 EAM_Dynamo_LiuAdams_1998_AlMg__MO_019873715786_000
EAM_Dynamo_LiuAdams_1998_AlMg__MO_019873715786_000
| Title
A single sentence description.
|
EAM potential (LAMMPS cubic hermite tabulation) for the Al-Mg system developed by Liu and Adams (1998) v000 |
|---|---|
| Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
|
EAM potential for the Al-Mg system developed by Liu and Adams (1998) using the force-matching method. The potential is designed to study segregation of Mg to grain boundaries in Al. |
| Species
The supported atomic species.
| Al, Mg |
| Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
|
None |
| Content Origin | NIST IPRP (https://www.ctcms.nist.gov/potentials/Al.html#Al-Mg). Note: The parameter file was modified replacing Fortran double precision notation with standard exponential notation. |
| Contributor |
Ellad B. Tadmor |
| Maintainer |
Ellad B. Tadmor |
| Developer |
XY Liu J. B. Adams |
| Published on KIM | 2018 |
| How to Cite |
This Model originally published in [1] is archived in OpenKIM [2-5]. [1] Liu X-Y, Adams JB. Grain-boundary segregation in Al–10%Mg alloys at hot working temperatures. Acta Materialia. 1998;46(10):3467–76. doi:10.1016/S1359-6454(98)00038-X — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information. [2] Liu XY, Adams JB. EAM potential (LAMMPS cubic hermite tabulation) for the Al-Mg system developed by Liu and Adams (1998) v000. OpenKIM; 2018. doi:10.25950/3f4d32ac [3] Foiles SM, Baskes MI, Daw MS, Plimpton SJ. EAM Model Driver for tabulated potentials with cubic Hermite spline interpolation as used in LAMMPS v005. OpenKIM; 2018. doi:10.25950/68defa36 [4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6 [5] Elliott RS, Tadmor EB. Knowledgebase of Interatomic Models (KIM) Application Programming Interface (API). OpenKIM; 2011. doi:10.25950/ff8f563a Click here to download the above citation in BibTeX format. |
| Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on. The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel. The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied. The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP). Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis. OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm. |
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information. 
97 Citations (65 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) A. de Vaucorbeil, C. Sinclair, and W. Poole, “Atomistic insights into cluster strengthening in aluminum alloys,” Materialia. 2018. link Times cited: 12 USED (high confidence) T. P. Matson and C. Schuh, “Atomistic Assessment of Solute-Solute Interactions during Grain Boundary Segregation,” Nanomaterials. 2021. link Times cited: 7 Abstract: Grain boundary solute segregation is becoming increasingly c… read more USED (high confidence) P. Polyakova, J. A. Pukhacheva, S. Shcherbinin, J. Baimova, and R. Mulyukov, “Fabrication of Magnesium-Aluminum Composites under High-Pressure Torsion: Atomistic Simulation,” Applied Sciences. 2021. link Times cited: 3 Abstract: The aluminum–magnesium (Al–Mg) composite materials possess a… read more USED (high confidence) P.-A. Geslin and D. Rodney, “Microelasticity model of random alloys. Part I: mean square displacements and stresses,” Journal of The Mechanics and Physics of Solids. 2021. link Times cited: 15 USED (high confidence) E. Christiansen, I. G. Ringdalen, R. Bjørge, C. Marioara, and R. Holmestad, “Multislice image simulations of sheared needle-like precipitates in an Al-Mg-Si alloy.,” Journal of microscopy. 2020. link Times cited: 2 Abstract: The image contrast of sheared needle-like β'' prec… read more USED (high confidence) G. Zu and S. Groh, “Effect of segregated alloying element on the intrinsic fracture behavior of Mg,” Theoretical and Applied Fracture Mechanics. 2016. link Times cited: 3 USED (high confidence) S. Chen, D. Rittel, and D. Mordehai, “A Percolative Deformation Process Between Nanograins Promotes Dynamic Shear Localization,” Materials Research Letters. 2015. link Times cited: 3 Abstract: Shear localization is an important failure mechanism in crys… read more USED (high confidence) Q. Wei, X.-Y. Liu, and A. Misra, “Observation of continuous and reversible bcc–fcc phase transformation in Ag/V multilayers,” Applied Physics Letters. 2011. link Times cited: 19 Abstract: A continuous and reversible bcc–fcc phase transformation via… read more USED (high confidence) N. Du, Y. Qi, P. Krajewski, and A. Bower, “The Effect of Solute Atoms on Aluminum Grain Boundary Sliding at Elevated Temperature,” Metallurgical and Materials Transactions A. 2011. link Times cited: 28 USED (high confidence) J. Uan and C.-C. Chang, “Gallium-induced magnesium enrichment on grain boundary and the gallium effect on degradation of tensile properties of aluminum alloys,” Metallurgical and Materials Transactions A. 2006. link Times cited: 14 USED (high confidence) A. Landa, P. Wynblatt, D. J. Siegel, J. B. Adams, O. Mryasov, and X. Liu, “DEVELOPMENT OF GLUE-TYPE POTENTIALS FOR THE Al-Pb SYSTEM: PHASE DIAGRAM CALCULATION,” Acta Materialia. 2000. link Times cited: 67 USED (high confidence) E. Christiansen et al., “Detailed investigation of the shearing mechanism of β’ precipitates in Al-Mg-Si alloys.” 2020. link Times cited: 7 Abstract: The mechanism behind shearing of β′′ precipitates in Al-Mg-S… read more USED (high confidence) A. Vaucorbeil, “On the origin of cluster strengthening in aluminum alloys.” 2015. link Times cited: 1 Abstract: Understanding the influence of atomic clusters formed during… read more USED (low confidence) G. Shen et al., “Effects of heat treatment processes on the mechanical properties, microstructure evolution, and strengthening mechanisms of Al–Mg–Zn–Cu alloy,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) V. Menon, S. Das, V. Gavini, and L. Qi, “Atomistic simulations and machine learning of solute grain boundary segregation in Mg alloys at finite temperatures,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) Y. Cui, K. Song, Y. Bao, Y. Zhu, Q. Liu, and P. Qian, “Effect of Cu and Mg co-segregation on the strength of the Al grain boundaries: A molecular dynamics simulation,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) H. Zhang, D. Dai, L. Yuan, H. Liu, and D. Gu, “Temperature gradient induced tough-brittle transition behavior of a high-strength Al-4.2Mg-0.4Sc-0.2Zr alloy fabricated by laser powder bed fusion additive manufacturing,” Additive Manufacturing. 2023. link Times cited: 1 USED (low confidence) B. Sboui, D. Rodney, and P.-A. Geslin, “Elastic modelling of lattice distortions in concentrated random alloys,” Acta Materialia. 2023. link Times cited: 1 USED (low confidence) T. Lei, E. C. Hessong, D. Gianola, and T. Rupert, “Binary nanocrystalline alloys with strong glass forming interfacial regions: Complexion stability, segregation competition, and diffusion pathways,” Materials Characterization. 2023. link Times cited: 0 USED (low confidence) S. Wei, Y. Xu, Z. Han, Z. Li, and L. Xu, “Effect of microwave melting and subsequent rolling on the corrosion behavior and electrochemical properties of an aluminum anode using response surface methodology,” Ceramics International. 2023. link Times cited: 0 USED (low confidence) J. F. Troncoso, Y. Hu, N. M. della Ventura, A. Sharma, X. Maeder, and V. Turlo, “Machine learning of twin/matrix interfaces from local stress field,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (low confidence) L. Zhang, Z.-H. Zhang, X. Zhang, and X. Huang, “Computational simulation of grain boundary segregation of solute atoms in nanocrystalline metals,” Journal of Materials Research and Technology. 2022. link Times cited: 4 USED (low confidence) F. Sasani, A. Taheri, and M. Pouranvari, “Correlation between microstructure and mechanical properties of AlMg6/CNT-Al composite produced by accumulative roll bonding process: Experimental and modelling analysis,” Materials Science and Engineering: A. 2022. link Times cited: 5 USED (low confidence) J. Qin, Z. Li, M. Ma, D. Yi, and B. Wang, “Diversity of intergranular corrosion and stress corrosion cracking for 5083 Al alloy with different grain sizes,” Transactions of Nonferrous Metals Society of China. 2022. link Times cited: 13 USED (low confidence) W. Ye, J. Hohl, M. Misra, Y. Liao, and L. Mushongera, “Grain boundary relaxation in doped nano-grained aluminum,” Materials Today Communications. 2021. link Times cited: 5 USED (low confidence) W. Ye, P. Kumar, M. Misra, and L. Mushongera, “Local damage in grain boundary stabilized nanocrystalline aluminum,” Materials Letters. 2021. link Times cited: 4 USED (low confidence) Y. Han, P. Chen, J. Zhu, H. Liu, and Y. Zhang, “Mechanical behavior of single layer MoS2 sheets with aligned defects under uniaxial tension,” Journal of Applied Physics. 2021. link Times cited: 6 Abstract: Compared with a single defect or randomly distributed defect… read more USED (low confidence) L. Zhao, W. Xia, H. Yan, J. Chen, and B. Su, “Effects of Zn Addition on Dynamic Recrystallization of High Strain Rate Rolled Al–Mg Sheets,” Metals and Materials International. 2021. link Times cited: 3 USED (low confidence) C. M. Andolina, J. G. Wright, N. Das, and W. Saidi, “Improved Al-Mg alloy surface segregation predictions with a machine learning atomistic potential,” Physical Review Materials. 2021. link Times cited: 14 Abstract: Various industrial/commercial applications use Al-Mg alloys,… read more USED (low confidence) M. Bian, X. Huang, and Y. Chino, “Solute segregation assisted grain boundary precipitation and its impact to ductility of a precipitation-hardenable magnesium alloy,” Materials Science and Engineering: A. 2021. link Times cited: 7 USED (low confidence) L. Zhao et al., “High ductility and strong work-hardening behavior of Zn modified as-hot-rolled Al–Mg sheets,” Journal of Alloys and Compounds. 2021. link Times cited: 10 USED (low confidence) R. Shi, “Nonisothermal Dissolution Kinetics on Mg17Al12 Intermetallic in Mg-Al Alloys,” ChemRN: Molecular Modelling (Topic). 2021. link Times cited: 13 Abstract: In this work, nonisothermal dissolution of intermetallic Mg1… read more USED (low confidence) L.-F. Zhu, J. Janssen, S. Ishibashi, F. Körmann, B. Grabowski, and J. Neugebauer, “A fully automated approach to calculate the melting temperature of elemental crystals,” Computational Materials Science. 2021. link Times cited: 17 USED (low confidence) A. Kazemi and S. Yang, “Effects of magnesium dopants on grain boundary migration in aluminum-magnesium alloys,” Computational Materials Science. 2020. link Times cited: 30 USED (low confidence) M. Wagih and C. Schuh, “Grain boundary segregation beyond the dilute limit: Separating the two contributions of site spectrality and solute interactions,” Acta Materialia. 2020. link Times cited: 38 USED (low confidence) Y. Ding et al., “Nucleation and evolution of β phase and corresponding intergranular corrosion transition at 100–230 °C in 5083 alloy containing Er and Zr,” Materials & Design. 2019. link Times cited: 18 USED (low confidence) H. Dong, Y. Wang, L. Shi, P. Li, and S. Li, “Influence of cyclic non-isothermal heat treatment on microstructure, mechanical property and corrosion behavior of Al–Zn–Mg alloy,” Materials Research Express. 2019. link Times cited: 4 Abstract: The influence of cyclic non-isothermal heat treatment times … read more USED (low confidence) A. Devaraj et al., “Grain boundary segregation and intermetallic precipitation in coarsening resistant nanocrystalline aluminum alloys,” Acta Materialia. 2019. link Times cited: 76 USED (low confidence) Z. Li, J. Wang, and W. Liu, “Basal 〈a〉 dislocation-1¯011 contraction twin interactions in magnesium,” Computational Materials Science. 2018. link Times cited: 11 USED (low confidence) H. Fan, A. Ngan, K. Gan, and J. El-Awady, “Origin of double-peak precipitation hardening in metallic alloys,” International Journal of Plasticity. 2018. link Times cited: 44 USED (low confidence) Y. Yang, P. Zhao, L. Zou, and R. Fan, “Bake Precipitation Behavior in AA5182 Sheet for Can End Stock,” Materials Science Forum. 2018. link Times cited: 2 Abstract: By means of Vickers hardness tester, optical microscope (OM)… read more USED (low confidence) P. Lejček, M. Šob, and V. Paidar, “Interfacial segregation and grain boundary embrittlement: An overview and critical assessment of experimental data and calculated results,” Progress in Materials Science. 2017. link Times cited: 146 USED (low confidence) M. Gong et al., “Effects of concurrent grain boundary and surface segregation on the final stage of sintering: the case of Lanthanum doped yttria-stabilized zirconia,” Journal of Materials Science & Technology. 2017. link Times cited: 17 USED (low confidence) Y. S. Buranova et al., “Al3(Sc,Zr)-based precipitates in Al–Mg alloy: Effect of severe deformation,” Acta Materialia. 2017. link Times cited: 127 USED (low confidence) L. E. Kar’kina, I. N. Kar’kin, A. R. Kuznetsov, I. Razumov, P. Korzhavyi, and Y. Gornostyrev, “Solute-grain boundary interaction and segregation formation in Al : First principles calculations and molecular dynamics modeling,” Computational Materials Science. 2016. link Times cited: 33 USED (low confidence) R.-feng Zhang, S. Knight, R. L. Holtz, R. Goswami, C. Davies, and N. Birbilis, “A Survey of Sensitization in 5xxx Series Aluminum Alloys,” Corrosion. 2016. link Times cited: 90 Abstract: The 5xxx series (Al-Mg-based) aluminum alloys suffer from in… read more USED (low confidence) X. Sauvage, N. Enikeev, R. Valiev, Y. Nasedkina, and M. Murashkin, “Atomic-scale analysis of the segregation and precipitation mechanisms in a severely deformed Al–Mg alloy,” Acta Materialia. 2014. link Times cited: 198 USED (low confidence) S.-jun Zhang, O. Kontsevoi, A. Freeman, and G. Olson, “Cohesion enhancing effect of magnesium in aluminum grain boundary: A first-principles determination,” Applied Physics Letters. 2012. link Times cited: 15 Abstract: The effect of magnesium on grain boundary cohesion in alumin… read more USED (low confidence) M. Mostafaei and M. Kazeminezhad, “Hot deformation behavior of hot extruded Al–6Mg alloy,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2012. link Times cited: 58 USED (low confidence) U. Çaydaş and A. Hasçalik, “Effect of Mg on Mechanical Properties of Al-Mg Alloys,” Practical Metallography. 2010. link Times cited: 2 Abstract: In this study, the effect of Mg content on the mechanical pr… read more USED (low confidence) L. Tan and T. Allen, “Effect of thermomechanical treatment on the corrosion of AA5083,” Corrosion Science. 2010. link Times cited: 134 USED (low confidence) R. C. Picu and Z. Xu, “Vacancy concentration in Al–Mg solid solutions,” Scripta Materialia. 2007. link Times cited: 11 USED (low confidence) L. Wang and H. Liu, “The microstructural evolution of Al12Mg17 alloy during the quenching processes,” Journal of Non-crystalline Solids. 2006. link Times cited: 14 USED (low confidence) Z. Xu and R. C. Picu, “Dislocation–solute cluster interaction in Al–Mg binary alloys,” Modelling and Simulation in Materials Science and Engineering. 2006. link Times cited: 21 Abstract: The close-range interaction of dislocations and solute clust… read more USED (low confidence) C. Shet, N. Chandra, and S. Namilae, “Defect–Defect Interaction in Carbon Nanotubes under Mechanical Loading,” Mechanics of Advanced Materials and Structures. 2005. link Times cited: 7 Abstract: Topological defects are formed in carbon nanotubes (CNTs) du… read more USED (low confidence) N. Chandra, S. Namilae, and C. Shet, “Local elastic properties of carbon nanotubes in the presence of Stone-Wales defects,” Physical Review B. 2004. link Times cited: 186 Abstract: Carbon nanotubes are being contemplated as reinforcements fo… read more USED (low confidence) R. C. Picu and D. Zhang, “Atomistic study of pipe diffusion in Al–Mg alloys,” Acta Materialia. 2004. link Times cited: 149 USED (low confidence) S. Namilae, N. Chandra, and T. Nieh, “Atomistic simulation of grain boundary sliding in pure and magnesium doped aluminum bicrystals,” Scripta Materialia. 2002. link Times cited: 58 USED (low confidence) T. Lenosky et al., “Highly optimized empirical potential model of silicon,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 145 Abstract: We fit an empirical potential for silicon using the modified… read more USED (low confidence) X.-Y. Liu, C.-L. Liu, and L. J. Borucki, “A new investigation of copper’s role in enhancing Al-Cu interconnect electromigration resistance from an atomistic view,” Acta Materialia. 1999. link Times cited: 44 USED (low confidence) J. F. Troncoso and V. Turlo, “Evaluating the applicability of classical and neural network interatomic potentials for modeling body centered cubic polymorph of magnesium,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 2 Abstract: Magnesium (Mg) is one of the most abundant metallic elements… read more USED (low confidence) S. Groh and M. K. Nahhas, “Modeling Dislocation in Binary Magnesium-Based Alloys Using Atomistic Method,” Handbook of Mechanics of Materials. 2019. link Times cited: 1 USED (low confidence) C. Picu and D. Zhang, “Multiscale Modeling of Solute Bulk Diffusion at Dislocation Cores,” International Journal for Multiscale Computational Engineering. 2009. link Times cited: 1 USED (low confidence) J. Vetrano, C. Henager, and E. Simonen, “Role of vacancies and solute atoms on grain boundary sliding,” MRS Proceedings. 1999. link Times cited: 0 Abstract: It is necessary for grain boundary dislocations to slide and… read more NOT USED (low confidence) D. Zhou, X. Zhang, and D. Zhang, “Making strong Al(Mg)-Al3Mg2 composites,” Materialia. 2021. link Times cited: 6 NOT USED (low confidence) K. Zhang, M. Fan, Y. Liu, J. Schroers, M. Shattuck, and C. O’Hern, “Beyond packing of hard spheres: The effects of core softness, non-additivity, intermediate-range repulsion, and many-body interactions on the glass-forming ability of bulk metallic glasses.,” The Journal of chemical physics. 2015. link Times cited: 16 Abstract: When a liquid is cooled well below its melting temperature a… read more NOT USED (high confidence) P.-A. Geslin, A. Rida, and D. Rodney, “Microelasticity model of random alloys. Part II: displacement and stress correlations,” Journal of The Mechanics and Physics of Solids. 2021. link Times cited: 16 NOT USED (high confidence) L. Zhao, H. Yan, J. Chen, W. Xia, and B. Su, “Effects of Zn content on damping behaviours of Al–Mg alloys,” Materials Science and Technology. 2021. link Times cited: 1 Abstract: Effects of Zn content (0–1.5, in wt.%) on damping capacities… read more NOT USED (high confidence) W. Jiang, Y. Zhang, L. Zhang, and H. Wang, “Accurate Deep Potential model for the Al–Cu–Mg alloy in the full concentration space*,” arXiv: Materials Science. 2020. link Times cited: 24 Abstract: Combining first-principles accuracy and empirical-potential … read more NOT USED (high confidence) R. K. Koju and Y. Mishin, “Atomistic Study of Grain-Boundary Segregation and Grain-Boundary Diffusion in Al-Mg Alloys,” EngRN: Metals & Alloys (Topic). 2020. link Times cited: 60 Abstract: Mg grain boundary (GB) segregation and GB diffusion can impa… read more NOT USED (high confidence) P. Parajuli et al., “Misorientation dependence grain boundary complexions in <111> symmetric tilt Al grain boundaries,” Acta Materialia. 2019. link Times cited: 10 NOT USED (high confidence) A. K. Gupta et al., “Precipitate-induced nonlinearities of diffusion along grain boundaries in Al-based alloys,” Physical Review Materials. 2018. link Times cited: 11 NOT USED (high confidence) P. Parajuli, R. Mendoza-Cruz, A. Hurtado-Macías, U. Santiago, and M. Yacamán, “A Direct Observation of Ordered Structures Induced by Cu Segregation at Grain Boundaries of Al 7075 Alloys,” physica status solidi (a). 2018. link Times cited: 7 Abstract: The experimental investigation of the atomic‐scale structure… read more NOT USED (high confidence) W. Xiao, J. W. Wang, L. Sun, X. Li, Z. H. Li, and L. Wang, “Theoretical investigation of the strengthening mechanism and precipitation evolution in high strength Al-Zn-Mg alloys.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 4 Abstract: Density-functional theory calculations have been performed t… read more NOT USED (high confidence) L. Hale, “Comparing Modeling Predictions of Aluminum Edge Dislocations: Semidiscrete Variational Peierls–Nabarro Versus Atomistics,” JOM. 2018. link Times cited: 7 NOT USED (high confidence) F. Cao, Y.-zheng Jiang, T. Hu, and D. Yin, “Correlation of grain boundary extra free volume with vacancy and solute segregation at grain boundaries: a case study for Al,” Philosophical Magazine. 2018. link Times cited: 38 Abstract: Grain boundary extra free volume (GB EFV) can be considered … read more NOT USED (high confidence) D. Zhao, O. Løvvik, K. Marthinsen, and Y. Li, “Segregation of Mg, Cu and their effects on the strength of Al Σ5 (210)[001] symmetrical tilt grain boundary,” Acta Materialia. 2018. link Times cited: 86 NOT USED (high confidence) S. Groh, “Modified embedded-atom potential for B2-MgAg,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 5 Abstract: Interatomic potentials for pure Ag and Mg–Ag alloy have been… read more NOT USED (high confidence) A. Kumamoto et al., “Atomic structures of a liquid-phase bonded metal/nitride heterointerface,” Scientific Reports. 2016. link Times cited: 14 NOT USED (high confidence) H. Wang, M. Kohyama, S. Tanaka, and Y. Shiihara, “First-principles study of Si and Mg segregation in grain boundaries in Al and Cu: application of local-energy decomposition,” Journal of Materials Science. 2015. link Times cited: 27 NOT USED (high confidence) S. Jin, N. Tao, K. Marthinsen, and Y. Li, “Deformation of an Al–7Mg alloy with extensive structural micro-segregations during dynamic plastic deformation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 25 NOT USED (high confidence) A. Halap, T. Radetić, M. Popović, and E. Romhanji, “Influence of the Thermomechanical Treatment on the Intergranular Corrosion Susceptibility of Zn-Modified Al-5.1 Wt Pct Mg-0.7 Wt Pct Mn Alloy Sheet,” Metallurgical and Materials Transactions A. 2014. link Times cited: 12 NOT USED (high confidence) D. K. Xu, P. Rometsch, L. Li, L. Shen, and N. Birbilis, “Critical conditions for the occurrence of quench cracking in an Al–Zn–Mg–Cu alloy,” Journal of Materials Science. 2014. link Times cited: 5 NOT USED (high confidence) M. Liu et al., “Structure and mechanical properties of nanostructured Al–Mg alloys processed by severe plastic deformation,” Journal of Materials Science. 2013. link Times cited: 44 NOT USED (high confidence) X. Sauvage, A. Ganeev, Y. Ivanisenko, N. Enikeev, M. Murashkin, and R. Valiev, “Grain Boundary Segregation in UFG Alloys Processed by Severe Plastic Deformation,” Advanced Engineering Materials. 2012. link Times cited: 80 Abstract: Grain boundary (GB) segregations were investigated by atom p… read more NOT USED (high confidence) X. Sauvage, G. Wilde, S. Divinski, Z. Horita, and R. Valiev, “Grain boundaries in ultrafine grained materials processed by severe plastic deformation and related phenomena,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2012. link Times cited: 424 NOT USED (high confidence) B. Jelinek et al., “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Physical Review B. 2011. link Times cited: 218 Abstract: A set of modified embedded-atom method (MEAM) potentials for… read more NOT USED (high confidence) Y. Tian, B. Long, and C. Wang, “Finite element analysis of electromigration reliability in copper chip interconnect,” 2010 11th International Conference on Electronic Packaging Technology & High Density Packaging. 2010. link Times cited: 2 Abstract: To discuss the electromigration reliability of the copper ch… read more NOT USED (high confidence) X. Liu, F. Ercolessi, and J. B. Adams, “Aluminium interatomic potential from density functional theory calculations with improved stacking fault energy,” Modelling and Simulation in Materials Science and Engineering. 2004. link Times cited: 147 Abstract: A new Al potential with improved stacking fault energy is co… read more NOT USED (high confidence) D. J. Siegel, L. Hector, and J. B. Adams, “Adhesion, stability, and bonding at metal/metal-carbide interfaces: Al/WC,” Surface Science. 2002. link Times cited: 201 NOT USED (high confidence) R. H. Jones, D. Baer, M. Danielson, and J. Vetrano, “Role of Mg in the stress corrosion cracking of an Al-Mg alloy,” Metallurgical and Materials Transactions A. 2001. link Times cited: 167 NOT USED (high confidence) S. Karewar, N. Gupta, S. Groh, E. Martinez, A. Caro, and S. G. Srinivasan, “Effect of Li on the deformation mechanisms of nanocrystalline hexagonal close packed magnesium,” Computational Materials Science. 2017. link Times cited: 19 NOT USED (high confidence) E. Niebles, J. Unfried, and J. E. Torres, “METODOLOGÍAS PARA EL ESTUDIO DE SOLDABILIDAD EN UNIONES SOLDADAS.” 2014. link Times cited: 0 Abstract: A new methodology that describes relevant aspects of microst… read more NOT USED (high confidence) D. Zhang and R. C. Picu, “Solute clustering in Al–Mg binary alloys,” Modelling and Simulation in Materials Science and Engineering. 2004. link Times cited: 18 Abstract: Clustering of Mg in Al–Mg binary alloys is studied by means … read more NOT USED (definite) Y. Hu and T. Rupert, “Atomistic modeling of interfacial segregation and structural transitions in ternary alloys,” Journal of Materials Science. 2018. link Times cited: 19 |
| Funding | Not available |
| Short KIM ID
The unique KIM identifier code.
| MO_019873715786_000 |
| Extended KIM ID
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
| EAM_Dynamo_LiuAdams_1998_AlMg__MO_019873715786_000 |
| DOI |
10.25950/3f4d32ac https://doi.org/10.25950/3f4d32ac https://commons.datacite.org/doi.org/10.25950/3f4d32ac |
| KIM Item Type
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
| Portable Model using Model Driver EAM_Dynamo__MD_120291908751_005 |
| Driver | EAM_Dynamo__MD_120291908751_005 |
| KIM API Version | 2.0 |
| Potential Type | eam |
| Grade | Name | Category | Brief Description | Full Results | Aux File(s) |
|---|---|---|---|---|---|
| P | vc-species-supported-as-stated | mandatory | The model supports all species it claims to support; see full description. |
Results | Files |
| P | vc-periodicity-support | mandatory | Periodic boundary conditions are handled correctly; see full description. |
Results | Files |
| P | vc-permutation-symmetry | mandatory | Total energy and forces are unchanged when swapping atoms of the same species; see full description. |
Results | Files |
| B | vc-forces-numerical-derivative | consistency | Forces computed by the model agree with numerical derivatives of the energy; see full description. |
Results | Files |
| F | vc-dimer-continuity-c1 | informational | The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description. |
Results | Files |
| P | vc-objectivity | informational | Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description. |
Results | Files |
| P | vc-inversion-symmetry | informational | Total energy is unchanged and forces change sign when inverting a configuration through the origin; see full description. |
Results | Files |
| N/A | vc-memory-leak | informational | The model code does not have memory leaks (i.e. it releases all allocated memory at the end); see full description. |
Results | Files |
| P | vc-thread-safe | mandatory | The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description. |
Results | Files |
| P | vc-unit-conversion | mandatory | The model is able to correctly convert its energy and/or forces to different unit sets; see full description. |
Results | Files |
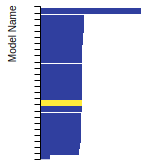
BCC Lattice Constant
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.

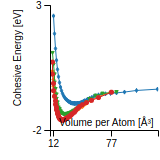
Cohesive Energy Graph
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.

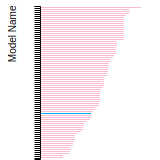
Diamond Lattice Constant
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.

Dislocation Core Energies
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
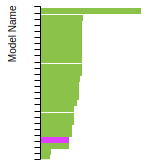
(No matching species)FCC Elastic Constants
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
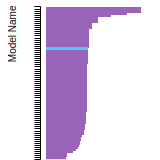

FCC Lattice Constant
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
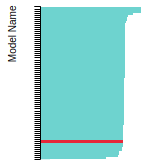

FCC Stacking Fault Energies
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
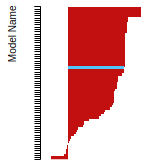
FCC Surface Energies
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
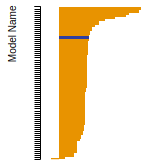
SC Lattice Constant
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.

Cubic Crystal Basic Properties Table
Species: AlSpecies: Mg
Creators:
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/64cb38c5
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Cohesive energy versus lattice constant curve for bcc Al v004 | view | 2870 | |
| Cohesive energy versus lattice constant curve for bcc Mg v004 | view | 2347 | |
| Cohesive energy versus lattice constant curve for diamond Al v004 | view | 3168 | |
| Cohesive energy versus lattice constant curve for diamond Mg v004 | view | 2586 | |
| Cohesive energy versus lattice constant curve for fcc Al v004 | view | 2268 | |
| Cohesive energy versus lattice constant curve for fcc Mg v004 | view | 2650 | |
| Cohesive energy versus lattice constant curve for sc Al v004 | view | 2576 | |
| Cohesive energy versus lattice constant curve for sc Mg v004 | view | 2298 |
Creators:
Contributor: ilia
Publication Year: 2024
DOI: https://doi.org/10.25950/888f9943
Computes the elastic constants for an arbitrary crystal. A robust computational protocol is used, attempting multiple methods and step sizes to achieve an acceptably low error in numerical differentiation and deviation from material symmetry. The crystal structure is specified using the AFLOW prototype designation as part of the Crystal Genome testing framework. In addition, the distance from the obtained elasticity tensor to the nearest isotropic tensor is computed.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for AlMg in AFLOW crystal prototype A12B17_cI58_217_g_acg at zero temperature and pressure v000 | view | 175779 |
Creators: Junhao Li and Ellad Tadmor
Contributor: tadmor
Publication Year: 2019
DOI: https://doi.org/10.25950/5853fb8f
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for bcc Al at zero temperature v006 | view | 4926 | |
| Elastic constants for bcc Mg at zero temperature v006 | view | 2175 | |
| Elastic constants for fcc Al at zero temperature v006 | view | 2015 | |
| Elastic constants for fcc Mg at zero temperature v006 | view | 1599 | |
| Elastic constants for sc Al at zero temperature v006 | view | 6526 | |
| Elastic constants for sc Mg at zero temperature v006 | view | 1791 |
Creators: Junhao Li
Contributor: jl2922
Publication Year: 2019
DOI: https://doi.org/10.25950/d794c746
Computes the elastic constants for hcp crystals by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for hcp Al at zero temperature v004 | view | 1560 | |
| Elastic constants for hcp Mg at zero temperature v004 | view | 1878 |
Creators:
Contributor: ilia
Publication Year: 2024
DOI: https://doi.org/10.25950/2f2c4ad3
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Creators:
Contributor: brunnels
Publication Year: 2022
DOI: https://doi.org/10.25950/2c59c9d6
Computes grain boundary energy for a range of tilt angles given a crystal structure, tilt axis, and material.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Relaxed energy as a function of tilt angle for a 100 symmetric tilt grain boundary in fcc Al v003 | view | 6104507 | |
| Relaxed energy as a function of tilt angle for a 110 symmetric tilt grain boundary in fcc Al v001 | view | 19639193 | |
| Relaxed energy as a function of tilt angle for a 111 symmetric tilt grain boundary in fcc Al v001 | view | 16733078 | |
| Relaxed energy as a function of tilt angle for a 112 symmetric tilt grain boundary in fcc Al v001 | view | 69243176 |
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/2765e3bf
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium zero-temperature lattice constant for bcc Al v007 | view | 1535 | |
| Equilibrium zero-temperature lattice constant for bcc Mg v007 | view | 1695 | |
| Equilibrium zero-temperature lattice constant for diamond Al v007 | view | 2271 | |
| Equilibrium zero-temperature lattice constant for diamond Mg v007 | view | 4063 | |
| Equilibrium zero-temperature lattice constant for fcc Al v007 | view | 4095 | |
| Equilibrium zero-temperature lattice constant for fcc Mg v007 | view | 3839 | |
| Equilibrium zero-temperature lattice constant for sc Al v007 | view | 1919 | |
| Equilibrium zero-temperature lattice constant for sc Mg v007 | view | 2175 |
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/c339ca32
Calculates lattice constant of hexagonal bulk structures at zero temperature and pressure by using simplex minimization to minimize the potential energy.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium lattice constants for hcp Al v005 | view | 14740 | |
| Equilibrium lattice constants for hcp Mg v005 | view | 25310 |
Creators:
Contributor: mjwen
Publication Year: 2024
DOI: https://doi.org/10.25950/9d9822ec
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Linear thermal expansion coefficient of fcc Al at 293.15 K under a pressure of 0 MPa v002 | view | 1003889 |
Creators: Matt Bierbaum
Contributor: mattbierbaum
Publication Year: 2019
DOI: https://doi.org/10.25950/64f4999b
Calculates the phonon dispersion relations for fcc lattices and records the results as curves.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Phonon dispersion relations for fcc Al v004 | view | 51982 |
Creators:
Contributor: SubrahmanyamPattamatta
Publication Year: 2019
DOI: https://doi.org/10.25950/b4cfaf9a
Intrinsic and extrinsic stacking fault energies, unstable stacking fault energy, unstable twinning energy, stacking fault energy as a function of fractional displacement, and gamma surface for a monoatomic FCC lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Stacking and twinning fault energies for fcc Al v002 | view | 8286239 |
Creators: Matt Bierbaum
Contributor: mattbierbaum
Publication Year: 2019
DOI: https://doi.org/10.25950/6c43a4e6
Calculates the surface energy of several high symmetry surfaces and produces a broken-bond model fit. In latex form, the fit equations are given by:
E_{FCC} (\vec{n}) = p_1 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (2 ( |x+ 2y + z| + |x+2y-z| + |x-2y + z| + |x-2y-z| + |2x+y+z| + |2x+y-z| +|2x-y+z| +|2x-y-z| +|x+y+2z| +|x+y-2z| +|x-y+2z| +|x-y-2z| ) + c
E_{BCC} (\vec{n}) = p_1 (6 \left( | x+y+z| + |x+y-z| + |-x+y-z| + |x-y+z| \right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) +c.
In Python, these two fits take the following form:
def BrokenBondFCC(params, index):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*(abs(x+2*y+z) + abs(x+2*y-z) +abs(x-2*y+z) +abs(x-2*y-z) + abs(2*x+y+z) +abs(2*x+y-z) +abs(2*x-y+z) +abs(2*x-y-z) + abs(x+y+2*z) +abs(x+y-2*z) +abs(x-y+2*z) +abs(x-y-2*z))+params[3]
def BrokenBondBCC(params, x, y, z):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*6*(abs(x+y+z) + abs(x-y-z) + abs(x-y+z) + abs(x+y-z)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[3]
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Broken-bond fit of high-symmetry surface energies in fcc Al v004 | view | 29046 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/fca89cea
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Monovacancy formation energy and relaxation volume for fcc Al | view | 347930 | |
| Monovacancy formation energy and relaxation volume for hcp Mg | view | 309132 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/c27ba3cd
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Vacancy formation and migration energy for fcc Al | view | 1569221 | |
| Vacancy formation and migration energy for hcp Mg | view | 706757 |
| Test | Error Categories | Link to Error page |
|---|---|---|
| Elastic constants for diamond Al at zero temperature v001 | other | view |
| Elastic constants for diamond Mg at zero temperature v001 | other | view |
EquilibriumCrystalStructure__TD_457028483760_000
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium crystal structure and energy for AlMg in AFLOW crystal prototype A12B17_cI58_217_g_acg v000 | other | view |
| Equilibrium crystal structure and energy for AlMg in AFLOW crystal prototype A67B41_cP108_221_aeh2il_cfgm v000 | other | view |
LatticeConstantCubicEnergy__TD_475411767977_006
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium zero-temperature lattice constant for bcc Mg | other | view |
| Equilibrium zero-temperature lattice constant for diamond Al | other | view |
| Equilibrium zero-temperature lattice constant for diamond Mg | other | view |
LatticeConstantHexagonalEnergy__TD_942334626465_004
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium lattice constants for hcp Al | other | view |
| Equilibrium lattice constants for hcp Mg | other | view |
| EAM_Dynamo_LiuAdams_1998_AlMg__MO_019873715786_000.txz | Tar+XZ | Linux and OS X archive |
| EAM_Dynamo_LiuAdams_1998_AlMg__MO_019873715786_000.zip | Zip | Windows archive |
This Model requires a Model Driver. Archives for the Model Driver EAM_Dynamo__MD_120291908751_005 appear below.
| EAM_Dynamo__MD_120291908751_005.txz | Tar+XZ | Linux and OS X archive |
| EAM_Dynamo__MD_120291908751_005.zip | Zip | Windows archive |