Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
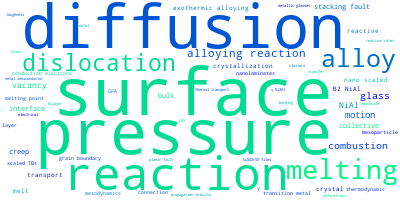
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
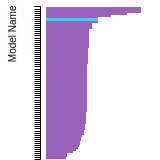
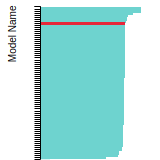
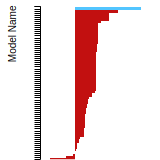
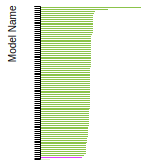
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
254 Citations (172 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) V. Turlo and T. Rupert, “Discovery of a Wide Variety of Linear Complexions in Face Centered Cubic Alloys,” MatSciRN: Other Mechanical Properties & Deformation of Materials (Topic). 2019. link Times cited: 1 Abstract: Linear complexions are defect states that have been recently… read moreAbstract: Linear complexions are defect states that have been recently discovered along dislocations in body centered cubic Fe-based alloys. In this work, we use atomistic simulations to extend this concept and explore segregation-driven structural transitions at dislocations in face centered cubic alloys. We discovered a variety of stable, nanoscale-size structural and chemical states, which are confined near dislocations and can be classified as linear complexions. Depending on the alloy system and thermodynamic conditions, such new states can preserve, partially modify, or completely replace the original defects they were born at. By considering different temperatures and compositions, we construct linear complexion diagrams that are similar to bulk phase diagrams, defining the important conditions for complexion formation while also specifying an expected complexion size and type. Several notable new complexion types were discovered here: (1) nanoparticle arrays comprised of L12 phases in Ni-Fe, Ni-Al, and Al-Zr, (2) replacement of stacking faults with layered complexions comprised of (111) planes from the Cu5Zr intermetallic phase in Cu-Zr, (3) platelet arrays comprised of two-dimensional Guinier-Preston zones in Al-Cu, and finally (4) coexistence of multiple linear complexions containing both Guinier-Preston zones and L12 phases in ternary Al-Cu-Zr. All of these new complexion states are expected to alter material properties and affect the stability of the dislocations themselves, offering a unique opportunity for future materials design. read less USED (high confidence) D. Singh, V. Turlo, D. Gianola, and T. Rupert, “Linear complexions directly modify dislocation motion in face-centered cubic alloys,” Materials Science and Engineering: A. 2022. link Times cited: 1 USED (high confidence) Y.-C. Hu and H. Tanaka, “Revealing the role of liquid preordering in crystallisation of supercooled liquids,” Nature Communications. 2022. link Times cited: 19 USED (high confidence) Y.-C. Hu and H. Tanaka, “Physical origin of glass formation from multicomponent systems,” Science Advances. 2020. link Times cited: 31 Abstract: We show that locally favored structural and chemical orders … read moreAbstract: We show that locally favored structural and chemical orders in melt control the glass-forming ability of metallic alloys. The origin of glass formation is one of the most fundamental issues in glass science. The glass-forming ability (GFA) of multicomponent systems, such as metallic glasses and phase-change materials, can be enormously changed by slight modifications of the constituted elements and compositions. However, its physical origin remains mostly unknown. Here, by molecular dynamics simulations, we study three model metallic systems with distinct GFA. We find that they have a similar driving force of crystallization, but a different liquid-crystal interface tension, indicating that the latter dominates the GFA. Furthermore, we show that the interface tension is determined by nontrivial coupling between structural and compositional orderings and affects crystal growth. These facts indicate that the classical theories of crystallization need critical modifications by considering local ordering effects. Our findings provide fresh insight into the physical control of GFA of metallic alloys and the switching speed of phase-change materials without relying on experience. read less USED (high confidence) Z. Sun, B. Liu, C. He, L. Xie, and Q. Peng, “Shift of Creep Mechanism in Nanocrystalline NiAl Alloy,” Materials. 2019. link Times cited: 6 Abstract: We have examined the effects of temperature, stress, and gra… read moreAbstract: We have examined the effects of temperature, stress, and grain size on the creep process including creep strain, crystal structure, dislocations and diffusions of nanocrystalline NiAl alloy through molecular dynamics simulations. A smaller grain size accelerates the creep process due to the large volume fraction of grain boundaries. Higher temperatures and stress levels also speed up this process in terms of dislocation changes and atom diffusion. In both primary creep and steady-state creep stages, atomic diffusion at the grain boundary could be seen and the dislocation density increased gradually, indicating that the creep mechanism at these stages is Coble creep controlled by grain boundary diffusion while accompanied by dislocation nucleation. When the model enters the tertiary creep stage, it can be observed that the diffusion of atoms in the grain boundary and in the crystal and the dislocation density gradually decreases, implying that the creep mechanisms at this stage are Coble creep, controlled by grain boundary diffusion, and Nabarro–Herring creep, controlled by lattice diffusion. read less USED (high confidence) B. Witbeck, J. Sink, and D. Spearot, “Influence of vacancy defect concentration on the combustion of reactive Ni/Al nanolaminates,” Journal of Applied Physics. 2018. link Times cited: 13 Abstract: Self-propagating reactions in Ni/Al nanolaminates have been … read moreAbstract: Self-propagating reactions in Ni/Al nanolaminates have been widely studied for their high combustion temperatures surpassing 1900 K and rapid combustion wave speeds exceeding 10 m/s. These combustion characteristics have motivated unique industrial applications, such as soldering of electrical components, and possible military applications. Unfortunately, there is a limited understanding of the effect of lattice defects on combustion characteristics. This work explores the effect of vacancy concentration on the combustion rate and peak temperature of reactive Ni/Al nanolaminates. Increasing vacancy concentration increases both reaction rates and peak reaction temperatures. For the reaction rate, vacancy concentration effects are shown to be interdependent with bilayer thickness, initial temperature, and hydrostatic pressure. The effects on reaction peak temperature are independent of these other system parameters. A new method for mapping vacancy and composition profiles is presented to demonstrate the formation and migration of vacancies during the self-propagating reaction.Self-propagating reactions in Ni/Al nanolaminates have been widely studied for their high combustion temperatures surpassing 1900 K and rapid combustion wave speeds exceeding 10 m/s. These combustion characteristics have motivated unique industrial applications, such as soldering of electrical components, and possible military applications. Unfortunately, there is a limited understanding of the effect of lattice defects on combustion characteristics. This work explores the effect of vacancy concentration on the combustion rate and peak temperature of reactive Ni/Al nanolaminates. Increasing vacancy concentration increases both reaction rates and peak reaction temperatures. For the reaction rate, vacancy concentration effects are shown to be interdependent with bilayer thickness, initial temperature, and hydrostatic pressure. The effects on reaction peak temperature are independent of these other system parameters. A new method for mapping vacancy and composition profiles is presented to demonstrate the fo... read less USED (high confidence) T. Ahmed, W. Wang, R. Kozubski, Z.-kui Liu, I. Belova, and G. Murch, “Interdiffusion and thermotransport in Ni–Al liquid alloys,” Philosophical Magazine. 2018. link Times cited: 10 Abstract: ABSTRACT In this paper, we present extensive self-consistent… read moreAbstract: ABSTRACT In this paper, we present extensive self-consistent results of molecular dynamics (MD) simulations of diffusion and thermotransport properties of Ni–Al liquid alloys. We develop a new formalism that allows easy connection between results of the MD simulations and the real experiments. In addition, this formalism can be extended to the case of ternary and higher component liquid alloys. We focus on the temperature and composition dependence of the self-diffusion coefficients, interdiffusion coefficients, thermodynamic factor, Manning factor and the reduced heat of transport. The two latter quantities both represent measures of the off-diagonal Onsager phenomenological coefficients. The Manning factor and the reduced heat of transport can be related to experimentally obtainable quantities provided the thermodynamic factor is available. The simulation results for the reduced heat of transport show that for all compositions, in the presence of a temperature gradient, Ni tends to migrate to the cold end. This is in agreement with an available experimental study for a Ni21.5Al78.5 melt (only qualitative result is available so far). read less USED (high confidence) C. Tang and P. Harrowell, “Composition susceptibility and the role of one, two, and three-body interactions in glass forming alloys: Cu50Zr50 vs Ni50Al50.,” The Journal of chemical physics. 2018. link Times cited: 1 Abstract: In this paper, we compare the composition fluctuations and i… read moreAbstract: In this paper, we compare the composition fluctuations and interaction potentials of a good metallic glass former, Cu50Zr50, and a poor glass former, Ni50Al50. The Bhatia-Thornton correlation functions are calculated. Motivated by the observation of chemical ordering at the NiAl surface, we derive a new property, R^cn(q), corresponding to the linear susceptibility of concentration to a perturbation in density. We present a direct comparison of the potentials for the two model alloys using a 2nd order density expansion, and establish that the one-body energy plays a crucial role in stabilizing the crystal relative to the liquid in both alloys but that the three-body contribution to the heat of fusion is significantly larger in NiAl than CuZr. read less USED (high confidence) J. Liu, E. Tennessen, J. Miao, Y. Huang, J. Rondinelli, and H. Heinz, “Understanding Chemical Bonding in Alloys and the Representation in Atomistic Simulations,” The Journal of Physical Chemistry C. 2018. link Times cited: 29 Abstract: Alloys are widely used in catalysts and structural materials… read moreAbstract: Alloys are widely used in catalysts and structural materials. The nature of chemical bonding and the origin of alloy formation energies, defect energies, and interfacial properties have not been well understood to date but are critical to material performance. In this contribution, we explain the polar nature of chemical bonding and an implementation in classical and reactive atomistic simulations to understand such properties more quantitatively. Electronegativity differences between metal atoms lead to polar bonding, and exothermic alloy formation energies are related to charge transfer between the different elements. These differences can be quantified by atomic charges using pairwise charge increments, determined by matching the computed alloy formation energy to experimentally measured alloy formation energies using pair potentials for the pure metals. The polar character of alloys is comparable to organic molecules and partially ionic minerals, for example, AlNi and AlNi3 alloys assume significant a... read less USED (high confidence) M. Ma’zdziarz, J. Rojek, and S. Nosewicz, “Molecular dynamics study of self-diffusion in stoichiometric B2-NiAl crystals,” Philosophical Magazine. 2018. link Times cited: 5 Abstract: ABSTRACT Self-diffusion parameters in stoichiometric B2-NiAl… read moreAbstract: ABSTRACT Self-diffusion parameters in stoichiometric B2-NiAl solid state crystals were estimated by molecular statics/dynamics simulations with the study of required simulation time to stabilise diffusivity results. An extrapolation procedure to improve the diffusion simulation results was proposed. Calculations of volume diffusivity for the B2 type NiAl in the 1224–1699 K temperature range were performed using the embedded-atom-model potential. The results obtained here are in much better agreement with the experimental results than the theoretical estimates obtained with other methods. read less USED (high confidence) H. Chen, B. Qu, D. Li, R. Zhou, and B. Zhang, “Atomic structure and dynamics properties of Cu50Zr50 films,” Journal of Applied Physics. 2018. link Times cited: 6 Abstract: In this paper, the structural and dynamic properties of Cu50… read moreAbstract: In this paper, the structural and dynamic properties of Cu50Zr50 films are investigated by molecular dynamics simulations. Our results show that the dynamics of the surface atoms are much faster than those of the bulk. Especially, the diffusion coefficient of the surface atoms is about forty times larger than that of the bulk at 600 K, which qualitatively agrees with the experimental results. Meanwhile, we find that the population of the icosahedral (-like) clusters in the surface region is obviously higher than that of the bulk, which prevents the surface from crystallization. A new method to determine the string-like collective atomic motion is introduced in the paper, and it suggests a possible connection between the glass formation ability and collective atomic motion. By using the method, the effects of surface on collective motion are illustrated. Our results show that the string-like collective atomic motion of surface atoms is weakened while that of the interior atoms is strengthened. The studies clearly explain the effects of surface on the structural and dynamic properties of Cu50Zr50 films from the atomic scale.In this paper, the structural and dynamic properties of Cu50Zr50 films are investigated by molecular dynamics simulations. Our results show that the dynamics of the surface atoms are much faster than those of the bulk. Especially, the diffusion coefficient of the surface atoms is about forty times larger than that of the bulk at 600 K, which qualitatively agrees with the experimental results. Meanwhile, we find that the population of the icosahedral (-like) clusters in the surface region is obviously higher than that of the bulk, which prevents the surface from crystallization. A new method to determine the string-like collective atomic motion is introduced in the paper, and it suggests a possible connection between the glass formation ability and collective atomic motion. By using the method, the effects of surface on collective motion are illustrated. Our results show that the string-like collective atomic motion of surface atoms is weakened while that of the interior atoms is strengthened. The studies ... read less USED (high confidence) E. Levchenko, T. Ahmed, and A. Evteev, “Composition dependence of diffusion and thermotransport in Ni-Al melts: A step towards molecular dynamics assisted databases,” Acta Materialia. 2017. link Times cited: 21 USED (high confidence) W. Tucker and P. Schelling, “Thermodiffusion in liquid binary alloys computed from molecular-dynamics simulation and the Green-Kubo formalism,” Computational Materials Science. 2016. link Times cited: 8 USED (high confidence) S. An, J. Li, Y. Li, S. Li, Q. Wang, and B.-xin Liu, “Two-step crystal growth mechanism during crystallization of an undercooled Ni50Al50 alloy,” Scientific Reports. 2016. link Times cited: 21 USED (high confidence) A. Rogachev et al., “Combustion in reactive multilayer Ni/Al nanofoils: Experiments and molecular dynamic simulation,” Combustion and Flame. 2016. link Times cited: 68 USED (high confidence) K. Dayal, “A Multiscale Atomistic Method for Long-Range Electrical Interactions with Application to Multiphysics Calculations in Functional Materials.” 2016. link Times cited: 0 Abstract: : Modern technology is continually evolving and pushing the … read moreAbstract: : Modern technology is continually evolving and pushing the limits of our understanding of materials. This is especially true at the smallest length scales, where phenomenological and experimental data is challenging to acquire. Computational methodologies are becoming more important in understanding the physics of materials at these small length scales. Multiscale methods, in particular, are playing a prominent role in understanding the importance of defects and how they effect properties. We focus specifically on the Quasicontinuum Method (QC). We extend the local Cauchy-Born QC to long-range Coulombic interactions. We then use the method to simulate electrical and mechanical loading in an ionic solid. read less USED (high confidence) S. Sedighi, D. Kirk, C. V. Singh, and S. Thorpe, “Investigating the atomic level influencing factors of glass forming ability in NiAl and CuZr metallic glasses.,” The Journal of chemical physics. 2015. link Times cited: 9 Abstract: Bulk metallic glasses are a relatively new class of amorphou… read moreAbstract: Bulk metallic glasses are a relatively new class of amorphous metal alloy which possess unique mechanical and magnetic properties. The specific concentrations and combinations of alloy elements needed to prevent crystallization during melt quenching remains poorly understood. A correlation between atomic properties that can explain some of the previously identified glass forming ability (GFA) anomalies of the NiAl and CuZr systems has been identified, with these findings likely extensible to other transition metal-transition metal and transition metal-metalloid (TM-M) alloy classes as a whole. In this work, molecular dynamics simulation methods are utilized to study thermodynamic, kinetic, and structural properties of equiatomic CuZr and NiAl metallic glasses in an attempt to further understand the underlying connections between glass forming ability, nature of atomic level bonding, short and medium range ordering, and the evolution of structure and relaxation properties in the disordered phase. The anomalous breakdown of the fragility parameter as a useful GFA indicator in TM-M alloy systems is addressed through an in-depth investigation of bulk stiffness properties and the evolution of (pseudo)Gruneisen parameters over the quench domain, with the efficacy of other common glass forming ability indicators similarly being analyzed through direct computation in respective CuZr and NiAl systems. Comparison of fractional liquid-crystal density differences in the two systems revealed 2-3 times higher values for the NiAl system, providing further support for its efficacy as a general purpose GFA indicator. read less USED (high confidence) K.-H. Lin and A. Strachan, “Role of direct electron-phonon coupling across metal-semiconductor interfaces in thermal transport via molecular dynamics.,” The Journal of chemical physics. 2015. link Times cited: 6 Abstract: Motivated by significant interest in metal-semiconductor and… read moreAbstract: Motivated by significant interest in metal-semiconductor and metal-insulator interfaces and superlattices for energy conversion applications, we developed a molecular dynamics-based model that captures the thermal transport role of conduction electrons in metals and heat transport across these types of interface. Key features of our model, denoted eleDID (electronic version of dynamics with implicit degrees of freedom), are the natural description of interfaces and free surfaces and the ability to control the spatial extent of electron-phonon (e-ph) coupling. Non-local e-ph coupling enables the energy of conduction electrons to be transferred directly to the semiconductor/insulator phonons (as opposed to having to first couple to the phonons in the metal). We characterize the effect of the spatial e-ph coupling range on interface resistance by simulating heat transport through a metal-semiconductor interface to mimic the conditions of ultrafast laser heating experiments. Direct energy transfer from the conduction electrons to the semiconductor phonons not only decreases interfacial resistance but also increases the ballistic transport behavior in the semiconductor layer. These results provide new insight for experiments designed to characterize e-ph coupling and thermal transport at the metal-semiconductor/insulator interfaces. read less USED (high confidence) W.-jin Zhang, Y. Peng, and Z.-L. Liu, “Molecular dynamics simulations of the melting curve of NiAl alloy under pressure,” AIP Advances. 2014. link Times cited: 22 Abstract: The melting curve of B2-NiAl alloy under pressure has been i… read moreAbstract: The melting curve of B2-NiAl alloy under pressure has been investigated using molecular dynamics technique and the embedded atom method (EAM) potential. The melting temperatures were determined with two approaches, the one-phase and the two-phase methods. The first one simulates a homogeneous melting, while the second one involves a heterogeneous melting of materials. Both approaches reduce the superheating effectively and their results are close to each other at the applied pressures. By fitting the well-known Simon equation to our melting data, we yielded the melting curves for NiAl: 1783(1 + P/9.801)0.298 (one-phase approach), 1850(1 + P/12.806)0.357 (two-phase approach). The good agreement of the resulting equation of states and the zero-pressure melting point (calc., 1850 ± 25 K, exp., 1911 K) with experiment proved the correctness of these results. These melting data complemented the absence of experimental high-pressure melting of NiAl. To check the transferability of this EAM potential, we have also predicted the melting curves of pure nickel and pure aluminum. Results show the calculated melting point of Nickel agrees well with experiment at zero pressure, while the melting point of aluminum is slightly higher than experiment. read less USED (high confidence) K.-H. Lin, B. Holian, T. Germann, and A. Strachan, “Mesodynamics with implicit degrees of freedom.,” The Journal of chemical physics. 2014. link Times cited: 19 Abstract: Mesoscale phenomena--involving a level of description betwee… read moreAbstract: Mesoscale phenomena--involving a level of description between the finest atomistic scale and the macroscopic continuum--can be studied by a variation on the usual atomistic-level molecular dynamics (MD) simulation technique. In mesodynamics, the mass points, rather than being atoms, are mesoscopic in size, for instance, representing the centers of mass of polycrystalline grains or molecules. In order to reproduce many of the overall features of fully atomistic MD, which is inherently more expensive, the equations of motion in mesodynamics must be derivable from an interaction potential that is faithful to the compressive equation of state, as well as to tensile de-cohesion that occurs along the boundaries of the mesoscale units. Moreover, mesodynamics differs from Newton's equations of motion in that dissipation--the exchange of energy between mesoparticles and their internal degrees of freedom (DoFs)--must be described, and so should the transfer of energy between the internal modes of neighboring mesoparticles. We present a formulation where energy transfer between the internal modes of a mesoparticle and its external center-of-mass DoFs occurs in the phase space of mesoparticle coordinates, rather than momenta, resulting in a Galilean invariant formulation that conserves total linear momentum and energy (including the energy internal to the mesoparticles). We show that this approach can be used to describe, in addition to mesoscale problems, conduction electrons in atomic-level simulations of metals, and we demonstrate applications of mesodynamics to shockwave propagation and thermal transport. read less USED (high confidence) F. Delogu, “Ignition of an exothermal reaction by collision between Al and Ni crystals,” Journal of Applied Physics. 2011. link Times cited: 11 Abstract: Classical molecular dynamics methods have been used to inves… read moreAbstract: Classical molecular dynamics methods have been used to investigate the atomic-scale dynamics of collisions between two Al and Ni crystals with rough surfaces. The crystals were approached along the direction perpendicular to the surfaces and simultaneously displaced along the direction parallel to them at relative velocities in the range between 1 and 10 nm ns−1. The mechanical stresses operating at collision determine a local deformation of Al and Ni lattices, accompanied by a significant temperature rise. As the Al melting point is reached, the Al crystal partially melts and Ni atoms start dissolving into the molten phase. The significant heat of mixing liberated further promotes the Al melting and the Ni dissolution processes. In the absence of neighboring Al-Ni interfaces, the heat dissipation processes and the limited rate of Ni dissolution gradually lead to the extinction of the reactive behavior. Conversely, the presence of Al-Ni interfaces in the vicinity of the Al-Ni one formed by collision permi... read less USED (high confidence) H.-Z. Wu and S. Zhao, “Molecular dynamics study of the response of nanostructured Al/Ni clad particles system under thermal loading.,” The journal of physical chemistry. A. 2011. link Times cited: 5 Abstract: Molecular dynamics simulations are used to study the exother… read moreAbstract: Molecular dynamics simulations are used to study the exothermic alloying reactions by imposing a thermal loading on a local area of nanostructured Al/Ni clad particles. The combustion parameters, such as particles size, density, and ignition temperature, are characterized. Reducing the size of Al/Ni clad particles makes the propagation velocity of reaction front increase but lowers both the adiabatic combustion temperature and pressure of the system. However, increasing either mass density or ignition temperature makes the propagation velocity of reaction front increase and raises the adiabatic temperature and pressure as well. We estimate the propagation velocity of the chemical reaction front to range from 35.70 to 44.06 m/s. read less USED (high confidence) C. Becker, F. Tavazza, and L. Levine, “Implications of the choice of interatomic potential on calculated planar faults and surface properties in nickel,” Philosophical Magazine. 2011. link Times cited: 15 Abstract: With the increasing use of molecular simulation to understan… read moreAbstract: With the increasing use of molecular simulation to understand deformation mechanisms in transition metals, it is important to assess how well the simulations reproduce physical behavior both near equilibrium and under more extreme conditions. In particular, it is important to examine whether simulations predict unusual deformation paths that are competitive with those experimentally observed. In this work we compare generalized planar fault energy landscapes and surface energies for various interatomic potentials with those from density functional theory calculations to examine how well these more complicated planar faults and surface energies are captured and whether any deformations are energetically competitive with the {111}⟨112⟩ slip observed in FCC crystals. To do this we examine not just the (111) fault orientation, but also the (100), (110), (210), (211), (311), and (331) orientations to test behavior outside of the fitting range of the interatomic potentials. We find that the shape of the (111)[11 ] stacking fault energy curve varies significantly with potential, with the ratio of unstable to stable stacking fault energies ranging from 1.22 to 14.07, and some deformation paths for non-(111) orientations give activation barriers less than 50% higher than the unstable stacking fault energies. These are important considerations when choosing an interatomic potential for deformation simulations. read less USED (high confidence) N. S. Weingarten and B. Rice, “A molecular dynamics study of the role of relative melting temperatures in reactive Ni/Al nanolaminates,” Journal of Physics: Condensed Matter. 2011. link Times cited: 38 Abstract: Molecular dynamics (MD) simulations using a recently develop… read moreAbstract: Molecular dynamics (MD) simulations using a recently developed first-principles-based embedded-atom-method (EAM) potential are used to simulate the exothermic alloying reactions of a Ni/Al bilayer initially equilibrated at 1200 K. Simulations are performed in the isobaric–isoenthalpic (NPH) ensemble, which provides insight into the influence of pressure on atomic mixing and the subsequent alloying reaction. For pressures lower than 8 GPa, the mechanism of mixing is the same: as mixing and reaction occur at the interface, the heat generated first melts the Al layer, and subsequent mixing leads to further heat generation after which the Ni layer melts, leading to additional mixing until the alloying reactions are completed. However, for simulations at pressures higher than 8 GPa, the reaction does not occur within the time interval of the simulation. The results will be compared with our previous simulations of a Ni/Al bilayer using a different interatomic potential, which predicts substantially different pressure-dependent melting behavior of the pure components. This comparative study suggests that pressure-dependent melting behavior of components of reactive materials can be used to influence reaction rates and mechanisms. read less USED (high confidence) X. Hu, L. Liu, Y. Zhang, G. Lu, and T.-min Wang, “Energy investigation of effects of O on mechanical properties of NiAl intermetallics,” Journal of Physics: Condensed Matter. 2011. link Times cited: 15 Abstract: We have investigated effects of O on mechanical properties o… read moreAbstract: We have investigated effects of O on mechanical properties of NiAl by calculating the cleavage energy (γC) and the unstable stacking fault energy (γus) using a first-principles method. O is shown to reduce γC/γus for the [001](110) and [100](001) slip systems, indicating that the presence of O should be associated with the ductility reduction of NiAl. Further, γC/γus of the NiAl–O system can be increased by Cr, suggesting the possibility to suppress the negative effect of O via alloying elements. read less USED (high confidence) A. Biborski, L. Zosiak, R. Kozubski, R. Sot, and V. Pierron-Bohnes, “Semi-grand canonical Monte Carlo simulation of ternary bcc lattice-gas decomposition: Vacancy formation correlated with B2 atomic ordering in A–B intermetallics,” Intermetallics. 2010. link Times cited: 14 USED (high confidence) H. Zhou, S. Qu, and W. Yang, “Toughening by nano-scaled twin boundaries in nanocrystals,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 62 Abstract: Joint enhancement on strength and toughness provides a cutti… read moreAbstract: Joint enhancement on strength and toughness provides a cutting-edge research frontier for metals and alloys. Conventional strengthening methods typically lead to suppressed ductility and fracture toughness. In this study, large-scale atomic simulation on the fracture process is performed featuring nanocrystals embedded with nano-scaled twin boundaries (TBs). Four toughening mechanisms by nano-scaled TBs are identified: (i) crack blunting through dislocation accommodation along the nano-scaled TBs; (ii) crack deflection in a manner of intragranular propagation; (iii) daughter crack formation along the nano-scaled TBs that further enhances the toughness and (iv) curved TB planes owing to an excessive pileup of geometrically necessary dislocations. These toughening mechanisms jointly dictate the mechanical behavior of nano-structured materials, and provide insights into the application of nano-scaled TBs with an aim to simultaneously obtain enhanced strength and toughness. New approaches to introduce these coherent internal defects into the nanostructure of crystalline materials are also proposed. read less USED (high confidence) N. S. Weingarten, W. Mattson, and B. Rice, “Determination of the pressure dependent melting temperatures of Al and Ni using molecular dynamics,” Journal of Applied Physics. 2009. link Times cited: 33 Abstract: We present the results of a molecular dynamics simulation st… read moreAbstract: We present the results of a molecular dynamics simulation study designed to calculate the melting temperatures of pure nickel and pure aluminum at various system pressures using an embedded atom method type potential. The melting points are determined using a two-phase coexistence method, where the liquid and solid phases are modeled simultaneously at a fixed pressure and temperature, allowing us to bracket the value within a desired range of accuracy. The values obtained for the melting points of aluminum are consistently higher than expected based on experiment, while those for nickel are lower. Other thermal properties of aluminum and nickel were determined in order to fit the melting temperature data into a standard theoretical framework. Also, planar material defects, such as twin boundaries and stacking faults, were observed in crystals grown from the melt, occurring more often in aluminum systems than in nickel. Planar defect energies were calculated for both systems in order to explain these obser... read less USED (high confidence) N. S. Weingarten, W. Mattson, A. D. Yau, T. Weihs, and B. Rice, “A molecular dynamics study of the role of pressure on the response of reactive materials to thermal initiation,” Journal of Applied Physics. 2009. link Times cited: 47 Abstract: To elucidate the mechanisms of energy release in a reacting … read moreAbstract: To elucidate the mechanisms of energy release in a reacting nickel/aluminum bilayer, we simulate the exothermic alloying reactions using both microcanonical and isoenthalpic-isobaric molecular dynamics simulations and an embedded-atom method type potential. The mechanism of the mixing consists of a sequence of steps in which mixing and reaction first occurs at the interface; the resulting heat generated from the mixing then melts the Al layer; subsequent mixing leads to further heat generation after which the Ni layer melts. The mixing continues until the alloying reactions are completed. The results indicate that pressure has a significant influence on the rates of atomic mixing and alloying reactions. Local pressures and temperatures within the individual layers at the time of melting are calculated, and these results are compared with the pressure-dependent melting curves determined for pure Al and pure Ni using this interaction potential. read less USED (high confidence) S. Zhao, T. Germann, and A. Strachan, “Atomistic simulations of shock-induced alloying reactions in Ni/Al nanolaminates.,” The Journal of chemical physics. 2006. link Times cited: 83 Abstract: We employ molecular dynamics simulations with a first princi… read moreAbstract: We employ molecular dynamics simulations with a first principles-based many body potential to characterize the exothermic alloying reactions of nanostructured Ni/Al multilayers induced by shock loading. We introduce a novel technique that captures both the initial shock transit as well as the subsequent longer-time-scale Ni3Al alloy formation. Initially, the softer Al layers are shock heated to a higher temperature than the harder Ni layers as a result of a series of shock reflections from the impedance-mismatched interfaces. Once initiated, the highly exothermic alloying reactions can propagate in a self-sustained manner by mass and thermal diffusion. We also characterize the role of voids on the initiation of alloying. The interaction of the shock wave with the voids leads not only to significant local heating (hot spots) but also directly aids the intermixing between Al and Ni; both of these phenomena contribute to a significant acceleration of the alloying reactions. read less USED (high confidence) Y. Amouyal, E. Rabkin, and Y. Mishin, “Correlation between grain boundary energy and geometry in Ni-rich NiAl,” Acta Materialia. 2005. link Times cited: 55 USED (high confidence) A. Suzuki and Y. Mishin, “Atomic mechanisms of grain boundary diffusion: Low versus high temperatures,” Journal of Materials Science. 2005. link Times cited: 128 USED (high confidence) J. A. Brown and Y. Mishin, “Segregation and structural transformations at Σ = 3 grain boundaries in NiAl: A Monte-Carlo study,” Acta Materialia. 2005. link Times cited: 17 USED (high confidence) S. Das, J. Horbach, M. Koza, S. M. Chatoth, and A. Meyer, “Influence of chemical short-range order on atomic diffusion in Al–Ni melts,” Applied Physics Letters. 2005. link Times cited: 100 Abstract: We use inelastic neutron scattering and molecular dynamics s… read moreAbstract: We use inelastic neutron scattering and molecular dynamics simulation to investigate the chemical short-range order (CSRO), visible through prepeaks in the structure factors, and its relation to self-diffusion in Al–Ni melts. As a function of composition at 1795K, Ni self-diffusion coefficients from experiment and simulation exhibit a nonlinear dependence with a pronounced increase on the Al-rich side. This comes along with a change in CSRO with increasing Al content that is related to a more dense packing of the atoms in Ni-rich Al–Ni systems. read less USED (high confidence) M. Prasad and T. Sinno, “Feature activated molecular dynamics: an efficient approach for atomistic simulation of solid-state aggregation phenomena.,” The Journal of chemical physics. 2004. link Times cited: 3 Abstract: An efficient approach is presented for performing efficient … read moreAbstract: An efficient approach is presented for performing efficient molecular dynamics simulations of solute aggregation in crystalline solids. The method dynamically divides the total simulation space into "active" regions centered about each minority species, in which regular molecular dynamics is performed. The number, size, and shape of these regions is updated periodically based on the distribution of solute atoms within the overall simulation cell. The remainder of the system is essentially static except for periodic rescaling of the entire simulation cell in order to balance the pressure between the isolated molecular dynamics regions. The method is shown to be accurate and robust for the Environment-Dependant Interatomic Potential (EDIP) for silicon and an Embedded Atom Method potential (EAM) for copper. Several tests are performed beginning with the diffusion of a single vacancy all the way to large-scale simulations of vacancy clustering. In both material systems, the predicted evolutions agree closely with the results of standard molecular dynamics simulations. Computationally, the method is demonstrated to scale almost linearly with the concentration of solute atoms, but is essentially independent of the total system size. This scaling behavior allows for the full dynamical simulation of aggregation under conditions that are more experimentally realizable than would be possible with standard molecular dynamics. read less USED (high confidence) N. Papanicolaou, H. Chamati, G. Evangelakis, and D. Papaconstantopoulos, “Second-moment interatomic potential for Al, Ni and Ni-Al alloys, and molecular dynamics application,” Computational Materials Science. 2003. link Times cited: 37 USED (high confidence) J.-ping Du, C.-yu Wang, C.-yu Wang, and T. Yu, “Construction and application of multi-element EAM potential (Ni–Al–Re) in γ/γ′ Ni-based single crystal superalloys,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 44 Abstract: Based on experiments and first-principles calculations, a Ni… read moreAbstract: Based on experiments and first-principles calculations, a Ni–Al–Re system embedded atom method (EAM) potential is constructed for the γ(Ni)/γ′(Ni3Al) superalloy. The contribution of the inner elastic constants is considered in the fitting of Re with a hexagonal close-packed structure. Using this potential, point defects, planar defects and lattice misfit of γ(Ni) and γ′(Ni3Al) are investigated. The interaction between Re and the misfit dislocation of the γ(Ni)/γ′(Ni3Al) system is also calculated. We conclude that the embedding energy has an important effect on the properties of the alloys, such as the planar fault energies of Ni3Al, by considering the relationship between the charge transfer calculated from first-principles, the elastic constants of Ni3Al and the host electron density of the EAM potential. The multi-element potential predicts that Re does not form clusters in γ(Ni), which is consistent with recent experiments and first-principles calculations. read less USED (high confidence) X. Liu, J. Gu, Y. Shen, J. Li, and C. Chen, “Lattice dynamical finite-element method,” Acta Materialia. 2010. link Times cited: 24 USED (low confidence) C.-han Yang, F.-yao Wu, and S. Lin, “Phase equilibria, thermodynamic assessment, and the mechanical property of B2 phase of the Al-Ga-Ni ternary system,” Intermetallics. 2024. link Times cited: 0 USED (low confidence) Y. Li and W. Qiang, “Compositional effects on the antiphase boundary energies in B2-type NiTi and NiTi-based high-entropy intermetallics,” Materials Chemistry and Physics. 2023. link Times cited: 0 USED (low confidence) D. Singh, D. Gianola, and T. Rupert, “Dislocation breakaway from nanoparticle array linear complexions: Plasticity mechanisms and strength scaling laws,” Materialia. 2023. link Times cited: 0 USED (low confidence) H. Mes-adi, R. Herbazi, M. Lablali, K. Saadouni, and M. Mazroui, “NiAl (0 0 1) terminated surface effect on the growth of the Al thin film,” Computational Materials Science. 2023. link Times cited: 3 USED (low confidence) E. Yousefi et al., “Dynamics of intermetallics formation in the Al/Ni reactive wetting system,” Materialia. 2023. link Times cited: 1 USED (low confidence) H. Zhang, C. Luo, Z. Zheng, and Y. Han, “Effects of size ratio on particle packing in binary glasses,” Acta Materialia. 2023. link Times cited: 1 USED (low confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (low confidence) R. Rozas, V. Ankudinov, and P. Galenko, “Kinetics of rapid growth and melting of Al50Ni50 alloying crystals: phase field theory versus atomistic simulations revisited,” Journal of Physics: Condensed Matter. 2022. link Times cited: 3 Abstract: A revised study of the growth and melting of crystals in con… read moreAbstract: A revised study of the growth and melting of crystals in congruently melting Al50Ni50 alloy is carried out by molecular dynamics (MDs) and phase field (PF) methods. An embedded atom method (EAM) potential of Purja Pun and Mishin (2009 Phil. Mag. 89 3245) is used to estimate the material’s properties (density, enthalpy, and self-diffusion) of the B2 crystalline and liquid phases of the alloy. Using the same EAM potential, the melting temperature, density, and diffusion coefficient become well comparable with experimental data in contrast with previous works where other potentials were used. In the new revision of MD data, the kinetics of melting and solidification are quantitatively evaluated by the ‘crystal-liquid interface velocity–undercooling’ relationship exhibiting the well-known bell-shaped kinetic curve. The traveling wave solution of the kinetic PF model as well as the hodograph equation of the solid-liquid interface quantitatively describe the ‘velocity–undercooling’ relationship obtained in the MD simulation in the whole range of investigated temperatures for melting and growth of Al50Ni50 crystals. read less USED (low confidence) T. Gao et al., “Molecular dynamics simulations on the connectivity of topologically close-packed clusters in TiAl3 alloys,” Physica Scripta. 2022. link Times cited: 0 Abstract: Presently, there has been increasing attention on TiAl3, whi… read moreAbstract: Presently, there has been increasing attention on TiAl3, which is commonly used for fabricating power aviation devices owing to its good oxidation resistance and outstanding mechanical properties in high-temperature. As the microstructures determine the macroscopic properties of a material, we investigated the connectivity of icosahedral central atoms in TiAl3 using conventional methods in this study. The topologically close-packed (TCP) structures are present in supercooled liquids, metallic glasses, and metallic liquids. They are intrinsic to liquid metals and are an essential character of the structure in metallic glasses (MGs). However, because of the lack of the concept of connectivity of the TCP structures, we investigated connectivity from the icosahedral central atoms to TCP structures, and a formula was proposed to calculate the connectivity of the TCP structures. Based on the results, low temperatures and cooling rates are conducive to generate high connectivity between icosahedral central atoms and TCP structures. The proposed formula can characterize the connectivity of the TCP structures. These findings open new opportunities for conducting research on the connectivity of clusters in binary alloys. read less USED (low confidence) S. Kolli et al., “Fracture mechanisms of Ni-Al interfaces – A nanoscale view,” Materials Today Communications. 2022. link Times cited: 1 USED (low confidence) Y.-F. Wu, W. Yu, and S. Shen, “Strain-modulated initial oxidation of Al(1-)Ni alloy surface,” Applied Surface Science. 2022. link Times cited: 2 USED (low confidence) Y. Xu, G. Wang, P. Qian, and Y. Su, “Element segregation and thermal stability of Ni–Rh nanoparticles,” Journal of Solid State Chemistry. 2022. link Times cited: 6 USED (low confidence) Y. Li and W. Qiang, “Ordered structure and solute-suppressed atomic ordering in iron-cobalt alloys,” Computational Materials Science. 2022. link Times cited: 8 USED (low confidence) W. Li, X. Peng, A. Ngan, and J. El-Awady, “Surface energies and relaxation of NiCoCr and NiFeX (X = Cu, Co or Cr) equiatomic multiprincipal element alloys from first principles calculations,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 0 Abstract: First principles calculations of the energies and relaxation… read moreAbstract: First principles calculations of the energies and relaxation of unreconstructed low-index surfaces, i.e. (001), (011) and (111) surfaces, in NiCoCr and NiFeX (X = Cu, Co or Cr) equiatomic multi-principal element alloys (MPEAs) are presented. The calculations were conducted for 12-layer slabs represented by special quasi-random supercells using the projector augmented wave method within the generalized gradient approximation. While experimental predictions are unavailable for comparison, the calculated surface energies agree fairly well with those from thermodynamic modeling and a bond-cutting model. In addition, the calculations unveil an important surface structure, namely, that the topmost surface layer is in contraction except for the (001) surface of NiFeCr alloy, the next layer below is in extension, and the bulk spacing is gradually recovered from the subsequent layers down. Additionally, the surface contraction is the most pronounced on the (011) plane, being about 4%–10% relative to the bulk spacings. The results presented here can provide an understanding of surface-controlled phenomena such as corrosion, catalytic activities and fracture properties in these equiatomic MPEAs. read less USED (low confidence) Y. L. Li, X. Cheng, W. Duan, and W. Qiang, “Improved ductility by coupled motion of grain boundaries in nanocrystalline B2-FeCo alloys,” Computational Materials Science. 2021. link Times cited: 7 USED (low confidence) M. Wagih and C. Schuh, “Thermodynamics and design of nanocrystalline alloys using grain boundary segregation spectra,” Acta Materialia. 2021. link Times cited: 20 USED (low confidence) Q. Yang, H. Liu, and H. Peng, “Crystal growth in deeply undercooled Ni50Al50: Signature of the ordering sequence at the interface.,” The Journal of chemical physics. 2021. link Times cited: 7 Abstract: Crystal growth of the intermetallic alloy, Ni50Al50, is inve… read moreAbstract: Crystal growth of the intermetallic alloy, Ni50Al50, is investigated by molecular dynamics simulations with two different interatomic potentials. The calculated growth rate can be captured by the Wilson-Frenkel or Broughton-Gilmer-Jackson model at small undercoolings but deviates from the theory at deep undercoolings. Failure of the theory is found to be correlated with the dynamic processes that emerged at the interface, but not apparently with the static interface structure. The chemical segregation of Ni and Al atoms occurs before the geometrical ordering upon crystallization at small undercoolings. In contrast, the geometrical ordering precedes the chemical one at deep undercoolings. These two ordering processes show a collapsed time evolution at the crossover temperature consistent with the onset of the theoretical deviation. We rationalize the delayed chemical segregation behavior by the collective atomic motion, which is characterized by the super-Arrhenius transition of the temperature-dependent diffusivity and structural relaxation time at the crossover point. read less USED (low confidence) Q. Yin, Y. Lian, R. Wu, L. Gao, S.-Q. Chen, and Z. Wen, “Effect of the potential function and strain rate on mechanical behavior of the single crystal Ni-based alloys: A molecular dynamics study*,” Chinese Physics B. 2021. link Times cited: 1 USED (low confidence) J. Li et al., “Tensile mechanical performance of Al/Ni dissimilar metals bonded by self-propagating exothermic reaction based on molecular dynamics simulation,” Materials today communications. 2021. link Times cited: 3 USED (low confidence) J. Huo, K. Wang, Y. Wang, P. Qian, C. Ji, and Y. Su, “Mechanical and thermodynamic properties study of Al-based binary and ternary solid solutions using the pseudoatomic potential method,” Intermetallics. 2020. link Times cited: 1 USED (low confidence) M. Zaenudin, M. Mohammed, A. Gamayel, and A. Sunardi, “Atomistic investigation of diffusion welding of dissimilar materials through molecular dynamics simulation.” 2020. link Times cited: 0 Abstract: Molecular dynamics simulation of diffusion welding between a… read moreAbstract: Molecular dynamics simulation of diffusion welding between aluminum and nickel has been performed and the tensile behavior of the as-welded material has been examined. Two slabs Al-Ni are being contacted directly to introduce diffusion between the two with certain parameters. Diffusion welding was performed at a temperature of 500 K, at the pressure of 10 MPa in the x-direction, and a holding time of 200 ps. The tensile test is performed at a strain value of 2.64e9/s. The results have shown that the displacement of atoms during diffusion welding significantly occurs from the beginning until it reaches about 10 ps. However, the movement of atoms experiencing a plateau stage and less atomic exchange is occurring afterward. nickel slab that has a better strength was able to maintain its fcc crystalline structure during diffusion welding and tensile test than aluminum which has lower strength. The higher melting point of Ni promoting better resistance in the interface to be deformed, while the lower melting point of Al has been promoting better bonding between the two that is promoted by the defects that occur at the interface. During the tensile test, the lower strength of Al made it randomly deformed and crack seems to occur. However, failure of the diffusion-welded Al-Ni has occurred around the interface. The ultimate tensile strength of the diffusion-welded Al-Ni reaches up to 4.166 GPa at a strain value of 0.07128. read less USED (low confidence) N. Esakkiraja, A. K. Gupta, V. Jayaram, T. Hickel, S. Divinski, and A. Paul, “Diffusion, defects and understanding the growth of a multicomponent interdiffusion zone between Pt-modified B2 NiAl bond coat and single crystal superalloy,” Acta Materialia. 2020. link Times cited: 26 USED (low confidence) L. Wang, K. C. Lai, L. Huang, J. Evans, and Y. Han, “Low-index surface energies, cleavage energies, and surface relaxations for crystalline NiAl from first-principles calculations,” Surface Science. 2020. link Times cited: 16 USED (low confidence) G. Wang, Y. Xu, P. Qian, and Y. Su, “Vacancy concentration of films and nanoparticles,” Computational Materials Science. 2020. link Times cited: 8 USED (low confidence) A. Bhattacharya, K. Mondal, C. Upadhyay, and S. Sangal, “A phase-field investigation of the effect of grain-boundary diffusion on austenite to ferrite transformation,” Computational Materials Science. 2020. link Times cited: 7 USED (low confidence) G. Wang, Y. Xu, P. Qian, and Y. Su, “The effects of size and shape on the structural and thermal stability of platinum nanoparticles,” Computational Materials Science. 2019. link Times cited: 13 USED (low confidence) V. Jordan and I. Shmakov, “The emergence of a heterostructure of the intermetallic phases in the process of SH-synthesis simulation in a nonstoichiometric nanoscale layered Ti-Al system,” Journal of Physics: Conference Series. 2019. link Times cited: 1 Abstract: Computational experiments (CEs) have been carried out to sim… read moreAbstract: Computational experiments (CEs) have been carried out to simulate the propagation of the combustion wave of the SH-synthesis process in a package of alternating nanosized layers of crystal lattices of Ti and Al atoms by the «molecular dynamics» method. By means the LAMMPS package performing parallel computing the computational experiments (CEs) have been carried out. The “embedded atom” model (EAM) for the interatomic interaction potential was used. The two structures of Ti-Al system with various quantities of atoms and nonstoichiometric ratios have been studied. The sets of temperature and density profiles along the layered structure at successive instants of time (up to 16 ns) have been calculated. In addition, the corresponding sets of vertical cross-sections of the distributions of atoms and various types of elementary cells (fcc, hcp, bcc, etc.) have been calculated. The emergence of heterostructure of intermetallic phases in the Ti-Al system with 453974 atoms and the nonstoichiometric ratio of 1.23 has been detected. read less USED (low confidence) D. Nguyen-Trong, K. Pham-Huu, and P. Nguyen-Tri, “Simulation on the Factors Affecting the Crystallization Process of FeNi Alloy by Molecular Dynamics,” ACS Omega. 2019. link Times cited: 24 Abstract: This paper investigates the crystallization process of FeNi … read moreAbstract: This paper investigates the crystallization process of FeNi alloys with different impurity concentrations of Ni(x) [x = 10% (Fe90Ni10), 20% (Fe80Ni20), 30% (Fe70Ni30), 40% (Fe60Ni40), and 50% (Fe50Ni50)] at temperature (T) = 300 K and Fe70Ni30 at heating rates of 4 × 1012, 4 × 1013, and 4 × 1014 K/s at different temperatures, T = 300, 400, 500, 600, 700, 900, 1100, and 1300 K. Molecular dynamics models with the Sutton–Chen embedded interaction potential and recirculating boundary conditions are used to calculate the molecular parameters of alloys, such as radial distribution function, total energy of the system (Etot), size (l), and crystallization temperature (through the relationship between Etot and T). The common neighborhood analysis method is used to confirm the theoretical results of crystallization for Fe–Fe, Fe–Ni, and Ni–Ni. The annealing process did not have an effect on the crystallization process of FeNi alloys. The effect of Ni content, heating rate, and annealing time on structural unit numbers, such as face-centered cubic, hexagonal close-packed, blocked cubic center, and amorphous, and the crystallization process of FeNi alloys is also investigated. read less USED (low confidence) V. Turlo and T. Rupert, “Prediction of a wide variety of linear complexions in face centered cubic alloys,” Acta Materialia. 2019. link Times cited: 11 USED (low confidence) J. Wang, J. Liang, Z. Wen, and Z. Yue, “Atomic simulation of void location effect on the void growth in nickel-based single crystal,” Computational Materials Science. 2019. link Times cited: 17 USED (low confidence) J. Wang, C. Chang, K. Song, L. Wang, and Y. Pan, “Short-range ordering in metallic supercooled liquids and glasses,” Journal of Alloys and Compounds. 2019. link Times cited: 11 USED (low confidence) G. Wang and Y. Xu, “Embedded-atom potential for Ni-Al alloy,” IOP Conference Series: Materials Science and Engineering. 2018. link Times cited: 1 Abstract: We construct a new embedded-atom potential for Ni-Al system.… read moreAbstract: We construct a new embedded-atom potential for Ni-Al system. The lattice constants, cohesive energies, elastic constants, vacancy formation energies, stacking fault energies and equations of state of Ni and Al are included in the fitting process of potentials for pure Ni and pure Al. The cross-interaction potential is fitted to the lattice constants, cohesive energies, elastic constants of Ni3Al and NiAl and the energies of (100) APB and (111) SISF of Ni3Al. The potential accurately reproduces the various physical properties of Ni, Al, Ni3Al and NiAl phases. The results of the new embedded-atom potential and other embedded-atom potentials are compared and discussed in detail. read less USED (low confidence) V. Jordan and I. Shmakov, “The study of microstructure and propagation of the combustion wave of SHS in nanodimensional multilayer systems of Ni-Al with using molecular-dynamic simulation,” Journal of Physics: Conference Series. 2018. link Times cited: 0 Abstract: Computational experiments (CEs) on the molecular-dynamic sim… read moreAbstract: Computational experiments (CEs) on the molecular-dynamic simulation of the propagation of the combustion wave of the SH-synthesis process in a stack of alternating layers of nanoscale crystal lattices of Ni and Al atoms have been carried out. In the calculations are used two versions of the interatomic interaction potential in the “embedded atom model” (EAM) and the LAMMPS package taking into account parallel computings. As the results of computational experiments for two varieties of the EAM potential, a family of temperature profiles along the layers of the structure at successive instants of time (up to 40 ns) and the corresponding set of microsections (snapshots) of the layered structure are given. For large values of the initial heating temperature and the stoichiometric ratio of Ni and Al atoms in the SHS sample, the computational experiments were confirmed the heterogeneous SHS-reaction mechanism, referred to in scientific publications as “mosaic-dissolution-precipitation”. In addition, when under such conditions a combustion wave of SHS passes through, the effect of heat localization is observed with the establishment of a higher temperature in the final region of the sample compared with the initial region, as a result of which on the “plateau” of the temperature profile corresponding to 16 ns a smooth “dip” is observed. Then, at subsequent times, there is a continuation of combustion (more precisely, after combustion) in the inverse direction with the equalization of the temperature plateau corresponding to a higher temperature value of the final region of the SHS-sample. read less USED (low confidence) H. N. Pishkenari, F. S. Yousefi, and A. Taghibakhshi, “Determination of surface properties and elastic constants of FCC metals: a comparison among different EAM potentials in thin film and bulk scale,” Materials Research Express. 2018. link Times cited: 22 Abstract: Three independent elastic constants C11, C12, and C44 were c… read moreAbstract: Three independent elastic constants C11, C12, and C44 were calculated and compared using available potentials of eight different metals with FCC crystal structure; Gold, Silver, Copper, Nickel, Platinum, Palladium, Aluminum and Lead. In order to calculate the elastic constants, the second derivative of the energy density of each system was calculated with respect to different directions of strains. Each set of the elastic constants of the metals in bulk scale was compared with experimental results, and the average relative error was for each was calculated and compared with other available potentials. Then, using the Voigt-Reuss-Hill method, approximated values for Young and shear moduli and Poisson’s ratio of the FCC metals in the bulk scale were found for each potential. Furthermore, to observe the surface effects on the metals in nanoscale, surface elastic constants of the thin films of the metals have been calculated. In the study of the thin films of materials in nanoscale, the number of surface atoms is considerable compared to all atoms of the object. This leads to an increase in the surface effects, which influence the elastic properties. By considering this fact and employing related definitions and equations, the properties of the thin films of the metals were calculated, and the surface effects for different crystallographic directions were compared. Subsequently, in some cases, comparisons among characteristics of the metals in the thin film and bulk material were made. read less USED (low confidence) N. Dũng, “Influence of impurity concentration, atomic number, temperature and tempering time on microstructure and phase transformation of Ni1−xFex (x = 0.1, 0.3, 0.5) nanoparticles,” Modern Physics Letters B. 2018. link Times cited: 17 Abstract: The influence of the concentration of impurity Fe in nanopar… read moreAbstract: The influence of the concentration of impurity Fe in nanoparticles Ni1−xFex with x = 0.1, 0.3 and 0.5 at T = 300 K; 4000 atoms, 5324 atoms, 6912 atoms and 8788 atoms at T = 300 K; 6912 atoms at T =... read less USED (low confidence) F. Baras and O. Politano, “Epitaxial growth of the intermetallic compound NiAl on low-index Ni surfaces in Ni/Al reactive multilayer nanofoils,” Acta Materialia. 2018. link Times cited: 20 USED (low confidence) S. Yalameha and A. Vaez, “Ab-initio thermodynamic and elastic properties of AlNi and AlNi3 intermetallic compounds,” International Journal of Modern Physics B. 2018. link Times cited: 15 Abstract: In this paper, thermodynamic and elastic properties of the A… read moreAbstract: In this paper, thermodynamic and elastic properties of the AlNi and AlNi3 were investigated using density functional theory (DFT). The full-potential linearized augmented plane-wave (APW) in the framework of the generalized gradient approximation as used as implemented in the Wien2k package. The temperature dependence of thermal expansion coefficient, bulk modulus and heat capacity in a wide range of temperature (0–1600 K) were investigated. The calculated elastic properties of the compounds show that both intermetallic compounds of AlNi and AlNi3 have surprisingly negative Poisson’s ratio (NPR). The results were compared with other experimental and computational data. read less USED (low confidence) C. Tang and P. Harrowell, “Chemical ordering and crystal nucleation at the liquid surface: A comparison of Cu50Zr50 and Ni50Al50 alloys.,” The Journal of chemical physics. 2018. link Times cited: 6 Abstract: We study the influence of the liquid-vapor surface on the cr… read moreAbstract: We study the influence of the liquid-vapor surface on the crystallization kinetics of supercooled metal alloys. While a good glass former, Cu50Zr50, shows no evidence of surface enhancement of crystallization, Ni50Al50 exhibits an increased rate of crystallization due to heterogeneous nucleation at the free liquid surface. The difference in the compositional fluctuations at the interface is proposed as the explanation of the distinction between the two alloys. Specifically, we observe compositional ordering at the surface of Ni50Al50, while the Cu50Zr50 alloy only exhibits a diffuse adsorption of the Cu at the interface. We argue that the general difference in composition susceptibilities at planar surfaces represents an important factor in understanding the difference in the glass forming ability of the two alloys. read less USED (low confidence) A. Hassani, A. Makan, K. Sbiaai, A. Tabyaoui, and A. Hasnaoui, “Incidence energy effect and impact assessment during homoepitaxial growth of nickel on (001), (111) and (110) surfaces,” Thin Solid Films. 2017. link Times cited: 21 USED (low confidence) J. Ding, M. Asta, and R. Ritchie, “On the question of fractal packing structure in metallic glasses,” Proceedings of the National Academy of Sciences. 2017. link Times cited: 29 Abstract: Significance Our work clearly demonstrates a lack of fractal… read moreAbstract: Significance Our work clearly demonstrates a lack of fractal structure in metallic glasses, considering the packing of all atoms or solute-centered clusters. This finding clarifies the long-standing debate over the packing structure of metallic glasses from the perspective of fractal models; it is thus of significance for the community of researchers working with amorphous solids. Moreover, our percolation analysis of metallic glasses, revealing their cluster structure and percolation threshold, provides an important and powerful theoretical framework to describe the properties of amorphous alloys, such as the glass transition and mechanical deformation. This work addresses the long-standing debate over fractal models of packing structure in metallic glasses (MGs). Through detailed fractal and percolation analyses of MG structures, derived from simulations spanning a range of compositions and quenching rates, we conclude that there is no fractal atomic-level structure associated with the packing of all atoms or solute-centered clusters. The results are in contradiction with conclusions derived from previous studies based on analyses of shifts in radial distribution function and structure factor peaks associated with volume changes induced by pressure and compositional variations. The interpretation of such shifts is shown to be challenged by the heterogeneous nature of MG structure and deformation at the atomic scale. Moreover, our analysis in the present work illustrates clearly the percolation theory applied to MGs, for example, the percolation threshold and characteristics of percolation clusters formed by subsets of atoms, which can have important consequences for structure–property relationships in these amorphous materials. read less USED (low confidence) P. Kürnsteiner, M. B. Wilms, A. Weisheit, P. Barriobero-Vila, E. Jägle, and D. Raabe, “Massive nanoprecipitation in an Fe-19Ni-xAl maraging steel triggered by the intrinsic heat treatment during laser metal deposition,” Acta Materialia. 2017. link Times cited: 201 USED (low confidence) F. Hadef, “Synthesis and disordering of B2 TM-Al (TM = Fe, Ni, Co) intermetallic alloys by high energy ball milling: A review,” Powder Technology. 2017. link Times cited: 31 USED (low confidence) R. Ramakrishnan and R. Sankarasubramanian, “Crystal-melt kinetic coefficients of Ni3Al,” Acta Materialia. 2017. link Times cited: 18 USED (low confidence) A. Hassani, A. Makan, K. Sbiaai, A. Tabyaoui, and A. Hasnaoui, “THE INFLUENCE OF THE SURFACE ORIENTATION ON THE MORPHOLOGY DURING HOMOEPITAXIAL GROWTH OF NICKEL BY MOLECULAR DYNAMICS SIMULATION,” Surface Review and Letters. 2017. link Times cited: 17 Abstract: Homoepitaxial growth film for (001), (110) and (111) Ni subs… read moreAbstract: Homoepitaxial growth film for (001), (110) and (111) Ni substrates is investigated by means of molecular dynamics (MD) simulation. Embedded atom method (EAM) is considered to represent the interaction potential between nickel atoms. The simulation is performed at 300K using an incident energy of 0.06eV. In this study, the deposition process is performed periodically and the period, n, is relative to a perfect layer filling. The coverage rate of the actual expected level, L(n), can be considered a determinant for thin-film growth of nickel. The L(n) level is the most filled level during the deposition on (001) substrate, while it is the less filled one in the case of (111) substrate. Moreover, the upper level is the one which is responsible for the surface roughness and the appearance time of an upper layer may also be a factor influencing the surface roughness. The deposition on (111) substrate induces the most rigorous surface with a rapid appearance time of the upper layers. The L(n−1) layers are almost completely filled for all substrates. The L(n−2) and lower layers are completely filled for (001) and (110) substrates while for (111) substrate the completely filled layers are L(n−3) and lower ones. read less USED (low confidence) Q.-N. Fan, C.-yu Wang, and T. Yu, “Construction of ternary Ni–Al–Ta potential and its application in the effect of Ta on [1 1 0] edge dislocation slipping in γ′(Ni3Al),” Computational Materials Science. 2016. link Times cited: 2 USED (low confidence) K. Xiong, H. Lu, and J. Gu, “Atomistic simulations of the nanoindentation-induced incipient plasticity in Ni3Al crystal,” Computational Materials Science. 2016. link Times cited: 38 USED (low confidence) O. Politano and F. Baras, “Molecular dynamics simulations of self-propagating reactions in Ni–Al multilayer nanofoils,” Journal of Alloys and Compounds. 2015. link Times cited: 23 USED (low confidence) B.-H. Wu, J. Zhou, C. Xue, and H. Liu, “Molecular dynamics simulation of the deposition and annealing of NiAl film on Ni substrate,” Applied Surface Science. 2015. link Times cited: 32 USED (low confidence) V. Turlo, O. Politano, and F. Baras, “Dissolution process at solid/liquid interface in nanometric metallic multilayers: Molecular dynamics simulations versus diffusion modeling,” Acta Materialia. 2015. link Times cited: 40 USED (low confidence) Y.-C. Hu, F. Li, M. Li, H. Bai, and W.-chao Wang, “Five-fold symmetry as indicator of dynamic arrest in metallic glass-forming liquids,” Nature Communications. 2015. link Times cited: 190 USED (low confidence) A. Hassani, A. Makan, K. Sbiaai, A. Tabyaoui, and A. Hasnaoui, “Molecular dynamics study of growth and interface structure during aluminum deposition on Ni(1 0 0) substrate,” Applied Surface Science. 2015. link Times cited: 29 USED (low confidence) S. Kalidindi, J. A. Gomberg, Z. Trautt, and C. Becker, “Application of data science tools to quantify and distinguish between structures and models in molecular dynamics datasets,” Nanotechnology. 2015. link Times cited: 39 Abstract: Structure quantification is key to successful mining and ext… read moreAbstract: Structure quantification is key to successful mining and extraction of core materials knowledge from both multiscale simulations as well as multiscale experiments. The main challenge stems from the need to transform the inherently high dimensional representations demanded by the rich hierarchical material structure into useful, high value, low dimensional representations. In this paper, we develop and demonstrate the merits of a data-driven approach for addressing this challenge at the atomic scale. The approach presented here is built on prior successes demonstrated for mesoscale representations of material internal structure, and involves three main steps: (i) digital representation of the material structure, (ii) extraction of a comprehensive set of structure measures using the framework of n-point spatial correlations, and (iii) identification of data-driven low dimensional measures using principal component analyses. These novel protocols, applied on an ensemble of structure datasets output from molecular dynamics (MD) simulations, have successfully classified the datasets based on several model input parameters such as the interatomic potential and the temperature used in the MD simulations. read less USED (low confidence) J. M. Ortiz-Roldán et al., “Thermostructural behaviour of Ni-Cr materials: modelling of bulk and nanoparticle systems.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 15 Abstract: The thermostructural properties of Ni-Cr materials, as bulk … read moreAbstract: The thermostructural properties of Ni-Cr materials, as bulk and nanoparticle (NP) systems, have been predicted with a newly developed interatomic potential, for Ni/Cr ratios from 100/0 to 60/40. The potential, which has been fitted using experimental data and further validated using Density Functional Theory (DFT), describes correctly the variation with temperature of lattice parameters and the coefficient of thermal expansion, from 100 K to 1000 K. Using this potential, we have performed Molecular Dynamics (MD) simulations on bulk Ni-Cr alloys of various compositions, for which no experimental data are available. Similarly, NPs with diameters of 3, 5, 7, and 10 nm were studied. We found a very rapid convergence of NP properties with the size of the systems, showing already the 5 nm NPs with a thermostructural behaviour similar to the bulk. MD simulations of two 5 nm NPs show very little sintering and thermally induced damage, for temperatures between 300 K and 1000 K, suggesting that materials formed by agglomeration of Ni-Cr NPs meet the thermostructural stability requirements for catalysis applications. read less USED (low confidence) X.-yuan Yang, Y. Wu, and F. Liu, “MOLECULAR DYNAMICS SIMULATION OF THE SIZE EFFECT ON THE ELASTIC PROPERTIES OF THE B2-NiAl NANOFILM,” Surface Review and Letters. 2015. link Times cited: 0 Abstract: In the paper, molecular dynamics simulation with the modifie… read moreAbstract: In the paper, molecular dynamics simulation with the modified analytical embedded atom method (MAEAM) is applied to study the size effect on the elastic properties of the B2-NiAl nanofilm. The simulation results indicate that there is a critical thickness, which is about 5.38 nm, to distinguish the size dependence of the elastic properties of the nanofilm. On the one hand, these properties, such as the averaged cohesive energy and the bulk modulus, change evidently as the size is smaller than the critical thickness and the change tendency is tightly controlled by the surface atom composition. On the other hand, as the nanofilm size exceeds the critical one, the calculated values of the elastic properties are almost independent of the film thickness. Relatively, the bulk modulus magnitude of the nanofilm is apparently larger than that of the corresponding bulk material. Finally, the inherent mechanisms of the size impacting on the elastic properties of the B2-NiAl nanofilm have been discussed in more detail. The strengthening effect of the bulk modulus results from the smaller multilayer relaxation of the interlayer distance as compared to those of the bulk materials. read less USED (low confidence) S. Wilson, K. Gunawardana, and M. Mendelev, “Solid-liquid interface free energies of pure bcc metals and B2 phases.,” The Journal of chemical physics. 2015. link Times cited: 32 Abstract: The solid-liquid interface (SLI) free energy was determined … read moreAbstract: The solid-liquid interface (SLI) free energy was determined from molecular dynamics (MD) simulation for several body centered cubic (bcc) metals and B2 metallic compounds (space group: Pm3̄m; prototype: CsCl). In order to include a bcc metal with a low melting temperature in our study, a semi-empirical potential was developed for Na. Two additional synthetic "Na" potentials were also developed to explore the effect of liquid structure and latent heat on the SLI free energy. The obtained MD data were compared with the empirical Turnbull, Laird, and Ewing relations. All three relations are found to predict the general trend observed in the MD data for bcc metals obtained within the present study. However, only the Laird and Ewing relations are able to predict the trend obtained within the sequence of "Na" potentials. The Laird relation provides the best prediction for our MD data and other MD data for bcc metals taken from the literature. Overall, the Laird relation also agrees well with our B2 data but requires a proportionality constant that is substantially different from the bcc case. It also fails to explain a considerable difference between the SLI free energies of some B2 phases which have nearly the same melting temperature. In contrast, this difference is satisfactorily described by the Ewing relation. Moreover, the Ewing relation obtained from the bcc dataset also provides a reasonable description of the B2 data. read less USED (low confidence) P. Sowa et al., “Semigrand Canonical and Kinetic Monte Carlo simulations of binary B2-ordered nano-films with triple defects,” Intermetallics. 2014. link Times cited: 0 USED (low confidence) A. Evteev, E. Levchenko, I. Belova, R. Abdank-Kozubski, Z.-kui Liu, and G. Murch, “Theoretical Study of the Heat of Transport in a Liquid Ni50Al50 Alloy: Green-Kubo Approach,” Diffusion Foundations. 2014. link Times cited: 6 Abstract: We analyse the formalism of transport in a binary system esp… read moreAbstract: We analyse the formalism of transport in a binary system especially focussing on a detailed consideration of the heat of transport parameter characterizing diffusion driven by a temperature gradient. We introduce the reduced heat of transport parameter Qc*' which characterizes part of the interdiffusion flux that is proportional to the temperature gradient. In an isothermal system Qc*' represents the reduced heat flow (pure heat conduction) consequent upon unit interdiffusion flux. We demonstrate that Qc*' is independent of reference frame and is practically useful for direct comparison of simulation and experimental data from different sources obtained in different reference frames. Then, we use equilibrium molecular dynamics simulations in conjunction with the Green-Kubo formalism to study the heat transport properties of a model of the liquid Ni50Al50 alloy at three state points within the temperature range 1500 – 4000 K. Our results predict that in the liquid Ni50Al50 alloy in the presence of a temperature gradient Ni tends to diffuse from the cold end to the hot end whilst Al tends to diffuse from the hot end to the cold end. read less USED (low confidence) A. Biborski, R. Abdank-Kozubski, and V. Pierron-Bohnes, “‘Order-Order’ Kinetics in Triple-Defect B2-Ordered Binary Intermetallics: Kinetic Monte Carlo Simulation,” Diffusion Foundations. 2014. link Times cited: 3 Abstract: Triple-defect formation in B2-ordered binary A-B intermetall… read moreAbstract: Triple-defect formation in B2-ordered binary A-B intermetallic compounds results fromthe asymmetry between the formation energies of A- and B-antisite defects. Chemical disorderingin such systems is strictly correlated with vacancy formation, which is the reason for usually veryhigh vacancy concentration. Consequently, Kinetic Monte Carlo (KMC) simulation of processes occurringin the triple-defect systems and controlled by atomic migration via vacancy mechanism mustinvolve complete vacancy thermodynamics – i.e. the simulated system must contain the equilibriumtemperature-dependent number of vacancies. The fully consistent approach based on two differentMonte Carlo techniques has been applied in the present study. The AB intermetallic was modelled withan Ising-type Hamiltonian and KMC simulated for “order-order” kinetics with temperature-dependentequilibrium number of vacancies previously determined by means of Semi Grand Canonical MonteCarlo (SGCMC) simulations. The procedure required in addition the determination of saddle -pointenergies assigned to particular atomic jumps to nn vacancies. Their values were estimated in relationto the nn pair-interaction energies with reference to Molecular Statics simulations performed for NiAlsystem with EAM energetics. The results elucidated the role of triple-defect formation as the atomisticscaleorigin of the experimentally observed surprisingly low rate of the “order-order” kinetics in bulkNiAl. read less USED (low confidence) V. K. Sutrakar, A. C. Pillai, and D. Mahapatra, “Orientation, Size, and Temperature Dependent Ductile Brittle Transition in NiAl Nanowire under Tensile Loading - A Molecular Dynamics Study,” Defence Science Journal. 2014. link Times cited: 1 Abstract: In the present paper, thermo-mechanical response of B2-NiAl … read moreAbstract: In the present paper, thermo-mechanical response of B2-NiAl nanowire along the , , and orientations has been studied using molecular dynamics simulations. Nanowire with cross-sectional dimensions of ~20x20 A2, ~25x25 A2, and ~30x30 A2 and temperature range of 10 K-900 K has been considered. A Combined effect of size, orientation, and temperature on the stress-strain behavior under uniaxial tensile loading has been presented. It has been observed that oriented NiAl nanowire that is energetically most stable gives highest yield stress which further reduces with and orientations. A remarkable ductile brittle transition (DBT) with an increase in temperature has also been reported for all the orientations considered in the present study. The DBT observed for the nanowire has also been compared with the reported DBT of bulk B2-NiAl obtained from experiments. Alternate technique has also been proposed to increase the toughness of a given material especially at lower temperature regions, i.e. below DBT. Defence Science Journal, 2014, 64(2), pp. 179-185. DOI: http://dx.doi.org/10.14429/dsj.64.4310 read less USED (low confidence) X. Wang and S. Shen, “Effects of temperature and strain on thermal properties of Ni/Al laminated structure,” Computational Materials Science. 2014. link Times cited: 12 USED (low confidence) A. Rogachev et al., “Structure evolution and reaction mechanism in the Ni/Al reactive multilayer nanofoils,” Acta Materialia. 2014. link Times cited: 86 USED (low confidence) D. Tingaud and R. Besson, “Point defects and diffusion in ordered alloys: An ab initio study of the effect of vibrations,” Intermetallics. 2014. link Times cited: 6 USED (low confidence) N. Huynh, C. Lu, G. Michal, and A. K. Tieu, “A Misorientation Dependent Criterion of Crack Opening in FCC Single Crystal,” Materials Science Forum. 2013. link Times cited: 0 Abstract: This paper proposes a criterion for crack opening in FCC sin… read moreAbstract: This paper proposes a criterion for crack opening in FCC single crystals based on analyses of lattice orientation and interface energy of two adjacent crystals in a crystal plasticity finite element model (CPFEM). It also demonstrates the implementation of the criterion in Abaqus/Standard to simulate crack initiation and propagation in single-edged notch single crystal aluminium samples. Elements in the FEM mesh that have crystalline structures satisfying the crack opening criterion are removed from the mesh at the end of every loading step and FEM analyses are restarted on the new mesh in the next loading step. Removed elements effectively act as voids in the material due to crack nucleation. Similarly, the coalescence of newly removed elements at the end of a loading step with the existent ones simulates crack growth in the material. Two advantages of this approach are noted. Firstly, crack nucleation and its subsequent growth in the material is simulated solely based on lattice evolution history in the material without any presumptions of crack paths or regions where cracks are likely to occur. Secondly, as the criterion for crack nucleation is evaluated based on, and thus changes with, the lattice evolution during loading, a predefined energy criterion for crack opening, which could be erroneous, is avoided. Preliminary results of void nucleation and void growth around the notch tip in Cube and Brass oriented samples using CPFEM modelling appear to agree with molecular dynamics simulations of void growth in FCC single crystals. read less USED (low confidence) A. Ovrutsky and A. Prokhoda, “Particularities of nucleation and growth of the B2-phase: Results of simulations for the Al50Ni50 alloy,” Computational Materials Science. 2013. link Times cited: 6 USED (low confidence) H. Fu, Z. Hou, J. Fu, and Y. Ma, “Elastic anisotropy and phonon focusing in NiAl: Atomic study,” Intermetallics. 2013. link Times cited: 7 USED (low confidence) J. Michalka, P. McIntyre, and J. Gezelter, “Molecular Dynamics Simulations of the Surface Reconstructions of Pt(557) and Au(557) under Exposure to CO,” Journal of Physical Chemistry C. 2013. link Times cited: 5 Abstract: The mechanism and dynamics of surface reconstructions of Pt(… read moreAbstract: The mechanism and dynamics of surface reconstructions of Pt(557) and Au(557) exposed to various coverages of carbon monoxide (CO) were investigated using molecular dynamics simulations. Metal–CO interactions were parametrized from experimental data and plane-wave density functional theory (DFT) calculations. The large difference in binding strengths of the Pt–CO and Au–CO interactions was found to play a significant role in step-edge stability and adatom diffusion constants. Various mechanisms for CO-mediated step wandering and step doubling were investigated on the Pt(557) surface. We find that the energetics of CO adsorbed to the surface can explain the step-doubling reconstruction observed on Pt(557) and the lack of such a reconstruction on the Au(557) surface. However, more complicated reconstructions into triangular clusters that have been seen in recent experiments were not observed in these simulations. read less USED (low confidence) C. Tang and P. Harrowell, “Anomalously slow crystal growth of the glass-forming alloy CuZr.,” Nature materials. 2013. link Times cited: 178 USED (low confidence) F. Ma, K. Xu, and P. Chu, “Surface-induced structural transformation in nanowires,” Materials Science & Engineering R-reports. 2013. link Times cited: 24 USED (low confidence) O. Politano, F. Baras, A. Mukasyan, S. Vadchenko, and A. Rogachev, “Microstructure development during NiAl intermetallic synthesis in reactive Ni–Al nanolayers: Numerical investigations vs. TEM observations,” Surface & Coatings Technology. 2013. link Times cited: 43 USED (low confidence) Z. Hu, J. Zhang, Y. Yan, J. Yan, and T. Sun, “Molecular dynamics simulation of tensile behavior of diffusion bonded Ni/Al nanowires,” Journal of Mechanical Science and Technology. 2013. link Times cited: 12 USED (low confidence) Z. Hu, J. Zhang, Y. Yan, J. Yan, and T. Sun, “Molecular dynamics simulation of tensile behavior of diffusion bonded Ni/Al nanowires,” Journal of Mechanical Science and Technology. 2013. link Times cited: 0 USED (low confidence) A. Evteev, E. Levchenko, I. Belova, and G. Murch, “Molecular dynamics simulation of surface segregation, diffusion and reaction phenomena in equiatomic Ni-Al systems,” The Physics of Metals and Metallography. 2012. link Times cited: 8 USED (low confidence) S. Skirlo and M. Demkowicz, “The role of thermal spike compactness in radiation-induced disordering and Frenkel pair production in Ni3Al,” Scripta Materialia. 2012. link Times cited: 15 USED (low confidence) R. Rozas, R. Rozas, P. Kuhn, and J. Horbach, “Particle-Based Computer Simulation of Crystal Nucleation and Growth Kinetics in Undercooled Melts.” 2012. link Times cited: 2 USED (low confidence) H. Zhou, L. Zhang, and S. Qu, “Temperature effect on critical shear stress for twin boundary migration,” Computational Materials Science. 2012. link Times cited: 10 USED (low confidence) M. Horstemeyer, “Case Study: Conducting a Structural Scale Metal Forming Finite Element Analysis Starting from Electronics Structures Calculations Using ICME Tools.” 2012. link Times cited: 0 USED (low confidence) A. Linde, O. Politano, and F. Baras, “Study of the Reactive Dynamics of Nanometric Metallic Multilayers Using Molecular Dynamics : The Al-Ni System,” Defect and Diffusion Forum. 2012. link Times cited: 2 Abstract: A molecular dynamics study of a layered Ni-Al-Ni system is d… read moreAbstract: A molecular dynamics study of a layered Ni-Al-Ni system is developed using an embedded atom method potential. The specific geometry is designed to model a Ni-Al nanometric metallic multilayer. The system is initially thermalized at the fixed temperature of 600 K. We first observe the interdiffusion of Ni and Al at the interfaces, which is followed by the spontaneous phase formation of B2-NiAl in the Al layer. The solid-state reaction is associated with a rapid system's heating which further enhances the diffusion processes. NiAl phase is organized in small regions separated by grain boundaries. This study confirms the hypothesis of a layer-by-layer development of the new phase. For longer times, the temperature is notably higher (> 1000 K) and the system may partly lose some its B2-NiAl microstructure in favor of the formation of Ni3Al in L12 configuration. This work shows the spontaneous development of a real exothermic solid-state reaction in metallic nanosystems mostly constituted by interfaces. read less USED (low confidence) H. Fu, X. Li, W. Liu, Y. Ma, T. Gao, and X. Hong, “Electronic and dynamical properties of NiAl studied from first principles,” Intermetallics. 2011. link Times cited: 61 USED (low confidence) J. Wang et al., “First-principles calculations of binary Al compounds: Enthalpies of formation and elastic properties,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2011. link Times cited: 77 USED (low confidence) E. Levchenko, A. Evteev, I. Belova, and G. Murch, “Molecular dynamics determination of the time–temperature–transformation diagram for crystallization of an undercooled liquid Ni50Al50 alloy,” Acta Materialia. 2011. link Times cited: 24 USED (low confidence) N. Barnard, S. Brown, F. Devred, J. Bakker, B. Nieuwenhuys, and N. Adkins, “A quantitative investigation of the structure of Raney-Ni catalyst material using both computer simulation and experimental measurements,” Journal of Catalysis. 2011. link Times cited: 19 USED (low confidence) Y. Wen and J.-min Zhang, “Studies of the structure and the atomic diffusion properties of the Σ = 5 [001] twist GB in B2-type intermetallic compound NiAl,” Canadian Journal of Physics. 2011. link Times cited: 1 Abstract: The structural properties, the formation energies and the at… read moreAbstract: The structural properties, the formation energies and the atoms' diffusion behaviors by vacancy mechanism near the S = 5 (001) twist GB of the B2-type intermetallic compound NiAl have been investigated by using the modified analyt- ical embedded-atom method and a molecular dynamics simulation. Both the largest displacement and rippling effect occur at the first layer near the GB. The Ni vacancies at uncoincident sites are most easily formed on the first and second layers of the Ni- and Al-terminations, respectively. Furthermore, the Ni vacancy at an uncoincident site on the second layer of the Al-termination tends to migrate to the coincident Ni site of the first layer of the Ni-termination along a six-step jump path. The Ni vacancies at either the coincident or uncoincident site of the first layer tend to migrate in the first layer and finally return to their original site. Therefore, there is a collective tendency for the Ni vacancies to appear in the GB without local disorder. read less USED (low confidence) A. Evteev, E. Levchenko, F. A. Hagel, I. Belova, and G. Murch, “Molecular dynamics study of reaction pathways in an Al-coated Ni nanoparticle,” Intermetallics. 2011. link Times cited: 16 USED (low confidence) A. Evteev, E. Levchenko, I. Belova, and G. Murch, “Molecular dynamics simulation of diffusion in a (110) B2-NiAl film,” Intermetallics. 2011. link Times cited: 16 USED (low confidence) A. Hussain, S. Aryal, P. Rulis, M. A. Choudhry, J. Chen, and W. Ching, “Ab initio electronic structure calculations and optical properties of ordered and disordered Ni3Al,” Journal of Alloys and Compounds. 2011. link Times cited: 13 USED (low confidence) A. Ovrutsky, V. Bashev, and A. Prokhoda, “Modeling of initial crystallization in the alloys Al–10Ni and Al–5Ni–2.7Y at high undercoolings,” Computational Materials Science. 2011. link Times cited: 4 USED (low confidence) S. Qu, G. Wang, H. Zhou, and Z. Huang, “Can nanoscale twin boundaries serve as dislocation sources in single crystals,” Computational Materials Science. 2011. link Times cited: 16 USED (low confidence) Q. Xu and A. V. der Ven, “The effect of large vacancy concentration on intrinsic and interdiffusion coefficients: A first-principle study of B2-NiAl,” Acta Materialia. 2011. link Times cited: 10 USED (low confidence) J. Fan, “Applications of Atomistic Simulation in Ceramics and Metals.” 2010. link Times cited: 0 USED (low confidence) E. Levchenko, A. Evteev, D. R. Beck, I. Belova, and G. Murch, “Molecular dynamics simulation of the thermophysical properties of an undercooled liquid Ni50Al50 alloy,” Computational Materials Science. 2010. link Times cited: 20 USED (low confidence) E. Levchenko, A. Evteev, I. Belova, and G. Murch, “Molecular dynamics study of density, surface energy and self-diffusion in a liquid Ni50Al50 alloy,” Computational Materials Science. 2010. link Times cited: 15 USED (low confidence) V. K. Sutrakar and D. Mahapatra, “Asymmetry in structural and thermo-mechanical behavior of intermetallic NiAl nanowire under tensile/compressive loading: A molecular dynamics study,” Intermetallics. 2010. link Times cited: 20 USED (low confidence) D. Tingaud, F. Nardou, and R. Besson, “Critical effect of local pressure on jump frequencies in intermetallics,” Scripta Materialia. 2010. link Times cited: 1 USED (low confidence) V. K. Sutrakar and D. Mahapatra, “Superplasticity in intermetallic NiAl nanowires via atomistic simulations,” Materials Letters. 2010. link Times cited: 14 USED (low confidence) H. Yildirim, A. Kara, T. Rahman, R. Heid, and K. Bohnen, “Surface vibrational thermodynamics from ab initio calculations for fcc(1 0 0),” Surface Science. 2010. link Times cited: 5 USED (low confidence) T. Kogita, M. Kohyama, and Y. Kido, “Structure and dynamics of NiAl(110) studied by high-resolution ion scattering combined with density functional calculations,” Physical Review B. 2009. link Times cited: 3 Abstract: The surface relaxation and the rumpling of the top and the 2… read moreAbstract: The surface relaxation and the rumpling of the top and the 2nd layer together with the mean thermal vibration amplitudes of NiAl(110) are determined by high-resolution medium energy ion scattering (MEIS) with an excellent depth resolution of typically ±0.01 A. We also perform classical molecular dynamics (MD) simulations employing the embedded atom method and the first principles calculations using the VASP (Vienna Ab-initio Simulation Package) code. The results obtained by MEIS are compared with the theoretical predictions and experimental analysis reported so far. Interestingly, the present MEIS analysis observes slightly expanded relaxation 12 e Δ , which is supported by the present MD and VASP calculations and by X-ray diffraction analysis, whereas other experimental and theoretical analyses give contracted relaxation. The root mean square thermal vibration amplitude of the bulk Ni atoms is determined to be 0.10±0.005 A, which agrees well with the value of 0.097 A derived from the phonon dispersion relation calculated from VASP. A slightly enhanced thermal vibration amplitude of the top layer Ni in the surface normal direction observed is consistent with the MD simulation. 1 Department of Physics, Ritsumeikan University, Kusatsu, Shiga-ken 525-8577, Japan 2 Advanced Industrial Science and Technology, AIST, Ikeda, Osaka 563-8577, Japan read less USED (low confidence) X. Hu et al., “Effect of O impurity on structure and mechanical properties of NiAl intermetallics: A first-principles study,” Intermetallics. 2009. link Times cited: 37 USED (low confidence) Q. Xu and A. V. der Ven, “First-principles investigation of migration barriers and point defect complexes in B2–NiAl,” Intermetallics. 2009. link Times cited: 38 USED (low confidence) H. Fu, D.-hua Li, F. Peng, T. Gao, and X. Cheng, “Ab initio calculations of elastic constants and thermodynamic properties of NiAl under high pressures,” Computational Materials Science. 2008. link Times cited: 309 USED (low confidence) A. Hamid, “Study on the electron momentum density distribution and the Fermi surface of the ordered inter-metallic phase B2-NiAl,” Solid State Communications. 2008. link Times cited: 1 USED (low confidence) S. Das, J. Horbach, and T. Voigtmann, “Structural relaxation in a binary metallic melt: Molecular dynamics computer simulation of undercooled Al 80 Ni 20,” Physical Review B. 2008. link Times cited: 45 Abstract: Molecular dynamics computer simulations are performed to stu… read moreAbstract: Molecular dynamics computer simulations are performed to study structure and structural relaxation in the glassforming metallic alloy Al 80 Ni 20 . The interactions between the particles are modeled by an effective potential of the embedded atom type. Our model of Al 80 Ni 20 exhibits chemical short-range order (CSRO) that is reflected in a broad prepeak around a wave number of 1.8 A −1 in the partial static structure factor for the Ni-Ni correlations. The CSRO is due to the preference of Ni atoms to have Al rather than Ni atoms as nearest neighbors. By analyzing incoherent and coherent intermediate scattering functions as well as self-diffusion constants and shear viscosity, we discuss how the chemical ordering is reflected in the dynamics of the deeply undercooled melt. The q dependence of the alpha relaxation time as well as the Debye-Waller factor for the Al-Al correlations show oscillations at the location of the prepeak in the partial static structure factor for the Ni-Ni correlations. The latter feature of the Debye-Waller factor is well reproduced by a calculation in the framework of the mode coupling theory (MCT) of the glass transition, using the partial static structure factors from the simulation as input. We also check the validity of the Stokes-Einstein-Sutherland formula that relates the self-diffusion coefficients with the shear viscosity. We show that it breaks down already far above the mode coupling critical temperature Tc. The failure of the Stokes-Einstein-Sutherland relation is not related to the specific chemical ordering in Al 80 Ni 20 . read less USED (low confidence) O. Semenova, “Ordering phenomena and modelling of pair interactions in Ni3Ga compound,” Journal of Alloys and Compounds. 2008. link Times cited: 2 USED (low confidence) N. Lazarev, C. Abromeit, R. Schäublin, and R. Gotthardt, “Atomic-scale simulation of martensitic phase transformations in NiAl,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2008. link Times cited: 14 USED (low confidence) O. Semenova and R. Krachler, “A statistical–thermodynamic model for ordering phenomena in thin film intermetallic structures,” Thin Solid Films. 2008. link Times cited: 4 USED (low confidence) S. Zhao, T. Germann, and A. Strachan, “Molecular Dynamics Characterization of the Response of Ni/Al Nanolaminates Under Dynamic Loading,” Journal of Propulsion and Power. 2007. link Times cited: 8 Abstract: We use a recently developed molecular dynamics method with a… read moreAbstract: We use a recently developed molecular dynamics method with an accurate, first-principles-based force field to study shock propagation in Ni/Al nanolaminates and the induced (highly exothermic) chemical reactions. We characterize both perfect nanolaminates and specimens containing small (4-nm diameter) voids. The new method enables the accurate description of both the nonequilibrium shock-loading process and the long time evolution of the shocked material, providing an atomic-level picture of the complex interplay between the mechanical, thermal, and chemical processes that govern the behavior of the metastable composites. We shock the nanolaminates in the direction normal to the Ni/Al interfaces, leading to multiple wave reflections, due to the elastic mismatch between Ni and Al; this leads to the Al layers having a higher temperature during the early stages of the process. In the perfect nanolaminates, the chemical reactions start at the interfaces closest to the impact plane and then propagate through the material. A rapid increase in the rate of chemical reactions (3Ni + Al → Ni 3 Al) is observed following the melting of the Ni and Al layers. We estimate the propagation velocity of the chemical front to be about 200 m/s. The porous samples exhibit much faster energy-release rates, due to the mechanical intermixing of Al and Ni caused by shock-induced pore collapse and the higher shock temperatures. read less USED (low confidence) S. Yu, C.-yu Wang, T. Yu, and J. Cai, “Self-diffusion in the intermetallic compounds NiAl and Ni3Al: An embedded atom method study,” Physica B-condensed Matter. 2007. link Times cited: 39 USED (low confidence) K. Moriguchi and M. Igarashi, “Correlation between lattice-strain energetics and melting properties: Molecular dynamics and lattice dynamics using EAM models of Al,” Physical Review B. 2006. link Times cited: 14 USED (low confidence) Y. Mishin, “Atomistic Computer Modeling of Intermetallic Alloys,” Materials Science Forum. 2005. link Times cited: 3 Abstract: The paper gives a brief overview of our recent work on atomi… read moreAbstract: The paper gives a brief overview of our recent work on atomistic computer modeling of ordered intermetallic compounds of the Ni-Al and Ti-Al systems. Atomic interactions in these systems are modeled by semi-empirical potentials fit to experimental and first-principles data. The methodology includes a large variety of techniques ranging from harmonic lattice dynamics to molecular dynamics and Monte Carlo simulations. The properties studied include lattice characteristics (elastic constants, phonons, thermal expansion), point-defect properties, atomic diffusion, generalized stacking faults, dislocations, surfaces, grain boundaries, interphase boundaries, and phase diagrams. We especially emphasize the recent progress in the understanding of diffusion mechanisms in NiAl and TiAl, calculation of stacking fault energies in Ni3Al in relation to dislocation behavior, and calculations of / 0 interface boundaries in Ni-Al alloys. read less USED (low confidence) R. Krachler and H. Ipser, “Triple-defect complexes in theB2intermetallic compound NiAl,” Physical Review B. 2004. link Times cited: 17 USED (low confidence) P. Hazzledine and Y. Sun, “The interaction and concentration of vacancies in NiAl,” Intermetallics. 2004. link Times cited: 2 USED (low confidence) Y. Mishin, “Atomistic modeling of the γ and γ’-phases of the Ni-Al system,” Acta Materialia. 2004. link Times cited: 395 USED (low confidence) A. Lozovoi and Y. Mishin, “Point defects in NiAl: The effect of lattice vibrations,” Physical Review B. 2003. link Times cited: 32 Abstract: We investigate the effect of atomic vibrations on point defe… read moreAbstract: We investigate the effect of atomic vibrations on point defect free energies and equilibrium concentrations in the B2 NiAl compound using the quasiharmonic approximation in combination with a recently developed embedded-atom potential. The entropy term appears to be the dominant contribution to the Gibbs free energies of point defects. Vibrational entropies of the main defect complexes: triple-Ni defect, exchange defect, divacancy, and even of the interbranch-Al defect turn out to be positive in the whole range of temperatures studied here (0\char21{}1700 K). This leads to an increase in the concentrations of all four types of point defects in Ni-rich and stoichiometric NiAl. On the Al-rich side, the effect of lattice vibrations is to shift the minimum on the vacancy concentration versus temperature curve towards lower temperatures. The effect of zero-point vibrations is shown to be too small to affect the type of constitutional defects in NiAl. The constitutional defects remain nickel antisite atoms on the Ni-rich side and nickel vacancies on the Al-rich side. read less USED (low confidence) Y. Sun and N. Yang, “The onset and blocking of slip in NiAl,” Acta Materialia. 2003. link Times cited: 5 USED (low confidence) R. Kozubski, D. Kmiec, E. Partyka, and M. Danielewski, “Orderorder kinetics in Ni 50.5Al 49.5 single crystal,” Intermetallics. 2003. link Times cited: 19 USED (low confidence) R. Zope and Y. Mishin, “Interatomic potentials for atomistic simulations of the Ti-Al system,” Physical Review B. 2003. link Times cited: 477 Abstract: Semiempirical interatomic potentials have been developed for… read moreAbstract: Semiempirical interatomic potentials have been developed for Al, $\ensuremath{\alpha}\ensuremath{-}\mathrm{Ti},$ and $\ensuremath{\gamma}\ensuremath{-}\mathrm{TiAl}$ within the embedded atom method (EAM) formalism by fitting to a large database of experimental as well as ab initio data. The ab initio calculations were performed by the linearized augmented plane wave (LAPW) method within the density functional theory to obtain the equations of state for a number of crystal structures of the Ti-Al system. Some of the calculated LAPW energies were used for fitting the potentials while others for examining their quality. The potentials correctly predict the equilibrium crystal structures of the phases and accurately reproduce their basic lattice properties. The potentials are applied to calculate the energies of point defects, surfaces, and planar faults in the equilibrium structures. Unlike earlier EAM potentials for the Ti-Al system, the proposed potentials provide a reasonable description of the lattice thermal expansion, demonstrating their usefulness for molecular-dynamics and Monte Carlo simulations at high temperatures. The energy along the tetragonal deformation path (Bain transformation) in $\ensuremath{\gamma}\ensuremath{-}\mathrm{TiAl}$ calculated with the EAM potential is in fairly good agreement with LAPW calculations. Equilibrium point defect concentrations in $\ensuremath{\gamma}\ensuremath{-}\mathrm{TiAl}$ are studied using the EAM potential. It is found that antisite defects strongly dominate over vacancies at all compositions around stoichiometry, indicating that $\ensuremath{\gamma}\ensuremath{-}\mathrm{TiAl}$ is an antisite disorder compound, in agreement with experimental data. read less USED (low confidence) X. Xie and Y. Mishin, “Monte Carlo simulation of grain boundary segregation and decohesion in NiAl,” Acta Materialia. 2002. link Times cited: 24 USED (low confidence) G. Wang, Y. Xu, P. Qian, and Y. Su, “ADP potential for the Au-Rh system and its application in element segregation of nanoparticles,” Computational Materials Science. 2021. link Times cited: 6 USED (low confidence) J. Tang et al., “Activation volume dominated diffusivity of Ni50Al50 melt under extreme conditions,” Computational Materials Science. 2020. link Times cited: 6 USED (low confidence) V. Jordan and I. Shmakov, “Thermal and Microstructural Analysis of Intermetallide Synthesis in the Ni-Al Layered-Block Atomic Structure Based on the Computer-Aided Simulation of SHS.” 2020. link Times cited: 1 USED (low confidence) H. Xiang et al., “Shock-induced stacking fault pyramids in Ni/Al multilayers,” Applied Surface Science. 2018. link Times cited: 25 USED (low confidence) E. Karchevskaya, D. Minakov, and P. Levashov, “Quantum molecular dynamics simulation of structural and thermodynamic properties of NiAl,” Journal of Physics: Conference Series. 2018. link Times cited: 0 Abstract: In this work, structural and thermodynamic properties of a s… read moreAbstract: In this work, structural and thermodynamic properties of a solid and liquid Ni–Al compound are studied using an ab initio method of quantum molecular dynamics (QMD). Simulations were carried out in 700–3000 K temperature range at atmospheric pressure. Radial distribution functions are analyzed to determine the presence of Ni–Al chemical bonds. Diffusion coefficients for individual components are also calculated. Another goal of this work is the investigation of the reaction propagation in thermally-initiated Ni–Al foils. For this purpose, we performed QMD simulations of Ni–Al layers in the microcanonical ensemble. An exothermic reaction between the solid Ni–Al layers is observed in our simulations at temperature less than the melting temperatures of the components. read less USED (low confidence) A. Matsushita, S. Takamoto, A. Hatano, and S. Izumi, “Development of Hybrid Method Using Ab initio and Classical Molecular Dynamics for Calculating the Thermal Expansion Coefficient of Alloys at High Temperature,” Journal of The Society of Materials Science, Japan. 2018. link Times cited: 0 Abstract: In order to predict the thermal expansion coefficient (TEC),… read moreAbstract: In order to predict the thermal expansion coefficient (TEC), quasiharmonic approximation based on the ab initio electronic structure calculation is an effective and conventional scheme. However, it is known that the deviation due to anharmonic effect arises at high temperature. Although Molecular Dynamics (MD) naturally includes the anharmonic effect, classical MD is lacking in accuracy because of its empirical interatomic potential and ab initio MD is not applicable because of its high computational cost. In this paper, we have proposed a new hybrid method using ab initio electronic structure calculation and classical molecular dynamics for calculating TEC of alloys at high temperature. Our method is non-empirical, highly accurate and computationally inexpensive. The method consists of three steps. Firstly, various snapshots of the atomic coordination at a certain temperature are sampled by classical MD. Secondly, physical properties of each snapshot are calculated by ab initio electronic structure calculation. Finally, by analyzing them statistically, the equilibrium volume is computed. TEC is obtained by repeating these steps at different temperatures. We have calculated the TECs of Al, Ni3Al, and NiAl. Results show good agreements with experimental results. Our method enables us to improve the conventional quasiharmonic approximation and obtain accurate TEC at high temperature through the incorporation of anharmonic effect. read less USED (low confidence) S. Divinski, “Defects and Diffusion in Ordered Compounds.” 2017. link Times cited: 10 USED (low confidence) Y. Yang, “Effects of Size and Coalescence on the Interfacial Dynamics of Nanoparticles: A Molecular Dynamics Study.” 2015. link Times cited: 0 USED (low confidence) Q.-N. Fan, C.-yu Wang, T. Yu, and J.-ping Du, “A ternary Ni–Al–W EAM potential for Ni-based single crystal superalloys,” Physica B-condensed Matter. 2015. link Times cited: 11 USED (low confidence) A. Ovrutsky, A. Prokhoda, and M. Rasshchupkyna, “Modern Simulations by the Molecular Dynamics Method.” 2014. link Times cited: 0 USED (low confidence) I. Egry, “Structure and Properties of Molten Metals.” 2014. link Times cited: 7 USED (low confidence) A. Evteev, E. Levchenko, I. Belova, and G. Murch, “Computer simulation of diffusion and reaction in metallic nanoparticles.” 2012. link Times cited: 0 USED (low confidence) A. Ovrutsky and A. Prokhoda, “Peculiarities of crystallization at high undercooling: Analysis of the simulation data for aluminum,” Journal of Crystal Growth. 2011. link Times cited: 4 USED (low confidence) K. Koshiyama and K. Shintani, “Atomistic study of the mechanical properties of metallic-glass nanowires,” MRS Proceedings. 2011. link Times cited: 0 Abstract: Melt-growth simulations based on the molecular-dynamics meth… read moreAbstract: Melt-growth simulations based on the molecular-dynamics method for both the Cu-Zr and Ni-Al crystalline nanowires of B2 structure are performed to produce metallic-glass nanowires of amorphous structure. Next, tensile deformations of these nanowires are simulated at various temperatures. For the sake of comparison, Cu-Zr and Ni-Al crystalline nanowires of B2 structure are also elongated. It is revealed that the tensile strength of the metallic-glass nanowires is third or fourth of the tensile strength of the crystalline nanowires. Increasing tensile strain, the Cu-Zr crystalline nanowires of B2 structure change their structure twice, whereas the metallic-glass nanowires only decrease their thicknesses locally, and necking takes place. read less USED (low confidence) E. Levchenko, A. Evteev, D. Riley, I. Belova, and G. E. Murch, “Molecular dynamics simulation of the alloying reaction in Al-coated Ni nanoparticle,” Computational Materials Science. 2010. link Times cited: 61 USED (low confidence) K. Binder, J. Horbach, and M. Hawlitzky, “Molecular Dynamics Simulations of Glassforming Network Fluids,” MRS Proceedings. 2007. link Times cited: 0 Abstract: Molecular Dynamics simulations of molten oxides, such as flu… read moreAbstract: Molecular Dynamics simulations of molten oxides, such as fluid silicon dioxide and germanium dioxide, based on simple classical pair potentials, are compared with corresponding Car-Parrinello “ab initio” Molecular Dynamics (CPMD) work and with experiment. It is shown that CPMD provides a significantly better account for properties on short length scales, but classical MD is still indispensable to deal with larger scales of length and time. The behavior of the mean square displacement of the particles as well as the incoherent intermediate scattering function is compatible with a mode coupling description, at least at very high temperatures, while the diffusion constants show a crossover to Arrhenius behavior near the mode coupling critical temperature of these systems. Finally, the results for the network forming liquids are compared to those from simulations of binary metallic alloys such as Al 80 Ni 20 , which form a structure similar to densely packed hard spheres. read less USED (low confidence) Y. Mishin, “Atomistic Computer Simulation of Diffusion.” 2005. link Times cited: 7 USED (low confidence) K. Nordlund and R. Averback, “Point Defects in Metals.” 2005. link Times cited: 2 NOT USED (low confidence) L.-F. Zhu, J. Janssen, S. Ishibashi, F. Körmann, B. Grabowski, and J. Neugebauer, “A fully automated approach to calculate the melting temperature of elemental crystals,” Computational Materials Science. 2021. link Times cited: 17 NOT USED (low confidence) N. Ren, L. Hu, L. Wang, and P. Guan, “Revealing a hidden dynamic signature of the non-Arrhenius crossover in metallic glass-forming liquids,” Scripta Materialia. 2020. link Times cited: 12 NOT USED (low confidence) V. Jordan and I. Shmakov, “The influence of the ignition conditions of the SH-synthesis of intermetallic compounds on the combustion parameters of the Ti-15.82wt.%Al composition: computer simulation and computing experiments,” Journal of Physics: Conference Series. 2019. link Times cited: 0 Abstract: Computational experiments (CEs) have been carried out to sim… read moreAbstract: Computational experiments (CEs) have been carried out to simulate the propagation of the combustion wave of the SH-synthesis process in a package of alternating layers of nanoscale crystal lattices of Ti and Al atoms by molecular dynamics method. The interatomic interaction potential in the embedded atom model (EAM) was used in the LAMMPS package. Using the LAMMPS configuration with parallel computing, the following results of CEs were obtained: sets of temperature profiles along the layers of the structure at successive instants of time (up to 16 ns) and the corresponding sets of snapshots (vertical cross-sections of the atomic arrangement along the layers), as well as a tables with the number and percentage of the content of various types of elementary cells (fcc, hcp, bcc, other) at the same instants of time. The influence of the initiation’s conditions of the SH-synthesis of intermetallic compounds on the combustion parameters of the nanoscale layered Ti-15.82wt.%Al composition was showed. The ignition conditions of SHS that are provided the stable motion of the combustion wave in the SHS-sample were determined. And the ignition conditions of SHS with dominance of either TiAl or Ti3Al phase formation in the SHS products was also determined. read less NOT USED (low confidence) J. Fürnkranz, “Publication list,” Journal of Physics: Conference Series. 2019. link Times cited: 10 Abstract: PUBLICATION LIST of VICTOR MANUEL VILLANUEVA SANDOVAL availa… read moreAbstract: PUBLICATION LIST of VICTOR MANUEL VILLANUEVA SANDOVAL available in this PDF. read less NOT USED (low confidence) H. Bhattarai, K. E. Newman, and J. Gezelter, “Polarizable potentials for metals: The density readjusting embedded atom method (DR-EAM),” Physical Review B. 2019. link Times cited: 6 Abstract: In simulations of metallic interfaces, a critical aspect of … read moreAbstract: In simulations of metallic interfaces, a critical aspect of metallic behavior is missing from the some of the most widely used classical molecular dynamics force fields. We present a modification of the embedded atom method (EAM) which allows for electronic polarization of the metal by treating the valence density around each atom as a fluctuating dynamical quantity. The densities are represented by a set of additional fluctuating variables (and their conjugate momenta) which are propagated along with the nuclear coordinates. This ``density readjusting EAM'' (DR-EAM) preserves nearly all of the useful qualities of traditional EAM, including bulk elastic properties and surface energies. However, it also allows valence electron density to migrate through the metal in response to external perturbations. We show that DR-EAM can successfully model polarization in response to external charges, capturing the image charge effect in atomistic simulations. DR-EAM also captures some of the behavior of metals in the presence of uniform electric fields, predicting surface charging and shielding internal to the metal. We further show that it predicts charge transfer between the constituent atoms in alloys, leading to novel predictions about unit cell geometries in layered $\mathrm{L}{1}_{0}$ structures. read less NOT USED (low confidence) K. Choudhary, A. Biacchi, S. Ghosh, L. Hale, A. Walker, and F. Tavazza, “High-throughput assessment of vacancy formation and surface energies of materials using classical force-fields,” Journal of Physics: Condensed Matter. 2018. link Times cited: 16 Abstract: In this work, we present an open access database for surface… read moreAbstract: In this work, we present an open access database for surface and vacancy-formation energies using classical force-fields (FFs). These quantities are essential in understanding diffusion behavior, nanoparticle formation and catalytic activities. FFs are often designed for a specific application, hence, this database allows the user to understand whether a FF is suitable for investigating particular defect and surface-related material properties. The FF results are compared to density functional theory and experimental data whenever applicable for validation. At present, we have 17 506 surface energies and 1000 vacancy formation energies calculation in our database and the database is still growing. All the data generated, and the computational tools used, are shared publicly at the following websites: www.ctcms.nist.gov/~knc6/periodic.html, https://jarvis.nist.gov and https://github.com/usnistgov/jarvis. Approximations used during the high-throughput calculations are clearly mentioned. Using some of the example cases, we show how our data can be used to directly compare different FFs for a material and to interpret experimental findings such as using Wulff construction for predicting equilibrium shape of nanoparticles. Similarly, the vacancy formation energies data can be useful in understanding diffusion related properties. read less NOT USED (low confidence) Z. Trautt, F. Tavazza, and C. Becker, “Facilitating the selection and creation of accurate interatomic potentials with robust tools and characterization,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 14 Abstract: The Materials Genome Initiative seeks to significantly decre… read moreAbstract: The Materials Genome Initiative seeks to significantly decrease the cost and time of development and integration of new materials. Within the domain of atomistic simulations, several roadblocks stand in the way of reaching this goal. While the NIST Interatomic Potentials Repository hosts numerous interatomic potentials (force fields), researchers cannot immediately determine the best choice(s) for their use case. Researchers developing new potentials, specifically those in restricted environments, lack a comprehensive portfolio of efficient tools capable of calculating and archiving the properties of their potentials. This paper elucidates one solution to these problems, which uses Python-based scripts that are suitable for rapid property evaluation and human knowledge transfer. Calculation results are visible on the repository website, which reduces the time required to select an interatomic potential for a specific use case. Furthermore, property evaluation scripts are being integrated with modern platforms to improve discoverability and access of materials property data. To demonstrate these scripts and features, we will discuss the automation of stacking fault energy calculations and their application to additional elements. While the calculation methodology was developed previously, we are using it here as a case study in simulation automation and property calculations. We demonstrate how the use of Python scripts allows for rapid calculation in a more easily managed way where the calculations can be modified, and the results presented in user-friendly and concise ways. Additionally, the methods can be incorporated into other efforts, such as openKIM. read less NOT USED (low confidence) M. Zachariah, “Understanding and Quantifying the Reactivity of Energetic NanoParticles and NanoComposites.” 2015. link Times cited: 0 Abstract: : The focus of this work was to under stand quantitatively, … read moreAbstract: : The focus of this work was to under stand quantitatively, the nature of the reactivity of nanoparticles and nanocomposites for energetic materials applications. Our approach took two thrusts. 1. Single Particle Kinetics 2. Ensemble Fuel/Oxide Nanocomposite Kinetics. Our goal was to: A. Explore the size resolved reactivity of nanoparticles. B. Explain the behavior using phenomenological modeling and compare with bulk materials. C. Explore condensed state kinetics using a new type of mass-spectrometry tool. Approach Use novel diagnostic tools to probe reactivity of nanocomposites and tease out mechanisms: Including advanced ion-mobility and mass spectrometry tools. Conduct bulk powder measurements in combustion bombs and wires to extract combustion time scales and the role of mixtures and stoichiometry on burning. Use new high heating rate electron microscopy to visualize condensed state reactions between nanocomposites. Conduct Molecular Dynamics simulations to understand properties of nanoparticles. read less NOT USED (low confidence) E. Tadmor and R. E. Miller, “Modeling Materials: Continuum, Atomistic and Multiscale Techniques.” 2011. link Times cited: 395 Abstract: 1. Introduction Part I. Continuum Mechanics and Thermodynami… read moreAbstract: 1. Introduction Part I. Continuum Mechanics and Thermodynamics: 2. Essential continuum mechanics and thermodynamics Part II. Atomistics: 3. Lattices and crystal structures 4. Quantum mechanics of materials 5. Empirical atomistic models of materials 6. Molecular statics Part III. Atomistic Foundations of Continuum Concepts: 7. Classical equilibrium statistical mechanics 8. Microscopic expressions for continuum fields 9. Molecular dynamics Part IV. Multiscale Methods: 10. What is multiscale modeling? 11. Atomistic constitutive relations for multilattice crystals 12. Atomistic/continuum coupling: static methods 13. Atomistic/continuum coupling: finite temperature and dynamics Appendix References Index. read less NOT USED (low confidence) R. Neugebauer, R. Wertheim, and U. Semmler, “THE ATOMIC FINITE ELEMENT METHOD AS A BRIDGE BETWEEN MOLECULAR DYNAMICS AND CONTINUUM MECHANICS,” Journal of Multiscale Modelling. 2011. link Times cited: 6 Abstract: On cutting tools for high performance cutting (HPC) processe… read moreAbstract: On cutting tools for high performance cutting (HPC) processes or for hard-to-cut materials, there is an increased importance in so-called superlattice coatings with hundreds of layers each of which is only a few nanometers in thickness. Homogeneity or average material properties based on the properties of single layers are not valid in these dimensions any more. Consequently, continuum mechanical material models cannot be used for modeling the behavior of nanolayers. Therefore, the interaction potentials between the single atoms should be considered. A new, so-called atomic finite element method (AFEM) is presented. In the AFEM the interatomic bonds are modeled as nonlinear spring elements. The AFEM is the connection between the molecular dynamics (MD) method and the crystal plasticity FEM (CPFEM). The MD simulates the atomic deposition process. The CPFEM considers the behavior of anisotropic crystals using the continuum mechanical FEM. On one side, the atomic structure data simulated by MD defines the interface to AFEM. On the other side, the boundary conditions (displacements and tractions) of the AFEM model are interpolated from the CPFEM simulations. In AFEM, the lattice deformation, the crack and dislocation behavior can be simulated and calculated at the nanometer scale. read less NOT USED (low confidence) V. Tatarenko, O. Sobol, D. Leonov, Y. A. Kunyts’kyy, and S. Bokoch, “Statistical Thermodynamics and Physical Kinetics of Structural Changes of Quasi-Binary Solid Solutions Based on the Close-Packed Simple Lattices (According to the Data About Evolution of a Pattern of Scattering of Waves of Various Kinds).” 2011. link Times cited: 2 NOT USED (low confidence) A. V. der Ven, J. Thomas, Q. Xu, and J. Bhattacharya, “Linking the electronic structure of solids to their thermodynamic and kinetic properties,” Math. Comput. Simul. 2010. link Times cited: 154 NOT USED (low confidence) O. Semenova, R. Krachler, and H. Ipser, “A generalized defect correlation model for B2 compounds,” Solid State Sciences. 2008. link Times cited: 14 NOT USED (low confidence) M. Michelon and A. Antonelli, “Nonphysical thermodynamical phases in L12 intermetallic alloys from semiempirical tight-binding potentials,” Computational Materials Science. 2008. link Times cited: 4 NOT USED (low confidence) С. Волегов, Р. М. Герасимов, and Р. П. Давлятшин, “MODELS OF MOLECULAR DYNAMICS: A REVIEW OF EAM-POTENTIALS. PART 2. POTENTIALS FOR MULTI-COMPONENT SYSTEMS.” 2018. link Times cited: 1 Abstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликова… read moreAbstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликована: 29 июня 2018 г. В статье представлена вторая часть обзора современных подходов и работ, посвященных построению потенциалов межатомного взаимодействия с использованием методологии погруженного атома (EAM-потенциалы). Эта часть обзора посвящена одной из наиболее остро стоящих проблем в молекулярной динамике – вопросам построения потенциалов, которые были бы пригодны для описания структуры и физико-механических свойств многокомпонентных (в первую очередь – бинарных и тернарных) материалов. Отмечены первые работы, в которых предлагались подходы к построению функций перекрестного взаимодействия для сплавов никеля и меди – как с использованием методологии EAM, так и несколько отличающийся по процедуре построения потенциал типа Финисса-Синклера. Рассматриваются работы, в которых производится сопоставление различных подходов к построению потенциалов, а также к процедуре идентификации их параметров на примере одних и тех же многокомпонентных систем (типа Al-Ni или Cu-Au). Кроме того, особый интерес представляют некоторые тернарные системы, например Fe–Ni–Cr, W–H– He или U–Mo–Xe, которые являются ключевыми для материалов атомной энергетики и которые в последние годы активно изучаются как возможные материалы для использования в термоядерных ректорах. Приведены примеры работ, в которых предлагаются и исследуются потенциалы для описания многокомпонентных систем, пригодных для использования в аэрокосмической промышленности и изготовленных прежде всего на основе никеля. Рассмотрены результаты исследований различных интерметаллических соединений, отмечены работы, в которых при помощи построенного EAM потенциала удалось количественно точно описать фазовые диаграммы соединений и вычислить характеристики фазовых переходов. read less NOT USED (low confidence) A. Ovrutsky, A. Prokhoda, and M. Rasshchupkyna, “Simulation Techniques for Atomic Systems.” 2014. link Times cited: 3 NOT USED (low confidence) Y. Mishin, “Interatomic Potentials for Metals.” 2005. link Times cited: 41 NOT USED (high confidence) H. Juárez et al., “Modeling of surface phenomena of liquid Al–Ni alloys using molecular dynamics,” Scientific Reports. 2023. link Times cited: 0 NOT USED (high confidence) V. Zalizniak, O. A. Zolotov, and K. A. Sidorov, “Interatomic potential for metal diborides,” Molecular Simulation. 2021. link Times cited: 0 Abstract: ABSTRACT Parameters of the embedded atom method inter-atomic… read moreAbstract: ABSTRACT Parameters of the embedded atom method inter-atomic potential for metal diborides MB2 (M = Al, Mg, Mo, Hf, Nb, Sc, Ti, Y, Zr, V) are presented in this paper. The potential parameters were determined empirically by fitting to a first-principles and experimental data of the equilibrium lattice constant, cohesion energy and bulk modulus of metal diborides. The potential provides a good representation of the desired properties of considered metal diborides. To test the applicability of the proposed potential to molecular dynamics simulation of metal diborides, the specific heat capacities of TiB2 and ZrB2 were determined. The proposed potential is intended for use in molecular dynamics simulations of metal diboride nanostructures. read less NOT USED (high confidence) H. Bhattarai, K. E. Newman, and J. Gezelter, “The role of polarizability in the interfacial thermal conductance at the gold-water interface.,” The Journal of chemical physics. 2020. link Times cited: 3 Abstract: We have studied the interfacial thermal conductance, G, of t… read moreAbstract: We have studied the interfacial thermal conductance, G, of the flat Au(111)-water interface using non-equilibrium molecular dynamics simulations. We utilized two metal models, one based on the embedded atom method (EAM) and the other including metallic polarizability via a density readjusting EAM. These were combined with three popular water models, SPC/E, TIP4P, and TIP4P-FQ, to understand the role of polarizability in the thermal transport process. A thermal flux was introduced using velocity shearing and scaling reverse non-equilibrium molecular dynamics, and transport coefficients were measured by calculating the resulting thermal gradients and temperature differences at the interface. Our primary finding is that the computed interfacial thermal conductance between a bare metal interface and water increases when polarizability is taken into account in the metal model. Additional work to understand the origin of the conductance difference points to changes in the local ordering of the water molecules in the first two layers of water above the metal surface. Vibrational densities of states on both sides of the interface exhibit interesting frequency modulation close to the surface but no obvious differences due to metal polarizability. read less NOT USED (high confidence) D. Farkas and A. Caro, “Model interatomic potentials for Fe–Ni–Cr–Co–Al high-entropy alloys,” Journal of Materials Research. 2020. link Times cited: 76 Abstract: A set of embedded atom model (EAM) interatomic potentials wa… read moreAbstract: A set of embedded atom model (EAM) interatomic potentials was developed to represent highly idealized face-centered cubic (FCC) mixtures of Fe–Ni–Cr–Co–Al at near-equiatomic compositions. Potential functions for the transition metals and their crossed interactions are taken from our previous work for Fe–Ni–Cr–Co–Cu [D. Farkas and A. Caro: J. Mater. Res. 33 (19), 3218–3225, 2018], while cross-pair interactions involving Al were developed using a mix of the component pair functions fitted to known intermetallic properties. The resulting heats of mixing of all binary equiatomic random FCC mixtures not containing Al is low, but significant short-range ordering appears in those containing Al, driven by a large atomic size difference. The potentials are utilized to predict the relative stability of FCC quinary mixtures, as well as ordered L1_2 and B2 phases as a function of Al content. These predictions are in qualitative agreement with experiments. This interatomic potential set is developed to resemble but not model precisely the properties of this complex system, aiming at providing a tool to explore the consequences of the addition of a large size-misfit component into a high entropy mixture that develops multiphase microstructures. read less NOT USED (high confidence) M. Wagih, P. M. Larsen, and C. Schuh, “Learning grain boundary segregation energy spectra in polycrystals,” Nature Communications. 2020. link Times cited: 62 NOT USED (high confidence) V. Jordan, И. В. Иванович, I. Shmakov, and Ш. И. Александрович, “Computer molecular-dynamic simulation of shs microkinetics in the atomic structure with a checkerboard-like arrangement of nanoscale blocks of Ni and Al atoms,” Yugra State University Bulletin. 2020. link Times cited: 0 Abstract: The article presents the results of computer simulation of t… read moreAbstract: The article presents the results of computer simulation of the propagation of the combustion wave of "self-propagating high-temperature synthesis (SHS)" process in an atomic layered structure. In each layer of the structure, nanosized blocks of two types alternate: a block of the first type is composed as a packet of unit cells of Ni atoms, and a block of the second type is composed of a packet of elementary cells of Al atoms. In each pair of layers adjacent to each other, sequences of alternating blocks of two types are shifted relative to each other by one block, so the full layered structure of the layers with alternating blocks in them is associated with a chessboard pattern. Computer simulation of SHS in such a structure was carried out using the LAMMPS software package taking into account parallel computations, which uses the molecular dynamics method and the interatomic interaction potential in the embedded atom" model (EAM). In addition to the LAMMPS package, the authors implemented program procedures for calculating the temperature and density profiles of the substance along the motion direction of the SHS combustion wave front, which made it possible to carry out temperature analysis of the SHS microkinetics (to estimate the velocity of the combustion wave front) and recognition of intermetallic phases in the reaction volume of the Ni-Al system when using the OVITO package. read less NOT USED (high confidence) S. O. Kart, H. H. Kart, and T. Çagin, “Atomic-scale insights into structural and thermodynamic stability of spherical Al@Ni and Ni@Al core–shell nanoparticles,” Journal of Nanoparticle Research. 2020. link Times cited: 6 NOT USED (high confidence) C. Zhang, F.-yang Tian, and X. Ni, “First-principles investigation on ideal strength of B2 NiAl and NiTi alloys,” Chinese Physics B. 2020. link Times cited: 2 NOT USED (high confidence) J.-yu Yang, Y. Zhang, Y. Liu, W. Hu, and X. Dai, “A comparative atomic simulation study of the configurations in M-Al (M = Mg, Ni, and Fe) nanoalloys: influence of alloying ability, surface energy, atomic radius, and atomic arrangement,” Journal of Nanoparticle Research. 2020. link Times cited: 3 NOT USED (high confidence) W. Jian, D. Hui, and D. Lau, “Nanoengineering in biomedicine: Current development and future perspectives,” Nanotechnology Reviews. 2020. link Times cited: 35 Abstract: Recent advances in biomedicine largely rely on the developme… read moreAbstract: Recent advances in biomedicine largely rely on the development in nanoengineering. As the access to unique properties in biomaterials is not readily available from traditional techniques, the nanoengineering becomes an effective approach for research and development, by which the performance as well as the functionalities of biomaterials has been greatly improved and enriched. This review focuses on the main materials used in biomedicine, including metallic materials, polymers, and nanocomposites, as well as the major applications of nanoengineering in developing biomedical treatments and techniques. Research that provides an in-depth understanding of material properties and efficient enhancement of material performance using molecular dynamics simulations from the nanoengineering perspective are discussed. The advanced techniques which facilitate nanoengineering in biomedical applications are also presented to inspire further improvement in the future. Furthermore, the potential challenges of nanoengineering in biomedicine are evaluated by summarizing concerned issues and possible solutions. Graphical abstract read less NOT USED (high confidence) J. Betlej, P. Sowa, R. Kozubski, G. Murch, and I. Belova, “Self-diffusion in a triple-defect A-B binary system: Monte Carlo simulation,” Computational Materials Science. 2019. link Times cited: 5 NOT USED (high confidence) U. Sarder et al., “Mass and thermal transport in liquid Cu-Ag alloys,” Philosophical Magazine. 2018. link Times cited: 10 Abstract: ABSTRACT In this paper, the diffusion, thermodynamic and the… read moreAbstract: ABSTRACT In this paper, the diffusion, thermodynamic and thermotransport properties in Cu–Ag liquid alloys are extensively investigated with molecular dynamics over a wide composition and temperature range. The simulations are performed with the most reliable EAM potential. The Green-Kubo formalism is employed for calculating transport properties. It is found that the reduced heat of transport in Cu–Ag is very small (about 0.10 eV in absolute value) and almost temperature independent. Further it is found that the interdiffusion coefficient together with both self-diffusion coefficients are almost composition independent. In Cu–Ag, the thermodynamic factor is found to be less than unity whereas the Manning factor is greater than unity (with significant composition and temperature dependence) and their product is very close to 1. read less NOT USED (high confidence) F. Baras, V. Turlo, O. Politano, S. Vadchenko, A. Rogachev, and A. Mukasyan, “SHS in Ni/Al Nanofoils: A Review of Experiments and Molecular Dynamics Simulations,” Advanced Engineering Materials. 2018. link Times cited: 38 Abstract: Non‐isothermal processes in nanometric metallic multilayers … read moreAbstract: Non‐isothermal processes in nanometric metallic multilayers are reviewed, both experimentally and theoretically. The Ni/Al nanofoil is considered as a model system. On the one hand, the experimental methods of elaboration and analysis are presented and, on the other hand, the modeling approach at the macroscopic and atomic scale. The basic experimental features are reported together with recent achievements. Molecular dynamics investigation of the reactivity of Ni/Al systems is reported for bulk systems and nanosystems including nanoparticles, nanowires, nanofilms, and multilayers. The focus is on atomic‐scale modeling versus experiments. Molecular dynamics approaches allow us to elucidate the mechanisms of non‐isothermal processes occurring in nanoscale systems, such as phase transformations and self‐propagation reactions. read less NOT USED (high confidence) L. Hale, “Comparing Modeling Predictions of Aluminum Edge Dislocations: Semidiscrete Variational Peierls–Nabarro Versus Atomistics,” JOM. 2018. link Times cited: 7 NOT USED (high confidence) J.-yu Yang, W. Hu, and X. Dai, “Composition and Size Dependence of Alloying in Ni–Al Nanoparticles With Icosahedral and Rhombohedral Configurations: An Atomic Simulation Study,” physica status solidi (b). 2017. link Times cited: 0 Abstract: The surface segregation and alloying of Ni–Al nanoparticles … read moreAbstract: The surface segregation and alloying of Ni–Al nanoparticles with icosahedral and rhombohedral configurations are studied via Monte Carlo simulations based on the analytic embedded atom model. The simulation shows that the alloying between Ni and Al at nanolevel is beneficial for all the studied nanoparticles. For the Ni–Al nanoalloy with icosahedral configuration, the excess energy is negative for all the compositions. This result indicates an energetic preference for the alloying of Ni and Al. The minimum excess energy decreases with increasing nanoalloy size because of the low surface–volume ratio. The compositions of Ni corresponding to the minimum excess energy increase linearly from 51.5 to 60.5% with increasing nanoalloy size. Among all the considered icosahedra and rhombohedra at the same size, the energy of the rhombohedra at the composition of 1:1, which are arranged on B2 crystal, is the smallest. With increasing nanoparticle size, the excess energies of the icosahedra at the 3:1 composition and the rhombohedra at the 1:1 composition increase. By contrast, the excess energy of the icosahedra at the 1:3 composition decreases. The result can be attributed to the competition between the Al surface segregation and alloying between Ni and Al. read less NOT USED (high confidence) V. Turlo, F. Baras, and O. Politano, “Comparative study of embedded-atom methods applied to the reactivity in the Ni–Al system,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 23 Abstract: Structural, thermodynamic, atomic and thermal transport prop… read moreAbstract: Structural, thermodynamic, atomic and thermal transport properties of solid and liquid phases of the Ni–Al system were studied by means of MD simulations using three embedded-atom method (EAM) potentials developed by Mishin and colleagues (Mishin et al 2002 Phys. Rev. B 65 224114; Mishin 2004 Acta Mater. 52 145167; Purja Pun and Mishin 2009 Phil. Mag. 89 32453267). The extracted properties (lattice parameter, enthalpy, heat capacity, mass diffusivity and thermal conductivity) were compared with experimental data. The limitations of EAM potentials for studying different aspects of reactivity were assessed for each potential separately. read less NOT USED (high confidence) M. D. Grapes and T. Weihs, “Exploring the reaction mechanism in self-propagating Al/Ni multilayers by adding inert material,” Combustion and Flame. 2016. link Times cited: 31 NOT USED (high confidence) E. Levchenko et al., “Influence of the interatomic potential on thermotransport in binary liquid alloys: case study on NiAl,” Philosophical Magazine. 2016. link Times cited: 14 Abstract: Equilibrium molecular dynamics simulation in conjunction wit… read moreAbstract: Equilibrium molecular dynamics simulation in conjunction with the Green-Kubo formalism is employed to study the transport properties of a model Ni50Al50 melt with the embedded-atom method potential developed in [G.P. Purja Pun, Y. Mishin, Phil. Mag., 2009, 89, 3245]. The principal objective of the work is to quantitatively characterise and analyse thermotransport in the system, i.e. diffusion driven by a temperature gradient. In addition, direct phenomenological coefficients for mass and thermal transport are also evaluated and analysed in the process. Furthermore, the results obtained are compared with previously published data for a different model of Ni50Al50 melt with an alternative embedded-atom method potential for the alloy as well as with experiment where possible. It is found that both potentials are able to consistently predict both direct transport coefficients over a wide temperature range. However, these two potentials are found to be inconsistent in characterising the cross-coupled heat and mass transport, predicting even different directions (sign) of the heat of thermotransport. The origin of this difference is discussed in the paper in detail. read less NOT USED (high confidence) V. M. Bezpal’chuk, S. Marchenko, O. Rymar, O. O. Bogatyryov, and A. Gusak, “Problem of a Choice of the First Phase in Reaction between Nanofilms of Nickel and Aluminium,” Metallofizika I Noveishie Tekhnologii. 2016. link Times cited: 0 NOT USED (high confidence) J. Michalka, A. P. Latham, and J. Gezelter, “CO-Induced Restructuring on Stepped Pt Surfaces: A Molecular Dynamics Study,” Journal of Physical Chemistry C. 2016. link Times cited: 7 Abstract: The effects of plateau width and step-edge kinking on carbon… read moreAbstract: The effects of plateau width and step-edge kinking on carbon monoxide (CO)-induced restructuring of platinum surfaces were explored using molecular dynamics (MD) simulations. Platinum crystals displaying four different vicinal surfaces [(321), (765), (112), and (557)] were constructed and exposed to partial coverages of carbon monoxide. Platinum–CO interactions were fit to recent experimental data and density functional theory (DFT) calculations, providing a classical interaction model that captures the atop binding preference on Pt. The differences in Pt–Pt binding strength between edge atoms on the various facets were found to play a significant role in step-edge wandering and reconstruction events. Because the mechanism for step doubling relies on a stochastic meeting of two wandering edges, the widths of the plateaus on the original surfaces were also found to play a role in these reconstructions. On the Pt(321) surfaces, the CO adsorbate was found to assist in reordering the kinked step edges into st... read less NOT USED (high confidence) G. P. P. Pun, V. Yamakov, and Y. Mishin, “Interatomic potential for the ternary Ni–Al–Co system and application to atomistic modeling of the B2–L10 martensitic transformation,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 80 Abstract: Ni–Al–Co is a promising system for ferromagnetic shape memor… read moreAbstract: Ni–Al–Co is a promising system for ferromagnetic shape memory applications. This paper reports on the development of a ternary embedded-atom potential for this system by fitting to experimental and first-principles data. Reasonably good agreement is achieved for physical properties between values predicted by the potential and values known from experiment and/or first-principles calculations. The potential reproduces basic features of the martensitic phase transformation from the B2-ordered high-temperature phase to a tetragonal CuAu-ordered low-temperature phase. The compositional and temperature ranges of this transformation and the martensite microstructure predicted by the potential compare well with existing experimental data. These results indicate that the proposed potential can be used for simulations of the shape memory effect in the Ni–Al–Co system. read less NOT USED (high confidence) A. Evteev, E. Levchenko, L. Momenzadeh, Y. Sohn, I. Belova, and G. Murch, “Molecular dynamics study of phonon-mediated thermal transport in a Ni50Al50 melt: case analysis of the influence of the process on the kinetics of solidification,” Philosophical Magazine. 2015. link Times cited: 12 Abstract: The phonon-mediated contribution to the thermal transport pr… read moreAbstract: The phonon-mediated contribution to the thermal transport properties of liquid NiAl alloy is investigated in detail over a wide temperature range. The calculations are performed in the framework of equilibrium molecular dynamics making use of the Green–Kubo formalism and one of the most reliable embedded-atom method potentials for the intermetallic alloy. The phonon-mediated contribution to the thermal conductivity of the liquid alloy is calculated at equilibrium as well as for the steady state. The relative magnitude of the thermal conductivity decrease induced by the transition to the steady state is estimated to be less than 2% below 2000 K and less than 1% at 3000 and 4000 K. It is also found that the phonon-mediated contribution to the thermal conductivity of the liquid alloy can be accurately estimated (well within 1%) on the basis of an approximation which invokes the straightforwardly accessible microscopic expression for the total heat flux without demanding calculations of the partial enthalpies needed for the precise evolution of the reduced heat flux (pure heat conduction). On the basis of these calculations, the correspondence between the experimentally observed and modelled kinetics of solidification due to a difference in thermal conductivity is discussed. read less NOT USED (high confidence) A. Evteev, E. Levchenko, I. Belova, R. Kozubski, Z.-kui Liu, and G. Murch, “Thermotransport in binary system: case study on Ni50Al50 melt,” Philosophical Magazine. 2014. link Times cited: 21 Abstract: The formalism of thermotransport in a binary system is analy… read moreAbstract: The formalism of thermotransport in a binary system is analysed. Focus is put on a detailed consideration of the heat of transport parameter characterizing diffusion driven by a temperature gradient. We introduce the reduced heat of transport parameter , which characterizes part of the interdiffusion flux that is proportional to the temperature gradient. In an isothermal system represents the reduced heat flow (pure heat conduction) consequent upon unit interdiffusion flux. It is demonstrated that is independent of reference frame and is useful in a practical way for direct comparison of simulation and experimental data from different sources obtained in different reference frames. In the case study of the liquid Ni50Al50 alloy, we use equilibrium molecular dynamics simulations in conjunction with the Green–Kubo formalism to evaluate the heat transport properties of the model within the temperature range of 1500–4000 K. Our results predict that in the presence of a temperature gradient Ni tends to diffuse from the cold end to the hot end whilst Al tends to diffuse from the hot end to the cold end. read less NOT USED (high confidence) J.-ping Du, C.-yu Wang, and T. Yu, “The ternary Ni-Al-Co embedded-atom-method potential for gamma/gamma ’ Ni-based single-crystal superalloys: Construction and application,” Chinese Physics B. 2014. link Times cited: 11 Abstract: An Ni—Al—Co system embedded-atom-method potential is constru… read moreAbstract: An Ni—Al—Co system embedded-atom-method potential is constructed for the γ(Ni)/γ'(Ni3Al) superalloy based on experiments and first-principles calculations. The stacking fault energies (SFEs) of the Ni(Co, Al) random solid solutions are calculated as a function of the concentrations of Co and Al. The calculated SFEs decrease with increasing concentrations of Co and Al, which is consistent with the experimental results. The embedding energy term in the present potential has an important influence on the SFEs of the random solid solutions. The cross-slip processes of a screw dislocation in homogenous Ni(Co) solid solutions are simulated using the present potential and the nudged elastic band method. The cross-slip activation energies increase with increasing Co concentration, which implies that the creep resistance of γ(Ni) may be improved by the addition of Co. read less NOT USED (high confidence) J. Rogal et al., “Perspectives on point defect thermodynamics,” physica status solidi (b). 2014. link Times cited: 54 Abstract: We review and discuss methods for including the role of poin… read moreAbstract: We review and discuss methods for including the role of point defects in calculations of the free energy, composition and phase stability of elements and compounds. Our principle aim is to explain and to reconcile, with examples, the perspectives on this problem that are often strikingly different between exponents of CALPHAD, and others working in the overlapping fields of physics, chemistry and materials science. Current methodologies described here include the compound energy formalism of CALPHAD, besides the rather different but related canonical and grand‐canonical formalisms. We show how the calculation of appropriate defect formation energies should be formulated, how they are included in the different formalisms and in turn how these yield equilibrium defect concentrations and their contribution to free energies and chemical potentials. Furthermore, we briefly review the current state‐of‐the‐art and challenges in determining point defect properties from first‐principles calculations as well as from experimental measurements. read less NOT USED (high confidence) C. Becker, F. Tavazza, Z. Trautt, and R. B. D. Macedo, “Considerations for choosing and using force fields and interatomic potentials in materials science and engineering,” Current Opinion in Solid State & Materials Science. 2013. link Times cited: 196 NOT USED (high confidence) H. Kwak, Y. Shin, A. V. van Duin, and A. Vasenkov, “Ab initio based multiscale modeling of alloy surface segregation,” Journal of Physics: Condensed Matter. 2012. link Times cited: 8 Abstract: A fully integrated ab initio based multiscale model for anal… read moreAbstract: A fully integrated ab initio based multiscale model for analysis of segregation at alloy surfaces is presented. Major components of the model include a structure-energy analysis from the first-principles density functional theory (DFT), a Monte Carlo/molecular dynamics (MC/MD) hybrid simulation scheme for atomic transport, and a reactive force field formalism that binds the two. The multiscale model accurately describes the atomic transport processes in a multi-component alloy system at finite temperature, and is capable of providing quantitative predictions for surface compositions. The validity of the model was demonstrated by investigating the temperature-dependent segregation behavior of B2 FeAl binary alloy surfaces with a detailed description of the segregation mechanism. Based on the model’s prediction capabilities, potential extension of the model to the analysis of systems undergoing rapid chemical reactions is discussed. read less NOT USED (high confidence) C.-H. Zhang, H. Shuo, S. Jiang, and N.-xian Chen, “Chen’s lattice inversion embedded-atom method for Ni-Al alloy,” Chinese Physics B. 2012. link Times cited: 13 Abstract: The structural properties, the enthalpies of formation, and … read moreAbstract: The structural properties, the enthalpies of formation, and the mechanical properties of some Ni–Al intermetallic compounds (NiAl, Ni3Al, NiAl3, Ni5Al3, Ni3Al4) are studied by using Chen’s lattice inversion embedded-atom method (CLI-EAM). Our calculated lattice parameters and cohesive energies of Ni–Al compounds are consistent with the experimental and the other EAM results. The results of enthalpy of formation indicate a strong chemical interaction between Ni and Al in the intermetallic compounds. Through analyzing the alloy elastic constants, we find that all the Ni–Al intermetallic compounds discussed are mechanically stable. The bulk moduli of the compounds increase with the increasing Ni concentration. Our results also suggest that NiAl, Ni3Al, NiAl3, and Ni5Al3 are ductile materials with lower ratios of shear modulus to bulk modulus; while Ni3Al4 is brittle with a higher ratio. read less NOT USED (high confidence) O. Semenova and R. Krachler, “A Statistical‐Thermodynamic Modeling of Ordering Phenomena in Binary Intermetallic B2‐ and L12‐Structures,” Advanced Engineering Materials. 2012. link Times cited: 0 Abstract: Statistical‐thermodynamic models for the description of orde… read moreAbstract: Statistical‐thermodynamic models for the description of ordering in non‐stoichiometric intermetallic phases can be very helpful for the design of new functional materials for various purposes. Here, we present a review of methods for describing B2 (BCC) and L12 (FCC) – type binary alloys. The free energy of binary alloys where ordered structures are formed, is evaluated by statistical approaches employing applications of the Bragg–Williams–Gorsky mean‐field approximation. In the original mean field method, the degree of order in the neighborhood of a given site in the crystal is determined by the average state of order throughout the macroscopic crystal. Actually, the force tending to produce long‐range order depends on the fluctuations in the configuration, an effect which is neglected in the mean field approximation but is taken under consideration, e.g., in the Quasi‐Chemical Approach (QCA) by Guggenheim and its advanced applications, in various sophisticated treatments using the Cluster Variation Method (CVM) originally developed by Kikuchi, as well as in the Defect Correlation Model (DCM). The latter model is a less sophisticated method based on the simultaneous description of short‐range order and long‐range order by use of properly defined non‐overlapping clusters. All these methods allow the “pure” mean‐field results to be substantially refined, particularly at high temperatures, and for alloys with large deviations from the stoichiometric composition. Defect concentration‐alloy composition diagrams and activity‐alloy composition diagrams are shown for several methods with focus on the QCA and DCM approaches. The calculated results are compared with each other and with experimental data on thermodynamic and structural properties from the literature. read less NOT USED (high confidence) S. Izvekov and B. Rice, “Free-energy based pair-additive potentials for bulk Ni-Al systems: application to study Ni-Al reactive alloying.,” The Journal of chemical physics. 2012. link Times cited: 11 Abstract: We present new numerical pair-additive Al, Ni, and Al-Ni pot… read moreAbstract: We present new numerical pair-additive Al, Ni, and Al-Ni potentials by force-matching (FM) ionic force and virial data from single (bulk liquid) phase ab initio molecular dynamics (MD) simulations using the Born-Oppenheimer method. The potentials are represented by piece-wise functions (splines) and, therefore, are not constrained to a particular choice of analytical functional form. The FM method with virial constraint naturally yields a potential which maps out the ionic free-energy surface of the reference ensemble. To further improve the free energetics of the FM ensemble, the FM procedure is modified to bias the potentials to reproduce the experimental melting temperatures of the reference (FCC-Al, FCC-Ni, B2-NiAl) phases, the only macroscopic data included in the fitting set. The performance of the resultant potentials in simulating bulk metallic phases is then evaluated. The new model is applied to perform MD simulations of self-propagating exothermic reaction in Ni-Al bilayers at P = 0-5 GPa initiated at T = 1300 K. Consistent with experimental observations, the new model describes realistically a sequence of peritectic phase transformations throughout the reaction and at a realistic rate. The reaction proceeds through interlayer diffusion of Al and Ni atoms at the interface with formation of B2-NiAl in the Al melt. Such material responses have, in the past, been proven to be difficult to observe with then-existing potentials. read less NOT USED (high confidence) M. He and S. Li, “An embedded atom hyperelastic constitutive model and multiscale cohesive finite element method,” Computational Mechanics. 2012. link Times cited: 30 NOT USED (high confidence) O. Semenova and R. Krachler, “Quasi chemical and defect correlation models for intermetallic compounds with B2-structure: new applications,” Journal of Materials Science. 2012. link Times cited: 1 NOT USED (high confidence) M. He and S. Li, “An embedded atom hyperelastic constitutive model and multiscale cohesive finite element method,” Computational Mechanics. 2011. link Times cited: 0 NOT USED (high confidence) J. C. Crone, J. Knap, P. Chung, and B. Rice, “Role of microstructure in initiation of Ni-Al reactive multilayers,” Applied Physics Letters. 2011. link Times cited: 34 Abstract: Through molecular dynamics simulations, the effects of micro… read moreAbstract: Through molecular dynamics simulations, the effects of microstructure on reaction initiation are studied in nickel-aluminum (Ni–Al) reactive multilayers. Ni–Al multilayer systems of varying misfit strain and layer thickness are created and the ignition temperature is estimated by heating and thermalizing over small temperature increments until a reaction is observed. Results show that ignition temperatures drop significantly with increasing misfit strain. Our results indicate that the sensitivity of reactive multilayers can be controlled, in part, by microstructure, with changes of the order of 350 K. read less NOT USED (high confidence) X. Hu, X. Liu, Z. Xu, J. Liang, and T.-min Wang, “First-principles investigation of the effects of B impurities on the mechanical properties of NiAl intermetallics,” Science China Physics, Mechanics and Astronomy. 2011. link Times cited: 5 NOT USED (high confidence) J. Zhang, Z. Chen, Y. Lu, M.-yi Zhang, and Y. Wang, “Microscopic phase field study of the antisite defect of Ni3Al in binary Ni-Al alloys,” Science China Physics, Mechanics and Astronomy. 2010. link Times cited: 1 NOT USED (high confidence) B. Henz, T. Hawa, and M. Zachariah, “Molecular Dynamics Simulation of the Kinetic Reaction of Nickel and Aluminum Nanoparticles.” 2010. link Times cited: 1 Abstract: : Molecular dynamics simulations are used to simulate the ki… read moreAbstract: : Molecular dynamics simulations are used to simulate the kinetic reaction of nickel (Ni) and aluminum (Al) particles at the nanometer scale. The effect of particle size on reaction time and temperature for separate nanoparticles is considered as a model system for a powder metallurgy system. Coated nanoparticles in the form of Ni-coated Al nanoparticles and Al-coated Ni nanoparticles are also analyzed as a model for nanoparticles embedded within a matrix. The differences in melting temperature and phase change behavior, e.g., the volumetric expansion of Al between Al and Ni is expected to produce differing results for the coated nanoparticle systems. For instance, the volumetric expansion of Al upon melting is expected to produce large tensile stresses and possibly rupture in the Ni shell for Ni-coated Al. Simulation results showed that the sintering time for separate and coated nanoparticles was nearly linearly dependent upon the number of atoms or volume of the sintering nanoparticles. We also found that nanoparticle size and surface energy was an important factor in determining the adiabatic reaction temperature for both systems at nanoparticle sizes of less than 10 nm in diameter. read less NOT USED (high confidence) Y. Mishin, M. Asta, and J. Li, “Atomistic modeling of interfaces and their impact on microstructure and properties,” Acta Materialia. 2010. link Times cited: 418 NOT USED (high confidence) H. Zhou and S. Qu, “The effect of nanoscale twin boundaries on fracture toughness in nanocrystalline Ni,” Nanotechnology. 2010. link Times cited: 54 Abstract: Nanoscale twin boundaries (TBs) were recently reported to be… read moreAbstract: Nanoscale twin boundaries (TBs) were recently reported to be capable of enhancing the fracture toughness of nanocrystalline (nc) metals. The present study aims to investigate the toughening effects of nanoscale TBs in nc Ni by using molecular dynamics (MD) simulation. It is shown that the presence of embedded nanoscale TBs facilitates the accommodation of dislocations through partial dislocation motion along TBs, resulting in improved fracture toughness. Moreover, crack propagation is observed to be intragranular in a nanotwinned sample, concurrent with nucleation of nanovoids in the intersections of TBs and grain boundaries (GBs). read less NOT USED (high confidence) Y. Zhou and A. Strachan, “Thermal conduction in molecular materials using coarse grain dynamics: role of mass diffusion and quantum corrections for molecular dynamics simulations.,” The Journal of chemical physics. 2009. link Times cited: 12 Abstract: We use a mesodynamical method, denoted dynamics with implici… read moreAbstract: We use a mesodynamical method, denoted dynamics with implicit degrees of freedom (DID), to characterize thermal transport in a model molecular crystal below and above its melting temperature. DID represents groups of atoms (molecules in this case) using mesoparticles and the thermal role of the intramolecular degrees of freedom (DoFs) are described implicitly using their specific heat. We focus on the role of these intramolecular DoFs on thermal transport. We find that thermal conductivity is independent of intramolecular specific heat for solid samples and a linear relationship between the two quantities in liquid samples with the coefficient of proportionality being the mass diffusivity of the mesoparticles. As the temperature of the liquids is increased, thermal conductivity exhibits an increased sensitivity with respect to the specific heat of the internal DoFs due to the enhanced molecular mobility. Based on these results, we propose a simple method to incorporate quantum corrections to thermal conductivity obtained from nonequilibrium molecular dynamics simulations of molecular liquids. Our results also provide insight into the development of thermally accurate coarse grain models of soft materials. read less NOT USED (high confidence) G. P. P. Pun and Y. Mishin, “Development of an interatomic potential for the Ni-Al system,” Philosophical Magazine. 2009. link Times cited: 341 Abstract: We construct an interatomic potential for the Ni-Al system w… read moreAbstract: We construct an interatomic potential for the Ni-Al system within the embedded-atom method formalism. The potential is based on previously developed accurate potentials for pure Ni and Al. The cross-interactions are fitted to experimental cohesive energy, lattice parameter and elastic constants of B2-NiAl, as well as to ab initio formation energies of several real or imaginary intermetallic compounds with different crystal structures and chemical compositions. The potential accurately reproduces a variety of physical properties of the NiAl and Ni3Al phases, and shows reasonable agreement with experimental and ab initio data for phase stability across the Ni-Al phase diagram. Most of the properties reproduced by the new potential were not involved in the fitting process, which demonstrates its excellent transferability. Advantages and certain weaknesses of the new potential in comparison with other existing potentials are discussed in detail. The potential is expected to be especially suitable for simulations of heterophase interfaces and mechanical behavior of Ni-Al alloys. read less NOT USED (high confidence) A. Evteev, E. Levchenko, D. Riley, I. Belova, and G. Murch, “Reaction of a Ni-coated Al nanoparticle to form B2-NiAl: A molecular dynamics study,” Philosophical Magazine Letters. 2009. link Times cited: 29 Abstract: The kinetic reaction in a Ni-coated Al nanoparticle with equ… read moreAbstract: The kinetic reaction in a Ni-coated Al nanoparticle with equi-atomic fractions and diameter of approximately 4.5 nm is studied by means of molecular dynamics simulation, using a potential of the embedded atom type to model the interatomic interactions. First, the large driving force for the alloying of Ni and Al initiates solid state amorphization of the nanoparticle with the formation of Ni50Al50 amorphous alloy. Amorphization makes intermixing of the components much easier compared to the crystalline state. The average rate of penetration of Ni atoms can be estimated to be about two times higher than Al atoms, whilst the total rate of inter-penetration can be estimated to be of the order of 10−2 m/s. The heat of the intermixing with the formation of Ni50Al50 amorphous alloy can be estimated at approximately −0.34 eV/at. Next, the crystallization of the Ni50Al50 amorphous alloy into B2-NiAl ordered crystal structure is observed. The heat of the crystallization can be estimated as approximately −0.08 eV/at. Then, the B2-NiAl ordered nanoparticle melts at a temperature of approximately 1500 K. It is shown that, for the alloying reaction in the initial Ni-coated Al nanoparticle, the ignition temperature can be as low as approximately 200 K, while the adiabatic temperature for the reaction is below the melting temperature of the nanoparticle with the B2-NiAl ordered structure. read less NOT USED (high confidence) D. Shi, B. Wen, R. Melnik, S. Yao, and T. Li, “First-principles studies of Al–Ni intermetallic compounds,” Journal of Solid State Chemistry. 2009. link Times cited: 154 NOT USED (high confidence) P. Scott and R. W. Smith, “Estimation of the solute diffusion coefficient of a dilute liquid alloy: static structure factor and isothermal compressibility estimates obtained using the rational function approximation of the radial distribution,” Journal of Physics: Condensed Matter. 2009. link Times cited: 1 Abstract: A simple method for estimating the mass diffusion coefficien… read moreAbstract: A simple method for estimating the mass diffusion coefficient of a dilute binary liquid alloy that sequentially uses experimental data for the static structure factor and isothermal susceptibility of the solvent is presented, as well as another using the static structure factor alone and a method using the isothermal susceptibility alone. A fourth method that simultaneously uses the static structure factor and isothermal susceptibility is also noted. Of significance is the fact that these methods do not require information about the interatomic potential. Stability with respect to weights in the optimization process employed has been established and is reported, as well as some indication of the upper limits on the applicable solute concentration. Comparisons are made with results from a high quality capillary experiment for Pb 1 wt% Au liquid alloy performed in microgravity, and with velocity autocorrelation estimates derived from molecular dynamics simulation. The results suggest that the capillary experiments are influenced by reverse diffusion of the solvent, and actually measure an average of the mass diffusion coefficients, Dij, weighted by the equilibrium concentrations of the solvent, x1, and solute, x2, defined by The three methods are required to provide upper and lower estimates for the mixed solvent–solute diffusion coefficient, which is not directly accessible from the experimental data, and demonstrate agreement with the experiment via Dtot. read less NOT USED (high confidence) B. Henz, T. Hawa, and M. Zachariah, “Molecular dynamics simulation of the kinetic sintering of Ni and Al nanoparticles,” Molecular Simulation. 2009. link Times cited: 47 Abstract: The kinetic sintering of Ni and Al nanoparticles is consider… read moreAbstract: The kinetic sintering of Ni and Al nanoparticles is considered using molecular dynamics simulations. We report on the effects of nanoparticle size on sintering temperature and time, with results showing that surface energy has a slight effect on both results. The effect of surface energy on combustion temperature is limited to nanoparticles of less than 10 nm in diameter. An analysis of the various alloys formed during sintering gives insight into the reaction process. The formation of Al-rich compounds is observed initially with a final equilibration and rapid formation of the eutectic alloy immediately preceded by melting of the Ni nanoparticle. We have observed that nanoparticle size and surface energy are both important factors in determining the adiabatic reaction temperature for this material system at nanoparticle sizes of less than 10 nm in diameter. read less NOT USED (high confidence) V. K. Sutrakar and D. Mahapatra, “Stress-induced phase transformation and pseudo-elastic/pseudo-plastic recovery in intermetallic Ni–Al nanowires,” Nanotechnology. 2009. link Times cited: 16 Abstract: Extensive molecular dynamics (MD) simulations have been perf… read moreAbstract: Extensive molecular dynamics (MD) simulations have been performed in a B2-NiAl nanowire using an embedded atom method (EAM) potential. We show a stress induced -centered-tetragonal (BCT) phase transformation and a novel temperature and cross-section dependent pseudo-elastic/pseudo-plastic recovery from such an unstable BCT phase with a recoverable strain of ∼30% as compared to 5–8% in polycrystalline materials. Such a temperature and cross-section dependent pseudo-elastic/pseudo-plastic strain recovery can be useful in various interesting applications of shape memory and strain sensing in nanoscale devices. Effects of size, temperature, and strain rate on the structural and mechanical properties have also been analyzed in detail. For a given size of the nanowire the yield stress of both the B2 and the BCT phases is found to decrease with increasing temperature, whereas for a given temperature and strain rate the yield stress of both the B2 and the BCT phase is found to increase with increase in the cross-sectional dimensions of the nanowire. A constant elastic modulus of ∼80 GPa of the B2 phase is observed in the temperature range of 200–500 K for nanowires of cross-sectional dimensions in the range of 17.22–28.712 Å, whereas the elastic modulus of the BCT phase shows a decreasing trend with an increase in the temperature. read less NOT USED (high confidence) B. Henz, T. Hawa, and M. Zachariah, “Molecular dynamics simulation of the energetic reaction between Ni and Al nanoparticles,” Journal of Applied Physics. 2009. link Times cited: 55 Abstract: Molecular dynamics simulations are used to simulate the ener… read moreAbstract: Molecular dynamics simulations are used to simulate the energetic reaction of Ni and Al particles at the nanometer scale. The effect of particle size on reaction time and temperature for separate nanoparticles has been considered as a model system for a powder metallurgy system. Coated nanoparticles in the form of Ni-coated Al nanoparticles and Al-coated Ni nanoparticles are also analyzed as a model for nanoparticles embedded within a matrix. The differences in melting temperature and phase change behavior, e.g., the volumetric expansion of Al between Al and Ni, are expected to produce differing results for the coated nanoparticle systems. For instance, the volumetric expansion of Al upon melting is expected to produce large tensile stresses and possibly rupture in the Ni shell for Ni-coated Al. Simulation results show that the sintering time for separate and coated nanoparticles is nearly linearly dependent on the number of atoms or volume of the sintering nanoparticles. We have also found that nanoparti... read less NOT USED (high confidence) R. W. Smith, P. Scott, and B. Szpunar, “Solute Diffusion in Nonionic Liquids—Effects of Gravity,” Annals of the New York Academy of Sciences. 2009. link Times cited: 4 Abstract: We have been engaged in examining the influence of gravity o… read moreAbstract: We have been engaged in examining the influence of gravity on the results of experiments to measure the variation of solute diffusion coefficients (D) with temperature (T) in fused metals and semimetals since our first STS flights in 1992. These early experiments, conducted with the in situ g‐jitter of the shuttle, showed the near‐parabolic variation of D with T reported by others. However, with the aid of the Canadian Space Agency's microgravity isolation mount (MIM) to isolate the diffusion facility from the existing g‐jitter of the Russian space station MIR, we showed that in all the alloy systems and over the temperature range studied, D increased linearly with T. If the isolating system was deactivated, then the more familiar parabolic relationship appeared. We have always assumed that the values of D measured using the MIM would be closer to the intrinsic values for the alloy system considered; to test this contention, we have been involved in two modeling activities. The first has been to estimate the effects of g‐jitter‐level disturbances on solute distributions in long capillary diffusion couples. The second has been to conduct various molecular dynamics modeling studies of solute diffusion. This paper presents results of these studies. read less NOT USED (high confidence) R. Besson, A. Legris, D. Connétable, and P. Maugis, “Atomic-scale study of low-temperature equilibria in iron-rich Al-C-Fe,” Physical Review B. 2008. link Times cited: 10 Abstract: The capability of the thermodynamic approach based on the in… read moreAbstract: The capability of the thermodynamic approach based on the independent point defect approximation to describe low-temperature phase equilibria is investigated and applied to the Al-C-Fe system. The method gives a reasonable description of the multicomponent and multisublattice Fe-rich corner and evidences numerous peculiarities concerning the ordered phases as well as the density-functional-theory (DFT) energy models. The study of Fe3Al(-C), revealing strong defect-induced instabilities, rules out the LDA, SLDA and GGA schemes and leaves (spin-polarized) SGGA as the only valid one. C stabilizes L12 Fe3Al with respect to D03, which justifies the fcc-type structure of the kappa Fe3AlC compound. The present work also helps in justifying the experimentally observed depletion of C in the kappa phase. Finally, a correct description of both Fe3C and kappa requires inclusion of interstitial carbon at low temperature, emphasizing the unexpected importance of interstitial defects in ordered phases. read less NOT USED (high confidence) J. C. Zhou, W. Li, and J. B. Zhu, “Theoretical study of Ni–Al nanoalloy clusters using particle swarm optimisation algorithm,” Materials Science and Technology. 2008. link Times cited: 5 Abstract: The global structural optimisations for Ni–Al nanoalloy clus… read moreAbstract: The global structural optimisations for Ni–Al nanoalloy clusters at different compositions have been investigated using particle swarm optimisation combined with simulated annealing method. The second moment approximation of the tight binding potential has been used in describing the interatomic interactions. Some stable structures were obtained for NixAlx(x=1–8), Ni3xAlx(x=1–4) and NixAl3x(x=1–4) nanoalloy clusters. The simulation results show that the lowest energy isomers of nanoalloy clusters with the approximate composition 'NiAl, Ni3Al and NiAl3' generally have structures based on icosahedral packing. It is confirmed that segregation is favoured for Ni–Al nanoalloy clusters, with the surface becoming richer in Al and the core becoming richer in Ni. read less NOT USED (high confidence) T. Radchenko and V. Tatarenko, “Fe–Ni Alloys at High Pressures and Temperatures: Statistical Thermodynamics and Kinetics of the L1_2 or D0_19 Atomic Order.” 2008. link Times cited: 19 NOT USED (high confidence) J.-min Zhang, D.-D. Wang, G. X. Chen, and K. Xu, “Surface structure and energy of B2 type intermetallic compound NiAl,” Applied Surface Science. 2008. link Times cited: 8 NOT USED (high confidence) Y. Zhou, B. Anglin, and A. Strachan, “Phonon thermal conductivity in nanolaminated composite metals via molecular dynamics.,” The Journal of chemical physics. 2007. link Times cited: 42 Abstract: We use nonequilibrium molecular dynamics to characterize the… read moreAbstract: We use nonequilibrium molecular dynamics to characterize the phonon contribution to thermal conduction of Al nanostructures and the role of interfaces in metallic nanocomposites. We characterize the lattice thermal conductivity of pure Al samples as a function of size and temperature from which we obtain, using kinetic theory, the temperature dependence of the phonon mean free path. We also calculated the thermal conductivity of AlAl* and AlNi nanolaminate composites (where Al* differs from Al only in its mass) for various periodic sizes and compositions as well as the associated interfacial thermal resistivities (ITRs). We find that simple, additive models provide good estimates of the thermal conductivities of the nanocomposites in terms of those of the individual components and interfaces if size effects on the behavior of the individual components are considered. The additive models provide important insight to the decrease in thermal conductivity of the nanolaminates as their periodicity (thickness of a bilayer) is reduced to a size comparable with the phonon mean free path and break down when this characteristic size is reduced further. At this point the system can be regarded as homogeneous and the conductivity increases with decreasing periodicity of the laminates. We also observe that the ITR depends on the direction of the heat flux; this is the first molecular level characterization of such thermal diode behavior in a realistic three dimensional material. read less NOT USED (high confidence) Y. Mishin and A. Lozovoi, “Angular-dependent interatomic potential for tantalum,” Acta Materialia. 2006. link Times cited: 70 NOT USED (high confidence) N. Lazarev, C. Abromeit, R. Schäublin, and R. Gotthardt, “Temperature-controlled martensitic phase transformations in a model NiAl alloy,” Journal of Applied Physics. 2006. link Times cited: 12 Abstract: Reversible martensitic phase transformations in a partially … read moreAbstract: Reversible martensitic phase transformations in a partially disordered Ni–Al alloy within the composition range from 60to65at.% of Ni are investigated using molecular dynamics simulation. During a complete temperature cycle a wide hysteresis in enthalpy, volume, and shape of the simulated crystals is observed. The temperature T0 of the phase transformation is found from the calculated free energy evolution. To investigate the atomic-scale development during the phase transformation a local order parameter is defined which is based on a combined method of Voronoy tessellation [J. Reine Angew. Math. 134, 198 (1908)] with common-neighbor analysis. This local order parameter allows us to get a detailed localized picture of nucleation and growth of the new phases. Both homogeneous formation of the new phase and heterogeneous nucleation are observed. The velocity of new phase growth front is estimated. read less NOT USED (high confidence) K. M. Carling, W. Glover, H. Gunaydin, T. A. Mitchell, and E. Carter, “Comparison of S, Pt, and Hf adsorption on NiAl(1 1 0),” Surface Science. 2006. link Times cited: 24 NOT USED (high confidence) Y. Mishin, M. Mehl, D. Papaconstantopoulos, and D. Papaconstantopoulos, “Phase stability in the Fe–Ni system: Investigation by first-principles calculations and atomistic simulations,” Acta Materialia. 2005. link Times cited: 261 NOT USED (high confidence) H. H. Kart, M. Tomak, and T. Çagin, “Thermal and mechanical properties of Cu–Au intermetallic alloys,” Modelling and Simulation in Materials Science and Engineering. 2005. link Times cited: 41 Abstract: The thermal and mechanical properties of Cu, Au pure metals … read moreAbstract: The thermal and mechanical properties of Cu, Au pure metals and their ordered intermetallic alloys of Cu3Au(L12), CuAu(L10) and CuAu3(L12) are studied by using the molecular dynamics simulation. The effects of temperature and concentration on the physical properties of CuxAu1−x are analysed. Sutton–Chen (SC) and quantum Sutton–Chen (Q-SC) many-body potentials are used. The simulation results such as cohesive energy, density, elastic constants, bulk modulus, heat capacity, thermal expansion, melting points and phonon dispersion curves are in good agreement with the available experimental data at the various temperatures. Q-SC potential parameter results are usually closer to experimental values than the ones predicted from SC potential parameters. read less NOT USED (high confidence) G. Ackland, X. Huang, and K. Rabe, “First-principles thermodynamics of transition metals: W, NiAl, and PdTi,” Physical Review B. 2003. link Times cited: 27 Abstract: We apply the pseudopotential density-functional-perturbation… read moreAbstract: We apply the pseudopotential density-functional-perturbation theory approach along with the quasiharmonic approximation to calculate the thermal expansion of tungsten and two important metallic alloys NiAl and PdTi. We derive the theory for anisotropic crystal structures and test the approximation that the anisotropic effects of thermal expansion are equivalent to negative pressure-this simplifies the calculation enormously for complex structures. Throughout, we find excellent agreement with experimental results. read less NOT USED (high confidence) Y. Mishin, A. Lozovoi, and A. Alavi, “Evaluation of diffusion mechanisms in NiAl by embedded-atom and first-principles calculations,” Physical Review B. 2003. link Times cited: 75 Abstract: The energetics of Ni vacancy jumps in the intermetallic comp… read moreAbstract: The energetics of Ni vacancy jumps in the intermetallic compound NiAl are studied by combining embedded-atom and first-principles calculations. The embedded-atom potential used in this work is fit to both experimental and first-principles data and provides an accurate description of point defect energies and vacancy jump barriers in NiAl. Some of the embedded-atom results reported here, are independently verified by plane-wave pseudopotential calculations. The results suggest that the atomic configuration produced by a nearest-neighbor jump of a Ni vacancy is mechanically unstable. Because of this instability, the vacancy implements two sequential nearest-neighbor jumps as one collective, two-atom transition. Such collective jumps initiate and complete six-jump vacancy cycles of a Ni vacancy, which are shown to occur by either four or three vacancy jumps. Next-nearest-neighbor vacancy jumps are shown to have diffusion rates comparable to experimental ones at the stoichiometric composition, suggesting that this is an important diffusion mechanism in NiAl. read less NOT USED (definite) D. M. Riffe, J. D. Christensen, and R. B. Wilson, “Vibrational dynamics within the embedded-atom-method formalism and the relationship to Born–von-Kármán force constants,” Journal of Physics: Condensed Matter. 2018. link Times cited: 1 Abstract: We derive expressions for the dynamical matrix of a crystall… read moreAbstract: We derive expressions for the dynamical matrix of a crystalline solid with total potential energy described by an embedded-atom-method (EAM) potential. We make no assumptions regarding the number of atoms per unit cell. These equations can be used for calculating both bulk phonon modes as well the modes of a slab of material, which is useful for the study of surface phonons. We further discuss simplifications that occur in cubic lattices with one atom per unit cell. The relationship of Born–von-Kármán (BvK) force constants—which are readily extracted from experimental vibrational dispersion curves—to the EAM potential energy is discussed. In particular, we derive equations for BvK force constants for bcc and fcc lattices in terms of the functions that define an EAM model. The EAM—BvK relationship is useful for assessing the suitability of a particular EAM potential for describing vibrational spectra, which we illustrate using vibrational data from the bcc metals K and Fe and the fcc metal Au. read less NOT USED (definite) H. Ding, B. K. Medasani, W. Chen, K. Persson, M. Haranczyk, and M. Asta, “PyDII: A python framework for computing equilibrium intrinsic point defect concentrations and extrinsic solute site preferences in intermetallic compounds,” Comput. Phys. Commun. 2015. link Times cited: 20 NOT USED (definite) Z. W. Wu, M. Li, W. Wang, and K. Liu, “Hidden topological order and its correlation with glass-forming ability in metallic glasses,” Nature Communications. 2015. link Times cited: 111 NOT USED (definite) B. Sadigh, P. Erhart, A. Stukowski, and A. Caro, “Composition-dependent interatomic potentials: A systematic approach to modelling multicomponent alloys,” Philosophical Magazine. 2009. link Times cited: 16 Abstract: We propose a simple scheme to construct composition-dependen… read moreAbstract: We propose a simple scheme to construct composition-dependent interatomic potentials for multicomponent systems that, when superposed onto the potentials for the pure elements, can reproduce not only the heat of mixing of the solid solution in the entire concentration range but also the energetics of a wider range of configurations including intermetallic phases. We show that an expansion in cluster interactions provides a way to systematically increase the accuracy of the model, and that it is straightforward to generalise this procedure to multicomponent systems. Concentration-dependent interatomic potentials can be built upon almost any type of potential for the pure elements including embedded atom method (EAM), modified EAM, bond-order, and Stillinger–Weber type potentials. In general, composition-dependent N-body terms in the total energy lead to explicit (N + 1)-body forces, which potentially render them computationally expensive. We present an algorithm that overcomes this problem and that can speed up the calculation of the forces for composition-dependent pair potentials in such a way as to make them computationally comparable in efficiency and scaling behaviour to standard EAM potentials. We also discuss the implementation in Monte Carlo simulations. Finally, we exemplarily review the composition-dependent EAM model for the Fe–Cr system. read less NOT USED (definite) B. Muite and O. U. Salman, “Computations of geometrically linear and nonlinear Ginzburg-Landau mo dels for martensitic pattern formation.” 2009. link Times cited: 4 Abstract: Computations show that a two dimensional geometrically nonli… read moreAbstract: Computations show that a two dimensional geometrically nonlinear Ginzburg-Landau model with inertia exhibits long lived metastable states, that have martensite domains with split tips and bent needles similar to those observed in NiAl. In comparison, the geometrically linear model quickly relaxes to states with twins which extend all the way across the sample and have only short lived tip splitting and needle bending. read less |
 EAM_Dynamo_MishinMehlPapaconstantopoulos_2002_NiAl__MO_109933561507_005
EAM_Dynamo_MishinMehlPapaconstantopoulos_2002_NiAl__MO_109933561507_005