Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
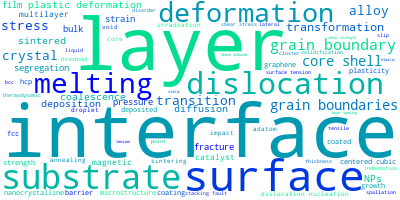
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
1334 Citations (1028 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) Q. Liu, Y. Zhang, and P. Qian, “Molecular dynamics study on the thermodynamic stability and structural evolution of crown-jewel structured PdCu nanoalloys,” RSC Advances. 2023. link Times cited: 0 Abstract: The novel crown-jewel (CJ) structured PdCu nanoalloys have a… read moreAbstract: The novel crown-jewel (CJ) structured PdCu nanoalloys have attracted considerable interest in high-performance single-atom catalysis. The characteristics of demanding high-temperature calcination in the synthesis of these samples disable us from experimentally understanding the details of the thermal evolution behavior of PdCu nanoclusters during the heating process. In this work, by analyses of potential energy surface, bond order parameter, and radial distribution function, we have theoretically studied the thermodynamic stabilities and structural evolution of Pd-decorated Cu-based CJ nanoclusters with various compositions and sizes by molecular dynamics simulations. PdCu nanoclusters undergo a cuboctahedral (Cubo) to icosahedral (Ico) structural transformation before melting. This transformation is size- and Pd-composition dependent. The small size and high Pd-composition of PdCu nanoclusters facilitate this transformation. In addition, we find that the surface and interface effects of clusters have an important impact on the structural transformation and Cubo–Ico structural transformation is strongly related to the release of excess energy. read less USED (high confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (high confidence) I. Winter, T. Oppelstrup, T. Frolov, and R. Rudd, “Characterization and Visualization of Grain Boundary Disconnections,” SSRN Electronic Journal. 2022. link Times cited: 1 Abstract: We introduce a method to visualize dislocations along grain … read moreAbstract: We introduce a method to visualize dislocations along grain boundaries at the atomic level. It uses an atomic-level Nye tensor, representing the dislocation density. To calculate the Nye tensor at grain boundaries, we extend the Hartley-Mishin strain gradient calculation to the displacement shift complete lattice. We show that the method is effective in visualizing disconnections and the dislocation content of grain boundary phase junctions in bodycentered cubic tungsten, as well as face-centered cubic copper. In addition, we use the method to characterize the morphology of a two-dimensional grain boundary phase nucleus in a symmetric tilt grain boundary in tungsten. This method can be applied to both bulk dislocations and grain boundary disconnections, which makes it ideal for studying the interactions and reactions of bulk dislocations with grain boundaries, and grain boundary disconnections. read less USED (high confidence) S. Chen et al., “Simultaneously enhancing the ultimate strength and ductility of high-entropy alloys via short-range ordering,” Nature Communications. 2021. link Times cited: 110 USED (high confidence) A. Ravichandran, M. Mehta, A. Woodworth, and J. Lawson, “Molecular dynamics simulations of ultrafast radiation induced melting at metal–semiconductor interfaces,” Journal of Applied Physics. 2021. link Times cited: 3 Abstract: Metal–semiconductor contacts in silicon carbide (SiC) diodes… read moreAbstract: Metal–semiconductor contacts in silicon carbide (SiC) diodes endure damages at the interface when exposed to harsh radiation environments. Due to the rapid rise in temperature and ultrafast cooling that follows the radiation impact, the structural properties of the materials can be altered through melting, recrystallization, and amorphization. A detailed understanding of the material failure modes at the interface is lacking, specifically at the nanoscale. We use molecular simulations to investigate the ultrafast melting at tungsten (W)–SiC interfaces following radiation damage and apply deep learning techniques to track the transient evolution of the local molecular structures. We show that W near the radiation track undergoes melting and, eventually, most of it recrystallizes with a noticeable degree of undercooling, while SiC is rendered permanently amorphous. The observation of local undercooling in W films is important as it can affect the device performance even before the bulk melting temperature of the material is reached. We also show that at high temperatures, the interface undergoes a fracture-like failure. The results presented here are significant in understating the different failure modes of SiC diode materials. read less USED (high confidence) S. Goel et al., “Horizons of modern molecular dynamics simulation in digitalized solid freeform fabrication with advanced materials,” Materials Today Chemistry. 2020. link Times cited: 17 USED (high confidence) M. Su, Q. Deng, M. An, and L. Liu, “Plastic deformation mechanism transition of Ti/Ni nanolaminate with pre-existing crack: Molecular dynamics study,” Chinese Physics B. 2020. link Times cited: 2 USED (high confidence) B. Chen, S. Li, H. Zong, X. Ding, J. Sun, and E. Ma, “Unusual activated processes controlling dislocation motion in body-centered-cubic high-entropy alloys,” Proceedings of the National Academy of Sciences of the United States of America. 2020. link Times cited: 90 Abstract: Significance This work demonstrates dislocation dynamics in … read moreAbstract: Significance This work demonstrates dislocation dynamics in body-centered-cubic (BCC) high-entropy alloys (HEAs). The local composition inhomogeneity in these concentrated solutions leads to an unconventional rate-controlling mechanism that governs dislocation mobility. The activated process becomes nanoscale detrapping, of kinks on screw dislocations and of trapped segments of edge dislocations, presenting an activation barrier of similar magnitude for both dislocation types. Their sluggish mobility explains the elevated strength and strain hardening in BCC HEAs. Atomistic simulations of dislocation mobility reveal that body-centered cubic (BCC) high-entropy alloys (HEAs) are distinctly different from traditional BCC metals. HEAs are concentrated solutions in which composition fluctuation is almost inevitable. The resultant inhomogeneities, while locally promoting kink nucleation on screw dislocations, trap them against propagation with an appreciable energy barrier, replacing kink nucleation as the rate-limiting mechanism. Edge dislocations encounter a similar activated process of nanoscale segment detrapping, with comparable activation barrier. As a result, the mobility of edge dislocations, and hence their contribution to strength, becomes comparable to screw dislocations. read less USED (high confidence) K. Yashiro, “Deformation mode analysis by eigenvectors of atomic elastic stiffness in static uniaxial tension of various fcc, bcc, and hcp metals,” AIP Advances. 2020. link Times cited: 4 Abstract: In order to clarify the physical meaning of the eigenvector … read moreAbstract: In order to clarify the physical meaning of the eigenvector of the atomic elastic stiffness matrix, Bija=Δσia/Δej, static calculations of uniaxial tension are performed on various fcc, bcc, and hcp metals with four different embedded atom method (EAM) potentials. Many fcc metals show instability for the constant volume mode, or the eigenvector of (Δexx, Δeyy, Δezz) = (±1, ∓1, 0), under the [001] tension. Bcc also loses resistance against other constant volume mode, (Δexx, Δeyy, Δezz) = (±1, ±1, ∓2), in the [001] tension. Hcp shows shear modes Δγyz and Δγzx under the [0001] tension, which correspond to atom migration by dislocation on the slip plane. Similar shear modes appear in the [111] tension of fcc and [110] tension of bcc. Hcp also changes the mode to constant volume and shear in the [1¯010] tension, which imply the deformation in the pyramidal and prismatic planes. read less USED (high confidence) C. Guan, X. Lv, Z. Han, and C. Chen, “The wetting characteristics of aluminum droplets on rough surfaces with molecular dynamics simulations.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 12 Abstract: In the present study, the impact of surface roughness on the… read moreAbstract: In the present study, the impact of surface roughness on the wettability behavior of Al droplets has been investigated via molecular dynamics (MD) simulations. In this work, amorphous carbon (AC) and graphite substrates with different depths and widths were considered. The results show that the increased width of grooves causes the transition of the wetting state from Cassie to Wenzel. Thermodynamic property analysis results indicate that the solid-liquid adhesion and the work done for the removal of the Al droplet from the solid surface decrease as the roughness increases. However, the adhesion in the Wenzel wetting state is better than that in the Cassie wetting state. Therefore, the contact angle increases with the increased roughness in the Cassie wetting systems, while in the Wenzel wetting systems, the contact angle is less than that in other rough systems. In addition, due to the heterogeneity of the surfaces, the density of Al droplets in the solid-liquid interface is decreased with the increased roughness. The anisotropic spreading of Al liquid can be explained by the MSD curves along the X and Y directions. read less USED (high confidence) D. Zeng, C. Liu, J. Jiang, and J. Fan, “Langevin dynamics with energy dissipation on periodic lattice structures modeling as an equivalent two-body method for atom-surface interactions,” Materials Research Express. 2019. link Times cited: 0 Abstract: In this work, we propose a classical equivalent two-body (ET… read moreAbstract: In this work, we propose a classical equivalent two-body (ETB) model that can capture more detailed dynamic features arising from energy dissipation and atom oscillations, by introducing the Langevin equation of a harmonic oscillator. The trapping probability, scattering angle and the residence time of Ar interacting with Pt (111) and W (110) surfaces predicted by the ETB model agree well with the measured experimental data or molecular dynamics simulations. Moreover, the ETB model is also used to study the influence of oscillating and dissipating properties on the thermal accommodation coefficients and rainbow scattering of gas atoms colliding with the surface. It is found that the dependence of energy accommodation coefficients and rainbow scattering on the oscillating and dissipating parameters shows nonlinear behaviors, and the associated mechanisms are disclosed. ETB model further provides the possibility to explore the physics beyond the existing two-body models for a better description of energy transferring during atom surface interactions. read less USED (high confidence) X. Lv, C. Guan, Z. Han, C. Chen, and Q. Sun, “Coalescence and wetting mechanism of Al droplets on different types of carbon for developing wettable cathodes: a molecular dynamics simulation.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 0 Abstract: So far, there have been few studies on the microscopic wetti… read moreAbstract: So far, there have been few studies on the microscopic wetting behavior of aluminum liquid on cathode surfaces, which is critical for developing wettable cathode materials. In the present study, an investigation on the coalescence and wetting mechanism of Al droplets on different carbonaceous substrates has been performed via molecular dynamics (MD) simulation for developing wettable cathodes. The growth rate of liquid bridge, the mean squared displacement, the balanced contact angle, and the time of full coalescence were calculated to describe the coalescence and wetting of the Al droplets. The results illustrate the sequence of full coalescence time for the Al droplets: DG < HCNT < VCNT ≈ AC and the corresponding balanced contact angles were 47.98°, 53.32°, 55.02°, and 63.12°, respectively. Furthermore, the presence of defects on DG will increase the time of coalescence and the contact angle but the directions of defects have little influence. The free energy analysis indicates that the defects reduce the solid-liquid interaction and the work done for removing the Al droplet from the substrates so that the wettability is weaker than that for perfect graphene, which also explains the balanced wettability of Al droplets on the other substrates. In addition, the surface roughness increases the contact angle of Al liquid on AC (from 62° to 113°-120°) and hence, the wettability is changed from good to poor. In general, our results can improve the understanding of the wetting of AC and graphene by Al liquid at the atomic level, which can provide direction and theoretical guidance for further research on wettable cathodes. read less USED (high confidence) A. Khomenko, M. Zakharov, and B. Persson, “Frictional Anisotropy of Al, Pt, and Pd Nanoparticles on a Graphene Substrate,” Tribology Letters. 2019. link Times cited: 3 USED (high confidence) Z.-yong Liu, B. He, X. Qu, L.-B. Niu, R. Li, and F. Wang, “Energetics and diffusion of point defects in Au/Ag metals: A molecular dynamics study,” Chinese Physics B. 2019. link Times cited: 2 USED (high confidence) W. Nöhring, W. Nöhring, and W. Curtin, “Correlation of microdistortions with misfit volumes in High Entropy Alloys,” Scripta Materialia. 2019. link Times cited: 28 USED (high confidence) R. Li, F. Li, D. Xin, J.-jun Luo, S. Chen, and Y. Zhang, “A molecular dynamics simulation of energetics and diffusion of point defects in a Au–Ag alloy,” Bulletin of Materials Science. 2019. link Times cited: 2 USED (high confidence) J. Chen, H. Zhang, and H. Xiang, “Atomistic modelling of interface structure and deformation mechanisms in the Al/GaN multilayer under compression,” Molecular Simulation. 2019. link Times cited: 3 Abstract: ABSTRACT The properties of ohmic contact and thermal boundar… read moreAbstract: ABSTRACT The properties of ohmic contact and thermal boundary conductance between Al and GaN have been studied extensively, but the interface structures and deformation mechanisms in the Al/GaN multilayer can be rarely found in literatures. By molecular dynamics (MD) simulations, we systematically studied the interface structures and structural deformations in the Al/GaN multilayer. Two kinds of interface structures are identified according to the different terminal surfaces of GaN; glide-set terminal interface and shuffle-set terminal interface. Further analysis shows that interface has the maximum stress and misfit lines have the maximum stress values, which serve as the dislocation sources in the Al layer due to the larger stress in the interface. The mechanical responses of the Al/GaN multilayer exhibit a minor stage and some distinctive drops in the stress–strain curve. The first stage is associated with the dislocation nucleation from the interface. Upon further compression, more slip systems appear in the Al layer and dislocation nucleation in GaN could induce drops in curves. Meanwhile, the multiplications of dislocations cause strain hardening behaviours. read less USED (high confidence) M. An, H. Y. Song, Q. Deng, M. Su, and Y. M. Liu, “Influence of interface with mismatch dislocations on mechanical properties of Ti/Al nanolaminate,” Journal of Applied Physics. 2019. link Times cited: 10 Abstract: As a representative boundary, interphase-interface may affec… read moreAbstract: As a representative boundary, interphase-interface may affect the strength or ductility of multilayered composites dramatically. However, the effect of the interface with mismatch dislocations on the mechanical behavior of multilayered composites is still not clear. In the present work, we performed molecular dynamics simulations to investigate the effect of interface structures and layer spacing on the mechanical properties of the Ti/Al nanolaminate. The results indicate that there are two transitions of the plastic deformation mechanism in the Ti layer with the increase of layer spacing in the sample with a coherent interface. The plastic deformation mechanism evolves from one that is dominated by dislocation to the phase transformation from the hcp-Ti to the fcc-Ti mode, which transfers to the dislocation slip deformation again. For the samples with an incoherent interface, the plastic deformation is dominated by the transformation from hcp-Ti to fcc-Ti, regardless of the variation of layer spacing, while the plastic deformations in the Al layers are mainly dislocations confined in the layer slip in the samples with both coherent and incoherent interfaces. When the layer spacing is larger than 6.6 nm, an obvious second hardening is observed due to the superior dislocation storage ability of the Ti/Al laminate with the incoherent interface. Meanwhile, extraordinary ductility is obtained when optimal layer spacing is employed in the Ti/Al laminate. Moreover, the phase transformation mechanism of hcp-Ti to bcc-Ti has also been explicated in the present work. The general conclusions derived from this work may provide a guideline for the design of high-performance Ti/Al multilayer and alloy devices.As a representative boundary, interphase-interface may affect the strength or ductility of multilayered composites dramatically. However, the effect of the interface with mismatch dislocations on the mechanical behavior of multilayered composites is still not clear. In the present work, we performed molecular dynamics simulations to investigate the effect of interface structures and layer spacing on the mechanical properties of the Ti/Al nanolaminate. The results indicate that there are two transitions of the plastic deformation mechanism in the Ti layer with the increase of layer spacing in the sample with a coherent interface. The plastic deformation mechanism evolves from one that is dominated by dislocation to the phase transformation from the hcp-Ti to the fcc-Ti mode, which transfers to the dislocation slip deformation again. For the samples with an incoherent interface, the plastic deformation is dominated by the transformation from hcp-Ti to fcc-Ti, regardless of the variation of layer spacing, wh... read less USED (high confidence) H. Lin, T. Li, and H. Li, “Molecular dynamics study on the heterogeneous nucleation of liquid Al-Cu alloys on different kinds of copper substrates.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 5 Abstract: Al-Cu alloys are widely used in aeronautics and aerospace en… read moreAbstract: Al-Cu alloys are widely used in aeronautics and aerospace engineering because they exhibit outstanding performance. However, their casting characteristics are poor. Heterogeneous nucleation plays a significant role in controlling crystal growth. MD simulations are performed to explore the heterogeneous nucleation of liquid Al-Cu on a copper substrate including the heat-up process, preservation process, and freezing process. The simulation results show that the Al-Cu melt becomes layered at the liquid-solid interface and tends to be ordered in the structure because of the induced effect from the substrate. The crystal structure information is found to be gradually delivered from the substrate to the liquid, and this transmission capacity of information decays with increasing distance from the substrate. The liquid Al-Cu alloy with high copper content frozen on the single substrate tends to form a perfect crystal structure more easily, but the Al-Cu melt with low copper content between two copper substrates tends to form an arch-shaped structure, which can disappear when the copper content reaches a specific proportion. Moreover, different angles of the grooved substrates also affect the heterogeneous nucleation of the Al-Cu melt and its solidified structure. Our findings provide new insights into the defect and structural changes during heterogeneous nucleation, and our findings also promote leading-edge studies, which can provide new ideas to mechanical research. read less USED (high confidence) X. W. Zhou, M. E. Foster, and R. Sills, “An Fe‐Ni‐Cr embedded atom method potential for austenitic and ferritic systems,” Journal of Computational Chemistry. 2018. link Times cited: 65 Abstract: Fe‐Ni‐Cr stainless‐steels are important structural materials… read moreAbstract: Fe‐Ni‐Cr stainless‐steels are important structural materials because of their superior strength and corrosion resistance. Atomistic studies of mechanical properties of stainless‐steels, however, have been limited by the lack of high‐fidelity interatomic potentials. Here using density functional theory as a guide, we have developed a new Fe‐Ni‐Cr embedded atom method potential. We demonstrate that our potential enables stable molecular dynamics simulations of stainless‐steel alloys at high temperatures, accurately reproduces the stacking fault energy—known to strongly influence the mode of plastic deformation (e.g., twinning vs. dislocation glide vs. cross‐slip)—of these alloys over a range of compositions, and gives reasonable elastic constants, energies, and volumes for various compositions. The latter are pertinent for determining short‐range order and solute strengthening effects. Our results suggest that our potential is suitable for studying mechanical properties of austenitic and ferritic stainless‐steels which have vast implementation in the scientific and industrial communities. Published 2018. This article is a U.S. Government work and is in the public domain in the USA. read less USED (high confidence) J. Balogh et al., “Asymmetric alloy formation at the Fe-on-Ti and Ti-on-Fe interfaces,” Journal of Physics: Condensed Matter. 2018. link Times cited: 3 Abstract: The Fe-on-Ti and Ti-on-Fe interfaces were studied experiment… read moreAbstract: The Fe-on-Ti and Ti-on-Fe interfaces were studied experimentally by Mössbauer spectroscopy (MS), transmission electron microscopy (TEM) and x-ray reflectometry (XRR) on Ti/Fe/Ti trilayers grown on Si(1 1 1) substrates by vacuum evaporation. The nanoscale structure and composition were explored in cross sections using TEM, the layer structure and the interface widths by specular x-ray reflectometry. MS was applied to identify the interface alloy phases and to determine the pure and alloyed Fe layer fractions. The experimental results were compared with molecular dynamics (MD) simulations of layer growth on Fe or Ti underlayers of different orientations. The concentration distributions provided by MD simulations show an asymmetry at the interfaces in the layer growth direction. The transition is atomically sharp at the Ti-on-Fe interface for the (0 0 1) and (1 1 0) crystallographic orientations of the Fe underlayer, while it spreads over a few atomic layers for Fe(1 1 1) underlayer and for all studied Ti underlayer orientations at the Fe-on-Ti interface. MS and XRR data on Ti/Fe/Ti trilayers confirm the asymmetry between the bottom and top Fe interface, but the inferred interface widths considerable exceed those deduced from the MD simulations. read less USED (high confidence) Z. Zhang, P. Chen, and F. Qin, “Molecular dynamic simulation of grain size and work temperature effect on mechanical properties of polycrystalline copper,” 2018 19th International Conference on Electronic Packaging Technology (ICEPT). 2018. link Times cited: 2 Abstract: Polycrystalline copper is considered by the industries to be… read moreAbstract: Polycrystalline copper is considered by the industries to be the best TSV electroplating material and other interconnected structure material because of its ultra-low resistivity, high conductivity, low electro-migration rate and good compatibility with the multilayer interconnects process. Its mechanical properties are completely different from the bulk monocrystalline copper, these properties are critically vital to evaluate the thermomechanical reliability of TSV and interconnected structure in 3D packaging. To investigate the effect of the work temperature and grain size on the mechanical properties of polycrystalline Cu, The MD simulations of uniaxial tensile test are performed. The results showed that the elastic modulus gradually increases with the augment of the mean grain size, and the corresponding flow stress concurrently increases, and the flow stress is proportional to the square-root of the grain size, which satisfies the inverse-Hall-Petch relation. It also turned that the elastic modulus decreases with increasing of ambient temperatures, the flow stress is negatively correlated with the temperature. From all the tensile simulation tests, it was confirmed that the mechanism of plastic deformation for polycrystalline copper with 4.65–9.31nm grain sizes is mainly the grain’s rotation and grain- boundary sliding, the dislocation nucleation and migration is no longer the dominant factor of plastic deformation. read less USED (high confidence) F. Rahmani, J. Jeon, S. Jiang, and S. Nouranian, “Melting and solidification behavior of Cu/Al and Ti/Al bimetallic core/shell nanoparticles during additive manufacturing by molecular dynamics simulation,” Journal of Nanoparticle Research. 2018. link Times cited: 28 USED (high confidence) P. Zhu and R. Li, “Study of Nanoscale Friction Behaviors of Graphene on Gold Substrates Using Molecular Dynamics,” Nanoscale Research Letters. 2018. link Times cited: 15 USED (high confidence) M. Sun et al., “Enhanced inter-diffusion of immiscible elements Fe/Cu at the interface of FeZr/CuZr amorphous multilayers,” Materials Research Letters. 2018. link Times cited: 11 Abstract: ABSTRACT Fe75Zr25/Cu64Zr36 amorphous multilayers were prepar… read moreAbstract: ABSTRACT Fe75Zr25/Cu64Zr36 amorphous multilayers were prepared by magnetron sputtering. Atom probe tomography was employed to analyze the atomic inter-diffusion at the interface of the multilayers before and after annealing (573 K, 60 min). An unexpected enhanced inter-diffusion of the immiscible elements Fe and Cu was detected at the interface of the multilayers. As the inter-diffusion in amorphous multilayers is much faster than that in the crystalline counterparts, this process may open a way to manipulate or create amorphous multilayers with new properties. This idea agrees with the observation of the variation of magnetic properties of Fe75Zr25/Cu64Zr36 amorphous multilayers. GRAPHICAL ABSTRACT IMPACT STATEMENT This paper reveals the enhanced atomic inter-diffusion at the interface of amorphous materials, and may open a way to manipulate or create amorphous multilayers with new properties. read less USED (high confidence) Yuan-Yuan 圆圆 Tian 田, Jia 甲 Li 李, Ze-Ying 泽英 Hu 胡, Zhi-Peng 志鹏 Wang 王, and Q. Fang 方, “Molecular dynamics study of plastic deformation mechanism in Cu/Ag multilayers,” Chinese Physics B. 2017. link Times cited: 4 Abstract: The plastic deformation mechanism of Cu/Ag multilayers is in… read moreAbstract: The plastic deformation mechanism of Cu/Ag multilayers is investigated by molecular dynamics (MD) simulation in a nanoindentation process. The result shows that due to the interface barrier, the dislocations pile-up at the interface and then the plastic deformation of the Ag matrix occurs due to the nucleation and emission of dislocations from the interface and the dislocation propagation through the interface. In addition, it is found that the incipient plastic deformation of Cu/Ag multilayers is postponed, compared with that of bulk single-crystal Cu. The plastic deformation of Cu/Ag multilayers is affected by the lattice mismatch more than by the difference in stacking fault energy (SFE) between Cu and Ag. The dislocation pile-up at the interface is determined by the obstruction of the mismatch dislocation network and the attraction of the image force. Furthermore, this work provides a basis for further understanding and tailoring metal multilayers with good mechanical properties, which may facilitate the design and development of multilayer materials with low cost production strategies. read less USED (high confidence) T. Liang, D. Zhou, Z. Wu, and P. Shi, “Size-dependent melting modes and behaviors of Ag nanoparticles: a molecular dynamics study,” Nanotechnology. 2017. link Times cited: 25 Abstract: The size-dependent melting behaviors and mechanisms of Ag na… read moreAbstract: The size-dependent melting behaviors and mechanisms of Ag nanoparticles (NPs) with diameters of 3.5–16 nm were investigated by molecular dynamics (MD). Two distinct melting modes, non-premelting and premelting with transition ranges of about 7–8 nm, for Ag NPs were demonstrated via the evolution of distribution and transition of atomic physical states during annealing. The small Ag NPs (3.5–7 nm) melt abruptly without a stable liquid shell before the melting point, which is characterized as non-premelting. A solid-solid crystal transformation is conducted through the migration of adatoms on the surface of Ag NPs with diameters of 3.5–6 nm before the initial melting, which is mainly responsible for slightly increasing the melting point of Ag NPs. On the other hand, surface premelting of Ag NPs with diameters of 8–16 nm propagates from the outer shell to the inner core with initial anisotropy and late isotropy as the temperature increases, and the close-packed facets {111} melt by a side-consumed way which is responsible for facets {111} melting in advance relative to the crystallographic plane {111}. Once a stable liquid shell is formed, its size-independent minimum thickness is obtained, and a three-layer structure of atomic physical states is set up. Lastly, the theory of point defect-pair (vacancy-interstitial) severing as the mechanism of formation and movement of the solid-liquid interface was also confirmed. Our study provides a basic understanding and theoretical guidance for the research, production and application of Ag NPs. read less USED (high confidence) T. Li, Z. Wang, Y. Duan, J. Li, and H. Li, “Molecular dynamics study on the formation of self-organized core/shell structures in the Pb alloy at the nanoscale,” RSC Advances. 2017. link Times cited: 10 Abstract: Herein, we report a self-organized core/shell (CS) structure… read moreAbstract: Herein, we report a self-organized core/shell (CS) structure consisting of an Al core and a Pb shell in the liquid Pb alloys at a constant temperature. This is contrary to a believed opinion that the thermal gradient acts as the only driving force for the formation of this kind of structure. The results show that its forming ability greatly depends on the composition and temperature. Importantly, when the alloys are placed in the confined space, a structure of Al–Pb–Al sandwich construction and a completely different CS structure composed of a Pb core and an Al shell are obtained; this suggests a new strategy for controlling the phase-separated structures at the nanoscale. More interestingly, an abnormal phenomenon in the low Pb alloys with the amorphous thin Pb shell and the crystal Al core is observed after solidification, which does not occur in the high Pb alloys that just possess a secondary wrapped structure that never appears in the liquid state. The result of this study may help to shed light on understanding the formation of this core/shell structure and controlling it at an atomic view that have a potential application in the fabrication of these structural materials through nanotechnology. read less USED (high confidence) E. S. Cho et al., “Hierarchically Controlled Inside‐Out Doping of Mg Nanocomposites for Moderate Temperature Hydrogen Storage,” Advanced Functional Materials. 2017. link Times cited: 69 Abstract: Demand for pragmatic alternatives to carbon‐intensive fossil… read moreAbstract: Demand for pragmatic alternatives to carbon‐intensive fossil fuels is growing more strident. Hydrogen represents an ideal zero‐carbon clean energy carrier with high energy density. For hydrogen fuel to compete with alternatives, safe and high capacity storage materials that are readily cycled are imperative. Here, development of such a material, comprised of nickel‐doped Mg nanocrystals encapsulated by molecular‐sieving reduced graphene oxide (rGO) layers, is reported. While most work on advanced hydrogen storage composites to date endeavor to explore either nanosizing or addition of carbon materials as secondary additives individually, methods to enable both are pioneered: “dual‐channel” doping combines the benefits of two different modalities of enhancement. Specifically, both external (rGO strain) and internal (Ni doping) mechanisms are used to efficiently promote both hydriding and dehydriding processes of Mg nanocrystals, simultaneously achieving high hydrogen storage capacity (6.5 wt% in the total composite) and excellent kinetics while maintaining robustness. Furthermore, hydrogen uptake is remarkably accomplished at room temperature and also under 1 bar—as observed during in situ measurements—which is a substantial advance for a reversible metal hydride material. The realization of three complementary functional components in one material breaks new ground in metal hydrides and makes solid‐state materials viable candidates for hydrogen‐fueled applications. read less USED (high confidence) P. Quaino et al., “Understanding the structure and reactivity of NiCu nanoparticles: an atomistic model.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 7 Abstract: The structure of bimetallic NiCu nanoparticles (NP) is inves… read moreAbstract: The structure of bimetallic NiCu nanoparticles (NP) is investigated as a function of their composition and size by means of Lattice MonteCarlo (LMC) and molecular dynamics (MD) simulations. According to our results, copper segregation takes place at any composition of the particles. We found that this feature is not size-dependent. In contrast, nickel segregation depends on the NP size. When the size increases, Ni atoms tend to remain in the vicinity of the surface and deeper. For smaller NPs, Ni atoms are located at the surface as well. Our results also showed that most of the metal atoms segregated at the surface area were found to decorate edges and/or form islands. Our findings agree qualitatively with the experimental data found in the literature. In addition, we comment on the reactivity of these nanoparticles. read less USED (high confidence) C. Sabater, W. Dednam, M. R. Calvo, M. Fern’andez, C. Untiedt, and M. Caturla, “Role of first-neighbor geometry in the electronic and mechanical properties of atomic contacts,” Physical Review B. 2017. link Times cited: 10 Abstract: This work has been funded from the Spanish Ministerio de Edu… read moreAbstract: This work has been funded from the Spanish Ministerio de Educacion y Ciencia through Grants No. FIS2013-47328 and No. MAT2016-78625 and the Conselleria d’Educacio, Investigacio, Cultura i Esport de la Generalitat Valenciana, PROMETEO/2017/139. C.S. gratefully acknowledges financial support from SEPE Servicio Publico de Empleo Estatal. W.D. acknowledges funding from the National Research Foundation of South Africa through the Innovation Doctoral scholarship programme, Grant UID 102574. read less USED (high confidence) G. Li, H. Wu, H. Luo, Z. Chen, A. Tay, and W. Zhu, “Diffusion behavior of Cu/Ta heterogeneous interface under high temperature and high strain: An atomistic investigation,” AIP Advances. 2017. link Times cited: 6 Abstract: Three-dimensional (3D) integration technology using Cu inter… read moreAbstract: Three-dimensional (3D) integration technology using Cu interconnections has emerged as a promising solution to improve the performance of silicon microelectronic devices. However, Cu diffuses into SiO2 and requires a barrier layer such as Ta to ensure acceptable reliability. In this paper, the effects of temperature and strain normal to the interface on the inter-diffusion of Cu and Ta at annealing conditions are investigated using a molecular dynamics (MD) technique with embedded atomic method (EAM) potentials. Under thermal annealing conditions without strain, it is found that a Cu-rich diffusion region approximately 2 nm thick is formed at 1000 K after 10 ns of annealing. Ta is capable of diffusing into the interior of Cu but Cu hardly diffuses into the inner lattice of Ta. At the Cu side near the interface an amorphous structure is formed due to the process of diffusion. The diffusion activation energy of Cu and Ta are found to be 0.9769 and 0.586 eV, respectively. However, when a strain is applied, a... read less USED (high confidence) Y. Wei and S. Peng, “The stress-velocity relationship of twinning partial dislocations and the phonon-based physical interpretation,” Science China Physics, Mechanics & Astronomy. 2017. link Times cited: 14 USED (high confidence) J. Wang, J. Bian, X. Niu, and G. Wang, “A universal method to calculate the surface energy density of spherical surfaces in crystals,” Acta Mechanica Sinica. 2017. link Times cited: 6 USED (high confidence) A. Georgi et al., “Tuning the Pseudospin Polarization of Graphene by a Pseudomagnetic Field.,” Nano letters. 2016. link Times cited: 104 Abstract: One of the intriguing characteristics of honeycomb lattices … read moreAbstract: One of the intriguing characteristics of honeycomb lattices is the appearance of a pseudomagnetic field as a result of mechanical deformation. In the case of graphene, the Landau quantization resulting from this pseudomagnetic field has been measured using scanning tunneling microscopy. Here we show that a signature of the pseudomagnetic field is a local sublattice symmetry breaking observable as a redistribution of the local density of states. This can be interpreted as a polarization of graphene's pseudospin due to a strain induced pseudomagnetic field, in analogy to the alignment of a real spin in a magnetic field. We reveal this sublattice symmetry breaking by tunably straining graphene using the tip of a scanning tunneling microscope. The tip locally lifts the graphene membrane from a SiO2 support, as visible by an increased slope of the I(z) curves. The amount of lifting is consistent with molecular dynamics calculations, which reveal a deformed graphene area under the tip in the shape of a Gaussian. The pseudomagnetic field induced by the deformation becomes visible as a sublattice symmetry breaking which scales with the lifting height of the strained deformation and therefore with the pseudomagnetic field strength. Its magnitude is quantitatively reproduced by analytic and tight-binding models, revealing fields of 1000 T. These results might be the starting point for an effective THz valley filter, as a basic element of valleytronics. read less USED (high confidence) J. Wang, J. Bian, X. Niu, and G. Wang, “A universal method to calculate the surface energy density of spherical surfaces in crystals,” Acta Mechanica Sinica. 2016. link Times cited: 0 USED (high confidence) T. Li, W. Wu, and H. Li, “Coalescence behavior of liquid immiscible metal drops in two-wall confinement.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 19 Abstract: Molecular dynamics (MD) simulations are performed to investi… read moreAbstract: Molecular dynamics (MD) simulations are performed to investigate the coalescence of the liquid Al and Pb drops in the graphene (G) walls and pillared-graphene (PG) walls. The confining walls can affect the coalescence dynamics of two adjacent films by restricting the movement of one of the metal drops; however, the coalescence behavior is different in the G-walls and the PG-walls. Two un-contacted films can still merge into one bigger drop because of the restricting effect of the walls, in which the movement of the Pb drop plays a predominant role. The coalescence time decreases with the decrease of the confining space. Our findings demonstrate that the coalescence dynamics can be controlled by tuning the confining space or wall surface. read less USED (high confidence) H. Li, W. Wu, and K. Zhang, “Structural and Dynamical Properties of Metallic Glassy Films.” 2016. link Times cited: 0 Abstract: In this chapter, a series of molecular dynamics simulations … read moreAbstract: In this chapter, a series of molecular dynamics simulations have been carried out to explore structural and dynamical features of monatomic liquid metallic films during rapid cooling. Results show a semi‐ordered inhomogeneous morphology containing crystal‐like and disordered regions. The icosahedron contributes to nucleation through the synergy with other short‐range ordered structures and participates in crystal growth via assimilation, but the pinning effect should be overcome. The second‐peak splitting in pair correlation functions is found as the result of a statistical average of crystal‐ like and disordered structural regions, not just the amorphous structure. The splitting can be viewed as a prototype of crystal‐like peaks exhibiting distorted and vestigial features. Besides, we use the parameter P(a, τ, ν) for predicting both local structural order and motion propensity. The fraction of crystalline clusters follows a negative power‐law scaling with the cooling rate increasing, which is the inverse of P(a, τ, ν). read less USED (high confidence) L. Wan, X.-xiang Yu, X. Zhou, and G. Thompson, “Interrelationship of in situ growth stress evolution and phase transformations in Ti/W multilayered thin films,” Journal of Applied Physics. 2016. link Times cited: 8 Abstract: This paper addresses the in situ growth stress evolution and… read moreAbstract: This paper addresses the in situ growth stress evolution and phase transformation of bcc to hcp Ti in Ti/W multilayered thin films. A series of equal layer thicknesses from 20 nm to 1 nm were deposited. As the bilayer thickness reduced, the overall film stress became less compressive until the Ti transformed from hcp (at the larger layer thicknesses) to bcc in the 1 nm/1 nm multilayer. The pseudomorphic bcc stabilization resulted in a recovery of the compressive stress to values near that for the bulk phase stabilized for the 5 nm/5 nm multilayer. A discernable change in stress slope was noted for the bcc to hcp Ti transition as a function of Ti layer thickness. The stress states for each film, during film growth, are rationalized by the lattice matching of the phase with the growth surface. These results are coupled to a molecular dynamics deposition simulation which revealed good agreement with the experimentally observed transformation thickness. read less USED (high confidence) X. Zhou et al., “Heterogeneous nucleation of Al melt in symmetrical or asymmetrical confined nanoslits.,” Nanoscale. 2016. link Times cited: 5 Abstract: MD simulations are performed to study the solidification of … read moreAbstract: MD simulations are performed to study the solidification of Al melt in confined nanoslits (NSs) constructed by identical or different substrates, as well as on Fe substrates. Compared to the single substrate, the confined NS could promote the crystallization of Al melt, and its size has a significant impact on the solidified structure. In symmetrical NSs, liquid Al atoms would stack based on the atomic arrangement mode of the substrate, however in asymmetrical confined NSs, the atomic arrangement mode of liquid Al is governed by the constitution of asymmetrical substrates. Specifically, for the NS formed by Fe(110) and Fe(111) substrates, the induced region from the Fe(110) substrate is much bigger than that from Fe(111). Moreover, the freezing of liquid Al in asymmetrical NSs constructed from copper and iron has also been studied. These results throw light on heterogeneous nucleation in confined space. read less USED (high confidence) K. Ito, Y. Ichikawa, and K. Ogawa, “Experimental and Numerical Analyses on the Deposition Behavior of Spherical Aluminum Particles in the Cold-Spray-Emulated High-Velocity Impact Process,” Materials Transactions. 2016. link Times cited: 4 Abstract: Understanding the deposition mechanism of ne solid particles… read moreAbstract: Understanding the deposition mechanism of ne solid particles is essential for the effective use of the cold spray (CS) technique, which is used to synthesize dense and thick metallic coatings. As such, in this study, the deposition behaviors of spherical pure aluminum particles were investigated in detail in order to understand the deposition mechanism; these particles had a diameter of 1 mm and were deposited on ve metallic substrate materials, during the CS-emulated high-velocity impact process. A single particle impact testing system, which is a modi ed single-stage light gas gun, was used to evaluate the deposition process. This evaluation con rmed that the critical velocities of Al particles vary signi cantly with the substrate material. In order to identify the dominant factors, the bonding energies, rebound velocities, plastic deformation experienced by the particles and substrates, and removability of the natural oxide lms were evaluated. The results revealed that the critical velocities increased signi cantly with increasing Ar sputtering time required for complete removal of the natural oxide lm; this time represents the removability of the lm. This result con rms that, of the factors considered, the removability of the natural oxide lm exerts the most in uence on Al particle deposition on metallic substrates. [doi:10.2320/matertrans.T-M2016803] read less USED (high confidence) A. Dutta, A. Raychaudhuri, and T. Saha‐Dasgupta, “Plasticity-mediated collapse and recrystallization in hollow copper nanowires: a molecular dynamics simulation,” Beilstein Journal of Nanotechnology. 2016. link Times cited: 2 Abstract: We study the thermal stability of hollow copper nanowires us… read moreAbstract: We study the thermal stability of hollow copper nanowires using molecular dynamics simulation. We find that the plasticity-mediated structural evolution leads to transformation of the initial hollow structure to a solid wire. The process involves three distinct stages, namely, collapse, recrystallization and slow recovery. We calculate the time scales associated with different stages of the evolution process. Our findings suggest a plasticity-mediated mechanism of collapse and recrystallization. This contradicts the prevailing notion of diffusion driven transport of vacancies from the interior to outer surface being responsible for collapse, which would involve much longer time scales as compared to the plasticity-based mechanism. read less USED (high confidence) M. Warrier, U. Bhardwaj, H. Hemani, R. Schneider, A. Mutzke, and M. C. Valsakumar, “Statistical study of defects caused by primary knock-on atoms in fcc Cu and bcc W using molecular dynamics,” Journal of Nuclear Materials. 2015. link Times cited: 20 USED (high confidence) A. Mayer and P. N. Mayer, “Strength of solid and molten aluminum under dynamic tension,” JETP Letters. 2015. link Times cited: 12 USED (high confidence) P. N. Mayer and A. Mayer, “Model of fracture of metal melts and the strength of melts under dynamic conditions,” Journal of Experimental and Theoretical Physics. 2015. link Times cited: 6 USED (high confidence) M. Yang, J.-G. Xu, H. Song, and Y.-G. Zhang, “Effects of tilt interface boundary on mechanical properties of Cu/Ni nanoscale metallic multilayer composites,” Chinese Physics B. 2015. link Times cited: 2 Abstract: The effect of tilt interfaces and layer thickness of Cu/Ni m… read moreAbstract: The effect of tilt interfaces and layer thickness of Cu/Ni multilayer nanowires on the deformation mechanism are investigated by molecular dynamics simulations. The results indicate that the plasticity of the sample with a 45° tilt angle is much better than the others. The yield stress is found to decrease with increasing the tilt angle and it reaches its lowest value at 33°. Then as the tilt angle continues to increase, the yield strength increases. Furthermore, the studies show that with the decrease of layer thickness, the yield strength gradually decreases. The study also reveals that these different deformation behaviors are associated with the glide of dislocation. read less USED (high confidence) A. Mayer and P. N. Mayer, “Continuum model of tensile fracture of metal melts and its application to a problem of high-current electron irradiation of metals,” Journal of Applied Physics. 2015. link Times cited: 36 Abstract: A continuum model of the metal melt fracture is formulated o… read moreAbstract: A continuum model of the metal melt fracture is formulated on the basis of the continuum mechanics and theory of metastable liquid. A character of temperature and strain rate dependences of the tensile strength that is predicted by the continuum model is verified, and parameters of the model are fitted with the use of the results of the molecular dynamics simulations for ultra-high strain rates (≥1–10/ns). A comparison with experimental data from literature is also presented for Al and Ni melts. Using the continuum model, the dynamic tensile strength of initially uniform melts of Al, Cu, Ni, Fe, Ti, and Pb within a wide range of strain rates (from 1–10/ms to 100/ns) and temperatures (from melting temperature up to 70–80% of critical temperature) is calculated. The model is applied to numerical investigation of a problem of the high-current electron irradiation of Al, Cu, and Fe targets. read less USED (high confidence) V. Borysiuk and I. Lyashenko, “Atomistic simulation of the melting behavior of the Au-Ag nanoparticles with core-shell structure,” 2015 IEEE 35th International Conference on Electronics and Nanotechnology (ELNANO). 2015. link Times cited: 4 Abstract: Melting of the Au-Ag nanoparticle with core-shell structure … read moreAbstract: Melting of the Au-Ag nanoparticle with core-shell structure was investigated by molecular dynamic simulation. Structural behavior of the nanoparticle was investigated in the range of temperature from 300 to 1300K. To detect the melting point Lindemann index was calculated during the simulation in the whole temperature range. It is shown that the melting of the Au-Ag nanoparticle under consideration occurs at the temperature of about 900 K. To detect the structural changes the radial distribution function was computed before and after melting. It is shown that the core-shell structure of the nanoparticle is not preserved due to the increased diffusion. read less USED (high confidence) A. Dongare, “Quasi-coarse-grained dynamics: modelling of metallic materials at mesoscales,” Philosophical Magazine. 2014. link Times cited: 26 Abstract: A computationally efficient modelling method called quasi-co… read moreAbstract: A computationally efficient modelling method called quasi-coarse-grained dynamics (QCGD) is developed to expand the capabilities of molecular dynamics (MD) simulations to model behaviour of metallic materials at the mesoscales. This mesoscale method is based on solving the equations of motion for a chosen set of representative atoms from an atomistic microstructure and using scaling relationships for the atomic-scale interatomic potentials in MD simulations to define the interactions between representative atoms. The scaling relationships retain the atomic-scale degrees of freedom and therefore energetics of the representative atoms as would be predicted in MD simulations. The total energetics of the system is retained by scaling the energetics and the atomic-scale degrees of freedom of these representative atoms to account for the missing atoms in the microstructure. This scaling of the energetics renders improved time steps for the QCGD simulations. The success of the QCGD method is demonstrated by the prediction of the structural energetics, high-temperature thermodynamics, deformation behaviour of interfaces, phase transformation behaviour, plastic deformation behaviour, heat generation during plastic deformation, as well as the wave propagation behaviour, as would be predicted using MD simulations for a reduced number of representative atoms. The reduced number of atoms and the improved time steps enables the modelling of metallic materials at the mesoscale in extreme environments. read less USED (high confidence) A. Bolesta and V. Fomin, “Molecular dynamics simulation of polycrystalline copper,” Journal of Applied Mechanics and Technical Physics. 2014. link Times cited: 11 USED (high confidence) A. Bolesta and V. Fomin, “Molecular dynamics simulation of polycrystalline copper,” Journal of Applied Mechanics and Technical Physics. 2014. link Times cited: 0 USED (high confidence) O. Sharia, J. Holzgrafe, N. Park, and G. Henkelman, “Rare event molecular dynamics simulations of plasma induced surface ablation.,” The Journal of chemical physics. 2014. link Times cited: 2 Abstract: The interaction of thermal Ar plasma particles with Si and W… read moreAbstract: The interaction of thermal Ar plasma particles with Si and W surfaces is modeled using classical molecular dynamics (MD) simulations. At plasma energies above the threshold for ablation, the ablation yield can be calculated directly from MD. For plasma energies below threshold, the ablation yield becomes exponentially low, and direct MD simulations are inefficient. Instead, we propose an integration method where the yield is calculated as a function of the Ar incident kinetic energy. Subsequent integration with a Boltzmann distribution at the temperature of interest gives the thermal ablation yield. At low plasma temperatures, the ablation yield follows an Arrhenius form in which the activation energy is shown to be the threshold energy for ablation. Interestingly, equilibrium material properties, including the surface and bulk cohesive energy, are not good predictors of the threshold energy for ablation. The surface vacancy formation energy is better, but is still not a quantitative predictor. An analysis of the trajectories near threshold shows that ablation occurs by different mechanisms on different material surfaces, and both the mechanism and the binding of surface atoms determine the threshold energy. read less USED (high confidence) F. Dai and W.-Z. Zhang, “Identification of Secondary Dislocations by Singular Value Decomposition of the Nye Tensor,” Acta Metallurgica Sinica (English Letters). 2014. link Times cited: 7 USED (high confidence) A. Bolesta and V. Fomin, “Phase transition behind a shock front in polycrystalline copper,” Doklady Physics. 2014. link Times cited: 7 USED (high confidence) F. Ulomek and V. Mohles, “Molecular dynamics simulations of grain boundary mobility in Al, Cu and γ-Fe using a symmetrical driving force,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 13 Abstract: We present a new artificial driving force for the determinat… read moreAbstract: We present a new artificial driving force for the determination of grain boundary mobility by molecular dynamics. The new driving force is a symmetric version of the synthetic driving force formerly introduced by Janssens et al 2006 Nature Mater. 5 124–7. The new version depends on two orientation parameters instead of one. We analyze the advantages and disadvantages of these two driving force methods. Grain boundary mobilities are simulated for eight symmetric CSL tilt grain boundaries in Al, Cu and γ-Fe, and two MD potentials for each of these materials. Boundary conditions are kept as similar as possible to show the influence of the different materials and to compare to the influence of the different MD potential types on simulated GB mobilities. We find that the newly introduced artificial driving force is a slight improvement, but it cannot remove the shortcomings of the original approach. Also, it is found that the differences in calculated MD mobilities between different materials are of the same order as those between different MD potentials of any one element. Sources for such differences are identified and classified by severity. read less USED (high confidence) M. Park, K. C. Alexander, and C. Schuh, “Diffusion of tungsten in chromium: Experiments and atomistic modeling,” Journal of Alloys and Compounds. 2014. link Times cited: 12 USED (high confidence) K. A. Bukreeva, A. Iskandarov, S. Dmitriev, Y. Umeno, and R. Mulyukov, “Theoretical shear strength of FCC and HCP metals,” Physics of the Solid State. 2014. link Times cited: 12 USED (high confidence) X.-Y. Li, Y. He, Y. Wang, J. Dong, and H. Li, “Dewetting Properties of Metallic Liquid Film on Nanopillared Graphene,” Scientific Reports. 2014. link Times cited: 23 USED (high confidence) M. H. Ulz and S. Moran, “Optimal kernel shape and bandwidth for atomistic support of continuum stress,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 6 Abstract: The treatment of atomistic scale interactions via molecular … read moreAbstract: The treatment of atomistic scale interactions via molecular dynamics simulations has recently found favour for multiscale modelling within engineering. The estimation of stress at a continuum point on the atomistic scale requires a pre-defined kernel function. This kernel function derives the stress at a continuum point by averaging the contribution from atoms within a region surrounding the continuum point. This averaging volume, and therefore the associated stress at a continuum point, is highly dependent on the bandwidth and shape of the kernel. In this paper we propose an effective and entirely data-driven strategy for simultaneously computing the optimal shape and bandwidth for the kernel. We thoroughly evaluate our proposed approach on copper using three classical elasticity problems. Our evaluation yields three key findings: firstly, our technique can provide a physically meaningful estimation of kernel bandwidth; secondly, we show that a uniform kernel is preferred, thereby justifying the default selection of this kernel shape in future work; and thirdly, we can reliably estimate both of these attributes in a data-driven manner, obtaining values that lead to an accurate estimation of the stress at a continuum point. read less USED (high confidence) C. R. Freeze, X. Ji, A. Kingon, and D. Irving, “Impact of Joule heating, roughness, and contaminants on the relative hardness of polycrystalline gold,” Journal of Physics: Condensed Matter. 2013. link Times cited: 0 Abstract: Asperities play a central role in the mechanical and electri… read moreAbstract: Asperities play a central role in the mechanical and electrical properties of contacting surfaces. Changes in trends of uniaxial compression of an asperity tip in contact with a polycrystalline substrate as a function of substrate geometry, compressive stress and applied voltage are investigated here by implementation of a coupled continuum and atomistic approach. Surprisingly, an unmodified Au polycrystalline substrate is found to be softer than one containing a void for conditions of high stress and an applied voltage of 0.2 V. This is explained in terms of the temperature distribution and weakening of Au as a function of temperature. The findings in this communication are important to the design of materials for electrical contacts because applied conditions may play a role in reversing relative hardness of the materials for conditions experienced during operation. read less USED (high confidence) Yuan-Yuan 圆圆 Ju 巨, Q. Zhang 张, Zi-Zheng 自正 Gong 龚, and Guang-Fu 广富 Ji 姬, “Molecular dynamics simulation of self-diffusion coefficients for liquid metals,” Chinese Physics B. 2013. link Times cited: 25 Abstract: The temperature-dependent coefficients of self-diffusion for… read moreAbstract: The temperature-dependent coefficients of self-diffusion for liquid metals are simulated by molecular dynamics methods based on the embedded-atom-method (EAM) potential function. The simulated results show that a good inverse linear relation exists between the natural logarithm of self-diffusion coefficients and temperature, though the results in the literature vary somewhat, due to the employment of different potential functions. The estimated activation energy of liquid metals obtained by fitting the Arrhenius formula is close to the experimental data. The temperature-dependent shear-viscosities obtained from the Stokes—Einstein relation in conjunction with the results of molecular dynamics simulation are generally consistent with other values in the literature. read less USED (high confidence) M. H. Ulz and S. Moran, “A Gaussian mixture modelling approach to the data-driven estimation of atomistic support for continuum stress,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 7 Abstract: Recent developments in multiscale modelling include the trea… read moreAbstract: Recent developments in multiscale modelling include the treatment of atomistic scale interactions via molecular dynamics simulations. The atomistic stress definition at a given continuum point contains a space-averaging volume over nearby atoms to provide an averaged macroscopic stress measure. Previous work on atomistic stress measures introduce the size of this volume as an a priori given parameter. In this contribution we let the atomistic data speak for itself by hypothesizing that the influence between atoms can be effectively estimated from their relative spatial position and stress. Atoms with highly similar spatial position and stress should therefore be contained within the same space-averaging volume. We motivate the application of Gaussian mixture modelling as a principled probabilistic means of estimating this similarity directly from the atomistic data. This model enables the discovery of homogeneous sub-populations of atoms in an entirely data-driven manner. The size of the space-averaging volume then follows naturally from the average maximum extent of the sub-populations. Furthermore, we demonstrate how the model can be used to compute the stress at arbitrary continuum points. Thorough evaluation is conducted on a numerical example of an edge dislocation in a single crystal. We find that our results are in excellent agreement with the corresponding analytical solution. read less USED (high confidence) R. Deák and Z. Néda, “Kinetic Monte Carlo approach for triangular‐shaped Pt islands on Pt(111) surfaces,” physica status solidi (b). 2012. link Times cited: 8 Abstract: Formation of triangular‐shaped Pt adatom islands on a Pt(111… read moreAbstract: Formation of triangular‐shaped Pt adatom islands on a Pt(111) surface is investigated using a kinetic Monte Carlo approach. The energy barriers are calculated with the generalized embedded atom method and the nudged elastic band approach. The numerical results reveal that the preferential orientation of the triangles cannot be explained solely by the differences in the diffusion coefficients of the atoms along the topologically non‐equivalent edges of the islands. For a self‐consistent explanation of the triangle orientations, one has to examine the topological and energetic details of the diffusion paths for all the edge diffusion processes, kink‐formation or kink‐breaking events, and corner to edge jumps. read less USED (high confidence) G. Li, Q. Wang, T. Liu, K. Wang, C.-li Wu, and J. He, “Controllable irregular phenomenon induced by Ag atomic segregation on the melting of (AgPd)561 bimetallic clusters,” Journal of Nanoparticle Research. 2012. link Times cited: 4 USED (high confidence) C. Sabater, C. Untiedt, J. Palacios, and M. Caturla, “Mechanical annealing of metallic electrodes at the atomic scale.,” Physical review letters. 2012. link Times cited: 32 Abstract: The process of creating an atomically defined and robust met… read moreAbstract: The process of creating an atomically defined and robust metallic tip is described and quantified using measurements of contact conductance between gold electrodes and numerical simulations. Our experiments show how the same conductance behavior can be obtained for hundreds of cycles of formation and rupture of the nanocontact by limiting the indentation depth between the two electrodes up to a conductance value of approximately 5G0 in the case of gold. This phenomenon is rationalized using molecular dynamics simulations together with density functional theory transport calculations which show how, after repeated indentations (mechanical annealing), the two metallic electrodes are shaped into tips of reproducible structure. These results provide a crucial insight into fundamental aspects relevant to nanotribology or scanning probe microscopies. read less USED (high confidence) L. Meng, X. Peng, K. W. Zhang, C. Tang, and J. Zhong, “Structural phase transitions of FeCo and FeNi nanoparticles: A molecular dynamics study,” Journal of Applied Physics. 2012. link Times cited: 23 Abstract: We have investigated the structural phase transition of FeCo… read moreAbstract: We have investigated the structural phase transition of FeCo and FeNi nanoparticles by molecular dynamics (MD) simulation using the generalized embedded atom potential (GEAM). It is found that the phase varies with the atomic compositions and annealing processes. By using the Honeycutt and Andersen index (HA index), bond order parameters (BOP) and pair correlation function (PCF), we found that a BCC to defective icosahedra phase transition occurs in the FeCo nanoparticle when Co composition is increased to about 60 at %. In the FeNi nanoparticle, three phases have been identified, namely, the BCC phase, the mixed BCC/FCC phase, and the multilayer defective icosahedral phase, which correspond to the Ni compositions of 0–20 at %, 20–70 at %, and 70–100 at %, respectively. Our simulations have well reproduced the phase transition points and most of the phases observed in recent experiments. read less USED (high confidence) M. Hakamada, F. Hirashima, M. Takahashi, T. Nakazawa, and M. Mabuchi, “Large-strain-induced magnetic properties of Co electrodeposited on nanoporous Au,” Journal of Applied Physics. 2011. link Times cited: 5 Abstract: Nanostructured Co with large lattice extension and contracti… read moreAbstract: Nanostructured Co with large lattice extension and contraction was produced by electrodepositing Co on nanoporous Au. The Co deposited showed a low magnetic saturation of 76 emu/g and a high coercivity of 462 Oe. First-principles calculations showed that the magnetic moment of a Co atom is significantly decreased by lattice contraction. Therefore, the noteworthy magnetic properties of the Co deposited are attributed to the large lattice strain. Also, molecular dynamics simulation showed that the lattice extension and contraction of about 10% are generated in the overall Co crystal. This is in agreement with the experimental results of HRTEM observation. The constraint of the movement of Co atoms by the concave structure of nanoporous Au leads to a wide spread of large strain region. read less USED (high confidence) L. Meng, X. Peng, C. Tang, K. W. Zhang, G. M. Stocks, and J. Zhong, “A quasicore-shell structure of FeCo and FeNi nanoparticles,” Journal of Applied Physics. 2010. link Times cited: 4 Abstract: Based on semiempirical generalized embedded atom method (GEA… read moreAbstract: Based on semiempirical generalized embedded atom method (GEAM), we carried out molecular dynamics and Monte Carlo simulations to study the structural properties of FeCo and FeNi nanoparticles. It is found that these two kinds of nanoparticles possess a new stable quasicore-shell structure, no matter whether they are in molten or condensed state and whether they are prepared by annealing or quenching. In FeCo (FeNi) nanoparticles of various sizes and atom compositions, the quasicore-shell structure is always preferred, with the shell formed only by Fe atoms and the core formed by randomly distributed Co(Ni) and Fe atoms. We have also investigated the formation mechanism of the quasicore-shell structure by energy difference analysis of the pure and doped icosahedra structure of FeCo and FeNi nanoparticles. read less USED (high confidence) X. Xiao, “Irregular phenomenon induced by Ag atomic segregation during the heating process of Ag—Pd cluster,” Chinese Physics B. 2010. link Times cited: 7 Abstract: This paper studies the melting of icosahedral Ag—Pd bimetall… read moreAbstract: This paper studies the melting of icosahedral Ag—Pd bimetallic clusters by using molecular dynamics with the embedded atom method. It finds that the mixed Ag—Pd cluster shows an irregular phenomenon before melting, i.e., the atomic energy decreases with the increase of temperature. It indicates that the segregation of Ag atoms results in this phenomenon by analysing atomic radius distribution. Since the surface energy of Ag is lower than that of Pd, this leads to the result that the decreased energy by the Ag atomic segregation is larger than the increased energy by the heating. This provides a new method to obtain irregular thermodynamic properties. read less USED (high confidence) A. Zarefy et al., “Influence of Co layer thickness on the structural and magnetic properties of (\rm Pt/\rm Co_t_\rm Co)_\rm 3 /\rm Pt_t_\rm Pt/\rm IrMn multilayers,” Journal of Physics D. 2010. link Times cited: 18 Abstract: The correlated effects of the insertion of a Pt spacer betwe… read moreAbstract: The correlated effects of the insertion of a Pt spacer between ferromagnetic and antiferromagnetic layers and of the variation of the Co layers' thickness on the structural and magnetic properties of multilayers have been studied. Samples with n = 1 and 7, tCo = 0.4 and 0.6 nm, tPt = 0 and 0.4 nm have been investigated by tomographic atom probe and superconducting quantum interference device magnetometry. For spacer-free samples (tPt = 0), the structural investigation shows that when tCo = 0.4 nm, Mn and Ir atoms diffuse deeply into the (Pt/Co)3 multilayers. In contrast for tCo = 0.6 nm, the Mn and Ir diffusion is much reduced. Because Pt acts as a barrier against the Mn and Ir diffusion, this difference is less pronounced in samples with Pt insertion. The hysteresis loops shapes, the exchange bias fields and the saturation magnetization values were correlated with the structural properties of these samples and discussed, taking into account the susceptibility, exchange stiffness and perpendicular magnetic anisotropy. read less USED (high confidence) D. Ivanov, Z. Lin, B. Rethfeld, G. O’Connor, T. Glynn, and L. Zhigilei, “Nanocrystalline structure of nanobump generated by localized photoexcitation of metal film,” Journal of Applied Physics. 2010. link Times cited: 60 Abstract: The extreme cooling rates in material processing can be achi… read moreAbstract: The extreme cooling rates in material processing can be achieved in a number of current and emerging femtosecond laser techniques capable of highly localized energy deposition. The mechanisms of rapid solidification of a nanoscale region of a metal film transiently melted by a localized photoexcitation are investigated in a large-scale atomistic simulation. The small size of the melted region, steep temperature gradients, and fast two-dimensional electron heat conduction result in the cooling rate exceeding 1013 K/s and create conditions for deep undercooling of the melt. The velocity of the liquid/crystal interface rises up to the maximum value of ∼80 m/s during the initial stage of the cooling process and stays approximately constant as the temperature of the melted region continues to decrease. When the temperature drops down to the level of ∼0.6Tm, a massive homogeneous nucleation of the crystal phase inside the undercooled liquid region takes place and prevents the undercooled liquid from reaching th... read less USED (high confidence) S. Hara, T. Kumagai, S. Izumi, and S. Sakai, “Multiscale analysis on the onset of nanoindentation-induced delamination: Effect of high-modulus Ru overlayer,” Acta Materialia. 2009. link Times cited: 7 USED (high confidence) S.-G. Lee and Y.-C. Chung, “The early stage of deposition process for Fe–Cu magnetic multilayer systems: molecular dynamics simulation,” Journal of Physics D: Applied Physics. 2009. link Times cited: 9 Abstract: The deposition behaviour for a Fe–Cu magnetic multilayer sys… read moreAbstract: The deposition behaviour for a Fe–Cu magnetic multilayer system in an early stage of the deposition process was investigated by molecular dynamics (MD) simulation. Specifically, the steering effect was quantitatively investigated through extensive measurements of the trajectory, deposition flux and force of atoms near the artificially structured Fe or Cu step positioned on the Cu(0 0 1) or Fe(0 0 1) surface. Near the step edges of the planar structure at a low incident energy of 0.1 eV, the steering effect for the case of Fe/Cu(0 0 1) was observed more significantly than for Cu/Fe(0 0 1). Additionally, the mechanism of down-diffusion from the step was discussed and the corresponding energetic was calculated using the molecular statics method. read less USED (high confidence) G. Li, Q. Wang, K. Wang, T. Liu, D. Li, and J. He, “Composition, concentration and configuration dependence of the icosahedral transformations in Cu-based bimetallic clusters,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 15 Abstract: The dependence of icosahedral transformations on the composi… read moreAbstract: The dependence of icosahedral transformations on the composition, concentration and configuration in Cu-based bimetallic clusters was studied by using molecular dynamics with the embedded atom method. The results show that the transformation is strongly related to the release of excess energy and can be controlled by tuning the composition, concentration and configuration. The transformation can be easily induced by the addition of Co while it is difficult in the case of Ni. The transformation temperature Ttrans decreases with the increase in Co concentration and increases with the increase in Ni concentration. The transformation can be controlled by fabricating different configurations and distributions of the composition. read less USED (high confidence) A. Zarefy et al., “Characterization of (Pt2 nm/Co0.4 nm)3/Ptx/IrMn7 nm multilayers by tomographic atom probe: On the role of a Pt spacer,” Journal of Applied Physics. 2009. link Times cited: 9 Abstract: Structural investigation of Ta3 nm/[(Pt2 nm/Co0.4 nm)3/Ptx/I… read moreAbstract: Structural investigation of Ta3 nm/[(Pt2 nm/Co0.4 nm)3/Ptx/IrMn7 nm]7/Pt10 nm multilayers without (x=0 nm) and with (x=0.4 nm) a Pt spacer has been performed by laser-assisted tomographic atom probe. Without a Pt spacer a strong intermixing is observed at the Co/IrMn interface. In the multilayer containing a Pt spacer the Co/Pt/IrMn interface is very weakly intermixed. It thus appears that the Pt spacer acts as a diffusion barrier that prevents the Ir and Mn atoms from diffusing into the Co layer. The consequences of this effect on the magnetic properties are discussed. The exchange bias field and the anisotropy direction of these two multilayers are analyzed and correlated with the structural investigation. read less USED (high confidence) W. Qiang, L. Guojian, L. Dong-gang, L. Xiao, and H. Jicheng, “Evolution of three-shell onion-like and core-shell structures in (AgCo)201 bimetallic clusters,” Chinese Physics B. 2009. link Times cited: 13 Abstract: This paper studies the structural evolution of (AgCo)201 clu… read moreAbstract: This paper studies the structural evolution of (AgCo)201 clusters with different Co concentrations under various temperature conditions by using molecular dynamics with the embedded atom method. The most stable position for Co atoms in the cluster is the subsurface layer at low temperature (lower than 200 K for the Ag200Co1 cluster). The position changes to the core layer with the increase of temperature, but there is an energy barrier in the middle layer. This makes the Ag–Co cluster form an Ag–Co–Ag three-shell onion-like configuration. When the temperature is high enough [higher than 800 K for (AgCo)201 clusters with 50% Co], Co atoms can obtain enough energy to overcome the energy barrier and the cluster forms an Ag–Co core-shell configuration. Amorphization for the onion-like and core-shell clusters is induced by the large lattice misfit at Ag–Co interfaces. The structural evolution in the Ag–Co cluster is related to the release of excess energy. read less USED (high confidence) L. Guojian, W. Qiang, L. Tie, L. Dong-gang, L. Xiao, and H. Jicheng, “Molecular Dynamics Simulation of Icosahedral Transformations in Solid Cu—Co Clusters,” Chinese Physics Letters. 2009. link Times cited: 7 Abstract: We study the icosahedral transformations of solid Cu–Co clus… read moreAbstract: We study the icosahedral transformations of solid Cu–Co clusters with different initial configurations by using molecular dynamics with the embedded atom method. It is found that the formation of symmetric icosahedral cluster is strongly related to the atomic number and initial configuration. The transformation originates from the surface into the interior of the cluster and is a structural change which is rapid and diffusionless. The icosahedral clusters with any composition and configuration, such as core-shell or three-shell cluster, can be prepared by the means of solid-solid phase transition in bimetallic clusters. read less USED (high confidence) S.-G. Lee and Y.-C. Chung, “Molecular-dynamics investigation of the surface characteristics of Fe–Cu magnetic thin-film layers,” Journal of Vacuum Science and Technology. 2008. link Times cited: 7 Abstract: Using molecular dynamics simulation, the structural characte… read moreAbstract: Using molecular dynamics simulation, the structural characteristics of Fe and Cu thin films grown on Cu and Fe(001) substrates, respectively, were investigated with respect to the incident energy of adatoms and substrate temperature. In the case of Cu on Fe(001), no surface alloying at the interface was observed in the early stage of thin-film deposition, and growth generally followed the layer-by-layer growth mode. For Fe on a Cu(001) surface, a mixture confined to a single atomic layer at the Cu(001) surface was found to form at room temperature while films showed island growth. The steering effect due to atomic attraction was also observed at low incident energy, resulting in a rougher surface. Fe/Cu(001) growth changed to a layer-by-layer mode for an incident energy of 6 eV. The different aspects of surface morphology between Fe/Cu(001) and Cu/Fe(001) systems were explained in terms of surface free energy and impact cascade diffusion. read less USED (high confidence) Z.-H. Hong, S. Hwang, and T. Fang, “Effect of substrate temperature and deposition rate on alloyzation for Co or Fe onto Cu(001) substrate,” Journal of Applied Physics. 2008. link Times cited: 12 Abstract: The mixing situation of Fe or Co atoms implanting onto Cu(00… read moreAbstract: The mixing situation of Fe or Co atoms implanting onto Cu(001) substrate is investigated with regard to substrate temperature and deposition rate by molecular dynamics. The tight-binding-second-momentum-approach many-body potential is used to model the atomic interaction. The results indicate that the morphology of the layer is under epitaxial growth as the substrate temperature is 700 or 1000 K, while it is not epitaxial at the substrate temperature of 300 or 450 K. The quality of epitaxial film is better when the substrate temperature is increased. The intermixing at the deposited layers becomes clear as the substrate temperature increases. It also indicates that there are more Co atoms penetrating into the substrate than the Fe atoms, regardless of the substrate temperature. Hence, one could say that the interface mixing of Co and Cu atoms is better than that of Fe and Cu atoms. When the deposition rate is raised from 5 to 10 atoms/ps, there is no increase in the interface mixing at both systems except... read less USED (high confidence) D. Ivanov, B. Rethfeld, G. O’Connor, T. Glynn, A. Volkov, and L. Zhigilei, “The mechanism of nanobump formation in femtosecond pulse laser nanostructuring of thin metal films,” Applied Physics A. 2008. link Times cited: 97 USED (high confidence) O. A. Oviedo, E. Leiva, and M. Mariscal, “Diffusion mechanisms taking place at the early stages of cobalt deposition on Au(111),” Journal of Physics: Condensed Matter. 2008. link Times cited: 4 Abstract: In the present work a detailed atomic-level analysis of some… read moreAbstract: In the present work a detailed atomic-level analysis of some of the main diffusion mechanisms which take place during cobalt adatom deposition are studied within atom dynamics (AD) and the nudged elastic band (NEB) method. Our computer simulations reveal a very fast exchange between Co and Au atoms when the deposit is a single cobalt adatom. However, when the nucleus size increases, a decrease in the exchange probability is observed. Activation energies for different transitions are obtained using AD in combination with the NEB method. read less USED (high confidence) C. Srivastava, J. Balasubramanian, C. Turner, J. Wiest, H. Bagaria, and G. Thompson, “Formation mechanism and composition distribution of FePt nanoparticles,” Journal of Applied Physics. 2007. link Times cited: 26 Abstract: Self-assembled FePt nanoparticle arrays are candidate struct… read moreAbstract: Self-assembled FePt nanoparticle arrays are candidate structures for ultrahigh density magnetic storage media. One of the factors limiting their application to this technology is particle-to-particle compositional variation. This variation will affect the A1 to L10 transformation as well as the magnetic properties of the nanoparticles. In the present study, an analysis is provided for the formation mechanism of these nanoparticles when synthesized by the superhydride reduction method. Additionally, a comparison is provided of the composition distributions of nanoparticles synthesized by the thermal decomposition of Fe(CO)5 and the reduction of FeCl2 by superhydride. The latter process produced a much narrower composition distribution. A thermodynamic analysis of the mechanism is described in terms of free energy perturbation Monte Carlo simulations. read less USED (high confidence) R. Evans, F. Dorfbauer, O. Myrasov, O. Chubykalo-Fesenko, T. Schrefl, and R. Chantrell, “The Effects of Surface Coating on the Structural and Magnetic Properties of CoAg Core-Shell Nanoparticles,” IEEE Transactions on Magnetics. 2007. link Times cited: 7 Abstract: We have investigated the impact of Ag surface coating on the… read moreAbstract: We have investigated the impact of Ag surface coating on the microstructure of CoAg core-shell nanoparticles, and the consequences for the magnetic properties. Atomic structures were simulated using a molecular dynamics approach utilizing the embedded atom method. The magnetic properties were then simulated using an atomistic approach using a classical spin Hamiltonian, taking into account the long-range nature, atomic separation and directional and phase dependence of the exchange interactions in Co. For pure cobalt nanoparticles with a diameter less than approximately 3 nm the internal crystal structure showed multiple twinned regions and the morphology of an icosahedron. The addition of a monolayer silver coating alters the internal cobalt crystal structure to a regular planar form along the cubic [111] direction with a mixture of face centred cubic (fcc) and hexagonal close packed (hcp) atomic arrangements. The local atomic environment was used to assign anisotropies to the atoms on a site by site basis. Moreover, the capping layer influences the shape of the particle and yielded a morphology similar to that of a truncated octahedron. Taking into account the effects of surface anisotropy in addition gives an overall picture of anisotropy for CoAg nanoparticles. We present the results of calculations estimating the energy barrier to magnetization reversal and the intrinsic coercivity for these particles read less USED (high confidence) X. W. Zhou and H. Wadley, “A potential for simulating the atomic assembly of cubic AB compounds,” Computational Materials Science. 2007. link Times cited: 7 USED (high confidence) J. Quan, X. W. Zhou, L. He, R. Hull, and H. Wadley, “Low energy ion assisted deposition of Ta/Cu films,” Journal of Applied Physics. 2007. link Times cited: 11 Abstract: A combination of molecular dynamics simulations and experime… read moreAbstract: A combination of molecular dynamics simulations and experiments has been used to investigate the use of various low energy ion assisted vapor deposition approaches for controlling the interfacial structures of a model copper∕tantalum multilayer system. Films were grown using argon ion beam assistance with either a fixed or modulated ion energy during metal deposition. The effect of sequential ion assistance (after layer’s deposition) was also investigated. The argon ion energy was varied between 0 and 50eV and the effect on the atomic scale structure of Ta∕Cu film interfaces and the film electrical resistivity were studied. The use of simultaneous argon ion assistance with an ion energy of ∼10eV and an ion∕metal atom flux ratio of ∼6 resulted in atomically sharp interfaces with little intermixing, consistent with simulation predictions. Ion impacts in this range activated surface atom jumping and promoted a step flow film growth mode. Higher energies were also successful at interface flattening, but they ... read less USED (high confidence) Z. Lin and L. Zhigilei, “Thermal excitation of d band electrons in Au: implications for laser-induced phase transformations,” SPIE High-Power Laser Ablation. 2006. link Times cited: 55 Abstract: The temperature dependences of the electron heat capacity an… read moreAbstract: The temperature dependences of the electron heat capacity and the electron-phonon coupling factor are investigated for Au based on the electron density of states obtained from ab initio electronic structure calculations. Thermal excitation of d band electrons leads to a significant (up to an order of magnitude) increase in the electronphonon coupling factor and makes a considerable contribution to the electron heat capacity in the range of electron temperatures typically realized in femtosecond laser material processing applications. Simulations performed with a combined atomistic-continuum method demonstrate that the increase in the strength of the electron-phonon coupling at high electron temperatures leads to a faster lattice heating, generation of stronger thermoelastic stresses, and a significant decrease in the time of the onset of the melting process. The timescale of the melting process predicted in the simulation accounting for the thermal excitation of d band electrons is in excellent agreement with the results of recent time-resolved electron diffraction experiments. A simulation performed with commonly used approximations of a constant electron-phonon coupling factor and a linear temperature dependence of the electron heat capacity, on the other hand, significantly overpredicts the time of the beginning of the melting process, supporting the importance of the electron density of states effects and thermal excitation of lower band electrons for realistic modeling of femtosecond pulse laser processing. read less USED (high confidence) N. Lümmen and T. Kraska, “Molecular dynamics investigation of homogeneous nucleation and cluster growth of platinum clusters from supersaturated vapour,” Nanotechnology. 2005. link Times cited: 36 Abstract: The formation of platinum nanoparticles from a supersaturate… read moreAbstract: The formation of platinum nanoparticles from a supersaturated vapour phase is investigated by molecular dynamics simulations. Argon is added as carrier gas, removing the condensation heat from the nucleating system. The interactions between the platinum atoms are modelled by the multi-body embedded atom method. The nucleation rates in highly supersaturated systems are estimated as well as properties of the critical clusters from the nucleation theorems. Furthermore, the coalescence of platinum nanoparticles is investigated and a coalescence activated structural transition in platinum nanoparticles is described. read less USED (high confidence) D. Murdick, X. W. Zhou, H. Wadley, and D. Nguyen-Manh, “Predicting surface free energies with interatomic potentials and electron counting,” Journal of Physics: Condensed Matter. 2005. link Times cited: 15 Abstract: Current interatomic potentials for compound semiconductors, … read moreAbstract: Current interatomic potentials for compound semiconductors, such as GaAs, fail to correctly predict the ab initio calculated and experimentally observed surface reconstructions. These potentials do not address the electron occupancies of dangling bonds associated with surface atoms and their well established role in the formation of low-energy surfaces. The electron counting rule helps account for the electron distribution among covalent and dangling bonds, which, when applied to GaAs surfaces, requires the arsenic dangling bonds to be fully occupied and the gallium dangling bonds to be empty. A simple method for linking this electron counting constraint with interatomic potentials is proposed and used to investigate energetics of the atomic scale structures of the GaAs(001) surface using molecular statics methods. read less USED (high confidence) S. D. Chen, A. Soh, and F. Ke, “Molecular Dynamics Modeling of Diffusion Bonding,” Scripta Materialia. 2005. link Times cited: 56 USED (high confidence) L. Zhigilei, D. Ivanov, E. Leveugle, B. Sadigh, and E. Bringa, “Computer modeling of laser melting and spallation of metal targets,” SPIE High-Power Laser Ablation. 2004. link Times cited: 59 Abstract: The mechanisms of melting and photomechanical damage/spallat… read moreAbstract: The mechanisms of melting and photomechanical damage/spallation occurring under extreme superheating/deformation rate conditions realized in short pulse laser processing are investigated in a computational study performed with a hybrid atomistic-continuum model. The model combines classical molecular dynamics method for simulation of non-equilibrium processes of lattice superheating and fast phase transformations with a continuum description of the laser excitation and subsequent relaxation of the conduction band electrons. The kinetics and microscopic mechanisms of melting are investigated in simulations of laser interaction with free-standing Ni films and bulk targets. A significant reduction of the overheating required for the initiation of homogeneous melting is observed and attributed to the relaxation of laser-induced stresses, which leads to the uniaxial expansion and associated anisotropic lattice distortions. The evolution of photomechanical damage is investigated in a large-scale simulation of laser spallation of a 100 nm Ni film. The evolution of photomechanical damage is observed to take place in two stages, the initial stage of void nucleation and growth, when both the number of voids and the range of void sizes are increasing, followed by the void coarsening, coalescence and percolation, when large voids grow at the expense of the decreasing population of small voids. In both regimes the size distributions of voids are found to be well described by the power law with an exponent gradually increasing with time. A good agreement of the results obtained for the evolution of photomechanical damage in a metal film with earlier results reported for laser spallation of molecular systems and shock-induced back spallation in metals suggests that the observed processes of void nucleation, growth and coalescence may reflect general characteristics of the dynamic fracture at high deformation rates. read less USED (high confidence) S.-P. Kim, S.-C. Lee, K.-R. Lee, and Y.-C. Chung, “Atomic Mixing Behavior of Co/Al(001) vs. Al/fcc-Co(001): Molecular Dynamics Simulation,” Journal of Electroceramics. 2004. link Times cited: 10 USED (high confidence) E. Leveugle, D. Ivanov, and L. Zhigilei, “Photomechanical spallation of molecular and metal targets: molecular dynamics study,” Applied Physics A. 2004. link Times cited: 201 USED (high confidence) M. Trochet, N. Mousseau, L. Béland, and G. Henkelman, “Off-Lattice Kinetic Monte Carlo Methods,” Handbook of Materials Modeling. 2020. link Times cited: 13 USED (high confidence) E. T. Karim, C. Wu, and L. Zhigilei, “Molecular Dynamics Simulations of Laser-Materials Interactions: General and Material-Specific Mechanisms of Material Removal and Generation of Crystal Defects.” 2014. link Times cited: 11 USED (high confidence) H. Miura, K. Suzuki, Y. Sasaki, T. Sano, and N. Murata, “High Temperature Damage of Ni-Base Superalloy Caused by the Change of Microtexture due to the Strain-Induced Anisotropic Diffusion of Component Elements.” 2011. link Times cited: 1 Abstract: In order to assure the reliability of advanced gas turbine s… read moreAbstract: In order to assure the reliability of advanced gas turbine systems, it is very important to evaluate the damage of high temperature materials such as Ni-base superalloys under creep and fatigue conditions quantitatively. Since the micro texture of the gamma-prime (γ′ ) phase was found to vary during the creep damage process, it is possible, therefore, to evaluate the creep damage of this material quantitatively by measuring the change of the micro texture. The mechanism of the directional coarsening of γ′ phasesof Ni-base superalloy under uni-axial strain at high temperatures, which is called rafting, was analyzed by using molecular dynamics (MD) analysis. The stress-induced anisotropic diffusion of Al atoms perpendicular to the finely dispersed γ/γ′ interface in the superalloy was observed clearly in a Ni(001)/Ni3 Al(001) interface structure. The stress-induced anisotropic diffusion was validated by experiment using the stacked thin films structures which consisted of the (001) face-centered cubic (FCC) interface. The reduction of the diffusion of Al atoms perpendicular to the interface is thus, effective for improving the creep and fatigue resistance of the alloy. It was also found by MD analysis that the dopant elements in the superalloy also affected the strain-induced diffusion of Al atoms. Both palladium and tantalum were effective elements which restrain Al atoms from moving around the interface under the applied stress, while titanium and tungsten accelerated the strain-induced anisotropic diffusion, and thus, the rafting phenomenon.Copyright © 2011 by ASME read less USED (high confidence) K. Suzuki, H. Ito, T. Inoue, and H. Miura, “Creep Damage Process of Ni-Base Superalloy Caused by Stress-Induced Anisotropic Atomic Diffusion,” Journal of Solid Mechanics and Materials Engineering. 2009. link Times cited: 7 Abstract: In order to make clear the mechanism of the directional coar… read moreAbstract: In order to make clear the mechanism of the directional coarsening of γ' phases (rafting) of Ni-base superalloy under uni-axial strain, molecular dynamics (MD) analysis was applied to analyze the effect of strain on the diffusion characteristics around the interface between different materials. In a Ni (001)/Al (001) interface structure, the stress induced diffusion of Al atoms perpendicular to the interface was found. The stress induced anisotropic diffusion of Al was also found in a Ni (001)/Ni3Al (001) interface. These results imply that it is highly possible the rafting occurs predominantly by the stress induced anisotropic diffusion of Al atoms. read less USED (high confidence) S. Hara, S. Izumi, S. Sakai, Y. Eguchi, and T. Iwasaki, “Simulations of an Interface Crack Nucleation During Nanoindentaion : Molecular Dynamics and Finite Element Coupling Approach,” MRS Proceedings. 2008. link Times cited: 1 Abstract: We carried out the nanoindentation simulations for the Ru (s… read moreAbstract: We carried out the nanoindentation simulations for the Ru (superlayer) / Cu (film) / SiO2 (substrate) system using the finite temperature MD-FEM coupling method. The calculations are performed for the different adhesion energies of Cu/SiO2 ranging from 0.2 to 0.6 J/m 2 . During loading, it was found that the interfacial crack nucleation occurs at three to four times the contact radius, driven by the tensile stress acted on the Cu/SiO2 interface. We also show that the asymmetric defect behavior have a great effect on giving birth to the crack nucleation. The observation of our simulation indicates that the mechanism of the crack nucleation strongly depends on the interfacial bonding energy. read less USED (high confidence) W. Guang-hai, P. Hui, K. Fu-jiu, X. Meng-Fen, and B. Yilong, “Study Of Mechanical Properties Of Amorphous Copper With Molecular Dynamics Simulation,” Chinese Physics B. 2008. link Times cited: 8 Abstract: The formation and mechanical properties of amorphous copper … read moreAbstract: The formation and mechanical properties of amorphous copper are studied using molecular dynamics simulation. The simulations of tension and shearing show that more pronounced plasticity is found under shearing, compared to tension. Apparent strain hardening and strain rate effect are observed. Interestingly, the variations of number density of atoms during deformation indicate free volume creation, especially under higher strain rate. In particular, it is found that shear induced dilatation does appear in the amorphous metal. read less USED (high confidence) O. Tucker, D. Ivanov, L. Zhigilei, R. E. Johnson, and E. Bringa, “Molecular dynamics simulation of sputtering from a cylindrical track: EAM versus pair potentials,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 22 USED (high confidence) V. Zhakhovsky et al., “Shock-induced melting and crystallization in titanium irradiated by ultrashort laser pulse,” Physics of Fluids. 2023. link Times cited: 4 Abstract: Modification of titanium microstructure after propagation of… read moreAbstract: Modification of titanium microstructure after propagation of a melting shock wave (SW) generated by a femtosecond laser pulse is investigated experimentally and analyzed using hydrodynamic and atomistic simulations. Scanning and transmission electron microscopy with analysis of microdiffraction is used to determine the microstructure of modified subsurface layers of titanium. We found that two layers are modified beneath the surface. A top surface polycrystalline layer of nanoscale grains is formed from shock-molten material via rapid crystallization. In a deeper subsurface layer, where the shock-induced melting changes into plastic deformation due to attenuation of SW, the grain structure of solid is considerably affected, which results in a grain size distribution differing from that in the intact titanium. Molecular dynamics simulation of single-crystal titanium reveals that the SW front continues to melt even after its temperature drops below the melting curve Tm(P). The enormous shear stress of ∼12 GPa generated in a narrow SW front leads to free slip of atomic planes, collapse of the crystal lattice, and formation of a supercooled metastable melt. Such melt crystallizes in an unloading tail of SW. The mechanical melting ceases after drop in the shear stress giving rise to the shock-induced plastic deformation. The last process triggers a long-term rearrangement of atomic structures in solid. The overall depth of modified layers is limited by SW attenuation to the Hugoniot elastic limit and can reach several micrometers. The obtained results reveal the basic physical mechanisms of surface hardening of metals by ultrashort laser pulses. read less USED (high confidence) M. Wang, Y. Zeng, Y. Chen, and S. Zhang, “Crystal structure effect on metallic mechanical properties of under tension stress: molecular dynamics study,” 22nd International Scientific Conference Engineering for Rural Development Proceedings. 2023. link Times cited: 0 Abstract: In the paper, the tensile mechanical properties and deformat… read moreAbstract: In the paper, the tensile mechanical properties and deformation behaviour of different crystal structures (face-centered cubic (FCC) metal ~Al and closely packed hexagonal (HCP) metal ~Ti) were studied using molecular dynamics calculation through the LAMMPS module of Materials and Processes Simulations (MAPS) software, and the effect of microstructure on their mechanical properties was explored from the atomic level, in order to reveal the deformation mechanism of different lattice types of metals at the atomic scale. The simulation results using MAPS showed that the elastic modulus and the tensile strength of Al was calculated with 45.0 GPa and 6.2 GPa, respectively, the flow stress of Al has a stable value of around 0.69-1.29 GPa. And the elastic modulus and the tensile strength of Ti was calculated with 73.1 GPa and 8.5 GPa, respectively. According to calculation by Ovite, the occurrence of dislocations in Ti was later than that in Al, indicating that the strain energy that can be accumulated in the Ti lattice of HCP structure was higher and the ability to resist deformation was greater. As the periodic boundary conditions were used in all three directions of the two model, there was no macroscopic breaking phenomenon. To sum up, the lattice of HCP structure (~Ti) can accumulate higher strain energy and have greater deformation resistance ability than that of FCC structure (~Al), which was the reason for the elastic modulus and tensile strength of Ti higher than those of Al. read less USED (high confidence) W. H. Oo, M. Baykara, and H. Gao, “A Computational Study of Cluster Dynamics in Structural Lubricity: Role of Cluster Rotation,” Tribology Letters. 2023. link Times cited: 0 USED (high confidence) G. Anand, “GAASP: Genetic Algorithm-Based Atomistic Sampling Protocol for High-Entropy Materials,” Materials and Manufacturing Processes. 2023. link Times cited: 1 Abstract: ABSTRACT High-entropy materials are composed of multiple ele… read moreAbstract: ABSTRACT High-entropy materials are composed of multiple elements on comparatively simpler lattices. Due to the multi-component nature of such materials, atomic-scale sampling is computationally expensive due to the combinatorial complexity. This study proposes a genetic algorithm-based methodology for sampling such complex chemically disordered materials. Genetic Algorithm-based Atomistic Sampling Protocol (GAASP) variants can generate low as well as high-energy structures. GAASP low-energy variant in conjugation with metropolis criteria avoids premature convergence as well as ensures detailed balance condition. GAASP can be employed to generate low-energy structures for thermodynamic predictions, and diverse structures can be generated for machine-learning applications. read less USED (high confidence) E. Toulkeridou, J. Kioseoglou, and P. Grammatikopoulos, “On the melting point depression, coalescence, and chemical ordering of bimetallic nanoparticles: the miscible Ni–Pt system,” Nanoscale Advances. 2022. link Times cited: 1 Abstract: Among the properties that distinguish nanoparticles (NPs) fr… read moreAbstract: Among the properties that distinguish nanoparticles (NPs) from their bulk counterparts is their lower melting points. It is also common knowledge that relatively low melting points enhance the coalescence of (usually) nascent nanoclusters toward larger NPs. Finally, it is well established that the chemical ordering of bi- (or multi-) metallic NPs can have a profound effect on their physical and chemical properties, dictating their potential applications. With these three considerations in mind, we investigated the coalescence mechanisms for Ni and Pt NPs of various configurations using classical molecular dynamics (MD) computer simulations. Benchmarking the coalescence process, we identified a steeper melting point depression for Pt than for Ni, which indicates a reversal in the order of melting for same-size NPs of the two elements. This reversal, also evident in the nano-phase diagram thermodynamically constructed using the regular solution model, may be useful for utilising NP coalescence as a means to design and engineer non-equilibrium NPs via gas-phase synthesis. Indeed, our MD simulations revealed different coalescence mechanisms at play depending on the conditions, leading to segregated chemical orderings such as quasi-Janus core-satellite, or core–(partial) shell NPs, despite the expected theoretical tendency for elemental mixing. read less USED (high confidence) I. Novikov, O. Kovalyova, A. Shapeev, and M. Hodapp, “AI-accelerated materials informatics method for the discovery of ductile alloys,” Journal of Materials Research. 2022. link Times cited: 3 Abstract: In computational materials science, a common means for predi… read moreAbstract: In computational materials science, a common means for predicting macroscopic (e.g., mechanical) properties of an alloy is to define a model using combinations of descriptors that depend on some material properties (elastic constants, misfit volumes, etc.), representative for the macroscopic behavior. The material properties are usually computed using special quasi-random structures, in tandem with density functional theory (DFT). However, DFT scales cubically with the number of atoms and is thus impractical for a screening over many alloy compositions. Here, we present a novel methodology which combines modeling approaches and machine-learning interatomic potentials. Machine-learning interatomic potentials are orders of magnitude faster than DFT, while achieving similar accuracy, allowing for a predictive and tractable high-throughput screening over the whole alloy space. The proposed methodology is illustrated by predicting the room temperature ductility of the medium-entropy alloy Mo–Nb–Ta. Graphical abstract read less USED (high confidence) I. M. P. Espinosa, T. Jacobs, and A. Martini, “Atomistic Simulations of the Elastic Compression of Platinum Nanoparticles,” Nanoscale Research Letters. 2022. link Times cited: 6 USED (high confidence) H. Tatsumi, C. Kao, and H. Nishikawa, “Solid-state bonding behavior between surface-nanostructured Cu and Au: a molecular dynamics simulation,” Scientific Reports. 2022. link Times cited: 1 USED (high confidence) H. Jo et al., “Direct strain correlations at the single-atom level in three-dimensional core-shell interface structures,” Nature Communications. 2022. link Times cited: 8 USED (high confidence) E. Pervolarakis, G. Tritsaris, P. Rosakis, and I. Remediakis, “Machine Learning for the edge energies of high symmetry Au nanoparticles,” Surface Science. 2022. link Times cited: 1 USED (high confidence) M. Dupraz et al., “Imaging the facet surface strain state of supported multi-faceted Pt nanoparticles during reaction,” Nature Communications. 2022. link Times cited: 8 USED (high confidence) H. Gao and M. Müser, “Structural lubricity of physisorbed gold clusters on graphite and its breakdown: Role of boundary conditions and contact lines,” Frontiers in Chemistry. 2022. link Times cited: 1 Abstract: The sliding motion of gold slabs adsorbed on a graphite subs… read moreAbstract: The sliding motion of gold slabs adsorbed on a graphite substrate is simulated using molecular dynamics. The central quantity of interest is the mean lateral force, that is, the kinetic friction rather than the maximum lateral forces, which correlates with the static friction. For most setups, we find Stokesian damping to resist sliding. However, velocity-insensitive (Coulomb) friction is observed for finite-width slabs sliding parallel to the armchair direction if the bottom-most layer of the three graphite layers is kept at zero stress rather than at zero displacement. Although the resulting kinetic friction remains much below the noise produced by the erratic fluctuations of (conservative) forces typical for structurally lubric contacts, the nature of the instabilities leading to Coulomb friction could be characterized as quasi-discontinuous dynamics of the Moiré patterns formed by the normal displacements near a propagating contact line. It appears that the interaction of graphite with the second gold layer is responsible for the symmetry break occurring at the interface when a contact line moves parallel to the armchair rather than to the zigzag direction. read less USED (high confidence) Z. Sun, J. Zhang, G. Xin, L. Xie, L. Yang, and Q. Peng, “Tensile mechanical properties of CoCrFeNiTiAl high entropy alloy via molecular dynamics simulations,” Intermetallics. 2022. link Times cited: 18 USED (high confidence) A. Rogachev et al., “Mechanical alloying in the Co-Fe-Ni powder mixture: Experimental study and molecular dynamics simulation,” Powder Technology. 2022. link Times cited: 6 USED (high confidence) L. Zhang et al., “Equivariant analytical mapping of first principles Hamiltonians to accurate and transferable materials models,” npj Computational Materials. 2021. link Times cited: 26 USED (high confidence) A. Sharma et al., “Pseudoelastic deformation in Mo-based refractory multi-principal element alloys,” Acta Materialia. 2021. link Times cited: 11 USED (high confidence) B. Lim et al., “A convolutional neural network for defect classification in Bragg coherent X-ray diffraction,” npj Computational Materials. 2021. link Times cited: 6 USED (high confidence) A. Abdelmawla, T. Phan, L. Xiong, and A. Bastawros, “A combined experimental and computational analysis on how material interface mediates plastic flow in amorphous/crystalline composites,” Journal of Materials Research. 2021. link Times cited: 3 Abstract: In this work, we study the deformation behavior in amorphous… read moreAbstract: In this work, we study the deformation behavior in amorphous/crystalline metallic composites (A/C-MCs) through nanoindentation experiments and molecular dynamic (MD) simulations. The atomic deformation processes in both crystalline (C-) and amorphous (A-) phases near the amorphous-crystalline interface (ACI) are investigated and correlated with the material’s overall constitutive behavior at the microscale. Our major findings are (i) the ACIs enable a co-deformation of the A- and C-phases through “stiffening” the soft phases but “softening” the stiff phases in A/C-MCs through different micro-mechanisms; (ii) there exists an ACI-induced transition zone with a thickness of ~ 10 nm; (iii) the strong coupling between shear transformation zones (STZs) and dislocations can be quantified through carefully designed indentation experiments and simulations; and (iv) the nanoscale MD-simulation-predicted mechanisms can be mapped to the “pop-in” or “excursion” events on the force–indentation depth curves extracted from microscale experiments, although there is a length-scale gap in between. read less USED (high confidence) K. Bang, B. C. Yeo, D. Kim, S. Han, and H. M. Lee, “Accelerated mapping of electronic density of states patterns of metallic nanoparticles via machine-learning,” Scientific Reports. 2021. link Times cited: 0 USED (high confidence) I. M. P. Espinosa, T. Jacobs, and A. Martini, “Evaluation of Force Fields for Molecular Dynamics Simulations of Platinum in Bulk and Nanoparticle Forms.,” Journal of chemical theory and computation. 2021. link Times cited: 7 Abstract: Understanding the size- and shape-dependent properties of pl… read moreAbstract: Understanding the size- and shape-dependent properties of platinum nanoparticles is critical for enabling the design of nanoparticle-based applications with optimal and potentially tunable functionality. Toward this goal, we evaluated nine different empirical potentials with the purpose of accurately modeling faceted platinum nanoparticles using molecular dynamics simulation. First, the potentials were evaluated by computing bulk and surface properties-surface energy, lattice constant, stiffness constants, and the equation of state-and comparing these to prior experimental measurements and quantum mechanics calculations. Then, the potentials were assessed in terms of the stability of cubic and icosahedral nanoparticles with faces in the {100} and {111} planes, respectively. Although none of the force fields predicts all the evaluated properties with perfect accuracy, one potential-the embedded atom method formalism with a specific parameter set-was identified as best able to model platinum in both bulk and nanoparticle forms. read less USED (high confidence) J. Ge et al., “Rapid fabrication of complex nanostructures using room-temperature ultrasonic nanoimprinting,” Nature Communications. 2021. link Times cited: 16 USED (high confidence) I. Karkin, L. Karkina, Y. Gornostyrev, and P. Korzhavyi, “Effect of Ni and Al on the Decomposition Kinetics and Stability of Cu-Enriched Precipitates in Fe–Cu–Ni–Al Alloys: Results of MD + MC Simulation,” Physics of Metals and Metallography. 2021. link Times cited: 2 USED (high confidence) G. Bucci, K. Gadelrab, and W. Carter, “Mesoscale Model for Ostwald Ripening of Catalyst Nanoparticles,” Journal of The Electrochemical Society. 2021. link Times cited: 5 USED (high confidence) T. Brink, E. Milanese, and J. Molinari, “Effect of wear particles and roughness on nanoscale friction,” Physical Review Materials. 2021. link Times cited: 9 Abstract: Frictional contacts lead to the formation of a surface layer… read moreAbstract: Frictional contacts lead to the formation of a surface layer called the third body, consisting of wear particles and structures resulting from their agglomerates. Its behavior and properties at the nanoscale control the macroscopic tribological performance. It is known that wear particles and surface topography evolve with time and mutually influence one another. However, the formation of the mature third body is largely uncharted territory and the properties of its early stages are unknown. Here we show how a third body initially consisting of particles acting as roller bearings transitions into a shear-band-like state by forming adhesive bridges between the particles. Using large-scale atomistic simulations on a brittle model material, we find that this transition is controlled by the growth and increasing disorganization of the particles with increasing sliding distance. Sliding resistance and wear rate are at first controlled by the surface roughness, but upon agglomeration wear stagnates and friction becomes solely dependent on the real contact area in accordance with the plasticity theory of contact by Bowden and Tabor. read less USED (high confidence) R. Allaire, L. Kondic, L. Cummings, P. Rack, and M. Fuentes-Cabrera, “The Role of Phase Separation on Rayleigh-Plateau Type Instabilities in Alloys,” The Journal of Physical Chemistry C. 2021. link Times cited: 6 Abstract: Classical molecular dynamics (MD) simulations are used to in… read moreAbstract: Classical molecular dynamics (MD) simulations are used to investigate the role of phase separation (PS) on the Rayleigh-Plateau (RP) instability. Ni–Ag bulk structures are created at temperatures (... read less USED (high confidence) C. Liang et al., “Influence of load orientations with respect to twin boundaries on the deformation behaviors of high-entropy alloy nanocrystals,” MRS Bulletin. 2020. link Times cited: 8 Abstract: The orientation between twin boundary (TB) and loading direc… read moreAbstract: The orientation between twin boundary (TB) and loading direction may play an intriguing role in the deformation behaviors of twinned metallic materials. In this aspect, its essential effect on the high-entropy alloy (HEA) nanocrystals is elusive. Attention herein is focused on the atomicscaled deformation mechanisms and fracture behaviors of HEA nanocrystals containing twins of even smaller spacings via a combined approach of in situ tensile tests inside a high-resolution transmission electron microscope and molecular dynamics simulations. The results indicate that the deformation mechanisms (especially dislocation activities) of HEA nanocrystals depend on the load orientation with respect to TBs. Because of the low activation energy and uneven local composition of HEA, the surface acts as an effective dislocation source and, together with Schmid factor, dominate the activated dislocation slip system. The load orientation-dependent TB-dislocation interactions may transform the type of fracture from semi-brittle to ductile. Our results indicate that the deformation mechanisms and the types of fracture in HEA nanocrystals can be controlled by changing the orientation. The nanotwinned materials attract extensive attention because of their exceptional combination of high strength and ductility. The deep understanding of the role of twin will bring advances to design new strong and ductile materials. The orientation between twin boundary (TB) and loading direction plays an intriguing role in the deformation behaviors. In this work, we use a novel method, in situ transmission electron microscope (TEM) melting, to produce nanotwin with special load/TB orientation. The strong load/TB orientation dependences of deformation mechanisms and fracture modes are revealed via in situ TEM strain and molecular dynamics simulations. The free surface together with Schmid factor dominate the activated dislocation slip system. This work points out an alternative route in designing advanced twin-inducedplasticity materials by controlling load/TB orientations. read less USED (high confidence) S. T. Oyinbo and T. Jen, “Molecular Dynamics Simulation of Dislocation Plasticity Mechanism of Nanoscale Ductile Materials in the Cold Gas Dynamic Spray Process,” Coatings. 2020. link Times cited: 12 Abstract: The dislocation plasticity of ductile materials in a dynamic… read moreAbstract: The dislocation plasticity of ductile materials in a dynamic process of cold gas spraying is a relatively new research topic. This paper offers an insight into the microstructure and dislocation mechanism of the coating using simulations of molecular dynamics (MD) because of the short MD simulation time scales. The nano-scale deposition of ductile materials onto a deformable copper substrate has been investigated in accordance with the material combination and impact velocities in the particle/substrate interfacial region. To examine the jetting mechanisms in a range of process parameters, rigorous analyses of the developments in pressure, temperature, dislocation plasticity, and microstructure are investigated. The pressure wave propagation’s critical function was identified by the molecular dynamics’ simulations in particle jet initiation, i.e., exterior material flow to the periphery of the particle and substrate interface. The initiation of jet occurs at the point of shock waves interact with the particle/substrate periphery and leads to localization of the metal softening in this region. In particular, our findings indicate that the initial particle velocity significantly influences the interactions between the material particles and the substrate surface, yielding various atomic strain and temperature distribution, processes of microstructure evolution, and the development of dislocation density in the particle/substrate interfacial zone for particles with various impact velocities. The dislocation density in the particle/substrate interface area is observed to grow much more quickly during the impact phase of Ni and Cu particles and the evolution of the microstructure for particles at varying initial impact velocities is very different. read less USED (high confidence) S. Luan, S. Yu, C. Gui, and S. Zhou, “Atomic-scale structural evolution and welding deformations of laser welded joints in Ag nanowire connectors on homogeneous substrates,” Japanese Journal of Applied Physics. 2020. link Times cited: 3 Abstract: The formation mechanism and the influence of laser power on … read moreAbstract: The formation mechanism and the influence of laser power on welding deformations of welded joints in Ag nanowire connectors were investigated using molecular dynamic simulations. Simulation results revealed that some regions of voids in the welded joint were filled due to the heat in the heat affected zone. The laser irradiation with the appropriate power can decrease dislocations and phase transitions generated by cold welding. The welding strength possessed an upward trend with the increase in laser power. With the increase of the laser power, the shear resistance of the welded joint improved. However, the welded joint after the laser with the excessive power weakened. Furthermore, by comparing the laser welding and the thermal annealing welding, we found that the laser welded joint possesses fewer amorphous atoms, phase transitions, dislocations, residual stress, and possibility of the shear deformation via simulation. read less USED (high confidence) X. W. Zhou, “Thermodynamic analysis of dissociation of periodic dislocation dipoles in isotropic crystals,” RSC Advances. 2020. link Times cited: 2 Abstract: In the past, experimentally observed dislocations were often… read moreAbstract: In the past, experimentally observed dislocations were often interpreted using an isolated dislocation assumption because the effect of background dislocation density was difficult to evaluate. Contrarily, dislocations caused by atomistic simulations under periodic boundary conditions can be better interpreted because linear elastic theory has been developed to address the effect of periodic dislocation array in the literature. However, this elastic theory has been developed only for perfect dislocations, but not for dissociated dislocations. The periodic boundary conditions may significantly change the dissociation energy of dislocations and stacking fault width, which in turn, change the deformation phenomena observed in simulations. To enable materials scientists to understand the dislocation behavior under the periodic boundary conditions, we use isotropic elastic theory to analyze the thermodynamics of dissociated periodic dislocations with an arbitrary dislocation character angle. Analytical expressions for force, stacking fault width, and energies are presented in the study. Results obtained from the periodic dislocation array were compared with those obtained from isolated dislocations to shed light on the interpretation of experimentally observed and simulated dislocations. read less USED (high confidence) X. Shi et al., “Nanoscale Mapping of Heterogeneous Strain and Defects in Individual Magnetic Nanocrystals,” Crystals. 2020. link Times cited: 4 Abstract: We map the three-dimensional strain heterogeneity within a s… read moreAbstract: We map the three-dimensional strain heterogeneity within a single core-shell Ni nanoparticle using Bragg coherent diffractive imaging. We report the direct observation of both uniform displacements and strain within the crystalline core Ni region. We identify non-uniform displacements and dislocation morphologies across the core–shell interface, and within the outer shell at the nanoscale. By tracking individual dislocation lines in the outer shell region, and comparing the relative orientation between the Burgers vector and dislocation lines, we identify full and partial dislocations. The full dislocations are consistent with elasticity theory in the vicinity of a dislocation while the partial dislocations deviate from this theory. We utilize atomistic computations and Landau–Lifshitz–Gilbert simulation and density functional theory to confirm the equilibrium shape of the particle and the nature of the (111) displacement field obtained from Bragg coherent diffraction imaging (BCDI) experiments. This displacement field distribution within the core-region of the Ni nanoparticle provides a uniform distribution of magnetization in the core region. We observe that the absence of dislocations within the core-regions correlates with a uniform distribution of magnetization projections. Our findings suggest that the imaging of defects using BCDI could be of significant importance for giant magnetoresistance devices, like hard disk-drive read heads, where the presence of dislocations can affect magnetic domain wall pinning and coercivity. read less USED (high confidence) D. Utt et al., “The origin of jerky dislocation motion in high-entropy alloys,” Nature Communications. 2020. link Times cited: 20 USED (high confidence) K. Bang, B. C. Yeo, D. Kim, S. Han, and H.-M. Lee, “Accelerated mapping of electronic density of states patterns of metallic nanoparticles via machine-learning,” Scientific Reports. 2020. link Times cited: 10 USED (high confidence) Z. Zhao and G. Lu, “Bimetallenes for selective electrocatalytic conversion of CO2: a first-principles study,” Journal of Materials Chemistry. 2020. link Times cited: 6 Abstract: Two-dimensional (2D) materials are full of surprises and fas… read moreAbstract: Two-dimensional (2D) materials are full of surprises and fascinating potential. Motivated by a recent discovery that sub-nanometer PdMo bimetallenes can realize exceptional performance in the oxygen reduction reaction [Nature 2019, 574, 81–85], we explore the potential of 2D bimetallenes for catalyzing the CO2 electroreduction reaction (CO2RR). Following extensive first-principles calculations on more than a hundred bimetallenes, we identify 17 Cu- and Ag-based bimetallenes, which are highly active and selective toward the formation of formic acid and simultaneously suppress the competing hydrogen evolution reaction. Equally important, we find that CO2RR products via intermediates of COOH and CO are disfavored on these bimetallenes. Although surface strains are developed on the bimetallenes, their contribution to the catalytic activities is moderate as compared to that of the alloying effect. This work opens the door to future applications of bimetallenes as active and selective catalysts for the CO2RR. read less USED (high confidence) S. Menon, G. D. Leines, R. Drautz, and J. Rogal, “Role of pre-ordered liquid in the selection mechanism of crystal polymorphs during nucleation.,” The Journal of chemical physics. 2020. link Times cited: 8 Abstract: We investigate the atomistic mechanism of homogeneous nuclea… read moreAbstract: We investigate the atomistic mechanism of homogeneous nucleation during solidification in molybdenum employing transition path sampling. The mechanism is characterized by the formation of a pre-structured region of high bond-orientational order in the supercooled liquid followed by the emergence of the crystalline bulk phase within the center of the growing solid cluster. This precursor plays a crucial role in the process as it provides a diffusive interface between the liquid and crystalline core, which lowers the interfacial free energy and facilitates the formation of the bulk phase. Furthermore, the structural features of the pre-ordered regions are distinct from the liquid and solid phases and preselect the specific polymorph that nucleates. The similarity in the nucleation mechanism of Mo with that of metals that exhibit different crystalline bulk phases indicates that the formation of a precursor is a general feature observed in these materials. The strong influence of the structural characteristics of the precursors on the final crystalline bulk phase demonstrates that for the investigated system, polymorph selection takes place in the very early stages of nucleation. read less USED (high confidence) J. Liang et al., “Biaxial Strains Mediated Oxygen Reduction Electrocatalysis on Fenton Reaction Resistant L10‐PtZn Fuel Cell Cathode,” Advanced Energy Materials. 2020. link Times cited: 91 Abstract: PtM alloy catalysts (e.g., PtFe, PtCo), especially in an int… read moreAbstract: PtM alloy catalysts (e.g., PtFe, PtCo), especially in an intermetallic L10 structure, have attracted considerable interest due to their respectable activity and stability for the oxygen reduction reaction (ORR) in proton exchange membrane fuel cells (PEMFCs). However, metal‐catalyzed formation of ·OH from H2O2 (i.e., Fenton reaction) by Fe‐ or Co‐containing catalysts causes severe degradation of PEM/catalyst layers, hindering the prospects of commercial applications. Zinc is known as an antioxidant in Fenton reaction, but is rarely alloyed with Pt owing to its relatively negative redox potential. Here, sub‐4 nm intermetallic L10‐PtZn nanoparticles (NPs) are synthesized as high‐performance PEMFC cathode catalysts. In PEMFC tests, the L10‐PtZn cathode achieves outstanding activity (0.52 A mgPt−1 at 0.9 ViR‐free, and peak power density of 2.00 W cm−2) and stability (only 16.6% loss in mass activity after 30 000 voltage cycles), exceeding the U.S. DOE 2020 targets and most of the reported ORR catalysts. Density function theory calculations reveal that biaxial strains developed upon the disorder‐order (A1L10) transition of PtZn NPs would modulate the surface PtPt distances and optimize PtO binding for ORR activity enhancement, while the increased vacancy formation energy of Zn atoms in an ordered structure accounts for the improved stability. read less USED (high confidence) J. Meiser and H. Urbassek, “α ↔ γ phase transformation in iron: comparative study of the influence of the interatomic interaction potential,” Modelling and Simulation in Materials Science and Engineering. 2020. link Times cited: 6 Abstract: Only few available interatomic interaction potentials implem… read moreAbstract: Only few available interatomic interaction potentials implement the α ↔ γ phase transformation in iron by featuring a stable low-temperature bcc and high-temperature fcc lattice structure. Among these are the potentials by Meyer and Entel (1998 Phys. Rev. B 57 5140), by Müller et al (2007 J. Phys.: Condens. Matter 19 326220) and by Lee et al (2012 J. Phys.: Condens. Matter 24 225404). We study how these potentials model the phase transformation during heating and cooling; in order to help initiating the transformation, the simulation volume contains a grain boundary. For the martensitic transformation occurring on cooling an fcc structure, we additionally study two potentials that only implement a stable bcc structure of iron, by Zhou et al (2004 Phys. Rev. B 69 144113) and by Mendelev et al (2003 Philos. Mag. 83 3977). We find that not only the transition temperature depends on the potential, but that also the height of the energy barrier between fcc and bcc phase governs whether the transformation takes place at all. In addition, details of the emerging microstructure depend on the potential, such as the fcc/hcp fraction formed in the α → γ transformation, or the twinning induced in and the lattice orientation of the bcc phase in the γ → α transformation. read less USED (high confidence) N. Lanzillo, L. Clevenger, R. Robison, and D. Edelstein, “Structural and transport properties of Cu/Ta(N)/Cu interfaces in vertical interconnects,” Journal of Applied Physics. 2020. link Times cited: 4 Abstract: We use first-principles calculations to investigate the stru… read moreAbstract: We use first-principles calculations to investigate the structural and transport properties of various Cu/Ta(N)/Cu interface stacks, which are representative of the metal interfaces located at the bottom of vertical interconnects in state-of-the-art back-end-of-line technology. In particular, we consider approximately 2-nm thick layers of several different Ta-based barrier layers sandwiched between two Cu(111) layers, including TaN, α-Ta, β-Ta, and a bilayer TaN/ α-Ta structure. Our results highlight that the bilayer Cu/TaN/ α-Ta/Cu structure shows both an attractive combination of low electrical resistance and superior dielectric adhesion. We also find that inelastic phonon transport across the interface structures is largely determined by the frequency overlap of the bulk-like phonon density of states of each metal layer. Our results are fed into a simple interconnect performance benchmarking model based on a single-driver signal wire, where we find that metal barrier optimization can result in a net 2.5% stage delay reduction without comprising reliability. read less USED (high confidence) P. Gupta, K. Vaduganathan, and N. Yedla, “Elevated Temperature Compression Behavior of Al–Cu50Zr50 Nano-laminates,” Transactions of the Indian Institute of Metals. 2020. link Times cited: 0 USED (high confidence) S. Subedi, L. Morrissey, S. M. Handrigan, and S. Nakhla, “The effect of many-body potential type and parameterisation on the accuracy of predicting mechanical properties of aluminium using molecular dynamics,” Molecular Simulation. 2020. link Times cited: 8 Abstract: ABSTRACT As opposed to traditional laboratory testing, Molec… read moreAbstract: ABSTRACT As opposed to traditional laboratory testing, Molecular Dynamics (MD) offers an atomistic scale method to estimate the mechanical properties of metals. However, there is limited literature that shows the effect of interatomic potentials when determining mechanical properties. Hence, the present research was conducted to investigate the accuracy of various interatomic potentials in estimating mechanical properties of aluminium. Several types of potentials, including Embedded Atom Method (EAM), Modified EAM (MEAM) and Reactive Force Field (ReaxFF) were compared with available experimental data for pure aluminium to determine the most accurate interatomic potential. A uniaxial tensile test was performed at room temperature using MD simulations for nanoscale aluminium. Results demonstrated that those potentials parameterised with elastic constants at physically realisable temperatures were consistently more accurate. Overall, the Mishin et al. EAM potential was the most accurate when compared to single-crystal experimental values. Regardless of the potential type, the error was significantly higher for those potentials that did not consider elastic constants during development. In brief, the application of the interatomic potentials to estimate mechanical properties of a nanoscale aluminium was investigated. read less USED (high confidence) S. Peng, Y. Wei, and H. Gao, “Nanoscale precipitates as sustainable dislocation sources for enhanced ductility and high strength,” Proceedings of the National Academy of Sciences of the United States of America. 2020. link Times cited: 79 Abstract: Significance Precipitates in a material are traditionally th… read moreAbstract: Significance Precipitates in a material are traditionally thought of as dislocation obstacles that lead to elevated strength and reduced ductility. In contrast, recent experiments suggest that nanoscale precipitates facilitate both high strength and large ductility. To help resolve this apparent paradox, here we reveal that nanoprecipitates provide a unique type of dislocation sources that are activated at sufficiently high stress levels and render uniform plasticity by simultaneously serving as efficient dislocation sources and obstacles to dislocation motion, giving rise to sustained deformability. The findings can guide development of next generation of materials such as multiple-element alloys with precipitate engineering. Traditionally, precipitates in a material are thought to serve as obstacles to dislocation glide and cause hardening of the material. This conventional wisdom, however, fails to explain recent discoveries of ultrahigh-strength and large-ductility materials with a high density of nanoscale precipitates, as obstacles to dislocation glide often lead to high stress concentration and even microcracks, a cause of progressive strain localization and the origin of the strength–ductility conflict. Here we reveal that nanoprecipitates provide a unique type of sustainable dislocation sources at sufficiently high stress, and that a dense dispersion of nanoprecipitates simultaneously serve as dislocation sources and obstacles, leading to a sustainable and self-hardening deformation mechanism for enhanced ductility and high strength. The condition to achieve sustainable dislocation nucleation from a nanoprecipitate is governed by the lattice mismatch between the precipitate and matrix, with stress comparable to the recently reported high strength in metals with large amount of nanoscale precipitates. It is also shown that the combination of Orowan’s precipitate hardening model and our critical condition for dislocation nucleation at a nanoprecipitate immediately provides a criterion to select precipitate size and spacing in material design. The findings reported here thus may help establish a foundation for strength–ductility optimization through densely dispersed nanoprecipitates in multiple-element alloy systems. read less USED (high confidence) J. Feng et al., “Shock-induced consolidation of tungsten nanoparticles—A molecular dynamics approach,” Journal of Applied Physics. 2020. link Times cited: 5 Abstract: Shock-induced consolidation of tungsten nanoparticles to for… read moreAbstract: Shock-induced consolidation of tungsten nanoparticles to form a bulk material was modeled using molecular dynamics simulation. By arranging the nanoparticles in a three-dimensional model of body-centered cubic super-lattice, the calculated shock velocity-particle velocity Hugoniot data are in good agreement with the experiments. Three states, including solid-undensified, solid-densified, and liquid-densified, can be sequentially obtained with the increase of the impact velocity. It is due to the flow deformation at the particle surface that densifies the cavity, and the high pressure and temperature that join the particles together. Melting is not a necessary factor for shock consolidation. Based on whether or not melting takes place, the consolidation mechanisms are liquid-diffusion welding or solid-pressure welding.Shock-induced consolidation of tungsten nanoparticles to form a bulk material was modeled using molecular dynamics simulation. By arranging the nanoparticles in a three-dimensional model of body-centered cubic super-lattice, the calculated shock velocity-particle velocity Hugoniot data are in good agreement with the experiments. Three states, including solid-undensified, solid-densified, and liquid-densified, can be sequentially obtained with the increase of the impact velocity. It is due to the flow deformation at the particle surface that densifies the cavity, and the high pressure and temperature that join the particles together. Melting is not a necessary factor for shock consolidation. Based on whether or not melting takes place, the consolidation mechanisms are liquid-diffusion welding or solid-pressure welding. read less USED (high confidence) Y. Wang, J. Li, W. Lu, F. Yuan, and X. Wu, “Enhanced co-deformation of a heterogeneous nanolayered Cu/Ni composite,” Journal of Applied Physics. 2019. link Times cited: 8 Abstract: Nanolayered metallic composites have attracted intensive sci… read moreAbstract: Nanolayered metallic composites have attracted intensive scientific interests due to their ultrahigh strength. However, the deformation incompatibility among the component layers with high mechanical contrast leads to extremely low tensile ductility in the nanolayered composites, which is a great setback for their engineering applications. Here, by molecular dynamics simulations, we show that a heterogeneous nanolayered design by combining 2.5 nm and 24 nm Cu/Ni bilayers in a composite in an appropriate way can promote the dislocation activity of the hard phase, i.e., the Ni layers. In the new heterogeneous structure, each 24 nm Cu or Ni layer is coated on both surfaces by one 2.5 nm Cu/Ni bilayer. The simulations show that the dislocations in the 24 nm Ni layers can nucleate and glide almost synchronously with those in the 24 nm Cu layers. The enhanced dislocation activities are attributed to the presence of the 2.5 nm Cu layer that can promote the dislocation nucleation and motion in the 24 nm Ni layer by forming more nodes in the dislocation network of the interface.Nanolayered metallic composites have attracted intensive scientific interests due to their ultrahigh strength. However, the deformation incompatibility among the component layers with high mechanical contrast leads to extremely low tensile ductility in the nanolayered composites, which is a great setback for their engineering applications. Here, by molecular dynamics simulations, we show that a heterogeneous nanolayered design by combining 2.5 nm and 24 nm Cu/Ni bilayers in a composite in an appropriate way can promote the dislocation activity of the hard phase, i.e., the Ni layers. In the new heterogeneous structure, each 24 nm Cu or Ni layer is coated on both surfaces by one 2.5 nm Cu/Ni bilayer. The simulations show that the dislocations in the 24 nm Ni layers can nucleate and glide almost synchronously with those in the 24 nm Cu layers. The enhanced dislocation activities are attributed to the presence of the 2.5 nm Cu layer that can promote the dislocation nucleation and motion in the 24 nm Ni layer ... read less USED (high confidence) J. Ding et al., “Thick grain boundary induced strengthening in nanocrystalline Ni alloy.,” Nanoscale. 2019. link Times cited: 25 Abstract: Grain refinement has been extensively used to strengthen met… read moreAbstract: Grain refinement has been extensively used to strengthen metallic materials for decades. Grain boundaries act as effective barriers to the transmission of dislocations, consequently leading to strengthening. Conventional grain boundaries have a thickness of 1-2 atomic layers, typically ∼0.5 nm for most metallic materials. Here, we report, however, the formation of ∼3 nm thick grain boundaries in a nanocrystalline Ni alloy. In situ micropillar compression studies coupled with molecular dynamics simulations suggest that the thick grain boundaries are stronger barriers than conventional grain boundaries to the transmission of dislocations. This study provides a fresh perspective for the design of high strength, deformable nanostructured metallic materials. read less USED (high confidence) S. Zhao, “On the role of heterogeneity in concentrated solid-solution alloys in enhancing their irradiation resistance,” Journal of Materials Research. 2019. link Times cited: 14 Abstract: Concentrated solid-solution alloys (CSAs) demonstrate excell… read moreAbstract: Concentrated solid-solution alloys (CSAs) demonstrate excellent mechanical properties and promising irradiation resistance depending on their compositions. Existing experimental and simulation results indicate that their heterogeneous structures induced by the random arrangement of different elements are one of the most important reasons responsible for their outstanding properties. Nevertheless, the details of this heterogeneity remain unclear. Specifically, which properties induced by heterogeneity are most relevant to their irradiation response? In this work, we scrutinize the role of heterogeneity in CSAs played in damage evolution in different aspects through atomistic simulations, including lattice misfit, thermodynamic mixing, point defect energetics, point defect diffusion, and dislocation properties. Our results reveal that structural parameters, such as lattice misfit and enthalpy of mixing, are generally not suitable to assess their irradiation response under cascade conditions. Instead, atomic-level defect properties are the keys to understand defect evolution in CSAs. Therefore, tuning chemical disorder to tailor defect properties is a possible way to further improve the irradiation performance of CSAs. read less USED (high confidence) V. Samsonov et al., “Complex Approach to Atomistic Simulation of the Size Dependences of the Temperature and the Heat of Melting of Co Nanoparticles: Molecular Dynamics and Monte Carlo Method,” Journal of Surface Investigation: X-ray, Synchrotron and Neutron Techniques. 2019. link Times cited: 2 USED (high confidence) P. Zakharov, M. Starostenkov, E. Korznikova, A. Eremin, I. Lutsenko, and S. V. Dmitriev, “Excitation of Soliton-Type Waves in Crystals of the A3B Stoichiometry,” Physics of the Solid State. 2019. link Times cited: 8 USED (high confidence) A. Ghafarollahi, F. Maresca, and W. Curtin, “Solute/screw dislocation interaction energy parameter for strengthening in bcc dilute to high entropy alloys,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 25 Abstract: Strengthening, i.e. increased stress required to move a disl… read moreAbstract: Strengthening, i.e. increased stress required to move a dislocation, in dilute or complex alloys arises from the totality of the interaction energies between the solutes and an individual dislocation. Prevailing theories for strengthening in bcc alloys consider only solute interactions in the core of the screw dislocation while computations suggest longer-range interactions. Here, a full statistical solute/screw interaction energy parameter relevant for predicting strengthening in random bcc alloys is presented. The parameter is valid for any number of constituent atoms and at any concentrations, thus including the range from dilute binary alloys to high-entropy alloys. The interaction energy parameter is then calculated for many bcc alloys in the Nb–Ta-V–Ti–Zr family using the Zhou-Johnson EAM potentials to demonstrate the spatial range of solutes contributing to this key quantity and to assess accuracy of previous simplified models. The interaction energy parameter is found to converge if solutes out to sixth neighbors are included while the simplified models are generally not very accurate. A recently-proposed correlation between solute/dislocation interaction energy and the solute/[111]/6 unstable stacking fault (USF) interaction energy is then assessed in detail. A very good correlation is found between the full interaction energy parameter introduced here and the solute/USF interaction energy. This points toward a simplified approach to estimating the interaction energy parameter using first-principles methods. read less USED (high confidence) J. Liang et al., “Tungsten-Doped L10-PtCo Ultrasmall Nanoparticles as High-Performance Fuel Cell Cathode.,” Angewandte Chemie. 2019. link Times cited: 112 Abstract: The commercialization of proton exchange membrane fuel cells… read moreAbstract: The commercialization of proton exchange membrane fuel cells (PEMFCs) relies on highly active and stable electrocatalysts for oxygen reduction reaction (ORR) in acid media. The most successful catalysts for this reaction are nanostructured Pt-alloy with a Pt-skin. Here, we report the synthesis of ultrasmall and ordered L1 0 -PtCo nanoparticle ORR catalysts further doped with a few percent of transition metals (i.e., W, Ga, and Zn). Compared to commercial Pt/C catalyst, the L1 0 -W-PtCo/C catalyst shows significant improvement in both initial activity and high-temperature stability. Importantly, the L1 0 -W-PtCo/C catalyst achieves high activity and stability in the PEMFC after 50k voltage cycles at 80 °C, superior to the DOE 2020 targets. Extended X-ray absorption fine structure analysis and density functional theory calculations reveal that W doping not only stabilizes the ordered intermetallic structure, but also tunes the Pt-Pt distances in such a way to optimize the binding energy between Pt and O intermediates on the surface. This work demonstrates a new strategy of stabilizing the intermetallic nanoparticles with transition metal doping to improve the performance of electrocatalysts. read less USED (high confidence) Y. Chen, S. Shabanov, and D. McDowell, “Concurrent atomistic-continuum modeling of crystalline materials,” Journal of Applied Physics. 2019. link Times cited: 31 Abstract: In this work, we present a concurrent atomistic-continuum (C… read moreAbstract: In this work, we present a concurrent atomistic-continuum (CAC) method for modeling and simulation of crystalline materials. The CAC formulation extends the Irving-Kirkwood procedure for deriving transport equations and fluxes for homogenized molecular systems to that for polyatomic crystalline materials by employing a concurrent two-level description of the structure and dynamics of crystals. A multiscale representation of conservation laws is formulated, as a direct consequence of Newton's second law, in terms of instantaneous expressions of unit cell-averaged quantities using the mathematical theory of distributions. Finite element (FE) solutions to the conservation equations, as well as fluxes and temperature in the FE representation, are introduced, followed by numerical examples of the atomic-scale structure of interfaces, dynamics of fracture and dislocations, and phonon thermal transport across grain boundaries. In addition to providing a methodology for concurrent multiscale simulation of transport processes under a single theoretical framework, the CAC formulation can also be used to compute fluxes (stress and heat flux) in atomistic and coarse-grained atomistic simulations.In this work, we present a concurrent atomistic-continuum (CAC) method for modeling and simulation of crystalline materials. The CAC formulation extends the Irving-Kirkwood procedure for deriving transport equations and fluxes for homogenized molecular systems to that for polyatomic crystalline materials by employing a concurrent two-level description of the structure and dynamics of crystals. A multiscale representation of conservation laws is formulated, as a direct consequence of Newton's second law, in terms of instantaneous expressions of unit cell-averaged quantities using the mathematical theory of distributions. Finite element (FE) solutions to the conservation equations, as well as fluxes and temperature in the FE representation, are introduced, followed by numerical examples of the atomic-scale structure of interfaces, dynamics of fracture and dislocations, and phonon thermal transport across grain boundaries. In addition to providing a methodology for concurrent multiscale simulation of transpo... read less USED (high confidence) R. Singh, P. Gupta, and N. Yedla, “Single-crystal Al–Cu50Zr50 metallic glass cold welds: tensile and creep behaviour,” Molecular Simulation. 2019. link Times cited: 10 Abstract: ABSTRACT Tensile and creep properties of dissimilar cold wel… read moreAbstract: ABSTRACT Tensile and creep properties of dissimilar cold weld joints (Al (metal)–Cu50Zr50 (metallic glass)) are investigated using molecular dynamics simulations. Embedded atom method potential is used to model the interactions between Al–Cu–Zr atoms. Cold welding is carried out at three different velocities (20, 30 and 40 m/s) and for three interferences (0.4, 1.3 and 2.3 nm). The strength of the welded joints is measured using the tensile test carried out at a strain rate of 1.5 × 109/s. Structure studies by radial distribution function analysis indicate amorphisation of Al in the weld regions. Tensile studies show that the maximum strength is obtained in the sample that is welded for 1.3 nm interference. Creep studies carried out over range of stresses (200–350 MPa) and temperatures (200–500 K) show very short primary creep and significant steady-state creep. The stress exponent n has two values; at lower stress, n = 1.2, and at higher stress, n = 4.06, respectively. The deformation mechanisms are observed to be slip by Shockley partial dislocation and by twinning in Al region. The icosahedral cluster population in metallic glass decreases as the temperature increases and contributes to large plastic strain. read less USED (high confidence) G. Li, Y.-bao Wang, K. Wang, M. Xiang, and J. Chen, “Shock induced plasticity and phase transition in single crystal lead by molecular dynamics simulations,” Journal of Applied Physics. 2019. link Times cited: 7 Abstract: Shock-induced plasticity and phase transition in single crys… read moreAbstract: Shock-induced plasticity and phase transition in single crystal lead are investigated by nonequilibrium molecular dynamics simulations. Under dynamic shock loading, the appearance of plasticity in materials precedes that of phase transition. Plasticity mainly causes two effects: one is that plasticity has a significant relaxation effect on shear stress, and the other is that deformation twinning serves as important nucleation sites for the phase transition. This twinning is caused by mutual impediments among different cross-slips and {111} slips. There are three main stages in the dynamic phase transition process of lead: fcc → bcc-like phase transition, plasticity, and hcp phase formation and growth. Moreover, phase transition has a more significant relaxation effect on shear stress, which relaxes the shear stress to a minimum value. The spall strength of lead decreases as the shock intensity increases, but its rate of decrease under different shock intensities is different. Plasticity, especially phase transition, would obviously result in a lower rate of decrease in spall strength.Shock-induced plasticity and phase transition in single crystal lead are investigated by nonequilibrium molecular dynamics simulations. Under dynamic shock loading, the appearance of plasticity in materials precedes that of phase transition. Plasticity mainly causes two effects: one is that plasticity has a significant relaxation effect on shear stress, and the other is that deformation twinning serves as important nucleation sites for the phase transition. This twinning is caused by mutual impediments among different cross-slips and {111} slips. There are three main stages in the dynamic phase transition process of lead: fcc → bcc-like phase transition, plasticity, and hcp phase formation and growth. Moreover, phase transition has a more significant relaxation effect on shear stress, which relaxes the shear stress to a minimum value. The spall strength of lead decreases as the shock intensity increases, but its rate of decrease under different shock intensities is different. Plasticity, especially phase ... read less USED (high confidence) P. He et al., “A Grain Boundary Regulates the Friction Behaviors between Graphene and a Gold Substrate,” Crystals. 2019. link Times cited: 7 Abstract: The nanofriction of graphene is critical for its broad appli… read moreAbstract: The nanofriction of graphene is critical for its broad applications as a lubricant and in flexible electronics. Herein, using a Au substrate as an example, we have investigated the effect of the grain boundary on the nanofriction of graphene by means of molecular dynamics simulations. We have systematically examined the coupling effects of the grain boundary with different mechanical pressures, velocities, temperatures, contact areas, and relative rotation angles on nanofriction. It is revealed that grain boundaries could reduce the friction between graphene and the gold substrate with a small deformation of the latter. Large lateral forces were observed under severe deformation around the grain boundary. The fluctuation of lateral forces was bigger on surfaces with grain boundaries than that on single-crystal surfaces. Friction forces induced by the armchair grain boundaries was smaller than those by the zigzag grain boundaries. read less USED (high confidence) M. Schnedlitz et al., “Effects of the Core Location on the Structural Stability of Ni–Au Core–Shell Nanoparticles,” The Journal of Physical Chemistry. C, Nanomaterials and Interfaces. 2019. link Times cited: 19 Abstract: Structural changes of Ni–Au core–shell nanoparticles with in… read moreAbstract: Structural changes of Ni–Au core–shell nanoparticles with increasing temperature are studied at atomic resolution. The bimetallic clusters, synthesized in superfluid helium droplets, show a centralized Ni core, which is an intrinsic feature of the growth process inside helium. After deposition on SiNx, the nanoparticles undergo a programmed temperature treatment in vacuum combined with an in situ transmission electron microscopy study of structural changes. We observe not only full alloying far below the actual melting temperature, but also a significantly higher stability of core–shell structures with decentralized Ni cores. Explanations are provided by large-scale molecular dynamics simulations on model structures consisting of up to 3000 metal atoms. Two entirely different diffusion processes can be identified for both types of core–shell structures, strikingly illustrating how localized, atomic features can still dictate the overall behavior of a nanometer-sized particle. read less USED (high confidence) R. Allaire et al., “Surface, Interface, and Temperature Effects on the Phase Separation and Nanoparticle Self Assembly of Bi-Metallic Ni0.5Ag0.5: A Molecular Dynamics Study,” Nanomaterials. 2019. link Times cited: 7 Abstract: Classical molecular dynamics (MD) simulations were used to i… read moreAbstract: Classical molecular dynamics (MD) simulations were used to investigate how free surfaces, as well as supporting substrates, affect phase separation in a NiAg alloy. Bulk samples, droplets, and droplets deposited on a graphene substrate were investigated at temperatures that spanned regions of interest in the bulk NiAg phase diagram, i.e., miscible and immiscible liquid, liquid-crystal, and crystal-crystal regions. Using MD simulations to cool down a bulk sample from 3000 K to 800 K, it was found that phase separation below 2400 K takes place in agreement with the phase diagram. When free surface effects were introduced, phase separation was accompanied by a core-shell transformation: spherical droplets created from the bulk samples became core-shell nanoparticles with a shell made mostly of Ag atoms and a core made of Ni atoms. When such droplets were deposited on a graphene substrate, the phase separation was accompanied by Ni layering at the graphene interface and Ag at the vacuum interface. Thus, it should be possible to create NiAg core-shell and layer-like nanostructures by quenching liquid NiAg samples on tailored substrates. Furthermore, interesting bimetallic nanoparticle morphologies might be tuned via control of the surface and interface energies and chemical instabilities of the system. read less USED (high confidence) V. Samsonov, I. Talyzin, A. Kartoshkin, and M. V. Samsonov, “Prediction of Segregation in Binary Metal Nanoparticles: Thermodynamic and Atomistic Simulations,” Physics of Metals and Metallography. 2019. link Times cited: 5 USED (high confidence) J. Yu, H. Xie, F. Yin, and T. Yu, “Face-centered-cubic to body-centered-cubic phase transformation of Cu nanoplate under [100] tensile loading,” Philosophical Magazine. 2019. link Times cited: 1 Abstract: ABSTRACT Molecular dynamics simulation was used to stretch C… read moreAbstract: ABSTRACT Molecular dynamics simulation was used to stretch Cu nanoplates along its [100] direction at various strain rates and temperatures. Under high strain rate and beyond the elastic limit, the Cu nanoplates underwent an unusual deformation mechanism with expansion along free surface lateral direction and contraction along the other lateral direction, which leaded to the face-centred-cubic phase transforming into unstressed body-centred-cubic phase. Under low strain rate, the deformation of the nanoplate went back to well-known dislocation mechanism. The face-centred-cubic to body-centred-cubic phase transformation mechanism was further discussed in terms of elastic stability theory and free surface stress effect. read less USED (high confidence) S. Rogachev, A. Rogachev, and M. Alymov, “Estimating the Critical Glass Transition Rate of Pure Metals Using Molecular Dynamic Modeling,” Doklady Physics. 2019. link Times cited: 3 USED (high confidence) R. Arifin, M. Malyadi, Munaji, G. A. Buntoro, and Darminto, “Pressure dependence of the structure of liquid NiTi: a molecular dynamics study,” Journal of Physics: Condensed Matter. 2019. link Times cited: 4 Abstract: We evaluate the structure of liquid NiTi under various press… read moreAbstract: We evaluate the structure of liquid NiTi under various pressures from 0 GPa to 40 GPa in the atomic level using molecular dynamics simulations. The structure factor and radial distribution function are used to investigate the general structural change of the system. Further identification of the local structures is examined by the bond-angle method and bond-angle distribution analysis. From our results, we found that the count of the local structure of fcc, hcp, bcc, and icosahedral short-range order monotonically increase when the pressures increase. We also observed in our results that the size of the local cluster grows as the pressure increases, and the long-range connectivity of the quasi-crystal is achieved at high pressure. read less USED (high confidence) J. Zhou et al., “Observing crystal nucleation in four dimensions using atomic electron tomography,” Nature. 2019. link Times cited: 198 USED (high confidence) J. Ramade et al., “Environmental Plasmonic Spectroscopy of Silver–Iron Nanoparticles: Chemical Ordering under Oxidizing and Reducing Conditions,” The Journal of Physical Chemistry C. 2019. link Times cited: 7 Abstract: The chemical structure and the localized surface plasmon res… read moreAbstract: The chemical structure and the localized surface plasmon resonance (LSPR) of size-selected AgxFe1–x (x = 0.5, 0.8) nanoparticles (NPs), produced by laser vaporization, were investigated experimenta... read less USED (high confidence) D. Ishikawa et al., “Analysis of Bonding Interfaces of Pressureless-sintered Cu on Metallization Layers,” 2019 International Conference on Electronics Packaging (ICEP). 2019. link Times cited: 3 Abstract: this paper describes thermal stabilities (573 K for 8 h) of … read moreAbstract: this paper describes thermal stabilities (573 K for 8 h) of pressureless-sintered Copper (Cu) on four kinds of top metallization layers (Ni, Cu, Ag, and Au) by experiments. Evolutions of sintering process of Cu nanoparticles and diffusion coefficients of interfaces between a bulk Cu layer and metallization layers were also evaluated by molecular dynamics (MD) simulations. After aging at 573 K for 8 h in terms of bonding samples, the shear strengths of sintered Cu on Ni and Cu layer increased, whereas those of sintered Cu on Ag and Au layer decreased. It was confirmed that interdiffusion occurred in the interfaces between sintered Cu layer and Ag layer or Au layer by energy dispersive X-ray spectroscopy (EDX), which increased the porosities of sintered Cu near the interfaces. The increases of interfacial porosities on sintered Cu/Ag and sintered Cu/Au decreased the shear strengths. In contrast, the porosities near the interface between sintered Cu layer and Ni layer or Cu layer hardly changed after aging. MD simulations revealed that Kirkendall voids were promoted by higher interdiffusion coefficients and higher ratio of intrinsic diffusion coefficients between a bulk Cu layer and metallization layers, which consequently increased the porosities of sintered Cu near the interfaces. The interdiffusion coefficients, which seem to have a correlation with the shear strengths of sintered Cu, can be used as an index value to find metallization layers that are suitable for the sintered Cu layer by calculations of MD simulations. read less USED (high confidence) Q. Li, J. Zhang, H. Tang, H. Ye, and Y. Zheng, “Regulating the mechanical properties of nanocrystalline nickel via molybdenum segregation: an atomistic study,” Nanotechnology. 2019. link Times cited: 9 Abstract: The effects of segregation of impurity molybdenum (Mo) atoms… read moreAbstract: The effects of segregation of impurity molybdenum (Mo) atoms on the tensile mechanical properties of nanocrystalline nickel (Ni) are investigated with molecular dynamics simulation. The results show that the segregation of Mo atoms induces an obvious increase in the elastic modulus and strength of nanocrystalline Ni, and the strengthening effect is more significant with smaller grain size. When the grain size decreases below a critical value, at which the softening occurs in non-segregated Ni-Mo alloy, no evident softening phenomenon is observed in Mo-segregated systems. Furthermore, based on a bicrystal configuration, it is found that Mo atoms segregating to the grain boundary reduce the energy and mobility of the grain boundary, increasing the grain boundary stability and thus accommodating the strengthening. The present findings will shed light on the fabrication of high strength nanocrystalline materials by controlling the segregation of atoms. read less USED (high confidence) I. N. Kar’kin, L. E. Kar’kina, Y. Gornostyrev, and A. P. Korzhavyi, “Kinetics of Early Decomposition Stages in Diluted bcc Fe–Сu–Ni–Al Alloy: MC+MD Simulation,” Physics of the Solid State. 2019. link Times cited: 4 USED (high confidence) T. Wang et al., “Sub‐6 nm Fully Ordered L10‐Pt–Ni–Co Nanoparticles Enhance Oxygen Reduction via Co Doping Induced Ferromagnetism Enhancement and Optimized Surface Strain,” Advanced Energy Materials. 2019. link Times cited: 100 Abstract: Engineering the crystal structure of Pt–M (M = transition me… read moreAbstract: Engineering the crystal structure of Pt–M (M = transition metal) nanoalloys to chemically ordered ones has drawn increasing attention in oxygen reduction reaction (ORR) electrocatalysis due to their high resistance against M etching in acid. Although Pt–Ni alloy nanoparticles (NPs) have demonstrated respectable initial ORR activity in acid, their stability remains a big challenge due to the fast etching of Ni. In this work, sub‐6 nm monodisperse chemically ordered L10‐Pt–Ni–Co NPs are synthesized for the first time by employing a bifunctional core/shell Pt/NiCoOx precursor, which could provide abundant O‐vacancies for facilitated Pt/Ni/Co atom diffusion and prevent NP sintering during thermal annealing. Further, Co doping is found to remarkably enhance the ferromagnetism (room temperature coercivity reaching 2.1 kOe) and the consequent chemical ordering of L10‐Pt–Ni NPs. As a result, the best‐performing carbon supported L10‐PtNi0.8Co0.2 catalyst reveals a half‐wave potential (E1/2) of 0.951 V versus reversible hydrogen electrode in 0.1 m HClO4 with 23‐times enhancement in mass activity over the commercial Pt/C catalyst along with much improved stability. Density functional theory (DFT) calculations suggest that the L10‐PtNi0.8Co0.2 core could tune the surface strain of the Pt shell toward optimized Pt–O binding energy and facilitated reaction rate, thereby improving the ORR electrocatalysis. read less USED (high confidence) E. Levo, F. Granberg, D. Utt, K. Albe, K. Nordlund, and F. Djurabekova, “Radiation stability of nanocrystalline single-phase multicomponent alloys,” Journal of Materials Research. 2019. link Times cited: 9 Abstract: In search of materials with better properties, polycrystalli… read moreAbstract: In search of materials with better properties, polycrystalline materials are often found to be superior to their respective single crystalline counterparts. Reduction of grain size in polycrystalline materials can drastically alter the properties of materials. When the grain sizes reach the nanometer scale, the improved mechanical response of the materials make them attractive in many applications. Multicomponent solid-solution alloys have shown to have a higher radiation tolerance compared with pure materials. Combining these advantages, we investigate the radiation tolerance of nanocrystalline multicomponent alloys. We find that these alloys withstand a much higher irradiation dose, compared with nanocrystalline Ni, before the nanocrystallinity is lost. Some of the investigated alloys managed to keep their nanocrystallinity for twice the irradiation dose as pure Ni. read less USED (high confidence) F. Maresca and W. Curtin, “Mechanistic origin of high strength in refractory BCC high entropy alloys up to 1900K,” Acta Materialia. 2019. link Times cited: 219 USED (high confidence) J. Maldonis et al., “StructOpt: A modular materials structure optimization suite incorporating experimental data and simulated energies,” Computational Materials Science. 2019. link Times cited: 6 USED (high confidence) Y. Yildiz and M. Kırca, “Compression and shear behavior of ultrathin coated nanoporous gold: A molecular dynamics study,” Journal of Applied Physics. 2018. link Times cited: 4 Abstract: This paper investigates the compressive and shear properties… read moreAbstract: This paper investigates the compressive and shear properties of nanoporous gold (np-Au) coated with different ultrathin metallic materials (i.e., platinum and silver) via molecular dynamics simulations. Atomistic models used for the geometric representation of coated and uncoated np-Au structures are generated through a modeling technique based on the Voronoi tessellation method. Three different coating thickness values are used to examine the role of thickness for the coating performance under compressive and shear loading by comparing the mechanical characteristics of the atomistic models such as Young's modulus, yield, and ultimate strengths. Moreover, adaptive common neighbor analyses are carried out by monitoring the evolution of the crystal structure of the specimens during the loading process. In this way, the deformation mechanisms of coated and uncoated nanoporous specimens are identified thoroughly. As a key finding from the simulation results, it is observed that the mechanical properties of np-Au are crucially dependent on the type of the coating material. However, a significant improvement on the toughness within the plastic regime is demonstrated for all types of coating materials and loading conditions.This paper investigates the compressive and shear properties of nanoporous gold (np-Au) coated with different ultrathin metallic materials (i.e., platinum and silver) via molecular dynamics simulations. Atomistic models used for the geometric representation of coated and uncoated np-Au structures are generated through a modeling technique based on the Voronoi tessellation method. Three different coating thickness values are used to examine the role of thickness for the coating performance under compressive and shear loading by comparing the mechanical characteristics of the atomistic models such as Young's modulus, yield, and ultimate strengths. Moreover, adaptive common neighbor analyses are carried out by monitoring the evolution of the crystal structure of the specimens during the loading process. In this way, the deformation mechanisms of coated and uncoated nanoporous specimens are identified thoroughly. As a key finding from the simulation results, it is observed that the mechanical properties of np... read less USED (high confidence) V. Samsonov, I. Talyzin, A. Kartoshkin, and S. Vasilyev, “Surface segregation in binary Cu–Ni and Au–Co nanoalloys and the core–shell structure stability/instability: thermodynamic and atomistic simulations,” Applied Nanoscience. 2018. link Times cited: 26 USED (high confidence) G. Arora, K. Rawat, and D. Aidhy, “Effect of atomic order/disorder on Cr segregation in Ni-Fe alloys,” Journal of Applied Physics. 2018. link Times cited: 4 Abstract: Recent irradiation experiments on concentrated random solid … read moreAbstract: Recent irradiation experiments on concentrated random solid solution alloys (CSAs) show that some CSAs can undergo disorder-to-order transition, i.e., the atoms that are initially randomly distributed on a face centered cubic crystal lattice undergo ordering (e.g., L10 or L12) due to irradiation. In this work, we elucidate that the atomic structure could affect the segregation properties of grain boundaries. While working on Ni and Ni-Fe alloys, from static atomistic simulations on 138 grain boundaries, we show that despite identical alloy composition, Cr segregation is higher in the disordered structures compared to ordered structures in both Ni0.50Fe0.50 and Ni0.75Fe0.25 systems. We also show that grain boundary (GB) energy could act as a descriptor for impurity segregation. We illustrate that there is a direct correlation between Cr segregation and grain boundary energy, i.e., segregation increases with the increase in the GB energy. Such correlation is observed in pure Ni and in the Ni-Fe alloys studied in this work.Recent irradiation experiments on concentrated random solid solution alloys (CSAs) show that some CSAs can undergo disorder-to-order transition, i.e., the atoms that are initially randomly distributed on a face centered cubic crystal lattice undergo ordering (e.g., L10 or L12) due to irradiation. In this work, we elucidate that the atomic structure could affect the segregation properties of grain boundaries. While working on Ni and Ni-Fe alloys, from static atomistic simulations on 138 grain boundaries, we show that despite identical alloy composition, Cr segregation is higher in the disordered structures compared to ordered structures in both Ni0.50Fe0.50 and Ni0.75Fe0.25 systems. We also show that grain boundary (GB) energy could act as a descriptor for impurity segregation. We illustrate that there is a direct correlation between Cr segregation and grain boundary energy, i.e., segregation increases with the increase in the GB energy. Such correlation is observed in pure Ni and in the Ni-Fe alloys studi... read less USED (high confidence) D. Ishikawa et al., “Copper Die-Bonding Sinter Paste: Sintering and Bonding Properties,” 2018 7th Electronic System-Integration Technology Conference (ESTC). 2018. link Times cited: 13 Abstract: this paper describes the sintering properties and bonding pr… read moreAbstract: this paper describes the sintering properties and bonding properties of copper (Cu) die-bonding sinter paste for power devices operating at high temperatures. The Cu paste can be sintered pressure less in 100% H2 or under pressure in 100% N2 atmospheres. The as-sintered density, thermal conductivity and resistivity of pressure less-sintered Cu (in 100% H2, 300 °C, 1 h) is found to be 7S%, 180 Wm^-1K^-1 and 4.3 $\mu\Omega\cdot cm$, respectively. The pressurelesssintered Cu has higher 0.2% proof stress than the pressure-sintered Ag (sintered density =87%, in air, 300 °C 10 MPa, 10min) as a comparison material in a three-point bending test. The die-shear strength of appropriate pressurelesssintered Cu on four different metal adherends (Cu, Ni, Ag and Au) was 30 MPa or higher. The die-shear strength of pressure- sintered Cu in 100% N2 was 36 MPa or higher. A thermal cycle tolerance of 1000 cycles or greater was shown in a power device test package which was bonded using the pressurelesssintered Cu and encapsulated with an epoxy molding compound. The Cu sinter paste can be used as a reliable die-bonding material for power modules operating at high temperatures. read less USED (high confidence) Y. Liu, Y. Liu, and J. Luo, “Atomic Scale Simulation on the Fracture Mechanism of Black Phosphorus Monolayer under Indentation,” Nanomaterials. 2018. link Times cited: 3 Abstract: Molecular dynamics simulations on the indentation process of… read moreAbstract: Molecular dynamics simulations on the indentation process of freestanding and Pt(111)-supported black phosphorus (BP) monolayer were conducted to study the fracture mechanism of the membrane. For the freestanding BP monolayer, crack grows firstly along armchair direction and then zigzag direction during the indentation process. Whereas, for the Pt(111)-supported BP monolayer, crack growth shows no obvious directionality, with irregular distribution of crack tips. Further study on stress distribution shows that maximum normal stress component at elastic stage is in zigzag direction for the freestanding BP monolayer, and in vertical direction for the Pt(111)-supported BP monolayer. As BP monolayer is remarkably anisotropic for in-plane mechanical properties and homogeneous for out-of-plane mechanical properties, the difference of stress state may be a key reason for the different fracture behavior in these two cases. These findings may help to understand the failure mechanism of BP, when applied in nano-devices. read less USED (high confidence) S. Guo, M. Wang, Y. Zhang, X. Lin, and W. Huang, “Region selectivity of nanometer scale crystallization behavior in metallic glass,” Journal of Materials Science. 2018. link Times cited: 0 USED (high confidence) A. Gola and L. Pastewka, “Embedded atom method potential for studying mechanical properties of binary Cu–Au alloys,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 13 Abstract: We present an embedded atom method (EAM) potential for the b… read moreAbstract: We present an embedded atom method (EAM) potential for the binary Cu–Au system. The unary phases are described by two well-tested unary EAM potentials for Cu and Au. We fitted the interaction between Cu and Au to experimental properties of the binary intermetallic phases Cu3Au, CuAu and CuAu3. Particular attention has been paid to reproducing stacking fault energies in order to obtain a potential suitable for studying deformation in this binary system. The resulting energies, lattice constant, elastic properties and melting points are in good agreement with available experimental data. We use nested sampling to show that our potential reproduces the phase boundaries between intermetallic phases and the disordered face-centered cubic solid solution. We benchmark our potential against four popular Cu–Au EAM parameterizations and density-functional theory calculations. read less USED (high confidence) H. Zhou, J. Li, Y. Xian, R. Wu, G. Hu, and R. Xia, “Molecular dynamics study on cold-welding of 3D nanoporous composite structures.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 14 Abstract: Nanoporous metals are a class of novel nanomaterials with po… read moreAbstract: Nanoporous metals are a class of novel nanomaterials with potential applications in many fields. Herein, we demonstrate the cold-welding mechanism of nanoporous metals with various combinations using molecular dynamics simulations. This study shows that it is possible to cold-weld two nanoporous metals to form a novel composite material. The influence of temperature, in the range of 300-900 K, on the mechanical properties of the resultant composite material was investigated. With an increase in temperature, the weld stress and the mechanical strength of the nanoporous structures significantly decreased as an increase in disorder magnitude was observed. These results could lead to bottom-up nanofabrication and nanoassembly of combined nanoporous metals for high mechanical performance. read less USED (high confidence) W. Li, K. Li, K. Fan, D.-xing Zhang, and W.-dong Wang, “Temperature and Pressure Dependences of the Elastic Properties of Tantalum Single Crystals Under <100> Tensile Loading: A Molecular Dynamics Study,” Nanoscale Research Letters. 2018. link Times cited: 10 USED (high confidence) J. Lee, X. Hu, A. Voevodin, A. Martini, and D. Berman, “Effect of Substrate Support on Dynamic Graphene/Metal Electrical Contacts,” Micromachines. 2018. link Times cited: 9 Abstract: Recent advances in graphene and other two-dimensional (2D) m… read moreAbstract: Recent advances in graphene and other two-dimensional (2D) material synthesis and characterization have led to their use in emerging technologies, including flexible electronics. However, a major challenge is electrical contact stability, especially under mechanical straining or dynamic loading, which can be important for 2D material use in microelectromechanical systems. In this letter, we investigate the stability of dynamic electrical contacts at a graphene/metal interface using atomic force microscopy (AFM), under static conditions with variable normal loads and under sliding conditions with variable speeds. Our results demonstrate that contact resistance depends on the nature of the graphene support, specifically whether the graphene is free-standing or supported by a substrate, as well as on the contact load and sliding velocity. The results of the dynamic AFM experiments are corroborated by simulations, which show that the presence of a stiff substrate, increased load, and reduced sliding velocity lead to a more stable low-resistance contact. read less USED (high confidence) A. Klemenz, A. Gola, M. Moseler, and L. Pastewka, “Contact mechanics of graphene-covered metal surfaces,” Applied Physics Letters. 2018. link Times cited: 16 Abstract: We carry out molecular statics simulations of the indentatio… read moreAbstract: We carry out molecular statics simulations of the indentation of bare and graphene-covered Pt (111) surfaces with smooth and rough indenters of radius 1.5 to 10 nm. Our simulations show that the plastic yield of bare surfaces strongly depends on atomic-scale indenter roughness such as terraces or amorphous disorder. Covering surfaces with graphene regularizes this response to the results obtained for ideally smooth indenters. Our results suggest that graphene monolayers and other 2D materials mitigate the effect of roughness, which could be exploited to improve the fidelity of experiments that probe the mechanical properties of interfaces. read less USED (high confidence) S. Yang, N. Zhou, H. Zheng, S. Ong, and J. Luo, “First-Order Interfacial Transformations with a Critical Point: Breaking the Symmetry at a Symmetric Tilt Grain Boundary.,” Physical review letters. 2018. link Times cited: 48 Abstract: First-order interfacial phaselike transformations that break… read moreAbstract: First-order interfacial phaselike transformations that break the mirror symmetry of the symmetric ∑5 (210) tilt grain boundary (GB) are discovered by combining a modified genetic algorithm with hybrid Monte Carlo and molecular dynamics simulations. Density functional theory calculations confirm this prediction. This first-order coupled structural and adsorption transformation, which produces two variants of asymmetric bilayers, vanishes at an interfacial critical point. A GB complexion (phase) diagram is constructed via semigrand canonical ensemble atomistic simulations for the first time. read less USED (high confidence) W. Gao et al., “Dynamics of Transformation from Platinum Icosahedral Nanoparticles to Larger FCC Crystal at Millisecond Time Resolution,” Scientific Reports. 2017. link Times cited: 6 USED (high confidence) N. Miyazawa, T. Yamaoka, M. Hakamada, and M. Mabuchi, “Atomistic study of inelastic deformation in aluminium grain boundary fractures,” Philosophical Magazine Letters. 2017. link Times cited: 3 Abstract: The plastic work of fracture in a deformable solid has been … read moreAbstract: The plastic work of fracture in a deformable solid has been believed to be related to only the ideal brittle fracture energy. However, additional factors affecting the plastic work must also exist because the plastic work is a path function. In the present work, first-principles calculations and molecular dynamics simulations of tensile tests were performed on Σ3(1 1)[1 1 0] and Σ11(1 3)[1 1 0] symmetric tilt aluminium grain boundaries, where the grain boundary energy of the Σ11(1 3) grain boundary is higher than that of the Σ3(1 1)[1 1 0] gain boundary. The calculations showed that, although the ideal brittle fracture energy for the Σ11(1 3) grain boundary was almost the same as that for the Σ3(1 1) grain boundary, the plastic work for the former was larger than that for the latter, resulting in a larger fracture energy for the Σ11(1 3) grain boundary. Local inelastic deformation occurred around the atoms with high internal energy at the grain boundary for the Σ11(1 3) grain boundary. It is therefore suggested that the plastic work is a function of both the grain boundary energy and the ideal brittle fracture energy. read less USED (high confidence) X. Hu and A. Martini, “Atomistic simulations of contact area and conductance at nanoscale interfaces.,” Nanoscale. 2017. link Times cited: 7 Abstract: Atomistic simulations were used to study conductance across … read moreAbstract: Atomistic simulations were used to study conductance across the interface between a nanoscale gold probe and a graphite surface with a step edge. Conductance on the graphite terrace was observed to increase with load and be approximately proportional to contact area calculated from the positions of atoms in the interface. The relationship between area and conductance was further explored by varying the position of the contact relative to the location of the graphite step edge. These simulations reproduced a previously-reported current dip at step edges measured experimentally and the trend was explained by changes in both contact area and the distribution of distances between atoms in the interface. The novel approach reported here provides a foundation for future studies of the fundamental relationships between conductance, load and surface topography at the atomic scale. read less USED (high confidence) Y. Yildiz and M. Kırca, “Effects of ultrathin coating on the tensile behavior of nanoporous gold,” Journal of Applied Physics. 2017. link Times cited: 5 Abstract: In this study, the mechanical properties of nanoporous gold … read moreAbstract: In this study, the mechanical properties of nanoporous gold (np-Au) coated with different ultrathin metallic materials (i.e., platinum and silver) are studied through molecular dynamics simulations. A new atomistic modelling technique, which is based on the Voronoi tessellation method providing periodic atomistic specimens, is used for the geometric representation of np-Au structure. Three different coating thickness values are used to examine the role of thickness on the coating performance under tensile loading at a constant strain rate. Several parameters, including Young's modulus, yield, and ultimate strengths, are utilized to compare the mechanical characteristics of coated and uncoated np-Au specimens. Moreover, adaptive common neighbor analyses are performed on the specimens for the purpose of understanding the deformation mechanisms of coated and uncoated nanoporous specimens comprehensively by monitoring the microstructural evolution of the crystal structure of the specimens within the deformati... read less USED (high confidence) C. Wang, C. Li, J. Han, L. Yan, B. Deng, and X. Liu, “The pressure-temperature phase diagram of pure Co based on first-principles calculations.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 2 Abstract: We optimized the high pressure-temperature phase diagram of … read moreAbstract: We optimized the high pressure-temperature phase diagram of pure Co up to the liquidus temperature and 120 GPa, based on thermodynamic properties calculated using first-principles. The Gibbs energy for each phase was evaluated in the framework of a quasiharmonic approximation, with a consideration of the thermal electronic contribution at finite temperatures. Particularly, the liquidus temperature, as a function of pressure, was determined using classical Molecular Dynamics simulations. Our results in this work successfully integrated experimental observations and the previous theoretical predictions. The critical solid phase transitions of ε → γf and ε → β were clarified using the Gibbs energy as a function of pressure and temperature. In addition, the magnetism of β above 70 GPa was verified to be nonmagnetic. The difference between γp and β, which was unclear before, has been illustrated to be associated with the magnetic transformation from the paramagnetic state of the γp phase to the nonmagnetic state of the β phase rather than the structural transformation. read less USED (high confidence) O. Waseda et al., “Stability of nanocrystalline Ni-based alloys: coupling Monte Carlo and molecular dynamics simulations,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 5 Abstract: The thermal stability of nanocrystalline Ni due to small add… read moreAbstract: The thermal stability of nanocrystalline Ni due to small additions of Mo or W (up to 1 at%) was investigated in computer simulations by means of a combined Monte Carlo (MC)/molecular dynamics (MD) two-steps approach. In the first step, energy-biased on-lattice MC revealed segregation of the alloying elements to grain boundaries. However, the condition for the thermodynamic stability of these nanocrystalline Ni alloys (zero grain boundary energy) was not fulfilled. Subsequently, MD simulations were carried out for up to 0.5 μs at 1000 K. At this temperature, grain growth was hindered for minimum global concentrations of 0.5 at% W and 0.7 at% Mo, thus preserving most of the nanocrystalline structure. This is in clear contrast to a pure Ni model system, for which the transformation into a monocrystal was observed in MD simulations within 0.2 μs at the same temperature. These results suggest that grain boundary segregation of low-soluble alloying elements in low-alloyed systems can produce high-temperature metastable nanocrystalline materials. MD simulations carried out at 1200 K for 1 at% Mo/W showed significant grain boundary migration accompanied by some degree of solute diffusion, thus providing additional evidence that solute drag mostly contributed to the nanostructure stability observed at lower temperature. read less USED (high confidence) A. Takahashi, A. Seko, and I. Tanaka, “Conceptual and practical bases for the high accuracy of machine learning interatomic potential,” arXiv: Materials Science. 2017. link Times cited: 29 Abstract: Machine learning interatomic potentials (MLIPs) based on a l… read moreAbstract: Machine learning interatomic potentials (MLIPs) based on a large dataset obtained by density functional theory (DFT) calculation have been developed recently. This study gives both conceptual and practical bases for the high accuracy of MLIPs, although MLIPs have been considered to be simply an accurate black-box description of atomic energy. We also construct the most accurate MLIP of the elemental Ti ever reported using a linearized MLIP framework and many angular-dependent descriptors, which also corresponds to a generalization of the modified embedded atom method (MEAM) potential. read less USED (high confidence) E. Levo, F. Granberg, C. Fridlund, K. Nordlund, and F. Djurabekova, “Radiation damage buildup and dislocation evolution in Ni and equiatomic multicomponent Ni-based alloys,” Journal of Nuclear Materials. 2017. link Times cited: 67 USED (high confidence) J. Velasco, A. Concustell, E. Pineda, and D. Crespo, “Plastic deformation induced anisotropy in metallic glasses: A molecular dynamics study,” Journal of Alloys and Compounds. 2017. link Times cited: 9 USED (high confidence) W. Kim and E. Tadmor, “Accelerated quasicontinuum: a practical perspective on hyper-QC with application to nanoindentation,” Philosophical Magazine. 2017. link Times cited: 8 Abstract: Hyper-QC is a multiscale method based on the quasicontinuum … read moreAbstract: Hyper-QC is a multiscale method based on the quasicontinuum (QC) method in which time is accelerated using hyperdynamics through the addition of a suitable bias potential. This paper describes the practical details of implementing and carrying out hyper-QC simulations and introduces a novel mechanism-based bias potential for deformation processes in face-centred cubic (fcc) systems. The factors limiting the maximum achievable acceleration are discussed. The method is demonstrated for nanoindentation into a thin film of single crystal fcc nickel at near experimental loading rates. Speed up factors as high as 10,000 are achieved. The simulations reveal a thermally activated dislocation nucleation mechanism with a logarithmic dependence on temperature and indenter velocity in agreement with a theoretical model. read less USED (high confidence) G. Agarwal and A. Dongare, “Modeling the thermodynamic behavior and shock response of Ti systems at the atomic scales and the mesoscales,” Journal of Materials Science. 2017. link Times cited: 20 USED (high confidence) Y. Zhang, G. Tucker, and J. Trelewicz, “Stress-assisted grain growth in nanocrystalline metals: Grain boundary mediated mechanisms and stabilization through alloying,” Acta Materialia. 2017. link Times cited: 64 USED (high confidence) S. Zhang, K. Nordlund, F. Djurabekova, F. Granberg, Y. Zhang, and T. S. Wang, “Radiation damage buildup by athermal defect reactions in nickel and concentrated nickel alloys,” Materials Research Letters. 2017. link Times cited: 34 Abstract: ABSTRACT We develop a new method using binary collision appr… read moreAbstract: ABSTRACT We develop a new method using binary collision approximation simulating the Rutherford backscattering spectrometry in channeling conditions (RBS/C) from molecular dynamics atom coordinates of irradiated cells. The approach allows comparing experimental and simulated RBS/C signals as a function of depth without fitting parameters. The simulated RBS/C spectra of irradiated Ni and concentrated solid solution alloys (CSAs, NiFe and NiCoCr) show a good agreement with the experimental results. The good agreement indicates the damage evolution under damage overlap conditions in Ni and CSAs at room temperature is dominated by defect recombination and migration induced by irradiation rather than activated thermally. GRAPHICAL ABSTRACT IMPACT STATEMENT A new method simulating the Rutherford backscattering Spectrometry in channeling conditions (RBS/C) was proposed. The RBS/C simulations reveal that the radiation damage buildup in Ni, NiFe and NiCoCr was dominated by athermal defect reactions. read less USED (high confidence) L. Chen, J. Fan, and H. Gong, “Phase transition and mechanical properties of tungsten nanomaterials from molecular dynamic simulation,” Journal of Nanoparticle Research. 2017. link Times cited: 17 USED (high confidence) Y. Yildiz and M. Kırca, “Atomistic simulation of Voronoi-based coated nanoporous metals,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 12 Abstract: In this study, a new method developed for the generation of … read moreAbstract: In this study, a new method developed for the generation of periodic atomistic models of coated and uncoated nanoporous metals (NPMs) is presented by examining the thermodynamic stability of coated nanoporous structures. The proposed method is mainly based on the Voronoi tessellation technique, which provides the ability to control cross-sectional dimension and slenderness of ligaments as well as the thickness of coating. By the utilization of the method, molecular dynamic (MD) simulations of randomly structured NPMs with coating can be performed efficiently in order to investigate their physical characteristics. In this context, for the purpose of demonstrating the functionality of the method, sample atomistic models of Au/Pt NPMs are generated and the effects of coating and porosity on the thermodynamic stability are investigated by using MD simulations. In addition to that, uniaxial tensile loading simulations are performed via MD technique to validate the nanoporous models by comparing the effective Young’s modulus values with the results from literature. Based on the results, while it is demonstrated that coating the nanoporous structures slightly decreases the structural stability causing atomistic configurational changes, it is also shown that the stability of the atomistic models is higher at lower porosities. Furthermore, adaptive common neighbour analysis is also performed to identify the stabilized atomistic structure after the coating process, which provides direct foresights for the mechanical behaviour of coated nanoporous structures. read less USED (high confidence) C. Dai, L. Balogh, Z. Yao, and M. Daymond, “The habit plane of 〈a〉-type dislocation loops in α-zirconium: an atomistic study,” Philosophical Magazine. 2017. link Times cited: 15 Abstract: We use both a model of dislocation energy and molecular dyna… read moreAbstract: We use both a model of dislocation energy and molecular dynamics (MD) simulations to explore the habit planes of 〈a〉-type dislocation loops, while cascade simulations are produced to investigate the effect of irradiation on those loops. Vacancy and interstitial loops are artificially created on perfect prism planes in MD, and they reorient to their preferred habit planes during a relaxation stage. The statistics presented in stereographic projections show that the preferred habit planes are close to the prism plane , consistent with experimental data from the literature. We also confirm that the angle between the Burgers vector and the loop’s plane is a useful parameter when identifying the stability of 〈a〉-type dislocation loops. read less USED (high confidence) F. Granberg, F. Djurabekova, E. Levo, and K. Nordlund, “Damage buildup and edge dislocation mobility in equiatomic multicomponent alloys,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 23 USED (high confidence) Z. Chen, X. Zhang, and G. Lu, “Multiscale Computational Design of Core/Shell Nanoparticles for Oxygen Reduction Reaction,” Journal of Physical Chemistry C. 2017. link Times cited: 22 Abstract: We propose a multiscale computational framework to design co… read moreAbstract: We propose a multiscale computational framework to design core/shell nanoparticles (NPs) for oxygen reduction reaction (ORR). Essential to the framework are linear scaling relations between oxygen adsorption energy and surface strain, which can be determined for NP facets and edges from first-principles and multiscale QM/MM calculations, respectively. Based on the linear scaling relations and a microkinetic model, we can estimate ORR rates as a function of surface strain on core/shell NPs. Employing the multiscale framework, we have systematically examined the ORR activity on Pd-based core/shell NPs as a function of their shape, size, shell thickness, and alloy composition of the core. Three NP shapes—icosahedron, octahedron, and truncated octahedron—are explored, and the truncated octahedron is found to be the most active and the icosahedron is the least active. NixPd1–x@Pd NPs with high Ni concentrations and thin shells could exhibit higher ORR rates than the pure Pt(111) surface and/or Pt NPs. AgxPd1–x... read less USED (high confidence) J. Shao, P. Wang, and A. He, “Influence of shock pressure and profile on the microjetting from a grooved Pb surface,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 14 Abstract: This work investigates the shock-induced microjetting from a… read moreAbstract: This work investigates the shock-induced microjetting from a grooved surface (10 nm, 120 degree) of low-melting metal Pb with molecular dynamics simulations. The microjetting processes under surface/release melting conditions are presented in detail, and some properties on the microjet mass and velocity are revealed for different shock pressure and profile cases. It is found that the increase of microjet mass with shock pressure experiences three stages: rapid increase (solid phase), slowdown increase (release melting) and almost no increase (shock melting). For all cases, the ratio of the maximal jetting velocity to the surface velocity approximately keeps a constant (1.5–1.55), but this value undergoes a degree of exponential decay with time for the solid release cases. In addition, the temperature of the microjet is found to be always above the melting point (zero pressure) and keep a continuous increase towards the microjet tip. When introducing slow decaying profiles, the microjet mass begins to increase with the decay rate, which is dominated by the deformation of bubble during pull-back. When the decay rate becomes fast enough, the microspall occurs as expected, meanwhile the microjet appears to reduce because of the shock energy reduction. But that cannot cut off the microjet completely. The velocity distribution along the loading direction shows two linear regions corresponding to the microspall and microjet, and the latter seems to have a greater velocity gradient. read less USED (high confidence) B. Balasubramanian et al., “Structure and magnetism of new rare-earth-free intermetallic compounds: Fe3+xCo3-xTi2(0 ≤ x ≤ 3),” APL Materials. 2016. link Times cited: 6 Abstract: We report the fabrication of a set of new rare-earth-free ma… read moreAbstract: We report the fabrication of a set of new rare-earth-free magnetic compounds, which form the Fe3Co3Ti2-type hexagonal structure with P-6m2 symmetry. Neutron powder diffraction shows a significant Fe/Co anti-site mixing in the Fe3Co3Ti2 structure, which has a strong effect on the magnetocrystalline anisotropy as revealed by first-principle calculations. Increasing substitution of Fe atoms for Co in the Fe3Co3Ti2 lattice leads to the formation of Fe4Co2Ti2, Fe5CoTi, and Fe6Ti2 with significantly improved permanent-magnet properties. A high magnetic anisotropy (13.0 Mergs/cm3) and saturation magnetic polarization (11.4 kG) are achieved at 10 K by altering the atomic arrangements and decreasing Fe/Co occupancy disorder. read less USED (high confidence) N. Argibay, M. Chandross, S. Cheng, and J. R. Michael, “Linking microstructural evolution and macro-scale friction behavior in metals,” Journal of Materials Science. 2016. link Times cited: 76 USED (high confidence) Q. Li, C. Huang, Y. Liang, T. Fu, and T. Peng, “Molecular Dynamics Simulation of Nanoindentation of Cu/Au Thin Films at Different Temperatures,” Journal of Nanomaterials. 2016. link Times cited: 12 Abstract: Two methods, deposition method and ideal modeling based on l… read moreAbstract: Two methods, deposition method and ideal modeling based on lattice constant, are used to prepare three modulation periods’ 1.8 nm Cu/3.6 nm Au, 2.7 nm Cu/2.7 nm Au, and 3.6 nm Cu/1.8 nm Au thin films for nanoindentation at different temperatures. The results show that the temperature will weaken the hardness of thin films. The deposition method and the formation of coherent interface will result in a lot of defects in thin films. These defects can reduce the residual stress in the thin films which is caused by the external force. The proposed system will provide potential benefits in designing the microstructures for thin films. read less USED (high confidence) G. Ren, S.-wen Zhang, R. Hong, T. Tang, and Y. Chen, “Influence of shockwave profile on ejecta from shocked Pb surface: Atomistic calculations,” Chinese Physics B. 2016. link Times cited: 8 Abstract: We conduct molecular dynamics simulations of the ejection pr… read moreAbstract: We conduct molecular dynamics simulations of the ejection process from a grooved Pb surface subjected to supported and unsupported shock waves with various shock-breakout pressures (P SB) inducing a solid–liquid phase transition upon shock or release. It is found that the total ejecta mass changing with P SB under a supported shock reveals a similar trend with that under an unsupported shock and the former is always less than the latter at the same P SB. The origin of such a discrepancy could be unraveled that for an unsupported shock, a larger velocity difference between the jet tip and its bottom at an early stage of jet formation results in more serious damage, and therefore a greater amount of ejected particles are produced. The cumulative areal density distributions also display the discrepancy. In addition, we discuss the difference of these simulated results compared to the experimental findings. read less USED (high confidence) Z.-L. Liu, J.-S. Sun, R. Li, X.-L. Zhang, and L. Cai, “Comparative Study on Two Melting Simulation Methods: Melting Curve of Gold,” Communications in Theoretical Physics. 2016. link Times cited: 9 Abstract: Melting simulation methods are of crucial importance to dete… read moreAbstract: Melting simulation methods are of crucial importance to determining melting temperature of materials efficiently. A high-efficiency melting simulation method saves much simulation time and computational resources. To compare the efficiency of our newly developed shock melting (SM) method with that of the well-established two-phase (TP) method, we calculate the high-pressure melting curve of Au using the two methods based on the optimally selected interatomic potentials. Although we only use 640 atoms to determine the melting temperature of Au in the SM method, the resulting melting curve accords very well with the results from the TP method using much more atoms. Thus, this shows that a much smaller system size in SM method can still achieve a fully converged melting curve compared with the TP method, implying the robustness and efficiency of the SM method. read less USED (high confidence) J. Zhang et al., “Crystal structure and magnetic properties of new Fe3Co3X2 (X = Ti, Nb) intermetallic compounds,” Journal of Physics D: Applied Physics. 2016. link Times cited: 8 Abstract: The structure and magnetic properties of new magnetic Fe3Co3… read moreAbstract: The structure and magnetic properties of new magnetic Fe3Co3X2 (X = Ti, Nb) compounds are studied by genetic algorithm, first-principles density functional theory (DFT) calculations, and experiments. The atomic structure of a hexagonal structure with P-6m2 symmetry is determined. The simulated x-ray diffraction (XRD) spectra of the P-6m2 structures agree well with experimental XRD data for both Fe3Co3Ti2 and Fe3Co3Nb2. The magnetic properties of these structures as well as the effect of the disorder of Fe and Co on their magnetic properties are also investigated. The magnetocrystalline anisotropy energy is found to be very sensitive to the occupancy disorder between Fe and Co. read less USED (high confidence) S. Alkan, P. Chowdhury, H. Sehitoglu, R. Rateick, and H. Maier, “Role of nanotwins on fatigue crack growth resistance - Experiments and theory,” International Journal of Fatigue. 2016. link Times cited: 34 USED (high confidence) T. Fu et al., “MD simulation of effect of crystal orientations and substrate temperature on growth of Cu/Ni bilayer films,” Applied Physics A. 2016. link Times cited: 28 USED (high confidence) E. Bourasseau, P. Malfreyt, and A. Ghoufi, “Surface tension and long range corrections of cylindrical interfaces.,” The Journal of chemical physics. 2015. link Times cited: 5 Abstract: The calculation of the surface tension of curved interfaces … read moreAbstract: The calculation of the surface tension of curved interfaces has been deeply investigated from molecular simulation during this last past decade. Recently, the thermodynamic Test-Area (TA) approach has been extended to the calculation of surface tension of curved interfaces. In the case of the cylindrical vapour-liquid interfaces of water and Lennard-Jones fluids, it was shown that the surface tension was independent of the curvature of the interface. In addition, the surface tension of the cylindrical interface is higher than that of the planar interface. Molecular simulations of cylindrical interfaces have been so far performed (i) by using a shifted potential, (ii) by means of large cutoff without periodic boundary conditions, or (iii) by ignoring the long range corrections to the surface tension due to the difficulty to estimate them. Indeed, unlike the planar interfaces there are no available operational expressions to consider the tail corrections to the surface tension of cylindrical interfaces. We propose here to develop the long range corrections of the surface tension for cylindrical interfaces by using the non-exponential TA (TA2) method. We also extend the formulation of the Mecke-Winkelmann corrections initially developed for planar surfaces to cylindrical interfaces. We complete this study by the calculation of the surface tension of cylindrical surfaces of liquid tin and copper using the embedded atom model potentials. read less USED (high confidence) S. Hong and T. Rahman, “Geometric and electronic structure and magnetic properties of Fe-Au nanoalloys: insights from ab initio calculations.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 10 Abstract: We have performed density functional theory (DFT) based calc… read moreAbstract: We have performed density functional theory (DFT) based calculations of Fe-Au nanoalloys containing 113 atoms, Fe(x)Au(113-x) (x = 23, 56, 90), to determine their preferred geometric structure and the ensuing electronic structural and magnetic properties. We find that these nanoalloys prefer the formation of a core-shell structure and the Fe core maintains almost a constant magnetic moment of ∼2.8 μ(B) regardless of the Fe content, which is 27% enhancement from the bulk value and in qualitative agreement with some previous results. The local magnetic moment of Fe atoms is well correlated with the local coordination of the Fe atoms. Furthermore, the enhancement of the magnetic moment may be traced to charge depletion from the Fe atoms in the core to the Au atoms in the shell. The preference for the core-shell structure over one with segregated Fe and Au parts could be the low surface tension at the Fe-Au interface, which is larger for the core-shell structure, and can be attributed to strong Fe-Au interfacial interaction as a result of large charge transfer at the interface. read less USED (high confidence) K. Zhou, T. Zhang, and Z. Wang, “Positron lifetime calculation for possible defects in nanocrystalline copper,” Physica Scripta. 2015. link Times cited: 7 Abstract: Structural models for dislocation, vacancy clusters, twin bo… read moreAbstract: Structural models for dislocation, vacancy clusters, twin boundary, stacking fault and nanocrystalline sample are constructed using copper as a model material. Positron lifetimes and momentum distributions of annihilating electron–positron pairs are calculated for these structural models. The calculated results indicate that the dislocation, twin boundary and stacking fault are shallow traps to positrons. The dislocation associated with monovacancies gives rise to a positron lifetime similar to that of monovacancies. The calculated positron lifetimes of the nanocrystalline copper show no dependence on the mean grain size. The as-constructed nanocrystalline samples contain vacancy clusters in grain boundaries, and positrons are localized by the vacancy clusters. However after relaxation the samples show only other two kinds of free volumes: one is the interatomic space in grain boundaries which is a shallow trap to positrons; the other is similar to a monovacancy. The latter contributes a positron lifetime of about 163 ps. This kind of free volume is not only observed in grain boundaries but also in the regions near grain boundaries. Positron lifetime calculation combined with the momentum distribution calculation is useful to identify the defect in the nanocrystalline Cu. read less USED (high confidence) C. Chen, Q. Tang, and T.-C. Wang, “A new thermo-elasto-plasticity constitutive theory for polycrystalline metals,” Acta Mechanica Sinica. 2015. link Times cited: 4 USED (high confidence) X. Zhao, L. Ke, M. C. Nguyen, C. Wang, and K. Ho, “Structures and magnetic properties of Co-Zr-B magnets studied by first-principles calculations,” arXiv: Materials Science. 2015. link Times cited: 14 Abstract: The structures and magnetic properties of the Co-Zr-B alloys… read moreAbstract: The structures and magnetic properties of the Co-Zr-B alloys near the Co5Zr composition were studied using adaptive genetic algorithm and first-principles calculations to guide further experimental effort on optimizing their magnetic performances. Through extensive structural searches, we constructed the contour maps of the energetics and magnetic moments of the Co-Zr-B magnet alloys as a function of composition. We found that the Co-Zr-B system exhibits the same structural motif as the "Co11Zr2" polymorphs, which plays a key role in achieving high coercivity. Boron atoms can either substitute selective cobalt atoms or occupy the interstitial sites. First-principles calculation shows that the magnetocrystalline anisotropy energies can be significantly improved through proper boron doping. read less USED (high confidence) D. Xu, M. Hook, and M. Mayer, “Molecular dynamics study of nano-scale Ag surface electromigration and effect of Pd coating layer,” 14th IEEE International Conference on Nanotechnology. 2014. link Times cited: 0 Abstract: Ag is the most conductive metal but is vulnerable to electro… read moreAbstract: Ag is the most conductive metal but is vulnerable to electromigration (EM), which can limit its application in e.g. microelectronics. Molecular dynamics (MD) is used to simulate the migrating behavior of an Ag surface by adding an extra directional force on each atom. The migration of Ag atoms is found to be limited to the topmost 4 (002) lattice planes in the first 40 ns while atoms in the crystal bulk remain oscillating around their equilibrium positions. A Pd coating layer is shown to be a protection from EM for an Ag surface. After adding a layer of 9 Pd (002) lattice planes, the same procedure is repeated with different forces. No migration happens until the extra directional force become so large that all atoms of the model end up moving freely. The MD model presented in this paper can lead to an understanding of EM at the atomic scale and a guideline for potential reliability improvement of microelectronics by coating technology. read less USED (high confidence) B. Li, G. Xing, H. Wang, and R. Wang, “Tailoring characteristic thermal stability of Ni-Au binary nanocrystals via structure and composition engineering: theoretical insights into structural evolution and atomic inter-diffusion,” AIP Advances. 2014. link Times cited: 3 Abstract: We report on the structural evolution and atomic inter-diffu… read moreAbstract: We report on the structural evolution and atomic inter-diffusion characteristics of the bimetallic Ni-Au nanocrystals (NCs) by molecular dynamics simulations studies. Our results reveal that the thermal stability dynamics of Ni-Au NCs strongly depends on the atomic configurations. By engineering the structural construction with Ni:Au = 1:1 atomic composition, compared with core-shell Au@Ni and alloy NCs, the melting point of core-shell Ni@Au NCs is significantly enhanced up to 1215 K. Unexpectedly, with atomic ratio of Au:Ni= 1:9, the melting process initiates from the atoms in the shell of Ni@Au and alloy NCs, while starts from the core of Au@Ni NCs. The corresponding features and evolution process of structural motifs, mixing and segregation are illustrated via a series of dynamic simulations videos. Moreover, our results revealed that the face centered cubic phase Au0.75Ni0.25 favorably stabilizes in NCs form but does not exist in the bulk counterpart, which elucidates the anomalies of previously repor... read less USED (high confidence) G. Ren, Y. Chen, T. Tang, and Q. Li, “Ejecta production from shocked Pb surface via molecular dynamics,” Journal of Applied Physics. 2014. link Times cited: 28 Abstract: Molecular dynamics simulations are employed to examine the r… read moreAbstract: Molecular dynamics simulations are employed to examine the relation between ejecta production and shock-breakout pressure for single crystal Pb subjected to a decaying shockwave loading. To better understand the physical mechanism of ejecta formation, a surface with multiple triangular grooves representing the imperfections left from machining finish is taken into consideration. It is found that the ejecta volume density distribution displays a smooth nature and the amount of ejecta increases significantly after melting on release or shock. Additionally, the ejecta particle mass distribution is captured by a power law scaling, revealing the self-similarity. These results are in reasonable agreement with the characteristics of experimentally diagnosed findings. read less USED (high confidence) A. Dutta, S. Chatterjee, A. Raychaudhuri, A. Moitra, and T. Saha‐Dasgupta, “In-silico investigation of Rayleigh instability in ultra-thin copper nanowire in premelting regime,” Journal of Applied Physics. 2014. link Times cited: 9 Abstract: Motivated by the recent experimental reports, we explore the… read moreAbstract: Motivated by the recent experimental reports, we explore the formation of Rayleigh-like instability in metallic nanowires during the solid state annealing, a concept originally introduced for liquid columns. Our molecular dynamics study using realistic interatomic potential reveals instability induced pattern formation at temperatures even below the melting temperature of the wire, in accordance with the experimental observations. We find that this is driven by the surface diffusion, which causes plastic slips in the system initiating necking in the nanowire. We further find the surface dominated mass-transport is of subdiffusive nature with time exponent less than unity. Our study provides an atomistic perspective of the instability formation in nanostructured solid phase. read less USED (high confidence) S.-Y. Chen, Z. Wu, and K. Liu, “Atomic diffusion across Ni 50 Ti 50 —Cu explosive welding interface: Diffusion layer thickness and atomic concentration distribution,” Chinese Physics B. 2014. link Times cited: 6 Abstract: Molecular dynamics simulations are carried out to study atom… read moreAbstract: Molecular dynamics simulations are carried out to study atomic diffusion in the explosive welding process of Ni50Ti50—Cu (at.%). By using a hybrid method which combines molecular dynamics simulation and classical diffusion theory, the thickness of the diffusion layer and the atomic concentration distribution across the welding interface are obtained. The results indicate that the concentration distribution curves at different times have a geometric similarity. According to the geometric similarity, the atomic concentration distribution at any time in explosive welding can be calculated. Ni50Ti50—Cu explosive welding and scanning electron microscope experiments are done to verify the results. The simulation results and the experimental results are in good agreement. read less USED (high confidence) D. Oliver et al., “One-to-one spatially matched experiment and atomistic simulations of nanometre-scale indentation,” Nanotechnology. 2014. link Times cited: 21 Abstract: We have carried out nanoindentation studies of gold in which… read moreAbstract: We have carried out nanoindentation studies of gold in which the indenter is atomically characterized by field-ion microscopy and the scale of deformation is sufficiently small to be directly compared with atomistic simulations. We find that many features of the experiment are correctly reproduced by molecular dynamics simulations, in some cases only when an atomically rough indenter rather than a smooth repulsive-potential indenter is used. Heterogeneous nucleation of dislocations is found to take place at surface defect sites. Using input from atomistic simulations, a model of indentation based on stochastic transitions between continuum elastic–plastic states is developed, which accurately predicts the size distributions of plastic ‘pop-in’ events and their dependence on tip geometry. read less USED (high confidence) X. Zhang and G. Lu, “Computational Design of Core/Shell Nanoparticles for Oxygen Reduction Reactions.,” The journal of physical chemistry letters. 2014. link Times cited: 69 Abstract: A computational strategy to design core/shell nanoparticle c… read moreAbstract: A computational strategy to design core/shell nanoparticle catalysts for oxygen reduction reactions (ORRs) is proposed based on multiscale modeling. Using a quantum mechanics/molecular mechanics (QM/MM) coupling method, we have studied the ORR on Pt-Cu core/shell nanoparticles with the size ranging from 3 to 8 nm. We have calculated the oxygen adsorption energy on the nanoparticle surface (a descriptor for ORR activity) as a function of the nanoparticle size and thickness of the Pt shell. We find that the Pt-Cu core/shell nanoparticles exhibit higher ORR activities than flat Pt(111) surfaces, consistent with experimental observations. We predict that the diameter of the core/shell nanoparticles should be larger than 7 nm to reach the peak of ORR activities. By examining the effects of ligand, quantum confinement, and surface strain, we confirm that the strain plays the dominant role on ORR activities for the core/shell nanoparticles. A universal relation between the surface strain and the oxygen adsorption energy is established based on which one can computationally screen and design core/shell nanoparticle catalysts for superior ORR activities. read less USED (high confidence) N. Inogamov et al., “Electron‐Ion Relaxation, Phase Transitions, and Surface Nano‐Structuring Produced by Ultrashort Laser Pulses in Metals,” Contributions to Plasma Physics. 2013. link Times cited: 39 Abstract: Fundamental physical phenomena in metals irradiated by ultra… read moreAbstract: Fundamental physical phenomena in metals irradiated by ultrashort laser pulses with absorbed fluences higher than few tens of mJ/cm2 are investigated. For those fluences, laser‐produced electron distribution function relaxes to equilibrium Fermi distribution with electron temperature Te within a short time of 10‐100 fs. Because the electron subsystem has Te highly exceeding much the ion subsystem temperature Ti the well‐known twotemperature hydrodynamic model (2T‐HD) is used to evaluate heat propagation associated with hot conductive electron diffusion and electron‐ion energy exchange. The model coefficients of electron heat conductivity κ (ϱ, Te, Ti) and electron‐ion coupling parameter α (ϱ, Te) together with 2T equation of state E (ϱ, Te, Ti) and P (ϱ, Te, Ti) are calculated. read less USED (high confidence) H. Somekawa, T. Inoue, and K. Tsuzaki, “Effect of solute atoms on fracture toughness in dilute magnesium alloys,” Philosophical Magazine. 2013. link Times cited: 32 Abstract: The effect of alloying elements on the toughness and the fra… read moreAbstract: The effect of alloying elements on the toughness and the fracture behaviour was investigated on seven kinds of Mg-0.3 at.% X (X = Ag, Al, Ca, Pb, Sn, Y and Zn) alloys with a grain size of 3–5 μm. The fracture toughness and fracture behaviour in magnesium alloys were closely related to the segregation energy. The Mg–Al and –Zn alloys that had small segregation energy showed high toughness and ductile fracture in most regions, while the Mg–Ca alloy with large segregation energy exhibited low toughness and intergranular fracture. These different tendencies resulted from solute segregation at grain boundaries (GBs). The change in the lattice parameter ratio was the influential material parameter regardless of whether the GB embrittlement was for enhancement or suppression. read less USED (high confidence) A. Suiker and B. Thijsse, “Nucleation, kinetics and morphology of displacive phase transformations in iron,” Journal of The Mechanics and Physics of Solids. 2013. link Times cited: 13 USED (high confidence) D. Matsunaka, Y. Ohnishi, and Y. Shibutani, “Effects of Stacking Fault Energy on Fundamental Deformation Modes in Single Crystalline Magnesium by Molecular Dynamics Simulations,” Materials Transactions. 2013. link Times cited: 6 Abstract: In order to investigate effects of stacking fault energies (… read moreAbstract: In order to investigate effects of stacking fault energies (SFEs) on fundamental deformation modes of slips and deformation twinnings in magnesium, we carried out molecular dynamics simulations of shear deformations for the deformation modes with two kinds of many-body interatomic potentials. The SFEs of the basal and second-pyramidal planes are lower for a generalized embedded atom method (GEAM) potential than for an embedded atom method (EAM) potential. While the basal slip quite easily occurs and the prism dislocation is activated, the first-pyramidal slip and the second-pyramidal slip are hard to be operated. However, for the GEAM simulations, the second-pyramidal slip was activated due to reduction of the second-pyramidal SFE. Additionally, the reduction of the SFEs suppresses nucleation of the f10 11g twin in the bf10 11g 2 shearing direction. The relative order of the other fundamental deformation modes in the critical shear stress is qualitatively maintained despite the reduction of the SFEs. [doi:10.2320/matertrans.MAW201311] read less USED (high confidence) M. Xiang, H. Hu, J. Chen, and Y. Liao, “Molecular dynamics studies of thermal dissipation during shock induced spalling,” Journal of Applied Physics. 2013. link Times cited: 19 Abstract: Under shock loadings, the temperature of materials may vary … read moreAbstract: Under shock loadings, the temperature of materials may vary dramatically during deformation and fracture processes. Thus, thermal effect is important for constructing dynamical failure models. Existing works on thermal dissipation effects are mostly from meso- to macro-scale levels based on phenomenological assumptions. The main purpose of the present work is to provide several atomistic scale perspectives about thermal dissipation during spall fracture by nonequilibrium molecular dynamics simulations on single-crystalline and nanocrystalline Pb. The simulations show that temperature arising starts from the vicinity of voids during spalling. The thermal dissipation rate in void nucleation stage is much higher than that in the later growth and coalescence stages. Both classical spallation and micro-spallation are taken into account. Classical spallation is corresponding to spallation phenomenon where materials keep in solid state during shock compression and release stages, while micro-spallation is corres... read less USED (high confidence) M. Yuasa, M. Hakamada, H. Nakano, M. Mabuchi, and Y. Chino, “Softening due to disordered grain boundaries in nanocrystalline Co,” Journal of Physics: Condensed Matter. 2013. link Times cited: 2 Abstract: Nanocrystalline Co consisting of fcc and hcp phases was proc… read moreAbstract: Nanocrystalline Co consisting of fcc and hcp phases was processed by electrodeposition, and its mechanical properties were investigated by hardness tests. In addition, high-resolution transmission electron microscopy observations and molecular dynamics (MD) simulations were performed to investigate the grain boundary structure and dislocation nucleation from the grain boundaries. A large amount of disorders existed at the grain boundaries and stacking faults were formed from the grain boundaries in the as-deposited Co specimen. The as-deposited specimen showed a lower hardness than did the annealed specimen, although the grain size of the former was smaller than that of the latter. The activation volume of the as-deposited specimen (=1.5b3) was lower than that of the annealed specimen (=50b3), thus indicating that nucleation of dislocations from grain boundaries is more active in the as-deposited specimen than in the annealed specimens. The MD simulations showed that dislocation nucleation was closely related to a change in the defect structures at the boundary. Therefore, it is suggested that a significant amount of defects enhance changes in the defect structures at the boundary, resulting in softening of the as-deposited specimen. read less USED (high confidence) D. Matsunaka, A. Kanoh, and Y. Shibutani, “Energetic Analysis of Deformation Twins and Twinning Dislocations in Magnesium,” Materials Transactions. 2013. link Times cited: 7 Abstract: 平成25年4月1日より,従来の大阪大学大学院工学研究科附属原 子分子イオン制御理工学センターが改組となり,新たなアトミッ… read moreAbstract: 平成25年4月1日より,従来の大阪大学大学院工学研究科附属原 子分子イオン制御理工学センターが改組となり,新たなアトミッ クデザイン研究センターとなりました. 当センターでは, i) 原子・分子構造からの材料・構造・機能設計を意図した 研究に重点 ii) シミュレーションベースト・エンジニアリングの積極的な 推進 iii) 産業応用に直結させたプロトタイプリサーチに重点 をセンター趣旨の3本柱と考えております. 従来のセンターで構築してきましたアトミックテクノロジーを さらに発展させ,「原子・分子からのものづくり」をモットーと して,デザインの観点を加味した先端ものづくりのセンターとし て邁進していきたいと思っております. 当センターでの成果や関連事項を迅速に皆様にお知らせするた めに,このニュースレター(CAMT Newsletter)を配信させていた だくことにいたしました. センターのホームページも立ち上げましたので,お時間のある 時にご覧いただければ幸いです. 今後とも,関係各位の皆様方からのご指導,ご鞭撻のほど,よ ろしくお願い申し上げます. 目次 read less USED (high confidence) M. Xiang, H. Hu, J. Chen, and Y. Long, “Molecular dynamics simulations of micro-spallation of single crystal lead,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 54 Abstract: We present a molecular dynamics (MD) study of the micro-spal… read moreAbstract: We present a molecular dynamics (MD) study of the micro-spallation of lead (Pb), which corresponds to damage and liquid fragment ejection following the reflection of a strong shock wave on the free surface of the target. First, the Hugoniot and melting curves of Pb are derived by equilibrium MD simulations, and the potential function is validated by comparing these curves with experimental results. Then nonequilibrium MD simulations are conducted to study the dynamical processes of micro-spallation. Damage and ejection processes are analyzed by a binning analysis and direct observations of atom configurations. Comparisons with classical spallation simulations or experiments are made where necessary. It is found that damages in classical spallation and micro-spallation are both dominated by cavitation, i.e. nucleation and the growth and coalescence of voids. The main difference in the cavitation process of classical and micro-spallation lies in the amount and spatial distribution of void nucleation sites. Different properties in dynamical stress evolutions between micro-spallation and classical spallation are also discussed. In addition, the properties of the surface micro-spall are found to be different from those of interior micro-spall particles in some shock intensity regimes. Factors that cause such differences are studied by analyzing in detail the thermodynamics paths of different parts of the shocked target. read less USED (high confidence) M. Xiang, H. Hu, and J. Chen, “Spalling and melting in nanocrystalline Pb under shock loading: Molecular dynamics studies,” Journal of Applied Physics. 2013. link Times cited: 49 Abstract: The mechanisms of spalling and melting in nanocrystalline Pb… read moreAbstract: The mechanisms of spalling and melting in nanocrystalline Pb under shock loading are studied by molecular dynamics simulations. A wide range of shock intensity is conducted with the lowest one just above the threshold of solid spallation, and the highest one higher than the threshold of compression melting. The spallation mechanism is dominated by cavitation, i.e., nucleation, growth, and coalescence of voids. Our results show that grain boundaries have significant influences on spalling behaviors in cases of classical spallation and releasing melting. In these cases, cavitation and melting both start on grain boundaries, and they display mutual promotion: melting makes the voids nucleate at smaller tensile stress, and void growth speeds melting. Influences of microstructure, strain rate, and temperature on spall strength are qualitatively discussed. Due to grain boundary effects, the spall strength of nanocrystalline Pb varies slowly with the shock intensity in cases of classical spallation. In cases of ... read less USED (high confidence) C. Weinberger, G. Tucker, and S. Foiles, “Peierls potential of screw dislocations in bcc transition metals: Predictions from density functional theory,” Physical Review B. 2013. link Times cited: 81 Abstract: It is well known that screw dislocation motion dominates the… read moreAbstract: It is well known that screw dislocation motion dominates the plastic deformation in body-centered-cubic metals at low temperatures. The nature of the nonplanar structure of screw dislocations gives rise to high lattice friction, which results in strong temperature and strain rate dependence of plastic flow. Thus the nature of the Peierls potential, which is responsible for the high lattice resistance, is an important physical property of the material. However, current empirical potentials give a complicated picture of the Peierls potential. Here, we investigate the nature of the Peierls potential using density functional theory in the bcc transition metals. The results show that the shape of the Peierls potential is sinusoidal for every material investigated. Furthermore, we show that the magnitude of the potential scales strongly with the energy per unit length of the screw dislocation in the material. read less USED (high confidence) T. Ogura, M. Nishimura, H. Tatsumi, W. Takahara, and A. Hirose, “Interfacial Bonding Behavior between Silver Nanoparticles and Gold Substrate Using Molecular Dynamics Simulation,” Materials Transactions. 2012. link Times cited: 14 Abstract: Molecular dynamics (MD) simulation was applied to the sinter… read moreAbstract: Molecular dynamics (MD) simulation was applied to the sintering behavior of silver nanoparticles on a gold substrate in order to elucidate the sintering mechanism of the nanoparticles on the substrate. The simulation revealed that silver atoms from 1 and 2nm nanoparticles migrated freely because of their larger surface energy and then epitaxially reoriented to the gold substrate so as to reduce grain boundary energy. The silver nanoparticles were more spread out on the (011) gold substrate than on the (001) substrate, indicating that substrates with larger surface energy induce greater spreading rates. Consideration of the competition of neck growth and epitaxial growth in sintering of nanoparticles revealed that reduction of surface energy is the predominant driving force in the initiation of sintering of silver nanoparticles, and that the reduction of grain boundary energy is subsequently consequential. [doi:10.2320/matertrans.MB201201] read less USED (high confidence) D. Oliver et al., “Conductivity of an atomically defined metallic interface,” Proceedings of the National Academy of Sciences. 2012. link Times cited: 25 Abstract: A mechanically formed electrical nanocontact between gold an… read moreAbstract: A mechanically formed electrical nanocontact between gold and tungsten is a prototypical junction between metals with dissimilar electronic structure. Through atomically characterized nanoindentation experiments and first-principles quantum transport calculations, we find that the ballistic conduction across this intermetallic interface is drastically reduced because of the fundamental mismatch between s wave-like modes of electron conduction in the gold and d wave-like modes in the tungsten. The mechanical formation of the junction introduces defects and disorder, which act as an additional source of conduction losses and increase junction resistance by up to an order of magnitude. These findings apply to nanoelectronics and semiconductor device design. The technique that we use is very broadly applicable to molecular electronics, nanoscale contact mechanics, and scanning tunneling microscopy. read less USED (high confidence) M. Barisik and A. Beskok, “Boundary treatment effects on molecular dynamics simulations of interface thermal resistance,” J. Comput. Phys. 2012. link Times cited: 51 USED (high confidence) E. Huang et al., “Plastic Deformation of a Nano‐Precipitate Strengthened Ni‐Base Alloy Investigated by Complementary In Situ Neutron Diffraction Measurements and Molecular‐Dynamics Simulations,” Advanced Engineering Materials. 2012. link Times cited: 13 Abstract: In situ neutron‐diffraction experiments at the spallation ne… read moreAbstract: In situ neutron‐diffraction experiments at the spallation neutron source, simultaneously illuminating the diffraction of the matrix and the strengthening nano precipitates, allow the determination of their plastic deformation. An irreversible neutron‐diffraction‐profile evolution of the nano precipitates is observed. However, there is no conclusive trend of the nano‐precipitate peak‐width evolution subjected to the greater stress levels. Hence, in the present work, molecular‐dynamics simulations are applied to reveal the deformation mechanisms of the nano precipitate and its interaction with the surrounding matrix. The microstructure size, dislocation content, and structural parameters of the nano precipitates, quantified by X‐ray, transmission electron microscopy, and small‐angle neutron scattering, are used as the simulation input and reference. The simulation results show that there are two competing deformation mechanisms, which lead to the fluctuation of the nano‐precipitate‐diffraction widths, occurring during the higher plastic deformation stages. read less USED (high confidence) C. Liu, X.-R. Chen, C. Xu, L. Cai, and F. Jing, “Melting curves and entropy of fusion of body-centered cubic tungsten under pressure,” Journal of Applied Physics. 2012. link Times cited: 27 Abstract: The melting curves and entropy of fusion of body-centered cu… read moreAbstract: The melting curves and entropy of fusion of body-centered cubic (bcc) tungsten (W) under pressure are investigated via molecular dynamics (MD) simulations with extended Finnis-Sinclair (EFS) potential. The zero pressure melting point obtained is better than other theoretical results by MD simulations with the embedded-atom-method (EAM), Finnis-Sinclair (FS) and modified EAM potentials, and by ab initio MD simulations. Our radial distribution function and running coordination number analyses indicate that apart from the expected increase in disorder, the main change on going from solid to liquid is thus a slight decrease in coordination number. Our entropy of fusion of W during melting, Delta S, at zero pressure, 7.619 J/mol.K, is in good agreement with the experimental and other theoretical data. We found that, with the increasing pressure, the entropy of fusion Delta S decreases fast first and then oscillates with pressure; when the pressure is higher than 100 GPa, the entropy of fusion Delta S is about 6.575 +/- 0.086 J/mol.K, which shows less pressure effect. (C) 2012 American Institute of Physics. [http://dx.doi.org/10.1063/1.4733947] read less USED (high confidence) A. Singh, N. Iyer, and S. Harimkar, “Prediction of glass forming ability and glass forming range for electrodeposited binary Co-W alloys,” Journal of Applied Physics. 2012. link Times cited: 7 Abstract: This paper reports on the transition of crystalline to amorp… read moreAbstract: This paper reports on the transition of crystalline to amorphous structure with increasing W content in electrodeposited binary Co-W alloys. The glass forming ability of the electrodeposited Co-based alloys, including Co-W alloy, is analyzed using deep eutectic criteria based on reduced liquidus temperature and relative composition ratio. MD simulations based on solid solution models indicated glass forming ability above 20 at. % W for Co-rich Co-W alloys, which is in good agreement with the experimental observations. read less USED (high confidence) Z. Pereira and E. Silva, “Cold Welding of Gold and Silver Nanowires: A Molecular Dynamics Study,” Journal of Physical Chemistry C. 2011. link Times cited: 92 Abstract: Recently a new possibility of welding was experimentally sho… read moreAbstract: Recently a new possibility of welding was experimentally shown in the case of gold nanowires (NWs) at ambient temperatures, without need of additional heat and with low pressures, called cold weldi... read less USED (high confidence) Y. Kobayashi, Y. Doi, and A. Nakatani, “Strain Dependence of Formation Mechanism of Growth Layer in Molecular Beam Epitaxy of Gallium Nitride,” Japanese Journal of Applied Physics. 2010. link Times cited: 2 Abstract: In this study, the epitaxial growth of gallium nitride (GaN)… read moreAbstract: In this study, the epitaxial growth of gallium nitride (GaN) on a GaN substrate is investigated by a molecular dynamics (MD) method. Furthermore, the difference between the surface diffusion of atoms of a strained substrate and an unstrained substrate is examined. From the results of this examination, it is found that the diffusion characteristic in the unstrained case is higher than that in the strained case. Therefore, in the unstrained case, GaN grows layer-by-layer. On the other hand, in the strained case, multiple layers of GaN grow simultaneously. Furthermore, it is also found that the wurtzite structure of GaN differs between the strained case and the unstrained case. read less USED (high confidence) Y. Umeno, T. Shimada, and T. Kitamura, “Dislocation nucleation in a thin Cu film from molecular dynamics simulations: Instability activation by thermal fluctuations,” Physical Review B. 2010. link Times cited: 14 Abstract: To elucidate the mechanism responsible for structural instab… read moreAbstract: To elucidate the mechanism responsible for structural instability at the atomic level, atomistic modeling simulation of tension in a Cu thin film containing a notch was performed using an embedded-atom method potential and dislocation nucleation was observed. Mechanical stability during tension was analyzed by solving the eigenvalue problem of the Hessian matrix taking into account all the degrees of freedom of the atoms in the system. Since an eigenvalue designates the curvature of the potential energy landscape in the direction of the corresponding eigenvector, which indicates a deformation mode, the system is unstable under vanishing temperature at the critical strain (cid:1) (cid:1) c (cid:2) when any eigenvalue is zero or negative. At a strain smaller than (cid:1) c where all the eigenvalues are positive, atomic fluctuations due to finite temperature may cause structural instability. We found that the path of activated instability (cid:1) dislocation emission from the notch (cid:2) could be written with a linear combination of the eigenvectors having small eigenvalues obtained under a corresponding external strain at zero temperature. The energy landscape has a much lower hill along the mixed-mode path than along any single-mode paths. In a molecular dynamics simulation under finite temperature, components of deformation modes having small eigenvalues fluctuate at low frequency, which dominate the activation of instability. DOI: 10. read less USED (high confidence) E.-K. Lee, H. Choi, S.-G. Lee, and Y.-C. Chung, “Energetics of Pb heterostructures formation on the Cu (111) in the early stage of the deposition process,” Journal of Applied Physics. 2010. link Times cited: 3 Abstract: The structural and self-assembling characteristic of Pb hete… read moreAbstract: The structural and self-assembling characteristic of Pb heterostructures on the Cu (111) substrate in the early stage of the deposition process were investigated using a molecular dynamics simulation and density functional theory. The Pb islands formed on the Cu (111) surface were observed to diffuse actively in lateral directions following the layer-by-layer growth mode. A heptameric hexagonal island was found to be most stable under highly nonequilibrium conditions. This result can be explained by the tendency of Pb heterostructures, which have minimum surface energy, to have the maximum number of Pb–Pb bondings. In addition, the atomic binding energy, the surface diffusion coefficient prefactor, and the surface diffusion energy barrier for Pb adatoms were quantitatively calculated according to various shapes of Pb islands to determine the stability of the corresponding island. read less USED (high confidence) B.-H. Kim and Y.-C. Chung, “Molecular dynamics simulation of the thin film deposition of Co/Cu(111) with Pb surfactant,” Journal of Applied Physics. 2009. link Times cited: 9 Abstract: Using molecular dynamics simulation, the effect of Pb surfac… read moreAbstract: Using molecular dynamics simulation, the effect of Pb surfactant for the thin film growth of Co atoms on Cu(111) substrate was investigated. Specifically, the behavior of Co atoms being deposited on Cu(111) substrate with predeposited Pb layer was extensively investigated and compared with the case of without Pb layer to explain the effect of Pb surfactant. It was observed that Pb layer was floating during the Co deposition. It was, quantitatively, found that Pb surfactant played an important role in suppression of active diffusion of Co atoms, which was accomplished by the increase in the surface diffusion barrier energy. The energy change in the deposited Co adatom on the Cu(111) substrate with predeposited Pb layer showed that the approaching Co adatom penetrated into the Pb layer; then, the Co adatom settled down on the Cu(111) substrate. Consequently, Pb atoms around Co adatom suppressed the further diffusion of Co adatom. read less USED (high confidence) S.-I. I. Kim, K.-R. Lee, Y. Chung, M. Sahashi, and Y.-geun Kim, “Molecular dynamics simulation study of deposition and annealing behaviors of Al atoms on Cu surface,” Journal of Applied Physics. 2009. link Times cited: 5 Abstract: Deposition and annealing behaviors of Al atoms on rough Cu (… read moreAbstract: Deposition and annealing behaviors of Al atoms on rough Cu (111) surface were investigated on the atomic scale by three-dimensional classical molecular dynamics simulation. The rough Cu surface was modeled by depositing 5 ML of Cu on Ta (011) substrate. Most Al atoms deposited on the rough Cu surface placed on the atomic steps, preserving the major features of the surface during Al deposition. This behavior was discussed in terms of the smaller barrier of the surface diffusion than Ehrlich–Schwoebel barrier of Al on Cu (111) surface. By annealing at 700 K, significant intermixing between Al and Cu rapidly occurs with decrease in the surface roughness. This behavior reveals that the exchange process of Al with substrate Cu dominates during the initial stage of high temperature annealing. read less USED (high confidence) B.-H. Kim and Y.-C. Chung, “Atomic-Level Investigation for Surface Characteristics in a Co-Cu Multilayer System: Molecular Dynamics Simulation,” IEEE Transactions on Magnetics. 2008. link Times cited: 4 Abstract: Using molecular dynamics simulation, the growth behavior of … read moreAbstract: Using molecular dynamics simulation, the growth behavior of Co atoms on Cu substrates for two different crystallographic orientations, (001) and (111), was extensively investigated. The surface roughness became noticeably higher during 20-monolayer (ML) deposition of Co thin-film on the Cu(111) substrate compared to the case of the Cu(001) substrate. It was found that the high diffusivity of Co adatoms on Cu(111) enhanced the atomic lateral movement, and the deposited Co adatoms could be agglomerated easily. The different growth behavior could be successfully explained in terms of the lateral atomic displacement and local acceleration energy. read less USED (high confidence) L. Wu, Y. Zhu, H. Wang, and M. Li, “Crystal–melt coexistence in fcc and bcc metals: a molecular-dynamics study of kinetic coefficients,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 5 Abstract: As a sequel to the previous paper on the calculation of the … read moreAbstract: As a sequel to the previous paper on the calculation of the crystal–melt interface free energy (2021 Materialia 15 100962), here we report the results on the kinetic coefficients using molecular dynamics simulations performed on six fcc metals and four bcc metals with the intention to compare the crystal structural influence. We found that the calculated kinetic coefficients are well described by the model by Broughton, Gilmer and Jackson (1982 Phys. Rev. Lett. 49 1496), and in particular, they exhibit varying degrees of anisotropy. We reveal that the anisotropies are related to the fluctuation of the crystal–melt interfaces, which causes the increase of the actual interface area in melting or solidification. The kinetic coefficients always display asymmetry between the solidification and melting process, and the difference is much more pronounced for the (111) interfaces in fcc metals which have the highest anisotropy. We found that the atomic mechanisms of the kinetic behaviors of these interfaces are closely related to the formation of twin-crystal domains during solidification, which delays the solidification process and consequently causes a decrease in the calculated kinetic coefficients. read less USED (high confidence) S. Oyinbo, T. Jen, S. A. Aasa, O. Abegunde, and Y. Zhu, “Development of palladium nanoparticles deposition on a copper substrate using a molecular dynamic (MD) simulation: a cold gas dynamic spray process,” Manufacturing Review. 2020. link Times cited: 6 Abstract: The objective of this study is to create an ultra-thin palla… read moreAbstract: The objective of this study is to create an ultra-thin palladium foil with a molecular dynamic (MD) simulation technique on a copper substrate surface. The layer formed onto the surface consists of a singular 3D palladium (Pd) nanoparticle structure which, by the cold gas dynamic spray (CGDS) technique, is especially incorporated into the low-cost copper substrate. Pd and Cu have been chosen for their possible hydrogen separation technology applications. The nanoparticles were deposited to the substrate surface with an initial velocity ranging from 500 to 1500 m/s. The particle radius was 1 to 4 nm and an angle of impact of 90° at room temperature of 300 K, in order to evaluate changes in the conduct of deformation caused by effects of size. The deformation mechanisms study revealed that the particle and substrate interface is subject to the interfacial jet formation and adiabatic softening resulting in a uniform layering. However, shear instabilities at high impact speeds were confirmed by the evolution of von Mises shear strain, temperature evolution and plastic strain. The results of this study can be used to further our existing knowledge in the complex spraying processes of cold gas dynamic spray technology. read less USED (high confidence) D. Ishikawa et al., “Bonding Strength of Cu Sinter Die-Bonding Paste on Ni, Cu, Ag, and Au Surfaces under Pressureless Bonding Process,” Transactions of The Japan Institute of Electronics Packaging. 2020. link Times cited: 6 Abstract: Herein we report on a copper (Cu) sinter die-bonding paste t… read moreAbstract: Herein we report on a copper (Cu) sinter die-bonding paste that sinters under pressureless bonding conditions for power devices operating at high temperatures. The shear strengths of pressureless-sintered Cu on four different metallization layers (Ni, Cu, Ag, and Au) were studied by experiment. The sintering behavior of Cu nanoparticles and diffusion coefficients at interfaces between a bulk Cu layer and the metallization layers was also evaluated by molecular dynamics (MD) simulation. After aging (573 K, 8 h), the shear strength of the Cu sintered to the Ni and Cu layers increased, whereas that of Cu sintered to the Ag and Au layers decreased. The interdiffusion at the interfaces between the sintered Cu layer and the Ag or Au layer increased the interfacial porosity of sintered Cu, which decreased the shear strengths in the sintered Cu/Ag and Cu/Au systems. In contrast, the interfacial porosity between the sintered Cu and the Ni or Cu layer hardly changed after aging. MD simulations revealed that Kirkendall voids were promoted by higher interdiffusion coefficients and a higher ratio of intrinsic diffusion coefficients between a bulk Cu layer and an Ag or Au layer. This consequently increased the porosity of Cu sintered near the interfaces. read less USED (high confidence) S. Xu, J. Rigelesaiyin, L. Xiong, Y. Chen, and D. McDowell, “Generalized Continua Concepts in Coarse-Graining Atomistic Simulations.” 2018. link Times cited: 12 USED (high confidence) L. Xie, P. Brault, A. Thomann, X. Yang, Y. Zhang, and G. Shang, “Molecular dynamics simulation of Al–Co–Cr–Cu–Fe–Ni high entropy alloy thin film growth,” Intermetallics. 2016. link Times cited: 64 USED (high confidence) X. Zhao, “Accelerating materials discovery and design: computational study of the structure and properties of materials.” 2015. link Times cited: 0 Abstract: This thesis summarizes our efforts to study the structure an… read moreAbstract: This thesis summarizes our efforts to study the structure and properties of materials computationally. The adaptive genetic algorithm (AGA) developed by us to predict crystal/surface/interface structures is presented. Applications of AGA to a variety of systems, such as non-rare earth magnetic materials, ultra-hard transition metal borides and SrTiO3 grain boundaries, are discussed. We demonstrated by AGA the capability of solving crystal structures with more than 100 atoms per unit cell and rapidly accessing the structures and phase stabilities of different compositions in multicomponent systems. We also introduced a motif-network scheme to study the complex crystal structures in silicate cathodes. In addition, we explored different computational methods for atomistic simulations of materials behavior, such as Monte Carlo modeling of the alnico magnets. read less USED (high confidence) T. Ogura, M. Nishimura, H. Tatsumi, N. Takeda, W. Takahara, and A. Hirose, “Evaluation of Interfacial Bonding Utilizing Ag2O-Derived Silver Nanoparticles Using TEM Observation and Molecular Dynamics Simulation,” The Open Surface Science Journal. 2010. link Times cited: 10 Abstract: The interfacial bonding utilizing Ag2O-derived silver nanopa… read moreAbstract: The interfacial bonding utilizing Ag2O-derived silver nanoparticles was evaluated using TEM observation and molecular dynamics simulation. The TEM observation reveals that the crystal orientation of the sintered silver corresponded to that of the gold substrate. This is considered that the epitaxial layer of silver was formed through in-situ formation of silver nanoparticles from Ag2O paste, and oriented in the direction of the gold crystal. MD simulation successfully recreated the sintering behavior of silver nanoparticles and the gold substrate. The simulation results clearly showed the epitaxial layers of silver atoms were formed on the substrate. The existence of the closed pore indicates the acceleration of the sintering between nanoparticles and the gold substrate to minimize the total sum of surface energy and grain boundary energy. read less USED (high confidence) D. Schebarchov, “Mechanisms in Carbon Nanotube Growth: Modelling and Molecular Dynamics Simulations.” 2010. link Times cited: 0 Abstract: A selection of nanoscale processes is studied theoretically,… read moreAbstract: A selection of nanoscale processes is studied theoretically, with the aim of identifying themechanisms that could lead to selective carbon nanotube (CNT) growth. Only mechanisms relevant to catalytic chemical vapour deposition (CVD) are considered. The selected processes are analysed with classical molecular dynamics (MD) simulations and continuum modelling. The melting and pre-melting behaviour of supported nickel catalyst particles is investigated. Favourable epitaxy between a nanoparticle and the substrate is shown to significantly raise themelting point of the particle. It is also demonstrated that substrate binding can induce solid-solid transformations, whilst the epitaxy may even determine the orientation of individual crystal planes in supported catalysts. These findings suggest that the substrate crystal structure alone can potentially be used to manipulate the properties of catalyst particles and, hence, influence the structure of CNTs. The first attempt at modelling catalyst dewetting, a process where the catalyst unbinds from the inner walls of a nucleating nanotube, is presented. It is argued that understanding this process and gaining control over itmay lead to better selectivity in CNT growth. Two mutually exclusive dewetting mechanisms, namely cap lift-off and capillary withdrawal, are identified and then modelled as elastocapillary phenomena. The modelling yields an upper bound on the diameter of CNTs that can stem from a catalyst particle of a given size. It is also demonstrated that cap lift-off is sensitive to cap topology, suggesting that it may be possible to link catalyst characteristics to the structural properties of nucleating CNTs. However, a clear link to the chiral vector remains elusive. It is shown that particle size, as well as binding affinity, plays a critical role in capillary absorption and withdrawal of catalyst nanoparticles. This size dependence is explored in detail, revealing interesting ramifications to the statics and dynamics of capillary-driven flows at the nanoscale. The findings bear significant implications for our understanding of CNT growth from catalyst particles, whilst also suggesting new nanofluidic applications and methods for fabricating composite metal-CNT materials. read less USED (high confidence) N. Matsumoto, R. Matsumoto, and N. Miyazaki, “Estimation of shear-banding resistance in metallic glass containing nano-crystalline particles,” Journal of Non-crystalline Solids. 2009. link Times cited: 8 USED (low confidence) Y. Gao et al., “Investigation of interfacial matching between 3C-SiC substrate crystals and its surface layer deposited Cu elements using molecular dynamics simulations,” Surfaces and Interfaces. 2023. link Times cited: 0 USED (low confidence) Y. Gao et al., “Investigation of deformation mechanism of SiC–CuNi composite thin film material nanochannels by molecular dynamics simulation,” Results in Physics. 2023. link Times cited: 0 USED (low confidence) A. M. Kazakov, G. Korznikova, I. I. Tuvalev, A. A. Izosimov, and E. A. Korznikova, “The Effect of Copper–Graphene Composite Architecture on Thermal Transport Efficiency,” Materials. 2023. link Times cited: 0 Abstract: This paper presents the results of molecular dynamic modelin… read moreAbstract: This paper presents the results of molecular dynamic modeling, revealing that inserting confined graphene layers into copper crystal reduces the thermal conductivity of the whole composite, and the coefficient of thermal conductivity κ decreases upon an increase in the number of graphene layers. The injection of one, two, and three layers of 15 nm graphene leads to a change in the coefficient of thermal conductivity from 380 W/(m·K) down to 205.9, 179.1, and 163.6 W/(m·K), respectively. Decreasing the length of graphene layers leads to a decrease in the density of defects on which heat is dissipated. With one, two, and three layers of 8 nm graphene, the coefficient of thermal conductivity of the composite is equal to 272.6, 246.8, and 240.8 W/(m·K), appropriately. Meanwhile the introduction of an infinite graphene layer results in the growth of κ to 414.2–803.3 W/(m·K). read less USED (low confidence) J. Wang, N. Lu, and C.-H. Zhang, “Study of high temperature mechanical properties and irradiation behavior of Zr3Al alloy by Chen’s Lattice Inversion EAM,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) G. Balbus, S. I. Rao, O. Senkov, and E. Payton, “Orientation dependent plasticity of the refractory multi-principal element alloy MoNbTi investigated via micropillar compression,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) Z. Yan, J. Zhao, R. Liu, B. Liu, Y.-lin Shao, and M. Liu, “An insight into sintering mechanisms of silicon carbide nanoparticles with additives using MD simulation,” Powder Technology. 2023. link Times cited: 0 USED (low confidence) J. Li, S. Ren, B. Liu, P. K. Liaw, and Q. Fang, “Influence of chemistry and temperature on mechanical behavior and deformation mechanisms of refractory high-entropy alloys: an integrated simulation-modeling analysis,” Acta Mechanica Sinica. 2023. link Times cited: 0 USED (low confidence) S. Liu and S. Nambu, “A multi-scale study on the effect of interfacial microstructure on interfacial strength and fracture behaviour in Fe/Ni bonded interfaces,” Materials Science and Engineering: A. 2023. link Times cited: 1 USED (low confidence) Y. Wu and J. Shao, “Unraveling Anisotropy in Crystalline Orientation under Shock-Induced Dynamic Responses in High-Entropy Alloy Co25Ni25Fe25Al7.5Cu17.5,” Nanomaterials. 2023. link Times cited: 0 Abstract: Shock-induced plastic deformation and spall damage in the si… read moreAbstract: Shock-induced plastic deformation and spall damage in the single-crystalline FCC Co25Ni25Fe25Al7.5Cu17.5 high-entropy alloy (HEA) under varying shock intensities were systematically investigated using large-scale molecular dynamics simulations. The study reveals the significant influence of crystalline orientation on the deformation mechanism and spall damage. Specifically, the shock wave velocities in the [110] and [111] directions are significantly higher than that in the [001] direction, resulting in a two-zone elastic-plastic shock wave structure observed in the [110] and [111] samples, while only a single-wave structure is found in the [001] sample. The plastic deformation is dominated by the FCC to BCC transformation following the Bain path and a small amount of stacking faults during the compression stage in the [001] sample, whereas it depends on the stacking faults induced by Shockley dislocation motion in the [110] and [111] samples. The stacking faults and phase transformation in the [001] sample exhibit high reversibility under release effects, while extensive dislocations are present in the [110] and [111] samples after release. Interestingly, tension-strain-induced FCC to BCC phase transformation is observed in the [001] sample during the release stage, resulting in increased spall strength compared to the [110] and [111] samples. The spall strength estimated from both bulk and free surface velocity history shows reasonable consistency. Additionally, the spall strength remains stable with increasing shock intensities. The study discusses in detail the shock wave propagation, microstructure change, and spall damage evolution. Overall, our comprehensive studies provide deep insights into the deformation and fracture mechanisms of Co25Ni25Fe25Al7.5Cu17.5 HEA under shock loading, contributing to a better understanding of dynamic deformation under extreme environments. read less USED (low confidence) B. Aroboto, S. Chen, T. Hsu, B. C. Wood, Y. Jiao, and J. Chapman, “Universal and interpretable classification of atomistic structural transitions via unsupervised graph learning,” Applied Physics Letters. 2023. link Times cited: 0 Abstract: Materials processing often occurs under extreme dynamic cond… read moreAbstract: Materials processing often occurs under extreme dynamic conditions leading to a multitude of unique structural environments. These structural environments generally occur at high temperatures and/or high pressures, often under non-equilibrium conditions, which results in drastic changes in the material's structure over time. Computational techniques, such as molecular dynamics simulations, can probe the atomic regime under these extreme conditions. However, characterizing the resulting diverse atomistic structures as a material undergoes extreme changes in its structure has proved challenging due to the inherently non-linear relationship between structures as large-scale changes occur. Here, we introduce SODAS++, a universal graph neural network framework, that can accurately and intuitively quantify the atomistic structural evolution corresponding to the transition between any two arbitrary phases. We showcase SODAS++ for both solid–solid and solid–liquid transitions for systems of increasing geometric and chemical complexity, such as colloidal systems, elemental Al, rutile and amorphous TiO2, and the non-stoichiometric ternary alloy Ag26Au5Cu19. We show that SODAS++ can accurately quantify all transitions in a physically interpretable manner, showcasing the power of unsupervised graph neural network encodings for capturing the complex and non-linear pathway, a material's structure takes as it evolves. read less USED (low confidence) S. Liu and S. Nambu, “Effects of interfacial energy on interfacial strength and work of adhesion in bcc-Fe tilt interfaces: A molecular dynamic study,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) T. Liu et al., “Orientation-dependent phase transition pathways of single-crystal nickel over large shock range,” International Journal of Mechanical Sciences. 2023. link Times cited: 0 USED (low confidence) A. Bayazitov, A. Semenov, and S. V. Dmitriev, “Simulation of the Dynamics of Supersonic N-Crowdions in fcc Lead and Nickel,” Micro. 2023. link Times cited: 0 Abstract: In the case where an interstitial atom is located in a close… read moreAbstract: In the case where an interstitial atom is located in a close-packed atomic row of the crystal lattice, it is called a crowdion. Crowdions play an important role in the processes of mass and energy transfer resulting from irradiation, severe plastic deformation, ion implantation, plasma and laser processing, etc. In this work, supersonic N-crowdions (N=1, 2) in fcc lattices of lead and nickel are studied by the method of molecular dynamics. Modeling shows that the propagation distance of a supersonic 2-crowdion in lead at a high initial velocity is less than that of a supersonic 1-crowdion. In other fcc metals studied, including nickel, supersonic 2-crowdions have a longer propagation distance than 1-crowdions. The relatively short propagation distance of supersonic 2-crowdions in lead is due to their instability and rapid transformation into supersonic 1-crowdions. This feature of the dynamics of supersonic N-crowdions in lead explains its high radiation-shielding properties. read less USED (low confidence) T. Li, H. Zong, F. Zhao, X. Ding, T. Lookman, and J. Sun, “Effects of atomic size mismatch on glass transition decoupling in high-entropy metallic glasses,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) P. Hopur, W. Chen, Y. Zhou, J. Zhou, and T. Wang, “The Correlation among the Atomic Structure, Electronic Valence Band and Properties of Zr-Cu-Al-Ag Bulk Metallic Glasses,” Metals. 2023. link Times cited: 0 Abstract: Investigating the relationship between the glass-forming abi… read moreAbstract: Investigating the relationship between the glass-forming ability (GFA), mechanical properties, and structure of metallic glasses is crucial to understanding the nature of the metallic glass state. In this study, the correlation among the atomic structure, electronic valence band, and properties have been studied using Zr50Cu44.5−xAl5.5Agx (x = 0, 1.5, 3 at.%) bulk metallic glasses (BMGs). The results reveal that through the micro-addition of Ag, the GFA of Zr50Cu44.5Al5.5 BMG can be enhanced; meanwhile, the critical diameter of Zr50Cu44.5Al5.5 glass rods increases from approximately 2.5 mm to 5.0 mm with the addition of 3% Ag. Through the addition of Ag, the thermal stability of Zr50Cu44.5Al5.5 BMG is improved, and the proportion of icosahedral-like clusters increases. The plasticity of the Zr50Cu44.5−xAl5.5Agx (x = 0, 1.5, 3 at.%) BMGs decreased from 4.6% to 0.8% with the addition of Ag. The valence band spectrum of the Zr50Cu44.5−xAl5.5Agx (x = 0, 1.5, 3 at.%) BMGs indicates that with the addition of Ag, the p-d hybridization near the Fermi level is enhanced, and the binding energy will move to a lower value. read less USED (low confidence) Y. Zhang et al., “Effect of radiation damage on liquid Pb corrosion at the Fe/Pb solid-liquid interface,” Journal of Nuclear Materials. 2023. link Times cited: 0 USED (low confidence) L. Liu, R. Wang, M. Su, M. An, and Z. Wu, “Size dependence for mechanical properties of Ti/Cu multilayered nanowire with a semi-coherent interface,” Journal of Physics: Conference Series. 2023. link Times cited: 0 Abstract: Metallic multilayered nanowires have a wide application pros… read moreAbstract: Metallic multilayered nanowires have a wide application prospect in micro-nano devices because of their superior physical and chemical properties and microstructure designability. Size effects on the tensile behaviors of Ti/Cu multilayered nanowires are investigated by molecular dynamic simulations. Aspect ratios of 1:4, 1:3, 1:2, 1:1, 1:0.75, and 1:0.67 and sectional dimensions of 3, 4, 5, 6, and 7 nm are adopted to construct nanowires with different sizes. Simulation results indicate that the strength of Ti/Cu nanowires decreases with the decrease of aspect ratio in the large aspect ratio range (>1:2) and all simulated sectional dimension ranges, showing a reverse Hall-Petch effect. The Hall-Petch law can only be satisfied in a small aspect ratio range (<1:2). Deformation mechanism transition is found in the critical aspect ratio of 1:2. When the aspect ratio is larger than 1:2, crystalline phases of Ti and Cu layers dominate the plastic deformation of Ti/Cu nanowires. Crystal phases and interface both bear plastic deformation when the aspect ratio is smaller than 1:2. Interface is an important factor in the strength and deformation of Ti/Cu nanowires. The variation of interface fraction and interaction between interface and dislocation motion determine the tendency of strength variation for Ti/Cu nanowires. read less USED (low confidence) V. Borysiuk, I. Lyashenko, and V. Popov, “Thermal Stability and Melting Dynamics of Bimetallic Au@Pt@Au Core-Shell Nanoparticles,” Sensors (Basel, Switzerland). 2023. link Times cited: 1 Abstract: Thermal stability is an important feature of the materials u… read moreAbstract: Thermal stability is an important feature of the materials used as components and parts of sensors and other devices of nanoelectronics. Here we report the results of the computational study of the thermal stability of the triple layered Au@Pt@Au core-shell nanoparticles, which are promising materials for H2O2 bi-directional sensing. A distinct feature of the considered sample is the raspberry-like shape, due to the presence of Au nanoprotuberances on its surface. The thermal stability and melting of the samples were studied within classical molecular dynamics simulations. Interatomic forces were computed within the embedded atom method. To investigate the thermal properties of Au@Pt@Au nanoparticles, structural parameters such as Lindemann indexes, radial distribution functions, linear distributions of concentration, and atomistic configurations were calculated. As the performed simulations showed, the raspberry-like structure of the nanoparticle was preserved up to approximately 600 K, while the general core-shell structure was maintained up to approximately 900 K. At higher temperatures, the destruction of the initial fcc crystal structure and core-shell composition was observed for both considered samples. As Au@Pt@Au nanoparticles demonstrated high sensing performance due to their unique structure, the obtained results may be useful for the further design and fabrication of the nanoelectronic devices that are required to work within a certain range of temperatures. read less USED (low confidence) T. Shu, N. Hu, F. Liu, and G. Cheng, “Nanoparticles induced intragranular and dislocation substructures in powder bed fusion for strengthening of high-entropy-alloy,” Materials Science and Engineering: A. 2023. link Times cited: 2 USED (low confidence) O. Senkov et al., “Mechanical Properties of an Al10Nb20Ta15Ti30V5Zr20 A2/B2 Refractory Superalloy and Its Constituent Phases,” Acta Materialia. 2023. link Times cited: 1 USED (low confidence) J. Li, Y.-F. Wu, Z. Bai, W. Yu, and S. Shen, “Nanostructure-property relation of Σ5 grain boundary in HfNbZrTi high-entropy alloy under shear,” Journal of Materials Science. 2023. link Times cited: 3 USED (low confidence) “The structure and energy of symmetric tilt grain boundaries in tungsten,” Journal of Nuclear Materials. 2023. link Times cited: 1 USED (low confidence) Z. Yu et al., “Cu/oriented-carbon nanotubes composite with ultralow thermal contact resistance,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) R. Kumar, M. K. Gupta, S. Rai, and V. Panwar, “Grain size responsive uniaxial tensile behavior of polycrystalline nanocopper under different temperatures and strain rates,” Multidiscipline Modeling in Materials and Structures. 2023. link Times cited: 0 Abstract: PurposeThe changes in tensile behavior of polycrystalline na… read moreAbstract: PurposeThe changes in tensile behavior of polycrystalline nanocopper lattice with changes in temperature, average grain size (AGS) and strain rate, have been explored. The existence of a critical AGS has also been observed which shows that the Hall–Petch relationship behaves inversely.Design/methodology/approachNanoscale deformation of polycrystalline nanocopper has been done in this study with the help of an embedded atom method (EAM) potential. Voronoi construction method has been employed for creating four polycrystals of nanocopper with different sizes. Statistical analysis has been used to examine the observations with emphasis on the polycrystal size effect on melting point temperature.FindingsThe study has found that the key stress values (i.e. elastic modulus, yield stress and ultimate tensile stress) are significantly influenced by the considered parameters. The increase in strain rate is observed to have an increasing impact on mechanical properties, whereas the increase in temperature degrades the mechanical properties. In-depth analysis of the deformation mechanism has been studied to deliver real-time visualization of grain boundary motion.Originality/valueThis study provides the relationship between required grain size variations for consecutive possible variations in mechanical properties and may help to reduce the trial processes in the synthesis of polycrystalline copper based on different temperatures and strain rates. read less USED (low confidence) O. Senkov, S. Rao, D. Miracle, A. E. Mann, and A. Yousefiani, “Effect of Re and Al additions on the microstructure and mechanical properties of Nb-18Ti-12W alloy,” Materials Science and Engineering: A. 2023. link Times cited: 1 USED (low confidence) F. Wang, L. Li, H. Tang, X. Wang, and Y. Hu, “Damping of aluminum-matrix composite reinforced by carbon nanotube: Multiscale modeling and characteristics,” Science China Technological Sciences. 2023. link Times cited: 4 USED (low confidence) B. Wu and Y. Sun, “Normal Contact Analysis Between Two Self-affine Fractal Surfaces at the Nanoscale by Molecular Dynamics Simulations,” Tribology Letters. 2023. link Times cited: 3 USED (low confidence) Y. Hu et al., “Nanoindentation characteristics of nanocrystalline tungsten via atomistic simulation,” Philosophical Magazine. 2023. link Times cited: 0 Abstract: ABSTRACT Molecular dynamics simulations are performed to exp… read moreAbstract: ABSTRACT Molecular dynamics simulations are performed to explore nanoindentation characteristics of tungsten, and the influences of grain size, indenter velocity, indenter size, and temperature are discussed. The results illustrate that the hardness reduces as the grain size (5.00 ∼ 24.62 nm) decreases. There is no phase change observed during the whole deformation process. For monocrystalline W, the dislocation nucleation and propagation dominate the deformation mechanisms. Differently, the primary deformation mode of nanocrystalline W is the grain split and motion of GBs. Dislocations primarily nucleate below the contact surface of the indenter and substrate and then glide in the grain core. The monocrystalline W has better pattern-forming ability than nanocrystalline. Besides, the pattern-forming ability of nanocrystalline W is negatively correlated with the average grain size (5.00 ∼ 24.62 nm). The von Mises stress is mainly concentrated in the interface between the indenter and substrate, the dislocation area for monocrystalline, and grain boundaries for nanocrystalline. The indentation force and hardness are positively correlated with indenter radius size (30 ∼ 80 Å), negatively correlated with temperature (10 ∼ 1500 K), and insensitive to the indenter velocity when velocity is lower than 3.0 Å /ps (300 m/s). read less USED (low confidence) W. Bao, Z.-liang Wang, B. Hu, and D. Tang, “Thermal transport across graphene/GaN and MoS2/GaN interfaces,” International Journal of Heat and Mass Transfer. 2023. link Times cited: 2 USED (low confidence) I. Lobzenko, Y. Shiihara, H. Mori, and T. Tsuru, “Influence of group IV element on basic mechanical properties of BCC medium-entropy alloys using machine-learning potentials,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) Q. Pei, J. Guo, A. Suwardi, and G. Zhang, “Insights into interfacial thermal conductance in Bi2Te3-based systems for thermoelectrics,” Materials Today Physics. 2023. link Times cited: 5 USED (low confidence) O. Senkov, S. Rao, G. Balbus, R. Wheeler, and E. Payton, “Microstructure and deformation behavior of MoReW in the temperature range from 25°C to 1500°C,” Materialia. 2023. link Times cited: 3 USED (low confidence) Y.-hong Niu, D. Zhao, B. Zhu, S. Wang, Z. Wang, and H. Zhao, “Investigations on the role of chemical short-range order in the tensile deformation of FCC Co30Fe16.67Ni36.67Ti16.67 high-entropy alloys via Monte Carlo and molecular dynamics hybrid simulations,” Computational Materials Science. 2022. link Times cited: 4 USED (low confidence) H. He, S. Ma, and S. Wang, “Effect of Grain Boundary Atomic Density and Temperature on <110> Symmetric Tilt Grain Boundaries in Tungsten: An Atomistic Study,” Journal of Nuclear Materials. 2022. link Times cited: 1 USED (low confidence) S. Xu, W. Jian, and I. Beyerlein, “Ideal simple shear strengths of two HfNbTaTi-based quinary refractory multi-principal element alloys,” APL Materials. 2022. link Times cited: 4 Abstract: Atomistic simulations are employed to investigate chemical s… read moreAbstract: Atomistic simulations are employed to investigate chemical short-range ordering in two body-centered cubic refractory multi-principal element alloys, HfMoNbTaTi and HfNbTaTiZr, and its influence on their ideal simple shear strengths. Both the alias and affine shear strengths are analyzed on the {110} and {112} planes in the two opposing [Formula: see text] directions. In both quinary alloys, local ordering of NbNb, TaTa, HfNb, HfTa, and NbTa is preferred as the annealing temperature decreases from 900 to 300 K. The pair that achieves the highest degree of local ordering is TiTi in HfMoNbTaTi and HfTi in HfNbTaTiZr. Subject to the affine shear, these alloys yield by first phase transformation at the most likely pairs followed by deformation twinning at those sites. read less USED (low confidence) P. Fan, N. K. Katiyar, X. W. Zhou, and S. Goel, “Uniaxial pulling and nano-scratching of a newly synthesized high entropy alloy,” APL Materials. 2022. link Times cited: 7 Abstract: Multicomponent alloys possessing nanocrystalline structure, … read moreAbstract: Multicomponent alloys possessing nanocrystalline structure, often alluded to as Cantor alloys or high entropy alloys (HEAs), continue to attract the great attention of the research community. It has been suggested that about 64 elements in the periodic table can be mixed in various compositions to synthesize as many as ∼108 different types of HEA alloys. Nanomechanics of HEAs combining experimental and atomic simulations are rather scarce in the literature, which was a major motivation behind this work. In this spirit, a novel high-entropy alloy (Ni25Cu18.75Fe25Co25Al6.25) was synthesized using the arc melting method, which followed a joint simulation and experimental effort to investigate dislocation-mediated plastic mechanisms leading to side flow, pileup, and crystal defects formed in the sub-surface of the HEA during and after the scratch process. The major types of crystal defects associated with the plastic deformation of the crystalline face-centered cubic structure of HEA were 2,3,4-hcp layered such as defect coordination structures, coherent ∑3 twin boundary, and ∑11 fault or tilt boundary, in combination with Stair rods, Hirth locks, Frank partials, and Lomer–Cottrell locks. Moreover, 1/6 <112> Shockley, with exceptionally larger dislocation loops, was seen to be the transporter of stacking faults deeper into the substrate than the location of the applied cutting load. The (100) orientation showed the highest value for the kinetic coefficient of friction but the least amount of cutting stress and cutting temperature during HEA deformation, suggesting that this orientation is better than the other orientations for improved contact-mode manufacturing. read less USED (low confidence) S. Xu, X. Fan, C. Gu, Y. Shi, D. J. Singh, and W. Zheng, “Molecular Dynamics Simulation of Bubble Growth under Surface of Tungsten under Helium Irradiation,” Journal of Nuclear Materials. 2022. link Times cited: 0 USED (low confidence) M. Gounzari, Y. Belkassmi, A. Kotri, M. Bouzelmad, and L. E. Maimouni, “Mechanical characterization of Nanoporous two-dimensional Ti3C2 MXene membranes,” Chinese Journal of Physics. 2022. link Times cited: 3 USED (low confidence) A. Meo, P. Chureemart, R. Chantrell, and J. Chureemart, “The role of interfacial intermixing on HAMR dynamics in bilayer media,” Journal of Physics: Condensed Matter. 2022. link Times cited: 0 Abstract: We use an atomistic spin model to simulate FePt-based bilaye… read moreAbstract: We use an atomistic spin model to simulate FePt-based bilayers for heat assisted magnetic recording (HAMR) devices and investigate the effect of various degrees intermixing that might arise throughout the fabrication, growth and annealing processes, as well as different interlayer exchange couplings, on HAMR magnetisation dynamics. Intermixing can impact the device functionality, but interestingly does not deteriorate the properties of the system. Our results suggest that modest intermixing can prove beneficial and yield an improvement in the magnetisation dynamics for HAMR processes, also relaxing the requirement for weak exchange coupling between the layers. Therefore, we propose that a certain intermixing across the interface could be engineered in the fabrication process to improve HAMR technology further. read less USED (low confidence) J. Liu, X. Zhang, Y. He, Z. Zhao, M. Xia, and Y. Hu, “Suspended water droplet confined laser shock processing at elevated temperatures,” International Journal of Machine Tools and Manufacture. 2022. link Times cited: 5 USED (low confidence) D. Yang, B. Chen, S. Li, X. Ding, and J. Sun, “Effect of local lattice distortion on the core structure of edge dislocation in NbMoTaW multi-principal element alloys and the subsystems,” Materials Science and Engineering: A. 2022. link Times cited: 4 USED (low confidence) Y. Lei, R. Zhou, and B. Zhang, “The influence of atomic delocalization on dynamic behavior in Ce-Ni metallic melts,” Computational Materials Science. 2022. link Times cited: 0 USED (low confidence) C. Zhao, Y. Lin, and X. Wu, “Molecular Dynamics Study on Wetting Characteristics of Lead Droplet on Iron Surface at High Temperatures,” Materials Today Communications. 2022. link Times cited: 3 USED (low confidence) J. Luo et al., “Strengthening nanocrystalline immiscible bimetallic composite by high-entropy effect,” Composites Part B: Engineering. 2022. link Times cited: 3 USED (low confidence) S. Chen, T. Wang, X. Li, Y. Cheng, G. Zhang, and H. Gao, “Short-Range Ordering and its Impact on Thermodynamic Property of High-Entropy Alloys,” Acta Materialia. 2022. link Times cited: 15 USED (low confidence) Y. Chen et al., “Development of a Hybrid Intelligent Process Model for Micro-Electro Discharge Machining Using the TTM-MDS and Gaussian Process Regression,” Micromachines. 2022. link Times cited: 3 Abstract: This paper proposed a hybrid intelligent process model, base… read moreAbstract: This paper proposed a hybrid intelligent process model, based on a hybrid model combining the two-temperature model (TTM) and molecular dynamics simulation (MDS) (TTM-MDS). Combined atomistic-continuum modeling of short-pulse laser melting and disintegration of metal films [Physical Review B, 68, (064114):1–22.], and Gaussian process regression (GPR), for micro-electrical discharge machining (micro-EDM) were also used. A model of single-spark micro-EDM process has been constructed based on TTM-MDS model to predict the removed depth (RD) and material removal rate (MRR). Then, a GPR model was proposed to establish the relationship between input process parameters (energy area density and pulse-on duration) and the process responses (RD and MRR) for micro-EDM machining. The GPR model was trained, tested, and tuned using the data generated from the numerical simulations. Through the GPR model, it was found that micro-EDM process responses can be accurately predicted for the chosen process conditions. Therefore, the hybrid intelligent model proposed in this paper can be used for a micro-EDM process to predict the performance. read less USED (low confidence) P. Malakar, M. A. R. Anan, M. Islam, M. S. H. Thakur, and S. Mojumder, “Atomistic study of coreshell and functionally graded nanospheres under compressive loading,” International Journal of Mechanical Sciences. 2022. link Times cited: 2 USED (low confidence) J. Chapman, T. Hsu, X. Chen, T. Heo, and B. Wood, “Quantifying disorder one atom at a time using an interpretable graph neural network paradigm,” Nature Communications. 2022. link Times cited: 2 USED (low confidence) M. Wang, S. Jiang, D. Sun, Y. Zhang, and B. Yan, “Molecular dynamics simulation of mechanical behavior and phase transformation of nanocrystalline NiTi shape memory alloy with gradient structure,” Computational Materials Science. 2022. link Times cited: 7 USED (low confidence) T. Shi et al., “Distinct point defect behaviours in body-centered cubic medium-entropy alloy NbZrTi induced by severe lattice distortion,” Acta Materialia. 2022. link Times cited: 34 USED (low confidence) S. Nisany and D. Mordehai, “A Multiple Site Type Nucleation Model and Its Application to the Probabilistic Strength of Pd Nanowires,” Metals. 2022. link Times cited: 4 Abstract: Pristine specimens yield plastically under high loads by nuc… read moreAbstract: Pristine specimens yield plastically under high loads by nucleating dislocations. Since dislocation nucleation is a thermally activated process, the so-called nucleation-controlled plasticity is probabilistic rather than deterministic, and the distribution of the yield strengths depends on the activation parameters to nucleate. In this work, we develop a model to predict the strength distribution in nucleation-controlled plasticity when there are multiple nucleation site types. We then apply the model to molecular dynamics (MD) simulations of Pd nanowires under tension. We found that in Pd nanowires with a rhombic cross-section, nucleation starts from the edges, either with the acute or the obtuse cross-section angles, with a probability that is temperature-dependent. We show that the distribution of the nucleation strain is approximately normal for tensile loading at a constant strain rate. We apply the proposed model and extract the activation parameters for site types from both site types. With additional nudged elastic bands simulations, we propose that the activation entropy, in this case, has a negligible contribution. Additionally, the free-energy barriers obey a power-law with strain, with different exponents, which corresponds to the non-linear elastic deformation of the nanowires. This multiple site type nucleation model is not subjected only to two site types and can be extended to a more complex scenario like specimen with rough surfaces which has a distribution of nucleation sites with different conditions to nucleate dislocations. read less USED (low confidence) S. Xu, S. Chavoshi, and Y. Su, “On calculations of basic structural parameters in multi-principal element alloys using small atomistic models,” Computational Materials Science. 2022. link Times cited: 9 USED (low confidence) S. Shcherbinin, K. Krylova, G. Chechin, E. Soboleva, and S. Dmitriev, “Delocalized nonlinear vibrational modes in fcc metals,” Commun. Nonlinear Sci. Numer. Simul. 2022. link Times cited: 11 USED (low confidence) T. Fu et al., “Twin boundary migration and reactions with stacking fault tetrahedron in Cu and CoCrCuFeNi high-entropy alloy,” Journal of Materials Research and Technology. 2022. link Times cited: 10 USED (low confidence) P. Wang, Q. Cao, Y. Lan, H. Zhu, S. Liu, and P. Qing, “Nanostructuring enforced sandwich-tubular CNT-Cu interconnects,” Composite Structures. 2021. link Times cited: 1 USED (low confidence) M. Wang, Y. Zhang, and S. Jiang, “Atomic Simulation of Crystallographic Orientation Effect on Void Shrinkage and Collapse in Single-Crystal Copper under Shock Compression,” Journal of Materials Engineering and Performance. 2021. link Times cited: 0 USED (low confidence) J. Chapman and N. Goldman, “Quantifying the atomistic free-volume morphology of materials with graph theory,” Computational Materials Science. 2021. link Times cited: 0 USED (low confidence) A. Yamada, “Classical electronic and molecular dynamics simulation for optical response of metal system.,” The Journal of chemical physics. 2021. link Times cited: 3 Abstract: An extended molecular dynamics simulation that incorporates … read moreAbstract: An extended molecular dynamics simulation that incorporates classical free electron dynamics in the framework of the force-field model has been developed to enable us to describe the optical response of metal materials under the visible light electric field. In the simulation, dynamical atomic point charges follow equations of motion of classical free electrons that include Coulomb interactions with the oscillating field and surrounding atomic sites and collision effects from nearby electrons and ions. This scheme allows us to simulate an interacting system of metals with molecules using an ordinary polarizable force-field and preserves energy conservation in the case without applying an external electric field. As the first applications, we show that the presented simulation accurately reproduces (i) the classical image potential in a metal-charge interaction system and (ii) the dielectric function of bulk metal. We also demonstrate (iii) calculations of absorption spectra of metal nano-particles with and without a water solvent at room temperature, showing reasonable red-shift by the solvent effect, and (iv) plasmon resonant excitation of the metal nano-particle in solution under the visible light pulse and succeeding energy relaxation of the absorbed light energy from electrons to atoms on the metal and to the water solvent. Our attempt thus opens the possibility to expand the force-field based molecular dynamics simulation to an alternative tool for optical-related fields. read less USED (low confidence) H. Li, L. Zhao, Y. Yang, H. Zong, and X. Ding, “Improving radiation-tolerance of bcc multi-principal element alloys by tailoring compositional heterogeneities,” Journal of Nuclear Materials. 2021. link Times cited: 7 USED (low confidence) V. Samsonov, I. Talyzin, A. Kartoshkin, S. Vasilyev, and M. Alymov, “On the problem of stability/instability of bimetallic core-shell nanostructures: Molecular dynamics and thermodynamic simulations,” Computational Materials Science. 2021. link Times cited: 7 USED (low confidence) R. Romero, S. Xu, W. Jian, I. Beyerlein, and C. Ramana, “Atomistic simulations of the local slip resistances in four refractory multi-principal element alloys,” International Journal of Plasticity. 2021. link Times cited: 23 USED (low confidence) O. Bachurina and A. A. Kudreyko, “Two-component localized vibrational modes in fcc metals,” The European Physical Journal B. 2021. link Times cited: 4 USED (low confidence) A. Kedharnath, R. Kapoor, and A. Sarkar, “Classical molecular dynamics simulations of the deformation of metals under uniaxial monotonic loading: A review,” Computers & Structures. 2021. link Times cited: 16 USED (low confidence) T. Ruan, B. Wang, Y. Li, and C. Xu, “Atomistic insight into the solid-solid phase transitions in iron nanotube: A molecular dynamics study,” Materials Today Communications. 2021. link Times cited: 1 USED (low confidence) L. Wang et al., “Influences of strain rate, Al concentration and grain heterogeneity on mechanical behavior of CoNiFeAlxCu1-x high-entropy alloys: a molecular dynamics simulation,” Journal of materials research and technology. 2021. link Times cited: 17 USED (low confidence) E. Antillon, C. Woodward, S. Rao, and B. Akdim, “Chemical short range order strengthening in BCC complex concentrated alloys,” Acta Materialia. 2021. link Times cited: 35 USED (low confidence) M. Su, Q. Deng, L. Liu, L.-Y. Chen, H. He, and Y. Miao, “Molecular dynamics study on mechanical behaviors of Ti/Ni nanolaminate with a pre-existing void,” Nano Materials Science. 2021. link Times cited: 2 USED (low confidence) X. Chen et al., “Machine learning enhanced empirical potentials for metals and alloys,” Comput. Phys. Commun. 2021. link Times cited: 5 USED (low confidence) M. Su, Q. Deng, M. An, L. Liu, and L.-Y. Chen, “Role of amorphous layer and interfaces on the tensile behaviors of triple-phase Ti/Ni nanolaminates: A molecular dynamics study,” Journal of Alloys and Compounds. 2021. link Times cited: 6 USED (low confidence) X. Li, S. Peng, X. Zhang, X. Jiang, and Q. Wang, “Microscopic and macroscopic analyses of the interaction mechanism between defect growth and dislocation emission in single‐crystal aluminum,” Fatigue & Fracture of Engineering Materials & Structures. 2021. link Times cited: 7 USED (low confidence) Q. Wang, L. Li, and M. Yang, “Melting characteristics and dynamic evolution of microstructure of single-crystal gold film irradiated by laser under the non-Fourier effect,” Molecular Crystals and Liquid Crystals. 2021. link Times cited: 0 Abstract: The process of ultra-thin single crystal gold film irradiate… read moreAbstract: The process of ultra-thin single crystal gold film irradiated by femtosecond laser is simulated by the combination of improved two-temperature model (TTM) and molecular dynamics (MD). The results indicate that under the non-Fourier effect, the gold film shows special melting characteristics and spalling mechanism. The phenomena of homogeneous melting and pore spalling of gold film are successfully distinguished by curing rate method and atomic density method respectively. Due to the tiny thickness of the gold film studied, the damage mechanism of the gold film irradiated by laser has also changed. read less USED (low confidence) S. Matsunaga, “Structure and transport phenomena of non-aqueous electrolyte in Al ion batteries -Study by molecular dynamics-,” PROCEEDINGS OF THE 10TH INTERNATIONAL ADVANCES IN APPLIED PHYSICS AND MATERIALS SCIENCE CONGRESS & EXHIBITION. 2021. link Times cited: 1 Abstract: Molecular dynamics (MD) simulation of Al ions was performed … read moreAbstract: Molecular dynamics (MD) simulation of Al ions was performed as a model of Al ion battery using 1-Ethyl-3-methylimidazolium chloride ([EMIm]Cl) as an electrolyte. It is one of the safe secondary battery candidates that can replace Li-ion battery. The structure and charge of the EMIm ions used in the MD simulation were calculated in advance by Gaussian09 based on the density functional theory (DFT). The pair distribution function, the mean square displacement, and frequency dependent diffusion coefficient are obtained to examine structure, diffusion coefficient, and vibrational spectra. The significant large vibrational peak of Al has been obtained, which may be attributed to the cage effect of Al ion in the [EMIm]Cl electrolyte. read less USED (low confidence) J. P. Esquillo, “Flipped classroom instructional strategy in teaching mathematics to grade five learners: Basis for video-assisted instructional materials,” International Journal of Research Studies in Education. 2021. link Times cited: 0 Abstract: This study is an experimental research that aimed to determi… read moreAbstract: This study is an experimental research that aimed to determine the effectiveness of the Flipped Classroom Instructional Strategies in teaching Mathematics 5. The control group, comprising of 34 pupils, and the experimental group composing of 35 pupils was tasked to provide their age, and sex. The conventional teaching method taught to the control group, while the Flipped Classroom Approach taught for a period of two (2) weeks to the experimental group. Surveyed students are in appropriate age for their current grade level. The study is dominated by male respondents. Respondents from control and experimental group have low to average performance during the pre- test. Performances of control group and experimental group have increased. Though, experimental group has higher post-test score rating than control group. Pre-test scores of both groups do not have significant difference. Also, it depicts that the ability of respondents on both control and experimental is almost at the same level. An intervention should be used to improve their performance. Post-test scores of both groups have significant difference. Although there is an increase on the performance of the control group, experimental group has performed even better. The use of flipped classroom instructional strategies in teaching Mathematics is deemed effective. read less USED (low confidence) V. Sorkin, S. Chen, T. L. Tan, Z. Yu, M. Man, and Y.-W. Zhang, “First-principles-based high-throughput computation for high entropy alloys with short range order,” Journal of Alloys and Compounds. 2021. link Times cited: 12 USED (low confidence) L. Zhao, H. Zong, X. Ding, and T. Lookman, “Anomalous dislocation core structure in shock compressed bcc high-entropy alloys,” Acta Materialia. 2021. link Times cited: 38 USED (low confidence) X. Wang, S. Xu, W. Jian, X.-G. Li, Y. Su, and I. Beyerlein, “Generalized stacking fault energies and Peierls stresses in refractory body-centered cubic metals from machine learning-based interatomic potentials,” Computational Materials Science. 2021. link Times cited: 30 USED (low confidence) P. Wang, Q. Cao, H. Wang, S. Liu, Y. Chen, and Q. Peng, “CNT-sandwiched copper composites as super thermal conductors for heat management,” Physica E-low-dimensional Systems & Nanostructures. 2021. link Times cited: 6 USED (low confidence) X.-song Huang et al., “Atomistic simulation of chemical short-range order in HfNbTaZr high entropy alloy based on a newly-developed interatomic potential,” Materials & Design. 2021. link Times cited: 55 USED (low confidence) Y. Hu, J. Leng, and T. Chang, “Mechanosensing of a Graphene Flake on a Bent Beam,” Journal of Applied Mechanics. 2021. link Times cited: 1 Abstract:
The ability of mechanosensing is essential for intelligent… read moreAbstract:
The ability of mechanosensing is essential for intelligent systems. Here we show by molecular dynamics (MD) simulations that a graphene flake on a bent beam exhibits amazing mechanosensing behavior, termed flexotaxis. The graphene flake can perceive the beam bending gradient which indeed leads to a gradient of atomic density that produces a driving force on the flake toward the direction of increasing density. An analytical model is developed to further confirm the mechanism, and the simulation results can be well reproduced by the model. Our findings may have general implications not only for the potential applications of graphene as sensing elements in nanoscale intelligent devices but also for the exploration of mechanosensing capability of other two-dimensional materials. read less USED (low confidence) E. Mak, B. Yin, and W. Curtin, “A ductility criterion for bcc high entropy alloys,” Journal of the Mechanics and Physics of Solids. 2021. link Times cited: 44 USED (low confidence) Q. Jia et al., “Sintering mechanism of Ag-Pd nanoalloy film for power electronic packaging,” Applied Surface Science. 2021. link Times cited: 11 USED (low confidence) S. Chen et al., “Chemical-Affinity Disparity and Exclusivity Drive Atomic Segregation, Short-Range Ordering, and Cluster Formation in High-Entropy Alloys,” Acta Materialia. 2021. link Times cited: 35 USED (low confidence) S.-wei Liu, J. Yin, and Y. Zhao, “Revealing twinning from triple lines in nanocrystalline copper via molecular dynamics simulation and experimental observation,” Journal of materials research and technology. 2021. link Times cited: 2 USED (low confidence) S. Rao, C. Woodward, B. Akdim, O. Senkov, and D. Miracle, “Theory of solid solution strengthening of BCC Chemically Complex Alloys,” Acta Materialia. 2021. link Times cited: 43 USED (low confidence) N. Bertin, W. Cai, S. Aubry, and V. Bulatov, “Core energies of dislocations in bcc metals,” Physical Review Materials. 2021. link Times cited: 5 Abstract: Accurate methods and an efficient workflow for computing and… read moreAbstract: Accurate methods and an efficient workflow for computing and documenting dislocation core energies are developed and applied to $\frac{1}{2}\ensuremath{\langle}111\ensuremath{\rangle}$ and $\ensuremath{\langle}100\ensuremath{\rangle}$ dislocations in five body-centered cubic (bcc) metals W, Ta, V, Mo, and $\ensuremath{\alpha}$-Fe represented by 13 model interatomic potentials. For each dislocation type, dislocation core energies are extracted for a large number of dislocation characters thoroughly sampling the entire 2-space of crystallographic line orientations of the bcc lattice. Of particular interest, core energies of the $\frac{1}{2}\ensuremath{\langle}111\ensuremath{\rangle}{110}$ dislocations are found to be distinctly asymmetric with respect to the sign of the character angle, whereas core energies of $\ensuremath{\langle}100\ensuremath{\rangle}{110}$ junction dislocations exhibit marked cusps for line orientations vicinal to the closed-packed $\ensuremath{\langle}111\ensuremath{\rangle}$ directions. Our findings furnish substantial insights for developing accurate models of dislocation core energies employed in mesoscale dislocation dynamics simulations of crystal plasticity. read less USED (low confidence) S. Corthay et al., “Elevated-temperature high-strength h-BN-doped Al2014 and Al7075 composites: Experimental and theoretical insights,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2021. link Times cited: 10 USED (low confidence) M. He, E. T. Karim, M. Shugaev, and L. Zhigilei, “Atomistic simulation of the generation of vacancies in rapid crystallization of metals,” Acta Materialia. 2021. link Times cited: 7 USED (low confidence) J. M. Molina and T. White, “A molecular dynamics study of laser-excited gold,” Matter and Radiation at Extremes. 2021. link Times cited: 7 Abstract: The structural evolution of laser-excited systems of gold ha… read moreAbstract: The structural evolution of laser-excited systems of gold has previously been measured through ultrafast MeV electron diffraction. However, there has been a long-standing inability of atomistic simulations to provide a consistent picture of the melting process, leading to large discrepancies between the predicted threshold energy density for complete melting, as well as the transition between heterogeneous and homogeneous melting. We make use of two-temperature classical molecular dynamics simulations utilizing three highly successful interatomic potentials and reproduce electron diffraction data presented by Mo et al. [Science 360, 1451–1455 (2018)]. We recreate the experimental electron diffraction data, employing both a constant and temperature-dependent electron–ion equilibration rate. In all cases, we are able to match time-resolved electron diffraction data, and find consistency between atomistic simulations and experiments, only by allowing laser energy to be transported away from the interaction region. This additional energy-loss pathway, which scales strongly with laser fluence, we attribute to hot electrons leaving the target on a timescale commensurate with melting. read less USED (low confidence) Z. Sun, F. Dai, and W.-Z. Zhang, “A molecular dynamics study of dislocation-interphase boundary interactions in FCC/BCC phase transformation system,” Computational Materials Science. 2020. link Times cited: 3 USED (low confidence) H. Feng et al., “A molecular dynamics investigation into deformation mechanism of nanotwinned Cu/high entropy alloy FeCoCrNi nanolaminates,” Surface & Coatings Technology. 2020. link Times cited: 8 USED (low confidence) C. Yang, M. Zhang, and L. Qi, “Grain boundary structure search by using an evolutionary algorithm with effective mutation methods,” Computational Materials Science. 2020. link Times cited: 11 USED (low confidence) S. Starikov, I. Gordeev, Y. Lysogorskiy, L. Kolotova, and S. Makarov, “Optimized interatomic potential for study of structure and phase transitions in Si-Au and Si-Al systems,” Computational Materials Science. 2020. link Times cited: 19 USED (low confidence) O. Senkov, S. Rao, T. Butler, T. Daboiku, and K. Chaput, “Microstructure and properties of Nb-Mo-Zr based refractory alloys,” International Journal of Refractory Metals & Hard Materials. 2020. link Times cited: 21 USED (low confidence) S. Si et al., “Enhancing resistance to radiation hardening and radiation thermal conductivity degradation by tungsten/graphene interface engineering,” Journal of Nuclear Materials. 2020. link Times cited: 9 USED (low confidence) O. Bachurina and A. Kudreyko, “Two-dimensional discrete breathers in fcc metals,” Computational Materials Science. 2020. link Times cited: 15 USED (low confidence) Y. Wu, W. Yu, and S. Shen, “Strain hardening and embrittlement of Al crystal with a surface oxidized void,” Mechanics of Materials. 2020. link Times cited: 3 USED (low confidence) Q. Liu et al., “Influence of composition and size on the thermodynamic stability and structural evolution of CuAlNi nanoclusters,” Progress in Natural Science: Materials International. 2020. link Times cited: 6 USED (low confidence) Y. Tao et al., “The enhancement of heat conduction across the metal/graphite interface treated with a focused ion beam.,” Nanoscale. 2020. link Times cited: 6 Abstract: In this work, we report a convenient and efficient approach … read moreAbstract: In this work, we report a convenient and efficient approach to improve heat conduction across the metal/graphite interface. It is demonstrated that the interfacial thermal conductance between Al and graphite can be enhanced by a factor of ∼5 after milling the graphite with a focused ion beam. Such enhancement is attributed to the decreased Fermi level of the milled graphite compared with the pristine counterpart. Once graphite is milled with the focused ion beam, surface defects are formed that induce the redistribution of electrons at the interface between Al and graphite. The formation of enormous dipoles on the milled graphite/Al interface leads to the conversion of the interfacial interaction from physisorption to chemisorption, which is beneficial for phonon transmission across the interface. Based on the measured Fermi level difference, the non-equilibrium Green's function method predicts that the interfacial interaction strength in the Al/milled graphite is increased 4-fold compared with Al/pristine graphite, which causes the increase of the interfacial thermal conductance. Our theoretical model also predicts that the interfacial thermal conductance does not increase monotonically with the interaction strength. Once the interaction strength exceeds a critical value, the interface thermal conductance will decrease. read less USED (low confidence) P. Wang, Y. Bu, J. Liu, Q. Li, H. Wang, and W. Yang, “Atomic deformation mechanism and interface toughening in metastable high entropy alloy,” Materials Today. 2020. link Times cited: 41 USED (low confidence) K. C. Katakam and N. Yedla, “Irradiation studies on nano-scale single crystal copper by molecular dynamics simulation,” IOP Conference Series: Materials Science and Engineering. 2020. link Times cited: 0 Abstract: We report molecular dynamics (MD) simulation studies on the … read moreAbstract: We report molecular dynamics (MD) simulation studies on the structure and tensile properties (temperature = 300 K and strain rate = 1010 s−1) of irradiated copper single crystals (101.8 Å× 50.54Å × 50.54Å) oriented along the [100] and [110] crystallographic orientations. Primary knock-on atom (PKA) method is used for radiation (0.5 keV to 5 keV). Structural studies indicate liquid like regions near the PKA region and are more in [110] orientation. The self-diffusivity (D) of copper in the PKA regions is found to increase from 4.05 × 10−5 m2/s at 1 keV irradiation energy to 27.36 × 10−5 at 5 keV irradiation energy in [110] orientation. However, no such relationship is observed in the [100] orientation. Vacancies, self-interstitials, and dislocations are more prominent in [100] orientation (dislocation density = 1 × 1016 m−2). Young’s modulus and yield strength decrease due to the formation of defects as anticipated. read less USED (low confidence) R. Singh and V. Sharma, “Molecular dynamics study of tensile behaviour for cold and linear friction welded single crystal tungsten.,” Journal of molecular graphics & modelling. 2020. link Times cited: 9 USED (low confidence) O. Senkov, S. Rao, T. Butler, T. Daboiku, and K. Chaput, “Effect of Fe additions on the microstructure and properties of Nb-Mo-Ti alloys,” International Journal of Refractory Metals and Hard Materials. 2020. link Times cited: 13 USED (low confidence) L. Li et al., “Effects of temperature and strain rate on plastic deformation mechanisms of nanocrystalline high-entropy alloys,” Intermetallics. 2020. link Times cited: 42 USED (low confidence) M. Bahramyan, R. T. Mousavian, and D. Brabazon, “Study of the plastic deformation mechanism of TRIP–TWIP high entropy alloys at the atomic level,” International Journal of Plasticity. 2020. link Times cited: 55 USED (low confidence) F. Zhu, Z. Guo, and T. Chang, “Nanoscale continuous cyclic motion driven by a stable thermal field,” Applied Materials Today. 2020. link Times cited: 8 USED (low confidence) F. Z. Dai, Z. Sun, and W.-Z. Zhang, “From coherent to semicoherent—Evolution of precipitation crystallography in an fcc/bcc system,” Acta Materialia. 2020. link Times cited: 10 USED (low confidence) W. Li, S. Rao, Q. Wang, H. Fan, J. Yang, and J. El-Awady, “Core structure and mobility of edge dislocations in face-centered-cubic chemically complex NiCoFe and NiCoFeCu equiatomic solid-solution alloys,” Materialia. 2020. link Times cited: 18 USED (low confidence) R. Fang, W. Wang, L. Guo, K. Zhang, X. Zhang, and H. Li, “Atomic insight into the solidification of Cu melt confined in graphene nanoslits,” Journal of Crystal Growth. 2020. link Times cited: 10 USED (low confidence) O. Senkov, J. Couzinié, S. Rao, V. Soni, and R. Banerjee, “Temperature dependent deformation behavior and strengthening mechanisms in a low density refractory high entropy alloy Al10Nb15Ta5Ti30Zr40,” Materialia. 2020. link Times cited: 43 USED (low confidence) X. Zhu and X. Cheng, “Symmetric tilt grain boundary evolution during the growth of copper thin films: Molecular dynamics simulation,” Physica B-condensed Matter. 2020. link Times cited: 1 USED (low confidence) S. Chen, Z. Aitken, Z. Wu, Z. Yu, R. Banerjee, and Y.-W. Zhang, “Hall-Petch and inverse Hall-Petch relations in high-entropy CoNiFeAlxCu1-x alloys,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2020. link Times cited: 83 USED (low confidence) J. Leng, Y. Hu, and T. Chang, “Nanoscale directional motion by angustotaxis.,” Nanoscale. 2019. link Times cited: 5 Abstract: Directing motion of a nanoscale object on solid surfaces, in… read moreAbstract: Directing motion of a nanoscale object on solid surfaces, in particular in an intrinsic way, is crucial for many aspects of nanotechnology applications. Here we report a novel intrinsic mechanism for nanoscale directional motion, termed angustotaxis, where a wide single walled carbon nanotube in a tapered channel drives itself toward the narrower end of the channel. The underlying physics of angustotaxis is attributed to the lower system potential when the nanotube is at a narrower region of the channel due to the increased contact area between the nanotube and the channel. Angustotaxis could lead to promising routes not only for nanoscale energy conversion from van der Waals potential to mechanical work, but also for mass transport like surface cleaning. read less USED (low confidence) X. Lu, D. Laughlin, and J. Zhu, “On the conditions for ordered hexagonal mm2 Co3Pt,” Journal of Magnetism and Magnetic Materials. 2019. link Times cited: 0 USED (low confidence) S. Mishra, S. Maiti, B. S. Dwadasi, and B. Rai, “Realistic microstructure evolution of complex Ta-Nb-Hf-Zr high-entropy alloys by simulation techniques,” Scientific Reports. 2019. link Times cited: 22 USED (low confidence) O. Senkov, S. Rao, T. Butler, and K. Chaput, “Ductile Nb alloys with reduced density and cost,” Journal of Alloys and Compounds. 2019. link Times cited: 42 USED (low confidence) L. Liu, Q. Deng, M. Su, M. An, and R. Wang, “Strain rate and temperature effects on tensile behavior of Ti/Al multilayered nanowire: A molecular dynamics study,” Superlattices and Microstructures. 2019. link Times cited: 10 USED (low confidence) J. Nandy, N. Yedla, P. Gupta, H. Sarangi, and S. Sahoo, “Sintering of AlSi10Mg particles in direct metal laser sintering process: A molecular dynamics simulation study,” Materials Chemistry and Physics. 2019. link Times cited: 43 USED (low confidence) J. Fu et al., “Molecular dynamics simulations of high-energy radiation damage in W and W–Re alloys,” Journal of Nuclear Materials. 2019. link Times cited: 33 USED (low confidence) X. W. Zhou, “Impact of Molecular Dynamics Simulations on Research and Development of Semiconductor Materials,” MRS Advances. 2019. link Times cited: 2 Abstract: Atomic scale defects critically limit performance of semicon… read moreAbstract: Atomic scale defects critically limit performance of semiconductor materials. To improve materials, defect effects and defect formation mechanisms must be understood. In this paper, we demonstrate multiple examples where molecular dynamics simulations have effectively addressed these issues that were not well addressed in prior experiments. In the first case, we report our recent progress on modelling graphene growth, where we found that defects in graphene are created around periphery of islands throughout graphene growth, not just in regions where graphene islands impinge as believed previously. In the second case, we report our recent progress on modelling TlBr, where we discovered that under an electric field, edge dislocations in TlBr migrate in both slip and climb directions. The climb motion ejects extensive vacancies that can cause the rapid aging of the material seen in experiments. In the third case, we discovered that the growth of InGaN films on (0001) surfaces suffers from a serious polymorphism problem that creates enormous amounts of defects. Growth on (1120) surfaces, on the other hand, results in single crystalline wurtzite films without any of these defects. In the fourth case, we first used simulations to derive dislocation energies that do not possess any noticeable statistical errors, and then used these error-free methods to discover possible misuse of misfit dislocation theory in past thin film studies. Finally, we highlight the significance of molecular dynamics simulations in reducing defects in the design space of nanostructures. read less USED (low confidence) Y. Wang et al., “Hydrogen distribution induced screw dislocation core spreading in tungsten,” Journal of Nuclear Materials. 2019. link Times cited: 5 USED (low confidence) S. Feng et al., “Tuning deformation behavior of Cu0.5CoNiCrAl high-entropy alloy via cooling rate gradient: An atomistic study,” Intermetallics. 2019. link Times cited: 10 USED (low confidence) W. Li, H. Fan, J.-J. Tang, Q. Wang, X. Zhang, and J. El-Awady, “Effects of alloying on deformation twinning in high entropy alloys,” Materials Science and Engineering: A. 2019. link Times cited: 33 USED (low confidence) M. Akdeniz and A. Mekhrabov, “Size dependent stability and surface energy of amorphous FePt nanoalloy,” Journal of Alloys and Compounds. 2019. link Times cited: 4 USED (low confidence) P. Gupta and A. Kumar, “Effect of surface elasticity on extensional and torsional stiffnesses of isotropic circular nanorods,” Mathematics and Mechanics of Solids. 2019. link Times cited: 12 Abstract: We present a continuum formulation to obtain the effects of … read moreAbstract: We present a continuum formulation to obtain the effects of surface residual stress and surface elastic constants on extensional and torsional stiffnesses of isotropic circular nanorods. Analytical expressions of axial force, twisting moment, and extensional and torsional stiffnesses are obtained. Unlike the case of rectangular nanorods, we show that the stiffnesses of circular nanorods also depend on surface residual stress components. This is attributed to non-zero surface curvature inherent in circular nanorods. We further normalize these expressions and analyze their asymptotic limits in the limit of the nanorod’s radius approaching both zero and infinity, corresponding to surface-dominated and bulk-dominated regimes, respectively. Finally, we use the recently proposed helical Cauchy–Born rule and perform molecular statics calculations to obtain axial force, twisting moment, and stiffnesses of the tungsten nanorod. The tungsten material is selected since its bulk crystal exhibits isotropy in the stress-free state. The results from molecular statics calculations are shown to match the derived continuum formulas accurately. read less USED (low confidence) O. Bachurina, “Plane and plane-radial discrete breathers in fcc metals,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 16 Abstract: Discrete breather (DB) is a time-periodic, spatially localiz… read moreAbstract: Discrete breather (DB) is a time-periodic, spatially localized vibrational mode in a perfect nonlinear lattice. In this study, two new types of DBs are reported in fcc metals (Al, Cu and Ni), based on the molecular dynamics simulations using standard embedded atom method interatomic potentials. All calculations are performed at a zero temperature in a three-dimensional computational cell with the use of periodic boundary conditions. A plane DB is excited in a single (111) atomic plane by displacing the atoms from their equilibrium lattice sites according to a specific pattern corresponding to a delocalized vibrational mode in a two-dimensional triangular lattice. This plane DB is delocalized in two dimensions and localized in one dimension, normal to the excited (111) plane. It is shown that in all studied metals the plane DBs have maximal lifetimes of 17–22 ps in the range of initial amplitudes of 0.15–0.30 Å. Herewith the studied mode demonstrates a hard type of nonlinearity, i.e. its frequency increases with amplitude. The second new type of DB is the plane-radial DB obtained by imposing a radial localizing function on the plane DB. This disk-type DB is localized in all three dimensions. The time evolution of the atomic displacement amplitudes and the kinetic energy are studied. The DBs of this type can exist for 9 and 4 ps in Cu and Ni, respectively, and then they decay by dissipating their vibrational energy onto neighboring atoms. The long-lived plane-radial DB in Al is not found. read less USED (low confidence) S. Rao, E. Antillon, C. Woodward, B. Akdim, T. Parthasarathy, and O. Senkov, “Solution hardening in body-centered cubic quaternary alloys interpreted using Suzuki’s kink-solute interaction model,” Scripta Materialia. 2019. link Times cited: 40 USED (low confidence) S. Rao, B. Akdim, E. Antillon, C. Woodward, T. Parthasarathy, and O. Senkov, “Modeling solution hardening in BCC refractory complex concentrated alloys: NbTiZr, Nb1.5TiZr0.5 and Nb0.5TiZr1.5,” Acta Materialia. 2019. link Times cited: 93 USED (low confidence) Q. Xiong, T. Kitamura, and Z. Li, “Nanocrystallization in single-crystal copper under laser shock compression: A molecular dynamics study,” Materials Science and Engineering: A. 2019. link Times cited: 26 USED (low confidence) O. Bachurina, “Linear discrete breather in fcc metals,” Computational Materials Science. 2019. link Times cited: 21 USED (low confidence) D. Zhang and S. Chaudhuri, “Solidification dynamics and microstructure evolution in nanocrystalline cobalt,” Computational Materials Science. 2019. link Times cited: 8 USED (low confidence) M. H. Ulz, “A Continuum-on-Atomistic Framework with Bi-Stable Elements for the Computation of Minimum Free Energy Paths,” Mechanics of Solids. 2019. link Times cited: 0 USED (low confidence) H. Bhattarai, K. E. Newman, and J. Gezelter, “Polarizable potentials for metals: The density readjusting embedded atom method (DR-EAM),” Physical Review B. 2019. link Times cited: 6 Abstract: In simulations of metallic interfaces, a critical aspect of … read moreAbstract: In simulations of metallic interfaces, a critical aspect of metallic behavior is missing from the some of the most widely used classical molecular dynamics force fields. We present a modification of the embedded atom method (EAM) which allows for electronic polarization of the metal by treating the valence density around each atom as a fluctuating dynamical quantity. The densities are represented by a set of additional fluctuating variables (and their conjugate momenta) which are propagated along with the nuclear coordinates. This ``density readjusting EAM'' (DR-EAM) preserves nearly all of the useful qualities of traditional EAM, including bulk elastic properties and surface energies. However, it also allows valence electron density to migrate through the metal in response to external perturbations. We show that DR-EAM can successfully model polarization in response to external charges, capturing the image charge effect in atomistic simulations. DR-EAM also captures some of the behavior of metals in the presence of uniform electric fields, predicting surface charging and shielding internal to the metal. We further show that it predicts charge transfer between the constituent atoms in alloys, leading to novel predictions about unit cell geometries in layered $\mathrm{L}{1}_{0}$ structures. read less USED (low confidence) M. S. Beni, T. H. Tan, and K. Yu, “Atomistic modeling of pileup process in metal deposition manufacture,” Results in Physics. 2019. link Times cited: 3 USED (low confidence) V. Borysiuk and V. Mochalin, “Thermal stability of two-dimensional titanium carbides Ti_n+1C_n (MXenes) from classical molecular dynamics simulations,” MRS Communications. 2019. link Times cited: 25 Abstract: We report the classical molecular dynamics (MD) study of the… read moreAbstract: We report the classical molecular dynamics (MD) study of thermal stability of three two-dimensional (2D) titanium carbides Ti_2C, Ti_3C_2, and Ti_4C_3 (MXenes). Thermal properties of 2D nanomaterials are of fundamental importance and raise particular interest due to their potential applications in nanoelectronics. To investigate the behavior of Ti_ n +1C_n MXenes during heating, structural parameters such as Lindemann indexes, radial distribution functions, and atomistic configurations were calculated. The analysis of MD data allowed us to obtain approximate values of MXene degradation temperatures that are 1050, 1500, and 1700 K for Ti_2C, Ti_3C_2, and Ti_3C_4 MXenes, respectively. read less USED (low confidence) A. Hassani et al., “Statistical investigations of the film-substrate interface during aluminum deposition on Ni(111) by molecular dynamics simulation,” Superlattices and Microstructures. 2019. link Times cited: 9 USED (low confidence) M. Su, Q. Deng, M. An, L. Liu, and C. Ma, “Molecular dynamics study of the tensile behaviors of Ti(0 0 0 1)/Ni(1 1 1) multilayered nanowires,” Computational Materials Science. 2019. link Times cited: 13 USED (low confidence) A. R. Hinkle, W. Nöhring, R. J. Leute, T. Junge, and L. Pastewka, “The emergence of small-scale self-affine surface roughness from deformation,” Science Advances. 2019. link Times cited: 47 Abstract: When solids are deformed plastically, they develop surface r… read moreAbstract: When solids are deformed plastically, they develop surface roughness because plastic flow is not laminar at small scales. Most natural and man-made surfaces appear to be rough on many length scales. There is presently no unifying theory of the origin of roughness or the self-affine nature of surface topography. One likely contributor to the formation of roughness is deformation, which underlies many processes that shape surfaces such as machining, fracture, and wear. Using molecular dynamics, we simulate the biaxial compression of single-crystal Au, the high-entropy alloy Ni36.67Co30Fe16.67Ti16.67, and amorphous Cu50Zr50 and show that even surfaces of homogeneous materials develop a self-affine structure. By characterizing subsurface deformation, we connect the self-affinity of the surface to the spatial correlation of deformation events occurring within the bulk and present scaling relations for the evolution of roughness with strain. These results open routes toward interpreting and engineering roughness profiles. read less USED (low confidence) P. Zhu and J. Li, “Investigation into the Realization of a Single Atomic Layer Removal in Nanoscale Mechanical Machining of Single Crystalline Copper,” Springer Tracts in Mechanical Engineering. 2018. link Times cited: 0 USED (low confidence) Y. Gao, G. Li, Y. Piao, S. Liu, S.-bin Liu, and Q. Wang, “Size-dependent cuboctahedron-icosahedron transformations of Co-based bimetallic by molecular dynamics simulation,” Materials Letters. 2018. link Times cited: 2 USED (low confidence) X. Cui and S. Ringer, “On the nexus between atom probe microscopy and density functional theory simulations,” Materials Characterization. 2018. link Times cited: 18 USED (low confidence) P. N. Mayer and A. Mayer, “Size distribution of pores in metal melts at non-equilibrium cavitation and further stretching, and similarity with the spall fracture of solids,” International Journal of Heat and Mass Transfer. 2018. link Times cited: 18 USED (low confidence) H. N. Pishkenari, F. S. Yousefi, and A. Taghibakhshi, “Determination of surface properties and elastic constants of FCC metals: a comparison among different EAM potentials in thin film and bulk scale,” Materials Research Express. 2018. link Times cited: 22 Abstract: Three independent elastic constants C11, C12, and C44 were c… read moreAbstract: Three independent elastic constants C11, C12, and C44 were calculated and compared using available potentials of eight different metals with FCC crystal structure; Gold, Silver, Copper, Nickel, Platinum, Palladium, Aluminum and Lead. In order to calculate the elastic constants, the second derivative of the energy density of each system was calculated with respect to different directions of strains. Each set of the elastic constants of the metals in bulk scale was compared with experimental results, and the average relative error was for each was calculated and compared with other available potentials. Then, using the Voigt-Reuss-Hill method, approximated values for Young and shear moduli and Poisson’s ratio of the FCC metals in the bulk scale were found for each potential. Furthermore, to observe the surface effects on the metals in nanoscale, surface elastic constants of the thin films of the metals have been calculated. In the study of the thin films of materials in nanoscale, the number of surface atoms is considerable compared to all atoms of the object. This leads to an increase in the surface effects, which influence the elastic properties. By considering this fact and employing related definitions and equations, the properties of the thin films of the metals were calculated, and the surface effects for different crystallographic directions were compared. Subsequently, in some cases, comparisons among characteristics of the metals in the thin film and bulk material were made. read less USED (low confidence) A. V. Khomenkc, M. Zakharov, K. Khomenko, Y. V. Khyzhnya, and P. Trofimenko, “Atomistic Modeling of Friction Force Dependence on Contact Area of Metallic Nanoparticles on Graphene,” 2018 IEEE 8th International Conference Nanomaterials: Application & Properties (NAP). 2018. link Times cited: 4 Abstract: We compare the calculation of the contact area between the p… read moreAbstract: We compare the calculation of the contact area between the palladium and aluminum nanoparticles and graphene substrate by an ellipse and by the distance between atoms. Linear approximations of shear stress depending on the contact area of different metals were compared. It has been observed that aluminum nanoparticles are spherical and palladium nanoparticles have an ellipsoid shape, because the metal-metal interaction in palladium is stronger than that of aluminum. read less USED (low confidence) T. Sipkens and K. Daun, “Effect of Surface Interatomic Potential on Thermal Accommodation Coefficients Derived from Molecular Dynamics,” The Journal of Physical Chemistry C. 2018. link Times cited: 14 Abstract: This work investigates how the interatomic surface potential… read moreAbstract: This work investigates how the interatomic surface potential influences molecular dynamics (MD)-derived thermal accommodation coefficients (TACs). Iron, copper, and silicon surfaces are considered over a range of temperatures that include their melting points. Several classes of potentials are reviewed, including two-body, three-body, and bond-order force fields. MD-derived densities and visualization of the surfaces are used to explain the differences in the parameterizations of these potentials within the context of gas–surface scattering. Finally, TACs are predicted for a range of gas–surface combinations, and recommended values of the TAC are selected that take into account the robustness and uncertainties of each of the considered parameterizations. Further, it is observed that there is a significant change in the TAC about phase changes that must be taken into account for applications with a large range of surface temperatures. read less USED (low confidence) S. Gerstl, ScopeM, and E. Zurich, “Atom Probe Tomography,” Practical Metallography. 2018. link Times cited: 26 Abstract: In den letzten zwei Jahrzehnten übte die Atomsondentomograph… read moreAbstract: In den letzten zwei Jahrzehnten übte die Atomsondentomographie (Atom Probe Tomography, APT) erheblichen Einfluss auf den Bereich der Materialwissenschaft und in jüngster Zeit auch darüber hinaus aus [1]. Es handelt sich um eine Kombination aus Mikroskop und Massenspektrometer, durch die 3D-Daten einer großen Bandbreite an Feststoffen in atomarem Maßstab bereitgestellt werden können. Die Bezeichnung „Atomsonde“ entstand ursprünglich in Anlehnung an die Erfindung der Elektronenmikrosonde zu dieser Zeit, da ihr Erfinder, Erwin Müller, die Tatsache betonen wollte, dass dieses Instrument faktisch Einzelatomempfindlichkeit erreichen konnte. Der Begriff beschreibt die Erfindung nicht vollumfänglich, da nicht wirklich eine Sonde zum Einsatz kommt, sondern vielmehr ein elektrisches Feld, das oft mit einer Wärmeeinwirkung durch einen Laser kombiniert wird, um Atome von einer scharfen Spitze zu extrahieren und sie in Richtung eines für einzelne Atome empReceived: May 25, 2018 Accepted: June 13, 2018 read less USED (low confidence) B. Faria, C. Guarda, N. Silvestre, J. Lopes, and D. Galhofo, “Strength and failure mechanisms of cnt-reinforced copper nanocomposite,” Composites Part B: Engineering. 2018. link Times cited: 40 USED (low confidence) R. Sachan et al., “Radiation-induced extreme elastic and inelastic interactions in concentrated solid solutions,” Materials & Design. 2018. link Times cited: 13 USED (low confidence) M. Jafary-Zadeh, Z. Aitken, R. Tavakoli, and Y.-W. Zhang, “On the controllability of phase formation in rapid solidification of high entropy alloys,” Journal of Alloys and Compounds. 2018. link Times cited: 26 USED (low confidence) J. Liu, C.-J. Zhao, and W.-qiang Lu, “Molecular dynamics simulation on the corrosion characteristics of iron in liquid lead,” Annals of Nuclear Energy. 2018. link Times cited: 11 USED (low confidence) Q. Guo and G. Thompson, “Evolution of in situ growth stresses and interfacial structure in phase changing Cu/V multilayered thin films,” Acta Materialia. 2018. link Times cited: 6 USED (low confidence) T. Frolov, Q. Zhu, T. Oppelstrup, J. Marian, and R. Rudd, “Structures and transitions in bcc tungsten grain boundaries and their role in the absorption of point defects,” Acta Materialia. 2018. link Times cited: 48 USED (low confidence) J. Li, Q. Fang, B. Liu, and Y. Liu, “Transformation induced softening and plasticity in high entropy alloys,” Acta Materialia. 2018. link Times cited: 155 USED (low confidence) V. Borysiuk, V. Mochalin, and Y. Gogotsi, “Bending Rigidity of Two-Dimensional Titanium Carbide (MXene) Nanoribbons: A Molecular Dynamics Study,” Computational Materials Science. 2018. link Times cited: 114 USED (low confidence) L. Lu et al., “Molecular dynamics simulation of effects of interface imperfections and modulation periods on Cu/Ta multilayers,” Computational Materials Science. 2018. link Times cited: 37 USED (low confidence) X. W. Zhou, R. Jones, and K. Chu, “Polymorphic improvement of Stillinger-Weber potential for InGaN,” Journal of Applied Physics. 2017. link Times cited: 4 Abstract: A Stillinger-Weber potential is computationally very efficie… read moreAbstract: A Stillinger-Weber potential is computationally very efficient for molecular dynamics simulations. Despite its simple mathematical form, the Stillinger-Weber potential can be easily parameterized to ensure that crystal structures with tetrahedral bond angles (e.g., diamond-cubic, zinc-blende, and wurtzite) are stable and have the lowest energy. As a result, the Stillinger-Weber potential has been widely used to study a variety of semiconductor elements and alloys. When studying an A-B binary system, however, the Stillinger-Weber potential is associated with two major drawbacks. First, it significantly overestimates the elastic constants of elements A and B, limiting its use for systems involving both compounds and elements (e.g., an A/AB multilayer). Second, it prescribes equal energy for zinc-blende and wurtzite crystals, limiting its use for compounds with large stacking fault energies. Here, we utilize the polymorphic potential style recently implemented in LAMMPS to develop a modified Stillinger-Weber potential for InGaN that overcomes these two problems.A Stillinger-Weber potential is computationally very efficient for molecular dynamics simulations. Despite its simple mathematical form, the Stillinger-Weber potential can be easily parameterized to ensure that crystal structures with tetrahedral bond angles (e.g., diamond-cubic, zinc-blende, and wurtzite) are stable and have the lowest energy. As a result, the Stillinger-Weber potential has been widely used to study a variety of semiconductor elements and alloys. When studying an A-B binary system, however, the Stillinger-Weber potential is associated with two major drawbacks. First, it significantly overestimates the elastic constants of elements A and B, limiting its use for systems involving both compounds and elements (e.g., an A/AB multilayer). Second, it prescribes equal energy for zinc-blende and wurtzite crystals, limiting its use for compounds with large stacking fault energies. Here, we utilize the polymorphic potential style recently implemented in LAMMPS to develop a modified Stillinger-Weber... read less USED (low confidence) F. Xu, F. Fang, and X. Zhang, “Hard particle effect on surface generation in nano-cutting,” Applied Surface Science. 2017. link Times cited: 34 USED (low confidence) J.-S. Kim, D. Seol, J. Ji, H.-S. Jang, Y. Kim, and B.-J. Lee, “Second nearest-neighbor modified embedded-atom method interatomic potentials for the Pt-M (M = Al, Co, Cu, Mo, Ni, Ti, V) binary systems,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2017. link Times cited: 31 USED (low confidence) Y. Wang, Q. Li, C. Li, G. Shu, B. Xu, and W. Liu, “Dislocation core structures of tungsten with dilute solute hydrogen,” Journal of Nuclear Materials. 2017. link Times cited: 3 USED (low confidence) T. Frolov et al., “Grain boundary phases in bcc metals.,” Nanoscale. 2017. link Times cited: 52 Abstract: We report a computational discovery of novel grain boundary … read moreAbstract: We report a computational discovery of novel grain boundary structures and multiple grain boundary phases in elemental body-centered cubic (bcc) metals represented by tungsten, tantalum and molybdenum. While grain boundary structures created by the γ-surface method as a union of two perfect half crystals have been studied extensively, it is known that the method has limitations and does not always predict the correct ground states. Herein, we use a newly developed computational tool, based on evolutionary algorithms, to perform a grand-canonical search of high-angle symmetric tilt and twist boundaries, and we find new ground states and multiple phases that cannot be described using the conventional structural unit model. We use molecular dynamics (MD) simulations to demonstrate that the new structures can coexist at finite temperature in a closed system, confirming that these are examples of different grain boundary phases. The new ground state is confirmed by first-principles calculations. read less USED (low confidence) A. Hassani, A. Makan, K. Sbiaai, A. Tabyaoui, and A. Hasnaoui, “Incidence energy effect and impact assessment during homoepitaxial growth of nickel on (001), (111) and (110) surfaces,” Thin Solid Films. 2017. link Times cited: 21 USED (low confidence) Z. Zhang et al., “Ultrahigh hardness on a face-centered cubic metal,” Applied Surface Science. 2017. link Times cited: 39 USED (low confidence) M. Kozłowski, D. Scopece, J. Janczak-Rusch, L. Jeurgens, R. Abdank-Kozubski, and D. Passerone, “Validation of an Embedded-Atom Copper Classical Potential via Bulk and Nanostructure Simulations,” Diffusion Foundations. 2017. link Times cited: 0 Abstract: The validation of classical potentials for describing multic… read moreAbstract: The validation of classical potentials for describing multicomponent materials in complex geometries and their high temperature structural modifications (disordering and melting) requires to verify both a faithful description of the individual phases and a convincing scheme for the mixed interactions, like it is the case of the embedded atom scheme. The present paper addresses the former task for an embedded atom potential for copper, namely the widely adopted parametrization by Zhou, through application to bulk, surface and nanocluster systems. It is found that the melting point is underestimated by 200 degrees with respect to experiment, but structural and temperature-dependent properties are otherwise faithfully reproduced. read less USED (low confidence) M. An, Q. Deng, Y. Li, H. Y. Song, M. Su, and J. Cai, “Molecular dynamics study of tension-compression asymmetry of nanocrystal α-Ti with stacking fault,” Materials & Design. 2017. link Times cited: 32 USED (low confidence) 肖绪洋 and 陈润平, “Study of Structure and Melting for (AgCo) 561 Nanoclusters during Heating Process,” Material Sciences. 2017. link Times cited: 0 Abstract: 本文采用分子动力学方法模拟Ag及其掺杂团簇升温过程,通过平均原子势能曲线、团簇快照图分析团簇的结构和熔化行为。研究发现A… read moreAbstract: 本文采用分子动力学方法模拟Ag及其掺杂团簇升温过程,通过平均原子势能曲线、团簇快照图分析团簇的结构和熔化行为。研究发现Ag团簇在熔化前出现二十面体结构转变,转变温度随团簇尺寸减小而降低。在(AgCo)561掺杂团簇中,Co原子的数量和位置对团簇结构和性质有重要作用:掺杂Co原子促进(AgCo)561团簇的二十面体结构转变,引起转变温度降低,且掺杂原子数越多二十面体结构转变温度越低;中心掺杂团簇的二十面体结构转变温度最高,而外层掺杂可诱导出无序态异常结构,导致团簇升温无二十面体结构转变;掺杂对团簇熔点有影响,掺杂导致团簇熔点降低,但在中心位置掺杂55个Co原子引起熔点升高。 In this work, the Ag and doping clusters during the heating processes were studied using molecular dynamics simulations. Based on the potential-temperature curves, the snapshots are obtained and the structural transitions and melting behavior are analyzed. Our results show an icosahedral structure change during the heating processes of Ag clusters and the temperature of structure change is decreased with the size reduction of clusters. For doping Ag clusters, we found that the structure and property were influenced by the number and doping position of Co atoms in (AgCo)561 cluster. The structure change temperature of icosahedral is decreased with the increasing Co atoms and increased with center position doping by Co atoms. For a special case, there is an irregular structure by doping Co atoms in outer layer of clusters and the icosahedral structure change is disappear in this cluster. And the melting temperature is decreased by doping Co atoms, but the higher melting temperature is obtained in Co55Ag506 by Co center doping. read less USED (low confidence) J. Gruber, X. W. Zhou, R. Jones, S. R. Lee, and G. Tucker, “Molecular dynamics studies of defect formation during heteroepitaxial growth of InGaN alloys on (0001) GaN surfaces.,” Journal of applied physics. 2017. link Times cited: 25 Abstract: We investigate the formation of extended defects during mole… read moreAbstract: We investigate the formation of extended defects during molecular-dynamics (MD) simulations of GaN and InGaN growth on (0001) and ([Formula: see text]) wurtzite-GaN surfaces. The simulated growths are conducted on an atypically large scale by sequentially injecting nearly a million individual vapor-phase atoms towards a fixed GaN surface; we apply time-and-position-dependent boundary constraints that vary the ensemble treatments of the vapor-phase, the near-surface solid-phase, and the bulk-like regions of the growing layer. The simulations employ newly optimized Stillinger-Weber In-Ga-N-system potentials, wherein multiple binary and ternary structures are included in the underlying density-functional-theory training sets, allowing improved treatment of In-Ga-related atomic interactions. To examine the effect of growth conditions, we study a matrix of >30 different MD-growth simulations for a range of In x Ga 1-x N-alloy compositions (0 ≤ x ≤ 0.4) and homologous growth temperatures [0.50 ≤ T/T*m (x) ≤ 0.90], where T*m (x) is the simulated melting point. Growths conducted on polar (0001) GaN substrates exhibit the formation of various extended defects including stacking faults/polymorphism, associated domain boundaries, surface roughness, dislocations, and voids. In contrast, selected growths conducted on semi-polar ([Formula: see text]) GaN, where the wurtzite-phase stacking sequence is revealed at the surface, exhibit the formation of far fewer stacking faults. We discuss variations in the defect formation with the MD growth conditions, and we compare the resulting simulated films to existing experimental observations in InGaN/GaN. While the palette of defects observed by MD closely resembles those observed in the past experiments, further work is needed to achieve truly predictive large-scale simulations of InGaN/GaN crystal growth using MD methodologies. read less USED (low confidence) X. Dong and Y. Shin, “Multi-scale modeling of thermal conductivity of SiC-reinforced aluminum metal matrix composite,” Journal of Composite Materials. 2017. link Times cited: 9 Abstract: High thermal conductivity is one important factor in the sel… read moreAbstract: High thermal conductivity is one important factor in the selection or development of ceramics or composite materials. Predicting the thermal conductivity would be useful to the design and application of such materials. In this paper, a multi-scale model is developed to predict the effective thermal conductivity in SiC particle-reinforced aluminum metal matrix composite. A coupled two-temperature molecular dynamics model is used to calculate the thermal conductivity of the Al/SiC interface. The electronic effects on the interfacial thermal conductivity are studied. A homogenized finite element model with embedded thin interfacial elements is used to predict the properties of bulk materials, considering the microstructure. The effects of temperatures, SiC particle sizes, and volume fractions on the thermal conductivity are also studied. A good agreement is found between prediction results and experimental measurements. The successful prediction of thermal conductivity could help a better understanding and an improvement of thermal transport within composites and ceramics. read less USED (low confidence) S. Bukkuru, U. Bhardwaj, M. Warrier, A. Rao, and M. C. Valsakumar, “Identifying self-interstitials of bcc and fcc crystals in molecular dynamics,” Journal of Nuclear Materials. 2017. link Times cited: 6 USED (low confidence) M. An, Q. Deng, M. Su, H. Y. Song, and Y. L. Li, “Dependence of deformation mechanisms on layer spacing in multilayered Ti/Al composite,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2017. link Times cited: 20 USED (low confidence) P. Brault et al., “Molecular dynamics simulations of ternary PtxPdyAuz fuel cell nanocatalyst growth,” International Journal of Hydrogen Energy. 2016. link Times cited: 8 USED (low confidence) M. Jafary-Zadeh, R. Tavakoli, D. Srolovitz, and Y.-W. Zhang, “Thermally induced failure mechanism transition and its correlation with short-range order evolution in metallic glasses,” Extreme Mechanics Letters. 2016. link Times cited: 23 USED (low confidence) M. Tavakol, M. Mahnama, and R. Naghdabadi, “Shock wave sintering of Al/SiC metal matrix nano-composites: A molecular dynamics study,” Computational Materials Science. 2016. link Times cited: 28 USED (low confidence) H. Aboulfadl, I. Gallino, R. Busch, and F. Mücklich, “Atomic scale analysis of phase formation and diffusion kinetics in Ag/Al multilayer thin films,” Journal of Applied Physics. 2016. link Times cited: 16 Abstract: Thin films generally exhibit unusual kinetics leading to che… read moreAbstract: Thin films generally exhibit unusual kinetics leading to chemical reactions far from equilibrium conditions. Binary metallic multilayer thin films with miscible elements show some similar behaviors with respect to interdiffusion and phase formation mechanisms. Interfacial density, lattice defects, internal stresses, layer morphologies and deposition conditions strongly control the mass transport between the individual layers. In the present work, Ag/Al multilayer thin films are used as a simple model system, in which the effects of the sputtering power and the bilayer period thickness on the interdiffusion and film reactions are investigated. Multilayers deposited by DC magnetron sputtering undergo calorimetric and microstructural analyses. In particular, atom probe tomography is extensively used to provide quantitative information on concentration gradients, grain boundary segregations, and reaction mechanisms. The magnitude of interdiffusion was found to be inversely proportional to the period thickness... read less USED (low confidence) T.-E. Fan, T. Liu, J.-W. Zheng, G. Shao, and Y. Wen, “Structure and stability of Fe-Pt bimetallic nanoparticles: Initial structure, composition and shape effects,” Journal of Alloys and Compounds. 2016. link Times cited: 12 USED (low confidence) G. Anand, R. Goodall, and C. Freeman, “Role of configurational entropy in body-centred cubic or face-centred cubic phase formation in high entropy alloys,” Scripta Materialia. 2016. link Times cited: 56 USED (low confidence) S. Bukkuru, U. Bhardwaj, A. Rao, and M. Warrier, “Identifying Interstitials in MD Simulations — Max Space Clustering Method,” Journal of Physics: Conference Series. 2016. link Times cited: 1 Abstract: Molecular Dynamics (MD) simulations are suitable to study th… read moreAbstract: Molecular Dynamics (MD) simulations are suitable to study the details of atomistic processes. Identification of interstitials is one of the main difficulties in the analysis of defect dynamics. To identify the interstitials, different methods use different criteria. All the methods, except the Wigner-Seitz method, assume some input parameters which may lead to an erroneous number of interstitials. We describe an unsupervised clustering algorithm, called Max Space clustering method, to identify interstitials without assuming any input parameters. We show that the algorithm is suited for identifying interstitials at high temperatures when amplitude of atomic oscillations become larger, a fact ignored by other models. read less USED (low confidence) T. Li, J. Li, L. Wang, Y. Duan, and H. Li, “Coalescence of Immiscible Liquid Metal Drop on Graphene,” Scientific Reports. 2016. link Times cited: 34 USED (low confidence) X. Zhou, T. Kaub, R. Martens, and G. Thompson, “Influence of Fe(Cr) miscibility on thin film grain size and stress,” Thin Solid Films. 2016. link Times cited: 17 USED (low confidence) P. Zhu and F. Fang, “Study of the minimum depth of material removal in nanoscale mechanical machining of single crystalline copper,” Computational Materials Science. 2016. link Times cited: 27 USED (low confidence) S. M. Rassoulinejad-Mousavi, Y. Mao, and Y. Zhang, “Evaluation of Copper, Aluminum and Nickel Interatomic Potentials on Predicting the Elastic Properties,” arXiv: Computational Physics. 2016. link Times cited: 63 Abstract: Choice of appropriate force field is one of the main concern… read moreAbstract: Choice of appropriate force field is one of the main concerns of any atomistic simulation that needs to be seriously considered in order to yield reliable results. Since, investigations on mechanical behavior of materials at micro/nanoscale has been becoming much more widespread, it is necessary to determine an adequate potential which accurately models the interaction of the atoms for desired applications. In this framework, reliability of multiple embedded atom method based interatomic potentials for predicting the elastic properties was investigated. Assessments were carried out for different copper, aluminum and nickel interatomic potentials at room temperature which is considered as the most applicable case. Examined force fields for the three species were taken from online repositories of National Institute of Standards and Technology (NIST), as well as the Sandia National Laboratories, the LAMMPS database. Using molecular dynamic simulations, the three independent elastic constants, C11, C12 and C44 were found for Cu, Al and Ni cubic single crystals. Voigt-Reuss-Hill approximation was then implemented to convert elastic constants of the single crystals into isotropic polycrystalline elastic moduli including Bulk, Shear and Young's modulus as well as Poisson's ratio. Simulation results from massive molecular dynamic were compared with available experimental data in the literature to justify the robustness of each potential for each species. Eventually, accurate interatomic potentials have been recommended for finding each of the elastic properties of the pure species. Exactitude of the elastic properties was found to be sensitive to the choice of the force fields. Those potentials were fitted for a specific compound may not necessarily work accurately for all the existing pure species. read less USED (low confidence) B. Ma, Q. Rao, and Y. He, “Molecular dynamics simulation of temperature effect on tensile mechanical properties of single crystal tungsten nanowire,” Computational Materials Science. 2016. link Times cited: 33 USED (low confidence) V. V. Pogorelko and A. Mayer, “Influence of titanium and magnesium nanoinclusions on the strength of aluminum at high-rate tension: Molecular dynamics simulations,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2016. link Times cited: 27 USED (low confidence) Y. Wang, C. Li, B. Xu, and W. Liu, “Hydrogen‐Induced Core Structures Change of Screw and Edge Dislocations in Tungsten.” 2016. link Times cited: 0 USED (low confidence) T. Fu, X. Peng, C. Feng, Y. Zhao, and Z. Wang, “MD simulation of growth of Pd on Cu (1 1 1) and Cu on Pd (1 1 1) substrates,” Applied Surface Science. 2015. link Times cited: 19 USED (low confidence) A. Mayer, P. N. Mayer, V. Krasnikov, and D. S. Voronin, “Multi-scale model of the dynamic fracture of molten and solid metals,” Journal of Physics: Conference Series. 2015. link Times cited: 5 Abstract: A multi-scale model of the tensile fracture of metal melts i… read moreAbstract: A multi-scale model of the tensile fracture of metal melts is developed based on a combination of molecular dynamics (MD) simulations and continuum description of kinetics and dynamics of voids; the model considerably extends the time and length scales of MD. Nucleation of voids due to thermal fluctuations is taken into account. Growth of a void in melts is well described by the Rayleigh-Plesset equation, while in the case of a solid metal we propose a dislocation-based model of the void growth. Based on the MD simulations, we investigate the nucleation rates in the uniform monocrystalline metals and metal melts, dynamics of pre-existing voids and compare them with the continuum model (equations of nucleation and growth). Using of the literature data on the surface tension and viscosity of melts allows us to get a correspondence between the continuum description and MD. With the use of the model, we calculated the strength of the uniform melts of Al, Cu, Ni, Pb, Fe and Ti within a wide range of strain rates (from 103-104 to 109-1011 s-1) and temperatures (from melting temperature to 70-80% of critical temperature). Calculations show that the tensile strength of homogeneous melts decreases slowly with the strain rate decrease. As a result, within the range of strain rates of 106-108 s-1, a homogeneous nucleation mode can be realized, in which the dynamic strength of a melt can be comparable to, or even higher than the strength of a solid metal. read less USED (low confidence) V. Krasnikov and A. Mayer, “Plasticity driven growth of nanovoids and strength of aluminum at high rate tension: Molecular dynamics simulations and continuum modeling,” International Journal of Plasticity. 2015. link Times cited: 62 USED (low confidence) S. Maiti and W. Steurer, “Structural-disorder and its effect on the mechanical properties in single-phase TaNbHfZr high-entropy alloys,” arXiv: Materials Science. 2015. link Times cited: 224 USED (low confidence) V. Turlo, O. Politano, and F. Baras, “Dissolution process at solid/liquid interface in nanometric metallic multilayers: Molecular dynamics simulations versus diffusion modeling,” Acta Materialia. 2015. link Times cited: 40 USED (low confidence) X.-Y. Li et al., “Wettability and Coalescence of Cu Droplets Subjected to Two-Wall Confinement,” Scientific Reports. 2015. link Times cited: 16 USED (low confidence) M. M. Sichani and D. Spearot, “A molecular dynamics study of the role of grain size and orientation on compression of nanocrystalline Cu during shock,” Computational Materials Science. 2015. link Times cited: 25 USED (low confidence) E. S. Wise, M. Liu, and T. Miller, “Sputtering of cubic metal crystals by low-energy xenon-ions,” Computational Materials Science. 2015. link Times cited: 5 USED (low confidence) X. Zhou, W. Wu, Y. He, Y. Li, L. Wang, and H. Li, “Theoretical study of the pathway to heterogeneous nucleation of liquid copper on the groove substrate with different wedge angles.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 4 Abstract: Heterogeneous nucleation is of significance in controlling t… read moreAbstract: Heterogeneous nucleation is of significance in controlling the crystal growth, but the key route is still limited. Simulations are performed to explore the microscopic details of how the wedge substrate spreads its structural information to a growing crystal and further affects the solidification process. The simulation results show that, owing to the induced effect from the substrate, the copper atoms become layered at the liquid-solid interface in a "V"-shaped pattern and tend to form a twin crystal. The structural information delivery of the substrate decays with the distance away from the substrate, and the final solidified structure would gradually recover its inherent structure. Interestingly, the wedge angle of 90° seems to be an exception at which the solidified structure exhibits a perfect crystal due to the nearly perfect match with the Cu atoms. Moreover, the cooling rate and the atomic structure of the substrate are also found to have striking correlations with the final structures. These research results are favorable for a better understanding of the inherent relation between the solidified structure and the substrate. read less USED (low confidence) C. Huang, Q. Wang, and Z. Rao, “Thermal conductivity prediction of copper hollow nanowire,” International Journal of Thermal Sciences. 2015. link Times cited: 16 USED (low confidence) W. Wu, L. Zhang, H. Ren, K. Zhang, H. Li, and Y. He, “Synergy and pinning effects in a monatomic liquid film in confined conditions.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 4 Abstract: The freezing behavior of a monatomic liquid film in confined… read moreAbstract: The freezing behavior of a monatomic liquid film in confined conditions during rapid cooling was studied by molecular dynamics simulations. We illustrated the synergy and pinning effects of the local icosahedral order during freezing. Our results show that the icosahedron contributes to nucleation through the synergy with other short-range ordered structures and participates in crystal growth via assimilation, but the pinning effect should be overcome when crystals grow. Furthermore, a semi-ordered morphology with maze-like nano-patterns emerged due to the cooperation between the synergy effect and the pinning effect. Our findings shed light on the correlation between the local icosahedral order and the crystalline medium-range order, providing a better understanding of the rapid solidification. read less USED (low confidence) L. Meng, L. Wang, S. Wang, and Y. Qi, “Relating glass formation to eutectic composition in Cu100-xZrx alloy by molecular dynamics simulation,” Physics and Chemistry of Liquids. 2015. link Times cited: 1 Abstract: The relationship between eutectic compositions and glass-for… read moreAbstract: The relationship between eutectic compositions and glass-forming ability (GFA) in Cu100-xZrx (x = 8–83) system is studied by molecular dynamic (MD) simulation based on the embedded atom method (EAM). Local maxima of GFA occur in Cu56Zr44, Cu47Zr53 and Cu31Zr69 alloys, close to eutectic points in Cu-Zr binary phase diagram. The structural parameters indicate that the heterogeneous Cu-Zr pairs exhibit stronger and more stable interaction than that of homogeneous pairs. The small peak with a negative amplitude ahead of the main peak in SCuZr(Q) also implies the preference of Cu-Zr pairs. The avoidance of Cu-Cu neighbours and Zr-Zr neighbours, which correspond to the prepeak prior to the main peak of SCuCu(Q) and SZrZr(Q), leads to structural order on intermediate length scales. Perfect and defect icosahedra constitute medium range order (MRO) clusters by sharing atoms, which promotes the glass formation. read less USED (low confidence) I. Ikhtiar, “Temperature dependence of magneto-transport properties in Co 2 Fe(Ga 0.5 Ge 0.5 )/Cu lateral spin valves,” The Japan Society of Applied Physics. 2015. link Times cited: 9 Abstract: The non-local spin signals of Co2Fe(Ga0.5Ge0.5)/Cu lateral s… read moreAbstract: The non-local spin signals of Co2Fe(Ga0.5Ge0.5)/Cu lateral spin valves with sub-micron size dimensions were measured with varying temperatures. The non-local spin signal reaches 54 mΩ at 4 K, while it degrades down to 13 mΩ at room temperature. Analysis based on the one-dimensional spin diffusion model clarifies the dominant source for degrading of the spin signal is suppression of the spin diffusion length in Cu, not the spin polarization, indicating Co2Fe(Ga0.5Ge0.5) keeps half-metallic nature even at room temperature. The temperature dependence of non-local spin signal was found to exhibit a downturn at 36 K. The presence of magnetic impurities, detrimental effect of which becomes more pronounced for diffusive transport in long Cu wires, is suggested to cause the observed downturn in non-local spin signals. read less USED (low confidence) M. Werner, A. Crossley, and C. Johnston, “Characterization of Nanostructured Materials.” 2015. link Times cited: 2 USED (low confidence) W. Dednam et al., “Modeling contact formation between atomic-sized gold tips via molecular dynamics,” Journal of Physics: Conference Series. 2015. link Times cited: 7 Abstract: The formation and rupture of atomic-sized contacts is modell… read moreAbstract: The formation and rupture of atomic-sized contacts is modelled by means of molecular dynamics simulations. Such nano-contacts are realized in scanning tunnelling microscope and mechanically controlled break junction experiments. These instruments routinely measure the conductance across the nano-sized electrodes as they are brought into contact and separated, permitting conductance traces to be recorded that are plots of conductance versus the distance between the electrodes. One interesting feature of the conductance traces is that for some metals and geometric configurations a jump in the value of the conductance is observed right before contact between the electrodes, a phenomenon known as jump-to-contact. This paper considers, from a computational point of view, the dynamics of contact between two gold nano-electrodes. Repeated indentation of the two surfaces on each other is performed in two crystallographic orientations of face-centred cubic gold, namely (001) and (111). Ultimately, the intention is to identify the structures at the atomic level at the moment of first contact between the surfaces, since the value of the conductance is related to the minimum cross-section in the contact region. Conductance values obtained in this way are determined using first principles electronic transport calculations, with atomic configurations taken from the molecular dynamics simulations serving as input structures. read less USED (low confidence) L. Sun, D. Shi, C. Guo, X. Xiao, and J. Bai, “A Molecular Dynamics Study on the Physical Properties of (CuPd)147 Alloy Nanoparticles,” Applied Mechanics and Materials. 2015. link Times cited: 3 Abstract: The melting of CuPd bimetallic clusters was studied by using… read moreAbstract: The melting of CuPd bimetallic clusters was studied by using molecular dynamics with the embedded-atom method. The results show that the same number of Cu atoms with Pd clusters, their temperature curve of clusters and the melting point value can be induced by the process of Cu atomic segregation to appear big differences. According to the analysis of HA index and clusters snapshot image, the singular phenomenon is caused by hierarchical differences between Cu atomic distances from the inner surface segregation. It is concluded that the difference of the melting point of Cu-Pd bimetallic clusters provides an effective method of controllable preparation. read less USED (low confidence) M. Moody et al., “Atomically resolved tomography to directly inform simulations for structure–property relationships,” Nature Communications. 2014. link Times cited: 55 USED (low confidence) L. Wan, X.-xiang Yu, and G. Thompson, “Phase stability and in situ growth stresses in Ti/Nb thin films,” Acta Materialia. 2014. link Times cited: 15 USED (low confidence) J. Guo, G. Zhang, Y. Huang, W. Ming, M. Liu, and H. Huang, “Investigation of the removing process of cathode material in micro-EDM using an atomistic-continuum model,” Applied Surface Science. 2014. link Times cited: 23 USED (low confidence) Z. Pan, L. Kecskes, and Q. Wei, “The nature behind the preferentially embrittling effect of impurities on the ductility of tungsten,” Computational Materials Science. 2014. link Times cited: 20 USED (low confidence) C. A. Huang, C. Chen, C. C. Chen, T. Kelly, and H. Lin, “Microstructure analysis of a Cr–Ni multilayer pulse-electroplated in a bath containing trivalent chromium and divalent nickel ions,” Surface & Coatings Technology. 2014. link Times cited: 9 USED (low confidence) S.-W. Ren, J. Sun, and Y. Hao, “Analysis of structures and melting properties of the 19-atom Ni–Co clusters,” International Journal of Modern Physics B. 2014. link Times cited: 6 Abstract: In this paper, by using the classical molecular dynamics met… read moreAbstract: In this paper, by using the classical molecular dynamics method and the GEAM potential, the geometric structure and the melting properties of the 19-atom Ni–Co clusters with different compositions are studied. It is found that all the clusters have the double icosahedron structures although some of the structures are slightly deformed. With the increase of the temperature, a pre-melting phenomenon is observed. The pre-melting temperatures of the pure cobalt and nickel clusters are very close. But on the whole, the pre-melting temperature decreases with the increase of the number of the nickel atom for the mixed clusters. The effects of the substitution atoms on the melting temperature of the clusters are similar to that on the pre-melting temperature although there are some oscillations in the decrease process. The mechanism of these findings are also investigated and analyzed. read less USED (low confidence) I. Shin and E. Carter, “Simulations of dislocation mobility in magnesium from first principles,” International Journal of Plasticity. 2014. link Times cited: 47 USED (low confidence) Y. Sun, Y. Sun, P. Yang, and H. Li, “Cluster distribution entropy in Ti50Cu50 and Ti50Cu45Ni5 metallic glasses,” Journal of Non-crystalline Solids. 2014. link Times cited: 2 USED (low confidence) P. Wen, G. Tao, and P. Zhou, “Molecular Dynamics Simulation of Two-Phase Structures of Copper Formed by Laser Grooving,” Applied Mechanics and Materials. 2014. link Times cited: 0 Abstract: It is a concerned problem that what influences of cooling ra… read moreAbstract: It is a concerned problem that what influences of cooling rate on evolution of microstructures of the treated and untreated Cu are during solidification process after Cu’s being melted by laser grooving. Based on the embedded-atom method, molecular dynamics simulations have been performed on the cooling processes of liquid metal Cu by using crystal-liquid configuration method at six different cooling rates. Through the two-phase structural features, pair correlation function, average atomic energy and mean square displacement analysis, it is found that the cooling rate plays a critical role on the solidification process to the liquid Cu. The liquid Cu becomes its crystalline state during relatively low cooling rate, while the glass transition of the liquid Cu is performed with the relatively high cooling rate. The solidification process of the liquid Cu has effect on the solid crystal Cu and leads to form the indented interface between two-phase structures. read less USED (low confidence) L. Hale, J. Zimmerman, and C. Weinberger, “Simulations of bcc tantalum screw dislocations: Why classical inter-atomic potentials predict 1 1 2 slip,” Computational Materials Science. 2014. link Times cited: 27 USED (low confidence) T. Troev, N. Nankov, and T. Yoshiie, “Simulation of defects in fusion plasma first wall materials.” 2014. link Times cited: 2 Abstract: Numerical calculations of radiation damages in beryllium, al… read moreAbstract: Numerical calculations of radiation damages in beryllium, alpha-iron and tungsten irradiated by fusion neutrons were performed using molecular dynamics (MD) simulations. The displacement cascades efficiency has been calculated using the Norgett-Robinson-Torrens (NRT) formula, the universal pair-potential of Ziegler-Biersack-Littmark (ZBL) and the EAM inter-atomic potential. The pair potential overestimates the defects production by a factor of 2. The ZBL pair potential results and the EAM are comparable at higher primary knock-on atom (PKA) energies (E > 100 keV). We found that the most common types of defects are single vacancies, di-vacancies, interstitials and small number of interstitial clusters. On the bases of calculated results, the behavior of vacancies, empty nano-voids and nano-voids with hydrogen and helium were discussed. read less USED (low confidence) L. Xie, P. Brault, J. Bauchire, A. Thomann, and L. Bedra, “Molecular dynamics simulations of clusters and thin film growth in the context of plasma sputtering deposition,” Journal of Physics D: Applied Physics. 2014. link Times cited: 40 Abstract: Carrying out molecular dynamics (MD) simulations is a releva… read moreAbstract: Carrying out molecular dynamics (MD) simulations is a relevant way to understand growth phenomena at the atomic scale. Initial conditions are defined for reproducing deposition conditions of plasma sputtering experiments. Two case studies are developed to highlight the implementation of MD simulations in the context of plasma sputtering deposition: ZrxCu1−x metallic glass and AlCoCrCuFeNi high entropy alloy thin films deposited onto silicon. Effects of depositing atom kinetic energies and atomic composition are studied in order to predict the evolution of morphologies and atomic structure of MD grown thin films. Experimental and simulated x-ray diffraction patterns are compared. read less USED (low confidence) B. Liu and Q. Hsu, “Study on bonding phenomena of high strength aluminium alloy in nano-scale analysis,” Materials Research Innovations. 2014. link Times cited: 3 Abstract: The bonding properties of three kinds of multi-alloy materia… read moreAbstract: The bonding properties of three kinds of multi-alloy materials Al0·9Cu0·1, Al0·9Cu0·035Mg0·065 and Al0·9Cu0·03Mg0·06Fe0·01 in nano-scale are studied. The molecular dynamics method and the embedded atom potential were used to simulate the bonding properties in nano-scale under the assumption of the single crystal structure of face centred cubic and the motion of each atom in the system dominated by Newton's laws of motion. The total energy of the simulated system includes the molecular potential energy and embedded atom potential. The aluminium alloy is divided into upper and lower blocks while the former moves downward to the latter maintained steady during simulation. Simulation comprises three stages: compressing, holding and stretching. In the first two simulation stages, simulation results depict that in stretching stage the multilayer slips occur along (111) surfaces, and the final plastic deformation leads to strain hardening and the fracture of ductile shear. read less USED (low confidence) J. Sun, “The Structures and Melting Properties of the Al-W Mixed Clusters,” Applied Mechanics and Materials. 2014. link Times cited: 0 Abstract: Through the classical molecular dynamics, the melting proper… read moreAbstract: Through the classical molecular dynamics, the melting properties of the the tungsten and aluminum mixed clusters are explored. By using simulated annealing the structure of the mixed cluster is obtained. It is found that the Al atoms tend to locate on the surface of the clusters. The melting behaviors of the clusters are analyzed in detail by the atom-resolved Lindemann index. The curves of Lindemann index show that the melting process of the can experience three steps which is not common in the melting process of the nanoclusters. read less USED (low confidence) X. Xiao, R. Chen, and H. Huang, “Influence of Ag Atoms on Structural Evolution of Pd-Ag Bimetallic Clusters during Heating Processes,” Advanced Materials Research. 2014. link Times cited: 0 Abstract: In bimetallic clusters, the component and position of doping… read moreAbstract: In bimetallic clusters, the component and position of doping atoms can influence on structures and properties. It may induce irregular physical and chemical properties by tuning the doping atoms. In this study, (PdAg)309 clusters with different contents and positions of doping Ag atoms have been investigated by molecular dynamics simulation based on the embedded atom method. It is found that the structure of (PdAg)309 clusters transformed from truncated octahedron to icosahedron, and melting in heating processes from 200 K to 1500 K. Results indicated that the temperature of structural transformation and melting are strongly related to the number and position of doping Ag atoms. The melting point decreased with the increased Ag atoms, but there is irregular by doping Ag atoms at different position. The structural transformed temperature from truncated octahedron to icosahedron decreased with the increasing Ag atoms and the outer doping position. This means that the effect of doping Ag atoms can be used to tune the special structures of bimetallic clusters. read less USED (low confidence) H. Huang, Z. Cheng, X. Xiao, J. H. Xia, and T. Liu, “The Structure Transition of (AgCu)309 Clusters during the Freezing Process: A Molecular Dynamics Simulation,” Applied Mechanics and Materials. 2014. link Times cited: 0 Abstract: In bimetallic cluster, research on the frozen structure with… read moreAbstract: In bimetallic cluster, research on the frozen structure with the changing concentration plays an important role in exploring new structural materials. This paper studies the freezing processes of (AgCu)309 clusters with different Ag concentrations. The results indicated that the structural transformation was strongly related to concentration. It was found that the frozen structures were changed form icosahedron, hcp and fcc-hcp with the change of Ag concentration. The frozen structures were formed icosahedral for the clusters with Ag concentration at 10%, 20%, 30%, and the pure Ag309. For the clusters with Ag content at 40%, 50%, 60%, 70%, and 80%, the frozen structures were formed defect icosahedral. It was also found that the frozen structure have hcp character for the pure Cu309 cluster. Meanwhile, the frozen structure of (AgCu)309 with 90% Ag concentration was formed fcc-hcp structure. The segregation effects of the Ag-Cu are the key reason for the structural transformation. read less USED (low confidence) X. Yang and A. Hassanein, “Atomic scale calculations of tungsten surface binding energy and beryllium-induced tungsten sputtering,” Applied Surface Science. 2014. link Times cited: 26 USED (low confidence) E. Wimmer et al., “Atomistic Simulations as Part of an Integrated Computational Environment for Structural Design.” 2014. link Times cited: 0 Abstract: Atomistic simulations add a new and fundamental dimension to… read moreAbstract: Atomistic simulations add a new and fundamental dimension to structural design by integrating development, optimization, and life-time predictions of materials with the well established macroscopic simulations based on classical theories. This integration has now become possible due to the availability of advanced computational approaches and atomistic simulation software environments such as MedeA combined with ready access to massive compute power. The current capabilities will be illustrated by specific examples including the quantitative prediction of structural and elastic properties as a function of temperature for materials ranging from metallic alloys to thermoset polymers; the precipitation of new phases either for precipitation hardening or as undesired phenomenon leading to embrittlement, for example in Ni-Cr alloys; the effect of alloying elements and impurities on microstructures due to modifications in the strength of grain boundaries; the stress-strain behavior of amorphous boron; the bonding strength of interfaces; and the prediction of transport coefficients including diffusion and thermal conductivity. This contribution will conclude with an analysis of the issues related to the integration of these quantitative atomistic capabilities in the structural design process and it will outline a path forward. read less USED (low confidence) X. Xiao, D. Shi, J. Xia, and Z. Cheng, “INFLUENCE OF INITIAL TEMPERATURES ON THE COOLING OF Ag–Pd BIMETALLIC CLUSTERS VIA MOLECULAR DYNAMICS SIMULATION,” NANO. 2013. link Times cited: 1 Abstract: Atomic segregation in bimetallic clusters can influence the … read moreAbstract: Atomic segregation in bimetallic clusters can influence the surface nucleation and also the structures of clusters. It is important to study the effect of atomic segregation on the structure. In this study, initial cooling temperatures were used to tune the atomic segregation ability. Molecular dynamics simulation with an embedded atom method was used to study the relationship between the structure and atomic segregation. It was found that the higher the initial cooling temperature, the more obvious the Ag atomic segregation. When the clusters cooled down from 800 K and 600 K, the clusters formed a mixed icosahedron due to the weak atomic segregation ability. When the initial cooling temperature is higher than 1200 K, all the Ag atoms segregated to the surface layer, the clusters formed a Pd–Ag-core–shell chemical ordering. For 1200 K initial temperature, the structure is decahedral. The clusters formed an fcc structure when the clusters cooled down from 2000 K, 1800 K, and 1500 K. When the clusters cooled down from 860 K and 900 K, not all the Ag atoms segregated to the surface layer, the clusters formed a core–shell chemical ordering with a mixed Pd–Ag core. But the structure of 860 K is twinned of icosahedron and decahedron and that of 900 K is decahedral. This means that the chemical orderings and structures were influenced by the atomic segregation. read less USED (low confidence) K. Suzuki, T. Sano, and H. Miura, “Effect of Alloying Elements on Creep and Fatigue Damage of Ni-Base Superalloy Caused by Strain-Induced Anisotropic Diffusion.” 2013. link Times cited: 0 Abstract: In order to make clear the mechanism of the directional coar… read moreAbstract: In order to make clear the mechanism of the directional coarsening (rafting) of γ′ phases in Ni-base superalloys under uni-axial tensile strain, molecular dynamics (MD) analysis was applied to investigate effects of alloying elements on diffusion characteristics around the interface between the γ phase and the γ′ phase. In this study, a simple interface structure model corresponding to the γ/γ′ interface, which consisted of Ni as γ and Ni3Al as γ′ structure, was used to analyze the diffusion properties of Ni and Al atoms under tensile strain. The strain-induced anisotropic diffusion of Al atoms perpendicular to the interface between the Ni(001) layer and the Ni3Al(001) layer was observed in the MD simulation, suggesting that the strain-induced anisotropic diffusion of Al atoms in γ′ phase is one of the dominant factors of the kinetics of the rafting during creep damage. The effect of alloying elements in the Ni-base superalloy on the strain-induced anisotropic diffusion of Al atoms was also analyzed. Both the atomic radius and the binding energy with Al and Ni of the alloying element are the dominant factors that change the strain-induced diffusion of Al atoms in the Ni-base super-alloy.Copyright © 2013 by ASME read less USED (low confidence) 黄维 and 梁工英, “Ag-Cu共晶合金非晶转变及晶化的分子动力学模拟 A Molecular Dynamics Study on Amorphous Formation and Crystallization of Ag-Cu Eutectic Alloys,” Applied physics. 2013. link Times cited: 0 Abstract: 本文采用分子动力学方法研究了Ag-Cu共晶合金的结晶过程。依靠体系内能变化、公共近邻分析和原子可视化技术对Ag-Cu共晶… read moreAbstract: 本文采用分子动力学方法研究了Ag-Cu共晶合金的结晶过程。依靠体系内能变化、公共近邻分析和原子可视化技术对Ag-Cu共晶合金的结构演变进行了分析。模拟结果表明,在10%~40%Cu范围内,随着Cu含量的增加,合金形成非晶结构的临界冷却速度减小,玻璃化转变温度增加。公共近邻分析的结果表明,在非晶晶化前的阶段,表征非晶结构的1551、1541和1431键对占据主要部分,但也存在少量的bcc和fcc晶核团簇。其中,表征正二十面体团簇的1551键对,随着Cu含量的增加而增加,表征缺陷二十面体团簇的1541和1431键对则相应减少,这表明,随着Cu含量的增加非晶体系的稳定性增加。与此同时,表征晶体相结构的键对随Cu含量的增加都有所减少,其中,bcc晶核的数量减少较小,但fcc晶核的数量却有较大的下降,从而使非晶晶化的形核核心减少,这也从结构上解释了非晶合金的玻璃化转变温度随成分增加而增加的现象。在非晶晶化以后,虽然体系以fcc结构为主,但仍存在一些无序的非晶态结构,这些结构存在于共晶两相界面,随Cu含量的增加而增加。利用可视化技术,我们可以发现,随着Cu含量的增加,Ag-Cu合金的共晶组织存在从固溶体形、网状共晶到层状共晶的演变。 The crystallization of Ag-Cu alloys was studied by molecular dynamics method (MD) with embedded atom potential (EAM). The structural developments of Ag-Cu alloys were analyzed based on the variations of internal energy, common neighbor analysis (CNA), and atomic visualization technique. The simulation results showed that with the increase in Cu composition, the critical cooling rate of amorphous formation decreased and the glass-transition temperature of amorphous increased under the same heating rate. The results of CNA showed that the amorphous structure is main, and a few crystal clusters (bcc and fcc) are in it. Among the amorphous bond pairs, the pairs 1551 increases with the increase in Cu composition, which is the character of regular icosahedrons cluster. Meanwhile, the pairs 1541 and 1431, which are the character of defect icosahedrons clusters, reduce correspondingly. These show that the stability of amorphous increases. With the increase in Cu composition, the pairs 1441, 1661 and 1421 are all reduced, the 1441 and 1661 decrease little and the 1421 decreases great, which implies that the nuclei reduce during the crystallization of amorphous and the glass-transition temperature increases. After crystallization, the fcc structure is dominant but there are a few defect icosahedrons clusters between the eutectic boundaries. Moreover, the eutectic structure of Ag-Cu alloys can be transformed from solid solution, net-like into the lamellar morphologies with composition during the solidification and crystallization. read less USED (low confidence) A. Ruffini, J. Durinck, J. Colin, C. Coupeau, and J. Grilhé, “Buckling-induced dislocation emission in thin films on substrates,” International Journal of Solids and Structures. 2013. link Times cited: 9 USED (low confidence) R. Marchal, A. Genest, S. Krüger, and N. Rösch, “Structure of Pd/Au Alloy Nanoparticles from a Density Functional Theory-Based Embedded-Atom Potential,” Journal of Physical Chemistry C. 2013. link Times cited: 23 Abstract: On the basis of DFT results for a set of representative bulk… read moreAbstract: On the basis of DFT results for a set of representative bulk, surface, and cluster systems, we determined a new embedded-atom potential for Pd/Au alloys. This embedded-atom approach accurately reproduces DFT properties of such alloy systems. We applied this potential in Monte Carlo simulations to study the effects of temperature and composition on the structure and bonding of Pd/Au alloy nanoparticles up to 5 nm in diameter. We characterized the structure of those particles by evaluating the gold concentration at the surface and in the interior, the coordination numbers of atoms, the nature of Pd entities at the surface, and the number of suggested active sites for vinyl-acetate formation. read less USED (low confidence) A. Fraile, S. Cuesta-López, R. Iglesias, A. Caro, and J. Perlado, “Atomistic molecular point of view for liquid lead and lithium in Nuclear Fusion technology,” Journal of Nuclear Materials. 2013. link Times cited: 16 USED (low confidence) A. M. Igoshkin, I. Golovnev, and V. Fomin, “Molecular dynamics analysis of the substrate temperature effect on thermomechanical characteristics of vapor deposited nanofilms,” Physical Mesomechanics. 2013. link Times cited: 1 USED (low confidence) T. Fang, L. Wang, and Y. Qi, “Molecular dynamics simulation of crystal growth of undercooled liquid Co,” Physica B-condensed Matter. 2013. link Times cited: 9 USED (low confidence) 黄维 and 梁工英, “金属银凝固与非晶晶化过程的分子动力学模拟 A Study on Solidification and Amorphous Crystallization of Metal Ag by Molecular Dynamics Simulation,” Applied physics. 2013. link Times cited: 0 Abstract: 文章采用分子动力学方法,以金属银为对象,模拟了面心立方金属的凝固和非晶化过程,借助于体系的内能变化、径向分布函数(RDF… read moreAbstract: 文章采用分子动力学方法,以金属银为对象,模拟了面心立方金属的凝固和非晶化过程,借助于体系的内能变化、径向分布函数(RDF)、公共近邻分析(CNA)、原子可视化技术对凝固和晶化形核过程中的结构演变细节进行了描述。模拟结果表明,液态金属中就存在11% bcc结构的短程有序集团,随着液态金属不断过冷,bcc集团逐渐增加,在凝固前达到最高的31%,在非晶形成时,bcc集团大约有24%。当最近邻原子距离相同时,bcc结构单位原子所占体积大于fcc结构,有利于在液态金属和非晶中存在。这些bcc结构作为结晶时的非自发形核的核心,与周围的无序原子一起迅速转变为fcc结构和少量的hcp结构。在过冷液体和非晶系统中并没有发现fcc和hcp的结构,fcc结构为凝固和非晶晶化的很短周期中迅速转变而成。 The melting and solidification of Ag was simulated by molecular dynamics method. The structural transfor- mation of Ag during the metal solidification and amorphous crystallization was analyzed based on the variations of the internal energy, radial distribution function (RDF), common neighbor analysis (CNA), and atomic visualization technique. The simulation results showed that the embryonic crystals similar to body-centered cubic (bcc-like) structure (about 11%) already exist in the liquid metal, the content of bcc increases with cooling and it is up to 31% near the solidifying. About 24% bcc structure has in the amorphous structure. The volume per atom of bcc is larger than that of fcc, which is beneficial in the cooling liquid metal and amorphous structure. In the nucleation process, bcc-like embryonic crystals can be used as the nucleus with disorder atoms near the bcc to transform fcc crystal. There is no fcc and hcp structure in the cooling melt and amorphous structure. They are formed in the solidification directly. read less USED (low confidence) A. Ruffini, J. Durinck, J. Colin, C. Coupeau, and J. Grilhé, “Interface step-induced thin-film delamination and buckling,” Acta Materialia. 2013. link Times cited: 12 USED (low confidence) Y. He, H. Li, Y. Li, K. Zhang, Y. Jiang, and X. Bian, “Atomic insight into copper nanostructures nucleation on bending graphene.,” Physical chemistry chemical physics : PCCP. 2013. link Times cited: 12 Abstract: Some findings in heterogeneous nucleation that the structura… read moreAbstract: Some findings in heterogeneous nucleation that the structural features of a growing crystal are usually inherited from the heterogeneous nucleus, although attracting more and more attention, are not yet well understood. Here we report numerical simulations of copper nucleation on bending graphene (BG) to explore the microscopic details of how the curved surface influences the freezing structure of the liquid metal. The simulation result clearly shows that copper atoms become layered at the solid-liquid interface in a "C"-shaped pattern resembling the BG. This kind of shape control decays with increasing distance from the wall and the outmost layers transform into twin crystal composed of two fcc wedges. It is found that the final structures have striking correlations with the curvature radius, central angle and arc length of the BG. Our study would provide an opportunity for comprehensive and satisfactory understanding of the heterogeneous nucleation on curved surfaces. read less USED (low confidence) H. Tang, M. Bai, Y. Dou, Q. Ran, and G. Lo, “Computer simulations of laser-induced melting of aluminum,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2013. link Times cited: 4 USED (low confidence) J. H. Xia, Z. Cheng, and X. Xiao, “Molecular Dynamics Simulation of Structural Changes of Ag965 Clusters during Freezing,” Advanced Materials Research. 2013. link Times cited: 0 Abstract: The structural transitions of Ag965 clusters during two diff… read moreAbstract: The structural transitions of Ag965 clusters during two different quenching processes (Q1:1.0×1014 K/s, Q2: 1.0×1012 K/s) were studied using molecular dynamics simulations. This work gives the structure properties including the variations with temperature of pair-correlation function, bond-angle distribution function, bond pairs and bond orientational order parameters in both rapid quenching processes. Our results indicated that the liquid Ag965 was frozen into amorphous structure at 100 K under the quenching condition Q1. While the liquid Ag965 transformed to hexagonal close-packed (hcp) phase at the temperature 100 K under the quenching condition Q2.These instructions give you basic guidelines for preparing papers for conference proceedings. read less USED (low confidence) K. Blobaum et al., “Characterization of Blistering and Delamination in Depleted Uranium Hohlraums,” Fusion Science and Technology. 2013. link Times cited: 6 Abstract: Blistering and delamination are the primary failure mechanis… read moreAbstract: Blistering and delamination are the primary failure mechanisms during the processing of depleted uranium (DU) hohlraums. These hohlraums consist of a sputter-deposited DU layer sandwiched between two sputter-deposited layers of gold; a final thick gold layer is electrodeposited on the exterior. The hohlraum is deposited on a copper-coated aluminum mandrel; the Al and Cu are removed with chemical etching after the gold and DU layers are deposited. After the mandrel is removed, blistering and delamination are observed on the interiors of some hohlraums, particularly at the radius region. It is hypothesized that blisters are caused by pinholes in the copper and gold layers; etchant leaking through these holes reaches the DU layer and causes it to oxidize, resulting in a blister. Depending on the residual stress in the deposited layers, blistering can initiate larger-scale delamination at layer interfaces. Scanning electron microscopy indicates that inhomogeneities in the machined aluminum mandrel are replicated in the sputter-deposited copper layer. Furthermore, the Cu layer exhibits columnar growth with pinholes that likely allow etchant to come in contact with the gold layer. Any inhomogeneities or pinholes in this initial gold layer then become nucleation sites for blistering. Using a focused ion beam system to etch through the gold layer and extract a cross-sectional sample for transmission electron microscopy, amorphous, intermixed layers at the gold/DU interfaces are observed. Nanometer-sized bubbles in the sputtered and electrodeposited gold layers are also present. Characterization of the morphology and composition of the deposited layers is the first step in determining modifications to processing parameters, with the goal of attaining a significant improvement in hohlraum yield. read less USED (low confidence) K. Zhang, H. Li, L. Li, and X. Bian, “Why does the second peak of pair correlation functions split in quasi-two-dimensional disordered films?,” Applied Physics Letters. 2013. link Times cited: 20 Abstract: Molecular dynamics simulation has been performed to study th… read moreAbstract: Molecular dynamics simulation has been performed to study the splitting of the second peak in pair correlation functions of quasi-two-dimensional disordered film. A quasi-two-dimensional inhomogeneous structural model, which contains both crystal-like and disordered regions, supports the hypothesis that the splitting of the second peak is result of a statistical average of crystal-like and disordered structural regions in the system, not just the amorphous structure. The second-peak splitting can be viewed as a prototype of the crystal-like peak exhibiting distorted and vestigial features. read less USED (low confidence) O. Politano, F. Baras, A. Mukasyan, S. Vadchenko, and A. Rogachev, “Microstructure development during NiAl intermetallic synthesis in reactive Ni–Al nanolayers: Numerical investigations vs. TEM observations,” Surface & Coatings Technology. 2013. link Times cited: 43 USED (low confidence) V. Bocchetti and H. Diep, “Monte Carlo simulation of melting and lattice relaxation of the (111) surface of silver,” Surface Science. 2013. link Times cited: 13 USED (low confidence) P. White, “Molecular dynamic modelling of fatigue crack growth in aluminium using LEFM boundary conditions,” International Journal of Fatigue. 2012. link Times cited: 27 USED (low confidence) Z. Wang, H. Chen, and L. Zhang, “Atomistic simulation of self-diffusion and interfacial diffusion of liquid lead,” Journal of Non-crystalline Solids. 2012. link Times cited: 9 USED (low confidence) G. Li, Q. Wang, Y. Cao, K. Wang, J. Du, and J. He, “Icosahedral-Decahedral Transformation in the (PdAg) 309 Cluster Induced by Ag Atomic Segregation,” Cmc-computers Materials & Continua. 2012. link Times cited: 3 Abstract: This paper studies the influence of Ag atomic segregation on… read moreAbstract: This paper studies the influence of Ag atomic segregation on the structural evolutions of the mixed (PdAg)309clusters during the heating processes by using molecular dynamics with a general embedded atom method. The results show that the Ag atomic segregation makes the cluster exhibit a segregate-melting stage in which the energy does not monotonic increase with the increase of temperature. In this stage, the cluster first transforms to form a disorder structure from the initial icosahedron and then a decahedron. By comparing with the cases in the pure Pd309, Ag309, and core-shell (PdAg)309, it is found that the icosahedral-decahedral transformation is induced by the Ag atomic segregation. read less USED (low confidence) S. D. Chen, Y. Zhou, and A. Soh, “Molecular dynamics simulations of mechanical properties for Cu(0 0 1)/Ni(0 0 1) twist boundaries,” Computational Materials Science. 2012. link Times cited: 25 USED (low confidence) A. Ruffini, J. Durinck, J. Colin, C. Coupeau, and J. Grilhé, “Effects of sliding on interface delamination during thin film buckling,” Scripta Materialia. 2012. link Times cited: 15 USED (low confidence) S.-J. Lin, C. Wu, T. Fang, and L.-M. Kuo, “Effects of layered structure, composition, and annealing on nanoformed Au–Cu rods using molecular dynamics simulation,” Computational Materials Science. 2012. link Times cited: 7 USED (low confidence) D. Cereceda et al., “Assessment of interatomic potentials for atomistic analysis of static and dynamic properties of screw dislocations in W,” Journal of Physics: Condensed Matter. 2012. link Times cited: 50 Abstract: Screw dislocations in bcc metals display non-planar cores at… read moreAbstract: Screw dislocations in bcc metals display non-planar cores at zero temperature which result in high lattice friction and thermally-activated strain rate behavior. In bcc W, electronic structure molecular statics calculations reveal a compact, non-degenerate core with an associated Peierls stress between 1.7 and 2.8 GPa. However, a full picture of the dynamic behavior of dislocations can only be gained by using more efficient atomistic simulations based on semiempirical interatomic potentials. In this paper we assess the suitability of five different potentials in terms of static properties relevant to screw dislocations in pure W. Moreover, we perform molecular dynamics simulations of stress-assisted glide using all five potentials to study the dynamic behavior of screw dislocations under shear stress. Dislocations are seen to display thermally-activated motion in most of the applied stress range, with a gradual transition to a viscous damping regime at high stresses. We find that one potential predicts a core transformation from compact to dissociated at finite temperature that affects the energetics of kink-pair production and impacts the mechanism of motion. We conclude that a modified embedded-atom potential achieves the best compromise in terms of static and dynamic screw dislocation properties, although at an expense of about ten-fold compared to central potentials. read less USED (low confidence) X. Xiao, Z. Cheng, and J. H. Xia, “MOLECULAR DYNAMICS SIMULATION STUDY OF ATOMIC SEGREGATION OF (PdPt)147 DURING THE HEATING PROCESS,” Modern Physics Letters B. 2012. link Times cited: 3 Abstract: Research on the influence of alloy concentration and distrib… read moreAbstract: Research on the influence of alloy concentration and distribution on bimetallic cluster plays a key role in exploring new structural material. This paper studies the melting process of icosahedral bimetallic cluster (PdPt)147 with different Pt concentrations and different atomic distributions by using molecular dynamics with an embedded atom method. The results indicate that the mixed Pd–Pt cluster shows an irregular phenomenon between 580 and 630 K, i.e. the atomic energy decreases with the increase of temperature. This is because the surface energy of Pd is lower than that of Pt; the decreased energy due to Pd atomic segregation is larger than the increased energy due to heating during the segregation process. In addition, the temperature of Pd atomic segregation is strongly related to Pt concentration. This leads to that Pd atoms prefer to remain on the surface even after the cluster melted. read less USED (low confidence) E. Pastukhov, N. I. Sidorov, A. A. Vostrjakov, and V. Chentsov, “Molecular Dynamic Simulation of Short Order and Hydrogen Diffusion in the Disordered Metal Systems.” 2012. link Times cited: 0 Abstract: Main concepts of Hydrogen permeability (HP) mechanism for th… read moreAbstract: Main concepts of Hydrogen permeability (HP) mechanism for the pure crystal metals are already stated. There are well-founded theoretical models and numerous experimental researches. As far as disordered systems (in which Hydrogen solubility is much more, than in crystal samples) are concerned, such works appear to be comparatively recent and rare. Particularly, they are devoted to Hydrogen interaction of with amorphous structures. Deficiency of similar researches is caused by thermo-temporal instability of amorphous materials structure and properties. read less USED (low confidence) E. Pastukhov, A. A. Vostrjakov, N. I. Sidorov, and V. Chentsov, “Hydrogen and Electric Field Effect on Iron Impurities Removal from Molten Zirconium,” Defect and Diffusion Forum. 2012. link Times cited: 2 Abstract: Hydrogen and iron diffusion coefficients in molten zirconium… read moreAbstract: Hydrogen and iron diffusion coefficients in molten zirconium have been calculated using a molecular dynamics (МD) model. The molecular dynamics method using micro-canonical (NVT) ensemble was used to analyze iron and zirconium diffusion coefficient dependence on electric field intensity and the presence of hydrogen in the zirconium melt. Results obtained are compared to the literature data on impurity removal in plasma-arc zirconium melting in the presence of hydrogen as well as in electron-beam and vacuum-arc melting. The limiting stage of iron removal from the melt is established. The contribution of the electric field to iron removal is estimated. We carried out systematization of the DHMe data for Zr, Nb, and Ta. An Arrhenius equation analysis for DHMe and its extrapolation to the premelting zone taking into account the Gorsky effect was carried out too. The analysis enabled the estimation of DHMe for the temperature interval where experiment encounters difficulties. read less USED (low confidence) A. Ruffini, J. Durinck, J. Colin, C. Coupeau, and J. Grilhé, “Gliding at interface during thin film buckling: A coupled atomistic/elastic approach,” Acta Materialia. 2012. link Times cited: 16 USED (low confidence) A. Takeuchi and A. Inoue, “Molecular Dynamics Simulations Based on Plastic Crystal Model with Introducing United Atom Scheme Demonstrated for Zr2Ni Metallic Glass,” Materials Science Forum. 2012. link Times cited: 0 Abstract: Molecular dynamics (MD) simulations were performed for a Zr2… read moreAbstract: Molecular dynamics (MD) simulations were performed for a Zr2Ni alloy by referring to crystallographic features of a metastable Zr2Ni phase. Simulation method was identical to our previous studies named plastic crystal model (PCM), which includes crystallographic operations for an intermetallic compound in terms of the random rotations of hypothetical clusters around their center of gravity and subsequent annealing at a low temperature. On the basis of MD-PCM, the present study considers an additional refinement named united atom scheme (UAS) on the motions of atoms in the hypothetical clusters. In MD-PCM-UAS, Dreiding potential was assigned for atomic bonds in a cluster whereas Generalized Embedded Atom Method potential for the other atomic pairs. The simulation results by MD-PCM-UAS yield a liquid-like structure. However, annealing did not cause subsequent structural relaxation, which differs from the results by MD-PCM and conventional MD simulations. Further simulations based on MD-PCM-UAS were performed for a nanostructure comprising clusters and glue atoms, leading to the best fit with the experimental data. read less USED (low confidence) J. Li and Z.-qiang Han, “Calculation of Melting Curve of Aluminum under Pressure through Molecular Dynamics Simulations,” Advanced Materials Research. 2011. link Times cited: 0 Abstract: The melting curve is an important thermodynamic property in … read moreAbstract: The melting curve is an important thermodynamic property in studies of solid-liquid phase transitions. It can be calculated via molecular dynamics simulations. We simulated the melting process of pure Al with three methods, the heat-until-it-melts (HUM) method, the two-phase method and the hysteresis method. The results calculated via HUM method is approximately 20% higher than experiment data while the results calculated via two-phase method and hysteresis method are in good agreement with experiment data. read less USED (low confidence) C. Soontrapa and Y. Chen, “Molecular dynamics potentials in magnetite (Fe3O4) modeling,” Computational Materials Science. 2011. link Times cited: 14 USED (low confidence) Y. H. Park and I. Hijazi, “Simple analytic embedded atom potential for FCC materials,” International Journal of Microstructure and Materials Properties. 2011. link Times cited: 5 Abstract: A simple empirical embedded-atom potential that includes a l… read moreAbstract: A simple empirical embedded-atom potential that includes a long range force is developed for FCC metals. The potential parameters of this model are determined by fitting lattice constant, three elastic constants, cohesive energy, and vacancy formation energy using an optimisation technique (Arora, 2007). Parameters for Cu, Ag, Au, Al, Ni, Pd and Pt have been obtained. The obtained parameters are used to calculate bulk modulus, divacancy formation energy, and melting point. The predicted values are in good agreement with experimental results. The predicted total energy as a function of lattice parameter is also in good agreement with the equation of state of Rose et al. read less USED (low confidence) S.-J. Lin, C. Wu, T. Fang, and L.-M. Kuo, “Effects of forging temperature and velocity on nano-forming process using molecular dynamics simulation,” Computational Materials Science. 2011. link Times cited: 16 USED (low confidence) X. W. Zhou, F. Doty, and P. Yang, “Atomistic simulation study of atomic size effects on B1 (NaCl), B2 (CsCl), and B3 (zinc-blende) crystal stability of binary ionic compounds,” Computational Materials Science. 2011. link Times cited: 15 USED (low confidence) X. Jing, Z. Liu, H. Wei, and K. Yao, “The influences of the local impact site and incident energy on the transport behaviors of single copper atom onto Cu (0 0 1) surface,” Applied Surface Science. 2011. link Times cited: 3 USED (low confidence) D. Larson, T. Prosa, B. Geiser, and W. Egelhoff, “Effect of analysis direction on the measurement of interfacial mixing in thin metal layers with atom probe tomography.,” Ultramicroscopy. 2011. link Times cited: 39 USED (low confidence) M. Kodzuka, T. Ohkubo, and K. Hono, “Laser assisted atom probe analysis of thin film on insulating substrate.,” Ultramicroscopy. 2011. link Times cited: 6 USED (low confidence) G. Li, Q. Wang, Y. Cao, J. Du, and J. He, “Size and composition dependence of the frozen structures in Co-based bimetallic clusters.” 2011. link Times cited: 7 Abstract: This Letter studies the size-dependent freezing of Co, Co–Ni… read moreAbstract: This Letter studies the size-dependent freezing of Co, Co–Ni, and Co–Cu clusters by using molecular dynamics with embedded atom method. Size effect occurs in these three types of clusters. The clusters with large sizes always freeze to form their bulk-like structures. However, the frozen structures for small sizes are generally related to their compositions. The icosahedral clusters are formed for Co clusters (for ⩽ 3.2 nm diameter) and also for Co–Ni clusters but at a larger size range (for ⩽ 4.08 nm ). Upon the Co–Cu clusters, decahedral structure is obtained for small size (for 2.47 nm). The released energy induced the structural transformation plays a key role in the frozen structures. These results indicate that the preformed clusters with special structures can be tuned by controlling their compositions and sizes. read less USED (low confidence) T. Nagase, K. Takizawa, and Y. Umakoshi, “Electron-irradiation-induced solid-state amorphization in supersaturated Ni–Zr solid solutions,” Intermetallics. 2011. link Times cited: 9 USED (low confidence) E. A. Pastukhov, N. I. Sidorov, A. A. Vostrjakov, and V. Chentsov, “Diffusion of Hydrogen in Amorphous Ni-Zr Alloys,” Defect and Diffusion Forum. 2011. link Times cited: 0 Abstract: Amorphous alloys Ni64Zr36 and Ni36Zr64 structure and hydroge… read moreAbstract: Amorphous alloys Ni64Zr36 and Ni36Zr64 structure and hydrogen mobility are researched by the molecular dynamics method. The analysis of structure factors and partial distribution functions of atoms revealed hydrogen affects the short order parameters of the disordered systems. Diffusion coefficients of hydrogen are shown to depend on its concentration. read less USED (low confidence) T. Troev, N. Nankov, and T. Yoshiie, “Simulation of displacement cascades in tungsten irradiated by fusion neutrons,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2011. link Times cited: 42 USED (low confidence) J. Fan, “Applications of Atomistic Simulation in Ceramics and Metals.” 2010. link Times cited: 0 USED (low confidence) M. Hu, K. Giapis, J. Goicochea, and D. Poulikakos, “Surface segregation of bimetallic alloys in nanoscale confinement,” Applied Physics Letters. 2010. link Times cited: 9 Abstract: The surface segregation of Pt atoms in liquid bimetallic all… read moreAbstract: The surface segregation of Pt atoms in liquid bimetallic alloys confined in carbon nanotube cavities was studied using molecular dynamics simulations. Considerable enrichment in the Pt-atom surface density was found to occur in Pt alloys, when the complementary metal has surface energy higher than Pt and simultaneously metal-wall interaction strength lower than that of Pt with the confining wall. The results suggest that solidification of liquid binary alloys in nanochannels could produce core-shell nanorods with the shell enriched in one of the components for catalytic and other applications. read less USED (low confidence) C. Tourek and S. Sundararajan, “Atom Scale Characterization of the Near Apex Region of an Atomic Force Microscope Tip,” Microscopy and Microanalysis. 2010. link Times cited: 3 Abstract: Three-dimensional atom probe tomography (APT) is successfull… read moreAbstract: Three-dimensional atom probe tomography (APT) is successfully used to analyze the near-apex regions of an atomic force microscope (AFM) tip. Atom scale material structure and chemistry from APT analysis for standard silicon AFM tips and silicon AFM tips coated with a thin film of Cu is presented. Comparison of the thin film data with that observed using transmission electron microscopy indicates that APT can be reliably used to investigate the material structure and chemistry of the apex of an AFM tip at near atomic scales. read less USED (low confidence) G. Schmitz, “Nanoanalysis by Atom Probe Tomography,” Nanotechnology. 2010. link Times cited: 5 Abstract: Atom probe tomography is a most advanced microscopic analysi… read moreAbstract: Atom probe tomography is a most advanced microscopic analysis technique based on the principle of field ion microscopy. Atoms are individually desorbed from the specimen, identified and localized. From the measured data, the three-dimensional atomic structure of nanometer-sized specimens is reconstructed numerically. Significant improvements in the total volume of analysis, in the range of materials that can be investigated, and in specimen preparation have recently broadened the range of possible applications of this method decisively. In this chapter, the physical basis of this exciting nanoanalytical tool is presented in some detail, in order that the reader can appreciate the advantages and limitations of the method. The design of actual instruments is described, and the physics of field desorption and volume reconstruction algorithms are discussed. Application of the method to relevant problems of material physics in nanotechnology is illustrated with case studies.
Keywords:
atom probe tomography;
field evaporation;
3D-volume reconstruction;
chemical mapping;
thin-film reaction;
magnetic sensors;
interfaces;
grain boundaries read less USED (low confidence) M. Wakeda and Y. Shibutani, “Icosahedral clustering with medium-range order and local elastic properties of amorphous metals,” Acta Materialia. 2010. link Times cited: 104 USED (low confidence) A. Zarefy et al., “Interface characterization of a [Pt/Co)] 3 /IrMn multilayer by laser-assisted tomographic atom probe,” Journal of Magnetism and Magnetic Materials. 2010. link Times cited: 2 USED (low confidence) M. Yuasa, T. Nakazawa, and M. Mabuchi, “Atomic simulation of grain boundary sliding in Co/Cu two-phase bicrystals,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2010. link Times cited: 10 USED (low confidence) T. Nagase, K. Takizawa, M. Wakeda, Y. Shibutani, and Y. Umakoshi, “Electron-irradiation-induced solid-state amorphization caused by thermal relaxation of lattice defects,” Intermetallics. 2010. link Times cited: 9 USED (low confidence) E. A. Pastukhov, A. A. Vostrjakov, N. I. Sidorov, and V. Chentsov, “Molecular Dynamics Calculation of Hydrogen and Iron Diffusion in Molten Tantalum under an Electric Field,” Defect and Diffusion Forum. 2010. link Times cited: 5 Abstract: The molecular dynamics (MD) method for analysis of the elect… read moreAbstract: The molecular dynamics (MD) method for analysis of the electric field intensity affecting iron impurities removal from tantalum in the presence of hydrogen is used. The radial distribution functions and diffusivities of hydrogen and iron atoms in a tantalum melt at 3400K under an electric field and without are obtained. An electric field imposed on liquid tantalum with an iron impurity increases the tantalum diffusivity more considerably than for iron atoms. Hydrogen introduction into the MD - cell without an electric field gives a much lower increase of the tantalum diffusivity, but a more considerable increase for iron. Simultaneous imposing an electric field and hydrogen introduction into the MD – cell keeps the tantalum and iron diffusivities at the level of the effect of an electric field. Thus the imposition of an electric field is a main parameter of the increase of the iron diffusivity. read less USED (low confidence) A. Takeuchi, K. Yubuta, A. Yavari, and A. Inoue, “Zr60Al15(Ni,Cu)25 noncrystalline alloys created by referring to ionic arrangements of a garnet structure with molecular dynamics simulations based on a plastic crystal model,” Intermetallics. 2010. link Times cited: 0 USED (low confidence) J. Pang, M. Tan, and K. M. Liew, “Computational Study of Tensile Deformation of a Constrained Nanoscale Metallic Glass,” International Journal of Modern Physics B. 2010. link Times cited: 1 Abstract: In this study a nanometer-sized metallic glass (nano-MG) Ti5… read moreAbstract: In this study a nanometer-sized metallic glass (nano-MG) Ti50Cu50 was generated with constrained atoms at both ends and was extended until fracture under a tensile load by molecular dynamics simulation using the general embedded-atom model (GEAM) potential. Totally different mechanical behavior was observed in the nano-MG, such as strain hardening and necking, both of which have been discovered in a few real and simulated MGs and can be related to the generation of shear transformation zones (STZs). A dramatic drop in Young's modulus was found due to the surface effect. Such effect results from the large fraction of surface atoms which have a different surrounding configuration from bulk atoms. At fracture the nano-MG breaks by atomic separation as reported in metal nanowires. The fracture strain is as large as about 120%, indicating that nano-MGs are intrinsically ductile. read less USED (low confidence) J. Pang, M. Tan, A. Jarfors, and P. D. Chuang, “Glass Formation and Structural Study of Ti50Cu50 Alloy by Molecular Dynamics,” Materials Science Forum. 2010. link Times cited: 2 Abstract: Ti-based metallic glasses (MGs) due to their relative low de… read moreAbstract: Ti-based metallic glasses (MGs) due to their relative low densities exhibit ultrahigh specific characteristics. In this article the glass-forming behavior and atomic structure of Ti50Cu50 MG were investigated through molecular dynamics simulation (MDS) using the general embedded-atom method (GEAM) potential. As observed experimentally, simulated Ti50Cu50 alloy undergoes three states on quenching: (i) equilibrium liquid; (ii) supercooled liquid and (iii) glassy solid. The atomic configuration of the glass was analysed based on the radial distribution function (RDF) and Voronoi tessellation (VT). It was found that there exist a variety of polyhedral units in Ti50Cu50 MG, where distorted icosohedral and bcc clusters are dominant. read less USED (low confidence) G. Li, Q. Wang, D. Li, X. Lu, and J. He, “Size and composition effects on the melting of bimetallic Cu–Ni clusters studied via molecular dynamics simulation,” Materials Chemistry and Physics. 2009. link Times cited: 46 USED (low confidence) N. Das, K. Suzuki, K. Ogawa, and T. Shoji, “Early stage SCC initiation analysis of fcc Fe―Cr―Ni ternary alloy at 288 °C: A quantum chemical molecular dynamics approach,” Corrosion Science. 2009. link Times cited: 60 USED (low confidence) Y. Shibuta, S. Takamoto, and T. Suzuki, “Dependence of the grain boundary energy on the alloy composition in the bcc iron–chromium alloy: A molecular dynamics study,” Computational Materials Science. 2009. link Times cited: 33 USED (low confidence) G. Ziegenhain, H. Urbassek, and A. Hartmaier, “Influence of crystal anisotropy on elastic deformation and onset of plasticity in nanoindentation – a simulational study,” Journal of Applied Physics. 2008. link Times cited: 133 Abstract: Using molecular-dynamics simulation we simulate nanoindentat… read moreAbstract: Using molecular-dynamics simulation we simulate nanoindentation into the three principal surfaces—the (100), (110), and (111) surface—of Cu and Al. In the elastic regime, the simulation data agree fairly well with the linear elastic theory of indentation into an elastically anisotropic substrate. With increasing indentation depth, the effect of pressure hardening becomes visible. When the critical stress for dislocation nucleation is reached, even the elastically isotropic Al shows a strong dependence of the force-displacement curves on the surface orientation. After the load drop, when plasticity has set in, the influence of the surface orientation is lost, and the contact pressure (hardness) becomes independent of the surface orientation. read less USED (low confidence) G. Li, Q. Wang, D. Li, X. Lu, and J. He, “Structure evolution during the cooling and coalesced cooling processes of Cu–Co bimetallic clusters,” Physics Letters A. 2008. link Times cited: 40 USED (low confidence) A. Upadhyay, N. Inogamov, B. Rethfeld, and H. Urbassek, “Ablation by ultrashort laser pulses: Atomistic and thermodynamic analysis of the processes at the ablation threshold,” Physical Review B. 2008. link Times cited: 88 Abstract: Ultrafast laser irradiation of solids may ablate material of… read moreAbstract: Ultrafast laser irradiation of solids may ablate material off the surface. We study this process for thin films using molecular-dynamics simulation and thermodynamic analysis. Both metals and Lennard-Jones (LJ) materials are studied. We find that despite the large difference in thermodynamical properties between these two classes of materials--e.g., for aluminum versus LJ the ratio T{sub c}/T{sub tr} of critical to triple-point temperature differs by more than a factor of 4--the values of the ablation threshold energy E{sub abl} normalized to the cohesion energy, {epsilon}{sub abl}=E{sub abl}/E{sub coh}, are surprisingly universal: all are near 0.3 with {+-}30% scattering. The difference in the ratio T{sub c}/T{sub tr} means that for metals the melting threshold {epsilon}{sub m} is low, {epsilon}{sub m} {epsilon}{sub abl}. This thermodynamical consideration gives a simple explanation for the difference between metals and LJ. It explains why despite the universality in {epsilon}{sub abl}, metals thermomechanically ablate always from the liquid state. This is opposite to LJ materials, which (near threshold) ablate from the solid state. Furthermore, we find that immediately below the ablation threshold, the formation of large voids (cavitation) in the irradiated material leads to a strong temporary expansion on amore » very slow time scale. This feature is easily distinguished from the acoustic oscillations governing the material response at smaller intensities, on the one hand, and the ablation occurring at larger intensities, on the other hand. This finding allows us to explain the puzzle of huge surface excursions found in experiments at near-threshold laser irradiation.« less read less USED (low confidence) S. Sankaranarayanan and S. Ramanathan, “Molecular dynamics simulation study of nanoscale passive oxide growth on Ni-Al alloy surfaces at low temperatures,” Physical Review B. 2008. link Times cited: 41 Abstract: Oxidation kinetics of Ni-Al (100) alloy surface is investiga… read moreAbstract: Oxidation kinetics of Ni-Al (100) alloy surface is investigated at low temperatures (300-600 K) and at different gas pressures using molecular dynamics (MD) simulations with dynamic charge transfer between atoms. Monte Carlo simulations employing the bond order simulation model are used to generate the surface segregated minimum energy initial alloy configurations for use in the MD simulations. In the simulated temperature-pressure-composition regime for Ni-Al alloys, we find that the oxide growth curves follow a logarithmic law beyond an initial transient regime. The oxidation rates for Ni-Al alloys were found to decrease with increasing Ni composition. Structure and dynamical correlations in the metal/oxide/gas environments are used to gain insights into the evolution and morphology of the growing oxide film. Oxidation of Ni-Al alloys is characterized by the absence of Ni-O bond formation. Oxide films formed on the various simulated metal surfaces are amorphous in nature and have a limiting thickness ranging from {approx}1.7 nm for pure Al to 1.1 nm for 15% Ni-Al surfaces. Oxide scale analysis indicates significant charge transfer as well as variation in the morphology and structure of the oxide film formed on pure Al and 5% Ni-Al alloy. For oxide scales thicker than 1 nm, the oxidemore » structure in case of pure Al exhibits a mixed tetrahedral (AlO{sub 4}{approx}37%) and octahedral (AlO{sub 6}{approx}19%) environment, whereas the oxide scale on Ni-Al alloy surface is almost entirely composed of tetrahedral environment (AlO{sub 4}{approx}60%) with very little AlO{sub 6} ( 100 ps) occurs in case of 5% Ni-Al alloy. The oxide growth on both pure Al and Ni-Al alloy surfaces occurs by inward anion and outward cation diffusions. The cation diffusion in both the cases is similar, whereas the anion diffusion in case of 5% Ni-Al is 25% lower than pure Al, thereby resulting in reduced self-limiting thickness of oxide scale on the alloy surface. The simulation findings agree well with previously reported experimental observations of oxidation on Ni-Al alloy surface.« less read less USED (low confidence) S.-G. Lee and Y.-C. Chung, “Atomic Investigation of Fe–Cu Magnetic Thin Films by Molecular Dynamics Simulation,” Japanese Journal of Applied Physics. 2007. link Times cited: 1 Abstract: The deposition behavior of an Fe–Cu magnetic metallic multil… read moreAbstract: The deposition behavior of an Fe–Cu magnetic metallic multilayer system was investigated at the atomic level using molecular dynamics simulation. It was found that a mixture confined to a single atomic layer at the Cu(001) surface was formed at room temperature. The Fe–Cu system shows mixing characteristics such as layer coverage function and mixing length, that are significantly different from those of the Fe–Al metallic system. The different intermixing behavior could be successfully explained in terms of cohesive energy and atomic size mismatch. read less USED (low confidence) S.-G. Lee and Y.-C. Chung, “Atomic-level investigation of Al and Ni thin film growth on Ni(1 1 1) surface: Molecular dynamics simulation,” Applied Surface Science. 2007. link Times cited: 19 USED (low confidence) M. Mariscal, E. Leiva, and S. A. Dassie, “Computer simulation of electrochemical nanostructuring induced by supersaturation conditions,” Journal of Electroanalytical Chemistry. 2007. link Times cited: 5 USED (low confidence) F. Dorfbauer, R. Evans, M. Kirschner, O. Chubykalo-Fesenko, R. Chantrell, and T. Schrefl, “Effects of surface anisotropy on the energy barrier in cobalt–silver core–shell nanoparticles,” Journal of Magnetism and Magnetic Materials. 2007. link Times cited: 13 USED (low confidence) K. Sugita, M. Matsumoto, M. Mizuno, H. Araki, and Y. Shirai, “Atomic structure and positron lifetime in the metallic glass Zr55Cu30Ni5Al10,” Physica Status Solidi (c). 2007. link Times cited: 1 Abstract: Zr-based metallic glasses have superior characteristics such… read moreAbstract: Zr-based metallic glasses have superior characteristics such as mechanical strength, corrosion resistance and precision casting ability. From positron lifetime measurements, a negative temperature dependence of the mean positron lifetime above room temperature was reported and ascribed to the presence of shallow traps. However, the trapping sites remain unknown under the present circumstances. To get a further understanding of the experimental positron lifetime value, the positron density distribution and the lifetime in the Zr-based metallic glass Zr55Cu30Ni5Al10 have been calculated. The calculation shows that the positrons are annihilated inhomogeneously and most positrons are annihilated preferentially around Cu/Al. These results indicate that the positron lifetime not exactly reflects the total free volume. (© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim) read less USED (low confidence) K. Sugita, M. Mizuno, H. Araki, and Y. Shirai, “Molecular Dynamics Study of Glass-Forming Ability of Zr-Based Metallic Glasses,” Materials Transactions. 2007. link Times cited: 3 Abstract: A new method to estimate the stability of supercooled liquid… read moreAbstract: A new method to estimate the stability of supercooled liquid based on the temperature dependence of the free volume fraction, which is obtained by a molecular dynamics simulation with no empirical data was proposed. The molecular dynamics simulations for some Zr-based metallic glasses, Zr55Cu30Ni5Al10 ,Z r 67Ni33 and Zr67Cu33 in Zr-Cu-Ni-Al system were performed. The features of the first peaks in calculated radial density functions well correspond to the experimental results by XRD and EXAFS spectroscope. The free volume fractions at 0 K in the quenched amorphous, Fquenched, and in ‘‘fully relaxed’’ supercooled liquid states, Frelaxed, are evaluated by the fitting of the temperature vs freevolume-fraction curve obtained by a molecular dynamics simulation. The calculated normalized free volume fraction Nfree, defined as Fquenched=Frelaxed, shows similar tendency with other experimental Trg. criterion. The temperature dependence of the free volume fraction in a supercooled liquid state governs the quenched free volume in the amorphous phase, which shows that the local structures in supercooled liquid or liquid phases are especially important for the glass formation. [doi:10.2320/matertrans.MF200608] read less USED (low confidence) T. Kumagai, D. Nikkuni, S. Hara, S. Izumi, and S. Sakai, “Development of Interatomic Potential for Zr-Ni Amorphous Systems,” Materials Transactions. 2007. link Times cited: 7 Abstract: This study develops a way of determining the interatomic pot… read moreAbstract: This study develops a way of determining the interatomic potential of Zr-Ni using an embedded atom method for binary systems that can reproduce the material properties of its amorphous states. In order to ensure the robustness of the developed interatomic potential, the potential energies and lattice constants of Zr crystals, Ni crystals, and Zr-Ni binary crystals that involve a wide range of local atomic environments are employed for fitting. The elastic properties of some such crystals are also employed. In addition, in order to reproduce Zr-Ni amorphous properties, the radial distribution function of Zr70Ni30 amorphous structures and the defect formation energies of Zr-Ni structures are employed. By fitting to a portion of the material properties that requires relatively little computation time, optimization using genetic algorithms is carried out as a first step. As a result, several potential parameter sets are generated. The final potential parameter set, which can reproduce all the material properties used for fitting, is selected from them. The developed potential can reproduce the material properties used for fitting which involve the radial distribution function of the Zr70Ni30 amorphous structure. [doi:10.2320/matertrans.MF200602] read less USED (low confidence) A. Upadhyay and H. Urbassek, “Response of ultrathin metal films to ultrafast laser irradiation: A comparative molecular-dynamics study.” 2007. link Times cited: 10 Abstract: Using molecular-dynamics computer simulation, we study the m… read moreAbstract: Using molecular-dynamics computer simulation, we study the materials processes in ultrathin metal films induced by ultrafast laser irradiation. We investigate four different metals (Al, Cu, Ti, W), which vary widely in their cohesive energy, melting temperature, bulk modulus, and crystal structure. Despite these variations, we find that the same materials processes are induced in these films: With increasing laser fluence, the film melts, voids are formed, the film tears (spallation), and finally fragments to form a multitude of clusters. When the energy transfer starting the process is scaled to the cohesive energy of the material, the thresholds of these processes adopt similar – but not identical – values. read less USED (low confidence) Z. Lin and L. Zhigilei, “Time-resolved diffraction profiles and structural dynamics of Ni film under short laser pulse irradiation.” 2007. link Times cited: 11 Abstract: The evolution of the diffraction profiles during the fast th… read moreAbstract: The evolution of the diffraction profiles during the fast thermoelastic deformation and structural transformations induced in a thin Ni film by short pulse laser irradiation is investigated in molecular dynamics simulations. Fast disappearance of the diffraction peaks characteristic for the initial crystal structure is related to the homogeneous nucleation and growth of liquid regions inside the overheated crystal. Transient thermoelastic deformation of the film prior to melting is reflected in shifts and splittings of the diffraction peaks, providing an opportunity for experimental probing of the ultrafast deformations. read less USED (low confidence) J. Quan, X. W. Zhou, and H. Wadley, “Low-energy ion-assisted control of interfacial structures in metallic multilayers,” Journal of Crystal Growth. 2007. link Times cited: 8 USED (low confidence) A. Grenier et al., “Structural investigation of TbCo2/Fe magnetostrictive thin films by tomographic atom probe and Mössbauer spectrometry,” Journal of Magnetism and Magnetic Materials. 2007. link Times cited: 6 USED (low confidence) D. Cheng, Z. Yan, and L. Yan, “Misfit dislocation network in Cu/Ni multilayers and its behaviors during scratching,” Thin Solid Films. 2007. link Times cited: 37 USED (low confidence) T. Kumagai, S. Hara, S. Izumi, and S. Sakai, “Development of a Method for Making Interatomic Potential: An Application to Metallic Systems,” Materials Science Forum. 2007. link Times cited: 4 Abstract: A method for making interatomic potentials is proposed and i… read moreAbstract: A method for making interatomic potentials is proposed and is applied to Cu-Zr-Hf-Ni- Al bulk-metallic-glass systems. The method consists of three steps. Firstly, potential function form is determined so that bonding nature can be described. Secondly, materials properties used for fitting are selected so that the potential has enough robustness. Here, it is noted that materials properties must be added in accordance with the purpose of the study. Finally, potential parameters have been optimized using global-search procedure. Developed potential well reproduces material properties of them. read less USED (low confidence) R. Drautz, X. W. Zhou, D. Murdick, B. Gillespie, H. Wadley, and D. Pettifor, “Analytic bond-order potentials for modelling the growth of semiconductor thin films,” Progress in Materials Science. 2007. link Times cited: 28 USED (low confidence) M. Caffio, G. Rovida, and A. Atrei, “Structural investigation of the c(√2 × 5√2)R45° surface alloy formed by Ti deposition on Cu(0 0 1),” Surface Science. 2007. link Times cited: 3 USED (low confidence) N. Shamsutdinov, A. Böttger, and F. Tichelaar, “The effect of Cu interlayers on grain size and stress in sputtered Fe–Cu multilayered thin films,” Scripta Materialia. 2006. link Times cited: 19 USED (low confidence) F. Dorfbauer et al., “Nanostructure calculation of CoAg core-shell clusters,” Journal of Applied Physics. 2006. link Times cited: 35 Abstract: Detailed studies of the structure of magnetic nanoclusters a… read moreAbstract: Detailed studies of the structure of magnetic nanoclusters are crucial for understanding their magnetic properties. We have investigated the structure of CoxAg1−x nanoparticles by means of molecular dynamics simulations utilizing the embedded atom method. Starting from a completely random distribution of Co and Ag atoms, the clusters were heated up to 1300K and subsequently cooled down. The size of the resulting particles was 2.8nm (864 atoms). A clear segregation of the Ag atoms on the surface of the Co core was obtained. read less USED (low confidence) R. Evans et al., “The influence of shape and structure on the Curie temperature of Fe and Co nanoparticles,” Journal of Applied Physics. 2006. link Times cited: 22 Abstract: We have investigated the effect of lattice fluctuations on t… read moreAbstract: We have investigated the effect of lattice fluctuations on the magnetic properties of nanoparticles of Fe and Co. Atomic structures were simulated using a molecular-dynamic approach, with the system slowly cooled into the ordered phase. The magnetic properties were then simulated using an atomistic approach using a classical spin Hamiltonian taking into account the long-range nature and atomic separation dependence of the exchange. The magnetic properties are found to be affected by both the particle shape and the lattice fluctuations. For a perfectly ordered lattice we find that a spherical particle has a larger magnetization for a given temperature than a cube containing the same number of atoms. We have also studied the effect of lattice fluctuations. This involves a comparison of M(T) for two cases, firstly, a nanoparticle with a fixed lattice corresponding to the low-temperature annealed state (T=20K), and secondly a nanoparticle with a lattice structure equilibrated at the temperature T, the latter ... read less USED (low confidence) W. Schmickler, K. Pötting, and M. Mariscal, “A new simulation model for electrochemical metal deposition,” Chemical Physics. 2006. link Times cited: 18 USED (low confidence) B. Remington et al., “Materials science under extreme conditions of pressure and strain rate,” Metallurgical and Materials Transactions A. 2004. link Times cited: 75 USED (low confidence) D. Larson, A. Petford-Long, Y. Ma, and A. Cerezo, “Information storage materials: nanoscale characterisation by three-dimensional atom probe analysis,” Acta Materialia. 2004. link Times cited: 83 USED (low confidence) D. Beke, Z. Erdélyi, I. Szabó, and C. Cserháti, “Nanoscale Effects in Diffusion,” Journal of Metastable and Nanocrystalline Materials. 2004. link Times cited: 10 Abstract: Diffusion in nanostructures has many challenging features ev… read moreAbstract: Diffusion in nanostructures has many challenging features even if the role of structural defects (dislocations, phaseor grain-boundaries) can be neglected. This can be the case for diffusion in amorphous materials, in epitaxial, highly ideal thin films or multilayers, in dissolution of thin films or in kinetics of surface segregation, where diffusion along short circuits can be ignored and only fundamental difficulties, related to nanoscale effects, raise. Different examples for diffusion in such materials will be given with special emphasis on the validity limit of the continuum approach especially if the diffusion coefficient depends strongly on composition (nonlinearity). It was shown recently by us that even for ideal systems (like Cu/Ni) this non-linearity leads to linear shift of an originally sharp interface (instead of the parabolic behaviour) and to sharpening of an initially diffuse interface. The sharpening takes place, even if the effects of stresses (built in mismatch, thermal and diffusional stresses) are taken into account. Furthermore deviations from the parabolic law are present in phase separating systems as well, but here the interplay of the strong composition dependence and the phase separation tendency results in a more complex behaviour. These deviations are typical nanoeffects i.e. they diminish with time; in the examples investigated here after dissolution of several hundred atomic planes the parabolic law already fulfills. read less USED (low confidence) M. Mao et al., “Sub-milli-Torr magnetron sputter deposition of magnetic thin films,” Digest of INTERMAG 2003. International Magnetics Conference (Cat. No.03CH37401). 2003. link Times cited: 2 Abstract: In this paper, we evaluate the single films stress of variou… read moreAbstract: In this paper, we evaluate the single films stress of various component layers in spin-valve or magnetic tunnel junction multilayers and the magnetic thickness loss due to interlayer atomic mixing in the magnetic thin films deposited by sub-milli-Torr magnetron sputtering with that from ion beam sputtering. read less USED (low confidence) A. Petford-Long and P. Shang, “High resolution structural and magnetic imaging,” Journal of Magnetism and Magnetic Materials. 2002. link Times cited: 1 USED (low confidence) O. Chirayutthanasak et al., “Universal function for grain boundary energies in bcc metals,” Scripta Materialia. 2024. link Times cited: 0 USED (low confidence) S. Xu, A. A. Mamun, S. Mu, and Y. Su, “Uniaxial deformation of nanowires in 16 refractory multi-principal element alloys,” Journal of Alloys and Compounds. 2023. link Times cited: 1 USED (low confidence) X.-T. Vu, V.-H. Nguyen, T.-V. Tran, Q. Nguyen, and D.-Q. Doan, “Mechanical characteristics and deformation behavior of Al polycrystal reinforced with SiC particles,” Journal of Physics and Chemistry of Solids. 2023. link Times cited: 1 USED (low confidence) Z. Zhang, Z. Yang, C. Wang, Q. Zhang, S. Zheng, and W. Xu, “Mechanisms of femtosecond laser ablation of Ni3Al: Molecular dynamics study,” Optics and Laser Technology. 2021. link Times cited: 15 USED (low confidence) B. Natalich, Y. O. Kravchenko, O. Maksakova, and V. Borysiuk, “Size-Dependent Melting Behavior of Silver Nanoparticles: A Molecular Dynamics Study,” Springer Proceedings in Physics. 2020. link Times cited: 0 USED (low confidence) M. Rajput, P. V. Subhash, and R. Srinivasan, “Displacement damage study in tungsten and iron for fusion neutron irradiation,” Fusion Engineering and Design. 2020. link Times cited: 6 USED (low confidence) F. Z. Dai, Z. Sun, and W.-Z. Zhang, “From Coherent to Semi-Coherent — Evolution of Precipitation Crystallography in an fcc/bcc System,” EnergyRN: Electrochemical Energy Engineering (EnergyRN) (Topic). 2019. link Times cited: 0 Abstract: A comprehensive knowledge about how crystallographic feature… read moreAbstract: A comprehensive knowledge about how crystallographic features are developed at the early stage of a phase transformation in an anisotropic misfit system (e.g., bcc/fcc) is limited, which impedes the deep understanding and quantitative modeling of microstructure evolution. In this work, we simulated coherency loss process during the early growth of a Cr precipitate (bcc) in a Cu matrix (fcc) by the combination of Monte Carlo and molecular dynamics. The simulation results reveal the fine details in the evolution of the crystallographic features, including the orientation relationship (OR) between two phases, the precipitate morphology, and interfacial dislocation structure at the early stage of the precipitate growth. Generation of a dominant set of dislocations is the governing event during the evolution process. The selection of the dominant dislocations was rationalized based on the minimization of interfacial energy in the major facet containing a single set of these dislocations. The initial OR developed between the coherent precipitate and the matrix is near the Nishiyama-Wassermann OR, and it changes discretely in response to the early generation of the dominant dislocations, towards the ideal OR corresponding to the O-line condition, near the Kurdjumov-Sachs OR. This tendency reflects the experimental observations in a Cu-Cr system, and provides helpful insight into the evolution of crystallographic features in physical reality. read less USED (low confidence) S. Ratanaphan, T. Boonkird, R. Sarochawikasit, H. Beladi, K. Barmak, and G. Rohrer, “Atomistic simulations of grain boundary energies in tungsten,” Materials Letters. 2017. link Times cited: 16 USED (low confidence) H. Koguchi and Y. Tanaka, “Analysis of singular stress field in nanoscaled dissimilar material joints using the anisotropic elasticity theory considering interface properties.” 2017. link Times cited: 0 Abstract: In recent advanced semiconductor products, elements of thick… read moreAbstract: In recent advanced semiconductor products, elements of thickness and width are frequently in nanoscale. As the size of elements reduces to nanometer, a ratio of surface to volume increases. Surface properties, such as surface stresses and surface elasticity, influence on the distributions of bulk stresses near the surface. In the present study, the singular stress at the corner in an anisotropic two-dimensional multi-wedge joint consisting of three kinds of material is analyzed using molecular statics (MS) method and the anisotropic elasticity theory with a boundary condition considering interface properties. The joints are composed of Ni, Au and Cu. Several joints with different wedge angles at the interface are analyzed. Distributions of atomic stress calculated by the MS method are compared with those by the theory. In the results of the MS analysis, stress jumps exist at the interface in the circumferential direction of the stress distribution around the stress singular point. It is supposed that these are attributed to the influence of interface stresses. Then, interface stresses and elastic properties at the interfaces are investigated using the MS method. The obtained interface properties are used in the stress analysis using the Stroh’s formalism. Finally, the theoretical analysis considering the interface properties can be expressed the jumps of stress at the interfaces. read less USED (low confidence) L. Hai, H. Jie, Z. Zhi-xuan, and M. Zhao-xia, “Atomistic Simulations of Elastic-Plastic Deformation in Nickel Single Crystal under Shock Loading,” Procedia Engineering. 2017. link Times cited: 3 USED (low confidence) G. Anand and P. Chattopadhyay, “Computational Design of Microstructure: An Overview.” 2016. link Times cited: 0 Abstract: During the last couple of decades, treatment of microstructu… read moreAbstract: During the last couple of decades, treatment of microstructure in materials science has been shifted from the diagnostic to design paradigm. Design of microstructure is inherently complex problems due to non linear spatial and temporal interaction of composition and parameters leading to the target properties. In most of the cases, different properties are reciprocally correlated i.e., improvement of one lead to the degradation of other. Also, the design of microstructure is a multiscale problem, as the knowledge of phenomena at range of scales from electronic to mesoscale is required for precise compositionmicrostructure-property determination. In the view of above, present chapter provides the introduction to computationally driven microstructure engineering in the framework of constitutive length scale in microstructure design. The important issues pertaining to design such as phase stability and interfaces has been explained. Additionally, the bird-eye view of various computational techniques in order of length scale has been introduced, with an aim to present the picture of combination of various techniques for solving microstructural design problems under various scenarios. read less USED (low confidence) G. Li, Q. Wang, and K. Wang, “Atomic behavior and structural evolution of alloy nanoparticles during thermodynamic processes.” 2015. link Times cited: 0 USED (low confidence) K. Suzuki, M. Ochi, and H. Miura, “Stress-induced anisotropic diffusion of component elements in stacked thin-film multi-layer structures.” 2015. link Times cited: 0 Abstract: The mechanism of the directional coarsening (rafting) of γ&a… read moreAbstract: The mechanism of the directional coarsening (rafting) of γ' phases of Ni-base super-alloy under uniaxial strain at high temperatures was investigated by molecular dynamics (MD) analysis of stacked thin-film multi-layer structures. The strain-induced anisotropic diffusion of constituent atoms perpendicular to the interface was observed clearly in Ni(001)/Ni 3 Al(001) and Ni(001)/Cu(001) interfaces. In contrast, no strain-induced anisotropic diffusion occurred in the interface structure consisted of the face-centered cubic (FCC) metals with (111) crystallographic orientation. The estimated strain-induced anisotropic diffusion was validated by experiment using the Ni/Cu stacked thin films structures. read less USED (low confidence) M. Yalcin, A. Mekhrabov, and M. Akdeniz, “Effects of Nanoparticle Geometry and Temperature on the Structural Evolution in FeCo Nanoalloys,” Acta Physica Polonica A. 2014. link Times cited: 3 USED (low confidence) B. Shen et al., “Molecular dynamics simulation studies of structural and dynamical properties of rapidly quenched Al,” Journal of Non-crystalline Solids. 2014. link Times cited: 23 USED (low confidence) T. Nagase et al., “MeV Electron Irradiation Induced Solid-State Amorphization (SSA) in B2 Intermetallic Compounds,” Journal of The Society of Materials Science, Japan. 2013. link Times cited: 2 Abstract: 1 緒 言 多くの金属間化合物は,フレンケルペアの導入だけで 容易に固相アモルファス化 (Solid-State Amo… read moreAbstract: 1 緒 言 多くの金属間化合物は,フレンケルペアの導入だけで 容易に固相アモルファス化 (Solid-State Amorphization, SSA) することが知られている.1)MeV電子照射による固 相アモルファス化に関する系統的な実験研究 1)および理 論研究 2)によって,この現象が材料において普遍的におこ る一般的な現象であることが明らかとなっている. Okamotoらは,カウツマンパラドックスを考慮し,固相ア モルファス化を理想ガラス転移温度 (Ideal glass transition temperature, TK) 以下における機械的融解 (Mechanical Melting) 現象であると考え,Generalized Lindemann Melting (GLM) Criterionを提唱した.2) 近年,Bulk Metallic Glass (BMG) が開発され,その工 学的応用と同時にガラス転移 (Glass-to-Liquid transition) の研究が進展している.現実に観察されるガラス転移は 一次の相転移であるが,二次で発現すれば理想ガラス (Ideal glass) 状態を経由すると考えられる.固相アモル ファス化すなわち結晶-ガラス転移 (Crystal-to-Glass transition) の場合も,二次の相転移で発現すれば理想ガ ラスが形成される.ガラス転移・固相アモルファス化は 一次相転移であるが,いずれも二次的に発現すれば理想 ガラス状態を経るという共通点を考えると,この両者に は大きな相関が存在する.固相アモルファス化現象の解 明は,ガラス転移・理想ガラスの根源を明らかにするこ とにつながり,結果としてバルク金属ガラスの開発およ び材料科学に大きく貢献する.そこで本研究では,固相 アモルファス化現象の中でも,実験的に最も二次に近い 形で相転移を誘起できると考えられる高圧電子顕微鏡MeV電子照射法 2)に注目し,MeV電子照射誘起固相ア モルファス化現象の未解明点である,B2化合物における 固相アモルファス化発現とマルテンサイト変態の相関関 係について調べた. 2 実験方法・計算方法 照射に用いた TiNiおよび Ti (Ni, Fe)合金インゴット は,純金属を所定の組成になるよう秤量し,アーク溶解 法により作製した.照射用試料は,このインゴットある B2化合物の電子照射誘起固相アモルファス化現象 永 瀬 丈 嗣 佐々木 淳 志 滝 澤 和 也 安 田 弘 行 寺 井 智 之 福 田 隆 掛 下 知 行 譯 田 真 人 渋 谷 陽 二 保 田 英 洋 森 博太郎 渋 谷 陽 二 MeV Electron Irradiation Induced Solid-State Amorphization (SSA) in B2 Intermetallic Compounds read less USED (low confidence) M. Yuasa, Y. Chino, and M. Mabuchi, “Atomic simulations of GB sliding in pure and segregated bicrystals,” MRS Proceedings. 2013. link Times cited: 1 Abstract: Grain boundary (GB) sliding is an important deformation mode… read moreAbstract: Grain boundary (GB) sliding is an important deformation mode in polycrystals, and it has been extensively investigated, for example, there are many studies on influences of the atomic geometry in the GB region. However, it is important to investigate GB sliding from the electronic structure of GB for deeper understandings of the sliding mechanisms. In the present work, we investigated the GBs sliding in pure and segregated bicrystals with classical molecular dynamics (MD) simulations and first-principles calculations. It is accepted that the sliding rate is affected by the GB energy. However, there was no correlation between the sliding rate and the GB energy in either the pure or the segregated bicrystals. First-principles calculations revealed that the sliding rate calculated by the MD simulations increases with decreasing minimum charge density at the bond critical point in the GB. This held in both the pure and segregated bicrystals. It seems that the sliding rate depends on atomic movement at the minimum charge density sites. read less USED (low confidence) D. Seif and N. Ghoniem, “Dislocation Bias Calculations in Metals Using a Combined Finite-Element Rate-Theory Approach.” 2013. link Times cited: 2 USED (low confidence) J. Liang et al., “Correlating conductance and structure of silver nano-contacts created by jump-to-contact STM break junction,” Journal of Electroanalytical Chemistry. 2013. link Times cited: 9 USED (low confidence) T. Kumagai and S. Izumi, “Development of a Software to Optimize Parameters of Interatomic Potentials for Solid Systems,” Transactions of the Japan Society of Mechanical Engineers. A. 2011. link Times cited: 9 Abstract: Generally, it is difficult to develop practical interatomic … read moreAbstract: Generally, it is difficult to develop practical interatomic potentials which can be used in classical molecular dynamics calculations, since a method for development had not been established. In order to solve such problems, we had proposed a framework to develop interatomic potentials. However, the framework had not been widely used due to the difficulty of coding a process to optimize potential-parameters, which was involved in the framework. In this work, a practical software to optimize potential-parameters for solid systems, was developed based on real-coded genetic algorithm (GA). Fitness (=optimization-function) in GA for potential-parameter optimization could be calculated in practical time, and a concept of the corresponded module in GA-code was proposed. The developed software has an extensible structure: a user can define a new crystal-structure, or a new potential function to use in potential-parameter optimization by defining a corresponded class for that in separated header files. Convenient interfacial class to calculate equilibrium material properties of crystals and that to calculate material properties of user-inputted atomic-structures were prepared to define optimization-function. Several sample input files and header files were available for easy starting. As a result of distribution, several effective interatomic potentials were developed by using the software. As a sample of the development of interatomic potential, Tersoff-type potential for SiO2 systems were shown. read less USED (low confidence) H. Saeed, S. Izumi, S. Hara, and S. Sakai, “Reaction Pathway Analysis of Homogeneous Dislocation Nucleation in a Perfect Molybdenum Crystal,” MRS Proceedings. 2011. link Times cited: 1 Abstract: Reaction pathway analysis was carried out for homogeneous di… read moreAbstract: Reaction pathway analysis was carried out for homogeneous dislocation nucleation in perfect crystal Mo. The reaction sampling method employed was based on the Nudged Elastic Band algorithm and other extended schemes. Results obtained were compared with corresponding results for Cu and Si. The stress range for activation energies less than 5 eV is found to be considerably higher for Mo than those for Cu as well as Si. Stresses in excess of 12 GPa make homogeneous dislocation nucleation in Mo an unrealistic transition. The results also show the dislocation cores under this stress range to be diffused, with shear displacement of particles being considerably less than the Burgers vector. Depending on the applied stress, displacement of extra slip-plane atoms can be considerable in Mo. This is in contrast to Cu, in which dislocation nucleation is essentially a two-plane phenomenon. read less USED (low confidence) L. Yuan-yuan, J. Guo-bin, Y. Bin, and L. Dachun, “Molecular Dynamics Simulation of Thermodynamic Properties for Pb-Au Alloys,” Rare Metal Materials and Engineering. 2011. link Times cited: 2 USED (low confidence) J. Greene, “Thin Film Nucleation, Growth, and Microstructural Evolution: An Atomic Scale View.” 2010. link Times cited: 54 USED (low confidence) Y. Shibuta, “Numerical Approach to the Phase Transformation of Iron and Related Properties at the Interface by Molecular Simulation.” 2009. link Times cited: 0 USED (low confidence) Y. Gan and Z. Chen, “A Study on the Collapse of Self-Similar Hardening Behavior of Nanostructures,” International Journal for Multiscale Computational Engineering. 2009. link Times cited: 1 USED (low confidence) A. Takeuchi, K. Yubuta, and A. Inoue, “Cluster packed structures in bulk metallic glasses created from BCC derivative compounds,” Journal of Physics: Conference Series. 2009. link Times cited: 5 Abstract: Molecular dynamics simulations based on plastic crystal mode… read moreAbstract: Molecular dynamics simulations based on plastic crystal model were performed for the Fe69.0Nb10.3B20.7 and Zr57.1Ni28.6Al14.3 alloys to investigate their forming ability and features of the noncrystalline structures. The Fe69.0Nb10.3B20.7 and Zr57.1Ni28.6Al14.3 alloys were created from C6Cr23 and Ti4Ni2O structures, respectively, by assuming the presence of hypothetical clusters in the crystalline structures under a treatment that the clusters and glue atoms are regarded as the components. The analysis with total pair-distribution function, g(r), revealed that these alloys have high tendencies to form noncrystalline structures while their partial g(r) profiles for pairs among components showed the presence of medium range order. Cluster packed structures from bcc derivative compounds cause high forming ability of noncrystalline structure and the medium range order in the noncrystalline structure. read less USED (low confidence) C. Engin and H. Urbassek, “Molecular-dynamics investigation of the fcc → bcc phase transformation in Fe,” Computational Materials Science. 2008. link Times cited: 49 USED (low confidence) K. Sugita, M. Mizuno, H. Araki, and Y. Shirai, “Chapter 13 Electronic Structure and Bonding in Amorphous Zr67Ni33 and Zr67Cu33,” Advances in Quantum Chemistry. 2008. link Times cited: 1 USED (low confidence) S. Wen, R. Zong, F. Zeng, Y. Gao, and F. Pan, “Evaluating modulus and hardness enhancement in evaporated Cu/W multilayers,” Acta Materialia. 2007. link Times cited: 94 USED (low confidence) N. Lümmen, B. Fischer, and T. Kraska, “Homogeneous nucleation and growth from highly supersaturated vapor by molecular dynamics simulation.” 2007. link Times cited: 2 USED (low confidence) N. Nakamura, H. Ogi, R. Tarumi, T. Yasui, T. Ono, and M. Hirao, “Strain-Dependent Elastic Constants and Magnetic Anisotropy of Co/Pt Superlattice Thin Films,” Journal of The Society of Materials Science, Japan. 2005. link Times cited: 0 Abstract: In this paper, the contribution of magnetoelastic-anisotropy… read moreAbstract: In this paper, the contribution of magnetoelastic-anisotropy effect on perpendicular-magnetic anisotropy of Co/Pt superlattice thin films is studied considering the strain-dependent elastic constants. We measured the effective-magnetic-anisotropy energy changing the Pt-layer thickness (2 to 20 A) keeping the Co-layer thickness unchanged (4 A). The effective-magnetic-anisotropy energy changed depending on the Pt-layer thickness and showed a maximum near dPt=12 A. This result is explained by calculating the magnetoelastic-anisotropy energy considering the change of the elastic constants caused by the lattice misfit. We measured anisotropic elastic constants of Co/Pt superlattice thin films by resonance-ultrasound-spectroscopy method coupled with laser-Doppler interferometry and the picosecond laser-ultrasound method. The measured elastic constants showed elastic anisotropy between the in-plane and out-of-plane directions. They agreed with those predicted from the strain-dependent elastic-constant model. The elastic constants show correlations with the perpendicular-magnetic anisotropy. read less USED (low confidence) G. Thompson, M. K. Miller, R. Banerjee, and H. Fraser, “Atom Probe Tomography Characterization of Thin Film Multilayers,” MRS Proceedings. 2003. link Times cited: 0 Abstract: As the individual layer of Ti is reduced in thickness in a T… read moreAbstract: As the individual layer of Ti is reduced in thickness in a Ti/Nb multilayered thin film structure, the Ti layer can undergo a change in phase stability from hcp to bcc . Due to its high spatial resolution, atom probe tomography (APT) is ideally suited to characterize the compositional variations in such thin film structures. A series of hcp Ti / bcc Nb and bcc Ti / bcc Nb multilayers have been sputtered deposited and prepared as APT specimens using a Focus Ion Beam (FIB) milling procedure. The APT results have shown a substantial interdiffusion of Nb into the bcc Ti layers to a pseudo-equilibrium concentration of approximately Ti-20at%Nb while maintaining a compositionally modulated interface. In contrast, the hcp Ti layers indicated little Nb interdiffusion within the layers. Thermodynamic volumetric free energy modeling has indicated that this unexpected Nb interdiffusion facilitated the bcc phase stability. The coupling of APT results to the pseudomorphic bcc Ti phase demonstrates the capability APT has in quantifying the compositional characteristics in these types of multilayered nanocomposite systems. read less USED (low confidence) Y. Mahmood, M. S. Daw, M. Chandross, and F. Abdeljawad, “Universal trends in computed grain boundary energies of FCC metals,” Scripta Materialia. 2024. link Times cited: 0 USED (low confidence) C. Xue, B. Gao, T. Han, C. Che, Z. Chu, and L. Tuo, “Dislocation evolution mechanism of plastic deformation process of AZ31 magnesium alloy with different grain size,” Computational Materials Science. 2024. link Times cited: 0 USED (low confidence) P.-A. Geslin, “Modeling of solid solution strengthening in FCC alloys: Atomistic simulations, statistical models and elastic continuous approaches,” Computational Materials Science. 2024. link Times cited: 0 USED (low confidence) Z.-yu Zhou, Q. Zheng, Y. Li, C. Ding, G.-J. Peng, and Z.-Y. Piao, “Research on the mechanism of the two-dimensional ultrasonic surface burnishing process to enhance the wear resistance for aluminum alloy,” Friction. 2023. link Times cited: 3 USED (low confidence) T. He, X. Li, Y. Qi, M. Zhao, and M. Feng, “Molecular dynamics simulation of primary irradiation damage in Ti-6Al-4V alloys,” Nuclear Engineering and Technology. 2023. link Times cited: 0 USED (low confidence) Q. Zhang et al., “Room-temperature super-elongation in high-entropy alloy nanopillars,” Nature Communications. 2023. link Times cited: 1 USED (low confidence) Y. Li, H. Wang, L. Weng, B. Tu, and M. Lei, “Wetting and spreading of AgCuTi on Fe substrate at high temperatures: A molecular dynamics study,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) H. Hu, T. Fu, C. Li, and X. Peng, “Temperature- and internal structural size-dependent strength of nanotwinned face-centered cubic metals,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) A. Khoei, M. R. Seddighian, and A. R. Sameti, “Machine learning-based multiscale framework for mechanical behavior of nano-crystalline structures,” International Journal of Mechanical Sciences. 2023. link Times cited: 0 USED (low confidence) X. Zeng, C. He, X. Li, and Q. Hu, “Study on the Stability of Cu-Ni Cluster Components and the Effect of Strain on Its Structure,” Materials. 2023. link Times cited: 0 Abstract: Solute clusters are one of the important mechanisms of irrad… read moreAbstract: Solute clusters are one of the important mechanisms of irradiation embrittlement of ferritic steels. It is of great significance to study the stability of solute clusters in ferritic steels and their effects on the mechanical properties of the materials. Molecular dynamics was used to study the binding energy, defect energy, and interaction energy of 2 nm-diameter Cu-Ni clusters in the ferritic lattice, which have six categories of Cu-Ni clusters, such as the pure Cu cluster, the core–shell structural cluster with one layer to four layers of Ni atoms and the pure Ni cluster. It was found that Cu-Ni clusters have lower energy advantages than pure Ni clusters. Through shear strain simulation of the three clusters, the structure of 2 nm diameter clusters does not undergo phase transformation. The number of slip systems and the length of dislocation lines in the cluster system are positively correlated with the magnitude of the critical stress of material plastic deformation. read less USED (low confidence) S. Shuang, Y. Liang, X. Zhang, F. Yuan, G. Kang, and X. Zhang, “Impact of local chemical ordering on deformation mechanisms in single-crystalline CuNiCoFe high-entropy alloys: a molecular dynamics study,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 0 Abstract: High-entropy alloys (HEAs), composed of multiple constituent… read moreAbstract: High-entropy alloys (HEAs), composed of multiple constituent elements with concentrations ranging from 5% to 35%, have been considered ideal solid solution of multi-principal elements. However, recent experimental and computational studies have demonstrated that complex enthalpic interactions among constituents lead to a wide variety of local chemical ordering (LCO) at lower temperatures. HEAs containing Cu typically decompose by forming of Cu-rich phases during annealing, thus affecting mechanical properties. In this study, CuNiCoFe HEA was chosen as a model with a tendency for Cu segregation at low temperatures. The formation of LCO and its impact on the deformation behaviors in the single-crystalline CuNiCoFe HEA were studied via molecular dynamics simulations. Our results demonstrate that CuNiCoFe HEA decomposes by Cu clustering, in agreement with prior experimental and computational studies, owing to insufficient configuration entropy to compete against the mixing enthalpy at lower temperatures. A softening in ultimate stress in the LCO models was observed compared to the random solid solution models. The softening is due to the lower unstable stacking fault energy, which determines the nucleation event of dislocations, thereby rationalizing the dislocation nucleation in the Cu-rich regions and the softening of the overall ultimate strength in the LCO models. Additionally, the inhomogeneous FCC–BCC transformation is closely associated with concentration inhomogeneity. CuNiCoFe HEA with LCO can be regarded as composites, consisting of clusters with different properties. Consequently, concentration inhomogeneity induced by LCO profoundly impacts the mechanical properties and deformation behaviors of the HEA. This study provides insights into the effect of LCO on the mechanical properties of CuNiCoFe HEAs, which is crucial for developing HEAs with tailored properties for specific applications. read less USED (low confidence) S. Chen et al., “Ideal plasticity and shape memory of nanolamellar high-entropy alloys,” Science Advances. 2023. link Times cited: 1 Abstract: Understanding the relationship among elemental compositions,… read moreAbstract: Understanding the relationship among elemental compositions, nanolamellar microstructures, and mechanical properties enables the rational design of high-entropy alloys (HEAs). Here, we construct nanolamellar AlxCoCuFeNi HEAs with alternating high– and low–Al concentration layers and explore their mechanical properties using a combination of molecular dynamic simulation and density functional theory calculation. Our results show that the HEAs with nanolamellar structures exhibit ideal plastic behavior during uniaxial tensile loading, a feature not observed in homogeneous HEAs. This remarkable ideal plasticity is attributed to the unique deformation mechanisms of phase transformation coupled with dislocation nucleation and propagation in the high–Al concentration layers and the confinement and slip-blocking effect of the low–Al concentration layers. Unexpectedly, this ideal plasticity is fully reversible upon unloading, leading to a remarkable shape memory effect. Our work highlights the importance of nanolamellar structures in controlling the mechanical and functional properties of HEAs and presents a fascinating route for the design of HEAs for both functional and structural applications. read less USED (low confidence) M. Gastaldo, J. Varillas, Á. Rodríguez, M. Velický, O. Frank, and M. Kalbáč, “Tunable strain and bandgap in subcritical-sized MoS2 nanobubbles,” npj 2D Materials and Applications. 2023. link Times cited: 0 USED (low confidence) S. Meguid, S. I. Kundalwal, and A. Alian, “Role played by phonon drag on accuracy of MD simulations of nanowires due to deficiently selected strain rates,” International Journal of Mechanics and Materials in Design. 2023. link Times cited: 0 USED (low confidence) M. Dias, P. Carvalho, A. Gonçalves, E. Alves, and J. B. Correia, “Hybrid molecular dynamic Monte Carlo simulation and experimental production of a multi-component Cu–Fe–Ni–Mo–W alloy,” Intermetallics. 2023. link Times cited: 1 USED (low confidence) X. Guo et al., “Effects of He-ion irradiation on microstructures of low activation Ti-Ta-V alloy from atomic simulations and irradiation experiments,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2023. link Times cited: 0 USED (low confidence) X. Chen, Y. Xie, K. Wang, Z. Wang, and Y. Huang, “Construction of a new n-body potential and multi-scale investigations of the direct alloying behaviors for immiscible W/Cu system,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) A. Prada, F. Valencia, M. Ramírez, A. Varas, and J. Rogan, “Synthesis of hollow bimetallic nanoparticles from Ultrafast Laser Irradiation: An atomistic simulation study,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) K. Wang, Y. Yan, Y. Xiong, S. Zhao, D. Chen, and K. Woller, “Enhanced radiation resistance of W-based HEA under helium-ion irradiation conditions,” Journal of Nuclear Materials. 2023. link Times cited: 0 USED (low confidence) T. Fedyaeva, S. Mathesan, A. Bisht, Z. Liang, D. Mordehai, and E. Rabkin, “The effects of composition and microstructure on compressive strength of Ag-Au nanoparticles,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) Y. Li, L. Weng, H. Wang, B. Tu, and M. Lei, “Wetting and spreading behavior of AgCuTi on Ti substrate: A molecular dynamics study,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) X. Liu, R. Xing, H. Zhai, P. Lu, G. Wang, and C. Cheng, “Atomistic simulations of compressive response and deformation mechanisms of body-centered-cubic AlCrFeCoNi high-entropy alloys,” Physica B: Condensed Matter. 2023. link Times cited: 0 USED (low confidence) J. Li, J. Li, Y. Chen, L. Dai, and L. Chen, “On the strain rate sensitivity of mechanical properties of nanoporous gold: Temperature effect,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) J. Niu, R. Hu, X. Luo, Z. Gao, and P. Li, “Study on the Microscopic Compression Deformation Mechanism of Nanocrystalline Single-phase Gold Platinum Alloy,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) P. Fan, N. K. Katiyar, M. Arshad, M. Bai, H. Mao, and S. Goel, “Anisotropic plasticity mechanisms in a newly synthesised High Entropy Alloy investigated using atomic simulations and nanoindentation experiments,” Journal of Alloys and Compounds. 2023. link Times cited: 0 USED (low confidence) Y. Zhao et al., “Molecular dynamics study on surface effect in ultrasonic vibration assisted upsetting of monocrystalline copper,” Journal of Materials Processing Technology. 2023. link Times cited: 0 USED (low confidence) J. Shi et al., “Alloy strengthening toward improving mechanical and tribological performances of CuxNi100-x/Ta nano multilayer materials,” Tribology International. 2023. link Times cited: 0 USED (low confidence) G. Zhao et al., “Understanding the role of engineered cluster evolution in enhancing Ag layer growth on oxides,” Applied Surface Science. 2023. link Times cited: 0 USED (low confidence) X. Liu and W. A. Curtin, “Atomistic simulations reveal strength reductions due to short-range order in alloys,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) X. Qin, Y. Liang, and J. Gu, “Effects of Stress State, Crack—γ/γ′ Phase Interface Relative Locations and Orientations on the Deformation and Crack Propagation Behaviors of the Ni-Based Superalloy—A Molecular Dynamics Study,” Crystals. 2023. link Times cited: 0 Abstract: In this study, we systematically investigate the influence o… read moreAbstract: In this study, we systematically investigate the influence of stress states, relative locations, and orientations of crack—γ/γ′ phase interfaces on the deformation and crack propagation behaviors of the Ni-based superalloy through molecular dynamics simulations. The stress state with high stress triaxiality will impede the plastic deformation process of the system, thereby promoting brittle crack propagation within the system. But the stress state of low stress triaxiality results in obvious plastic deformation and plastic crack propagation behaviors of the system. The deformation system with cracks located in both the γ and γ′ phase exhibits the slowest growth rate, regardless of applied stress states. Additionally, the deformation process demonstrates prominent plastic behavior. For the deformation system with cracks perpendicular to the γ/γ′ phase interface, the γ/γ′ phase interface will hinder the crack propagation. Our research provides interesting observations on deformation and crack propagation behaviors at an atomic level and at a nano-scale which are important for understanding deformation and fracture behaviors at a macroscopic scale for the Ni-based superalloy. read less USED (low confidence) F. Duan et al., “Anisotropic Mechanical Properties and Deformation Mechanisms of Nanotwinned Ni and Ni Alloys With Extremely Fine Twin Boundary Spacings,” Acta Materialia. 2023. link Times cited: 2 USED (low confidence) T. Cao et al., “Dynamic deformation behaviors and mechanisms of CoCrFeNi high-entropy alloys,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) S. G. Bierschenk, M. F. Becker, and D. Kovar, “Effect of an Oxide Layer on High Velocity Impact of Tantalum Particles Characterized using Molecular Dynamics,” Applied Surface Science. 2023. link Times cited: 0 USED (low confidence) S. Chen et al., “Crack Tip Dislocation Activity in Refractory High-Entropy Alloys,” International Journal of Mechanical Sciences. 2023. link Times cited: 0 USED (low confidence) X. Wang, R. Xu, M. Zhou, S. Matharage, Y. Zhou, and Z. Wang, “Effects of the graphene/metal interface on elastic properties of Cu and W matrices: Molecular dynamics simulation,” Mechanics of Materials. 2023. link Times cited: 0 USED (low confidence) A. A. Mamun, S. Xu, X.-G. Li, and Y. Su, “Comparing interatomic potentials in calculating basic structural parameters and Peierls stress in tungsten-based random binary alloys,” Physica Scripta. 2023. link Times cited: 0 Abstract: The field of machine learning-based interatomic potentials (… read moreAbstract: The field of machine learning-based interatomic potentials (ML-IAPs) has seen increasing development in recent years. In this work, we compare three widely used ML-IAPs–the moment tensor potential (MTP), the spectral neighbor analysis potential (SNAP), and the tabulated Gaussian approximation potential (tabGAP)with a conventional non-ML-IAP, the embedded atom method (EAM) potential. We evaluated these potentials on the basis of their accuracy and efficiency in determining basic structural parameters and Peierls stress under equivalent conditions. Three tungsten (W)-based alloys (Mo-W, Nb-W, and Ta-W) are considered, and their lattice parameter, formation energy, elastic tensor, and Peierls stress of edge dislocation are calculated. Compared with DFT results, MTP demonstrates the highest accuracy in predicting the lattice parameter and the best computational efficiency among the three ML-IAPs, while tabGAP accurately predicts two independent elastic constants, C 11 and C 12. Despite being the slowest, SNAP shows the highest accuracy in predicting the third independent elastic constant C 44 and its Peierls stress value is comparable to that based on MTP. read less USED (low confidence) D. Kim et al., “Investigation of Effect of Platinum Nanoparticle Shape on Oxygen Transport in PEMFC Catalyst Layer Using Molecular Dynamics Simulation,” ACS Omega. 2023. link Times cited: 0 Abstract: For the widespread adoption of polymer electrolyte membrane … read moreAbstract: For the widespread adoption of polymer electrolyte membrane fuel cells, it is compelling to investigate the influence of the Pt nanoparticle shapes on the electrocatalytic activity. In this study, a catalyst layer was modeled by incorporating four types of Pt nanoparticles: tetrahedron, cube, octahedron, and truncated octahedron, to investigate the relationship between the shapes of the nanoparticles and their impact on the oxygen transport properties using molecular dynamics simulations. The results of our study reveal that the free volume, which has a substantial impact on the oxygen transport properties, exhibited higher values in the sequence of the tetrahedron, cube, octahedron, and truncated octahedron model. The difference in free volume following the formation of less dense ionomers was also related to the surface adsorption of Pt nanoparticles. Consequently, this led to an improved facilitation of oxygen transport. To clarify the dependence of the oxygen transport on the shape of the Pt nanoparticles in detail, we analyzed the structural properties of different Pt shapes by dividing the Pt nanoparticle regions into corners, edges, and facets. Examination of the structural properties showed that the structure of the ionomer depended not only on the shape of the Pt nanoparticles but also on the number of corners and edges in the upper and side regions of the Pt nanoparticles. read less USED (low confidence) C. Zhang et al., “Local structures and undercooling ability of Zr–Ti melts,” Journal of Non-Crystalline Solids. 2023. link Times cited: 1 USED (low confidence) Z. Zhang, Q. Huang, and H. Zhou, “High-entropy alloy nanocrystals with low-angle grain boundary for superb plastic deformability and recoverability,” International Journal of Plasticity. 2023. link Times cited: 3 USED (low confidence) Y.-C. Wu and J. Shao, “FCC-BCC Phase Transformation Induced Simultaneous Enhancement of Tensile Strength and Ductility at High Strain Rate in High-Entropy Alloy,” International Journal of Plasticity. 2023. link Times cited: 3 USED (low confidence) W. Liu, J. Wang, G. Xia, and Z. Li, “Thermophoresis of nanoparticles in the transition regime,” Physics of Fluids. 2023. link Times cited: 0 Abstract: The thermophoresis of nanoparticles suspended in gas is inve… read moreAbstract: The thermophoresis of nanoparticles suspended in gas is investigated in the transition regime by molecular dynamics simulations. It is found that there exists significant discrepancy between the simulation results and the theoretical predictions for the thermophoretic force, which is attributed to the adsorption of gas molecules on nanoparticles and the gas–particle non-rigid body collisions. By using the effective particle radius, the simulation results and Talbot et al.'s equation could agree with each other in the transition regime. In addition, the effect of the finite system size of the molecular dynamics simulations is non-negligible, and the simulation results modified by effective particle radius can coincide with Phillips' equation quite well. Therefore, for particles of a few nanometers, the non-rigid body collision effect and the adsorption of gas molecules and the effective radius of the nanoparticle under strong gas–particle coupling should be taken into account in the theoretical model. The investigation presented in this paper provides guidance for the application of nanoparticles in aerosol science. read less USED (low confidence) S. Wang, X. Cai, Z. Wang, J. Ju, J. Zhou, and F. Xue, “The inhibition mechanism of liquid metal embrittlement cracks in the Fe–Cu system by Al: atomistic simulations and calculations,” Journal of Materials Science. 2023. link Times cited: 0 USED (low confidence) T. Zhou, Y. Wu, and J.-J. You, “Evolution of the shape and microstructure of body-centered cubic seeds during Cu melt solidification,” Chemical Physics Letters. 2023. link Times cited: 0 USED (low confidence) Y. Cheng et al., “Excellent impact resistance of multilayer metallic glass films subjected to micro-ballistic impact by overcoming dynamic size effects,” Extreme Mechanics Letters. 2023. link Times cited: 0 USED (low confidence) A. Roy, D. Senor, D. Edwards, A. Casella, and R. Devanathan, “Insights into radiation resistance of titanium alloys from displacement cascade simulations,” Journal of Nuclear Materials. 2023. link Times cited: 0 USED (low confidence) C. Barr et al., “Autonomous healing of fatigue cracks via cold welding,” Nature. 2023. link Times cited: 5 USED (low confidence) X. Chen, Y. Xie, and Y. Huang, “Multi-scale simulations of the mechanical behaviors of the W-Cu joint interface with a diffusion layer,” Journal of Molecular Modeling. 2023. link Times cited: 0 USED (low confidence) V. Samsonov, A. Romanov, I. Talyzin, A. Lutsay, D. Zhigunov, and V. Puytov, “Puzzles of Surface Segregation in Binary Pt–Pd Nanoparticles: Molecular Dynamics and Thermodynamic Simulations,” Metals. 2023. link Times cited: 1 Abstract: Up till now, there have been extremely contradictory opinion… read moreAbstract: Up till now, there have been extremely contradictory opinions and inadequate results concerning surface segregation in binary platinum–palladium (Pt–Pd) nanoparticles, including the problems regarding segregating components, as well as the size and temperature dependences of segregation. Taking into account such a situation, we investigated the surface segregation in Pt–Pd nanoparticles by combining atomistic (molecular dynamics) and thermodynamic simulations. For molecular dynamics experiments, the well-known program LAMMPS and the embedded atom method were employed. In the course of the atomistic simulations, two different sets of parameterizations for the Pt–Pt, Pd–Pd, and Pt–Pd interatomic interaction potentials were used. The thermodynamic simulation was based on solving the Butler equation by employing several successive approximations. The results obtained via atomistic simulation and thermodynamic simulation on the basis of the Butler equation were compared with each other, as well as with predictions that were based on the Langmuir–McLean equation and some experimental data. Both simulation methods (atomistic and thermodynamic) predicted the surface segregation of Pd, which diminishes with the nanoparticle size and with increasing temperature. Our simulation results do not confirm the predictions of some authors on surface segregation inversion, i.e., the reversal from the surface segregation of Pd to the surface segregation of Pt when diminishing the nanoparticle size. read less USED (low confidence) Y. Fang et al., “Structural prediction of Fe-Mg-O compounds at super-Earth’s pressures,” Physical Review Materials. 2023. link Times cited: 0 Abstract: Terrestrial exoplanets are of great interest for being simul… read moreAbstract: Terrestrial exoplanets are of great interest for being simultaneously similar to and different from Earth. Their compositions are likely comparable to those of solar-terrestrial objects, but their internal pressures and temperatures can vary significantly with their masses/sizes. The most abundant non-volatile elements are O, Mg, Si, Fe, Al, and Ca, and there has been much recent progress in understanding the nature of magnesium silicates up to and beyond ~3 TPa. However, a critical element, Fe, has yet to be systematically included in materials discovery studies of potential terrestrial planet-forming phases at ultra-high pressures. Here, using the adaptive genetic algorithm (AGA) crystal structure prediction method, we predict several unreported stable crystalline phases in the binary Fe-Mg and ternary Fe-Mg-O systems up to pressures of 3 TPa. The analysis of the local packing motifs of the low-enthalpy Fe-Mg-O phases reveals that the Fe-Mg-O system favors a BCC motif under ultra-high pressures regardless of chemical composition. Besides, oxygen enrichment is conducive to lowering the enthalpies of the Fe-Mg-O phases. Our results extend the current knowledge of structural information of the Fe-Mg-O system to exoplanet pressures. read less USED (low confidence) Z. Zhen-yu, Z. Qiu-yang, L. Yu, J. Zhi-guo, Y. Zhi-peng, and P. Zhong-yu, “Effect of ultrasonic vibration on the deformation through indentation test and molecular dynamics simulation,” Mechanics of Materials. 2023. link Times cited: 0 USED (low confidence) C. Tsai, C.-H. Tung, C.-C. Chen, and S.-Y. Chang, “Chemical Complexity-Changed Deformation Behavior of NiTi-Based B2 Low- to High-Entropy Intermetallic Compounds: Atomistic Simulations,” Journal of Alloys and Compounds. 2023. link Times cited: 0 USED (low confidence) R. Bunting, F. Wodaczek, T. Torabi, and B. Cheng, “Reactivity of Single-Atom Alloy Nanoparticles: Modeling the Dehydrogenation of Propane,” Journal of the American Chemical Society. 2023. link Times cited: 3 Abstract: Physical catalysts often have multiple sites where reactions… read moreAbstract: Physical catalysts often have multiple sites where reactions can take place. One prominent example is single-atom alloys, where the reactive dopant atoms can preferentially locate in the bulk or at different sites on the surface of the nanoparticle. However, ab initio modeling of catalysts usually only considers one site of the catalyst, neglecting the effects of multiple sites. Here, nanoparticles of copper doped with single-atom rhodium or palladium are modeled for the dehydrogenation of propane. Single-atom alloy nanoparticles are simulated at 400–600 K, using machine learning potentials trained on density functional theory calculations, and then the occupation of different single-atom active sites is identified using a similarity kernel. Further, the turnover frequency for all possible sites is calculated for propane dehydrogenation to propene through microkinetic modeling using density functional theory calculations. The total turnover frequencies of the whole nanoparticle are then described from both the population and the individual turnover frequency of each site. Under operating conditions, rhodium as a dopant is found to almost exclusively occupy (111) surface sites while palladium as a dopant occupies a greater variety of facets. Undercoordinated dopant surface sites are found to tend to be more reactive for propane dehydrogenation compared to the (111) surface. It is found that considering the dynamics of the single-atom alloy nanoparticle has a profound effect on the calculated catalytic activity of single-atom alloys by several orders of magnitude. read less USED (low confidence) O. Bialas et al., “Laser assisted fabrication of mechanochemically robust Ti3Au intermetallic at Au-Ti interface,” Engineering Science and Technology, an International Journal. 2023. link Times cited: 1 USED (low confidence) J. Zhou, Y. Lu, C. Wang, D. Feng, H. Zhang, and Y. Li, “Molecular dynamics study of graphene-coated reinforced tribomechanical properties: Hard versus soft substrates,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) Y. Li and W. Qiang, “Defect properties of a body-centered cubic equiatomic TiVZrTa high-entropy alloy from atomistic simulations,” Journal of Physics: Condensed Matter. 2023. link Times cited: 1 Abstract: TiVZrTa high-entropy alloys (HEAs) have been experimentally … read moreAbstract: TiVZrTa high-entropy alloys (HEAs) have been experimentally proven to exhibit excellent irradiation tolerance. In this work, defect energies and evolution were studied to reveal the underlying mechanisms of the excellent irradiation tolerance in TiVZrTa HEA via molecular statics calculations and molecular dynamics simulations. The atomic size mismatch of TiVZrTa is ∼6%, suggesting a larger lattice distortion compared to most face-centered cubic and body-centered cubic M/HEAs. Compared to pure Ta and V, smaller vacancy formation and migration energies with large energy spreads lead to higher equilibrium vacancy concentration and faster vacancy diffusion via low-energy migration paths. Vacancies in TiVZrTa have weaker abilities to form large vacancy clusters and prefer to form small clusters, indicating excellent resistance to radiation swelling. The formation energies of different types of dumbbells in TiVZrTa show significant differences and have large energy spreads. The binding abilities of interstitials in TiVZrTa are weaker compared to that in pure Ta and V. In TiVZrTa, fast vacancy diffusion and slow interstitial diffusion result in closer mobilities of vacancies and interstitials, significantly promoting point defect recombination. We further studied the effects of short-range ordered structures (SROs) on defect diffusion and evolution. SROs in TiVZrTa can effectively lead to higher fractions of defect recombination and fewer surviving defects. Our findings provide a comprehensive understanding of the underlying mechanisms of the high irradiation tolerance in body-centered cubic HEAs with large lattice distortion and suggest SROs are beneficial microstructures for enhancing irradiation tolerance. read less USED (low confidence) T. E. hafi et al., “Microstructural and mechanical behaviors of Nickel pure metallic glass investigated by molecular dynamics simulations,” 2023 3rd International Conference on Innovative Research in Applied Science, Engineering and Technology (IRASET). 2023. link Times cited: 0 Abstract: The purpose of this study is to examine the microstructural … read moreAbstract: The purpose of this study is to examine the microstructural properties of Ni pure metallic glass and to uncover how the system reacts when subjected to mechanical pressure during tensile testing. Molecular dynamics simulations, in conjunction with the embedded-atom approach, were used to carry out the investigation. The local structure of the Ni-monatomic metallic glass was examined by analyzing structure parameters such as the radial distribution function and Voronoi tessellation. The results show that the distorted icosahedra <0,1,10,2> and <0,2,8,4> are the most significant structures in the Ni system. Furthermore, the study reveals that mechanical testing has an impact on the local structures. In fact, the tensile testing decreased the significance of short-range order by reducing the fraction of icosahedra and icosahedra-like structures. read less USED (low confidence) V. Samsonov, I. Talyzin, S. Vasilyev, V. Puytov, and A. A. Romanov, “On surface pre-melting of metallic nanoparticles: molecular dynamics study,” Journal of Nanoparticle Research. 2023. link Times cited: 0 USED (low confidence) H. Huang, L. Ma, T. Liu, B. Cai, H. Li, and Q. Peng, “Atomistic simulations of defect clustering evolution in heavily irradiated Ti35 alloy,” Vacuum. 2023. link Times cited: 0 USED (low confidence) X. Zhang et al., “Effects of lattice distortion and chemical short-range ordering on the incipient behavior of Ti-based multi-principal element alloys: MD simulations and DFT calculations,” International Journal of Plasticity. 2023. link Times cited: 4 USED (low confidence) K. C. Katakam, S. R. Gorja, and N. Yedla, “Experiment and atomistic simulation of uniaxial compression of Ni–W single-crystal alloy,” Materials Today: Proceedings. 2023. link Times cited: 0 USED (low confidence) Y.-F. Wu, W. Yu, and S. Shen, “Developing a variable charge potential for Hf/Nb/Ta/Ti/Zr/O system via machine learning global optimization,” Materials & Design. 2023. link Times cited: 1 USED (low confidence) H. Xu et al., “Integrated Effect of Aging and Heavy Ion Radiation on FeNiCrAl Duplex Alloy for Accident-tolerant Fuel Cladding,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) Y. Hu, S. Ding, J. Xu, Y. Zhang, W. Wu, and R. Xia, “Anisotropic orientation dependent shock wave responses of monocrystalline molybdenum,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) H. Tatsumi, C. Kao, and H. Nishikawa, “Molecular Dynamics Simulation of Cu-Cu Solid-State Bonding under Various Bonding Parameters,” 2023 International Conference on Electronics Packaging (ICEP). 2023. link Times cited: 0 Abstract: A solid-state bonding technology, particularly Cu-Cu bonding… read moreAbstract: A solid-state bonding technology, particularly Cu-Cu bonding, has attracted attention to overcome crucial drawbacks of conventional Sn-based solders in the latest three-dimensional integrated circuits. In this study, the effects of bonding parameters on its bonding behavior were investigated by an atomistic-scale simulation method, molecular dynamics (MD) simulation. MD simulations with various bonding temperatures and interfacial crystalline structures were performed focusing on interfacial densification behavior. The results indicated that the interfacial densification rapidly occurred with increasing the bonding temperature. It was also suggested that the combination of crystal orientations of the bonding surfaces constituting the interface would be important for its bondability. read less USED (low confidence) D. Ishikawa, H. Nakako, T. Blank, H. Wurst, and F. Steiner, “Adhesive Strength and Diffusion at Interfaces between Sintered Cu Layer and Metal Surfaces (Cu, Ag, Au, Pd, Pt, Ni, NiPx, and NiBx),” 2023 International Conference on Electronics Packaging (ICEP). 2023. link Times cited: 0 Abstract: Die-bonding technology using a sintering reaction of Cu nano… read moreAbstract: Die-bonding technology using a sintering reaction of Cu nanoparticles at low temperatures is an attractive and important joint method for power devices operating at higher temperatures. We report the adhesive strengths of mechanical pressure-assisted sintered Cu (sCu) onto eight different metal surfaces (Cu, Ag, Au, Pd, Pt, Ni, NiPx, and NiBx). The die-shear strengths (τb) of sCu on the metal platings under sintering temperatures (260-300 °C) in N2 were measured. As the sintering temperatures increased, the τb on the Cu, Ag, Ni, NiPx, and NiBx increased, whereas those on the Au, Pd, and Pt decreased. The MD simulations imply that the diffused couples (Au, Pd, Pt) with high a ratio of intrinsic diffusion coefficients can deteriorate the shear strengths and the self- and non-diffusion couples (Cu, Ni, Ag) can keep the high shear strengths. read less USED (low confidence) L. Liting, L. Xin, and Z. Hongyu, “An explanation for the effect of Au surface finish on the quality of sintered Ag-Au joints,” Applied Surface Science. 2023. link Times cited: 0 USED (low confidence) H. Wang et al., “Strain rate sensitivity in Zr-based metallic glass: Experiments and molecular dynamics study,” Journal of Non-Crystalline Solids. 2023. link Times cited: 1 USED (low confidence) S. Mukesh and N. Lanzillo, “A Multiscale Simulation Study of the Structural Integrity of Damascene Interconnects in Advanced Technology Nodes,” IEEE Transactions on Electron Devices. 2023. link Times cited: 0 Abstract: The structural stability of tight-pitched (18 nm and below) … read moreAbstract: The structural stability of tight-pitched (18 nm and below) damascene interconnects for back-end-of-line (BEOL) technologies are analyzed using force-field-based molecular dynamics simulations and finite-element modeling. At these pitches surface energy-dominated effects come into the picture, which lead to structural instability. The candidate metals analyzed are beyond copper (Cu) interconnect metals-ruthenium (Ru), cobalt (Co), and tungsten (W); and Cu is analyzed for reference. Cohesive traction and normal bonding energy are calculated using force-field- based molecular dynamics simulations and then fed as input to a finite-element analysis (FEA) tool, where their dependence on the physical dimensions of the interconnect lines is studied. The parameters studied for the BEOL structures are sidewall angle, aspect ratio, the internal stress of the metal, and modulus of elasticity of the dielectric material around the metal to understand the sensitivity of these parameters to the structural stability of the interconnects. We observe that a lower aspect ratio and higher modulus of elasticity of the dielectric results in stable structures whereas, intrinsic stress of the metal and side wall angle have a minor impact on the overall stability. The stability is analyzed at the seed-layer deposition step and based on this study, Co is the most stable alternate metal amongst Ru, Co, and W. read less USED (low confidence) Y. Xiong, J. Zhang, S. Ma, S. Huang, B. Xu, and S. Zhao, “Multiscale modeling of irradiation-induced defect evolution in BCC multi principal element alloys,” Journal of Alloys and Compounds. 2023. link Times cited: 1 USED (low confidence) K. Wang, X. Chen, S. Huang, X. Chen, Z. Wang, and Y. Huang, “Diffusion Behavior Determined by the New N-Body Potential in Highly Immiscible W/Cu System Through Molecular Dynamics Simulations,” SSRN Electronic Journal. 2023. link Times cited: 2 USED (low confidence) J. Ren et al., “Atomistic Simulation of Tribology Behaviors Of Ti-Based Feconiti High Entropy Alloy Coating During Nanoscratching,” SSRN Electronic Journal. 2023. link Times cited: 1 USED (low confidence) H. Zou, Y. Feng, X. Zhang, T. Ohara, and L. Qiu, “Enhancing mechanism of CNT-CNT interface by metal nanoparticle and nanowire effect on the inside and outside of CNT,” International Journal of Thermal Sciences. 2023. link Times cited: 2 USED (low confidence) Y. Liao, P. Li, J. Jia, T. Tao, J. Chen, and M. Xiang, “Molecular dynamics simulation of Au-Ag nanowires under tensile loading,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) N. Fominykh, V. Nikolskiy, and V. Stegailov, “Atomistic model of an oxide film in contact with a liquid metal coolant: Defects concentrations and chemical potentials of dissolved Fe–O,” Computational Materials Science. 2023. link Times cited: 5 USED (low confidence) V.-T. Nguyen and G. A. V. Phan, “Atomistic insight into welding silver nanowires and interfacial characteristics of the welded zone,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) T. E. hafi, O. Bajjou, H. Jabraoui, J. Louafi, M. Mazroui, and Y. Lachtioui, “Effects of cooling rate on the glass formation process and the microstructural evolution of Silver mono-component metallic glass,” Chemical Physics. 2023. link Times cited: 7 USED (low confidence) Y. Li, D. Yang, and W. Qiang, “Atomistic simulations of enhanced irradiation resistance and defect properties in body-centered cubic W-V-Cr and W-Ta-V alloys,” Journal of Alloys and Compounds. 2023. link Times cited: 2 USED (low confidence) M. Kateb, J. Gudmundsson, and S. Ingvarsson, “Epitaxial growth and characterization of (001) [NiFe/M]20 (M = Cu, CuPt and Pt) superlattices,” Surfaces and Interfaces. 2023. link Times cited: 0 USED (low confidence) X. Li, W. Zhang, and L. Miao, “Research on diffusion wear mechanism of WC tool cutting Al2024 based on MD,” The International Journal of Advanced Manufacturing Technology. 2023. link Times cited: 0 USED (low confidence) G. Dai, S. Wu, X. Huang, M. Wang, and R. Wu, “Research on the coating formation of Al-induced electroless plating on metallic surfaces,” Journal of Materials Science. 2023. link Times cited: 3 USED (low confidence) C. Zhang et al., “Strong and ductile refractory high-entropy alloys with super formability,” Acta Materialia. 2023. link Times cited: 12 USED (low confidence) Y. Hu et al., “Tensile responses of polycrystalline Mo via molecular dynamics simulation: Grain size and temperature effects,” Materials Chemistry and Physics. 2023. link Times cited: 8 USED (low confidence) T. Cheng et al., “Enhanced resistance to helium irradiations through unusual interaction between high-entropy-alloy and helium,” Acta Materialia. 2023. link Times cited: 7 USED (low confidence) F. Tan, L. Li, J. Li, B. Liu, P. Liaw, and Q. Fang, “Multiscale modelling of irradiation damage behavior in high entropy alloys,” Advanced Powder Materials. 2023. link Times cited: 3 USED (low confidence) Z. Sun, Y. Ma, Y. He, Q. Chang, B. Zhang, and L. Zhang, “Phase Transition Induced Low Temperature Diffusion Bonding of Zr-4 Alloy Using a Pure Ti Interlayer,” Journal of Alloys and Compounds. 2023. link Times cited: 1 USED (low confidence) H. Huang, X. Yuan, L. Ma, J. Lin, G. Zhang, and B. Cai, “Atomistic simulations of defect accumulation and evolution in heavily irradiated titanium for nuclear-powered spacecraft,” Nuclear Engineering and Technology. 2023. link Times cited: 0 USED (low confidence) A. Farahvash, M. Agrawal, A. Peterson, and A. Willard, “Modeling Surface Vibrations and Their Role in Molecular Adsorption: A Generalized Langevin Approach.,” Journal of chemical theory and computation. 2023. link Times cited: 0 Abstract: The atomic vibrations of a solid surface can play a signific… read moreAbstract: The atomic vibrations of a solid surface can play a significant role in the reactions of surface-bound molecules, as well as their adsorption and desorption. Relevant phonon modes can involve the collective motion of atoms over a wide array of length scales. In this paper, we demonstrate how the generalized Langevin equation can be utilized to describe these collective motions weighted by their coupling to individual sites. Our approach builds upon the generalized Langevin oscillator (GLO) model originally developed by Tully. We extend the GLO by deriving parameters from atomistic simulation data. We apply this approach to study the memory kernel of a model platinum surface and demonstrate that the memory kernel has a bimodal form due to coupling to both low-energy acoustic modes and high-energy modes near the Debye frequency. The same bimodal form was observed across a wide variety of solids of different elemental compositions, surface structures, and solvation states. By studying how these dominant modes depend on the simulation size, we argue that the acoustic modes are frozen in the limit of macroscopic lattices. By simulating periodically replicated slabs of various sizes, we quantify the influence of phonon confinement effects in the memory kernel and their concomitant effect on simulated sticking coefficients. read less USED (low confidence) B. Dong et al., “A CALPHAD-MD coupled method to reveal the strengthening mechanism in precipitation-strengthening Cu-Ni-Al alloy with evolving microstructures,” International Journal of Plasticity. 2023. link Times cited: 0 USED (low confidence) C. Liu, X. Zhu, L. Ye, Z. Tong, and X. Li, “Directional transport and coalescence behavior on Titanium-Tantalum alloy surface: insights from experiment and molecular dynamics simulation,” Tribology International. 2023. link Times cited: 0 USED (low confidence) Y. Hu, J. Xu, L. Su, Y. Zhang, S. Ding, and R. Xia, “Atomistic simulations of mechanical characteristics dependency on relative density, grain size, and temperature of nanoporous tungsten,” Physica Scripta. 2022. link Times cited: 0 Abstract: A series of atomistic simulations are adopted to explore the… read moreAbstract: A series of atomistic simulations are adopted to explore the influences of relative density, grain size, and temperature on the tensile characteristics of nanoporous tungsten (NPW). Results illustrate that the dominant mechanism of deformation for monocrystalline NPW is the combination of twin boundaries (TBs) migration and 1/2 〈111〉 dislocation movement. The relative density, which has a positive relationship with stiffness and strength, significantly affects the mechanical properties of NPW. With relative density growing from 0.30 to 0.60, Young’s modulus, UTS, and yield strength of monocrystalline NPW increase from 18.55, 0.65, and 0.45 GPa to 93.78, 2.93, and 2.59 GPa, respectively. Young’s modulus and relative density have a quadratic relationship, meaning that the dominant deformation is the bending deformation of ligaments during the elastic stage. The scaling law for yield strength reveals that the axial yielding of ligaments dominates the yielding behavior of NPW. The relationship between mean grain size (5.00 ∼ 17.07 nm) and strength follows the reverse Hall-Petch relation. Besides, the effect of temperature on mechanical characteristics is discussed. With the increase of temperature from 10 K to 1500 K, Young’s modulus of monocrystalline NPW and nanocrystalline NPW (d = 5.00, 10.99, and 17.07 nm) decrease from 69.24, 51.73, 61.08, and 63.75 GPa to 48.98, 34.77, 44.65, and 49.05 GPa. The findings systematically reveal the mechanical properties of NPW under tension and provide guidance for its application. read less USED (low confidence) L. C. Thijs et al., “On the surface chemisorption of oxidizing fine iron particles: Insights gained from molecular dynamics simulations,” Combustion and Flame. 2022. link Times cited: 4 USED (low confidence) O. Kushnerov, “Computer simulation of AlCoCuFeNi high-entropy alloy thin film deposition and crystallization,” Journal of Physics and Electronics. 2022. link Times cited: 0 Abstract: The processes of deposition and crystallization of high-entr… read moreAbstract: The processes of deposition and crystallization of high-entropy AlCoCuFeNi alloy thin film on a substrate of silicon (100) are studied by classical molecular dynamics simulation. Total simulation time reaches 50 ns. The embedded atom model is used to describe the interaction among Al–Co–Cu–Ni–Fe. Interaction between the atoms of Al, Co, Cu, Fe, Ni, and the Si substrate is described using the Lennard–Jones potential, and the interaction between the silicon atoms was modeled using the Stillinger–Weber potential. It is found that at the first stage of deposition small clusters are formed and the process of crystallization starts after ~ 5 ns of simulation, at the characteristic sizes of clusters of about 2 nm. At the end of the simulation, after the 50 ns of modeling, the simulated film contains a face-centered cubic phase, a body-centered cubic phase, a hexagonal close-packed phase, and an amorphous phase. An analysis of the radial distribution of atoms makes it possible to determine the distances between the nearest neighbors and estimate the lattice parameters of these phases. read less USED (low confidence) A.-S. Tran, D.-Q. Doan, and V. Chu, “Molecular simulation study on mechanical properties and elastic recovery of nanoimprinted CuAgAu metallic glasses,” Journal of Non-Crystalline Solids. 2022. link Times cited: 4 USED (low confidence) S. Ahmad, T. Brink, C. Liebscher, and G. Dehm, “Microstates and defects of incoherent Σ3 [111] twin boundaries in aluminum,” Acta Materialia. 2022. link Times cited: 4 USED (low confidence) X.-W. Chen, R. Huang, and Y. Wen, “Phase segregation in bimetallic nanodroplets,” Journal of Materials Science. 2022. link Times cited: 0 USED (low confidence) Z.-H. Li, C. Lu, A. Shi, S. Zhao, B. Ou, and N. Wei, “A Multi-Scale Study on Deformation and Failure Process of Metallic Structures in Extreme Environment,” International Journal of Molecular Sciences. 2022. link Times cited: 0 Abstract: It is a macro-micro model study for defect initiation, growt… read moreAbstract: It is a macro-micro model study for defect initiation, growth and crack propagation of metallic truss structure under high engine temperature and pressure conditions during the reentry atmosphere. Till now, the multi-scale simulation methods for these processes are still unclear. We explore the deformation and failure processes from macroscale to nanoscale using the Gas-Kinetic Unified Algorithm (GKUA) and all-atomic, molecular dynamic (MD) simulation method. The behaviors of the dislocations, defect evolution and crack propagation until failure for Aluminum-Magnesium (Al-Mg) alloy are considered with the different temperature background and strain fields. The results of distributions of temperature and strain field in the aerodynamic environment obtained by molecular dynamics simulations are in good agreement with those obtained from the macroscopic Boltzmann method. Compared to the tensile loading, the alloy structure is more sensitive to compression loading. The polycrystalline Al-Mg alloy has higher yield strength with a larger grain size. It is due to the translation of plastic deformation mode from grain boundary (GB) sliding to dislocation slip and the accumulation of dislocation line. Our findings have paved a new way to analyze and predict the metallic structural failure by micro-scale analysis under the aerodynamic thermal extreme environment of the reentry spacecraft on service expiration. read less USED (low confidence) L. Zepeda-Ruiz, “Melting temperature, critical nucleus size, and interfacial free energy in single FCC metals — A Molecular Dynamics study of liquid–solid phase equilibria,” Journal of Crystal Growth. 2022. link Times cited: 0 USED (low confidence) P. Wu and Y. Yang, “Melting characteristics and strain-based mechanical characterization of single metal nanoparticles,” Journal of Nanoparticle Research. 2022. link Times cited: 0 USED (low confidence) Y. Li, D. Yang, and W. Qiang, “Abnormal notch brittleness induced by short-range ordering in low-cobalt iron-cobalt alloys under tensile and impact loading:A combined experimental and molecular dynamics simulation study,” Journal of Alloys and Compounds. 2022. link Times cited: 2 USED (low confidence) K. Jiang et al., “Abnormal hardening and amorphization in an FCC high entropy alloy under extreme uniaxial tension,” International Journal of Plasticity. 2022. link Times cited: 14 USED (low confidence) X. Yue, Z. Chen, and J. Liu, “Investigation of subsurface defects structural evolution in nano electro machining of copper,” Materials Today Communications. 2022. link Times cited: 0 USED (low confidence) B. Gu, W. Wang, S. Shen, Z. Chen, and H. Ma, “Investigation of formation and breakage mechanism of microweld of typical wire-bonding materials via molecular dynamics simulation,” MRS Communications. 2022. link Times cited: 3 Abstract: Wire bonding is the most popular first-level interconnection… read moreAbstract: Wire bonding is the most popular first-level interconnection technology used in traditional electronic packaging but still needs fundamental research. This work investigated the formation and breakage mechanism of wire-bonding microwelds in typical wire-bonding material pairs by analyzing the loading force curve, total energy curve, atomic morphology, and von Mises stress. The longer the range of the high attractive force of the atoms of a material, the earlier the real contact between the wire and substrate occurs, which means the bonding is completed earlier. The higher stiffness of the material strengthens the weld and causes damage during the bonding process. Graphical abstract read less USED (low confidence) K. Sikdar, A. Mahata, B. Roy, and D. Roy, “Thermokinetic stabilisation of nanocrystalline Cu by ternary approach,” Philosophical Magazine. 2022. link Times cited: 0 Abstract: ABSTRACT Nanocrystalline alloy design with the synergistic c… read moreAbstract: ABSTRACT Nanocrystalline alloy design with the synergistic contribution of ‘thermodynamic’ and ‘kinetic’ stabilisation mechanism leads to much superior microstructural stabilisation at elevated temperatures. Ternary Cu98.5W1Zr0.5 (at. %) alloy, synthesised by mechanical milling under cryogenic temperature followed by consolidation through hot pressing at 550°C, has been examined to access the potential of their concurrence. A meager drop in hardness (∼0.5 GPa) confirms the stability of the alloy up to 800°C. The effect of alloy addition has been studied in terms of microstructure alteration, measured by X-ray diffraction, transmission electron microscopy, and Molecular dynamics (MD) simulation. In addition, the shear punch test (SPT) has been employed to assess the mechanical property of the consolidated alloy. Results suggest that the current approach provides a framework en route to designing bulk nanostructured alloys adapting the bottom-up method. read less USED (low confidence) J. J. Wang, J. Gong, A. McGaughey, and D. Segal, “Simulations of heat transport in single-molecule junctions: Investigations of the thermal diode effect.,” The Journal of chemical physics. 2022. link Times cited: 4 Abstract: With the objective of understanding microscopic principles g… read moreAbstract: With the objective of understanding microscopic principles governing thermal energy flow in nanojunctions, we study phononic heat transport through metal-molecule-metal junctions using classical molecular dynamics (MD) simulations. Considering a single-molecule gold-alkanedithiol-gold junction, we first focus on aspects of method development and compare two techniques for calculating thermal conductance: (i) The Reverse Nonequilibrium MD (RNEMD) method, where heat is inputted and extracted at a constant rate from opposite metals. In this case, the thermal conductance is calculated from the nonequilibrium temperature profile that is created at the junction. (ii) The Approach-to-Equilibrium MD (AEMD) method, with the thermal conductance of the junction obtained from the equilibration dynamics of the metals. In both methods, simulations of alkane chains of a growing size display an approximate length-independence of the thermal conductance, with calculated values matching computational and experimental studies. The RNEMD and AEMD methods offer different insights, and we discuss their benefits and shortcomings. Assessing the potential application of molecular junctions as thermal diodes, alkane junctions are made spatially asymmetric by modifying their contact regions with the bulk, either by using distinct endgroups or by replacing one of the Au contacts with Ag. Anharmonicity is built into the system within the molecular force-field. We find that, while the temperature profile strongly varies (compared with the gold-alkanedithiol-gold junctions) due to these structural modifications, the thermal diode effect is inconsequential in these systems-unless one goes to very large thermal biases. This finding suggests that one should seek molecules with considerable internal anharmonic effects for developing nonlinear thermal devices. read less USED (low confidence) R. Song et al., “Ultrafine nanoporous intermetallic catalysts by high-temperature liquid metal dealloying for electrochemical hydrogen production,” Nature Communications. 2022. link Times cited: 24 USED (low confidence) Z. Shi, Y.-X. Shen, D. Peng, Y. C. Jiang, and H. Gong, “Fundamental effect of lead on mechanical properties of iron from a constructed iron-lead potential,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) S. Luo, J. Luo, Q.-liang Kang, Z. Li, and G. Wang, “Molecular dynamics simulation and experimental verification of the crystallization behavior of a Ti–Zr–Ni alloy prepared by SPS low temperature liquid phase sintering,” Vacuum. 2022. link Times cited: 1 USED (low confidence) Y. Lei et al., “An Embedded-Atom Method Potential for studying the properties of Fe-Pb solid-liquid interface,” Journal of Nuclear Materials. 2022. link Times cited: 1 USED (low confidence) Y. Guo et al., “In situ observation of atomic-scale processes accomplishing grain rotation at mixed grain boundaries,” Acta Materialia. 2022. link Times cited: 6 USED (low confidence) V.-T. Nguyen and T. Fang, “Temperature and vibration-assisted effects in nanoimprint gold: An atomistic study,” Materials Chemistry and Physics. 2022. link Times cited: 3 USED (low confidence) L. Zhang, G. Csányi, E. Giessen, and F. Maresca, “Atomistic fracture in bcc iron revealed by active learning of Gaussian approximation potential,” npj Computational Materials. 2022. link Times cited: 3 USED (low confidence) A. Hegde, E. Weiss, W. Windl, H. Najm, and C. Safta, “Bayesian calibration of interatomic potentials for binary alloys,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) W. Liu, Z. Li, X. Bai, Y. Ma, and C. Liang, “A unified model for yield strength and plastic behavior of nanovoid evolution in tungsten based on molecular dynamics simulations,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) B. Yao, Z. R. Liu, and R. F. Zhang, “EAPOTc: An integrated empirical interatomic potential optimization platform for compound solids,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) Y. Li, Z. Huang, K.-yan Wang, D. Yang, F. Ding, and W. Qiang, “Abnormally fast crack propagation induced by short-range ordering in iron-cobalt alloys: A combined experiments and molecular dynamics simulations,” Journal of Alloys and Compounds. 2022. link Times cited: 3 USED (low confidence) P. Ou, Z. Cao, J. Rong, and X. Yu, “Molecular Dynamics Study on the Welding Behavior in Dissimilar TC4-TA17 Titanium Alloys,” Materials. 2022. link Times cited: 3 Abstract: Titanium alloys have become the material of choice for marin… read moreAbstract: Titanium alloys have become the material of choice for marine parts manufacturing due to their high specific strength and excellent resistance to seawater corrosion. However, it is still challenging for a single titanium alloy to meet the comprehensive specifications of a structural component. In this study, we have applied a molecular dynamics approach to simulate the aging phase transformation, K-TIG welding process, and mechanical properties of the TC4-TA17 (Ti6Al4V-Ti4Al2V) alloy. The results show that during the aging phase transformation process, changes in the structure of the titanium alloys are mainly manifested in the precipitation of a new phase from the sub-stable β-phase, and after the state stabilization, the α-phase content reaches 45%. Moreover, during the melting and diffusion process of TC4-TA17, aluminum atoms near the interface diffuse, followed by titanium atoms, while relatively few vanadium atoms are involved in the diffusion. Finally, the results of tensile simulations of the TC4-TA17 alloy after welding showed that stress values can reach up to 9.07 GPa and that the mechanical properties of the alloy in the weld zone are better than those of the single alloys under the same conditions. This study will provide theoretical support for the optimization of process parameters for TC4-TA17 alloy welding. read less USED (low confidence) X. Li, Y. Shi, J. An, J. Chen, and T. Chen, “The research on the essence of unsteady mechanical behavior of Fe14.6Ni (at%) nanocrystalline elastocaloric refrigeration alloy through molecular dynamics simulation,” Journal of Materials Research and Technology. 2022. link Times cited: 1 USED (low confidence) Y. Zhang et al., “Atomistic modeling of surface and grain boundary dislocation nucleation in FCC metals,” Acta Materialia. 2022. link Times cited: 11 USED (low confidence) T.-X. Bui, T. Fang, and C.-I. Lee, “Deformation and machining mechanism of nanocrystalline NiCoCrFe high entropy alloys,” Journal of Alloys and Compounds. 2022. link Times cited: 8 USED (low confidence) Y. Kashyrina, A. S. Muratov, V. Kazimirov, and O. S. Roik, “X-ray diffraction study and molecular dynamic simulation of liquid Al-Cu alloys: a new data and interatomic potentials comparison,” Journal of Molecular Modeling. 2022. link Times cited: 0 USED (low confidence) M. K. Gupta, V. Panwar, and R. Mahapatra, “Computational analysis of mechanical behavior and potential energy of thermoresponsive copper-tantalum nanoalloy,” Journal of Molecular Modeling. 2022. link Times cited: 1 USED (low confidence) G. Dai, S. Wu, X. Huang, M.-jie Wang, and X. Teng, “Thermal diffusion behavior of Fe/Cu/Ni multilayer coatings: a molecular dynamics study,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 1 Abstract: In this paper, the thermal diffusion behavior of Fe/Cu/Ni mu… read moreAbstract: In this paper, the thermal diffusion behavior of Fe/Cu/Ni multilayer coatings was investigated by molecular dynamics. The results show that the Fe, Cu, and Ni elements can diffuse each other at 1250 K. Meanwhile, the intrinsic diffusion coefficients and interdiffusion coefficients of the Fe, Cu, and Ni were calculated. Besides, the diffusion mechanism for high melting-point elements of Fe and Ni at 1250 K was analyzed in the paper. According to the simulation result, the Fe and Ni lattices were disturbed by the active Cu particles. Fe and Ni particles at higher energies may move out of their original positions and migrate into the Cu lattice randomly. Thus, the Fe and Ni elements were involved in the thermal diffusion. This can be confirmed by the decrease of the peak and the disappearance of the secondary peak in the radial distribution function curves. However, the position of the curve peaks did not change. Thus, the lattice structure was still maintained during the whole diffusion process. The thermal diffusion of the three elements was carried out by particle substitution at the lattice positions. It was a solid phase diffusion process. Furthermore, there was a clear particle diffusion asymmetry at the original interface of the element. It was consistent with the diffusion asymmetry of diffusion-couple experiments. The primary reason for this diffusion asymmetry was the difference in the interaction potential of the three elements. This asymmetry was ultimately reflected in the intrinsic diffusion coefficient and the interdiffusion coefficient of each element. For the Fe–Cu–Ni ternary system, the largest diffusion coefficient was copper and the smallest was iron at 1250 K. read less USED (low confidence) S. Han, G. Barcaro, A. Fortunelli, S. Lysgaard, T. Vegge, and H. Hansen, “Unfolding the structural stability of nanoalloys via symmetry-constrained genetic algorithm and neural network potential,” npj Computational Materials. 2022. link Times cited: 5 USED (low confidence) Y. Zhao et al., “Molecular dynamics study of acoustic softening effect in ultrasonic vibration assisted tension of monocrystalline/polycrystalline coppers,” Journal of Materials Processing Technology. 2022. link Times cited: 9 USED (low confidence) X. Liu et al., “Atomistic understanding of incipient plasticity in BCC refractory high entropy alloys,” Journal of Alloys and Compounds. 2022. link Times cited: 16 USED (low confidence) X. Li, Y. Shi, T.-Y. Chen, S. Wang, and K. Fan, “Study on Sintering Mechanism and Mechanical Properties of Fe–Ni Elastocaloric Refrigeration Alloy through Molecular Dynamics Simulation,” Materials Today Communications. 2022. link Times cited: 4 USED (low confidence) S. Nasiri, G. Yang, E. Spiecker, and Q. Li, “An Improved Approach to Manufacture Carbon Nanotube Reinforced Magnesium AZ91 Composites with Increased Strength and Ductility,” Metals. 2022. link Times cited: 3 Abstract: Multiwalled carbon nanotubes (MWCNTs) are decorated with Pt … read moreAbstract: Multiwalled carbon nanotubes (MWCNTs) are decorated with Pt nanoparticles by a “layer-by-layer” approach using poly (sodium 4-styrene sulfonate) (PSS) and poly (diallyl dimethylammonium chloride) (PDDA). Transmission electron microscopy (TEM) images and Energy Dispersive X-ray (EDX) analysis of the samples confirm Pt deposition on surfaces of CNTs. Dispersibility and dispersion stability of MWCNTs in the solvents are enhanced when MWCNTs are coated with Pt nanoparticles. Mg AZ91 composites reinforced with MWCNTs are then produced by a melt stirring process. Compression tests of the composites show that adding 0.05% wt Pt-coated MWCNTs in AZ91 improves the composite’s mechanical properties compared to the pure AZ91 and pristine MWCNT/AZ91. Fracture surface analysis of the composite using a scanning electron microscope (SEM) shows individual pulled out MWCNTs in the case of the Pt-coated MWCNT/AZ91 composites. This finding can be attributed to the uniform dispersion of Pt-coated MWCNTs in Mg due to the improved wettability of Pt-coated MWCNTs in Mg melts. The study of the pull-out behaviour of pristine and Pt-coated CNTs from an Mg matrix using molecular dynamics simulation supports this interpretation. read less USED (low confidence) P. Hiremath, S. Melin, E. Bitzek, and P. Olsson, “Effects of interatomic potential on fracture behaviour in single- and bicrystalline tungsten,” Computational Materials Science. 2022. link Times cited: 14 USED (low confidence) C. Xue et al., “Molecular dynamics study on the effect of temperature on HCP→FCC phase transition of magnesium alloy,” Journal of Magnesium and Alloys. 2022. link Times cited: 3 USED (low confidence) C. Baruffi and W. Curtin, “Theory of spontaneous grain boundary roughening in high entropy alloys,” Acta Materialia. 2022. link Times cited: 7 USED (low confidence) J. Wang et al., “Deformation evolution of Cu/Ta nanoscale multilayer during nanoindentation by a molecular dynamics study,” Surface and Coatings Technology. 2022. link Times cited: 9 USED (low confidence) J. H. Lee, H. Kang, S.-D. Yim, Y. Sohn, and S. G. Lee, “Revelation of transport properties of ultra-thin ionomer films in catalyst layer of polymer electrolyte membrane fuel cells using molecular dynamics,” Applied Surface Science. 2022. link Times cited: 7 USED (low confidence) M. Celtek, “Atomic structure of Cu60Ti20Zr20 metallic glass under high pressures,” Intermetallics. 2022. link Times cited: 4 USED (low confidence) Q. Jia et al., “Effects of structure relaxation and surface oxidation on nanoscopic wear behaviors of metallic glass,” Acta Materialia. 2022. link Times cited: 54 USED (low confidence) S. Kwon, S. Y. Lee, H. J. Kim, S.-D. Yim, Y. Sohn, and S. G. Lee, “Multiscale simulation approach to investigate the binder distribution in catalyst layers of high-temperature polymer electrolyte membrane fuel cells,” Scientific Reports. 2022. link Times cited: 1 USED (low confidence) Q. Zhang et al., “Size effects and plastic deformation mechanisms in single-crystalline CoCrFeNi micro/nanopillars,” Journal of the Mechanics and Physics of Solids. 2022. link Times cited: 21 USED (low confidence) M. Surana, T. Ahmed, and N. Admal, “Interface mechanics of 2D materials on metal substrates,” Journal of the Mechanics and Physics of Solids. 2022. link Times cited: 1 USED (low confidence) Y. Xu, G. Wang, P. Qian, and Y. Su, “Element segregation and thermal stability of Ni–Rh nanoparticles,” Journal of Solid State Chemistry. 2022. link Times cited: 6 USED (low confidence) C. Dahale, S. G. Srinivasan, S. Mishra, S. Maiti, and B. Rai, “Surface Segregation in AgAuCuPdPt High Entropy Alloy: Insights From Molecular Simulations,” Molecular Systems Design & Engineering. 2022. link Times cited: 1 Abstract: High entropy alloys (HEAs) are emerging as a novel class of … read moreAbstract: High entropy alloys (HEAs) are emerging as a novel class of superior catalysts for diverse chemical conversions. The activity of a catalyst is intimately related to the composition and atomic structure at its surface. In this work, we used embedded atom (EAM) potential based Monte Carlo – Molecular Dynamics simulations to study surface segregation in the equimolar AuAgCuPdPt HEA, that was recently shown to be an efficient catalyst for CO2 electrochemical reduction. Firstly, EAM potentials were extensively validated against experimental segregation data for several different binary and ternary compositions. Subsequently, simulations on the HEA were carried out for four different surface orientations, spherical and cubical nanoparticles, to obtain detailed structural and concentration profiles normal to the surface. In all cases, Ag atoms were found to preferentially segregate to the surface while the subsurface layer mainly consisted of Au atoms. No Pt atoms were found on the surface layer for all systems. A detailed analysis neighborhood of each surface site revealed that the atoms formed a finite number of chemically unique clusters. The percentage of chemically unique sites were larger for elements with lower concentration at the surface. Together with the physical diversity surrounding each site, the enrichment of one or more element(s) at the surface also increased its number of unique catalytically active sites. Results from our work suggest that HEAs are prone to surface segregation and such effects must be taken into consideration while modeling the surface chemistry of these materials. read less USED (low confidence) K. Yashiro, K. Suzuki, and K. Naito, “Ab-initio DFT Calculations on Elastic Coefficients, (001) Surface Energy, Stability Limit of Pure Metals and Separation Energy of Bimetal Interface,” Journal of the Society of Materials Science, Japan. 2022. link Times cited: 1 Abstract: Ab-initio density functional theory (DFT) calculations on th… read moreAbstract: Ab-initio density functional theory (DFT) calculations on the elastic coefficients at no-load equilibrium, (001) surface energy and stability limit under the [001] static tension are implemented for eight fcc, four bcc and four hcp metals. The elastic coefficients and surface energy are compared with DFT database of Materials Project, Zhou’s EAM potential, and experimental data available. The stability limit is evaluated with the eigenvalues of the elastic stiffness matrix, B ij = ∆ σ i / ∆ ε j . Here ∆ σ i and ∆ ε j are the change in the stress and strain in Voigt notation. Most elements show instability for the eigenvalues of η Born (= B 11 − B 12 ) or η spinodal of the 3x3 partial matrix of B ij . Pt and W show instability for shear component B 44 while Fe does for B 66 . Then the separation energy of bimetal interface for 120 combinations of these 16 metal elements are estimated from the energy deference of adhered/separated interface in a supercell of stacked unit lattices. read less USED (low confidence) G. Kamalakshi, P. Pant, and M. Gururajan, “Deformation behaviour of Cu and Cu–Al in the dislocation starved regime: A molecular dynamics study,” Computational Materials Science. 2022. link Times cited: 4 USED (low confidence) L. Chang, X. Liu, J. Zhao, and C.-yu Zhou, “Effect of interatomic potential on modelling fracture behavior in hcp titanium: A molecular dynamics study,” Journal of Materials Research and Technology. 2022. link Times cited: 3 USED (low confidence) R. Bodlos, V. Fotopoulos, J. Spitaler, A. Shluger, and L. Romaner, “Energies and structures of Cu/Nb and Cu/W interfaces from density functional theory and semi-empirical calculations,” Materialia. 2022. link Times cited: 9 USED (low confidence) J. Shi et al., “Nanoscratching-induced plastic deformation mechanism and tribology behavior of Cu/Ta bilayer and multilayer by a molecular dynamics study,” Applied Surface Science. 2022. link Times cited: 11 USED (low confidence) A. Roy, R. Devanathan, D. D. Johnson, and G. Balasubramanian, “Grain-Size Effects on the Deformation in Nanocrystalline Multi-Principal Element Alloy,” Computational Materials Science eJournal. 2022. link Times cited: 9 Abstract: Multi-principal element alloys (MPEAs) continue to garner&nb… read moreAbstract: Multi-principal element alloys (MPEAs) continue to garner interest due to their remarkable mechanical properties, especially at elevated temperatures. Here, we examine a representative nanocrystalline refractory MPEA and identify a crossover from a Hall-Petch to inverse-Hall-Petch relation. While the considered MPEA predominantly assumes a single-phase BCC lattice, the presence of grain boundaries imparts amorphous phase distributions that increase with decreasing grain size (i.e., increasing grain boundary volume fraction). Using molecular dynamics simulations, we find that the yield strength of the MPEA increases with decreasing average grain size, but below a critical grain size < 23.2 nm the yield strength decreases. This change in the deformation behavior is driven by the transition from dislocation slip to grain-boundary slip as the predominant mechanism. Our results reveal that the change from Hall-Petch to inverse-Hall-Petch regime is correlated to dislocation stacking at the grain boundary when dislocation density reaches a maximum. read less USED (low confidence) I. M. P. Espinosa, S. Azadehranjbar, R. Ding, A. Baker, T. Jacobs, and A. Martini, “Platinum nanoparticle compression: Combining in situ TEM and atomistic modeling,” Applied Physics Letters. 2022. link Times cited: 6 USED (low confidence) L. Yang, Y.-X. Shen, S. Mi, J. Fan, and H. Gong, “Phase stability and mechanical property of W–Cu solid solutions from a newly derived W–Cu potential,” Physica B: Condensed Matter. 2022. link Times cited: 5 USED (low confidence) J. Li, Y. Hu, Y. Zhang, and R. Xia, “Optimum atomic concentration in structurally disordered nanoporous Pt–Co alloys with the strongest mechanical properties,” Microporous and Mesoporous Materials. 2022. link Times cited: 4 USED (low confidence) H. He, S. Ma, and S. Wang, “Survey of Grain Boundary Energies in Tungsten and Beta-Titanium at High Temperature,” Materials. 2021. link Times cited: 1 Abstract: Heat treatment is a necessary means to obtain desired proper… read moreAbstract: Heat treatment is a necessary means to obtain desired properties for most of the materials. Thus, the grain boundary (GB) phenomena observed in experiments actually reflect the GB behaviors at relatively high temperature to some extent. In this work, 405 different GBs were systematically constructed for body-centered cubic (BCC) metals and the grain boundary energies (GBEs) of these GBs were calculated with molecular dynamics for W at 2400 K and β-Ti at 1300 K and by means of molecular statics for Mo and W at 0 K. It was found that high temperature may result in the GB complexion transitions for some GBs, such as the Σ11{332}{332} of W. Moreover, the relationships between GBEs and sin(θ) can be described by the functions of the same type for different GB sets having the same misorientation axis, where θ is the angle between the misorientation axis and the GB plane. Generally, the GBs tend to have lower GBE when sin(θ) is equal to 0. However, the GB sets with the <110> misorientation axis have the lowest GBE when sin(θ) is close to 1. Another discovery is that the local hexagonal-close packed α phase is more likely to form at the GBs with the lattice misorientations of 38.9°/<110>, 50.5°/<110>, 59.0°/<110> and 60.0°/<111> for β-Ti at 1300 K. read less USED (low confidence) S. Zhao, “Effects of local elemental ordering on defect-grain boundary interactions in high-entropy alloys,” Journal of Alloys and Compounds. 2021. link Times cited: 19 USED (low confidence) Z. Jian et al., “Shock-induced plasticity and phase transformation in single crystal magnesium: an interatomic potential and non-equilibrium molecular dynamics simulations,” Journal of Physics: Condensed Matter. 2021. link Times cited: 8 Abstract: An effective and reliable Finnis–Sinclair (FS) type potentia… read moreAbstract: An effective and reliable Finnis–Sinclair (FS) type potential is developed for large-scale molecular dynamics (MD) simulations of plasticity and phase transition of magnesium (Mg) single crystals under high-pressure shock loading. The shock-wave profiles exhibit a split elastic–inelastic wave in the [0001]HCP shock orientation and a three-wave structure in the [10-10]HCP and [-12-10]HCP directions, namely, an elastic precursor, a followed plastic front, and a phase-transition front. The shock Hugoniot of the particle velocity (U p) vs the shock velocity (U s) of Mg single crystals in three shock directions under low shock strength reveals apparent anisotropy, which vanishes with increasing shock strength. For the [0001]HCP shock direction, the amorphization caused by strong atomic strain plays an important role in the phase transition and allows for the phase transition from an isotropic stressed state to the product phase. The reorientation in the shock directions [10-10]HCP and [-12-10]HCP, as the primary plasticity deformation, leads to the compressed hexagonal close-packed (HCP) phase and reduces the phase-transition threshold pressure. The phase-transition pathway in the shock direction [0001]HCP includes a preferential contraction strain along the [0001]HCP direction, a tension along [-12-10]HCP direction, an effective contraction and shear along the [10-10]HCP direction. For the [10-10]HCP and [-12-10]HCP shock directions, the phase-transition pathway consists of two steps: a reorientation and the subsequent transition from the reorientation hexagonal close-packed phase (RHCP) to the body-centered cubic (BCC). The orientation relationships between HCP and BCC are (0001)HCP ⟨-12-10⟩HCP // {110}BCC ⟨001⟩BCC. Due to different slipping directions during the phase transition, three variants of the product phase are observed in the shocked samples, accompanied by three kinds of typical coherent twin-grain boundaries between the variants. The results indicate that the highly concentrated shear stress leads to the crystal lattice instability in the elastic precursor, and the plasticity or the phase transition relaxed the shear stress. read less USED (low confidence) G. Singh, A. Waas, and V. Sundararaghavan, “Understanding defect structures in nanoscale metal additive manufacturing via molecular dynamics,” Computational Materials Science. 2021. link Times cited: 11 USED (low confidence) S. Gowthaman and T. Jagadeesha, “Study on the effect of temperature and strain rate on Ni2FeCrCuAl high entropy alloy: a molecular dynamics study,” Engineering Research Express. 2021. link Times cited: 1 Abstract: High entropy alloy has offered significant attention in vari… read moreAbstract: High entropy alloy has offered significant attention in various material science applications, due to its excellent material features. In this investigation, the mechanical characteristics of Ni2FeCrCuAl High Entropy Alloy (HEA) have been examined under variable temperature and strain rates to analyze its influence over the material features of high entropy alloy through Molecular Dynamics (MD) simulation and it is stated that the formation of various point defects and dislocations are the major cause for the augmentation of tensile deformation which impacts the tensile behavior of high entropy alloy. Moreover, the Radial Distribution Function (RDF) has been examined throughout tensile deformation, to investigate the impact of applied stress over the de-bonding of various atoms and it is found that the strain rate has a greater beneficial impact over the material feature trailed by the temperature outcome, owed to its superior impact on the formation of point defects and shear strain during tensile characterization. read less USED (low confidence) C. Liu, X. Zhu, X. Li, and Q. Shi, “Investigation on sintering processes and mechanical properties of Ti–Ta alloys by molecular dynamics simulation.,” Powder Technology. 2021. link Times cited: 14 USED (low confidence) H. He, S. Ma, and S. Wang, “Molecular dynamics investigation on tilt grain boundary energies of beta-titanium and tungsten at high temperature,” Materials Research Express. 2021. link Times cited: 2 Abstract: The grain boundary energies (GBEs) of symmetric tilt grain b… read moreAbstract: The grain boundary energies (GBEs) of symmetric tilt grain boundaries (STGBs) and asymmetric tilt grain boundaries (ATGBs) for W at 0 and 2400 K and β-Ti at 1300 K were calculated by means of molecular statics and dynamics simulations to investigate the effects of temperature on GBE and the relationships between GBEs and grain boundary (GB) planes. Generally, the variation trends of GBE with the tilt angle are similar for the three cases, when the tilt axis is specified. It is of course that these similarities result from their similar GB microstructures in most cases. However, the variation trends of GBE with tilt angle are somewhat different between β-Ti at 1300 K and W at 2400 K for STGBs with <100> and <110> tilt axes. This difference mainly stems from the following two reasons: firstly, the GB microstructures of W at 2400 K and β-Ti at 1300 K are different for some STGBs; secondly, the atoms at the STGB of β-Ti at 1300 K tend to evolve into the local ω- or α-like structures distributed at the STGB for some STGBs with <110> tilt axis, which makes the corresponding STGBs more stable, thereby decreasing the GBEs. Furthermore, a geometric parameter θ, the angle between the misorientation axis and the GB plane, was defined to explore the relationships between GBEs and GB planes. It was found that the relationships between GBEs and GB planes can be described by some simple functions of sin(θ) for the GBs with definite lattice misorientation, which can well explain and predict the preferred GB planes for the GBs having the same lattice misorientation. Our calculations not only extend the investigation of GBs to higher temperature, but also deepen the understanding on the temperature contributions to the microstructure evolution at GBs and on the relationships between GBEs and possible geometric parameters. read less USED (low confidence) O. Uche and M. Sidorick, “A Comparative Analysis of the Vibrational and Structural Properties of Nearly Incommensurate Overlayer Systems,” Surface Science. 2021. link Times cited: 0 USED (low confidence) Q. Li, J. Zhang, H. Tang, H. W. Zhang, H. Ye, and Y. Zheng, “Influence of Mo Segregation at Grain Boundaries on the High Temperature Creep Behavior of Ni-Mo Alloys: An Atomistic Study,” Materials. 2021. link Times cited: 0 Abstract: Based on molecular dynamics simulations, the creep behaviors… read moreAbstract: Based on molecular dynamics simulations, the creep behaviors of nanocrystalline Ni before and after the segregation of Mo atoms at grain boundaries are comparatively investigated with the influences of external stress, grain size, temperature, and the concentration of Mo atoms taken into consideration. The results show that the creep strain rate of nanocrystalline Ni decreases significantly after the segregation of Mo atoms at grain boundaries due to the increase of the activation energy. The creep mechanisms corresponding to low, medium, and high stress states are respectively diffusion, grain boundary slip and dislocation activities based on the analysis of stress exponent and grain size exponent for both pure Ni and segregated Ni-Mo samples. Importantly, the influence of external stress and grain size on the creep strain rate of segregated Ni-Mo samples agrees well with the classical Bird-Dorn-Mukherjee model. The results also show that segregation has little effect on the creep process dominated by lattice diffusion. However, it can effectively reduce the strain rate of the creep deformation dominated by grain boundary behaviors and dislocation activities, where the creep rate decreases when increasing the concentration of Mo atoms at grain boundaries within a certain range. read less USED (low confidence) F. Zhang et al., “Magnetocaloric Effect in the (Mn,Fe)2(P,Si) System: From Bulk to Nano,” Acta Materialia. 2021. link Times cited: 15 USED (low confidence) I. Lutsenko, P. Zakharov, M. Starostenkov, S. Dmitriev, and E. Korznikova, “Stability of supratransmission waves in a crystal of A3B stoichiometry upon interaction with single dislocations,” Journal of Physics: Conference Series. 2021. link Times cited: 2 Abstract: Supratransmission waves are stable objects that can exist in… read moreAbstract: Supratransmission waves are stable objects that can exist in different discrete environments. In this paper, we consider the interaction of such waves with single edge dislocations of various configurations in a crystal with A3B stoichiometry. The model was a Pt3Al crystal, the potential obtained by the embedded atom method was used to describe the interaction of its atoms. Quantitative characteristics of the wave were obtained before and after the interaction. It is found that the degree of energy dissipation by dislocations depends on the mutual orientation of the wave front and the extra plane of the dislocation. Numerical estimates are made for four different configurations. The results obtained can be useful in studying the propagation of soliton-type waves in defect crystals of various compositions. read less USED (low confidence) S. Zhao, “Role of chemical disorder and local ordering on defect evolution in high-entropy alloys,” Physical Review Materials. 2021. link Times cited: 15 Abstract: High-entropy alloys (HEAs) have stimulated great interest du… read moreAbstract: High-entropy alloys (HEAs) have stimulated great interest due to their remarkable mechanical and irradiation performance. Experiments suggest that delayed defect evolution in HEAs, compared to conventional metals and dilute alloys, is the main reason for their improved irradiation resistance. However, the mechanism responsible for the observation remains elusive. Here we show that the potential energy landscape of defects under the influence of random arrangement of different species is the reason for the delayed defect evolution. We arrive at the conclusion by investigating the diffusion of defects and defect clusters under three cases: the averaged-atom model, random model, and the model with local short-range ordering. Our results suggest that, compared to the average model, the chemical fluctuation inherent in HEAs can suppress interstitial motion more than vacancy motion. The effects are more pronounced when SRO develops. For defect clusters, the chemical disorder can reduce their jump frequencies significantly and enhance correlation effects, leading to suppressed defect motion. Notably, we find that with SRO, such defect motion can be entirely trapped in local regions. This work demonstrates that chemical fluctuations and SRO are the main reason responsible for the suppressed defect evolution in HEAs, which dictates a promising way to improve the irradiation performance of HEAs through manipulating its chemical disorder states, such as local ordering. read less USED (low confidence) H. Lang et al., “Superior lubrication and electrical stability of graphene as highly effective solid lubricant at sliding electrical contact interface,” Carbon. 2021. link Times cited: 21 USED (low confidence) S. Zhao, Y. Xiong, S.-hui Ma, J. Zhang, B. Xu, and J. Kai, “Defect accumulation and evolution in refractory multi-principal element alloys,” Acta Materialia. 2021. link Times cited: 30 USED (low confidence) Z. Liang, Y. Jiang, X. Gong, and H. Gong, “Atomistic modelling of the immiscible Fe–Bi system from a constructed bond order potential,” Journal of Physics: Condensed Matter. 2021. link Times cited: 2 Abstract: An analytical bond-order potential (BOP) of Fe–Bi has been c… read moreAbstract: An analytical bond-order potential (BOP) of Fe–Bi has been constructed and has been validated to have a better performance than the Fe–Bi potentials already published in the literature. Molecular dynamics simulations based on this BOP has been then conducted to investigate the ground-state properties of Bi, structural stability of the Fe–Bi binary system, and the effect of Bi on mechanical properties of BCC Fe. It is found that the present BOP could accurately predict the ground-state A7 structure of Bi and its structural parameters, and that a uniform amorphous structure of Fe100−x Bi x could be formed when Bi is located in the composition range of 26 ⩽ x < 70. In addition, simulations also reveal that the addition of a very small percentage of Bi would cause a considerable decrease of tensile strength and critical strain of BCC Fe upon uniaxial tensile loading. The obtained results are in nice agreement with similar experimental observations in the literature. read less USED (low confidence) D. Roy, S. Pal, C. Tiwary, A. Gupta, P. N. Babu, and R. Mitra, “Stable nanocrystalline structure attainment and strength enhancement of Cu base alloy using bi-modal distributed tungsten dispersoids,” Philosophical Magazine. 2021. link Times cited: 4 Abstract: In this study, an experimental and atomic-scale simulationba… read moreAbstract: In this study, an experimental and atomic-scale simulationbased investigation has been performed to investigate the possible stability of the nano-crystalline structure and thereby considerable strength at a higher temperature in the case of synthesised Cu alloy along with logical understanding. Dispersions of high melting BCC metal (1 at % W) in nanostructured Cu is achieved using conveniently scalable cryomilling followed by hot pressing (at 550 °C). The thermal stability (till 800 °C) of grain size in synthesised nano-crystalline Cu alloy has been examined through X-ray diffraction (XRD) and Transmission Electron Microscopy (TEM). The nano-sized W particles dispersed at the Cu matrix grain boundaries, restricting grain growth by Zener pinning even after annealing was carried out at 800 °C. The hot-pressed pellets of nanocrystalline Cu99W1 alloy with a nearly uniform distribution of W particles have exhibited higher hardness than pure Cu and increased in strength and strain to failure up to 10% and 46%, respectively. The improvement in mechanical properties is further rationalised by the Molecular Dynamics (MD) based simulation findings. ARTICLE HISTORY Received 6 December 2020 Accepted 26 September 2021 read less USED (low confidence) Y. Lin, S. Lu, and P. H. Teng, “Metallurgical Behavior of Amorphous Alloy during Annealing at Different Temperatures via Molecular Dynamics Simulation,” Key Engineering Materials. 2021. link Times cited: 0 Abstract: The mechanism of annealing-induced amorphization of metallic… read moreAbstract: The mechanism of annealing-induced amorphization of metallic glass is investigated in this study via molecular dynamics simulation. Spherical nucleuses of Cu–Ni–Al alloy with a face-centered cubic structure are embedded to simulate nanograins in Cu–Ni–Al amorphous alloy; subsequently, the material is annealed at different temperatures. The results show that the critical radius for nucleation at temperatures above the glass transition temperature (Tg) affected the behavior, grain growth, and annihilation of nanograins in the Cu–Ni–Al amorphous alloy during annealing. When the temperature increased, the critical radius for nucleation increased as well. This causes the small nanograins to annihilate quickly and the large nanograins to develop rapidly. When the annealing temperature is higher than Tg, part of the crystal nuclei, which is smaller than the critical radius, can be eliminated. The crystallinity of the metallic glass decreased, and the minimum crystallinity is attained after a period of annealing simulation. Subsequently, as the residual effective nanograins began developing, the crystallinity of the amorphous metal increased again. Therefore, the annealing duration time is critical to the crystallinity of the amorphous alloy after annealing. read less USED (low confidence) Y. He et al., “Atomistic observation on diffusion-mediated friction between single-asperity contacts,” Nature Materials. 2021. link Times cited: 15 USED (low confidence) J. Peng et al., “The predicted rate-dependent deformation behaviour and multistage strain hardening in a model heterostructured body-centered cubic high entropy alloy,” International Journal of Plasticity. 2021. link Times cited: 26 USED (low confidence) Y. L. Li, X. Cheng, W. Duan, and W. Qiang, “Improved ductility by coupled motion of grain boundaries in nanocrystalline B2-FeCo alloys,” Computational Materials Science. 2021. link Times cited: 7 USED (low confidence) B. Goh and J. Choi, “Mechanical evaluation of bidirectional surface deformation in contact between nanometer-sized carbon particle and copper substrate: A molecular dynamics approach,” Surfaces and Interfaces. 2021. link Times cited: 3 USED (low confidence) T. Zeng, F. Li, and Y. Huang, “Construction of an n-Body Potential for Revealing the Atomic Mechanism for Direct Alloying of Immiscible Tungsten and Copper,” Materials. 2021. link Times cited: 7 Abstract: W-Cu laminated composites are critical materials used to con… read moreAbstract: W-Cu laminated composites are critical materials used to construct nuclear fusion reactors, and it is very important to obtain direct alloying between W and Cu at the W/Cu interfaces of the composites. Our previous experimental studies showed that it is possible to overcome the immiscibility between W and Cu and obtain direct alloying when the alloying temperature is close to the melting point of Cu. Because the W-Cu interatomic potentials published thus far cannot accurately reproduce the alloying behaviors of immiscible W and Cu, an interatomic potential suitable for the W-Cu system has been constructed in the present study. Based on this potential, direct alloying between W and Cu at high temperature has been verified, and the corresponding diffusion mechanism has been studied, through molecular dynamics (MD) simulations. The results indicate that the formation of an amorphous Cu layer at the W/Cu interface plays a critical role in alloying because it allows Cu atoms to diffuse into W. The simulation results for direct alloying between W and Cu can be verified by experimental results and transmission electron microscopy observations. This indicates that the constructed W-Cu potential can correctly model the high-temperature performance of the W-Cu system and the diffusion mechanism of direct alloying between W and Cu. read less USED (low confidence) M. Wagih and C. Schuh, “Thermodynamics and design of nanocrystalline alloys using grain boundary segregation spectra,” Acta Materialia. 2021. link Times cited: 20 USED (low confidence) E. Levo, F. Granberg, K. Nordlund, and F. Djurabekova, “Temperature effect on irradiation damage in equiatomic multi-component alloys,” Computational Materials Science. 2021. link Times cited: 5 USED (low confidence) B. Yao, Z. Liu, and R. Zhang, “EAPOTs: An integrated empirical interatomic potential optimization platform for single elemental solids,” Computational Materials Science. 2021. link Times cited: 3 USED (low confidence) E. Torres, “Atomistic study of the structure and deformation behavior of symmetric tilt grain boundaries in α-zirconium,” Computational Materials Science. 2021. link Times cited: 4 USED (low confidence) S. Starikov and D. Smirnova, “Optimized interatomic potential for atomistic simulation of Zr-Nb alloy,” Computational Materials Science. 2021. link Times cited: 15 USED (low confidence) A. H. M. Faisal and C. Weinberger, “Modeling twin boundary structures in body centered cubic transition metals,” Computational Materials Science. 2021. link Times cited: 6 USED (low confidence) V. Samsonov et al., “Factors of the Stability/Instability of Bimetallic Core–Shell Nanostructures,” Bulletin of the Russian Academy of Sciences: Physics. 2021. link Times cited: 3 USED (low confidence) Q. Yang, C. Xue, Z. Chu, Y. Li, L. Ma, and H. Gao, “Molecular dynamics study on the relationship between phase transition mechanism and loading direction of AZ31,” Scientific Reports. 2021. link Times cited: 1 USED (low confidence) P. Sreeramagiri, A. Roy, and G. Balasubramanian, “Effect of Cooling Rate on the Phase Formation of AlCoCrFeNi High-Entropy Alloy,” Journal of Phase Equilibria and Diffusion. 2021. link Times cited: 18 USED (low confidence) M. Celtek, S. Sengul, U. Domekeli, and V. Guder, “Dynamical and structural properties of metallic liquid and glass Zr48Cu36Ag8Al8 alloy studied by molecular dynamics simulation,” Journal of Non-crystalline Solids. 2021. link Times cited: 12 USED (low confidence) P. Wurm and M. H. Ulz, “A hybrid static–dynamic continuum approach for concurrent atomistic‐to‐continuum methods,” International Journal for Numerical Methods in Engineering. 2021. link Times cited: 0 Abstract: Concurrent atomistic‐to‐continuum methods commonly employ ei… read moreAbstract: Concurrent atomistic‐to‐continuum methods commonly employ either a dynamic or a quasi‐static continuum model. Both of these approaches have advantages and drawbacks which render their applicability problem‐specific. We present a new hybrid static–dynamic continuum model, which combines the advantages of both approaches while potentially removing the drawbacks. This numerical approach is an innovative superposition of a dynamic and a quasi‐static subproblem and, thus, is limited to linear elastic continua. We apply the approach to a prototypical representative of the concurrent atomistic‐to‐continuum methods and namely, the coupled atomistic and discrete dislocation method. We present three numerical examples to compare the performance of the dynamic, quasi‐static and hybrid continuum model. read less USED (low confidence) Y. Wang and J. Li, “Strengthening Cu/Ni nanolayered composites by introducing thin Ag interlayers: A molecular dynamics simulation study,” Journal of Applied Physics. 2021. link Times cited: 8 Abstract: Experiments have shown that the ultrahigh strength of nanola… read moreAbstract: Experiments have shown that the ultrahigh strength of nanolayered metallic composites originates from their high-density interfaces of special characteristics. Hence, the modulation of interface structures becomes an effective route to enhance the mechanical performance of the nanolayered composites. One of the general ways to tune the interfacial feature is to introduce interlayers of several nanometers among constituent layers, such as amorphous (disordered) and crystalline (ordered) interlayers. Here, the deformation of a Cu/Ni layered composite with Ag interlayers of different thicknesses was simulated by molecular dynamics simulations. Our simulations show that the yield stress of 25 nm Cu/25 nm Ni nanolayered composites with Ag interlayers can be significantly enhanced, i.e., it can be 56.4% higher than that of their counterparts without interlayers. We also found that the yield strength of the new composites can be maximized by selecting an appropriate thickness for the Ag interlayer. The optimum interlayer thickness is 2.1 nm in tension and 4.2 nm for compression. It is revealed that the extra strength results from the alleviation of stress concentration by stimulating abundant interfacial dislocations at the Cu–Ag and Ag–Ni interfaces. These findings show that the introduction of additional interlayers is a new route to design stronger nanolayered metallic composites. read less USED (low confidence) N. Eom, M. Messing, J. Johansson, and K. Deppert, “Sintering Mechanism of Core@Shell Metal@Metal Oxide Nanoparticles,” The Journal of Physical Chemistry C. 2021. link Times cited: 14 USED (low confidence) L. Zhang, “Modeling the Effect of Crystallographic Orientations on Coalescing and Sintering for Two Ti Nanoparticles with Equal Size at Atomic Scale,” Journal of Materials Engineering and Performance. 2021. link Times cited: 2 USED (low confidence) M. Li, Q. Hou, J. Cui, M. Qiu, A. Yang, and M. Zhou, “Atomistic simulations of helium behavior at the Cu(111)/W(110) interface,” Journal of Nuclear Materials. 2021. link Times cited: 3 USED (low confidence) Z. Zhang et al., “Molecular dynamics-guided quality improvement in the femtosecond laser percussion drilling of microholes using a two-stage pulse energy process,” Optics & Laser Technology. 2021. link Times cited: 9 USED (low confidence) Z. Zhen-yu, Z. Qiu-yang, D. Cong, Y. Ju-yu, P. Guang-jian, and P. Zhong-yu, “Research on the promotion mechanism of surface burnishing process by two-dimensional ultrasonic vibration,” Journal of Materials Research and Technology. 2021. link Times cited: 15 USED (low confidence) S. Starikov et al., “Angular-dependent interatomic potential for large-scale atomistic simulation of iron: Development and comprehensive comparison with existing interatomic models,” Physical Review Materials. 2021. link Times cited: 16 Abstract: The development of classical interatomic potential for iron … read moreAbstract: The development of classical interatomic potential for iron is a quite demanding task with a long history background. A new interatomic potential for simulation of iron was created with a focus on description of crystal defects properties. In contrast with previous studies, here the potential development was based on force-matching method that requires only ab initio data as reference values. To verify our model, we studied various features of body-centered-cubic iron including the properties of point defects (vacancy and self-interstitial atom), the Peierls energy barrier for dislocations (screw and mix types), and the formation energies of planar defects (surfaces, grain boundaries, and stacking fault). The verification also implies thorough comparison of a potential with 11 other interatomic potentials reported in literature. This potential correctly reproduces the largest number of iron characteristics which ensures its advantage and wider applicability range compared to the other considered classical potentials. Here application of the model is illustrated by estimation of self-diffusion coefficients and the calculation of fcc lattice properties at high temperature. read less USED (low confidence) S. Gao et al., “Core-shell PdAu nanocluster catalysts to suppress sulfur poisoning.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 2 Abstract: Reducing sulfur poisoning is significant for maintaining the… read moreAbstract: Reducing sulfur poisoning is significant for maintaining the catalytic efficiency and durability of heterogeneous catalysts. We screened PdAu nanoclusters with specific Pd : Au ratios based on Monte Carlo simulations and then carried out density functional calculations to reveal how to reduce sulfur poisoning via alloying. Among various nanoclusters, the core-shell structure Pd13Au42 (Pd@Au) exhibits a low adsorption energy of SO2 (-0.67 eV), comparable with O2 (-0.45 eV) and lower than CO (-1.25 eV), thus avoiding sulfur poisoning during the CO catalytic oxidation. Fundamentally, the weak adsorption of SO2 originates from the negative d-band center of the shell and delocalized charge distribution near the Fermi level, due to the appropriate charge transfer from the core to shell. Core-shell nanoclusters with a different core (Ni, Cu, Ag, Pt) and a Pd@Au slab model were further constructed to validate and extend the results. These findings provide insights into designing core-shell catalysts to suppress sulfur poisoning while optimizing catalytic behaviors. read less USED (low confidence) T. Zhou, X. Gao, Z. Ma, H. Chang, T. Shen, and Z. Wang, “Atomistic simulation of α-Fe(100)-lead-bismuth eutectic (LBE) solid-liquid interface,” Journal of Nuclear Materials. 2021. link Times cited: 5 USED (low confidence) D. K. Das, A. Mallick, and S. Singh, “Estimating thermal properties of plumbene by multiscale modeling using molecular dynamics simulation technique,” Mechanics of Advanced Materials and Structures. 2021. link Times cited: 3 Abstract: To estimate the thermal properties of plumbene under differe… read moreAbstract: To estimate the thermal properties of plumbene under different temperatures, a plumbene sheet is developed by multi-scale modeling. Plumbene sheets of variable sample sizes are also investigated to obtain a broader view of thermal properties of the material for useful application in several engineering fields. Melting point, specific heat at constant volume and pressure, heat of fusion and the coefficient of linear and surface expansion within a temperature range of 318–398 K are also determined. Present applied techniques will guide experimental approaches for designing of plumbene sheets with specific thermophysical properties for targeted applications. read less USED (low confidence) S. Kwon, H. Kang, Y. Sohn, J. Lee, S.-M. Shim, and S. G. Lee, “Molecular dynamics simulation study on the effect of perfluorosulfonic acid side chains on oxygen permeation in hydrated ionomers of PEMFCs,” Scientific Reports. 2021. link Times cited: 13 USED (low confidence) X. Zhou, S. He, and J. Marian, “Cross-kinks control screw dislocation strength in equiatomic bcc refractory alloys,” Acta Materialia. 2021. link Times cited: 21 USED (low confidence) S. Kwon, S. Y. Lee, H. J. Kim, S. Jang, and S. G. Lee, “Distribution characteristics of phosphoric acid and PTFE binder on Pt/C surfaces in high-temperature polymer electrolyte membrane fuel cells: Molecular dynamics simulation approach,” International Journal of Hydrogen Energy. 2021. link Times cited: 7 USED (low confidence) A. Roy, J. Munshi, and G. Balasubramanian, “Low energy atomic traps sluggardize the diffusion in compositionally complex refractory alloys,” Intermetallics. 2021. link Times cited: 20 USED (low confidence) K. Nakamura, K. Yashiro, and K. Naito, “Molecular dynamics simulation on (001) interfacial fracture of Fe/Ni and Fe/Pd/Ni and deformation mode analysis by eigenvector of atomic elastic stiffness matrix.” 2021. link Times cited: 1 USED (low confidence) J. Tian, Q. Feng, J. Zheng, X. Liu, and W. Zhou, “Radiation damage buildup and basal vacancy cluster formation in hcp zirconium: A molecular dynamics study,” Journal of Nuclear Materials. 2021. link Times cited: 10 USED (low confidence) A. Sharma et al., “Pseudoelastic Deformation in Refractory (MoW) 85 Zr 7.5(TaTi) 7.5 High-Entropy Alloy,” Materials Engineering eJournal. 2021. link Times cited: 2 Abstract: Phase diagrams supported by density functional theory method… read moreAbstract: Phase diagrams supported by density functional theory methods can be crucial for designing high-entropy alloys (HEAs). We present phase and property analysis of refractory quinary (MoW) xZr y(TaTi) 1-x-y HEAs from combined Calculation of Phase Diagram (CALPHAD) and density-functional theory results, supplemented by molecular dynamics (MD) simulations. Our analysis indicates a Mo-W-rich region of this quinary system has a stable single-phase body-centered-cubic (bcc). The (MoW) 85Zr 7.5(TaTi) 7.5 was down-selected based on temperature-dependent CALPHAD phase diagram analysis and MD predicted elastic behavior that reveals twinning-assisted pseudoelastic behavior in this refractory HEA. While mostly unexplored in bcc crystals, twinning is a fundamental deformation mechanism that competes against dislocation slip in crystalline solids. This alloy shows identical cyclic deformation characteristics during uniaxial <100> loading, i.e., the pseudoelasticity is isotropic in loading direction. Additionally, a temperature increase from 77 to 1,500 K enhances the elastic strain recovery in load-unload cycles, offering possibly control to tune the pseudoelastic behavior. read less USED (low confidence) L. Wu, H. Wang, Y. Zhu, and M. Li, “Crystal-melt coexistence in FCC and BCC metals: A molecular-dynamics study of crystal-melt interface free energies,” Materialia. 2021. link Times cited: 6 USED (low confidence) H. Xie, T. Ma, T. Yu, and F. Yin, “Body-centered-cubic to face-centered-cubic phase transformation of iron under compressive loading along [100] direction,” Materials today communications. 2021. link Times cited: 1 USED (low confidence) O. Uche, H. G. Le, and L. B. Brunner, “Size-selective, rapid dynamics of large, hetero-epitaxial islands on fcc(0 0 1) surfaces,” Computational Materials Science. 2021. link Times cited: 1 USED (low confidence) H. Zuo, S. Cao, and Q. Yin, “Molecular dynamics study of alloying process of Cu–Au nanoparticles with different heating rates,” International Journal of Modern Physics B. 2021. link Times cited: 0 Abstract: In this paper, molecular dynamics (MD) simulation is utilize… read moreAbstract: In this paper, molecular dynamics (MD) simulation is utilized for the investigation of impact of heating rates on Au and Cu nanoparticles alloying process. Aggregation of contacted nanoparticles experiences three stages due to the contacting, while the alloying process can be distinguished into five regimes because of the contacting and melting. Different heating rates result in different contact temperatures. The decrease of the potential energy can be observed when the temperature reaches the melting temperature. When the temperature reaches the melting point, shrinkage ratio and relative gyration radius have drastic changes during the alloying process. It is shown that heating rates have an apparent effect on the shrinkage ratio and the relative gyration radius during the fusing process, and the shrinkage ratio and the relative gyration radius of Au and Cu alloying systems with lower heating rates have relative larger increasing ratio and decreasing ratio, respectively. read less USED (low confidence) X. Yang et al., “Molecular dynamics modeling of mechanical and tribological properties of additively manufactured AlCoCrFe high entropy alloy coating on aluminum substrate,” Materials Chemistry and Physics. 2021. link Times cited: 19 USED (low confidence) Y. Tang, H. Pan, and D. Y. Li, “Contribution of cold-work to the wear resistance of materials and its limitation – A study combining molecular dynamics modeling and experimental investigation,” Wear. 2021. link Times cited: 13 USED (low confidence) U. Domekeli, “A molecular dynamic study of the effects of high pressure on the structure formation of liquid metallic Ti62Cu38 alloy during rapid solidification,” Computational Materials Science. 2021. link Times cited: 9 USED (low confidence) R. Su et al., “Ultra-high strength and plasticity mediated by partial dislocations and defect networks: Part II: Layer thickness effect,” Acta Materialia. 2021. link Times cited: 7 USED (low confidence) L.-F. Zhu, J. Janssen, S. Ishibashi, F. Körmann, B. Grabowski, and J. Neugebauer, “A fully automated approach to calculate the melting temperature of elemental crystals,” Computational Materials Science. 2021. link Times cited: 17 USED (low confidence) S. Guo, Y. Zhang, H. Chen, X. Li, and M. Wang, “Impact of local chemical order on the structure evolution of dual-phase high-entropy alloy during solidification process,” Vacuum. 2021. link Times cited: 9 USED (low confidence) B.-H. Kim, S. Kim, J. Kang, Y.-C. Chung, K.-S. Kim, and K.-R. Lee, “Ion irradiation induced surface composition modulation in equiatomic binary alloys,” Applied Surface Science. 2021. link Times cited: 0 USED (low confidence) S. R. Pulagam, S. Kumari, and A. Dutta, “Core-structure and lattice friction of twinning dislocation in platinum,” Materials Today: Proceedings. 2021. link Times cited: 0 USED (low confidence) S. Oyinbo, T. Jen, and P. Oviroh, “Atomistic simulation of the preheating treatment and crystal orientation influences in the hardness of cold gas dynamic sprayed nickel coating unto the copper substrate,” Materials Today: Proceedings. 2021. link Times cited: 0 USED (low confidence) S. Kwon, H. Kang, Y. Sohn, J. Lee, S.-M. Shim, and S. G. Lee, “Effect of perfluorosulfonic acid side chains on oxygen permeation in hydrated ionomers of PEMFCs: Molecular dynamics simulation approach.” 2020. link Times cited: 1 Abstract:
We prepared two types of perfluorosulfonic acid (PFSA) ion… read moreAbstract:
We prepared two types of perfluorosulfonic acid (PFSA) ionomers with Aquivion (short side chain) and Nafion (long side chain) on a Pt surface and varied their water contents (2.92 ≤ λ ≤ 13.83) to calculate the solubility and permeability of O2 in hydrated PFSA ionomers on a Pt surface using full atomistic molecular dynamics (MD) simulations. The solubility and permeability of O2 molecules in hydrated Nafion ionomers were greater than those of O2 molecules in hydrated Aquivion ionomers at the same water content, indicating that the permeation of O2 molecules in the ionomers is affected not only by the diffusion coefficient of O2 but also by the solubility of O2. Notably, O2 molecules are more densely distributed in regions where water and hydronium ions have a lower density in hydrated Pt/PFSA ionomers. Radial distribution function (RDF) analysis was performed to investigate where O2 molecules preferentially dissolve in PFSA ionomers on a Pt surface. The results showed that O2 molecules preferentially dissolved between hydrophilic and hydrophobic regions in a hydrated ionomer. The RDF analysis was performed to provide details of the O2 location in hydrated PFSA ionomers on a Pt surface to evaluate the influence of O2 solubility in ionomers with side chains of different lengths. The coordination number of C(center)–O(O2) and O(side chain)–O(O2) pairs in hydrated Nafion ionomers was higher than that of the same pairs in hydrated Aquivion ionomers with the same water content. Our investigation provides detailed information about the properties of O2 molecules in different PFSA ionomers on a Pt surface and with various water contents, potentially enabling the design of better-performing PFSA ionomers for use in polymer electrolyte membrane fuel cells. read less USED (low confidence) D. Chakravarty et al., “Ultrahigh transverse rupture strength in tungsten-based nanocomposites with minimal lattice misfit and dual microstructure,” International Journal of Refractory Metals & Hard Materials. 2020. link Times cited: 2 USED (low confidence) S. Luan, Q. Zhao, C. Gui, and S. Zhou, “Welding deformations of welded joints between 1D Ag nanowire connectors and 3D substrates: a molecular dynamics study,” Japanese Journal of Applied Physics. 2020. link Times cited: 2 Abstract: In order to enrich the understanding of the relationship bet… read moreAbstract: In order to enrich the understanding of the relationship between 1D and 3D Ag nanomaterials in welding deformation, molecular dynamics simulations were performed to study a common structure of welded joints in Ag nanowire (NW) connectors on Ag substrates. The effects of the overlapping length, welding temperature and NW diameter on welding strength, dislocation and atomic strain were investigated, with the aim of understanding welding deformations of welded joints. With the increase in the overlapping length, welding temperature and NW diameter, the welding strength increases while the increment decreases. Dislocations can be reduced by increasing the overlapping lengths, NW diameters and annealing time. Moreover, the welded joint performance in shear strength could be improved by performing thermal annealing or decreasing NW diameters. The coordination number, residual stress and energy variation have also been analyzed to explain the above phenomenon. This work can provide guidance for the welding of nanomaterials with different dimensions. read less USED (low confidence) X. Wang, X. Wang, Z. Wang, Y. Guo, and Y. Wang, “Enhancing mechanism of interfacial metal element on the thermal transport across Cu-graphene interfaces revealed by molecular dynamics simulations,” Materials today communications. 2020. link Times cited: 13 USED (low confidence) V. Güder and M. Çeltek, “CuTi Nanotellerinin Germe Oranı ve Boyuta Bağlı Mekanik Davranışı.” 2020. link Times cited: 0 Abstract: Bu calisma, farkli boyutlarda CuTi (B11) kristal nanotelleri… read moreAbstract: Bu calisma, farkli boyutlarda CuTi (B11) kristal nanotellerinin [001] yonundeki esneklik-kirilma mekanizmasini ve deformasyonunu gozlemek icin ayrintili bir analiz sunmaktadir. Germe orani ve boyut gibi degiskenlerin nanotelin mekanik ozellikleri uzerine etkileri etkilesmelerin gomulu atom potansiyeli ile tanimlandigi molekuler dinamik benzetimleri ile incelenmistir. Uygulanan dis degiskenlerin CuTi nanotellerinin elastik ve plastik deformasyonlari uzerindeki etkileri iki temel baslik altinda ozetlenmistir. Nanotelin elastik tepkisinin yuksek germe orani ve kucuk boyut ile arttigi gozlenmistir. Elastisite Modulunun germe orani ile de karakterize edilebilmesine ragmen nanotel boyutu istenen dayaniklilik mekanizmasini belirlemede daha etkin role sahiptir. Diger yandan, dusuk germe orani ve kucuk boyutun CuTi nanotellerin kirilma dayanimini ve esnekligini azalttigi izlenmistir. read less USED (low confidence) Y. Yang, Y. Liang, J. Bi, Y. Bai, S. He, and B. Li, “The wetting characteristics of molten Ag-Cu-Au on Cu substrates: a molecular dynamics study.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 6 Abstract: Ag-Cu-Au ternary alloys are promising solder materials for w… read moreAbstract: Ag-Cu-Au ternary alloys are promising solder materials for wire bonding. Limited experimental studies on Ag-Cu-Au materials can be found due to the high cost of gold. In this study, face-centered-cubic Cu(100), Cu(111), and Cu(110) substrates wetted by molten Ag45Cu42Au13 were investigated via molecular dynamics (MD). As demonstrated by melting simulation results, the Ag45Cu42Au13 alloy has a lower melting temperature compared to the eutectic alloy, Ag60Cu40. MD methods were also used to investigate the dissolutive characteristics of Ag45Cu42Au13/Cu wetting. Density profiles and contact angles show an increase in wettability in the Ag45Cu42Au13/Cu(100) wetting system. For molten Ag60Cu40 and Ag45Cu42Au13 the spreading behavior on Cu(100) shows a promoted tendency, which contrasts with both Cu(111) and Cu(110). Solid-liquid adhesion is indicative of the comparative spreading degrees. The contact angles and PMF analysis of wetting behaviors on rough and smooth Cu substrates illustrate that solid-liquid adhesion in Wenzel states is stronger than in Cassie wetting states. read less USED (low confidence) S. A. Etesami, M. Laradji, and E. Asadi, “Reliability of molecular dynamics interatomic potentials for modeling of titanium in additive manufacturing processes,” Computational Materials Science. 2020. link Times cited: 5 USED (low confidence) S. Sawarkar and A. Chatterjee, “Decision tree driven construction of rate constant models: Identifying the ‘top-N’ environment atoms that influence surface diffusion barriers in Ag, Cu, Ni, Pd and Pt,” Computational Materials Science. 2020. link Times cited: 7 USED (low confidence) C. Singh, K. C. Katakam, G. Katakareddi, and N. Yedla, “Creep behavior of polycrystalline Al (metal)-Cu50Zr50 (metallic glass) cold welds,” Materials Today: Proceedings. 2020. link Times cited: 2 USED (low confidence) K. Yashiro, S. Tsuchiya, and K. Naito, “Atomic elastic stiffness analysis to predict twinning in Fe single crystal under shear,” Computational Materials Science. 2020. link Times cited: 3 USED (low confidence) J. Li, H.-W. Chen, Q. Fang, C. Jiang, Y. Liu, and P. Liaw, “Unraveling the dislocation–precipitate interactions in high-entropy alloys,” International Journal of Plasticity. 2020. link Times cited: 68 USED (low confidence) Ş. Safaltın and S. Gürmen, “Molecular dynamics simulation of size, temperature, heating and cooling rates on structural formation of Ag-Cu-Ni ternary nanoparticles (Ag34-Cu33-Ni33),” Computational Materials Science. 2020. link Times cited: 8 USED (low confidence) K. Kowalczyk-Gajewska and M. Ma’zdziarz, “Elastic properties of nanocrystalline materials of hexagonal symmetry: The core-shell model and atomistic estimates,” International Journal of Engineering Science. 2020. link Times cited: 5 USED (low confidence) J. Chapman and R. Ramprasad, “Nanoscale Modeling of Surface Phenomena in Aluminum Using Machine Learning Force Fields,” The Journal of Physical Chemistry C. 2020. link Times cited: 7 Abstract: The study of nano-scale surface phenomena is essential in un… read moreAbstract: The study of nano-scale surface phenomena is essential in understanding the physical processes that aid in technologically relevant applications, such as catalysis, material growth, and failure nuc... read less USED (low confidence) S. Xu, E. Hwang, W. Jian, Y. Su, and I. Beyerlein, “Atomistic calculations of the generalized stacking fault energies in two refractory multi-principal element alloys,” Intermetallics. 2020. link Times cited: 39 USED (low confidence) Z. Cao et al., “Strong and plastic metallic composites with nanolayered architectures,” Acta Materialia. 2020. link Times cited: 26 USED (low confidence) D. Chen et al., “Effects of minor alloying addition on He bubble formation in the irradiated FeCoNiCr-based high-entropy alloys,” Journal of Nuclear Materials. 2020. link Times cited: 13 USED (low confidence) S. Nag and W. Curtin, “Effect of solute-solute interactions on strengthening of random alloys from dilute to high entropy alloys,” Acta Materialia. 2020. link Times cited: 28 USED (low confidence) H. Rui et al., “Effect of lead atom penetration on displacement cascade in bcc iron studied by molecular dynamics simulation,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2020. link Times cited: 1 USED (low confidence) M. Daly, A. Kumar, C. V. Singh, and G. Hibbard, “On the competition between nucleation and thickening in deformation twinning of face-centered cubic metals,” International Journal of Plasticity. 2020. link Times cited: 8 USED (low confidence) A. Giwa, Z. Aitken, P. Liaw, Y.-W. Zhang, and J. Greer, “Effect of temperature on small-scale deformation of individual face-centered-cubic and body-centered-cubic phases of an Al0.7CoCrFeNi high-entropy alloy,” Materials & Design. 2020. link Times cited: 15 USED (low confidence) V. Guder and S. Sengul, “Tensile strength and failure mechanism of hcp zirconium nanowires: Effect of diameter, temperature and strain rate,” Computational Materials Science. 2020. link Times cited: 11 USED (low confidence) X. Chen, X. Gao, Y. Zhao, D. Lin, W. Chu, and H. Song, “TensorAlloy: An automatic atomistic neural network program for alloys,” Comput. Phys. Commun. 2020. link Times cited: 10 USED (low confidence) E. Antillon, C. Woodward, S. Rao, B. Akdim, and T. Parthasarathy, “Chemical Short Range Order Strengthening in a Model FCC High Entropy Alloy,” Acta Materialia. 2020. link Times cited: 131 Abstract: In order to understand the role of chemical short-range orde… read moreAbstract: In order to understand the role of chemical short-range order on deformation mechanisms in FCC compositionally complex alloys, a random model alloy (Co30-Fe16.67-Ni36.67-Ti16.67) is annealed at various temperatures using Hybrid Molecular-dynamics/Monte-Carlo simulations. The simulations produce significant chemical short-range order (CSRO) that increases with decreasing annealing temperature. Annealing tends to homogenize regions of high enthalpy due to: (1) chemical species redistributing into more compact configurations, and (2) pairs of atoms forming chemical bonds that lower the overall energy of the system; the composition explored here shows significant amount of ordering in Ti-Fe pairs with respect to random distributions as described by pairwise (EAM) potentials due to Johnson and Zhou. An energy topology approach is used to assess the local strengthening behavior in random solid solutions and annealed systems, where an interesting interplay is observed between misfit components and chemical short-range order affecting the overall critical resolved shear stress. The role of short-range order on the critical yield stress is quantified and compared with current solid solution models. Finally, we propose and validate an extension to the Labusch-Varvenne class of high-concentration solid-solution analytic models that incorporates the effects of chemical short range order. read less USED (low confidence) M. Guo et al., “Structural optimization and melting behavior investigation of Pd-Ag bimetallic nanoparticles by molecular simulations,” Computational Materials Science. 2020. link Times cited: 2 USED (low confidence) Y. Lachtioui, M. Kbirou, K. Saadouni, M. Sajieddine, and M. Mazroui, “Glass formation and structure evolution in the rapidly solidified monatomic metallic liquid Pt under high pressure,” Chemical Physics. 2020. link Times cited: 11 USED (low confidence) M. Bahramyan, R. T. Mousavian, and D. Brabazon, “Determination of atomic-scale structure and compressive behavior of solidified AlxCrCoFeCuNi high entropy alloys,” International Journal of Mechanical Sciences. 2020. link Times cited: 33 USED (low confidence) S. Sengul, “Evolution of local structure during melting of Zr0.7Pd0.3 nanowires by molecular dynamics simulations,” Vacuum. 2020. link Times cited: 4 USED (low confidence) A. Fourmont, S. L. Gallet, O. Politano, C. Desgranges, and F. Baras, “Effects of planetary ball milling on AlCoCrFeNi high entropy alloys prepared by Spark Plasma Sintering: Experiments and molecular dynamics study,” Journal of Alloys and Compounds. 2020. link Times cited: 46 USED (low confidence) A. Alian, E. Mahdi, and S. Meguid, “Coupled molecular dynamics-Monte Carlo modeling of gold nanowire surface fasteners,” Applied Surface Science. 2020. link Times cited: 3 USED (low confidence) C.-C. Yen et al., “Lattice distortion effect on elastic anisotropy of high entropy alloys,” Journal of Alloys and Compounds. 2020. link Times cited: 25 USED (low confidence) J. Li, Y. Zhang, C. Tian, H. Zhou, G. Hu, and R. Xia, “Structurally ordered nanoporous Pt–Co alloys with enhanced mechanical behaviors in tension,” Microporous and Mesoporous Materials. 2020. link Times cited: 18 USED (low confidence) Z. Wu, M. Li, S. Tian, and L. Zhang, “Molecular dynamics simulation of sintering of Cu and Au nanoparticles,” International Journal of Modern Physics B. 2020. link Times cited: 2 Abstract: Particle coalescence has wide applications in nature and ind… read moreAbstract: Particle coalescence has wide applications in nature and industry. In this study, molecular dynamics (MDs) simulations were employed to examine the sintering of Cu and Au nanoparticles, as well as ... read less USED (low confidence) C. Liang, D. Wang, Z. Wang, X. Ding, and Y. Wang, “Revealing the Atomistic Mechanisms of Strain Glass Transition in Ferroelastics,” EngRN: Materials Chemistry (Topic). 2020. link Times cited: 11 Abstract: As a new ferroelastic state, strain glass has attracted a lo… read moreAbstract: As a new ferroelastic state, strain glass has attracted a lot of recent attentions and, most importantly, strain glass transitions (SGTs) could underpin many phenomena that have puzzled the physics community for decades, including the quasi-linear superelasticity and Invar and Elinvar anomalies. However, there has been a lack of fundamental understanding at the atomistic level beyond the phenomenological Landau theory. In this paper, we propose a way to obtain quantitatively the continuous strain/stress fields distribution caused by point defects through molecular statics calculations by incorporating a Gaussian probability distribution function. By using the quantitative strain/stress fields distribution to inform phase field simulations, we reproduce quantitatively the experimentally observed critical defect concentrations separating the normal martensitic phase transition from SGTs at different temperatures and critical temperatures for spontaneous strain glass to martensitic transition at different defect concentrations. Based on percolation theory, we demonstrate how the strain network created by point defects with a critical concentration regulates the nucleation and growth of martensitic domains, suppresses autocatalysis by strain frustration, and changes the sharp first-order martensitic transformation into a continuous SGT. A general temperature- and defect-concentration-dependent percolation criterion is formulated for accurate prediction of SGT, which could enable high throughput computations for systematic search of new strain glass systems using simply molecular static calculations. read less USED (low confidence) J. Chapman, R. Batra, and R. Ramprasad, “Machine learning models for the prediction of energy, forces, and stresses for Platinum,” Computational Materials Science. 2020. link Times cited: 18 USED (low confidence) R. T. Mousavian et al., “Development of BMG-B2 nanocomposite structure in HAZ during laser surface processing of ZrCuNiAlTi bulk metallic glasses,” Applied Surface Science. 2020. link Times cited: 6 USED (low confidence) D. Utt, A. Stukowski, and K. Albe, “Grain boundary structure and mobility in high-entropy alloys: A comparative molecular dynamics study on a Σ11 symmetrical tilt grain boundary in face-centered cubic CuNiCoFe,” Acta Materialia. 2020. link Times cited: 55 USED (low confidence) X. Lv, C. Guan, Z. Han, H.-bo Zhang, and Q. Liu, “The wetting characteristics of copper droplets on tungsten surfaces on atomic scale: A molecular dynamics simulation,” Computational Materials Science. 2020. link Times cited: 11 USED (low confidence) G. Wang, Y. Xu, P. Qian, and Y. Su, “Vacancy concentration of films and nanoparticles,” Computational Materials Science. 2020. link Times cited: 8 USED (low confidence) C. Cheng et al., “Development and application of EAM potentials for Ti, Al and Nb with enhanced planar fault energy of Ti,” Computational Materials Science. 2020. link Times cited: 4 USED (low confidence) R. Su et al., “Ultra-high strength and plasticity mediated by partial dislocations and defect networks: Part I: Texture effect,” Acta Materialia. 2020. link Times cited: 19 USED (low confidence) P. Gupta and N. Yedla, “Temperature and Loading Rate Effect on the Load-Displacement Response of Metal-Metallic Glass (Al-Cu50Zr50) Layered Structure during Nano-Indentation,” Materials Science Forum. 2020. link Times cited: 1 Abstract: Molecular dynamics (MD) simulations of metal-metallic glass … read moreAbstract: Molecular dynamics (MD) simulations of metal-metallic glass (Al-Cu50Zr50) multilayer during nanoindentation is carried out to investigate the load-displacement response, mechanical properties and deformation mechanisms. The indentation study is carried out at temperatures in the range of cryogenic to room temperature (10 K-300 K). The indenter speeds are varied between 0.5-5 Å/ps to study the effect of loading rate. The interaction between Al-Cu-Zr atoms are defined by EAM (Embedded Atom Method) potential. A sample size of 200 Å × 200 Å × 200 Å (in x y z-direction) comprising of 538538 atoms is used for nanoindentation. P P S boundary condition (BC) in x y z direction and NVT ensemble are used. We observed a peak load of 117 nN, at a temperature of 10 K with a loading rate of 5 Å/ps. We found that as the loading rate increase, the peak load also increases. As anticipated, the increase in temperature decreases the strength of the multilayer. The atomic displacement vector plots reveal that MG act as hurdles to the movement of dislocations nucleated at the interface. read less USED (low confidence) D. Smirnova et al., “Atomistic description of self-diffusion in molybdenum: A comparative theoretical study of non-Arrhenius behavior,” Physical Review Materials. 2020. link Times cited: 16 Abstract: According to experimental observations, the temperature depe… read moreAbstract: According to experimental observations, the temperature dependence of self-diffusion coefficient in most body-centered cubic metals (bcc) exhibits non-Arrhenius behavior. The origin of this behavio ... read less USED (low confidence) C. Yu et al., “Highly Efficient AuPd Catalyst for Synthesizing Polybenzoxazole with Controlled Polymerization.” 2019. link Times cited: 5 USED (low confidence) R. Su et al., “Phase transformation induced plasticity in high-strength hexagonal close packed Co with stacking faults,” Scripta Materialia. 2019. link Times cited: 22 USED (low confidence) P. Zakharov, A. Cherednichenko, M. Starostenkov, A. Eremin, and A. Markidonov, “Nonlinear high-amplitude excitations of the CuPt7 crystal lattice: a molecular dynamics study,” Journal of Physics: Conference Series. 2019. link Times cited: 0 Abstract: This article analyzes the structure and properties of the Cu… read moreAbstract: This article analyzes the structure and properties of the CuPt7 crystal for the possible existence of non-linear high-amplitude oscillations of the light alloy component. The densities of the phonon states of a crystal are calculated and analyzed for its different structures. It is demonstrated that the phonon system of a crystal has a narrow forbidden zone. The dependences of the possibility of the existence of high-amplitude oscillations on the initial conditions are revealed. It is shown that such oscillations are possible with the initial deviation of the Cu atom from the equilibrium position by more than 0.95 Å for FCC CuPt7. For such oscillators, their lifetime, the amount of stored energy, and other characteristics were obtained, which make it possible to characterize nonlinear high-amplitude lattice vibrations. read less USED (low confidence) M. Pengyan et al., “Quantitative investigation on sink strength of nano-grain boundary for irradiation resistance,” Journal of Nuclear Materials. 2019. link Times cited: 15 USED (low confidence) X. Yang, X.-guo Zeng, H. Chen, Y. Wang, L. He, and F. Wang, “Molecular dynamics investigation on complete Mie-Grüneisen equation of state: Al and Pb as prototypes,” Journal of Alloys and Compounds. 2019. link Times cited: 13 USED (low confidence) T. Babuska et al., “Achieving high strength and ductility in traditionally brittle soft magnetic intermetallics via additive manufacturing,” Acta Materialia. 2019. link Times cited: 40 USED (low confidence) N. Ahmad, M. Bon, D. Passerone, and R. Erni, “Template Assisted In Situ Synthesis of Ag@Au Bimetallic Nanostructures Employing Liquid Phase Transmission Electron Microscopy.,” ACS nano. 2019. link Times cited: 20 Abstract: Noble metal nanostructure synthesis via seed-mediated route … read moreAbstract: Noble metal nanostructure synthesis via seed-mediated route is a widely adopted strategy for a plethora of nanocrystal systems. Ag@Au core-shell nanostructures are radiolytically grown in real-time using in situ liquid-cell (scanning) transmission electron microscopy (LCTEM). Here we employ a capping agent, dimethyl-amine (DMA) and a coordinating complex, potassium iodide (KI) in an organic solvent (methanol) in order to: 1) slow down the reaction kinetics to observe mechanistic insights into the overgrowth pro-cess, 2) shift the growth regime from galvanic-replacement mode to direct synthesis mode resulting in the conventional synthesis of Ag@Au core-shell structures. A theoretical approach based on classical simulations complements our experiments providing further insight on the growth modes. In particular, we focus on the shape evolution and chemical ordering, as currently there is an insufficient understanding regarding mixed composition phases at interfaces of alloys even with well-known miscibilities. Furthermore, the comparison of theoretical and experimental data reveals that the final morphology of these nanoalloys is not simply a function of crystallinity of the underlying seed structure but instead is readily modified by extrinsic parameters such as additives, capping agent and modulation of surface energies of exposed crystal surfaces by the encapsulating solvent. The impact of these additional parameters is systematically investigated using an empirical approach in light of ab-initio simulations. read less USED (low confidence) O. I. Kushnerov, “AlCoCuFeNi high–entropy alloy nanoparticle melting and solidification: a classical molecular dynamics simulation study,” Journal of Physics and Electronics. 2019. link Times cited: 5 Abstract: The processes of melting and solidification of AlCoCuFeNi na… read moreAbstract: The processes of melting and solidification of AlCoCuFeNi nanoparticle of about 10 nm is studied by molecular dynamics simulation at three different cooling rates (1∙1011 K/s, 1∙1012 K/s, and 1∙1013 K/s) using the embedded atom model (EAM) potential. The melting and crystallization of the nanoparticle are characterized by studying the temperature dependence of the potential energy. The adaptive common neighbor analysis (CNA) is performed and the radial distribution function (RDF) is calculated to determine the structure and lattice parameters of phases of the AlCoCuFeNi nanoparticle. It is shown that the final structure of the investigated nanoparticle changes from amorphous to crystalline with decreasing of the rate of cooling, and the temperature hysteresis takes place during the melting and crystallization of AlCoCuFeNi HEA nanoparticle. read less USED (low confidence) J. Méndez and M. Ponga, “MXE: A package for simulating long-term diffusive mass transport phenomena in nanoscale systems,” Comput. Phys. Commun. 2019. link Times cited: 8 USED (low confidence) S. Fazeli and S. K. Sadrnezhaad, “Molecular dynamics simulation of plastic deformation and interfacial delamination of NiTi/Ag bilayer by cyclic-nanoindentation: Effects of crystallographic orientation of substrate,” Computational Materials Science. 2019. link Times cited: 9 USED (low confidence) M. Luo et al., “PdMo bimetallene for oxygen reduction catalysis,” Nature. 2019. link Times cited: 690 USED (low confidence) K. Li et al., “Determination of the accuracy and reliability of molecular dynamics simulations in estimating the melting point of iron: Roles of interaction potentials and initial system configurations,” Journal of Molecular Liquids. 2019. link Times cited: 8 USED (low confidence) S. He, W. Ecker, R. Pippan, and V. Razumovskiy, “Hydrogen-enhanced decohesion mechanism of the special Σ5(0 1 2)[1 0 0] grain boundary in Ni with Mo and C solutes,” Computational Materials Science. 2019. link Times cited: 32 USED (low confidence) M. R. Vazirisereshk, S. A. Sumaiya, A. Martini, and M. Baykara, “Measurement of electrical contact resistance at nanoscale gold-graphite interfaces,” Applied Physics Letters. 2019. link Times cited: 8 Abstract: An approach to measuring electrical contact resistance as a … read moreAbstract: An approach to measuring electrical contact resistance as a direct function of the true contact size at the nanoscale is presented. The approach involves conductive atomic force microscopy (C-AFM) measurements performed on a sample system comprising atomically flat interfaces (up to several hundreds of nanometers in lateral size) formed between gold islands and a highly oriented pyrolytic graphite (HOPG) substrate. The method overcomes issues associated with traditional C-AFM such that conduction can be correlated with a measurable true, conductive contact area. Proof-of-principle experiments performed on gold islands of varying size point toward an increasing contribution of the island-HOPG junction to the measured total resistance with decreasing island size. Atomistic simulations complement and elucidate experimental results, revealing the maximum island size below which the electrical contact resistance at the island-HOPG junction can be feasibly extracted from the measured total resistance. read less USED (low confidence) K. Kowalczyk-Gajewska and M. Maździarz, “Effective stiffness tensor of nanocrystalline materials of cubic symmetry: The core-shell model and atomistic estimates,” International Journal of Engineering Science. 2019. link Times cited: 5 USED (low confidence) L. Zhang, G.-W. Hong, and S. Cai, “Molecular dynamics simulation of aggregation of monocrystal and polycrystal copper nanoparticles,” International Journal of Modern Physics B. 2019. link Times cited: 4 Abstract: Molecular dynamics simulations were employed to investigate … read moreAbstract: Molecular dynamics simulations were employed to investigate the aggregation of monocrystal and polycrystal nanoparticles. The lattice structure, displacement vector, potential energy, shrinkage ratio, relative gyration radius and mean square displacement of the two systems are compared. The results indicate that the aggregation of polycrystal nanoparticles is more drastic than that of monocrystal nanoparticles. Besides, the polycrystal nanoparticles are found contacted and melted at lower-temperature than that of monocrystal nanoparticles. The reason for all these phenomena is that there is additional surface energy in the grain boundary of polycrystal nanoparticles. read less USED (low confidence) R. Arifin et al., “Glassy NiTi produced with different cooling times: Structural investigation using molecular dynamics simulations,” Results in physics. 2019. link Times cited: 3 USED (low confidence) J. Li et al., “Tuning the mechanical behavior of high-entropy alloys via controlling cooling rates,” Materials Science and Engineering: A. 2019. link Times cited: 32 USED (low confidence) X. Wang, K. Li, Y. Zhu, W. Li, and W. Wang, “Molecular Dynamics Study on Mechanical Properties of Nanocrystalline tantalum,” 2019 IEEE 19th International Conference on Nanotechnology (IEEE-NANO). 2019. link Times cited: 1 Abstract: The study of nanocrystalline(NC) polycrystals is a hot topic… read moreAbstract: The study of nanocrystalline(NC) polycrystals is a hot topic, and the study of nanomaterial properties by molecular dynamics has become the first choice for many researchers. The purpose of this paper is to simulate the tensile tests of single and polycrystalline tantalum by molecular dynamics(MD) to obtain its mechanical properties. Firstly, the Ravelo-EAM potential was used to conduct tensile tests on tantalum in the <100> direction. Secondly, it can be seen that the elastic modulus E100 decreases with the temperature gradually increases from 1 K to 1500 K according to the simulation results. Finally, the Hall-Petch(H-P) effect based on grain size is verified from the tensile test of polycrystalline tantalum. read less USED (low confidence) V. Cruz and J. Enrique., “Estudio de la inhomogeneidad elástica en vidrios metalicos en la mesoescala.” 2019. link Times cited: 0 Abstract: Metallic glasses are amorphous solids produced by rapid cool… read moreAbstract: Metallic glasses are amorphous solids produced by rapid cooling, with disordered atomic structure and lacking long-range order. This structural disorder makes them to show mechanical properties different from those observed in a crystalline solid.
Metallic glasses are ideally isotropic, but they can become anisotropic during the manufacturing process or as a result of non-homogeneous plastic deformation, creep, etc. Experimental studies showed remnant anisotropy in amorphous PdSiCuP under a homogeneous deformation regime at temperatures close to the glass transition (Tg). In this thesis we studied the remnant anisotropy induced by shear in two amorphous systems by using molecular dynamics simulation. The Cu13Ni34Pd53 system was chosen to approach the PdCuSiP compositions, while the Zr46Cu46Al8 system is an excellent metallic glass former.
Amorphous systems were obtained by fast cooling from the liquid and subsequent thermal annealing. Both systems were then sheared in the [100] direction at a deformation speed of 10^10 s^(-1), and returned to its original form at the same speed. Shearing simulations were performed in isothermal-isobaric conditions at different temperatures. The resulting states were examined using the directional pair distribution function (d-PDF), calculated from the distribution function of interatomic distances in planes perpendicular to the selected axis.
Remnant anisotropy was detected in both systems after a deformation-recovery cycle. Cu13Ni34Pd53 displays remnant anisotropy below the glass transition at 0 and 300 K after the shear process, and again at 700 K after the full deformation-recovery cycle.
The intensity of anisotropy in the d-PDF of Zr46Cu46Al8 is lower than in Cu13Ni34Pd53, possibly due to the different atomic radii. However, it is found in all the studied temperature range, with decreasing intensity with temperature.
Anisotropy is concentrated in the [110] planes and [1(-1)0], corresponding to the planes of maximum and minimum shear respectively. This result indicates that remnant anisotropy is highly directional and can go unnoticed if not searched for. It also states that the shear process is essentially symmetrical with respect to the plane [110].
The shear and recovery process induces the creation of free volume, falling into the category of rejuvenation processes discussed in the literature. Counterintuitively, the process of free volume creation is accompanied by a reduction of the distance of the first maximum of the pair distribution function g (r), indicating a decrease of the most likely distance (mode) between first neighbors. However, this decrease in mode is accompanied by an increase of the width of the first peak, increasing the standard deviation of the distribution of distances between first neighbors. This explains the apparent contradiction between the increase in the volume and reduction of the most probable distance between first neighbors. Simultaneously, the second coordination sphere is more homogenous after the deformation and recovery process. As a result, medium range order is increased.
The discrepancy between the results obtained between Cu13Ni34Pd53 and Zr46Cu46Al8 may be due to the fact that the potential used in the simulations of Zr46Cu46Al8 is more representative of the behavior of metallic glasses. As a result, we hypothesize that the presence of remnant anisotropy after mechanical deformation could be a general feature in metallic glasses.
The computational cost of this study was very high, since it was necessary to simulate million-atom systems to avoid that results were dependent on the size of the simulation box.
Los vidrios metálicos son sólidos amorfos producidos por enfriamiento rápido, con estructura atómica desordenada y sin orden de largo alcance. Este desorden estructural les proporciona propiedades mecánicas diferentes de las observadas en un sólido cristalino. Idealmente los vidrios metálicos son isotrópicos pero pueden convertirse en anisotrópicos durante el proceso de fabricación o como consecuencia de una deformación plástica no homogénea, fluencia, etc. Estudios experimentales mostraron anisotropía remanente en PdSiCuP amorfo bajo un régimen de deformación homogéneo a temperaturas cercanas a la transición vítrea (Tg). En esta tesis estudiamos la anisotropía remanente inducida por cizalla en dos sistemas amorfos mediante simulación por dinámica molecular. El sistema Cu13Ni34Pd53 se eligió intentando aproximarse a las composiciones PdCuSiP. Por otra parte, el sistema Zr46Cu46Al8 es un excelente formador de vidrios metálicos. Tras la obtención del sistema amorfo por enfriamiento rápido y equilibrado térmico ambos modelos fueron sometidos a deformación por cizalladura en la dirección [100], a una velocidad de deformación de 10^10 s^(-1), y retornados a su forma original con la misma velocidad, en condiciones isotérmicas-isobáricas a diferentes temperaturas. Los estados obtenidos fueron examinados utilizando la función de distribución par direccional (dPDF), calculada a partir de la distribución de distancias interatómicas en los planos perpendiculares al eje escogido. En ambos sistemas se detectó anisotropía remanente después de un ciclo deformación- recuperación. En Cu13Ni34Pd53 se observa anisotropía remanente por debajo de la transición vítrea en 0 y 300 K al final del proceso de cizalla, y nuevamente en el estado deformado a 700 K. En Zr46Cu46Al8, la intensidad de la anisotropía determinada en las d-PDF es menor que en Cu13Ni34Pd53, posiblemente debido a los radios atómicos, pero se observa en todo el rango de temperaturas estudiadas con intensidad decreciente con la temperatura. La anisotropía se concentra en los planos [110] y [1(-1)0], correspondientes a los planos de máxima y mínima cizalla respectivamente. Este resultado indica que la anisotropía remanente es altamente direccional y puede pasar desapercibida si no se busca específicamente. Asimismo, se observa que el proceso de cizalla es esencialmente simétrico con respecto al plano [1(-1)0]. El proceso de cizalla y recuperación induce la creación de volumen libre, entrando dentro de la categoría de los procesos de rejuvenecimiento analizados en la literatura. Contraintuitivamente, el proceso de creación de volumen libre va acompañando de una reducción de la distancia del primer máximo de la función de distribución de pares g(r), indicando una disminución de la distancia más probable (moda) entre primeros vecinos. Sin embargo, esta disminución de la moda va acompañada de un aumento de la anchura del primer pico, aumentando la desviación estándard de la distribución de distancias entre primeros vecinos. Esto explica la aparente contradicción entre el aumento del volumen y la reducción de la distancia más probable entre primeros vecinos. Simultáneamente, la segunda esfera de coordinación es más homogénea después del proceso de deformación y recuperación. En consecuencia, se produce un aumento del orden a media distancia. La discrepancia entre los resultados obtenidos entre Cu13Ni34Pd53 y Zr46Cu46Al8 puede deberse a que el potencial en Zr46Cu46Al8 es más representativo del comportamiento de los vidrios metálicos. Consecuentemente, la presencia de anisotropía remanente después de deformación mecánica podría ser una característica general en los vidrios metálicos. El coste computacional de este estudio ha sido muy alto, puesto que ha sido necesario simular sistemas con muchos átomos para evitar que los resultados obtenidos fuesen dependientes del tamaño de la caja de simulación utilizadas read less USED (low confidence) M. Liu, M. Seita, T. Duong, W. Kuo, and M. Demkowicz, “Preferential corrosion of coherent twin boundaries in pure nickel under cathodic charging,” Physical Review Materials. 2019. link Times cited: 6 Abstract: We describe an investigation of microstructure in pure nicke… read moreAbstract: We describe an investigation of microstructure in pure nickel (Ni) after cathodic charging in aqueous electrolyte. Intergranular corrosion occurs on the sample surface and takes the form of long trenches along coherent twin boundaries (CTBs): sites that have often been considered especially resistant to corrosion. Integrating electron backscatter diffraction and x-ray computed tomography, we show that the trenches are formed by the growth and eventual overlap of isolated conical cavities, which, in turn, are aligned with <110>-type directions within CTB planes. The <110>-type orientation is consistent with cavity initiation at high-energy, symmetric incoherent twin boundary facets within CTB planes. Our observations show that, far from being always corrosion resistant, CTBs are especially susceptible to intergranular corrosion under some conditions. read less USED (low confidence) P. Gupta and N. Yedla, “Tensile-compression loading and pre-strain effects on the evolution of stacking fault tetrahedra, dislocation density, and free volume in crystal-amorphous thin film interface: A large-scale molecular dynamics study,” Journal of Non-Crystalline Solids. 2019. link Times cited: 3 USED (low confidence) M. Celtek, “The effect of atomic concentration on the structural evolution of Zr100-xCox alloys during rapid solidification process,” Journal of Non-Crystalline Solids. 2019. link Times cited: 16 USED (low confidence) Z. Islam, H. Gao, and A. Haque, “Synergy of elastic strain energy and electron wind force on thin film grain growth at room temperature,” Materials Characterization. 2019. link Times cited: 13 USED (low confidence) J. Wang, A. Agrawal, and K. Flores, “Are hints about glass forming ability hidden in the liquid structure?,” Acta Materialia. 2019. link Times cited: 16 USED (low confidence) D. Spearot, L. Capolungo, and C. Tomé, “Shear-driven motion of Mg
101¯2
twin boundaries via disconnection terrace nucleation, growth, and coalescence,” Physical Review Materials. 2019. link Times cited: 15 USED (low confidence) M. Bon, N. Ahmad, R. Erni, and D. Passerone, “Reliability of two embedded atom models for the description of Ag@Au nanoalloys,” The Journal of Chemical Physics. 2019. link Times cited: 7 Abstract: The validation of embedded atom models (EAM) for modelling n… read moreAbstract: The validation of embedded atom models (EAM) for modelling nanoalloys requires to verify both a faithful description of the individual phases and a convincing scheme for the mixed interactions. In this work, we present a systematic benchmarking of two widely adopted EAM parameterizations, i.e. by Foiles [S. M. Foiles et al. Phys. Rev. B 33, 7983 (1986)] and by Zhou [X. W. Zhou et al. Phys. Rev. B, 69, 144113 (2004)] with density functional theory calculations for the description of processes at Ag@Au nanoalloys surfaces and nanoclusters. read less USED (low confidence) J. Shao, C. Wang, P. Wang, A. He, and F.-guo Zhang, “Atomistic simulations and modeling analysis on the spall damage in lead induced by decaying shock,” Mechanics of Materials. 2019. link Times cited: 20 USED (low confidence) S. Guo et al., “Research on the crystallization behavior occurred in the process of preparing bulk metallic glass with selective laser melting,” Materials Research Express. 2019. link Times cited: 3 Abstract: The crystallization behavior occurred in the heat affected z… read moreAbstract: The crystallization behavior occurred in the heat affected zone in the process of bulk metallic glasses preparation with selective laser melting has been studied with molecular dynamics simulation. Cyclical heating conditions are applied with three different temperature variation rates in the simulation cases, while the heating rate is ten times of the cooling rate in each case. The results reveal that the newly formed crystal cluster has more Ni atoms than the parent phase. When the cooling rate is about one third of the critical cooling rate for the formation of metallic glass, the crystallization behaviors happen in the first cooling process, which is controlled alternately by growth mechanism and nucleation mechanism. In the following heating process, the numbers of both crystal clusters and crystal atoms first increase and then decrease. When the cooling rate is about two fifths of the critical cooling rate, the crystallization behaviors happen in the second cooling process. When the cooling rate increases to about two thirds of the critical cooling rate, massive crystallization behavior is not observed, showing that the crystallization occurred in the HAZ in the process of preparing BMGs with SLM can be suppressed with a critical temperature variation rate. read less USED (low confidence) Z. Aitken and Y.-W. Zhang, “Revealing the deformation twinning nucleation mechanism of BCC HEAs,” MRS Communications. 2019. link Times cited: 14 Abstract: Deformation twinning has been frequently observed in body-ce… read moreAbstract: Deformation twinning has been frequently observed in body-centered cubic (BCC) high entropy alloys (HEAs), however, the underlying mechanism remains elusive. We perform molecular dynamics simulations on a representative BCC HEA nanopillar under high-symmetry compression, describe atomic details of deformation twinning, and propose a mechanism of twin nucleation from the surface. We find that twinned regions are formed by partial dislocations and that chemical heterogeneity can reduce local fault energy and promote stacking faults and twins. These results help to understand the propensity for stacking fault formation and twinning in HEAs and may guide the design of novel HEAs through control of active twinning mechanisms. read less USED (low confidence) X. Zhou, X.-xiang Yu, D. Jacobson, and G. Thompson, “A molecular dynamics study on stress generation during thin film growth,” Applied Surface Science. 2019. link Times cited: 25 USED (low confidence) X. Kong et al., “Stronger and more failure-resistant with three-dimensional serrated bimetal interfaces,” Acta Materialia. 2019. link Times cited: 30 USED (low confidence) S. Rogachev, O. Politano, F. Baras, and A. Rogachev, “Explosive crystallization in amorphous CuTi thin films: a molecular dynamics study,” Journal of Non-Crystalline Solids. 2019. link Times cited: 8 USED (low confidence) R. Arifin, M. Malyadi, Munaji, G. A. Buntoro, and Darminto, “Evaluation of melting behaviour of Nickel, Titanium, and NiTi alloy using EAM and MEAM type potential,” Journal of Physics: Conference Series. 2019. link Times cited: 6 Abstract: The atomic level study of NiTi alloy at high temperature is … read moreAbstract: The atomic level study of NiTi alloy at high temperature is very important to understand the mechanism of NiTi fabrication, in partial the process during the hot working. In the atomic investigation using molecular dynamics simulation, the use of the interatomic potential greatly affects the results. Therefore, the suitability of the interatomic potential applied in some specific condition has to be examined. In our previous work, we have tested the performance of EAM and MEAM potential to reproduce the lattice constant of NiTi alloy. Our previous results have shown that the MEAM potential work better than the EAM potential. In this research, we further investigate the performance of EAM and MEAM type potential to describe the melting behavior of nickel, titanium, and NiTi alloy. We find from the current result that the accuracy of the MEAM potential is better than EAM potential in high temperature MD simulations. read less USED (low confidence) S. A. Etesami, M. Laradji, and E. Asadi, “Transferability of interatomic potentials in predicting the temperature dependency of elastic constants for titanium, zirconium and magnesium,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 4 Abstract: We present our investigation of the current state of the art… read moreAbstract: We present our investigation of the current state of the art for the transferability of molecular dynamics (MD) interatomic potentials for high temperature simulations of material processes in terms of elastic constants. With the current advancement of computer power, nanoscale computational models such as MD have the potential to accelerate optimization and development of high temperature material processes provided a robust and transferable interatomic potential. Temperature dependency of elastic constants, despite the low temperature elastic constants, is not commonly used as one of the target material properties to develop interatomic potentials for metals; thus, it is a reliable index to determine the transferability of interatomic potentials for high temperature simulations. We consider all five independent elastic constants and their temperature dependency as an index for our evaluations of available interatomic potentials for titanium (Ti), zirconium (Zr), and magnesium (Mg) as representative metals with a relatively complex crystal structure (hcp). The calculated elastic constants and their deviation from their corresponding experimental values are presented. We provide a through discussion on the transferability of each potential and summarize with the most suitable potentials for high temperature material process simulations for each considered material. read less USED (low confidence) S. M. T. Mousavi, H. Zhou, G. Zou, and H. Gao, “Transition from source- to stress-controlled plasticity in nanotwinned materials below a softening temperature,” npj Computational Materials. 2019. link Times cited: 16 USED (low confidence) P. Tsai and Y. Jeng, “Coalescence and epitaxial self-assembly of Cu nanoparticles on graphene surface: A molecular dynamics study,” Computational Materials Science. 2019. link Times cited: 17 USED (low confidence) H. Fan, A. Ngan, K. Gan, and J. El-Awady, “Origin of double-peak precipitation hardening in metallic alloys,” International Journal of Plasticity. 2018. link Times cited: 44 USED (low confidence) R. Yan et al., “Structural and mechanical properties of homogeneous solid-liquid interface of Al modelled with COMB3 potential,” Computational Materials Science. 2018. link Times cited: 9 USED (low confidence) A. Giwa, Z. Aitken, M. Jafary-Zadeh, P. Liaw, Y.-W. Zhang, and J. Greer, “Effect of Temperature on Small-Scale Deformation of Individual FCC and BCC Phases of Al0.7CoCrFeNi High Entropy Alloy,” MatSciRN: Lightweight Alloys (Topic). 2018. link Times cited: 0 Abstract: High Entropy Alloys (HEAs) represent an important class of s… read moreAbstract: High Entropy Alloys (HEAs) represent an important class of structural materials because of their high strength, ductility, and thermal stability due to the solid solution nature of the multi-component metallic system. Understanding the mechanical response of isolated phases (FCC and BCC) of a dual-phase HEA is integral to understanding mechanical properties of these special alloys in bulk. We investigate the compressive response of single crystalline cylinders with diameters between 400 nm and 2 µm excised from individual grains within FCC and BCC phases of Al0.7CoCrFeNi HEA at 295 K, 143 K and 40 K. Micro-compression experiments were conducted in an in-situ SEM equipped with a custom-constructed cryogenic setup; FCC samples had a [324] crystallographic orientation, and those extracted from the BCC phase had a [001] orientation. We observed a "smaller is stronger" size effect in the yield strength as a function of pillar diameter, D, of both alloy phases for all temperatures, τ_y ∝D^m with a power law exponent, m, decreasing from -0.68 at 295K to -0.47 at 143K to -0.38 at 40K for FCC phase, and remaining constant at ~-0.33 for all temperatures for the BCC phase. We also observed reduced work hardening rates and more extensive strain bursts during deformation at lower temperatures in all samples. All deformed FCC samples contained multiple parallel slip offsets for all pillar sizes and temperatures; compressed BCC pillars had wavy slip traces, which are evidence of multiple intersecting slip systems. Transmission Electron Microscopy (TEM) microstructural analysis of the compressed FCC samples reveals parallel slip lines and distorted slip planes, while compressed BCC samples contained entangled dislocation networks, as well as several twinned regions in samples deformed at 40 K. Molecular dynamics (MD) simulations of representative FCC and BCC HEA compressions reveal that deformation in FCC HEAs is dominated by nucleation and propagation of partial dislocations along parallel slip planes but by partial dislocation/twinning in the BCC HEA at all temperatures. Simulations also predict a decrease in stacking fault energy with increased alloying. For example, a reduction in the stable stacking fault energy of the FCC HEA up to 55% with respect to pure constituents is observed. This reduction in stable stacking fault energy may drive the observed deformation mechanisms. We also discuss theories of low-temperature strengthening in HEAs, compare them to our experimental data and assess how they manifest in the observed temperature-dependent size effect. read less USED (low confidence) Y. Liao, M. Xiang, G. Li, K. Wang, X. Zhang, and J. Chen, “Molecular dynamics studies on energy dissipation and void collapse in graded nanoporous nickel under shock compression,” Mechanics of Materials. 2018. link Times cited: 26 USED (low confidence) M. Jagannath, S. N. Divi, and A. Chatterjee, “Kinetic Map for Destabilization of Pt-Skin Au Nanoparticles via Atomic Scale Rearrangements,” The Journal of Physical Chemistry C. 2018. link Times cited: 7 Abstract: A commonly used strategy to enhance the mass activity of Pt-… read moreAbstract: A commonly used strategy to enhance the mass activity of Pt-based catalysts involves the synthesis of Au nanoparticles (NPs) with a monolayer-thick Pt-skin layer. The synergistic effect of Au and Pt results in a higher catalytic activity and better Pt utilization. However, the stability of the Pt-skin layer is questionable as our recent equilibrium Monte Carlo simulations predict that eventually the surface Pt is replaced by Au. The role of Au during destabilization of Pt-skin in vacuum and solution is investigated with the help of molecular dynamics. Different starting Au–Pt arrangements are studied mimicking various NP synthesis approaches. Beyond a critical number of atoms in a Pt cluster, the ideal Pt monolayer rapidly transforms to a three-dimensional (3D) Pt cluster. This is supported by our model predicting transition from the Pt monolayer to Volmer–Weber growth in the Au–Pt system. At room temperature, Pt atoms move into the subsurface layer at second timescales mainly via the exchange mechanism i... read less USED (low confidence) S. Huang, Z. Zhang, C. Xu, Z. Zhu, J. Cui, and B. Wang, “Origin and evolution of a fivefold twin on the surface of a nickel alloy,” Materials Letters. 2018. link Times cited: 8 USED (low confidence) R. Su et al., “Deformation mechanisms in FCC Co dominated by high-density stacking faults,” Materials Science and Engineering: A. 2018. link Times cited: 25 USED (low confidence) Z. Wang et al., “Local chemical ordering within the incubation period as a trigger for nanocrystallization of a highly supercooled Ti-based liquid,” Materials & Design. 2018. link Times cited: 15 USED (low confidence) Z. Pan, V. Borovikov, M. Mendelev, and F. Sansoz, “Development of a semi-empirical potential for simulation of Ni solute segregation into grain boundaries in Ag,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 19 Abstract: An Ag–Ni semi-empirical potential was developed to simulate … read moreAbstract: An Ag–Ni semi-empirical potential was developed to simulate the segregation of Ni solutes at Ag grain boundaries (GBs). The potential combines a new Ag potential fitted to correctly reproduce the stable and unstable stacking fault energies in this metal and the existing Ni potential from Mendelev et al (2012 Phil. Mag. 92 4454–69). The Ag–Ni cross potential functions were fitted to ab initio data on the liquid structure of the Ag80Ni20 alloy to properly incorporate the Ag–Ni interaction at small atomic separations, and to the Ni segregation energies at different sites within a high-energy Σ9 (221) symmetric tilt GB. By deploying this potential with hybrid Monte Carlo/molecular dynamics simulations, it was found that heterogeneous segregation and clustering of Ni atoms at GBs and twin boundary defects occur at low Ni concentrations, 1 and 2 at%. This behavior is profoundly different from the homogeneous interfacial dispersion generally observed for the Cu segregation in Ag. A GB transformation to amorphous intergranular films was found to prevail at higher Ni concentrations (10 at%). The developed potential opens new opportunities for studying the selective segregation behavior of Ni solutes in interface-hardened Ag metals and its effect on plasticity. read less USED (low confidence) G. Ma, J. Fan, and H. Gong, “Mechanical behavior of Cu-W interface systems upon tensile loading from molecular dynamics simulations,” Computational Materials Science. 2018. link Times cited: 37 USED (low confidence) V. Imandi, M. Jagannath, and A. Chatterjee, “Role of solvent in metal-on-metal surface diffusion: A case for rational solvent selection for materials synthesis,” Surface Science. 2018. link Times cited: 7 USED (low confidence) G.-U. Jeong, C. S. Park, H.-S. Do, S.-M. Park, and B.-J. Lee, “Second nearest-neighbor modified embedded-atom method interatomic potentials for the Pd-M (M = Al, Co, Cu, Fe, Mo, Ni, Ti) binary systems,” Calphad. 2018. link Times cited: 12 USED (low confidence) G. Sun, J. Xu, and P. Harrowell, “The mechanism of the ultrafast crystal growth of pure metals from their melts,” Nature Materials. 2018. link Times cited: 59 USED (low confidence) D. K. Das, J. Sarkar, and S. K. Singh, “Effect of sample size, temperature and strain velocity on mechanical properties of plumbene by tensile loading along longitudinal direction: A molecular dynamics study,” Computational Materials Science. 2018. link Times cited: 21 USED (low confidence) B. T. Susi and J. Tu, “Molecular dynamics simulations of the wetting behavior of carbon nanotubes in liquid copper,” Computers & Fluids. 2018. link Times cited: 1 USED (low confidence) T. A. Jui, P. Bose, T. Rakib, S. Mojumder, and M. Motalab, “Shear based analysis of nickel nano-plate by molecular dynamics simulations.” 2018. link Times cited: 0 Abstract: The determination of shear based properties of Nickel (Ni) h… read moreAbstract: The determination of shear based properties of Nickel (Ni) has a great importance since it is more likely to fail by shear than tension due to its ductile nature. It also features a wide variety of applications in structure, thin film, tubes, and plates due to its unique thermal and electrical properties. Molecular Dynamics Simulations were performed on Ni nano-plate subjected to shear loading to study the effect of voids in the structure using embedded atom method (EAM) potential. The shear stress-strain behavior was observed for Ni nano-plate with voids of 1.0 nm, 1.5 nm, and 2.0 nm radius. Snapshots taken at different strains show the formation of slip planes, crack propagation, and dislocation activity. Simulation results show that the modulus of rupture decreases with the increase of void radius due to more dislocation activity for larger void. Lastly, the effect of different void size on the shear modulus of rigidity is also incorporated.The determination of shear based properties of Nickel (Ni) has a great importance since it is more likely to fail by shear than tension due to its ductile nature. It also features a wide variety of applications in structure, thin film, tubes, and plates due to its unique thermal and electrical properties. Molecular Dynamics Simulations were performed on Ni nano-plate subjected to shear loading to study the effect of voids in the structure using embedded atom method (EAM) potential. The shear stress-strain behavior was observed for Ni nano-plate with voids of 1.0 nm, 1.5 nm, and 2.0 nm radius. Snapshots taken at different strains show the formation of slip planes, crack propagation, and dislocation activity. Simulation results show that the modulus of rupture decreases with the increase of void radius due to more dislocation activity for larger void. Lastly, the effect of different void size on the shear modulus of rigidity is also incorporated. read less USED (low confidence) I. V. Chepkasov, M. Visotin, E. A. Kovaleva, A. Manakhov, V. S. Baidyshev, and Z. Popov, “Stability and Electronic Properties of PtPd Nanoparticles via MD and DFT Calculations,” The Journal of Physical Chemistry C. 2018. link Times cited: 15 Abstract: The structural as well as electronic properties of PtPd nano… read moreAbstract: The structural as well as electronic properties of PtPd nanoparticles (NPs) were investigated by using molecular dynamics simulations and density functional theory calculations. A wide range of NPs of different sizes (from 1.5 to 4 nm), structures (core–shell, alloy, Janus), and compositions were taken into consideration. It was shown that PtPd NPs of less than ∼2.0 nm are prone to structural transformations to icosahedral (Ih) shape, regardless of their initial structure and composition. On the other hand, for NPs of size ∼2.5 nm, the increase of temperature up to 700–900 K leads to structural changes only for compositions close to 40% Pt, which corresponds to energetic minimum for Pt@Pd NPs. The Ih form of Pd@Pt NPs with monolayer thickness of Pt on the surface appears to have the most negatively charged surface which makes this kind of NPs the best candidate for catalysis application. read less USED (low confidence) K. Kushnir and A. Ostapovets, “Variability of Twin Boundary Structure in Computer Simulations of Tensile Twins in Magnesium,” Defect and Diffusion Forum. 2018. link Times cited: 3 Abstract: Variety of interatomic potentials for magnesium can be found… read moreAbstract: Variety of interatomic potentials for magnesium can be found in the literature. Result of computer simulations can be slightly different depending on used potential. Particularly, twin boundary structure with the lowest energy can be different in a frame of different models. Comparison of several popular embedded-atom method potentials is provided. It is shown that either reflection or glide structure of twin boundary has the lowest energy for different potentials. read less USED (low confidence) X.-G. Li, C. Hu, C. Chen, Z. Deng, J. Luo, and S. Ong, “Quantum-accurate spectral neighbor analysis potential models for Ni-Mo binary alloys and fcc metals,” Physical Review B. 2018. link Times cited: 61 Abstract: In recent years, efficient interatomic potentials approachin… read moreAbstract: In recent years, efficient interatomic potentials approaching the accuracy of density functional theory (DFT) calculations have been developed using rigorous atomic descriptors satisfying strict invariances, for example, for translation, rotation, permutation of homonuclear atoms, among others. In this paper, we generalize the spectral neighbor analysis potential (SNAP) model to bcc-fcc binary alloy systems. We demonstrate that machine-learned SNAP models can yield significant improvements even over the well-established high-performing embedded atom method (EAM) and modified EAM potentials for fcc Cu and Ni. We also report on the development of a SNAP model for the fcc Ni-bcc Mo binary system by machine learning a carefully constructed large computed data set of elemental and intermetallic compounds. We demonstrate that this binary Ni-Mo SNAP model can achieve excellent agreement with experiments in the prediction of a Ni-Mo phase diagram as well as near-DFT accuracy in the prediction of many key properties, such as elastic constants, formation energies, melting points, etc., across the entire binary composition range. In contrast, the existing Ni-Mo EAM has significant errors in the prediction of the phase diagram and completely fails in binary compounds. This paper provides a systematic model development process for multicomponent alloy systems, including an efficient procedure to optimize the hyperparameters in the model fitting, and paves the way for long-time large-scale simulations of such systems. read less USED (low confidence) Z. Dong and J. Wang, “Experimental and molecular dynamics investigation of a sessile dislocation core structure in Au,” Ferroelectrics. 2018. link Times cited: 0 Abstract: ABSTRACT A novel type of sessile dislocation core structure … read moreAbstract: ABSTRACT A novel type of sessile dislocation core structure and a 45° glissile dislocation are obtained in Au using high-resolution transmission electron microscopy. The formation mechanism of the sessile core structure is studied by molecular dynamics simulation. The 45° dislocation has a strong mobility on the {100} slip plane. Depending on the applied strain, two 45° dislocations with same sign screw components repelled each other; two 45° dislocations with screw components of opposite signs attract each other to form this type of sessile structure. Under a large applied strain, the sessile structure remained stable and motionless. read less USED (low confidence) T.-H. Lan, T. Ragab, and C. Basaran, “Electron-phonon scattering and Joule heating in copper at extreme cold temperatures,” Computational Materials Science. 2018. link Times cited: 3 USED (low confidence) S. Saha and M. Motalab, “Nature of creep deformation in nanocrystalline Tungsten,” Computational Materials Science. 2018. link Times cited: 18 USED (low confidence) A. Gola, P. Gumbsch, and L. Pastewka, “Atomic-scale simulation of structure and mechanical properties of Cu1−xAgx|Ni multilayer systems,” Acta Materialia. 2018. link Times cited: 17 USED (low confidence) P. Reyes et al., “The stability of hollow nanoparticles and the simulation temperature ramp,” Inorganic chemistry frontiers. 2018. link Times cited: 6 Abstract: Hollow nanoparticles (hNPs) are of interest because their la… read moreAbstract: Hollow nanoparticles (hNPs) are of interest because their large cavities and small thickness give rise to a large surface to volume ratio. However, in general they are not in equilibrium and far from their global energy minimum, which often makes them unstable against perturbations. In fact, a temperature increase can induce a structural collapse into a nanoparticle, and consequently a loss of their unique properties. This problem has been studied by means of molecular dynamics (MD) simulations, but without emphasis on the speed of the temperature increase. Here we explore how the temperature variation, and the rate at which it is varied in MD simulations, determines the final conformation of the hNPs. In particular, we show how different temperature ramps determine the final shape of Pt hNPs that initially have an external radius between 0.7 and 24 nm, and an internal radius between 0.19 and 2.4 nm. In addition, we also perform the simulations of other similar metals like Ag and Au. Our results indicate that the temperature ramp strongly modifies the final hNP shape, even at ambient temperature. In fact, a rapid temperature increase leads to the formation of stacking faults and twin boundaries which are not generated by a slower temperature increase. Quantitative criteria are established and they indicate that the stacking fault energy is the dominant parameter. read less USED (low confidence) D. Chakraborty, A. Harms, M. A. Ullah, W. J. Weber, and D. Aidhy, “Effect of atomic order/disorder on vacancy clustering in concentrated NiFe alloys,” Computational Materials Science. 2018. link Times cited: 7 USED (low confidence) K. Choudhary, A. Biacchi, S. Ghosh, L. Hale, A. Walker, and F. Tavazza, “High-throughput assessment of vacancy formation and surface energies of materials using classical force-fields,” Journal of Physics: Condensed Matter. 2018. link Times cited: 16 Abstract: In this work, we present an open access database for surface… read moreAbstract: In this work, we present an open access database for surface and vacancy-formation energies using classical force-fields (FFs). These quantities are essential in understanding diffusion behavior, nanoparticle formation and catalytic activities. FFs are often designed for a specific application, hence, this database allows the user to understand whether a FF is suitable for investigating particular defect and surface-related material properties. The FF results are compared to density functional theory and experimental data whenever applicable for validation. At present, we have 17 506 surface energies and 1000 vacancy formation energies calculation in our database and the database is still growing. All the data generated, and the computational tools used, are shared publicly at the following websites: www.ctcms.nist.gov/~knc6/periodic.html, https://jarvis.nist.gov and https://github.com/usnistgov/jarvis. Approximations used during the high-throughput calculations are clearly mentioned. Using some of the example cases, we show how our data can be used to directly compare different FFs for a material and to interpret experimental findings such as using Wulff construction for predicting equilibrium shape of nanoparticles. Similarly, the vacancy formation energies data can be useful in understanding diffusion related properties. read less USED (low confidence) Y. Gao, M. Takahashi, C. Cavallotti, and G. Raos, “Molecular dynamics simulation of metallic impurity diffusion in liquid lead-bismuth eutectic (LBE),” Journal of Nuclear Materials. 2018. link Times cited: 13 USED (low confidence) S. Bukkuru, U. Bhardwaj, K. S. Rao, A. Rao, M. Warrier, and M. C. Valsakumar, “Kinetics of self-interstitial migration in bcc and fcc transition metals,” Materials Research Express. 2018. link Times cited: 11 Abstract: Radiation damage is a multi-scale phenomenon. A thorough und… read moreAbstract: Radiation damage is a multi-scale phenomenon. A thorough understanding of diffusivities and the migration energies of defects is a pre-requisite to quantify the after-effects of irradiation. We investigate the thermally activated mobility of self-interstitial atom (SIA) in bcc transition metals Fe, Mo, Nb and fcc transition metals Ag, Cu, Ni, Pt using molecular dynamics (MD) simulations. The self-interstitial diffusion involves various mechanisms such as interstitialcy, dumbbell or crowdion mechanisms. Max-Space Clustering (MSC) method has been employed to identify the interstitial and its configuration over a wide range of temperature. The self-interstitial diffusion is Arrhenius like, however, there is a slight deviation at high temperatures. The migration energies, pre-exponential factors of diffusion and jump-correlation factors, obtained from these simulations can be used as inputs to Monte Carlo simulations of defect transport. The jump-correlation factor shows the degree of preference of rectilinear or rotational jumps. We obtain the average jump-correlation factor of 1.4 for bcc metals and 0.44 for fcc metals. It indicates that rectilinear jumps are preferred in bcc metals and rotational jumps are preferred in fcc metals. read less USED (low confidence) D. Yadav, P. Gupta, and N. Yedla, “Nano-Indentation of Aluminium Reinforced Metallic Glass Composites: A Molecular Dynamics Study,” IOP Conference Series: Materials Science and Engineering. 2018. link Times cited: 1 Abstract: Molecular dynamics (MD) simulations are performed for nanoin… read moreAbstract: Molecular dynamics (MD) simulations are performed for nanoindentation on metal (Al)-metallic glass (Cu50Zr50) reinforced composites to investigate the mechanical properties and the effects of volume percentage on behavior of the load-displacement curves. The interaction among Al-Cu-Zr is modelled using a EAM (Embedded Atom Method) potential. Simulation box size of 100 Å (x) × 100 Å (y) × 100 Å (z) is modelled for investigating the properties of the sintered models by altering the volume percentage on the scale of 5%-20%. Nanoindentation is done along y-direction with a spherical diamond indenter at temperature of 300 K with constant indentation speed of 100 m/s. NVT ensemble is used with a timestep of 0.002 ps. Investigations on the effect of volume percentage show that as volume percentage of Metallic Glass (MG) increases, the corresponding Load required to penetrate inside the sample also increases. As a result of this Hardness also increase as volume percentage varies from 5% to 20%. read less USED (low confidence) X.-guo Zeng, T. Han, Y. Guo, and F. Wang, “Molecular dynamics modeling of crack propagation in titanium alloys by using an experiment-based Monte Carlo model,” Engineering Fracture Mechanics. 2017. link Times cited: 15 USED (low confidence) C. Liu, C. Xu, Y. Cheng, X.-R. Chen, and L. Cai, “Molecular dynamics studies of body-centered cubic tungsten during melting under pressure,” Chinese Journal of Physics. 2017. link Times cited: 10 USED (low confidence) G. Chen, C. Wang, and P. Zhang, “Molecular dynamics simulation of the coalescence and melting process of Au and Cu nano-clusters,” International Journal of Modern Physics B. 2017. link Times cited: 2 Abstract: Molecular dynamic (MD) method is used to study the coalescen… read moreAbstract: Molecular dynamic (MD) method is used to study the coalescence and fusing process of Au and Cu nanoclusters. The results show that shear deformation, surface and interface diffusion play important role in different stages of all simulation procedure. In most cases, shear deformation produces the twin boundary or/and stacking fault in particles by particle rotation and slide. The angle between the {111} of Au and Cu particles decrease with increasing temperature, which promotes the formation of the stable interface. Furthermore, the coalescence point and melting temperature increase as cluster diameter increases. For the other cases, there are no particle rotation and slide phenomenon in the elevating temperature process because the stable interface can be formed by forming twin boundaries once two particles contact. read less USED (low confidence) Y. Hu, J. Schuler, and T. Rupert, “Identifying interatomic potentials for the accurate modeling of interfacial segregation and structural transitions,” Computational Materials Science. 2017. link Times cited: 16 USED (low confidence) K. Duan, L. Li, Y. Hu, and X. Wang, “Damping characteristic of Ni-coated carbon nanotube/copper composite,” Materials & Design. 2017. link Times cited: 30 USED (low confidence) P. Gupta and N. Yedla, “Dislocation and Structural Studies at Metal–Metallic Glass Interface at Low Temperature,” Journal of Materials Engineering and Performance. 2017. link Times cited: 13 USED (low confidence) P. Singh et al., “Design of high-strength refractory complex solid-solution alloys,” npj Computational Materials. 2017. link Times cited: 67 USED (low confidence) S. Saha, M. Motalab, and M. Mahboob, “Investigation on mechanical properties of polycrystalline W nanowire,” Computational Materials Science. 2017. link Times cited: 24 USED (low confidence) K. Barmak, J. Liu, L. Harlan, P. Xiao, J. R. Duncan, and G. Henkelman, “Transformation of topologically close-packed β-W to body-centered cubic α-W: Comparison of experiments and computations.,” The Journal of chemical physics. 2017. link Times cited: 21 Abstract: The enthalpy and activation energy for the transformation of… read moreAbstract: The enthalpy and activation energy for the transformation of the metastable form of tungsten, β-W, which has the topologically close-packed A15 structure (space group Pm3¯n), to equilibrium α-W, which is body-centered cubic (A2, space group Im3¯m), was measured using differential scanning calorimetry. The β-W films were 1 μm-thick and were prepared by sputter deposition in argon with a small amount of nitrogen. The transformation enthalpy was measured as -8.3 ± 0.4 kJ/mol (-86 ± 4 meV/atom) and the transformation activation energy as 2.2 ± 0.1 eV. The measured enthalpy was found to agree well with the difference in energies of α and β tungsten computed using density functional theory, which gave a value of -82 meV/atom for the transformation enthalpy. A calculated concerted transformation mechanism with a barrier of 0.4 eV/atom, in which all the atoms in an A15 unit cell transform into A2, was found to be inconsistent with the experimentally measured activation energy for any critical nucleus larger than two A2 unit cells. Larger calculations of eight A15 unit cells spontaneously relax to a mechanism in which part of the supercell first transforms from A15 to A2, creating a phase boundary, before the remaining A15 transforms into the A2 phase. Both calculations indicate that a nucleation and growth mechanism is favored over a concerted transformation. More consistent with the experimental activation energy was that of a calculated local transformation mechanism at the A15-A2 phase boundary, computed as 1.7 eV using molecular dynamics simulations. This calculated phase transformation mechanism involves collective rearrangements of W atoms in the disordered interface separating the A15 and A2 phases. read less USED (low confidence) L. Zhang, C. Lu, G. Michal, G. Deng, and K. Tieu, “The formation and destruction of stacking fault tetrahedron in fcc metals: A molecular dynamics study,” Scripta Materialia. 2017. link Times cited: 37 USED (low confidence) Q. Li et al., “Molecular dynamics simulations of aggregation of copper nanoparticles with different heating rates,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 53 USED (low confidence) P. Saidi, C. Dai, T. Power, Z. Yao, and M. Daymond, “An embedded atom method interatomic potential for the zirconium-iron system,” Computational Materials Science. 2017. link Times cited: 5 USED (low confidence) M. Celtek, S. Sengul, and U. Domekeli, “Glass formation and structural properties of Zr50Cu50-xAlx bulk metallic glasses investigated by molecular dynamics simulations,” Intermetallics. 2017. link Times cited: 35 USED (low confidence) C. Gang, W. Chuanjie, and Z. Peng, “The role of interface in uniaxial tensile process of nano-scale bilayer Cu/Ni,” Computational Materials Science. 2017. link Times cited: 10 USED (low confidence) L. Koch et al., “Local segregation versus irradiation effects in high-entropy alloys : Steady-state conditions in a driven system,” Journal of Applied Physics. 2017. link Times cited: 61 Abstract: We study order transitions and defect formation in a model h… read moreAbstract: We study order transitions and defect formation in a model high-entropy alloy (CuNiCoFe) under ion irradiation by means of molecular dynamics simulations. Using a hybrid Monte-Carlo/molecular dynamics scheme, a model alloy is generated which is thermodynamically stabilized by configurational entropy at elevated temperatures, but partly decomposes at lower temperatures by copper precipitation. Both the high-entropy and the multiphase sample are then subjected to simulated particle irradiation. The damage accumulation is analyzed and compared to an elemental Ni reference system. The results reveal that the high-entropy alloy—independent of the initial configuration—installs a certain fraction of short-range order even under particle irradiation. Moreover, the results provide evidence that defect accumulation is reduced in the high-entropy alloy. This is because the reduced mobility of point defects leads to a steady state of defect creation and annihilation. The lattice defects generated by irradiation are ... read less USED (low confidence) K. Zhou, B. Liu, S. Shao, and Y. Yao, “Molecular dynamics simulations of tension–compression asymmetry in nanocrystalline copper,” Physics Letters A. 2017. link Times cited: 33 USED (low confidence) G. Brunetto and A. Martini, “Atomistic description of coupled thermal-mechanical stresses on a gold/HOPG nanocontact,” Computational Materials Science. 2017. link Times cited: 3 USED (low confidence) K. Duan, L. Li, Y. Hu, and X. Wang, “Enhanced interfacial strength of carbon nanotube/copper nanocomposites via Ni-coating: Molecular-dynamics insights,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 31 USED (low confidence) W. Choi, Y. Kim, D. Seol, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for the Co-Cr, Co-Fe, Co-Mn, Cr-Mn and Mn-Ni binary systems,” Computational Materials Science. 2017. link Times cited: 62 USED (low confidence) S. Guo, M. Wang, Z. Zhao, Y. Zhang, X. Lin, and W. Huang, “Molecular dynamics simulation on the micro-structural evolution in heat-affected zone during the preparation of bulk metallic glasses with selective laser melting,” Journal of Alloys and Compounds. 2017. link Times cited: 23 USED (low confidence) S. Xu, L. Xiong, Y. Chen, and D. McDowell, “Comparing EAM Potentials to Model Slip Transfer of Sequential Mixed Character Dislocations Across Two Symmetric Tilt Grain Boundaries in Ni,” JOM. 2017. link Times cited: 36 USED (low confidence) K. Zhou and T. Zhang, “Positron Lifetime Calculation for Plastic Deformed Nanocrystalline Copper,” Defect and Diffusion Forum. 2017. link Times cited: 0 Abstract: Positron lifetime calculation has been performed on a comput… read moreAbstract: Positron lifetime calculation has been performed on a computer-generated nanocrystalline copper with a mean grain size of 9.1 nm during its deformation. For the undeformed and deformed nanocrystalline copper, calculated positron lifetimes are around 157 ps which come from the positron annihilation in the free volume in grain boundaries. Due to the grain-boundary deformation mechanism, no vacancies or vacancy clusters will be induced in grains during the plastic deformation of the nanocrystalline copper, which is different to the deformation of the conventional polycrystal. From this point of view, in-situ positron annihilation measurements can provide important experimental information on the deformation mechanism of nanocrystalline metals. read less USED (low confidence) Munaji, Sudarno, D. L. Purwaningroom, and R. Arifin, “Performance of EAM and MEAM Potential for NiTi Alloys: A Comparative Study,” IOP Conference Series: Materials Science and Engineering. 2017. link Times cited: 3 Abstract: NiTi alloys is one of the unique materials exhibiting shape … read moreAbstract: NiTi alloys is one of the unique materials exhibiting shape memory effect. The martensitic transformation is the main reason for their behaviour, which is very sensitive to the heat treatment and the ratio of Ni-Ti atoms. The study of the NiTi alloys behaviour at the atomic level is indispensable to elucidate the mechanism of the martensitic transformation under the specific conditions. Molecular dynamics simulation is widely used in this kind of study. The results of the molecular dynamics simulation depend on the selection of the interatomic potential. This study is aimed to evaluate the performances of the standard EAM potentials of Zhou et al. and the modified EAM of Ko et al. by means of obtaining the more accurate lattice constant in comparison with the experimental value. These interatomic potentials are also tested to reproduce the recrystallization behaviour below the melting temperature. We found that the high accuracy of the lattice constant for NiTi alloy system could be achieved by employing MEAM potential of Ko et el. However, the EAM potential by Zhou et al. gives the rapid recrystallization of NiTi alloys at 1100 K. These results indicate that the MEAM potential of Ko et. al. shows the better performance at low temperature simulation. read less USED (low confidence) S. Fazeli, M. Vahedpour, and S. Sadrnezhaad, “What is the copper thin film thickness effect on thermal properties of NiTi/Cu bi-layer?,” Materials Research Express. 2017. link Times cited: 0 Abstract: Molecular dynamics (MD) simulation was used to study of ther… read moreAbstract: Molecular dynamics (MD) simulation was used to study of thermal properties of NiTi/Cu. Embedded atom method (EAM) potentials for describing of inter-atomic interaction and Nose–Hoover thermostat and barostat are employed. The melting of the bi-layers was considered by studying the temperature dependence of the cohesive energy and mean square displacement. To highlight the differences between bi-layers with various copper layer thickness, the effect of copper film thickness on thermal properties containing the cohesive energy, melting point, isobaric heat capacity and latent heat of fusion was estimated. The results show that thermal properties of bi-layer systems are higher than that of their corresponding of pure NiTi. But, these properties of bi-layer systems approximately are independent of copper film thicknesses. The mean square displacement (MSD) results show that, the diffusion coefficients enhance upon increasing of copper film thickness in a linear performance. read less USED (low confidence) K. Mackenchery and A. Dongare, “Shock Hugoniot behavior of single crystal titanium using atomistic simulations.” 2017. link Times cited: 4 Abstract: Atomistic shock simulations are performed for single crystal… read moreAbstract: Atomistic shock simulations are performed for single crystal titanium using four different interatomic potentials at impact velocities ranging from 0.5 km/s to 2.0 km/s. These potentials comprise of three parameterizations in the formulation of the embedded atom method and one formulation of the modified embedded atom method. The capability of the potentials to model the shock deformation and failure behavior is investigated by computing the shock hugoniot response of titanium and comparing to existing experimental data. In addition, the capability to reproduce the shock induced alpha (α) to omega (ω) phase transformation seen in Ti is investigated. The shock wave structure is discussed and the velocities for the elastic, plastic and the α-ω phase transformation waves are calculated for all the interatomic potentials considered. read less USED (low confidence) S. Fazeli, M. Vahedpour, and S. Sadrnezhaad, “Comparison of the mechanical properties of NiTi/Cu bilayer by nanoindentation and tensile test: molecular dynamics simulation,” Materials Research Express. 2016. link Times cited: 5 Abstract: Molecular dynamics simulation was used to study of mechanica… read moreAbstract: Molecular dynamics simulation was used to study of mechanical properties of NiTi/Cu bilayer by nanoindentation and tensile testing. A comparison has been made among mechanical properties measured and plastic deformation process at different copper thickness during nanoindnetation and tensile test of the samples. Embedded atom method potentials for describing of inter-atomic interaction and Nose–Hoover thermostat and barostat are employed in the simulation at 400 K. The results showed that as the copper film thickness decreased, the maximum load and hardness values increased during nanoindetation. Saha and Nix model is used to describe reduced young’s modulus behaviour of the bilayer system through nanoindentation. A good agreement among calculated reduced elastic modulus by nanoindentation test and young’s modulus behaviour via tensile test have been obtained. The ‘incoherent interface’ in both of nanoindentation test and tensile testing is one of the governing factors for the dislocation propagation, which resulted in significant strengthening of the bilayer. It was observed that during tensile test, only copper layers were necked and fractured in all of samples. However, the present study seeks to examine the effect of film thickness on the free energy values that is obtained using Jarzynski’s equality during nanoindentation. As the copper film thickness was decreased, the free energy difference increased. According to both techniques, the thin film copper thickness provides lower number of nucleation locations resulting in the higher value of yield strength, hardness and free energy difference during nanoindenation. Mechanical properties of bilayer systems are improved with decreasing of copper film thickness. However, it specifies that strengths of all bilayer systems have prominent increase in young’s modulus in compared to the pure NiTi. read less USED (low confidence) Q. Li, T. Fu, T. Peng, X. Peng, C. Liu, and X. Shi, “Coalescence of Cu contacted nanoparticles with different heating rates: A molecular dynamics study,” International Journal of Modern Physics B. 2016. link Times cited: 18 Abstract: The coalescence, the initial stage of sintering, of two cont… read moreAbstract: The coalescence, the initial stage of sintering, of two contacted Cu nanoparticles is investigated under different heating rates of 700, 350 and 233 K/ns. The nanoparticles coalesced rapidly at the initial stage when the temperature of the system is low. Then, the nanoparticles collided softly in an equilibrium period. After the system was increased to a high temperature, the shrinkage ratio, gyration radius and atoms’ diffusion started to change dramatically. The lower heating rate can result in smaller shrinkage ratio, larger gyration radius and diffusion of atoms. However, the growth of sintering neck is hardly influenced by the heating rate. The results provide a theoretical guidance for the fundamental understanding and potential application regarding nanoparticle sintering. read less USED (low confidence) D. H. Chung, H. Guk, S.-H. Choi, and D. Kim, “Wettability of Ag nanocluster on Cu-Ni alloys: A computational approach,” Journal of Alloys and Compounds. 2016. link Times cited: 5 USED (low confidence) N. N. Kulkarni and A. Chatterjee, “Capturing local atomic environment dependence of activation barriers in metals using cluster expansion models,” Journal of Physics: Conference Series. 2016. link Times cited: 3 Abstract: It is well known that surface diffusion in metals can procee… read moreAbstract: It is well known that surface diffusion in metals can proceed via multiple mechanisms, such as hop, exchange and other types of concerted moves. However, the manner in which kinetic rates associated with a mechanism can depend sensitively on local atomic environment is relatively less understood. We describe recent attempts in our research group to capture the atomic environment dependence using the cluster expansion model (CEM). In particular, we focus on hop and exchange moves at the (001) surface in homoepitaxy, and show that while CEM can work remarkably well in most cases, it can sometimes provide inaccurate predictions for concerted moves. read less USED (low confidence) Z. Zhang et al., “A novel approach to fabricating a nanotwinned surface on a ternary nickel alloy,” Materials & Design. 2016. link Times cited: 30 USED (low confidence) P. Gupta, S. Pal, and N. Yedla, “Molecular dynamics based cohesive zone modeling of Al (metal)–Cu50Zr50 (metallic glass) interfacial mechanical behavior and investigation of dissipative mechanisms,” Materials & Design. 2016. link Times cited: 56 USED (low confidence) C. Dai, L. Balogh, Z. Yao, and M. Daymond, “Atomistic simulations of the formation of -component dislocation loops in α-zirconium,” Journal of Nuclear Materials. 2016. link Times cited: 26 USED (low confidence) S. An, J. Li, Y. Li, S. Li, Q. Wang, and B.-xin Liu, “Two-step crystal growth mechanism during crystallization of an undercooled Ni50Al50 alloy,” Scientific Reports. 2016. link Times cited: 21 USED (low confidence) A. Patra, M. Meraj, S. Pal, N. Yedla, and S. K. Karak, “Experimental and atomistic simulation based study of W based alloys synthesized by mechanical alloying,” International Journal of Refractory Metals & Hard Materials. 2016. link Times cited: 25 USED (low confidence) V. Imandi and A. Chatterjee, “Estimating Arrhenius parameters using temperature programmed molecular dynamics.,” The Journal of chemical physics. 2016. link Times cited: 24 Abstract: Kinetic rates at different temperatures and the associated A… read moreAbstract: Kinetic rates at different temperatures and the associated Arrhenius parameters, whenever Arrhenius law is obeyed, are efficiently estimated by applying maximum likelihood analysis to waiting times collected using the temperature programmed molecular dynamics method. When transitions involving many activated pathways are available in the dataset, their rates may be calculated using the same collection of waiting times. Arrhenius behaviour is ascertained by comparing rates at the sampled temperatures with ones from the Arrhenius expression. Three prototype systems with corrugated energy landscapes, namely, solvated alanine dipeptide, diffusion at the metal-solvent interphase, and lithium diffusion in silicon, are studied to highlight various aspects of the method. The method becomes particularly appealing when the Arrhenius parameters can be used to find rates at low temperatures where transitions are rare. Systematic coarse-graining of states can further extend the time scales accessible to the method. Good estimates for the rate parameters are obtained with 500-1000 waiting times. read less USED (low confidence) S. Guo et al., “Molecular dynamics simulation of melt structure evolution during cooling process for lead on nickel substrate surface,” Materials Letters. 2016. link Times cited: 4 USED (low confidence) K. Wang, J.-fang Liu, and Q. Chen, “Palladium clusters deposited on the heterogeneous substrates,” Applied Surface Science. 2016. link Times cited: 4 USED (low confidence) K. Sebeck, C. Shao, and J. Kieffer, “Alkane-Metal Interfacial Structure and Elastic Properties by Molecular Dynamics Simulation.,” ACS applied materials & interfaces. 2016. link Times cited: 9 Abstract: The structure of amorphous materials near the interface with… read moreAbstract: The structure of amorphous materials near the interface with an ordered substrate can be affected by various characteristics of the adjoining phases, such as the lattice spacing of the adherent surface, polymer chain length, and adhesive strength. To discern the influence of each of these factors, four FCC metal lattices are examined for three chain lengths of n-alkane and van der Waals interfacial interactions are controlled by adjusting the Lennard-Jones 12-6 potential parameters. The role of interaction strength is investigated for a single chain length and substrate combination. Four nanoconfined systems are also analyzed in terms of their mechanical strength. A strong layering effect is observed near the interface for all systems. The distinctiveness of polymer layering, i.e., the maximum density and spatial extent, exhibits a logarithmic dependence on the interaction strength between polymer and substrate. Congruency with the substrate lattice parameter further enhances this effect. Moreover, the elastic modulus of the alkane phase as a function of layer thickness indicates that the effects of ordering within the structure extend beyond the immediately obvious interfacial region. read less USED (low confidence) G. Chen, C. Wang, and P. Zhang, “The equilibrium crystallisation process of non-crystalline Cu3Au,” Physics and Chemistry of Liquids. 2016. link Times cited: 0 Abstract: ABSTRACT The equilibrium crystallisation of non-crystalline … read moreAbstract: ABSTRACT The equilibrium crystallisation of non-crystalline Cu3Au has been studied by molecular dynamics method. The results of structure analysis show that the crystallisation kinetics of non-crystalline Cu3Au in the isothermal process is in agreement with the Avrami equation in the temperature range between 400 and 850 K. The time–temperature transformation diagram of crystallisation process presents a typical C-curve with nose temperature about 650 K. In the crystallisation process, there are also many metastable clusters produced in the disordered structure. The metastable polyhedron structure can promote the nucleation and growth process of crystal. read less USED (low confidence) G. Dai, B. Wang, S. Xu, Y. Lu, and Y. Shen, “Side-to-Side Cold Welding for Controllable Nanogap Formation from ‘Dumbbell’ Ultrathin Gold Nanorods.,” ACS applied materials & interfaces. 2016. link Times cited: 15 Abstract: Cold welding has been regarded as a promising bottom-up nano… read moreAbstract: Cold welding has been regarded as a promising bottom-up nanofabrication technique because of its ability to join metallic nanostructures at room temperature with low applied stress and without introducing damage. Usually, the cold welding process can be done instantaneously for ultrathin nanowires (diameter <10 nm) in "head-to-head" joining. Here, we demonstrate that "dumbbell" shaped ultrathin gold nanorods can be cold welded in the "side-to-side" mode in a highly controllable manner and can form an extremely small nanogap via a relatively slow welding process (up to tens of minutes, allowing various functional applications). By combining in situ high-resolution transmission electron microscopic analysis and molecular dynamic simulations, we further reveal the underlying mechanism for this "side-to-side" welding process as being dominated by atom kinetics instead of thermodynamics, which provides critical insights into three-dimensional nanosystem integration as well as the building of functional nanodevices. read less USED (low confidence) M. Xiang, J. Chen, and R. Su, “Spalling behaviors of Pb induced by ramp-wave-loading: Effects of the loading rise time studied by molecular dynamics simulations,” Computational Materials Science. 2016. link Times cited: 18 USED (low confidence) M. Meraj and S. Pal, “Deformation of Ni20W20Cu20Fe20Mo20 high entropy alloy for tensile followed by compressive and compressive followed by tensile loading: A molecular dynamics simulation based study,” IOP Conference Series: Materials Science and Engineering. 2016. link Times cited: 6 Abstract: In this paper, molecular dynamics (MD) simulations based stu… read moreAbstract: In this paper, molecular dynamics (MD) simulations based study on deformation behavior during uniaxial tension followed by compression and compression followed by tension after 0.6 pre-strain for Ni20W20Cu20Fe20Mo20 high entropy alloy (20 at. % each element) single crystals has been reported. This MD simulation is carried out at strain rate of 108 s-1 and at the temperature of -10°C. The influence of observed nano twin on deformation behaviour for such two types of loading process (i.e. tensile followed by compressive and compressive followed by tensile) has been investigated thoroughly. It is found that the dominant deformation mechanism is twin for tensile forward loading in Ni20W20Cu20Fe20Mo20 high entropy alloy single crystal, whereas atomic diffusion is the dominating factor for deformation behaviour in compressive reverse loading direction of high entropy alloy. read less USED (low confidence) P. Gupta and N. Yedla, “High Temperature Mechanical Behavior of Aluminum- Cu50Zr50Metallic Glass Interface,” IOP Conference Series: Materials Science and Engineering. 2016. link Times cited: 1 Abstract: Molecular dynamics (MD) simulations are carried out to deter… read moreAbstract: Molecular dynamics (MD) simulations are carried out to determine interface strength between aluminum (metal) and Cu50Zr50 (metallic glass) at temperature of 500 K and at strain rate of 108 s-1. Simulation box of size 100 Å (x) × 110 Å (y) × 50 Å (z) is used for the above studies. At first Al-Cu50Zr50 crystalline interface model is built with the base layer-Al of 50 A and the top layer-Cu50Zr50 of 55 Å along y-direction. Later Cu50Zr50 metallic glass is obtained by quenching at a cooling rate of 4 x 1012 Ks-1. NPT ensemble is used in metallic glass preparation simulation. The interface model is then equilibrated at 300 K for 500 ps to relieve the internal stresses. EAM (Embedded Atom Method) potential is used for modelling the interaction between Al-Cu-Zr atoms. The interface strength of Al-Cu50Zr50 model interface is determined by applying load in the directions normal (mode-I) and parallel (mode-II) to the interface. NVT ensemble is used for the deformation studies. In mode-I for perfect and cracked interface, the interface fractures in the Al-region via necking. Sticking of the Al-atoms to the metallic glass is observed in both the loading conditions. Also, multiple voids are nucleated at the interface. read less USED (low confidence) J. Wang, S. Chen, K. Cui, D. Li, and D. Chen, “Approach and Coalescence of Gold Nanoparticles Driven by Surface Thermodynamic Fluctuations and Atomic Interaction Forces.,” ACS nano. 2016. link Times cited: 57 Abstract: The approach and coalescence behavior of gold nanoparticles … read moreAbstract: The approach and coalescence behavior of gold nanoparticles on a silicon surface were investigated by experiments and molecular dynamics simulations. By analyzing the behavior of the atoms in the nanoparticles in the simulations, it was found that the atoms in a single isolated nanoparticle randomly fluctuated and that the surface atoms showed greater fluctuation. The fluctuation increased as the temperature increased. When there were two or more neighboring nanoparticles, the fluctuating surface atoms of the nanoparticles "flowed" toward the neighboring nanoparticle because of atomic interaction forces between the nanoparticles. With the surface atoms "flowing", the gold nanoparticles approached and finally coalesced. The simulation results were in good agreement with the experimental results. It can be concluded that surface thermodynamic fluctuations and atomic interaction forces are the causes of the approach and coalescence behavior of the gold nanoparticles. read less USED (low confidence) C. Liu, C. Xu, Y. Cheng, X.-R. Chen, and L. Cai, “Melting curves and structural properties of tantalum from the modified-Z method,” Journal of Applied Physics. 2015. link Times cited: 6 Abstract: The melting curves and structural properties of tantalum (Ta… read moreAbstract: The melting curves and structural properties of tantalum (Ta) are investigated by molecular dynamics simulations combining with potential model developed by Ravelo et al. [Phys. Rev. B 88, 134101 (2013)]. Before calculations, five potentials are systematically compared with their abilities of producing reasonable compressional and equilibrium mechanical properties of Ta. We have improved the modified-Z method introduced by Wang et al. [J. Appl. Phys. 114, 163514 (2013)] by increasing the sizes in Lx and Ly of the rectangular parallelepiped box (Lx = Ly ≪ Lz). The influences of size and aspect ratio of the simulation box to melting curves are also fully tested. The structural differences between solid and liquid are detected by number density and local-order parameters Q6. Moreover, the atoms' diffusion with simulation time, defects, and vacancies formations in the sample are all studied by comparing situations in solid, solid-liquid coexistence, and liquid state. read less USED (low confidence) Q. Li, X. Peng, T. Peng, Q. Tang, X. Zhang, and C. Huang, “Molecular dynamics simulation of Cu/Au thin films under temperature gradient,” Applied Surface Science. 2015. link Times cited: 30 USED (low confidence) A. Prokhoda, “Formation of multiply twinned nanoparticles of pure (Al, Cu, Ni) metals during crystallization: Results of molecular dynamics simulation,” 2015 International Young Scientists Forum on Applied Physics (YSF). 2015. link Times cited: 0 Abstract: Structures of simulated metals (Al, Cu, Ni) obtained in resu… read moreAbstract: Structures of simulated metals (Al, Cu, Ni) obtained in result of isothermal annealing after quick cooling to certain temperatures are studied in detail. Obtained large enough structures heave a core skeleton from hcp-planes in the form of icosahedron that already are named “Ih-fractal” (tetrahedra with internal fcc structure are divided one from another by twinning hcp-planes). Rows from elementary decahedra in structures of hcp-planes give the basis of new 60 twinning planes for forming new sectors at growing of such formation. New rows from decahedra are appearing in the places of intersections of the secondary twinning planes, and a new family of twinning planes is forming. read less USED (low confidence) T. Zhang, K. Zhou, and Z. Chen, “Strain rate effect on plastic deformation of nanocrystalline copper investigated by molecular dynamics,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 47 USED (low confidence) S. Solhjoo and A. Vakis, “Definition and detection of contact in atomistic simulations,” Computational Materials Science. 2015. link Times cited: 17 USED (low confidence) Y. Zhao, X. Peng, T. Fu, R. Sun, C. Feng, and Z. Wang, “MD simulation of nanoindentation on (001) and (111) surfaces of Ag–Ni multilayers,” Physica E-low-dimensional Systems & Nanostructures. 2015. link Times cited: 39 USED (low confidence) A. Spitale, M. A. Perez, S. Mejía-Rosales, M. Yacamán, and M. Mariscal, “Gold-palladium core@shell nanoalloys: experiments and simulations.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 12 Abstract: In this work, we report a facile synthesis route, structural… read moreAbstract: In this work, we report a facile synthesis route, structural characterization, and full atomistic simulations of gold-palladium nanoalloys. Through aberration corrected-STEM, UV-vis spectroscopy and EDS chemical analysis, we were able to determine that Au(core)-Pd(shell) bimetallic nanoparticles were formed. Using different computational approaches, we were capable of establishing how the size of the core and the thickness of the shell will affect the thermodynamic stability of several core-shell nanoalloys. Finally, grand canonical simulations using different sampling procedures were used to study the growth mechanism of Pd atoms on Au seeds of different shapes. read less USED (low confidence) G. Jiang et al., “Core/Shell Face-Centered Tetragonal FePd/Pd Nanoparticles as an Efficient Non-Pt Catalyst for the Oxygen Reduction Reaction.,” ACS nano. 2015. link Times cited: 151 Abstract: We report the synthesis of core/shell face-centered tetragon… read moreAbstract: We report the synthesis of core/shell face-centered tetragonal (fct)-FePd/Pd nanoparticles (NPs) via reductive annealing of core/shell Pd/Fe3O4 NPs followed by temperature-controlled Fe etching in acetic acid. Among three different kinds of core/shell FePd/Pd NPs studied (FePd core at ∼8 nm and Pd shell at 0.27, 0.65, or 0.81 nm), the fct-FePd/Pd-0.65 NPs are the most efficient catalyst for the oxygen reduction reaction (ORR) in 0.1 M HClO4 with Pt-like activity and durability. This enhanced ORR catalysis arises from the desired Pd lattice compression in the 0.65 nm Pd shell induced by the fct-FePd core. Our study offers a general approach to enhance Pd catalysis in acid for ORR. read less USED (low confidence) D. Louzguine-Luzgin et al., “Structural changes in liquid Fe and Fe–B alloy on cooling,” Journal of Molecular Liquids. 2015. link Times cited: 11 USED (low confidence) S. Kalidindi, J. A. Gomberg, Z. Trautt, and C. Becker, “Application of data science tools to quantify and distinguish between structures and models in molecular dynamics datasets,” Nanotechnology. 2015. link Times cited: 39 Abstract: Structure quantification is key to successful mining and ext… read moreAbstract: Structure quantification is key to successful mining and extraction of core materials knowledge from both multiscale simulations as well as multiscale experiments. The main challenge stems from the need to transform the inherently high dimensional representations demanded by the rich hierarchical material structure into useful, high value, low dimensional representations. In this paper, we develop and demonstrate the merits of a data-driven approach for addressing this challenge at the atomic scale. The approach presented here is built on prior successes demonstrated for mesoscale representations of material internal structure, and involves three main steps: (i) digital representation of the material structure, (ii) extraction of a comprehensive set of structure measures using the framework of n-point spatial correlations, and (iii) identification of data-driven low dimensional measures using principal component analyses. These novel protocols, applied on an ensemble of structure datasets output from molecular dynamics (MD) simulations, have successfully classified the datasets based on several model input parameters such as the interatomic potential and the temperature used in the MD simulations. read less USED (low confidence) V. Podryga and S. Polyakov, “3D molecular dynamic simulation of thermodynamic equilibrium problem for heated nickel.” 2015. link Times cited: 6 Abstract: This work is devoted to molecular dynamic modeling of the th… read moreAbstract: This work is devoted to molecular dynamic modeling of the thermal impact processes on the metal sample consisting of nickel atoms. For the solution of this problem, a continuous mathematical model on the basis of the classical Newton mechanics equations has been used; a numerical method based on the Verlet scheme has been chosen; a parallel algorithm has been offered, and its realization within the MPI and OpenMP technologies has been executed. By means of the developed parallel program, the investigation of thermodynamic equilibrium of nickel atoms’ system under the conditions of heating a sample to desired temperature has been executed. In numerical experiments both optimum parameters of calculation procedure and physical parameters of analyzed process have been defined. The obtained numerical results are well corresponding to known theoretical and experimental data. read less USED (low confidence) S. Solhjoo and A. Vakis, “Single asperity nanocontacts: Comparison between molecular dynamics simulations and continuum mechanics models,” Computational Materials Science. 2015. link Times cited: 32 USED (low confidence) Z. W. Wu, M. Li, W. Wang, and K. Liu, “Hidden topological order and its correlation with glass-forming ability in metallic glasses,” Nature Communications. 2015. link Times cited: 111 USED (low confidence) K. Gaminchev and H. Chamati, “Dynamic stability of Fe under high pressure,” Journal of Physics: Conference Series. 2014. link Times cited: 1 Abstract: We study the dynamic stability of bcc iron at both high pres… read moreAbstract: We study the dynamic stability of bcc iron at both high pressure and temperature via Molecular Dynamics in conjunction with three different interatomic potentials constructed within the embedded-atom method. We computed the phonon dispersions, the phonon density of states, as well as the radial distribution functions. It is found that these quantities exhibit different behaviours depending on the potential used. Furthermore it is revealed that the simulated sample remains dynamically stable over a wide range of temperature and pressure for all potentials. read less USED (low confidence) X. Li, X.-B. Yang, and Y.-J. Zhao, “Quasilattice-Conserved Optimization of the Atomic Structure of Decagonal Ael-Co-Ni Quasicrystals,” Chinese Physics Letters. 2014. link Times cited: 0 Abstract: The detailed atomic structure of quasicrystals has been an o… read moreAbstract: The detailed atomic structure of quasicrystals has been an open problem for decades. Here we present a quasilattice-conserved optimization method (quasi-OPT), under particular quasiperiodic boundary conditions. As the atomic coordinates are described by basic cells and quasilattices, we are able to maintain the self-similarity characteristics of qusicrystals with the atomic structure of the boundary region updated timely following the relaxing region. Exemplified with the study of decagonal Al-Co-Ni (d-Al-Co-Ni), we propose a more stable atomic structure model based on Penrose quasilattice and our quasi-OPT simulations. In particular, rectangle-triangle rules are suggested for the local atomic structures of d-Al-Co-Ni quasicrystals. read less USED (low confidence) A. Klemenz, L. Pastewka, S. Balakrishna, A. Caron, R. Bennewitz, and M. Moseler, “Atomic scale mechanisms of friction reduction and wear protection by graphene.,” Nano letters. 2014. link Times cited: 203 Abstract: We study nanoindentation and scratching of graphene-covered … read moreAbstract: We study nanoindentation and scratching of graphene-covered Pt(111) surfaces in computer simulations and experiments. We find elastic response at low load, plastic deformation of Pt below the graphene at intermediate load, and eventual rupture of the graphene at high load. Friction remains low in the first two regimes, but jumps to values also found for bare Pt(111) surfaces upon graphene rupture. While graphene substantially enhances the load carrying capacity of the Pt substrate, the substrate's intrinsic hardness and friction are recovered upon graphene rupture. read less USED (low confidence) K. Zhou, B. Liu, Y. Yao, and K. Zhong, “Effects of grain size and shape on mechanical properties of nanocrystalline copper investigated by molecular dynamics,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2014. link Times cited: 81 USED (low confidence) W.-jin Zhang, Z.-L. Liu, and Y. Peng, “Molecular dynamics simulations of the melting curves and nucleation of nickel under pressure,” Physica B-condensed Matter. 2014. link Times cited: 11 USED (low confidence) B. Ma, Q. Rao, and Y. He, “Effect of crystal orientation on tensile mechanical properties of single-crystal tungsten nanowire,” Transactions of Nonferrous Metals Society of China. 2014. link Times cited: 26 USED (low confidence) H. Su, X. Fang, X. Feng, and B. Yan, “The Temperature-Dependent Strength of Metals: Theory and Experimental Validation,” Journal of Applied Mechanics. 2014. link Times cited: 9 Abstract: In this work, we propose a strength theory as a function of … read moreAbstract: In this work, we propose a strength theory as a function of temperature and state of stresses for metals. Based on the fracture in the hydrostatic stress, we derived a generalized strength model, in which the fracture strength decreases almost linearly with the increasing of the temperature. Furthermore this generalized strength model was extended to the general state of stresses by replacing the equivalent hydrostatic stresses with the temperature effect based on the general thermodynamics principles. Molecular dynamics (MD) simulation was also conducted to simulate the fracture evolution at high temperature and to explain the mechanism of temperature-dependent strength at atomic scale. The proposed model was also verified by experiment of Mo-10Cu alloy at elevated temperature. read less USED (low confidence) Y. Zhou and K. Fichthorn, “Internal Stress-Induced Orthorhombic Phase in 5-Fold-Twinned Noble Metal Nanowires,” Journal of Physical Chemistry C. 2014. link Times cited: 28 Abstract: We use atomistic simulations to show that 5-fold-twinned nan… read moreAbstract: We use atomistic simulations to show that 5-fold-twinned nanowires of several face-centered cubic metals (Ag, Au, Cu, and Pd) exhibit an overall body-centered orthorhombic phase resulting from the large internal stress associated with their twinned structure. The distribution of atomic stress in the nanowires confirms the existence of a disclination at the 5-fold axis, in addition to an anisotropic distortion of the lattice. We find that two regions of the nanowire are highly stressed: local stress maxima are distributed in the shape of leaflets running along each twin boundary, as well as in semicircular regions near the free surfaces. The large elastic strain energy associated with the distortion may be partially released via the formation and propagation of partial dislocations, which are restricted to a single subunit of the nanowire. Our calculations are in line with experiment and indicate the complex ways in which the structures of these metal nanocrystals can depend on their shape. read less USED (low confidence) T. Zientarski and D. Chocyk, “Strain and structure in nano Ag films deposited on Au: Molecular dynamics simulation,” Applied Surface Science. 2014. link Times cited: 9 USED (low confidence) T. Zientarski and D. Chocyk, “Structure and stress in Cu/Au and Fe/Au systems: A molecular dynamics study,” Thin Solid Films. 2014. link Times cited: 12 USED (low confidence) Y. Qi, L. Wang, and T. He, “Structure and Dynamical Relaxation in Undercooled Liquid of Cu50Co50 with Miscible Gap,” Advanced Materials Research. 2014. link Times cited: 0 Abstract: Molecular dynamics simulation has been performed to explore … read moreAbstract: Molecular dynamics simulation has been performed to explore the structural and dynamical properties of undercooled Cu50Co50 melt based upon the embedded atom method (EAM), namely due to Zhou. With the reduction of temperature, liquid phase separation phenomenon is more obvious. The simulated coordination number (CN) of liquid Cu50Co50 indicate that no stronger interaction of heterogenic atom pairs than those of the same atom pairs happens in melts, and Cu50Co50 melt is close to an weak demixing system. And the Bhatia-Thornton (B-T) structure factor gets the same result. Temperature dependence of mean squared displacement (MSD) shows very weak glass formation behavior of Cu50Co50. Our work discusses that the relationship between the properties and L-L phase separation in undercooled liquid of Cu50Co50. read less USED (low confidence) W.-jin Zhang, Y. Peng, and Z.-L. Liu, “Molecular dynamics study of melting curve, entropy of fusion and solid-liquid interfacial energy of cobalt under pressure,” Physica B-condensed Matter. 2014. link Times cited: 10 USED (low confidence) T. Fang, L. Wang, and Y. Qi, “Solid–liquid interface growth of Cu50Ni50 under deep undercoolings,” Physics and Chemistry of Liquids. 2014. link Times cited: 5 Abstract: Molecular dynamics simulations have been performed to explor… read moreAbstract: Molecular dynamics simulations have been performed to explore interface growth of liquid Cu50Ni50 alloy by using an embedded atom potential, namely due to Zhou. The simulated melting temperature is 1585 K in agreement well with the experimental value of 1600 K. The calculated interface velocity increases with decreasing temperature ranging from 1585 K to 1100 K, where the calculated values are a little higher than the experimental ones at higher temperatures, and in agreement with the experimental ones at lower temperatures; while the calculated interface velocity decreases with decreasing temperature lower than 1100 K. The activation energy of atom is 0.0048 eV, almost close to zero under deep undercoolings, although the crystal growth still proceeds with the speed ranging from 50 m s−1 to 10 m s−1. The crystal growth of Cu50Ni50 is not controlled by diffusion mechanism under deep undercoolings. read less USED (low confidence) K. Zhou, B. Liu, Y. Yao, and K. Zhong, “Grain coarsening in nanocrystalline copper with very small grain size during tensile deformation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2014. link Times cited: 9 USED (low confidence) C. Gang, Z. Peng, and H. Liu, “Molecular Dynamics Simulation of Solidification of Pd-Ni Clusters with Different Nickel Content,” Advances in Materials Science and Engineering. 2014. link Times cited: 4 Abstract: Molecular dynamics simulation has been performed for investi… read moreAbstract: Molecular dynamics simulation has been performed for investigating the glass transition of Pd-Ni alloy nanoparticles in the solidification process. The results showed that the Pd-Ni nanoparticles with composition far from pure metal should form amorphous structure more easily, which is in accordance with the results of the thermodynamic calculation. There are some regular and distorted fivefold symmetry in the amorphous Pd-Ni alloy nanoparticles. The nanoclusters with bigger difference value between formation enthalpies of solutions and glasses will transform to glass more easily than the other Pd-Ni alloy nanoclusters. read less USED (low confidence) H. Lee and V. Tomar, “Understanding the influence of grain boundary thickness variation on the mechanical strength of a nickel-doped tungsten grain boundary,” International Journal of Plasticity. 2014. link Times cited: 14 USED (low confidence) Y. Umeno, W. Nöhring, A. Iskandarov, and E. Bitzek, “Atomistic Model Analysis of Local and Global Instabilities in Crystals at Finite Temperature,” Key Engineering Materials. 2013. link Times cited: 0 Abstract: There have been a lot of studies dedicated to structural ins… read moreAbstract: There have been a lot of studies dedicated to structural instability in solids. For local instability, theoretical (ideal) strength of crystals has been extensively studied with ab initio calculations. Global instability taking into account the collective motion of atoms involved in deformation has also been investigated. However, these studies have usually been done at 0 K and little has been understood about the effect of temperature. In this study, we demonstrate computational approaches to the effect of temperature on local and global instabilities. Ideal shear strength (ISS) of silicon at finite temperatures is calculated by molecular dynamics (MD) simulations with an empirical potential. ISS is obtained as a function of temperature. Our results imply that, unlike metals, the reduction in ISS by temperature cannot be estimated simply by taking into account thermal expansion of volume. In addition, global instability for dislocation nucleation in a Cu thin film model under tension is investigated. We first evaluated instability modes at 0 K with increasing strain, and then performed MD simulations at 50 K. After the nucleation of a partial dislocation, the second dislocation can be one to create a twin or one to create another partial dislocation. These different deformations can be understood as the competition of latent instability modes that have relatively small eigenvalues. read less USED (low confidence) M. C. Nguyen, J.-H. Choi, X. Zhao, C. Wang, Z. Zhang, and K. Ho, “New layered structures of cuprous chalcogenides as thin film solar cell materials: Cu2Te and Cu2Se.,” Physical review letters. 2013. link Times cited: 79 Abstract: The stable crystal structures of two cuprous chalcogenides o… read moreAbstract: The stable crystal structures of two cuprous chalcogenides of Cu2X (X=Te or Se) are predicted using an adaptive genetic algorithm in combination with first-principles density functional theory calculations. Both systems are found to prefer a unique and previously unrecognized layered structure, with the total energies much lower than all structures proposed in the literature so far. The newly discovered structures are further shown to be dynamically and mechanically stable, and possess electronic properties consistent with existing experimental observations. In particular, their layered nature is expected to prevail over other structural forms at the interfaces of thin-film solar cells, and knowledge about the precise atomic structures of the interfaces is a prerequisite for achieving long-term stability and high efficiency of CdTe and Cu(In,Ga)Se2 solar cells. read less USED (low confidence) Y. Qi, L. Wang, and T. Fang, “Demixing behaviour in binary Cu-Co melt,” Physics and Chemistry of Liquids. 2013. link Times cited: 8 Abstract: Molecular dynamics simulation has been performed to explore … read moreAbstract: Molecular dynamics simulation has been performed to explore the structure, thermodynamics and dynamics properties of Cu-Co melt based upon embedded atom method (EAM). The pair correlation function of liquid Cu50Co50 show stronger interaction of homogeneous atom pairs. The coordination number (CN) of Cu-Cu and Co-Co in Cu50Co50 melt is a little higher than that of Cu-Co at temperature of 1800 K. The calculated enthalpy of mixing is positive in the whole concentration range and S CC(q) increases sharply at lower q, which are the typical features of dense fluid that exhibits phase segregation tendency. The interdiffusion coefficient shows same concentration dependence as that of demixing alloy. Our work indicates that Cu-Co melt exhibits weak demixing behaviour even at temperatures greater than those of bimodal curve. read less USED (low confidence) H. Somekawa and T. Mukai, “Molecular dynamics simulation of grain boundary plasticity in magnesium and solid-solution magnesium alloys,” Computational Materials Science. 2013. link Times cited: 19 USED (low confidence) M. Xiang, H. Hu, and J. Chen, “Molecular Dynamics Studies of Spalling and Melting in Shocked Nanocrystalline Pb,” Key Engineering Materials. 2013. link Times cited: 2 Abstract: The mechanisms of spalling and melting in nanocrystalline Pb… read moreAbstract: The mechanisms of spalling and melting in nanocrystalline Pb under shock loading are studied by molecular dynamics simulations. Our results show that grain boundaries have significant influences on spalling behaviors in cases of classical spallation and releasing melting. In these cases, cavitation and melting both start on grain boundaries, and they display mutual promotion: melting makes the voids nucleate at smaller tensile stress, and void growth speeds melting. Due to grain boundary effects, the spall strength of nanocrystalline Pb varies slowly with the shock intensity in cases of classical spallation. In cases of releasing melting and compression melting, spall strength of both single-crystalline and nanocrystalline Pb drops dramatically as shock intensity increases. read less USED (low confidence) T. Fang, L. Wang, and Y. Qi, “Structural, Thermodynamics and Dynamics Properties of Fe-Ni Melts with Different EAM Models,” Advanced Materials Research. 2013. link Times cited: 1 Abstract: Molecular dynamics (MD) simulation has been performed to exp… read moreAbstract: Molecular dynamics (MD) simulation has been performed to explore the microstructure, thermodynamics and dynamics properties of liquid Fe-Ni alloy based upon two different embedded atom method (EAM) models. The calculated PCFs with two EAM models are good agreement with the experimental values. While the calculated Scc (q) of Bhatia-Thornton (B-T) structure factor (SF) shows different behavior: a sharp increasing and a small one at lower q from G. Bonnys model and Zhous model respectively. The mixing of enthalpy with G. Bonnys EAM is positive in the whole concentration range. While the different mixing behavior with a slightly negative mixing of enthalpy based on Zhous model, which is consistent with the experimental results, is observed. Density and diffusion coefficients of liquid Fe-Ni as a function of composition show the same tendency based on both G. Bonnys model and Zhous model. In this work, Fe-Ni melts show different mixing behavior based on the two different EAM models. read less USED (low confidence) R. Marchal, I. V. Yudanov, A. Matveev, and N. Rösch, “Scalable properties of metal clusters: A comparative DFT study of ionic-core treatments,” Chemical Physics Letters. 2013. link Times cited: 9 USED (low confidence) S. Verma, T. Rehman, and A. Chatterjee, “A cluster expansion model for rate constants of surface diffusion processes on Ag, Al, Cu, Ni, Pd and Pt(100) surfaces,” Surface Science. 2013. link Times cited: 30 USED (low confidence) H. Lou et al., “Negative expansions of interatomic distances in metallic melts,” Proceedings of the National Academy of Sciences. 2013. link Times cited: 105 Abstract: When a material is heated, generally, it dilates. Here, we f… read moreAbstract: When a material is heated, generally, it dilates. Here, we find a general trend that the average distance between a center atom and atoms in the first nearest-neighbor shell contracts for several metallic melts upon heating. Using synchrotron X-ray diffraction technique and molecular dynamics simulations, we elucidate that this anomaly is caused by the redistribution of polyhedral clusters affected by temperature. In metallic melts, the high-coordinated polyhedra are inclined to evolve into low-coordinated ones with increasing temperature. As the coordination number decreases, the average atomic distance between a center atom and atoms in the first shell of polyhedral clusters is reduced. This phenomenon is a ubiquitous feature for metallic melts consisting of various-sized polyhedra. This finding sheds light on the understanding of atomic structures and thermal behavior of disordered materials and will trigger more experimental and theoretical studies of liquids, amorphous alloys, glasses, and casting temperature effect on solidification process of crystalline materials. read less USED (low confidence) C. Ni, H. Ding, C. Li, L. Kong, and X. Jin, “Pseudo-elasticity and ultra-high recoverable strain in cobalt nanowire: A molecular dynamics study,” Scripta Materialia. 2013. link Times cited: 8 USED (low confidence) V. Borovikov, X. Tang, D. Perez, X. Bai, B. Uberuaga, and A. Voter, “Influence of point defects on grain boundary mobility in bcc tungsten,” Journal of Physics: Condensed Matter. 2013. link Times cited: 21 Abstract: Atomistic computer simulations were performed to study the i… read moreAbstract: Atomistic computer simulations were performed to study the influence of radiation-induced damage on grain boundary (GB) sliding processes in bcc tungsten (W), the divertor material in the ITER tokamak and the leading candidate for the first wall material in future fusion reactors. In particular, we calculated the average sliding-friction force as a function of the number of point defects introduced into the GB for a number of symmetric tilt GBs. In all cases the average sliding-friction force at fixed shear strain rate depends on the number of point defects introduced into the GB, and in many cases introduction of these defects reduces the average sliding-friction force by roughly an order of magnitude. We have also observed that as the number of interstitials in the GB is varied, the direction of the coupled GB motion sometimes reverses, causing the GB to migrate in the opposite direction under the same applied shear stress. This could be important in the microstructural evolution of polycrystalline W under the harsh radiation environment in a fusion reactor, in which high internal stresses are present and frequent collision cascades generate interstitials and vacancies. read less USED (low confidence) P. Armstrong and W. Peukert, “Size effects in the elastic deformation behavior of metallic nanoparticles,” Journal of Nanoparticle Research. 2012. link Times cited: 26 USED (low confidence) A. Thompson et al., “Automated Algorithms for Quantum-Level Accuracy in Atomistic Simulations: LDRD Final Report.” 2012. link Times cited: 3 Abstract: This report summarizes the result of LDRD project 12-0395, t… read moreAbstract: This report summarizes the result of LDRD project 12-0395, titled “Automated Algorithms for Quantum-level Accuracy in Atomistic Simulations.” During the course of this LDRD, we have developed an interatomic potential for solids and liquids called Spectral Neighbor Analysis Potential (SNAP). The SNAP potential has a very general form and uses machine-learning techniques to reproduce the energies, forces, and stress tensors of a large set of small configurations of atoms, which are obtained using high-accuracy quantum electronic structure (QM) calculations. The local environment of each atom is characterized by a set of bispectrum components of the local neighbor density projected on to a basis of hyperspherical harmonics in four dimensions. The SNAP coefficients are determined using weighted least-squares linear regression against the full QM training set. This allows the SNAP potential to be fit in a robust, automated manner to large QM data sets using many bispectrum components. The calculation of the bispectrum components and the SNAP potential are implemented in the LAMMPS parallel molecular dynamics code. Global optimization methods in the DAKOTA software package are used to seek out good choices of hyperparameters that define the overall structure of the SNAP potential. FitSnap.py, a Python-based software package interfacing to both LAMMPS and DAKOTA is used to formulate the linear regression problem, solve it, and analyze the accuracy of the resultant SNAP potential. We describe a SNAP potential for tantalum that accurately reproduces a variety of solid and liquid properties. Most significantly, in contrast to existing tantalum potentials, SNAP correctly predicts the Peierls barrier for screw dislocation motion. We also present results from SNAP potentials generated for indium phosphide (InP) and silica (SiO2). We describe efficient algorithms for calculating SNAP forces and energies in molecular dynamics simulations using massively parallel computers and advanced processor architectures. Finally, we briefly describe the MSM method for efficient calculation of electrostatic interactions on massively parallel computers. read less USED (low confidence) W. Dong, H.-K. Kim, W. Ko, B.-M. Lee, and B.-J. Lee, “Atomistic modeling of pure Co and Co–Al system,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2012. link Times cited: 42 USED (low confidence) S.-G. Lee, H. Choi, and Y.-C. Chung, “Molecular dynamics simulation of film growth characterization of Fe and Cu on Cu(111) surface in the early stages of the deposition process,” Current Applied Physics. 2011. link Times cited: 6 USED (low confidence) H. Choi, E.-K. Lee, and Y.-C. Chung, “Surface diffusion coefficient determination by uniaxial tensile strain in Pb/Cu(111) surface systems,” Current Applied Physics. 2011. link Times cited: 4 USED (low confidence) S. Garruchet, O. Politano, P. Arnoux, and V. Vignal, “Numerical Studies of the Diffusion Processes and First Step Oxidation in Nickel-Oxygen Systems by Variable Charge Molecular Dynamics,” Defect and Diffusion Forum. 2010. link Times cited: 1 Abstract: Variable charge molecular dynamic simulations have been perf… read moreAbstract: Variable charge molecular dynamic simulations have been performed to study the diffusion mechanisms of oxygen atoms (O) in nickel (Ni) in the temperature range 950-1600 K and the very first steps of oxidation of monocrystalline nickel surfaces at 300 K and 950 K. The oxygen diffusivity can be well described by an Arrhenius law over the temperature range considered. The oxygen diffusion coefficient has been analysed and values of Ea = 1.99 eV for the activation energy and D0 = 39 cm2.s-1 for the pre-exponential factor were obtained. The first steps growth of the oxide layer show that after the dissociative chemisorption of the oxygen molecules on nickel surface, the oxidation leads to an island growth mode as observed experimentally. read less USED (low confidence) S. Garruchet, O. Politano, P. Arnoux, and V. Vignal, “Diffusion of oxygen in nickel: A variable charge molecular dynamics study,” Solid State Communications. 2010. link Times cited: 40 USED (low confidence) D. Schebarchov, S. C. Hendy, S. C. Hendy, and W. Polak, “Molecular dynamics study of the melting of a supported 887-atom Pd decahedron,” Journal of Physics: Condensed Matter. 2009. link Times cited: 13 Abstract: We employ classical molecular dynamics simulations to invest… read moreAbstract: We employ classical molecular dynamics simulations to investigate the melting behaviour of a decahedral Pd887 cluster on a single layer of graphite (graphene). The interaction between Pd atoms is modelled with an embedded-atom potential, while the adhesion of Pd atoms to the substrate is approximated with a Lennard-Jones potential. We find that the decahedral structure persists at temperatures close to the melting point, but that just below the melting transition, the cluster accommodates to the substrate by means of complete melting and then recrystallization into an fcc structure. These structural changes are in qualitative agreement with recently proposed models, and they verify the existence of an energy barrier preventing softly deposited clusters from ‘wetting’ the substrate at temperatures below the melting point. read less USED (low confidence) D. Schebarchov and S. Hendy, “Dynamics of capillary absorption of droplets by carbon nanotubes.,” Physical review. E, Statistical, nonlinear, and soft matter physics. 2008. link Times cited: 26 Abstract: We consider the capillary absorption of liquid metal droplet… read moreAbstract: We consider the capillary absorption of liquid metal droplets by carbon nanotubes using molecular dynamics simulations and the steady-state flow model due to Marmur [A. Marmur, J. Colloid Interface Sci. 122, 209 (1988)]. We find an exact solution to Marmur's evolution equation for the height of the absorbed liquid column as a function of time, and show that this reproduces the dynamics observed in the simulations well. The simulations show that the flow of the metal exhibits a large degree of slippage at the tube walls, with slip lengths of up to 10 nm depending on the wettability of the nanotube. The results support the use of the Lucas-Washburn approach for modeling capillary absorption at the nanoscale. read less USED (low confidence) D. Schebarchov and S. Hendy, “Capillary absorption of metal nanodroplets by single-wall carbon nanotubes.,” Nano letters. 2008. link Times cited: 56 Abstract: We present a simple model that demonstrates the possibility … read moreAbstract: We present a simple model that demonstrates the possibility of capillary absorption of nonwetting liquid nanoparticles by carbon nanotubes (CNTs) assisted by the action of the Laplace pressure due to the droplet surface tension. We test this model with molecular dynamics simulation and find excellent agreement with the theory, which shows that for a given nanotube radius there is a critical size below which a metal droplet will be absorbed. The model also explains recent observations of capillary absorption of nonwetting Cu nanodroplets by carbon nanotubes. This finding has implications for our understanding of the growth of CNTs from metal catalyst particles and suggests new methods for fabricating composite metal-CNT materials. read less USED (low confidence) Y. Kudryavtsev et al., “Evolution of the magnetic properties ofCo2MnGaHeusler alloy films: From amorphous to ordered films,” Physical Review B. 2007. link Times cited: 26 USED (low confidence) H. Yuasa, H. Fukuzawa, and H. Iwasaki, “CPP–GMR of spin valves with CoxFe1−x alloy,” Journal of Magnetism and Magnetic Materials. 2005. link Times cited: 15 USED (low confidence) I. Galanakis, “Towards half-metallic interfaces: Co2CrAl/InP contacts,” Journal of Physics: Condensed Matter. 2004. link Times cited: 51 Abstract: Although the interest in half-metallic Heusler alloys, likel… read moreAbstract: Although the interest in half-metallic Heusler alloys, likely to be usable in spintronic applications, has grown considerably, their interfaces with semiconductors show very low spin polarization. I identify mechanisms which can keep high spin polarization at the interface (more than 80% of the electrons at the Fermi level of majority spin) although the half-metallicity is lost. The large enhancement of the Cr moment at the interface between a CrAl-terminated Co2CrAl (001) spacer and the InP(001) semiconductor weakens the effect of the interface states, resulting in this high spin polarization. On the other hand, the Co2CrAl/InP interfaces made up by a Co layer and either an In or a P one show a severe decrease of the Co spin moment, but Cr in the subinterface layer is bulklike and the resulting spin polarization is similar to that of the CrAl-based interfaces. read less USED (low confidence) Q. Zheng, Z.-an Tian, T. Gao, Y.-chao Liang, Q. Chen, and Q. Xie, “Effect of graphene on solid–liquid coexistence in Cu nanodroplets,” Applied Surface Science. 2023. link Times cited: 0 USED (low confidence) L. Xie, G. Wu, Q. Peng, and W. R. Wang, “Molecular dynamics simulation on the dissolution and diffusion characteristics of FeCrAl alloy in liquid LBE,” Annals of Nuclear Energy. 2023. link Times cited: 1 USED (low confidence) H. Zou, Y. Feng, and L. Qiu, “Excellent heat transfer enhancement of CNT-metal interface by loading carbyne and metal nanowire into CNT,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 9 USED (low confidence) S. Xu, Y. Su, W. Jian, and I. Beyerlein, “Local slip resistances in equal-molar MoNbTi multi-principal element alloy,” Acta Materialia. 2021. link Times cited: 48 USED (low confidence) A. Panda et al., “Molecular dynamics studies on formation of stacking fault tetrahedra in FCC metals,” Computational Materials Science. 2021. link Times cited: 11 USED (low confidence) V. Podryga and S. Polyakov, “Correction of Boundary Conditions in Micromodels by Molecular Dynamic Method.” 2021. link Times cited: 0 USED (low confidence) D. Mishra, S. K. Badjena, and S. Pal, “A Comparative Nanoindentation Study on HEA Coated FCC Metals and Stacking Fault Tetrahedra Evolution in HEA Coated Single Crystal Al: A MD Simulation Study,” Springer Proceedings in Materials. 2021. link Times cited: 0 USED (low confidence) S. N. Divi and A. Chatterjee, “Investigating Factors Affecting Mixing Patterns in Ternary Metal Alloy Nanoparticles.” 2020. link Times cited: 0 USED (low confidence) F. Maresca and W. Curtin, “Theory of screw dislocation strengthening in random BCC alloys from dilute to ‘High-Entropy’ alloys,” Acta Materialia. 2020. link Times cited: 123 USED (low confidence) Y. Cui, K. Ma, Z.-ying Chen, J. Yang, Z. Geng, and J. Zeng, “Atomic-level insights into strain effect on p-nitrophenol reduction via Au@Pd core–shell nanocubes as an ideal platform,” Journal of Catalysis. 2020. link Times cited: 27 USED (low confidence) W. K. Kim and E. Tadmor, “Temporal Acceleration in Coupled Continuum-Atomistic Methods,” Handbook of Materials Modeling. 2020. link Times cited: 0 USED (low confidence) D. Ishikawa et al., “Bonding Abilities of Pressure-assisted Sintered Copper for Die-Bonding of Large Chips,” Transactions of The Japan Institute of Electronics Packaging. 2020. link Times cited: 1 Abstract: This paper describes sintering properties and bonding abilit… read moreAbstract: This paper describes sintering properties and bonding abilities of mechanical pressure-assisted sintered copper (pressure-sintered Cu) in N2 atmosphere for power devices with large chips. The pressure-sintered Cu showed equal or improved sintering and bonding properties compared with pressureless-sintered Cu and pressure-sintered silver (Ag). The bonding abilities of pressure-sintered Cu on two different Ag plating (Ti/Ni/Ag, Ti/Ni/Au/Ag) were also investigated. The Cu pressure-sintered at 2 MPa and 300°C for 5 min was successfully bonded to 10 × 10 mm chips with Ti/ Ni/Ag. Thus, pressure-sintered Cu has a potential as a reliable die-bonding material for power devices with large chips. read less USED (low confidence) A. Nikonov, “Molecular dynamic study of the mechanical properties of TiAlTaN coating on a titanium substrate subjected to scratching.” 2020. link Times cited: 0 USED (low confidence) A. Tran and Y. Wang, “Reliable molecular dynamics simulations for intrusive uncertainty quantification using generalized interval analysis.” 2020. link Times cited: 0 USED (low confidence) X. Duan et al., “Development of a pair potential for Ta-He system,” Computational Materials Science. 2019. link Times cited: 3 USED (low confidence) W. Li, J.-J. Tang, Q. Wang, and H. Fan, “Molecular Dynamics Simulations on the Mechanical Behavior of AlCoCrCu0.5FeNi High-Entropy Alloy Nanopillars,” TMS 2019 148th Annual Meeting & Exhibition Supplemental Proceedings. 2019. link Times cited: 4 USED (low confidence) S. Groh and M. K. Nahhas, “Modeling Dislocation in Binary Magnesium-Based Alloys Using Atomistic Method,” Handbook of Mechanics of Materials. 2019. link Times cited: 1 USED (low confidence) P. Zakharov, E. Korznikova, S. Dmitriev, E. Ekomasov, and K. Zhou, “Surface discrete breathers in Pt3Al intermetallic alloy,” Surface Science. 2019. link Times cited: 36 USED (low confidence) H. Pan, T. Lü, J. Zheng, and S. Lin, “Calculation and Simulation of the Ground State Properties of Copper-Nickel Alloys.” 2019. link Times cited: 0 Abstract: : In radiation damage research, copper, nickel, and their al… read moreAbstract: : In radiation damage research, copper, nickel, and their alloys are widely used model systems for face-centered–cubic (FCC) metals. The ground-states properties of the ordered and disordered alloys are studied by using the molecular dynamics (MD) and first-principles calculations. For copper-nickel alloys, the equilibrium properties have been predicted by large-scale atomic/molecular massively parallel simulator (LAMMPS), Abinit and WIEN2k, thermodynamics is calculated by LAMMPS and Abinit. In order to investigate the disordered alloys, special quasirandom structures (SQS) and fractional function are adopted in LAMMPS, and their capabilities are demonstrated to predict the properties of disordered alloys. For both the ordered and disordered alloys, the lattice constants in agreement with the Vegard's law are predicted and the bulk moduli present the deviations with respect to the experimental values; with the increasing weight concentration of nickel, the equilibrium volumes reduce and the bulk moduli increase. The calculated cohesive energy of copper and nickel are consistent with the experimental values; the cohesive energies of ordered alloys are predicted. The energies of formation of disordered alloys are always lower than those of ordered alloys. read less USED (low confidence) A. Eremin, P. Zakharov, N. А. Manakov, and M. Starostenkov, “ON THE QUESTION ON THE STATISTICAL COMPARISON OF ACCURATE DISCRETE BREEZERS WITH QUASI-BREEZE MODEL SOLUTIONS OF A3B STEHOMETRY CRYSTAL.” 2018. link Times cited: 0 Abstract: ВЕСТНИК ОРЕНБУРГСКОГО ГОСУДАРСТВЕННОГО УНИВЕРСИТЕТА 2018 No … read moreAbstract: ВЕСТНИК ОРЕНБУРГСКОГО ГОСУДАРСТВЕННОГО УНИВЕРСИТЕТА 2018 No 5 (217) Дискретный бризер (ДБ) представляет собой нелинейные локализованные незатухающие колебания большой амплитуды атомов идеального кристалла [1]. Стоит отметить, что для большинства моделей реальных кристаллов, имеет смысл говорить не о ДБ, а о квазибризерах или квазидискретных бризерах (КДБ) [2], в силу отсутствия возможности задания идеальных начальных условий для всех атомов, участвующих в колебаниях. Данное ограничение приводит к конечному времени жизни КДБ и разбросу частот атомов входящих в состав дискретного бризера. При этом в работе [2] говориться, что присутствие в решении членов с малыми амплитудами, частоты которых отличаются от основной бризерной частоты, не является причиной потери их устойчивости. Тем не менее, КДБ имеют ограниченное время жизни в реальных моделях кристаллов, которое зависит от начальной конфигурации атомов и свойств рассматриваемого материала. УДК 538.913 Ерёмин А.М., Захаров П.В., Манаков Н.А., Старостенков М.Д. Алтайский государственный гуманитарно-педагогический университет им. В.М. Шукшина, г. Бийск, Россия Оренбургский государственный университет, г. Оренбург, Россия Алтайский государственный технический университет им. И.И. Ползунова, г. Барнаул, Россия read less USED (low confidence) D. Chocyk and T. Zientarski, “Molecular dynamics simulation of Ni thin films on Cu and Au under nanoindentation,” Vacuum. 2018. link Times cited: 14 USED (low confidence) D. Okumura, M. Otsuka, and Y. Shibutani, “Mechanism of Plastic Deformation in a Magnesium Nanotwinned Structure by Molecular Dynamics Simulations,” Journal of The Society of Materials Science, Japan. 2018. link Times cited: 0 Abstract: In this study, we perform molecular dynamics simulations to … read moreAbstract: In this study, we perform molecular dynamics simulations to investigate plastic deformation modes of a magnesium (Mg) nanotwinned structure. Periodic units including (1011) twin boundaries (TBs) are analyzed using an embedded atom method potential. Equal spaces between the TBs are assumed at the initial state and the space is parametrized in the range between about 5 nm and 30 nm. It is found that plastic deformation is triggered by the slip along a (1011) twinning plane near a TB, and that this event induces two different deformation modes depending on the space, i.e., the migration of the TBs and the evolution of double twinning, which leads to void nucleation and polycrystallization. The plastic deformation provided by the two different modes is quantitatively verified from geometric analysis. As the space decreases, the migration of the TBs is superior to the evolution of double twinning. read less USED (low confidence) A. Matsushita, S. Takamoto, A. Hatano, and S. Izumi, “Development of Hybrid Method Using Ab initio and Classical Molecular Dynamics for Calculating the Thermal Expansion Coefficient of Alloys at High Temperature,” Journal of The Society of Materials Science, Japan. 2018. link Times cited: 0 Abstract: In order to predict the thermal expansion coefficient (TEC),… read moreAbstract: In order to predict the thermal expansion coefficient (TEC), quasiharmonic approximation based on the ab initio electronic structure calculation is an effective and conventional scheme. However, it is known that the deviation due to anharmonic effect arises at high temperature. Although Molecular Dynamics (MD) naturally includes the anharmonic effect, classical MD is lacking in accuracy because of its empirical interatomic potential and ab initio MD is not applicable because of its high computational cost. In this paper, we have proposed a new hybrid method using ab initio electronic structure calculation and classical molecular dynamics for calculating TEC of alloys at high temperature. Our method is non-empirical, highly accurate and computationally inexpensive. The method consists of three steps. Firstly, various snapshots of the atomic coordination at a certain temperature are sampled by classical MD. Secondly, physical properties of each snapshot are calculated by ab initio electronic structure calculation. Finally, by analyzing them statistically, the equilibrium volume is computed. TEC is obtained by repeating these steps at different temperatures. We have calculated the TECs of Al, Ni3Al, and NiAl. Results show good agreements with experimental results. Our method enables us to improve the conventional quasiharmonic approximation and obtain accurate TEC at high temperature through the incorporation of anharmonic effect. read less USED (low confidence) P. Chowdhury and H. Sehitoglu, “Atomistic Fault Energetics and Critical Stress Prediction for fcc and bcc Twinning : Recent Progress AQ 3.” 2017. link Times cited: 10 Abstract: 15 This article recounts recent advances on the atomistic mo… read moreAbstract: 15 This article recounts recent advances on the atomistic modeling of twinning in bcc and fcc alloy. Specifically, we have reviewed: (i) the experimental evidence of twinningdominated deformation in singleand multigrain microstructures (ii) calculation of generalized planar fault energy (GPFE) landscapes, and (iii) the prediction of critical friction stresses to initiate twinning-governed plasticity (e.g., twin nucleation, twin–slip and twin–twin interactions). Possible avenues for further research are outlined. [DOI: 10.1115/1.4038673] read less USED (low confidence) J. Krishan, P. Gupta, K. Vaduganathan, and N. Yedla, “Superplastic Pd50Pt50 monocrystalline bimetallic alloy nanowire: a molecular dynamics simulation study,” Revue De Metallurgie-cahiers D Informations Techniques. 2017. link Times cited: 2 Abstract: We report uniaxial tensile studies of monocrystalline cylind… read moreAbstract: We report uniaxial tensile studies of monocrystalline cylindrical Pd50 Pt50 bimetallic alloy nanowire (NW) (11.67 A diameter × 101.14 A length) subjected to strain rates of 1%, 3%, 5%, 8% and 10% ps−1 and temperatures of 100 K, 300 K, 500 K and 700 K using molecular dynamics simulations. The tensile test is carried out along y -axis [0 1 0] with periodic boundary conditions and non-periodic along x -axis and z -axis. Dissipative mechanisms such as dislocations, partial dislocations and stair rod dislocations are also identified during deformation. It is found that NW is sensitive to strain rates and temperatures and exhibits superplastic behavior with strain >100% at higher strain rates. Young's modulus is found to be sensitive to strain rate and temperature and is about 25% less that of bulk value at strain rates >5% ps−1 and at 300 K. Yield strength increases linearly with strain rate and decreases with increase in temperature as expected. Crystalline to amorphous-phase transformations take place at strain rates >5% ps−1 and could be the reason behind large plastic strain. The calculated mechanical properties of NWs will be useful in devices applicable to sensing and catalysis. read less USED (low confidence) P. Gupta and N. Yedla, “Strain Rate and Temperature Effects on the Strength and Dissipative Mechanisms in Al-Cu50Zr50 Interface Model: Molecular Dynamics Simulation Study ☆,” Procedia Engineering. 2017. link Times cited: 10 USED (low confidence) D. Matsunaka, Y. Shibutani, and Y. Ohnishi, “Molecular Dynamics Analyses of Fracture Toughness of Magnesium,” Journal of The Society of Materials Science, Japan. 2016. link Times cited: 1 Abstract: Crack propagation in Magnesium was investigated using molecu… read moreAbstract: Crack propagation in Magnesium was investigated using molecular dynamics (MD) simulations. By using a disk-shaped atomic cell containing a crack under the anisotropic linear elastic displacement, the deformation field around the crack tip was first atomistically resolved. In order to consider effects of surface energy on crack propagation, two kinds of interatomic potentials were adopted. For an embedded atom method (EAM) potential, basal and first-pyramidal surfaces are favorable cleavage planes due to relatively low surface energies. Thus, MD simulations using the EAM showed unstable crack propagation on those planes. For the other generalized embedded atom method (GEAM) potential which provides the higher surface energies instead, the basal plane has the larger critical stress intensity factor by the Griffith’s formula because it is proportional to square root of surface energy. Consequently, the dislocation nucleation and deformation twin nucleation were occurred in the vicinity of the crack tip. The other MD analyses of defect interaction between a crack and twin boundaries (TBs) were also carried out. Twinning dislocations of (101�2) twin were generated by reaction with the basal dislocations emitted from the crack tip, and thus (101�2) TB easily moved. On the other hand, a void or deformation twin was generated along the boundary region where the dislocations from the crack tip piled up, and then the crack propagation near (101�1) TB arose. read less USED (low confidence) Y. Umeno, “Analysis of Atomistic Scale Instability of Dislocation Nucleation from Interfaces and Surface Steps,” Journal of Solid Mechanics and Materials Engineering. 2012. link Times cited: 0 Abstract: To better understand the mechanism of structural instability… read moreAbstract: To better understand the mechanism of structural instability at the atomistic level, we have proposed a rigorous method of instability mode analysis by solving the eivgenvalue problem of the Hessian dynamical matrix and applied it to dislocation nucleation from surface steps and interfaces. In dislocation nucleation from an interface edge, relation between instability at a low but nonzero temperature and instability modes for vanishing temperature is investigated. An instability mode corresponding to an eigenvector that has a small eigenvalue is activated by thermal fluctuation, resulting in deformation indicated by the eigenvector. This is in contrast to the case of dislocation nucleation from a surface notch, where instability occurrs along a mixed mode. In the case of dislocation nucleation from surface steps, we discuss the area of influence from a step by examining the distribution of eigenvectors utilizing the quasicontinuum (QC) method. read less USED (low confidence) A. Iskandarov, S. Dmitriev, and Y. Umeno, “On Accurate Approach for Molecular Dynamics Study of Ideal Strength at Elevated Temperature,” Journal of Solid Mechanics and Materials Engineering. 2012. link Times cited: 3 Abstract: Influence of temperature on ideal shear strength (ISS), τc, … read moreAbstract: Influence of temperature on ideal shear strength (ISS), τc, of two fcc metals (Al and Cu) was studied by means of molecular dynamics simulations. To get reliable results we investigated influence of parameters of the applied Parrinello-Rahman stress control method and implemented damping of simulation cell fluctuations to avoid occurrence of structural instability assisted by too high fluctuations. The damping successfully reduces strain and stress fluctuations during simulations if the damping factor is specified properly. We also investigated simulation cell size effect to evaluate minimal number of atoms providing reliable results in order to reduce computational efforts and estimate the possibility of applying ab initio calculations. Recently developed embedded atom method (EAM) interatomic potentials for both metals were also examined to find most appropriate for our study. EAM potential developed by Zope et al. and Mishin et al. were revealed to be most suitable for Al and Cu, respectively. It is essential to choose appropriate simulation parameters and interatomic potentials for the valid evaluation of ISS at elevated temperatures. We find almost linear decrease in ideal strength with increasing temperature for [112](111) shear deformation, while critical strain decreases in a nonlinear manner. At room temperature, reduction of shear strength for Al(Cu) is less than 35%(25%) compared to that at 0 K. read less USED (low confidence) Y. Kobayashi, Y. Doi, and A. Nakatani, “Study of Temperature Dependence of Island Formation and Structure of Gallium Nitride in MBE Growth,” journal of the Japan Society for Testing Materials. 2010. link Times cited: 0 Abstract: Homoepitaxial growth on gallium nitride substrate is simulat… read moreAbstract: Homoepitaxial growth on gallium nitride substrate is simulated by molecular dynamics (MD) method. Crystal growth configuration is evaluated qualitatively according to the atomic configuration and the radial distribution function. Moreover, crystal structure is evaluated quantitatively by considering variance of local atomic density as a monitoring index. As a result, we found that the growth layer is formed two-dimensionally and like film formation when the substrate temperature is high. It is found that the dynamics of crystal growth can be understood from not only variance value but also gradient of temporal evolution of variance. read less NOT USED (low confidence) P. Liu, S. Chen, Q. Pei, Z. Aitken, W. Li, and Y.-W. Zhang, “Simultaneously enhancing the tensile strength and ductility of high entropy alloys by nanoscale precipitates/fillers,” APL Materials. 2023. link Times cited: 1 Abstract: High entropy alloys (HEAs) in the solid solution (SS) phase … read moreAbstract: High entropy alloys (HEAs) in the solid solution (SS) phase have attracted much attention due to their novel strengthening mechanisms. Recent studies have shown that introducing nanoscale precipitates/fillers can further strengthen the SS HEAs. In this work, we performed large-scale molecular dynamics simulations of AlxCoCuFeNi HEAs filled with randomly distributed AlNi3 nanoparticles. The effects of AlNi3 particle size and volume fraction, the chemical composition of the HEA matrix, and temperature on the mechanical properties, deformation, and failure behavior of the composite are systematically investigated. Our simulations show that, remarkably, the AlNi3 nanoparticles can simultaneously enhance the ultimate tensile strength and ultimate tensile strain of the composite. The underlying mechanism is that the AlNi3 nanoparticles greatly suppressed the phase change and dislocation appearance in the HEA matrix, resulting in a delayed material failure during the deformation. We also find that Young’s modulus, ultimate tensile strength, and ultimate tensile strain follow the lower-bound of the rule of mixtures and further present the underlying reason for this lower-bound relation. The present work not only provides insights into the mechanical properties, deformation, and failure behavior of AlNi3 nanoparticle-reinforced AlxCoCuFeNi HEAs but is also useful for guiding the rational design of HEAs for engineering applications. read less NOT USED (low confidence) O. Bachurina, R. Murzaev, S. Shcherbinin, A. A. Kudreyko, S. V. Dmitriev, and D. Bachurin, “Multi-component delocalized nonlinear vibrational modes in nickel,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 0 Abstract: Delocalized nonlinear vibrational modes (DNVMs) are relative… read moreAbstract: Delocalized nonlinear vibrational modes (DNVMs) are relatively new dynamical objects that can be used for testing interatomic potentials and for classification and finding new types of discrete breathers. In this work, for the first time, multi-component DNVMs in a single crystal of fcc nickel are studied using molecular dynamics method. Previously discovered two one-component DNVMs are used to construct and investigate properties of all possible two- and three-component superpositions. A quasi-periodic energy exchange between components in multi-component DNVMs is described. If the amplitudes of the one-component DNVMs in a superposition differ by less than four times, then an equivalent energy exchange between them is observed. Otherwise, an unequal energy exchange takes place, i.e. when the high-amplitude component gives up only a part of its energy leading to a slight increase in the amplitude of another component. The DNVMs consisting of two- and three-components have a lifetime of more than 10 ps as long as the initial atomic amplitudes do not exceed 0.08 Å. An increase in the initial amplitude leads to a substantial decrease in the lifetime due to the rapidly developing modulational instability. Some superpositions of modes with the same initial amplitudes of the components can transform into a one-component DNVM, while others remain multi-component ones. The results obtained in this work demonstrate the existence of multi-component DNVMs, being a superposition of two or three components, which significantly expands our understanding of their dynamics in an fcc lattice. read less NOT USED (low confidence) B. Zhu, D. Zhao, Y.-hong Niu, Z. Zhang, and H. Zhao, “The short-range ordering and atomic segregation in various phases of high-entropy alloy during the solidification process,” Materials & Design. 2023. link Times cited: 1 NOT USED (low confidence) O. Senkov, T. Daboiku, T. Butler, S. Rao, and E. Payton, “Microstructure, compression properties, and oxidation behavior of Hf-25Ta-5Me alloys (Me is Mo, Nb, W, 0.5Mo + 0.5 W, Cr, or Zr),” International Journal of Refractory Metals and Hard Materials. 2022. link Times cited: 4 NOT USED (low confidence) N. Kamanina, A. Toikka, and D. Kvashnin, “Nanostructuration Impact on the Basic Properties of the Materials: Novel Composite Carbon Nanotubes on a Copper Surface,” Journal of Composites Science. 2022. link Times cited: 2 Abstract: Copper is important material that is widely applicable in th… read moreAbstract: Copper is important material that is widely applicable in the electric and electronic industries. Nevertheless, in some circumstances, it is highly desirable to improve its properties. Therefore, combination of materials of various composition and properties attracts scientific and industrial society. Here, the composite based on carbon nanotubes (CNTs) on a Cu surface was fabricated using laser-oriented deposition (LOD) technique and studied. Examination of the novel composite showed that its reflectance was decreased, the microhardness was increased, and wetting of the surface exhibited higher hydrophobicity. A molecular dynamic simulation showed that the penetration depth increases with nanotube diameter decrease and growth of the acceleration rate. Topography observations made via AFM images revealed a dense thin film with an almost-homogeneous distribution of CNTs, with several locations with irregular thickness addressing the different lengths of CNTs. read less NOT USED (low confidence) J. Yuan et al., “Remarkable cryogenic strengthening and toughening in nano-coherent CoCrFeNiTi0.2 high-entropy alloys via energetically-tuning polymorphous precipitates,” Materials Science and Engineering: A. 2022. link Times cited: 15 NOT USED (low confidence) E. George, W. Curtin, and C. Tasan, “High entropy alloys: A focused review of mechanical properties and deformation mechanisms,” Acta Materialia. 2020. link Times cited: 774 NOT USED (low confidence) T. D. Huan, R. Batra, J. Chapman, C. Kim, A. Chandrasekaran, and R. Ramprasad, “Iterative-Learning Strategy for the Development of Application-Specific Atomistic Force Fields,” The Journal of Physical Chemistry C. 2019. link Times cited: 18 Abstract: Emerging data-driven approaches in materials science have tr… read moreAbstract: Emerging data-driven approaches in materials science have triggered the development of numerous machine-learning force fields. In practice, they are constructed by training a statistical model on a reference database to predict potential energy and/or atomic forces. Although most of the force fields can accurately recover the properties of the training set, some of them are becoming useful for actual molecular dynamics simulations. In this work, we employ a simple iterative-learning strategy for the development of machine-learning force fields targeted at specific simulations (applications). The strategy involves (1) preparing and fingerprinting a diverse reference database of atomic configurations and forces, (2) generating a pool of machine-learning force fields by learning the reference data, (3) validating the force fields against a series of targeted applications, and (4) selectively and recursively improving the force fields that are unsuitable for a given application while keeping their performance... read less NOT USED (low confidence) K. Choudhary, F. Y. Congo, T. Liang, C. Becker, R. Hennig, and F. Tavazza, “Evaluation and comparison of classical interatomic potentials through a user-friendly interactive web-interface,” Scientific Data. 2017. link Times cited: 21 NOT USED (low confidence) P. Wurm and M. H. Ulz, “A stochastic approximation approach to improve the convergence behavior of hierarchical atomistic-to-continuum multiscale models,” Journal of The Mechanics and Physics of Solids. 2016. link Times cited: 6 NOT USED (low confidence) M. H. Ulz, “A Multiscale Molecular Dynamics Method for Isothermal Dynamic Problems using the Seamless Heterogeneous Multiscale Method,” Computer Methods in Applied Mechanics and Engineering. 2015. link Times cited: 11 NOT USED (low confidence) 徐建刚 and 张云光, “层厚度和晶界‘阶梯型’缺陷对铜–镍复合薄膜的力学性能的分子动力学模拟研究,” Applied physics. 2015. link Times cited: 0 Abstract: 本文采用分子动力学方法研究了层厚度和“阶梯型”晶界缺陷对铜–镍纳米薄膜的力学性能的影响。模拟结果表明,随着层厚度的增加,… read moreAbstract: 本文采用分子动力学方法研究了层厚度和“阶梯型”晶界缺陷对铜–镍纳米薄膜的力学性能的影响。模拟结果表明,随着层厚度的增加,薄膜的应力逐渐增大,这是因为材料的层厚度越大,材料存储位错的能力就越强,及屈服强度越高。除此之外,研究结果发现晶界存在“阶梯型”缺陷降低了晶界对于位错传播的阻碍作用,使得铜–镍纳米薄膜屈服强度降低。 The effect of layer thickness and interfacial defect with steps of copper-nickel multilayer thin film on deformation mechanism is investigated by molecular dynamics simulations. The results indicate the yield stress is found to increase with increasing layer thickness. The result is mainly due to the fact that the room for dislocation storage can be affected by the changes of layer thickness. Furthermore, the studies show that interfacial defect with steps dominates interfacial barrier effect, resulting in the lowest yield stress. read less NOT USED (low confidence) J. Cheng et al., “A review of ultrafast laser materials micromachining,” Optics and Laser Technology. 2013. link Times cited: 329 NOT USED (low confidence) S. Sankaranarayanan, R. Subbaraman, and S. Ramanathan, “Considerations on ultra-high frequency electric field effects on oxygen vacancy concentration in oxide thin films.,” Physical chemistry chemical physics : PCCP. 2012. link Times cited: 0 Abstract: Atomistic simulations employing dynamic charge transfer betw… read moreAbstract: Atomistic simulations employing dynamic charge transfer between atoms are used to investigate ultra-thin oxide growth on Al(100) metal substrates in the presence of an ac electric field. In the range of 1-10 GHz frequencies, the enhancement in oxidation kinetics by ∼12% over natural oxidation can be explained by the Cabrera-Mott mechanism. At field frequencies approaching 0.1-1 THz, however, we observe a dramatic lowering of the kinetics of oxygen incorporation by ∼35% compared to the maximum oxidation achieved, which results in oxygen non-stoichiometry near the oxide-gas interface (O/Al ≈ 1.0). This is attributed to oxygen desorption from the oxide surface. These results suggest a general strategy to tune oxygen concentration at oxide surfaces using ac electric fields that could be of interest in diverse fields related to surface chemistry and applications such as tunnel barriers, thin dielectrics and oxide interfaces. read less NOT USED (low confidence) K. Nalepka and K. Nalepka, “Efficient approach to metal/metal oxide interfaces within variable charge model,” The European Physical Journal B. 2012. link Times cited: 8 NOT USED (low confidence) M. Tsuchiya, S. Sankaranarayanan, and S. Ramanathan, “Photon-assisted oxidation and oxide thin film synthesis: A review,” Progress in Materials Science. 2009. link Times cited: 68 NOT USED (low confidence) X. W. Zhou and F. Doty, “Embedded-ion method: An analytical energy-conserving charge-transfer interatomic potential and its application to the La-Br system,” Physical Review B. 2008. link Times cited: 30 NOT USED (low confidence) A. Cerezo, P. Clifton, S. Lozano-Perez, P. Panayi, G. Sha, and G. Smith, “Overview: Recent Progress in Three-Dimensional Atom Probe Instruments and Applications,” Microscopy and Microanalysis. 2007. link Times cited: 46 Abstract: Over the last few years there have been significant developm… read moreAbstract: Over the last few years there have been significant developments in the field of three-dimensional atom probe (3DAP) analysis. This article reviews some of the technical compromises that have led to different instrument designs and the recent improvements in performance. An instrument has now been developed, based around a novel reflectron configuration combining both energy compensation and focusing elements, that yields a large field of view and very high mass resolution. The use of laser pulsing in the 3DAP, together with developments in specimen preparation methods using a focused ion-beam instrument, have led to a significant widening in the range of materials science problems that can be addressed with the 3DAP. Recent studies of semiconductor materials and devices are described. read less NOT USED (low confidence) D. Larson, “Atom probe characterization of nanomagnetic materials,” Thin Solid Films. 2006. link Times cited: 22 NOT USED (low confidence) X. W. Zhou, D. Murdick, and H. Wadley, “An electron counting modification to potentials for covalently bonded surfaces,” Journal of Applied Physics. 2006. link Times cited: 4 Abstract: The surface structure of covalently bonded semiconductor mat… read moreAbstract: The surface structure of covalently bonded semiconductor materials undergoes reconstructions that are driven by electron redistribution between dangling and interatom bonds. Conventional interatomic potentials account for neither this electron redistribution nor its effects upon the atomic structure of surfaces. We have utilized an electron counting analysis to develop a surface interatomic potential that captures many of the effects of electron redistribution upon the surface structures of covalently bonded materials. The contributions from this potential decrease rapidly to zero beneath a surface. As a result, this surface potential can be added to many interatomic potentials for covalent materials without affecting its predictions of bulk properties such as cohesive energy, lattice parameters, and elastic constants. We demonstrate the approach by combining the surface potential with a recently proposed bond order potential and use it in a molecular statics simulation of the atomic reconstruction of a w... read less NOT USED (low confidence) A. Petford-Long et al., “The Role of Atomic Scale Investigation in the Development of Nanoscale Materials for Information Storage Applications,” Microscopy and Microanalysis. 2004. link Times cited: 3 Abstract: It is well established that the response of devices based on… read moreAbstract: It is well established that the response of devices based on the giant magnetoresistance (GMR) effect depends critically on film microstructure, with parameters such as interfacial abruptness, the roughness and waviness of the layers, and grain size being crucial. Such devices have applications in information storage systems, and are therefore of great technological interest as well as being of fundamental scientific interest. The layers must be studied at high spatial resolution if the microstructural parameters are to be characterized with sufficient detail to enable the effects of fabrication conditions on properties to be understood, and the techniques of high resolution electron microscopy, transmission electron microscopy chemical mapping, and atom probe microanalysis are ideally suited. This article describes the application of these techniques to a range of materials including spin valves, spin tunnel junctions, and GMR multilayers. read less NOT USED (low confidence) J. L. Yuan et al., “Remarkable Cryogenic Strengthening and Toughening in Nano-Coherent CoCrFeNiTi 0.2 High-Entropy Alloys via Energetically-Tuning Polymorphous Precipitates,” Acta Materialia. 2021. link Times cited: 1 Abstract: In the present work, three kinds of precipitates with differ… read moreAbstract: In the present work, three kinds of precipitates with different morphologies, structures, sizes, and volume fractions were obtained via energetically-tuning the microstructures of the nano-precipitated CoCrFeNiTi0.2 high-entropy alloy (HEA). Subjected to the heavy cold rolling immediately after homogeneous precipitation, L12 structured spherical nano-particles with an average size of 16.5 nm rapidly grow into 200 nm-sized spherical ones due to Ostwald ripening. On the other hand, superfluous mechanical energy storage energetically facilitates the phase transformation from spherical L12 to rod-shaped D024 structures for initially formed nano-precipitates. Besides, some other newly formed nano-precipitates with an average size of 6.5 nm are available, originating from heavily plastically deformed-induced nucleated sites. Multi-scale precipitates interact with dislocations in different ways. The strengthening provided by dislocations cutting through smaller nano-particles and bypassing grown ones account for 57.7% and 42.3% of precipitation strengthening, respectively, while rod-shaped precipitates can act as equivalent interfaces to hinder dislocation movement. Their synergistic effect has achieved remarkable strengthening and toughening. Specially, dislocation slips dominate at 298 K, while stacking faults (SFs) assist plastic deformation at 77 K. Compared with 298 K, the yield strength (YS) and ultimate tensile strength (UTS) of the current HEAs at 77 K are increased by 38.9% and 38.2% to 1 GPa and 1.5 GPa, respectively, and the tensile strain is slightly increased to 35% instead of loss, realizing excellent strength and plasticity combination. Theoretically established strengthening models agree well room-temperature and cryogenic yield strengths experimentally. Moreover, the tensile elongation is effectively predicted by the Whitehouse-Clyne model. This strengthening strategy of energetically-tuning polymorphous precipitates provides the basic guidance to develop high-performance nano-precipitated alloys. The current strengthening and plasticity models can be employed to well predict the mechanical properties of such kinds of alloys at cryogenic temperatures. read less NOT USED (low confidence) H. Mcarthur and D. Spalding, “STRUCTURE OF MATERIALS.” 2020. link Times cited: 50 Abstract: The behavior of Tin+1AlCn nanolaminates with n = 1, 2, 3 tha… read moreAbstract: The behavior of Tin+1AlCn nanolaminates with n = 1, 2, 3 that undergo a tensile deformation has been simulated using classical molecular dynamics methods. While calculating interatomic forces, a combination of twoand three-body potentials together with the embedded-atom method is applied. The stress-strain curves and the approximate values of the elastic moduli for the researched samples are calculated. The strain rate effect on the fracture dynamics is considered, and the corresponding atomistic configurations of examined samples are built. read less NOT USED (low confidence) M. H. Ulz, “Coupling the finite element method and molecular dynamics in the framework of the heterogeneous multiscale method for quasi-static isothermal problems,” Journal of The Mechanics and Physics of Solids. 2015. link Times cited: 22 NOT USED (low confidence) J. Wang et al., “Investigation of the Micromechanical Behavior of a Ti68Nb7Ta3Zr4Mo18 (at.%) High-Entropy Alloy,” Materials. 2023. link Times cited: 0 Abstract: Intense research efforts are focused on the development of a… read moreAbstract: Intense research efforts are focused on the development of advanced high-entropy alloys intended for premium aerospace components and other applications, where high strength and good formability are crucial. The mechanical properties of these alloys are closely related to the phase transformation, dislocation evolution, and grain size, and these factors are affected by the deformation temperature. The response of the retained austenite to strain-induced martensitic transformation at various temperatures was studied in an advanced Ti68Nb7Ta3Zr4Mo18 (at.%) high-entropy alloy via molecular dynamics simulation. It was found that the Ti68Nb7Ta3Zr4Mo18 alloy changes from a single crystal to a polycrystal during the tensile process, and the transition of the Ti68Nb7Ta3Zr4Mo18 (at.%) high-entropy alloy from the BCC phase to the FCC phase occurs. At high temperatures and low strain rates, grain boundary slip is the main deformation mechanism, and at low temperatures and high strain rates, dislocation slip replaces grain boundary slip as the dominant deformation mechanism, which improves the strength of the alloy. Moreover, when the grain size is too small, the strength of the alloy decreases, which does not satisfy the fine grain strengthening theory and shows an inverse Hall–Petch relationship. This study offers a new compositional window for the additive manufactured lightweight high-strength material categories for various applications including the aerospace industry. read less NOT USED (low confidence) J. A. Vita and D. Trinkle, “Exploring the necessary complexity of interatomic potentials,” Computational Materials Science. 2021. link Times cited: 8 NOT USED (low confidence) C. M. Andolina, M. Bon, D. Passerone, and W. Saidi, “Robust, Multi-Length-Scale, Machine Learning Potential for Ag–Au Bimetallic Alloys from Clusters to Bulk Materials,” The Journal of Physical Chemistry C. 2021. link Times cited: 22 NOT USED (low confidence) B. R. S. Kouamé et al., “Insights on the unique electro-catalytic behavior of PtBi/C materials,” Electrochimica Acta. 2020. link Times cited: 15 NOT USED (low confidence) Y. Zuo et al., “A Performance and Cost Assessment of Machine Learning Interatomic Potentials.,” The journal of physical chemistry. A. 2019. link Times cited: 413 Abstract: Machine learning of the quantitative relationship between lo… read moreAbstract: Machine learning of the quantitative relationship between local environment descriptors and the potential energy surface of a system of atoms has emerged as a new frontier in the development of interatomic potentials (IAPs). Here, we present a comprehensive evaluation of ML-IAPs based on four local environment descriptors --- atom-centered symmetry functions (ACSF), smooth overlap of atomic positions (SOAP), the Spectral Neighbor Analysis Potential (SNAP) bispectrum components, and moment tensors --- using a diverse data set generated using high-throughput density functional theory (DFT) calculations. The data set comprising bcc (Li, Mo) and fcc (Cu, Ni) metals and diamond group IV semiconductors (Si, Ge) is chosen to span a range of crystal structures and bonding. All descriptors studied show excellent performance in predicting energies and forces far surpassing that of classical IAPs, as well as predicting properties such as elastic constants and phonon dispersion curves. We observe a general trade-off between accuracy and the degrees of freedom of each model, and consequently computational cost. We will discuss these trade-offs in the context of model selection for molecular dynamics and other applications. read less NOT USED (low confidence) X. Zhao, C. Wang, M. Kim, and K. Ho, “Fe-Cluster Compounds of Chalcogenides: Candidates for Rare-Earth-Free Permanent Magnet and Magnetic Nodal-Line Topological Material.,” Inorganic chemistry. 2017. link Times cited: 3 Abstract: Fe-cluster-based crystal structures are predicted for chalco… read moreAbstract: Fe-cluster-based crystal structures are predicted for chalcogenides Fe3X4 (X = S, Se, Te) using an adaptive genetic algorithm. Topologically different from the well-studied layered structures of iron chalcogenides, the newly predicted structures consist of Fe clusters that are either separated by the chalcogen atoms or connected via sharing of the vertex Fe atoms. Using first-principles calculations, we demonstrate that these structures have competitive or even lower formation energies than the experimentally synthesized Fe3X4 compounds and exhibit interesting magnetic and electronic properties. In particular, we show that Fe3Te4 can be a good candidate as a rare-earth-free permanent magnet and Fe3S4 can be a magnetic nodal-line topological material. read less NOT USED (low confidence) P. Chowdhury, H. Sehitoglu, and R. Rateick, “Recent advances in modeling fatigue cracks at microscale in the presence of high density coherent twin interfaces,” Current Opinion in Solid State & Materials Science. 2016. link Times cited: 26 NOT USED (low confidence) Z. Trautt, F. Tavazza, and C. Becker, “Facilitating the selection and creation of accurate interatomic potentials with robust tools and characterization,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 14 Abstract: The Materials Genome Initiative seeks to significantly decre… read moreAbstract: The Materials Genome Initiative seeks to significantly decrease the cost and time of development and integration of new materials. Within the domain of atomistic simulations, several roadblocks stand in the way of reaching this goal. While the NIST Interatomic Potentials Repository hosts numerous interatomic potentials (force fields), researchers cannot immediately determine the best choice(s) for their use case. Researchers developing new potentials, specifically those in restricted environments, lack a comprehensive portfolio of efficient tools capable of calculating and archiving the properties of their potentials. This paper elucidates one solution to these problems, which uses Python-based scripts that are suitable for rapid property evaluation and human knowledge transfer. Calculation results are visible on the repository website, which reduces the time required to select an interatomic potential for a specific use case. Furthermore, property evaluation scripts are being integrated with modern platforms to improve discoverability and access of materials property data. To demonstrate these scripts and features, we will discuss the automation of stacking fault energy calculations and their application to additional elements. While the calculation methodology was developed previously, we are using it here as a case study in simulation automation and property calculations. We demonstrate how the use of Python scripts allows for rapid calculation in a more easily managed way where the calculations can be modified, and the results presented in user-friendly and concise ways. Additionally, the methods can be incorporated into other efforts, such as openKIM. read less NOT USED (low confidence) S. Guo et al., “Nanocatalyst superior to Pt for oxygen reduction reactions: the case of core/shell Ag(Au)/CuPd nanoparticles.,” Journal of the American Chemical Society. 2014. link Times cited: 156 Abstract: Controlling the electronic structure and surface strain of a… read moreAbstract: Controlling the electronic structure and surface strain of a nanoparticle catalyst has become an important strategy to tune and to optimize its catalytic efficiency for a chemical reaction. Using density functional theory (DFT) calculations, we predicted that core/shell M/CuPd (M = Ag, Au) NPs with a 0.8 or 1.2 nm CuPd2 shell have similar but optimal surface strain and composition and may surpass Pt in catalyzing oxygen reduction reactions. We synthesized monodisperse M/CuPd NPs by the coreduction of palladium acetylacetonate and copper acetylacetonate in the presence of Ag (or Au) nanoparticles with controlled shell thicknesses of 0.4, 0.75, and 1.1 nm and CuPd compositions and evaluated their catalysis for the oxygen reduction reaction in 0.1 M KOH solution. As predicted, our Ag/Cu37Pd63 and Au/Cu40Pd60 catalysts with 0.75 and 1.1 nm shells were more efficient catalysts than the commercial Pt catalyst (Fuel Cells Store), with their mass activity reaching 0.20 A/mg of noble metal at -0.1 V vs Ag/AgCl (4 M KCl); this was over 3 times higher than that (0.06 A/mg Pt) from the commercial Pt. These Ag(Au)/CuPd nanoparticles are promising non-Pt catalysts for oxygen reduction reactions. read less NOT USED (low confidence) A. Kusne et al., “On-the-fly machine-learning for high-throughput experiments: search for rare-earth-free permanent magnets,” Scientific Reports. 2014. link Times cited: 215 NOT USED (low confidence) J. F. Troncoso and V. Turlo, “Evaluating the applicability of classical and neural network interatomic potentials for modeling body centered cubic polymorph of magnesium,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 2 Abstract: Magnesium (Mg) is one of the most abundant metallic elements… read moreAbstract: Magnesium (Mg) is one of the most abundant metallic elements in nature and presents attractive mechanical properties in the industry. Particularly, it has a low density and relatively high strength/weight and stiffness/weight ratios, which make it one of the most attractive lightweight metals. However, the huge potential of Mg is restricted by its low ductility, associated with its hexagonal close packed (hcp) structure. This problem can be solved if Mg adopts the body centered cubic (bcc) structure, which is stable at high pressure or in confinement with stiff bcc metals like Nb. Molecular dynamics method is a magnificent tool to study material’s structure and deformation mechanisms at the atomic level, however, requiring accurate interatomic potentials. The majority of the interatomic potentials available in the literature for Mg have only been fitted to the properties of its stable hcp phase. In the present work, we perform systematic study of applicability of currently available Mg potentials to modeling the properties of metastable bcc polymorph of Mg, taking into account cohesive energy curves, elastic constants, stacking fault energies, and phonon dispersion curves. We conclude that the modified embedded atom method (MEAM) potentials are the most suitable for investigating bcc Mg in Mg/Nb nano-composites, while the properties of high-pressure bcc Mg would be better modeled by neural network interatomic potentials after different local atomic environments corresponding to bcc Mg being included into the fitting database. read less NOT USED (low confidence) С. Волегов, Р. М. Герасимов, and Р. П. Давлятшин, “MODELS OF MOLECULAR DYNAMICS: A REVIEW OF EAM-POTENTIALS. PART 2. POTENTIALS FOR MULTI-COMPONENT SYSTEMS.” 2018. link Times cited: 1 Abstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликова… read moreAbstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликована: 29 июня 2018 г. В статье представлена вторая часть обзора современных подходов и работ, посвященных построению потенциалов межатомного взаимодействия с использованием методологии погруженного атома (EAM-потенциалы). Эта часть обзора посвящена одной из наиболее остро стоящих проблем в молекулярной динамике – вопросам построения потенциалов, которые были бы пригодны для описания структуры и физико-механических свойств многокомпонентных (в первую очередь – бинарных и тернарных) материалов. Отмечены первые работы, в которых предлагались подходы к построению функций перекрестного взаимодействия для сплавов никеля и меди – как с использованием методологии EAM, так и несколько отличающийся по процедуре построения потенциал типа Финисса-Синклера. Рассматриваются работы, в которых производится сопоставление различных подходов к построению потенциалов, а также к процедуре идентификации их параметров на примере одних и тех же многокомпонентных систем (типа Al-Ni или Cu-Au). Кроме того, особый интерес представляют некоторые тернарные системы, например Fe–Ni–Cr, W–H– He или U–Mo–Xe, которые являются ключевыми для материалов атомной энергетики и которые в последние годы активно изучаются как возможные материалы для использования в термоядерных ректорах. Приведены примеры работ, в которых предлагаются и исследуются потенциалы для описания многокомпонентных систем, пригодных для использования в аэрокосмической промышленности и изготовленных прежде всего на основе никеля. Рассмотрены результаты исследований различных интерметаллических соединений, отмечены работы, в которых при помощи построенного EAM потенциала удалось количественно точно описать фазовые диаграммы соединений и вычислить характеристики фазовых переходов. read less NOT USED (low confidence) В. О. Подрыга, “МНОГОМАСШТАБНЫЙ ПОДХОД К ТРЕХМЕРНОМУ РАСЧЕТУ ТЕЧЕНИЙ ГАЗОВ И ИХ СМЕСЕЙ В МИКРОКАНАЛАХ ТЕХНИЧЕСКИХ СИСТЕМ.” 2016. link Times cited: 2 Abstract: Работа посвящена трехмерному моделированию течений газов в м… read moreAbstract: Работа посвящена трехмерному моделированию течений газов в микроканалах сложных технических систем. Предложен многомасштабный подход, сочетающий решения уравнений квазигазодинамики (КГД) и молекулярной динамики (МД). Представлена параллельная реализация подхода, основанная на методах расщепления по физическим процессам и разделения областей. Реализация ориентирована на использование вычислительных систем с центральной и гибридной архитектурами. Расчеты показали устойчивость численного алгоритма. С его помощью методами МД были получены основные коэффициентные зависимости для КГД системы, проверен переход от МД к КГД и обратно, произведен расчет трехмерного течения. Полученные результаты подтвердили эффективность разработанного подхода. read less NOT USED (high confidence) J. Kloppenburg, L. Pártay, H. J’onsson, and M. A. Caro, “A general-purpose machine learning Pt interatomic potential for an accurate description of bulk, surfaces, and nanoparticles.,” The Journal of chemical physics. 2023. link Times cited: 6 Abstract: A Gaussian approximation machine learning interatomic potent… read moreAbstract: A Gaussian approximation machine learning interatomic potential for platinum is presented. It has been trained on density-functional theory (DFT) data computed for bulk, surfaces, and nanostructured platinum, in particular nanoparticles. Across the range of tested properties, which include bulk elasticity, surface energetics, and nanoparticle stability, this potential shows excellent transferability and agreement with DFT, providing state-of-the-art accuracy at a low computational cost. We showcase the possibilities for modeling of Pt systems enabled by this potential with two examples: the pressure-temperature phase diagram of Pt calculated using nested sampling and a study of the spontaneous crystallization of a large Pt nanoparticle based on classical dynamics simulations over several nanoseconds. read less NOT USED (high confidence) G. S. Dhaliwal, P. Nair, and C. V. Singh, “Machine learned interatomic potentials using random features,” npj Computational Materials. 2022. link Times cited: 10 NOT USED (high confidence) P. Brault, M. Ji, D. Sciacqua, F. Poncin‐Epaillard, J. Berndt, and E. Kovačević, “Insight into acetylene plasma deposition using molecular dynamics simulations,” Plasma Processes and Polymers. 2021. link Times cited: 4 NOT USED (high confidence) S. Zhang et al., “Defect agglomeration induces a reduction in radiation damage resistance of In-rich In x Ga1−x N,” Journal of Physics D: Applied Physics. 2021. link Times cited: 4 Abstract: To investigate the reason for the reduction in damage resist… read moreAbstract: To investigate the reason for the reduction in damage resistance of In x Ga1−x N with increasing indium (In) content, we used molecular dynamics methods to simulate the threshold displacement energies, the individual recoil damage and the overlapping cascade processes in In x Ga1−x N (x = 0.3, 0.5, 0.7) during ion implantation. The average threshold displacement energy of In x Ga1−x N decreases a little (from 41.0 eV to 34.6 eV) as the In content increases (from 0.3 to 0.7) and the number of defects produced by individual cascades increases less than 30% with increasing In content (from 0.3 to 0.7), while the overlapping cascade simulations showed that with In content increasing the dynamic annealing processes in cascades were significantly suppressed. Thus, the suppression of dynamic annealing in the cascades is the main reason for the reduction of damage resistance of In x Ga1−x N by adding In content. The analysis of defect distribution during overlapping cascades showed that defects in In-rich In x Ga1−x N (x = 0.7) agglomerate more rapidly as the irradiation dose increases and are likely to form large clusters, which are harder to anneal during cascade evolution. Therefore, the suppression of dynamic annealing in In-rich In x Ga1−x N can be attributed to the rapid agglomeration of defects with the irradiation dose. read less NOT USED (high confidence) G. Xiao, K. Song, Y. He, W. Wang, Y. Zhang, and W. Dai, “Prediction and experimental research of abrasive belt grinding residual stress for titanium alloy based on analytical method,” The International Journal of Advanced Manufacturing Technology. 2021. link Times cited: 35 NOT USED (high confidence) C. Zhang, J. Zhou, and W. Xie, “Incident-angle dependence of deformation characteristics of aluminum surface under low-energy xenon-ion impact,” Journal of Physics: Conference Series. 2021. link Times cited: 0 Abstract: Ion thruster is a revolution technology with potential appli… read moreAbstract: Ion thruster is a revolution technology with potential applications in space mission but the thruster’s operation lifetime is limited by the sputtering from thruster components. In this work, molecular dynamic simulations are performed to explore the dependence of deformation characteristics of an aluminum surface on incident angle and kinetic energy under low-energy xenon-ion impact. The fraction of non-12-coordinated atoms is used to quantitatively characterize the microstructural evolution and defect density levels. It is found that defect density level has a linear relation with incident energy, and there exists a critical incident angle around 20°, at which the aluminum surface has the maximum defect density level. In addition, a collision model is developed to theoretically reveal the physical mechanisms behind the dependence. Our findings may helpful in developing long endurance electric propulsion devices for practical applications. read less NOT USED (high confidence) V. Borysiuk, “Механічні властивості наноламінатів Tin + 1AlCn: дослідження методами молекулярної динаміки,” Ukrainian Journal of Physics. 2020. link Times cited: 0 Abstract: Проведено моделювання поведiнки наноламiнатiв Tin+1AlCn з n … read moreAbstract: Проведено моделювання поведiнки наноламiнатiв Tin+1AlCn з n = 1, 2, 3 при деформацiї розтягнення на основi методiв класичної молекулярної динамiки. Для розрахункiв сил мiжатомної взаємодiї в дослiджуваних зразках був використаний пiдхiд iз комбiнацiєю парного та тричастинкового потенцiалiв i моделi зануреного атома. Для розглянутих зразкiв розраховано кривi навантаження та наближенi значення механiчних параметрiв, а саме модулiв пружностi. Дослiджено вплив швидкостi деформацiї на динамiку руйнування, а також побудовано вiдповiднi атомiстичнi конфiгурацiї дослiджуваних зразкiв. read less NOT USED (high confidence) W. Dednam, C. Sabater, O. Tal, J. Palacios, A. E. Botha, and M. Caturla, “Refined electron-spin transport model for single-element ferromagnetic systems: Application to nickel nanocontacts,” Physical Review B. 2020. link Times cited: 5 Abstract: W. Dednam ,1,2,* C. Sabater ,3 O. Tal ,3 J. J. Palacios ,4 A… read moreAbstract: W. Dednam ,1,2,* C. Sabater ,3 O. Tal ,3 J. J. Palacios ,4 A. E. Botha ,2 and M. J. Caturla 1 1Departamento de Física Aplicada and Unidad Asociada CSIC, Universidad de Alicante, Campus de San Vicente del Raspeig, E-03690 Alicante, Spain 2Department of Physics, Unisa Science Campus, University of South Africa, Johannesburg 1710, South Africa 3Chemical Physics Department, Weizmann Institute of Science, 76100 Rehovot, Israel 4Departamento de Física de la Materia Condensada, Condensed Matter Physics Center (IFIMAC) and Instituto Nicolás Cabrera, Universidad Autónoma de Madrid, Madrid E-28049, Spain read less NOT USED (high confidence) H. Bhattarai, K. E. Newman, and J. Gezelter, “The role of polarizability in the interfacial thermal conductance at the gold-water interface.,” The Journal of chemical physics. 2020. link Times cited: 3 Abstract: We have studied the interfacial thermal conductance, G, of t… read moreAbstract: We have studied the interfacial thermal conductance, G, of the flat Au(111)-water interface using non-equilibrium molecular dynamics simulations. We utilized two metal models, one based on the embedded atom method (EAM) and the other including metallic polarizability via a density readjusting EAM. These were combined with three popular water models, SPC/E, TIP4P, and TIP4P-FQ, to understand the role of polarizability in the thermal transport process. A thermal flux was introduced using velocity shearing and scaling reverse non-equilibrium molecular dynamics, and transport coefficients were measured by calculating the resulting thermal gradients and temperature differences at the interface. Our primary finding is that the computed interfacial thermal conductance between a bare metal interface and water increases when polarizability is taken into account in the metal model. Additional work to understand the origin of the conductance difference points to changes in the local ordering of the water molecules in the first two layers of water above the metal surface. Vibrational densities of states on both sides of the interface exhibit interesting frequency modulation close to the surface but no obvious differences due to metal polarizability. read less NOT USED (high confidence) M. An 安, M. Su 宿, Qiong 琼 Deng 邓, H. Song 宋, C. Wang 王, and Yu 玉 Shang 尚, “Anisotropic plasticity of nanocrystalline Ti: A molecular dynamics simulation,” Chinese Physics B. 2020. link Times cited: 5 Abstract: Using molecular dynamics simulations, the plastic deformatio… read moreAbstract: Using molecular dynamics simulations, the plastic deformation behavior of nanocrytalline Ti has been investigated under tension and compression normal to the {0001}, {1¯010} , and { 1¯21¯0 } planes. The results indicate that the plastic deformation strongly depends on crystal orientation and loading directions. Under tension normal to basal plane, the deformation mechanism is mainly the grain reorientation and the subsequent deformation twinning. Under compression, the transformation of hexagonal-close packed (HCP)-Ti to face-centered cubic (FCC)-Ti dominates the deformation. When loading is normal to the prismatic planes (both {1¯010} and {1¯21¯0} ), the deformation mechanism is primarily the phase transformation among HCP, body-centered cubic (BCC), and FCC structures, regardless of loading mode. The orientation relations (OR) of {0001}HCP║{111}FCC and 〈1¯210〉HCP||〈110〉FCC , and {101¯0}HCP||{11¯0}FCC and 〈0001〉HCP||〈010〉FCC between the HCP and FCC phases have been observed in the present work. For the transformation of HCP → BCC → HCP, the OR is 0001α1||{110}β||{101¯0}α2 (HCP phase before the critical strain is defined as α1-Ti, BCC phase is defined as β-Ti, and the HCP phase after the critical strain is defined as α2-Ti). Energy evolution during the various loading processes further shows the plastic anisotropy of nanocrystalline Ti is determined by the stacking order of the atoms. The results in the present work will promote the in-depth study of the plastic deformation mechanism of HCP materials. read less NOT USED (high confidence) P. Wang, Q. Cao, H. Wang, Y. Nie, S. Liu, and Q. Peng, “Fivefold enhancement of yield and toughness of copper nanowires via coating carbon nanotubes,” Nanotechnology. 2019. link Times cited: 5 Abstract: Carbon nanotubes are outstanding reinforcements owing to the… read moreAbstract: Carbon nanotubes are outstanding reinforcements owing to their unparallel strength, while their effects on the copper nanowire are still not fully understood, hampering their broad applications. Herein, we have investigated the tensile behaviors of the nanocomposite-wire of carbon nanotube-copper using molecular dynamic simulations. For the nanocomposite, both the coated and embedded carbon nanotubes increase the Young’s modulus, fracture stress and toughness of the copper nanowire. A reinforcement of over fivefold in both yield strength (5.3 times) and toughness (5.1 times) has been achieved when the carbon nanotubes are coated on the copper nanowires, as well as 1.7 times in the Young’s modulus. Higher temperatures and lower loading rates reduce the reinforcement. read less NOT USED (high confidence) P. Brault, C. Coutanceau, A. Caillard, and S. Baranton, “Pt3MeAu (Me = Ni, Cu) Fuel Cell Nanocatalyst Growth, Shapes, and Efficiency: A Molecular Dynamics Simulation Approach,” The Journal of Physical Chemistry C. 2019. link Times cited: 5 Abstract: The formation of the ternary Pt3NiAu and Pt3CuAu nanostructu… read moreAbstract: The formation of the ternary Pt3NiAu and Pt3CuAu nanostructures is examined using molecular dynamics simulations in the context of free (i.e., unsupported) cluster growth in an unreactive atmospher... read less NOT USED (high confidence) S. Pozdnyakov, A. Oganov, E. Mazhnik, A. Mazitov, and I. Kruglov, “Fast general two- and three-body interatomic potential,” Physical Review B. 2019. link Times cited: 6 Abstract: We introduce a new class of machine learning interatomic pot… read moreAbstract: We introduce a new class of machine learning interatomic potentials - fast General Two- and Three-body Potential (GTTP) which are as fast as conventional empirical potentials and require computational time that remains constant with increasing fitting flexibility. GTTP does not contain any assumptions about functional form of two- and three-body interactions. These interactions can be modeled arbitrarily accurately potentially by thousands of parameters not affecting resulting computational cost. Time complexity is O(1) per every considered pair or triple of atoms. The fitting procedure is reduced to simple linear regression on ab initio calculated energies and forces and leads to effective two- and three-body potential which reproduces quantum many-body interactions as accurately as possible. Our potential can be made continuously differentiable any number of times at the expense of increased computational time. We made a number of performance tests on one-, two- and three-component systems. Flexibility of the introduced approach makes the potential transferable in terms of size and type of atomic systems. We show, that trained on randomly generated structures with just 8 atoms in the unit cell, it significantly outperforms common empirical interatomic potentials in the study of large systems, such as grain boundaries in polycrystalline materials. read less NOT USED (high confidence) A. Khomenko, M. Zakharov, B. Persson, Y. V. Khyzhnya, P. Trofymenko, and D. Logvinenko, “Correlation Anisotropy of Friction Force During Sliding of Metallic Nanoparticles on Graphene,” 2019 IEEE 9th International Conference Nanomaterials: Applications & Properties (NAP). 2019. link Times cited: 0 Abstract: The research of temperature properties and correlation of fr… read moreAbstract: The research of temperature properties and correlation of friction force of metallic nanoparticles on graphene is represented. Modeling was led by molecular dynamics method through CUDA-technology. Calculations have been performed for aluminum, palladium, cobalt and platinum nanoclusters containing from 10000 to 20000 atoms at the temperatures from 300 K to 1400 K. Radial distribution function is built to analyze the structural changes of nanoparticles. We discuss the strong influence of the types of metals on the form of functions describing the behavior of nanoparticles during motion. read less NOT USED (high confidence) M. Yan, H.-gui Huang, M. Wang, L. Ye, and J.-na Sun, “Multi-scale Simulation on Bonding Mechanism of Solid-Liquid Cast-Rolling of Cu/Al Cladding Strip based on FEM and MD,” Journal of Wuhan University of Technology-Mater. Sci. Ed. 2019. link Times cited: 0 NOT USED (high confidence) A. M. Igoshkin, “Molecular Dynamics Study of the Deposition of Palladium-Silver Films on a Silver Substrate,” Journal of Structural Chemistry. 2019. link Times cited: 0 NOT USED (high confidence) K. Yashiro, “Characteristics of atomic elastic stiffness at GSF energy surface, edge and screw dislocation cores in fcc, bcc and hcp metals,” AIP Advances. 2019. link Times cited: 3 Abstract: Basic characteristics of 6 × 6 matrix of atomic elastic stif… read moreAbstract: Basic characteristics of 6 × 6 matrix of atomic elastic stiffness (AES), Bija=Δσia/Δej, or the deformation resistance at each atom point, are discussed first in static analyses of generalized stacking fault (GSF) energy surface for 8 fcc, 4 bcc and 4 hcp metals with Zhou’s EAM potential. For hcp metals, the stress–strain peak along the GSF path exactly coincides with the point where the AES loses the resistance showing negative 1st eigenvalue ηa(1), or the solution of BijaΔej=ηaΔei=Δσia; however, all fcc and 2 bcc (Mo and W) never have negative ηa(1) along the GSF path. Fe and Ta transiently show ηa(1) < 0 while they also have positive ηa(1) at the GSF energy peak. Then we performed MD simulations of edge and screw dislocation dipoles in a periodic slab cell of typical elements of fcc, bcc and hcp; and discussed the eigenvalue and the corresponding eigenvector {Δexx, Δeyy, Δezz, Δγyz, Δγzx, Δγxy} of the dislocation cores. As expected from the results of the GSF analyses, dislocation cores in fcc Ni have no ηa(1) < 0 atoms, even in their glide process under external shear loading. Bcc Fe and hcp Co definitely have ηa(1) < 0 atoms in the dislocation cores and their migration direction can be visualized by the maximum shear direction of the strain tensor of the corresponding eigenvector.Basic characteristics of 6 × 6 matrix of atomic elastic stiffness (AES), Bija=Δσia/Δej, or the deformation resistance at each atom point, are discussed first in static analyses of generalized stacking fault (GSF) energy surface for 8 fcc, 4 bcc and 4 hcp metals with Zhou’s EAM potential. For hcp metals, the stress–strain peak along the GSF path exactly coincides with the point where the AES loses the resistance showing negative 1st eigenvalue ηa(1), or the solution of BijaΔej=ηaΔei=Δσia; however, all fcc and 2 bcc (Mo and W) never have negative ηa(1) along the GSF path. Fe and Ta transiently show ηa(1) < 0 while they also have positive ηa(1) at the GSF energy peak. Then we performed MD simulations of edge and screw dislocation dipoles in a periodic slab cell of typical elements of fcc, bcc and hcp; and discussed the eigenvalue and the corresponding eigenvector {Δexx, Δeyy, Δezz, Δγyz, Δγzx, Δγxy} of the dislocation cores. As expected from the results of the GSF analyses, dislocation cores in fcc Ni have n... read less NOT USED (high confidence) P. Chen, Z. Zhang, C. Liu, T. An, H. Yu, and F. Qin, “Temperature and grain size dependences of mechanical properties of nanocrystalline copper by molecular dynamics simulation,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 18 Abstract: Nanocrystalline copper (Cu) is considered to be one of the b… read moreAbstract: Nanocrystalline copper (Cu) is considered to be one of the best interconnected material in integration circuit (IC) industry, because of its ultra-low resistivity and high mechanical stability. Mechanical properties of nanocrystalline Cu are completely different from those of bulk monocrystalline Cu. These properties are of high importance in the assessment of the thermo-mechanical reliability of the interconnected IC structure. To investigate the effects of the grain sizes and temperature on the mechanical properties of nanocrystalline Cu, molecular dynamics simulations of uniaxial tensile test are performed in this study. The results show that the elastic modulus of nanocrystalline Cu with grain sizes of 4.65–12.41 nm gradually increases with the increase of the mean grain sizes, the corresponding flow stress concurrently increases, and the flow stress is proportional to the square-root of the grain size, which satisfies the inverse-Hall–Petch relation. Furthermore, the elastic modulus linearly decreases with the increase of temperature. The coupled effect of the flow stress, strain rate and temperature were elaborated by the Arrhenius hyperbolic sinusoidal model. Meanwhile, the deformation activation energy of nanocrystalline Cu for various grain sizes were obtained. All of the tensile simulation tests confirmed that the mechanism of plastic deformation for nanocrystalline Cu with 4.65–12.41 nm grain sizes is mainly specific to the grain boundary sliding and grain rotation. The dislocation nucleation and migration, which is usually the deformation mechanism of plasticity for macroscopic materials, is no longer the dominant factor for nanocrystalline Cu. read less NOT USED (high confidence) J. Meiser and H. Urbassek, “Effect of Alloying Elements on the α-γ Phase Transformation in Iron,” Materials. 2019. link Times cited: 2 Abstract: Small concentrations of alloying elements can modify the α-γ… read moreAbstract: Small concentrations of alloying elements can modify the α-γ phase transition temperature Tc of Fe. We study this effect using an atomistic model based on a set of many-body interaction potentials for iron and several alloying elements. Free-energy calculations based on perturbation theory allow us to determine the change in Tc introduced by the alloying element. The resulting changes are in semi-quantitative agreement with experiment. The effect is traced back to the shape of the pair potential describing the interaction between the Fe and the alloying atom. read less NOT USED (high confidence) S. J. Knauss, L. N. Guevara, and M. Atwater, “Enhanced Performance of Bimetallic Co-Pd Catalysts Prepared by Mechanical Alloying,” Metals. 2019. link Times cited: 2 Abstract: Bimetallic catalysts can provide enhanced performance, and C… read moreAbstract: Bimetallic catalysts can provide enhanced performance, and Co-based catalysts in particular have been studied in various respects for their activity in the deposition of carbon nanofibers (CNFs). The majority of studies on CNF catalysis use co-precipitation to create alloys, but recent work has demonstrated the suitability of mechanical alloying (MA) by ball milling to reduce cost and increase catalytic activity. This work establishes the unique ability of MA to control the microstructure to produce bimetallic composites, which retain distinct metallic phases that improve catalytic activity. It is demonstrated that Co-Pd alloys reach a maximum in catalytic activity at an intermediate time of mechanical activation, where 30 min of milling outperformed samples milled for 5, 15, 60, and 240 min at a reaction temperature of 550 °C and a 1:4 C2H4:H2 reactant ratio. This indicates there is benefit to retaining the metals in distinct phases in close proximity. Ball milling provides a relatively simple and scalable method to achieve these unique microstructures, and in the optimal condition tested here, the activity toward carbon deposition is increased fourfold over prior work. Furthermore, the minimum temperature for deposition is also reduced. The characteristics of these materials, the effects of milling and annealing, and the underlying mechanisms of deposition are discussed. read less NOT USED (high confidence) Y. Lysogorskiy, T. Hammerschmidt, J. Janssen, J. Neugebauer, and R. Drautz, “Transferability of interatomic potentials for molybdenum and silicon,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 14 Abstract: Interatomic potentials are widely used in computational mate… read moreAbstract: Interatomic potentials are widely used in computational materials science, in particular for simulations that are too computationally expensive for density functional theory (DFT). Most interatomic potentials have a limited application range and often there is very limited information available regarding their performance for specific simulations. We carried out high-throughput calculations for molybdenum and silicon with DFT and a number of interatomic potentials. We compare the DFT reference calculations and experimental data to the predictions of the interatomic potentials. We focus on a large number of basic materials properties, including the cohesive energy, atomic volume, elastic coefficients, vibrational properties, thermodynamic properties, surface energies and vacancy formation energies, which enables a detailed discussion of the performance of the different potentials. We further analyze correlations between properties as obtained from DFT calculations and how interatomic potentials reproduce these correlations, and suggest a general measure for quantifying the accuracy and transferability of an interatomic potential. From our analysis we do not establish a clearcut ranking of the potentials as each potential has its strengths and weaknesses. It is therefore essential to assess the properties of a potential carefully before application of the potential in a specific simulation. The data presented here will be useful for selecting a potential for simulations of Mo or Si. read less NOT USED (high confidence) M. He, C. Wu, M. Shugaev, G. Samolyuk, and L. Zhigilei, “Computational Study of Short-Pulse Laser-Induced Generation of Crystal Defects in Ni-Based Single-Phase Binary Solid–Solution Alloys,” The Journal of Physical Chemistry C. 2019. link Times cited: 20 Abstract: Single-phase concentrated solid–solution alloys exhibit enha… read moreAbstract: Single-phase concentrated solid–solution alloys exhibit enhanced mechanical characteristics and radiation damage resistance, making them promising candidate materials for applications involving an exposure to rapid localized energy deposition. In this paper, we use large-scale atomistic modeling to investigate the mechanisms of the generation of vacancies, dislocations, stacking faults, and twin boundaries in Ni, Ni50Fe50, Ni80Fe20, and Ni80Cr20 targets irradiated by short laser pulses in the regime of melting and resolidification. The decrease in the thermal conductivity and strengthening of the electron–phonon coupling due to the intrinsic chemical disorder in the solid-solution alloys are found to have important implications on localization of the energy deposition and generation of thermoelastic stresses. The interaction of the laser-induced stress waves with the melting front is found to play a key role in roughening of the crystal–liquid interface and generation of dislocations upon the solidificati... read less NOT USED (high confidence) X. Zhu and Y. Lu, “Growth of beryllium thin films on beryllium (0001) surface: Influence of incident energy and incident angle by molecular dynamics simulation,” Journal of Applied Physics. 2018. link Times cited: 5 Abstract: The morphology and microstructure of metallic thin films syn… read moreAbstract: The morphology and microstructure of metallic thin films synthesized by magnetron sputtering deposition are sensitive to incident energy and incident angle. The role of incident energy and incident angle in films’ morphology evolution of the beryllium thin films’ growth on beryllium (0001) surface was studied by molecular dynamics simulations. The analytical bond order potential was used to represent the interatomic interactions, and the common neighbor analysis algorithm for crystal structures was used for the structural characterization of the simulated films. It is found that when the incident energy is between 1 eV and 20 eV, the increased incident energy is beneficial to grow uniform crystal films and, when the incident energy is greater than 15 eV, the interstitial atoms formed inside the films. Furthermore, under the small incident angle conditions, the morphology of a smooth surface was formed, which means that the vertical incident conditions are desired for the growth of high quality films. In short, vertically inserted atoms with hyperthermal energy (5–10 eV) are more propitious for the growth of perfect crystal Be thin films. The obtained results can be used to guide the experiment. read less NOT USED (high confidence) S. Jin et al., “Colossal grain growth yields single-crystal metal foils by contact-free annealing,” Science. 2018. link Times cited: 141 Abstract: Turning many into one Single-crystal metal foils are valuabl… read moreAbstract: Turning many into one Single-crystal metal foils are valuable for their surface properties that allow for synthesis of materials like graphene. Jin et al. present a strategy for creating colossal single-crystal metal foils called “contact-free annealing” (see the Perspective by Rollett). The method relies on hanging and heating commercially available, inexpensive, cold-rolled metal foils. Almost as if by magic, the polycrystalline grains rotate and anneal into a large single-crystal sheet with a specific crystal orientation. The strategy allows for the creation of much larger and much cheaper single-crystal metal foils. Science, this issue p. 1021; see also p. 996 Contact-free annealing allows for the synthesis of large single-crystal metal foils. Single-crystal metals have distinctive properties owing to the absence of grain boundaries and strong anisotropy. Commercial single-crystal metals are usually synthesized by bulk crystal growth or by deposition of thin films onto substrates, and they are expensive and small. We prepared extremely large single-crystal metal foils by “contact-free annealing” from commercial polycrystalline foils. The colossal grain growth (up to 32 square centimeters) is achieved by minimizing contact stresses, resulting in a preferred in-plane and out-of-plane crystal orientation, and is driven by surface energy minimization during the rotation of the crystal lattice followed by “consumption” of neighboring grains. Industrial-scale production of single-crystal metal foils is possible as a result of this discovery. read less NOT USED (high confidence) A. Khomenko, M. Zakharov, D. Boyko, and B. Persson, “Atomistic modeling of tribological properties of Pd and Al nanoparticles on a graphene surface,” Beilstein Journal of Nanotechnology. 2018. link Times cited: 7 Abstract: Background: The frictional properties of nanoparticles have … read moreAbstract: Background: The frictional properties of nanoparticles have been studied to gain insight into the fundamental origin of sliding friction. Results: Using molecular dynamics we investigate frictional properties of aluminum and palladium nanoparticles deposited on a graphene layer. We study the time evolution of the total momentum of the system, the total and potential energies, the temperature, the velocity and position of the center of mass, the dimensions of the nanoparticle, and the friction and substrate forces acting on the particle. We also study how the friction force depends on the nanoparticle–graphene contact area and the temperature. Conclusion: The tribological properties of nanoparticles strongly depend on the materials. The particles move in an irregular (saw-like) manner. The averaged friction force depends nearly linearly on the contact area and non-monotonously on temperature. We observe ordered crystalline domains of atoms at the bottom surface of the metal particles, but the peaks of radial distribution function are blurred indicating that the nanoparticles are amorphous or polycrystalline. read less NOT USED (high confidence) X. W. Zhou, D. Ward, and M. E. Foster, “A bond-order potential for the Al–Cu–H ternary system,” New Journal of Chemistry. 2018. link Times cited: 13 Abstract: Al-Based Al–Cu alloys have a very high strength to density r… read moreAbstract: Al-Based Al–Cu alloys have a very high strength to density ratio, and are therefore important materials for transportation systems including vehicles and aircrafts. These alloys also appear to have a high resistance to hydrogen embrittlement, and as a result, are being explored for hydrogen related applications. To enable fundamental studies of mechanical behavior of Al–Cu alloys under hydrogen environments, we have developed an Al–Cu–H bond-order potential according to the formalism implemented in the molecular dynamics code LAMMPS. Our potential not only fits well to properties of a variety of elemental and compound configurations (with coordination varying from 1 to 12) including small clusters, bulk lattices, defects, and surfaces, but also passes stringent molecular dynamics simulation tests that sample chaotic configurations. Careful studies verified that this Al–Cu–H potential predicts structural property trends close to experimental results and quantum-mechanical calculations; in addition, it properly captures Al–Cu, Al–H, and Cu–H phase diagrams and enables simulations of H2 dissociation, chemisorption, and absorption on Al–Cu surfaces. read less NOT USED (high confidence) A. Khomenko, D. Boyko, M. Zakharov, K. Khomenko, and Y. V. Khyzhnya, “Molecular dynamics of aluminum nanoparticles friction on graphene,” 2017 IEEE 7th International Conference Nanomaterials: Application & Properties (NAP). 2017. link Times cited: 4 Abstract: Using molecular dynamics method and CUDA-technology for soft… read moreAbstract: Using molecular dynamics method and CUDA-technology for software creation, we investigate tribological properties observed when aluminum atoms are deposited on a graphene layer. During the modeling of the shear of nanoparticles, consisting of aluminum atoms, various parameters are measured the total momentum of the system, the total and potential energies, the temperature, the velocity and position of the center of mass, the dimensions of the nanoparticle, friction and substrate forces acting on the particle. We make a conclusion that tribological properties of nanoparticles strongly depend on materials. Detected heterogeneous form of dependence of substrate force is saw-like. The power dependence of averaged friction force on contact area was obtained. Since peaks of radial distribution function are blurred consequently nanoparticle has amorphous or polycrystalline form. read less NOT USED (high confidence) G. Henkelman, “Atomistic Simulations of Activated Processes in Materials,” Annual Review of Materials Research. 2017. link Times cited: 31 Abstract: Activated processes in materials are important for many of t… read moreAbstract: Activated processes in materials are important for many of the properties that make them function. Batteries and catalysts are examples for which understanding how the component materials function on a timescale of milliseconds to seconds is critical to making improvements in a rational way. Modeling materials over these long timescales, relative to the timescale of atomic vibrations, is one of the grand challenges of the field. Transition state theory is central to bridging this timescale gap, and in the materials community, the harmonic approximation and the determination of saddle points to quantify reaction rates are ubiquitous. In this review, single- and double-ended methods for saddle point finding are discussed, as well as how finding saddle points can be used in the adaptive kinetic Monte Carlo method to model materials properties on the timescale of activated processes. Applications of surface diffusion and chemistry, phase boundary migration, and solid-solid phase transitions are presented. read less NOT USED (high confidence) V. Borysiuk, “Atomistic simulation of two-dimensional titanium carbide Ti2C fracture under uniform tensile strain,” 2017 IEEE 37th International Conference on Electronics and Nanotechnology (ELNANO). 2017. link Times cited: 1 Abstract: The paper presents the results of the computer simulation of… read moreAbstract: The paper presents the results of the computer simulation of the failure dynamic of two-dimensional (or graphene like) titanium carbide Ti2C. The behavior of the sample under uniform tensile strain is studied within classical molecular dynamic (MD) simulation technique. Atomic configurations of the Ti2C nanosheet were computed for different values of the tensile strain. Performed simulation shows that cracks in the sample occur at strain of 0.07. Elastic constant of the Ti2C estimated from the strain-stress curve during uniform tension agrees with reported earlier values. read less NOT USED (high confidence) B. Gault et al., “A nexus between 3D atomistic data hybrids derived from atom probe microscopy and computational materials science: A new analysis of solute clustering in Al-alloys,” Scripta Materialia. 2017. link Times cited: 20 NOT USED (high confidence) S. Rao et al., “Atomistic simulations of dislocations in a model BCC multicomponent concentrated solid solution alloy,” Acta Materialia. 2016. link Times cited: 160 NOT USED (high confidence) D. Bai, J. Sun, W. Chen, and D. Du, “Molecular dynamics simulation of the diffusion behaviour between Co and Ti and its effect on the wear of WC/Co tools when titanium alloy is machined,” Ceramics International. 2016. link Times cited: 32 NOT USED (high confidence) E. Watkins et al., “Neutron reflectometry investigations of interfacial structures of Ti/TiN layers deposited by magnetron sputtering,” Thin Solid Films. 2016. link Times cited: 10 NOT USED (high confidence) L. Wan and G. Thompson, “BCC stabilization and growth stress behavior in Ti/V multilayers,” Thin Solid Films. 2016. link Times cited: 3 NOT USED (high confidence) X. W. Zhou, D. Ward, and M. E. Foster, “An analytical bond-order potential for the aluminum copper binary system,” Journal of Alloys and Compounds. 2016. link Times cited: 38 NOT USED (high confidence) J. Davoodi, S. Dadashi, and M. Yarifard, “Molecular dynamics simulations of the melting of Al–Ni nanowires,” Philosophical Magazine. 2016. link Times cited: 6 Abstract: Molecular dynamics (MD) simulations were performed to invest… read moreAbstract: Molecular dynamics (MD) simulations were performed to investigate the influence of nickel (Ni) composition and nanowire thickness on the thermal properties of Al-x%Ni (at%) nanowires using the embedded atom model (EAM) potential. The melting of the nanowire was characterised by studying the temperature dependence of the cohesive energy and mean square displacement. The effect of the nanowire thickness on the cohesive energy, melting temperature, heat capacity as well as latent heat was studied in canonical ensemble. Moreover, the crystal stability of Al, Al-20%Ni, Al-40%Ni, Al-60%Ni, Al-80%Ni, Al3Ni, Ni3Al and Ni nanowires was studied at different temperatures using mean square displacement and cohesive energy. read less NOT USED (high confidence) K. Wang, W. Zhu, S. Xiao, J. Chen, and W. Hu, “A new embedded-atom method approach based on the pth moment approximation,” Journal of Physics: Condensed Matter. 2016. link Times cited: 5 Abstract: Large scale atomistic simulations with suitable interatomic … read moreAbstract: Large scale atomistic simulations with suitable interatomic potentials are widely employed by scientists or engineers of different areas. The quick generation of high-quality interatomic potentials is urgently needed. This largely relies on the developments of potential construction methods and algorithms in this area. Quantities of interatomic potential models have been proposed and parameterized with various methods, such as the analytic method, the force-matching approach and multi-object optimization method, in order to make the potentials more transferable. Without apparently lowering the precision for describing the target system, potentials of fewer fitting parameters (FPs) are somewhat more physically reasonable. Thus, studying methods to reduce the FP number is helpful in understanding the underlying physics of simulated systems and improving the precision of potential models. In this work, we propose an embedded-atom method (EAM) potential model consisting of a new manybody term based on the pth moment approximation to the tight binding theory and the general transformation invariance of EAM potentials, and an energy modification term represented by pairwise interactions. The pairwise interactions are evaluated by an analytic-numerical scheme without the need to know their functional forms a priori. By constructing three potentials of aluminum and comparing them with a commonly used EAM potential model, several wonderful results are obtained. First, without losing the precision of potentials, our potential of aluminum has fewer potential parameters and a smaller cutoff distance when compared with some constantly-used potentials of aluminum. This is because several physical quantities, usually serving as target quantities to match in other potentials, seem to be uniquely dependent on quantities contained in our basic reference database within the new potential model. Second, a key empirical parameter in the embedding term of the commonly used EAM model is found to be related to the effective order of moments of local density of states. This may provide a way to improve the precision of EAM potentials further through more precise approximations to tight binding theory. In addition, some critical details about construction procedures are discussed. read less NOT USED (high confidence) T. Liu, T.-E. Fan, J.-W. Zheng, G. Shao, Q. Sun, and Y. Wen, “Structural optimization of Fe nanoclusters based on multi-populations differential evolution algorithm,” Journal of Nanoparticle Research. 2016. link Times cited: 6 NOT USED (high confidence) I. A. Alhafez, C. Ruestes, Y. Gao, and H. Urbassek, “Nanoindentation of hcp metals: a comparative simulation study of the evolution of dislocation networks,” Nanotechnology. 2016. link Times cited: 66 Abstract: Using molecular dynamics simulation, we study the nanoindent… read moreAbstract: Using molecular dynamics simulation, we study the nanoindentation of three hcp metals: Mg, Ti, and Zr. Both the basal and two prismatic surface planes are considered. We focus on the characterization of the plasticity generated in the crystal. The similarities to, and the differences from, the behavior of the more commonly investigated fcc and bcc metals are highlighted. We find that hcp metals show a larger variety than the fcc and bcc metals studied up until now. The prolific emission of prismatic loops can lead to extended plastic zones. The size of the plastic zone is quantified by the ratio f of the plastic zone radius to the radius of the contact area. We find values of between 1.6 (an almost collapsed zone) and >5; in the latter case, complex dislocation networks build up which are extended in the direction of easy glide. read less NOT USED (high confidence) A. Prakash et al., “Atom probe informed simulations of dislocation-precipitate interactions reveal the importance of local interface curvature,” Acta Materialia. 2015. link Times cited: 73 NOT USED (high confidence) V. Borysiuk, V. Mochalin, and Y. Gogotsi, “Molecular dynamic study of the mechanical properties of two-dimensional titanium carbides Tin+1Cn (MXenes),” Nanotechnology. 2015. link Times cited: 190 Abstract: Two-dimensional materials beyond graphene are attracting muc… read moreAbstract: Two-dimensional materials beyond graphene are attracting much attention. Recently discovered 2D carbides and nitrides (MXenes) have shown very attractive electrical and electrochemical properties, but their mechanical properties have not been characterized yet. There are neither experimental measurements reported in the literature nor predictions of strength or fracture modes for single-layer MXenes. The mechanical properties of two-dimensional titanium carbides were investigated in this study using classical molecular dynamics. Young’s modulus was calculated from the linear part of strain–stress curves obtained under tensile deformation of the samples. Strain-rate effects were observed for all Tin+1Cn samples. From the radial distribution function, it is found that the structure of the simulated samples is preserved during the deformation process. Calculated values of the elastic constants are in good agreement with published DFT data. read less NOT USED (high confidence) R. Xu et al., “Three-dimensional coordinates of individual atoms in materials revealed by electron tomography.,” Nature materials. 2015. link Times cited: 180 NOT USED (high confidence) H. Ding, Y. Lu, Y. Xie, H. Liu, and H. Schaefer, “The Energy Difference between the Triply-Bridged and All-Terminal Structures of Co₄(CO)₁₂, Rh₄(CO)₁₂, and Ir₄(CO)₁₂: A Difficult Test for Conventional Density Functional Methods.,” Journal of chemical theory and computation. 2015. link Times cited: 2 Abstract: The M4(CO)12 molecules Co4(CO)12, Rh4(CO)12, and Ir4(CO)12 h… read moreAbstract: The M4(CO)12 molecules Co4(CO)12, Rh4(CO)12, and Ir4(CO)12 have two low-lying structures, the all-terminal structure with Td symmetry and the triply bridged structure with C3v symmetry. A total of 45 density functional theory (DFT) methods have been used to predict structures and vibrational frequencies for Co4(CO)12, Rh4(CO)12, and Ir4(CO)12. The different DFT methods show a broad range of energy differences ΔE = E(Td) - E(C3v). For Rh4(CO)12, none of the 45 DFT predictions is within 11 kcal/mol of the 2005 experimental value of 5.1 ± 0.6 kcal/mol reported by Allian and Garland (Dalton Trans. 2005, 1957 - 1965). For the challenging Ir4(CO)12 molecule, 21 DFT methods predict the correct Td structure, while 24 DFT methods predict the C3v structure to lie lower in energy. This research reveals many peculiar problems in the computation of the vibrational frequencies for the all-terminal structure. read less NOT USED (high confidence) X. W. Zhou, D. Ward, M. Foster, and J. Zimmerman, “An analytical bond-order potential for the copper–hydrogen binary system,” Journal of Materials Science. 2015. link Times cited: 18 NOT USED (high confidence) G. Bonny, P. Grigorev, P. Grigorev, and D. Terentyev, “On the binding of nanometric hydrogen–helium clusters in tungsten,” Journal of Physics: Condensed Matter. 2014. link Times cited: 75 Abstract: In this work we developed an embedded atom method potential … read moreAbstract: In this work we developed an embedded atom method potential for large scale atomistic simulations in the ternary tungsten–hydrogen–helium (W–H–He) system, focusing on applications in the fusion research domain. Following available ab initio data, the potential reproduces key interactions between H, He and point defects in W and utilizes the most recent potential for matrix W. The potential is applied to assess the thermal stability of various H–He complexes of sizes too large for ab initio techniques. The results show that the dissociation of H–He clusters stabilized by vacancies will occur primarily by emission of hydrogen atoms and then by break-up of V–He complexes, indicating that H–He interaction does influence the release of hydrogen. read less NOT USED (high confidence) P. Norris, “Modeling Interfacial Thermal Boundary Conductance of Engineered Interfaces.” 2014. link Times cited: 2 Abstract: : The hypothesis that motivates this grant--that material ca… read moreAbstract: : The hypothesis that motivates this grant--that material can be added at an interface to enhance its effective thermal conductance--was first explored using simulations. Our primary tool was the classical molecular dynamics (MD) method. The first part of the simulation work demonstrated that an interfacial film can enhance conductance in simple systems. The second part laid the groundwork to extend those simulations to more complex material systems. To theoretically investigate the phonon transport underlying the conductance trends observed in our simulations, we used various theoretical approaches to understand fundamental phonon transport in interfacial structures: (1) semi-empirical methods such as the DMM, (2) the wavelet transform applied to MD simulations, (3) Green's functions, and (4) the interfering particle model (IPM). Finally, the findings from simulations and theoretical analysis were used to design a series of experimental measurements of hBD at interfaces with varying thicknesses of interfacial films. The findings have been published in the 16 archival journal papers and 3 conference proceeding papers cited here, not including publications still under preparation. Each of these publications acknowledged funding from the AFOSR. read less NOT USED (high confidence) N. Plyusnin, V. M. Il’yashchenko, S. A. Kitan, W. C. Lin, and C. Kuo, “Influence of the thermal power of a Fe atomic flux on the formation of Cu/Fe nanofilms on a Si(001) substrate,” Technical Physics. 2014. link Times cited: 0 NOT USED (high confidence) G. Bonny, D. Terentyev, A. Bakaev, P. Grigorev, and D. V. Neck, “Many-body central force potentials for tungsten,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 79 Abstract: Tungsten and tungsten-based alloys are the primary candidate… read moreAbstract: Tungsten and tungsten-based alloys are the primary candidate materials for plasma facing components in fusion reactors. The exposure to high-energy radiation, however, severely degrades the performance and lifetime limits of the in-vessel components. In an effort to better understand the mechanisms driving the materials' degradation at the atomic level, large-scale atomistic simulations are performed to complement experimental investigations. At the core of such simulations lies the interatomic potential, on which all subsequent results hinge. In this work we review 19 central force many-body potentials and benchmark their performance against experiments and density functional theory (DFT) calculations. As basic features we consider the relative lattice stability, elastic constants and point-defect properties. In addition, we also investigate extended lattice defects, namely: free surfaces, symmetric tilt grain boundaries, the 1/2〈1 1 1〉{1 1 0} and 1/2〈1 1 1〉 {1 1 2} stacking fault energy profiles and the 1/2〈1 1 1〉 screw dislocation core. We also provide the Peierls stress for the 1/2〈1 1 1〉 edge and screw dislocations as well as the glide path of the latter at zero Kelvin. The presented results serve as an initial guide and reference list for both the modelling of atomically-driven phenomena in bcc tungsten, and the further development of its potentials. read less NOT USED (high confidence) P. Xiao, D. Sheppard, J. Rogal, and G. Henkelman, “Solid-state dimer method for calculating solid-solid phase transitions.,” The Journal of chemical physics. 2014. link Times cited: 95 Abstract: The dimer method is a minimum mode following algorithm for f… read moreAbstract: The dimer method is a minimum mode following algorithm for finding saddle points on a potential energy surface of atomic systems. Here, the dimer method is extended to include the cell degrees of freedom for periodic solid-state systems. Using this method, reaction pathways of solid-solid phase transitions can be determined without having to specify the final state structure or reaction mechanism. Example calculations include concerted phase transitions between CdSe polymorphs and a nucleation and growth mechanism for the A15 to BCC transition in Mo. read less NOT USED (high confidence) K. Akmaldinov et al., “Mixing antiferromagnets to tune NiFe-[IrMn/FeMn] interfacial spin-glasses, grains thermal stability, and related exchange bias properties,” Journal of Applied Physics. 2014. link Times cited: 10 Abstract: Spintronics devices and in particular thermally assisted mag… read moreAbstract: Spintronics devices and in particular thermally assisted magnetic random access memories require a wide range of ferromagnetic/antiferromagnetic (F/AF) exchange bias (EB) properties and subsequently of AF materials to fulfil diverse functionality requirements for the reference and storage. For the reference layer, large EB energies and high blocking temperature (TB) are required. In contrast, for the storage layer, mostly moderate TB are needed. One of the present issues is to find a storage layer with properties intermediate between those of IrMn and FeMn and in particular: (i) with a TB larger than FeMn for better stability at rest-T but lower than IrMn to reduce power consumption at write-T and (ii) with improved magnetic interfacial quality, i.e., with reduced interfacial glassy character for lower properties dispersions. To address this issue, the EB properties of F/AF based stacks were studied for various mixed [IrMn/FeMn] AFs. In addition to EB loop shifts, the F/AF magnetic interfacial qualities a... read less NOT USED (high confidence) A. Fraile, S. Cuesta-L’opez, A. Caro, D. Schwen, and J. Perlado, “Interatomic potential for the compound-forming Li-Pb liquid alloy,” Journal of Nuclear Materials. 2013. link Times cited: 13 NOT USED (high confidence) H. Diep, V. Bocchetti, D.-T. Hoang, and V. T. Ngo, “Theory and Simulation of Magnetic Materials: Physics at Phase Frontiers,” Journal of Physics: Conference Series. 2013. link Times cited: 4 Abstract: The combination of theory and simulation is necessary in the… read moreAbstract: The combination of theory and simulation is necessary in the investigation of properties of complex systems where each method alone cannot do the task properly. Theory needs simulation to test ideas and to check approximations. Simulation needs theory for modeling and for understanding results coming out from computers. In this review, we give recent examples to illustrate this necessary combination in a few domains of interest such as frustrated spin systems, surface magnetism, spin transport and melting. Frustrated spin systems have been intensively studied for more than 30 years. Surface effects in magnetic materials have been widely investigated also in the last three decades. These fields are closely related to each other and their spectacular development is due to numerous applications. We confine ourselves to theoretical developments and numerical simulations on these subjects with emphasis on spectacular effects occurring at frontiers of different phases. read less NOT USED (high confidence) P. Zhang and D. Trinkle, “Database optimization for empirical interatomic potential models,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 8 Abstract: Weighted least squares fitting to a database of quantum mech… read moreAbstract: Weighted least squares fitting to a database of quantum mechanical calculations can determine the optimal parameters of empirical potential models. While algorithms exist to provide optimal potential parameters for a given fitting database of structures with corresponding energy-related predictions and to estimate prediction errors using Bayesian sampling, defining an optimal fitting database based on potential predictions remains elusive. A testing set of structures and energy-related predictions provides an empirical measure of potential transferability. Here, we propose an objective function for fitting databases based on testing set errors. The objective function allows the optimization of the weights in a fitting database, the assessment of the adding or removing of structures in the fitting database, or the comparison of two different fitting databases. To showcase this technique, we consider an example Lennard-Jones potential for Ti, where modeling multiple complicated crystal structures is difficult for a radial pair potential. The algorithm finds different optimal fitting databases, depending on the objective function of potential prediction error for a testing set. read less NOT USED (high confidence) K. Akmaldinov, S. Auffret, I. Joumard, B. Dieny, and V. Baltz, “Benefit of inserting a (Cu/Pt) intermixing dual barrier for the blocking temperature distribution of exchange biased Co/(Cu/Pt)/IrMn stacks,” Applied Physics Letters. 2013. link Times cited: 16 Abstract: Exchange bias based spintronics devices involve ferromagneti… read moreAbstract: Exchange bias based spintronics devices involve ferromagnetic/antiferromagnetic interfaces and concomitant layers intermixing. As a consequence, interfacial spin-glass-like phases with reduced properties and increased dispersions form and lower the device performance. It is therefore necessary to limit intermixing by introduction of diffusion barriers. One of the major difficulties is that the barrier must be inert. This paper uses blocking temperature distributions to quantify the interfacial quality of Co/IrMn based stacks. Inserting a (Cu/Pt) dual barrier fulfils the manifold requirements of limiting Co-Mn, Co-Pt, and Cu-Mn intermixing, which takes place when using either no or single Pt and Cu barriers, respectively. read less NOT USED (high confidence) B. E. Gaddy, A. Kingon, and D. Irving, “Effects of alloying and local order in AuNi contacts for Ohmic radio frequency micro electro mechanical systems switches via multi-scale simulation,” Journal of Applied Physics. 2013. link Times cited: 3 Abstract: Ohmic RF-MEMS switches hold much promise for low power wirel… read moreAbstract: Ohmic RF-MEMS switches hold much promise for low power wireless communication, but long-term degradation currently plagues their reliable use. Failure in these devices occurs at the contact and is complicated by the fact that the same asperities that bear the mechanical load are also important to the flow of electrical current needed for signal processing. Materials selection holds the key to overcoming the barriers that prevent widespread use. Current efforts in materials selection have been based on the material's (or alloy's) ability to resist oxidation as well as its room-temperature properties, such as hardness and electrical conductivity. No ideal solution has yet been found via this route. This may be due, in part, to the fact that the in-use changes to the local environment of the asperity are not included in the selection criteria. For example, Joule heating would be expected to raise the local temperature of the asperity and impose a non-equilibrium thermal gradient in the same region expected t... read less NOT USED (high confidence) H. Wang, L. Zepeda-Ruiz, G. Gilmer, and M. Upmanyu, “Atomistics of vapour–liquid–solid nanowire growth,” Nature Communications. 2013. link Times cited: 89 NOT USED (high confidence) C. Sabater, M. Caturla, J. Palacios, and C. Untiedt, “Understanding the structure of the first atomic contact in gold,” Nanoscale Research Letters. 2013. link Times cited: 11 NOT USED (high confidence) Y. Shin, H. Kwak, C. Zou, A. Vasenkov, and A. V. van Duin, “Development and validation of a ReaxFF reactive force field for Fe/Al/Ni alloys: molecular dynamics study of elastic constants, diffusion, and segregation.,” The journal of physical chemistry. A. 2012. link Times cited: 59 Abstract: We have developed a ReaxFF force field for Fe/Al/Ni binary a… read moreAbstract: We have developed a ReaxFF force field for Fe/Al/Ni binary alloys based on quantum mechanical (QM) calculations. In addition to the various bulk phases of the binary alloys, the (100), (110) and (111) surface energies and adatom binding energies were included in the training set for the force field parametrization of the Fe/Al/Ni binary alloys. To validate these optimized force fields, we studied (i) elastic constants of the binary alloys at finite temperatures, (ii) diffusivity of alloy components in Al/Ni alloy, and (iii) segregation on the binary alloy surfaces. First, we calculated linear elastic constants of FeAl, FeNi(3), and Ni(3)Al in the temperature range 300 to 1100 K. The temperature dependences of the elastic constants of these three alloys, showing a decrease in C(11), C(12), and C(44) as temperature increases, were in good agreement with the experimental results. We also performed ReaxFF molecular dynamics (MD) simulations for Al or Ni diffusion in the system modeled as Al/Ni mixed layers with the linear composition gradients. At 1000 K, Al diffusivity at the pure Al end was 2 orders of magnitude larger than that in the Al trace layers, probably explaining the nature of different diffusion behavior between molten metals and alloys. However, the diffusivity of Ni at the pure Ni end was only slightly larger than that in the Ni trace layers at the system temperature much lower than the melting temperature of Ni. Third, we investigated the surface segregation in L1(2)-Fe(3)Al, Fe(3)Ni, and Ni(3)Al clusters at high temperature (2500 K). From the analysis of composition distribution of the alloy components from the bulk to the surface layer, it was found that the degree of segregation depended on the chemical composition of the alloy. Al surface segregation occurred most strongly in Fe(3)Al, whereas it occurred most weakly in Ni(3)Al. These results may support the segregation mechanism that surface segregation results from the interplay between the energetic stability of the ordered bulk phase and the surface reconstruction. In addition, the surface segregation induced the depletion layers of segregating metal species (Al in Fe(3)Al and Ni(3)Al, and Ni in Fe(3)Ni) next to the segregation layers. These simulation results qualitatively agreed with early experimental observations of segregation in Fe/Al/Ni binary alloys. read less NOT USED (high confidence) F. Letellier et al., “Effects of sputter-deposition-induced and post-deposition thermally activated intermixing on the exchange bias properties of [Pt/Co]×3/(Pt)/IrMn films,” Journal of Physics D: Applied Physics. 2012. link Times cited: 10 Abstract: The effects of sputter-deposition-induced and post-depositio… read moreAbstract: The effects of sputter-deposition-induced and post-deposition thermally activated layer intermixing on the exchange bias properties of [Pt/Co]x3/Pt(tPt)/IrMn films with out-of-plane anisotropy and deposited under a perpendicular magnetic field are investigated. The consequences of the intermixing on the magnetic properties are correlated with atom probe structural investigations. We observe that Co–Mn and Co–Pt intermixing are already present in the as-deposited state. The intermixing is more pronounced for Pt on Co (∼50%) than for Mn on Co (∼10%). It is observed that annealing up to 200 °C does not result in noticeable further diffusion of Mn in Co. In contrast, it significantly accentuates the Co–Pt initial intermixing. This enhanced intermixing leads to a 40% reduction in the exchange bias field. We measured that this reduction is not primarily due to changes in effective anisotropy or in saturation magnetization. This is possibly ascribed to a decrease in the mean interfacial moment and exchange stiffness both due to the observed reduction in Co content within the Co sublayers. read less NOT USED (high confidence) T. Lee, M. Baskes, S. Valone, and J. Doll, “Atomistic modeling of thermodynamic equilibrium and polymorphism of iron,” Journal of Physics: Condensed Matter. 2012. link Times cited: 52 Abstract: We develop two new modified embedded-atom method (MEAM) pote… read moreAbstract: We develop two new modified embedded-atom method (MEAM) potentials for elemental iron, intended to reproduce the experimental phase stability with respect to both temperature and pressure. These simple interatomic potentials are fitted to a wide variety of material properties of bcc iron in close agreement with experiments. Numerous defect properties of bcc iron and bulk properties of the two close-packed structures calculated with these models are in reasonable agreement with the available first-principles calculations and experiments. Performance at finite temperatures of these models has also been examined using Monte Carlo simulations. We attempt to reproduce the experimental iron polymorphism at finite temperature by means of free energy computations, similar to the procedure previously pursued by Müller et al (2007 J. Phys.: Condens. Matter 19 326220), and re-examine the adequacy of the conclusion drawn in the study by addressing two critical aspects missing in their analysis: (i) the stability of the hcp structure relative to the bcc and fcc structures and (ii) the compatibility between the temperature and pressure dependences of the phase stability. Using two MEAM potentials, we are able to represent all of the observed structural phase transitions in iron. We discuss that the correct reproductions of the phase stability among three crystal structures of iron with respect to both temperature and pressure are incompatible with each other due to the lack of magnetic effects in this class of empirical interatomic potential models. The MEAM potentials developed in this study correctly predict, in the bcc structure, the self-interstitial in the 〈110〉 orientation to be the most stable configuration, and the screw dislocation to have a non-degenerate core structure, in contrast to many embedded-atom method potentials for bcc iron in the literature. read less NOT USED (high confidence) M. Benoudia et al., “Thermoelasticity and interdiffusion in CuNi multilayers,” Physical Review B. 2012. link Times cited: 7 Abstract: X-ray scattering experiments on coherent CuNi multilayers ar… read moreAbstract: X-ray scattering experiments on coherent CuNi multilayers are performed at various annealing temperatures. First, we show that the classical thermoelasticity theory can be applied in such nanosamples to link composition and strain fields at intermediate temperature. Second, when interdiffusion takes place at higher temperature, the satellite peaks measured at different annealing times indicate the presence of a layer-by-layer interdiffusion mode controlled by the asymmetry of the atomic mobilities in this system. read less NOT USED (high confidence) J. Pang, M. Tan, and K. M. Liew, “Structural evolution of Ti50Cu50 on rapid cooling by molecular dynamics simulation,” Applied Physics A. 2012. link Times cited: 4 NOT USED (high confidence) A. Fraile, S. Cuesta-López, and J. Perlado, “Molecular Dynamics Simulations of Lead and Lithium in Liquid Phase,” Fusion Science and Technology. 2012. link Times cited: 3 Abstract: Pb17Li is today a reference breeder material in diverse fusi… read moreAbstract: Pb17Li is today a reference breeder material in diverse fusion R&D programs worldwide. Extracting dynamic and structural properties of liquid LiPb mixtures via molecular dynamics simulations, represent a crucial step for multiscale modeling efforts in order to understand the suitability of this compound for future Nuclear Fusion technologies. At present a Li-Pb cross potential is not available in the literature. Here we present our first results on the validation of two semi-empirical potentials for Li and Pb in liquid phase. Our results represent the establishment of a solid base as a previous but crucial step to implement a LiPb cross potential. Structural and thermodynamical analyses confirm that the implemented potentials for Li and Pb are realistic to simulate both elements in the liquid phase. read less NOT USED (high confidence) R. Deák, Z. Néda, and P. Barna, “A kinetic Monte Carlo approach for self-diffusion of Pt Atom Clusters on a Pt(111) Surface,” Communications in Computational Physics. 2011. link Times cited: 4 Abstract: A lattice Kinetic Monte Carlo (KMC) approach is considered t… read moreAbstract: A lattice Kinetic Monte Carlo (KMC) approach is considered to study the statistical properties of the diffusion of Pt atom clusters on a Pt(111) surface. The interatomic potential experienced by the diffusing atoms is calculated by the embedded atom method and the hopping barrier for the allowed atomic movements are calculated using the Nudged Elastic Band method. The diffusion coefficient is computed for various cluster sizes and system temperatures. The obtained results are in agreement with the ones obtained in previous experimental and theoretical works. A simple scaling argument is proposed for the size dependence of the diffusion coefficient’s prefactor. A detailed statistical analysis of the event by event KMC dynamics reveals two important and co-existing mechanisms for the diffusion of the cluster’s center of mass. At low temperatures (below T =400K) the dominating mechanism responsible for the displacement of the cluster’s center of mass is the periphery (or edge) diffusion of the atoms. At high temperatures (above T = 800K) the dissociation and recombination of the clusters becomes more and more important. PACS: 05.10.-a, 68.35.Fx, 68.43.Jk read less NOT USED (high confidence) A. Hannour, R. Lardé, M. Jean, J. Bran, P. Pareige, and J. L. Breton, “Atomic-scale investigation and magnetic properties of Cu80Co20 nanowires,” Journal of Applied Physics. 2011. link Times cited: 3 Abstract: Cu80Co20 granular alloy nanowires were synthesized by electr… read moreAbstract: Cu80Co20 granular alloy nanowires were synthesized by electrodeposition method and investigated by x-ray diffraction (XRD), Laser Assisted Wide Angle Tomographic Atom Probe (LAWATAP), and SQUID magnetometry. XRD results reveal the existence of a fcc Cu matrix and fcc Co-rich nanograins, with a preferred orientation along the [200] direction (perpendicular to the substrate surface). The Co-rich nanograins could be coherent with the Cu matrix. 3D reconstructions of a nano-sized volume, obtained by LAWATAP, reveal the heterogeneous aspect of the Cu80Co20 nanowires: Co-rich nanoclusters with size between 2 and 10 nm are detected, and the presence of Cu and Co oxides is evidenced. Magnetization measurements indicate that the Co-rich nanoclusters are superparamagnetic, with a blocking temperature that extends up to, at least, room temperature. The presence of ferromagnetic domains at room temperature indicates that some Co-rich nanoclusters are correlated within a volume that corresponds to a so-called interact... read less NOT USED (high confidence) B. Jelinek et al., “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Physical Review B. 2011. link Times cited: 218 Abstract: A set of modified embedded-atom method (MEAM) potentials for… read moreAbstract: A set of modified embedded-atom method (MEAM) potentials for the interactions between Al, Si, Mg, Cu, and Fe was developed from a combination of each element's MEAM potential in order to study metal alloying. Previously published MEAM parameters of single elements have been improved for better agreement to the generalized stacking fault energy (GSFE) curves when compared with ab initio generated GSFE curves. The MEAM parameters for element pairs were constructed based on the structural and elastic properties of element pairs in the NaCl reference structure garnered from ab initio calculations, with adjustment to reproduce the ab initio heat of formation of the most stable binary compounds. The new MEAM potentials were validated by comparing the formation energies of defects, equilibrium volumes, elastic moduli, and heat of formation for several binary compounds with ab initio simulations and experiments. Single elements in their ground-state crystal structure were subjected to heating to test the potentials at elevated temperatures. An Al potential was modified to avoid formation of an unphysical solid structure at high temperatures. The thermal expansion coefficient of a compound with the composition of AA 6061 alloy was evaluated and compared with experimental values. MEAM potential tests performed in this work, utilizing the universal atomistic simulation environment (ASE), are distributed to facilitate reproducibility of the results. read less NOT USED (high confidence) Z.-Y. Zeng, C.-E. Hu, L. Cai, X.-R. Chen, and F. Jing, “Molecular dynamics study of the melting curve of NiTi alloy under pressure,” Journal of Applied Physics. 2011. link Times cited: 19 Abstract: The melting curve of NiTi alloy was predicted by using molec… read moreAbstract: The melting curve of NiTi alloy was predicted by using molecular dynamics simulations combining with the embedded atom model potential. The calculated thermal equation of state consists well with our previous results obtained from quasiharmonic Debye approximation. Fitting the well-known Simon form to our Tm data yields the melting curves for NiTi: 1850(1 + P/21.938)0.328 (for one-phase method) and 1575(1 + P/7.476)0.305 (for two-phase method). The two-phase simulations can effectively eliminate the superheating in one-phase simulations. At 1 bar, the melting temperature of NiTi is 1575 ± 25 K and the corresponding melting slope is 64 K/GPa. read less NOT USED (high confidence) X.-J. Yuan, N. Chen, J. Shen, and W. Hu, “Embedded-atom-method interatomic potentials from lattice inversion,” Journal of Physics: Condensed Matter. 2010. link Times cited: 26 Abstract: The present work develops a physically reliable procedure fo… read moreAbstract: The present work develops a physically reliable procedure for building the embedded-atom-method (EAM) interatomic potentials for the metals with fcc, bcc and hcp structures. This is mainly based on Chen–Möbius lattice inversion (Chen et al 1997 Phys. Rev. E 55 R5) and first-principles calculations. Following Baskes (Baskes et al 2007 Phys. Rev. B 75 094113), this new version of the EAM eliminates all of the prior arbitrary choices in the determination of the atomic electron density and pair potential functions. Parameterizing the universal form deduced from the calculations within the density-functional scheme for homogeneous electron gas as the embedding function, the new-type EAM potentials for Cu, Fe and Ti metals have successfully been constructed by considering interatomic interactions up to the fifth neighbor, the third neighbor and the seventh neighbor, respectively. The predictions of elastic constants, structural energy difference, vacancy formation energy and migration energy, activation energy of vacancy diffusion, latent heat of melting and relative volume change on melting all satisfactorily agree with the experimental results available or first-principles calculations. The predicted surface energies for low-index crystal faces and the melting point are in agreement with the experimental data to the same extent as those calculated by other EAM-type potentials such as the FBD-EAM, 2NN MEAM and MS-EAM. In addition, the order among the predicted low-index surface energies is also consistent with the experimental information. read less NOT USED (high confidence) Y. Liu, G. Jia, and B. Yang, “Molecular dynamics simulation on diffusion properties of Pb-Mg alloy,” Science China Technological Sciences. 2010. link Times cited: 8 NOT USED (high confidence) A. A. Vostryakov, N. I. Sidorov, E. Pastukhov, and V. Lisin, “Influence of the electric field strength and hydrogen on the rate of removal of iron atom impurities in refining remelting of tantalum,” Russian Metallurgy (Metally). 2010. link Times cited: 0 NOT USED (high confidence) G. Jia, Y. Liu, B. Yang, and D. Liu, “Molecular dynamics simulation on thermodynamic properties of Pb-Ag alloys,” Rare Metals. 2010. link Times cited: 5 NOT USED (high confidence) G. Li, Q. Wang, T. Liu, K. Wang, and J. He, “Molecular Dynamics Simulation of the Melting and Coalescence in the Mixed Cu–Ni Nanoclusters,” Journal of Cluster Science. 2010. link Times cited: 16 NOT USED (high confidence) S. Hara, S. Izumi, and S. Sakai, “Reaction pathway analysis for dislocation nucleation from a Ni surface step,” Journal of Applied Physics. 2009. link Times cited: 19 Abstract: Threshold strain required for a thermally activated dislocat… read moreAbstract: Threshold strain required for a thermally activated dislocation nucleation from a Ni surface step has been measured using an atomistic-based reaction pathway analysis. We show that the saddle-point configuration and the stress-dependent activation energy are strongly influenced by the presence of a surface step. Our results provide insight into the previous experimental findings concerning the mechanism on a coherency loss at the Ni∕Cu(001) interface. We conclude that the coherency strain caused by a lattice mismatch between Ni and Cu does not yield a sufficient driving force for the dislocation nucleation. read less NOT USED (high confidence) S. Sankaranarayanan, E. Kaxiras, and S. Ramanathan, “Electric field tuning of oxygen stoichiometry at oxide surfaces: molecular dynamics simulations studies of zirconia,” Energy and Environmental Science. 2009. link Times cited: 29 Abstract: Ultra-thin metal-oxides such as zirconia have tremendous tec… read moreAbstract: Ultra-thin metal-oxides such as zirconia have tremendous technological applications such as electrolyte membranes for advanced solid oxide fuel cells, fuel cladding material for light water nuclear reactors, pressure tube materials for heavy water nuclear reactors and corrosion resistant coatings. Oxide non-stoichiometry is an important factor which significantly affects their functional properties and applicability. Here, we report on the ability to athermally control oxygen non-stoichiometry in ultra-thin zirconia films through local electric field perturbations from simulations. Variable charge molecular dynamics simulations indicate significantly enhanced oxidation kinetics on Zr (0001) substrate in the presence of an electric field. Natural oxidation with no field resulted in an amorphous oxide scale with a self limiting thickness of ∼10 A which increased to ∼17–26 A for applied electric fields of 1–10 MV/cm. Electric field (∼107 V/cm) lowers the activation energy barrier for ionic migration through the oxide film and leads to significantly increased oxygen incorporation into the oxide film. Activation energy barrier for oxidation decreased from 1.13 eV with no field to 0.08 eV for an applied field of 10 MV/cm. This manifests itself in the form of dramatic density and stoichiometry improvements of the grown ultra-thin oxide film, as indicated by the calculated structural and dynamical correlation functions. Oxide stoichiometry (O/Zr ratio) for natural oxidation was 1.42 indicative of a sub-stoichiometric and oxygen deficient oxide which increased to near stoichiometric value of 1.86 for 10 MV/cm field assisted oxidation. The simulation findings agree well with previously reported experimental observations. Our results demonstrate a pathway to athermally control oxygen concentration in near-surface regions that is of great importance to technologies utilizing ultra-thin oxides ranging from catalysis, energy and electronic device technologies. read less NOT USED (high confidence) L. Hale, X. W. Zhou, J. Zimmerman, N. Moody, R. Ballarini, and W. Gerberich, “Molecular dynamics simulation of delamination of a stiff, body-centered-cubic crystalline film from a compliant Si substrate,” Journal of Applied Physics. 2009. link Times cited: 7 Abstract: Compliant substrate technology offers an effective approach … read moreAbstract: Compliant substrate technology offers an effective approach to grow high-quality multilayered films, of importance to microelectronics and microelectromechanical systems devices. By using a thin, soft substrate to relieve the mismatch strain of an epitaxial film, the critical thickness of misfit dislocation formation in the overlayer is effectively increased. Experiments have indicated that stiff films deposited onto Si substrates can delaminate at the interface. However, the atomic mechanisms of the deformation and the fracture of the films have not been well studied. Here, we have applied molecular dynamics simulations to study the delamination of a stiff body-centered-cubic crystalline film from a compliant Si substrate due to tensile loading. The observed mechanical behavior is shown to be relatively independent of small changes in temperature, loading rate, and system size. Fracture occurs at the interface between the two materials resulting in nearly atomically clean surfaces. Dislocations are seen ... read less NOT USED (high confidence) D. Larson, A. Cerezo, J. Juraszek, K. Hono, and G. Schmitz, “Atom-Probe Tomographic Studies of Thin Films and Multilayers,” MRS Bulletin. 2009. link Times cited: 28 Abstract: This article reviews investigations of the growth and reacti… read moreAbstract: This article reviews investigations of the growth and reactions within thin metal and oxide films using atom-probe tomography. Included in this review are (1) studies of interfacial and growth reactions in magnetoresistive metallic, metal/oxide, and magnetic magnetostrictive multilayers; (2) comparison of selected portions of these results to simulated film growth using molecular dynamics; and (3) study of the origin of room-temperature ferromagnetism in dilute magnetic semiconductors. Information of this type is useful in order to understand the formation and thermal evolution of thin films (and to compare to theory and modeling) and, ultimately, to permit further optimization of devices based on thin films. read less NOT USED (high confidence) H. Miura, K. Suzuki, H. Ito, and T. Inoue, “Creep and Fatigue Damages of Ni-Base-Superalloy Caused by Strain-Induced Anisotropic Diffusion of Component Elements,” Key Engineering Materials. 2009. link Times cited: 2 Abstract: The mechanism of the directional coarsening of ' phase… read moreAbstract: The mechanism of the directional coarsening of ' phases (rafting) of Ni-base superalloy under an uni-axial strain was analyzed by molecular dynamics (MD) analysis. The stress-induced anisotropic diffusion of Al atoms perpendicular to the interface was observed clearly in a Ni(001)/Ni3Al(001) interface structure, The reduction of the diffusion of Al atoms perpendicular to the interface is thus, effective for improving the creep and fatigue resistance of the alloy. It was also found that the dopant elements in the superalloy also affected the strain-induced diffusion of Al atoms. Pd was one of the most effective elements which restrain Al atoms from moving around the interface. read less NOT USED (high confidence) A. Dongare, L. Zhigilei, A. Rajendran, and B. Lamattina, “Interatomic potentials for atomic scale modeling of metal–matrix ceramic particle reinforced nanocomposites,” Composites Part B-engineering. 2009. link Times cited: 15 NOT USED (high confidence) B. Shan et al., “First-principles-based embedded atom method for PdAu nanoparticles,” Physical Review B. 2009. link Times cited: 48 Abstract: One of the key problems in studying alloy nanoparticle catal… read moreAbstract: One of the key problems in studying alloy nanoparticle catalysis is their surface morphology and segregation behavior. We have developed an accurate embedded atom method (EAM) potential and employed it in the simulation of PdAu metal alloy nanoparticles. The potential was parameterized based on an extensive set of density-functional-theory (DFT) calculations of metal clusters in addition to bulk-alloy properties. The EAM potential accurately reproduces DFT energies of both bulk PdAu alloys and small nanoparticles. We utilized the developed EAM potential in a Monte Carlo simulation of PdAu nanoparticles ranging from 55-atom $(\ensuremath{\sim}1\text{ }\text{nm})$ to 5083-atom particles $(\ensuremath{\sim}4.5\text{ }\text{nm})$. The effects of different factors (particle size, temperature, and composition ratios) on the segregation behavior of PdAu alloy are examined. Our simulation results quantitatively reveal the extent of surface segregation and a strong dependence of surface morphology on the nanoparticle size. read less NOT USED (high confidence) W. Chen, D. Nam, J. Lu, K. West, and S. Wolf, “Effects of target bias voltage in magnetic tunnel junctions grown by ion beam deposition,” Journal of Applied Physics. 2009. link Times cited: 7 Abstract: Magnetic tunnel junctions (MTJs) with an AlOx barrier were f… read moreAbstract: Magnetic tunnel junctions (MTJs) with an AlOx barrier were fabricated by a biased target ion beam deposition (BTIBD) sputtering technique using a low energy ion source (0–50 eV) and voltage biased targets. The BTIBD system applies a bias voltage directly onto the desired targets, providing enough sputtering energy and avoiding overspill contamination during film deposition. The successful deposition of AlOx-MTJs demonstrated the capability of the BTIBD to make multilayer structures with good film quality. MTJ thin film surface roughness and intermixing between layers are among the key problems leading to low tunneling magnetoresistance (TMR) performance. Here we study the effects of bias voltage on MTJ properties via the measurements of the Neel coupling field and TMR. We suggest that a lower bias voltage reduces the intermixing that occurs when a top CoFe free layer is deposited on an AlOx barrier, but produces relatively high surface roughness. On the other hand, higher energy deposition enhances both i... read less NOT USED (high confidence) C. M. Behrens and A. Armaou, “Optimal design and operation of a multiscale GaAs/AlAs deposition process,” 2009 American Control Conference. 2009. link Times cited: 0 Abstract: We consider optimal operating conditions of a gallium arseni… read moreAbstract: We consider optimal operating conditions of a gallium arsenide/aluminum arsenide (GaAs/AlAs) deposition process with objectives of uniform deposition of the film heterostructure across the wafer surface at the macroscopic scale and the interfacial uniformity of the GaAs/AlAs heterostructure at the microscopic scale. We use a finite element solver to determine macroscale solutions and kinetic Monte Carlo (kMC) techniques to determine the mesoscale solution of the problem.We characterize the interfaces between species and apply the simulation methodology to a multiscale optimization framework to minimize the interfacial step densities while also minimizing temperature, annealing time, and maximizing thickness uniformity. The design variables are temperature, annealing time, precursor concentration, and input velocity. In order to reduce the prohibitive computational expense of the function evaluations, we employ an in situ adaptive tabulation scheme around the mesoscale inputs. The resulting optimization problem combined with this methodology becomes computationally tractable, and is able to increase the thickness uniformity and maintain low interfacial step densit read less NOT USED (high confidence) L. Lechevallier et al., “Structural analysis and magnetic properties of (Pt/Co) 3 /Pt tPt /IrMn multilayers,” Physical Review B. 2009. link Times cited: 20 Abstract: Structural and magnetic investigations of ${\text{Ta}}_{3\te… read moreAbstract: Structural and magnetic investigations of ${\text{Ta}}_{3\text{ }\text{nm}}/{[{({\text{Pt}}_{2\text{ }\text{nm}}/{\text{Co}}_{0.4\text{ }\text{nm}})}_{3}/{\text{Pt}}_{t\text{Pt}}/{\text{IrMn}}_{7\text{ }\text{nm}}]}_{7}/{\text{Pt}}_{10\text{ }\text{nm}}$ multilayers with ${t}_{\text{Pt}}=0$ and ${t}_{\text{Pt}}=0.4\text{ }\text{nm}$ have been carried out using x-ray reflectometry, tomographic atom probe, and superconducting quantum interference device magnetometry. The structural investigations show that the Co/IrMn interface is more diffuse in the absence of the Pt spacer. The consequences on the magnetic properties are discussed. The exchange-bias field and the anisotropy direction of these two specimens are analyzed in comparison to ${\text{Ta}}_{3\text{ }\text{nm}}/[{({\text{Pt}}_{2\text{ }\text{nm}}/{\text{Co}}_{0.4\text{ }\text{nm}})}_{3}/{\text{Pt}}_{t\text{Pt}}/{\text{IrMn}}_{7\text{ }\text{nm}}]/{\text{Pt}}_{10\text{ }\text{nm}}$ multilayers containing only one ferromagnetic/antiferromagnetic repeat and correlated with the structural investigations. read less NOT USED (high confidence) R. Lardé et al., “Structural analysis of a (Pt/Co)3/IrMn multilayer: Investigation of sub-nanometric layers by tomographic atom probe,” Journal of Applied Physics. 2009. link Times cited: 25 Abstract: The structure of a Ta3 nm/[(Pt2 nm/Co0.4 nm)3/IrMn7 nm]7/Pt1… read moreAbstract: The structure of a Ta3 nm/[(Pt2 nm/Co0.4 nm)3/IrMn7 nm]7/Pt10 nm multilayer exhibiting perpendicular exchange bias has been investigated by x-ray reflectometry and laser-assisted tomographic atom probe (LATAP). A strong intermixing at the Co/IrMn interface is pointed out by x-ray reflectometry, this interface being more diffuse than the IrMn/Pt interface. A direct observation of this intermixing at the atomic scale is obtained thanks to the LATAP in real space. The three-dimensional reconstructions reveal the atomic planes in the Pt layers and the Pt–Co intermixing in the (Pt/Co)3 multilayer. The analysis of the concentration profiles allows to determine the chemical composition of the Co subnanometric layers; thus providing for the first time an accurate structural characterization of such layers leading to an estimation of their thickness, roughness, atomic concentration and width of their interfaces. read less NOT USED (high confidence) S. Sankaranarayanan and S. Ramanathan, “On the Low-Temperature Oxidation and Ultrathin Oxide Growth on Zirconium in the Presence of Atomic Oxygen: A Modeling Study,” Journal of Physical Chemistry C. 2008. link Times cited: 22 Abstract: Variable charge molecular dynamics simulations are used to i… read moreAbstract: Variable charge molecular dynamics simulations are used to investigate oxidation kinetics and nanoscale oxide growth on Zr(0001) surfaces due to atomic and molecular oxygen. Oxidation kinetics using atomic oxygen were significantly enhanced and resulted in an oxide scale with self-limiting thickness of ∼20−22 A in the simulated low-temperature range of 300−600 K. This represented a 2-fold increase over molecular oxygen. Enhanced oxidation kinetics resulted from lowering of activation energy barrier by 0.46 eV in the case of atomic oxygen. Structural and dynamic correlations indicate that the oxide growth is primarily inward in both cases with oxygen diffusivities being ∼88% higher for oxidation in atomic oxygen. Our analysis indicated the formation of an oxide film with O/Zr ratio varying from 1.74 to 1.92 in the 300−600 K range in the presence of atomic oxygen, whereas that formed by natural oxidation was substoichiometric and oxygen deficient with O/Zr values varying from 1.42 to 1.57 in the 300−600 K t... read less NOT USED (high confidence) Z. Lin, R. A. Johnson, and L. Zhigilei, “Computational study of the generation of crystal defects in a bcc metal target irradiated by short laser pulses,” Physical Review B. 2008. link Times cited: 93 Abstract: The generation of crystal defects in a Cr target irradiated … read moreAbstract: The generation of crystal defects in a Cr target irradiated by a short, 200 fs, laser pulse is investigated in computer simulations performed with a computational model that combines the classical molecular dynamics method with a continuum description of the laser excitation of conduction band electrons, electron-phonon coupling, and electron heat conduction. Interatomic interactions are described by the embedded atom method EAM potential with a parametrization designed for Cr. The potential is tested by comparing the properties of the EAM Cr material with experimental data and predictions of density functional theory calculations. The simulations are performed at laser fluences close to the threshold for surface melting. Fast temperature variation and strong thermoelastic stresses produced by the laser pulse are causing surface melting and epitaxial resolidification, transient appearance of a high density of stacking faults along the 110 planes, and generation of a large number of point defects vacancies and self-interstitials. The stacking faults appear as a result of internal shifts in the crystal undergoing a rapid uniaxial expansion in the direction normal to the irradiated surface. The stacking faults are unstable and disappear shortly after the laser-induced tensile stress wave leaves the surface region of the target. Thermally activated generation of vacancy-interstitial pairs during the initial temperature spike and quick escape of highly mobile self-interstitials to the melting front or the free surface of the target, along with the formation of vacancies at the solid-liquid interface during the fast resolidification process, result in a high density of vacancies, on the order of 10 3 per lattice site, created in the surface region of the target. The strong supersaturation of vacancies can be related to the incubation effect in multipulse laser ablation/damage and should play an important role in mixing/alloying of multicomponent or composite targets. read less NOT USED (high confidence) C. Engin, L. Sandoval, and H. Urbassek, “Characterization of Fe potentials with respect to the stability of the bcc and fcc phase,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 64 Abstract: By calculating free energies, several published interatomic … read moreAbstract: By calculating free energies, several published interatomic interaction potentials for iron are investigated with respect to the stability of the low-temperature bcc phase and the high-temperature fcc phase. These are empirical many-body potentials for use in atomistic simulation. We find that in all of these potentials—except one—the bcc phase is the stable crystal structure for all temperatures up to the melting point. However, several potentials exhibit a metastable fcc phase in the sense that the fcc structure corresponds to a local minimum of the free energy. read less NOT USED (high confidence) X. W. Zhou, J. Zimmerman, B. M. Wong, and J. Hoyt, “An embedded-atom method interatomic potential for Pd–H alloys,” Journal of Materials Research. 2008. link Times cited: 84 Abstract: Palladium hydrides have important applications. However, the… read moreAbstract: Palladium hydrides have important applications. However, the complex Pd–H alloy system presents a formidable challenge to developing accurate computational models. In particular, the separation of a Pd–H system to dilute (α) and concentrated (β) phases is a central phenomenon, but the capability of interatomic potentials to display this phase miscibility gap has been lacking. We have extended an existing palladium embedded-atom method potential to construct a new Pd–H embedded-atom method potential by normalizing the elemental embedding energy and electron density functions. The developed Pd–H potential reasonably well predicts the lattice constants, cohesive energies, and elastic constants for palladium, hydrogen, and PdH_x phases with a variety of compositions. It ensures the correct hydrogen interstitial sites within the hydrides and predicts the phase miscibility gap. Preliminary molecular dynamics simulations using this potential show the correct phase stability, hydrogen diffusion mechanism, and mechanical response of the Pd–H system. read less NOT USED (high confidence) S. P. Kim, S.-C. Lee, K.-R. Lee, and Y.-C. Chung, “Asymmetric surface intermixing during thin-film growth in the Co–Al system: Role of local acceleration of the deposited atoms,” Acta Materialia. 2008. link Times cited: 15 NOT USED (high confidence) E. Webb and X. W. Zhou, “Atomically engineering Cu/Ta interfaces.” 2007. link Times cited: 0 Abstract: This report summarizes the major research and development ac… read moreAbstract: This report summarizes the major research and development accomplishments for the late start LDRD project (investment area: Enable Predictive Simulation) entitled 'Atomically Engineering Cu/Ta Interfaces'. Two ultimate goals of the project are: (a) use atomistic simulation to explore important atomistic assembly mechanisms during growth of Cu/Ta multilayers; and (b) develop a non-continuum model that has sufficient fidelity and computational efficiency for use as a design tool. Chapters 2 and 3 are essentially two papers that address respectively these two goals. In chapter 2, molecular dynamics simulations were used to study the growth of Cu films on (010) bcc Ta and Cu{sub x}Ta{sub 1-x} alloy films on (111) fcc Cu. The results indicated that fcc crystalline Cu films with a (111) texture are always formed when Cu is grown on Ta. The Cu films are always polycrystalline even when the Ta substrate is single crystalline. These polycrystalline films are composed of grains with only two different orientations, which are separated by either orientational grain boundaries or misfit dislocations. Periodic misfit dislocations and stacking fault bands are observed. The Cu film surface roughness was found to decrease with increasing adatom energy. Due to a Cu surface segregation effect, the Cu{sub x}Ta{sub 1-x}more » films deposited on Cu always have a higher Cu composition than that used in the vapor mixture. When Cu and Ta compositions in the films are comparable, amorphous structures may form. The fundamental origins for all these phenomena have been studied in terms of crystallography and interatomic interactions. In chapter 3, a simplified computational method, diffusional Monte Carlo (dMC) method, was developed to address long time kinetic processes of materials. Long time kinetic processes usually involve material transport by diffusion. The corresponding microstructural evolution of materials can be analyzed by kinetic Monte Carlo simulation methods, which essentially simulate structural evolution by tracing each atomic jump. However, if the simulation is carried out at a high temperature, or a jump mechanism with a very low energy barrier is encountered, the jump frequency may approach the atom vibration frequency, and the computational efficiency of the kinetic Monte Carlo method rapidly decreases to that of a molecular dynamics simulation. The diffusional Monte Carlo method addresses the net effects of many atom jumps over a finite duration, kinetically controlled process. First, atom migration due to both random and non-random jumps is discussed. The concept of dMC is then introduced for random jump diffusion. The validity of the method is demonstrated using several diffusion cases in one-, two- and three-dimensional spaces, including the dissolution of spinodal structures. The application of the non-random diffusion theory to spinodal decomposition is also demonstrated.« less read less NOT USED (high confidence) M. Francis, M. Neurock, X. W. Zhou, J. Quan, H. Wadley, and E. Webb, “Atomic assembly of Cu/Ta multilayers: Surface roughness, grain structure, misfit dislocations, and amorphization,” Journal of Applied Physics. 2007. link Times cited: 23 Abstract: Molecular dynamics simulations and selected experiments have… read moreAbstract: Molecular dynamics simulations and selected experiments have been carried out to study the growth of Cu films on (010) bcc Ta and the deposition of CuxTa1−x alloy films on (111) fcc Cu. They indicate that fcc Cu films with a (111) texture are always formed when Cu is deposited on Ta surfaces. These films are polycrystalline even when the Ta substrate is single crystalline. The grains have one of two different orientations and are separated by either orientational or misfit dislocations. Periodic misfit dislocations and stacking faults develop within these grains to release structure difference induced misfit strain energy. The Cu film surface roughness was found to decrease with increase in the adatom energy for deposition. When CuxTa1−x is deposited on Ta, the films always have a higher Cu composition than that of the vapor mixture. This arises from a surface segregation phenomenon. When the Cu and Ta fractions in the films are comparable, amorphous structures form. The fundamental origins for the segreg... read less NOT USED (high confidence) X. W. Zhou, H. Wadley, and D. Wang, “Transient hole formation during the growth of thin metal oxide layers,” Computational Materials Science. 2007. link Times cited: 12 NOT USED (high confidence) H. Hwang et al., “Asymmetric intermixing in a Co–Al thin film system: An investigation using coaxial impact collision ion scattering spectroscopy,” Journal of Applied Physics. 2007. link Times cited: 4 Abstract: Surface structure evolution during atomic deposition in a Co… read moreAbstract: Surface structure evolution during atomic deposition in a Co–Al system was investigated using coaxial impact collision ion scattering spectroscopy (CAICISS). Half monolayer of Al and Co atoms were deposited on Co(0001) and Al(001) single crystal surfaces, respectively, in an ultrahigh-vacuum environment. CAICISS analysis of the deposited surface revealed an asymmetric interfacial reaction, as predicted by previous molecular dynamics simulations. Al atoms deposited on a Co substrate are placed on the surface with no interatomic intermixing. In contrast, significant surface intermixing with the deposited Co atoms occurs on the Al(001) substrate, resulting in the formation of a CoAl intermetallic surface layer of B2 structure. These asymmetric features would be important to the understanding of the structural evolution of thin film multilayers. read less NOT USED (high confidence) S. Chen, F. Ke, M. Zhou, and Y.-long Bai, “Atomistic investigation of the effects of temperature and surface roughness on diffusion bonding between Cu and Al,” Acta Materialia. 2007. link Times cited: 144 NOT USED (high confidence) J. Quan, S. Wolf, and H. Wadley, “Low energy ion beam assisted deposition of a spin valve,” Journal of Applied Physics. 2007. link Times cited: 16 Abstract: The spin dependent electron transport in giant magnetoresist… read moreAbstract: The spin dependent electron transport in giant magnetoresistive (GMR) multilayers is significantly affected by the atomic scale structure of their interfaces. Devices with atomically flat and chemically sharp interfaces are preferred for magnetic sensor and memory applications. Recent atomic simulations of the atom-by-atom assembly of these devices indicate that near optimal interfacial structures can be created using low energy, ion assisted vapor deposition techniques with ion energies in the 5–10eV range. A recently developed biased target ion beam deposition system has been used to experimentally test this hypothesis. Prototypical Ta∕NiFe∕Co∕Cu∕Co∕FeMn∕Cu spin valve structures were first grown using (simultaneous) argon ion assistance during deposition of the Co∕Cu∕Co trilayer part of the spin valve multilayer. Assisting ion energies of around 10eV resulted in structures with a 30% higher magnetoresistance ratio and significantly reduced coupling field compared to samples grown with no ion assistance ... read less NOT USED (high confidence) X. W. Zhou and H. Wadley, “A potential for simulating the atomic assembly of cubic elements,” Computational Materials Science. 2007. link Times cited: 10 NOT USED (high confidence) T. Kelly and M. K. Miller, “Invited review article: Atom probe tomography.,” The Review of scientific instruments. 2007. link Times cited: 725 Abstract: The technique of atom probe tomography (APT) is reviewed wit… read moreAbstract: The technique of atom probe tomography (APT) is reviewed with an emphasis on illustrating what is possible with the technique both now and in the future. APT delivers the highest spatial resolution (sub-0.3-nm) three-dimensional compositional information of any microscopy technique. Recently, APT has changed dramatically with new hardware configurations that greatly simplify the technique and improve the rate of data acquisition. In addition, new methods have been developed to fabricate suitable specimens from new classes of materials. Applications of APT have expanded from structural metals and alloys to thin multilayer films on planar substrates, dielectric films, semiconducting structures and devices, and ceramic materials. This trend toward a broader range of materials and applications is likely to continue. read less NOT USED (high confidence) X. W. Zhou et al., “Atomic Assembly of Thin Film Materials,” Materials Science Forum. 2007. link Times cited: 0 Abstract: The atomic-scale structures and properties of thin films are… read moreAbstract: The atomic-scale structures and properties of thin films are critically determined by the various kinetic processes activated during their atomic assembly. Molecular dynamics simulations of growth allow these kinetic processes to be realistically addressed at a timescale that is difficult to reach using ab initio calculations. The newest approaches have begun to enable the growth simulation to be applied for a wide range of materials. Embedded atom method potentials can be successfully used to simulate the growth of closely packed metal multilayers. Modified charge transfer ionic + embedded atom method potentials are transferable between metallic and ionic materials and have been used to simulate the growth of metal oxides on metals. New analytical bond order potentials are now enabling significantly improved molecular dynamics simulations of semiconductor growth. Selected simulations are used to demonstrate the insights that can be gained about growth processes at surfaces. read less NOT USED (high confidence) J. Quan, X. Zhou, and H. Wadley, “Atomic assembly of metal surfaces and interfaces,” Surface Science. 2006. link Times cited: 17 NOT USED (high confidence) D. Reis, K. Gaffney, G. Gilmer, and B. Torralva, “Ultrafast Dynamics of Laser-Excited Solids,” MRS Bulletin. 2006. link Times cited: 18 Abstract: We discuss recent experimental and theoretical results on ul… read moreAbstract: We discuss recent experimental and theoretical results on ultrafast materials dynamics. Intense, femtosecond lasers can deposit energy in a time that is short compared with relaxation processes and can generate extremely large carrier densities that drive bond softening, nonthermal melting, and ablation. In particular, we present optical experiments on electronic softening of coherent phonons in bismuth and x-ray experiments on ultrafast disordering in indium antimonide that probe the bonding of the lattice under successively higher carrier concentrations. We review a number of molecular dynamics simulations and their assumptions, which address nonthermal melting. Large-scale molecular dynamics simulations elucidate the role of void formation in laser ablation. read less NOT USED (high confidence) J. Quan, X. W. Zhou, and H. Wadley, “Low energy ion assisted atomic assembly of metallic superlattices,” Surface Science. 2006. link Times cited: 16 NOT USED (high confidence) Z. Lin and L. Zhigilei, “Time-resolved diffraction profiles and atomic dynamics in short-pulse laser-induced structural transformations: Molecular dynamics study,” Physical Review B. 2006. link Times cited: 93 Abstract: The diffraction profiles and density correlation functions a… read moreAbstract: The diffraction profiles and density correlation functions are calculated for transient atomic configurations generated in molecular dynamics simulations of a $20\phantom{\rule{0.3em}{0ex}}\mathrm{nm}$ Au film irradiated with $200\phantom{\rule{0.3em}{0ex}}\mathrm{fs}$ laser pulses of different intensity. The results of the calculations provide an opportunity to directly relate the detailed information on the atomic-level structural rearrangements available from the simulations to the diffraction spectra measured in time-resolved x-ray and electron diffraction experiments. Three processes are found to be responsible for the evolution of the diffraction profiles. During the first several picoseconds after the laser excitation, the decrease of the intensity of the diffraction peaks is largely due to the increasing amplitude of thermal atomic vibrations and can be well described by the Debye-Waller factor. The effect of thermoelastic deformation of the film prior to melting is reflected in shifts and splittings of the diffraction peaks, providing an opportunity for experimental probing of the ultrafast deformations. Finally, the onset of the melting process results in complete disappearance of the crystalline diffraction peaks. The homogeneous nucleation of a large number of liquid regions throughout the film is found to be more effective in reducing long-range correlations in atomic positions and diminishing the diffraction peaks as compared to the heterogeneous melting by melting front propagation. For the same fraction of atoms retaining the local crystalline environment, the diffraction peaks are more pronounced in heterogeneous melting. A detailed analysis of the real space correlations in atomic positions is also performed and the atomic-level picture behind the experimentally observed fast disappearance of the correlation peak corresponding to the second nearest neighbors in the fcc lattice during the laser heating and melting processes is revealed. read less NOT USED (high confidence) L. Péter et al., “Temperature dependence of giant magnetoresistance and magnetic properties in electrodeposited Co − Cu ∕ Cu multilayers: The role of superparamagnetic regions,” Physical Review B. 2006. link Times cited: 34 Abstract: We have shown recently that both the magnetization and the m… read moreAbstract: We have shown recently that both the magnetization and the magnetoresistance of electrodeposited $\mathrm{Co}\text{\ensuremath{-}}\mathrm{Cu}∕\mathrm{Cu}$ multilayers can be decomposed by assuming the presence of both ferromagnetic (FM) and superparamagnetic (SPM) regions in the magnetic layers. In the present work, for two selected samples, one with a large SPM and another one with a large FM contribution to the giant magnetoresistance, low temperature magnetic and magnetoresistance measurements were performed in order to reveal the evolution of the FM and SPM terms with temperature. The average apparent magnetic moment of the SPM regions deduced from the two sets of data showed a good agreement. The role of electrochemical processes in the formation of the SPM regions is discussed. An attempt has also been made to elaborate on some models for the spatial distribution of the constituent elements (Co and Cu) leading to the occurrence of SPM regions. The results are discussed also in the framework of interacting SPM regions. read less NOT USED (high confidence) P. F. Ladwig et al., “Intermixing and phase separation at the atomic scale in Co-rich (Co,Fe) and Cu multilayered nanostructures,” Applied Physics Letters. 2005. link Times cited: 10 Abstract: Despite the fact that Co-rich (Co,Fe) alloys and Cu are immi… read moreAbstract: Despite the fact that Co-rich (Co,Fe) alloys and Cu are immiscible materials in bulk form, evidence of thermally induced mixing at the atomic scale has been observed in thin-film multilayers of (Co,Fe) and Cu. However, long term anneals at lower temperatures produced a breakup of the multilayers into a two-phase mixture of (Co,Fe) and Cu particles. The observations were made with the use of the three-dimensional atom probe technique, with supporting evidence from differential scanning calorimetry and x-ray diffraction. Besides their scientific importance, these results are of interest where these (Co,Fe) and Cu thin films are used to produce the giant magnetoresistive effect. read less NOT USED (high confidence) X. W. Zhou, “Analytical and numerical calculations of interatomic forces and stresses,” Molecular Simulation. 2005. link Times cited: 0 Abstract: Atomistic simulation methods such as molecular dynamics requ… read moreAbstract: Atomistic simulation methods such as molecular dynamics require an efficient calculation of interatomic forces and stresses from pre–defined interatomic potentials. Both analytical and numerical approaches can be used to do this. Analytical approach directly calculates forces and stresses using analytical formulae, and can therefore yield accurate results. However, the force and stress expressions may become extremely complicated as the complexity level of the potential increases, resulting in a prolonged development cycle to implement new potentials. Numerical approach uses finite difference method to evaluate forces and stresses through simple calculation of energies at selected perturbations of crystal configurations. The method can be quickly implemented and tested for any potentials. However, it may result in significant numerical errors. We have compared analytical and numerical calculations of interatomic forces and stresses in molecular dynamics, and identified the conditions where numerical method can be successfully used without significant errors. read less NOT USED (high confidence) L. Zhigilei and D. Ivanov, “Channels of energy redistribution in short-pulse laser interactions with metal targets,” Applied Surface Science. 2005. link Times cited: 47 NOT USED (high confidence) X. W. Zhou, H. Wadley, and S. Sainathan, “Low energy sputtering of nickel by normally incident xenon ions,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 19 NOT USED (high confidence) X. Zhou and H. Wadley, “A charge transfer ionic–embedded atom method potential for the O–Al–Ni–Co–Fe system,” Journal of Physics: Condensed Matter. 2005. link Times cited: 47 Abstract: Magnetic tunnel junctions (MTJs) require the growth of a thi… read moreAbstract: Magnetic tunnel junctions (MTJs) require the growth of a thin (∼20 Å) dielectric metal oxide layer, such as Al2O3, on a ferromagnetic metal layer, such as Co, CoFe, or CoNiFe. The atomic assembly mechanisms that combine to form a uniformly thin metal oxide layer on these metal surfaces are not well understood. The application of molecular dynamics simulations to the growth of metal and metal oxide multilayers that involve more than one metal element has not been possible using the conventional interatomic potentials. A recently proposed modified charge transfer ionic–embedded atom method potential appears to correctly enable the charge transfer between oxygen and numerous metal elements to be modelled in a format amenable for molecular dynamics studies. Here we parametrize this charge transfer ionic–embedded atom method potential for the quinternary O–Al–Ni–Co–Fe system so that a direct molecular dynamics simulation of the growth of the tunnelling magnetoresistive multilayers can be realized. read less NOT USED (high confidence) X. W. Zhou and H. Wadley, “Atomistic simulation of AlOx magnetic tunnel junction growth,” Physical Review B. 2005. link Times cited: 29 Abstract: The tunneling magnetoresistance sTMRd of thin dielectric tun… read moreAbstract: The tunneling magnetoresistance sTMRd of thin dielectric tunnel barriers that are sandwiched between pairs of ferromagnetic metal thin films is highly sensitive to the barrier layer atomic scale thickness, the uniformity in thickness, and the composition. Widely used AlO x barriers are formed by the oxidation of 1–2 nm thick aluminum layers vapor deposited onto one of the ferromagnetic metal electrodes. The device is completed by vapor depositing the second ferromagnetic layer upon the oxide. Efforts to increase the TMR and tunneling conductance by reducing the thickness of the barrier have been successful until the aluminum layer thickness is decreased below,1 nm whereupon the TMR disappears. The TMR loss is thought to occur because the oxide layer becomes discontinuous leading to regions of metal contact across the barrier layer in the completed device. Using a molecular dynamics simulation technique combined with a recently developed charge transfer potential for metal alloy oxides, we have investigated the atomistic scale phenomena responsible for the disruption of the oxide film’s continuity. We show that discontinuous oxides always form during the oxidation of ,0.6 nm thick crystalline aluminum films ons111d Ni65Co20Fe15 single-crystal layers even when the precursor aluminum layer is continuous and of uniform thickness. The discontinuous mechanism of oxidation is shown to result from a surface-tension-driven dewetting as aluminum is converted to an amorphous oxide. The phenomenon establishes a lower limit of about 1 nm for the thickness of an aluminum oxide tunnel barrier fabricated by oxidation ons111d single-crystal Ni 65Co20Fe15 surfaces. read less NOT USED (high confidence) D. Ivanov and L. Zhigilei, “Combined atomistic-continuum model for simulation of laser interaction with metals: application in the calculation of melting thresholds in Ni targets of varying thickness,” Applied Physics A. 2004. link Times cited: 54 NOT USED (high confidence) X. W. Zhou, R. Johnson, and H. Wadley, “Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers,” Physical Review B. 2004. link Times cited: 936 Abstract: Recent molecular dynamics simulations of the growth of $[{\m… read moreAbstract: Recent molecular dynamics simulations of the growth of $[{\mathrm{Ni}}_{0.8}{\mathrm{Fe}}_{0.2}/\mathrm{Au}]$ multilayers have revealed the formation of misfit-strain-reducing dislocation structures very similar to those observed experimentally. Here we report similar simulations showing the formation of edge dislocations near the interfaces of vapor-deposited (111) [NiFe/CoFe/Cu] multilayers. Unlike misfit dislocations that accommodate lattice mismatch, the dislocation structures observed here increase the mismatch strain energy. Stop-action observations of the dynamically evolving atomic structures indicate that during deposition on the (111) surface of a fcc lattice, adatoms may occupy either fcc sites or hcp sites. This results in the random formation of fcc and hcp domains, with dislocations at the domain boundaries. These dislocations enable atoms to undergo a shift from fcc to hcp sites, or vice versa. These shifts lead to missing atoms, and therefore a later deposited layer can have missing planes compared to a previously deposited layer. This dislocation formation mechanism can create tensile stress in fcc films. The probability that such dislocations are formed was found to quickly diminish under energetic deposition conditions. read less NOT USED (high confidence) X. W. Zhou and H. Wadley, “Misfit dislocations in gold/Permalloy multilayers,” Philosophical Magazine. 2004. link Times cited: 16 Abstract: Several groups have reported the misfit dislocation structur… read moreAbstract: Several groups have reported the misfit dislocation structures in Au/Ni0.8Fe0.2 multilayers where the lattice parameter misfit is very large. To explore the factors controlling such structures, molecular dynamics simulations have been used to simulate the vapour-phase growth of (111)-oriented Au/Ni0.8Fe0.2 multilayers. The simulations revealed the formation of misfit dislocations at both the gold-on-Ni0.8Fe0.2 and the Ni0.8Fe0.2-on-gold interfaces. The dislocation configuration and density were found to be in good agreement with previously reported high-resolution transmission electron microscopy observations. Additional atomic-scale simulations of a model nickel–gold system indicated that dislocations are nucleated as the first nickel layer is deposited on gold. These dislocations have an (a/6)⟨112⟩ Burgers vector, typical of a Shockley partial dislocation. Each dislocation creates an extra {220} plane in the smaller lattice parameter nickel layer. These misfit-type dislocations effectively relieve misfit strain. The results also indicated that the dislocation structure is insensitive to the energy of the depositing atoms. Manipulation of the deposition processes is therefore unlikely to reduce this component of the defect population. read less NOT USED (high confidence) D. Ivanov and L. Zhigilei, “Combined atomistic-continuum modeling of short-pulse laser melting and disintegration of metal films,” Physical Review B. 2003. link Times cited: 616 Abstract: The kinetics and microscopic mechanisms of laser melting and… read moreAbstract: The kinetics and microscopic mechanisms of laser melting and disintegration of thin Ni and Au films irradiated by a short, from 200 fs to 150 ps, laser pulse are investigated in a coupled atomistic-continuum computational model. The model provides a detailed atomic-level description of fast nonequilibrium processes of laser melting and film disintegration and, at the same time, ensures an adequate description of the laser light absorption by the conduction band electrons, the energy transfer to the lattice due to the electron-phonon coupling, and the fast electron heat conduction in metals. The interplay of two competing processes, the propagation of the liquid-crystal interfaces (melting fronts) from the external surfaces of the film and homogeneous nucleation and growth of liquid regions inside the crystal, is found to be responsible for melting of metal films irradiated by laser pulses at fluences close to the melting threshold. The relative contributions of the homogeneous and heterogeneous melting mechanisms are defined by the laser fluence, pulse duration, and the strength of the electron-phonon coupling. At high laser fluences, significantly exceeding the threshold for the melting onset, a collapse of the crystal structure overheated above the limit of crystal stability takes place simultaneously in the whole overheated region within \ensuremath{\sim}2 ps, skipping the intermediate liquid-crystal coexistence stage. Under conditions of the inertial stress confinement, realized in the case of short $\ensuremath{\tau}l~10\mathrm{ps}$ laser pulses and strong electron-phonon coupling (Ni films), the dynamics of the relaxation of the laser-induced pressure has a profound effect on the temperature distribution in the irradiated films as well as on both homogeneous and heterogeneous melting processes. Anisotropic lattice distortions and stress gradients associated with the relaxation of the laser-induced pressure destabilize the crystal lattice, reduce the overheating required for the initiation of homogeneous melting down to $T\ensuremath{\approx}{1.05T}_{m},$ and expand the range of pulse durations for which homogeneous melting is observed in 50 nm Ni films up to \ensuremath{\sim}150 ps. High tensile stresses generated in the middle of an irradiated film can also lead to the mechanical disintegration of the film. read less NOT USED (high confidence) S. Takamoto, S. Izumi, T. Nakata, S. Sakai, S. Oinuma, and Y. Nakatani, “Analytical method for estimating the thermal expansion coefficient of metals at high temperature,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 2 Abstract: In this paper, we propose an analytical method for estimatin… read moreAbstract: In this paper, we propose an analytical method for estimating the thermal expansion coefficient (TEC) of metals at high-temperature ranges. Although the conventional method based on quasiharmonic approximation (QHA) shows good results at low temperatures, anharmonic effects caused by large-amplitude thermal vibrations reduces its accuracy at high temperatures. Molecular dynamics (MD) naturally includes the anharmonic effect. However, since the computational cost of MD is relatively high, in order to make an interatomic potential capable of reproducing TEC, an analytical method is essential. In our method, analytical formulation of the radial distribution function (RDF) at finite temperature realizes the estimation of the TEC. Each peak of the RDF is approximated by the Gaussian distribution. The average and variance of the Gaussian distribution are formulated by decomposing the fluctuation of interatomic distance into independent elastic waves. We incorporated two significant anharmonic effects into the method. One is the increase in the averaged interatomic distance caused by large amplitude vibration. The second is the variation in the frequency of elastic waves. As a result, the TECs of fcc and bcc crystals estimated by our method show good agreement with those of MD. Our method enables us to make an interatomic potential that reproduces the TEC at high temperature. We developed the GEAM potential for nickel. The TEC of the fitted potential showed good agreement with experimental data from room temperature to 1000 K. As compared with the original potential, it was found that the third derivative of the wide-range curve was modified, while the zeroth, first and second derivatives were unchanged. This result supports the conventional theory of solid state physics. We believe our analytical method and developed interatomic potential will contribute to future high-temperature material development. read less NOT USED (high confidence) M. H. Ulz, K. Mandadapu, and P. Papadopoulos, “On the estimation of spatial averaging volume for determining stress using atomistic methods,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 16 Abstract: The estimation of stress at a continuum point from the atomi… read moreAbstract: The estimation of stress at a continuum point from the atomistic scale requires volume averaging over a region that contains this point. A hypothesis is put forth to obtain a lower bound for the size of this region based on an analogy to the Ising model. This hypothesis is tested on copper using two classical elasticity problems. read less NOT USED (high confidence) C. Tourek, “Application of atom probe tomography to the investigation of atomic force microscope tips and interfacial phenomena.” 2012. link Times cited: 1 Abstract: This dissertation demonstrates the research effort to extend… read moreAbstract: This dissertation demonstrates the research effort to extend the analytical capabilities of atomic force microscopy (AFM) by utilizing atom probe tomography (APT) to investigate AFM tips for the first time. Novel techniques were developed to enable successful analysis of various AFM tips and layers created on the tips. Initial investigations focused on comparing results obtained from as received Si AFM tips to a standard Si APT sample. It was found that by using careful alignment and stiffer cantilevers (>25N/m) AFM tips are readily analyzable by APT. The techniques were then applied to commercially available coated Si AFM tips and high aspect ratio metallic needle tips to demonstrate the broad applicability. A new method to calibrate the optical sensitivity of AFM cantilevers was developed to eliminate damage to the AFM tip. Finally the new calibration method was utilized in the investigation of transferred material from AFM sliding experiments. It was found that the transferred material was readily detected and characterized. The successful results demonstrate the ability of APT application to AFM tips enhancing the information from AFM experiments and opening new areas of investigation. read less NOT USED (high confidence) L. Zhigilei et al., “Atomic/Molecular-Level Simulations of Laser–Materials Interactions.” 2010. link Times cited: 33 NOT USED (high confidence) H. Wadley, X. W. Zhou, and W. Butler, “Atomic Assembly of Magnetoresistive Multilayers.” 2008. link Times cited: 4 NOT USED (high confidence) L. Zhigilei, Z. Lin, and D. Ivanov, “MOLECULAR DYNAMICS STUDY OF SHORT-PULSE LASER MELTING, RECRYSTALLIZATION, SPALLATION, AND ABLATION OF METAL TARGETS.” 2006. link Times cited: 4 Abstract: A hybrid computational model combining classical molecular d… read moreAbstract: A hybrid computational model combining classical molecular dynamics method for simulation of fast nonequilibrium phase transformations with a continuum description of the laser excitation and subsequent relaxation of the conduction band electrons is developed. The model is applied for a systematic computational investigation of the mechanisms of short pulse laser interaction with bulk metal targets. The material response to laser irradiation is investigated in three regimes corresponding to the melting and resolidification of the surface region of the target, photomechanical spallation of a single or multiple layers/droplets, and ablation driven by the thermodynamic driving forces. The conditions leading to the transitions between the different regimes and the atomic-level characteristics of the involved processes are established. read less NOT USED (high confidence) X.-G. Li, S. Xu, Q. Zhang, S. Liu, and J. Shuai, “Complex strengthening mechanisms in nanocrystalline Ni-Mo alloys revealed by a machine-learning interatomic potential,” Journal of Alloys and Compounds. 2023. link Times cited: 2 NOT USED (high confidence) C. Baruffi, F. Maresca, and W. Curtin, “Screw vs. edge dislocation strengthening in body-centered-cubic high entropy alloys and implications for guided alloy design,” MRS Communications. 2022. link Times cited: 5 Abstract: Body-centered-cubic (BCC) high entropy alloys (HEAs) can sho… read moreAbstract: Body-centered-cubic (BCC) high entropy alloys (HEAs) can show exceptionally high strength up to high temperatures. Mechanistic theories are needed to guide alloy discovery within the immense multicomponent HEA compositional space. Here, two new theories for strengthening as controlled by screw and edge dislocations, respectively, are applied to predict the yield stresses of a range of BCC alloys over a wide range of temperatures. Results show that the screw theory, with one fitting parameter, can capture experiments in many dilute and non-dilute alloys while the parameter-free edge theory agrees with experiments in non-dilute alloys having a sufficiently large misfit parameter. These results indicate a transition in single-phase alloy strengthening from traditional screw dominance to edge dominance with increasing misfit that is enabled in complex non-dilute alloys. These results point to the use of the edge theory to guide design of high-temperature alloys in the non-dilute range. Graphical abstract read less NOT USED (high confidence) B. Feng, J. Liu, Y. Zeng, and L. Fan, “Atomistic Insights into the Heat Conductance Across the Interfaces between Erythritol and Different Metals: A Non-Equilibrium Molecular Dynamics Study,” SSRN Electronic Journal. 2022. link Times cited: 0 NOT USED (high confidence) Q. Yang et al., “Effect of deformation conditions on compression phase transformation of AZ31,” Nanotechnology Reviews. 2022. link Times cited: 0 Abstract: In this article, the compression simulation of AZ31 magnesiu… read moreAbstract: In this article, the compression simulation of AZ31 magnesium alloy is simulated by the molecular dynamics method. The effects of loading mode, temperature, and strain rate on the compression behavior are analyzed. The lattice distortion, mechanical behavior, structural evolution, and dislocation evolution in the compression process are deeply analyzed, and the results of different loading modes are obtained. The hexagonal close-packed (HCP) → face-centered cubic (FCC) phase transformation mechanism of AZ31 magnesium alloy during compression at temperature and strain rate, which is related to the mechanical behavior, has been studied completely. This article perfects the research on the compression behavior of magnesium alloys, excavates the application potential of magnesium alloys, and provides a new idea for improving the processing technology and developing high-performance magnesium alloys. read less NOT USED (high confidence) F. Gitzhofer, J. Aluha, P.-O. Langlois, F. Barandehfard, T. Ntho, and N. Abatzoglou, “Proven Anti-Wetting Properties of Molybdenum Tested for High-Temperature Corrosion-Resistance with Potential Application in the Aluminum Industry,” Materials. 2021. link Times cited: 0 Abstract: The behavior of Mo in contact with molten Al was modelled by… read moreAbstract: The behavior of Mo in contact with molten Al was modelled by classical molecular dynamics (CMD) simulation of a pure Mo solid in contact with molten Al at 1200 K using the Materials Studio®. Results showed that no reaction or cross diffusion of atoms occurs at the Mo(s)–Al(l) interface, and that molten Al atoms exhibit an epitaxial alignment with the exposed solid Mo crystal morphology. Furthermore, the two phases {Mo(s) and Al(l)} are predicted to interact with weak van der Waals forces and give interfacial energy of about 203 mJ/m2. Surface energy measurements by the sessile drop experiment using the van Oss–Chaudhury–Good (VCG) theory established a Mo(s)–Al(l) interface energy equivalent to 54 mJ/m2, which supports the weak van der Waals interaction. The corrosion resistance of a high purity (99.97%) Mo block was then tested in a molten alloy of 5% Mg mixed in Al (Al-5 wt.%Mg) at 1123 K for 96 h, using the ALCAN’s standard “immersion” test, and the results are presented. No Mo was found to be dissolved in the molten Al-Mg alloy. However, a 20% mass loss in the Mo block was due to intergranular corrosion scissoring the Mo block in the ALCAN test, but not as a result of the reaction of pure Mo with the molten Al-Mg alloy. It was observed that the Al-Mg alloy did not stick to the Mo block. read less NOT USED (high confidence) P. Zakharov, S. Dmitriev, and E. Korznikova, “Molecular dynamic analysis of energy transport in a Pt3Al crystal under the impact in the spectrum gap frequency,” Letters on Materials. 2021. link Times cited: 2 Abstract: Сonstant search for new mechanisms of energy transport assoc… read moreAbstract: Сonstant search for new mechanisms of energy transport associated with a decrease in losses results in the active investigation of the possibilities of generating mobile excitations at frequencies outside the phonon spectrum. The effect of energy transfer at frequencies outside the band, possible in case if the excitation amplitude exceeds the critical value, was previously actively studied in reduced dimension systems. In this work, for the first time, the possibility and features of energy transfer at frequencies outside the phonon spectrum of a crystal are studied on the Pt3Al biatomic lattice, the corresponding excitations in which are created through the implementation of harmonic vibrations at frequencies lying in the phonon spectrum gap. It was found that as a result of the initial impact, the parameters of which lie in the band gap, at the first stage, discrete breathers are formed on aluminum atoms lying in the immediate vicinity of the impact zone. The threshold value of the energy required to excite breathers, determined by the amplitude and frequency of exposure, decreases as the frequency approaches the boundary of the slit in the spectrum. At the second stage, after the end of the harmonic action, a soliton-like wave is formed, moving along the crystal with an average speed of 4.5 km / s. The wave velocity is practically independent of the initial disturbance amplitude. The shape and amplitude of the moving wave are weakly dependent on the initial frequency. Significant differences in the profile and a tendency to bimodality were revealed only when exposed to frequencies close to the lower limit of the spectrum. The results obtained contribute to a fundamental understanding of the features of the propagation of elastic disturbances in nonlinear media in case of forbidden zones region impacts. read less NOT USED (high confidence) L. A. Mistryukova, N. P. Kryuchkov, V. Mantsevich, A. Sapelkin, and S. Yurchenko, “Interpolation method for crystals with many-body interactions,” Physical Review B. 2021. link Times cited: 0 Abstract: We propose an interpolation scheme to describe pair correlat… read moreAbstract: We propose an interpolation scheme to describe pair correlations in crystals with many-body interactions that requires only information on relative displacements for the nearest neighbours and in the long range. Using crystalline Ni as a test case, the scheme is shown to deliver the functional form for the radial distribution function at least as well as molecular dynamics simulations. The results provide a fast route for verification of interatomic potentials and study of many-body interactions using a combination of x-ray scattering and x-ray absorption spectroscopy. read less NOT USED (high confidence) R. E. Kubilay, A. Ghafarollahi, F. Maresca, and W. Curtin, “High energy barriers for edge dislocation motion in body-centered cubic high entropy alloys,” npj Computational Materials. 2021. link Times cited: 19 NOT USED (high confidence) M. G. Urazaliev, M. E. Stupak, and V. Popov, “Structure and Energy of Symmetric Tilt Boundaries with the 〈110〉 Axis in Ni and the Energy of Formation of Vacancies in Grain Boundaries,” Physics of Metals and Metallography. 2021. link Times cited: 4 NOT USED (high confidence) M. Çeltek, “Saf Kalsiyum Elementinin Isıtma Sürecinin Moleküler Dinamik Benzetim Yöntemi ile İncelenmesi,” Bitlis Eren Üniversitesi Fen Bilimleri Dergisi. 2021. link Times cited: 0 Abstract: In the study, the structural and some physical properties of… read moreAbstract: In the study, the structural and some physical properties of pure calcium (Ca) during the heating process were investigated by classical molecular dynamic (MD) simulations method by using the embedded atom method (EAM) and tight-binding (TB) many body potentials. During this process, energy-, lattice-parameter and densitytemperature curves were used to see the changes in physical parameters depending on temperature. In addition, the evolution of the atomic structure of the system was investigated using different analysis methods such as the pair distribution function, the structure factor and the Honeycutt-Andersen (HA) method. The results obtained for both potentials were compared with appropriate experimental and other MD simulation results reported in the literature and discussed together. It has been observed that the EAM potential in a wide temperature range produces more successful results than the TB potential. HA results showed that especially 1541 and 1551 type quintet clusters and 1431 type quartet clusters play more effective roles in the melting process of the system. read less NOT USED (high confidence) A.-S. Tran, “Control of plastic deformation in Cu50Ta50 metallic glass by insertion of Cu crystalline cores,” Physica Scripta. 2021. link Times cited: 3 Abstract: The tensile characteristics and deformation mechanisms of Cu… read moreAbstract: The tensile characteristics and deformation mechanisms of Cu-Ta metallic glasses with the insertion of Cu crystalline cores are investigated using molecular dynamics (MD) simulations. The effects of different Cu crystalline core diameters (D Cu ), experiment temperatures (T), and Cu crystalline core numbers (N) are studied. The results show that the plasticity of the Cu-Ta MGs is significantly improved by inserting Cu crystalline cores. The Shockley dislocations (<112>) make up the majority, and the FCC structures mainly transform into the HCP structures in the Cu crystalline cores. As increasing D Cu , the shear transformation zones (STZs) form more severely, the fraction of atoms with the high shear strain increases, and the tensile strength reduces. As increasing T, the STZs formation is fainter and most intense at 100 K, the fraction of atoms with the shear strain greater than 0.5 (f0.5) and the tensile strength reduce, while the fraction of atoms with the shear strain greater than 0.3 (f0.3) increases. As changing N, the STZs formations in the samples with the N = 2 and 8 are more pronounced, the f0.5 of the samples with the N = 1 and 8 are lower than those in the other cases, and the tensile strength reduces as the N increases. read less NOT USED (high confidence) Q. Yang, C. Xue, Z. Chu, Y. Li, and L. Ma, “Molecular dynamics simulation of the effect of solute atoms on the compression of magnesium alloy,” Applied Physics A. 2021. link Times cited: 3 NOT USED (high confidence) B. Li, J. Li, X. Su, and Y. Cui, “Molecular dynamics study on structural and atomic evolution between Au and Ni nanoparticles through coalescence,” Scientific Reports. 2021. link Times cited: 2 NOT USED (high confidence) Y. Xie and S. Li, “Finite temperature atomistic‐informed crystal plasticity finite element modeling of single crystal tantalum ( α ‐Ta) at micron scale,” International Journal for Numerical Methods in Engineering. 2021. link Times cited: 8 Abstract: In this work, we have developed a temperature‐dependent high… read moreAbstract: In this work, we have developed a temperature‐dependent higher‐order Cauchy–Born (THCB) rule for multiscale crystal defect dynamics (MCDD) of crystalline solids based on the harmonic approximation. As a template, we employed the THCB rule to develop an atomistic‐informed constitutive model for the body‐centered cubic (BCC) single crystal tantalum ( α ‐Ta). Considering the effect of strain gradients in different process zone elements, the corresponding higher order stress are used to model crystal plasticity of single crystal α ‐Ta. Different from face‐centered cubic crystals, BCC crystals are strongly influenced by temperature. It is shown in this article that the developed finite temperature atomistic‐informed crystal plasticity finite element method is able to capture the temperature‐dependent dislocation substructure and hence crystal plastic deformation. The main contributions and novelties of the present work are highlighted by following findings: (1) A THCB rule and an atomistic‐informed strain gradient theory have been developed, and the corresponding temperature‐related higher‐order stress and elastic tensor formulations are derived; (2) The finite temperature MCDD provides an atomistic‐informed crystal plasticity finite element method that can simulate anisotropic crystal plasticity in any orientation within stereographic triangle at micron scale and above; (3) The developed MCDD is able to capture the non‐Schmid effects of BCC single crystal tantalum ( α ‐Ta); (4) The developed MCDD is able to capture the size effect of single crystal plasticity; and (5) The finite temperature MCDD can simulate the temperature dependent dislocation substructure, and it captures cross‐slip in single crystal tantalum at low temperature (∼20 K) and captures dislocation cell structure at high temperature (∼500 K). read less NOT USED (high confidence) H. Guo, L. Zhang, Q. Zhu, C. Wang, G. Chen, and P. Zhang, “Molecular Dynamics Simulation of the Coalescence and Melting Process of Cu and Ag Nanoparticles,” Advances in Condensed Matter Physics. 2021. link Times cited: 0 Abstract: The coalescence and melting process of different sizes and a… read moreAbstract: The coalescence and melting process of different sizes and arrangements of Ag and Cu nanoparticles is studied through the molecular dynamics (MD) method. The results show that the twin boundary or stacking fault formation and atomic diffusion of the nanoparticles play an important role in the different stages of the heating process. At the beginning of the simulation, Cu and Ag nanoparticles will contact to each other in a very short time. As the temperature goes up, Cu and Ag nanoparticles may generate stacking fault or twin boundary to stabilize the interface structure. When the temperature reaches a critical value, the atoms gain a strong ability to diffuse and eventually melt into one liquid sphere. The coalescence point and melting temperature increase as cluster diameter increases. Moreover, the arrangement of Cu and Ag nanoparticles has a certain effect on the stability of the initial joint interface, which will affect subsequent coalescence and melting behavior. read less NOT USED (high confidence) K. Yashiro, “Molecular dynamics study on atomic elastic stiffness at mode I crack along bi-metal interface,” Philosophical Transactions of the Royal Society A. 2021. link Times cited: 3 Abstract: Propagation of mode I crack along bi-metal (001) interfaces … read moreAbstract: Propagation of mode I crack along bi-metal (001) interfaces of Fe/W, Fe/Ni, Fe/Co and Ti/Mg is simulated by molecular dynamics and discussed with the eigenvalue/vector of the atomic elastic stiffness, Bija=Δσia/Δεj, and surface energy. The crack does not propagate at the interface but in the adjacent phase of smaller surface energy, except in Fe/Ni. The 1st eigenvalue ηa(1), or the solution of BijaΔεj=ηaΔεi of each atom, clarifies the difference of ‘soft/hard’ of both phases at the onset of crack propagation. In the case of Fe/Ni, the ηa(1) of Ni atoms remarkably decreases in the Fe/Ni bi-metal structure, even though Ni has higher ηa(1) than Fe at no-load perfect lattices. Thus the rupture occurs in the Ni side even though the Ni has slightly higher (001) surface energy than Fe. Deformation modes at the crack propagation are also visualized by the eigenvector of ηa(1) < 0 unstable atoms. This article is part of the theme issue ‘Fracture dynamics of solid materials: from particles to the globe’. read less NOT USED (high confidence) R. Wang et al., “Prediction of crystal structures and motifs in the Fe–Mg–O system at Earth’s core pressures,” New Journal of Physics. 2021. link Times cited: 2 Abstract: Fe, Mg, and O are among the most abundant elements in terres… read moreAbstract: Fe, Mg, and O are among the most abundant elements in terrestrial planets. While the behavior of the Fe–O, Mg–O, and Fe–Mg binary systems under pressure have been investigated, there are still very few studies of the Fe–Mg–O ternary system at relevant Earth’s core and super-Earth’s mantle pressures. Here, we use the adaptive genetic algorithm (AGA) to study ternary Fe x Mg y O z phases in a wide range of stoichiometries at 200 GPa and 350 GPa. We discovered three dynamically stable phases with stoichiometries FeMg2O4, Fe2MgO4, and FeMg3O4 with lower enthalpy than any known combination of Fe–Mg–O high-pressure compounds at 350 GPa. With the discovery of these phases, we construct the Fe–Mg–O ternary convex hull. We further clarify the composition- and pressure-dependence of structural motifs with the analysis of the AGA-found stable and metastable structures. Analysis of binary and ternary stable phases suggest that O, Mg, or both could stabilize a BCC iron alloy at inner core pressures. read less NOT USED (high confidence) R. Singh et al., “Neural-network model for force prediction in multi-principal-element alloys,” Computational Materials Science. 2021. link Times cited: 3 NOT USED (high confidence) Y. Chen, Z. Guan, Y. Yao, and H. Wang, “Tuning nanoscale adhesive contact behavior to a near ideal Hertzian state via graphene coverage,” arXiv: Applied Physics. 2020. link Times cited: 4 NOT USED (high confidence) M. Wagih, P. M. Larsen, and C. Schuh, “Learning grain boundary segregation energy spectra in polycrystals,” Nature Communications. 2020. link Times cited: 62 NOT USED (high confidence) S. Kumari and A. Dutta, “Nucleation of twinning dislocation loops in fcc metals,” arXiv: Materials Science. 2020. link Times cited: 5 NOT USED (high confidence) M. Dupraz, S. Leake, and M. Richard, “Bragg coherent imaging of nanoprecipitates: role of superstructure reflections,” Journal of Applied Crystallography. 2020. link Times cited: 0 Abstract: Coherent precipitation of ordered phases is responsible for … read moreAbstract: Coherent precipitation of ordered phases is responsible for providing exceptional high-temperature mechanical properties in a wide range of compositionally complex alloys. Ordered phases are also essential to enhance the magnetic or catalytic properties of alloyed nanoparticles. The present work aims to demonstrate the relevance of Bragg coherent diffraction imaging (BCDI) for studying bulk and thin-film samples or isolated nanoparticles containing coherent nanoprecipitates/ordered phases. The structures of crystals of a few tens of nanometres in size are modelled with realistic interatomic potentials and are relaxed after introduction of coherent ordered nanoprecipitates. Diffraction patterns from fundamental and superstructure reflections are calculated in the kinematic approximation and used as input to retrieve the strain fields using algorithmic inversion. First, the case of single nanoprecipitates is tackled and it is shown that the strain field distribution from the ordered phase is retrieved very accurately. Then, the influence of the order parameter S on the strain field retrieved from the superstructure reflections is investigated. A very accurate strain distribution can be retrieved for partially ordered phases with large and inhomogeneous strains. Subsequently, the relevance of BCDI is evaluated for the study of systems containing many precipitates, and it is demonstrated that the technique is relevant for such systems. Finally, the experimental feasibility of using BCDI to image ordered phases is discussed in the light of the new possibilities offered by fourth-generation synchrotron sources. read less NOT USED (high confidence) S. Combettes et al., “How interface properties control the equilibrium shape of core-shell Fe-Au and Fe-Ag nanoparticles.,” Nanoscale. 2020. link Times cited: 6 Abstract: While combining two metals in the same nanoparticle can lead… read moreAbstract: While combining two metals in the same nanoparticle can lead to remarkable novel applications, the resulting structure in terms of crystallinity and shape remains difficult to predict. It is thus essential to provide a detailed atomistic picture of the underlying growth processes. In the present work we address the case of core-shell Fe-Au and Fe-Ag nanoparticles. Interface properties between Fe and the noble metals Au and Ag, computed using DFT, were used to parameterize Fe-Au and Fe-Ag pairwise interactions in combination with available many-body potentials for the pure elements. The growth of Au or Ag shells on nanometric Fe cores with prescribed shapes was then modelled by means of Monte Carlo simulations. The shape of the obtained Fe-Au nanoparticles is found to strongly evolve with the amount of metal deposited on the Fe core, a transition from the polyhedral Wulff shape of bare iron to a cubic shape taking place as the amount of deposited gold exceeds two monolayers. In striking contrast, the growth of silver proceeds in a much more anisotropic, Janus-like way and with a lesser dependence on the iron core shape. In both cases, the predicted morphologies are found to be in good agreement with experimental observations in which the nanoparticles are grown by physical deposition methods. Understanding the origin of these differences, which can be traced back to subtle variations in the electronic structure of the Au/Fe and Ag/Fe interfaces, should further contribute to the better design of core-shell bimetallic nanoparticles. read less NOT USED (high confidence) W. Jiang, Y. Zhang, L. Zhang, and H. Wang, “Accurate Deep Potential model for the Al–Cu–Mg alloy in the full concentration space*,” arXiv: Materials Science. 2020. link Times cited: 24 Abstract: Combining first-principles accuracy and empirical-potential … read moreAbstract: Combining first-principles accuracy and empirical-potential efficiency for the description of the potential energy surface (PES) is the philosopher's stone for unraveling the nature of matter via atomistic simulation. This has been particularly challenging for multi-component alloy systems due to the complex and non-linear nature of the associated PES. In this work, we develop an accurate PES model for the Al-Cu-Mg system by employing Deep Potential (DP), a neural network based representation of the PES, and DP Generator (DP-GEN), a concurrent-learning scheme that generates a compact set of ab initio data for training. The resulting DP model gives predictions consistent with first-principles calculations for various binary and ternary systems on their fundamental energetic and mechanical properties, including formation energy, equilibrium volume, equation of state, interstitial energy, vacancy and surface formation energy, as well as elastic moduli. Extensive benchmark shows that the DP model is ready and will be useful for atomistic modeling of the Al-Cu-Mg system within the full range of concentration. read less NOT USED (high confidence) Y. Zhang, C. Hu, and B. Jiang, “Accelerating atomistic simulations with piecewise machine-learned ab Initio potentials at a classical force field-like cost.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 12 Abstract: Recently, machine learning methods have become easy-to-use t… read moreAbstract: Recently, machine learning methods have become easy-to-use tools for constructing high-dimensional interatomic potentials with ab initio accuracy. Although machine-learned interatomic potentials are generally orders of magnitude faster than first-principles calculations, they remain much slower than classical force fields, at the price of using more complex structural descriptors. To bridge this efficiency gap, we propose an embedded atom neural network approach with simple piecewise switching function-based descriptors, resulting in a favorable linear scaling with the number of neighbor atoms. Numerical examples validate that this piecewise machine-learning model can be over an order of magnitude faster than various popular machine-learned potentials with comparable accuracy for both metallic and covalent materials, approaching the speed of the fastest embedded atom method (i.e. several μs per atom per CPU core). The extreme efficiency of this approach promises its potential in first-principles atomistic simulations of very large systems and/or in a long timescale. read less NOT USED (high confidence) H.-W. Chen, Q. Fang, K. Zhou, Y. Liu, and J. Li, “Unraveling atomic-scale crystallization and microstructural evolution of a selective laser melted FeCrNi medium-entropy alloy,” CrystEngComm. 2020. link Times cited: 14 Abstract: Selective laser melting (SLM) provides flexibility to manufa… read moreAbstract: Selective laser melting (SLM) provides flexibility to manufacture components with complex structures. However, the unexpected crystallization and the dynamical microstructural evolution at the atomic scale still remains unknown during the SLM process. Here, the crystallization and formation mechanism of an equimolar FeCrNi medium-entropy alloy (MEA) with excellent mechanical properties prepared by SLM is studied via molecular dynamics (MD) simulations. The surface morphology, atomic microstructural evolution, and Cr elemental segregation are investigated during the crystallization process. The crystallization and microstructural characteristics are clearly observed. A large number of stacking faults take place at the boundary of the melting pool, but some stacking faults extend to the center region due to the thermal gradient effect. A segregation phenomenon of a nanoscale Cr-rich cluster occurs in the FeCrNi MEA to reveal the dynamic precipitation mechanism during the SLM process. Furthermore, higher energy density and lower scanning speed would promote the segregation and even form a mesh connected structure. The current result provides an insight into the crystallization and formation mechanism of microstructures to develop advanced alloys with high strength and toughness via the reasonable choice of SLM parameters. read less NOT USED (high confidence) J. Chapman and R. Ramprasad, “Predicting the dynamic behavior of the mechanical properties of platinum with machine learning.,” The Journal of chemical physics. 2020. link Times cited: 2 Abstract: Over the last few decades, computational tools have been ins… read moreAbstract: Over the last few decades, computational tools have been instrumental in understanding the behavior of materials at the nano-meter length scale. Until recently, these tools have been dominated by two levels of theory: quantum mechanics (QM) based methods and semi-empirical/classical methods. The former are time-intensive but accurate and versatile, while the latter methods are fast but are significantly limited in veracity, versatility, and transferability. Recently, machine learning (ML) methods have shown the potential to bridge the gap between these two chasms due to their (i) low cost, (ii) accuracy, (iii) transferability, and (iv) ability to be iteratively improved. In this work, we further extend the scope of ML for atomistic simulations by capturing the temperature dependence of the mechanical and structural properties of bulk platinum through molecular dynamics simulations. We compare our results directly with experiments, showcasing that ML methods can be used to accurately capture large-scale materials phenomena that are out of reach of QM calculations. We also compare our predictions with those of a reliable embedded atom method potential. We conclude this work by discussing how ML methods can be used to push the boundaries of nano-scale materials research by bridging the gap between QM and experimental methods. read less NOT USED (high confidence) S. Nasiri et al., “Multilayer Structures of Graphene and Pt Nanoparticles: A Multiscale Computational Study,” Advanced Engineering Materials. 2020. link Times cited: 5 Abstract: Multiscale simulation study results of multilayer structures… read moreAbstract: Multiscale simulation study results of multilayer structures consisting of graphene sheets with embedded Pt nanoparticles is reported. Density functional theory is used to understand the energetics of Pt–graphene interfaces and provide reference data for the parameterization of a Pt–graphene interaction potential. Molecular dynamics simulations then provide the conformation and energetics of graphene sheets with embedded Pt nanoparticles of varying density, form, and size. These results are interpreted using a continuum mechanical model of sheet deformation, and serve to parameterize a meso‐scale Monte Carlo model to investigate the question under which conditions the free volume around the Pt nanoparticles forms a percolating cluster, such that the structures can be used in catalytic applications. This article is concluded with a discussion of potential applications of such multilayer structures. read less NOT USED (high confidence) E. Antillon and M. Ghazisaeidi, “Efficient determination of solid-state phase equilibrium with the multicell Monte Carlo method.,” Physical review. E. 2020. link Times cited: 5 Abstract: Building on our previously introduced multicell Monte Carlo … read moreAbstract: Building on our previously introduced multicell Monte Carlo (MC)^{2} method for modeling phase coexistence, this paper provides important improvements for efficient determination of phase equilibria in solids. The (MC)^{2} method uses multiple cells, representing possible phases. Mass transfer between cells is modeled virtually by solving the mass balance equation after the composition of each cell is changed arbitrarily. However, searching for the minimum free energy during this process poses a practical problem. The solution to the mass balance equation is not unique away from equilibrium, and consequently the algorithm is in risk of getting trapped in nonequilibrium solutions. Therefore, a proper stopping condition for (MC)^{2} is currently lacking. In this work, we introduce a consistency check via a predictor-corrector algorithm to penalize solutions that do not satisfy a necessary condition for equivalence of chemical potentials and steer the system toward finding equilibrium. The most general acceptance criteria for (MC)^{2} is derived starting from the isothermal-isobaric Gibbs ensemble for mixtures. Using this ensemble, translational MC moves are added to include vibrational excitations as well as volume MC moves to ensure the condition of constant pressure and temperature entirely with a MC approach, without relying on any other method for relaxation of these degrees of freedom. As a proof of concept the method is applied to two binary alloys with miscibility gaps and a model quaternary alloy, using classical interatomic potentials. read less NOT USED (high confidence) H.-hong Jia, D. Bao, Y. Zhang, and S. Du, “Structural and thermal stabilities of Au@Ag core-shell nanoparticles and their arrays: A molecular dynamics simulation,” Chinese Physics B. 2020. link Times cited: 3 NOT USED (high confidence) P. Gupta, K. C. Katakam, G. Katakareddi, and N. Yedla, “Crack and its interaction with defects in Al coated with Cu50Zr50 metallic glass thin film: an MD simulation study,” Journal of Molecular Modeling. 2020. link Times cited: 2 NOT USED (high confidence) H. Feng et al., “Indentation-induced plastic behaviour of nanotwinned Cu/high entropy alloy FeCoCrNi nanolaminate: an atomic simulation,” RSC Advances. 2020. link Times cited: 17 Abstract: Using large-scale molecular dynamics (MD) simulations, the e… read moreAbstract: Using large-scale molecular dynamics (MD) simulations, the effects of interface and layer number in the nanoindentation response of experimentally observed nanotwinned Cu/high entropy alloy (HEA) FeCoCrNi nanolaminate are studied. The dislocations are nucleated and emitted, which are more limited to the first twinning layer > second twinning layer > HEA layer. The stacking fault strengthening is pronounced due to the obvious difference of stacking fault energy between Cu and HEA, which can be rarely observed from the previous work in traditional alloys and metals. After the indentation induced deformation, the nanotwinned Cu/HEA FeCoCrNi nanolaminates for different layer numbers generate a mass of Shockley partial dislocations to cause the good plasticity, attributed to the strong strain gradient effect. The strong layer number and interface structure effects found here can provide insight for the design of advanced nanolaminate with high strength and good plasticity. read less NOT USED (high confidence) R. Wang et al., “Theoretical search for possible Li–Ni–B crystal structures using an adaptive genetic algorithm,” Journal of Applied Physics. 2020. link Times cited: 8 Abstract: The structural diversity of rare-earth and transition metal … read moreAbstract: The structural diversity of rare-earth and transition metal borides indicates that alkali-transition metal borides (A-T-B) show tremendous promise in exhibiting a variety of crystal structures with different dimensionalities of T-B frameworks. On the other hand, the A-T-B ternary systems are severely underexplored because of the synthetic challenges associated with their preparation. Accurate and efficient computational predictions of low-energy stable and metastable phases can identify the optimal compositions of the hypothetical compounds in the A-T-B systems to guide the synthesis. In this work, we have computationally discovered several new phases in the Li–Ni–B ternary system. The newly discovered LiNiB, Li2Ni3B, and Li2NiB phases expand the existing theoretical database, and the convex-hull surface of Li–Ni–B has been re-constructed. The lowest energy structure of the LiNiB compound has been found by an adaptive genetic algorithm with layered motif, which matches with the experimentally determined structure. According to our electrochemical calculations, LiNiB and another predicted layered Li2NiB compounds have great potential as anode materials for lithium batteries. The Li2Ni3B compound with the space group P4332 was predicted to crystallize in a cubic structure composed of distorted octahedral units of BNi6, which is isostructural to two noncentrosymmetric superconductors Li2Pd3B and Li2Pt3B. While we were unable to experimentally confirm the Li2Ni3B compound utilizing the hydride synthetic route, attempts to synthesize this compound by alternate methods remain highly desirable, considering its potential superconducting properties. read less NOT USED (high confidence) Y.-heng Zhang, Y. Xu, and H. Chu, “Size effect of layer thickness on stress fields due to interface core-spreading dislocation arrays in multilayers,” Science China Technological Sciences. 2020. link Times cited: 1 NOT USED (high confidence) Y. J. Shen, L. C. Liu, S. Mi, H. Gong, and S. F. Zhou, “Construction of an n-body Fe–Cu potential and its application in atomistic modeling of Fe–Cu solid solutions,” Journal of Applied Physics. 2020. link Times cited: 6 Abstract: By means of the embedded-atom method, a Fe–Cu potential has … read moreAbstract: By means of the embedded-atom method, a Fe–Cu potential has been constructed through a newly mathematic form of cross potential. The newly constructed Fe–Cu potential has demonstrated to be more reliable than the five reported Fe–Cu potentials. Based on the Fe–Cu potential, the mechanical and thermodynamic properties and the structural stability of Fe–Cu solid solutions in the whole composition range are derived by molecular dynamics simulation. It is found that the heat of formation curves of the FexCu100 − x solid solutions with body-centered-cubic (BCC) and face-centered-cubic (FCC) structures intersect at the point of x = 65, implying that FexCu100 − x solid solutions with FCC and BCC structures are thermodynamically stable when 0 ≤ x ≤ 65 and 65 < x ≤ 100, respectively. In addition, the derived lattice constants, structural stability, elastic constants, elastic moduli, heat capacity, and coefficients of thermal expansion of Fe–Cu solid solutions from the new Fe–Cu potential agree well with the data of the experiments, first-principles calculation, and the Miedema model.By means of the embedded-atom method, a Fe–Cu potential has been constructed through a newly mathematic form of cross potential. The newly constructed Fe–Cu potential has demonstrated to be more reliable than the five reported Fe–Cu potentials. Based on the Fe–Cu potential, the mechanical and thermodynamic properties and the structural stability of Fe–Cu solid solutions in the whole composition range are derived by molecular dynamics simulation. It is found that the heat of formation curves of the FexCu100 − x solid solutions with body-centered-cubic (BCC) and face-centered-cubic (FCC) structures intersect at the point of x = 65, implying that FexCu100 − x solid solutions with FCC and BCC structures are thermodynamically stable when 0 ≤ x ≤ 65 and 65 < x ≤ 100, respectively. In addition, the derived lattice constants, structural stability, elastic constants, elastic moduli, heat capacity, and coefficients of thermal expansion of Fe–Cu solid solutions from the new Fe–Cu potential agree well with the data o... read less NOT USED (high confidence) J. Jeon, S. Jiang, F. Rahmani, and S. Nouranian, “Molecular dynamics study of temperature and heating rate–dependent sintering of titanium nanoparticles and its influence on the sequent tension tests of the formed particle-chain products,” Journal of Nanoparticle Research. 2020. link Times cited: 6 NOT USED (high confidence) O. I. Kushnerov, “Molecular dynamics simulation of the solidification of AlCoCuFeNi high–entropy alloy nanowire.” 2019. link Times cited: 0 Abstract: Molecular dynamics simulation of the solidification behavior… read moreAbstract: Molecular dynamics simulation of the solidification behavior of AlCoCuFeNi nanowire was carried out basing on the embedded atom potential with different cooling rates (1∙10, 1∙10, and 1∙10 K/s). To simulate an infinite nanowire, a periodical boundary condition along the nanowire axis direction was applied. The crystallization of the nanowire was characterized by studying the temperature dependence of the potential energy. The adaptive common neighbor analysis (CNA) was performed and the radial distribution function (RDF) was calculated to determine the structure and lattice parameters of phases of the AlCoCuFeNi nanowire. It has been shown that the final structure of investigated nanoparticle changes from amorphous to crystalline with decreasing of the rate of cooling. read less NOT USED (high confidence) M. Çeltek, S. Şengül, and U. Domekeli, “Hızlı Soğutma Sürecinde Dörtlü Zr48Cu36Ag8Al8 İri Hacimli Metalik Camının Atomik Yapısının Gelişimi,” Süleyman Demirel Üniversitesi Fen Bilimleri Enstitüsü Dergisi. 2019. link Times cited: 6 Abstract: Calismamizda Zr 48 Cu 36 Al 8 Ag 8 dortlu alasiminin atomik … read moreAbstract: Calismamizda Zr 48 Cu 36 Al 8 Ag 8 dortlu alasiminin atomik yapisi ve cam olusturma sureci molekuler dinamik simulasyon ile gomulu atom metodu kullanilarak arastirildi. Cam gecis surecini ve sicakliga bagli atomik yapi gelisimini arastirmak icin ortalama hacim-sicaklik egrisi, ciftler dagilim fonksiyonu (PDF) ve cift analiz metodu kullanildi. 300 K’de hesaplanan toplam PDF, g(r ), daha once rapor edilen deneysel g(r) ile iyi bir uyum saglamaktadir. Ote yandan ortalama hacim-sicaklik grafiginden yararlanilarak belirlenen cam gecis sicakligi da deneysel degerlerle birbirlerine yakindir. Zr-Zr ve Cu-Cu ciftlerinin kismi PDF'lerin pikleri sicaklik dususune bagli olarak normal bir artis egilimi gosterirken, Al-Al ve Ag-Ag ciftleri ise cok yuksek pikler ureterek anormal bir davranis sergilemektedir. Bu davranisin nedeninin simulasyon hucresindeki Al ve Ag atomlarinin topaklanmasi oldugu gorulmustur. Sistemin mikro yapisi incelendiginde ise kisa menzil duzenin gostergesi olan 1431, 1541 ve 1551 bagli ciftlerinin butun sicakliklarda baskin oldugu gozlenmistir. Azalan sicaklikla birlikte ozellikle ideal ikosahedral duzeni temsil eden 1551 bagli ciftlerinin oraninin artmasi sistemin kisa menzil duzeninin geliserek artmaya devam ettigini gostermektedir. read less NOT USED (high confidence) F. Ojaghnezhad and H. Shodja, “Second strain gradient theory in orthogonal curvilinear coordinates: Prediction of the relaxation of a solid nanosphere and embedded spherical nanocavity,” Applied Mathematical Modelling. 2019. link Times cited: 5 NOT USED (high confidence) Y. Cui, Y. Ju, and S. Meguid, “Atomistic treatment of periodic gold nanowire array nanofasteners under shear loading,” Nanotechnology. 2019. link Times cited: 5 Abstract: Comprehensive molecular dynamics simulations are conducted t… read moreAbstract: Comprehensive molecular dynamics simulations are conducted to unravel the mechanics and mechanisms associated with the strength and fracture behavior of a highly ordered gold nanowire (Au-NW) array of a pair of nanofasteners (nanoconnectors) under externally applied shear strain. Large-scale atomic/molecular massively parallel simulator (LAMMPS and embedded atom method were adopted to model the atomic interactions of a number of neighboring nanofasteners. This was affected via the use of a periodic simulation box around a pair of highly ordered nanotube arrays to minimize the cost of the computations. Energy minimization using a conjugate gradient algorithm was first performed and followed by atomic relaxation to achieve an equilibrated configuration under the canonical ensemble of constant temperature and volume. The relaxed equilibrated configuration of the nanofastener was then subjected to an externally applied shear strain γ x y at a rate of γ ̇ x y = 0.1 per nanosecond under the canonical ensemble. Our results reveal the importance of the morphology and the overlap depth of the mating nanowire arrays upon the mechanical and fracture behavior of the nanofastener under shear loading. Our work also disclosed the phenomenon of multiple contacts of some displaced nanowires with their neighbors even after their fracture leading to multiple cold-welds with added redundancy to the nanofastener. Finally, in this research, we identified the locations of dislocation emissions and the resulting fracture processes that govern the mechanical integrity and ultimately the functionality of the Au-NW connector. The proposed highly ordered alignment, as conceived numerically herein, can yield a peak stress two to three times higher than that corresponding to a random alignment reported in a previous study. The Au-NW connector also exhibited resistance to fracture, even in cases where small overlap depth is considered in joint bonding. The nanoconnector was also tested at high temperatures (up to 450 K). Our results show that the rising temperature only leads to a minor reduction in the load transmitted by the nanoconnector. read less NOT USED (high confidence) G. Singh, K. Kumar, and R. K. Moudgil, “Alloying-induced spin Seebeck effect and spin figure of merit in Pt-based bimetallic atomic wires of noble metals.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 2 Abstract: We have investigated using first principles the occurrence a… read moreAbstract: We have investigated using first principles the occurrence and tunability of the spin Seebeck effect in Pt-based bimetallic wires of noble metals (viz. XPt, X = Cu, Ag, and Au) modelled in linear, ladder, and double zigzag (DZZ) topologies. The spin figure of merit ZsT and its charge counterpart ZcT are calculated by considering both electronic and phononic contributions to the thermal conductance. For this endeavour, we have employed the Landauer-Büttiker approach, with the requisite electron τel(E) and phonon τph(E) transmission functions obtained using the non-equilibrium Green's function approach, based on density functional theory and the general utility lattice program, respectively. We find that alloying and/or topological tailoring bring in quantitative as well as qualitative changes in the transport properties. Unlike the pristine wires, τel(E) now depends (except for the CuPt wire in the ladder topology) markedly on spin, thus resulting in an unequal current in the two spin channels and hence a non-zero spin Seebeck coefficient Ss. Remarkably, the AgPt wire in the linear topology and all wires in the DZZ1 and DZZ3 configurations of the DZZ topology exhibit half-metallic conduction, with a sizable gap in the density of ↑-spin states at the Fermi level. Alloying also introduces energy gap(s) in the phonon density of states. Consequently, we find a significant reduction in the electronic and phononic thermal conductance. Interestingly, Ss is of the same order as its charge counterpart Sc, and both can be tuned via the topology and chemical potential μ of the electrodes. This together with the reduced thermal conductance results in a significantly high room-temperature ZcT (∼33) and ZsT (∼25) at μ ∼ 0.24 eV in the AgPt wire in the DZZ3 topology, with about a ten-fold increase in ZcT as compared to the pristine wires. Furthermore, there exists a characteristic μ in some bimetallic wires for which Sc approaches zero, but Ss remains quite appreciable. This result arises due to the emergence of bipolar thermal conduction, which under criticality makes S↑ + S↓ = 0. Importantly, such a situation may be exploited to generate a pure thermal spin voltage. Our results can be explicated on the basis of changes in the electronic band structure and phononic spectra due to alloying and topological effects. read less NOT USED (high confidence) S. Mi, H. Gong, and J. Fan, “Structural stability and mechanical property of Fe-W solid solutions from a constructed Fe-W potential,” Journal of Applied Physics. 2019. link Times cited: 15 Abstract: An Fe-W potential has been constructed by means of the embed… read moreAbstract: An Fe-W potential has been constructed by means of the embedded-atom method and has proven to be more realistic than the three published Fe-W potentials in the literature. Based on the constructed Fe-W potential, molecular dynamic simulation has been used to reveal structural stability, thermodynamic properties, and mechanical properties of BCC Fe-W solid solutions within the entire composition range. It is found that the Fe-W interaction in BCC Fe-W solid solutions should be weak and attractive with small and negative heats of formation, which agree well with those from the thermodynamic Miedema model and could clarify the controversy regarding heats of formation of Fe-W solid solutions in the literature. In addition, the derived coefficient of thermal expansion, heat capacity, elastic constants, and elastic moduli of Fe-W solid solutions from the present Fe-W potential are in good agreement with the corresponding data from ab initio calculation or experiments in the literature.An Fe-W potential has been constructed by means of the embedded-atom method and has proven to be more realistic than the three published Fe-W potentials in the literature. Based on the constructed Fe-W potential, molecular dynamic simulation has been used to reveal structural stability, thermodynamic properties, and mechanical properties of BCC Fe-W solid solutions within the entire composition range. It is found that the Fe-W interaction in BCC Fe-W solid solutions should be weak and attractive with small and negative heats of formation, which agree well with those from the thermodynamic Miedema model and could clarify the controversy regarding heats of formation of Fe-W solid solutions in the literature. In addition, the derived coefficient of thermal expansion, heat capacity, elastic constants, and elastic moduli of Fe-W solid solutions from the present Fe-W potential are in good agreement with the corresponding data from ab initio calculation or experiments in the literature. read less NOT USED (high confidence) Y. Su, S. Xu, and I. Beyerlein, “Density functional theory calculations of generalized stacking fault energy surfaces for eight face-centered cubic transition metals,” Journal of Applied Physics. 2019. link Times cited: 38 Abstract: In this work, we use density functional theory to calculate … read moreAbstract: In this work, we use density functional theory to calculate the entire generalized stacking fault energy (GSFE) surface for eight transition metals with a face-centered cubic structure: Ag, Au, Cu, Ir, Ni, Pd, Pt, and Rh. Analysis of the ⟨ 112 ⟩ GSFE curves finds that the displacements corresponding to the unstable stacking fault energy are larger than the ideal value for all eight metals except Ag and Cu. Over the entire surface, Pt is found to not possess well-defined local maxima or minima, suggesting spreading in favor of dissociation of the dislocation core, unlike the other seven metals. Our calculations also reveal that at a large ⟨ 112 ⟩ displacement, where atoms on two {111} adjacent planes are aligned, an anomalous local minimum occurs for Ir and Rh. The oddity is explained by relatively large, localized atomic displacements that take place in the two metals to accommodate the alignment that do not occur in the other six metals. In addition to the fully calculated surfaces, we characterize a continuous 11-term Fourier-series function, which provides a particularly excellent representation of the GSFE surfaces for Ag, Au, Cu, Ni, and Pd. read less NOT USED (high confidence) М. Д. Старостенков, П. В. Захаров, and Н. Н. Медведев, “Дискретные бризеры в ГЦК и ГЦТ-кристаллах,” Izvestiya of Altai State University. 2019. link Times cited: 0 Abstract: Обзор посвящен исследованию дискретных бризеров в кристаллах… read moreAbstract: Обзор посвящен исследованию дискретных бризеров в кристаллах стехиометрии A3B, AB и в моноатомных кристаллах. Дана краткая история развития концепции дискретных бризеров в кристаллах, рассмотрены возможные пути дальнейших исследований. Приведены данные, свидетельствующие о том, что для существования дискретных бризеров необходимо наличие свойств дискретности и нелинейности исследуемой системы.
В рассматриваемых работах исследование дискретных бризеров осуществлялось методом молекулярной динамики с использованием как парных, так и многочастичных потенциалов.
На примере кристаллов Pt3Al и CuAu показано, что дискретные бризеры с жестким типом нелинейности способны сосредоточивать энергию порядка нескольких электронвольт, при этом они являются мобильными и могут перемещаться по кристаллу вдоль плотноупакованных направлений. Выявлены два механизма возбуждения дискретных бризеров с мягким типом нелинейности в кристалле стехиометрии А3В потоком частиц. Продемонстрировано, что внешние поля, осциллирующие с частотой вне фононного спектра кристалла А3В, могут являться причиной возбуждения бризеров с мягким типом нелинейности вблизи его поверхности. Для металлов Pt, Au, Ni, Pd, Cu показана зависимость продолжительности жизни ДБ от их коэффициента Пуассона. Приведены амплитудно-частотные характеристики и зависимости времени жизни дискретных бризеров от начальных параметров возбуждения. read less NOT USED (high confidence) P. Chen, Z. Zhang, and F. Qin, “Study of interfacial tensile and shear strength for Cu/Ta interface by molecular dynamic simulation,” 2019 20th International Conference on Electronic Packaging Technology(ICEPT). 2019. link Times cited: 0 Abstract: Three-dimensional integration technology using TSV interconn… read moreAbstract: Three-dimensional integration technology using TSV interconnections has emerged as a promising solution to improve the performance of microelectronic devices. It is necessary to study the effect of strain rate and work temperature on the interfacial strength of Cu/Ta interface for microelectronic devices reliability. In this work, to investigate the effect of the temperature and strain rate on the Cu/Ta interfacial properties, a series of large-scale molecular dynamic (MD) simulations were performed. The stress-strain curves and the deformation processes of Cu/Ta interface were obtained. The results showed that the interfacial tensile strength and interfacial shear strength of Cu(010)/Ta(010) interface are 5.56 GPa and 0.465 GPa, respectively, when the strain rate is 109/s and temperature is 300 K. And the location of failure for Cu/Ta interface is closer to the monocrystalline Cu parts. Then, the effects of strain rate and temperature on the interfacial tensile and shear strength were discussed, and the results indicated that the interfacial strength decrease with the decrease of strain rate from 109/s to 107/s. Similarly, the strongest correlation was seen to be between the interfacial strength and temperature, it can be seen that the interfacial tensile and shear strength decrease with increasing of the temperature. read less NOT USED (high confidence) G. Po, N. Admal, and B. Svendsen, “Non-local Thermoelasticity Based on Equilibrium Statistical Thermodynamics,” Journal of Elasticity. 2019. link Times cited: 1 NOT USED (high confidence) M. Çeltek, Dömekeli̇ Ünal, and S. Şengül, “Moleküler Dinamik Benzetim Yöntemi ile Isıtma İşlemi Sırasında Platin Metalinin Yapısal Gelişimi ve Erime Noktası Üzerine Atomlar-arası Potansiyel Etkisinin Araştırılması,” Bitlis Eren Üniversitesi Fen Bilimleri Dergisi. 2019. link Times cited: 5 Abstract: Bu calismada, farkli atomlar-arasi potansiyeller ve klasik m… read moreAbstract: Bu calismada, farkli atomlar-arasi potansiyeller ve klasik molekuler dinamik (MD) benzetimleri kullanilarak, yuzey merkezli kubik (fcc) yapiya sahip platin (Pt) elementinin taban durumdan baslayip erime noktasinin hemen uzerindeki bir sicaklik araligindaki fiziksel ozellikleri ve atomik yapisinin devinimi detayli bir sekilde incelenmistir. Artan sicakliga bagli olarak kati sistemin yapisal gelisimini analiz etmek ve erime noktasini belirlemek icin ciftler dagilim fonksiyonu (PDF), enerji-sicaklik (E-T), orgu parametresi-sicaklik (a-T), dogrusal termal genlesme katsayi-sicaklik (CLTE-T) egrileri ve cift analiz yontemi gibi analiz yontemleri kullanildi. Tum potansiyeller icin MD benzetiminin sonuclarinin analizinden elde edilen veriler daha once rapor edilen deneysel veya teorik verilerle karsilastirilmis ve tartisilmistir. Farkli atomlar-arasi potansiyellerle elde edilen sonuclar cogunlukla birbirleri ile tutarli olmasina ragmen, farklilik gosterdikleri noktalar da bulunmaktadir. Ozellikle, sistemin erime noktasinin belirlenmesi konusunda, her bir potansiyelin farkli erime sicakliklari urettigi gozlenmistir. Tum potansiyel enerji fonksiyonlarinda ortak olarak, artan sicaklikla birlikte fcc yapiyi temsil eden 1421 bagli ciftlerinin sayisi azalmis ve bu ciftlerin buyuk bir bolumunun ozellikle kusurlu icosahedra ( deficos ) ve kusurlu fcc yapiyi temsil eden 1541 ve 1431 bagli ciftlerine donustugu gorulmustur. Pt elementi icin burada ele alinan potansiyellerin bazilari dusuk bazilari ise yuksek sicaklik araligindaki fiziksel ozellikleri aciklamada basarili olurken, Sheng ve arkadaslari tarafindan one surulen gomulu atom metot potansiyeli (EAM1) ve siki-bagli (TB) potansiyelinin saf Pt elementinin genis sicaklik olceginde ele alinan tum ozelliklerini aciklamada digerlerine gore daha basarili oldugu gorulmustur. read less NOT USED (high confidence) P. Zakharov, M. Starostenkov, and A. Eremin, “Energy transport in an A3B crystal with intense external exposure at frequencies outside the crystal spectrum,” IOP Conference Series: Materials Science and Engineering. 2019. link Times cited: 1 Abstract: The molecular dynamics method is used to consider the effect… read moreAbstract: The molecular dynamics method is used to consider the effect of energy transport in an A3B stoichiometry crystal, using Pt3Al as an example, which consists in the transfer of energy at frequencies outside the phonon spectrum of the crystal. This effect is called the effect of nonlinear supratransmission. The model was a bulk face-centered cubic crystal, the atoms of which interacted through a multi-particle potential, obtained by the immersed atom method. Different forms of oscillation of the region of external influence are considered. The possibility of transporting energy from the crystal surface into the depths by means of excitation of quasi-breathers near the impact area and their subsequent destruction in a crystal and dissipation of energy, stored on them, is shown. The quasi-breathers most intensely occurred near the impact region with a sinusoidal waveform. The results obtained indicate that the contribution of quasi-breathers to the energy transfer through the crystal, increases with increasing exposure amplitude. The minimum amplitude of the external influence, at which this effect was observed, is established. The results of the study can be useful in creating materials with predetermined properties, through various intensive external influences. read less NOT USED (high confidence) W. Wei, L. Chen, H. Gong, and J. Fan, “Strain-stress relationship and dislocation evolution of W–Cu bilayers from a constructed n-body W–Cu potential,” Journal of Physics: Condensed Matter. 2019. link Times cited: 24 Abstract: An n-body W–Cu potential is constructed under the framework … read moreAbstract: An n-body W–Cu potential is constructed under the framework of the embedded-atom method by means of a proposed function of the cross potential. This W–Cu potential is realistic to reproduce mechanical property and structural stability of WCu solid solutions within the entire composition range, and has better performances than the three W–Cu potentials already published in the literature. Based on this W–Cu potential, molecular dynamics simulation is conducted to reveal the mechanical property and dislocation evolution of the bilayer structure between pure W and W0.7Cu0.3 solid solution. It is found that the formation of the interface improves the strength of the W0.7Cu0.3 solid solutions along tensile loading perpendicular to the interface, as the interface impedes the evolution of the dislocation lines from the W0.7Cu0.3 solid solutions to the W part. Simulation also reveals that the interface has an important effect to significantly reduce the tensile strength and critical strain of W along the tensile loading parallel to the interface, which is intrinsically due to the slip of the edge or screw dislocations at low strains as a result of the lattice mismatch. read less NOT USED (high confidence) A. Vorontsov, A. E. Korenchenko, and B. Gelchinski, “Analysis of Stability of Small Metal Clusters during Metal Vapor Condensation,” High Temperature. 2019. link Times cited: 4 NOT USED (high confidence) Y.-heng Zhang, Y. Xu, and H. Chu, “Size effect of layer thickness on stress fields due to interface core-spreading dislocation arrays in multilayers,” Science China Technological Sciences. 2019. link Times cited: 0 NOT USED (high confidence) A. Flor, J. Feliu, C. K. Tsung, and P. Scardi, “Vibrational Properties of Pd Nanocubes,” Nanomaterials. 2019. link Times cited: 4 Abstract: The atomic disorder and the vibrational properties of Pd nan… read moreAbstract: The atomic disorder and the vibrational properties of Pd nanocubes have been studied through a combined use of X-ray diffraction and molecular dynamics simulations. The latter show that the trend of the mean square relative displacement as a function of the radius of the coordination shells is characteristic of the nanoparticle shape and can be described by a combined model: A correlated Debye model for the thermal displacement and a parametric expression for the static disorder. This combined model, supplemented by results of line profile analysis of the diffraction patterns collected at different temperatures (100, 200, and 300 K) can explain the observed increase in the Debye–Waller coefficient, and shed light on the effect of the finite domain size and of the atomic disorder on the vibrational properties of metal nanocrystals. read less NOT USED (high confidence) C. Dai, F. Long, P. Saidi, L. Béland, Z. Yao, and M. Daymond, “Primary damage production in the presence of extended defects and growth of vacancy-type dislocation loops in hcp zirconium,” Physical Review Materials. 2019. link Times cited: 13 Abstract: Production rates in long-term predictive radiation damage ac… read moreAbstract: Production rates in long-term predictive radiation damage accumulation models are generally considered independent of the material's microstructure for reactor components. In this study, the effect of pre-existing microstructural elements on primary damage production in alpha-Zr -- and vice-versa -- is assessed by molecular dynamics (MD) simulations. a-type dislocation loops, c-component dislocation loops and a tilt grain boundary (GB) were considered. Primary damage production is reduced in the presence of all these microstructural elements, and clustering behavior is dependent on the microstructure. Collision cascades do not cause a-type loop growth or shrinkage, but they cause c-component loop shrinkage. Cascades in the presence of the GBs produce more vacancies than interstitials. This result, as well as other theoretical, MD and experimental evidence, confirm that vacancy loops will grow in the vacancy supersaturated environment near GBs. Distinct temperature-dependent growth regimes are identified. Also, MD reveals cascade-induced events where a-type vacancy loops are absorbed by GBs. Fe segregation at the loops inhibits this cascade-induced absorption. read less NOT USED (high confidence) Z. Aitken, V. Sorkin, and Y.-W. Zhang, “Atomistic modeling of nanoscale plasticity in high-entropy alloys,” Journal of Materials Research. 2019. link Times cited: 32 Abstract: Lattice structures, defect structures, and deformation mecha… read moreAbstract: Lattice structures, defect structures, and deformation mechanisms of high-entropy alloys (HEAs) have been studied using atomistic simulations to explain their remarkable mechanical properties. These atomistic simulation techniques, such as first-principles calculations and molecular dynamics allow atomistic-level resolution of structure, defect configuration, and energetics. Following the structure–property paradigm, such understandings can be useful for guiding the design of high-performance HEAs. Although there have been a number of atomistic studies on HEAs, there is no comprehensive review on the state-of-the-art techniques and results of atomistic simulations of HEAs. This article is intended to fill the gap, providing an overview of the state-of-the-art atomistic simulations on HEAs. In particular, we discuss how atomistic simulations can elucidate the nanoscale mechanisms of plasticity underlying the outstanding properties of HEAs, and further present a list of interesting problems for forthcoming atomistic simulations of HEAs. read less NOT USED (high confidence) H. Ke and I. Mastorakos, “Deformation behavior of core–shell nanowire structures with coherent and semi-coherent interfaces,” Journal of Materials Research. 2019. link Times cited: 6 Abstract: The mechanical properties of core–shell bimetallic composite… read moreAbstract: The mechanical properties of core–shell bimetallic composite nanowires, forming the bases of nanoporous metallic foams, have been investigated and compared with pure metal nanowires using molecular dynamics simulations. In the current study, pure copper and gold nanowires under uniaxial loading were tested at room temperature and compared to composite nanowires of the same materials (core) with a nickel coating (shell). The core radius ranged from 1 to 15 nm, and the shell thickness ranged from 0.1 to 5 nm. The tension strain was performed along the [001] direction under room temperature. Both coherent and semi-coherent composite nanowires were studied, and the effect of coating layer thickness was investigated. The strengthening mechanisms of the core–shell structures due to the presence of the two different types of interfaces were investigated for various nickel thicknesses. The atomistic simulation results revealed that the addition of the nickel shell strengthens the structure when the layer thickness exceeds a critical value. read less NOT USED (high confidence) A. Cherednichenko, P. Zakharov, M. Starostenkov, M. O. Sysoeva, and A. Eremin, “Nonlinear supratransmission in a Pt3Al crystal at intense external influence,” Computer Research and Modeling. 2019. link Times cited: 10 NOT USED (high confidence) D. Mishra, M. Meraj, S. K. Badjena, and S. Pal, “Structural evolution and dislocation behaviour study during nanoindentation of Mo20W20Co20Ta20Zr20 high entropy alloy coated Ni single crystal using molecular dynamic simulation,” Molecular Simulation. 2019. link Times cited: 16 Abstract: ABSTRACT In this present study, deformation behaviour of Mo2… read moreAbstract: ABSTRACT In this present study, deformation behaviour of Mo20W20Co20Ta20Zr20 high entropy alloy (HEA) coated single crystal (SC) nickel (Ni) subjected to nanoindentation test have been investigated to study the mechanical properties and underlying mechanism during nanoindentation test using molecular dynamics (MD) simulation with embedded atom method (EAM) potential. Centro-Symmetry Parameter (CSP) Analysis and Radial Distribution Function (RDF) plots are obtained to get insight of structural evolution during nanoindentation and thereby determine the underlying physics of deformation. During nanoindention test Stacking faults (SFs) formation, dislocation generation, dislocation loops, Lomer–Cottrell (LC) lock and Hirth lock formation due to dislocation-dislocation interaction are observed. At higher indentation depth, formation of dislocation loops is augmented, which indicates nanoindentation deformation is found to be Stacking Fault dominated deformation. The accumulation and relaxation of shear stress near indenter tip at the time of deformation process under nanoindentation test causes the variation of dislocation density, strain hardening, and plastic deformation, which is influenced by the formation of dislocation barriers (LC and Hirth locks) and dislocation loops (shear and prismatic loops). read less NOT USED (high confidence) L. Zhang, Y. Shibuta, X. Huang, C. Lu, and M. Liu, “Grain boundary induced deformation mechanisms in nanocrystalline Al by molecular dynamics simulation: From interatomic potential perspective,” Computational Materials Science. 2019. link Times cited: 39 NOT USED (high confidence) A. Eremin, P. Zakharov, M. Starostenkov, and A. S. Vdovin, “ANALYSIS OF STATISTICAL CHARACTERISTICS OF QUASI-BREATHER IN MONOATOMIC FCC METALS Au, Cu, Ni, Pd AND Pt,” Конденсированные среды и межфазные границы. 2018. link Times cited: 1 Abstract: The molecular dynamics method is used to calculate and analy… read moreAbstract: The molecular dynamics method is used to calculate and analyze the statistical characteristics of a quasi-breather with a hard type of nonlinearity in monoatomic FCC metals, for example, Cu, Au, Pt, Ni and Pd. Within the framework of this model, the following statistical characteristics and dependencies were calculated for quasi-breathers: a grouped statistical series of absolute and relative frequencies, a polygon of absolute and relative frequencies, a histogram of relative frequencies, an empirical distribution function, an estimate of the mathematical expectation and variance of the original sample. The densities of phonon states are calculated for all crystals. Statistics allow you to understand the causes of the destruction of breathers and more fully describe the process of their dissipation of energy.
ACKNOWLEDGMENTS
For AME, PVZ, the study was carried out with the financial support of the Russian Federal Property Fund and the Altai Territory within the framework of the scientific project No. 18-42-220002; The MDS is grateful to the Ministry of Education and Science of the basic part of the state task, project No. 3.4820.2017/BC.
read less NOT USED (high confidence) K. Wang, W. Zhu, M. Xiang, Y. Xu, G. Li, and J. Chen, “Improved embedded-atom model potentials of Pb at high pressure: application to investigations of plasticity and phase transition under extreme conditions,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 10 Abstract: Local stress relaxation mechanisms of crystals are a long-st… read moreAbstract: Local stress relaxation mechanisms of crystals are a long-standing interest in the field of materials physics. Constantly encountered inelastic deformation mechanisms in metals under dynamic loadings, such as dislocation, deformation twinning and phase transition, have been extensively discussed separately or as some of their combinations. Recently, virtual melting is found to be a dominant local stress relaxation mechanism under extreme strain rates. However, these deformation mechanisms have never been investigated in the same metal at an atomic level due to the lack of high pressure interatomic potentials. In this work, an embedded-atom model potential of Pb is developed and tested for high pressure applications. The developed potential of Pb could not only reproduce many energetic, elastic and defective properties at ambient conditions well, but also correctly describe face-centered cubic (fcc)-hexagonal close packed (hcp) and hcp-body-centered cubic phase transition of Pb under high pressures. Shock Hugoniot, as well as equation of states for fcc and hcp phase, also agrees well with the literature ones up to more than 100 GPa. With the developed potential, non-equilibrium molecular dynamic simulations are conducted to investigate dynamic behaviors of Pb single crystal under ramp-shock compressions. Depending on applied strain rates, dislocation-mediated plasticity, phase transition and virtual melting, constantly reported by experiments or theoretical investigations, are observed in our results. Additionally, a new phase transition mechanism of Pb subjected to the ramp compressions is uncovered. read less NOT USED (high confidence) L. Zhang, “Studying Stability of Atom Packing for Ti Nanoparticles on Heating by Molecular Dynamics Simulations,” Advanced Engineering Materials. 2018. link Times cited: 11 Abstract: Molecular dynamics simulations using an embedded atom method… read moreAbstract: Molecular dynamics simulations using an embedded atom method (EAM) potential shows that melting behaviors of Ti nanoparticles are strongly dependent on their size. For the particles having the diameter of less than 2.5 nm, their structures are preferred with the icosahedron of geometric shell closures, and there are multi‐structures’ transitions. With the increase in size, while most atoms in the particles can hold their HCP packing patterns, there exist movements and structural rearrangements of the atoms in the surface. At a high temperature, the accumulation of structural disorder can quickly extend into the entire particle, which resembles the melting of bulk Ti. In the HCP particles with sizes of less than 4 nm, the surface atoms still have important influence on their melting. read less NOT USED (high confidence) Y.-C. Su, S. Jiang, Y. Gan, Z. Chen, and J.-M. Lu, “Investigation of the mechanical responses of copper nanowires based on molecular dynamics and coarse-grained molecular dynamics,” Computational Particle Mechanics. 2018. link Times cited: 4 NOT USED (high confidence) V. Samsonov, A. G. Bembel,’ A. Kartoshkin, S. Vasilyev, and I. Talyzin, “Molecular dynamics and thermodynamic simulations of segregation phenomena in binary metal nanoparticles,” Journal of Thermal Analysis and Calorimetry. 2018. link Times cited: 22 NOT USED (high confidence) P. Delafrouz and H. N. Pishkenari, “Coarse-graining models for molecular dynamics simulations of FCC metals,” Journal of Theoretical and Applied Mechanics. 2018. link Times cited: 6 Abstract: In this paper, four coarse-graining (CG) models are proposed… read moreAbstract: In this paper, four coarse-graining (CG) models are proposed to accelerate molecular dynamics
simulations of FCC metals. To this aim, at first, a proper map between beads of the
CG models and atoms of the all-atom (AA) system is assigned, afterwards mass of the beads
and the parameters of the CG models are determined in a manner that the CG models and
the original all-atom model have the same physical properties. To evaluate and compare
precision of these four CG models, different static and dynamic simulations are conducted.
The results show that these CG models are at least 4 times faster than the AA model, while
their errors are less than 1 percent. read less NOT USED (high confidence) P. Gupta and N. Yedla, “Deformation Behavior and Fracture of Al-CuZr Nano-Laminates: A Molecular Dynamics Simulation Study,” Proceedings of the 7th International Conference on Fracture Fatigue and Wear. 2018. link Times cited: 1 NOT USED (high confidence) J. Choi, S. Yoo, S. Song, J. S. Park, and K. Kang, “Molecular dynamics study of Hugoniot relation in shocked nickel single crystal,” Journal of Mechanical Science and Technology. 2018. link Times cited: 0 NOT USED (high confidence) J. Choi, S. Yoo, S. Song, J. Park, and K. Kang, “Molecular dynamics study of Hugoniot relation in shocked nickel single crystal,” Journal of Mechanical Science and Technology. 2018. link Times cited: 8 NOT USED (high confidence) S. Rogachev, O. Politano, F. Baras, and A. Rogachev, “Molecular Dynamics Simulation of Self-Propagating Thermal Waves in Amorphous Cu50Ti50 Films and Thin Cu/Ti Sandwiches,” International Journal of Self-Propagating High-Temperature Synthesis. 2018. link Times cited: 0 NOT USED (high confidence) A. Leino et al., “GeV ion irradiation of NiFe and NiCo: Insights from MD simulations and experiments,” Acta Materialia. 2018. link Times cited: 24 NOT USED (high confidence) L. Hale, Z. Trautt, and C. Becker, “Evaluating variability with atomistic simulations: the effect of potential and calculation methodology on the modeling of lattice and elastic constants,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 40 Abstract: Atomistic simulations using classical interatomic potentials… read moreAbstract: Atomistic simulations using classical interatomic potentials are powerful investigative tools linking atomic structures to dynamic properties and behaviors. It is well known that different interatomic potentials produce different results, thus making it necessary to characterize potentials based on how they predict basic properties. Doing so makes it possible to compare existing interatomic models in order to select those best suited for specific use cases, and to identify any limitations of the models that may lead to unrealistic responses. While the methods for obtaining many of these properties are often thought of as simple calculations, there are many underlying aspects that can lead to variability in the reported property values. For instance, multiple methods may exist for computing the same property and values may be sensitive to certain simulation parameters. Here, we introduce a new high-throughput computational framework that encodes various simulation methodologies as Python calculation scripts. Three distinct methods for evaluating the lattice and elastic constants of bulk crystal structures are implemented and used to evaluate the properties across 120 interatomic potentials, 18 crystal prototypes, and all possible combinations of unique lattice site and elemental model pairings. Analysis of the results reveals which potentials and crystal prototypes are sensitive to the calculation methods and parameters, and it assists with the verification of potentials, methods, and molecular dynamics software. The results, calculation scripts, and computational infrastructure are self-contained and openly available to support researchers in performing meaningful simulations. read less NOT USED (high confidence) Y. Liu, Y. Liu, T. Ma, and J. Luo, “Atomic Scale Simulation on the Anti-Pressure and Friction Reduction Mechanisms of MoS2 Monolayer,” Materials. 2018. link Times cited: 9 Abstract: MoS2 nanosheets can be used as solid lubricants or additives… read moreAbstract: MoS2 nanosheets can be used as solid lubricants or additives of lubricating oils to reduce friction and resist wear. However, the atomic scale mechanism still needs to be illustrated. Herein, molecular simulations on the indentation and scratching process of MoS2 monolayer supported by Pt(111) surface were conducted to study the anti-pressure and friction reduction mechanisms of the MoS2 monolayer. Three deformation stages of Pt-supported MoS2 monolayer were found during the indentation process: elastic deformation, plastic deformation and finally, complete rupture. The MoS2 monolayer showed an excellent friction reduction effect at the first two stages, as a result of enhanced load bearing capacity and reduced deformation degree of the substrate. Unlike graphene, rupture of the Pt-supported MoS2 monolayer was related primarily to out-of-plane compression of the monolayer. These results provide a new insight into the relationship between the mechanical properties and lubrication properties of 2D materials. read less NOT USED (high confidence) F. Gao et al., “Modulation period dependent mechanical properties of Cu/Fe metallic multilayered films,” AIP Advances. 2018. link Times cited: 2 Abstract: We performed molecular dynamics simulations for uniaxial ten… read moreAbstract: We performed molecular dynamics simulations for uniaxial tension of Cu/Fe nano-multilayered films with different modulation periods (λ) using Kurdjumov-Sachs (K-S) relationship to investigate their interfacial morphologies and the effects of λ on their mechanical properties. It shows that the mismatch dislocation lines at the interfaces are periodic. At the Cu side of the interface, triangles distribute with the smallest period; while at the Fe side, parallelograms distribute with the smallest period. There are two yield points, except the case of λ=2.10 nm where there is only one yield point, in the stress-strain curve of Cu/Fe multilayer film under tension, corresponding to the sequential nucleation of dislocations in the Cu and Fe layers from interface, respectively. We further found that there is a critical modulation period, λc, and the flow stress decreases with the increases of λ if λ > λc while increases with the increases of λ if λ λc while increases with the increases of λ if λ < λc. The possible mechanisms are discussed. read less NOT USED (high confidence) P. Chowdhury and H. Sehitoglu, “Atomistic Energetics and Critical Twinning Stress Prediction in Face and Body Centered Cubic Metals: Recent Progress,” Journal of Engineering Materials and Technology-transactions of The Asme. 2018. link Times cited: 19 Abstract: This paper recounts recent advances on the atomistic modelin… read moreAbstract: This paper recounts recent advances on the atomistic modeling of twinning in bodycentered cubic (bcc) and face-centered cubic (fcc) alloy. Specifically, we have reviewed: (i) the experimental evidence of twinning-dominated deformation in singleand multigrain microstructures, (ii) calculation of generalized planar fault energy (GPFE) landscapes, and (iii) the prediction of critical friction stresses to initiate twinning-governed plasticity (e.g., twin nucleation, twin–slip and twin–twin interactions). Possible avenues for further research are outlined. [DOI: 10.1115/1.4038673] read less NOT USED (high confidence) A. Halder, L. Curtiss, A. Fortunelli, and Š. Vajda, “Perspective: Size selected clusters for catalysis and electrochemistry.,” The Journal of chemical physics. 2018. link Times cited: 76 Abstract: Size-selected clusters containing a handful of atoms may pos… read moreAbstract: Size-selected clusters containing a handful of atoms may possess noble catalytic properties different from nano-sized or bulk catalysts. Size- and composition-selected clusters can also serve as models of the catalytic active site, where an addition or removal of a single atom can have a dramatic effect on their activity and selectivity. In this perspective, we provide an overview of studies performed under both ultra-high vacuum and realistic reaction conditions aimed at the interrogation, characterization, and understanding of the performance of supported size-selected clusters in heterogeneous and electrochemical reactions, which address the effects of cluster size, cluster composition, cluster-support interactions, and reaction conditions, the key parameters for the understanding and control of catalyst functionality. Computational modeling based on density functional theory sampling of local minima and energy barriers or ab initio molecular dynamics simulations is an integral part of this research by providing fundamental understanding of the catalytic processes at the atomic level, as well as by predicting new materials compositions which can be validated in experiments. Finally, we discuss approaches which aim at the scale up of the production of well-defined clusters for use in real world applications. read less NOT USED (high confidence) L. Mones, C. Ortner, and G. Csányi, “Preconditioners for the geometry optimisation and saddle point search of molecular systems,” Scientific Reports. 2018. link Times cited: 12 NOT USED (high confidence) S. M. Rassoulinejad-Mousavi and Y. Zhang, “Interatomic Potentials Transferability for Molecular Simulations: A Comparative Study for Platinum, Gold and Silver,” Scientific Reports. 2018. link Times cited: 33 NOT USED (high confidence) A. Shirinyan, G. Wilde, and Y. Bilogorodskyy, “Solidification loops in the phase diagram of nanoscale alloy particles: from a specific example towards a general vision,” Journal of Materials Science. 2018. link Times cited: 19 NOT USED (high confidence) V. S. Baidyshev, Y. Gafner, S. Gafner, and L. Redel, “Thermal stability of Pt nanoclusters interacting to carbon sublattice,” Physics of the Solid State. 2017. link Times cited: 4 NOT USED (high confidence) S. N. Divi, G. Agrahari, S. Kadulkar, S. Kumar, and A. Chatterjee, “Improved prediction of heat of mixing and segregation in metallic alloys using tunable mixing rule for embedded atom method,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 17 Abstract: Capturing segregation behavior in metal alloy nanoparticles … read moreAbstract: Capturing segregation behavior in metal alloy nanoparticles accurately using computer simulations is contingent upon the availability of high-fidelity interatomic potentials. The embedded atom method (EAM) potential is a widely trusted interatomic potential form used with pure metals and their alloys. When limited experimental data is available, the A-B EAM cross-interaction potential for metal alloys AxB1−x are often constructed from pure metal A and B potentials by employing a pre-defined ‘mixing rule’ without any adjustable parameters. While this approach is convenient, we show that for AuPt, NiPt, AgAu, AgPd, AuNi, NiPd, PtPd and AuPd such mixing rules may not even yield the correct alloy properties, e.g., heats of mixing, that are closely related to the segregation behavior. A general theoretical formulation based on scaling invariance arguments is introduced that addresses this issue by tuning the mixing rule to better describe alloy properties. Starting with an existing pure metal EAM potential that is used extensively in literature, we find that the mixing rule fitted to heats of mixing for metal solutions usually provides good estimates of segregation energies, lattice parameters and cohesive energy, as well as equilibrium distribution of metals within a nanoparticle using Monte Carlo simulations. While the tunable mixing rule generally performs better than non-adjustable mixing rules, the use of the tunable mixing rule may still require some caution. For e.g., in Pt–Ni system we find that the segregation behavior can deviate from the experimentally observed one at Ni-rich compositions. Despite this the overall results suggest that the same approach may be useful for developing improved cross-potentials with other existing pure metal EAM potentials as well. As a further test of our approach, mixing rule estimated from binary data is used to calculate heat of mixing in AuPdPt, AuNiPd, AuPtNi, AgAuPd and NiPtPd. Excellent agreement with experiments is observed for AuPdPt. read less NOT USED (high confidence) T. He et al., “Inflating hollow nanocrystals through a repeated Kirkendall cavitation process,” Nature Communications. 2017. link Times cited: 114 NOT USED (high confidence) A. Takahashi, A. Seko, and I. Tanaka, “Linearized machine-learning interatomic potentials for non-magnetic elemental metals: Limitation of pairwise descriptors and trend of predictive power.,” The Journal of chemical physics. 2017. link Times cited: 20 Abstract: Machine-learning interatomic potential (MLIP) has been of gr… read moreAbstract: Machine-learning interatomic potential (MLIP) has been of growing interest as a useful method to describe the energetics of systems of interest. In the present study, we examine the accuracy of linearized pairwise MLIPs and angular-dependent MLIPs for 31 elemental metals. Using all of the optimal MLIPs for 31 elemental metals, we show the robustness of the linearized frameworks, the general trend of the predictive power of MLIPs, and the limitation of pairwise MLIPs. As a result, we obtain accurate MLIPs for all 31 elements using the same linearized framework. This indicates that the use of numerous descriptors is the most important practical feature for constructing MLIPs with high accuracy. An accurate MLIP can be constructed using only pairwise descriptors for most non-transition metals, whereas it is very important to consider angular-dependent descriptors when expressing interatomic interactions of transition metals. read less NOT USED (high confidence) I. Chepkasov, Y. Gafner, M. A. Vysotin, and L. Redel, “A study of melting of various types of Pt–Pd nanoparticles,” Physics of the Solid State. 2017. link Times cited: 10 NOT USED (high confidence) Y. Zhang, S. Zhao, W. J. Weber, K. Nordlund, F. Granberg, and F. Djurabekova, “Atomic-level heterogeneity and defect dynamics in concentrated solid-solution alloys,” Current Opinion in Solid State & Materials Science. 2017. link Times cited: 142 NOT USED (high confidence) L. Chen, Q. Wang, and L. Xiong, “Molecular dynamics study on structure stability, lattice variation, and melting behavior of silver nanoparticles,” Journal of Nanoparticle Research. 2017. link Times cited: 3 NOT USED (high confidence) T. Otieno and K. Abou-El-Hossein, “Molecular dynamics analysis of nanomachining of rapidly solidified aluminium,” The International Journal of Advanced Manufacturing Technology. 2017. link Times cited: 0 NOT USED (high confidence) L. Hale and C. Becker, “Vacancy dissociation in body-centered cubic screw dislocation cores,” Computational Materials Science. 2017. link Times cited: 9 NOT USED (high confidence) P. Zakharov, A. Eremin, M. Starostenkov, and I. S. Lucenko, “Analysis of Statistical Characteristics of Quasi-Breather with Soft-Type of Nonlinearity in the Crystals of A3B Stoichiometry,” Key Engineering Materials. 2017. link Times cited: 7 Abstract: The research of quasi-breather statistical characteristics i… read moreAbstract: The research of quasi-breather statistical characteristics in the model crystal of A3B stoichiometry is conducted by means of molecular dynamics method in the paper by the example of Pt3Al. The phonon spectrum of this model crystal, the dependences of mean-square deviation, the coefficient of variation and the average frequency of the model quasi-breather on the time of its existence are obtained. The statistical data analysis allows for the conclusion that a quasi-breather model solution slightly differs from the exact breather corresponding to it in the model under consideration using the interatomic potential obtained by means of embedded atom method (EAM). read less NOT USED (high confidence) C. Chen, Z. Deng, R. Tran, H. Tang, I. Chu, and S. Ong, “Accurate Force Field for Molybdenum by Machine Learning Large Materials Data,” arXiv: Computational Physics. 2017. link Times cited: 94 Abstract: In this work, we present a highly accurate spectral neighbor… read moreAbstract: In this work, we present a highly accurate spectral neighbor analysis potential (SNAP) model for molybdenum (Mo) developed through the rigorous application of machine learning techniques on large materials data sets. Despite Mo's importance as a structural metal, existing force fields for Mo based on the embedded atom and modified embedded atom methods still do not provide satisfactory accuracy on many properties. We will show that by fitting to the energies, forces and stress tensors of a large density functional theory (DFT)-computed dataset on a diverse set of Mo structures, a Mo SNAP model can be developed that achieves close to DFT accuracy in the prediction of a broad range of properties, including energies, forces, stresses, elastic constants, melting point, phonon spectra, surface energies, grain boundary energies, etc. We will outline a systematic model development process, which includes a rigorous approach to structural selection based on principal component analysis, as well as a differential evolution algorithm for optimizing the hyperparameters in the model fitting so that both the model error and the property prediction error can be simultaneously lowered. We expect that this newly developed Mo SNAP model will find broad applications in large-scale, long-time scale simulations. read less NOT USED (high confidence) Y. Afkham, M. Bahramyan, R. T. Mousavian, and D. Brabazon, “Tensile properties of AlCrCoFeCuNi glassy alloys: A molecular dynamics simulation study,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2017. link Times cited: 41 NOT USED (high confidence) Y. Jiang, J. Luo, and Y. Wu, “The validation and preference among different EAM potentials to describe the solid–liquid transition of aluminum,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 6 Abstract: Empirical potential is vital to the classic atomic simulatio… read moreAbstract: Empirical potential is vital to the classic atomic simulation, especially for the study of phase transitions, as well as the solid-interface. In this paper, we attempt to set up a uniform procedure for the validation among different potentials before the formal simulation study of phase transitions of metals. Two main steps are involved: (1) the prediction of the structures of both solid and liquid phases and their mutual transitions, i.e. melting and crystallization; (2) the prediction of vital thermodynamic (the equilibrium melting point at ambient pressure) and dynamic properties (the degrees of superheating and undercooling). We applied this procedure to the testing of seven published embedded-atom potentials (MKBA (Mendelev et al 2008 Philos. Mag. 88 1723), MFMP (Mishin et al 1999 Phys. Rev. B 59 3393), MDSL (Sturgeon and Laird 2000 Phys. Rev. B 62 14720), ZM (Zope and Mishin 2003 Phys. Rev. B 68 024102), LEA (Liu et al 2004 Model. Simul. Mater. Sci. Eng. 12 665), WKG (Winey et al 2009 Model. Simul. Mater. Sci. Eng. 17 055004) and ZJW (Zhou et al 2004 Phys. Rev. B 69 144113)) for the description of the solid–liquid transition of Al. All the predictions of structure, melting point and superheating/undercooling degrees were compared with the experiments or theoretical calculations. Then, two of them, MKBA and MDSL, were proven suitable for the study of the solid–liquid transition of Al while the residuals were unqualified. However, potential MKBA is more accurate to predict the structures of solid and liquid, while MDSL works a little better in the thermodynamic and dynamic predictions of solid–liquid transitions. read less NOT USED (high confidence) Z. Zhang, S. Huang, L. Chen, Z. Zhu, and D. Guo, “Formation mechanism of fivefold deformation twins in a face-centered cubic alloy,” Scientific Reports. 2017. link Times cited: 12 NOT USED (high confidence) J. Zhu, P. Quarterman, and J. Wang, “Molecular dynamic simulation study of plasma etching L10 FePt media in embedded mask patterning (EMP) process,” AIP Advances. 2017. link Times cited: 0 Abstract: Plasma etching process of single-crystal L10-FePt media [H. … read moreAbstract: Plasma etching process of single-crystal L10-FePt media [H. Wang et al., Appl. Phys. Lett. 102(5) (2013)] is studied using molecular dynamic simulation. Embedded-Atom Method [M. S. Daw and M. I. Baskes, Phy. Rev. B 29, 6443 (1984); X. W. Zhou, R. A. Johnson and H. N. G. Wadley, Phy. Rev. B 69, 144113 (2004)] is used to calculate the interatomic potential within atoms in FePt alloy, and ZBL potential [J.F. Ziegler, J. P. Biersack and U. Littmark, “The Stopping and Range of Ions in Matter,” Volume 1, Pergamon,1985] in comparison with conventional Lennard-Jones “12-6” potential is applied to interactions between etching gas ions and metal atoms. It is shown the post-etch structure defects can include amorphized surface layer and lattice interstitial point defects that caused by etchant ions passed through the surface layer. We show that the amorphized or damaged FePt lattice surface layer (or “magnetic dead-layer”) thickness after etching increases with ion energy for Ar ion impacts, but significantly small ... read less NOT USED (high confidence) F. Calvo, N. Combe, J. Morillo, and M. Benoit, “Modeling Iron–Gold Nanoparticles Using a Dedicated Semi-Empirical Potential: Application to the Stability of Core–Shell Structures,” Journal of Physical Chemistry C. 2017. link Times cited: 16 Abstract: Core–shell nanoparticles made from iron embedded in gold hav… read moreAbstract: Core–shell nanoparticles made from iron embedded in gold have a strong potential interest in nanomedicine, the Au shell providing an efficient biocompatible coating for the magnetic Fe core. With the aim of determining theoretically the equilibrium morphologies of Fe–Au nanoparticles in a broad size range and with different compositions, a semiempirical many-body Fe–Au potential was designed combining well-established models for the pure metals and introducing dedicated contributions for the interactions involving mixed elements. The potential was parametrized against various energetic properties involving impurities, intermetallics, and finite clusters obtained using density functional calculations in the generalized gradient approximation. The potential was tested to investigate Fe–Au nanoparticles near equiconcentration containing about 1000–2000 atoms at finite temperature using parallel tempering Monte Carlo simulations initiated from the core–shell structure. The core–shell nanoparticles are found t... read less NOT USED (high confidence) Y.-hua Zhou, R. Smith, S. Kenny, and A. L. Lloyd, “Development of an empirical interatomic potential for the Ag–Ti system,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 5 NOT USED (high confidence) A. Tran and Y. Wang, “Reliable Molecular Dynamics: Uncertainty quantification using interval analysis in molecular dynamics simulation,” Computational Materials Science. 2017. link Times cited: 17 NOT USED (high confidence) S. N. Divi and A. Chatterjee, “Understanding Segregation Behavior in AuPt, NiPt, and AgAu Bimetallic Nanoparticles Using Distribution Coefficients,” Journal of Physical Chemistry C. 2016. link Times cited: 21 Abstract: Bimetallic nanoparticles (BNPs) often possess peculiar segre… read moreAbstract: Bimetallic nanoparticles (BNPs) often possess peculiar segregation behavior as the particle size, composition, shape, and temperature are varied. However, a thermodynamic model for this phenomenon has been lacking thus far. We show for the first time that the distribution of metal species within a nanoparticle can be adequately captured in terms of distribution coefficients calculated for the facets, facet edges, and bulk regions. Thermodynamic relations for the distribution coefficients are derived. Only m distribution coefficients from the m(m – 1) distribution coefficients are independent, where m denotes the number of regions. The theory is applied to AuPt, NiPt, and AuAg BNPs. Distribution coefficients are calculated at 400 and 600 K using Monte Carlo (MC) simulations of varying BNP sizes and compositions. A wide range of mixing behavior from alloying to partial or full segregation and core–shell to onion-like structures can be observed. A key finding is that the distribution coefficients are indepen... read less NOT USED (high confidence) K. Sasikumar et al., “Evolutionary Optimization of a Charge Transfer Ionic Potential Model for Ta/Ta-Oxide Heterointerfaces,” Chemistry of Materials. 2016. link Times cited: 21 Abstract: Heterostructures of tantalum and its oxide are of tremendous… read moreAbstract: Heterostructures of tantalum and its oxide are of tremendous technological interest for myriad technological applications, including electronics, thermal management, catalysis, and biochemistry. In particular, local oxygen stoichiometry variation in TaOx memristors comprising thermodynamically stable metallic (Ta) and insulating oxide (Ta2O5) have been shown to result in fast switching on the subnanosecond time scale over a billion cycles. This rapid switching opens up the potential for advanced functional platforms such as stateful logic operations and neuromorphic computation. Despite its broad importance, an atomistic scale understanding of oxygen stoichiometry variation across Ta/TaOx heterointerfaces, such as during early stages of oxidation and oxide growth, is not well understood. This is mainly due to the lack of a unified interatomic potential model for tantalum oxides that can accurately describe metallic (Ta) and ionic (TaOx) as well as mixed (Ta/TaOx interfaces) bonding environments simultaneo... read less NOT USED (high confidence) E. P. Estevez, R. Burganova, and Y. V. Lysogorskii, “Computer Simulation of the Elastic Properties of Titanium Alloys for Medical Applications,” Journal of Engineering Physics and Thermophysics. 2016. link Times cited: 1 NOT USED (high confidence) T. Zientarski and D. Chocyk, “Stress induced grain boundaries in thin Co layer deposited on Au and Cu,” Applied Physics A. 2016. link Times cited: 5 NOT USED (high confidence) E. P. Estevez, R. Burganova, and Y. V. Lysogorskii, “Computer Simulation of the Elastic Properties of Titanium Alloys for Medical Applications,” Journal of Engineering Physics and Thermophysics. 2016. link Times cited: 0 NOT USED (high confidence) A. Sharma, P. Singh, D. D. Johnson, P. Liaw, and G. Balasubramanian, “Atomistic clustering-ordering and high-strain deformation of an Al0.1CrCoFeNi high-entropy alloy,” Scientific Reports. 2016. link Times cited: 86 NOT USED (high confidence) S. Groh, “Modified embedded-atom potential for B2-MgAg,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 5 Abstract: Interatomic potentials for pure Ag and Mg–Ag alloy have been… read moreAbstract: Interatomic potentials for pure Ag and Mg–Ag alloy have been developed in the framework of the second nearest-neighbors modified embedded-atom method (MEAM). The validity and the transferability of the Ag potential were obtained by calculating physical, mechanical, thermal, and dislocation related properties. Since the {1 1 1}-generalized stacking fault energy curves obtained from first-principle calculations was used to develop the Ag potential, the critical resolved shear stress to move screw dislocations in Ag single crystal is in good agreement with the experimental data. By combining the ability of the potential to predict the surface energies with its accuracy in describing dislocation properties, the potential is thought to be a predictive model for analyzing the fracture properties of Ag. In addition, the performance of the potential was tested under dynamics conditions by predicting the melting temperature, where a good agreement with experimental value was found. The Ag-MEAM potential was then coupled to an existing Mg-MEAM potential to describe the properties of the binary system MgAg. While the heat of formation, the elastic constants, and the (1 1 0) γ-surface of the MgAg compound in the B2 phase were used to parameterize the potential, heat of formation for MgAg alloys with different stoichiometry, thermal properties of the B2-MgAg compound, as well as dislocation related properties in B2-MgAg compound were tested to validate the transferability of the potential. The heat of formation of Mg5Ag2, MgAg, and MgAg3, the elastic constants and the thermal properties of B2-MgAg obtained with the proposed potential align with first-principles and experimental data. In addition, the core structure of both 〈0 0 1〉 and 〈1 1 1〉 dislocations in {1 1 0} are in agreement with theoretical predictions, and the magnitudes of the critical resolved shear stress obtained at 0 K for both slip systems partially validate the slip behavior of B2-MgAg. Furthermore, the interaction between silver solute element and dislocations from the basal plane is correctly captured by the potential. read less NOT USED (high confidence) N. Admal, J. Marian, and G. Po, “The atomistic representation of first strain-gradient elastic tensors,” Journal of The Mechanics and Physics of Solids. 2016. link Times cited: 36 NOT USED (high confidence) Gui-Jun 贵钧 Cheng 程, B. Fu 付, Q. Hou 侯, X. Zhou 周, and Jun 俊 Wang 汪, “Diffusion behavior of helium in titanium and the effect of grain boundaries revealed by molecular dynamics simulation,” Chinese Physics B. 2016. link Times cited: 11 Abstract: The microstructures of titanium (Ti), an attractive tritium … read moreAbstract: The microstructures of titanium (Ti), an attractive tritium (T) storage material, will affect the evolution process of the retained helium (He). Understanding the diffusion behavior of He at the atomic scale is crucial for the mechanism of material degradation. The novel diffusion behavior of He has been reported by molecular dynamics (MD) simulation for the bulk hcp-Ti system and the system with grain boundary (GB). It is observed that the diffusion of He in the bulk hcp-Ti is significantly anisotropic (the diffusion coefficient of the [0001] direction is higher than that of the basal plane), as represented by the different migration energies. Different from convention, the GB accelerates the diffusion of He in one direction but not in the other. It is observed that a twin boundary (TB) can serve as an effective trapped region for He. The TB accelerates diffusion of He in the direction perpendicular to the twinning direction (TD), while it decelerates the diffusion in the TD. This finding is attributable to the change of diffusion path caused by the distortion of the local favorable site for He and the change of its number in the TB region. read less NOT USED (high confidence) P. Chowdhury, H. Sehitoglu, H. Maier, and R. Rateick, “Strength prediction in NiCo alloys - The role of composition and nanotwins,” International Journal of Plasticity. 2016. link Times cited: 58 NOT USED (high confidence) F. Granberg et al., “Mechanism of Radiation Damage Reduction in Equiatomic Multicomponent Single Phase Alloys.,” Physical review letters. 2016. link Times cited: 313 Abstract: Recently a new class of metal alloys, of single-phase multic… read moreAbstract: Recently a new class of metal alloys, of single-phase multicomponent composition at roughly equal atomic concentrations ("equiatomic"), have been shown to exhibit promising mechanical, magnetic, and corrosion resistance properties, in particular, at high temperatures. These features make them potential candidates for components of next-generation nuclear reactors and other high-radiation environments that will involve high temperatures combined with corrosive environments and extreme radiation exposure. In spite of a wide range of recent studies of many important properties of these alloys, their radiation tolerance at high doses remains unexplored. In this work, a combination of experimental and modeling efforts reveals a substantial reduction of damage accumulation under prolonged irradiation in single-phase NiFe and NiCoCr alloys compared to elemental Ni. This effect is explained by reduced dislocation mobility, which leads to slower growth of large dislocation structures. Moreover, there is no observable phase separation, ordering, or amorphization, pointing to a high phase stability of this class of alloys. read less NOT USED (high confidence) S. Baik et al., “Atomic‐Scale Structural and Chemical Study of Columnar and Multilayer Re–Ni Electrodeposited Thermal Barrier Coating ,” Advanced Engineering Materials. 2016. link Times cited: 15 Abstract: Rhenium alloys exhibit a unique combination of chemical, phy… read moreAbstract: Rhenium alloys exhibit a unique combination of chemical, physical, and mechanical properties that makes them attractive for a variety of applications. Herein, we present atomic‐scale structural and atomic part‐per‐million level three‐dimensional (3D) chemical characterization of a Re–Ni coating, combining aberration‐corrected scanning transmission electron microscopy (STEM) and atom‐probe tomography (APT). A unique combination of a columnar and multilayer structure is formed by single‐bath dc‐electroplating and is reported here for the first time. Alternating thicker Re‐rich and thinner Ni‐rich layers support a mechanism in which Ni acts as a reducing agent. The multilayers exhibit hetero‐epitaxial growth resulting in high residual shear stresses that lead to formation of corrugated interfaces and an outer layer with mud‐cracks. read less NOT USED (high confidence) M. Barisik and A. Beskok, “Interface Resistance and Thermal Transport in Nano-Confined Liquids.” 2016. link Times cited: 0 Abstract: Miniaturization of microelectronic device components and the… read moreAbstract: Miniaturization of microelectronic device components and the development of nano– electro– mechanical systems require advanced understanding of thermal transport in nano-materials and devices, where the atomic nature of matter becomes important and the validity of well-known continuum approximations becomes questionable [1]. In the case of semiconductors and insulators, heat is carried primarily by vibrations in the crystal lattice known as phonons. Phonon transport is classically studied by lattice dynamics based on harmonic wave theory in the frequency space. However, the anharmonic behaviors forming in a crystal structure cannot be described with this theory. Alternatively, the coupled motions of the atoms in real space can be modeled by molecular dynamics (MD), which provides the natural formation and transport of phonons via vibrations in the crystal lattice. Hence, MD has been widely employed to model phonon transfer in nanostructures and channels [2,3]. The performance and reliability of aforementioned devices strongly depend on the removal of heat either to the ambient or to a coolant. In such cases, phonon transport observed at the interfaces of nanoscale device components and surrounding/confined fluid, or at the interfaces of suspended nanoparticles and fluid medium in nano-fluidic coolants plays a critical role. At such interfaces, heat transfer is interrupted with a temperature jump due to the deficiency in overlap between phonon dispersions of dissimilar materials. Classical theories considering specular or diffuse phonon scattering predict the upper or lower limits of interface thermal resistance (ITR), while a detailed investigation of intermolecular interactions is needed to resolve interface phonon scattering mechanisms. In this chapter, we present interface phonon transfer at the molecular level, and investigate the validity of continuum hypothesis and Fourier’s law in nano-channels. First, we focus on the conventional ways of using MD for heat transport problems. Most of the previous MD research sandwiched a liquid domain between two solid walls and induced heat flux by fixing the wall CONTENTS read less NOT USED (high confidence) K. Wang, J.-fang Liu, Q. Chen, W. Sun, A. Ni, and C. Zhang, “Molecular dynamics simulation of impact of palladium clusters on the zirconium substrate,” Russian Journal of Physical Chemistry A. 2016. link Times cited: 1 NOT USED (high confidence) G. P. P. Pun, K. Darling, L. Kecskes, and Y. Mishin, “Angular-dependent interatomic potential for the Cu–Ta system and its application to structural stability of nano-crystalline alloys,” Acta Materialia. 2015. link Times cited: 92 NOT USED (high confidence) P. Brault and E. Neyts, “Molecular dynamics simulations of supported metal nanocatalyst formation by plasma sputtering,” Catalysis Today. 2015. link Times cited: 26 NOT USED (high confidence) V. Podryga and S. Polyakov, “Molecular dynamic simulation of thermodynamic equilibrium establishment in nickel,” Mathematical Models and Computer Simulations. 2015. link Times cited: 13 NOT USED (high confidence) K. Zhang, M. Fan, Y. Liu, J. Schroers, M. Shattuck, and C. O’Hern, “Beyond packing of hard spheres: The effects of core softness, non-additivity, intermediate-range repulsion, and many-body interactions on the glass-forming ability of bulk metallic glasses.,” The Journal of chemical physics. 2015. link Times cited: 16 Abstract: When a liquid is cooled well below its melting temperature a… read moreAbstract: When a liquid is cooled well below its melting temperature at a rate that exceeds the critical cooling rate Rc, the crystalline state is bypassed and a metastable, amorphous glassy state forms instead. Rc (or the corresponding critical casting thickness dc) characterizes the glass-forming ability (GFA) of each material. While silica is an excellent glass-former with small Rc < 10(-2) K/s, pure metals and most alloys are typically poor glass-formers with large Rc > 10(10) K/s. Only in the past thirty years have bulk metallic glasses (BMGs) been identified with Rc approaching that for silica. Recent simulations have shown that simple, hard-sphere models are able to identify the atomic size ratio and number fraction regime where BMGs exist with critical cooling rates more than 13 orders of magnitude smaller than those for pure metals. However, there are a number of other features of interatomic potentials beyond hard-core interactions. How do these other features affect the glass-forming ability of BMGs? In this manuscript, we perform molecular dynamics simulations to determine how variations in the softness and non-additivity of the repulsive core and form of the interatomic pair potential at intermediate distances affect the GFA of binary alloys. These variations in the interatomic pair potential allow us to introduce geometric frustration and change the crystal phases that compete with glass formation. We also investigate the effect of tuning the strength of the many-body interactions from zero to the full embedded atom model on the GFA for pure metals. We then employ the full embedded atom model for binary BMGs and show that hard-core interactions play the dominant role in setting the GFA of alloys, while other features of the interatomic potential only change the GFA by one to two orders of magnitude. Despite their perturbative effect, understanding the detailed form of the intermetallic potential is important for designing BMGs with cm or greater casting thickness. read less NOT USED (high confidence) V. Podryga and S. Polyakov, “Molecular dynamic simulation of thermodynamic equilibrium establishment in nickel,” Mathematical Models and Computer Simulations. 2015. link Times cited: 0 NOT USED (high confidence) P. Moseley and W. A. Curtin, “Computational Design of Strain in Core-Shell Nanoparticles for Optimizing Catalytic Activity.,” Nano letters. 2015. link Times cited: 78 Abstract: Surface strains in core-shell nanoparticles modify catalytic… read moreAbstract: Surface strains in core-shell nanoparticles modify catalytic activity. Here, a continuum-based strategy enables accurate surface-strain-based screening and design of core-shell systems using minimal input as a means to enhance catalytic activity. The approach is validated here for Pt shells on Cu(x)Pt(1-x) cores and used to interpret experimental results on the oxygen reduction reaction in the same system. The analysis shows that precise control of particle sizes and shell thicknesses is required to achieve peak activity, rationalizing the limited increases in activity observed in experiments. The method is also applied to core-shell nanorods to demonstrate its wide applicability. read less NOT USED (high confidence) T. Brink, M. Peterlechner, H. Rosner, K. Albe, and G. Wilde, “Influence of Crystalline Nanoprecipitates on Shear-Band Propagation in Cu-Zr Based Metallic Glasses,” arXiv: Materials Science. 2015. link Times cited: 48 Abstract: The interaction of shear bands with crystalline nanoprecipit… read moreAbstract: The interaction of shear bands with crystalline nanoprecipitates in Cu-Zr-based metallic glasses is investigated by a combination of high-resolution TEM imaging and molecular-dynamics computer simulations. Our results reveal different interaction mechanisms: Shear bands can dissolve precipitates, can wrap around crystalline obstacles, or can be blocked depending on size and density of the precipitates. If the crystalline phase has a low yield strength, we also observe slip transfer through the precipitate. Based on the computational results and experimental findings, a qualitative mechanism map is proposed that categorizes the various processes as a function of the critical stress for dislocation nucleation, precipitate size, and distance. read less NOT USED (high confidence) J. Velasco‐Vélez et al., “The structure of interfacial water on gold electrodes studied by x-ray absorption spectroscopy,” Science. 2014. link Times cited: 349 Abstract: The molecular structure of the electrical double layer deter… read moreAbstract: The molecular structure of the electrical double layer determines the chemistry in all electrochemical processes. Using x-ray absorption spectroscopy (XAS), we probed the structure of water near gold electrodes and its bias dependence. Electron yield XAS detected at the gold electrode revealed that the interfacial water molecules have a different structure from those in the bulk. First principles calculations revealed that ~50% of the molecules lie flat on the surface with saturated hydrogen bonds and another substantial fraction with broken hydrogen bonds that do not contribute to the XAS spectrum because their core-excited states are delocalized by coupling with the gold substrate. At negative bias, the population of flat-lying molecules with broken hydrogen bonds increases, producing a spectrum similar to that of bulk water. The water double-layer structure at an electrode changed from ordered to disordered when the applied bias was switched. Dissecting the electrical double layer The structure of water within a nanometer of an electrode surface is known as the electrical double layer. This layer creates a strong electrical field that can affect electrochemical reactions. Velasco-Velez et al. explored the structure of the electrical double layer at a bare gold electrode. With no applied potential and at positive potentials, the layer is highly structured (resembling ice) with few dangling hydrogen bonds. However, at negative potentials, the layer was more like bulk water, and half of the water molecules lie flat on the surface. Science, this issue p. 831 read less NOT USED (high confidence) E. Bourasseau, G. Filippini, A. Ghoufi, and P. Malfreyt, “Calculation of the surface tension of pure tin from atomistic simulations of liquid–vapour systems,” Molecular Physics. 2014. link Times cited: 8 Abstract: Monte Carlo simulations of heterogeneous systems of tin at l… read moreAbstract: Monte Carlo simulations of heterogeneous systems of tin at liquid–vapour equilibrium have been performed at several temperatures from 600 to 1500 K, using a modified embedded atom model potential. Surface tension of the corresponding planar interfaces has been evaluated using the test area method. Calculation results are in good agreement with experiments presenting a maximum deviation of 10% from experiments. In addition, the Monte Carlo simulations provide a temperature coefficient (the derivative of the surface tension in regard with temperature) in reasonable agreement with the experimental coefficient. read less NOT USED (high confidence) A. Thompson, L. Swiler, C. Trott, S. Foiles, and G. Tucker, “Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials,” J. Comput. Phys. 2014. link Times cited: 589 NOT USED (high confidence) G. Filippini, E. Bourasseau, A. Ghoufi, F. Goujon, and P. Malfreyt, “Communication: Slab thickness dependence of the surface tension: toward a criterion of liquid sheets stability.,” The Journal of chemical physics. 2014. link Times cited: 9 Abstract: Microscopic Monte Carlo simulations of liquid sheets of copp… read moreAbstract: Microscopic Monte Carlo simulations of liquid sheets of copper and tin have been performed in order to study the dependence of the surface tension on the thickness of the sheet. It results that the surface tension is constant with the thickness as long as the sheet remains in one piece. When the sheet is getting thinner, holes start to appear, and the calculated surface tension rapidly decreases with thickness until the sheet becomes totally unstable and forms a cylinder. We assume here that this decrease is not due to a confinement effect as proposed by Werth et al. [Physica A 392, 2359 (2013)] on Lennard-Jones systems, but to the appearance of holes that reduces the energy cost of the surface modification. We also show in this work that a link can be established between the stability of the sheet and the local fluctuations of the surface position, which directly depends on the value of the surface tension. Finally, we complete this study by investigating systems interacting through different forms of Lennard-Jones potentials to check if similar conclusions can be drawn. read less NOT USED (high confidence) S. Khanal et al., “Synthesis, characterization, and growth simulations of Cu–Pt bimetallic nanoclusters,” Beilstein Journal of Nanotechnology. 2014. link Times cited: 17 Abstract: Summary Highly monodispersed Cu–Pt bimetallic nanoclusters w… read moreAbstract: Summary Highly monodispersed Cu–Pt bimetallic nanoclusters were synthesized by a facile synthesis approach. Analysis of transmission electron microscopy (TEM) and spherical aberration (C s)-corrected scanning transmission electron microscopy (STEM) images shows that the average diameter of the Cu–Pt nanoclusters is 3.0 ± 1.0 nm. The high angle annular dark field (HAADF-STEM) images, intensity profiles, and energy dispersive X-ray spectroscopy (EDX) line scans, allowed us to study the distribution of Cu and Pt with atomistic resolution, finding that Pt is embedded randomly in the Cu lattice. A novel simulation method is applied to study the growth mechanism, which shows the formation of alloy structures in good agreement with the experimental evidence. The findings give insight into the formation mechanism of the nanosized Cu–Pt bimetallic catalysts. read less NOT USED (high confidence) F. Z. Dai and W.-Z. Zhang, “A simple method for constructing a reliable initial atomic configuration of a general interface for energy calculation,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 9 Abstract: Interfacial energy calculated with an atomistic model is ver… read moreAbstract: Interfacial energy calculated with an atomistic model is very sensitive to adding atoms to or removing atoms from the interface, especially when the interface has a general orientation. Therefore, it is crucial to construct an appropriate initial atomic configuration in order to obtain a reliable value for the interfacial energy from atomistic simulations. In this work, a simple method is proposed for constructing the atomic configuration of a general interface under the condition that the interface is virtually free of interstitial and vacancy. The validity of the method is demonstrated by using it to calculate the equilibrium morphology of precipitates with interfaces in irrational orientations, which shows good agreement with experimental observations. read less NOT USED (high confidence) B. Onat and S. Durukanoğlu, “An optimized interatomic potential for Cu–Ni alloys with the embedded-atom method,” Journal of Physics: Condensed Matter. 2014. link Times cited: 86 Abstract: We have developed a semi-empirical and many-body type model … read moreAbstract: We have developed a semi-empirical and many-body type model potential using a modified charge density profile for Cu–Ni alloys based on the embedded-atom method (EAM) formalism with an improved optimization technique. The potential is determined by fitting to experimental and first-principles data for Cu, Ni and Cu–Ni binary compounds, such as lattice constants, cohesive energies, bulk modulus, elastic constants, diatomic bond lengths and bond energies. The generated potentials were tested by computing a variety of properties of pure elements and the alloy of Cu, Ni: the melting points, alloy mixing enthalpy, lattice specific heat, equilibrium lattice structures, vacancy formation and interstitial formation energies, and various diffusion barriers on the (100) and (111) surfaces of Cu and Ni. read less NOT USED (high confidence) M. Grouchko et al., “Correction: Corrigendum: Merging of metal nanoparticles driven by selective wettability of silver nanostructures,” Nature Communications. 2014. link Times cited: 62 NOT USED (high confidence) M. Grouchko et al., “Merging of metal nanoparticles driven by selective wettability of silver nanostructures,” Nature Communications. 2014. link Times cited: 10 NOT USED (high confidence) L. Xie, P. Brault, A. Thomann, and J. Bauchire, “AlCoCrCuFeNi high entropy alloy cluster growth and annealing on silicon: A classical molecular dynamics simulation study,” Applied Surface Science. 2013. link Times cited: 102 NOT USED (high confidence) J. Yang, C. Mao, X. Li, and C. Liu, “On the Cauchy-Born approximation at finite temperature for alloys,” Discrete & Continuous Dynamical Systems - B. 2013. link Times cited: 4 NOT USED (high confidence) F. Dai and W.-Z. Zhang, “A systematic study on the interfacial energy of O-line interfaces in fcc/bcc systems,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 11 Abstract: Habit planes between face-centered cubic (fcc)/body-centered… read moreAbstract: Habit planes between face-centered cubic (fcc)/body-centered cubic (bcc) phases usually exhibit irrational orientations, which often agree with the O-line criterion. Previously, energy calculation was made to test whether the habit planes were energetically favorable, but the values of the energy were found very sensitive to the initial atomic configuration in an irrationally orientated interface. In this paper, under the O-line condition, simple selection criteria are proposed to define and remove interfacial interstitials and vacancies in the initial atomic configuration. The criteria are proved to be effective in obtaining robust energy results. Interfacial energies of two types of O-line interfaces in fcc/bcc systems are calculated following the criteria. The observed transformation crystallography of precipitates in Ni–Cr and Cu–Cr systems can be explained consistently as the irrational habit plane in each system is associated with the lowest energy O-line interface. read less NOT USED (high confidence) D. Hatomi, N. Ohnishi, and M. Nishikino, “Molecular dynamics simulation of cluster formation in femtosecond laser ablation,” Optics & Photonics - Optical Engineering + Applications. 2013. link Times cited: 0 Abstract: Short-period laser ablation of a platinum solid target was i… read moreAbstract: Short-period laser ablation of a platinum solid target was investigated through three-dimensional classical molecular dynamics simulations using the embedded atom method potential. The platinum target was ablated by an ultrashort-pulse laser with three different fluences near the ablation threshold and single 100-fs pulse. Although each laser fluence causes melting and evaporation of the target surface, ablation processes are morphologically different. When the laser fluence is just above the ablation threshold, the surface layer of the solid target breaks away, and so-called spallation occurs. With the moderate laser fluence, homogeneous nucleation of nano-sized clusters takes place in the liquidized layer at the surface, resulting in the homogenization in the emitted cluster size, while the surface layer fragments and vaporizes with the higher fluence. Moreover, in the spallation regime, the recreated surface has nano-sized roughness and is formed after the surface oscillates with a rv20-ns period. This inherent roughness formation may be a seed of the nano-sized regular structure observed by past experiments with repetitive pulses. read less NOT USED (high confidence) E. Bourasseau, A.-A. Homman, O. Durand, A. Ghoufi, and P. Malfreyt, “Calculation of the surface tension of liquid copper from atomistic Monte Carlo simulations,” The European Physical Journal B. 2013. link Times cited: 25 NOT USED (high confidence) L. Xie, P. Brault, A. Thomann, and L. Bedra, “Molecular dynamic simulation of binary ZrxCu100-x metallic glass thin film growth,” Applied Surface Science. 2013. link Times cited: 47 NOT USED (high confidence) V. Borovikov, X. Tang, D. Perez, X. Bai, B. Uberuaga, and A. Voter, “Coupled motion of grain boundaries in bcc tungsten as a possible radiation-damage healing mechanism under fusion reactor conditions,” Nuclear Fusion. 2013. link Times cited: 36 Abstract: As a potential first-wall fusion reactor material, tungsten … read moreAbstract: As a potential first-wall fusion reactor material, tungsten will be subjected to high radiation flux and extreme mechanical stress. We propose that under these conditions, coupled grain boundary (GB) motion, in some cases enhanced by interstitial loading, can lead to a radiation-damage healing mechanism, in which a large stress activates coupled GB motion, and the GB sweeps up the defects, such as voids and vacancies, as it passes through the material. The stress-induced mobility characteristics of a number of GBs in tungsten are examined to investigate the likelihood of this scenario. read less NOT USED (high confidence) R. Matsumoto, M. Uranagase, and N. Miyazaki, “Molecular Dynamics Analyses of Deformation Behavior of Long-Period-Stacking-Ordered Structures,” Materials Transactions. 2013. link Times cited: 34 Abstract: Magnesium alloys containing long-period-stacking ordered (LP… read moreAbstract: Magnesium alloys containing long-period-stacking ordered (LPSO) phases have attracted considerable attention because they have been reported to exhibit excellent mechanical properties, including high strength and reasonable ductility. It is thought that the LPSO phase plays a critical role in producing these favorable mechanical properties. We analyze the deformation behavior of the LPSO phases with different stacking sequences using molecular dynamics simulations. To highlight the specific deformation behavior of the LPSO phases, we also perform deformation analyses of hexagonal-close-packed and face-centered-cubic (FCC) structures. We focus on the influence of the stacking order rather than the segregated atoms around the FCC-structured layers, and we model an LPSO structure by single element composition where the interatomic interaction is described by a smoothed Lennard-Jones potential. Our simulations indicate that an LPSO structure with a shorter stacking sequence tends to exhibit a higher compressive flow stress, because FCC-structured layers inhibit twinning deformations and non-basal slips. Kinking deformation is observed for an LPSO structure when both compression and shear deformation are present. It is shown that the first-order pyramidal-hcþ ai dislocation disarranges the stacking of an LPSO structure and leaves behind many lattice defects. In addition, those lattice defects activate numerous basal slips. Finally, basal dislocations arrange in a line and generate a misorientation angle. Furthermore, this angle originates the compressive deformation. We also observed some prismatic-hai dislocations and cross slips to the basal plane. These results suggest the importance of non-basal slips for kinking deformation. [doi:10.2320/matertrans.MI201211] read less NOT USED (high confidence) L. Hale, B. M. Wong, J. Zimmerman, and X. Zhou, “Atomistic potentials for palladium–silver hydrides,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 27 Abstract: New embedded-atom method potentials for the ternary palladiu… read moreAbstract: New embedded-atom method potentials for the ternary palladium–silver–hydrogen system are developed by extending a previously developed palladium–hydrogen potential. The ternary potentials accurately capture the heat of mixing and structural properties associated with solid solution alloys of palladium–silver. Stable hydrides are produced with properties that smoothly transition across the compositions. Additions of silver to palladium are predicted to alter the properties of the hydrides by decreasing the miscibility gap and increasing the likelihood of hydrogen atoms occupying tetrahedral interstitial sites over octahedral interstitial sites. read less NOT USED (high confidence) Y. Shim and J. Amar, “Shape transitions in strained Cu islands on Ni(100): kinetics versus energetics.,” Physical review letters. 2012. link Times cited: 7 Abstract: We examine the ramified islands observed in submonolayer Cu/… read moreAbstract: We examine the ramified islands observed in submonolayer Cu/Ni(100) growth. Our results indicate that the strain-energy contribution to the dependence of island energy on shape is surprisingly weak. In contrast, our accelerated dynamics simulations indicate that unexpected concerted popout processes occurring at step edges may be responsible. Kinetic Monte Carlo (KMC) simulations which include these processes produce island shapes which are very similar to those observed in experiment. These results suggest that the shape transition is of kinetic origin but is strongly mediated by strain. read less NOT USED (high confidence) L. Wang and A. van de Walle, “Ab initio calculations of the melting temperatures of refractory bcc metals.,” Physical chemistry chemical physics : PCCP. 2012. link Times cited: 13 Abstract: We present ab initio calculations of the melting temperature… read moreAbstract: We present ab initio calculations of the melting temperatures for bcc metals Nb, Ta and W. The calculations combine phase coexistence molecular dynamics (MD) simulations using classical embedded-atom method potentials and ab initio density functional theory free energy corrections. The calculated melting temperatures for Nb, Ta and W are, respectively, within 3%, 4%, and 7% of the experimental values. We compare the melting temperatures to those obtained from direct ab initio molecular dynamics simulations and see if they are in excellent agreement with each other. The small remaining discrepancies with experiment are thus likely due to inherent limitations associated with exchange-correlation energy approximations within density-functional theory. read less NOT USED (high confidence) C. Felser, H. Elmers, and G. Fecher, “The Properties of Co2Cr1-xFexAl Heusler Compounds,” Lecture Notes in Physics. 2005. link Times cited: 7 NOT USED (high confidence) I. Galanakis and P. Dederichs, “Half-metallicity and Slater-Pauling behavior in the ferromagnetic Heusler alloys,” Lecture Notes in Physics. 2004. link Times cited: 37 NOT USED (high confidence) S. Koch, “Development of RF-MEAM interaction potentials for Fe-Y.” 2019. link Times cited: 0 Abstract: Der Fokus dieser Arbeit lag zunachst auf einer simulationsge… read moreAbstract: Der Fokus dieser Arbeit lag zunachst auf einer simulationsgestutzen Untersuchung uber die Entsteh- ungsmechanismen von Oxidteilchen in ODS-Stahlen. Hierbei bilden empirische Wechselwirkungs- potenziale von Eisen-Yttrium-Sauerstoff (Fe-Y-O) die Grundlage fur eine Beschreibung dieser Oxid- teilchen-Bildungs-Prozesse in Molekulardynamik (MD) Simulationen, die auch Eigenschaften von Versetzungen und anderen Bestrahlungs-Panomenen detailiert zur weiteren Aufklarung behandeln konnen.
Zu diesem Zweck ist das speziell auf die Simulation zugeschnittene Anfitten der o.g. MD Potenziale (hier fur Fe-Y-O) notwendig. Hierzu dienen die zuvor durchgefuhrten ab-initio (DFT) Rechnungen als Daten- referenzgrundlage (z.B. von Phasen oder Defekten) zur Optimierung der Potenzialparameter wahrend des Anfittens, um ein moglichst exaktes MD Potenzial zu erzeugen, dass die ab-initio Daten auf groseren MD Skalen detailgetreu abbildet. Im ersten Drittel dieses Projektes wurden mehrere Potenziale fur die einzelnen Metall-Komponenten, Fe-Fe und Y-Y, erzeugt. Dabei stellte sich heraus, dass etablierte Standardmethoden nicht in der Lage sind genaue Fe-Y Potenziale als Teillosung fur das Fe-Y-O Problem zu erzeugen. Dabei wurde eine Kombination aus dem (M)EAM Modell und zur Optimierung eine LSM gestutzte Software (POTFIT) genutzt. Die Komplexitat des Problems liegt in den richtungsabhangigen Atombindungen, die die hier entwickelten fortgeschrittenen Simulations- und Fitmethoden benotigen.
Im ersten Schritt von drei Schritten (chapter 3) wurden zunachst einmal die Defizite der Standard-Fittechniken evaluiert, indem die wahrend des Fitting-Prozesses gefundenen Parametersets im EAM Formalismus mit der flexiblen Software POTFIT auf ihre Eignung hin grundlich untersucht worden sind. Die hierfur genutzten Fitfunktionen wurden ursprunglich Anfang 2000 von Zhou und Wadley entwickelt. Hierbei liegt die Ursache fur die dann entdeckte Parameterset-Problematik darin, dass zur Beschreibung des Fe-Y Systems das Model aus drei Potentialkomponenten besteht: Fe-Fe, Y-Y und Fe-Y. Fur diese einzelnen Komponenten sind die Potentialparameter erfolgreich angefittet worden mit Bezug zur Gitterkonstante und Bindungs- bzw. Kohasionsenergie (beides mit 1% Genauigkeit bezgl. DFT Rechnungen) sowie zu allen elastischen Konstanten (5% Genauigkeit bezgl. Experimente). All dies unter Zuhilfenahme von Parametersuchraum-beschrankenden Techniken, die zur Einhaltung der oben genannten Eigenschaften dienen und urspurnglich von Johnson & Oh sind. Selbst kompliziertere Defekteigenschaften, wie Zwischengitter- und Leerstellenbildungsenergien wurden erfolgreich angefittet. Das hier entwickelte EAM Potenzial fur Y-Y ist z.B. in der Lage bei Eigenzwischengitteratomen die basal oktaedrische Position von Zwischengitteratomen (ZA) im Yttrium hcp-Gitter als Grundzustand und die Transition eines jeden ZAs aus einer anderen Position, wie zuvor in DFT berechnet, zu reproduzieren.
Zur Bildung des angestrebten Fe-Y Potenzials wurden diese beiden Komponenten, Fe-Fe und Y-Y, zum weiteren Fitten in dem weitgefacherten und komplexen Fe-Y Potzenzialsuchraum genutzt. Die Parametersets wurden mit sogenannten hier entwickelten Hauptparameter (Key Driver) systematisch untersucht. Ein flexibleres Konzept statt der starreren Universal Binding Relations in Abhangigkeit von der Rose Gleichung. Dieser Hauptparameter zeigte eindeutig, dass die Nutzung der Rose Gleichung zur Parametersuchraum-Minimierung den Suchraum dahingehend einschrankt, sodass ein akkurates Anfitten der hier genutzten 900 DFT Datensets nicht mehr moglich ist. Allerdings ist die Orientierung im Parametersuchraum mit dieser Rose Gleichung bei standardmasigen Optimierungsmethoden (wie LSM) unabdingbar, da ohne diese die benotigten globalen Optima fur die Parameter nicht auffindbar sind.
Als aufklarendes Testverfahren zur weiteren Ergrundung dieser Problematik und Prufung zur Eignung fur Fe-Y Potenziale und den anschliesenden Simulationen diente der Versuch, 9 verschiedene Bindungs-energien von Yttrium-Leerstellenclustern mit ansteigender Leerstellenzahl zu reproduzieren. Dieser Test konnte von diesen Potenzialen nur teilweise erfullt werden und wurde auf die fehlende Beschreibung der Bindungswinkelabhangigkeit im Modell zuruckgefuhrt. Die Erweiterung von EAM durch MEAM mit Winkelabhangigkeit ist jedoch keineswegs eine zufriedenstellende Losung, da MEAM alternativlos auf der irrefuhrenden Rose Gleichung beruht. Daher war die Benutzung des ubersichtlicheren EAM Typs aus zwei Grunden nutzlich: 1. MEAM braucht die Rose Gleichung um diesen komplexen Formalismus zu beherrschen mit denselben Problemen wie in EAM, aber dieses grundlegende Problem ist in MEAM deutlich schwerer zu identifizieren als in EAM. 2. Die mit EAM gefundenen, angefitteten Parameter sind eine hervorragende Startparameter-Grundlage fur den verbesserten darauffolgenden RF-MEAM Typ.
Im zweiten Schritt wurde das Problem aus dem ersten Schritt gelost, indem ein modifizierter MEAM Spezialtyp im referenzlosen Format (RF-MEAM) angewandt worden ist. Im Gegensatz zum herkommlichen MEAM wird hier die Rose Gleichung durch mehr DFT Daten und insbesondere einer intelligenteren Machine Learning ahnlichen Genetic Algorithmus (GA) Optimiertechnik ersetzt, die allerdings eine bedachte Startparameterwahl vorraussetzt, womit Schritt 1 wieder ins Spiel kommt. Die genutzte fortgeschrittene MEAMfit Software, die per GA funktioniert, wurde zwischen 2016 und 2017 funktionierend eigens dafur implementiert. Mit den in Schritt 1 gefitteten Parametern und Set-Auswahltechniken konnten die weiterfuhrenden Fits mit optimalen Startparametern durchgefuhrt werden.
Auf dieser Stufe waren diese Fits mit der speziell verbesserten Technik in der Lage ein detailgetreues Fe-Y Potenzial zu generieren, das sowohl alle Phasen (Fe2Y, Fe3Y, Fe5Y, Fe23Y6 und Fe17Y2 sowohl als auch reines Fe und Y) als auch die gesamte Defektdatenbasis mit einer durchschnittlichen Abweichung von ≈11% erfolgreich abbildet. Zusatzlich bestatigend zu dieser allgemeinen Ubereinstimmung wurde konsequenterweise der in Schritt 1 entwickelte Test hervorragend mit einmaliger Genauigkeit bestanden, mit max. 5% Abweichung von den komlexen o.g. Y-Leerstellen Bildungsenergien. Allerdings konnte ein systematischer Fehlertrend aufgespurt werden, der Schwachen in der Fe-Fe Komponente offenbarte. Als Folge dessen wurde umgehend diese Komponente durch ein anderes etabliertes Fe-Fe Potenzial von G. Ackland mit einer extrem genauen Schmelztemperatur (nur 3% Abweichung vom Exp.) ausgetauscht. Mit diesem genauen Potenzial konnte zum ersten Mal die Clusterbildung von gelosten Yttrium Atomen in einer Eisenschmelze erfolgreich per MD Simulation auf atomarer Ebene nachgestellt werden oberhalb von 1750 K. Temperaturen darunter hatten eine Ausscheidungsbildung von Y mit sehr geringer Y-Loslichkeit (<0.1%) in Ubereinstimmung mit den Experimenten zur Folge. Dies wurde durch den Pot. Typ A ermoglicht, der aber die energetische Reihenfolge bei den Fe-Y Phasen nicht ganz genau einhalt. Typ B hingegen halt diese ein, dort fehlt aber die Y-Clusterbildungsneigung. Durch den gebotenen Praxisbezug zur Metallurgie mussen die Loslichkeit und Clusterbildung gleichzeitig in der Simulation genau reproduzierbar sein, was aber weder Typ A noch B kann, was zum Typ A/B Dilemma fuhrt.
Dieses Typ A/B Dilemma (Phasen oder Defekt Genauigkeit) fuhrt zum letzten dritten Schritt (chapter 5). Darin ist zusatzlich die Strukturaufklarung von der Fe17Y2 Phase mit Vergleichen zu exp. EXAFS Spektren unserer Kollaborationspartner vom ISSP (Riga) enthalten. Diese Aufklarung dient auch dazu die fehlenden magnetischen Abhangigkeiten im Potenzial zu kompensieren, da die Phasenreihenfolge mit sehr feinen Energieunterschieden wohl stark von magnetischen Wechselwirkungen gepragt ist. Obwohl Potenzial Typ B diesen (Magnetismus) nicht direkt beachtet, ist es in der Lage das tatsachlich gemessene EXAFS Spektrum grostenteils genau wiederzugeben. Allerdings offenbart eine einzige ausgepragte Phasenverschiebung, dass die angenommene hcp Struktur durch eine unterschwellige rhombohedrale Komponente, die sporadisch in der c-Gitterrichtung auftritt, korrigiert werden muss. AIMD (DFT) Berechnungen in Kooperation mit der University of Edinburgh bestatigen dies und zeigen sogar, dass magnetische Wechselwirkungen diese Strukturmischung stabilisieren. Endgultig bestatigt werden konnte dies mit der genauen EXAFS Spektren Reproduktion mit dem durch AIMD verbesserten nochmals gefitteten Potenzialtyp B, der als neuer Typ C durch AIMD indirekt den Einfluss der magnetischen Wechselwirkungen mit einschliest. Diese erstmalige nahezu deckungsgleiche MD Simulation eines EXAFS Spektrums von einem komplexen metallischen Alloy, hier Fe-Y, stellt eine bisher unerreichte Verbesserung dar. Schlieslich lost Typ C das Typ A/B Dilemma und ernoglicht eine genaue gleichzeitige MD Modellierung von Phasen- und Defekten in Fe-Y – ein Durchbruch in der MD-Potenzialentwicklung. read less NOT USED (high confidence) В. О. Подрыга, V. O. Podryga, Е. В. Вихров, E. V. Vikhrov, С. В. Поляков, and S. Polyakov, “Молекулярно-динамический расчет макропараметров технических газов на примере аргона, азота, водорода и метана,” Математическое моделирование. 2019. link Times cited: 1 NOT USED (high confidence) D. Mishra, M. Meraj, S. K. Badjena, and S. Pal, “Dislocation Interaction and V-Shaped Growth of the Distorted Structure During Nanoindentation of Cu20Ni20Al20Co20Fe20 (high-entropy alloy)-Coated Copper: A Molecular Dynamics Simulation-Based Study,” Transactions of the Indian Institute of Metals. 2019. link Times cited: 14 NOT USED (high confidence) Y.-hua Zhou, A. L. Lloyd, R. Smith, and S. Kenny, “Modelling thin film growth in the Ag–Ti system,” Surface Science. 2019. link Times cited: 2 NOT USED (high confidence) T. Otieno and K. Abou-El-Hossein, “Molecular dynamics analysis of nanomachining of rapidly solidified aluminium,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 7 NOT USED (high confidence) X. Hu, T. Gao, Y. Li, L. Ren, X. Luo, and Q. Xie, “Properties of Icosahedral Clusters and Medium-Range Order in AlCuZr Alloy,” DEStech Transactions on Engineering and Technology Research. 2017. link Times cited: 0 Abstract: The evolution characteristics of icosahedral clusters during… read moreAbstract: The evolution characteristics of icosahedral clusters during the rapid solidification of Al10Cu50Zr40 alloy under cooling rates of 1.0 × 1011 K/s are investigated based on molecular dynamics simulations. The structural properties of short-range order and medium-range order of Al10Cu50Zr40 alloy are analyzed by several structural characterization methods. It is found that the icosahedral cluster is the dominant short range order structure. Simultaneously, the medium -range order structural evolutions are described in detail by quantitative method and visualization technology during the rapid solidification. The results reveal that the medium-range order icosahedral clusters are bonding with each other by chain, triangle, tetrahedral and their combination structures, and have excellent structural stability and configural continuity with the temperature decreasing. The medium-range order clusters that consist of , and interlocked with each other and built the densely packed network structures. read less NOT USED (high confidence) A. Nemati, H. N. Pishkenari, A. Meghdari, and S. Sohrabpour, “Directing the diffusive motion of fullerene-based nanocars using nonplanar gold surfaces.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 19 Abstract: A new method for guiding the motion of fullerene and fullere… read moreAbstract: A new method for guiding the motion of fullerene and fullerene-based nanocars is introduced in this paper. The effects of non-flat substrates on the motion of C60, a nanocar and a nanotruck are investigated at different conditions and temperatures. Their behavior is studied using two different approaches: analyzing the variation in potential energy and conducting all-atom classical molecular dynamics simulations. This paper proposes that the use of a stepped substrate will make their motion more predictable and controllable. The results of the simulations show that C60 stays on the top side of the step and cannot jump over the step at temperatures of 400 K and lower. However, at temperatures of 500 K and higher, C60 has sufficient energy to travel to the down side of the step. C60 attaches to the edge and moves just alongside of the edge when it is on the down side of the step. The edge also restricts the motion of C60 alongside the edge and reduces its range of motion. By considering the motion of C60, the general behavior of the nanocar and nanotruck is predictable. The nanocar stays on the top side of the step at temperatures of 400 K and less; at 500 K and higher temperatures, its wheels jump off the edge, and its range of motion is restricted. The relatively rigid chassis of the nanotruck does not allow the free individual motion of the wheels. As a result, the entire nanotruck stays on the top side of the step, even at 600 K. A pathway with the desired route can be fabricated for the motion of C60 and nanocars using the method presented in this paper. This represents a step towards the directional motion of C60 and nanocars. read less NOT USED (high confidence) V. Podryga, Y. N. Karamzin, T. Kudryashova, and S. Polyakov, “MULTISCALE SIMULATION OF THREE-DIMENSIONAL UNSTEADY GAS FLOWS IN MICROCHANNELS OF TECHNICAL SYSTEMS.” 2016. link Times cited: 10 Abstract: Abstract. The work is devoted to modeling the gas flows in m… read moreAbstract: Abstract. The work is devoted to modeling the gas flows in microchannels of technical systems in conditions of multiscale computational domain. As an example, the problem of nitrogen flow in nickel microchannel for three-dimensional geometry is considered. General attention is paid to the calculation of gaseous medium macroparameters considering the molecular processes that occur in the gas flow and on the walls of the microchannel. The difference in the scales of the computational domain (the length of the channel, cross section of the channel, the free path of the molecules, the thickness of the boundary layer) and near-surface interaction of the gas with the metal lead to the necessity taking into account the relief and the properties of the microchannel at the molecular level. As a result, the mathematical model of the research flow can not be fully formulated within the framework of the macroscopic approach. For decision of the problem multiscale approach combining the solution of a quasigasdynamic (QGD) equations and correction of gasdynamic parameters by molecular dynamics method (MD), is used. QGD system of equations is solved by method of finite volumes. The MD system of equations is applied within each control volume and is solved by Verlet scheme. In MD calculations particles interactions are described by the potentials determining the basic properties of the components of the considered system. Parallel implementation of the approach is based on method of splitting into physical processes and separation of areas. Algorithms are focused on the use of computer systems with central and hybrid architectures. Calculations showed that the overall numerical algorithm is resistant to the use of data for the flow correction obtained by the MD calculations. With the help of MD methods basic coefficient relations for QGD system were obtained, the transitions from MD to QGD and back were checked, three-dimensional calculation of the nitrogen flow in the nickel microchannel was produced. The results confirmed the efficiency of the developed approach. read less NOT USED (high confidence) V. O. Podryga and S. Polyakov, “Molecular dynamic calculation of gas macroparameters in the stream and on the boundary,” Keldysh Institute Preprints. 2016. link Times cited: 9 NOT USED (high confidence) I. Galanakis, “Theory of Heusler and Full-Heusler Compounds.” 2016. link Times cited: 34 NOT USED (high confidence) J. S. Gibson, S. G. Srinivasan, M. Baskes, R. E. Miller, and A. K. Wilson, “A multi-state modified embedded atom method potential for titanium,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 3 Abstract: The continuing search for broadly applicable, predictive, an… read moreAbstract: The continuing search for broadly applicable, predictive, and unique potential functions led to the invention of the multi-state modified embedded atom method (MS-MEAM) (Baskes et al 2007 Phys. Rev. B 75 094113). MS-MEAM replaced almost all of the prior arbitrary choices of the MEAM electron densities, embedding energy, pair potential, and angular screening functions by using first-principles computations of energy/volume relationships for multiple reference crystal structures and transformation paths connecting those reference structures. This strategy reasonably captured diverse interactions between atoms with variable coordinations in a face-centered-cubic (fcc)-stable copper system. However, a straightforward application of the original MS-MEAM framework to model technologically useful hexagonal-close-packed (hcp) metals proved elusive. This work describes the development of an hcp-stable/fcc-metastable MS-MEAM to model titanium by introducing a new angular function within the background electron density description. This critical insight enables the titanium MS-MEAM potential to reproduce first principles computations of reference structures and transformation paths extremely well. Importantly, it predicts lattice and elastic constants, defect energetics, and dynamics of non-ideal hcp and liquid titanium in good agreement with first principles computations and corresponding experiments, and often better than the three well-known literature models used as a benchmark. The titanium MS-MEAM has been made available in the Knowledgebase of Interatomic Models (https://openkim.org/) (Tadmor et al 2011 JOM 63 17). read less NOT USED (high confidence) C. Gang, Z. Peng, and L. Wei, “Analysis of Pd-Ni nanobelts melting process using molecular dynamics simulation,” Journal of Nanomaterials. 2013. link Times cited: 6 Abstract: The melting process of Pd-Ni alloy nanobelts with different … read moreAbstract: The melting process of Pd-Ni alloy nanobelts with different Ni atom content has been simulated by molecular dynamic (MD) method. The radial distribution function, the Lindemann index, and pair analysis method were used to characterize Pd-Ni nanobelt models in simulation. The results indicate that the melting temperature of Pd-Ni nanobelt with composition far from pure metal was lower than that of othermodels, and the breaking point of the nanobelt can be illustrated by the Lindemann index. Pair analysis indicates that the number of FCC pairs will decrease and almost disappear at melting point with increasing temperature. The melting points of Pd-Ni alloy nanobelts were also calculated by thermodynamic method, and the results were close to that obtained by MD simulation. read less NOT USED (high confidence) H. Jeong, A. Caruso, and C. Borca, “Surface segregation and compositional instability at the surface of half-metal ferromagnets and related compounds,” Lecture Notes in Physics. 2005. link Times cited: 2 |
 EAM_Dynamo_ZhouJohnsonWadley_2004_W__MO_524392058194_005
EAM_Dynamo_ZhouJohnsonWadley_2004_W__MO_524392058194_005