Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
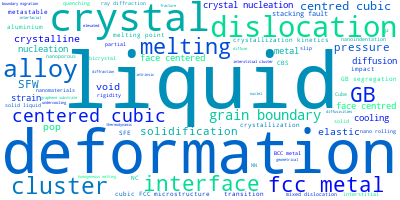
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
341 Citations (267 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) M. Wang, Y. Zeng, Y. Chen, and S. Zhang, “Crystal structure effect on metallic mechanical properties of under tension stress: molecular dynamics study,” 22nd International Scientific Conference Engineering for Rural Development Proceedings. 2023. link Times cited: 0 Abstract: In the paper, the tensile mechanical properties and deformat… read moreAbstract: In the paper, the tensile mechanical properties and deformation behaviour of different crystal structures (face-centered cubic (FCC) metal ~Al and closely packed hexagonal (HCP) metal ~Ti) were studied using molecular dynamics calculation through the LAMMPS module of Materials and Processes Simulations (MAPS) software, and the effect of microstructure on their mechanical properties was explored from the atomic level, in order to reveal the deformation mechanism of different lattice types of metals at the atomic scale. The simulation results using MAPS showed that the elastic modulus and the tensile strength of Al was calculated with 45.0 GPa and 6.2 GPa, respectively, the flow stress of Al has a stable value of around 0.69-1.29 GPa. And the elastic modulus and the tensile strength of Ti was calculated with 73.1 GPa and 8.5 GPa, respectively. According to calculation by Ovite, the occurrence of dislocations in Ti was later than that in Al, indicating that the strain energy that can be accumulated in the Ti lattice of HCP structure was higher and the ability to resist deformation was greater. As the periodic boundary conditions were used in all three directions of the two model, there was no macroscopic breaking phenomenon. To sum up, the lattice of HCP structure (~Ti) can accumulate higher strain energy and have greater deformation resistance ability than that of FCC structure (~Al), which was the reason for the elastic modulus and tensile strength of Ti higher than those of Al. read less USED (high confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (high confidence) Z. Yang and M. Buehler, “Linking atomic structural defects to mesoscale properties in crystalline solids using graph neural networks,” npj Computational Materials. 2022. link Times cited: 11 USED (high confidence) I. Chesser and Y. Mishin, “Point-defect avalanches mediate grain boundary diffusion,” Communications Materials. 2022. link Times cited: 7 USED (high confidence) R. Dsouza, L. Huber, B. Grabowski, and J. Neugebauer, “Approximating the impact of nuclear quantum effects on thermodynamic properties of crystalline solids by temperature remapping,” Physical Review B. 2022. link Times cited: 1 Abstract: When computing finite-temperature properties of materials wit… read moreAbstract: When computing finite-temperature properties of materials with atomistic simulations, nuclear quantum effects are often neglected or approximated at the quasiharmonic level. The inclusion of these effects beyond this level using approaches like the path integral method is often not feasible due to their large computational effort. We discuss and evaluate the performance of a temperature-remapping approach that links the finite-temperature quantum system to its best classical surrogate via a temperature map. This map, which is constructed using the internal energies of classical and quantum harmonic oscillators, is shown to accurately capture the impact of quantum effects on thermodynamic properties at an additional cost that is negligible compared to classical molecular dynamics simulations. Results from this approach show excellent agreement with previously reported path integral Monte Carlo simulation results for diamond cubic carbon and silicon. The approach is also shown to work well for obtaining thermodynamic properties of light metals and for the prediction of the fcc to bcc phase transition in calcium. read less USED (high confidence) G. Ananthakrishnan et al., “Graphene-mediated stabilization of surface facets on metal substrates,” Journal of Applied Physics. 2021. link Times cited: 5 USED (high confidence) W. Velilla-Díaz, L. Ricardo, A. Palencia, and H. R. Zambrano, “Fracture Toughness Estimation of Single-Crystal Aluminum at Nanoscale,” Nanomaterials. 2021. link Times cited: 6 Abstract: In this publication, molecular dynamics simulations are used… read moreAbstract: In this publication, molecular dynamics simulations are used to investigate the fracture behavior of single-crystal aluminum. The stress intensity factor is estimated by means of four different methods, the accuracy is assessed for each approach and the fracture toughness is estimated. The proposed methodology is also applied to estimate the fracture toughness for graphene and diamond using published data from other scientific articles. The obtained fracture toughness for the single-crystal aluminum is compared with other nanomaterials that have similar microstructures. Dislocation emission during the fracture simulation of the cracked nano-crystal of aluminum is analyzed to study the fracture behavior. Brittle fracture behavior is the predominant failure mode for the nanomaterials studied in this research. read less USED (high confidence) Y. Li, P. Peng, D. Xu, and R. Yang, “Identification of critical nuclei in the rapid solidification via configuration heredity,” Journal of Physics: Condensed Matter. 2021. link Times cited: 2 Abstract: The identification and characterization of critical nuclei i… read moreAbstract: The identification and characterization of critical nuclei is a long-standing issue in the rapid solidification of metals and alloys. An ambiguous description for their sizes and shapes used to lead to an overestimation or underestimation of homogeneous nucleation rates IT in the framework of classical nucleation theory (CNT). In this paper, a unique method able to distinguish the critical nucleus from numerous embryos is put forward on the basis of configuration heredities of clusters during rapid solidifications. As this technique is applied to analyze the formation and evolution of various fcc-Al single crystal clusters in a large-scale molecular dynamics simulation system, it is found that the size n c and geometrical configuration of critical nuclei as well as their liquid–solid interfacial structure can be determined directly. For the present deep super-cooled system with an undercooling of Tm=0.42Tmcal , the average size of critical nuclei is demonstrated to be nc̄≈26 , but most of which are non-spherical lamellae. Also, their liquid–solid interfaces are revealed to be not an fcc-liquid duplex-phase interface but an fcc/hcp-liquid multi-phase structure. These findings shed some lights on the CNT, and a good agreement with previous simulations and experiments in IT indicates this technique can be used to explore the early-stage of nucleation from atomistic levels. read less USED (high confidence) D. Louzguine-Luzgin and A. Bazlov, “Crystallization of FCC and BCC Liquid Metals Studied by Molecular Dynamics Simulation,” Metals. 2020. link Times cited: 17 Abstract: The atomic structure variations on cooling, vitrification an… read moreAbstract: The atomic structure variations on cooling, vitrification and crystallization processes in liquid metals face centered cubic (FCC) Cu are simulated in the present work in comparison with body centered cubic (BCC) Fe. The process is done on continuous cooling and isothermal annealing using a classical molecular-dynamics computer simulation procedure with an embedded-atom method potential at constant pressure. The structural changes are monitored with direct structure observation in the simulation cells containing from about 100 k to 1 M atoms. The crystallization process is analyzed under isothermal conditions by monitoring density and energy variation as a function of time. A common-neighbor cluster analysis is performed. The results of thermodynamic calculations on estimating the energy barrier for crystal nucleation and a critical nucleus size are compared with those obtained from simulation. The differences in crystallization of an FCC and a BCC metal are discussed. read less USED (high confidence) V. Ankudinov, “Approximation of correlation functions in phase‐field crystal model by machine learning approach,” Mathematical Methods in the Applied Sciences. 2020. link Times cited: 1 Abstract: Two different phase‐field crystal (PFC) free excess energy e… read moreAbstract: Two different phase‐field crystal (PFC) free excess energy expansions have been analyzed with respect to the order of a truncation term and to the accuracy of a pair correlation functions fitting. The coefficients of the correlation function in PFC model was found for the aluminum crystal based on the molecular dynamics data. A machine learning (ML) approach has been utilized to construct a neural network (NN) for the PFC approximation of correlation functions. The effective iteratomic potentials of the embedded atom model were used as an input data for NN training. Obtained NN predicts the coefficients of the correlation functions with the potentials data as input. Predictions were studied and verified with respect to the properties of NN and parameters of ML. read less USED (high confidence) A. Antropov, “Diffusion of Nanobubbles in fcc Aluminum,” JETP Letters. 2020. link Times cited: 3 USED (high confidence) R. K. Koju and Y. Mishin, “Atomistic Study of Grain-Boundary Segregation and Grain-Boundary Diffusion in Al-Mg Alloys,” EngRN: Metals & Alloys (Topic). 2020. link Times cited: 60 Abstract: Mg grain boundary (GB) segregation and GB diffusion can impa… read moreAbstract: Mg grain boundary (GB) segregation and GB diffusion can impact the processing and properties of Al-Mg alloys. Yet, Mg GB diffusion in Al has not been measured experimentally or predicted by simulations. We apply atomistic computer simulations to predict the amount and the free energy of Mg GB segregation, and the impact of segregation on GB diffusion of both alloy components. At low temperatures, Mg atoms segregated to a tilt GB form clusters with highly anisotropic shapes. Mg diffuses in Al GBs slower than Al itself, and both components diffuse slowly in comparison with Al GB self-diffusion. Thus, Mg segregation significantly reduces the rate of mass transport along GBs in Al-Mg alloys. The reduced atomic mobility can be responsible for the improved stability of the microstructure at elevated temperatures. read less USED (high confidence) Y. Wu, K. Zhang, J. Xiao, Y. Jiang, and L. Lv, “Conjugated bilayer structure of the homogeneous solid-liquid interface of metals.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 1 Abstract: The concept of "interfacial region" has long preva… read moreAbstract: The concept of "interfacial region" has long prevailed for over half century for describing the homogeneous solid-liquid (SL) interface of metals, but its intrinsic structure is still unclear due to the homogeneity. In this study, we reveal, for the first time, the intrinsic structure of these homogeneous SL interfaces consisting of two conjugated monoatomic layers of interfacial solid (IS) and interfacial liquid (IL) with a certain degree of corrugation via molecular dynamics simulations. We named it as the conjugated bilayer structure (CBS). In the framework of CBS, only the IS + IL bilayer plays stepwise transition roles from the solid to the liquid, which defines the four-terrace nature of the interface and act simultaneously as the boundaries of the bilateral bulk phases. The inherent diffuse nature of the "interfacial region" is proven originating from the corrugation of the IS + IL bilayer and its four-terrace nature. More importantly, the CBS also explains that the interfacial free energy originates mainly from the increase in the potential energy of the IS layer relative to its counterpart bulk solid instead of the previously argued entropy loss of the liquid phase. After all these verifications and interpretations, the CBS was verified as the intrinsic structure of the homogeneous SL interface of metals. Accordingly, we argue that the concept of CBS also resolves the volume-bearing flaw of the "interfacial region" concept and can definitively locate the intrinsic surface according to the capillary wave theory. read less USED (high confidence) A. Abu-Odeh, M. Cottura, and M. Asta, “Insights into dislocation climb efficiency in FCC metals from atomistic simulations,” Acta Materialia. 2020. link Times cited: 15 USED (high confidence) L.-F. Zhu, F. Körmann, A. Ruban, J. Neugebauer, and B. Grabowski, “Performance of the standard exchange-correlation functionals in predicting melting properties fully from first principles: Application to Al and magnetic Ni,” Physical Review B. 2020. link Times cited: 10 Abstract: We apply the efficient two-optimized references thermodynami… read moreAbstract: We apply the efficient two-optimized references thermodynamic integration using Langevin dynamics method [Phys. Rev. B 96, 224202 (2017)2469-995010.1103/PhysRevB.96.224202] to calculate highly accurate melting properties of Al and magnetic Ni from first principles. For Ni we carefully investigate the impact of magnetism on the liquid and solid free energies including longitudinal spin fluctuations and the reverse influence of atomic vibrations on magnetic properties. We show that magnetic fluctuations are effectively canceling out for both phases and are thus not altering the predicted melting temperature. For both elements, the generalized gradient approximation (GGA) and the local-density approximation (LDA) are used for the exchange-correlation functional revealing a reliable ab initio confidence interval capturing the respective experimental melting point, enthalpy of fusion, and entropy of fusion. read less USED (high confidence) S. Adibi and J. Wilkerson, “Evolving structure–property relationships in metals with nonequilibrium concentrations of vacancies,” Journal of Applied Physics. 2020. link Times cited: 14 Abstract: Here, we use molecular dynamics simulations as a tool to inv… read moreAbstract: Here, we use molecular dynamics simulations as a tool to investigate vacancy clustering in pure aluminum single crystals. A 1% superconcentration of single vacancies are randomly introduced into an otherwise perfect lattice, and the system is allowed to evolve for 500 ns at an elevated temperature of 728 K. Under these conditions, the individual vacancies rapidly agglomerate into larger clusters to reduce their overall energy. The systems are then subject to mechanical deformation to failure. The results of a total of 35 molecular dynamics simulations are reported. The mechanical behavior of these systems is found to be highly sensitive to the vacancy cluster microstructure, with the largest cluster size being most closely correlated with the cavitation strength. Since the largest cluster size evolves, an interesting time–structure–property coupling governs the behavior of these supersaturated metals. Despite the idealizations of the microstructure and loading conditions, we find a remarkably favorable agreement with laser-driven spall experiments. read less USED (high confidence) N. Zhou, K. Elkhodary, X. Huang, S. Tang, and Y. Li, “Dislocation structure and dynamics govern pop-in modes of nanoindentation on single-crystal metals,” Philosophical Magazine. 2020. link Times cited: 14 Abstract: ABSTRACT There are two types of pop-in mode that have been w… read moreAbstract: ABSTRACT There are two types of pop-in mode that have been widely observed in nanoindentation experiments: the single pop-in, and the successive pop-in modes. Here we employ the molecular dynamics (MD) modelling to simulate nanoindentation for three face-centred cubic (FCC) metals, including Al, Cu and Ni, and two body-centred cubic (BCC) metals, such as Fe and Ta. We aim to examine the deformation mechanisms underlying these pop-in modes. Our simulation results indicate that the dislocation structures formed in single crystals during nanoindentation are mainly composed of half prismatic dislocation loops. These half prismatic dislocation loops in FCC metals are primarily constituted of extended dislocations. Lomer–Cottrell locks that result from the interactions between these extended dislocations can resist the slipping of half dislocation loops. These locks can build up the elastic energy that is needed to activate the nucleation of new half dislocation loops. A repetition of this sequence results in successive pop-in events in Al and other FCC metals. Conversely, the half prismatic dislocation loops that form in BCC metals after first pop-in are prone to slip into the bulk, which sustains plastic indentation process after first pop-in and prevents subsequent pop-ins. We thus conclude that pop-in modes are correlated with lattice structures during nanoindentation, regardless of their crystal orientations. read less USED (high confidence) S. Subedi, L. Morrissey, S. M. Handrigan, and S. Nakhla, “The effect of many-body potential type and parameterisation on the accuracy of predicting mechanical properties of aluminium using molecular dynamics,” Molecular Simulation. 2020. link Times cited: 8 Abstract: ABSTRACT As opposed to traditional laboratory testing, Molec… read moreAbstract: ABSTRACT As opposed to traditional laboratory testing, Molecular Dynamics (MD) offers an atomistic scale method to estimate the mechanical properties of metals. However, there is limited literature that shows the effect of interatomic potentials when determining mechanical properties. Hence, the present research was conducted to investigate the accuracy of various interatomic potentials in estimating mechanical properties of aluminium. Several types of potentials, including Embedded Atom Method (EAM), Modified EAM (MEAM) and Reactive Force Field (ReaxFF) were compared with available experimental data for pure aluminium to determine the most accurate interatomic potential. A uniaxial tensile test was performed at room temperature using MD simulations for nanoscale aluminium. Results demonstrated that those potentials parameterised with elastic constants at physically realisable temperatures were consistently more accurate. Overall, the Mishin et al. EAM potential was the most accurate when compared to single-crystal experimental values. Regardless of the potential type, the error was significantly higher for those potentials that did not consider elastic constants during development. In brief, the application of the interatomic potentials to estimate mechanical properties of a nanoscale aluminium was investigated. read less USED (high confidence) P. Parajuli et al., “Misorientation dependence grain boundary complexions in <111> symmetric tilt Al grain boundaries,” Acta Materialia. 2019. link Times cited: 10 USED (high confidence) D. Xie et al., “Controlled growth of single-crystalline metal nanowires via thermomigration across a nanoscale junction,” Nature Communications. 2019. link Times cited: 16 USED (high confidence) J. Yu, H. Xie, F. Yin, and T. Yu, “Face-centered-cubic to body-centered-cubic phase transformation of Cu nanoplate under [100] tensile loading,” Philosophical Magazine. 2019. link Times cited: 1 Abstract: ABSTRACT Molecular dynamics simulation was used to stretch C… read moreAbstract: ABSTRACT Molecular dynamics simulation was used to stretch Cu nanoplates along its [100] direction at various strain rates and temperatures. Under high strain rate and beyond the elastic limit, the Cu nanoplates underwent an unusual deformation mechanism with expansion along free surface lateral direction and contraction along the other lateral direction, which leaded to the face-centred-cubic phase transforming into unstressed body-centred-cubic phase. Under low strain rate, the deformation of the nanoplate went back to well-known dislocation mechanism. The face-centred-cubic to body-centred-cubic phase transformation mechanism was further discussed in terms of elastic stability theory and free surface stress effect. read less USED (high confidence) S. Hayakawa, K. Doihara, T. Okita, M. Itakura, M. Aichi, and K. Suzuki, “Screw dislocation–spherical void interactions in fcc metals and their dependence on stacking fault energy,” Journal of Materials Science. 2019. link Times cited: 17 USED (high confidence) A. Hernandez, A. Balasubramanian, F. Yuan, S. Mason, and T. Mueller, “Fast, accurate, and transferable many-body interatomic potentials by symbolic regression,” npj Computational Materials. 2019. link Times cited: 51 USED (high confidence) K. V. Reddy and S. Pal, “Structural evolution and dislocation behaviour during nano-rolling process of FCC metals: A molecular dynamics simulation based investigation,” Journal of Applied Physics. 2019. link Times cited: 11 Abstract: Though the structural properties of nanomaterials are signif… read moreAbstract: Though the structural properties of nanomaterials are significantly influenced by the rolling process, the deformation mechanism at an atomic level is unknown. In this study, molecular dynamics simulations have been applied to investigate the deformation mechanism and structural evolution of single crystal Al and Cu specimens during the room temperature nano-rolling process. Also, the effect of crystallographic orientation and stacking fault energy (SFE) on the nano-rolling behaviour is analyzed. Results from dislocation analysis show that all the Cu specimens have higher dislocation densities when compared with that of the Al specimens. This is attributed to lower SFE of Cu specimens that exhibit a limited recovery process and affect the dislocation mobility causing higher dislocation densities. This phenomenon is also confirmed through atomic strain analysis which has shown a higher volume fraction of slip bands in the Cube oriented Cu specimen when compared with the Cube oriented Al specimen. On the other hand, Brass- and Copper-oriented Al and Cu specimens show strain imbalance between the lower and upper sections of the specimen due to the different slip mechanism, which causes a lag between the movement of both sections and consequent bending of the specimen.Though the structural properties of nanomaterials are significantly influenced by the rolling process, the deformation mechanism at an atomic level is unknown. In this study, molecular dynamics simulations have been applied to investigate the deformation mechanism and structural evolution of single crystal Al and Cu specimens during the room temperature nano-rolling process. Also, the effect of crystallographic orientation and stacking fault energy (SFE) on the nano-rolling behaviour is analyzed. Results from dislocation analysis show that all the Cu specimens have higher dislocation densities when compared with that of the Al specimens. This is attributed to lower SFE of Cu specimens that exhibit a limited recovery process and affect the dislocation mobility causing higher dislocation densities. This phenomenon is also confirmed through atomic strain analysis which has shown a higher volume fraction of slip bands in the Cube oriented Cu specimen when compared with the Cube oriented Al specimen. On the ot... read less USED (high confidence) G. Po, N. Admal, and M. Lazar, “The Green tensor of Mindlin’s anisotropic first strain gradient elasticity,” Materials Theory. 2019. link Times cited: 11 USED (high confidence) S. Khan, X. D. Wang, Q. Cao, D. X. Zhang, and J. Jiang, “Pressure-induced structural change and nucleation in liquid aluminum,” Journal of Applied Physics. 2018. link Times cited: 2 Abstract: The relationship between the atomic structure and dynamics o… read moreAbstract: The relationship between the atomic structure and dynamics of liquid aluminum (Al) has been studied at 1500 K as a function of pressure via ab initio molecular dynamics simulations. The origin of the structural evolution is unveiled by various techniques. The structure factor and the mean square displacement data indicate the fortuity of a crystalline-like phase at 25 GPa, first evolving into the metastable body-centered cubic-like local order, followed by face-centered cubic, which is different from the crystallization mechanism proposed for the liquid Al by Desgranges and Delhommelle [J. Chem. Phys. 127, 144509 (2007)]. The three-dimensional structural analysis demonstrates the concentration of distorted icosahedron-like clusters, e.g., Voronoi and , which are closely correlated with crystal nucleation and growth. Crystallization in the liquid Al is distinctly correlated with the bond orientational order (Q6) fluctuations, instead of density fluctuations, where the temperature or pressure comparison on the local atomic structure in the liquid Al and degree of crystallization is also elucidated. The electronic structure study reveals that at ambient pressure, some valence electrons are already localized, showing a strong tendency of electron pairing with each other in the interstitial regions.The relationship between the atomic structure and dynamics of liquid aluminum (Al) has been studied at 1500 K as a function of pressure via ab initio molecular dynamics simulations. The origin of the structural evolution is unveiled by various techniques. The structure factor and the mean square displacement data indicate the fortuity of a crystalline-like phase at 25 GPa, first evolving into the metastable body-centered cubic-like local order, followed by face-centered cubic, which is different from the crystallization mechanism proposed for the liquid Al by Desgranges and Delhommelle [J. Chem. Phys. 127, 144509 (2007)]. The three-dimensional structural analysis demonstrates the concentration of distorted icosahedron-like clusters, e.g., Voronoi and , which are closely correlated with crystal nucleation and growth. Crystallization in the liquid Al is distinctly correlated with the bond orientational order (Q6) fluctuations, instead of density fluctuations, where the temperature o... read less USED (high confidence) D. R. Garcia, Z. Zhang, B. Linke, and H. Urbassek, “Molecular dynamics simulations of single grain pure aluminum in a vice fixture for nanomanufacturing applications,” CIRP Journal of Manufacturing Science and Technology. 2018. link Times cited: 2 USED (high confidence) H. Zhou, J. Li, Y. Xian, G. Hu, X. Li, and R. Xia, “Nanoscale Assembly of Copper Bearing-Sleeve via Cold-Welding: A Molecular Dynamics Study,” Nanomaterials. 2018. link Times cited: 10 Abstract: A bearing is an important component in contemporary machiner… read moreAbstract: A bearing is an important component in contemporary machinery and equipment, whose main function is to support the mechanical rotator, reduce the friction coefficient during its movement, and guarantee the turning accuracy. However, assembly of a nanoscale bearing and sleeve is a challenging process for micro-nano mechanical manufacturers. Hence, we show the cold-welding mechanism of a copper nanobearing-nanosleeve via molecular dynamic simulations. We demonstrate that it is feasible to assemble a bearing and sleeve at the nanoscale to form a stable mechanism. The effect of temperature in the range of 150 to 750 K is investigated. As the temperature rises, the mechanical strength and the weld stress of the welded structures markedly decrease, accompanied by the observation of increasing disorder magnitude. This comparison study is believed to facilitate future mechanical processing and structural nano-assembly of metallic elements for better mechanical performance. read less USED (high confidence) Y.-Y. Zhang, H. Niu, G. Piccini, D. Mendels, and M. Parrinello, “Improving collective variables: The case of crystallization.,” The Journal of chemical physics. 2018. link Times cited: 36 Abstract: Several enhanced sampling methods, such as umbrella sampling… read moreAbstract: Several enhanced sampling methods, such as umbrella sampling or metadynamics, rely on the identification of an appropriate set of collective variables. Recently two methods have been proposed to alleviate the task of determining efficient collective variables. One is based on linear discriminant analysis; the other is based on a variational approach to conformational dynamics and uses time-lagged independent component analysis. In this paper, we compare the performance of these two approaches in the study of the homogeneous crystallization of two simple metals. We focus on Na and Al and search for the most efficient collective variables that can be expressed as a linear combination of X-ray diffraction peak intensities. We find that the performances of the two methods are very similar. Wherever the different metastable states are well-separated, the method based on linear discriminant analysis, based on its harmonic version, is to be preferred because simpler to implement and less computationally demanding. The variational approach, however, has the potential to discover the existence of different metastable states. read less USED (high confidence) J. Cho, J. Molinari, W. Curtin, and G. Anciaux, “The coupled atomistic/discrete-dislocation method in 3d. Part III: Dynamics of hybrid dislocations,” Journal of the Mechanics and Physics of Solids. 2018. link Times cited: 24 USED (high confidence) L. Wu et al., “Calculation of solid–liquid interfacial free energy and its anisotropy in undercooled system,” Rare Metals. 2018. link Times cited: 6 USED (high confidence) K. Dang and D. Spearot, “Pressure Dependence of the Peierls Stress in Aluminum,” JOM. 2018. link Times cited: 11 USED (high confidence) L. Zhang, C. Lu, and Y. Shibuta, “Shear response of grain boundaries with metastable structures by molecular dynamics simulations,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 18 Abstract: Grain boundaries (GBs) can play a role as the favored locati… read moreAbstract: Grain boundaries (GBs) can play a role as the favored locations to annihilate point defects, such as interstitial atoms and vacancies. It is thus highly probable that different boundary structures can be simultaneously present in equilibrium with each other in the same GB, and thus the GB achieves a metastable state. However, the structural transition and deformation mechanism of such GBs are currently not well understood. In this work, molecular dynamics simulations were carried out to study the multiple structures of a Σ5(310)/[001] GB in bicrystal Al and to investigate the effect of structural multiplicity on the mechanical and kinetic properties of such a GB. Different GB structures were obtained by changing the starting atomic configuration of the bicrystal model, and the GB structures had significantly different atomic density. For the Σ5(310) GB with metastable structures, GB sliding was the dominant mechanism at a low temperature (T = 10 K) under shear stress. The sliding mechanism resulted from the uncoordinated transformation of the inhomogeneous structural units. The nucleation of voids was observed during GB sliding at the low temperature, and the voids subsequently evolved to a nanocrack at the boundary plane. Increasing the temperature can induce the structural transition of local GB structures and can change their overall kinetic properties. GB migration with occasional GB sliding dominated the deformation mechanism at elevated temperatures (T = 300 and 600 K), and the migration process of the metastable GB structures is closely related to the thermally assisted diffusion mechanism. read less USED (high confidence) Z. Zhang and H. Urbassek, “Dislocations penetrating an Al/Si interface,” AIP Advances. 2017. link Times cited: 8 Abstract: We study indentation of a nanolayered material consisting of… read moreAbstract: We study indentation of a nanolayered material consisting of a Si top layer above an Al substrate, using molecular dynamics simulation. We focus on the activity of Si dislocations upon reaching the interface. We find that passage of the dislocations through the interface is possible, if the slip systems of the two crystals are aligned. Upon absorption at the interface, the Si dislocations generate slip which leads to 1-monolayer deep interface pits with well-defined steps; on the Al side dislocations and stacking fault planes are generated, which are pinned to the interface pit. For interfaces with not well aligned slip systems, the passage of dislocations is strongly suppressed. However, still interface pits, albeit with less well defined contours, and stacking fault planes aligned with the interface are created. read less USED (high confidence) S. Chandra, M. K. Samal, N. Kumar, V. Chavan, and S. Raghunathan, “An atomistic modelling and statistical analysis study of crack–void interaction in Aluminum,” Philosophical Magazine Letters. 2017. link Times cited: 4 Abstract: The interaction between a brittle crack and pre-existing voi… read moreAbstract: The interaction between a brittle crack and pre-existing void in front of the crack tip was studied in the realm of molecular dynamics simulations at the nanoscale in face centred cubic Al within the framework of embedded-atom method. The results provide corroborative evidence of the fact that presence of a void in front of a running crack deters further crack growth. By performing an extensive series of simulations with different void sizes and crack-void distances coupled with statistical analysis, it has been found that (1) major role of voids is to decrease the fracture stress with increasing void size, in addition to slight increase in strain at which the crack growth occurs and the consequent strain to fracture, (2) fracture stress for a constant void size follows a sinusoidal distribution by varying the crack-void distance and (3) it is the void size that is most crucial in dictating the fracture properties of the material, rather than the void placement. read less USED (high confidence) L. Zhao, G. Bokas, J. Perepezko, and I. Szlufarska, “Nucleation kinetics in Al-Sm metallic glasses,” Acta Materialia. 2017. link Times cited: 24 USED (high confidence) Y. Sun et al., “Overcoming the Time Limitation in Molecular Dynamics Simulation of Crystal Nucleation: A Persistent-Embryo Approach.,” Physical review letters. 2017. link Times cited: 36 Abstract: The crystal nucleation from liquid in most cases is too rare… read moreAbstract: The crystal nucleation from liquid in most cases is too rare to be accessed within the limited time scales of the conventional molecular dynamics (MD) simulation. Here, we developed a "persistent embryo" method to facilitate crystal nucleation in MD simulations by preventing small crystal embryos from melting using external spring forces. We applied this method to the pure Ni case for a moderate undercooling where no nucleation can be observed in the conventional MD simulation, and obtained nucleation rate in good agreement with the experimental data. Moreover, the method is applied to simulate an even more sluggish event: the nucleation of the B2 phase in a strong glass-forming Cu-Zr alloy. The nucleation rate was found to be 8 orders of magnitude smaller than Ni at the same undercooling, which well explains the good glass formability of the alloy. Thus, our work opens a new avenue to study solidification under realistic experimental conditions via atomistic computer simulation. read less USED (high confidence) T. Metspalu et al., “Cu self-sputtering MD simulations for 0.1-5 keV ions at elevated temperatures,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 5 USED (high confidence) S. Kumar and S. Das, “A triaxial tensile deformation-induced nanoporous structure of aluminium: estimation of surface area, solid volume, and dimensionless aspect ratio.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 4 Abstract: Nanoporous aluminium has great importance for large scale pr… read moreAbstract: Nanoporous aluminium has great importance for large scale production of automobile and aerospace spare parts due to its lightweight and non-corrosive nature. It is also suitable for various packaging applications of edible things, electronic components, and medicines. We have used triaxial tensile deformation methodology to create a nanoporous structure of aluminium using molecular dynamics simulation. The surface area and solid volume have been calculated to characterize the 3-D nanoporous structure of aluminium. We have quantitatively characterized the growth and coalescences of the nanoporous structure via estimation of the number of nanopores, nanopore diameters, and dimensionless aspect-ratios (surface area to volume ratio). A high aspect ratio indicates a large number of tiny nanopores in the 3-D nanoporous structure of aluminium. We have found that crystalline aluminium (under ambient condition) significantly depicts a smaller aspect ratio as compared to amorphous aluminium during triaxial tensile deformation. We believe that the results of this study will provide new understanding to the researchers for the design and characterization of nanoporous metals. read less USED (high confidence) N. Burbery, R. Das, and G. Ferguson, “Mobility of dissociated mixed dislocations under an Escaig stress,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 4 Abstract: In FCC metals, the structure of a mixed dislocation core con… read moreAbstract: In FCC metals, the structure of a mixed dislocation core consists of two Shockley Partials which have different screw and edge character. The interactions between the partial dislocations can influence the stacking fault width (SFW). The SFW can also be manipulated by controlling the non-glide component of the total shear stress within the glide plane, commonly referred to as the ‘Escaig stress’, or τe. Molecular dynamics simulations were used to reproduce the dynamic behaviour of the atomistic core and stacking fault of a moving mixed 30° dislocation in copper and with several magnitudes of τe stress. Results showed that the τ e must be relatively large to cause a significant effect on the SFW and that once the SFW is changed it also has a corresponding effect on the drag coefficient for a dislocation moving at steady-state. The reduction of the SFW, to the extent that the partial dislocations come within close proximity (i.e., partially merge into an imperfect full dislocation), changed the linear curve-fit of the stress–velocity curve and could be associated with a ‘quasi-Peierls barrier’ effect. The SFW was also shown to change under a pure glide stress without the addition of a τe stress when the velocity approached the supersonic limit, and caused an increase of the SFW in one direction and a reduction of the SFW in the other direction. This result demonstrates a unique characteristic of mixed dislocations which cannot be observed in the traditionally representative core structures of pure screw and pure edge dislocations because of their perfect symmetry. Surprisingly, the effects of the glide stress on the SFW were similar in magnitude to the effects of τ e , and also were found to have a corresponding effect on the drag coefficient. The unique characteristics observed are summarised in terms of a unified equation for the force equilibrium at steady-state, which considers the drag coefficient of the two partials as independent functions of the velocities. This relationship is rationalised in terms of a drag force imbalance between the two partials of a mixed 30° dislocation, caused by a slightly higher drag force in the ‘mildly non planar’ 0° partial than in the 60° partial. read less USED (high confidence) S. Kumar, “Effect of applied force and atomic organization of copper on its adhesion to a graphene substrate,” RSC Advances. 2017. link Times cited: 12 Abstract: Copper/graphene composites are lightweight and possess many … read moreAbstract: Copper/graphene composites are lightweight and possess many attractive properties such as improved mechanical, electrical, and thermal properties. The organization of copper atoms at the copper/graphene interface highly influences the abovementioned properties. In this study, the organization of copper atoms and applied force-induced desorption of copper from a graphene substrate were studied via molecular dynamics (MD) simulation. The copper atoms were organized in face-centred cubic (fcc) and hexagonal close-packed (hcp) lattices over the graphene substrate. However, at the copper/graphene interface, copper atoms were organized in the {111} facet of the fcc lattice. The applied force-induced desorption of copper atoms from a graphene substrate was studied at high temperature (T = 1000 K). A critical force was required to be exceeded before the detachment of copper atoms from the substrate. It was found that a higher critical force was required to remove copper atoms from the graphene substrate in the z-direction (perpendicular to the substrate) compared to that in the x-direction. The outcome of this study may provide useful scientific information about the metal/graphene interface properties, which will help enhance the performance of graphene-based metallic nanocomposites. read less USED (high confidence) L. Zhang, C. Lu, G. Michal, K. Tieu, X. Zhao, and G. Deng, “Influence of temperature and local structure on the shear‐coupled grain boundary migration,” physica status solidi (b). 2017. link Times cited: 18 Abstract: Grain boundary migration plays a significant role on the def… read moreAbstract: Grain boundary migration plays a significant role on the deformation and mechanical properties of ultrafine‐grained and nanocrystalline materials. How does the temperature and local structure effect on the behavior of grain boundary migration has not been well understood. In this study, molecular dynamics simulations are carried out to examine the shear‐coupled grain boundary migration in bicrystal Al with various Σ5[001](310) GB structures at a wide range of temperatures. The simulation result reveals that the shear‐coupled behavior of grain boundary migration is sensitive to the temperature and local boundary structure. The increased temperature can facilitate GB structural transformation and, therefore, change its coupling mode. read less USED (high confidence) J. Cho, J. Molinari, and G. Anciaux, “Mobility law of dislocations with several character angles and temperatures in FCC Aluminum,” International Journal of Plasticity. 2017. link Times cited: 75 USED (high confidence) M. Bahramyan, R. T. Mousavian, and D. Brabazon, “Molecular dynamic simulation of edge dislocation-void interaction in pure Al and Al-Mg alloy,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2016. link Times cited: 14 USED (high confidence) B. Cheng and J. Trelewicz, “Mechanistic coupling of dislocation and shear transformation zone plasticity in crystalline-amorphous nanolaminates,” Acta Materialia. 2016. link Times cited: 51 USED (high confidence) R. Rezaei, C. Deng, H. Tavakoli-Anbaran, and M. Shariati, “Deformation twinning-mediated pseudoelasticity in metal–graphene nanolayered membrane,” Philosophical Magazine Letters. 2016. link Times cited: 29 Abstract: In this study, we investigated the deformation behaviour of … read moreAbstract: In this study, we investigated the deformation behaviour of metal–graphene nanolayered composites for five face-centred cubic metals under compression using molecular dynamics simulations. It was found that by increasing the thickness of the individual metal layers, the composite strength increased, while the deformation mechanism changed from buckling to deformation twining in Cu, Au and Ag, which was absent in the monolithic form of those metals of the same orientation and size. The deformation twinning was found to be enabled by the graphene layer, which introduced pseudoelasticity and shape memory effects in the nanolayered membrane with more than 15% recoverable compressive strain. read less USED (high confidence) R. Babicheva et al., “Elastic moduli of nanocrystalline binary Al alloys with Fe, Co, Ti, Mg and Pb alloying elements,” Philosophical Magazine. 2016. link Times cited: 13 Abstract: The paper studies the elastic moduli of nanocrystalline (NC)… read moreAbstract: The paper studies the elastic moduli of nanocrystalline (NC) Al and NC binary Al–X alloys (X is Fe, Co, Ti, Mg or Pb) by using molecular dynamics simulations. X atoms in the alloys are either segregated to grain boundaries (GBs) or distributed randomly as in disordered solid solution. At 0 K, the rigidity of the alloys increases with decrease in atomic radii of the alloying elements. An addition of Fe, Co or Ti to the NC Al leads to increase in the Young’s E and shear μ moduli, while an alloying with Pb decreases them. The elastic moduli of the alloys depend on a distribution of the alloying elements. The alloys with the random distribution of Fe or Ti demonstrate larger E and μ than those for the corresponding alloys with GB segregations, while the rigidity of the Al–Co alloy is higher for the case of the GB segregations. The moduli E and μ for polycrystalline aggregates of Al and Al–X alloys with randomly distributed X atoms are estimated based on the elastic constants of corresponding single-crystals according to the Voigt-Reuss-Hill approximation, which neglects the contribution of GBs to the rigidity. The results show that GBs in NC materials noticeably reduce their rigidity. Furthermore, the temperature dependence of μ for the NC Al–X alloys is analyzed. Only the Al–Co alloy with GB segregations shows the decrease in μ to the lowest extent in the temperature range of 0–600 K in comparison with the NC pure Al. read less USED (high confidence) J. Han et al., “Abnormal correlation between phase transformation and cooling rate for pure metals,” Scientific Reports. 2016. link Times cited: 25 USED (high confidence) M. Aramfard and C. Deng, “Interaction of shear-coupled grain boundary motion with crack: Crack healing, grain boundary decohesion, and sub-grain formation,” Journal of Applied Physics. 2016. link Times cited: 14 Abstract: Stress-driven grain boundary motion is one of the main mecha… read moreAbstract: Stress-driven grain boundary motion is one of the main mechanisms responsible for microstructural evolution in polycrystalline metals during deformation. In this research, the interaction of shear-coupled grain boundary motion (SCGBM) in face-centered cubic metals with crack, which is a common type of structural defects in engineering materials, has been studied by using molecular dynamics simulations in simple bicrystal models. The influences of different parameters such as metal type, temperature, grain boundary structure, and crack geometry have been examined systematically. Three types of microstructural evolution have been identified under different circumstances, namely, crack healing, grain boundary decohesion, and sub-grain formation. The underlying atomistic mechanisms for each type of SCGBM-crack interaction, particularly grain boundary decohesion and crack healing, have also been examined. It is found that crack healing is generally favoured during the SCGBM-crack interaction at relatively high... read less USED (high confidence) C. Li, X. Han, Y. Luan, and J.-G. Li, “Structural origin underlying the effect of cooling rate on solidification point,” Chinese Physics B. 2015. link Times cited: 2 Abstract: Solidification behaviors of liquid aluminum at different coo… read moreAbstract: Solidification behaviors of liquid aluminum at different cooling rates were examined via classical molecular dynamics simulation with an embedded atom method potential. The results demonstrate that solidification point decreases with increasing cooling rate. To explain this phenomenon, solid-like cluster in liquid was analyzed by the structural analysis method of bond order parameters. The results reveal that the size of the largest solid-like cluster in deeply undercooled liquid decreases with the increase of cooling rate, which can provide a structural interpretation to the above phenomenon. read less USED (high confidence) Y. Kim et al., “Effect of a high angle grain boundary on deformation behavior of Al nanopillars,” Scripta Materialia. 2015. link Times cited: 34 USED (high confidence) J. Cho, T. Junge, J. Molinari, and G. Anciaux, “Toward a 3D coupled atomistic and discrete dislocation dynamics simulation: dislocation core structures and Peierls stresses with several character angles in FCC aluminum,” Advanced Modeling and Simulation in Engineering Sciences. 2015. link Times cited: 35 USED (high confidence) Z. Noori, M. Panjepour, and M. Ahmadian, “Study of the effect of grain size on melting temperature of Al nanocrystals by molecular dynamics simulation,” Journal of Materials Research. 2015. link Times cited: 12 Abstract: This research is devoted to the study of the effect of grain… read moreAbstract: This research is devoted to the study of the effect of grain size and structural disorders on the melting behavior of Al nanocrystals under nonequilibrium conditions. The results indicate that T _m is constant and similar to T _m of perfect crystal for nanocrystals of 14 nm and higher. But, by a decrease in the grain size, T _m is significantly reduced. In addition, by further decrease in the size of the grain up to about three times the value of Al-lattice parameter, the behavior of the melt will be similar to the amorphous phase. Since it seems that these behaviors are related to high percentage of grain boundaries in nanocrystalline materials, the structural disorders of the atoms in different regions of nanocrystalline samples are separately studied during heating. The results show that premelting of boundary regions causes the melting process of nanostructure materials to be done within one temperature limit instead of at one temperature point. read less USED (high confidence) S. Wilson and M. Mendelev, “Dependence of solid–liquid interface free energy on liquid structure,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 17 Abstract: The Turnbull relation is widely believed to enable predictio… read moreAbstract: The Turnbull relation is widely believed to enable prediction of solid–liquid interface (SLI) free energies from measurements of the latent heat and the solid density. Ewing proposed an additional contribution to the SLI free energy to account for variations in liquid structure near the interface. In the present study, molecular dynamics (MD) simulations were performed to investigate whether SLI free energy depends on liquid structure. Analysis of the MD simulation data for 11 fcc metals demonstrated that the Turnbull relation is only a rough approximation for highly ordered liquids, whereas much better agreement is observed with Ewing's theory. A modification to Ewing's relation is proposed in this study that was found to provide excellent agreement with MD simulation data. read less USED (high confidence) V. Pisarev, “Nonclassical nucleation kinetics in the crystallization of a supercooled melt,” Russian Journal of Physical Chemistry A. 2014. link Times cited: 11 USED (high confidence) M. Kramer, M. Mendelev, and M. Asta, “Structure of liquid Al and Al67Mg33 alloy: comparison between experiment and simulation,” Philosophical Magazine. 2014. link Times cited: 9 Abstract: We report data on the structure of liquid Al and an Al67Mg33… read moreAbstract: We report data on the structure of liquid Al and an Al67Mg33 alloy obtained from state-of-the-art X-ray diffraction experiments and ab initio molecular dynamics (AIMD) simulations. To facilitate a direct comparison between these data, we develop a method to elongate the AIMD pair correlation function in order to obtain reliable AIMD structure factors. The comparison reveals an appreciable level of discrepancy between experimental and AIMD liquid structures, with the latter being consistently more ordered than the former at the same temperature. The discrepancy noted in this study is estimated to have significant implications for simulation-based calculations of liquid transport properties and solid–liquid interface kinetic properties. read less USED (high confidence) Q.-J. Hong and A. van de Walle, “Solid-liquid coexistence in small systems: A statistical method to calculate melting temperatures.,” The Journal of chemical physics. 2013. link Times cited: 49 Abstract: We propose an efficient and accurate scheme to calculate the… read moreAbstract: We propose an efficient and accurate scheme to calculate the melting point (MP) of materials. This method is based on the statistical analysis of small-size coexistence molecular dynamics simulations. It eliminates the risk of metastable superheated solid in the fast-heating method, while also significantly reducing the computer cost relative to the traditional large-scale coexistence method. Using empirical potentials, we validate the method and systematically study the finite-size effect on the calculated MPs. The method converges to the exact result in the limit of large system size. An accuracy within 100 K in MP is usually achieved when simulation contains more than 100 atoms. Density functional theory examples of tantalum, high-pressure sodium, and ionic material NaCl are shown to demonstrate the accuracy and flexibility of the method in its practical applications. The method serves as a promising approach for large-scale automated material screening in which the MP is a design criterion. read less USED (high confidence) V. Péron-Lührs, A. Jérusalem, F. Sansoz, L. Stainier, and L. Noels, “A two-scale model predicting the mechanical behavior of nanocrystalline solids,” Journal of The Mechanics and Physics of Solids. 2013. link Times cited: 14 USED (high confidence) B. Cheng and A. Ngan, “Thermally induced solid-solid structural transition of copper nanoparticles through direct geometrical conversion.,” The Journal of chemical physics. 2013. link Times cited: 12 Abstract: Molecular dynamics simulations of small Cu nanoparticles usi… read moreAbstract: Molecular dynamics simulations of small Cu nanoparticles using three different interatomic potentials at rising temperature indicate that small nanoparticles can undergo solid-solid structural transitions through a direct geometrical conversion route. The direct geometrical conversion can happen for cuboctahedral nanoparticles, which turn into an icosahedra shape: one diagonal of the square faces contracts, and the faces are folded along the diagonal to give rise to two equilateral triangles. The transition is a kinetic process that cannot be fully explained through an energetic point of view. It has low activation energy and fast reaction time in the simulations. The transition mechanism is via the transmission of shear waves initiated from the particle surface and does not involve dislocation activity. read less USED (high confidence) M. Mendelev and A. King, “The interactions of self-interstitials with twin boundaries,” Philosophical Magazine. 2013. link Times cited: 57 Abstract: A new mechanism of adsorption of self-interstitials onto twi… read moreAbstract: A new mechanism of adsorption of self-interstitials onto twin boundaries (TB) in face-centred cubic (fcc) metals is identified using molecular dynamics simulations. In this mechanism, self-interstitials are arranged in the twin boundary plane forming a previously unknown kind of self-interstitial cluster. The self-interstitial cluster in the twin boundary is bounded by lines of atoms under high hydrostatic pressure while the pressure inside the cluster is much smaller. The atoms in the middle of the cluster have hcp short range order rather than fcc. However, if a new self-interstitial cluster forms in the middle of a pre-existing one, then the atoms in the middle of the new cluster will have regular twin boundary coordination. As a consequence of the formation of self-interstitial clusters inside each other, TB can be powerful, non-saturating sinks for self-interstitials. read less USED (high confidence) V. Pisarev, “Determination of free energy of the crystal-melt interface,” High Temperature. 2012. link Times cited: 4 USED (high confidence) J. Hoyt and A. A. Potter, “A Molecular Dynamics Simulation Study of the Cavitation Pressure in Liquid Al,” Metallurgical and Materials Transactions A. 2012. link Times cited: 15 USED (high confidence) G. Norman and V. Pisarev, “Molecular dynamics analysis of the crystallization of an overcooled aluminum melt,” Russian Journal of Physical Chemistry A. 2012. link Times cited: 12 USED (high confidence) A. M. Nieves, C. Y. Chuang, and T. Sinno, “Inherent structure analysis of defect thermodynamics and melting in silicon,” Molecular Simulation. 2012. link Times cited: 3 Abstract: The inherent structure landscapes associated with various ty… read moreAbstract: The inherent structure landscapes associated with various types of microstructure in crystalline silicon are probed within the empirical Environment-Dependent Interatomic Potential. Inherent structure sampling is carried out using a combination of molecular dynamics simulation and energy minimisation. Several isolated point defect clusters are analysed first, followed by the more complex phenomenon of crystal melting. In the latter case, both homogeneous and surface-driven melting phenomena are considered, and selected comparisons are made to face-centered cubic aluminium represented by an embedded-atom potential. It is shown that inherent structure theory provides a useful framework for consolidating various aspects of microstructure thermodynamics in crystals, and also naturally allows for the treatment of more complex situations such as applied mechanical fields. read less USED (high confidence) M. Mendelev, “Molecular dynamics simulation of solidification and devitrification in a one-component system,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 16 Abstract: A specially designed semi-empirical potential of the Finnis–… read moreAbstract: A specially designed semi-empirical potential of the Finnis–Sinclair type was used to simulate the phase transformation in a disordered one-component system. The potential provides that the face-centered cubic (fcc) phase is the most stable phase in the system below the melting temperature, Tm; however, the potential does not lead to the fcc nucleation during molecular dynamics (MD) simulation, allowing studying the liquid–glass transformation. The potential also allows studying the fcc-liquid and fcc-glass interface migration. It was found that the liquid–glass transformation described by this potential is of the first order. The Wilson–Frenkel theory of the solid–liquid interface (SLI) migration satisfactory describes the results of the MD simulation in the temperature interval from 0.55Tm to Tm while the Broughton–Gilmer–Jackson theory is less accurate in describing the temperature dependence of the SLI velocity in the same temperature interval. Below 0.55Tm, the results of the MD simulation strongly depend on how the disordered phase model was prepared and none of the existing theories is capable of reproducing the temperature dependence of the interface velocity. read less USED (high confidence) K. Kang, J. Wang, and I. Beyerlein, “Atomic structure variations of mechanically stable fcc-bcc interfaces,” Journal of Applied Physics. 2012. link Times cited: 77 Abstract: It has recently been shown that under severe plastic deforma… read moreAbstract: It has recently been shown that under severe plastic deformation processing bi-metal fcc/bcc composites develop a mechanically stable heterophase interface that joins the {112}fcc//{112}bcc planes in the Kurdjumov-Sachs orientation relationship. In this article, we study variations in the relaxed equilibrium atomic structure of this interface with changes in fcc stacking fault energy (SFE) and lattice mismatch between the two crystals. Using molecular statics/dynamics simulations for three fcc/bcc systems, Cu-Nb, Al-Fe, and Al-Nb, we find that the number of distinct sets of intrinsic interfacial dislocations and their core structures vary significantly among these three systems. The impact of these atomic-scale structural differences on interfacial properties is demonstrated through their interactions with point defects. The interfaces studied here are shown to exhibit a wide variation in ability, ranging from being a poor to an excellent sink for vacancies. read less USED (high confidence) A. M. Nieves and T. Sinno, “An enthalpy landscape view of homogeneous melting in crystals.,” The Journal of chemical physics. 2011. link Times cited: 10 Abstract: A detailed analysis of homogeneous melting in crystalline ma… read moreAbstract: A detailed analysis of homogeneous melting in crystalline materials modeled by empirical interatomic potentials is presented using the theory of inherent structures. We show that the homogeneous melting of a perfect, infinite crystalline material can be inferred directly from the growth exponent of the inherent structure density-of-states distribution expressed as a function of formation enthalpy. Interestingly, this growth is already established by the presence of very few homogeneously nucleated point defects in the form of Frenkel pairs. This finding supports the notion that homogeneous melting is appropriately defined in terms of a one-phase theory and does not require detailed consideration of the liquid phase. We then apply this framework to the study of applied hydrostatic compression on homogeneous melting and show that the inherent structure analysis used here is able to capture the correct pressure-dependence for two crystalline materials, namely silicon and aluminum. The coupling between the melting temperature and applied pressure arises through the distribution of formation volumes for the various inherent structures. read less USED (high confidence) M. Mendelev, M. J. Rahman, J. Hoyt, and M. Asta, “Molecular-dynamics study of solid–liquid interface migration in fcc metals,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 85 Abstract: In order to establish a link between various structural and … read moreAbstract: In order to establish a link between various structural and kinetic properties of metals and the crystal–melt interfacial mobility, free-solidification molecular-dynamics simulations have been performed for a total of nine embedded atom method interatomic potentials describing pure Al, Cu and Ni. To fully explore the space of materials properties three new potentials have been developed. The new potentials are based on a previous description of Al, but in each case the liquid structure, the melting point and/or the latent heat are varied considerably. The kinetic coefficient, μ, for all systems has been compared with several theoretical predictions. It is found that at temperatures close to the melting point the magnitude of μ correlates well with the value of the diffusion coefficient in the liquid. read less USED (high confidence) M. Mendelev and M. Kramer, “Reliability of methods of computer simulation of structure of amorphous alloys,” Journal of Applied Physics. 2010. link Times cited: 16 Abstract: We took a model created by the molecular dynamics (MD) simul… read moreAbstract: We took a model created by the molecular dynamics (MD) simulation with a semiempirical potential as a target system and explored how its amorphous structure and a few other properties depend on the simulation method. We found that if the cooling rate is too high, 1013–1014 K/s, the system has no time to adjust its structure to the change in temperature/density. Since this cooling corresponds to a typical ab initio MD simulation, this brings into doubt that an equilibrium glass structure can be obtained using ab initio MD simulation. We also used the target partial pair correlation functions (PPCFs) to explore a possibility to create the atomic models from diffraction data alone. We were able to create models with the PPCFs, which nearly coincided with the target ones. Nevertheless, we found that the potential energy of the quenched states and the distribution of the Voronoi polyhedra in the models created from PPCFs were different than the target quantities. This study shows that reverse Monte Carlo techn... read less USED (high confidence) M. Mendelev, M. J. Kramer, R. T. Ott, and D. Sordelet, “Molecular dynamics simulation of diffusion in supercooled Cu–Zr alloys,” Philosophical Magazine. 2009. link Times cited: 60 Abstract: Molecular dynamics (MD) simulations of diffusion in Cu–Zr al… read moreAbstract: Molecular dynamics (MD) simulations of diffusion in Cu–Zr alloys in their liquid and supercooled liquid states were performed using a recently developed Finnis–Sinclair many-body interatomic potential. To help assess how well the interatomic potential describes the energetics of the Cu–Zr system, the liquid structure determined by MD simulations was compared with wide-angle X-ray scattering measurements of the liquid structure for a Cu64.5Zr35.5 alloy. Diffusion was examined as a function of composition, pressure and temperature. The simulations reveal that the diffusion exhibits strong compositional dependence, with both species exhibiting minimum diffusivities at ∼70% Cu. Moreover, the MD simulations show that the activation volumes for Zr and Cu atoms exhibit a maximum near 70% Cu. Evidence is obtained that the glass transition temperature also changes strongly with composition, thereby contributing to the diffusion behaviour. The relationship between this minimum in diffusion and the apparent best glass-forming composition in the Cu–Zr system is discussed. read less USED (high confidence) M. Mendelev, R. Ott, M. Heggen, M. Feuerebacher, M. Kramer, and D. Sordelet, “Deformation behavior of an amorphous Cu64.5Zr35.5 alloy: A combined computer simulation and experimental study,” Journal of Applied Physics. 2008. link Times cited: 21 Abstract: Molecular dynamics (MD) simulations were performed to examin… read moreAbstract: Molecular dynamics (MD) simulations were performed to examine the temperature-dependent elastic properties and high-temperature deformation behavior of a Cu64.5Zr35.5 amorphous alloy. From the simulations we find that the elastic constants of the amorphous solid and supercooled liquid exhibit an approximately linear temperature dependence. The predicted temperature dependence of the Young’s modulus for the amorphous solid obtained from the MD simulations is in good agreement with experimental measurements using dynamic mechanical analysis. Furthermore, the high-temperature plastic deformation behavior determined by MD simulations is qualitatively in good agreement with results from plastic deformation experiments performed on 1 mm diameter Cu64.5Zr35.5 metallic glass rods at 698 K. Notably, the MD simulations reveal that the flow softening regime of the stress-strain curve corresponds to an increase in the free volume in the atomic structure. Moreover, the simulations indicate that the atomic mobility sig... read less USED (high confidence) L. Wu, Y. Zhu, H. Wang, and M. Li, “Crystal–melt coexistence in fcc and bcc metals: a molecular-dynamics study of kinetic coefficients,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 5 Abstract: As a sequel to the previous paper on the calculation of the … read moreAbstract: As a sequel to the previous paper on the calculation of the crystal–melt interface free energy (2021 Materialia 15 100962), here we report the results on the kinetic coefficients using molecular dynamics simulations performed on six fcc metals and four bcc metals with the intention to compare the crystal structural influence. We found that the calculated kinetic coefficients are well described by the model by Broughton, Gilmer and Jackson (1982 Phys. Rev. Lett. 49 1496), and in particular, they exhibit varying degrees of anisotropy. We reveal that the anisotropies are related to the fluctuation of the crystal–melt interfaces, which causes the increase of the actual interface area in melting or solidification. The kinetic coefficients always display asymmetry between the solidification and melting process, and the difference is much more pronounced for the (111) interfaces in fcc metals which have the highest anisotropy. We found that the atomic mechanisms of the kinetic behaviors of these interfaces are closely related to the formation of twin-crystal domains during solidification, which delays the solidification process and consequently causes a decrease in the calculated kinetic coefficients. read less USED (high confidence) S. Huang, J. Wang, and C. Zhou, “Deformation of Heterogeneous Nanocrystalline Lamella with a Preexisting Crack,” JOM. 2017. link Times cited: 4 USED (high confidence) C. Li, X. Han, Y. Luan, and J.-G. Li, “Abnormal breakdown of Stokes–Einstein relation in liquid aluminium*,” Chinese Physics B. 2017. link Times cited: 5 Abstract: We present the results of systematic molecular dynamics simu… read moreAbstract: We present the results of systematic molecular dynamics simulations of pure aluminium melt with a well-accepted embedded atom potential. The structure and dynamics were calculated over a wide temperature range, and the calculated results (including the pair correlation function, self-diffusion coefficient, and viscosity) agree well with the available experimental observations. The calculated data were used to examine the Stokes–Einstein relation (SER). The results indicate that the SER begins to break down at a temperature (~1090 K) which is well above the equilibrium melting point (912.5 K). This high-temperature breakdown is confirmed by the evolution of dynamics heterogeneity, which is characterised by the non-Gaussian parameter α . The maximum value of α , α , increases at an accelerating rate as the temperature falls below . The development of α was found to be related to the liquid structure change evidenced by local five-fold symmetry. Accordingly, we suggest that this high-temperature breakdown of SER has a structural origin. The results of this study are expected to make researchers reconsider the applicability of SER and promote greater understanding of the relationship between dynamics and structure. read less USED (high confidence) T. Junge, “Modelling Plasticity in Nanoscale Contact.” 2014. link Times cited: 6 Abstract: The problem of mechanical contact is a truly multiscale one.… read moreAbstract: The problem of mechanical contact is a truly multiscale one. Atomistic effects that violate continuum theory dominate the deformations of contacting asperities, while the interactions between distant asperities occur through long-range elasticity. This thesis concentrates on the numerical modelling of nanoscale frictional contact between crystalline metals by using both single-scale atomistic methods and improving concurrent multiscale methods. A novel approach to quantify frictional work and the energy associated with plastic activity in \md simulations is presented. In combination with a statistical criterion to determine the significance of simulation box size, microstructure and sliding rate effects on the frictional quantities such as the friction coefficient and stored plastic energies, the method is used in a large parametric molecular dynamics study of single-asperity nanoscratch on monocrystalline and polycrystalline aluminium substrates. Some fundamental differences in the friction mechanisms between monocrystalline and polycrystalline substrates are presented. The study shows the limitations of single-scale modelling and highlights the importance of developing appropriate multiscale methods for nanoscale plasticity. One such method is the Coupled Atomistics and Discrete Dislocations (CADD), which previously only existed for two-dimensional problems. A three-dimensional version of the CADD method is presented theoretically as well as a detailed practical road map for its efficient implementation. The foundations of three-dimensional CADD are presented using practical test cases. CADD avoids ghost forces at the coupling interfaces through displacement-coupling. I reveal that such displacement-coupling methods generally exhibit an inherent dynamic instability which makes them particularly ill suited for finite temperature calculations, despite their wide use. The instability is analysed in detail. Multiple remedies to manage it are discussed and a fundamental solution to the underlying problem is presented in the form of a new coupling method. read less USED (low confidence) S. M. Handrigan and S. Nakhla, “Generation of viable nanocrystalline structures using the melt-cool method: the influence of force field selection,” Philosophical Magazine. 2023. link Times cited: 0 USED (low confidence) Y. Jin et al., “Design and catalytic performance of Cu/?-Al2O3?for electrocatalytic methanol oxidation reaction by using density functional theory and molecular dynamics,” Applied Surface Science. 2023. link Times cited: 0 USED (low confidence) W. Velilla-Díaz and H. R. Zambrano, “Effects of Grain Boundary Misorientation Angle on the Mechanical Behavior of Al Bicrystals,” Nanomaterials. 2023. link Times cited: 0 Abstract: This research article explores the effect of grain boundary … read moreAbstract: This research article explores the effect of grain boundary (GB) misorientation on the mechanical behavior of aluminum (Al) bicrystals by means of molecular dynamics (MD) simulations. The effect of GB misorientation on the mechanical properties, fracture resistance, and crack propagation are evaluated under monotonic and cyclic load conditions. The J-integral and the crack tip opening displacement (CTOD) are assessed to establish the effect of the GB misorientation angle on the fracture resistance. The simulations reveal that the misorientation angle plays a significant role in the mechanical response of Al bicrystals. The results also evidence a gradual change in the mechanical behavior from brittle to ductile as the misorientation angle is increased. read less USED (low confidence) G. S. Jung, S. Lee, and J. Y. Choi, “Data Distillation for Neural Network Potentials toward Foundational Dataset,” ArXiv. 2023. link Times cited: 0 Abstract: Machine learning (ML) techniques and atomistic modeling have… read moreAbstract: Machine learning (ML) techniques and atomistic modeling have rapidly transformed materials design and discovery. Specifically, generative models can swiftly propose promising materials for targeted applications. However, the predicted properties of materials through the generative models often do not match with calculated properties through ab initio calculations. This discrepancy can arise because the generated coordinates are not fully relaxed, whereas the many properties are derived from relaxed structures. Neural network-based potentials (NNPs) can expedite the process by providing relaxed structures from the initially generated ones. Nevertheless, acquiring data to train NNPs for this purpose can be extremely challenging as it needs to encompass previously unknown structures. This study utilized extended ensemble molecular dynamics (MD) to secure a broad range of liquid- and solid-phase configurations in one of the metallic systems, nickel. Then, we could significantly reduce them through active learning without losing much accuracy. We found that the NNP trained from the distilled data could predict different energy-minimized closed-pack crystal structures even though those structures were not explicitly part of the initial data. Furthermore, the data can be translated to other metallic systems (aluminum and niobium), without repeating the sampling and distillation processes. Our approach to data acquisition and distillation has demonstrated the potential to expedite NNP development and enhance materials design and discovery by integrating generative models. read less USED (low confidence) H. Duan, Y. Yang, Y. Ma, and P. He, “The growth progress of Nb films on Cu: a molecular dynamics simulation,” Radiation Detection Technology and Methods. 2023. link Times cited: 0 USED (low confidence) J. Shi et al., “Double-Shock Compression Pathways from Diamond to BC8 Carbon.,” Physical review letters. 2023. link Times cited: 0 Abstract: Carbon is one of the most important elements for both indust… read moreAbstract: Carbon is one of the most important elements for both industrial applications and fundamental research, including life, physics, chemistry, materials, and even planetary science. Although theoretical predictions on the transition from diamond to the BC8 (Ia3[over ¯]) carbon were made more than thirty years ago, after tremendous experimental efforts, direct evidence for the existence of BC8 carbon is still lacking. In this study, a machine learning potential was developed for high-pressure carbon fitted from first-principles calculations, which exhibited great capabilities in modeling the melting and Hugoniot line. Using the molecular dynamics based on this machine learning potential, we designed a thermodynamic pathway that is achievable for the double shock compression experiment to obtain the elusive BC8 carbon. Diamond was compressed up to 584 GPa after the first shock at 20.5 km/s. Subsequently, in the second shock compression at 24.8 or 25.0 km/s, diamond was compressed to a supercooled liquid and then solidified to BC8 in around 1 ns. Furthermore, the critical nucleus size and nucleation rate of BC8 were calculated, which are crucial for nano-second x-ray diffraction measurements to observe BC8 carbon during shock compressions. The key to obtaining BC8 carbon lies in the formation of liquid at a sufficient supercooling. Our work provides a feasible pathway by which the long-sought BC8 phase of carbon can be reached in experiments. read less USED (low confidence) Y.-qi Jiang, J. Lv, W. He, and P. Peng, “Shed light on new growth pattern in rapid solidified Ta metal: MD simulation, DFT calculation and CALYPSO search,” Vacuum. 2023. link Times cited: 3 USED (low confidence) A. Swamy, D. Dolce, and P. Choudhury, “Atomistic Insight into Dendrite Growth Orientation Transition in Al-Cu Alloy,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) J. Nan, X. He, X. Qu, J. Wei, and Z. Zhang, “Effect of Ar pressure on the wettability of copper droplet on graphite substrate by molecular dynamics simulation,” Applied Physics A. 2023. link Times cited: 0 USED (low confidence) M. Tahani, E. Postek, and T. Sadowski, “Investigating the Influence of Diffusion on the Cohesive Zone Model of the SiC/Al Composite Interface,” Molecules. 2023. link Times cited: 1 Abstract: Modeling metal matrix composites in finite element software … read moreAbstract: Modeling metal matrix composites in finite element software requires incorporating a cohesive zone model (CZM) to represent the interface between the constituent materials. The CZM determines the behavior of traction–separation (T–S) in this region. Specifically, when a diffusion zone is formed due to heat treatment, it becomes challenging to determine experimentally the equivalent mechanical properties of the interface. Additionally, understanding the influence of heat treatment and the creation of a diffusion zone on the T–S law is crucial. In this study, the molecular dynamics approach was employed to investigate the effect of the diffusion region formation, resulting from heat treatment, on the T–S law at the interface of a SiC/Al composite in tensile, shear, and mixed-mode loadings. It was found that the formation of a diffusion layer led to an increase in tensile and shear strengths and work of separation compared with the interfaces without heat treatment. However, the elastic and shear moduli were not significantly affected by the creation of the diffusion layer. Moreover, the numerical findings indicated that the shear strength in the diffusion region was higher when compared with the shear strength of the {111} slip plane within the fcc aluminum component of the composite material. Therefore, in the diffusion region, crack propagation did not occur in the pure shear loading case; however, shear sliding was observed at the aluminum atomic layers. read less USED (low confidence) Z. Hao, X. Xu, and Y. Fan, “Deformation mechanisms in the cutting process of SiCp/Al composites using the molecular dynamics (MD) approach,” Proceedings of the Institution of Mechanical Engineers, Part B: Journal of Engineering Manufacture. 2023. link Times cited: 0 Abstract: In order to clarify the reasons for the emergence of problem… read moreAbstract: In order to clarify the reasons for the emergence of problems such as high machining difficulty and poor machining surface quality encountered in the machining process of SiCp/Al composites, this paper presents a simulation study of the deformation mechanism occurring during the cutting process of SiCp/Al (SiC particle-reinforced Al matrix composites) by using the molecular dynamics (MD) method. The mechanism of strengthening and hardening occurring during cutting of SiCp/Al composites is discussed in terms of the slip expansion of dislocations and the interaction between different types of dislocations. It is found that the presence of SiC balls hinders the slip and expansion of dislocations in the Al (aluminum) matrix, resulting in the strengthening of the Al matrix between the SiC (silicon carbide) balls, mainly because the dislocations in the workpiece interact with each other during the cutting process to form dislocation tangles, stacking layer dislocations and dislocation locks, which hinder the slip and expansion of dislocations and thus increase the strength of the Al matrix. By analyzing the stress distribution inside the workpiece during the cutting process, it is found that the stress inside the workpiece is mainly concentrated in the area where the tool flank face is in contact with the workpiece, which is the main reason for the occurrence of working hardening in SiCp/Al composites. read less USED (low confidence) A. Kartamyshev, A. Lipnitskii, V. Maksimenko, A. V. Vyazmin, I. Nelasov, and D. Poletaev, “N-body potential for simulating lattice defects and diffusion in copper,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) A. S. Al-Awad, L. Batet, and L. Sedano, “Parametrization of embedded-atom method potential for liquid lithium and lead-lithium eutectic alloy,” Journal of Nuclear Materials. 2023. link Times cited: 0 USED (low confidence) Y.-qi Jiang and P. Peng, “The influence of Engineering strain rates on the atomic structure, crack propagation and cavitation formation in Al based metallic glass,” Journal of Materials Research and Technology. 2023. link Times cited: 2 USED (low confidence) Y. Zhang et al., “Study of Nanoscale Microprotrusions on Metal Electrode Surfaces Under High Electric Fields,” IEEE Transactions on Plasma Science. 2023. link Times cited: 0 Abstract: Microprotrusions under high electric fields are considered t… read moreAbstract: Microprotrusions under high electric fields are considered to be sources of metal vapor and microplasma on metal electrode surfaces and may even initiate vacuum breakdown in vacuum gaps. The mechanism of the phenomena has been studied for a long time. However, the dynamic evolution processes of microprotrusions under high electric fields considering the influence of the material properties are still not clear. The objective of this article is to study the dynamic evolution processes of the nanoscale microprotrusions on Cu and Cr electrode surfaces under high electric fields based on atomistic modeling. With considering the electron emission heating, surface charge, Coulomb, and electric field forces, a 3-D numerical model is established by coupling molecular dynamics (MD) and finite difference method (FDM), for simulating the dynamic evolution processes of the microprotrusions under high electric field. Furthermore, the influence of material properties on the dynamic evolution processes is discussed and compared between Cu and Cr. The simulation results show that the heating effect of the electron emission induced by an intense electric field could lead the microprotrusions to localized melting and subsequent elongation and may finally generate metal vapor in the vacuum gap. In addition, the material properties have a significant influence on the field-induced dynamic evolution processes of microprotrusions. read less USED (low confidence) P. Lafourcade et al., “Robust crystal structure identification at extreme conditions using a density-independent spectral descriptor and supervised learning,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) A. Allera, A. Goryaeva, P. Lafourcade, J. Maillet, and M. Marinica, “Neighbors Map: An efficient atomic descriptor for structural analysis,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) W. Wciślik and S. Lipiec, “Voids Development in Metals: Numerical Modelling,” Materials. 2023. link Times cited: 1 Abstract: The article is a continuation of two previous review papers … read moreAbstract: The article is a continuation of two previous review papers on the fracture mechanism of structural metals through the nucleation, growth and coalescence of voids. In the present paper, the literature on the numerical modelling of void nucleation and development has been reviewed. The scope of the work does not include porous material models and their numerical implementation. As part of the discussion on void initiation, nucleation around second phase particles and nucleation as an effect of the discontinuity of the crystal structure were discussed separately. The basic void cell models, finite element method (FEM) models of periodically distributed particles/voids and models based on the results of the observations of the actual microstructure of materials have been characterised. Basic issues related to the application of the cohesive approach in void nucleation modelling have been considered. A separate issue is the characteristics of atomistic simulations and peridynamic modelling, which have been developed in recent years. Numerical approaches to modelling the growth and coalescence of voids are described, with particular emphasis on the influence of the stress state and strain localisation. Basic conclusions from the simulation are presented, pointing to the contribution of FEM modelling to the understanding of microstructural phenomena leading to ductile fracture. read less USED (low confidence) O. Klimanova, T. Miryashkin, and A. Shapeev, “Accurate melting point prediction through autonomous physics-informed learning,” Physical Review B. 2023. link Times cited: 0 Abstract: We present an algorithm for computing melting points by auto… read moreAbstract: We present an algorithm for computing melting points by autonomously learning from coexistence simulations in the NPT ensemble. Given the interatomic interaction model, the method makes decisions regarding the number of atoms and temperature at which to conduct simulations, and based on the collected data predicts the melting point along with the uncertainty, which can be systematically improved with more data. We demonstrate how incorporating physical models of the solid-liquid coexistence evolution enhances the algorithm's accuracy and enables optimal decision-making to effectively reduce predictive uncertainty. To validate our approach, we compare the results of 20 melting point calculations from the literature to the results of our calculations, all conducted with same interatomic potentials. Remarkably, we observe significant deviations in about one-third of the cases, underscoring the need for accurate and reliable algorithms for materials property calculations. read less USED (low confidence) I. D. Arellano-Ramírez, E. A. H. Ladino, and E. Restrepo‐Parra, “Effect of aluminum nanoparticle size on phase transitions: a molecular dynamics study,” Indian Journal of Physics. 2023. link Times cited: 0 USED (low confidence) M. P. Hazarika, A. Tripathi, and S. N. Chakraborty, “Two-temperature molecular dynamics simulation study of copper thin film irradiation with femtosecond and picosecond laser pulses,” Journal of Laser Applications. 2023. link Times cited: 0 Abstract: Metal targets irradiated with laser pulses have a wide range… read moreAbstract: Metal targets irradiated with laser pulses have a wide range of applications in thin film preparation, nanomaterial synthesis, bio-medical imaging, and metal ablation. Here, using two-temperature model based molecular dynamics simulation, we investigate laser mediated ablation in copper. Ablation of the film starts with the formation of voids within it. This void forming mechanism at low laser fluences ([Formula: see text] mJ/cm[Formula: see text]) is studied using both picosecond and femtosecond pulses. At the same fluence, shorter laser pulse transfers more energy to the atoms generating temperatures greater than the melting temperature of the crystal. This increases the kinetic energy of the atoms and they start vibrating with different velocities. If these vibrations cross a threshold of 5 Å per picosecond (500 m/s), voids and faults start appearing in the system. At the same fluence, higher concentration of voids are also created at a faster rate with the femtosecond pulse. read less USED (low confidence) Z. Yu et al., “Phase transformation behavior of aluminum under high hydrostatic pressure: A molecular dynamics study,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) X. Gao, N. Li, Z. Song, K. Wu, Y. Cheng, and B.-yang Xiao, “Atomic structure evolution and linear regression fitting models for pre-breakdown electric field strength of FCC, BCC and HCP metal nano-emitters under high electric field from PIC-ED–MD simulations,” Journal of Physics D: Applied Physics. 2023. link Times cited: 1 Abstract: Multi-scale and multi-physics simulations are carried out fo… read moreAbstract: Multi-scale and multi-physics simulations are carried out for nano-emitters consisting of FCC (Al, Cu and Au), BCC (V, Mo and W) and HCP (Ti, Zn and Zr) metals, using hybrid electrodynamics coupled with molecular dynamics-particle in cell simulations (PIC-ED–MD). We show that the tilting of the nano-emitter at low temperature and small electric field (E-field) is mainly caused either by the movement of partial dislocations at the apex of the nanotip or by the elastic local distortions of atomic registries away from their ideal lattice sites (FCC/BCC/HCP). At high E-field, the intense resistive heating due to the strong electron emission leads to the direct melting of the apex of nano-emitters. For nano-emitters consisting of low melting point metals such as Al, Zn and Au, the thermal runaway is driven by the elongation, thinning and necking of the molten region. Meanwhile, the elongation, thinning and sharpening produce the nano-protrusion at the apex of metal nano-emitters, and the detachment of atoms or atomic clusters from the nano-protrusion mainly contributes to the thermal runaway event for refractor metals such as Ti, Zr, Mo and W. The critical E-field strength of metal nano-emitters is found to be strongly correlated with structural parameters (atomic coordination number of liquid and equilibrium lattice constant), thermodynamic quantities (cohesive energy and enthalpy of evaporation) and phase transition temperatures (melting point and boiling point). These correlations enable us to establish either single-variable linear fitting models or multi-variable linear regression models to predict the critical E-field value for metal nano-emitters with good credibility. read less USED (low confidence) N. Kondratyuk, R. Ryltsev, V. Ankudinov, and N. Chtchelkatchev, “First-principles calculations of the viscosity in multicomponent metallic melts: Al-Cu-Ni as a test case,” Journal of Molecular Liquids. 2023. link Times cited: 4 USED (low confidence) C. Guarda, B. Faria, N. Silvestre, and J. C. C. Lopes, “Influence of Matrix Recrystallization and Nanofiller Porosity on the Interfacial Properties of Holey Graphene-Aluminium Nanocomposites,” Composite Structures. 2023. link Times cited: 2 USED (low confidence) L. Safina and E. A. Rozhnova, “MOLECULAR DYNAMICS SIMULATION OF THE DEFORMATION BEHAVIOR OF THE GRAPHENE/Al COMPOSITE,” Journal of Structural Chemistry. 2023. link Times cited: 2 USED (low confidence) I. Chesser, R. K. Koju, A. Vellore, and Y. Mishin, “Atomistic modeling of metal-nonmetal interphase boundary diffusion,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) B. Medina and R. Fernández, “Material Behavior Around the Fsw/Fsp Tool Described by Molecular Dynamics,” SSRN Electronic Journal. 2022. link Times cited: 0 Abstract: Friction stir welding and processing (FSW/FSP) involves seve… read moreAbstract: Friction stir welding and processing (FSW/FSP) involves severe plastic deformation of metals or polymers at high temperature around a rotating tool. The material’s flow is usually modelled by FEM using a complex combination of thermomechanical and friction models. However, the description of the behavior of the first atomic layers in contact with the tool cannot be undertaken by continuum mechanics modelling such as FEM. Among the available simulation techniques, molecular dynamics (MD) where friction and heat are generated by material layers’ relative movement, allows the simulation of the behavior of the first atomic layers of the work piece in contact with the tool. In this work, in aluminum, the effect of temperature and advancing and rotating speeds on FSW/FSP material’s flow and crystallography in the vicinity of the tool are discussed. The data analyzed demonstrate that a normalization of the weld-pitch parameter by the pin radius allows obtaining reliable heat input, momentum, and temperatures typical of this critical region in the FSW/FSP processes by MD. The results show that MD provide reliable data as an input for the FEM in a multiscale FSW/FSP modelling. read less USED (low confidence) X. Chen, W. Fan, W. Jiang, D. Lin, Z. Wang, and S. Jiang, “Effects of Pressure on Homogeneous Nucleation and Growth during Isothermal Solidification in Pure Al: A Molecular Dynamics Simulation Study,” Metals. 2022. link Times cited: 1 Abstract: Effects of different pressures on the isothermal-solidificat… read moreAbstract: Effects of different pressures on the isothermal-solidification process of pure Al were studied by molecular dynamics (MD) simulation using the embedded-atom method (EAM). Al was first subjected to a rapid-cooling process, and then it was annealed under different pressures conditions. Mean first-passage times (MFPT) method, Johnson-Mehl-Avrami (JMA) law, and X-ray diffraction (XRD) simulation analysis method were used to qualify the solidification- kinetic processing. Nucleation rate, critical-nucleus size, Avrami exponent, growth exponent, and crystallite size were calculated. Results show that the nucleation rate increases as the pressure increases. The change of critical-nucleation size is not obvious as the pressure increases. With the pressure increasing, growth exponent decreases, indicative of decreased grain-growth rate. It was also found that with the pressure increasing, the Avrami exponent decreases, indicating that the increased pressure has an effect on growth modes during solidification, which changes from three-dimensional growth to one-dimensional growth. Results of XRD simulation shows that with pressure increasing, crystallite size decreases. read less USED (low confidence) Z. Hou et al., “Effect of Twin Spacing on the Mechanical Behavior and Deformation Mechanism of Nanotwinned Al,” SSRN Electronic Journal. 2022. link Times cited: 3 USED (low confidence) A. B. Sivak, D. N. Demidov, and P. A. Sivak, “Diffusion Characteristics of Self-Point Defects in Copper: Molecular Dynamic Study,” Physics of Atomic Nuclei. 2022. link Times cited: 1 USED (low confidence) S. Ahmad, T. Brink, C. Liebscher, and G. Dehm, “Microstates and defects of incoherent Σ3 [111] twin boundaries in aluminum,” Acta Materialia. 2022. link Times cited: 4 USED (low confidence) A. Antropov and V. Stegailov, “Helium bubbles diffusion in aluminum: influence of gas pressure,” Journal of Nuclear Materials. 2022. link Times cited: 3 USED (low confidence) Z. Liu, Y. Li, P. Peng, and K. Dong, “An exact measurement of nucleation incubation times in isothermal crystallizations of liquid metal Al via configuration heredity,” Journal of Crystal Growth. 2022. link Times cited: 0 USED (low confidence) D. Louzguine-Luzgin, “Structural Changes in Metallic Glass-Forming Liquids on Cooling and Subsequent Vitrification in Relationship with Their Properties,” Materials. 2022. link Times cited: 13 Abstract: The present review is related to the studies of structural c… read moreAbstract: The present review is related to the studies of structural changes observed in metallic glass-forming liquids on cooling and subsequent vitrification in terms of radial distribution function and its analogues. These structural changes are discussed in relationship with liquid’s properties, especially the relaxation time and viscosity. These changes are found to be directly responsible for liquid fragility: deviation of the temperature dependence of viscosity of a supercooled liquid from the Arrhenius equation through modification of the activation energy for viscous flow. Further studies of this phenomenon are necessary to provide direct mathematical correlation between the atomic structure and properties. read less USED (low confidence) X. Chen et al., “Effects of Cooling Rate on the Solidification Process of Pure Metal Al: Molecular Dynamics Simulations Based on the MFPT Method,” Metals. 2022. link Times cited: 3 Abstract: Isothermal solidification process of pure metal Al was studi… read moreAbstract: Isothermal solidification process of pure metal Al was studied by molecular dynamics (MD) simulation using EAM potential. The effects of different cooling rates on the isothermal solidification process of metallic Al were studied. Al was first subjected to a rapid cooling process, and then it was annealing under isothermal conditions. The mean first-passage times (MFPT) method and Johnson-Mehl-Avrami (JMA) law were used to qualify the solidification kinetic processing, and the nucleation rate, critical nucleus size, Avrami exponent and growth exponent of grains were calculated. Results show that the nucleation rate and critical size decrease as the cooling rate increases. Also, an increase in the cooling rate leads to the increase of grain growth rate. At all investigated cooling rates, nucleation and growth processes are in the typical three-dimensional growth mode. read less USED (low confidence) A. Y. Morkina, D. Bachurin, S. Dmitriev, A. S. Semenov, and E. Korznikova, “Modulational Instability of Delocalized Modes in fcc Copper,” Materials. 2022. link Times cited: 4 Abstract: Delocalized nonlinear vibrational modes (DNVMs) are exact so… read moreAbstract: Delocalized nonlinear vibrational modes (DNVMs) are exact solutions of the equations of motion, and therefore, DNVMs exist at any vibration amplitude and do not depend on interaction potentials. For the first time, modulation instability of four one-component three-dimensional DNVMs is studied in a single crystal of fcc copper with the use of methods of molecular dynamics. DNVMs frequencies, evolution of stresses, kinetic and potential energies, and heat capacity depending on the oscillation amplitudes are analyzed. It is found that all four DNVMs are characterized by a hard-type anharmonicity. Modulation instability of DNVMs results in a formation of chaotic discrete breathers (DBs) with frequency above the upper edge of the phonon spectrum of the crystal. The lifetime of chaotic DBs is found to be in the range of 30–100 ps. At low-oscillation frequencies, longer-lived DBs are formed. The growth of modulation instability leads to an increase in mechanical stresses and a decrease in the heat capacity of the crystal. The results obtained in this work enrich our understanding of the influence of the modulation instability of DNVMs on the properties of metals. read less USED (low confidence) M. Wagih and C. Schuh, “Learning Grain-Boundary Segregation: From First Principles to Polycrystals.,” Physical review letters. 2022. link Times cited: 14 Abstract: The segregation of solute atoms at grain boundaries (GBs) ca… read moreAbstract: The segregation of solute atoms at grain boundaries (GBs) can strongly impact the structural and functional properties of polycrystals. Yet, due to the limited availability of simulation tools to study polycrystals at the atomistic scale (i.e., interatomic potentials), there is a minimal understanding of the variation of solute segregation tendencies across the very complex space of GB microenvironments and the large range of alloys in which it can occur. Here, we develop an algorithmic framework that can directly learn the full spectrum of segregation energies for a metal solute atom in a metal polycrystal from ab initio methods, bypassing the need for alloy interatomic potentials. This framework offers a pathway to a comprehensive catalog of GB solute segregation with quantum accuracy, for the entire alloy space. As an initial demonstration in this pursuit, we build an extensive GB segregation database for aluminum-based alloys across the periodic table, including dozens of alloys for which there are substantially no prior data. read less USED (low confidence) C. Guarda, B. Faria, N. Silvestre, J. Lopes, and N. Pugno, “Melted and recrystallized holey-graphene-reinforced aluminum composites: Structure, elasticity and strength,” Composite Structures. 2022. link Times cited: 4 USED (low confidence) L. Wu et al., “A new method for computing the anisotropic free energy of the crystal-melt interface,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) J. Li, Y. Huang, Y. Zhou, and F. Zhu, “Molecular Dynamics Study of Compressive Properties and Atomistic Behavior of Boron Nitride Nanosheets Reinforced in Aluminum Matrix Composites,” JOM. 2022. link Times cited: 2 USED (low confidence) B. Kong, D. Wen, L. Wang, L. Wang, S. Wang, and T. Xiao, “Atomic-Scale Study of Grain Boundary Evolution in the Abrasive Wear of An Al–Li Alloy,” Transactions of the Indian Institute of Metals. 2022. link Times cited: 0 USED (low confidence) Y. Li, Z. Liu, P. Peng, and K. Dong, “Spinodal limits of supercooled liquid Al deduced from configuration heredity of crystal clusters,” Computational Materials Science. 2022. link Times cited: 2 USED (low confidence) D. Trong, V. C. Long, and Ș. Ţălu, “Molecular Dynamics Simulation of Bulk Cu Material under Various Factors,” Applied Sciences. 2022. link Times cited: 4 Abstract: In this paper, the molecular dynamics (MD) method was used t… read moreAbstract: In this paper, the molecular dynamics (MD) method was used to study the influence of factors of bulk Cu material, such as the effect of the number of atoms (N) at temperature (T), T = 300 K, temperature T, and annealing time (t) with Cu5324 on the structure properties, phase transition, and glass temperature Tg of the bulk Cu material. The obtained results showed that the glass transition temperature (Tg) of the bulk Cu material was Tg = 652 K; the length of the link for Cu-Cu had a negligible change; r = 2.475 Å; and four types of structures, FCC, HCP, BCC, Amor, always existed. With increasing the temperature the FCC, HCP, and BCC decrease, and Amorphous (Amor) increases. With an increasing number of atoms and annealing time, the FCC, HCP, and BCC increased, and Amor decreased. The simulated results showed that there was a great influence of factors on the structure found the gradient change, phase transition, and successful determination of the glass temperature point above Tg of the bulk Cu material. On the basis of these results, essential support will be provided for future studies on mechanical, optical, and electronic properties. read less USED (low confidence) Q. Li et al., “Investigation of Grain Boundary Content on Crack Propagation Behavior of Nanocrystalline Al by Molecular Dynamics Simulation,” physica status solidi (b). 2022. link Times cited: 0 Abstract: The nanocrystalline metal material has been an investigation… read moreAbstract: The nanocrystalline metal material has been an investigation hotspot due to its excellent mechanical property. The high content of grain boundaries (GBs) in microstructure is the key factor affecting its fracture behavior during service. Therefore, it is essential to investigate the effect mechanism of nanoscale GB on crack propagation. In this study, four molecular dynamics (MD) models of nanocrystalline aluminum (Al) with different GB contents are established. The results show that the high‐content GBs in polycrystals can increase the toughness and absorb energy, reducing the risk of brittle crack propagation. The presence of GB can reduce the stress concentration of the crack, and the dislocation emission from the crack tip can be absorbed by the front GB. The microstructure with high‐content GBs can actuate more plastic deformation mechanisms such as multiple slips, GB slip, and migration. The region with more GBs can induce a more even deformation of the whole microstructure by means of dislocation emission and GB migration to the region with fewer GBs. The purpose of this study is to provide a rational mechanism reference for the failure of nanocrystalline metal material. read less USED (low confidence) S. R. Pulagam and A. Dutta, “Peierls–Nabarro modeling of twinning dislocations in fcc metals,” Computational Materials Science. 2022. link Times cited: 3 USED (low confidence) A. Arkundato, F. Monado, I. Sugihartono, A. K. Rivai, and Z. Su’ud, “Diffusion coefficient calculation of iron in liquid lead using molecular dynamics method with new mixing rule for Lennard-Jones potential parameters,” Kuwait Journal of Science. 2022. link Times cited: 0 Abstract: The diffusion coefficient data of materials are crucial for … read moreAbstract: The diffusion coefficient data of materials are crucial for several applications, and can be calculated theoretically up to considerable accuracies. Using molecular dynamics simulation it is possible to compute this property for several conditions as temperature and pressure. The corrosion phenomena of steel types in the fast nuclear reactor can be correlated and studied based on the the diffusion process of iron atoms that dissolve into a liquid lead coolant via molecular dynamics methods using certain potential energy. A widely type of the interatomic interaction potential of materials is the Lennard-Jones potential. Regarding this potential, for a pair of different elements A and B, we can determine the potential parameter ( read less USED (low confidence) M. I. Ojovan and D. Louzguine-Luzgin, “On Structural Rearrangements during the Vitrification of Molten Copper,” Materials. 2022. link Times cited: 11 Abstract: We utilise displacement analysis of Cu-atoms between the che… read moreAbstract: We utilise displacement analysis of Cu-atoms between the chemical bond-centred Voronoi polyhedrons to reveal structural changes at the glass transition. We confirm that the disordered congruent bond lattice of Cu loses its rigidity above the glass transition temperature (Tg) in line with Kantor–Webman theorem due to percolation via configurons (broken Cu-Cu chemical bonds). We reveal that the amorphous Cu has the Tg = 794 ± 10 K at the cooling rate q = 1 × 1013 K/s and that the determination of Tg based on analysis of first sharp diffraction minimum (FDSM) is sharper compared with classical Wendt–Abraham empirical criterion. read less USED (low confidence) A. Susila, E. Handoko, E. Budi, and A. S. Budi, “Influence of thickness on heat treatment from 300 to 1100 K of aluminum thin film,” Journal of Physics: Conference Series. 2022. link Times cited: 0 Abstract: We investigated the influence of Aluminum thin film thicknes… read moreAbstract: We investigated the influence of Aluminum thin film thickness during heat treatment. The films with thickness of 5 nm, 10 nm and 15 nm were heated from room temperature up to above melting point with same heating rate. Molecular Dynamics (MD) simulation is employed to study the behaviour of the thin film where the atoms were followed based on its trajectories. Thin film thickness gives significance contribution to the mechanism of the melting. Smaller thickness suffered strong pressure oscillation while the thin film temperature is increases. Local crystal structure confirmed the transformation of the system from crystalline state to melting state. read less USED (low confidence) R. Fahdiran, I. Sugihartono, M. Delina, T. Prayitno, Sunaryo, and E. Budi, “Influence of heating and cooling rates on thermodynamic properties of aluminum thin film from 300 to 1100 K,” Journal of Physics: Conference Series. 2022. link Times cited: 0 Abstract: Temperature and pressure evolution due to heating and coolin… read moreAbstract: Temperature and pressure evolution due to heating and cooling in the range of 300 to 1100 K of Aluminum thin film with thickness 10 nm were investigated based on Molecular Dynamics (MD) simulation. Pressure evolution shows that heating and cooling rates with comparison of 3:2:1 provides significant contribution on melting and recrystallization of the system. The oscillation of the pressure is the strongest at the highest heating rate which indicate that the system collapses stronger than the lower rate. It is responsible for the destruction of the structure correlated to the elevation of the temperature. While for recrystallization, the analysis on pressure oscillations confirmed the influence of the rates. Analysis based on local crystal structure indicated that at T = 1100 K, all the systems are melted. read less USED (low confidence) R. Fahdiran, I. Sugihartono, M. Delina, T. Prayitno, Sunaryo, and H. Nasbey, “Structure evolution due to heat treatment of aluminum nanoparticle with different sizes: a molecular dynamics study,” Journal of Physics: Conference Series. 2022. link Times cited: 1 Abstract: In this study, we performed Molecular Dynamics (MD) Simulati… read moreAbstract: In this study, we performed Molecular Dynamics (MD) Simulation to investigate the significance of diameter during heat treatment of Aluminum nanoparticle. Structure information along with thermodynamics evolution is analyzed to explain the significance. Different sizes of nanoparticle diameter, i.e., 5 nm, 10 nm, and 15 nm, were investigated with same heating rate. The results show that smallest diameter is suffered for total melting, while for larger nanoparticle small fractions remains. Thermodynamics information indicated the relation that smaller diameter will experience faster pressure oscillations period. While for larger system, the period is longer, but the pressure value become stronger. Structure analysis confirmed that the nanoparticles are melted at the end of the simulation. read less USED (low confidence) A. Susila, E. Handoko, A. S. Budi, and H. Nasbey, “Thermodynamic and structure properties of aluminum nanoparticle due to heat treatment: a molecular dynamics study,” Journal of Physics: Conference Series. 2022. link Times cited: 1 Abstract: Thermodynamic and structure evolution of Aluminum nanopartic… read moreAbstract: Thermodynamic and structure evolution of Aluminum nanoparticle with diameter of 10 nm are investigated due to heat treatment by means of stepping heat and compared it to sudden heat. Molecular Dynamics (MD) simulation is employed to track the trajectories of each atom and its surrounding to define thermodynamic properties, e.g., temperature and pressure. The temperature evolution gives clear different profile for both cases, while pressure profile strongly evident different mechanism of melting. From thermodynamics point of view, the nanoparticle suffered different final state for both heating methods. Structure analysis later confirmed that for stepping heat the nanoparticle is melted while for sudden heat it is only partially melted. read less USED (low confidence) Z. Yan, B. Xu, J. Li, and L. Kong, “Defect-mediated crystal growth from deeply undercooled melts,” Computational Materials Science. 2022. link Times cited: 2 USED (low confidence) S. Shcherbinin, K. Krylova, G. Chechin, E. Soboleva, and S. Dmitriev, “Delocalized nonlinear vibrational modes in fcc metals,” Commun. Nonlinear Sci. Numer. Simul. 2022. link Times cited: 11 USED (low confidence) X. Luo et al., “The near-surface microstructural evolution and the influence of Si particles during nanoscratching of nanocrystalline Al,” Applied Surface Science. 2022. link Times cited: 2 USED (low confidence) L.-li Zhou, J. Pan, L. Lang, Z. Tian, Y. Mo, and K. Dong, “Atomic structure evolutions and mechanisms of the crystallization pathway of liquid Al during rapid cooling,” RSC Advances. 2021. link Times cited: 6 Abstract: The solidification of pure aluminum has been studied by a la… read moreAbstract: The solidification of pure aluminum has been studied by a large-scale molecular dynamic simulation. The potential energy, position D, height H, and width W of the first peak and valley of PDF curves, and the local structures were investigated. It was found that the FCC-crystallization ability of pure Al is so strong that still local crystal regions exist in the amorphized solid. As the temperature decreases, besides the counter-intuitive increase in Dp (D of the first peak), Hp increases monotonically; Wp, Dv, and Hv decrease monotonically; only Wv first decreases and then increases. They all change critically when phase transition happens. After the nucleation, orientation-disordered HCP-regions, as the grain boundaries or defects of FCC crystals, rapidly transform into FCC structures, and then the surviving HCP-regions regularize into few parallel layers or orientation-disordered HCP-regions. If parallel layers result in dislocation pinning, structural evolution terminates; otherwise, it continues. These findings will have a positive impact on the development of the solidification and nucleation theory. read less USED (low confidence) Y. Mo et al., “Competition between TCP and crystalline clusters during phase transition of rapidly super-cooled aluminum,” Journal of Non-Crystalline Solids. 2021. link Times cited: 5 USED (low confidence) W. Velilla-Díaz and H. R. Zambrano, “Crack Length Effect on the Fracture Behavior of Single-Crystals and Bi-Crystals of Aluminum,” Nanomaterials. 2021. link Times cited: 6 Abstract: Molecular dynamics simulations of cracked nanocrystals of al… read moreAbstract: Molecular dynamics simulations of cracked nanocrystals of aluminum were performed in order to investigate the crack length and grain boundary effects. Atomistic models of single-crystals and bi-crystals were built considering 11 different crack lengths. Novel approaches based on fracture mechanics concepts were proposed to predict the crack length effect on single-crystals and bi-crystals. The results showed that the effect of the grain boundary on the fracture resistance was beneficial increasing the fracture toughness almost four times for bi-crystals. read less USED (low confidence) S. Mondal and A. Dutta, “Atomistic design of nanocrystalline samples: A Bayesian approach,” Materials Letters. 2021. link Times cited: 1 USED (low confidence) A. Kedharnath, R. Kapoor, and A. Sarkar, “Classical molecular dynamics simulations of the deformation of metals under uniaxial monotonic loading: A review,” Computers & Structures. 2021. link Times cited: 16 USED (low confidence) S. Becker, E. Devijver, R. Molinier, and N. Jakse, “Unsupervised topological learning approach of crystal nucleation,” Scientific Reports. 2021. link Times cited: 5 USED (low confidence) J. Li, Y. Huang, B. Zeng, C. Feng, and F. Zhu, “Mechanical Response of BNNS-reinforced Aluminum Composites under Uniaxial Compression,” 2021 22nd International Conference on Electronic Packaging Technology (ICEPT). 2021. link Times cited: 0 Abstract: Boron nitride nanosheet (BNNS) has been widely used as the r… read moreAbstract: Boron nitride nanosheet (BNNS) has been widely used as the reinforcing material in metal matrix composites because of excellent thermal, electrical and mechanical properties. Chirality directions of boron nitride nanosheet include armchair and zigzag. In this paper, the influence of temperature on the mechanical reponses of aluminum (Al) and BNNS/Al composite under uniaxial compression loads haa been investigated by molecular dynamics (MD) simulations. MD simulation results show that there is a negative relationship between temperature and ultimate stress of the above material, and ultimate stress decreases as the temperatue increases. The relationship between critical strain and temperature is the same response. In addition, BNNS plays a significant role in improving ultimate stress and Young's modulus of the nanocomposite, despite its small volume fraction. However, compression loads along different chiral directions can also result in some diversities of mechanical response. For different temperature conditions, the ultimate stress of zigzag BNNS/Al composite is bigger than others in general, and armchair BNNS/Al composite shows the better temperature stability. read less USED (low confidence) S. Adibi and J. Wilkerson, “Time–temperature superposition for cavitation resistance of metals with nonequilibrium vacancy concentrations,” Extreme Mechanics Letters. 2021. link Times cited: 2 USED (low confidence) S. Terayama, Y. Iwase, S. Hayakawa, T. Okita, M. Itakura, and K. Suzuki, “Molecular dynamic simulations evaluating the effect of the stacking fault energy on defect formations in face-centered cubic metals subjected to high-energy particle irradiation,” Computational Materials Science. 2021. link Times cited: 9 USED (low confidence) H. Song and M. Mendelev, “Molecular Dynamics Study of Mechanism of Solid–Liquid Interface Migration and Defect Formation in Al3Sm Alloy,” JOM. 2021. link Times cited: 2 USED (low confidence) Y. Sun, F. Zhang, M. Mendelev, R. Wentzcovitch, and K. Ho, “Two-step nucleation of the Earth’s inner core,” Proceedings of the National Academy of Sciences of the United States of America. 2021. link Times cited: 17 Abstract: Significance Understanding the formation of the Earth’s inne… read moreAbstract: Significance Understanding the formation of the Earth’s inner core is essential to understanding the geodynamo and Earth's history. However, recent attempts to explain the initial solidification of the inner core have been unsuccessful. The supercooling necessary to form hcp iron is unrealistically large and creates the “inner core nucleation paradox.” Our work demonstrates that molten iron can crystallize via a two-step nucleation process involving the intermediate bcc phase under core conditions. This mechanism significantly reduces the required undercooling necessary to nucleate solid iron. This work also suggests that bcc and hcp iron have similar free energies at pressures near the inner core center. The Earth's inner core started forming when molten iron cooled below the melting point. However, the nucleation mechanism, which is a necessary step of crystallization, has not been well understood. Recent studies have found that it requires an unrealistic degree of undercooling to nucleate the stable, hexagonal, close-packed (hcp) phase of iron that is unlikely to be reached under core conditions and age. This contradiction is referred to as the inner core nucleation paradox. Using a persistent embryo method and molecular dynamics simulations, we demonstrate that the metastable, body-centered, cubic (bcc) phase of iron has a much higher nucleation rate than does the hcp phase under inner core conditions. Thus, the bcc nucleation is likely to be the first step of inner core formation, instead of direct nucleation of the hcp phase. This mechanism reduces the required undercooling of iron nucleation, which provides a key factor in solving the inner core nucleation paradox. The two-step nucleation scenario of the inner core also opens an avenue for understanding the structure and anisotropy of the present inner core. read less USED (low confidence) S. Konorev, R. Kozubski, M. Albrecht, and I. Vladymyrskyi, “Self-diffusion of Fe and Pt in L1-Ordered FePt: Molecular Dynamics simulation,” Computational Materials Science. 2021. link Times cited: 4 USED (low confidence) L. Wu, H. Wang, Y. Zhu, and M. Li, “Crystal-melt coexistence in FCC and BCC metals: A molecular-dynamics study of crystal-melt interface free energies,” Materialia. 2021. link Times cited: 6 USED (low confidence) S. Zhang, D. He, P. Huang, and F. Wang, “Moiré pattern at graphene/Al (111) interface: Experiment and simulation,” Materials & Design. 2021. link Times cited: 5 USED (low confidence) L.-F. Zhu, J. Janssen, S. Ishibashi, F. Körmann, B. Grabowski, and J. Neugebauer, “A fully automated approach to calculate the melting temperature of elemental crystals,” Computational Materials Science. 2021. link Times cited: 17 USED (low confidence) M. Papanikolaou and K. Salonitis, “Grain size effects on nanocutting behaviour modelling based on molecular dynamics simulations,” Applied Surface Science. 2021. link Times cited: 16 USED (low confidence) X. W. Zhou and M. E. Foster, “Character angle effects on dissociated dislocation core energy in aluminum.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 4 Abstract: Dislocation core energy is an important property in material… read moreAbstract: Dislocation core energy is an important property in materials mechanics but can only be obtained from atomistic simulations. Periodic boundary conditions are ideally suited for atomistic calculations of dislocation energies but have faced two major challenges. First, viable methods to extract core energies from atomistic data of total energies have been developed only for non-dissociated dislocations whereas realistic dislocations are often dissociated into partials. Second, core energy is a function of dislocation character angle. This functional dependence can only be revealed through calculations at a variety of character angles. This requires both additional computational resources and a robust method to implement arbitrary character angles. Here a new procedure has been developed to overcome both challenges. By applying this approach, we have calculated 22 core energies of dissociated dislocations in aluminium over the entire character angle range between 0° and 90°. In addition to the discrete core energy data for dissociated dislocations, we found that core energy can be approximated by a continuous function of character angle. Specifically, our dissociated dislocation core energies have been well fitted to a polynomial Sinoidal function of character angle. We have also discovered that there exists a critical system dimension below which dislocation core energies cannot be calculated due to dislocation transformation. read less USED (low confidence) M. He, E. T. Karim, M. Shugaev, and L. Zhigilei, “Atomistic simulation of the generation of vacancies in rapid crystallization of metals,” Acta Materialia. 2021. link Times cited: 7 USED (low confidence) V. Ankudinov, K. Elder, and P. K. Galenko, “Traveling waves of the solidification and melting of cubic crystal lattices.,” Physical review. E. 2020. link Times cited: 8 Abstract: Using the phase field crystal model (PFC model), an analysis… read moreAbstract: Using the phase field crystal model (PFC model), an analysis of slow and fast dynamics of solid-liquid interfaces in solidification and melting processes is presented. Dynamical regimes for cubic lattices invading metastable liquids (solidification) and liquids propagating into metastable crystals (melting) are described in terms of the evolving amplitudes of the density field. Dynamical equations are obtained for body-centered cubic (bcc) and face-centered cubic (fcc) crystal lattices in one- and two-mode approximations. A universal form of the amplitude equations is obtained for the three-dimensional dynamics for different crystal lattices and crystallographic directions. Dynamics of the amplitude's propagation for different lattices and PFC mode's approximations is qualitatively compared. The traveling-wave velocity is quantitatively compared with data of molecular dynamics simulation previously obtained by Mendelev et al. [Modell. Simul. Mater. Sci. Eng. 18, 074002 (2010)MSMEEU0965-039310.1088/0965-0393/18/7/074002] for solidification and melting of the aluminum fcc lattice. read less USED (low confidence) S. Starikov, I. Gordeev, Y. Lysogorskiy, L. Kolotova, and S. Makarov, “Optimized interatomic potential for study of structure and phase transitions in Si-Au and Si-Al systems,” Computational Materials Science. 2020. link Times cited: 19 USED (low confidence) M. Li, Q. Hou, J. Cui, M. Qiu, and A. Yang, “Atomistic simulations of helium clustering in copper,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2020. link Times cited: 3 USED (low confidence) P. N. Babu, C. Becquart, and S. Pal, “Molecular dynamics simulation-based study of creep–ratcheting behavior of nanocrystalline aluminum,” Applied Nanoscience. 2020. link Times cited: 11 USED (low confidence) S. Kurian and R. Mirzaeifar, “Selective laser melting of aluminum nano-powder particles, a molecular dynamics study,” Additive manufacturing. 2020. link Times cited: 30 USED (low confidence) J. Gabriel et al., “Uncertainty Quantification in Atomistic Modeling of Metals and Its Effect on Mesoscale and Continuum Modeling: A Review,” JOM. 2020. link Times cited: 6 USED (low confidence) Y. Wu, T. Zhou, R. Yu, Q. Lai, H. Wang, and J. You, “A New Crystallization Pattern of Nested Tetrahedral Lamellar Structure for the Face-Centered Cubic Metals with Low Stacking Fault Energy,” MatSciRN: Other Materials Performance (Topic). 2020. link Times cited: 7 Abstract: It's well known that the lamellar (LAM) and five-fold-t… read moreAbstract: It's well known that the lamellar (LAM) and five-fold-twinning (FFT) structures are two primary patterns during the early crystallization stage of FCC (face-centered cubic) metals. In this letter, we add a new third one, i.e. a nested tetrahedral lamellar (NTL) structure. In common with LAM and FFT, NTL is also a mixture of FCC and HCP (hexagonal close-packed). Differently, NTL has four tetrahedral growth orientations corresponding to four cellular bulges and a central core of tetrahedral shells nested one-by-one. This NTL acts also as the precursor of FFT and it has great differences in growth morphology and kinetics with LAM. read less USED (low confidence) R. Yan, S. Ma, W. Sun, T. Jing, and H. Dong, “The solid–liquid interface free energy of Al: A comparison between molecular dynamics calculations and experimental measurements,” Computational Materials Science. 2020. link Times cited: 8 USED (low confidence) M. Shi, N. Admal, and E. Tadmor, “Noise filtering in atomistic stress calculations for crystalline materials,” Journal of The Mechanics and Physics of Solids. 2020. link Times cited: 2 USED (low confidence) S. Xu, J. Mianroodi, A. Hunter, B. Svendsen, and I. Beyerlein, “Comparative modeling of the disregistry and Peierls stress for dissociated edge and screw dislocations in Al,” International Journal of Plasticity. 2020. link Times cited: 32 USED (low confidence) B. Faria, C. Guarda, N. Silvestre, and J. Lopes, “Aluminum composites reinforced by γ-graphynes: The effect of nanofillers porosity and shape on crystal growth and composite strengthening,” Computational Materials Science. 2020. link Times cited: 6 USED (low confidence) L. He, F. Wang, X.-guo Zeng, X. Yang, and Z. Qi, “Atomic insights into shock-induced spallation of single-crystal aluminum through molecular dynamics modeling,” Mechanics of Materials. 2020. link Times cited: 25 USED (low confidence) V. Reshetniak and A. Aborkin, “Aluminum–Carbon Interaction at the Aluminum–Graphene and Aluminum–Graphite Interfaces,” Journal of Experimental and Theoretical Physics. 2020. link Times cited: 8 USED (low confidence) X. Zhou, X. Liu, J. Lei, and Q. Yang, “Atomic simulations of the formation of twist grain boundary and mechanical properties of graphene/aluminum nanolaminated composites,” Computational Materials Science. 2020. link Times cited: 18 USED (low confidence) M. M. Rahman, N. Sakib, A. Ashikuzzaman, and M. F. Alam, “Vertically twinned aluminum nano-pillars under tensile loading: a molecular dynamics study,” Materials Research Express. 2020. link Times cited: 0 Abstract: Nano twinned FCC materials show superior properties comparat… read moreAbstract: Nano twinned FCC materials show superior properties comparatively to their single crystal counterparts. The properties of nano-twinned materials are possessed by the interactions of dislocations with the coherent twin boundaries (TBs). In this paper, we describe the fabrication of arrays of vertically aligned aluminum nano-pillars that contain different number of TBs (different twin boundary spacing) and no grain boundaries or other microstructural features. We have investigated the influence of twin boundary (TB) spacing on the mechanical responses of individual nano-pillars under tensile loading. The investigation fabricated with molecular dynamics (MD) simulation reveals that, the yield strength is dependent on number of vertical twins. Yield strength increases with increasing number of twins upto a critical value and then starts to decrease with further increment of twin numbers. An increase of ductility was also found as a result of immobilized dislocation. The deformation process was nucleated by spontaneous dislocation buds and eventually turned into mature partial dislocations. The simulation was done until fracture to give an insight about dislocation behavior. read less USED (low confidence) G. Plummer and G. Tucker, “Bond-order potentials for theTi3AlC2andTi3SiC2MAX phases,” Physical Review B. 2019. link Times cited: 12 USED (low confidence) F. Lv, P. Liu, H. Qi, J. Liu, R. Sun, and W. Wang, “The early stage of the thermal pulse explosions of aluminum nanowires under different energy deposition levels,” Computational Materials Science. 2019. link Times cited: 12 USED (low confidence) T. Wejrzanowski, J. Lipecka, J. Janczak-Rusch, and M. Lewandowska, “Al-Si/AlN nanomultilayered systems with reduced melting point: Experiments and simulations,” Applied Surface Science. 2019. link Times cited: 7 USED (low confidence) A. Vlasova, “Deformation behaviour of aluminium nanocrystals under shock-wave loading,” Journal of Physics: Conference Series. 2019. link Times cited: 0 Abstract: Nonequilibrium process of shock-wave loading of aluminum nan… read moreAbstract: Nonequilibrium process of shock-wave loading of aluminum nanocrystals were investigated numerically under uniaxial stress conditions using molecular dynamics simulations (MD). Deformation curves, time dependence of strain rate and the change in potential energy were constructed. Calculated properties of aluminium using embedded atom interatomic potential are compared with each other. Relationship between the calculated characteristics are discussed. The behaviour of deformation curves for two different aluminium nanosrystals is explained. The problems of modeling the deformation hardening of aluminum nanocrystals are considered. read less USED (low confidence) I. Mitiche, O. Lamrous, S. Makhlouf, F. Marchetti, and N. Laidani, “Effect of the interface layer vibration modes in enhancing thermal conductivity of nanofluids.,” Physical review. E. 2019. link Times cited: 11 Abstract: The present paper reports on an investigation of the effect … read moreAbstract: The present paper reports on an investigation of the effect of the interface layers in enhancing thermal conductivity of Cu-Ar nanofluids. The approach is based on linear response theory combined with equilibrium molecular dynamics simulations. For a wettability parameter of 1.4 and volume fraction of 5.8%, simulation results show enhancements in thermal conductivity as high as 50%. Among others, the most salient result concerns the contribution of the vibration modes of liquid Ar atoms around Cu nanoparticles (NPs) in enhancing thermal conductivity of the nanofluid. Our findings reveal that these vibration modes coincide on a large domain of frequencies (10-50ps^{-1}) with those of Cu atoms of the NPs. The enhancement of the thermal conductivity was explained by the increase of vibrational mean-free paths. read less USED (low confidence) S. Huang, I. Beyerlein, and C. Zhou, “Unusual size effects from tilted twin boundaries in nano-twinned metals,” Extreme Mechanics Letters. 2019. link Times cited: 4 USED (low confidence) H. Peng et al., “Chemical effect on the structural and dynamical properties in Zr-Ni-Al liquids,” Physical Review B. 2019. link Times cited: 9 Abstract: We develop an embedded-atom method (EAM) model to perform cl… read moreAbstract: We develop an embedded-atom method (EAM) model to perform classical molecular-dynamics computer simulations of a model of Zr-Ni-Al ternary melts, based on the existing binary ones. The EAM potential is validated against a broad range of experimental data for the liquid melt, including both static-structure factors and dynamical data on the mass-transport coefficients. We use our simulation model to address the structural and dynamical changes induced by a systematic replacement of Zr by Al in ${\mathrm{Zr}}_{75\ensuremath{-}x}{\mathrm{Ni}}_{25}{\mathrm{Al}}_{x}\phantom{\rule{4pt}{0ex}}(x=0--30)$ ternary alloys. We find strong chemical-ordering effects exhibited as the locally preferred structure when the Al-concentration ${c}_{\text{Al}}$ is increased. Along with the chemical effects, effective-power-law relations are found between the self-diffusion coefficients in the melts, with an exponent that monotonically decreases with increasing Al concentration. The associated Stokes-Einstein relation between diffusivity and viscosity breaks down at higher temperature upon Al addition. We also address the influence of Al admixture on the vibrational spectrum of the melt. With increasing ${c}_{\text{Al}}$, sound waves move faster, and an optical vibrational mode is found. read less USED (low confidence) W. Velilla-Díaz, A. Pacheco-Sanjuán, and H. R. Zambrano, “The role of the grain boundary in the fracture toughness of aluminum bicrystal,” Computational Materials Science. 2019. link Times cited: 9 USED (low confidence) S. Kumar, S. K. Pattanayek, and S. Das, “Reactivity-Controlled Aggregation of Graphene Nanoflakes in Aluminum Matrix: Atomistic Molecular Dynamics Simulation,” The Journal of Physical Chemistry C. 2019. link Times cited: 10 Abstract: Aluminum graphene nanoflakes composite depicts many useful p… read moreAbstract: Aluminum graphene nanoflakes composite depicts many useful properties such as excellent mechanical strength, lightweight, high electrical, thermal properties, etc. Aggregation and dispersion of graphene nanoflakes in aluminum matrix highly influence the above-mentioned properties. In this paper, aggregation of graphene nanoflakes in aluminum matrix has been studied using molecular dynamics simulation. During simulations, adaptive intermolecular reactive empirical bond order (AIREBO) and embedded atom method force field were used for graphene nanoflakes and aluminum, respectively. AIREBO potential is capable of reproducing sp2–sp2 (covalent) bond formation or breaking between the reactive edge of graphene nanoflakes. The reactive edges of graphene nanoflakes form covalent bond with the neighboring graphene that produces a unique interconnected network in aluminum matrix. However, reactivity of graphene edge exclusively depends on the interfacial interaction between graphene and aluminum. Further, interfaci... read less USED (low confidence) B. Bokstein, A. Rodin, A. Itckovich, and L. Klinger, “Segregation and Phase Transitions in Grain Boundaries,” Diffusion Foundations. 2019. link Times cited: 1 Abstract: The paper is devoted to some properties of grain boundaries:… read moreAbstract: The paper is devoted to some properties of grain boundaries: Segregation and concentration phase transitions – two important consequences of atomic interactions in grain boundaries. Except of a short description the Gibbs method of surface excesses and grain boundary segregation isotherms with the limited number of segregation sites in grain boundary, the paper concentrates on the effects of complexes formation, including thermodynamic and computer modeling, and concentration phase transition in the grain boundaries in systems with restricted solubility and intermediate compounds. read less USED (low confidence) F. Lv, P. Liu, H. Qi, and J. Liu, “Molecular dynamics simulations on the effect of energy deposition rate on the electrical explosion of metal nanowires,” Computational Materials Science. 2019. link Times cited: 8 USED (low confidence) K. Dang, D. Bamney, K. Bootsita, L. Capolungo, and D. Spearot, “Mobility of dislocations in Aluminum: Faceting and asymmetry during nanoscale dislocation shear loop expansion,” Acta Materialia. 2019. link Times cited: 33 USED (low confidence) S. An et al., “Common mechanism for controlling polymorph selection during crystallization in supercooled metallic liquids,” Acta Materialia. 2018. link Times cited: 16 USED (low confidence) R. Yan et al., “Structural and mechanical properties of homogeneous solid-liquid interface of Al modelled with COMB3 potential,” Computational Materials Science. 2018. link Times cited: 9 USED (low confidence) Y. Sun, F. Zhang, H. Song, M. Mendelev, C. Wang, and K. Ho, “Temperature dependence of the solid-liquid interface free energy of Ni and Al from molecular dynamics simulation of nucleation.,” The Journal of chemical physics. 2018. link Times cited: 16 Abstract: The temperature dependence of the solid-liquid interfacial f… read moreAbstract: The temperature dependence of the solid-liquid interfacial free energy, γ, is investigated for Al and Ni at the undercooled temperature regime based on a recently developed persistent-embryo method. The atomistic description of the nucleus shape is obtained from molecular dynamics simulations. The computed γ shows a linear dependence on the temperature. The values of γ extrapolated to the melting temperature agree well with previous data obtained by the capillary fluctuation method. Using the temperature dependence of γ, we estimate the nucleation free energy barrier in a wide temperature range from the classical nucleation theory. The obtained data agree very well with the results from the brute-force molecular dynamics simulations. read less USED (low confidence) H. N. Pishkenari, F. S. Yousefi, and A. Taghibakhshi, “Determination of surface properties and elastic constants of FCC metals: a comparison among different EAM potentials in thin film and bulk scale,” Materials Research Express. 2018. link Times cited: 22 Abstract: Three independent elastic constants C11, C12, and C44 were c… read moreAbstract: Three independent elastic constants C11, C12, and C44 were calculated and compared using available potentials of eight different metals with FCC crystal structure; Gold, Silver, Copper, Nickel, Platinum, Palladium, Aluminum and Lead. In order to calculate the elastic constants, the second derivative of the energy density of each system was calculated with respect to different directions of strains. Each set of the elastic constants of the metals in bulk scale was compared with experimental results, and the average relative error was for each was calculated and compared with other available potentials. Then, using the Voigt-Reuss-Hill method, approximated values for Young and shear moduli and Poisson’s ratio of the FCC metals in the bulk scale were found for each potential. Furthermore, to observe the surface effects on the metals in nanoscale, surface elastic constants of the thin films of the metals have been calculated. In the study of the thin films of materials in nanoscale, the number of surface atoms is considerable compared to all atoms of the object. This leads to an increase in the surface effects, which influence the elastic properties. By considering this fact and employing related definitions and equations, the properties of the thin films of the metals were calculated, and the surface effects for different crystallographic directions were compared. Subsequently, in some cases, comparisons among characteristics of the metals in the thin film and bulk material were made. read less USED (low confidence) S. An, R. Su, S. Zhao, J.-bo Liu, B.-xin Liu, and P. Guan, “Ultrasmall nanoparticles inducing order-to-disorder transition,” Physical Review B. 2018. link Times cited: 4 Abstract: The vitrification of single-element metallic glass remains a… read moreAbstract: The vitrification of single-element metallic glass remains a challenging research topic and attracts a great deal of attention. It is reported that the propensity for formation of the amorphous phase increases as the size of the system decreases in experiments. However, the thermodynamic contributions to this phenomenon are still elusive. Here, we study the size effects on the thermodynamic properties of the Cu nanoparticles in both crystalline and amorphous phases using the Frenkel-Ladd methods. Smaller particles show stronger size dependences on these quantities. From the perspective of free-energy difference between the amorphous phase and the crystalline phase, the thermodynamic driving force for vitrification of single-element metallic liquids catches up with that for crystallization dramatically, given the particle diameter smaller than a critical value (i.e., $\ensuremath{\sim}4$ nm for Cu), which provides a thermodynamic interpretation for the size dependence of the glass-forming ability. read less USED (low confidence) D. Yi et al., “What Drives Metal-Surface Step Bunching in Graphene Chemical Vapor Deposition?,” Physical review letters. 2018. link Times cited: 35 Abstract: Compressive strain relaxation of a chemical vapor deposition… read moreAbstract: Compressive strain relaxation of a chemical vapor deposition (CVD) grown graphene overlayer has been considered to be the main driving force behind metal surface step bunching (SB) in CVD graphene growth. Here, by combining theoretical studies with experimental observations, we prove that the SB can occur even in the absence of a compressive strain, is enabled by the rapid diffusion of metal adatoms beneath the graphene and is driven by the release of the bending energy of the graphene overlayer in the vicinity of steps. Based on this new understanding, we explain a number of experimental observations such as the temperature dependence of SB, and how SB depends on the thickness of the graphene film. This study also shows that SB is a general phenomenon that can occur in all substrates covered by films of two-dimensional (2D) materials. read less USED (low confidence) Z. Cong and S. Lee, “Study of mechanical behavior of BNNT-reinforced aluminum composites using molecular dynamics simulations,” Composite Structures. 2018. link Times cited: 43 USED (low confidence) H. Y. Song, M. Wang, Q. Deng, and Y. L. Li, “Deformation mode transitions in Cu 50 Zr 50 amorphous/Cu crystalline nanomultilayer: A molecular dynamics study,” Journal of Non-Crystalline Solids. 2018. link Times cited: 21 USED (low confidence) S. P. Patil, S. H. Chilakamarri, and B. Markert, “A novel nonlinear nano-scale wear law for metallic brake pads.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 7 Abstract: In the present work, molecular dynamics simulations were car… read moreAbstract: In the present work, molecular dynamics simulations were carried out to investigate the temperature distribution as well as the fundamental friction characteristics such as the coefficient of friction and wear in a disc-pad braking system. A wide range of constant velocity loadings was applied on metallic brake pads made of aluminium, copper and iron with different rotating speeds of a diamond-like carbon brake disc. The average temperature of Newtonian atoms and the coefficient of friction of the brake pad were investigated. The resulting relationship of the average temperature with the speed of the disc as well as the applied loading velocity can be described by power laws. The quantitative description of the volume lost from the brake pads was investigated, and it was found that the volume lost increases linearly with the sliding distance. Our results show that Archard's linear wear law is not applicable to a wide range of normal loads, e.g., in cases of low normal load where the wear rate was increased considerably and in cases of high load where there was a possibility of severe wear. In this work, a new formula for the brake pad wear in a disc brake assembly is proposed, which displays a power law relationship between the lost volume of the metallic brake pads per unit sliding distance and the applied normal load with an exponent of 0.62 ± 0.02. This work provides new insights into the fundamental understanding of the wear mechanism at the nano-scale leading to a new bottom-up wear law for metallic brake pads. read less USED (low confidence) S. Chandra, M. K. Samal, V. Chavan, and S. Raghunathan, “An atomistic study of crack-void interaction in aluminum.” 2018. link Times cited: 0 Abstract: The interaction between a brittle crack and pre-existing voi… read moreAbstract: The interaction between a brittle crack and pre-existing void was investigated using molecular dynamics simulations in face centered cubic Al. The results reveal that a pre-existing void offers considerable resistance to crack growth. By changing the void radius, it has been found that increase in void size leads to an increase in strain to fracture. Consequently, this work highlights the importance of larger voids in impacting the fracture response of materials that fail in a potentially brittle manner.The interaction between a brittle crack and pre-existing void was investigated using molecular dynamics simulations in face centered cubic Al. The results reveal that a pre-existing void offers considerable resistance to crack growth. By changing the void radius, it has been found that increase in void size leads to an increase in strain to fracture. Consequently, this work highlights the importance of larger voids in impacting the fracture response of materials that fail in a potentially brittle manner. read less USED (low confidence) K. Choudhary, A. Biacchi, S. Ghosh, L. Hale, A. Walker, and F. Tavazza, “High-throughput assessment of vacancy formation and surface energies of materials using classical force-fields,” Journal of Physics: Condensed Matter. 2018. link Times cited: 16 Abstract: In this work, we present an open access database for surface… read moreAbstract: In this work, we present an open access database for surface and vacancy-formation energies using classical force-fields (FFs). These quantities are essential in understanding diffusion behavior, nanoparticle formation and catalytic activities. FFs are often designed for a specific application, hence, this database allows the user to understand whether a FF is suitable for investigating particular defect and surface-related material properties. The FF results are compared to density functional theory and experimental data whenever applicable for validation. At present, we have 17 506 surface energies and 1000 vacancy formation energies calculation in our database and the database is still growing. All the data generated, and the computational tools used, are shared publicly at the following websites: www.ctcms.nist.gov/~knc6/periodic.html, https://jarvis.nist.gov and https://github.com/usnistgov/jarvis. Approximations used during the high-throughput calculations are clearly mentioned. Using some of the example cases, we show how our data can be used to directly compare different FFs for a material and to interpret experimental findings such as using Wulff construction for predicting equilibrium shape of nanoparticles. Similarly, the vacancy formation energies data can be useful in understanding diffusion related properties. read less USED (low confidence) S. Kumar, “Graphene Engendered aluminium crystal growth and mechanical properties of its composite: An atomistic investigation,” Materials Chemistry and Physics. 2018. link Times cited: 29 USED (low confidence) Z. Zhang and H. Urbassek, “Dislocation-based strengthening mechanisms in metal-matrix nanocomposites: a molecular dynamics study of the influence of reinforcement shape in the Al-Si system,” Computational Materials Science. 2018. link Times cited: 20 USED (low confidence) T. A. Sharp, S. L. Thomas, E. D. Cubuk, S. Schoenholz, D. Srolovitz, and A. J. Liu, “Machine learning determination of atomic dynamics at grain boundaries,” Proceedings of the National Academy of Sciences. 2018. link Times cited: 63 Abstract: Significance A machine learning method is used to analyze th… read moreAbstract: Significance A machine learning method is used to analyze the atomic structures that rearrange within the grain boundaries of polycrystals. The method readily separates the atomic structures into those that rarely rearrange and those that often rearrange. The likelihood of an atom rearranging under a thermal fluctuation is correlated with free volume and potential energy but is not entirely attributable to those quantities. A machine-learned quantity allows estimation of the energy barrier to rearrangement for particular atoms. The grain boundary atoms that rearrange most have more possible rearrangement trajectories rather than much-reduced energy barriers, as in bulk glasses. The work suggests that polycrystal plasticity can be studied in part from the local atomic structural environments without traditional classification of microstructure. In polycrystalline materials, grain boundaries are sites of enhanced atomic motion, but the complexity of the atomic structures within a grain boundary network makes it difficult to link the structure and atomic dynamics. Here, we use a machine learning technique to establish a connection between local structure and dynamics of these materials. Following previous work on bulk glassy materials, we define a purely structural quantity (softness) that captures the propensity of an atom to rearrange. This approach correctly identifies crystalline regions, stacking faults, and twin boundaries as having low likelihood of atomic rearrangements while finding a large variability within high-energy grain boundaries. As has been found in glasses, the probability that atoms of a given softness will rearrange is nearly Arrhenius. This indicates a well-defined energy barrier as well as a well-defined prefactor for the Arrhenius form for atoms of a given softness. The decrease in the prefactor for low-softness atoms indicates that variations in entropy exhibit a dominant influence on the atomic dynamics in grain boundaries. read less USED (low confidence) A. Itckovich, M. Mendelev, A. Rodin, and B. Bokstein, “Effect of Atomic Complexes Formation in Grain Boundaries on Grain Boundary Diffusion,” Defect and Diffusion Forum. 2018. link Times cited: 7 Abstract: The peculiarities of grain boundary diffusion in Cu connecte… read moreAbstract: The peculiarities of grain boundary diffusion in Cu connected with the effect of atomic pairs formation in grain boundaries (GB) were studied using the molecular dynamics (MD) simulation. In present study Cu GB selfdiffusion was simulated with the use of semi-empirical potential. Besides, the ‘heterodiffusion’ simulation was performed with the artificially addеd energy of interaction (E) between identical atoms in arbitrary chosen pairs. To obtain reliable data on the mean square displacements (MSD) the simulation cell, consisted about three hundreds thousands atoms and two symmetrical GBs Σ5 (001)(012), was used. 70 pairs of identical Cu atoms in GBs, bonded into pairs, were chosen as initial state. Energy of interaction was varied between 0 and - 0.5eV/atomThe results obtained for selfdiffusion are in a good agreement with experimental results and other results of computer simulation. Two main effects for heterodiffusion are under discussion. The first is atomic exchange between GB zone and adjacent lattice zone, where the mobility of the atoms decreases significantly. As a result, the MSD decrease. Another effect is connected with attraction between the “marked” atoms, which leads to formation of relatively stable complexes and the MSD also decreases. The results obtained involve also dependence the number of the stable pairs on time and temperature and show the possibility of pairs to condense into ternary, quarterly and more numerous complexes. read less USED (low confidence) S. Chandra, M. K. Samal, V. Chavan, and S. Raghunathan, “Hierarchical multiscale modeling of plasticity in copper: From single crystals to polycrystalline aggregates,” International Journal of Plasticity. 2018. link Times cited: 45 USED (low confidence) A. Kohnert, M. Cusentino, and B. Wirth, “Molecular statics calculations of the biases and point defect capture volumes of small cavities,” Journal of Nuclear Materials. 2018. link Times cited: 16 USED (low confidence) S. Kumar, “Graphene engendered 2-D structural morphology of aluminium atoms: Molecular dynamics simulation study,” Materials Chemistry and Physics. 2017. link Times cited: 18 USED (low confidence) P. Wang and H. Wang, “Meta-Atom Molecular Dynamics for Studying Material Property Dependent Deformation Mechanisms of Alloys,” Journal of Applied Mechanics. 2017. link Times cited: 12 USED (low confidence) D. Kryzhevich, K. Zolnikov, and A. Korchuganov, “Features of structural changes in the near-surface aluminum layer under various schemes of ion implantation.” 2017. link Times cited: 0 Abstract: The molecular dynamics simulation of structural rearrangemen… read moreAbstract: The molecular dynamics simulation of structural rearrangements in the surface layer of aluminum samples under ion implantation of various intensities was carried out. The features of the internal structure and the crystallographic orientation of the irradiated crystallite were taken into account. To describe the interatomic interaction many-body potentials obtained in the framework of the embedded atom method were used. Irradiation of the {100} surface results in much less number of formed defects than irradiation of the {110} and {111} ones. When irradiating surfaces with beams of relatively low energy grains remain unchanged in the surface region and the formation of stacking faults was not observed. At a high intensity of irradiation, the near-surface layer of the crystallite melts. In the absence of heat removal, the centers of crystallization become grains lying on the boundary of the solid and liquid phases. Those grains increase due to the adjustment of the atoms of the liquid phase to their lattic... read less USED (low confidence) M. Kozłowski, D. Scopece, J. Janczak-Rusch, L. Jeurgens, R. Abdank-Kozubski, and D. Passerone, “Validation of an Embedded-Atom Copper Classical Potential via Bulk and Nanostructure Simulations,” Diffusion Foundations. 2017. link Times cited: 0 Abstract: The validation of classical potentials for describing multic… read moreAbstract: The validation of classical potentials for describing multicomponent materials in complex geometries and their high temperature structural modifications (disordering and melting) requires to verify both a faithful description of the individual phases and a convincing scheme for the mixed interactions, like it is the case of the embedded atom scheme. The present paper addresses the former task for an embedded atom potential for copper, namely the widely adopted parametrization by Zhou, through application to bulk, surface and nanocluster systems. It is found that the melting point is underestimated by 200 degrees with respect to experiment, but structural and temperature-dependent properties are otherwise faithfully reproduced. read less USED (low confidence) N. Burbery, R. Das, and W. Ferguson, “Dynamic behaviour of mixed dislocations in FCC metals under multi-oriented loading with molecular dynamics simulations,” Computational Materials Science. 2017. link Times cited: 10 USED (low confidence) C. Ruestes, E. Bringa, Y. Gao, and H. Urbassek, “Molecular Dynamics Modeling of Nanoindentation.” 2017. link Times cited: 35 USED (low confidence) H. Y. Song, J. Xu, Y. G. Zhang, S. Li, D. Wang, and Y. L. Li, “Molecular dynamics study of deformation behavior of crystalline Cu/amorphous Cu50Zr50 nanolaminates,” Materials & Design. 2017. link Times cited: 58 USED (low confidence) M. SamsonovVladimir, G. BembelAlexei, PopovIlya, A. VasilyevSergey, and TalyzinIgor, “Solid-state wetting at the nanoscale: molecular dynamics and surface diffusion approach,” Surface Innovations. 2017. link Times cited: 7 Abstract: The authors simulated spreading of solid copper (Cu) and gol… read moreAbstract: The authors simulated spreading of solid copper (Cu) and gold (Au) nanoparticles (5–7 nm size) on the (100) face of the same metal by using molecular dynamics. Then, the results obtained for the co... read less USED (low confidence) P. Piaggi and M. Parrinello, “Entropy based fingerprint for local crystalline order.,” The Journal of chemical physics. 2017. link Times cited: 87 Abstract: We introduce a new fingerprint that allows distinguishing be… read moreAbstract: We introduce a new fingerprint that allows distinguishing between liquid-like and solid-like atomic environments. This fingerprint is based on an approximate expression for the entropy projected on individual atoms. When combined with local enthalpy, this fingerprint acquires an even finer resolution and it is capable of discriminating between different crystal structures. read less USED (low confidence) F. Ye, H. Xv, J. Liu, and K. Tong, “Effects of Uniaxial Strain on the Structures of Vacancy Clusters in FCC Metals,” Materials Science Forum. 2017. link Times cited: 1 Abstract: The effects of [001] uniaxial strain on the stable structure… read moreAbstract: The effects of [001] uniaxial strain on the stable structures and structural evolution of vacancy clusters in fcc metals, Cu, Ni, Al and Fe, have been studied and compared. Under uniaxial strain, the clusters in all these metals tend to align parallel or perpendicular to the strain axis under tensile or compressive strain. Moreover, both the body cluster and the {001} planar cluster become the dominant types. In addition, the stacking fault tetrahedron cluster becomes another dominant type in Al under compressive strain. The cluster structures in Fe are disordered under strain possibly because the pure fcc Fe is thermodynamically unstable under the current simulation condition. read less USED (low confidence) X. Zhou and J. Song, “Effect of local stress on hydrogen segregation at grain boundaries in metals,” Materials Letters. 2017. link Times cited: 19 USED (low confidence) D. S. Kryzhevich, A. Korchuganov, K. Zolnikov, and S. Y. Korostelev, “Features of plastic deformation nucleation in the elastically loaded aluminium crystallites during irradiation,” Journal of Physics: Conference Series. 2017. link Times cited: 0 Abstract: The simulation of peculiarities of the surface layer reconst… read moreAbstract: The simulation of peculiarities of the surface layer reconstruction in the crystallites of aluminum after the ion bombardment and the copper film irradiated by the electron beam is carried out. The performed calculations are based on the molecular dynamics method. It is shown that the orientation of the irradiated surface and preliminary elastic deformation have a significant impact on features of atomic structure formation in the ion-modified layer in aluminum. Weak structural changes in the surface layer are observed at the irradiation of the {100} surfaces. Sufficiently great number of stacking faults is formed under irradiation of the {111} and {110} surfaces. It is shown that heating by the electron beam of the {110} surface in the copper film leads to the formation of stacking faults. It is shown that the preliminary elastic deformation of the material lowers the energy of the irradiation, at which formation of structural defects and fragmentation of crystallites of aluminum and copper will take place. read less USED (low confidence) S. Paul, S. Kumar, and S. Tarafder, “Effect of loading conditions on nucleation of nano void and failure of nanocrystalline aluminum: An atomistic investigation,” Engineering Fracture Mechanics. 2017. link Times cited: 16 USED (low confidence) S. Chandra, N. Kumar, M. K. Samal, V. Chavan, and S. Raghunathan, “An atomistic insight into the fracture behavior of bicrystal aluminum containing twist grain boundaries,” Computational Materials Science. 2017. link Times cited: 25 USED (low confidence) J. Xu, M. Xiang, B. Dang, and Z. Jian, “Relation of cooling rate, undercooling and structure for rapid solidification of iron melt,” Computational Materials Science. 2017. link Times cited: 17 USED (low confidence) D. Kryzhevich, A. Korchuganov, K. Zolnikov, and S. Psakhie, “Features of structural changes in aluminum specimens with various crystallographic orientation under ion irradiation.” 2016. link Times cited: 0 Abstract: Atomic structure changes of surface layers of aluminum cryst… read moreAbstract: Atomic structure changes of surface layers of aluminum crystallites after ion bombardment are studied. The molecular dynamics simulation is used for the investigation of structural changes in irradiated crystallites. The results of calculations show that orientation of the irradiated surface and preliminary elastic deformation has a significant impact on features of formation of the atomic structure in the ion-modified layer. The minimal changes in the surface layer are observed at the irradiation of surfaces with {100} lattice planes. A sufficiently great number of stacking faults was formed under irradiation of {111} and {110} surfaces. On the base of the simulation, it can be expected that the process of the surface structure fragmentation is most favorable for cases when surfaces with the {111} and {110} lattice planes are irradiated. An increase in the degree of preliminary elastic deformation can reduce the energy of incident particles, which is necessary for fragmentation of the surface structure. read less USED (low confidence) V. Borovikov, M. Mendelev, and A. King, “Effects of stable and unstable stacking fault energy on dislocation nucleation in nano-crystalline metals,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 50 Abstract: Dislocation nucleation from grain boundaries (GB) can contro… read moreAbstract: Dislocation nucleation from grain boundaries (GB) can control plastic deformation in nano-crystalline metals under certain conditions, but little is known about what controls dislocation nucleation, because when data from different materials are compared, the variations of many interacting properties tend to obscure the effects of any single property. In this study, we seek clarification by applying a unique capability of semi-empirical potentials in molecular dynamics simulations: the potentials can be modified such that all significant material properties but one, are kept constant. Using a set of potentials developed to isolate the effects of stacking fault energy, we show that for a given grain boundary, loading orientation and strain rate, the yield stress depends linearly on both the stable and unstable stacking fault energies. The coefficients of proportionality depend on the GB structure and the value of the yield stress is related to the density of the E structural units in the GB. While the impact of the stable stacking fault energy is easy to understand, the unstable stacking fault energy requires more elucidation and we provide a framework for understanding how it affects the nucleation and propagation process. read less USED (low confidence) N. Lopanitsyna and A. Kuksin, “Nucleation and the spall strength of liquid metals,” Journal of Physics: Conference Series. 2016. link Times cited: 2 Abstract: This article presents calculation of the nucleation rate for… read moreAbstract: This article presents calculation of the nucleation rate for liquid metals (Al, Fe, Mo) based on molecular dynamic simulation for embedded atom method (EAM) potentials. The dependence of nucleation rate on pressure and temperature could be approximated accurately in the form of classical nucleation theory taking into account surface tension dependency on pore radius σ = σ0/(1 + 2δ/r), where σ—surface tension, δ—the Tolman length. Basing on the results of the calculations, we have developed a model allowing calculating the spall strength of liquid metals under tension using such parameters as surface tension, viscosity, which could be measured experimentally. The obtained results for Mo and Al are consistent with experimental data and direct MD calculations at strain rates approx. 1010-1011 s-1. read less USED (low confidence) N. Burbery, R. Das, and W. Ferguson, “Thermo-kinetic mechanisms for grain boundary structure multiplicity, thermal instability and defect interactions,” Materials Chemistry and Physics. 2016. link Times cited: 9 USED (low confidence) P. White, S. Barter, and N. Medhekar, “Hydrogen induced amorphisation around nanocracks in aluminium,” Engineering Fracture Mechanics. 2016. link Times cited: 10 USED (low confidence) R. Mishra and R. Lalneihpuii, “Test of the universal scaling law for square well liquid metals,” Journal of Non-crystalline Solids. 2016. link Times cited: 6 USED (low confidence) N. Gunkelmann, Y. Rosandi, C. Ruestes, E. Bringa, and H. Urbassek, “Compaction and plasticity in nanofoams induced by shock waves: A molecular dynamics study,” Computational Materials Science. 2016. link Times cited: 24 USED (low confidence) R. Aghababaei, D. Warner, and J. Molinari, “Critical length scale controls adhesive wear mechanisms,” Nature Communications. 2016. link Times cited: 201 USED (low confidence) W. Fang, H. Xie, F. Yin, J. Li, D. Khan, and F. Qian, “Molecular dynamics simulation of grain boundary geometry on crack propagation of bi-crystal aluminum,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2016. link Times cited: 39 USED (low confidence) S. M. Rassoulinejad-Mousavi, Y. Mao, and Y. Zhang, “Evaluation of Copper, Aluminum and Nickel Interatomic Potentials on Predicting the Elastic Properties,” arXiv: Computational Physics. 2016. link Times cited: 63 Abstract: Choice of appropriate force field is one of the main concern… read moreAbstract: Choice of appropriate force field is one of the main concerns of any atomistic simulation that needs to be seriously considered in order to yield reliable results. Since, investigations on mechanical behavior of materials at micro/nanoscale has been becoming much more widespread, it is necessary to determine an adequate potential which accurately models the interaction of the atoms for desired applications. In this framework, reliability of multiple embedded atom method based interatomic potentials for predicting the elastic properties was investigated. Assessments were carried out for different copper, aluminum and nickel interatomic potentials at room temperature which is considered as the most applicable case. Examined force fields for the three species were taken from online repositories of National Institute of Standards and Technology (NIST), as well as the Sandia National Laboratories, the LAMMPS database. Using molecular dynamic simulations, the three independent elastic constants, C11, C12 and C44 were found for Cu, Al and Ni cubic single crystals. Voigt-Reuss-Hill approximation was then implemented to convert elastic constants of the single crystals into isotropic polycrystalline elastic moduli including Bulk, Shear and Young's modulus as well as Poisson's ratio. Simulation results from massive molecular dynamic were compared with available experimental data in the literature to justify the robustness of each potential for each species. Eventually, accurate interatomic potentials have been recommended for finding each of the elastic properties of the pure species. Exactitude of the elastic properties was found to be sensitive to the choice of the force fields. Those potentials were fitted for a specific compound may not necessarily work accurately for all the existing pure species. read less USED (low confidence) F. Ye, J. Liu, K. Tong, Z. Li, H. Che, and M. Lei, “Effects of uniaxial strain on stability and structural evolution of vacancy clusters in copper,” Computational Materials Science. 2016. link Times cited: 5 USED (low confidence) R. Babicheva et al., “Effect of grain boundary segregation on the deformation mechanisms and mechanical properties of nanocrystalline binary aluminum alloys,” Computational Materials Science. 2016. link Times cited: 49 USED (low confidence) L.-N. Wu, C. Li, B.-ye Xu, Q. Li, and W. Liu, “Study of the Temperature Effects on Solid‐Liquid Anisotropic Interfacial Energy.” 2016. link Times cited: 0 USED (low confidence) L. Wu, B. Xu, Q. L. Li, and W. Liu, “Self-instability of finite sized solid-liquid interfaces,” Scientific Reports. 2015. link Times cited: 6 USED (low confidence) T. Wang, C. Begau, G. Sutmann, and A. Hartmaier, “Large scale Molecular Dynamics simulation of microstructure formation during thermal spraying of pure copper,” Surface & Coatings Technology. 2015. link Times cited: 15 USED (low confidence) Z. Hou et al., “Atomic dynamics of grain boundaries in bulk nanocrystalline aluminium: A molecular dynamics simulation study,” Computational Materials Science. 2015. link Times cited: 10 USED (low confidence) E. S. Wise, M. Liu, and T. Miller, “Sputtering of cubic metal crystals by low-energy xenon-ions,” Computational Materials Science. 2015. link Times cited: 5 USED (low confidence) S. Kalidindi, J. A. Gomberg, Z. Trautt, and C. Becker, “Application of data science tools to quantify and distinguish between structures and models in molecular dynamics datasets,” Nanotechnology. 2015. link Times cited: 39 Abstract: Structure quantification is key to successful mining and ext… read moreAbstract: Structure quantification is key to successful mining and extraction of core materials knowledge from both multiscale simulations as well as multiscale experiments. The main challenge stems from the need to transform the inherently high dimensional representations demanded by the rich hierarchical material structure into useful, high value, low dimensional representations. In this paper, we develop and demonstrate the merits of a data-driven approach for addressing this challenge at the atomic scale. The approach presented here is built on prior successes demonstrated for mesoscale representations of material internal structure, and involves three main steps: (i) digital representation of the material structure, (ii) extraction of a comprehensive set of structure measures using the framework of n-point spatial correlations, and (iii) identification of data-driven low dimensional measures using principal component analyses. These novel protocols, applied on an ensemble of structure datasets output from molecular dynamics (MD) simulations, have successfully classified the datasets based on several model input parameters such as the interatomic potential and the temperature used in the MD simulations. read less USED (low confidence) L. Pei et al., “Brittle versus ductile fracture behaviour in nanotwinned FCC crystals,” Materials Letters. 2015. link Times cited: 15 USED (low confidence) T. Junge, G. Anciaux, and J. Molinari, “Dynamic stability of displacement-based atomistic/continuum coupling methods,” Journal of The Mechanics and Physics of Solids. 2015. link Times cited: 12 USED (low confidence) Z. Hou, Z.-an Tian, R.-su Liu, K. Dong, and A. Yu, “Formation mechanism of bulk nanocrystalline aluminium with multiply twinned grains by liquid quenching: A molecular dynamics simulation study,” Computational Materials Science. 2015. link Times cited: 31 USED (low confidence) Y. Gao, C. Ruestes, D. Tramontina, and H. Urbassek, “Comparative simulation study of the structure of the plastic zone produced by nanoindentation,” Journal of The Mechanics and Physics of Solids. 2015. link Times cited: 111 USED (low confidence) A. Murin and I. Shabanova, “Comparative study of local atomic structure of liquid and supercooled Cu, Ni, And Au,” Surface and Interface Analysis. 2014. link Times cited: 1 Abstract: In the present paper, the results of the molecular‐dynamics … read moreAbstract: In the present paper, the results of the molecular‐dynamics simulation of Ni, Cu and Au in liquid and supercooled liquid states are displayed. The potentials of interatomic interaction within the framework of the embedded‐atom method are used to generate realistic atomic configurations. The structural analysis of the cluster structure has been conducted with the use of the bond orientational order parameter. In contrast with previous findings, we take into the account 5d‐metal (Au) and using the same approach for simulation of different liquid metals. It is shown that the local icosahedral order is present, and it enhances at supercooling of the melts of the metals under discussion. The results are insensitive to the model describing interatomic bonds and the size of the system. Such behavior is most general from the topological perspective for d‐metals with close‐packed premelting structure. Copyright © 2014 John Wiley & Sons, Ltd. read less USED (low confidence) Z. Hou, Z.-an Tian, Y. Mo, and R.-su Liu, “Local atomic structures in grain boundaries of bulk nanocrystalline aluminium: A molecular dynamics simulation study,” Computational Materials Science. 2014. link Times cited: 15 USED (low confidence) Y. Sun et al., “‘Crystal Genes’ in Metallic Liquids and Glasses,” Scientific Reports. 2014. link Times cited: 59 USED (low confidence) Y. Zhou and K. Fichthorn, “Internal Stress-Induced Orthorhombic Phase in 5-Fold-Twinned Noble Metal Nanowires,” Journal of Physical Chemistry C. 2014. link Times cited: 28 Abstract: We use atomistic simulations to show that 5-fold-twinned nan… read moreAbstract: We use atomistic simulations to show that 5-fold-twinned nanowires of several face-centered cubic metals (Ag, Au, Cu, and Pd) exhibit an overall body-centered orthorhombic phase resulting from the large internal stress associated with their twinned structure. The distribution of atomic stress in the nanowires confirms the existence of a disclination at the 5-fold axis, in addition to an anisotropic distortion of the lattice. We find that two regions of the nanowire are highly stressed: local stress maxima are distributed in the shape of leaflets running along each twin boundary, as well as in semicircular regions near the free surfaces. The large elastic strain energy associated with the distortion may be partially released via the formation and propagation of partial dislocations, which are restricted to a single subunit of the nanowire. Our calculations are in line with experiment and indicate the complex ways in which the structures of these metal nanocrystals can depend on their shape. read less USED (low confidence) Y. Rosandi and H. Urbassek, “Ablation of a nanostructured metal surface by ultrashort X-ray pulses,” Applied Surface Science. 2014. link Times cited: 4 USED (low confidence) T. Okita, K. Asari, S. Fujita, and M. Itakura, “Effect of the Stacking Fault Energy on Interactions between an Edge Dislocation and a Spherical Void in FCC Metals at Various Spatial Geometries,” Fusion Science and Technology. 2014. link Times cited: 10 Abstract: Molecular dynamics simulations were conducted using six inte… read moreAbstract: Molecular dynamics simulations were conducted using six interatomic potentials for face-centered cubic metals that differed only in the stacking fault energies (SFEs). We investigated the effects of the SFE on interactions between an edge dislocation and a void of 4.0 nm diameter at 13 intersection positions. In the high SFE, most interaction morphologies at the depinning are such that the two partial dislocations reverse into the perfect dislocation locally at the void interface. In contrast, in the low SFE, the partial dislocations are depinned individually from the void with some certain time lag. The critical resolved shear stress (CRSS) is not symmetrical about the center of the void. CRSS is higher when the center of the void is located not on the glide plane, but in the compressive side of the edge dislocation. In some cases for these conditions, climb motion is observed, which further increases CRSS. The probability of climb motion occurrence is higher with higher SFE. In lower SFE, climb motion occurs temporarily, followed by the disappearance of jog by dislocation releasing several vacancies inside of the void. CRSS is higher with higher SFE for all the intersection positions. read less USED (low confidence) L. Sun, X. He, J. Wang, and J. Lu, “Deformation and failure mechanisms of nanotwinned copper films with a pre-existing crack,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2014. link Times cited: 22 USED (low confidence) E. Asadi, M. A. Zaeem, A. Moitra, and M. Tschopp, “Effect of vacancy defects on generalized stacking fault energy of fcc metals,” Journal of Physics: Condensed Matter. 2014. link Times cited: 28 Abstract: Molecular dynamics (MD) and density functional theory (DFT) … read moreAbstract: Molecular dynamics (MD) and density functional theory (DFT) studies were performed to investigate the influence of vacancy defects on generalized stacking fault (GSF) energy of fcc metals. MEAM and EAM potentials were used for MD simulations, and DFT calculations were performed to test the accuracy of different common parameter sets for MEAM and EAM potentials in predicting GSF with different fractions of vacancy defects. Vacancy defects were placed at the stacking fault plane or at nearby atomic layers. The effect of vacancy defects at the stacking fault plane and the plane directly underneath of it was dominant compared to the effect of vacancies at other adjacent planes. The effects of vacancy fraction, the distance between vacancies, and lateral relaxation of atoms on the GSF curves with vacancy defects were investigated. A very similar variation of normalized SFEs with respect to vacancy fractions were observed for Ni and Cu. MEAM potentials qualitatively captured the effect of vacancies on GSF. read less USED (low confidence) T. Junge, J. Molinari, and G. Anciaux, “Plastic activity in nanoscratch molecular dynamics simulations of pure aluminum.” 2014. link Times cited: 59 USED (low confidence) K. Asari, Ø. Hetland, S. Fujita, M. Itakura, and T. Okita, “The effect of stacking fault energy on interactions between an edge dislocation and a spherical void by molecular dynamics simulations,” Journal of Nuclear Materials. 2013. link Times cited: 24 USED (low confidence) X. Zheng, H. Zhu, A. K. Tieu, and B. Kosasih, “A molecular dynamics simulation of 3D rough lubricated contact,” Tribology International. 2013. link Times cited: 42 USED (low confidence) Yuan-Yuan 圆圆 Ju 巨, Q. Zhang 张, Zi-Zheng 自正 Gong 龚, and Guang-Fu 广富 Ji 姬, “Molecular dynamics simulation of self-diffusion coefficients for liquid metals,” Chinese Physics B. 2013. link Times cited: 25 Abstract: The temperature-dependent coefficients of self-diffusion for… read moreAbstract: The temperature-dependent coefficients of self-diffusion for liquid metals are simulated by molecular dynamics methods based on the embedded-atom-method (EAM) potential function. The simulated results show that a good inverse linear relation exists between the natural logarithm of self-diffusion coefficients and temperature, though the results in the literature vary somewhat, due to the employment of different potential functions. The estimated activation energy of liquid metals obtained by fitting the Arrhenius formula is close to the experimental data. The temperature-dependent shear-viscosities obtained from the Stokes—Einstein relation in conjunction with the results of molecular dynamics simulation are generally consistent with other values in the literature. read less USED (low confidence) P. White, “Molecular dynamic modelling of fatigue crack growth in aluminium using LEFM boundary conditions,” International Journal of Fatigue. 2012. link Times cited: 27 USED (low confidence) M. Asta, “Computational Investigations of Solid-Liquid Interfaces.” 2011. link Times cited: 0 Abstract: In a variety of materials synthesis and processing contexts,… read moreAbstract: In a variety of materials synthesis and processing contexts, atomistic processes at heterophase interfaces play a critical role governing defect formation, growth morphologies, and microstructure evolution. Accurate knowledge of interfacial structure, free energies, mobilities and segregation coefficients are critical for predictive modeling of microstructure evolution, yet direct experimental measurement of these fundamental interfacial properties remains elusive in many cases. In this project first-principles calculations were combined with molecular-dynamics (MD) and Monte-Carlo (MC) simulations, to investigate the atomic-scale structural and dynamical properties of heterophase interfaces, and the relationship between these properties and the calculated thermodynamic and kinetic parameters that influence the evolution of phase transformation structures at nanometer to micron length scales. The topics investigated in this project were motivated primarily by phenomena associated with solidification processing of metals and alloys, and the main focus of the work was thus on solid-liquid interfaces and high-temperature grain boundaries. Additional efforts involved first-principles calculations of coherent solid-solid heterophase interfaces, where a close collaboration with researchers at the National Center for Electron Microscopy was undertaken to understand the evolution of novel core-shell precipitate microstructures in aluminum alloys. read less USED (low confidence) B. Jelinek et al., “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Physical Review B. 2011. link Times cited: 218 Abstract: A set of modified embedded-atom method (MEAM) potentials for… read moreAbstract: A set of modified embedded-atom method (MEAM) potentials for the interactions between Al, Si, Mg, Cu, and Fe was developed from a combination of each element's MEAM potential in order to study metal alloying. Previously published MEAM parameters of single elements have been improved for better agreement to the generalized stacking fault energy (GSFE) curves when compared with ab initio generated GSFE curves. The MEAM parameters for element pairs were constructed based on the structural and elastic properties of element pairs in the NaCl reference structure garnered from ab initio calculations, with adjustment to reproduce the ab initio heat of formation of the most stable binary compounds. The new MEAM potentials were validated by comparing the formation energies of defects, equilibrium volumes, elastic moduli, and heat of formation for several binary compounds with ab initio simulations and experiments. Single elements in their ground-state crystal structure were subjected to heating to test the potentials at elevated temperatures. An Al potential was modified to avoid formation of an unphysical solid structure at high temperatures. The thermal expansion coefficient of a compound with the composition of AA 6061 alloy was evaluated and compared with experimental values. MEAM potential tests performed in this work, utilizing the universal atomistic simulation environment (ASE), are distributed to facilitate reproducibility of the results. read less USED (low confidence) J. Fan, “Basics of Atomistic Simulation.” 2010. link Times cited: 1 USED (low confidence) E. Holm, D. Olmsted, and S. Foiles, “Comparing grain boundary energies in face-centered cubic metals: Al, Au, Cu and Ni,” Scripta Materialia. 2010. link Times cited: 124 USED (low confidence) B. Qin and W. Lai, “A Molecular Dynamics Study of an hcp-to-fco Martensitic Transformation and Its Crystallographic Correlation in a Ti Lattice, upon Dissolution of Cu Atoms,” Journal of the Physical Society of Japan. 2010. link Times cited: 1 Abstract: A hexagonal close-packed to face-centered orthorhombic (hcp-… read moreAbstract: A hexagonal close-packed to face-centered orthorhombic (hcp-to-fco) transformation in a Ti lattice, upon dissolution of Cu atoms, is observed by molecular dynamics simulations based on a proven realistic many-body Cu–Ti potential. It was found that the phase transformation takes place through two main simultaneous actions, i.e., lattice elongation along the close-packed directions and relative movement or slide between adjacent close-packed planes along the directions. By analyzing the crystallographic relationship between the hcp, bcc (body-centered cubic), and fco structures, it is proposed that the phase transformation occurs through a reverse mechanism, for the body-centered cubic to hexagonal closed-packed (bcc-to-hcp) phase transformation, together with lattice constant adjustments. read less USED (low confidence) B. Qin and W. Lai, “Crystalline to Amorphous Transition and Relative Stability of Amorphous Phase versus Solid Solution in the Cu–Ti System Studied by Molecular Dynamics Simulations,” Journal of the Physical Society of Japan. 2010. link Times cited: 3 Abstract: A relative stability of solid solutions versus amorphous pha… read moreAbstract: A relative stability of solid solutions versus amorphous phases in the Cu–Ti system has been studied by molecular dynamics simulations using an n -body potential. The simulations have shown that for the Cu x Ti 100- x alloys, a spontaneous crystalline to amorphous transition takes place in the central composition region and that the energy of amorphous phases is lower than the solid solutions at 22≤ x ≤71 at. % owing to a fast energy drop of the former than the latter, in accordance with the thermodynamics analysis and experimental results. The detailed structural analysis showed that for the Cu-rich side, with increasing the Ti concentration, both the total average coordination number and the short range order chemically favoring unlike bonds are better allowed and more pronounced in the amorphous state than in the fcc solid solution. Therefore, the relative stability of both phases revealed by difference of energy is correlated with the structure change of the amorphous phase. read less USED (low confidence) Z. Wu, Y.-W. Zhang, and D. Srolovitz, “Grain boundary finite length faceting,” Acta Materialia. 2009. link Times cited: 26 USED (low confidence) L. Hung and E. Carter, “Accurate simulations of metals at the mesoscale: Explicit treatment of 1 million atoms with quantum mechanics,” Chemical Physics Letters. 2009. link Times cited: 97 USED (low confidence) M. Mendelev and Y. Mishin, “Molecular dynamics study of self-diffusion in bcc Fe,” Physical Review B. 2009. link Times cited: 99 Abstract: A semiempirical interatomic potential for Fe was used to cal… read moreAbstract: A semiempirical interatomic potential for Fe was used to calculate the diffusivity in bcc Fe assuming the vacancy and interstitial mechanisms of self-diffusion. Point-defect concentrations and diffusivities were obtained directly from molecular dynamics (MD) simulations. It was found that self-diffusion in bcc Fe is controlled by the vacancy mechanism at all temperatures. This result is due to the fact that the equilibrium vacancy concentration is always much larger than the equilibrium interstitial concentration. The predominance of the equilibrium vacancy concentration over the interstitial concentration is explained by the lower vacancy-formation energy at low temperatures and high vacancy-formation entropy at high temperatures. The calculated diffusivity is in good agreement with experimental data. The MD simulations were also used to test the quasiharmonic (QH) approximation for point-defect calculations. It was found that the QH approximation can considerably underestimate variations in point-defect characteristics with temperature. read less USED (low confidence) M. Mendelev, R. Ott, M. Kramer, and D. Sordelet, “Determining strain in amorphous alloys: Uncertainties with analyzing structural changes during deformation,” Journal of Applied Physics. 2009. link Times cited: 1 Abstract: Molecular dynamics simulations were utilized to test the rel… read moreAbstract: Molecular dynamics simulations were utilized to test the reliability of strain values obtained from diffraction data for noncrystalline alloys. We found that in the case of a one-component system, the strain value obtained from the pair correlation functions underestimates the actual value because of a small degree of atomic relaxations, which minimize the effects of the applied deformation. In the case of multicomponent systems, the different pairs are affected by applied deformation to different extents; moreover, this implies that the strain value determined from diffraction data should depend on the type of scattering. read less USED (low confidence) S. Y. Korostelev, E. E. Slyadnikov, and I. Turchanovsky, “The resistance of amorphous metals to thermal effects. Molecular dynamics modeling,” PHYSICAL MESOMECHANICS OF CONDENSED MATTER: Physical Principles of Multiscale Structure Formation and the Mechanisms of Nonlinear Behavior: MESO2022. 2023. link Times cited: 0 USED (low confidence) I. Shepelev and E. Korznikova, “Dependence of the supersonic propagation of 2-crowdions on the stacking fault energy in FCC metals,” MATHEMATICS EDUCATION AND LEARNING. 2022. link Times cited: 0 USED (low confidence) S. Kim, H. Hwang, and J. Huh, “Phase-field simulations of dendritic morphologies in hot-dip galvanized Zn-Al coatings,” Computational Materials Science. 2021. link Times cited: 3 USED (low confidence) Z. Yan, H. Sheng, E. Ma, B. Xu, J. Li, and L. Kong, “Intermediate structural evolution preceding growing BCC crystal interface in deeply undercooled monatomic metallic liquids,” Acta Materialia. 2021. link Times cited: 6 USED (low confidence) A. Tran and Y. Wang, “Reliable molecular dynamics simulations for intrusive uncertainty quantification using generalized interval analysis.” 2020. link Times cited: 0 USED (low confidence) L.-X. Liu et al., “Mechanical behaviour of rapidly solidified aluminium with multiple twinned nanograins: A molecular dynamics simulation study,” Computational Materials Science. 2019. link Times cited: 2 USED (low confidence) L. Zou et al., “Dislocation nucleation facilitated by atomic segregation.,” Nature materials. 2018. link Times cited: 87 USED (low confidence) D. S. Kryzhevich, K. Zolnikov, and A. Korchuganov, “Atomic mechanisms of grain structure restructuring in surface of aluminum during ion implantation,” Journal of Physics: Conference Series. 2018. link Times cited: 0 Abstract: The molecular dynamics study of features of structural rearr… read moreAbstract: The molecular dynamics study of features of structural rearrangements in the surface layer of the aluminum crystallite under ion irradiation was carried out. It is shown that the number of generated defects in the crystallite depends on the irradiation dose and the crystallographic orientation of the irradiated surface. It is revealed that significantly more defects were formed at the irradiation of {111} and {110} surfaces than of the {100} surface. At low irradiation doses the stacking faults were formed only in some grains of the nanocrystalline material. The higher irradiation doses led to grain structure changes in the surface layer. This is connected with melting and crystallization of the surface layer. The grains in the region of the boundary of liquid and crystal phases were centers of crystallization of the liquid surface layer. read less USED (low confidence) A. Matsushita, S. Takamoto, A. Hatano, and S. Izumi, “Development of Hybrid Method Using Ab initio and Classical Molecular Dynamics for Calculating the Thermal Expansion Coefficient of Alloys at High Temperature,” Journal of The Society of Materials Science, Japan. 2018. link Times cited: 0 Abstract: In order to predict the thermal expansion coefficient (TEC),… read moreAbstract: In order to predict the thermal expansion coefficient (TEC), quasiharmonic approximation based on the ab initio electronic structure calculation is an effective and conventional scheme. However, it is known that the deviation due to anharmonic effect arises at high temperature. Although Molecular Dynamics (MD) naturally includes the anharmonic effect, classical MD is lacking in accuracy because of its empirical interatomic potential and ab initio MD is not applicable because of its high computational cost. In this paper, we have proposed a new hybrid method using ab initio electronic structure calculation and classical molecular dynamics for calculating TEC of alloys at high temperature. Our method is non-empirical, highly accurate and computationally inexpensive. The method consists of three steps. Firstly, various snapshots of the atomic coordination at a certain temperature are sampled by classical MD. Secondly, physical properties of each snapshot are calculated by ab initio electronic structure calculation. Finally, by analyzing them statistically, the equilibrium volume is computed. TEC is obtained by repeating these steps at different temperatures. We have calculated the TECs of Al, Ni3Al, and NiAl. Results show good agreements with experimental results. Our method enables us to improve the conventional quasiharmonic approximation and obtain accurate TEC at high temperature through the incorporation of anharmonic effect. read less USED (low confidence) D. Kryzhevich, A. Korchuganov, and K. Zolnikov, “Modification of grain structure of the near-surface layer in aluminum under high energy impact.” 2017. link Times cited: 0 USED (low confidence) R. Ferrando, “Theoretical and computational methods for nanoalloy structure and thermodynamics.” 2016. link Times cited: 5 USED (low confidence) A. Ovrutsky, A. Prokhoda, and M. Rasshchupkyna, “Modern Simulations by the Molecular Dynamics Method.” 2014. link Times cited: 0 USED (low confidence) T. Junge and J. Molinari, “Molecular dynamics nano-scratching of aluminium: a novel quantitative energy-based analysis method,” Procedia IUTAM. 2012. link Times cited: 14 USED (low confidence) A. Iskandarov, S. Dmitriev, and Y. Umeno, “On Accurate Approach for Molecular Dynamics Study of Ideal Strength at Elevated Temperature,” Journal of Solid Mechanics and Materials Engineering. 2012. link Times cited: 3 Abstract: Influence of temperature on ideal shear strength (ISS), τc, … read moreAbstract: Influence of temperature on ideal shear strength (ISS), τc, of two fcc metals (Al and Cu) was studied by means of molecular dynamics simulations. To get reliable results we investigated influence of parameters of the applied Parrinello-Rahman stress control method and implemented damping of simulation cell fluctuations to avoid occurrence of structural instability assisted by too high fluctuations. The damping successfully reduces strain and stress fluctuations during simulations if the damping factor is specified properly. We also investigated simulation cell size effect to evaluate minimal number of atoms providing reliable results in order to reduce computational efforts and estimate the possibility of applying ab initio calculations. Recently developed embedded atom method (EAM) interatomic potentials for both metals were also examined to find most appropriate for our study. EAM potential developed by Zope et al. and Mishin et al. were revealed to be most suitable for Al and Cu, respectively. It is essential to choose appropriate simulation parameters and interatomic potentials for the valid evaluation of ISS at elevated temperatures. We find almost linear decrease in ideal strength with increasing temperature for [112](111) shear deformation, while critical strain decreases in a nonlinear manner. At room temperature, reduction of shear strength for Al(Cu) is less than 35%(25%) compared to that at 0 K. read less USED (low confidence) A. Ovrutsky and A. Prokhoda, “Peculiarities of crystallization at high undercooling: Analysis of the simulation data for aluminum,” Journal of Crystal Growth. 2011. link Times cited: 4 NOT USED (low confidence) A. Khoei, M. R. Seddighian, and A. R. Sameti, “Machine learning-based multiscale framework for mechanical behavior of nano-crystalline structures,” International Journal of Mechanical Sciences. 2023. link Times cited: 0 NOT USED (low confidence) B. Yao, Z. R. Liu, D. Legut, and R. F. Zhang, “Hybrid potential model with high feasibility and flexibility for metallic and covalent solids,” Physical Review B. 2023. link Times cited: 0 NOT USED (low confidence) K. Dang, J. Chen, B. Rodgers, and S. Fensin, “LAVA 1.0: A general-purpose python toolkit for calculation of material properties with LAMMPS and VASP,” Comput. Phys. Commun. 2023. link Times cited: 2 NOT USED (low confidence) M. Cioni, D. Polino, D. Rapetti, L. Pesce, M. D. Piane, and G. Pavan, “Innate dynamics and identity crisis of a metal surface unveiled by machine learning of atomic environments.,” The Journal of chemical physics. 2022. link Times cited: 5 Abstract: Metals are traditionally considered hard matter. However, it… read moreAbstract: Metals are traditionally considered hard matter. However, it is well known that their atomic lattices may become dynamic and undergo reconfigurations even well below the melting temperature. The innate atomic dynamics of metals is directly related to their bulk and surface properties. Understanding their complex structural dynamics is, thus, important for many applications but is not easy. Here, we report deep-potential molecular dynamics simulations allowing to resolve at an atomic resolution the complex dynamics of various types of copper (Cu) surfaces, used as an example, near the Hüttig (∼1/3 of melting) temperature. The development of deep neural network potential trained on density functional theory calculations provides a dynamically accurate force field that we use to simulate large atomistic models of different Cu surface types. A combination of high-dimensional structural descriptors and unsupervized machine learning allows identifying and tracking all the atomic environments (AEs) emerging in the surfaces at finite temperatures. We can directly observe how AEs that are non-native in a specific (ideal) surface, but that are, instead, typical of other surface types, continuously emerge/disappear in that surface in relevant regimes in dynamic equilibrium with the native ones. Our analyses allow estimating the lifetime of all the AEs populating these Cu surfaces and to reconstruct their dynamic interconversions networks. This reveals the elusive identity of these metal surfaces, which preserve their identity only in part and in part transform into something else under relevant conditions. This also proposes a concept of "statistical identity" for metal surfaces, which is key to understanding their behaviors and properties. read less NOT USED (low confidence) I. Toda-Caraballo, J. Wróbel, and D. Nguyen-Manh, “Generalized universal equation of states for magnetic materials: A novel formulation for an interatomic potential in Fe,” Physical Review Materials. 2022. link Times cited: 0 NOT USED (low confidence) M. Kulichenko et al., “The Rise of Neural Networks for Materials and Chemical Dynamics.,” The journal of physical chemistry letters. 2021. link Times cited: 35 Abstract: Machine learning (ML) is quickly becoming a premier tool for… read moreAbstract: Machine learning (ML) is quickly becoming a premier tool for modeling chemical processes and materials. ML-based force fields, trained on large data sets of high-quality electron structure calculations, are particularly attractive due their unique combination of computational efficiency and physical accuracy. This Perspective summarizes some recent advances in the development of neural network-based interatomic potentials. Designing high-quality training data sets is crucial to overall model accuracy. One strategy is active learning, in which new data are automatically collected for atomic configurations that produce large ML uncertainties. Another strategy is to use the highest levels of quantum theory possible. Transfer learning allows training to a data set of mixed fidelity. A model initially trained to a large data set of density functional theory calculations can be significantly improved by retraining to a relatively small data set of expensive coupled cluster theory calculations. These advances are exemplified by applications to molecules and materials. read less NOT USED (low confidence) S. Mohammadi, A. Montazeri, and H. Urbassek, “Geometrical aspects of nanofillers influence the tribological performance of Al-based nanocomposites,” Wear. 2020. link Times cited: 16 NOT USED (low confidence) Z. Trautt, F. Tavazza, and C. Becker, “Facilitating the selection and creation of accurate interatomic potentials with robust tools and characterization,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 14 Abstract: The Materials Genome Initiative seeks to significantly decre… read moreAbstract: The Materials Genome Initiative seeks to significantly decrease the cost and time of development and integration of new materials. Within the domain of atomistic simulations, several roadblocks stand in the way of reaching this goal. While the NIST Interatomic Potentials Repository hosts numerous interatomic potentials (force fields), researchers cannot immediately determine the best choice(s) for their use case. Researchers developing new potentials, specifically those in restricted environments, lack a comprehensive portfolio of efficient tools capable of calculating and archiving the properties of their potentials. This paper elucidates one solution to these problems, which uses Python-based scripts that are suitable for rapid property evaluation and human knowledge transfer. Calculation results are visible on the repository website, which reduces the time required to select an interatomic potential for a specific use case. Furthermore, property evaluation scripts are being integrated with modern platforms to improve discoverability and access of materials property data. To demonstrate these scripts and features, we will discuss the automation of stacking fault energy calculations and their application to additional elements. While the calculation methodology was developed previously, we are using it here as a case study in simulation automation and property calculations. We demonstrate how the use of Python scripts allows for rapid calculation in a more easily managed way where the calculations can be modified, and the results presented in user-friendly and concise ways. Additionally, the methods can be incorporated into other efforts, such as openKIM. read less NOT USED (low confidence) С. Волегов, Р. М. Герасимов, and Р. П. Давлятшин, “MODELS OF MOLECULAR DYNAMICS: A REVIEW OF EAM-POTENTIALS. PART 2. POTENTIALS FOR MULTI-COMPONENT SYSTEMS.” 2018. link Times cited: 1 Abstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликова… read moreAbstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликована: 29 июня 2018 г. В статье представлена вторая часть обзора современных подходов и работ, посвященных построению потенциалов межатомного взаимодействия с использованием методологии погруженного атома (EAM-потенциалы). Эта часть обзора посвящена одной из наиболее остро стоящих проблем в молекулярной динамике – вопросам построения потенциалов, которые были бы пригодны для описания структуры и физико-механических свойств многокомпонентных (в первую очередь – бинарных и тернарных) материалов. Отмечены первые работы, в которых предлагались подходы к построению функций перекрестного взаимодействия для сплавов никеля и меди – как с использованием методологии EAM, так и несколько отличающийся по процедуре построения потенциал типа Финисса-Синклера. Рассматриваются работы, в которых производится сопоставление различных подходов к построению потенциалов, а также к процедуре идентификации их параметров на примере одних и тех же многокомпонентных систем (типа Al-Ni или Cu-Au). Кроме того, особый интерес представляют некоторые тернарные системы, например Fe–Ni–Cr, W–H– He или U–Mo–Xe, которые являются ключевыми для материалов атомной энергетики и которые в последние годы активно изучаются как возможные материалы для использования в термоядерных ректорах. Приведены примеры работ, в которых предлагаются и исследуются потенциалы для описания многокомпонентных систем, пригодных для использования в аэрокосмической промышленности и изготовленных прежде всего на основе никеля. Рассмотрены результаты исследований различных интерметаллических соединений, отмечены работы, в которых при помощи построенного EAM потенциала удалось количественно точно описать фазовые диаграммы соединений и вычислить характеристики фазовых переходов. read less NOT USED (high confidence) A. Goryaeva et al., “Compact A15 Frank-Kasper nano-phases at the origin of dislocation loops in face-centred cubic metals,” Nature Communications. 2023. link Times cited: 2 NOT USED (high confidence) Y. L. Müller, L. Jeurgens, A. Antušek, and V. Turlo, “Atomistic Assessment of Melting Point Depression and Enhanced Interfacial Diffusion of Cu in Confinement with AlN.,” ACS applied materials & interfaces. 2022. link Times cited: 3 Abstract: The continuing trend in heterogeneous integration (i.e., min… read moreAbstract: The continuing trend in heterogeneous integration (i.e., miniaturization and diversification of devices and components) requires a fundamental understanding of the phase stability and diffusivity of nanoconfined metals in functional nanoarchitectures, such as nanomultilayers (NMLs). Nanoconfinement effects, such as interfacial melting and anomalous fast interfacial diffusion, offer promising engineering tools to enhance the reaction kinetics at low temperatures for targeted applications in the fields of joining, solid-state batteries, and low-temperature sintering technologies. In the present study, the phase stability and atomic mobility of confined metals in Cu/AlN NMLs were investigated by molecular dynamics, with the interatomic potential compared to the ab initio calculations of the Cu/AlN interface adhesion energy. Simulations of the structural evolution of Cu/AlN nanomultilayers upon heating in dependence on the Cu nanolayer thickness demonstrate the occurrence of interfacial premelting, a melting point depression, as well as extraordinary fast solid-state diffusion of confined Cu atoms along the defective heterogeneous interfaces. The model predictions rationalize recent experimental observations of premelting and anomalous fast interface diffusion of nanoconfined metals in nanostructured Cu/AlN brazing fillers at strikingly low temperatures. read less NOT USED (high confidence) V. Vardanyan, B. Linke, and H. Urbassek, “Distortion of a polycrystalline Al bar in a vice fixture: molecular dynamics analysis of grain movement and rotation,” The International Journal of Advanced Manufacturing Technology. 2021. link Times cited: 0 NOT USED (high confidence) Z. Yin, P. Zhu, and B. Li, “Study of Nanoscale Wear of SiC/Al Nanocomposites Using Molecular Dynamics Simulations,” Tribology Letters. 2021. link Times cited: 29 NOT USED (high confidence) L. Morrissey and S. Nakhla, “Considerations when calculating the mechanical properties of single crystals and bulk polycrystals from molecular dynamics simulations,” Molecular Simulation. 2020. link Times cited: 4 Abstract: ABSTRACT The choice of a proper interatomic potential is cri… read moreAbstract: ABSTRACT The choice of a proper interatomic potential is critical to obtaining accurate and realistic molecular dynamics results. However, previous studies that have tested the suitability of a potential to predict mechanical properties often do so using elastic constants from a triaxial stress state that ignores Poisson’s effect. While this method is suitable it is not consistent with macroscale experimental methods and cannot provide the complete loading behaviour. Further, there is a lack of knowledge as to whether accuracy in predicting elastic constants from a fixed volume condition indicates accuracy for elastic moduli from uniaxial tensile simulations. Moreover, those studies that did account for Poisson’s effect studied only one crystal orientation and thus assumed potential accuracy is independent of crystal orientation. Results from the current study demonstrated that accuracy of a potential is dependent on the crystal direction. Further, the most accurate potentials for elastic constants calculated using a fixed volume condition were not necessarily the most accurate at predicting elastic moduli from a physically realisable tension test. Finally, the Voigt Reuss Hill (VRH) method was shown to accurately predict polycrystalline mechanical properties from single crystal data as a function of temperature. read less NOT USED (high confidence) Y. Chen, Z. Guan, Y. Yao, and H. Wang, “Tuning nanoscale adhesive contact behavior to a near ideal Hertzian state via graphene coverage,” arXiv: Applied Physics. 2020. link Times cited: 4 NOT USED (high confidence) A. Goryaeva, C. Lapointe, C. Dai, J. Dérès, J. Maillet, and M. Marinica, “Reinforcing materials modelling by encoding the structures of defects in crystalline solids into distortion scores,” Nature Communications. 2020. link Times cited: 27 NOT USED (high confidence) V. Vijayaraghavan and L. Zhang, “Tensile Properties of Boron Nitride-Carbon Nanosheet-Reinforced Aluminum Nanocomposites Using Molecular Dynamics Simulation,” JOM. 2020. link Times cited: 9 NOT USED (high confidence) V. Vardanyan, Z. Zhang, I. A. Alhafez, and H. Urbassek, “Cutting of Al/Si bilayer systems: molecular dynamics study of twinning, phase transformation, and cracking,” The International Journal of Advanced Manufacturing Technology. 2020. link Times cited: 10 NOT USED (high confidence) M. Mendelev et al., “Development of a semi-empirical potential suitable for molecular dynamics simulation of vitrification in Cu-Zr alloys.,” The Journal of chemical physics. 2019. link Times cited: 50 Abstract: The fast increase in available computation power allowed us … read moreAbstract: The fast increase in available computation power allowed us to decrease the cooling rate in molecular dynamics (MD) simulation of vitrification by several orders of magnitude. While the reliability of the MD simulation should obviously benefit from this increase in the computational power, in some cases, it led to unexpected results. In particular, Ryltsev et al. [J. Chem. Phys. 149, 164502 (2018)] found that the most popular potentials for the Cu-Zr and Cu-Zr-Al alloys from Mendelev et al. [Philos. Mag. 89, 967 (2009)] and Cheng et al. [Phys. Rev. Lett. 102, 245501 (2009)] do not actually describe good glass forming systems but in contradiction with experiment predict rather fast crystallization of the Cu64.5Zr35.5 alloy which is the well-known example of bulk metallic glasses. In this paper, we present a new Cu-Zr semiempirical potential suitable to simulate vitrification. No crystal nucleation was observed in MD simulation using this potential in the concentration range from 75% to 5% of Zr. Since the new potential leads to about the same liquid structure and viscosity as the Cu-Zr potential from Mendelev et al. [Philos. Mag. 89, 967 (2009)] which failed to describe the good glass formability, our study clearly shows that no reliable conclusions about the glass formability can be deduced based solely on the analysis of the liquid properties and a nucleation/crystal growth study should be performed to address this question. read less NOT USED (high confidence) Y. Hong, B. Hou, H. Jiang, and J. Zhang, “Machine learning and artificial neural network accelerated computational discoveries in materials science,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2019. link Times cited: 55 Abstract: Artificial intelligence (AI) has been referred to as the “fo… read moreAbstract: Artificial intelligence (AI) has been referred to as the “fourth paradigm of science,” and as part of a coherent toolbox of data‐driven approaches, machine learning (ML) dramatically accelerates the computational discoveries. As the machinery for ML algorithms matures, significant advances have been made not only by the mainstream AI researchers, but also those work in computational materials science. The number of ML and artificial neural network (ANN) applications in the computational materials science is growing at an astounding rate. This perspective briefly reviews the state‐of‐the‐art progress in some supervised and unsupervised methods with their respective applications. The characteristics of primary ML and ANN algorithms are first described. Then, the most critical applications of AI in computational materials science such as empirical interatomic potential development, ML‐based potential, property predictions, and molecular discoveries using generative adversarial networks (GAN) are comprehensively reviewed. The central ideas underlying these ML applications are discussed, and future directions for integrating ML with computational materials science are given. Finally, a discussion on the applicability and limitations of current ML techniques and the remaining challenges are summarized. read less NOT USED (high confidence) H. He, Y. Rong, and L. Zhang, “Molecular dynamics studies on the sintering and mechanical behaviors of graphene nanoplatelet reinforced aluminum matrix composites,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 15 Abstract: The sintering process of graphene nanoplatelet (GNP) reinfor… read moreAbstract: The sintering process of graphene nanoplatelet (GNP) reinforced aluminum matrix composite powder was simulated by molecular dynamics method. The effects of Al nanoparticle size and sintering temperature on sintering behavior were studied. Uniaxial tensile simulation was applied to study the mechanical properties of sintered composites. The results show that the nanoparticle size and sintering temperature have significant effects on the sintering behavior of the composites. Smaller size nanoparticle system has lower melting point, which requires lower sintering temperature. Larger size particle system requires longer sintering time and higher sintering temperature. At lower temperatures, the main coalescence mechanisms of nanoparticle systems are surface diffusion and grain boundary diffusion. When the temperature is close to the melting point, volume diffusion and surface diffusion dominate. Tensile simulation results of sintered composites show that the addition of GNP can greatly improve the mechanical properties of the composites. Dislocation reinforcement and stress transfer are the main reinforcement mechanisms. read less NOT USED (high confidence) Y. Zhu, L. Abdulmajeid, and K. K. Hauser, “A Data-driven Approach for Fast Simulation of Robot Locomotion on Granular Media,” 2019 International Conference on Robotics and Automation (ICRA). 2019. link Times cited: 10 Abstract: In this paper, we propose a semi-empirical approach for simu… read moreAbstract: In this paper, we propose a semi-empirical approach for simulating robot locomotion on granular media. We first develop a contact model based on the stick-slip behavior between rigid objects and granular grains, which is then learned through running extensive experiments. The contact model represents all possible contact wrenches that the granular substrate can provide as a convex volume, which our method formulates as constraints in an optimization-based contact force solver. During simulation, granular substrates are treated as rigid objects that allow penetration and the contact solver solves for wrenches that maximize frictional dissipation. We show that our method is able to simulate plausible interaction response with several granular media at interactive rates. read less NOT USED (high confidence) H. Wang, X. Guo, L. Zhang, H. Wang, and J. Xue, “Deep learning inter-atomic potential model for accurate irradiation damage simulations,” Applied Physics Letters. 2019. link Times cited: 32 Abstract: We propose a hybrid scheme that interpolates smoothly the Zi… read moreAbstract: We propose a hybrid scheme that interpolates smoothly the Ziegler-Biersack-Littmark (ZBL) screened nuclear repulsion potential with a newly developed deep learning potential energy model. The resulting DP-ZBL model can not only provide overall good performance on the predictions of near-equilibrium material properties but also capture the right physics when atoms are extremely close to each other, an event that frequently happens in computational simulations of irradiation damage events. We applied this scheme to the simulation of the irradiation damage processes in the face-centered-cubic aluminium system, and found better descriptions in terms of the defect formation energy, evolution of collision cascades, displacement threshold energy, and residual point defects, than the widely-adopted ZBL modified embedded atom method potentials and its variants. Our work provides a reliable and feasible scheme to accurately simulate the irradiation damage processes and opens up new opportunities to solve the predicament of lacking accurate potentials for enormous newly-discovered materials in the irradiation effect field. read less NOT USED (high confidence) K. Belouarda, S. Trady, K. Saadouni, and M. Mazroui, “Influence of mechanical tensile and compression tests under high strain rate on structural properties of copper monatomic metallic glass,” The European Physical Journal B. 2019. link Times cited: 8 NOT USED (high confidence) Y. Sun, F. Zhang, H. Song, M. Mendelev, C. Wang, and K. Ho, “Competitive B2 and B33 Nucleation during Solidification of Ni50Zr50 Alloy: Molecular Dynamics Simulation and Classical Nucleation Theory,” The Journal of Physical Chemistry C. 2019. link Times cited: 4 Abstract: We investigated the homogenous nucleation of the stoichiomet… read moreAbstract: We investigated the homogenous nucleation of the stoichiometric B2 and B33 phases in the Ni50Zr50 alloy using the persistent embryo method and the classical nucleation theory. The two phases become very close competitors at large supercoolings, which is consistent with the experimental observations. In the case of the B2 phase, the linear temperature dependence of the solid-liquid interface (SLI) free energy extrapolated to the melting temperature leads to the same value as the one obtained from the capillarity fluctuation method (CFM). In the case of the B33 phases, the SLI free energy is also a linear function of temperature at large supercoolings but the extrapolation to the melting temperature leads to a value which is considerably different from the CFM value. This is consistent with the large anisotropy of the SLI properties of the B33 phase nearby the melting temperature observed in the simulation of the nominally flat interface migration. read less NOT USED (high confidence) J. Maldonis et al., “StructOpt: A modular materials structure optimization suite incorporating experimental data and simulated energies,” Computational Materials Science. 2019. link Times cited: 6 NOT USED (high confidence) L. Zhang, Y. Shibuta, X. Huang, C. Lu, and M. Liu, “Grain boundary induced deformation mechanisms in nanocrystalline Al by molecular dynamics simulation: From interatomic potential perspective,” Computational Materials Science. 2019. link Times cited: 39 NOT USED (high confidence) X. Mei, W. Mohamed, and J. Eapen, “Approach to local thermodynamic equilibrium and the evolution to a glassy core following neutron/ion radiation impact,” Philosophical Magazine. 2018. link Times cited: 2 Abstract: ABSTRACT Using molecular dynamics simulations and statistica… read moreAbstract: ABSTRACT Using molecular dynamics simulations and statistical-mechanical metrics, we make quantitative predictions on the local thermodynamic and dynamic states following an ion or neutron impact in three materials – copper, silicon and solid argon. Through a two-energy distribution, we first capture the non-equilibrium temperature evolution and the approach to the local thermal equilibrium in three generic stages. By examining the time-resolved van Hove self-correlator, we then demonstrate that the impact core of all the three materials shows the dynamic characteristics of a jammed or glassy state. We delineate a dynamic atom-hopping mechanism that attests to a rapid defect recovery stage in copper; silicon, on the contrary, accommodates only small displacements which resist recovery. The dissimilitude between copper with a close-packed structure and silicon with an open network structure is further drawn out through an isoconfigurational analysis of displacements, which shows a compact dendritic-like condensation front for the mobile atoms in copper through atom hopping. In contrast, silicon portrays larger-scale spatial oscillations of dynamically separated regions, which appear to be a precursor to dynamic lattice instability and eventual amorphisation. read less NOT USED (high confidence) Z. Zhang, I. A. Alhafez, and H. Urbassek, “Scratching an Al/Si Interface: Molecular Dynamics Study of a Composite Material,” Tribology Letters. 2018. link Times cited: 20 NOT USED (high confidence) K. Doihara, T. Okita, M. Itakura, M. Aichi, and K. Suzuki, “Atomic simulations to evaluate effects of stacking fault energy on interactions between edge dislocation and spherical void in face-centred cubic metals,” Philosophical Magazine. 2018. link Times cited: 21 Abstract: In this study, molecular dynamics simulations were performed… read moreAbstract: In this study, molecular dynamics simulations were performed to elucidate the effects of stacking fault energy (SFE) on the physical interactions between an edge dislocation and a spherical void in the crystal structure of face-centred cubic metals at various temperatures and for different void sizes. Four different types of interaction morphologies were observed, in which (1) two partial dislocations detached from the void separately, and the maximum stress corresponded to the detachment of the trailing partial; (2) two partial dislocations detached from the void separately, and the maximum stress corresponded to the detachment of the leading partial; (3) the partial dislocations detached from the void almost simultaneously without jog formation; and (4) the partial dislocations detached from the void almost simultaneously with jog formation. With an increase in void size or SFE, the interaction morphology changed in the above-mentioned order. It was observed that the magnitude of the critical resolved shear stress (CRSS) and its dependence on the SFE were determined by these interaction morphologies. The value of the CRSS in the case of interaction morphology (1) is almost equal to an analytical one based on the linear elasticity by employing the Burgers vector of a single partial dislocation. The maximum value of the CRSS is also obtained by the analytical model with the Burgers vector of the two partial dislocations. read less NOT USED (high confidence) L. Hale, Z. Trautt, and C. Becker, “Evaluating variability with atomistic simulations: the effect of potential and calculation methodology on the modeling of lattice and elastic constants,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 40 Abstract: Atomistic simulations using classical interatomic potentials… read moreAbstract: Atomistic simulations using classical interatomic potentials are powerful investigative tools linking atomic structures to dynamic properties and behaviors. It is well known that different interatomic potentials produce different results, thus making it necessary to characterize potentials based on how they predict basic properties. Doing so makes it possible to compare existing interatomic models in order to select those best suited for specific use cases, and to identify any limitations of the models that may lead to unrealistic responses. While the methods for obtaining many of these properties are often thought of as simple calculations, there are many underlying aspects that can lead to variability in the reported property values. For instance, multiple methods may exist for computing the same property and values may be sensitive to certain simulation parameters. Here, we introduce a new high-throughput computational framework that encodes various simulation methodologies as Python calculation scripts. Three distinct methods for evaluating the lattice and elastic constants of bulk crystal structures are implemented and used to evaluate the properties across 120 interatomic potentials, 18 crystal prototypes, and all possible combinations of unique lattice site and elemental model pairings. Analysis of the results reveals which potentials and crystal prototypes are sensitive to the calculation methods and parameters, and it assists with the verification of potentials, methods, and molecular dynamics software. The results, calculation scripts, and computational infrastructure are self-contained and openly available to support researchers in performing meaningful simulations. read less NOT USED (high confidence) L. Hale, “Comparing Modeling Predictions of Aluminum Edge Dislocations: Semidiscrete Variational Peierls–Nabarro Versus Atomistics,” JOM. 2018. link Times cited: 7 NOT USED (high confidence) X. W. Zhou, D. Ward, and M. E. Foster, “A bond-order potential for the Al–Cu–H ternary system,” New Journal of Chemistry. 2018. link Times cited: 13 Abstract: Al-Based Al–Cu alloys have a very high strength to density r… read moreAbstract: Al-Based Al–Cu alloys have a very high strength to density ratio, and are therefore important materials for transportation systems including vehicles and aircrafts. These alloys also appear to have a high resistance to hydrogen embrittlement, and as a result, are being explored for hydrogen related applications. To enable fundamental studies of mechanical behavior of Al–Cu alloys under hydrogen environments, we have developed an Al–Cu–H bond-order potential according to the formalism implemented in the molecular dynamics code LAMMPS. Our potential not only fits well to properties of a variety of elemental and compound configurations (with coordination varying from 1 to 12) including small clusters, bulk lattices, defects, and surfaces, but also passes stringent molecular dynamics simulation tests that sample chaotic configurations. Careful studies verified that this Al–Cu–H potential predicts structural property trends close to experimental results and quantum-mechanical calculations; in addition, it properly captures Al–Cu, Al–H, and Cu–H phase diagrams and enables simulations of H2 dissociation, chemisorption, and absorption on Al–Cu surfaces. read less NOT USED (high confidence) I. A. Alhafez, C. Ruestes, and H. Urbassek, “Size of the Plastic Zone Produced by Nanoscratching,” Tribology Letters. 2018. link Times cited: 21 NOT USED (high confidence) Z. Zhang and H. Urbassek, “Comparative Study of Interatomic Interaction Potentials for Describing Indentation into Si Using Molecular Dynamics Simulation,” Applied Mechanics and Materials. 2017. link Times cited: 5 Abstract: We compare the performance of three interatomic interaction … read moreAbstract: We compare the performance of three interatomic interaction potentials for describing the evolution of plasticity and phase transformations in Si: the well established Stillinger-Weber potential, a recent modification used in the description of Al/Si composites, and a modification of the well known Tersoff potential. We show that the generation of dislocations and the evolution of plasticity are well described by the Stillinger-Weber potential and its modification, while the phase transformation to the high-pressure bct5 modification and the subsequent amorphization are better included in the modified Tersoff potential. read less NOT USED (high confidence) A. Fedorov, A. V. Shul’gin, and S. Lavruk, “Investigation of the physical properties of iron nanoparticles in the course of the melting and solidification,” Physics of Metals and Metallography. 2017. link Times cited: 6 NOT USED (high confidence) A. Mahata, M. A. Zaeem, and M. Baskes, “Understanding homogeneous nucleation in solidification of aluminum by molecular dynamics simulations,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 65 Abstract: Homogeneous nucleation from aluminum (Al) melt was investiga… read moreAbstract: Homogeneous nucleation from aluminum (Al) melt was investigated by million-atom molecular dynamics simulations utilizing the second nearest neighbor modified embedded atom method potentials. The natural spontaneous homogenous nucleation from the Al melt was produced without any influence of pressure, free surface effects and impurities. Initially isothermal crystal nucleation from undercooled melt was studied at different constant temperatures, and later superheated Al melt was quenched with different cooling rates. The crystal structure of nuclei, critical nucleus size, critical temperature for homogenous nucleation, induction time, and nucleation rate were determined. The quenching simulations clearly revealed three temperature regimes: sub-critical nucleation, super-critical nucleation, and solid-state grain growth regimes. The main crystalline phase was identified as face-centered cubic, but a hexagonal close-packed (hcp) and an amorphous solid phase were also detected. The hcp phase was created due to the formation of stacking faults during solidification of Al melt. By slowing down the cooling rate, the volume fraction of hcp and amorphous phases decreased. After the box was completely solid, grain growth was simulated and the grain growth exponent was determined for different annealing temperatures. read less NOT USED (high confidence) Y. Jiang, J. Luo, and Y. Wu, “The validation and preference among different EAM potentials to describe the solid–liquid transition of aluminum,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 6 Abstract: Empirical potential is vital to the classic atomic simulatio… read moreAbstract: Empirical potential is vital to the classic atomic simulation, especially for the study of phase transitions, as well as the solid-interface. In this paper, we attempt to set up a uniform procedure for the validation among different potentials before the formal simulation study of phase transitions of metals. Two main steps are involved: (1) the prediction of the structures of both solid and liquid phases and their mutual transitions, i.e. melting and crystallization; (2) the prediction of vital thermodynamic (the equilibrium melting point at ambient pressure) and dynamic properties (the degrees of superheating and undercooling). We applied this procedure to the testing of seven published embedded-atom potentials (MKBA (Mendelev et al 2008 Philos. Mag. 88 1723), MFMP (Mishin et al 1999 Phys. Rev. B 59 3393), MDSL (Sturgeon and Laird 2000 Phys. Rev. B 62 14720), ZM (Zope and Mishin 2003 Phys. Rev. B 68 024102), LEA (Liu et al 2004 Model. Simul. Mater. Sci. Eng. 12 665), WKG (Winey et al 2009 Model. Simul. Mater. Sci. Eng. 17 055004) and ZJW (Zhou et al 2004 Phys. Rev. B 69 144113)) for the description of the solid–liquid transition of Al. All the predictions of structure, melting point and superheating/undercooling degrees were compared with the experiments or theoretical calculations. Then, two of them, MKBA and MDSL, were proven suitable for the study of the solid–liquid transition of Al while the residuals were unqualified. However, potential MKBA is more accurate to predict the structures of solid and liquid, while MDSL works a little better in the thermodynamic and dynamic predictions of solid–liquid transitions. read less NOT USED (high confidence) N. T. Brown, E. Martínez, and J. Qu, “Interfacial free energy and stiffness of aluminum during rapid solidification,” Acta Materialia. 2017. link Times cited: 12 NOT USED (high confidence) A. Akbarzadeh, Y. Cui, and Z. Chen, “Thermal wave: from nonlocal continuum to molecular dynamics,” RSC Advances. 2017. link Times cited: 24 Abstract: It is well known that the continuum model of Fourier's … read moreAbstract: It is well known that the continuum model of Fourier's law of heat conduction violates the relativity theory, admits an instantaneous thermal response, and assumes a quasi-equilibrium thermodynamic condition. Transient heat transport, however, is a non-equilibrium phenomenon with a finite thermal wave speed for applications involving very low temperatures, extremely high temperature gradients, and ballistic heat transfers. Hyperbolic and phase-lag heat conduction models have enabled detection of the finite thermal wave speed in heat transport. To accommodate effects of thermomass and size-dependency of thermophysical properties on nano/microscale heat transport and to remove the theoretical singularity of temperature gradients across the thermal wavefront, a nonlocal, fractional-order, three-phase-lag heat conduction is introduced. The model is capable of simulating heat conduction phenomena in multiple spatio-temporal scales. To confirm the existence of thermal waves in nano/microscale heat transport, a molecular dynamics simulation is implemented for the heat transfer within a nanoscale copper slab. Correlating thermal responses in continuum and atomistic scales sheds light on the effect of length scale, fractional order, and phase-lags in multiscale heat transport. The multiscale simulation is of practical importance for microelectromechanical system design, photothermal techniques, and ultrafast laser-assisted processing of advanced materials. read less NOT USED (high confidence) A. Tran and Y. Wang, “Reliable Molecular Dynamics: Uncertainty quantification using interval analysis in molecular dynamics simulation,” Computational Materials Science. 2017. link Times cited: 17 NOT USED (high confidence) V. Krasnikov and A. Mayer, “Melting of aluminum with ideal or defect lattice: Molecular dynamics simulations with accounting of electronic heat conductivity,” Journal of Physics: Conference Series. 2016. link Times cited: 2 Abstract: In this work, the atomistic simulations of rapid melting of … read moreAbstract: In this work, the atomistic simulations of rapid melting of aluminum are performed. We use the two-temperature approach separately describing the ionic and electronic subsystems of crystal. Both ideal and defect states of initial lattice are considered. The dependence of melting temperature on pressure is investigated in the simulations of thermal equilibrium establishment in the systems with plate interphase boundaries. Non-equilibrium melting of aluminum is studied in simulations with the constant rate of heat energy supply. The maximal temperatures of overheated material before complete melting are obtained in dependence of energy supply rate. Presence of initial defects of lattice substantially decreases the overheating of material. Electronic heat conductivity significantly accelerates the thermal equilibrium establishment in systems with interphase boundaries and decreases the drop of temperature after beginning of melting in the systems with constant rate of heating. read less NOT USED (high confidence) M. Mendelev, T. L. Underwood, and G. Ackland, “Development of an interatomic potential for the simulation of defects, plasticity, and phase transformations in titanium.,” The Journal of chemical physics. 2016. link Times cited: 122 Abstract: New interatomic potentials describing defects, plasticity, a… read moreAbstract: New interatomic potentials describing defects, plasticity, and high temperature phase transitions for Ti are presented. Fitting the martensitic hcp-bcc phase transformation temperature requires an efficient and accurate method to determine it. We apply a molecular dynamics method based on determination of the melting temperature of competing solid phases, and Gibbs-Helmholtz integration, and a lattice-switch Monte Carlo method: these agree on the hcp-bcc transformation temperatures to within 2 K. We were able to develop embedded atom potentials which give a good fit to either low or high temperature data, but not both. The first developed potential (Ti1) reproduces the hcp-bcc transformation and melting temperatures and is suitable for the simulation of phase transitions and bcc Ti. Two other potentials (Ti2 and Ti3) correctly describe defect properties and can be used to simulate plasticity or radiation damage in hcp Ti. The fact that a single embedded atom method potential cannot describe both low and high temperature phases may be attributed to neglect of electronic degrees of freedom, notably bcc has a much higher electronic entropy. A temperature-dependent potential obtained from the combination of potentials Ti1 and Ti2 may be used to simulate Ti properties at any temperature. read less NOT USED (high confidence) X. W. Zhou, D. Ward, and M. E. Foster, “An analytical bond-order potential for the aluminum copper binary system,” Journal of Alloys and Compounds. 2016. link Times cited: 38 NOT USED (high confidence) N. Admal, J. Marian, and G. Po, “The atomistic representation of first strain-gradient elastic tensors,” Journal of The Mechanics and Physics of Solids. 2016. link Times cited: 36 NOT USED (high confidence) Z. Hou, K. Dong, Z. Tian, R. Liu, Z. Wang, and J. G. Wang, “Cooling rate dependence of solidification for liquid aluminium: a large-scale molecular dynamics simulation study.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 62 Abstract: The effect of the cooling rate on the solidification process… read moreAbstract: The effect of the cooling rate on the solidification process of liquid aluminium is studied using a large-scale molecular dynamics method. It is found that there are various types of short-range order (SRO) structures in the liquid, among which the icosahedral (ICO)-like structures are dominant. These SRO structures are in dynamic fluctuation and transform each other. The effect of the cooling rate on the microstructure is very weak at high temperatures and in supercooled liquids, and it appears only below the liquid-solid transition temperature. Fast cooling rates favour the formation of amorphous structures with ICO-like features, while slow cooling rates favour the formation of FCC crystalline structures. Furthermore, FCC and HCP structures can coexist in crystalline structures. It is also found that nanocrystalline aluminium can be achieved at appropriate cooling rates, and its formation mechanism is thoroughly investigated by tracing the evolution of nanoclusters. The arrangement of FCC and HCP atoms in the nanograins displays various twinned structures as observed using visualization analysis, which is different from the layering or phase separation structures observed in the solidification of Lennard-Jones fluids and some metal liquids. read less NOT USED (high confidence) M. Skarlinski and D. Quesnel, “Effect of native oxide layers on copper thin-film tensile properties: A reactive molecular dynamics study,” Journal of Applied Physics. 2015. link Times cited: 8 Abstract: Metal-oxide layers are likely to be present on metallic nano… read moreAbstract: Metal-oxide layers are likely to be present on metallic nano-structures due to either environmental exposure during use, or high temperature processing techniques such as annealing. It is well known that nano-structured metals have vastly different mechanical properties from bulk metals; however, difficulties in modeling the transition between metallic and ionic bonding have prevented the computational investigation of the effects of oxide surface layers. Newly developed charge-optimized many body [Liang et al., Mater. Sci. Eng., R 74, 255 (2013)] potentials are used to perform fully reactive molecular dynamics simulations which elucidate the effects that metal-oxide layers have on the mechanical properties of a copper thin-film. Simulated tensile tests are performed on thin-films while using different strain-rates, temperatures, and oxide thicknesses to evaluate changes in yield stress, modulus, and failure mechanisms. Findings indicate that copper-thin film mechanical properties are strongly affected by... read less NOT USED (high confidence) J. Jalkanen and M. Müser, “Systematic analysis and modification of embedded-atom potentials: case study of copper,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 10 Abstract: In this study, we evaluate the functionals of different embe… read moreAbstract: In this study, we evaluate the functionals of different embedded-atom methods (EAM) by fitting their free parameters to ab-initio results for copper. Our emphasis lies on testing the transferability of the potentials between systems which vary in their spatial dimension and geometry. The model structures encompass zero-dimensional clusters, one-dimensional chains, two-dimensional tilings, and three-dimensional bulk systems. To avoid having to mimic charge transfer, which is outside the scope of conventional EAM potentials, we focus on structures, in which all atoms are symmetrically equivalent. We find that the simple, four-parameter Gupta EAM potential is overall satisfactory. Adding complexity to it decreases the errors on our set of structures only by marginal amounts, unless EAM is modified to depend also on density gradients, higher-order derivatives, or related terms. All tested conventional EAM functions reveal similar problems: the binding energy of closed-packed systems is overestimated in comparison to open or planar geometries, and structures with small coordination tend to be too rigid. These deficiencies can be fixed in terms of a systematically modified embedded-atom method (SMEAM), which reproduces DFT results on bond lengths, binding energies, and stiffnesses or bulk moduli by typically O(1%), O(5%), and O(15%) accuracy, respectively. SMEAM also predicts the radial distribution function of liquid copper quite accurately. Yet, it does not overcome the difficulty to reproduce the elastic tensor elements of a hypothetical diamond lattice. read less NOT USED (high confidence) L. Wu, B. Xu, Q. Li, W. Liu, and M. Li, “Anisotropic crystal–melt interfacial energy and stiffness of aluminum,” Journal of Materials Research. 2015. link Times cited: 21 Abstract: The crystal–melt interfacial free energy is an important qua… read moreAbstract: The crystal–melt interfacial free energy is an important quantity governing many kinetic phenomena including solidification and crystal growth. Although general calculation methods are available, it is still difficult to obtain the interfacial energies that differ only slightly due to anisotropy. Here, we report such a calculation of Al crystal–melt interfacial energy based on the general framework of the capillary fluctuation method (CFM). The subtle dependence of both the melting temperature and interfacial free energy at melting temperature on the crystal interface orientation was examined. For Al, the average melting temperature is obtained at 934.79 ± 5 K and the orientationally averaged mean interfacial free energy is 98.35 mJ/m^2. In addition, the anisotropy of the interfacial free energy is found weak, nevertheless with the values ranked as γ_100 > γ_110 > γ_111. read less NOT USED (high confidence) M. Mendelev et al., “Development of interatomic potentials appropriate for simulation of devitrification of Al90Sm10 alloy,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 59 Abstract: A semi-empirical potential for the Al90Sm10 alloy is present… read moreAbstract: A semi-empirical potential for the Al90Sm10 alloy is presented. The potential provides satisfactory reproduction of pure Al properties, the formation energies of a set of Al–Sm crystal phases with Sm content about 10%, and the structure of the liquid Al90Sm10 alloy. During molecular dynamics simulation in which the liquid alloy is cooled at a rate of 1010 K s−1, the developed potential produces a glass structure with lower ab initio energy than that produced by ab initio molecular dynamics (AIMD) itself using a typical AIMD cooling rate of 8·1013 K s−1. Based on these facts the developed potential should be suitable for simulations of phase transformations in the Al90Sm10 alloy. read less NOT USED (high confidence) T. Niiyama and T. Shimokawa, “Atomistic mechanisms of intermittent plasticity in metals: dislocation avalanches and defect cluster pinning.,” Physical review. E, Statistical, nonlinear, and soft matter physics. 2015. link Times cited: 15 Abstract: Intermittent plastic deformation in crystals with power-law … read moreAbstract: Intermittent plastic deformation in crystals with power-law behaviors has been reported in previous experimental studies. The power-law behavior is reminiscent of self-organized criticality, and mesoscopic models have been proposed that describe this behavior in crystals. In this paper, we show that intermittent plasticity in metals under tensile deformation can be observed in molecular dynamics models, using embedded atom method potentials for Ni, Cu, and Al. Power-law behaviors of stress drop and waiting time of plastic deformation events are observed. It is shown that power-law behavior is due to dislocation avalanche motions in Cu and Ni. A different mechanism of dislocation pinning is found in Al. These different stress relaxation mechanisms give different power-law exponents. We propose a probabilistic model to describe the novel dislocation motion in Al and analytically deduce the power-law behavior. read less NOT USED (high confidence) V. Borovikov, M. Mendelev, A. King, and R. LeSar, “Effect of stacking fault energy on mechanism of plastic deformation in nanotwinned FCC metals,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 53 Abstract: Starting from a semi-empirical potential designed for Cu, we… read moreAbstract: Starting from a semi-empirical potential designed for Cu, we have developed a series of potentials that provide essentially constant values of all significant (calculated) materials properties except for the intrinsic stacking fault energy, which varies over a range that encompasses the lowest and highest values observed in nature. These potentials were employed in molecular dynamics (MD) simulations to investigate how stacking fault energy affects the mechanical behavior of nanotwinned face-centered cubic (FCC) materials. The results indicate that properties such as yield strength and microstructural stability do not vary systematically with stacking fault energy, but rather fall into two distinct regimes corresponding to ‘low’ and ‘high’ stacking fault energies. read less NOT USED (high confidence) K. Choudhary et al., “Charge optimized many-body potential for aluminum,” Journal of Physics: Condensed Matter. 2014. link Times cited: 19 Abstract: An interatomic potential for Al is developed within the thir… read moreAbstract: An interatomic potential for Al is developed within the third generation of the charge optimized many-body (COMB3) formalism. The database used for the parameterization of the potential consists of experimental data and the results of first-principles and quantum chemical calculations. The potential exhibits reasonable agreement with cohesive energy, lattice parameters, elastic constants, bulk and shear modulus, surface energies, stacking fault energies, point defect formation energies, and the phase order of metallic Al from experiments and density functional theory. In addition, the predicted phonon dispersion is in good agreement with the experimental data and first-principles calculations. Importantly for the prediction of the mechanical behavior, the unstable stacking fault energetics along the direction on the (1 1 1) plane are similar to those obtained from first-principles calculations. The polycrsytal when strained shows responses that are physical and the overall behavior is consistent with experimental observations. read less NOT USED (high confidence) P. Saidi, T. Frolov, J. Hoyt, and M. Asta, “An angular embedded atom method interatomic potential for the aluminum–silicon system,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 19 Abstract: A modified version of the Stillinger–Weber (SW) interatomic … read moreAbstract: A modified version of the Stillinger–Weber (SW) interatomic potential for pure Si has been developed. In contrast to the original SW form, the modified version allows one to grow diamond cubic crystal structures from the melt at high temperatures. Now, the modified SW potential has been combined with an embedded atom (EAM) description of pure Al developed by Mendelev et al to formulate an Al–Si binary potential of the angular EAM type. The Al–Si potential reproduces quite well the experimental enthalpy of mixing in the liquid. It also predicts an Al–Si phase diagram with a eutectic concentration for the liquid that agrees with experimental values within 4 at% and a eutectic temperature that differs from experimental values by just 13 K. read less NOT USED (high confidence) D. Belashchenko, “Computer simulation of liquid metals,” Physics—Uspekhi. 2013. link Times cited: 84 Abstract: Methods for and the results of the computer simulation of li… read moreAbstract: Methods for and the results of the computer simulation of liquid metals are reviewed. Two basic methods, classical molecular dynamics with known interparticle potentials and the ab initio method, are considered. Most attention is given to the simulated results obtained using the embedded atom model (EAM). The thermodynamic, structural, and diffusion properties of liquid metal models under normal and extreme (shock) pressure conditions are considered. Liquid-metal simulated results for the Groups I–IV elements, a number of transition metals, and some binary systems (Fe–C, Fe–S) are examined. Possibilities for the simulation to account for the thermal contribution of delocalized electrons to energy and pressure are considered. Solidification features of supercooled metals are also discussed. read less NOT USED (high confidence) H. Shodja, M. Tabatabaei, A. Ostadhossein, and L. Pahlevani, “Elastic fields of interacting point defects within an ultra-thin fcc film bonded to a rigid substrate,” Central European Journal of Engineering. 2013. link Times cited: 4 Abstract: Certain physical and mechanical phenomena within ultra-thin … read moreAbstract: Certain physical and mechanical phenomena within ultra-thin face-centered cubic (fcc) films containing common types of interacting point defects are addressed. An atomic-scale lattice statics in conjunction with many-body interatomic potentials suitable for binary systems is conducted to analyze the effects of the depth on the: (1) formation energy and layer-by-layer displacements due to the presence of vacancy-octahedral self-interstitial atom (OSIA) ensemble, and (2) elastic fields as well as the free surface shape in the case of vacancy-dopant interaction. Moreover, the effects of the inter-defect spacing for various depths are also examined. To ensure reasonable accuracy and numerical convergence, the atomic interaction up to the second-nearest neighbor is considered. read less NOT USED (high confidence) D. Belashchenko, “Computer simulation of copper and silver under shock compression conditions,” Inorganic Materials. 2013. link Times cited: 2 NOT USED (high confidence) M. Mendelev, M. Kramer, S. Hao, K. Ho, and C. Z. Wang, “Development of interatomic potentials appropriate for simulation of liquid and glass properties of NiZr2 alloy,” Philosophical Magazine. 2012. link Times cited: 116 Abstract: A new interatomic potential for the Ni–Zr system is presente… read moreAbstract: A new interatomic potential for the Ni–Zr system is presented. This potential was developed specifically to match experimental scattering data from Ni, Zr and NiZr2 liquids. Both ab initio and published thermodynamic data were used to optimise the potential to study the liquid and amorphous structure of the NiZr2 alloy. This potential has the C 16 phase, being more stable than C 11b phase in the NiZr2 alloy, consistent with experiments. The potential leads to the correct glass structure in the molecular dynamics simulation and, therefore, can be used to study the liquid–glass transformation in the NiZr2 alloy. read less NOT USED (high confidence) Q.-J. Hong and A. van de Walle, “Direct first-principles chemical potential calculations of liquids.,” The Journal of chemical physics. 2012. link Times cited: 14 Abstract: We propose a scheme that drastically improves the efficiency… read moreAbstract: We propose a scheme that drastically improves the efficiency of Widom's particle insertion method by efficiently sampling cavities while calculating the integrals providing the chemical potentials of a physical system. This idea enables us to calculate chemical potentials of liquids directly from first-principles without the help of any reference system, which is necessary in the commonly used thermodynamic integration method. As an example, we apply our scheme, combined with the density functional formalism, to the calculation of the chemical potential of liquid copper. The calculated chemical potential is further used to locate the melting temperature. The calculated results closely agree with experiments. read less NOT USED (high confidence) D. Belashchenko, “Embedded atom method potentials for liquid copper and silver,” Inorganic Materials. 2012. link Times cited: 7 NOT USED (high confidence) I. Morozov, A. Kazennov, R. Bystryi, G. Norman, V. Pisarev, and V. Stegailov, “Molecular dynamics simulations of the relaxation processes in the condensed matter on GPUs,” Comput. Phys. Commun. 2011. link Times cited: 47 NOT USED (high confidence) X. J. Han and H. Schober, “Transport properties and Stokes-Einstein relation in a computer-simulated glass-forming Cu 33 . 3 Zr 66 . 7 melt,” Physical Review B. 2011. link Times cited: 66 Abstract: Molecular dynamics simulation with a modified embedded atom … read moreAbstract: Molecular dynamics simulation with a modified embedded atom potential was used to study transport properties and the Stokes-Einstein relation of a glass-forming Cu${}_{33.3}$Zr${}_{66.7}$ metallic melt. Upon cooling, at high temperatures, the self-diffusion coefficients of the two species evolve nearly parallel, whereas they diverge below 1600 K. The viscosity as function of temperature is calculated from the Green-Kubo equation. The critical temperature of mode coupling theory ${T}_{\mathrm{c}}$ is found as 1030 K, from both the transport properties and the \ensuremath{\alpha}-relaxation time. It is found that the Stokes-Einstein relation between viscosity and diffusivity breaks down at around 1600 K, far above ${T}_{\mathrm{c}}$ and even above the melting temperature. The temperature dependence of the effective diameter in the Stokes-Einstein relation correlates closely with the first derivative of the ratio of the self-diffusion coefficients of the two components. We propose that the onset of Stokes-Einstein relation breakdown could be predicted quantitatively by the divergence behavior of diffusion coefficients, and the breakdown of Stokes-Einstein relation is ascribed to the sudden increase of the dynamic heterogeneity. read less NOT USED (high confidence) B. Qin and W. Lai, “Metallic glass-forming composition range of the Cu–Zr–Ti ternary system determined by molecular dynamics simulations with many-body potentials,” Journal of Materials Research. 2011. link Times cited: 8 Abstract: An n -body Cu–Zr–Ti potential is constructed and applied to … read moreAbstract: An n -body Cu–Zr–Ti potential is constructed and applied to evaluate a glass-forming composition range (GFR) of the Cu–Zr–Ti ternary system by molecular dynamics simulations using a solid-solution model, which is formed via random substitution of solvent atoms by a certain number of solute atoms. It is found that the GFR of the Cu–Zr–Ti ternary system is located within an approximate distorted quadrilateral composition region, in which the solid solutions are unstable and spontaneously collapse to form amorphous phases. The compositions of the four vertexes of the distorted quadrilateral are determined to be Cu_22Zr_78Ti_0, Cu_24Zr_0Ti_76, Cu_56Zr_0Ti_44, and Cu_72Zr_28Ti_0, respectively. In addition, the simulation results are in good agreement with the experimental observations and compatible with some empirical rules. read less NOT USED (high confidence) C. Becker and M. J. Kramer, “Atomistic comparison of volume-dependent melt properties from four models of aluminum,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 30 Abstract: With the increasing use of simulations in materials research… read moreAbstract: With the increasing use of simulations in materials research and design, it is important to quantify the differences between, and accuracy of, models used in these simulations. Here we present the results of such a comparison for four embedded-atom models of aluminum that were optimized to have good liquid properties, particularly the melting temperatures. The effects of temperature and volume are systematically examined in the melts for bulk thermodynamic quantities, pair correlation functions and structure factors and diffusion coefficients for each interatomic potential. Where possible, these are then compared with experimental values. We find quantitative differences in the properties determined from the various interatomic potentials despite the fact that they were fit with similar sets of data. read less NOT USED (high confidence) M. Mendelev et al., “Experimental and computer simulation determination of the structural changes occurring through the liquid–glass transition in Cu–Zr alloys,” Philosophical Magazine. 2010. link Times cited: 44 Abstract: Molecular dynamics (MD) simulations were performed of the st… read moreAbstract: Molecular dynamics (MD) simulations were performed of the structural changes occurring through the liquid–glass transition in Cu–Zr alloys. The total scattering functions (TSF), and their associated primary diffuse scattering peak positions (K p), heights (K h) and full-widths at half maximum (K FWHM) were used as metrics to compare the simulations to high-energy X-ray scattering data. The residuals of difference between the model and experimental TSFs are ∼0.03 for the liquids and about 0.07 for the glasses. Over the compositional range studied, Zr1− x Cu x (0.1 ≤ x ≤ 0.9), K p, K h and K FWHM show a strong dependence on composition and temperature. The simulation and experimental data correlate well between each other. MD simulation revealed that the Cu–Zr bonds undergo the largest changes during cooling of the liquid, whereas the Cu–Cu bonds change the least. Changes in the partial-pair correlations are more readily seen in the second and third shells. The Voronoi polyhedra (VP) in glasses are dominated by only a few select types that are compositionally dependent. The relative concentrations of the dominant VPs rapidly change in their relative proportion in the deeply undercooled liquid. The experimentally determined region of best glass formability, x Cu ∼ 65%, shows the largest temperature dependent changes for the deeply undercooled liquid in the MD simulation. This region also exhibits very strong temperature dependence for the diffusivity and the total energy of the system. These data point to a strong topological change in the best glass-forming alloys and a concurrent change in the VP chemistry in the deeply undercooled liquid. read less NOT USED (high confidence) Y. Mishin, M. Asta, and J. Li, “Atomistic modeling of interfaces and their impact on microstructure and properties,” Acta Materialia. 2010. link Times cited: 418 NOT USED (high confidence) M. Mendelev and B. Bokstein, “Molecular dynamics study of self-diffusion in Zr,” Philosophical Magazine. 2010. link Times cited: 56 Abstract: We employed a recently developed semi-empirical Zr potential… read moreAbstract: We employed a recently developed semi-empirical Zr potential to determine the diffusivities in hcp and bcc Zr via molecular dynamics simulation. The point defect concentration was determined directly from molecular dynamics (MD) simulation rather than from theoretical methods using T = 0 calculations. Our MD simulation indicates that the diffusion proceeds via the interstitial mechanism in hcp Zr, and both vacancy and interstitial mechanisms contribute to diffusivity in bcc Zr. The agreement with the experimental data is excellent for hcp Zr and rather good for bcc Zr at high temperatures, but there is considerable disagreement at low temperatures. read less NOT USED (high confidence) M. Mendelev, M. Asta, M. J. Rahman, and J. Hoyt, “Development of interatomic potentials appropriate for simulation of solid–liquid interface properties in Al–Mg alloys,” Philosophical Magazine. 2009. link Times cited: 126 Abstract: Different approaches are analyzed for construction of semi-e… read moreAbstract: Different approaches are analyzed for construction of semi-empirical potentials for binary alloys, focusing specifically on the capability of these potentials to describe solid–liquid phase equilibria, as a pre-requisite to studies of solidification phenomena. Fitting ab initio compound data does not ensure correct reproduction of the dilute solid-solution formation energy, and explicit inclusion of this quantity in the potential development procedure does not guarantee that the potential will predict the correct solid–liquid phase diagram. Therefore, we conclude that fitting only to solid phase properties, as is done in most potential development procedures, generally is not sufficient to develop a semi-empirical potential suitable for the simulation of solidification. A method is proposed for the incorporation of data for liquid solution energies in the potential development procedure, and a new semi-empirical potential developed suitable for simulations of dilute alloys of Mg in Al. The potential correctly reproduces both zero-temperature solid properties and solidus and liquid lines on the Al-rich part of the Al–Mg phase diagram. read less NOT USED (high confidence) M. Mendelev, M. Kramer, R. Ott, D. Sordelet, D. Yagodin, and P. Popel,’ “Development of suitable interatomic potentials for simulation of liquid and amorphous Cu–Zr alloys,” Philosophical Magazine. 2009. link Times cited: 334 Abstract: We present a new semi-empirical potential suitable for molec… read moreAbstract: We present a new semi-empirical potential suitable for molecular dynamics simulations of liquid and amorphous Cu–Zr alloys. To provide input data for developing the potential, new experimental measurements of the structure factors for amorphous Cu64.5Zr35.5 alloy were performed. In this work, we propose a new method to include diffraction data in the potential development procedure, which also includes fitting to first-principles and liquid density and enthalpy of mixing data. To refine the new potential, we used first-principles and liquid enthalpy of mixing data published earlier combined with the densities of liquid Cu64.5Zr35.5 measured over a range of temperatures. We show that the potential predicts a liquid-to-glass transition temperature that agrees reasonably well with experimental data. Finally, we compare the new potential with two previously developed semi-empirical potentials for Cu–Zr alloys and examine their comparative and contrasting descriptions of structure and properties for Cu64.5Zr35.5 liquids and glasses. read less NOT USED (high confidence) J.-Q. Zhu, X. Liu, Z. Wang, and Q. Yang, “Wrinkles-assisted nanocrystalline formation and mechanical properties of wrinkled graphene/aluminum matrix composites,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 3 Abstract: The graphene-reinforced metal matrix composites (Gr/MMCs), p… read moreAbstract: The graphene-reinforced metal matrix composites (Gr/MMCs), prepared by additive manufacturing technology, render a wide range of promising applications. The graphene sheets in Gr/MMCs may get wrinkled during preparation, which renders a significant influence on crystalline texture and deformation behavior. Herein, molecular dynamics models of wrinkled graphene/aluminum (W-Gr/Al) composites are established to study the effect of graphene wrinkle angle on crystal texture of the metallic matrix during the sintering process and mechanical properties of the resulting composites. The results indicate that the sintering temperature significantly affects the molding quality of W-Gr/Al composites, promoting the transformation of sintering mechanism. Furthermore, new wrinkles appear on initially-wrinkled graphene surfaces during the sintering process, resulting in grain boundaries and twin boundaries (TBs), which facilitate the refinement of Al grains. Moreover, uniaxial compression tests reveal that the W-Gr/Al composites sintered at 900 K exhibit the highest Young’s modulus and yield strength. It has been demonstrated that the enhancement effect of composite originates from the skeleton network, consisting of wrinkled graphene and TBs. These results provide significant guidance for the design and development of Gr/MMCs. read less NOT USED (high confidence) D. Sun, “Proliferation of Twinning in Metals: Application to Magnesium Alloys.” 2018. link Times cited: 2 Abstract: In the search for new alloys with a great strength-to-weight… read moreAbstract: In the search for new alloys with a great strength-to-weight ratio, magnesium has emerged at the forefront. With a strength rivaling that of steel and aluminum alloys --- materials which are deployed widely in real world applications today --- but only a fraction of the density, magnesium holds great promise in a variety of next-generation applications. Unfortunately, the widespread adoption of magnesium is hindered by the fact that it fails in a brittle fashion, which is undesirable when it comes to plastic deformation mechanisms. Consequently, one must design magnesium alloys to navigate around this shortcoming and fail in a more ductile fashion. However, such designs are not possible without a thorough understanding of the underlying mechanisms of deformation in magnesium, which is somewhat contested at the moment. In addition to slip, which is one of the dominant mechanisms in metallic alloys, a mechanism known as twinning is also present, especially in hexagonal close-packed (HCP) materials such as magnesium. Twinning involves the reorientation of the material lattice about a planar discontinuity and has been shown as one of the preferred mechanisms by which magnesium accommodates out-of-plane deformation. Unfortunately, twinning is not particularly well-understood in magnesium, and needs to be addressed before progress can be made in materials design. In particular, though two specific modes of twinning have been acknowledged, various works in the literature have identified a host of additional modes, many of which have been cast aside as "anomalous" observations. To this end, we introduce a new framework for predicting the modes by which a material can twin, for any given material. Focusing on magnesium, we begin our investigation by introducing a kinematic framework that predicts novel twin configurations, cataloging these twins modes by their planar normal and twinning shear. We then subject the predicted twin modes to a series of atomistic simulations, primarily in molecular statics but with supplementary calculations using density functional theory, giving us insight on both the energy of the twin interface and barriers to formation. We then perform a stress analysis and identify the twin modes which are most likely to be activated, thus finding the ones most likely to affect the yield surface of magnesium. Over the course of our investigation, we show that many different modes actually participate on the yield surface of magnesium; the two classical modes which are accepted by the community are confirmed, but many additional modes --- some of which are close to modes which have been previously regarded as anomalies --- are also observed. We also perform some extensional work, showing the flexibility of our framework in predicting twins in other materials and in other environments and highlighting the complicated nature of twinning, especially in HCP materials. read less NOT USED (high confidence) Z. Zhang and H. Urbassek, “Indentation into an Al/Si composite: enhanced dislocation mobility at interface,” Journal of Materials Science. 2017. link Times cited: 21 NOT USED (high confidence) P. Wang et al., “Atomistic simulation for deforming complex alloys with application toward TWIP steel and associated physical insights,” Journal of The Mechanics and Physics of Solids. 2017. link Times cited: 42 NOT USED (high confidence) C. Tackes, “Thermal analysis of undercooled metallic liquids by electromagnetic levitation drop calorimetry.” 2013. link Times cited: 2 Abstract: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . … read moreAbstract: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xvi CHAPTER read less |
 EAM_Dynamo_MendelevKramerBecker_2008_Cu__MO_945691923444_005
EAM_Dynamo_MendelevKramerBecker_2008_Cu__MO_945691923444_005