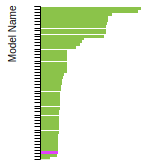
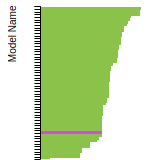
Current potential: EDIP_LAMMPS_JiangMorganSzlufarska_2012_SiC__MO_667792548433_000
Deep Citation determination:
Does the citing paper use the current potential to generate results displayed in the paper?
Provide us with identifying information so that we know you are not a bot (you will not be added to a mailing list):
Title
A single sentence description.
EDIP model for SiC developed by Jiang, Morgan, and Szlufarska (2012) v000
Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
This is an EDIP potential originally implemented in LAMMPS by Chao Jiang for the purpose of searching for the ground state of small carbon interstitial clusters in SiC.
Species
The supported atomic species.
C, Si
Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
This Model originally published in [1] is archived in OpenKIM [2-5].
[1] Jiang C, Morgan D, Szlufarska I. Carbon tri-interstitial defect: A model for the D_\mathrmII center. Phys Rev B [Internet]. 2012Oct;86(14):144118. Available from: https://link.aps.org/doi/10.1103/PhysRevB.86.144118 doi:10.1103/PhysRevB.86.144118 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information.
[2] Jiang C, Morgan D, Szlufarska I. EDIP model for SiC developed by Jiang, Morgan, and Szlufarska (2012) v000. OpenKIM; 2022. doi:10.25950/bce89e8e
[3] Afshar Y, Ferraro L, Jiang C, McSweeney SJ, Justo JF, Bazant MZ, et al. The environment-dependent interatomic potential (EDIP) potential v000. OpenKIM; 2021. doi:10.25950/a6a67b9f
[4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
27 Citations (16 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) L. He et al., “Atomic Resolution Imaging of Black Spot Defects in Ion Irradiated Silicon Carbide,” Microscopy and Microanalysis. 2015. link Times cited: 1
Abstract: Silicon carbide is of great interest as a nuclear fuel cladd… read more
Abstract: Silicon carbide is of great interest as a nuclear fuel cladding material. At relatively low irradiation temperatures (< 1000 C) and doses (< 10 dpa or displacements per atom), the major irradiation induced defects are black spot defects (BSD), which appear as nanometer scale black spots in bright field transmission electron microscopy (TEM) images [1,2]. BSDs are associated with radiation-induced swelling [1]. The detailed internal structure of BSD is unknown. We are working towards understanding the structures and evolution of BSD by combining high resolution scanning transmission electron microscopy (STEM) and defect structure modeling [3]. read less
USED (low confidence) S. Leroch, R. Stella, A. Hössinger, and L. Filipovic, “Molecular dynamics study of Al implantation in 4H-SiC,” 2023 International Conference on Simulation of Semiconductor Processes and Devices (SISPAD). 2023. link Times cited: 0
Abstract: We have performed a molecular dynamics study of Al-implantat… read more
Abstract: We have performed a molecular dynamics study of Al-implantation in 4H-SiC while investigating the types of defects produced and their quantity depending on the implantation temperature and dose. The damage to the SiC lattice should be minimized to facilitate the subsequent Al activation during annealing. Using the empirical Gao-Weber potential, together with a recently proposed Morse potential for the Al-SiC interaction, we show that implantation at elevated temperatures considerably reduces the creation of amorphous pockets and extended defect clusters. In a follow-up annealing study we aim to provide a correlation between dose/implantation temperature and the Al activation rate to give guidance for future fabrication of SiC devices. read less
USED (low confidence) Y. Huang, Y. Zhou, J. Li, and F. Zhu, “Understanding the role of surface mechanical properties in SiC surface machining,” Materials Science in Semiconductor Processing. 2023. link Times cited: 0
USED (low confidence) Y. Liu et al., “Deep learning inter-atomic potential for irradiation damage in 3C-SiC,” Computational Materials Science. 2023. link Times cited: 0
USED (low confidence) K. Wu et al., “Vapor Deposition Growth of Sic Crystal on 4h-Sic Substrate by Molecular Dynamics Simulation,” SSRN Electronic Journal. 2023. link Times cited: 1
Abstract: Due to the lack of appropriate experimental methods for imag… read more
Abstract: Due to the lack of appropriate experimental methods for imaging the evolution of the microstructure of materials at the growth conditions, our understanding of the physical behavior of crystal growth and defect formation during the vapor deposition growth of SiC crystals is still rather limited. In the present work, the vapor deposition growth of SiC crystal on a 4H-SiC substrate has been investigated by the molecular dynamics (MD) computer simulation method. Three different lattice planes of 4H-SiC ((0001), (112-0) and (1-100)) were selected as the surface of the substrate, and three different temperatures for substrate (2200 K, 2300 K and 2400 K) were used in growth simulations. The characteristics of the formation of different polytypes of SiC and dislocations in the grown crystals were examined. The results show that the SiC crystals were grown by a subsurface nucleation and growth mode in the vapor deposition process. For substrates with (0001) plane as the surface, the 3C-SiC single crystal was obtained in the deposited thin film. For substrates with (112-0) or (1-100) plane as the surface, the 4H-SiC single crystal was obtained instead. The temperature of the substrate was found to have a significant effect on the dislocation density generated in the grown crystals. The mechanism for the formation of Frank partial dislocations during the growth of SiC crystals has been analyzed, for which the importance of the diffusivity of atoms on the surface layer in growth has been highlighted, and it gives a good explanation of the temperature effect on dislocation formation in the grown crystals. These results can be helpful for experimental vapor deposition growth of SiC single crystals and epitaxial layers of high quality. read less
USED (low confidence) S. Zhao, G. Ran, F. Gao, S. Ma, D. Cui, and G. Yang, “Rotation and migration behavior of self-interstitial atoms in 3C-SiC: A comprehensive ab initio study,” Journal of Nuclear Materials. 2021. link Times cited: 1
USED (low confidence) Y. Huang, M. Wang, J. Li, and F. Zhu, “Effect of abrasive particle shape on the development of silicon substrate during nano-grinding,” Computational Materials Science. 2021. link Times cited: 14
USED (low confidence) S. Zhao, G. Ran, F. Gao, and S. Ma, “Investigation of Migration and Transformation of Self-Interstitial Atoms in 3C-SiC via DFT Calculations,” AMI: Scripta Materialia. 2021. link Times cited: 0
Abstract: First-principles calculations were carried out to investigat… read more
Abstract: First-principles calculations were carried out to investigate the stable and potential metastable structure of self-interstitial atoms (SIAs) in 3C-SiC. The most stable C and Si interstitials we obtained are Ci-C and Sii-Si split interstitials, respectively. In addition, a serious of metastable structures Ci-C (x = 0.63 ~ 0.81), Ci-C , Ci-Si , Sii-Si and Sii-Si have been observed. The migration and transformation of these SIAs were systematically studied. The minimum migration barrier for the full space rotation of C and Si interstitial are 0.49 eV and 0.73 eV, respectively. For the long-distance migration of SIAs, the minimum migration energy for C and Si interstitials is 0.71 eV and 0.73 eV, the corresponding migration paths are Ci-C(2 ‾5‾ 2) ↔ Ci-Si(1 1 6) ↔ Ci-C(‾5‾ 2 2), and Sii-Si ↔ SiTC ↔ Sii-Si . read less
USED (low confidence) B. Yang, H. Yang, T. Li, J. Yang, and P. Yang, “Thermal transport at 6H-SiC/graphene buffer layer/GaN heterogeneous interface,” Applied Surface Science. 2021. link Times cited: 16
USED (low confidence) J. Luo, C. Zhou, Y. Cheng, and L. Liu, “Assessing the EDIP potential for atomic simulation of carbon diffusion, segregation and solubility in silicon melt,” Journal of Crystal Growth. 2020. link Times cited: 2
USED (low confidence) D. Sprouster, L. Snead, E. Dooryhee, S. Ghose, T. Koyanagi, and Y. Katoh, “Pair distribution function analysis of neutron-irradiated silicon carbide,” Journal of Nuclear Materials. 2019. link Times cited: 4
USED (low confidence) C. Liu, J. Xi, and I. Szlufarska, “Sensitivity of SiC Grain Boundaries to Oxidation,” The Journal of Physical Chemistry C. 2019. link Times cited: 13
Abstract: Molecular dynamics simulations of dry oxidation of bicrystal… read more
Abstract: Molecular dynamics simulations of dry oxidation of bicrystals with incoherent and coherent grain boundaries (GBs) in 3C–SiC are performed at 2000 K and the results are compared to oxidation of single-crystal SiC. Oxidation near incoherent GBs is found faster than that in single crystals and in coherent GBs, whereas oxidation of coherent GBs is comparable to that of single crystals. The accelerated oxidation near incoherent GBs is attributed to strain and the presence of under-coordinated Si within the GB region, both of which reduce the positive charge on silicon atoms, making them more reactive with oxygen. Although atoms with similar properties are found in dislocation cores of coherent GBs, dislocation cores are isolated from each other by crystalline regions, which in turn control the rate of oxidation. read less
USED (low confidence) I. P. Vali, P. Shetty, M. Mahesha, V. Sathe, D. Phase, and R. Choudhary, “Structural and optical studies of gamma irradiated N-doped 4H-SiC,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2019. link Times cited: 9
USED (low confidence) K. Skrobas, S. Stelmakh, S. Gierlotka, and B. Palosz, “A model of density waves in atomic structure of nanodiamond by molecular dynamics simulations,” Diamond and Related Materials. 2019. link Times cited: 10
USED (low confidence) X. Ma et al., “Graphitization resistance determines super hardness of lonsdaleite, nanotwinned and nanopolycrystalline diamond,” Carbon. 2018. link Times cited: 25
USED (low confidence) J. Xi et al., “Evolution of Defects and Defect Clusters in β-SiC Irradiated at High Temperature,” Fusion Science and Technology. 2014. link Times cited: 11
Abstract: A molecular dynamics study has been performed to investigate… read more
Abstract: A molecular dynamics study has been performed to investigate the generation and evolution of damage states in irradiated β-SiC at high temperature. It is found that most of the C antisites (SiC) are created during the early collisional phase, while the Si antisites (CSi) are significantly produced during the thermal spike phase. A modified near-neighbor point defect density (NPDD) is introduced to study the spatial aggregation of different defects during the displacement cascades, and feature of defect clusters evolution is analyzed in details. The dominated types of vacancy clusters after the displacement cascades are two- and three-size chainlike ones. And the vacancy NPDD (V-NPDD) decreases as the recoil energy increases. Furthermore, after the thermal spike phase, there is an additional annealing process during which the interstitials and antisites turn into defect clusters, respectively. read less
NOT USED (low confidence) X. Jiang, H. Sun, K. Choudhary, H. Zhuang, and Q. Nian, “Interpretable Ensemble Learning for Materials Property Prediction with Classical Interatomic Potentials: Carbon as an Example,” ArXiv. 2023. link Times cited: 0
Abstract: Machine learning (ML) is widely used to explore crystal mate… read more
Abstract: Machine learning (ML) is widely used to explore crystal materials and predict their properties. However, the training is time-consuming for deep-learning models, and the regression process is a black box that is hard to interpret. Also, the preprocess to transfer a crystal structure into the input of ML, called descriptor, needs to be designed carefully. To efficiently predict important properties of materials, we propose an approach based on ensemble learning consisting of regression trees to predict formation energy and elastic constants based on small-size datasets of carbon allotropes as an example. Without using any descriptor, the inputs are the properties calculated by molecular dynamics with 9 different classical interatomic potentials. Overall, the results from ensemble learning are more accurate than those from classical interatomic potentials, and ensemble learning can capture the relatively accurate properties from the 9 classical potentials as criteria for predicting the final properties. read less
NOT USED (low confidence) A. Rohskopf, S. Wyant, K. Gordiz, H. R. Seyf, M. G. Muraleedharan, and A. Henry, “Fast & accurate interatomic potentials for describing thermal vibrations,” Computational Materials Science. 2020. link Times cited: 7
NOT USED (high confidence) Y. Xie, J. Vandermause, S. Ramakers, N. Protik, A. Johansson, and B. Kozinsky, “Uncertainty-aware molecular dynamics from Bayesian active learning for phase transformations and thermal transport in SiC,” npj Computational Materials. 2022. link Times cited: 14
NOT USED (high confidence) C.-gen Qian, B. Mclean, D. Hedman, and F. Ding, “A comprehensive assessment of empirical potentials for carbon materials,” APL Materials. 2021. link Times cited: 22
Abstract: Carbon materials and their unique properties have been exten… read more
Abstract: Carbon materials and their unique properties have been extensively studied by molecular dynamics, thanks to the wide range of available carbon bond order potentials (CBOPs). Recently, with the increase in popularity of machine learning (ML), potentials such as Gaussian approximation potential (GAP), trained using ML, can accurately predict results for carbon. However, selecting the right potential is crucial as each performs differently for different carbon allotropes, and these differences can lead to inaccurate results. This work compares the widely used CBOPs and the GAP-20 ML potential with density functional theory results, including lattice constants, cohesive energies, defect formation energies, van der Waals interactions, thermal stabilities, and mechanical properties for different carbon allotropes. We find that GAP-20 can more accurately predict the structure, defect properties, and formation energies for a variety of crystalline phase carbon compared to CBOPs. Importantly, GAP-20 can simulate the thermal stability of C60 and the fracture of carbon nanotubes and graphene accurately, where CBOPs struggle. However, similar to CBOPs, GAP-20 is unable to accurately account for van der Waals interactions. Despite this, we find that GAP-20 outperforms all CBOPs assessed here and is at present the most suitable potential for studying thermal and mechanical properties for pristine and defective carbon. read less
NOT USED (high confidence) Y. Lysogorskiy, T. Hammerschmidt, J. Janssen, J. Neugebauer, and R. Drautz, “Transferability of interatomic potentials for molybdenum and silicon,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 14
Abstract: Interatomic potentials are widely used in computational mate… read more
Abstract: Interatomic potentials are widely used in computational materials science, in particular for simulations that are too computationally expensive for density functional theory (DFT). Most interatomic potentials have a limited application range and often there is very limited information available regarding their performance for specific simulations. We carried out high-throughput calculations for molybdenum and silicon with DFT and a number of interatomic potentials. We compare the DFT reference calculations and experimental data to the predictions of the interatomic potentials. We focus on a large number of basic materials properties, including the cohesive energy, atomic volume, elastic coefficients, vibrational properties, thermodynamic properties, surface energies and vacancy formation energies, which enables a detailed discussion of the performance of the different potentials. We further analyze correlations between properties as obtained from DFT calculations and how interatomic potentials reproduce these correlations, and suggest a general measure for quantifying the accuracy and transferability of an interatomic potential. From our analysis we do not establish a clearcut ranking of the potentials as each potential has its strengths and weaknesses. It is therefore essential to assess the properties of a potential carefully before application of the potential in a specific simulation. The data presented here will be useful for selecting a potential for simulations of Mo or Si. read less
NOT USED (high confidence) C. Liu and I. Szlufarska, “Distribution of defect clusters in the primary damage of ion irradiated 3C-SiC,” Journal of Nuclear Materials. 2018. link Times cited: 21
NOT USED (high confidence) H. Ko, A. Kaczmarowski, I. Szlufarska, and D. Morgan, “Data for: Optimization of self-interstitial clusters in 3C-SiC with Genetic Algorithm.” 2017. link Times cited: 9
NOT USED (high confidence) C. Liu et al., “Evolution of small defect clusters in ion-irradiated 3C-SiC: Combined cluster dynamics modeling and experimental study,” Acta Materialia. 2017. link Times cited: 54
NOT USED (high confidence) A. Kaczmarowski, S. Yang, I. Szlufarska, and D. Morgan, “Genetic algorithm optimization of defect clusters in crystalline materials,” Computational Materials Science. 2015. link Times cited: 24
NOT USED (high confidence) H. Jiang, C. Jiang, D. Morgan, and I. Szlufarska, “Accelerated atomistic simulation study on the stability and mobility of carbon tri-interstitial cluster in cubic SiC,” arXiv: Materials Science. 2014. link Times cited: 19
NOT USED (high confidence) C. Jiang, D. Morgan, and I. Szlufarska, “Structures and stabilities of small carbon interstitial clusters in cubic silicon carbide,” Acta Materialia. 2014. link Times cited: 19
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
The letter grade A was assigned because the normalized error in the computation was 1.13719e-10 compared with a machine precision of 2.22045e-16. The letter grade was based on 'score=log10(error/eps)', with ranges A=[0, 7.5], B=(7.5, 10.0], C=(10.0, 12.5], D=(12.5, 15.0), F>15.0. 'A' is the best grade, and 'F' indicates failure.
vc-forces-numerical-derivative
consistency
Forces computed by the model agree with numerical derivatives of the energy; see full description.
The model is C^1 continuous. This means that the model has continuous energy and continuous first derivative.
vc-dimer-continuity-c1
informational
The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description.
Model energy and forces are invariant with respect to rigid-body motion (translation and rotation) for all configurations the model was able to compute.
vc-objectivity
informational
Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description.
All threads give identical results for tested case. Model appears to be thread-safe.
vc-thread-safe
mandatory
The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description.
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
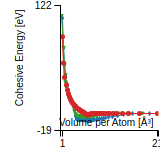
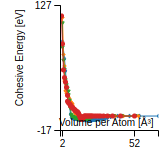
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Given an xyz file corresponding to a finite cluster of atoms, this Test Driver computes the total potential energy and atomic forces on the configuration. The positions are then relaxed using conjugate gradient minimization and the final positions and forces are recorded. These results are primarily of interest for training machine-learning algorithms.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the elastic constants for an arbitrary crystal. A robust computational protocol is used, attempting multiple methods and step sizes to achieve an acceptably low error in numerical differentiation and deviation from material symmetry. The crystal structure is specified using the AFLOW prototype designation as part of the Crystal Genome testing framework. In addition, the distance from the obtained elasticity tensor to the nearest isotropic tensor is computed.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the elastic constants for an arbitrary crystal. A robust computational protocol is used, attempting multiple methods and step sizes to achieve an acceptably low error in numerical differentiation and deviation from material symmetry. The crystal structure is specified using the AFLOW prototype designation as part of the Crystal Genome testing framework. In addition, the distance from the obtained elasticity tensor to the nearest isotropic tensor is computed.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Given atomic species and structure type (graphene-like, 2H, or 1T) of a 2D hexagonal monolayer crystal, as well as an initial guess at the lattice spacing, this Test Driver calculates the equilibrium lattice spacing and cohesive energy using Polak-Ribiere conjugate gradient minimization in LAMMPS
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Given an extended xyz file corresponding to a non-orthogonal periodic box of atoms, use LAMMPS to compute the total potential energy and atomic forces.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
 EDIP_LAMMPS_JiangMorganSzlufarska_2012_SiC__MO_667792548433_000
EDIP_LAMMPS_JiangMorganSzlufarska_2012_SiC__MO_667792548433_000