 MEAM_LAMMPS_LeeLeeKim_2006_FeN__MO_432861766738_002
MEAM_LAMMPS_LeeLeeKim_2006_FeN__MO_432861766738_002
| Title
A single sentence description.
|
MEAM Potential for the Fe-N system developed by Lee, Lee and Kim. (2006) v002 |
|---|---|
| Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
|
The potential parameters were determined by fitting to the dilute heat of solution and migration energy of nitrogen atoms, the vacancy–nitrogen binding energy and its configuration in body-centered cubic iron, and the enthalpy of formation and lattice parameter of Fe4N. The potential reproduces very well the known physical properties of nitrogen as an interstitial solute element in body- and face-centered cubic iron and of various nitrides. In the original paper (Lee et al., Acta Materialia, 54(17), 2006), the similarity and difference between nitrogen and carbon as equally important interstitial elements in iron are also examined. The applicability of the potential to atomistic approaches for investigating interactions between nitrogen atoms and other defects such as vacancies, dislocations, and grain boundaries, and also for investigating the effects of nitrogen on various deformation and mechanical behaviors of iron is demonstrated. |
| Species
The supported atomic species.
| Fe, N |
| Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
|
None |
| Content Origin | http://cmse.postech.ac.kr/home_2nnmeam |
| Contributor |
Hyeon-Seok Do |
| Maintainer |
Hyeon-Seok Do |
| Developer |
Byeong-Joo Lee Tae-Ho Lee Sung-Joon Kim |
| Published on KIM | 2023 |
| How to Cite |
This Model originally published in [1] is archived in OpenKIM [2-5]. [1] Lee B-J, Lee T-H, Kim S-J. A modified embedded-atom method interatomic potential for the Fe–N system: a comparative study with the Fe–C system. Acta materialia. 2006;54(17):4597–607. doi:10.1016/j.actamat.2006.06.003 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information. [2] Lee B-J, Lee T-H, Kim S-J. MEAM Potential for the Fe-N system developed by Lee, Lee and Kim. (2006) v002. OpenKIM; 2023. doi:10.25950/22f2df58 [3] Afshar Y, Hütter S, Rudd RE, Stukowski A, Tipton WW, Trinkle DR, et al. The modified embedded atom method (MEAM) potential v002. OpenKIM; 2023. doi:10.25950/ee5eba52 [4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6 [5] Elliott RS, Tadmor EB. Knowledgebase of Interatomic Models (KIM) Application Programming Interface (API). OpenKIM; 2011. doi:10.25950/ff8f563a Click here to download the above citation in BibTeX format. |
| Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on. The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel. The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied. The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP). Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis. OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm. |
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information. 
70 Citations (44 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) X. Wan et al., “Iron atom–cluster interactions increase activity and improve durability in Fe–N–C fuel cells,” Nature Communications. 2022. link Times cited: 84 USED (high confidence) A. Karimi and M. Auinger, “Nitrogen Diffusion in Vacancy-Rich Ferrite and Austenite, from First Principles to Applications,” EngRN: Process Engineering (Topic). 2021. link Times cited: 1 Abstract: This work contains a systematic study of the diffusion of ni… read more USED (high confidence) A. T. AlMotasem, M. Posselt, and T. Polcar, “Deformation Behavior of Nanocrystalline Body-Centered Cubic Iron with Segregated, Foreign Interstitial: A Molecular Dynamics Study,” Materials. 2020. link Times cited: 3 Abstract: In the present work, modified embedded atom potential and la… read more USED (high confidence) J. Kong, D. Du, A. Song, F. Zhang, and W. Huang, “Surface Physical and Chemical Modification of Pure Iron by Using Atmospheric Pressure Plasma Treatment,” Materials. 2020. link Times cited: 2 Abstract: To investigate the mechanism of surface modification of pure… read more USED (high confidence) J. Wang et al., “Greater diffusion rate of carbon atoms from nonlinear migration in micro-cell and spatially heterogeneous stable states in FCC iron,” Journal of Materials Science. 2018. link Times cited: 2 USED (high confidence) M. Mohammadzadeh and R. Mohammadzadeh, “Effect of interstitial and substitution alloying elements on the intrinsic stacking fault energy of nanocrystalline fcc-iron by atomistic simulation study,” Applied Physics A. 2017. link Times cited: 13 USED (high confidence) F. Ulomek and V. Mohles, “Molecular dynamics simulations of grain boundary mobility in Al, Cu and γ-Fe using a symmetrical driving force,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 13 Abstract: We present a new artificial driving force for the determinat… read more USED (high confidence) M. H. Wu, X. H. Liu, J. F. Gu, and Z. H. Jin, “First-principles simulations of iron with nitrogen: from surface adsorption to bulk diffusion,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 14 Abstract: Adsorption, absorption and diffusion pathways of nitrogen ar… read more USED (low confidence) M. Billah, M. S. Rabbi, K. A. Rahman, and P. Acar, “Temperature and strain rate dependent tensile properties of Titanium carbide/nitride MXenes,” Materials Chemistry and Physics. 2023. link Times cited: 0 USED (low confidence) S. Risal et al., “Development of the RF-MEAM Interatomic Potential for the Fe-C System to Study the Temperature-Dependent Elastic Properties,” Materials. 2023. link Times cited: 0 Abstract: One of the major impediments to the computational investigat… read more USED (low confidence) L. Liu, G. Wang, Y. Xiao, Z. Zhao, and Z. Yang, “Molecular dynamics simulation of Cr-N clusters formation in high nitrogen austenitic stainless steel,” Scripta Materialia. 2023. link Times cited: 0 USED (low confidence) J. Zhu and J. P. Wang, “Simulation of thermal decomposition of γ′-Fe4N using molecular dynamics method,” AIP Advances. 2023. link Times cited: 0 Abstract: α″-Fe16N2 is a promising environmentally friendly rare-earth… read more USED (low confidence) S. Manda, S. Kumar, K. Pal, A. Bhattacharyya, A. Panwar, and I. Samajdar, “Correction: Snoek-Dominated Internal Friction Response in bcc Steel: Relating Experiments With a Multi-scale Atomistic Computational Framework,” Metallurgical and Materials Transactions A. 2022. link Times cited: 1 USED (low confidence) R. Salloom, M. Baskes, and S. G. Srinivasan, “Atomic level simulations of the phase stability and stacking fault energy of FeCoCrMnSi high entropy alloy,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 4 Abstract: High entropy alloys (HEAs) have many promising properties be… read more USED (low confidence) A. S. M. Miraz, N. Dhariwal, W. Meng, B. Ramachandran, and C. Wick, “Development and application of interatomic potentials to study the stability and shear strength of Ti/TiN and Cu/TiN interfaces,” Materials & Design. 2020. link Times cited: 15 USED (low confidence) I. Aslam et al., “Thermodynamic and kinetic behavior of low-alloy steels: An atomic level study using an Fe-Mn-Si-C modified embedded atom method (MEAM) potential,” Materialia. 2019. link Times cited: 12 USED (low confidence) S. Ding and X.-qiang Wang, “A systematic study on the MEAM interatomic potentials of the transition metal nitrides TMNs (TM=Ti, V, Cr, Fe) binary systems,” Journal of Alloys and Compounds. 2019. link Times cited: 10 USED (low confidence) K. Tong, F. Ye, and Y. K. Wang, “Short-range ordered structure and phase stability of supersaturated nitrided layer on austenitic stainless steel,” Acta Materialia. 2019. link Times cited: 15 USED (low confidence) Y. Cheng et al., “Dynamic and structural heterogeneity in undercooled miscible and immiscible metallic liquid,” Journal of Alloys and Compounds. 2019. link Times cited: 4 USED (low confidence) N. Razmara and R. Mohammadzadeh, “Effect of nitrogen content on the crack growth behavior in the Fe-N alloy at high temperatures via molecular dynamics simulations,” Theoretical and Applied Fracture Mechanics. 2018. link Times cited: 4 USED (low confidence) F. Ye, K. Tong, Y. K. Wang, Z. Li, and F. Zhou, “First-principles study of interaction between vacancies and nitrogen atoms in fcc iron,” Computational Materials Science. 2018. link Times cited: 8 USED (low confidence) P. Liu, X. Han, D. Sun, and Q. Wang, “Development and application of a ternary Ti-Al-N interatomic potential for Ti2AlN/TiAl composite,” Journal of Alloys and Compounds. 2018. link Times cited: 20 USED (low confidence) K. M. Wang, H. Wu, C. Wang, and X. Li, “Study on Preparation and MD Simulation of Nano Fe4N,” NANO. 2017. link Times cited: 2 Abstract: Nano Fe4N powder has been successfully prepared by the combi… read more USED (low confidence) R. Mohammadzadeh, N. Razmara, and F. Razmara, “Molecular dynamics study of strain-induced diffusivity of nitrogen in pure iron nanocrystalline,” Physica A-statistical Mechanics and Its Applications. 2016. link Times cited: 3 USED (low confidence) M. Mohammadzadeh and R. Mohammadzadeh, “WITHDRAWN: Molecular dynamics study on the effect of interstitial and substitutional alloying elements on stacking fault energies of fcc iron,” Physica B-condensed Matter. 2016. link Times cited: 0 USED (low confidence) F. Ye, C. Yin, K. Tong, C. Zhang, and W. Liu, “Structural evolution of vacancy clusters by combination of cluster units in alpha-iron,” Materials Research Innovations. 2014. link Times cited: 0 Abstract: The development of vacancy clusters in α-Fe is essential for… read more USED (low confidence) D. Sangiovanni, D. Edström, L. Hultman, I. Petrov, J. Greene, and V. Chirita, “Ab initio and classical molecular dynamics simulations of N2 desorption from TiN(001) surfaces,” Surface Science. 2014. link Times cited: 52 USED (low confidence) R. Neugebauer, R. Wertheim, and U. Semmler, “THE ATOMIC FINITE ELEMENT METHOD AS A BRIDGE BETWEEN MOLECULAR DYNAMICS AND CONTINUUM MECHANICS,” Journal of Multiscale Modelling. 2011. link Times cited: 6 Abstract: On cutting tools for high performance cutting (HPC) processe… read more USED (low confidence) A. L. Nikolaev and T. Kurennykh, “On the interaction between radiation-induced defects and foreign interstitial atoms in α-iron,” Journal of Nuclear Materials. 2011. link Times cited: 14 USED (low confidence) X. Zhen, L. Yuan, D. Shan, and B. Guo, “A molecular dynamics simulation of TiN film growth on TiN(0 0 1),” Computational Materials Science. 2011. link Times cited: 22 USED (low confidence) B.-J. Lee, W. Ko, H.-K. Kim, and E.-H. Kim, “The modified embedded-atom method interatomic potentials and recent progress in atomistic simulations,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2010. link Times cited: 138 USED (low confidence) Y.-M. Kim, N. Kim, and B.-J. Lee, “Atomistic Modeling of pure Mg and Mg―Al systems,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2009. link Times cited: 117 USED (low confidence) H. Yu and F.-jiu Sun, “A modified embedded atom method interatomic potential for the Ti―N system,” Physica B-condensed Matter. 2009. link Times cited: 9 USED (low confidence) S. R. Hosseini and F. Ashrafizadeh, “Accurate measurement and evaluation of the nitrogen depth profile in plasma nitrided iron,” Vacuum. 2009. link Times cited: 24 USED (low confidence) I. Sa and B.-J. Lee, “Modified embedded-atom method interatomic potentials for the Fe-Nb and Fe-Ti binary systems,” Scripta Materialia. 2008. link Times cited: 47 USED (low confidence) H.-K. Kim, W. Jung, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for the Fe–Ti–C and Fe–Ti–N ternary systems,” Acta Materialia. 2008. link Times cited: 120 USED (low confidence) E.-H. Kim, Y.-H. Shin, and B.-J. Lee, “A modified embedded-atom method interatomic potential for Germanium,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2008. link Times cited: 85 USED (low confidence) J. Jang, B.-J. Lee, and J.-H. Hong, “Influence of Cu, Cr and C on the irradiation defect in Fe: A molecular dynamics simulation study,” Journal of Nuclear Materials. 2008. link Times cited: 14 USED (low confidence) Y.-M. Kim and B.-J. Lee, “A modified embedded-atom method interatomic potential for the Cu–Zr system,” Journal of Materials Research. 2004. link Times cited: 65 USED (low confidence) J. Zhu, G. Guo, and J. P. Wang, “Study of γ′-F4N Annealing Process Through Molecular Dynamics Modeling,” TMS 2022 151st Annual Meeting & Exhibition Supplemental Proceedings. 2022. link Times cited: 0 USED (low confidence) Z. Xu, Q. Zeng, L. Yuan, Y. Qin, M. Chen, and D. Shan, “Molecular dynamics study of the interactions of incident N or Ti atoms with the TiN(001) surface,” Applied Surface Science. 2016. link Times cited: 17 USED (low confidence) W. Joost, “Modeling the Influence of Phase Boundaries and Oxygen Interstitials on the Nucleation and Growth of Deformation Twins in the Alpha-Phase of Titanium Alloys.” 2015. link Times cited: 0 Abstract: Title of dissertation: MODELING THE INFLUENCE OF PHASE BOUND… read more USED (low confidence) T. Prasanthi, C. Sudha, S. Saroja, and M. Vijayalakshmi, “Simulation of nitrogen diffusion in Ni vis-à-vis Fe – Identification of better structural material for neutron detectors,” Results in physics. 2014. link Times cited: 3 USED (low confidence) Y.-M. Kim, Y.-H. Shin, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for pure Mn and the Fe–Mn system,” Acta Materialia. 2009. link Times cited: 64 NOT USED (low confidence) Y. Lei et al., “An Embedded-Atom Method Potential for studying the properties of Fe-Pb solid-liquid interface,” Journal of Nuclear Materials. 2022. link Times cited: 1 NOT USED (low confidence) Y. Sun, H. Wang, Z. He, B. Qiao, and X. Chen, “Role of initial stage nitridation on the mechanical properties of an α-Fe(100) nanofilm in NH3.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 2 Abstract: Nitrogen is one of the most significant non-native interstit… read more NOT USED (low confidence) K. Hyodo, S. Munetoh, T. Tsuchiyama, and S. Takaki, “Empirical interatomic potential for Fe-N binary system based on Finnis–Sinclair potential,” Computational Materials Science. 2020. link Times cited: 7 NOT USED (low confidence) X. Chong, Y.-hua Jiang, and J. Feng, “Exploring the intrinsic ductile metastable Fe-C compounds: Complex chemical bonds, anisotropic elasticity and variable thermal expansion,” Journal of Alloys and Compounds. 2018. link Times cited: 27 NOT USED (low confidence) B. Narayanan et al., “Development of a Modified Embedded Atom Force Field for Zirconium Nitride Using Multi-Objective Evolutionary Optimization,” Journal of Physical Chemistry C. 2016. link Times cited: 23 Abstract: Zirconium nitride (ZrN) exhibits exceptional mechanical, che… read more NOT USED (low confidence) B. Liu, H. Zhang, J. Tao, Z. R. Liu, X. Chen, and Y. Zhang, “Development of a second-nearest-neighbor modified embedded atom method potential for silicon–phosphorus binary system,” Computational Materials Science. 2016. link Times cited: 8 NOT USED (high confidence) M. Zacate, “Modified embedded-atom method potential for cadmium,” Hyperfine Interactions. 2019. link Times cited: 0 NOT USED (high confidence) P. Srinivasan, A. Duff, T. Mellan, M. Sluiter, L. Nicola, and A. Simone, “The effectiveness of reference-free modified embedded atom method potentials demonstrated for NiTi and NbMoTaW,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 15 Abstract: One of the effective potentials that has proven to be very v… read more NOT USED (high confidence) G. Almyras, D. Sangiovanni, and K. Sarakinos, “Semi-Empirical Force-Field Model for the Ti1−xAlxN (0 ≤ x ≤ 1) System,” Materials. 2019. link Times cited: 20 Abstract: We present a modified embedded atom method (MEAM) semi-empir… read more NOT USED (high confidence) D. Dickel, C. Barrett, R. Cariño, M. Baskes, and M. Horstemeyer, “Mechanical instabilities in the modeling of phase transitions of titanium,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 17 Abstract: In this paper, we demonstrate that previously observed β to … read more NOT USED (high confidence) S. Liu et al., “Refinement effect of TiC on ferrite by molecular statics/dynamics simulations and first-principles calculations,” Journal of Alloys and Compounds. 2018. link Times cited: 3 NOT USED (high confidence) K. Tong, F. Ye, M. Gao, M. Lei, and C. Zhang, “Interatomic potential for Fe–Cr–Ni–N system based on the second nearest-neighbor modified embedded-atom method,” Molecular Simulation. 2016. link Times cited: 7 Abstract: The interatomic potential for Fe–Cr–Ni–N system based on the… read more NOT USED (high confidence) C. P. Chui, W. Liu, Y. Xu, and Y. Zhou, “Molecular Dynamics Simulation of Iron — A Review.” 2015. link Times cited: 3 Abstract: Molecular dynamics (MD) is a technique of atomistic simulati… read more NOT USED (high confidence) M. H. Wu, X. Liu, J. Gu, and Z. Jin, “DFT study of nitrogen–vacancy complexions in (fcc) Fe,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 9 Abstract: Formation energies of nitrogen (N)/vacancy (v) monomers, N–N… read more NOT USED (high confidence) K. Henriksson, C. Björkas, and K. Nordlund, “Atomistic simulations of stainless steels: a many-body potential for the Fe–Cr–C system,” Journal of Physics: Condensed Matter. 2013. link Times cited: 62 Abstract: Stainless steels found in real-world applications usually ha… read more NOT USED (high confidence) Y. You and M. Yan, “Many-body potential for nitrogen in α-iron,” Philosophical Magazine Letters. 2012. link Times cited: 3 Abstract: N atom is one of the most frequent foreign interstitial atom… read more NOT USED (high confidence) J. Kang, B. Hosseinkhani, and P. Rivera-Diaz-Del-Castillo, “Rolling contact fatigue in bearings: multiscale overview,” Materials Science and Technology. 2012. link Times cited: 77 Abstract: For over a century, rolling contact fatigue in bearings has … read more NOT USED (high confidence) S. R. Hosseini, F. Ashrafizadeh, and A. Kermanpur, “Analytical–experimental approach for accurate depth profile evaluation of diffusion zone in plasma nitrided iron,” Surface Engineering. 2011. link Times cited: 3 Abstract: Nitrogen depth profile of diffusion zone in plasma nitrided … read more NOT USED (high confidence) H.-K. Kim, W. Jung, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for the Nb-C, Nb-N, Fe-Nb-C, and Fe-Nb-N systems,” Journal of Materials Research. 2010. link Times cited: 21 Abstract: Modified embedded-atom method (MEAM) interatomic potentials … read more NOT USED (high confidence) B.-J. Lee, “A Semi-Empirical Atomistic Approach in Materials Research,” Journal of Phase Equilibria and Diffusion. 2009. link Times cited: 3 NOT USED (high confidence) E. C. Do, Y.-H. Shin, and B.-J. Lee, “Atomistic modeling of III–V nitrides: modified embedded-atom method interatomic potentials for GaN, InN and Ga1−xInxN,” Journal of Physics: Condensed Matter. 2009. link Times cited: 25 Abstract: Modified embedded-atom method (MEAM) interatomic potentials … read more NOT USED (high confidence) L. A. Bol’shov, V. Bogdanov, and V. A. Gorbunov, “Thermodynamic analysis of the data on the solubility of nitrogen in gamma iron at a high pressure,” The Physics of Metals and Metallography. 2009. link Times cited: 2 NOT USED (high confidence) B.-J. Lee, J. Shim, and J. Kwon, “Effects of crystallographic structure and Cr on the rate of void nucleation in BCC Fe: An atomistic simulation study,” Metals and Materials International. 2008. link Times cited: 1 Abstract: Atomistic Monte Carlo simulations based on modified embedded… read more NOT USED (high confidence) S. Valone and M. Baskes, “Self-Irradiation Cascade Simulations in Plutonium Metal: Model Behavior at High Energy,” Journal of Computer-Aided Materials Design. 2007. link Times cited: 10 NOT USED (high confidence) N. Razmara and R. Mohammadzadeh, “Molecular dynamics study of nitrogen diffusion in nanocrystalline iron,” Journal of Molecular Modeling. 2016. link Times cited: 6 NOT USED (high confidence) W. Joost, S. Ankem, and M. Kuklja, “A modified embedded atom method potential for the titanium–oxygen system,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 16 Abstract: Small concentrations of impurity atoms can affect the behavi… read more |
| Funding | Not available |
| Short KIM ID
The unique KIM identifier code.
| MO_432861766738_002 |
| Extended KIM ID
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
| MEAM_LAMMPS_LeeLeeKim_2006_FeN__MO_432861766738_002 |
| DOI |
10.25950/22f2df58 https://doi.org/10.25950/22f2df58 https://commons.datacite.org/doi.org/10.25950/22f2df58 |
| KIM Item Type
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
| Portable Model using Model Driver MEAM_LAMMPS__MD_249792265679_002 |
| Driver | MEAM_LAMMPS__MD_249792265679_002 |
| KIM API Version | 2.2 |
| Potential Type | meam |
| Previous Version | MEAM_LAMMPS_LeeLeeKim_2006_FeN__MO_432861766738_001 |
| Grade | Name | Category | Brief Description | Full Results | Aux File(s) |
|---|---|---|---|---|---|
| P | vc-species-supported-as-stated | mandatory | The model supports all species it claims to support; see full description. |
Results | Files |
| P | vc-periodicity-support | mandatory | Periodic boundary conditions are handled correctly; see full description. |
Results | Files |
| P | vc-permutation-symmetry | mandatory | Total energy and forces are unchanged when swapping atoms of the same species; see full description. |
Results | Files |
| A | vc-forces-numerical-derivative | consistency | Forces computed by the model agree with numerical derivatives of the energy; see full description. |
Results | Files |
| F | vc-dimer-continuity-c1 | informational | The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description. |
Results | Files |
| P | vc-objectivity | informational | Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description. |
Results | Files |
| P | vc-inversion-symmetry | informational | Total energy is unchanged and forces change sign when inverting a configuration through the origin; see full description. |
Results | Files |
| P | vc-memory-leak | informational | The model code does not have memory leaks (i.e. it releases all allocated memory at the end); see full description. |
Results | Files |
| P | vc-thread-safe | mandatory | The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description. |
Results | Files |
| P | vc-unit-conversion | mandatory | The model is able to correctly convert its energy and/or forces to different unit sets; see full description. |
Results | Files |
BCC Lattice Constant
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Cohesive Energy Graph
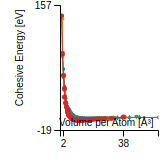
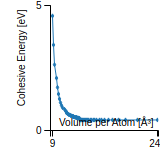
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
Diamond Lattice Constant
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
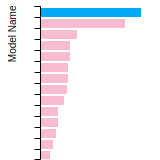
Dislocation Core Energies
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
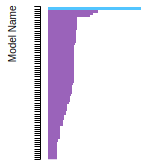
(No matching species)FCC Elastic Constants
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
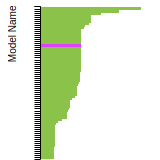
FCC Lattice Constant
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.

FCC Stacking Fault Energies
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
(No matching species)FCC Surface Energies
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
(No matching species)SC Lattice Constant
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Cubic Crystal Basic Properties Table
Species: FeSpecies: N
Creators:
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/64cb38c5
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Cohesive energy versus lattice constant curve for bcc Fe v004 | view | 7141 | |
| Cohesive energy versus lattice constant curve for diamond Fe v004 | view | 6674 | |
| Cohesive energy versus lattice constant curve for diamond N v004 | view | 7141 | |
| Cohesive energy versus lattice constant curve for fcc Fe v004 | view | 7320 | |
| Cohesive energy versus lattice constant curve for sc Fe v004 | view | 6604 |
Creators: Junhao Li and Ellad Tadmor
Contributor: tadmor
Publication Year: 2019
DOI: https://doi.org/10.25950/5853fb8f
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for bcc Fe at zero temperature v006 | view | 46627 | |
| Elastic constants for diamond Fe at zero temperature v001 | view | 47681 | |
| Elastic constants for diamond N at zero temperature v001 | view | 59268 | |
| Elastic constants for fcc Fe at zero temperature v006 | view | 44025 | |
| Elastic constants for sc Fe at zero temperature v006 | view | 24000 |
Creators:
Contributor: ilia
Publication Year: 2024
DOI: https://doi.org/10.25950/2f2c4ad3
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Creators:
Contributor: brunnels
Publication Year: 2022
DOI: https://doi.org/10.25950/2c59c9d6
Computes grain boundary energy for a range of tilt angles given a crystal structure, tilt axis, and material.
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/2765e3bf
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium zero-temperature lattice constant for bcc Fe v007 | view | 13258 | |
| Equilibrium zero-temperature lattice constant for diamond Fe v007 | view | 13109 | |
| Equilibrium zero-temperature lattice constant for diamond N v007 | view | 13099 | |
| Equilibrium zero-temperature lattice constant for fcc Fe v007 | view | 11338 | |
| Equilibrium zero-temperature lattice constant for sc Fe v007 | view | 10969 |
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/c339ca32
Calculates lattice constant of hexagonal bulk structures at zero temperature and pressure by using simplex minimization to minimize the potential energy.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium lattice constants for hcp Fe v005 | view | 113817 | |
| Equilibrium lattice constants for hcp N v005 | view | 118750 |
Creators:
Contributor: mjwen
Publication Year: 2024
DOI: https://doi.org/10.25950/9d9822ec
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Linear thermal expansion coefficient of bcc Fe at 293.15 K under a pressure of 0 MPa v002 | view | 1506349 |
Creators: Matt Bierbaum
Contributor: mattbierbaum
Publication Year: 2019
DOI: https://doi.org/10.25950/6c43a4e6
Calculates the surface energy of several high symmetry surfaces and produces a broken-bond model fit. In latex form, the fit equations are given by:
E_{FCC} (\vec{n}) = p_1 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (2 ( |x+ 2y + z| + |x+2y-z| + |x-2y + z| + |x-2y-z| + |2x+y+z| + |2x+y-z| +|2x-y+z| +|2x-y-z| +|x+y+2z| +|x+y-2z| +|x-y+2z| +|x-y-2z| ) + c
E_{BCC} (\vec{n}) = p_1 (6 \left( | x+y+z| + |x+y-z| + |-x+y-z| + |x-y+z| \right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) +c.
In Python, these two fits take the following form:
def BrokenBondFCC(params, index):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*(abs(x+2*y+z) + abs(x+2*y-z) +abs(x-2*y+z) +abs(x-2*y-z) + abs(2*x+y+z) +abs(2*x+y-z) +abs(2*x-y+z) +abs(2*x-y-z) + abs(x+y+2*z) +abs(x+y-2*z) +abs(x-y+2*z) +abs(x-y-2*z))+params[3]
def BrokenBondBCC(params, x, y, z):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*6*(abs(x+y+z) + abs(x-y-z) + abs(x-y+z) + abs(x+y-z)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[3]
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Broken-bond fit of high-symmetry surface energies in bcc Fe v004 | view | 65902 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/fca89cea
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Monovacancy formation energy and relaxation volume for bcc Fe | view | 299635 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/c27ba3cd
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Vacancy formation and migration energy for bcc Fe | view | 4206896 |
| Test | Error Categories | Link to Error page |
|---|---|---|
| Elastic constants for hcp Fe at zero temperature v004 | other | view |
| Elastic constants for hcp N at zero temperature v004 | other | view |
EquilibriumCrystalStructure__TD_457028483760_002
GrainBoundaryCubicCrystalSymmetricTiltRelaxedEnergyVsAngle__TD_410381120771_003
| Test | Error Categories | Link to Error page |
|---|---|---|
| Relaxed energy as a function of tilt angle for a 112 symmetric tilt grain boundary in fcc Fe v001 | other | view |
LatticeConstantCubicEnergy__TD_475411767977_007
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium zero-temperature lattice constant for bcc N v007 | other | view |
| Equilibrium zero-temperature lattice constant for fcc N v007 | other | view |
| Equilibrium zero-temperature lattice constant for sc N v007 | other | view |
SurfaceEnergyCubicCrystalBrokenBondFit__TD_955413365818_004
| Test | Error Categories | Link to Error page |
|---|---|---|
| Broken-bond fit of high-symmetry surface energies in bcc Fe v004 | other | view |
| MEAM_LAMMPS_LeeLeeKim_2006_FeN__MO_432861766738_002.txz | Tar+XZ | Linux and OS X archive |
| MEAM_LAMMPS_LeeLeeKim_2006_FeN__MO_432861766738_002.zip | Zip | Windows archive |
This Model requires a Model Driver. Archives for the Model Driver MEAM_LAMMPS__MD_249792265679_002 appear below.
| MEAM_LAMMPS__MD_249792265679_002.txz | Tar+XZ | Linux and OS X archive |
| MEAM_LAMMPS__MD_249792265679_002.zip | Zip | Windows archive |
Parameter Choices in the SW Potential
In the original Stillinger-Weber paper (SW85: PRB 31:5262, 1985) it is stated that in order to obtain the correct “atomization energy” (cohesive energy) the following choice for epsilon must be made (see Eqn. (2.9) in [SW85]):
epsilon = 50 kcal/mol = 3.4723E-12 erg/atom
Unfortunately, there appears to be an error in the unit conversion here. (There is also an indeterminacy associated with the “kcal” unit which can mean different things.) The kcal and erg values have led to two different values for epsilon being used in articles that cite [SW85].
(a) If the kcal number is selected (assuming that S&W meant the thermochemical kcal unit), then epsilon = 2.1682 eV. This can be seen from the following conversion (which uses NIST conversion factors):
(50 kcal_th/mol)/(6.02214129 mol^-1) = 8.30269461E-23 kcal_th
8.30269461E-23 kcal_th x 4.184E+03 (J/kcal_th) = 3.47384742E-19 J
3.47384742E-19 J x 6.24150934E+18 (eV/J) = 2.168205112 eV
(b) If the erg number is selected, then epsilon = 2.1672 eV, which follows from:
3.4723E-12 erg x 6.24150934E+11 (eV/erg) = 2.16723929 eV
As noted above, both values have been used in simulations that cite the original [SW85] paper. However, it appears that S&W intended to use the 50 kcal_th/mol value since they refer to this number more than once in the paper. (See for example discussion after Eqn. (8.1) in [SW85].) Therefore we argue that the appropriate choice is
epsilon = 8.30269461E-23 kcal_th
or in eV units (reduced to 5-digit significant digits):
epsilon = 2.1682 eV
Note that in the Si.sw parameterization for this potential distributed with LAMMPS, a value of epsilon=2.1683 is used, which appears to be due to slightly different unit conversion.
Another source of confusion is that in [SW85] the potential is fitted to an incorrect value for the cohesive energy of silicon. This is corrected by Balamane in a 1992 paper. See https://openkim.org/cite/MO_113686039439_004 for more details.
SW Functions
See the Stillinger-Weber Model driver page (linked above) for the definition of the model and its functions. The graphs below are for the Stillinger-Weber parametrization for silicon.
The graph of function
The contour plot of the function
Login to edit Wiki content