 DUNN_WenTadmor_2019v2_C__MO_956135237832_000
DUNN_WenTadmor_2019v2_C__MO_956135237832_000
| Title
A single sentence description.
|
Dropout uncertainty neural network (DUNN) potential for condensed-matter carbon systems developed by Wen and Tadmor (2019) v000 |
|---|---|
| Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
|
A dropout uncertainty neural network (DUNN) potential for condensed-matter carbon systems with a dropout ratio of 0.2. This is an ensemble model consisting of 100 different network structures obtained by dropout. Before dropout, there are three hidden layers each containing 128 neurons; each neuron in the hidden layers has probability 0.2 of being removed from the network. By default, the model will run in the 'mean' mode where the output energy, forces, and virial are obtained by averaging over the 100 ensembles. If desired, one can set the 'active_member_id' to '0' to use the fully-connected structure or to '1, 2, ..., 100' to use a single ensemble member. When multiple ensemble members are used, the ensemble average of the energy and forces are what are ultimately returned for a given configuration. |
| Species
The supported atomic species.
| C |
| Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
|
None |
| Contributor |
Mingjian Wen |
| Maintainer |
Mingjian Wen |
| Developer |
Mingjian Wen Ellad B. Tadmor |
| Published on KIM | 2019 |
| How to Cite |
This Model originally published in [1] is archived in OpenKIM [2-5]. [1] Wen M, Tadmor EB. Uncertainty quantification in molecular simulations with dropout neural network potentials. npj Computational Materials. 2020;6(1). doi:10.1038/s41524-020-00390-8 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information. [2] Wen M, Tadmor EB. Dropout uncertainty neural network (DUNN) potential for condensed-matter carbon systems developed by Wen and Tadmor (2019) v000. OpenKIM; 2019. doi:10.25950/5cdb2c9f [3] Wen M, Tadmor EB. A dropout uncertainty neural network (DUNN) model driver v000. OpenKIM; 2019. doi:10.25950/9573ca43 [4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6 [5] Elliott RS, Tadmor EB. Knowledgebase of Interatomic Models (KIM) Application Programming Interface (API). OpenKIM; 2011. doi:10.25950/ff8f563a Click here to download the above citation in BibTeX format. |
| Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on. The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel. The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied. The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP). Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis. OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm. |
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information. 
39 Citations (6 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (low confidence) D. Huo et al., “Evaluation of pre-neutron-emission mass distributions in induced fission of typical actinides based on Monte Carlo dropout neural network,” The European Physical Journal A. 2023. link Times cited: 0 USED (low confidence) A. Catalysis et al., “Atomistic Insights into the Oxidation of Flat and Stepped Platinum Surfaces Using Large-Scale Machine Learning Potential-Based Grand-Canonical Monte Carlo,” ACS Catalysis. 2022. link Times cited: 7 Abstract: : Understanding catalyst surface structure changes under rea… read more USED (low confidence) D. Varivoda, R. Dong, S. S. Omee, and J. Hu, “Materials Property Prediction with Uncertainty Quantification: A Benchmark Study,” ArXiv. 2022. link Times cited: 9 Abstract: Uncertainty quantification (UQ) has increasing importance in… read more USED (low confidence) W. Li and C.-G. Yang, “Thermal transport properties of monolayer GeS and SnS: A comparative study based on machine learning and SW interatomic potential models,” AIP Advances. 2022. link Times cited: 5 Abstract: Phonon transport properties of two-dimensional materials can… read more USED (low confidence) M. Tsitsvero, “Learning inducing points and uncertainty on molecular data,” ArXiv. 2022. link Times cited: 0 Abstract: Uncertainty control and scalability to large datasets are th… read more USED (low confidence) T. Swinburne, “Uncertainty and anharmonicity in thermally activated dynamics,” Computational Materials Science. 2021. link Times cited: 4 NOT USED (low confidence) H. Sandström, M. Rissanen, J. Rousu, and P. Rinke, “Data-Driven Compound Identification in Atmospheric Mass Spectrometry.,” Advanced science. 2023. link Times cited: 0 Abstract: Aerosol particles found in the atmosphere affect the climate… read more NOT USED (low confidence) C. Hong et al., “Applications and training sets of machine learning potentials,” Science and Technology of Advanced Materials: Methods. 2023. link Times cited: 0 Abstract: ABSTRACT Recently, machine learning potentials (MLPs) have b… read more NOT USED (low confidence) J. A. Vita et al., “ColabFit exchange: Open-access datasets for data-driven interatomic potentials.,” The Journal of chemical physics. 2023. link Times cited: 2 Abstract: Data-driven interatomic potentials (IPs) trained on large co… read more NOT USED (low confidence) T. Rensmeyer, B. Craig, D. Kramer, and O. Niggemann, “High Accuracy Uncertainty-Aware Interatomic Force Modeling with Equivariant Bayesian Neural Networks,” ArXiv. 2023. link Times cited: 1 Abstract: Even though Bayesian neural networks offer a promising frame… read more NOT USED (low confidence) M. C. Venetos, M. Wen, and K. Persson, “Machine Learning Full NMR Chemical Shift Tensors of Silicon Oxides with Equivariant Graph Neural Networks,” The Journal of Physical Chemistry. a. 2023. link Times cited: 1 Abstract: The nuclear magnetic resonance (NMR) chemical shift tensor i… read more NOT USED (low confidence) L. O. AGBOLADE et al., “Recent advances in density functional theory approach for optoelectronics properties of graphene,” Heliyon. 2023. link Times cited: 1 NOT USED (low confidence) M. Wen, E. Spotte-Smith, S. M. Blau, M. J. McDermott, A. Krishnapriyan, and K. Persson, “Chemical reaction networks and opportunities for machine learning,” Nature Computational Science. 2023. link Times cited: 11 NOT USED (low confidence) S. Thaler, G. Doehner, and J. Zavadlav, “Scalable Bayesian Uncertainty Quantification for Neural Network Potentials: Promise and Pitfalls,” Journal of chemical theory and computation. 2022. link Times cited: 3 Abstract: Neural network (NN) potentials promise highly accurate molec… read more NOT USED (low confidence) Z. Shui, D. S. Karls, M. Wen, I. A. Nikiforov, E. Tadmor, and G. Karypis, “Injecting Domain Knowledge from Empirical Interatomic Potentials to Neural Networks for Predicting Material Properties,” ArXiv. 2022. link Times cited: 2 Abstract: For decades, atomistic modeling has played a crucial role in… read more NOT USED (low confidence) Y. Hu, J. Musielewicz, Z. W. Ulissi, and A. Medford, “Robust and scalable uncertainty estimation with conformal prediction for machine-learned interatomic potentials,” Machine Learning: Science and Technology. 2022. link Times cited: 14 Abstract: Uncertainty quantification (UQ) is important to machine lear… read more NOT USED (low confidence) Z. Fan et al., “GPUMD: A package for constructing accurate machine-learned potentials and performing highly efficient atomistic simulations.,” The Journal of chemical physics. 2022. link Times cited: 46 Abstract: We present our latest advancements of machine-learned potent… read more NOT USED (low confidence) M. Müser, S. Sukhomlinov, and L. Pastewka, “Interatomic potentials: achievements and challenges,” Advances in Physics: X. 2022. link Times cited: 12 Abstract: ABSTRACT Interatomic potentials approximate the potential en… read more NOT USED (low confidence) A. B. Li, L. Miroshnik, B. Rummel, G. Balakrishnan, S. Han, and T. Sinno, “A unified theory of free energy functionals and applications to diffusion,” Proceedings of the National Academy of Sciences of the United States of America. 2022. link Times cited: 3 Abstract: Significance The free energy functional is a central compone… read more NOT USED (low confidence) A. Thompson et al., “LAMMPS - A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales,” Computer Physics Communications. 2021. link Times cited: 2377 NOT USED (low confidence) J. Xu, X. Cao, and P. Hu, “Perspective on computational reaction prediction using machine learning methods in heterogeneous catalysis.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 24 Abstract: Heterogeneous catalysis plays a significant role in the mode… read more NOT USED (low confidence) A. M. Miksch, T. Morawietz, J. Kästner, A. Urban, and N. Artrith, “Strategies for the construction of machine-learning potentials for accurate and efficient atomic-scale simulations,” Machine Learning: Science and Technology. 2021. link Times cited: 45 Abstract: Recent advances in machine-learning interatomic potentials h… read more NOT USED (low confidence) X. Liu, Q. Wang, and J. Zhang, “Machine Learning Interatomic Force Fields for Carbon Allotropic Materials.” 2021. link Times cited: 0 NOT USED (high confidence) Y. Liu, X. He, and Y. Mo, “Discrepancies and error evaluation metrics for machine learning interatomic potentials,” npj Computational Materials. 2023. link Times cited: 1 NOT USED (high confidence) H. Zhai and J. Yeo, “Multiscale mechanics of thermal gradient coupled graphene fracture: A molecular dynamics study,” International Journal of Applied Mechanics. 2022. link Times cited: 2 Abstract: The thermo-mechanical coupling mechanism of graphene fractur… read more NOT USED (high confidence) A. J. W. Zhu, S. L. Batzner, A. Musaelian, and B. Kozinsky, “Fast Uncertainty Estimates in Deep Learning Interatomic Potentials,” The Journal of chemical physics. 2022. link Times cited: 15 Abstract: Deep learning has emerged as a promising paradigm to give ac… read more NOT USED (high confidence) K. S. Csizi and M. Reiher, “Universal QM/MM approaches for general nanoscale applications,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2022. link Times cited: 6 Abstract: Quantum mechanics/molecular mechanics (QM/MM) hybrid models … read more NOT USED (high confidence) S.-H. Lee, V. Olevano, and B. Sklénard, “A generalizable, uncertainty-aware neural network potential for GeSbTe with Monte Carlo dropout,” Solid-State Electronics. 2022. link Times cited: 2 NOT USED (high confidence) Y. Duan, M. N. Ridao, M. Eaton, and M. Bluck, “Non-intrusive semi-analytical uncertainty quantification using Bayesian quadrature with application to CFD simulations,” International Journal of Heat and Fluid Flow. 2022. link Times cited: 1 NOT USED (high confidence) Y. Kurniawan et al., “Bayesian, frequentist, and information geometric approaches to parametric uncertainty quantification of classical empirical interatomic potentials.,” The Journal of chemical physics. 2021. link Times cited: 6 Abstract: In this paper, we consider the problem of quantifying parame… read more NOT USED (high confidence) L. Fiedler, K. Shah, M. Bussmann, and A. Cangi, “Deep dive into machine learning density functional theory for materials science and chemistry,” Physical Review Materials. 2021. link Times cited: 18 Abstract: With the growth of computational resources, the scope of ele… read more NOT USED (high confidence) D. Wang et al., “A hybrid framework for improving uncertainty quantification in deep learning-based QSAR regression modeling,” Journal of Cheminformatics. 2021. link Times cited: 15 NOT USED (high confidence) Y. Duan, J. S. Ahn, M. D. Eaton, and M. Bluck, “Quantification of the uncertainty within a SAS-SST simulation caused by the unknown high-wavenumber damping factor,” Nuclear Engineering and Design. 2021. link Times cited: 2 NOT USED (high confidence) L. Kahle and F. Zipoli, “Quality of uncertainty estimates from neural network potential ensembles.,” Physical review. E. 2021. link Times cited: 11 Abstract: Neural network potentials (NNPs) combine the computational e… read more NOT USED (high confidence) M. Wen, Y. Afshar, R. Elliott, and E. Tadmor, “KLIFF: A framework to develop physics-based and machine learning interatomic potentials,” Comput. Phys. Commun. 2021. link Times cited: 12 NOT USED (high confidence) C.-gen Qian, B. Mclean, D. Hedman, and F. Ding, “A comprehensive assessment of empirical potentials for carbon materials,” APL Materials. 2021. link Times cited: 22 Abstract: Carbon materials and their unique properties have been exten… read more NOT USED (high confidence) M. Buze, T. Woolley, and L. A. Mihai, “A stochastic framework for atomistic fracture,” SIAM J. Appl. Math. 2021. link Times cited: 0 Abstract: We present a stochastic modeling framework for atomistic pro… read more NOT USED (high confidence) M. Wen and E. Tadmor, “Hybrid neural network potential for multilayer graphene,” Physical Review B. 2019. link Times cited: 40 Abstract: Monolayer and multilayer graphene are promising materials fo… read more NOT USED (high confidence) H. Fan, M. Ferianc, Z. Que, X. Niu, M. L. Rodrigues, and W. Luk, “Accelerating Bayesian Neural Networks via Algorithmic and Hardware Optimizations,” IEEE Transactions on Parallel and Distributed Systems. 2022. link Times cited: 4 Abstract: Bayesian neural networks (BayesNNs) have demonstrated their … read more |
| Funding | Not available |
| Short KIM ID
The unique KIM identifier code.
| MO_956135237832_000 |
| Extended KIM ID
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
| DUNN_WenTadmor_2019v2_C__MO_956135237832_000 |
| DOI |
10.25950/5cdb2c9f https://doi.org/10.25950/5cdb2c9f https://commons.datacite.org/doi.org/10.25950/5cdb2c9f |
| KIM Item Type
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
| Portable Model using Model Driver DUNN__MD_292677547454_000 |
| Driver | DUNN__MD_292677547454_000 |
| KIM API Version | 2.0 |
| Potential Type | dunn |
| Grade | Name | Category | Brief Description | Full Results | Aux File(s) |
|---|---|---|---|---|---|
| P | vc-periodicity-support | mandatory | Periodic boundary conditions are handled correctly; see full description. |
Results | Files |
| P | vc-dimer-continuity-c1 | informational | The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description. |
Results | Files |
| P | vc-memory-leak | informational | The model code does not have memory leaks (i.e. it releases all allocated memory at the end); see full description. |
Results | Files |
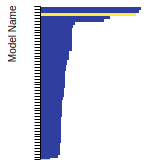
BCC Lattice Constant
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
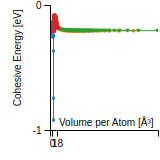
Cohesive Energy Graph
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
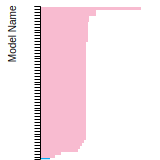
Diamond Lattice Constant
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Dislocation Core Energies
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
(No matching species)FCC Elastic Constants
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
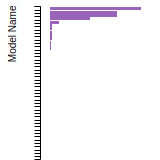
FCC Lattice Constant
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
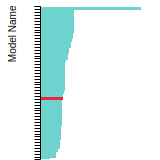
FCC Stacking Fault Energies
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
(No matching species)FCC Surface Energies
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
(No matching species)SC Lattice Constant
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
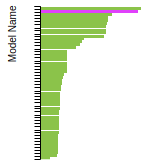
Cubic Crystal Basic Properties Table
Species: CCreators:
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/64cb38c5
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Cohesive energy versus lattice constant curve for bcc C v004 | view | 17982 | |
| Cohesive energy versus lattice constant curve for diamond C v004 | view | 1451579 | |
| Cohesive energy versus lattice constant curve for fcc C v004 | view | 152983 | |
| Cohesive energy versus lattice constant curve for sc C v004 | view | 16451 |
Creators: Junhao Li and Ellad Tadmor
Contributor: tadmor
Publication Year: 2019
DOI: https://doi.org/10.25950/5853fb8f
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for bcc C at zero temperature v006 | view | 43124 | |
| Elastic constants for diamond C at zero temperature v001 | view | 2867666 | |
| Elastic constants for fcc C at zero temperature v006 | view | 138737 | |
| Elastic constants for sc C at zero temperature v006 | view | 47559 |
Creators:
Contributor: ilia
Publication Year: 2024
DOI: https://doi.org/10.25950/2f2c4ad3
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/2765e3bf
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium zero-temperature lattice constant for bcc C v007 | view | 40626 | |
| Equilibrium zero-temperature lattice constant for diamond C v007 | view | 730404 | |
| Equilibrium zero-temperature lattice constant for fcc C v007 | view | 57408 | |
| Equilibrium zero-temperature lattice constant for sc C v007 | view | 32025 |
EquilibriumCrystalStructure__TD_457028483760_002
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium crystal structure and energy for C in AFLOW crystal prototype A_oC16_65_mn v002 | other | view |
| Equilibrium crystal structure and energy for C in AFLOW crystal prototype A_oC8_67_m v002 | other | view |
| DUNN_WenTadmor_2019v2_C__MO_956135237832_000.txz | Tar+XZ | Linux and OS X archive |
| DUNN_WenTadmor_2019v2_C__MO_956135237832_000.zip | Zip | Windows archive |
This Model requires a Model Driver. Archives for the Model Driver DUNN__MD_292677547454_000 appear below.
| DUNN__MD_292677547454_000.txz | Tar+XZ | Linux and OS X archive |
| DUNN__MD_292677547454_000.zip | Zip | Windows archive |