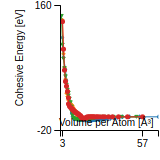
Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
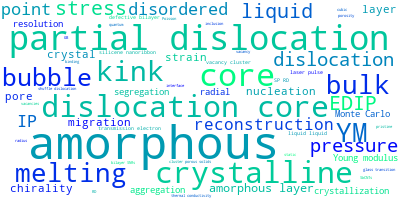
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
333 Citations (156 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) M. Wen, J. Li, P. Brommer, R. Elliott, J. Sethna, and E. Tadmor, “A KIM-compliant potfit for fitting sloppy interatomic potentials: application to the EDIP model for silicon,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 16 Abstract: Fitted interatomic potentials are widely used in atomistic s… read moreAbstract: Fitted interatomic potentials are widely used in atomistic simulations thanks to their ability to compute the energy and forces on atoms quickly. However, the simulation results crucially depend on the quality of the potential being used. Force matching is a method aimed at constructing reliable and transferable interatomic potentials by matching the forces computed by the potential as closely as possible, with those obtained from first principles calculations. The potfit program is an implementation of the force-matching method that optimizes the potential parameters using a global minimization algorithm followed by a local minimization polish. We extended potfit in two ways. First, we adapted the code to be compliant with the KIM Application Programming Interface (API) standard (part of the Knowledgebase of Interatomic Models project). This makes it possible to use potfit to fit many KIM potential models, not just those prebuilt into the potfit code. Second, we incorporated the geodesic Levenberg–Marquardt (LM) minimization algorithm into potfit as a new local minimization algorithm. The extended potfit was tested by generating a training set using the KIM environment-dependent interatomic potential (EDIP) model for silicon and using potfit to recover the potential parameters from different initial guesses. The results show that EDIP is a ‘sloppy model’ in the sense that its predictions are insensitive to some of its parameters, which makes fitting more difficult. We find that the geodesic LM algorithm is particularly efficient for this case. The extended potfit code is the first step in developing a KIM-based fitting framework for interatomic potentials for bulk and two-dimensional materials. The code is available for download via https://www.potfit.net. read less USED (definite) E. Holmström et al., “Dependence of short and intermediate-range order on preparation in experimental and modeled pure a-Si,” Journal of Non-crystalline Solids. 2016. link Times cited: 16 USED (definite) J. Samela, S. Norris, K. Nordlund, and M. Aziz, “Optimization of large amorphous silicon and silica structures for molecular dynamics simulations of energetic impacts,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2011. link Times cited: 10 USED (definite) T. Zhu, J. Li, and S. Yip, “Atomistic characterization of three-dimensional lattice trapping barriers to brittle fracture,” Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2006. link Times cited: 39 Abstract: We present a detailed account of an atomistic study of three… read moreAbstract: We present a detailed account of an atomistic study of three-dimensional lattice trapping barriers to brittle fracture in Si. By means of a prototypical interatomic potential model, we map out the molecular details of the evolution of atomically sharp cracks in the (111) cleavage plane with straight crack fronts along the and directions, respectively. The thermally activated processes of bond rupturing along the crack front are quantitatively characterized using a reaction pathway sampling scheme. The calculated minimum energy paths reveal a mechanism of kink-pair formation and migration in facilitating the crack front advancement. We show that the physical origin of directional anisotropy in cleavage crack propagation can be attributed to a difference in the kink-pair formation energy for different crack orientations. The effects of interatomic potentials are delineated by comparing the Stillinger–Weber model with an environment-dependent model. read less USED (high confidence) Y. Kurniawan et al., “Extending OpenKIM with an Uncertainty Quantification Toolkit for Molecular Modeling,” 2022 IEEE 18th International Conference on e-Science (e-Science). 2022. link Times cited: 0 Abstract: Atomistic simulations are an important tool in materials mod… read moreAbstract: Atomistic simulations are an important tool in materials modeling. Interatomic potentials (IPs) are at the heart of such molecular models, and the accuracy of a model's predictions depends strongly on the choice of IP. Uncertainty quantification (UQ) is an emerging tool for assessing the reliability of atomistic simulations. The Open Knowledgebase of Interatomic Models (OpenKIM) is a cyberinfrastructure project whose goal is to collect and standardize the study of IPs to enable transparent, reproducible research. Part of the OpenKIM framework is the Python package, KIM-based Learning-Integrated Fitting Framework (KLIFF), that provides tools for fitting parameters in an IP to data. This paper introduces a UQ toolbox extension to KLIFF. We focus on two sources of uncertainty: variations in parameters and inadequacy of the functional form of the IP. Our implementation uses parallel-tempered Markov chain Monte Carlo (PTMCMC), adjusting the sampling temperature to estimate the uncertainty due to the functional form of the IP. We demonstrate on a Stillinger–Weber potential that makes predictions for the atomic energies and forces for silicon in a diamond configuration. Finally, we highlight some potential subtleties in applying and using these tools with recommendations for practitioners and IP developers. read less USED (high confidence) Y. Shi and I. Szlufarska, “Effect of growth twins on strength and microstructural evolution of nanocrystalline aluminum,” Journal of Materials Science. 2021. link Times cited: 2 USED (high confidence) M. Chávez-Castillo, M. Rodríguez-Meza, and L. Meza-Montes, “Mechanical response of bilayer silicene nanoribbons under uniaxial tension,” RSC Advances. 2018. link Times cited: 3 Abstract: Understanding the behaviour of nanoscale systems is of great… read moreAbstract: Understanding the behaviour of nanoscale systems is of great importance to tailor their properties. To this aim, we investigate the Young's modulus (YM) of defect-free and defective armchair bilayer silicene nanoribbons (SNRs), at room temperature, as a function of length and distance between layers. In this study, we perform molecular dynamics simulations using the environment-dependent interatomic potential to describe the interaction of the Si atoms. We show that the Young's modulus of pristine and defective bilayer SNRs increases with the ribbon length exhibiting size dependence. In general, YM of defective bilayer SNRs is smaller than the value obtained for the defect-free case, as a result of the number of missing bonds. In all cases, as the interlayer distance increases YM decreases and the buckling increases. It is shown that the YM exhibits a quadratic interlayer distance dependence. Finally, when only one layer has a mono-vacancy defect, the atomic stress distribution of the pristine layer is affected by the presence of the vacancy. This effect can be considered as a “ghost vacancy” since the deterioration of the pristine layer is similar to that shown by the defective one. These results show that YM of pristine and defective bilayer SNRs could be tailored for a given length and interlayer distance. It is also found that the fracture stress and the fracture strain of defective bilayers are both smaller than those obtained for the defect-free ones. read less USED (high confidence) N. Winter, M. Becton, L. Zhang, and X. Wang, “Effects of pore design on mechanical properties of nanoporous silicon,” Acta Materialia. 2017. link Times cited: 23 USED (high confidence) X. Cartoixà, R. Dettori, C. Melis, L. Colombo, and R. Rurali, “Thermal transport in porous Si nanowires from approach-to-equilibrium molecular dynamics calculations,” Applied Physics Letters. 2016. link Times cited: 21 Abstract: We study thermal transport in porous Si nanowires (SiNWs) by… read moreAbstract: We study thermal transport in porous Si nanowires (SiNWs) by means of approach-to-equilibrium molecular dynamics simulations. We show that the presence of pores greatly reduces the thermal conductivity, κ, of the SiNWs as long mean free path phonons are suppressed. We address explicitly the dependence of κ on different features of the pore topology—such as the porosity and the pore diameter—and on the nanowire (NW) geometry—diameter and length. We use the results of the molecular dynamics calculations to tune an effective model, which is capable of capturing the dependence of κ on porosity and NW diameter. The model illustrates the failure of Matthiessen's rule to describe the coupling between boundary and pore scattering, which we account for by the inclusion of an additional empirical term. read less USED (high confidence) D. T. Ho, S. D. Park, S. Y. Kwon, T. Han, and S. Y. Kim, “Negative Poisson’s ratio in cubic materials along principal directions,” physica status solidi (b). 2016. link Times cited: 33 Abstract: This report employed molecular statics simulation and densit… read moreAbstract: This report employed molecular statics simulation and density‐functional‐theory calculation to study the Poisson's ratios of face‐centered‐cubic materials. We provide numerical and theoretical evidences to show that cubic materials can exhibit auxetic behavior in a principal direction under proper loading conditions. When a stress perpendicular to the loading direction is applied, cubic materials can exhibit a negative Poisson's ratio at finite strain. The negative Poisson's ratio behavior, including its direction and value, is highly dependent on the direction and magnitude of the transversely applied stresses. As a result, we show that it is possible to tune the direction and magnitude of the negative Poisson's ratio behavior of cubic materials by controlling the transverse loadings. read less USED (high confidence) L. Pizzagalli, “Atomistic modeling of the dissociation of a screw dislocation in silicon,” Journal of Materials Science. 2016. link Times cited: 8 USED (high confidence) M. Chávez-Castillo, M. Rodríguez-Meza, and L. Meza-Montes, “Size, vacancy and temperature effects on Young’s modulus of silicene nanoribbons,” RSC Advances. 2015. link Times cited: 20 Abstract: We report results on the Young’s modulus (YM) of defect-free… read moreAbstract: We report results on the Young’s modulus (YM) of defect-free and defective silicene nanoribbons (SNRs) as a function of length and temperature. In this study, we perform molecular dynamics simulations using the Environment-Dependent Interatomic Potential (EDIP) to describe the interaction of the Si atoms. We find that the YM of pristine and defective SNRs increases with the ribbon length in both chirality directions. It is shown that the YM of defective SNRs exhibits a complex dependence on the combinations of vacancies. With respect to temperature, we find that YM for SNRs with and without vacancy defects shows nonlinear behavior and it could be tailored for a given length and chirality. read less USED (high confidence) C. Melis, L. Colombo, and G. Mana, “Lattice strain at c-Si surfaces: a density functional theory calculation,” Metrologia. 2014. link Times cited: 13 Abstract: The measurement of the Avogadro constant by counting Si atom… read moreAbstract: The measurement of the Avogadro constant by counting Si atoms is based on the assumption that Si balls of about 94 mm diameter have a perfect crystal structure up to the outermost atom layers. This is not the case because of the surface relaxation and reconstruction, the possible presence of an amorphous layer, and the oxidation process due to the interaction with the ambient. This paper gives the results of density functional calculations of the strain components orthogonal to crystal surface in a number of configurations likely found in real samples. read less USED (high confidence) C. Melis, R. Dettori, S. Vandermeulen, and L. Colombo, “Calculating thermal conductivity in a transient conduction regime: theory and implementation,” The European Physical Journal B. 2014. link Times cited: 56 USED (high confidence) Y. Jing, Y. Sun, H. Niu, and J. Shen, “Atomistic simulations on the mechanical properties of silicene nanoribbons under uniaxial tension,” physica status solidi (b). 2013. link Times cited: 37 Abstract: The mechanical properties of silicene are investigated using… read moreAbstract: The mechanical properties of silicene are investigated using ab initio calculation and molecular dynamics simulations with different empirical potentials. The simulation results show that the calculated Young's modulus of bulk silicene with EDIP model is consistent with the ab initio calculations. The chirality has a significant effect on the critical strain and stress of bulk silicene under uniaxial tension. In addition, the Young's modulus depends strongly on the chirality and size of the silicene nanoribbon due to the edge effects. The fracture process of a silicene nanoribbon is also studied. read less USED (high confidence) K. Zhong, Q. Meng, and W. Zhao, “A new stable core structure of 60° shuffle dislocation in silicon and associated mobility behavior,” physica status solidi (b). 2012. link Times cited: 2 Abstract: We performed density‐functional theory simulations and obtai… read moreAbstract: We performed density‐functional theory simulations and obtained a new stable reconfiguration of shuffle 60° dislocation in silicon. The configuration is characterized by a complex of a shuffle 60° dislocation and an additional bond defect. Its mobility is discussed by determining the critical shear strain for dislocation glide. The critical shear strain is 10.6% without thermal activation, and decreases to 7.9% at a temperature ranging from 100 to 600 K. It can take part in the process of plastic deformation by emitting a common glissile shuffle 60° dislocation under high stress at low temperature. read less USED (high confidence) C.-F. Wang, Z. Wang, Q. Meng, and Z. Wang, “Dynamic properties of reconstruction defect on 90° partial dislocation in Si,” physica status solidi (b). 2012. link Times cited: 2 Abstract: The molecular dynamics (MD) and nudged elastic band (NEB) me… read moreAbstract: The molecular dynamics (MD) and nudged elastic band (NEB) methods are employed to investigate the dynamic properties of the reconstruction defect (RD) on 90° partial dislocation in Si. This involves the motion of RD in single‐period (SP‐RD) and double‐period (DP‐RD) structures. When the temperature is lower than 1100 K, the migration processes and velocities of SP‐RD can be simply observed. It is found that SP‐RD is remarkably mobile, which is essentially determined by its structural characteristics. At relatively higher temperature, the previous prediction that SP‐RD may act as the nucleating center of a double kink is proved. All these MD results are in good agreement with the calculated barriers. The migration of DP‐RD is carried out by the motion of left–right kink RD complex (LR‐RD) and right–left kink RD complex (RL‐RD). Their motion sequences are described in detail and it is also found that the dangling bonds make the movement of the two complexes easier. read less USED (high confidence) K. Garcez and A. Antonelli, “Pressure effects on the transitions between disordered phases in supercooled liquid silicon.,” The Journal of chemical physics. 2011. link Times cited: 10 Abstract: We investigate the pressure effects on the transitions betwe… read moreAbstract: We investigate the pressure effects on the transitions between the disordered phases in supercooled liquid silicon through Monte Carlo simulations and efficient methods to compute free energies. Our calculations, using an environment dependent interatomic potential for Si, indicate that at zero pressure the liquid-liquid phase transition, between the high density liquid and the low density liquid, occurs at a temperature 325K below melting. We found that the liquid-liquid transition temperature decreases with increasing pressure, following the liquid-solid coexistence curve. As pressure increases, the liquid-liquid coexistence curve approaches the region where the glass transition between the low density liquid and the low density amorphous takes place. Above 5 GPa, our calculations show that the liquid-liquid transition is suppressed by the glassy dynamics of the system. We also found that above 5 GPa, the glass transition temperature is lower than that at lower pressures, suggesting that under these conditions the glass transition occurs between the high density liquid and the high density amorphous. read less USED (high confidence) S. Ryu, C. Weinberger, M. Baskes, and W. Cai, “Improved modified embedded-atom method potentials for gold and silicon,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 47 Abstract: The modified embedded-atom method interatomic potentials for… read moreAbstract: The modified embedded-atom method interatomic potentials for pure gold and pure silicon are improved in their melting point and latent heat predictions, by modifying the multi-body screening function and the equation of state function. The fitting of the new parameters requires rapid calculations of melting point and latent heat, which are enabled by efficient free-energy methods. The results provide the basis for constructing a cross-potential that will be fitted to the binary gold–silicon phase diagram. read less USED (high confidence) M. Beaufort, L. Pizzagalli, A. Gandy, E. Oliviero, D. Eyidi, and S. Donnelly, “Solid-phase epitaxial regrowth of amorphous silicon containing helium bubbles,” Journal of Applied Physics. 2008. link Times cited: 15 Abstract: Transmission electron microscopy has been used to study proc… read moreAbstract: Transmission electron microscopy has been used to study processes occurring when a layer of amorphous silicon (a-Si) containing helium-filled cavities buried in crystalline silicon (c-Si) recrystallizes by solid phase epitaxial growth (SPEG). The buried layer was formed in (100) silicon by means of bombardment with 150 keV Li ions with the bubbles resulting from subsequent implantation of 80 keV He ions; the energies being chosen to ensure that the resulting bubble distribution was entirely contained within the amorphous layer. The presence of bubbles in a-Si undergoing SPEG at a (100) interface with c-Si has previously been observed to give rise to the formation of microtwin lamellas, assumed to nucleate at the bubble surfaces; however, the present work indicates clearly that, in fact during SPEG, many microtwins nucleate remote from the bubbles. There is also an apparent interaction between the amorphous-crystalline (a-c) interface and the bubbles, in which the bubbles seem to be swept by the moving interface. The paper will discuss possible mechanisms for both phenomena, in terms of interstitial defects for the nucleation of microtwins and in terms of enhanced bubble mobility in a-Si for the apparent interaction between the a-c interface and the bubbles. read less USED (high confidence) L. Tian and X. Wang, “Pulsed Laser-Induced Rapid Surface Cooling and Amorphization,” Japanese Journal of Applied Physics. 2008. link Times cited: 2 Abstract: In this work, hybrid atomistic-macroscale simulation is cond… read moreAbstract: In this work, hybrid atomistic-macroscale simulation is conducted to explore the crystallization and amorphization of Si surface in the situation of fast melting and solidification induced by ultrafast laser heating and heat conduction. Our work is focused on investigating the relationship between the amorphization threshold (Ec) and the laser pulse width (tg). An empirical correlation Ec=448.76×tg0.56 is obtained to relate the critical fluence to the laser pulse width. By exploring the microstructure of the amorphous and crystalline state of Si, a sharp interface of about 0.6 nm thickness is observed between the amorphous layer and the crystalline Si. The relationship between the final thickness of amorphous layer and the fluence of the laser pulse is further studied in this work. Employing laser pulses with full width at half maximum (FWHM) equal to 6.67 ns, the formation and recrystallization processes of a 12-nm-thick amorphous layer is investigated. read less USED (high confidence) L. Pizzagalli, A. Pedersen, A. Arnaldsson, H. J’onsson, and P. Beauchamp, “Theoretical study of kinks on screw dislocation in silicon,” Physical Review B. 2008. link Times cited: 43 Abstract: Theoretical calculations of the structure, formation and mig… read moreAbstract: Theoretical calculations of the structure, formation and migration of kinks on a non-dissociated screw dislocation in silicon have been carried out using density functional theory calculations as well as calculations based on interatomic potential functions. The results show that the structure of a single kink is characterized by a narrow core and highly stretched bonds between some of the atoms. The formation energy of a single kink ranges from 0.9 to 1.36 eV, and is of the same order as that for kinks on partial dislocations. However, the kinks migrate almost freely along the line of an undissociated dislocation unlike what is found for partial dislocations. The effect of stress has also been investigated in order to compare with previous silicon deformation experiments which have been carried out at low temperature and high stress. The energy barrier associated with the formation of a stable kink pair becomes as low as 0.65 eV for an applied stress on the order of 1 GPa, indicating that displacements of screw dislocations likely occur via thermally activated formation of kink pairs at room temperature. read less USED (high confidence) Y. Kowaki, A. Harada, F. Shimojo, and K. Hoshino, “Radius dependence of the melting temperature of single-walled carbon nanotubes: molecular-dynamics simulations,” Journal of Physics: Condensed Matter. 2007. link Times cited: 20 Abstract: We have investigated the radius dependence of the melting te… read moreAbstract: We have investigated the radius dependence of the melting temperature of single-walled carbon nanotubes (SWCNTs) by classical molecular-dynamics (MD) simulations using the environment-dependent interatomic potential (EDIP) proposed by Marks. Here we define the ‘melting temperature’ as a temperature at which there occurs a thermal instability of SWCNTs. We have carried out molecular-dynamics simulations at several temperatures for carbon nanotubes with various radii and estimated the ‘melting temperature’ based on the temperature dependence of the radial distribution functions, mean-square displacements and atomic configurations. It is shown that the ‘melting temperature’ of SWCNTs decreases with decreasing radius. The origin of this radius dependence of the melting temperature of SWCNTs is discussed in relation to the stability of SWCNTs energetically based on the strain energy of carbon nanotubes. read less USED (high confidence) G. Opletal, T. Petersen, I. Snook, and D. McCulloch, “Modeling of structure and porosity in amorphous silicon systems using Monte Carlo methods.,” The Journal of chemical physics. 2007. link Times cited: 20 Abstract: Porous solids are very important from a scientific point of … read moreAbstract: Porous solids are very important from a scientific point of view as they provide a medium in which to study the behavior of confined fluids. Although some porous solids have a well defined pore geometry such as zeolites, many porous solids lack crystalline order and are usually described as amorphous. The description of the pore geometry in such structures is very difficult. The authors develop a modeling approach using a Monte Carlo algorithm to simulate porosity within amorphous systems based on constraints for the internal volume and surface area. To illustrate this approach, a model of microporous amorphous silicon is presented. Structural aspects of the porous model are then compared against hybrid reverse Monte Carlo simulations of nonporous amorphous silicon and published results from the literature. It is found that coordination defects are predominately located at the pore surface walls. read less USED (high confidence) L. Sun and J. Murthy, “Domain size effects in molecular dynamics simulation of phonon transport in silicon,” Applied Physics Letters. 2006. link Times cited: 59 Abstract: Molecular dynamics (MD) simulation is employed to compute th… read moreAbstract: Molecular dynamics (MD) simulation is employed to compute thermal conductivity and dispersion curves for bulk silicon using the environment dependent interatomic potential. Thermal conductivity simulations using the Green-Kubo method are found to converge to the bulk value with 216 atoms or more. Computed values in the 300–1000K range compare well with experiment. MD results are analyzed to obtain phonon dispersion curves along the [100] direction and compare well with those using the dynamical matrix approach. It is found that bulk thermal properties may be computed using MD in relatively small domains provided that the dominant energy-containing wavelengths are well resolved. read less USED (high confidence) J. C. H. Spence⊥ et al., “Imaging dislocation cores – the way forward,” Philosophical Magazine. 2006. link Times cited: 27 Abstract: Although the sub-angstrom resolution of the modern transmiss… read moreAbstract: Although the sub-angstrom resolution of the modern transmission electron microscope (TEM) has made major contributions to defect structure analysis in many fields (such as oxides, interfaces, nanoparticles and superconductors) it has yielded little direct information on the core structure of dislocations. We suggest that “forbidden reflection” lattice images recorded in an ultra-high vacuum TEM in projections normal to the dislocation line could provide interpretable images of cores at atomic resolution. These could answer crucial questions, such as the nature of kinks, core reconstruction and periodicity, the nature of obstacles, and help distinguish obstacle theories of kink motion from the secondary Peierls–valley Hirth–Lothe theory. We give experimental forbidden reflection images and a new image obtained from silicon under UHV conditions with atomically smooth surfaces, whose preparation did not anneal out all dislocations. We also show experimental coherent nanodiffraction patterns and scanning transmission electron microscope (STEM) images recorded with the beam parallel to the core, so that core reconstruction can be expected to introduce a “half-order” Laue zone ring. We discuss the contribution that energy-loss spectroscopy from dislocation cores can be expected to make if a nanoprobe beam is used. read less USED (high confidence) G. Lulli et al., “Investigation of heavily damaged ion implanted Si by atomistic simulation of Rutherford backscattering channeling spectra,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 5 USED (high confidence) Y. Mo, M. Bazant, and E. Kaxiras, “Sulfur point defects in crystalline and amorphous silicon,” Physical Review B. 2004. link Times cited: 44 Abstract: We present first-principles calculations for the behavior of… read moreAbstract: We present first-principles calculations for the behavior of sulfur point defects in crystalline and amorphous silicon structures. By introducing the sulfur point defects at various representative positions in the samples, including substitutional and interstitial sites in the crystal and fourfold coordinated or miscoordinated sites (dangling bond and floating bond sites ) in the amorphous, we analyze the energetics in detail and determine the most stable structures. Two important conclusions we draw are: (a) in crystalline Si, the S defects form pairs in which the two S atoms are energetically bound but not covalently bonded; (b) in amorphous Si, they preferentially occupy threefold coordinated sites, even when the starting configuration has higher coordination (four- or fivefold). The implications of these results for the electronic structure of sulfur-doped Si samples are also analyzed in the context of the present calculations. read less USED (high confidence) M. Prasad and T. Sinno, “Feature activated molecular dynamics: an efficient approach for atomistic simulation of solid-state aggregation phenomena.,” The Journal of chemical physics. 2004. link Times cited: 3 Abstract: An efficient approach is presented for performing efficient … read moreAbstract: An efficient approach is presented for performing efficient molecular dynamics simulations of solute aggregation in crystalline solids. The method dynamically divides the total simulation space into "active" regions centered about each minority species, in which regular molecular dynamics is performed. The number, size, and shape of these regions is updated periodically based on the distribution of solute atoms within the overall simulation cell. The remainder of the system is essentially static except for periodic rescaling of the entire simulation cell in order to balance the pressure between the isolated molecular dynamics regions. The method is shown to be accurate and robust for the Environment-Dependant Interatomic Potential (EDIP) for silicon and an Embedded Atom Method potential (EAM) for copper. Several tests are performed beginning with the diffusion of a single vacancy all the way to large-scale simulations of vacancy clustering. In both material systems, the predicted evolutions agree closely with the results of standard molecular dynamics simulations. Computationally, the method is demonstrated to scale almost linearly with the concentration of solute atoms, but is essentially independent of the total system size. This scaling behavior allows for the full dynamical simulation of aggregation under conditions that are more experimentally realizable than would be possible with standard molecular dynamics. read less USED (high confidence) J. Feldman, N. Bernstein, D. Papaconstantopoulos, and M. Mehl, “Tight-binding study of structure and vibrations of amorphous silicon,” Physical Review B. 2004. link Times cited: 11 Abstract: We present a tight-binding calculation that, for the first t… read moreAbstract: We present a tight-binding calculation that, for the first time, accurately describes the structural, vibrational and elastic properties of amorphous silicon. We compute the interatomic force constants and find an unphysical feature of the Stillinger-Weber empirical potential that correlates with a much noted error in the radial distribution function associated with that potential. We also find that the intrinsic first peak of the radial distribution function is asymmetric, contrary to usual assumptions made in the analysis of diffraction data. We use our results for the normal mode frequencies and polarization vectors to obtain the zero-point broadening effect on the radial distribution function, enabling us to directly compare theory and a high resolution x-ray diffraction experiment. read less USED (high confidence) T. Sinno and M. Prasad, “Internally consistent verification of mean-field models for aggregation using large-scale molecular dynamics,” Molecular Physics. 2004. link Times cited: 3 Abstract: The underlying atomistic mechanisms that govern vacancy aggr… read moreAbstract: The underlying atomistic mechanisms that govern vacancy aggregation in crystalline silicon are probed using a parametrically consistent, two-scale approach. The essential ingredient in this framework is a direct, quantitative comparison between the predictions of atomistic and continuum simulations for the transient size distribution of vacancy clusters. The former is carried out with parallel molecular dynamics simulation of a silicon system containing 215 000 atoms and 1000 vacancies. The continuum model is based on a sequence of coupled Master equations and is parametrized based on the same empirical potential used to perform the atomistic aggregation simulation. An excellent representation of the cluster size distribution can be obtained with consistent parameters only if the relevant physical mechanisms are captured correctly. The inclusion of vacancy cluster diffusion and a model to capture the dynamic nature of cluster morphology at high temperature are necessary to reproduce the results of the large-scale atomistic simulation. Finally, the continuum model is used to investigate cluster evolution for longer times, which are relevant for process simulation of defect-optimized silicon substrates for microelectronic device fabrication. read less USED (high confidence) R. Dash, P. Voyles, J. Gibson, M. Treacy, and P. Keblinski, “A quantitative measure of medium-range order in amorphous materials from transmission electron micrographs,” Journal of Physics: Condensed Matter. 2003. link Times cited: 33 Abstract: We propose an extension to the technique of fluctuation elec… read moreAbstract: We propose an extension to the technique of fluctuation electron microscopy that quantitatively measures a medium-range order correlation length in amorphous materials. In both simulated images from computer-generated paracrystalline amorphous silicon models and experimental images of amorphous silicon, we find that the spatial autocorrelation function of dark-field transmission electron micrographs of amorphous materials exhibits a simple exponential decay. The decay length measures a nanometre-scale structural correlation length in the sample, although it also depends on the microscope resolution. We also propose a new interpretation of the fluctuation microscopy image variance in terms of fluctuations in local atomic pair distribution functions. read less USED (high confidence) M. Prasad and T. Sinno, “Internally consistent approach for modeling solid-state aggregation. II. Mean-field representation of atomistic processes,” Physical Review B. 2003. link Times cited: 20 Abstract: A detailed continuum (mean-field) model is presented that ca… read moreAbstract: A detailed continuum (mean-field) model is presented that captures quantitatively the evolution of a vacancy cluster size distribution in crystalline silicon simulated directly by large-scale parallel molecular dynamics. The continuum model is parametrized entirely using the results of atomistic simulations based on the same empirical potential used to perform the atomistic aggregation simulation, leading to an internally consistent comparison across the two scales. It is found that an excellent representation of all measured components of the cluster size distribution can be obtained with consistent parameters only if the assumed physical mechanisms are captured correctly. In particular, the inclusion of vacancy cluster diffusion and a model to capture the dynamic nature of cluster morphology at high temperature are necessary to reproduce the results of the large-scale atomistic simulation. Dynamic clusters with large capture volumes at high temperature, which are the result of rapid cluster shape fluctuations, are shown to be larger than would be expected from static analyses, leading to substantial enhancement of the nucleation rate. Based on these results, it is shown that a parametrically consistent atomistic-continuum comparison can be used as a sensitive framework for formulating accurate continuum models of complex phenomena such as defect aggregation in solids. read less USED (high confidence) J. F. Justo, R. W. Nunes, and L. Assali, “Microscopic structure of the 90 ◦ and 30 ◦ partial dislocations in gallium arsenide,” Journal of Physics: Condensed Matter. 2002. link Times cited: 8 Abstract: We performed a theoretical investigation on the atomic struc… read moreAbstract: We performed a theoretical investigation on the atomic structure of {111} glide partial dislocations in gallium arsenide. The calculations were carried out using ab initio total energy methods, based on the density functional theory and the pseudopotential model. We addressed the microscopic structure of the 90° partial and the 30° partial dislocations. Our results show that the atomic configurations of the dislocation cores are similar to those proposed for the same dislocations in non-polar semiconductors. For the 90° partial, the double-period reconstruction is energetically more favourable than the single-period reconstruction. In addition, we computed the interaction of intrinsic defects with the dislocation cores. read less USED (high confidence) A. Antonelli, J. F. Justo, and A. Fazzio, “Arsenic segregation, pairing and mobility on the cores of partial dislocations in silicon,” Journal of Physics: Condensed Matter. 2002. link Times cited: 3 Abstract: We studied the effects of arsenic on properties of dislocati… read moreAbstract: We studied the effects of arsenic on properties of dislocations in silicon. The theoretical investigation was carried out using ab initio total energy methods, based on the density functional theory. We find that the interaction of an arsenic impurity in the crystal with a dislocation results in a charge exchange, driving the dislocation core to a negative charge state. This interaction is essentially electrostatic and attractive, and leads to arsenic segregation. Although arsenic segregation to the core is energetically favourable, formation of arsenic pairs inside the core is energetically unfavourable. We also investigated the role of vacancies in arsenic diffusion inside the dislocation core. read less USED (high confidence) S. Nakhmanson and N. Mousseau, “Crystallization study of model tetrahedral semiconductors,” Journal of Physics: Condensed Matter. 2002. link Times cited: 13 Abstract: The microscopic mechanisms leading to crystallization are no… read moreAbstract: The microscopic mechanisms leading to crystallization are not yet fully understood. This is due, in part, to the lack of atomistic as well as interatomic interaction models for a wide range of materials that can lead to crystallization on a computer-simulation timescale, i.e. < 100 ns. While the nucleation in close-packed systems has been extensively studied, there are almost no numerical results for covalent tetrahedral semiconductors. We present here the simulation results of crystallization from the liquid and amorphous states of a 1000-atom model of silicon, described with a modified Stillinger?Weber potential. With this potential, it is possible to crystallize the model in as little as a few nanoseconds, which opens a door to detailed studies of the nucleation processes in covalent systems. Using topological analysis, we also present a first characterization of the structural fluctuations of the nucleation centres in this system and give a rough estimate for the critical size of these centres. read less USED (high confidence) A. Antonelli, J. F. Justo, and A. Fazzio, “Interaction of As impurities with 30° partial dislocations in Si: An ab initio investigation,” Journal of Applied Physics. 2002. link Times cited: 11 Abstract: We investigated through ab initio total energy calculations … read moreAbstract: We investigated through ab initio total energy calculations the interaction of arsenic impurities with the core of a 30° partial dislocation in silicon. It was found that when an arsenic atom sits in a crystalline position near the dislocation core, there is charge transfer from the arsenic towards the dislocation core. As a result, the arsenic becomes positively charged and the core negatively charged. The results indicate that the structural changes around the impurity are very small in both environments, namely, the crystal and the dislocation core. In this scenario, the interaction between arsenic and the core is essentially electrostatic, which eventually leads to arsenic segregation. The segregation energy was found to be as large as 0.5 eV/atom. Additionally, it was found that arsenic pairing inside the core is not energetically favorable. read less USED (high confidence) N. Mousseau and G. Barkema, “Fast bond-transposition algorithms for generating covalent amorphous structures,” Current Opinion in Solid State & Materials Science. 2001. link Times cited: 8 USED (high confidence) J. F. Justo and L. Assali, “Reconstruction defects on partial dislocations in semiconductors,” Applied Physics Letters. 2001. link Times cited: 4 Abstract: Using ab initio total energy calculations, we investigated t… read moreAbstract: Using ab initio total energy calculations, we investigated the structural and electronic properties of reconstruction defects, or antiphase defects, in the core of a 30° partial dislocation in silicon and gallium arsenide. In GaAs, we identified two different reconstruction defects in the dislocation cores, corresponding to a Ga undercoordinated atom, and an As undercoordinated atom. Formation energies of these reconstruction defects were compared to experimental results on the concentration of electrically active centers in deformed semiconducting materials. read less USED (high confidence) J. F. Justo, M. Koning, W. Cai, and V. Bulatov, “Point defect interaction with dislocations in silicon,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2001. link Times cited: 8 USED (high confidence) W. Cai, V. Bulatov, and S. Yip, “Kinetic Monte Carlo method for dislocation glide in silicon,” Journal of Computer-Aided Materials Design. 1999. link Times cited: 20 USED (high confidence) M. de Koning, A. Antonelli, and S. Yip, “Reversible scaling: Optimized free-energy determination using atomistic simulation techniques,” Journal of Computer-Aided Materials Design. 1999. link Times cited: 2 USED (high confidence) A. Lopez-Cazalilla et al., “Simulation of redistributive and erosive effects in a-Si under Ar+ irradiation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2018. link Times cited: 14 USED (high confidence) S. Dorfman, D. Fuks, L. A. C. Malbouisson, K. C. Mundim, and D. Ellis, “Influence of many‐body interactions on resistance of a grain boundary with respect to a sliding shift,” International Journal of Quantum Chemistry. 2002. link Times cited: 4 Abstract: We performed nonempirical simulations of the properties of t… read moreAbstract: We performed nonempirical simulations of the properties of the tungsten Σ3(111) grain boundary (GB) with a boron atom and demonstrate the influence of many-body interactions on the resistance of the GB with respect to sliding. We also studied the propagation of relaxations in the vicinity of the GB. The many-body interatomic potentials (IP) used in these simulations were obtained with the recursion procedure from ab initio total energy calculations. At each step of the slip process, the equilibrium positions of the atoms near GB were calculated with the generalized simulated annealing technique. It was demonstrated how the sliding shift influences the penetration of the elastic field inside the grain. © 2002 Wiley Periodicals, Inc. Int J Quantum Chem, 2002 read less USED (low confidence) L. Dai et al., “Mechanism of phase transition from OLCs with different structures to nPCD at high temperature and high pressure,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) E. Mørtsell, D. Zhao, A. Autruffe, Y. Chen, M. Sabatino, and Y. Li, “The Nature of a Low Angle Grain-Boundary in a Si Bi-Crystal with Added Fe Impurities,” SSRN Electronic Journal. 2023. link Times cited: 0 USED (low confidence) A. Lopez-Cazalilla, K. Nordlund, and F. Djurabekova, “Formation of parallel and perpendicular ripples on solid amorphous surfaces by ion beam-driven atomic flow on and under the surface,” Physical Review Materials. 2023. link Times cited: 1 USED (low confidence) A. Galashev, “Numerical simulation of functioning a silicene anode of a lithium-ion battery,” J. Comput. Sci. 2022. link Times cited: 3 USED (low confidence) N. J. Corrente, E. L. Hinks, A. Kasera, R. Gough, P. Ravikovitch, and A. Neimark, “Modeling adsorption of simple fluids and hydrocarbons on nanoporous carbons,” Carbon. 2022. link Times cited: 2 USED (low confidence) G. G. Vidable, R. González, F. Valencia, N. Amigo, D. Tramontina, and E. Bringa, “Simulations of plasticity in diamond nanoparticles showing ultrahigh strength,” Diamond and Related Materials. 2022. link Times cited: 8 USED (low confidence) Y. Liu, W. Wan, Q. Li, Z. Xiong, C. Tang, and L. Zhou, “Revisiting the Rate-Dependent Mechanical Response of Typical Silicon Structures via Molecular Dynamics,” Nanomaterials. 2022. link Times cited: 1 Abstract: Strain rate is a critical parameter in the mechanical applic… read moreAbstract: Strain rate is a critical parameter in the mechanical application of nano-devices. A comparative atomistic study on both perfect monocrystalline silicon crystal and silicon nanowire was performed to investigate how the strain rate affects the mechanical response of these silicon structures. Using a rate response model, the strain rate sensitivity and the critical strain rate of two structures were given. The rate-dependent dislocation activities in the fracture process were also discussed, from which the dislocation nucleation and motion were found to play an important role in the low strain rate deformations. Finally, through the comparison of five equivalent stresses, the von Mises stress was verified as a robust yield criterion of the two silicon structures under the strain rate effects. read less USED (low confidence) W. Wan, C. Tang, J. Zhang, and L. Zhou, “General Molecular Dynamics Approach to Understand the Mechanical Anisotropy of Monocrystalline Silicon under the Nanoscale Effects of Point Defect,” Nanomaterials. 2021. link Times cited: 7 Abstract: Mechanical anisotropy and point defects would greatly affect… read moreAbstract: Mechanical anisotropy and point defects would greatly affect the product quality while producing silicon wafers via diamond-wire cutting. For three major orientations concerned in wafer production, their mechanical performances under the nanoscale effects of a point defect were systematically investigated through molecular dynamics methods. The results indicated anisotropic mechanical performance with fracture phenomena in the uniaxial deformation process of monocrystalline silicon. Exponential reduction caused by the point defect has been demonstrated for some properties like yield strength and elastic strain energy release. Dislocation analysis suggested that the slip of dislocations appeared and created hexagonal diamond structures with stacking faults in the [100] orientation. Meanwhile, no dislocation was observed in [110] and [111] orientations. Visualization of atomic stress proved that the extreme stress regions of the simulation models exhibited different geometric and numerical characteristics due to the mechanical anisotropy. Moreover, the regional evolution of stress concentration and crystal fracture were interrelated and mutually promoted. This article contributes to the research towards the mechanical and fracture anisotropy of monocrystalline silicon. read less USED (low confidence) Y. Wang et al., “Tension–compression asymmetry in amorphous silicon,” Nature Materials. 2021. link Times cited: 27 USED (low confidence) K. Talaat, M. El-Genk, and B. Cowen, “Extrapolation of thermal conductivity in non-equilibrium molecular dynamics simulations to bulk scale,” International Communications in Heat and Mass Transfer. 2020. link Times cited: 2 USED (low confidence) Y. Shi and I. Szlufarska, “Wear-induced microstructural evolution of nanocrystalline aluminum and the role of zirconium dopants,” Acta Materialia. 2020. link Times cited: 18 USED (low confidence) T. C. Sagar, V. Chinthapenta, and M. Horstemeyer, “Effect of defect guided out-of-plane deformations on the mechanical properties of graphene,” Fullerenes, Nanotubes and Carbon Nanostructures. 2020. link Times cited: 5 Abstract: In this paper, nanoscale mechanical properties and failure b… read moreAbstract: In this paper, nanoscale mechanical properties and failure behavior of graphene with Stone-Wales defect concentration were investigated using molecular dynamics simulations with the latest ReaxFFC-2013 potential that can accurately capture bond breakages of graphitic compounds. The choice of interatomic potential plays an essential role in capturing the deformation mechanism accurately. Stable configuration of two-dimensional graphene experiences out-of-plane deformation leading to ripples and wrinkles in graphene. It is observed that the mechanical properties such as Young’s modulus, ultimate tensile strength, and the fracture strain are dependent on the out-of-plane deformation, temperature, defect concentration, defect orientation, defect layout and loading configuration. It is observed that the post transient phase non-homogenous ripples and wrinkles influence the mechanical properties at low and high defect concentrations, respectively. read less USED (low confidence) M. Alam, L. Lymperakis, and J. Neugebauer, “Phase diagram of grain boundary facet and line junctions in silicon,” Physical Review Materials. 2020. link Times cited: 1 Abstract: The presence of facets and line junctions connecting facets … read moreAbstract: The presence of facets and line junctions connecting facets on grain boundaries (GBs) has a strong impact on the properties of structural, functional, and optoelectronic materials: They govern the mobility of interfaces, the segregation of impurities, as well the electronic properties. In the present paper, we employ density-functional theory and modified embedded atom method calculations to systematically investigate the energetics and thermodynamic stability of these defects. As a prototype system, we consider (cid:2) 3 tilt GBs in Si. By analyzing the energetics of different faceted GBs, we derive a diagram that describes and predicts the reconstruction of these extended defects as a function of facet length and boundary inclination angle. The phase diagram sheds light upon the fundamental mechanisms causing GB faceting phenomena. It demonstrates that the properties of faceting are not determined solely by anisotropic GB energies but by a complex interplay between geometry and microstructure, boundary energies as well as long-range strain interactions. read less USED (low confidence) F. Ojaghnezhad and H. Shodja, “Mechanics of carbon-coated silicon nanowire via second strain gradient theory,” European Journal of Mechanics A-solids. 2020. link Times cited: 6 USED (low confidence) S. N. H. Eliassen, J. Friis, I. G. Ringdalen, N. Mousseau, M. Trochet, and Y. Li, “Atomistic approach to simulate kink migration and kink-pair formation in silicon: The kinetic activation-relaxation technique,” Physical Review B. 2019. link Times cited: 0 Abstract: The energy conversion efficiency of solar cells based on mul… read moreAbstract: The energy conversion efficiency of solar cells based on multicrystalline silicon is greatly deteriorated by dislocations. However, an in-depth understanding on the dislocation motion dynamics down to atomic scale is still lacking. In this paper, we propose a novel atomistic approach to simulate the kink migration and kink-pair formation which govern dislocation motion in silicon, namely the kinetic activation-relax technique (k-ART). With this method, long timescale events can be simulated and complex energy landscapes can be explored. Four mechanisms for kink migration are observed, with total activation energy of 0.16, 0.25, 0.32, and 0.25 eV. New nontrivial kink structures that participate in kink migration are identified due to the open-ended search algorithm for saddle points in k-ART. In addition, a new pathway for kink-pair formation, with a minimum activation energy of 1.11 eV is discovered. The effect of shear stress on kink migration is also investigated. It shows that shear stress shifts the energy barriers of available events to lower energies, resulting in a change of the preferred kink-migration mechanism and a reduction of kink-pair formation energy. read less USED (low confidence) H. Nguyen, “Graphene layer of hybrid graphene/hexagonal boron nitride model upon heating,” Carbon Letters. 2019. link Times cited: 9 USED (low confidence) J. Zhang, J. Yang, D. Hou, and Q. Ding, “Molecular dynamics study on calcium aluminosilicate hydrate at elevated temperatures: Structure, dynamics and mechanical properties,” Materials Chemistry and Physics. 2019. link Times cited: 14 USED (low confidence) H. Nguyen and T. T. Hanh, “Melting process of zigzag boron nitride nanoribbon,” Physica E: Low-dimensional Systems and Nanostructures. 2019. link Times cited: 4 USED (low confidence) T. Sipkens and K. Daun, “Effect of Surface Interatomic Potential on Thermal Accommodation Coefficients Derived from Molecular Dynamics,” The Journal of Physical Chemistry C. 2018. link Times cited: 14 Abstract: This work investigates how the interatomic surface potential… read moreAbstract: This work investigates how the interatomic surface potential influences molecular dynamics (MD)-derived thermal accommodation coefficients (TACs). Iron, copper, and silicon surfaces are considered over a range of temperatures that include their melting points. Several classes of potentials are reviewed, including two-body, three-body, and bond-order force fields. MD-derived densities and visualization of the surfaces are used to explain the differences in the parameterizations of these potentials within the context of gas–surface scattering. Finally, TACs are predicted for a range of gas–surface combinations, and recommended values of the TAC are selected that take into account the robustness and uncertainties of each of the considered parameterizations. Further, it is observed that there is a significant change in the TAC about phase changes that must be taken into account for applications with a large range of surface temperatures. read less USED (low confidence) F. González-Cataldo, F. Corvacho, and G. Gutiérrez, “Melting curve of Si by means of the Z-method,” Journal of Physics: Conference Series. 2018. link Times cited: 1 Abstract: The melting curve of silicon is investigated through classic… read moreAbstract: The melting curve of silicon is investigated through classical molecular dynamics simulations. We explore pressures from 0 to 20 GPa using the EDIP, Stillinger-Weber, and Tersoff interactomic potentials. Using the Z method, we demonstrate that the predicted melting temperature Tm can be significantly overestimated, depending on the potential chosen. Our results show that none of the potentials explored is able to reproduce the experimental melting curve of silicon by means of the Z-method. However, the EDIP potential does predict the change in the Clapeyron slope, associated with the diamond to β-tin phase transition. read less USED (low confidence) R. Atta-Fynn and P. Biswas, “Nearly defect-free dynamical models of disordered solids: The case of amorphous silicon.,” The Journal of chemical physics. 2018. link Times cited: 17 Abstract: It is widely accepted in the materials modeling community th… read moreAbstract: It is widely accepted in the materials modeling community that defect-free realistic networks of amorphous silicon cannot be prepared by quenching from a molten state of silicon using classical or ab initio molecular-dynamics (MD) simulations. In this work, we address this long-standing problem by producing nearly defect-free ultra-large models of amorphous silicon, consisting of up to half a million atoms, using classical MD simulations. The structural, topological, electronic, and vibrational properties of the models are presented and compared with experimental data. A comparison of the models with those obtained from using the modified Wooten-Winer-Weaire bond-switching algorithm shows that the models are on par with the latter, which were generated via event-based total-energy relaxations of atomistic networks in the configuration space. The MD models produced in this work represent the highest quality of amorphous-silicon networks so far reported in the literature using MD simulations. read less USED (low confidence) H. N. Pishkenari and S. Rezaei, “Characterization of silicon surface elastic constants based on different interatomic potentials,” Thin Solid Films. 2017. link Times cited: 7 USED (low confidence) H. N. Pishkenari, E. Mohagheghian, and A. Rasouli, “Molecular dynamics study of the thermal expansion coefficient of silicon,” Physics Letters A. 2016. link Times cited: 23 USED (low confidence) O. Strickson, “Numerical constitutive modelling for continuum mechanics simulation.” 2016. link Times cited: 0 USED (low confidence) H. N. Pishkenari, B. Afsharmanesh, and F. Tajaddodianfar, “Continuum models calibrated with atomistic simulations for the transverse vibrations of silicon nanowires,” International Journal of Engineering Science. 2016. link Times cited: 27 USED (low confidence) M. Ganchenkova et al., “Influence of the ab-initio calculation parameters on prediction of energy of point defects in silicon,” Modern Electronic Materials. 2015. link Times cited: 7 USED (low confidence) N. S. Mikhaleva, M. Visotin, Z. Popov, A. Kuzubov, and A. Fedorov, “Ab initio and empirical modeling of lithium atoms penetration into silicon,” Computational Materials Science. 2015. link Times cited: 4 USED (low confidence) P. Hecquet, “Subcritical damping of SA step energy on Si(001) vicinals by lowering terrace stress,” Surface Science. 2015. link Times cited: 0 USED (low confidence) P. Hecquet, “Interaction energy between dipole lines applied on symmetric (2 × 1) reconstructed Si(001),” Surface Science. 2014. link Times cited: 1 USED (low confidence) G. Fugallo and A. Mattoni, “Thermally induced recrystallization of textured hydrogenated nanocrystalline silicon,” Physical Review B. 2014. link Times cited: 18 USED (low confidence) P. Hecquet, “Surface stresses on symmetric (2 × 1) reconstructed Si(001) calculated from surface energy variations,” Surface Science. 2013. link Times cited: 3 USED (low confidence) C. D. Cruz, P. Chantrenne, and X. Kleber, “Molecular Dynamics Simulations and Kapitza Conductance Prediction of Si/Au Systems Using the New Full 2NN MEAM Si/Au Cross-Potential,” Journal of Heat Transfer-transactions of The Asme. 2012. link Times cited: 10 Abstract: Superlattices made by superposing dielectric and metal nanol… read moreAbstract: Superlattices made by superposing dielectric and metal nanolayers are of great interest as their small size restricts the thermal energy carrier mean free path, decreasing the thermal conductivity and thereby increasing the thermoelectric figure of merit. It is, therefore, essential to predict their thermal conductivity. Potentials for Au and Si are discussed, and the potential of second nearest-neighbor modified embedded atom method (2NN MEAM) is chosen as being the best for simulating heat transfer in Si/Au systems. Full 2NN MEAM Si/Au cross-potential parameterization is developed, and the results are compared with ab initio calculations to test its ability to reproduce local density approximation (LDA) calculations. Volume-constant (NVT) molecular dynamics simulations are performed to deposit Au atoms on an Si substrate by physical vapor deposition, and the results of the intermixing zone are in good agreement with the Cahn and Hilliard theory. Nonequilibrium molecular dynamics simulations are performed for an average temperature of 300 K to determine the Kapitza conductance of Si/Au systems, and the obtained value of 158 MW/m 2 K is in good agreement with the results of Komarov for Au deposited on isotopically pure Si- 28 and natural Si, with values ranging between 133 and 182 MW/m2 K. read less USED (low confidence) H. Whitlow and S. Nakagawa, “Ordering effects in extreme high-resolution depth profiling with MeV ion beams,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2012. link Times cited: 0 USED (low confidence) E. Holmstrom, J. Kotakoski, L. Lechner, U. Kaiser, and K. Nordlund, “Atomic-scale effects behind structural instabilities in Si lamellae during ion beam thinning,” AIP Advances. 2012. link Times cited: 9 Abstract: The rise of nanotechnology has created an ever-increasing ne… read moreAbstract: The rise of nanotechnology has created an ever-increasing need to probe structures on the atomic scale, to which transmission electron microscopy has largely been the answer. Currently, the only way to efficiently thin arbitrary bulk samples into thin lamellae in preparation for this technique is to use a focused ion beam (FIB). Unfortunately, the established FIB thinning method is limited to producing samples of thickness above ∼20 nm. Using atomistic simulations alongside experiments, we show that this is due to effects from finite ion beam sharpness at low milling energies combined with atomic-scale effects at high energies which lead to shrinkage of the lamella. Specifically, we show that attaining thickness below 26 nm using a milling energy of 30 keV is fundamentally prevented by atomistic effects at the top edge of the lamella. Our results also explain the success of a recently proposed alternative FIB thinning method, which is free of the limitations of the conventional approach due to the absence... read less USED (low confidence) D. Daisenberger et al., “Polyamorphic amorphous silicon at high pressure: raman and spatially resolved X-ray scattering and molecular dynamics studies.,” The journal of physical chemistry. B. 2011. link Times cited: 34 Abstract: We studied the low-frequency Raman and X-ray scattering beha… read moreAbstract: We studied the low-frequency Raman and X-ray scattering behavior of amorphous silicon (a-Si) at high pressure throughout the range where the density-driven polyamorphic transformation between the low-density amorphous (LDA) semiconductor and a novel metallic high-density amorphous (HDA) polyamorph occurs. The experimental data were analyzed with the aid of molecular dynamics (MD) simulations using the Stillinger-Weber potential. The heat capacity of a-Si obtained from the low pressure Raman data exhibits non Debye-like behavior, but the effect is small, and our data support the conclusion that no boson peak is present. The high-pressure Raman data show the presence of a distinct low frequency band for the HDA polyamorph in agreement with ab initio MD simulations. Spatially resolved synchrotron X-ray diffraction was used to study the high pressure behavior of the a-Si sample throughout the LDA-HDA transition range without interference by crystallization events. The X-ray data were analyzed using an iterative refinement strategy to extract real-space structural information. The appearance of the first diffraction peak (FDP) in the scattering function S(Q) is discussed in terms of the void structure determined from Voronoi analysis of the MD simulation data. read less USED (low confidence) C.-ying Wang, Z. Wang, Q. Meng, C. Li, and H.-wei Zheng, “Atomic simulation of the dynamic properties for a double period structure with 90° partial dislocation in Si,” Superlattices and Microstructures. 2011. link Times cited: 3 USED (low confidence) C.-ying Wang, Z. Wang, and Q. Meng, “Comparative study of the empirical interatomic potentials and density-functional simulations of divacancy and hexavacancy in silicon,” Physica B-condensed Matter. 2011. link Times cited: 3 USED (low confidence) J. Rabier and L. Pizzagalli, “Dislocation dipole annihilation in diamond and silicon,” Journal of Physics: Conference Series. 2011. link Times cited: 5 Abstract: The mechanism of dislocation dipole annihilation has been in… read moreAbstract: The mechanism of dislocation dipole annihilation has been investigated in C and Si using atomistic calculations with the aim of studying their annihilation by-products. It is shown, in C as well as in Si, that dipole annihilation yields debris that can be depicted as a cluster of vacancies, or alternately by two internal free surfaces. These defects have no strain field and can hardly be seen using usual TEM techniques. This suggests that the brown colouration of diamond could be due to microstructures resulting from deformation mechanisms associated with dipole formation and their annihilation rather than to a climb mechanism and vacancy aggregation. In silicon where a number of dipoles have been evidenced by TEM when dislocation trails are found, such debris could be the missing link responsible for the observation of strong chemical reactivity and electrical activity in the wake of moving dislocations. read less USED (low confidence) L. Pizzagalli, J. Godet, J. Guénolé, and S. Brochard, “Dislocation cores in silicon: new aspects from numerical simulations,” Journal of Physics: Conference Series. 2011. link Times cited: 9 Abstract: Recent theoretical investigations of the properties of dislo… read moreAbstract: Recent theoretical investigations of the properties of dislocation cores in silicon are reviewed. New results, obtained from numerical simulations for the non-dissociated screw and 60° dislocations, are presented and discussed in relation with experiments. read less USED (low confidence) S. Giordano, A. Mattoni, and L. Colombo, “Brittle Fracture: From Elasticity Theory to Atomistic Simulations,” Reviews in Computational Chemistry. 2010. link Times cited: 13 Abstract: Understanding the mechanical properties of materials with th… read moreAbstract: Understanding the mechanical properties of materials with theory traditionally has been done by using continuum methods, ranging from elastic theory (in both linear and nonlinear regimes), to plastic theory, and to fracture mechanics. The computational counterpart of continuum modeling is represented by finite element analysis. Continuum theories have been extremely successful, as proved by the tremendous achievements reached in structural design of buildings, ships, bridges, air-/space crafts, nuclear reactors, and so on. Overall this represents the core of theoretical and computational solid mechanics. In the last 20 years or so, the technological rush toward nano-sized systems has forced researchers to investigate mechanical phenomena at a length scale in which matter no longer can be considered as a continuum. This is the case, for instance, of investigating the crack-related features in a material displaying elastic or structural complexity (or, equivalently, inhomogeneity or disorder) at the nanoscale. This problem of atomic-scale granularity immediately seems to be prohibitive for (standard) solid mechanics. To better elaborate on this read less USED (low confidence) M. Amsler and S. Goedecker, “Crystal structure prediction using the minima hopping method.,” The Journal of chemical physics. 2010. link Times cited: 219 Abstract: A structure prediction method is presented based on the mini… read moreAbstract: A structure prediction method is presented based on the minima hopping method. To escape local minima, moves on the configurational enthalpy surface are performed by variable cell shape molecular dynamics. To optimize the escape steps the initial atomic and cell velocities are aligned to low curvature directions of the current local minimum. The method is applied to both silicon crystals and well-studied binary Lennard-Jones mixtures. For the latter new putative ground state structures are presented. It is shown that a high success rate is achieved and a reliable prediction of unknown ground state structures is possible. read less USED (low confidence) E. Lampin, C. Priester, C. Krzeminski, and L. Magaud, “Graphene buffer layer on Si-terminated SiC studied with an empirical interatomic potential,” Journal of Applied Physics. 2010. link Times cited: 23 Abstract: The atomistic structure of the graphenebuffer layer on Si-te… read moreAbstract: The atomistic structure of the graphenebuffer layer on Si-terminated SiC is investigated using a modified version of the environment-dependent interatomic potential. The determination of the equilibrium state by the conjuguate gradients method suffers from a complex multiple-minima energy surface. The initial configuration is therefore modified to set the system in specific valleys of the energy surface. The solution of minimal energy forms a hexagonal pattern composed of stuck regions separated by unbonded rods that release the misfit with the SiC surface. The structure presents the experimental symmetries and a global agreement with an ab initio calculation. It is therefore expected that the interatomic potential could be used in classical molecular dynamics calculations to study the graphene growth. read less USED (low confidence) Y. Jing and Q. Meng, “Molecular dynamics simulations of the mechanical properties of crystalline/amorphous silicon core/shell nanowires,” Physica B-condensed Matter. 2010. link Times cited: 32 USED (low confidence) C. Li, Q. Meng, and K. Zhong, “ATOMISTIC STUDY OF THE STRENGTH AND ELASTIC CONSTANTS OF PERFECT AND DEFECTED SILICON,” International Journal of Modern Physics B. 2009. link Times cited: 6 Abstract: The effects of vacancies on the strength and elastic constan… read moreAbstract: The effects of vacancies on the strength and elastic constants of silicon, such as Young's modulus and Poisson's ratio are investigated using the molecular dynamics simulations with the Stillinger–Weber potential. The defected crystalline cells contain randomly generated defect distributions in the simulation models. The ideal strength is found to be 33.6 GPa at the strain 0.26. The Young's modulus and Poisson's ratio is 148 GPa and 0.252, respectively. It is found that the strength decreases as the point defect fraction increases, and the variation of the strength versus the point defect fraction coincides with a decaying exponential function. In addition, vacancies are shown to reduce the elastic constants. In general, the elastic constants of silicon vary linearly versus the defect fraction. read less USED (low confidence) A. Pedersen, L. Pizzagalli, and H. Jónsson, “Finding mechanism of transitions in complex systems: formation and migration of dislocation kinks in a silicon crystal,” Journal of Physics: Condensed Matter. 2009. link Times cited: 21 Abstract: We demonstrate how a saddle point search method can be used … read moreAbstract: We demonstrate how a saddle point search method can be used to study dislocation mobility in a covalent material—a non-trivial transition mechanism in a complex system. Repeated saddle point searches have been carried out by using the minimum mode following algorithm and dimer method in combination with several empirical potential functions for silicon in order to determine the mechanisms for the creation and migration of kinks on a non-dissociated screw dislocation in a silicon crystal. For the environment-dependent interatomic potential, three possible kink migration processes have been identified with activation energies of 0.17, 0.25, and 0.33 eV. The Lenosky potential gives a single, low energy migration mechanism with an activation energy of 0.07 eV, in good agreement with density functional theory results. The kink formation mechanism determined using this potential has an activation barrier of 1.2 eV. Calculations were also carried out with the Tersoff potential, Stillinger–Weber potential and Bolding–Andersen potential. The various potential functions give quite different results for the kink structure and the mechanism of transition. read less USED (low confidence) Y. Jing, Q. Meng, and W. Zhao, “Molecular dynamics simulations of the tensile and melting behaviours of silicon nanowires,” Physica E-low-dimensional Systems & Nanostructures. 2009. link Times cited: 30 USED (low confidence) C.-ying Wang, Q. Meng, K. Zhong, and Z. Yang, “Atomic simulations of the dynamic properties of the30°partial dislocation in Si crystal,” Physical Review B. 2008. link Times cited: 9 USED (low confidence) C. Li, Q. Meng, K. Zhong, and C.-ying Wang, “Computer simulation of the 60° dislocation interaction with vacancy cluster in silicon,” Physical Review B. 2008. link Times cited: 11 Abstract: In the current work, the interaction of the 60\ifmmode^\circ… read moreAbstract: In the current work, the interaction of the 60\ifmmode^\circ\else\textdegree\fi{} shuffle dislocation with the vacancy cluster under applied shear stress in silicon crystal is studied via the molecular dynamics method. Stillinger-Weber (SW) potential and environment-dependent interatomic potential (EDIP) are used to calculate the interatomic forces. Simulation results show that at low shear stress, the dislocation is pinned by a vacancy cluster. With the stress level increased to a certain critical value ${\ensuremath{\sigma}}_{l}$, the dislocation can overcome the pinning and get through. It is found that ${\ensuremath{\sigma}}_{l}$ reaches its maximum at a transition temperature, which is about $350\phantom{\rule{0.3em}{0ex}}\mathrm{K}$. Also revealed in the simulations is a generalized dislocation dissociation that a 60\ifmmode^\circ\else\textdegree\fi{} dislocation, while interacting with a vacancy cluster, can result in 30\ifmmode^\circ\else\textdegree\fi{} and 90\ifmmode^\circ\else\textdegree\fi{} partial dislocations when the applied shear stress reaches another critical value ${\ensuremath{\sigma}}_{h}$. The two resultant partial dislocations are separated by an intrinsic stacking fault. Unlike ${\ensuremath{\sigma}}_{l}$, ${\ensuremath{\sigma}}_{h}$ keeps decreasing at temperatures higher than $400\phantom{\rule{0.3em}{0ex}}\mathrm{K}$ and remains a constant at lower temperatures. read less USED (low confidence) P. Heino, “Dispersion and thermal resistivity in silicon nanofilms
by molecular dynamics,” The European Physical Journal B. 2007. link Times cited: 45 USED (low confidence) H. Whitlow and S. Nakagawa, “Low-energy primary knock on atom damage distributions near MeV proton beams focused to nanometre dimensions,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 14 USED (low confidence) A. Chehaidar and T. Chermiti, “Microstructural analysis of nanoporous paracrystalline atomistic models of amorphous silicon,” Journal of Non-crystalline Solids. 2007. link Times cited: 7 USED (low confidence) T. Kumagai, S. Izumi, S. Hara, and S. Sakai, “Development of bond-order potentials that can reproduce the elastic constants and melting point of silicon for classical molecular dynamics simulation,” Computational Materials Science. 2007. link Times cited: 148 USED (low confidence) C. R. Miranda, K. V. Tretiakov, and S. Scandolo, “A computational study of elastic properties of disordered systems with voids,” Journal of Non-crystalline Solids. 2006. link Times cited: 9 USED (low confidence) H. Wilson, S. Prawer, P. Spizzirri, D. Jamieson, N. Stavrias, and D. Mckenzie, “P-2 dimer implantation in silicon: A molecular dynamics study,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2006. link Times cited: 11 USED (low confidence) A. Chehaidar and H. Khelifi, “Microstructural analysis of paracrystalline atomistic models of amorphous silicon,” Journal of Non-crystalline Solids. 2006. link Times cited: 1 USED (low confidence) M. Bachlechner et al., “Mechanisms of pit formation at strained crystallineSi(111)∕Si3N4(0001)interfaces: Molecular-dynamics simulations,” Physical Review B. 2006. link Times cited: 8 USED (low confidence) Z. Huang, X. Wang, and C. Lawrence, “Molecular Dynamics Simulation of Thermal Transport in Single-Wall Silicon Nanotubes.” 2006. link Times cited: 0 USED (low confidence) M. Bachlechner, J. Y. Zhang, Y.-F. Wang, J. Schiffbauer, S. Knudsen, and D. Korakakis, “Molecular dynamics simulations of the mechanical strength of Si/ Si 3 N 4 interfaces,” Physical Review B. 2005. link Times cited: 8 Abstract: Molecular dynamics simulations are performed on parallel com… read moreAbstract: Molecular dynamics simulations are performed on parallel computers to investigate the crystalline $\mathrm{Si}(111)∕{\mathrm{Si}}_{3}{\mathrm{N}}_{4}(0001)$ interface that is modeled as an eight-component system. The average total energy per particle and the average kinetic energy per particle of the subsystems are monitored during the preparation of the system. The Young's modulus of the interface is compared with that of the silicon part alone and that of the silicon-nitride film, respectively. The results for one extended simulation feature a crack in the silicon-nitride film and dislocated atoms in silicon below the crack. Simulations at rates of strain ranging from $0.00125\phantom{\rule{0.3em}{0ex}}\text{to}\phantom{\rule{0.3em}{0ex}}0.05\phantom{\rule{0.3em}{0ex}}{\mathrm{ps}}^{\ensuremath{-}1}$ show that for lower strain rates, the systems stretched faster reach their ultimate strength at a higher strain value than those that were stretched more slowly. At the highest strain rates, however, the failure mechanisms change qualitatively indicative of a more ductile behavior. read less USED (low confidence) T. Petersen, I. Snook, I. Yarovsky, and D. McCulloch, “Monte Carlo based modeling of carbon nanostructured surfaces,” Physical Review B. 2005. link Times cited: 11 Abstract: We have developed Monte Carlo based algorithms to produce re… read moreAbstract: We have developed Monte Carlo based algorithms to produce realistic models of complex carbon nanostructures with nontrivial curvature, including fullerene junctions between nanotubes. The models are constructed by first analytically defining curved surfaces and then optimizing the configuration of carbon atoms on these surfaces using a realistic interatomic potential. We illustrate our method by generating several previously proposed and also new types of structures, which all show realistic physical properties. Our method is not limited to these structures and can be used to generate large models of nanostructured materials with complex surface geometries and porous structure on the nano-scale. read less USED (low confidence) Q. Lu and B. Bhattacharya, “The role of atomistic simulations in probing the small-scale aspects of fracture—a case study on a single-walled carbon nanotube,” Engineering Fracture Mechanics. 2005. link Times cited: 71 USED (low confidence) X. W. Zhou, “Analytical and numerical calculations of interatomic forces and stresses,” Molecular Simulation. 2005. link Times cited: 0 Abstract: Atomistic simulation methods such as molecular dynamics requ… read moreAbstract: Atomistic simulation methods such as molecular dynamics require an efficient calculation of interatomic forces and stresses from pre–defined interatomic potentials. Both analytical and numerical approaches can be used to do this. Analytical approach directly calculates forces and stresses using analytical formulae, and can therefore yield accurate results. However, the force and stress expressions may become extremely complicated as the complexity level of the potential increases, resulting in a prolonged development cycle to implement new potentials. Numerical approach uses finite difference method to evaluate forces and stresses through simple calculation of energies at selected perturbations of crystal configurations. The method can be quickly implemented and tested for any potentials. However, it may result in significant numerical errors. We have compared analytical and numerical calculations of interatomic forces and stresses in molecular dynamics, and identified the conditions where numerical method can be successfully used without significant errors. read less USED (low confidence) N. Marks, “Thin film deposition of tetrahedral amorphous carbon: a molecular dynamics study,” Diamond and Related Materials. 2005. link Times cited: 56 USED (low confidence) M. Posselt, F. Gao, and D. Zwicker, “Atomistic Study of the Migration of Di- and Tri-Interstitials in Silicon,” Physical Review B. 2005. link Times cited: 40 Abstract: A comprehensive study on the migration of di- and tri-inters… read moreAbstract: A comprehensive study on the migration of di- and tri-interstitials in silicon is performed using classical molecular dynamics simulations with a Stillinger-Weber potential. At first the structures and energetics of the di- and the tri-interstitial are investigated, and the accuracy of the interatomic potential is tested by comparing the results with literature data obtained by tight-binding and density-functional-theory calculations. Then the migration is investigated for temperatures between 800 and 1600 K. Very long simulation times, large computational cells and different initial conditions are considered. The defect diffusivity, the self-diffusion coefficient per defect and the corresponding effective migration barriers are calculated. Compared to a mono-interstitial, the di-interstitial migrates faster, whereas the tri-interstitial diffuses slower. The mobility of the di- and the mono-interstitial is higher than the mobility of the lattice atoms during the diffusion of these defects. On the other hand, the tri-interstitial mobility is lower than the corresponding atomic mobility. The migration mechanism of the di-interstitial shows a pronounced dependence on the temperature. At low temperature a high mobility on zigzag-like lines along a axis within a {l_brace}110{r_brace} plane is found, whereas the change between equivalent directions or equivalent {l_brace}110{r_brace} planes occurs seldomly and requires a long simulationmore » time, but the rate of directional change increases with increasing temperature. During the diffusion within {l_brace}110{r_brace} planes the di-interstitial moves like a wave packet so that the atomic mobility is lower than that of the defect. On the other hand, the change between equivalent {l_brace}110{r_brace} migration planes is characterized by frequent atomic rearrangements. The visual analysis of the tri-interstitial diffusion reveals complex migration mechanisms and a high atomic mobility. The diffusivities and effective migration barriers obtained are compared with the few data from the literature. The implications of the present results for the explanation of experimental data on defect evolution and migration are discussed.« less read less USED (low confidence) M. Bianconi, E. Albertazzi, S. Balboni, L. Colombo, G. Lulli, and A. Satta, “Channeling characterization of defects in silicon: an atomistic approach,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 15 USED (low confidence) G. Pearce, N. Marks, D. Mckenzie, and M. Bilek, “Molecular dynamics simulation of the thermal spike in amorphous carbon thin films,” Diamond and Related Materials. 2005. link Times cited: 20 USED (low confidence) J. Feldman and N. Bernstein, “Vibrational spectroscopy of an amorphous∕crystalline sandwich structure for silicon: Numerical results,” Physical Review B. 2004. link Times cited: 19 USED (low confidence) S. Izumi, S. Hara, T. Kumagai, and S. Sakai, “A method for calculating surface stress and surface elastic constants by molecular dynamics: application to the surface of crystal and amorphous silicon,” Thin Solid Films. 2004. link Times cited: 65 USED (low confidence) M. Bianconi, E. Albertazzi, S. Balboni, and G. Lulli, “Analysis of ion implanted silicon by RBS-channeling: influence of the damage model,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2004. link Times cited: 3 USED (low confidence) R. W. Nunes, L. Assali, and J. F. Justo, “Ab initio investigations on the dislocation core properties in zinc-blende semiconductors,” Computational Materials Science. 2004. link Times cited: 4 USED (low confidence) N. Marks, J. Bell, G. Pearce, D. Mckenzie, and M. Bilek, “Atomistic simulation of energy and temperature effects in the deposition and implantation of amorphous carbon thin films,” Diamond and Related Materials. 2003. link Times cited: 28 USED (low confidence) R. Rurali and E. Hernández, “Trocadero: a multiple-algorithm multiple-model atomistic simulation program,” Computational Materials Science. 2003. link Times cited: 61 USED (low confidence) C. R. S. Silva, “Optimizing Metropolis Monte Carlo simulations of semiconductors,” Computer Physics Communications. 2003. link Times cited: 7 USED (low confidence) L. Pizzagalli, P. Beauchamp, and J. Rabier, “Undissociated screw dislocations in silicon: Calculations of core structure and energy,” Philosophical Magazine. 2003. link Times cited: 47 Abstract: The stability of the perfect screw dislocation in silicon ha… read moreAbstract: The stability of the perfect screw dislocation in silicon has been investigated using both classical potentials and first-principles calculations. Although a recent study by Koizumi et al. stated that the stable screw dislocation was located in both the 'shuffle' and the 'glide' sets of {111} planes, it is shown that this result depends on the classical potential used, and that the most stable configuration belongs to the 'shuffle' set only, in the centre of one (amp;1tilde;01) hexagon. We also investigated the stability of an sp2 hybridization in the core of the dislocation, obtained for one metastable configuration in the 'glide' set. The core structures are characterized in several ways, with a description of the three-dimensional structure, differential displacement maps and derivatives of the disregistry. read less USED (low confidence) C. Bishop and W. Carter, “Relating atomistic grain boundary simulation results to the phase-field model,” Computational Materials Science. 2002. link Times cited: 27 USED (low confidence) J. Feldman, “Calculations of the generalized dynamic structure factor for amorphous silicon,” Journal of Non-crystalline Solids. 2002. link Times cited: 3 USED (low confidence) S. Dorfman, D. Ellis, K. C. Mundim, V. Liubich, and D. Fuks, “Many‐Body Ab Initio Potentials in Simulations of Grain Boundary Sliding and Decohesion in Metals,” Advanced Engineering Materials. 2002. link Times cited: 1 Abstract: A direct scheme for theoretical study of sliding and decohes… read moreAbstract: A direct scheme for theoretical study of sliding and decohesion properties of the grain boundaries (GB) in metals is presented here. This approach combines ab initio calculations and Monte Carlo (MC) simulations with non-empirical many-body (MB) potentials. The authors studied the propagation of the elastic field in the vicinity of the GB and show how the sliding or decohesion shifts influence the penetration of the elastic field inside the grain. read less USED (low confidence) F. Finkemeier and W. Niessen, “Reply to ‘Comment on ‘Boson peak in amorphous silicon: A numerical study’ ,’” Physical Review B. 2002. link Times cited: 7 Abstract: Nakhmanson, Drabold, and Mousseau [Phys. Rev. B 66, 087201 (… read moreAbstract: Nakhmanson, Drabold, and Mousseau [Phys. Rev. B 66, 087201 (2002)], preceding paper criticized the model of the present authors [F. Finkemeier and W. von Niessen, Phys. Rev. B 63, 235204 (2001)] which was used for an explanation of the Boson peak in $a\ensuremath{-}\mathrm{Si}.$ NDM criticized the generation of the model and the potential used by us. The low-frequency vibrational density of states $g(\ensuremath{\omega})$ of $a\ensuremath{-}\mathrm{Si}$ are reinvestigated with the help of an improved structural model using the modified Stillinger-Weber potential. The previously described deviation from the Debye ${\ensuremath{\omega}}^{2}$ behavior is shown to be an artifact caused by an unrealistic high defect concentration. Nevertheless the improved model, which possesses a strongly decreased number of defects, still shows the existence of additional low-frequency modes compared to the crystal, which could be part of an explanation of the boson peak arising already in the harmonic approximation. read less USED (low confidence) V. Burlakov, Y. Tsukahara, G. Briggs, and A. Sutton, “Simulation of growth of porous SiOx nanostructures,” Materials Science and Technology. 2002. link Times cited: 3 Abstract: Monte Carlo simulation of deposition of non-stoichiometric a… read moreAbstract: Monte Carlo simulation of deposition of non-stoichiometric amorphous SiOx nanolayers from vapour phase onto a polymer surface is reported. The model developed is based on the network properties of silica and takes into account dangling bonds arising during the real process of deposition. The model is validated via comparison of the radial and bond angle distribution functions for the simulated Si and SiO2 structures with those obtained from experiment for bulk materials. Porosity of the simulated amorphous layer is characterised by the relative volume of pores and the ratio of the pore surfaces to the pore volume. It was found that porosity strongly depends on nucleation sites density (NSD) on the polymer substrate. At NSD lower than 1 nm-2 the porosity may reach as much as 30% of the layer volume, while at NSD higher than 4 nm-2 it decreases down to 3/7%. Possible implications of the obtained results are discussed. read less USED (low confidence) J. Nord, K. Nordlund, and J. Keinonen, “Molecular dynamics simulation of ion-beam-amorphization of Si, Ge and GaAs,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2002. link Times cited: 24 USED (low confidence) N. Marks, “Modelling diamond-like carbon with the environment-dependent interaction potential,” Journal of Physics: Condensed Matter. 2002. link Times cited: 97 Abstract: The environment-dependent interaction potential is a transfe… read moreAbstract: The environment-dependent interaction potential is a transferable empirical potential for carbon which is well suited for studying disordered systems. Ab initio data are used to motivate and parametrize the functional form, which includes environment-dependence in the pair and triple terms, and a generalized aspherical coordination describing dihedral rotation and non-bonded π-repulsion. Simulations of liquid carbon compare very favourably with Car-Parrinello calculations, while amorphous networks generated by liquid quench have properties superior to Tersoff, Brenner and orthogonal tight-binding calculations. The efficiency of the method enables the first simulations of tetrahedral amorphous carbon by deposition, and a new model for the formation of diamond-like bonding is presented. read less USED (low confidence) A. Antonelli, J. F. Justo, and A. Fazzio, “Dopant interaction with a dislocation in silicon: local and non-local effects,” Physica B-condensed Matter. 2001. link Times cited: 1 USED (low confidence) X. P. Xie, M. Liang, Z. M. Choo, and S. Li, “A COMPARATIVE SIMULATION STUDY OF SILICON (001) SURFACE RECONSTRUCTION USING DIFFERENT INTERATOMIC POTENTIALS,” Surface Review and Letters. 2001. link Times cited: 3 Abstract: We have performed a comparative study of Si(001) surface rec… read moreAbstract: We have performed a comparative study of Si(001) surface reconstruction employing molecular dynamics simulation using the interatomic potentials of Stillinger–Weber, Tersoff and Bazant–Kaxiras. Simulations were carried out for temperatures at 300 K and 1000 K using each of these three potentials. At 300 K, the three potentials were found to generate surface features comprising mainly the simple (2 × 1) reconstruction. At 1000 K, more complex reconstruction similar to the p(2 × 2) and c(2 × 2) patterns was observed on the surfaces of Stillinger–Weber and Tersoff crystals while the surface generated on Bazant–Kaxiras crystal is characterized by disorderliness with no identifiable pattern of reconstruction. read less USED (low confidence) S. Dorfman, K. C. Mundim, V. Liubich, and D. Fuks, “Sliding and decohesion of Σ3〈111〉 grain boundary in tungsten: Monte Carlo simulations with many-body ab initio potentials,” Journal of Applied Physics. 2001. link Times cited: 6 Abstract: We perform atomistic simulations of the properties of the Σ3… read moreAbstract: We perform atomistic simulations of the properties of the Σ3〈111〉 grain boundary (GB) in W and demonstrate the influence of many-body interactions on the resistance of the grain boundary with respect to sliding and decohesion shifts. The distribution of the elastic field in the vicinity of the GB is considered. The interatomic potentials used in these simulations are obtained from ab initio total energy calculations using a recursion procedure to extract interatomic potentials. read less USED (low confidence) M. Mazzarolo, L. Colombo, G. Lulli, and E. Albertazzi, “Low-energy recoils in crystalline silicon: Quantum simulations,” Physical Review B. 2001. link Times cited: 19 USED (low confidence) R. Vink, G. Barkema, W. F. Weg, and N. Mousseau, “Fitting the Stillinger–Weber potential to amorphous silicon,” Journal of Non-crystalline Solids. 2001. link Times cited: 137 USED (low confidence) S. Pizzini, M. Acciarri, E. Leoni, and A. Donne, “About the D1 and D2 Dislocation Luminescence and Its Correlation with Oxygen Segregation,” Physica Status Solidi B-basic Solid State Physics. 2000. link Times cited: 34 Abstract: It is well known that the dislocation luminescence (DL) in s… read moreAbstract: It is well known that the dislocation luminescence (DL) in silicon consists of four main bands, conventionally labelled D1 to D4, where E 1 = 0.807 eV, E 2 = 0.874 eV, E 3 = 0.95 eV and E 4 = 0.99 eV, of which the D1 is considered of interest for optoelectronic devices working in the third window of optical communications. Although DL has been the subject of a number of investigations in the last twenty years, still some questions remain open, concerning both the origin of the dislocation luminescence and its intrinsic or extrinsic nature. We report in this paper the results of a combination of complementary dislocation generation processes (deformation and oxide segregation) and characterisation procedures (photoluminescence and surface photovoltage), which give a strong evidence that the Dl band is correlated in a very complex manner with the presence of optically active silicon self-interstitials and oxygen clusters. read less USED (low confidence) L. Brambilla, L. Colombo, V. Rosato, and F. Cleri, “Solid–liquid interface velocity and diffusivity in laser-melt amorphous silicon,” Applied Physics Letters. 2000. link Times cited: 27 Abstract: We studied the microscopic kinetics of the amorphous-liquid … read moreAbstract: We studied the microscopic kinetics of the amorphous-liquid interface in supercooled laser-melt silicon by means of molecular dynamics computer simulations. The interface velocity was obtained as a function of temperature by direct simulation of the interface motion in an amorphous-liquid model system. The temperature dependence of the kinetic prefactor was extracted from the interface velocity function and compared to the values of self-diffusivity obtained from independent molecular dynamics simulations of bulk amorphous Si. The kinetic prefactor for interfacial diffusion shows a distinctly non-Arrhenius behavior which is attributed to Fulcher–Vogel kinetics in the supercooled liquid. read less USED (low confidence) T. Sinno, E. Dornberger, W. Ammon, R. A. Brown, and F. Dupret, “Defect engineering of Czochralski single-crystal silicon,” Materials Science & Engineering R-reports. 2000. link Times cited: 112 USED (low confidence) C. Herrero, “Path-integral Monte Carlo study of amorphous silicon,” Journal of Non-crystalline Solids. 2000. link Times cited: 8 USED (low confidence) M. Mäki-Jaskari, K. Kaski, and A. Kuronen, “Simulations of crack initiation in silicon,” Computational Materials Science. 2000. link Times cited: 6 USED (low confidence) S. Nakhmanson and D. A. Drabold, “Computer simulation of low-energy excitations in amorphous silicon with voids,” Journal of Non-crystalline Solids. 1999. link Times cited: 15 USED (low confidence) J. F. Justo, V. Bulatov, and S. Yip, “DISLOCATION CORE RECONSTRUCTION AND ITS EFFECT ON DISLOCATION MOBILITY IN SILICON,” Journal of Applied Physics. 1999. link Times cited: 31 Abstract: Through atomistic calculations of kink nucleation and migrat… read moreAbstract: Through atomistic calculations of kink nucleation and migration in the core of partial dislocations in silicon we demonstrate that symmetry-breaking structural reconstructions will strongly affect dislocation mobility. Core reconstructions give rise to multiple kink species, and, relative to kinks in an unreconstructed dislocation, an increase in kink formation and migration energies. These factors provide additional resistance to dislocation motion which scales with the energy reconstruction. Our results indicate that the observed variations of dislocation mobility in going from elemental to IV–IV, and further to III–V and II–VI zinc-blende semiconductors, can be attributed in part to the weakening of core reconstruction across the series. read less USED (low confidence) E. Kaxiras and S. Yip, “Modelling and simulation of solids,” Current Opinion in Solid State & Materials Science. 1998. link Times cited: 14 USED (low confidence) Y. Shi and I. Szlufarska, “Simulations of Wear-Induced Microstructural Evolution in Nanocrystalline Aluminum.” 2021. link Times cited: 0 USED (low confidence) B. Gargeya and S. Pal, “Influence of specimen size and strain rate on tensile deformation and fracture behavior of single-layer Silicene,” Materials Today: Proceedings. 2019. link Times cited: 0 USED (low confidence) A. Kiselev, “Molecular dynamics simulations of laser ablation in covalent materials.” 2017. link Times cited: 1 Abstract: 15 Deutsche Zusammenfassung 18 I. Theoretical background 27… read moreAbstract: 15 Deutsche Zusammenfassung 18 I. Theoretical background 27 read less USED (low confidence) E. Lampin, “Recrystallization of Silicon by Classical Molecular Dynamics.” 2015. link Times cited: 0 USED (low confidence) M. Ganchenkova and R. Nieminen, “Mechanical Properties of Silicon Microstructures.” 2015. link Times cited: 4 USED (low confidence) T. Sinno, “Atomistic Calculation of Defect Thermodynamics in Crystalline Silicon.” 2015. link Times cited: 0 USED (low confidence) R. Khanna and V. Sahajwalla, “Atomistic Simulations of Properties and Phenomena at High Temperatures.” 2014. link Times cited: 3 USED (low confidence) P. K. Soin, A. Horsfield, and D. Nguyen-Manh, “Magnetic Tight-Binding Simulations of Defects in Iron,” MRS Proceedings. 2011. link Times cited: 0 USED (low confidence) H. Lai, S. M. Sea, H. Kennel, and S. Dunham, “Molecular Dynamics Modeling of Stress and Orientation Dependence of Solid Phase Epitaxial Regrowth,” MRS Proceedings. 2010. link Times cited: 2 USED (low confidence) E. Holmström, A. Krasheninnikov, and K. Nordlund, “Quantum and Classical Molecular Dynamics Studies of the Threshold Displacement Energy in Si Bulk and Nanowires,” MRS Proceedings. 2009. link Times cited: 6 USED (low confidence) L. Tian and X. Wang, “Pulsed Laser Heating-induced Surface Rapid Cooling and Amorphization,” MRS Proceedings. 2008. link Times cited: 0 USED (low confidence) J. A. Pascual-Gutiérrez, J. Murthy, and R. Viskanta, “Physical Properties of Confined Silicon Structures Using EDIP.” 2006. link Times cited: 0 Abstract: Physically confined structures such as thin films and nanowi… read moreAbstract: Physically confined structures such as thin films and nanowires are becoming increasingly important in the energy and electronics sectors. This has resulted from the ability to tailor nanostructures to yield physical properties that are significantly different from bulk. The main focus of this work is to examine how physical confinement in one and two dimensions affects the phonon wave vector spectrum within the first Brillouin zone of silicon thin films and silicon nanowires. Dispersion curves as well as density of states (DOS) are obtained using the dynamical matrix approach and a harmonic approximation to the three-body environmentally-dependent interatomic potential (EDIP). It is also shown how these changes in the phonon spectrum for both films and wires affect the volumetric specific heat with respect to bulk. The simulations are carried out assuming ideal free-standing boundary conditions. It is shown that confinement effects on the phonon specific heat are only important below 5 mm for both silicon films and wires.Copyright © 2006 by ASME read less USED (low confidence) P. Kratzer, “Atomistic Simulations of Processes at Surfaces.” 2004. link Times cited: 1 USED (low confidence) E. J. Albenze, L. A. Matejik, N. F. Fynan, and P. Clancy, “Prediction of the Interface Response Functions for Amorphous and Crystalline Phases of Silicon and Germanium.” 2003. link Times cited: 1 USED (low confidence) J. F. Justo, A. Antonelli, and A. Fazzio, “The energetics of dislocation cores in semiconductors and their role on dislocation mobility,” Physica B-condensed Matter. 2001. link Times cited: 4 USED (low confidence) N. Bernstein and D. Hess, “Multiscale Simulations of Brittle Fracture and the Quantum-Mechanical Nature of Bonding in Silicon,” MRS Proceedings. 2000. link Times cited: 5 USED (low confidence) V. Burlakov, Y. Tsukahara, G. Briggs, and A. Sutton, “Monte Carlo Simulation of Vapour Deposition of Nonstoichiometric Amorphous Silica,” MRS Proceedings. 2000. link Times cited: 1 USED (low confidence) M. M. Buneat, P. Fastenko, and S. Dunham, “Atomistic Simulations of Damage Evolution in Silicon,” MRS Proceedings. 1999. link Times cited: 3 Abstract: We have studied the damage annealing process using kinetic l… read moreAbstract: We have studied the damage annealing process using kinetic lattice Monte Carlo (KLMC) and molecular dynamics (MD) with initial damage distribution from Monte Carlo ion implant simulations. MD calculations find a long range interstitial vacancy interaction, as also seen in previous tight-binding molecular dynamics (TBMD) simulations. 1 The influence of the long range interaction as well as the initial spatial correlations present in the implant damage are then analyzed with KLMC in the form of corrections to the +1 model. We find that both long range interactions and the initial spatial correlations are significant at low doses, while the effects disappear at high doses. read less USED (low confidence) M. Koning, A. Antonelli, M. Bazant, E. Kaxiras, and J. F. Justo, “Unstable Stacking Fault Free Energies in Silicon through Empirical Modeling,” MRS Proceedings. 1998. link Times cited: 0 NOT USED (low confidence) X. Jiang, H. Sun, K. Choudhary, H. Zhuang, and Q. Nian, “Interpretable Ensemble Learning for Materials Property Prediction with Classical Interatomic Potentials: Carbon as an Example,” ArXiv. 2023. link Times cited: 0 Abstract: Machine learning (ML) is widely used to explore crystal mate… read moreAbstract: Machine learning (ML) is widely used to explore crystal materials and predict their properties. However, the training is time-consuming for deep-learning models, and the regression process is a black box that is hard to interpret. Also, the preprocess to transfer a crystal structure into the input of ML, called descriptor, needs to be designed carefully. To efficiently predict important properties of materials, we propose an approach based on ensemble learning consisting of regression trees to predict formation energy and elastic constants based on small-size datasets of carbon allotropes as an example. Without using any descriptor, the inputs are the properties calculated by molecular dynamics with 9 different classical interatomic potentials. Overall, the results from ensemble learning are more accurate than those from classical interatomic potentials, and ensemble learning can capture the relatively accurate properties from the 9 classical potentials as criteria for predicting the final properties. read less NOT USED (low confidence) M. Maździarz, “Transferability of interatomic potentials for silicene,” Beilstein Journal of Nanotechnology. 2023. link Times cited: 1 Abstract: The ability of various interatomic potentials to reproduce t… read moreAbstract: The ability of various interatomic potentials to reproduce the properties of silicene, that is, 2D single-layer silicon, polymorphs was examined. Structural and mechanical properties of flat, low-buckled, trigonal dumbbell, honeycomb dumbbell, and large honeycomb dumbbell silicene phases, were obtained using density functional theory and molecular statics calculations with Tersoff, MEAM, Stillinger–Weber, EDIP, ReaxFF, COMB, and machine-learning-based interatomic potentials. A quantitative systematic comparison and a discussion of the results obtained are reported. read less NOT USED (low confidence) H. Niu et al., “A machine-learning interatomic potential to understand primary radiation damage of silicon,” Computational Materials Science. 2023. link Times cited: 3 NOT USED (low confidence) D. Vizoso, G. Subhash, K. Rajan, and R. Dingreville, “Connecting Vibrational Spectroscopy to Atomic Structure via Supervised Manifold Learning: Beyond Peak Analysis,” Chemistry of Materials. 2023. link Times cited: 1 Abstract: Vibrational spectroscopy is a nondestructive technique commo… read moreAbstract: Vibrational spectroscopy is a nondestructive technique commonly used in chemical and physical analyses to determine atomic structures and associated properties. However, the evaluation and interpretation of spectroscopic profiles based on human-identifiable peaks can be difficult and convoluted. To address this challenge, we present a reliable protocol based on supervised manifold learning techniques meant to connect vibrational spectra to a variety of complex and diverse atomic structure configurations. As an illustration, we examined a large database of virtual vibrational spectroscopy profiles generated from atomistic simulations for silicon structures subjected to different stress, amorphization, and disordering states. We evaluated representative features in those spectra via various linear and nonlinear dimensionality reduction techniques and used the reduced representation of those features with decision trees to correlate them with structural information unavailable through classical human-identifiable peak analysis. We show that our trained model accurately (over 97% accuracy) and robustly (insensitive to noise) disentangles the contribution from the different material states, hence demonstrating a comprehensive decoding of spectroscopic profiles beyond classical (human-identifiable) peak analysis. read less NOT USED (low confidence) J. Singh and S. Singh, “A review on Machine learning aspect in physics and mechanics of glasses,” Materials Science and Engineering: B. 2022. link Times cited: 10 NOT USED (low confidence) A. M. Barboza, L. C. R. Aliaga, D. Faria, and I. Bastos, “Bilayer Graphene Kirigami,” SSRN Electronic Journal. 2022. link Times cited: 1 NOT USED (low confidence) L. J. Lewis, “Fifty years of amorphous silicon models : the end of the story?,” Journal of Non-Crystalline Solids. 2022. link Times cited: 8 NOT USED (low confidence) X. Qian and R. Yang, “Machine learning for predicting thermal transport properties of solids,” Materials Science and Engineering: R: Reports. 2021. link Times cited: 34 NOT USED (low confidence) S. Yoo, B. Lee, and K. Kang, “Density functional theory study of the mechanical behavior of silicene and development of a Tersoff interatomic potential model tailored for elastic behavior,” Nanotechnology. 2021. link Times cited: 8 Abstract: Silicene, a graphene-like 2D material made from Si atoms, ha… read moreAbstract: Silicene, a graphene-like 2D material made from Si atoms, has been fabricated and studied for its promising applications in micro/nanoelectronics. For the reliable function of silicene devices, it is important to investigate silicene’s mechanical properties. In this study, the authors conducted density functional theory (DFT) simulations of mechanical tests of silicene and investigated the elastic modulus and mechanical response such as structural transformation. In addition, the authors optimized the Tersoff potential parameters using a gradient-based minimization with a grid search method in hyperdimensional parameter space, to match the DFT calculation results in the elastic regime. With the new parameter set, the elastic moduli of silicene in the zigzag (ZZ) and armchair (AC) directions were computed with molecular statics (MS) simulations and compared with those of other Si interatomic potential models and DFT results. In addition, uniaxial tensile tests along the ZZ and AC directions were performed to examine how far the Tersoff model is transferable with our new parameter set to describe the nonlinear mechanical behavior of silicene. The results of uniaxial tensile tests suggest that the angle penalty function in the Tersoff model needs to be modified and that the stress–strain curve predicted with this modification shows improvement compared to the original function. read less NOT USED (low confidence) Y. Lysogorskiy et al., “Performant implementation of the atomic cluster expansion (PACE) and application to copper and silicon,” npj Computational Materials. 2021. link Times cited: 84 NOT USED (low confidence) A. Rohskopf, S. Wyant, K. Gordiz, H. R. Seyf, M. G. Muraleedharan, and A. Henry, “Fast & accurate interatomic potentials for describing thermal vibrations,” Computational Materials Science. 2020. link Times cited: 7 NOT USED (low confidence) S. Gelin, A. Champagne-Ruel, and N. Mousseau, “Enthalpy-entropy compensation of atomic diffusion originates from softening of low frequency phonons,” Nature Communications. 2020. link Times cited: 16 NOT USED (low confidence) V. Tikare, R. Devanathan, and M. Caturla, “List of Authors,” 2019 Panhellenic Conference on Electronics & Telecommunications (PACET). 2019. link Times cited: 0 NOT USED (low confidence) J. V. Michelin, L. G. Gonçalves, and J. Rino, “On the transferability of interaction potentials for condensed phases of silicon,” Journal of Molecular Liquids. 2019. link Times cited: 6 NOT USED (low confidence) J. Harrison, J. Schall, S. Maskey, P. Mikulski, M. T. Knippenberg, and B. Morrow, “Review of force fields and intermolecular potentials used in atomistic computational materials research,” Applied Physics Reviews. 2018. link Times cited: 99 Abstract: Molecular simulation is a powerful computational tool for a … read moreAbstract: Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferability. When selecting a potential energy function for use in molecular simulations, the accuracy, transferability, and computational speed must all be considered. In this focused review, some of the more commonly used potential energy functions for molecular simulations are reviewed with an eye toward presenting their general forms, strengths, and weaknesses.Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferabilit... read less NOT USED (low confidence) A. Antidormi, X. Cartoixà, and L. Colombo, “Nature of microscopic heat carriers in nanoporous silicon,” Physical Review Materials. 2018. link Times cited: 6 Abstract: We performed a systematic analysis of the vibrational modes … read moreAbstract: We performed a systematic analysis of the vibrational modes in nanoporous silicon for different values of porosity, separating them into extended modes (diffusons and propagons) and localized vibrations (locons). By calculating the density of states, the participation ratio, and the systems' dispersion curves, the spatial character of each mode as well as the effect of porosity on the thermal conductivity have been investigated. An increase of porosity is shown to promote the existence of increasingly localized modes on one side, and the progressive transformation of propagons to diffusons on the other. Finally, we provide evidence of the sizable contribution of locons to thermal transport found in large porosity samples and discuss the mechanism of energy transfer in terms of mode-mode autocorrelations and cross-correlations. read less NOT USED (low confidence) A. Bartók, J. Kermode, N. Bernstein, and G. Csányi, “Machine Learning a General-Purpose Interatomic Potential for Silicon,” Physical Review X. 2018. link Times cited: 291 Abstract: The success of first principles electronic structure calcula… read moreAbstract: The success of first principles electronic structure calculation for predictive modeling in chemistry, solid state physics, and materials science is constrained by the limitations on simulated length and time scales due to computational cost and its scaling. Techniques based on machine learning ideas for interpolating the Born-Oppenheimer potential energy surface without explicitly describing electrons have recently shown great promise, but accurately and efficiently fitting the physically relevant space of configurations has remained a challenging goal. Here we present a Gaussian Approximation Potential for silicon that achieves this milestone, accurately reproducing density functional theory reference results for a wide range of observable properties, including crystal, liquid, and amorphous bulk phases, as well as point, line, and plane defects. We demonstrate that this new potential enables calculations that would be extremely expensive with a first principles electronic structure method, such as finite temperature phase boundary lines, self-diffusivity in the liquid, formation of the amorphous by slow quench, and dynamic brittle fracture. We show that the uncertainty quantification inherent to the Gaussian process regression framework gives a qualitative estimate of the potential's accuracy for a given atomic configuration. The success of this model shows that it is indeed possible to create a useful machine-learning-based interatomic potential that comprehensively describes a material, and serves as a template for the development of such models in the future. read less NOT USED (low confidence) N. Admal, J. Marian, and G. Po, “The atomistic representation of first strain-gradient elastic tensors,” Journal of The Mechanics and Physics of Solids. 2016. link Times cited: 36 NOT USED (low confidence) B. Liu, H. Zhang, J. Tao, X. Chen, and Y.-A. Zhang, “Comparative investigation of a newly optimized modified embedded atom method potential with other potentials for silicon,” Computational Materials Science. 2015. link Times cited: 7 NOT USED (low confidence) S. Eldridge, A. Waterland, M. Seltzer, J. Appavoo, and A. Joshi, “Towards General-Purpose Neural Network Computing,” 2015 International Conference on Parallel Architecture and Compilation (PACT). 2015. link Times cited: 22 Abstract: Machine learning is becoming pervasive, decades of research … read moreAbstract: Machine learning is becoming pervasive, decades of research in neural network computation is now being leveraged to learn patterns in data and perform computations that are difficult to express using standard programming approaches. Recent work has demonstrated that custom hardware accelerators for neural network processing can outperform software implementations in both performance and power consumption. However, there is neither an agreed-upon interface to neural network accelerators nor a consensus on neural network hardware implementations. We present a generic set of software/hardware extensions, X-FILES, that allow for the general-purpose integration of feedforward and feedback neural network computation in applications. The interface is independent of the network type, configuration, and implementation. Using these proposed extensions, we demonstrate and evaluate an example dynamically allocated, multi-context neural network accelerator architecture, DANA. We show that the combination of X-FILES and our hardware prototype, DANA, enables generic support and increased throughput for neural-network-based computation in multi-threaded scenarios. read less NOT USED (low confidence) S. Pizzini, “Point Defects in Semiconductors.” 2015. link Times cited: 20 NOT USED (low confidence) S. Pizzini, “Extended Defects in Semiconductors and Their Interactions with Point Defects and Impurities.” 2015. link Times cited: 0 NOT USED (low confidence) A. Afzali and S. Maghsoodlou, “Atomistic Simulations Investigation in Nanoscience: A Detailed Review.” 2015. link Times cited: 0 NOT USED (low confidence) J. Behler, “Representing potential energy surfaces by high-dimensional neural network potentials,” Journal of Physics: Condensed Matter. 2014. link Times cited: 293 Abstract: The development of interatomic potentials employing artifici… read moreAbstract: The development of interatomic potentials employing artificial neural networks has seen tremendous progress in recent years. While until recently the applicability of neural network potentials (NNPs) has been restricted to low-dimensional systems, this limitation has now been overcome and high-dimensional NNPs can be used in large-scale molecular dynamics simulations of thousands of atoms. NNPs are constructed by adjusting a set of parameters using data from electronic structure calculations, and in many cases energies and forces can be obtained with very high accuracy. Therefore, NNP-based simulation results are often very close to those gained by a direct application of first-principles methods. In this review, the basic methodology of high-dimensional NNPs will be presented with a special focus on the scope and the remaining limitations of this approach. The development of NNPs requires substantial computational effort as typically thousands of reference calculations are required. Still, if the problem to be studied involves very large systems or long simulation times this overhead is regained quickly. Further, the method is still limited to systems containing about three or four chemical elements due to the rapidly increasing complexity of the configuration space, although many atoms of each species can be present. Due to the ability of NNPs to describe even extremely complex atomic configurations with excellent accuracy irrespective of the nature of the atomic interactions, they represent a general and therefore widely applicable technique, e.g. for addressing problems in materials science, for investigating properties of interfaces, and for studying solvation processes. read less NOT USED (low confidence) J. Thibault, J. Rouviere, and A. Bourret, “Grain Boundaries in Semiconductors,” Materials Science and Technology. 2013. link Times cited: 2 Abstract: The sections in this article are
Introduction
Grain Bo… read moreAbstract: The sections in this article are
Introduction
Grain Boundary Structure: Concepts and Tools
Grain Boundary Definitions
Geometrical Concepts
Dislocation Model
Primary Dislocation Network
Secondary Dislocation Network
Stress Field Associated with Grain Boundaries
Structural Unit Descriptions
Stick and Ball Structural Units
Energetic Structural Units
Algebraic Structural Units
Structural Units and Dislocations/Disclinations
The Limits of the Structural Unit Descriptions
Computer Simulation Techniques
Methods
Boundary Conditions
Interaction Laws
Experimental Techniques
Grain Boundary Structure: Experience and Simulation Results
Silicon and Germanium
Tilt Grain Boundaries
Twist Grain Boundaries
Diamond
SiC
GaAs
GaN
AlN
NiO
Comments on Grain Boundary Structures
Electrical Properties of Grain Boundaries
Introduction
Electrical Effects Induced by Grain Boundaries
Electronic States Associated with a Grain Boundary
Potential Barrier and Transport Properties
Dynamic Properties and Recombination Properties
Experimental Methods for Measuring the Grain Boundary Electrical Activity
Methods Based on Transport
Transient Methods
Correlation Between Electrical Activity and Structure
Transport Experiments in Bicrystals
Transient Properties Measured on Bicrystals
Emission and Capture Properties of Silicon and Germanium Grain Boundaries
Polycrystalline Silicon
Intrinsic or Extrinsic Origin of Electrical Activity of Grain Boundaries
Impurity Segregation and Precipitation Induced by Grain Boundaries
Introduction
Dopant Elements
Oxygen and Sulfur
Transition Elements
Copper
Nickel
Iron
Conclusions
Mechanical Properties of Grain Boundaries in Semiconductors
Introduction
Interaction Between Dislocations and Grain Boundaries
Dislocation Absorption
Dislocation Transmission Across Grain Boundaries
Grain Boundaries as a Dislocation Source
Grain Boundary Dislocation Movement
Physical Consequences
Grain Boundary Migration
Recovery of the Grain Boundary Structure and Cavitation
Deformation Modelling
Conclusions read less NOT USED (low confidence) C. Koch, “Conventional and Advanced Electron Transmission Microscopy.” 2012. link Times cited: 1 NOT USED (low confidence) A. R. Jivani and A. R. Jani, “ELASTIC PROPERTIES OF GROUP IV SEMICONDUCTORS BY PSEUDOPOTENTIAL APPROACH,” International Journal of Modern Physics B. 2011. link Times cited: 4 Abstract: The higher-order perturbation theory based on pseudopotentia… read moreAbstract: The higher-order perturbation theory based on pseudopotential approach is used to investigate few elastic and vibrational properties of Group IV semiconductors. The homogeneous deformation method is used to calculate elastic constants. To consider electron-ion interactions, our own proposed potential is employed to investigate such properties of Si, Ge and α-Sn. The potential contains only single parameter and its value is determined by fitting experimental value of bulk modulus. The calculated physical properties like elastic constants, pressure derivatives of the bulk modulus, pressure derivatives of elastic constants, Young's modulus and Poisson's ratio etc., of Si, Ge and α-Sn are in good agreement with available experimental and other available theoretical results. The deviation of the present calculations using our potential with respect to experimental data is found to be less than 10% in most of the calculated physical properties. To consider the exchange and correlation effect, five different local-field correction functions are incorporated in the present investigations. From the present study, it can be observed that incorporation of local-field correction and the covalent-correction term are important in such type of investigations. read less NOT USED (low confidence) M. Timonova and B. Thijsse, “Thermodynamic properties and phase transitions of silicon using a new MEAM potential,” Computational Materials Science. 2010. link Times cited: 13 NOT USED (low confidence) A. Pedersen and H. Jónsson, “Distributed implementation of the adaptive kinetic Monte Carlo method,” Math. Comput. Simul. 2010. link Times cited: 18 NOT USED (low confidence) S. Ghasemi et al., “The energy landscape of silicon systems and its description by force fields, tight binding schemes, density functional methods and Quantum Monte Carlo methods,” arXiv: Computational Physics. 2009. link Times cited: 27 Abstract: The accuracy of the energy landscape of silicon systems obta… read moreAbstract: The accuracy of the energy landscape of silicon systems obtained from various density functional methods, a tight binding scheme and force fields is studied. Quantum Monte Carlo results serve as quasi exact reference values. In addition to the well known accuracy of DFT methods for geometric ground states and metastable configurations we find that DFT methods give a similar accuracy for transition states and thus a good overall description of the energy landscape. On the other hand, force fields give a very poor description of the landscape that are in most cases too rugged and contain many fake local minima and saddle points or ones that have the wrong height. read less NOT USED (low confidence) P. Schelling, “Phase behavior and kinetics of a new bond-order potential for silicon,” Computational Materials Science. 2008. link Times cited: 24 NOT USED (low confidence) D. Fleetwood, “Selected High-Impact Journal Articles on Defects in Microelectronic Materials and Devices: Selected High-Impact Journal Articles on Defects in Microelectronic Materials and Devices.” 2008. link Times cited: 3 NOT USED (low confidence) A. Mattoni and L. Colombo, “Crystallization kinetics of mixed amorphous-crystalline nanosystems,” Physical Review B. 2008. link Times cited: 16 NOT USED (low confidence) E.-H. Kim, Y.-H. Shin, and B.-J. Lee, “A modified embedded-atom method interatomic potential for Germanium,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2008. link Times cited: 86 NOT USED (low confidence) D. Broido, M. Malorny, G. Birner, N. Mingo, and D. Stewart, “Intrinsic lattice thermal conductivity of semiconductors from first principles,” Applied Physics Letters. 2007. link Times cited: 698 Abstract: The original version of this article may be found at the App… read moreAbstract: The original version of this article may be found at the Applied Physics Letters website:
http://dx.doi.org/10.1063/1.2822891
Copyright (2007) American Institute of Physics read less NOT USED (low confidence) S. Munetoh, T. Motooka, K. Moriguchi, and A. Shintani, “Interatomic potential for Si–O systems using Tersoff parameterization,” Computational Materials Science. 2007. link Times cited: 382 NOT USED (low confidence) Y. Yoshimoto, “Extended multicanonical method combined with thermodynamically optimized potential: application to the liquid-crystal transition of silicon.,” The Journal of chemical physics. 2006. link Times cited: 12 Abstract: A novel method is proposed to study first-order phase transi… read moreAbstract: A novel method is proposed to study first-order phase transition in real materials. It is applied to the liquid-crystal transition of silicon successfully. It consists of two parts: a direct simulation of the transition by an extended multicanonical ensemble with an order parameter defined with structure factors that characterize the transition, and optimization of a model interatomic potential in terms of the ensemble from an accurate one. These provide a principle to project a first-principles approach on a model-based approach conserving thermodynamic properties of multiple phases. read less NOT USED (low confidence) S. Pizzini et al., “Nanocrystalline silicon films as multifunctional material for optoelectronic and photovoltaic applications,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 2006. link Times cited: 27 NOT USED (low confidence) S. Billeter, A. Curioni, D. Fischer, and W. Andreoni, “Ab initio derived augmented Tersoff potential for silicon oxynitride compounds and their interfaces with silicon,” Physical Review B. 2006. link Times cited: 42 Abstract: Coordination-dependent interatomic potentials are proposed f… read moreAbstract: Coordination-dependent interatomic potentials are proposed for silicon oxides and oxynitrides\char22{}also hydrogenated ones\char22{}with a functional form based on the widely used Tersoff silicon potential. They are intended for an accurate sampling of the configurational space of realistic silicon oxynitride systems and their interfaces with silicon, including defects and changes of oxidation states. The parameters, which are given in the text, are obtained by simultaneously mapping forces and energies onto the results of density-functional-theory calculations performed for a set of diverse systems and configurations and a wide composition range. Application to a larger set of systems and configurations shows the transferability of these augmented Tersoff potentials and their validity in predicting bulk lattice parameters, energetics of defect relaxation, and vibrational spectra. read less NOT USED (low confidence) J. Los, L. Ghiringhelli, E. Meijer, and A. Fasolino, “Improved long-range reactive bond-order potential for carbon. I. Construction (Correction on vol 72, pg 214102, 2005),” Acta Crystallographica Section B-structural Science. 2005. link Times cited: 181 Abstract: We present LCBOPII, an improvement of the long-range carbon … read moreAbstract: We present LCBOPII, an improvement of the long-range carbon bond-order potential (LCBOP) by Los and Fasolino [Phys. Rev. B 68, 024107 (2003)]. LCBOPII contains a coordination dependent medium range term for bond distances between 1.7 and $4\phantom{\rule{0.3em}{0ex}}\mathrm{\AA{}}$, meant to reproduce the dissociation energy curves for single, double, and triple bonds and improve the reactive properties as well as the description of the liquid and of low coordinated phases. Other features of LCBOPII are a coordination dependent angular correlation, a correction for antibonding states, and a conjugation dependent torsional interaction based on ab initio calculations of the torsional barriers for a set of molecular configurations. We present results for the geometry and energetics of the graphite-to-diamond transformation and of the vacancy in diamond and graphite as well as the prediction of the energy barrier of the 5-77-5 defect in graphite and graphene for which ab initio results are available only for unsuitably small samples. In the accompanying paper (Ghiringhelli et al., Phys. Rev. B 72, 214103 (2005) we use LCBOPII to evaluate several properties, including the equation of state, of liquid carbon. read less NOT USED (low confidence) N. Voulgarakis, G. Hadjisavvas, P. Kelires, and G. Tsironis, “Computational investigation of intrinsic localization in crystalline Si,” Physical Review B. 2004. link Times cited: 41 Abstract: We investigate numerically existence and dynamical propertie… read moreAbstract: We investigate numerically existence and dynamical properties of intrinsic localization in crystalline silicon through the use of interatomic Tersoff force fields. We find a band of intrinsic localized modes (discrete breathers) each with lifetime of at least 60 ps in the spectral region 548-578 cm - 1 , located just above the zone end phonon frequency calculated at 536 cm - 1 . The localized modes extend to more than second neighbors and involve pair central-atom compressions in the range from 6.1 % to 8.6% of the covalent bond length per atom. Finite temperature simulations show that they remain robust to room temperatures or higher with a typical lifetime equal to 6 ps. read less NOT USED (low confidence) N. Bernstein and D. Hess, “Lattice trapping barriers to brittle fracture.,” Physical review letters. 2003. link Times cited: 138 Abstract: We present a multiscale simulation of a crack in silicon und… read moreAbstract: We present a multiscale simulation of a crack in silicon under tensile loading that is consistent with experiment; fracture is brittle with a modest lattice-trapping energy barrier to crack propagation. Our multiscale molecular-dynamics simulation has a tight-binding description of bonding near the crack tip embedded in an empirical-potential (EP) region. Forces on atoms in the tight-binding region are computed using a Green's function method. Comparing our multiscale simulation with EP simulations shows that the EP models severely overestimate lattice trapping, explaining the failure of the Griffith criterion and the dramatic differences in crack morphology. A two-length-scale model for the lattice-trapping energy barrier correctly predicts the critical load for brittle fracture. We argue that lattice trapping plays an important role in the brittle-to-ductile transition. read less NOT USED (low confidence) G. Hobler and G. Otto, “Status and open problems in modeling of as-implanted damage in silicon,” Materials Science in Semiconductor Processing. 2003. link Times cited: 64 NOT USED (low confidence) N. Mousseau, G. Barkema, and S. Nakhmanson, “Recent developments in the study of continuous random networks,” Philosophical Magazine B. 2002. link Times cited: 11 Abstract: We report on recent progress in the development of new techn… read moreAbstract: We report on recent progress in the development of new techniques to generate high-quality models of continuous random networks, which are used as models for elemental and binary tetrahedral semiconductors such as amorphous Si and amorphous GaAs. The availability of such models has allowed us to look at a number of outstanding issues regarding their electronic properties, the fundamental role of defects and dynamics. We describe briefly our modifications made to the Wooten-Winer-Weaire bond-switching algorithm, allowing us to produce low-strain amorphous and paracrystalline networks of up to 10000 atoms. Then some of the structural and electronic properties of these models are presented. We also discuss briefly some recent results on the recrystallization of amorphous networks. read less NOT USED (low confidence) J. F. Justo and L. Assali, “Electrically active centers in partial dislocations in semiconductors,” Physica B-condensed Matter. 2001. link Times cited: 4 NOT USED (low confidence) J. F. Justo, A. Antonelli, and A. Fazzio, “Dislocation core properties in semiconductors,” Solid State Communications. 2001. link Times cited: 20 NOT USED (low confidence) V. Ásgeirsson and H. Jónsson, “Exploring Potential Energy Surfaces with Saddle Point Searches,” Handbook of Materials Modeling. 2020. link Times cited: 11 NOT USED (low confidence) P. Szarek and A. Tachibana, “Contemporary analysis of the influence of adsorbents on the structure, stability, and reactivity of main group nanoparticles using regional density functional theory,” Harnessing Nanoscale Surface Interactions. 2019. link Times cited: 0 NOT USED (low confidence) K. N. Clayton, J. Khor, and S. Wereley, “Rapid Electrokinetic Patterning and Its Applications.” 2015. link Times cited: 1 NOT USED (low confidence) N. Marks, “Amorphous Carbon and Related Materials.” 2010. link Times cited: 7 NOT USED (low confidence) R. W. Nunes and J. F. Justo, “Silicon Nanowires: From Empirical to First Principles Modeling.” 2010. link Times cited: 0 NOT USED (low confidence) M. Ganchenkova and R. Nieminen, “Chapter Eleven – Mechanical Properties of Silicon Microstructures.” 2010. link Times cited: 3 NOT USED (low confidence) R. Khare, S. L. Mielke, J. T. Paci, S. Zhang, G. Schatz, and T. Belytschko, “Two quantum mechanical/molecular mechanical coupling schemes appropriate for fracture mechanics studies.” 2007. link Times cited: 10 Abstract: Many coupled quantum mechanical/molecular mechanical (QM/MM)… read moreAbstract: Many coupled quantum mechanical/molecular mechanical (QM/MM) methods employ disjoint sub domains for the MM and QM regions together with link atoms to ameliorate the effects of severing covalent bonds that straddle the QM/MM interface. In the context of simulations of mechanical properties, this can be problematic because the interactions bet ween the subdomains are then modeled by bonds involving link atoms and such bonds typically do not closely resemble those of the original system. In this paper we consider two coupling schemes that employ overlapping domains. The first is the ONIOM schem e of Morokuma et al. that includes an MM treatment of the entire system together with QM corrections for key subdomains. The second is a new approach that we will refer to as the overlapping domain link atom (ODLA) method. This method involves only a min imal overlap between the QM and MM subdomains. One important advantage of the ODLA scheme as compared to the ONIOM method is that, within the region that is treated entirely by QM methods, chemical interactions can be modeled for which reliable MM potenti als are unavailable. Results of fracture studies of defected graphene sheets obtained with the ONIOM and ODLA methods are compared to benchmark results obtained by an entirely QM treatment. Both coupling methods perform well and the two coupling methods display very close agreement. read less NOT USED (low confidence) J. F. Justo, “Modeling Covalent Bond with Interatomic Potentials.” 2005. link Times cited: 1 NOT USED (low confidence) E. Kaxiras and S. Yip, “Introduction: Atomistic Nature of Materials.” 2005. link Times cited: 2 NOT USED (low confidence) C. L. Allred, J. Borenstein, M. Weinberg, X. Yuan, M. Bazant, and L. Hobbs, “Defect-Induced Shifts in the Elastic Constants of Silicon,” MRS Proceedings. 2002. link Times cited: 1 Abstract: As MEMS devices become ever more sensitive, even slight shif… read moreAbstract: As MEMS devices become ever more sensitive, even slight shifts in materials properties can be detrimental to device performance. Radiation-induced defects can change both the dimensions and mechanical properties of MEMS materials, which will be of concern to designers of MEMS for applications involving radiation exposure, such as those in a reactor environment or in space. We have performed atomistic simulations of the effect that defects and amorphous regions, such as could be produced by radiation damage, have on the elastic constants of silicon. We have then applied the results of the elastic constant shift calculations to a hypothetical MEMS device, and calculated the difference that would be generated by this effect. read less NOT USED (high confidence) M. Qamar, M. Mrovec, Y. Lysogorskiy, A. Bochkarev, and R. Drautz, “Atomic Cluster Expansion for Quantum-Accurate Large-Scale Simulations of Carbon.,” Journal of chemical theory and computation. 2022. link Times cited: 17 Abstract: We present an atomic cluster expansion (ACE) for carbon that… read moreAbstract: We present an atomic cluster expansion (ACE) for carbon that improves over available classical and machine learning potentials. The ACE is parametrized from an exhaustive set of important carbon structures over extended volume and energy ranges, computed using density functional theory (DFT). Rigorous validation reveals that ACE accurately predicts a broad range of properties of both crystalline and amorphous carbon phases while being several orders of magnitude more computationally efficient than available machine learning models. We demonstrate the predictive power of ACE on three distinct applications: brittle crack propagation in diamond, the evolution of amorphous carbon structures at different densities and quench rates, and the nucleation and growth of fullerene clusters under high-pressure and high-temperature conditions. read less NOT USED (high confidence) Z. El-Machachi, M. Wilson, and V. L. Deringer, “Exploring the configurational space of amorphous graphene with machine-learned atomic energies,” Chemical Science. 2022. link Times cited: 4 Abstract: Two-dimensionally extended amorphous carbon (“amorphous grap… read moreAbstract: Two-dimensionally extended amorphous carbon (“amorphous graphene”) is a prototype system for disorder in 2D, showing a rich and complex configurational space that is yet to be fully understood. Here we explore the nature of amorphous graphene with an atomistic machine-learning (ML) model. We create structural models by introducing defects into ordered graphene through Monte-Carlo bond switching, defining acceptance criteria using the machine-learned local, atomic energies associated with a defect, as well as the nearest-neighbor (NN) environments. We find that physically meaningful structural models arise from ML atomic energies in this way, ranging from continuous random networks to paracrystalline structures. Our results show that ML atomic energies can be used to guide Monte-Carlo structural searches in principle, and that their predictions of local stability can be linked to short- and medium-range order in amorphous graphene. We expect that the former point will be relevant more generally to the study of amorphous materials, and that the latter has wider implications for the interpretation of ML potential models. read less NOT USED (high confidence) Y. Kurniawan et al., “Bayesian, frequentist, and information geometric approaches to parametric uncertainty quantification of classical empirical interatomic potentials.,” The Journal of chemical physics. 2021. link Times cited: 6 Abstract: In this paper, we consider the problem of quantifying parame… read moreAbstract: In this paper, we consider the problem of quantifying parametric uncertainty in classical empirical interatomic potentials (IPs) using both Bayesian (Markov Chain Monte Carlo) and frequentist (profile likelihood) methods. We interface these tools with the Open Knowledgebase of Interatomic Models and study three models based on the Lennard-Jones, Morse, and Stillinger-Weber potentials. We confirm that IPs are typically sloppy, i.e., insensitive to coordinated changes in some parameter combinations. Because the inverse problem in such models is ill-conditioned, parameters are unidentifiable. This presents challenges for traditional statistical methods, as we demonstrate and interpret within both Bayesian and frequentist frameworks. We use information geometry to illuminate the underlying cause of this phenomenon and show that IPs have global properties similar to those of sloppy models from fields, such as systems biology, power systems, and critical phenomena. IPs correspond to bounded manifolds with a hierarchy of widths, leading to low effective dimensionality in the model. We show how information geometry can motivate new, natural parameterizations that improve the stability and interpretation of uncertainty quantification analysis and further suggest simplified, less-sloppy models. read less NOT USED (high confidence) M. Wen, Y. Afshar, R. Elliott, and E. Tadmor, “KLIFF: A framework to develop physics-based and machine learning interatomic potentials,” Comput. Phys. Commun. 2021. link Times cited: 12 NOT USED (high confidence) F. Dai, D. Zhao, and L. Zhang, “Atomic Simulations of Packing Structures, Local Stress and Mechanical Properties for One Silicon Lattice with Single Vacancy on Heating,” Materials. 2021. link Times cited: 2 Abstract: The effect of vacancy defects on the structure and mechanica… read moreAbstract: The effect of vacancy defects on the structure and mechanical properties of semiconductor silicon materials is of great significance to the development of novel microelectronic materials and the processes of semiconductor sensors. In this paper, molecular dynamics is used to simulate the atomic packing structure, local stress evolution and mechanical properties of a perfect lattice and silicon crystal with a single vacancy defect on heating. In addition, their influences on the change in Young’s modulus are also analyzed. The atomic simulations show that in the lower temperature range, the existence of vacancy defects reduces the Young’s modulus of the silicon lattice. With the increase in temperature, the local stress distribution of the atoms in the lattice changes due to the migration of the vacancy. At high temperatures, the Young’s modulus of the silicon lattice changes in anisotropic patterns. For the lattice with the vacancy, when the temperature is higher than 1500 K, the number and degree of distortion in the lattice increase significantly, the obvious single vacancy and its adjacent atoms contracting inward structure disappears and the defects in the lattice present complex patterns. By applying uniaxial tensile force, it can be found that the temperature has a significant effect on the elasticity–plasticity behaviors of the Si lattice with the vacancy. read less NOT USED (high confidence) C.-gen Qian, B. Mclean, D. Hedman, and F. Ding, “A comprehensive assessment of empirical potentials for carbon materials,” APL Materials. 2021. link Times cited: 22 Abstract: Carbon materials and their unique properties have been exten… read moreAbstract: Carbon materials and their unique properties have been extensively studied by molecular dynamics, thanks to the wide range of available carbon bond order potentials (CBOPs). Recently, with the increase in popularity of machine learning (ML), potentials such as Gaussian approximation potential (GAP), trained using ML, can accurately predict results for carbon. However, selecting the right potential is crucial as each performs differently for different carbon allotropes, and these differences can lead to inaccurate results. This work compares the widely used CBOPs and the GAP-20 ML potential with density functional theory results, including lattice constants, cohesive energies, defect formation energies, van der Waals interactions, thermal stabilities, and mechanical properties for different carbon allotropes. We find that GAP-20 can more accurately predict the structure, defect properties, and formation energies for a variety of crystalline phase carbon compared to CBOPs. Importantly, GAP-20 can simulate the thermal stability of C60 and the fracture of carbon nanotubes and graphene accurately, where CBOPs struggle. However, similar to CBOPs, GAP-20 is unable to accurately account for van der Waals interactions. Despite this, we find that GAP-20 outperforms all CBOPs assessed here and is at present the most suitable potential for studying thermal and mechanical properties for pristine and defective carbon. read less NOT USED (high confidence) J. Weinreich, M. Paleico, and J. Behler, “Properties of α-Brass Nanoparticles II: Structure and Composition,” The Journal of Physical Chemistry C. 2021. link Times cited: 4 Abstract: Nanoparticles have become increasingly interesting for a wid… read moreAbstract: Nanoparticles have become increasingly interesting for a wide range of applications, because in principle it is possible to tailor their properties by controlling size, shape and composition. One of these applications is heterogeneous catalysis, and a fundamental understanding of the structural details of the nanoparticles is essential for any knowledge-based improvement of reactivity and selectivity. In this work we investigate the atomic structure of brass nanoparticles containing up to 5000 atoms as a typical example for a binary alloy consisting of Cu and Zn. As systems of this size are too large for electronic structure calculations, in our simulations we use a recently parametrized machine learning potential providing close to density functional theory accuracy. This potential is employed for a structural characterization as a function of chemical composition by various types of simulations like Monte Carlo in the Semi-Grand Canonical Ensemble and simulated annealing molecular dynamics. Our analysis reveals that the distribution of both elements in the nanoparticles is inhomogeneous, and zinc accumulates in the outermost layer, while the first subsurface layer shows an enrichment of copper. Only for high zinc concentrations alloying can be found in the interior of the nanoparticles, and regular patterns corresponding to crystalline bulk phases of $\alpha$-brass can then be observed. The surfaces of the investigated clusters exhibit well-ordered single-crystal facets, which can give rise to grain boundaries inside the clusters. The melting temperature of the nanoparticles is found to decrease with increasing zinc-atom fraction, a trend which is well-known also for the bulk phase diagram of brass. read less NOT USED (high confidence) M. Haro et al., “Nano-vault architecture mitigates stress in silicon-based anodes for lithium-ion batteries,” Communications Materials. 2021. link Times cited: 8 NOT USED (high confidence) X. Tian and M. Chen, “Descriptor selection for predicting interfacial thermal resistance by machine learning methods,” Scientific Reports. 2021. link Times cited: 5 NOT USED (high confidence) A. B. Svechnikov, “Slow Ion Channeling in Monocrystalline Silicon,” Physics of the Solid State. 2020. link Times cited: 1 NOT USED (high confidence) I. Nevliudov, M. Omarov, and O. Chala, “MATHEMATICAL MODEL OF THE DEVELOPMENT OF MANUFACTURING DEFECTS IN THE SURFACE LAYER OF SUBSTRATES OF MOEMS’ FUNCTIONAL COMPONENTS,” Eskişehir Technical University Journal of Science and Technology A - Applied Sciences and Engineering. 2020. link Times cited: 1 Abstract: A mathematical model of the development of manufacturing def… read moreAbstract: A mathematical model of the development of manufacturing defects, with the prediction of the random component of the model in the substrates of functional components of MOEMS, which are made of semiconductors, in particular, silicon, are developed in the article. The main manufacturing defects that arise in the surface layer of the substrates of the MOEMS functional components taking into account the technological processes of their production and dynamic processes were used when developing the model. The developed mathematical model takes into account the occurrence of a random component of the model with its predictive ability. The possibility of such control is the basis for the development of the scientific direction of technology and equipment for the production of semiconductors, materials and electronic devices - defect engineering, based on the management and forecasting of defect formation processes. read less NOT USED (high confidence) H. Moosavian and H. Shodja, “Mindlin–Eringen anisotropic micromorphic elasticity and lattice dynamics representation,” Philosophical Magazine. 2020. link Times cited: 9 Abstract: ABSTRACT To account for certain essential features of materi… read moreAbstract: ABSTRACT To account for certain essential features of material such as dispersive behaviour and optical branches in dispersion curves, a fundamental departure from classical elasticity to polar theories is required. Among the polar theories, micromorphic elasticity of appropriate grades and anisotropy is capable of capturing these physical phenomena completely. In the mathematical framework of micromorphic elasticity, in addition to the traditional elastic constants, some additional constants are introduced in the pertinent governing equations of motion. A precise evaluation of the numerical values of the aforementioned elastic constants in the realm of the experimentations poses serious difficulties. Thus this paper aims to provide a remedy as how to determine the micromorphic elastic constants theoretically in terms of the atomic force constants and lattice parameters of the crystalline solid with general anisotropy. In this treatment capture of the discrete nature of matter becomes an essential factor. To this end, the discrete lattice dynamics equations of a crystal are related to the pertinent anisotropic micromorphic equations of motion. This approach allows incorporating the symmetry groups of the crystals within lattice dynamics equations conveniently. For the illustration of the current theoretical developments, the micromorphic elastic constants of diamond and silicon crystals are computed in conjunction with ab initio density functional perturbation theory (DFPT). Moreover, the longitudinal and transverse optical and acoustic branches pertinent to [100] and [110] directions are presented. The accuracy of the results is verified by comparing the dispersion curves derived from the micromorphic theory, those of available experiments, and those directly obtained from DFPT calculations. read less NOT USED (high confidence) R. Li, E. Lee, and T. Luo, “A unified deep neural network potential capable of predicting thermal conductivity of silicon in different phases,” arXiv: Materials Science. 2019. link Times cited: 47 NOT USED (high confidence) M. Amsler et al., “FLAME: A library of atomistic modeling environments,” Comput. Phys. Commun. 2019. link Times cited: 19 NOT USED (high confidence) V. Kuryliuk, O. Nepochatyi, P. Chantrenne, D. Lacroix, and M. Isaiev, “Thermal conductivity of strained silicon: Molecular dynamics insight and kinetic theory approach,” Journal of Applied Physics. 2019. link Times cited: 15 Abstract: In this work, we investigated tensile and compression forces… read moreAbstract: In this work, we investigated tensile and compression forces effect on the thermal conductivity of silicon. We used equilibrium molecular dynamics approach for the evaluation of thermal conductivity considering different interatomic potentials. More specifically, we tested Stillinger-Weber, Tersoff, Environment-Dependent Interatomic Potential and Modified Embedded Atom Method potentials for the description of silicon atom motion under different strain and temperature conditions. Additionally, we extracted phonon density of states and dispersion curves from molecular dynamics simulations. These data were used for direct calculations of thermal conductivity considering the kinetic theory approach. Comparison of molecular dynamics and kinetic theory simulations results as a function of strain and temperature allowed us to investigate the different factors affecting the thermal conductivity of strained silicon. read less NOT USED (high confidence) Y. Lysogorskiy, T. Hammerschmidt, J. Janssen, J. Neugebauer, and R. Drautz, “Transferability of interatomic potentials for molybdenum and silicon,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 14 Abstract: Interatomic potentials are widely used in computational mate… read moreAbstract: Interatomic potentials are widely used in computational materials science, in particular for simulations that are too computationally expensive for density functional theory (DFT). Most interatomic potentials have a limited application range and often there is very limited information available regarding their performance for specific simulations. We carried out high-throughput calculations for molybdenum and silicon with DFT and a number of interatomic potentials. We compare the DFT reference calculations and experimental data to the predictions of the interatomic potentials. We focus on a large number of basic materials properties, including the cohesive energy, atomic volume, elastic coefficients, vibrational properties, thermodynamic properties, surface energies and vacancy formation energies, which enables a detailed discussion of the performance of the different potentials. We further analyze correlations between properties as obtained from DFT calculations and how interatomic potentials reproduce these correlations, and suggest a general measure for quantifying the accuracy and transferability of an interatomic potential. From our analysis we do not establish a clearcut ranking of the potentials as each potential has its strengths and weaknesses. It is therefore essential to assess the properties of a potential carefully before application of the potential in a specific simulation. The data presented here will be useful for selecting a potential for simulations of Mo or Si. read less NOT USED (high confidence) D. Prasad and N. Mitra, “An atomistic study of phase transition in cubic diamond Si single crystal subjected to static compression,” Computational Materials Science. 2019. link Times cited: 7 NOT USED (high confidence) L. Martín, I. Santos, P. López, L. Marqués, M. Aboy, and L. Pelaz, “Modeling SiGe Through Classical Molecular Dynamics Simulations: Chasing an Appropriate Empirical Potential,” 2018 Spanish Conference on Electron Devices (CDE). 2018. link Times cited: 2 Abstract: We used classical molecular dynamics simulations to reproduc… read moreAbstract: We used classical molecular dynamics simulations to reproduce basic properties of Si, Ge and SiGe using different empirical potentials available in the literature. The empirical potential that offered the better compromise with experimental data was used to study the surface stability of these materials. We considered the (100), $(100)2\times 1$ and (111) surfaces, and we found the processing temperature range to avoid the structural degradation of studied surfaces. read less NOT USED (high confidence) B. Lorenzi et al., “Phonon Scattering in Silicon by Multiple Morphological Defects: A Multiscale Analysis,” Journal of Electronic Materials. 2018. link Times cited: 11 NOT USED (high confidence) A. Lopez-Cazalilla et al., “Pattern formation on ion-irradiated Si surface at energies where sputtering is negligible,” Journal of Applied Physics. 2018. link Times cited: 25 Abstract: The effect of low energy irradiation, where the sputtering i… read moreAbstract: The effect of low energy irradiation, where the sputtering is imperceptible, has not been deeply studied in the pattern formation. In this work, we want to address this question by analyzing the nanoscale topography formation on a Si surface, which is irradiated at room temperature by Ar+ ions near the displacement threshold energy, for incidence angles ranging from 0° to 85°. The transition from the smooth to ripple patterned surface, i.e., the stability/instability bifurcation angle is observed at 55°, whereas the ripples with their wave-vector is parallel to the ion beam projection in the angular window of 60°–70°, and with 90° rotation with respect to the ion beam projection at the grazing angles of incidence. A similar irradiation setup has been simulated by means of molecular dynamics, which made it possible, first, to quantify the effect of the irradiation in terms of erosion and redistribution using sequential irradiation and, second, to evaluate the ripple wavelength using the crater function formalism. The ripple formation results can be solely attributed to the mass redistribution based mechanism, as erosion due to ion sputtering near or above the threshold energy is practically negligible. read less NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 38 NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 0 NOT USED (high confidence) M. El-Genk, K. Talaat, and B. Cowen, “Thermal conductivity of silicon using reverse non-equilibrium molecular dynamics,” Journal of Applied Physics. 2018. link Times cited: 13 Abstract: Simulations are performed using the reverse non-equilibrium … read moreAbstract: Simulations are performed using the reverse non-equilibrium molecular dynamics (rNEMD) method and the Stillinger-Weber (SW) potential to determine the input parameters for achieving ±1% convergence of the calculated thermal conductivity of silicon. These parameters are then used to investigate the effects of the interatomic potentials of SW, Tersoff II, Environment Dependent Interatomic Potential (EDIP), Second Nearest Neighbor, Modified Embedded-Atom Method (MEAM), and Highly Optimized Empirical Potential MEAM on determining the bulk thermal conductivity as a function of temperature (400–1000 K). At temperatures > 400 K, data collection and swap periods of 15 ns and 150 fs, system size ≥6 × 6 UC2 and system lengths ≥192 UC are adequate for ±1% convergence with all potentials, regardless of the time step size (0.1–0.5 fs). This is also true at 400 K, except for the SW potential, which requires a data collection period ≥30 ns. The calculated bulk thermal conductivities using the rNEMD method and the EDIP potential are close to, but lower than experimental values. The 10% difference at 400 K increases gradually to 20% at 1000 K.Simulations are performed using the reverse non-equilibrium molecular dynamics (rNEMD) method and the Stillinger-Weber (SW) potential to determine the input parameters for achieving ±1% convergence of the calculated thermal conductivity of silicon. These parameters are then used to investigate the effects of the interatomic potentials of SW, Tersoff II, Environment Dependent Interatomic Potential (EDIP), Second Nearest Neighbor, Modified Embedded-Atom Method (MEAM), and Highly Optimized Empirical Potential MEAM on determining the bulk thermal conductivity as a function of temperature (400–1000 K). At temperatures > 400 K, data collection and swap periods of 15 ns and 150 fs, system size ≥6 × 6 UC2 and system lengths ≥192 UC are adequate for ±1% convergence with all potentials, regardless of the time step size (0.1–0.5 fs). This is also true at 400 K, except for the SW potential, which requires a data collection period ≥30 ns. The calculated bulk thermal conductivities using the rNEMD method and the EDIP... read less NOT USED (high confidence) J. Behler, “Hochdimensionale neuronale Netze für Potentialhyperflächen großer molekularer und kondensierter Systeme,” Angewandte Chemie. 2017. link Times cited: 9 Abstract: Moderne Simulationstechniken haben heute ein Niveau erreicht… read moreAbstract: Moderne Simulationstechniken haben heute ein Niveau erreicht, das es ermoglicht, ein breites Spektrum von Problemen in der Chemie und in den Materialwissenschaften zu untersuchen. Trotz der Entwicklung immer leistungsfahigerer Hardware ist die Anwendung von Elektronenstrukturmethoden, die Vorhersagen ohne Ruckgriff auf experimentelle Daten ermoglichen, jedoch noch immer auf kleine Systeme begrenzt, und in absehbarer Zukunft ist keine Besserung dieser Situation zu erwarten. Um auch komplexe Systeme auf atomarer Ebene verstehen zu konnen, ist die Entwicklung von effizienteren und gleichzeitig zuverlassigen atomistischen Potentialen in den letzten Jahren immer mehr in den Fokus geruckt. Ein vielversprechender neuer Ansatz ist die Nutzung von maschinellem Lernen (ML) zur Beschreibung der atomaren Wechselwirkungen. Nach einem Trainingsprozess mit Elektronstrukturdaten konnen solche ML-Potentiale Computersimulationen um mehrere Grosenordnungen beschleunigen, wahrend die quantenmechanische Genauigkeit erhalten bleibt. Anhand einer wichtigen Klasse von ML-Potentialen, die auf kunstlichen neuronalen Netzen basiert, werden in diesem Aufsatz die Grundideen, die Anwendbarkeit und die offenen Fragen dieses Ansatzes diskutiert. read less NOT USED (high confidence) J. Behler, “First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems.,” Angewandte Chemie. 2017. link Times cited: 409 Abstract: Modern simulation techniques have reached a level of maturit… read moreAbstract: Modern simulation techniques have reached a level of maturity which allows a wide range of problems in chemistry and materials science to be addressed. Unfortunately, the application of first principles methods with predictive power is still limited to rather small systems, and despite the rapid evolution of computer hardware no fundamental change in this situation can be expected. Consequently, the development of more efficient but equally reliable atomistic potentials to reach an atomic level understanding of complex systems has received considerable attention in recent years. A promising new development has been the introduction of machine learning (ML) methods to describe the atomic interactions. Once trained with electronic structure data, ML potentials can accelerate computer simulations by several orders of magnitude, while preserving quantum mechanical accuracy. This Review considers the methodology of an important class of ML potentials that employs artificial neural networks. read less NOT USED (high confidence) F. Lehnert and S. G. Mayr, “Nanoporous amorphous Ge-Si alloys - unraveling the physics behind ion beam induced morphogenesis.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 2 Abstract: Despite a high technical relevance and 35 years of observati… read moreAbstract: Despite a high technical relevance and 35 years of observation, self-organized morphogenesis of nanoporous sponge-like amorphous structures during exposure of selected covalent materials to energetic ions is still insufficiently understood. Due to the presence and absence of these effects in amorphous Ge and Si, respectively, the Ge-Si alloy system constitutes an ideal testbed to track down the underlying physics at the atomic scale. This is realized within the present study by a combination of tailored experiments and extensive molecular dynamics computer modeling. The swelling capabilities of a variety of interaction potentials for the Ge-Si system and its elemental constituents are scrutinized with respect to the experimental observations and related to relevant physical properties of the model systems. This allows to identify defect kinetics in combination with a moderate radiation induced fluidity as key ingredients for nanopore morphogenesis. Cast in a simple quantitative model, it enables to account for both experimental as well as computational results, thus paving the way for a design by understanding approach in synthesis. read less NOT USED (high confidence) E. Dontsova and R. Ballarini, “Atomistic modeling of the fracture toughness of silicon and silicon-silicon interfaces,” International Journal of Fracture. 2017. link Times cited: 6 NOT USED (high confidence) J.-W. Jiang and Y.-P. Zhou, “Parameterization of Stillinger-Weber Potential for Two- Dimensional Atomic Crystals,” arXiv: Materials Science. 2017. link Times cited: 51 Abstract: We parametrize the Stillinger-Weber potential for 156 two-di… read moreAbstract: We parametrize the Stillinger-Weber potential for 156 two-dimensional atomic crystals. Parameters for the Stillinger-Weber potential are obtained from the valence force field model following the analytic approach (Nanotechnology 26, 315706 (2015)), in which the valence force constants are determined by the phonon spectrum. The Stillinger-Weber potential is an efficient nonlinear interaction, and is applicable for numerical simulations of nonlinear physical or mechanical processes. The supplemental resources for all simulations in the present work are available online in Ref. 1, including a fortran code to generate crystals' structures, files for molecular dynamics simulations using LAMMPS, files for phonon calculations with the Stillinger-Weber potential using GULP, and files for phonon calculations with the valence force field model using GULP. read less NOT USED (high confidence) G. P. P. Pun and Y. Mishin, “Optimized interatomic potential for silicon and its application to thermal stability of silicene,” Physical Review B. 2017. link Times cited: 35 Abstract: An optimized interatomic potential has been constructed for … read moreAbstract: An optimized interatomic potential has been constructed for silicon using a modified Tersoff model. The potential reproduces a wide range of properties of Si and improves over existing potentials with respect to point defect structures and energies, surface energies and reconstructions, thermal expansion, melting temperature, and other properties. The proposed potential is compared with three other potentials from the literature. The potentials demonstrate reasonable agreement with first-principles binding energies of small Si clusters as well as single-layer and bilayer silicenes. The four potentials are used to evaluate the thermal stability of free-standing silicenes in the form of nanoribbons, nanoflakes, and nanotubes. While single-layer silicene is found to be mechanically stable at zero Kelvin, it is predicted to become unstable and collapse at room temperature. By contrast, the bilayer silicene demonstrates a larger bending rigidity and remains stable at and even above room temperature. The results suggest that bilayer silicene might exist in a free-standing form at ambient conditions. read less NOT USED (high confidence) C. Tomas, I. Suarez-Martinez, and N. Marks, “Graphitization of amorphous carbons: A comparative study of interatomic potentials,” Carbon. 2016. link Times cited: 160 NOT USED (high confidence) T. Sanyal and S. Shella, “Coarse-grained models using local-density potentials optimized with the relative entropy: Application to implicit solvation.,” The Journal of chemical physics. 2016. link Times cited: 75 Abstract: Bottom-up multiscale techniques are frequently used to devel… read moreAbstract: Bottom-up multiscale techniques are frequently used to develop coarse-grained (CG) models for simulations at extended length and time scales but are often limited by a compromise between computational efficiency and accuracy. The conventional approach to CG nonbonded interactions uses pair potentials which, while computationally efficient, can neglect the inherently multibody contributions of the local environment of a site to its energy, due to degrees of freedom that were coarse-grained out. This effect often causes the CG potential to depend strongly on the overall system density, composition, or other properties, which limits its transferability to states other than the one at which it was parameterized. Here, we propose to incorporate multibody effects into CG potentials through additional nonbonded terms, beyond pair interactions, that depend in a mean-field manner on local densities of different atomic species. This approach is analogous to embedded atom and bond-order models that seek to capture multibody electronic effects in metallic systems. We show that the relative entropy coarse-graining framework offers a systematic route to parameterizing such local density potentials. We then characterize this approach in the development of implicit solvation strategies for interactions between model hydrophobes in an aqueous environment. read less NOT USED (high confidence) D. Kaiser, S. Ghosh, S. Han, and T. Sinno, “Modeling and simulation of compositional engineering in SiGe films using patterned stress fields.” 2016. link Times cited: 2 Abstract: Semiconductor alloys such as silicon–germanium (SiGe) offer … read moreAbstract: Semiconductor alloys such as silicon–germanium (SiGe) offer attractive environments for engineering quantum-confined structures that are the basis for a host of current and future optoelectronic devices. Although vertical stacking of such structures is routinely achieved via heteroepitaxy, lateral manipulation has proven much more challenging. We have recently demonstrated that a patterned elastic stress field applied, with an array of nanoscale indenters, to an initially compositionally uniform SiGe substrate will drive atomic interdiffusion leading to compositional patterns in the near-surface region of the substrate. While this approach may offer a potentially efficient and robust pathway to producing laterally ordered arrays of quantum-confined structures, optimizing it with respect to the various process parameters, such as indenter array geometry, annealing history, and SiGe substrate thickness and composition, is highly challenging. Here, a mesoscopic model based on coarse-grained lattice kinetic Monte Carlo simulation is presented that describes quantitatively the atomic interdiffusion processes in SiGe alloy films subjected to applied stress. We first show that the model provides predictions that are quantitatively consistent with experimental measurements. Then, the model is used to investigate the impact of several process parameters such as indenter shape and pitch. We find that certain indenter configurations produce compositional patterns that are favorable for engineering lateral arrays of quantum-confined structures. read less NOT USED (high confidence) T. Ito, T. Akiyama, and K. Nakamura, “Systematic approach to developing empirical interatomic potentials for III–N semiconductors,” Japanese Journal of Applied Physics. 2016. link Times cited: 5 Abstract: A systematic approach to the derivation of empirical interat… read moreAbstract: A systematic approach to the derivation of empirical interatomic potentials is developed for III–N semiconductors with the aid of ab initio calculations. The parameter values of empirical potential based on bond order potential are determined by reproducing the cohesive energy differences among 3-fold coordinated hexagonal, 4-fold coordinated zinc blende, wurtzite, and 6-fold coordinated rocksalt structures in BN, AlN, GaN, and InN. The bond order p is successfully introduced as a function of the coordination number Z in the form of p = a exp(−bZn) if Z ≤ 4 and p = (4/Z)α if Z ≥ 4 in empirical interatomic potential. Moreover, the energy difference between wurtzite and zinc blende structures can be successfully evaluated by considering interaction beyond the second-nearest neighbors as a function of ionicity. This approach is feasible for developing empirical interatomic potentials applicable to a system consisting of poorly coordinated atoms at surfaces and interfaces including nanostructures. read less NOT USED (high confidence) S. Bringuier, V. Manga, K. Runge, P. Deymier, and K. Muralidharan, “An atomic scale characterization of coupled grain boundary motion in silicon bicrystals,” Philosophical Magazine. 2015. link Times cited: 7 Abstract: The mechanical response of symmetric tilt grain boundaries (… read moreAbstract: The mechanical response of symmetric tilt grain boundaries (GBs) in silicon bicrystals under shear loading are characterized using molecular dynamics simulations. It is seen that under shear, high-angle GBs namely Σ5 and Σ13 having a rotation axis [0 0 1] demonstrate coupled GB motion, such that the displacement of grains parallel to the GB interface is accompanied by normal GB motion. An atomic-scale characterization revealed that concerted rotations of silicon tetrahedra within the GB are the primary mechanisms leading to the coupled GB motion. Interestingly, so far, this phenomenon has only been examined in detail for metallic systems. A distinguishing feature of the coupled GB motion observed for the silicon symmetric tilt bicrystals as compared to metallic bicrystals is the fact that in the absence of shear, spontaneous coupled motion is not observed at high temperatures. read less NOT USED (high confidence) J. Harrison, M. Fallet, K. E. Ryan, B. L. Mooney, M. T. Knippenberg, and J. Schall, “Recent developments and simulations utilizing bond-order potentials,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 12 Abstract: Bond-order potentials (BOPs) have been used successfully in … read moreAbstract: Bond-order potentials (BOPs) have been used successfully in simulations of a wide range of processes. A brief overview of bond-order potentials is provided which focuses on the reactive empirical bond-order (REBO) potential for hydrocarbons (Brenner et al 2002 J. Phys.: Condens. Matter 14 783) and the large number of useful potentials it has spawned. Two specific extensions of the REBO potential that make use of its formalism are discussed. First, the 2B-SiCH potential (Schall and Harrison 2013 J. Phys. Chem. C 117 1323) makes the appropriate changes to the hydrocarbon REBO potential so that three atom types, Si, C, and H, can be modeled. Second, we recently added the electronegative element O, along with the associated charge terms, to the adaptive intermolecular REBO (AIREBO) potential (Stuart et al 2000 J. Chem. Phys. 112 6472). The resulting qAIREBO potential (Knippenberg et al 2012 J. Chem. Phys. 136 164701) makes use of the bond-order potential/split-charge (BOP/SQE) equilibration method (Mikulski et al 2009 J. Chem. Phys. 131 241105) and the Lagrangian approach to charge dynamics (Rick et al 1994 J. Chem. Phys. 101 6141). The integration of these two techniques allows for atomic charges to evolve with time during MD simulations: as a result, chemical reactions can be modeled in C-, O-, and H-containing systems. The usefulness of the 2B-SiCH potential for tribological investigations is demonstrated in molecular dynamics (MD) simulations of axisymmetric tips composed of Si and SiC placed in sliding contact with diamond(1 1 1) surfaces with varying amounts of hydrogen termination. The qAIREBO potential is used to investigate confinement of sub-monolayer coverages of water between nanostructured surfaces. read less NOT USED (high confidence) L. Sang, V. V. Hoang, and D. T. N. Tranh, “Melting of crystalline Si nanoparticle investigated by simulation,” The European Physical Journal D. 2015. link Times cited: 6 NOT USED (high confidence) P. Dagenais, L. J. Lewis, and S. Roorda, “Dominant structural defects in amorphous silicon,” Journal of Physics: Condensed Matter. 2015. link Times cited: 1 Abstract: The nature of disorder in amorphous silicon (a-Si) is explor… read moreAbstract: The nature of disorder in amorphous silicon (a-Si) is explored by investigating the spatial arrangement and energies of coordination defects in a numerical model. Spatial correlations between structural defects are examined on the basis of a parameter that quantifies the probability for two sites to share a bond. Pentacoordinated atoms are found to be the dominant coordination defects. They show a tendency to cluster, and about 17% of them are linked through three-membered rings. As for tricoordinated sites, they are less numerous, and tend to be distant by at least two bond lengths. Typical local geometries associated to under and overcoordinated atoms are extracted from the model and described using partial bond angle distributions. An estimate of the formation energies of structural defects is provided. Using molecular-dynamics calculations, we simulate the implantation of high-energy atoms in the initial structure in order to study the effect of relaxation on the coordination defects and their environments. read less NOT USED (high confidence) C. D. Latham, A. J. McKenna, T. Trevethan, M. Heggie, M. Rayson, and P. Briddon, “On the validity of empirical potentials for simulating radiation damage in graphite: a benchmark,” Journal of Physics: Condensed Matter. 2015. link Times cited: 23 Abstract: In this work, the ability of methods based on empirical pote… read moreAbstract: In this work, the ability of methods based on empirical potentials to simulate the effects of radiation damage in graphite is examined by comparing results for point defects, found using ab initio calculations based on density functional theory (DFT), with those given by two state of the art potentials: the Environment-Dependent Interatomic Potential (EDIP) and the Adaptive Intermolecular Reactive Empirical Bond Order potential (AIREBO). Formation energies for the interstitial, the vacancy and the Stone–Wales (5775) defect are all reasonably close to DFT values. Both EDIP and AIREBO can thus be suitable for the prompt defects in a cascade, for example. Both potentials suffer from arefacts. One is the pinch defect, where two α-atoms adopt a fourfold-coordinated sp3 configuration, that forms a cross-link between neighbouring graphene sheets. Another, for AIREBO only, is that its ground state vacancy structure is close to the transition state found by DFT for migration. The EDIP fails to reproduce the ground state self-interstitial structure given by DFT, but has nearly the same formation energy. Also, for both potentials, the energy barriers that control diffusion and the evolution of a damage cascade, are not well reproduced. In particular the EDIP gives a barrier to removal of the Stone–Wales defect as 0.9 eV against DFT's 4.5 eV. The suite of defect structures used is provided as supplementary information as a benchmark set for future potentials. read less NOT USED (high confidence) V. Lacatena, M. Haras, J. Robillard, S. Monfray, T. Skotnicki, and E. Dubois, “Toward quantitative modeling of silicon phononic thermocrystals,” Applied Physics Letters. 2015. link Times cited: 13 Abstract: The wealth of technological patterning technologies of deca-… read moreAbstract: The wealth of technological patterning technologies of deca-nanometer resolution brings opportunities to artificially modulate thermal transport properties. A promising example is given by the recent concepts of "thermocrystals" or "nanophononic crystals" that introduce regular nano-scale inclusions using a pitch scale in between the thermal phonons mean free path and the electron mean free path. In such structures, the lattice thermal conductivity is reduced down to two orders of magnitude with respect to its bulk value. Beyond the promise held by these materials to overcome the well-known “electron crystal-phonon glass” dilemma faced in thermoelectrics, the quantitative prediction of their thermal conductivity poses a challenge. This work paves the way toward understanding and designing silicon nanophononic membranes by means of molecular dynamics simulation. Several systems are studied in order to distinguish the shape contribution from bulk, ultra-thin membranes (8 to 15 nm), 2D phononic crystals, and finally 2D phononic membranes. After having discussed the equilibrium properties of these structures from 300 K to 400 K, the Green-Kubo methodology is used to quantify the thermal conductivity. The results account for several experimental trends and models. It is confirmed that the thin-film geometry as well as the phononic structure act towards a reduction of the thermal conductivity. The further decrease in the phononic engineered membrane clearly demonstrates that both phenomena are cumulative. Finally, limitations of the model and further perspectives are discussed. read less NOT USED (high confidence) K. Garcez and A. Antonelli, “Polyamorphism in tetrahedral substances: Similarities between silicon and ice.,” The Journal of chemical physics. 2015. link Times cited: 1 Abstract: Tetrahedral substances, such as silicon, water, germanium, a… read moreAbstract: Tetrahedral substances, such as silicon, water, germanium, and silica, share various unusual phase behaviors. Among them, the so-called polyamorphism, i.e., the existence of more than one amorphous form, has been intensively investigated in the last three decades. In this work, we study the metastable relations between amorphous states of silicon in a wide range of pressures, using Monte Carlo simulations. Our results indicate that the two amorphous forms of silicon at high pressures, the high density amorphous (HDA) and the very high density amorphous (VHDA), can be decompressed from high pressure (∼20 GPa) down to the tensile regime, where both convert into the same low density amorphous. Such behavior is also observed in ice. While at high pressure (∼20 GPa), HDA is less stable than VHDA, at the pressure of 10 GPa both forms exhibit similar stability. On the other hand, at much lower pressure (∼5 GPa), HDA and VHDA are no longer the most stable forms, and, upon isobaric annealing, an even less dense form of amorphous silicon emerges, the expanded high density amorphous, again in close similarity to what occurs in ice. read less NOT USED (high confidence) W.-L. Lv and A. Henry, “Direct calculation of modal contributions to thermal conductivity via Green–Kubo modal analysis,” New Journal of Physics. 2015. link Times cited: 115 Abstract: We derived a new method for direct calculation of the modal … read moreAbstract: We derived a new method for direct calculation of the modal contributions to thermal conductivity, which is termed Green–Kubo modal analysis (GKMA). The GKMA method combines the lattice dynamics formalism with the Green–Kubo formula for thermal conductivity, such that the thermal conductivity becomes a direct summation of modal contributions, where one need not define the phonon velocity. As a result, the GKMA method can be applied to any material/group of atoms, where the atoms vibrate around stable equilibrium positions, which includes non-stoichiometric compounds, random alloys, amorphous materials and even rigid molecules. By using molecular dynamics simulations to obtain the time history of each mode’s contribution to the heat current, one naturally includes anharmonicity to full order and can obtain insight into the interactions between different modes through the cross-correlations. As an example, we applied the GMKA method to crystalline and amorphous silicon. The modal contributions at each frequency result from the analysis and thereby allow one to apply a quantum correction to the mode heat capacity to determine the temperature dependence of thermal conductivity. The predicted temperature dependent thermal conductivity for amorphous silicon shows the best agreement with experiments to date. The GKMA method provides new insight into the nature of phonon transport, as it casts the problem in terms of mode–mode correlation instead of scattering, and provides a general unified formalism that can be used to understand phonon–phonon interactions in essentially any class of materials or structures where the atoms vibrate around stable equilibrium sites. read less NOT USED (high confidence) G. P. Srivastava, “Tuning phonon properties in thermoelectric materials,” Reports on Progress in Physics. 2015. link Times cited: 21 Abstract: This review article presents a discussion of theoretical pro… read moreAbstract: This review article presents a discussion of theoretical progress made over the past several decades towards our understanding of thermoelectric properties of materials. Particular emphasis is placed upon describing recent progress in ‘tuning’ phonon properties of nanocomposite materials for gaining enhancement of the thermoelectric figure of merit. read less NOT USED (high confidence) M. Park, I. Lee, and Y.-S. Kim, “Lattice thermal conductivity of crystalline and amorphous silicon with and without isotopic effects from the ballistic to diffusive thermal transport regime,” Journal of Applied Physics. 2014. link Times cited: 23 Abstract: Thermal conductivity of a material is an important physical … read moreAbstract: Thermal conductivity of a material is an important physical parameter in electronic and thermal devices, and as the device size shrinks down, its length-dependence becomes unable to be neglected. Even in micrometer scale devices, materials having a long mean free path of phonons, such as crystalline silicon (Si), exhibit a strong length dependence of the thermal conductivities that spans from the ballistic to diffusive thermal transport regime. In this work, through non-equilibrium molecular-dynamics (NEMD) simulations up to 17 μm in length, the lattice thermal conductivities are explicitly calculated for crystalline Si and up to 2 μm for amorphous Si. The Boltzmann transport equation (BTE) is solved within a frequency-dependent relaxation time approximation, and the calculated lattice thermal conductivities in the BTE are found to be in good agreement with the values obtained in the NEMD. The isotopic effects on the length-dependent lattice thermal conductivities are also investigated both in the crystalline and amorphous Si. read less NOT USED (high confidence) S. Chill et al., “EON: software for long time simulations of atomic scale systems,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 56 Abstract: The EON software is designed for simulations of the state-to… read moreAbstract: The EON software is designed for simulations of the state-to-state evolution of atomic scale systems over timescales greatly exceeding that of direct classical dynamics. States are defined as collections of atomic configurations from which a minimization of the potential energy gives the same inherent structure. The time evolution is assumed to be governed by rare events, where transitions between states are uncorrelated and infrequent compared with the timescale of atomic vibrations. Several methods for calculating the state-to-state evolution have been implemented in EON, including parallel replica dynamics, hyperdynamics and adaptive kinetic Monte Carlo. Global optimization methods, including simulated annealing, basin hopping and minima hopping are also implemented. The software has a client/server architecture where the computationally intensive evaluations of the interatomic interactions are calculated on the client-side and the state-to-state evolution is managed by the server. The client supports optimization for different computer architectures to maximize computational efficiency. The server is written in Python so that developers have access to the high-level functionality without delving into the computationally intensive components. Communication between the server and clients is abstracted so that calculations can be deployed on a single machine, clusters using a queuing system, large parallel computers using a message passing interface, or within a distributed computing environment. A generic interface to the evaluation of the interatomic interactions is defined so that empirical potentials, such as in LAMMPS, and density functional theory as implemented in VASP and GPAW can be used interchangeably. Examples are given to demonstrate the range of systems that can be modeled, including surface diffusion and island ripening of adsorbed atoms on metal surfaces, molecular diffusion on the surface of ice and global structural optimization of nanoparticles. read less NOT USED (high confidence) K. Garcez and A. Antonelli, “Pressure-induced Transformations In Amorphous Silicon: A Computational Study,” Journal of Applied Physics. 2014. link Times cited: 2 Abstract: We study the transformations between amorphous phases of Si … read moreAbstract: We study the transformations between amorphous phases of Si through molecular simulations using the environment dependent interatomic potential (EDIP) for Si. Our results show that upon pressure, the material undergoes a transformation from the low density amorphous (LDA) Si to the high density amorphous (HDA) Si. This transformation can be reversed by decompressing the material. This process, however, exhibits clear hysteresis, suggesting that the transformation LDA ↔ HDA is first-order like. The HDA phase is predominantly five-fold coordinated, whereas the LDA phase is the normal tetrahedrally bonded amorphous Si. The HDA phase at 400 K and 20 GPa was submitted to an isobaric annealing up to 800 K, resulting in a denser amorphous phase, which is structurally distinct from the HDA phase. Our results also show that the atomic volume and structure of this new amorphous phase are identical to those of the glass obtained by an isobaric quenching of the liquid in equilibrium at 2000 K and 20 GPa down to 400 K... read less NOT USED (high confidence) P. Käshammer and T. Sinno, “Interactions of twin boundaries with intrinsic point defects and carbon in silicon,” Journal of Applied Physics. 2013. link Times cited: 22 Abstract: Although multicrystalline silicon (mc-Si) is currently the m… read moreAbstract: Although multicrystalline silicon (mc-Si) is currently the most widely used material for fabricating photovoltaic cells, its electrical properties remain limited by several types of defects, which interact in complex ways that are not yet fully understood. A particularly important phenomenon is the interaction between grain boundaries and intrinsic point defects or impurity atoms, such as carbon, oxygen, nitrogen, and various types of metals. Here, we use empirical molecular dynamics to study the interactions of Σ3{111}, Σ9{221}, and Σ27{552} twin boundaries, which account for over 50% of all grain boundaries in mc-Si, with self-interstitials, vacancies, and substitutional carbon atoms. It is shown that twin boundary-point defect interaction energies increase with twinning order and that they are predominantly attractive. We also find that twin boundary interactions with substitutional carbon are highly spatially heterogeneous, exhibiting alternating repulsive-attractive regions that correlate strongly wi... read less NOT USED (high confidence) J. R. Srour and J. W. Palko, “Displacement Damage Effects in Irradiated Semiconductor Devices,” IEEE Transactions on Nuclear Science. 2013. link Times cited: 154 Abstract: A review of radiation-induced displacement damage effects in… read moreAbstract: A review of radiation-induced displacement damage effects in semiconductor devices is presented, with emphasis placed on silicon technology. The history of displacement damage studies is summarized, and damage production mechanisms are discussed. Properties of defect clusters and isolated defects are addressed. Displacement damage effects in materials and devices are considered, including effects produced in silicon particle detectors, visible imaging arrays, and solar cells. Additional topics examined include NIEL scaling, carrier concentration changes, random telegraph signals, radiation hardness assurance, and simulation methods for displacement damage. Areas needing further study are noted. read less NOT USED (high confidence) S. Pizzini, “Silicon Science and Technology as the Background of the Current and Future Knowledge Society.” 2012. link Times cited: 2 Abstract: This introductory chapter aims to present the unique potenti… read moreAbstract: This introductory chapter aims to present the unique potentialities of semiconductor silicon as the substrate or the component of a variety of devices that support the development of the society in which we live today and where our sons and daughters will live, hopefully, tomorrow; taking, however, as known all the very basic physics concerning the electronic and optical properties of semiconductor silicon as well as the basic concepts concerning silicon devices [1–7]. Also, considering the number of issues that should be taken into consideration to enlighten this critical role of silicon, only a few of these, selected in a very personal, and possibly not entirely objective, manner will be discussed in full detail. The discussion will start from the thermonuclear synthesis of silicon and will end with the properties and applications of silicon nanodots and nanowires studied today in research labs worldwide, with the consideration that silicon’s uniqueness derives from its specific structural, physical and chemical properties, which make elemental silicon readily obtainable from widely diffused raw materials and directly suitable for technological applications in microelectronics, optoelectronics and photovoltaics, without neglecting high-power devices, chemical sensors and radiation detectors. The analysis will be focused on the variety of its structural forms, which range from single crystal towards microcrystalline, nanocrystalline and amorphous, with a discontinuous change of properties that, in fact, allow a multiplicity of applications. read less NOT USED (high confidence) H. Lai, S. Cea, H. Kennel, and S. Dunham, “Molecular dynamics modeling of solid phase epitaxial regrowth,” Journal of Applied Physics. 2012. link Times cited: 3 Abstract: Solid phase epitaxial regrowth (SPER) is of great technologi… read moreAbstract: Solid phase epitaxial regrowth (SPER) is of great technological importance in semiconductor device fabrication. A better understanding and accurately modeling of its behavior are vital to the design of fabrication processes and the improvement of the device performance. In this paper, SPER was modeled by molecular dynamics (MD) with Tersoff potential. Extensive MD simulations were conducted to study the dependence of SPER rate on temperature, growth orientation, pressure, and uniaxial stress. The simulation data were fitted to empirical formula, and the results were compared with experimental data. It was concluded that MD with Tersoff potential can qualitatively describe the SPER process. For a more quantitatively accurate model, larger simulation systems and a better interatomic potential are needed. read less NOT USED (high confidence) F. Zirkelbach, B. Stritzker, K. Nordlund, J. Lindner, W. Schmidt, and E. Rauls, “Combined ab initio and classical potential simulation study on silicon carbide precipitation in silicon,” Physical Review B. 2011. link Times cited: 22 Abstract: Atomistic simulations on the silicon carbide precipitation i… read moreAbstract: Atomistic simulations on the silicon carbide precipitation in bulk silicon employing both, classical potential and first-principles methods are presented. The calculations aim at a comprehensive, microscopic understanding of the precipitation mechanism in the context of controversial discussions in the literature. For the quantum-mechanical treatment, basic processes assumed in the precipitation process are calculated in feasible systems of small size. The migration mechanism of a carbon 〈1 0 0〉 interstitial and silicon 〈11 0〉 self-interstitial in otherwise defect-free silicon are investigated using density functional theory calculations. The influence of a nearby vacancy, another carbon interstitial and a substitutional defect as well as a silicon self-interstitial has been investigated systematically. Interactions of various combinations of defects have been characterized including a couple of selected migration pathways within these configurations. Most of the investigated pairs of defects tend to agglomerate allowing for a reduction in strain. The formation of structures involving strong carbon–carbon bonds turns out to be very unlikely. In contrast, substitutional carbon occurs in all probability. A long range capture radius has been observed for pairs of interstitial carbon as well as interstitial carbon and vacancies. A rather small capture radius is predicted for substitutional carbon and silicon self-interstitials. Initial assumptions regarding the precipitation mechanism of silicon carbide in bulk silicon are established and conformability to experimental findings is discussed. Furthermore, results of the accurate first-principles calculations on defects and carbon diffusion in silicon are compared to results of classical potential simulations revealing significant limitations of the latter method. An approach to work around this problem is proposed. Finally, results of the classical potential molecular dynamics simulations of large systems are examined, which reinforce previous assumptions and give further insight into basic processes involved in the silicon carbide transition. read less NOT USED (high confidence) C. D. Cruz, K. Termentzidis, P. Chantrenne, and X. Kleber, “Molecular dynamics simulations for the prediction of thermal conductivity of bulk silicon and silicon nanowires: Influence of interatomic potentials and boundary conditions,” Journal of Applied Physics. 2011. link Times cited: 62 Abstract: The reliability of molecular dynamics (MD) results depends s… read moreAbstract: The reliability of molecular dynamics (MD) results depends strongly on the choice of interatomic potentials and simulation conditions. Five interatomic potentials have been evaluated for heat transfer MD simulations of silicon, based on the description of the harmonic (dispersion curves) and anharmonic (linear thermal expansion) properties. The best interatomic potential is the second nearest-neighbor modified embedded atom method potential followed by the Stillinger-Weber, and then the Tersoff III. However, the prediction of the bulk silicon thermal conductivity leads to the conclusion that the Tersoff III potential gives the best results for isotopically pure silicon at high temperatures. The thermal conductivity of silicon nanowires as a function of cross-section and length is calculated, and the influence of the boundary conditions is studied for those five potentials. read less NOT USED (high confidence) J. Los, C. Bichara, and R. Pellenq, “Tight binding within the fourth moment approximation: Efficient implementation and application to liquid Ni droplet diffusion on graphene.” 2011. link Times cited: 9 Abstract: (Received 8 February 2011; revised manuscript received 13 Ma… read moreAbstract: (Received 8 February 2011; revised manuscript received 13 May 2011; published 31 August 2011)Application of the fourth moment approximation (FMA) to the local density of states within a tight bindingdescription to build a reactive, interatomic interaction potential for use in large scale molecular simulations,is a logical and significant step forward to improve the second moment approximation, standing at the basisof several, widely used (semi-)empirical interatomic interaction models. In this paper we present a sufficientlydetailed description of the FMA and its technical implications, containing the essential elements for an efficientimplementationinasimulationcode.Usingarecent,existingFMA-basedmodelforC-Nisystems,weinvestigatedthesizedependenceofthediffusionofaliquidNiclusteronagraphenesheetandfindapowerlawdependenceofthediffusionconstantontheclustersize(numberofclusteratoms)withanexponentverycloseto−2 read less NOT USED (high confidence) C. Krzeminski, Q. Brulin, V. Cuny, E. Lecat, E. Lampin, and F. Cleri, “Molecular dynamics simulation of the recrystallization of amorphous Si layers: Comprehensive study of the dependence of the recrystallization velocity on the interatomic potential,” arXiv: Computational Physics. 2011. link Times cited: 37 Abstract: The molecular dynamics method is applied to simulate the rec… read moreAbstract: The molecular dynamics method is applied to simulate the recrystallization of an amorphous/crystalline silicon interface. The atomic structure of the amorphous material is constructed with the method of Wooten, Winer, and Weaire. The amorphous on crystalline stack is annealed afterward on a wide range of temperature and time using five different interatomic potentials: Stillinger-Weber, Tersoff, EDIP, SW115, and Lenosky. The simulations are exploited to systematically extract the recrystallization velocity. A strong dependency of the results on the interatomic potential is evidenced and explained by the capability of some potentials (Tersoff and SW115) to correctly handle the amorphous structure, while other potentials (Stillinger-Weber, EDIP, and Lenosky) lead to the melting of the amorphous. Consequently, the interatomic potentials are classified according to their ability to simulate the solid or the liquid phase epitaxy. read less NOT USED (high confidence) A. Furmanchuk, O. Isayev, T. Dinadayalane, and J. Leszczynski, “Car-parrinello molecular dynamics simulations of tensile tests on Si〈001〉 nanowires,” Journal of Physical Chemistry C. 2011. link Times cited: 5 Abstract: Theoretical simulations of tensile tests on Si⟨001⟩ nanowire… read moreAbstract: Theoretical simulations of tensile tests on Si⟨001⟩ nanowires have been carried out using Car–Parrinello molecular dynamics. H-passivation was used to model experimentally occurring passivation in Si nanowires. First-principle molecular dynamics simulations at ambient temperature reveal the governing role of size, overall shape, and composition of the surface layer for the mechanical properties. Our results indicate that SiH2 groups in the outer layer and the octahedral shape of the wire soften Young’s modulus and allow wire to handle larger transverse strains than SiH groups in wires with the tetrahedral shape. The importance of the overall shape of the wire has been discussed by comparing the behavior of surface layers of {100} and {110} facets. The presence of the {100} facets helps to relax the transverse strain during tension. On the basis of changes in structural parameters, we have presented the schematic motion of Si atoms in core and surface layers before the fracture appeared. read less NOT USED (high confidence) P. K. Soin, P. K. Soin, A. Horsfield, and D. Nguyen-Manh, “Efficient self-consistency for magnetic tight binding,” Comput. Phys. Commun. 2011. link Times cited: 12 NOT USED (high confidence) T. Kunze, S. Gemming, M. Posselt, and G. Seifert, “Tribological Aspects of Carbon-Based Nanocoatings – Theory and Simulation,” Zeitschrift für Physikalische Chemie. 2011. link Times cited: 2 Abstract: Nanocoatings have the potential to improve the surface prope… read moreAbstract: Nanocoatings have the potential to improve the surface properties of various materials. They are of extreme importance for surfaces in sliding contact such as highly stressed engine parts. Here, nanocoatings have to be optimized with respect to low friction properties and a high wear resistance to enhance the energetic and environmental efficiency. An important example are diamond-like carbon (DLC) films, which exhibit high mechanical stability depending on their deposition process. We present an introduction to this field of tribology by giving a short overview on DLC films, on the influence of lubricants from a theoretical point of view and in a broader sense, on basic principles of modeling tribological processes with molecular dynamic methods. read less NOT USED (high confidence) F. Zirkelbach, B. Stritzker, K. Nordlund, J. Lindner, W. Schmidt, and E. Rauls, “Defects in carbon implanted silicon calculated by classical potentials and first-principles methods,” Physical Review B. 2010. link Times cited: 7 Abstract: A comparative theoretical investigation of carbon interstiti… read moreAbstract: A comparative theoretical investigation of carbon interstitials in silicon is presented. Calculations using classical potentials are compared to first-principles density-functional theory calculations of the geometries, formation, and activation energies of the carbon dumbbell interstitial, showing the importance of a quantummechanical description of this system. In contrast to previous studies, the present first-principles calculations of the interstitial carbon migration path yield an activation energy that excellently matches the experiment. The bond-centered interstitial configuration shows a net magnetization of two electrons, illustrating the need for spin-polarized calculations. read less NOT USED (high confidence) J. Guénolé, J. Godet, and L. Pizzagalli, “Determination of activation parameters for the core transformation of the screw dislocation in silicon,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 13 Abstract: The non-dissociated screw dislocation in a model covalent ma… read moreAbstract: The non-dissociated screw dislocation in a model covalent material like silicon is known to exist in three possible stable core configurations. We performed calculations combining the nudged elastic band technique and a semi-empirical description in order to determine mechanisms and activation parameters for transforming one core into another. Our results showed that a glide core is necessarily reconstructed, since the energy barrier for reconstruction is easily overcome by thermal activation. Conversely, a transformation between a shuffle and a glide core appears unlikely at low temperature, which raises questions about the existence of the double-period glide configuration. read less NOT USED (high confidence) A. Kerrache, N. Mousseau, and L. J. Lewis, “Amorphous silicon under mechanical shear deformations: Shear velocity and temperature effects,” Physical Review B. 2010. link Times cited: 8 Abstract: Mechanical shear deformations lead, in some cases, to effect… read moreAbstract: Mechanical shear deformations lead, in some cases, to effects similar to those resulting from ion irradiation. Here we characterize the effects of shear velocity and temperature on amorphous silicon (\aSi) modelled using classical molecular dynamics simulations based on the empirical Environment Dependent Inter-atomic Potential (EDIP). With increasing shear velocity at low temperature, we find a systematic increase in the internal strain leading to the rapid appearance of structural defects (5-fold coordinated atoms). The impacts of externally applied strain can be almost fully compensated by increasing the temperature, allowing the system to respond more rapidly to the deformation. In particular, we find opposite power-law relations between the temperature and the shear velocity and the deformation energy. The spatial distribution of defects is also found to strongly depend on temperature and strain velocity. For low temperature or high shear velocity, defects are concentrated in a few atomic layers near the center of the cell while, with increasing temperature or decreasing shear velocity, they spread slowly throughout the full simulation cell. This complex behavior can be related to the structure of the energy landscape and the existence of a continuous energy-barrier distribution. read less NOT USED (high confidence) K. Jarolimek, G. de Wijs, R. A. de Groot, and M. Zeman, “Structural models of a‐Si:H with a low defect concentration: A first‐principles molecular dynamics study,” physica status solidi (a). 2010. link Times cited: 1 Abstract: We present a theoretical study of hydrogenated amorphous sil… read moreAbstract: We present a theoretical study of hydrogenated amorphous silicon (a‐Si:H) with a device quality hydrogen concentration of 11 at%. We used a first principle, parameters‐free method. The interaction between the atoms was treated quantum mechanically within the density functional theory approximation. Amorphous structures were prepared by cooling from the liquid phase. When using a cooling rate of 0.02 K/fs defect‐free structures were prepared. All silicon atoms were fourfold coordinated and there were no defect states in the band gap. The calculated short range order showed a good agreement with available neutron scattering measurements. We further calculated the formation energy of dangling bonds (DBs; threefold coordinated Si atom) in all three charge states (negative, neutral, and positive) as a function of the Fermi energy. Interestingly, the DB correlation energies can have both positive and negative values. read less NOT USED (high confidence) G. Lucas, M. Bertolus, and L. Pizzagalli, “An environment-dependent interatomic potential for silicon carbide: calculation of bulk properties, high-pressure phases, point and extended defects, and amorphous structures,” Journal of Physics: Condensed Matter. 2010. link Times cited: 41 Abstract: An interatomic potential has been developed to describe inte… read moreAbstract: An interatomic potential has been developed to describe interactions in silicon, carbon and silicon carbide, based on the environment-dependent interatomic potential (EDIP) (Bazant et al 1997 Phys. Rev. B 56 8542). The functional form of the original EDIP has been generalized and two sets of parameters have been proposed. Tests with these two potentials have been performed for many properties of SiC, including bulk properties, high-pressure phases, point and extended defects, and amorphous structures. One parameter set allows us to keep the original EDIP formulation for silicon, and is shown to be well suited for modelling irradiation-induced effects in silicon carbide, with a very good description of point defects and of the disordered phase. The other set, including a new parametrization for silicon, has been shown to be efficient for modelling point and extended defects, as well as high-pressure phases. read less NOT USED (high confidence) J. A. Pascual-Gutiérrez, J. Murthy, and R. Viskanta, “Thermal conductivity and phonon transport properties of silicon using perturbation theory and the environment-dependent interatomic potential,” Journal of Applied Physics. 2009. link Times cited: 35 Abstract: Silicon thermal conductivities are obtained from the solutio… read moreAbstract: Silicon thermal conductivities are obtained from the solution of the linearized phonon Boltzmann transport equation without the use of any parameter-fitting. Perturbation theory is used to compute the strength of three-phonon and isotope scattering mechanisms. Matrix elements based on Fermi’s golden rule are computed exactly without assuming either average or mode-dependent Grueisen parameters, and with no underlying assumptions of crystal isotropy. The environment-dependent interatomic potential is employed to describe the interatomic force constants and the perturbing Hamiltonians. A detailed methodology to accurately find three-phonon processes satisfying energy- and momentum-conservation rules is also described. Bulk silicon thermal conductivity values are computed across a range of temperatures and shown to match experimental data very well. It is found that about two-thirds of the heat transport in bulk silicon may be attributed to transverse acoustic modes. Effective relaxation times and mean free ... read less NOT USED (high confidence) M. Zhao, M. Iron, P. Staszewski, N. E. Schultz, R. Valero, and D. Truhlar, “Valence-Bond Order (VBO): A New Approach to Modeling Reactive Potential Energy Surfaces for Complex Systems, Materials, and Nanoparticles.,” Journal of chemical theory and computation. 2009. link Times cited: 12 Abstract: The extension of molecular mechanics to reactive systems, me… read moreAbstract: The extension of molecular mechanics to reactive systems, metals, and covalently bonded clusters with variable coordination numbers requires new functional forms beyond those popular for organic chemistry and biomolecules. Here we present a new scheme for reactive molecular mechanics, which is denoted as the valence-bond order model, for approximating reactive potential energy surfaces in large molecules, clusters, nanoparticles, solids, and other condensed-phase materials, especially those containing metals. The model is motivated by a moment approximation to tight binding molecular orbital theory, and we test how well one can approximate potential energy surfaces with a very simple functional form involving only interatomic distances with no explicit dependence on bond angles or dihedral angles. For large systems the computational requirements scale linearly with system size, and no diagonalizations or iterations are required; thus the method is well suited to large-scale simulations. The method is illustrated here by developing a force field for particles and solids composed of aluminum and hydrogen. The parameters were optimized against both interaction energies and relative interaction energies. The method performs well for pure aluminum clusters, nanoparticles, and bulk lattices and reasonably well for pure hydrogen clusters; the mean unsigned error per atom for the aluminum-hydrogen clusters is 0.1 eV/atom. read less NOT USED (high confidence) Q. Meng and Q. Wang, “Molecular dynamics simulation of annihilation of 60° dislocations in Si crystals,” physica status solidi (b). 2009. link Times cited: 1 Abstract: The annihilation of two 60° shuffle dislocations is studied … read moreAbstract: The annihilation of two 60° shuffle dislocations is studied via the molecular dynamics method. The Stillinger–Weber (SW) potential and environment‐dependent interatomic potential (EDIP) are used to describe the atomic interactions. The simulation results show that the complete annihilation of the 60° dislocations takes place only when the two dislocations lie on the same slip plane. The annihilation process may occur without external shear stress when the temperature is higher than a critical value. It is found that the critical temperature increases exponentially as a function of distance between the two dislocations. Also revealed in this simulation is an incomplete annihilation occurring when the distance between the slip planes of the two dislocations is less than about 1 nm. If the distance between the two slip planes is larger than about 1 nm, the dislocations will glide on their own slip planes as if no interaction exists between them. (© 2009 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) S. Schönborn, S. Goedecker, S. Roy, and A. Oganov, “The performance of minima hopping and evolutionary algorithms for cluster structure prediction.,” The Journal of chemical physics. 2008. link Times cited: 84 Abstract: We compare evolutionary algorithms with minima hopping for g… read moreAbstract: We compare evolutionary algorithms with minima hopping for global optimization in the field of cluster structure prediction. We introduce a new average offspring recombination operator and compare it with previously used operators. Minima hopping is improved with a softening method and a stronger feedback mechanism. Test systems are atomic clusters with Lennard-Jones interaction as well as silicon and gold clusters described by force fields. The improved minima hopping is found to be well-suited to all these homoatomic problems. The evolutionary algorithm is more efficient for systems with compact and symmetric ground states, including LJ(150), but it fails for systems with very complex energy landscapes and asymmetric ground states, such as LJ(75) and silicon clusters with more than 30 atoms. Both successes and failures of the evolutionary algorithm suggest ways for its improvement. read less NOT USED (high confidence) X. Wang, Z. Huang, T. Wang, Y. Tang, and X. Zeng, “Structure and thermophysical properties of single- wall Si nanotubes,” Physica B-condensed Matter. 2008. link Times cited: 15 NOT USED (high confidence) J. Schall, G. Gao, and J. Harrison, “Elastic constants of silicon materials calculated as a function of temperature using a parametrization of the second-generation reactive empirical bond-order potential,” Physical Review B. 2008. link Times cited: 48 Abstract: A parametrization for silicon is presented that is based on … read moreAbstract: A parametrization for silicon is presented that is based on the second-generation reactive empirical bondorder REBO formalism Brenner, Shenderova, Harrison, Stuart, Ni, and Sinnott J. Phys.: Condens. Matter 14, 783 2002 . Because it shares the same analytic form as Brenner’s second-generation REBO, this new potential is a step toward a single potential that can model many atom systems that contain C, Si, and H, where bond breaking and bond making are important. The widespread use of Brenner’s REBO potential, its ability to model both zero-Kelvin elastic constants of diamond and the temperature dependence of the elastic constants, and the existence of parameters for many atom types were the motivating factors for obtaining this parametrization for Si. While Si-C-H classical bond-order potentials do exist, they are based on Brenner’s original formalism. This new parametrization is validated by examining the structure and stability of a large number of crystalline silicon structures, by examining the relaxation energies of point defects, the energies of silicon surfaces, the effects of adatoms on surface energies, and the structures of both liquid silicon and amorphous silicon. Finally, the elastic constants of diamond-cubic and amorphous silicon between 0 and 1100 K are calculated with this new parametrization and compared to values calculated using a previously published potential. read less NOT USED (high confidence) J. Kermode, “Multiscale hybrid simulation of brittle fracture.” 2008. link Times cited: 5 NOT USED (high confidence) J. Samela and K. Nordlund, “Emergence of non-linear effects in nanocluster collision cascades in amorphous silicon,” New Journal of Physics. 2008. link Times cited: 11 Abstract: Cluster ion beams create considerably more damage in silicon… read moreAbstract: Cluster ion beams create considerably more damage in silicon and other substrates and eject more material than single ions that deposit at the same kinetic energy on the substrate. The mechanisms that causes the non-linear growth of damage and sputtering are interesting from the point of view of both basic materials research and industrial applications. Using classical molecular dynamics, we analyse the dynamics of collision cascades that are induced in amorphous silicon by small noble gas nanoclusters. We show that the sputtering and other non-linear effects emerge due to the high-energy density induced in a relatively small region in the substrate during the cluster stopping phase and because of the timing of consequent events that dissipate the energy over a larger volume of the substrate. read less NOT USED (high confidence) J. A. Pascual-Gutiérrez, J. Murthy, and R. Viskanta, “Limits of size confinement in silicon thin films and wires,” Journal of Applied Physics. 2007. link Times cited: 15 Abstract: Physically confined structures such as thin films and nanowi… read moreAbstract: Physically confined structures such as thin films and nanowires are becoming increasingly important in nanoscale energy conversion and nanoelectronics. The main focus of this work is to determine the size threshold below which the volumetric specific heat and group velocity of one- and two-dimensionally confined silicon nanostructures begin to differ significantly with respect to bulk silicon and to quantify these changes. The dynamical matrix approach subject to free-standing boundary conditions is employed to determine the phonon normal modes of vibration of the structures. The environment-dependent interatomic potential under the harmonic approximation is used to model interatomic forces. We find that above 10nm thickness, silicon [111]-films yield specific heats and group velocities which exhibit size-invariant behavior; for [111]-silicon nanowires, the limit is approximately 5nm. Moreover, we show that computed phonon group velocities using the dynamical matrix approach are affected by geometry-speci... read less NOT USED (high confidence) S. L. Mielke, T. Belytschko, and G. Schatz, “Nanoscale fracture mechanics.,” Annual review of physical chemistry. 2007. link Times cited: 42 Abstract: Theoretical calculations on undefected nanoscale materials p… read moreAbstract: Theoretical calculations on undefected nanoscale materials predict impressive mechanical properties. In this review we summarize the status of experimental efforts to directly measure the fracture strengths of inorganic and carbon nanotubes and discuss possible explanations for the deviations between the predicted and observed values. We also summarize the role of theory in understanding the molecular-level origin of these deviations. In particular, we consider the role of defects such as vacancies, Stone-Wales defects, adatoms and ad-dimers, chemical functionalization, and oxidative pitting. read less NOT USED (high confidence) B. Yang and V. Tewary, “Multiscale modeling of point defects in Si-Ge(001) quantum wells,” Physical Review B. 2007. link Times cited: 8 Abstract: A computationally efficient hybrid Green's function (GF… read moreAbstract: A computationally efficient hybrid Green's function (GF) technique is developed for multiscale modeling of point defects in a trilayer lattice system that links seamlessly the length scales from lattice (subnanometers) to continuum (bulk). The model accounts for the discrete structure of the lattice including nonlinear effects at the atomistic level and full elastic anisotropy at the continuum level. The model is applied to calculate the discrete core structure of point defects (vacancies and substitutional impurities) in Si-Ge(001) quantum wells (QWs) that are of contemporary technological interest. Numerical results are presented for the short range and long range lattice distortions and strains in the lattice caused by the defects and their formation energy and Kanzaki forces that are basic characteristics of the defects. The continuum and the lattice GFs of the material system are used to link the different length scales, which enables us to model the point defects and extended defects such as the quantum well in a unified formalism. Nonlinear effects in the core of the point defects are taken into account by using an iterative scheme. The Tersoff potential is used to set up the lattice structure, compute the unrelaxed forces and force constants in the lattice, andmore » derive the elastic constants required for the continuum GF. It is found that the overall elastic properties of the material and the properties of defects vary considerably when the material is strained from the bulk to the QW state. This change in the defect properties is very significant and can provide a characteristic signature of the defect. For example, in the case of a single vacancy in Ge, the strain reverses the sign of the relaxation volume. It is also found that the defect properties, such as the defect core structures, change abruptly across a Ge/Si interface. The transition occurs over a region extending from two to four lattice constants, depending upon the defect species.« less read less NOT USED (high confidence) R. D. Menezes, J. F. Justo, and L. Assali, “Energetics of silicon nanowires: a molecular dynamics investigation,” physica status solidi (a). 2007. link Times cited: 4 Abstract: Silicon nanowires, with the 〈100〉 and 〈110〉 growth direction… read moreAbstract: Silicon nanowires, with the 〈100〉 and 〈110〉 growth directions and at several surface facet configurations, were investigated by molecular dynamics simulations. We considered three commonly used interatomic potentials for silicon, and tested the reliability of each model to describe silicon nanowires. We find that, for each growth direction, the facet family plays a central role on the nanowire energy, which follows a universal scaling law as a function of the nanowire perimeter. Those results were discussed in the context of recent experimental and ab initio data. (© 2007 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) L. Sun, C. Le, F. Saied, and J. Murthy, “Performance of a Parallel Molecular Dynamics Program for Computation of Thermal Properties,” Numerical Heat Transfer, Part B: Fundamentals. 2007. link Times cited: 5 Abstract: The parallel performance of classical molecular dynamics sim… read moreAbstract: The parallel performance of classical molecular dynamics simulations of the thermal properties of solid-state materials is evaluated. Computations are validated by predicting the bulk silicon thermal conductivity as a function of temperature. The performance of the computational algorithm and software are tested on three different architectures, including the IBM BlueGene, the IBM Power 4 +, and an Intel Xeon Linux cluster, corresponding to different combinations of processor speeds, communications bandwidth, and latency. Two popular three-body potentials used for silicon simulation are evaluated and compared. In addition, the popular Lennard-Jones potential is used to investigate to role of cutoff distance on parallel performance. read less NOT USED (high confidence) R. Drautz, X. W. Zhou, D. Murdick, B. Gillespie, H. Wadley, and D. Pettifor, “Analytic bond-order potentials for modelling the growth of semiconductor thin films,” Progress in Materials Science. 2007. link Times cited: 28 NOT USED (high confidence) J. Samela, K. Nordlund, J. Keinonen, and V. Popok, “Comparison of silicon potentials for cluster bombardment simulations,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 26 NOT USED (high confidence) Q. Tang and Y. Yao, “The Kapitza Resistance Across Grain Boundary by Molecular Dynamics Simulation,” Nanoscale and Microscale Thermophysical Engineering. 2006. link Times cited: 6 Abstract: Nonequilibrium molecular dynamics (NEMD) simulations are per… read moreAbstract: Nonequilibrium molecular dynamics (NEMD) simulations are performed to calculate thermal boundary resistance that arises from heat flow across Si grain boundary. The environment-dependent interatomic potential (EDIP) on crystal silicon is adopted as a model system. The issues are related to nonlinear response, local thermal equilibrium, and statistical averaging. The tilt grain boundaries Σ5 and Σ13 are simulated, and the values of thermal boundary resistance by nonequilibrium molecular dynamics are compared with those by Maiti et al. (Solid State Communications, vol. 102, 1997). Using the disperse relation of EDIP potential, an average transmission coefficient of thermal conductivity across boundary is calculated. read less NOT USED (high confidence) C. Ni and J. Murthy, “Multiscale Stress Analysis Using EDIP Silicon,” Thermal and Thermomechanical Proceedings 10th Intersociety Conference on Phenomena in Electronics Systems, 2006. ITHERM 2006. 2006. link Times cited: 1 Abstract: It is necessary to be able to seamlessly meld molecular dyna… read moreAbstract: It is necessary to be able to seamlessly meld molecular dynamics (MD) and continuum stress analysis in many engineering situations. In this paper, we developed a finite volume method (FVM) to solve the continuum stress problem, while the MD simulation employs the environmentally dependent inter-atomic potential (EDIP), a three-body potential suitable for modeling silicon. Fixed-free and fixed-fixed bar bending problems are simulated by both the FVM method and the MD EDIP simulation. The results from the two computations are seen to match well for a cantilever of 32.136times5.43times5.43 nm3. A one-way multiscale analysis is then performed . Here, the FVM simulation is carried out in the whole domain, while the MD simulation is performed in a sub-domain, with an overlap region in which MD and continuum models are both employed. Material velocities computed by the continuum solution are interpolated to the atomic positions in the MD solver, but velocities from the MD solution do not influence the continuum solution. The method is applied to the beam bending problem and a reasonable match with the pure MD, pure continuum and the multiscale simulation is found read less NOT USED (high confidence) T. Fujii and Y. Akiniwa, “Molecular dynamics analysis for fracture behaviour of single crystal silicon thin film with micro notch,” Modelling and Simulation in Materials Science and Engineering. 2006. link Times cited: 18 Abstract: Thin films of single crystal silicon are widely required in … read moreAbstract: Thin films of single crystal silicon are widely required in many applications of semiconductor devices and micro-electro-mechanical systems. In this study, molecular dynamics simulation is conducted to investigate the effect of notch depth on fracture strength. The specimen size is about 10 nm × 5 nm, and the periodic boundary condition is applied for thin films. The loading directions are [100], [110] and [111] direction. Brittle fracture occurs on the plane perpendicular to the loading direction. The stress intensity factor at the onset of the crack propagation increases with increasing notch depth. When the notch depth is longer than 2 nm, the stress intensity factor is nearly constant. At the onset of the crack propagation, the stress at the notch root becomes constant irrespective of the notch depth. read less NOT USED (high confidence) G. Lulli, E. Albertazzi, S. Balboni, and L. Colombo, “Defect-induced homogeneous amorphization of silicon: the role of defect structure and population,” Journal of Physics: Condensed Matter. 2006. link Times cited: 7 Abstract: Molecular dynamics based on the environment-dependent intera… read moreAbstract: Molecular dynamics based on the environment-dependent interatomic potential is used to investigate the influence of the nature and distribution of defects on solid state, homogeneous amorphization of Si. To this end, different kinds of defects, including single interstitials and vacancies (both uncorrelated and correlated distributions), bond defects, and small interstitial and vacancy clusters, have been considered. It is shown that the threshold defect concentration for amorphization depends on the defect type, and, in the case of single defects, on the degree of correlation between interstitial and vacancy distributions. The threshold varies within the interval [0.18–0.28] atomic fraction, the upper value corresponding to the case of bond defects, the lower to the uncorrelated distributions of single split interstitials plus compensating vacancies. read less NOT USED (high confidence) R. Drautz, D. Murdick, D. Nguyen-Manh, X. W. Zhou, H. Wadley, and D. Pettifor, “Analytic bond-order potential for predicting structural trends across the sp-valent elements,” Physical Review B. 2005. link Times cited: 48 Abstract: An analytic interatomic bond-order potential BOP is derived … read moreAbstract: An analytic interatomic bond-order potential BOP is derived that depends explicitly on the group number of the sp-valent element. This is achieved by generalizing the previously published BOP for group-IV elements by extrapolating from half-full occupancy using a simple envelope function for the upper bound of the bond order. This interatomic potential predicts the structural trends across the sp-valent elements that are found by our tight-binding reference calculations and observed by experiment. Unlike empirical interatomic potentials this theoretically derived BOP includes the valence-dependent character of the bonding naturally within its remit. read less NOT USED (high confidence) S. Kapur, M. Prasad, J. Crocker, and T. Sinno, “Role of configurational entropy in the thermodynamics of clusters of point defects in crystalline solids,” Physical Review B. 2005. link Times cited: 36 Abstract: Received 28 March 2005; revised manuscript received 24 May 2… read moreAbstract: Received 28 March 2005; revised manuscript received 24 May 2005; published 20 July 2005The internal configurational entropy of point defect clusters in crystalline silicon is studied in detail byanalyzing their potential energy landscapes. Both on-lattice and off-lattice calculation approaches are employedto demonstrate the importance of off-lattice configurational states that arise due to a large number of inherentstructures local minima in the energy landscape generated by the interatomic potential function. The resultingcluster configurational entropy of formation is shown to exhibit behavior that is qualitatively similar to thatobserved in supercooled liquids and amorphous solids and substantially alters the thermodynamic properties ofpoint defect clusters in crystals at high temperature. This behavior is shown to be independent of interatomicpotential and cluster type, and suggests that defects in crystals at high temperature should be generally de-scribed by a quasicontinuous collection of nondegenerate states rather than as a single ground state structure.The modified thermodynamic properties of vacancy clusters at high temperature are found to explain a long-standing discrepancy between simulation predictions and experimental measurements of vacancy aggregationdynamics in silicon.DOI: 10.1103/PhysRevB.72.014119 PACS number s : 61.72.Bb, 61.72.Qq read less NOT USED (high confidence) N. Lorente, R. Rurali, and H. Tang, “Single-molecule manipulation and chemistry with the STM,” Journal of Physics: Condensed Matter. 2005. link Times cited: 59 Abstract: We review recent theoretical work on the manipulation of sin… read moreAbstract: We review recent theoretical work on the manipulation of single molecules with scanning probes, in particular the scanning tunnelling microscope (STM). The aim of theories and simulations is to account for the processes, ideally at a quantitative level, that permit the controlled manipulation of matter at the atomic scale in adsorbed molecular systems. In order to achieve this, simulations rely on total energy and electronic structure calculations where a trade-off is made between the size of the system and the accuracy of the calculation. This first stage of the calculation yields the basic quantities used for the second stage: the evaluation of the coupled electron–nuclear dynamics. This second stage is a formidable task and many approximations are involved. In this review, we will present some of the customary approximations regarding the theoretical study of mechanical and inelastic manipulations. Mechanical manipulations use the interaction between the acting probe (usually a metallic tip) and the targeted adsorbate. We review recent results in the field of adsorbate mechanical manipulations and explain how manipulations can be effected by using the interaction between the probe’s tip and certain molecular groups of complex chemisorbed molecular systems. On the other hand, inelastic manipulations use the tunnelling current to convey energy with sub-ångström precision. This current can excite localized vibrations that can induce measurable variations of the tunnelling conductance, hence providing a means of detecting single-molecule vibrations. This current can also inject energy in a few reaction coordinates. Recently, the possibility of vibrational selective manipulations of NH3/Cu(100) has been experimentally demonstrated. The theory presented here addresses the actual pathways accessed when the molecule is excited by the tunnelling current from an STM. read less NOT USED (high confidence) Y. Long, N. Chen, and W. Zhang, “Pair potentials for a metal–ceramic interface by inversion of adhesive energy,” Journal of Physics: Condensed Matter. 2005. link Times cited: 46 Abstract: A concise and general formula is introduced to obtain ab ini… read moreAbstract: A concise and general formula is introduced to obtain ab initio pair potentials between atoms across a metal–ceramic interface by inversion of the adhesive energies of the interface. Derivation of interfacial potentials ΦAg−Mg and ΦAg−O from ab initio adhesive energies is performed by applying the formula to the Ag/MgO(001) interface. Transferability of these potentials at Ag/MgO(100), Ag/MgO(110) and Ag/MgO(111) interfaces is discussed. read less NOT USED (high confidence) P. Erhart and K. Albe, “Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide,” Physical Review B. 2005. link Times cited: 462 Abstract: We present an analytical bond-order potential for silicon, c… read moreAbstract: We present an analytical bond-order potential for silicon, carbon, and silicon carbide that has been optimized by a systematic fitting scheme. The functional form is adopted from a preceding work {\}Phys. Rev. B 65, 195124 (2002) and is built on three independently fitted potentials for Si-Si, C-C, and Si-C interaction. For elemental silicon and carbon, the potential perfectly reproduces elastic properties and agrees very well with first-principles results for high-pressure phases. The formation enthalpies of point defects are reasonably reproduced. In the case of silicon stuctural features of the melt agree nicely with data taken from literature. For silicon carbide the dimer as well as the solid phases B1, B2, and B3 were considered. Again, elastic properties are very well reproduced including internal relaxations under shear. Comparison with first-principles data on point defect formation enthalpies shows fair agreement. The successful validation of the potentials for configurations ranging from the molecular to the bulk regime indicates the transferability of the potential model and makes it a good choice for atomistic simulations that sample a large configuration space. read less NOT USED (high confidence) E. J. Albenze and P. Clancy, “Interface Response Functions for Amorphous and Crystalline Si and the Implications for Explosive Crystallization,” Molecular Simulation. 2005. link Times cited: 12 Abstract: Interface response functions (IRFs) for amorphous and crysta… read moreAbstract: Interface response functions (IRFs) for amorphous and crystalline forms of Si have been determined for several empirical atomic-scale models using Molecular Dynamics and compared to available experimental results fitted to a Wilson-Frenkel equation form. Stillinger–Weber (SW), the environment-dependent intermolecular potential (EDIP), and a version of the modified embedded atom method (MEAM) models were found to produce unacceptable representations of the IRFs of both solid phases; they were either unable to predict the amorphous melting point and/or the maximum solidification velocity. The best of these models was judged to be the SW potential, known to produce a very accurate IRF for crystalline silicon. Increasing the strength of the three-body term by up to 25% above that of the original SW potential improves the prediction of the melting characteristics of the amorphous phase. Above this limit, liquid phase properties are impaired. The resultant IRFs provide an important backdrop to understand the kinetics of explosive crystallization (EC) processes, as we shall show in comparison to recent experimental data on the EC of amorphous Ge. [A. Chojnacka and M.O. Thompson, in Growth, Evolution and Properties of Surfaces, Thin Films and Self-Organized Structures, edited by S.C. Moss, D.B. Poker, D. Ila, (Mat. Res. Soc. Symp. Proc. 648, Warrendale, PA 2001) p. P11.12.1–8]. We also provide evidence that homogeneous melting within the bulk of the amorphous material competes with heterogeneous melting at the planar amorphous/liquid interface. read less NOT USED (high confidence) C. L. Allred, X. Yuan, M. Bazant, and L. Hobbs, “Elastic constants of defected and amorphous silicon with the environment-dependent interatomic potential,” Physical Review B. 2004. link Times cited: 31 Abstract: The elastic constants of a wide range of models of defected … read moreAbstract: The elastic constants of a wide range of models of defected crystalline and amorphous silicon are calculated, using the environment-dependent interatomic potential (EDIP). The defected crystalline simulation cells contain randomly generated defect distributions. An extensive characterization of point defects is performed, including structure, energy and influence on elastic constants. Three important conclusions are drawn. (1) Defects have independent effects on the elastic constants of silicon up to (at least) a defect concentration of 0.3%. (2) The linear effect of Frenkel pairs on the Young's modulus of silicon is -1653 GPa per defect fraction. (3) 17 different point defect types cause a very similar decrease in the Young's modulus: -(0.28{+-}0.05)% when calculated in isolation using a 1728-atom cell. These principles will be very useful for predicting the effect of radiation damage on the elastic modulus of silicon in the typical case in which point-defect concentrations can be estimated, but the exact distribution and species of defects is unknown. We also study amorphous samples generated in quenching the liquid with EDIP, including an ideal structure of perfect fourfold coordination, samples with threefold and fivefold coordinated defects, one with a nanovoid, and one with an amorphous inclusion in a crystalline matrix.more » In the last case, a useful finding is that the change in the Young's modulus is simply related to the volume fraction of amorphous material, as has also been observed by experiment.« less read less NOT USED (high confidence) Q. Tang, “A molecular dynamics simulation: the effect of finite size on the thermal conductivity in a single crystal silicon,” Molecular Physics. 2004. link Times cited: 22 Abstract: Non-equilibrium molecular dynamics (NEMD) simulations are pe… read moreAbstract: Non-equilibrium molecular dynamics (NEMD) simulations are performed to calculate thermal conductivity. The environment-dependent interatomic potential (EDIP) potential on crystal silicon is adopted as a model system. The issues are related to nonlinear response, local thermal equilibrium and statistical averaging. The simulation results by non-equilibrium molecular dynamics show that the calculated thermal conductivity decreases almost linearly as the film thickness reduced at the nanometre scale. The effect of size on the thermal conductivity is also obtained by a theoretic analysis of the kinetic theory and formulas of the heat capacity. The analysis reveals that the contributions of phonon mean free path (MFP) and phonon number in a finite cell to thermal conductivity are very important. read less NOT USED (high confidence) S. Kapur, M. Prasad, and T. Sinno, “Carbon-Mediated Aggregation of Self-Interstitials in Silicon.” 2004. link Times cited: 12 Abstract: The carbon-mediated aggregation of silicon self-interstitial… read moreAbstract: The carbon-mediated aggregation of silicon self-interstitials is investigated with large-scale parallel molecular dynamics. The presence of carbon in the silicon matrix is shown to lead to concentration-dependent self-interstitial cluster pinning, dramatically reducing cluster coalescence and thereby inhibiting the nucleation process. The extent of cluster pinning increases with cluster size for the range of cluster sizes observed in the simulation. The direct effect of carbon on single self-interstitials is shown to be of secondary importance, and the concentration of single self-interstitials as a function of time is essentially unchanged in the presence of carbon. A quasi-single-component mean-field interpretation of the atomistic simulation results is proposed and further confirms these conclusions. Based on these results, it is suggested that the experimentally observed effect of carbon on transient-enhanced diffusion of boron could be due to the direct interaction between carbon atoms and self-interstitial clusters. read less NOT USED (high confidence) C. R. Miranda and A. Antonelli, “Transitions between disordered phases in supercooled liquid silicon.,” The Journal of chemical physics. 2004. link Times cited: 32 Abstract: We have investigated the transitions between disordered phas… read moreAbstract: We have investigated the transitions between disordered phases in supercooled liquid silicon using computer simulations. The thermodynamic properties were directly obtained from the free energy, which was computed using the recently proposed reversible scaling method. The calculated free energies of the crystalline and liquid phases of silicon at zero pressure, obtained using the environment dependent interatomic potential, are in excellent agreement with the available experimental data. The results show that, at zero pressure, a weak first-order liquid-liquid transition occurs at 1135 K and a continuous liquid-amorphous transition takes place at 843 K. These results are consistent with the existence of a second critical point for the liquid-liquid transition at a negative pressure. read less NOT USED (high confidence) D. Pettifor, M. Finnis, D. Nguyen-Manh, D. Murdick, X. W. Zhou, and H. Wadley, “Analytic bond-order potentials for multicomponent systems,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2004. link Times cited: 53 NOT USED (high confidence) P. Biswas, R. Atta-Fynn, and D. A. Drabold, “Reverse Monte Carlo modeling of amorphous silicon,” Physical Review B. 2004. link Times cited: 71 Abstract: An implementation of the Reverse Monte Carlo algorithm is pr… read moreAbstract: An implementation of the Reverse Monte Carlo algorithm is presented for the study of amorphous tetrahedral semiconductors. By taking into account a number of constraints that describe the tetrahedral bonding geometry along with the radial distribution function, we construct a model of amorphous silicon using the reverse monte carlo technique. Starting from a completely random configuration, we generate a model of amorphous silicon containing 500 atoms closely reproducing the experimental static structure factor and bond angle distribution and in improved agreement with electronic properties. Comparison is made to existing Reverse Monte Carlo models, and the importance of suitable constraints beside experimental data is stressed. read less NOT USED (high confidence) J. Godet, L. Pizzagalli, S. Brochard, and P. Beauchamp, “Comparison between classical potentials and ab initio methods for silicon under large shear,” Journal of Physics: Condensed Matter. 2003. link Times cited: 47 Abstract: The homogeneous shear of the {111} planes along the directio… read moreAbstract: The homogeneous shear of the {111} planes along the direction of bulk silicon has been investigated using ab initio techniques, to better understand the strain properties of both shuffle and glide set planes. Similar calculations have been done with three empirical potentials, Stillinger–Weber, Tersoff and EDIP, in order to find the one giving the best results under large shear strains. The generalized stacking fault energies have also been calculated with these potentials to complement this study. It turns out that the Stillinger–Weber potential better reproduces the ab initio results, for the smoothness and the amplitude of the energy variation as well as the localization of shear in the shuffle set. read less NOT USED (high confidence) M. Prasad and T. Sinno, “Internally Consistent Approach for Modeling Solid-State Aggregation: I. Atomistic Calculations of Vacancy Clustering in Silicon,” Physical Review B. 2003. link Times cited: 33 Abstract: A computational framework is presented for describing the nu… read moreAbstract: A computational framework is presented for describing the nucleation and growth of vacancy clusters in crystalline silicon. The overall approach is based on a parametrically consistent comparison between two representations of the process in order to provide a systematic method for probing the details of atomic mechanisms responsible for aggregation. In this paper, the atomistic component of the overall framework is presented. First, a detailed set of targeted atomistic simulations are described that characterize fully the thermodynamic and transport properties of vacancy clusters over a wide range of sizes. It is shown that cluster diffusion is surprisingly favorable because of the availability of multiple, almost degenerate configurations. A single large-scale parallel molecular dynamics simulation is then used to compute directly the evolution of the vacancy cluster size distribution in a supersaturated system initially containing 1000 uniformly distributed vacancies in a host lattice of 216 000 Si atoms at 1600 K. The results of this simulation are interpreted in the context of mean-field scaling theory based on the observed power-law evolution of the size distribution moments. It is shown that the molecular dynamics results for aggregation of vacancy clusters, particularly the evolution of the average cluster size, can be very well represented by a highly simplified mean-field model. A direct comparison to a detailed continuum model is made in a subsequent article. read less NOT USED (high confidence) J. Los and A. Fasolino, “Intrinsic long-range bond-order potential for carbon: Performance in Monte Carlo simulations of graphitization,” Physical Review B. 2003. link Times cited: 233 Abstract: We propose a bond order potential for carbon with built-in l… read moreAbstract: We propose a bond order potential for carbon with built-in long-range interactions. The potential is defined as the sum of an angular and coordination dependent short-range part accounting for the strong covalent interactions and a radial long-range part describing the weak interactions responsible, e.g., for the interplanar binding in graphite. The short-range part is a Brenner type of potential, with several modifications introduced to get an improved description of elastic properties and conjugation. Contrary to previous long-range extensions of existing bond order potentials, we prevent the loss of accuracy by compensating for the additional long-range interactions by an appropriate parametrization of the short-range part. We also provide a short-range bond order potential. In Monte Carlo simulations our potential gives a good description of the diamond to graphite transformation. For thin (111) slabs graphitization proceeds perpendicular to the surface as found in ab initio simulations, whereas for thick layers we find that graphitization occurs layer by layer. read less NOT USED (high confidence) C. R. Miranda, R. W. Nunes, and A. Antonelli, “Temperature effects on dislocation core energies in silicon and germanium,” Physical Review B. 2003. link Times cited: 14 Abstract: Temperature effects on the energetics of the 90° partial dis… read moreAbstract: Temperature effects on the energetics of the 90° partial dislocation in silicon and germanium are investigated, using nonequilibrium methods to estimate free energies, coupled with Monte Carlo simulations. Atomic interactions are described by Tersoff and environment-dependent interatomic potentials. Our results indicate that the vibrational entropy has the effect of increasing the difference in free energy between the two possible reconstructions of the 90° partial, namely, the single-period and the double-period geometries. This effect further increases the energetic stability of the double-period reconstruction at high temperatures. The results also indicate that anharmonic effects may play an important role in determining the structural properties of these defects in the high-temperature regime. read less NOT USED (high confidence) L. Pizzagalli, P. Beauchamp, and J. Rabier, “Stability and core structure of undissociated screw dislocations in group IV materials investigated by means of atomistic calculations,” Journal of Physics: Condensed Matter. 2002. link Times cited: 11 Abstract: We have examined the various possible configurations for an … read moreAbstract: We have examined the various possible configurations for an undissociated screw dislocation in group IV materials (Ge, Si, 3C-SiC, diamond) by means of semi-empirical atomistic calculations. A complete structural characterization and a determination of the relative stability are performed. We found that, in contrast to the case for Ge and Si, a geometry with the presence of sp 2 atoms in th ec ore is the most stable structure for 3C-SiC and diamond. This yields a stable screw dislocation configuration i nt he ‘shuffle’ set for Si and Ge, and in th e‘ glide’ set for 3C-SiC and diamond. read less NOT USED (high confidence) S. Nakhmanson, D. A. Drabold, and N. Mousseau, “Comment on ‘Boson peak in amorphous silicon: A numerical study,’” Physical Review B. 2002. link Times cited: 4 Abstract: (Dated: July 27, 2001) Based on molecular-dynamics simulatio… read moreAbstract: (Dated: July 27, 2001) Based on molecular-dynamics simulations using the Stillinger-Weber interatomic potential, Finkemeier and von Niessen recently proposed that the presence of the Boson peak in a–Si can be attributed to coordination defects [Phys. Rev. B 63, 235204 (2001)] and claimed agreement with earlier simulation results for models of a–Si with voids [Phys. Rev. B 61, 5376 (2000)]. In this Comment we clarify this issue and suggest that (i) the atomistic models of Finkemeier and von Niessen do not represent realistic amorphous silicon and (ii) the results for the models with voids do not support the hypothesis that coordination defects are the main cause of the appearance of the Boson peak in this material. read less NOT USED (high confidence) P. Keblinski, M. Bazant, R. Dash, and M. Treacy, “Thermodynamic behavior of a model covalent material described by the environment-dependent interatomic potential,” Physical Review B. 2002. link Times cited: 38 Abstract: Using molecular-dynamics simulations we study the thermodyna… read moreAbstract: Using molecular-dynamics simulations we study the thermodynamic behavior of a single-component covalent material described by the recently proposed environment-dependent interatomic potential (EDIP). The parametrization of EDIP for silicon exhibits a range of unusual properties typically found in more complex materials, such as the existence of two structurally distinct disordered phases, a density increase upon melting of the low-temperature amorphous phase, and negative thermal-expansion coefficients for both the crystal (at high temperatures) and the amorphous phase (at all temperatures). Structural differences between the two disordered phases also lead to a first-order transition between them, which suggests the existence of a second critical point, as is believed to exist for amorphous forms of frozen water. For EDIP-Si, however, the unusual behavior is associated not only with the open nature of tetrahedral bonding but also with a competition between fourfold (covalent) and fivefold (metallic) coordination. The unusual behavior of the model and its unique ability to simulate the liquid/amorphous transition on molecular-dynamics time scales make it a suitable prototype for fundamental studies of anomalous thermodynamics in disordered systems. read less NOT USED (high confidence) N. C. Cooper, M. S. Fagan, C. Goringe, N. Marks, and D. Mckenzie, “Surface structure and sputtering in amorphous carbon thin films: a tight-binding study of film deposition,” Journal of Physics: Condensed Matter. 2002. link Times cited: 22 Abstract: A tight-binding simulation of the atom-by-atom deposition of… read moreAbstract: A tight-binding simulation of the atom-by-atom deposition of amorphous carbon (a-C) at 100 eV incident energy is presented. More than 500 atoms were deposited. Chains are observed to form on the surface, some of which are sputtered. The good agreement with the experimental sputter frequency data and observation that all such clusters are linear provides strong support for the existence of these chains and the direct emission model of sputtering. The bulk of the grown film is a-C with a tetrahedral bonding fraction of 20%. Experiments have shown that at this incident energy of 100 eV, tetrahedral a-C is the preferred structural form rather than the a-C produced by this simulation. This discrepancy is attributed to the short range of the interatomic potential. read less NOT USED (high confidence) M. Ishimaru, “Atomistic simulations of structural relaxation processes in amorphous silicon,” Journal of Applied Physics. 2002. link Times cited: 36 Abstract: Structural relaxation processes in amorphous silicon (a-Si) … read moreAbstract: Structural relaxation processes in amorphous silicon (a-Si) have been examined by molecular-dynamics (MD) simulations using the Tersoff interatomic potential. The a-Si networks generated by rapid quenching from liquid Si were annealed. Structural changes due to the relaxation of a-Si networks were observed. The present MD simulations reproduce well experimental measurements of changes in radial distribution functions, static structure factors, bond angle distributions, and phonon densities of states due to structural relaxation. read less NOT USED (high confidence) S. Dorfman, V. Liubich, D. Fuks, and K. C. Mundim, “Simulations of decohesion and slip of the Σ3⟨111⟩ grain boundary in tungsten with non-empirically derived interatomic potentials: the influence of boron interstitials,” Journal of Physics: Condensed Matter. 2001. link Times cited: 12 Abstract: Monte Carlo atomistic simulations of the properties of Σ3111… read moreAbstract: Monte Carlo atomistic simulations of the properties of Σ3111 grain boundaries in W are carried out. We demonstrate the influence of boron additive on the resistance of the grain boundary with respect to different shifts. The interatomic potentials used in these simulations are obtained from ab initio total-energy calculations. These calculations are performed in the framework of density functional theory in the coherent potential approximation. A recursion procedure for extracting A-B-type interatomic potentials is suggested. read less NOT USED (high confidence) M. Ishimaru, “Molecular-dynamics study on atomistic structures of amorphous silicon,” Journal of Physics: Condensed Matter. 2001. link Times cited: 20 Abstract: Structural characteristics of amorphous silicon (a-Si) have … read moreAbstract: Structural characteristics of amorphous silicon (a-Si) have been examined by molecular-dynamics calculations using the Tersoff interatomic potential. It was confirmed that the computer-generated atomic configurations reproduce well the structural and dynamical properties of a-Si obtained experimentally. The a-Si networks contained two types of structural defect: threefold coordinated Si atoms (dangling bonds) and fivefold coordinated ones (floating bonds). The average bond length increased with the coordination number. Bond angles were distributed around 120° for the threefold coordinations, suggesting the existence of the atomic clusters constructed by sp2 bonding. On the other hand, they had peaks at ~60° and 90° for the fivefold coordinated atoms. Partial radial distribution functions revealed that the floating bonds have a tendency to cluster in the a-Si network. read less NOT USED (high confidence) V. Bulatov et al., “Parameter-free modelling of dislocation motion: The case of silicon,” Philosophical Magazine A. 2001. link Times cited: 69 Abstract: In silicon and other materials with a high Peierls potential… read moreAbstract: In silicon and other materials with a high Peierls potential. dislocation motion takes place by nucleation and propagation of kink pairs. The rates of these unit processes are complex unknown functions of interatomic interactions in the dislocation core, stress and temperature. This work is an attempt to develop a quantitative physical description of dislocation motion in silicon based on understanding of the core structure and the energetics of core mechanisms of mobility. Atomistic simulations reveal multiple and complex kink mechanisms of dislocation translation; however, this complexity can be rationalized through the analysis of a straight kink-free dislocation, based on symmetry-breaking arguments. Further reduction is achieved by observing that the energetics of kink mechanisms is scaled by a single parameter, the energy required to break a bond in the core. To obtain accurate values of this energy we perform density functional calculations that lead us to conclude that the low mobility of the 30° dislocation results from its high bond-breaking energy. Armed with the knowledge of kink mechanisms, we develop a kinetic Monte Carlo model that makes direct use of the atomistic data as the material-defining input and predicts the dislocation velocity on the length and time scales accessible to experiments. This provides the connection between the atomistic aspects of the dislocation core and the mobility behaviour of single dislocations. read less NOT USED (high confidence) J. F. Justo, A. Fazzio, and A. Antonelli, “Dislocation core reconstruction in zinc-blende semiconductors,” Journal of Physics: Condensed Matter. 2000. link Times cited: 15 Abstract: Using ab initio total-energy calculations, we computed core … read moreAbstract: Using ab initio total-energy calculations, we computed core reconstruction energies of partial dislocations in zinc-blende semiconductors. The reconstruction energy of 30° partials was found to scale almost linearly with the experimental activation energy of 60° dislocations. The electronic structure of a dislocation shows that in an unreconstructed core, the gap states comprise a half-filled one-dimensional band, which splits up into bonding and antibonding states upon reconstruction. The energy states which lie in the electronic gap come from the core of β-partials, while those related to α-partials remain resonant in the valence band. read less NOT USED (high confidence) M. Bazant and B. Trout, “A method to extract potentials from the temperature dependence of Langmuir constants for clathrate-hydrates,” Physica A-statistical Mechanics and Its Applications. 2000. link Times cited: 29 NOT USED (high confidence) T. Lenosky et al., “Highly optimized empirical potential model of silicon,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 145 Abstract: We fit an empirical potential for silicon using the modified… read moreAbstract: We fit an empirical potential for silicon using the modified embedded atom (MEAM) functional form, which contains a nonlinear function of a sum of pairwise and three-body terms. The three-body term is similar to the Stillinger-Weber form. We parametrized our model using five cubic splines, each with 10 fitting parameters, and fitted the parameters to a large database using the force-matching method. Our model provides a reasonable description of energetics for all atomic coordinations, Z, from the dimer (Z = 1) to fcc and hcp (Z = 12). It accurately reproduces phonons and elastic constants, as well as point defect energetics. It also provides a good description of reconstruction energetics for both the 30° and 90° partial dislocations. Unlike previous models, our model accurately predicts formation energies and geometries of interstitial complexes - small clusters, interstitial-chain and planar {311} defects. read less NOT USED (high confidence) A. Kawamoto, J. Jameson, K. Cho, and R. Dutton, “Challenges for atomic scale modeling in alternative gate stack engineering,” IEEE Transactions on Electron Devices. 2000. link Times cited: 24 Abstract: We review the challenges for atomic scale modeling of altern… read moreAbstract: We review the challenges for atomic scale modeling of alternative gate dielectric stacks. We begin by highlighting recent achievements of state-of-the-art atomistic simulations of the Si-SiO/sub 2/ system, showing how such calculations have elucidated the microscopic origins of several important experimental phenomena. For the benefit of readers who may be unfamiliar with the simulation tools, we overview and compare the relevant methods. We then describe the difficulties encountered in extending these approaches to investigate high-k dielectric stacks, pointing out exciting research directions aimed at overcoming these challenges. We conclude by presenting a roadmap of computational goals for atomic scale modeling of alternative gate dielectrics. read less NOT USED (high confidence) K. Scheerschmidt, D. Conrad, A. Belov, and D. Timpel, “Enhanced semi-empirical potentials in molecular dynamics simulations of wafer bonding,” Materials Science in Semiconductor Processing. 2000. link Times cited: 4 NOT USED (high confidence) C. Herrero, “Quantum atomic dynamics in amorphous silicon; a path-integral Monte Carlo simulation,” Journal of Physics: Condensed Matter. 2000. link Times cited: 10 Abstract: The quantum dynamics of atoms in amorphous silicon has been … read moreAbstract: The quantum dynamics of atoms in amorphous silicon has been addressed by using path-integral Monte Carlo simulations. Structural results (radial distribution functions) found from these simulations agree well with experimental data. We study the quantum delocalization of the silicon atoms around their equilibrium positions. This delocalization is larger for coordination defects (fivefold-coordinated Si atoms). Correlations in the atomic displacements are analysed as a function of the interatomic distance and compared with those derived from classical Monte Carlo simulations. At high temperatures, the classical limit is recovered. Our results are also compared with those derived from similar quantum simulations for crystalline silicon. Structural disorder favours a larger vibrational amplitude for the atoms in amorphous silicon. read less NOT USED (high confidence) M. Schaible, “Empirical Molecular Dynamics Modeling of Silicon and Silicon Dioxide: A Review,” Critical Reviews in Solid State and Materials Sciences. 1999. link Times cited: 28 Abstract: A number of computational methods have been developed over t… read moreAbstract: A number of computational methods have been developed over the last 40 years to simulate the behavior of solid materials with small dimensions. On the macro-scale, Finite Element analysis calculates mechanical stress on micron-sized cantilevers and motors. On the atomic level, newer ab initio methods compute nuclear and electronic behavior of hundred atom models with unprecedented rigor. By implementing the laws of classic mechanics, empirical Molecular Dynamics (MD) programs help bridge these two computational extremes. MD identifies nonelectronic, particle motion for large 100,000 atom cells with good success. MD derives both equilibrium and nonequilibrium properties for many complex condensed regimes; quantitatively (and qualitatively) reaffirms empirical data; aids discovery of new materials processing techniques, and helps predict unknown physical phenomena often only observable under extreme environmental settings. One material of great technical importance to the semiconductor industry is silicon (... read less NOT USED (high confidence) S. Myers and D. Follstaedt, “FORCES BETWEEN CAVITIES AND DISLOCATIONS AND THEIR INFLUENCE ON SEMICONDUCTOR MICROSTRUCTURES,” Journal of Applied Physics. 1999. link Times cited: 16 Abstract: An approximate continuum method for computing the energy of … read moreAbstract: An approximate continuum method for computing the energy of interaction between cavities and strain fields in complex configurations is described and tested by comparison with results for simple, exactly solvable cases. The method is then used to examine semiquantitatively the effective forces between cavities and screw and edge dislocations, taking into account the effects of surface tension and pressurized gas within the cavity. The discussion encompasses not only local interactions involving individual cavities, but also the combined forces acting upon dislocations in the vicinity of multiple cavities and simultaneously within range of external-surface image forces. The results are used to interpret a range of observed microstructures in semiconductors and to assess the possible exploitation of cavity–dislocation binding for dislocation control in Si–Ge heteroepitaxial structures. read less NOT USED (high confidence) D.-K. Cao, “Pair distribution functions in molecular dynamics simulations of interfaces.” 2020. link Times cited: 0 Abstract: Pair Distribution Functions in Molecular Dynamics Simulation… read moreAbstract: Pair Distribution Functions in Molecular Dynamics Simulations of Interface by Deng Cao Thin films of silicon nitride on silicon are well suited materials for many applications including photovoltaics. Large-scale molecular-dynamics simulations of silicon/silicon nitride interfaces under externally applied tensile strain are performed in an attempt to improve understanding of this interface. The simulations reveal stress release in form of fracture, slip, pit formation, and interface phase transition under high stress condition. The silicon/silicon nitride interface is described as an eight-component system thereby offering valuable information in some of the thirty-six different pair distribution functions. We find that fracture in silicon nitride, with a centerpiece breaking off the sides, is reflected in a return to the original height of the first peak of the Si-N pair distribution function indicating that this centerpiece is essentially unstretched. Slip and pit formation in silicon as well as formation of domains of two different interface phases are identified by additional peaks in the pair distribution functions at and across the interface. Understanding selective pair distribution functions calculated at various stages of a particular simulation offer the opportunity to analyze structural and mechanical failure of large systems without knowing the detailed properties of individual atoms in the system. In particular, the occurrence of peaks reflecting new interatomic distances allows early predictions of failure. read less NOT USED (high confidence) M. Timonova and B. Thijsse, “Optimizing the MEAM potential for silicon,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 25 Abstract: By applying simulated annealing techniques we fit the modifi… read moreAbstract: By applying simulated annealing techniques we fit the modified embedded atom method (MEAM) potential to a database of ab initio energies for silicon and construct an improved parametrization of this potential. In addition, we introduce a new, reference-free version of the MEAM potential. This MEAM version is also fitted to the silicon data and shows an even better agreement, although the improvement is modest. Finally, we investigate whether increasing the number of different angular terms in the MEAM potential from 3 to 4 will lead to a better potential. The aim of this work is to determine a broad-ranged potential, one that is reliable in many different low- and high-energy atomic geometries in silicon crystals, molecules, near defects and under strain. To verify this, the performance of the new potentials is tested in different circumstances that were not explicitly included in the fit: relaxed defect energies, thermal expansion, melting temperature and liquid silicon. The new MEAM parametrizations found in this work, called MEAM-M and RF-MEAM, are shown to be overall more accurate than previous potentials—although a few defect energies are exceptions—and we recommend them for future work. The melting temperatures are closer to the experiment than those of other MEAM potentials, but they are still too high. read less NOT USED (high confidence) M. Prasad and T. Sinno, “Feature Activated Molecular Dynamics: Parallelization and Application to Systems with Globally Varying Mechanical Fields,” Journal of Computer-Aided Materials Design. 2005. link Times cited: 1 NOT USED (high confidence) K. Scheerschmidt, “Molecular dynamics simulations of wafer bonding,” MRS Proceedings. 2001. link Times cited: 0 Abstract: Molecular dynamics simulations using empirical potentials ha… read moreAbstract: Molecular dynamics simulations using empirical potentials have been employed to describe atomic inte ractions at interfaces created by the macroscopic wafer bonding process. Investigating perfect o r distorted s urfaces of different s emiconductor materials as well as of silica enables one to study the elementary processes and the resulting defects at the interfaces, and to characterize the ability of the potentials used. T wist r otation due to misalignment and bonding over steps influence strongly the bondability of larger areas. Empirical potentials developed by the bond o rder tight-binding approximation incl ude -bonds and yield enhanced interface structures, energies, and transferability to new materials systems. read less NOT USED (definite) A. Rohskopf, H. Seyf, K. Gordiz, T. Tadano, and A. Henry, “Empirical interatomic potentials optimized for phonon properties,” npj Computational Materials. 2017. link Times cited: 35 NOT USED (definite) S. Chavoshi, S. Xu, and X. Luo, “Dislocation-mediated plasticity in silicon during nanometric cutting : a molecular dynamics simulation study materials science in semiconductor processing,” Materials Science in Semiconductor Processing. 2016. link Times cited: 50 NOT USED (definite) I. Suarez-Martinez, P. Higginbottom, and N. Marks, “Molecular dynamics simulations of the transformation of carbon peapods into double-walled carbon nanotubes,” Carbon. 2010. link Times cited: 15 NOT USED (definite) H. J. Christie, M. Robinson, D. L. Roach, D. Ross, I. Suarez-Martinez, and N. Marks, “Simulating radiation damage cascades in graphite,” Carbon. 2015. link Times cited: 48 NOT USED (definite) “Realistic time-scale fully atomistic simulations of surface nucleation of dislocations in pristine nanopillars,” arXiv: Materials Science. 2012. link Times cited: 0 Abstract: We use our recently proposed accelerated dynamics algorithm … read moreAbstract: We use our recently proposed accelerated dynamics algorithm (Tiwary and van de Walle, 2011) to calculate temperature and stress dependence of activation free energy for surface nucleation of dislocations in pristine Gold nanopillars under realistic loads. While maintaining fully atomistic resolution, we achieve the fraction of a second time-scale regime. We find that the activation free energy depends significantly and non-linearly on the driving force (stress or strain) and temperature, leading to very high activation entropies. We also perform compression tests on Gold nanopillars for strain-rates varying between 7 orders of magnitudes, reaching as low as 10^3/s. Our calculations bring out the perils of high strain-rate Molecular Dynamics calculations: we find that while the failure mechanism for compression of Gold nanopillars remains the same across the entire strain-rate range, the elastic limit (defined as stress for nucleation of the first dislocation) depends significantly on the strain-rate. We also propose a new methodology that overcomes some of the limits in our original accelerated dynamics scheme (and accelerated dynamics methods in general). We lay out our methods in sufficient details so as to be used for understanding and predicting deformation mechanism under realistic driving forces for various problems. read less |
 EDIP_JustoBazantKaxiras_1998_Si__MO_958932894036_002
EDIP_JustoBazantKaxiras_1998_Si__MO_958932894036_002