Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
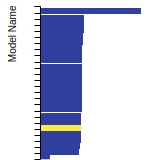
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.

306 Citations (239 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) X. Yang, S. Xu, and Q. Chi, “Plastic Deformation Behavior of Bi-Crystal Magnesium Nanopillars with a 101¯2 Twin Boundary under Compression: Molecular Dynamics Simulations,” Materials. 2019. link Times cited: 2 Abstract: In this study, molecular dynamics simulations were performed… read moreAbstract: In this study, molecular dynamics simulations were performed to study the uniaxial compression deformation of bi-crystal magnesium nanopillars with a {101¯2} twin boundary (TB). The generation and evolution process of internal defects of magnesium nanopillars were analyzed in detail. Simulation results showed that the initial deformation mechanism was mainly caused by the migration of the twin boundary, and the transformation of TB into (basal/prismatic) B/P interface was observed. After that, basal slip as well as pyramidal slip nucleated during the plastic deformation process. Moreover, a competition mechanism between twin boundary migration and basal slip was found. Basal slip can inhibit the migration of the twin boundary, and {101¯1}〈101¯2〉 twins appear at a certain high strain level (ε = 0.104). In addition, Schmid factor (SF) analysis was conducted to understand the activations of deformation modes. read less USED (definite) Y. Tang, “Uncovering the inertia of dislocation motion and negative mechanical response in crystals,” Scientific Reports. 2018. link Times cited: 19 USED (definite) D. Sun, M. Ponga, K. Bhattacharya, and M. Ortiz, “Proliferation of twinning in hexagonal close-packed metals: Application to magnesium,” Journal of The Mechanics and Physics of Solids. 2017. link Times cited: 26 USED (definite) E. Lazar, J. Han, and D. Srolovitz, “Topological framework for local structure analysis in condensed matter,” Proceedings of the National Academy of Sciences. 2015. link Times cited: 97 Abstract: Significance Richard Feynman famously described the hypothes… read moreAbstract: Significance Richard Feynman famously described the hypothesis “All things are made of atoms” as among the most significant of all scientific knowledge. How atoms are arranged in “things” is an interesting and natural question. However, aside from perfect crystals and ideal gases, understanding these arrangements in an insightful yet tractable manner is challenging. We introduce a unified mathematical framework for classifying and identifying local structure in imperfect condensed matter systems using Voronoi topology. This versatile approach enables visualization and analysis of a wide range of complex atomic systems, including highly defected solids and glass-forming liquids. The proposed framework presents a new perspective into the structure of discrete systems of particles, ordered and disordered alike. Physical systems are frequently modeled as sets of points in space, each representing the position of an atom, molecule, or mesoscale particle. As many properties of such systems depend on the underlying ordering of their constituent particles, understanding that structure is a primary objective of condensed matter research. Although perfect crystals are fully described by a set of translation and basis vectors, real-world materials are never perfect, as thermal vibrations and defects introduce significant deviation from ideal order. Meanwhile, liquids and glasses present yet more complexity. A complete understanding of structure thus remains a central, open problem. Here we propose a unified mathematical framework, based on the topology of the Voronoi cell of a particle, for classifying local structure in ordered and disordered systems that is powerful and practical. We explain the underlying reason why this topological description of local structure is better suited for structural analysis than continuous descriptions. We demonstrate the connection of this approach to the behavior of physical systems and explore how crystalline structure is compromised at elevated temperatures. We also illustrate potential applications to identifying defects in plastically deformed polycrystals at high temperatures, automating analysis of complex structures, and characterizing general disordered systems. read less USED (definite) A. Ostapovets and A. Serra, “Slip dislocation and twin nucleation mechanisms in hcp metals,” Journal of Materials Science. 2016. link Times cited: 45 USED (high confidence) B. Liu et al., “Rejuvenation of plasticity via deformation graining in magnesium,” Nature Communications. 2022. link Times cited: 24 USED (high confidence) B. Liu et al., “Rejuvenation of plasticity via deformation graining in magnesium,” Nature Communications. 2022. link Times cited: 0 USED (high confidence) N. Li, N. Ding, J. Zhou, L. Liu, F. Zaïri, and Y. Yang, “Mechanical properties and deformation behavior of the magnesium crystal with nano-cracks,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 1 Abstract: Understanding how intrinsic defects impact magnesium (Mg) cr… read moreAbstract: Understanding how intrinsic defects impact magnesium (Mg) crystals mechanics is of prime importance for engineering applications. In this work, the mechanical performance of Mg crystals with cracks at the nanoscale was studied using molecular dynamics method. Influence of the nano-crack type and size on the deformation behavior of Mg crystals was analyzed in details. The obtained results show that the mechanical properties of Mg crystals decrease with the increase of the nano-crack length (perpendicular to the tensile direction). However, the yield stress of Mg crystals is enhanced by increasing the nano-crack width (parallel to the tensile direction) while the nano-crack length remains unchanged. The effect of temperature on Young’s modulus of Mg crystals is weak along z-axis, while Young’s modulus along y-axis is clearly temperature-dependent. The yield stress of Mg crystals decreases with increasing temperature. The appearance of twins is the main deformation mechanism in Mg crystal along z-axis while the deformation begins with the formation of a prismatic slip along y-axis. The results obtained in this work would provide useful information for further mechanical properties regulation of Mg crystals. read less USED (high confidence) Z.-C. Ma, X. Tang, Y. Mao, and Y.-F. Guo, “The Plastic Deformation Mechanisms of hcp Single Crystals with Different Orientations: Molecular Dynamics Simulations,” Materials. 2021. link Times cited: 7 Abstract: The deformation mechanisms of Mg, Zr, and Ti single crystals… read moreAbstract: The deformation mechanisms of Mg, Zr, and Ti single crystals with different orientations are systematically studied by using molecular dynamics simulations. The affecting factors for the plasticity of hexagonal close-packed (hcp) metals are investigated. The results show that the basal dislocation, prismatic dislocation, and pyramidal dislocation are activated in Mg, Zr, and Ti single crystals. The prior slip system is determined by the combined effect of the Schmid factor and the critical resolved shear stresses (CRSS). Twinning plays a crucial role during plastic deformation since basal and prismatic slips are limited. The 101¯2 twinning is popularly observed in Mg, Zr, and Ti due to its low CRSS. The 101¯1 twin appears in Mg and Ti, but not in Zr because of the high CRSS. The stress-induced hcp-fcc phase transformation occurs in Ti, which is achieved by successive glide of Shockley partial dislocations on basal planes. More types of plastic deformation mechanisms (including the cross-slip, double twins, and hcp-fcc phase transformation) are activated in Ti than in Mg and Zr. Multiple deformation mechanisms coordinate with each other, resulting in the higher strength and good ductility of Ti. The simulation results agree well with the related experimental observation. read less USED (high confidence) R. K. Koju and Y. Mishin, “Atomistic Study of Grain-Boundary Segregation and Grain-Boundary Diffusion in Al-Mg Alloys,” EngRN: Metals & Alloys (Topic). 2020. link Times cited: 60 Abstract: Mg grain boundary (GB) segregation and GB diffusion can impa… read moreAbstract: Mg grain boundary (GB) segregation and GB diffusion can impact the processing and properties of Al-Mg alloys. Yet, Mg GB diffusion in Al has not been measured experimentally or predicted by simulations. We apply atomistic computer simulations to predict the amount and the free energy of Mg GB segregation, and the impact of segregation on GB diffusion of both alloy components. At low temperatures, Mg atoms segregated to a tilt GB form clusters with highly anisotropic shapes. Mg diffuses in Al GBs slower than Al itself, and both components diffuse slowly in comparison with Al GB self-diffusion. Thus, Mg segregation significantly reduces the rate of mass transport along GBs in Al-Mg alloys. The reduced atomic mobility can be responsible for the improved stability of the microstructure at elevated temperatures. read less USED (high confidence) Y. Zhu, H. Zhang, S. Xu, and J. Nie, “The β1 Triad-Related Configurations in a Mg-RE Alloy,” Metallurgical and Materials Transactions A. 2020. link Times cited: 2 USED (high confidence) S. Song, Y. Wang, Y. Wang, X. Wang, and Z. Zhang, “The effect of tension twin on the dynamic recrystallization behavior in polycrystal magnesium by atomistic simulation,” Applied Physics A. 2020. link Times cited: 6 USED (high confidence) A. Vlasova, “Simulation of Uniaxial Deformation of Magnesium Nanocrystals of ‘Rigid’ and ‘Soft’ Orientations,” Physics of the Solid State. 2020. link Times cited: 1 USED (high confidence) S. Song, Y. Wang, Y. Wang, X. Wang, and Z. Zhang, “The effect of tension twin on the dynamic recrystallization behavior in polycrystal magnesium by atomistic simulation,” Applied Physics A. 2020. link Times cited: 0 USED (high confidence) S. Kavousi, B. R. Novak, M. Baskes, M. A. Zaeem, and D. Moldovan, “Modified embedded-atom method potential for high-temperature crystal-melt properties of Ti–Ni alloys and its application to phase field simulation of solidification,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 21 Abstract: We developed new interatomic potentials, based on the second… read moreAbstract: We developed new interatomic potentials, based on the second nearest-neighbor modified embedded-atom method (2NN-MEAM) formalism, for Ti, Ni, and the binary Ti–Ni system. These potentials were fit to melting points, latent heats, the binary phase diagrams for the Ti rich and Ni rich regions, and the liquid phase enthalpy of mixing for binary alloys, therefore they are particularly suited for calculations of crystal-melt (CM) interface thermodynamic and transport properties. The accuracy of the potentials for pure Ti and pure Ni were tested against both 0 K and high temperature properties by comparing various properties obtained from experiments or density functional theory calculations including structural properties, elastic constants, point-defect properties, surface energies, temperatures and enthalpies of phase transformations, and diffusivity and viscosity in the liquid phase. The fitted binary potential for Ti–Ni was also tested against various non-fitted properties at 0 K and high temperatures including lattice parameters, formation energies of different intermetallic compounds, and the temperature dependence of liquid density at various concentrations. The CM interfacial free energies obtained from simulations, based on the newly developed Ti–Ni potential, show that the bcc alloys tend to have smaller anisotropy compared with fcc alloys which is consistent with the finding from the previous studies comparing single component bcc and fcc materials. Moreover, the interfacial free energy and its anisotropy for Ti-2 atom% Ni were also used to parameterize a 2D phase field (PF) model utilized in solidification simulations. The PF simulation predictions of microstructure development during solidification are in good agreement with a geometric model for dendrite primary arm spacing. read less USED (high confidence) C. Xu, L. Yuan, D. Shan, and B. Guo, “The influence of lamellar twins on deformation mechanism in nanocrystalline magnesium under uniaxial compression,” Journal of Materials Science. 2019. link Times cited: 4 USED (high confidence) S. Song, Y. Wang, Y. Wang, and X. Wang, “Atomistic Simulation on the Twin Boundary Migration in Mg under Shear Deformation,” Materials. 2019. link Times cited: 3 Abstract: In this paper, the {101¯2} twinning and detwinning was studi… read moreAbstract: In this paper, the {101¯2} twinning and detwinning was studied by molecular dynamics simulation under different shear directions and strain rates. The results showed that the twin was thickened under [1¯011] shear direction and shrunken with shearing in the opposite direction. The critical resolved shear stress of {101¯2} twin boundary migration increased with the increase of the strain rate. By analyzing the atom’s displacement, it was concluded that the {101¯2} twin migration was achieved by both the shear and the atomic shuffling. Every atom would be affected by the shear, and different shear directions would cause opposite move directions, which led to twinning or detwinning. The atom shuffling was only used for adjusting the glide twin boundary and mirror-symmetric twin boundary structure evolution. read less USED (high confidence) S. Wang, H. Pan, P. Wang, and F.-guo Zhang, “Microstructural evolution of single-crystal magnesium under elevated temperature and ultra-high strain rate,” Journal of Applied Physics. 2019. link Times cited: 6 Abstract: Despite numerous studies of the deformation behavior of magn… read moreAbstract: Despite numerous studies of the deformation behavior of magnesium (Mg), its microstructural evolution at different temperatures and strain rates remains largely unexplored. In this paper, the evolution of dislocations and amorphous regions in single-crystal Mg under compressive loading along the c-axis is investigated using molecular dynamics simulations, and temperature and strain-rate dependence of the microstructural evolution is revealed. At a strain rate of 107 s−1, the dislocations are low in density, and they slip and evolve unevenly as the strain in the single crystal increases. Consequently, the stress in the single crystal varies in a zigzag manner with increasing strain. The dislocation density is higher at strain rates of 108 s−1 and 109 s−1, resulting in relatively smooth deformation and stress–strain curves. At a strain rate of 1010 s−1, the amorphous regions achieve a very high fraction during deformation, contributing to softening and smoother deformation of the single crystal. The fraction of amorphous regions also increases with increasing temperature, which is an important cause of the temperature softening effect. Furthermore, the initiation of dislocations and amorphous regions is also studied at different strain rates and temperatures.Despite numerous studies of the deformation behavior of magnesium (Mg), its microstructural evolution at different temperatures and strain rates remains largely unexplored. In this paper, the evolution of dislocations and amorphous regions in single-crystal Mg under compressive loading along the c-axis is investigated using molecular dynamics simulations, and temperature and strain-rate dependence of the microstructural evolution is revealed. At a strain rate of 107 s−1, the dislocations are low in density, and they slip and evolve unevenly as the strain in the single crystal increases. Consequently, the stress in the single crystal varies in a zigzag manner with increasing strain. The dislocation density is higher at strain rates of 108 s−1 and 109 s−1, resulting in relatively smooth deformation and stress–strain curves. At a strain rate of 1010 s−1, the amorphous regions achieve a very high fraction during deformation, contributing to softening and smoother deformation of the single crystal. The fractio... read less USED (high confidence) C. Xu, L. Yuan, R. Shivpuri, D. Shan, and B. Guo, “Role of misorientation angle in twinning and dislocation slip for nano Mg bicrystals with [2-1-10] symmetric tilt grain boundaries under uniaxial compression and tension,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 7 Abstract: Atomistic modeling is employed to investigate the role of di… read moreAbstract: Atomistic modeling is employed to investigate the role of different mechanisms in the plastic response of Mg bicrystal with [2-1-10] symmetric tilt grain boundary (STGB). Excess potential energies for 30 stable STGBs with different misorientation angles are used to different models. The structure of [2-1-10] STGB consists of a base plane and an array of intrinsic grain boundary dislocations (GBDs). The STGB structures varying with the misorientation angle influence the deformation mode, and nucleation and propagation of twins and basal dislocations in the bicrystal models. Uniaxial compression and tension are imposed on 14 bicrystal models containing STGBs under a strain rate of 1 × 108 s−1 at 300 K. For the hcp lattice, non-symmetry of compressive and tensile response is demonstrated. Dislocation nucleation prefers to occur from the GBDs where intrinsic stacking fault facets may nucleate prior to the dislocation emission. Through reaction with (01-11) twin boundary (TB), the basal dislocation from matrix is dissociated into a residual dislocation and a pyramidal dislocation which can glide along the pyramidal plane of twin. While the basal dislocation reacting with (01-13) TB is dissociated into some serrated facets. read less USED (high confidence) A. Ostapovets and A. Sheikh-Ali, “Misorientation dependence of atomic structure and energy of symmetric tilt boundaries in magnesium,” Philosophical Magazine. 2018. link Times cited: 6 Abstract: ABSTRACT Twelve symmetric tilt boundaries in magnesium spann… read moreAbstract: ABSTRACT Twelve symmetric tilt boundaries in magnesium spanning misorientation range 78.29° ≤ θ ≤ 145.85° are investigated with molecular dynamics simulation at 0 K, using an embedded-atom method. Three favoured boundaries are identified in this range: 78.29°, 116.88° and 145.85°. Boundary structures intermediate between the first two boundaries are predicted by the structural unit model, which cannot be applied, however, to the misorientation range 116.88° < θ < 145.85°. A sharp transition between the structural units of the 116.88° and 145.85° boundaries is observed within the narrow misorientation range 135° < θ < 138° at or close to the energy maximum. The transition is obviously caused by avoidance of long-range compatibility stresses between these units. GRAPHICAL ABSTRACT read less USED (high confidence) A. Vlasova and A. Nikonov, “Formation of Dislocations and Twins As a Result of Uniaxial Compression of Magnesium Single Crystals: Molecular Dynamics Simulation,” Crystallography Reports. 2018. link Times cited: 5 USED (high confidence) H. Yang, E. Goudeli, and C. J. Hogan, “Condensation and dissociation rates for gas phase metal clusters from molecular dynamics trajectory calculations.,” The Journal of chemical physics. 2018. link Times cited: 23 Abstract: In gas phase synthesis systems, clusters form and grow via c… read moreAbstract: In gas phase synthesis systems, clusters form and grow via condensation, in which a monomer binds to an existing cluster. While a hard-sphere equation is frequently used to predict the condensation rate coefficient, this equation neglects the influences of potential interactions and cluster internal energy on the condensation process. Here, we present a collision rate theory-molecular dynamics simulation approach to calculate condensation probabilities and condensation rate coefficients. We use this approach to examine atomic condensation onto 6-56-atom Au and Mg clusters. The probability of condensation depends upon the initial relative velocity (v) between atom and cluster and the initial impact parameter (b). In all cases, there is a well-defined region of b-v space where condensation is highly probable, and outside of which the condensation probability drops to zero. For Au clusters with more than 10 atoms, we find that at gas temperatures in the 300-1200 K range, the condensation rate coefficient exceeds the hard-sphere rate coefficient by a factor of 1.5-2.0. Conversely, for Au clusters with 10 or fewer atoms and for 14- and 28-atom Mg clusters, as cluster equilibration temperature increases, the condensation rate coefficient drops to values below the hard-sphere rate coefficient. Calculations also yield the self-dissociation rate coefficient, which is found to vary considerably with gas temperature. Finally, calculations results reveal that grazing (high b) atom-cluster collisions at elevated velocity (>1000 m s-1) can result in the colliding atom rebounding (bounce) from the cluster surface or binding while another atom dissociates (replacement). The presented method can be applied in developing rate equations to predict material formation and growth rates in vapor phase systems. read less USED (high confidence) Z. Yang, L. Zhang, M. Chisholm, X. Zhou, H. Ye, and S. Pennycook, “Precipitation of binary quasicrystals along dislocations,” Nature Communications. 2018. link Times cited: 36 USED (high confidence) S. Xu, Y. Su, and S. Chavoshi, “Deformation of periodic nanovoid structures in Mg single crystals,” Materials Research Express. 2018. link Times cited: 19 Abstract: Large scale molecular dynamics (MD) simulations in Mg single… read moreAbstract: Large scale molecular dynamics (MD) simulations in Mg single crystal containing periodic cylindrical voids subject to uniaxial tension along the z direction are carried out. Models with different initial void sizes and crystallographic orientations are explored using two interatomic potentials. It is found that (i) a larger initial void always leads to a lower yield stress, in agreement with an analytic prediction; (ii) in the model with x [ 1 ¯ 100 ] – y [ 0001 ] – z [ 11 2 ¯ 0 ] orientations, the two potentials predict different types of tension twins and phase transformation; (iii) in the model with x [ 0001 ] – y [ 11 2 ¯ 0 ] – z [ 1 ¯ 100 ] orientations, the two potentials identically predict the nucleation of edge dislocations on the prismatic plane, which then glide away from the void, resulting in extrusions at the void surface; in the case of the smallest initial void, these surface extrusions pinch the void into two voids. Besides bringing new physical understanding of the nanovoid structures, our work highlights the variability and uncertainty in MD simulations arising from the interatomic potential, an issue relatively lightly addressed in the literature to date. read less USED (high confidence) H. Fan, J.-J. Tang, X. Tian, Q. Wang, X. Tian, and J. El-Awady, “Core structures and mobility of ⟨c⟩ dislocations in magnesium,” Scripta Materialia. 2017. link Times cited: 21 USED (high confidence) A. Ostapovets and O. Vatazhuk, “Peierls barriers of a-type edge and screw dislocations moving on basal and prismatic planes in magnesium,” Low Temperature Physics. 2017. link Times cited: 8 Abstract: Exact shape of Peierls barriers are calculated for ⟨a⟩ edge … read moreAbstract: Exact shape of Peierls barriers are calculated for ⟨a⟩ edge and screw dislocation gliding on basal and prismatic planes in magnesium by using of several popular interatomic potentials. Comparison of these potentials is performed in order to describe their abilities and limitations. Stability of different types of dislocation cores are analyzed as well as their mutual transformations during dislocation slip. It was found that the Peierls stresses and barrier height are dependent on core type. It was concluded that transformations of dislocation cores along minimal energy paths have to be taken into account for development of analytical models of the slip in magnesium. The results are compared with available first-principles calculations. read less USED (high confidence) Z. Pei et al., “Atomic structures of twin boundaries in hexagonal close-packed metallic crystals with particular focus on Mg,” npj Computational Materials. 2017. link Times cited: 33 USED (high confidence) T. Mukhopadhyay, A. Mahata, S. Dey, and S. Adhikari, “Probabilistic Analysis and Design of HCP Nanowires: An Efficient Surrogate Based Molecular Dynamics Simulation Approach,” Journal of Materials Science & Technology. 2016. link Times cited: 41 USED (high confidence) G. Zu and S. Groh, “Effect of segregated alloying element on the intrinsic fracture behavior of Mg,” Theoretical and Applied Fracture Mechanics. 2016. link Times cited: 3 USED (high confidence) G. Agarwal and A. Dongare, “Shock wave propagation and spall failure in single crystal Mg at atomic scales,” Journal of Applied Physics. 2016. link Times cited: 43 Abstract: Large scale molecular dynamics (MD) simulations are carried … read moreAbstract: Large scale molecular dynamics (MD) simulations are carried out to investigate the wave propagation and failure behavior of single crystal Mg under shock loading conditions. The embedded atom method interatomic potential, used to model the Mg systems, is first validated by comparing the predicted Hugoniot behavior with that observed using experiments. The first simulations are carried out to investigate the effect of loading orientation on the wave propagation and failure behavior by shock loading the system along the [0001] direction (c-axis) and the [101¯0] direction using a piston velocity of 1500 m/s. The spall strength (peak tensile pressure prior to failure) is predicted to be higher for loading along the [101¯0] direction than that predicted for loading along the [0001] direction. To investigate the effect of shock pressure on the failure behavior and spall strength of the metal, the MD simulations are carried out using piston velocities of 500 m/s, 1000 m/s, 1500 m/s, and 2000 m/s for loading alon... read less USED (high confidence) E. Asadi and M. A. Zaeem, “The anisotropy of hexagonal close-packed and liquid interface free energy using molecular dynamics simulations based on modified embedded-atom method,” Acta Materialia. 2016. link Times cited: 37 USED (high confidence) Q. Zu, Y.-F. Guo, S. Xu, X. Tang, and Y. Wang, “Molecular Dynamics Simulations of the Orientation Effect on the Initial Plastic Deformation of Magnesium Single Crystals,” Acta Metallurgica Sinica (English Letters). 2016. link Times cited: 19 USED (high confidence) H. Fan and J. El-Awady, “Molecular Dynamics Simulations of Orientation Effects During Tension, Compression, and Bending Deformations of Magnesium Nanocrystals,” Journal of Applied Mechanics. 2015. link Times cited: 48 Abstract: The deformation modes in magnesium nanocrystals during uniax… read moreAbstract: The deformation modes in magnesium nanocrystals during uniaxial tension, uniaxial compression, and pure bending are investigated using molecular dynamics (MD) simulations at room temperature. For each loading condition, the crystal orientation effects are studied by increasing the crystal c-axis orientation angle θ relative to the loading direction from 0 deg to 90 deg by a 15 deg increment. The simulation results reveal a number of different deformation modes and an obvious tension–compression asymmetry in magnesium nanocrystals. As the c-axis is rotated away from the tension loading direction, the deformation mode at yielding changes from tension twinning (θ ≤ 45 deg) to compression twinning (θ > 45 deg). For compression loading, yielding is dominated by only dislocation slip on the pyramidal (θ 60 deg) planes. The nucleation stress in general decreases with increasing θ for both uniaxial tension and uniaxial compression loadings. For pure bending simulations, the yielding is mostly controlled by the weaker deformation mode between the compressive and tensile sides. The bending nucleation stress also decreases as the c-axis deviates away from the loading direction. read less USED (high confidence) H. Fan and J. El-Awady, “Towards resolving the anonymity of Pyramidal slip in magnesium,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 66 USED (high confidence) M. Itakura, H. Kaburaki, M. Yamaguchi, and T. Tsuru, “Atomistic study on the cross-slip process of a screw dislocation in magnesium,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 15 Abstract: The cross-slip process of a screw dislocation from the ba… read moreAbstract: The cross-slip process of a screw dislocation from the basal to the prismatic plane in magnesium was studied using the density functional theory and the molecular dynamics calculations. An atomistic method for calculating the total Peierls energy map has been devised to track the transition path of a dissociated and/or constricted screw dislocation in the cross-slip process. The barrier of a screw dislocation from the basal to the prismatic plane is estimated by the density functional theory for the first time to be 61.4±2.0 ?> meV per Burgers vector length. The activation enthalpy for the cross slip is calculated using a line tension model based on the density functional theory to be 1.4–1.7 eV, which is in reasonable agreement with experiments. On the basis of the results, the effect of temperature on the cross-slip process of the dissociated screw dislocation on the basal plane is studied in detail using the molecular dynamics method with the embedded-atom-method (EAM) interatomic potential, in which the critical resolved shear stress for the cross slip is evaluated. It is confirmed that the bowed-out dislocation line on the prismatic plane consists of slightly dissociated rectilinear segments with connecting jogs at low temperatures and, as the temperature rises, the curved dislocation line becomes smooth with many segments. The motion of an dislocation on the prismatic plane is jerky in the low temperature region, while it is retarded by the formation of the largely dissociated plateau segment above the room temperature. A large reduction of the critical shear stress for the cross slip is obtained when the screw dislocation interacts with a hard-sphere particle placed on the basal plane in the low temperature region. read less USED (high confidence) Y. Tang and J. El-Awady, “Highly anisotropic slip-behavior of pyramidal I 〈c+a〉 dislocations in hexagonal close-packed magnesium,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2014. link Times cited: 35 USED (high confidence) Y. Tang and J. El-Awady, “Formation and slip of pyramidal dislocations in hexagonal close-packed magnesium single crystals,” Acta Materialia. 2014. link Times cited: 147 USED (high confidence) M. Liao, B. Li, and M. Horstemeyer, “Interaction Between Basal Slip and a Mg17Al12 Precipitate in Magnesium,” Metallurgical and Materials Transactions A. 2014. link Times cited: 29 USED (high confidence) D. Matsunaka, Y. Ohnishi, and Y. Shibutani, “Effects of Stacking Fault Energy on Fundamental Deformation Modes in Single Crystalline Magnesium by Molecular Dynamics Simulations,” Materials Transactions. 2013. link Times cited: 6 Abstract: In order to investigate effects of stacking fault energies (… read moreAbstract: In order to investigate effects of stacking fault energies (SFEs) on fundamental deformation modes of slips and deformation twinnings in magnesium, we carried out molecular dynamics simulations of shear deformations for the deformation modes with two kinds of many-body interatomic potentials. The SFEs of the basal and second-pyramidal planes are lower for a generalized embedded atom method (GEAM) potential than for an embedded atom method (EAM) potential. While the basal slip quite easily occurs and the prism dislocation is activated, the first-pyramidal slip and the second-pyramidal slip are hard to be operated. However, for the GEAM simulations, the second-pyramidal slip was activated due to reduction of the second-pyramidal SFE. Additionally, the reduction of the SFEs suppresses nucleation of the f10 11g twin in the bf10 11g 2 shearing direction. The relative order of the other fundamental deformation modes in the critical shear stress is qualitatively maintained despite the reduction of the SFEs. [doi:10.2320/matertrans.MAW201311] read less USED (high confidence) M. A. Bhatia and K. Solanki, “Energetics of vacancy segregation to symmetric tilt grain boundaries in hexagonal closed pack materials,” Journal of Applied Physics. 2013. link Times cited: 34 Abstract: Molecular static simulations of 190 symmetric tilt grain bou… read moreAbstract: Molecular static simulations of 190 symmetric tilt grain boundaries in hexagonal closed pack metals were used to understand the energetics of vacancy segregation, which is important for designing stable interfaces in harsh environments. Simulation results show that the local arrangements of grain boundaries and the resulting structural units have a significant influence on the magnitude of vacancy binding energies, and the site-to-site variation within each boundary is substantial. Comparing the vacancy binding energies for each site in different c/a ratio materials shows that the binding energy increases significantly with an increase in c/a ratio. For example, in the [12¯10] tilt axis, Ti and Zr with c/a = 1.5811 have a lower vacancy binding energy than the Mg with c/a = 1.6299. Furthermore, when the grain boundary energies of all 190 boundaries in all three elements are plotted against the vacancy binding energies of the same boundaries, a highly negative correlation (r = −0.7144) is revealed that has ... read less USED (high confidence) Y. Zhang, L. Zhou, and H.-C. Huang, “Size dependence of twin formation energy of metallic nanowires,” International Journal of Smart and Nano Materials. 2013. link Times cited: 3 Abstract: Twin formation energy is an intrinsic quantity for bulk crys… read moreAbstract: Twin formation energy is an intrinsic quantity for bulk crystals. At the nanoscale, the twin formation energy of covalent SiC nanowires goes up with decreasing dimension. In contrast, this article reports that the twin formation energy of metallic nanowires goes down with decreasing dimension. This result is based on classical molecular statics simulations of four representative metals. Cu and Al represent face-centered cubic (FCC) metals of low and high twin formation energies. Ta represents a body-centered cubic (BCC) metal, and Mg represents a hexagonal close-packed (HCP) metal. For all the four metals, the dependence of twin formation energy on size correlates with lower twin formation energy near surfaces, according to atomic-level analysis. Based on this atomic-level insight, the authors propose a core–shell model that reveals the twin formation energy as inversely proportional to the diameter of nanowires. This dependence is in agreement with the results of molecular statics simulations. read less USED (high confidence) M. Uranagase, S. Kamigaki, R. Matsumoto, and N. Miyazaki, “Activation Free Energy of Nucleation of a Dislocation Pair in Magnesium,” Materials Transactions. 2013. link Times cited: 7 Abstract: Kink deformation is one of the possible principal deformatio… read moreAbstract: Kink deformation is one of the possible principal deformation modes of alloys with a long-period stacking ordered structure under compression parallel to the basal plane. In this deformation, dislocation pairs are massively nucleated, and these dislocations align in a line to form kink bands. In this study, we investigated the nucleation of a dislocation pair in a pure magnesium single crystal by molecular dynamics simulations. We also evaluated the activation free energy of nucleation of a dislocation pair and investigated the dependence of the activation free energy on the applied shear stress and temperature. [doi:10.2320/matertrans.MI201213] read less USED (high confidence) A. Luque, M. Ghazisaeidi, and W. A. Curtin, “Deformation modes in magnesium (0 0 0 1) and single crystals: simulations versus experiments,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 43 Abstract: Magnesium is an excellent candidate as lightweight structura… read moreAbstract: Magnesium is an excellent candidate as lightweight structural material, but has strong plastic anisotropy, and the activation of, operation of, and competition between different slip and twinning systems remain active areas of research. Here, the nucleation of twinning and basal slip in Mg single-crystalline nanopillars are studied using molecular dynamics over a range of strain rates allowing for reasonable extrapolation to experimental rates. Deformation along the [0 0 0 1] direction shows tension and compression twinning at stresses predicted to be ∼1400 and ∼1700 MPa at a strain rate of 10−3 s−1. Moreover, twin nuclei are shown to be absolutely stable only above 1170 MPa. No evidence of nanotwinning is found and the twin-growth velocities are very fast (∼400 m s−1). These results do not support recently proposed mechanisms for nanotwinning. Deformation along the direction shows basal dislocation nucleation at stresses of 1000–1300 MPa in tension and 670–900 MPa in compression, at experimental strain rates, with one EAM potential exhibiting compression/tension asymmetry. Size effects are observed between pillars of diameters between 5 and 10 nm, which are attributable to surface stress effects driving nucleation and expected to be irrelevant at experimental pillar sizes (200 nm and above). Overall, most of the observed deformation mechanisms mirror those found in experiments but the stress levels, even when extrapolated to experimental strain rates, remain well above those found in micro- and nanopillar experiments. This indicates that deformation in the experimental specimens is controlled by the motion of pre-existing dislocations or is associated with significant stress concentrations due to surface defects. read less USED (high confidence) T. Tsuru, Y. Udagawa, M. Yamaguchi, M. Itakura, H. Kaburaki, and Y. Kaji, “Solution softening in magnesium alloys: the effect of solid solutions on the dislocation core structure and nonbasal slip,” Journal of Physics: Condensed Matter. 2013. link Times cited: 56 Abstract: There is a pressing need to improve the ductility of magnesi… read moreAbstract: There is a pressing need to improve the ductility of magnesium alloys so that they can be applied as lightweight structural materials. In this study, a mechanism for enhancing the ductility of magnesium alloys has been pursued using the atomistic method. The generalized stacking fault (GSF) energies for basal and prismatic planes in magnesium were calculated by using density functional theory, and the effect of the GSF energy on the dislocation core structures was examined using a semidiscrete variational Peierls–Nabarro model. Yttrium was found to have an anomalous influence on the solution softening owing to a reduction in the GSF energy gradient. read less USED (high confidence) J. Mosler and M. Homayonifar, “Variational constitutive updates for microstructure evolution in hcp metals,” GAMM‐Mitteilungen. 2012. link Times cited: 2 Abstract: Magnesium and its alloys are promising materials for lightwe… read moreAbstract: Magnesium and its alloys are promising materials for lightweight applications. Unfortunately, the macroscopic formability of such materials is relatively poor at room temperature and these metals are characterized by a complex mechanical response. This response is a result of the interplay between different deformation modes at the microscale. Since magnesium is a material showing a hexagonal close‐packed (hcp) structure of the underlying atomic lattice, plasticity caused by dislocations and deformation‐induced twinning are the most relevant deformation modes. Within the present paper, two different recently advocated modeling approaches suitable for capturing such modes at the microscale are analyzed. It is shown that both models can be rewritten into a variationally consistent format where every aspect is naturally driven by energy minimization. In addition to this already known feature, it turns out that both models are based on the same minimization problem. The difference between the models results from different constraints enforced within the variational principle. For getting further insight into the interaction between dislocations and twinning interfaces, accompanying atomistic simulations based on molecular dynamics are also performed. The results of such simulations enter the micromechanical model through the initial plastic deformation within the twinned phase (© 2011 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less USED (high confidence) J. Yasi, L. Hector, and D. Trinkle, “Prediction of thermal cross-slip stress in magnesium alloys from a geometric interaction model,” Acta Materialia. 2012. link Times cited: 102 USED (high confidence) J. Yasi et al., “Prediction of thermal cross-slip stress in magnesium alloys from direct first-principles data,” Acta Materialia. 2011. link Times cited: 98 USED (high confidence) T. Nogaret, W. Curtin, J. Yasi, L. Hector, and D. Trinkle, “Atomistic study of edge and screw (c + a) dislocations in magnesium,” Acta Materialia. 2010. link Times cited: 113 USED (high confidence) S. Groh, E. Marin, M. Horstemeyer, and D. Bammann, “Dislocation motion in magnesium: a study by molecular statics and molecular dynamics,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 104 Abstract: The motion of dislocations with Burgers' vector lying o… read moreAbstract: The motion of dislocations with Burgers' vector lying on the basal, prismatic and pyramidal slip planes in pure magnesium was investigated numerically under static and dynamic loading conditions. The analysis of the dislocation core structures revealed that the basal slip system was the most favorable energetically, and therefore a dislocation loop cannot extend on the pyramidal slip plane, because screw dislocations were not stable in this slip plane. In agreement with experimental data, a strong anisotropy between slip systems was observed. In both the basal and the prismatic slip planes, the dislocation velocity is consistent with phonon drag theory. In addition, the edge dislocation velocity was always larger than the screw dislocation velocity independent of the slip system, while the dislocation velocity on the prismatic slip plane was always lower than the dislocation velocity on the basal plane regardless of the dislocation character. read less USED (high confidence) I. Steinbach, “Phase-field models in materials science,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 988 Abstract: The phase-field method is reviewed against its historical an… read moreAbstract: The phase-field method is reviewed against its historical and theoretical background. Starting from Van der Waals considerations on the structure of interfaces in materials the concept of the phase-field method is developed along historical lines. Basic relations are summarized in a comprehensive way. Special emphasis is given to the multi-phase-field method with extension to elastic interactions and fluid flow which allows one to treat multi-grain multi-phase structures in multicomponent materials. Examples are collected demonstrating the applicability of the different variants of the phase-field method in different fields of materials science. read less USED (high confidence) J. Tang and J. Yao, “Molecular dynamics study on the deformation of void single crystal magnesium under uniaxial stress,” Journal of Physics: Conference Series. 2021. link Times cited: 0 Abstract: The uniaxial tension and compression process of single cryst… read moreAbstract: The uniaxial tension and compression process of single crystal magnesium model with voids along [0001] direction was simulated by using embedded atom potential and molecular dynamics method, and the micro plastic deformation mechanism of voids under tension and compression was studied. The results show that the elastic modulus of the single crystal magnesium model under compression is greater than the elastic modulus under tension, indicating that compression deformation is more difficult; In the process of plastic deformation, the dislocation, stacking fault and twin will be produced in the single crystal magnesium model under tension and compression, but the emission mechanism of dislocation is different. Tension will make the dislocation slip to the edge of the model along the direction of 45 ° and produce four symmetrical slip bands, while compression will produce an annular defect band near the cavity; In addition, the stacking fault area and twin type produced are also different. This asymmetry is mainly caused by the different initial deformation mechanisms under the two loading conditions. read less USED (high confidence) C. Grégoire, “Dynamic behaviour of nano-sized voids in hexagonal close-packed materials.” 2018. link Times cited: 0 Abstract: The dynamic behaviour and failure mechanisms of nano-sized v… read moreAbstract: The dynamic behaviour and failure mechanisms of nano-sized voids in single crystals is studied for three hexagonal close-packed materials by means of molecular dynamics simulations. Our study reveals that in Magnesium the response is highly anisotropic leading to a brittle to ductile transition in the failure modes under different load orientations. This transition is accompanied by different mechanisms of deformation and is associated with the anisotropic HCP lattice structure of Mg and the associated barrier for dislocation motion. Remarkably, brittle failure is observed when external loads produce a high stress triaxiality while the response is more ductile when the stress triaxiality decreases. On the other hand, the failure in other two hexagonal close-packed materials studied in this work, i.e, Titanium and Zirconium, is more ductile, in high contrast with the brittle failure observed in Magnesium. We find that this difference is due to the fact that nano-sized voids in Titanium and Zirconium emit substantially more dislocations than Magnesium, allowing for large displacements of the atoms and plastic work, including non-basal planes. Based on our findings, we postulate that this brittle failure in Magnesium is due to a competition between dislocations emission in the basal plane and crack propagation in non-basal planes. Thus, we propose to use the ratio between unstable stacking fault and surface energy in these materials to assess the tendency of hexagonal close-packed materials and alloys to fail under brittle or ductile modes. Using this ratio, we critically identify the low surface energy of Mg as responsible for this brittle behaviour and recommend that Mg-based alloys with large surface energies can lead to better performance for dynamic applications. The fundamental mechanisms observed, therefore, explain the low spall strength of Mg and suggest the possibility of manipulating some mechanisms to increase ductility and spall strength of new lightweight Mg alloys. read less USED (high confidence) S. Karewar, N. Gupta, S. Groh, E. Martinez, A. Caro, and S. G. Srinivasan, “Effect of Li on the deformation mechanisms of nanocrystalline hexagonal close packed magnesium,” Computational Materials Science. 2017. link Times cited: 19 USED (high confidence) I. Shin and E. Carter, “Orbital-free density functional theory simulations of dislocations in magnesium,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 46 Abstract: Metal plasticity is controlled by nucleation and motion of d… read moreAbstract: Metal plasticity is controlled by nucleation and motion of dislocations. Key metrics determining the ease of these two events are stacking fault energies (SFEs) and dislocation structures. Here we study screw and edge dislocation structures on the basal, prismatic and pyramidal planes in hexagonal-close-packed magnesium (Mg) using orbital-free density functional theory (OFDFT) in order to gain insight into plastic deformation mechanisms in Mg. The accuracy of the method is first benchmarked against the more accurate Kohn–Sham DFT (KSDFT) with emphasis on testing OFDFT's main approximations, i.e. the kinetic energy density functional and the bulk-derived local pseudopotential by comparing predicted equilibrium bulk energies, elastic constants and various SFEs. Then we compare generalized SFEs for the basal, prismatic and pyramidal slip systems calculated by OFDFT versus two mainstream counterparts, KSDFT and the classical potential embedded atom method (EAM). The latter produces spurious minima along the generalized SFE surface on the prismatic plane whereas OFDFT agrees with qualitative experimental observations. Thereafter, we optimize isolated dislocation structures within periodic cells containing a few thousand atoms. We predict that on the basal plane, the screw and edge dislocations separate into partial dislocations with widths of ∼12 and ∼24 Å, respectively. Screw dislocations on the prismatic and pyramidal planes preferentially cross-slip and dissociate on the basal plane although a local minimum exists for a dissociated prismatic screw dislocation with widths of ⩾∼5 Å. By contrast, the edge dislocations on prismatic and pyramidal planes are predicted to remain undissociated. Such cross-slip behavior of screw dislocations is not reproduced by EAM simulations. We propose that the propensity for screw dislocations to remain on or cross-slip to Mg's basal plane, along with the compact nature of edge dislocations on non-basal planes, is likely to be responsible for its limited ductility. read less USED (low confidence) J. S. Lee et al., “Atomistic investigation into the formation of axial weak twins during the compression of single-crystal Mg nanopillars,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) Y. Sun and Y.-gui Chen, “Atomistic modeling of Mg-Al-Zn solid–liquid interfacial free energy,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) S. Yang et al., “Complex hexagonal close-packed dendritic growth during alloy solidification by graphics processing unit-accelerated three-dimensional phase-field simulations: demo for Mg–Gd alloy,” Rare Metals. 2023. link Times cited: 1 USED (low confidence) K. Yu et al., “Twin nucleation from disconnection-dense sites between stacking fault pairs in a random defect network,” Materialia. 2023. link Times cited: 1 USED (low confidence) X. Lai et al., “Plastic deformation response during crack propagation in Mg bicrystals with twin boundaries,” Journal of Materials Research and Technology. 2023. link Times cited: 1 USED (low confidence) M.-wu Wu, B. Tian, A. Zhang, Z. Guo, and S. Xiong, “Phase-field lattice-Boltzmann study on dendritic growth of hcp metals under gravity-driven natural convection,” Transactions of Nonferrous Metals Society of China. 2023. link Times cited: 0 USED (low confidence) Y. Q. Tang, A. Kumar, D. L. Chen, D. Y. Li, Q. Y. Li, and W. Li, “Bauschinger effect on wear of cold-worked Cu and Mg – A study combining molecular dynamics modeling and experimental investigation,” Wear. 2023. link Times cited: 0 USED (low confidence) S. Oyinbo, S. Singhaneka, and R. Matsumoto, “Exploring the basal/prismatic slip transfer at grain boundaries in magnesium: A molecular dynamic simulation,” Vacuum. 2023. link Times cited: 0 USED (low confidence) S. Lee, H.-S. Kang, and D. Bae, “Molecular Dynamics Study on Crack Propagation in Al Containing Mg–Si Clusters Formed during Natural Aging,” Materials. 2023. link Times cited: 2 Abstract: The crack propagation behavior of Al containing Mg–Si cluste… read moreAbstract: The crack propagation behavior of Al containing Mg–Si clusters is investigated using molecular dynamics (MD) simulations to demonstrate the relationship between the natural aging time in Al–Si–Mg alloys and ductility. Experimental results show that the elongation at failure decreases with natural aging. There are few studies on the relationship between natural aging and ductility because of the difficult observation of Mg–Si clusters. To solve the difficulty, cracked Al containing Mg–Si clusters of varying sizes are assumed for the MD simulations. A larger Mg–Si cluster in Al results in earlier crack opening and dislocation emission. Moreover, as the Mg–Si cluster size increases, the stress near the crack tip becomes more concentrated. This causes rapid crack propagation, a similar effect to that of crack tip sharpening. As a result of long-term natural aging, the cracks expand rapidly. The influence of geometry is also investigated. Crack lengthening and thickness reduction negatively impact the fracture toughness, with the former having a larger impact than the latter. Although there are several discrepancies in the practical deformation conditions, the simulation results can help to more thoroughly understand natural aging in Al–Si–Mg alloys. read less USED (low confidence) M. Li, Q. Guo, L. Chen, L. Li, H. Hou, and Y.-hong Zhao, “Microstructure and properties of graphene nanoplatelets reinforced AZ91D matrix composites prepared by electromagnetic stirring casting,” Journal of Materials Research and Technology. 2022. link Times cited: 70 USED (low confidence) P. Goswami, M. Gupta, and S. Pal, “Atomistic assessment of structural evolution for magnesium during hypervelocity nanoprojectile penetration,” Journal of Molecular Modeling. 2022. link Times cited: 0 USED (low confidence) C. Ma, C. Xue, Z. Chu, Q. Yang, S. Li, and B.-H. Yang, “Effect of tensile rate on structural transformation and dislocation of magnesium single crystal based on molecular dynamics,” Materials Today Communications. 2022. link Times cited: 1 USED (low confidence) Y. Wang et al., “Simulation of Microstructure Evolution in Mg Alloys by Phase-Field Methods: A Review,” Crystals. 2022. link Times cited: 0 Abstract: Microstructure is one of the vital factors that determine th… read moreAbstract: Microstructure is one of the vital factors that determine the mechanical properties of magnesium (Mg) alloys. However, traditional microstructure characterization methods hardly satisfy the needs of tracking the morphological evolution of Mg alloys. With the rapid development of computer simulation, using the phase-field method to simulate the evolution of microstructures in Mg alloys has become the new norm. This article provides a review of the applications of the phase-field method in Mg alloys. First, classic phase-field models and the derived multi-phase and polycrystalline phase-field models are reviewed, then a review of the twin and solid-state phase transition phase-field models was undertaken, and the research progress of phase-field simulation in the solidification, recrystallization, and solid-state phase transformation of Mg alloys, were gradually introduced. In addition, unresolved problems of phase-field simulation were summarized, and the possible direction of future studies on phase-field simulation in Mg alloys field were discussed. read less USED (low confidence) H. Issa, A. Taherizadeh, and A. Maleki, “Atomistic study of the effect of grain size and reinforcement particle on mechanical behavior of magnesium / silica nanocomposite,” Materialia. 2022. link Times cited: 2 USED (low confidence) D. Fang, D. Fang, T. Chai, X. Lin, Y. Lian, and K. Jin, “Correlation of C Rystal Orientation, Crystal Morphology and Mechanical Properties of Directionally Solidified Mg-Xgd Alloys,” SSRN Electronic Journal. 2022. link Times cited: 6 USED (low confidence) Y. Dan and D. Trinkle, “First-principles core energies of isolated basal and prism screw dislocations in magnesium,” Materials Research Letters. 2022. link Times cited: 4 Abstract: We use first-principles energy density method (EDM) to calcu… read moreAbstract: We use first-principles energy density method (EDM) to calculate atomic energies for isolated -type basal and prism screw dislocation cores in Mg and compute line energies and core energy differences. The atomic energy distribution in the dislocations reflect the slip in the cores and the elastic energy further afield. Line energies are computed by summing up atomic energies, from which core energies and energy differences are straightforward to determine. We compare our results with two different classical potentials. GRAPHICAL ABSTRACT IMPACT STATEMENT This work is the first exact, direct calculation of core energies of isolated dislocations using density functional theory via atomic energies, applied to core energy differences in magnesium. read less USED (low confidence) S. A. Etesami, M. Laradji, and E. Asadi, “The influence of Pb content on the interfacial free energy of solid Sn in eutectic Pb–Sn liquid mixtures using molecular dynamics simulations,” Molecular Simulation. 2022. link Times cited: 2 Abstract: ABSTRACT The solid–liquid interfacial free energy (γ) for bi… read moreAbstract: ABSTRACT The solid–liquid interfacial free energy (γ) for binary Pb–Sn systems at different concentrations and crystal orientations is calculated using molecular dynamics (MD) simulation that employs a modified-embedded atom method interatomic potential. First, the solid–liquid interface is constructed in MD simulations according to its calculated phase diagram. Then, the capillary fluctuation method (CFM) is employed for the calculation of the solid–liquid interfacial stiffness. A novel systematic approach is proposed that eliminates the error in the linear approximation of the CFM. The results show that orientation of the solid Sn crystal does not have a significant influence on the value of the stiffness, which explains the experimentally observed spherical shape of Sn particles in eutectic liquid Pb–Sn mixtures. The calculated γ for pure Sn is in good agreement with experimental counterparts. In addition, this study finds that γ slightly increases with increasing the concentration of Pb in the eutectic liquid Pb–Sn mixture, which agrees with available experimental data. read less USED (low confidence) C. Chen and J.-Y. Song, “A Combined Atomistic-Continuum Study on the Unfaulting of Single and Multi-layer Interstitial Dislocation Loops in Irradiated FCC and HCP Metals,” International Journal of Plasticity. 2022. link Times cited: 4 USED (low confidence) R. Verma, L. Král, and A. Ostapovets, “Faceting of twin interfaces in rolled pure magnesium,” Philosophical Magazine. 2022. link Times cited: 2 Abstract: ABSTRACT Recent studies proved that the faceting of twin bou… read moreAbstract: ABSTRACT Recent studies proved that the faceting of twin boundaries is a common feature in hcp materials. It was demonstrated that the asymmetrical facets in the most frequent and twin interfaces are the consequence of the twin boundary migration mechanism. We demonstrate that a similar mechanism is also applicable to less common twins, observed in pure magnesium rolled at room temperature. The migration of twin boundary is mediated by disconnections, which have Burgers vector and step height equal to four interplanar distances. The mechanism of asymmetrical facets nucleation is also demonstrated by using atomistic simulations. The simulations are supported by transmission electron microscopy (TEM) of asymmetrical basal-pyramidal interfaces of twins. read less USED (low confidence) U. Tewary et al., “The Origin of Graphite Morphology in Cast Iron,” Acta Materialia. 2022. link Times cited: 13 USED (low confidence) Z. Jian et al., “Shock-induced plasticity and phase transformation in single crystal magnesium: an interatomic potential and non-equilibrium molecular dynamics simulations,” Journal of Physics: Condensed Matter. 2021. link Times cited: 8 Abstract: An effective and reliable Finnis–Sinclair (FS) type potentia… read moreAbstract: An effective and reliable Finnis–Sinclair (FS) type potential is developed for large-scale molecular dynamics (MD) simulations of plasticity and phase transition of magnesium (Mg) single crystals under high-pressure shock loading. The shock-wave profiles exhibit a split elastic–inelastic wave in the [0001]HCP shock orientation and a three-wave structure in the [10-10]HCP and [-12-10]HCP directions, namely, an elastic precursor, a followed plastic front, and a phase-transition front. The shock Hugoniot of the particle velocity (U p) vs the shock velocity (U s) of Mg single crystals in three shock directions under low shock strength reveals apparent anisotropy, which vanishes with increasing shock strength. For the [0001]HCP shock direction, the amorphization caused by strong atomic strain plays an important role in the phase transition and allows for the phase transition from an isotropic stressed state to the product phase. The reorientation in the shock directions [10-10]HCP and [-12-10]HCP, as the primary plasticity deformation, leads to the compressed hexagonal close-packed (HCP) phase and reduces the phase-transition threshold pressure. The phase-transition pathway in the shock direction [0001]HCP includes a preferential contraction strain along the [0001]HCP direction, a tension along [-12-10]HCP direction, an effective contraction and shear along the [10-10]HCP direction. For the [10-10]HCP and [-12-10]HCP shock directions, the phase-transition pathway consists of two steps: a reorientation and the subsequent transition from the reorientation hexagonal close-packed phase (RHCP) to the body-centered cubic (BCC). The orientation relationships between HCP and BCC are (0001)HCP ⟨-12-10⟩HCP // {110}BCC ⟨001⟩BCC. Due to different slipping directions during the phase transition, three variants of the product phase are observed in the shocked samples, accompanied by three kinds of typical coherent twin-grain boundaries between the variants. The results indicate that the highly concentrated shear stress leads to the crystal lattice instability in the elastic precursor, and the plasticity or the phase transition relaxed the shear stress. read less USED (low confidence) M. S. Nitol, S. Mun, D. Dickel, and C. Barrett, “Unraveling Mg 〈c + a〉 slip using neural network potential,” Philosophical Magazine. 2021. link Times cited: 8 Abstract: ABSTRACT Magnesium (Mg) activates 〈c + a〉 dislocation slip o… read moreAbstract: ABSTRACT Magnesium (Mg) activates 〈c + a〉 dislocation slip on the second order pyramidal slip plane. This slip mode is very complex compared to other modes including several metastable structures. Due to the complexity and very similar energies of the different structures, reliably modelling this slip mode is challenging. The problem is exacerbated when considering alloying, in which a combination of 1st order and 2nd order pyramidal slip is usually observed. Motivated by the need for a high fidelity potential for Mg alloys, we have developed first a highly accuracy potential for pure Mg. The new potential shows better agreement with density functional theory and experimental calculations than previous interatomic potentials for Mg. With the help of this new potential, we demonstrate that the basal dissociated 〈c + a〉 core is not sessile, as previously thought, and that constant stress molecular dynamics demonstrate clear preference for the 2nd order pyramidal system over the 1st order system. read less USED (low confidence) W. Wang, W. Liu, X. Yang, R. Xu, and Q. Dai, “Multi-scale simulation of the dendrite growth during selective laser melting of rare earth magnesium alloy,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 0 Abstract: The solidification microstructure of the alloy fabricated by… read moreAbstract: The solidification microstructure of the alloy fabricated by the selective-laser-melting (SLM) process can significantly impact its mechanical properties. In this study, a multi-scale model which couples the macroscale model for thermal-fluid and microscale cellular automata (CA) was proposed to simulate the complex solidification evolution and the dendrite growth (from planar to cellular to dendritic growth) during the SLM process. The solid–liquid interface of CA was dispersed with the bilinear interpolation method. On that basis, the curvature was accurately determined, and the calculation result was well verified by employing the Kurz–Giovanola–Trivedi analytical solution. The dendrite morphology, solute distribution, and primary dendrite arm spacing during the solidification of the SLM molten pool were quantitatively analyzed with the proposed model, well consistent with the experiment. The distribution of the undercooling field and the concentration field at the tip of dendrites different orientations were analyzed, and the two competing growth mechanisms of converging and diverging growth were revealed. Moreover, the research also indicates that during the growth of dendrites, the result of dendrite competition is determined by the height of the dendrite tip position in the direction of the thermal gradient, while the distribution of the concentration field (symmetrical or asymmetric) at the tip of the dendrite critically impacted the competing growth form of dendrites. read less USED (low confidence) J.-J. Tang et al., “Interaction between dislocation loop and 101¯2 twin boundary in magnesium,” Journal of Nuclear Materials. 2021. link Times cited: 4 USED (low confidence) C. Ji, X. Cai, Z. Zhou, F. Dong, S. Liu, and B. Gao, “Effects of intermetallic compound layer thickness on the mechanical properties of silicon-copper interface,” Materials & Design. 2021. link Times cited: 6 USED (low confidence) A. Kedharnath, R. Kapoor, and A. Sarkar, “Classical molecular dynamics simulations of the deformation of metals under uniaxial monotonic loading: A review,” Computers & Structures. 2021. link Times cited: 16 USED (low confidence) Y. Wang et al., “Revealing the Diversity of Dendritic Morphology Evolution During Solidification of Magnesium Alloys using Synchrotron X-ray Imaging: A Review,” Acta Metallurgica Sinica (English Letters). 2021. link Times cited: 5 USED (low confidence) J. Varillas, J. Očenášek, J. Torner, and J. Alcalá, “Understanding imprint formation, plastic instabilities and hardness evolutions in FCC, BCC and HCP metal surfaces,” Acta Materialia. 2021. link Times cited: 25 USED (low confidence) L. Burakovsky, S. R. Baty, and D. Errandonea, “Ab initio phase diagram of silver,” Journal of Physics: Condensed Matter. 2021. link Times cited: 4 Abstract: Silver has been considered as one of the simple one-phase ma… read moreAbstract: Silver has been considered as one of the simple one-phase materials that do not exhibit high pressure or high temperature polymorphism. The solid phase of Ag at ambient conditions is face-centered cubic (fcc) one. However, very recently another solid phase of silver, body-centered cubic (bcc) one, was detected in shock-wave (SW) experiments, and a more sophisticated phase diagram of Ag with the two solid phases was published by Smirnov. In this work, using a suite of ab initio quantum molecular dynamics (QMD) simulations based on the Z methodology which combines both direct Z method for the simulation of melting curves and inverse Z method for the calculation of solid–solid phase boundaries, we refine the phase diagram of Smirnov. We calculate the melting curves of both fcc-Ag and bcc-Ag and obtain an equation for the fcc–bcc solid–solid phase transition boundary. We also obtain the thermal equation of state of Ag which is in agreement with experimental data and QMD simulations. We argue that, despite being a polymorphic rather than a simple one-phase material, silver can be considered as an SW standard. read less USED (low confidence) X. Shen, B. Yao, Z. R. Liu, D. Legut, H. J. Zhang, and R. Zhang, “Mechanistic insights into interface-facilitated dislocation nucleation and phase transformation at semicoherent bimetal interfaces,” International Journal of Plasticity. 2021. link Times cited: 11 USED (low confidence) X. Zhou, G. Jiang, S. Song, J. Li, and L. Liu, “Molecular dynamics simulation of enhanced interfacial cohesive behavior of Ni-coated MWCNT/Mg composites,” Composite Interfaces. 2021. link Times cited: 1 Abstract: ABSTRACT Debonding processes of different Ni-coated MWCNT/Mg… read moreAbstract: ABSTRACT Debonding processes of different Ni-coated MWCNT/Mg interface models were studied by molecular dynamics simulations, and the stress–displacement relationships of different interface models were obtained by fitting a modified exponential cohesive zone law for interface. In addition, the debonding behaviors of Ni-coated and uncoated MWCNT/Mg interface were compared. The results show that CNT wall numbers and inner diameters as well as interface orientations have significant effects on the Ni-coated and uncoated MWCNT/Mg interface cohesive-zone model parameters. Compared with the above two kinds of uncoated MWCNT/Mg interface models, the average values of the peak stress and the work of separation for the Ni-coated MWCNT/Mg interface models are both increased significantly, showing that the addition of a Ni-modified coating on the surface of MWCNTs can effectively enhance the adhesion capacity of the MWCNT/Mg interface. Graphical abstract read less USED (low confidence) Z. Hao, Z. Lou, and Y. Fan, “Study on staged work hardening mechanism of nickel-based single crystal alloy during atomic and close-to-atomic scale cutting,” Precision Engineering-journal of The International Societies for Precision Engineering and Nanotechnology. 2021. link Times cited: 20 USED (low confidence) X.-Y. Zhou, H. Fu, J.-hua Zhu, and X. Yang, “Atomistic simulations of the surface severe plastic deformation-induced grain refinement in polycrystalline magnesium: The effect of processing parameters,” Journal of Magnesium and Alloys. 2021. link Times cited: 7 USED (low confidence) L.-F. Zhu, J. Janssen, S. Ishibashi, F. Körmann, B. Grabowski, and J. Neugebauer, “A fully automated approach to calculate the melting temperature of elemental crystals,” Computational Materials Science. 2021. link Times cited: 17 USED (low confidence) A. Vlasova, “Parallel computing for the simulation of deformation of hexagonal close-packed crystal (HCP).” 2020. link Times cited: 0 Abstract: Modern materials science requires extensive development of p… read moreAbstract: Modern materials science requires extensive development of parallel computing algorithms on supercomputers to solve a wide range of fundamental and applied problems at the atomistic level. The accumulation of new results, along with trivial experimental data and theoretical models, is facilitated by a number of software packages developed for this purpose. The main characteristics of plastic deformation in the case of poly- and nanocrystals of magnesium were calculated using the method of molecular dynamics on the URAN supercomputer (Ural branch of the Russian Academy of Sciences). Stress–strain curves for magnesium nanocrystals under high-speed deformation are constructed. The results are compared with experimental data. Various dislocation structures responsible for different levels of strength and plasticity of the nanocrystals under consideration are given. read less USED (low confidence) Z. Zhang et al., “101¯2 twinning nucleation in magnesium assisted by alternative sweeping of partial dislocations via an intermediate precursor,” Journal of Magnesium and Alloys. 2020. link Times cited: 10 USED (low confidence) S. A. Etesami, M. Laradji, and E. Asadi, “Reliability of molecular dynamics interatomic potentials for modeling of titanium in additive manufacturing processes,” Computational Materials Science. 2020. link Times cited: 5 USED (low confidence) C. Xu, L. Yuan, D. Shan, and B. Guo, “1 0 −1 2 twin boundaries migration accompanied by void in magnesium,” Computational Materials Science. 2020. link Times cited: 4 USED (low confidence) G. Hu, C. Luo, L. Wu, Q. Tang, Z. Ren, and B. Xu, “Molecular dynamics simulation of solid/liquid interfacial energy of uranium,” Journal of Nuclear Materials. 2020. link Times cited: 7 USED (low confidence) C. Xue, H.-zhu Wang, Z. Chu, Y.-gui Li, and H. Gui, “Influence of pore size on the plastic deformation of c-axis-compressed magnesium single crystals,” Materials Research Express. 2020. link Times cited: 1 Abstract: The molecular dynamics method is used to establish a single … read moreAbstract: The molecular dynamics method is used to establish a single crystal model of magnesium with different void sizes. Uniaxial compression along the c-axis is carried out at 300 K. Combined with the stress–strain curve, potential energy curve and dislocation density curve of the four models, the compression mechanical energy and structural evolution process of a single crystal of magnesium with different hole sizes are analysed. Results show that when the radius of the single spherical void is large, the elastic modulus is small, the yield stress is low, the potential energy value is large, and the absolute value is small, such conditions facilitate deformation. When the hole radius is small, complete closure under c-axis compression requires minimal time and deformation. read less USED (low confidence) R. Yan, S. Ma, W. Sun, T. Jing, and H. Dong, “The solid–liquid interface free energy of Al: A comparison between molecular dynamics calculations and experimental measurements,” Computational Materials Science. 2020. link Times cited: 8 USED (low confidence) S. Yoshikawa and D. Matsunaka, “Defect nucleation from a pre-existing intrinsic I1 stacking fault in magnesium by molecular dynamics simulations,” Computational Materials Science. 2020. link Times cited: 3 USED (low confidence) R. Namakian, G. Voyiadjis, and P. Kwaśniak, “On the slip and twinning mechanisms on first order pyramidal plane of magnesium: Molecular dynamics simulations and first principal studies,” Materials & Design. 2020. link Times cited: 19 USED (low confidence) Z. Tang, Y. Chen, and W. Ye, “Calculation of Surface Properties of Cubic and Hexagonal Crystals through Molecular Statics Simulations,” Crystals. 2020. link Times cited: 8 Abstract: Surface property is an important factor that is widely consi… read moreAbstract: Surface property is an important factor that is widely considered in crystal growth and design. It is also found to play a critical role in changing the constitutive law seen in the classical elasticity theory for nanomaterials. Through molecular static simulations, this work presents the calculation of surface properties (surface energy density, surface stress and surface stiffness) of some typical cubic and hexagonal crystals: face-centered-cubic (FCC) pure metals (Cu, Ni, Pd and Ag), body-centered-cubic (BCC) pure metals (Mo and W), diamond Si, zincblende GaAs and GaN, hexagonal-close-packed (HCP) pure metals (Mg, Zr and Ti), and wurzite GaN. Sound agreements of the bulk and surface properties between this work and the literature are found. New results are first reported for the surface stiffness of BCC pure metals, surface stress and surface stiffness of HCP pure metals, Si, GaAs and GaN. Comparative studies of the surface properties are carried out to uncover trends in their behaviors. The results in this work could be helpful to the investigation of material properties and structure performances of crystals. read less USED (low confidence) D. Spearot, V. Taupin, K. Dang, and L. Capolungo, “Structure and kinetics of three-dimensional defects on the 101¯2 twin boundary in magnesium: Atomistic and phase-field simulations,” Mechanics of Materials. 2020. link Times cited: 14 USED (low confidence) B. Yao and R. F. Zhang, “AADIS: An atomistic analyzer for dislocation character and distribution,” Comput. Phys. Commun. 2020. link Times cited: 19 USED (low confidence) S. Wang, H. Pan, A. He, P. Wang, and F.-guo Zhang, “Amorphous structure in single-crystal magnesium under compression along the
c
axis with ultrahigh strain rate,” Physical Review B. 2019. link Times cited: 3 USED (low confidence) A. Vlasova, “Hardening of [0001]-magnesium nanocrystals: Molecular dynamics simulation,” PROCEEDINGS OF THE INTERNATIONAL CONFERENCE ON ADVANCED MATERIALS WITH HIERARCHICAL STRUCTURE FOR NEW TECHNOLOGIES AND RELIABLE STRUCTURES 2019. 2019. link Times cited: 1 USED (low confidence) X. Zhou, W. Bu, S. Song, F. Sansoz, and X. Huang, “Multiscale modeling of interfacial mechanical behaviours of SiC/Mg nanocomposites,” Materials & Design. 2019. link Times cited: 22 USED (low confidence) N. T. Brown, E. Martínez, and J. Qu, “Solid-liquid metal interface definition studies using capillary fluctuation method,” Computational Materials Science. 2019. link Times cited: 3 USED (low confidence) X. Wan, J. Zhang, X. Mo, and F. Pan, “3D atomic-scale growth characteristics of 10–12 twin in magnesium,” Journal of Magnesium and Alloys. 2019. link Times cited: 21 USED (low confidence) A. Ostapovets and A. Gornakova, “On faceting of 101¯1 and 101¯2 twin boundaries in hcp metals,” Materials Letters. 2019. link Times cited: 4 USED (low confidence) C. Dai, P. Saidi, Z. Yao, L. Béland, and M. Daymond, “Deformation-free nanotwin formation in zirconium and titanium,” Materials Letters. 2019. link Times cited: 8 USED (low confidence) B. Liu et al., “Large plasticity in magnesium mediated by pyramidal dislocations,” Science. 2019. link Times cited: 229 Abstract: Smaller but more ductile Poor ductility is one limiting fact… read moreAbstract: Smaller but more ductile Poor ductility is one limiting factor in widespread use of strong but lightweight magnesium alloys in cars, trains, and planes. The usual way to try to circumvent this poor ductility is by adding other elements, which can be costly. Liu et al. show that very small samples of pure magnesium are much more ductile than previously believed (see the Perspective by Proust). The small samples suppress the deformation twinning that causes fractures in larger samples. Avoiding this mechanism should allow development of high-ductility magnesium and other metal alloys. Science, this issue p. 73; see also p. 30 Submicrometer-sized samples of magnesium suppress deformation mechanisms that normally prevent good ductility. Lightweight magnesium alloys are attractive as structural materials for improving energy efficiency in applications such as weight reduction of transportation vehicles. One major obstacle for widespread applications is the limited ductility of magnesium, which has been attributed to 〈 c+a 〉 dislocations failing to accommodate plastic strain. We demonstrate, using in situ transmission electron microscope mechanical testing, that 〈 c+a 〉 dislocations of various characters can accommodate considerable plasticity through gliding on pyramidal planes. We found that submicrometer-size magnesium samples exhibit high plasticity that is far greater than for their bulk counterparts. Small crystal size usually brings high stress, which in turn activates more 〈 c+a 〉 dislocations in magnesium to accommodate plasticity, leading to both high strength and good plasticity. read less USED (low confidence) D. Spearot, L. Capolungo, and C. Tomé, “Shear-driven motion of Mg
101¯2
twin boundaries via disconnection terrace nucleation, growth, and coalescence,” Physical Review Materials. 2019. link Times cited: 15 USED (low confidence) L. B. Marinho, P. Filho, and V. Albuquerque, “Ultrasonic sensor signals and self organized mapping with nearest neighbors for the microstructural characterization of thermally-aged Inconel 625 alloy,” Comput. Ind. 2019. link Times cited: 5 USED (low confidence) Q. Li, Q. Li, and X. Jiao, “Recrystallization mechanism and activation energies of severely-deformed magnesium during annealing process,” Materialia. 2019. link Times cited: 12 USED (low confidence) M. Becker, J. Dantzig, M. Kolbe, S. T. Wiese, and F. Kargl, “Dendrite orientation transition in Al Ge alloys,” Acta Materialia. 2019. link Times cited: 39 USED (low confidence) J. Du et al., “Effect of additional solute elements (X= Al, Ca, Y, Ba, Sn, Gd and Zn) on crystallographic anisotropy during the dendritic growth of magnesium alloys,” Journal of Alloys and Compounds. 2019. link Times cited: 13 USED (low confidence) S. H. Zhang, D. Legut, and R. F. Zhang, “PNADIS: An automated Peierls-Nabarro analyzer for dislocation core structure and slip resistance,” Comput. Phys. Commun. 2018. link Times cited: 24 USED (low confidence) A. Vlasova, “Deformation features of magnesium [11̄01]- and [0001]-nanocrystals.” 2018. link Times cited: 1 USED (low confidence) A. Vlasova, “Deformation features of magnesium [11̅01]- and [0001]-nanocrystal with hydrogen and vacancies.” 2018. link Times cited: 0 USED (low confidence) H. Fan, Y. Zhu, and Q. Wang, “Effect of precipitate orientation on the twinning deformation in magnesium alloys,” Computational Materials Science. 2018. link Times cited: 19 USED (low confidence) J. Zhang, Y. Zhang, J. El-Awady, and Y. Tang, “The plausibility of dislocation slip on -12-11 planes in Mg,” Scripta Materialia. 2018. link Times cited: 6 USED (low confidence) Z. Pei, “An overview of modeling the stacking faults in lightweight and high-entropy alloys: Theory and application,” Materials Science and Engineering: A. 2018. link Times cited: 33 USED (low confidence) D. Smirnova, S. Starikov, and A. Vlasova, “New interatomic potential for simulation of pure magnesium and magnesium hydrides,” Computational Materials Science. 2018. link Times cited: 17 USED (low confidence) S. Shuai et al., “Synchrotron tomographic quantification of the influence of Zn concentration on dendritic growth in Mg-Zn alloys,” Acta Materialia. 2018. link Times cited: 42 USED (low confidence) J. Du, A. Zhang, Z. Guo, M. Yang, M. Li, and S. Xiong, “Mechanism of the growth pattern formation and three-dimensional morphological transition of hcp magnesium alloy dendrite,” Physical Review Materials. 2018. link Times cited: 22 USED (low confidence) K. Kushnir and A. Ostapovets, “Variability of Twin Boundary Structure in Computer Simulations of Tensile Twins in Magnesium,” Defect and Diffusion Forum. 2018. link Times cited: 3 Abstract: Variety of interatomic potentials for magnesium can be found… read moreAbstract: Variety of interatomic potentials for magnesium can be found in the literature. Result of computer simulations can be slightly different depending on used potential. Particularly, twin boundary structure with the lowest energy can be different in a frame of different models. Comparison of several popular embedded-atom method potentials is provided. It is shown that either reflection or glide structure of twin boundary has the lowest energy for different potentials. read less USED (low confidence) H. Fan, Y. Zhu, J. El-Awady, and D. Raabe, “Precipitation hardening effects on extension twinning in magnesium alloys,” International Journal of Plasticity. 2018. link Times cited: 86 USED (low confidence) D. Bigoni, D. Capuani, and D. Giarola, “Scattering of Waves by a Shear Band.” 2018. link Times cited: 0 USED (low confidence) A. Mayer, V. Krasnikov, and V. V. Pogorelko, “Limit of Ultra-high Strain Rates in Plastic Response of Metals.” 2018. link Times cited: 8 USED (low confidence) M. Mendelev et al., “Molecular dynamics simulation of the solid-liquid interface migration in terbium.,” The Journal of chemical physics. 2018. link Times cited: 16 Abstract: We developed a Tb embedded atom method potential which prope… read moreAbstract: We developed a Tb embedded atom method potential which properly reproduces the liquid structure obtained from the ab initio molecular dynamics simulation, the hexagonal close packed (hcp)-body-centered cubic (bcc) phase transformation, and melting temperatures. At least three crystal phases [hcp, face-centered cubic (fcc), and bcc] described by this potential can coexist with the liquid phase. Thus, the developed potential provides an excellent test bed for studies of the completive phase nucleation and growth in a single component system. The molecular dynamics simulation showed that all crystal phases can grow from the liquid phase close to their melting temperatures. However, in the cases of the hcp and fcc growth from the liquid phase at very large supercoolings, the bcc phase forms at the solid-liquid interface in the close packed orientations in spite of the fact that both hcp and fcc phases are more stable than the bcc phase at these temperatures. This bcc phase closes the hcp and fcc phase from the liquid such that the remaining liquid solidifies into the bcc phase. The initial hcp phase then slowly continues growing in expense of the bcc phase. read less USED (low confidence) G. Li, Y.-bao Wang, M. Xiang, Y. Liao, K. Wang, and J. Chen, “Shock response of nanoporous magnesium by molecular dynamics simulations,” International Journal of Mechanical Sciences. 2018. link Times cited: 44 USED (low confidence) G. Kurtuldu, K. F. Shamlaye, and J. F. Löffler, “Metastable quasicrystal-induced nucleation in a bulk glass-forming liquid,” Proceedings of the National Academy of Sciences of the United States of America. 2018. link Times cited: 34 Abstract: Significance A model alloy, Mg69Zn27Yb4, concurrently forms … read moreAbstract: Significance A model alloy, Mg69Zn27Yb4, concurrently forms bulk metallic glass, metastable quasicrystals (QCs), and crystalline approximant phases from the melt. We demonstrate that a transient QC phase nucleates first from the melt and subsequently transforms into an equilibrium approximant phase. This nucleation path is likely to be a general mechanism in metastable QC-forming systems. We observed a metastable-to-stable phase transformation when we deployed fast differential scanning calorimetry using the experimental strategy of interrupted cooling after the onset of crystallization followed by heating at ultrafast rates to “up-quench” the previously frozen structure. This strategy can yield the discovery of hidden transient phases that are key to understanding the crystallization behavior in metallic systems, polymers, biological solutions, and pharmaceutical substances. This study presents a unique Mg-based alloy composition in the Mg–Zn–Yb system which exhibits bulk metallic glass, metastable icosahedral quasicrystals (iQCs), and crystalline approximant phases in the as-cast condition. Microscopy revealed a smooth gradual transition from glass to QC. We also report the complete melting of a metastable eutectic phase mixture (including a QC phase), generated via suppression of the metastable-to-stable phase transition at high heating rates using fast differential scanning calorimetry (FDSC). The melting temperature and enthalpy of fusion of this phase mixture could be measured directly, which unambiguously proves its metastability in any temperature range. The kinetic pathway from liquid state to stable solid state (an approximant phase) minimizes the free-energy barrier for nucleation through an intermediate state (metastable QC phase) because of its low solid–liquid interfacial energy. At high undercooling of the liquid, where diffusion is limited, another approximant phase with near-liquid composition forms just above the glass-transition temperature. These experimental results shed light on the competition between metastable and stable crystals, and on glass formation via system frustration associated with the presence of several free-energy minima. read less USED (low confidence) M. Han, L. Li, and G. Zhao, “Shearing single crystal magnesium in the close-packed basal plane at different temperatures,” Physica B-condensed Matter. 2018. link Times cited: 0 USED (low confidence) H.-S. Jang, K.-M. Kim, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for pure Zn and Mg-Zn binary system,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2018. link Times cited: 23 USED (low confidence) A. Ostapovets and O. Vatazhuk, “Non-Schmid behavior of extended dislocations in computer simulations of magnesium,” Computational Materials Science. 2018. link Times cited: 9 USED (low confidence) T. Vo and B. H. Kim, “Physical origins of temperature continuity at an interface between a crystal and its melt.,” The Journal of chemical physics. 2018. link Times cited: 11 Abstract: We justify and discuss the physical origins for the assumpti… read moreAbstract: We justify and discuss the physical origins for the assumption of temperature continuity at crystal/melt interfaces by performing atomistic simulations. We additionally answer why the crystal/melt interfaces differ from the typical solid/liquid interfaces, which usually exhibit dissimilarities and a resulting temperature drop. We present results for pure silver modeled using the embedded-atom method and Lennard-Jones potential function and contrast the results with each other. We find that the temperature continuity at an interface between a crystal and its melt originates from the perfect vibrational coupling, which is caused by the interfacial structural diffusivity. This study provides fundamental insights into the heat transfer for cases of extremely large heat flux and thermal gradients occurring during rapid melting and solidification. The findings additionally determine the role of rough surfaces in manipulating the thermal conductance in nanodevices. read less USED (low confidence) J. Du, A. Zhang, Z. Guo, M. Yang, M. Li, and S. Xiong, “Atomistic Determination of Anisotropic Surface Energy-Associated Growth Patterns of Magnesium Alloy Dendrites,” ACS Omega. 2017. link Times cited: 26 Abstract: Because of the existence of anisotropic surface energy with … read moreAbstract: Because of the existence of anisotropic surface energy with respect to the hexagonal close-packed (hcp) lattice structure, magnesium alloy dendrite prefers to grow along certain crystallographic directions and exhibits a complex growth pattern. To disclose the underlying mechanism behind the three-dimensional (3-D) growth pattern of magnesium alloy dendrite, an anisotropy function was developed in light of the spherical harmonics and experimental findings. Relevant atomistic simulations based on density functional theory were then performed to determine the anisotropic surface energy along different crystallographic directions, and the corresponding anisotropic strength was quantified via the least-square regression. Results of phase field simulations showed that the proposed anisotropy function could satisfactorily describe the 3-D growth pattern of the α-Mg dendrite observed in the experiments. Our investigations shed great insight into understanding the pattern formation of the hcp magnesium alloy dendrite at an atomic level. read less USED (low confidence) Y. Wang et al., “Effect of cooling rates on the dendritic morphology transition of Mg–6Gd alloy by in situ X-ray radiography,” Journal of Materials Science & Technology. 2017. link Times cited: 24 USED (low confidence) A. Vlasova and A. G. Kesarev, “Simulation of uniaxial deformation of hexagonal crystals (Mg, Be).” 2017. link Times cited: 2 Abstract: Molecular dynamics (MD) simulations were performed for the n… read moreAbstract: Molecular dynamics (MD) simulations were performed for the nanocompression loading of nanocrystalline magnesium and beryllium modeled by an interatomic potential of the embedded atom method (EAM). It is shown that the main deformation modes are prismatic slip and twinning for magnesium, and only prismatic slip for beryllium. The formation of stable configurations of dislocation grids in magnesium and beryllium was observed. Dislocation networks are formed in the habit plane of the twin in a magnesium nanocrystall. Some dislocation reactions are suggested to explain the appearance of such networks. Shockley partial dislocations in a beryllium nanocrystall form grids in the slip plane. A strong anisotropy between slip systems was observed, which is in agreement with experimental data. read less USED (low confidence) J. Du, Z. Guo, M. Yang, and S. Xiong, “Growth pattern and orientation selection of magnesium alloy dendrite: From 3-D experimental characterization to theoretical atomistic simulation,” Materials today communications. 2017. link Times cited: 16 USED (low confidence) F. Jianglei et al., “Effects of solidification parameters on the growth direction of α phase in directionally solidified Ti-49Al alloy,” Intermetallics. 2017. link Times cited: 9 USED (low confidence) H. Somekawa, T. Tsuru, A. Singh, S. Miura, and C. Schuh, “Effect of crystal orientation on incipient plasticity during nanoindentation of magnesium,” Acta Materialia. 2017. link Times cited: 32 USED (low confidence) D. E. L. Ojos and J. Sort, “Nanoindentation Modeling: From Finite Element to Atomistic Simulations.” 2017. link Times cited: 1 USED (low confidence) C. Grégoire and M. Ponga, “Nanovoid failure in Magnesium under dynamic loads,” Acta Materialia. 2017. link Times cited: 11 USED (low confidence) H. Chu, H. Huang, and J. Wang, “Clustering on Magnesium Surfaces – Formation and Diffusion Energies,” Scientific Reports. 2017. link Times cited: 4 USED (low confidence) C. Qi, B. Xu, L. Kong, and J. Li, “Solid-liquid interfacial free energy and its anisotropy in the Cu-Ni binary system investigated by molecular dynamics simulations,” Journal of Alloys and Compounds. 2017. link Times cited: 21 USED (low confidence) E. Apfelbaum and V. S. Vorob’ev, “The application of the Zeno line similarities to alkaline earth metals,” Journal of Molecular Liquids. 2017. link Times cited: 9 USED (low confidence) G. Han, H. Wang, D. Lin, X. Y. Zhu, S. Hu, and H. Song, “Phase-field modeling of void anisotropic growth behavior in irradiated zirconium,” Computational Materials Science. 2017. link Times cited: 9 USED (low confidence) A. Kumar, B. Morrow, R. Mccabe, and I. Beyerlein, “An atomic-scale modeling and experimental study of 〈c+a〉 dislocations in Mg,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2017. link Times cited: 34 USED (low confidence) K. Yashiro, “Local lattice instability analysis on mode I crack tip in hcp-Mg: Unstable mode for crack propagation vs. dislocation emission,” Computational Materials Science. 2017. link Times cited: 20 USED (low confidence) D. Casari et al., “Morphological Transition of α-Mg Dendrites During Near-Isothermal Solidification of a Mg–Nd–Gd–Zn–Zr Casting Alloy.” 2017. link Times cited: 0 USED (low confidence) G. Agarwal and A. Dongare, “Atomistic study of shock Hugoniot of single crystal Mg.” 2017. link Times cited: 10 Abstract: Molecular Dynamics (MD) simulations are carried out to inves… read moreAbstract: Molecular Dynamics (MD) simulations are carried out to investigate the shock Hugoniot for single crystal Mg using a planar shock. The MD simulations were carried out using two embedded atom method (EAM) potentials for impact velocities in the range of 500 m/s – 2000 m/s. The anisotropic behavior of shock wave propagation with loading orientation of the crystal is investigated by computing the particle velocity profiles for impact along [0001], [112¯0] and [101¯0] orientations. A split two wave (elastic-plastic) structure above the Hugoniot elastic limit is observed for all the three orientations considered. The shock pressure and the shock velocity of the elastic and the plastic wave are computed as functions of impact velocity. The Hugoniot response predicted for both the potentials agrees very well with the experimental data and therefore suggests that these can be reliably used to model the shock response of single crystal Mg. read less USED (low confidence) H. Liu, X. Cheng, and N. Valanoor, “Universal Approach for Predicting Crystallography of Heterogeneous Epitaxial Nanocrystals with Multiple Orientation Relationships.,” ACS applied materials & interfaces. 2016. link Times cited: 4 Abstract: Heteroepitaxial nanocrystals are one of the most fundamental… read moreAbstract: Heteroepitaxial nanocrystals are one of the most fundamentally and technologically important classes of materials systems. The correlation between form, dictated by crystallographic features such as growth habit and direction, and function, in terms of the ultimate physio-chemical properties is well established, thus placing an onus on precise synthesis control of nanocrystal morphology. Yet, nanocrystal heteroepitaxy can be a frustrating, time-consuming iterative process, particularly during the initial stages of development. What is desired is a powerful predictive tool that is able to successfully predict not only the interface or habit plane, but also rationalize the occurrence of epitaxial growth complexities such as multiple orientation relationships (MORs) and high-index faceting planes for a diverse range of materials. Here we provide such a powerful approach that is based on an invariant deformation element (IDE) model, and fundamentally founded on the crystallography of diffusional phase transformations. We demonstrate its impact by detailed computations supported by transmission electron microscopy evidence, for an archetypical complex metal oxide nanocrystal system (that has up to five MORs for three differing growth orientations). The method is then applied to successfully explain growth for different materials ranging from metals to metal carbides to transition metal oxides, even in thin film form. Thus, this relatively simple yet powerful predictive guide significantly reduces the systemic inefficiencies of guesswork and blind growth. Ultimately it can be easily integrated with machine learning techniques toward reliable and efficient advanced nanomaterials fabrication. read less USED (low confidence) J. R. Espinosa, C. Navarro, E. Sanz, C. Valeriani, and C. Vega, “On the time required to freeze water.,” The Journal of chemical physics. 2016. link Times cited: 61 Abstract: By using the seeding technique the nucleation rate for the f… read moreAbstract: By using the seeding technique the nucleation rate for the formation of ice at room pressure will be estimated for the TIP4P/ICE model using longer runs and a smaller grid of temperatures than in the previous work. The growth rate of ice will be determined for TIP4P/ICE and for the mW model of water. Although TIP4P/ICE and mW have a similar melting point and melting enthalpy, they differ significantly in the dynamics of freezing. The nucleation rate of mW is lower than that of TIP4P/ICE due to its higher interfacial free energy. Experimental results for the nucleation rate of ice are between the predictions of these two models when obtained from the seeding technique, although closer to the predictions of TIP4P/ICE. The growth rate of ice for the mW model is four orders of magnitude larger than for TIP4P/ICE. Avrami's expression is used to estimate the crystallization time from the values of the nucleation and growth rates. For mW the minimum in the crystallization time is found at approximately 85 K below the melting point and its value is of about a few ns, in agreement with the results obtained from brute force simulations by Moore and Molinero. For the TIP4P/ICE the minimum is found at about 55 K below the melting point, but its value is about ten microseconds. This value is compatible with the minimum cooling rate required to avoid the formation of ice and obtaining a glass phase. The crossover from the nucleation controlled crystallization to the growth controlled crystallization will be discussed for systems of finite size. This crossover could explain the apparent discrepancy between the values of J obtained by different experimental groups for temperatures below 230 K and should be considered as an alternative hypothesis to the two previously suggested: internal pressure and/or surface freezing effects. A maximum in the compressibility was found for the TIP4P/ICE model in supercooled water. The relaxation time is much smaller than the crystallization time at the temperature at which this maximum occurs, so this maximum is a real thermodynamic feature of the model. At the temperature of minimum crystallization time, the crystallization time is larger than the relaxation time by just two orders of magnitude. read less USED (low confidence) A. Vlasova, A. Nikonov, A. Zhuravlev, and A. G. Kesarev, “Dislocation structure of the magnesium nanocrystal in uniaxial loading.” 2016. link Times cited: 2 Abstract: We report on molecular-dynamics (MD) simulations of compress… read moreAbstract: We report on molecular-dynamics (MD) simulations of compression loading of nanocrystalline magnesium modeled by the embedded-atom method (EAM) potential. It is shown that plastic deformation is by basal slip and (102) twinning. The formation of stable configurations of dislocation grids is observed. Some dislocation reactions are suggested to explain the occurrence of grids. The structure of the dislocation core is shown with the Burgers vector 1/18[04¯43]. read less USED (low confidence) D. Casari et al., “α-Mg primary phase formation and dendritic morphology transition in solidification of a Mg-Nd-Gd-Zn-Zr casting alloy,” Acta Materialia. 2016. link Times cited: 35 USED (low confidence) K. Matsubara, H. Kimizuka, and S. Ogata, “Formation of 112¯1 twins from I1-type stacking faults in Mg: A molecular dynamics study,” Computational Materials Science. 2016. link Times cited: 8 USED (low confidence) M. Yang, S. Xiong, and Z. Guo, “Effect of different solute additions on dendrite morphology and orientation selection in cast binary magnesium alloys,” Acta Materialia. 2016. link Times cited: 60 USED (low confidence) M. Uranagase and R. Matsumoto, “Tension–compression asymmetry in uniaxial deformation of a magnesium bicrystal with [1¯100] symmetric tilt grain boundary,” Computational Materials Science. 2016. link Times cited: 7 USED (low confidence) C. Barrett and R. Cariño, “The MEAM parameter calibration tool: an explicit methodology for hierarchical bridging between ab initio and atomistic scales,” Integrating Materials and Manufacturing Innovation. 2016. link Times cited: 8 USED (low confidence) R. Babicheva et al., “Effect of grain boundary segregation on the deformation mechanisms and mechanical properties of nanocrystalline binary aluminum alloys,” Computational Materials Science. 2016. link Times cited: 49 USED (low confidence) D. Buey and M. Ghazisaeidi, “Atomistic simulation of screw dislocation cross-slip in Mg,” Scripta Materialia. 2016. link Times cited: 22 USED (low confidence) H. Song and J. Hoyt, “A molecular dynamics study of heterogeneous nucleation at grain boundaries during solid-state phase transformations,” Computational Materials Science. 2016. link Times cited: 29 USED (low confidence) K. Xiong, Y. Zhang, and J. Gu, “Deformation Twinning in Hexagonal Close-Packed Single Crystals under Uniaxial Compression,” Materials Science Forum. 2016. link Times cited: 0 Abstract: In this paper, the uniaxial compression of Mg, Ti, Zr and Co… read moreAbstract: In this paper, the uniaxial compression of Mg, Ti, Zr and Co single crystals along the direction is performed by molecular dynamics (MD) to investigate the elastic-to-plastic transition in these hexagonal close-packed (hcp) metals. Two deformation twinning modes are observed in these simulations, including the twinning in Ti, Zr and Co and the [0001] twinning in Mg. The underlying atomistic mechanisms of these twinning modes are analyzed in detail. read less USED (low confidence) I. A. Alhafez and H. Urbassek, “Scratching of hcp metals: A molecular-dynamics study,” Computational Materials Science. 2016. link Times cited: 39 USED (low confidence) M. Uranagase and R. Matsumoto, “Effects of normal stresses on the homogeneous nucleation of a basal dislocation in magnesium,” Computational Materials Science. 2016. link Times cited: 2 USED (low confidence) P. Yi, R. Cammarata, and M. Falk, “Atomistic simulation of solid solution hardening in Mg/Al alloys: Examination of composition scaling and thermo-mechanical relationships,” Acta Materialia. 2016. link Times cited: 20 USED (low confidence) K. Matsubara, H. Kimizuka, and S. Ogata, “Long-range intercluster interactions of solute nanoprecipitates in Mg–Y alloys: A first-principles study,” Journal of Alloys and Compounds. 2016. link Times cited: 12 USED (low confidence) E. Asadi and M. A. Zaeem, “Predicting Solidification Properties of Magnesium by Molecular Dynamics Simulations.” 2016. link Times cited: 0 USED (low confidence) Y. Jiang, L. Lv, and Y. Wu, “Solid-Liquid Phase Transitions of FCC-Al and HCP-Mg Nanoparticles.” 2016. link Times cited: 1 USED (low confidence) X. Zhou, S. Song, L. Li, and R.-jie Zhang, “Molecular dynamics simulation for mechanical properties of magnesium matrix composites reinforced with nickel-coated single-walled carbon nanotubes,” Journal of Composite Materials. 2016. link Times cited: 17 Abstract: As the interfacial structure and bonding strength play an im… read moreAbstract: As the interfacial structure and bonding strength play an important role in determining the mechanical performance of carbon nanotube reinforced metal matrix composite, investigating the interfacial mechanical properties of surface modified carbon nanotube reinforced metal matrix composite becomes one of the key factors for the improvement. The mechanical behaviors of nickel-coated single-walled carbon nanotube reinforced magnesium matrix composites were investigated using molecular dynamics simulation method. The results show that the Young's modulus of the nickel-coated single-walled carbon nanotube/Mg composite is obviously larger than that of the uncoated single-walled carbon nanotube/Mg composite. The results also show that the interfacial bonding of single-walled carbon nanotube/Mg composite can be drastically increased by addition of nickel coating to improve the wettability of the nanotube surface and Mg matrix. Furthermore, the influences of nickel coating number on the interfacial bonding characteristics of single-walled carbon nanotube/Mg composites also were studied. For three types of nickel coating number, i.e. without nickel coating, with one layer of nickel and two layers of nickel, the final pullout interfacial bonding strength of the nickel-coated single-walled carbon nanotube from Mg matrix about are 3.9 and 11.9 times larger, respectively, than that of the uncoated single-walled carbon nanotube. The simulation results have proved that such interfaces can effectively transfer load between the nanotube and magnesium matrix in the carbon nanotube/Mg composite, and this will provide the theoretical and experimental basis for the interface mechanics design of the carbon nanotube reinforced composites. read less USED (low confidence) C. Ni, H. Ding, M. Asta, and X. Jin, “Computational study of symmetric tilt grain boundaries in Mg and Ti,” Scripta Materialia. 2015. link Times cited: 19 USED (low confidence) E. Hahn and M. Meyers, “Grain-size dependent mechanical behavior of nanocrystalline metals,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 162 USED (low confidence) A. Monas, O. Shchyglo, D. Höche, M. Tegeler, and I. Steinbach, “Dual-scale phase-field simulation of Mg-Al alloy solidification,” IOP Conference Series: Materials Science and Engineering. 2015. link Times cited: 6 Abstract: Phase-field simulations of the nucleation and growth of prim… read moreAbstract: Phase-field simulations of the nucleation and growth of primary α-Mg phase as well as secondary, β-phase of a Mg-Al alloy are presented. The nucleation model for α- and β-Mg phases is based on the “free growth model” by Greer et al.. After the α-Mg phase solidification we study a divorced eutectic growth of α- and β-Mg phases in a zoomed in melt channel between α-phase dendrites. The simulated cooling curves and final microstructures of α-grains are compared with experiments. In order to further enhance the resolution of the interdendritic region a high-performance computing approach has been used allowing significant simulation speed gain when using supercomputing facilities. read less USED (low confidence) E. Asadi, M. A. Zaeem, S. Nouranian, and M. Baskes, “Two-Phase Solid-Liquid Coexistence of Ni, Cu, and Al by Molecular Dynamics Simulations using the Modified Embedded-Atom Method,” Acta Materialia. 2015. link Times cited: 94 USED (low confidence) K. Kim, J. Jeon, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for Mg–X (X=Y, Sn, Ca) binary systems,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2015. link Times cited: 70 USED (low confidence) J. Xiao, R.-B. Li, and Y. Wu, “Ostwald’s Step Rule in the Crystallization of Supercooled Magnesium from Molecular Dynamic Simulation.” 2015. link Times cited: 0 USED (low confidence) A. Ostapovets, P. Molnár, and P. Lejček, “Boundary plane distribution for Σ13 grain boundaries in magnesium,” Materials Letters. 2014. link Times cited: 14 USED (low confidence) S. Groh, “Transformation of shear loop into prismatic loops during bypass of an array of impenetrable particles by edge dislocations,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2014. link Times cited: 15 USED (low confidence) I. Shin and E. Carter, “Simulations of dislocation mobility in magnesium from first principles,” International Journal of Plasticity. 2014. link Times cited: 47 USED (low confidence) Y.-F. Guo, S. Xu, X. Tang, Y. Wang, and S. Yip, “Twinnability of hcp metals at the nanoscale,” Journal of Applied Physics. 2014. link Times cited: 15 Abstract: Twinning is generally considered to be the primary deformati… read moreAbstract: Twinning is generally considered to be the primary deformation mechanism for hexagonal close-packed (hcp) metals due to their limited slip systems. Recent microcompression experiments point to strong size effects indicating that pyramidal slips can dominate in deformation under compression. We present analysis on the twinnability of an ideal hcp single crystal at the nanoscale. A criterion for deformation twinning is derived by considering the elastic lattice-rotation strain, and the result tested against molecular dynamics simulations of magnesium and titanium single crystals. We find ⟨c + a⟩ pyramidal slip dominates the compression deformation at the nanoscale, which is consistent with experimental observations on microcompression. This analysis gives an interpretation of size effects in deformation twinning, at the same time it provides an explanation for the so-called strength differential effect. read less USED (low confidence) R. Aghababaei and S. Joshi, “Micromechanics of tensile twinning in magnesium gleaned from molecular dynamics simulations,” Acta Materialia. 2014. link Times cited: 51 USED (low confidence) M. Ghazisaeidi, L. Hector, and W. Curtin, “First-principles core structures of edge and screw dislocations in Mg,” Scripta Materialia. 2014. link Times cited: 76 USED (low confidence) I. Shin and E. Carter, “First-principles simulations of plasticity in body-centered-cubic magnesium–lithium alloys,” Acta Materialia. 2014. link Times cited: 35 USED (low confidence) R.-B. Li and Y. Wu, “Nucleation and Growth of Nucleus in Supercooled Liquid Fe: A Molecular Dynamics Study.” 2014. link Times cited: 0 USED (low confidence) M. Liao, B. Li, and M. Horstemeyer, “Interaction between prismatic slip and a Mg17Al12 precipitate in magnesium,” Computational Materials Science. 2013. link Times cited: 29 USED (low confidence) A. Moitra, “Grain size effect on microstructural properties of 3D nanocrystalline magnesium under tensile deformation,” Computational Materials Science. 2013. link Times cited: 26 USED (low confidence) M. Tane, Y. Nagai, H. Kimizuka, K. Hagihara, and Y. Kawamura, “Elastic properties of an Mg-Zn-Y alloy single crystal with a long-period stacking-ordered structure,” Acta Materialia. 2013. link Times cited: 118 USED (low confidence) H. Zhou, X. Lin, M. Wang, and W. Huang, “Calculation of solid-liquid interfacial free energy of Cu by two different methods,” Journal of Crystal Growth. 2013. link Times cited: 11 USED (low confidence) A. Ostapovets and P. Molnár, “On the relationship between the ‘shuffling-dominated’ and ‘shear-dominated’ mechanisms for 101¯2 twinning in magnesium,” Scripta Materialia. 2013. link Times cited: 58 USED (low confidence) M. Liao, B. Li, and M. Horstemeyer, “Unstable dissociation of a prismatic dislocation in magnesium,” Scripta Materialia. 2013. link Times cited: 13 USED (low confidence) Z. Pei et al., “Ab initio and atomistic study of generalized stacking fault energies in Mg and Mg–Y alloys,” New Journal of Physics. 2013. link Times cited: 107 Abstract: Magnesium–yttrium alloys show significantly improved room te… read moreAbstract: Magnesium–yttrium alloys show significantly improved room temperature ductility when compared with pure Mg. We study this interesting phenomenon theoretically at the atomic scale employing quantum-mechanical (so-called ab initio) and atomistic modeling methods. Specifically, we have calculated generalized stacking fault energies for five slip systems in both elemental magnesium (Mg) and Mg–Y alloys using (i) density functional theory and (ii) a set of embedded-atom-method (EAM) potentials. These calculations predict that the addition of yttrium results in a reduction in the unstable stacking fault energy of basal slip systems. Specifically in the case of an I2 stacking fault, the predicted reduction of the stacking fault energy due to Y atoms was verified by experimental measurements. We find a similar reduction for the stable stacking fault energy of the non-basal slip system. On the other hand, other energies along this particular γ-surface profile increase with the addition of Y. In parallel to our quantum-mechanical calculations, we have also developed a new EAM Mg–Y potential and thoroughly tested its performance. The comparison of quantum-mechanical and atomistic results indicates that the new potential is suitable for future large-scale atomistic simulations. read less USED (low confidence) Q. Cao, P.-P. Wang, D.-hui Huang, Q. Li, F.-hou Wang, and L. Cai, “Pressure Dependence of Fusion Entropy and Fusion Volume of Six Metals,” Journal of Chemical & Engineering Data. 2013. link Times cited: 6 Abstract: Molecular dynamics simulations of the melting curves of six … read moreAbstract: Molecular dynamics simulations of the melting curves of six metals including Ag, Cu, Al, Mg, Ta, and Mo for the pressure range (0 to 15) GPa are reported. The melting curves of Ag, Cu, Al, and Mg fully confirm measurements and previous calculations. Meanwhile, the melting curves of Ta and Mo are consistent with previous calculations but diverge from laser-heated diamond-anvil cells values at high pressure. Our results suggest that the melting slope at 100 kPa is related to the electronic configuration of the element. In addition, the pressure dependence of fusion entropy and fusion volume are calculated up to 15 GPa. The overall fusion entropy is separated into topological entropy of fusion (ΔSD) due to the configuration change in melting and the volume entropy of fusion (ΔSV) due to the latent volume change in melting. Furthermore, we checked the R ln 2 rule under high pressure, according to which the value of ΔSD is a constant at ambient pressure. Result shows that the value of ΔSD is close to R ln 2 at... read less USED (low confidence) C. Barrett, H. Kadiri, and M. Tschopp, “Breakdown of the Schmid law in homogeneous and heterogeneous nucleation events of slip and twinning in magnesium,” Journal of The Mechanics and Physics of Solids. 2012. link Times cited: 116 USED (low confidence) L. Wang, T. Fang, and Y. Qi, “Crystal Growth of Ni on Liquid-Solid Interface,” Applied Mechanics and Materials. 2012. link Times cited: 0 Abstract: Molecular dynamics simulations have been performed to explor… read moreAbstract: Molecular dynamics simulations have been performed to explore the crystal growth of solid - liquid interface of pure Ni by using a potential of embedded atom (EAM) type. The solid-liquid interface is structured by liquid-solid-liquid, considering the (100) orientation. The crystal growth rates are determined by observing interfacial moving velocity, the calculated kinetic growth coefficient μ, which is defined as the ratio of kinetic growth velocity to the interface undercooled temperature, is 60cm/s/K. The melting temperature determined by time dependence of the volume per particle for different temperature is 1740 K, which is well agreement with experimental values and other simulated ones. read less USED (low confidence) J. Hoyt, M. Asta, and A. Karma, “Atomistic Simulations of Solute Trapping and Solute Drag.” 2012. link Times cited: 2 USED (low confidence) C. Barrett, M. Tschopp, and H. Kadiri, “Automated analysis of twins in hexagonal close-packed metals using molecular dynamics,” Scripta Materialia. 2012. link Times cited: 33 USED (low confidence) S. Gurevich, M. Amoorezaei, D. Montiel, and N. Provatas, “Evolution of microstructural length scales during solidification of magnesium alloys,” Acta Materialia. 2012. link Times cited: 13 USED (low confidence) P. Bavli, E. Polturak, and J. Adler, “Molecular dynamics study of melting of the hcp metal Mg,” Physical Review B. 2011. link Times cited: 13 Abstract: We present molecular dynamics simulations of the melting tra… read moreAbstract: We present molecular dynamics simulations of the melting transition of Mg, an hcp metal, using the potential developed by Sun et al. This study was motivated by the question of whether the hierarchy of premelting phenomena, found to occur between different facets of metals with an fcc or bcc structure, is also present in hcp metals. We first determined the structural and energetic properties of the effectively infinite solid with no boundaries. We then investigated the low-index surfaces of Mg, namely the c (0001), a (1010), and s (1011) facets. We found that as the temperature increases, the (1010) surface disorders first, followed by the (1011) surface, while the (0001) surface remains stable up to the melting temperature. The disorder spreads from the surface into the bulk, establishing a thin quasiliquid film in the surface region. We conclude that the effect of premelting phenomena is inversely proportional to the surface atomic density, being most pronounced at the a (1010) facet which has the lowest density. This conclusion is in line with the behavior found for fcc and bcc metals. read less USED (low confidence) M. Wang, J. J. Williams, L. Jiang, F. Carlo, T. Jing, and N. Chawla, “Dendritic morphology of α-Mg during the solidification of Mg-based alloys: 3D experimental characterization by X-ray synchrotron tomography and phase-field simulations,” Scripta Materialia. 2011. link Times cited: 48 USED (low confidence) T. Tang, S. Kim, J. Jordon, M. Horstemeyer, and P. T. Wang, “Atomistic simulations of fatigue crack growth and the associated fatigue crack tip stress evolution in magnesium single crystals,” Computational Materials Science. 2011. link Times cited: 28 USED (low confidence) V. D. Nguyen, Z. Hu, and P. Schall, “Single crystal growth and anisotropic crystal-fluid interfacial free energy in soft colloidal systems.,” Physical review. E, Statistical, nonlinear, and soft matter physics. 2011. link Times cited: 21 Abstract: We measure the anisotropy of the crystal-fluid interfacial f… read moreAbstract: We measure the anisotropy of the crystal-fluid interfacial free energy in soft colloidal systems. A temperature gradient is used to direct crystal nucleation and control the growth of large single crystals in order to achieve well-equilibrated crystal-fluid interfaces. Confocal microscopy is used to follow both the growth process and the equilibrium crystal-fluid interface at the particle scale: heterogeneous crystal nucleation, the advancing interface, and the stationary equilibrium interface. We use the measured growth velocity to determine the chemical potential difference between crystal and fluid phases. Well-equilibrated, large crystal-fluid interfaces are then used to determine the interfacial free energy and its anisotropy directly from thermally excited interface fluctuations. We find that while the measured average interfacial free energy is in good agreement with values found in simulations, the anisotropy is significantly larger than simulation values. Finally, we investigate the effect of impurities on the advancing interface. We determine the critical force needed to overcome impurity particles from the local interface curvature. read less USED (low confidence) A. A. Potter and J. Hoyt, “A molecular dynamics simulation study of the crystal–melt interfacial free energy and its anisotropy in the Cu–Ag–Au ternary system,” Journal of Crystal Growth. 2011. link Times cited: 28 USED (low confidence) D.-H. Kim, F. Ebrahimi, M. Manuel, J. Tulenko, and S. Phillpot, “Grain-boundary activated pyramidal dislocations in nano-textured Mg by molecular dynamics simulation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2011. link Times cited: 43 USED (low confidence) V. D. Nguyen, M. T. Dang, B. Weber, Z. Hu, and P. Schall, “Visualizing the Structural Solid–Liquid Transition at Colloidal Crystal/Fluid Interfaces,” Advanced Materials. 2011. link Times cited: 12 Abstract: The crystal/melt interface is central to the understanding o… read moreAbstract: The crystal/melt interface is central to the understanding of crystal nucleation and the morphological stability of crystal growth, but it is difficult to study experimentally. Micrometer-sized stimuli-dependent colloidal particles are assembled into large crystals using temperature fields. The structure of the colloidal crystal/melt interface is imaged directly in three dimensions. Interface reconstructions allow direct connection of the structure and interface free energy. read less USED (low confidence) C. Barrett, M. Tschopp, H. Kadiri, and B. Li, “Influence of Crystallographic Orientation on Twin Nucleation in Single Crystal Magnesium,” Magnesium Technology. 2011. link Times cited: 3 USED (low confidence) T. Tang, S. Kim, M. Horstemeyer, and P. T. Wang, “A molecular dynamics study of fracture behavior in magnesium single crystal,” Magnesium Technology. 2011. link Times cited: 4 USED (low confidence) D. Trinkle, J. Yasi, and L. Hector, “Predicting Mg Strength from First-Principles: Solid-Solution Strengthening, Softening, and Cross-Slip.” 2011. link Times cited: 2 USED (low confidence) K. Solanki, A. Moitra, and M. A. Bhatia, “Effect of Substituted Aluminum in Magnesium Tension Twin.” 2011. link Times cited: 2 USED (low confidence) M. Amoorezaei, S. Gurevich, and N. Provatas, “Dendritic Microstructure in Directional Solidification of Magnesium Alloys.” 2011. link Times cited: 1 USED (low confidence) J. Han, X. Su, Z. Jin, and Y. Zhu, “Basal‐Plane Stacking‐Fault Energies of Mg: A First‐Principles Study of Li‐ and Al‐Alloying Effects,” Scripta Materialia. 2011. link Times cited: 125 USED (low confidence) G. Caginalp and E. Esenturk, “Anisotropic phase field equations of arbitrary order,” Discrete and Continuous Dynamical Systems - Series S. 2010. link Times cited: 26 Abstract: We derive a set of higher order phase field equations using … read moreAbstract: We derive a set of higher order phase field equations using a microscopic
interaction Hamiltonian with detailed anisotropy in the interactions of the
form $a_{0}+\delta\sum_{n=1}^{N}{a_{n}\cos( 2n\theta) + b_{n}\sin( 2n\theta) }$ where $\theta$ is the angle with
respect to a fixed axis, and $\delta$ is a parameter. The Hamiltonian is
expanded using complex Fourier series, and leads to a free energy and phase
field equation with arbitrarily high order derivatives in the spatial
variable. Formal asymptotic analysis is performed on these phase field
equation in terms of the interface thickness in order to obtain the
interfacial conditions. One can capture $2N$-fold anisotropy by retaining at
least $2N^{th}$ degree phase field equation. We derive, in the limit of small
$\delta,$ the classical result $( T-T_{E} ) [s]_{E}=-\kappa
{\sigma( \theta ) + \sigma^{''}(
\theta) }$ where $T-T_{E}$ is the difference between the temperature
at the interface and the equilibrium temperature between phases, $[s]_{E}$ is
the entropy difference between phases, $\sigma$ is the surface tension and
$\kappa$ is the curvature. If there is only one mode in the anisotropy [i.e.,
the sum contains only one term: $A_{n}\cos( 2n\theta) $] then
this identity is exact (valid for any magnitude of $\delta$) if the surface
tension is interpreted as the sharp interface limit of excess free energy
obtained by the solution of the $2N^{th}$ degree differential equation. The
techniques rely on rewriting the sums of derivatives using complex variables
and combinatorial identities, and performing formal asymptotic analyses for
differential equations of arbitrary order. read less USED (low confidence) Y. Gao, Y. Yang, D. Sun, M. Asta, and J. Hoyt, “Molecular dynamics simulations of the crystal–melt interface mobility in HCP Mg and BCC Fe,” Journal of Crystal Growth. 2010. link Times cited: 28 USED (low confidence) T. Tang, S. Kim, and M. Horstemeyer, “Molecular dynamics simulations of void growth and coalescence in single crystal magnesium,” Acta Materialia. 2010. link Times cited: 90 USED (low confidence) L. Shen, G. Proust, and G. Ranzi, “An atomistic study of dislocation-solute interaction in Mg-Al alloys,” IOP Conference Series: Materials Science and Engineering. 2010. link Times cited: 6 Abstract: In this study, atomistic simulations are performed to study … read moreAbstract: In this study, atomistic simulations are performed to study the dislocation-Al solute effect on the strength of Mg alloys. At temperature T = 0 K, molecular mechanics simulations are carried out to investigate the effect of Al solute concentration on the Peierls stress of basal plane edge and screw dislocations. It is found that the Peierls stress will increase by at least one order of magnitude when 0.25 at% Al atoms are randomly introduced in the Mg alloys with an edge dislocation. Generally, the Peierls stress will increase with the increase of the Al concentration up to 8 at%. Furthermore, the interaction between the basal plane edge dislocation and Al solute at T = 300 K is studied using molecular dynamics. It appears from the simulations that the critical shear stress increases with the Al solute concentration. Comparing with the solute effect at T = 0 K, however, the critical shear stress at T = 300 K is lower since the kinetic energy of the atoms can help the dislocation conquer the energy barriers created by the Al atoms. read less USED (low confidence) J. Eiken, “Phase-Field Simulations of Dendritic Orientation Selection in Mg-Alloys with Hexagonal Anisotropy,” Materials Science Forum. 2010. link Times cited: 8 Abstract: A special feature of Mg solidification is the anisotropy of … read moreAbstract: A special feature of Mg solidification is the anisotropy of the hexagonal closed packed lattice, which under directional growth conditions causes a strong crystallographic texture. Although this primary growth texture is in technical processes masked by subsequent solid state processes, its understanding can be helpful for efficient microstructure optimization. The aim of the present work is to study the fundamental orientation selection mechanisms by numerical simulation. For this pur-pose, a phase-field model has been extended to allow for complex 3D anisotropic interfacial ener-gies and interfacial mobilities, calibrated by data from molecular dynamics studies. The model is first applied in 3D to Mg-6%Al, revealing two major stages of texture formation. Directly after nuc-leation, all grains with basal plane parallel to the gradient direction are selected. During further competitive growth, grains with <1120> closely aligned to the temperature gradient commonly pre-vail, but process dependent also other orientations of the basal plane (between <1120> and <1010>) may coexist. The latter phenomenon is investigated in detail in 2D for the ternary alloy AZ31. read less USED (low confidence) J. Eiken, “Phase-field simulation of microstructure formation in technical magnesium alloys,” International Journal of Materials Research. 2010. link Times cited: 29 Abstract: A phase-field model is presented which is specially tailored… read moreAbstract: A phase-field model is presented which is specially tailored for engineering-oriented application. It addresses multiphase, multicomponent alloys and considers lattice specific crystallographic anisotropy. Versatile application examples to magnesium alloys demonstrate the model's capability of handling important aspects of microstructure formation during solidification. Thermodynamic data is derived from a Calphad database for the Mg – Al – Zn – Ca – Mn system. First, orientation selection and texture evolution is studied under directional growth conditions. A new approach is proposed to model the anisotropy of the hexagonal close-packed magnesium phase. Subsequently, grain structure formation is simulated for technical Mg – Al-based alloys under equiaxed growth conditions. It is shown how the cooling rate and the addition of specific alloy elements affect the grain size. Finally, the solidification path and its effect on the precipitation of the secondary phase MgAl has been investigated. read less USED (low confidence) S. Groh, E. Marin, and M. Horstemeyer, “NANOSCALE VOID GROWTH IN MAGNESIUM: A MOLECULAR DYNAMICS STUDY,” International Journal of Applied Mechanics. 2010. link Times cited: 15 Abstract: Molecular dynamics calculations were carried out in single c… read moreAbstract: Molecular dynamics calculations were carried out in single crystal magnesium specimens to reveal the dependence of strain rate, temperature, and orientation of the crystal on damage evolution as defined by pore growth. Two specific crystallographic orientations [0001] and were examined. During a [0001] tensile test, twin boundaries developed at the void surface leading to a constraint on the crystallographic orientation. On the other hand, during the tensile deformation, emission of shear loops in the prismatic slip planes arose when void growth initiated. Furthermore, analysis of the damage components (nucleation, growth and coalescence) revealed that a large number of small voids nucleated that rapidly grew and fractured the specimens independent of the temperature and the strain rate. read less USED (low confidence) C. G. Johansen, H.-C. Huang, and T. Lu, “Diffusion and formation energies of adatoms and vacancies on magnesium surfaces,” Computational Materials Science. 2009. link Times cited: 27 USED (low confidence) H. Zhang, “Atomistic simulation of sliding of [101¯0] tilt grain boundaries in Mg,” Journal of Materials Research. 2009. link Times cited: 14 Abstract: A series of molecular dynamics simulations was performed to … read moreAbstract: A series of molecular dynamics simulations was performed to study grain boundary sliding of three types of [101¯0] tilt grain boundaries in a magnesium bicrystal. In particular, a near S11 twin boundary, an asymmetric near S11 twin boundary, and a ? = 40.3° general [101¯0] tilt grain boundary were studied. Simulations showed that grain boundary sliding (a rigid motion of two grains relative to each other along boundary plane) did not occur over the stress range applied; instead, coupled shear motion (grain boundary sliding induced boundary migration) was dominant. Although the measured coupling coefficient, the ratio of boundary tangential displacement to boundary normal displacement, was in good agreement with theoretical prediction, the detailed shear behavior was different, depending on types of grain boundary, magnitude of applied shear stress, and temperature. It was also noted that grain boundary twining was the predominant mechanism that allowed the coupled shear motion to occur in hexagonal close-packed (HCP) magnesium. read less USED (low confidence) M. Wang, T. Jing, and B. Liu, “Phase-field simulations of dendrite morphologies and selected evolution of primary α-Mg phases during the solidification of Mg-rich Mg-Al-based alloys,” Scripta Materialia. 2009. link Times cited: 26 USED (low confidence) J. Eiken, “Dendritic growth texture evolution in Mg-based alloys investigated by phase-field simulation,” International Journal of Cast Metals Research. 2009. link Times cited: 31 Abstract: Under directional growth conditions, a strong crystallograph… read moreAbstract: Under directional growth conditions, a strong crystallographic texture is observed in Mg-based alloys due to the anisotropy of the hcp-phase. The aim of the present work is to investigate the fundamental mechanisms of this growth texture evolution by microstructure simulations. For this purpose, a phase-field model has been extended to allow for complex 3D anisotropic interfacial energies and mobilities, calibrated by data from molecular dynamics studies. The model is first applied in 2D to AZ31 (Mg–3%Al–1%Zn). Bicrystal simulations are performed for a systematic study of grain interactions. Extended simulations are compared to experiments. Comprehensive texture evolution is investigated in 3D for Mg–6%Al. Texture is found to evolve mainly due to retarded growth in basal <0001> orientations. Grains with the fast growing <1120> orientation most closely aligned to the temperature gradient commonly prevail, but process dependent also other orientations of the basal plane (between <1120> and <1010>) may coexist. read less USED (low confidence) M. Mendelev and Y. Mishin, “Molecular dynamics study of self-diffusion in bcc Fe,” Physical Review B. 2009. link Times cited: 99 Abstract: A semiempirical interatomic potential for Fe was used to cal… read moreAbstract: A semiempirical interatomic potential for Fe was used to calculate the diffusivity in bcc Fe assuming the vacancy and interstitial mechanisms of self-diffusion. Point-defect concentrations and diffusivities were obtained directly from molecular dynamics (MD) simulations. It was found that self-diffusion in bcc Fe is controlled by the vacancy mechanism at all temperatures. This result is due to the fact that the equilibrium vacancy concentration is always much larger than the equilibrium interstitial concentration. The predominance of the equilibrium vacancy concentration over the interstitial concentration is explained by the lower vacancy-formation energy at low temperatures and high vacancy-formation entropy at high temperatures. The calculated diffusivity is in good agreement with experimental data. The MD simulations were also used to test the quasiharmonic (QH) approximation for point-defect calculations. It was found that the QH approximation can considerably underestimate variations in point-defect characteristics with temperature. read less USED (low confidence) M. Li, T. Tamura, and K. Miwa, “Controlling microstructures of AZ31 magnesium alloys by an electromagnetic vibration technique during solidification: From experimental observation to theoretical understanding,” Acta Materialia. 2007. link Times cited: 83 USED (low confidence) J. Zhou, Y.-F. Guo, J. Ren, and X. Tang, “The mechanism of hcp-bcc phase transformation in Mg single crystal under high pressure,” Scripta Materialia. 2023. link Times cited: 0 USED (low confidence) J. F. Troncoso and V. Turlo, “Evaluating the applicability of classical and neural network interatomic potentials for modeling body centered cubic polymorph of magnesium,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 2 Abstract: Magnesium (Mg) is one of the most abundant metallic elements… read moreAbstract: Magnesium (Mg) is one of the most abundant metallic elements in nature and presents attractive mechanical properties in the industry. Particularly, it has a low density and relatively high strength/weight and stiffness/weight ratios, which make it one of the most attractive lightweight metals. However, the huge potential of Mg is restricted by its low ductility, associated with its hexagonal close packed (hcp) structure. This problem can be solved if Mg adopts the body centered cubic (bcc) structure, which is stable at high pressure or in confinement with stiff bcc metals like Nb. Molecular dynamics method is a magnificent tool to study material’s structure and deformation mechanisms at the atomic level, however, requiring accurate interatomic potentials. The majority of the interatomic potentials available in the literature for Mg have only been fitted to the properties of its stable hcp phase. In the present work, we perform systematic study of applicability of currently available Mg potentials to modeling the properties of metastable bcc polymorph of Mg, taking into account cohesive energy curves, elastic constants, stacking fault energies, and phonon dispersion curves. We conclude that the modified embedded atom method (MEAM) potentials are the most suitable for investigating bcc Mg in Mg/Nb nano-composites, while the properties of high-pressure bcc Mg would be better modeled by neural network interatomic potentials after different local atomic environments corresponding to bcc Mg being included into the fitting database. read less USED (low confidence) Z. Xing, H. Fan, J.-J. Tang, B. Wang, and G. Kang, “Molecular dynamics simulation on the cyclic deformation of magnesium single crystals,” Computational Materials Science. 2021. link Times cited: 22 USED (low confidence) L. Jiang et al., “Twin nucleation from a single dislocation in hexagonal close-packed crystals,” Acta Materialia. 2021. link Times cited: 9 USED (low confidence) K. Dang, C. Tomé, and L. Capolungo, “Three-Dimensional Atomistic Simulations of $ \left{ 10\bar12 \right}$ Non-cozone Twin–Twin Interaction in Mg—Role of Twin Stability and Mobility.” 2021. link Times cited: 0 USED (low confidence) L. Zhou and W. He, “Mechanical Behavior and the Strengthening Mechanism of LSP-Induced Gradient Microstructure in Metal Materials.” 2021. link Times cited: 0 USED (low confidence) Q. Dong, P. Saidi, L. Béland, Z. Yao, C. Dai, and M. Daymond, “In situ TEM and multiscale study of dislocation loop formation in the vicinity of a grain boundary,” Journal of Nuclear Materials. 2020. link Times cited: 6 USED (low confidence) S. Groh and M. K. Nahhas, “Modeling Dislocation in Binary Magnesium-Based Alloys Using Atomistic Method,” Handbook of Mechanics of Materials. 2019. link Times cited: 1 USED (low confidence) M. Jahanshahi, A. Khoei, N. Heidarzadeh, and N. Jafarian, “A hierarchical thermo-mechanical multi-scale technique for modeling of edge dislocations in nano-crystalline structures,” Computational Materials Science. 2018. link Times cited: 13 USED (low confidence) D. Okumura, M. Otsuka, and Y. Shibutani, “Mechanism of Plastic Deformation in a Magnesium Nanotwinned Structure by Molecular Dynamics Simulations,” Journal of The Society of Materials Science, Japan. 2018. link Times cited: 0 Abstract: In this study, we perform molecular dynamics simulations to … read moreAbstract: In this study, we perform molecular dynamics simulations to investigate plastic deformation modes of a magnesium (Mg) nanotwinned structure. Periodic units including (1011) twin boundaries (TBs) are analyzed using an embedded atom method potential. Equal spaces between the TBs are assumed at the initial state and the space is parametrized in the range between about 5 nm and 30 nm. It is found that plastic deformation is triggered by the slip along a (1011) twinning plane near a TB, and that this event induces two different deformation modes depending on the space, i.e., the migration of the TBs and the evolution of double twinning, which leads to void nucleation and polycrystallization. The plastic deformation provided by the two different modes is quantitatively verified from geometric analysis. As the space decreases, the migration of the TBs is superior to the evolution of double twinning. read less USED (low confidence) P. Saidi and J. Hoyt, “Atomistic simulation of the step mobility at the Al–Si(1 1 1) crystal–melt interface using molecular dynamics,” Computational Materials Science. 2016. link Times cited: 8 USED (low confidence) M. Uranagase and R. Matsumoto, “Basal Dislocation Nucleated from a Free Surface of an Elliptic Cylindrical Mg Single Crystal,” Journal of The Society of Materials Science, Japan. 2016. link Times cited: 2 Abstract: Dislocation nucleation from free surfaces is one of the impo… read moreAbstract: Dislocation nucleation from free surfaces is one of the important process of plastic deformation of a size reduced pillar, in which pre-existing dislocations is not sufficient to take plastic deformation. In this work, nucleation of a basal dislocation from a free surface of an elliptic cylindrical Mg single crystal is considered. Molecular dynamics simulations of tensile deformation of crystals show that the nucleation site of a dislocation changes when the ratio between axes of the ellipse changes, though the critical resolved shear stress is almost the same. Dependence of the activation free energy of nucleation of a dislocation on the nucleation site is also studied by applying metadynamics method to the atomistic simulation. Then, we found that the activation free energy of nucleation of a dislocation lowers at the site where a dislocation is actually nucleated in molecular dynamics simulations. read less USED (low confidence) D. Matsunaka, Y. Shibutani, and Y. Ohnishi, “Molecular Dynamics Analyses of Fracture Toughness of Magnesium,” Journal of The Society of Materials Science, Japan. 2016. link Times cited: 1 Abstract: Crack propagation in Magnesium was investigated using molecu… read moreAbstract: Crack propagation in Magnesium was investigated using molecular dynamics (MD) simulations. By using a disk-shaped atomic cell containing a crack under the anisotropic linear elastic displacement, the deformation field around the crack tip was first atomistically resolved. In order to consider effects of surface energy on crack propagation, two kinds of interatomic potentials were adopted. For an embedded atom method (EAM) potential, basal and first-pyramidal surfaces are favorable cleavage planes due to relatively low surface energies. Thus, MD simulations using the EAM showed unstable crack propagation on those planes. For the other generalized embedded atom method (GEAM) potential which provides the higher surface energies instead, the basal plane has the larger critical stress intensity factor by the Griffith’s formula because it is proportional to square root of surface energy. Consequently, the dislocation nucleation and deformation twin nucleation were occurred in the vicinity of the crack tip. The other MD analyses of defect interaction between a crack and twin boundaries (TBs) were also carried out. Twinning dislocations of (101�2) twin were generated by reaction with the basal dislocations emitted from the crack tip, and thus (101�2) TB easily moved. On the other hand, a void or deformation twin was generated along the boundary region where the dislocations from the crack tip piled up, and then the crack propagation near (101�1) TB arose. read less USED (low confidence) C. Ni, H. Ding, and X. Jin, “Super-plasticity via secondary twinning in magnesium nanowire revealed by molecular dynamics simulations,” Computational Materials Science. 2016. link Times cited: 15 USED (low confidence) E. Asadi and M. A. Zaeem, “A Review of Quantitative Phase-Field Crystal Modeling of Solid–Liquid Structures,” JOM. 2015. link Times cited: 44 USED (low confidence) M. Uranagase, S. Kamigaki, and R. Matsumoto, “Analysis on Nucleation of a Dislocation Loop in a Magnesium Single Crystal -Approach from Atomistic Simulations-,” Journal of The Society of Materials Science, Japan. 2014. link Times cited: 2 Abstract: We performed atomistic simulations to demonstrate the nuclea… read moreAbstract: We performed atomistic simulations to demonstrate the nucleation of a dislocation loop for the basal and prismatic slips of a magnesium single crystal. The dislocation loop was successfully simulated by employing the constrained molecular dynamics method, which involves constraining the atomic motion above and below the activation site, keeping the distance between the domain centers constant in the slip direction. Then, the activation free energy for nucleation of a dislocation loop was evaluated using the thermodynamic integration method and the dependence of this nucleation on the ambient temperature and applied shear stress was studied. Moreover, we compared the activation free energy of nucleation from a free surface to that of homogeneous nucleation in order to investigate the effect of inhomogeneity on the activation free energy. read less USED (low confidence) S. Agnew, “Deformation mechanisms of magnesium alloys.” 2012. link Times cited: 58 Abstract: Abstract: This chapter discusses the deformation mechanisms … read moreAbstract: Abstract: This chapter discusses the deformation mechanisms responsible for the complex mechanical behaviors exhibited by Mg and its alloys. The use of new experimental (e.g. electron backscattered diffraction (EBSD) and in situ neutron diffraction) and computational (e.g. crystal plasticity, molecular dynamics, and electron density functional theory) approaches has recently provided a wealth of new understanding. These findings are enumerated for each of the basal and non-basal dislocation slip mechanisms as well as deformation twinning and kink banding. The impacts of alloy and microstructure design strategies to improve strength, ductility, formability, and tolerance for dynamic loading are discussed for the individual mechanisms. read less USED (low confidence) T. Tang, S. Kim, M. Horstemeyer, and P. T. Wang, “Atomistic modeling of crack growth in magnesium single crystal,” Engineering Fracture Mechanics. 2011. link Times cited: 27 NOT USED (low confidence) J. Jofré, A. Gheribi, and J. Harvey, “Development of a flexible quasi-harmonic-based approach for fast generation of self-consistent thermodynamic properties used in computational thermochemistry,” Calphad. 2023. link Times cited: 0 NOT USED (low confidence) A. Ostapovets and O. Vatazhuk, “Комп’ютерне моделювання впливу гідростатичного тиску на ковзання гвинтових дислокацій в Mg.” 2020. link Times cited: 0 Abstract: Atomistic modeling of hydrostatic pressure influence on crit… read moreAbstract: Atomistic modeling of hydrostatic pressure influence on critical resolved shear stress was performed for glide of screw dislocation in magnesium. It was found that application of pressure can change the resolved critical stress for basal and prismatic slip. The effect is dependent on dislocation core structure. It can be connected to the pressure dependence transient dilatation of the dislocation core. read less NOT USED (high confidence) M. Poul, L. Huber, E. Bitzek, and J. Neugebauer, “Systematic atomic structure datasets for machine learning potentials: Application to defects in magnesium,” Physical Review B. 2022. link Times cited: 3 Abstract: We present a physically motivated strategy for the construct… read moreAbstract: We present a physically motivated strategy for the construction of training sets for transferable machine learning interatomic potentials. It is based on a systematic exploration of all possible space groups in random crystal structures, together with deformations of cell shape, size, and atomic positions. The resulting potentials turn out to be unbiased and generically applicable to studies of bulk defects without including any defect structures in the training set or employing any additional Active Learning. Using this approach we construct transferable potentials for pure Magnesium that reproduce the properties of hexagonal closed packed (hcp) and body centered cubic (bcc) polymorphs very well. In the process we investigate how different types of training structures impact the properties and the predictive power of the resulting potential. read less NOT USED (high confidence) K. Dang, D. Blaschke, S. Fensin, and D. Luscher, “Limiting velocities and transonic dislocations in Mg,” Computational Materials Science. 2022. link Times cited: 5 NOT USED (high confidence) D. Tourret, H. Liu, and J. Llorca, “Phase-field modeling of microstructure evolution: Recent applications, perspectives and challenges,” Progress in Materials Science. 2021. link Times cited: 61 NOT USED (high confidence) M. Stricker, B. Yin, E. Mak, and W. Curtin, “Machine learning for metallurgy II. A neural-network potential for magnesium,” Physical Review Materials. 2020. link Times cited: 26 Abstract: Interatomic potentials are essential for studying fundamenta… read moreAbstract: Interatomic potentials are essential for studying fundamental mechanisms of deformation and failure in metals and alloys because the relevant defects (dislocations, cracks, etc.) are far above the scales accessible to first-principles studies. Existing potentials for non-fcc metals and nearly all alloys are, however, not sufficiently quantitative for many crucial phenomena. Here machine learning in the Behler-Parrinello neural-network framework is used to create a broadly applicable potential for pure hcp magnesium (Mg). Lightweight Mg and its alloys are technologically important while presenting a diverse range of slip systems and crystal surfaces relevant to both plasticity and fracture that present a significant challenge for any potential. The machine learning potential is trained on first-principles density-functional theory (DFT) computable metallurgically relevant properties and is then shown to well predict metallurgically crucial dislocation and crack structures and competing phenomena. Extensive comparisons to an existing very good modified embedded atom method potential are made. These results demonstrate that a single machine learning potential can represent the wide scope of phenomena required for metallurgical studies. The DFT database is openly available for use in any other machine learning method. The method is naturally extendable to alloys, which are necessary for engineering applications but where ductility and fracture are controlled by complex atomic-scale mechanisms that are not well predicted by existing potentials. read less NOT USED (high confidence) W. Jiang, Y. Zhang, L. Zhang, and H. Wang, “Accurate Deep Potential model for the Al–Cu–Mg alloy in the full concentration space*,” arXiv: Materials Science. 2020. link Times cited: 24 Abstract: Combining first-principles accuracy and empirical-potential … read moreAbstract: Combining first-principles accuracy and empirical-potential efficiency for the description of the potential energy surface (PES) is the philosopher's stone for unraveling the nature of matter via atomistic simulation. This has been particularly challenging for multi-component alloy systems due to the complex and non-linear nature of the associated PES. In this work, we develop an accurate PES model for the Al-Cu-Mg system by employing Deep Potential (DP), a neural network based representation of the PES, and DP Generator (DP-GEN), a concurrent-learning scheme that generates a compact set of ab initio data for training. The resulting DP model gives predictions consistent with first-principles calculations for various binary and ternary systems on their fundamental energetic and mechanical properties, including formation energy, equilibrium volume, equation of state, interstitial energy, vacancy and surface formation energy, as well as elastic moduli. Extensive benchmark shows that the DP model is ready and will be useful for atomistic modeling of the Al-Cu-Mg system within the full range of concentration. read less NOT USED (high confidence) J. Ombogo, A. H. Zahiri, T. Ma, and L. Cao, “Nucleation of 1012 Twins in Magnesium through Reversible Martensitic Phase Transformation,” Metals. 2020. link Times cited: 6 Abstract: We report the discovery of a rigorous nucleation mechanism f… read moreAbstract: We report the discovery of a rigorous nucleation mechanism for {101¯2} twins in hexagonal close-packed (hcp) magnesium through reversible hcp-tetragonal-hcp martensitic phase transformations with a metastable tetragonal phase as the intermediate state. Specifically, the parent hcp phase first transforms to a metastable tetragonal phase, which subsequently transforms to a twinned hcp phase. The evanescent nature of the tetragonal phase severely hinders its direct observation, while our carefully designed molecular dynamics simulations rigorously reveal the critical role of this metastable phase in the nucleation of {101¯2} twins in magnesium. Moreover, we prove that the reversible hcp-tetragonal-hcp phase transformations involved in the twinning process follow strict orientation relations between the parent hcp, intermediate tetragonal, and twin hcp phases. This phase transformation-mediated twinning mechanism is naturally compatible with the ultrafast twin growth speed. This work will be important for a better understanding of the twinning mechanism and thus the development of novel strategies for enhancing the ductility of magnesium alloys. read less NOT USED (high confidence) W. Kurz, M. Rappaz, and R. Trivedi, “Progress in modelling solidification microstructures in metals and alloys. Part II: dendrites from 2001 to 2018,” International Materials Reviews. 2020. link Times cited: 94 Abstract: ABSTRACT This is the first account of the history of modelli… read moreAbstract: ABSTRACT This is the first account of the history of modelling dendritic and cellular solidification. While Part I reviewed the progress up to the year 2000 [Kurz W, Fisher DJ, Trivedi R. Progress in modelling solidification microstructures in metals and alloys: dendrites from 1700 to 2000. Intern Mater Rev. 2019;64:311–354], Part II retraces our modelling capabilities developed during the early years of the present century. Advances in in-situ X-ray observations of solidification of metallic alloys are also presented. While the most important contributions are mentioned, the authors are aware that such a historical review must leave many worthy articles by the wayside. This overview considers dendrite tip growth and morphology, rapid solidification, melt flow, fragmentation, columnar-to-equiaxed transition, dendrite spacings, coalescence, grain competition, and cellular growth. Modelling across the length scales from nano- up to macroscopic solidification phenomena by massive phase field computations or multiscale approaches show the potential for the simulation of real processes such as additive manufacturing, single crystal casting, welding or advanced solidification processes. read less NOT USED (high confidence) Y. Wu, K. Zhang, J. Xiao, Y. Jiang, and L. Lv, “Conjugated bilayer structure of the homogeneous solid-liquid interface of metals.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 1 Abstract: The concept of "interfacial region" has long preva… read moreAbstract: The concept of "interfacial region" has long prevailed for over half century for describing the homogeneous solid-liquid (SL) interface of metals, but its intrinsic structure is still unclear due to the homogeneity. In this study, we reveal, for the first time, the intrinsic structure of these homogeneous SL interfaces consisting of two conjugated monoatomic layers of interfacial solid (IS) and interfacial liquid (IL) with a certain degree of corrugation via molecular dynamics simulations. We named it as the conjugated bilayer structure (CBS). In the framework of CBS, only the IS + IL bilayer plays stepwise transition roles from the solid to the liquid, which defines the four-terrace nature of the interface and act simultaneously as the boundaries of the bilateral bulk phases. The inherent diffuse nature of the "interfacial region" is proven originating from the corrugation of the IS + IL bilayer and its four-terrace nature. More importantly, the CBS also explains that the interfacial free energy originates mainly from the increase in the potential energy of the IS layer relative to its counterpart bulk solid instead of the previously argued entropy loss of the liquid phase. After all these verifications and interpretations, the CBS was verified as the intrinsic structure of the homogeneous SL interface of metals. Accordingly, we argue that the concept of CBS also resolves the volume-bearing flaw of the "interfacial region" concept and can definitively locate the intrinsic surface according to the capillary wave theory. read less NOT USED (high confidence) X. Shi, X. Feng, B. Zhang, Y. Sun, W. He, and L. Zhou, “Research on microstructure deformation mechanism of crack tip in titanium under tension along different orientations,” Molecular Simulation. 2020. link Times cited: 2 Abstract: ABSTRACT The deformation process of titanium with an initial… read moreAbstract: ABSTRACT The deformation process of titanium with an initial crack was simulated under uniaxial tension by the molecular dynamics method. The effect of [0001], and orientations on the microstructure deformation mechanisms at the crack tip was investigated using embedded atom method potentials. In the simulation, various deformation evidence were clearly observed, such as dislocation nucleation and movement, stacking faults formation, deformation twinning and phase transformation. The stress–strain curves showed that orientations had a greater influence on the plastic stage and less on the elastic stage. By applying uniaxial tension with a constant strain rate along different orientations, microstructure deformation at the crack tip was analysed. When loading along [0001] orientation, twinning was the main deformation mechanism and stacking fault occurred and expanded along slip system with the increase in strain. Applying uniaxial tension along orientation, an abnormal ‘secondary increase’ phenomenon was observed in the stress–strain curve. At the same time, dislocation loops appeared at the crack tip and they expanded, merged with the increase in strain. The massive phase transformation from HCP to BCC was observed in the model when loading along orientation, which was caused by multiple Shockley incomplete dislocations under the size limitation. read less NOT USED (high confidence) W. Zheng et al., “Real time imaging of two-dimensional iron oxide spherulite nanostructure formation,” Nano Research. 2019. link Times cited: 7 NOT USED (high confidence) A. Mahata and M. A. Zaeem, “Size effect in molecular dynamics simulation of nucleation process during solidification of pure metals: investigating modified embedded atom method interatomic potentials,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 8 Abstract: Due to the significant increase in computing power in recent… read moreAbstract: Due to the significant increase in computing power in recent years, the simulation size of atomistic methods for studying the nucleation process during solidification has been gradually increased, even to billion atom simulations (sub-micron length scale). But the question is how big of a model is required for size-independent and accurate simulations of the nucleation process during solidification? In this work, molecular dynamics simulations with model sizes ranging from ∼2000 to ∼8 million atoms were used to study nucleation during solidification. To draw general conclusions independent of crystal structures, the most advanced second nearest-neighbor modified embedded atom method interatomic potentials for Al (face-centered cubic), Fe (body-centered cubic), and Mg (hexagonal-close packed) were utilized for molecular dynamics simulations. We have analyzed several quantitative characteristics such as nucleation time, density of nuclei, nucleation rate, self-diffusion coefficient, and change in free energy during solidification. The results showed that by increasing the model size to about two million atoms, the simulations and measurable quantities become entirely independent of simulation cell size. The prediction of cell size required for size-independent computed data can considerably reduce the computational costs of atomistic simulations and at the same time increase the accuracy and reliability of the computational data. read less NOT USED (high confidence) Y. Sun, F. Zhang, H. Song, M. Mendelev, C. Wang, and K. Ho, “Competitive B2 and B33 Nucleation during Solidification of Ni50Zr50 Alloy: Molecular Dynamics Simulation and Classical Nucleation Theory,” The Journal of Physical Chemistry C. 2019. link Times cited: 4 Abstract: We investigated the homogenous nucleation of the stoichiomet… read moreAbstract: We investigated the homogenous nucleation of the stoichiometric B2 and B33 phases in the Ni50Zr50 alloy using the persistent embryo method and the classical nucleation theory. The two phases become very close competitors at large supercoolings, which is consistent with the experimental observations. In the case of the B2 phase, the linear temperature dependence of the solid-liquid interface (SLI) free energy extrapolated to the melting temperature leads to the same value as the one obtained from the capillarity fluctuation method (CFM). In the case of the B33 phases, the SLI free energy is also a linear function of temperature at large supercoolings but the extrapolation to the melting temperature leads to a value which is considerably different from the CFM value. This is consistent with the large anisotropy of the SLI properties of the B33 phase nearby the melting temperature observed in the simulation of the nominally flat interface migration. read less NOT USED (high confidence) D. Dickel and C. Barrett, “Methods for the determination of diffusionless transformation conditions from atomistic simulations,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 3 Abstract: The phase transition process between solid phases plays a cr… read moreAbstract: The phase transition process between solid phases plays a critical role in defining the microstructural characteristics of many metals and alloys. Therefore, accurate reproduction of phase transformations enables significant predictive abilities in material modeling which cannot be otherwise achieved. At the atomistic scale, phase transitions naturally occur in modeling as large numbers of atoms in a system relax to their equilibrium phase over a relatively long time scale. However, the accuracy of the simulations in predicting the equilibrium phase for a given pressure, temperature, and solute concentration are often inadequate. Sufficient calibration for a given atomistic potential to reliably reflect these properties is often not achieved because methods of determining the transformation face a number of limitations including high computational cost and sometimes poor accuracy of the results. Herein, we review the methods which have been used to determine equilibrium phase transition conditions at the discrete atom scale and compare their relative efficiency and efficacy. read less NOT USED (high confidence) S. A. Etesami, M. Laradji, and E. Asadi, “Transferability of interatomic potentials in predicting the temperature dependency of elastic constants for titanium, zirconium and magnesium,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 4 Abstract: We present our investigation of the current state of the art… read moreAbstract: We present our investigation of the current state of the art for the transferability of molecular dynamics (MD) interatomic potentials for high temperature simulations of material processes in terms of elastic constants. With the current advancement of computer power, nanoscale computational models such as MD have the potential to accelerate optimization and development of high temperature material processes provided a robust and transferable interatomic potential. Temperature dependency of elastic constants, despite the low temperature elastic constants, is not commonly used as one of the target material properties to develop interatomic potentials for metals; thus, it is a reliable index to determine the transferability of interatomic potentials for high temperature simulations. We consider all five independent elastic constants and their temperature dependency as an index for our evaluations of available interatomic potentials for titanium (Ti), zirconium (Zr), and magnesium (Mg) as representative metals with a relatively complex crystal structure (hcp). The calculated elastic constants and their deviation from their corresponding experimental values are presented. We provide a through discussion on the transferability of each potential and summarize with the most suitable potentials for high temperature material process simulations for each considered material. read less NOT USED (high confidence) X. Zhou, X. Liu, F. Sansoz, and M. Shen, “Molecular dynamics simulation on temperature and stain rate-dependent tensile response and failure behavior of Ni-coated CNT/Mg composites,” Applied Physics A. 2018. link Times cited: 13 NOT USED (high confidence) B. Zhang, L. Zhou, Y. Sun, W. He, and Y.-Z. Chen, “Molecular dynamics simulation of crack growth in pure titanium under uniaxial tension,” Molecular Simulation. 2018. link Times cited: 14 Abstract: ABSTRACT The analysis of crack growth in titanium was perfor… read moreAbstract: ABSTRACT The analysis of crack growth in titanium was performed using molecular dynamics simulation with Embedded Atom Method potentials. The effect of temperature and strain rate on the mechanism of crack growth and the change of microstructure were discussed. After setting an initial crack, the specimen was subjected to uniaxial tension strain up to the total strain level of 0.2 with a constant strain rate. During the period, the shape and the microstructure of crack tip as well as the stress–strain curves were monitored. In the simulation, the gather of voids and stress concentration leading to the crack growth occurred, which are in agreement with experimental results observed by transmission electron microscopy. The transformation from HCP to BCC also occurred at crack tip. The remarkable effect of temperature and strain rate on the growth direction and rate of stacking fault of crack tip was observed. Moreover, initial crack greatly lowered the tension yield point of pure titanium. In the stage of deformation, simulation results showed that loading strain rate and temperature strongly influenced peak stress point, which was increased by the low temperature and high strain, whereas the initial slope of the stress strain curve was independent of loading strain rate. read less NOT USED (high confidence) M. K. Nahhas and S. Groh, “Atomistic modeling of grain boundary behavior under shear conditions in magnesium and magnesium-based binary alloys,” Journal of Physics and Chemistry of Solids. 2018. link Times cited: 12 NOT USED (high confidence) J. Du, Z. Guo, A. Zhang, M. Yang, M. Li, and S. Xiong, “Correlation between crystallographic anisotropy and dendritic orientation selection of binary magnesium alloys,” Scientific Reports. 2017. link Times cited: 30 NOT USED (high confidence) A. Takahashi, A. Seko, and I. Tanaka, “Linearized machine-learning interatomic potentials for non-magnetic elemental metals: Limitation of pairwise descriptors and trend of predictive power.,” The Journal of chemical physics. 2017. link Times cited: 20 Abstract: Machine-learning interatomic potential (MLIP) has been of gr… read moreAbstract: Machine-learning interatomic potential (MLIP) has been of growing interest as a useful method to describe the energetics of systems of interest. In the present study, we examine the accuracy of linearized pairwise MLIPs and angular-dependent MLIPs for 31 elemental metals. Using all of the optimal MLIPs for 31 elemental metals, we show the robustness of the linearized frameworks, the general trend of the predictive power of MLIPs, and the limitation of pairwise MLIPs. As a result, we obtain accurate MLIPs for all 31 elements using the same linearized framework. This indicates that the use of numerous descriptors is the most important practical feature for constructing MLIPs with high accuracy. An accurate MLIP can be constructed using only pairwise descriptors for most non-transition metals, whereas it is very important to consider angular-dependent descriptors when expressing interatomic interactions of transition metals. read less NOT USED (high confidence) H. Liu and J. Nie, “Phase field simulation of microstructures of Mg and Al alloys,” Materials Science and Technology. 2017. link Times cited: 13 Abstract: ABSTRACT The phase field method has recently emerged as a po… read moreAbstract: ABSTRACT The phase field method has recently emerged as a powerful and versatile tool for mesoscale simulation of microstructure evolution in Mg and Al alloys. It permits the study of the evolution of arbitrary and complex microstructures without any presumption. This article provides a review of applications of the phase-field method in Mg and Al alloys. It covers the evolution of dendrites, the equilibrium shape of some key strengthening precipitates, the effect of pre-existing precipitates and dislocations on the distribution of precipitates, and strengthening effects caused by plate-shaped precipitates that are often encountered in Mg and Al alloys. To further improve the accuracy of the phase field simulation results and to further expand the phase field method to predict mechanical properties, the phase field method needs to be integrated with other methods to establish a multi-scale approach. This integration and its application on Mg and Al alloys are also reviewed. This paper is part of a thematic issue on Light Alloys. read less NOT USED (high confidence) P. Saidi, R. Freitas, T. Frolov, M. Asta, and J. Hoyt, “Free energy of steps at faceted (1 1 1) solid-liquid interfaces in the Si-Al system calculated using capillary fluctuation method,” Computational Materials Science. 2017. link Times cited: 8 NOT USED (high confidence) R. Feng, J. T. Lu, H. Y. Li, H. Cao, and Z. Rui, “Effect of the Microcrack Inclination Angle on Crack Propagation Behavior of TiAl Alloy,” Strength of Materials. 2017. link Times cited: 7 NOT USED (high confidence) M. Mendelev, T. L. Underwood, and G. Ackland, “Development of an interatomic potential for the simulation of defects, plasticity, and phase transformations in titanium.,” The Journal of chemical physics. 2016. link Times cited: 122 Abstract: New interatomic potentials describing defects, plasticity, a… read moreAbstract: New interatomic potentials describing defects, plasticity, and high temperature phase transitions for Ti are presented. Fitting the martensitic hcp-bcc phase transformation temperature requires an efficient and accurate method to determine it. We apply a molecular dynamics method based on determination of the melting temperature of competing solid phases, and Gibbs-Helmholtz integration, and a lattice-switch Monte Carlo method: these agree on the hcp-bcc transformation temperatures to within 2 K. We were able to develop embedded atom potentials which give a good fit to either low or high temperature data, but not both. The first developed potential (Ti1) reproduces the hcp-bcc transformation and melting temperatures and is suitable for the simulation of phase transitions and bcc Ti. Two other potentials (Ti2 and Ti3) correctly describe defect properties and can be used to simulate plasticity or radiation damage in hcp Ti. The fact that a single embedded atom method potential cannot describe both low and high temperature phases may be attributed to neglect of electronic degrees of freedom, notably bcc has a much higher electronic entropy. A temperature-dependent potential obtained from the combination of potentials Ti1 and Ti2 may be used to simulate Ti properties at any temperature. read less NOT USED (high confidence) T. L. Underwood and G. Ackland, “monteswitch : A package for evaluating solid-solid free energy differences via lattice-switch Monte Carlo,” Comput. Phys. Commun. 2016. link Times cited: 6 NOT USED (high confidence) T. Fan et al., “Application of the Peierls–Nabarro Model to Symmetric Tilt Low-Angle Grain Boundary with Full Dislocation in Pure Magnesium,” Acta Metallurgica Sinica (English Letters). 2016. link Times cited: 0 NOT USED (high confidence) S. Shuai et al., “Anomalous α-Mg Dendrite Growth During Directional Solidification of a Mg-Zn Alloy,” Metallurgical and Materials Transactions A. 2016. link Times cited: 12 NOT USED (high confidence) S. Shuai, E. Guo, Q. Zheng, M. Wang, T. Jing, and Y.-nan Fu, “Three-dimensional α-Mg dendritic morphology and branching structure transition in Mg-Zn alloys,” Materials Characterization. 2016. link Times cited: 24 NOT USED (high confidence) R. Babicheva et al., “Elastic moduli of nanocrystalline binary Al alloys with Fe, Co, Ti, Mg and Pb alloying elements,” Philosophical Magazine. 2016. link Times cited: 13 Abstract: The paper studies the elastic moduli of nanocrystalline (NC)… read moreAbstract: The paper studies the elastic moduli of nanocrystalline (NC) Al and NC binary Al–X alloys (X is Fe, Co, Ti, Mg or Pb) by using molecular dynamics simulations. X atoms in the alloys are either segregated to grain boundaries (GBs) or distributed randomly as in disordered solid solution. At 0 K, the rigidity of the alloys increases with decrease in atomic radii of the alloying elements. An addition of Fe, Co or Ti to the NC Al leads to increase in the Young’s E and shear μ moduli, while an alloying with Pb decreases them. The elastic moduli of the alloys depend on a distribution of the alloying elements. The alloys with the random distribution of Fe or Ti demonstrate larger E and μ than those for the corresponding alloys with GB segregations, while the rigidity of the Al–Co alloy is higher for the case of the GB segregations. The moduli E and μ for polycrystalline aggregates of Al and Al–X alloys with randomly distributed X atoms are estimated based on the elastic constants of corresponding single-crystals according to the Voigt-Reuss-Hill approximation, which neglects the contribution of GBs to the rigidity. The results show that GBs in NC materials noticeably reduce their rigidity. Furthermore, the temperature dependence of μ for the NC Al–X alloys is analyzed. Only the Al–Co alloy with GB segregations shows the decrease in μ to the lowest extent in the temperature range of 0–600 K in comparison with the NC pure Al. read less NOT USED (high confidence) S. Wilson and M. Mendelev, “A unified relation for the solid-liquid interface free energy of pure FCC, BCC, and HCP metals.,” The Journal of chemical physics. 2016. link Times cited: 37 Abstract: We study correlations between the solid-liquid interface (SL… read moreAbstract: We study correlations between the solid-liquid interface (SLI) free energy and bulk material properties (melting temperature, latent heat, and liquid structure) through the determination of SLI free energies for bcc and hcp metals from molecular dynamics (MD) simulation. Values obtained for the bcc metals in this study were compared to values predicted by the Turnbull, Laird, and Ewing relations on the basis of previously published MD simulation data. We found that of these three empirical relations, the Ewing relation better describes the MD simulation data. Moreover, whereas the original Ewing relation contains two constants for a particular crystal structure, we found that the first coefficient in the Ewing relation does not depend on crystal structure, taking a common value for all three phases, at least for the class of the systems described by embedded-atom method potentials (which are considered to provide a reasonable approximation for metals). read less NOT USED (high confidence) I. A. Alhafez, C. Ruestes, Y. Gao, and H. Urbassek, “Nanoindentation of hcp metals: a comparative simulation study of the evolution of dislocation networks,” Nanotechnology. 2016. link Times cited: 66 Abstract: Using molecular dynamics simulation, we study the nanoindent… read moreAbstract: Using molecular dynamics simulation, we study the nanoindentation of three hcp metals: Mg, Ti, and Zr. Both the basal and two prismatic surface planes are considered. We focus on the characterization of the plasticity generated in the crystal. The similarities to, and the differences from, the behavior of the more commonly investigated fcc and bcc metals are highlighted. We find that hcp metals show a larger variety than the fcc and bcc metals studied up until now. The prolific emission of prismatic loops can lead to extended plastic zones. The size of the plastic zone is quantified by the ratio f of the plastic zone radius to the radius of the contact area. We find values of between 1.6 (an almost collapsed zone) and >5; in the latter case, complex dislocation networks build up which are extended in the direction of easy glide. read less NOT USED (high confidence) Z. Wu, M. Francis, and W. Curtin, “Magnesium interatomic potential for simulating plasticity and fracture phenomena,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 128 Abstract: Magnesium has multiple dislocation and twinning systems with… read moreAbstract: Magnesium has multiple dislocation and twinning systems with starkly different properties, which make its plastic deformation strongly anisotropic and highly complex. Existing empirical interatomic potentials fail to capture the full scope of these properties, making current molecular statics and dynamics simulation results of limited quantitative and predictive use. Here, based on the work by Kim et al, a new modified embedded-atom method potential for magnesium is introduced and rigorously validated against existing ab initio, continuum theory and experimental results. The new potential satisfactorily reproduces all the necessary mechanical properties for plastic deformation, including the various generalized stacking fault energy surfaces, dislocations core structures, Peierls stresses, surface energies and basal plane cohesive strength. The capability of this potential to accurately describe all the important slip systems and fracture behavior makes it valuable for future realistic atomistic studies of general magnesium deformation and failure problems. read less NOT USED (high confidence) H. Song and J. Hoyt, “An atomistic simulation study of the crystallographic orientation relationships during the austenite to ferrite transformation in pure Fe,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 21 Abstract: Molecular dynamics (MD) simulations on a model of pure Fe ha… read moreAbstract: Molecular dynamics (MD) simulations on a model of pure Fe have been used in the investigation of solid-state nucleation of a body-centered-cubic (BCC) phase from a polycrystalline face-centered-cubic (FCC) matrix. A neighbor vector analysis (NVA) method has been introduced and it is shown how the NVA can be used to determine the misorientation of grain or interphase boundaries. In particular, the NVA was utilized to identify the orientation relationships (ORs) of several BCC nuclei and three special ORs were tested, namely the Kurdjumov-Sachs (KS), Nishiyama–Wassermann (NW) and Pitsch (P). From several quasi-2D simulations, it was found that all stable nuclei at grain boundaries formed at least one orientation relationship with the parent grains that was consistent with either the KS or NW relationship. Several initial MD simulation cells, which prohibited the formation of special ORs, were also examined and in these simulations no nucleation was observed after long run times. In addition, the {1 1 1}γ//{1 1 0}α ?> orientation was detected in all mobile phase boundaries. Consistent with experimental findings, these observations demonstrate the importance of this high coherency atomic plane during both the nucleation and growth process. The nucleation and phase boundary characteristics identified here may provide important insights into the nucleation rate and grain orientation of more general solid state nucleation processes. read less NOT USED (high confidence) C. Jian, W. Haipeng, and W. Bingbo, “Rapid Solidification Characteristics of Highly Undercooled Liquid Ni-Cu-Mo-Ge Quaternary Alloy under Electromagnetic Levitation Condition,” Conference on Industrial Electronics and Applications. 2015. link Times cited: 0 Abstract: The dendritic growth characteristics of undercooled liquid N… read moreAbstract: The dendritic growth characteristics of undercooled liquid Ni-5%Cu-5%Mo-5%Ge quaternary alloy were investigated by electromagnetic levitation method. The measured dendritic growth velocity of α-Ni phase increases with undercooling according to a power law relation, which attains a value of 28 m/s at the maximum undercooling of 321 K (0.19TL). The microstructure morphology appears as coarse dendrites at small undercoolings, while it is refined into equiaxed grains at substantial undercoolings. Furthermore, all the solute elements Cu, Mo and Ge exhibit a significant solute trapping effect during rapid dendritic growth. Keywords-dendritic growth; high undercooling; solute trapping. read less NOT USED (high confidence) L. Wu, B. Xu, Q. Li, W. Liu, and M. Li, “Anisotropic crystal–melt interfacial energy and stiffness of aluminum,” Journal of Materials Research. 2015. link Times cited: 21 Abstract: The crystal–melt interfacial free energy is an important qua… read moreAbstract: The crystal–melt interfacial free energy is an important quantity governing many kinetic phenomena including solidification and crystal growth. Although general calculation methods are available, it is still difficult to obtain the interfacial energies that differ only slightly due to anisotropy. Here, we report such a calculation of Al crystal–melt interfacial energy based on the general framework of the capillary fluctuation method (CFM). The subtle dependence of both the melting temperature and interfacial free energy at melting temperature on the crystal interface orientation was examined. For Al, the average melting temperature is obtained at 934.79 ± 5 K and the orientationally averaged mean interfacial free energy is 98.35 mJ/m^2. In addition, the anisotropy of the interfacial free energy is found weak, nevertheless with the values ranked as γ_100 > γ_110 > γ_111. read less NOT USED (high confidence) A. Monas, O. Shchyglo, S.-J. Kim, C. Yim, D. Höche, and I. Steinbach, “Divorced Eutectic Solidification of Mg-Al Alloys,” JOM. 2015. link Times cited: 19 NOT USED (high confidence) K. Kuribayashi, H. Kato, K. Nagayama, Y. Inatomi, and M. V. Kumar, “An experimental verification of a criterion for forming metastable phases in containerless solidification,” Journal of Applied Physics. 2015. link Times cited: 5 Abstract: On the thermodynamic condition for forming a metastable phas… read moreAbstract: On the thermodynamic condition for forming a metastable phase from undercooled melt in a containerless state, we had proposed a criterion that crystals will preferentially form if they have a smaller entropy of fusion than the entropy of fusion of equilibrium crystals (Kuribayashi et al., Mater. Sci. Eng., A 449–451, 675 (2007)). This criterion is proposed for being applied to materials that exhibit a faceted interface, such as semiconductors and oxides. However, no experimental data that support this criterion have been obtained. From this point, we used an aerodynamic levitator as a tool for forming metastable phases from undercooled melt and verified the above-mentioned criterion using LnFeO3 (Ln: lanthanide and Y) as the model material. In addition, the condition for double recalescence, which corresponds to forming metastable phases and stable phases, was discussed in terms of competitive 2D isomorphic nucleation of the metastable phase and 3D polymorphic nucleation of the stable phase. read less NOT USED (high confidence) J. Benet, L. G. Macdowell, and E. Sanz, “Interfacial free energy of the NaCl crystal-melt interface from capillary wave fluctuations.,” The Journal of chemical physics. 2015. link Times cited: 10 Abstract: In this work we study, by means of molecular dynamics simula… read moreAbstract: In this work we study, by means of molecular dynamics simulations, the solid-liquid interface of NaCl under coexistence conditions. By analysing capillary waves, we obtain the stiffness for different orientations of the solid and calculate the interfacial free energy by expanding the dependency of the interfacial free energy with the solid orientation in terms of cubic harmonics. We obtain an average value for the solid-fluid interfacial free energy of 89 ± 6 mN m(-1) that is consistent with previous results based on the measure of nucleation free energy barriers [Valeriani et al., J. Chem. Phys. 122, 194501 (2005)]. We analyse the influence of the simulation setup on interfacial properties and find that facets prepared as an elongated rectangular stripe give the same results as those prepared as squares for all cases but the 111 face. For some crystal orientations, we observe at small wave-vectors a behaviour not consistent with capillary wave theory and show that this behavior does not depend on the simulation setup. read less NOT USED (high confidence) S. Wilson, K. Gunawardana, and M. Mendelev, “Solid-liquid interface free energies of pure bcc metals and B2 phases.,” The Journal of chemical physics. 2015. link Times cited: 32 Abstract: The solid-liquid interface (SLI) free energy was determined … read moreAbstract: The solid-liquid interface (SLI) free energy was determined from molecular dynamics (MD) simulation for several body centered cubic (bcc) metals and B2 metallic compounds (space group: Pm3̄m; prototype: CsCl). In order to include a bcc metal with a low melting temperature in our study, a semi-empirical potential was developed for Na. Two additional synthetic "Na" potentials were also developed to explore the effect of liquid structure and latent heat on the SLI free energy. The obtained MD data were compared with the empirical Turnbull, Laird, and Ewing relations. All three relations are found to predict the general trend observed in the MD data for bcc metals obtained within the present study. However, only the Laird and Ewing relations are able to predict the trend obtained within the sequence of "Na" potentials. The Laird relation provides the best prediction for our MD data and other MD data for bcc metals taken from the literature. Overall, the Laird relation also agrees well with our B2 data but requires a proportionality constant that is substantially different from the bcc case. It also fails to explain a considerable difference between the SLI free energies of some B2 phases which have nearly the same melting temperature. In contrast, this difference is satisfactorily described by the Ewing relation. Moreover, the Ewing relation obtained from the bcc dataset also provides a reasonable description of the B2 data. read less NOT USED (high confidence) D. Quirinale, G. Rustan, S. Wilson, M. Kramer, A. Goldman, and M. Mendelev, “Appearance of metastable B2 phase during solidification of Ni50Zr50 alloy: electrostatic levitation and molecular dynamics simulation studies,” Journal of Physics: Condensed Matter. 2015. link Times cited: 24 Abstract: High-energy x-ray diffraction measurements of undercooled, e… read moreAbstract: High-energy x-ray diffraction measurements of undercooled, electrostatically levitated Ni50Zr50 liquid droplets were performed. The observed solidification pathway proceeded through the nucleation and growth of the metastable B2 phase, which persisted for several seconds before the rapid appearance of the stable B33 phase. This sequence is shown to be consistent with predictions from classical nucleation theory using data obtained from molecular dynamics (MD) simulations. A plausible mechanism for the B2–B33 transformation is proposed and investigated through further MD simulations. read less NOT USED (high confidence) L. Leclercq, L. Capolungo, and D. Rodney, “Atomic-Scale Comparison Between Twin Growth Mechanisms in Magnesium,” Materials Research Letters. 2014. link Times cited: 31 Abstract: The two most frequently observed twins in hexagonal close-pa… read moreAbstract: The two most frequently observed twins in hexagonal close-packed (HCP) Mg, and twins, have surprisingly different properties and morphologies, with twins appearing under higher stresses and being much thinner than twins. By considering the atomic-scale elementary properties of the twin interfaces and their disconnections, we show that (1) the transverse propagation of twins is hindered by the absence of low-energy mobile interfaces, whereas twins benefit from prismatic-basal interfaces and (2) the thickening of twins is slowed by higher energy barriers against both the nucleation and propagation of disconnections along their interfaces. read less NOT USED (high confidence) F. Podmaniczky, G. T’oth, T. Pusztai, and L. Gr’an’asy, “Free energy of the bcc–liquid interface and the Wulff shape as predicted by the phase-field crystal model,” Journal of Crystal Growth. 2014. link Times cited: 9 NOT USED (high confidence) L. Shen, “Molecular dynamics study of Al solute-dislocation interactions in Mg alloys,” Interaction and multiscale mechanics. 2013. link Times cited: 7 Abstract: In this study, atomistic simulations are performed to study … read moreAbstract: In this study, atomistic simulations are performed to study the effect of Al solute on the behaviour of edge dislocation in Mg alloys. After the dissociation of an Mg basal edge dislocation into two Shockley partials using molecular mechanics, the interaction between the dislocation and Al solute at different temperatures is studied using molecular dynamics. It appears from the simulations that the critical shear stress increases with the Al solute concentration. Comparing with the solute effect at T = 0 K, however, the critical shear stress at a finite temperature is lower since the kinetic energy of the atoms can help the dislocation conquer the energy barriers created by the Al atoms. The velocity of the edge dislocation decreases as the Al concentration increases when the external shear stress is relatively small regardless of temperature. The Al concentration effect on the dislocation velocity is not significant at very high shear stress level when the solute concentration is below 4.0 at%. Drag coefficient B increases with the Al concentration when the stress to temperature ratio is below 0.3 MPa/K, although the effect is more significant at low temperatures. read less NOT USED (high confidence) R. Matsumoto, M. Uranagase, and N. Miyazaki, “Molecular Dynamics Analyses of Deformation Behavior of Long-Period-Stacking-Ordered Structures,” Materials Transactions. 2013. link Times cited: 34 Abstract: Magnesium alloys containing long-period-stacking ordered (LP… read moreAbstract: Magnesium alloys containing long-period-stacking ordered (LPSO) phases have attracted considerable attention because they have been reported to exhibit excellent mechanical properties, including high strength and reasonable ductility. It is thought that the LPSO phase plays a critical role in producing these favorable mechanical properties. We analyze the deformation behavior of the LPSO phases with different stacking sequences using molecular dynamics simulations. To highlight the specific deformation behavior of the LPSO phases, we also perform deformation analyses of hexagonal-close-packed and face-centered-cubic (FCC) structures. We focus on the influence of the stacking order rather than the segregated atoms around the FCC-structured layers, and we model an LPSO structure by single element composition where the interatomic interaction is described by a smoothed Lennard-Jones potential. Our simulations indicate that an LPSO structure with a shorter stacking sequence tends to exhibit a higher compressive flow stress, because FCC-structured layers inhibit twinning deformations and non-basal slips. Kinking deformation is observed for an LPSO structure when both compression and shear deformation are present. It is shown that the first-order pyramidal-hcþ ai dislocation disarranges the stacking of an LPSO structure and leaves behind many lattice defects. In addition, those lattice defects activate numerous basal slips. Finally, basal dislocations arrange in a line and generate a misorientation angle. Furthermore, this angle originates the compressive deformation. We also observed some prismatic-hai dislocations and cross slips to the basal plane. These results suggest the importance of non-basal slips for kinking deformation. [doi:10.2320/matertrans.MI201211] read less NOT USED (high confidence) M. Paliwal and I. Jung, “Solid/Liquid Interfacial Energy of Mg-Al Alloys,” Metallurgical and Materials Transactions A. 2013. link Times cited: 7 NOT USED (high confidence) Z. Sun, A. López, and J. Schliemann, “Zero-field magnetization reversal of two-body Stoner particles with dipolar interaction,” Journal of Applied Physics. 2010. link Times cited: 6 Abstract: We investigate magnetization reversal in a system of two Sto… read moreAbstract: We investigate magnetization reversal in a system of two Stoner particles with uniaxial anisotropies both subject to a static and antiparallel magnetic field, and taking into account their mutual dipolar interaction. We identify an interesting regime of stable synchronized magnetic dynamics where the two particles are implementing a single information bit. Here a modified Stoner-Wohlfarth limit occurs which results in a dramatically lower critical switching field Hc (including Hc=0) and also a substantially shorter reversal time. Our analytical results are verified by numerical simulations and offer new technological perspectives regarding devices for information storage and/or fast magnetic response. read less NOT USED (high confidence) T. Tang, S. Kim, and M. Horstemeyer, “Fatigue crack growth in magnesium single crystals under cyclic loading: Molecular dynamics simulation,” Computational Materials Science. 2010. link Times cited: 92 NOT USED (high confidence) J. Hoyt, Z. Trautt, and M. Upmanyu, “Fluctuations in molecular dynamics simulations,” Math. Comput. Simul. 2010. link Times cited: 47 NOT USED (high confidence) M. Mendelev and B. Bokstein, “Molecular dynamics study of self-diffusion in Zr,” Philosophical Magazine. 2010. link Times cited: 56 Abstract: We employed a recently developed semi-empirical Zr potential… read moreAbstract: We employed a recently developed semi-empirical Zr potential to determine the diffusivities in hcp and bcc Zr via molecular dynamics simulation. The point defect concentration was determined directly from molecular dynamics (MD) simulation rather than from theoretical methods using T = 0 calculations. Our MD simulation indicates that the diffusion proceeds via the interstitial mechanism in hcp Zr, and both vacancy and interstitial mechanisms contribute to diffusivity in bcc Zr. The agreement with the experimental data is excellent for hcp Zr and rather good for bcc Zr at high temperatures, but there is considerable disagreement at low temperatures. read less NOT USED (high confidence) M. Mendelev, M. Asta, M. J. Rahman, and J. Hoyt, “Development of interatomic potentials appropriate for simulation of solid–liquid interface properties in Al–Mg alloys,” Philosophical Magazine. 2009. link Times cited: 126 Abstract: Different approaches are analyzed for construction of semi-e… read moreAbstract: Different approaches are analyzed for construction of semi-empirical potentials for binary alloys, focusing specifically on the capability of these potentials to describe solid–liquid phase equilibria, as a pre-requisite to studies of solidification phenomena. Fitting ab initio compound data does not ensure correct reproduction of the dilute solid-solution formation energy, and explicit inclusion of this quantity in the potential development procedure does not guarantee that the potential will predict the correct solid–liquid phase diagram. Therefore, we conclude that fitting only to solid phase properties, as is done in most potential development procedures, generally is not sufficient to develop a semi-empirical potential suitable for the simulation of solidification. A method is proposed for the incorporation of data for liquid solution energies in the potential development procedure, and a new semi-empirical potential developed suitable for simulations of dilute alloys of Mg in Al. The potential correctly reproduces both zero-temperature solid properties and solidus and liquid lines on the Al-rich part of the Al–Mg phase diagram. read less NOT USED (high confidence) J. Yasi, T. Nogaret, D. Trinkle, Y. Qi, L. G. Hector, and W. A. Curtin, “Basal and prism dislocation cores in magnesium: comparison of first-principles and embedded-atom-potential methods predictions,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 120 Abstract: The core structures of screw and edge dislocations on the ba… read moreAbstract: The core structures of screw and edge dislocations on the basal and prism planes in Mg, and the associated gamma surfaces, were studied using an ab initio method and the embedded-atom-method interatomic potentials developed by Sun et al and Liu et al. The ab initio calculations predict that the basal plane dislocations dissociate into partials split by 16.7 Å (edge) and 6.3 Å (screw), as compared with 14.3 Å and 12.7 Å (Sun and Liu edge), and 6.3 Å and 1.4 Å (Sun and Liu screw), with the Liu screw dislocation being metastable. In the prism plane, the screw and edge cores are compact and the edge core structures are all similar, while ab initio does not predict a stable prismatic screw in stress-free conditions. These results are qualitatively understood through an examination of the gamma surfaces for interplanar sliding on the basal and prism planes. The Peierls stresses at T = 0 K for basal slip are a few megapascals for the Sun potential, in agreement with experiments, but are ten times larger for the Liu potential. The Peierls stresses for prism slip are 10–40 MPa for both potentials. Overall, the dislocation core structures from ab initio are well represented by the Sun potential in all cases while the Liu potential shows some notable differences. These results suggest that the Sun potential is preferable for studying other dislocations in Mg, particularly the ⟨c + a⟩ dislocations, for which the core structures are much larger and not accessible by ab initio methods. read less NOT USED (high confidence) M. Mendelev, M. Kramer, C. Becker, and M. Asta, “Analysis of semi-empirical interatomic potentials appropriate for simulation of crystalline and liquid Al and Cu,” Philosophical Magazine. 2008. link Times cited: 365 Abstract: We investigate the application of embedded atom method (EAM)… read moreAbstract: We investigate the application of embedded atom method (EAM) interatomic potentials in the study of crystallization kinetics from deeply undercooled melts, focusing on the fcc metals Al and Cu. For this application, it is important that the EAM potential accurately reproduces melting properties and liquid structure, in addition to the crystalline properties most commonly fit in its development. To test the accuracy of previously published EAM potentials and to guide the development of new potential in this work, first-principles calculations have been performed and new experimental measurements of the Al and Cu liquid structure factors have been undertaken by X-ray diffraction. We demonstrate that the previously published EAM potentials predict a liquid structure that is too strongly ordered relative to measured diffraction data. We develop new EAM potentials for Al and Cu to improve the agreement with the first-principles and measured liquid diffraction data. Furthermore, we calculate liquid-phase diffusivities and find that this quantity correlates well with the liquid structure. Finally, we perform molecular dynamics simulations of crystal nucleation from the melt during quenching at constant cooling rate. We find that EAM potentials, which predict the same zero-temperature crystal properties but different liquid structures, can lead to quite different crystallization kinetics. More interestingly, we find that two potentials predicting very similar equilibrium solid and liquid properties can still produce very different crystallization kinetics under far-from-equilibrium conditions characteristic of the rapid quenching simulations employed here. read less NOT USED (high confidence) D. Buta, M. Asta, and J. Hoyt, “Kinetic coefficient of steps at the Si(111) crystal-melt interface from molecular dynamics simulations.,” The Journal of chemical physics. 2007. link Times cited: 64 Abstract: Nonequilibrium molecular dynamics simulations are applied to… read moreAbstract: Nonequilibrium molecular dynamics simulations are applied to the investigation of step-flow kinetics at crystal-melt interfaces of silicon, modeled with the Stillinger-Weber potential [Phys. Rev. B 31, 5262 (1985)]. Step kinetic coefficients are calculated from crystallization rates of interfaces that are vicinals of the faceted (111) orientation. These vicinal interfaces contain periodic arrays of bilayer steps, and they are observed to crystallize in a step-flow growth mode at undercoolings lower than 40 K. Kinetic coefficients for both [110] and [121] oriented steps are determined for several values of the average step separation, in the range of 7.7-62.4 A. The values of the step kinetic coefficients are shown to be highly isotropic, and are found to increase with increasing step separation until they saturate at step separations larger than approximately 50 A. The largest step kinetic coefficients are found to be in the range of 0.7-0.8 m(sK), values that are more than five times larger than the kinetic coefficient for the rough (100) crystal-melt interface in the same system. The dependence of step mobility on step separation and the relatively large value of the step kinetic coefficient are discussed in terms of available theoretical models for crystal growth kinetics from the melt. read less NOT USED (high confidence) M. Mendelev and G. Ackland, “Development of an interatomic potential for the simulation of phase transformations in zirconium,” Philosophical Magazine Letters. 2007. link Times cited: 266 Abstract: In recent years, some 30 studies have been published on the … read moreAbstract: In recent years, some 30 studies have been published on the molecular dynamics (MD) of zirconium, primarily of its twinning deformation and response to radiation damage. Its low thermal neutron absorption makes it uniquely suited for the latter application. Surprisingly, currently used interatomic potentials do not encapsulate the unique properties of Zr, namely its high stacking-fault energy, anomolous self-diffusion, melting and phase transformation under temperature and pressure (or alloying). Ab initio calculations have shown deficiencies in the description of point defects, both vacancies and interstitials, using existing interatomic potentials, deficiencies that can now be rectified by refitting. Here, we show the calculation of phase transitions self-consistently and present a potential for Zr that correctly reproduces the energetics of our extended database of ab initio configurations and high-temperature phase transitions. The potential has an analytic many-body form, making it suitable for existing large-scale MD codes. We also present a best-fit potential for the hcp structure and its defects. read less NOT USED (high confidence) T. Haxhimali, A. Karma, F. Gonzales, and M. Rappaz, “Orientation selection in dendritic evolution,” Nature Materials. 2006. link Times cited: 344 NOT USED (high confidence) S. Kavousi, “Combined Molecular Dynamics and Phase Field Simulation of Crystal Melt Interfacial Properties and Microstructure Evolution during Rapid Solidification of TI-NI Alloys.” 2019. link Times cited: 0 NOT USED (high confidence) D. Sun, “Proliferation of Twinning in Metals: Application to Magnesium Alloys.” 2018. link Times cited: 2 Abstract: In the search for new alloys with a great strength-to-weight… read moreAbstract: In the search for new alloys with a great strength-to-weight ratio, magnesium has emerged at the forefront. With a strength rivaling that of steel and aluminum alloys --- materials which are deployed widely in real world applications today --- but only a fraction of the density, magnesium holds great promise in a variety of next-generation applications. Unfortunately, the widespread adoption of magnesium is hindered by the fact that it fails in a brittle fashion, which is undesirable when it comes to plastic deformation mechanisms. Consequently, one must design magnesium alloys to navigate around this shortcoming and fail in a more ductile fashion. However, such designs are not possible without a thorough understanding of the underlying mechanisms of deformation in magnesium, which is somewhat contested at the moment. In addition to slip, which is one of the dominant mechanisms in metallic alloys, a mechanism known as twinning is also present, especially in hexagonal close-packed (HCP) materials such as magnesium. Twinning involves the reorientation of the material lattice about a planar discontinuity and has been shown as one of the preferred mechanisms by which magnesium accommodates out-of-plane deformation. Unfortunately, twinning is not particularly well-understood in magnesium, and needs to be addressed before progress can be made in materials design. In particular, though two specific modes of twinning have been acknowledged, various works in the literature have identified a host of additional modes, many of which have been cast aside as "anomalous" observations. To this end, we introduce a new framework for predicting the modes by which a material can twin, for any given material. Focusing on magnesium, we begin our investigation by introducing a kinematic framework that predicts novel twin configurations, cataloging these twins modes by their planar normal and twinning shear. We then subject the predicted twin modes to a series of atomistic simulations, primarily in molecular statics but with supplementary calculations using density functional theory, giving us insight on both the energy of the twin interface and barriers to formation. We then perform a stress analysis and identify the twin modes which are most likely to be activated, thus finding the ones most likely to affect the yield surface of magnesium. Over the course of our investigation, we show that many different modes actually participate on the yield surface of magnesium; the two classical modes which are accepted by the community are confirmed, but many additional modes --- some of which are close to modes which have been previously regarded as anomalies --- are also observed. We also perform some extensional work, showing the flexibility of our framework in predicting twins in other materials and in other environments and highlighting the complicated nature of twinning, especially in HCP materials. read less NOT USED (high confidence) Y. Sun et al., “Structural ordering at solid-liquid interfaces in Al-Sm system: A molecular-dynamics study,” Materials Letters. 2017. link Times cited: 17 NOT USED (high confidence) R. Weisburgh, “Scalable, Composable Operators for Defect Design and Analysis.” 2016. link Times cited: 0 Abstract: Title of thesis: SCALABLE, COMPOSABLE OPERATORS FOR DEFECT D… read moreAbstract: Title of thesis: SCALABLE, COMPOSABLE OPERATORS FOR DEFECT DESIGN AND ANALYSIS Rose Weisburgh, Master of Science, 2016 Thesis directed by: Professor Peter W Chung Department of Mechanical Engineering It is well understood that defects adversely affect the electro-mechanical properties of materials. Ideally, defect compositions of raw materials used in devices could be measured, but present technology in the field of atomic defect detection is either destructive in nature, or is unable to determine the precise atomic composition of materials. In the adjacent field of damage detection in large-scale truss networks, algorithms based on spectral measurements have successfully been employed to locate damaged members. Already similar principles have been applied to material lattices experimentally by using Raman Spectroscopy to qualitatively approximate defect densities within materials. However, the applications have largely been limited to surface defects or two-dimensional materials, and the host lattices and defect types are primarily studied anecdotally. This thesis details a numerical method for determining the precise phonon or vibration spectra of material lattices with defects, as it was originally presented in [1]. The dynamical matrices of lattices containing defects are calculated by introducing defects systematically into the dynamical matrices of pristine, defect-free lattices using linear operators. Each operation modifies or removes an individual bond or interaction. Complex defect configurations can be composed through reiterative application of the operators. The proposed methods may be applied to systems containing any interaction type or bond order, including space trusses and atomic lattices. The method is demonstrated by numerically determining the convergence rate of phonon properties in the dilute limit of a single point vacancy. Then the same methodology is applied to two-dimensional atomic lattices with central forces, two-dimension truss networks with distributed mass, as well as three-dimensional atomic lattices with non-linear many body potentials. In each example, the defect structure and properties are shown to alter the spectral properties of the materials. SCALABLE, COMPOSABLE OPERATORS FOR DEFECT DESIGN AND ANALYSIS read less NOT USED (high confidence) J. S. Gibson, S. G. Srinivasan, M. Baskes, R. E. Miller, and A. K. Wilson, “A multi-state modified embedded atom method potential for titanium,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 3 Abstract: The continuing search for broadly applicable, predictive, an… read moreAbstract: The continuing search for broadly applicable, predictive, and unique potential functions led to the invention of the multi-state modified embedded atom method (MS-MEAM) (Baskes et al 2007 Phys. Rev. B 75 094113). MS-MEAM replaced almost all of the prior arbitrary choices of the MEAM electron densities, embedding energy, pair potential, and angular screening functions by using first-principles computations of energy/volume relationships for multiple reference crystal structures and transformation paths connecting those reference structures. This strategy reasonably captured diverse interactions between atoms with variable coordinations in a face-centered-cubic (fcc)-stable copper system. However, a straightforward application of the original MS-MEAM framework to model technologically useful hexagonal-close-packed (hcp) metals proved elusive. This work describes the development of an hcp-stable/fcc-metastable MS-MEAM to model titanium by introducing a new angular function within the background electron density description. This critical insight enables the titanium MS-MEAM potential to reproduce first principles computations of reference structures and transformation paths extremely well. Importantly, it predicts lattice and elastic constants, defect energetics, and dynamics of non-ideal hcp and liquid titanium in good agreement with first principles computations and corresponding experiments, and often better than the three well-known literature models used as a benchmark. The titanium MS-MEAM has been made available in the Knowledgebase of Interatomic Models (https://openkim.org/) (Tadmor et al 2011 JOM 63 17). read less NOT USED (high confidence) M. Amoorezaei, S. Gurevich, and N. Provatas, “Orientation selection in solidification patterning,” Acta Materialia. 2012. link Times cited: 67 NOT USED (high confidence) P. Bavli and J. Adler, “Parallel codes for simulating elastic constants and melting in Ar and Mg,” Physics Procedia. 2010. link Times cited: 5 NOT USED (definite) H. Song, Y. Sun, F. Zhang, C. Wang, K. Ho, and M. Mendelev, “Nucleation of stoichiometric compounds from liquid: Role of the kinetic factor,” Physical Review Materials. 2017. link Times cited: 21 Abstract: While the role of the free energy barrier during nucleation … read moreAbstract: While the role of the free energy barrier during nucleation is a text-book subject the importance of the kinetic factor is frequently underestimated. We obtained both quantities from molecular dynamics (MD) simulations for the pure Ni and B2 phases in the Ni50Al50 and Cu50Zr50 alloys. The free-energy barrier was found to be higher in Ni but the nucleation rate is much lower in the Ni50Al50 alloy which was attributed to the ordered nature of the B2 phase. Since the Cu50Zr50 B2 phase can has even smaller fraction of the anti-site defects its nucleation is never observed in the MD simulation. read less NOT USED (definite) J. Benet, L. G. Macdowell, and E. Sanz, “A study of the ice-water interface using the TIP4P/2005 water model.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 41 Abstract: In this work we study the ice-water interface under coexiste… read moreAbstract: In this work we study the ice-water interface under coexistence conditions by means of molecular simulations using the TIP4P/2005 water model. Following the methodology proposed by Hoyt and co-workers [J. J. Hoyt, M. Asta and A. Karma, Phys. Rev. Lett., 2001, 86, 5530] we measure the interfacial free energy of ice with liquid water by analysing the spectrum of capillary fluctuations of the interface. We get an orientationally averaged interfacial free energy of 27(2) mN m(-1), in good agreement with a recent estimate obtained from simulation data of the size of critical clusters [E. Sanz, C. Vega, J. R. Espinosa, R. Caballero-Bernal, J. L. F. Abascal and C. Valeriani, J. Am. Chem. Soc., 2013, 135, 15008]. We also estimate the interfacial free energy of different planes and obtain 27(2), 28(2) and 28(2) mN m(-1) for the basal, the primary prismatic and the secondary prismatic planes respectively. Finally, we inspect the structure of the interface and find that its thickness is approximately 4-5 molecular diameters. Moreover, we find that when the basal plane is exposed to the fluid the interface alternates regions of cubic ice with regions of hexagonal ice. read less NOT USED (definite) Q. Yu, L. Qi, R. Mishra, J. Li, and A. Minor, “Reducing deformation anisotropy to achieve ultrahigh strength and ductility in Mg at the nanoscale,” Proceedings of the National Academy of Sciences. 2013. link Times cited: 109 Abstract: In mechanical deformation of crystalline materials, the crit… read moreAbstract: In mechanical deformation of crystalline materials, the critical resolved shear stress (CRSS; τCRSS) is the stress required to initiate movement of dislocations on a specific plane. In plastically anisotropic materials, such as Mg, τCRSS for different slip systems differs greatly, leading to relatively poor ductility and formability. However, τCRSS for all slip systems increases as the physical dimension of the sample decreases to approach eventually the ideal shear stresses of a material, which are much less anisotropic. Therefore, as the size of a sample gets smaller, the yield stress increases and τCRSS anisotropy decreases. Here, we use in situ transmission electron microscopy mechanical testing and atomistic simulations to demonstrate that τCRSS anisotropy can be significantly reduced in nanoscale Mg single crystals, where extremely high stresses (∼2 GPa) activate multiple deformation modes, resulting in a change from basal slip-dominated plasticity to a more homogeneous plasticity. Consequently, an abrupt and dramatic size-induced “brittle-to-ductile” transition occurs around 100 nm. This nanoscale change in the CRSS anisotropy demonstrates the powerful effect of size-related deformation mechanisms and should be a general feature in plastically anisotropic materials. read less NOT USED (definite) M. Wang, J. Williams, L. Jiang, F. Carlo, T. Jing, and N. Chawla, “Three Dimensional (3D) Microstructural Characterization and Quantitative Analysis of Solidified Microstructures in Magnesium-Based Alloys,” Metallography, Microstructure, and Analysis. 2012. link Times cited: 28 |
 EAM_Dynamo_SunMendelevBecker_2006_Mg__MO_848345414202_005
EAM_Dynamo_SunMendelevBecker_2006_Mg__MO_848345414202_005