems.” 1996. link Times cited: 0 NOT USED (low confidence) E. Alonso, M. Caturla, M. Tang, H. Huang, and T. D. Rubia, “Molecular dynamics simulation of cascade damage in gold,” MRS Proceedings. 1996. link Times cited: 5 Abstract: High-energy cascades have been simulated in gold using molec… read moreAbstract: High-energy cascades have been simulated in gold using molecular dynamics with a modified embedded atom method potential. The results show that both vacancy and interstitial clusters form with high probability as a result of intracascade processes. The formation of clusters has been interpreted in terms of the high pressures generated in the core of the cascade during the early stages. The authors provide evidence that correlation between interstitial and vacancy clustering exists. read less NOT USED (low confidence) B. Mutasa and D. Farkas, “Atomistic Structure of High Index Surfaces,” MRS Proceedings. 1996. link Times cited: 0 NOT USED (low confidence) D. Papaconstantopoulos and M. Mehl, “New Tight-Binding Methodology for Calculating Total Energies of Solids.” 1996. link Times cited: 1 NOT USED (low confidence) P. Gumbsch, “Atomistic Modeling of Failure Mechanisms.” 1996. link Times cited: 5 NOT USED (low confidence) D. Keffer, F. Streitz, and J. Mintmire, “Atomic-Scale Simulations of Structural Properties of Ceramics,” MRS Proceedings. 1996. link Times cited: 0 NOT USED (low confidence) T. Kitamura, K. Yashiro, and R. Ohtani, “Atomic Simulation on Deformation and Fracture of Microcomponent,” Advances in Engineering Plasticity and its Applications. 1996. link Times cited: 1 NOT USED (low confidence) R. Averback, M. Ghaly, and H. Zhu, “Surface effects during ion beam processing of materials,” MRS Proceedings. 1995. link Times cited: 2 Abstract: Microstructural changes of surfaces during ion implantation … read moreAbstract: Microstructural changes of surfaces during ion implantation have been investigated on the atomic level by molecular dynamics computer simulations. Unlike past surface studies, which have been focused on the problem of sputtering, the current work examines the effects of collective materials response on surface topography. Collective behavior has been noted for the crystal interior in the context of thermal spikes, but the authors show here that it can lead to far more dramatic consequences at the surface. The investigation includes implantation in several metals, but emphasizing Pt, Si and Ge. In addition, the study includes the first simulations of implantations of a metallic glass, CuTi, and amorphous Si. read less NOT USED (low confidence) P. Gumbsch, “On The Response Of Dynamic Cracks To Increasing Overload,” MRS Proceedings. 1995. link Times cited: 4 Abstract: One of the most interesting questions in the dynamics of bri… read moreAbstract: One of the most interesting questions in the dynamics of brittle fracture is how a running brittle crack responds to an overload, i.e. to a mechanical energy release rate larger than that due to the increase in surface energy of the two cleavage surfaces. To address this question, dynamically running cracks in different crystal lattices are modelled atomistically under the condition of constant energy release rate. Stable crack propagation as well as the onset of crack tip instabilities are studied. It will be shown that small overloads lead to stable crack propagation with steady state velocities which quickly reach the terminal velocity of about 0.4 of the Rayleigh wave speed upon increasing the overload. Further increasing the overload does not change the steady state velocity but significantly changes the energy dissipation process towards shock wave emission at the breaking of every single atomic bond. Eventually the perfectly brittle crack becomes unstable, which then leads to dislocation generation and to the production of cleavage steps. The onset of the instability as well as the terminal velocity are related to the non-linearity of the interatomic interaction. read less NOT USED (low confidence) F. Streitz and J. Mintmire, “Metal/oxide interfaces: an electrostatics-based model,” Composite Interfaces. 1994. link Times cited: 7 Abstract: We report on the development of a novel computational method… read moreAbstract: We report on the development of a novel computational method for molecular dynamics simulations which explicitly includes variable charge transfer between anions and cations. This method is found to be capable of describing the elastic properties, surface energies, and surface relaxation of crystalline metal-oxides accurately. We present results for a simulation of adhesive failure at a model metal/oxide heterophase interface between an aluminum (111) face and an α-alumina (0001) face. Our results indicate that this approach can provide physically realistic empirical potentials for future simulations on mixed metal/metal-oxide systems. read less NOT USED (low confidence) R. P. Messmer, “Computational materials science — a personal perspective of an industrial scientist,” Computational Materials Science. 1994. link Times cited: 6 NOT USED (low confidence) D. Paithankar, J. Talbot, and R. P. Andres, “Molecular Dynamics Simulation of the Elastic Deformation of Nanometer Diameter Metal Clusters,” MRS Proceedings. 1994. link Times cited: 0 NOT USED (low confidence) M.-L. Turi, R. Zugic, B. Szpunar, U. Erb, G. Palumbo, and V. Krstić, “Atomistic simulation of grain boundaries of the twin limited structure in Nisub 3Al,” MRS Proceedings. 1994. link Times cited: 0 Abstract: Embedded atom method molecular dynamics simulations of low {… read moreAbstract: Embedded atom method molecular dynamics simulations of low {Sigma} grain boundaries in Ni{sub 3}Al are presented. The results show that the grain boundary plane has a larger effect on grain boundary energy than the {Epsilon} value, rigid body translations and stoichiometry. Assessment of the energies of {Sigma}3{sup n} (n {>=} 1) grain boundaries in Ni{sub 3}Al for various grain boundary planes indicates that only the {Sigma}3 grain boundary is energetically preferred. The implications of this result for the development of the twin limited structure based on energetic considerations are discussed. read less NOT USED (low confidence) F. Streitz and J. Mintmire, “Atomic Scale Simulations of Tensile Failure in Metal Oxides,” MRS Proceedings. 1994. link Times cited: 1 Abstract: The authors describe atomic-scale simulations of the failure… read moreAbstract: The authors describe atomic-scale simulations of the failure under tensile load of an aluminum-alumina heterostructure, comparing the results with similar simulations of failure in metallic aluminum and the ceramic {alpha}-alumina. The simulations were performed using a novel computational method which explicitly includes variable charge transfer between cations and anions in an empirical potential. From their simulations they estimate the theoretical limit of yield stress for the interface to be approximately 2 GPa, at a strain of only a few percent. The theoretical limit for yield stress in {alpha}-alumina, for comparison, is about 45 GPa. read less NOT USED (low confidence) Y. Kogure, Y. Abe, O. Kouchi, and M. Doyama, “Determination of parameters in n-body interaction potentials by embedded function.” 1994. link Times cited: 0 NOT USED (low confidence) Y. Kogure, O. Kouchi, and M. Doyama, “Elastic and Lattice Dynamical Properties of Metals Studied by N-Body Potential,” MRS Proceedings. 1994. link Times cited: 1 Abstract: Higher order elastic constants and phonon dispersion relatio… read moreAbstract: Higher order elastic constants and phonon dispersion relation have been calculated by using the n-body potential based on the embedded atom method. Results of second- and third-order elastic constants for Cu, Ag, and Au crystal were compared with the experimental data and the Cauchy discrepancy was discussed. A result of phonon dispersion relation for Cu crystal was also shown. read less NOT USED (low confidence) J. Hafner, “Quantum Theory of Structure: Crystals and Quasicrystals, Melts and Glasses.” 1994. link Times cited: 0 NOT USED (low confidence) S. E. Wonchoba, W. P. Hu, and D. Truhlar, “Reaction Path Approach to Dynamics at a Gas-Solid Interface: Quantum Tunneling Effects for an Adatom on a non-rigid Metallic Surface.” 1994. link Times cited: 6 NOT USED (low confidence) C. L. Rohrer, “Interatomic potentials for Al-Cu-Ag solid solutions,” Modelling and Simulation in Materials Science and Engineering. 1994. link Times cited: 16 Abstract: A set of embedded atom method (EAM) interatomic potentials f… read moreAbstract: A set of embedded atom method (EAM) interatomic potentials for dilute Al-Cu-Ag solid solutions has been developed. These potentials reproduce the experimental elastic constants, stacking fault energies, vacancy formation energies, and migration energies of the pure metals, as well as the migration energies of Cu and Ag in Al and the dependence of host lattice parameter on solid solution composition. With these potentials, simulations can be carried out which may provide insight into such phenomena as interface segregation, the influence of composition on stacking faults, and dislocation-solute interactions in binary Al-Cu, Al-Ag, and Cu-Ag and ternary Al-Cu-Ag solid solutions. read less NOT USED (low confidence) F. Streitz and J. Mintmire, “Charge transfer and bonding in metallic oxides,” Journal of Adhesion Science and Technology. 1994. link Times cited: 27 Abstract: We discuss the development of interaction potentials which e… read moreAbstract: We discuss the development of interaction potentials which explicitly allow for charge transfer in metallic oxides. The charge transfer is calculated self-consistently using a charge equilibration approach, which allows the amount of charge transferred to respond to the electrostatic environment. We model the metal-metal, metal-oxygen, and oxygen-oxygen interactions with Rydberg function pair potentials. By fitting the Rydberg potential parameters to the elastic and structural constants of the material, we arrive at an efficient model for the simulation of metallic oxides. We demonstrate the applicability of the model by describing some preliminary results on the rutile phase of titanium dioxide. read less NOT USED (low confidence) J. Sprague and C. M. Gilmore, “Effects of atom energy on metal-on-metal film nucleation and growth,” Surface & Coatings Technology. 1994. link Times cited: 4 NOT USED (low confidence) G. Boisvert and L. J. Lewis, “Single-Atom Diffusion on Silver and Gold Surfaces,” MRS Proceedings. 1993. link Times cited: 1 Abstract: We present the results of a detailed Molecular-dynamics stud… read moreAbstract: We present the results of a detailed Molecular-dynamics study of single-atom diffusion on the surfaces Ag (100) and (111), and Au (111), using the embedded-atom method to describe the interactions between the atoms. We find that diffusion is Arrhenius-like up to temperatures corresponding to a large fraction of the activation energy. We demonstrate, in addition, that an excellent description of the rate of diffusion is provided by a simple transition-state theory, together with parameters that derive directly from the static potential-energy surface. The Model predicts very accurately the activation energies, while the prefactor for diffusion is obtained within a factor of 2, a discrepancy we attribute to the neglect, in the Model, of the details of the structure of the surface. At higher temperatures, diffusion becomes clearly non-Arrhenius, and the model fails. read less NOT USED (low confidence) G. Beltz and S. Schmauder, “A Multi-Plane Model for Defect Nucleation at Cracks,” MRS Proceedings. 1993. link Times cited: 2 Abstract: A mathematical model (2D) of dislocation generation at crack… read moreAbstract: A mathematical model (2D) of dislocation generation at cracks on interfaces is presented, which takes into account the role of slip processes on several slip planes in the vicinity of a crack. The work investigates the effects of other incipient dislocations on the nucleation and emission of the primary dislocation that emits first and is responsible for crack-tip blunting on atomic length scales. The modeling makes use of the recently-developed Peierls-Nabarro framework for dislocation nucleation. It is found that there is a moderate increase in the critical load necessary to emit a dislocation, when incipient slip activity is allowed to occur on the prolongation of the crack plane. Furthermore, the slip at the tip, the quantity which characterizes to what extent an incipient dislocation forms before it emits, decreases when the dual slip-plane model is used. Implications for the ductile versus brittle response of Ni are discussed. read less NOT USED (low confidence) J. R. Ray and R. J. Wolf, “New Ensembles for Constant-Chemical-Potential Simulations.” 1993. link Times cited: 0 NOT USED (low confidence) T. Tsong, “Studies of atomic processes useful for atomic manipulation with STM,” Materials Chemistry and Physics. 1993. link Times cited: 5 NOT USED (low confidence) J. Bernholc, D. J. Oh, D. Sullivan, and J. Yi, “Quantum Molecular Dynamics Studies of the Structure and Dynamics of Metal Clusters.” 1993. link Times cited: 0 NOT USED (low confidence) U. Landman, R. Barnett, H.-P. Cheng, C. L. Cleveland, and W. Luedtke, “Simulations of Materials: Clusters and Interfacial Junctions.” 1993. link Times cited: 3 NOT USED (low confidence) L. Zhao, R. Najafabadi, and D. J. Srolovtz, “An Atomistic Study of Surface Vacancy Diffusion,” MRS Proceedings. 1993. link Times cited: 0 Abstract: Diffusion of atoms and molecules on surfaces plays an import… read moreAbstract: Diffusion of atoms and molecules on surfaces plays an important role in the growth of thin films. In the present study, the surface vacancy diffusion on Cu and Ni (100) and (111) planes is investigated via atomistic simulations. This investigation is performed using the Embedded Atom Method (EAM) interatomic potentials and the finite temperature properties are determined within the local harmonic and quasiharmonic frameworks. This study helps reveal fundamentals of surface vacancy diffusion in the thin film growth. Our results show that the vacancy diffusion is important on (100) surface but it is not the dominant diffusion mechanism on (111) plane. read less NOT USED (low confidence) Y. Sasajima, S. Taya, and R. Yamamoto, “A Computation of Elastic Constant of Metallic Multilayered Film Using EAM Potential,” Materials Transactions Jim. 1993. link Times cited: 4 Abstract: Elastic constants of metallic multilayered film have been ca… read moreAbstract: Elastic constants of metallic multilayered film have been calculated using EAM (Embedded Atom Method) potential. Structural relaxation by variable cell molecular dynamics has been performed carefully to get fully relaxed model structure of (111) stacked multilayers of Au/Ni, Cu/Ni, Cu/Pd and Ag/Pd. We could not observe any supermodulus effect in the calculated samples read less NOT USED (low confidence) G. Gilmer, “CHAPTER 8 – Atomic-scale Models of Crystal Growth.” 1993. link Times cited: 5 NOT USED (low confidence) N. Miskovsky, T. Tsong, and C.-M. Wei, “Atomic Manipulation Using Field Evaporation.” 1993. link Times cited: 0 NOT USED (low confidence) B. Bolding and E. Carter, “Two-dimensional Metallic Adlayers: Dispersion Versus Island Formation.” 1993. link Times cited: 0 NOT USED (low confidence) U. Landman and W. Luedtke, “Consequences of Tip—Sample Interactions.” 1993. link Times cited: 11 NOT USED (low confidence) B. Szpunar, U. Erb, K. T. Aust, G. Palumbo, and L. J. Lewis, “The Influence of Grain-Boundary Structural Disorder on the Magnetic Properties of Nanocrystalline Nickel,” MRS Proceedings. 1993. link Times cited: 4 NOT USED (low confidence) M. Bernasconi, E. Tosatti, and E. Tosatti, “Reconstruction, disordering and roughening of metal surfaces,” Surface Science Reports. 1993. link Times cited: 45 NOT USED (low confidence) J. Rickman, R. Najafabadi, and D. Srolovitz, “Finite-Temperature Properties From a Single Zero-Temperature Energy Minimization,” MRS Proceedings. 1992. link Times cited: 0 NOT USED (low confidence) A. Taiwo, H. Yan, and G. Kalonji, “Structure and Elastic Properties of Ni/Cu and Ni/Au Multilayers,” MRS Proceedings. 1992. link Times cited: 2 NOT USED (low confidence) L. Wille and H. Dreyssé, “Computer Simulation of Freezing and Melting in Transition Metal Clusters.” 1992. link Times cited: 0 NOT USED (low confidence) T. Tombrello, “Molecular Dynamics Simulation of Cluster-Ion Impacts.” 1992. link Times cited: 0 NOT USED (low confidence) J. Rice, G. Beltz, and Y. Sun, “Peierls Framework for Dislocation Nucleation from a Crack Tip.” 1992. link Times cited: 55 NOT USED (low confidence) S. Foiles, “Interatomic Interactions for BCC Metals Based on the Low Order Moments of the Density of States,” MRS Proceedings. 1992. link Times cited: 4 Abstract: A model of the energetics of bcc transition metals based on … read moreAbstract: A model of the energetics of bcc transition metals based on the low-order moments of the electronic density of states is presented. The new feature of the model is an additional energy term related to the fourth moment of the density of states. This term reflects the coarse shape of the density of states. The model is tested by the computation of point defect properties, phonon dispersions, structural energy differences and surface properties. The results are compared to experiment, ab-initio calculations and other model interatomic potentials. The results indicate that the inclusion of the fourth moment term in the energy does not significantly improve the description of properties of the bulk bcc metals. However, the fourth moment term substantially improves the description of large deviations from the bcc bulk such as surfaces and alternative crystal structures. read less NOT USED (low confidence) F. Ruette, A. Sierraalta, and A. J. Hernández, “Quantum Mechanical Calculations of Chemical Interactions on Transition Metal Surfaces.” 1992. link Times cited: 3 NOT USED (low confidence) C. Liu and J. B. Adams, “Structure and diffusion of clusters on Ni surfaces,” Surface Science. 1992. link Times cited: 50 NOT USED (low confidence) Y. Sasajima, S. Taya, S. Ozawa, and R. Yamamoto, “Computer simulation study of film formation process.” 1992. link Times cited: 0 NOT USED (low confidence) M. Finnis, “Metal-ceramic cohesion and the image interaction,” Acta Metallurgica Et Materialia. 1992. link Times cited: 77 NOT USED (low confidence) J. Hirth, “Dislocation twist boundary field in cubic crystals,” Physica Scripta. 1992. link Times cited: 0 Abstract: The anisotropic elastic field of a {100} twist boundary in a… read moreAbstract: The anisotropic elastic field of a {100} twist boundary in a cubic crystal is determined. The resulting displacements are compared to experimental results. Several postulates of Lothe are verified. read less NOT USED (low confidence) U. Landman and W. Luedtke, “Atomistic processes of surface and interface formation,” Applied Surface Science. 1992. link Times cited: 7 NOT USED (low confidence) E. C. Sowa, A. Gonis, and X. Zhang, “The Real-Space Multiple-Scattering Theory: A First-Principles Method for the Computation of the Electronic Structure of Extended Defects.” 1992. link Times cited: 0 NOT USED (low confidence) Y. Beaudet, L. J. Lewis, and M. Persson, “Anharmonic Effects at the (100) and (110) Surfaces of Ni,” MRS Proceedings. 1992. link Times cited: 0 NOT USED (low confidence) C. W. Finley, J. Ferrante, and P. Abel, “Homonuclear Metallic Microclusters: Structure and Energetics.” 1992. link Times cited: 0 NOT USED (low confidence) J. Sprague and C. M. Gilmore, “Molecular Dynamics Simulations of Low-Energy Atom-Surface Interactions,” MRS Proceedings. 1992. link Times cited: 1 NOT USED (low confidence) P. Blandin, C. Massobrio, and J. Buttet, “Dynamics of Silver Dimer Deposition on Platinum Surfaces,” MRS Proceedings. 1992. link Times cited: 0 Abstract: We present molecular dynamics results on the diffusion prope… read moreAbstract: We present molecular dynamics results on the diffusion properties of Ag monomers and dimers on the (111) surface of Pt. In particular we focus on the behavior of dimers upon collision on the surface with impact energies of 0.5eV and 5eV. We found that the dimer has the largest probability of being found stable on the surface when the temperature is in the range 150K read less NOT USED (low confidence) U. Landman, W. Luedtke, and E. Ringer, “Paper I (I) Atomic Scale Mechanisms of Adhesion, Friction and Wear,” Tribology and Interface Engineering Series. 1992. link Times cited: 0 NOT USED (low confidence) M. Robinson, “Computer Simulation of Atomic Collision Processes in Solids,” MRS Proceedings. 1992. link Times cited: 2 Abstract: Computer simulation is a major tool for studying the interac… read moreAbstract: Computer simulation is a major tool for studying the interactions of swift ions with solids which underlie processes such as particle backscattering, ion implantation, radiation damage, and sputtering. Numerical models are classed as molecular dynamics or binary collision models, along with some intermediate types. Binary collision models are divided into those for crystalline targets and those for structureless ones. The foundations of such models are reviewed, including interatomic potentials, electron excitations, and relationships among the various types of codes. Some topics of current interest are summarized. read less NOT USED (low confidence) Y. S. Li, Y. Cai, and J. Newsam, “Structure and Growth of Small Palladium Clusters,” MRS Proceedings. 1992. link Times cited: 1 NOT USED (low confidence) J. Ferrante and G. Bozzolo, “Computational Techniques in Tribology and Material Science at the Atomic Level.” 1992. link Times cited: 2 NOT USED (low confidence) S. Foiles, “Atomistic Simulations of Surfaces and Interfaces.” 1992. link Times cited: 5 NOT USED (low confidence) L. Zhao, R. Najafabadl, and D. Srolovitz, “Methods for Determining Vacancy Formation Thermodynamic,” MRS Proceedings. 1992. link Times cited: 0 Abstract: The vacancy formation thermodynamics in six fcc metals Ag, A… read moreAbstract: The vacancy formation thermodynamics in six fcc metals Ag, Au, Cu, Ni, Pd and Pt are determined from atomistic simulations as a function of temperature. This investigation is performed using the Embedded Atom Method interatomic potentials and the finite temperature properties are determined within the local harmonic and the quasiharmonic frameworks. We find that the temperature dependence of the vacancy formation energy can make a significant contribution to the vacancy concentration at high temperatures. An additional goal of the present study is to evaluate the accuracy of the local harmonic method under circumstances in which the excess entropy associated with the formation of a defect is very small. Our data demonstrate that while the errors associated with determining the vacancy formation entropy in the local harmonic model are large, a simple extension to the local harmonic method yields thermodynamic properties comparable to that obtained in the quasiharmonic model, but with much higher computational efficiency. read less NOT USED (low confidence) J. R. Rice, “Dislocation nucleation from a crack tip : an analysis based on the Peierls concept,” Journal of The Mechanics and Physics of Solids. 1992. link Times cited: 26 NOT USED (low confidence) P. Bristowe, “The sensitivity of diffraction profiles to grain boundary segregation,” Scripta Metallurgica Et Materialia. 1991. link Times cited: 1 NOT USED (low confidence) K. Merkle and D. Wolf, “Correlations Between Grain Boundary Structure and Energy,” MRS Proceedings. 1991. link Times cited: 5 Abstract: High-resolution electron-microscopy (HREM) and computer simu… read moreAbstract: High-resolution electron-microscopy (HREM) and computer simulations of tilt grain boundaries (GBs) in Au are used to investigate correlations between atomic-scale GB structure and energy. The energies calculated for a variety of symmetric and asymmetric GBs suggest that asymmetric GB-plane orientations are often preferred over symmetric ones. The experimentally observed faceting behavior agrees with the computed energies. Computer simulations indicate general interrelations between GB energy and (i) volume expansion and (ii) the number of broken bonds per unit area of GB. These atomic-scale microstructural GB parameters, as evaluated from HREM observations, are compared to simulation results. read less NOT USED (low confidence) F. Ducastelle, “Tight-Binding Potentials.” 1991. link Times cited: 11 NOT USED (low confidence) R. Berry, “How We and Molecules Explore Molecular Landscapes.” 1991. link Times cited: 3 NOT USED (low confidence) J. Bernholc, J. Yi, and D. Sullivan, “Structural transitions in metal clusters,” Faraday Discussions. 1991. link Times cited: 23 Abstract: The methods for first-principles calculation of the structur… read moreAbstract: The methods for first-principles calculation of the structure and dynamics of clusters have now progressed to a point where clusters containing ca. 50 non-transition-metal atoms can be studied. As a paradigm, we studied the energetics of structural transformations in 13- and 55-atom Al clusters, which can assume both perfect icosahedral and cuboctahedral structures. Using the Car–Parrinello (quantum molecular dynamics) formalism, we found Al13 has a unique structure, a slightly distorted icosahedron, but Al55 has several inequivalent but energetically nearly degenerate structures. The degeneracy in Al55 is due to the short range of the effective interatomic interactions in a metallic cluster and should lead to floppiness at finite temperatures. A new accurate procedure for calculating ionization potentials (Ei) and electron affinities (Eea) within the Car–Parrinello formalism was developed and applied to Al clusters. Unfortunately, it appears that at least for some clusters, most notably for Al55, the Ei and Eea are very similar for different structural models of this cluster. A formulation and the first tests of a new multigrid-based method for real space electronic structure calculations are briefly described. This method should make possible calculations similar to the above for clusters containing transition metal and/or first-row atoms in the fairly near future. read less NOT USED (low confidence) J. B. Adams, W. G. Wolfer, S. Foiles, C. Rohlfing, and C. V. Siclen, “Theoretical Studies of Helium in Metals.” 1991. link Times cited: 6 NOT USED (low confidence) J. Sun and C. Rottman, “Reconstructions and Morphological Instabilities of FCC (110)Surfaces,” MRS Proceedings. 1991. link Times cited: 0 NOT USED (low confidence) A. Banerjea, J. Ferrante, and J. R. Smith, “Adhesion at metal interfaces.” 1991. link Times cited: 7 NOT USED (low confidence) J. Eckert, J. C. Holzer, C. Krill, and W. Johnson, “Synthesis And Characterization Of Ball-Milled Nanocrystalline Fcc Metals,” MRS Proceedings. 1991. link Times cited: 5 Abstract: Nanocrystalline fee metals (Al, Cu, Ni, Pd, Rh, Ir) have bee… read moreAbstract: Nanocrystalline fee metals (Al, Cu, Ni, Pd, Rh, Ir) have been prepared by ball milling. The development of the microstructure is investigated by x-ray diffraction, differential scanning calorimetry (DSC), and transmission electron microscopy (TEM). The final grain sizes range from 6 to 22 nm and scale with the melting point and the bulk modulus of the elements: metals with higher melting point and bulk modulus have a smaller final grain size. From this a general relation between the deformation mechanism during ball milling and the ultimate grain size achievable by this technique is inferred. With decreasing grain size the lattice strain is enhanced and deformation enthalpies of up to 40 % of the heat of fusion are stored in the material. The contributions of the lattice strain and of die excess enthalpy of the grain boundaries to the stored enthalpies are critically assessed. The kinetics of grain growth are investigated by mermal analysis. The activation energy for grain boundary migration is derived from a modified Kissinger analysis and estimates of the grain boundary enthalpy are given. read less NOT USED (low confidence) N. Moody and S. Foiles, “An Atomistic Study of the Equilibrium Segregation of Hydrogen to Tilt Boundaries in Nickel,” MRS Proceedings. 1991. link Times cited: 5 NOT USED (low confidence) P. Shewmon and S. A. Dregia, “Determination of Energy Minima for Dissimilar Metal Interfaces,” MRS Proceedings. 1991. link Times cited: 0 Abstract: Relative orientations which correspond to minimum energy can… read moreAbstract: Relative orientations which correspond to minimum energy can be found by particle rotation methods, both for particles on free surface and particles inside a solid. For common fcc metals (Ni,Ag,Cu,Ag) the minimum energy orientations predicted by Embedded Atom Method calculations correspond well with experimental observations. Epitaxial studies of growth on (001)Cu and (111)Cu show the observed orientation relationships of vapor deposited Ag and Au are consistent with EAM calculations and the limited particle rotation experiments available. read less NOT USED (low confidence) N. Moody and S. Foiles, “An Atomistic Study of Hydrogen Effects on the Fracture of Tilt Boundaries in Nickel.,” MRS Proceedings. 1991. link Times cited: 2 NOT USED (low confidence) N. Winograd and B. Garrison, “Surface Structure and Reaction Studies by Ion-Solid Collisions,” ChemInform. 1991. link Times cited: 4 NOT USED (low confidence) R. Averback and H. Hsieh, “Fundamental Aspects of Energetic Collisions Between Clusters of Atoms and Metal Surfaces,” MRS Proceedings. 1991. link Times cited: 1 NOT USED (low confidence) W. S. Lee, R. Outlaw, G. Hoflund, and M. Davidson, “Auger electron intensity variations in oxygen-charged silver,” Applied Surface Science. 1991. link Times cited: 2 NOT USED (low confidence) B. Legrand, M. Guillopé, J. S. Luo, and G. Tréglia, “Multilayer relaxation and reconstruction in bcc and fcc transition and noble metals,” Vacuum. 1990. link Times cited: 21 NOT USED (low confidence) C. English, S. M. Murphy, and J. M. Perks, “Radiation-induced segregation in metals,” Journal of the Chemical Society, Faraday Transactions. 1990. link Times cited: 19 Abstract: During fast-particle irradiation significant fluxes of point… read moreAbstract: During fast-particle irradiation significant fluxes of point defects can be set up adjacent to sinks such as surfaces or internal grain boundaries. In general, the different atomic species in an alloy move at different rates in response to these point-defect fluxes so that some species move towards sinks while others move away. This radiation-induced segregation causes significant changes in the local composition near sinks such as grain boundaries, and this can have important implications for the bulk properties of materials. In recent years this has been studied in some detail, particularly in structural materials used in nuclear reactor components. This paper describes recent advances in experimental techniques which have improved our understanding of radiation-induced segregation. The experimental evidence for the various different mechanisms for solute segregation are discussed, particularly for the cases of dilute and concentrated alloys. The role of accelerator-based experiments on high-purity model alloys is emphasised and the problems of understanding the behaviour in more complex alloys and steels is highlighted. Theoretical models for radiation-induced segregation behaviour in both dilute and concentrated alloys are reviewed, and their success in describing experimental data discussed. In the case of dilute alloys the work of Lidiard and co-workers on the kinetic theory of diffusion in dilute alloys has provided a firm theoretical foundation for the model of irradiation-induced segregation. Lastly, the directions for further work are indicated, including the need for a greater understanding of the role of interstitial point defects. read less NOT USED (low confidence) P. Gumbsch, M. Daw, S. Foiles, and H. Fischmeister, “Atomistic Structure and Composition of a Ag/Ni Interphase Boundary,” MRS Proceedings. 1990. link Times cited: 1 NOT USED (low confidence) B. Dodson, “Nonadiabatic bonding interactions in sub-keV ion-solid processes,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1990. link Times cited: 4 NOT USED (low confidence) H.-P. Cheng and R. Berry, “Surface Melting and Surface Diffusion on Clusters,” MRS Proceedings. 1990. link Times cited: 13 NOT USED (low confidence) B. Rice, B. Garrett, M. Koszykowski, S. Foiles, and M. Daw, “Kinetic isotope effects for hydrogen diffusion in bulk nickel and on nickel surfaces,” Journal of Chemical Physics. 1990. link Times cited: 28 Abstract: Diffusion coefficients for H, D, and T on a Ni(100) surface … read moreAbstract: Diffusion coefficients for H, D, and T on a Ni(100) surface and in bulk Ni are calculated using variational transition state theory with semiclassical ground‐state transmission coefficients using two potential energy surfaces obtained by the embedded atom method (EAM). The original EAM potential reproduces experimental bulk diffusion coefficients, but greatly overestimates the diffusion coefficients for H and D on Ni(100). By refining the empirical potential parameters, a new EAM potential is developed that accurately reproduces both the bulk and surface diffusion coefficients. The variational transition state theory calculations are used to analyze the unusually low (compared to gas phase) H/D kinetic isotope effects for diffusion in bulk and on the Ni(100) surface. For the temperature range for which experiments have been carried out, quantum mechanical tunneling contributes negligibly to the diffusion and, in these cases, the kinetic isotope effect is determined largely by the change in zero‐point ener... read less NOT USED (low confidence) M. Daw, “The Embedded Atom Method: A Review.” 1990. link Times cited: 19 NOT USED (low confidence) P. Bacher and P. Wynblatt, “Monte Carlo Modeling of Interphase Boundaries in Cu-Ag and Cu-Ag-Au Alloys,” MRS Proceedings. 1990. link Times cited: 1 Abstract: Monte Carlo simulation, in conjunction with the embedded ato… read moreAbstract: Monte Carlo simulation, in conjunction with the embedded atom method, has been used to model the composition and structure of a semicoherent (001) interphase boundary separating coexisting Cu-rich and Ag-rich phases in a binary Cu-Ag alloy. The results are compared with earlier simulations of the same boundary in a Cu-Ag-Au alloy, in which Au was found to segregate to the interface, and the boundary was found to be unstable with respect to break-up into {111} facets. The boundary in the binary system is also unstable to faceting, but displays both {100} as well as {111} facets. It is concluded that Au segregation in the ternary alloy plays an important role in stabilizing the {111} facets. The interplay between the misfit dislocations present at the interface, and the compositional features of the boundary are also discussed. read less NOT USED (low confidence) A. Carlsson, “Beyond Pair Potentials in Elemental Transition Metals and Semiconductors,” Journal of Physics C: Solid State Physics. 1990. link Times cited: 169 NOT USED (low confidence) G. Tichy, “Linear Surface Model and the Environment-Dependent Effective Pair Potential.” 1990. link Times cited: 2 NOT USED (low confidence) R. Johnson, “Implications of the Embedded-Atom Method Format.” 1990. link Times cited: 5 NOT USED (low confidence) S. Foiles, “Calculation of the Atomic Structure of Grain Boundaries in Metals and Alloys,” MRS Proceedings. 1990. link Times cited: 0 NOT USED (low confidence) D. Wolf and J. Lutsko, “COMPUTER SIMULATION OF ELASTIC AND STRUCTURAL ANOMALIES OF THIN FILMS AND SUPERLATTICES.” 1990. link Times cited: 2 NOT USED (low confidence) V. Heine and J. Hafner, “Volume and Pair Forces in Solids and Liquids.” 1990. link Times cited: 6 NOT USED (low confidence) C. Jansson and P. Morgen, “Factor analysis of d(NE)/dE Auger electron spectra of AuCu alloys: Surface composition during Ar+ ion bombardment and oxidation†,” Surface and Interface Analysis. 1990. link Times cited: 11 Abstract: Factor analysis is applied to d(NE)/dE Auger electron spectr… read moreAbstract: Factor analysis is applied to d(NE)/dE Auger electron spectra of a series of AuCu alloys (25% Au, 50% Au and 75% Au), including in the analysis the spectra of clean Au and Cu surfaces. Surface quantitation is obtained from the low-energy Au NOO and Cu LMM spectra of the alloys. The overlap of these spectra is resolved with factor analysis. An accuracy of the derived, relative surface concentrations of 1:100 is possible with this method, with a similar sensitivity to changes in surface composition. During Ar+ ion bombardment the surfaces of 25% and 50% Au alloys show no difference from the bulk concentrations, within ±1:100, from 1 to 3 keV Ar+ ion energy. For the 75% Au alloy, a slight Au enrichment is produced, when the energy increases from 1 to 3 keV. However, at 500 eV Ar+ ion energy, the surfaces become strongly enriched in Au, probably due to threshold effects for Au. Thus, no evidence for such strongly varying processes are found in molecular dynamics simulations of the mass effect in the sputtering process from a 50%:50% alloy. Factor analysis is also used to detect the presence of chemically affected spectral features during oxygen exposure at room temperature of the alloys and of pure Cu. Oxidation of the copper component is observed, producing at saturation a cuprous oxide with a total copper enrichment of the surface. read less NOT USED (low confidence) J. Boehm and R. Nieminen, “Dynamic Simulations of Dislocation Core Structures in Gold Using Many-Atom Interactions.” 1990. link Times cited: 0 NOT USED (low confidence) V. Vítek, G. Ackland, and J. Cserti, “Atomistic Modeling of Extended Defects in Metalic Alloys: Dislocations and Grain Boundaries in Ll2 Compounds,” MRS Proceedings. 1990. link Times cited: 32 Abstract: Extended defects, such as dislocations and grain boundaries,… read moreAbstract: Extended defects, such as dislocations and grain boundaries, control a wide variety of material properties and their atomic structure is often a governing factor. A necessary precursor for modeling of these structures is a suitable description of atomic interactions. We present here empirical many-body potentials for alloys which represent a very simple scheme for the evaluation of total energies and yet reflect correctly the basic physical features of the alloy systems modeled. As examples of atomistic studies we show results of calculations of the core structures of screw dislocations in Li2 compounds. The same potentials have also been used to calculate structures of grain boundaries in these compounds. The deformation and fracture behavior of L12 alloys is then discussed in the light of grain boundary and dislocation core studies. read less NOT USED (low confidence) B. Dodson, “Molecular dynamics modeling of vapor-phase and very-low-energy ion-beam crystal growth processes,” Critical Reviews in Solid State and Materials Sciences. 1990. link Times cited: 26 Abstract: The development of techniques capable of performing atomicsc… read moreAbstract: The development of techniques capable of performing atomicscale computer simulation of realistic materials science problems is a recent phenomenon. Although the initial attempts to utilize such simulation techniques began not long after introduction of the first digital computers, application was mainly limited to highly idealized statistical mechanics problems due to the computational complexity of more realistic problems. Such problems were generally studied in systems consisting of up to several hundred atoms interacting through simple pairwise potentials. These simulations were of great value in fundamental studies of melting and other phase transitions, the structure of simple classical and quantum liquids, and simple diffusion problems. read less NOT USED (low confidence) M. Manninen, R. Nieminen, and M. Puska, “Introduction to Many-Atom Interactions in Solids.” 1990. link Times cited: 0 NOT USED (low confidence) P. Taylor, “Atomic-scale simulation of adhesion between metallic surfaces,” MRS Proceedings. 1990. link Times cited: 0 Abstract: We have performed MD simulations of adhesive phenomena, on a… read moreAbstract: We have performed MD simulations of adhesive phenomena, on an atomic scale, between metals possessing both smooth and stepped-surfaces. Studies of adhesion between identical metals, consisting of either Au, Cu, or Ni, with (001) or (111) orientations, reveal the existence of adhesive avalanches as the bodies are brought to within a critical separation ({approximately}2 {angstrom}). That is, as the surfaces approach one another, one or both surface layers becomes unstable, and abruptly moves toward the other. This signals a transition from an initial system with two distinct surfaces to one possessing no identifiable surfaces. The presence of adhesive avalanches will pose difficulties in determining adhesive forces and energies by means of atomic force microscopy at sub-nanometer separations of probe tip and sample surface. 7 refs., 3 figs. read less NOT USED (low confidence) G. Campbell, S. Foiles, W. King, M. Rühle, and W. L. Wien, “Hrem Investigation of the Structure of the Σ5(210)/[001] Symmetric Tilt Grain Boundaries in Nb.,” MRS Proceedings. 1990. link Times cited: 0 NOT USED (low confidence) H. Y. Wang, R. Najafabadi, D. Srolovitz, and R. LeSar, “The Free Energy Simulation Approach to Grain Boundary Segregation In Cu-Ni,” MRS Proceedings. 1990. link Times cited: 0 NOT USED (low confidence) R.-J. Jhan and P. Bristowe, “Dynamical Simulation of the Motion of Curved Grain Boundaries Composed of Pyramidal-Shaped Ledges,” MRS Proceedings. 1990. link Times cited: 2 Abstract: A dynamical simulation of curved grain boundaries composed o… read moreAbstract: A dynamical simulation of curved grain boundaries composed of pyramidal-shaped ledges has shown that the boundaries can move by local conservative shuffles of atoms or groups of atoms such that one adjoining crystal grows at the expense of the other. In the model system studied, the shuffles often take the form of correlated rotational displacements about the axis normal to the boundary. The simulations provide support for the atomic mechanism proposed by Babcock and Balluffi to explain their observation of grain boundary migration without the participation of SGBDs. read less NOT USED (low confidence) J. R. Smith, T. Perry, and A. Banerjea, “New, Simple Approach to Defect Energies in Solids via Equivalent Crystals.” 1989. link Times cited: 1 NOT USED (low confidence) H. Hsieh, R. Averback, and R. Benedek, “Molecular Dynamics Simulations of Ionized Cluster Beam Deposition,” MRS Proceedings. 1989. link Times cited: 0 Abstract: Fully dynamical computer simulations have been used to study… read moreAbstract: Fully dynamical computer simulations have been used to study the physical mechanisms of ionized cluster beam deposition. Clusters containing 92 atoms were directed at surfaces with energies per cluster atom ranging from one sixth to three times the cohesive energy of the target. Simulation events employed either Lennard-Jones or Embedded Atom Method potentials. The atoms in the cluster appear to undergo local melting on impact with the substrate. Higher cluster energy increases the spreading of cluster atoms on the substrate and improves epitaxy, but it also increases interdiffusion and produces point defects. 6 refs., 2 figs. read less NOT USED (low confidence) A. Carlsson, “Angular Forces in Transition Metals and Diamond Structure Semiconductors.” 1989. link Times cited: 1 NOT USED (low confidence) M. Daw, “Embedded Atom Method: Many-Atom Description of Metallic Cohesion.” 1989. link Times cited: 15 NOT USED (low confidence) D. Wolf, J. Lutsko, and M. Kluge, “Physical Properties of Grain-Boundary Materials: Comparison of EAM and Central-Force Potentials.” 1989. link Times cited: 18 NOT USED (low confidence) T. Nguyen and S. Yip, “Molecular dynamics study of a bicrystal at elevated temperatures,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1989. link Times cited: 4 NOT USED (low confidence) G. Ackland and V. Vítek, “Application of Many-Body Potentials to Noble Metal Alloys.” 1989. link Times cited: 2 NOT USED (low confidence) J. B. Adams, W. G. Wolfer, and S. Foiles, “Elastic Properties of Grain Boundaries in Copper,” MRS Proceedings. 1989. link Times cited: 3 NOT USED (low confidence) E. C. Sowa, A. Gonis, and X.-G. Zhang, “The Electronic Structure pf Σ5 Grain Boundaries in CU,” MRS Proceedings. 1989. link Times cited: 1 NOT USED (low confidence) D. J. Oh and R. Johnson, “Embedded Atom Method Model for Close-Packed Metals.” 1989. link Times cited: 26 NOT USED (low confidence) J. Eridon, “Derivation of Many Body Potentials to Reproduce Elastic and Vibrational Qualities of FCC and BCC Metals.” 1989. link Times cited: 2 NOT USED (low confidence) J. B. Adams, S. Foiles, and W. G. Wolfer, “Self-Diffusion and Impurity Diffusion of FCC Metals Using the Embedded Atom Method.” 1989. link Times cited: 14 NOT USED (low confidence) R. Najafabadi and G. Kalonji, “A Computer Calorimetry Study of Segregation Free Energy: Cu in a Ni Grain Boundary,” MRS Proceedings. 1988. link Times cited: 1 NOT USED (low confidence) W. G. Hoover, C. G. Hoover, I. Stowers, and W. Siekhaus, “Interface Tribology Via Nonequilibrium Molecular Dynamics,” MRS Proceedings. 1988. link Times cited: 26 Abstract: By borrowing ideas from control theory, Nonequilibrium Molec… read moreAbstract: By borrowing ideas from control theory, Nonequilibrium Molecular Dynamics incorporates temperature, stress, and heat flux directly into atomistic, time-reversible, deterministic equations of motion. We are applying this technique to studies of surface indentation, surface cutting, friction, ablation, and condensation. Here we describe simulations of the indentation and cutting processes using two-dimensional crystals composed of a few thousand particles. read less NOT USED (low confidence) D. Lo, T. Tombrello, M. Shapiro, and D. E. Harrison, “Molecular Dynamics Simulation of Sputtering with Mmany-Body Interactions,” MRS Proceedings. 1988. link Times cited: 0 NOT USED (low confidence) C. M. Gilmore, J. Eridon, V. Provenzano, and J. Sprague, “An Embedded Atom Model of Epitaxy in Ni-Au and Ni-Pt Bicrystals,” MRS Proceedings. 1988. link Times cited: 1 Abstract: The embedded atom model was utilized to study the atomic dis… read moreAbstract: The embedded atom model was utilized to study the atomic displacements and the energy of (111) interfaces in Ni-Au and Ni-Pt bicrystals. A sphere of 459 atoms was utilized with the Ni (111) surface as a great circle through the crystal. The Au or Pt atoms were initially placed in perfect registry with the Ni atoms and then the crystal was allowed to relax to equilibrium positions. Shockley partial dislocations formed at the interface. The elastic strains were not in agreement with classical elasticity calculations. The simulation shows that it is easier to pull atoms apart than to push them together; and thus the larger gold lattice was strained less than the smaller nickel lattice even though the elastic modulus of nickel is greater than that of gold. The Ni and Pt crystals exhibited strong bonding at the interface; whereas, the Ni and Au crystals exhibited little evidence of interface bonding. This result is in agreement with alloy heats of solution and the phase diagrams of these alloys. read less NOT USED (low confidence) Y. Gauthier, “Structure and Composition of Alloy Surfaces by Low Energy Electron Diffraction,” Studies in Surface Science and Catalysis. 1988. link Times cited: 0 NOT USED (low confidence) H. Bonzel and K. Dückers, “Relationship Between Anisotropy of Specific Surface Free Energy and Surface Reconstruction.” 1988. link Times cited: 0 NOT USED (low confidence) P. Taylor and B. Dodson, “Nanometer-scale shock-front structure in metals,” MRS Proceedings. 1988. link Times cited: 0 Abstract: Molecular dynamics shock wave simulations have been performe… read moreAbstract: Molecular dynamics shock wave simulations have been performed, which for the first time include a realistic many-body description of the atomic interactions. The structural instabilities observed in the shock-front structure are dramatically influenced by the many-body effects of these atomic interactions. 11 refs., 2 figs. read less NOT USED (low confidence) M. Baskes, M. Daw, and S. Foiles, “The Embedded Atom Method: Theory and Application,” MRS Proceedings. 1988. link Times cited: 10 NOT USED (low confidence) J. Lutsko and D. Wolf, “A molecular dynamics study of grain boundary behavior at elevated temperatures using an embedded atom potential,” Scripta Metallurgica. 1988. link Times cited: 4 NOT USED (low confidence) P. Underhill, “An investigation of the anomalous surface phonon softening on noble metal 〈111〉 surfaces using the embedded atom method,” Surface Science. 1988. link Times cited: 7 NOT USED (low confidence) S. Foiles, M. Baskes, and M. Daw, “Atomistic Studies of Interfacial Structure and Properties,” MRS Proceedings. 1988. link Times cited: 7 Abstract: The computer simulation of the structure and fracture of int… read moreAbstract: The computer simulation of the structure and fracture of interfaces on an atomic scale requires a computationally efficient prescription for the total energy that is reliable both for small deviations from the bulk as well as for the free surfaces produced during fracture. The recently developed Embedded Atom Method is such a method. It will be briefly described and compared to traditional pair interaction approaches. In particular, it will be shown that the many-body effects inherent in the Embedded Atom Method are essential to correctly describe the experimentally observed surface reconstructions of Au surfaces. The necessary first step in simulating the fracture of an interface, such as a grain boundary, is the determination of the initial or equilibrium atomic configuration of the interface. Equilibrium Monte Carlo simulations using the Embedded Atom Method can determine this structure. This approach will be outlined and various results for grain boundary structure in fcc metals will be presented. The atomic structure of symmetric tilt boundaries is found to be significantly different from that deduced from energy minimization techniques. In addition, the Monte Carlo technique allows for the determination of thermal effects such as the vibrational amplitudes at the interface and the thermal expansion of the interface. read less NOT USED (low confidence) D. Steigerwald and P. Wynblatt, “Calculation of the anisotropy of equilibrium surface composition in metallic solid solutions using the embedded atom method,” Surface Science. 1988. link Times cited: 24 NOT USED (low confidence) M. Daw and S. Foiles, “Calculations of Structural Phases of Transition Metal Surfaces Using the Embedded Atom Method.” 1988. link Times cited: 0 NOT USED (low confidence) A. Zangwill and A. C. Redfield, “Structural selectivity in aluminium-transition-metal alloys,” Journal of Physics F: Metal Physics. 1988. link Times cited: 13 Abstract: The authors present a theory of structural selectivity in al… read moreAbstract: The authors present a theory of structural selectivity in aluminium-rich transition-metal alloys based on an embedded-atom approximation to density functional theory. With this method, which is fast and flexible enough to permit study of complex crystal structures, they discover that electron density requirements largely determine the local arrangement of Al atoms around a central transition metal atom. This fact, in turn, permits them to explain a number of observed crystallographic trends in these systems including the appearance of metastable icosahedral phases which exhibit five-fold diffraction patterns. read less NOT USED (low confidence) J. Ferrante and S. Pepper, “Fundamentals of tribology at the atomic level,” MRS Proceedings. 1988. link Times cited: 3 Abstract: Tribology, the science and engineering of solid surfaces in … read moreAbstract: Tribology, the science and engineering of solid surfaces in moving contact, is a field that encompasses many disciplines: solid state physics, chemistry, materials science, and mechanical engineering. In spite of the practical importance and maturity of the field, the fundamental understanding of basic phenomena has only recently been attacked. An attempt to define some of these problems and indicate some profitable directions for future research is presented. There are three broad classifications: (1) fluid properties (compression, rheology, additives and particulates); (2) material properties of the solids (deformation, defect formation and energy loss mechanisms); and (3) interfacial properties (adhesion, friction chemical reactions, and boundary films). Research in the categories has traditionally been approached by considering macroscopic material properties. Recent activity has shown that some issues can be approached at the atomic level: the atoms in the materials can be manipulated both experimentally and theoretically, and can produce results related to macroscopic phenomena. read less NOT USED (low confidence) K. Jacobsen and J. Nørskov, “Reconstruction of fcc(110) Surfaces.” 1988. link Times cited: 5 NOT USED (low confidence) D. J. Oh and R. Johnson, “A Semi-Empirical Potential for Graphite,” MRS Proceedings. 1988. link Times cited: 4 NOT USED (low confidence) J. Ferrante, G. Bozzolo, C. W. Finley, and A. Banerjea, “Interfacial adhesion - Theory and experiment,” MRS Proceedings. 1988. link Times cited: 14 Abstract: Adhesion, the binding of different materials at an interface… read moreAbstract: Adhesion, the binding of different materials at an interface, is of general interest to many branches of technology, e.g., microelectronics, tribology, manufacturing, construction, etc. However, there is a lack of fundamental understanding of such diverse interfaces. In addition, experimental techniques generally have practical objectives, such as the achievement of sufficient strength to sustain mechanical or thermal effects and/or have the proper electronic properties. In addition, the theoretical description of binding at interfaces is quite limited, and a proper data base for such theoretical analysis does not exist. This presentation will review both experimental and theoretical aspects of adhesion in nonpolymer materials. The objective will be to delineate the critical parameters needed, governing adhesion testing along with an outline of testing objectives. A distinction will be made between practical and fundamental objectives. Examples are given where interfacial bonding may govern experimental consideration. The present status of theory is presented along wiith recommendations for future progress and needs. read less NOT USED (low confidence) S. Foiles, “Calculation of the Structure of Au Grain Boundaries Using the Embedded Atom Method,” MRS Proceedings. 1988. link Times cited: 0 NOT USED (low confidence) M. Daw and M. Baskes, “Application of the Embedded Atom Method to Hydrogen Embrittlement.” 1987. link Times cited: 15 NOT USED (low confidence) S. A. Dregia, P. Wynblatt, and C. Bauer, “Epitaxy for Weakly Interacting Systems of Large Misfit,” MRS Proceedings. 1987. link Times cited: 9 NOT USED (low confidence) F. Ercolessi, A. Bartolini, M. Garofalo, M. Parrinello, and E. Tosatti, “Molecular Dynamics Studies of Gold Surfaces,” Physica Scripta. 1987. link Times cited: 11 Abstract: In the glue model the total cohesion of a metal is determine… read moreAbstract: In the glue model the total cohesion of a metal is determined by a pairwise atom-atom effective interaction plus a many-body force (the glue) which is introduced to ensure optimal coordination. Using parameters optimized for gold, we have studied the structural behaviour of the low index surfaces Au(100), Au(110) and Au(111). We have used a simulated annealing strategy based on molecular dynamics to search the lowest surface energy configuration. In all cases the optimal structures are found to be reconstructed, and remarkably similar to some experimentally suggested reconstruction models. The main driving mechanism is the formation of close-packed triangular surface layers favoured by the glue term. read less NOT USED (low confidence) R. Nieminen, “Dynamics of atoms in low-symmetry systems,” Physica Scripta. 1987. link Times cited: 3 Abstract: Recent ideas to extend the scope and applicability of large-… read moreAbstract: Recent ideas to extend the scope and applicability of large-scale computer simulation of condensed phases are discussed. These include (i) the use of simulated annealing and related methods in first-principles calculations and (ii) the development "effective-medium" and similar approximate approaches to interatomic interactions in low-symmetry situations. Examples of applications to molecular dynamics simulations are presented. read less NOT USED (low confidence) A. Voter and S.-ping Chen, “Accurate Interatomic Potentials for Ni, Al and Ni3Al,” MRS Proceedings. 1986. link Times cited: 529 Abstract: To obtain meaningful results from atomistic simulations of m… read moreAbstract: To obtain meaningful results from atomistic simulations of materials, the interatomic potentials must be capable of reproducing the thermodynamic properties of the system of interest. Pairwise potentials have known deficiencies that make them unsuitable for quantitative investigations of defective regions such as crack tips and free surfaces. Daw and Baskes [Phys. Rev. B 29, 6443 (1984)] have shown that including a local “volume” term for each atom gives the necessary many-body character without the severe computational dependence of explicit n-body potential terms. Using a similar approach, we have fit an interatomic potential to the Ni 3 Al alloy system. This potential can treat diatomic Ni 2 , diatomic Al 2 , fcc Ni, fcc Al and L1 2 Ni 3 Al on an equal footing. Details of the fitting procedure are presented, along with the calculation of some properties not included in the fit. read less NOT USED (low confidence) S. Foiles, “Calculation of the Surface Segregation of Pd-Cu, Pd-Ag, and Pd-Au Alloys,” MRS Proceedings. 1986. link Times cited: 11 Abstract: The surface composition of the (111) surfaces of the binary … read moreAbstract: The surface composition of the (111) surfaces of the binary alloys Pd-Cu, Pd-Ag, and Pd-Au have been computed by Monte Carlo computer simulation with the energetics determined by the Embedded Atom Method. Ag and Au are found to segregate to the first atomic layer of their alloys with Pd while for Pd-Cu alloys the degree of segregation is small but occurs mainly on the second atomic plane. The short-range order of the surfaces is addressed by studying the relative abundance of different compositions of nearest neighbor triangles on the (11) surfaces. The presence of triangles containing only atoms of one element is found to be suppressed by the short-range order. This result is shown to follow from the enhancement for these alloys of nearest neighbor pairs of atoms of different types. read less NOT USED (low confidence) S. Foiles, “Calculation of the Defect and Interface Properties of Ni 3 Al,” MRS Proceedings. 1986. link Times cited: 15 Abstract: The structure and energetics of point defects, surfaces and … read moreAbstract: The structure and energetics of point defects, surfaces and grain boundaries in Ni 3 A1 are investigated using the Embedded Atom Method. The approach is shown to reproduce the experimental phase diagram of the Ni-Al system and the elastic properties of Ni 3 AL. The vacancy and anti-site defect energies are calculated and used to predict the vacancy concentration as a function of bulk composition. The preferred geometries and energies of the low index surfaces are also computed. The equilibrium structure of certain ideal grain boundaries are computed by Monte Carlo computer simulations as a function of bulk composition. It is found that the boundaries act as a sink for anti-sitedefects and the degree of ordering at the boundaries is strongly affected by the bulk composition. The cohesive energy of grain boundaries in Ni 3 A1 is computed and is found to be comparable to that for pure Ni. read less NOT USED (high confidence) S. Kumar et al., “Transferable interatomic potential for aluminum from ambient conditions to warm dense matter,” Physical Review Research. 2023. link Times cited: 0 Abstract: We present a study on the transport and materials properties… read moreAbstract: We present a study on the transport and materials properties of aluminum spanning from ambient to warm dense matter conditions using a machine-learned interatomic potential (ML-IAP). Prior research has utilized ML-IAPs to simulate phenomena in warm dense matter, but these potentials have often been calibrated for a narrow range of temperature and pressures. In contrast, we train a single ML-IAP over a wide range of temperatures, using density functional theory molecular dynamics (DFT-MD) data. Our approach overcomes computational limitations of DFT-MD simulations, enabling us to study transport and materials properties of matter at higher temperatures and longer time scales. We demonstrate the ML-IAP transferability across a wide range of temperatures using molecular-dynamics (MD) by examining the thermal conductivity, diffusion coefficient, viscosity, sound velocity, and ion-ion structure factor of aluminum up to about 60,000 K, where we find good agreement with previous theoretical data. read less NOT USED (high confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 NOT USED (high confidence) W. Guo, C. Wu, X. Meng, C. Luo, and Z. Lin, “Molecular dynamics simulation-based microstructure evolution and subsurface damage of Fe-Ni alloy grinding,” Journal of Intelligent Manufacturing and Special Equipment. 2022. link Times cited: 0 Abstract: PurposeMolecular dynamics is an emerging simulation techniqu… read moreAbstract: PurposeMolecular dynamics is an emerging simulation technique in the field of machining in recent years. Many researchers have tried to simulate different processing methods of various materials with the theory of molecular dynamics (MD), and some preliminary conclusions have been obtained. However, the application of MD simulation is more limited compared with traditional finite element model (FEM) simulation technique due to the complex modeling approach and long computation time. Therefore, more studies on the MD simulations are required to provide a reliable theoretical basis for the nanoscale interpretation of grinding process. This study investigates the crystal structures, dislocations, force, temperature and subsurface damage (SSD) in the grinding of iron-nickel alloy using MD analysis.Design/methodology/approach In this study the simulation model is established on the basis of the workpiece and single cubic boron nitride (CBN) grit with embedded atom method and Morse potentials describing the forces and energies between different atoms. The effects of grinding parameters on the material microstructure are studied based on the simulation results.FindingsWhen CBN grit goes through one of the grains, the arrangement of atoms within the grain will be disordered, but other grains will not be easily deformed due to the protection of the grain boundaries. Higher grinding speed and larger cutting depth can cause greater impact of grit on the atoms, and more body-centered cubic (BCC) structures will be destroyed. The dislocations will appear in grain boundaries due to the rearrangement of atoms in grinding. The increase of grinding speed results in the more transformation from BCC to amorphous structures.Originality/valueThis study is aimed to study the grinding of Fe-Ni alloy (maraging steel) with single grit through MD simulation method, and to reveal the microstructure evolution within the affected range of SSD layer in the workpiece. The simulation model of polycrystalline structure of Fe-Ni maraging steel and grinding process of single CBN grit is constructed based on the Voronoi algorithm. The atomic accumulation, transformation of crystal structures, evolution of dislocations as well as the generation of SSD are discussed according to the simulation results. read less NOT USED (high confidence) R. Bendimerad and E. Petro, “Molecular dynamics studies of ionic liquid-surface interactions for electrospray thrusters,” Journal of Electric Propulsion. 2022. link Times cited: 1 NOT USED (high confidence) H. Han, C. Lee, Y. Kim, J. Lee, S. Yoon, and B. Yoo, “The self-annealing phenomenon of electrodeposited nano-twin copper with high defect density,” Frontiers in Chemistry. 2022. link Times cited: 1 Abstract: Electroplated copper was prepared under typical conditions a… read moreAbstract: Electroplated copper was prepared under typical conditions and a high defect density to study the effect of the defects on its self-annealing phenomenon. Two conditions, grain growth and stress relaxation during self-annealing, were analyzed with electron backscattered diffraction and a high-resolution X-ray diffractometer. Abnormal grain growth was observed in both conditions; however, the grown crystal orientation differed. The direction and relative rate at which abnormal grain growth proceeds were specified through textured orientation, and the self-annealing mechanism was studied by observing the residual stress changes over time in the films using the sin2Ψ method. read less NOT USED (high confidence) A. D. Backer, S. V. Aert, C. Faes, E. A. Irmak, P. Nellist, and L. Jones, “Experimental reconstructions of 3D atomic structures from electron microscopy images using a Bayesian genetic algorithm,” npj Computational Materials. 2022. link Times cited: 2 NOT USED (high confidence) J. Qi, C. Oberdorfer, W. Windl, and E. Marquis, “Ab initio
simulation of field evaporation,” Physical Review Materials. 2022. link Times cited: 5 Abstract: A new simulation approach of field evaporation is presented.… read moreAbstract: A new simulation approach of field evaporation is presented. The model combines classical electrostatics with molecular dynamics (MD) simulations. Unlike previous atomic-level simulation approaches, our method does not rely on an evaporation criterion based on thermal activation theory, instead, electric-field-induced forces on atoms are explicitly calculated and added to the interatomic forces. Atoms then simply move according to the laws of classical molecular dynamics and are"evaporated"when the external force overcomes interatomic bonding. This approach thus makes no ad-hoc assumptions concerning evaporation fields and criteria, which makes the simulation fully physics-based and"ab-initio"apart from the interatomic potential. As proof of principle, we perform simulations to determine material dependent critical voltages which allow assessing the evaporation fields and the corresponding steady-state tip shapes in different metals. We also extract critical evaporation fields in elemental metals and sublimation energies in a high entropy alloy to have a more direct comparison with tabulated values. In contrast to previous approaches, we show that our method is able to successfully reproduce the enhanced zone lines observed in experimental field desorption patterns. We also demonstrate the need for careful selection of the interatomic potential by a comparative study for the example of Cu-Ni alloys. read less NOT USED (high confidence) X. Zhou and T. Suo, “Enhanced electroactive β-phase formation in electrospun poly (vinylidene fluoride-co-hexafluoropropylene) nanowires with gold nanoparticles,” Acta Mechanica Sinica. 2022. link Times cited: 4 NOT USED (high confidence) R. Jagatramka, C. Wang, and M. Daly, “An analytical method to quantify the statistics of energy landscapes in random solid solutions,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (high confidence) J. Gac, “Numerical Modelling of Formation of Highly Ordered Structured Micro- and Nanoparticles – A Review,” KONA Powder and Particle Journal. 2022. link Times cited: 2 Abstract: The aerosol particles play a significant role in the environ… read moreAbstract: The aerosol particles play a significant role in the environment and human health. They are also increasingly used in medicine (drug carriers), preparation (nanocatalysts) and many other fields. For these applications, the particles have to possess unique properties which arise directly from their structure and topology. Indeed, the functionality of the nanostructure particle is defined through its application, like chromatography, sensors, microelectronics, catalysis, and others. That is the reason why people are more and more interested in manufacturing structured particles. The structured particles are the particles with well-defined topological structure. Examples of such particles are porous particles, hollow particles (with the empty space inside), or multi-component particles with the segregation of components in the particle structure. Such particles usually have very interesting features, e.g. porous particles have a significantly larger surface area than the simple spherical particles with similar volume. The present paper contains a comprehensive review of the numerical simulation methods of the formation of highly ordered structured particles. The most important methods will be described in detail and their fields of application (with specific examples), advantages, limitations and information about their accuracy will be given. read less NOT USED (high confidence) D. Hu et al., “Insights into the high-sulphur aging of sintered silver nanoparticles: An experimental and ReaxFF study,” Corrosion Science. 2021. link Times cited: 2 NOT USED (high confidence) S. Ding, Y. Li, Y. Luo, Z. Wu, and X. Wang, “Modified Embedded-Atom Interatomic Potential Parameters of the Ti–Cr Binary and Ti–Cr–N Ternary Systems,” Frontiers in Chemistry. 2021. link Times cited: 1 Abstract: The second nearest-neighbor modified embedded-atom method (2… read moreAbstract: The second nearest-neighbor modified embedded-atom method (2NN MEAM) potential parameters of the Ti–Cr binary and Ti–Cr–N ternary systems are optimized in accordance with the 2NN MEAM method. The novel constructed potential parameters can well reproduce the multiple fundamental physical characteristics of binary and ternary systems and reasonably agree with the first-principles calculation or experimental data. Thus, the newly constructed 2NN MEAM potential parameters can be used for atomic simulations to determine the underlying principle of the hardness enhancement of TiN/CrN multilayered coatings. read less NOT USED (high confidence) P. Brault, M. Ji, D. Sciacqua, F. Poncin‐Epaillard, J. Berndt, and E. Kovačević, “Insight into acetylene plasma deposition using molecular dynamics simulations,” Plasma Processes and Polymers. 2021. link Times cited: 4 NOT USED (high confidence) L. A. Mistryukova, N. P. Kryuchkov, V. Mantsevich, A. Sapelkin, and S. Yurchenko, “Interpolation method for crystals with many-body interactions,” Physical Review B. 2021. link Times cited: 0 Abstract: We propose an interpolation scheme to describe pair correlat… read moreAbstract: We propose an interpolation scheme to describe pair correlations in crystals with many-body interactions that requires only information on relative displacements for the nearest neighbours and in the long range. Using crystalline Ni as a test case, the scheme is shown to deliver the functional form for the radial distribution function at least as well as molecular dynamics simulations. The results provide a fast route for verification of interatomic potentials and study of many-body interactions using a combination of x-ray scattering and x-ray absorption spectroscopy. read less NOT USED (high confidence) M. Gzik-Szumiata, T. Szumiata, D. Morozow, and R. Szewczyk, “Elementary, Atomic-Level Friction Processes in Systems with Metallic Inclusions—Systematic Simulations for a Wide Range of Local Pressures,” Materials. 2021. link Times cited: 0 Abstract: In this work, simulations of friction at the atomic level we… read moreAbstract: In this work, simulations of friction at the atomic level were performed to evaluate the influence of inclusions coming from metallic nanoadditives in the friction pair. The simple 2D model was applied considering appropriate values of Lennard–Jones potential parameters for given sets of interacting atoms. The real sliding pairs were replaced by effective equivalents consisting of several atoms. The calculations were based on the pseudo-static approximation. The simplicity of the model enabled to repeat the fast calculations in a very wide range of local pressures and for several types of atomic tribopairs. The performed simulations demonstrated a strong dependence of the coefficient of friction (COF) on the atomic environment of the atoms constituting a tribopair. It was confirmed theoretically that the Mo-Fe pair is characterized by lower atomic COF than Fe-Fe, Cu-Fe, and Ag-Fe pairs. This points to the great applicational potential of metallic molybdenum coating applications in tribological systems. Moreover, it was demonstrated that, although Cu-Cu and Ag-Ag pairs are characterized by relatively high COF, they lower the friction as inclusions in Fe surfaces. read less NOT USED (high confidence) M. G. Urazaliev, M. E. Stupak, and V. Popov, “Structure and Energy of Symmetric Tilt Boundaries with the 〈110〉 Axis in Ni and the Energy of Formation of Vacancies in Grain Boundaries,” Physics of Metals and Metallography. 2021. link Times cited: 4 NOT USED (high confidence) A. Drewienkiewicz, A. Żydek, M. Trybula, and J. Pstruś, “Atomic Level Insight into Wetting and Structure of Ag Droplet on Graphene Coated Copper Substrate—Molecular Dynamics versus Experiment,” Nanomaterials. 2021. link Times cited: 6 Abstract: Understanding the atomic-level phenomena occurring upon the … read moreAbstract: Understanding the atomic-level phenomena occurring upon the wetting of graphene-coated Cu with liquid Ag is pivotal for the description of the wetting phenomenon and the role of graphene as a diffusion barrier. We have performed molecular dynamics (MD) simulations and confronted with our present experimental results to characterize wetting behavior of graphene coated Cu surfaces. Perfect and defected graphene layers covering Cu surface were wetted with liquid Ag droplet at 1273 K. Structural and topological aspects are discussed to characterize structure of the liquid Ag droplet and a product of wetting reaction occurring on Cu/Gn and Cu/Gndef substrates, also including perfect graphene layer and a pure Cu surface. The obtained results reveal the importance of defects in graphene structure, which play a key role in wetting mechanism and the formation of AgCu alloy. As a consequence, we observe a change of the wetting behavior and topology of both bulk and adsorbed Ag atoms by using Voronoi analysis (VA). Despite the differences in time scale, atomistic simulations allowed us to catch the early stages of wetting, which are important for explaining the final stage of wetting delivered from experiment. Our findings reveal also graphene translucency to metal-metal interactions, observed in previous papers. read less NOT USED (high confidence) I. M. P. Espinosa, T. Jacobs, and A. Martini, “Evaluation of Force Fields for Molecular Dynamics Simulations of Platinum in Bulk and Nanoparticle Forms.,” Journal of chemical theory and computation. 2021. link Times cited: 7 Abstract: Understanding the size- and shape-dependent properties of pl… read moreAbstract: Understanding the size- and shape-dependent properties of platinum nanoparticles is critical for enabling the design of nanoparticle-based applications with optimal and potentially tunable functionality. Toward this goal, we evaluated nine different empirical potentials with the purpose of accurately modeling faceted platinum nanoparticles using molecular dynamics simulation. First, the potentials were evaluated by computing bulk and surface properties-surface energy, lattice constant, stiffness constants, and the equation of state-and comparing these to prior experimental measurements and quantum mechanics calculations. Then, the potentials were assessed in terms of the stability of cubic and icosahedral nanoparticles with faces in the {100} and {111} planes, respectively. Although none of the force fields predicts all the evaluated properties with perfect accuracy, one potential-the embedded atom method formalism with a specific parameter set-was identified as best able to model platinum in both bulk and nanoparticle forms. read less NOT USED (high confidence) A. Fedotov et al., “Theoretical Basis of Quantum-Mechanical Modeling of Functional Nanostructures,” Symmetry. 2021. link Times cited: 0 Abstract: The paper presents an analytical review of theoretical metho… read moreAbstract: The paper presents an analytical review of theoretical methods for modeling functional nanostructures. The main evolutionary changes in the approaches of quantum-mechanical modeling are described. The foundations of the first-principal theory are considered, including the stationery and time-dependent Schrödinger equations, wave functions, the form of writing energy operators, and the principles of solving equations. The idea and specifics of describing the motion and interaction of nuclei and electrons in the framework of the theory of the electron density functional are presented. Common approximations and approaches in the methods of quantum mechanics are presented, including the Born–Oppenheimer approximation, the Hartree–Fock approximation, the Thomas–Fermi theory, the Hohenberg–Kohn theorems, and the Kohn–Sham formalism. Various options for describing the exchange–correlation energy in the theory of the electron density functional are considered, such as the local density approximation, generalized and meta-generalized gradient approximations, and hybridization of the generalized gradient method. The development of methods of quantum mechanics to quantum molecular dynamics or the dynamics of Car–Parrinello is shown. The basic idea of combining classical molecular modeling with calculations of the electronic structure, which is reflected in the potentials of the embedded atom, is described. read less NOT USED (high confidence) H. Guo, L. Zhang, Q. Zhu, C. Wang, G. Chen, and P. Zhang, “Molecular Dynamics Simulation of the Coalescence and Melting Process of Cu and Ag Nanoparticles,” Advances in Condensed Matter Physics. 2021. link Times cited: 0 Abstract: The coalescence and melting process of different sizes and a… read moreAbstract: The coalescence and melting process of different sizes and arrangements of Ag and Cu nanoparticles is studied through the molecular dynamics (MD) method. The results show that the twin boundary or stacking fault formation and atomic diffusion of the nanoparticles play an important role in the different stages of the heating process. At the beginning of the simulation, Cu and Ag nanoparticles will contact to each other in a very short time. As the temperature goes up, Cu and Ag nanoparticles may generate stacking fault or twin boundary to stabilize the interface structure. When the temperature reaches a critical value, the atoms gain a strong ability to diffuse and eventually melt into one liquid sphere. The coalescence point and melting temperature increase as cluster diameter increases. Moreover, the arrangement of Cu and Ag nanoparticles has a certain effect on the stability of the initial joint interface, which will affect subsequent coalescence and melting behavior. read less NOT USED (high confidence) C. Jian-hao, Z. Qiu-yang, Z. Zhen-yu, D. Cong, and P. Zhong-yu, “Molecular dynamics simulation of monocrystalline copper nano-scratch process under the excitation of ultrasonic vibration,” Materials Research Express. 2021. link Times cited: 8 Abstract: In order to explore the mechanism of unidirectional ultrason… read moreAbstract: In order to explore the mechanism of unidirectional ultrasonic vibration-assisted machining from a microscopic point of view, the molecular dynamic (MD) simulation method is used to simulate the scratch process of monocrystalline copper under ultrasonic excitation. By comparing the simulation results of traditional scratching and ultrasonic vibration-assisted scratching, the influences of ultrasonic vibration on the surface morphology, the tangential force, and the evolution of the crystal’s internal defects are discussed. The results show that the ultrasonic vibration can improve the surface quality of the workpiece, reduce the tangential force, and reduce the energy consumption. Simultaneously, ultrasonic vibration promotes the interaction between dislocations, accelerates the annihilation of dislocations, effectively reduces work hardening caused by dislocation accumulation, and forms a large number of vacancies and interstitial atoms. read less NOT USED (high confidence) H.-S. Jin, P. Song, C.-G. Jon, and J.-C. Kim, “Thermodynamic properties of fcc metals using reparameterized MEAM potentials,” Indian Journal of Physics. 2021. link Times cited: 4 NOT USED (high confidence) L. Friedeheim, N. Bailey, and J. Dyre, “Effectively one-dimensional phase diagram of CuZr liquids and glasses,” Physical Review B. 2021. link Times cited: 2 Abstract: This paper presents computer simulations of CuxZr100−x (x = … read moreAbstract: This paper presents computer simulations of CuxZr100−x (x = 36, 50, 64) in the liquid and glass phases. The simulations are based on the effective-medium theory (EMT) potentials. We find good invariance of both structure and dynamics in reduced units along the isomorphs of the systems. The state points studied involve a density variation of almost a factor of two and temperatures going from 1500 K to above 4000 K for the liquids and from 500 K to above 1500 K for the glasses. For comparison, results are presented also for similar temperature variations along isochores, showing little invariance. In general for a binary system the phase diagram has three axes: composition, temperature and pressure (or density). When isomorphs are present, there are effectively only two axes, and for a fixed composition just one. We conclude that the thermodynamic phase diagram of this metallic glass former for a fixed composition is effectively one-dimensional in the sense that many physical properties are invariant along the same curves, implying that in order to investigate the phase diagram, it is only necessary to go across these curves. read less NOT USED (high confidence) K. Kasum, F. Mulyana, M. Zaenudin, A. Gamayel, and M. Mohammed, “Molecular Dynamics Simulation on Creep Mechanism of Nanocrystalline Cu-Ni Alloy.” 2021. link Times cited: 3 Abstract: Creep mechanism is an essential mechanism for material when … read moreAbstract: Creep mechanism is an essential mechanism for material when subjected to a high temperature and high pressure. It shows material ability during an extreme application to maintain its structure and properties, especially high pressure and temperature. This test is already done experimentally in many materials such as metallic alloys, various stainless steel, and composites. However, understanding the creep mechanism at the atomic level is challenging due to the instruments limitation. Still, the improvement of mechanical properties is expected can be done in such a group. In this work, the creep mechanism of the nanocrystalline Cu-Ni alloy is demonstrated in terms of molecular dynamics simulation. The result shows a significant impact on both temperature and pressure. The deformation supports the mechanisms as a result of the grain boundary diffusion. Quantitative analysis shows a more substantial difference in creep-rate at a higher temperature and pressure parameters. This study has successfully demonstrated the mechanism of creep at the atomic scale and may be used for improving the mechanical properties of the material. read less NOT USED (high confidence) Y. Chen, J. Xiong, and G. Zhang, “Generation mechanism of irregular microstructures on the machined surface in single-point diamond turning,” The International Journal of Advanced Manufacturing Technology. 2021. link Times cited: 6 NOT USED (high confidence) R. Wang et al., “Prediction of crystal structures and motifs in the Fe–Mg–O system at Earth’s core pressures,” New Journal of Physics. 2021. link Times cited: 2 Abstract: Fe, Mg, and O are among the most abundant elements in terres… read moreAbstract: Fe, Mg, and O are among the most abundant elements in terrestrial planets. While the behavior of the Fe–O, Mg–O, and Fe–Mg binary systems under pressure have been investigated, there are still very few studies of the Fe–Mg–O ternary system at relevant Earth’s core and super-Earth’s mantle pressures. Here, we use the adaptive genetic algorithm (AGA) to study ternary Fe x Mg y O z phases in a wide range of stoichiometries at 200 GPa and 350 GPa. We discovered three dynamically stable phases with stoichiometries FeMg2O4, Fe2MgO4, and FeMg3O4 with lower enthalpy than any known combination of Fe–Mg–O high-pressure compounds at 350 GPa. With the discovery of these phases, we construct the Fe–Mg–O ternary convex hull. We further clarify the composition- and pressure-dependence of structural motifs with the analysis of the AGA-found stable and metastable structures. Analysis of binary and ternary stable phases suggest that O, Mg, or both could stabilize a BCC iron alloy at inner core pressures. read less NOT USED (high confidence) K. Kanhaiya, S. Kim, W.-G. Im, and H. Heinz, “Accurate simulation of surfaces and interfaces of ten FCC metals and steel using Lennard–Jones potentials,” npj Computational Materials. 2021. link Times cited: 32 NOT USED (high confidence) S. Atlas, “Embedding Quantum Statistical Excitations in a Classical Force Field.,” The journal of physical chemistry. A. 2021. link Times cited: 3 Abstract: Quantum-mechanically driven charge polarization and charge t… read moreAbstract: Quantum-mechanically driven charge polarization and charge transfer are ubiquitous in biomolecular systems, controlling reaction rates, allosteric interactions, ligand-protein binding, membrane transport, and dynamically driven structural transformations. Molecular dynamics (MD) simulations of these processes require quantum mechanical (QM) information in order to accurately describe their reactive dynamics. However, current techniques-empirical force fields, subsystem approaches, ab initio MD, and machine learning-vary in their ability to achieve a consistent chemical description across multiple atom types, and at scale. Here we present a physics-based, atomistic force field, the ensemble DFT charge-transfer embedded-atom method, in which QM forces are described at a uniform level of theory across all atoms, avoiding the need for explicit solution of the Schrödinger equation or large, precomputed training data sets. Coupling between the electronic and atomistic length scales is effected through an ensemble density functional theory formulation of the embedded-atom method originally developed for elemental materials. Charge transfer is expressed in terms of ensembles of ionic state basis densities of individual atoms, and charge polarization, in terms of atomic excited-state basis densities. This provides a highly compact yet general representation of the force field, encompassing both local and system-wide effects. Charge rearrangement is realized through the evolution of ensemble weights, adjusted at each dynamical time step via chemical potential equalization. read less NOT USED (high confidence) B. Xu et al., “Scalable monolayer-functionalized nanointerface for thermal conductivity enhancement in copper/diamond composite,” Carbon. 2021. link Times cited: 14 NOT USED (high confidence) H. Bhattarai, K. E. Newman, and J. Gezelter, “The role of polarizability in the interfacial thermal conductance at the gold-water interface.,” The Journal of chemical physics. 2020. link Times cited: 3 Abstract: We have studied the interfacial thermal conductance, G, of t… read moreAbstract: We have studied the interfacial thermal conductance, G, of the flat Au(111)-water interface using non-equilibrium molecular dynamics simulations. We utilized two metal models, one based on the embedded atom method (EAM) and the other including metallic polarizability via a density readjusting EAM. These were combined with three popular water models, SPC/E, TIP4P, and TIP4P-FQ, to understand the role of polarizability in the thermal transport process. A thermal flux was introduced using velocity shearing and scaling reverse non-equilibrium molecular dynamics, and transport coefficients were measured by calculating the resulting thermal gradients and temperature differences at the interface. Our primary finding is that the computed interfacial thermal conductance between a bare metal interface and water increases when polarizability is taken into account in the metal model. Additional work to understand the origin of the conductance difference points to changes in the local ordering of the water molecules in the first two layers of water above the metal surface. Vibrational densities of states on both sides of the interface exhibit interesting frequency modulation close to the surface but no obvious differences due to metal polarizability. read less NOT USED (high confidence) S. Ono and T. Ito, “Theory of dynamical stability for two- and three-dimensional Lennard-Jones crystals,” Physical Review B. 2020. link Times cited: 6 Abstract: The dynamical stability of three-dimensional (3D) Lennard-Jo… read moreAbstract: The dynamical stability of three-dimensional (3D) Lennard-Jones (LJ) crystals has been studied for many years. The fcc and hcp structures are dynamically stable, while the bcc structure is stable only for long-range LJ potentials that are characterized by relatively small integer pairs $(m,n)$. Here, we study the dynamical stability of two-dimensional (2D) LJ crystals, where the planar hexagonal, the buckled honeycomb, and the buckled square structures are assumed. We demonstrate that the stability property of 2D and 3D LJ crystals can be classified into four groups depending on $(m,n)$. The instabilities of the planar hexagonal, the buckled square, and the bcc structures are investigated within analytical expressions. The structure-stability relationship between the LJ crystals and the elemental metals in the periodic table is also discussed. read less NOT USED (high confidence) W. E. Ernst and A. W. Hauser, “Metal clusters synthesized in helium droplets: structure and dynamics from experiment and theory.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 0 Abstract: Metal clusters have drawn continuous interest because of the… read moreAbstract: Metal clusters have drawn continuous interest because of their high potential for the assembly of matter with special properties that may significantly differ from the corresponding bulk. Controlled combination of particular elements in one nanoparticle can increase the options for the creation of new materials for photonic, catalytic, or electronic applications. Superfluid helium droplets provide confinement and ultralow temperature, i.e. an ideal environment for the atom-by-atom aggregation of a new nanoparticle. This perspective presents a review of the current research progress on the synthesis of tailored metal and metal oxide clusters including core-shell designs, their characterization within the helium droplet beam, deposition on various solid substrates, and analysis via surface diagnostics. Special attention is given to the thermal properties of mixed metal clusters and questions about alloy formation on the nanoscale. Experimental results are accompanied by theoretical approaches employing computational chemistry, molecular dynamics simulations and He density functional theory. read less NOT USED (high confidence) E. M. Gavilán-Arriazu, R. E. Giménez, and O. A. Pinto, “Structural surface and thermodynamics analysis of nanoparticles with defects.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 0 Abstract: In this work, we analyze the surface structure and thermodyn… read moreAbstract: In this work, we analyze the surface structure and thermodynamics regarding the decoration of nanoparticles with defects, using statistical calculations and Monte Carlo simulations in a complementary way. The main objective is to design and analyze a simple model as a general tool that can help the interpretation of results from more specific and complex models. In particular, we show how the presence of surface defects of the same nature as the nanoparticle induces different site distributions depending on different factors such as the density of defects, and the geometry and size of the considered nanoparticle. These distributions are analyzed for icosahedron nanoparticles of different sizes and densities of defects, and then are linked with Monte Carlo simulations to interpret the thermodynamic effects of the modified surfaces. Under low temperature or strong attractive interaction conditions, the details emerging from the defective surfaces were manifested as wide plateaus in the isotherm and peaks in the compressibility of the adlayer. Different situations were observed as the temperature increases, since the structural details gradually disappear from the thermodynamic measurements, until plateaus and compressibility peaks completely merge under high enough temperature conditions. Adsorption site distribution, adsorption isotherms, energy per site, compressibility of the adlayer, and other relevant properties are analyzed as a function of the number of defects and the chemical potential. In addition to the icosahedron, cuboctahedron and truncated octahedron geometries are also analyzed. read less NOT USED (high confidence) M. Masuduzzaman and B. H. Kim, “Scale Effects in Nanoscale Heat Transfer for Fourier’s Law in a Dissimilar Molecular Interface,” ACS Omega. 2020. link Times cited: 17 Abstract: The dependence of the heat transfer of a nanoscopic liquid c… read moreAbstract: The dependence of the heat transfer of a nanoscopic liquid channel residing at the solid–liquid interface is traditionally ascribed to the temperature jump, interfacial thermal resistance, wettability, and heat flux. Other contributions stemming from the channel width dependence such as the boundary position are typically ignored. Here, we conducted nonequilibrium molecular dynamics simulations to better understand the relation between channel width and boundary positions located at the solid–liquid interface. The system under investigation is a simple liquid confined between the solid from nanochannels of different sizes (3.27–7.35 nm). In this investigation, the existence of the correlation between the boundary position and the channel width is observed, which follows an exponential function. The thermal conductivity of the boundary positions is compared with the experimental value and Green–Kubo prediction to verify the actual boundary position. Atomistic simulation reveals that the solid–liquid boundary position, which matches the experimental value of thermal conductivity, varies with the channel width because of the intermolecular force and the phonon mismatch of the solid and the liquid. read less NOT USED (high confidence) B. Sun, W. Ouyang, J. Gu, C. Wang, J. Wang, and L. Mi, “Formation of Moiré superstructure of epitaxial graphene on Pt(111): A molecular dynamic simulation investigation,” Materials Chemistry and Physics. 2020. link Times cited: 5 NOT USED (high confidence) M. Dupraz, S. Leake, and M. Richard, “Bragg coherent imaging of nanoprecipitates: role of superstructure reflections,” Journal of Applied Crystallography. 2020. link Times cited: 0 Abstract: Coherent precipitation of ordered phases is responsible for … read moreAbstract: Coherent precipitation of ordered phases is responsible for providing exceptional high-temperature mechanical properties in a wide range of compositionally complex alloys. Ordered phases are also essential to enhance the magnetic or catalytic properties of alloyed nanoparticles. The present work aims to demonstrate the relevance of Bragg coherent diffraction imaging (BCDI) for studying bulk and thin-film samples or isolated nanoparticles containing coherent nanoprecipitates/ordered phases. The structures of crystals of a few tens of nanometres in size are modelled with realistic interatomic potentials and are relaxed after introduction of coherent ordered nanoprecipitates. Diffraction patterns from fundamental and superstructure reflections are calculated in the kinematic approximation and used as input to retrieve the strain fields using algorithmic inversion. First, the case of single nanoprecipitates is tackled and it is shown that the strain field distribution from the ordered phase is retrieved very accurately. Then, the influence of the order parameter S on the strain field retrieved from the superstructure reflections is investigated. A very accurate strain distribution can be retrieved for partially ordered phases with large and inhomogeneous strains. Subsequently, the relevance of BCDI is evaluated for the study of systems containing many precipitates, and it is demonstrated that the technique is relevant for such systems. Finally, the experimental feasibility of using BCDI to image ordered phases is discussed in the light of the new possibilities offered by fourth-generation synchrotron sources. read less NOT USED (high confidence) H. Li, Z.-F. Shao, R. Feng, Y. Qi, Q. Wu, and C. Lei, “Subsurface defect evolution and crystal-structure transformation of single-crystal copper in nanoscale combined machining,” Philosophical Magazine. 2020. link Times cited: 2 Abstract: ABSTRACT In the paper, molecular dynamics simulation is appl… read moreAbstract: ABSTRACT In the paper, molecular dynamics simulation is applied to study the evolution and distribution of subsurface defects during nanoscale machining process of single-crystal copper. The chip-removal mechanism and the machined-surface-generative mechanism are examined through analysis of the dislocation evolution and atomic migration of the workpieces. The findings show that under different stresses and temperatures, the difference of the binding energy leads to a zoned phenomenon in the chip. Owing to elastic deformation, some of the dislocations could be recovered and form surface steps; moreover, the work hardening of the workpiece can be achieved on account of generation of twin boundaries, Lomer-Cottrell dislocations, and stacking fault tetrahedra (SFT) by plastic deformation. A process of evolution of an immobile dislocation group containing stair-rod dislocations into SFT is discovered, which is different from the traditional Silcox-Hirsch mechanism. Furthermore, a growth oscillation phenomenon, which corresponding stacking fault planes growth and retraction during the formation of the stable SFT, is discussed. read less NOT USED (high confidence) J.-ping Du, W.-T. Geng, K. Arakawa, J. Li, and S. Ogata, “Hydrogen-Enhanced Vacancy Diffusion in Metals.,” The journal of physical chemistry letters. 2020. link Times cited: 23 Abstract: Vacancy diffusion is fundamental to materials science. Hydro… read moreAbstract: Vacancy diffusion is fundamental to materials science. Hydrogen atoms bind strongly to vacancies and are often believed to retard vacancy diffusion. Here, we use a potential-of-mean-force method to study the diffusion of vacancies in Cu and Pd. We find H atoms, instead of dragging, enhance the diffusivity of vacancies due to a positive hydrogen Gibbs excess at the saddle-point: that is, the migration saddle attracts more H than the vacancy ground state, characterized by an activation excess ΓHm ≈ 1 H, together with also-positive migration activation volume Ωm and activation entropy Sm. Thus, according to the Gibbs adsorption isotherm generalized to the activation path, a higher μH significantly lowers the migration free-energy barrier. This is verified by ab initio grand canonical Monte Carlo simulations and direct molecular dynamics simulations. This trend is believed to be generic for migrating dislocations, grain boundaries, and so on that also have a higher capacity for attracting H atoms due to a positive activation volume at the migration saddles. read less NOT USED (high confidence) N. Zhang, D. Zhang, H. Chen, G. Wang, and G. Yin, “Propionic acid–assisted surfactant-free synthesis of icosahedral Pt3Pd nanoparticles with enhanced electrochemical performance,” Ionics. 2020. link Times cited: 2 NOT USED (high confidence) V. Samsonov et al., “Melting temperature and binding energy of metal nanoparticles: size dependences, interrelation between them, and some correlations with structural stability of nanoclusters,” Journal of Nanoparticle Research. 2020. link Times cited: 11 NOT USED (high confidence) C. Li, S. Dang, P. Han, X. He, and X. Long, “Effect of Cr on the generalized stacking fault energy of impure doped Ni (111) surface: a first-principles study,” The European Physical Journal B. 2020. link Times cited: 3 NOT USED (high confidence) C. Richert and N. Huber, “A Review of Experimentally Informed Micromechanical Modeling of Nanoporous Metals: From Structural Descriptors to Predictive Structure–Property Relationships,” Materials. 2020. link Times cited: 22 Abstract: Nanoporous metals made by dealloying take the form of macros… read moreAbstract: Nanoporous metals made by dealloying take the form of macroscopic (mm- or cm-sized) porous bodies with a solid fraction of around 30%. The material exhibits a network structure of “ligaments” with an average ligament diameter that can be adjusted between 5 and 500 nm. Current research explores the use of nanoporous metals as functional materials with respect to electrochemical conversion and storage, bioanalytical and biomedical applications, and actuation and sensing. The mechanical behavior of the network structure provides the scope for fundamental research, particularly because of the high complexity originating from the randomness of the structure and the challenges arising from the nanosized ligaments, which can be accessed through an experiment only indirectly via the testing of the macroscopic properties. The strength of nanoscale ligaments increases systematically with decreasing size, and owing to the high surface-to-volume ratio their elastic and plastic properties can be additionally tuned by applying an electric potential. Therefore, nanoporous metals offer themselves as suitable model systems for exploring the structure–property relationships of complex interconnected microstructures as well as the basic mechanisms of the chemo-electro-mechanical coupling at interfaces. The micromechanical modeling of nanoporous metals is a rapidly growing field that strongly benefits from developments in computational methods, high-performance computing, and visualization techniques; it also benefits at the same time through advances in characterization techniques, including nanotomography, 3D image processing, and algorithms for geometrical and topological analysis. The review article collects articles on the structural characterization and micromechanical modeling of nanoporous metals and discusses the acquired understanding in the context of advancements in the experimental discipline. The concluding remarks are given in the form of a summary and an outline of future perspectives. read less NOT USED (high confidence) M. M. Rahman, M. Islam, and N. Anjum, “Investigation on mechanical behaviors of Cu-Ni binary alloy nanopillars: a molecular dynamics study,” Journal of Molecular Modeling. 2020. link Times cited: 3 NOT USED (high confidence) M. Bagheripoor and R. Klassen, “The effect of grain boundary on the local incipient plastic deformation of fcc metals during nanoindentation,” Journal of Applied Physics. 2020. link Times cited: 7 Abstract: The effect of grain boundaries (GBs) on deformation mechanis… read moreAbstract: The effect of grain boundaries (GBs) on deformation mechanisms becomes increasingly important as the volume of deformation reaches the submicrometer and nanoscale. The current work investigates the impact of grain boundaries on the incipient plasticity of small-scale deformations of fcc metals. For this purpose, the behavior of single and bi-crystal Au thin films during nanoindentation are studied, using large-scale atomistic simulations. Various symmetric ⟨110⟩ tilt GBs with a wide range of misorientation angles are included to analyze the effect of GB geometry on the nanoscale plasticity mechanisms. Potentially, GBs can act as a source, sink, or obstacle for lattice dislocation, depending on their geometry, energy level, and distance from the deformation zone. The role of the heterogeneous nucleation and emission of dislocations from GBs on the plasticity and hardness of bicrystals is analyzed. According to our results, the intrinsic free volume involved in the GB region is associated with dislocation nucleation at the GB. The volume of the plastic zone generated beneath the tip and the way it grows is strongly dependent on the GB structure. Dislocation nucleation occurs predominantly in the early stages of indentation at GBs with a dissociated interface structural unit, before the interaction of lattice dislocation and GB. Coherent twin boundaries display the lowest effect on the hardness. Based on our results, there is a strong correlation between the interfacial boundary energy and its effect on the bicrystal hardness. GBs with lower interfacial energy offer a higher barrier against slip transmission and nucleation at the GB. read less NOT USED (high confidence) C. Zhao, F. Liu, X. Kong, T. Yan, and F. Ding, “The wrinkle formation in graphene on transition metal substrate: a molecular dynamics study,” International Journal of Smart and Nano Materials. 2020. link Times cited: 8 Abstract: ABSTRACT To explore the mechanism of the wrinkle formation i… read moreAbstract: ABSTRACT To explore the mechanism of the wrinkle formation in graphene on transition metal substrate, a molecular dynamics (MD) simulation package that allows us to simulate systems of millions of atoms was developed. Via the MD simulation, we reveal the detailed kinetics of wrinkles formation on a Cu substrate under compressive strain, from nucleation to one-dimensional propagation and then the splitting of a large wrinkle to a few smaller ones, which is in good conformity with experimental observation. Further study reveals that both friction and the adhesion between graphene and Cu substrate are critical for the wrinkle formation and wrinkles can be easily formed with a lower frictional force and/or a smaller adhesion. Finally, we have shown that impurities in graphene or substrates can greatly facilitate the nucleation of wrinkles. The systematic exploration of the wrinkle formation in graphene on a substrate is expected to facilitate the experimental designs for the controllable synthesis of high-quality graphene. read less NOT USED (high confidence) A. Galashev and O. Rakhmanova, “Stability of a Two-Layer Silicene on a Nickel Substrate upon Intercalation of Graphite,” Glass Physics and Chemistry. 2020. link Times cited: 1 NOT USED (high confidence) A. Galashev, O. Rakhmanova, and A. Isakov, “Molecular Dynamic Behavior of Lithium Atoms in a Flat Silicene Pore on a Copper Substrate,” Russian Journal of Physical Chemistry B. 2020. link Times cited: 3 NOT USED (high confidence) D. Ma and L. Zhang, “Enhancement of interface thermal conductance between Cr–Ni alloy and dielectric via Cu nano-interlayer,” Journal of Physics: Condensed Matter. 2020. link Times cited: 10 Abstract: Facilitating interfacial thermal transport is highly desirab… read moreAbstract: Facilitating interfacial thermal transport is highly desirable for various engineering applications, such as improving heat dissipation in microelectronics and efficiency of electrothermal heating element. Here, the interface thermal conductances (ITCs) of Cr0.22Ni0.78/MgO and Cr0.22Ni0.78/Al2O3 interfaces are studied through the non-equilibrium molecular dynamics simulation. It is found that the two ITCs can be hugely enhanced by 3 and 2.4 times, respectively, with the introduction of Cu nano-interlayer of a thickness larger than 7.2 Å. The enhanced ratio is robust and shows weak dependence on temperature. Further vibrational spectral analysis and phonon transmission function reveal that the enhancement in ITC mainly originates from the boosting of inelastic phonon scattering, which is generally considered to contribute a small proportion to ITC. Here, the inelastic scattering contributes as high as 63% to the ITC of Cr0.22Ni0.78/Cu/MgO interface at 300 K. The findings provide an effective strategy to enhance ITC at a wide temperature range and advance our understanding of inelastic scattering in interfacial phonon transport. read less NOT USED (high confidence) J. Chapman and R. Ramprasad, “Predicting the dynamic behavior of the mechanical properties of platinum with machine learning.,” The Journal of chemical physics. 2020. link Times cited: 2 Abstract: Over the last few decades, computational tools have been ins… read moreAbstract: Over the last few decades, computational tools have been instrumental in understanding the behavior of materials at the nano-meter length scale. Until recently, these tools have been dominated by two levels of theory: quantum mechanics (QM) based methods and semi-empirical/classical methods. The former are time-intensive but accurate and versatile, while the latter methods are fast but are significantly limited in veracity, versatility, and transferability. Recently, machine learning (ML) methods have shown the potential to bridge the gap between these two chasms due to their (i) low cost, (ii) accuracy, (iii) transferability, and (iv) ability to be iteratively improved. In this work, we further extend the scope of ML for atomistic simulations by capturing the temperature dependence of the mechanical and structural properties of bulk platinum through molecular dynamics simulations. We compare our results directly with experiments, showcasing that ML methods can be used to accurately capture large-scale materials phenomena that are out of reach of QM calculations. We also compare our predictions with those of a reliable embedded atom method potential. We conclude this work by discussing how ML methods can be used to push the boundaries of nano-scale materials research by bridging the gap between QM and experimental methods. read less NOT USED (high confidence) J. Alberdi-Rodriguez, S. Acharya, T. Rahman, A. Arnau, and M. Gosálvez, “Dominant contributions to the apparent activation energy in two-dimensional submonolayer growth: comparison between Cu/Ni(111) and Ni/Cu(111),” Journal of Physics: Condensed Matter. 2020. link Times cited: 0 Abstract: For surface-mediated processes in general, such as epitaxial… read moreAbstract: For surface-mediated processes in general, such as epitaxial growth and heterogeneous catalysis, a constant slope in the Arrhenius diagram of the rate of interest, R, against inverse temperature, log R vs 1/T, is traditionally interpreted as the existence of a bottleneck elementary reaction (or rate-determining step), whereby the constant slope (or apparent activation energy, EappR) reflects the value of the energy barrier for that elementary reaction. In this study, we express EappR as a weighted average, where every term contains the traditional energy barrier for the corresponding elementary reaction plus an additional configurational term, while identifying each weight as the probability of executing the corresponding elementary reaction. Accordingly, the change in the leading (most probable) elementary reaction with the experimental conditions (e.g. temperature) is automatically captured and it is shown that a constant value of EappR is possible even if control shifts from one elementary reaction to another. To aid the presentation, we consider kinetic Monte Carlo simulations of submonolayer growth of Cu on Ni(111) and Ni on Cu(111) at constant deposition flux, including a large variety of single-atom, multi-atom and complete-island diffusion events. In addition to analysing the dominant contributions to the diffusion constant of the complete adparticle system (or tracer diffusivity) and its apparent activation energy as a function of both coverage and temperature for the two heteroepitaxial systems, their surface morphologies and island densities are also compared. read less NOT USED (high confidence) T. Loeffler, S. Manna, T. Patra, H. Chan, B. Narayanan, and S. Sankaranarayanan, “Active Learning A Neural Network Model For Gold Clusters & Bulk From Sparse First Principles Training Data,” ChemCatChem. 2020. link Times cited: 15 Abstract: Small metal clusters are of fundamental scientific interest … read moreAbstract: Small metal clusters are of fundamental scientific interest and of tremendous significance in catalysis. These nanoscale clusters display diverse geometries and structural motifs depending on the cluster size; a knowledge of this size‐dependent structural motifs and their dynamical evolution has been of longstanding interest. Given the high computational cost of first‐principles calculations, molecular modeling and atomistic simulations such as molecular dynamics (MD) has proven to be an important complementary tool to aid this understanding. Classical MD typically employ predefined functional forms which limits their ability to capture such complex size‐dependent structural and dynamical transformation. Neural Network (NN) based potentials represent flexible alternatives and in principle, well‐trained NN potentials can provide high level of flexibility, transferability and accuracy on‐par with the reference model used for training. A major challenge, however, is that NN models are interpolative and requires large quantities ( ∼104 or greater) of training data to ensure that the model adequately samples the energy landscape both near and far‐from‐equilibrium. A highly desirable goal is minimize the number of training data, especially if the underlying reference model is first‐principles based and hence expensive. Here, we introduce an active learning (AL) scheme that trains a NN model on‐the‐fly with minimal amount of first‐principles based training data. Our AL workflow is initiated with a sparse training dataset ( ∼ 1 to 5 data points) and is updated on‐the‐fly via a Nested Ensemble Monte Carlo scheme that iteratively queries the energy landscape in regions of failure and updates the training pool to improve the network performance. Using a representative system of gold clusters, we demonstrate that our AL workflow can train a NN with ∼ 500 total reference calculations. Using an extensive DFT test set of ∼1100 configurations, we show that our AL‐NN is able to accurately predict both the DFT energies and the forces for clusters of a myriad of different sizes. Our NN predictions are within 30 meV/atom and 40 meV/Å of the reference DFT calculations. Moreover, our AL‐NN model also adequately captures the various size‐dependent structural and dynamical properties of gold clusters in excellent agreement with DFT calculations and available experiments. We finally show that our AL‐NN model also captures bulk properties reasonably well, even though they were not included in the training data. read less NOT USED (high confidence) M. Mirakhory, M. M. Khatibi, and S. Sadeghzadeh, “Nanoparticle mass detection by single-layer triangular graphene sheets, the extraordinary geometry for detection of nanoparticles,” Journal of Nanoparticle Research. 2020. link Times cited: 4 NOT USED (high confidence) B. Zhang, Y. Liang, B. Liu, W. Liu, and Z. Liu, “Enhancing the Thermo-Mechanical Property of Polymer by Weaving and Mixing High Length–Diameter Ratio Filler,” Polymers. 2020. link Times cited: 1 Abstract: Improving thermo-mechanical characteristics of polymers can … read moreAbstract: Improving thermo-mechanical characteristics of polymers can efficiently promote their applications in heat exchangers and thermal management. However, a feasible way to enhance the thermo-mechanical property of bulk polymers at low filler content still remains to be explored. Here, we propose mixing high length-diameter ratio filler such as carbon nanotube (CNT), boron nitride (BN) nanotube, and copper (Cu) nanowire, in the woven polymer matrix to meet the purpose. Through molecular dynamics (MD) simulation, the thermal properties of three woven polymers including woven polyethylene (PE), woven poly (p-phenylene) (PPP), and woven polyacetylene (PA) are investigated. Besides, using woven PE as a polymer matrix, three polymer nanocomposites, namely PE-CNT, PE-BN, and PE-Cu, are constructed by mixing CNT, BN nanotube, and Cu nanowire respectively, whose thermo-mechanical characteristics are compared via MD simulation. Morphology and phonons spectra analysis are conducted to reveal the underlying mechanisms. Furthermore, impacts of electron-phonon coupling and electrical field on the thermal conductivity of PE-Cu are uncovered via two temperature model MD simulation. Classical theoretical models are modified to predict the effects of filler and matrix on the thermal conductivity of polymer nanocomposites. This work can provide useful guidelines for designing thermally conductive bulk polymers and polymer nanocomposites. read less NOT USED (high confidence) S. Ajori, H. Parsapour, R. Ansari, and S. Haghighi, “Effect of metallic nanowire encapsulation on the tensile behavior of single-walled carbon nanotubes: a molecular dynamics study,” The European Physical Journal D. 2020. link Times cited: 3 NOT USED (high confidence) Z. Tang, Y. Chen, and W. Ye, “Calculation of Surface Properties of Cubic and Hexagonal Crystals through Molecular Statics Simulations,” Crystals. 2020. link Times cited: 8 Abstract: Surface property is an important factor that is widely consi… read moreAbstract: Surface property is an important factor that is widely considered in crystal growth and design. It is also found to play a critical role in changing the constitutive law seen in the classical elasticity theory for nanomaterials. Through molecular static simulations, this work presents the calculation of surface properties (surface energy density, surface stress and surface stiffness) of some typical cubic and hexagonal crystals: face-centered-cubic (FCC) pure metals (Cu, Ni, Pd and Ag), body-centered-cubic (BCC) pure metals (Mo and W), diamond Si, zincblende GaAs and GaN, hexagonal-close-packed (HCP) pure metals (Mg, Zr and Ti), and wurzite GaN. Sound agreements of the bulk and surface properties between this work and the literature are found. New results are first reported for the surface stiffness of BCC pure metals, surface stress and surface stiffness of HCP pure metals, Si, GaAs and GaN. Comparative studies of the surface properties are carried out to uncover trends in their behaviors. The results in this work could be helpful to the investigation of material properties and structure performances of crystals. read less NOT USED (high confidence) S. Jindal and S. Bulusu, “Structural evolution in gold nanoparticles using artificial neural network based interatomic potentials.,” The Journal of chemical physics. 2020. link Times cited: 5 Abstract: Relativistic effects of gold make its behavior different fro… read moreAbstract: Relativistic effects of gold make its behavior different from other metals. Unlike silver and copper, gold does not require symmetrical structures as the stable entities. We present the evolution of gold from a cluster to a nanoparticle by considering a majority of stable structural possibilities. Here, an interatomic potential (artificial neural network), trained on quantum mechanical data comprising small to medium sized clusters, gives exceptional results for larger size clusters. We have explored the potential energy surface for "magic" number clusters 309, 561, and 923. This study reveals that these clusters are not completely symmetric, but they require a distorted symmetric core with amorphous layers of atoms over it. The amorphous geometries tend to be more stable in comparison to completely symmetric structures. The first ever gold cluster to hold an icosahedron-Au13 was identified at Au60 [S. Pande et al., J. Phys. Chem. Lett. 10, 1820 (2019)]. Through our study, we have found a plausible evolution of a symmetric core as the size of the nanoparticle increases. The stable cores were found at Au160, Au327, and Au571, which can be recognized as new magic numbers. Au923 is found to have a stable symmetric core of 147 atoms covered with layers of atoms that are not completely amorphous. This shows the preference of symmetric structures as the size of the nanoparticle increases (<3.3 nm). read less NOT USED (high confidence) A. Galashev and K. Ivanichkina, “Computer Study of Silicene Applicability in Electrochemical Devices,” Journal of Structural Chemistry. 2020. link Times cited: 6 NOT USED (high confidence) E. Giessen et al., “Roadmap on multiscale materials modeling,” Modelling and Simulation in Materials Science and Engineering. 2020. link Times cited: 91 Abstract: Modeling and simulation is transforming modern materials sci… read moreAbstract: Modeling and simulation is transforming modern materials science, becoming an important tool for the discovery of new materials and material phenomena, for gaining insight into the processes that govern materials behavior, and, increasingly, for quantitative predictions that can be used as part of a design tool in full partnership with experimental synthesis and characterization. Modeling and simulation is the essential bridge from good science to good engineering, spanning from fundamental understanding of materials behavior to deliberate design of new materials technologies leveraging new properties and processes. This Roadmap presents a broad overview of the extensive impact computational modeling has had in materials science in the past few decades, and offers focused perspectives on where the path forward lies as this rapidly expanding field evolves to meet the challenges of the next few decades. The Roadmap offers perspectives on advances within disciplines as diverse as phase field methods to model mesoscale behavior and molecular dynamics methods to deduce the fundamental atomic-scale dynamical processes governing materials response, to the challenges involved in the interdisciplinary research that tackles complex materials problems where the governing phenomena span different scales of materials behavior requiring multiscale approaches. The shift from understanding fundamental materials behavior to development of quantitative approaches to explain and predict experimental observations requires advances in the methods and practice in simulations for reproducibility and reliability, and interacting with a computational ecosystem that integrates new theory development, innovative applications, and an increasingly integrated software and computational infrastructure that takes advantage of the increasingly powerful computational methods and computing hardware. read less NOT USED (high confidence) C.-Y. Shih, M. Shugaev, C. Wu, and L. Zhigilei, “The effect of pulse duration on nanoparticle generation in pulsed laser ablation in liquids: insights from large-scale atomistic simulations.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 38 Abstract: The generation of colloidal solutions of chemically clean na… read moreAbstract: The generation of colloidal solutions of chemically clean nanoparticles through pulsed laser ablation in liquids (PLAL) has evolved into a thriving research field that impacts industrial applications. The complexity and multiscale nature of PLAL make it difficult to untangle the various processes involved in the generation of nanoparticles and establish the dependence of nanoparticle yield and size distribution on the irradiation parameters. Large-scale atomistic simulations have yielded important insights into the fundamental mechanisms of ultrashort (femtoseconds to tens of picoseconds) PLAL and provided a plausible explanation of the origin of the experimentally observed bimodal nanoparticle size distributions. In this paper, we extend the atomistic simulations to short (hundreds of picoseconds to nanoseconds) laser pulses and focus our attention on the effect of the pulse duration on the mechanisms responsible for the generation of nanoparticles at the initial dynamic stage of laser ablation. Three distinct nanoparticle generation mechanisms operating at different stages of the ablation process and in different parts of the emerging cavitation bubble are identified in the simulations. These mechanisms are (1) the formation of a thin transient metal layer at the interface between the ablation plume and water environment followed by its decomposition into large molten nanoparticles, (2) the nucleation, growth, and rapid cooling/solidification of small nanoparticles at the very front of the emerging cavitation bubble, above the transient interfacial metal layer, and (3) the spinodal decomposition of a part of the ablation plume located below the transient interfacial layer, leading to the formation of a large population of nanoparticles growing in a high-temperature environment through inter-particle collisions and coalescence. The coexistence of the three distinct mechanisms of the nanoparticle formation at the initial stage of the ablation process can be related to the broad nanoparticle size distributions commonly observed in nanosecond PLAL experiments. The strong dependence of the nanoparticle cooling and solidification rates on the location within the low-density metal-water mixing region has important implications for the long-term evolution of the nanoparticle size distribution, as well as for the ability to quench the nanoparticle growth or dope them by adding surface-active agents or doping elements to the liquid environment. read less NOT USED (high confidence) R. Wang et al., “Theoretical search for possible Li–Ni–B crystal structures using an adaptive genetic algorithm,” Journal of Applied Physics. 2020. link Times cited: 8 Abstract: The structural diversity of rare-earth and transition metal … read moreAbstract: The structural diversity of rare-earth and transition metal borides indicates that alkali-transition metal borides (A-T-B) show tremendous promise in exhibiting a variety of crystal structures with different dimensionalities of T-B frameworks. On the other hand, the A-T-B ternary systems are severely underexplored because of the synthetic challenges associated with their preparation. Accurate and efficient computational predictions of low-energy stable and metastable phases can identify the optimal compositions of the hypothetical compounds in the A-T-B systems to guide the synthesis. In this work, we have computationally discovered several new phases in the Li–Ni–B ternary system. The newly discovered LiNiB, Li2Ni3B, and Li2NiB phases expand the existing theoretical database, and the convex-hull surface of Li–Ni–B has been re-constructed. The lowest energy structure of the LiNiB compound has been found by an adaptive genetic algorithm with layered motif, which matches with the experimentally determined structure. According to our electrochemical calculations, LiNiB and another predicted layered Li2NiB compounds have great potential as anode materials for lithium batteries. The Li2Ni3B compound with the space group P4332 was predicted to crystallize in a cubic structure composed of distorted octahedral units of BNi6, which is isostructural to two noncentrosymmetric superconductors Li2Pd3B and Li2Pt3B. While we were unable to experimentally confirm the Li2Ni3B compound utilizing the hydride synthetic route, attempts to synthesize this compound by alternate methods remain highly desirable, considering its potential superconducting properties. read less NOT USED (high confidence) Á. Jász, Á. Rák, I. Ladjánszki, and G. Cserey, “Classical molecular dynamics on graphics processing unit architectures,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2020. link Times cited: 6 Abstract: Molecular dynamics (MD) has experienced a significant growth… read moreAbstract: Molecular dynamics (MD) has experienced a significant growth in the recent decades. Simulating systems consisting of hundreds of thousands of atoms is a routine task of computational chemistry researchers nowadays. Thanks to the straightforwardly parallelizable structure of the algorithms, the most promising method to speed‐up MD calculations is exploiting the large‐scale processing power offered by the parallel hardware architecture of graphics processing units or GPUs. Programming GPUs is becoming easier with general‐purpose GPU computing frameworks and higher levels of abstraction. In the recent years, implementing MD simulations on graphics processors has gained a large interest, with multiple popular software packages including some form of GPU‐acceleration support. Different approaches have been developed regarding various aspects of the algorithms, with important differences in the specific solutions. Focusing on published works in the field of classical MD, we describe the chosen implementation methods and algorithmic techniques used for porting to GPU, as well as how recent advances of GPU architectures will provide even more optimization possibilities in the future. read less NOT USED (high confidence) Y. Akkus, A. T. Gurer, and K. Bellur, “Drifting mass accommodation coefficients: in situ measurements from a steady state molecular dynamics setup,” Nanoscale and Microscale Thermophysical Engineering. 2020. link Times cited: 11 Abstract: ABSTRACT A fundamental understanding of the evaporation/cond… read moreAbstract: ABSTRACT A fundamental understanding of the evaporation/condensation phenomena is vital to many fields of science and engineering, yet there is many discrepancies in the usage of phase-change models and associated coefficients. First, a brief review of the kinetic theory of phase change is provided, and the mass accommodation coefficient (MAC, ) and its inconsistent definitions are discussed. The discussion focuses on the departure from equilibrium; represented as a macroscopic “drift” velocity. Then, a continuous flow, phase change driven molecular-dynamics setup is used to investigate steady-state condensation at a flat liquid-vapor interface of argon at various phase-change rates and temperatures to elucidate the effect of equilibrium departure. MAC is computed directly from the kinetic theory-based Hertz–Knudsen (H-K) and Schrage (exact and approximate) expressions without the need for a priori physical definitions, ad-hoc particle injection/removal, or particle counting. MAC values determined from the approximate and exact Schrage expressions ( and ) are between 0.8 and 0.9, while MAC values from the H-K expression ( ) are above unity for all cases tested. yield value closest to the results from transition state theory [J Chem Phys, 118, 1392–1399 (2003)]. The departure from equilibrium does not affect the value of but causes to vary drastically emphasizing the importance of a drift velocity correction. Additionally, equilibrium departure causes a nonuniform distribution in vapor properties. At the condensing interface, a local rise in vapor temperature and a drop in vapor density is observed when compared with the corresponding bulk values. When the deviation from bulk values are taken into account, all values of MAC including show a small yet noticeable difference that is both temperature and phase-change rate dependent. Graphical abstract read less NOT USED (high confidence) A. Galashev and K. Ivanichkina, “Silicene Anodes for Lithium-Ion Batteries on Metal Substrates,” Journal of The Electrochemical Society. 2020. link Times cited: 20 NOT USED (high confidence) N. Amadou, T. D. Rességuier, A. Dragon, and E. Brambrink, “Effects of orientation, lattice defects and temperature on plasticity and phase transition in ramp-compressed single crystal iron,” Computational Materials Science. 2020. link Times cited: 17 NOT USED (high confidence) L. K. Scarbath-Evers, R. Hammer, D. Golze, M. Brehm, D. Sebastiani, and W. Widdra, “From flat to tilted: gradual interfaces in organic thin film growth.,” Nanoscale. 2020. link Times cited: 5 Abstract: We investigate domain formation and local morphology of thin… read moreAbstract: We investigate domain formation and local morphology of thin films of α-sexithiophene (α-6T) on Au(100) beyond monolayer coverage by combining high resolution scanning tunneling microscopy (STM) experiments with electronic structure theory calculations and computational structure search. We report a layerwise growth of highly-ordered enantiopure domains. For the second and third layer, we show that the molecular orbitals of individual α-6T molecules can be well resolved by STM, providing access to detailed information on the molecular orientation. We find that already in the second layer the molecules abandon the flat adsorption structure of the monolayer and adopt a tilted conformation. Although the observed tilted arrangement resembles the orientation of α-6T in the bulk, the observed morphology does not yet correspond to a well-defined surface of the α-6T bulk structure. A similar behavior is found for the third layer indicating a growth mechanism where the bulk structure is gradually adopted over several layers. read less NOT USED (high confidence) D. Hurley, M. Jin, L. K. A. Jr, and R. Schley, “Scoping Studies to ascertain the change in the phase transition temperature of metallic fuels.” 2020. link Times cited: 0 NOT USED (high confidence) X. Wang, D. Venerus, I. Puri, and S. Murad, “On using the anisotropy in the thermal resistance of solid–fluid interfaces to more effectively cool nano-electronics,” Molecular Simulation. 2020. link Times cited: 1 Abstract: ABSTRACT As power-intensive electronic components are furthe… read moreAbstract: ABSTRACT As power-intensive electronic components are further miniaturised into nanodevices, their heat dissipation is a serious operational and safety concern. While nanochannels and nanofins are often used for facilitating heat dissipation, the liquid-solid interfaces that form (Kapitza resistance), become significant barriers to heat transfer. We demonstrate that the thermal resistance of these interfaces is strongly anisotropic. The resistance of an interface to heat transfer parallel to the interface (solid surface) is significantly smaller than the more well-known Kapitza resistance (associated with heat transfer across the interface – perpendicular to the solid surface) and is even lower than that of the bulk fluid. As a result, if devices are designed to dissipate heat parallel to an interface, heat dissipation can be significantly enhanced. Our studies are also able to explain the molecular basis of this observed anisotropy in interfacial resistance, which has hitherto remained unreported for solid–liquid interfaces. read less NOT USED (high confidence) Z. Wu, K. Tan, R. Zhang, Q. Wei, and Y. Lin, “Atomistic kinetic Monte Carlo—Embedded atom method simulation on growth and morphology of Cu–Zn–Sn precursor of Cu_2ZnSnS_4 solar cells,” Journal of Materials Research. 2020. link Times cited: 2 Abstract: An atomistic kinetic Monte Carlo coupled with the embedded-a… read moreAbstract: An atomistic kinetic Monte Carlo coupled with the embedded-atom method is used to simulate film growth and morphology evolution of a Cu–Zn–Sn precursor of Cu_2ZnSnS_4 solar cells by single-step electrodeposition. The deposition and diffusion events of three different metallic atoms are described by the simulation. Moreover, the multibody Cu–Zn–Sn potential is used to calculate diffusion barrier energy. The effects of process factors, including temperature and electrode potential, on the cross-section morphology and surface roughness are explored, while keeping the elemental composition ratios constant. The lowest roughness with the smoothest morphology is obtained at the optimal parameters. The distribution and transformation behaviors of cluster sizes are investigated to describe the alloy film growth process. Furthermore, the comparison between deposition events and diffusion events reveals that deposition events depend primarily on individual deposition rates of different metallic atoms, but diffusion events are mainly dependent on the interaction of metallic atoms. The film morphology evolution is visualized by three-dimensional configuration with increasing numbers of atoms, which suggests a competing mechanism between nucleation and growth of the thin film alloy. read less NOT USED (high confidence) X. Wang, C. Jameson, and S. Murad, “Interfacial Thermal Conductivity and Its Anisotropy,” Processes. 2019. link Times cited: 2 Abstract: There is a significant effort in miniaturizing nanodevices, … read moreAbstract: There is a significant effort in miniaturizing nanodevices, such as semi-conductors, currently underway. However, a major challenge that is a significant bottleneck is dissipating heat generated in these energy-intensive nanodevices. In addition to being a serious operational concern (high temperatures can interfere with their efficient operation), it is a serious safety concern, as has been documented in recent reports of explosions resulting from many such overheated devices. A significant barrier to heat dissipation is the interfacial films present in these nanodevices. These interfacial films generally are not an issue in macro-devices. The research presented in this paper was an attempt to understand these interfacial resistances at the molecular level, and present possibilities for enhancing the heat dissipation rates in interfaces. We demonstrated that the thermal resistances of these interfaces were strongly anisotropic; i.e., the resistance parallel to the interface was significantly smaller than the resistance perpendicular to the interface. While the latter is well-known—usually referred to as Kapitza resistance—the anisotropy and the parallel component have previously been investigated only for solid-solid interfaces. We used molecular dynamics simulations to investigate the density profiles at the interface as a function of temperature and temperature gradient, to reveal the underlying physics of the anisotropy of thermal conductivity at solid-liquid, liquid-liquid, and solid-solid interfaces. read less NOT USED (high confidence) L. Safina, J. Baimova, and R. Mulyukov, “Nickel nanoparticles inside carbon nanostructures: atomistic simulation,” Mechanics of Advanced Materials and Modern Processes. 2019. link Times cited: 17 NOT USED (high confidence) W. Xie and F. Fang, “Cutting-based single atomic layer removal mechanism of monocrystalline copper: edge radius effect,” Nanoscale Research Letters. 2019. link Times cited: 10 NOT USED (high confidence) G. Fernandez, “Abstract,” Journal of the ICRU. 2019. link Times cited: 0 Abstract: Dosimetry methods for use in dose assessment for individuals… read moreAbstract: Dosimetry methods for use in dose assessment for individuals following acute exposure to radiation are described. Primary methods include biodosimetry and physical dosimetry techniques, while additional supplementary methods are bioassays, neutron activation, and radiation field mapping. Biodosimetry methods include the established techniques of dicentric chromosome assay, cytokinesis-block micronucleus assay, translocation analysis by fluorescent in-situ hybridization, premature chromosome condensation, and the γ-H2AX assay. Emerging techniques include RNA expression-based, protein-based, and metabolomic-based assays. Physical dosimetry methods include electron paramagnetic resonance and the luminescence-based techniques of thermoluminescence and optically stimulated luminescence. Electron paramagnetic resonance methods are used to assess absorbed dose in biologically derived materials, such as bone, teeth, and keratinous tissue, as well as non-biologically derived materials such as sugars, glasses, and polymeric materials used in fabrics and other personal items. Thermoluminescence and optically stimulated luminescence techniques are used to assess absorbed dose in the components of personal electronics, along with other items such as plastic cards, fabrics, and clothing. There have also been similar efforts for teeth and dental repair ceramics. Since the above-listed techniques cannot distinguish between exposure to internal and external sources, bioassays may be used to assess exposure from internal contamination, including thyroid counting, chest counting, and excretion analysis methods. When a neutron exposure is expected, neutron activation analysis in blood, hair, or other non-biological items is useful. Radiation field mapping can be a useful method for determining locations where doses to individuals may be expected to be high and may complement radiation transport calculations performed for that purpose. Since immediate medical assessment is concerned with tissue reactions (deterministic effects), the quantity of interest for the above dosimetry methods is absorbed dose (expressed in gray). This Report concludes with a summary of the various methods and a brief discussion of the uses of such information in the aftermath of acute radiation exposure. read less NOT USED (high confidence) E. Jekal, “Atomistic Simulation for Rejuvenation of Cu-Zr Metallic Glasses by Strain.” 2019. link Times cited: 0 Abstract: Molecular dynamics simulations were carried out to investiga… read moreAbstract: Molecular dynamics simulations were carried out to investigate the atomic structure of a model Cu64Zr36 bulk metallic glass (BMG). It is found that the amount of icosahedral content of the system is significantly increased in a well relaxed structure. While we considered four connection types of vertex-, edge-, face-, and volume-sharing, the huge cluster in the relaxed samples mainly involve volume-type connection and exhibits a remarkable athermal plasticity that great stiffness and great yield strength compared to the as-quenched samples. In addition, the bond-angle distribution of annealed sample shows sharp peaks at specific bond angles which is an evidence of crystallized Lavesphase formed by icosahedral atoms, however the peaks are to be broaden after loading, which indicates decreasing amount of icosahedral content and their shape distortion. These results suggest that icosahedral content in a bulk metallic glasses plays a key role to determine the mechanical properties such as rigidity and maximum stress carrying capacity. read less NOT USED (high confidence) X. Wang, W.-S. Xu, H. Zhang, and J. Douglas, “Universal nature of dynamic heterogeneity in glass-forming liquids: A comparative study of metallic and polymeric glass-forming liquids.,” The Journal of chemical physics. 2019. link Times cited: 29 Abstract: Glass-formation is a ubiquitous phenomenon that is often obs… read moreAbstract: Glass-formation is a ubiquitous phenomenon that is often observed in a broad class of materials ranging from biological matter to commonly encountered synthetic polymer, as well as metallic and inorganic glass-forming (GF) materials. Despite the many regularities in the dynamical properties of GF materials, the structural origin of the universal dynamical properties of these materials has not yet been identified. Recent simulations of coarse-grained polymeric GF liquids have indicated the coexistence of clusters of mobile and immobile particles that appear to be directly linked, respectively, to the rate of molecular diffusion and structural relaxation. The present work examines the extent to which these distinct types of "dynamic heterogeneity" (DH) arise in metallic GF liquids (Cu-Zr, Ni-Nb, and Pd-Si alloys) having a vastly different molecular structure and chemistry. We first identified mobile and immobile particles and their transient clusters and found the DH in the metallic alloys to be remarkably similar in form to polymeric GF liquids, confirming the "universality" of the DH phenomenon. Furthermore, the lifetime of the mobile particle clusters was found to be directly related to the rate of diffusion in these materials, while the lifetime of immobile particles was found to be proportional to the structural relaxation time, providing some insight into the origin of decoupling in GF liquids. An examination of particles having a locally preferred atomic packing, and clusters of such particles, suggests that there is no one-to-one relation between these populations of particles so that an understanding of the origin of DH in terms of static fluid structure remains elusive. read less NOT USED (high confidence) P. Brault, C. Coutanceau, A. Caillard, and S. Baranton, “Pt3MeAu (Me = Ni, Cu) Fuel Cell Nanocatalyst Growth, Shapes, and Efficiency: A Molecular Dynamics Simulation Approach,” The Journal of Physical Chemistry C. 2019. link Times cited: 5 Abstract: The formation of the ternary Pt3NiAu and Pt3CuAu nanostructu… read moreAbstract: The formation of the ternary Pt3NiAu and Pt3CuAu nanostructures is examined using molecular dynamics simulations in the context of free (i.e., unsupported) cluster growth in an unreactive atmospher... read less NOT USED (high confidence) Z. Chang, R. Yang, and Y. Wei, “The linear-dependence of adhesion strength and adhesion range on temperature in soft membranes,” Journal of the Mechanics and Physics of Solids. 2019. link Times cited: 17 NOT USED (high confidence) L. Farsi and N. Deskins, “First principles analysis of surface dependent segregation in bimetallic alloys.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 20 Abstract: Stability is an important aspect of alloys, and proposed all… read moreAbstract: Stability is an important aspect of alloys, and proposed alloys may be unstable due to unfavorable atomic interactions. Segregation of an alloy may occur preferentially at specific exposed surfaces, which could affect the alloy's structure since certain surfaces may become enriched in certain elements. Using density functional theory (DFT), we modeled surface segregation in bimetallic alloys involving all transition metals doped in Pt, Pd, Ir, and Rh. We not only modeled common (111) surfaces of such alloys, but we also modeled (100), (110), and (210) facets of such alloys. Segregation is more preferred for early and late transition metals, with middle transition metals being most stable within the parent metal. We find these general trends in segregation energies for the parent metals: Pt > Rh > Pd > Ir. A comparison of different surfaces suggests no consistent trends across the different parent hosts, but segregation energies can vary up to 2 eV depending on the exposed surface. We also developed a statistical model to predict surface-dependent segregation energies. Our model is able to distinguish segregation at different surfaces (as opposed to generic segregation common in previous models), and agrees well with the DFT data. The present study provides valuable information about surface-dependent segregation and helps explain why certain alloy structures occur (e.g. core-shell). read less NOT USED (high confidence) N. Ahmad, M. Bon, D. Passerone, and R. Erni, “Template Assisted In Situ Synthesis of Ag@Au Bimetallic Nanostructures Employing Liquid Phase Transmission Electron Microscopy.,” ACS nano. 2019. link Times cited: 20 Abstract: Noble metal nanostructure synthesis via seed-mediated route … read moreAbstract: Noble metal nanostructure synthesis via seed-mediated route is a widely adopted strategy for a plethora of nanocrystal systems. Ag@Au core-shell nanostructures are radiolytically grown in real-time using in situ liquid-cell (scanning) transmission electron microscopy (LCTEM). Here we employ a capping agent, dimethyl-amine (DMA) and a coordinating complex, potassium iodide (KI) in an organic solvent (methanol) in order to: 1) slow down the reaction kinetics to observe mechanistic insights into the overgrowth pro-cess, 2) shift the growth regime from galvanic-replacement mode to direct synthesis mode resulting in the conventional synthesis of Ag@Au core-shell structures. A theoretical approach based on classical simulations complements our experiments providing further insight on the growth modes. In particular, we focus on the shape evolution and chemical ordering, as currently there is an insufficient understanding regarding mixed composition phases at interfaces of alloys even with well-known miscibilities. Furthermore, the comparison of theoretical and experimental data reveals that the final morphology of these nanoalloys is not simply a function of crystallinity of the underlying seed structure but instead is readily modified by extrinsic parameters such as additives, capping agent and modulation of surface energies of exposed crystal surfaces by the encapsulating solvent. The impact of these additional parameters is systematically investigated using an empirical approach in light of ab-initio simulations. read less NOT USED (high confidence) K. Huwig, V. Grigoryan, and M. Springborg, “Global Optimization of Li and Na Clusters: Application of a Modified Embedded Atom Method,” Journal of Cluster Science. 2019. link Times cited: 4 NOT USED (high confidence) A. Mahata and M. A. Zaeem, “Size effect in molecular dynamics simulation of nucleation process during solidification of pure metals: investigating modified embedded atom method interatomic potentials,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 8 Abstract: Due to the significant increase in computing power in recent… read moreAbstract: Due to the significant increase in computing power in recent years, the simulation size of atomistic methods for studying the nucleation process during solidification has been gradually increased, even to billion atom simulations (sub-micron length scale). But the question is how big of a model is required for size-independent and accurate simulations of the nucleation process during solidification? In this work, molecular dynamics simulations with model sizes ranging from ∼2000 to ∼8 million atoms were used to study nucleation during solidification. To draw general conclusions independent of crystal structures, the most advanced second nearest-neighbor modified embedded atom method interatomic potentials for Al (face-centered cubic), Fe (body-centered cubic), and Mg (hexagonal-close packed) were utilized for molecular dynamics simulations. We have analyzed several quantitative characteristics such as nucleation time, density of nuclei, nucleation rate, self-diffusion coefficient, and change in free energy during solidification. The results showed that by increasing the model size to about two million atoms, the simulations and measurable quantities become entirely independent of simulation cell size. The prediction of cell size required for size-independent computed data can considerably reduce the computational costs of atomistic simulations and at the same time increase the accuracy and reliability of the computational data. read less NOT USED (high confidence) Y. Su, S. Xu, and I. Beyerlein, “Density functional theory calculations of generalized stacking fault energy surfaces for eight face-centered cubic transition metals,” Journal of Applied Physics. 2019. link Times cited: 38 Abstract: In this work, we use density functional theory to calculate … read moreAbstract: In this work, we use density functional theory to calculate the entire generalized stacking fault energy (GSFE) surface for eight transition metals with a face-centered cubic structure: Ag, Au, Cu, Ir, Ni, Pd, Pt, and Rh. Analysis of the ⟨ 112 ⟩ GSFE curves finds that the displacements corresponding to the unstable stacking fault energy are larger than the ideal value for all eight metals except Ag and Cu. Over the entire surface, Pt is found to not possess well-defined local maxima or minima, suggesting spreading in favor of dissociation of the dislocation core, unlike the other seven metals. Our calculations also reveal that at a large ⟨ 112 ⟩ displacement, where atoms on two {111} adjacent planes are aligned, an anomalous local minimum occurs for Ir and Rh. The oddity is explained by relatively large, localized atomic displacements that take place in the two metals to accommodate the alignment that do not occur in the other six metals. In addition to the fully calculated surfaces, we characterize a continuous 11-term Fourier-series function, which provides a particularly excellent representation of the GSFE surfaces for Ag, Au, Cu, Ni, and Pd. read less NOT USED (high confidence) H. S. Huang, L. Ai, A. V. van Duin, M. Chen, and Y. Lü, “ReaxFF reactive force field for molecular dynamics simulations of liquid Cu and Zr metals.,” The Journal of chemical physics. 2019. link Times cited: 10 Abstract: We develop a ReaxFF reactive force field used for the molecu… read moreAbstract: We develop a ReaxFF reactive force field used for the molecular dynamics simulations of thermophysical properties of liquid Cu and Zr metals. The ReaxFF parameters are optimized by fitting to the first-principles density-functional calculations on the equations of state for bulk crystal structures and surface energies. To validate the force field, we compare the ReaxFF results with those from experiments and embedded-atom-method (EAM) potentials. We demonstrate that the present ReaxFF force field well represents structural characteristics and diffusion behaviors of elemental Cu and Zr up to high-temperature liquid regions. It reasonably reproduces the thermodynamic processes associated with crystal-liquid interface. In particular, the equilibrium melting temperatures show better agreement with experimental measurements than the results from EAM potentials. The ReaxFF reactive force field method exhibits a good transferability to the nonreactive processes of liquid systems. read less NOT USED (high confidence) J. Macko et al., “Hydrophobicity of Highly Ordered Nanorod Polycrystalline Nickel and Silver Surfaces,” Journal of Minerals and Materials Characterization and Engineering. 2019. link Times cited: 2 Abstract: Highly ordered nickel and silver nanorods arrays prepared by… read moreAbstract: Highly ordered nickel and silver nanorods arrays prepared by alumina template assisted electrodeposition were investigated to determine the effect of the array geometry on metal surface hydrophobicity and adhesion forces. The nanorod geometry, clustering and pinning were used to characterize surface hydrophobicity and its modulation. A contribution of metal crystallographic orientation to the surface energy was calculated. To characterize nanorod array surface properties and elucidate the source of the particle adhesion effects has been calculated. The dispersive components of surface tension γSD and surface polarizability ks, as surface features that markedly characterize hydrophobicity and adhesion, were calculated. The highest hydrophobicity was found for Ag nanorods with aspect ratio of 10 then Ni nanorods with aspect ratio 10. The same geometry of nanorods particles resulted in different surface hydrophobicity and it was ascribed to the orientation of Ag and Ni crystals formed on the top of nanorods. Due to high hydrophobicity nanorod array surfaces could be used as an antifouling surface in medicine to select areas on implant surface not to be colonized by cells and tissues. read less NOT USED (high confidence) A. Markidonov, D. Lubyanoi, V. Kovalenko, and M. Starostenkov, “Calculation of the Thermodynamic Characteristics of Fe–P System by the Molecular Dynamics Method,” Steel in Translation. 2019. link Times cited: 0 NOT USED (high confidence) H. Zhang, X. Wang, and J. Douglas, “Localization model description of diffusion and structural relaxation in superionic crystalline UO2.,” The Journal of chemical physics. 2019. link Times cited: 13 Abstract: We test the Localization Model (LM) prediction of a paramete… read moreAbstract: We test the Localization Model (LM) prediction of a parameter-free relationship between the α-structural relaxation time τα and the oxygen ion diffusion coefficient DO with the Debye-Waller factor ⟨u2⟩ for crystalline UO2 under superionic conditions where large anharmonic interactions lead to non-Arrhenius relaxation and high ion mobility. As in a previous study of structural relaxation in Cu-Zr metallic glass materials having a range of compositions, we find that the LM relationship between the picosecond atomic dynamics ("fast" beta relaxation) and the long-time structural relaxation time and oxygen ion diffusion coefficient holds to an excellent approximation without any free parameters over the full range of temperatures and pressures investigated in our simulations. read less NOT USED (high confidence) M. Azadeh, M. Kateb, and P. Marashi, “Determining phase transition using potential energy distribution and surface energy of Pd nanoparticles,” Computational Materials Science. 2019. link Times cited: 8 NOT USED (high confidence) J. Gu’enol’e et al., “Assessment and optimization of the fast inertial relaxation engine (fire) for energy minimization in atomistic simulations and its implementation in lammps,” Computational Materials Science. 2019. link Times cited: 89 NOT USED (high confidence) M. Shugaev and L. Zhigilei, “Thermodynamic analysis and atomistic modeling of subsurface cavitation in photomechanical spallation,” Computational Materials Science. 2019. link Times cited: 13 NOT USED (high confidence) R. H. Mirdha, P. Naskar, and P. Chaudhury, “Constructing transformation paths for conformational changes in (MgF2) n clusters using a stochastic procedure,” Molecular Physics. 2019. link Times cited: 0 Abstract: We present a stochastic optimisation procedure using simulat… read moreAbstract: We present a stochastic optimisation procedure using simulated annealing for studying conformational changes in clusters. The pathways for transformation between stable geometries through a transition state is studied for cluster sizes n=3−12. The work uses an objective function which exploits the sign of the eigenvalues of the Hessian matrix to locate transition states. Once it is located for a specific size it is examined if one minimum geometry can be converted to another through the transition state. The potential energy surface for the system is searched using the stochastic optimisation procedure of simulated annealing. The ruggedness of the surface makes the choice of the optimiser an ideal one. To complete the results the progress of the pathway is depicted as the function of the path coordinates at intermediate geometries. GRAPHICAL ABSTRACT read less NOT USED (high confidence) Z. Chen, Y. Cao, W. Tian, and Y. Wang, “Surface roughness analysis of Cu films deposited on Si substrates: A molecular dynamic analysis,” Journal of Applied Physics. 2019. link Times cited: 4 Abstract: Cu is a promising material to replace Al and Au in integrate… read moreAbstract: Cu is a promising material to replace Al and Au in integrated circuits and microscale devices because of its low electrical resistivity, high electromigration resistance, and low cost. However, surface roughness affects the contact resistance of these devices, especially when the device is on a microscale or nanoscale. This paper focuses on surface roughness analysis of Cu films deposited on Si substrates by molecular dynamic simulation based on the mechanism of physical vapor deposition. The effects of film thickness, deposition temperature, deposition interval, and reflow temperature on the surface roughness of Cu films are studied in detail. The simulation results show that the surface roughness can be improved by appropriate adjustments of these parameters. They also provide a foundation for further work on the deposition of Cu films on Si substrates.Cu is a promising material to replace Al and Au in integrated circuits and microscale devices because of its low electrical resistivity, high electromigration resistance, and low cost. However, surface roughness affects the contact resistance of these devices, especially when the device is on a microscale or nanoscale. This paper focuses on surface roughness analysis of Cu films deposited on Si substrates by molecular dynamic simulation based on the mechanism of physical vapor deposition. The effects of film thickness, deposition temperature, deposition interval, and reflow temperature on the surface roughness of Cu films are studied in detail. The simulation results show that the surface roughness can be improved by appropriate adjustments of these parameters. They also provide a foundation for further work on the deposition of Cu films on Si substrates. read less NOT USED (high confidence) W. Xie and F. Fang, “Cutting-Based Single Atomic Layer Removal Mechanism of Monocrystalline Copper: Atomic Sizing Effect,” Nanomanufacturing and Metrology. 2019. link Times cited: 19 NOT USED (high confidence) T. Roy, A. Sharma, D. Datta, and R. Balasubramaniam, “Molecular dynamics simulation of single discharge and dimensionless correlation with actual material removal in micro electrical discharge machining,” Molecular Simulation. 2019. link Times cited: 2 Abstract: ABSTRACT Shorter time pulses and length scales in micro EDM … read moreAbstract: ABSTRACT Shorter time pulses and length scales in micro EDM (MEDM) makes it difficult to observe the material removal phenomena taking place at very small zone during discharge. Since numerical or analytical model considers material to be uniform/isotropic, it cannot predict the discrete effects viz. crystal structure distortion due to discharge. Also, little attention has been given to the mechanism of material removal in MEDM at the atomistic level. Therefore, to understand the behaviour of material removal and crystal structure distortion due to single spark in MEDM, MD simulations have been carried out. Based on MD simulations, it was found that the percentage of material removed by vaporisation (∼40%) was higher at spark energies used in this model as compared to spark energies used by Wong et al. (20% at 23.50 µJ). Conversion from FCC crystal structure to amorphous was observed at the top surface of crater in both cases; the one with higher spark energy has marginally higher distortion indicating higher amorphisation. A new method is proposed based on which a dimensionless correlation was obtained between results of MDS and experiments which relates the ratio of specific material removal at higher spark energy to that at lower energy for both MDS and experiments. read less NOT USED (high confidence) T. Zhang, G. Wang, C. Wang, C. Tang, F. Zhang, and Y. Luo, “Effect of AuNP-AuNP vdW interaction on the mechanics and piezoresistivity of AuNP-polymer nanocomposite,” AIP Advances. 2019. link Times cited: 1 Abstract: Gold nanoparticle (AuNP)-polymer composite has attracted con… read moreAbstract: Gold nanoparticle (AuNP)-polymer composite has attracted considerable attention due to its high stretchability, metal-like electrical conductivity and substantial piezoresistivity. In the nanocomposite, the effect of the van der Waals (vdW) interaction increases greatly between AuNPs, which may significantly change its overall mechanical and electrical properties. In examining this issue, the nanocomposite with randomly distributed AuNPs is constructed by Monte Carlo method, tensile tests on the material are then performed via molecular dynamics simulations and finally, its piezo-resistivity is studied based on an effective resistor model. The effects of AuNP interaction are examined for the mechanics, dynamics and piezoresistivity of the nanocomposite by comparing the results obtained in the presence and absence of the vdW interaction. It is found that the AuNP attraction tends to hold the AuNPs together, leading to enhanced Young’s modulus, yield and fracture stress even at the low volume fraction 5% to 10% of AuNPs. The piezoresistive effect of the composite is also improved as the AuNP attraction substantially affects AuNP dynamics in large deformation. It is expected that similar effects of NP vdW interaction can also be obtained for the nanocomposites based on copper or silver NPs embedded in polymer.Gold nanoparticle (AuNP)-polymer composite has attracted considerable attention due to its high stretchability, metal-like electrical conductivity and substantial piezoresistivity. In the nanocomposite, the effect of the van der Waals (vdW) interaction increases greatly between AuNPs, which may significantly change its overall mechanical and electrical properties. In examining this issue, the nanocomposite with randomly distributed AuNPs is constructed by Monte Carlo method, tensile tests on the material are then performed via molecular dynamics simulations and finally, its piezo-resistivity is studied based on an effective resistor model. The effects of AuNP interaction are examined for the mechanics, dynamics and piezoresistivity of the nanocomposite by comparing the results obtained in the presence and absence of the vdW interaction. It is found that the AuNP attraction tends to hold the AuNPs together, leading to enhanced Young’s modulus, yield and fracture stress even at the low volume fraction 5% to... read less NOT USED (high confidence) V. Korolev, V. Samsonov, and P. Protsenko, “Molecular Dynamics Simulation of Unstable Equilibrium of a Spherical Nucleus for Determining the Interfacial Energy in a Pb–Cu Two-Component System,” Colloid Journal. 2019. link Times cited: 0 NOT USED (high confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “A molecular dynamics simulation of wear mechanism of diamond tool in nanoscale cutting of copper beryllium,” The International Journal of Advanced Manufacturing Technology. 2019. link Times cited: 33 NOT USED (high confidence) A. Vorontsov, A. E. Korenchenko, and B. Gelchinski, “Analysis of Stability of Small Metal Clusters during Metal Vapor Condensation,” High Temperature. 2019. link Times cited: 4 NOT USED (high confidence) M. Dardouri, K. Sbiaai, A. Hassani, A. Hasnaoui, Y. Boughaleb, and A. Arbaoui, “Silver monolayer formation on Cu(110) by kinetic Monte Carlo method,” The European Physical Journal Plus. 2019. link Times cited: 3 NOT USED (high confidence) P. Zhao, Y.-bo Guo, F. Zhang, Y. He, and Y. Yan, “Element proportion effect on internal stress from interfaces and other microstructural components in Cu–Pb alloys,” Molecular Simulation. 2019. link Times cited: 2 Abstract: ABSTRACT Polycrystalline materials like Cu–Pb alloy consist … read moreAbstract: ABSTRACT Polycrystalline materials like Cu–Pb alloy consist of four types of microstructural components, including grain cells, grain boundaries, triple junctions and vertex points, the mechanical properties of which governed by the atomic proportion of the alloy elements to a certain degree. The internal stresses from such microstructural components are quite different. Due to experimental limitations, the internal stresses from the alloy materials are difficult to measure directly, especially in the microstructural components. Here, we report a bottom-up approach using an atomistic calculation to obtain atomic properties in Cu-based alloy, as well as that in the microstructural components. The results reveal that a steep stress gradient exists at the interfaces of the alloy, which decreases significantly with the increase of the Pb. The defects evolution process in the alloy samples are investigated during tensile loading, revealing that the defect nucleation is delayed due to the decreasing von Mises stress gradient in the interfaces region as Pb increased. And the increased hydrostatic pressure in the interfaces regions, as a secondary factor can promote the defect nucleation. Among alloy samples with a grain size of 18.58 nm, that with 6.6 at.% Pb has minimal defects and the best mechanical properties. read less NOT USED (high confidence) P. Wynblatt, D. Chatain, A. Rollett, and U. Dahmen, “Origin of an unusual systematic variation in the heteroepitaxy of Ag on Ni – The roles of twinning and step alignment,” Acta Materialia. 2019. link Times cited: 6 NOT USED (high confidence) T. Fu, S. Zhang, J. Huang, D. Cai, J. Li, and J. Zhao, “Direct One‐pot Synthesis of Carbon Supported Ag‐Pt Alloy Nanoparticles as High Performance Electrocatalyst for Fuel Cell Application,” Fuel Cells. 2019. link Times cited: 7 Abstract: Ag‐Pt alloy material is one of the most potential candidates… read moreAbstract: Ag‐Pt alloy material is one of the most potential candidates to improve the catalytic activity of Pt‐based catalyst toward the oxygen reduction reaction (ORR), which can reduce the dosage of the high‐cost metal Pt used in the cathode of fuel cells. But it is hard to synthesis Ag‐Pt alloy with nanosize in an easy and cheap way. In this study, the Ag‐Pt alloy nanoparticles supported on carbon (Ag‐Pt/C) with different Ag at.% were prepared by a facile one‐pot approach with low costs. The XRD, TEM and XPS measurements were applied to verify the alloy structure of Ag‐Pt nanoparticles. These Ag‐Pt/C samples showed enhanced mass activities and catalytic stabilities for the ORR, which was up to two times higher than that of the commercial Pt/C catalyst at 0.9 V (vs. reversible hydrogen electrode). Furthermore, the membrane electrode assembly (MEA) made from the Ag‐Pt/C catalyst also showed a higher power density in H2‐O2 fuel cell test compared to the MEA made from the commercial Pt catalyst. These enhancements are attributed to the higher utilization of Pt and the regulated oxygen adsorption energy of Pt in the surface, which was confirmed using the density function theory calculation. read less NOT USED (high confidence) P. Paranjape, P. Gopal, and S. G. Srinivasan, “First-principles study of diffusion and interactions of hydrogen with silicon, phosphorus, and sulfur impurities in nickel,” Journal of Applied Physics. 2019. link Times cited: 4 Abstract: Using density functional theory (DFT), we systematically stu… read moreAbstract: Using density functional theory (DFT), we systematically study the effect of Si, P, and S impurities on the diffusion and binding of an H atom in a face-centered-cubic (FCC) Ni lattice. First, we quantify binding energies of an H atom to a vacancy, an impurity atom, and a vacancy-impurity atom defect pair. The energetics of H interactions show that a vacancy-impurity atom defect pair with larger binding energy traps the H atom more strongly and correlates with electronic bonding. Next, we study how the impurities influence diffusion of an H atom by using the Climbing Image Nudged Elastic band method to evaluate the Minimum Energy Path and the energy barrier for diffusion. The H atom preferentially diffuses between tetrahedral to octahedral (T-O) interstitial positions in pure Ni and when impurities are present. However, the activation energy significantly decreases from 0.95 eV in pure Ni to 0.47 eV, 0.52 eV, and 0.46 eV, respectively, in the presence of Si, P, and S impurities, which speeds up H diffusion. We rationalize this by comparing the bonding character of the saddle point configuration and changes in the electronic structure of Ni for each system. Notably, these analyses correlate the lower values of the activation energies to a local atomic strain in a Ni lattice. Our DFT study also validates the hypothesis of Berkowitz and Kane that P increases the H diffusion and, thereby, significantly increases H embrittlement susceptibility of Ni. We report a similar effect for Si and S impurities in Ni.Using density functional theory (DFT), we systematically study the effect of Si, P, and S impurities on the diffusion and binding of an H atom in a face-centered-cubic (FCC) Ni lattice. First, we quantify binding energies of an H atom to a vacancy, an impurity atom, and a vacancy-impurity atom defect pair. The energetics of H interactions show that a vacancy-impurity atom defect pair with larger binding energy traps the H atom more strongly and correlates with electronic bonding. Next, we study how the impurities influence diffusion of an H atom by using the Climbing Image Nudged Elastic band method to evaluate the Minimum Energy Path and the energy barrier for diffusion. The H atom preferentially diffuses between tetrahedral to octahedral (T-O) interstitial positions in pure Ni and when impurities are present. However, the activation energy significantly decreases from 0.95 eV in pure Ni to 0.47 eV, 0.52 eV, and 0.46 eV, respectively, in the presence of Si, P, and S impurities, which speeds up H diffusio... read less NOT USED (high confidence) H. Bhattarai, K. E. Newman, and J. Gezelter, “Polarizable potentials for metals: The density readjusting embedded atom method (DR-EAM),” Physical Review B. 2019. link Times cited: 6 Abstract: In simulations of metallic interfaces, a critical aspect of … read moreAbstract: In simulations of metallic interfaces, a critical aspect of metallic behavior is missing from the some of the most widely used classical molecular dynamics force fields. We present a modification of the embedded atom method (EAM) which allows for electronic polarization of the metal by treating the valence density around each atom as a fluctuating dynamical quantity. The densities are represented by a set of additional fluctuating variables (and their conjugate momenta) which are propagated along with the nuclear coordinates. This ``density readjusting EAM'' (DR-EAM) preserves nearly all of the useful qualities of traditional EAM, including bulk elastic properties and surface energies. However, it also allows valence electron density to migrate through the metal in response to external perturbations. We show that DR-EAM can successfully model polarization in response to external charges, capturing the image charge effect in atomistic simulations. DR-EAM also captures some of the behavior of metals in the presence of uniform electric fields, predicting surface charging and shielding internal to the metal. We further show that it predicts charge transfer between the constituent atoms in alloys, leading to novel predictions about unit cell geometries in layered $\mathrm{L}{1}_{0}$ structures. read less NOT USED (high confidence) V. Sunko et al., “Controlled Introduction of Defects to Delafossite Metals by Electron Irradiation,” Physical Review X. 2019. link Times cited: 21 Abstract: The delafossite metals PdCoO$_{2}$, PtCoO$_{2}$ and PdCrO$_{… read moreAbstract: The delafossite metals PdCoO$_{2}$, PtCoO$_{2}$ and PdCrO$_{2}$ are among the highest conductivity materials known, with low temperature mean free paths of tens of microns in the best as-grown single crystals. A key question is whether these very low resistive scattering rates result from strongly suppressed backscattering due to special features of the electronic structure, or are a consequence of highly unusual levels of crystalline perfection. We report the results of experiments in which high energy electron irradiation was used to introduce point disorder to the Pd and Pt layers in which the conduction occurs. We obtain the cross-section for formation of Frenkel pairs in absolute units, and cross-check our analysis with first principles calculations of the relevant atomic displacement energies. We observe an increase of resistivity that is linear in defect density with a slope consistent with scattering in the unitary limit. Our results enable us to deduce that the as-grown crystals contain extremely low levels of in-plane defects of approximately $0.001\%$. This confirms that crystalline perfection is the most important factor in realizing the long mean free paths, and highlights how unusual these delafossite metals are in comparison with the vast majority of other multi-component oxides and alloys. We discuss the implications of our findings for future materials research. read less NOT USED (high confidence) V. Grigoryan and M. Springborg, “Temperature and isomeric effects in nanoclusters.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 16 Abstract: The canonical thermodynamics of clusters is determined quant… read moreAbstract: The canonical thermodynamics of clusters is determined quantum mechanically in the general case of multiple minima of the potential energy surface (PES) using the superposition approximation. As an illustration of the consequences of our approach, we study in detail the thermodynamic properties of CuN clusters with N from 2 to 150 as a function of cluster size, temperature, and number of isomers. Thereby, for instance, solid-solid transition temperatures for several cluster sizes are determined. We show that the cluster vibrations have a strong impact on the stability of the clusters and that this can be observed not only at medium and high temperatures but also at low temperatures and even at T = 0 K. Thus, including zero-point corrections can change the relative energetic ordering of different isomers. This isomeric effect results in an oscillatory dependence of the heat capacity on cluster size at moderate and high temperatures. Moreover, for the identification of magic clusters at non-vanishing temperature, the Helmholtz free energy is analyzed as a function of cluster size and temperature within a one-, two-, and three-minima model of the PES. Thereby, we demonstrate that the concept of magic clusters is strongly temperature dependent so that in several cases clusters change from being magic or non-magic at T = 0 K to be the opposite at non-vanishing temperature. We emphasize that all these effects are not specific for copper clusters alone but can also be observed in other metal or semiconductor nanoclusters. read less NOT USED (high confidence) J. Shin et al., “Controlling dislocation nucleation-mediated plasticity in nanostructures via surface modification,” Acta Materialia. 2019. link Times cited: 35 NOT USED (high confidence) X. Zhang, H. Zhang, X.-C. Zheng, M. Walls, and Y. Peng, “A novel 96.5Sn3Cu0.5Mn nanosolder with enhanced wettability applied to nanosoldering of WO3 nanomaterial,” Nanotechnology. 2019. link Times cited: 3 Abstract: Nano-soldering relying on a sacrificial nanosolder, is a fle… read moreAbstract: Nano-soldering relying on a sacrificial nanosolder, is a flexible interconnection technique, having promising applications in joining nanosized functional materials; that is an essential step in the assembly of nano-devices. In a soldering, the wettability is important in the bonding of two nanomaterial, which determines the quality of the junction. Tungsten trioxide nanomaterial has unique characteristics such as electro-, opto-, gaso-chromic. To assemble this nanomaterial into functional nano-devices, a superior nanosolder is necessary. The conventional SnCu nanosolder has been chosen, but its wetting on WO3 is unsatisfactory. Here, our study indicates that the SnCu wettability on WO3 material has been improved greatly by adding minor manganese, in which the contact angle has a significant change from 73.2° to 41.7°. Then the wetting mechanism is investigated by observing the soldering interface. Lastly, a more robust and higher-reliable junction has been obtained by thermal soldering two individual WO3 nano-objects into a cross-shaped pattern. read less NOT USED (high confidence) H. Zhang, X. Wang, A. Chremos, and J. Douglas, “Superionic UO2: A model anharmonic crystalline material.,” The Journal of chemical physics. 2019. link Times cited: 28 Abstract: Crystalline materials at elevated temperatures and pressures… read moreAbstract: Crystalline materials at elevated temperatures and pressures can exhibit properties more reminiscent of simple liquids than ideal crystalline materials. Superionic crystalline materials having a liquidlike conductivity σ are particularly interesting for battery, fuel cell, and other energy applications, and we study UO2 as a prototypical superionic material since this material is widely studied given its commercial importance as reactor fuel. Using molecular dynamics, we first investigate basic thermodynamic and structural properties. We then quantify structural relaxation, dynamic heterogeneity, and average ion mobility. We find that the non-Arrhenius diffusion and structural relaxation time of this prototypical superionic material can be described in terms of a generalized activated transport model ("string model") in which the activation energy varies with the average string length. Our transport data can also be described equally well by an Adam-Gibbs model in which the excess entropy density of the crystalline material is estimated from specific heat and thermal expansion data, consistent with the average scale of stringlike collective motion scaling inversely with the excess entropy of the crystal. Strong differences in the temperature dependence of the interfacial mobility from nonionic materials are observed, and we suggest that this difference is due to the relatively high cohesive interaction of ionic materials. In summary, the study of superionic UO2 provides insight into the role of cooperative motion in enhancing ion mobility in ionic materials and offers design principles for the development of new superionic materials for use in diverse energy applications. read less NOT USED (high confidence) R. D. Kamachali, “Surface-Induced Phase Transition During Coalescence of Au Nanoparticles: A Molecular Dynamics Simulation Study,” arXiv: Materials Science. 2019. link Times cited: 1 Abstract: In this study, the melting and coalescence of Au nanoparticl… read moreAbstract: In this study, the melting and coalescence of Au nanoparticles were investigated using molecular dynamics simulation. The melting points of nanoparticles were calculated by studying the potential energy and Lindemann indices as a function of temperature. The simulations show that coalescence of two Au nanoparticles of the same size occurs at far lower temperatures than their corresponding melting temperature. For smaller nanoparticles, the difference between melting and coalescence temperature increases. Detailed analyses of the Lindemann indices and potential energy distribution across the nanoparticles show that the surface melting in nanoparticles begins at several hundred degrees below the melting point. This suggests that the coalescence is governed by the liquid-phase surface diffusion. Furthermore, the surface reduction during the coalescence accelerates its kinetics. It is found that for small enough particles and/or at elevated temperatures, the heat released due to the surface reduction result in a melting transition of the two attached nanoparticles. read less NOT USED (high confidence) M. Bonvalet, X. Sauvage, and D. Blavette, “Intragranular nucleation of tetrahedral precipitates and discontinuous precipitation in Cu-5wt%Ag,” Acta Materialia. 2019. link Times cited: 19 NOT USED (high confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “A molecular dynamics simulation of wear mechanism of diamond tool in nanoscale cutting of copper beryllium,” The International Journal of Advanced Manufacturing Technology. 2019. link Times cited: 0 NOT USED (high confidence) S. Borisova, G. Rusina, S. Eremeev, and E. Chulkov, “Phonons on Cu(001) surface covered by submonolayer alkali metals,” Journal of Physics: Condensed Matter. 2019. link Times cited: 1 Abstract: We present a theoretical investigation of the structural and… read moreAbstract: We present a theoretical investigation of the structural and vibrational properties of ordered 2D phases formed by the Li, Na and K atoms on the Cu surface. The lattice relaxation, phonon dispersions and polarization of vibrational modes as well as the local density of states are calculated using the embedded-atom method. The obtained structural parameters and vibrational frequencies are in close agreement with available experimental results. read less NOT USED (high confidence) X. Qiu, J. Mankowski, J. Dickens, A. Neuber, and R. P. Joshi, “Model evaluations of surface modification by energetic incident carbon atoms on graphene coated copper electrodes,” Physics of Plasmas. 2019. link Times cited: 1 NOT USED (high confidence) P. Spiering, M. Wijzenbroek, and M. Somers, “An improved static corrugation model.,” The Journal of chemical physics. 2018. link Times cited: 10 Abstract: Accurately describing surface temperature effects for the di… read moreAbstract: Accurately describing surface temperature effects for the dissociation of H2 on Cu(111) remains challenging. While Ab initio Molecular Dynamics (AIMD), the current state-of-the-art method for modelling such systems, can produce accurate results, it is computationally very expensive to use for extensive testing of, for example, density functionals. A chemically accurate static corrugation model for H2 and D2 on Cu(111) dissociation was made by introducing effective three-body interactions as well as an H2-bond dependence and fitting the model to density functional theory energies for 15 113 different configurations. Reaction probabilities and rovibrational (in)elastic scattering probabilities were computed and compared to experiments and other calculations. Theoretical and experimental results are in good agreement, except for the reaction of (v = 0, J = 0) H2 where both AIMD and the newly developed static corrugation model, both based on the same underlying density functional, predict a similar deviation from the experiment. read less NOT USED (high confidence) Y.-duan Xie and N. Dimitrov, “Highly Active and Durable CuxAu(1–x) Ultrathin-Film Catalysts for Nitrate Electroreduction Synthesized by Surface-Limited Redox Replacement,” ACS Omega. 2018. link Times cited: 12 Abstract: CuxAu(1–x) bimetallic ultrathin-film catalysts for nitrate e… read moreAbstract: CuxAu(1–x) bimetallic ultrathin-film catalysts for nitrate electroreduction have been synthesized using electrochemical atomic layer deposition by surface-limited redox replacement of Pb underpotentially deposited layer. Controlled by the ratio of [Cu2+] ions and [AuCl4–] complex in the deposition solution, the alloy film composition (atomic fraction, x in the range of 0.5–1) has been determined by X-ray photoelectron spectroscopy and indirectly estimated by anodic stripping voltammetry. The catalytic activity and durability of CuxAu(1–x) thin films, Cu thin film, and bulk Cu have been studied by one- and multiple-cycle voltammetry. The synthesized CuxAu(1–x) thin films feature up to two times higher nitrate electroreduction activity in acidic solution compared to bulk and thin-film Cu counterparts. Highest activity has been measured with a Cu0.70Au0.30 catalyst. Durability tests have demonstrated that Cu thin films undergo rapid deactivation losing 65% of its peak activity for 92 cycles, whereas Cu0.70Au0.30 catalysts lose only 45% of their top performance. The significantly better durability of alloy films can be attributed to effective resistance to poisoning and/or hindered dissolution of Cu active centers. It has been also found that both CuxAu(1–x) and pure Cu thin films show best electroreduction activity at lowest pH. read less NOT USED (high confidence) S. Mahmoud and N. Mousseau, “Long-time point defect diffusion in ordered nickel-based binary alloys: How small kinetic differences can lead to completely long-time structural evolution,” Materialia. 2018. link Times cited: 15 NOT USED (high confidence) B. Sun, H. Barrón, B. Wells, G. Opletal, and A. Barnard, “Correlating anisotropy and disorder with the surface structure of platinum nanoparticles.,” Nanoscale. 2018. link Times cited: 4 Abstract: Due to the competition between numerous physicochemical vari… read moreAbstract: Due to the competition between numerous physicochemical variables during formation and processing, platinum nanocatalysts typically contain a mixture of shapes, distributions of sizes, and a considerable degree of surface imperfection. Structural imperfection and sample polydispersivity are inevitable at scale, but accepting bulk and surface diversity as legitimate design features provides new opportunities for nanoparticle design. In recent years disorder and anisotropy have been embraced as useful design parameters but predicting the impact of uncontrollable imperfection a priori is challenging. In the present work we have created an ensemble of uniquely imperfect nanoparticles extracted from classical molecular dynamics trajectories and applied statistical filters to restrict the ensemble in ways that reflect common industrial design principles. We find that targeting different sizes and size distributions may be an effective way of promoting or suppressing internal disorder or crystallinity (as required), but the degree of anisotropy of the particle as a whole has a greater impact on the population of different types of surface ordering and active sites. These results indicate that tuning of disordered and anisotropic Pt nanoparticles is possible, but it is not as straightforward as geometrically ideal nanoparticles with a high degree of crystallinity. It is unlikely that a synthesis strategy could eliminate this diversity entirely, or ensure this type of structural complexity does not develop post-synthesis under operational conditions, but it may be possible to bias the formation of specific bulk structures and the surface anisotropy. read less NOT USED (high confidence) M. Kateb, M. Azadeh, P. Marashi, and S. Ingvarsson, “Size and shape-dependent melting mechanism of Pd nanoparticles,” Journal of Nanoparticle Research. 2018. link Times cited: 11 NOT USED (high confidence) S. Dokukin, S. Kolesnikov, A. Saletsky, and A. Klavsyuk, “Growth of the Pt/Cu(111) surface alloy: Self-learning kinetic Monte Carlo simulations,” Journal of Alloys and Compounds. 2018. link Times cited: 12 NOT USED (high confidence) J. Byggmästar, F. Granberg, and K. Nordlund, “Effects of the short-range repulsive potential on cascade damage in iron,” Journal of Nuclear Materials. 2018. link Times cited: 52 NOT USED (high confidence) Z. Chen et al., “Interatomic Potential in the Nonequilibrium Warm Dense Matter Regime.,” Physical review letters. 2018. link Times cited: 18 Abstract: We present a new measurement of lattice disassembly times in… read moreAbstract: We present a new measurement of lattice disassembly times in femtosecond-laser-heated polycrystalline Au nanofoils. The results are compared with molecular dynamics simulations incorporating a highly optimized, embedded-atom-method interatomic potential. For absorbed energy densities of 0.9-4.3 MJ/kg, the agreement between the experiment and simulation reveals a single-crystal-like behavior of homogeneous melting and corroborates the applicability of the interatomic potential in the nonequilibrium warm dense matter regime. For energy densities below 0.9 MJ/kg, the measurement is consistent with nanocrystal behavior where melting is initiated at the grain boundaries. read less NOT USED (high confidence) G. Bonny et al., “Classical interatomic potential for quaternary Ni–Fe–Cr–Pd solid solution alloys,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 9 Abstract: In this paper, we present a new quaternary interatomic poten… read moreAbstract: In this paper, we present a new quaternary interatomic potential for the NiFeCrPd system, which is an extension on the previous NiFeCr potential. Density functional theory is used to calculate the quantities to be fitted, with particular focus on the energetics of point defects with solutes, for the potential to be used towards understanding radiation damage properties. The potential thus will enable the modeling of multi-elemental solid solution alloys consisting of up to four elements. To test the potential, we have performed atomistic kinetic Monte Carlo simulations to investigate the effect of configurational entropy on the self-diffusion coefficients. The self-diffusion coefficients are found to increase with chemical complexity, contrary to the common postulation of sluggish diffusion in high entropy alloys (HEAs). In addition, we have performed molecular dynamics simulations to elucidate the effect of Pd on vacancy diffusion and clustering in pure Ni and binary alloys. In agreement with recent irradiation experiments, our simulations show that while large vacancy clusters, such as stacking fault tetrahedra, are formed in pure Ni, Ni–Fe and Ni–Cr systems, negligible vacancy clustering is observed in Ni–Pd systems, indicating a possible effect of Pd in reducing cluster sizes. We suggest that this potential will be useful for studying the defect evolution in multi-component HEAs. read less NOT USED (high confidence) F. Baras, V. Turlo, O. Politano, S. Vadchenko, A. Rogachev, and A. Mukasyan, “SHS in Ni/Al Nanofoils: A Review of Experiments and Molecular Dynamics Simulations,” Advanced Engineering Materials. 2018. link Times cited: 38 Abstract: Non‐isothermal processes in nanometric metallic multilayers … read moreAbstract: Non‐isothermal processes in nanometric metallic multilayers are reviewed, both experimentally and theoretically. The Ni/Al nanofoil is considered as a model system. On the one hand, the experimental methods of elaboration and analysis are presented and, on the other hand, the modeling approach at the macroscopic and atomic scale. The basic experimental features are reported together with recent achievements. Molecular dynamics investigation of the reactivity of Ni/Al systems is reported for bulk systems and nanosystems including nanoparticles, nanowires, nanofilms, and multilayers. The focus is on atomic‐scale modeling versus experiments. Molecular dynamics approaches allow us to elucidate the mechanisms of non‐isothermal processes occurring in nanoscale systems, such as phase transformations and self‐propagation reactions. read less NOT USED (high confidence) Z. Wu, S. Huang, J. Ding, W. Wang, and X. Luo, “Molecular dynamics simulation of cylindrical Richtmyer-Meshkov instability,” Science China Physics, Mechanics & Astronomy. 2018. link Times cited: 15 NOT USED (high confidence) A. Bacher, T. Schrøder, and J. Dyre, “The EXP pair-potential system. II. Fluid phase isomorphs.,” The Journal of chemical physics. 2018. link Times cited: 27 Abstract: This paper continues the investigation of the exponentially … read moreAbstract: This paper continues the investigation of the exponentially repulsive EXP pair-potential system of Paper I [A. K. Bacher et al., J. Chem. Phys. 149, 114501 (2018)] with a focus on isomorphs in the low-temperature gas and liquid phases. As expected from the EXP system's strong virial potential-energy correlations, the reduced-unit structure and dynamics are isomorph invariant to a good approximation. Three methods for generating isomorphs are compared: the small-step method that is exact in the limit of small density changes and two versions of the direct-isomorph-check method that allows for much larger density changes. Results from the latter two approximate methods are compared to those of the small-step method for each of the three isomorphs generated by 230 one percent density changes, covering one decade of density variation. Both approximate methods work well. read less NOT USED (high confidence) D. L. Mafra et al., “Ambient-pressure CVD of graphene on low-index Ni surfaces using methane: A combined experimental and first-principles study,” Physical Review Materials. 2018. link Times cited: 15 Abstract: Daniela L. Mafra,1 Jimena A. Olmos-Asar,2,3,* Fabio R. Negre… read moreAbstract: Daniela L. Mafra,1 Jimena A. Olmos-Asar,2,3,* Fabio R. Negreiros,2,3,† Alfonso Reina,4 Ki Kang Kim,5 Mildred S. Dresselhaus,1,6 Jing Kong,1 Gary J. Mankey,7,8 and Paulo T. Araujo7,8,9,‡ 1Department of Electrical Engineering and Computer Sciences, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA 2Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, Santo André, 09210-580 SP, Brazil 3INFIQC, CONICET, Departamento de Química Teórcia y Computacional, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, X5000HUA, Argentina 4Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA 5Department of Energy and Materials Engineering, Dongguk University-Seoul, Seoul, 04620, Republic of Korea 6Department of Physics, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA 7Department of Physics and Astronomy, The University of Alabama, Tuscaloosa, Alabama 35487, USA 8Center for Materials for Information Technology (MINT Center), The University of Alabama, Tuscaloosa, Alabama 35401, USA 9Natural Sciences Institute, Graduate Program in Physics Federal University of Para, Belem, PA 66075-110, Brazil read less NOT USED (high confidence) J. Cuny, N. Tarrat, F. Spiegelman, A. Huguenot, and M. Rapacioli, “Density-functional tight-binding approach for metal clusters, nanoparticles, surfaces and bulk: application to silver and gold,” Journal of Physics: Condensed Matter. 2018. link Times cited: 17 Abstract: Density-functional based tight-binding (DFTB) is an efficien… read moreAbstract: Density-functional based tight-binding (DFTB) is an efficient quantum mechanical method that can describe a variety of systems, going from organic and inorganic compounds to metallic and hybrid materials. The present topical review addresses the ability and performance of DFTB to investigate energetic, structural, spectroscopic and dynamical properties of gold and silver materials. After a brief overview of the theoretical basis of DFTB, its parametrization and its transferability, we report its past and recent applications to gold and silver systems, including small clusters, nanoparticles, bulk and surfaces, bare and interacting with various organic and inorganic compounds. The range of applications covered by those studies goes from plasmonics and molecular electronics, to energy conversion and surface chemistry. Finally, perspectives of DFTB in the field of gold and silver surfaces and NPs are outlined. read less NOT USED (high confidence) Z. A. Ahmatov, A. Gangapshev, V. Romanenko, A. Khokonov, and V. Kuzminov, “Low-Background Method of Isotope Markers for Measuring the Efficiency of Intercalation of Graphite by Potassium Atoms,” Physics of Particles and Nuclei. 2018. link Times cited: 0 NOT USED (high confidence) A. Kotri, E. E. koraychy, M. Mazroui, and I. Achik, “Hetero-diffusion of small clusters on Ag(111) surface,” The European Physical Journal Plus. 2018. link Times cited: 6 NOT USED (high confidence) P. Brault, “Multiscale Molecular Dynamics Simulation of Plasma Processing: Application to Plasma Sputtering,” Frontiers in Physics. 2018. link Times cited: 16 Abstract: Molecular dynamics is an atomistic tool that is able to trea… read moreAbstract: Molecular dynamics is an atomistic tool that is able to treat dynamics of atom/molecules/cluster assemblies mainly in the condensed and liquid phases. The goal of the present article is to provide a new methodology for describing all phenomena of plasma processing and beyond such as gas phase chemistry as well. Simulations of condensed matter and liquid processes by molecular dynamics are now readily accessible provided the interaction potentials are available, so quantitative parameters can be deduced as diffusion coefficient, … The situation is less clear for gas phase processes while they operate on larger space and time scales than for condensed phases and at lower specie densities. The present article is proposing a new methodology for describing plasma core interactions in taking into account experimental space and time scales. This is illustrated on a plasma sputtering process and deposition in a single simulation. read less NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 0 NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 38 NOT USED (high confidence) Y. Wang, R. Wang, C. Wang, and X. Yu, “AuNP-PE interface/phase and its effects on the tensile behaviour of AuNP-PE composites,” Journal of Applied Physics. 2018. link Times cited: 3 Abstract: A comprehensive study was conducted for a gold nanoparticle … read moreAbstract: A comprehensive study was conducted for a gold nanoparticle (AuNP)-polyethylene (PE) composite. Molecular dynamic (MD) simulations were employed to construct the AuNP-PE systems, achieve their constitutive relations, and measure their tensile properties. Specifically, the AuNP-PE interface/phase was studied via the mass density profile, and its effect was evaluated by comparing the composite with a pure PE matrix. These research studies were followed by the study of the fracture mechanisms and the size and volume fraction effects of AuNPs. Efforts were also made to reveal the underlying physics of the MD simulations. In the present work, an AuNP-PE interface and a densified PE interphase were achieved due to the AuNP-PE van der Waals interaction. Such an interface/phase is found to enhance the Young's modulus and yield stress but decrease the fracture strength and strain.A comprehensive study was conducted for a gold nanoparticle (AuNP)-polyethylene (PE) composite. Molecular dynamic (MD) simulations were employed to construct the AuNP-PE systems, achieve their constitutive relations, and measure their tensile properties. Specifically, the AuNP-PE interface/phase was studied via the mass density profile, and its effect was evaluated by comparing the composite with a pure PE matrix. These research studies were followed by the study of the fracture mechanisms and the size and volume fraction effects of AuNPs. Efforts were also made to reveal the underlying physics of the MD simulations. In the present work, an AuNP-PE interface and a densified PE interphase were achieved due to the AuNP-PE van der Waals interaction. Such an interface/phase is found to enhance the Young's modulus and yield stress but decrease the fracture strength and strain. read less NOT USED (high confidence) K. Gilroy, X. Yang, S. Xie, M. Zhao, D. Qin, and Y. Xia, “Shape‐Controlled Synthesis of Colloidal Metal Nanocrystals by Replicating the Surface Atomic Structure on the Seed,” Advanced Materials. 2018. link Times cited: 99 Abstract: Controlling the surface structure of metal nanocrystals whil… read moreAbstract: Controlling the surface structure of metal nanocrystals while maximizing the utilization efficiency of the atoms is a subject of great importance. An emerging strategy that has captured the attention of many research groups involves the conformal deposition of one metal as an ultrathin shell (typically 1–6 atomic layers) onto the surface of a seed made of another metal and covered by a set of well‐defined facets. This approach forces the deposited metal to faithfully replicate the surface atomic structure of the seed while at the same time serving to minimize the usage of the deposited metal. Here, the recent progress in this area is discussed and analyzed by focusing on the synthetic and mechanistic requisites necessary for achieving surface atomic replication of precious metals. Other related methods are discussed, including the one‐pot synthesis, electrochemical deposition, and skin‐layer formation through thermal annealing. To close, some of the synergies that arise when the thickness of the deposited shell is decreased controllably down to a few atomic layers are highlighted, along with how the control of thickness can be used to uncover the optimal physicochemical properties necessary for boosting the performance toward a range of catalytic reactions. read less NOT USED (high confidence) M. M. Blazhynska, A. Kyrychenko, and O. Kalugin, “Molecular dynamics simulation of the size-dependent morphological stability of cubic shape silver nanoparticles,” Molecular Simulation. 2018. link Times cited: 25 Abstract: The morphological stability of sharp-edged silver nanopartic… read moreAbstract: The morphological stability of sharp-edged silver nanoparticles is examined by the classical molecular dynamics (MD) simulations. The crystalline structure and the perfect fcc atom packing of a series of silver nanocubes (AgNC) of different sizes varying from 63 up to 1099 atoms are compared against quasi-spherical nanoparticles of the same sizes at temperature 303 K. Our MD simulations demonstrate that starting from the preformed perfect crystalline structures the cubic shape is preserved for AgNCs composed of 365–1099 atoms. Surprisingly, the rapid loss of the cubic shape morphology and transformation into the non-fcc-structure are found for smaller AgNCs composed of less than ~256 atoms. No such loss of the preformed crystalline structure is seen for quasi-spherical nanoparticles composed of 38–1007 atoms. The analysis of the temperature dependence and the binding energy of outermost Ag surface atoms suggests that the loss of the perfect cubic shape, rounding and smoothing of sharp edges and corners are driven by the tendency towards the increase in their coordination number. In addition, we revealed that AgNC1099 partially loses its sharp edges and corners in the aqueous environment; however, the polymer coating with poly(vinyl alcohol) (PVA) was able to preserve the well-defined cubic morphology. Finally, these results help improve the understanding of the role of surface capping agents in solution phase synthesis of Ag nanocubes. read less NOT USED (high confidence) Y. Long, B. He, W. Cui, X. Zhuang, and J. Twiefel, “Molecular Dynamics Simulation of Microwelds Formation and Breakage During Ultrasonic Copper Wire Bonding,” 2018 IEEE 68th Electronic Components and Technology Conference (ECTC). 2018. link Times cited: 3 Abstract: Nowadays ultrasonic (US) copper wire bonding gests more requ… read moreAbstract: Nowadays ultrasonic (US) copper wire bonding gests more required and applied in power electronics. Despite its large amounts of usage, the underlying bonding mechanisms are still unclear. Among them, the dynamic changes of microwelds are essential to the bonding process as the bonding quality and reliability are greatly influenced by the formed microwelds. In this work, the formation and breakage of microwelds during US copper wire bonding are analyzed by molecular dynamics simulation. Due to the limit of the computational expense, a small local interface consisting of ~40000 atoms is simulated. In the model, the copper substrate is fixed while the movement of the copper wire is imposed. Microwelds are first formed during the downwards moving of the wire and get enlarged with further vertical displacement. The formed microwelds can be broken due to the vibration while new microwelds can be formed in the meantime. Because of the formation, deformation and breakage of the microwelds, the surface roughness can be significantly changed and the vertical displacement is the most influential factor. Defects caused by the microwelds formation and breakage can be clearly observed in the simulation results. The achieved information has a high potential to enhance the bonding quality and reliability. read less NOT USED (high confidence) F. G. VanGessel, J. Peng, and P. Chung, “A review of computational phononics: the bulk, interfaces, and surfaces,” Journal of Materials Science. 2018. link Times cited: 22 NOT USED (high confidence) S. Acharya and T. Rahman, “Erratum to: Toward multiscale modeling of thin-film growth processes using SLKMC,” Journal of Materials Research. 2018. link Times cited: 3 Abstract: The paper by Acharya and Rahman1 was inadvertently published… read moreAbstract: The paper by Acharya and Rahman1 was inadvertently published in Volume 33, Issue 6 of Journal of Materials Research . The article is part of the Journal of Materials Research Focus Issue on Advanced Atomistic Algorithms in Materials Science and is referenced as such in the introduction to that issue.2 read less NOT USED (high confidence) X. W. Zhou, D. Ward, and M. E. Foster, “A bond-order potential for the Al–Cu–H ternary system,” New Journal of Chemistry. 2018. link Times cited: 13 Abstract: Al-Based Al–Cu alloys have a very high strength to density r… read moreAbstract: Al-Based Al–Cu alloys have a very high strength to density ratio, and are therefore important materials for transportation systems including vehicles and aircrafts. These alloys also appear to have a high resistance to hydrogen embrittlement, and as a result, are being explored for hydrogen related applications. To enable fundamental studies of mechanical behavior of Al–Cu alloys under hydrogen environments, we have developed an Al–Cu–H bond-order potential according to the formalism implemented in the molecular dynamics code LAMMPS. Our potential not only fits well to properties of a variety of elemental and compound configurations (with coordination varying from 1 to 12) including small clusters, bulk lattices, defects, and surfaces, but also passes stringent molecular dynamics simulation tests that sample chaotic configurations. Careful studies verified that this Al–Cu–H potential predicts structural property trends close to experimental results and quantum-mechanical calculations; in addition, it properly captures Al–Cu, Al–H, and Cu–H phase diagrams and enables simulations of H2 dissociation, chemisorption, and absorption on Al–Cu surfaces. read less NOT USED (high confidence) P. Chowdhury, “Frontiers of Theoretical Research on Shape Memory Alloys: A General Overview,” Shape Memory and Superelasticity. 2018. link Times cited: 16 NOT USED (high confidence) А. Векман, Б. Ф. Демьянов, and А. С. Драгунов, “Grain Boundary Simulation: the Role of the Interatomic Potential.” 2018. link Times cited: 0 Abstract: В обзоре проведено сравнение результатов моделирования атомн… read moreAbstract: В обзоре проведено сравнение результатов моделирования атомных структур различного уровня сложности с использованием парного потенциала Морзе и многочастичного потенциала Клери — Розато. Параметры потенциальных функций соответствовали алюминию. Выполнен расчет энергии идеального кристалла, структуры и энергии образования вакансии и специальной границы зерен (ГЗ) (013), а также проведено моделирование процесса зернограничной самодиффузии. Сравнительный анализ показал, что парные потенциалы в молекулярно-динамическом эксперименте дают качественно такие же результаты, как и многочастичные потенциалы. Энергии идеального кристалла, рассчитанные с учетом трех координационных сфер, совпадают. Позиции атомов в области вакансии различаются на величину, не превышающую 0,1 Å. Структура границы зерен не зависит от выбора потенциала: позиции атомов различаются не более чем на 0,1 Å, что составляет 2,5% от параметра решетки. Атомная структура хорошо совпадает с экспериментальными изображениями ГЗ. Моделирование процесса самодиффузии по ГЗ проведено в интервале температур от 600 К до температуры плавления. Аррениусовы зависимости имеют по два линейных участка, изменение наклона которых свидетельствует о смене механизма самодиффузии. Получены близкие значения температуры, при которой происходит смена механизмов диффузии при использовании разных потенциалов взаимодействия. Энергии активации самодиффузии имеют близкие значения. Ключевые слова: компьютерное моделирование, потенциал взаимодействия, граница зерен, молекулярная статика, молекулярная динамика. The review compares simulation results of different levels of atomic structure complexity with the use of the Morse pair potential and the Cleri-Rosato manybody potential. The parameters of the potential functions correspond to those of aluminum. We have calculated the perfect crystal energy, the structure and energy of vacancy forming, and the structure and energy of special grain boundary GB (013). Also, we have simulated the process of grain boundary self-diffusion. The comparative analysis has shown that pair potentials and many-body potentials have results of similar quality in the molecular dynamic experiment. The perfect crystal energies calculated with the consideration of three coordination spheres coincide. Atomic positions in the vacancy area differ by the value not exceeding 0.1 Å. A grain boundary structure does not depend on the potential choice as the difference between atomic positions does not exceed 0.1 Å which is 2.5% of the lattice parameter. The simulation of the self-diffusion process along GB has been performed in the temperature range from 600 K up to the melting point. Each Arrhenius plot has two linear parts. A change in the tilt of the Arrhenius plots is the proof of the change in the self-diffusion mechanism. We have obtained relatively similar temperature values at which diffusion mechanisms change with the use of different interaction potentials. The activation energies of self-diffusion have relatively similar values. read less NOT USED (high confidence) D. M. O. Steinhauser, “Multiscale Modeling and Simulation of Shock Wave-Induced Failure in Materials Science,” Springer Fachmedien Wiesbaden. 2018. link Times cited: 1 NOT USED (high confidence) A. Ahadi, P. Hansson, and S. Melin, “Effects of Voids in Tensile Single-Crystal Cu Nanobeams,” Molecular Dynamics. 2018. link Times cited: 0 Abstract: Molecular dynamic simulations of defect nano-sized beams of … read moreAbstract: Molecular dynamic simulations of defect nano-sized beams of single-crystal Cu, loaded in displacement controlled tension until rupture, have been performed. The defects are square-shaped, through-the-thickness voids of different sizes, placed centrally in the beams. Three different cross section sizes and two different crystallographic orientations are investigated. As expected, the sizes of the beam cross section and the void as well as the crystal orientation strongly influences both the elastic and the plastic behaviors of the beams. It was seen that the strain at plastic initiation increases with beam cross section size as well as with decreasing void size. It is further observed that the void deformed in different ways depending on cross section and void size. Sometimes void closure, leading to necking of the beam cross section followed by rupture occurred. In other cases the void elongated leading to that the two ligaments above and below the void ruptured independently. (Less) read less NOT USED (high confidence) Z. Liang and P. Keblinski, “Molecular simulation of steady-state evaporation and condensation in the presence of a non-condensable gas.,” The Journal of chemical physics. 2018. link Times cited: 29 Abstract: Using molecular dynamics simulations, we study evaporation a… read moreAbstract: Using molecular dynamics simulations, we study evaporation and condensation of fluid Ar in the presence of a non-condensable Ne gas in a nanochannel. The evaporation and condensation are driven by the temperature difference, ΔTL, between the evaporating and condensing liquid surfaces. The steady-state evaporation and condensation fluxes (JMD) are also affected by the Ne concentration, ρNe, and the nanochannel length. We find that across a wide range of ΔTL and ρNe, JMD is in good agreement with the prediction from Stefan's law and from Schrage relationships. Furthermore, for ΔTL less than ∼20% of the absolute average temperature, we find that both steady-state heat and mass fluxes are proportional to ΔTL. This allows us to determine the interfacial resistance to the heat and mass transfer and compare it with the corresponding resistances in the gas phase. In this context, we derive an analytical expression for the effective thermal conductivity of the gas region in the nanochannel and the mass transport interfacial resistance equivalent length, i.e., the length of the nanochannel for which the resistance to the mass flow is the same as the interfacial resistance to the mass flow. read less NOT USED (high confidence) J. Gordon and H. Kee, “Selective Phonon Damping in Topological Semimetals,” arXiv: Strongly Correlated Electrons. 2018. link Times cited: 1 Abstract: Topological semimetals are characterized by their intriguing… read moreAbstract: Topological semimetals are characterized by their intriguing Fermi surfaces (FSs) such as Weyl and Dirac points, or nodal FS, and their associated surface states. Among them, topological crystalline semimetals, in the presence of strong spin-orbit coupling, possess a nodal FS protected by non-symmorphic lattice symmetries. In particular, it was theoretically proposed that $\mathrm{SrIrO}_{3}$ exhibits a bulk nodal ring due to glide symmetries, as well as flat two-dimensional surface states related to chiral and mirror symmetries. However, due to the semimetallic nature of the bulk, direct observation of these surface states is difficult. Here we study the effect of flat-surface states on phonon modes for $\mathrm{SrIrO}_{3}$ side surfaces. We show that particular phonon modes, based on mirror symmetry, have qualitatively different damping mechanisms due to the surface states which could be used to infer their existence. Experimental techniques for such measurements are also discussed. read less NOT USED (high confidence) S. Mahmoud, M. Trochet, Ó. Restrepo, and N. Mousseau, “Study of point defects diffusion in nickel using kinetic activation-relaxation technique,” Acta Materialia. 2018. link Times cited: 21 NOT USED (high confidence) C. O’Brien, C. M. Barr, P. M. Price, K. Hattar, and S. Foiles, “Grain boundary phase transformations in PtAu and relevance to thermal stabilization of bulk nanocrystalline metals,” Journal of Materials Science. 2018. link Times cited: 61 NOT USED (high confidence) X. Zhang et al., “Theoretical analytical model of vacancy formation energy with simultaneous dependence on surface orientation, temperature, and material size,” Journal of Physics D: Applied Physics. 2018. link Times cited: 4 Abstract: From the perspectives of bond energy theory, the bond–order–… read moreAbstract: From the perspectives of bond energy theory, the bond–order–length–strength correlation mechanism, and the core–surface configuration for nanomaterials, a physics-based model, free of any adjustable parameters and simultaneously considering the coupling effects of surface orientation, temperature, and size on the vacancy formation energy of metal materials is developed. To confirm our present model, the temperature-dependent vacancy formation energies of six face-centered cubic metals and the size-dependent vacancy formation energies of gold particles are predicted, which are in reasonable agreement with the simulation results. In particular, the model can provide a convenient method to predict the temperature-dependent vacancy formation energy of nanomaterials with different surface orientations, and also can provide a new method to study the structural relaxation. The study shows that the size effect on the vacancy formation energy depends on the stronger bond energy in the surface layers compared with those in the core interior, and the temperature-dependent vacancy formation energy arises from cohesive energy weakening, with the opposite trend to that induced by size reduction. read less NOT USED (high confidence) T. Vo and B. H. Kim, “Physical origins of temperature continuity at an interface between a crystal and its melt.,” The Journal of chemical physics. 2018. link Times cited: 11 Abstract: We justify and discuss the physical origins for the assumpti… read moreAbstract: We justify and discuss the physical origins for the assumption of temperature continuity at crystal/melt interfaces by performing atomistic simulations. We additionally answer why the crystal/melt interfaces differ from the typical solid/liquid interfaces, which usually exhibit dissimilarities and a resulting temperature drop. We present results for pure silver modeled using the embedded-atom method and Lennard-Jones potential function and contrast the results with each other. We find that the temperature continuity at an interface between a crystal and its melt originates from the perfect vibrational coupling, which is caused by the interfacial structural diffusivity. This study provides fundamental insights into the heat transfer for cases of extremely large heat flux and thermal gradients occurring during rapid melting and solidification. The findings additionally determine the role of rough surfaces in manipulating the thermal conductance in nanodevices. read less NOT USED (high confidence) K. Sukuta, M. V. D. Bossche, A. Pedersen, and H. Jónsson, “Nanocluster structure deduced from AC-STEM images coupled to theoretical modelling.” 2017. link Times cited: 1 NOT USED (high confidence) M. Benlattar, E. E. koraychy, A. Kotri, and M. Mazroui, “Hetero-diffusion of Au epitaxy on stepped Ag(110) surface: Study of the jump rate and diffusion coefficient,” The European Physical Journal Plus. 2017. link Times cited: 2 NOT USED (high confidence) P. Umstätter and H. Urbassek, “Influence of Elastic Stiffness and Surface Adhesion on Bouncing of Nanoparticles,” Nanoscale Research Letters. 2017. link Times cited: 1 NOT USED (high confidence) B. Narayanan et al., “Machine learnt bond order potential to model metal-organic (Co-C) heterostructures.,” Nanoscale. 2017. link Times cited: 7 Abstract: A fundamental understanding of the inter-relationships betwe… read moreAbstract: A fundamental understanding of the inter-relationships between structure, morphology, atomic scale dynamics, chemistry, and physical properties of mixed metallic-covalent systems is essential to design novel functional materials for applications in flexible nano-electronics, energy storage and catalysis. To achieve such knowledge, it is imperative to develop robust and computationally efficient atomistic models that describe atomic interactions accurately within a single framework. Here, we present a unified Tersoff-Brenner type bond order potential (BOP) for a Co-C system, trained against lattice parameters, cohesive energies, equation of state, and elastic constants of different crystalline phases of cobalt as well as orthorhombic Co2C derived from density functional theory (DFT) calculations. The independent BOP parameters are determined using a combination of supervised machine learning (genetic algorithms) and local minimization via the simplex method. Our newly developed BOP accurately describes the structural, thermodynamic, mechanical, and surface properties of both the elemental components as well as the carbide phases, in excellent accordance with DFT calculations and experiments. Using our machine-learnt BOP potential, we performed large-scale molecular dynamics simulations to investigate the effect of metal/carbon concentration on the structure and mechanical properties of porous architectures obtained via self-assembly of cobalt nanoparticles and fullerene molecules. Such porous structures have implications in flexible electronics, where materials with high electrical conductivity and low elastic stiffness are desired. Using unsupervised machine learning (clustering), we identify the pore structure, pore-distribution, and metallic conduction pathways in self-assembled structures at different C/Co ratios. We find that as the C/Co ratio increases, the connectivity between the Co nanoparticles becomes limited, likely resulting in low electrical conductivity; on the other hand, such C-rich hybrid structures are highly flexible (i.e., low stiffness). The BOP model developed in this work is a valuable tool to investigate atomic scale processes, structure-property relationships, and temperature/pressure response of Co-C systems, as well as design organic-inorganic hybrid structures with a desired set of properties. read less NOT USED (high confidence) H. Zhou, W. Wu, R. Wu, G. Hu, and R. Xia, “Effects of various conditions in cold-welding of copper nanowires: A molecular dynamics study,” Journal of Applied Physics. 2017. link Times cited: 19 Abstract: Cold-welding possesses such desirable environment as low tem… read moreAbstract: Cold-welding possesses such desirable environment as low temperature and low applied stress, thus becoming the prime candidate for nanojointing and nanoassembly techniques. To explore the welding mechanism of nanoscale structures, here, molecular dynamics was performed on copper nanowires under different welding conditions and various original characteristics to obtain an atomic-level depiction of their cold-welding behavior. By analyzing the mechanical properties of as-welded nanowires, the relations between welding quality and welding variables are revealed and identified. This comparison study will be of great importance to future mechanical processing and structural assembly of metallic nanowires. read less NOT USED (high confidence) G. González‐Rubio et al., “Femtosecond laser reshaping yields gold nanorods with ultranarrow surface plasmon resonances,” Science. 2017. link Times cited: 199 Abstract: Laser-shaping nanoparticles For many applications of the pla… read moreAbstract: Laser-shaping nanoparticles For many applications of the plasmon resonances of metal nanoparticles, it is necessary to have narrow resonance lines. However, most methods for synthesizing nanoparticles create a distribution of sizes and shapes that broaden the resonance lines. González-Rubio et al. annealed gold nanorods dispersed in an aqueous solution of a surfactant with carefully tuned ultrafast (femtosecond) laser pulses. This approach reshaped the nanoparticles to create a near-uniform distribution with resonance lines nearly as sharp as for a single nanorod. Science, this issue p. 640 Heating nanoparticles with femtosecond laser pulses can reshape them and sharpen their plasmon resonances. The irradiation of gold nanorod colloids with a femtosecond laser can be tuned to induce controlled nanorod reshaping, yielding colloids with exceptionally narrow localized surface plasmon resonance bands. The process relies on a regime characterized by a gentle multishot reduction of the aspect ratio, whereas the rod shape and volume are barely affected. Successful reshaping can only occur within a narrow window of the heat dissipation rate: Low cooling rates lead to drastic morphological changes, and fast cooling has nearly no effect. Hence, a delicate balance must be achieved between irradiation fluence and surface density of the surfactant on the nanorods. This perfection process is appealing because it provides a simple, fast, reproducible, and scalable route toward gold nanorods with an optical response of exceptional quality, near the theoretical limit. read less NOT USED (high confidence) T. Jacobs and A. Martini, “Measuring and Understanding Contact Area at the Nanoscale: A Review,” Applied Mechanics Reviews. 2017. link Times cited: 80 Abstract: The size of the mechanical contact between nanoscale bodies … read moreAbstract: The size of the mechanical contact between nanoscale bodies that are pressed together under load has implications for adhesion, friction, and electrical and thermal transport at small scales. Yet, because the contact is buried between the two bodies, it is challenging to accurately measure the true contact area and to understand its dependence on load and material properties. Recent advancements in both experimental techniques and simulation methodologies have provided unprecedented insights into nanoscale contacts. This review provides a detailed look at the current understanding of nanocontacts. Experimental methods for determining contact area are discussed, including direct measurements using in situ electron microscopy, as well as indirect methods based on measurements of contact resistance, contact stiffness, lateral forces, and topography. Simulation techniques are also discussed, including the types of nanocontact modeling that has been performed and the various methods for extracting the magnitude of the contact area from a simulation. To describe and predict contact area, three different theories of nanoscale contact are reviewed: single-contact continuum mechanics; multi-contact continuum mechanics; and atomistic accounting. Representative results from nanoscale experimental and simulation investigations are presented in the context of these theories. Finally, the critical challenges are described, as well as the opportunities on the path to establishing a fundamental and actionable understanding of what it means to be “in contact” at the nanoscale. read less NOT USED (high confidence) I. Matrane, M. Mazroui, K. Sbiaai, A. Eddiai, and Y. Boughaleb, “Energy barriers of single-adatoms diffusion on unreconstructed and reconstructed (110) surfaces,” The European Physical Journal B. 2017. link Times cited: 5 NOT USED (high confidence) Y. Zhang and S. Jiang, “Molecular Dynamics Simulation of Crack Propagation in Nanoscale Polycrystal Nickel Based on Different Strain Rates.” 2017. link Times cited: 11 Abstract: Based on the strain rates of 2 × 108 s−1 and 2 × 1010 s−1, m… read moreAbstract: Based on the strain rates of 2 × 108 s−1 and 2 × 1010 s−1, molecular dynamics simulation was conducted so as to study mechanisms of crack propagation in nanoscale polycrystal nickel. The strain rate has an important effect on the mechanism of crack propagation in nanoscale polycrystal nickel. In the case of a higher strain rate, local non-3D-crystalline atoms are induced and Lomer-Cottrell locks are formed, which plays a critical role in crack initiation and propagation. Orientation difference between adjacent grains leads to the slipping of dislocations along the different directions, which results in the initiation of a void near the triple junction of grain boundaries and further contributes to accelerating the crack propagation. read less NOT USED (high confidence) L. Bulat, A. Ivanov, V. Osvenskiĭ, D. Pshenay-Severin, and A. Sorokin, “Thermal conductivity of Cu2Se taking into account the influence of mobile copper ions,” Physics of the Solid State. 2017. link Times cited: 3 NOT USED (high confidence) K. Chen, L. Wang, Y. Chen, and Q. Wang, “Molecular dynamics simulation of interfaces and microstructure evolution during high-speed sliding,” Numerical Heat Transfer, Part A: Applications. 2017. link Times cited: 4 Abstract: ABSTRACT Three-dimensional nonequilibrium molecular dynamics… read moreAbstract: ABSTRACT Three-dimensional nonequilibrium molecular dynamics simulations are performed to investigate the tribological characteristics of the high-speed sliding system. A sliding simulation model with two blocks is built. The friction force, the evolution of the structure of interface and the temperature profiles of the sliding system are obtained. The relationship among tribological characteristic, friction heat, and interface structure is studied. The influence of the sliding velocity is investigated. The velocity field of the interface is also considered. The theoretical analysis of the mixing layer is also built to study the flow and heat transfer characteristics in the interface. The results show that structure of the interface can greatly affect the friction force and temperature rising. The structure factors and the velocity field of the interface evidently suggest that atomic flow during sliding is similar to fluid flow, which is beneficial to reduce the friction force and heat dissipation. With the increase of sliding velocity, the thickness of the mixing layer increases, the steady state friction force decreases. The atom distribution and the radial distribution function of the interface indicate that the degree of short-range disorder of the mixing layer increases. read less NOT USED (high confidence) G. Benetti et al., “Bottom-Up Mechanical Nanometrology of Granular Ag Nanoparticles Thin Films,” Journal of Physical Chemistry C. 2017. link Times cited: 33 Abstract: Ultrathin metal nanoparticles coatings, synthesized by gas-p… read moreAbstract: Ultrathin metal nanoparticles coatings, synthesized by gas-phase deposition, are emerging as go-to materials in a variety of fields ranging from pathogens control and sensing to energy storage. Predicting their morphology and mechanical properties beyond a trial-and-error approach is a crucial issue limiting their exploitation in real-life applications. The morphology and mechanical properties of Ag nanoparticle ultrathin films, synthesized by supersonic cluster beam deposition, are here assessed adopting a bottom-up, multitechnique approach. A virtual film model is proposed merging high resolution scanning transmission electron microscopy, supersonic cluster beam dynamics, and molecular dynamics simulations. The model is validated against mechanical nanometrology measurements and is readily extendable to metals other than Ag. The virtual film is shown to be a flexible and reliable predictive tool to access morphology-dependent properties such as mesoscale gas-dynamics and elasticity of ultrathin films sy... read less NOT USED (high confidence) K. Gkagkas and V. Ponnuchamy, “The impact of coulombic interactions among polar molecules and metal substrates on flow and lubrication properties,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 2 Abstract: In the current work we present an extensive study on the imp… read moreAbstract: In the current work we present an extensive study on the impact of short- and long-range interactions between solids and liquids on the hydrodynamic and lubrication behaviour of a tribological system. We have implemented a coarse grain molecular dynamics description of two ionic liquids (ILs) as lubricants which are confined by two infinitely long flat iron solids and which are subjected to a shearing flow. The impact of surface polarizability and molecule geometry on the ion arrangement under shearing has been studied in detail. The results have revealed two regimes of lubrication, with a liquid phase being present under low normal loads, while solidification of the ILs, accompanied by a steep rise of normal forces and significant wall slip is observed at small plate-to-plate distances. read less NOT USED (high confidence) V. Turlo, F. Baras, and O. Politano, “Comparative study of embedded-atom methods applied to the reactivity in the Ni–Al system,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 23 Abstract: Structural, thermodynamic, atomic and thermal transport prop… read moreAbstract: Structural, thermodynamic, atomic and thermal transport properties of solid and liquid phases of the Ni–Al system were studied by means of MD simulations using three embedded-atom method (EAM) potentials developed by Mishin and colleagues (Mishin et al 2002 Phys. Rev. B 65 224114; Mishin 2004 Acta Mater. 52 145167; Purja Pun and Mishin 2009 Phil. Mag. 89 32453267). The extracted properties (lattice parameter, enthalpy, heat capacity, mass diffusivity and thermal conductivity) were compared with experimental data. The limitations of EAM potentials for studying different aspects of reactivity were assessed for each potential separately. read less NOT USED (high confidence) S. Huang and C. Zhou, “Modeling and Simulation of Nanoindentation,” JOM. 2017. link Times cited: 17 NOT USED (high confidence) J. Liu, Y. Zheng, and S. Hou, “Facile synthesis of Cu/Ni alloy nanospheres with tunable size and elemental ratio,” RSC Advances. 2017. link Times cited: 9 Abstract: We report a facile synthesis of copper/nickel (Cu/Ni) alloy … read moreAbstract: We report a facile synthesis of copper/nickel (Cu/Ni) alloy nanospheres in high purity and with tunable, well-controlled sizes and elemental ratios. The success of this synthesis relies on the use of one-pot, direct thermal reduction of a Cu and Ni precursor mixture in the presence of trioctylphosphine (TOP) and oleylamine (OAm) at an elevated temperature to form Cu/Ni alloy nanoparticles with a spherical shape and uniform size. Their sizes could be readily tuned in the range of 6.9–27.3 nm by simply varying the volume of TOP added to the reaction solution. The elemental ratio of Cu to Ni in resultant products was found to remain the same as that in the precursor, which offers a simple way to manipulate the composition of Cu/Ni alloy nanospheres. We also tested the catalytic performance of as-prepared Cu/Ni alloy nanospheres and evaluate the effect of size and elemental ratio using the reduction of 4-nitrophenol as the model reaction. The current strategy enables the size and composition controlled synthesis of Cu/Ni alloy nanomaterials and could find important use in the fabrication of other types of bimetallic alloy nanomaterials with desired sizes and compositions for catalytic purposes. read less NOT USED (high confidence) А. В. Гусев, С. И. Морозов, and А. Е. Чудаков, “Совместная поверхностная сегрегация серебра и олова в тройных сплавах Cu–Ag–Sn.” 2017. link Times cited: 0 Abstract: The study of Cu–Ag–Sn ternary alloys by the methods of tempe… read moreAbstract: The study of Cu–Ag–Sn ternary alloys by the methods of temperature-programmed desorption (TPD) and statistical modeling is с arried out. The results of the TPD experiment on the combined surface segregation of Ag and Sn atoms in dilute solutions of silver and tin in copper in polycrystalline and liquid states are presented. It was found out that the desorption of particles from the surface of the alloy occurs from regions enriched with this component. In addition, the silver atoms form, in the main, slightly layered islets. Data are also provided for computer simulation of the surface of these alloys by the Monte Carlo method in combination with the embedded atom method (EAM). Using the EAM method, it was possible to avoid a priori assumptions about the composition and structure of the alloy surface. Simulation showed that the binding energy of surface atoms is close to the experimental activation energies of thermal desorption for Cu and Ag atoms. But there is a significant difference for tin atoms. Secondly, in the modeling for the solid state of the Cu 98 Ag 1 Sn 1 alloy, the surface concentrations of Θ Ag ≈ Θ Sn = 30 at.%, which for Ag atoms are close to the results of the TPD experiment. In the melt, the degree of surface segregation is reduced to 8 at.% Ag and 14 at.% Sn. Both the TPD experiment and the simulation give an excess of 10 to 30 times the surface concentrations of Ag and Sn over bulk values, both in the solid and in the liquid state of the alloy. Third, computer models reveal surface structures in the form of zigzag chains of Sn atoms, which alternate with chains of Ag atoms. The profiles of tin concentrations in alloys confirm the hypothesis of a two-layered character of segregation of tin. read less NOT USED (high confidence) Y. Zhang and S. Jiang, “Investigation on dislocation-based mechanisms of void growth and coalescence in single crystal and nanotwinned nickels by molecular dynamics simulation,” Philosophical Magazine. 2017. link Times cited: 24 Abstract: Molecular dynamics simulations were conducted to elucidate d… read moreAbstract: Molecular dynamics simulations were conducted to elucidate dislocation mechanisms of the void growth and coalescence in single crystal and nanotwinned nickels subjected to uniaxial tension. The simulation results reveal that twin boundary is capable of decreasing the critical stress, suppressing the emission of dislocations and reducing the overall stiffness of the crystal. A size-scale dependence of critical stress is definitely illustrated through stress–strain response, where the larger void size leads to the lower critical stress and strain. It is the successive emissions of leading partials and the subsequent trailing partials that cause the atoms on the void surfaces to escape from the void surfaces continually, and consequently the voids grow to be larger and larger with increasing strain. The voids in the nanotwinned nickel coalesce earlier than those in the single crystal nickel even though the initiation of dislocations in the former is later than that in the latter. Void fraction remains a constant during elastic deformation, while it presents a linear increase with increasing strain during plastic deformation. Evolution of void fraction during void growth and coalescence is independent on void size. read less NOT USED (high confidence) T. Cheng, Y. Huang, H. Xiao, and W. Goddard, “Predicted Structures of the Active Sites Responsible for the Improved Reduction of Carbon Dioxide by Gold Nanoparticles.,” The journal of physical chemistry letters. 2017. link Times cited: 38 Abstract: Gold (Au) nanoparticles (NPs) are known experimentally to re… read moreAbstract: Gold (Au) nanoparticles (NPs) are known experimentally to reduce carbon dioxide (CO2) to carbon monoxide (CO), with far superior performance to Au foils. To obtain guidance in designing improved CO2 catalysts, we want to understand the nature of the active sites on Au NPs. Here, we employed multiscale atomistic simulations to computationally synthesize and characterize a 10 nm thick Au NP on a carbon nanotube (CNT) support, and then we located active sites from quantum mechanics (QM) calculations on 269 randomly selected sites. The standard scaling relation is that the formation energy of *COOH (ΔE*COOH) is proportional to the binding energy of *CO (Ebinding*CO); therefore, decreasing ΔE*COOH to boost the CO2 reduction reaction (CO2RR) causes an increase of Ebinding*CO that retards CO2RR. We show that the NPs have superior CO2RR because there are many sites at the twin boundaries that significantly break this scaling relation. read less NOT USED (high confidence) A. Fedorov, A. V. Shul’gin, and S. Lavruk, “Investigation of the physical properties of iron nanoparticles in the course of the melting and solidification,” Physics of Metals and Metallography. 2017. link Times cited: 6 NOT USED (high confidence) J. Li and J.-hua Li, “Large-scale image similarity search optimization based on multi-core architecture,” 2017 13th International Conference on Natural Computation, Fuzzy Systems and Knowledge Discovery (ICNC-FSKD). 2017. link Times cited: 0 Abstract: Web-based image search is a specialized data search used to … read moreAbstract: Web-based image search is a specialized data search used to find images from a database of digital images, which is a new usage model in search technology. Researchers are proactively working on image search development for new user experiences and are promoting a GPGPU solution to improve performance of image searches. The key image search algorithms are Union and Sort in typical image search application, which mainly contain compare operations (integer intensive) and heavy random memory access. To compare with GPGPU, we evaluate the performance of the Knights Corner (KNC) processor, which is the first generation of the Intel Many Integrated Core architecture(MIC). KNC is capable of modern GPU architectures. Besides a variety of floating point vector instructions, there are also many integer vector instructions supported like packed compare and gather/scatter which provided another choice to optimize Union and Sort on KNC. In this paper, based on the union and sort code from alibaba, we conducted several optimizations like cache blocking, utilize gather/scatter to optimize random memory access on Intel MIC architecture, further, we design an hybrid parallel paradigm to utilize all cores on the KNC processor. read less NOT USED (high confidence) T. Vo and B. H. Kim, “Molecular dynamics study of thermodynamic properties of nanoclusters for additive manufacturing,” International Journal of Precision Engineering and Manufacturing-Green Technology. 2017. link Times cited: 28 NOT USED (high confidence) C. V. D. Walt, J. Terblans, and H. Swart, “A study of diffusion, atom migration and segregation in Cu and Ag alloy bulk- and nanocrystals,” AIP Advances. 2017. link Times cited: 6 Abstract: Using the Sutton-Chen potential, Molecular Dynamics simulati… read moreAbstract: Using the Sutton-Chen potential, Molecular Dynamics simulations were done of Ag and Cu bulk and nanocrystals and the vacancy formation energy, migration energy, and diffusion activation energy were calculated. Values for Cu compared very well with literature, and Ag less so. The migration energy along a diffusion path was studied for different low index surface orientations. Using the mixed form of the potential for bimetallic interactions with a slight adjustment, the interactions between Ag and Cu were also simulated. Migration energy depth profiles along with segregation energies at different depths were studied. Surface segregation of Ag in Cu was successfully simulated and the calculated segregation values of a Ag atom in Cu compared well to literature values. read less NOT USED (high confidence) A. Antony et al., “Charge optimized many body (COMB) potentials for Pt and Au,” Journal of Physics: Condensed Matter. 2017. link Times cited: 12 Abstract: Interatomic potentials for Pt and Au are developed within th… read moreAbstract: Interatomic potentials for Pt and Au are developed within the third generation charge optimized many-body (COMB3) formalism. The potentials are capable of reproducing phase order, lattice constants, and elastic constants of Pt and Au systems as experimentally measured or calculated by density functional theory. We also fit defect formation energies, surface energies and stacking fault energies for Pt and Au metals. The resulting potentials are used to map a 2D contour of the gamma surface and simulate the tensile test of 16-grain polycrystalline Pt and Au structures at 300 K. The stress–strain behaviour is investigated and the primary slip systems {1 1 1}〈1 1¯ 0〉 are identified. In addition, we perform high temperature (1800 K for Au and 2300 K for Pt) molecular dynamics simulations of 30 nm Pt and Au truncated octahedron nanoparticles and examine morphological changes of each particle. We further calculate the activation energy barrier for surface diffusion during simulations of several nanoseconds and report energies of 0.62±0.16 eV for Pt and 1.44±0.06 eV for Au. This initial parameterization and application of the Pt and Au potentials demonstrates a starting point for the extension of these potentials to multicomponent systems within the COMB3 framework. read less NOT USED (high confidence) X. Yue and X. Yang, “Molecular dynamics simulation of single pulse discharge process: clarifying the function of pressure generated inside the melting area in EDM,” Molecular Simulation. 2017. link Times cited: 11 Abstract: Due to the unique and superior properties of using thermal e… read moreAbstract: Due to the unique and superior properties of using thermal energy to machine electrically conductive material regardless of its hardness, electrical discharge machining has been widely used and has become indispensable in the manufacturing industry. However, the incompleteness and imperfection of the fundamental theory behind it seriously hinders its further application and development. In this work, a single pulse discharge in deionised water was simulated to study the material removal motivity and mechanisms with molecular dynamics simulations. The results show that during the discharge process, there exists a decreasing pressure gradient along the depth direction towards the surface of the melting area. When the pressure gradient overcomes the atomic bonding forces, the melting material can be ablated from the electrode. Thus, it is not the case that the melting material cannot be removed during the discharge process; material removal occurs throughout the whole discharge process. In addition, it was found that the bulge is formed for two reasons: the main reason is the shearing flow of the melting material caused by the pressure gradient along the radial direction in the melting area, and the other is the accumulation of the spattering material from the opposite electrode. read less NOT USED (high confidence) P. Naskar, S. Talukder, and P. Chaudhury, “An adaptive mutation simulated annealing based investigation of Coulombic explosion and identification of dissociation patterns in (CO2)n2+ clusters.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 10 Abstract: In this communication, we would like to discuss the advantag… read moreAbstract: In this communication, we would like to discuss the advantages of adaptive mutation simulated annealing (AMSA) over standard simulated annealing (SA) in studying the Coulombic explosion of (CO2)n2+ clusters for n = 20-68, where 'n' is the size of the cluster. We have demonstrated how AMSA itself can overcome the predicaments which can arise in conventional SA and carry out the search for better results by adapting the parameters (only when needed) dynamically during the simulations so that the search process can come out of high energy basins and not go astray for better exploration and convergence, respectively. This technique also has in-built properties for getting more than one minimum in a single run. For a (CO2)n2+ cluster system we have found the critical limit to be n = 43, above which the attractive forces between individual units become greater in value than that of the large repulsive forces and the clusters stay intact as the energetically favoured isomers. This result is in good concurrence with earlier studies. Moreover, we have studied the fragmentation patterns for the entire size range and we have found fission type fragmentation as the favoured mechanism nearly for all sizes. read less NOT USED (high confidence) D. Zhang and T. R. Dhakal, “Shock waves simulated using the dual domain material point method combined with molecular dynamics,” J. Comput. Phys. 2017. link Times cited: 9 NOT USED (high confidence) G. M. Faccin, M. San-Miguel, J. Andrés, E. Longo, and E. Silva, “Computational Modeling for the Ag Nanoparticle Coalescence Process: A Case of Surface Plasmon Resonance,” Journal of Physical Chemistry C. 2017. link Times cited: 14 Abstract: Motivated by recent transmission electron microscopy (TEM) e… read moreAbstract: Motivated by recent transmission electron microscopy (TEM) experiments on α-Ag2WO4, the coalescence process of Ag nanoparticles (NPs) is investigated using molecular dynamics (MD) simulations. These Ag NPs are formed by irradiation of α-Ag2WO4 crystals by electrons from a TEM gun. This behavior can be considered as a clear example of surface plasmon resonance (SPR), in which Ag NP coalescence processes are controlled by dipole–dipole interaction forming larger clusters. The interactions between Ag NPs along the coalescence processes are studied using MD simulations with embedded atom method (EAM) effective potentials for Ag. With these choices of methods, coalescence is studied by addressing different scenarios for the interacting NPs, which all could possibly occur in experiments. read less NOT USED (high confidence) Y. H. Park and I. Hijazi, “Development of physics based analytical interatomic potential for palladium-hydride,” Journal of Molecular Modeling. 2017. link Times cited: 6 NOT USED (high confidence) C.-Y. Shih, C. Wu, M. Shugaev, and L. Zhigilei, “Atomistic modeling of nanoparticle generation in short pulse laser ablation of thin metal films in water.,” Journal of colloid and interface science. 2017. link Times cited: 124 NOT USED (high confidence) A. Akbarzadeh, Y. Cui, and Z. Chen, “Thermal wave: from nonlocal continuum to molecular dynamics,” RSC Advances. 2017. link Times cited: 24 Abstract: It is well known that the continuum model of Fourier's … read moreAbstract: It is well known that the continuum model of Fourier's law of heat conduction violates the relativity theory, admits an instantaneous thermal response, and assumes a quasi-equilibrium thermodynamic condition. Transient heat transport, however, is a non-equilibrium phenomenon with a finite thermal wave speed for applications involving very low temperatures, extremely high temperature gradients, and ballistic heat transfers. Hyperbolic and phase-lag heat conduction models have enabled detection of the finite thermal wave speed in heat transport. To accommodate effects of thermomass and size-dependency of thermophysical properties on nano/microscale heat transport and to remove the theoretical singularity of temperature gradients across the thermal wavefront, a nonlocal, fractional-order, three-phase-lag heat conduction is introduced. The model is capable of simulating heat conduction phenomena in multiple spatio-temporal scales. To confirm the existence of thermal waves in nano/microscale heat transport, a molecular dynamics simulation is implemented for the heat transfer within a nanoscale copper slab. Correlating thermal responses in continuum and atomistic scales sheds light on the effect of length scale, fractional order, and phase-lags in multiscale heat transport. The multiscale simulation is of practical importance for microelectromechanical system design, photothermal techniques, and ultrafast laser-assisted processing of advanced materials. read less NOT USED (high confidence) S. Mun, A. Bowman, S. Nouranian, S. Gwaltney, M. Baskes, and M. Horstemeyer, “Interatomic Potential for Hydrocarbons on the Basis of the Modified Embedded-Atom Method with Bond Order (MEAM-BO).,” The journal of physical chemistry. A. 2017. link Times cited: 18 Abstract: In this paper, we develop a new modified embedded atom metho… read moreAbstract: In this paper, we develop a new modified embedded atom method (MEAM) potential that includes the bond order (MEAM-BO) to describe the energetics of unsaturated hydrocarbons (double and triple carbon bonds) and also develop improved parameters for saturated hydrocarbons from those of our previous work. Such quantities like bond lengths, bond angles, and atomization energies at 0 K, dimer molecule interactions, rotational barriers, and the pressure-volume-temperature relationships of dense systems of small molecules give a comparable or more accurate property relative to experimental and first-principles data than the classical reactive force fields REBO and ReaxFF. Our extension of the MEAM potential for unsaturated hydrocarbons (MEAM-BO) is a step toward developing more reliable and accurate polymer simulations with their associated structure-property relationships, such as reactive multicomponent (organic/metal) systems, polymer-metal interfaces, and nanocomposites. When the constants for the BO are zero, MEAM-BO reduces to the original MEAM potential. As such, this MEAM-BO potential describing the interaction of organic materials with metals within the same MEAM formalism is a significant advancement for computational materials science. read less NOT USED (high confidence) S. Ryu, J. Yoon, H. Moon, B. Shong, H. Kim, and H. B. R. Lee, “Atomic layer deposition of 1D and 2D nickel nanostructures on graphite,” Nanotechnology. 2017. link Times cited: 10 Abstract: One-dimensional (1D) nanowires (NWs) and two-dimensional (2D… read moreAbstract: One-dimensional (1D) nanowires (NWs) and two-dimensional (2D) thin films of Ni were deposited on highly ordered pyrolytic graphite (HOPG) by atomic layer deposition (ALD), using NH3 as a counter reactant. Thermal ALD using NH3 gas forms 1D NWs along step edges, while NH3 plasma enables the deposition of a continuous 2D film over the whole surface. The lateral and vertical growth rates of the Ni NWs are numerically modeled as a function of the number of ALD cycles. Pretreatment with NH3 gas promotes selectivity in deposition by the reduction of oxygenated functionalities on the HOPG surface. On the other hand, NH3 plasma pretreatment generates surface nitrogen species, and results in a morphological change in the basal plane of graphite, leading to active nucleation across the surface during ALD. The effects of surface nitrogen species on the nucleation of ALD Ni were theoretically studied by density functional theory calculations. Our results suggest that the properties of Ni NWs, such as their density and width, and the formation of Ni thin films on carbon surfaces can be controlled by appropriate use of NH3. read less NOT USED (high confidence) C. Hu et al., “Kernel optimization for short-range molecular dynamics,” Comput. Phys. Commun. 2017. link Times cited: 18 NOT USED (high confidence) H. Barrón, G. Opletal, R. Tilley, and A. Barnard, “Predicting the role of seed morphology in the evolution of anisotropic nanocatalysts.,” Nanoscale. 2017. link Times cited: 7 Abstract: Controlling the structure of nanocrystals is an effective wa… read moreAbstract: Controlling the structure of nanocrystals is an effective way to tune their properties and improve performance in a wide variety of applications. However, the atomic pathways for achieving this goal are difficult to identify and exercise, due to competing kinetic and thermodynamic influences during formation. In particular, an understanding of how symmetry, and symmetry breaking, determine nanocrystal morphology would significantly advance our ability to produce nanomaterials with prescribed functions. In this study we present results of a detailed computational study into the atomic structure of platinum nanoparticles at early growth stages of formation, as a function of temperature and atomic deposition rates. We investigate the impact of different types of crystalline seeds and characterize the emergent structures via simulated High Resolution Transmission Electron Microscopy (HRTEM) images. We find that the choice of initial seed is an important driver for symmetry breaking, due to a combination of atomic deposition and etching on different seed facets. A mix of low index facets causes the formation of important surface defects, in addition to the absorption/adsorption of single atoms, which can be correlated with different catalytic reactions as the process perpetuates. These findings provide new insights into nanocrystal shape-control mechanisms and suggest new opportunities for future design of this important class of nanomaterials. read less NOT USED (high confidence) P. Li and K. Merz, “Metal Ion Modeling Using Classical Mechanics,” Chemical Reviews. 2017. link Times cited: 230 Abstract: Metal ions play significant roles in numerous fields includi… read moreAbstract: Metal ions play significant roles in numerous fields including chemistry, geochemistry, biochemistry, and materials science. With computational tools increasingly becoming important in chemical research, methods have emerged to effectively face the challenge of modeling metal ions in the gas, aqueous, and solid phases. Herein, we review both quantum and classical modeling strategies for metal ion-containing systems that have been developed over the past few decades. This Review focuses on classical metal ion modeling based on unpolarized models (including the nonbonded, bonded, cationic dummy atom, and combined models), polarizable models (e.g., the fluctuating charge, Drude oscillator, and the induced dipole models), the angular overlap model, and valence bond-based models. Quantum mechanical studies of metal ion-containing systems at the semiempirical, ab initio, and density functional levels of theory are reviewed as well with a particular focus on how these methods inform classical modeling efforts. Finally, conclusions and future prospects and directions are offered that will further enhance the classical modeling of metal ion-containing systems. read less NOT USED (high confidence) A. Tsukanov and S. Psakhie, “ADHESION EFFECTS WITHIN THE HARD MATTER – SOFT MATTER INTERFACE: MOLECULAR DYNAMICS.” 2016. link Times cited: 17 Abstract: In the present study three soft matter – hard matter systems… read moreAbstract: In the present study three soft matter – hard matter systems consisting of different nanomaterials and organic molecules were studied using the steered molecular dynamics approach in order to reveal regularities in the formation of organic-inorganic hybrids and the stability of multimolecular complexes, as well as to analyze the energy aspects of adhesion between bio-molecules and layered ceramics. The combined process free energy estimation (COPFEE) procedure was used for quantitative and qualitative assessment of the considered heterogeneous systems. Interaction of anionic and cationic amino acids with the surface of a [Mg4Al2(OH)122+ 2Cl–] layered double hydroxide (LDH) nanosheet was considered. In both cases, strong adhesion was observed despite the opposite signs of electric charge. The free energy of the aspartic amino acid anion, which has two deprotonated carboxylic groups, was determined to be –45 kJ/mol for adsorption on the LDH surface. For the cationic arginine, with only one carboxylic group and a positive net charge, the energy of adsorption was –26 kJ/mol, which is twice higher than that of chloride anion adsorption on the same cationic nanosheet. This fact clearly demonstrates the capability of “soft matter” species to adjust themselves and fit into the surface, minimizing energy of the system. The adsorption of protonated histamine, having no carboxylic groups, on a boehmite nanosheet is also energetically favorable, but the depth of free energy well is quite small at 3.6 kJ/mol. In the adsorbed state the protonated amino-group of histamine plays the role of proton donor, while the hydroxyl oxygens of the layered hydroxide have the role of proton acceptor, which is unusual. The obtained results represent a small step towards further understanding of the adhesion effects within the hard matter – soft matter contact zone. read less NOT USED (high confidence) J. Guénolé, A. Prakash, and E. Bitzek, “Influence of intrinsic strain on irradiation induced damage: the role of threshold displacement and surface binding energies,” Materials & Design. 2016. link Times cited: 10 NOT USED (high confidence) G. Wang et al., “Phase transitions and kinetic properties of gold nanoparticles confined between two-layer graphene nanosheets,” Journal of Physics and Chemistry of Solids. 2016. link Times cited: 2 NOT USED (high confidence) M. Korayem, R. N. Hefzabad, A. Homayooni, and H. Aslani, “Molecular dynamics simulation of nanomanipulation based on AFM in liquid ambient,” Applied Physics A. 2016. link Times cited: 6 NOT USED (high confidence) M. H. Korayem, R. N. Hefzabad, A. Homayooni, and H. Aslani, “Molecular dynamics simulation of nanomanipulation based on AFM in liquid ambient,” Applied Physics A. 2016. link Times cited: 0 NOT USED (high confidence) Y. Gong, Z. Zhu, Y. Zhou, and Y. Sun, “Research on the nanometric machining of a single crystal nickel via molecular dynamics simulation,” Science China Technological Sciences. 2016. link Times cited: 13 NOT USED (high confidence) Y. Gong, Z. Zhu, Y. Zhou, and Y. Sun, “Research on the nanometric machining of a single crystal nickel via molecular dynamics simulation,” Science China Technological Sciences. 2016. link Times cited: 0 NOT USED (high confidence) L. F. Oliveira et al., “Benchmarking Density Functional Based Tight-Binding for Silver and Gold Materials: From Small Clusters to Bulk.,” The journal of physical chemistry. A. 2016. link Times cited: 44 Abstract: We benchmark existing and improved self-consistent-charge de… read moreAbstract: We benchmark existing and improved self-consistent-charge density functional based tight-binding (SCC-DFTB) parameters for silver and gold clusters as well as for bulk materials. In the former case, our benchmarks focus on both the structural and energetic properties of small-size AgN and AuN clusters (N from 2 to 13), medium-size clusters with N = 20 and 55, and finally larger nanoparticles with N = 147, 309, and 561. For bulk materials, structural, energetics and elastic properties are discussed. We show that SCC-DFTB is quite satisfactory in reproducing essential differences between silver and gold aggregates, in particular their 2D-3D structural transitions, and their dependency upon cluster charge. SCC-DFTB is also in agreement with DFT and experiments in the medium-size regime regarding the energetic ordering of the different low-energy isomers and allows for an overall satisfactory treatment of bulk properties. A consistent convergence between the cohesive energies of the largest investigated nanoparticles and the bulk's is obtained. On the basis of our results for nanoparticles of increasing size, a two-parameter analytical extrapolation of the cohesive energy is proposed. This formula takes into account the reduction of the cohesive energy for undercoordinated surface sites and converges properly to the bulk cohesive energy. Values for the surface sites cohesive energies are also proposed. read less NOT USED (high confidence) F. Hosseinzadeh, F. Shirazian, R. Shahsavari, and A. Khoei, “Local density variation of gold nanoparticles in aquatic environments,” Physica E-low-dimensional Systems & Nanostructures. 2016. link Times cited: 3 NOT USED (high confidence) T. Zientarski and D. Chocyk, “Stress induced grain boundaries in thin Co layer deposited on Au and Cu,” Applied Physics A. 2016. link Times cited: 5 NOT USED (high confidence) I. Pilch et al., “Nanoparticle growth by collection of ions: orbital motion limited theory and collision-enhanced collection,” Journal of Physics D: Applied Physics. 2016. link Times cited: 5 Abstract: The growth of nanoparticles in plasma is modeled for situati… read moreAbstract: The growth of nanoparticles in plasma is modeled for situations where the growth is mainly due to the collection of ions of the growth material. The model is based on the classical orbit motion limited (OML) theory with the addition of a collision-enhanced collection (CEC) of ions. The limits for this type of model are assessed with respect to three processes that are not included: evaporation of the growth material, electron field emission, and thermionic emission of electrons. It is found that both evaporation and thermionic emission can be disregarded below a temperature that depends on the nanoparticle material and on the plasma parameters; for copper in our high-density plasma this limit is about 1200 K. Electron field emission can be disregarded above a critical nanoparticle radius, in our case around 1.4 nm. The model is benchmarked, with good agreement, to the growth of copper nanoparticles from a radius of 5 nm–20 nm in a pulsed power hollow cathode discharge. Ion collection by collisions contributes with approximately 10% of the total current to particle growth, in spite of the fact that the collision mean free path is four orders of magnitude longer than the nanoparticle radius. read less NOT USED (high confidence) O. Gatsenko, O. E. Zasymchuk, P. Tesel’ko, S. Stirenko, and Y. Gordienko, “Computer Modelling of Mechanism of Formation of Localized Synergetic Defect Substructures under Plastic Deformation of Metal Nanocrystals,” Metallofizika I Noveishie Tekhnologii. 2016. link Times cited: 5 NOT USED (high confidence) J. Seo et al., “Exfoliated Metal Oxide Nanosheets as Effective and Applicable Substrates for Atomically Dispersed Metal Nanoparticles with Tailorable Functionalities,” Advanced Materials Interfaces. 2016. link Times cited: 4 Abstract: An effective methodology to stabilize highly dispersed metal… read moreAbstract: An effective methodology to stabilize highly dispersed metal nanoparticles is developed by employing the exfoliated 2D metal oxide nanosheets with variable surface structures as substrates. The selection of appropriate crystal structure of titanate nanosheet is very crucial in stabilizing atomically dispersed Pt nanoparticles through the tuning of chemical interaction between Pt and titanate substrate. A theoretical study using density functional theory calculations confirms the significant influence of the crystal structure of layered titanate nanosheet on the crystal growth behavior of immobilized metal nanoparticle. As a consequence of the good dispersion of Pt nanoparticles, the Pt–trititanate nanohybrids with stronger interaction show much higher content of atomically dispersed Pt and better catalyst performances than do the Pt–lepidocrocite titanate ones. The applicability of the present method for other metal species is evidenced by the successful tuning of the crystal size and functionality of Au nanoparticles via immobilization on layered titanate nanosheets. The functionality of Au for surface‐enhanced Raman spectroscopy becomes improved by the anchoring on the lepidocrocite‐type titanate nanosheet. The present study underscores that the use of the metal oxide 2D nanosheets with appropriate surface structure as substrates is effective in tailoring the crystal growth and the functionalities of immobilized metal nanoparticles. read less NOT USED (high confidence) R. Rozas, Demi̇rag A., P. Toledo, and J. Horbach, “Thermophysical properties of liquid Ni around the melting temperature from molecular dynamics simulation,” Journal of Chemical Physics. 2016. link Times cited: 17 Abstract: Thermophysical properties of liquid nickel (Ni) around the m… read moreAbstract: Thermophysical properties of liquid nickel (Ni) around the melting temperature are investigated by means of classical molecular dynamics (MD) simulation, using three different embedded atom method potentials to model the interactions between the Ni atoms. Melting temperature, enthalpy, static structure factor, self-diffusion coefficient, shear viscosity, and thermal diffusivity are compared to recent experimental results. Using ab initio MD simulation, we also determine the static structure factor and the mean-squared displacement at the experimental melting point. For most of the properties, excellent agreement is found between experiment and simulation, provided the comparison relative to the corresponding melting temperature. We discuss the validity of the Hansen-Verlet criterion for the static structure factor as well as the Stokes-Einstein relation between self-diffusion coefficient and shear viscosity. The thermal diffusivity is extracted from the autocorrelation function of a wavenumber-dependent temperature fluctuation variable. read less NOT USED (high confidence) J. Michalka, A. P. Latham, and J. Gezelter, “CO-Induced Restructuring on Stepped Pt Surfaces: A Molecular Dynamics Study,” Journal of Physical Chemistry C. 2016. link Times cited: 7 Abstract: The effects of plateau width and step-edge kinking on carbon… read moreAbstract: The effects of plateau width and step-edge kinking on carbon monoxide (CO)-induced restructuring of platinum surfaces were explored using molecular dynamics (MD) simulations. Platinum crystals displaying four different vicinal surfaces [(321), (765), (112), and (557)] were constructed and exposed to partial coverages of carbon monoxide. Platinum–CO interactions were fit to recent experimental data and density functional theory (DFT) calculations, providing a classical interaction model that captures the atop binding preference on Pt. The differences in Pt–Pt binding strength between edge atoms on the various facets were found to play a significant role in step-edge wandering and reconstruction events. Because the mechanism for step doubling relies on a stochastic meeting of two wandering edges, the widths of the plateaus on the original surfaces were also found to play a role in these reconstructions. On the Pt(321) surfaces, the CO adsorbate was found to assist in reordering the kinked step edges into st... read less NOT USED (high confidence) G. Schusteritsch, T. Kühne, Z. Guo, and E. Kaxiras, “The effect of Ag, Pb and Bi impurities on grain boundary sliding and intergranular decohesion in Copper,” Philosophical Magazine. 2016. link Times cited: 8 Abstract: We investigate the changes in grain boundary sliding (GBS) a… read moreAbstract: We investigate the changes in grain boundary sliding (GBS) and intergranular decohesion in copper (Cu), due to the inclusion of bismuth (Bi), lead (Pb) and silver (Ag) substitutional impurity atoms at a symmetric tilt grain boundary (GB), using a first-principles concurrent multiscale approach. We first study the segregation behavior of the impurities by determining the impurity segregation energy in the vicinity of the GB. We find that the energetically preferred sites are on the GB plane. We investigate the intergranular decohesion of Cu by Bi and Pb impurities and compare this to the effect of Ag impurities by considering the work of separation, and the tensile strength, . Both and decrease in the presence of Bi and Pb impurities, indicating their great propensity for intergranular embrittlement, whilst the presence of Ag impurities has only a small effect. We consider GBS to assess the mechanical properties in nanocrystalline metals and find that all three impurities strongly inhibit GBS, with Ag having the biggest effect. This suggests that Ag has a strong effect on the mechanical properties of nanocrystalline Cu, even though its effect on the intergranular decohesion properties of coarse-grained Cu is not significant. read less NOT USED (high confidence) N. Admal, J. Marian, and G. Po, “The atomistic representation of first strain-gradient elastic tensors,” Journal of The Mechanics and Physics of Solids. 2016. link Times cited: 36 NOT USED (high confidence) H. Gu, J.-jiao Wang, and Z. Li, “Molecular Dynamics Simulation of Tensile Behavior on Ceramic Particles Reinforced Aluminum Matrix Nanocomposites,” International Journal of Materials Science and Applications. 2016. link Times cited: 9 Abstract: The mechanical properties and interfacial structures for alu… read moreAbstract: The mechanical properties and interfacial structures for aluminum matrix composites reinforced by nanometer-sized SiC-β particles has been studied using molecular dynamics (MD) simulation. The modified embedded atom methods, was implemented to describe the atomic interactions. The molecular model undergoes an annealing MD simulation from 300 K to 1000 K to reach its minimum energy point. Tensile tests were performed with periodic boundary conditions. The stress-strain relationship has been studied and elastic constants were predicted as well. The results were compared with those given by continuum-based finite element analysis (FEA) together with the experimental data available in the literatures. It showed that both the elastic modulus and yield stress were further strengthened due to the presence of the nano-particles. Also, it was found that the existing SiC nano-particles have an effect on the initial arrangement of Al atoms in such a manner: Al atoms were inclined to aggregate around the particle surface. Aluminum concentrations were also observed inside the SiC particles close to the surface. The depth of hybridization is uniform and planar. read less NOT USED (high confidence) Y. Cui and Z. Chen, “Material transport via the emission of shear loops during void growth: A molecular dynamics study,” Journal of Applied Physics. 2016. link Times cited: 22 Abstract: The growth of a nanovoid in single-crystal copper has been s… read moreAbstract: The growth of a nanovoid in single-crystal copper has been studied via molecular dynamics (MD) method. The objective is to build the correlation between material transport pattern and dislocation structures. MD results are examined by characterizing the material transport via the “relative displacement” of atoms, where the homogenous elastic deformation has been excluded. Through this novel approach, we are able to illustrate the feasibility of void growth induced by shear loops/curves. At a smaller scale, the formation and emission of shear loops/curves contribute to the local mass transport. At a larger scale, a new mechanism of void growth via frustum-like dislocation structure is revealed. A phenomenological description of void growth via frustum-like dislocation structure is also proposed. read less NOT USED (high confidence) Z. Chen and Z. Zhang, “A semi-analytical method to compute acoustic nonlinearity parameter of Cu, Ag and Au,” Rare Metals. 2016. link Times cited: 0 NOT USED (high confidence) Z. Zhu, Y. Gong, Y. Zhou, and Q. Gao, “Molecular dynamics simulation of single crystal Nickel nanometric machining,” Science China Technological Sciences. 2016. link Times cited: 17 NOT USED (high confidence) P. Brault, S. Chuon, and J. Bauchire, “Molecular Dynamics Simulations of Platinum Plasma Sputtering: A Comparative Case Study,” Frontiers in Physics. 2016. link Times cited: 12 Abstract: Molecular Dynamics simulations are carried out for investiga… read moreAbstract: Molecular Dynamics simulations are carried out for investigating atomic processes of platinum sputtering. Sputtered Pt atom energy distribution functions are determined at different sputtering argon ionenergies: 100, 500 and 1000 eV. Calculated energy distribution functions show a cross-over from Thompson theory to binary collision model when increasing argon ion energy and Pt atom sputtered energy. Implanted argon ion number is depending on its kinetic energy, while it is not the case in binary collision approximation. Finally sputtering yields are greater for Thompson theory than for binary collision model at low energy, but converge to the close values at high energy. read less NOT USED (high confidence) Z.-L. Liu, J.-S. Sun, R. Li, X.-L. Zhang, and L. Cai, “Comparative Study on Two Melting Simulation Methods: Melting Curve of Gold,” Communications in Theoretical Physics. 2016. link Times cited: 9 Abstract: Melting simulation methods are of crucial importance to dete… read moreAbstract: Melting simulation methods are of crucial importance to determining melting temperature of materials efficiently. A high-efficiency melting simulation method saves much simulation time and computational resources. To compare the efficiency of our newly developed shock melting (SM) method with that of the well-established two-phase (TP) method, we calculate the high-pressure melting curve of Au using the two methods based on the optimally selected interatomic potentials. Although we only use 640 atoms to determine the melting temperature of Au in the SM method, the resulting melting curve accords very well with the results from the TP method using much more atoms. Thus, this shows that a much smaller system size in SM method can still achieve a fully converged melting curve compared with the TP method, implying the robustness and efficiency of the SM method. read less NOT USED (high confidence) C. Turner, Y. Lei, and Y. Bao, “Modeling the atomistic growth behavior of gold nanoparticles in solution.,” Nanoscale. 2016. link Times cited: 16 Abstract: The properties of gold nanoparticles strongly depend on thei… read moreAbstract: The properties of gold nanoparticles strongly depend on their three-dimensional atomic structure, leading to an increased emphasis on controlling and predicting nanoparticle structural evolution during the synthesis process. In order to provide this atomistic-level insight and establish a link to the experimentally-observed growth behavior, a kinetic Monte Carlo simulation (KMC) approach is developed for capturing Au nanoparticle growth characteristics. The advantage of this approach is that, compared to traditional molecular dynamics simulations, the atomistic nanoparticle structural evolution can be tracked on time scales that approach the actual experiments. This has enabled several different comparisons against experimental benchmarks, and it has helped transition the KMC simulations from a hypothetical toy model into a more experimentally-relevant test-bed. The model is initially parameterized by performing a series of automated comparisons of Au nanoparticle growth curves versus the experimental observations, and then the refined model allows for detailed structural analysis of the nanoparticle growth behavior. Although the Au nanoparticles are roughly spherical, the maximum/minimum dimensions deviate from the average by approximately 12.5%, which is consistent with the corresponding experiments. Also, a surface texture analysis highlights the changes in the surface structure as a function of time. While the nanoparticles show similar surface structures throughout the growth process, there can be some significant differences during the initial growth at different synthesis conditions. read less NOT USED (high confidence) S. López-Moreno, J. Mejía‐López, F. Muñoz, A. Calles, and J. Morán‐López, “Energetics and the magnetic state of Mn2 adsorbed on Au(111): Dimer bond distance dependence,” Journal of Magnetism and Magnetic Materials. 2016. link Times cited: 4 NOT USED (high confidence) Y.-qing Xia, Y. Wu, T. Hang, J. Chang, and M. Li, “Electrodeposition of High Density Silver Nanosheets with Controllable Morphologies Served as Effective and Reproducible SERS Substrates.,” Langmuir : the ACS journal of surfaces and colloids. 2016. link Times cited: 22 Abstract: Silver nanosheets with a nanogap smaller than 10 nm and high… read moreAbstract: Silver nanosheets with a nanogap smaller than 10 nm and high reproducibility were constructed through simple and environmentally friendly electrodeposition method on copper plate. The sizes of the nanogaps can be varied from around 7 to 150 nm by adjusting the deposition time and current density. The nanosheets with different nanogaps exhibited varied surface-enhanced Raman scattering (SERS) properties due to electromagnetic mechanism (EM). The optimized high density silver nanosheets with a nanogap smaller than 10 nm showed effective SERS ability with an enhanced factor as high as 2.0 × 10(5). Furthermore, the formation mechanism of the nanosheets during the electrodeposition process has been investigated by discussing the influence of boric acid and current density. This method has proved to be applicable on different metal substrates, which exhibits the potential to be widely used in different fields. read less NOT USED (high confidence) B. Zhu et al., “Effects of vibration frequency on vibration-assisted nano-scratch process of mono-crystalline copper via molecular dynamics simulation,” AIP Advances. 2016. link Times cited: 26 Abstract: It has always been a critical issue to understand the materi… read moreAbstract: It has always been a critical issue to understand the material removal behavior of Vibration-Assisted Machining (VAM), especially on atomic level. To find out the effects of vibration frequency on material removal response, a three-dimensional molecular dynamics (MD) model has been established in this research to investigate the effects of scratched groove, crystal defects on the surface quality, comparing with the Von Mises shear strain and tangential force in simulations during nano-scratching process. Comparisons are made among the results of simulations from different vibration frequency with the same scratching feed, depth, amplitude and crystal orientation. Copper potential in this simulation is Embedded-Atom Method (EAM) potential. Interaction between copper and carbon atoms is Morse potential. Simulational results show that higher frequency can make groove smoother. Simulation with high frequency creates more dislocations to improve the machinability of copper specimen. The changing frequency does not have evident effects on Von Mises shear strain. Higher frequency can decrease the tangential force to reduce the consumption of cutting energy and tool wear. In conclusion, higher vibration frequency in VAM on mono-crystalline copper has positive effects on surface finish, machinablility and tool wear reduction. read less NOT USED (high confidence) M. C. Giménez, “Stochastic resonance phenomenon in Monte Carlo simulations of silver adsorbed on gold,” The European Physical Journal B. 2016. link Times cited: 6 NOT USED (high confidence) Esfahania, M. Nasr, M. Rostgaard, J. Henri, and B. Erdem, “Thermo-coupled Surface Cauchy-Born Theory: An Engineering Finite Element Approach to Modeling of Nanowire Thermomechanical Response,” Mechanics of Materials. 2016. link Times cited: 2 NOT USED (high confidence) C. Wu and L. Zhigilei, “Nanocrystalline and Polyicosahedral Structure of a Nanospike Generated on Metal Surface Irradiated by a Single Femtosecond Laser Pulse,” Journal of Physical Chemistry C. 2016. link Times cited: 50 Abstract: Short pulse laser irradiation of metal targets can trigger a… read moreAbstract: Short pulse laser irradiation of metal targets can trigger a cascade of highly nonequilibrium processes leading to the formation of unique surface structures of interest to various practical applications. In this paper, we report the results of a large-scale atomistic simulation predicting the generation of a ∼200 nm long frozen nanospike on the surface of a Ag target irradiated by a femtosecond laser pulse. The simulation provides detailed information on the mechanisms responsible for the formation of the nanospike and the processes that define its complex nanostructure. The competing contributions of the epitaxial regrowth of the solid part of the target and the homogeneous nucleation of new crystallites triggered by the strong undercooling of the liquid regions are found to produce a remarkable variability of the structural motifs coexisting in different regions of the frozen nanospike. The homogeneous solidification, in particular, proceeds along two distinct paths selected at the nucleation stage and... read less NOT USED (high confidence) D. Maciążek, M. Kański, L. Gaza, B. Garrison, and Z. Postawa, “Computer modeling of angular emission from Ag(100) and Mo(100) surfaces due to Arn cluster bombardment,” Journal of Vacuum Science & Technology. B. Nanotechnology and Microelectronics: Materials, Processing, Measurement, and Phenomena. 2016. link Times cited: 8 Abstract: Molecular dynamics computer simulations are employed to inve… read moreAbstract: Molecular dynamics computer simulations are employed to investigate the effect of projectile size and surface morphology on the angular emission stimulated by impact of Ar gas cluster projectiles. Argon clusters of sizes n = 10–1000 and kinetic energies of 10 and 20 keV Arn aimed at normal incidence are used to sputter Ag(100) and Mo(100) samples. The total sputtering yield is larger for Ag(100) than for Mo(100). The ratio of sputtering yields is inversely proportional to the ratio of sublimation energies of these solids for projectiles between Ar20 and Ar250. In both systems, the angular distributions are sensitive to both the projectile size and the surface roughness. The maximum of angular spectra shifts from direction normal to the surface toward off-normal direction with the increase in the projectile size. An opposite trend is observed with the increase in the surface roughness. Formation of a cloud composed of projectile atoms and the enhanced lateral material relocation caused by projectile latera... read less NOT USED (high confidence) L. Chang, A. Fisher, Z. Liu, and D. Cheng, “Highly sensitive and selective colorimetric detection of sulphide using Ag–Au nanoalloys: a DFT study,” RSC Advances. 2016. link Times cited: 5 Abstract: A density functional theory approach is applied to investiga… read moreAbstract: A density functional theory approach is applied to investigate the sensing mechanism for the colorimetric detection of sulphide (S) among sulphide species, such as S, SH, cysteine (Cys), and H2S, using Ag–Au nanoalloys. By exploring the adsorption of sulphide species on the Ag42Au13 and Ag55 clusters, it is found that the adsorption strength of those sulphide species on the Ag42Au13 cluster is stronger than those on the Ag55 cluster, corresponding to the higher sensitivity of the Ag42Au13 cluster compared with the Ag55 one for the colorimetric detection of sulphide species. In addition, it is found that the adsorption strength of the Ag42Au13 and Ag55 clusters towards sulphide species follows the order of S > SH > Cys > H2S, indicating that both the Ag42Au13 and Ag55 clusters possess high selectivity for the colorimetric detection of S over other sulphide species. By investigating the coverage effect of S on the Ag42Au13 cluster, it is found that increasing the coverage of S leads to the decrease of the adsorption strength. Our theoretical results are expected to provide new guidelines for rational design of more powerful adsorption-based colorimetric sensors for detecting S using Ag–Au nanoalloys. read less NOT USED (high confidence) S. Hartmann et al., “Experimental and computational studies on the role of surface functional groups in the mechanical behavior of interfaces between single-walled carbon nanotubes and metals,” Journal of Materials Science. 2016. link Times cited: 8 NOT USED (high confidence) M. Barisik and A. Beskok, “Interface Resistance and Thermal Transport in Nano-Confined Liquids.” 2016. link Times cited: 0 Abstract: Miniaturization of microelectronic device components and the… read moreAbstract: Miniaturization of microelectronic device components and the development of nano– electro– mechanical systems require advanced understanding of thermal transport in nano-materials and devices, where the atomic nature of matter becomes important and the validity of well-known continuum approximations becomes questionable [1]. In the case of semiconductors and insulators, heat is carried primarily by vibrations in the crystal lattice known as phonons. Phonon transport is classically studied by lattice dynamics based on harmonic wave theory in the frequency space. However, the anharmonic behaviors forming in a crystal structure cannot be described with this theory. Alternatively, the coupled motions of the atoms in real space can be modeled by molecular dynamics (MD), which provides the natural formation and transport of phonons via vibrations in the crystal lattice. Hence, MD has been widely employed to model phonon transfer in nanostructures and channels [2,3]. The performance and reliability of aforementioned devices strongly depend on the removal of heat either to the ambient or to a coolant. In such cases, phonon transport observed at the interfaces of nanoscale device components and surrounding/confined fluid, or at the interfaces of suspended nanoparticles and fluid medium in nano-fluidic coolants plays a critical role. At such interfaces, heat transfer is interrupted with a temperature jump due to the deficiency in overlap between phonon dispersions of dissimilar materials. Classical theories considering specular or diffuse phonon scattering predict the upper or lower limits of interface thermal resistance (ITR), while a detailed investigation of intermolecular interactions is needed to resolve interface phonon scattering mechanisms. In this chapter, we present interface phonon transfer at the molecular level, and investigate the validity of continuum hypothesis and Fourier’s law in nano-channels. First, we focus on the conventional ways of using MD for heat transport problems. Most of the previous MD research sandwiched a liquid domain between two solid walls and induced heat flux by fixing the wall CONTENTS read less NOT USED (high confidence) Y. Wang and Z. Xu, “Water Intercalation for Seamless, Electrically Insulating, and Thermally Transparent Interfaces.,” ACS applied materials & interfaces. 2016. link Times cited: 26 Abstract: The interface between functional nanostructures and host sub… read moreAbstract: The interface between functional nanostructures and host substrates is of pivotal importance in the design of their nanoelectronic applications because it conveys energy and information between the device and environment. We report here an interface-engineering approach to establish a seamless, electrically insulating, while thermally transparent interface between graphene and metal substrates by introducing water intercalation. Molecular dynamics simulations and first-principles calculations are performed to demonstrate this concept of design, showing that the presence of the interfacial water layer helps to unfold wrinkles formed in the graphene membrane, insulate the electronic coupling between graphene and the substrate, and elevate the interfacial thermal conductance. The findings here lay the ground for a new class of nanoelectronic setups through interface engineering, which could lead to significant improvement in the performance of nanodevices, such as the field-effect transistors. read less NOT USED (high confidence) M. C. Giménez, L. Reinaudi, and E. Leiva, “Monte Carlo simulation of elongating metallic nanowires in the presence of surfactants.,” The Journal of chemical physics. 2015. link Times cited: 4 Abstract: Nanowires of different metals undergoing elongation were stu… read moreAbstract: Nanowires of different metals undergoing elongation were studied by means of canonical Monte Carlo simulations and the embedded atom method representing the interatomic potentials. The presence of a surfactant medium was emulated by the introduction of an additional stabilization energy, represented by a parameter Q. Several values of the parameter Q and temperatures were analyzed. In general, it was observed for all studied metals that, as Q increases, there is a greater elongation before the nanowire breaks. In the case of silver, linear monatomic chains several atoms long formed at intermediate values of Q and low temperatures. Similar observations were made for the case of silver-gold alloys when the medium interacted selectively with Ag. read less NOT USED (high confidence) D. Watvisave, B. Puranik, and U. Bhandarkar, “A hybrid MD-DSMC coupling method to investigate flow characteristics of micro-devices,” J. Comput. Phys. 2015. link Times cited: 14 NOT USED (high confidence) Y. Wu, T. Hang, Z. Yu, J. Gu, and M. Li, “Quasi‐Periodical 3D Hierarchical Silver Nanosheets with Sub‐10 nm Nanogap Applied as an Effective and Applicable SERS Substrate,” Advanced Materials Interfaces. 2015. link Times cited: 7 Abstract: DOI: 10.1002/admi.201500359 effective SERS substrate. In the… read moreAbstract: DOI: 10.1002/admi.201500359 effective SERS substrate. In the following years, using electrochemical methods researchers successfully prepared various silver nanoplates on different metal substrates, such as silvernanojujubes,[15] silver dendrites,[14,16] silver nanosheets,[17] and silver nanoplates.[18] These methods have solved the “practical” issue, but these silver nanoplates always grow randomly on the substrate and thus lack tunability and will cause poor signal reproducibility. In order to get both abundant “hot” sites and good reproducibility, in recent years, researchers have tried to deposit a second structure of silver on periodic 1D nanostructure arrays, such as Si nanowires,[19] ZnO nanorods,[20] carbon nanotubes,[21] porous membranes,[22] and some biological structures[23] by several approaches, which include physical or chemical vapor deposition (PVD or CVD) and electrochemical deposition. The 3D structure, especially the hierarchical structure,[24] can form a 3D electromagnetic enhancement field due to the three different nanogaps,[20,25] by which high SERS “ability” can be realized.[26] However, this method relies heavily on the fabrication of the basic periodic arrays, which have to be prepared with a biology template, lithography technology, CVD, PVD, seed assistant thermal deposition, and other methods. These methods are not convenient or efficient for practical production. For example, in 2015, Cai and co-workers[27] reported a hierarchical silver nanoplate/ZnO nanocone arrays that can trace streptomycin sulfate by template plating, ion etching, Au sputtering and electrodeposition. Compared with the methods mentioned above, chemical deposition is an environmentally friendly, efficient, and low-cost method for preparing dual-scale structures on metal substrates. Herein, a new quasi-periodical silver sphere array comprised of abundant thin silver nanosheets (CCMS) has been easily fabricated in batches at low cost by chemically depositing silver nanosheets on copper cones. The deposition process is illustrated in Figure S1 (Supporting Information). The special cross linking phenomenon of the nanosheets growing on the tips of copper cones inspired study of the growth mechanism of silver nanosheets. This as-prepared CCMS silver film provided sensitive detection as low as 10−12 m of R6G, as well as a reproducible and recyclable detection, showed high SERS ability over ordinary 3D nanostructures due to the abundant “hot” sites of SERS, which originate from the densely packed silver nanosheets at the sharp corners and from the 3D hierarchical structures. At the same time, the finite element method (FEM) simulation will help shed light on the EM mechanism of the Raman enhancement originating from 3D hierarchical structures. To fabricate the 3D silver sphere with highly packed nanosheet, we employed quasi ordered copper cones with a Surface-enhanced Raman scattering (SERS) is considered as the most promising trace detection method since its first discovery on rough silver electrodes in 1974.[1] After years of development, it has exhibited great potential as a nondamaging single molecule analysis method applied in a wide range of areas, including biological labeling,[2] chemistry,[3] agriculture,[4] environmental science,[5] food safety,[6] and so forth.[7] The origin of the SERS effect is still in dispute but there is an agreement on an electromagnetic enhancement mechanism (EM)[8] due to the excitation of the localized surface plasmon resonance (LSPR) on the nanostructures with existance of nanogaps.[9] Enhancement efficiency and applicability are the two factors that evaluate the SERS substrate. The effeciency means the substrate can trace molecules sensitively, reproducibly, and precisely, while the applicability indicates the SERS substrate can be prepared easily at low cost. Silver nanoplates have attracted researchers’ attention due to the structure’s unique sharp corners, which can provide amplified LSPR properties compared to the traditional silver nano particles. Various kinds of silver nanoplates, such as nanocubes,[10] nanorods,[11] and nanotriangles,[12] have been obtained through a solution-phase method with the assistance of various surfactant capping agents, for instance, polymeric chains, micellar assemblies, coordinating molecules, biological reagents, and so on. However, the nanoplates in these suspensions easily aggregate, which causes the inactivation of the nanoplates, and the surfactant attached is hard to be removed, which will interfere with the detection result and therefore hinder its practical application to some degree. To prevent this problem, researchers have been trying to deposit silver nanoplates directly on substrates by electrochemical deposition. In 2007, Sun and Wiederrecht[13] deposited silver nanoplates on n-type GaAs without employing any surfactant or coordinating molecules in the synthesis. In 2009, Albert Gutés et al.[14] prepared silver dendrites from galvanic displacement on commercial aluminum foil as an read less NOT USED (high confidence) A. Walter et al., “X-ray photoemission analysis of clean and carbon monoxide-chemisorbed platinum(111) stepped surfaces using a curved crystal,” Nature Communications. 2015. link Times cited: 51 NOT USED (high confidence) B. Narayanan et al., “Describing the Diverse Geometries of Gold from Nanoclusters to Bulk—A First-Principles-Based Hybrid Bond-Order Potential,” Journal of Physical Chemistry C. 2015. link Times cited: 27 Abstract: Molecular dynamics simulations using empirical force fields … read moreAbstract: Molecular dynamics simulations using empirical force fields (EFFs) are crucial for gaining fundamental insights into atomic structure and long time scale dynamics of Au nanoclusters with far-reaching applications in energy and devices. This approach is thwarted by the failure of currently available EFFs in describing the size-dependent dimensionality and diverse geometries exhibited by Au clusters (e.g., planar structures, hollow cages, tubes, pyramids, space-filled structures). Here, we mitigate this issue by introducing a new hybrid bond-order potential (HyBOP), which accounts for (a) short-range interactions via Tersoff-type BOP terms that accurately treat bond directionality and (b) long-range dispersion effects by a scaled Lennard–Jones term whose contribution depends on the local atomic density. We optimized the independent parameters for our HyBOP using a global optimization scheme driven by genetic algorithms. Moreover, to ensure good transferability of these parameters across different length sca... read less NOT USED (high confidence) Y. Cui and Z. Chen, “Molecular dynamics modeling on the role of initial void geometry in a thin aluminum film under uniaxial tension,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 18 Abstract: The effect of initial void geometry on damage progression in… read moreAbstract: The effect of initial void geometry on damage progression in a thin aluminum film under uniaxial load is studied via molecular dynamics (MD) method. The embedded voids are with different initial geometries regarding shape, porosity and intervoid ligament distance (ILD). Major simulations are run upon twelve MD geometries with each containing 8–27 million atoms. The corresponding stress–strain relation is monitored during the microstructure evolution of the specimens. The critical stress to trigger the dislocation emission is found in line with the prediction of the Lubarda model. The simulation results reveal that the initial void geometry has substantial impact on the stress–strain relation especially for a specimen with larger initial porosity. read less NOT USED (high confidence) B. Prasai, A. R. Wilson, B. J. Wiley, Y. Ren, and V. Petkov, “On the road to metallic nanoparticles by rational design: bridging the gap between atomic-level theoretical modeling and reality by total scattering experiments.,” Nanoscale. 2015. link Times cited: 20 Abstract: The extent to which current theoretical modeling alone can r… read moreAbstract: The extent to which current theoretical modeling alone can reveal real-world metallic nanoparticles (NPs) at the atomic level was scrutinized and demonstrated to be insufficient and how it can be improved by using a pragmatic approach involving straightforward experiments is shown. In particular, 4 to 6 nm in size silica supported Au(100-x)Pd(x) (x = 30, 46 and 58) explored for catalytic applications is characterized structurally by total scattering experiments including high-energy synchrotron X-ray diffraction (XRD) coupled to atomic pair distribution function (PDF) analysis. Atomic-level models for the NPs are built by molecular dynamics simulations based on the archetypal for current theoretical modeling Sutton-Chen (SC) method. Models are matched against independent experimental data and are demonstrated to be inaccurate unless their theoretical foundation, i.e. the SC method, is supplemented with basic yet crucial information on the length and strength of metal-to-metal bonds and, when necessary, structural disorder in the actual NPs studied. An atomic PDF-based approach for accessing such information and implementing it in theoretical modeling is put forward. For completeness, the approach is concisely demonstrated on 15 nm in size water-dispersed Au particles explored for bio-medical applications and 16 nm in size hexane-dispersed Fe48Pd52 particles explored for magnetic applications as well. It is argued that when "tuned up" against experiments relevant to metals and alloys confined to nanoscale dimensions, such as total scattering coupled to atomic PDF analysis, rather than by mere intuition and/or against data for the respective solids, atomic-level theoretical modeling can provide a sound understanding of the synthesis-structure-property relationships in real-world metallic NPs. Ultimately this can help advance nanoscience and technology a step closer to producing metallic NPs by rational design. read less NOT USED (high confidence) L. Zosiak, C. Goyhenex, R. Kozubski, and G. Tréglia, “Electronic structure of CoPt based systems: from bulk to nanoalloys,” Journal of Physics: Condensed Matter. 2015. link Times cited: 4 Abstract: An accurate description of the local electronic structure is… read moreAbstract: An accurate description of the local electronic structure is necessary for guiding the design of materials with targeted properties in a controlled way. For complex materials like nanoalloys, self-consistent tight-binding calculations should be a good alternative to ab initio methods, for handling the most complex and large systems (hundreds to thousands of atoms), provided that these parameterized method is well founded from ab initio ones that they intend to replace. Ab initio calculations (density functional theory) enabled us to derive rules for charge distribution as a function of structural change and alloying effects in Co and Pt based systems, from bulk to nanoalloys. A general local neutrality rule per site, orbital and species was found. Based on it, self-consistent tight-binding calculations could be implemented and applied to CoPt nanoalloys. A very good agreement is obtained between tight-binding and DFT calculations in terms of local electronic structure. read less NOT USED (high confidence) S. R. Das et al., “Single-Layer Graphene as a Barrier Layer for Intense UV Laser-Induced Damages for Silver Nanowire Network.,” ACS nano. 2015. link Times cited: 52 Abstract: Single-layer graphene (SLG) has been proposed as the thinnes… read moreAbstract: Single-layer graphene (SLG) has been proposed as the thinnest protective/barrier layer for wide applications involving resistance to oxidation, corrosion, atomic/molecular diffusion, electromagnetic interference, and bacterial contamination. Functional metallic nanostructures have lower thermal stability than their bulk forms and are therefore susceptible to high energy photons. Here, we demonstrate that SLG can shield metallic nanostructures from intense laser radiation that would otherwise ablate them. By irradiation via a UV laser beam with nanosecond pulse width and a range of laser intensities (in millions of watt per cm(2)) onto a silver nanowire network, and conformally wrapping SLG on top of the nanowire network, we demonstrate that graphene "extracts and spreads" most of the thermal energy away from nanowire, thereby keeping it damage-free. Without graphene wrapping, the radiation would fragment the wires into smaller pieces and even decompose them into droplets. A systematic molecular dynamics simulation confirms the mechanism of SLG shielding. Consequently, particular damage-free and ablation-free laser-based nanomanufacturing of hybrid nanostructures might be sparked off by application of SLG on functional surfaces and nanofeatures. read less NOT USED (high confidence) P. Brault and E. Neyts, “Molecular dynamics simulations of supported metal nanocatalyst formation by plasma sputtering,” Catalysis Today. 2015. link Times cited: 26 NOT USED (high confidence) Z.-L. Liu, X.-L. Zhang, and L. Cai, “Shock melting method to determine melting curve by molecular dynamics: Cu, Pd, and Al.,” The Journal of chemical physics. 2015. link Times cited: 20 Abstract: A melting simulation method, the shock melting (SM) method, … read moreAbstract: A melting simulation method, the shock melting (SM) method, is proposed and proved to be able to determine the melting curves of materials accurately and efficiently. The SM method, which is based on the multi-scale shock technique, determines melting curves by preheating and/or prepressurizing materials before shock. This strategy was extensively verified using both classical and ab initio molecular dynamics (MD). First, the SM method yielded the same satisfactory melting curve of Cu with only 360 atoms using classical MD, compared to the results from the Z-method and the two-phase coexistence method. Then, it also produced a satisfactory melting curve of Pd with only 756 atoms. Finally, the SM method combined with ab initio MD cheaply achieved a good melting curve of Al with only 180 atoms, which agrees well with the experimental data and the calculated results from other methods. It turned out that the SM method is an alternative efficient method for calculating the melting curves of materials. read less NOT USED (high confidence) A. Hens, G. Biswas, and S. De, “Evaporation of water droplets on Pt-surface in presence of external electric field–A molecular dynamics study.,” The Journal of chemical physics. 2015. link Times cited: 23 Abstract: Evaporation of a sessile droplet on a hot solid substrate is… read moreAbstract: Evaporation of a sessile droplet on a hot solid substrate is an important problem in fluid mechanics. It is relevant to theoretical issues in heat transfer as well as several practical applications. This study investigates the spreading and evaporation of a nanoscale water droplet on a solid platinum surface. The major objective was to analyze the effect of an external electric field on these phenomena. Varying the intensity and direction of the external electric field, a series of molecular dynamics simulations were carried out to understand these phenomena at a molecular level. The results reveal that a horizontal electric field assists in droplet spreading, whereas a vertical electric field enhances the rate of evaporation for a certain range of field intensities. It also shows that the substrate temperature plays an important role in such processes. It is seen that the effect of an external electric field on droplet evaporation becomes significant at an intermediate range of surface temperatures and this effect is not clearly visible for either very high or very low range of surface temperatures. read less NOT USED (high confidence) B. M. Iskakov, K. B. Baigisova, and G. Bondarenko, “Determination of the vacancy migration energy in FCC metals with allowance for the relaxation of the nearest atoms,” Russian Metallurgy (Metally). 2015. link Times cited: 5 NOT USED (high confidence) L. Mancera and D. Benoit, “An alternative methodology to assess the quality of empirical potentials for small gold clusters,” Computational and Theoretical Chemistry. 2015. link Times cited: 4 NOT USED (high confidence) J.-feng Tang, L. Deng, S. Xiao, H. Deng, X. Zhang, and W. Hu, “Chemical Ordering and Surface Segregation in Cu–Pt Nanoalloys: The Synergetic Roles in the Formation of Multishell Structures,” Journal of Physical Chemistry C. 2015. link Times cited: 29 Abstract: We performed Monte Carlo simulations coupled with MAEAM pote… read moreAbstract: We performed Monte Carlo simulations coupled with MAEAM potentials to study the surface segregation and chemical ordering patterns in Cu–Pt nanoalloy particles for a broad range of sizes, shapes, composition, and temperature. It was found that both the Cu segregation on the surface and the chemical ordering in the core are the general rules and usually compete with each other. Surface segregation of Cu is enhanced with increasing particle size or surface openness or global Cu composition. Despite their different morphologies, most of the types of ordered phases in the core region are the same as bulk alloys. Due to the modification or suppression effects of surface segregation, the degrees of chemical ordering shift to the Pt-richer side and are more apparent in a large-sized particle. Particularly, at a narrow composition range, the multishell structures (onion-ring or multishell/maze-like core) form in (truncated) octahedrons, illustrating a subtle synergy between the segregated Cu {111} facets and the ... read less NOT USED (high confidence) N. Nakamura, N. Yoshimura, H. Ogi, and M. Hirao, “Formation of continuous metallic film on quartz studied by noncontact resonant ultrasound spectroscopy,” Journal of Applied Physics. 2015. link Times cited: 7 Abstract: Dynamics of continuous film formation of metallic films on q… read moreAbstract: Dynamics of continuous film formation of metallic films on quartz substrates is studied using an electrodeless resonance method. Bare quartz is used as a substrate, and a metallic film is deposited on it. We use antenna transmission technique to measure the evolution of resonance frequencies and internal friction of the substrate during and after deposition, and the morphological transition between discontinuous islands and a continuous film is detected. By comparison with atomic force microscopy images, we confirm that the frequency drop and the internal-friction peak that appear during deposition indicate this transition. We also find that Pt film shows unexpected morphology change after deposition. read less NOT USED (high confidence) S. Takahashi et al., “Oxygen reduction reaction activity and structural stability of Pt-Au nanoparticles prepared by arc-plasma deposition.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 29 Abstract: The oxygen reduction reaction (ORR) activity and durability … read moreAbstract: The oxygen reduction reaction (ORR) activity and durability of various Au(x)/Pt100 nanoparticles (where x is the atomic ratio of Au against Pt) are evaluated herein. The samples were fabricated on a highly-oriented pyrolytic graphite substrate at 773 K through sequential arc-plasma depositions of Pt and Au. The electrochemical hydrogen adsorption charges (electrochemical surface area), particularly the characteristic currents caused by the corner and edge sites of the Pt nanoparticles, decrease with increasing Au atomic ratio (x). In contrast, the specific ORR activities of the Au(x)/Pt100 samples were dependent on the atomic ratios of Pt and Au: the Au28/Pt100 sample showed the highest specific activity among all the investigated samples (x = 0-42). As for ORR durability evaluated by applying potential cycles between 0.6 and 1.0 V in oxygen-saturated 0.1 M HClO4, Au28/Pt100 was the most durable sample against the electrochemical potential cycles. The results clearly showed that the Au atoms located at coordinatively-unsaturated sites, e.g. at the corners or edges of the Pt nanoparticles, can improve the ORR durability by suppressing unsaturated-site-induced degradation of the Pt nanoparticles. read less NOT USED (high confidence) J. Ye, “Shape anisotropy and instability of holes formed during dewetting of single-crystal palladium and nickel films,” Journal of Vacuum Science and Technology. 2015. link Times cited: 4 Abstract: This study investigates the shape anisotropy and instability… read moreAbstract: This study investigates the shape anisotropy and instability of holes formed during dewetting of single-crystal palladium and nickel films. The length ratios of edges constrained to expose {111} facets to other edges of the holes are found to be greater in palladium than in nickel films. The pinch-off is found to occur earlier in palladium than in nickel films. These morphological differences are explained in terms of oxygen adsorption and its effect on the surface energy anisotropy of the film materials. read less NOT USED (high confidence) D. T. Ho, H. Kim, S. Y. Kwon, and S. Y. Kim, “Auxeticity of face‐centered cubic metal (001) nanoplates,” physica status solidi (b). 2015. link Times cited: 37 Abstract: We present the results of an atomistic study on the Poisson&… read moreAbstract: We present the results of an atomistic study on the Poisson's ratios of face‐centered cubic metal (001) nanoplates under tensile loading. Here, we find that the behavior of the Poisson's ratios of metal nanoplates is strongly dependent on the characteristics of a phase transformation that takes place in their bulk counterparts as well as on the amount of compressive stress induced in the nanoplates. In addition, we discuss the effects of the nanoplate thickness and temperature on the mechanical behavior of the nanoplates. As the thickness decreases, the amount of compressive stress increases. As a result, the metal nanoplates become more auxetic. Higher temperatures cause the phase transformation to occur sooner. Thus, strongly auxetic nanoplates can be obtained by raising the temperature. In addition to investigating the effects of the thickness and temperature, we compare the behaviors of the Poisson's ratios of (001) nanoplates of six different metals. Interestingly, the behaviors of the Poisson's ratios of the metal nanoplates differ, even though their corresponding bulks have similar and positive Poisson's ratios. This is because the six metals exhibit large differences in their surface stresses as well as in the critical strains for the phase transformation. read less NOT USED (high confidence) A. Weckman, B. Dem’yanov, and A. S. Dragunov, “Molecular-dynamic investigation of the interaction of vacancies with symmetrical tilt grain boundaries in aluminum,” The Physics of Metals and Metallography. 2015. link Times cited: 3 NOT USED (high confidence) J. Michalka and J. Gezelter, “Island Formation on Pt/Pd(557) Surface Alloys in the Presence of Adsorbed CO: A Molecular Dynamics Study,” Journal of Physical Chemistry C. 2015. link Times cited: 9 Abstract: Stepped surfaces of bimetallic Pt/Pd alloys were exposed to … read moreAbstract: Stepped surfaces of bimetallic Pt/Pd alloys were exposed to a range of coverages of adsorbed carbon monoxide (CO) using molecular dynamics (MD) simulations. Metal–CO interactions for both metals were parametrized from experimental data and density functional theory (DFT) calculations, providing classical potentials that capture the atop binding preference on Pt and the hollow/bridge preference on Pd. The MD simulations indicate significant restructuring in the surface alloy, with Pt-rich islands forming on the Pd substrate within 60 ns. The time dependence of the surface domain sizes and the dynamics of nearest-neighbor metal populations suggest that multilayer Pt islands form more rapidly in the presence of adsorbed CO. We find that the different binding preference of CO adsorbed to the two metals can help explain the observed stabilization of the Pd surface structures as well as the roughening of the Pt step edges. Because the CO acts to lower the surface energy of the Pt, we conclude that the mechanism... read less NOT USED (high confidence) I. M. Robertson et al., “Hydrogen Embrittlement Understood,” Metallurgical and Materials Transactions A. 2015. link Times cited: 422 NOT USED (high confidence) X. Ben and H. S. Park, “Surface Plasmon Resonance-Induced Stiffening of Silver Nanowires,” Scientific Reports. 2015. link Times cited: 2 NOT USED (high confidence) P. Wynblatt and D. Chatain, “Importance of interfacial step alignment in hetero-epitaxy and orientation relationships: the case of Ag equilibrated on Ni substrates. Part 1 computer simulations,” Journal of Materials Science. 2015. link Times cited: 11 NOT USED (high confidence) L. A. Merzhievskii, “Deformation models under intense dynamic loading (Review),” Combustion, Explosion, and Shock Waves. 2015. link Times cited: 11 NOT USED (high confidence) V. Zalizniak, O. A. Zolotov, В. Е. Зализняк, and О. А. Золотов, “Towards a Universal Embedded Atom Method Interatomic Potential for Pure Metals.” 2015. link Times cited: 6 Abstract: A new interatomic potential for metals based on the embedded… read moreAbstract: A new interatomic potential for metals based on the embedded atom method is proposed in this paper. Some approximation of electron density distribution is suggested from the basic principles of quantum mechanics. The functional form of the electron density distribution includes two adjustable parameters. The form of this distribution defines the pair potential and, in part, the form of embedding energy function. The parameters are determined empirically by fitting to the equilibrium lattice constant, cohesion energy, vacancy formation energy, low index surface energy and elastic constants. Potential parameters for 27 metals (10 fcc metals, 9 bcc metals and 8 hcp metals) are presented. Potential is expressed by simple functions and can be used in molecular dynamics simulations of large atomic systems. PACS: 34.20.Cf, 61.50.Ah read less NOT USED (high confidence) B. Buesser and S. Pratsinis, “Morphology and Crystallinity of Coalescing Nanosilver by Molecular Dynamics,” Journal of Physical Chemistry C. 2015. link Times cited: 38 Abstract: Sintering and its final stage of coalescence of silver nanop… read moreAbstract: Sintering and its final stage of coalescence of silver nanoparticles with various morphologies have been investigated in vacuo between 400 and 1000 K by molecular dynamics simulations using the embedded atom method (EAM). It was found that the Ag nanoparticle melting temperature increases with increasing particle size and approaches the bulk melting point of Ag for bigger particles (>10 nm) consistent with simulation literature and experimental data. The motion of surface and bulk Ag atoms within Ag nanoparticles is monitored closely during their sintering. Early on, the sintering or coalescence of nanoparticles is dominated by surface diffusion whereas a transition toward plastic flow sintering can be observed near their melting point. The sintering rate of straight nanoparticle chains is much slower than that of more compact structures. The formation of new crystal domains during Ag particle sintering is demonstrated for the first time to the best of our knowledge, and mechanisms leading to formation of... read less NOT USED (high confidence) X. Qiu, D. Xu, T. Lin, X. Yang, Y. Liu, and P. He, “Molecular dynamics simulation of the effect of carbon nanotube chirality on nano-joining with gold particle,” Materials Transactions. 2015. link Times cited: 0 Abstract: The behavior of gold atoms depending on the CNT chirality in… read moreAbstract: The behavior of gold atoms depending on the CNT chirality in a nanojoining process is studied by molecular dynamics simulation. The deformation regularity and the diffusing characteristic of the gold particle during the joining process, as well as the C-Au bonds distribution in the final joint are studied. Our results show that when joining with higher spirality CNT, gold particle tends to deform more. With the CNT more similar to armchair type, the gold particle as a whole displaces more. In the final joint, the total bonds number decreases from typical armchair CNT to typical zig-zag CNT. However, the bonds distribution in detail is irregular from joint to joint, which is the consequence of lattice structure of both materials. [doi:10.2320/matertrans.MI201403] read less NOT USED (high confidence) B. N. Vadgama, R. Jackson, and D. Harris, “Molecular scale analysis of dry sliding copper asperities,” Applied Nanoscience. 2015. link Times cited: 7 NOT USED (high confidence) I. M. Robertson et al., “Hydrogen Embrittlement Understood,” Metallurgical and Materials Transactions B. 2015. link Times cited: 281 NOT USED (high confidence) L. A. Merzhievskii, “Deformation models under intense dynamic loading (Review),” Combustion, Explosion, and Shock Waves. 2015. link Times cited: 0 NOT USED (high confidence) S. Li and W. Qi, “Unification of Two Different Melting Mechanisms of Nanovoids,” Journal of Physical Chemistry C. 2015. link Times cited: 7 Abstract: Void melting in solids is a very complicated process, while … read moreAbstract: Void melting in solids is a very complicated process, while the mechanism is far from understood. In this paper, we studied the void melting in Pd and Si solids using a molecular dynamics simulation method. It is found that there exist two different melting mechanisms for nanovoid, nucleation melting and non-nucleation melting; although void melting in Pd follows the former mechanism, that in Si follows the latter (unless the nanovoid size decreases to a critically small value). For nucleation melting, there will be liquid nucleate at the surface of the nanovoid, and then the liquid fills the void before the temperature reaches the melting point of the solids. For non-nucleation melting, there will be local stiffening around the nanovoid, and the nanovoid always exists until the whole matrix comes to melt. For these two different melting mechanisms, the inner surface atoms will behave totally differently. We find the most exciting thing to be that the two mechanisms can be well unified based on surface me... read less NOT USED (high confidence) X. W. Zhou, D. Ward, M. Foster, and J. Zimmerman, “An analytical bond-order potential for the copper–hydrogen binary system,” Journal of Materials Science. 2015. link Times cited: 18 NOT USED (high confidence) C. Wu, M. Christensen, J. Savolainen, P. Balling, and L. Zhigilei, “Generation of subsurface voids and a nanocrystalline surface layer in femtosecond laser irradiation of a single-crystal Ag target,” Physical Review B. 2015. link Times cited: 103 NOT USED (high confidence) G. Venturini, K. G. Wang, I. Romero, M. P. Ariza, and M. Ortiz, “Atomistic long-term simulation of heat and mass transport,” Journal of The Mechanics and Physics of Solids. 2014. link Times cited: 38 NOT USED (high confidence) X. Luo, Z. Tong, and Y. Liang, “Investigation of the shape transferability of nanoscale multi-tip diamond tools in the diamond turning of nanostructures,” Applied Surface Science. 2014. link Times cited: 32 NOT USED (high confidence) L. Barnard et al., “Atomistic modeling of the order-disorder phase transformation in the Ni 2 Cr model alloy,” Acta Materialia. 2014. link Times cited: 25 NOT USED (high confidence) D. Xu, M. Hook, and M. Mayer, “Molecular dynamics study of nano-scale Ag surface electromigration and effect of Pd coating layer,” 14th IEEE International Conference on Nanotechnology. 2014. link Times cited: 0 Abstract: Ag is the most conductive metal but is vulnerable to electro… read moreAbstract: Ag is the most conductive metal but is vulnerable to electromigration (EM), which can limit its application in e.g. microelectronics. Molecular dynamics (MD) is used to simulate the migrating behavior of an Ag surface by adding an extra directional force on each atom. The migration of Ag atoms is found to be limited to the topmost 4 (002) lattice planes in the first 40 ns while atoms in the crystal bulk remain oscillating around their equilibrium positions. A Pd coating layer is shown to be a protection from EM for an Ag surface. After adding a layer of 9 Pd (002) lattice planes, the same procedure is repeated with different forces. No migration happens until the extra directional force become so large that all atoms of the model end up moving freely. The MD model presented in this paper can lead to an understanding of EM at the atomic scale and a guideline for potential reliability improvement of microelectronics by coating technology. read less NOT USED (high confidence) X. Ben and H. S. Park, “Atomistic simulations of electric field effects on the Youngʼs modulus of metal nanowires,” Nanotechnology. 2014. link Times cited: 21 Abstract: We present a computational, atomistic study of electric fiel… read moreAbstract: We present a computational, atomistic study of electric field effects on the Youngʼs modulus of metal nanowires. The simulations are electromechanically coupled, where the mechanical forces on the atoms are obtained from realistic embedded atom method potentials, and where the electrostatic forces on the atoms are obtained using a point dipole electrostatic model that is modified to account for the different polarizability and bonding environment of surface atoms. By considering three different nanowire axial orientations ( ⟨ 100 ⟩ ?> , ⟨ 110 ⟩ ?> and ⟨ 111 ⟩ ?> ) of varying cross sectional sizes and aspect ratios, we find that the Youngʼs modulus of the nanowires differs from that predicted for the purely mechanical case due to the elimination of nonlinear elastic stiffening or softening effects due to the electric field-induced positive relaxation strain relative to the relaxed mechanical configuration. We further find that ⟨ 100 ⟩ ?> nanowires are most sensitive to the applied electric field, with Youngʼs moduli that can be increased more than 20% with increasing aspect ratio. Finally, while the orientation of the transverse surfaces does impact the Youngʼs modulus of the nanowires under applied electric field, the key factor controlling the magnitude of the stiffness change of the nanowires is the distance between atomic planes along the axial direction of the nanowire bulk. read less NOT USED (high confidence) B. Onat and S. Durukanoğlu, “The role of vibrations in thermodynamic properties of Cu-Ni alloys,” The European Physical Journal B. 2014. link Times cited: 8 NOT USED (high confidence) D. Fantauzzi, J. Bandlow, L. Sabo, J. Mueller, A. V. van Duin, and T. Jacob, “Development of a ReaxFF potential for Pt-O systems describing the energetics and dynamics of Pt-oxide formation.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 53 Abstract: ReaxFF force field parameters describing Pt-Pt and Pt-O inte… read moreAbstract: ReaxFF force field parameters describing Pt-Pt and Pt-O interactions have been developed and tested. The Pt-Pt parameters are shown to accurately account for the chemical nature, atomic structures and other materials properties of bulk platinum phases, low and high-index platinum surfaces and nanoclusters. The Pt-O parameters reliably describe bulk platinum oxides, as well as oxygen adsorption and oxide formation on Pt(111) terraces and the {111} and {100} steps connecting them. Good agreement between the force field and both density functional theory (DFT) calculations and experimental observations is demonstrated in the relative surface free energies of high symmetry Pt-O surface phases as a function of the oxygen chemical potential, making ReaxFF an ideal tool for more detailed investigations of more complex Pt-O surface structures. Validation for its application to studies of the kinetics and dynamics of surface oxide formation in the context of either molecular dynamics (MD) or Monte Carlo simulations are provided in part by a two-part investigation of oxygen diffusion on Pt(111), in which nudged elastic band (NEB) calculations and MD simulations are used to characterize diffusion processes and to determine the relevant diffusion coefficients and barriers. Finally, the power of the ReaxFF reactive force field approach in addressing surface structures well beyond the reach of routine DFT calculations is exhibited in a brief proof-of-concept study of oxygen adsorbate displacement within ordered overlayers. read less NOT USED (high confidence) C. Li, S. Dang, L. Wang, C.-li Zhang, and P. Han, “First-principles study of the effects of selected interstitial atoms on the generalized stacking fault energies, strength, and ductility of Ni,” Chinese Physics B. 2014. link Times cited: 6 Abstract: We analyze the influences of interstitial atoms on the gener… read moreAbstract: We analyze the influences of interstitial atoms on the generalized stacking fault energy (GSFE), strength, and ductility of Ni by first-principles calculations. Surface energies and GSFE curves are calculated for the 〈11〉 (111) and 〈10〉 (111) systems. Because of the anisotropy of the single crystal, the addition of interstitials tends to promote the strength of Ni by slipping along the 〈10〉 direction while facilitating plastic deformation by slipping along the 〈11〉 direction. There is a different impact on the mechanical behavior of Ni when the interstitials are located in the slip plane. The evaluation of the Rice criterion reveals that the addition of the interstitials H and O increases the brittleness in Ni and promotes the probability of cleavage fracture, while the addition of S and N tends to increase the ductility. Besides, P, H, and S have a negligible effect on the deformation tendency in Ni, while the tendency of partial dislocation is more prominent with the addition of N and O. The addition of interstitial atoms tends to increase the high-energy barrier γmax, thereby the second partial resulting from the dislocation tends to reside and move on to the next layer. read less NOT USED (high confidence) V. Mazhukin, A. V. Shapranov, A. Samokhin, and A. Ivochkin, “Modeling of explosive boiling of a thin film during homogeneous subnanosecond heating,” Mathematical Models and Computer Simulations. 2014. link Times cited: 1 NOT USED (high confidence) B. Ma, Q. Rao, and Y. He, “Effect of crystal orientation on tensile mechanical properties of single-crystal tungsten nanowire,” Transactions of Nonferrous Metals Society of China. 2014. link Times cited: 26 NOT USED (high confidence) C. Hu, M.-li Bai, J. Lv, P. Wang, L. Zhang, and X.-jie Li, “Molecular dynamics simulation of nanofluid’s flow behaviors in the near-wall model and main flow model,” Microfluidics and Nanofluidics. 2014. link Times cited: 27 NOT USED (high confidence) S. Wilson and M. Mendelev, “Dependence of solid–liquid interface free energy on liquid structure,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 17 Abstract: The Turnbull relation is widely believed to enable predictio… read moreAbstract: The Turnbull relation is widely believed to enable prediction of solid–liquid interface (SLI) free energies from measurements of the latent heat and the solid density. Ewing proposed an additional contribution to the SLI free energy to account for variations in liquid structure near the interface. In the present study, molecular dynamics (MD) simulations were performed to investigate whether SLI free energy depends on liquid structure. Analysis of the MD simulation data for 11 fcc metals demonstrated that the Turnbull relation is only a rough approximation for highly ordered liquids, whereas much better agreement is observed with Ewing's theory. A modification to Ewing's relation is proposed in this study that was found to provide excellent agreement with MD simulation data. read less NOT USED (high confidence) E. Cieren, L. Colombet, S. Pitoiset, and R. Namyst, “ExaStamp: A Parallel Framework for Molecular Dynamics on Heterogeneous Clusters,” Euro-Par Workshops. 2014. link Times cited: 8 NOT USED (high confidence) G. Kroes, M. Pavanello, M. Blanco-Rey, M. Alducin, and D. Auerbach, “Ab initio molecular dynamics calculations on scattering of hyperthermal H atoms from Cu(111) and Au(111).,” The Journal of chemical physics. 2014. link Times cited: 43 Abstract: Energy loss from the translational motion of an atom or mole… read moreAbstract: Energy loss from the translational motion of an atom or molecule impinging on a metal surface to the surface may determine whether the incident particle can trap on the surface, and whether it has enough energy left to react with another molecule present at the surface. Although this is relevant to heterogeneous catalysis, the relative extent to which energy loss of hot atoms takes place to phonons or electron-hole pair (ehp) excitation, and its dependence on the system's parameters, remain largely unknown. We address these questions for two systems that present an extreme case of the mass ratio of the incident atom to the surface atom, i.e., H + Cu(111) and H + Au(111), by presenting adiabatic ab initio molecular dynamics (AIMD) predictions of the energy loss and angular distributions for an incidence energy of 5 eV. The results are compared to the results of AIMDEFp calculations modeling energy loss to ehp excitation using an electronic friction ("EF") model applied to the AIMD trajectories, so that the energy loss to the electrons is calculated "post" ("p") the computation of the AIMD trajectory. The AIMD calculations predict average energy losses of 0.38 eV for Cu(111) and 0.13-0.14 eV for Au(111) for H-atoms that scatter from these surfaces without penetrating the surface. These energies closely correspond with energy losses predicted with Baule models, which is suggestive of structure scattering. The predicted adiabatic integral energy loss spectra (integrated over all final scattering angles) all display a lowest energy peak at an energy corresponding to approximately 80% of the average adiabatic energy loss for non-penetrative scattering. In the adiabatic limit, this suggests a way of determining the approximate average energy loss of non-penetratively scattered H-atoms from the integral energy loss spectrum of all scattered H-atoms. The AIMDEFp calculations predict that in each case the lowest energy loss peak should show additional energy loss in the range 0.2-0.3 eV due to ehp excitation, which should be possible to observe. The average non-adiabatic energy losses for non-penetrative scattering exceed the adiabatic losses to phonons by 0.9-1.0 eV. This suggests that for scattering of hyperthermal H-atoms from coinage metals the dominant energy dissipation channel should be to ehp excitation. These predictions can be tested by experiments that combine techniques for generating H-atom beams that are well resolved in translational energy and for detecting the scattered atoms with high energy-resolution. read less NOT USED (high confidence) D. Davydov, J.-P. Pelteret, and P. Steinmann, “Comparison of several staggered atomistic-to-continuum concurrent coupling strategies,” Computer Methods in Applied Mechanics and Engineering. 2014. link Times cited: 20 NOT USED (high confidence) Z. Tong, Y. Liang, X. Yang, and X. Luo, “Investigation on the thermal effects during nanometric cutting process while using nanoscale diamond tools,” The International Journal of Advanced Manufacturing Technology. 2014. link Times cited: 40 NOT USED (high confidence) Z. Tong, Y. Liang, X. Yang, and X. Luo, “Investigation on the thermal effects during nanometric cutting process while using nanoscale diamond tools,” The International Journal of Advanced Manufacturing Technology. 2014. link Times cited: 0 NOT USED (high confidence) B. M. Iskakov, K. B. Baigisova, and G. Bondarenko, “Determination of the vacancy migration energy in fcc metals using a modified embedded-atom method,” Russian Metallurgy (Metally). 2014. link Times cited: 3 NOT USED (high confidence) S. Kiselev, “Method of molecular dynamics in mechanics of deformable solids,” Journal of Applied Mechanics and Technical Physics. 2014. link Times cited: 8 NOT USED (high confidence) Q. Zhang et al., “Analytic Force Field for Clusters and Nanoparticles of Aluminum and Its Hydride,” Physical review applied. 2014. link Times cited: 0 Abstract: An analytic potential energy function is developed for simul… read moreAbstract: An analytic potential energy function is developed for simulating clusters and nanoparticles of aluminum and its hydride. An embedded atom method is used which modulates the background electron density as a function of the number of nearest neighbor atoms. The method is parameterized and tested using an extensive training set computed from first principles density functional theory. The potential energy function is found to be reliable for clusters of arbitrary size, shape, and composition ratio. The force field obtained from the analytic potential energy function is computationally efficient and well-suited for simulating large systems of aluminum and aluminum hydride particles. A proposed molecular dynamics simulation related to hydrogen storage technologies for onboard automotive applications is briefly discussed. read less NOT USED (high confidence) W. Yu and Z. Wang, “Interactions between edge lattice dislocations and Σ11 symmetrical tilt grain boundary: comparisons among several FCC metals and interatomic potentials,” Philosophical Magazine. 2014. link Times cited: 15 Abstract: Interactions between edge dislocations and a symmetrical til… read moreAbstract: Interactions between edge dislocations and a symmetrical tilt grain boundary (GB) in face-centred cubic metals of Ni and Al are studied via a quasicontinuum method (QCM). A variety of embedding atom method potentials are used, and the results are compared to previous studies of Cu [W.S. Yu, Z.Q. Wang, Acta Mater., 60 (2012) 5010]. Different potentials do not significantly affect the edge dislocation–GB interactions in these metals. Edge dislocations can easily transmit across grain boundaries in Ni and Cu, even for a single incoming dislocation. However, slip-transmission in Al occurs only after the GB absorbs many incoming dislocations. Stable nucleation of grain boundary dislocations (GBD) in Cu and Ni plays an important role in the slip-transmissions. The slip transmission in Al is found to be difficult due to the metastable nucleation of GBD. The incoming leading and trailing partials in Al are absorbed together by the GB because of the larger values of (, and are the shear modulus, magnitude of Burgers vector of a partial dislocation and the stable stacking fault (SF) energy, respectively). The parameter ( as the unstable SF energy) [Z.H. Jin et al., Acta. Mater. 56 (2008) 1126] incorporates and , and can be used to measure the slip transmission ability of an edge dislocation in these metals. It is also shown that certain loading conditions can help enhance the nucleation of GBDs and GBD dipoles in Al, such that the incoming, leading and trailing partial dislocations can be absorbed separately. read less NOT USED (high confidence) A. Altberg, G. Atiya, V. Mikhelashvili, G. Eisenstein, and W. Kaplan, “The equilibrium orientation relationship between Pt and SrTiO3 and its implication on Pt films deposited by physical vapor phase deposition,” Journal of Materials Science. 2014. link Times cited: 7 NOT USED (high confidence) S. Valone, S. Atlas, and M. Baskes, “Fragment Hamiltonian model potential for nickel: metallic character and defects in crystalline lattices,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 5 Abstract: The Fragment Hamiltonian (FH) model is introduced as the bas… read moreAbstract: The Fragment Hamiltonian (FH) model is introduced as the basis for a new class of atomistic potentials that may be viewed as generalizations of the embedded atom method (EAM) and related atomistic potentials. Many metals and alloys have been successfully modeled by this method and other related methods, but the nature of the metallic character in the models has been lost. Here we attempt to recover this character, at a qualitative level, by defining an embedding energy as a function of two variables through the FH model. One of these variables, called the ionicity, is associated with the established concept of background density in EAM models. The FH embedding energy is composed of two types of energies, one for energies of different states of an atom and the other for hopping energies that transform an atom from one state to another. A combination of the energies for the states of an atom yield a local gap energy that conforms to a generalized definition of the ‘Hubbard-U’ energy. The hopping energies compete with the gap energy to provide a notion of metallic behavior in an atomic-scale model. Lattices of nickel with different coordinations and spatial dimensions, elastic constants, energies for several types of defects in three-dimensional lattices and two surface energies are calculated to show the strengths and limitations of the current implementation and to explore their metallic character. read less NOT USED (high confidence) S. Kiselev, “Method of molecular dynamics in mechanics of deformable solids,” Journal of Applied Mechanics and Technical Physics. 2014. link Times cited: 1 NOT USED (high confidence) J. Petucci, C. LeBlond, and M. Karimi, “Molecular dynamics simulations of brittle fracture in fcc crystalline materials in the presence of defects,” Computational Materials Science. 2014. link Times cited: 24 NOT USED (high confidence) Z. Wang, L. Zhu, J. Wang, and F. Ding, “A multiscale approach to determine binding energy distribution on a strained surface.,” Nanoscale. 2014. link Times cited: 0 Abstract: A multiscale approach was developed by combining ab initio c… read moreAbstract: A multiscale approach was developed by combining ab initio calculations with classical molecular mechanics (MM) simulations to investigate the adsorption and diffusion of an adatom on a strained and/or defective surface. Specifically, the binding energy of the adatom was calculated as a function of the local substrate strain near the adsorption site by an ab initio method and the strain distribution of a large defective surface was calculated by the MM method. Then a map of the binding energy of the adatom on a large defective surface was derived by bridging the DFT calculated binding energy and the MM determined strain distribution. As an example, the approach is applied to explore the adsorption and diffusion of a carbon atom on the Ni(111) surfaces with dislocations and grain boundaries, respectively. This approach bridges models of different length scales and can be extended to systems with an uneven distribution of strain or curvature. read less NOT USED (high confidence) S. Hartmann et al., “Quantitative in-situ scanning electron microscope pull-out experiments and molecular dynamics simulations of carbon nanotubes embedded in palladium,” Journal of Applied Physics. 2014. link Times cited: 16 Abstract: In this paper, we present our results of experimental and nu… read moreAbstract: In this paper, we present our results of experimental and numerical pull-out tests on carbon nanotubes (CNTs) embedded in palladium. We prepared simple specimens by employing standard silicon wafers, physical vapor deposition of palladium and deposition of CNTs with a simple drop coating technique. An AFM cantilever with known stiffness connected to a nanomanipulation system was utilized inside a scanning electron microscope (SEM) as a force sensor to determine forces acting on a CNT during the pull-out process. SEM-images of the cantilever attached to a CNT have been evaluated for subsequent displacement steps with greyscale correlation to determine the cantilever deflection. We compare the experimentally obtained pull-out forces with values of numerical investigations by means of molecular dynamics and give interpretations for deviations according to material impurities or defects and their influence on the pull-out data. We find a very good agreement of force data from simulation and experiment, which is 17 nN and in the range of 10–61 nN, respectively. Our findings contribute to the ongoing research of the mechanical characterization of CNT-metal interfaces. This is of significant interest for the design of future mechanical sensors utilizing the intrinsic piezoresistive effect of CNTs or other future devices incorporating CNT-metal interfaces. read less NOT USED (high confidence) A. Ito, Y. Yoshimoto, S. Saito, A. Takayama, and H. Nakamura, “Molecular dynamics simulation of a helium bubble bursting on tungsten surfaces,” Physica Scripta. 2014. link Times cited: 43 Abstract: The bursting and expansion of helium bubbles near the surfac… read moreAbstract: The bursting and expansion of helium bubbles near the surface of a tungsten material were investigated by using a molecular dynamics (MD) simulation. These helium bubble processes are considered to be important in the formation mechanism of fuzzy tungsten nano-structures. The phase diagram of the occurrence of bursting and expansion of helium bubbles was obtained by our MD simulation. The results of the simulation indicate that a helium bubble with a radius of 1.0 nm needs a high pressure of several tens of GPa to burst near the surface and to expand the bubble structures under the surface to the scale of ten nanometers. Moreover, from the viewpoint of the dynamics, the results of the MD simulation imply that the concavities and convexities observed on the surface in the early stage of the formation of a tungsten fuzzy nano-structure are caused by the bursting of the helium bubble. read less NOT USED (high confidence) J. Liu, J. Song, and Y. Wei, “Size Effects of Elastic Modulus of FCC Metals Based on the Cauchy-Born Rule and Nanoplate Models,” Acta Mechanica Solida Sinica. 2014. link Times cited: 2 NOT USED (high confidence) M. S. Yaghmaee and R. Riahifar, “In the Search of Fundamental Inner Bond Strength of Solid Elements,” The Scientific World Journal. 2014. link Times cited: 3 Abstract: In order to understand the physics behind the surface proper… read moreAbstract: In order to understand the physics behind the surface properties and nano-scale phenomena, we are motivated first to investigate the inner bond strengths as well as the effect of number of neighboring atoms and their relative distance in addition to space positions (crystallography). Therefore, in order to study the effect of the nature of metallic bond on their physico-chemical properties, we first tried to investigate and introduce a mathematical model for transforming the bulk molar cohesion energy into microscopic bond strengths between atoms. Then an algorithm for estimating the nature of bond type including the materials properties and lattice scale “cutoff” has been proposed. This leads to a new fundamental energy scale free from the crystallography and number of atoms. The results of our model in case of fundamental energy scale of metals not only perfectly describe the inter relation between binding and melting phenomena but also adequately reproduce the bond strength for different bond types with respect to other estimations reported in literatures. The generalized algorithm and calculation methodology introduced here by us are suggested to be used for developing energy scale of bulk crystal materials to explain or predict any particular materials properties related to bond strengths of metallic elements. read less NOT USED (high confidence) B.-M. Lee and B.-J. Lee, “A Comparative Study on Hydrogen Diffusion in Amorphous and Crystalline Metals Using a Molecular Dynamics Simulation,” Metallurgical and Materials Transactions A. 2014. link Times cited: 35 NOT USED (high confidence) T. Milek, T. Döpper, C. Neiss, A. Görling, and D. Zahn, “Charge distribution analysis in Agnm+$ \mathbfAg_\mathbfn^\mathbfm+ $clusters: molecular modeling and DFT calculations,” Journal of Molecular Modeling. 2014. link Times cited: 4 NOT USED (high confidence) A. Evteev, L. Momenzadeh, E. Levchenko, I. Belova, and G. Murch, “Molecular dynamics prediction of phonon-mediated thermal conductivity of f.c.c. Cu,” Philosophical Magazine. 2014. link Times cited: 20 Abstract: The phonon-mediated thermal conductivity of f.c.c. Cu is inv… read moreAbstract: The phonon-mediated thermal conductivity of f.c.c. Cu is investigated in detail in the temperature range 40–1300 K. The calculations are performed in the framework of equilibrium molecular dynamics making use of the Green–Kubo formalism and one of the most reliable embedded-atom method potentials for Cu. It is found that the temporal decay of the heat current autocorrelation function (HCACF) of the Cu model at low and intermediate temperatures demonstrate a more complex behaviour than the two-stage decay observed previously for the f.c.c. Ar model. After the first stage of decay, it demonstrates a peak in the temperature range 40–800 K. A decomposition model of the HCACF is introduced. In the framework of that model we demonstrate that a classical description of the phonon thermal transport in the Cu model can be used down to around one quarter of the Debye temperature (about 90 K). Also, we find that above 300 K the thermal conductivity of the Cu model varies with temperature more rapidly than , following an exponent close to −1.4 in agreement with previous calculations on the Ar model. Phonon thermal conductivity of Cu is found to be about one order of magnitude higher than Ar. The phonon contribution to the total thermal conductivity of Cu can be estimated to be about 0.5% at 1300 K and about 10% at 90 K. read less NOT USED (high confidence) M. Liao, I. Chang, and F.-R. Chang, “Influence of film thickness and surface orientation on melting behaviors of copper nanofilms,” Journal of Materials Research. 2014. link Times cited: 4 Abstract: The effects of film thickness and surface orientation on mel… read moreAbstract: The effects of film thickness and surface orientation on melting behaviors of copper nanofilms were investigated by molecular dynamics simulations. A stepwise heating scheme was adopted to make sure that the nanofilms reached thermal equilibrium before further temperature increase. Melting of the nanofilms was monitored by examining the equilibrium potential energy, radial distribution function, and mean square displacement of the simulated nanofilms. From the simulation, the melting was observed to occur at a specific temperature within 1 K error, unlike the progressive melting process reported in the literature. The melted temperature and the latent heat of fusion of the nanofilms were found to increase with film thickness and approach the bulk value. The nanofilms with (111) surface have the highest melted temperature and the largest latent heat of fusion as compared to the ones with (001) and (011) surfaces, which could be explained by the lowest surface energy of (111) surface. read less NOT USED (high confidence) Z. Tong, Y. Liang, X. Jiang, and X. Luo, “An atomistic investigation on the mechanism of machining nanostructures when using single tip and multi-tip diamond tools,” Applied Surface Science. 2014. link Times cited: 60 NOT USED (high confidence) H. Zhang and J. Banfield, “Interatomic Coulombic interactions as the driving force for oriented attachment,” CrystEngComm. 2014. link Times cited: 71 Abstract: Growth of nano- and mesocrystals in both laboratory and natu… read moreAbstract: Growth of nano- and mesocrystals in both laboratory and natural environments can proceed via the oriented attachment (OA) pathway. However, the driving force for OA is controversial; surface energy reduction, van der Waals interaction, and/or dipole–dipole interactions have been proposed. Here, we analyzed the interaction energy of two approaching nanoparticles by comparing the magnitudes of Coulombic interactions from molecular energetic calculations, van der Waals interactions from the Hamaker formulation, and surface charge repulsion from the DLVO theory. The analyses were conducted for three materials: SiO2, ZnS and ZnO. Results show that in vacuum, or when two nanoparticles are in close proximity in an electrolyte solution, the strong intrinsic interatomic Coulombic interactions between two nanoparticles provide the primary physical driving force for OA. However, when two particles are far apart in a solution, the interatomic Coulombic interactions are screened and van der Waals interactions become the physical driving force (that superimposes onto the random force from the Brownian motion). In both vacuum and a solution, the energy change that occurs following an OA event (i.e., the thermodynamic driving force) comes largely from the interatomic Coulombic interactions arising from both the surface atoms (accounting for the surface energy reduction) and the atoms in interior of the nanoparticles. This energy change is crystallographic orientation-dependent. The findings of this study indicate the range of conditions under which interatomic Coulombic interactions provide the primary driving force for crystal growth by OA and highlight the effects of aqueous solution and ionic strength on the energetics of the process. read less NOT USED (high confidence) B. Onat and S. Durukanoğlu, “An optimized interatomic potential for Cu–Ni alloys with the embedded-atom method,” Journal of Physics: Condensed Matter. 2014. link Times cited: 86 Abstract: We have developed a semi-empirical and many-body type model … read moreAbstract: We have developed a semi-empirical and many-body type model potential using a modified charge density profile for Cu–Ni alloys based on the embedded-atom method (EAM) formalism with an improved optimization technique. The potential is determined by fitting to experimental and first-principles data for Cu, Ni and Cu–Ni binary compounds, such as lattice constants, cohesive energies, bulk modulus, elastic constants, diatomic bond lengths and bond energies. The generated potentials were tested by computing a variety of properties of pure elements and the alloy of Cu, Ni: the melting points, alloy mixing enthalpy, lattice specific heat, equilibrium lattice structures, vacancy formation and interstitial formation energies, and various diffusion barriers on the (100) and (111) surfaces of Cu and Ni. read less NOT USED (high confidence) M. Grouchko et al., “Correction: Corrigendum: Merging of metal nanoparticles driven by selective wettability of silver nanostructures,” Nature Communications. 2014. link Times cited: 62 NOT USED (high confidence) C. Becker et al., “Thermodynamic modelling of liquids: CALPHAD approaches and contributions from statistical physics,” physica status solidi (b). 2014. link Times cited: 32 Abstract: We describe current approaches to thermodynamic modelling of… read moreAbstract: We describe current approaches to thermodynamic modelling of liquids for the CALPHAD method, the use of available experimental methods and results in this type of modelling, and considerations in the use of atomic‐scale simulation methods to inform a CALPHAD approach. We begin with an overview of the formalism currently used in CALPHAD to describe the temperature dependence of the liquid Gibbs free energy and outline opportunities for improvement by reviewing the current physical understanding of the liquid. Brief descriptions of experimental methods for extracting high‐temperature data on liquids and the preparation of undercooled liquid samples are presented. Properties of a well‐determined substance, B2 O3, including the glass transition, are then discussed in detail to emphasize specific modelling requirements for the liquid. We then examine the two‐state model proposed for CALPHAD in detail and compare results with experiment and theory, where available. We further examine the contributions of atomic‐scale methods to the understanding of liquids and their potential for supplementing available data. We discuss molecular dynamics (MD) and Monte Carlo methods that employ atomic interactions from classical interatomic potentials, as well as contributions from ab initio MD. We conclude with a summary of our findings. read less NOT USED (high confidence) D. Belashchenko, “Computer simulation of liquid metals,” Physics—Uspekhi. 2013. link Times cited: 84 Abstract: Methods for and the results of the computer simulation of li… read moreAbstract: Methods for and the results of the computer simulation of liquid metals are reviewed. Two basic methods, classical molecular dynamics with known interparticle potentials and the ab initio method, are considered. Most attention is given to the simulated results obtained using the embedded atom model (EAM). The thermodynamic, structural, and diffusion properties of liquid metal models under normal and extreme (shock) pressure conditions are considered. Liquid-metal simulated results for the Groups I–IV elements, a number of transition metals, and some binary systems (Fe–C, Fe–S) are examined. Possibilities for the simulation to account for the thermal contribution of delocalized electrons to energy and pressure are considered. Solidification features of supercooled metals are also discussed. read less NOT USED (high confidence) A. Politano, G. Chiarello, G. Benedek, E. Chulkov, and P. Echenique, “Vibrational spectroscopy and theory of alkali metal adsorption and co-adsorption on single-crystal surfaces,” Surface Science Reports. 2013. link Times cited: 56 NOT USED (high confidence) D. Savio, “Nanoscale phenomena in lubrication : From atomistic simulations to their integration into continuous models.” 2013. link Times cited: 4 Abstract: The modern trends in lubrication aim at reducing the oil qua… read moreAbstract: The modern trends in lubrication aim at reducing the oil quantity in tribological applications. As a consequence, the film thickness in the contact zone decreases significantly and can reach the order of magnitude of a few nanometres. Hence, the surface separation is ensured by very few lubricant molecules. Atomistic simulations based on the Molecular Dynamics method are used to analyze the local behavior of these severely confined films. A particular attention is paid to the occurrence of wall slip: predictive models and analytical laws are formulated to quantify and predict this phenomenon as a function of the surface-lubricant pair or the local operating conditions in a contact interface. Then, the coupling between Molecular Dynamics simulations and macroscopic models is explored. The classical lubrication theory is modified to include slip effects characterized previously. This approach is employed to study an entire contact featuring a nano-confined lubricant in its center, showing a severe modification of the film thickness and friction. Finally, the lubricant quantity reduction is pushed to the limits up to the occurrence of local film breakdown and direct surface contact. In this scenario, atomistic simulations allow to understand the relationship between the configuration of the last fluid molecules in the contact and the local tribological behavior. read less NOT USED (high confidence) Y.-N. Wen, “Study of the surface relaxation and single vacancy formation in very thin Cu (001) film by using MAEAM,” Central European Journal of Physics. 2013. link Times cited: 4 Abstract: The surface relaxation and the formation of a single vacancy… read moreAbstract: The surface relaxation and the formation of a single vacancy in very thin Cu (001) film formed by 2 ∼ 14 atomic layers have been studied by using MAEAM and MD simulation. For the surface relaxtion, the highest surface energy is in the l = 2 atomic layers. The multilayer relaxation mainly occurs between the first two atomic layers, and the maximum contractive displacement is obtained in the very thin Cu (001) film formed by l = 3 atomic layers. For the vacancy formed in l′ = 1 of the very thin Cu (001) film formed by l = 2 ∼ 14 layers, the most difficult site in the film formed by l = 3 atomic layers. read less NOT USED (high confidence) A. Agrawal, R. Mishra, L. T. Ward, K. Flores, and W. Windl, “An embedded atom method potential of beryllium,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 15 Abstract: We present an embedded atom method (EAM) potential for hexag… read moreAbstract: We present an embedded atom method (EAM) potential for hexagonal beryllium, with a pair function in the form of a Morse potential and a Johnson embedding function with exponential electron density. The cohesive energy, elastic constants, lattice parameters and relaxed vacancy formation energy were used to fit the potential. The fitted-potential was validated by a comparison to first-principles and, wherever available, experimental results for the lattice energies of various crystal structures: vacancy cluster, interstitial formation and surface. Using a large cutoff distance of 5 Å, which includes interactions to approximately the eighth neighbor shell of beryllium, allows our potential to reproduce these quantities considerably better than previous EAM potentials. The accuracy obtained by our potential is similar to or in some cases even better than available modified EAM potentials, while being computationally less intensive. read less NOT USED (high confidence) J. Yang, C. Mao, X. Li, and C. Liu, “On the Cauchy-Born approximation at finite temperature for alloys,” Discrete & Continuous Dynamical Systems - B. 2013. link Times cited: 4 NOT USED (high confidence) A. Fraile, S. Cuesta-L’opez, A. Caro, D. Schwen, and J. Perlado, “Interatomic potential for the compound-forming Li-Pb liquid alloy,” Journal of Nuclear Materials. 2013. link Times cited: 13 NOT USED (high confidence) A. V. Fedorov and A. V. Shul’gin, “Complex modeling of melting of an aluminum nanoparticle,” Combustion, Explosion, and Shock Waves. 2013. link Times cited: 14 NOT USED (high confidence) P. Zhang and D. Trinkle, “Database optimization for empirical interatomic potential models,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 8 Abstract: Weighted least squares fitting to a database of quantum mech… read moreAbstract: Weighted least squares fitting to a database of quantum mechanical calculations can determine the optimal parameters of empirical potential models. While algorithms exist to provide optimal potential parameters for a given fitting database of structures with corresponding energy-related predictions and to estimate prediction errors using Bayesian sampling, defining an optimal fitting database based on potential predictions remains elusive. A testing set of structures and energy-related predictions provides an empirical measure of potential transferability. Here, we propose an objective function for fitting databases based on testing set errors. The objective function allows the optimization of the weights in a fitting database, the assessment of the adding or removing of structures in the fitting database, or the comparison of two different fitting databases. To showcase this technique, we consider an example Lennard-Jones potential for Ti, where modeling multiple complicated crystal structures is difficult for a radial pair potential. The algorithm finds different optimal fitting databases, depending on the objective function of potential prediction error for a testing set. read less NOT USED (high confidence) I. Merkulov, M. Yoon, and D. Geohegan, “How the shape of catalyst nanoparticles determines their crystallographic orientation during carbon nanofiber growth,” Carbon. 2013. link Times cited: 7 NOT USED (high confidence) H. Shuo, Z. Chuan-hui, S. Jing, and S. Jiang, “The influence of 3d-metal alloy additions on the elastic and thermodynamic properties of CuPd3,” Chinese Physics B. 2013. link Times cited: 2 Abstract: Embedded-atom method (EAM) potentials are used to investigat… read moreAbstract: Embedded-atom method (EAM) potentials are used to investigate the effects of alloying (e.g. 3d-metals) on the trends of elastic and thermodynamic properties for CuPd3 alloy. Our calculated lattice parameter, cohesive energy, and elastic constants of CuPd3 are consistent with the available experimental and theoretical data. The results of elastic constants indicate that all these alloys are mechanically stable. Further mechanical behavior analysis shows that the additions of Cr, Fe, Co, and Ni could improve the hardness of CuPd3 while V could well increase its ductility. Moreover, in order to evaluate the thermodynamic contribution of 3d-metals, the Debye temperature, phonon density of states, and vibrational entropy for CuMPd6 alloy are also investigated. read less NOT USED (high confidence) S. Jiang, Y. Zhang, Y. Gan, Z. Chen, and H. Peng, “Molecular dynamics study of neck growth in laser sintering of hollow silver nanoparticles with different heating rates,” Journal of Physics D: Applied Physics. 2013. link Times cited: 53 Abstract: Engineered hollow nanoparticles have exhibited their potenti… read moreAbstract: Engineered hollow nanoparticles have exhibited their potential in nanotechnology applications, but so far the investigation of the deformation mechanisms for these hollow particles during the sintering process has rarely been reported. Hence, a comparative study of both solid and hollow spherical silver nanoparticles with different sizes under different heating rates of laser sintering is conducted systematically in this paper, based on molecular dynamics simulations. An interesting phenomenon is observed where the temperature for fast neck growth shows an inverse trend in all the hollow nanoparticle pairs at an ultrahigh heating rate, which is quite different from that known in the solid particle cases. This finding implies that besides the size and heating rate, the nanoparticle geometry could also play an important role in the sintering process. At a low heating rate, the plastic deformation combined with structural reconfigurations induced by the lattice sliding in the hollow shells is found to be an important mechanism during the heating process. At an ultrahigh heating rate, the transition from fcc crystal directly to disordered structure from both outside and inside surfaces becomes more dominant than the structural reconfiguration with lattice defects, which is facilitated by the introduction of the inner free surfaces in hollow nanoparticles. The entire hollow particle pairs thus show an obvious tendency to coalesce and melt at a lower temperature level than with a low heating rate. read less NOT USED (high confidence) D. Lin, Y. Wang, S. Shang, Z. Lu, Z.-kui Liu, and X. Hui, “A new many-body potential with the second-moment approximation of tight-binding scheme for Hafnium,” Science China Physics, Mechanics and Astronomy. 2013. link Times cited: 1 NOT USED (high confidence) T. Senftle, R. Meyer, M. Janik, and A. V. van Duin, “Development of a ReaxFF potential for Pd∕O and application to palladium oxide formation.,” The Journal of chemical physics. 2013. link Times cited: 71 Abstract: Oxide formation on palladium surfaces impacts the activity a… read moreAbstract: Oxide formation on palladium surfaces impacts the activity and selectivity of Pd-based catalysts, which are widely employed under oxygen rich operating conditions. To investigate oxidation processes over Pd catalysts at time and length scales inaccessible to quantum based computational methods, we have developed a Pd∕O interaction potential for the ReaxFF reactive force field. The parameters of the ReaxFF potential were fit against an extensive set of quantum data for both bulk and surface properties. Using the resulting potential, we conducted molecular dynamics simulations of oxide formation on Pd(111), Pd(110), and Pd(100) surfaces. The results demonstrate good agreement with previous experimental observations; oxygen diffusion from the surface to the subsurface occurs faster on the Pd(110) surface than on the Pd(111) and Pd(100) surfaces under comparable conditions at high temperatures and pressures. Additionally, we developed a ReaxFF-based hybrid grand canonical Monte Carlo∕molecular dynamics (GC-MC∕MD) approach to assess the thermodynamic stability of oxide formations. This method is used to derive a theoretical phase diagram for the oxidation of Pd935 clusters in temperatures ranging from 300 K to 1300 K and oxygen pressures ranging from 10(-14) atm to 1 atm. We observe good agreement between experiment and ReaxFF, which validates the Pd∕O interaction potential and demonstrates the feasibility of the hybrid GC-MC∕MD method for deriving theoretical phase diagrams. This GC-MC∕MD method is novel to ReaxFF, and is well suited to studies of supported-metal-oxide catalysts, where the extent of oxidation in metal clusters can significantly influence catalytic activity, selectivity, and stability. read less NOT USED (high confidence) D. Lin, Y. Wang, S. Shang, Z. Lu, Z.-kui Liu, and X. Hui, “A new many-body potential with the second-moment approximation of tight-binding scheme for Hafnium,” Science China Physics, Mechanics and Astronomy. 2013. link Times cited: 0 NOT USED (high confidence) J. Michalka, P. McIntyre, and J. Gezelter, “Molecular Dynamics Simulations of the Surface Reconstructions of Pt(557) and Au(557) under Exposure to CO,” Journal of Physical Chemistry C. 2013. link Times cited: 5 Abstract: The mechanism and dynamics of surface reconstructions of Pt(… read moreAbstract: The mechanism and dynamics of surface reconstructions of Pt(557) and Au(557) exposed to various coverages of carbon monoxide (CO) were investigated using molecular dynamics simulations. Metal–CO interactions were parametrized from experimental data and plane-wave density functional theory (DFT) calculations. The large difference in binding strengths of the Pt–CO and Au–CO interactions was found to play a significant role in step-edge stability and adatom diffusion constants. Various mechanisms for CO-mediated step wandering and step doubling were investigated on the Pt(557) surface. We find that the energetics of CO adsorbed to the surface can explain the step-doubling reconstruction observed on Pt(557) and the lack of such a reconstruction on the Au(557) surface. However, more complicated reconstructions into triangular clusters that have been seen in recent experiments were not observed in these simulations. read less NOT USED (high confidence) N. Tsakiris and L. J. Lewis, “Phase diagram of aluminum from EAM potentials,” The European Physical Journal B. 2013. link Times cited: 2 NOT USED (high confidence) B. Eberhard and F. Haider, “Thorium in tungsten: construction of interatomic EAM potentials from ab initio data,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 0 Abstract: The interatomic interaction potential of tungsten and thoriu… read moreAbstract: The interatomic interaction potential of tungsten and thorium crystals and those of hypothetical tungsten and thorium alloys within the embedded atom approach are considered. The corresponding Ansatz functions are fitted against full potential linear augmented plane wave data of real tungsten- and thorium- and hypothetical tungsten-thorium-crystals. The result is interatomic potentials, ready for use within classical molecular dynamics schemes. A cross check of the resulting force scheme derived by comparison of ab initio and classical molecular dynamics data is provided. Furthermore, we used the potentials to calculate the phonon dispersion relations, which then serve as an additional check. read less NOT USED (high confidence) A. V. Fedorov and A. Shulgin, “Complex modeling of melting of an aluminum nanoparticle,” Combustion, Explosion, and Shock Waves. 2013. link Times cited: 1 NOT USED (high confidence) G. Rusina and E. Chulkov, “Phonons on the clean metal surfaces and in adsorption structures,” Russian Chemical Reviews. 2013. link Times cited: 15 Abstract: The state-of-the-art studies of the vibrational dynamics of … read moreAbstract: The state-of-the-art studies of the vibrational dynamics of clean metal surfaces and metal surface structures formed upon the sub-monolayer adsorption of the atoms of various elements are considered. A brief historical survey of the milestones of investigations of surface phonons is presented. The results of studies of the atomic structure and vibration characteristics of surfaces with low and high Miller indices and adsorption structures are analyzed. It is demonstrated that vicinal surfaces of FCC metals tend to exhibit specific vibrational modes located on the step and polarized along the step. Irrespective of the type and position of adsorption or the substrate structure, the phonon spectra of sub-monolayer adsorption structures always tend to display two modes for combined translational displacements of adatoms and for coupled vibrations of substrate atoms and adatoms polarized in the direction normal to the surface. The bibliography includes 202 references. read less NOT USED (high confidence) Y. Zheng, J. Chen, Q. Wu, and T. Tang, “Accelerating with 512-bit SIMD : A case study for molecular dynamics simulation on Intel’s Knights Corner,” 2013 Third International Conference on Communications and Information Technology (ICCIT). 2013. link Times cited: 1 Abstract: Multi/many-core design combined with wide vector extension h… read moreAbstract: Multi/many-core design combined with wide vector extension has become the mainstream of modern process architectures. Recently, Intel released Knights Corner, a many-core processor of Intel's Many Integrated Core (MIC) architecuture. Knights Corner comprises up to 62 cores, each supports 512-bit SIMD operation, that is, 8-way double precision floating-point vector operation. In this paper, to analyze the practical effect of the 512-bit SIMD extension, we port a molecular dynamics application onto the Knights Corner using SIMD intrinsics and then adopt optimizations such as loop unrolling and data prefetching. The experimental results demonstrate that our 512-bit SIMD implementation can achieve nearly ideal SIMD speedups (up to 7.69) over the non-SIMD version for the force computation task. read less NOT USED (high confidence) Y. Han, R. Li, Y. Ge, and J. Dong, “Growth of single-walled Ag and Cu nanotubes confined in carbon nanotubes, studied by molecular dynamics simulations,” Journal of Applied Physics. 2013. link Times cited: 7 Abstract: Growth of single-walled silver and copper nanotubes (Ag- and… read moreAbstract: Growth of single-walled silver and copper nanotubes (Ag- and Cu-SWNTs), confined in carbon nanotubes (CNTs), has been studied by using the classical molecular dynamics method. It is found that: (1) Four kinds of Ag-SWNTs, i.e., (3, 2), (4, 2), (4, 3), and (5, 3) ones, and five kinds of Cu-SWNTs, i.e., (3, 2), (4, 2), (4, 3), (4, 4), and (5, 3) ones, could be formed when the diameters of outside CNT containers are changed from 6.78 to 10.86 A. (2) The formation of the Ag- and Cu-SWNTs in confined CNTs is less sensitive to the CNTs' tube indices, but heavily influenced by the CNTs’ diameters. And the Ag- and Cu-SWNTs, formed in confined CNTs, are radially compressed, when the CNTs’ diameters are small. (3) The frequencies of the radial breathing modes of Ag- and Cu-SWNTs are approximately to vary linearly with the inverse tube diameters. read less NOT USED (high confidence) T. Rehman, M. Jaipal, A. Chatterjee, and A. Chatterjee, “A cluster expansion model for predicting activation barrier of atomic processes,” J. Comput. Phys. 2013. link Times cited: 26 NOT USED (high confidence) N. Artrith, B. Hiller, and J. Behler, “Neural network potentials for metals and oxides – First applications to copper clusters at zinc oxide,” physica status solidi (b). 2013. link Times cited: 107 Abstract: The development of reliable interatomic potentials for large… read moreAbstract: The development of reliable interatomic potentials for large‐scale molecular dynamics (MD) simulations of chemical processes at surfaces and interfaces is a formidable challenge because a wide range of atomic environments and very different types of bonding can be present. In recent years interatomic potentials based on artificial neural networks (NNs) have emerged offering an unbiased approach to the construction of potential energy surfaces (PESs) for systems that are difficult to describe by conventional potentials. Here, we review the basic properties of NN potentials and describe their construction for materials like metals and oxides. The accuracy and efficiency are demonstrated using copper and zinc oxide as benchmark systems. First results for a potential of the combined ternary CuZnO system aiming at the description of oxide‐supported copper clusters are reported. read less NOT USED (high confidence) H. Chen, Y. Yao, and S. Chen, “Adhesive contact between a graphene sheet and a nano-scale corrugated surface,” Journal of Physics D: Applied Physics. 2013. link Times cited: 22 Abstract: Adhesive contacts between graphene sheets and corrugated sur… read moreAbstract: Adhesive contacts between graphene sheets and corrugated surfaces are investigated. It is found that the final configuration between the graphene sheet and the substrate depends not only on the surface roughness of the substrate, but also on the length of graphene. A continuous transition, rather than a recent observation of ‘snap-through’ transition, is exhibited in our study. For a graphene sheet with a fixed length, it is easy to fully conform to the substrate of small roughness. Otherwise, the graphene sheet will remain flat on top of the corrugated substrate due to the unsatisfied bending energy or partially conform to the substrate due to the resistance of large interface friction. In order to reduce the effect of interface friction on the adhesive configuration, a new method, i.e. tilting the graphene sheet with a proper angle, is proposed. The tilting angle will significantly influence the final conformation of the adhesive interface. Some interesting types of behaviour are observed, such as rolling graphene, a double layer of graphene and fully adhesive contact, which is physically determined by the competition of thermal fluctuation and interfacial van der Waals interaction. read less NOT USED (high confidence) L. Zhang, H. Huang, H. Zhao, Z. Ma, Y. Yang, and X. Hu, “The evolution of machining-induced surface of single-crystal FCC copper via nanoindentation,” Nanoscale Research Letters. 2013. link Times cited: 19 NOT USED (high confidence) W. Song and S.-jin Zhao, “Development of the ReaxFF reactive force field for aluminum–molybdenum alloy,” Journal of Materials Research. 2013. link Times cited: 10 Abstract: We have developed a reactive force field within the ReaxFF f… read moreAbstract: We have developed a reactive force field within the ReaxFF framework to accurately describe reactions involving aluminum–molybdenum alloy, which are part parameters of Al–O–Mo ternary system metastable intermolecular composites. The parameters are optimized from a training set, whose data come from density functional theory (DFT) calculations and experimental value, such as heat of formation, geometry data, and equation of states, which are reproduced well by ReaxFF. Body-centered cubic molybdenum’s surface energy, vacancy formation, and two transformational paths, Bain and trigonal paths are calculated to validate the ReaxFF ability describing the defects and deformations. Some structures’ elastic constant and phonon are calculated by DFT and ReaxFF to predict the structures’ mechanics and kinetic stability. All those results indicate that the fitted parameters can describe the energy difference of various structures under various circumstances and generally represent the diffusion property but cannot reproduce the elasticity and phonon spectra so well. read less NOT USED (high confidence) M. Bhattacharya, A. Dutta, and P. Barat, “Quasistatic stick-slip of a dislocation core and the Frenkel-Kontorova chain,” Physical Review B. 2013. link Times cited: 1 Abstract: Department of Metallurgical and Materials Engineering,Jadavp… read moreAbstract: Department of Metallurgical and Materials Engineering,Jadavpur University, Kolkata 700032, India(Dated: April 23, 2013)By means of atomistic simulations, we demonstrate that a dislocation core exhibits intermittentquasistatic restructuring during incremental shear within the same Peierls valley. This can be re-garded as a stick-slip transition, which is also reproduced for a one-dimensional Frenkel-Kontorovachain under rigid boundary conditions. This occurs due to a discontinuous jump in an order pa-rameter of the system, which signi es the extent of region forbidden for the presence of particles inthe chain. The stick-slip phenomenon observed in the dislocation core is also shown to be reectedafter dimensionality reduction of the multidimensional atomic coordinates, which provides a basisfor comparison with the simple one-dimensional chain. read less NOT USED (high confidence) Z. Pei et al., “Ab initio and atomistic study of generalized stacking fault energies in Mg and Mg–Y alloys,” New Journal of Physics. 2013. link Times cited: 107 Abstract: Magnesium–yttrium alloys show significantly improved room te… read moreAbstract: Magnesium–yttrium alloys show significantly improved room temperature ductility when compared with pure Mg. We study this interesting phenomenon theoretically at the atomic scale employing quantum-mechanical (so-called ab initio) and atomistic modeling methods. Specifically, we have calculated generalized stacking fault energies for five slip systems in both elemental magnesium (Mg) and Mg–Y alloys using (i) density functional theory and (ii) a set of embedded-atom-method (EAM) potentials. These calculations predict that the addition of yttrium results in a reduction in the unstable stacking fault energy of basal slip systems. Specifically in the case of an I2 stacking fault, the predicted reduction of the stacking fault energy due to Y atoms was verified by experimental measurements. We find a similar reduction for the stable stacking fault energy of the non-basal slip system. On the other hand, other energies along this particular γ-surface profile increase with the addition of Y. In parallel to our quantum-mechanical calculations, we have also developed a new EAM Mg–Y potential and thoroughly tested its performance. The comparison of quantum-mechanical and atomistic results indicates that the new potential is suitable for future large-scale atomistic simulations. read less NOT USED (high confidence) J. Cai, Y. Wang, and Z.-wei Huang, “Effect of size on energy and elastic constants of Ni nanoparticles studied using the embedded-atom method,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 4 Abstract: Using the embedded-atom method potential, the energy and ela… read moreAbstract: Using the embedded-atom method potential, the energy and elastic constants of Ni nanoparticles are investigated as a function of size. It is found that a simple formula derived from the total energy can be used to explain the change characteristics of the size-dependent energy and C11 and C44 of Ni nanoparticles. The change characteristics of the size-dependent C12 are slightly different from the situation of C11 and C44 and they perhaps depend on its strain character when the nanoparticle is larger than ∼2.0 nm in size. It is also found that a transition occurs from a fast change of these size-dependent properties to a slow one. Such a transition occurs at ∼1.5 nm in size for the nanoparticles, i.e. the energy and elastic constants of Ni nanoparticles show approximately no change as long as their size is larger than 2 nm. It is also demonstrated that when the size of the nanoparticle is large enough (>20 nm) the calculated elastic constants and the cohesive energy are in agreement with those of their respective bulk counterpart. read less NOT USED (high confidence) D. Bang et al., “One-step electrochemical fabrication of vertically self-organized silver nanograss,” Journal of Materials Chemistry. 2013. link Times cited: 22 Abstract: Fabrication of one dimensional metal nanomaterials offers ma… read moreAbstract: Fabrication of one dimensional metal nanomaterials offers many beneficial aspects due to their unique size- and shape-dependent characteristics. However, facile fabrication of a robust one dimensional nanostructure has still remained a great challenge. Here, we developed a new synthetic route of one-step electrochemical deposition of silver nanograss without the assistance of a template. By applying an overpotential of −2.0 V (vs. Ag/AgCl) under aqueous alkaline conditions, silver nanograss with a slight tilt in a randomly oriented direction was spontaneously formed on the working electrode surface. Two applications that utilize advantageous features of this silver nanograss were demonstrated: (i) an efficient surface-enhanced Raman scattering substrate for a chemical sensor and (ii) an enzyme-less hydrogen peroxide sensor. Compared to silver nanowire arrays fabricated using anodized aluminum oxide (AAO) templates, the silver nanograss exhibited comparable hydrogen sensing due to its catalytic hydrogen peroxide reduction activity and produced a much stronger surface-enhanced Raman spectroscopy (SERS) signal due to its innate structure. read less NOT USED (high confidence) Y. Gafner, Z. V. Goloven’ko, and S. Gafner, “Formation of the structure of gold nanoclusters during crystallization,” Journal of Experimental and Theoretical Physics. 2013. link Times cited: 8 NOT USED (high confidence) Y. Dong, Q. Li, and A. Martini, “Molecular dynamics simulation of atomic friction: A review and guide,” Journal of Vacuum Science and Technology. 2013. link Times cited: 158 Abstract: This paper reviews recent progress in molecular dynamics sim… read moreAbstract: This paper reviews recent progress in molecular dynamics simulation of atomic-scale friction measured by an atomic force microscopy. Each section of the review focuses on an individual condition or parameter that affects atomic friction including materials, surfaces, compliance, contact area, normal load, temperature, and velocity. The role each parameter plays is described in the context of both experimental measurements and simulation predictions. In addition, the discussion includes an overview of the research community's current understanding of observed effects, guidelines for implementation of those effects in an atomistic simulation, and suggestions for future research to address open questions. Taken together, this review conveys the message that friction at the atomic scale is affected by many interrelated parameters and that the use of molecular dynamics simulation as a predictive tool can be accomplished only through careful model design. read less NOT USED (high confidence) D. Savio, N. Fillot, and P. Vergne, “A Molecular Dynamics Study of the Transition from Ultra-Thin Film Lubrication Toward Local Film Breakdown,” Tribology Letters. 2013. link Times cited: 37 NOT USED (high confidence) H. Su and Q. Tang, “MD simulations of loading rate dependence of detwinning deformation in nanocrystalline Ni,” Science China Physics, Mechanics and Astronomy. 2013. link Times cited: 5 NOT USED (high confidence) H. Su and Q. Tang, “MD simulations of loading rate dependence of detwinning deformation in nanocrystalline Ni,” Science China Physics, Mechanics and Astronomy. 2013. link Times cited: 0 NOT USED (high confidence) C. Sun et al., “In situ Evidence of Defect Cluster Absorption by Grain Boundaries in Kr Ion Irradiated Nanocrystalline Ni,” Metallurgical and Materials Transactions A. 2013. link Times cited: 98 NOT USED (high confidence) W. Hoover and C. G. Hoover, “Time-Symmetry Breaking in Hamiltonian Mechanics,” arXiv: Statistical Mechanics. 2013. link Times cited: 14 Abstract: Hamiltonian trajectories are strictly time-reversible. Any t… read moreAbstract: Hamiltonian trajectories are strictly time-reversible. Any time series of Hamiltonian coordinates {q} satisfying Hamilton's motion equations will likewise satisfy them when played "backwards", with the corresponding momenta changing signs : {+p} --> {-p}. Here we adopt Levesque and Verlet's precisely bit-reversible motion algorithm to ensure that the trajectory reversibility is exact, with the forward and backward sets of coordinates identical. Nevertheless, the associated instantaneous Lyapunov instability, or "sensitive dependence on initial conditions" of "chaotic" (or "Lyapunov unstable") bit-reversible coordinate trajectories can still exhibit an exponentially growing time-symmetry-breaking irreversibility. Surprisingly, the positive and negative exponents, as well as the forward and backward Lyapunov spectra, are usually not closely related, and so give four differing topological measures of "local" chaos. We have demonstrated this symmetry breaking for fluid shockwaves, for free expansions, and for chaotic molecular collisions. Here we illustrate and discuss this time-symmetry breaking for three statistical-mechanical systems, [1] a minimal (but still chaotic) one-body "cell model" with a four-dimensional phase space; [2] relatively small colliding crystallites, for which the whole Lyapunov spectrum is accessible; [3] a near-continuum inelastic collision of two larger 400-particle balls. In the last two of these pedagogical problems the two colliding bodies coalesce. The particles most prone to Lyapunov instability are dramatically different in the two time directions. Thus this Lyapunov-based symmetry breaking furnishes an interesting Arrow of Time. read less NOT USED (high confidence) J. Zhong, R. Shakiba, and J. B. Adams, “Molecular dynamics simulation of severe adhesive wear on a rough aluminum substrate,” Journal of Physics D: Applied Physics. 2013. link Times cited: 41 Abstract: Severe adhesive wear on a rough aluminum (Al) substrate is s… read moreAbstract: Severe adhesive wear on a rough aluminum (Al) substrate is simulated by a hard Lennard-Jones asperity impacting an Al-asperity at high speeds using molecular dynamics (MD). Multiple simulations investigate the effects of variations in the inter-asperity bonding, the geometric overlap between two asperities, the relative impact velocity and the starting temperature. The effect of these experimental variables on degree of adhesive wear and the temperature profiles are discussed, and a design of experiments method is used to help interpret the results. The results indicate that increasing the inter-asperity bonding, the geometric overlap and the starting temperature of two asperities will substantially increase the wear rate, while raising the impact velocity slightly decreases the wear rate. It is observed that the deformation mechanism involves local melting and the formation of a liquid like layer in the contact area between two asperities, and the amorphous deformation of the Al-asperity. read less NOT USED (high confidence) K. Yoshida, X. Zhang, A. Bright, K. Saitoh, and N. Tanaka, “Dynamic environmental transmission electron microscopy observation of platinum electrode catalyst deactivation in a proton-exchange-membrane fuel cell,” Nanotechnology. 2013. link Times cited: 28 Abstract: Spherical-aberration-corrected environmental transmission el… read moreAbstract: Spherical-aberration-corrected environmental transmission electron microscopy (AC-ETEM) was applied to study the catalytic activity of platinum/amorphous carbon electrode catalysts in proton-exchange-membrane fuel cells (PEMFCs). These electrode catalysts were characterized in different atmospheres, such as hydrogen and air, and a conventional high vacuum of 10−5 Pa. A high-speed charge coupled device camera was used to capture real-time movies to dynamically study the diffusion and reconstruction of nanoparticles with an information transfer down to 0.1 nm, a time resolution below 0.2 s and an acceleration voltage of 300 kV. With such high spatial and time resolution, AC-ETEM permits the visualization of surface-atom behaviour that dominates the coalescence and surface-reconstruction processes of the nanoparticles. To contribute to the development of robust PEMFC platinum/amorphous carbon electrode catalysts, the change in the specific surface area of platinum particles was evaluated in hydrogen and air atmospheres. The deactivation of such catalysts during cycle operation is a serious problem that must be resolved for the practical use of PEMFCs in real vehicles. In this paper, the mechanism for the deactivation of platinum/amorphous carbon electrode catalysts is discussed using the decay rate of the specific surface area of platinum particles, measured first in a vacuum and then in hydrogen and air atmospheres for comparison. read less NOT USED (high confidence) Z. Xiong and W. Wu, “Controllable Growth of Ni Nanocrystals Embedded in BaTiO 3 /SrTiO 3 Superlattices.” 2013. link Times cited: 0 Abstract: BaTiO3/SrTiO3 superlattices with embedded Ni nanocrystals (N… read moreAbstract: BaTiO3/SrTiO3 superlattices with embedded Ni nanocrystals (NCs) have been grown on SrTiO3 (001) substrate using laser molecular beam epitaxy (L-MBE). In situ reflection high-energy electron diffraction (RHEED) was employed to investigate the process of lattice strain in the self-organization of Ni NCs and the epitaxial growth of BaTiO3/SrTiO3 superlattices. The results indicated that the strain from large lattice mismatch drove the self-organization of Ni NCs. Also, the layer-by-layer growth of BaTiO3/SrTiO3 superlattices Keywords: Nanocrystal; Superlattices; Self-organization 1. Introduction Oxide artificial superlattices, especially (001) oriented BaTiOsuperlattices and the island growth of Ni NCs were controllable ac-curately. The fine alternation of the two processes would provide a possible route to engineer controllably the nano-composite microstructure. read less NOT USED (high confidence) V. Bocchetti and H. Diep, “Monte Carlo simulation of melting and lattice relaxation of the (111) surface of silver,” Surface Science. 2013. link Times cited: 13 NOT USED (high confidence) H. Kwak, Y. Shin, A. V. van Duin, and A. Vasenkov, “Ab initio based multiscale modeling of alloy surface segregation,” Journal of Physics: Condensed Matter. 2012. link Times cited: 8 Abstract: A fully integrated ab initio based multiscale model for anal… read moreAbstract: A fully integrated ab initio based multiscale model for analysis of segregation at alloy surfaces is presented. Major components of the model include a structure-energy analysis from the first-principles density functional theory (DFT), a Monte Carlo/molecular dynamics (MC/MD) hybrid simulation scheme for atomic transport, and a reactive force field formalism that binds the two. The multiscale model accurately describes the atomic transport processes in a multi-component alloy system at finite temperature, and is capable of providing quantitative predictions for surface compositions. The validity of the model was demonstrated by investigating the temperature-dependent segregation behavior of B2 FeAl binary alloy surfaces with a detailed description of the segregation mechanism. Based on the model’s prediction capabilities, potential extension of the model to the analysis of systems undergoing rapid chemical reactions is discussed. read less NOT USED (high confidence) M. Mendelev, M. Kramer, S. Hao, K. Ho, and C. Z. Wang, “Development of interatomic potentials appropriate for simulation of liquid and glass properties of NiZr2 alloy,” Philosophical Magazine. 2012. link Times cited: 116 Abstract: A new interatomic potential for the Ni–Zr system is presente… read moreAbstract: A new interatomic potential for the Ni–Zr system is presented. This potential was developed specifically to match experimental scattering data from Ni, Zr and NiZr2 liquids. Both ab initio and published thermodynamic data were used to optimise the potential to study the liquid and amorphous structure of the NiZr2 alloy. This potential has the C 16 phase, being more stable than C 11b phase in the NiZr2 alloy, consistent with experiments. The potential leads to the correct glass structure in the molecular dynamics simulation and, therefore, can be used to study the liquid–glass transformation in the NiZr2 alloy. read less NOT USED (high confidence) M. Imran, F. Hussain, M. Rashid, and S. Ahmad, “Molecular dynamics study of the mechanical characteristics of Ni/Cu bilayer using nanoindentation,” Chinese Physics B. 2012. link Times cited: 23 Abstract: In the present work, a three-dimensional molecular dynamics … read moreAbstract: In the present work, a three-dimensional molecular dynamics simulation is carried out to perform the nanoindentation experiment on Ni single crystal. The substrate indenter system is modeled using hybrid interatomic potentials including the many-body potential embedded atom method (EAM), and two-body morse potential. To simulate the indentation process, a spherical indenter (diameter = 80 Å, 1 Å=0.1 nm) is chosen. The results show that the mechanical behaviour of a monolithic Ni is not affected by crystalline orientation. To elucidate the effect of a heterogeneous interface, three bilayer interface systems are constructed, namely Ni(100)/Cu(111), Ni(110)/Cu(111), and Ni(111)/Cu(111). The simulations along these systems clearly describe that mechanical behaviour directly depends on the lattice mismatch. The interface with the smaller mismatch between the specified crystal planes is proved to be harder and vice versa. To describe the relationship between film thickness and interface effect, we choose various values of film thickness ranging from 20 Å to 50 Å to perform the nanoindentation experiment. It is observed that the interface is significant only for the relatively small thickness of film and the separation between interface and the indenter tip. It is shown that with the increase in film thickness, the mechanical behaviour of the film shifts more toward that of monolithic material. read less NOT USED (high confidence) J. Titantah and M. Karttunen, “Multiphase density functional theory parameterization of the interatomic potential for silver and gold,” The European Physical Journal B. 2012. link Times cited: 8 NOT USED (high confidence) H. Gong, W. Lu, L. Wang, and G. Li, “Cluster size and substrate temperature affecting thin film formation during copper cluster deposition on a Si (001) surface,” Chinese Physics B. 2012. link Times cited: 4 Abstract: The soft deposition of Cu clusters on a Si (001) surface was… read moreAbstract: The soft deposition of Cu clusters on a Si (001) surface was studied by molecular dynamics simulations. The embedded atom method, the Stillinger—Weber and the Lennar—Jones potentials were used to describe the interactions between the cluster atoms, between the substrate atoms, and between the cluster and the substrate atoms, respectively. The Cu13, Cu55, and Cu147 clusters were investigated at different substrate temperatures. We found that the substrate temperature had a significant effect on the Cu147 cluster. For smaller Cu13 and Cu55 clusters, the substrate temperature in the range of study appeared to have little effect on the mean center-of-mass height. The clusters showed better degrees of epitaxy at 800 K. With the same substrate temperature, the Cu55 cluster demonstrated the highest degree of epitaxy, followed by Cu147 and then Cu13 clusters. In addition, the Cu55 cluster showed the lowest mean center-of-mass height. These results suggested that the Cu55 cluster is a better choice for the thin-film formation among the clusters considered. Our studies may provide insight into the formation of desired Cu thin films on a Si substrate. read less NOT USED (high confidence) C. Henager, F. Gao, S. Hu, G. Lin, E. Bylaska, and N. Zabaras, “Simulating Interface Growth and Defect Generation in CZT – Simulation State of the Art and Known Gaps.” 2012. link Times cited: 1 Abstract: This one-year, study topic project will survey and investiga… read moreAbstract: This one-year, study topic project will survey and investigate the known state-of-the-art of modeling and simulation methods suitable for performing fine-scale, fully 3-D modeling, of the growth of CZT crystals at the melt-solid interface, and correlating physical growth and post-growth conditions with generation and incorporation of defects into the solid CZT crystal. In the course of this study, this project will also identify the critical gaps in our knowledge of modeling and simulation techniques in terms of what would be needed to be developed in order to perform accurate physical simulations of defect generation in melt-grown CZT. The transformational nature of this study will be, for the first time, an investigation of modeling and simulation methods for describing microstructural evolution during crystal growth and the identification of the critical gaps in our knowledge of such methods, which is recognized as having tremendous scientific impacts for future model developments in a wide variety of materials science areas. read less NOT USED (high confidence) M. Imran, F. Hussain, M. Rashid, and S. Ahmad, “Dynamic characteristics of nanoindentation in Ni: A molecular dynamics simulation study,” Chinese Physics B. 2012. link Times cited: 28 Abstract: In this work, three-dimensional molecular dynamics simulatio… read moreAbstract: In this work, three-dimensional molecular dynamics simulation is carried out to elucidate the nanoindentation behaviour of single crystal Ni. The substrate indenter system is modelled using hybrid interatomic potentials including the manybody potential (embedded atom method) and two-body Morse potential. The spherical indenter is chosen, and the simulation is performed for different loading rates from 10 m/s to 200 m/s. Results show that the maximum indentation load and hardness of the system increase with the increase of velocity. The effect of indenter size on the nanoindentation response is also analysed. It is found that the maximum indentation load is higher for the large indenter whereas the hardness is higher for the smaller indenter. Dynamic nanoindentation is carried out to investigate the behaviour of Ni substrate to multiple loading-unloading cycles. It is observed from the results that the increase in the number of loading unloading cycles reduces the maximum load and hardness of the Ni substrate. This is attributed to the decrease in recovery force due to defects and dislocations produced after each indentation cycle. read less NOT USED (high confidence) M. Hennes, J. Buchwald, and S. G. Mayr, “Structural properties of spherical Cu/Ni nanoparticles,” CrystEngComm. 2012. link Times cited: 6 Abstract: While Cu/Ni bulk alloys—partially in the presence of flat su… read moreAbstract: While Cu/Ni bulk alloys—partially in the presence of flat surfaces—have been extensively studied, less is known about the properties of Cu/Ni nanoparticles. In the present study we employ a combined Molecular-Dynamics/Metropolis-Monte-Carlo (MD/MMC) simulation approach to analyze equilibrium segregation profiles in Cu/Ni clusters. Special emphasis is put on the relative stability of different segregation patterns and the feasibility of technically meaningful core-shell (CS) nanocrystals. We show that, although the bulk system exhibits a miscibility gap below 630 K, spherical symmetric CS nanoparticles will not form even at low temperatures, where the clusters rather adopt a highly ordered state with janus-like core structure and a Cu surface segregation monolayer atop of Ni, thereby minimizing atomic level stresses. Our simulations, performed for various particle sizes, temperatures and cross checked by using different semiempirical potentials challenge existing theoretical models and shed new light on the energy landscape of this nanoscaled system. read less NOT USED (high confidence) J.-W. Jiang, A. Leach, K. Gall, H. S. Park, and T. Rabczuk, “A surface stacking fault energy approach to predicting defect nucleation in surface-dominated nanostructures,” Journal of The Mechanics and Physics of Solids. 2012. link Times cited: 30 NOT USED (high confidence) A. Ponrouch, S. Garbarino, E. Bertin, C. Andrei, G. Botton, and D. Guay, “Highly Porous and Preferentially Oriented 100 Platinum Nanowires and Thin Films,” Advanced Functional Materials. 2012. link Times cited: 50 Abstract: Highly {100} oriented Pt deposits were prepared by electrode… read moreAbstract: Highly {100} oriented Pt deposits were prepared by electrodeposition from a 10 mM HCl, 100 mM KCl and Na2PtCl6.xH2O electrolyte. The deposits were prepared in the form of thin films and array of nanowires. A qualitative assessment of the proportion of {100} oriented Pt surfaces was obtained through X‐ray diffraction measurements and cyclic voltammetry in 0.5 M H2SO4. The effect of the deposition potential, Edep, temperature of the electrolyte, Tdep, platinum salt concentration [Na2PtCl6.xH2O], and nature of the substrate were investigated. It was shown that the proportion of {100} oriented Pt surfaces reaches a maximum for Edep = ‐0.35 V vs SCE. Moreover, this proportion increases steadily as Tdep and [Na2PtCl6.xH2O] are decreased from 75 to 25 °C and from 2.5 to 0.25 mM, respectively. Scanning electron microscopy and high‐resolution transmission electron microscopy micrographs indicate that the more oriented samples are made of pine tree‐like structures that are effectively single crystals, and that the growth facets appear to be close to the {001} plane. This observation also clearly indicates that the plane exposed during the CV experiment is also {001}. As suggested by these micrographs, the films and nanowires are highly porous and roughness factors as large as 1000 were obtained on highly {100} oriented Pt nanowires. The predominance of {100} facets is attributed to their energetically favoured growth in the presence of hydrogen, and is shown to be significantly enhanced when the mass transport of Pt4+ is limited. Due to the predominance of {100} facets, the normalized electrocatalytic activity (μA cm−2Pt) for the electro‐oxidation of hydrazine and ammonia is higher than non‐oriented polycrystalline Pt by a factor of 4 and 2.7, respectively. read less NOT USED (high confidence) Y. Yuan et al., “The effects of electropulsing on the recrystallization behavior of rolled pure tungsten,” Journal of Materials Research. 2012. link Times cited: 19 Abstract: Electropulsing treatment (EPT) has been first applied to the… read moreAbstract: Electropulsing treatment (EPT) has been first applied to the recrystallization of a refractory metal—tungsten (W). We have three major observations: (i) the recrystallization temperature of a rolled pure W under EPT is ∼900 K higher than its conventional recrystallization temperature (1603 K); (ii) the time required for recrystallization is significantly reduced compared with that of conventional heat treatment (CHT); (iii) the recrystallized grains are also much finer than the ones under CHT. Based on quantitative analysis, we conclude that the huge increase of the recrystallization temperature of the rolled pure W under EPT is due to the high heating rate generated by EPT and high activation energy for vacancy diffusion of W, and the accelerated recrystallization and grain refinement have resulted from the coupling of thermal and electromigration effects of EPT at relatively high temperatures. read less NOT USED (high confidence) M. Backman, N. Juslin, and K. Nordlund, “Bond order potential for gold,” The European Physical Journal B. 2012. link Times cited: 11 NOT USED (high confidence) T. Yokoyama, “Path Integral Effective Classical Potential Method Applied to Anharmonicity and Quantum Effects in Thermal Expansion of Invar Alloy,” E-journal of Surface Science and Nanotechnology. 2012. link Times cited: 2 Abstract: Extended X-ray Absorption Fine Structure (EXAFS) spectroscop… read moreAbstract: Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy is a powerful experimental technique to investigate anharmonic vibrational properties of solids especially when one combines experimental EXAFS with quantum mechanical theoretical evaluations. In this article, the path-integral effective-classical-potential (PIECP) theory is applied to temperature dependence of EXAFS in a real system. The anharmonicity and quantum effects in the Invar alloy Fe64.6Ni35.4 that shows anomalously small thermal expansion are investigated. Experimental Fe and Ni K-edge EXAFS measurements and the computational PIECP simulations have been performed. It is experimentally revealed that the first nearest-neighbor (NN) shells around Fe show almost no thermal expansion, while those around Ni exhibit meaningful but smaller expansion than that of fcc Ni. At low temperature (<100 K), the vibrational quantum effect is found to play an essentially important role, which is confirmed by comparing the quantum mechanical simulations to the classical ones, the latter of which exhibit large (normal) thermal expansion at low temperature. It is also clarified that thermal expansion for the Ni-Ni and Ni-Fe pairs is noticeably suppressed, even though the Ni electronic state may not vary depending on the temperature. On the other hand, the anharmonicity (asymmetric distribution) clearly exist for all the first-NN shells as in the case of the normal thermal expansion system, where thermal expansion originates almost exclusively from the anharmonic interatomic potential. [DOI: 10.1380/ejssnt.2012.486] read less NOT USED (high confidence) H. Zhan, Y. T. Gu, and H. S. Park, “Beat phenomena in metal nanowires, and their implications for resonance-based elastic property measurements.,” Nanoscale. 2012. link Times cited: 32 Abstract: The elastic properties of 1D nanostructures such as nanowire… read moreAbstract: The elastic properties of 1D nanostructures such as nanowires are often measured experimentally through actuation of nanowires at their resonance frequency, and then relating the resonance frequency to the elastic stiffness using the elementary beam theory. In the present work, we utilize large scale molecular dynamics simulations to report a novel beat phenomenon in [110] oriented Ag nanowires. The beat phenomenon is found to arise from the asymmetry of the lattice spacing in the orthogonal elementary directions of [110] nanowires, i.e. the [110] and [001] directions, which results in two different principal moments of inertia. Because of this, actuations imposed along any other direction are found to decompose into two orthogonal vibrational components based on the actuation angle relative to these two elementary directions, with this phenomenon being generalizable to <110> FCC nanowires of different materials (Cu, Au, Ni, Pd and Pt). The beat phenomenon is explained using a discrete moment of inertia model based on the hard sphere assumption; the model is utilized to show that surface effects enhance the beat phenomenon, while effects are reduced with increasing nanowire cross-sectional size or aspect ratio. Most importantly, due to the existence of the beat phenomena, we demonstrate that in resonance experiments only a single frequency component is expected to be observed, particularly when the damping ratio is relatively large or very small. Furthermore, for a large range of actuation angles, the lower frequency is more likely to be detected than the higher one, which implies that experimental predictions of the Young's modulus obtained from resonance may in fact be under-predictions. The present study therefore has significant implications for experimental interpretations of the Young's modulus as obtained via resonance testing. read less NOT USED (high confidence) T. Stone and M. Horstemeyer, “Length scale effects of friction in particle compaction using atomistic simulations and a friction scaling model,” Journal of Nanoparticle Research. 2012. link Times cited: 3 NOT USED (high confidence) C. Binns et al., “Preparation of hydrosol suspensions of elemental and core–shell nanoparticles by co-deposition with water vapour from the gas-phase in ultra-high vacuum conditions,” Journal of Nanoparticle Research. 2012. link Times cited: 35 NOT USED (high confidence) H. Gong, W. Lu, L. Wang, G. Li, and S. Zhang, “The effects of substrate size and temperature on the deposition of Cu clusters on a Si substrate,” Journal of Applied Physics. 2012. link Times cited: 8 Abstract: The deposition of a Cu13 cluster on a Si (001) surface was s… read moreAbstract: The deposition of a Cu13 cluster on a Si (001) surface was studied by molecular dynamics simulations. Embedded atom method, Stillinger-Weber, and Lennar-Jones potentials were used to describe the interaction between cluster atoms, substrate atoms, and the cluster-substrate interaction. Quantitative characteristic parameters, such as kinetic energy of the cluster and the substrate, the degree of epitaxy, and the mean height of mass center of the cluster, were calculated to study the effect of substrate size and substrate temperature on cluster deposition. The substrate temperature was found to affect the degree of epitaxy at different substrate sizes. When the size ratio of the substrate and cluster is relatively small or large, the epitaxial degree was higher at 800 K than at 300 K. If the size of the substrate matches that of the cluster, the substrate temperature appeared to have minimum effect. For a given temperature, the substrate size was found to have no obvious effect on the degree of epitaxy or t... read less NOT USED (high confidence) J. Wu, L. Qi, H. You, A. Gross, J. Li, and H. Yang, “Icosahedral platinum alloy nanocrystals with enhanced electrocatalytic activities.,” Journal of the American Chemical Society. 2012. link Times cited: 452 Abstract: This communication describes the synthesis of Pt-M (M = Au, … read moreAbstract: This communication describes the synthesis of Pt-M (M = Au, Ni, Pd) icosahedral nanocrystals based on the gas reducing agent in liquid solution method. Both CO gas and organic surface capping agents play critical roles in stabilizing the icosahedral shape with {111} surfaces. Among the Pt-M alloy icosahedral nanocrystals generated, Pt(3)Ni had an impressive ORR specific activity of 1.83 mA/cm(2)(Pt) and 0.62 A/mg(Pt). Our results further show that the area-specific activity of icosahedral Pt(3)Ni catalysts was about 50% higher than that of the octahedral Pt(3)Ni catalysts (1.26 mA/cm(2)(Pt)), even though both shapes are bound by {111} facets. Density functional theory calculations and molecular dynamics simulations indicate that this improvement may arise from strain-induced electronic effects. read less NOT USED (high confidence) M. Lagos, P. A. Autreto, D. Galvão, and D. Ugarte, “Correlation between quantum conductance and atomic arrangement of atomic-size silver nanowires.” 2012. link Times cited: 11 Abstract: We have studied the effect of thermal effects on the structu… read moreAbstract: We have studied the effect of thermal effects on the structural and transport response of Ag atomic-size nanowires (NWs) generated by mechanical elongation. Our study involves both time-resolved atomic resolution transmission electron microscopy imaging and quantum conductance measurement using an ultra-high-vacuum mechanically controllable break junction. We have observed drastic changes in conductance and structural properties of Ag nanowires generated at different temperatures (150 and 300 K). By combining electron microscopy images, electronic transport measurements, and quantum transport calculations, we have been able to obtain a consistent correlation between the conductance and structural properties of Ag NWs. In particular, our study has revealed the formation of metastable rectangular rod-like Ag wire (3/3) along the [001] crystallographic direction, whose formation is enhanced. These results illustrate the high complexity of analyzing structural and quantum conductance behaviour of metal atomic... read less NOT USED (high confidence) R. Hong and J.-Y. Yang, “Molecular Dynamics Study on Enhanced Cu Coverage of Trench Filling with Low-Index Ta Surfaces,” Japanese Journal of Applied Physics. 2012. link Times cited: 0 Abstract: The Cu coverage of trench filling enhanced by different low-… read moreAbstract: The Cu coverage of trench filling enhanced by different low-index surfaces of tantalum in physical vapor deposition is studied by molecular dynamics simulation with the embedded atom method (EAM) as the interaction potential for the present alloy metal system. The deposition morphologies and bottom step coverage enhancement of trenches with three different aspect ratios are examined. It is found that the Cu adatom on Ta(110) with uniform and low surface diffusion barrier energy and that on Ta(111) with high surface energy lead to the improvement of the surface diffusion of Cu adatoms. The shadowing effect is inhibited on Ta(110) and Ta(111) such that the bottom step coverage of the trench is enhanced markedly at an early stage and the final coverage of trench filling is improved significantly. Also, the texture of deposition on the trench with Ta(110) has a uniform structure owing to the low surface energy, while that with Ta(111) has a nonuniform structure owing to the high surface energy on the sidewall. read less NOT USED (high confidence) D. Belashchenko, “Electron contribution to energy of alkali metals in the scheme of an embedded atom model,” High Temperature. 2012. link Times cited: 13 NOT USED (high confidence) H. Hou, R. Wang, J. Wang, X. Liu, G. Chen, and P. Huang, “An analytic bond-order potential for the Fe–Cu system,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 5 Abstract: An angular-dependent analytic bond-order potential (ABOP) fo… read moreAbstract: An angular-dependent analytic bond-order potential (ABOP) for copper and Fe–Cu system was developed, based on the ABOP of pure iron introduced by Müller et al (2007 J. Phys.: Condens. Matter 19 326220). The potential parameters for the present ABOP model of copper were determined by fitting to the experimental data of the basic properties of fcc Cu and ab initio calculated properties of bcc Cu. The model predicts the vacancy formation energy in good agreement with the experimental result, although no vacancy formation information was used in the fitting of the model parameters. The melting point of Cu is also properly reproduced. The Fe–Cu binary system was described by adding two independent cross parameters in the potential model. The cross parameters were fitted using the ab initio data of the formation energies and lattice parameters of fictitious Fe–Cu alloys. The potential was applied to investigate the point defects and small defect clusters in dilute Fe–Cu alloys. The results were compared with the ab initio data and the values obtained with other potentials. read less NOT USED (high confidence) Y. V. Suleimanov, “Surface Diffusion of Hydrogen on Ni(100) from Ring Polymer Molecular Dynamics,” Journal of Physical Chemistry C. 2012. link Times cited: 69 Abstract: In this paper, we present a detailed ring polymer molecular … read moreAbstract: In this paper, we present a detailed ring polymer molecular dynamics (RPMD) study of the diffusion of hydrogen on Ni(100) using the well-established embedded atom method (EAM) interaction potential. We pay particular attention to the effects of lattice motion, transition state recrossing, and multiple hops. We show that all these effects can be assessed within a unified theoretical framework using RPMD. First, we study the low-temperature regime where the diffusion coefficient can be calculated by the random walk model. The crossover from thermally activated diffusion to almost temperature-independent quantum diffusion is found at around 70 K, in agreement with earlier quantum instanton calculations. We show that the recrossings of the transition state dividing surface become significant only below the crossover temperature in our RPMD calculations. The lattice motion slightly increases the diffusion coefficient above and slightly decreases it below the crossover temperature. We also show that quantizing ... read less NOT USED (high confidence) C. Taylor, “Atomistic Modeling of Corrosion Events at the Interface between a Metal and Its Environment,” International Journal of Corrosion. 2012. link Times cited: 39 Abstract: Atomistic simulation is a powerful tool for probing the stru… read moreAbstract: Atomistic simulation is a powerful tool for probing the structure and properties of materials and the nature of chemical reactions. Corrosion is a complex process that involves chemical reactions occurring at the interface between a material and its environment and is, therefore, highly suited to study by atomistic modeling techniques. In this paper, the complex nature of corrosion processes and mechanisms is briefly reviewed. Various atomistic methods for exploring corrosion mechanisms are then described, and recent applications in the literature surveyed. Several instances of the application of atomistic modeling to corrosion science are then reviewed in detail, including studies of the metal-water interface, the reaction of water on electrified metallic interfaces, the dissolution of metal atoms from metallic surfaces, and the role of competitive adsorption in controlling the chemical nature and structure of a metallic surface. Some perspectives are then given concerning the future of atomistic modeling in the field of corrosion science. read less NOT USED (high confidence) X.-J. Yuan, N. Chen, and J. Shen, “Construction of embedded-atom-method interatomic potentials for alkaline metals (Li, Na, and K) by lattice inversion,” Chinese Physics B. 2012. link Times cited: 1 Abstract: The lattice-inversion embedded-atom-method interatomic poten… read moreAbstract: The lattice-inversion embedded-atom-method interatomic potential developed previously by us is extended to alkaline metals including Li, Na, and K. It is found that considering interatomic interactions between neighboring atoms of an appropriate distance is a matter of great significance in constructing accurate embedded-atom-method interatomic potentials, especially for the prediction of surface energy. The lattice-inversion embedded-atom-method interatomic potentials for Li, Na, and K are successfully constructed by taking the fourth-neighbor atoms into consideration. These angular-independent potentials markedly promote the accuracy of predicted surface energies, which agree well with experimental results. In addition, the predicted structural stability, elastic constants, formation and migration energies of vacancy, and activation energy of vacancy diffusion are in good agreement with available experimental data and first-principles calculations, and the equilibrium condition is satisfied. read less NOT USED (high confidence) S. I. Shah, G. Nandipati, A. Kara, and T. Rahman, “Extended pattern recognition scheme for self-learning kinetic Monte Carlo simulations,” Journal of Physics: Condensed Matter. 2012. link Times cited: 11 Abstract: We report the development of a pattern recognition scheme th… read moreAbstract: We report the development of a pattern recognition scheme that takes into account both fcc and hcp adsorption sites in performing self-learning kinetic Monte Carlo (SLKMC-II) simulations on the fcc(111) surface. In this scheme, the local environment of every under-coordinated atom in an island is uniquely identified by grouping fcc sites, hcp sites and top-layer substrate atoms around it into hexagonal rings. As the simulation progresses, all possible processes, including those such as shearing, reptation and concerted gliding, which may involve fcc–fcc, hcp–hcp and fcc–hcp moves are automatically found, and their energetics calculated on the fly. In this article we present the results of applying this new pattern recognition scheme to the self-diffusion of 9-atom islands (M9) on M(111), where M = Cu, Ag or Ni. read less NOT USED (high confidence) Q. Tang, “Effect of size on mechanical behavior of Au pillars by molecular dynamics study,” Science China Physics, Mechanics and Astronomy. 2012. link Times cited: 11 NOT USED (high confidence) Q. Tang, “Effect of size on mechanical behavior of Au pillars by molecular dynamics study,” Science China Physics, Mechanics and Astronomy. 2012. link Times cited: 0 NOT USED (high confidence) A. Oluwajobi, “Molecular Dynamics Simulation of Nanoscale Machining.” 2012. link Times cited: 6 Abstract: Product miniaturization is a major motivation for the develo… read moreAbstract: Product miniaturization is a major motivation for the development of ultra-precision technologies and processes which can achieve high form and excellent surface finish. Of all the available manufacturing approaches, mechanical nanometric machining is still a good option for machining complex 3D devices in a controllable way (Jackson, 2008). As the dimension goes down to the nanoscale, the machining phenomena take place in a limited region of tool-workpiece interface. At this length scale and interface, the material removal mechanisms are not fully understood, so more insight is needed, which on the long run will help to achieve high precision manufacturing with predictability, repeatability and productivity (Luo, 2004). At present, it is very difficult to observe the diverse microscopic physical phenomena occurring through experiments at the nanoscale (Rentsch, 2008). Subsequently, the other alternative is to explore available simulation techniques. Continuum mechanics approach is not adequate, as the point of interest/interface cannot be assumed to be homogeneous, but rather discrete, so, atomistic simulation methods are the suitable techniques for modelling at the nanoscale. read less NOT USED (high confidence) D. Belashchenko, “Computer simulation of the properties of liquid metals: Gallium, lead, and bismuth,” Russian Journal of Physical Chemistry A. 2012. link Times cited: 22 NOT USED (high confidence) M. He and S. Li, “An embedded atom hyperelastic constitutive model and multiscale cohesive finite element method,” Computational Mechanics. 2012. link Times cited: 30 NOT USED (high confidence) K. Yun et al., “Monte Carlo simulations of the structure of Pt-based bimetallic nanoparticles,” arXiv: Materials Science. 2012. link Times cited: 67 NOT USED (high confidence) M. Mariscal, J. Velázquez‐Salazar, and M. Yacamán, “Growth mechanism of nanoparticles: theoretical calculations and experimental results,” CrystEngComm. 2012. link Times cited: 24 Abstract: We report a combined theoretical and experimental study on t… read moreAbstract: We report a combined theoretical and experimental study on the growth mechanism of silver and gold nanoparticles. We introduce for the first time the grand-canonical Monte Carlo method to study the growth/dissolution steps of nanoparticle growth mediated by seeds. In particular we found that small changes on the chemical potential, i.e. the activity of the metal ions, produce significant changes on the size and morphologies of the nanoparticles. We have found very good agreements between simulated structures with those observed experimentally by means of scanning electron microscopy. read less NOT USED (high confidence) A. Fraile, S. Cuesta-López, and J. Perlado, “Molecular Dynamics Simulations of Lead and Lithium in Liquid Phase,” Fusion Science and Technology. 2012. link Times cited: 3 Abstract: Pb17Li is today a reference breeder material in diverse fusi… read moreAbstract: Pb17Li is today a reference breeder material in diverse fusion R&D programs worldwide. Extracting dynamic and structural properties of liquid LiPb mixtures via molecular dynamics simulations, represent a crucial step for multiscale modeling efforts in order to understand the suitability of this compound for future Nuclear Fusion technologies. At present a Li-Pb cross potential is not available in the literature. Here we present our first results on the validation of two semi-empirical potentials for Li and Pb in liquid phase. Our results represent the establishment of a solid base as a previous but crucial step to implement a LiPb cross potential. Structural and thermodynamical analyses confirm that the implemented potentials for Li and Pb are realistic to simulate both elements in the liquid phase. read less NOT USED (high confidence) B. Fu, Z. Li, and W. Liu, “Covalent electron density analysis and surface energy calculation of gold with the empirical electron surface model,” International Journal of Minerals, Metallurgy, and Materials. 2011. link Times cited: 5 NOT USED (high confidence) K. Kolluri and M. Demkowicz, “Coarsening by network restructuring in model nanoporous gold,” Acta Materialia. 2011. link Times cited: 77 NOT USED (high confidence) H. Zhan, Y. T. Gu, X.-Q. Feng, and P. Yarlagadda, “Numerical exploration of plastic deformation mechanisms of copper nanowires with surface defects.” 2011. link Times cited: 36 NOT USED (high confidence) X.-J. Yuan, N. Chen, and J. Shen, “Lattice-Inversion Embedded-Atom-Method Interatomic Potentials for Group-VA Transition Metals,” Chinese Physics Letters. 2011. link Times cited: 1 Abstract: The lattice-inversion embedded-atom-method (LI-EAM) interato… read moreAbstract: The lattice-inversion embedded-atom-method (LI-EAM) interatomic potential we developed previously [J. Phys.: Condens. Matter 22 (2010) 375503] is extended to group-VA transition metals (V, Nb and Ta). It is found that considering interatomic interactions up to appropriate-distance-neighbor atoms is crucial to constructing accurate EAM potentials, especially for the prediction of surface energy. The LI-EAM interatomic potentials for group-VA transition metals are successfully built by considering interatomic interactions up to the fifth neighbor atoms. These angular-independent potentials drastically promote the accuracy of the predicted surface energies, which match the experimental results well. read less NOT USED (high confidence) P. Wang, W. Chou, A. Nie, H. Yang, H. Yao, and H. Wang, “Molecular dynamics simulation on deformation mechanisms in body-centered-cubic molybdenum nanowires,” Journal of Applied Physics. 2011. link Times cited: 54 Abstract: A systematic study on the deformation mechanisms of molybden… read moreAbstract: A systematic study on the deformation mechanisms of molybdenum (Mo) nanowires (NWs) was conducted using molecular dynamics simulations. Both axial orientation and wire thickness were found to play important roles in determining the deformation pathways. In the NWs with orientation 〈110〉/{111}, full dislocation plasticity is referentially activated on {110} planes. For both 〈100〉/{110} and 〈100〉/{100} NWs, twinning is the dominant mechanism with {112} being the coherent twin boundaries. A progressive slip process leads to a uniform elongation of 41% and the 〈100〉 wire axis reorients to 〈110〉. For 〈100〉/{100} NWs, the reorientation mechanism ceases to operate when the diameter d 8 nm. The atomic chains are energetically preferred for ultrathin NWs after yielding due to the resemblance of the surface to the close-packed bcc planes, while multiple slip systems tend to be activated for larger NWs. Finally, a theoretical model is proposed to explain the underlying mechanism of size dependence of t... read less NOT USED (high confidence) P. T. Barton, M. Kalweit, D. Drikakis, and G. Ball, “Multi-scale analysis of high-speed dynamic friction,” Journal of Applied Physics. 2011. link Times cited: 2 Abstract: Friction occurring at the interface between dissimilar metal… read moreAbstract: Friction occurring at the interface between dissimilar metallic components as a result of high velocity impact or explosive loading can have a profound effect on the subsequent motion. A comprehensive understanding of the involved processes across a wide range of initial conditions remains outstanding. Dry sliding of single crystal silver on copper at high pressure is investigated for a range of sliding speeds using a multi-scale modelling method based upon the domain decomposition approach (molecular dynamics in the near interface region and continuum mechanics elsewhere). The transient solutions reveal detailed observations of the processes that lead to phenomena such as the growth of epitaxial layers of the softer material, shifting of the sliding interface due to formation of shear-bands, development of amorphous structures, and ultimately the resultant motion of the components. Analysis of the results also links these processes to the changes in the state of the material through growth of dislocations and thermal effects. read less NOT USED (high confidence) S. Angioletti-Uberti, “The solid–liquid interface free-energy of Pb: comparison of theory and experiments,” Journal of Physics: Condensed Matter. 2011. link Times cited: 11 Abstract: The solid–liquid interface free-energy γsl is a key paramete… read moreAbstract: The solid–liquid interface free-energy γsl is a key parameter controlling nucleation and growth during solidification and other phenomena. Different experimental techniques are available for its evaluation but results are often widely scattered and strongly depend on the technique used. One of the best examples in this sense is the case of elemental Pb, with values for γsl differing by as much as 150% between nucleation rate experiments and contact angle data. Even using simple many-body potentials, theoretical calculations of γsl can exceed this level of accuracy and thus be employed to assess the present experimental data. To this purpose, atomistic calculations are performed in conjunction with two different many-body potentials for Pb. These show good agreement with nucleation rate and depression of melting point experiments and support the case for a reassessment of the measurements reported from contact angle data. Possible sources of errors that might have affected these experiments are critically discussed, showing how the magnitude of the anisotropy in the interfacial free energies can be important in closing the gap between the different sets of experimental data. read less NOT USED (high confidence) D. Duffy, “Modelling materials for fusion power,” International Materials Reviews. 2011. link Times cited: 12 Abstract: Fusion has the potential for delivering safe, clean, low car… read moreAbstract: Fusion has the potential for delivering safe, clean, low carbon power; however, significant scientific and engineering hurdles must first be overcome. One such hurdle is the design of materials that will withstand the harsh conditions. The materials which line the vessel walls will experience exceptionally high heat and particle fluxes, which will gradually erode the materials and contaminate the plasma. The deuterium–tritium fusion reaction will produce high energy neutrons, which will create defects and transmutation reactions in the vessel walls. These defects, along with the transmutation gasses, evolve over time and change the microstructure and properties of the material. In order to design suitable materials for fusion, the radiation damage, and its evolution over time, must be understood and evaluated for a broad class of materials. Modelling has a vital role to play because it can provide details about processes that occur on length and timescales that are inaccessible to experiment. In this review, the challenges that face designers of fusion power plants are discussed. The modelling techniques that are used to model radiation effects are described and the links between modelling and experiment are discussed. The review concludes with a discussion about the future direction for fusion materials research. read less NOT USED (high confidence) M. Molayem, V. Grigoryan, and M. Springborg, “Global Minimum Structures and Magic Clusters of CumAgn Nanoalloys,” Journal of Physical Chemistry C. 2011. link Times cited: 57 Abstract: The putative global minimum structures of bimetallic CumAgn … read moreAbstract: The putative global minimum structures of bimetallic CumAgn nanoalloys for all (m,n) with N = m + n from 2 to 60 atoms have been determined. The embedded-atom method was used for the description of the interatomic interactions in combination with the basin-hopping algorithm for the global structural optimization. The obtained global-minimum structures are mostly based on icosahedra, polyicosahedra as well as 5-fold pancakes. But also truncated octahedral are found for some clusters with 38 atoms. The bond-order parameter reveals the formation of CucoreAgshell clusters. The analysis of energetic properties, through stability functions, the excess and the mixing energies, gives that the clusters with more Ag atoms are energetically more favorable. Moreover, from our analysis we identify the most stable stoichiometries as a function of N. Finally, the results of the present study are compared with similar results on NimAgn clusters. read less NOT USED (high confidence) D. Schopf, P. Brommer, B. Frigan, and H. Trebin, “Embedded atom method potentials for Al-Pd-Mn phases,” Physical Review B. 2011. link Times cited: 23 Abstract: A novel embedded atom method (EAM) potential for the Ξ phase… read moreAbstract: A novel embedded atom method (EAM) potential for the Ξ phases of Al-Pd-Mn has been determined with the force-matching method. Different combinations of analytic functions were tested for the pair and transfer part. The best results are obtained if one allows for oscillations on two different length scales. These potentials stabilize structure models of the Ξ phases and describe their energy with high accuracy. Simulations at temperatures up to 1200 K show very good agreement with ab initio results with respect to stability and dynamics of the system. read less NOT USED (high confidence) D. Belashchenko, A. V. Vorotyagin, and B. R. Gelchinsky, “Computer simulation of aluminum in the high-pressure range,” High Temperature. 2011. link Times cited: 9 NOT USED (high confidence) S.-K. Chien, Y.-T. Yang, and C.-K. Chen, “A molecular dynamics study of the mechanical properties of graphene nanoribbon-embedded gold composites.,” Nanoscale. 2011. link Times cited: 13 Abstract: Molecular dynamics simulations were performed to investigate… read moreAbstract: Molecular dynamics simulations were performed to investigate the mechanical properties of a single-crystal gold nanosheet and graphene nanoribbon-embedded gold (GNR/Au) composites for various embedded locations, temperatures, and lengths. The computational results show that the Young's modulus, tensile strength, and fracture strain of GNR/Au composites are much larger than those of pure gold. The mechanical properties of GNR/Au composites deteriorate drastically due to C-C bond breaking. Thermal fluctuation and an increase in length can decrease the mechanical properties of GNR/Au composites. read less NOT USED (high confidence) M. Salciccioli, M. Stamatakis, S. Caratzoulas, and D. Vlachos, “A review of multiscale modeling of metal-catalyzed reactions: Mechanism development for complexity and emergent behavior,” Chemical Engineering Science. 2011. link Times cited: 292 NOT USED (high confidence) M. He and S. Li, “An embedded atom hyperelastic constitutive model and multiscale cohesive finite element method,” Computational Mechanics. 2011. link Times cited: 0 NOT USED (high confidence) H. M. Khan and S.-G. Kim, “On the wear mechanism of thin nickel film during AFM-based scratching process using molecular dynamics,” Journal of Mechanical Science and Technology. 2011. link Times cited: 18 NOT USED (high confidence) J. Lloyd, J. Zimmerman, R. Jones, X. W. Zhou, and D. McDowell, “Finite element analysis of an atomistically derived cohesive model for brittle fracture,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 22 Abstract: In order to apply information from molecular dynamics (MD) s… read moreAbstract: In order to apply information from molecular dynamics (MD) simulations in problems governed by engineering length and time scales, a coarse graining methodology must be used. In previous work by Zhou et al (2009 Acta Mater. 57 4671–86), a traction-separation cohesive model was developed using results from MD simulations with atomistic-to-continuum measures of stress and displacement. Here, we implement this cohesive model within a combined finite element/cohesive surface element framework (referred to as a finite element approach or FEA), and examine the ability for the atomistically informed FEA to directly reproduce results from MD. We find that FEA shows close agreement of both stress and crack opening displacement profiles at the cohesive interface, although some differences do exist that can be attributed to the stochastic nature of finite temperature MD. The FEA methodology is then used to study slower loading rates that are computationally expensive for MD. We find that the crack growth process initially exhibits a rate-independent relationship between crack length and boundary displacement, followed by a rate-dependent regime where, at a given amount of boundary displacement, a lower applied strain rate produces a longer crack length. Our method is also extended to larger length scales by simulating a compact tension fracture-mechanics specimen with sub-micrometer dimensions. Such a simulation shows a computational speedup of approximately four orders of magnitude over conventional atomistic simulation, while exhibiting the expected fracture-mechanics response. Finally, differences between FEA and MD are explored with respect to ensemble and temperature effects in MD, and their impact on the cohesive model and crack growth behavior. These results enable us to make several recommendations to improve the methodology used to derive cohesive laws from MD simulations. In light of this work, which has critical implications for efforts to derive continuum laws from MD simulations, it is shown care must be taken when using a similar approach, and effects of ensemble, temperature and strain rate must be considered. read less NOT USED (high confidence) X. Li and M. Luskin, “Lattice Stability for Atomistic Chains Modeled by Local Approximations of the Embedded Atom Method,” arXiv: Numerical Analysis. 2011. link Times cited: 3 NOT USED (high confidence) A. Oluwajobi and X. Chen, “The effect of interatomic potentials on the molecular dynamics simulation of nanometric machining,” International Journal of Automation and Computing. 2011. link Times cited: 45 NOT USED (high confidence) T. Koido, K. Tomarikawa, S. Yonemura, and T. Tokumasu, “Molecular dynamics study of the effects of translational energy and incident angle on dissociation probability of hydrogen/deuterium molecules on Pt(111),” Journal of Applied Physics. 2011. link Times cited: 8 Abstract: The dissociation probabilities of H2 and D2 molecules on a P… read moreAbstract: The dissociation probabilities of H2 and D2 molecules on a Pt(111) surface with thermal motion were analyzed using the molecular dynamics (MD) method. The potential constructed using the embedded atom method was used as the interaction potential between a gas molecule and the surface. The effects of changing the translational energy and incident polar angle of D2 molecules impinging on a Pt(111) surface were analyzed using MD simulations. The effect of initial orientation, incident azimuthal angle, rotational energy of gas molecules, and the impinging points on the surface were averaged by setting the initial values in a random manner. When the molecules approach normal to the surface, the dissociation probability increases with the initial translational energy. At larger incident angles, the probability becomes smaller. The impinging processes were categorized in terms of reaching the chemisorption layer by analyzing the repulsion forces from the surface. The effective translational energies for impingem... read less NOT USED (high confidence) T. Klaver, D. Hepburn, and G. Ackland, “Defect and solute properties in dilute Fe-Cr-Ni austenitic alloys from first principles,” Performance Evaluation. 2011. link Times cited: 54 Abstract: We present results of an extensive set of first-principles d… read moreAbstract: We present results of an extensive set of first-principles density functional theory calculations of point defect formation, binding, and clustering energies in austenitic Fe with dilute concentrations of Cr and Ni solutes. A large number of possible collinear magnetic structures were investigated as appropriate reference states for austenite. We found that the antiferromagnetic single- and double-layer structures with tetragonal relaxation of the unit cell were the most suitable reference states and highlighted the inherent instabilities in the ferromagnetic states. Test calculations for the presence and influence of noncollinear magnetism were performed but proved mostly negative. We calculate the vacancy formation energy to be between 1.8 and 1.95 eV. Vacancy cluster binding was initially weak at 0.1 eV for divacancies but rapidly increased with additional vacancies. Clusters of up to six vacancies were studied and a highly stable octahedral cluster and stacking fault tetrahedron were found with total binding energies of 2.5 and 2.3 eV, respectively. The ?100? dumbbell was found to be the most stable self-interstitial with a formation energy of between 3.2 and 3.6 eV and was found to form strongly bound clusters, consistent with other fcc metals. Pair interaction models were found to be capable of capturing the trends in the defect cluster binding energy data. Solute-solute interactions were found to be weak in general, with a maximal positive binding of 0.1 eV found for Ni-Ni pairs and maximum repulsion found for Cr-Cr pairs of ?0.1 eV. Solute cluster binding was found to be consistent with a pair interaction model, with Ni-rich clusters being the most stable. Solute-defect interactions were consistent with Ni and Cr being modestly oversized and undersized solutes, respectively, which is exactly opposite to the experimentally derived size factors for Ni and Cr solutes in type 316 stainless steel and in the pure materials. Ni was found to bind to the vacancy and to the ?100? dumbbell in the tensile site by 0.1 eV and was repelled from mixed and compressive sites. In contrast, Cr showed a preferential binding to interstitials. Calculation of tracer diffusion coefficients found that Ni diffuses significantly more slowly than both Cr and Fe, which is consistent with the standard mechanism used to explain radiation-induced segregation effects in Fe-Cr-Ni austenitic alloys by vacancy-mediated diffusion. Comparison of our results with those for bcc Fe showed strong similarity for pure Fe and no correlation with dilute Ni and Cr. read less NOT USED (high confidence) J. Elliott, “Novel approaches to multiscale modelling in materials science,” International Materials Reviews. 2011. link Times cited: 133 Abstract: Computational modelling techniques are now widely employed i… read moreAbstract: Computational modelling techniques are now widely employed in materials science, due to recent advances in computing power and simulation methodologies, since they can enable rapid testing of theoretical predictions or understanding of complex experimental data at relatively low cost. However, many problems at the leading edge of materials science involve collective phenomena that occur over a range of time and length scales which are intrinsically difficult to capture in a single simulation. This review summarises some of the latest developments in multiscale modelling techniques over the past decade, as applied to selected problems in materials science and engineering, thereby motivating the reader to explore how such techniques might be applied in their own area of specialty. Methods for accelerating molecular dynamics by enhancement of kinetic barrier crossing, such as hyperdynamics and metadynamics, are discussed alongside mesoscale simulation techniques, such as dissipative particle dynamics or adaptive coarse graining, for enabling larger and longer simulations. The applications are mainly focused on simulations of microstructure and mechanical properties, and examples of surface diffusion in metals, radiation damage in ceramics, strengthening of nanocrystalline metals and alloys, crack propagation in brittle solids, polymer chain relaxation in nanocomposites and the control of nucleation in biomimetic materials are discussed. read less NOT USED (high confidence) H. Zhang, P. Kalvapalle, and J. Douglas, “String-like collective atomic motion in the melting and freezing of nanoparticles.,” The journal of physical chemistry. B. 2011. link Times cited: 30 Abstract: The melting of a solid represents a transition between a sol… read moreAbstract: The melting of a solid represents a transition between a solid state in which atoms are localized about fixed average crystal lattice positions to a fluid state that is characterized by relative atomic disorder and particle mobility so that the atoms wander around the material as a whole, impelled by the random thermal impulses of surrounding atoms. Despite the fundamental nature and practical importance of this particle delocalization transition, there is still no fundamental theory of melting and instead one often relies on the semi-phenomenological Lindemann-Gilvarry criterion to estimate roughly the melting point as an instability of the crystal lattice. Even the earliest simulations of melting in hexagonally packed hard discs by Alder and Wainwright indicated the active role of nonlocal collective atomic motions in the melting process, and here we utilize molecular dynamics (MD) simulation to determine whether the collective particle motion observed in melting has a similar geometrical form as those in recent studies of nanoparticle (NP) interfacial dynamics and the molecular dynamics of metastable glass-forming liquids. We indeed find string-like collective atomic motion in NP melting that is remarkably similar in form to the collective interfacial motions in NPs at equilibrium and to the collective motions found in the molecular dynamics of glass-forming liquids. We also find that the spatial localization and extent of string-like motion in the course of NP melting and freezing evolves with time in distinct ways. Specifically, the collective atomic motion propagates from the NP surface and from within the NP in melting and freezing, respectively, and the average string length varies smoothly with time during melting. In contrast, the string-like cooperative motion peaks in an intermediate stage of the freezing process, reflecting a general asymmetry in the dynamics of NP superheating and supercooling. read less NOT USED (high confidence) W. Ko, J. Shim, and B.-J. Lee, “Atomistic modeling of the Al–H and Ni–H systems,” Journal of Materials Research. 2011. link Times cited: 15 Abstract: Second nearest-neighbor modified embedded-atom method (MEAM)… read moreAbstract: Second nearest-neighbor modified embedded-atom method (MEAM) interatomic potentials for the Al–H and Ni–H binary systems have been developed on the basis of previously developed MEAM potentials of pure Al, Ni, and H. The potentials can describe various fundamental physical properties of the relevant binary alloys (structural, thermodynamic, defect, and dynamic properties of metastable hydrides or hydrogen in face-centered cubic solid solutions) in good agreement with experiments or first-principles calculations. The applicability of the present potentials to atomic level investigations of dynamic behavior of hydrogen atoms in metal membranes is also discussed. read less NOT USED (high confidence) D. Belashchenko, “Optimal algorithm for constructing the embedded atom method potential for liquid metals,” Inorganic Materials. 2011. link Times cited: 6 NOT USED (high confidence) S. J. Wang, X. Kuang, C. Lu, Y. Li, and Y. R. Zhao, “Geometries, stabilities, and electronic properties of Pt-group-doped gold clusters, their relationship to cluster size, and comparison with pure gold clusters.,” Physical chemistry chemical physics : PCCP. 2011. link Times cited: 23 Abstract: A systematic study of bimetallic Au(n)M(2) (n = 1-6, M = Ni,… read moreAbstract: A systematic study of bimetallic Au(n)M(2) (n = 1-6, M = Ni, Pd, and Pt) clusters is performed by using density functional theory at the B3LYP level. The geometric structures, relative stabilities, HOMO-LUMO gaps, natural charges and electronic magnetic moments of these clusters are investigated, and compared with pure gold clusters. The results indicate that the properties of Au(n)M(2) clusters for n = 1-3 diverge more from pure gold clusters, while those for n = 4-6 show good agreement with Au(n) clusters. The dissociation energies, the second-order difference of energies, and HOMO-LUMO energy gaps, exhibiting an odd-even alternation, indicate that the Au(4)M(2) clusters are the most stable structures for Au(n)M(2) (n = 1-6, M = Ni, Pd, and Pt) clusters. Moreover, we predict that the average atomic binding energies of these clusters should tend to a limit in the range 1.56-2.00 eV. read less NOT USED (high confidence) Z.-L. Liu, J. Yang, L. Cai, F. Jing, and D. Alfé, “Structural and thermodynamic properties of compressed palladium: Ab initio and molecular dynamics study,” Physical Review B. 2011. link Times cited: 30 Abstract: First-principles and classical molecular dynamics simulation… read moreAbstract: First-principles and classical molecular dynamics simulations have been performed to study the structural and thermodynamic properties of Pd under pressure. By comparing the Gibbs free energy, in the quasiharmonic approximation (QHA), of the face-centered cubic (fcc) phase with those of the hexagonal-close-packed (hcp) and body-centered-cubic (bcc) phases we found that the fcc phase is stable up to 500 GPa and 5000 K. The predicted high-temperature elastic constants of fcc Pd agree well with experiments. The phonon dispersion curves are obtained at various pressures. In contrast with experiments we did not observe any phonon anomalies in Pd. We reproduced the thermodynamic properties of Pd accurately by taking into account the electron and phonon contributions to the free energy of Pd. The obtained thermal expansion coefficient, Hugoniot curves, and specific heat capacity compare well with experiments. In particular, the excellent agreement of the thermal expansion coefficients with experiment supports the validity of the QHA for Pd at high temperatures. Our QHA-based Hugoniot curves also show good agreement with experiments and our dynamic shock simulations. Shocks along [100] produced amelting temperature with a superheating of 18.3% at 226 GPa, compared with our high-pressure melting curve of Pd from coexistence-phase simulations based on an embedded atom model. read less NOT USED (high confidence) H. Sheng, M. Kramer, A. Cadien, T. Fujita, and M. Chen, “Highly optimized embedded-atom-method potentials for fourteen fcc metals,” Physical Review B. 2011. link Times cited: 387 Abstract: Highly optimized embedded-atom-method (EAM) potentials have … read moreAbstract: Highly optimized embedded-atom-method (EAM) potentials have been developed for 14 face-centered-cubic (fcc) elements across the periodic table. The potentials were developed by fitting the potential-energy surface (PES) of each element derived from high-precision first-principles calculations. The as-derived potential-energy surfaces were shifted and scaled to match experimental reference data. In constructing the PES, a variety of properties of the elements were considered, including lattice dynamics, mechanical properties, thermal behavior, energetics of competing crystal structures, defects, deformation paths, liquid structures, and so forth. For each element, the constructed EAM potentials were tested against the experiment data pertaining to thermal expansion, melting, and liquid dynamics via molecular dynamics computer simulation. The as-developed potentials demonstrate high fidelity and robustness. Owing to their improved accuracy and wide applicability, the potentials are suitable for high-quality atomistic computer simulation of practical applications. read less NOT USED (high confidence) S. Koh, A. Saxena, W. V. van Driel, G. Q. Zhang, and R. Tummala, “Low cycle fatigue crack growth in nanostructure copper,” 2011 12th Intl. Conf. on Thermal, Mechanical & Multi-Physics Simulation and Experiments in Microelectronics and Microsystems. 2011. link Times cited: 2 Abstract: ITRS has predicted that integrated chip (IC) packages will h… read moreAbstract: ITRS has predicted that integrated chip (IC) packages will have interconnections with I/O pitch of 90 nm by the year 2018. Lead-based solder materials in flip chip technology will not be able to satisfy the thermal mechanical requirement these fine pitches. Of all the known interconnect technologies, nanostructure interconnects such as nanocrystalline Cu are the most promising technology to meet the high mechanical reliability and electrical requirements of next generation devices. However, there is a need to fully characterize their fatigue properties. In this research, numerical analysis has been employed to study the semi-elliptical crack growth and shape evolution in nanostructured interconnects subject to uniaxial fatigue loading. The results indicate that nanocrystalline copper is in fact a suitable candidate for ultra-fine pitch interconnects applications. This study also predicts that crack growth is a relatively small portion of the total fatigue life of interconnects under LCF conditions. Hence, crack initiation life is the main factor in determining the fatigue life of interconnects. read less NOT USED (high confidence) C. Vardeman, K. M. Stocker, and J. Gezelter, “The Langevin Hull: Constant pressure and temperature dynamics for non-periodic systems.,” Journal of chemical theory and computation. 2011. link Times cited: 11 Abstract: We have developed a new isobaric-isothermal (NPT) algorithm … read moreAbstract: We have developed a new isobaric-isothermal (NPT) algorithm which applies an external pressure to the facets comprising the convex hull surrounding the system. A Langevin thermostat is also applied to the facets to mimic contact with an external heat bath. This new method, the "Langevin Hull", can handle heterogeneous mixtures of materials with different compressibilities. These systems are problematic for traditional affine transform methods. The Langevin Hull does not suffer from the edge effects of boundary potential methods, and allows realistic treatment of both external pressure and thermal conductivity due to the presence of an implicit solvent. We apply this method to several different systems including bare metal nanoparticles, nanoparticles in an explicit solvent, as well as clusters of liquid water. The predicted mechanical properties of these systems are in good agreement with experimental data and previous simulation work. read less NOT USED (high confidence) M. Molayem, V. Grigoryan, and M. Springborg, “Theoretical Determination of the Most Stable Structures of NimAgn Bimetallic Nanoalloys,” Journal of Physical Chemistry C. 2011. link Times cited: 54 Abstract: The structure of the global total-energy minimum for all bim… read moreAbstract: The structure of the global total-energy minimum for all bimetallic NimAgn nanoalloys with m + n = N = 2−60 atoms has been identified theoretically by combining the embedded-atom model for the total-energy evaluation with the basin-hopping algorithm for global structure optimization. All global minima structures are found to be related to icosahedra and polyicosahedra, except for some Ni- or Ag-rich clusters for N = 38. Through a careful analysis of the total energy as a function of (m,n), various particularly stable structures can be identified. The results show in most cases that Ag-rich structures are more favored and stable compared with the Ni-rich structures. By analyzing the bond-order parameter and the radial distances, we can demonstrate the existence of core−shell structures with a partial segregation of Ag to the surface of the Ni−Ag clusters. read less NOT USED (high confidence) T. Tokumasu and D. Ito, “The dynamic effects on dissociation probability of H2–Pt(111) system by embedded atom method,” Journal of Applied Physics. 2011. link Times cited: 9 Abstract: The effects of the motion of atoms or molecules on the disso… read moreAbstract: The effects of the motion of atoms or molecules on the dissociation probability of the H2–Pt(111) system were analyzed by molecular dynamics. The embedded atom method (EAM) was used to model the interaction between a Pt(111) surface and an H2 molecule to consider the dependence of electron density. Initially, the EAM potential was constructed to express the characteristics of the system, such as the electron density or dissociation barrier at certain sites and orientations, as obtained by density functional theory (DFT). Using this potential, simulations of an H2 molecule impinging on a Pt(111) surface were performed, and the characteristics of the collision were observed. These simulations were performed many times, changing the orientation of the H2 molecule, and a dynamic dissociation probability at each site against impinging energy was obtained. On the other hand, a static dissociation probability was defined from the dissociation barrier of a hydrogen molecule obtained by the EAM potential. These re... read less NOT USED (high confidence) M. C. Giménez and W. Schmicker, “Monte Carlo simulation of nanowires of different metals and two-metal alloys.,” The Journal of chemical physics. 2011. link Times cited: 10 Abstract: Nanowires of different metals and two-metal alloys have been… read moreAbstract: Nanowires of different metals and two-metal alloys have been studied by means of canonical Monte Carlo simulations and the embedded atom method for the interatomic potentials. For nanowires of gold, a relatively stable three-atom-wide chain was observed. The presence of one-atom-wide linear atomic chains is not stable in any case. For two-metal alloy nanowires, the metal with a higher surface energy tends to locate in the inner region of the nanowire. read less NOT USED (high confidence) S. M. Amir, M. Guptaand, A. Gupta, J. Stahn, and A. Wildes, “Surfactant induced symmetric and thermally stable interfaces in Cu/Co multilayers,” Journal of Physics: Condensed Matter. 2011. link Times cited: 5 Abstract: In this work we studied Ag surfactant induced growth of Cu/C… read moreAbstract: In this work we studied Ag surfactant induced growth of Cu/Co multilayers. The Cu/Co multilayers were deposited using Ag surfactant by the ion beam sputtering technique. It was found that Ag surfactant balances the asymmetry between the surface free energies of Cu and Co. As a result, the Co-on-Cu and Cu-on-Co interfaces become sharp and symmetric and thereby improve the thermal stability of the multilayer. On the basis of obtained results, a mechanism leading to symmetric and stable interfaces in Cu/Co multilayers is discussed. read less NOT USED (high confidence) M. Gupta, S. M. Amir, A. Gupta, and J. Stahn, “Surfactant mediated growth of Ti/Ni multilayers,” Applied Physics Letters. 2011. link Times cited: 14 Abstract: The surfactant mediated growth of Ti/Ni multilayers is studi… read moreAbstract: The surfactant mediated growth of Ti/Ni multilayers is studied. They were prepared using ion beam sputtering at different adatom energies. It was found that the interface roughness decreased significantly when the multilayers were sputtered with Ag as surfactant at an ion energy of 0.75 keV. On the other hand, when the ion energy was increased to 1 keV, it resulted in enhanced intermixing at the interfaces and no appreciable effect of Ag surfactant could be observed. On the basis of the obtained results, the influence of adatom energy on the surfactant mediated growth mechanism is discussed. read less NOT USED (high confidence) M. Chassagne, M. Legros, and D. Rodney, “Atomic-scale simulation of screw dislocation/coherent twin boundary interaction in Al, Au, Cu and Ni,” Acta Materialia. 2011. link Times cited: 124 NOT USED (high confidence) W. Wu, Y.-F. Guo, Y. Wang, R. Müller, and D. Gross, “Molecular dynamics simulation of the structural evolution of misfit dislocation networks at γ/γ′ phase interfaces in Ni-based superalloys,” Philosophical Magazine. 2011. link Times cited: 52 Abstract: The structural evolution of misfit dislocation networks at γ… read moreAbstract: The structural evolution of misfit dislocation networks at γ/γ′ phase interfaces in Ni-based single crystal superalloys under tensile loading and temperatures is simulated by molecular dynamics. From the simulation, we find that, with increasing load or temperature, the patterns of dislocation networks on the (100), (110) and (111) phase interfaces change from regular to irregular or disappear. Under the same load and temperature, the dislocation networks of the different phase interfaces show different degrees and patterns of damage. The density and stability of the dislocation networks decrease with increasing temperature. When the interfacial dislocation networks become more regular, the γ/γ′ interfaces become more stable. The simulated results are supported by related experimental findings. Moreover, based on MD simulations, the averaged stress–strain responses for different phase interfaces under loading are presented. The results indicate that the combined influences of temperature and load play an important role for the structure evolution of misfit dislocation networks at γ/γ′ phase interfaces of Ni-based superalloys. read less NOT USED (high confidence) H. Wang, M. Stamatakis, D. Hansgen, S. Caratzoulas, and D. Vlachos, “Understanding mixing of Ni and Pt in the Ni/Pt(111) bimetallic catalyst via molecular simulation and experiments.,” The Journal of chemical physics. 2010. link Times cited: 18 Abstract: Molecular dynamics (MD) simulations employing embedded atom … read moreAbstract: Molecular dynamics (MD) simulations employing embedded atom method potentials and ultrahigh vacuum (UHV) experiments were carried out to study the mixing process between the Ni and Pt atoms in the Ni/Pt(111) bimetallic system. The barrier for a Ni atom to diffuse from the top surface to the subsurface layer is rather high (around 1.7 eV) as calculated using the nudged elastic band (NEB) method. Analysis of the relaxation dynamics of the Ni atoms showed that they undergo diffusive motion through a mechanism of correlated hops. At 600 K, all Ni atoms remain trapped on the top surface due to large diffusion barriers. At 900 K, the majority of Ni atoms diffuse to the second layer and at 1200 K diffusion to the bulk is observed. We also find that smaller Ni coverages and the presence of Pt steps facilitate the Ni-Pt mixing. By simulated annealing simulations, we found that in the mixed state, the Ni fraction oscillates between layers, with the second layer being Ni-richer at equilibrium. The simulation results at multiple time scales are consistent with the experimental data. read less NOT USED (high confidence) K. L. Joshi, A. Duin, and T. Jacob, “Development of a ReaxFF description of gold oxides and initial application to cold welding of partially oxidized gold surfaces,” Journal of Materials Chemistry. 2010. link Times cited: 30 Abstract: We present the ReaxFF reactive force field methodology for m… read moreAbstract: We present the ReaxFF reactive force field methodology for modeling a gold–oxygen binary system. The force field parameters were fitted against a data set including equations of state, heats of formation and binding energies derived from DFT calculations. The trained force field was then used to study the diffusion properties of oxygen on a gold surface. The diffusion study shows that oxygen atoms have a relatively low mobility on the gold surface. We also present a prospective application of this force field by performing molecular dynamics simulations studying the effect of oxidation level on contact strength of a cold welded joint. The results indicate that low levels of oxidation can significantly impact the joint cohesive energy. read less NOT USED (high confidence) E. H. Megchiche, C. Mijoule, and M. Amarouche, “First principles calculations of vacancy–vacancy interactions in nickel: thermal expansion effects,” Journal of Physics: Condensed Matter. 2010. link Times cited: 21 Abstract: The energetic properties of the divacancy defect in fcc nick… read moreAbstract: The energetic properties of the divacancy defect in fcc nickel are studied by ab initio calculations based on density functional theory. The formation and binding enthalpies of the divacancy in the first (1nn), second (2nn) and third (3nn) nearest-neighbor configurations are presented. Results show that the 1nn divacancy configuration is the most stable with a formation enthalpy H2vf of 2.71 eV and a small binding energy H2vb of 0.03 eV. In the 2nn configuration, the monovacancy–monovacancy interaction is repulsive, and it vanishes in the 3nn configuration. The migration process of the divacancy in its stable configuration is studied. We find that the divacancy migrates in the (111) plane by successive rotational steps of 60°. The corresponding migration enthalpy H2vm is predicted to be 0.59 eV, about half of that found for the monovacancy. For a better comparison of our results with high temperature experimental data, we have analyzed the effects of thermal expansion. Our results show that the inclusion of thermal expansion allows us to reproduce satisfactorily the experimental predictions. read less NOT USED (high confidence) G. Rusina, S. Borisova, and E. Chulkov, “Small Al clusters on the Cu(111) surface: Atomic relaxation and vibrational properties,” Russian Journal of Physical Chemistry A. 2010. link Times cited: 2 NOT USED (high confidence) C. Becker and M. J. Kramer, “Atomistic comparison of volume-dependent melt properties from four models of aluminum,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 30 Abstract: With the increasing use of simulations in materials research… read moreAbstract: With the increasing use of simulations in materials research and design, it is important to quantify the differences between, and accuracy of, models used in these simulations. Here we present the results of such a comparison for four embedded-atom models of aluminum that were optimized to have good liquid properties, particularly the melting temperatures. The effects of temperature and volume are systematically examined in the melts for bulk thermodynamic quantities, pair correlation functions and structure factors and diffusion coefficients for each interatomic potential. Where possible, these are then compared with experimental values. We find quantitative differences in the properties determined from the various interatomic potentials despite the fact that they were fit with similar sets of data. read less NOT USED (high confidence) X.-J. Yuan, N. Chen, J. Shen, and W. Hu, “Embedded-atom-method interatomic potentials from lattice inversion,” Journal of Physics: Condensed Matter. 2010. link Times cited: 26 Abstract: The present work develops a physically reliable procedure fo… read moreAbstract: The present work develops a physically reliable procedure for building the embedded-atom-method (EAM) interatomic potentials for the metals with fcc, bcc and hcp structures. This is mainly based on Chen–Möbius lattice inversion (Chen et al 1997 Phys. Rev. E 55 R5) and first-principles calculations. Following Baskes (Baskes et al 2007 Phys. Rev. B 75 094113), this new version of the EAM eliminates all of the prior arbitrary choices in the determination of the atomic electron density and pair potential functions. Parameterizing the universal form deduced from the calculations within the density-functional scheme for homogeneous electron gas as the embedding function, the new-type EAM potentials for Cu, Fe and Ti metals have successfully been constructed by considering interatomic interactions up to the fifth neighbor, the third neighbor and the seventh neighbor, respectively. The predictions of elastic constants, structural energy difference, vacancy formation energy and migration energy, activation energy of vacancy diffusion, latent heat of melting and relative volume change on melting all satisfactorily agree with the experimental results available or first-principles calculations. The predicted surface energies for low-index crystal faces and the melting point are in agreement with the experimental data to the same extent as those calculated by other EAM-type potentials such as the FBD-EAM, 2NN MEAM and MS-EAM. In addition, the order among the predicted low-index surface energies is also consistent with the experimental information. read less NOT USED (high confidence) X. Li and M. Luskin, “An Analysis of the Quasi-Nonlocal Quasicontinuum Approximation of the Embedded Atom Model,” arXiv: Numerical Analysis. 2010. link Times cited: 7 Abstract: The quasi-nonlocal quasicontinuum method (QNL) is a consiste… read moreAbstract: The quasi-nonlocal quasicontinuum method (QNL) is a consistent hybrid coupling method for atomistic and continuum models. Embedded atom models are empirical many-body potentials that are widely used for FCC metals such as copper and aluminum. In this paper, we consider the QNL method for EAM potentials, and we give a stability and error analysis for a chain with next-nearest neighbor interactions. We identify conditions for the pair potential, electron density function, and embedding function so that the lattice stability of the atomistic and the EAM-QNL models are asymptotically equal. read less NOT USED (high confidence) P. Zhu, Y.-zhong Hu, and H. Wang, “Atomistic simulations of the effect of a void on nanoindentation response of nickel,” Science China Physics, Mechanics and Astronomy. 2010. link Times cited: 9 NOT USED (high confidence) R. E. Jones, J. A. Zimmerman, J. Oswald, and T. Belytschko, “An atomistic J-integral at finite temperature based on Hardy estimates of continuum fields,” Journal of Physics: Condensed Matter. 2010. link Times cited: 38 Abstract: In this work we apply a material-frame, kernel-based estimat… read moreAbstract: In this work we apply a material-frame, kernel-based estimator of continuum fields to atomic data in order to estimate the J-integral for the analysis of an atomically sharp crack at finite temperatures. Instead of the potential energy appropriate for zero temperature calculations, we employ the quasi-harmonic free energy as an estimator of the Helmholtz free energy required by the Eshelby stress in isothermal conditions. We employ the simplest of the quasi-harmonic models, the local harmonic model of LeSar and co-workers, and verify that it is adequate for correction of the zero temperature J-integral expression for various deformation states for our Lennard-Jones test material. We show that this method has the properties of: consistency among the energy, stress and deformation fields; path independence of the contour integrals of the Eshelby stress; and excellent correlation with linear elastic fracture mechanics theory. read less NOT USED (high confidence) J. Feng, B. Xiao, L. Liu, J.-chao Chen, Y. Du, and R. Zhou, “Molecular dynamical simulation of the behavior of early precipitated stage in aging process in dilute Cu–Cr alloy,” Journal of Applied Physics. 2010. link Times cited: 2 Abstract: The aging behaviors of Cu–Cr alloys in the early stage at di… read moreAbstract: The aging behaviors of Cu–Cr alloys in the early stage at different temperatures are investigated by molecular dynamics simulations. First principles potentials are used for the interactions between Cu and Cr atoms. The initial behavior of precipitation is characterized by transmission electron microscope and electron energy disperse spectroscopy. The results showed that Cu–Cr supersaturated solid solution is thermodynamically unstable. The mean-square displacements of the atoms are used to describe the diffusivity. At room temperature, the atoms only show harmonic vibrations near the equilibrium positions. The mutual diffusion at 873 K is different from the unidirectional diffusion in low temperatures. The calculation shows that aging process is accelerated with increasing temperature, which is not only due to the lower diffusion activation energy of Cr at higher temperature, but also because Cu atoms are also participated in the aging process. When “aging” at 1073 K, the precipitation of Cr element is dissolved again into Cu matrix, which is an “over-aging” state of Cu–Cr alloy at high temperature. read less NOT USED (high confidence) T. Delph and J. Zimmerman, “Prediction of instabilities at the atomic scale,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 17 Abstract: Atomic-scale instabilities, in which atomic bonds are broken… read moreAbstract: Atomic-scale instabilities, in which atomic bonds are broken and reform as the body shifts into a lower-energy configuration, are responsible for a wide range of material behaviours of interest. Building upon previous work, we outline here the construction of a criterion for the prediction of such instabilities. The criterion is implemented within the context of the well-known embedded atom method family of interatomic potentials. We present two examples of the application of this criterion: oriented cavitation in an FCC crystal due to uniform triaxial stretching and dislocation nucleation due to nanoindentation of the (0 0 1) face of an FCC crystal. read less NOT USED (high confidence) G. Jia, Y. Liu, B. Yang, and D. Liu, “Molecular dynamics simulation on thermodynamic properties of Pb-Ag alloys,” Rare Metals. 2010. link Times cited: 5 NOT USED (high confidence) K. Aït-Mansour and O. Gröning, “Comment on ‘Ag organisation on Ni(111) surface’ [Surface Science 602 (2008) 2363],” Surface Science. 2010. link Times cited: 2 NOT USED (high confidence) T. Tokumasu and D. Ito, “A Molecular Dynamics Study for the Dissociation Phenomena of Gas Molecule on Metal Surface,” E-journal of Surface Science and Nanotechnology. 2010. link Times cited: 3 Abstract: Dissociation phenomena of gas molecule on a metal surface, e… read moreAbstract: Dissociation phenomena of gas molecule on a metal surface, especially, the effect of motion of a gas molecule on dissociation probability of the molecule on a metal surface was analyzed by Molecular Dynamics (MD) method. Platinum (111) surface and hydrogen were chosen as the metal surface and the gas molecule, respectively. Embedded Atom Method (EAM) was used to express the interaction potential as the functional of the electron density. Parameters or functions of the EAM potential were determined so that the characteristics of the interaction potential obtained by the EAM method were consistent with those obtained by Density Functional Theory (DFT). Dissociation probabilities at specific sites (top, brg and fcc) were obtained by MD method against impinging energy. On the other hand, the dissociation probabilities were determined from dissociation barriers of the sites and orientations of hydrogen molecule. These results were compared with each other and the effect of motion of atoms or molecules on dissociation probability was analyzed. [DOI: 10.1380/ejssnt.2010.211] read less NOT USED (high confidence) G. Bonny and R. Pasianot, “Gauge transformations to combine multi-component many-body interatomic potentials,” Philosophical Magazine Letters. 2010. link Times cited: 19 Abstract: Many-body interatomic potentials play an important role in a… read moreAbstract: Many-body interatomic potentials play an important role in atomistic modelling of materials. For pure elements it is known that there exist gauge transformations that can change the form of the potential functions without modifying its properties. These same transformations, however, fail when applied to alloys. Even though different research groups may use the same potentials to describe pure elements, the gauges employed for fitting alloys will generally be different. In this scenario, it is a priori impossible to merge them into one potential describing the combined system, and thus no advantage is taken from state-of-the-art developments in the literature. Here, we generalise the gauge transformations applied to pure species in order to leave the properties of alloys invariant. Based on these transformations, a strategy to merge potentials developed within different gauges is presented, aiming at the description of the combined system. Advantage of existing state-of-the-art potentials is so taken, thus focusing the efforts on fitting only the missing interactions. Such a procedure constitutes a helpful tool for the development of potentials targeted to alloys of increased complexity, while maintaining the description quality of their constituents. read less NOT USED (high confidence) A. Abdrashitov, D. Kryzhevich, K. Zolnikov, and S. Psakhie, “Simulation of nanoparticles with block structure formation by electric dispertion of metal wire,” Procedia Engineering. 2010. link Times cited: 7 NOT USED (high confidence) D. Kryzhevich, K. Zolnikov, and S. Psakhie, “Simulation of plastic deformation initiation in crystal materials under dynamic loading,” Procedia Engineering. 2010. link Times cited: 0 NOT USED (high confidence) E. Chulkov et al., “Electronic structure and excitations on clean and nanostructured metal surfaces,” The European Physical Journal B. 2010. link Times cited: 2 NOT USED (high confidence) K. Mirabbaszadehi, P. Nayebiii, S. Saramad, and E. Zaminpayma, “Molecular Dynamics Simulation of Al Energetic Nano Cluster Impact (ECI) onto the Surface.” 2010. link Times cited: 0 Abstract: O n the atomic scale, Molecular Dynamic (MD) Simulation of N… read moreAbstract: O n the atomic scale, Molecular Dynamic (MD) Simulation of Nano Al cluster impact on Al (100) substrate surface has been carried out for energies of 1-20 eV/atom to understand quantitatively the interaction mechanisms between the cluster atoms and the substrate atoms. The many body Embedded Atom Method (EAM) was used in this simulation. We investigated the maximum substrate temperature Tmax and t he time tmax within which this temperature is reached as a funct ion of cluster sizes. The temperature Tmax is l inearly proportional to both energy per atom and total cluster energy. For the constant energy per atom and the cluster size increase, the correlated collisions rapidly transferred energy to the substrate, and the time tmax a pproached a constant value. We investigated the temperature Tmax dependence on the total energy ET and the c luster size. We showed that the cluster implantation and sputtering atoms from the surface are affected by the cluster size and kinetic energy of the clusters. Finally, time dependence of the number Ndis of disordered a toms in the substrate was observed. read less NOT USED (high confidence) M. Taheri, J. Sebastian, B. Reed, D. Seidman, and A. Rollett, “Site-specific atomic scale analysis of solute segregation to a coincidence site lattice grain boundary.,” Ultramicroscopy. 2010. link Times cited: 29 NOT USED (high confidence) G. Benedek et al., “Theory of surface phonons at metal surfaces: recent advances,” Journal of Physics: Condensed Matter. 2010. link Times cited: 55 Abstract: Recent studies of the surface dynamics of Al(001) and Cu(111… read moreAbstract: Recent studies of the surface dynamics of Al(001) and Cu(111) based on density functional perturbation theory have substantiated the existence of subsurface optical phonon resonances of all three polarizations, thus confirming early predictions of the embedded-atom method. The hybridization of the shear-vertical optical resonance with the longitudinal acoustic phonon branch accounts for the ubiquitous anomalous acoustic resonance as an intrinsic feature of metal surfaces. The DFPT calculation of the phonon-induced surface charge density oscillations shows that helium atom scattering spectroscopy (HAS) can indeed probe subsurface resonances. This opens new perspectives to HAS for the measurement of subsurface phonon dispersion curves in thin films, as proved by recent HAS studies on Pb and Fe ultrathin films on copper. After discussing these recent advances, this paper briefly reviews other important trends of surface dynamics expressed in recent years. read less NOT USED (high confidence) Y. Mishin, M. Asta, and J. Li, “Atomistic modeling of interfaces and their impact on microstructure and properties,” Acta Materialia. 2010. link Times cited: 418 NOT USED (high confidence) H. Akbarzadeh, H. Abroshan, and G. Parsafar, “Surface free energy of platinum nanoparticles at zero pressure: A molecular dynamic study,” Solid State Communications. 2010. link Times cited: 13 NOT USED (high confidence) B. Szpunar and R. W. Smith, “A molecular dynamics simulation of the diffusion of the solute (Au) and the self-diffusion of the solvent (Cu) in a very dilute liquid Cu–Au solution,” Journal of Physics: Condensed Matter. 2010. link Times cited: 7 Abstract: The identification of the manner in which a solute diffusion… read moreAbstract: The identification of the manner in which a solute diffusion coefficient (D) might vary with temperature (T) in a fused metal or semimetal has led to considerable experimental study and some theoretical analysis. However, the conclusions of this work are inconsistent. In the present work, molecular dynamics studies of diffusion of a very dilute solute (Au) in liquid Cu are presented. Using the simple Enskog theory of diffusion, it is shown that the ratio of the diffusion constant of the solute to the diffusion constant of the solvent for a very dilute solution is approximately constant. This prediction is confirmed by molecular dynamics simulations although the values of ratios agree only within 20%–25%. In agreement with experiment, current simulations predict that within the usually investigated temperature range, the diffusion coefficient is linearly dependent on temperature. A very small contribution of parabolic behavior can only be observed for a temperature range much wider than that available for physical experiments due to materials limitations. read less NOT USED (high confidence) Y. Shim and J. Amar, “Complex behavior in a simple system: Low-temperature Ag/Ag(100) growth revisited,” Physical Review B. 2010. link Times cited: 7 Abstract: The experimentally observed nonmonotonic temperature depende… read moreAbstract: The experimentally observed nonmonotonic temperature dependence of the surface roughness in Ag/Ag(100) growth over the temperature range $T=55\char21{}180\text{ }\text{K}$ is examined. In general, we find that the surface roughness depends sensitively on a competition between a variety of low-barrier processes including downward funneling of depositing atoms, island relaxation via edge zipping and edge diffusion, atom attraction, and concerted interlayer diffusion at kinks. The short-range attraction of depositing atoms to microprotrusions also plays a crucial role in determining the surface roughness, especially at low temperature. By taking these processes into account in our simulations, good agreement with experiment is obtained over the entire temperature range. read less NOT USED (high confidence) P. Zhu, Y.-zhong Hu, and H. Wang, “Molecular dynamics simulations of atomic-scale friction in diamond-silver sliding system,” Chinese Science Bulletin. 2009. link Times cited: 8 NOT USED (high confidence) M. Steinhauser and S. Hiermaier, “A Review of Computational Methods in Materials Science: Examples from Shock-Wave and Polymer Physics,” International Journal of Molecular Sciences. 2009. link Times cited: 90 Abstract: This review discusses several computational methods used on … read moreAbstract: This review discusses several computational methods used on different length and time scales for the simulation of material behavior. First, the importance of physical modeling and its relation to computer simulation on multiscales is discussed. Then, computational methods used on different scales are shortly reviewed, before we focus on the molecular dynamics (MD) method. Here we survey in a tutorial-like fashion some key issues including several MD optimization techniques. Thereafter, computational examples for the capabilities of numerical simulations in materials research are discussed. We focus on recent results of shock wave simulations of a solid which are based on two different modeling approaches and we discuss their respective assets and drawbacks with a view to their application on multiscales. Then, the prospects of computer simulations on the molecular length scale using coarse-grained MD methods are covered by means of examples pertaining to complex topological polymer structures including star-polymers, biomacromolecules such as polyelectrolytes and polymers with intrinsic stiffness. This review ends by highlighting new emerging interdisciplinary applications of computational methods in the field of medical engineering where the application of concepts of polymer physics and of shock waves to biological systems holds a lot of promise for improving medical applications such as extracorporeal shock wave lithotripsy or tumor treatment. read less NOT USED (high confidence) B. Sadigh, P. Erhart, A. Stukowski, and A. Caro, “Composition-dependent interatomic potentials: A systematic approach to modelling multicomponent alloys,” Philosophical Magazine. 2009. link Times cited: 16 Abstract: We propose a simple scheme to construct composition-dependen… read moreAbstract: We propose a simple scheme to construct composition-dependent interatomic potentials for multicomponent systems that, when superposed onto the potentials for the pure elements, can reproduce not only the heat of mixing of the solid solution in the entire concentration range but also the energetics of a wider range of configurations including intermetallic phases. We show that an expansion in cluster interactions provides a way to systematically increase the accuracy of the model, and that it is straightforward to generalise this procedure to multicomponent systems. Concentration-dependent interatomic potentials can be built upon almost any type of potential for the pure elements including embedded atom method (EAM), modified EAM, bond-order, and Stillinger–Weber type potentials. In general, composition-dependent N-body terms in the total energy lead to explicit (N + 1)-body forces, which potentially render them computationally expensive. We present an algorithm that overcomes this problem and that can speed up the calculation of the forces for composition-dependent pair potentials in such a way as to make them computationally comparable in efficiency and scaling behaviour to standard EAM potentials. We also discuss the implementation in Monte Carlo simulations. Finally, we exemplarily review the composition-dependent EAM model for the Fe–Cr system. read less NOT USED (high confidence) G. Bonny, R. Pasianot, N. Castin, and L. Malerba, “Ternary Fe–Cu–Ni many-body potential to model reactor pressure vessel steels: First validation by simulated thermal annealing,” Philosophical Magazine. 2009. link Times cited: 206 Abstract: In recent years, the development of atomistic models dealing… read moreAbstract: In recent years, the development of atomistic models dealing with microstructure evolution and subsequent mechanical property change in reactor pressure vessel steels has been recognised as an important complement to experiments. In this framework, a literature study has shown the necessity of many-body interatomic potentials for multi-component alloys. In this paper, we develop a ternary many-body Fe–Cu–Ni potential for this purpose. As a first validation, we used it to perform a simulated thermal annealing study of the Fe–Cu and Fe–Cu–Ni alloys. Good qualitative agreement with experiments is found, although fully quantitative comparison proved impossible, due to limitations in the used simulation techniques. These limitations are also briefly discussed. read less NOT USED (high confidence) G. Bonny, R. Pasianot, and L. Malerba, “Fitting interatomic potentials consistent with thermodynamics: Fe, Cu, Ni and their alloys,” Philosophical Magazine. 2009. link Times cited: 24 Abstract: In computational materials science, many atomistic methods h… read moreAbstract: In computational materials science, many atomistic methods hinge on an interatomic potential to describe material properties. In alloys, besides a proper description of problem-specific properties, a reasonable reproduction of the experimental phase diagram by the potential is essential. In this framework, two complementary methods were developed to fit interatomic potentials to the thermodynamic properties of an alloy. The first method involves the zero Kelvin phase diagram and makes use of the concept of the configuration polyhedron. The second method involves phase boundaries at finite temperature and is based on the cluster variation method. As an example for both techniques, they are applied to the Fe–Cu, Fe–Ni and Cu–Ni systems. The resulting potentials are compared to those found in the literature and are found to reproduce the experimental phase diagram more consistently than the latter. read less NOT USED (high confidence) Z. Jing-xiang, L. Hui, S. Xi-gui, and Z. Jie, “Inverse Monte Carlo study on effective interaction potential of Ag Rh alloy from pair correlation functions,” Chinese Physics B. 2009. link Times cited: 0 Abstract: This paper presents an inverse Monte Carlo method to reconst… read moreAbstract: This paper presents an inverse Monte Carlo method to reconstruct pair interaction potential from pair correlation function. This approach adopts an iterative algorithm on interaction potential to fit known pair correlation function by compelling deviations of canonical average to meet with Hamiltonian parameters on a basis of statistical mechanism. The effective interaction potential between particles in liquid Ag–Rh alloys has been calculated with the inverse Monte Carlo method. It demonstrates an effective and simple way to obtain the effective potential of complex melt systems. read less NOT USED (high confidence) S. Psakhie, A. V. Zheleznyakov, I. Konovalenko, G. E. Rudenskii, and K. Zolnikov, “Influence of structure defects on behavior of unclosed crystal nanostructures,” Russian Physics Journal. 2009. link Times cited: 5 NOT USED (high confidence) N. Okamoto, Y. Fujii, H. Kurihara, and K. Kondo, “Effects of Microstructure of Deposited Sn Films and Orientation Index of Cu Foils on Sn Whisker Formation Using Substitutionally-Deposited Sn Films,” Materials Transactions. 2009. link Times cited: 4 Abstract: The effects of the orientation indexes of Cu foils, used as … read moreAbstract: The effects of the orientation indexes of Cu foils, used as a copper pattern for flexible printed circuits (FPC), and the grain size of substitutionally-deposited crystalline Sn films on Sn whisker formation were investigated. In particular, the relationship between the grain size of substitutionally-deposited Sn films and the structure of intermetallic compound deposits formed at the interface between the substitutionally-deposited Sn films and Cu foils, as a function of aging was examined. Two types of Cu foils were used as substrates in this study. One had granular-shaped grains 0.5~1.0μm in size while other had pillar-shaped grains about 5.0 μm in size. We called the former "with the gelatin additive" and the latter "with the Cl - ion additive" since we used gelatin and Cl- ions for the fabrication of Cu foils. In addition, single crystal Cu (100), (110), (11 1) samples were also used as substrates. Two types of the Sn deposition bath were used in this study, a hydrofluoroboric acid bath and an organic acid bath. The structures of the deposited Sn films and Cu foils were investigated using TEM and SEM. The number of whiskers formed on the Sn-deposited film increased with aging. The number of whiskers formed on the substitutionally-deposited Sn films using the hydrofluoroboric acid bath was larger than that formed using the organic acid bath. The number of whiskers formed on the deposited Sn films on the Cu single crystal (111) was larger than that formed on the deposited Sn films on the other single crystals. The mechanism of inhibiting whisker formation by heat treatment was also investigated. No whiskers were seen on the substitutionally-deposited Sn films with heat treatment. Analysis of TEM selected-area diffraction patterns obtained from the sample subjected to heat treatment indicated the presence of Cu 6 Sn 5 and Cu 3 Sn intermetallic compound deposits near the interface between the deposited Sn films and the Cu foils. The results suggest that the microstructures of the substrates strongly affect the Sn whisker formation on substitutionally-deposited Sn films. read less NOT USED (high confidence) D. Sen and M. Buehler, “Size and Geometry Effects on Flow Stress in Bioinspired de novo Metal‐matrix Nanocomposites,” Advanced Engineering Materials. 2009. link Times cited: 4 Abstract: Metal-based nanocomposites provide great potential for appli… read moreAbstract: Metal-based nanocomposites provide great potential for applications in high hardness and toughness material design. Potential applications include coatings for friction and wearresistant cutting tools, shock impact-dissipating structures, and other tribological applications where strong functional materials are the key to initiate further technological development. [1–4] Recent advances in the development of nanocomposite materials have suggested that a new paradigm of composite design might be to systematically engineer the nanostructural arrangement of components by designing their properties, interfaces, and geometry to tailor desired macroscopic functional properties. These efforts extend earlier studies of creating nanomaterials out of metallic constituents (e.g. nanowires, thin films) toward bulk materials. [5] However, the optimal choice of nanostructural arrangement of material constituents to maximize performance remains unknown, preventing us from systematically carrying out a bottom-up design approach. A variety of biological structural materials such as bone and nacre are known to feature a common ‘‘brick-and-mortar’’ structural motifs at the nanoscale, composed of material constituents with disparate properties (Fig. 1). [6–10] These universal nanostructures are seen to combine inferior building materials, soft protein, and brittle calcite or hydroxyapatite crystals to obtain structures with high strength and high toughness at biological scales. [11–13] Their improved properties have been attributed to their hierarchical structure, as well as their fundamental structural organization of constituting elements at the nanoscale. [14] The biological role of these materials is strongly related to load carrying and armor protection in nature. Based on their intriguing properties, these materials raise an important question whether their design strategies could provide directions for conventional structural engineering material design. However, for materials development, the use of proteins and platelets is not a viable option, because these materials are rather difficult to synthesize and engineer. Here, we propose an alternative approach, based on using metal–metal nanocomposites that utilize the material concepts identified from biological analogs as guiding principles in the design process. However, despite earlier studies, [15,16] the transferability of designs found in biological structures toward conventional metal and ceramic based composites remains an unresolved question, partly because the fundamental mechanisms of how structure and properties are related have not yet been explored. Specifically, the wide parameter space associated with different platelet shapes and orientations has not been described in the literature. read less NOT USED (high confidence) D. A. Thomas, Z. Lin, L. Zhigilei, E. Gurevich, S. Kittel, and R. Hergenröder, “Atomistic modeling of femtosecond laser-induced melting and atomic mixing in Au film - Cu substrate system,” Applied Surface Science. 2009. link Times cited: 36 NOT USED (high confidence) T. Järvi, A. Kuronen, K. Nordlund, and K. Albe, “Low energy cluster deposition of nanoalloys,” Journal of Applied Physics. 2009. link Times cited: 9 Abstract: Low energy deposition of metal alloy nanoclusters is studied… read moreAbstract: Low energy deposition of metal alloy nanoclusters is studied by molecular dynamics simulations. In a previous study, two mechanisms were introduced for epitaxial alignment of elemental clusters: The heating induced by the surface energy released upon impact and the thermally activated dislocation motion. In this study, these mechanisms are shown to dominate for Cu3Ag, Cu3Au, and Cu3Ni clusters as well. The question whether the alloyed nature of the system or the initial chemical ordering of the particles influences epitaxial alignment with a substrate is discussed. Chemical ordering is shown to have a negligible effect due to disordering occurring at the initial stages of deposition. read less NOT USED (high confidence) A. Dongare, L. Zhigilei, A. Rajendran, and B. Lamattina, “Interatomic potentials for atomic scale modeling of metal–matrix ceramic particle reinforced nanocomposites,” Composites Part B-engineering. 2009. link Times cited: 15 NOT USED (high confidence) P. Nayebi and E. Zaminpayma, “Crystallization of Liquid Gold Nanoparticles by Molecular Dynamics Simulation,” Journal of Cluster Science. 2009. link Times cited: 6 NOT USED (high confidence) M. Demkowicz and R. Hoagland, “Simulations of Collision Cascades in Cu–Nb Layered Composites Using an EAM Interatomic Potential.” 2009. link Times cited: 77 Abstract: The embedded atom method (EAM) is used to construct an inter… read moreAbstract: The embedded atom method (EAM) is used to construct an interatomic potential for modelling interfaces in Cu–Nb nanocomposites. Implementation of the Ziegler–Biersack–Littmark (ZBL) model for short-range interatomic interactions enables studies of response to ion bombardment. Collision cascades are modelled in fcc Cu, bcc Nb, and in Cu–Nb layered composites in the experimentally-observed Kurdjumov–Sachs (KS) orientation relation. The interfaces in these composites reduce the number of vacancies and interstitials created per keV of the primary knock-on atom (PKA) by 50–70% compared to fcc Cu or bcc Nb. read less NOT USED (high confidence) J.-min Zhang, F. Wang, and K. Xu, “Self-interstitial configuration in molybdenum studied by modified analytical embedded atom method,” Pramana. 2009. link Times cited: 0 NOT USED (high confidence) V. Král, “The Trajectory Dependent Scattering οf Li Ions by Al Substrate,” Acta Physica Polonica A. 2009. link Times cited: 0 Abstract: Scattering of ions by solids is a powerful tool for studying… read moreAbstract: Scattering of ions by solids is a powerful tool for studying of solid surfaces. Many techniques like secondary ion mass spectroscopy (SIMS) have been developed and they give at present the convincing information about solid surfaces. The purpose of this contribution is to give a description of trajectory dependent scattering of Li ions by Al surface. The trajectory dependent scattering has been reported in [1] for incident Na ions. They show that results of scattering depend on the place of surface by which is a ion scattered. The spectra obtained in these experiments allow to recognize the origin of outgoing ions. One can imagine this situation by a following reasoning. read less NOT USED (high confidence) Q. Peng, X. Zhang, and G. Lu, “Structure, mechanical and thermodynamic stability of vacancy clusters in Cu,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 15 Abstract: The atomic structure, mechanical and thermodynamic stability… read moreAbstract: The atomic structure, mechanical and thermodynamic stability of vacancy clusters in Cu are studied by atomistic simulations. The most stable atomic configuration of small vacancy clusters is determined. The mechanical stability of the vacancy clusters is examined by applying uniaxial and volumetric tensile strains to the system. The yield stress and yield strain of the system are significantly reduced compared with the perfect lattice. The dependence of vacancy formation and binding energy as a function of strain is explored and can be understood from the liquid-drop model. We find that the formation energy of the vacancy clusters decreases monotonically as a function of the uniaxial strain, while the formation energy increases first then decreases under the volumetric tensile strain. The thermodynamic stability of the vacancy clusters is analyzed by calculating the Helmholtz free binding energy and the total probability of dissociation of the vacancy clusters at 300 and 900 K under uniaxial and volumetric strains. We find that although most of the vacancy clusters appear to be thermodynamically stable, some of the intermediate sized clusters have a high probability of dissociation into smaller clusters. read less NOT USED (high confidence) E. C. Do, Y.-H. Shin, and B.-J. Lee, “Atomistic modeling of III–V nitrides: modified embedded-atom method interatomic potentials for GaN, InN and Ga1−xInxN,” Journal of Physics: Condensed Matter. 2009. link Times cited: 26 Abstract: Modified embedded-atom method (MEAM) interatomic potentials … read moreAbstract: Modified embedded-atom method (MEAM) interatomic potentials for the Ga–N and In–N binary and Ga–In–N ternary systems have been developed based on the previously developed potentials for Ga, In and N. The potentials can describe various physical properties (structural, elastic and defect properties) of both zinc-blende and wurtzite-type GaN and InN as well as those of constituent elements, in good agreement with experimental data or high-level calculations. The potential can also describe the structural behavior of Ga1−xInxN ternary nitrides reasonably well. The applicability of the potentials to atomistic investigations of atomic/nanoscale structural evolution in Ga1−xInxN multi-component nitrides during the deposition of constituent element atoms is discussed. read less NOT USED (high confidence) B. Shan et al., “First-principles-based embedded atom method for PdAu nanoparticles,” Physical Review B. 2009. link Times cited: 48 Abstract: One of the key problems in studying alloy nanoparticle catal… read moreAbstract: One of the key problems in studying alloy nanoparticle catalysis is their surface morphology and segregation behavior. We have developed an accurate embedded atom method (EAM) potential and employed it in the simulation of PdAu metal alloy nanoparticles. The potential was parameterized based on an extensive set of density-functional-theory (DFT) calculations of metal clusters in addition to bulk-alloy properties. The EAM potential accurately reproduces DFT energies of both bulk PdAu alloys and small nanoparticles. We utilized the developed EAM potential in a Monte Carlo simulation of PdAu nanoparticles ranging from 55-atom $(\ensuremath{\sim}1\text{ }\text{nm})$ to 5083-atom particles $(\ensuremath{\sim}4.5\text{ }\text{nm})$. The effects of different factors (particle size, temperature, and composition ratios) on the segregation behavior of PdAu alloy are examined. Our simulation results quantitatively reveal the extent of surface segregation and a strong dependence of surface morphology on the nanoparticle size. read less NOT USED (high confidence) S. Li, M. Sellers, C. Basaran, A. Schultz, and D. Kofke, “Lattice Strain Due to an Atomic Vacancy,” International Journal of Molecular Sciences. 2009. link Times cited: 42 Abstract: Volumetric strain can be divided into two parts: strain due … read moreAbstract: Volumetric strain can be divided into two parts: strain due to bond distance change and strain due to vacancy sources and sinks. In this paper, efforts are focused on studying the atomic lattice strain due to a vacancy in an FCC metal lattice with molecular dynamics simulation (MDS). The result has been compared with that from a continuum mechanics method. It is shown that using a continuum mechanics approach yields constitutive results similar to the ones obtained based purely on molecular dynamics considerations. read less NOT USED (high confidence) B. Feng, Z. Li, and X. Zhang, “Role of phonon in the thermal and electrical transports in metallic nanofilms,” Journal of Applied Physics. 2009. link Times cited: 33 Abstract: The electronic thermal transport in metallic nanofilms has b… read moreAbstract: The electronic thermal transport in metallic nanofilms has been extensively studied. There are, however, rare reports on the influence of phonon on the thermal and electrical transports in metallic films. In the present work, equilibrium molecular dynamics with embedded-atom method is used to investigate the lattice thermal conductivity in a single-crystalline copper film. The results show that the lattice contribution to the total thermal conductivity is relatively small, nevertheless enhanced compared to that in bulk copper. The low-dimensional phonon system in metallic films is characterized by the elastic continuum model. In addition to the traditional boundary scattering, the reduced phonon group velocity and reduced Debye temperature are taken into account to discuss the finite size effect on phonon transport and electronic thermal resistivity and electrical resistivity. read less NOT USED (high confidence) G. Yun and H. S. Park, “Surface stress effects on the bending properties of fcc metal nanowires,” Physical Review B. 2009. link Times cited: 89 Abstract: The major purpose of this work is to investigate surface str… read moreAbstract: The major purpose of this work is to investigate surface stress effects on the bending behavior and properties of 100 /100 gold nanowires with both fixed/fixed and fixed/free boundary conditions. The results are obtained through utilization of the recently developed surface Cauchy-Born model, which captures surface stress effects on the elastic properties of nanostructures through a three-dimensional, nonlinear finite element formulation. There are several interesting findings in the present work. First, we quantify the stress and displacement fields that result in the nanowires due to bending deformation. In doing so, we find that regardless of boundary condition, the stresses that are present in the nanowires due to deformation induced by surface stresses prior to any applied bending deformation dominate any stresses that are generated by the bending deformation unless very large 5% bending strains are applied. In contrast, when the stresses and displacements induced by surface stresses prior to bending are subtracted from the stress and displacement fields of the bent nanowires, we find that the bending stresses and displacements do match the solutions expected from bulk continuum beam theory, but only within the nanowire bulk, and not at the nanowire surfaces. Second, we find that the deformation induced by surface stresses also has a significant impact on the nanowire Young’s modulus that is extracted from the bending simulations, where a strong boundary-condition dependence is also found. By comparing all results to those that would be obtained using various linear surface-elastic theories, we demonstrate that a nonlinear, finite deformation formulation that captures changes in both bulk- and surfaceelastic properties resulting from surface stress-induced deformation is critical to reproducing the experimentally observed boundary-condition dependence in Young’s modulus of metal nanowires. Furthermore, we demonstrate that linear surface-elastic theories based solely on the surface energy erroneously predict an increase in Young’s modulus with decreasing nanowire size regardless of boundary condition. In contrast, while the linear surface-elastic theories based upon the Gurtin and Murdoch formalism can theoretically predict elastic softening with decreasing size, we demonstrate that, regardless of boundary condition, the stiffening due to the surface stress dominates the softening due to the surface stiffness for the range of nanowire geometries considered in the present work. Finally, we determine that the nanowire Young’s modulus is essentially identical when calculated via either bending or resonance for both boundary conditions, indicating that surface effects have a similar impact on the elastic properties of nanowires for both loading conditions. read less NOT USED (high confidence) T. Zhou, X. Yang, and C.-yao Chen, “Quasicontinuum simulation of single crystal nano-plate with a mixed-mode crack,” International Journal of Solids and Structures. 2009. link Times cited: 13 NOT USED (high confidence) F. Römer, S. Braun, and T. Kraska, “Development of an EAM potential for zinc and its application to the growth of nanoparticles.,” Physical chemistry chemical physics : PCCP. 2009. link Times cited: 10 Abstract: In the context of the investigation of particle formation, a… read moreAbstract: In the context of the investigation of particle formation, a potential model by means of the embedded atom method is developed for the hexagonal close packed metal zinc. This type of model includes many-body interactions caused by delocalised electrons in metals. The effective core charge as function of the distance is calculated here by an integral over the electron distribution function rather than fitting it to experimental data. In addition, the dimer potential is included in the parameterisation because we focus on the formation of nanoparticles from the vapour phase. With this potential model, the growth of zinc clusters consisting of 125 to 1000 atoms is investigated, which takes place at elevated temperatures in a liquid-like cluster state. The growing clusters are embedded in an argon carrier gas atmosphere which regulates the cluster temperature. The average thermal expansion of the clusters and the different lattice constants are analysed. For the determination of the cluster structure, the common-neighbour analysis method is extended to hexagonal close packed surface structures. During growth, small clusters with less than approximately 60 atoms develop transient icosahedral structure before transforming into hexagonal close-packed structure. The surface of the clusters exhibits a transformation from planes with high surface energy to the most stable ones. Besides ambiguous surface structures the final clusters are almost completely in an hexagonal close packed structure. read less NOT USED (high confidence) G. J. Soldano and M. Mariscal, “On the structural and mechanical properties of Fe-filled carbon nanotubes—a computer simulation approach,” Nanotechnology. 2009. link Times cited: 12 Abstract: The structural and mechanical properties of single-and multi… read moreAbstract: The structural and mechanical properties of single-and multi-walled carbon nanotubes filled with iron nanowires are studied using a recent parameterization of the modified embedded atom model. We have analyzed the effect of different crystal structures of iron (bcc and fcc) inside carbon nanotubes of different topographies. We have computed strain energy versus strain curves for pure systems: Fe nanowires, carbon and Fe-filled carbon nanotubes. A noticeable difference is found when these monatomic systems are joined to form iron-capped nanowires and where multi-layers of graphite are added to the nanotubes. read less NOT USED (high confidence) A. Lyalin, A. Hussien, A. Solov’yov, and W. Greiner, “Impurity effect on the melting of nickel clusters as seen via molecular dynamics simulations,” Physical Review B. 2009. link Times cited: 23 Abstract: We demonstrate that the addition of a carbon impurity leads … read moreAbstract: We demonstrate that the addition of a carbon impurity leads to significant changes in the thermodynamic properties of a ${\text{Ni}}_{147}$ cluster. The magnitude of the change induced is dependent on the parameters of the Ni-C interaction. Hence, thermodynamic properties of Ni clusters can be effectively tuned by the addition of a particular type of impurity. We also show that the presence of a carbon impurity considerably changes the mobility and diffusion of atoms in the Ni cluster at temperatures close to its melting point. The calculated diffusion coefficients of the carbon impurity in the Ni cluster can be used as a reliable estimate of the growth rate of carbon nanotubes. read less NOT USED (high confidence) T. Uehara, C. Asai, and N. Ohno, “Molecular dynamics simulation of shape memory behaviour using a multi-grain model,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 21 Abstract: Shape-memory behaviour in multi-grain material is simulated … read moreAbstract: Shape-memory behaviour in multi-grain material is simulated using a molecular dynamics method. An embedded-atom-method potential for NiAl alloy is applied, and a sequence of conditions including loading, unloading, heating and cooling is imposed. Two types of grain arrangement are used, and the deformation and shape recovery due to phase transformation are observed for both models. The stress–strain relation is revealed to draw a hysteresis loop, and the individual curves are smoother than those previously obtained from a single-crystal model. The deformation mechanism during loading is discussed using local structure analysis. Local deformation is initiated at the grain boundaries, and the deformed region propagates along the twin plane in the grain. The propagation is then obstructed by the grain boundaries, and a band pattern of the deformed area is formed. The influence of the grain shape and distribution, as well as the crystal orientation of each grain, on the deformation behaviour is also investigated. Qualitatively common features in the deformation mechanism and stress–strain relation are observed despite different grain distributions, while the critical values in stress vary, owing to the crystal orientations of the grains. read less NOT USED (high confidence) R. W. Smith, P. Scott, and B. Szpunar, “Solute Diffusion in Nonionic Liquids—Effects of Gravity,” Annals of the New York Academy of Sciences. 2009. link Times cited: 4 Abstract: We have been engaged in examining the influence of gravity o… read moreAbstract: We have been engaged in examining the influence of gravity on the results of experiments to measure the variation of solute diffusion coefficients (D) with temperature (T) in fused metals and semimetals since our first STS flights in 1992. These early experiments, conducted with the in situ g‐jitter of the shuttle, showed the near‐parabolic variation of D with T reported by others. However, with the aid of the Canadian Space Agency's microgravity isolation mount (MIM) to isolate the diffusion facility from the existing g‐jitter of the Russian space station MIR, we showed that in all the alloy systems and over the temperature range studied, D increased linearly with T. If the isolating system was deactivated, then the more familiar parabolic relationship appeared. We have always assumed that the values of D measured using the MIM would be closer to the intrinsic values for the alloy system considered; to test this contention, we have been involved in two modeling activities. The first has been to estimate the effects of g‐jitter‐level disturbances on solute distributions in long capillary diffusion couples. The second has been to conduct various molecular dynamics modeling studies of solute diffusion. This paper presents results of these studies. read less NOT USED (high confidence) J. Zhang, T. Sun, Y. Yan, Y. Liang, and S. Dong, “Molecular dynamics study of groove fabrication process using AFM-based nanometric cutting technique,” Applied Physics A. 2009. link Times cited: 44 NOT USED (high confidence) M. Stan, “MULTI-SCALE MODELS AND SIMULATIONS OF NUCLEAR FUELS,” Nuclear Engineering and Technology. 2009. link Times cited: 29 Abstract: Theory-based models and high performance simulations are bri… read moreAbstract: Theory-based models and high performance simulations are briefly reviewed starting with atomistic methods, such as Electronic Structure calculations, Molecular Dynamics, and Monte Carlo, continuing with meso-scale methods, such as Dislocation Dynamics and Phase Field, and ending with continuum methods that include Finite Element and Finite Volume. Special attention is paid to relating thermo-mechanical and chemical properties of the fuel to reactor parameters. By inserting atomistic models of point defects into continuum thermo-chemical calculations, a model of oxygen diffusivity in UO UO2+x is developed and used to predict point defect concentrations, oxygen diffusivity, and fuel stoichiometry at various temperatures and oxygen pressures. The simulations of coupled heat transfer and species diffusion demonstrate that including the dependence of thermal conductivity and density on composition can lead to changes in the calculated centerline temperature and thermal expansion displacements that exceed 5%. A review of advanced nuclear fuel performance codes reveals that the many codes are too dedicated to specific fuel forms and make excessive use of empirical correlations in describing properties of materials. The paper ends with a review of international collaborations and a list of lessons learned that includes the importance of education in creating a large pool of experts to cover all necessary theoretical, experimental, and computational tasks. read less NOT USED (high confidence) B. Onat, M. Konuk, S. Durukanoğlu, and G. Dereli, “Energetics and atomic relaxations of Cu nanowires: the effect of local strain and cross-sectional area,” Nanotechnology. 2009. link Times cited: 4 Abstract: We have calculated the activation energies for several singl… read moreAbstract: We have calculated the activation energies for several single atom and vacancy diffusion processes on Cu nanowires with the axial orientation of , using the nudged elastic band technique based on the interaction potential obtained from the embedded atom method. It is shown that the dimer-initiated local strain and its relief at the transition state have a significant effect on the characteristics of self-surface diffusion mechanisms on nanowires. Contrary to the case for cylindrical multishell-type Cu nanowires, the vacancy formation energy for rectangular nanowires is maximum in the core region and is nearly zero at the corner of the nanowire. In addition, the activation energy barriers for the vacancy diffusion processes taking place in the core region are found to be higher than those occurring near the corner of the nanowire. Our calculations further show that the vacancy diffusion processes taking place near the corner of the wire are dictated by the lower coordination of the surrounding atoms. From the structural investigation of nanowires, we have also established that multilayer relaxations for rectangular nanowires with smaller cross-sectional area cannot be defined. read less NOT USED (high confidence) D. Graves and P. Brault, “Molecular dynamics for low temperature plasma–surface interaction studies,” Journal of Physics D: Applied Physics. 2009. link Times cited: 112 Abstract: The mechanisms of physical and chemical interactions of low … read moreAbstract: The mechanisms of physical and chemical interactions of low temperature plasmas with surfaces can be fruitfully explored using molecular dynamics (MD) simulations. MD simulations follow the detailed motion of sets of interacting atoms through integration of atomic equations of motion, using inter-atomic potentials that can account for bond breaking and formation that result when energetic species from the plasma impact surfaces. This paper summarizes the current status of the technique for various applications of low temperature plasmas to material processing technologies. The method is reviewed, and commonly used inter-atomic potentials are described. Special attention is paid to the use of MD in understanding various representative applications, including tetrahedral amorphous carbon film deposition from energetic carbon ions, the interactions of radical species with amorphous hydrogenated silicon films, silicon nanoparticles in plasmas, and plasma etching. read less NOT USED (high confidence) G. Nandipati et al., “Parallel kinetic Monte Carlo simulations of Ag(111) island coarsening using a large database,” Journal of Physics: Condensed Matter. 2009. link Times cited: 29 Abstract: The results of parallel kinetic Monte Carlo (KMC) simulation… read moreAbstract: The results of parallel kinetic Monte Carlo (KMC) simulations of the room-temperature coarsening of Ag(111) islands carried out using a very large database obtained via self-learning KMC simulations are presented. Our results indicate that, while cluster diffusion and coalescence play an important role for small clusters and at very early times, at late time the coarsening proceeds via Ostwald ripening, i.e. large clusters grow while small clusters evaporate. In addition, an asymptotic analysis of our results for the average island size S(t) as a function of time t leads to a coarsening exponent n = 1/3 (where S(t)∼t2n), in good agreement with theoretical predictions. However, by comparing with simulations without concerted (multi-atom) moves, we also find that the inclusion of such moves significantly increases the average island size. Somewhat surprisingly we also find that, while the average island size increases during coarsening, the scaled island-size distribution does not change significantly. Our simulations were carried out both as a test of, and as an application of, a variety of different algorithms for parallel kinetic Monte Carlo including the recently developed optimistic synchronous relaxation (OSR) algorithm as well as the semi-rigorous synchronous sublattice (SL) algorithm. A variation of the OSR algorithm corresponding to optimistic synchronous relaxation with pseudo-rollback (OSRPR) is also proposed along with a method for improving the parallel efficiency and reducing the number of boundary events via dynamic boundary allocation (DBA). A variety of other methods for enhancing the efficiency of our simulations are also discussed. We note that, because of the relatively high temperature of our simulations, as well as the large range of energy barriers (ranging from 0.05 to 0.8 eV), developing an efficient algorithm for parallel KMC and/or SLKMC simulations is particularly challenging. However, by using DBA to minimize the number of boundary events, we have achieved significantly improved parallel efficiencies for the OSRPR and SL algorithms. Finally, we note that, among the three parallel algorithms which we have tested here, the semi-rigorous SL algorithm with DBA led to the highest parallel efficiencies. As a result, we have obtained reasonable parallel efficiencies in our simulations of room-temperature Ag(111) island coarsening for a small number of processors (e.g. Np = 2 and 4). Since the SL algorithm scales with system size for fixed processor size, we expect that comparable and/or even larger parallel efficiencies should be possible for parallel KMC and/or SLKMC simulations of larger systems with larger numbers of processors. read less NOT USED (high confidence) J. Rogan et al., “Diversity driven unbiased search of minimum energy cluster configurations,” Journal of Physics: Condensed Matter. 2009. link Times cited: 13 Abstract: The determination of the spatial distributions that atoms ad… read moreAbstract: The determination of the spatial distributions that atoms adopt to form condensed matter is a problem of crucial importance, since most physical properties depend on the atomic arrangement. This is especially relevant for clusters, where periodicity is nonexistent. Several optimization procedures have been implemented to tackle this problem, with ever increasing success. Here we put forward a search scheme which preserves as large a diversity as allowed by the use of phenomenological potentials, generating in an unbiased fashion a bank of configurations to be explored; a procedure we denominate diversity driven unbiased search (DDUS). It consists in the generation, using phenomenological potentials, of a data bank of putative minima rather than a single, or just a few, configurations which are based on the conformational space annealing method (CSA). All of the configurations in the bank are thereafter refined by means of DFT computations. Certainly, in spite of our efforts to generate a bank as diverse as possible, not all relevant structures might be included in it, since quantum effects are ignored. The procedure is applied to several examples of rhodium, palladium, silver, platinum and gold clusters, between 5 and 23 atoms in size. The main conclusion we reach is that unbiased search, among a significant number of candidates, quite often leads to rather unexpectedly low symmetry configurations, which turn out to be the lowest energy ones within our scheme. read less NOT USED (high confidence) G. Wu, G. Lu, C. García-Cervera, and E. Weinan, “Density-gradient-corrected embedded atom method,” Physical Review B. 2009. link Times cited: 9 Abstract: Through detailed comparisons between Embedded Atom Method (E… read moreAbstract: Through detailed comparisons between Embedded Atom Method (EAM) and first-principles calculations for Al, we find that EAM tends to fail when there are large electron density gradients present. We attribute the observed failures to the violation of the uniform density approximation (UDA) underlying EAM. To remedy the insufficiency of UDA, we propose a gradient-corrected EAM model which introduces gradient corrections to the embedding function in terms of exchange-correlation and kinetic energies. Based on the perturbation theory of "quasiatoms" and density functional theory, the new embedding function captures the essential physics missing in UDA, and paves the way for developing more transferable EAM potentials. With Voter-Chen EAM potential as an example, we show that the gradient corrections can significantly improve the transferability of the potential. read less NOT USED (high confidence) S. Maruyama, “Molecular Dynamics Method for Micro/Nano Systems.” 2009. link Times cited: 13 Abstract: Molecular dynamics simulations are becoming more important a… read moreAbstract: Molecular dynamics simulations are becoming more important and more practical for microscale and nanoscale heat transfer problems. For example, studies of basic mechanisms of heat transfer such as phase change demand the understanding of microscopic liquid-solid contact phenomena. The efficient heat transfer at a three-phase interface (evaporation and condensation of liquid on a solid surface) becomes the singular problem in the macroscopic treatment. The nucleation theory of liquid droplets in vapor or of vapor bubbles in liquid sometimes needs to take account of nuclei of the size of molecular clusters. The effect of the surfactant on the heat and mass transfer through liquid-vapor interface is also an example of the direct effect of molecular scale phenomena on the macroscopic heat and mass transfer. Even though there has been much effort of extending our macroscopic analysis to extremely microscopic conditions in space (micrometer and nanometer scales), time (microseconds, nanoseconds and picoseconds), and rate (extremely high heat flux), there are certain limitations in the extrapolations. Hence, the bottom-up approach from molecular level is strongly anticipated. On the other hand, recent advances in microscale and nanoscale heat transfer and in nanotechnology require the detailed understandings of phase change and heat and mass transfer in nanometer and micrometer scale regimes. The chemical engineering processes to generate nanoscale structures such as carbon nanotubes or mesoporous silica structures are examples. The wetting of liquid or absorption is also important since the adhesive force is extremely important for micro/nano system and the creation of extremely large surface area is possible with nanoscale structures. The use of molecular dynamics simulations is straightforward for such a nanoscale system. Here, again, it is important to compare such nanoscale phenomena with macroscopic phenomena, because an analogy to the macroscopic system is often an important strategy in understanding a nanoscale phenomenon. Important physics intrinsic to a nanoscale system is usually found through the rational comparison 4 with a macroscopic system. In this chapter, one of the promising numerical techniques, the classical molecular dynamics method, is overviewed with a special emphasis on applications to inter-phase and heat transfer problems. The molecular dynamics methods have long been used and are well developed as a tool in statistical mechanics and physical chemistry [1, 2]. However, it is a new challenge to extend the method to the spatial and temporal scales of macroscopic heat transfer phenomena [3-6]. On the other hand, the thin film technology related … read less NOT USED (high confidence) H. H. Wu and D. Trinkle, “Cu/Ag EAM potential optimized for heteroepitaxial diffusion from ab initio data,” Computational Materials Science. 2009. link Times cited: 92 Abstract: A binary embedded-atom method (EAM) potential is optimized f… read moreAbstract: A binary embedded-atom method (EAM) potential is optimized for Cu on Ag(1 1 1) by fitting to ab initio data. The fitting database consists of DFT calculations of Cu monomers and dimers on Ag(1 1 1), specifically their relative energies, adatom heights, and dimer separations. We start from the Mishin Cu–Ag EAM potential and first modify the Cu–Ag pair potential to match the FCC/HCP site energy difference then include Cu–Cu pair potential optimization for the entire database. The potential generated from this optimization method gives better agreement to DFT calculations of Cu monomers, dimers, and trimers than previous EAMs as well as a SEAM optimized potential. In trimer calculations, the optimized potential produces the DFT relative energy between FCC and HCP trimers, though a different ground state is predicted. We use the optimized potential to calculate diffusion barriers for Cu monomers, dimers, and trimers. The predicted monomer barrier is the same as DFT, while experimental barriers for monomers and dimers are lower than predicted here. We attribute the difference with experiment to the overestimation of surface adsorption energies by DFT and a simple correction is presented. Our results show that this optimization method is suitable for other heteroepitaxial systems; and that the optimized Cu–Ag EAM can be applied in the study of larger Cu islands on Ag(1 1 1). read less NOT USED (high confidence) A. Gautam and J. Howe, “In situ TEM study of Au–Cu alloy nanoparticle migration and coalescence,” Journal of Materials Science. 2009. link Times cited: 8 NOT USED (high confidence) J. Song and D. Srolovitz, “Mechanism for material transfer in asperity contact,” Journal of Applied Physics. 2008. link Times cited: 31 Abstract: We perform a series of molecular dynamics simulations of asp… read moreAbstract: We perform a series of molecular dynamics simulations of asperity contact and separation in a model metallic system for both symmetric and asymmetric asperity geometries, for loading in the [001], [110], and [111] directions, and for systems with different works of adhesion Γ. We examine contact morphology evolution, force-displacement relations, and the quantity of atoms transferred from one surface to the other NT upon separation with a focus on underlying physical mechanisms that control these. We find that there is a critical work of adhesion, below which no plastic deformation occurs on contact separation and a higher one in which plastic deformation occurs but no material transfer occurs. We interpret these within a model for dislocation nucleation at the crack tip. We observe abrupt changes in the amount of material transferred with increasing work of adhesion that represent thresholds for changes in deformation mechanisms. These depend on the geometry of the contact and the crystallographic orientation relative to the loading direction.We perform a series of molecular dynamics simulations of asperity contact and separation in a model metallic system for both symmetric and asymmetric asperity geometries, for loading in the [001], [110], and [111] directions, and for systems with different works of adhesion Γ. We examine contact morphology evolution, force-displacement relations, and the quantity of atoms transferred from one surface to the other NT upon separation with a focus on underlying physical mechanisms that control these. We find that there is a critical work of adhesion, below which no plastic deformation occurs on contact separation and a higher one in which plastic deformation occurs but no material transfer occurs. We interpret these within a model for dislocation nucleation at the crack tip. We observe abrupt changes in the amount of material transferred with increasing work of adhesion that represent thresholds for changes in deformation mechanisms. These depend on the geometry of the contact and the crystallographic orient... read less NOT USED (high confidence) L. Pan, H. Lee, and C. Lu, “Melting behaviours of nickel nanorods,” The European Physical Journal D. 2008. link Times cited: 6 NOT USED (high confidence) W. Yao and N. Wang, “Monte Carlo simulation of specific heat of liquid Ni–Mo alloys,” Journal of Physics D: Applied Physics. 2008. link Times cited: 4 Abstract: The Monte Carlo method with embedded-atom method (EAM) poten… read moreAbstract: The Monte Carlo method with embedded-atom method (EAM) potential is applied to simulate the specific heat Cp of a liquid Ni–Mo binary alloy system. The simulated Cp value of liquid Ni at the melting temperature is 22.79 J mol−1 K−1, indicating that the simulation method and EAM parameters in simulation are acceptable. The simulated temperature coefficient of the specific heat for liquid Ni is −2.10 × 10−2 J mol−1 K−2. Based on the relationship between system energy and temperature, the various specific heats of liquid Ni–Mo alloys under different undercooling and compositions were determined. The dependence of the specific heat of liquid Ni–Mo alloys on the composition and undercooling is discussed. read less NOT USED (high confidence) J. Wang, R. Hoagland, J. Hirth, and A. Misra, “Atomistic modeling of the interaction of glide dislocations with ‘weak’ interfaces,” Acta Materialia. 2008. link Times cited: 244 NOT USED (high confidence) G. Grochola, I. Snook, and S. Russo, “Influence of substrate morphology on the growth of gold nanoparticles.,” The Journal of chemical physics. 2008. link Times cited: 6 Abstract: We have simulated the vacuum deposition and subsequent growt… read moreAbstract: We have simulated the vacuum deposition and subsequent growth of gold nanoparticles on various substrates in order to explore the effects that substrate morphology has on the resultant morphology of gold nanoparticles. The substrates and conditions explored included, the three low index faces, namely, (111), (100), and (110) for both fcc and bcc crystalline substrate structures, including various substrate lattice constants and temperatures. Firstly, we cataloged the major nanoparticle morphologies produced overall. While some substrates were found to produce a mixture of the main nanoparticle morphologies we were successful in identifying certain substrates and temperature conditions for which only I(h), D(h), or certain fcc crystalline nanoparticles can be grown almost exclusively. The substrate characteristics, temperature conditions, and governing growth dynamics are analyzed. We shed light on the balance between substrate influences and vacuum growth tendencies. From observations we can speculate that a substrate alters both the free energy stability of gold nanoparticles and/or the free energy barriers to transformation between certain morphologies. As such we find that substrates are an effective tool in templating the selective growth of desired nanoparticles or surface nanostructures. read less NOT USED (high confidence) Z. Wang, T. Gu, T. Tada, and S. Watanabe, “Excess-silver-induced bridge formation in a silver sulfide atomic switch,” Applied Physics Letters. 2008. link Times cited: 41 Abstract: Structural properties and electron transport of a Ag2S atomi… read moreAbstract: Structural properties and electron transport of a Ag2S atomic switch composed of Ag–Ag2S–Ag heterostructure are investigated by nonequilibrium Green’s function calculations considering the effect of excess Ag in the Ag2S layer. In addition to confirming experimentally the formation of the Ag bridge inside Ag2S, the bridge is found to consist of units having a structure similar to that of the Ag (111) face in the bulk Ag. The analyses of Mulliken population, transmission spectra, and current-voltage characteristics reveal that the bridge has a conductive and metallic nature. read less NOT USED (high confidence) R. N. Salaway, P. Hopkins, P. Norris, and R. Stevens, “Phonon Contribution to Thermal Boundary Conductance at Metal Interfaces Using Embedded Atom Method Simulations,” International Journal of Thermophysics. 2008. link Times cited: 12 NOT USED (high confidence) J. Paul and S. Narasimhan, “Effect of coordination on bond properties: A first principles study,” Bulletin of Materials Science. 2008. link Times cited: 9 NOT USED (high confidence) H. A. Wu and X. X. Wang, “An atomistic-continuum inhomogeneous material model for the elastic bending of metal nanocantilevers,” Adv. Eng. Softw. 2008. link Times cited: 7 NOT USED (high confidence) C. Acharya, D. I. Sullivan, and C. Turner, “Characterizing the Interaction of Pt and PtRu Clusters with Boron-Doped, Nitrogen-Doped, and Activated Carbon : Density Functional Theory Calculations and Parameterization,” Journal of Physical Chemistry C. 2008. link Times cited: 57 Abstract: Previous density functional theory calculations of Pt and Pt… read moreAbstract: Previous density functional theory calculations of Pt and PtRu clusters on carbon supports have shown that the adsorption energies of these metal clusters increase substantially with substitutional boron defects in the carbon lattice. Here, the stability of metal clusters is further probed with substitutional nitrogen defects and surface functional groups. Also, the dynamics of Pt and Ru atoms on pure and boron-doped carbon are studied as a function of temperature using ab initio molecular dynamics (AIMD) simulations. Although the time scale accessible is limited, the AIMD simulations show reduced mobility on the boron-doped surface. In order to calculate additional dynamic properties of the systems, such as diffusion coefficients, the motion of the metal clusters should be studied for much longer periods of time, which can be accomplished by performing classical molecular dynamics (MD) simulations. Thus, we have parametrized our electronic structure calculations to an analytical Lennard-Jones (LJ) potent... read less NOT USED (high confidence) H. Yildirim, A. Kara, and T. Rahman, “Structural, vibrational and thermodynamic properties of AgnCu34−n nanoparticles,” Journal of Physics: Condensed Matter. 2008. link Times cited: 15 Abstract: We report results of a systematic study of structural, vibra… read moreAbstract: We report results of a systematic study of structural, vibrational and thermodynamical properties of 34-atom bimetallic nanoparticles from the AgnCu34−n family using model interaction potentials as derived from the embedded atom method and invoking the harmonic approximation of lattice dynamics. Systematic trends in the bond length and dynamical properties can be explained largely from arguments based on local coordination and elemental environment. Thus an increase in the number of silver atoms in a given neighborhood introduces a monotonic increase in bond length, while an increase of the copper content does the reverse. Moreover, for the bond lengths of the lowest-coordinated (six and eight) copper atoms with their nearest neighbors (Cu atoms), we find that the nanoparticles divide into two groups with the average bond length either close to (∼2.58 Å) or smaller than (∼2.48 Å) that in bulk copper, accompanied by characteristic features in their vibrational density of states. For the entire set of nanoparticles, we find vibrational modes above the bulk bands of copper/silver. We trace a blue shift in the high-frequency end of the spectrum that occurs as the number of copper atoms increases in the nanoparticles, leading to shrinkage of the bond lengths from those in the bulk. The vibrational densities of states at the low-frequency end of the spectrum scale linearly with frequency as for single-element nanoparticles, with a more pronounced effect for these nanoalloys. The Debye temperature is found to be about one-third of that of the bulk for pure copper and silver nanoparticles, with a non-linear increase as copper atoms increase in the nanoalloy. read less NOT USED (high confidence) J. Harrison, J. Schall, M. T. Knippenberg, G. Gao, and P. Mikulski, “Elucidating atomic-scale friction using molecular dynamics and specialized analysis techniques,” Journal of Physics: Condensed Matter. 2008. link Times cited: 51 Abstract: Because all quantities associated with a given atom are know… read moreAbstract: Because all quantities associated with a given atom are known as a function of time, molecular dynamics simulations can provide unparalleled insight into dynamic processes. Many quantities calculated from simulations can be directly compared to experimental values, while others provide information not available from experiment. For example, the tilt and methyl angles of chains within a self-assembled monolayer and the amount of hydrogen in a diamond-like carbon (DLC) film are measurable in an experiment. In contrast, the atomic contact force on a single substrate atom, i.e., the force on that atom due to the tip atoms only, and the changes in hybridization of a carbon atom within a DLC film during sliding are not quantities that are currently obtainable from experiments. Herein, the computation of many quantities, including the ones discussed above, and the unique insights that they provided into compression, friction, and wear are discussed. read less NOT USED (high confidence) J. Zimmerman, R. Jones, and J. Templeton, “A material frame approach for evaluating continuum variables in atomistic simulations,” J. Comput. Phys. 2008. link Times cited: 71 NOT USED (high confidence) I. Chang and M. S. Yeh, “An atomistic study of nanosprings,” Journal of Applied Physics. 2008. link Times cited: 7 Abstract: Molecular statics method incorporating minimum energy concep… read moreAbstract: Molecular statics method incorporating minimum energy concept was employed to study the one-dimensional copper nanospring with faced-center-cubic crystal structure. Various geometric sizes (wire diameter, radius, pitch), numbers of turns and crystal orientations of nanosprings were systematically modeled to investigate the size dependence of elastic properties. It was observed that as the wire diameter increases and the radius and number of turns decrease, the nanospring stiffness would increase irrespective of the crystal orientations. Moreover, the elastic constants of nanosprings would become larger while the pitches become smaller for almost all the crystal orientations. Also the simulation results were compared to the predictions based on continuum theory in order to clarify whether the classical theory could apply to nanosprings. read less NOT USED (high confidence) E. Sanville, A. Bholoa, R. Smith, and S. Kenny, “Silicon potentials investigated using density functional theory fitted neural networks,” Journal of Physics: Condensed Matter. 2008. link Times cited: 34 Abstract: We present a method for fitting neural networks to geometric… read moreAbstract: We present a method for fitting neural networks to geometric and energetic data sets. We then apply this method by fitting a neural network to a set of data generated using the local density approximation for systems composed entirely of silicon. In order to generate atomic potential energy data, we use the Bader analysis scheme to partition the total system energy among the constituent atoms. We then demonstrate the transferability of the neural network potential by fitting to various bulk, surface, and cluster systems. read less NOT USED (high confidence) B. Garrison and Z. Postawa, “Computational view of surface based organic mass spectrometry.,” Mass spectrometry reviews. 2008. link Times cited: 125 Abstract: Surface based mass spectrometric approaches fill an importan… read moreAbstract: Surface based mass spectrometric approaches fill an important niche in the mass analysis portfolio of tools. The particular niche depends on both the underlying physics and chemistry of molecule ejection as well as experimental characteristics. In this article, we use molecular dynamics computer simulations to elucidate the fundamental processes giving rise to ejection of organic molecules in atomic and cluster secondary ion mass spectrometry (SIMS), massive cluster impact (MCI) mass spectrometry, and matrix-assisted laser desorption ionization (MALDI) mass spectrometry. This review is aimed at graduate students and experimental researchers. read less NOT USED (high confidence) O. A. Oviedo, E. Leiva, and M. Mariscal, “Diffusion mechanisms taking place at the early stages of cobalt deposition on Au(111),” Journal of Physics: Condensed Matter. 2008. link Times cited: 4 Abstract: In the present work a detailed atomic-level analysis of some… read moreAbstract: In the present work a detailed atomic-level analysis of some of the main diffusion mechanisms which take place during cobalt adatom deposition are studied within atom dynamics (AD) and the nudged elastic band (NEB) method. Our computer simulations reveal a very fast exchange between Co and Au atoms when the deposit is a single cobalt adatom. However, when the nucleus size increases, a decrease in the exchange probability is observed. Activation energies for different transitions are obtained using AD in combination with the NEB method. read less NOT USED (high confidence) S. Koh, R. Tummala, A. Saxena, and P. Selvam, “A numerical analysis of crack growth and morphology evolution in chip-to-packages nano-interconnections,” 2008 58th Electronic Components and Technology Conference. 2008. link Times cited: 2 Abstract: The International Technology Roadmap for semiconductors (ITR… read moreAbstract: The International Technology Roadmap for semiconductors (ITRS) has predicted that by the year 2007, integrated chip (IC) packages will contain feature sizes of 65 nm and an I/O pitch for the die-to-package interconnects approaching 80 mum. These will reduce even further in the next five years. The current approach of using surface mount technology and flip chip are mainly solder based and the lead and lead-free solder interconnects are known to fail mechanically as the pitch is reduced from 200 mum down to lower levels due to the thermal mismatch between the substrate and the chip. Although compliant interconnection could solve some of the mechanical issues, it is done at the expense of the electric performance. The PRC at Georgia Institute of Technology is proposing re-workable copper based nano-interconnections as a new interconnection paradigm as the next step beyond lead-free solders for future low-cost, high performance and high reliability packages. However, very limited data is published about the fatigue life of nano-crystalline materials and specifically those of nano-crystalline copper. It is important to predict crack growth as it can aid the understanding of the useful life of the IC-packages' interconnections. Multiple mechanisms may be responsible for crack initiation, but eventually most dominant fatigue cracks form a surface crack, which often have a semi-elliptical shape. Hence, the fatigue crack growth life predictions in this study are based on the assumption of elliptical and semi-elliptical cracks being initiated in the nano-interconnections. In this study, numerical analysis using the J-integral stress intensity parameter, in conjunction with experimental fatigue crack growth data, has been employed to study semi-elliptical crack growth and morphology evolution in nano-interconnection subject to uniaxial fatigue loading in linear-elastic conditions. The results indicate that a J-integral finite element analysis, using the loading portion of the fatigue cycle, in conjunction with known rates of fatigue crack growth can approximate surface crack morphology evolution. This study also predicts that the long crack growth is a relatively small portion of the total fatigue life of the material for the experimental LCF conditions. Hence, initiation of the cracks in the interconnection is the main criterion used to predict its fatigue life. read less NOT USED (high confidence) J. Rogan et al., “The structure and properties of small Pd clusters,” Nanotechnology. 2008. link Times cited: 18 Abstract: The zero-temperature minimal energy structure of small free-… read moreAbstract: The zero-temperature minimal energy structure of small free-standing Pd clusters (14≤N≤21, where N is the number of atoms in the cluster), their characteristics and their magnetic configurations are investigated. Results obtained using five different phenomenological many-body potentials (implemented in combination with a genetic algorithm search) are refined by means of various density functional theory (DFT) techniques. The agreement and differences between the results obtained with our procedure, using these five potentials, are displayed in detail. While phenomenological potentials yield values that approach the minimal energies of larger clusters, as compared with DFT results, they fail to predict the right symmetry group for some of the clusters with N>14. We find that the minimal energy configurations are not necessarily associated with high symmetry of the atomic arrangement. Actually, several cases of previously overlooked low symmetry structures turn out to have lower energies than more symmetric ones. read less NOT USED (high confidence) M. Mendelev, M. Kramer, C. Becker, and M. Asta, “Analysis of semi-empirical interatomic potentials appropriate for simulation of crystalline and liquid Al and Cu,” Philosophical Magazine. 2008. link Times cited: 365 Abstract: We investigate the application of embedded atom method (EAM)… read moreAbstract: We investigate the application of embedded atom method (EAM) interatomic potentials in the study of crystallization kinetics from deeply undercooled melts, focusing on the fcc metals Al and Cu. For this application, it is important that the EAM potential accurately reproduces melting properties and liquid structure, in addition to the crystalline properties most commonly fit in its development. To test the accuracy of previously published EAM potentials and to guide the development of new potential in this work, first-principles calculations have been performed and new experimental measurements of the Al and Cu liquid structure factors have been undertaken by X-ray diffraction. We demonstrate that the previously published EAM potentials predict a liquid structure that is too strongly ordered relative to measured diffraction data. We develop new EAM potentials for Al and Cu to improve the agreement with the first-principles and measured liquid diffraction data. Furthermore, we calculate liquid-phase diffusivities and find that this quantity correlates well with the liquid structure. Finally, we perform molecular dynamics simulations of crystal nucleation from the melt during quenching at constant cooling rate. We find that EAM potentials, which predict the same zero-temperature crystal properties but different liquid structures, can lead to quite different crystallization kinetics. More interestingly, we find that two potentials predicting very similar equilibrium solid and liquid properties can still produce very different crystallization kinetics under far-from-equilibrium conditions characteristic of the rapid quenching simulations employed here. read less NOT USED (high confidence) K. Mirabbaszadeh, E. Zaminpayma, P. Nayebi, and S. Saramad, “Large-Scale Molecular Dynamics Simulations of Energetic Ni Nanocluster Impact onto the Surface,” Journal of Cluster Science. 2008. link Times cited: 5 NOT USED (high confidence) L. Bianchettin et al., “Core level shifts of undercoordinated Pt atoms.,” The Journal of chemical physics. 2008. link Times cited: 36 Abstract: We present the results of high-energy resolution core level … read moreAbstract: We present the results of high-energy resolution core level photoelectron spectroscopy experiments paralleled by density functional theory calculations to investigate the electronic structure of highly undercoordinated Pt atoms adsorbed on Pt(111) and its correlation with chemical activity. Pt4f(7/2) core level binding energies corresponding to atoms in different configurations are shown to be very sensitive not only to the local atomic coordination number but also to the interatomic bond lengths. Our results are rationalized by introducing an indicator, the effective coordination, which includes both contributions. The calculated energy center of the valence 5d-band density of states, which is a well known depicter of the surface chemical reactivity, shows a noteworthy correlation with the Pt4f(7/2) core level shifts and with the effective coordination. read less NOT USED (high confidence) S. Eremeev, G. Rusina, S. Borisova, and E. Chulkov, “Electron-phonon interaction in the quantum well state of the 1 ML Na/Cu(111) system,” Physics of the Solid State. 2008. link Times cited: 11 NOT USED (high confidence) X. W. Zhou, J. Zimmerman, B. M. Wong, and J. Hoyt, “An embedded-atom method interatomic potential for Pd–H alloys,” Journal of Materials Research. 2008. link Times cited: 84 Abstract: Palladium hydrides have important applications. However, the… read moreAbstract: Palladium hydrides have important applications. However, the complex Pd–H alloy system presents a formidable challenge to developing accurate computational models. In particular, the separation of a Pd–H system to dilute (α) and concentrated (β) phases is a central phenomenon, but the capability of interatomic potentials to display this phase miscibility gap has been lacking. We have extended an existing palladium embedded-atom method potential to construct a new Pd–H embedded-atom method potential by normalizing the elemental embedding energy and electron density functions. The developed Pd–H potential reasonably well predicts the lattice constants, cohesive energies, and elastic constants for palladium, hydrogen, and PdH_x phases with a variety of compositions. It ensures the correct hydrogen interstitial sites within the hydrides and predicts the phase miscibility gap. Preliminary molecular dynamics simulations using this potential show the correct phase stability, hydrogen diffusion mechanism, and mechanical response of the Pd–H system. read less NOT USED (high confidence) D. Belashchenko, N. Kravchunovskaya, and O. Ostrovski, “Properties of iron under Earth’s core conditions: Molecular dynamics simulation with an embedded-atom method potential,” Inorganic Materials. 2008. link Times cited: 2 NOT USED (high confidence) R. McEntire and Y. L. Shen, “Parametric variations of the interatomic potential in atomistic analysis of nano-scale metal plasticity,” International Journal of Mechanics and Materials in Design. 2008. link Times cited: 2 NOT USED (high confidence) M. Hou, O. Melikhova, and S. Pisov, “Mechanical properties of bimetallic crystalline and nanostructured nanowires.,” Faraday discussions. 2008. link Times cited: 3 Abstract: Nanowires are basic components of interconnects at the nanos… read moreAbstract: Nanowires are basic components of interconnects at the nanoscale level in electronic as well as in electromechanical devices. Presently, there is a fast growing interest in their synthesis as well as in their mechanical testing. Focused ion beams now allow machining pillars with diameters as small as a few tens of nanometres and nanoindenter systems allow measuring strains at the atomic scale and compressive stresses up to the 10 GPa range. Such pillars typically contain less than millions of atoms, which makes their modelling and the modelling of their mechanical properties at the atomic scale realistic. A few Molecular Dynamics studies are presently available, discussing deformation mechanisms in thin narrow crystalline nanowires, but the literature about nanoalloy wires and nanostructured wires, as they can be synthesized from clusters, is almost non-existent. In the latter, the dislocation activity may be inhibited, leading to specific mechanical properties. By means of large scale computations, we use Ni3A1 to discuss the mechanical properties of crystalline and nanostructured nanowires. We also compare wires to their bulk counterparts. Both isothermal and isoenergetic whereby mechanical work converts into heat in the system-deformation mechanisms are considered. The comparison between pair correlation functions, stress distributions, configuration analysis and strain stress relations capture most of the stress-induced evolution mechanisms of nanowires with different diameters and structures, including elastic properties, dislocation activity, grain rotation and boundary motion, local melting, superplasticity and fracture. A structural transition which may be martensitic is predicted for the first time at the nanoscale level, suggesting possible shape memory properties of nanoalloy nanowires. read less NOT USED (high confidence) F. Ma, S. Ma, K. Xu, and P. Chu, “Surface Stability of Platinum Nanoparticles Surrounded by High-Index Facets,” Journal of Physical Chemistry C. 2008. link Times cited: 10 Abstract: We have investigated the thermal stability of surface struct… read moreAbstract: We have investigated the thermal stability of surface structures of tetrahexahedral platinum nanoparticles surrounded by high-index facets such as (730), (210), (310), and (520) by molecular dynamic simulation. The instantaneous atomic configurations, mean-squared displacement, radical distribution function, and average energy per atom are studied. At the lower temperature range, that is, 0∼860 K, both surface atoms and interior atoms oscillate around their equilibrium positions slightly and the high-index surface steps at which chemical reactions usually take place are maintained regularly. If the temperature is raised above 860 K, surface diffusion becomes dramatic and those surface steps disappear, though the general shape of the nanoparticles is still retained to a high temperature of 1100 K or so. Based on this study, the improved catalysis of Pt nanoparticles can be maintained only at temperatures lower than 860 K. read less NOT USED (high confidence) M. Springborg et al., “Theoretical Studies of Structural, Energetic, and Electronic Properties of Clusters,” Zeitschrift für Physikalische Chemie. 2008. link Times cited: 3 Abstract: Size in combination with low symmetry makes theoretical stud… read moreAbstract: Size in combination with low symmetry makes theoretical studies of the properties of clusters a challenge. This is in particular the case when the studies also shall identify the structures of the lowest total energy. We discuss here various methods for calculating the structural, energetic, and electronic properties of nanoparticles, emphasizing that the computational method always should be chosen carefully according to the scientific questions that shall be addressed. Therefore, different approximate methods for calculating the total energy of a given structure are discussed, including the embedded-atom method and a parameterized density-functional method. Moreover, different approaches for choosing/determining the structures are presented, including an Aufbau/Abbau method and genetic algorithms. In order to illustrate the approaches we present results from calculations on metallic and semiconducting nanoparticles as well as on nanostructured HAlO. read less NOT USED (high confidence) C. Vardeman and J. Gezelter, “Simulations of Laser-Induced Glass Formation in Ag−Cu Nanoparticles,” Journal of Physical Chemistry C. 2008. link Times cited: 8 Abstract: Using molecular dynamics simulations, we have simulated the … read moreAbstract: Using molecular dynamics simulations, we have simulated the rapid cooling experienced by bimetallic nanoparticles following laser excitation at the plasmon resonance and find evidence that glassy beads, specifically Ag−Cu bimetallic particles at the eutectic composition (60% Ag, 40% Cu), can be formed during these experiments. The bimetallic nanoparticles are embedded in an implicit solvent with a viscosity tuned to yield cooling curves that match the experimental cooling behavior as closely as possible. Because the nanoparticles have a large surface-to-volume ratio, experimentally realistic cooling rates are accessible via relatively short simulations. The presence of glassy structural features was verified using bond orientational order parameters that are sensitive to the formation of local icosahedral ordering in condensed phases. As the particles cool from the liquid droplet state into glassy beads, a silver-rich monolayer develops on the outer surface and local icosahedra can develop around the silv... read less NOT USED (high confidence) B. Henz, T. Hawa, and M. Zachariah, “Mechano-chemical stability of gold nanoparticles coated with alkanethiolate SAMs.,” Langmuir : the ACS journal of surfaces and colloids. 2008. link Times cited: 32 Abstract: Molecular dynamics simulations are used to probe the structu… read moreAbstract: Molecular dynamics simulations are used to probe the structure and stability of alkanethiolate self-assembled monolayers (SAMs) on gold nanoparticles. We observed that the surface of gold nanoparticles becomes highly corrugated by the adsorption of the SAMs. Furthermore, as the temperature is increased, the SAMs dissolve into the gold nanoparticle, creating a liquid mixture at temperatures much lower than the melting temperature of the gold nanoparticle. By analyzing the mechanical and chemical properties of gold nanoparticles at temperatures below the melting point of gold, with different SAM chain lengths and surface coverage properties, we determined that the system is metastable. The model and computational results that provide support for this hypothesis are presented. read less NOT USED (high confidence) R. Dingreville, A. Kulkarni, M. Zhou, and J. Qu, “A semi-analytical method for quantifying the size-dependent elasticity of nanostructures,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 46 Abstract: In this paper, a semi-analytical method is developed to comp… read moreAbstract: In this paper, a semi-analytical method is developed to compute the elastic stiffness of nanostructures such as nanowires, nanotubes and nanofilms. Compared with existing methods for such computations, this new method is more accurate and significantly reduces the computational time. It is based on the Taylor series expansion of an interatomic potential about the relaxed state of a nanostructure and implicitly accounts for the effects of shape, size and surface of the nanostructures. To analyze the applicability and accuracy of this method, as a case study, calculations are carried out to quantify the size dependence of the elastic moduli of nanofilms and nanowires with [0 0 1], [1 1 0] and [1 1 1] crystallographic growth orientations for groups 10 and 11 transition metals (Cu, Ni, Pd and Ag). The results are in excellent agreement with data in the literature and reveal consistent trends among the materials analyzed. read less NOT USED (high confidence) K. Henriksson, K. Nordlund, and J. Keinonen, “Annihilation of craters : Molecular dynamic simulations on a silver surface,” Physical Review B. 2007. link Times cited: 4 Abstract: The ability of silver cluster ions containing 13 atoms to fi… read moreAbstract: The ability of silver cluster ions containing 13 atoms to fill in a preexisting crater with a radius of about 28 A ring on a silver (001) target has been investigated using molecular dynamics simulations and the molecular-dynamics-Monte Carlo corrected effective medium potential. The largest lateral distance r between crater and ion was about three times the radius of the preexisting crater, namely, 75 A ring . The results reveal that when r 60 A ring the preexisting crater is partially filled in, and for other distances there is a net growth of the crater. The lattice damage created by the cluster ions, the total sputtering yield, the cluster sputtering yield, and simulated transmission electron microscopy images of the irradiated targets are also presented. read less NOT USED (high confidence) Y. Shao-horn, W. Sheng, S.-C. Chen, P. J. Ferreira, E. F. Holby, and D. Morgan, “Instability of Supported Platinum Nanoparticles in Low-Temperature Fuel Cells,” Topics in Catalysis. 2007. link Times cited: 810 NOT USED (high confidence) G. Grochola, I. Snook, and S. Russo, “Computational modeling of nanorod growth.,” The Journal of chemical physics. 2007. link Times cited: 53 Abstract: In this computational study, we used molecular dynamics and … read moreAbstract: In this computational study, we used molecular dynamics and the embedded atom method to successfully reproduce the growth of gold nanorod morphologies from starting spherical seeds in the presence of model surfactants. The surfactant model was developed through extensive systematic attempts aimed at inducing nonisotropic nanoparticle growth in strictly isotropic computational growth environments. The aim of this study was to identify key properties of the surfactants which were most important for the successful anisotropic growth of nanorods. The observed surface and collective dynamics of surfactants shed light on the likely growth phenomena of real nanoprods. These phenomena include the initial thermodynamically driven selective adsorption, segregation, and orientation of the surfactant groups on specific crystallographic surfaces of spherical nanoparticle seeds and the kinetic elongation of unstable surfaces due to growth inhibiting surfactants on those surfaces. Interestingly, the model not only reproduced the growth of nearly all known nanorod morphologies when starting from an initial fcc or fivefold seed but also reproduced the experimentally observed failure of nanorod growth when starting from spherical nanoparticles such as the I(h) morphology or morphologies containing a single twinning plane. Nanorod morphologies observed in this work included fivefold nanorods, fcc crystalline nanorods in the [100] direction and [112] directions and the more exotic "dumbell-like" nanorods. Non-nanorod morphologies observed included the I(h) and the nanoprism morphology. Some of the key properties of the most successful surfactants seemed to be suggestive of the important but little understood role played by silver ions in the growth process of real nanorods. read less NOT USED (high confidence) J.-min Zhang, B. Wang, and K. Xu, “Surface segregation of the metal impurity to the (100) surface of fcc metals,” Pramana. 2007. link Times cited: 5 NOT USED (high confidence) F. Ma, S. L. Ma, K. Xu, and P. Chu, “Activation volume and incipient plastic deformation of uniaxially-loaded gold nanowires at very high strain rates,” Nanotechnology. 2007. link Times cited: 8 Abstract: Uniaxial tensile loading is investigated by the molecular dy… read moreAbstract: Uniaxial tensile loading is investigated by the molecular dynamic (MD) method on Au nanowires at ultra-high strain rates. The activation volume is used to comprehensively characterize the incipient plastic deformation during this process. For lower strain rates such as 6.287 × 108 s−1, the moving velocity, V, of the atom planes due to uniaxial loading is two orders of magnitude smaller than the phonon wave propagation speed, V0, and the coherence between atoms is always maintained with a larger activation volume. In this case, plastic deformation is initiated mainly by collective atomic slipping, and thus only lower flow stress is needed. On the other hand, for higher strain rates such as 6.287 × 1010 s−1, V is elevated to the same magnitude as V0, the atom coherence is broken, their individual behavior is dominated by extraordinarily small activation volume, and atom diffusion becomes the main mechanism for plastic deformation. As a result, the yield strength is improved substantially. A higher temperature may weaken this strain-rate-dependent mechanical behavior because of the enhanced atom activity. read less NOT USED (high confidence) P. Mitev, G. Evangelakis, E. Kaxiras, and E. Kaxiras, “Embedded atom method potentials employing a faithful density representation,” Modelling and Simulation in Materials Science and Engineering. 2007. link Times cited: 9 Abstract: We present an approach for deriving embedded atom method ene… read moreAbstract: We present an approach for deriving embedded atom method energy functionals which employs a faithful representation of the valence electron that reproduces ab initio electronic structure calculations. This approach offers the possibility of improved accuracy and versatility over existing methods. Moreover, the approach has a distinct advantage for coupling to more accurate methods in the context of multiscale schemes. The embedding function is based on first breaking down the electronic density to individual atomic contributions and then designing an interatomic function which captures the interaction between the atomic contributions towards formation of the interatomic bonds. We use Al as a prototypical metallic solid to illustrate the application of the method and we employ density functional theory (DFT) to calculate the electronic charge densities and energies for determining the values of fitting parameters. We validate the approach by reproducing adequately experimental data for the cohesive energy, bulk modulus, elastic constants and dynamical properties at finite temperatures, obtained by molecular dynamics simulations. read less NOT USED (high confidence) D. B. Pedersen, S. Wang, E. J. S. Duncan, and S. Liang, “Adsorbate−Induced Diffusion of Ag and Au Atoms Out of the Cores of Ag@Au, Au@Ag, and Ag@AgI Core−Shell Nanoparticles,” Journal of Physical Chemistry C. 2007. link Times cited: 32 Abstract: Au@Ag and Ag@Au core−shell nanoparticles were synthesized us… read moreAbstract: Au@Ag and Ag@Au core−shell nanoparticles were synthesized using sequential laser ablation of metal targets in water. The resulting core−shell particles were then exposed to iodine or 1,6−hexanedithiol. For the Ag@Au + I2 → Au@AgI and Au@Ag + dithiol → Ag@Au−dithiol reactions, inversion of the core−shell structures resulted. These findings demonstrate that adsorbates can influence the core atoms through an intervening shell, and it is possible to use core−shell systems as vehicles for delivering reactivity on demand. Changing the shell material affords an opportunity to vary core susceptibility. Au@AgI nanoparticles, for example, did not react with 1,6−hexanedithiol. These results are most consistent with a charge−transfer-induced diffusion mechanism, analogous to the Cabrera−Mott model used for oxidation reactions of surfaces. The rate of uptake of iodine by Ag nanoparticles, measured with a quartz crystal microbalance, displayed kinetics that could be modeled using the same charge−transfer model. read less NOT USED (high confidence) I. Chang and Y.-C. Chen, “Is the molecular statics method suitable for the study of nanomaterials? A study case of nanowires,” Nanotechnology. 2007. link Times cited: 14 Abstract: Both molecular statics and molecular dynamics methods were e… read moreAbstract: Both molecular statics and molecular dynamics methods were employed to study the mechanical properties of copper nanowires. The size effect on both elastic and plastic properties of square cross-sectional nanowire was examined and compared systematically using two molecular approaches. It was found consistently from both molecular methods that the elastic and plastic properties of nanowires depend on the lateral size of nanowires. As the lateral size of nanowires decreases, the values of Young’s modulus decrease and dislocation nucleation stresses increase. However, it was shown that the dislocation nucleation stress would be significantly influenced by the axial periodic length of the nanowire model using the molecular statics method while molecular dynamics simulations at two distinct temperatures (0.01 and 300 K) did not show the same dependence. It was concluded that molecular statics as an energy minimization numerical scheme is quite insensitive to the instability of atomic structure especially without thermal fluctuation and might not be a suitable tool for studying the behaviour of nanomaterials beyond the elastic limit. read less NOT USED (high confidence) B. Aguilar, J. C. Flores, A. Coronado, and H.-C. Huang, “Atom diffusion of small Cu clusters across facet–facet barriers over Cu1 1 1 surfaces,” Modelling and Simulation in Materials Science and Engineering. 2007. link Times cited: 8 Abstract: The activation energy for atom diffusion across steps and fa… read moreAbstract: The activation energy for atom diffusion across steps and facets is a key factor in the surface structure formation during thin films deposition processes. In this work, we calculate facet–facet barriers for atom diffusion of small Cu clusters (up to eight atoms) from {1 1 1} facets to {1 1 1} (or {1 0 0}) facets. In each case we focus only on the atom-by-atom diffusion process because this mechanism was previously found as being energetically the most favourable. Our results show that, for all clusters considered, the diffusion barriers become independent of the facet size for three or more atomic layers. In addition, clusters bigger than trimers present similar lowest facet–facet barriers, except for heptamers which present the highest activation energy. In general, for single steps, diffusion from {1 1 1} to {1 1 1} presents smaller lowest and highest energy barriers than diffusion from {1 1 1} to {1 0 0}. But for multiple steps (facets), both cases show similar behaviours. Finally, it is worth noting that all these barriers are significantly greater than the corresponding concerted cluster diffusion over a plane. read less NOT USED (high confidence) S. Deng, J. Liu, N. Liang, and J. Zhang, “Validation of component assembly model and extension to plasticity,” Theoretical and Applied Fracture Mechanics. 2007. link Times cited: 5 NOT USED (high confidence) L. Yakovenkova and L. E. Kar’kina, “Theoretical and experimental study of processes of deformation and fracture in single-crystal Ti3Al oriented for the basal slip,” The Physics of Metals and Metallography. 2007. link Times cited: 3 NOT USED (high confidence) E. Kasabova, D. Alamanova, M. Springborg, and V. Grigoryan, “Deposition of Ni 13 and Cu 13 clusters on Ni(111) and Cu(111) surfaces,” The European Physical Journal D. 2007. link Times cited: 1 NOT USED (high confidence) R. Evans, F. Dorfbauer, O. Myrasov, O. Chubykalo-Fesenko, T. Schrefl, and R. Chantrell, “The Effects of Surface Coating on the Structural and Magnetic Properties of CoAg Core-Shell Nanoparticles,” IEEE Transactions on Magnetics. 2007. link Times cited: 7 Abstract: We have investigated the impact of Ag surface coating on the… read moreAbstract: We have investigated the impact of Ag surface coating on the microstructure of CoAg core-shell nanoparticles, and the consequences for the magnetic properties. Atomic structures were simulated using a molecular dynamics approach utilizing the embedded atom method. The magnetic properties were then simulated using an atomistic approach using a classical spin Hamiltonian, taking into account the long-range nature, atomic separation and directional and phase dependence of the exchange interactions in Co. For pure cobalt nanoparticles with a diameter less than approximately 3 nm the internal crystal structure showed multiple twinned regions and the morphology of an icosahedron. The addition of a monolayer silver coating alters the internal cobalt crystal structure to a regular planar form along the cubic [111] direction with a mixture of face centred cubic (fcc) and hexagonal close packed (hcp) atomic arrangements. The local atomic environment was used to assign anisotropies to the atoms on a site by site basis. Moreover, the capping layer influences the shape of the particle and yielded a morphology similar to that of a truncated octahedron. Taking into account the effects of surface anisotropy in addition gives an overall picture of anisotropy for CoAg nanoparticles. We present the results of calculations estimating the energy barrier to magnetization reversal and the intrinsic coercivity for these particles read less NOT USED (high confidence) A. Baraldi et al., “Highly under-coordinated atoms at Rh surfaces: interplay of strain and coordination effects on core level shift,” New Journal of Physics. 2007. link Times cited: 38 Abstract: The electronic structure of highly under-coordinated Rh atom… read moreAbstract: The electronic structure of highly under-coordinated Rh atoms, namely adatoms and ad-dimers, on homo-metallic surfaces has been probed by combining high-energy resolution core level photoelectron spectroscopy and density functional theory calculations. The Rh3d5/2 core level shifts are shown to be proportional to the number of Rh nearest-neighbours (n = 3, 4 and 5). A more refined analysis shows that the energy position of the different core level components is correlated with the calculated changes of the individual inter-atomic bond length and to the energy changes of the d-band centre, which is known to be a reliable descriptor of local chemical reactivity. read less NOT USED (high confidence) F. Ma and K. Xu, “Size-dependent structural phase transition of face-centered-cubic metal nanowires,” Journal of Materials Research. 2007. link Times cited: 5 Abstract: Taking Au as an example, we have investigated the epitaxial … read moreAbstract: Taking Au as an example, we have investigated the epitaxial bain paths of 〈001〉 oriented face-centered-cubic metal nanowires. It demonstrates that there are one stable and one metastable phase, having the lattice constant ratio c/a of about 0.6 and 1.0, respectively. Even without any external stimuli, the surface-tension-induced intrinsic stress in the interior may drive the nanowires to phase transform spontaneously for surface-energy minimization. However, this structural transition depends on the feature sizes of the nanowires. Specifically, only when the cross-section areas are reduced to 4.147 nm^2 or so can the surface energy and the intrinsic stress satisfy the thermodynamic and kinetic conditions simultaneously. read less NOT USED (high confidence) S. Chen, F. Ke, M. Zhou, and Y.-long Bai, “Atomistic investigation of the effects of temperature and surface roughness on diffusion bonding between Cu and Al,” Acta Materialia. 2007. link Times cited: 144 NOT USED (high confidence) G. Grochola, S. Russo, and I. Snook, “On morphologies of gold nanoparticles grown from molecular dynamics simulation.,” The Journal of chemical physics. 2007. link Times cited: 29 Abstract: The authors use a newly fitted gold embedded atom method pot… read moreAbstract: The authors use a newly fitted gold embedded atom method potential to simulate the initial nucleation, coalescence, and kinetic growth process of vapor synthesized gold nanoparticles. Overall the population statistics obtained in this work seemed to mirror closely recent experimental HREM observations by Koga and Sugawara [Surf. Sci. 529, 23 (2003)] of inert gas synthesized nanoparticles, in the types of nanoparticles produced and qualitatively in their observance ratio. Our results strongly indicated that early stage coalescence (sintering) events and lower temperatures are the mainly responsible for the occurrence of the Dh and fcc based morphologies, while "ideal" atom by atom growth conditions produced the Ih morphology almost exclusively. These results provide a possible explanation as to why the Dh to Ih occurrence ratio increases as a function of nanoparticle size as observed by Koga and Sugawara. read less NOT USED (high confidence) C. Cheng and X. Xu, “Molecular Dynamics Calculation of Critical Point of Nickel,” International Journal of Thermophysics. 2007. link Times cited: 17 NOT USED (high confidence) X. W. Zhou and H. Wadley, “A potential for simulating the atomic assembly of cubic elements,” Computational Materials Science. 2007. link Times cited: 10 NOT USED (high confidence) T. Järvi, A. Kuronen, K. Meinander, K. Nordlund, and K. Albe, “Contact epitaxy by deposition of Cu, Ag, Au, Pt, and Ni nanoclusters on (100) surfaces: Size limits and mechanisms,” Physical Review B. 2007. link Times cited: 41 Abstract: Low-energy deposition of individual metal clusters (6--2000 … read moreAbstract: Low-energy deposition of individual metal clusters (6--2000 atoms) on a (100) surface is studied for copper, nickel, platinum, silver, and gold by means of molecular-dynamics simulations. For different temperatures ranging from $0\phantom{\rule{0.3em}{0ex}}\text{to}\phantom{\rule{0.3em}{0ex}}750\phantom{\rule{0.3em}{0ex}}\mathrm{K}$ we determine the maximum size of clusters that will achieve complete contact epitaxy upon deposition. The results show that two mechanisms contribute to epitaxial alignment. For the smallest cluster sizes, the heat of adsorption released at the interface will immediately (ps time scales) allow the cluster to melt and become epitaxial by resolidification. This effect gives roughly the same limit for all elements studied. On longer (ns) time scales, the clusters can align epitaxially by thermally actived motion of twinning dislocations. This mechanism leads to much higher limits of epitaxy than the resolidification process. Moreover, the resulting limits differ significantly between the elements. read less NOT USED (high confidence) J. Li, X. Dai, T. Wang, and B. Liu, “A binomial truncation function proposed for the second-moment approximation of tight-binding potential and application in the ternary Ni–Hf–Ti system,” Journal of Physics: Condensed Matter. 2007. link Times cited: 38 Abstract: We propose a two-parameter binomial truncation function for … read moreAbstract: We propose a two-parameter binomial truncation function for the second-moment approximation of the tight-binding (TB-SMA) interatomic potential and illustrate in detail the procedure of constructing the potentials for binary and ternary transition metal systems. For the ternary Ni–Hf–Ti system, the lattice constants, cohesion energies, elastic constants and bulk moduli of six binary compounds, i.e. L12 Ni3Hf, NiHf3, Ni3Ti, NiTi3, Hf3Ti and HfTi3, are firstly acquired by ab initio calculations and then employed to derive the binomial-truncated TB-SMA Ni–Hf–Ti potential. Applying the ab initio derived Ni–Hf–Ti potential, the lattice constants, cohesive energy, elastic constants and bulk moduli of another six binary compounds, i.e. D03 NiHf3, NiTi3 HfTi3, and B2 NiHf, NiTi, HfTi, and two ternary compounds, i.e. C1b NiHfTi, L21 Ni2HfTi, are calculated, respectively. It is found that, for the eight binary compounds studied, the calculated lattice constants and cohesion energies are in excellent agreement with those directly acquired from ab initio calculations and that the elastic constants and bulk moduli calculated from the potential are also qualitatively consistent with the results from ab initio calculations. read less NOT USED (high confidence) B. Onat, S. Durukanoğlu, and H. Dağ, “A Parallel Implementation: Real Space Green’s Function Technique,” The International Journal of High Performance Computing Applications. 2007. link Times cited: 0 Abstract: We develop an MPI-based parallel algorithm to implement the … read moreAbstract: We develop an MPI-based parallel algorithm to implement the real space Green’s function technique for calculating the vibrational density of states corresponding to a solid. The Hamiltonian describing the interactions between the atoms within the system is obtained from the embedded atom method. The parallel implementation speeds up calculation by an order of magnitude. The parallel implementation details and results are presented in this paper. read less NOT USED (high confidence) N. Lümmen and T. Kraska, “Homogeneous nucleation and growth
in iron-platinum vapour investigated by molecular dynamics simulation,” The European Physical Journal D. 2007. link Times cited: 18 NOT USED (high confidence) K. Henriksson, K. Nordlund, and J. Keinonen, “Crater annihilation on silver by cluster ion impacts,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 10 NOT USED (high confidence) M. C. Giménez and E. Albano, “Dynamic response of silver monolayers adsorbed on gold(100) upon an oscillatory variation of the chemical potential : a monte carlo simulation study,” Journal of Physical Chemistry C. 2007. link Times cited: 9 Abstract: On the basis that the underpotential electrochemical deposit… read moreAbstract: On the basis that the underpotential electrochemical deposition of Ag atoms on the Au(100) surface exhibits sharp first-order phase transitions at well-defined values of the (coexistence) chemical potential (μcoex), we performed extensive simulations aimed at investigating the hysteretic dynamic behavior of the system close to coexistence upon the application of a periodic signal of the form μ(t) = μcoex + μo sin(2Πt/τ), where μo and τ are the amplitude and the period of the sweep, respectively. For relatively short periods and small enough amplitudes, the system becomes trapped either at low or high Ag coverage states, as shown by recording hysteresis loops. This scenario is identified as dynamically ordered states (DOS) such that the relaxation time (τrelax) of the corresponding metastable state obeys τrelax > τ. On the other hand, by properly increasing μo and/or τ, one finds that the Ag coverage gently follows the external drive (here τrelax < τ) and the system is said to enter into dynamically disord... read less NOT USED (high confidence) A. J. Francis and P. Salvador, “Crystal orientation and surface morphology of face-centered-cubic metal thin films deposited upon single-crystal ceramic substrates using pulsed laser deposition,” Journal of Materials Research. 2007. link Times cited: 36 Abstract: Cu, Pt, Ag, and Au were deposited on (100)-oriented ceramic … read moreAbstract: Cu, Pt, Ag, and Au were deposited on (100)-oriented ceramic substrates (SrTiO_3, LaAlO_3, and MgO). Over a wide range of temperatures (room temperature to 600 °C), Cu films were (100)-oriented and exhibited cube-on-cube epitaxy. Epitaxial Pt(100) films were obtained only at high temperature; oriented Pt(111) films were obtained at lower temperatures. Ag and Au were never obtained as purely (100)-oriented samples, although the amount of (100)-film increased with increasing temperature. Three-dimensional islands formed for all metals at higher temperatures, while flatter film surfaces developed at lower temperatures. At any given temperature, the surface roughness of films on SrTiO_3(100) increased in the order Pt < Cu < Au < Ag. The variations in film structural characteristics are described well by considering the metals’ (i) surface/interfacial energies, (ii) surface energy anisotropies, and (iii) surface diffusion coefficients. Flat, epitaxial growth is promoted by low-energy interfaces, low surface energy anisotropies, and slow surface diffusion. read less NOT USED (high confidence) S. Prudhomme, P. Bauman, and P. J. Tinsley Oden, “Error Control for Molecular Statics Problems,” International Journal for Multiscale Computational Engineering. 2006. link Times cited: 78 Abstract: In this paper, we present an extension of goal-oriented erro… read moreAbstract: In this paper, we present an extension of goal-oriented error estimation and adaptation to the simulation of multiscale problems of molecular statics. Computable error estimates for the quasicontinuum method are developed with respect to specific quantities of interest, and an adaptive strategy based on these estimates is proposed for error control. The theoretical results are illustrated on a nanoindentation problem in which the quantity of interest is the force acting on the indenter. The promising capability of such error estimates and adaptive procedure for the solution of multiscale problems is demonstrated on numerical examples. read less NOT USED (high confidence) S. Deng, J. Liu, J. Zhang, and N. Liang, “Component assembling model and its application to quasi-brittle damage,” Theoretical and Applied Fracture Mechanics. 2006. link Times cited: 8 NOT USED (high confidence) M. Karimi, T. Roarty, and T. Kaplan, “Molecular dynamics simulations of crack propagation in Ni with defects,” Modelling and Simulation in Materials Science and Engineering. 2006. link Times cited: 37 Abstract: A series of molecular dynamics simulations using the embedde… read moreAbstract: A series of molecular dynamics simulations using the embedded atom method is conducted to investigate crack propagation under mode I loading in a Ni single crystal with and without defects. The crack system (0 0 1)[1 0 0] in a slab of 160 000 atoms was studied. Defects consisting of lines of vacancies were introduced near the crack tip. Critical loads and strain energy distributions around the crack tip are obtained. Our results indicate that the critical strain necessary for crack propagation is dependent on the defect configuration and can either increase or decrease relative to the defect-free system. read less NOT USED (high confidence) D. Yu, H. Bonzel, and M. Scheffler, “Orientation-dependent surface and step energies of Pb from first principles,” Physical Review B. 2006. link Times cited: 14 Abstract: The orientation-dependent surface energies of 35 low-index a… read moreAbstract: The orientation-dependent surface energies of 35 low-index and vicinal Pb surface orientations, located in the [001], $[\overline{1}10]$, and $[01\overline{1}]$ zones, have been calculated by density-functional theory within the local-density approximation. The highest surface energy anisotropies in these zones are at the (210), (110), and (311) directions. Surface relaxation decreases the surface energy anisotropy significantly. For misorientations smaller than 12\ifmmode^\circ\else\textdegree\fi{} the (projected) surface energy in a given zone increases linearly with step density, while curvature is found at higher misorientations, indicative of repulsive step-step interactions. These results are fully consistent with the orientation-dependent surface energy predicted by the statistical mechanics of the terrace-step-kink model of vicinal surfaces. The step formation energies and surface and step relaxation energies are derived and analyzed. There is good agreement with available experimental data. The calculated surface energies in eV/atom correlate linearly with the number of broken surface bonds. Deviations from perfect linearity are found to be essential for a proper description of the equilibrium crystal shape of Pb. read less NOT USED (high confidence) Q. Bian, S. K. Bose, and R. Shukla, “Vibrational and thermodynamic properties of metals from a model embedded-atom potential,” Journal of Physics and Chemistry of Solids. 2006. link Times cited: 35 NOT USED (high confidence) H. S. Park, K. Gall, and J. Zimmerman, “Deformation of FCC nanowires by twinning and slip,” Journal of The Mechanics and Physics of Solids. 2006. link Times cited: 299 NOT USED (high confidence) E. Yildirim and Z. B. Guvenc, “Differences in melting behaviours of disordered and symmetric clusters: AuN(N = 54–56),” Modelling and Simulation in Materials Science and Engineering. 2006. link Times cited: 15 Abstract: We have investigated the melting behaviours of disordered an… read moreAbstract: We have investigated the melting behaviours of disordered and symmetric gold clusters (AuN, N = 54–56) by means of molecular dynamics simulations. We have found that there is no single isolated lowest energy structure for this size of Au clusters. Instead there are many nearly degenerate disordered low lying structures. The melting behaviours of these disordered structures showed that the melting occurs as a gradual process in which initially, behaviours of the surface and the inner atoms are quite different from each other, and they do not mix until the beginning of the melting. On the other hand, the symmetric forms of the AuN (N = 54–56) present different melting behaviours from those of the corresponding disordered structures. Their melting occurs suddenly, i.e. over a very short temperature interval. During the heating of these symmetric forms no phase changes occur until the melting temperature at which melting occurs as a collective motion of all the atoms in the cluster. On the other hand, the phase changes in the disordered structures take place as a result of both collective motions of all the atoms in the cluster, and as local displacements of the atoms. read less NOT USED (high confidence) T. Uehara, T. Tamai, and N. Ohno, “Molecular Dynamics Simulations of Shape-Memory Behavior Based on Martensite Transformation and Shear Deformation,” Jsme International Journal Series A-solid Mechanics and Material Engineering. 2006. link Times cited: 10 Abstract: Molecular dynamics simulations of the shape-memory effect ar… read moreAbstract: Molecular dynamics simulations of the shape-memory effect are carried out to investigate the atomistic behavior during deformation and shape-recovery processes. The embedded-atom-method potential function and parameters for Ni-Al alloy are applied. The initial configurations of atoms are set on the lattice points of the martensite structure, in which the distribution of the variant orientation is limited to the two-dimensional direction for simplicity. When the shear load is imposed toward the x direction, parallel to the variant interface, the deformation of the variants occurs, and finally, all variants settle into the uniform orientation. The deformed state is maintained after the load is released, and the original shape is recovered through heating and cooling processes because of phase transformation to bcc and martensite. In the loading process, the stress-strain curve exhibits a zigzag shape consisting of repeated stress increase and abrupt release. The interval of the stress peaks is revealed to be smaller as the model size becomes larger. Deformation observed in variant layers seems to occur at the same time at every points in the layer for a small model. However, the simulation with a large model indicates a nucleation and propagation behavior in each layer. read less NOT USED (high confidence) S. Psakh’e, K. P. Zol’nikov, D. S. Kryzhevich, and A. Lipnitskii, “Nucleation of structural defects in materials with a perfect crystal lattice by thermal fluctuations under dynamic loading,” Combustion, Explosion and Shock Waves. 2006. link Times cited: 0 NOT USED (high confidence) S. Valone and S. Atlas, “Electron correlation, reference states and empirical potentials,” Philosophical Magazine. 2006. link Times cited: 17 Abstract: Reference states as used in the physical sciences fall into … read moreAbstract: Reference states as used in the physical sciences fall into three main categories: simplified interactions, special limits or cases, and special symmetries or configurations. Regardless of the category, the general behaviour of a system is described as deviations from the specific behaviour of some reference state. After briefly reviewing examples from each of the categories, we more closely examine the role of reference states in constructing empirical atomistic potential energy surfaces. Although not universally used for parameterizing empirical potentials, we argue that the approach deserves more consideration based on the success of the embedded atom method (EAM), and its variants. We view a substantial part of the success of EAM as due to its use of a reference state. We further argue that one role of the reference state is to introduce correlation energy in a fundamental way. We take advantage of these characteristics in deriving a generalization of EAM that permits a description of charge transfer (CT-EAM). The generalization is based on a rigorous analysis of a valence bond model. The CT-EAM model introduces the charge of the reference state in a role that is analogous to the reference-state energy in the original EAM. The reference-state charge enables the model to switch between linear and quadratic dependence of the energy on charge, during a chemical reaction, as demanded by fundamental results from density functional theory. read less NOT USED (high confidence) E. Lobanov and D. Belashchenko, “Computer simulation of the coagulation of supersaturated Pb-Al solutions,” Inorganic Materials. 2006. link Times cited: 1 NOT USED (high confidence) M. S. Wu, K. Zhou, and A. A. Nazarov, “Stability and relaxation mechanisms of a wedge disclination in an HCP bicrystalline nanowire,” Modelling and Simulation in Materials Science and Engineering. 2006. link Times cited: 25 Abstract: The concept of critical disclination strength (ωc) in HCP bi… read moreAbstract: The concept of critical disclination strength (ωc) in HCP bicrystalline nanowires is developed on the basis of atomistic simulations using the modified embedded atom method. Disclinated titanium nanowires are created atomistically by combining two bicrystals with different misorientations along the tilt axis. Relaxations via molecular dynamic simulations of the atomic configurations at zero temperature result in either a stable or an unstable disclination, giving rise to the concept of ωc. An empirical size effect law for ωc is developed for nanowires with diameters between 10 and 20 nm, and extrapolation of this law to larger diameters yields predictions consistent with experimental data for plastically deformed titanium. As the disclination strength increases beyond ωc, various relaxation mechanisms are predicted depending on the grain boundary structures. For disclinations created by inserting a bicrystal into a reference one with B.B grain boundary structures, the three predicted relaxation mechanisms are basal plane and grain boundary cleavage, basal plane and grain boundary cleavage as well as new HCP grain nucleation ahead of the grain boundary crack and prismatic plane and grain boundary cleavage. read less NOT USED (high confidence) Y. Osetsky, D. Rodney, and D. Bacon, “Atomic-scale study of dislocation–stacking fault tetrahedron interactions. Part I: mechanisms,” Philosophical Magazine. 2006. link Times cited: 95 Abstract: Stacking fault tetrahedra (SFTs) are formed under irradiatio… read moreAbstract: Stacking fault tetrahedra (SFTs) are formed under irradiation in fcc metals and alloys. The high number density of SFTs observed suggests that they should contribute to radiation-induced hardening and, therefore, be taken into account when estimating mechanical property changes of irradiated materials. The key issue in this is to describe the interaction between a moving dislocation and an individual SFT, which is distinguished by a small physical size of the order of ∼1–10 nm. We have performed atomistic simulations of edge and screw dislocations interacting with SFTs of different sizes at different temperatures and strain rates. Five possible interaction outcomes have been identified, involving either partial absorption, or shearing or restoration of SFTs. The mechanisms that give rise to these processes are described and their dependence on interaction parameters, such as SFT size, dislocation–SFT geometry, temperature and stress/strain rate are determined. Mechanisms that help to explain the formation of defect-free channels cleared by gliding dislocations, as observed experimentally, are also discussed. Hardening due to the various mechanisms and their dependence on loading conditions will be presented in a following paper (Part II). read less NOT USED (high confidence) V. Tomar and M. Zhou, “Classical molecular-dynamics potential for the mechanical strength of nanocrystalline composite fccAl+α−Fe2O3,” Physical Review B. 2006. link Times cited: 26 Abstract: A classical molecular-dynamics potential for analyzing mecha… read moreAbstract: A classical molecular-dynamics potential for analyzing mechanical deformation in the -Fe2O3+fcc-Al material system is developed. The potential includes an embedded atom method cluster functional, a Morsetype pair function, and a second-order electrostatic interaction function. It is fitted to the lattice constants, elastic constants, and cohesive energies of fcc Al, bcc Fe, -Fe2O3, -Al2O3, and B2-FeAl, accounting for the fact that mixtures of Al and Fe2O3 are chemically reactive and deformation may cause the formation of these components as reaction products or intermediates. To obtain close approximations of the behavior of mixtures with any combination of the atomic elements, the potential is formulated and fitted such that the Al-Al, Fe-Fe, Al-Fe, O-O, Fe-O, and Al-O interactions are accounted for in an explicit and interdependent manner. In addition to being fitted to the lattice constants, elastic constants, and cohesive energies, the potential gives predictions of the surface and stacking fault energies for the crystalline components that compare well with the predictions of established potentials in the literature for the corresponding crystalline components. The potential is applied to analyze quasistatic tensile deformation in nanocrystalline Al, in nanocrystalline Fe2O3, and in nanocrystalline Al+Fe2O3 composites. Application of the potential to nanocrystalline Al reveals the features of mechanical deformation, such as the formation of unit dislocations, flow strength approaching ideal shear strength, and the Hall-Petch relationships, that are in close agreement with experiments and with the predictions of established potentials for Al in the literature. Analyses of deformation in nanocrystalline Fe2O3 and in nanocrystalline Al+Fe2O3 composites point to the possibility that the strength of the nanocomposites can only be calculated using the mixture theory if the average grain size is above a critical value. Below the critical grain size, an accurate account of interfacial stresses is important to the prediction of the strength. For composites with grain sizes above the critical value, the observed dependence of strength on volume fraction is in agreement with experimental observations. read less NOT USED (high confidence) X. Dai, Y. Kong, J. H. Li, and B. X. Liu, “Extended Finnis–Sinclair potential for bcc and fcc metals and alloys,” Journal of Physics: Condensed Matter. 2006. link Times cited: 138 Abstract: We propose an extended Finnis–Sinclair (FS) potential by ext… read moreAbstract: We propose an extended Finnis–Sinclair (FS) potential by extending the repulsive term into a sextic polynomial for enhancing the repulsive interaction and adding a quartic term to describe the electronic density function. It turns out that for bcc metals the proposed potential not only overcomes the ‘soft’ behaviour of the original FS potential, but also performs better than the modified FS one by Ackland et al, and that for fcc metals the proposed potential is able to reproduce the lattice constants, cohesive energies, elastic constant, vacancy formation energies, equations of state, pressure–volume relationships, melting points and melting heats. Moreover, for some fcc–bcc systems, e.g. the Ag–refractory metal systems, the lattice constants, cohesive energies and elastic constants of some alloys are reproduced by the proposed potential and are quite compatible with those directly determined by ab initio calculations. read less NOT USED (high confidence) B. Lee and K. Cho, “Extended embedded-atom method for platinum nanoparticles,” Surface Science. 2006. link Times cited: 19 NOT USED (high confidence) D. K. Yu, H. Bonzel, and M. Scheffler, “The stability of vicinal surfaces and the equilibrium crystal shape of Pb by first principles theory,” New Journal of Physics. 2006. link Times cited: 19 Abstract: The orientation-dependent surface energies of fcc Pb for mor… read moreAbstract: The orientation-dependent surface energies of fcc Pb for more than 30 vicinal orientations, distributed over the [110] and [001] zones of the stereographic triangle, have been studied by density-functional theory. For bulk-truncated structures almost all vicinal surfaces are found to be unstable and would facet into (111) and (100) orientations. However, after surface relaxation, all vicinal surfaces are stable relative to faceting into (111) and (100) orientations. There are also regions of relaxed vicinal surfaces which will facet into nearby stable vicinal surfaces. Overall, surface relaxation significantly affects the equilibrium crystal shape (ECS) of Pb. In both the [110] and [001] crystallographic zones the (110), (112), (221), and (023) facets are found on the ECS only after relaxation, in addition to (111) and (100). This result is in agreement with the experimental ECS of Pb at 353 K. Step formation energies for various vicinal orientations are estimated from facet diameters of the theoretical ECS and compared with experimental data. read less NOT USED (high confidence) D. Belashchenko, “Embedded atom model for liquid metals: Liquid iron,” Russian Journal of Physical Chemistry. 2006. link Times cited: 21 NOT USED (high confidence) K. Nordlund, J. Wallenius, and L. Malerba, “Molecular dynamics simulations of threshold displacement energies in Fe,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2006. link Times cited: 164 NOT USED (high confidence) D. Alamanova, V. Grigoryan, and M. Springborg, “Theoretical Study of Structure and Energetics of Gold Clusters with the EAM Method,” Zeitschrift für Physikalische Chemie. 2006. link Times cited: 11 Abstract: Using the embedded-atom method as developed by Voter and Che… read moreAbstract: Using the embedded-atom method as developed by Voter and Chen in combination with the variable metric/quasi-Newton and our own Aufbau/Abbau methods, we have identified the three most stable isomers of AuN clusters with N up to 150. For the first time clusters with tetrahedral symmetry are found to form the ground states of Au17 and Au34. The Au54icosahedron without a central atom and the Au146decahedron are found to be particularly stable, whereas the highly symmetric second and third Mackay icosahedra that could have been obtained for N = 55 and 147, respectively, do not correspond to the particularly stable structures. The three lowest-lying isomers of Au55 and Au147 are low-symmetrical structures. Various structural and energetic properties are analysed, such as stability function, occurrence of magic-sized clusters, construction of icosahedral and fcc shells, and cluster growth. read less NOT USED (high confidence) N. K. Sahoo, S. Thakur, R. Tokas, and M. Senthilkumar, “Correlation of atomic force–distance microscopy and spectrophotometric techniques in the analysis of optical multilayer spectral aging process,” Thin Solid Films. 2006. link Times cited: 3 NOT USED (high confidence) D. Belashchenko and O. I. Ostrovskii, “The embedded atom model for liquid metals: Liquid gallium and bismuth,” Russian Journal of Physical Chemistry. 2006. link Times cited: 23 NOT USED (high confidence) W. Benten, H. Freund, and M. Dähne, “Plasmonen in einzelnen oxidgetragenen Edelmetallpartikeln.” 2006. link Times cited: 0 Abstract: Plasmon excitations in nanoscopic metal particles lead to an… read moreAbstract: Plasmon excitations in nanoscopic metal particles lead to an element specific resonant behaviour in their optical properties, in particular for noble metals. This behaviour is used here to gain information on the alloying process of silver and gold. Optical properties of single supported clusters are obtained from scanning tunneling microscopy (STM) combined with optical emission spectroscopy. In the experiments, the STM tip is placed above a chosen cluster, which is excited by electron injection followed by photon emission. The emitted light is analysed using a spectrograph and detected by a CCD chip. The 5-10 nm sized clusters are deposited on an ultrathin, ordered alumina film on a NiAl(110) single crystal. The optical spectra are compared to model calculations based on Mie theory. From this comparison, the observed light emission can be attributed to the radiative decay of the perpendicular (1,0)-mode of a Mie plasmon resonance. It was further shown that simultaneous deposition of the two noble metals, silver and gold, leads to a complete mixing of both materials in the single particles. By successive deposition, clusters grow in a shell-core structure, whereby the metal deposited first forms the core. For gold-shell/silver-core clusters a strong intermixing of core and shell materials is found, explained by a higher mobility of the silver atoms. The relaxation dynamics of plasmons in single noble metal particles is analysed employing time-resolved two-photon photoemission by means of femtosecond laser pulses and photoemission electron microscopy. With this newly implemented approach, 500 nm sized silver particles on a silicon wafer are analysed. Plasmon modes oriented perpendicular (1,0) and parallel (1,1) to the sample surface can be excited by varying the polarisation direction of the laser pulses. Excitation of the (1,0)-mode results in a much stronger photoemission yield due to the resonant excitation of this plasmon mode. Furthermore, the photoemission signal varies in intensity and line width for the different particles, which is due to their size and shape distributions leading to different resonance properties. The experiments presented here demonstrate the possibility to perform optical emission spectroscopy on individual supported metal clusters and to characterise their optical properties regarding size, shape and chemical composition. Furthermore, the implemented time-resolved photoemission electron microscopy allows an investigation of plasmon excitations in single metal particles in space and time. read less NOT USED (high confidence) S. Debiaggi, M. Koning, and A. M. Monti, “Theoretical study of the thermodynamic and kinetic properties of self-interstitials in aluminum and nickel,” Physical Review B. 2006. link Times cited: 10 Abstract: The formation thermodynamics and migration properties of sel… read moreAbstract: The formation thermodynamics and migration properties of self-interstitials in aluminum and nickel are investigated as a function of temperature using atomistic simulation techniques and embedded-atom-type interatomic potentials. Molecular dynamics and nonequilibrium free-energy techniques are employed to investigate anharmonic effects on the ${H}_{O}$ $⟨100⟩$ dumbbell formation properties. The equilibrium concentration of this defect is compared to those of vacancies and divacancies. The results are then analyzed in the framework of the interstitialcy model, according to which very high vibrational formation entropies should be expected for self-interstitials at high temperatures. The kinetics of self-interstitial migration is also investigated using different atomistic techniques, revealing the simultaneous activity of more than one distinct interstitial configuration as the temperature increases. read less NOT USED (high confidence) A. Gross, “Reactivity of Bimetallic Systems Studied from First Principles,” Topics in Catalysis. 2006. link Times cited: 168 NOT USED (high confidence) C. Ghosh, D. J. Liu, C. Jenks, P. Thiel, and J. Evans, “Modelling of the energetics and kinetics of Al deposition on 5-fold Al-rich quasicrystal surfaces,” Philosophical Magazine. 2006. link Times cited: 10 Abstract: We determine potential energy surfaces for the binding of Al… read moreAbstract: We determine potential energy surfaces for the binding of Al adatoms on 5-fold AlPdMn and AlCuFe quasicrystal surfaces. Appropriate geometric models are used to select physical surface terminations. The interaction between the Al adsorbate and the substrate is described using Lennard–Jones pair-wise interactions, with parameters chosen to fit ab initio energetics for Al on relevant single crystal substrates. We identify a ‘disordered-bond-network’ (DBN) of local adsorption sites, which includes deep ‘starfish’ and ‘incomplete starfish’ ensembles. Our primary interest is in the kinetics of deposition and aggregation of Al atoms, and the possible formation of pseudomorphic starfish islands at starfish ensembles (observed for Al deposition on AlCuFe). The deposition process is modeled within the framework of a DBN lattice-gas model, wherein we specify rates for deposition and for hopping between neighboring sites of the DBN, as well as Al–Al adsorbate interactions, which stabilize islands. We, thus, present a picture for the formation of starfish islands subject to the competition between various deep sites for Al adatoms. read less NOT USED (high confidence) A. J. Francis, Y. Cao, and P. Salvador, “Epitaxial growth of Cu(100) and Pt(100) thin films on perovskite substrates,” Thin Solid Films. 2006. link Times cited: 29 NOT USED (high confidence) N. Bailey, J. Schiøtz, and K. Jacobsen, “Atomistic simulation study of the shear-band deformation mechanism in Mg-Cu metallic glasses,” Physical Review B. 2006. link Times cited: 81 Abstract: We have simulated plastic deformation of a model Mg-Cu metal… read moreAbstract: We have simulated plastic deformation of a model Mg-Cu metallic glass in order to study shear banding. In uniaxial tension, we find a necking instability occurs rather than shear banding. We can force the latter to occur by deforming in plane strain, forbidding the change of length in one of the transverse directions. Furthermore, in most of the simulations a notch is used to initiate shear bands, which lie at a 45 deg. angle to the tensile loading direction. The shear bands are characterized by the Falk and Langer local measure of plastic deformation D{sub min}{sup 2}, averaged here over volumes containing many atoms. The D{sub min}{sup 2} profile has a peak whose width is around 10 nm; this width is largely independent of the strain rate. Most of the simulations were, at least nominally, at 100 K, about T{sub g}/3 for this system. The development of the shear bands takes a few tens of ps, once plastic flow has started, more or less independent of strain rate. The shear bands can also be characterized using a correlation function defined in terms of D{sub min}{sup 2}, which, moreover, can detect incipient shear bands in cases where they do not fullymore » form. By averaging the kinetic energy over small regions, the local temperature can be calculated, and this is seen to be higher in the shear bands by about 50-100 K. Increases in temperature appear to initiate from interactions of the shear bands with the free surfaces and with each other, and are delayed somewhat with respect to the localization of plastic flow itself. We observe a slight decrease in density, up to 1%, within the shear band, which is consistent with notions of increased free volume or disorder within a plastically deforming amorphous material.« less read less NOT USED (high confidence) S. Q. Wang and H. Ye, “Theoretical studies of solid-solid interfaces,” Current Opinion in Solid State & Materials Science. 2006. link Times cited: 67 NOT USED (high confidence) G. Riveros, S. Green, A. Cortés, H. Gómez, R. Marotti, and E. Dalchiele, “Silver nanowire arrays electrochemically grown into nanoporous anodic alumina templates,” Nanotechnology. 2006. link Times cited: 105 Abstract: Silver nanowire arrays with high aspect ratios have been pre… read moreAbstract: Silver nanowire arrays with high aspect ratios have been prepared using potentiostatic electrodeposition within the confined nanochannels of a commercial porous anodic aluminium oxide template. The nucleation and growth processes are intensively studied by current versus time transients. Scanning electron microscopy results show that the nanowires have a highly anisotropic structure with diameters and lengths of 170 nm and 58 µm, respectively, which coincide with the dimensions of the template used. Structural characterization using x-ray diffraction shows that the Ag nanowires are highly crystalline, and those obtained at higher overpotentials present a very strong [220] preferred crystallographic orientation. The optical properties of the silver nanowires embedded in the alumina template show a clear edge close to 320 nm, that is an expected value for a silver–alumina composite material. read less NOT USED (high confidence) O. Straten, Y. Zhu, J. Rullan, K. Dunn, and A. Kaloyeros, “Study of copper-refractory metal interfaces via solid-state wetting for emerging nanoscale interconnect applications,” Journal of Materials Research. 2006. link Times cited: 8 Abstract: Solid-state wetting experiments were carried out to derive t… read moreAbstract: Solid-state wetting experiments were carried out to derive the work of adhesion (adhesion energy) of pertinent Cu/liner interfaces via the Young–Dupré equation using contact-angle measurements of the Cu equilibrium crystal shape on Ta and TaN_x liners. Four types of liner surfaces were examined: untreated sputtered Ta (uSp-Ta), untreated sputtered TaN_x (uSp-TaN), untreated atomic layer deposited (ALD) TaN_x (uALD-TaN), and indium surfactant-treated ALD TaN_x (tALD-TaN). All Cu-liner stacks were subsequently annealed at 600 °C for 48 h in a forming gas (95% Ar/5% H_2) ambient. For Cu/uSp-Ta, the work of adhesion was found to be 2170 mJ/m^2, corresponding to an average contact angle of 74°, while for Cu/uSp-TaN, the work of adhesion amounted to 1850 mJ/m^2 for an average contact angle of 85°. Alternatively, the work of adhesion for Cu/uALD-TaN was determined to be 1850 mJ/m^2, corresponding to an average contact angle of 85°, while for Cu/tALD-TaN, the work of adhesion was 2280 mJ/m^2, at an average contact angle of 70°. These findings indicate that the highest degree of surface wetting occurs for the indium surfactant-treated ALD TaN_x. It is thus suggested that surfactant treatment causes a reduction in the energy barrier to Cu nucleation, resulting in an enhancement in Cu wetting characteristics and a more uniform concentration of Cu nucleation sites. A critical potential outcome is the formation of atomically smooth Cu-liner interfaces with enhanced adhesion characteristics. read less NOT USED (high confidence) P. Olsson, J. Wallenius, C. Domain, K. Nordlund, and L. Malerba, “Two-band modeling of α -prime phase formation in Fe-Cr,” Physical Review B. 2005. link Times cited: 187 Abstract: We have developed a two-band model of Fe-Cr, fitted to prope… read moreAbstract: We have developed a two-band model of Fe-Cr, fitted to properties of the ferromagnetic alloy. Fitting many-body functionals to the calculated mixing enthalpy of the alloy and the mixed interstitial ... read less NOT USED (high confidence) N. Lümmen and T. Kraska, “Molecular dynamics investigation of homogeneous nucleation and cluster growth of platinum clusters from supersaturated vapour,” Nanotechnology. 2005. link Times cited: 36 Abstract: The formation of platinum nanoparticles from a supersaturate… read moreAbstract: The formation of platinum nanoparticles from a supersaturated vapour phase is investigated by molecular dynamics simulations. Argon is added as carrier gas, removing the condensation heat from the nucleating system. The interactions between the platinum atoms are modelled by the multi-body embedded atom method. The nucleation rates in highly supersaturated systems are estimated as well as properties of the critical clusters from the nucleation theorems. Furthermore, the coalescence of platinum nanoparticles is investigated and a coalescence activated structural transition in platinum nanoparticles is described. read less NOT USED (high confidence) M. Atis, H. Aktaş, and Z. B. Güvenç, “Structures and melting of AgN (N = 7, 12–14) clusters,” Modelling and Simulation in Materials Science and Engineering. 2005. link Times cited: 7 Abstract: We have studied the most stable structures of AgN (N = 7, 12… read moreAbstract: We have studied the most stable structures of AgN (N = 7, 12, 13, 14) clusters and investigated their binding energies and melting behaviours using molecular dynamics and thermal quenching methods based on the Voter–Chen version of the embedded atom method. The melting like transition is described in terms of atom resolved root-mean-square bond length fluctuations, specific heats, coordination numbers and short-time averaged temperatures. Some of these diagnostic tools are also used for the overall behaviour of the clusters. The results are compared with the relevant literature. read less NOT USED (high confidence) F. Mehmood, A. Kara, and T. Rahman, “First principles study of the electronic and geometric structure of Cu(5 3 2),” Surface Science. 2005. link Times cited: 10 NOT USED (high confidence) G. Grochola, S. Russo, and I. Snook, “On fitting a gold embedded atom method potential using the force matching method.,” The Journal of chemical physics. 2005. link Times cited: 232 Abstract: We fit a new gold embedded atom method (EAM) potential using… read moreAbstract: We fit a new gold embedded atom method (EAM) potential using an improved force matching methodology which included fitting to high-temperature solid lattice constants and liquid densities. The new potential shows a good overall improvement in agreement to the experimental lattice constants, elastic constants, stacking fault energy, radial distribution function, and fcc/hcp/bcc lattice energy differences over previous potentials by Foiles, Baskes, and Daw (FBD) [Phys. Rev. B 33, 7983 (1986)] Johnson [Phys. Rev. B 37, 3924 (1988)], and the glue model potential by Ercolessi et al. [Philos. Mag. A 50, 213 (1988)]. Surface energy was improved slightly as compared to potentials by FBD and Johnson but as a result vacancy formation energy is slightly inferior as compared to the same potentials. The results obtained here for gold suggest for other metal species that further overall improvements in potentials may still be possible within the EAM framework with an improved fitting methodology. On the other hand, we also explore the limitations of the EAM framework by attempting a brute force fit to all properties exactly which was found to be unsuccessful. The main conflict in such a brute force fit was between the surface energy and the liquid lattice constant where both could not be fitted identically. By intentionally using a very large number of spline sections for the pair potential, electron-density function, and embedding energy function, we eliminated a lack of functional freedom as a possible cause of this conflict and hence can conclude that it must result from a fundamental limitation in the EAM framework. read less NOT USED (high confidence) S. Seel, J. Hoyt, E. Webb, and J. Zimmerman, “Modeling metallic island coalescence stress via adhesive contact between surfaces,” Physical Review B. 2005. link Times cited: 14 Abstract: Tensile stress generation associated with island coalescence… read moreAbstract: Tensile stress generation associated with island coalescence is almost universally observed in thin films that grow via the Volmer-Weber mode. The commonly accepted mechanism for the origin of this tensile stress is a process driven by the reduction in surface energy at the expense of the strain energy associated with the deformation of coalescing islands during grain boundary formation. In the present work, we have performed molecular statics calculations using an embedded atom interatomic potential to obtain a functional form of the interfacial energy vs distance between two closely spaced free surfaces. The sum of interfacial energy plus strain energy provides a measure of the total system energy as a function of island separation. Depending on the initial separation between islands, we find that in cases where coalescence is thermodynamically favored, gap closure can occur either spontaneously or be kinetically limited due to an energetic barrier. Atomistic simulations of island coalescence using conjugate gradient energy minimization calculations agree well with the predicted stress as a function of island size from our model of spontaneous coalescence. Molecular dynamics simulations of island coalescence demonstrate that only modest barriers to coalescence can be overcome at room temperature. A comparison with thermally activated coalescence results at room temperature reveals that existing coalescence models significantly overestimate the magnitude of the stress resulting from island coalescence. read less NOT USED (high confidence) Z. Bangwei, E. Taglauer, S. Xiaolin, H. Wangyu, and D. Hui-qiu, “Simulation calculations of surface segregation for Au–Cu alloys using an analytic embedded atom model,” physica status solidi (a). 2005. link Times cited: 7 Abstract: The key problem for applying the Monte Carlo (MC) method to … read moreAbstract: The key problem for applying the Monte Carlo (MC) method to segregation lies in the selection of an appropriate energy model for the simulations. Zhang et al. proposed a modified analytic embedded atomic method (MAEAM), which has been applied to a variety of fundamental problems in metals and alloys. We used this MAEAM and MC method for the simulations of segregation of AuCu and Au3Cu alloys. For the (100) surface, the calculations show that Au is enriched in the 1st layer (70 at% for AuCu, 99 at% for Au3Cu), while Cu is enriched in the 2nd layer (50–62 at% for AuCu, 35–48 at% for Au3Cu). The composition profiles are generally oscillating. For the (111) surface, we also found Au to be enriched in the 1st layer. However, Cu is not enriched in the 2nd layer, it reaches the bulk composition from 2nd to 4th layer. Au increases from 92 to 97 at% and 99 to 100 at% for Au3Cu(111) and (100), respectively, when the temperature varies from 1000 to 200 K, which is basically in agreement with the measurements for Au3Cu(100) by Taglauer et al. However, the Au concentration does not change for the AuCu alloy in the same temperature range. We also calculate the segregation energy, the simulation results agree qualitatively with the experimental data available. Our results demonstrate that the MAEAM model provides an effective means for simulating the segregation of alloys. (© 2005 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) C. Cheng and X. Xu, “Mechanisms of decomposition of metal during femtosecond laser ablation,” Physical Review B. 2005. link Times cited: 149 Abstract: The mechanisms of decomposition of a metal (nickel) during f… read moreAbstract: The mechanisms of decomposition of a metal (nickel) during femtosecond laser ablation are studied using molecular dynamics simulations. It is found that phase explosion is responsible for gas bubble generation and the subsequent material removal at lower laser fluences. The phase explosion process occurs as combined results of heating, thermal expansion, and the propagation of tensile stress wave induced by the laser pulse. When the laser fluence is higher, it is revealed that critical point phase separation plays an important role in material removal. read less NOT USED (high confidence) A. Coronado and H.-C. Huang, “Facet-facet barriers on Cu{111} surfaces for Cu dimers,” Cmes-computer Modeling in Engineering & Sciences. 2005. link Times cited: 3 Abstract: Nanostructure fabrication or surface pro- cessingingeneral i… read moreAbstract: Nanostructure fabrication or surface pro- cessingingeneral ispredominantlykinetics-limited. One ofthekineticsfactors issurfacediffusion,which involves intricate interplay between the diffusing atoms and sub- strate atoms. On Cu{111} surfaces, both adatoms and dimers diffuse very fast. Recent studies have shown that adatoms encounter a large facet-facet barrier, even thoughtheirconventionalEhrlich-Schwoebelbarriers are small. This work examines the facet-facet diffusion bar- riers of dimers. Our resultsshow that a dimer prefers dif- fusion through atom-by-atom mechanism, having a bar- rierof 0.52 eV from {111}to {111}facet and a barrier of 0.55 eV from {111} to {100} facet. When the two atoms in a dimer diffuse simultaneously, the barrier is 0.97 eV from {111} to {111} facet and 0.62 eV from {111} to {100} facet. read less NOT USED (high confidence) J. Samela, J. Kotakoski, K. Nordlund, and J. Keinonen, “A quantitative and comparative study of sputtering yields in Au,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 56 NOT USED (high confidence) J. Kohanoff, A. Caro, and M. Finnis, “An isothermal-isobaric Langevin thermostat for simulating nanoparticles under pressure: application to Au clusters.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2005. link Times cited: 11 Abstract: We present a method for simulating clusters or molecules sub… read moreAbstract: We present a method for simulating clusters or molecules subjected to an external pressure, which is exerted by a pressure-transmitting medium. It is based on the canonical Langevin thermostat, but extended in such a way that the Brownian forces are allowed to operate only from the region exterior to the cluster. We show that the frictional force of the Langevin thermostat is linked to the pressure of the reservoir in a unique way, and that this property manifests itself when the particle it acts upon is not pointlike but has finite dimensions. By choosing appropriately the strength of the random forces and the friction coefficient, both temperature and pressure can be controlled independently. We illustrate the capabilities of this new method by calculating the compressibility of small gold clusters under pressure. read less NOT USED (high confidence) M. Jiang, K. Oikawa, and T. Ikeshoji, “Molecular-dynamic simulations of martensitic transformation of cobalt,” Metallurgical and Materials Transactions A. 2005. link Times cited: 18 NOT USED (high confidence) J. Petersen and S. G. Mayr, “Smoothening of internal phase boundaries by ion bombardment,” Journal of Applied Physics. 2005. link Times cited: 1 Abstract: The impact of heavy-ion irradiation on the morphology of bil… read moreAbstract: The impact of heavy-ion irradiation on the morphology of bilayers, which are composed of two immiscible metals, is investigated with the help of experiments and molecular-dynamics computer simulations. Using the model system Ag∕Ni, our main focus lies on the interface roughness of the Ag∕Ni phase boundary and its evolution in the course of ion bombardment. The mechanism which underlies these structural changes is identified as radiation-induced viscous flow—in combination with thermodynamic driving forces due to interface stress. read less NOT USED (high confidence) T. Okazawa, F. Takeuchi, and Y. Kido, “Enhanced and correlated thermal vibrations of Cu(111) and Ni(111) surfaces,” Physical Review B. 2005. link Times cited: 17 Abstract: Enhanced and correlated thermal vibrations are studied for C… read moreAbstract: Enhanced and correlated thermal vibrations are studied for Cu(111) and Ni(111) by high-resolution medium energy ion scattering (MEIS) using the ion shadowing effect. We also perform molecular dynamics (MD) simulations based on the embedded atom method (EAM). The MEIS analysis reveals a slight contraction of $0.011\ifmmode\pm\else\textpm\fi{}0.003$ and $0.007\ifmmode\pm\else\textpm\fi{}0.003\phantom{\rule{0.3em}{0ex}}\mathrm{\AA{}}$ for Cu(111) and Ni(111), respectively, for the first interlayer distance without any surface reconstruction. The root mean square (rms) bulk thermal vibration amplitude and thermal vibration amplitudes (TVAs) of the top-layer atoms in the surface normal and lateral directions, respectively, are determined to be $0.085\ifmmode\pm\else\textpm\fi{}0.005$, ${0.141}_{\ensuremath{-}0.005}^{+0.010}$, and ${0.094}_{\ensuremath{-}0.005}^{+0.008}\phantom{\rule{0.3em}{0ex}}\mathrm{\AA{}}$ for Cu(111) and $0.068\ifmmode\pm\else\textpm\fi{}0.005$, ${0.098}_{\ensuremath{-}0.005}^{+0.010}$, and ${0.074}_{\ensuremath{-}0.005}^{+0.008}\phantom{\rule{0.3em}{0ex}}\mathrm{\AA{}}$ for Ni(111). We also observe strong correlations between the nearest-neighbor atoms in the [110] string and determine the correlation coefficients to be $+0.24\ifmmode\pm\else\textpm\fi{}0.05$ and $+0.20\ifmmode\pm\else\textpm\fi{}0.05$ for Cu(111) and Ni(111), respectively, for the motion perpendicular to the [110] axis. The present MEIS result is basically in agreement with the MD simulations using the EAM potential proposed by Foiles, Baske, and Daw [Phys. Rev. B 33, 7983 (1986)] rather than that approximated by Oh and Johnson [J. Mater. Res. 3, 471 (1988)]. read less NOT USED (high confidence) H. Yildirim, A. Kara, S. Durukanoğlu, and T. Rahman, “Calculated pre-exponential factors and energetics for adatom hopping on terraces and steps of Cu(1 0 0) and Cu(1 1 0),” Surface Science. 2005. link Times cited: 37 NOT USED (high confidence) V. Grigoryan, D. Alamanova, and M. Springborg, “Structure and energetics of nickel, copper, and gold clusters,” The European Physical Journal D - Atomic, Molecular, Optical and Plasma Physics. 2005. link Times cited: 30 NOT USED (high confidence) X. W. Zhou, H. Wadley, and S. Sainathan, “Low energy sputtering of nickel by normally incident xenon ions,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 19 NOT USED (high confidence) S. Eremeev and A. Potekaev, “Effective Many-Body Interatomic Potentials in Molecular Dynamic Simulations,” Russian Physics Journal. 2005. link Times cited: 3 NOT USED (high confidence) P. Wynblatt and Z. Shi, “Relation between grain boundary segregation and grain boundary character in FCC alloys,” Journal of Materials Science. 2005. link Times cited: 35 NOT USED (high confidence) S. D. Chen, A. Soh, and F. Ke, “Molecular Dynamics Modeling of Diffusion Bonding,” Scripta Materialia. 2005. link Times cited: 56 NOT USED (high confidence) V. Král, “Molecular dynamics study of the sputtering of Al cluster by Ar and Kr atoms,” Czechoslovak Journal of Physics. 2005. link Times cited: 1 NOT USED (high confidence) N. Lorente, R. Rurali, and H. Tang, “Single-molecule manipulation and chemistry with the STM,” Journal of Physics: Condensed Matter. 2005. link Times cited: 59 Abstract: We review recent theoretical work on the manipulation of sin… read moreAbstract: We review recent theoretical work on the manipulation of single molecules with scanning probes, in particular the scanning tunnelling microscope (STM). The aim of theories and simulations is to account for the processes, ideally at a quantitative level, that permit the controlled manipulation of matter at the atomic scale in adsorbed molecular systems. In order to achieve this, simulations rely on total energy and electronic structure calculations where a trade-off is made between the size of the system and the accuracy of the calculation. This first stage of the calculation yields the basic quantities used for the second stage: the evaluation of the coupled electron–nuclear dynamics. This second stage is a formidable task and many approximations are involved. In this review, we will present some of the customary approximations regarding the theoretical study of mechanical and inelastic manipulations. Mechanical manipulations use the interaction between the acting probe (usually a metallic tip) and the targeted adsorbate. We review recent results in the field of adsorbate mechanical manipulations and explain how manipulations can be effected by using the interaction between the probe’s tip and certain molecular groups of complex chemisorbed molecular systems. On the other hand, inelastic manipulations use the tunnelling current to convey energy with sub-ångström precision. This current can excite localized vibrations that can induce measurable variations of the tunnelling conductance, hence providing a means of detecting single-molecule vibrations. This current can also inject energy in a few reaction coordinates. Recently, the possibility of vibrational selective manipulations of NH3/Cu(100) has been experimentally demonstrated. The theory presented here addresses the actual pathways accessed when the molecule is excited by the tunnelling current from an STM. read less NOT USED (high confidence) Z. G. Yang, T.-J. Wang, S. Zhong, G. Yang, and G. Wang, “Coherent interfacial energy and composition profiles in ternary FCC systems by a discrete lattice plane analysis,” Journal of Materials Science. 2005. link Times cited: 4 NOT USED (high confidence) M. Meineke, C. Vardeman, T. Lin, C. J. Fennell, and J. Gezelter, “OOPSE: An object‐oriented parallel simulation engine for molecular dynamics,” Journal of Computational Chemistry. 2005. link Times cited: 32 Abstract: OOPSE is a new molecular dynamics simulation program that is… read moreAbstract: OOPSE is a new molecular dynamics simulation program that is capable of efficiently integrating equations of motion for atom types with orientational degrees of freedom (e.g. “sticky” atoms and point dipoles). Transition metals can also be simulated using the embedded atom method (EAM) potential included in the code. Parallel simulations are carried out using the force‐based decomposition method. Simulations are specified using a very simple C‐based meta‐data language. A number of advanced integrators are included, and the basic integrator for orientational dynamics provides substantial improvements over older quaternion‐based schemes. © 2004 Wiley Periodicals, Inc. J Comput Chem 26: 252–271, 2005 read less NOT USED (high confidence) T. Rahman, A. Kara, A. Karim, and O. Trushin, “Cluster Diffusion and Coalescence on Metal Surfaces: applications of a Self-learning Kinetic Monte-Carlo method,” MRS Proceedings. 2005. link Times cited: 3 Abstract: The Kinetic Monte Carlo (KMC) method has become an important… read moreAbstract: The Kinetic Monte Carlo (KMC) method has become an important tool for examination of phenomena like surface diffusion and thin film growth because of its ability to carry out simulations for time scales that are relevant to experiments. But the method generally has limited predictive power because of its reliance on predetermined atomic events and their energetics as input. We present a novel method, within the lattice gas model in which we combine standard KMC with automatic generation of a table of microscopic events, facilitated by a pattern recognition scheme. Each time the system encounters a new configuration, the algorithm initiates a procedure for saddle point search around a given energy minimum. Nontrivial paths are thus selected and the fully characterized transition path is permanently recorded in a database for future usage. The system thus automatically builds up all possible single and multiple atom processes that it needs for a sustained simulation. Application of the method to the examination of the diffusion of 2-dimensional adatom clusters on Cu(111) displays the key role played by specific diffusion processes and also reveals the presence of a number of multiple atom processes, whose importance is found to decrease with increasing cluster size and decreasing surface temperature. Similarly, the rate limiting steps in the coalescence of adatom islands are determined. Results are compared with those from experiments where available and with those from KMC simulations based on a fixed catalogue of diffusion processes. read less NOT USED (high confidence) M. Mendelev, D. Srolovitz, G. Ackland, and S. Han, “Effect of Fe segregation on the migration of a non-symmetric ∑5 tilt grain boundary in Al,” Journal of Materials Research. 2005. link Times cited: 106 Abstract: We present an analysis, based upon atomistic simulation data… read moreAbstract: We present an analysis, based upon atomistic simulation data, of the effect of Fe impurities on grain boundary migration in Al. The first step is the development of a new interatomic potential for Fe in Al. This potential provides an accurate description of Al–Fe liquid diffraction data and the bulk diffusivity of Fe in Al. We use this potential to determine the physical parameters in the Cahn–Lücke–Stüwe (CLS) model for the effect of impurities on grain boundary mobility. These include the heat of segregation of Fe to grain boundaries in Al and the diffusivity of Fe in Al. Using the simulation-parameterized CLS model, we predict the grain boundary mobility in Al in the presence of Fe as a function of temperature and Fe concentration. The order of magnitude and the trends in the mobility from the simulations are in agreement with existing experimental results. read less NOT USED (high confidence) J. Schall and D. Brenner, “Atomistic simulation of the influence of pre-existing stress on the interpretation of nanoindentation data,” Journal of Materials Research. 2004. link Times cited: 43 Abstract: By using molecular dynamics simulations, we have accurately … read moreAbstract: By using molecular dynamics simulations, we have accurately determined the true contact area during plastic indentation of materials under an applied in-plane stress. We found that the mean pressure calculated from the true contact area varied slightly with applied pre-stress with higher values in compression than in tension and that the modulus calculated from the true contact area is essentially independent of the press-stress level in the substrate. These findings are largely consistent with the findings of Tsui, Pharr, and Oliver. On the other hand, if the contact area is estimated from approximate formulae, the contact area is underestimated and shows a strong dependence on the pre-stress level. When it is used to determine mean pressure and modulus, the empirically determined area leads to large errors. Our simulations demonstrate that this phenomenon, first reported for macroscale hardness measurements dating back to 1932, also exists at the nanometer-scale contact areas, apparently scaling over 10 orders of magnitude in contact area, from ~mm^2 to ~100 nm^2. read less NOT USED (high confidence) Y. G. Xu, K. Behdinan, and Z. Fawaz, “Molecular dynamics calculation of the J-integral fracture criterion for nano-sized crystals,” International Journal of Fracture. 2004. link Times cited: 21 NOT USED (high confidence) G. Rusina, S. Borisova, S. Eremeev, I. Sklyadneva, and E. Chulkov, “Vibrational modes on the
$Al\,(111) - (\sqrt 3 \times \sqrt 3 )R30^∘- Na$
surface,” Russian Physics Journal. 2004. link Times cited: 0 NOT USED (high confidence) J. Wang, H.-C. Huang, and T. Cale, “Diffusion barriers on Cu surfaces and near steps,” Modelling and Simulation in Materials Science and Engineering. 2004. link Times cited: 60 Abstract: This paper reports a molecular statics study of Cu surface d… read moreAbstract: This paper reports a molecular statics study of Cu surface diffusion barriers, particularly the facet–facet and step–facet barriers. The study focuses on two high-symmetry surfaces or facets, Cu{111} and Cu{100}. Our results show that these two barriers are distinct from conventional step barriers and are independent of facet size once it is beyond three atomic layers. Usually, the facet–facet barrier is substantially larger than diffusion barriers on flat surfaces or down monolayer steps, and the step–facet barrier is substantially larger than diffusion barriers along or across monolayer steps. Exceptions do exist. When two Cu{100} facets are involved, the two barriers decrease as the size of the ending facet increases from one layer to two layers, and then increase from two to three (or more) layers. As a result of the large facet–facet and step–facet barriers, surfaces of Cu thin films are of the order of 100 nm. The small facet–facet and step–facet barriers between two Cu{100} facets, when the ending facet is two to three layers, make it difficult to form another Cu{100} facet near one Cu{100} facet. For the same reason, nanowires along 100/{100} on the Cu{100} are unlikely, while nanowires along 110/{111} are feasible. read less NOT USED (high confidence) V. Grigoryan and M. Springborg, “Structural and energetic properties of nickel clusters: 2 ≤ N ≤ 150,” Physical Review B. 2004. link Times cited: 78 Abstract: The four most stable structures of ${\mathrm{Ni}}_{N}$ clust… read moreAbstract: The four most stable structures of ${\mathrm{Ni}}_{N}$ clusters with $N$ from 2 to 150 have been determined using a combination of the embedded-atom method in the version of Daw, Baskes, and Foiles, the variable metric/quasi-Newton method, and our own Aufbau/Abbau method. A systematic study of energetics, structure, growth, and stability of also larger clusters has been carried through without more or less severe assumptions on the initial geometries in the structure optimization, on the symmetry, or on bond lengths. It is shown that cluster growth is predominantly icosahedral with islands of fcc, tetrahedral and decahedral growth. For the first time in unbiased computations it is found that ${\mathrm{Ni}}_{147}$ is the multilayer (third Mackay) icosahedron. Further, we point to an enhanced ability of fcc clusters to compete with the icosahedral and decahedral structures in the vicinity of $N=79$. In addition, it is shown that conversion from the hcp/anti-Mackay kind of icosahedral growth to the fcc/Mackay one occurs within a transition layer including several cluster sizes. Moreover, we present and apply different analytical tools in studying structural and energetic properties of such a large class of clusters. These include means for identifying the overall shape, the occurrence of atomic shells, the similarity of the clusters with, e.g., fragments of the fcc crystal or of a large icosahedral cluster, and a way of analysing whether the $N$-atom cluster can be considered constructed from the $(N\ensuremath{-}1)$-atom one by adding an extra atom. In addition, we compare in detail with results from chemical-probe experiment. Maybe the most central result is that first for clusters with $N$ above 80 general trends can be identified. read less NOT USED (high confidence) B.-J. Lee, J. C. Lee, Y.-C. Kim, and S. hak Lee, “Behavior of amorphous materials under hydrostatic pressures: A molecular dynamics simulation study,” Metals and Materials International. 2004. link Times cited: 32 NOT USED (high confidence) M. Haftel, N. Bernstein, M. Mehl, and D. Papaconstantopoulos, “Interlayer surface relaxations and energies of fcc metal surfaces by a tight-binding method,” Physical Review B. 2004. link Times cited: 21 Abstract: The authors examine the interlayer surface relaxations and s… read moreAbstract: The authors examine the interlayer surface relaxations and surface energies for the low-index faces of fcc $\mathrm{Ni}$, $\mathrm{Pd}$, $\mathrm{Rh}$, $\mathrm{Pt}$, $\mathrm{Au}$, and $\mathrm{Ir}$ using the Naval Research Laboratory (NRL) tight-binding (TB) method. We compare the TB calculations, utilizing self-consistent charge transfer, with experimental measurements, density functional theory (DFT) calculations, and semiempirical methods. We find that for these metals the NRL-TB method largely reproduces the trends with respect to the exposed face and periodic table position obtained in DFT calculations and experimental measurements. We find that the inclusion of self-consistency in the TB surface calculations is essential for obtaining this agreement, as the TB calculations without it predict large first interlayer expansions for many of these surfaces. We also examine the energetics and relaxations of the $2\ifmmode\times\else\texttimes\fi{}1$ (011) missing row reconstruction for these metals. The TB method predicts that, in agreement with experiment, $\mathrm{Au}$ and $\mathrm{Pt}$ undergo this reconstruction, while $\mathrm{Ni}$, $\mathrm{Pd}$, and $\mathrm{Rh}$ do not, but predicts the $\mathrm{Ir}$ ground state structure to be unreconstructed $1\ifmmode\times\else\texttimes\fi{}1$, opposite to experiment. The interatomic relaxations of the (011) missing row structure for $\mathrm{Pt}$, $\mathrm{Au}$, and $\mathrm{Ir}$ are in good agreement with DFT calculations and experiment. Finally, we analyze the bonding characteristics of these metals using a decomposition of the TB total energy over neighboring atoms and angular momentum character. read less NOT USED (high confidence) Y. Wang, S. Teitel, and C. Dellago, “Melting of icosahedral gold nanoclusters from molecular dynamics simulations.,” The Journal of chemical physics. 2004. link Times cited: 201 Abstract: Molecular dynamics simulations show that gold clusters with … read moreAbstract: Molecular dynamics simulations show that gold clusters with about 600-3000 atoms crystallize into a Mackay icosahedron upon cooling from the liquid. A detailed surface analysis shows that the facets on the surface of the Mackay icosahedral gold clusters soften but do not premelt below the bulk melting temperature. This softening is found to be due to the increasing mobility of vertex and edge atoms with temperature, which leads to inter-layer and intra-layer diffusion, and a shrinkage of the average facet size, so that the average shape of the cluster is nearly spherical at melting. read less NOT USED (high confidence) V. Shenoy, “Mechanics at small scales,” SPIE Optics + Photonics. 2004. link Times cited: 0 Abstract: This paper presents a short overview of the methods used for… read moreAbstract: This paper presents a short overview of the methods used for the study of mechanics at small scales. The key issue to be tackled is the presence of multiple scales starting from the atomic scale. The methods outlined include continuum, atomistic and mixed methods. read less NOT USED (high confidence) R. F. Zhang, L. Kong, H. Gong, and B. Liu, “Comparative study of metastable phase formation in the immiscible Cu–W system by ab initio calculation and n-body potential,” Journal of Physics: Condensed Matter. 2004. link Times cited: 20 Abstract: The lattice constants and cohesive energies of some possible… read moreAbstract: The lattice constants and cohesive energies of some possible metastable Cu–W compounds are obtained by ab initio calculation and the formation of a metastable phase at Cu75W25 is predicted for the equilibrium immiscible Cu–W system. The prediction is in agreement with the fact that a metastable hcp phase was indeed observed in the Cu75W25 multilayer films upon ion beam mixing. Furthermore, some of the ab initio calculated properties are used in deriving an n-body Cu–W potential under the embedded atom method. The constructed Cu–W potential is then used to predict the phase stability of the metastable Cu–W phases over the entire composition and the prediction is also supported by some experimental observations. read less NOT USED (high confidence) J. Zimmerman, E. B. WebbIII, J. J. Hoyt, R. Jones, P. A. Klein, and D. Bammann, “Calculation of stress in atomistic simulation,” Modelling and Simulation in Materials Science and Engineering. 2004. link Times cited: 401 Abstract: Atomistic simulation is a useful method for studying materia… read moreAbstract: Atomistic simulation is a useful method for studying material science phenomena. Examination of the state of a simulated material and the determination of its mechanical properties is accomplished by inspecting the stress field within the material. However, stress is inherently a continuum concept and has been proven difficult to define in a physically reasonable manner at the atomic scale. In this paper, an expression for continuum mechanical stress in atomistic systems derived by Hardy is compared with the expression for atomic stress taken from the virial theorem. Hardy's stress expression is evaluated at a fixed spatial point and uses a localization function to dictate how nearby atoms contribute to the stress at that point; thereby performing a local spatial averaging. For systems subjected to deformation, finite temperature, or both, the Hardy description of stress as a function of increasing characteristic volume displays a quicker convergence to values expected from continuum theory than volume averages of the local virial stress. Results are presented on extending Hardy's spatial averaging technique to include temporal averaging for finite temperature systems. Finally, the behaviour of Hardy's expression near a free surface is examined, and is found to be consistent with the mechanical definition for stress. read less NOT USED (high confidence) K. Shintani, Y. Taniguchi, and S. Kameoka, “Molecular-dynamics analysis of morphological evolution of softly deposited Au nanoclusters,” Journal of Applied Physics. 2004. link Times cited: 12 Abstract: The initial period following deposition of soft-landing Au c… read moreAbstract: The initial period following deposition of soft-landing Au clusters is investigated by classical molecular-dynamics simulation. The embedded-atom method potential is adopted for the interaction between Au atoms. Clusters of specified sizes are cut out of the bulk crystal structure. Whether a cluster equilibrated at a given temperature is in a solid state or in a liquid state is judged by tracking the trajectory of an atom in the cluster and by examining the radial distribution function. The deposition simulation reveals that there is an energy barrier in the morphological accommodation of a cluster to the substrate if the cluster is crystalline before deposition, and is equilibrated at a temperature different from that of the substrate. On the other hand, there is no energy barrier in the morphological accommodation of a cluster that is in a liquid state before deposition. Exceptionally, a crystalline cluster that is nearly at a melting temperature can accommodate itself smoothly to the substrate maintain... read less NOT USED (high confidence) E. Webb, J. Hoyt, G. Grest, and D. Heine, “Atomistic simulations of reactive wetting in metallic systems,” Journal of Materials Science. 2004. link Times cited: 24 NOT USED (high confidence) P. Jensen, A. Clement, and L. J. Lewis, “Diffusion of nanoclusters,” Computational Materials Science. 2004. link Times cited: 19 NOT USED (high confidence) X. W. Zhou, R. Johnson, and H. Wadley, “Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers,” Physical Review B. 2004. link Times cited: 936 Abstract: Recent molecular dynamics simulations of the growth of $[{\m… read moreAbstract: Recent molecular dynamics simulations of the growth of $[{\mathrm{Ni}}_{0.8}{\mathrm{Fe}}_{0.2}/\mathrm{Au}]$ multilayers have revealed the formation of misfit-strain-reducing dislocation structures very similar to those observed experimentally. Here we report similar simulations showing the formation of edge dislocations near the interfaces of vapor-deposited (111) [NiFe/CoFe/Cu] multilayers. Unlike misfit dislocations that accommodate lattice mismatch, the dislocation structures observed here increase the mismatch strain energy. Stop-action observations of the dynamically evolving atomic structures indicate that during deposition on the (111) surface of a fcc lattice, adatoms may occupy either fcc sites or hcp sites. This results in the random formation of fcc and hcp domains, with dislocations at the domain boundaries. These dislocations enable atoms to undergo a shift from fcc to hcp sites, or vice versa. These shifts lead to missing atoms, and therefore a later deposited layer can have missing planes compared to a previously deposited layer. This dislocation formation mechanism can create tensile stress in fcc films. The probability that such dislocations are formed was found to quickly diminish under energetic deposition conditions. read less NOT USED (high confidence) G. Wang, M. A. V. Hove, P. N. Ross, and M. Baskes, “Monte Carlo simulations of segregation in Pt-Ni catalyst nanoparticles.,” The Journal of chemical physics. 2004. link Times cited: 128 Abstract: We have investigated the segregation of Pt atoms in the surf… read moreAbstract: We have investigated the segregation of Pt atoms in the surfaces of Pt-Ni nanoparticles, using modified embedded atom method potentials and the Monte Carlo method. The nanoparticles are constructed with disordered fcc configurations at two fixed overall concentrations (50 at. % Pt and 75 at. % Pt). We use octahedral and cubo-octahedral nanoparticles terminated by {111} and {100} facets to examine the extent of the Pt segregation to the nanoparticle surfaces at T=600 K. The model particles contain between 586 and 4033 atoms (particle size ranging from 2.5 to 5 nm). Our results imply that a complete {100}-facet reconstruction could make the cubo-octahendral Pt-Ni nanoparticles most energetically favorable. We predict that at 600 K due to segregation the equilibrium cubo-octahedral Pt50Ni50 nanoparticles with fewer than 1289 atoms and Pt75Ni25 nanoparticles with fewer than 4033 atoms would achieve a surface-sandwich structure, in which the Pt atoms are enriched in the outermost and third atomic shells while the Ni atoms are enriched in the second atomic shell. We also find that, due to an order-disorder transition, the Pt50Ni50 cubo-octahedral nanoparticles containing more than 2406 atoms would form a core-shell structure with a Pt-enriched surface and a Pt-deficient homogenous core. read less NOT USED (high confidence) J. J. Hoyt, A. Karma, M. Asta, and D. Sun, “From atoms to dendrites,” JOM. 2004. link Times cited: 13 NOT USED (high confidence) N. Lümmen and T. Kraska, “Investigation of the formation of iron nanoparticles from the gas phase by molecular dynamics simulation,” Nanotechnology. 2004. link Times cited: 72 Abstract: The formation of iron particles from the supersaturated gas … read moreAbstract: The formation of iron particles from the supersaturated gas phase is investigated by molecular dynamics simulation. The atomic interaction is modelled with a recent parameterization of the embedded atom method which is able to describe the bcc phase of bulk iron. The influence of the state conditions such as temperature and density on the growth mechanisms of the iron particles is analysed. With the common neighbour analysis method the structural changes of the particles over the course of the growth process is traced. Furthermore, the surface fraction is investigated as an order parameter for the morphology of the particles. It appears that in the early state of the growth process the atomic structure is dominated by icosahedral structures which are, over the course of the growth process, transformed into other structures such as fcc or hcp. Iron atoms in the bcc structure were not found for small particles. The structural reorganization depends mainly on the temperature of the system and on the magnitude of the heat removal from the system. The results are in agreement with experimental and other theoretical investigations on the structure of iron nanoparticles. read less NOT USED (high confidence) Q. Liu and J. Zhuang, “Simulation of dimer diffusion on metal fcc (001) surfaces by molecular dynamics,” Science in China Series E: Technological Sciences. 2004. link Times cited: 1 Abstract: We study systematically the dimer diffusion on metal fcc (00… read moreAbstract: We study systematically the dimer diffusion on metal fcc (001) surfaces through molecular dynamics calculations based on embedded atom method. Besides the conventional hopping and exchange mechanisms, some other interesting diffusion mechanisms are observed, such as the exchange rotation mechanism, the cooperative hopping mechanism, and the cooperative exchange mechanism. On the different kinds of metal surfaces, we find that not only the dominant diffusion mechanism but also the physical model for the exchange mechanism is different. read less NOT USED (high confidence) X. W. Zhou and H. Wadley, “Misfit dislocations in gold/Permalloy multilayers,” Philosophical Magazine. 2004. link Times cited: 16 Abstract: Several groups have reported the misfit dislocation structur… read moreAbstract: Several groups have reported the misfit dislocation structures in Au/Ni0.8Fe0.2 multilayers where the lattice parameter misfit is very large. To explore the factors controlling such structures, molecular dynamics simulations have been used to simulate the vapour-phase growth of (111)-oriented Au/Ni0.8Fe0.2 multilayers. The simulations revealed the formation of misfit dislocations at both the gold-on-Ni0.8Fe0.2 and the Ni0.8Fe0.2-on-gold interfaces. The dislocation configuration and density were found to be in good agreement with previously reported high-resolution transmission electron microscopy observations. Additional atomic-scale simulations of a model nickel–gold system indicated that dislocations are nucleated as the first nickel layer is deposited on gold. These dislocations have an (a/6)⟨112⟩ Burgers vector, typical of a Shockley partial dislocation. Each dislocation creates an extra {220} plane in the smaller lattice parameter nickel layer. These misfit-type dislocations effectively relieve misfit strain. The results also indicated that the dislocation structure is insensitive to the energy of the depositing atoms. Manipulation of the deposition processes is therefore unlikely to reduce this component of the defect population. read less NOT USED (high confidence) T. Rahman, A. Kara, and S. Durukanoğlu, “Structural relaxations, vibrational dynamics and thermodynamics of vicinal surfaces,” Journal of Physics: Condensed Matter. 2003. link Times cited: 25 Abstract: We present here a summary of the calculated structural, dyna… read moreAbstract: We present here a summary of the calculated structural, dynamical and thermodynamical properties of a number of vicinal surfaces of fcc metals with the aim of identifying trends in their characteristics. In general, multilayer relaxations indicate a contraction in the bonds between surface atoms except for that of the least undercoordinated surface atom ('corner') whose bond length with the nearest neighbour bulk atom displays an impressive expansion. Electronic structure calculations show a rearrangement of charge densities near the steps and the layer resolved density of states highlights the characteristics of the undercoordinated atoms. The frequencies of localized vibrational modes on stepped surfaces point to both softening and stiffening of specific force constants, which lead to enhancement of modes in both the lower and higher ends of the frequency spectrum in the vibrational densities of states. Contributions to the surface vibrational entropy from undercoordinated atoms are found to depend strongly on their atomic coordination and play an important role in determining the step excess free energy for certain step geometries. Comparisons of results are made with available experimental data. read less NOT USED (high confidence) W. Fan and X. Gong, “Simulation of Ni cluster diffusion on Au(1 1 0)-(1 × 2) surface,” Applied Surface Science. 2003. link Times cited: 4 NOT USED (high confidence) T. Kitamura, Y. Umeno, and N. Tsuji, “Internal atomic stress near ?5 tilt grain boundary in aluminium under tension,” Modelling and Simulation in Materials Science and Engineering. 2003. link Times cited: 4 Abstract: It is important to clarify the mechanical properties of inho… read moreAbstract: It is important to clarify the mechanical properties of inhomogeneous structures from the viewpoint of atomic scale to understand the mechanical behaviour of microscopic materials. Thus, the mechanical properties of local regions with inhomogeneous and non-uniform structure have been intensively investigated on the basis of the atomic stress and the elastic constant. In this study, in order to examine the mechanical behaviour of local regions under external load, we conduct atomic tensile simulations on a Σ5-(013) tilt grain boundary in aluminium using the many-body effective medium theory potential function. The structure of the grain boundary consists of two types of atomic layers, one of which has a higher elastic constant near the grain boundary and the other has a lower one. Although the grain boundary consists of identical atomic species, the local region near the grain boundary displays the mechanical properties similar to those of a composite material. read less NOT USED (high confidence) H. Avcı, M. Çivi, Z. B. Güvenç, and J. Jellinek, “Collisionless fragmentation of non-rotating Nin (n = 4-14) clusters: a molecular dynamics study,” Journal of Physics B. 2003. link Times cited: 2 Abstract: Collisionless fragmentation of non-rotating Nin (n = 4–14) c… read moreAbstract: Collisionless fragmentation of non-rotating Nin (n = 4–14) clusters is studied using micro-canonical molecular dynamics (MD) computer simulations. The clusters are modelled by an embedded-atom potential energy surface. The distributions of the channel-specific fragmentation probabilities, and the global and channel-specific fragmentation rate constants are computed and analysed as functions of the internal energy and size of the clusters. The trends derived from the dynamical calculations are compared to the fragmentation energy patterns, those of the Rice–Ramsperger–Kassel (RRK) and statistical approaches. The rate constants are an order of magnitude smaller for the RRK model than with both the MD and transition-state theory approaches. The results are also compared with the other multi-channel fragmentation works. read less NOT USED (high confidence) Z. Chvoj, C. Ghosh, T. Rahman, and M. Tringides, “Prefactors for interlayer diffusion on Ag/Ag(111),” Journal of Physics: Condensed Matter. 2003. link Times cited: 3 Abstract: Interlayer diffusion controls the transfer of atoms between … read moreAbstract: Interlayer diffusion controls the transfer of atoms between layers in a film and is the key factor determining whether growth is layer-by-layer or two-dimensional. Analysis of recent experimental data on Ag/Ag(111) taken at two sets of temperatures provides similar values for the step edge barrier ?Es but not for the ratio of the prefactors ?s/?t where ?t is the prefactor for diffusion on a terrace and ?s the prefactor at a step edge site. A prefactor ratio larger than 1, 1$>?s/?t > 1, is extracted from the measurements at low temperature (T ?s/?t > 1, in good agreement with the low temperature experiments. read less NOT USED (high confidence) I. Sklyadneva, G. Rusina, and E. Chulkov, “Vibrational properties ofCu(100)−c(2×2)−Pdsurface and subsurface alloys,” Physical Review B. 2003. link Times cited: 9 Abstract: Using interaction potentials from the embedded-atom method w… read moreAbstract: Using interaction potentials from the embedded-atom method we investigated the structural and vibrational properties of a Cu(100)-c(2 × 2)-Pd surface alloy and an underlayer c(2 × 2) alloy with a mixed CuPd second layer. The calculated surface phonon frequencies are in agreement with the experimental values obtained by electron energy-loss spectroscopy. From the calculated local phonon densities of states we find that surface effects are most pronounced in the first two layers for both systems studied. The results also indicate a very strong Pd-Cu bonding accompanied by a weaker bonding of the Cu surface atoms to their nearest neighbors. This has considerable influence on the surface phonon frequencies. read less NOT USED (high confidence) I. Choi, C. Whang, and C. Hwang, “Surface-induced phase separation in Pd–Ag alloy: the case opposite to surface alloying,” Journal of Physics: Condensed Matter. 2003. link Times cited: 8 Abstract: Since Pd–Ag alloys constitute a well known prototype binary … read moreAbstract: Since Pd–Ag alloys constitute a well known prototype binary alloy system, phase separation at the surface was not expected. However, we have shown direct evidence of surface-induced phase separation in Pd–Ag alloys using photoemission spectroscopy. This phenomenon is just the opposite of surface alloying between two miscible elements. The miscibility gap in this alloy near the surface is clearly observed in our core level spectra. The characteristics of this surface-induced phase separation have been probed over various composition ranges using x-ray photoelectron spectroscopy. read less NOT USED (high confidence) J. Jiménez‐Sáez, J. Domínguez-Vázquez, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “Molecular dynamics study of a Ni/Cu(001) interface,” Nanotechnology. 2003. link Times cited: 5 Abstract: The Ni/Cu(001) metallic interface shows interesting magnetic… read moreAbstract: The Ni/Cu(001) metallic interface shows interesting magnetic properties due to the lattice misfit. Its components exhibit a misfit of 2.6% in their lattice parameters. Hence, the growing thin film–substrate interface is strained. We are interested exclusively in the solid phase formation effects; therefore, the growth kinetics effects will be avoided. This work is focused on the analysis of atomic distances and deformations in this system and on the calculation of the interface energy. It is shown how the stabilization on an atomic scale of different Ni nanocrystals set down on top of a large enough Cu(001) crystal is achieved. The adjustment between the lattice parameters of the Ni clusters on the Cu substrate is analysed. Specially, changes in the atomic distances at the interface are quantified. The main result is that the anisotropy of the structural matching causes a cubic lattice to become a tetragonal one. In addition, we carry out the energetic analysis of this interface. read less NOT USED (high confidence) Y. Chui and K. Y. Chan, “Analyses of surface and core atoms in a platinum nanoparticle,” Physical Chemistry Chemical Physics. 2003. link Times cited: 16 Abstract: Molecular dynamics simulations of a platinum nanocluster con… read moreAbstract: Molecular dynamics simulations of a platinum nanocluster consisting 250 atoms were performed at different temperatures between 70 K and 298 K. The semi-empirical, many-body Sutton–Chen (SC) potential was used to model the interatomic interaction in the metallic system. Regions of core or bulk-like atoms and surface atoms can be defined from analyses of structures, atomic coordination, and the local density function of atoms as defined in the SC potential. The core atoms in the nanoparticle behave as bulk-like metal atoms with a predominant face centered cubic (fcc) packing. The interface between surface atoms and core atoms is marked by a peak in the local density function and corresponds to near surface atoms. The near surface atoms and surface atoms prefer a hexagonal closed packing (hcp). The temperature and size effects on structures of the nanoparticle and the dynamics of the surface region and the core region are discussed. read less NOT USED (high confidence) V. Grigoryan and M. Springborg, “Structure and energetics of Ni clusters with up to 150 atoms,” Chemical Physics Letters. 2003. link Times cited: 46 NOT USED (high confidence) A. Hasmy, P. Serena, and E. A. M. Dagger, “Molecular Dynamics Simulations for Metallic Nanosystems,” Molecular Simulation. 2003. link Times cited: 1 Abstract: Nanotechnology is a crucial field for future scientific deve… read moreAbstract: Nanotechnology is a crucial field for future scientific development where many different disciplines meet. Computational modelization of nanometer-sized structures is a key issue in this development because (i) it allows a considerable saving of resources and costly experimental setups intended to fabricate nanometric test devices and (ii) nowadays the study of nanometric sized systems is feasible with thoroughly designed computational codes and relatively low cost computational resources. This article describes how molecular dynamics simulations, in combination with potentials obtained in the framework of the embedded atom method, are able to describe the properties of two systems of interest for the development of future nanoelectronic devices: metallic nanowires and metallic nanofilms. Our results show that nanowire stretching results in a series of well-defined geometric structures (shells) and that thin films experiment a crystallographic phase transition for a decreasing number of layers. In both cases, good agreement with experiments is found. read less NOT USED (high confidence) G. Zhou and J. C. Yang, “Initial oxidation kinetics of copper (1 1 0) film investigated by in situ UHV-TEM,” Surface Science. 2003. link Times cited: 68 NOT USED (high confidence) J. Peltola, K. Nordlund, and J. Keinonen, “Heat spike effect on the straggling of cluster implants,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 26 NOT USED (high confidence) V.Sorkin, E. Polturak, and J. Adler, “Molecular dynamics study of melting of the bcc metal vanadium. I. Mechanical melting,” Physical Review B. 2003. link Times cited: 49 Abstract: We present molecular dynamics simulations of the homogeneous… read moreAbstract: We present molecular dynamics simulations of the homogeneous (mechanical) melting transition of a bcc metal, vanadium. We study both the nominally perfect crystal and one that includes point defects. According to the Borncriterion, a solid cannot be expanded above a critical volume, at which a "rigidity catastrophe" occurs. This catastrophe is caused by the vanishing of the elastic shear modulus. We found that this critical volume is independent of the route by which it is reached, whether by heating the crystal or by adding interstitials at a constant temperature which expand the lattice. Overall, these results are similar to what was found previously for an fcc metal, copper. The simulations establish a phase diagram of the mechanical melting temperature as a function of the concentration of interstitials. Our results show that the Born model of melting applies to bcc metals in both the nominally perfect state and the case where point defects are present. read less NOT USED (high confidence) Z. B. Güvenç and D. Güvenç, “Hydrogen recombination on a mixed adsorption layer at saturation on a metal surface: H → (D + H)sat + Ni(100),” Surface Science. 2003. link Times cited: 9 NOT USED (high confidence) I. A. Wojciechowski and B. Garrison, “Theoretical study of mechanisms responsible for emission of highly excited metal atoms,” Surface Science. 2003. link Times cited: 10 NOT USED (high confidence) J. A. Alonso and M. López, “Growth Ability and Stability Indices of Clusters,” Journal of Cluster Science. 2003. link Times cited: 7 NOT USED (high confidence) G. Rusina, I. Sklyadneva, and E. Chulkov, “Vibrational states of the Cu(100) surfaces with nickel adlayers,” Physics of the Solid State. 2003. link Times cited: 1 NOT USED (high confidence) Y. G. Xu and G. Liu, “Fitting interatomic potentials using molecular dynamics simulations and inter-generation projection genetic algorithm,” Journal of Micromechanics and Microengineering. 2003. link Times cited: 11 Abstract: In this paper we propose a new algorithm to fit interatomic … read moreAbstract: In this paper we propose a new algorithm to fit interatomic potentials. In the new algorithm, molecular dynamics simulations are applied to calculate the material properties which are used to match the experimental data during the fitting procedure. This includes the effect of atom relaxations in fitting calculations. An inter-generation projection genetic algorithm is used to optimize the fitting parameters until the error between the calculated and experimental material properties is within tolerance. This leads to a global optimal solution. The new algorithm significantly improves the accuracy and transferability of the fitted potential. It has been demonstrated by a numerical example of fitting potential of nickel. read less NOT USED (high confidence) J. Hoyt, J. W. Garvin, E. B. Webb, and M. Asta, “An embedded atom method interatomic potential for the Cu–Pb system,” Modelling and Simulation in Materials Science and Engineering. 2003. link Times cited: 57 Abstract: A simple procedure is used to formulate a Cu–Pb pair interac… read moreAbstract: A simple procedure is used to formulate a Cu–Pb pair interaction function within the embedded atom (EAM) method framework. Embedding, density and pair functions for pure Cu and pure Pb are taken from previously published EAM studies. Optimization of the Cu–Pb potential was achieved by comparing with experiment the computed heats of mixing for Cu–Pb liquid alloys and the equilibrium phase diagram, the latter being determined via a thermodynamic integration technique. The topology of the temperature-composition phase diagram computed with this EAM potential is consistent with experiment and features a liquid–liquid miscibility gap, low solubility of Pb in solid Cu and a monotectic reaction at approximately 1012 K. read less NOT USED (high confidence) T. Kitamura and Y. Umeno, “Validity of effective medium theory for aluminium under tension,” Modelling and Simulation in Materials Science and Engineering. 2003. link Times cited: 14 Abstract: Reliability of the potential functions under the condition f… read moreAbstract: Reliability of the potential functions under the condition far from equilibrium states, which is called transferability, is an important factor in the simulations of materials with nanoscopic complex structure under high stress condition. However, it has not been sufficiently investigated because it is difficult to get precise experimental data in such conditions. In this paper, simulations are conducted for aluminium bulk, grain boundary of aluminium and atomic chain under high strain using the potential function of the effective medium theory (EMT) as well as ab initio calculations in order to clarify the validity of EMT. In the cases of single crystal and the grain boundary under tensile strain, the results obtained from the EMT potential agree well with those obtained by ab initio analysis. However, the EMT cannot be applied to the atomic chain because the distribution of charge density differs significantly from that in the bulk. read less NOT USED (high confidence) A. Al-Rawi, A. Kara, and T. Rahman, “Comparative study of anharmonicity: Ni(111), Cu(111), and Ag(111),” Physical Review B. 2002. link Times cited: 20 Abstract: We present a comparative study of the structure and the dyna… read moreAbstract: We present a comparative study of the structure and the dynamics of the most close packed surface of Ni, Cu, and Ag from near room temperature up to 0.9T m , using molecular dynamics simulations and interaction potentials from the embedded atom method. Calculated shifts in the surface phonon frequencies, the broadening of their linewidths, and the variations in the mean square vibrational amplitudes of surface atoms, as a function of temperature, indicate that anharmonic effects are small on these surfaces. The surface thermal expansion of these three (111) surfaces is also found to be smaller than that of the respective (100) and (110) surfaces. Additionally, we do not find any premelting or pronounced disordering on these surfaces, in the temperature range considered. read less NOT USED (high confidence) W. Hu and F. Masahiro, “The application of the analytic embedded atom potentials to alkali metals,” Modelling and Simulation in Materials Science and Engineering. 2002. link Times cited: 40 Abstract: Analytic modified embedded atom method type many-body potent… read moreAbstract: Analytic modified embedded atom method type many-body potentials have been constructed for alkali metals. The bcc lattice is shown to be energetically most stable when compared with fcc and ideal hcp structures. The phonon dispersion curves, density of states, Debye temperature, heat capacity, surface energy and thermal expansion properties of these metals are calculated, which are comparable to experimental and other theoretical results. The properties of point defects, such as vacancy, divacancy, and self-interstitial atoms, have also been evaluated with these potentials, and the diffusion mechanism in alkali metals are discussed. read less NOT USED (high confidence) C. Rountree, R. Kalia, E. Lidorikis, A. Nakano, L. Brutzel, and P. Vashishta, “ATOMISTIC ASPECTS OF CRACK PROPAGATION IN BRITTLE MATERIALS: Multimillion Atom Molecular Dynamics Simulations,” Annual Review of Materials Research. 2002. link Times cited: 173 Abstract: ▪ Abstract Atomistic aspects of dynamic fracture in a variet… read moreAbstract: ▪ Abstract Atomistic aspects of dynamic fracture in a variety of brittle crystalline, amorphous, nanophase, and nanocomposite materials are reviewed. Molecular dynamics (MD) simulations, ranging from a million to 1.5 billion atoms, are performed on massively parallel computers using highly efficient multiresolution algorithms. These simulations shed new light on (a) branching, deflection, and arrest of cracks; (b) growth of nanoscale pores ahead of the crack and how pores coalesce with the crack to cause fracture; and (c) the influence of these mechanisms on the morphology of fracture surfaces. Recent advances in novel multiscale simulation schemes combining quantum mechanical, molecular dynamics, and finite-element approaches and the use of these hybrid approaches in the study of crack propagation are also discussed. read less NOT USED (high confidence) J. Rogan, R. Ramírez, A. Romero, and M. Kiwi, “Rearrangement collisions between gold clusters,” The European Physical Journal D - Atomic, Molecular, Optical and Plasma Physics. 2002. link Times cited: 21 NOT USED (high confidence) S.-P. Ping and P. Balbuena, “Platinum nanoclusters on graphite substrates: a molecular dynamics study,” Molecular Physics. 2002. link Times cited: 56 Abstract: Molecular dynamics simulations are used to investigate the s… read moreAbstract: Molecular dynamics simulations are used to investigate the shape and structure evolution of single platinum clusters of cubic and spherical shape containing 256 and 260 atoms, respectively, deposited on a static graphite substrate. The evolution is monitored at variable temperature, and as a function of metal-substrate interactions at constant temperature. The Pt-Pt interactions are modelled with the many-body Sutton-Chen potential, whereas a Lennard-Jones potential is used to describe the Pt-C interactions. Heating and cooling curves calculated between 200 K and 1800 K are used to determine solid-solid and solid-liquid transitions. Structural changes are detected through analyses of density profiles and diffusion coefficients. A clear analogy is observed between temperature-induced wetting phenomena and those resulting from enhancement of the metal-substrate interactions. read less NOT USED (high confidence) O. Trushin, E. Granato, S. Ying, P. Salo, and T. Ala‐Nissila, “Mechanisms of dislocation nucleation in strained epitaxial layers,” Physica Status Solidi B-basic Solid State Physics. 2002. link Times cited: 46 Abstract: Using molecular static simulations we have studied possible … read moreAbstract: Using molecular static simulations we have studied possible mechanisms of stress relaxation with misfit dislocation nucleation in strained heteroepitaxial layers. Two-dimensional models of atomic systems with Lennard-Jones type potential were considered. Combination of total energy minimization with spherical repulsion and nudged elastic band calculation allowed us to study possible transitions between an initial coherent interface state and a final state with a single dislocation. Different transition paths involving either successive relaxation of layers or concerted sliding along atomic rows have been identified as possible mechanisms with low activation energies. The role of these mechanisms in heteroepitaxial thin film growth and possible implementations in three dimensions are discussed. read less NOT USED (high confidence) T. I. Mazilova and I. M. Mikhailovskii, “Erosion of the field emitter surface exposed to low-energy ions,” Technical Physics. 2002. link Times cited: 2 NOT USED (high confidence) T. Rahman, J. Spangler, and A. Al-Rawi, “Theoretical studies of the surface phonon linewidth,” Journal of Physics: Condensed Matter. 2002. link Times cited: 9 Abstract: A brief review of two theoretical methods for calculating th… read moreAbstract: A brief review of two theoretical methods for calculating the linewidth of surface phonons is presented with specific application to the low-Miller-index surfaces of Ag, Cu, and Al. In the quasi-analytic method, linewidths are calculated by treating the cubic term of a reliable interatomic potential in first-order, time-dependent perturbation theory. In molecular dynamics?(MD) simulations, linewidths are obtained from appropriate correlation functions and include the fully anharmonic contribution of the interaction potential. Surface relaxations and phonon frequencies calculated in the harmonic approximation, using selected many-body interatomic potentials, are found to be in good agreement with results from experimental data and ab initio calculations. At 300?K, the surface phonon linewidths range approximately between?1 and 0.1?THz, and are found to be in reasonable agreement with the values deduced from experiments. Some disagreement is found between results from perturbative methods and MD simulations. For the low-Miller-index surfaces of Ni, available results from MD simulations also summarized. read less NOT USED (high confidence) J. Jiménez‐Sáez, J. Domínguez-Vázquez, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “A molecular dynamics study of an Au/Cu(001) interface,” Nanotechnology. 2002. link Times cited: 9 Abstract: This work focuses on the analysis of atomic distances and de… read moreAbstract: This work focuses on the analysis of atomic distances and deformations in an Au/Cu(001) metallic interface and on the calculation of the energy of this interface. We study the possible adaptation of the atomic distances at the interface of two crystals with a considerable difference between their lattice parameters, such as found in Au and Cu. These crystals have a misfit of 12.8% of such parameters. Hence, the growing thin film-substrate interface is strained. We show how the relaxation of different substrate-cluster structures (a few monolayers) takes place on an atomic scale. We find that pseudomorphic growth is only possible when the system is a Cu cluster on top of an Au substrate. In the opposite case, Au on a Cu substrate, the system relaxes generating a network of dislocations. In particular, mean changes in the lattice parameters at the interface are quantified. In addition, we carry out the energetic analysis of these systems, which is of great interest to describe local properties such as electrical conduction. read less NOT USED (high confidence) J. Miguel, J. Camarero, and R. Miranda, “Studies of surface diffusion and growth on Cu(111) by means of thermal energy atom scattering,” Journal of Physics: Condensed Matter. 2002. link Times cited: 11 Abstract: Some of the special characteristics of thermal energy atom s… read moreAbstract: Some of the special characteristics of thermal energy atom scattering make it a uniquely adapted technique for investigations on diffusion and growth. In this work we review some recent results obtained during homoepitaxial and heteroepitaxial growth, both on bare substrates and assisted by a surfactant layer. We describe the fundamentals of the technique and the method of analysis, putting special emphasis on the information that can be obtained about important physical parameters such as Ehrlich-Schwoebel barriers, surface and edge diffusion, step bunching, and interdiffusion at heteroepitaxial interfaces. We also show examples of how diffusion and growth can be tailored by different means, such as using surfactants. read less NOT USED (high confidence) V. Kamyshenko, V. Kartuzov, V. Shevchenko, and W. Gooch, “Effects of Lattice Relaxation and Limiting Shear Stress of fcc Metals at High-Rate Deformation,” Strength of Materials. 2002. link Times cited: 0 NOT USED (high confidence) T. Ala‐Nissila, R. Ferrando, and S. Ying, “Collective and single particle diffusion on surfaces,” Advances in Physics. 2002. link Times cited: 431 Abstract: We review in this article the current theoretical understand… read moreAbstract: We review in this article the current theoretical understanding of collective and single particle diffusion on surfaces and how it relates to the existing experimental data. We begin with a brief survey of the experimental techniques that have been employed for the measurement of the surface diffusion coefficients. This is followed by a section on the basic concepts involved in this field. In particular, we wish to clarify the relation between jump or exchange motion on microscopic length scales, and the diffusion coefficients which can be defined properly only in the long length and time scales. The central role in this is played by the memory effects. We also discuss the concept of diffusion under nonequilibrium conditions. In the third section, a variety of different theoretical approaches that have been employed in studying surface diffusion such as first principles calculations, transition state theory, the Langevin equation, Monte Carlo and molecular dynamics simulations, and path integral formalism are presented. These first three sections form an introduction to the field of surface diffusion. Section 4 contains subsections that discuss surface diffusion for various systems which have been investigated both experimentally and theoretically. The focus here is not so much on specific systems but rather on important issues concerning diffusion measurements or calculations. Examples include the influence of steps, diffusion in systems undergoing phase transitions, and the role of correlation and memory effects. Obviously, the choice of topics here reflects the interest and expertise of the authors and is by no means exhaustive. Nevertheless, these topics form a collection of issues that are under active investigation, with many important open questions remaining. read less NOT USED (high confidence) L. Hanley and S. Sinnott, “The growth and modification of materials via ion-surface processing,” Surface Science. 2002. link Times cited: 82 NOT USED (high confidence) T. Vegge and K. Jacobsen, “Atomistic simulations of dislocation processes in copper,” Journal of Physics: Condensed Matter. 2002. link Times cited: 38 Abstract: We discuss atomistic simulations of dislocation processes in… read moreAbstract: We discuss atomistic simulations of dislocation processes in copper based on effective medium theory interatomic potentials. Results on screw dislocation structures and processes are reviewed with particular focus on point defect mobilities and processes involving cross slip. For example, the stability of screw dislocation dipoles is discussed. We show that the presence of jogs will strongly influence cross slip barriers and dipole stability. We furthermore present some new results on jogged edge dislocations and edge dislocation dipoles. The jogs are found to be extended, and simulations of vacancy controlled climb show the jogs to climb easily in their extended form. The stability of small vacancy dipoles is discussed and it is seen that the introduction of jogs may lead to the formation of Z-type faulted vacancy dipoles. read less NOT USED (high confidence) Y. G. Yang, R. Johnson, and H. Wadley, “Kinetic Monte Carlo simulation of heterometal epitaxial deposition,” Surface Science. 2002. link Times cited: 17 NOT USED (high confidence) F. Baletto, R. Ferrando, A. Fortunelli, F. Montalenti, and C. Mottet, “Crossover among structural motifs in transition and noble-metal clusters,” Journal of Chemical Physics. 2002. link Times cited: 346 Abstract: The energetics of nanoclusters is investigated for five diff… read moreAbstract: The energetics of nanoclusters is investigated for five different metals (Ag, Cu, Au, Pd, and Pt) by means of quenched molecular dynamics simulations. Results are obtained for two different semiempirical potentials. Three different structural motifs are considered: icosahedra (Ih), decahedra (Dh), and truncated octahedra (TO). The crossover sizes among structural motifs are directly calculated, considering cluster up to sizes N≃40 000. For all the systems considered, it is found that icosahedra are favored at small sizes, decahedra at intermediate sizes, and truncated octahedra at large sizes. However, the crossover sizes depend strongly on the metal: in Cu, the icosahedral interval is rather large, and it is followed by a very wide decahedral window; on the contrary, in Au, the icosahedral interval is practically absent, and the decahedral window is narrow. The other metals display intermediate behaviors, Ag being close to Cu, and Pd and Pt being close to Au. A simple criterion, which is based on the rat... read less NOT USED (high confidence) C. Ghosh, A. Kara, and T. Rahman, “Theoretical aspects of vertical and lateral manipulation of atoms,” Surface Science. 2001. link Times cited: 15 NOT USED (high confidence) I. Galanakis, N. Papanikolaou, and P. Dederichs, “Applicability of the broken-bond rule to the surface energy of the fcc metals,” Surface Science. 2001. link Times cited: 147 NOT USED (high confidence) X. W. Zhou and H. Wadley, “Mechanisms of inert gas impact induced interlayer mixing in metal multilayers grown by sputter deposition,” Journal of Applied Physics. 2001. link Times cited: 9 Abstract: Control of interfacial roughness and chemical mixing is crit… read moreAbstract: Control of interfacial roughness and chemical mixing is critical in nanomaterials. For example, multilayers composed of ∼20 A conductive layer sandwiched between two ∼50 A ferromagnetic layers can exhibit giant magnetoresistance (GMR). This property has caused a tremendous recent increase in hard disk storage capacity, and can potentially result in a new generation of nonvolatile magnetic random access memories. It has been established that good GMR properties can be obtained when the interfacial roughness and interlayer mixing of these multilayers are low. However, flat interfaces in nanoscale multilayers are not thermodynamically stable, and cannot be obtained using thermal energy deposition processes such as molecular-beam epitaxy. Hyperthermal energy sputter deposition techniques using either plasma or ion-beam gun are able to create nonequilibrium flat interfaces, and have been shown to produce better GMR multilayers. In these processes, however, inert gas ions or neutrals with energies between 50 an... read less NOT USED (high confidence) Y. G. Yang, X. W. Zhou, R. A. Johnson, and H. Wadley, “MONTE CARLO SIMULATION OF HYPERTHERMAL PHYSICAL VAPOR DEPOSITION,” Acta Materialia. 2001. link Times cited: 33 NOT USED (high confidence) Z. Ovadyahu, “Electron-electron correlations in non-equilibrium hopping transport,” Philosophical Magazine B. 2001. link Times cited: 3 Abstract: Electron-electron interaction effects are expected to be pro… read moreAbstract: Electron-electron interaction effects are expected to be prominent in the insulating regime because of lack of screening. One of the well-known consequences of interactions is the Coulomb gap, first discussed by Pollak. The contribution of these effects however, is not always readily recognized in the near-equilibrium properties of hopping systems. Here we review some examples of transport properties of such systems driven far from equilibrium and discuss the role played by interactions. read less NOT USED (high confidence) S. Dorfman, V. Liubich, D. Fuks, and K. C. Mundim, “Simulations of decohesion and slip of the Σ3⟨111⟩ grain boundary in tungsten with non-empirically derived interatomic potentials: the influence of boron interstitials,” Journal of Physics: Condensed Matter. 2001. link Times cited: 12 Abstract: Monte Carlo atomistic simulations of the properties of Σ3111… read moreAbstract: Monte Carlo atomistic simulations of the properties of Σ3111 grain boundaries in W are carried out. We demonstrate the influence of boron additive on the resistance of the grain boundary with respect to different shifts. The interatomic potentials used in these simulations are obtained from ab initio total-energy calculations. These calculations are performed in the framework of density functional theory in the coherent potential approximation. A recursion procedure for extracting A-B-type interatomic potentials is suggested. read less NOT USED (high confidence) H. Lei, “Melting of free copper clusters,” Journal of Physics: Condensed Matter. 2001. link Times cited: 23 Abstract: Extensive molecular-dynamics simulations of melting processe… read moreAbstract: Extensive molecular-dynamics simulations of melting processes of copper clusters, Cu55, Cu147 and Cu309 with closed-shell icosahedral structure, were performed using an environment-dependent classical tight-binding potential. The results reveal that the cluster may transit completely from pure solid to pure liquid phase with temperature via a dynamics-coexistence (DC) process, depending upon the latent heat and vibrational entropy. In the DC regime, no other than the first-order solid-liquid phase transition with time is related to the specific structure of the cluster. read less NOT USED (high confidence) S. Foiles and J. Hoyt, “Computer Simulation of Bubble Growth in Metals Due to He.” 2001. link Times cited: 18 Abstract: Atomistic simulations of the growth of helium bubbles in met… read moreAbstract: Atomistic simulations of the growth of helium bubbles in metals are performed. The metal is represented by embedded atom method potentials for palladium. The helium bubbles are treated via an expanding repulsive spherical potential within the metal lattice. The simulations predict bubble pressures that decrease monotonically with increasing helium to metal ratios. The swelling of the material associated with the bubble growth is also computed. It is found that the rate of swelling increases with increasing helium to metal ratio consistent with experimental observations on the swelling of metal tritides. Finally, the detailed defect structure due to the bubble growth was investigated. Dislocation networks are observed to form that connect the bubbles. Unlike early model assumptions, prismatic loops between the bubbles are not retained. These predictions are compared to available experimental evidence. read less NOT USED (high confidence) D. Chase, M. Manning, J. A. Morgan, G. Nathanson, and R. Gerber, “Argon scattering from liquid indium: Simulations with embedded atom potentials and experiment,” Journal of Chemical Physics. 2000. link Times cited: 21 Abstract: An embedded-atom type potential for liquid indium is develop… read moreAbstract: An embedded-atom type potential for liquid indium is developed by fitting bulk liquid thermodynamic and structural data. An empirical pairwise Ar–In interaction is also proposed. Molecular-dynamics simulations of argon scattering from liquid indium are carried out and compared with molecular beam scattering data. Very good agreement is found between the experimental and theoretical angular and energy scattering distributions. This supports the potential functions used. Implications for the atomic-scale structure of liquid In and for gas–surface energy transfer are briefly discussed. read less NOT USED (high confidence) G. Barrera, R. Tendler, and E. P. Isoardi, “Structure and energetics of Cu-Au alloys,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 21 Abstract: The structures and energetics of Cu-Au alloys over a wide ra… read moreAbstract: The structures and energetics of Cu-Au alloys over a wide range of temperatures are studied using a combination of quasi-harmonic (QH) lattice dynamics and Monte Carlo (MC) simulations at constant temperature and constant pressure. The many-body potential used is fitted to room-temperature experimental data taking vibrational contributions into account. Transitions to the disordered phases are studied using MC simulations in which not only anisotropic deformation of the unit cell and atomic movements are allowed, but also exchange of atoms of different type is explicitly considered. Our calculations reproduce all characteristic features of the order-disorder transitions, including the characteristic peaks in the plots of heat capacity as a function of temperature. read less NOT USED (high confidence) J. Zimmerman, H. Gao, and F. F. Abraham, “Generalized stacking fault energies for embedded atom FCC metals,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 284 Abstract: Atomistic calculations for the 112 -generalized stacking fau… read moreAbstract: Atomistic calculations for the 112 -generalized stacking fault (GSF) energy curve are performed for various embedded atom models of FCC metals. Models include those by Voter and Chen; Angelo, Moody and Baskes; Oh and Johnson; Mishin and Farkas; and Ercolessi and Adams. The resulting curves show similar characteristics but vary in their agreement with the experimental estimates of the intrinsic stacking fault energy, sf , and with density functional theory (DFT) calculations of the GSF curve. These curves are used to obtain estimates of the unstable stacking fault energy, us , a quantity used in a criterion for dislocation nucleation. Curves for nickel and copper models show the theoretically expected skewed sinusoidal shape; however, several of the aluminium models produce an irregularly shaped GSF curve. Copper and aluminium values for us are underestimates of calculations from DFT, although some of the nickel models produce a value matching one of the available DFT results. Values for sf are either fitted to, or underestimate, the measured results. For use in simulations, the authors recommend using the Voter and Chen potential for copper, and either the Angelo, Moody and Baskes potential or the Voter and Chen potential for nickel. None of the potentials model aluminium well, indicating the need for a more-advanced empirical potential. read less NOT USED (high confidence) Y.-S. Kim and D. Choi, “Microscopic study for the behavior of grain boundary using molecular dynamics,” Metals and Materials. 2000. link Times cited: 3 NOT USED (high confidence) J. A. Alonso, “Electronic and atomic structure, and magnetism of transition-metal clusters.,” Chemical reviews. 2000. link Times cited: 422 NOT USED (high confidence) Y. Leng, G. Yang, Y.-zhong Hu, and L. Zheng, “Computer experiments on nano-indentation: A molecular dynamics approach to the elasto-plastic contact of metal copper,” Journal of Materials Science. 2000. link Times cited: 24 NOT USED (high confidence) M. Michailov and N. Georgiev, “Monte Carlo simulations of the interface layer in Pb/Cu(110); a tight-binding-model calculation,” Journal of Physics: Condensed Matter. 1999. link Times cited: 3 Abstract: Monte Carlo simulations with energies computed via the tight… read moreAbstract: Monte Carlo simulations with energies computed via the tight-binding model are applied in studying the equilibrium properties of the interface layer in the system Pb/Cu(110). In a submonolayer range below 0.5 ML (ML monolayer), the model based on the coverage-dependent interactions reveals a lattice-gas random adsorption, an equilibrium adatom-substrate intermixing and formation of a centred c(2 × 2) phase. The process of intermixing is found to be coverage and temperature dependent. At high adatom concentration, 0.75-0.80 ML, as a result of very subtle competition between the strain energy and the energy gain due to mixing, the system forms a succession of commensurate unidimensional p(n × 1) structures. The results are in line with those recently obtained from scanning tunnelling microscopy and thermal energy atom scattering observations. read less NOT USED (high confidence) R. Longo, O. Diéguez, C. Rey, and L. J. Gallego, “Embedded atom model calculations of the structures of small Ni clusters and of a full Ni monolayer on the (001) surface of Al,” The European Physical Journal D - Atomic, Molecular, Optical and Plasma Physics. 1999. link Times cited: 3 NOT USED (high confidence) K. P. Zol’nikov, T. Uvarov, A. Lipnitskii, D. Saraev, and S. Psakh’e, “Characteristics of cleavage fracture during interaction of nonlinear waves with the free surface of a copper single crystal,” Technical Physics Letters. 1999. link Times cited: 3 NOT USED (high confidence) Y. Devyatko, S. Rogozhkin, V. I. Troyan, E. Gusev, and T. Gustafsson, “Vacancy mechanism of the anomalous behavior of surface atoms at elevated temperatures,” Journal of Experimental and Theoretical Physics. 1999. link Times cited: 2 NOT USED (high confidence) M. C. Giménez, M. D. Pópolo, and E. Leiva, “Monte Carlo simulation for the formation and growth of low dimensionality phases during underpotential deposition of Ag on Au(100),” Electrochimica Acta. 1999. link Times cited: 33 NOT USED (high confidence) H. Avcı, M. Çivi, Z. B. Güvenç, and J. Jellinek, “Fragmentation of a Non-Rotating Ni19 Cluster: A Molecular Dynamics Study,” Mathematical & Computational Applications. 1999. link Times cited: 1 Abstract: Collisionless fragmentation of a non-rotating Ni19 cluster i… read moreAbstract: Collisionless fragmentation of a non-rotating Ni19 cluster is studied using constant-energy molecular dynamics computer simulations. The cluster is modelled by an embedded atom model (EAM) energy surface. Distribution of the channel-specific fragmentation probabilities, and global rate constants are computed and analyzed as functions of the internal energy of the cluster. The results are compared with those obtained using the RRK statistical approach, and also compared with the other multi-channel fragmentation work. read less NOT USED (high confidence) V. Shah and L. Yang, “Nanometre fcc clusters versus bulk bcc alloy: The structure of Cu-Pd catalysts,” Philosophical Magazine. 1999. link Times cited: 16 Abstract: We present a simple method to improve the accuracy of the ca… read moreAbstract: We present a simple method to improve the accuracy of the calculated heat of mixing for the Cu-Pd alloy within the formalism of the molecular dynamics/ Monte Carlo-corrected effective medium (MD/MC-CEM) theory by adding a fitted Morse potential to the pair interaction between Cu and Pd atoms. This leads to a much better agreement between the theoretical and experimental values of heats of mixing for five different compositions of the Cu-Pd alloy in the bulk phases. Using this newly fitted model, we have performed simulations on CuPd clusters consisting of 50–10000 atoms with fee and bcc structures. Our calculations show that in the range of cluster sizes of several thousand atoms, the fee structure is energetically favoured over the bcc structure. We estimate an approximate size for the fee to bcc (CsCl, known bulk structure for CuPd) transition in these clusters to be around 10000 atoms. Additionally, we have also performed calculations of the X-ray diffraction patterns of a variety of cluster g... read less NOT USED (high confidence) L. P. Ford, P. Blowers, and R. Masel, “Role of steps and kinks in catalytic activity,” Journal of Vacuum Science and Technology. 1999. link Times cited: 11 Abstract: In the literature, there is the idea that steps and kinks ar… read moreAbstract: In the literature, there is the idea that steps and kinks are the active sites for chemical reactions, but the experimental data are far from convincing. In this article we see if there is a correlation between step atom density, van Hardeveld and Hartog coordination numbers, or the electronic coordination number and reactivity for a number of simple decomposition and hydrogenolysis reactions on platinum as measured by temperature-programmed desorption. We have examined reactions of ethylene, nitric oxide, and methanol on (111), (110)-(1×1), (110)-(2×1), (100)-hex, (100)-(1×1), (210), (511), and (331) platinum surfaces. We have done a statistical analysis of our data to see if any of the correlations are non-negligible. We find that, in general, stepped surfaces have different reactivity than close-packed planes, but some stepped surfaces are more active than Pt(111) while others are less active than Pt(111). There are negligible correlations between step atom density and catalytic activity for our reacti... read less NOT USED (high confidence) S. Psakh’e, K. P. Zol’nikov, R. I. Kadryov, G. E. Rudenskii, and D. Saraev, “Interaction of solitary pulses produced by high-rate loading with a free surface,” Combustion, Explosion and Shock Waves. 1999. link Times cited: 0 NOT USED (high confidence) X. W. Zhou and H. Wadley, “Hyperthermal vapor deposition of copper: reflection and resputtering effects,” Surface Science. 1999. link Times cited: 51 NOT USED (high confidence) G. Bozzolo, J. Ferrante, R. Noebe, B. Good, F. Honecy, and P. Abel, “Surface segregation in multicomponent systems: Modeling of surface alloys and alloy surfaces,” Computational Materials Science. 1999. link Times cited: 66 NOT USED (high confidence) M. Ghaly, K. Nordlund, and R. Averback, “Molecular dynamics investigations of surface damage produced by kiloelectronvolt self-bombardment of solids,” Philosophical Magazine. 1999. link Times cited: 271 Abstract: Molecular dynamics computer simulations were employed to stu… read moreAbstract: Molecular dynamics computer simulations were employed to study damage production mechanisms at solid surfaces during bombardment with kiloelectronvolt ions. Three separate mechanisms are identified: ballistic damage, viscous flow and microexplosions. Ballistic damage is created by the direct knock-on of atoms onto the surface as described within the binary collision approximation. Viscous flow refers to local melting and the forced flow of liquid onto the surface, and microexplosions occur when the high pressures in cascades lead to rupturing of the nearby surface. The relative importance of each mechanism depends on several parameters: atomic mass, melting temperature, atomic density, structure and atomic bonding of the target, and the mass and energy of the projectile. The simulations were performed for Pt. Au, Cu, Ni and Ge self-atom bombardment. Cascades in the interior of the targets were also examined to provide a comparison for the surface events. In addition several events of 4.5keV Ne an... read less NOT USED (high confidence) R. A. McCoy and Y. Deng, “Parallel Particle Simulations of Thin-Film Deposition,” The International Journal of High Performance Computing Applications. 1999. link Times cited: 8 Abstract: Thin-film growth by sputter deposition is a manufacturing pr… read moreAbstract: Thin-film growth by sputter deposition is a manufacturing process that is well suited for study by particle simulation methods. The authors report on the development of a high performance, parallel, molecular-dynamics software package that simulates atomic metal systems under sputter deposition conditions. The package combines advanced techniques for parallel molecular dynamics with specialized schemes for the simulation of sputtered atoms impinging on thin films and substrates. The features of the package include asynchronous message passing, dynamic load balancing, mechanisms for data caching, and efficient memory management. For classical, semiempirical force calculations, the authors employ a modified version of the embedded-atom method with improved efficiency. Enhancements for the simulation of sputter deposition include an adjustable temperature control algorithm, the detection and ray tracing of emitted particles, and a Langevin localization procedure that restricts the dynamics computations to regions undergoing kinetic energy transfer. The authors describe in detail the features of the package, discuss its performance behavior, and also present some results from sputter deposition simulations. read less NOT USED (high confidence) M. Gungor and D. Maroudas, “Theoretical analysis of electromigration-induced failure of metallic thin films due to transgranular void propagation,” Journal of Applied Physics. 1999. link Times cited: 99 Abstract: Failure of metallic thin films driven by electromigration is… read moreAbstract: Failure of metallic thin films driven by electromigration is among the most challenging materials reliability problems in microelectronics toward ultra-large-scale integration. One of the most serious failure mechanisms in thin films with bamboo grain structure is the propagation of transgranular voids, which may lead to open-circuit failure. In this article, a comprehensive theoretical analysis is presented of the complex nonlinear dynamics of transgranular voids in metallic thin films as determined by capillarity-driven surface diffusion coupled with drift induced by electromigration. Our analysis is based on self-consistent dynamical simulations of void morphological evolution and it is aided by the conclusions of an approximate linear stability theory. Our simulations emphasize that the strong dependence of surface diffusivity on void surface orientation, the strength of the applied electric field, and the void size play important roles in the dynamics of the voids. The simulations predict void faceti... read less NOT USED (high confidence) D. Udler and D. Seidman, “Monte Carlo Simulation of the Concentration Dependence of Segregation at Vicinal Grain Boundaries,” Interface Science. 1998. link Times cited: 7 NOT USED (high confidence) M. Alemany, C. Rey, and L. J. Gallego, “A molecular dynamics study of the thermodynamic properties of liquid Ni using the Voter and Chen version of the embedded atom model,” Journal of Chemical Physics. 1998. link Times cited: 8 Abstract: Using the Voter and Chen version of the embedded atom model,… read moreAbstract: Using the Voter and Chen version of the embedded atom model, we performed molecular dynamics simulations to compute the thermodynamic properties of liquid Ni up to 3000 K, i.e., well above the melting temperature. Our results show good general agreement with available experimental data. Comparison between simulated and experimental heat capacities requires subtraction from the latter of the electronic contribution, which for liquid transition metals is usually an order of magnitude greater than for simple metals. read less NOT USED (high confidence) V. Kuznetsov, K. Tsai, and T. Turkebaev, “Calculation of thermodynamic properties of the Ni-Al alloys in normal conditions and under pressure,” Journal of Physics: Condensed Matter. 1998. link Times cited: 1 Abstract: We present an electron density model functional method (EDMF… read moreAbstract: We present an electron density model functional method (EDMF) based on the embedded atom theory and a pseudopotential approach to study thermodynamic properties and stability of binary metallic alloys. Test calculations of the pure Ni, Al and their alloys demonstrate the efficiency and accuracy of the method. The ground-state properties of ordered compounds and a disordered FCC phase in the Ni-Al system were obtained in normal conditions (P = 0) and under pressure. The results obtained at P = 0 are in quite good agreement with experimental data. Calculation under pressure predicts a change of compound stability with increasing pressure. read less NOT USED (high confidence) M. Alemany, C. Rey, and L. J. Gallego, “Transport coefficients of liquid transition metals: A computer simulation study using the embedded atom model,” Journal of Chemical Physics. 1998. link Times cited: 33 Abstract: Using the Voter and Chen version of the embedded atom model,… read moreAbstract: Using the Voter and Chen version of the embedded atom model, we carried out molecular dynamics simulations to compute the diffusion constants and shear viscosities of liquid Pd, Pt, Cu, Ag, and Au as representative of single-particle and collective dynamic properties, respectively. The generally good agreement between the calculated values and available experimental data is evidence that the Voter and Chen embedded atom model allows a reliable description of the dynamic properties of liquid transition metals in spite of being derived from solid-state data and the properties of the diatomic molecule. The discrepancy between the reported experimental diffusion constant of liquid Cu and the values calculated in this work, together with the consistency of these latter, suggests that the reported experimental value may be in error. read less NOT USED (high confidence) X. W. Zhou and H. Wadley, “Atomistic simulations of the vapor deposition of Ni/Cu/Ni multilayers: The effects of adatom incident energy,” Journal of Applied Physics. 1998. link Times cited: 117 Abstract: Vapor deposited multilayers consisting of a low electrical r… read moreAbstract: Vapor deposited multilayers consisting of a low electrical resistivity conductor sandwiched between ferromagnetic metals such as cobalt or nickel-iron alloys sometimes exhibit giant magnetoresistance (GMR). The GMR properties of these films are a sensitive function of structure and defects in the films and therefore depend upon the processing conditions used for their synthesis. A three-dimensional molecular dynamics method has been developed to simulate the [111] growth of model Ni/Cu/Ni multilayers and was used to investigate the role of vapor atom impact energy upon the film structure and defects. High incident atom energies were found to lower interfacial roughness but promoted intermixing by an atomic exchange mechanism. Low incident energies reduced intermixing, but resulted in films with rough, defective interfaces. The simulations identified an intermediate incident energy between 1 and 2 eV that resulted in both low roughness and intermixing, and an anticipated large GMR effect. The simulation me... read less NOT USED (high confidence) R. E. Miller, E. Tadmor, R. Phillips, and M. Ortiz, “Quasicontinuum simulation of fracture at the atomic scale,” Modelling and Simulation in Materials Science and Engineering. 1998. link Times cited: 238 Abstract: We study the problem of atomic scale fracture using the rece… read moreAbstract: We study the problem of atomic scale fracture using the recently developed quasicontinuum method in which there is a systematic thinning of the atomic-level degrees of freedom in regions where they are not needed. Fracture is considered in two distinct settings. First, a study is made of cracks in single crystals, and second, we consider a crack advancing towards a grain boundary (GB) in its path. In the investigation of single crystal fracture, we evaluate the competition between simple cleavage and crack-tip dislocation emission. In addition, we examine the ability of analytic models to correctly predict fracture behaviour, and find that the existing analytical treatments are too restrictive in their treatment of nonlinearity near the crack tip. In the study of GB-crack interactions, we have found a number of interesting deformation mechanisms which attend the advance of the crack. These include the migration of the GB, the emission of dislocations from the GB, and deflection of the crack front along the GB itself. In each case, these mechanisms are rationalized on the basis of continuum mechanics arguments. read less NOT USED (high confidence) B. Jiang, X. Liu, S. Zou, J. Sun, and J. Wang, “Atomistic computer study on Mg segregation in the Ni_3Al grain boundary,” Journal of Materials Research. 1998. link Times cited: 8 Abstract: The embedded atom method (EAM) was applied to calculate the … read moreAbstract: The embedded atom method (EAM) was applied to calculate the energy on Mg doping in polycrystalline Ni_3Al. The EAM predicted the energy of Mg in Al site in grain boundary is lower than that of Mg in Ni site and much lower than that of Mg in Al or Ni site in bulk and in free surface. It means that Mg would segregate to grain boundary rather than bulk and free surface and Mg will favor to be the substitute of Al rather than of Ni in grain boundary. These results were consistent with the experiments that Mg segregated to grain boundaries with Al depletion and Ni enrichment. read less NOT USED (high confidence) S. S. Pohlong and P. N. Ram, “Analytic embedded atom method potentials for face-centered cubic metals,” Journal of Materials Research. 1998. link Times cited: 15 Abstract: The universal form of embedding function suggested by Banerj… read moreAbstract: The universal form of embedding function suggested by Banerjea and Smith together with a pair-potential of the Morse form are used to obtain embedded atom method (EAM) potentials for fcc metals: Cu, Ag, Au, Ni, Pd, and Pt. The potential parameters are determined by fitting to the Cauchy pressure ( C _12 − C _44)/2, shear constant G _V = ( C _11 − C _12 + 3 C _44)/5, and C _44, the cohesive energy and the vacancy formation energy. The obtained parameters are utilized to calculate the unrelaxed divacancy binding energy and the unrelaxed surface energies of three low-index planes. The calculated quantities are in reasonable agreement with the experimental values except perhaps the divacancy energy in a few cases. In a further application, lattice dynamics of these metals are discussed using the present EAM potentials. On comparison with experimental phonons, the agreement is good for Cu, Ag, and Ni, while in the other three metals, Au, Pd, and Pt, the agreement is not so good. The phonon spectra are in reasonable agreement with the earlier calculations. The frequency spectrum and the mean square displacement of an atom in Cu are in agreement with the experiment and other calculated results. read less NOT USED (high confidence) T. Muramoto, K. Yorizane, and Y. Yamamura, “Angular distribution of cluster atoms due to cluster impact,” 1998 International Conference on Ion Implantation Technology. Proceedings (Cat. No.98EX144). 1998. link Times cited: 0 Abstract: The high-density effect on the angular distribution of penet… read moreAbstract: The high-density effect on the angular distribution of penetrating cluster atoms due to cluster impacts on solid surfaces has been studied by molecular dynamics simulations. MD simulations are performed for (Cu)/sub 201//spl rarr/Pt[111] and (Pt)/sub 201//spl rarr/Cu[111] where the cluster energies are changed from 5 eV to 50 keV per atom. It is found that the high-density formation is strongly dependent on the cluster energy and the mass ratio between a cluster atom and target atom and that the angular distributions of cluster atom are enhanced near /spl theta/=90/spl deg/ for 5 eV/atom Cu cluster. read less NOT USED (high confidence) J. Kuhalainen, M. Manninen, and E. Kautto, “Argon and neon trapping near copper surfaces,” Journal of Physics: Condensed Matter. 1998. link Times cited: 0 Abstract: The binding energies of argon and neon impurities trapped be… read moreAbstract: The binding energies of argon and neon impurities trapped below copper surfaces have been studied using a two-component effective-medium theory. The impurities are placed at various interstitial and substitutional sites and at divacancies within the first few surface layers, and the local energy minima as well as diffusion paths have been calculated taking into account full relaxation of the copper atoms. The results differ clearly from those obtained for bulk copper and are different for different surfaces. The results suggest that argon and neon are diffusing out from copper via a vacancy mechanism, with the help of a divacancy, or the impurity-vacancy pair dissociates clearly below the surface layer. read less NOT USED (high confidence) P. Gumbsch, “The accommodation of lattice mismatch in Ag/Ni heterophase boundaries,” Journal of Phase Equilibria. 1997. link Times cited: 5 NOT USED (high confidence) T. Mattsson, G. Wahnström, L. Bengtsson, and B. Hammer, “Quantum-mechanical calculation of H on Ni(001) using a model potential based on first-principles calculations,” Physical Review B. 1997. link Times cited: 41 Abstract: Hydrogen diffusion on metal surfaces has been a subject of g… read moreAbstract: Hydrogen diffusion on metal surfaces has been a subject of great interest, both theoretically and experimentally, due to the pronounced quantum behavior at low temperatures. read less NOT USED (high confidence) V. Paidar, A. Larere, and L. Priester, “Exponential many-body potentials and elastic constants of fcc transition metals,” Modelling and Simulation in Materials Science and Engineering. 1997. link Times cited: 6 Abstract: The elastic constants of fcc transition metals are studied a… read moreAbstract: The elastic constants of fcc transition metals are studied as functions of the exponential parameters of a many-body potential based on the second moment approximation of the d-electron density of states. It is shown that this model gives a reasonable agreement with the experimental values not only for all three independent elastic constants but also for the vacancy formation energy for Cu, Ag, Au and Ni. Less satisfactory results are obtained for Ir, Rh, Pt and Pd. read less NOT USED (high confidence) C. Massobrio and B. Nacer, “Collisional dynamics of Ag19 on Pd(100): a molecular dynamics study,” Zeitschrift für Physik D Atoms,Molecules and Clusters. 1997. link Times cited: 5 NOT USED (high confidence) R. Zugic, B. Szpunar, V. Krstić, and U. Erb, “Effect of porosity on the elastic response of brittle materials: An embedded-atom method approach,” Philosophical Magazine. 1997. link Times cited: 18 Abstract: The values for Young's modulus of porous single-crystal… read moreAbstract: The values for Young's modulus of porous single-crystal Ni in the [100] and [111] directions are computed using the embedded-atom method (EAM). Both pore volume fraction and pore size (defined by ratio S/R of the flaw size to pore radius) are varied. A reduction in Young's modulus with increasing pore volume fraction and with increasing S/R ratio is observed in the EAM simulations, in good agreement with a recent theoretical model proposed by Krstic and Erickson. A porous Σ = 5, [100] grain boundary also demonstrates a marked reduction in Young's modulus compared with the pore-free Σ = 5, [100] grain boundary. These results suggest that recent literature values demonstrating greatly reduced Young's modulus for some nanocrystalline materials (compared with conventional polycrystalline materials) may be a consequence of residual porosity in the material. Poisson's ratio is calculated for aligned pores with stress applied in the [100] direction. The crack-opening displacement is qualitatively and qu... read less NOT USED (high confidence) X. W. Zhou, R. Johnson, and H. Wadley, “A MOLECULAR DYNAMICS STUDY OF NICKEL VAPOR DEPOSITION: TEMPERATURE, INCIDENT ANGLE, AND ADATOM ENERGY EFFECTS,” Acta Materialia. 1997. link Times cited: 79 NOT USED (high confidence) J. Lu and J. Szpunar, “Applications of the embedded-atom method to glass formation and crystallization of liquid and glass transition-metal nickel,” Philosophical Magazine. 1997. link Times cited: 33 Abstract: The embedded-atom method (EAM) has been applied to investiga… read moreAbstract: The embedded-atom method (EAM) has been applied to investigate the effect of different cooling rates on the glass formation and crystallization processes in supercooled metallic liquid nickel. The crystallization of metallic glass as a function of increasing temperatures was also studied, using the constant-pressure molecular-dynamics simulation. The results indicate that the agreements between the calculated and experimental pair distribution functions and atomic volumes for the liquid nickel are quite good. The microstructure is greatly affected by the quenching rate. The non-equilibrium phase obtained in a supercooled liquid through a fast quenching process is a metallic glass, and the equilibrium phase resulting from slow cooling rate is a fcc-type crystal phase. Metallic glass is unstable during quenching and, with an increase in temperature, crystallization of the metallic glass occurs. The crystal structure resulting from the crystallization process is a fcc-type structure. The calculated ... read less NOT USED (high confidence) J. E. Angelo and M. Baskes, “Interfacial studies using the EAM and MEAM,” Interface Science. 1997. link Times cited: 27 NOT USED (high confidence) D. Kim, J. Doll, and J. Gubernatis, “The quantum dynamics of interfacial hydrogen: Path integral maximum entropy calculation of adsorbate vibrational line shapes for the H/Ni(111) system,” Journal of Chemical Physics. 1997. link Times cited: 35 Abstract: Vibrational line shapes for a hydrogen atom on an embedded a… read moreAbstract: Vibrational line shapes for a hydrogen atom on an embedded atom model (EAM) of the Ni(111) surface are extracted from path integral Monte Carlo data. Maximum entropy methods are utilized to stabilize this inversion. Our results indicate that anharmonic effects are significant, particularly for vibrational motion parallel to the surface. Unlike their normal mode analogs, calculated quantum line shapes for the EAM potential predict the correct ordering of vibrational features corresponding to parallel and perpendicular adsorbate motion. read less NOT USED (high confidence) M. Katagiri et al., “The dynamics of surfaces of metallic and monolayer systems: an embedded-atom molecular dynamics study,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1996. link Times cited: 3 NOT USED (high confidence) A. Wucher and B. Garrison, “Cluster formation in sputtering: A molecular dynamics study using the MD/MC-corrected effective medium potential,” Journal of Chemical Physics. 1996. link Times cited: 67 Abstract: We report on a molecular dynamics simulation of cluster emis… read moreAbstract: We report on a molecular dynamics simulation of cluster emission during sputtering of metals using a new many‐body potential developed by DePristo and co‐workers. For the specific case of silver as a sample target material, it is shown that this potential allows a much more realistic description of small clusters than the EAM potential used in our previous work. While this has a relatively large effect on the relative abundance of clusters within the total flux of sputtered material, other cluster properties like kinetic energy distributions and internal excitation are found to be less affected. By comparison with corresponding experimental data, we conclude that the formation of sputtered silver clusters can now be almost quantitatively modeled by the simulation. read less NOT USED (high confidence) C.-L. Liu, “Diffusion mechanisms at metallic grain boundaries,” Journal of Computer-Aided Materials Design. 1996. link Times cited: 0 NOT USED (high confidence) J. Schön and M. Jansen, “Auf dem Weg zur Syntheseplanung in der Festkörperchemie: Vorhersage existenzfähiger Strukturkandidaten mit Verfahren zur globalen Strukturoptimierung,” Angewandte Chemie. 1996. link Times cited: 56 Abstract: Wir stellen hier eine Methode vor, mit der im Prinzip die Ex… read moreAbstract: Wir stellen hier eine Methode vor, mit der im Prinzip die Existenz und Struktur von (meta)stabilen Festkorperverbindungen vorhergesagt werden kann. Sie beruht auf der Erkundung der Energiefunktion des betrachteten chemischen Systems mit Verfahren, die zunachst eine globale und schlieslich eine lokale Optimierung ermoglichen. Das hierfur entwickelte Programmsystem ist modular aufgebaut. Die Hauptelemente sind eine Reihe von Routinen fur globale und lokale Minimierungen sowie Algorithmen zur Untersuchung der Phasenraumstruktur in der Nahe lokaler Minima der potentiellen Energie und zur Analyse und Charakterisierung der Strukturkandidaten. Derzeit behandeln wir damit ionische Verbindungen, wobei die Energiefunktion zunachst mit Hilfe empirischer Potentiale berechnet wird und danach die Ergebnisse mit einem Hartree-Fock-Algorithmus verfeinert werden. Die globale Optimierung erfolgt mit einem stochastischen «Simulated-Annealing»-Algorithmus, die lokale Minimierung verwendet einen stochastischen «Quench» oder ein Gradientenverfahren. Die Umgebungen der lokalen Minima werden mit dem «Threshold»-Algorithmus untersucht. Wir zeigen Ergebnisse dieses Ansatzes an binaren Mischphasen der Edelgase sowie an binaren und ternaren ionischen Systemen. Diese schliesen einige bislang noch nicht synthetisierte Substanzen ein, die jedoch kinetisch stabil sein sollten, z.B. weitere Alkalimetallnitride neben Li3N sowie Ca3SiBr2 und SrTi2O5. read less NOT USED (high confidence) P. Scagnetti, R. Nagem, G. Sandri, and T. Bifano, “Stress and Strain Analysis in Molecular Dynamics Simulation of Solids,” Journal of Applied Mechanics. 1996. link Times cited: 7 Abstract: Molecular dynamics simulation of solid materials is applied … read moreAbstract: Molecular dynamics simulation of solid materials is applied to a two-dimensional indentation problem. Methods are presented for calculating the stress and strain tensors at interior points within the model. The stress calculations corresponding to the elastic deformation portion of the indentation process are compared with an analytical continuum solution. Stress calculations are also presented for the plastic deformation portion of the indentation process. The methods are derived independently so that future work can be directed at determining the constitutive relationship between stress and strain throughout the development of plastic flow and fracture in a solid material. read less NOT USED (high confidence) M. J. López, P. A. Marcos, and J. A. Alonso, “Structural and dynamical properties of Cu–Au bimetallic clusters,” Journal of Chemical Physics. 1996. link Times cited: 102 Abstract: The effect of alloying on the structural and thermal propert… read moreAbstract: The effect of alloying on the structural and thermal properties of Cun−xAux (n=13,14) clusters is investigated by constant energy Molecular Dynamics simulations. The interactions between the atoms in the clusters are mimicked by a many‐body (Gupta‐like) potential based on the second moment approximation to the tight‐binding model. The minimum energy structures and the lowest‐lying isomers of the pure and mixed clusters are obtained by thermal quenching. We find icosahedral‐like ground state structures for the 13‐ and 14‐atom clusters and for all the concentrations, the only exception being Au14 which has C6v symmetry. Mixed structures are preferred over the segregated ones. The lowest‐lying isomers of the binary clusters are the permutational ones, i.e., isomers having the same underlying geometry as the ground state structure and different relative arrangement of the unlike atoms in the atomic positions of the geometry. However, presence of these low lying permutational isomers does not affect the gross ... read less NOT USED (high confidence) F. Zypman and J. Ferrante, “Tight-binding surface correction to the embedded-atom method embedding function,” Journal of Physics: Condensed Matter. 1995. link Times cited: 2 Abstract: The embedded-atom method (EAM) has been highly successful in… read moreAbstract: The embedded-atom method (EAM) has been highly successful in predicting many properties of fcc metals. However, it is known to underestimate surface energies by as much as 40 to 50%. This suggests that it would be interesting to explore the possibility of obtaining a surface correction to the embedding energy. In this paper, the functional form for a surface embedding function, Fsurface, for the embedded-atom method (EAM) is proposed. The existence of a different F for surface atoms than for bulk atoms stems from the fact that the presence of a surface modifies the energy band structure of the solid. In order to study this change, we used the tight-binding method, that provides the ingredients needed to obtain an explicit expression for the relevant quantities. By comparing the energies of the EAM and tight binding for a surface-terminated bulk, we obtain a correction to the EAM embedding function and the EAM energy for the system. In order to quantify our result we apply it to the lower-index surface planes of Ag and Pd by adjusting our tight-binding parameters with known, available first-principles results for the (111) plane. We then predict the surface energies for the (100) and (110) planes with our method and show an improvement over using the bulk embedding function as compared with first-principles values. read less NOT USED (high confidence) P. Fay, J. R. Ray, and R. J. Wolf, “Detailed balance methods for chemical potential determination,” Journal of Chemical Physics. 1995. link Times cited: 2 Abstract: In earlier work [J. Chem. Phys. 100, 2154 (1994)] we present… read moreAbstract: In earlier work [J. Chem. Phys. 100, 2154 (1994)] we presented a new method of determining the chemical potential in Monte Carlo or molecular dynamics simulations which makes use of a detailed balance method (DBM). In the present paper we present results of a careful study of this method applied to liquid palladium down to and below the zero‐pressure melting temperature. A new surface detailed balance method (SDBM) is introduced, which is much more efficient near and below the melting temperature where the original method becomes too inefficient to determine an accurate value of the chemical potential. We also present results where the new surface detailed balance method is used to determine the chemical potential of the solid phase at a number of different temperatures. read less NOT USED (high confidence) J. Rousset et al., “Study of bimetallic Pd–Pt clusters in both free and supported phases,” Journal of Chemical Physics. 1995. link Times cited: 68 Abstract: We study PdPt bimetallic clusters in both free and supported… read moreAbstract: We study PdPt bimetallic clusters in both free and supported phases. These clusters have been produced with a laser vaporization source. Free clusters directly produced by the source are studied by time of flight mass spectrometry and photofragmentation technique. We observed a sequential evaporation of Pd atoms in the mixed clusters consistent with a palladium segregation process. This tendency has been also observed on supported particles from which the structure and the composition are determined by high resolution transmission electron microscopy and energy dispersive x‐ray analysis. A main result is that each particle has the composition of the massic rod vaporized in the source. The supported particles are well crystallized and exhibit truncated octahedron shapes. Experimental observations are well explained using a modified tight binding model. Indeed, within this model, we found that the equilibrium shape is strongly related to the variation of the cohesive energy with atomic coordination number. ... read less NOT USED (high confidence) M. Baskes, X. Sha, J. E. Angelo, and N. Moody, “COMMENT: Trapping of hydrogen to lattice defects in nickel,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 309 Abstract: This paper addresses the energy associated with the trapping… read moreAbstract: This paper addresses the energy associated with the trapping of hydrogen to defects in a nickel lattice. Several dislocations and grain boundaries which occur in nickel are studied. The dislocations include an edge, a screw, and a Lomer dislocation in the locked configuration, i.e. a Lomer-Cottrell lock (LCL). For both the edge and screw dislocations, the maximum trap site energy is approximately 0.1 eV occurring in the region where the lattice is in tension approximately 3-4 angstroms from the dislocation core. For the Lomer-Cottrell lock, the maximum binding energy is 0.33 eV and is located at the core of the a/6(110) dislocation. Several low-index coincident site lattice grain boundaries are investigated, specifically the Sigma 3(112), Sigma 9(221) and Sigma 11(113) tilt boundaries. The boundaries all show a maximum binding energy of approximately 0.25 eV at the tilt boundary. Relaxation of the boundary structures produces an asymmetric atomic structure for both the Sigma 3 and Sigma 9 boundaries and a symmetric structure for the Sigma 11 tilt boundary. The results of this study can be compared to recent experimental studies showing that the activation energy for hydrogen-initiated failure is approximately 0.3-0.4 eV in the Fe-based superalloy IN903. From the results of this comparison it can be concluded that the embrittlement process is likely associated with the trapping of hydrogen to grain boundaries and Lomer-Cottrell locks. read less NOT USED (high confidence) G. Bozzolo and J. Ferrante, “Bulk properties of Ni3Al (γ′) with Cu and Au additions,” Journal of Computer-Aided Materials Design. 1995. link Times cited: 14 NOT USED (high confidence) D. Udler and D. Seidman, “Solute-atom segregation/structure relations at high-angle (002) twist boundaries in dilute Ni−Pt alloys,” Interface Science. 1995. link Times cited: 15 NOT USED (high confidence) R. Taylor, C. L. Brummel, N. Winograd, B. Garrison, and J. Vickerman, “Molecular desorption in bombardment mass spectrometries,” Chemical Physics Letters. 1995. link Times cited: 15 NOT USED (high confidence) H. Fritsche, “Particle Size Effects in Embedded Atom Approximation Analytical and Numerical Investigations of Ni, Pd, Cu, Ag, and Au,” Zeitschrift für Physikalische Chemie. 1995. link Times cited: 4 Abstract: atom approximation of the total energy, analytical formulae … read moreAbstract: atom approximation of the total energy, analytical formulae have been derived for the particle size dependence of the cohesive energy, the surface energy, and the isotropie surface stress. The agreement with numerical results is very well for particles with more than 10 atoms. A comparison with similar results of van der Waals particles and with particles of ion pairs suggests, that the form of the derived expressions is mainly determined by geometrical factors of the particles, but the values of the coefficients strongly depend on the specific interactions inside the particle. read less NOT USED (high confidence) W. G. Hoover, T. Pierce, C. G. Hoover, J. O. Shugart, C. M. Stein, and A. L. Edwards, “Molecular dynamics, smoothed-particle applied mechanics, and irreversibility,” Computers & Mathematics With Applications. 1994. link Times cited: 25 NOT USED (high confidence) D. Seidman, B. Krakauer, and D. Udler, “Atomic scale studies of solute-atom segregation at grain boundaries: Experiments and simulations☆,” Journal of Physics and Chemistry of Solids. 1994. link Times cited: 55 NOT USED (high confidence) M. Baskes, “The Modified Embedded Atom Method.” 1994. link Times cited: 4 Abstract: Recent modifications have been made to generalize the Embedd… read moreAbstract: Recent modifications have been made to generalize the Embedded Atom Method (EAM) to describe bonding in diverse materials. By including angular dependence of the electron density in an empirical way, the Modified Embedded Atom Method (MEAM) has been able to reproduce the basic energetic and structural properties of 45 elements. This method is ideal for examining interfacial behavior of dissimilar materials. This paper explains in detail the derivation of the method, shows how parameters of MEAM are determined directly from experiment or first principles calculations, and examine the quality of the reproduction of the database. Materials with fcc, bcc, hcp, and diamond cubic crystal structure are discussed. A few simple examples of the application of the MEAM to surfaces and interfaces are presented. Calculations of pullout of a SiC fiber in a diamond matrix as a function of applied stress show nonuniform deformation of the fiber. read less NOT USED (high confidence) J. Gui et al., “Embedded-atom method study of the effect of the order degree on the lattice parameters of Cu-based shape memory alloys,” Journal of Physics: Condensed Matter. 1994. link Times cited: 18 Abstract: Using the embedded-atom method, the variation in the total e… read moreAbstract: Using the embedded-atom method, the variation in the total energies and lattice parameters with the degree of long-range order has been calculated for the parent and 18R1 martensite phases for several Cu-Zn-Al and Cu-Al-Ni shape memory alloys. It was found that the bond angle phi , i.e. the angle between lines connecting the nearest neighbours in the basal plane of the martensite, increases with increase in degree SB2 of B2-type order for both Cu-Zn-Al and Cu-Al-Ni alloys. When the degree SL2 of L21-type order increases, the bond angle phi increases for the Cu-Zn-Al shape memory alloys, but it decreases for the Cu-Al-Ni shape memory alloys. This result agrees well with experiment. read less NOT USED (high confidence) J. M. Montejano-Carrizales, M. Iñiguez, and J. Alonso, “Evolution of the structural stability of large Cu, Ni, Pd, and Ag clusters with size: An analysis within the embedded atom method,” Journal of Cluster Science. 1994. link Times cited: 15 NOT USED (high confidence) D. Udler and D. Seidman, “Solute-atom segregation at (002) twist boundaries in dilute NiPt alloys: Structural/chemical relations,” Acta Metallurgica Et Materialia. 1994. link Times cited: 23 NOT USED (high confidence) S. Swaminarayan, R. Najafabadi, and D. Srolovitz, “Polycrystalline surface properties from spherical crystallites: Ag, Au, Cu and Pt,” Surface Science. 1994. link Times cited: 28 NOT USED (high confidence) D. Wolf and J. Jaszczak, “Tailored elastic behavior of multilayers through controlled interface structure,” Journal of Computer-Aided Materials Design. 1994. link Times cited: 20 NOT USED (high confidence) Z. Bangwei and O. Yijang, “Calculations of the thermodynamic properties for binary hcp alloys with simple embedded atom method model,” Zeitschrift für Physik B Condensed Matter. 1993. link Times cited: 3 NOT USED (high confidence) Y. Sun, G. Beltz, and J. Rice, “Estimates from atomic models of tension-shear coupling in dislocation nucleation from a crack tip,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1993. link Times cited: 139 NOT USED (high confidence) L. Zhao, R. Najafabadi, and D. Srolovitz, “Finite temperature vacancy formation thermodynamics: local harmonic and quasiharmonic studies,” Modelling and Simulation in Materials Science and Engineering. 1993. link Times cited: 30 Abstract: The vacancy formation thermodynamics in six FCC metals Ag, A… read moreAbstract: The vacancy formation thermodynamics in six FCC metals Ag, Au, Cu, Ni, Pd and Pt are determined from atomistic simulations as a function of temperature. The investigation is performed using the embedded atom method interatomic potentials and the finite temperature properties are determined within the local harmonic and the quasiharmonic frameworks. The temperature dependence of the vacancy formation free energy, entropy, enthalpy and vacancy formation volume are determined. The authors find that the temperature dependence of the vacancy formation energy can make a significant contribution to the vacancy concentration at high temperatures. An additional goal of the study is to evaluate the accuracy of the local harmonic method under circumstances in which the excess entropy associated with the formation of a defect is very small. The data demonstrate that while the errors associated with determining the vacancy formation entropy in the local harmonic model are large, a simple extension to the local harmonic method yields thermodynamic properties comparable to that obtained in the quasiharmonic model, but with much higher computational efficiency. read less NOT USED (high confidence) D. Vlachos, L. Schmidt, and R. Aris, “Comparison of small metal clusters: Ni, Pd, Pt, Cu, Ag, Au,” Zeitschrift für Physik D Atoms, Molecules and Clusters. 1993. link Times cited: 12 NOT USED (high confidence) M. Daw, S. Foiles, and M. Baskes, “The embedded-atom method: a review of theory and applications,” Materials Science Reports. 1993. link Times cited: 1221 NOT USED (high confidence) C. C. Chen, D. Paithankar, J. Talbot, and R. P. Andres, “Molecular dynamics simulation of gold cluster collisions,” Zeitschrift für Physik D Atoms, Molecules and Clusters. 1993. link Times cited: 3 NOT USED (high confidence) B. Rice, C. Murthy, and B. Garrett, “Effects of surface structure and of embedded-atom pair functionals on adatom diffusion on fcc metallic surfaces,” Surface Science. 1992. link Times cited: 4 NOT USED (high confidence) A. Sachdev, R. Masel, and J. B. Adams, “An investigation of the shape of small platinum clusters using the embedded atom method,” Catalysis Letters. 1992. link Times cited: 10 NOT USED (high confidence) A. Guellil and J. B. Adams, “The application of the analytic embedded atom method to bcc metals and alloys,” Journal of Materials Research. 1992. link Times cited: 115 Abstract: Johnson and Oh have recently developed Embedded Atom Method … read moreAbstract: Johnson and Oh have recently developed Embedded Atom Method potentials for bcc metals (Na, Li, K, V, Nb, Ta, Mo, W, Fe). The predictive power of these potentials was first tested by calculating vacancy formation and migration energies. Due to the results of these calculations, some of the functions were slightly modified to improve their fit to vacancy properties. The modified potentials were then used to calculate phonon dispersion curves, surface relaxations, surface energies, and thermal expansion. In addition, Johnson’s alloy model, which works well for fcc metals, was applied to the bcc metals to predict dilute heats of solution. read less NOT USED (high confidence) J. Idiodi, “The Separable Potential Method A Unified Approach to Various Lattice Models,” Physica Status Solidi B-basic Solid State Physics. 1991. link Times cited: 1 Abstract: This work proposes the separable potential method (SPM) as a… read moreAbstract: This work proposes the separable potential method (SPM) as a unified treatment of a class of lattice models. The richness in structure of the unified approach is demonstrated through contact with some of the most important lattice models in the literature, the embedded-atom method (EAM) being one of such models. As a practical application of SPM some fairly extensive results for f.c.c. metallic copper are reported.
Ce travail propose le «separable potential method (SPM)» pour le traitement unifie d'une classe des modeles lattice. La richesse en structure de cette approche est mise en evidence en comparant avec des modeles les plus importants trouves dans la litterature tel que l' «embedded atom method (EAM)». Des nombreuses resultats pour c.f.c. Cu sont presentes pour demontrer l'application de la SPM. read less NOT USED (high confidence) R. Najafabadi, D. Srolovitz, and R. LeSar, “Thermodynamic and structural properties of [001] twist boundaries in gold,” Journal of Materials Research. 1991. link Times cited: 33 Abstract: We have employed the Local Harmonic (LH) model and the Embed… read moreAbstract: We have employed the Local Harmonic (LH) model and the Embedded Atom Method (EAM) to examine the structural and thermodynamic properties of a series of twelve [001] twist boundaries in gold for temperatures between 0 K and 700 K. For the majority of the grain boundary misorientations, metastable structures were observed with grain boundary energies that were typically less than 0.1% larger than the stable structures. Four of the twelve grain boundaries underwent first order structural phase transitions as seen by the crossing of the free energy versus temperature curves for the competing structures. Relatively small cusps or inflections in the grain boundary free energy versus misorientation curves were observed at Σ5 (36.87°) and Σ13 (22.62°) at low temperatures, at Σ13 (22.62°) and Σ17 (28.07°) at intermediate temperatures, and at Σ5 (36.87°) and Σ17 (28.07°) at elevated temperatures. A maximum in the grain boundary entropy versus misorientation was observed at Σ17 (28.07°) for all temperatures, and local minima were observed at Σ5 (36.87°) at low temperature and in Σ13 (22.62°) at high temperature. The excess volume associated with the grain boundary shows a roughly linear dependence on grain boundary free energy at each temperature examined. The room-temperature mean-square vibrational amplitude is approximately 25% larger than that for the bulk at the (002) plane adjacent to the boundary and decays to within 2% of the bulk value by the second (002) plane from the boundary. The room-temperature mean-square vibrational amplitude is dominated by the in-plane (parallel to the grain boundary) vibrations at the (002) plane nearest the grain boundary. read less NOT USED (high confidence) R. LeSar, R. Najafabadi, and D. Srolovitz, “Thermodynamics of solid and liquid embedded‐atom‐method metals: A variational study,” Journal of Chemical Physics. 1991. link Times cited: 40 Abstract: We present results of variational calculations of the Helmho… read moreAbstract: We present results of variational calculations of the Helmholtz free energy and the thermodynamic properties of a series of metallic liquids and solids (Ag, Au, Cu, Ni, Pd, Pt) described by embedded‐atom‐method potentials. For the solids, we use a variational procedure based on an Einstein‐model reference state. The free energies of liquids are calculated with an approximate variational method proposed by Ross. At the respective melting points, the present results for the Helmholtz free energy are within about 1% of the results of accurate Monte Carlo (MC) calculations with the same interaction potentials, both for the fluid and the solid. The average error in the melting points calculated with the present procedure relative to Monte Carlo results is about 7.5%. The internal energies and entropies are compared to MC results, and show, in general, good agreement. read less NOT USED (high confidence) M. Zoli, “Anharmonic lattice vibrations in palladium,” Journal of Physics: Condensed Matter. 1990. link Times cited: 6 Abstract: Macroscopic and microscopic effects of anharmonic lattice vi… read moreAbstract: Macroscopic and microscopic effects of anharmonic lattice vibrations in Pd have been studied. The interatomic forces are modelled according to a phenomenological potential which includes three-body angular forces in the harmonic part. The cubic and quartic force constants are fitted to the experimental linear thermal expansion coefficient and constant-pressure specific heat, respectively. Thermoelastic properties are evaluated in quasi-harmonic approximation while caloric properties require computation of constant-volume anharmonic contributions. The phonon lineshifts and linewidths have been calculated along the principal symmetry directions, in the framework of the second-order perturbation theory. The room-temperature experimental anomalies in the ( xi xi 0)T1 branch are reproduced by the theory. read less NOT USED (high confidence) D. Wolf, “A broken‐bond model for grain boundaries in face‐centered cubic metals,” Journal of Applied Physics. 1990. link Times cited: 53 Abstract: The interrelation between the number of nearest‐neighbor ato… read moreAbstract: The interrelation between the number of nearest‐neighbor atomic bonds broken upon formation of a grain boundary in an fcc metal and the related zero‐temperature boundary energy is investigated by atomistic simulation. Using both a Lennard–Jones and an embedded‐atom‐method potential, the structures and energies of symmetrical and asymmetrical tilt and twist boundaries are determined. As in free surfaces, a practically linear relationship between the nearest‐neighbor miscoordination per unit area of the grain boundary and the related interface energy is obtained. The so‐called random‐boundary model, in which the interactions across the interface are assumed to be entirely randomized, is shown to provide a basis for understanding the role of broken bonds in both high‐angle grain boundaries and free surfaces, thus naturally permitting the analysis of ideal cleavage‐fracture energies. A detailed study of low‐angle boundaries shows that only the dislocation cores—but not their strain fields—give rise to broken ... read less NOT USED (high confidence) A. Caro, M. Victoria, and R. Averback, “Threshold displacement and interstitial-atom formation energies in Ni_3Al,” Journal of Materials Research. 1990. link Times cited: 44 Abstract: Threshold displacement energies for atomic displacements alo… read moreAbstract: Threshold displacement energies for atomic displacements along <110>, <100>, and <111> directions, and formation enthalpies of several symmetric interstitial atom configurations were calculated for Ni_3Al by computer simulation using “embedded atom method” potentials. The Ni–Ni (100) dumbbell in the plane containing only Ni atoms has the lowest interstitial-atom enthalpy although the enthalpies of other configurations are similar. Interstitial configurations involving Al atoms all have much higher enthalpies. The anisotropy of the threshold energies in Ni_3Al is similar to pure metals and no significant difference in threshold energy was observed for <110> replacement chains in rows containing all Ni atoms or alternating Ni–Al atoms. Various metastable interstitial atom configurations were observed, including crowd-ions. In addition, the spontaneous recombination volume for some configurations can be much smaller than in pure metals. The consequences of these results for radiation induced segregation and amorphization are discussed. read less NOT USED (high confidence) D. Wolf, “Correlation between energy and volume expansion for grain boundaries in FCC metals,” Scripta Metallurgica. 1989. link Times cited: 81 NOT USED (high confidence) V. Mazhukin, O. Koroleva, M. Demin, and A. A. Aleksashkina, “Atomistic modeling of the properties of gold in the region of phase transitions of the first order,” Mathematica Montisnigri. 2022. link Times cited: 0 Abstract: The article presents the results of atomistic modeling of th… read moreAbstract: The article presents the results of atomistic modeling of the equilibrium thermophysical properties of gold in a wide temperature range (T~ 0.3–3.50 kK), covering the regions of first-order phase transitions of melting and evaporation. The temperature dependences of the density, linear size of the sample, coefficient of linear expansion, enthalpy, and heat capacity are determined. The obtained dependences of the properties of gold are approximated by polynomials of low degrees. There is an acceptable agreement between the obtained characteristics of gold and the experimental data. Numerical and graphic information on the obtained properties and results of comparison with experimental data is presented. read less NOT USED (high confidence) S. Taran, H. Yıldırım, and H. Arslan, “Structural and magnetic properties of polyicosahedral Ni–Pt–Cu ternary nanoalloys,” Journal of Physics B: Atomic, Molecular and Optical Physics. 2021. link Times cited: 2 Abstract: In this study, a series of simulations were carried out to s… read moreAbstract: In this study, a series of simulations were carried out to study the effects of size, composition and geometric structure on the structural and magnetic properties of ternary Ni–Pt–Cu nanoalloys. Different sizes and compositions were considered to compare the structural stability and magnetic behavior of Ni2Pt n Cu17-n (n = 0–17), Ni3Pt n Cu20-n (n = 0–20) and Ni4Pt n Cu22-n (n = 0–22) nanoalloy systems. We performed combinations of Gupta and density functional theory (DFT) simulations to check the validity of atomistic potentials against DFT. We calculated the excess energy to analyze the relative stability of Ni–Pt–Cu nanoalloys. The most negative excess energy values at the Gupta level are obtained in the compositions Ni2Pt5Cu12 in Ni2PtnCu17-n, Ni3Pt8Cu12 in Ni3PtnCu20-n and Ni4Pt9Cu13 in Ni4Pt n Cu22-n nanoalloys. While the most stable compositions Ni2Pt5Cu12 and Ni3Pt8Cu12 at the Gupta level do not agree with those obtained at the DFT level, the lowest energy values were obtained in the same composition Ni4Pt9Cu13 of 26-atom trimetallic nanoalloys at the Gupta and DFT levels. It was also found that most of the atoms on surface sites suffer tensile strain with substitution of Pt atoms. We have also investigated the total magnetic moments of the nanoalloys. In addition, the correlation of the local magnetic moments and charges of the atoms in these systems were discussed. It was found that total magnetic moments of trimetallic nanoalloys follow an almost linear dependence on the Pt concentration, despite the small concentration of Ni atoms and the weak magnetic properties of Pt atoms. read less NOT USED (high confidence) C. Galvin, R. Grimes, and P. Burr, “A molecular dynamics method to identify the liquidus and solidus in a binary phase diagram,” Computational Materials Science. 2021. link Times cited: 6 NOT USED (high confidence) T. Yokoyama, “Thermal expansion of FeNi Invar and zinc-blende CdTe from the view point of local structure,” Microstructures. 2021. link Times cited: 6 Abstract: Thermal expansion of FeNi Invar and zinc-blende CdTe was inv… read moreAbstract: Thermal expansion of FeNi Invar and zinc-blende CdTe was investigated from the view point of local structure using the extended x-ray absorption fine structure (EXAFS) spectroscopic data and the path-integral effective classical potential (PIECP) Monte Carlo computational simulations. In this Review article, first the quantum statistical perturbation theory is intuitively described to see different character concerning thermal expansion between the quantum and classical theories. The diatomic Br2 molecule is employed as a simple example. Second, the PIECP theory is briefly described to note advantages and disadvantages of this simulation technique. Historical background is also discussed for the EXAFS investigation of thermal expansion based on the quantum statistical theories. The results of the FeNi Invar alloy are then summarized. The origin of zero thermal expansion in the FeNi alloy is ascribed to the so-called Invar effect that implies the variation of the electronic structure of Fe atoms depending on temperature. Zero thermal expansion at low temperature is however found to originate from the vibrational quantum effect. It is also noted that the interatomic distances of Fe-Fe, Fe-Ni, and Ni-Ni pairs are slightly but meaningfully different from each other, although the alloy exhibit a simple fcc crystal. Such a pairdependent difference is also true for thermal expansion and we will discuss thermal expansion from the local point of view, which is interestingly different from the lattice thermal expansion significantly. Finally, the results of the zinc blende (or diamond) structure are presented. Although the origin of negative thermal expansion in these tetrahedral crystals is known as a result of classical vibrational anomaly within the Newton dynamics theory, the quantum statistical simulation is found to be essential to reproduce the negative thermal expansion of CdTe. It is emphasized that the vibrational quantum effect and classical anharmonicity are of great importance for the understanding of low-temperature thermal expansion as well as the elastic constants. Page 2 Yokoyama et al . Microstructures 2021;1: I http://dx.doi.org/10.20517/microstructures.2021.001 read less NOT USED (high confidence) M. Demin, O. Koroleva, A. A. Aleksashkina, and V. Mazhukin, “Molecular-dynamic modeling of thermophysical properties of phonon subsystem of copper in wide temperature range.” 2020. link Times cited: 7 Abstract: Copper is a noble metal and has unique properties, due to wh… read moreAbstract: Copper is a noble metal and has unique properties, due to which it is widely used in scientific research, industrial production and, more recently, in the problems of biomedicine. Using the molecular dynamics method, a series of calculations was performed to determine the lattice thermophysical properties of copper in a wide temperature range of 300K ≤ T ≤ 5800K. In the calculations, special attention is paid to the melting-crystallization and near-critical regions, in which cardinal changes in the thermophysical properties of the substance occur. The temperature dependences of the specific heat Cp(T), thermal conductivity κ(T), and density ρ(T) were among the studied characteristics of the phonon subsystem of Cu. Molecular dynamic modeling was carried out using the potential of the embedded atom method (EAM). A comparison of the results with the results of experiments and alternative calculations showed a good agreement. The obtained calculation results were approximated by polynomials of low degrees.. read less NOT USED (high confidence) D. Spearot, R. Dingreville, and C. O’Brien, “Atomistic Simulation Techniques to Model Hydrogen Segregation and Hydrogen Embrittlement in Metallic Materials,” Handbook of Mechanics of Materials. 2019. link Times cited: 5 NOT USED (high confidence) M. Demin, V. Mazhukin, and A. A. Aleksashkina, “Molecular dynamic calculation of lattice thermal conductivity of gold in the melting-crystallization region,” Mathematica Montisnigri. 2019. link Times cited: 6 Abstract: Of all the metals, gold is the most well-known and widely us… read moreAbstract: Of all the metals, gold is the most well-known and widely used material in scientific research, industrial production and, more recently, in biomedicine problems. In the temperature range 300 ≤ T ≤ 2000 K, including the region of the melting – crystallization phase transition, the results of modeling the phonon thermal conductivity of gold are presented. Phonon thermal conductivity plays an important role in modeling the mechanisms of interaction of pulsed laser radiation with gold in the framework of the two-temperature continuum model. In the region of the phase transition, overheating-undercooling of the solid phase occurs, the substance changes its structure. These phenomena are associated with changes in the phonon subsystem of gold, therefore, for mathematical modeling of heatingcooling, it is necessary to know the characteristic of heat transfer as the thermal conductivity of the phonon subsystem of gold. Obtaining the temperature dependence of phonon thermal conductivity in such a wide temperature range from experiment is problematic. In this work, phonon thermal conductivity was obtained by the direct non-equilibrium method in the framework of molecular dynamics modeling using the EAM potential. read less NOT USED (high confidence) C. Grégoire, “Dynamic behaviour of nano-sized voids in hexagonal close-packed materials.” 2018. link Times cited: 0 Abstract: The dynamic behaviour and failure mechanisms of nano-sized v… read moreAbstract: The dynamic behaviour and failure mechanisms of nano-sized voids in single crystals is studied for three hexagonal close-packed materials by means of molecular dynamics simulations. Our study reveals that in Magnesium the response is highly anisotropic leading to a brittle to ductile transition in the failure modes under different load orientations. This transition is accompanied by different mechanisms of deformation and is associated with the anisotropic HCP lattice structure of Mg and the associated barrier for dislocation motion. Remarkably, brittle failure is observed when external loads produce a high stress triaxiality while the response is more ductile when the stress triaxiality decreases. On the other hand, the failure in other two hexagonal close-packed materials studied in this work, i.e, Titanium and Zirconium, is more ductile, in high contrast with the brittle failure observed in Magnesium. We find that this difference is due to the fact that nano-sized voids in Titanium and Zirconium emit substantially more dislocations than Magnesium, allowing for large displacements of the atoms and plastic work, including non-basal planes. Based on our findings, we postulate that this brittle failure in Magnesium is due to a competition between dislocations emission in the basal plane and crack propagation in non-basal planes. Thus, we propose to use the ratio between unstable stacking fault and surface energy in these materials to assess the tendency of hexagonal close-packed materials and alloys to fail under brittle or ductile modes. Using this ratio, we critically identify the low surface energy of Mg as responsible for this brittle behaviour and recommend that Mg-based alloys with large surface energies can lead to better performance for dynamic applications. The fundamental mechanisms observed, therefore, explain the low spall strength of Mg and suggest the possibility of manipulating some mechanisms to increase ductility and spall strength of new lightweight Mg alloys. read less NOT USED (high confidence) A. Nemati, H. N. Pishkenari, A. Meghdari, and S. Sohrabpour, “Directing the diffusive motion of fullerene-based nanocars using nonplanar gold surfaces.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 19 Abstract: A new method for guiding the motion of fullerene and fullere… read moreAbstract: A new method for guiding the motion of fullerene and fullerene-based nanocars is introduced in this paper. The effects of non-flat substrates on the motion of C60, a nanocar and a nanotruck are investigated at different conditions and temperatures. Their behavior is studied using two different approaches: analyzing the variation in potential energy and conducting all-atom classical molecular dynamics simulations. This paper proposes that the use of a stepped substrate will make their motion more predictable and controllable. The results of the simulations show that C60 stays on the top side of the step and cannot jump over the step at temperatures of 400 K and lower. However, at temperatures of 500 K and higher, C60 has sufficient energy to travel to the down side of the step. C60 attaches to the edge and moves just alongside of the edge when it is on the down side of the step. The edge also restricts the motion of C60 alongside the edge and reduces its range of motion. By considering the motion of C60, the general behavior of the nanocar and nanotruck is predictable. The nanocar stays on the top side of the step at temperatures of 400 K and less; at 500 K and higher temperatures, its wheels jump off the edge, and its range of motion is restricted. The relatively rigid chassis of the nanotruck does not allow the free individual motion of the wheels. As a result, the entire nanotruck stays on the top side of the step, even at 600 K. A pathway with the desired route can be fabricated for the motion of C60 and nanocars using the method presented in this paper. This represents a step towards the directional motion of C60 and nanocars. read less NOT USED (high confidence) S. Lee, “Molecular dynamics study of the separation behavior at the interface between PVDF binder and copper current collector,” Journal of Nanomaterials. 2016. link Times cited: 26 Abstract: In Li-ion batteries, the mechanical strengths at the interfa… read moreAbstract: In Li-ion batteries, the mechanical strengths at the interfaces of binder/particle and binder/current collector play an important role in maintaining the mechanical integrity of the composite electrode. In this work, the separation behaviors between polyvinylidene fluoride (PVDF) binders and copper current collectors are studied in the opening and sliding modes using molecular dynamics (MD) simulations. The simulation shows that the separation occurs inside the PVDF rather than at the interface due to the strong adhesion between PVDF and copper. This fracture behavior is different from the behavior of the PVDF/graphite basal plane that shows a clear separation at the interface. The results suggest that the adhesion strength of the PVDF/copper is stronger than that of the PVDF/graphite basal plane. The methodology used in MD simulation can directly evaluate the adhesion strength at the interfaces of various materials between binders, substrates, and particles at the atomic scales. The proposed method can therefore provide a guideline for the design of the electrode in order to enhance the mechanical integrity for better battery performance. read less NOT USED (high confidence) R. Weisburgh, “Scalable, Composable Operators for Defect Design and Analysis.” 2016. link Times cited: 0 Abstract: Title of thesis: SCALABLE, COMPOSABLE OPERATORS FOR DEFECT D… read moreAbstract: Title of thesis: SCALABLE, COMPOSABLE OPERATORS FOR DEFECT DESIGN AND ANALYSIS Rose Weisburgh, Master of Science, 2016 Thesis directed by: Professor Peter W Chung Department of Mechanical Engineering It is well understood that defects adversely affect the electro-mechanical properties of materials. Ideally, defect compositions of raw materials used in devices could be measured, but present technology in the field of atomic defect detection is either destructive in nature, or is unable to determine the precise atomic composition of materials. In the adjacent field of damage detection in large-scale truss networks, algorithms based on spectral measurements have successfully been employed to locate damaged members. Already similar principles have been applied to material lattices experimentally by using Raman Spectroscopy to qualitatively approximate defect densities within materials. However, the applications have largely been limited to surface defects or two-dimensional materials, and the host lattices and defect types are primarily studied anecdotally. This thesis details a numerical method for determining the precise phonon or vibration spectra of material lattices with defects, as it was originally presented in [1]. The dynamical matrices of lattices containing defects are calculated by introducing defects systematically into the dynamical matrices of pristine, defect-free lattices using linear operators. Each operation modifies or removes an individual bond or interaction. Complex defect configurations can be composed through reiterative application of the operators. The proposed methods may be applied to systems containing any interaction type or bond order, including space trusses and atomic lattices. The method is demonstrated by numerically determining the convergence rate of phonon properties in the dilute limit of a single point vacancy. Then the same methodology is applied to two-dimensional atomic lattices with central forces, two-dimension truss networks with distributed mass, as well as three-dimensional atomic lattices with non-linear many body potentials. In each example, the defect structure and properties are shown to alter the spectral properties of the materials. SCALABLE, COMPOSABLE OPERATORS FOR DEFECT DESIGN AND ANALYSIS read less NOT USED (high confidence) H. Barrón, G. Opletal, R. Tilley, and A. Barnard, “Dynamic evolution of specific catalytic sites on Pt nanoparticles,” Catalysis Science & Technology. 2016. link Times cited: 15 Abstract: Platinum nanoparticles are widely used catalysts in many imp… read moreAbstract: Platinum nanoparticles are widely used catalysts in many important industrial applications, in the chemical, petrochemical, automotive and energy sectors. Due to the extremely high cost and the limited abundance of platinum, improving the efficiency of platinum-based nanocatalysts is key to the economic development of these materials, as well as being a challenge for basic research. Ultimately we seek to increase the active surfaces area, per unit volume, and to preserve the activity and selectivity over the functional lifetime of the product. In this work the formation of platinum nanoparticles is investigated by molecular dynamic simulations under different conditions of temperature and atomic deposition rates to identify the conditions that give rise to a greater density of different types of surface active sites. By tuning the growth conditions we obtained highly non-equilibrium morphologies with branches that expose larger surface areas that are consistent with experimental observations. The results are also used to clarify the relationship between growth conditions, surface structure and catalytic functionality based on a simple surface defect classification model, which differentiates between CO oxidation reactions, hydrogen evolution reactions (HER) and hydrogen oxidation reactions (HOR). read less NOT USED (high confidence) K. Tan et al., “Film Morphology and Growth Evolution of Copper Electrodeposition in Stacking Cu2ZnSnS4(CZTS) Precursor: A Kinetic Monte Carlo-Embedded Atom Method Simulation,” Journal of The Electrochemical Society. 2016. link Times cited: 3 NOT USED (high confidence) J. S. Gibson, S. G. Srinivasan, M. Baskes, R. E. Miller, and A. K. Wilson, “A multi-state modified embedded atom method potential for titanium,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 3 Abstract: The continuing search for broadly applicable, predictive, an… read moreAbstract: The continuing search for broadly applicable, predictive, and unique potential functions led to the invention of the multi-state modified embedded atom method (MS-MEAM) (Baskes et al 2007 Phys. Rev. B 75 094113). MS-MEAM replaced almost all of the prior arbitrary choices of the MEAM electron densities, embedding energy, pair potential, and angular screening functions by using first-principles computations of energy/volume relationships for multiple reference crystal structures and transformation paths connecting those reference structures. This strategy reasonably captured diverse interactions between atoms with variable coordinations in a face-centered-cubic (fcc)-stable copper system. However, a straightforward application of the original MS-MEAM framework to model technologically useful hexagonal-close-packed (hcp) metals proved elusive. This work describes the development of an hcp-stable/fcc-metastable MS-MEAM to model titanium by introducing a new angular function within the background electron density description. This critical insight enables the titanium MS-MEAM potential to reproduce first principles computations of reference structures and transformation paths extremely well. Importantly, it predicts lattice and elastic constants, defect energetics, and dynamics of non-ideal hcp and liquid titanium in good agreement with first principles computations and corresponding experiments, and often better than the three well-known literature models used as a benchmark. The titanium MS-MEAM has been made available in the Knowledgebase of Interatomic Models (https://openkim.org/) (Tadmor et al 2011 JOM 63 17). read less NOT USED (high confidence) A. Chatzopoulos, “Numerical Simulations of Metal-Oxides.” 2015. link Times cited: 0 Abstract: Oxides like silicates, alumina or periclase, are materials w… read moreAbstract: Oxides like silicates, alumina or periclase, are materials with significant properties and are therefore investigated extensively in experiment and in theory. The aim of this PhD thesis was to propose and further to develop methods, which make molecular dynamic simulations of oxides with large particle numbers and for long simulation times possible.
The work consists of three parts. In the first one the already existing methods for simulating oxides will be discussed, while in the second one their methodological progress will be presented. The third chapter is solely reserved for the phenomenon of flexoelectricity, which has been discovered during the visualization of the crack propagation in alumina.
Oxide, wie z.B. Silikate, Korund oder Periklas, sind bedeutende Funktionswerkstoffe und werden daher experimentell wie theoretisch intensiv untersucht. Ziel dieser Dissertation war es, Verfahren vorzustellen und derart zu optimieren, dass sie Molekulardynamiksimulationen von Oxiden mit grosen Teilchenzahlen und uber lange Zeiten ermoglichen.
Die Arbeit gliedert sich dabei in drei Bereiche. Im ersten Teil wird auf die einzelnen bereits vorhandenen Methoden zur Simulation von Oxiden eingegangen, im zweiten Kapitel deren Verbesserung vorgestellt. Der dritte Bereich widmet sich ausschlieslich dem Phanomen der Flexoelektrizitat, welche durch die geschickte Visualisierung der Rissausbreitung in Korund entdeckt wurde. read less NOT USED (high confidence) L. Xie et al., “Efficient amorphous platinum catalyst cluster growth on porous carbon: A combined Molecular Dynamics and experimental study,” Applied Catalysis B-environmental. 2015. link Times cited: 29 NOT USED (high confidence) P. Zhu, Y.-zhong Hu, T. Ma, R. Li, and H. Wang, “Atomic simulations of effects of contact size and interfacial interaction strength on superlubricity in incommensurate sliding interface,” Applied Physics A. 2015. link Times cited: 5 NOT USED (high confidence) J. Hutter, M. Iannuzzi, F. Schiffmann, and J. VandeVondele, “cp2k: atomistic simulations of condensed matter systems,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2014. link Times cited: 1976 Abstract: cp2k has become a versatile open‐source tool for the simulat… read moreAbstract: cp2k has become a versatile open‐source tool for the simulation of complex systems on the nanometer scale. It allows for sampling and exploring potential energy surfaces that can be computed using a variety of empirical and first principles models. Excellent performance for electronic structure calculations is achieved using novel algorithms implemented for modern and massively parallel hardware. This review briefly summarizes the main capabilities and illustrates with recent applications the science cp2k has enabled in the field of atomistic simulation. WIREs Comput Mol Sci 2014, 4:15–25. doi: 10.1002/wcms.1159 read less NOT USED (high confidence) P. Seleson, M. Parks, and M. Gunzburger, “Peridynamic State-Based Models and the Embedded-Atom Model,” Communications in Computational Physics. 2014. link Times cited: 27 Abstract: We investigate connections betweennonlocal continuum models … read moreAbstract: We investigate connections betweennonlocal continuum models andmolec- ular dynamics. A continuous upscaling of molecular dynamics models of the form of the embedded-atom model is presented, providing means for simulating molecular dynamics systems at greatly reduced cost. Results are presented for structured and structureless material models, supported by computational experiments. The nonlocal continuum models are shown to be instances of the state-based peridynamics theory. Connections relating multibody peridynamic models and upscaled nonlocal contin- uum models are derived. read less NOT USED (high confidence) J.-yu Yang, W. Hu, and J.-feng Tang, “Effect of incident energy on the configuration of Fe–Al nanoparticles, a molecular dynamics simulation of impact deposition,” RSC Advances. 2014. link Times cited: 10 Abstract: The impact deposition of Al (or Fe) atoms on the rhombohedro… read moreAbstract: The impact deposition of Al (or Fe) atoms on the rhombohedron of Fe (or the truncated octahedron of Al) nanoparticles is investigated by performing a molecular dynamics simulation using the embedded atom method. These simulations are performed in different incident energies (from 10 eV to 50 eV). The dependence of the incident energy of deposited atoms on the growth configurations of Fe–Al nanoparticles is analyzed. For the deposition of Al atoms on the Fe nanoparticle, some Al atoms are incorporated into the Fe core as the incident energy of Al increases. A nanoparticle configuration with Fe-core and Al-shell is usually observed at all incident energies considered. In this case, the substrate Fe atoms and the deposited Al atoms are arranged in body-centered cubic configuration. For the impact deposition of Fe atoms on the Al nanoparticle, an onion-like nanoparticle is observed at incident energy of 10 eV. A configuration with Al-shell and alloyed Fe–Al core is obtained as the incident energy increases. This study proposes a method of artificially controlling nanoalloy configuration. read less NOT USED (high confidence) Y. Zhao, “Surface stress detection and mechanism study with microcantilever based sensor for biomolecular monolayers.” 2014. link Times cited: 4 Abstract: Specific aims of this study are to investigate the mechanism… read moreAbstract: Specific aims of this study are to investigate the mechanism that governs the surface stress generation with hybridization of single stranded DNA (ssDNA) molecules immobilized on micro-cantilevers. The hybridization of DNA on cantilever surfaces leads to configurational change, charge redistribution, and steric hindrance between neighboring hybridized molecules, which result in surface stress change and measureable cantilever deformation. Differential interferometer with two adjacent micro-cantilevers (a sensing/reference pair) was investigated to measure the cantilever deformation. The sensing principle is that binding/reaction of specific chemical or biological species on the sensing cantilever transduces to mechanical deformation. The differential bending of the sensing cantilever respect to the reference cantilever ensures that measured response is insensitive to environmental disturbances. In order to improve the sensitivity for sensing system, new approach of immobilization was utilized to enhance the deformation of the cantilever surface. Immobilization of receptor molecules was modified to use ssDNA with thiol-groups on both 3’ and 5’ ends; therefore both ends of the ssDNA molecules were immobilized to the gold surface and cause stronger surface interactions. To confirm the improvement of the sensitivity of the system, surface stress change associated with hybridization of ssDNA and malachite green-aptamer binding was measured. To explore the mechanism under the surface stress change associated with ssDNA hybridization. A general beam bending model was established based on the minimization of the total energy of the system. The energy consisted of the bending energy of the cantilever and the in-film energy due to the hybridization of ssDNA on the surface. Different stages of read less NOT USED (high confidence) S. K. Biring, R. Sharma, and P. Chaudhury, “A new adaptive mutation simulated annealing algorithm: application to the study of pure and mixed Pt–Pd clusters,” Journal of Mathematical Chemistry. 2013. link Times cited: 10 NOT USED (high confidence) G. Tréglia, C. Goyhenex, C. Mottet, C. Legrand, and F. Ducastelle, “Electronic Structure of Nanoalloys: A Guide of Useful Concepts and Tools.” 2012. link Times cited: 2 NOT USED (high confidence) S.-H. Cheng and C. Sun, “Applicability of continuum fracture mechanics in atomistic systems.” 2011. link Times cited: 2 Abstract: Stress intensity factor is one of the most significant fract… read moreAbstract: Stress intensity factor is one of the most significant fracture parameters in linear elastic fracture mechanics (LEFM). Due to its simplicity, many researchers directly employed this concept to explain their results from molecular simulation. However, stress intensity factor defines the amplitude of the singular stress, which is the product of continuum elasticity. Under atomistic systems without the stress singularity, the concept of stress intensity factor must be examined. In addition, the difficulty of studying the stress intensity factor in atomistic systems may be traced back to the ambiguous definition of the local atomistic stress. In this study, the definition of the local virial stress is adopted. Subsequently, through the consideration of K-dominance, the approximated stress intensity factor based on the atomistic stress can be projected within a reasonable region. Moreover, the influence of cutting interatomic bonds to create traction free crack surfaces and the critical stress intensity factor is also discussed.Copyright © 2011 by ASME read less NOT USED (high confidence) F. Munoz et al., “Collisions between a single gold atom and 13 atom gold clusters: an ab initio approach,” The European Physical Journal D. 2011. link Times cited: 8 NOT USED (high confidence) P. Zhu, Y.-zhong Hu, T. Ma, and H. Wang, “Molecular Dynamics Study on Friction Due to Ploughing and Adhesion in Nanometric Scratching Process,” Tribology Letters. 2011. link Times cited: 89 NOT USED (high confidence) T. Luther, “Adaptation of atomistic and continuum methods for multiscale simulation of quasi-brittle intergranular damage.” 2010. link Times cited: 0 Abstract: The numerical simulation of damage using phenomenological mo… read moreAbstract: The numerical simulation of damage using phenomenological models on the macroscale was state of the art for many decades. However, such models are not able to capture the complex nature of damage, which simultaneously proceeds on multiple length scales. Furthermore, these phenomenological models usually contain damage parameters, which are physically not interpretable. Consequently, a reasonable experimental determination of these parameters is often impossible. In the last twenty years, the ongoing advance in computational capacities provided new opportunities for more and more detailed studies of the microstructural damage behavior. Today, multiphase models with several million degrees of freedom enable for the numerical simulation of micro-damage phenomena in naturally heterogeneous materials. Therewith, the application of multiscale concepts for the numerical investigation of the complex nature of damage can be realized. The presented thesis contributes to a hierarchical multiscale strategy for the simulation of brittle intergranular damage in polycrystalline materials, for example aluminum. The numerical investigation of physical damage phenomena on an atomistic microscale and the integration of these physically based information into damage models on the continuum meso- and macroscale is intended. Therefore, numerical methods for the damage analysis on the micro- and mesoscale including the scale transfer are presented and the transition to the macroscale is discussed. The investigation of brittle intergranular damage on the microscale is realized by the application of the nonlocal Quasicontinuum method, which fully describes the material behavior by atomistic potential functions, but reduces the number of atomic degrees of freedom by introducing kinematic couplings. Since this promising method is applied only by a limited group of researchers for special problems, necessary improvements have been realized in an own parallelized implementation of the 3D nonlocal Quasicontinuum method. The aim of this implementation was to develop and combine robust and efficient algorithms for a general use of the Quasicontinuum method, and therewith to allow for the atomistic damage analysis in arbitrary grain boundary configurations. The implementation is applied in analyses of brittle intergranular damage in ideal and nonideal grain boundary models of FCC aluminum, considering arbitrary misorientations. From the microscale simulations traction separation laws are derived, which describe grain boundary decohesion on the mesoscale. Traction separation laws are part of cohesive zone models to simulate the brittle interface decohesion in heterogeneous polycrystal structures. 2D and 3D mesoscale models are presented, which are able to reproduce crack initiation and propagation along cohesive interfaces in polycrystals. An improved Voronoi algorithm is developed in 2D to generate polycrystal material structures based on arbitrary distribution functions of grain size. The new model is more flexible in representing realistic grain size distributions. Further improvements of the 2D model are realized by the implementation and application of an orthotropic material model with Hill plasticity criterion to grains. The 2D and 3D polycrystal models are applied to analyze crack initiation and propagation in statically loaded samples of aluminum on the mesoscale without the necessity of initial damage definition. read less NOT USED (high confidence) D. Schebarchov, “Mechanisms in Carbon Nanotube Growth: Modelling and Molecular Dynamics Simulations.” 2010. link Times cited: 0 Abstract: A selection of nanoscale processes is studied theoretically,… read moreAbstract: A selection of nanoscale processes is studied theoretically, with the aim of identifying themechanisms that could lead to selective carbon nanotube (CNT) growth. Only mechanisms relevant to catalytic chemical vapour deposition (CVD) are considered. The selected processes are analysed with classical molecular dynamics (MD) simulations and continuum modelling. The melting and pre-melting behaviour of supported nickel catalyst particles is investigated. Favourable epitaxy between a nanoparticle and the substrate is shown to significantly raise themelting point of the particle. It is also demonstrated that substrate binding can induce solid-solid transformations, whilst the epitaxy may even determine the orientation of individual crystal planes in supported catalysts. These findings suggest that the substrate crystal structure alone can potentially be used to manipulate the properties of catalyst particles and, hence, influence the structure of CNTs. The first attempt at modelling catalyst dewetting, a process where the catalyst unbinds from the inner walls of a nucleating nanotube, is presented. It is argued that understanding this process and gaining control over itmay lead to better selectivity in CNT growth. Two mutually exclusive dewetting mechanisms, namely cap lift-off and capillary withdrawal, are identified and then modelled as elastocapillary phenomena. The modelling yields an upper bound on the diameter of CNTs that can stem from a catalyst particle of a given size. It is also demonstrated that cap lift-off is sensitive to cap topology, suggesting that it may be possible to link catalyst characteristics to the structural properties of nucleating CNTs. However, a clear link to the chiral vector remains elusive. It is shown that particle size, as well as binding affinity, plays a critical role in capillary absorption and withdrawal of catalyst nanoparticles. This size dependence is explored in detail, revealing interesting ramifications to the statics and dynamics of capillary-driven flows at the nanoscale. The findings bear significant implications for our understanding of CNT growth from catalyst particles, whilst also suggesting new nanofluidic applications and methods for fabricating composite metal-CNT materials. read less NOT USED (high confidence) X. Tang, H. Lü, Q. Zhang, S. Zhong, and Y. Lin, “A comparing study on the evolution of Pd/Ni (1 0 0) and Pt/Ni(1 0 0) heteroepitaxial systems,” Physica B-condensed Matter. 2010. link Times cited: 1 NOT USED (high confidence) D. Bachurin and P. Gumbsch, “Molecular Dynamics Study of Plastic Deformation of Nanocrystalline Palladium,” International Conference on High Performance Computing. 2010. link Times cited: 2 NOT USED (high confidence) S.-E. Bae and A. Gewirth, “Differential reactivity of Cu(111) and Cu(100) during nitrate reduction in acid electrolyte.,” Faraday discussions. 2008. link Times cited: 29 Abstract: The interactions of nitrate with Cu(100) and Cu(111) in acid… read moreAbstract: The interactions of nitrate with Cu(100) and Cu(111) in acidic solution are studied by cyclic voltammetry (CV) and in situ electrochemical scanning tunneling microscopy (EC-STM). CV results show that reduction of nitrate on Cu(111) commences at 0.0 V vs. Ag/AgCl while the corresponding potential is -0.3 V on Cu(100). EC-STM images show that the terrace of both Cu(111) and Cu(100) are atomically flat at potentials more negative than -0.7 V. The Cu(100) surface exhibits flat terraces throughout the entire cathodic potential range. Close to OCP, step edges start to corrode. In contrast to Cu(100), the first layer of Cu(111) is converted to an atomically rough and defected surface-associated with nascent surface oxidation at potentials positive of -0.7 V. This surface oxidation is correlated with nitrate reduction. read less NOT USED (high confidence) M. Jia, Y. Lai, Z. Tian, and Y.-xiang Liu, “Calculation of the surface free energy of fcc copper nanoparticles,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 31 Abstract: Using molecular dynamics simulations with the modified analy… read moreAbstract: Using molecular dynamics simulations with the modified analytic embedded-atom method we calculate the Gibbs free energy and surface free energy for fcc Cu bulk, and further obtain the Gibbs free energy of nanoparticles. Based on the Gibbs free energy of nanoparticles, we have investigated the heat capacity of copper nanoparticles. Calculation results indicate that the Gibbs free energy and the heat capacity of nanoparticles can be divided into two parts: bulk quantity and surface quantity. The molar heat capacity of the bulk sample is lower compared with the molar heat capacity of nanoparticles, and this difference increases with the decrease in the particle size. It is also observed that the size effect on the thermodynamic properties of Cu nanoparticles is not really significant until the particle is less than about 20 nm. It is the surface atoms that decide the size effect on the thermodynamic properties of nanoparticles. read less NOT USED (high confidence) H. Wadley, X. W. Zhou, and W. Butler, “Atomic Assembly of Magnetoresistive Multilayers.” 2008. link Times cited: 4 NOT USED (high confidence) Y. Ouyang, H. Chen, and X. Zhong, “Ab initio studies of small AlmFen clusters,” Theoretical Chemistry Accounts. 2006. link Times cited: 8 NOT USED (high confidence) J. Schall, D. Brenner, A. Kelkar, and R. Gupta, “Continuum and Atomistic Modeling of Thin Films Subjected to Nanoindentation.” 2005. link Times cited: 0 NOT USED (high confidence) Y.-S. Kim, C.-I. Kim, J. Park, and K. Na, “Molecular dynamic study for nanopatterning using atomic force microscopy,” Metallurgical and Materials Transactions A. 2005. link Times cited: 8 NOT USED (high confidence) J. A. Zimmerman, E. B. WebbIII, J. J. Hoyt, R. E. Jones, P. A. Klein, and D. J. Bammann, “Calculation of stress in atomistic simulation,” Modelling and Simulation in Materials Science and Engineering. 2004. link Times cited: 3 Abstract: Atomistic simulation is a useful method for studying materia… read moreAbstract: Atomistic simulation is a useful method for studying material science phenomena. Examination of the state of a simulated material and the determination of its mechanical properties is accomplished by inspecting the stress field within the material. However, stress is inherently a continuum concept and has been proven difficult to define in a physically reasonable manner at the atomic scale. In this paper, an expression for continuum mechanical stress in atomistic systems derived by Hardy is compared with the expression for atomic stress taken from the virial theorem. Hardy's stress expression is evaluated at a fixed spatial point and uses a localization function to dictate how nearby atoms contribute to the stress at that point; thereby performing a local spatial averaging. For systems subjected to deformation, finite temperature, or both, the Hardy description of stress as a function of increasing characteristic volume displays a quicker convergence to values expected from continuum theory than volume averages of the local virial stress. Results are presented on extending Hardy's spatial averaging technique to include temporal averaging for finite temperature systems. Finally, the behaviour of Hardy's expression near a free surface is examined, and is found to be consistent with the mechanical definition for stress. read less NOT USED (high confidence) Y. G. Xu and G. R. Liu, “Fitting interatomic potentials using molecular dynamics simulations and inter-generation projection genetic algorithm,” Journal of Micromechanics and Microengineering. 2003. link Times cited: 0 Abstract: In this paper we propose a new algorithm to fit interatomic … read moreAbstract: In this paper we propose a new algorithm to fit interatomic potentials. In the new algorithm, molecular dynamics simulations are applied to calculate the material properties which are used to match the experimental data during the fitting procedure. This includes the effect of atom relaxations in fitting calculations. An inter-generation projection genetic algorithm is used to optimize the fitting parameters until the error between the calculated and experimental material properties is within tolerance. This leads to a global optimal solution. The new algorithm significantly improves the accuracy and transferability of the fitted potential. It has been demonstrated by a numerical example of fitting potential of nickel. read less NOT USED (high confidence) E. Bitzek et al., “Recent Developments in IMD: Interactions for Covalent and Metallic Systems.” 2001. link Times cited: 11 NOT USED (high confidence) J. A. Zimmerman, H. Gao, and F. F. Abraham, “Generalized stacking fault energies for embedded atom FCC metals,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 27 Abstract: Atomistic calculations for the 112 -generalized stacking fau… read moreAbstract: Atomistic calculations for the 112 -generalized stacking fault (GSF) energy curve are performed for various embedded atom models of FCC metals. Models include those by Voter and Chen; Angelo, Moody and Baskes; Oh and Johnson; Mishin and Farkas; and Ercolessi and Adams. The resulting curves show similar characteristics but vary in their agreement with the experimental estimates of the intrinsic stacking fault energy, sf , and with density functional theory (DFT) calculations of the GSF curve. These curves are used to obtain estimates of the unstable stacking fault energy, us , a quantity used in a criterion for dislocation nucleation. Curves for nickel and copper models show the theoretically expected skewed sinusoidal shape; however, several of the aluminium models produce an irregularly shaped GSF curve. Copper and aluminium values for us are underestimates of calculations from DFT, although some of the nickel models produce a value matching one of the available DFT results. Values for sf are either fitted to, or underestimate, the measured results. For use in simulations, the authors recommend using the Voter and Chen potential for copper, and either the Angelo, Moody and Baskes potential or the Voter and Chen potential for nickel. None of the potentials model aluminium well, indicating the need for a more-advanced empirical potential. read less NOT USED (high confidence) H. Fritsche, H. Müller, and B. Fehrensen, “Formation of Superclusters from Metallic Clusters,” Zeitschrift für Physikalische Chemie. 1997. link Times cited: 3 NOT USED (high confidence) H. Fritsche, “Surface Induced Changes of the Size and of the Shape of Small Cuboctahedral Particles,” Zeitschrift für Physikalische Chemie. 1995. link Times cited: 3 NOT USED (high confidence) J. Alonso, “Density Functional Theory of the Structure of Bimetallic Clusters,” Physica Scripta. 1994. link Times cited: 3 Abstract: Density Functional Theory (DFT) is used to study the structu… read moreAbstract: Density Functional Theory (DFT) is used to study the structure of bimetallic clusters. Aggregates formed by two or three different alkali elements are first considered and the Kohn-Sham version of DFT is employed to analyse mixing and segregation properties, and their influence on the collective electronic response of the cluster. Then Cu-Ni clusters are chosen as an example of non simple metals. In this case the embedded-atom method, rooted on DFT, is used to study the structure of these clusters. read less NOT USED (high confidence) K. Y. Lee, H. Schober, and W. A. Oates, “Pair Potential and Pair Functional Models of Metal-Hydrogen Alloys*,” Zeitschrift für Physikalische Chemie. 1993. link Times cited: 1 NOT USED (high confidence) S. Plimpton and B. Hendrickson, “Parallel Molecular Dynamics With the Embedded Atom Method,” MRS Proceedings. 1992. link Times cited: 65 Abstract: Parallel computing offers new capabilities for using molecul… read moreAbstract: Parallel computing offers new capabilities for using molecular dynamics (MD) to simulate larger numbers of atoms and longer time scales. In this paper we discuss two methods we have used to implement the embedded atom method (EAM) formalism for molecular dynamics on multiple-instruction/multiple-data (MIMD) parallel computers. The first method (atom-decomposition) is simple and suitable for small numbers of atoms. The second method (force-decomposition) is new and is particularly appropriate for the EAM because all the computations are between pairs of atoms. Both methods have the advantage of not requiring any geometric information about the physical domain being simulated. We present timing results for the two parallel methods on a benchmark EAM problem and briefly indicate how the methods can be used in other kinds of materials MD simulations. read less NOT USED (high confidence) T. Raeker and A. Depristo, “Theory of chemical bonding based on the atom–homogeneous electron gas system,” International Reviews in Physical Chemistry. 1991. link Times cited: 85 Abstract: We review recent developments in the theory of chemical bond… read moreAbstract: We review recent developments in the theory of chemical bonding based upon replacement of an N-atom system by N individual systems each consisting of an atom embedded in a homogeneous electron gas. These theories include the corrected effective medium and effective-medium-based methods, which are either first principle or semi-empirical, as well as the embedded atom and related methods (e.g. the “glue” and Finnis-Sinclair methods), which are totally empirical. These methods can provide an accurate description of metal-metal interactions for simple or transition metals with weak d bonding, including homogeneous and heterogeneous systems. They also can describe the binding of non-metallic atoms to metals. A number of these methods are efficient enough computationally to be used in molecular dynamics and/or Monte Carlo simulations of systems with many thousands of atoms. read less |
 EAM_Dynamo_FoilesBaskesDaw_1986Universal3_Pt__MO_757342646688_000
EAM_Dynamo_FoilesBaskesDaw_1986Universal3_Pt__MO_757342646688_000