Current potential: MEAM_LAMMPS_DuLenoskyHennig_2011_Si__MO_883726743759_002
Deep Citation determination:
Does the citing paper use the current potential to generate results displayed in the paper?
Provide us with identifying information so that we know you are not a bot (you will not be added to a mailing list):
Title
A single sentence description.
Spline-based MEAM potential for Si system developed by Du et al. (2011) v002
Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
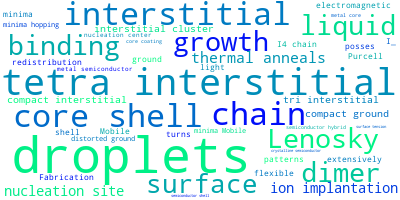
Mobile single interstitials can grow into extended interstitial defect structures during thermal anneals following ion implantation. The silicon tetra-interstitials present an important intermediate structure that can either provide a chain-like nucleation site for extended structures or form a highly stable compact interstitial cluster preventing further growth. In this paper, dimer searches using the tight-binding (TB) model by Lenosky et al. and density functional calculations show that the compact ground-state I-4(a) and the I-4-chain are surrounded by high-lying neighboring local minima. To furthermore explore the phase space of tetra-interstitial structures an empirical potential is optimized to a database of silicon defect structures. The minima hopping method combined with this potential extensively searches the energy landscape of tetra-interstitials and discovers several new low energy I-4 structures. The second lowest-energy I-4 structure turns out to be a distorted ground-state tri-interstitial bound with a single interstitial, which confirms that the ground-state tri-interstitial may serve as a nucleation center for the extended defects in silicon.
Species
The supported atomic species.
Si
Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
This Model originally published in [1] is archived in OpenKIM [2-5].
[1] Du YA, Lenosky TJ, Hennig RG, Goedecker S, Wilkins JW. Energy landscape of silicon tetra-interstitials using an optimized classical potential. physica status solidi (b). 2011;248(9):2050–5. doi:10.1002/pssb.201147137 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information.
[2] Du Y, Lenosky T, Hennig RG, Goedecker S, Wilkins JW. Spline-based MEAM potential for Si system developed by Du et al. (2011) v002. OpenKIM; 2023. doi:10.25950/53b24ae5
[3] Afshar Y, Hütter S, Rudd RE, Stukowski A, Tipton WW, Trinkle DR, et al. The modified embedded atom method (MEAM) potential v002. OpenKIM; 2023. doi:10.25950/ee5eba52
[4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
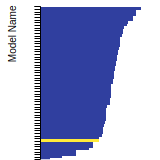
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
19 Citations (7 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) Y. Lysogorskiy et al., “Performant implementation of the atomic cluster expansion (PACE) and application to copper and silicon,” npj Computational Materials. 2021. link Times cited: 84
USED (high confidence) H. Zhang et al., “Fracture of a silicon nanowire at ultra-large elastic strain,” Acta Mechanica. 2017. link Times cited: 12
USED (high confidence) H. Zhang et al., “Fracture of a silicon nanowire at ultra-large elastic strain,” Acta Mechanica. 2017. link Times cited: 0
USED (high confidence) G. Zograf et al., “Modeling of formation mechanism and optical properties of Si/Au core-shell nanoparticles,” 2016 Days on Diffraction (DD). 2016. link Times cited: 2
Abstract: Fabrication of metal/semiconductor (“hybrid”) nanoparticles … read more
Abstract: Fabrication of metal/semiconductor (“hybrid”) nanoparticles is still a challenge due to the absence of methods of a metal core coating by crystalline semiconductor shell. We propose a novel principle of formation of a core-shell nanoparticle made of liquid silicon and gold droplets. Molecular dynamics simulations of the droplets behavior demonstrates that the core-shell structure with a gold core and a silicon shell can be formed if the droplets remain in the liquid state until final material redistribution. The main driven force in this process is surface tension governed by surface energies of the droplets, where silicon tends to cover gold due to lower surface energy. Taking into account this mechanism of core-shell nanoparticles formation, we provide numerical modelling, which demonstrates the resulting nanoparticle posses enhanced local electromagnetic field, high Purcell factor and flexible power patterns of scattered light. read less
USED (low confidence) X. H. Huang, Y.-J. Hu, and Q. An, “Locking of Screw Dislocations in Silicon due to Core Structure Transformation,” The Journal of Physical Chemistry C. 2021. link Times cited: 2
USED (low confidence) Y. Huang, M. Wang, J. Li, and F. Zhu, “Effect of abrasive particle shape on the development of silicon substrate during nano-grinding,” Computational Materials Science. 2021. link Times cited: 14
USED (low confidence) Q. Liu et al., “A semi-empirical fracture model for silicon cleavage fracture and its molecular dynamics study,” Theoretical and Applied Fracture Mechanics. 2019. link Times cited: 8
NOT USED (low confidence) M. Maździarz, “Transferability of interatomic potentials for silicene,” Beilstein Journal of Nanotechnology. 2023. link Times cited: 1
Abstract: The ability of various interatomic potentials to reproduce t… read more
Abstract: The ability of various interatomic potentials to reproduce the properties of silicene, that is, 2D single-layer silicon, polymorphs was examined. Structural and mechanical properties of flat, low-buckled, trigonal dumbbell, honeycomb dumbbell, and large honeycomb dumbbell silicene phases, were obtained using density functional theory and molecular statics calculations with Tersoff, MEAM, Stillinger–Weber, EDIP, ReaxFF, COMB, and machine-learning-based interatomic potentials. A quantitative systematic comparison and a discussion of the results obtained are reported. read less
NOT USED (low confidence) D. Vizoso, G. Subhash, K. Rajan, and R. Dingreville, “Connecting Vibrational Spectroscopy to Atomic Structure via Supervised Manifold Learning: Beyond Peak Analysis,” Chemistry of Materials. 2023. link Times cited: 1
Abstract: Vibrational spectroscopy is a nondestructive technique commo… read more
Abstract: Vibrational spectroscopy is a nondestructive technique commonly used in chemical and physical analyses to determine atomic structures and associated properties. However, the evaluation and interpretation of spectroscopic profiles based on human-identifiable peaks can be difficult and convoluted. To address this challenge, we present a reliable protocol based on supervised manifold learning techniques meant to connect vibrational spectra to a variety of complex and diverse atomic structure configurations. As an illustration, we examined a large database of virtual vibrational spectroscopy profiles generated from atomistic simulations for silicon structures subjected to different stress, amorphization, and disordering states. We evaluated representative features in those spectra via various linear and nonlinear dimensionality reduction techniques and used the reduced representation of those features with decision trees to correlate them with structural information unavailable through classical human-identifiable peak analysis. We show that our trained model accurately (over 97% accuracy) and robustly (insensitive to noise) disentangles the contribution from the different material states, hence demonstrating a comprehensive decoding of spectroscopic profiles beyond classical (human-identifiable) peak analysis. read less
NOT USED (low confidence) J. V. Michelin, L. G. Gonçalves, and J. Rino, “On the transferability of interaction potentials for condensed phases of silicon,” Journal of Molecular Liquids. 2019. link Times cited: 6
NOT USED (low confidence) G. V. Rudorff, C. Wehmeyer, and D. Sebastiani, “Efficient implementation and application of the artificial bee colony algorithm to low-dimensional optimization problems,” Comput. Phys. Commun. 2014. link Times cited: 3
NOT USED (high confidence) A. Jay et al., “Finding reaction pathways and transition states: r-ARTn and d-ARTn as an efficient and versatile alternative to string approaches.,” Journal of chemical theory and computation. 2020. link Times cited: 15
Abstract: Finding transition states and diffusion pathways is essentia… read more
Abstract: Finding transition states and diffusion pathways is essential to understand the evolution of materials and chemical reactions. Such characterization is hampered by the heavy computation costs associated with exploring energy landscapes at ab-initio accuracy. Here, we revisit the activation-relaxation technique (ARTn) to considerably reduce its costs when used with density functional theory (DFT) and propose three adapted versions of the algorithm to efficiently (i) explore the energy landscape of complex materials with the nowledge of a single minimum (ARTn); (ii) identify a transition state when two minima or a guess transition state are given (refining ART or r-ART) and (iii) reconstruct complex pathways between two given states (directed ART or d-ART). We show the application of these three variants on benchmark examples and on various complex defects in silicon. For the later, the presented improvements to ART leads to much more precise transition states while being two to six times faster than the commonly used string methods such as the Climbing Image Nudged Elastic Band method (CI-NEB). read less
NOT USED (high confidence) Y. Lysogorskiy, T. Hammerschmidt, J. Janssen, J. Neugebauer, and R. Drautz, “Transferability of interatomic potentials for molybdenum and silicon,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 14
Abstract: Interatomic potentials are widely used in computational mate… read more
Abstract: Interatomic potentials are widely used in computational materials science, in particular for simulations that are too computationally expensive for density functional theory (DFT). Most interatomic potentials have a limited application range and often there is very limited information available regarding their performance for specific simulations. We carried out high-throughput calculations for molybdenum and silicon with DFT and a number of interatomic potentials. We compare the DFT reference calculations and experimental data to the predictions of the interatomic potentials. We focus on a large number of basic materials properties, including the cohesive energy, atomic volume, elastic coefficients, vibrational properties, thermodynamic properties, surface energies and vacancy formation energies, which enables a detailed discussion of the performance of the different potentials. We further analyze correlations between properties as obtained from DFT calculations and how interatomic potentials reproduce these correlations, and suggest a general measure for quantifying the accuracy and transferability of an interatomic potential. From our analysis we do not establish a clearcut ranking of the potentials as each potential has its strengths and weaknesses. It is therefore essential to assess the properties of a potential carefully before application of the potential in a specific simulation. The data presented here will be useful for selecting a potential for simulations of Mo or Si. read less
NOT USED (high confidence) E. Dontsova and R. Ballarini, “Atomistic modeling of the fracture toughness of silicon and silicon-silicon interfaces,” International Journal of Fracture. 2017. link Times cited: 6
NOT USED (high confidence) S. Winczewski, J. Dziedzic, and J. Rybicki, “Central-force decomposition of spline-based modified embedded atom method potential,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 0
Abstract: Central-force decompositions are fundamental to the calculat… read more
Abstract: Central-force decompositions are fundamental to the calculation of stress fields in atomic systems by means of Hardy stress. We derive expressions for a central-force decomposition of the spline-based modified embedded atom method (s-MEAM) potential. The expressions are subsequently simplified to a form that can be readily used in molecular-dynamics simulations, enabling the calculation of the spatial distribution of stress in systems treated with this novel class of empirical potentials. We briefly discuss the properties of the obtained decomposition and highlight further computational techniques that can be expected to benefit from the results of this work. To demonstrate the practicability of the derived expressions, we apply them to calculate stress fields due to an edge dislocation in bcc Mo, comparing their predictions to those of linear elasticity theory. read less
NOT USED (high confidence) S. Goel, A. Kovalchenko, A. Stukowski, and G. Cross, “Influence of microstructure on the cutting behaviour of silicon,” Acta Materialia. 2016. link Times cited: 143
NOT USED (high confidence) S. Bringuier, V. Manga, K. Runge, P. Deymier, and K. Muralidharan, “An atomic scale characterization of coupled grain boundary motion in silicon bicrystals,” Philosophical Magazine. 2015. link Times cited: 7
Abstract: The mechanical response of symmetric tilt grain boundaries (… read more
Abstract: The mechanical response of symmetric tilt grain boundaries (GBs) in silicon bicrystals under shear loading are characterized using molecular dynamics simulations. It is seen that under shear, high-angle GBs namely Σ5 and Σ13 having a rotation axis [0 0 1] demonstrate coupled GB motion, such that the displacement of grains parallel to the GB interface is accompanied by normal GB motion. An atomic-scale characterization revealed that concerted rotations of silicon tetrahedra within the GB are the primary mechanisms leading to the coupled GB motion. Interestingly, so far, this phenomenon has only been examined in detail for metallic systems. A distinguishing feature of the coupled GB motion observed for the silicon symmetric tilt bicrystals as compared to metallic bicrystals is the fact that in the absence of shear, spontaneous coupled motion is not observed at high temperatures. read less
NOT USED (high confidence) P. Käshammer and T. Sinno, “Interactions of twin boundaries with intrinsic point defects and carbon in silicon,” Journal of Applied Physics. 2013. link Times cited: 22
Abstract: Although multicrystalline silicon (mc-Si) is currently the m… read more
Abstract: Although multicrystalline silicon (mc-Si) is currently the most widely used material for fabricating photovoltaic cells, its electrical properties remain limited by several types of defects, which interact in complex ways that are not yet fully understood. A particularly important phenomenon is the interaction between grain boundaries and intrinsic point defects or impurity atoms, such as carbon, oxygen, nitrogen, and various types of metals. Here, we use empirical molecular dynamics to study the interactions of Σ3{111}, Σ9{221}, and Σ27{552} twin boundaries, which account for over 50% of all grain boundaries in mc-Si, with self-interstitials, vacancies, and substitutional carbon atoms. It is shown that twin boundary-point defect interaction energies increase with twinning order and that they are predominantly attractive. We also find that twin boundary interactions with substitutional carbon are highly spatially heterogeneous, exhibiting alternating repulsive-attractive regions that correlate strongly wi... read less
NOT USED (high confidence) H. Park et al., “Ab initio based empirical potential used to study the mechanical properties of molybdenum,” Physical Review B. 2012. link Times cited: 70
Abstract: Density-functional theory energies, forces, and elastic cons… read more
Abstract: Density-functional theory energies, forces, and elastic constants determine the parametrization of an empirical, modified embedded-atom method potential for molybdenum. The accuracy and transferability of the potential are verified by comparison to experimental and density-functional data for point defects, phonons, thermal expansion, surface and stacking fault energies, and ideal shear strength. Searching the energy landscape predicted by the potential using a genetic algorithm verifies that it reproduces not only the correct bcc ground state of molybdenum but also all low-energy metastable phases. The potential is also applicable to the study of plastic deformation and used to compute energies, core structures, and Peierls stresses of screw and edge dislocations. Molybdenum's high strength and high-temperature stability make this refractory metal very attractive for use in advanced process technologies. The motion of dislocations is generally accepted to be responsible for the complex deformation behavior of this transition metal. 1-8 In recent years progress has been made on the description of the properties of screw dislocations using density-functional theory (DFT), tight- binding calculations, and empirical potentials. 9-19 However, DFT and tight-binding techniques are limited to small system sizes, which is problematic due to the long-range strain field of dislocations, and current empirical potentials lack the required accuracy for the description of the dislocation structure. Simulations of dislocation motion and interactions require efficient interatomic potentials which accurately describe the dislocation energies, core structures, and motion. In this work we develop an empirical potential for Mo which predicts the ideal shear strength, generalized stacking fault en- ergies, energies of dislocations, and the Peierls stress and core structure of the � 111� /2 screw dislocation. The potential form is given by the modified embedded-atom method (MEAM) and the potential parameters are optimized usingabinitio energies, lattice parameters, forces, and elastic constants. Section II describes the calculations for the DFT database, the functional form of the MEAM potential, and the optimization of the potential parameters to the DFT database. The accuracy of the potential for structural, elastic, and defect properties is verified in Sec. III by comparison to DFT results and experiments. A genetic algorithm search of the energy landscape of the MEAM potential confirms that the potential reproduces the correct bcc ground state and predicts several low-energy metastable structures whose energies agree well with DFT results. Results of the MEAM potential for formation energies of point defects, phonon dispersion, thermal expansion, surface energies, ideal shear strength, and generalized stacking faults for the MEAM potential closely match DFT results and available experimental data. In Sec. IV we apply the potential to determine energies and Peierls stresses of the screw and edge dislocation in bcc Mo. The results show that the MEAM potential accurately describes the structural and mechanical properties of Mo and should be applicable to simulate the motion of dislocations and the plastic deformation of Mo. read less
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
The letter grade A was assigned because the normalized error in the computation was 6.61348e-09 compared with a machine precision of 2.22045e-16. The letter grade was based on 'score=log10(error/eps)', with ranges A=[0, 7.5], B=(7.5, 10.0], C=(10.0, 12.5], D=(12.5, 15.0), F>15.0. 'A' is the best grade, and 'F' indicates failure.
vc-forces-numerical-derivative
consistency
Forces computed by the model agree with numerical derivatives of the energy; see full description.
Unable to perform verification check due to an error.
vc-dimer-continuity-c1
informational
The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description.
Model energy and forces are invariant with respect to rigid-body motion (translation and rotation) for all configurations the model was able to compute.
vc-objectivity
informational
Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description.
All threads give identical results for tested case. Model appears to be thread-safe.
vc-thread-safe
mandatory
The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description.
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
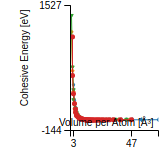
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Given an xyz file corresponding to a finite cluster of atoms, this Test Driver computes the total potential energy and atomic forces on the configuration. The positions are then relaxed using conjugate gradient minimization and the final positions and forces are recorded. These results are primarily of interest for training machine-learning algorithms.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Calculates lattice constant of hexagonal bulk structures at zero temperature and pressure by using simplex minimization to minimize the potential energy.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Given an extended xyz file corresponding to a non-orthogonal periodic box of atoms, use LAMMPS to compute the total potential energy and atomic forces.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
 MEAM_LAMMPS_DuLenoskyHennig_2011_Si__MO_883726743759_002
MEAM_LAMMPS_DuLenoskyHennig_2011_Si__MO_883726743759_002