Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
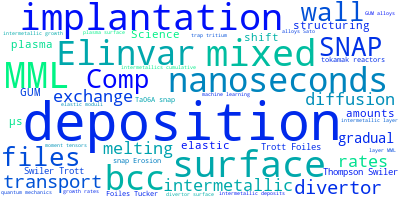
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
517 Citations (16 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) Q. Wang and L. Zhang, “Inverse design of glass structure with deep graph neural networks,” Nature Communications. 2021. link Times cited: 27 USED (high confidence) M. Cusentino, M. A. Wood, and A. Thompson, “Beryllium-driven structural evolution at the divertor surface,” Nuclear Fusion. 2021. link Times cited: 4 Abstract: Erosion of the beryllium first wall material in tokamak reac… read moreAbstract: Erosion of the beryllium first wall material in tokamak reactors has been shown to result in transport and deposition on the tungsten divertor. Experimental studies of beryllium implantation in tungsten indicate that mixed W–Be intermetallic deposits can form, which have lower melting temperatures than tungsten and can trap tritium at higher rates. To better understand the formation and growth rate of these intermetallics, cumulative molecular dynamics (MD) simulations of both high and low energy beryllium deposition in tungsten were performed. In both cases, a W–Be mixed material layer (MML) emerged at the surface within several nanoseconds, either through energetic implantation or a thermally-activated exchange mechanism, respectively. While some ordering of the material into intermetallics occurred, fully ordered structures did not emerge from the deposition simulations. Targeted MD simulations of the MML to further study the rate of Be diffusion and intermetallic growth rates indicate that for both cases, the gradual re-structuring of the material into an ordered intermetallic layer is beyond accessible MD time scales(⩽1 μs). However, the rapid formation of the MML within nanoseconds indicates that beryllium deposition can influence other plasma species interactions at the surface and begin to alter the tungsten material properties. Therefore, beryllium deposition on the divertor surface, even in small amounts, is likely to cause significant changes in plasma-surface interactions and will need to be considered in future studies. read less USED (high confidence) H. Mirhosseini, H. Tahmasbi, S. Kuchana, S. Ghasemi, and T. Kuhne, “An automated approach for developing neural network interatomic potentials with FLAME,” Computational Materials Science. 2021. link Times cited: 7 USED (high confidence) A. Shapeev, E. Podryabinkin, K. Gubaev, F. Tasn’adi, and I. Abrikosov, “Elinvar effect in β-Ti simulated by on-the-fly trained moment tensor potential,” New Journal of Physics. 2020. link Times cited: 20 Abstract: A combination of quantum mechanics calculations with machine… read moreAbstract: A combination of quantum mechanics calculations with machine learning techniques can lead to a paradigm shift in our ability to predict materials properties from first principles. Here we show that on-the-fly training of an interatomic potential described through moment tensors provides the same accuracy as state-of-the-art ab initio molecular dynamics in predicting high-temperature elastic properties of materials with two orders of magnitude less computational effort. Using the technique, we investigate high-temperature bcc phase of titanium and predict very weak, Elinvar, temperature dependence of its elastic moduli, similar to the behavior of the so-called GUM Ti-based alloys (Sato et al 2003 Science 300 464). Given the fact that GUM alloys have complex chemical compositions and operate at room temperature, Elinvar properties of elemental bcc-Ti observed in the wide temperature interval 1100–1700 K is unique. read less USED (high confidence) E. Hahn, S. Fensin, T. Germann, and M. Meyers, “Symmetric tilt boundaries in body-centered cubic tantalum,” Scripta Materialia. 2016. link Times cited: 40 USED (low confidence) T. Swinburne, “Coarse-Graining and Forecasting Atomic Material Simulations with Descriptors,” Physical Review Letters. 2023. link Times cited: 0 USED (low confidence) N. Amadou and T. de Rességuier, “Phase transformations and plasticity in single-crystal iron from shock compression to spall fracture,” Physical Review B. 2023. link Times cited: 0 USED (low confidence) N. Bertin, R. Carson, V. V. Bulatov, J. Lind, and M. Nelms, “Crystal plasticity model of BCC metals from large-scale MD simulations,” Acta Materialia. 2023. link Times cited: 1 USED (low confidence) J. Jiang et al., “Amorphous Zirconia-doped Tantala modeling and simulations using explicit multi-element spectral neighbor analysis machine learning potentials (EME-SNAP),” Physical Review Materials. 2023. link Times cited: 0 USED (low confidence) W. Sha, X. Dai, S. Chen, B. Yin, and F. Guo, “Phonon thermal transport in two-dimensional PbTe monolayers via extensive molecular dynamics simulations with a neuroevolution potential,” Materials Today Physics. 2023. link Times cited: 1 USED (low confidence) K. Nguyen-Cong et al., “Billion atom molecular dynamics simulations of carbon at extreme conditions and experimental time and length scales,” SC21: International Conference for High Performance Computing, Networking, Storage and Analysis. 2021. link Times cited: 23 Abstract: Billion atom molecular dynamics (MD) using quantum-accurate … read moreAbstract: Billion atom molecular dynamics (MD) using quantum-accurate machine-learning Spectral Neighbor Analysis Potential (SNAP) observed long-sought high pressure BC8 phase of carbon at extreme pressure (12 Mbar) and temperature (5,000 K). 24-hour, 4650 node production simulation on OLCF Summit demonstrated an unprecedented scaling and unmatched real-world performance of SNAP MD while sampling 1 nanosecond of physical time. Efficient implementation of SNAP force kernel in LAMMPS using the Kokkos CUDA backend on NVIDIA GPUs combined with excellent strong scaling (better than 97% parallel efficiency) enabled a peak computing rate of 50.0 PFLOPs (24.9% of theoretical peak) for a 20 billion atom MD simulation on the full Summit machine (27,900 GPUs). The peak MD performance of 6.21 Matom-steps/node-s is 22.9 times greater than a previous record for quantum-accurate MD. Near perfect weak scaling of SNAP MD highlights its excellent potential to advance the frontier of quantum-accurate MD to trillion atom simulations on upcoming exascale platforms. KEYWORDS molecular dynamics, machine-learning interatomic potentials, car-bon, extreme conditions read less USED (low confidence) P. Avraam et al., “Crystal plasticity finite element simulation of lattice rotation and x-ray diffraction during laser shock compression of tantalum,” Physical Review Materials. 2021. link Times cited: 2 Abstract: Wehrenberg et. al. [Nature 550 496 (2017)] used ultrafast in… read moreAbstract: Wehrenberg et. al. [Nature 550 496 (2017)] used ultrafast in situ x-ray diffraction at the LCLS x-ray free-electron laser facility to measure large lattice rotations resulting from slip and deformation twinning in shock-compressed laser-driven [110] fibre textured tantalum polycrystal. We employ a crystal plasticity finite element method model, with slip kinetics based closely on the isotropic dislocation-based Livermore Multiscale Model [Barton et. al., J. Appl. Phys. 109 (2011)], to analyse this experiment. We elucidate the link between the degree of lattice rotation and the kinetics of plasticity, demonstrating that a transition occurs at shock pressures of $\sim$27 GPa, between a regime of relatively slow kinetics, resulting in a balanced pattern of slip system activation and therefore relatively small net lattice rotation, and a regime of fast kinetics, due to the onset of nucleation, resulting in a lop-sided pattern of deformation-system activation and therefore large net lattice rotations. We demonstrate a good fit between this model and experimental x-ray diffraction data of lattice rotation, and show that this data is constraining of deformation kinetics. read less USED (low confidence) R. Dong, A. Lunghi, and S. Sanvito, “Stiffness and Atomic-Scale Friction in Superlubricant MoS2 Bilayers,” The Journal of Physical Chemistry Letters. 2021. link Times cited: 0 Abstract: Molecular dynamics simulations, performed with chemically ac… read moreAbstract: Molecular dynamics simulations, performed with chemically accurate ab initio machine-learning force fields, are used to demonstrate that layer stiffness has profound effects on the superlubricant state of two-dimensional van der Waals heterostructures. We engineer bilayers of different rigidity but identical interlayer sliding energy surface and show that a 2-fold increase in the intralayer stiffness reduces the friction by a factor of ∼6. Two sliding regimes as a function of the sliding velocity are found. At a low velocity, the heat generated by the motion is efficiently exchanged between the layers and the friction is independent of the layer order. In contrast, at a high velocity, the friction heat flux cannot be exchanged fast enough and a buildup of significant temperature gradients between the layers is observed. In this situation, the temperature profile depends on whether the slider is softer than the substrate. read less USED (low confidence) H. Kim, N. Mathew, D. Luscher, and A. Hunter, “Phase field dislocation dynamics (PFDD) modeling of non-Schmid behavior in BCC metals informed by atomistic simulations,” Journal of the Mechanics and Physics of Solids. 2021. link Times cited: 8 USED (low confidence) A. K. Gupta, X. Zhou, G. Thompson, and G. Tucker, “Role of grain boundary character and its evolution on interfacial solute segregation behavior in nanocrystalline Ni-P,” Acta Materialia. 2020. link Times cited: 32 USED (low confidence) X. Gu and C. Y. Zhao, “Thermal conductivity of single-layer MoS2(1−x)Se2x alloys from molecular dynamics simulations with a machine-learning-based interatomic potential,” Computational Materials Science. 2019. link Times cited: 47 NOT USED (low confidence) M. Hodapp, “Machine learning is funny but physics makes the money: How machine-learning potentials can advance computer-aided materials design in metallurgy,” Computational Materials Science. 2024. link Times cited: 0 NOT USED (low confidence) Z. Wang, X. Liu, H. Chen, T. Yang, and Y. He, “Exploring Multi-Fidelity Data in Materials Science: Challenges, Applications, and Optimized Learning Strategies,” Applied Sciences. 2023. link Times cited: 0 Abstract: Machine learning techniques offer tremendous potential for o… read moreAbstract: Machine learning techniques offer tremendous potential for optimizing resource allocation in solving real-world problems. However, the emergence of multi-fidelity data introduces new challenges. This paper offers an overview of the definition, applications, data preprocessing methodologies, and learning approaches associated with multi-fidelity data. To validate the algorithms, we examine three widely-used learning methods relevant to multi-fidelity data through the design of multi-fidelity datasets that encompass various types of noise. As we expected, employing multi-fidelity data learning methods yields better results compared to solely using high-fidelity data learning methods. Additionally, considering the inherent various types of noise within datasets, the comprehensive correction strategy proves to be the most effective. Moreover, multi-fidelity learning methods facilitate effective decision-making processes by enabling the combination of datasets from various sources. They extract knowledge from lower fidelity data, improving model accuracy compared to models solely relying on high-fidelity data. read less NOT USED (low confidence) V. Eyert, J. Wormald, W. A. Curtin, and E. Wimmer, “Machine-learned interatomic potentials: Recent developments and prospective applications,” Journal of Materials Research. 2023. link Times cited: 0 NOT USED (low confidence) K. Li et al., “Next-Generation Vitrimers Design through Theoretical Understanding and Computational Simulations.,” Advanced science. 2023. link Times cited: 0 Abstract: Vitrimers are an innovative class of polymers that boast a r… read moreAbstract: Vitrimers are an innovative class of polymers that boast a remarkable fusion of mechanical and dynamic features, complemented by the added benefit of end-of-life recyclability. This extraordinary blend of properties makes them highly attractive for a variety of applications, such as the automotive sector, soft robotics, and the aerospace industry. At their core, vitrimer materials consist of crosslinked covalent networks that have the ability to dynamically reorganize in response to external factors, including temperature changes, pressure variations, or shifts in pH levels. In this review, the aim is to delve into the latest advancements in the theoretical understanding and computational design of vitrimers. The review begins by offering an overview of the fundamental principles that underlie the behavior of these materials, encompassing their structures, dynamic behavior, and reaction mechanisms. Subsequently, recent progress in the computational design of vitrimers is explored, with a focus on the employment of molecular dynamics (MD)/Monte Carlo (MC) simulations and density functional theory (DFT) calculations. Last, the existing challenges and prospective directions for this field are critically analyzed, emphasizing the necessity for additional theoretical and computational advancements, coupled with experimental validation. read less NOT USED (low confidence) W. Yang, J. Ye, P. Bi, B. Huang, L. Chen, and Y. Yi, “Mechanical properties of Mo-Re alloy based on first-principles and machine learning potential function,” Materials Today Communications. 2023. link Times cited: 0 NOT USED (low confidence) O. Shayestehpour and S. Zahn, “Efficient Molecular Dynamics Simulations of Deep Eutectic Solvents with First-Principles Accuracy Using Machine Learning Interatomic Potentials.,” Journal of chemical theory and computation. 2023. link Times cited: 0 Abstract: In recent years, deep eutectic solvents emerged as highly tu… read moreAbstract: In recent years, deep eutectic solvents emerged as highly tunable and ecofriendly alternatives to common organic solvents and liquid electrolytes. In the present work, the ability of machine learning (ML) interatomic potentials for molecular dynamics (MD) simulations of these liquids is explored, showcasing a trained neural network potential for a 1:2 ratio mixture of choline chloride and urea (reline). Using the ML potentials trained on density functional theory data, MD simulations for large systems of thousands of atoms and nanosecond-long time scales are feasible at a fraction of the computational cost of the target first-principles simulations. The obtained structural and dynamical properties of reline from MD simulations using our machine learning models are in good agreement with the first-principles MD simulations and experimental results. Running a single MD simulation is highlighted as a general shortcoming of typical first-principles studies if the dynamic properties are investigated. Furthermore, velocity cross-correlation functions are employed to study the collective dynamics of the molecular components in reline. read less NOT USED (low confidence) P. Richard et al., “Ab Initio Phase Diagram of Gold in Extreme Conditions.,” Physical review letters. 2023. link Times cited: 0 Abstract: A phase diagram of gold is proposed in the [0; 1000] GPa and… read moreAbstract: A phase diagram of gold is proposed in the [0; 1000] GPa and [0; 10 000] K ranges of pressure and temperature, respectively, topologically modified with respect to previous predictions. Using finite-temperature ab initio simulations and nonequilibirum thermodynamic integration, both accelerated by machine learning, we evaluate the Gibbs free energies of three solid phases previously proposed. At room temperature, the face-centered cubic (fcc) phase is stable up to ∼500 GPa whereas the body-centered cubic (bcc) phase only appears above 1 TPa. At higher temperature, we do not highlight any fcc-bcc transition line between 200 and 400 GPa, in agreement with ramp-compressed experiments. The present results only disclose a bcc domain around 140-235 GPa and 6000-8000 K, consistent with the triple point recently found in shock experiments. We demonstrate that this re-stabilization of the bcc phase at high temperature is due to anharmonic effects. read less NOT USED (low confidence) A. S. Kotykhov, K. Gubaev, M. Hodapp, C. Tantardini, A. Shapeev, and I. S. Novikov, “Constrained DFT-based magnetic machine-learning potentials for magnetic alloys: a case study of Fe–Al,” Scientific Reports. 2023. link Times cited: 1 NOT USED (low confidence) S. Atchley et al., “Frontier: Exploring Exascale,” Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. 2023. link Times cited: 0 Abstract: As the US Department of Energy (DOE) computing facilities be… read moreAbstract: As the US Department of Energy (DOE) computing facilities began deploying petascale systems in 2008, DOE was already setting its sights on exascale. In that year, DARPA published a report on the feasibility of reaching exascale. The report authors identified several key challenges in the pursuit of exascale including power, memory, concurrency, and resiliency. That report informed the DOE's computing strategy for reaching exascale. With the deployment of Oak Ridge National Laboratory's Frontier supercomputer, we have officially entered the exascale era. In this paper, we discuss Frontier's architecture, how it addresses those challenges, and describe some early application results from Oak Ridge Leadership Computing Facility's Center of Excellence and the Exascale Computing Project. read less NOT USED (low confidence) J. Xia, Y. Zhang, and B. Jiang, “Accuracy Assessment of Atomistic Neural Network Potentials: The Impact of Cutoff Radius and Message Passing.,” The journal of physical chemistry. A. 2023. link Times cited: 0 Abstract: Atomistic neural network potentials have achieved great succ… read moreAbstract: Atomistic neural network potentials have achieved great success in accelerating atomistic simulations in complicated systems in recent years. They are typically based on the atomic decomposition of total properties, truncating the interatomic correlations to a local environment within a given cutoff radius. A more recently developed message passing (MP) neural network framework can, in principle, incorporate nonlocal effects through iteratively correlating some atoms outside the cutoff sphere with atoms inside, a process referred to as MP. However, how the model accuracy depends on the cutoff radius and the MP process has rarely been discussed. In this work, we investigate this dependence using a recursively embedded atom neural network method that possesses both local and MP features, in two representative systems: liquid H2O and solid Al2O3. We focus on how these settings influence predictions for structural and vibrational properties, namely, radial distribution functions (RDFs) and vibrational density of states (VDOSs). We find that while MP lowers test errors of energy and forces in general, it may not improve the prediction for RDFs and/or VDOSs if direct interatomic correlations in the local environment are insufficiently described. A cutoff radius exceeding the first neighbor shell is necessary, beyond which involving MP quickly enhances the model accuracy until convergence. This is a potentially more efficient way to increase the model accuracy than directly increasing the cutoff radius, especially with more memory savings in the GPU implementation. Our findings also suggest that using the mean test error as the measure of the model accuracy alone is inadequate. read less NOT USED (low confidence) Y.-B. Shi, Y. Chen, H. Dong, H. Wang, and P. Qian, “Investigation of phase transition, mechanical behavior and lattice thermal conductivity of halogen perovskites using machine learning interatomic potentials.,” Physical chemistry chemical physics : PCCP. 2023. link Times cited: 0 Abstract: Using a machine learning (ML) approach to fit DFT data, inte… read moreAbstract: Using a machine learning (ML) approach to fit DFT data, interatomic potentials have been successfully extracted. In this study, the phase transition, mechanical behavior and lattice thermal conductivity are investigated for halogen perovskites using NEP-based MD simulations in a large supercell including 16 000 atoms, which breaks through the size and temperature effects in DFT. A clear phase transition from orthorhombic (γ) → tetragonal (β) → cubic (α) is observed during the heating process. During the cooling process, CsPbCl3 and CsPbBr3 exhibit perfect reversible behavior, while CsPbI3 only undergoes a phase transition from α to β. Then, the key mechanical parameters, including Poisson's ratio, tensile strength, critical strain and bulk modulus, are predicted. The thermal conductivity is also investigated using the NEP-based MD simulations. At room temperature, they exhibit extremely low thermal conductivity. The predicted results are compared with the experimental results, and the rationality of ML potentials has been confirmed. read less NOT USED (low confidence) S. D. Griesemer, Y. Xia, and C. Wolverton, “Accelerating the prediction of stable materials with machine learning,” Nature Computational Science. 2023. link Times cited: 0 NOT USED (low confidence) S.-M. Qi, T. Bo, L. Zhang, Z. Chai, and W.-Q. Shi, “Machine-Learning-Driven Simulations on Microstructure, Thermodynamic Properties, and Transport Properties of LiCl-KCl-LiF Molten Salt,” Artificial Intelligence Chemistry. 2023. link Times cited: 0 NOT USED (low confidence) X. Lei, W. Ye, J. Montoya, T. Mueller, L. Hung, and J. Hummelshoej, “The Role of Reference Points in Machine-Learned Atomistic Simulation Models,” ArXiv. 2023. link Times cited: 0 Abstract: This paper introduces the Chemical Environment Modeling Theo… read moreAbstract: This paper introduces the Chemical Environment Modeling Theory (CEMT), a novel, generalized framework designed to overcome the limitations inherent in traditional atom-centered Machine Learning Force Field (MLFF) models, widely used in atomistic simulations of chemical systems. CEMT demonstrated enhanced flexibility and adaptability by allowing reference points to exist anywhere within the modeled domain and thus, enabling the study of various model architectures. Utilizing Gaussian Multipole (GMP) featurization functions, several models with different reference point sets, including finite difference grid-centered and bond-centered models, were tested to analyze the variance in capabilities intrinsic to models built on distinct reference points. The results underscore the potential of non-atom-centered reference points in force training, revealing variations in prediction accuracy, inference speed and learning efficiency. Finally, a unique connection between CEMT and real-space orbital-free finite element Density Functional Theory (FE-DFT) is established, and the implications include the enhancement of data efficiency and robustness. It allows the leveraging of spatially-resolved energy densities and charge densities from FE-DFT calculations, as well as serving as a pivotal step towards integrating known quantum-mechanical laws into the architecture of ML models. read less NOT USED (low confidence) C. T. Nguyen, D. T. Ho, and S. Y. Kim, “An Enhanced Sampling Approach for Computing the Free Energy of Solid Surface and Solid–Liquid Interface,” Advanced Theory and Simulations. 2023. link Times cited: 0 Abstract: Free energies of a solid surface and a solid–liquid interfac… read moreAbstract: Free energies of a solid surface and a solid–liquid interface play significant roles in thermodynamics. Due to the limited availability of experimental data, computational methods offer effective alternatives for calculating these properties. This study adopts advanced frameworks of the logarithmic mean force dynamics method to present an enhanced sampling approach for the calculation of the free energy at different temperatures. To achieve this, the free energy profile is constructed along with a pre‐established collective variable within the melting transition and cleavage processes. The values of the solid surface and solid–liquid interface free energies are then extrapolated from the excess free energy related to the formation and persistence of the solid surface or the solid–liquid interface. Furthermore, this methodology is employed to calculate the temperature dependence of the free energy measurements for the (100) and (110) surfaces and interfaces of Cu. It is shown that this methodology is robust and readily applicable in contemporary models of atomic interactions and various systems. read less NOT USED (low confidence) X. Chen et al., “TensorMD: Scalable Tensor-Diagram based Machine Learning Interatomic Potential on Heterogeneous Many-Core Processors,” ArXiv. 2023. link Times cited: 0 Abstract: Molecular dynamics simulations have emerged as a potent tool… read moreAbstract: Molecular dynamics simulations have emerged as a potent tool for investigating the physical properties and kinetic behaviors of materials at the atomic scale, particularly in extreme conditions. Ab initio accuracy is now achievable with machine learning based interatomic potentials. With recent advancements in high-performance computing, highly accurate and large-scale simulations become feasible. This study introduces TensorMD, a new machine learning interatomic potential (MLIP) model that integrates physical principles and tensor diagrams. The tensor formalism provides a more efficient computation and greater flexibility for use with other scientific codes. Additionally, we proposed several portable optimization strategies and developed a highly optimized version for the new Sunway supercomputer. Our optimized TensorMD can achieve unprecedented performance on the new Sunway, enabling simulations of up to 52 billion atoms with a time-to-solution of 31 ps/step/atom, setting new records for HPC + AI + MD. read less NOT USED (low confidence) R. K. Raju, S. Sivakumar, X. Wang, and Z. W. Ulissi, “Cluster-MLP: An Active Learning Genetic Algorithm Framework for Accelerated Discovery of Global Minimum Configurations of Pure and Alloyed Nanoclusters,” Journal of Chemical Information and Modeling. 2023. link Times cited: 0 Abstract: Structural characterization of nanoclusters is one of the ma… read moreAbstract: Structural characterization of nanoclusters is one of the major challenges in nanocluster modeling owing to the multitude of possible configurations of arrangement of cluster atoms. The genetic algorithm (GA), a class of evolutionary algorithms based on the principles of natural evolution, is a commonly employed search method for locating the global minimum configuration of nanoclusters. Although a GA search at the DFT level is required for the accurate description of a potential energy surface to arrive at the correct global minimum configuration of nanoclusters, computationally expensive DFT evaluation of the significantly larger number of cluster geometries limits its practicability. Recently, machine learning potentials (MLP) that are learned from DFT calculations gained significant attention as computationally cheap alternative options that provide DFT level accuracy. As the accuracy of the MLP predictions is dependent on the quality and quantity of the training DFT data, active learning (AL) strategies have gained significant momentum to bypass the need of large and representative training data. In this application note, we present Cluster-MLP, an on-the-fly active learning genetic algorithm framework that employs the Flare++ machine learning potential (MLP) for accelerating the GA search for global minima of pure and alloyed nanoclusters. We have used a modified version the Birmingham parallel genetic algorithm (BPGA) for the nanocluster GA search which is then incorporated into distributed evolutionary algorithms in Python (DEAP), an evolutionary computational framework for fast prototyping or technical experiments. We have shown that the incorporation of the AL framework in the BPGA significantly reduced the computationally expensive DFT calculations. Moreover, we have shown that both the AL-GA and DFT-GA predict the same global minima for all the clusters we tested. read less NOT USED (low confidence) C. Hong et al., “Applications and training sets of machine learning potentials,” Science and Technology of Advanced Materials: Methods. 2023. link Times cited: 0 Abstract: ABSTRACT Recently, machine learning potentials (MLPs) have b… read moreAbstract: ABSTRACT Recently, machine learning potentials (MLPs) have been attracting interest as an alternative to the computationally expensive density-functional theory (DFT) calculations. The data-driven approach in MLPs requires carefully curated training datasets, which define the valid domain of simulations. Therefore, acquiring training datasets that comprehensively span the domain of the desired simulations is important. In this review, we attempt to set guidelines for the systematic construction of training datasets according to target simulations. To this end, we extensively analyze the training sets in previous literature according to four application types: thermal properties, diffusion properties, structure prediction, and chemical reactions. In each application, we summarize characteristic reference structures and discuss specific parameters for DFT calculations such as MD conditions. We hope this review serves as a comprehensive guide for researchers and practitioners aiming to harness the capabilities of MLPs in material simulations. IMPACT STATEMENT This review reports on the selection of training sets for machine learning potentials tailored to their specific applications, which is currently not standardized in the rapidly evolving field. read less NOT USED (low confidence) M. Muniz, R. Car, and A. Panagiotopoulos, “Neural Network Water Model Based on the MB-Pol Many-Body Potential.,” The journal of physical chemistry. B. 2023. link Times cited: 0 Abstract: The MB-pol many-body potential accurately predicts many prop… read moreAbstract: The MB-pol many-body potential accurately predicts many properties of water, including cluster, liquid phase, and vapor-liquid equilibrium properties, but its high computational cost can make applying it in large-scale simulations quite challenging. In order to address this limitation, we developed a "deep potential" neural network (DPMD) model based on the MB-pol potential for water. We find that a DPMD model trained on mostly liquid configurations yields a good description of the bulk liquid phase but severely underpredicts vapor-liquid coexistence densities. By contrast, adding cluster configurations to the neural network training set leads to a good agreement for the vapor coexistence densities. Liquid phase densities under supercooled conditions are also represented well, even though they were not included in the training set. These results confirm that neural network models can combine accuracy and transferability if sufficient attention is given to the construction of a representative training set for the target system. read less NOT USED (low confidence) J.-R. Hill and W. Mannstadt, “Machine-learned potentials for eucryptite: A systematic comparison,” Journal of Materials Research. 2023. link Times cited: 1 NOT USED (low confidence) N. V. Maletin, V. Dremov, and I. I. Klebanov, “On the possibility of using quantum annealers to solve problems of computational materials science,” Laser Physics Letters. 2023. link Times cited: 1 Abstract: A promising area of application of quantum computing is comp… read moreAbstract: A promising area of application of quantum computing is computational materials science. In addition to the actively discussed use of quantum computers as simulators for modeling quantum systems, the possibility of using quantum computing to solve the problems of determining the parameters of model multiparameter potentials of intermolecular interaction is of great interest. Especially attractive for these purposes is the method of quantum annealing, as currently the most developed quantum computing technology to solve complex optimization problems. As a first step, the paper presents the algorithms developed for determining the parameters of two classical potentials—Lennard-Jones and Buckingham, designed for implementation on a quantum annealer. We demonstrate mathematical methods for the development of such algorithms. One of them seems to be worthy for the further development and promising for solving more complex problems. Also, we evaluate the scalability of the presented algorithms and justify the possibility of their practical implementation on the current version of the D-Wave quantum annealer. read less NOT USED (low confidence) J. A. Vita and D. Trinkle, “Spline-based neural network interatomic potentials: blending classical and machine learning models,” ArXiv. 2023. link Times cited: 0 Abstract: While machine learning (ML) interatomic potentials (IPs) are… read moreAbstract: While machine learning (ML) interatomic potentials (IPs) are able to achieve accuracies nearing the level of noise inherent in the first-principles data to which they are trained, it remains to be shown if their increased complexities are strictly necessary for constructing high-quality IPs. In this work, we introduce a new MLIP framework which blends the simplicity of spline-based MEAM (s-MEAM) potentials with the flexibility of a neural network (NN) architecture. The proposed framework, which we call the spline-based neural network potential (s-NNP), is a simplified version of the traditional NNP that can be used to describe complex datasets in a computationally efficient manner. We demonstrate how this framework can be used to probe the boundary between classical and ML IPs, highlighting the benefits of key architectural changes. Furthermore, we show that using spline filters for encoding atomic environments results in a readily interpreted embedding layer which can be coupled with modifications to the NN to incorporate expected physical behaviors and improve overall interpretability. Finally, we test the flexibility of the spline filters, observing that they can be shared across multiple chemical systems in order to provide a convenient reference point from which to begin performing cross-system analyses. read less NOT USED (low confidence) H. Zhou, D. Dickel, and C. D. Barrett, “Improving stability and transferability of machine learned interatomic potentials using physically informed bounding potentials,” Journal of Materials Research. 2023. link Times cited: 1 NOT USED (low confidence) D. G. Kizzire et al., “Modified embedded atom method interatomic potential for FCC γ-cerium,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) M. C. Barry, J. R. Gissinger, M. Chandross, K. Wise, S. Kalidindi, and S. Kumar, “Voxelized atomic structure framework for materials design and discovery,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) B. Sharma, Y. S. Teh, B. Sadigh, S. Hamel, V. Bulatov, and A. Samanta, “Development of an interatomic potential for the W–Ta system,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) D. Xu, Q. Zhang, X. Huo, Y. Wang, and M. Yang, “Advances in data‐assisted high‐throughput computations for material design,” Materials Genome Engineering Advances. 2023. link Times cited: 1 Abstract: Extensive trial and error in the variable space is the main … read moreAbstract: Extensive trial and error in the variable space is the main cause of low efficiency and high cost in material development. The experimental tasks can be reduced significantly in the case that the variable space is narrowed down by reliable computer simulations. Because of their numerous variables in material design, however, the variable space is still too large to be accessed thoroughly even with a computational approach. High‐throughput computations (HTC) make it possible to complete a material screening in a large space by replacing the conventionally manual and sequential operations with automatic, robust, and concurrent streamlines. The efficiency of HTC, which is one of the pillars of materials genome engineering, has been verified in many studies, but its applications are still limited by demanding computational costs. Introduction of data mining and artificial intelligence into HTC has become an effective approach to solve the problem. In the past years, many studies have focused on the development and application of HTC and data combined approaches, which is considered as a new paradigm in computational materials science. This review focuses on the main advances in the field of data‐assisted HTC for material research and development and provides our outlook on its future development. read less NOT USED (low confidence) W. C. Witt et al., “ACEpotentials.jl: A Julia implementation of the atomic cluster expansion.,” The Journal of chemical physics. 2023. link Times cited: 2 Abstract: We introduce ACEpotentials.jl, a Julia-language software pac… read moreAbstract: We introduce ACEpotentials.jl, a Julia-language software package that constructs interatomic potentials from quantum mechanical reference data using the Atomic Cluster Expansion [R. Drautz, Phys. Rev. B 99, 014104 (2019)]. As the latter provides a complete description of atomic environments, including invariance to overall translation and rotation as well as permutation of like atoms, the resulting potentials are systematically improvable and data efficient. Furthermore, the descriptor's expressiveness enables use of a linear model, facilitating rapid evaluation and straightforward application of Bayesian techniques for active learning. We summarize the capabilities of ACEpotentials.jl and demonstrate its strengths (simplicity, interpretability, robustness, performance) on a selection of prototypical atomistic modelling workflows. read less NOT USED (low confidence) X.-Y. Wang et al., “Deep neural network potential for simulating hydrogen blistering in tungsten,” Physical Review Materials. 2023. link Times cited: 0 NOT USED (low confidence) A. Mishra, K. Dang, E. M. Kober, S. Fensin, and N. Mathew, “Role of microscopic degrees of freedom in mechanical response of bicrystal nanopillars,” Materials Research Letters. 2023. link Times cited: 0 Abstract: This study investigated the high-strain rate deformation of … read moreAbstract: This study investigated the high-strain rate deformation of bicrystal Cu nanopillars, using atomistic simulations. Nanopillars with minimum grain boundary energy were deformed to investigate the role of macroscopic degrees of freedom, finding that geometric parameters (Schmid factor) influence the stress–strain response. The deformation of metastable grain boundaries (GBs) revealed that in addition to geometric parameters, the response was also governed by the local atomic arrangement at the boundary, dictating the dislocation-GB interactions. These findings shed light on the response of nanopillars as a function of GBs and show the importance of both macroscopic and microscopic degrees of freedom on the mechanical response. GRAPHICAL ABSTRACT IMPACT STATEMENT Metastable states, an often ignored aspect of GB structure, is shown to have a strong influence on dislocation-GB interactions; shedding new light on mechanical response of realistic GBs. read less NOT USED (low confidence) Y. Si et al., “Atomistic determination of Peierls barriers of dislocation glide in nickel,” Journal of the Mechanics and Physics of Solids. 2023. link Times cited: 2 NOT USED (low confidence) S. A. Mousavi and A. Montazeri, “Predicting mechanical properties of defective h-BN nanosheets using Data-Driven models,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) L. Kývala and C. Dellago, “Optimizing the architecture of Behler-Parrinello neural network potentials.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: The architecture of neural network potentials is typically o… read moreAbstract: The architecture of neural network potentials is typically optimized at the beginning of the training process and remains unchanged throughout. Here, we investigate the accuracy of Behler-Parrinello neural network potentials for varying training set sizes. Using the QM9 and 3BPA datasets, we show that adjusting the network architecture according to the training set size improves the accuracy significantly. We demonstrate that both an insufficient and an excessive number of fitting parameters can have a detrimental impact on the accuracy of the neural network potential. Furthermore, we investigate the influences of descriptor complexity, neural network depth, and activation function on the model's performance. We find that for the neural network potentials studied here, two hidden layers yield the best accuracy and that unbounded activation functions outperform bounded ones. read less NOT USED (low confidence) A. A. Mamun, S. Xu, X.-G. Li, and Y. Su, “Comparing interatomic potentials in calculating basic structural parameters and Peierls stress in tungsten-based random binary alloys,” Physica Scripta. 2023. link Times cited: 0 Abstract: The field of machine learning-based interatomic potentials (… read moreAbstract: The field of machine learning-based interatomic potentials (ML-IAPs) has seen increasing development in recent years. In this work, we compare three widely used ML-IAPs–the moment tensor potential (MTP), the spectral neighbor analysis potential (SNAP), and the tabulated Gaussian approximation potential (tabGAP)with a conventional non-ML-IAP, the embedded atom method (EAM) potential. We evaluated these potentials on the basis of their accuracy and efficiency in determining basic structural parameters and Peierls stress under equivalent conditions. Three tungsten (W)-based alloys (Mo-W, Nb-W, and Ta-W) are considered, and their lattice parameter, formation energy, elastic tensor, and Peierls stress of edge dislocation are calculated. Compared with DFT results, MTP demonstrates the highest accuracy in predicting the lattice parameter and the best computational efficiency among the three ML-IAPs, while tabGAP accurately predicts two independent elastic constants, C 11 and C 12. Despite being the slowest, SNAP shows the highest accuracy in predicting the third independent elastic constant C 44 and its Peierls stress value is comparable to that based on MTP. read less NOT USED (low confidence) K. K. Huguenin-Dumittan, P. Loche, H. Ni, and M. Ceriotti, “Physics-Inspired Equivariant Descriptors of Nonbonded Interactions,” The Journal of Physical Chemistry Letters. 2023. link Times cited: 3 Abstract: One essential ingredient in many machine learning (ML) based… read moreAbstract: One essential ingredient in many machine learning (ML) based methods for atomistic modeling of materials and molecules is the use of locality. While allowing better system-size scaling, this systematically neglects long-range (LR) effects such as electrostatic or dispersion interactions. We present an extension of the long distance equivariant (LODE) framework that can handle diverse LR interactions in a consistent way and seamlessly integrates with preexisting methods by building new sets of atom centered features. We provide a direct physical interpretation of these using the multipole expansion, which allows for simpler and more efficient implementations. The framework is applied to simple toy systems as proof of concept and a heterogeneous set of molecular dimers to push the method to its limits. By generalizing LODE to arbitrary asymptotic behaviors, we provide a coherent approach to treat arbitrary two- and many-body nonbonded interactions in the data-driven modeling of matter. read less NOT USED (low confidence) T. Han, J. Li, L. Liu, F.-Q. Li, and L.-W. Wang, “Accuracy evaluation of different machine learning force field features,” New Journal of Physics. 2023. link Times cited: 1 Abstract: Predicting energies and forces using machine learning force … read moreAbstract: Predicting energies and forces using machine learning force field (MLFF) depends on accurate descriptions (features) of chemical environment. Despite the numerous features proposed, there is a lack of controlled comparison among them for their universality and accuracy. In this work, we compared several commonly used feature types for their ability to describe physical systems. These different feature types include cosine feature, Gaussian feature, moment tensor potential (MTP) feature, spectral neighbor analysis potential feature, simplified smooth deep potential with Chebyshev polynomials feature and Gaussian polynomials feature, and atomic cluster expansion feature. We evaluated the training root mean square error (RMSE) for the atomic group energy, total energy, and force using linear regression model regarding to the density functional theory results. We applied these MLFF models to an amorphous sulfur system and carbon systems, and the fitting results show that MTP feature can yield the smallest RMSE results compared with other feature types for either sulfur system or carbon system in the disordered atomic configurations. Moreover, as an extending test of other systems, the MTP feature combined with linear regression model can also reproduce similar quantities along the ab initio molecular dynamics trajectory as represented by Cu systems. Our results are helpful in selecting the proper features for the MLFF development. read less NOT USED (low confidence) J. Yang, Z. Chen, H. Sun, and A. Samanta, “Graph-EAM: An Interpretable and Efficient Graph Neural Network Potential Framework.,” Journal of chemical theory and computation. 2023. link Times cited: 0 Abstract: The development of deep learning interatomic potentials has … read moreAbstract: The development of deep learning interatomic potentials has enabled efficient and accurate computations in quantum chemistry and materials science, circumventing computationally expensive ab initio calculations. However, the huge number of learnable parameters in deep learning models and their complex architectures hinder physical interpretability and affect the robustness of the derived potential. In this work, we propose graph-EAM, a lightweight graph neural network (GNN) inspired by the empirical embedded atom method to model the interatomic potential of single-element structures. Four material systems: platinum, niobium, silicon, and amorphous-carbon, for which quantum simulation data sets are publicly available, are examined to demonstrate that graph-EAM can achieve high energy and force prediction accuracy─comparable or better than existing state-of-the-art machine learning models─with much fewer parameters. It is also shown that the explicit inclusion of the angular information via three-body atomic density increases the prediction accuracy. The accuracy and efficiency of potentials obtained from graph-EAM can help accelerate the molecular dynamics simulation. read less NOT USED (low confidence) W. Yu et al., “High-Accuracy Machine-Learned Interatomic Potentials for the Phase Change Material Ge3Sb6Te5,” Chemistry of Materials. 2023. link Times cited: 0 NOT USED (low confidence) C. Ortner, “On the Atomic Cluster Expansion: interatomic potentials and beyond.” 2023. link Times cited: 0 Abstract: The Atomic Cluster Expansion (ACE) [R. Drautz, Phys. Rev. B,… read moreAbstract: The Atomic Cluster Expansion (ACE) [R. Drautz, Phys. Rev. B, 99:014104 (2019)] provides a systematically improvable, universal descriptor for the environment of an atom that is invariant to permutation, translation and rotation. ACE is being used extensively in newly emerging interatomic potentials based on machine learning. This commentary discusses the ACE framework and its potential impact. read less NOT USED (low confidence) J. Hueckelheim and J. Doerfert, “ORAQL — Optimistic Responses to Alias Queries in LLVM,” Proceedings of the 52nd International Conference on Parallel Processing. 2023. link Times cited: 1 Abstract: Alias analysis (AA) is a prerequisite for many compiler opti… read moreAbstract: Alias analysis (AA) is a prerequisite for many compiler optimizations, which are crucial for performance especially for parallel and scientific software. AA is the subject of ongoing research, and compilers can in practice only approximate the alias information of a given program. In this paper we investigate the extent to which performance in high-performance computing (HPC) applications could be improved if better AA were available in LLVM, one of the most widely used compilers today. To this end we present ORAQL, an optimistic (rather than conservative) AA pass for LLVM that determines AA queries that cannot be answered conclusively by existing techniques, and systematically explores which queries can be answered no-alias without breaking user-provided tests. While ORAQL does not result in provably correct programs and therefore should not be used to compile production code, it allows us to estimate the gap between current and ideal performance. By determining the AA queries that cause the majority of this gap, ORAQL may also guide developers toward beneficial modifications to AA or to HPC programs. Our results show that the performance of HPC proxy applications across multiple programming languages and parallel programming models is not severely limited by AA when compiled with LLVM, although we show performance gains for some applications. read less NOT USED (low confidence) M. Riera et al., “MBX: A many-body energy and force calculator for data-driven many-body simulations.,” The Journal of chemical physics. 2023. link Times cited: 18 Abstract: Many-Body eXpansion (MBX) is a C++ library that implements m… read moreAbstract: Many-Body eXpansion (MBX) is a C++ library that implements many-body potential energy functions (PEFs) within the "many-body energy" (MB-nrg) formalism. MB-nrg PEFs integrate an underlying polarizable model with explicit machine-learned representations of many-body interactions to achieve chemical accuracy from the gas to the condensed phases. MBX can be employed either as a stand-alone package or as an energy/force engine that can be integrated with generic software for molecular dynamics and Monte Carlo simulations. MBX is parallelized internally using Open Multi-Processing and can utilize Message Passing Interface when available in interfaced molecular simulation software. MBX enables classical and quantum molecular simulations with MB-nrg PEFs, as well as hybrid simulations that combine conventional force fields and MB-nrg PEFs, for diverse systems ranging from small gas-phase clusters to aqueous solutions and molecular fluids to biomolecular systems and metal-organic frameworks. read less NOT USED (low confidence) P. Lafourcade, J. Maillet, J. Roche, M. Sakano, B. Hamilton, and A. Strachan, “Multiscale Reactive Model for 1,3,5-Triamino-2,4,6-trinitrobenzene Inferred by Reactive MD Simulations and Unsupervised Learning,” The Journal of Physical Chemistry C. 2023. link Times cited: 1 NOT USED (low confidence) A. Duff, R. Sakidja, H. C. Walker, R. Ewings, and D. Voneshen, “Automated potential development workflow: Application to BaZrO3,” Comput. Phys. Commun. 2023. link Times cited: 0 NOT USED (low confidence) A. Anand, S.-J. Liu, and C. V. Singh, “Recent advances in computational design of structural multi-principal element alloys,” iScience. 2023. link Times cited: 1 NOT USED (low confidence) P. Ouyang et al., “Atomic Local Ordering and Alloying Effects on the Mg3(Sb1-xBix)2 Thermoelectric Material.,” ACS applied materials & interfaces. 2023. link Times cited: 1 Abstract: Mg3(Sb1-xBix)2 alloy has been extensively studied in the las… read moreAbstract: Mg3(Sb1-xBix)2 alloy has been extensively studied in the last 5 years due to its exceptional thermoelectric (TE) performance. The absence of accurate force field for inorganic alloy compounds presents great challenges for computational studies. Here, we explore the atomic microstructure, thermal, and elastic properties of the Mg3(Sb1-xBix)2 alloy at different solution concentrations through atomic simulations with a highly accurate machine learning interatomic potential (ML-IAP). We find atomic local ordering in the optimized structure with the Bi-Bi pair inclined to join adjacent layers and Sb-Sb pair preferring to stay within the same layer. The thermal conductivity changes with the solution concentrations can be correctly predicted through ML-IAP-based molecular dynamics simulations. Spectral thermal conductance analysis shows that the continuous movement of low-frequency peak to high frequency is responsible for the reduction of the thermal conductivity upon alloying. Elastic calculations reveal that similar to the thermal conductivity, solid solution alloying can reduce the overall elastic properties at both Mg3Sb2 and Mg3Bi2 ends, while anisotropic behavior is clearly observed with linear interpolation relationship upon alloying along the interlayer direction and nonlinearity along the intralayer direction. Although the atomic local ordering shows little effects on the properties of the Mg3(Sb1-xBix)2 alloy with only two alloying elements, it possesses potential important impacts on multiprincipal element inorganic TE alloys. This work provides a recipe for computational studies on the TE alloy systems and thus can accelerate the discovery and optimization of TE materials with high TE performance. read less NOT USED (low confidence) T. K. Stenczel et al., “Machine-learned acceleration for molecular dynamics in CASTEP.,” The Journal of chemical physics. 2023. link Times cited: 2 Abstract: Machine learning (ML) methods are of rapidly growing interes… read moreAbstract: Machine learning (ML) methods are of rapidly growing interest for materials modeling, and yet, the use of ML interatomic potentials for new systems is often more demanding than that of established density-functional theory (DFT) packages. Here, we describe computational methodology to combine the CASTEP first-principles simulation software with the on-the-fly fitting and evaluation of ML interatomic potential models. Our approach is based on regular checking against DFT reference data, which provides a direct measure of the accuracy of the evolving ML model. We discuss the general framework and the specific solutions implemented, and we present an example application to high-temperature molecular-dynamics simulations of carbon nanostructures. The code is freely available for academic research. read less NOT USED (low confidence) X. Jiang, H. Sun, K. Choudhary, H. Zhuang, and Q. Nian, “Interpretable Ensemble Learning for Materials Property Prediction with Classical Interatomic Potentials: Carbon as an Example,” ArXiv. 2023. link Times cited: 0 Abstract: Machine learning (ML) is widely used to explore crystal mate… read moreAbstract: Machine learning (ML) is widely used to explore crystal materials and predict their properties. However, the training is time-consuming for deep-learning models, and the regression process is a black box that is hard to interpret. Also, the preprocess to transfer a crystal structure into the input of ML, called descriptor, needs to be designed carefully. To efficiently predict important properties of materials, we propose an approach based on ensemble learning consisting of regression trees to predict formation energy and elastic constants based on small-size datasets of carbon allotropes as an example. Without using any descriptor, the inputs are the properties calculated by molecular dynamics with 9 different classical interatomic potentials. Overall, the results from ensemble learning are more accurate than those from classical interatomic potentials, and ensemble learning can capture the relatively accurate properties from the 9 classical potentials as criteria for predicting the final properties. read less NOT USED (low confidence) M. Cusentino, E. Sikorski, M. McCarthy, A. Thompson, and M. A. Wood, “Dynamic formation of preferentially lattice oriented, self trapped hydrogen clusters,” Materials Research Express. 2023. link Times cited: 0 Abstract: A series of MD and DFT simulations were performed to investi… read moreAbstract: A series of MD and DFT simulations were performed to investigate hydrogen self-clustering and retention in tungsten. Using a newly develop machine learned interatomic potential, spontaneous formation of hydrogen platelets was observed after implanting low-energy hydrogen into tungsten at high fluxes and temperatures. The platelets formed along low miller index orientations and neighboring tetrahedral and octahedral sites and could grow to over 50 atoms in size. High temperatures above 600 K and high hydrogen concentrations were needed to observe significant platelet formation. A critical platelet size of six hydrogen atoms was needed for long term stability. Platelets smaller than this were found to be thermally unstable within a few nanoseconds. To verify these observations, characteristic platelets from the MD simulations were simulated using large-scale DFT. DFT corroborated the MD results in that large platelets were also found to be dynamically stable for five or more hydrogen atoms. The LDOS from the DFT simulated platelets indicated that hydrogen atoms, particularly at the periphery of the platelet, were found to be at least as stable as hydrogen atoms in bulk tungsten. In addition, electrons were found to be localized around hydrogen atoms in the platelet itself and that hydrogen atoms up to 4.2 Å away within the platelet were found to share charge suggesting that the hydrogen atoms are interacting across longer distances than previously suggested. These results reveal a self-clustering mechanisms for hydrogen within tungsten in the absence of radiation induced or microstructural defects that could be a precursor to blistering and potentially explain the experimentally observed high hydrogen retention particularly in the near surface region. read less NOT USED (low confidence) Z. Ma and Z. Pan, “Efficient machine learning of solute segregation energy based on physics-informed features,” Scientific Reports. 2023. link Times cited: 0 NOT USED (low confidence) D. F. T. du Toit and V. L. Deringer, “Cross-platform hyperparameter optimization for machine learning interatomic potentials.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: Machine-learning (ML)-based interatomic potentials are incre… read moreAbstract: Machine-learning (ML)-based interatomic potentials are increasingly popular in material modeling, enabling highly accurate simulations with thousands and millions of atoms. However, the performance of machine-learned potentials depends strongly on the choice of hyperparameters-that is, of those parameters that are set before the model encounters data. This problem is particularly acute where hyperparameters have no intuitive physical interpretation and where the corresponding optimization space is large. Here, we describe an openly available Python package that facilitates hyperparameter optimization across different ML potential fitting frameworks. We discuss methodological aspects relating to the optimization itself and to the selection of validation data, and we show example applications. We expect this package to become part of a wider computational framework to speed up the mainstream adaptation of ML potentials in the physical sciences. read less NOT USED (low confidence) J. Guo et al., “AL4GAP: Active learning workflow for generating DFT-SCAN accurate machine-learning potentials for combinatorial molten salt mixtures.,” The Journal of chemical physics. 2023. link Times cited: 1 Abstract: Machine learning interatomic potentials have emerged as a po… read moreAbstract: Machine learning interatomic potentials have emerged as a powerful tool for bypassing the spatiotemporal limitations of ab initio simulations, but major challenges remain in their efficient parameterization. We present AL4GAP, an ensemble active learning software workflow for generating multicomposition Gaussian approximation potentials (GAP) for arbitrary molten salt mixtures. The workflow capabilities include: (1) setting up user-defined combinatorial chemical spaces of charge neutral mixtures of arbitrary molten mixtures spanning 11 cations (Li, Na, K, Rb, Cs, Mg, Ca, Sr, Ba and two heavy species, Nd, and Th) and 4 anions (F, Cl, Br, and I), (2) configurational sampling using low-cost empirical parameterizations, (3) active learning for down-selecting configurational samples for single point density functional theory calculations at the level of Strongly Constrained and Appropriately Normed (SCAN) exchange-correlation functional, and (4) Bayesian optimization for hyperparameter tuning of two-body and many-body GAP models. We apply the AL4GAP workflow to showcase high throughput generation of five independent GAP models for multicomposition binary-mixture melts, each of increasing complexity with respect to charge valency and electronic structure, namely: LiCl-KCl, NaCl-CaCl2, KCl-NdCl3, CaCl2-NdCl3, and KCl-ThCl4. Our results indicate that GAP models can accurately predict structure for diverse molten salt mixture with density functional theory (DFT)-SCAN accuracy, capturing the intermediate range ordering characteristic of the multivalent cationic melts. read less NOT USED (low confidence) D. J. Liu and J. Evans, “Fluorine spillover for ceria- vs silica-supported palladium nanoparticles: A MD study using machine learning potentials.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: Supported metallic nanoparticles play a central role in cata… read moreAbstract: Supported metallic nanoparticles play a central role in catalysis. However, predictive modeling is particularly challenging due to the structural and dynamic complexity of the nanoparticle and its interface with the support, given that the sizes of interest are often well beyond those accessible via traditional ab initio methods. With recent advances in machine learning, it is now feasible to perform MD simulations with potentials retaining near-density-functional theory (DFT) accuracy, which can elucidate the growth and relaxation of supported metal nanoparticles, as well as reactions on those catalysts, at temperatures and time scales approaching those relevant to experiments. Furthermore, the surfaces of the support materials can also be modeled realistically through simulated annealing to include effects such as defects and amorphous structures. We study the adsorption of fluorine atoms on ceria and silica supported palladium nanoparticles using machine learning potential trained by DFT data using the DeePMD framework. We show defects on ceria and Pd/ceria interfaces are crucial for the initial adsorption of fluorine, while the interplay between Pd and ceria and the reverse oxygen migration from ceria to Pd control spillover of fluorine from Pd to ceria at later stages. In contrast, silica supports do not induce fluorine spillover from Pd particles. read less NOT USED (low confidence) P. Lafourcade et al., “Robust crystal structure identification at extreme conditions using a density-independent spectral descriptor and supervised learning,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) Q. Mao, M. Feng, X. Jiang, Y. Ren, K. Luo, and A. V. van Duin, “Classical and reactive molecular dynamics: Principles and applications in combustion and energy systems,” Progress in Energy and Combustion Science. 2023. link Times cited: 10 NOT USED (low confidence) W. Chen, L. Li, Q. Zhu, and H. Zhuang, “Chemical short-range order in complex concentrated alloys,” MRS Bulletin. 2023. link Times cited: 1 Abstract: Complex concentrated alloys (CCAs) have drawn immense attent… read moreAbstract: Complex concentrated alloys (CCAs) have drawn immense attention from the materials research community and beyond. Because the vast compositional and structural degrees of freedom in CCAs can lead to novel properties (e.g., structural and functional) with a wide range of applications, the structure–property relationships of CCAs are of critical interest. One salient feature in the atomic structures of CCAs is the presence of chemical short-range ordering (CSRO). Understanding the roles of CSRO on properties, especially phase stability, requires joint efforts from experimental and computational approaches. In this article, we first briefly survey the most recent experimental efforts in identifying and characterizing CSRO of various CCAs. We then focus on the theoretical and computational techniques that have been deployed to investigate the CSRO effects. These computational methods include density functional theory (DFT), molecular dynamics (MD), and statistical mechanics methods such as cluster expansions and machine learning methods such as creating transferable interatomic potentials. Finally, we outline the challenges and future directions of CSRO research in CCAs. read less NOT USED (low confidence) O. Klimanova, T. Miryashkin, and A. Shapeev, “Accurate melting point prediction through autonomous physics-informed learning,” Physical Review B. 2023. link Times cited: 0 Abstract: We present an algorithm for computing melting points by auto… read moreAbstract: We present an algorithm for computing melting points by autonomously learning from coexistence simulations in the NPT ensemble. Given the interatomic interaction model, the method makes decisions regarding the number of atoms and temperature at which to conduct simulations, and based on the collected data predicts the melting point along with the uncertainty, which can be systematically improved with more data. We demonstrate how incorporating physical models of the solid-liquid coexistence evolution enhances the algorithm's accuracy and enables optimal decision-making to effectively reduce predictive uncertainty. To validate our approach, we compare the results of 20 melting point calculations from the literature to the results of our calculations, all conducted with same interatomic potentials. Remarkably, we observe significant deviations in about one-third of the cases, underscoring the need for accurate and reliable algorithms for materials property calculations. read less NOT USED (low confidence) P. T. Salzbrenner et al., “Developments and further applications of ephemeral data derived potentials.,” The Journal of chemical physics. 2023. link Times cited: 1 Abstract: Machine-learned interatomic potentials are fast becoming an … read moreAbstract: Machine-learned interatomic potentials are fast becoming an indispensable tool in computational materials science. One approach is the ephemeral data-derived potential (EDDP), which was designed to accelerate atomistic structure prediction. The EDDP is simple and cost-efficient. It relies on training data generated in small unit cells and is fit using a lightweight neural network, leading to smooth interactions which exhibit the robust transferability essential for structure prediction. Here, we present a variety of applications of EDDPs, enabled by recent developments of the open-source EDDP software. New features include interfaces to phonon and molecular dynamics codes, as well as deployment of the ensemble deviation for estimating the confidence in EDDP predictions. Through case studies ranging from elemental carbon and lead to the binary scandium hydride and the ternary zinc cyanide, we demonstrate that EDDPs can be trained to cover wide ranges of pressures and stoichiometries, and used to evaluate phonons, phase diagrams, superionicity, and thermal expansion. These developments complement continued success in accelerated structure prediction. read less NOT USED (low confidence) L. Fiedler, N. Modine, K. D. Miller, and A. Cangi, “Machine learning the electronic structure of matter across temperatures,” Physical Review B. 2023. link Times cited: 0 Abstract: We introduce machine learning (ML) models that predict the e… read moreAbstract: We introduce machine learning (ML) models that predict the electronic structure of materials across a wide temperature range. Our models employ neural networks and are trained on density functional theory (DFT) data. Unlike most other ML models that use DFT data, our models directly predict the local density of states (LDOS) of the electronic structure. This provides several advantages, including access to multiple observables such as the electronic density and electronic total free energy. Moreover, our models account for both the electronic and ionic temperatures independently, making them ideal for applications like laser-heating of matter. We validate the efficacy of our LDOS-based models on a metallic test system. They accurately capture energetic effects induced by variations in ionic and electronic temperatures over a broad temperature range, even when trained on a subset of these temperatures. These findings open up exciting opportunities for investigating the electronic structure of materials under both ambient and extreme conditions. read less NOT USED (low confidence) H. Chikuma, G. Takasao, T. Wada, P. Chammingkwan, J. Behler, and T. Taniike, “Accelerating Non-Empirical Structure Determination of Ziegler–Natta Catalysts with a High-Dimensional Neural Network Potential,” The Journal of Physical Chemistry C. 2023. link Times cited: 0 NOT USED (low confidence) J. Tang, G. Li, Q. Wang, J. Zheng, L. Cheng, and R. Guo, “Effect of Four-Phonon Scattering on Anisotropic Thermal Transport in Bulk Hexagonal Boron Nitride by Machine Learning Interatomic Potential,” SSRN Electronic Journal. 2023. link Times cited: 3 NOT USED (low confidence) K. Shimizu, R. Otsuka, M. Hara, E. Minamitani, and S. Watanabe, “Prediction of Born effective charges using neural network to study ion migration under electric fields: applications to crystalline and amorphous Li3PO4,” Science and Technology of Advanced Materials: Methods. 2023. link Times cited: 0 Abstract: ABSTRACT Understanding ionic behaviour under external electr… read moreAbstract: ABSTRACT Understanding ionic behaviour under external electric fields is crucial to develop electronic and energy-related devices using ion transport. In this study, we propose a neural network (NN) model to predict the Born effective charges of ions along an axis parallel to an applied electric field from atomic structures. The proposed NN model is applied to Li3PO4 as a prototype. The prediction error of the constructed NN model is 0.0376 $e$e/atom. In combination with an NN interatomic potential, molecular dynamics (MD) simulations are performed under a uniform electric field of 0.1 V/Å, whereby an enhanced mean square displacement of Li along the electric field is obtained, which seems physically reasonable. In addition, the external forces along the direction perpendicular to the electric field, originating from the off-diagonal terms of the Born effective charges, are found to have a nonnegligible effect on Li migration. Finally, additional MD simulations are performed to examine the Li motion in an amorphous structure. The results reveal that Li migration occurs in various areas despite the absence of explicitly introduced defects, which may be attributed to the susceptibility of the Li ions in the local minima to the electric field. We expect that the proposed NN method can be applied to any ionic material, thereby leading to atomic-scale elucidation of ion behaviour under electric fields. GRAPHICAL ABSTRACT IMPACT STATEMENT This study introduces a new computational scheme for analysing ion behaviour in solids under electric fields, through the development of a neural network model to predict the Born effective charges. read less NOT USED (low confidence) J. Bañuelos et al., “Oxide- and Silicate-Water Interfaces and Their Roles in Technology and the Environment.,” Chemical reviews. 2023. link Times cited: 8 Abstract: Interfacial reactions drive all elemental cycling on Earth a… read moreAbstract: Interfacial reactions drive all elemental cycling on Earth and play pivotal roles in human activities such as agriculture, water purification, energy production and storage, environmental contaminant remediation, and nuclear waste repository management. The onset of the 21st century marked the beginning of a more detailed understanding of mineral aqueous interfaces enabled by advances in techniques that use tunable high-flux focused ultrafast laser and X-ray sources to provide near-atomic measurement resolution, as well as by nanofabrication approaches that enable transmission electron microscopy in a liquid cell. This leap into atomic- and nanometer-scale measurements has uncovered scale-dependent phenomena whose reaction thermodynamics, kinetics, and pathways deviate from previous observations made on larger systems. A second key advance is new experimental evidence for what scientists hypothesized but could not test previously, namely, interfacial chemical reactions are frequently driven by "anomalies" or "non-idealities" such as defects, nanoconfinement, and other nontypical chemical structures. Third, progress in computational chemistry has yielded new insights that allow a move beyond simple schematics, leading to a molecular model of these complex interfaces. In combination with surface-sensitive measurements, we have gained knowledge of the interfacial structure and dynamics, including the underlying solid surface and the immediately adjacent water and aqueous ions, enabling a better definition of what constitutes the oxide- and silicate-water interfaces. This critical review discusses how science progresses from understanding ideal solid-water interfaces to more realistic systems, focusing on accomplishments in the last 20 years and identifying challenges and future opportunities for the community to address. We anticipate that the next 20 years will focus on understanding and predicting dynamic transient and reactive structures over greater spatial and temporal ranges as well as systems of greater structural and chemical complexity. Closer collaborations of theoretical and experimental experts across disciplines will continue to be critical to achieving this great aspiration. read less NOT USED (low confidence) A. K. A. Kandy, K. Rossi, A. Raulin-Foissac, G. Laurens, and J. Lam, “Comparing transferability in neural network approaches and linear models for machine-learning interaction potentials,” Physical Review B. 2023. link Times cited: 4 Abstract: Atomic simulations using machine learning interatomic potent… read moreAbstract: Atomic simulations using machine learning interatomic potential (MLIP) have gained a lot of popularity owing to their accuracy in comparison to conventional empirical potentials. However, the transferability of MLIP to systems outside the training-set poses a significant challenge. Here, we compare the transferability of three MLIP approaches: i) Neural Network Potentials (NNP), ii) Physical LassoLars Interactions Potential (PLIP) and iii) Linear Potentials with Belher-Parrinello descriptors, trained over a small but diverse configuration of zinc oxide polymorphs. We compared the obtained models with density functional theory reference results for physical properties including bulk lattice parameters, surface energies, and vibrational density of states and showed the superiority of both NNP and PLIP models. However, the NNP model performed poorly when compared to the other two linear models for the structural optimization of nanoparticles and molecular dynamics simulation of liquid phases, which are systems outside the training-set. While providing less accurate prediction for solid Zinc Oxides phases, both linear models appear more transferable than NNP when testing for nanoscale systems and liquid phases. Our results are finally rationalized by a combination of different statistical analysis including spread in force evaluation, information imbalance, convex hull calculation and density in descriptor space. read less NOT USED (low confidence) P.-Y. Yang, Y.-H. Chiang, C. Pao, and C.-C. Chang, “Hybrid Machine Learning-Enabled Potential Energy Model for Atomistic Simulation of Lithium Intercalation into Graphite from Plating to Overlithiation.,” Journal of chemical theory and computation. 2023. link Times cited: 0 Abstract: Graphite is one of the most widely used negative electrode m… read moreAbstract: Graphite is one of the most widely used negative electrode materials for lithium ion batteries (LIBs). However, because of the rapid growth of demands pursuing higher energy density and charging rates, comprehensive insights into the lithium intercalation and plating processes are critical for further boosting the potential of graphite electrodes. Herein, by utilizing the dihedral-angle-corrected registry-dependent potential (DRIP) (Wen et al., Phys. Rev. B 2018, 98, 235404), the Ziegler-Biersack-Littmark (ZBL) potential (Ziegler and Biersack, Astrophysics, Chemistry, and Condensed Matter; 1985, pp 93-129), and the machine learning-based spectral neighbor analysis (SNAP) potential (Thompson et al., J. Comput, Phys. 2015, 285, 316-330), we have successfully trained a hybrid machine learning-enabled potential energy model capable of simulating a wide spectrum of lithium intercalation scenario from plating to overlithiation. Our extensive atomistic simulations reveal the trapping of intercalated lithium atoms close to the graphite edges due to high hopping barriers, resulting in lithium plating. Furthermore, we report a stable dense graphite intercalation compound (GIC) LiC4 with a theoretical capacity of 558 mAh/g, wherein lithium atoms occupy alternating upper/lower graphene hollow sites with a nearest Li-Li distance of 2.8 Å. Surprisingly, following the same lithium insertion manner would allow the nearest Li-Li distance to be retained until the capacity reaches 845.2 mAh/g, corresponding to a GIC of LiC2.6. Hence, the present study demonstrates that the hybrid machine learning approach could further extend the scope of machine learning energy models, allowing us to investigate the lithium intercalation into graphite over a wide range of intercalation capacity to unveil the underlying mechanisms of lithium plating, diffusion, and discovery of new dense GICs for advanced LIBs with high charging rates and high energy densities. read less NOT USED (low confidence) B. Kozinsky, A. Musaelian, A. Johansson, and S. L. Batzner, “Scaling the Leading Accuracy of Deep Equivariant Models to Biomolecular Simulations of Realistic Size,” Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. 2023. link Times cited: 10 Abstract: This work brings the leading accuracy, sample efficiency, an… read moreAbstract: This work brings the leading accuracy, sample efficiency, and robustness of deep equivariant neural networks to the extreme computational scale. This is achieved through a combination of innovative model architecture, massive parallelization, and models and implementations optimized for efficient GPU utilization. The resulting Allegro architecture bridges the accuracy-speed tradeoff of atomistic simulations and enables description of dynamics in structures of unprecedented complexity at quantum fidelity. To illustrate the scalability of Allegro, we perform nanoseconds-long stable simulations of protein dynamics and scale up to a 44-million atom structure of a complete, all-atom, explicitly solvated HIV capsid on the Perlmutter supercomputer. We demonstrate excellent strong scaling up to 100 million atoms and 70% weak scaling to 5120 A100 GPUs. read less NOT USED (low confidence) J. Zeng et al., “DeePMD-kit v2: A software package for deep potential models,” The Journal of Chemical Physics. 2023. link Times cited: 22 Abstract: DeePMD-kit is a powerful open-source software package that f… read moreAbstract: DeePMD-kit is a powerful open-source software package that facilitates molecular dynamics simulations using machine learning potentials known as Deep Potential (DP) models. This package, which was released in 2017, has been widely used in the fields of physics, chemistry, biology, and material science for studying atomistic systems. The current version of DeePMD-kit offers numerous advanced features, such as DeepPot-SE, attention-based and hybrid descriptors, the ability to fit tensile properties, type embedding, model deviation, DP-range correction, DP long range, graphics processing unit support for customized operators, model compression, non-von Neumann molecular dynamics, and improved usability, including documentation, compiled binary packages, graphical user interfaces, and application programming interfaces. This article presents an overview of the current major version of the DeePMD-kit package, highlighting its features and technical details. Additionally, this article presents a comprehensive procedure for conducting molecular dynamics as a representative application, benchmarks the accuracy and efficiency of different models, and discusses ongoing developments. read less NOT USED (low confidence) A. M. Lewis, P. Lazzaroni, and M. Rossi, “Predicting the electronic density response of condensed-phase systems to electric field perturbations.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: We present a local and transferable machine-learning approac… read moreAbstract: We present a local and transferable machine-learning approach capable of predicting the real-space density response of both molecules and periodic systems to homogeneous electric fields. The new method, Symmetry-Adapted Learning of Three-dimensional Electron Responses (SALTER), builds on the symmetry-adapted Gaussian process regression symmetry-adapted learning of three-dimensional electron densities framework. SALTER requires only a small, but necessary, modification to the descriptors used to represent the atomic environments. We present the performance of the method on isolated water molecules, bulk water, and a naphthalene crystal. Root mean square errors of the predicted density response lie at or below 10% with barely more than 100 training structures. Derived polarizability tensors and even Raman spectra further derived from these tensors show good agreement with those calculated directly from quantum mechanical methods. Therefore, SALTER shows excellent performance when predicting derived quantities, while retaining all of the information contained in the full electronic response. Thus, this method is capable of predicting vector fields in a chemical context and serves as a landmark for further developments. read less NOT USED (low confidence) G. Jung, H. Myung, and S. Irle, “Artificial neural network potentials for mechanics and fracture dynamics of two-dimensional crystals,” Machine Learning: Science and Technology. 2023. link Times cited: 1 Abstract: Understanding the mechanics and failure of materials at the … read moreAbstract: Understanding the mechanics and failure of materials at the nanoscale is critical for their engineering and applications. The accurate atomistic modeling of brittle failure with crack propagation in covalent crystals requires a quantum mechanics-based description of individual bond-breaking events. Artificial neural network potentials (NNPs) have emerged to overcome the traditional, physics-based modeling tradeoff between accuracy and accessible time and length scales. Previous studies have shown successful applications of NNPs for describing the structure and dynamics of molecular systems and amorphous or liquid phases of materials. However, their application to deformation and failure processes in materials is still uncommon. In this study, we discuss the apparent limitations of NNPs for the description of deformation and fracture under loadings and propose a way to generate and select training data for their employment in simulations of deformation and fracture simulations of crystals. We applied the proposed approach to 2D crystalline graphene, utilizing the density-functional tight-binding method for more efficient and extensive data generation in place of density functional theory. Then, we explored how the data selection affects the accuracy of the developed artificial NNPs. It revealed that NNP’s reliability should not only be measured based on the total energy and atomic force comparisons for reference structures but also utilize comparisons for physical properties, e.g. stress–strain curves and geometric deformation. In sharp contrast to popular reactive bond order potentials, our optimized NNP predicts straight crack propagation in graphene along both armchair and zigzag (ZZ) lattice directions, as well as higher fracture toughness of ZZ edge direction. Our study provides significant insight into crack propagation mechanisms on atomic scales and highlights strategies for NNP developments of broader materials. read less NOT USED (low confidence) J. Tang, G. Li, Q. Wang, J. Zheng, L. Cheng, and R. Guo, “Competition between phonon-vacancy and four-phonon scattering in cubic boron arsenide by machine learning interatomic potential,” Physical Review Materials. 2023. link Times cited: 1 NOT USED (low confidence) B. Mortazavi, X. Zhuang, T. Rabczuk, and A. Shapeev, “Atomistic modeling of the mechanical properties: the rise of machine learning interatomic potentials.,” Materials horizons. 2023. link Times cited: 9 Abstract: Since the birth of the concept of machine learning interatom… read moreAbstract: Since the birth of the concept of machine learning interatomic potentials (MLIPs) in 2007, a growing interest has been developed in the replacement of empirical interatomic potentials (EIPs) with MLIPs, in order to conduct more accurate and reliable molecular dynamics calculations. As an exciting novel progress, in the last couple of years the applications of MLIPs have been extended towards the analysis of mechanical and failure responses, providing novel opportunities not heretofore efficiently achievable, neither by EIPs nor by density functional theory (DFT) calculations. In this minireview, we first briefly discuss the basic concepts of MLIPs and outline popular strategies for developing a MLIP. Next, by considering several examples of recent studies, the robustness of MLIPs in the analysis of the mechanical properties will be highlighted, and their advantages over EIP and DFT methods will be emphasized. MLIPs furthermore offer astonishing capabilities to combine the robustness of the DFT method with continuum mechanics, enabling the first-principles multiscale modeling of mechanical properties of nanostructures at the continuum level. Last but not least, the common challenges of MLIP-based molecular dynamics simulations of mechanical properties are outlined and suggestions for future investigations are proposed. read less NOT USED (low confidence) M. S. Nitol, K. Dang, S. Fensin, M. Baskes, D. Dickel, and C. Barrett, “Hybrid interatomic potential for Sn,” Physical Review Materials. 2023. link Times cited: 2 NOT USED (low confidence) M. Zhang, K. Hibi, and J. Inoue, “GPU-accelerated artificial neural network potential for molecular dynamics simulation,” Comput. Phys. Commun. 2023. link Times cited: 3 NOT USED (low confidence) V. Briganti and A. Lunghi, “Efficient generation of stable linear machine-learning force fields with uncertainty-aware active learning,” Machine Learning: Science and Technology. 2023. link Times cited: 1 Abstract: Machine-learning (ML) force fields (FFs) enable an accurate … read moreAbstract: Machine-learning (ML) force fields (FFs) enable an accurate and universal description of the potential energy surface of molecules and materials on the basis of a training set of ab initio data. However, large-scale applications of these methods rest on the possibility to train accurate ML models with a small number of ab initio data. In this respect, active-learning (AL) strategies, where the training set is self-generated by the model itself, combined with linear ML models are particularly promising. In this work, we explore an AL strategy based on linear regression and able to predict the model’s uncertainty on predictions for molecular configurations not sampled by the training set, thus providing a straightforward recipe for the extension of the latter. We apply this strategy to the spectral neighbor analysis potential and show that only tens of ab initio simulations of atomic forces are required to generate FFs for room-temperature molecular dynamics at or close to chemical accuracy and which stability can be systematically improved by the user at modest computational expenses. Moreover, the method does not necessitate any conformational pre-sampling, thus requiring minimal user intervention and parametrization. read less NOT USED (low confidence) D. Khan, S. Heinen, and O. A. von Lilienfeld, “Kernel based quantum machine learning at record rate: Many-body distribution functionals as compact representations.,” The Journal of chemical physics. 2023. link Times cited: 2 Abstract: The feature vector mapping used to represent chemical system… read moreAbstract: The feature vector mapping used to represent chemical systems is a key factor governing the superior data efficiency of kernel based quantum machine learning (QML) models applicable throughout chemical compound space. Unfortunately, the most accurate representations require a high dimensional feature mapping, thereby imposing a considerable computational burden on model training and use. We introduce compact yet accurate, linear scaling QML representations based on atomic Gaussian many-body distribution functionals (MBDF) and their derivatives. Weighted density functions of MBDF values are used as global representations that are constant in size, i.e., invariant with respect to the number of atoms. We report predictive performance and training data efficiency that is competitive with state-of-the-art for two diverse datasets of organic molecules, QM9 and QMugs. Generalization capability has been investigated for atomization energies, highest occupied molecular orbital-lowest unoccupied molecular orbital eigenvalues and gap, internal energies at 0 K, zero point vibrational energies, dipole moment norm, static isotropic polarizability, and heat capacity as encoded in QM9. MBDF based QM9 performance lowers the optimal Pareto front spanned between sampling and training cost to compute node minutes, effectively sampling chemical compound space with chemical accuracy at a sampling rate of ∼48 molecules per core second. read less NOT USED (low confidence) S. Nikolov, P. Nieves, A. Thompson, M. Wood, and J. Tranchida, “Temperature dependence of magnetic anisotropy and magnetoelasticity from classical spin-lattice calculations,” Physical Review B. 2023. link Times cited: 0 NOT USED (low confidence) M. Kulichenko et al., “Uncertainty-driven dynamics for active learning of interatomic potentials,” Nature Computational Science. 2023. link Times cited: 22 NOT USED (low confidence) L. O. AGBOLADE et al., “Recent advances in density functional theory approach for optoelectronics properties of graphene,” Heliyon. 2023. link Times cited: 1 NOT USED (low confidence) F. Wang and J. Cheng, “Understanding the solvation structures of glyme-based electrolytes by machine learning molecular dynamics,” Chinese Journal of Structural Chemistry. 2023. link Times cited: 0 NOT USED (low confidence) X. Fan, X. Wen, Y.-B. Zhuang, and J. Cheng, “Molecular insight into the GaP(110)–water interface using machine learning accelerated molecular dynamics,” Journal of Energy Chemistry. 2023. link Times cited: 7 NOT USED (low confidence) A. Ferrari, F. Körmann, M. Asta, and J. Neugebauer, “Simulating short-range order in compositionally complex materials,” Nature Computational Science. 2023. link Times cited: 5 NOT USED (low confidence) S. Pozdnyakov, M. J. Willatt, A. Bartók, C. Ortner, G. Csányi, and M. Ceriotti, “Completeness of Atomic Structure Representations,” ArXiv. 2023. link Times cited: 7 Abstract: In this paper, we address the challenge of obtaining a compr… read moreAbstract: In this paper, we address the challenge of obtaining a comprehensive and symmetric representation of point particle groups, such as atoms in a molecule, which is crucial in physics and theoretical chemistry. The problem has become even more important with the widespread adoption of machine-learning techniques in science, as it underpins the capacity of models to accurately reproduce physical relationships while being consistent with fundamental symmetries and conservation laws. However, the descriptors that are commonly used to represent point clouds -- most notably those adopted to describe matter at the atomic scale -- are unable to distinguish between special arrangements of particles. This makes it impossible to machine learn their properties. Frameworks that are provably complete exist but are only so in the limit in which they simultaneously describe the mutual relationship between all atoms, which is impractical. We present a novel approach to construct descriptors of finite correlations based on the relative arrangement of particle triplets, which can be employed to create symmetry-adapted models with universal approximation capabilities. Our strategy is demonstrated on a class of atomic arrangements that are specifically built to defy a broad class of conventional symmetric descriptors, showcasing its potential for addressing their limitations. read less NOT USED (low confidence) Y. Wang et al., “High‐Entropy Perovskites for Energy Conversion and Storage: Design, Synthesis, and Potential Applications,” Small Methods. 2023. link Times cited: 10 Abstract: Perovskites have shown tremendous promise as functional mate… read moreAbstract: Perovskites have shown tremendous promise as functional materials for several energy conversion and storage technologies, including rechargeable batteries, (electro)catalysts, fuel cells, and solar cells. Due to their excellent operational stability and performance, high‐entropy perovskites (HEPs) have emerged as a new type of perovskite framework. Herein, this work reviews the recent progress in the development of HEPs, including synthesis methods and applications. Effective strategies for the design of HEPs through atomistic computations are also surveyed. Finally, an outlook of this field provides guidance for the development of new and improved HEPs. read less NOT USED (low confidence) D. M. Anstine and O. Isayev, “Machine Learning Interatomic Potentials and Long-Range Physics,” The Journal of Physical Chemistry. a. 2023. link Times cited: 12 Abstract: Advances in machine learned interatomic potentials (MLIPs), … read moreAbstract: Advances in machine learned interatomic potentials (MLIPs), such as those using neural networks, have resulted in short-range models that can infer interaction energies with near ab initio accuracy and orders of magnitude reduced computational cost. For many atom systems, including macromolecules, biomolecules, and condensed matter, model accuracy can become reliant on the description of short- and long-range physical interactions. The latter terms can be difficult to incorporate into an MLIP framework. Recent research has produced numerous models with considerations for nonlocal electrostatic and dispersion interactions, leading to a large range of applications that can be addressed using MLIPs. In light of this, we present a Perspective focused on key methodologies and models being used where the presence of nonlocal physics and chemistry are crucial for describing system properties. The strategies covered include MLIPs augmented with dispersion corrections, electrostatics calculated with charges predicted from atomic environment descriptors, the use of self-consistency and message passing iterations to propagated nonlocal system information, and charges obtained via equilibration schemes. We aim to provide a pointed discussion to support the development of machine learning-based interatomic potentials for systems where contributions from only nearsighted terms are deficient. read less NOT USED (low confidence) J. López-Zorrilla, X. Aretxabaleta, I. W. Yeu, I. Etxebarria, H. Manzano, and N. Artrith, “ænet-PyTorch: A GPU-supported implementation for machine learning atomic potentials training.,” The Journal of chemical physics. 2023. link Times cited: 4 Abstract: In this work, we present ænet-PyTorch, a PyTorch-based imple… read moreAbstract: In this work, we present ænet-PyTorch, a PyTorch-based implementation for training artificial neural network-based machine learning interatomic potentials. Developed as an extension of the atomic energy network (ænet), ænet-PyTorch provides access to all the tools included in ænet for the application and usage of the potentials. The package has been designed as an alternative to the internal training capabilities of ænet, leveraging the power of graphic processing units to facilitate direct training on forces in addition to energies. This leads to a substantial reduction of the training time by one to two orders of magnitude compared to the central processing unit implementation, enabling direct training on forces for systems beyond small molecules. Here, we demonstrate the main features of ænet-PyTorch and show its performance on open databases. Our results show that training on all the force information within a dataset is not necessary, and including between 10% and 20% of the force information is sufficient to achieve optimally accurate interatomic potentials with the least computational resources. read less NOT USED (low confidence) B. Hamilton, P. Yoo, M. Sakano, M. M. Islam, and A. Strachan, “High-pressure and temperature neural network reactive force field for energetic materials.,” The Journal of chemical physics. 2023. link Times cited: 2 Abstract: Reactive force fields for molecular dynamics have enabled a … read moreAbstract: Reactive force fields for molecular dynamics have enabled a wide range of studies in numerous material classes. These force fields are computationally inexpensive compared with electronic structure calculations and allow for simulations of millions of atoms. However, the accuracy of traditional force fields is limited by their functional forms, preventing continual refinement and improvement. Therefore, we develop a neural network-based reactive interatomic potential for the prediction of the mechanical, thermal, and chemical responses of energetic materials at extreme conditions. The training set is expanded in an automatic iterative approach and consists of various CHNO materials and their reactions under ambient and shock-loading conditions. This new potential shows improved accuracy over the current state-of-the-art force fields for a wide range of properties such as detonation performance, decomposition product formation, and vibrational spectra under ambient and shock-loading conditions. read less NOT USED (low confidence) I. Lobzenko, Y. Shiihara, H. Mori, and T. Tsuru, “Influence of group IV element on basic mechanical properties of BCC medium-entropy alloys using machine-learning potentials,” Computational Materials Science. 2023. link Times cited: 1 NOT USED (low confidence) T. Mou et al., “Bridging the complexity gap in computational heterogeneous catalysis with machine learning,” Nature Catalysis. 2023. link Times cited: 17 NOT USED (low confidence) A. Weerasinghe, E. Martínez, B. Wirth, and D. Maroudas, “Molecular-Dynamics Analysis of the Mechanical Behavior of Plasma-Facing Tungsten.,” ACS applied materials & interfaces. 2023. link Times cited: 1 Abstract: We report a systematic computational analysis of the mechani… read moreAbstract: We report a systematic computational analysis of the mechanical behavior of plasma-facing component (PFC) tungsten focusing on the impact of void and helium (He) bubble defects on the mechanical response beyond the elastic regime. Specifically, we explore the effects of porosity and He atomic fraction on the mechanical properties and structural response of PFC tungsten, at varying temperature and bubble size. We find that the Young modulus of defective tungsten undergoes substantial softening that follows an exponential scaling relation as a function of matrix porosity and He atomic content. Beyond the elastic regime, our high strain rate simulations reveal that the presence of nanoscale spherical defects (empty voids and He bubbles) reduces the yield strength of tungsten in a monotonically decreasing fashion, obeying an exponential scaling relation as a function of tungsten matrix porosity and He concentration. Our detailed analysis of the structural response of PFC tungsten near the yield point reveals that yielding is initiated by emission of dislocation loops from bubble/matrix interfaces, mainly 1/2⟨111⟩ shear loops, followed by gliding and growth of these loops and reactions to form ⟨100⟩ dislocations. Furthermore, dislocation gliding on the ⟨111⟩{211} twin systems nucleates 1/6⟨111⟩ twin regions in the tungsten matrix. These dynamical processes reduce the stress in the matrix substantially. Subsequent dislocation interactions and depletion of the twin phases via nucleation and propagation of detwinning partials lead the tungsten matrix to a next deformation stage characterized by stress increase during applied straining. Our structural analysis reveals that the depletion of twin boundaries (areal defects) is strongly impacted by the density of He bubbles at higher porosities. After the initial stress relief upon yielding, increase in the dislocation density in conjunction with decrease in the areal defect density facilitates the initiation of dislocation-driven deformation mechanisms in the PFC crystal. read less NOT USED (low confidence) A. Seko, “Tutorial: Systematic development of polynomial machine learning potentials for elemental and alloy systems,” Journal of Applied Physics. 2023. link Times cited: 0 Abstract: Machine learning potentials (MLPs) developed from extensive … read moreAbstract: Machine learning potentials (MLPs) developed from extensive datasets constructed from density functional theory calculations have become increasingly appealing to many researchers. This paper presents a framework of polynomial-based MLPs, called polynomial MLPs. The systematic development of accurate and computationally efficient polynomial MLPs for many elemental and binary alloy systems and their predictive powers for various properties are also demonstrated. Consequently, many polynomial MLPs are available in a repository website [A. Seko, Polynomial Machine Learning Potential Repository at Kyoto University, https://sekocha.github.io ]. The repository will help many scientists perform accurate and efficient large-scale atomistic simulations and crystal structure searches. read less NOT USED (low confidence) M. Wen, E. Spotte-Smith, S. M. Blau, M. J. McDermott, A. Krishnapriyan, and K. Persson, “Chemical reaction networks and opportunities for machine learning,” Nature Computational Science. 2023. link Times cited: 11 NOT USED (low confidence) K. Dang, J. Chen, B. Rodgers, and S. Fensin, “LAVA 1.0: A general-purpose python toolkit for calculation of material properties with LAMMPS and VASP,” Comput. Phys. Commun. 2023. link Times cited: 2 NOT USED (low confidence) O. Gorynina, F. Legoll, T. Lelièvre, and D. Perez, “Combining machine-learned and empirical force fields with the parareal algorithm: application to the diffusion of atomistic defects,” ArXiv. 2022. link Times cited: 1 Abstract: We numerically investigate an adaptive version of the parare… read moreAbstract: We numerically investigate an adaptive version of the parareal algorithm in the context of molecular dynamics. This adaptive variant has been originally introduced in [F. Legoll, T. Lelievre and U. Sharma, SISC 2022]. We focus here on test cases of physical interest where the dynamics of the system is modelled by the Langevin equation and is simulated using the molecular dynamics software LAMMPS. In this work, the parareal algorithm uses a family of machine-learning spectral neighbor analysis potentials (SNAP) as fine, reference, potentials and embedded-atom method potentials (EAM) as coarse potentials. We consider a self-interstitial atom in a tungsten lattice and compute the average residence time of the system in metastable states. Our numerical results demonstrate significant computational gains using the adaptive parareal algorithm in comparison to a sequential integration of the Langevin dynamics. We also identify a large regime of numerical parameters for which statistical accuracy is reached without being a consequence of trajectorial accuracy. read less NOT USED (low confidence) H. Deng, J. Comer, and B. Liu, “A high-dimensional neural network potential for molecular dynamics simulations of condensed phase nickel and phase transitions,” Molecular Simulation. 2022. link Times cited: 0 Abstract: ABSTRACT A high-dimensional neural network interatomic poten… read moreAbstract: ABSTRACT A high-dimensional neural network interatomic potential was developed and used in molecular dynamics simulations of condensed phase Ni and Ni systems with liquid–solid phase coexistence. The reference data set was generated by sampling the potential energy surface over a broad temperature-pressure domain using ab initio MD simulations to train a unified potential. Excellent agreement was achieved between bulk face-centred cubic nickel thermal expansion simulations and relevant experimental data. The same potential also yields accurate structures and diffusivities in the liquid state. The phase transition between liquid and solid phases was simulated using the two-phase interface method. The predicted melting point temperature is within a few kelvins of the literature value. The general methodology could be applied to describe crystals with much more complex phase behaviours. read less NOT USED (low confidence) M. Chigaev et al., “Lightweight and effective tensor sensitivity for atomistic neural networks.,” The Journal of chemical physics. 2022. link Times cited: 3 Abstract: Atomistic machine learning focuses on the creation of models… read moreAbstract: Atomistic machine learning focuses on the creation of models that obey fundamental symmetries of atomistic configurations, such as permutation, translation, and rotation invariances. In many of these schemes, translation and rotation invariance are achieved by building on scalar invariants, e.g., distances between atom pairs. There is growing interest in molecular representations that work internally with higher rank rotational tensors, e.g., vector displacements between atoms, and tensor products thereof. Here, we present a framework for extending the Hierarchically Interacting Particle Neural Network (HIP-NN) with Tensor Sensitivity information (HIP-NN-TS) from each local atomic environment. Crucially, the method employs a weight tying strategy that allows direct incorporation of many-body information while adding very few model parameters. We show that HIP-NN-TS is more accurate than HIP-NN, with negligible increase in parameter count, for several datasets and network sizes. As the dataset becomes more complex, tensor sensitivities provide greater improvements to model accuracy. In particular, HIP-NN-TS achieves a record mean absolute error of 0.927 kcalmol for conformational energy variation on the challenging COMP6 benchmark, which includes a broad set of organic molecules. We also compare the computational performance of HIP-NN-TS to HIP-NN and other models in the literature. read less NOT USED (low confidence) E. Sikorski, M. Cusentino, M. J. McCarthy, J. Tranchida, M. A. Wood, and A. Thompson, “Machine learned interatomic potential for dispersion strengthened plasma facing components.,” The Journal of chemical physics. 2022. link Times cited: 5 Abstract: Tungsten (W) is a material of choice for the divertor materi… read moreAbstract: Tungsten (W) is a material of choice for the divertor material due to its high melting temperature, thermal conductivity, and sputtering threshold. However, W has a very high brittle-to-ductile transition temperature, and at fusion reactor temperatures (≥1000 K), it may undergo recrystallization and grain growth. Dispersion-strengthening W with zirconium carbide (ZrC) can improve ductility and limit grain growth, but much of the effects of the dispersoids on microstructural evolution and thermomechanical properties at high temperatures are still unknown. We present a machine learned Spectral Neighbor Analysis Potential for W-ZrC that can now be used to study these materials. In order to construct a potential suitable for large-scale atomistic simulations at fusion reactor temperatures, it is necessary to train on ab initio data generated for a diverse set of structures, chemical environments, and temperatures. Further accuracy and stability tests of the potential were achieved using objective functions for both material properties and high temperature stability. Validation of lattice parameters, surface energies, bulk moduli, and thermal expansion is confirmed on the optimized potential. Tensile tests of W/ZrC bicrystals show that although the W(110)-ZrC(111) C-terminated bicrystal has the highest ultimate tensile strength (UTS) at room temperature, observed strength decreases with increasing temperature. At 2500 K, the terminating C layer diffuses into the W, resulting in a weaker W-Zr interface. Meanwhile, the W(110)-ZrC(111) Zr-terminated bicrystal has the highest UTS at 2500 K. read less NOT USED (low confidence) H. Yu, L. Hong, S. Chen, X. Gong, and H. Xiang, “Capturing long-range interaction with reciprocal space neural network,” ArXiv. 2022. link Times cited: 4 Abstract: : Machine Learning (ML) interatomic models and potentials ha… read moreAbstract: : Machine Learning (ML) interatomic models and potentials have been widely employed in simulations of materials. Long-range interactions often dominate in some ionic systems whose dynamics behavior is significantly influenced. However, the long-range effect such as Coulomb and Van der Wales potential is not considered in most ML interatomic potentials. To address this issue, we put forward a method that can take long-range effects into account for most ML local interatomic models with the reciprocal space neural network. The structure information in real space is firstly transformed into reciprocal space and then encoded into a reciprocal space potential or a global descriptor with full atomic interactions. The reciprocal space potential and descriptor keep full invariance of Euclidean symmetry and choice of the cell. Benefiting from the reciprocal-space information, ML interatomic models can be extended to describe the long-range potential including not only Coulomb but any other long-range interaction. A model NaCl system considering Coulomb interaction and the Ga x N y system with defects are applied to illustrate the advantage of our approach. At the same time, our approach helps to improve the prediction accuracy of some global properties such as the band gap where the full atomic interaction beyond local atomic environments plays a very important role. In summary, our work has expanded the read less NOT USED (low confidence) J. L. A. Gardner, Z. Beaulieu, and V. L. Deringer, “Synthetic data enable experiments in atomistic machine learning,” ArXiv. 2022. link Times cited: 3 Abstract: Machine-learning models are increasingly used to predict pro… read moreAbstract: Machine-learning models are increasingly used to predict properties of atoms in chemical systems. There have been major advances in developing descriptors and regression frameworks for this task, typically starting from... read less NOT USED (low confidence) J. D. Morrow, J. L. A. Gardner, and V. L. Deringer, “How to validate machine-learned interatomic potentials.,” The Journal of chemical physics. 2022. link Times cited: 12 Abstract: Machine learning (ML) approaches enable large-scale atomisti… read moreAbstract: Machine learning (ML) approaches enable large-scale atomistic simulations with near-quantum-mechanical accuracy. With the growing availability of these methods, there arises a need for careful validation, particularly for physically agnostic models-that is, for potentials that extract the nature of atomic interactions from reference data. Here, we review the basic principles behind ML potentials and their validation for atomic-scale material modeling. We discuss the best practice in defining error metrics based on numerical performance, as well as physically guided validation. We give specific recommendations that we hope will be useful for the wider community, including those researchers who intend to use ML potentials for materials "off the shelf." read less NOT USED (low confidence) H. Hirai, T. Iizawa, T. Tamura, M. Karasuyama, R. Kobayashi, and T. Hirose, “Machine-learning-based prediction of first-principles XANES spectra for amorphous materials,” Physical Review Materials. 2022. link Times cited: 1 NOT USED (low confidence) Y. Zhang, Q. Lin, and B. Jiang, “Atomistic neural network representations for chemical dynamics simulations of molecular, condensed phase, and interfacial systems: Efficiency, representability, and generalization,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2022. link Times cited: 10 Abstract: Machine learning techniques have been widely applied in many… read moreAbstract: Machine learning techniques have been widely applied in many fields of chemistry, physics, biology, and materials science. One of the most fruitful applications is machine learning of the complicated multidimensional function of potential energy or related electronic properties from discrete quantum chemical data. In particular, substantial efforts have been dedicated to developing various atomistic neural network (AtNN) representations, which refer to a family of methods expressing the targeted physical quantity as a sum of atomic components represented by atomic NNs. This class of approaches not only fully preserves the physical symmetry of the system but also scales linearly with respect to the size of a system, enabling accurate and efficient chemical dynamics and spectroscopic simulations in complicated systems and even a number of variably sized systems across the phases. In this review, we discuss different strategies in developing highly efficient and representable AtNN potentials, and in generalizing these scalar AtNN models to learn vectorial and tensorial quantities with the correct rotational equivariance. We also review active learning algorithms to generate practical AtNN models and present selected examples of AtNN applications in gas‐surface systems to demonstrate their capabilities of accurately representing both molecular systems and condensed phase systems. We conclude this review by pointing out remaining challenges for the further development of more reliable, transferable, and scalable AtNN representations in more application scenarios. read less NOT USED (low confidence) A. Weerasinghe, B. Wirth, and D. Maroudas, “Thermal expansion of plasma-exposed tungsten,” Journal of Applied Physics. 2022. link Times cited: 1 Abstract: We report results from a systematic analysis of thermal expa… read moreAbstract: We report results from a systematic analysis of thermal expansion of plasma-exposed tungsten based on molecular-dynamics simulations using models of tungsten with distributions of helium (He) bubbles in the tungsten matrix. We distinguish between two approaches of filling the bubbles with He, where the amount of He in the bubble can or cannot vary with temperature. In the former case, the thermal expansion coefficient decreases monotonically with the porosity and He content of the tungsten matrix, while in the latter case, the thermal expansivity increases monotonically with increasing porosity and He content. The latter condition, where the He content in the bubble is determined at the implantation temperature and remains constant with varying temperature in the tungsten matrix, is consistent with He species transport in tungsten used as a plasma-facing component (PFC) in nuclear fusion reactors and implies the development of biaxial compressive thermal strains in the PFC material that contribute to accelerating the growth of a nanostructure on PFC tungsten surfaces. Our analysis advances the fundamental understanding of thermal expansion in PFC tungsten and contributes to the development of a thermophysical property database for properly incorporating effects of realistic heat loads into modeling the dynamical response of PFC tungsten under fusion reactor operating conditions. read less NOT USED (low confidence) D. Wang, Z. Wu, and X. Deng, “Thermal Conductivity of Hydrous Wadsleyite Determined by Non‐Equilibrium Molecular Dynamics Based on Machine Learning,” Geophysical Research Letters. 2022. link Times cited: 2 Abstract: The thermal conductivity of minerals is a fundamental parame… read moreAbstract: The thermal conductivity of minerals is a fundamental parameter in understanding the evolution and dynamics of the Earth. Wadsleyite, the major mineral in the mantle transition zone (MTZ), can contain abundant water. However, how water affects its thermal conductivity remains unknown. Here, we predicted the thermal conductivity of dry and hydrous wadsleyite at high pressure and temperature (P‐T) by combining non‐equilibrium molecular dynamics and machine learning potential trained with data from first‐principles calculations. We found that the thermal conductivity of wadsleyite is anisotropic and is reduced by ∼10% in the P‐T conditions of the MTZ by the presence of 0.81 wt.% water. The heat flow toward the slab tends to follow the direction with the lowest thermal conductivity due to the lattice‐preferred orientation of wadsleyite and olivine. Both hydration and thermal‐conductivity anisotropy slow down the heating of slabs, allowing hydrous minerals and metastable olivine to survive in the deeper mantle. read less NOT USED (low confidence) K. Pitike and W. Setyawan, “Accurate Fe–He machine learning potential for studying He effects in BCC-Fe,” Journal of Nuclear Materials. 2022. link Times cited: 1 NOT USED (low confidence) F. Wu et al., “Deep learning interatomic potential for Ca-O system at high pressure,” Physical Review Materials. 2022. link Times cited: 3 NOT USED (low confidence) A. Hernandez and T. Mueller, “Generalizability of Functional Forms for Interatomic Potential Models Discovered by Symbolic Regression,” ArXiv. 2022. link Times cited: 0 Abstract: In recent years there has been great progress in the use of … read moreAbstract: In recent years there has been great progress in the use of machine learning algorithms to develop interatomic potential models. Machine-learned potential models are typically orders of magnitude faster than density functional theory but also orders of magnitude slower than physics-derived models such as the embedded atom method. In our previous work, we used symbolic regression to develop fast, accurate and transferrable interatomic potential models for copper with novel functional forms that resemble those of the embedded atom method. To determine the extent to which the success of these forms was specific to copper, here we explore the generalizability of these models to other face-centered cubic transition metals and analyze their out-of-sample performance on several material properties. We found that these forms work particularly well on elements that are chemically similar to copper. When compared to optimized Sutton-Chen models, which have similar complexity, the functional forms discovered using symbolic regression perform better across all elements considered except gold where they have a similar performance. They perform similarly to a moderately more complex embedded atom form on properties on which they were trained, and they are more accurate on average on other properties. We attribute this improved generalized accuracy to the relative simplicity of the models discovered using symbolic regression. The genetic programming models are found to outperform other models from the literature about 50% of the time in a variety of property predictions, with about 1/10th the model complexity on average. We discuss the implications of these results to the broader application of symbolic regression to the development of new potentials and highlight how models discovered for one element can be used to seed new searches for different elements. read less NOT USED (low confidence) A. Lunghi and S. Sanvito, “Computational design of magnetic molecules and their environment using quantum chemistry, machine learning and multiscale simulations,” Nature Reviews Chemistry. 2022. link Times cited: 14 NOT USED (low confidence) Z. Zhang, L. Chen, J. Guo, X. Duan, B. Shan, and X. Duan, “Strategy to consider element distribution when constructing training datasets for developing machine learning potentials of alloys based on a Monte-Carlo-like method,” Physical Review B. 2022. link Times cited: 1 NOT USED (low confidence) M. L. H. Chandrappa, J. Qi, C. Chen, S. Banerjee, and S. Ong, “Thermodynamics and Kinetics of the Cathode–Electrolyte Interface in All-Solid-State Li–S Batteries,” Journal of the American Chemical Society. 2022. link Times cited: 9 Abstract: Lithium–sulfur batteries (LSBs) are among the most promising… read moreAbstract: Lithium–sulfur batteries (LSBs) are among the most promising energy storage technologies due to the low cost and high abundance of S. However, the issue of polysulfide shuttling with its corresponding capacity fading is a major impediment to its commercialization. Replacing traditional liquid electrolytes with solid-state electrolytes (SEs) is a potential solution. Here, we present a comprehensive study of the thermodynamics and kinetics of the cathode–electrolyte interface in all-solid-state LSBs using density functional theory based calculations and a machine learning interatomic potential. We find that among the major solid electrolyte chemistries (oxides, sulfides, nitrides, and halides), sulfide SEs are generally predicted to be the most stable against the S8 cathode, while the other SE chemistries are predicted to be highly electrochemically unstable. If the use of other SE chemistries is desired for other reasons, several binary and ternary sulfides (e.g., LiAlS2, Sc2S3, Y2S3) are predicted to be excellent buffer layers. Finally, an accurate moment tensor potential to study the S8|β-Li3PS4 interface was developed using an active learning approach. Molecular dynamics (MD) simulations of large interface models (>1000s atoms) revealed that the most stable Li3PS4(100) surface tends to form interfaces with S8 with 2D channels and lower activation barriers for Li diffusion. These results provide critical new insights into the cathode–electrolyte interface design for next-generation all-solid-state LSBs. read less NOT USED (low confidence) L. Wu et al., “Machine learning accelerated carbon neutrality research using big data—from predictive models to interatomic potentials,” Science China Technological Sciences. 2022. link Times cited: 2 NOT USED (low confidence) E. F. Bull-Vulpe, M. Riera, S. Bore, and F. Paesani, “Data-Driven Many-Body Potential Energy Functions for Generic Molecules: Linear Alkanes as a Proof-of-Concept Application.,” Journal of chemical theory and computation. 2022. link Times cited: 7 Abstract: We present a generalization of the many-body energy (MB-nrg)… read moreAbstract: We present a generalization of the many-body energy (MB-nrg) theoretical/computational framework that enables the development of data-driven potential energy functions (PEFs) for generic covalently bonded molecules, with arbitrary quantum mechanical accuracy. The "nearsightedness of electronic matter" is exploited to define monomers as "natural building blocks" on the basis of their distinct chemical identity. The energy of generic molecules is then expressed as a sum of individual many-body energies of incrementally larger subsystems. The MB-nrg PEFs represent the low-order n-body energies, with n = 1-4, using permutationally invariant polynomials derived from electronic structure data carried out at an arbitrary quantum mechanical level of theory, while all higher-order n-body terms (n > 4) are represented by a classical many-body polarization term. As a proof-of-concept application of the general MB-nrg framework, we present MB-nrg PEFs for linear alkanes. The MB-nrg PEFs are shown to accurately reproduce reference energies, harmonic frequencies, and potential energy scans of alkanes, independently of their length. Since, by construction, the MB-nrg framework introduced here can be applied to generic covalently bonded molecules, we envision future computer simulations of complex molecular systems using data-driven MB-nrg PEFs, with arbitrary quantum mechanical accuracy. read less NOT USED (low confidence) M. A. Caro, “Machine learning based modeling of disordered elemental semiconductors: understanding the atomic structure of a-Si and a-C,” Semiconductor Science and Technology. 2022. link Times cited: 1 Abstract: Disordered elemental semiconductors, most notably a-C and a-… read moreAbstract: Disordered elemental semiconductors, most notably a-C and a-Si, are ubiquitous in a myriad of different applications. These exploit their unique mechanical and electronic properties. In the past couple of decades, density functional theory (DFT) and other quantum mechanics-based computational simulation techniques have been successful at delivering a detailed understanding of the atomic and electronic structure of crystalline semiconductors. Unfortunately, the complex structure of disordered semiconductors sets the time and length scales required for DFT simulation of these materials out of reach. In recent years, machine learning (ML) approaches to atomistic modeling have been developed that provide an accurate approximation of the DFT potential energy surface for a small fraction of the computational time. These ML approaches have now reached maturity and are starting to deliver the first conclusive insights into some of the missing details surrounding the intricate atomic structure of disordered semiconductors. In this Topical Review we give a brief introduction to ML atomistic modeling and its application to amorphous semiconductors. We then take a look at how ML simulations have been used to improve our current understanding of the atomic structure of a-C and a-Si. read less NOT USED (low confidence) X. Liu, J. Zhang, and Z. Pei, “Machine Learning for High-entropy Alloys: Progress, Challenges and Opportunities,” Progress in Materials Science. 2022. link Times cited: 41 NOT USED (low confidence) D. P. Lara, H. Correa, and J. Diosa, “Molecular Dynamics Study of Structural and Transport Properties of Silver Iodide Using Effective Charges,” Molecules. 2022. link Times cited: 0 Abstract: The superionic conductor, solid state, and body-centered cub… read moreAbstract: The superionic conductor, solid state, and body-centered cubic structure, silver iodide at room temperature, has been studied via molecular dynamics simulations. The calculated results using pairwise Coulomb-Buckingham potential, zero pressure on the sample, a semi-rigid model system of 1000 Ag and 1000 I ions, (NVE) as a statistical ensemble, and an effective charge of Z=0.63 for the pairs Ag-Ag and I-I, were found to be consistent with experimental data and one study using Z=0.60, different potential, and simulation software. For the pair Ag-I, there is a discrepancy due to the high silver ion diffusion. The calculated value of the diffusion constant of the silver ion is greater than iodide ion. The dynamic transport properties (mean square displacement, velocity autocorrelation function) results indicated typical behavior reported by other authors, using different potentials in their DM simulations for iodine and silver ions. read less NOT USED (low confidence) N. Fedik et al., “Extending machine learning beyond interatomic potentials for predicting molecular properties,” Nature Reviews Chemistry. 2022. link Times cited: 30 NOT USED (low confidence) H. Kurban, M. Kurban, and M. M. Dalkilic, “Rapidly predicting Kohn–Sham total energy using data-centric AI,” Scientific Reports. 2022. link Times cited: 2 NOT USED (low confidence) K. Shimizu, Y. Dou, E. Arguelles, T. Moriya, E. Minamitani, and S. Watanabe, “Using neural network potentials to study defect formation and phonon properties of nitrogen vacancies with multiple charge states in GaN,” Physical Review B. 2022. link Times cited: 0 NOT USED (low confidence) A. Hegde, E. Weiss, W. Windl, H. Najm, and C. Safta, “Bayesian calibration of interatomic potentials for binary alloys,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) S. Sharma et al., “Machine Learning Methods for Multiscale Physics and Urban Engineering Problems,” Entropy. 2022. link Times cited: 0 Abstract: We present an overview of four challenging research areas in… read moreAbstract: We present an overview of four challenging research areas in multiscale physics and engineering as well as four data science topics that may be developed for addressing these challenges. We focus on multiscale spatiotemporal problems in light of the importance of understanding the accompanying scientific processes and engineering ideas, where “multiscale” refers to concurrent, non-trivial and coupled models over scales separated by orders of magnitude in either space, time, energy, momenta, or any other relevant parameter. Specifically, we consider problems where the data may be obtained at various resolutions; analyzing such data and constructing coupled models led to open research questions in various applications of data science. Numeric studies are reported for one of the data science techniques discussed here for illustration, namely, on approximate Bayesian computations. read less NOT USED (low confidence) A. Sundar, J. Yu, L. Qi, and M. N. Cinbiz, “High temperature stability and transport characteristics of hydrogen in alumina via multiscale computation,” International Journal of Hydrogen Energy. 2022. link Times cited: 1 NOT USED (low confidence) J. Guo et al., “Composition-transferable machine learning potential for LiCl-KCl molten salts validated by high-energy x-ray diffraction,” Physical Review B. 2022. link Times cited: 3 NOT USED (low confidence) P. A. Santos-Flórez et al., “Short-range order and its impacts on the BCC MoNbTaW multi-principal element alloy by the machine-learning potential,” Acta Materialia. 2022. link Times cited: 1 NOT USED (low confidence) S. Klawohn, J. Kermode, and A. P. Bart’ok, “Massively parallel fitting of Gaussian approximation potentials,” Machine Learning: Science and Technology. 2022. link Times cited: 2 Abstract: We present a data-parallel software package for fitting Gaus… read moreAbstract: We present a data-parallel software package for fitting Gaussian approximation potentials (GAPs) on multiple nodes using the ScaLAPACK library with MPI and OpenMP. Until now the maximum training set size for GAP models has been limited by the available memory on a single compute node. In our new implementation, descriptor evaluation is carried out in parallel with no communication requirement. The subsequent linear solve required to determine the model coefficients is parallelised with ScaLAPACK. Our approach scales to thousands of cores, lifting the memory limitation and also delivering substantial speedups. This development expands the applicability of the GAP approach to more complex systems as well as opening up opportunities for efficiently embedding GAP model fitting within higher-level workflows such as committee models or hyperparameter optimisation. read less NOT USED (low confidence) B. Bishnoi, “Lagrangian Density Space-Time Deep Neural Network Topology,” ArXiv. 2022. link Times cited: 1 Abstract: As a network-based functional approximator, we have proposed… read moreAbstract: As a network-based functional approximator, we have proposed a"Lagrangian Density Space-Time Deep Neural Networks"(LDDNN) topology. It is qualified for unsupervised training and learning to predict the dynamics of underlying physical science governed phenomena. The prototypical network respects the fundamental conservation laws of nature through the succinctly described Lagrangian and Hamiltonian density of the system by a given data-set of generalized nonlinear partial differential equations. The objective is to parameterize the Lagrangian density over a neural network and directly learn from it through data instead of hand-crafting an exact time-dependent"Action solution"of Lagrangian density for the physical system. With this novel approach, can understand and open up the information inference aspect of the"Black-box deep machine learning representation"for the physical dynamics of nature by constructing custom-tailored network interconnect topologies, activation, and loss/cost functions based on the underlying physical differential operators. This article will discuss statistical physics interpretation of neural networks in the Lagrangian and Hamiltonian domains. read less NOT USED (low confidence) E. Minamitani, I. Obayashi, K. Shimizu, and S. Watanabe, “Persistent homology-based descriptor for machine-learning potential of amorphous structures.,” The Journal of chemical physics. 2022. link Times cited: 0 Abstract: High-accuracy prediction of the physical properties of amorp… read moreAbstract: High-accuracy prediction of the physical properties of amorphous materials is challenging in condensed-matter physics. A promising method to achieve this is machine-learning potentials, which is an alternative to computationally demanding ab initio calculations. When applying machine-learning potentials, the construction of descriptors to represent atomic configurations is crucial. These descriptors should be invariant to symmetry operations. Handcrafted representations using a smooth overlap of atomic positions and graph neural networks (GNN) are examples of methods used for constructing symmetry-invariant descriptors. In this study, we propose a novel descriptor based on a persistence diagram (PD), a two-dimensional representation of persistent homology (PH). First, we demonstrated that the normalized two-dimensional histogram obtained from PD could predict the average energy per atom of amorphous carbon at various densities, even when using a simple model. Second, an analysis of the dimensional reduction results of the descriptor spaces revealed that PH can be used to construct descriptors with characteristics similar to those of a latent space in a GNN. These results indicate that PH is a promising method for constructing descriptors suitable for machine-learning potentials without hyperparameter tuning and deep-learning techniques. read less NOT USED (low confidence) M. D. K. Jones, J. Dawson, S. Campbell, V. Barrioz, L. D. Whalley, and Y. Qu, “Modelling Interfaces in Thin-Film Photovoltaic Devices,” Frontiers in Chemistry. 2022. link Times cited: 2 Abstract: Developing effective device architectures for energy technol… read moreAbstract: Developing effective device architectures for energy technologies—such as solar cells, rechargeable batteries or fuel cells—does not only depend on the performance of a single material, but on the performance of multiple materials working together. A key part of this is understanding the behaviour at the interfaces between these materials. In the context of a solar cell, efficient charge transport across the interface is a pre-requisite for devices with high conversion efficiencies. There are several methods that can be used to simulate interfaces, each with an in-built set of approximations, limitations and length-scales. These methods range from those that consider only composition (e.g. data-driven approaches) to continuum device models (e.g. drift-diffusion models using the Poisson equation) and ab-initio atomistic models (developed using e.g. density functional theory). Here we present an introduction to interface models at various levels of theory, highlighting the capabilities and limitations of each. In addition, we discuss several of the various physical and chemical processes at a heterojunction interface, highlighting the complex nature of the problem and the challenges it presents for theory and simulation. read less NOT USED (low confidence) I. Batatia, D. Kov’acs, G. Simm, C. Ortner, and G. Csányi, “MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields,” ArXiv. 2022. link Times cited: 102 Abstract: Creating fast and accurate force fields is a long-standing c… read moreAbstract: Creating fast and accurate force fields is a long-standing challenge in computational chemistry and materials science. Recently, several equivariant message passing neural networks (MPNNs) have been shown to outperform models built using other approaches in terms of accuracy. However, most MPNNs suffer from high computational cost and poor scalability. We propose that these limitations arise because MPNNs only pass two-body messages leading to a direct relationship between the number of layers and the expressivity of the network. In this work, we introduce MACE, a new equivariant MPNN model that uses higher body order messages. In particular, we show that using four-body messages reduces the required number of message passing iterations to just two, resulting in a fast and highly parallelizable model, reaching or exceeding state-of-the-art accuracy on the rMD17, 3BPA, and AcAc benchmark tasks. We also demonstrate that using higher order messages leads to an improved steepness of the learning curves. read less NOT USED (low confidence) N. Wilson, D. Willhelm, X. Qian, R. Arróyave, and X. Qian, “Batch active learning for accelerating the development of interatomic potentials,” Computational Materials Science. 2022. link Times cited: 5 NOT USED (low confidence) T. Ishiyama, T. Imajo, T. Suemasu, and K. Toko, “Machine learning of fake micrographs for automated analysis of crystal growth process,” Science and Technology of Advanced Materials: Methods. 2022. link Times cited: 3 Abstract: ABSTRACT Material informatics is being applied to crystal en… read moreAbstract: ABSTRACT Material informatics is being applied to crystal engineering, which is a core technology in electronics. Micrographs particularly provide important insights; however, they have not benefited significantly from material informatics because of the efforts required to acquire huge numbers of data. Herein, we propose a fast and automated analysis technique for micrographs showing the crystallization process of semiconductor thin films. We automatically generated fake micrographs and trained the crystal domain recognition capability on 10 different machine learning models. Experimentally obtained micrographs were analyzed using the developed model, which correctly determined the domain size and nuclei density. The activation energies required for growth and nucleation were determined from the lateral growth velocity and nucleation frequency, the variations of which were smaller than those measured by humans. Therefore, the proposed analysis framework not only reduces the time required to derive the crystal growth properties, but also enables a high accuracy without human subjectivity. GRAPHICAL ABSTRACT read less NOT USED (low confidence) N. Yao, X. Chen, Z. Fu, and Q. Zhang, “Applying Classical, Ab Initio, and Machine-Learning Molecular Dynamics Simulations to the Liquid Electrolyte for Rechargeable Batteries.,” Chemical reviews. 2022. link Times cited: 77 Abstract: Rechargeable batteries have become indispensable implements … read moreAbstract: Rechargeable batteries have become indispensable implements in our daily life and are considered a promising technology to construct sustainable energy systems in the future. The liquid electrolyte is one of the most important parts of a battery and is extremely critical in stabilizing the electrode-electrolyte interfaces and constructing safe and long-life-span batteries. Tremendous efforts have been devoted to developing new electrolyte solvents, salts, additives, and recipes, where molecular dynamics (MD) simulations play an increasingly important role in exploring electrolyte structures, physicochemical properties such as ionic conductivity, and interfacial reaction mechanisms. This review affords an overview of applying MD simulations in the study of liquid electrolytes for rechargeable batteries. First, the fundamentals and recent theoretical progress in three-class MD simulations are summarized, including classical, ab initio, and machine-learning MD simulations (section 2). Next, the application of MD simulations to the exploration of liquid electrolytes, including probing bulk and interfacial structures (section 3), deriving macroscopic properties such as ionic conductivity and dielectric constant of electrolytes (section 4), and revealing the electrode-electrolyte interfacial reaction mechanisms (section 5), are sequentially presented. Finally, a general conclusion and an insightful perspective on current challenges and future directions in applying MD simulations to liquid electrolytes are provided. Machine-learning technologies are highlighted to figure out these challenging issues facing MD simulations and electrolyte research and promote the rational design of advanced electrolytes for next-generation rechargeable batteries. read less NOT USED (low confidence) F. Wu et al., “Lattice inversion potential with neural network corrections for metallic systems,” Computational Materials Science. 2022. link Times cited: 0 NOT USED (low confidence) J. Doerfert et al., “Co-Designing an OpenMP GPU Runtime and Optimizations for Near-Zero Overhead Execution,” 2022 IEEE International Parallel and Distributed Processing Symposium (IPDPS). 2022. link Times cited: 8 Abstract: GPU accelerators are ubiquitous in modern HPC systems. To pr… read moreAbstract: GPU accelerators are ubiquitous in modern HPC systems. To program them, users have the choice between vendor-specific, native programming models, such as CUDA, which provide simple parallelism semantics with minimal runtime support, or portable alternatives, such as OpenMP, which offer rich parallel semantics and feature an extensive runtime library to support execution. While the operations of such a runtime can easily limit performance and drain resources, it was to some degree regarded an unavoidable overhead. In this work we present a co-design methodology for optimizing applications using a specifically crafted OpenMP GPU runtime such that most use cases induce near-zero overhead. Specifically, our approach exposes runtime semantics and state to the compiler such that optimization effectively eliminating abstractions and runtime state from the final binary. With the help of user provided assumptions we can further optimize common patterns that otherwise increase resource consumption. We evaluated our prototype build on top of the LLVM/OpenMP GPU offloading infrastructure with multiple HPC proxy applications and benchmarks. Comparison of CUDA, the original OpenMP runtime, and our co-designed alternative show that, by our approach, performance is significantly improved and resource consumption is significantly lowered. Oftentimes we can closely match the CUDA implementation without sacrificing the versatility and portability of OpenMP. read less NOT USED (low confidence) A. Mirzoev, B. Gelchinski, and A. A. Rempel, “Neural Network Prediction of Interatomic Interaction in Multielement Substances and High-Entropy Alloys: A Review,” Doklady Physical Chemistry. 2022. link Times cited: 2 NOT USED (low confidence) H. Kino, H. Dam, T. Miyake, and R. Mizoguchi, “Function Decomposition Tree with Causality-First Perspective and Systematic Description of Problems in Materials Informatics,” ArXiv. 2022. link Times cited: 0 Abstract: As interdisciplinary science is flourishing because of materi… read moreAbstract: As interdisciplinary science is flourishing because of materials informatics and additional factors; a systematic way is required for expressing knowledge and facilitating communication between scientists in various fields. A function decomposition tree is such a representation, but domain scientists face difficulty in constructing it. Thus, this study cites the general problems encountered by beginners in generating function decomposition trees and proposes a new function decomposition representation method based on a causality-first perspective for resolution of these problems. The causality-first decomposition tree was obtained from a workflow expressed according to the processing sequence. Moreover, we developed a program that performed automatic conversion using the features of the causality-first decomposition trees. The proposed method was applied to materials informatics to demonstrate the systematic representation of expert knowledge and its usefullness. read less NOT USED (low confidence) A. Mishra et al., “Virtual texture analysis to investigate the deformation mechanisms in metal microstructures at the atomic scale,” Journal of Materials Science. 2022. link Times cited: 8 NOT USED (low confidence) M. Müser, S. Sukhomlinov, and L. Pastewka, “Interatomic potentials: achievements and challenges,” Advances in Physics: X. 2022. link Times cited: 12 Abstract: ABSTRACT Interatomic potentials approximate the potential en… read moreAbstract: ABSTRACT Interatomic potentials approximate the potential energy of atoms as a function of their coordinates. Their main application is the effective simulation of many-atom systems. Here, we review empirical interatomic potentials designed to reproduce elastic properties, defect energies, bond breaking, bond formation, and even redox reactions. We discuss popular two-body potentials, embedded-atom models for metals, bond-order potentials for covalently bonded systems, polarizable potentials including charge-transfer approaches for ionic systems and quantum-Drude oscillator models mimicking higher-order and many-body dispersion. Particular emphasis is laid on the question what constraints ensue from the functional form of a potential, e.g., in what way Cauchy relations for elastic tensor elements can be violated and what this entails for the ratio of defect and cohesive energies, or why the ratio of boiling to melting temperature tends to be large for potentials describing metals but small for short-ranged pair potentials. The review is meant to be pedagogical rather than encyclopedic. This is why we highlight potentials with functional forms sufficiently simple to remain amenable to analytical treatments. Our main objective is to provide a stimulus for how existing approaches can be advanced or meaningfully combined to extent the scope of simulations based on empirical potentials. Graphical abstract read less NOT USED (low confidence) A. Musaelian et al., “Learning local equivariant representations for large-scale atomistic dynamics,” Nature Communications. 2022. link Times cited: 135 NOT USED (low confidence) Y. Gao, T. P. Mishra, S. H. Bo, G. Gautam, and P. Canepa, “Design and Characterization of Host Frameworks for Facile Magnesium Transport,” Annual Review of Materials Research. 2022. link Times cited: 9 Abstract: The development of inexpensive batteries based on magnesium … read moreAbstract: The development of inexpensive batteries based on magnesium (Mg) chemistry will contribute remarkably toward developing high-energy-density storage systems that can be used worldwide. Significant challenges remain in developing practical Mg batteries, the chief of which is designing materials that can provide facile transport of Mg. In this review, we cover the experimental and theoretical methods that can be used to quantify Mg mobility in a variety of host frameworks, the specific transport quantities that each technique is designed to measure or calculate, and some practical examples of their applications. We then list the unique challenges faced by different experimental and computational techniques in probing Mg ion transport in materials. This review concludes with an outlook on the directions that the scientific community could soon pursue as we strive to construct a pragmatic Mg battery. Expected final online publication date for the Annual Review of Materials Research, Volume 52 is July 2022. Please see http://www.annualreviews.org/page/journal/pubdates for revised estimates. read less NOT USED (low confidence) A. Eldridge, A. Rodriguez, M. Hu, and J. Hu, “Genetic programming-based learning of carbon interatomic potential for materials discovery,” ArXiv. 2022. link Times cited: 1 Abstract: Efficient and accurate interatomic potential functions are cr… read moreAbstract: Efficient and accurate interatomic potential functions are critical to computational study of materials while searching for structures with desired properties. Traditionally, potential functions or energy landscapes are designed by experts based on theoretical or heuristic knowledge. Here, we propose a new approach to leverage strongly typed parallel genetic programming (GP) for potential function discovery. We use a multi-objective evolutionary algorithm with NSGA-III selection to optimize individual age, fitness, and complexity through symbolic regression. With a DFT dataset of 863 unique carbon allotrope configurations drawn from 858 carbon structures, the generated potentials are able to predict total energies within ± 7 . 70 eV at low computational cost while generalizing well across multiple carbon structures. Our code is open source and available at http://www.github. com/usccolumbia/mlpotential . read less NOT USED (low confidence) T. Miyagawa, K. Mori, N. Kato, and A. Yonezu, “Development of neural network potential for MD simulation and its application to TiN,” Computational Materials Science. 2022. link Times cited: 3 NOT USED (low confidence) J. Byggmästar, K. Nordlund, and F. Djurabekova, “Simple machine-learned interatomic potentials for complex alloys,” Physical Review Materials. 2022. link Times cited: 5 Abstract: Developing data-driven machine-learning interatomic potentia… read moreAbstract: Developing data-driven machine-learning interatomic potentials for materials containing many elements becomes increasingly challenging due to the vast configuration space that must be sampled by the training data. We study the learning rates and achievable accuracy of machine-learning interatomic potentials for many-element alloys with different combinations of descriptors for the local atomic environments. We show that for a five-element alloy system, potentials using simple low-dimensional descriptors can reach meV/atom-accuracy with modestly sized training datasets, significantly outperforming the high-dimensional SOAP descriptor in data efficiency, accuracy, and speed. In particular, we develop a computationally fast machine-learned and tabulated Gaussian approximation potential (tabGAP) for Mo–Nb–Ta–V–W alloys with a combination of two-body, three-body, and a new simple scalar many-body density descriptor based on the embedded atom method. read less NOT USED (low confidence) V. Sharma and D. Datta, “Developing Potential Energy Surfaces for Graphene-based 2D-3D Interfaces from Modified High Dimensional Neural Networks for Applications in Energy Storage,” Journal of Electrochemical Energy Conversion and Storage. 2022. link Times cited: 3 Abstract:
Designing a new heterostructure electrode has many challen… read moreAbstract:
Designing a new heterostructure electrode has many challenges associated with interface engineering. Demanding simulation resources and lack of heterostructure databases continue to be a barrier to understanding the chemistry and mechanics of complex interfaces using simulations. Mixed-dimensional heterostructures composed of two-dimensional (2D) and three-dimensional (3D) materials are undisputed next-generation materials for engineered devices due to their changeable properties. The present work computationally investigates the interface between 2D graphene and 3D tin (Sn) systems with density functional theory (DFT) method. It uses computationally demanding simulation data to develop machine learning (ML) based potential energy surfaces (PES). The approach to developing PES for complex interface systems in the light of limited data and transferability of such models has been discussed. To develop PES for graphene-tin interface systems, high dimensional neural networks (HDNN) are used that rely on atom-centered symmetry function to represent structural information. HDNN are modified to train on the total energies of the interface system rather than atomic energies. The performance of modified HDNN trained on 5789 interface structures of graphene|Sn is tested on new interfaces of the same material pair with varying levels of structural deviations from the training dataset. Root mean squared error (RMSE) for test interfaces fall in the range of 0.01 – 0.45 eV/atom, depending on the structural deviations from the reference training dataset. By avoiding incorrect decomposition of total energy into atomic energies, modified HDNN model is shown to obtain higher accuracy and transferability despite limited dataset. Improved accuracy in ML-based modeling approach promises cost-effective means of designing interfaces in heterostructure energy storage systems with higher cycle life and stability. read less NOT USED (low confidence) H. Wei, H. Bao, and X. Ruan, “Perspective: predicting and optimizing thermal transport properties with machine learning methods,” Energy and AI. 2022. link Times cited: 19 NOT USED (low confidence) W. Qian, X. Xue, J. Liu, and C. Zhang, “Molecular Forcefield Methods for Describing Energetic Molecular Crystals: A Review,” Molecules. 2022. link Times cited: 6 Abstract: Energetic molecular crystals are widely applied for military… read moreAbstract: Energetic molecular crystals are widely applied for military and civilian purposes, and molecular forcefields (FF) are indispensable for treating the microscopic issues therein. This article reviews the three types of molecular FFs that are applied widely for describing energetic crystals—classic FFs, consistent FFs, and reactive FFs (ReaxFF). The basic principle of each type of FF is briefed and compared, with the application introduced, predicting polymorph, morphology, thermodynamics, vibration spectra, thermal property, mechanics, and reactivity. Finally, the advantages and disadvantages of these FFs are summarized, and some directions of future development are suggested. read less NOT USED (low confidence) W. Yang et al., “Exploring the Effects of Ionic Defects on the Stability of CsPbI3 with a Deep Learning Potential.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2022. link Times cited: 9 Abstract: Inorganic metal halide perovskites, such as CsPbI3 , have re… read moreAbstract: Inorganic metal halide perovskites, such as CsPbI3 , have recently drawn extensive attention due to their excellent optical properties and high photoelectric efficiencies. However, the structural instability originating from inherent ionic defects leads to a sharp drop in the photoelectric efficiency, which significantly limits their applications in solar cells. The instability induced by ionic defects remains unresolved due to its complicated reaction process. Herein, to explore the effects of ionic defects on stability, we develop a deep learning potential for a CsPbI3 ternary system based upon density functional theory (DFT) calculated data for large-scale molecular dynamics (MD) simulations. By exploring 2.4 million configurations, of which 7,730 structures are used for the training set, the deep learning potential shows an accuracy approaching DFT-level. Furthermore, MD simulations with a 5,000-atom system and a one nanosecond timeframe are performed to explore the effects of bulk and surface defects on the stability of CsPbI3 . This deep learning potential based MD simulation provides solid evidence together with the derived radial distribution functions, simulated diffraction of X-rays, instability temperature, molecular trajectory, and coordination number for revealing the instability mechanism of CsPbI3 . Among bulk defects, Cs defects have the most significant influence on the stability of CsPbI3 with a defect tolerance concentration of 0.32 %, followed by Pb and I defects. With regards to surface defects, Cs defects have the largest impact on the stability of CsPbI3 when the defect concentration is less than 15 %, whereas Pb defects act play a dominant role for defect concentrations exceeding 20 %. Most importantly, this machine-learning-based MD simulation strategy provides a new avenue to explore the ionic defect effects on the stability of perovskite-like materials, laying a theoretical foundation for the design of stable perovskite materials. read less NOT USED (low confidence) R. Fabregat et al., “Local Kernel Regression and Neural Network Approaches to the Conformational Landscapes of Oligopeptides,” Journal of Chemical Theory and Computation. 2022. link Times cited: 4 Abstract: The application of machine learning to theoretical chemistry… read moreAbstract: The application of machine learning to theoretical chemistry has made it possible to combine the accuracy of quantum chemical energetics with the thorough sampling of finite-temperature fluctuations. To reach this goal, a diverse set of methods has been proposed, ranging from simple linear models to kernel regression and highly nonlinear neural networks. Here we apply two widely different approaches to the same, challenging problem: the sampling of the conformational landscape of polypeptides at finite temperature. We develop a local kernel regression (LKR) coupled with a supervised sparsity method and compare it with a more established approach based on Behler-Parrinello type neural networks. In the context of the LKR, we discuss how the supervised selection of the reference pool of environments is crucial to achieve accurate potential energy surfaces at a competitive computational cost and leverage the locality of the model to infer which chemical environments are poorly described by the DFTB baseline. We then discuss the relative merits of the two frameworks and perform Hamiltonian-reservoir replica-exchange Monte Carlo sampling and metadynamics simulations, respectively, to demonstrate that both frameworks can achieve converged and transferable sampling of the conformational landscape of complex and flexible biomolecules with comparable accuracy and computational cost. read less NOT USED (low confidence) C. Chen and S. Ong, “A universal graph deep learning interatomic potential for the periodic table,” Nature Computational Science. 2022. link Times cited: 98 NOT USED (low confidence) J. Guo et al., “A Composition-Transferable Machine Learning Potential for LiCl-KCl Molten Salts Validated by HEXRD.” 2022. link Times cited: 0 Abstract: Unraveling the liquid structure of multi-component molten sa… read moreAbstract: Unraveling the liquid structure of multi-component molten salts is challenging due to the difficulty in conducting and interpreting high temperature diffraction experiments. Motivated by this challenge, we developed composition-transferable Gaussian Approximation Potentials (GAP) for molten LiCl-KCl. A DFT-SCAN accurate GAP is active learned from only ~1100 training configurations drawn from 10 unique mixture compositions enriched with metadynamics. The GAP-computed structures show strong agreement across HEXRD experiments, including for a eutectic not explicitly included in model training, thereby opening the possibility for composition discovery. read less NOT USED (low confidence) Z. Wei, C. Zhang, Y. Kan, Y. Zhang, and Y. Chen, “Developing machine learning potential for classical molecular dynamics simulation with superior phonon properties,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) Y. Wang et al., “Machine-learning interatomic potential for radiation damage effects in bcc-iron,” Computational Materials Science. 2022. link Times cited: 7 NOT USED (low confidence) C. Loyola, S. Davis, and J. Peralta, “Nonequilibrium, highly inhomogeneous melting in the microcanonical ensemble,” Physica A: Statistical Mechanics and its Applications. 2022. link Times cited: 0 NOT USED (low confidence) A. Bochkarev, Y. Lysogorskiy, S. Menon, M. Qamar, M. Mrovec, and R. Drautz, “Efficient parametrization of the atomic cluster expansion,” Physical Review Materials. 2022. link Times cited: 19 NOT USED (low confidence) A. Pandey, J. Gigax, and R. Pokharel, “Machine Learning Interatomic Potential for High-Throughput Screening of High-Entropy Alloys,” JOM. 2022. link Times cited: 1 NOT USED (low confidence) Z. Guo et al., “Extending the limit of molecular dynamics with ab initio accuracy to 10 billion atoms,” Proceedings of the 27th ACM SIGPLAN Symposium on Principles and Practice of Parallel Programming. 2022. link Times cited: 19 Abstract: High-performance computing, together with a neural network m… read moreAbstract: High-performance computing, together with a neural network model trained from data generated with first-principles methods, has greatly boosted applications of ab initio molecular dynamics in terms of spatial and temporal scales on modern supercomputers. Previous state-of-the-art can achieve 1 -- 2 nanoseconds molecular dynamics simulation per day for 100-million atoms on the entire Summit supercomputer. In this paper, we have significantly reduced the memory footprint and computational time by a comprehensive approach with both algorithmic and system innovations. The neural network model is compressed by model tabulation, kernel fusion, and redundancy removal. Then optimizations such as acceleration of customized kernel, tabulation of activation function, MPI+OpenMP parallelization are implemented on GPU and ARM architectures. Testing results of the copper system show that the optimized code can scale up to the entire machine of both Fugaku and Summit, and the corresponding system size can be extended by a factor of 134 to an unprecedented 17 billion atoms. The strong scaling of a 13.5-million atom copper system shows that the time-to-solution can be 7 times faster, reaching 11.2 nanoseconds per day. This work opens the door for unprecedentedly large-scale molecular dynamics simulations based on ab initio accuracy and can be potentially utilized in studying more realistic applications such as mechanical properties of metals, semiconductor devices, batteries, etc. The optimization techniques detailed in this paper also provide insight for relevant high-performance computing applications. read less NOT USED (low confidence) E. Huang et al., “Machine-learning and high-throughput studies for high-entropy materials,” Materials Science and Engineering: R: Reports. 2022. link Times cited: 39 NOT USED (low confidence) S. Raghunathan and U. Priyakumar, “Molecular representations for machine learning applications in chemistry,” International Journal of Quantum Chemistry. 2021. link Times cited: 21 NOT USED (low confidence) L. Zhang, H. Wang, M. Muniz, A. Panagiotopoulos, R. Car, and W. E, “A deep potential model with long-range electrostatic interactions.,” The Journal of chemical physics. 2021. link Times cited: 42 Abstract: Machine learning models for the potential energy of multi-at… read moreAbstract: Machine learning models for the potential energy of multi-atomic systems, such as the deep potential (DP) model, make molecular simulations with the accuracy of quantum mechanical density functional theory possible at a cost only moderately higher than that of empirical force fields. However, the majority of these models lack explicit long-range interactions and fail to describe properties that derive from the Coulombic tail of the forces. To overcome this limitation, we extend the DP model by approximating the long-range electrostatic interaction between ions (nuclei + core electrons) and valence electrons with that of distributions of spherical Gaussian charges located at ionic and electronic sites. The latter are rigorously defined in terms of the centers of the maximally localized Wannier distributions, whose dependence on the local atomic environment is modeled accurately by a deep neural network. In the DP long-range (DPLR) model, the electrostatic energy of the Gaussian charge system is added to short-range interactions that are represented as in the standard DP model. The resulting potential energy surface is smooth and possesses analytical forces and virial. Missing effects in the standard DP scheme are recovered, improving on accuracy and predictive power. By including long-range electrostatics, DPLR correctly extrapolates to large systems the potential energy surface learned from quantum mechanical calculations on smaller systems. We illustrate the approach with three examples: the potential energy profile of the water dimer, the free energy of interaction of a water molecule with a liquid water slab, and the phonon dispersion curves of the NaCl crystal. read less NOT USED (low confidence) S. Arabha, Z. S. Aghbolagh, K. Ghorbani, S. M. Hatam-Lee, and A. Rajabpour, “Recent advances in lattice thermal conductivity calculation using machine-learning interatomic potentials,” Journal of Applied Physics. 2021. link Times cited: 17 NOT USED (low confidence) J. A. Vita and D. Trinkle, “Exploring the necessary complexity of interatomic potentials,” Computational Materials Science. 2021. link Times cited: 8 NOT USED (low confidence) S. Wyant, A. Rohskopf, and A. Henry, “Machine learned interatomic potentials for modeling interfacial heat transport in Ge/GaAs,” Computational Materials Science. 2021. link Times cited: 4 NOT USED (low confidence) J. D. Morrow and V. L. Deringer, “Indirect learning and physically guided validation of interatomic potential models.,” The Journal of chemical physics. 2021. link Times cited: 3 Abstract: Machine learning (ML) based interatomic potentials are emerg… read moreAbstract: Machine learning (ML) based interatomic potentials are emerging tools for material simulations, but require a trade-off between accuracy and speed. Here, we show how one can use one ML potential model to train another: we use an accurate, but more computationally expensive model to generate reference data (locations and labels) for a series of much faster potentials. Without the need for quantum-mechanical reference computations at the secondary stage, extensive reference datasets can be easily generated, and we find that this improves the quality of fast potentials with less flexible functional forms. We apply the technique to disordered silicon, including a simulation of vitrification and polycrystalline grain formation under pressure with a system size of a million atoms. Our work provides conceptual insight into the ML of interatomic potential models and suggests a route toward accelerated simulations of condensed-phase systems. read less NOT USED (low confidence) J. Gao, X. Luo, F. Fang, and J. Sun, “Fundamentals of atomic and close-to-atomic scale manufacturing:A review,” International Journal of Extreme Manufacturing. 2021. link Times cited: 42 Abstract:
Atomic and Close-to-atomic Scale Manufacturing (ACSM) repr… read moreAbstract:
Atomic and Close-to-atomic Scale Manufacturing (ACSM) represents techniques for manufacturing high-end products in various fields, including future-generation computing, communication, energy and medical devices and materials. In this paper, the theoretical boundary between ACSM and classical manufacturing is identified after a thorough discussion of quantum mechanics and their effects on manufacturing. The physical origins of atomic interactions and energy beams-matter interactions are revealed from the point view of quantum mechanics. The mechanisms that dominate several key ACSM processes are introduced, and a current numerical study on these processes is reviewed. A comparison of current ACSM processes is performed in terms of dominant interactions, representative processes, resolution and modelling methods. Future fundamental research is proposed for establishing new approaches for modelling ACSM, material selection or preparation and control of manufacturing tools and environments. This paper is by no means comprehensive, but provides a starting point for further systematic investigation of ACSM fundamentals to support and accelerate its industrial scale implementation in the near future. read less NOT USED (low confidence) C. J. Leverant, J. A. Harvey, T. Alam, and J. Greathouse, “Machine Learning Self-Diffusion Prediction for Lennard-Jones Fluids in Pores,” The Journal of Physical Chemistry C. 2021. link Times cited: 10 NOT USED (low confidence) C. Daley et al., “Non-Recurring Engineering (NRE) Best Practices: A Case Study with the NERSC/NVIDIA OpenMP Contract,” SC21: International Conference for High Performance Computing, Networking, Storage and Analysis. 2021. link Times cited: 1 Abstract: The NERSC supercomputer, Perlmutter, consists of AMD CPUs an… read moreAbstract: The NERSC supercomputer, Perlmutter, consists of AMD CPUs and NVIDIA GPUs. NERSC users expect to be able to use OpenMP to take advantage of the highly capable GPUs. This paper describes how NERSC/NVIDIA constructed a Non-Recurring Engineering (NRE) contract to add OpenMP GPU-offload support to the NVIDIA HPC compilers. The paper describes how the contract incorporated the strengths of both parties and encouraged collaboration to improve the quality of the final deliverable. We include our best practices and how this particular contract took into account emerging OpenMP specifications, NERSC workload requirements, and how to use OpenMP most efficiently on GPU hardware. This paper includes OpenMP application performance results obtained with the NVIDIA compilers distributed in the NVIDIA HPC SDK. read less NOT USED (low confidence) K. Xie et al., “Neural network potential for Zr–Rh system by machine learning,” Journal of Physics: Condensed Matter. 2021. link Times cited: 3 Abstract: Zr–Rh metallic glass has enabled its many applications in ve… read moreAbstract: Zr–Rh metallic glass has enabled its many applications in vehicle parts, sports equipment and so on due to its outstanding performance in mechanical property, but the knowledge of the microstructure determining the superb mechanical property remains yet insufficient. Here, we develop a deep neural network potential of Zr–Rh system by using machine learning, which breaks the dilemma between the accuracy and efficiency in molecular dynamics simulations, and greatly improves the simulation scale in both space and time. The results show that the structural features obtained from the neural network method are in good agreement with the cases in ab initio molecular dynamics simulations. Furthermore, we build a large model of 5400 atoms to explore the influences of simulated size and cooling rate on the melt-quenching process of Zr77Rh23. Our study lays a foundation for exploring the complex structures in amorphous Zr77Rh23, which is of great significance for the design and practical application. read less NOT USED (low confidence) D. E. Farache, J. C. Verduzco, Z. D. McClure, S. Desai, and A. Strachan, “Active learning and molecular dynamics simulations to find high melting temperature alloys,” Computational Materials Science. 2021. link Times cited: 9 NOT USED (low confidence) P. Liu, C. Verdi, F. Karsai, and G. Kresse, “Phase transitions of zirconia: Machine-learned force fields beyond density functional theory,” Physical Review B. 2021. link Times cited: 12 Abstract: We present an approach to generate machine-learned force fie… read moreAbstract: We present an approach to generate machine-learned force fields (MLFF) with beyond density functional theory (DFT) accuracy. Our approach combines on-the-fly active learning and $\Delta$-machine learning in order to generate an MLFF for zirconia based on the random phase approximation (RPA). Specifically, an MLFF trained on-the-fly during DFT based molecular dynamics simulations is corrected by another MLFF that is trained on the differences between RPA and DFT calculated energies, forces and stress tensors. Thanks to the relatively smooth nature of the differences, the expensive RPA calculations are performed only on a small number of representative structures of small unit cells. These structures are determined by a singular value decomposition rank compression of the kernel matrix with low spatial resolution. This dramatically reduces the computational cost and allows us to generate an MLFF fully capable of reproducing high-level quantum-mechanical calculations beyond DFT. We carefully validate our approach and demonstrate its success in studying the phase transitions of zirconia. read less NOT USED (low confidence) A. Thompson et al., “LAMMPS - A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales,” Computer Physics Communications. 2021. link Times cited: 2377 NOT USED (low confidence) X. Qian and R. Yang, “Machine learning for predicting thermal transport properties of solids,” Materials Science and Engineering: R: Reports. 2021. link Times cited: 34 NOT USED (low confidence) J. Roberts, J. R. S. Bursten, and C. Risko, “Genetic Algorithms and Machine Learning for Predicting Surface Composition, Structure, and Chemistry: A Historical Perspective and Assessment,” Chemistry of Materials. 2021. link Times cited: 7 NOT USED (low confidence) V. L. Deringer, A. Bartók, N. Bernstein, D. Wilkins, M. Ceriotti, and G. Csányi, “Gaussian Process Regression for Materials and Molecules,” Chemical Reviews. 2021. link Times cited: 316 Abstract: We provide an introduction to Gaussian process regression (G… read moreAbstract: We provide an introduction to Gaussian process regression (GPR) machine-learning methods in computational materials science and chemistry. The focus of the present review is on the regression of atomistic properties: in particular, on the construction of interatomic potentials, or force fields, in the Gaussian Approximation Potential (GAP) framework; beyond this, we also discuss the fitting of arbitrary scalar, vectorial, and tensorial quantities. Methodological aspects of reference data generation, representation, and regression, as well as the question of how a data-driven model may be validated, are reviewed and critically discussed. A survey of applications to a variety of research questions in chemistry and materials science illustrates the rapid growth in the field. A vision is outlined for the development of the methodology in the years to come. read less NOT USED (low confidence) X. Chen et al., “Machine learning enhanced empirical potentials for metals and alloys,” Comput. Phys. Commun. 2021. link Times cited: 5 NOT USED (low confidence) H. Min et al., “Development of an interatomic potential for Fe-He by neural network,” Computational Materials Science. 2021. link Times cited: 2 NOT USED (low confidence) S. Ignatov, S. N. Belyaev, S. Panteleev, and A. Masunov, “How Many Isomers Do Metallic Clusters Have? Case of Magnesium Clusters of up to 55 Atoms.,” The journal of physical chemistry. A. 2021. link Times cited: 4 Abstract: About 9000 structures of magnesium clusters Mgn (n = 2-13) g… read moreAbstract: About 9000 structures of magnesium clusters Mgn (n = 2-13) generated via different methods were optimized at the DFT levels in order to estimate the number of all possible stable structures that can exist for the given cluster size (∼820,000 PES points were explored in total). It was found that the number of possible cluster isomers N quickly grows with a number of atoms n; however, it is significantly lower than the number of possible nonisomorphic graph structures, which can be drawn for the given n. At the DFT potential energy surface, we found only 543 local minima corresponding to the isomers of Mg2-Mg13. The number of isomers obtained in the DFT optimizations grows with n approximately as n4, whereas the N values extrapolated to the infinite generation process grow as n8. The cluster geometries obtained from the global DFT optimization were then used to adjust two empirical potentials of Gupta type (GP) and modified Sutton-Chen type (SCG3) describing the interactions between the magnesium atoms. Using these potentials, the extensive sets of structures Mg2-Mg55 (up to 30,000 clusters for each n) were optimized to obtain the dependence of the cluster isomer count on n in the continuous range of n = 2-30 and for selected n up to n = 55. It was found that the SCG3 potential, which is closer to the DFT results, gives a number of possible isomers growing as approximately n8.9, whereas GP potential results in the n4.3 dependence. read less NOT USED (low confidence) E. F. Bull-Vulpe, M. Riera, A. Goetz, and F. Paesani, “MB-Fit: Software infrastructure for data-driven many-body potential energy functions.,” The Journal of chemical physics. 2021. link Times cited: 20 Abstract: Many-body potential energy functions (MB-PEFs), which integr… read moreAbstract: Many-body potential energy functions (MB-PEFs), which integrate data-driven representations of many-body short-range quantum mechanical interactions with physics-based representations of many-body polarization and long-range interactions, have recently been shown to provide high accuracy in the description of molecular interactions from the gas to the condensed phase. Here, we present MB-Fit, a software infrastructure for the automated development of MB-PEFs for generic molecules within the TTM-nrg (Thole-type model energy) and MB-nrg (many-body energy) theoretical frameworks. Besides providing all the necessary computational tools for generating TTM-nrg and MB-nrg PEFs, MB-Fit provides a seamless interface with the MBX software, a many-body energy and force calculator for computer simulations. Given the demonstrated accuracy of the MB-PEFs, particularly within the MB-nrg framework, we believe that MB-Fit will enable routine predictive computer simulations of generic (small) molecules in the gas, liquid, and solid phases, including, but not limited to, the modeling of quantum isomeric equilibria in molecular clusters, solvation processes, molecular crystals, and phase diagrams. read less NOT USED (low confidence) M. Kulichenko et al., “The Rise of Neural Networks for Materials and Chemical Dynamics.,” The journal of physical chemistry letters. 2021. link Times cited: 35 Abstract: Machine learning (ML) is quickly becoming a premier tool for… read moreAbstract: Machine learning (ML) is quickly becoming a premier tool for modeling chemical processes and materials. ML-based force fields, trained on large data sets of high-quality electron structure calculations, are particularly attractive due their unique combination of computational efficiency and physical accuracy. This Perspective summarizes some recent advances in the development of neural network-based interatomic potentials. Designing high-quality training data sets is crucial to overall model accuracy. One strategy is active learning, in which new data are automatically collected for atomic configurations that produce large ML uncertainties. Another strategy is to use the highest levels of quantum theory possible. Transfer learning allows training to a data set of mixed fidelity. A model initially trained to a large data set of density functional theory calculations can be significantly improved by retraining to a relatively small data set of expensive coupled cluster theory calculations. These advances are exemplified by applications to molecules and materials. read less NOT USED (low confidence) Z. Cheng, J. Du, L. Zhang, J. Ma, W. Li, and S. Li, “Building Machine Learning Force Fields of Proteins with Fragment-Based Approach and Data Transfer.” 2021. link Times cited: 1 Abstract: We combined our generalized energy-based fragmentation (GEBF… read moreAbstract: We combined our generalized energy-based fragmentation (GEBF) approach and transfer learning technique to construct machine learning force fields (MLFFs) for proteins only from quantum mechanics (QM) calculations of small subsystems. Using a kernel-based model called Gaussian Approximation Potential (GAP), our protocol can automatically generate training sets with high efficiency. To facilitate the construction of training sets for various proteins, a protein’s data library is created to store all data of subsystems generated from trained proteins. With this data library, for a new protein only its subsystems with new topological types are required for the construction of the corresponding training set. With two polypeptides, 4ZNN and 1XQ8 segment, as examples, we demonstrate that GEBF-MLFFs can be constructed by either kernel methods or neural network methods with full QM quality. Therefore, the present work provides an effi-cient and systematic way to build force fields for biological systems like proteins with QM accuracy. read less NOT USED (low confidence) S.-H. Guan, C. Shang, and Z. Liu, “Structure and Dynamics of Energy Materials from Machine Learning Simulations: A Topical Review,” Chinese Journal of Chemistry. 2021. link Times cited: 4 Abstract: Energy materials featuring the capability to store and relea… read moreAbstract: Energy materials featuring the capability to store and release chemical energy reversibly involve generally complex geometrical structures with multiple elements. It has been a great challenge to establish the quantitative relationship between the structure of materials and their dynamic physicochemical properties. In recent years, machine learning (ML) technique has demonstrated its great power in accelerating the research on energy materials. This topical review introduces the key ingredients and typical applications of ML to energy materials. We mainly focus on the ML based atomic simulation via ML potentials in different architectures/implementations, including high dimensional neural networks (HDNN), Gaussian approximation potential (GAP), moment tensor potentials (MTP) and stochastic surface walking global optimization with global neural network potential (SSW-NN) method. Three cases studies, namely, Si, LiC and LiTiO systems, are presented to demonstrate the ability of ML simulation in assessing the thermodynamics and kinetics of complex material systems. We highlight that the SSW-NN method provides an automated solution for global potential energy surface data collection, ML potential construction and ML simulation, which boosts the current ability for large-scale atomic simulation and thus holds the great promise for fast property evaluation and material discovery. read less NOT USED (low confidence) M. Gilbert et al., “Perspectives on multiscale modelling and experiments to accelerate materials development for fusion,” Journal of Nuclear Materials. 2021. link Times cited: 33 NOT USED (low confidence) G. Pilania, “Machine learning in materials science: From explainable predictions to autonomous design,” Computational Materials Science. 2021. link Times cited: 85 NOT USED (low confidence) S. Fujii and A. Seko, “Structure and lattice thermal conductivity of grain boundaries in silicon by using machine learning potential and molecular dynamics,” Computational Materials Science. 2021. link Times cited: 8 NOT USED (low confidence) T. Kwon, H.-W. Song, S. Woo, J.-H. Kim, and B. Sung, “The estimation of the second virial coefficients of He and N2 based on neural network potentials with quantum mechanical calculations,” Chemical Physics. 2021. link Times cited: 2 NOT USED (low confidence) A. Glielmo, B. Husic, A. Rodriguez, C. Clementi, F. Noé, and A. Laio, “Unsupervised Learning Methods for Molecular Simulation Data,” Chemical Reviews. 2021. link Times cited: 138 Abstract: Unsupervised learning is becoming an essential tool to analy… read moreAbstract: Unsupervised learning is becoming an essential tool to analyze the increasingly large amounts of data produced by atomistic and molecular simulations, in material science, solid state physics, biophysics, and biochemistry. In this Review, we provide a comprehensive overview of the methods of unsupervised learning that have been most commonly used to investigate simulation data and indicate likely directions for further developments in the field. In particular, we discuss feature representation of molecular systems and present state-of-the-art algorithms of dimensionality reduction, density estimation, and clustering, and kinetic models. We divide our discussion into self-contained sections, each discussing a specific method. In each section, we briefly touch upon the mathematical and algorithmic foundations of the method, highlight its strengths and limitations, and describe the specific ways in which it has been used-or can be used-to analyze molecular simulation data. read less NOT USED (low confidence) X. Wang, S. Xu, W. Jian, X.-G. Li, Y. Su, and I. Beyerlein, “Generalized stacking fault energies and Peierls stresses in refractory body-centered cubic metals from machine learning-based interatomic potentials,” Computational Materials Science. 2021. link Times cited: 30 NOT USED (low confidence) M. Liu, Y. Yang, and J. Kitchin, “Semi-grand canonical Monte Carlo simulation of the acrolein induced surface segregation and aggregation of AgPd with machine learning surrogate models.,” The Journal of chemical physics. 2021. link Times cited: 7 Abstract: The single atom alloy of AgPd has been found to be a promisi… read moreAbstract: The single atom alloy of AgPd has been found to be a promising catalyst for the selective hydrogenation of acrolein. It is also known that the formation of Pd islands on the surface will greatly reduce the selectivity of the reaction. As a result, the surface segregation and aggregation of Pd on the AgPd surface under reaction conditions of selective hydrogenation of acrolein are of great interest. In this work, we lay out a workflow that can predict the surface segregation and aggregation of Pd on a FCC(111) AgPd surface with and without the presence of acrolein. We use machine learning surrogate models to predict the AgPd bulk energy, AgPd slab energy, and acrolein adsorption energy on AgPd slabs. Then, we use the semi-grand canonical Monte Carlo simulation to predict the surface segregation and aggregation under different bulk Pd concentrations. Under vacuum conditions, our method predicts that only trace amount of Pd will exist on the surface at Pd bulk concentrations less than 20%. However, with the presence of acrolein, Pd will start to aggregate as dimers on the surface at Pd bulk concentrations as low as 6.5%. read less NOT USED (low confidence) Z. Cheng, J. Du, L. Zhang, J. Ma, W. Li, and S. Li, “Building Machine Learning Force Fields of Proteins with Fragment-Based Approach and Transfer Learning,” ChemRxiv. 2021. link Times cited: 0 Abstract: Molecular dynamic
(MD) simulation plays an essential role in… read moreAbstract: Molecular dynamic
(MD) simulation plays an essential role in understanding protein functions at
atomic level. At present, MD simulations on proteins are mainly based on classical
force fields. However, the accuracy of classical force fields for proteins is
still insufficient for accurate descriptions of their structures and dynamical
properties. Here we present a novel protocol to construct machine learning
force field (MLFF) for a given protein with full quantum mechanics (QM)
accuracy. In this protocol, the energy of the target system is obtained by
fitting energies of its various subsystems constructed with the generalized
energy-based fragmentation (GEBF) approach. To facilitate the construction of MLFF
for various proteins, a protein’s data library is created to store all data of
subsystems generated from trained proteins. With this protein’s data library,
for a new protein only its subsystems with new topological types are required
for the construction of the corresponding MLFF. This protocol is illustrated
with two polypeptides, 4ZNN and 1XQ8 segment, as examples. The energies
and forces predicted from this MLFF are in good agreement with those from
density functional theory calculations, and dihedral angle distributions from
GEBF-MLFF MD simulations can also well reproduce those from ab initio MD
simulations. Therefore, this
GEBF-ML protocol is expected to be an efficient and systematic way to build
force fields for proteins and other biological systems with QM accuracy. read less NOT USED (low confidence) D. Rosenberger, J. S. Smith, and A. Garcia, “Modeling of Peptides with Classical and Novel Machine Learning Force Fields: A Comparison.,” The journal of physical chemistry. B. 2021. link Times cited: 19 Abstract: The replacement of classical force fields (FFs) with novel n… read moreAbstract: The replacement of classical force fields (FFs) with novel neural-network-based frameworks is an emergent topic in molecular dynamics (MD) simulations. In contrast to classical FFs, which have proven their capability to provide insights into complex soft matter systems at an atomistic resolution, the machine learning (ML) potentials have yet to demonstrate their applicability for soft materials. However, the underlying philosophy, which is learning the energy of an atom in its surrounding chemical environment, makes this approach a promising tool. In particular for the exploration of novel chemical compounds, which have not been considered in the original parametrization of classical FFs. In this article, we study the performance of the ANI-2x ML model and compare the results with those of two classical FFs, namely, CHARMM27 and the GROMOS96 43a1 FF. We explore the performance of these FFs for bulk water and two model peptides, trialanine and a 9-mer of the α-aminoisobutyric acid, in vacuum and water. The results for water describe a highly ordered water structure, with a structure similar to those using ab initio molecular dynamics simulations. The energy landscape of the peptides described by Ramachandran maps show secondary structure basins similar to those of the classical FFs but differ in the position and relative stability of the basins. Details of the sampled structures show a divergent performance of the different models, which can be related either to the short-ranged nature of the ML potentials or to shortcomings of the underlying data set used for training. These findings highlight the current state of the applicability of ANI-2x ML potential for MD simulations of soft matter systems. Simultaneously, they provide insights for future improvements of current ML potentials. read less NOT USED (low confidence) Z. Aitken, V. Sorkin, Z. Yu, S. Chen, Z. Wu, and Y.-W. Zhang, “Modified embedded-atom method potentials for the plasticity and fracture behaviors of unary fcc metals,” Physical Review B. 2021. link Times cited: 5 NOT USED (low confidence) G. Sivaraman et al., “Automated Development of Molten Salt Machine Learning Potentials: Application to LiCl.,” The journal of physical chemistry letters. 2021. link Times cited: 24 Abstract: The in silico modeling of molten salts is critical for emerg… read moreAbstract: The in silico modeling of molten salts is critical for emerging "carbon-free" energy applications but is inhibited by the cost of quantum mechanically treating the high polarizabilities of molten salts. Here, we integrate configurational sampling using classical force fields with active learning to automate and accelerate the generation of Gaussian approximation potentials (GAP) for molten salts. This methodology reduces the number of expensive ab initio evaluations required for training set generation to O(100), enabling the facile parametrization of a molten LiCl GAP model that exhibits a 19 000-fold speedup relative to AIMD. The developed molten LiCl GAP model is applied to sample extended spatiotemporal scales, permitting new physical insights into molten LiCl's coordination structure as well as experimentally validated predictions of structures, densities, self-diffusion constants, and ionic conductivities. The developed methodology significantly lowers the barrier to the in silico understanding and design of molten salts across the periodic table. read less NOT USED (low confidence) Y. Lysogorskiy et al., “Performant implementation of the atomic cluster expansion (PACE) and application to copper and silicon,” npj Computational Materials. 2021. link Times cited: 84 NOT USED (low confidence) S. L. Batzner et al., “SE(3)-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials,” ArXiv. 2021. link Times cited: 30 Abstract:
This work presents Neural Equivariant Interatomic Potentia… read moreAbstract:
This work presents Neural Equivariant Interatomic Potentials (NequIP), a SE(3)-equivariant neural network approach for learning interatomic potentials from ab-initio calculations for molecular dynamics simulations. While most contemporary symmetry-aware models use invariant convolutions and only act on scalars, NequIP employs SE(3)-equivariant convolutions for interactions of geometric tensors, resulting in a more information-rich and faithful representation of atomic environments. The method achieves state-of-the-art accuracy on a challenging set of diverse molecules and materials while exhibiting remarkable data efficiency. NequIP outperforms existing models with up to three orders of magnitude fewer training data, challenging the widely held belief that deep neural networks require massive training sets. The high data efficiency of the method allows for the construction of accurate potentials using high-order quantum chemical level of theory as reference and enables high-fidelity molecular dynamics simulations over long time scales. read less NOT USED (low confidence) J. Keith et al., “Combining Machine Learning and Computational Chemistry for Predictive Insights Into Chemical Systems,” Chemical Reviews. 2021. link Times cited: 224 Abstract: Machine learning models are poised to make a transformative … read moreAbstract: Machine learning models are poised to make a transformative impact on chemical sciences by dramatically accelerating computational algorithms and amplifying insights available from computational chemistry methods. However, achieving this requires a confluence and coaction of expertise in computer science and physical sciences. This Review is written for new and experienced researchers working at the intersection of both fields. We first provide concise tutorials of computational chemistry and machine learning methods, showing how insights involving both can be achieved. We follow with a critical review of noteworthy applications that demonstrate how computational chemistry and machine learning can be used together to provide insightful (and useful) predictions in molecular and materials modeling, retrosyntheses, catalysis, and drug design. read less NOT USED (low confidence) Q. Tong et al., “Machine learning metadynamics simulation of reconstructive phase transition,” Physical Review B. 2021. link Times cited: 5 Abstract: Simulating reconstructive phase transition requires an accur… read moreAbstract: Simulating reconstructive phase transition requires an accurate description of potential energy surface (PES). Density-functional-theory (DFT) based molecular dynamics can achieve the desired accuracy but it is computationally unfeasible for large systems and/or long simulation times. Here we introduce an approach that combines the metadynamics simulation and machine learning representation of PES at the accuracy close to the DFT calculations, but with the computational cost several orders of magnitude less, and scaling with system size approximately linear. The high accuracy of the method is demonstrated in the simulation of pressure-induced $B4\text{\ensuremath{-}}B1$ phase transition in gallium nitride (GaN). The large-scale simulation using a 4096-atom simulation box reveals the phase transition with excellent detail, revealing different simulated transition paths under particular stress conditions. With well-trained machine learning potentials, this method can be easily applied to all types of systems for accurate scalable simulations of solid-solid reconstructive phase transition. read less NOT USED (low confidence) G. Pan, J. Ding, Y. Du, D.-J. Lee, and Y. Lu, “A DFT accurate machine learning description of molten ZnCl2 and its mixtures: 2. Potential development and properties prediction of ZnCl2-NaCl-KCl ternary salt for CSP,” Computational Materials Science. 2021. link Times cited: 20 NOT USED (low confidence) A. M. Miksch, T. Morawietz, J. Kästner, A. Urban, and N. Artrith, “Strategies for the construction of machine-learning potentials for accurate and efficient atomic-scale simulations,” Machine Learning: Science and Technology. 2021. link Times cited: 45 Abstract: Recent advances in machine-learning interatomic potentials h… read moreAbstract: Recent advances in machine-learning interatomic potentials have enabled the efficient modeling of complex atomistic systems with an accuracy that is comparable to that of conventional quantum-mechanics based methods. At the same time, the construction of new machine-learning potentials can seem a daunting task, as it involves data-science techniques that are not yet common in chemistry and materials science. Here, we provide a tutorial-style overview of strategies and best practices for the construction of artificial neural network (ANN) potentials. We illustrate the most important aspects of (a) data collection, (b) model selection, (c) training and validation, and (d) testing and refinement of ANN potentials on the basis of practical examples. Current research in the areas of active learning and delta learning are also discussed in the context of ANN potentials. This tutorial review aims at equipping computational chemists and materials scientists with the required background knowledge for ANN potential construction and application, with the intention to accelerate the adoption of the method, so that it can facilitate exciting research that would otherwise be challenging with conventional strategies. read less NOT USED (low confidence) S. Watanabe et al., “High-dimensional neural network atomic potentials for examining energy materials: some recent simulations,” Journal of Physics: Energy. 2020. link Times cited: 16 Abstract: Owing to their simultaneous accuracy and computational effic… read moreAbstract: Owing to their simultaneous accuracy and computational efficiency, interatomic potentials machine-learned using first-principles calculation data are promising for investigating phenomena closely related to atomic motion in various energy materials. We have been working with one type of these potentials, high-dimensional (HD) neural network potentials (NNPs), and their applications, but we realized that our current understanding of HD NNPs, e.g. the meaning of the atomic energy mapping, remained insufficient, and that tuning their prediction performance for different target properties/phenomena often requires much trial and error. In this article, we illustrate the usefulness of NNPs through our studies on ion migration and thermal transport in energy and related materials. We also share our experiences with data sampling and training strategies and discuss the meaning of atomic energy mapping in HD NNPs. read less NOT USED (low confidence) L. Stanek, R. Clay, M. Dharma-wardana, M. Wood, K. Beckwith, and M. Murillo, “Efficacy of the radial pair potential approximation for molecular dynamics simulations of dense plasmas,” arXiv: Plasma Physics. 2020. link Times cited: 12 Abstract: Macroscopic simulations of dense plasmas rely on detailed mi… read moreAbstract: Macroscopic simulations of dense plasmas rely on detailed microscopic information that can be computationally expensive and is difficult to verify experimentally. In this work, we delineate the accuracy boundary between microscale simulation methods by comparing Kohn-Sham density functional theory molecular dynamics (KS-MD) and radial pair potential molecular dynamics (RPP- MD) for a range of elements, temperature, and density. By extracting the optimal RPP from KS-MD data using force-matching, we constrain its functional form and dismiss classes of potentials that assume a constant power law for small interparticle distances. Our results show excellent agreement between RPP-MD and KS-MD for multiple metrics of accuracy at temperatures of only a few electron volts. The use of RPPs offers orders of magnitude decrease in computational cost and indicates that three-body potentials are not required beyond temperatures of a few eV. Due to its efficiency, the validated RPP-MD provides an avenue for reducing errors due to finite-size effects that can be on the order of $\sim20\%$. read less NOT USED (low confidence) Y. Shaidu, E. Kucukbenli, R. Lot, F. Pellegrini, E. Kaxiras, and S. de Gironcoli, “A systematic approach to generating accurate neural network potentials: the case of carbon,” npj Computational Materials. 2020. link Times cited: 18 NOT USED (low confidence) R. Batra, L. Song, and R. Ramprasad, “Emerging materials intelligence ecosystems propelled by machine learning,” Nature Reviews Materials. 2020. link Times cited: 121 NOT USED (low confidence) T. Tamura and M. Karasuyama, “Prediction of formation energies of large-scale disordered systems via active-learning-based executions of
ab initio
local-energy calculations: A case study on a Fe random grain boundary model with millions of atoms,” Physical Review Materials. 2020. link Times cited: 0 Abstract: We have developed a method that can analyze large random gra… read moreAbstract: We have developed a method that can analyze large random grain boundary (GB) models with the accuracy of density functional theory (DFT) calculations using active learning. It is assumed that the atomic energy is represented by the linear regression of the atomic structural descriptor. The atomic energy is obtained through DFT calculations using a small cell extracted from a huge GB model, called replica DFT atomic energy. The uncertainty reduction (UR) approach in active learning is used to efficiently collect the training data for the atomic energy. In this approach, atomic energy is not required to search for candidate points; therefore, sequential DFT calculations are not required. This approach is suitable for massively parallel computers that can execute a large number of jobs simultaneously. In this study, we demonstrate the prediction of the atomic energy of a Fe random GB model containing one million atoms using the UR approach, and we show that the prediction error decreases more rapidly compared with random sampling. We conclude that the UR approach with replica DFT atomic energy is useful for modeling huge GBs and will be essential for modeling other structural defects. read less NOT USED (low confidence) I. A. Balyakin, S. Rempel, R. Ryltsev, and A. Rempel, “Deep machine learning interatomic potential for liquid silica.,” Physical review. E. 2020. link Times cited: 16 Abstract: The use of machine learning to develop neural network potent… read moreAbstract: The use of machine learning to develop neural network potentials (NNP) representing the interatomic potential energy surface allows us to achieve an optimal balance between accuracy and efficiency in computer simulation of materials. A key point in developing such potentials is the preparation of a training dataset of ab initio trajectories. Here we apply a deep potential molecular dynamics (DeePMD) approach to develop NNP for silica, which is the representative glassformer widely used as a model system for simulating network-forming liquids and glasses. We show that the use of a relatively small training dataset of high-temperature ab initio configurations is enough to fabricate NNP, which describes well both structural and dynamical properties of liquid silica. In particular, we calculate the pair correlation functions, angular distribution function, velocity autocorrelation functions, vibrational density of states, and mean-square displacement and reveal a close agreement with ab initio data. We show that NNP allows us to expand significantly the time-space scales achievable in simulations and thus calculating dynamical and transport properties with more accuracy than that for ab initio methods. We find that developed NNP allows us to describe the structure of the glassy silica with satisfactory accuracy even though no low-temperature configurations were included in the training procedure. The results obtained open up prospects for simulating structural and dynamical properties of liquids and glasses via NNP. read less NOT USED (low confidence) A. Rohskopf, S. Wyant, K. Gordiz, H. R. Seyf, M. G. Muraleedharan, and A. Henry, “Fast & accurate interatomic potentials for describing thermal vibrations,” Computational Materials Science. 2020. link Times cited: 7 NOT USED (low confidence) M. Sakano et al., “Unsupervised Learning-Based Multiscale Model of Thermochemistry in 1,3,5-Trinitro-1,3,5-triazinane (RDX).,” The journal of physical chemistry. A. 2020. link Times cited: 33 Abstract: The response of high-energy-density materials to thermal or … read moreAbstract: The response of high-energy-density materials to thermal or mechanical insults involves coupled thermal, mechanical, and chemical processes with disparate temporal and spatial scales that no single model can capture. Therefore, we developed a multiscale model for 1,3,5-trinitro-1,3,5-triazinane, RDX, where a continuum description is informed by reactive and nonreactive molecular dynamics (MD) simulations to describe chemical reactions and thermal transport. Reactive MD simulations under homogeneous isothermal and adiabatic conditions are used to develop a reduced-order chemical kinetics model. Coarse graining is done using unsupervised learning via non-negative matrix factorization. Importantly, the components resulting from the analysis can be interpreted as reactants, intermediates, and products, which allows us to write kinetics equations for their evolution. The kinetics parameters are obtained from isothermal MD simulations over a wide temperature range, 1200-3000 K, and the heat evolved is calibrated from adiabatic simulations. We validate the continuum model against MD simulations by comparing the evolution of a cylindrical hotspot 10 nm in diameter. We find excellent agreement in the time evolution of the hotspot temperature fields both in cases where quenching is observed and at higher temperatures for which the hotspot transitions into a deflagration wave. The validated continuum model is then used to assess the criticality of hotspots involving scales beyond the reach of atomistic simulations that are relevant to detonation initiation. read less NOT USED (low confidence) J. Yoon and Z. W. Ulissi, “Differentiable Optimization for the Prediction of Ground State Structures (DOGSS).,” Physical review letters. 2020. link Times cited: 7 Abstract: Ground state or relaxed inorganic structures are the startin… read moreAbstract: Ground state or relaxed inorganic structures are the starting point for most computational materials science or surface science analyses. Many of these structure relaxations represent systematic changes to the structure, but there are currently no general methods to improve the initial structure guess based on past calculations. Here we present a method to directly predict the ground state configuration using differentiable optimization and graph neural networks to learn the properties of a simple harmonic force field that approximates the ground state structure and properties. We demonstrate this flexible open source tool for improving the initial configurations for large datasets of inorganic multicomponent surface relaxations across 32 elements and the relaxation of adsorbates (H and CO) on these surfaces. Using these improved initial configurations reduces the expensive adsorbate-covered surface relaxations by approximately 50% and is complementary to other approaches to accelerate the relaxation process. read less NOT USED (low confidence) M. C. Barry, K. Wise, S. Kalidindi, and S. Kumar, “Voxelized Atomic Structure Potentials: Predicting Atomic Forces with the Accuracy of Quantum Mechanics Using Convolutional Neural Networks.,” The journal of physical chemistry letters. 2020. link Times cited: 9 Abstract: This paper introduces Voxelized Atomic Structure (VASt) pote… read moreAbstract: This paper introduces Voxelized Atomic Structure (VASt) potentials as a machine learning (ML) framework for developing interatomic potentials. The VASt framework utilizes a voxelized representation of the atomic structure directly as the input to a convolutional neural network (CNN). This allows for high fidelity representations of highly complex and diverse spatial arrangements of the atomic environments of interest. The CNN implicitly establishes the low-dimensional features needed to correlate each atomic neighborhood to its net atomic force. The selection of the salient features of the atomic structure (i.e., feature engineering) in the VASt framework is implicit, comprehensive, automated, scalable, and highly efficient. The calibrated convolutional layers learn the complex spatial relationships and multibody interactions that govern the physics of atomic systems with remarkable fidelity. We show that VASt potentials predict highly accurate forces on two phases of silicon carbide and the thermal conductivity of silicon over a range of isotropic strain. read less NOT USED (low confidence) Q. Tong et al., “Combining Machine Learning Potential and Structure Prediction for Accelerated Materials Design and Discovery.,” The journal of physical chemistry letters. 2020. link Times cited: 33 Abstract: Theoretical structure prediction method via quantum mechanic… read moreAbstract: Theoretical structure prediction method via quantum mechanical atomistic simulations such as density functional theory (DFT), solely based on chemical composition, already becomes a routine tool to determine the structures of physical and chemical systems, e.g. solids and clusters. However, the application of DFT to more realistic simulations, to a large extent, is impeded owing to the unfavourable scaling of the computational cost with respective to the system size. During recent years, machine learning potential (MLP) method has been rapidly rising as an accurate and efficient tool for atomistic simulations. In this Perspective, we provide an introduction on the basic principles and advantages for the combination of structure prediction and MLP, as well as challenges and opportunities along this promising direction. read less NOT USED (low confidence) P. Kang, C. Shang, and Z. Liu, “Large-Scale Atomic Simulation via Machine Learning Potentials Constructed by Global Potential Energy Surface Exploration.,” Accounts of chemical research. 2020. link Times cited: 52 Abstract: ConspectusAtomic simulations based on quantum mechanics (QM)… read moreAbstract: ConspectusAtomic simulations based on quantum mechanics (QM) calculations have entered into the tool box of chemists over the past few decades, facilitating an understanding of a wide range of chemistry problems, from structure characterization to reactivity determination. Due to the poor scaling and high computational cost intrinsic to QM calculations, one has to either sacrifice accuracy or time when performing large-scale atomic simulations. The battle to find a better compromise between accuracy and speed has been central to the development of new theoretical methods.The recent advances of machine-learning (ML)-based large-scale atomic simulations has shown great promise to the benefit of many branches of chemistry. Instead of solving the Schrödinger equation directly, ML-based simulations rely on a large data set of accurate potential energy surfaces (PESs) and complex numerical models to predict the total energy. These simulations feature both a high speed and a high accuracy for computing large systems. Due to the lack of a physical foundation in numerical models, ML models are often frustrated in their predictivity and robustness, which are key to applications. Focusing on these concerns, here we overview the recent advances in ML methodologies for atomic simulations on three key aspects. Namely, the generation of a representative data set, the extensity of ML models, and the continuity of data representation. While global optimization methods are the natural choice for building a representative data set, the stochastic surface walking method is shown to provide the desired PES sampling for both minima and transition regions on the PES. The current ML models generally utilize local geometrical descriptors as an input and consider the total energy as the sum of atomic energies. There are many flavors of data descriptors and ML models, but the applications for material and reaction predictions are still limited, not least because of the difficulty to train the associated vast global data sets. We show that our recently designed power-type structure descriptors together with a feed-forward neural network (NN) model are compatible with highly complex global PES data, which has led to a large family of global NN (G-NN) potentials.Two recent applications of G-NN potentials in material and reaction simulations are selected to illustrate how ML-based atomic simulations can help the discovery of new materials and reactions. read less NOT USED (low confidence) C. Pham, R. Lindsey, L. Fried, and N. Goldman, “Calculation of the detonation state of HN3 with quantum accuracy.,” The Journal of chemical physics. 2020. link Times cited: 14 Abstract: HN3 is a unique liquid energetic material that exhibits ultr… read moreAbstract: HN3 is a unique liquid energetic material that exhibits ultrafast detonation chemistry and a transition to metallic states during detonation. We combine the Chebyshev interaction model for efficient simulation (ChIMES) many-body reactive force field and the extended-Lagrangian multiscale shock technique molecular dynamics method to calculate the detonation properties of HN3 with the accuracy of Kohn-Sham density-functional theory. ChIMES is based on a Chebyshev polynomial expansion and can accurately reproduce density-functional theory molecular dynamics (DFT-MD) simulations for a wide range of unreactive and decomposition conditions of liquid HN3. We show that addition of random displacement configurations and the energies of gas-phase equilibrium products in the training set allows ChIMES to efficiently explore the complex potential energy surface. Schemes for selecting force field parameters and the inclusion of stress tensor and energy data in the training set are examined. Structural and dynamical properties and chemistry predictions for the resulting models are benchmarked against DFT-MD. We demonstrate that the inclusion of explicit four-body energy terms is necessary to capture the potential energy surface across a wide range of conditions. Our results generally retain the accuracy of DFT-MD while yielding a high degree of computational efficiency, allowing simulations to approach orders of magnitude larger time and spatial scales. The techniques and recipes for MD model creation we present allow for direct simulation of nanosecond shock compression experiments and calculation of the detonation properties of materials with the accuracy of Kohn-Sham density-functional theory. read less NOT USED (low confidence) M. Paleico and J. Behler, “A bin and hash method for analyzing reference data and descriptors in machine learning potentials,” Machine Learning: Science and Technology. 2020. link Times cited: 1 Abstract: In recent years the development of machine learning potentia… read moreAbstract: In recent years the development of machine learning potentials (MLPs) has become a very active field of research. Numerous approaches have been proposed, which allow one to perform extended simulations of large systems at a small fraction of the computational costs of electronic structure calculations. The key to the success of modern MLPs is the close-to first principles quality description of the atomic interactions. This accuracy is reached by using very flexible functional forms in combination with high-level reference data from electronic structure calculations. These data sets can include up to hundreds of thousands of structures covering millions of atomic environments to ensure that all relevant features of the potential energy surface are well represented. The handling of such large data sets is nowadays becoming one of the main challenges in the construction of MLPs. In this paper we present a method, the bin-and-hash (BAH) algorithm, to overcome this problem by enabling the efficient identification and comparison of large numbers of multidimensional vectors. Such vectors emerge in multiple contexts in the construction of MLPs. Examples are the comparison of local atomic environments to identify and avoid unnecessary redundant information in the reference data sets that is costly in terms of both the electronic structure calculations as well as the training process, the assessment of the quality of the descriptors used as structural fingerprints in many types of MLPs, and the detection of possibly unreliable data points. The BAH algorithm is illustrated for the example of high-dimensional neural network potentials using atom-centered symmetry functions for the geometrical description of the atomic environments, but the method is general and can be combined with any current type of MLP. read less NOT USED (low confidence) V. L. Deringer, “Modelling and understanding battery materials with machine-learning-driven atomistic simulations,” Journal of Physics: Energy. 2020. link Times cited: 49 Abstract: The realistic computer modelling of battery materials is an … read moreAbstract: The realistic computer modelling of battery materials is an important research goal, with open questions ranging from atomic-scale structure and dynamics to macroscopic phenomena. Quantum-mechanical methods offer high accuracy and predictive power in small-scale atomistic simulations, but they quickly reach their limits when complex electrochemical systems are to be studied—for example, when structural disorder or even fully amorphous phases are present, or when reactions take place at the interface between electrodes and electrolytes. In this Perspective, it is argued that emerging machine learning based interatomic potentials are promising tools for studying battery materials on the atomistic and nanometre length scales, affording quantum-mechanical accuracy yet being many orders of magnitude faster, and thereby extending the capabilities of current battery modelling methodology. Initial applications to solid-state electrolyte and anode materials in lithium-ion batteries are highlighted, and future directions and possible synergies with experiments are discussed. read less NOT USED (low confidence) H. Ghorbanfekr, J. Behler, and F. Peeters, “Insights into Water Permeation through hBN Nanocapillaries by Ab Initio Machine Learning Molecular Dynamics Simulations.,” The journal of physical chemistry letters. 2020. link Times cited: 29 Abstract: Water permeation between stacked layers of hBN sheets formin… read moreAbstract: Water permeation between stacked layers of hBN sheets forming 2D-nanochannels is investigated using large-scale ab initio-quality molecular dynamics simulations. A high-dimensional neural network potential trained on density-functional theory calculations is employed. We simulate water in van der Waals nanocapillaries and study the impact of nanometric confinement on the structure and dynamics of water using both equilibrium and non-equilibrium methods. At an interlayer distance of 10.2 Å confinement induces a first-order phase transition resulting in a well-defined AA-stacked bilayer of hexagonal ice. In contrast for h<9 Å, the 2D water monolayer consists of a mixture of different locally ordered patterns of squares, pentagons, and hexagons. We found a significant change in the transport properties of confined water particularly for monolayer water where the water-solid friction coefficient decreases to half and the diffusion coefficient increases by a factor of four as compared to bulk water. Accordingly, the slip-velocity is found to increase under confinement and we found that the overall permeation is dominated by monolayer water adjacent to the hBN membranes at extreme confinements. We conclude that monolayer water in addition to bilayer ice has a major contribution to water transport through 2D nanochannels. read less NOT USED (low confidence) R. Jinnouchi, K. Miwa, F. Karsai, G. Kresse, and R. Asahi, “On-the-Fly Active Learning of Interatomic Potentials for Large-Scale Atomistic Simulations.,” The journal of physical chemistry letters. 2020. link Times cited: 79 Abstract: The on-the-fly generation of machine-learning force fields b… read moreAbstract: The on-the-fly generation of machine-learning force fields by active-learning schemes attracts a great deal of attention in the community of atomistic simulations. The algorithms allow the machine to self-learn an interatomic potential and construct machine-learned models on the fly during simulations. State-of-the-art query strategies allow the machine to judge whether new structures are out of the training data set or not. Only when the machine judges the necessity of updating the data set with the new structures are first-principles calculations carried out. Otherwise, the yet available machine-learned model is used to update the atomic positions. In this manner, most of the first-principles calculations are bypassed during training, and overall, simulations are accelerated by several orders of magnitude while retaining almost first-principles accuracy. In this Perspective, after describing essential components of the active-learning algorithms, we demonstrate the power of the schemes by presenting recent applications. read less NOT USED (low confidence) H. Yanxon, D. Zagaceta, B. Tang, D. Matteson, and Q. Zhu, “PyXtal_FF: a python library for automated force field generation,” Machine Learning: Science and Technology. 2020. link Times cited: 15 Abstract: We present PyXtal_FF—a package based on Python programming l… read moreAbstract: We present PyXtal_FF—a package based on Python programming language—for developing machine learning potentials (MLPs). The aim of PyXtal_FF is to promote the application of atomistic simulations through providing several choices of atom-centered descriptors and machine learning regressions in one platform. Based on the given choice of descriptors (including the atom-centered symmetry functions, embedded atom density, SO4 bispectrum, and smooth SO3 power spectrum), PyXtal_FF can train MLPs with either generalized linear regression or neural network models, by simultaneously minimizing the errors of energy/forces/stress tensors in comparison with the data from ab-initio simulations. The trained MLP model from PyXtal_FF is interfaced with the Atomic Simulation Environment (ASE) package, which allows different types of light-weight simulations such as geometry optimization, molecular dynamics simulation, and physical properties prediction. Finally, we will illustrate the performance of PyXtal_FF by applying it to investigate several material systems, including the bulk SiO2, high entropy alloy NbMoTaW, and elemental Pt for general purposes. Full documentation of PyXtal_FF is available at https://pyxtal-ff.readthedocs.io. read less NOT USED (low confidence) R. Lindsey, L. Fried, N. Goldman, and S. Bastea, “Active learning for robust, high-complexity reactive atomistic simulations.,” The Journal of chemical physics. 2020. link Times cited: 19 Abstract: Machine learned reactive force fields based on polynomial ex… read moreAbstract: Machine learned reactive force fields based on polynomial expansions have been shown to be highly effective for describing simulations involving reactive materials. Nevertheless, the highly flexible nature of these models can give rise to a large number of candidate parameters for complicated systems. In these cases, reliable parameterization requires a well-formed training set, which can be difficult to achieve through standard iterative fitting methods. Here, we present an active learning approach based on cluster analysis and inspired by Shannon information theory to enable semi-automated generation of informative training sets and robust machine learned force fields. The use of this tool is demonstrated for development of a model based on linear combinations of Chebyshev polynomials explicitly describing up to four-body interactions, for a chemically and structurally diverse system of C/O under extreme conditions. We show that this flexible training database management approach enables development of models exhibiting excellent agreement with Kohn-Sham density functional theory in terms of structure, dynamics, and speciation. read less NOT USED (low confidence) M. Liu and J. Kitchin, “SingleNN: Modified Behler–Parrinello Neural Network with Shared Weights for Atomistic Simulations with Transferability,” Journal of Physical Chemistry C. 2020. link Times cited: 18 Abstract: In this article, we introduce the SingleNN, which is a modif… read moreAbstract: In this article, we introduce the SingleNN, which is a modified version of the Behler–Parrinello Neural Network (BPNN) where the neural networks for the prediction of atomic energy for different el... read less NOT USED (low confidence) R. Alexander et al., “Interatomic potentials for irradiation-induced defects in iron,” Journal of Nuclear Materials. 2020. link Times cited: 13 NOT USED (low confidence) R. Jadrich and J. Leiding, “Accelerating ab Initio Simulation via Nested Monte Carlo and Machine Learned Reference Potentials.,” The journal of physical chemistry. B. 2020. link Times cited: 9 Abstract: As a corollary of the rapid advances in computing, ab initio… read moreAbstract: As a corollary of the rapid advances in computing, ab initio simulation is playing an increasingly important role in modeling materials at the atomic scale. Two strategies are possible, ab initio Monte Carlo (AIMC) and molecular dynamics (AIMD) simulation. The former benefits from exact sampling from the correct thermodynamic distribution, while the latter is typically more efficient with its collective all-atom coordinate updates. Here, using a relatively simple test model comprised of Helium and Argon, we show that AIMC can be brought up to, and even above, the performance levels of AIMD via a hybrid nested sampling / machine learning (ML) strategy. Here, ML provides an accurate classical reference potential (up to three-body explicit interactions) that can pilot long collective Monte Carlo moves that are accepted or rejected in toto ala nested Monte Carlo (NMC); this is in contrast to the single move nature of a naive implementation. Our proposed method only requires a small up front expense from evaluating the ab initio energies and forces of O(100) random configurations for training. Importantly, our method does not totally rely on the trained, assuredly imperfect, interaction. We show that high performance and exact sampling at the desired level of theory can be realized even when the trained interaction has appreciable differences from the ab initio potential. Remarkably, at the highest levels of performance realized via our approach, a pair of statistically uncorrelated atomic configurations can be generated with O(1) ab initio calculations. read less NOT USED (low confidence) D. Zagaceta, H. Yanxon, and Q. Zhu, “Spectral neural network potentials for binary alloys,” Journal of Applied Physics. 2020. link Times cited: 4 Abstract: In this work, we present a numerical implementation to compu… read moreAbstract: In this work, we present a numerical implementation to compute the atom centered descriptors introduced by Bartok et al (Phys. Rev. B, 87, 184115, 2013) based on the harmonic analysis of the atomic neighbor density function. Specifically, we focus on two types of descriptors, the smooth SO(3) power spectrum with the explicit inclusion of a radial basis and the SO(4) bispectrum obtained through mapping the radial component onto a polar angle of a four dimensional hypersphere. With these descriptors, various interatomic potentials for binary Ni-Mo alloys are obtained based on linear and neural network regression models. Numerical experiments suggest that both descriptors produce similar results in terms of accuracy. For linear regression, the smooth SO(3) power spectrum is superior to the SO(4) bispectrum when a large band limit is used. In neural network regression, a better accuracy can be achieved with even less number of expansion components for both descriptors. As such, we demonstrate that spectral neural network potentials are feasible choices for large scale atomistic simulation. read less NOT USED (low confidence) X. Gao, F. Ramezanghorbani, O. Isayev, J. S. Smith, and A. Roitberg, “TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials,” Journal of chemical information and modeling. 2020. link Times cited: 127 Abstract: This paper presents TorchANI, a PyTorch based software for t… read moreAbstract: This paper presents TorchANI, a PyTorch based software for training/inference of ANI (ANAKIN-ME) deep learning models to obtain potential energy surfaces and other physical properties of molecular systems. ANI is an accurate neural network potential originally implemented using C++/CUDA in a program called NeuroChem. Compared with NeuroChem, TorchANI has a design emphasis on being light weight, user friendly, cross platform, and easy to read and modify for fast prototyping, while allowing acceptable sacrice on running performance. Because the computation of atomic environmental vectors (AEVs) and atomic neural networks are all implemented using PyTorch operators, TorchANI is able to use PyTorch's autograd engine to automatically compute analytical forces and Hessian matrices, as well as do force training without additional codes required. TorchANI is open-source and freely available on GitHub: https://github.com/aiqm/torchani. read less NOT USED (low confidence) W. Li and Y. Ando, “Effect of local structural disorder on lithium diffusion behavior in amorphous silicon,” Physical Review Materials. 2020. link Times cited: 7 Abstract: Lithium-ion batteries with amorphous silicon ($a$-Si) anodes… read moreAbstract: Lithium-ion batteries with amorphous silicon ($a$-Si) anodes exhibit very high theoretical energy capacity, with the lithium kinetic transport having the most crucial effect on the battery performance. In this study, the lithium diffusion pathways in a series of large-scale $a$-Si models (512 atoms) with various extents of structural order are calculated using the machine learning interatomic potential. Then, the Li diffusion behavior in different atomistic environments is estimated from the transient state theory. The Li diffusion activation energy is observed to be lower (higher) in an ordered (disordered) local environment. The activation energy varies within the range of 1.21--1.46 eV, which agrees well with experimental measurements, 1.38--1.46 eV. Our simulations also show that Li diffusion is enhanced at higher Li concentration, which is consistent with experimental observations. The effects of structural disorder and Li concentration can be explained by the ``trap'' mechanism. Finally, we show that the sources of Li diffusion traps are dangling bonds and large voids in the $a$-Si matrix with the help of first-principles calculations. Our work provides insight into the Li diffusion mechanism, which is beneficial for improving the performance of $a$-Si anodes for lithium-ion batteries. In addition, we demonstrate the significant dependence of the ion transport behavior on the local atomic environment, which will be useful for future theoretical studies of technologically important amorphous materials beyond Si. read less NOT USED (low confidence) M. Hodapp and A. Shapeev, “In operando active learning of interatomic interaction during large-scale simulations,” Machine Learning: Science and Technology. 2020. link Times cited: 17 Abstract: A well-known drawback of state-of-the-art machine-learning i… read moreAbstract: A well-known drawback of state-of-the-art machine-learning interatomic potentials is their poor ability to extrapolate beyond the training domain. For small-scale problems with tens to hundreds of atoms this can be solved by using active learning which is able to select atomic configurations on which a potential attempts extrapolation and add them to the ab initio-computed training set. In this sense an active learning algorithm can be viewed as an on-the-fly interpolation of an ab initio model. For large-scale problems, possibly involving tens of thousands of atoms, this is not feasible because one cannot afford even a single density functional theory (DFT) computation with such a large number of atoms. This work marks a new milestone toward fully automatic ab initio-accurate large-scale atomistic simulations. We develop an active learning algorithm that identifies local subregions of the simulation region where the potential extrapolates. Then the algorithm constructs periodic configurations out of these local, non-periodic subregions, sufficiently small to be computable with plane-wave DFT codes, in order to obtain accurate ab initio energies. We benchmark our algorithm on the problem of screw dislocation motion in bcc tungsten and show that our algorithm reaches ab initio accuracy, down to typical magnitudes of numerical noise in DFT codes. We show that our algorithm reproduces material properties such as core structure, Peierls barrier, and Peierls stress. This unleashes new capabilities for computational materials science toward applications which have currently been out of scope if approached solely by ab initio methods. read less NOT USED (low confidence) L. Xiang, X. Zeng, X. Huang, and G. Li, “The application of artificial neural-network potentials for flexoelectricity: Performance for anatase-type TiO2,” Physics Letters A. 2020. link Times cited: 3 NOT USED (low confidence) G. Pilania, P. Balachandran, J. Gubernatis, and T. Lookman, “Data-Based Methods for Materials Design and Discovery: Basic Ideas and General Methods.” 2020. link Times cited: 11 Abstract: Machine learning methods are changing the way we design and … read moreAbstract: Machine learning methods are changing the way we design and discover new materials. This book provides an overview of approaches successfully used in addressing materials problems (alloys,... read less NOT USED (low confidence) X. Xie, K. A. Persson, and D. W. Small, “Incorporating Electronic Information into Machine Learning Potential Energy Surfaces via Approaching the Ground-State Electronic Energy as a Function of Atom-Based Electronic Populations.,” Journal of chemical theory and computation. 2020. link Times cited: 37 Abstract: Machine Learning (ML) approximations to Density Functional T… read moreAbstract: Machine Learning (ML) approximations to Density Functional Theory (DFT) potential energy surfaces (PESs) are showing great promise for reducing the computational cost of accurate molecular simulations, but at present they are not applicable to varying electronic states, and in particular, they are not well suited for molecular systems in which the local electronic structure is sensitive to the medium to long-range electronic environment. With this issue as the focal point, we present a new Machine Learning approach called "BpopNN" for obtaining efficient approximations to DFT PESs. Conceptually, the methodology is based on approaching the true DFT energy as a function of electron populations on atoms; in practice, this is realized with available density functionals and constrained DFT (CDFT). The new approach creates approximations to this function with neural networks. These approximations thereby incorporate electronic information naturally into a ML approach, and optimizing the model energy with respect to populations allows the electronic terms to self-consistently adapt to the environment, as in DFT. We confirm the effectiveness of this approach with a variety of calculations on LinHn clusters. read less NOT USED (low confidence) R. Drautz, “Atomic cluster expansion of scalar, vectorial, and tensorial properties including magnetism and charge transfer,” Physical Review B. 2020. link Times cited: 55 Abstract: The atomic cluster expansion (Drautz, Phys. Rev. B 99, 01410… read moreAbstract: The atomic cluster expansion (Drautz, Phys. Rev. B 99, 014104 (2019)) is extended in two ways, the modelling of vectorial and tensorial atomic properties and the inclusion of atomic degrees of freedom in addition to the positions of the atoms. In particular, atomic species, magnetic moments and charges are attached to the atomic positions and an atomic cluster expansion that includes the different degrees of freedom on equal footing is derived. Expressions for the efficient evaluation of forces and torques are given. Relations to other methods are discussed. read less NOT USED (low confidence) A. Lunghi and S. Sanvito, “Surfing Multiple Conformation-Property Landscapes via Machine Learning: Designing Single-Ion Magnetic Anisotropy,” The Journal of Physical Chemistry C. 2020. link Times cited: 23 Abstract: Computational statistical disciplines, such as machine learn… read moreAbstract: Computational statistical disciplines, such as machine learning, are leading to a paradigm shift in the way we conceive the design of new compounds, offering a way to directly design the best compound for specific applications. This approach, known as reverse engineering, requires the construction of models able to efficiently predict continuous structure-property maps. Here we show that machine-learning offers such a possibility by designing a model that predicts both the energy and magnetic properties as function of the molecular structure of single-ion magnet. This model is then used to explore the molecular conformational landscapes in search of structures that maximise magnetic anisotropy. We find that a 5% change in one of the coordination angles leads to a ∼50% increase in the anisotropy. This approach can be applied to any structure-property relation and paves the way for a machine-learning-driven optimization of chemical compounds. read less NOT USED (low confidence) C. Devereux et al., “Extending the applicability of the ANI deep learning molecular potential to Sulfur and Halogens.,” Journal of chemical theory and computation. 2020. link Times cited: 141 Abstract: Machine learning (ML) methods have become powerful, predicti… read moreAbstract: Machine learning (ML) methods have become powerful, predictive tools in a wide range of applications, such as facial recognition and autonomous vehicles. In the sciences, computational chemists and physicists have been using ML for the prediction of physical phenomena, such as atomistic potential energy surfaces and reaction pathways. Transferable ML potentials, such as ANI-1x, have been developed with the goal of accurately simulating organic molecules containing the chemical elements H, C, N, and O. Here we provide an extension of the ANI-1x model. The new model, dubbed ANI-2x, is trained to three additional chemical elements: S, F, and Cl. Additionally, ANI-2x underwent torsional refinement training to better predict molecular torsion profiles. These new features open a wide range of new applications within organic chemistry and drug development. These seven elements (H, C, N, O, F, Cl, S) make up ~90% of drug like molecules. To show that these additions do not sacrifice accuracy, we have tested this model across a range of organic molecules and applications, including the COMP6 benchmark, dihedral rotations, conformer scoring, and non-bonded interactions. ANI-2x is shown to accurately predict molecular energies compared to DFT with a ~106 factor speedup and a negligible slowdown compared to ANI-1x. The resulting model is a valuable tool for drug development that can potentially replace both quantum calculations and classical force fields for myriad applications. read less NOT USED (low confidence) A. Singh and Y. Li, “Uncertainty Management and Reduction of Machine Learning Potential,” AIAA Scitech 2021 Forum. 2020. link Times cited: 0 NOT USED (low confidence) S. Ignatov, A. Razuvaev, A. S. Loginova, and A. Masunov, “Global Structure Optimization of Pt Clusters Based on the Modified Empirical Potentials, Calibrated using Density Functional Theory,” The Journal of Physical Chemistry C. 2019. link Times cited: 7 Abstract: The geometry of platinum clusters Ptn (n = 2–10, 13, 19, 24,… read moreAbstract: The geometry of platinum clusters Ptn (n = 2–10, 13, 19, 24, 38, 55, and 75) was optimized at the UBLYP/CRENBS, UBPW91/CRENBS, UBPW91/LANL2DZ, and UPBE0/LANL08 density functional theory (DFT) level... read less NOT USED (low confidence) M. Jafary-Zadeh, K. Khoo, R. Laskowski, P. S. Branicio, and A. Shapeev, “Applying a machine learning interatomic potential to unravel the effects of local lattice distortion on the elastic properties of multi-principal element alloys,” Journal of Alloys and Compounds. 2019. link Times cited: 39 NOT USED (low confidence) A. S. Christensen, L. A. Bratholm, F. A. Faber, and O. A. von Lilienfeld, “FCHL revisited: Faster and more accurate quantum machine learning.,” The Journal of chemical physics. 2019. link Times cited: 201 Abstract: We introduce the FCHL19 representation for atomic environmen… read moreAbstract: We introduce the FCHL19 representation for atomic environments in molecules or condensed-phase systems. Machine learning models based on FCHL19 are able to yield predictions of atomic forces and energies of query compounds with chemical accuracy on the scale of milliseconds. FCHL19 is a revision of our previous work [F. A. Faber et al., J. Chem. Phys. 148, 241717 (2018)] where the representation is discretized and the individual features are rigorously optimized using Monte Carlo optimization. Combined with a Gaussian kernel function that incorporates elemental screening, chemical accuracy is reached for energy learning on the QM7b and QM9 datasets after training for minutes and hours, respectively. The model also shows good performance for non-bonded interactions in the condensed phase for a set of water clusters with a mean absolute error (MAE) binding energy error of less than 0.1 kcal/mol/molecule after training on 3200 samples. For force learning on the MD17 dataset, our optimized model similarly displays state-of-the-art accuracy with a regressor based on Gaussian process regression. When the revised FCHL19 representation is combined with the operator quantum machine learning regressor, forces and energies can be predicted in only a few milliseconds per atom. The model presented herein is fast and lightweight enough for use in general chemistry problems as well as molecular dynamics simulations. read less NOT USED (low confidence) C. Schran, J. Behler, and D. Marx, “Automated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground,” Journal of chemical theory and computation. 2019. link Times cited: 72 Abstract: Highly accurate potential energy surfaces are of key interes… read moreAbstract: Highly accurate potential energy surfaces are of key interest for the detailed understanding and predictive modeling of chemical systems. In recent years, several new types of force fields, which are based on machine learning algorithms and fitted to ab initio reference calculations, have been introduced to meet this requirement. Here we show how high-dimensional neural network potentials can be employed to automatically generate the potential energy surface of finite sized clusters at coupled cluster accuracy, namely CCSD(T*)-F12a/aug-cc-pVTZ. The developed automated procedure utilizes the established intrinsic properties of the model such that the configurations for the training set are selected in an unbiased and efficient way to minimize the computational effort of expensive reference calculations. These ideas are applied to protonated water clusters from the hydronium cation, H3O+, up to the tetramer, H9O4+, and lead to a common potential energy surface that describes all these systems at essentially converged coupled cluster accuracy with a fitting error of 0.06 kJ/mol per atom. The fit is validated in detail for all clusters up to the tetramer and yields reliable results not only for stationary points, but also for reaction pathways, intermediate configurations, as well as different sampling techniques. Per design the NNPs constructed in this fashion can handle very different conditions including the quantum nature of the nuclei and enhanced sampling techniques covering very low as well as high temperatures. This enables fast and exhaustive exploration of the targeted protonated water clusters with essentially converged interactions. In addition, the automated process will allow one to tackle finite systems much beyond the present case. read less NOT USED (low confidence) I. Kruglov et al., “Machine Learning Interatomic Potentials for Global Optimization and Molecular Dynamics Simulation,” Materials Informatics. 2019. link Times cited: 3 NOT USED (low confidence) A. Goryaeva, J. Maillet, and M. Marinica, “Towards better efficiency of interatomic linear machine learning potentials,” Computational Materials Science. 2019. link Times cited: 34 NOT USED (low confidence) Y. Zuo et al., “A Performance and Cost Assessment of Machine Learning Interatomic Potentials.,” The journal of physical chemistry. A. 2019. link Times cited: 413 Abstract: Machine learning of the quantitative relationship between lo… read moreAbstract: Machine learning of the quantitative relationship between local environment descriptors and the potential energy surface of a system of atoms has emerged as a new frontier in the development of interatomic potentials (IAPs). Here, we present a comprehensive evaluation of ML-IAPs based on four local environment descriptors --- atom-centered symmetry functions (ACSF), smooth overlap of atomic positions (SOAP), the Spectral Neighbor Analysis Potential (SNAP) bispectrum components, and moment tensors --- using a diverse data set generated using high-throughput density functional theory (DFT) calculations. The data set comprising bcc (Li, Mo) and fcc (Cu, Ni) metals and diamond group IV semiconductors (Si, Ge) is chosen to span a range of crystal structures and bonding. All descriptors studied show excellent performance in predicting energies and forces far surpassing that of classical IAPs, as well as predicting properties such as elastic constants and phonon dispersion curves. We observe a general trade-off between accuracy and the degrees of freedom of each model, and consequently computational cost. We will discuss these trade-offs in the context of model selection for molecular dynamics and other applications. read less NOT USED (low confidence) G. R. Schleder, A. C. Padilha, C. M. Acosta, M. Costa, and A. Fazzio, “From DFT to machine learning: recent approaches to materials science–a review,” Journal of Physics: Materials. 2019. link Times cited: 414 Abstract: Recent advances in experimental and computational methods ar… read moreAbstract: Recent advances in experimental and computational methods are increasing the quantity and complexity of generated data. This massive amount of raw data needs to be stored and interpreted in order to advance the materials science field. Identifying correlations and patterns from large amounts of complex data is being performed by machine learning algorithms for decades. Recently, the materials science community started to invest in these methodologies to extract knowledge and insights from the accumulated data. This review follows a logical sequence starting from density functional theory as the representative instance of electronic structure methods, to the subsequent high-throughput approach, used to generate large amounts of data. Ultimately, data-driven strategies which include data mining, screening, and machine learning techniques, employ the data generated. We show how these approaches to modern computational materials science are being used to uncover complexities and design novel materials with enhanced properties. Finally, we point to the present research problems, challenges, and potential future perspectives of this new exciting field. read less NOT USED (low confidence) A. Lunghi and S. Sanvito, “A unified picture of the covalent bond within quantum-accurate force fields: From organic molecules to metallic complexes’ reactivity,” Science Advances. 2019. link Times cited: 30 Abstract: Machine learning atomic potentials can universally describe … read moreAbstract: Machine learning atomic potentials can universally describe chemical bonds in both organic and organometallic compounds. Computational studies of chemical processes taking place over extended size and time scales are inaccessible by electronic structure theories and can be tackled only by atomistic models such as force fields. These have evolved over the years to describe the most diverse systems. However, as we improve the performance of a force field for a particular physical/chemical situation, we are also moving away from a unified description. Here, we demonstrate that a unified picture of the covalent bond is achievable within the framework of machine learning–based force fields. Ridge regression, together with a representation of the atomic environment in terms of bispectrum components, can be used to map a general potential energy surface for molecular systems at chemical accuracy. This protocol sets the ground for the generation of an accurate and universal class of potentials for both organic and organometallic compounds with no specific assumptions on the chemistry involved. read less NOT USED (low confidence) S. Patala, “Understanding grain boundaries – The role of crystallography, structural descriptors and machine learning,” Computational Materials Science. 2019. link Times cited: 28 NOT USED (low confidence) A. Singraber, T. Morawietz, J. Behler, and C. Dellago, “Parallel Multistream Training of High-Dimensional Neural Network Potentials.,” Journal of chemical theory and computation. 2019. link Times cited: 111 Abstract: Over the past years high-dimensional neural network potentia… read moreAbstract: Over the past years high-dimensional neural network potentials (HDNNPs), fitted to accurately reproduce ab initio potential energy surfaces, have become a powerful tool in chemistry, physics and materials science. Here, we focus on the training of the neural networks that lies at the heart of the HDNNP method. We present an efficient approach for optimizing the weight parameters of the neural network via multistream Kalman filtering, using potential energies and forces as reference data. In this procedure, the choice of the free parameters of the Kalman filter can have a significant impact on the fit quality. Carrying out a large parameter study, we determine optimal settings and demonstrate how to optimize training results of HDNNPs. Moreover, we illustrate our HDNNP training approach by revisiting previously presented fits for water and developing a new potential for copper sulfide. This material, accessible in computer simulations so far only via first-principles methods, forms a particularly complex solid structure at low temperatures and undergoes a phase transition to a superionic state upon heating. Analyzing MD simulations carried out with the Cu2S HDNNP, we confirm that the underlying ab initio reference method indeed reproduces this behavior. read less NOT USED (low confidence) S. Ong, “Accelerating materials science with high-throughput computations and machine learning,” Computational Materials Science. 2019. link Times cited: 66 NOT USED (low confidence) A. Hernandez, A. Balasubramanian, F. Yuan, S. Mason, and T. Mueller, “Fast, accurate, and transferable many-body interatomic potentials by symbolic regression,” npj Computational Materials. 2019. link Times cited: 51 NOT USED (low confidence) H. Wang, X. Guo, L. Zhang, H. Wang, and J. Xue, “Deep learning inter-atomic potential model for accurate irradiation damage simulations,” Applied Physics Letters. 2019. link Times cited: 32 Abstract: We propose a hybrid scheme that interpolates smoothly the Zi… read moreAbstract: We propose a hybrid scheme that interpolates smoothly the Ziegler-Biersack-Littmark (ZBL) screened nuclear repulsion potential with a newly developed deep learning potential energy model. The resulting DP-ZBL model can not only provide overall good performance on the predictions of near-equilibrium material properties but also capture the right physics when atoms are extremely close to each other, an event that frequently happens in computational simulations of irradiation damage events. We applied this scheme to the simulation of the irradiation damage processes in the face-centered-cubic aluminium system, and found better descriptions in terms of the defect formation energy, evolution of collision cascades, displacement threshold energy, and residual point defects, than the widely-adopted ZBL modified embedded atom method potentials and its variants. Our work provides a reliable and feasible scheme to accurately simulate the irradiation damage processes and opens up new opportunities to solve the predicament of lacking accurate potentials for enormous newly-discovered materials in the irradiation effect field. read less NOT USED (low confidence) A. Singraber, J. Behler, and C. Dellago, “Library-Based LAMMPS Implementation of High-Dimensional Neural Network Potentials.,” Journal of chemical theory and computation. 2019. link Times cited: 155 Abstract: Neural networks and other machine learning approaches have b… read moreAbstract: Neural networks and other machine learning approaches have been successfully used to accurately represent atomic interaction potentials derived from computationally demanding electronic structure calculations. Due to their low computational cost, such representations open the possibility for large scale reactive molecular dynamics simulations of processes with bonding situations that cannot be described accurately with traditional empirical force fields. Here, we present a library of functions developed for the implementation of neural network potentials. Written in C++, this library incorporates several strategies resulting in a very high efficiency of neural network potential-energy and force evaluations. Based on this library, we have developed an implementation of the neural network potential within the molecular dynamics package LAMMPS and demonstrate its performance using liquid water as a test system. read less NOT USED (low confidence) R. Drautz, “Atomic cluster expansion for accurate and transferable interatomic potentials,” Physical Review B. 2019. link Times cited: 260 NOT USED (low confidence) C. Chen, W. Ye, Y. Zuo, C. Zheng, and S. Ong, “Graph Networks as a Universal Machine Learning Framework for Molecules and Crystals,” Chemistry of Materials. 2018. link Times cited: 602 Abstract: Graph networks are a new machine learning (ML) paradigm that… read moreAbstract: Graph networks are a new machine learning (ML) paradigm that supports both relational reasoning and combinatorial generalization. Here, we develop, for the first time, universal MatErials Graph Network (MEGNet) models for accurate property prediction in both molecules and crystals. We demonstrate that our MEGNet models significantly outperform prior ML models in 11 out of 13 properties of the QM9 molecule data set. Furthermore, a single-task unified MEGNet model can accurately predict the internal energy at 0 K and room temperature, enthalpy and Gibbs free energy, with temperature, pressure and entropy being global state inputs. Similarly, we show that MEGNet models trained on $\sim 60,000$ crystals in the Materials Project substantially outperform prior ML models in the prediction of the formation energies, band gaps and elastic moduli of crystals, achieving better than DFT accuracy over a much larger data set. Such MEGNet models are highly interpretable, and well-established periodic chemical trends can be extracted from the elemental embeddings. Finally, we demonstrate the transfer learning of elemental embeddings from a property model trained on a larger data set (formation energies) to accelerate the training of property models with smaller amounts of data (band gaps and elastic moduli) read less NOT USED (low confidence) W. Li and Y. Ando, “Comparison of different machine learning models for the prediction of forces in copper and silicon dioxide.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 17 Abstract: Recently, the machine learning (ML) force field has emerged … read moreAbstract: Recently, the machine learning (ML) force field has emerged as a powerful atomic simulation approach because of its high accuracy and low computational cost. However, there have been relatively fewer applications to multicomponent materials. In this study, we construct and compare ML force fields for both an elemental material (Cu) and binary material (SiO2) with varied inputs and regression models. The atomic environments are described by structural fingerprints that take into account the bond angle, and then, different ML techniques, including linear regression, a neural network and a mixture model method, are used to learn the structure-force relationship. We found that using angular structural fingerprints and a mixture model method significantly improves the accuracy of ML force fields. Additionally, we discuss an effective structural fingerprint auto-selection method based on the least absolute shrinkage and selection operator and the genetic algorithm. The atomic simulations conducted for ML force fields are in excellent agreement with ab initio calculations. As a result of the simulation with our ML force field for the structural and vibrational properties of amorphous SiO2, simulated annealing with a slow cooling rate improved the ring statistics in the amorphous structure and the phonon density of states. read less NOT USED (low confidence) J. M. Alred, K. V. Bets, Y. Xie, and B. Yakobson, “Machine learning electron density in sulfur crosslinked carbon nanotubes,” Composites Science and Technology. 2018. link Times cited: 37 NOT USED (low confidence) B. Nebgen et al., “Transferable Dynamic Molecular Charge Assignment Using Deep Neural Networks.,” Journal of chemical theory and computation. 2018. link Times cited: 77 Abstract: The ability to accurately and efficiently compute quantum-me… read moreAbstract: The ability to accurately and efficiently compute quantum-mechanical partial atomistic charges has many practical applications, such as calculations of IR spectra, analysis of chemical bonding, and classical force field parametrization. Machine learning (ML) techniques provide a possible avenue for the efficient prediction of atomic partial charges. Modern ML advances in the prediction of molecular energies [i.e., the hierarchical interacting particle neural network (HIP-NN)] has provided the necessary model framework and architecture to predict transferable, extensible, and conformationally dynamic atomic partial charges based on reference density functional theory (DFT) simulations. Utilizing HIP-NN, we show that ML charge prediction can be highly accurate over a wide range of molecules (both small and large) across a variety of charge partitioning schemes such as the Hirshfeld, CM5, MSK, and NBO methods. To demonstrate transferability and size extensibility, we compare ML results with reference DFT calculations on the COMP6 benchmark, achieving errors of 0.004e- (elementary charge). This is remarkable since this benchmark contains two proteins that are multiple times larger than the largest molecules in the training set. An application of our atomic charge predictions on nonequilibrium geometries is the generation of IR spectra for organic molecules from dynamical trajectories on a variety of organic molecules, which show good agreement with calculated IR spectra with reference method. Critically, HIP-NN charge predictions are many orders of magnitude faster than direct DFT calculations. These combined results provide further evidence that ML (specifically HIP-NN) provides a pathway to greatly increase the range of feasible simulations while retaining quantum-level accuracy. read less NOT USED (low confidence) K. Gubaev, E. Podryabinkin, G. Hart, and A. Shapeev, “Accelerating high-throughput searches for new alloys with active learning of interatomic potentials,” Computational Materials Science. 2018. link Times cited: 208 NOT USED (low confidence) M. Reveil and P. Clancy, “Classification of spatially resolved molecular fingerprints for machine learning applications and development of a codebase for their implementation.” 2018. link Times cited: 10 Abstract: Direct mapping between material structures and properties fo… read moreAbstract: Direct mapping between material structures and properties for various classes of materials is often the ultimate goal of materials researchers. Recent progress in the field of machine learning has created a unique path to develop such mappings based on empirical data. This new opportunity warranted the need for the development of advanced structural representations suitable for use with current machine learning algorithms. A number of such representations termed “molecular fingerprints” or descriptors have been proposed over the years for this purpose. In this paper, we introduce a classification framework to better explain and interpret existing fingerprinting schemes in the literature, with a focus on those with spatial resolution. We then present the implementation of SEING, a new codebase to computing those fingerprints, and we demonstrate its capabilities by building k-nearest neighbor (k-NN) models for force prediction that achieve a generalization accuracy of 0.1 meV A−1 and an R2 score as high as 0.99 at testing. Our results indicate that simple and generally overlooked k-NN models could be very promising compared to approaches such as neural networks, Gaussian processes, and support vector machines, which are more commonly used for machine learning-based predictions in computational materials science. read less NOT USED (low confidence) P. C. Myint and J. Belof, “Corrigendum: Rapid freezing of water under dynamic compression (2018 J. Phys.: Condens. Matter 30 233002),” Journal of Physics: Condensed Matter. 2018. link Times cited: 1 NOT USED (low confidence) M. Wood and A. Thompson, “Extending the accuracy of the SNAP interatomic potential form.,” The Journal of chemical physics. 2017. link Times cited: 130 Abstract: The Spectral Neighbor Analysis Potential (SNAP) is a classic… read moreAbstract: The Spectral Neighbor Analysis Potential (SNAP) is a classical interatomic potential that expresses the energy of each atom as a linear function of selected bispectrum components of the neighbor atoms. An extension of the SNAP form is proposed that includes quadratic terms in the bispectrum components. The extension is shown to provide a large increase in accuracy relative to the linear form, while incurring only a modest increase in computational cost. The mathematical structure of the quadratic SNAP form is similar to the embedded atom method (EAM), with the SNAP bispectrum components serving as counterparts to the two-body density functions in EAM. The effectiveness of the new form is demonstrated using an extensive set of training data for tantalum structures. Similar to artificial neural network potentials, the quadratic SNAP form requires substantially more training data in order to prevent overfitting. The quality of this new potential form is measured through a robust cross-validation analysis. read less NOT USED (low confidence) R. Lindsey, L. Fried, and N. Goldman, “ChIMES: A Force Matched Potential with Explicit Three-Body Interactions for Molten Carbon.,” Journal of chemical theory and computation. 2017. link Times cited: 50 Abstract: We present a new force field and development scheme for atom… read moreAbstract: We present a new force field and development scheme for atomistic simulations of materials under extreme conditions. These models, which explicitly include two- and three-body interactions, are generated by fitting linear combinations of Chebyshev polynomials through force matching to trajectories from Kohn-Sham density functional theory (DFT). We apply our method to liquid carbon near the diamond/graphite/liquid triple point and at higher densities and temperatures, where metallization and many-body effects may be substantial. We show that explicit inclusion of three-body interaction terms allows our model to yield improved descriptions of both dynamic and structural properties over previous empirical potential efforts, while exhibiting transferability to nearby state points. The simplicity of our functional form and subsequent efficiency of parameter determination allow for extension of DFT to experimental time and length scales while retaining most of its accuracy. read less NOT USED (low confidence) A. Thompson and C. Trott, “A Brief Description of the Kokkos implementation of the SNAP potential in ExaMiniMD.” 2017. link Times cited: 8 Abstract: Within the EXAALT project, the SNAP [1] approach is being us… read moreAbstract: Within the EXAALT project, the SNAP [1] approach is being used to develop high accuracy potentials for use in large-scale long-time molecular dynamics simulations of materials behavior. In particular, we have developed a new SNAP potential that is suitable for describing the interplay between helium atoms and vacancies in high-temperature tungsten[2]. This model is now being used to study plasma-surface interactions in nuclear fusion reactors for energy production. The high-accuracy of SNAP potentials comes at the price of increased computational cost per atom and increased computational complexity. The increased cost is mitigated by improvements in strong scaling that can be achieved using advanced algorithms [3]. read less NOT USED (low confidence) I. Kruglov, O. Sergeev, A. Yanilkin, and A. Oganov, “Energy-free machine learning force field for aluminum,” Scientific Reports. 2017. link Times cited: 51 NOT USED (low confidence) L. Zhang, J. Han, H. Wang, R. Car, and E. Weinan, “Deep Potential Molecular Dynamics: a scalable model with the accuracy of quantum mechanics,” Physical review letters. 2017. link Times cited: 785 Abstract: We introduce a scheme for molecular simulations, the deep po… read moreAbstract: We introduce a scheme for molecular simulations, the deep potential molecular dynamics (DPMD) method, based on a many-body potential and interatomic forces generated by a carefully crafted deep neural network trained with ab initio data. The neural network model preserves all the natural symmetries in the problem. It is first-principles based in the sense that there are no ad hoc components aside from the network model. We show that the proposed scheme provides an efficient and accurate protocol in a variety of systems, including bulk materials and molecules. In all these cases, DPMD gives results that are essentially indistinguishable from the original data, at a cost that scales linearly with system size. read less NOT USED (low confidence) L. T. Ward and C. Wolverton, “Atomistic calculations and materials informatics: A review,” Current Opinion in Solid State & Materials Science. 2017. link Times cited: 168 NOT USED (low confidence) G. Ferré, T. Haut, and K. Barros, “Learning molecular energies using localized graph kernels.,” The Journal of chemical physics. 2016. link Times cited: 50 Abstract: Recent machine learning methods make it possible to model po… read moreAbstract: Recent machine learning methods make it possible to model potential energy of atomic configurations with chemical-level accuracy (as calculated from ab initio calculations) and at speeds suitable for molecular dynamics simulation. Best performance is achieved when the known physical constraints are encoded in the machine learning models. For example, the atomic energy is invariant under global translations and rotations; it is also invariant to permutations of same-species atoms. Although simple to state, these symmetries are complicated to encode into machine learning algorithms. In this paper, we present a machine learning approach based on graph theory that naturally incorporates translation, rotation, and permutation symmetries. Specifically, we use a random walk graph kernel to measure the similarity of two adjacency matrices, each of which represents a local atomic environment. This Graph Approximated Energy (GRAPE) approach is flexible and admits many possible extensions. We benchmark a simple version of GRAPE by predicting atomization energies on a standard dataset of organic molecules. read less NOT USED (low confidence) K. Sarkar, M. Topsakal, N. Holzwarth, and R. Wentzcovitch, “Evolutionary optimization of PAW data-sets for accurate high pressure simulations,” J. Comput. Phys. 2016. link Times cited: 8 NOT USED (low confidence) N. Artrith and A. Urban, “An implementation of artificial neural-network potentials for atomistic materials simulations: Performance for TiO2,” Computational Materials Science. 2016. link Times cited: 350 NOT USED (low confidence) M. Shaughnessy and R. E. Jones, “Efficient Use of an Adapting Database of Ab Initio Calculations To Generate Accurate Newtonian Dynamics.,” Journal of chemical theory and computation. 2016. link Times cited: 3 Abstract: We develop and demonstrate a method to efficiently use densi… read moreAbstract: We develop and demonstrate a method to efficiently use density functional calculations to drive classical dynamics of complex atomic and molecular systems. The method has the potential to scale to systems and time scales unreachable with current ab initio molecular dynamics schemes. It relies on an adapting dataset of independently computed Hellmann-Feynman forces for atomic configurations endowed with a distance metric. The metric on configurations enables fast database lookup and robust interpolation of the stored forces. We discuss mechanisms for the database to adapt to the needs of the evolving dynamics, while maintaining accuracy, and other extensions of the basic algorithm. read less NOT USED (low confidence) H. Wilson, “Efficient ab initio free energy calculations by classically assisted trajectory sampling,” Comput. Phys. Commun. 2015. link Times cited: 1 NOT USED (low confidence) F. Bigi and M. Ceriotti, “Fast evaluation of real spherical harmonics and their derivatives in Cartesian coordinates,” ArXiv. 2023. link Times cited: 0 Abstract: Spherical harmonics provide a smooth, orthogonal, and symmet… read moreAbstract: Spherical harmonics provide a smooth, orthogonal, and symmetry-adapted basis to expand functions on a sphere, and they are used routinely in computer graphics, signal processing and different fields of science, from geology to quantum chemistry. More recently, spherical harmonics have become a key com-ponent of rotationally equivariant models for geometric deep learning, where they are used in combination with distance-dependent functions to describe the distribution of neighbors within local spherical environments within a point cloud. We present a fast and elegant algorithm for the evaluation of the real-valued spherical harmonics. Our construction integrates many of the desirable features of existing schemes and allows to compute Cartesian derivatives in a numerically stable and computationally efficient manner. We provide an efficient C implementation of the proposed algorithm, along with easy-to-use Python bindings. read less NOT USED (low confidence) F. Wang et al., “Atomic-scale simulations in multi-component alloys and compounds: A review on advances in interatomic potential,” Journal of Materials Science & Technology. 2023. link Times cited: 9 NOT USED (low confidence) Z. Lu et al., “Materials genome strategy for metallic glasses,” Journal of Materials Science & Technology. 2023. link Times cited: 1 NOT USED (low confidence) Y. Wang et al., “Anisotropic Thermal Transport in Chalcogenide Perovskite CaZrS3 from Machine Learning Interatomic Potential,” Engineered Science. 2023. link Times cited: 0 Abstract: Chalcogenide perovskites are being actively considered for p… read moreAbstract: Chalcogenide perovskites are being actively considered for photovoltaic, optoelectronic, and thermoelectric applications due to their high carrier mobility, strong light absorption, long-term stability, and environment-friendliness. For all these applications, thermal properties play a key role in determining the performance and lifetime of perovskite systems. In this work, we have developed a machine-learning Gaussian approximation potential to study the structural and thermal transport properties of chalcogenide perovskite CaZrS 3 . We show that the GAP achieves a DFT-level accuracy in describing both cubic and orthorhombic CaZrS 3 , with 2-4 orders of magnitude reduced computational cost. Specifically, we applied the GAP to predict the lattice thermal conductivities ( κ L ) and phonon properties of orthorhombic CaZrS 3 from 200 to 900 K by considering four-phonon processes. Compared to its counterpart CaZrSe 3 , the CaZrS 3 exhibits comparably low but relatively more anisotropic κ L mainly due to its strong anharmonicity and anisotropic group velocities. Specifically, its thermal conductivities along the a-and c-axis are close and notably lower than that along the b -axis. Optical phonons contribute as high as nearly half of the total thermal conductivity throughout the entire temperature range. Particularly, we observe non-* read less NOT USED (low confidence) M. Cusentino et al., “Molecular dynamics of high pressure tin phases: Empirical and machine learned interatomic potentials,” SHOCK COMPRESSION OF CONDENSED MATTER - 2022: Proceedings of the Conference of the American Physical Society Topical Group on Shock Compression of Condensed Matter. 2023. link Times cited: 0 NOT USED (low confidence) I. A. Balyakin and S. Sadovnikov, “Deep learning potential for superionic phase of Ag2S,” Computational Materials Science. 2022. link Times cited: 9 NOT USED (low confidence) G. Sivaraman et al., “A Combined Machine Learning and High-Energy X-ray Diffraction Approach to Understanding Liquid and Amorphous Metal Oxides,” Journal of the Physical Society of Japan. 2022. link Times cited: 2 NOT USED (low confidence) D. Zhang, H. Bi, F.-Z. Dai, W. Jiang, L. Zhang, and H. Wang, “DPA-1: Pretraining of Attention-based Deep Potential Model for Molecular Simulation,” ArXiv. 2022. link Times cited: 8 Abstract: Machine learning assisted modeling of the inter-atomic poten… read moreAbstract: Machine learning assisted modeling of the inter-atomic potential energy surface (PES) is revolutionizing the field of molecular simulation. With the accumulation of high-quality electronic structure data, a model that can be pretrained on all available data and finetuned on downstream tasks with a small additional effort would bring the field to a new stage. Here we propose DPA-1, a Deep Potential model with a novel attention mechanism, which is highly effective for representing the conformation and chemical spaces of atomic systems and learning the PES. We tested DPA-1 on a number of systems and observed superior performance compared with existing benchmarks. When pretrained on large-scale datasets containing 56 elements, DPA-1 can be successfully applied to various downstream tasks with a great improvement of sample efficiency. Surprisingly, for different elements, the learned type embedding parameters form a spiral in the latent space and have a natural correspondence with their positions on the periodic table, showing interesting interpretability of the pretrained DPA-1 model. read less NOT USED (low confidence) R. T. Evans et al., “Optimizing GPU-Enhanced HPC System and Cloud Procurements for Scientific Workloads,” Information Security Conference. 2021. link Times cited: 3 NOT USED (low confidence) C. Becquart, N. Mousseau, and C. Domain, “Atomistic Kinetic Monte Carlo and Solute Effects,” Handbook of Materials Modeling. 2020. link Times cited: 3 NOT USED (low confidence) J. Willman et al., “Quantum accurate SNAP carbon potential for MD shock simulations,” SHOCK COMPRESSION OF CONDENSED MATTER - 2019: Proceedings of the Conference of the American Physical Society Topical Group on Shock Compression of Condensed Matter. 2020. link Times cited: 6 Abstract: . We present a new quantum accurate Spectral Neighbor Analys… read moreAbstract: . We present a new quantum accurate Spectral Neighbor Analysis Potential (SNAP) machine-learning potential for simulating carbon under extreme conditions of dynamic compression (pressures up to 1 TPa and temperatures up to 10,000 K). The development of SNAP potential involves (1) the generation of the training database comprised of the consistent and meaningful set of first-principles DFT (Density Functional Theory) data for carbon materials at high pressure and temperature; (2) the robust and physically guided training of the SNAP parameters on first-principles data involving statistical data analysis; and (3) the validation of the SNAP potential in MD simulations of carbon at high PT conditions. The excellent performance of quadratic SNAP potential is demonstrated by simulating the radial distribution functions at high pressure-temperature conditions and melt curve of diamond, which were found in good agreement with DFT. read less NOT USED (low confidence) N. A. Mehta, R. Gayatri, Y. Ghadar, C. Knight, and J. Deslippe, “Evaluating Performance Portability of OpenMP for SNAP on NVIDIA, Intel, and AMD GPUs Using the Roofline Methodology,” WACCPD@SC. 2020. link Times cited: 4 NOT USED (low confidence) A. Tran, D. Liu, L. He-Bitoun, and Y. Wang, “Data-driven acceleration of first-principles saddle point and local minimum search based on scalable Gaussian processes.” 2020. link Times cited: 2 NOT USED (low confidence) R. Lindsey, M. Kroonblawd, L. Fried, and N. Goldman, “Force Matching Approaches to Extend Density Functional Theory to Large Time and Length Scales,” Computational Approaches for Chemistry Under Extreme Conditions. 2019. link Times cited: 6 NOT USED (low confidence) W. Xiao, Y. Li, and P. Wang, “Uncertainty Quantification of Atomistic Materials Simulation with Machine Learning Potentials.” 2018. link Times cited: 4 NOT USED (low confidence) J. Maillet, C. Denoual, and G. Csányi, “Machine-Learning Based Potential For Iron: Plasticity and Phase Transition,” Bulletin of the American Physical Society. 2018. link Times cited: 8 NOT USED (high confidence) A. M. Tokita and J. Behler, “How to train a neural network potential,” The Journal of Chemical Physics. 2023. link Times cited: 4 Abstract: The introduction of modern Machine Learning Potentials (MLPs… read moreAbstract: The introduction of modern Machine Learning Potentials (MLPs) has led to a paradigm change in the development of potential energy surfaces for atomistic simulations. By providing efficient access to energies and forces, they allow us to perform large-scale simulations of extended systems, which are not directly accessible by demanding first-principles methods. In these simulations, MLPs can reach the accuracy of electronic structure calculations, provided that they have been properly trained and validated using a suitable set of reference data. Due to their highly flexible functional form, the construction of MLPs has to be done with great care. In this Tutorial, we describe the necessary key steps for training reliable MLPs, from data generation via training to final validation. The procedure, which is illustrated for the example of a high-dimensional neural network potential, is general and applicable to many types of MLPs. read less NOT USED (high confidence) P. Plettenberg, B. Bauerhenne, and M. E. Garcia, “Neural network interatomic potential for laser-excited materials,” Communications Materials. 2023. link Times cited: 1 NOT USED (high confidence) K. Gubaev, V. Zaverkin, P. Srinivasan, A. Duff, J. Kästner, and B. Grabowski, “Performance of two complementary machine-learned potentials in modelling chemically complex systems,” npj Computational Materials. 2023. link Times cited: 1 NOT USED (high confidence) Y. Liu, X. He, and Y. Mo, “Discrepancies and error evaluation metrics for machine learning interatomic potentials,” npj Computational Materials. 2023. link Times cited: 1 NOT USED (high confidence) A. Rohskopf et al., “Exploring model complexity in machine learned potentials for simulated properties,” Journal of Materials Research. 2023. link Times cited: 1 Abstract: Machine learning (ML) enables the development of interatomic… read moreAbstract: Machine learning (ML) enables the development of interatomic potentials with the accuracy of first principles methods while retaining the speed and parallel efficiency of empirical potentials. While ML potentials traditionally use atom-centered descriptors as inputs, different models such as linear regression and neural networks map descriptors to atomic energies and forces. This begs the question: what is the improvement in accuracy due to model complexity irrespective of descriptors? We curate three datasets to investigate this question in terms of ab initio energy and force errors: (1) solid and liquid silicon, (2) gallium nitride, and (3) the superionic conductor Li$$_{10}$$
10
Ge(PS$$_{6}$$
6
)$$_{2}$$
2
(LGPS). We further investigate how these errors affect simulated properties and verify if the improvement in fitting errors corresponds to measurable improvement in property prediction. By assessing different models, we observe correlations between fitting quantity (e.g. atomic force) error and simulated property error with respect to ab initio values.
Graphical abstract read less NOT USED (high confidence) T. W. Ko, J. A. Finkler, S. Goedecker, and J. Behler, “Accurate Fourth-Generation Machine Learning Potentials by Electrostatic Embedding.,” Journal of chemical theory and computation. 2023. link Times cited: 3 Abstract: In recent years, significant progress has been made in the d… read moreAbstract: In recent years, significant progress has been made in the development of machine learning potentials (MLPs) for atomistic simulations with applications in many fields from chemistry to materials science. While most current MLPs are based on environment-dependent atomic energies, the limitations of this locality approximation can be overcome, e.g., in fourth-generation MLPs, which incorporate long-range electrostatic interactions based on an equilibrated global charge distribution. Apart from the considered interactions, the quality of MLPs crucially depends on the information available about the system, i.e., the descriptors. In this work we show that including─in addition to structural information─the electrostatic potential arising from the charge distribution in the atomic environments significantly improves the quality and transferability of the potentials. Moreover, the extended descriptor allows current limitations of two- and three-body based feature vectors to be overcome regarding artificially degenerate atomic environments. The capabilities of such an electrostatically embedded fourth-generation high-dimensional neural network potential (ee4G-HDNNP), which is further augmented by pairwise interactions, are demonstrated for NaCl as a benchmark system. Employing a data set containing only neutral and negatively charged NaCl clusters, even small energy differences between different cluster geometries can be resolved, and the potential shows an impressive transferability to positively charged clusters as well as the melt. read less NOT USED (high confidence) E. Podryabinkin, K. Garifullin, A. Shapeev, and I. Novikov, “MLIP-3: Active learning on atomic environments with moment tensor potentials.,” The Journal of chemical physics. 2023. link Times cited: 5 Abstract: Nowadays, academic research relies not only on sharing with … read moreAbstract: Nowadays, academic research relies not only on sharing with the academic community the scientific results obtained by research groups while studying certain phenomena but also on sharing computer codes developed within the community. In the field of atomistic modeling, these were software packages for classical atomistic modeling, and later for quantum-mechanical modeling; currently, with the fast growth of the field of machine-learning potentials, the packages implement such potentials. In this paper, we present the MLIP-3 package for constructing moment tensor potentials and performing their active training. This package builds on the MLIP-2 package [Novikov et al., "The MLIP package: moment tensor potentials with MPI and active learning," Mach. Learn.: Sci. Technol., 2(2), 025002 (2020)], however, with a number of improvements, including active learning on atomic neighborhoods of a possibly large atomistic simulation. read less NOT USED (high confidence) S. Kumar et al., “Transferable interatomic potential for aluminum from ambient conditions to warm dense matter,” Physical Review Research. 2023. link Times cited: 0 Abstract: We present a study on the transport and materials properties… read moreAbstract: We present a study on the transport and materials properties of aluminum spanning from ambient to warm dense matter conditions using a machine-learned interatomic potential (ML-IAP). Prior research has utilized ML-IAPs to simulate phenomena in warm dense matter, but these potentials have often been calibrated for a narrow range of temperature and pressures. In contrast, we train a single ML-IAP over a wide range of temperatures, using density functional theory molecular dynamics (DFT-MD) data. Our approach overcomes computational limitations of DFT-MD simulations, enabling us to study transport and materials properties of matter at higher temperatures and longer time scales. We demonstrate the ML-IAP transferability across a wide range of temperatures using molecular-dynamics (MD) by examining the thermal conductivity, diffusion coefficient, viscosity, sound velocity, and ion-ion structure factor of aluminum up to about 60,000 K, where we find good agreement with previous theoretical data. read less NOT USED (high confidence) A. Diggs et al., “Hydrogen-induced degradation dynamics in silicon heterojunction solar cells via machine learning,” Communications Materials. 2023. link Times cited: 1 NOT USED (high confidence) A. Rohskopf et al., “FitSNAP: Atomistic machine learning with LAMMPS,” J. Open Source Softw. 2023. link Times cited: 12 NOT USED (high confidence) M. Minotakis, H. Rossignol, M. Cobelli, and S. Sanvito, “Machine-learning surrogate model for accelerating the search of stable ternary alloys,” Physical Review Materials. 2023. link Times cited: 1 Abstract: The prediction of phase diagrams in the search for new phase… read moreAbstract: The prediction of phase diagrams in the search for new phases is a complex and computationally intensive task. Density functional theory provides, in many situations, the desired accuracy, but its throughput becomes prohibitively limited as the number of species involved grows, even when used with local and semi-local functionals. Here, we explore the possibility of integrating machine-learning models in the workflow for the construction of ternary convex hull diagrams. In particular, we train a set of spectral neighbour-analysis potentials (SNAPs) over readily available binary phases and we establish whether this is good enough to predict the energies of novel ternaries. Such a strategy does not require any new calculations specific for the construction of the model, but just avails of data stored in binary-phase-diagram repositories. We find that a so-constructed SNAP is capable of accurate total-energy estimates for ternary phases close to the equilibrium geometry but, in general, is not able to perform atomic relaxation. This is because during a typical relaxation path a given phase traverses regions in the parameter space poorly represented by the training set. Different metrics are then investigated to assess how an unknown structure is well described by a given SNAP model, and we find that the standard deviation of an ensemble of SNAPs provides a fast and non-specie-specific metric. read less NOT USED (high confidence) J. M. Goff, Y. Zhang, C. Negre, A. Rohskopf, and A. Niklasson, “Shadow Molecular Dynamics and Atomic Cluster Expansions for Flexible Charge Models.,” Journal of chemical theory and computation. 2023. link Times cited: 0 Abstract: A shadow molecular dynamics scheme for flexible charge model… read moreAbstract: A shadow molecular dynamics scheme for flexible charge models is presented where the shadow Born-Oppenheimer potential is derived from a coarse-grained approximation of range-separated density functional theory. The interatomic potential, including the atomic electronegativities and the charge-independent short-range part of the potential and force terms, is modeled by the linear atomic cluster expansion (ACE), which provides a computationally efficient alternative to many machine learning methods. The shadow molecular dynamics scheme is based on extended Lagrangian (XL) Born-Oppenheimer molecular dynamics (BOMD) [Eur. Phys. J. B 2021, 94, 164]. XL-BOMD provides stable dynamics while avoiding the costly computational overhead associated with solving an all-to-all system of equations, which normally is required to determine the relaxed electronic ground state prior to each force evaluation. To demonstrate the proposed shadow molecular dynamics scheme for flexible charge models using atomic cluster expansion, we emulate the dynamics generated from self-consistent charge density functional tight-binding (SCC-DFTB) theory using a second-order charge equilibration (QEq) model. The charge-independent potentials and electronegativities of the QEq model are trained for a supercell of uranium oxide (UO2) and a molecular system of liquid water. The combined ACE+XL-QEq molecular dynamics simulations are stable over a wide range of temperatures both for the oxide and for the molecular systems and provide a precise sampling of the Born-Oppenheimer potential energy surfaces. Accurate ground Coulomb energies are produced by the ACE-based electronegativity model during an NVE simulation of UO2, predicted to be within 1 meV of those from SCC-DFTB on average during comparable simulations. read less NOT USED (high confidence) X.-G. Li, S. Xu, Q. Zhang, S. Liu, and J. Shuai, “Complex strengthening mechanisms in nanocrystalline Ni-Mo alloys revealed by a machine-learning interatomic potential,” Journal of Alloys and Compounds. 2023. link Times cited: 2 NOT USED (high confidence) F. Bigi, G. Fraux, N. Browning, and M. Ceriotti, “Fast evaluation of spherical harmonics with sphericart.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: Spherical harmonics provide a smooth, orthogonal, and symmet… read moreAbstract: Spherical harmonics provide a smooth, orthogonal, and symmetry-adapted basis to expand functions on a sphere, and they are used routinely in physical and theoretical chemistry as well as in different fields of science and technology, from geology and atmospheric sciences to signal processing and computer graphics. More recently, they have become a key component of rotationally equivariant models in geometric machine learning, including applications to atomic-scale modeling of molecules and materials. We present an elegant and efficient algorithm for the evaluation of the real-valued spherical harmonics. Our construction features many of the desirable properties of existing schemes and allows us to compute Cartesian derivatives in a numerically stable and computationally efficient manner. To facilitate usage, we implement this algorithm in sphericart, a fast C++ library that also provides C bindings, a Python API, and a PyTorch implementation that includes a GPU kernel. read less NOT USED (high confidence) P. Grigorev, A. Goryaeva, M. Marinica, J. Kermode, and T. Swinburne, “Calculation of dislocation binding to helium-vacancy defects in tungsten using hybrid ab initio-machine learning methods,” Acta Materialia. 2023. link Times cited: 6 NOT USED (high confidence) S. Ignatov and A. Masunov, “Unexpected polarization properties of sub-nanosized magnesium clusters,” RSC Advances. 2023. link Times cited: 0 Abstract: The isotropic electrostatic polarizability (IEP) of sub-nano… read moreAbstract: The isotropic electrostatic polarizability (IEP) of sub-nanosized magnesium clusters Mg2–Mg32 was studied in an extensive set comprising 1237 structurally unique isomers. These isomers were found in the course of the global search for the potential energy surface minima of the magnesium clusters at the BP86/6-31G(d) level. The calculation of the polarizability at the same DFT level reveals an unexpected property of the IEP: the linear correlation between the polarizability of the most favorable isomers (and only them) and the cluster nuclearity n. Moreover, for each n, the most stable cluster isomer demonstrates nearly minimal IEP value among all found isomers of a given nuclearity. Surprisingly, these observed features are independent of the cluster structures which are quite different. We hypothesize that the energetic favorability of a cluster structure is related to their low polarizability. Apparently, the atoms forming the cluster tend to arrange themselves in such a way as to provide the most compact distribution of the cluster electron density. A possible explanation of the observed trends, their significance for cluster structure prediction, and the practical applications are discussed. read less NOT USED (high confidence) E. Sanscartier, F. Saint-Denis, K.-’E. Bolduc, and N. Mousseau, “Evaluating approaches for on-the-fly machine learning interatomic potentials for activated mechanisms sampling with the activation-relaxation technique nouveau.,” The Journal of chemical physics. 2023. link Times cited: 1 Abstract: In the last few years, much effort has gone into developing … read moreAbstract: In the last few years, much effort has gone into developing general machine-learning potentials capable of describing interactions for a wide range of structures and phases. Yet, as attention turns to more complex materials, including alloys and disordered and heterogeneous systems, the challenge of providing reliable descriptions for all possible environments becomes ever more costly. In this work, we evaluate the benefits of using specific vs general potentials for the study of activated mechanisms in solid-state materials. More specifically, we test three machine-learning fitting approaches using the moment-tensor potential to reproduce a reference potential when exploring the energy landscape around a vacancy in Stillinger-Weber silicon crystal and silicon-germanium zincblende structures using the activation-relaxation technique nouveau (ARTn). We find that a targeted on-the-fly approach specific to and integrated into ARTn generates the highest precision on the energetics and geometry of activated barriers while remaining cost-effective. This approach expands the types of problems that can be addressed with high-accuracy ML potential. read less NOT USED (high confidence) J. Xi, Y. Shi, V. Pronskikh, F. Pellemoine, D. Morgan, and I. Szlufarska, “Atomistic simulations of He bubbles in Beryllium,” Journal of Nuclear Materials. 2023. link Times cited: 0 NOT USED (high confidence) N. Nguyen, “Fast proper orthogonal descriptors for many-body interatomic potentials,” Physical Review B. 2022. link Times cited: 1 Abstract: The development of differentiable invariant descriptors for … read moreAbstract: The development of differentiable invariant descriptors for accurate representations of atomic environments plays a central role in the success of interatomic potentials for chemistry and materials science. We introduce a method to generate fast proper orthogonal descriptors for the construction of many-body interatomic potentials and discuss its relation to exising empirical and machine learning interatomic potentials. A traditional way of implementing the proper orthogonal descriptors has a computational complexity that scales exponentially with the body order in terms of the number of neighbors. We present an algorithm to compute the proper orthogonal descriptors with a computational complexity that scales linearly with the number of neighbors irrespective of the body order. We show that our method can enable a more efficient implementation for a number of existing potentials and provide a scalable systematic framework to construct new many-body potentials. The new potentials are demonstrated on a data set of density functional theory calculations for Tantalum and compared with other interatomic potentials. read less NOT USED (high confidence) Y. Lysogorskiy, A. Bochkarev, M. Mrovec, and R. Drautz, “Active learning strategies for atomic cluster expansion models,” Physical Review Materials. 2022. link Times cited: 7 Abstract: The atomic cluster expansion (ACE) was proposed recently as … read moreAbstract: The atomic cluster expansion (ACE) was proposed recently as a new class of data-driven interatomic potentials with a formally complete basis set. Since the development of any interatomic potential requires a careful selection of training data and thorough validation, an automation of the construction of the training dataset as well as an indication of a model's uncertainty are highly desirable. In this work, we compare the performance of two approaches for uncertainty indication of ACE models based on the D-optimality criterion and ensemble learning. While both approaches show comparable predictions, the extrapolation grade based on the D-optimality (MaxVol algorithm) is more computationally efficient. In addition, the extrapolation grade indicator enables an active exploration of new structures, opening the way to the automated discovery of rare-event configurations. We demonstrate that active learning is also applicable to explore local atomic environments from large-scale MD simulations. read less NOT USED (high confidence) S. Hu et al., “RLEKF: An Optimizer for Deep Potential with Ab Initio Accuracy,” AAAI Conference on Artificial Intelligence. 2022. link Times cited: 0 Abstract: It is imperative to accelerate the training of neural networ… read moreAbstract: It is imperative to accelerate the training of neural network force field such as Deep Potential, which usually requires thousands of images based on first-principles calculation and a couple of days to generate an accurate potential energy surface. To this end, we propose a novel optimizer named reorganized layer extended Kalman filtering (RLEKF), an optimized version of global extended Kalman filtering (GEKF) with a strategy of splitting big and gathering small layers to overcome the O(N^2) computational cost of GEKF. This strategy provides an approximation of the dense weights error covariance matrix with a sparse diagonal block matrix for GEKF. We implement both RLEKF and the baseline Adam in our alphaDynamics package and numerical experiments are performed on 13 unbiased datasets. Overall, RLEKF converges faster with slightly better accuracy. For example, a test on a typical system, bulk copper, shows that RLEKF converges faster by both the number of training epochs (x11.67) and wall-clock time (x1.19). Besides, we theoretically prove that the updates of weights converge and thus are against the gradient exploding problem. Experimental results verify that RLEKF is not sensitive to the initialization of weights. The RLEKF sheds light on other AI-for-science applications where training a large neural network (with tons of thousands parameters) is a bottleneck. read less NOT USED (high confidence) M. Dharma-wardana, L. Stanek, and M. Murillo, “Yukawa-Friedel-tail pair potentials for warm dense matter applications.,” Physical review. E. 2022. link Times cited: 3 Abstract: Accurate equations of state (EOS) and plasma transport prope… read moreAbstract: Accurate equations of state (EOS) and plasma transport properties are essential for numerical simulations of warm dense matter encountered in many high-energy-density situations. Molecular dynamics (MD) is a simulation method that generates EOS and transport data using an externally provided potential to dynamically evolve the particles without further reference to the electrons. To minimize computational cost, pair potentials needed in MD may be obtained from the neutral-pseudoatom (NPA) approach, a form of single-ion density functional theory (DFT), where many-ion effects are included via ion-ion correlation functionals. Standard N-ion DFT-MD provides pair potentials via the force matching technique but at much greater computational cost. Here we propose a simple analytic model for pair potentials with physically meaningful parameters based on a Yukawa form with a thermally damped Friedel tail (YFT) applicable to systems containing free electrons. The YFT model accurately fits NPA pair potentials or the nonparametric force-matched potentials from N-ion DFT-MD, showing excellent agreement for a wide range of conditions. The YFT form provides accurate extrapolations of the NPA or force-matched potentials for small and large particle separations within a physical model. Our method can be adopted to treat plasma mixtures, allowing for large-scale simulations of multispecies warm dense matter. read less NOT USED (high confidence) G. Anand et al., “Exploiting Machine Learning in Multiscale Modelling of Materials,” Journal of The Institution of Engineers (India): Series D. 2022. link Times cited: 1 NOT USED (high confidence) A. J. W. Zhu, S. L. Batzner, A. Musaelian, and B. Kozinsky, “Fast Uncertainty Estimates in Deep Learning Interatomic Potentials,” The Journal of chemical physics. 2022. link Times cited: 15 Abstract: Deep learning has emerged as a promising paradigm to give ac… read moreAbstract: Deep learning has emerged as a promising paradigm to give access to highly accurate predictions of molecular and material properties. A common short-coming shared by current approaches, however, is that neural networks only give point estimates of their predictions and do not come with predictive uncertainties associated with these estimates. Existing uncertainty quantification efforts have primarily leveraged the standard deviation of predictions across an ensemble of independently trained neural networks. This incurs a large computational overhead in both training and prediction, resulting in order-of-magnitude more expensive predictions. Here, we propose a method to estimate the predictive uncertainty based on a single neural network without the need for an ensemble. This allows us to obtain uncertainty estimates with virtually no additional computational overhead over standard training and inference. We demonstrate that the quality of the uncertainty estimates matches those obtained from deep ensembles. We further examine the uncertainty estimates of our methods and deep ensembles across the configuration space of our test system and compare the uncertainties to the potential energy surface. Finally, we study the efficacy of the method in an active learning setting and find the results to match an ensemble-based strategy at order-of-magnitude reduced computational cost. read less NOT USED (high confidence) T. Suzudo, K. Ebihara, T. Tsuru, and H. Mori, “Cleavages along 110 in bcc iron emit dislocations from the curved crack fronts,” Scientific Reports. 2022. link Times cited: 5 NOT USED (high confidence) S. Attarian, D. Morgan, and I. Szlufarska, “Thermophysical properties of FLiBe using moment tensor potentials,” Journal of Molecular Liquids. 2022. link Times cited: 2 NOT USED (high confidence) L. Fiedler et al., “Predicting electronic structures at any length scale with machine learning,” npj Computational Materials. 2022. link Times cited: 4 NOT USED (high confidence) I. Novikov, O. Kovalyova, A. Shapeev, and M. Hodapp, “AI-accelerated materials informatics method for the discovery of ductile alloys,” Journal of Materials Research. 2022. link Times cited: 3 Abstract: In computational materials science, a common means for predi… read moreAbstract: In computational materials science, a common means for predicting macroscopic (e.g., mechanical) properties of an alloy is to define a model using combinations of descriptors that depend on some material properties (elastic constants, misfit volumes, etc.), representative for the macroscopic behavior. The material properties are usually computed using special quasi-random structures, in tandem with density functional theory (DFT). However, DFT scales cubically with the number of atoms and is thus impractical for a screening over many alloy compositions. Here, we present a novel methodology which combines modeling approaches and machine-learning interatomic potentials. Machine-learning interatomic potentials are orders of magnitude faster than DFT, while achieving similar accuracy, allowing for a predictive and tractable high-throughput screening over the whole alloy space. The proposed methodology is illustrated by predicting the room temperature ductility of the medium-entropy alloy Mo–Nb–Ta. Graphical abstract read less NOT USED (high confidence) C. van der Oord, M. Sachs, D. Kov’acs, C. Ortner, and G. Csányi, “Hyperactive learning for data-driven interatomic potentials,” npj Computational Materials. 2022. link Times cited: 17 NOT USED (high confidence) M. Bianchini, V. Lacivita, D. Seo, and H. Kim, “Advances and challenges in multiscale characterizations and analyses for battery materials,” Journal of Materials Research. 2022. link Times cited: 0 Abstract: Rechargeable ion batteries are efficient energy storage devi… read moreAbstract: Rechargeable ion batteries are efficient energy storage devices widely employed in portable to large-scale applications such as electric vehicles and grids. Electrochemical reactions within batteries are complex phenomena, and they are strongly dependent on the battery materials and systems used. These electrochemical reactions often include detrimental irreversible reactions at various length scales from atomic- to macro-scales, which ultimately determine the overall electrochemical behavior of the system. Understanding such reaction mechanisms is a critical component to improve battery performance. To help this effort, this review article discusses recent advances and remaining challenges in both computational and experimental approaches to better understand dynamic electrochemical reactions in batteries across multiple length scales. Important related findings from this focus issue will also be highlighted. The aim of our focus issue is to contribute to the battery community towards having better understanding of complex reactions occurring in battery devices and of computational and experimental methods to investigate them. Graphical abstract read less NOT USED (high confidence) J. P. Darby et al., “Tensor-reduced atomic density representations,” Physical review letters. 2022. link Times cited: 11 Abstract: Density-based representations of atomic environments that ar… read moreAbstract: Density-based representations of atomic environments that are invariant under Euclidean symmetries have become a widely used tool in the machine learning of interatomic potentials, broader data-driven atomistic modeling, and the visualization and analysis of material datasets. The standard mechanism used to incorporate chemical element information is to create separate densities for each element and form tensor products between them. This leads to a steep scaling in the size of the representation as the number of elements increases. Graph neural networks, which do not explicitly use density representations, escape this scaling by mapping the chemical element information into a fixed dimensional space in a learnable way. By exploiting symmetry, we recast this approach as tensor factorization of the standard neighbour-density-based descriptors and, using a new notation, identify connections to existing compression algorithms. In doing so, we form compact tensor-reduced representation of the local atomic environment whose size does not depend on the number of chemical elements, is systematically convergable, and therefore remains applicable to a wide range of data analysis and regression tasks. read less NOT USED (high confidence) M. Domina, U. Patil, M. Cobelli, and S. Sanvito, “Cluster expansion constructed over Jacobi-Legendre polynomials for accurate force fields,” Physical Review B. 2022. link Times cited: 2 Abstract: We introduce a compact cluster expansion method, constructed… read moreAbstract: We introduce a compact cluster expansion method, constructed over Jacobi and Legendre polynomials, to generate highly accurate and flexible machine-learning force fields. The constituent many-body contributions are separated, interpretable and adaptable to replicate the physical knowledge of the system. In fact, the flexibility introduced by the use of the Jacobi polynomials allows us to impose, in a natural way, constrains and symmetries to the cluster expansion. This has the effect of reducing the number of parameters needed for the fit and of enforcing desired behaviours of the potential. For instance, we show that our Jacobi-Legendre cluster expansion can be designed to generate potentials with a repulsive tail at short inter-atomic distances, without the need of imposing any external function. Our method is here continuously compared with available machine-learning potential schemes, such as the atomic cluster expansion and potentials built over the bispectrum. As an example we construct a Jacobi-Legendre potential for carbon, by training a slim and accurate model capable of describing crystalline graphite and diamond, as well as liquid and amorphous elemental carbon. read less NOT USED (high confidence) N. Nguyen and A. Rohskopf, “Proper orthogonal descriptors for efficient and accurate interatomic potentials,” J. Comput. Phys. 2022. link Times cited: 6 NOT USED (high confidence) B. Burlacu, M. Kommenda, G. Kronberger, S. M. Winkler, and M. Affenzeller, “Symbolic Regression in Materials Science: Discovering Interatomic Potentials from Data,” ArXiv. 2022. link Times cited: 2 NOT USED (high confidence) L. Fiedler et al., “Accelerating equilibration in first-principles molecular dynamics with orbital-free density functional theory,” Physical Review Research. 2022. link Times cited: 7 Abstract: We introduce a practical hybrid approach that combines orbit… read moreAbstract: We introduce a practical hybrid approach that combines orbital-free density functional theory (DFT) with Kohn-Sham DFT for speeding up first-principles molecular dynamics simulations. Equilibrated ionic configurations are generated using orbital-free DFT for subsequent Kohn-Sham DFT molecular dynamics. This leads to a massive reduction of the simulation time without any sacrifice in accuracy. We assess this finding across systems of different sizes and temperature, up to the warm dense matter regime. To that end, we use the cosine distance between the time series of radial distribution functions representing the ionic configurations. Likewise, we show that the equilibrated ionic configurations from this hybrid approach significantly enhance the accuracy of machine-learning models that replace Kohn-Sham DFT. Our hybrid scheme enables systematic first-principles simulations of warm dense matter that are otherwise hampered by the large numbers of atoms and the prevalent high temperatures. Moreover, our finding provides an additional motivation for developing kinetic and noninteracting free energy functionals for orbital-free DFT. read less NOT USED (high confidence) P. A. Santos-Flórez, H. Yanxon, B. Kang, Y. Yao, and Q. Zhu, “Size-Dependent Nucleation in Crystal Phase Transition from Machine Learning Metadynamics.,” Physical review letters. 2022. link Times cited: 1 Abstract: In this Letter, we present a framework that combines machine… read moreAbstract: In this Letter, we present a framework that combines machine learning potential (MLP) and metadynamics to investigate solid-solid phase transition. Based on the spectral descriptors and neural networks regression, we develop a scalable MLP model to warrant an accurate interpolation of the energy surface where two phases coexist. Applying it to the simulation of B4-B1 phase transition of GaN under 50 GPa with different model sizes, we observe sequential change of the phase transition mechanism from collective modes to nucleation and growths. When the size is at or below 128 000 atoms, the nucleation and growth appear to follow a preferred direction. At larger sizes, the nuclei occur at multiple sites simultaneously and grow to microstructures by passing the critical size. The observed change of the atomistic mechanism manifests the importance of statistical sampling with large system size in phase transition modeling. read less NOT USED (high confidence) Z. Fan et al., “GPUMD: A package for constructing accurate machine-learned potentials and performing highly efficient atomistic simulations.,” The Journal of chemical physics. 2022. link Times cited: 46 Abstract: We present our latest advancements of machine-learned potent… read moreAbstract: We present our latest advancements of machine-learned potentials (MLPs) based on the neuroevolution potential (NEP) framework introduced in Fan et al. [Phys. Rev. B 104, 104309 (2021)] and their implementation in the open-source package gpumd. We increase the accuracy of NEP models both by improving the radial functions in the atomic-environment descriptor using a linear combination of Chebyshev basis functions and by extending the angular descriptor with some four-body and five-body contributions as in the atomic cluster expansion approach. We also detail our efficient implementation of the NEP approach in graphics processing units as well as our workflow for the construction of NEP models and demonstrate their application in large-scale atomistic simulations. By comparing to state-of-the-art MLPs, we show that the NEP approach not only achieves above-average accuracy but also is far more computationally efficient. These results demonstrate that the gpumd package is a promising tool for solving challenging problems requiring highly accurate, large-scale atomistic simulations. To enable the construction of MLPs using a minimal training set, we propose an active-learning scheme based on the latent space of a pre-trained NEP model. Finally, we introduce three separate Python packages, viz., gpyumd, calorine, and pynep, that enable the integration of gpumd into Python workflows. read less NOT USED (high confidence) J. T. Willman et al., “Machine learning interatomic potential for simulations of carbon at extreme conditions,” Physical Review B. 2022. link Times cited: 10 Abstract: A Spectral Neighbor Analysis (SNAP) machine learning interat… read moreAbstract: A Spectral Neighbor Analysis (SNAP) machine learning interatomic potential (MLIP) has been developed for simulations of carbon at extreme pressures (up to 5 TPa) and temperatures (up to 20,000 K). This was achieved using a large database of experimentally relevant quantum molecular dynamics (QMD) data, training the SNAP potential using a robust machine learning methodology, and performing extensive validation against QMD and experimental data. The resultant carbon MLIP demonstrates unprecedented accuracy and transferability in predicting the carbon phase diagram, melting curves of crystalline phases, and the shock Hugoniot, all within 3% of QMD. By achieving quantum accuracy and efficient implementation on leadership class high performance computing systems, SNAP advances frontiers of classical MD simulations by enabling atomic-scale insights at experimental time and length scales. read less NOT USED (high confidence) L. C. Erhard, J. Rohrer, K. Albe, and V. L. Deringer, “A machine-learned interatomic potential for silica and its relation to empirical models,” npj Computational Materials. 2022. link Times cited: 32 NOT USED (high confidence) T. Lee et al., “Atomic-scale origin of the low grain-boundary resistance in perovskite solid electrolyte Li0.375Sr0.4375Ta0.75Zr0.25O3,” Nature Communications. 2022. link Times cited: 9 NOT USED (high confidence) M. J. Waters and J. Rondinelli, “Benchmarking structural evolution methods for training of machine learned interatomic potentials,” Journal of Physics: Condensed Matter. 2022. link Times cited: 1 Abstract: When creating training data for machine-learned interatomic … read moreAbstract: When creating training data for machine-learned interatomic potentials (MLIPs), it is common to create initial structures and evolve them using molecular dynamics (MD) to sample a larger configuration space. We benchmark two other modalities of evolving structures, contour exploration (CE) and dimer-method (DM) searches against MD for their ability to produce diverse and robust density functional theory training data sets for MLIPs. We also discuss the generation of initial structures which are either from known structures or from random structures in detail to further formalize the structure-sourcing processes in the future. The polymorph-rich zirconium-oxygen composition space is used as a rigorous benchmark system for comparing the performance of MLIPs trained on structures generated from these structural evolution methods. Using Behler–Parrinello neural networks as our MLIP models, we find that CE and the DM searches are generally superior to MD in terms of spatial descriptor diversity and statistical accuracy. read less NOT USED (high confidence) Y. Xie, J. Vandermause, S. Ramakers, N. Protik, A. Johansson, and B. Kozinsky, “Uncertainty-aware molecular dynamics from Bayesian active learning for phase transformations and thermal transport in SiC,” npj Computational Materials. 2022. link Times cited: 14 NOT USED (high confidence) J. D. Lee et al., “Dilute Alloys Based on Au, Ag, or Cu for Efficient Catalysis: From Synthesis to Active Sites.,” Chemical reviews. 2022. link Times cited: 34 Abstract: The development of new catalyst materials for energy-efficie… read moreAbstract: The development of new catalyst materials for energy-efficient chemical synthesis is critical as over 80% of industrial processes rely on catalysts, with many of the most energy-intensive processes specifically using heterogeneous catalysis. Catalytic performance is a complex interplay of phenomena involving temperature, pressure, gas composition, surface composition, and structure over multiple length and time scales. In response to this complexity, the integrated approach to heterogeneous dilute alloy catalysis reviewed here brings together materials synthesis, mechanistic surface chemistry, reaction kinetics, in situ and operando characterization, and theoretical calculations in a coordinated effort to develop design principles to predict and improve catalytic selectivity. Dilute alloy catalysts─in which isolated atoms or small ensembles of the minority metal on the host metal lead to enhanced reactivity while retaining selectivity─are particularly promising as selective catalysts. Several dilute alloy materials using Au, Ag, and Cu as the majority host element, including more recently introduced support-free nanoporous metals and oxide-supported nanoparticle "raspberry colloid templated (RCT)" materials, are reviewed for selective oxidation and hydrogenation reactions. Progress in understanding how such dilute alloy catalysts can be used to enhance selectivity of key synthetic reactions is reviewed, including quantitative scaling from model studies to catalytic conditions. The dynamic evolution of catalyst structure and composition studied in surface science and catalytic conditions and their relationship to catalytic function are also discussed, followed by advanced characterization and theoretical modeling that have been developed to determine the distribution of minority metal atoms at or near the surface. The integrated approach demonstrates the success of bridging the divide between fundamental knowledge and design of catalytic processes in complex catalytic systems, which can accelerate the development of new and efficient catalytic processes. read less NOT USED (high confidence) M. Domina, M. Cobelli, and S. Sanvito, “Spectral neighbor representation for vector fields: Machine learning potentials including spin,” Physical Review B. 2022. link Times cited: 8 Abstract: We introduce a translational and rotational invariant local … read moreAbstract: We introduce a translational and rotational invariant local representation for vector fields, which can be employed in the construction of machine-learning energy models of solids and molecules. This allows us to describe, on the same footing, the energy fluctuations due to the atomic motion, the longitudinal and transverse excitations of the vector field, and their mutual interplay. The formalism can then be applied to physical systems where the total energy is determined by a vector density, as in the case of magnetism. Our representation is constructed over the power spectrum of the combined angular momentum describing the local atomic positions and the vector field, and can be used in conjunction with different machine-learning schemes and data taken from accurate ab initio electronic structure theories. We demonstrate the descriptive power of our representation for a range of classical spin Hamiltonian and machine-learning algorithms. In particular, we construct energy models based on both linear Ridge regression, as in conventional spectral neighbour analysis potentials, and gaussian approximation. These are both built to represent a Heisenberg-type Hamiltonian including a longitudinal energy term and spin-lattice coupling. read less NOT USED (high confidence) C. Ling, “A review of the recent progress in battery informatics,” npj Computational Materials. 2022. link Times cited: 35 NOT USED (high confidence) J. Wu, E. Zhou, Z. Qin, X. Zhang, and G. Qin, “Accessing negative Poisson’s ratio of graphene by machine learning interatomic potentials,” Nanotechnology. 2022. link Times cited: 2 Abstract: The negative Poisson’s ratio (NPR) is a novel property of ma… read moreAbstract: The negative Poisson’s ratio (NPR) is a novel property of materials, which enhances the mechanical feature and creates a wide range of application prospects in lots of fields, such as aerospace, electronics, medicine, etc. Fundamental understanding on the mechanism underlying NPR plays an important role in designing advanced mechanical functional materials. However, with different methods used, the origin of NPR is found different and conflicting with each other, for instance, in the representative graphene. In this study, based on machine learning technique, we constructed a moment tensor potential for molecular dynamics (MD) simulations of graphene. By analyzing the evolution of key geometries, the increase of bond angle is found to be responsible for the NPR of graphene instead of bond length. The results on the origin of NPR are well consistent with the start-of-art first-principles, which amend the results from MD simulations using classic empirical potentials. Our study facilitates the understanding on the origin of NPR of graphene and paves the way to improve the accuracy of MD simulations being comparable to first-principle calculations. Our study would also promote the applications of machine learning interatomic potentials in multiscale simulations of functional materials. read less NOT USED (high confidence) V. H. A. Nguyen and A. Lunghi, “Predicting tensorial molecular properties with equivariant machine learning models,” Physical Review B. 2022. link Times cited: 12 Abstract: Embedding molecular symmetries into machine-learning models … read moreAbstract: Embedding molecular symmetries into machine-learning models is key for efficient learning of chemico-physical scalar properties, but little evidence on how to extend the same strategy to tensorial quantities exists. Here we formulate a scalable equivariant machine-learning model based on local atomic environment descriptors. We apply it to a series of molecules and show that accurate predictions can be achieved for a comprehensive list of dielectric and magnetic tensorial properties of different ranks. These results show that equivariant models are a promising platform to extend the scope of machine learning in materials modelling. read less NOT USED (high confidence) R. Beckmann, F. Brieuc, C. Schran, and D. Marx, “Infrared Spectra at Coupled Cluster Accuracy from Neural Network Representations.,” Journal of chemical theory and computation. 2022. link Times cited: 11 Abstract: Infrared spectroscopy is key to elucidating molecular struct… read moreAbstract: Infrared spectroscopy is key to elucidating molecular structures, monitoring reactions, and observing conformational changes, while providing information on both structural and dynamical properties. This makes the accurate prediction of infrared spectra based on first-principle theories a highly desirable pursuit. Molecular dynamics simulations have proven to be a particularly powerful approach for this task, albeit requiring the computation of energies, forces and dipole moments for a large number of molecular configurations as a function of time. This explains why highly accurate first-principles methods, such as coupled cluster theory, have so far been inapplicable for the prediction of fully anharmonic vibrational spectra of large systems at finite temperatures. Here, we push cutting-edge machine learning techniques forward by using neural network representations of energies, forces, and in particular dipoles to predict such infrared spectra fully at "gold standard" coupled cluster accuracy as demonstrated for protonated water clusters as large as the protonated water hexamer, in its extended Zundel configuration. Furthermore, we show that this methodology can be used beyond the scope of the data considered during the development of the neural network models, allowing for the computation of finite-temperature infrared spectra of large systems inaccessible to explicit coupled cluster calculations. This substantially expands the hitherto existing limits of accuracy, speed, and system size for theoretical spectroscopy and opens up a multitude of avenues for the prediction of vibrational spectra and the understanding of complex intra- and intermolecular couplings. read less NOT USED (high confidence) M. Cobelli, P. Cahalane, and S. Sanvito, “Inversion of the chemical environment representations.” 2022. link Times cited: 2 Abstract: Machine-learning generative methods for material design are … read moreAbstract: Machine-learning generative methods for material design are constructed by representing a given chemical structure, either a solid or a molecule, over appropriate atomic features, generally called structural descriptors. These must be fully descriptive of the system, must facilitate the training process and must be invertible, so that one can extract the atomic configurations corresponding to the output of the model. In general, this last requirement is not automatically satisfied by the most efficient structural descriptors, namely the representation is not directly invertible. Such drawback severely limits our freedom of choice in selecting the most appropriate descriptors for the problem, and thus our flexibility to construct generative models. In this work, we present a general optimization method capable of inverting any local many-body descriptor of the chemical environment, back to a cartesian representation. The algorithm is then implemented together with the bispectrum representation of the local structure and demonstrated for a number of molecules. The scheme presented here, thus, represents a general approach to the inversion of structural descriptors, enabling the construction of efficient structural generative models. read less NOT USED (high confidence) P. Nieves, J. Tranchida, S. Nikolov, A. Fraile, and D. Legut, “Atomistic simulations of magnetoelastic effects on sound velocity.” 2022. link Times cited: 1 Abstract: P. Nieves1,∗ J. Tranchida2, S. Nikolov3, A. Fraile4, and D. … read moreAbstract: P. Nieves1,∗ J. Tranchida2, S. Nikolov3, A. Fraile4, and D. Legut1 1 IT4Innovations, VŠB Technical University of Ostrava, 17. listopadu 2172/15, 70800 Ostrava-Poruba, Czech Republic 2 CEA, DES/IRESNE/DEC, 13018 Saint Paul Lès Durance, France 3 Computational Multiscale Department, Sandia National Laboratories, P.O. Box 5800, MS 1322, 87185 Albuquerque, NM, United States and 4 Nuclear Futures Institute, Bangor University, Bangor, LL57 1UT, United Kingdom of Great Britain and Northern Ireland (Dated: January 26, 2022) read less NOT USED (high confidence) D. M. de Oca Zapiain, M. Wood, N. Lubbers, C. Z. Pereyra, A. Thompson, and D. Perez, “Training data selection for accuracy and transferability of interatomic potentials,” npj Computational Materials. 2022. link Times cited: 16 NOT USED (high confidence) G. S. Dhaliwal, P. Nair, and C. V. Singh, “Machine learned interatomic potentials using random features,” npj Computational Materials. 2022. link Times cited: 10 NOT USED (high confidence) P. Zhang and G. Chern, “Machine learning nonequilibrium electron forces for spin dynamics of itinerant magnets,” npj Computational Materials. 2021. link Times cited: 4 NOT USED (high confidence) C. Zeni, A. Anelli, A. Glielmo, and K. Rossi, “Exploring the robust extrapolation of high-dimensional machine learning potentials,” Physical Review B. 2021. link Times cited: 9 Abstract: We show that, contrary to popular assumptions, predictions f… read moreAbstract: We show that, contrary to popular assumptions, predictions from machine learning potentials built upon high-dimensional atom-density representations almost exclusively occur in regions of the representation space which lie outside the convex hull defined by the training set points. We then propose a perspective to rationalize the domain of robust extrapolation and accurate prediction of atomistic machine learning potentials in terms of the probability density induced by training points in the representation space. read less NOT USED (high confidence) H. Euchner and A. Gross, “Atomistic modeling of Li- and post-Li-ion batteries,” Physical Review Materials. 2021. link Times cited: 6 Abstract: Alkali metal ion batteries, and in particular Li–ion batteri… read moreAbstract: Alkali metal ion batteries, and in particular Li–ion batteries, have become a key technology for current and future energy storage, already nowadays powering many devices of our daily lives. Due to the inherent complexity of batteries and their components, the use of computational approaches on all length and time scales has been largely evolving within recent years. Gaining insight in complex processes or predicting new materials for specific applications are two of the main perspectives computational studies can offer, making them a indispensable tool of modern material science and hence battery research. After a short introduction to battery technology, this review will first focus on the theoretical concepts that underlie the functioning of Li– and post–Li–ion batteries. This will be followed by a discussion of the most prominent computational methods and their applications, currently available for the investigation of battery materials on an atomistic scale. read less NOT USED (high confidence) Z. Chen, F. Bononi, C. A. Sievers, W. Kong, and D. Donadio, “UV-Visible Absorption Spectra of Solvated Molecules by Quantum Chemical Machine Learning.,” Journal of chemical theory and computation. 2021. link Times cited: 6 Abstract: Predicting UV-visible absorption spectra is essential to und… read moreAbstract: Predicting UV-visible absorption spectra is essential to understand photochemical processes and design energy materials. Quantum chemical methods can deliver accurate calculations of UV-visible absorption spectra, but they are computationally expensive, especially for large systems or when one computes line shapes from thermal averages. Here, we present an approach to predict UV-visible absorption spectra of solvated aromatic molecules by quantum chemistry (QC) and machine learning (ML). We show that a ML model, trained on the high-level QC calculation of the excitation energy of a set of aromatic molecules, can accurately predict the line shape of the lowest-energy UV-visible absorption band of several related molecules with less than 0.1 eV deviation with respect to reference experimental spectra. Applying linear decomposition analysis on the excitation energies, we unveil that our ML models probe vertical excitations of these aromatic molecules primarily by learning the atomic environment of their phenyl rings, which align with the physical origin of the π →π* electronic transition. Our study provides an effective workflow that combines ML with quantum chemical methods to accelerate the calculations of UV-visible absorption spectra for various molecular systems. read less NOT USED (high confidence) W.-Z. L. S.-Z. Yu et al., “Machine‐learning‐based interatomic potentials for advanced manufacturing,” International Journal of Mechanical System Dynamics. 2021. link Times cited: 2 Abstract: This paper summarizes the progress of machine‐learning‐based… read moreAbstract: This paper summarizes the progress of machine‐learning‐based interatomic potentials and their applications in advanced manufacturing. Interatomic potential is essential for classical molecular dynamics. The advancements made in machine learning (ML) have enabled the development of fast interatomic potential with ab initio accuracy. The accelerated atomic simulation can greatly transform the design principle of manufacturing technology. The most widely used supervised and unsupervised ML methods are summarized and compared. Then, the emerging interatomic models based on ML are discussed: Gaussian approximation potential, spectral neighbor analysis potential, deep potential molecular dynamics, SCHNET, hierarchically interacting particle neural network, and fast learning of atomistic rare events. read less NOT USED (high confidence) L. Gigli, M. Veit, M. Kotiuga, G. Pizzi, N. Marzari, and M. Ceriotti, “Thermodynamics and dielectric response of BaTiO3 by data-driven modeling,” npj Computational Materials. 2021. link Times cited: 12 NOT USED (high confidence) T. Hullar et al., “Enhanced photodegradation of dimethoxybenzene isomers in/on ice compared to in aqueous solution,” Atmospheric Chemistry and Physics. 2021. link Times cited: 5 Abstract: Abstract. Photochemical reactions of contaminants in snow an… read moreAbstract: Abstract. Photochemical reactions of contaminants in snow and ice can be important sources and sinks for various organic and inorganic compounds. Snow contaminants can be found in the bulk ice matrix, in internal liquid-like regions (LLRs), or in quasi-liquid layers (QLLs) at the air-ice interface, where they can readily exchange with the firn air. Some studies have reported that direct photochemical reactions occur faster in LLRs and QLLs than in aqueous solution, while others have found similar rates. Here, we measure the photodegradation rate constants of the three dimethoxybenzene isomers under varying experimental conditions, including in aqueous solution, in LLRs, and at the air-ice interface of nature-identical snow. Relative to aqueous solution, we find modest photodegradation enhancements (3- and 6-fold) in LLRs for two of the isomers, and larger enhancements (15- to 30-fold) at the air-ice interface for all three isomers. We use computational modeling to assess the impact of light absorbance changes on photodegradation rate enhancements at the interface. We find small (2–5 nm) bathochromic (red) absorbance shifts at the interface relative to in solution, which increases light absorption, but this factor only accounts for less than 50 % of the measured rate constant enhancements. The major factor responsible for photodegradation rate enhancements at the air-ice interface appears to be more efficient photodecay: estimated dimethoxybenzene quantum yields are 6- to 24-fold larger at the interface compared to in aqueous solution and account for the majority (51–96 %) of the observed enhancements. Using a hypothetical model compound with an assumed Gaussian-shaped absorbance peak, we find that a shift in the peak to higher or lower wavelengths can have a minor to substantial impact on photodecay rate constants, depending on the original location of the peak and the magnitude of the shift. Changes in other peak properties at the air-ice interface, such as peak width and height (i.e., molar absorptivity) can also impact rates of light absorption and direct photodecay.
read less NOT USED (high confidence) R. Ryltsev and N. Chtchelkatchev, “Deep machine learning potentials for multicomponent metallic melts: development, predictability and compositional transferability,” Journal of Molecular Liquids. 2021. link Times cited: 23 NOT USED (high confidence) A. Goryaeva et al., “Efficient and transferable machine learning potentials for the simulation of crystal defects in bcc Fe and W,” Physical Review Materials. 2021. link Times cited: 20 NOT USED (high confidence) L. Fiedler, K. Shah, M. Bussmann, and A. Cangi, “Deep dive into machine learning density functional theory for materials science and chemistry,” Physical Review Materials. 2021. link Times cited: 18 Abstract: With the growth of computational resources, the scope of ele… read moreAbstract: With the growth of computational resources, the scope of electronic structure simulations has increased greatly. Artificial intelligence and robust data analysis hold the promise to accelerate large-scale simulations and their analysis to hitherto unattainable scales. Machine learning is a rapidly growing field for the processing of such complex datasets. It has recently gained traction in the domain of electronic structure simulations, where density functional theory takes the prominent role of the most widely used electronic structure method. Thus, DFT calculations represent one of the largest loads on academic high-performance computing systems across the world. Accelerating these with machine learning can reduce the resources required and enables simulations of larger systems. Hence, the combination of density functional theory and machine learning has the potential to rapidly advance electronic structure applications such as in-silico materials discovery and the search for new chemical reaction pathways. We provide the theoretical background of both density functional theory and machine learning on a generally accessible level. This serves as the basis of our comprehensive review including research articles up to December 2020 in chemistry and materials science that employ machine-learning techniques. In our analysis, we categorize the body of research into main threads and extract impactful results. We conclude our review with an outlook on exciting research directions in terms of a citation analysis. read less NOT USED (high confidence) H. Yu et al., “Complex Spin Hamiltonian Represented by Artificial Neural Network,” ArXiv. 2021. link Times cited: 12 Abstract: The effective spin Hamiltonian method is widely adopted to s… read moreAbstract: The effective spin Hamiltonian method is widely adopted to simulate and understand the behavior of magnetism. However, the magnetic interactions of some systems, such as itinerant magnets, are too complex to be described by any explicit function, which prevents an accurate description of magnetism in such systems. Here, we put forward a machine learning (ML) approach, applying an artificial neural network (ANN) and a local spin descriptor to develop effective spin potentials for any form of interaction. The constructed Hamiltonians include an explicit Heisenberg part and an implicit non-linear ANN part. Such a method successfully reproduces artificially constructed models and also sufficiently describe the itinerant magnetism of bulk Fe3GeTe2. Our work paves a new way for investigating complex magnetic phenomena (e.g., skyrmions) of magnetic materials. read less NOT USED (high confidence) S. Xie, M. Rupp, and R. Hennig, “Ultra-fast interpretable machine-learning potentials,” npj Computational Materials. 2021. link Times cited: 9 NOT USED (high confidence) Z. Fan, “Improving the accuracy of the neuroevolution machine learning potential for multi-component systems,” Journal of Physics: Condensed Matter. 2021. link Times cited: 16 Abstract: In a previous paper Fan et al (2021 Phys. Rev. B 104, 104309… read moreAbstract: In a previous paper Fan et al (2021 Phys. Rev. B 104, 104309), we developed the neuroevolution potential (NEP), a framework of training neural network based machine-learning potentials using a natural evolution strategy and performing molecular dynamics (MD) simulations using the trained potentials. The atom-environment descriptor in NEP was constructed based on a set of radial and angular functions. For multi-component systems, all the radial functions between two atoms are multiplied by some fixed factors that depend on the types of the two atoms only. In this paper, we introduce an improved descriptor for multi-component systems, in which different radial functions are multiplied by different factors that are also optimized during the training process, and show that it can significantly improve the regression accuracy without increasing the computational cost in MD simulations. read less NOT USED (high confidence) J. Goniakowski, S. Menon, G. Laurens, and J. Lam, “Nonclassical Nucleation of Zinc Oxide from a Physically Motivated Machine-Learning Approach,” The Journal of Physical Chemistry C. 2021. link Times cited: 3 Abstract: Observing non-classical nucleation pathways remains challeng… read moreAbstract: Observing non-classical nucleation pathways remains challenging in simulations of complex materials with technological interests. This is because it requires very accurate force fields that can capture the whole complexity of their underlying interatomic interactions and an advanced structural analysis. Here, we first report the construction of a machine-learning force field for zinc oxide interactions using the Physical LassoLars Interaction Potentials approach which allows us to be predictive even for untrained structures. Then, we carried out freezing simulations from a liquid and observed the crystal formation with atomistic precision. Our results, which are analyzed using a data-driven approach based on bond order parameters, demonstrate the presence of both prenucleation clusters and two-step nucleation scenarios thus retrieving seminal predictions of non-classical nucleation pathways made on much simpler models. read less NOT USED (high confidence) Y. Wang et al., “Accelerated prediction of atomically precise cluster structures using on-the-fly machine learning,” npj Computational Materials. 2021. link Times cited: 4 NOT USED (high confidence) S. Yin et al., “Atomistic simulations of dislocation mobility in refractory high-entropy alloys and the effect of chemical short-range order,” Nature Communications. 2021. link Times cited: 121 NOT USED (high confidence) M. Wen, Y. Afshar, R. Elliott, and E. Tadmor, “KLIFF: A framework to develop physics-based and machine learning interatomic potentials,” Comput. Phys. Commun. 2021. link Times cited: 12 NOT USED (high confidence) A. Niklasson, “Extended Lagrangian Born–Oppenheimer molecular dynamics: from density functional theory to charge relaxation models,” The European Physical Journal B. 2021. link Times cited: 5 NOT USED (high confidence) A. Clark et al., “The Middle Science: Traversing Scale In Complex Many-Body Systems,” ACS Central Science. 2021. link Times cited: 10 Abstract: A roadmap is developed that integrates simulation methodolog… read moreAbstract: A roadmap is developed that integrates simulation methodology and data science methods to target new theories that traverse the multiple length- and time-scale features of many-body phenomena. read less NOT USED (high confidence) M. S. Chen, T. Morawietz, H. Mori, T. Markland, and N. Artrith, “AENET-LAMMPS and AENET-TINKER: Interfaces for accurate and efficient molecular dynamics simulations with machine learning potentials.,” The Journal of chemical physics. 2021. link Times cited: 13 Abstract: Machine-learning potentials (MLPs) trained on data from quan… read moreAbstract: Machine-learning potentials (MLPs) trained on data from quantum-mechanics based first-principles methods can approach the accuracy of the reference method at a fraction of the computational cost. To facilitate efficient MLP-based molecular dynamics and Monte Carlo simulations, an integration of the MLPs with sampling software is needed. Here, we develop two interfaces that link the atomic energy network (ænet) MLP package with the popular sampling packages TINKER and LAMMPS. The three packages, ænet, TINKER, and LAMMPS, are free and open-source software that enable, in combination, accurate simulations of large and complex systems with low computational cost that scales linearly with the number of atoms. Scaling tests show that the parallel efficiency of the ænet-TINKER interface is nearly optimal but is limited to shared-memory systems. The ænet-LAMMPS interface achieves excellent parallel efficiency on highly parallel distributed-memory systems and benefits from the highly optimized neighbor list implemented in LAMMPS. We demonstrate the utility of the two MLP interfaces for two relevant example applications: the investigation of diffusion phenomena in liquid water and the equilibration of nanostructured amorphous battery materials. read less NOT USED (high confidence) Z. Fan et al., “Neuroevolution machine learning potentials: Combining high accuracy and low cost in atomistic simulations and application to heat transport,” Physical Review B. 2021. link Times cited: 42 Abstract: We develop a neuroevolution-potential (NEP) framework for ge… read moreAbstract: We develop a neuroevolution-potential (NEP) framework for generating neural network based machine-learning potentials. They are trained using an evolutionary strategy for performing large-scale molecular dynamics (MD) simulations. A descriptor of the atomic environment is constructed based on Chebyshev and Legendre polynomials. The method is implemented in graphic processing units within the open-source GPUMD package, which can attain a computational speed over $10^7$ atom-step per second using one Nvidia Tesla V100. Furthermore, per-atom heat current is available in NEP, which paves the way for efficient and accurate MD simulations of heat transport in materials with strong phonon anharmonicity or spatial disorder, which usually cannot be accurately treated either with traditional empirical potentials or with perturbative methods. read less NOT USED (high confidence) H. Liu, X. Qian, H. Bao, C. Zhao, and X. Gu, “High-temperature phonon transport properties of SnSe from machine-learning interatomic potential,” Journal of Physics: Condensed Matter. 2021. link Times cited: 18 Abstract: As a promising thermoelectric material, tin selenide (SnSe) … read moreAbstract: As a promising thermoelectric material, tin selenide (SnSe) is of relatively low thermal conductivity. However, the phonon transport mechanisms in SnSe are not fully understood due to the complex phase transition, dynamical instability, and strong anharmonicity. In this work, we perform molecular dynamics simulations with a machine-learning interatomic potential to explore the thermal transport properties of SnSe at different temperatures. The developed interatomic potential is parameterized using the framework of moment tensor potential, exhibiting satisfactory predictions on temperature-dependent lattice constants and phonon dispersion, as well as phase transition temperature. From equilibrium molecular dynamics simulations, we obtained the thermal conductivity tensor from 200 K to 900 K. The origins of temperature-dependent thermal conductivity anisotropy and the roles of four-phonon scatterings are identified. The obtained interatomic potential can be utilized to study the mechanical and thermal properties of SnSe and related nanostructures in a wide range of temperatures. read less NOT USED (high confidence) M. Hodapp and A. Shapeev, “Machine-learning potentials enable predictive and tractable high-throughput screening of random alloys,” Physical Review Materials. 2021. link Times cited: 7 Abstract: We present an automated procedure for computing stacking fau… read moreAbstract: We present an automated procedure for computing stacking fault energies in random alloys from large-scale simulations using moment tensor potentials (MTPs) with the accuracy of density functional theory (DFT). To that end, we develop an algorithm for training MTPs on random alloys. In the first step, our algorithm constructs a set of ~10000 or more training candidate configurations with 50-100 atoms that are representative for the atomic neighborhoods occurring in the large-scale simulation. In the second step, we use active learning to reduce this set to ~100 most distinct configurations - for which DFT energies and forces are computed and on which the potential is ultimately trained. We validate our algorithm for the MoNbTa medium-entropy alloy by showing that the MTP reproduces the DFT $\frac{1}{4}[111]$ unstable stacking fault energy over the entire compositional space up to a few percent. Contrary to state-of-the-art methods, e.g., the coherent potential approximation (CPA) or special quasi-random structures (SQSs), our algorithm naturally accounts for relaxation, is not limited by DFT cell sizes, and opens opportunities to efficiently investigate follow-up problems, such as chemical ordering. In a broader sense, our algorithm can be easily modified to compute related properties of random alloys, for instance, misfit volumes, or grain boundary energies. Moreover, it forms the basis for an efficient construction of MTPs to be used in large-scale simulations of multicomponent systems. read less NOT USED (high confidence) E. Kocer, T. W. Ko, and J. Behler, “Neural Network Potentials: A Concise Overview of Methods.,” Annual review of physical chemistry. 2021. link Times cited: 59 Abstract: In the past two decades, machine learning potentials (MLPs) … read moreAbstract: In the past two decades, machine learning potentials (MLPs) have reached a level of maturity that now enables applications to large-scale atomistic simulations of a wide range of systems in chemistry, physics, and materials science. Different machine learning algorithms have been used with great success in the construction of these MLPs. In this review, we discuss an important group of MLPs relying on artificial neural networks to establish a mapping from the atomic structure to the potential energy. In spite of this common feature, there are important conceptual differences among MLPs, which concern the dimensionality of the systems, the inclusion of long-range electrostatic interactions, global phenomena like nonlocal charge transfer, and the type of descriptor used to represent the atomic structure, which can be either predefined or learnable. A concise overview is given along with a discussion of the open challenges in the field. Expected final online publication date for the Annual Review of Physical Chemistry, Volume 73 is April 2022. Please see http://www.annualreviews.org/page/journal/pubdates for revised estimates. read less NOT USED (high confidence) I. Demiroglu, Y. Karaaslan, T. Kocabaş, M. Keçeli, Á. Vázquez-Mayagoitia, and C. Sevik, “Computation of the Thermal Expansion Coefficient of Graphene with Gaussian Approximation Potentials,” The Journal of Physical Chemistry C. 2021. link Times cited: 5 NOT USED (high confidence) E. Wright et al., “Refactoring the MPS/University of Chicago Radiative MHD (MURaM) model for GPU/CPU performance portability using OpenACC directives,” Proceedings of the Platform for Advanced Scientific Computing Conference. 2021. link Times cited: 3 Abstract: The MURaM (Max Planck University of Chicago Radiative MHD) c… read moreAbstract: The MURaM (Max Planck University of Chicago Radiative MHD) code is a solar atmosphere radiative MHD model that has been broadly applied to solar phenomena ranging from quiet to active sun, including eruptive events such as flares and coronal mass ejections. The treatment of physics is sufficiently realistic to allow for the synthesis of emission from visible light to extreme UV and X-rays, which is critical for a detailed comparison with available and future multi-wavelength observations. This component relies critically on the radiation transport solver (RTS) of MURaM; the most computationally intensive component of the code. The benefits of accelerating RTS are multiple fold: A faster RTS allows for the regular use of the more expensive multi-band radiation transport needed for comparison with observations, and this will pave the way for the acceleration of ongoing improvements in RTS that are critical for simulations of the solar chromosphere. We present challenges and strategies to accelerate a multi-physics, multi-band MURaM using a directive-based programming model, OpenACC in order to maintain a single source code across CPUs and GPUs. Results for a 2883 test problem show that MURaM with the optimized RTS routine achieves 1.73x speedup using a single NVIDIA V100 GPU over a fully subscribed 40-core Intel Skylake CPU node and with respect to the number of simulation points (in millions) per second, a single NVIDIA V100 GPU is equivalent to 69 Skylake cores. We also measure parallel performance on up to 96 GPUs and present weak and strong scaling results. read less NOT USED (high confidence) S. Mniszewski et al., “Enabling particle applications for exascale computing platforms,” The International Journal of High Performance Computing Applications. 2021. link Times cited: 18 Abstract: The Exascale Computing Project (ECP) is invested in co-desig… read moreAbstract: The Exascale Computing Project (ECP) is invested in co-design to assure that key applications are ready for exascale computing. Within ECP, the Co-design Center for Particle Applications (CoPA) is addressing challenges faced by particle-based applications across four “sub-motifs”: short-range particle–particle interactions (e.g., those which often dominate molecular dynamics (MD) and smoothed particle hydrodynamics (SPH) methods), long-range particle–particle interactions (e.g., electrostatic MD and gravitational N-body), particle-in-cell (PIC) methods, and linear-scaling electronic structure and quantum molecular dynamics (QMD) algorithms. Our crosscutting co-designed technologies fall into two categories: proxy applications (or “apps”) and libraries. Proxy apps are vehicles used to evaluate the viability of incorporating various types of algorithms, data structures, and architecture-specific optimizations and the associated trade-offs; examples include ExaMiniMD, CabanaMD, CabanaPIC, and ExaSP2. Libraries are modular instantiations that multiple applications can utilize or be built upon; CoPA has developed the Cabana particle library, PROGRESS/BML libraries for QMD, and the SWFFT and fftMPI parallel FFT libraries. Success is measured by identifiable “lessons learned” that are translated either directly into parent production application codes or into libraries, with demonstrated performance and/or productivity improvement. The libraries and their use in CoPA’s ECP application partner codes are also addressed. read less NOT USED (high confidence) J. Behler and G. Csányi, “Machine learning potentials for extended systems: a perspective,” The European Physical Journal B. 2021. link Times cited: 66 NOT USED (high confidence) A. Romualdi and G. Marchetti, “Machine learning S-wave scattering phase shifts bypassing the radial Schrödinger equation,” The European Physical Journal B. 2021. link Times cited: 1 NOT USED (high confidence) J. Byggmästar, K. Nordlund, and F. Djurabekova, “Modeling refractory high-entropy alloys with efficient machine-learned interatomic potentials: Defects and segregation,” Physical Review B. 2021. link Times cited: 32 Abstract: We develop a fast and accurate machine-learned interatomic p… read moreAbstract: We develop a fast and accurate machine-learned interatomic potential for the Mo–Nb–Ta–V–W quinary system and use it to study segregation and radiation damage in the body-centred cubic refractory high-entropy alloy MoNbTaVW. In the bulk alloy, we observe clear ordering of mainly Mo–Ta and V–W binaries at low temperatures. In damaged crystals, our simulations reveal clear segregation of vanadium, the smallest atom in the alloy, to compressed interstitial-rich regions like radiation-induced dislocation loops. Vanadium also dominates the population of single selfinterstitial atoms. In contrast, due to its larger size and low surface energy, niobium segregates to spacious regions like the inner surfaces of voids. When annealing samples with supersaturated concentrations of defects, we find that in complete contrast to W, interstitial atoms in MoNbTaVW cluster to create only small (∼ 1 nm) experimentally invisible dislocation loops enriched by vanadium. By comparison to W, we explain this by the reduced but three-dimensional migration of interstitials, the immobility of dislocation loops, and the increased mobility of vacancies in the high-entropy alloy, which together promote defect recombination over clustering. read less NOT USED (high confidence) D. Unruh, R. V. Meidanshahi, S. Goodnick, G. Csányi, and G. T. Zim’anyi, “Gaussian approximation potential for amorphous Si : H,” Physical Review Materials. 2021. link Times cited: 6 Abstract: Hydrogenation of amorphous silicon (a-Si:H) is critical for … read moreAbstract: Hydrogenation of amorphous silicon (a-Si:H) is critical for reducing defect densities, passivating mid-gap states and surfaces, and improving photoconductivity in silicon-based electro-optical devices. Modelling the atomic scale structure of this material is critical to understanding these processes, which in turn is needed to describe c-Si/a-Si:H heterjunctions that are at the heart of the modern solar cells with world record efficiency. Density functional theory (DFT) studies achieve the required high accuracy but are limited to moderate system sizes a hundred atoms or so by their high computational cost. Simulations of amorphous materials in particular have been hindered by this high cost because large structural models are required to capture the medium range order that is characteristic of such materials. Empirical potential models are much faster, but their accuracy is not sufficient to correctly describe the frustrated local structure. Data driven, “machine learned” interatomic potentials have broken this impasse, and have been highly successful in describing a variety of amorphous materials in their elemental phase. Here we extend the Gaussian approximation potential (GAP) for silicon by incorporating the interaction with hydrogen, thereby significantly improving the degree of realism with which amorphous silicon can be modelled. We show that our Si:H GAP enables the simulation of hydrogenated silicon with an accuracy very close to DFT, but with computational expense and run times reduced by several orders of magnitude for large structures. We demonstrate the capabilities of the Si:H GAP by creating models of hydrogenated liquid and amorphous silicon, and showing that their energies, forces and stresses are in excellent agreement with DFT results, and their structure as captured by bond and angle distributions, with both DFT and experiments. read less NOT USED (high confidence) H. Guo, Q. Wang, A. Stuke, A. Urban, and N. Artrith, “Accelerated Atomistic Modeling of Solid-State Battery Materials With Machine Learning,” Frontiers in Energy Research. 2021. link Times cited: 26 Abstract: Materials for solid-state batteries often exhibit complex ch… read moreAbstract: Materials for solid-state batteries often exhibit complex chemical compositions, defects, and disorder, making both experimental characterization and direct modeling with first principles methods challenging. Machine learning (ML) has proven versatile for accelerating or circumventing first-principles calculations, thereby facilitating the modeling of materials properties that are otherwise hard to access. ML potentials trained on accurate first principles data enable computationally efficient linear-scaling atomistic simulations with an accuracy close to the reference method. ML-based property-prediction and inverse design techniques are powerful for the computational search for new materials. Here, we give an overview of recent methodological advancements of ML techniques for atomic-scale modeling and materials design. We review applications to materials for solid-state batteries, including electrodes, solid electrolytes, coatings, and the complex interfaces involved. read less NOT USED (high confidence) J. Vandermause, Y. Xie, J. Lim, C. J. Owen, and B. Kozinsky, “Active learning of reactive Bayesian force fields applied to heterogeneous catalysis dynamics of H/Pt,” Nature Communications. 2021. link Times cited: 31 NOT USED (high confidence) M. Knudson and M. Desjarlais, “Interplay of high-precision shock wave experiments with first-principles theory to explore molecular systems at extreme conditions: A perspective,” Journal of Applied Physics. 2021. link Times cited: 3 Abstract: Conventional methods for probing molecular changes in conden… read moreAbstract: Conventional methods for probing molecular changes in condensed matter systems, such as electronic and vibrational spectroscopy, are difficult to implement at the extreme conditions associated with dynamic compression experiments. This is particularly true for experiments in the multimegabar regime; to achieve the requisite energy density to produce such pressures, sample sizes are necessarily quite small and experimental timescales are, therefore, extremely short. Furthermore, these extreme pressure conditions also result in high temperatures and, therefore, significant thermal emission even in the visible to infrared regime and in some cases render the sample opaque or reflective, thereby precluding bulk spectroscopy techniques, such as Raman scattering. These experimental challenges require a different approach to evaluating shock-induced changes at the molecular or atomic level in the multimegabar or the so-called warm dense matter regime. The past few decades have seen significant advances in the use of first-principles methods to investigate materials under extreme conditions, enabling these methods to become a powerful tool for exploring molecular systems at extreme conditions. Here, we discuss the construct of combining high-precision shock wave experiments with first-principles theory to explore molecular systems at extreme conditions. The results from high-fidelity dynamic compression experiments are used to evaluate first-principles theoretical frameworks and identify the framework that best reproduces experimental results in the regime of interest. That validated framework is then used to perform detailed simulations of the system of interest, providing unique insight into the response of the system at the molecular level. read less NOT USED (high confidence) O. T. Unke, S. Chmiela, M. Gastegger, K. T. Schütt, H. E. Sauceda, and K. Müller, “SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects,” Nature Communications. 2021. link Times cited: 130 NOT USED (high confidence) M. Eckhoff and J. Behler, “High-dimensional neural network potentials for magnetic systems using spin-dependent atom-centered symmetry functions,” npj Computational Materials. 2021. link Times cited: 29 NOT USED (high confidence) M. Uhrin, “Through the eyes of a descriptor: Constructing complete, invertible descriptions of atomic environments,” Physical Review B. 2021. link Times cited: 11 Abstract: In this work we apply methods for describing 3D images to th… read moreAbstract: In this work we apply methods for describing 3D images to the problem of encoding atomic environments in a way that is invariant to rotations, translations, and permutations of the atoms and, crucially, can be decoded back into the original environment modulo global orientation without the need for training a model. From the point of view of decoding, the descriptor is optimally complete and can be extended to arbitrary order, allowing for a systematic convergence of the fidelity of the description. In experiments on molecules ranging from 3 to 29 atoms in size, we demonstrate that positions can be decoded with a 96% success rate and positions plus species with a 60% rate of success, rising to 95% if a second fingerprint is used. In all cases, consistent recovery is observed for molecules with 14 or fewer atoms. Additionally, we evaluate the descriptor’s performance in predicting the energies and forces of bulk iron by means of a neural network model trained on DFT data, achieving root-mean-square deviations of 3.7 meV/atom and 0.19 eV/Å for energies and forces respectively. The combined ability to both decode and make property predictions from a representation that does not need to be learned lays the foundations for a novel way of building generative models that are tasked with solving the inverse problem of predicting atomic arrangements that are statistically likely to have certain desired properties. read less NOT USED (high confidence) G. Sivaraman et al., “Experimentally Driven Automated Machine-Learned Interatomic Potential for a Refractory Oxide.,” Physical review letters. 2021. link Times cited: 12 Abstract: Understanding the structure and properties of refractory oxi… read moreAbstract: Understanding the structure and properties of refractory oxides is critical for high temperature applications. In this work, a combined experimental and simulation approach uses an automated closed loop via an active learner, which is initialized by x-ray and neutron diffraction measurements, and sequentially improves a machine-learning model until the experimentally predetermined phase space is covered. A multiphase potential is generated for a canonical example of the archetypal refractory oxide, HfO_{2}, by drawing a minimum number of training configurations from room temperature to the liquid state at ∼2900 °C. The method significantly reduces model development time and human effort. read less NOT USED (high confidence) J. Weinreich, M. Paleico, and J. Behler, “Properties of α-Brass Nanoparticles II: Structure and Composition,” The Journal of Physical Chemistry C. 2021. link Times cited: 4 Abstract: Nanoparticles have become increasingly interesting for a wid… read moreAbstract: Nanoparticles have become increasingly interesting for a wide range of applications, because in principle it is possible to tailor their properties by controlling size, shape and composition. One of these applications is heterogeneous catalysis, and a fundamental understanding of the structural details of the nanoparticles is essential for any knowledge-based improvement of reactivity and selectivity. In this work we investigate the atomic structure of brass nanoparticles containing up to 5000 atoms as a typical example for a binary alloy consisting of Cu and Zn. As systems of this size are too large for electronic structure calculations, in our simulations we use a recently parametrized machine learning potential providing close to density functional theory accuracy. This potential is employed for a structural characterization as a function of chemical composition by various types of simulations like Monte Carlo in the Semi-Grand Canonical Ensemble and simulated annealing molecular dynamics. Our analysis reveals that the distribution of both elements in the nanoparticles is inhomogeneous, and zinc accumulates in the outermost layer, while the first subsurface layer shows an enrichment of copper. Only for high zinc concentrations alloying can be found in the interior of the nanoparticles, and regular patterns corresponding to crystalline bulk phases of $\alpha$-brass can then be observed. The surfaces of the investigated clusters exhibit well-ordered single-crystal facets, which can give rise to grain boundaries inside the clusters. The melting temperature of the nanoparticles is found to decrease with increasing zinc-atom fraction, a trend which is well-known also for the bulk phase diagram of brass. read less NOT USED (high confidence) Y. Xie, J. Vandermause, L. Sun, A. Cepellotti, and B. Kozinsky, “Bayesian force fields from active learning for simulation of inter-dimensional transformation of stanene,” npj Computational Materials. 2021. link Times cited: 35 NOT USED (high confidence) R. Jadrich, C. Ticknor, and J. Leiding, “First principles reactive simulation for equation of state prediction.,” The Journal of chemical physics. 2021. link Times cited: 3 Abstract: The high cost of density functional theory (DFT) has hithert… read moreAbstract: The high cost of density functional theory (DFT) has hitherto limited the ab initio prediction of the equation of state (EOS). In this article, we employ a combination of large scale computing, advanced simulation techniques, and smart data science strategies to provide an unprecedented ab initio performance analysis of the high explosive pentaerythritol tetranitrate (PETN). Comparison to both experiment and thermochemical predictions reveals important quantitative limitations of DFT for EOS prediction and thus the assessment of high explosives. In particular, we find that DFT predicts the energy of PETN detonation products to be systematically too high relative to the unreacted neat crystalline material, resulting in an underprediction of the detonation velocity, pressure, and temperature at the Chapman-Jouguet state. The energetic bias can be partially accounted for by high-level electronic structure calculations of the product molecules. We also demonstrate a modeling strategy for mapping chemical composition across a wide parameter space with limited numerical data, the results of which suggest additional molecular species to consider in thermochemical modeling. read less NOT USED (high confidence) K. Shimizu and S. Watanabe, “Applications of Interatomic Potentials Using Neural Network in Materials Science,” The Brain & Neural Networks. 2021. link Times cited: 0 Abstract: 概要 ビッグデータや機械学習の活用が注目を浴びているという点について,著者らが専門とする 物性物理学や材料科学の分野も例… read moreAbstract: 概要 ビッグデータや機械学習の活用が注目を浴びているという点について,著者らが専門とする 物性物理学や材料科学の分野も例外ではない.世界各所でデータベース作成の取り組みが加速 しているが,その背景には,密度汎関数理論に基づく第一原理計算によりミクロな物理量を精 度良く予測可能になったことがある.他方,材料中の欠陥やイオンの挙動,表面や界面で起こ る現象,非晶質材料など,多くの興味ある系での第一原理計算には非常に高い計算コストが必 要となる.この計算コストと精度との両立という問題の解決にも機械学習手法の活用が注目さ れている.著者らは,ニューラルネットワークを用いた機械学習によって原子間の相互作用を 第一原理計算と同等の精度で予測できると期待される,高次元ニューラルネットワークポテン シャルという手法を用いた材料研究を進めてきた.本稿では,著者らの応用事例を中心に,こ のニューラルネットワークポテンシャルを用いた材料科学研究について紹介する. read less NOT USED (high confidence) J. Qi et al., “Bridging the gap between simulated and experimental ionic conductivities in lithium superionic conductors,” Materials Today Physics. 2021. link Times cited: 33 NOT USED (high confidence) T. A. Young, T. Johnston-Wood, V. L. Deringer, and F. Duarte, “A transferable active-learning strategy for reactive molecular force fields,” Chemical Science. 2021. link Times cited: 18 Abstract: Predictive molecular simulations require fast, accurate and … read moreAbstract: Predictive molecular simulations require fast, accurate and reactive interatomic potentials. Machine learning offers a promising approach to construct such potentials by fitting energies and forces to high-level quantum-mechanical data, but doing so typically requires considerable human intervention and data volume. Here we show that, by leveraging hierarchical and active learning, accurate Gaussian Approximation Potential (GAP) models can be developed for diverse chemical systems in an autonomous manner, requiring only hundreds to a few thousand energy and gradient evaluations on a reference potential-energy surface. The approach uses separate intra- and inter-molecular fits and employs a prospective error metric to assess the accuracy of the potentials. We demonstrate applications to a range of molecular systems with relevance to computational organic chemistry: ranging from bulk solvents, a solvated metal ion and a metallocage onwards to chemical reactivity, including a bifurcating Diels–Alder reaction in the gas phase and non-equilibrium dynamics (a model SN2 reaction) in explicit solvent. The method provides a route to routinely generating machine-learned force fields for reactive molecular systems. read less NOT USED (high confidence) Y. Mishin, “Machine-Learning Interatomic Potentials for Materials Science,” Electrical Engineering eJournal. 2021. link Times cited: 103 NOT USED (high confidence) Y. Ouyang, C. Yu, G. Yan, and J. Chen, “Machine learning approach for the prediction and optimization of thermal transport properties,” Frontiers of Physics. 2021. link Times cited: 31 NOT USED (high confidence) D. Dickel, M. S. Nitol, and C. Barrett, “LAMMPS implementation of rapid artificial neural network derived interatomic potentials,” Computational Materials Science. 2021. link Times cited: 13 NOT USED (high confidence) P. Yoo, M. Sakano, S. Desai, M. M. Islam, P. Liao, and A. Strachan, “Neural network reactive force field for C, H, N, and O systems,” npj Computational Materials. 2021. link Times cited: 30 NOT USED (high confidence) F. Musil et al., “Efficient implementation of atom-density representations.,” The Journal of chemical physics. 2021. link Times cited: 37 Abstract: Physically motivated and mathematically robust atom-centered… read moreAbstract: Physically motivated and mathematically robust atom-centered representations of molecular structures are key to the success of modern atomistic machine learning. They lie at the foundation of a wide range of methods to predict the properties of both materials and molecules and to explore and visualize their chemical structures and compositions. Recently, it has become clear that many of the most effective representations share a fundamental formal connection. They can all be expressed as a discretization of n-body correlation functions of the local atom density, suggesting the opportunity of standardizing and, more importantly, optimizing their evaluation. We present an implementation, named librascal, whose modular design lends itself both to developing refinements to the density-based formalism and to rapid prototyping for new developments of rotationally equivariant atomistic representations. As an example, we discuss smooth overlap of atomic position (SOAP) features, perhaps the most widely used member of this family of representations, to show how the expansion of the local density can be optimized for any choice of radial basis sets. We discuss the representation in the context of a kernel ridge regression model, commonly used with SOAP features, and analyze how the computational effort scales for each of the individual steps of the calculation. By applying data reduction techniques in feature space, we show how to reduce the total computational cost by a factor of up to 4 without affecting the model's symmetry properties and without significantly impacting its accuracy. read less NOT USED (high confidence) S. Nikolov et al., “Data-driven magneto-elastic predictions with scalable classical spin-lattice dynamics,” npj Computational Materials. 2021. link Times cited: 25 NOT USED (high confidence) Y.-S. Lin, G. P. P. Pun, and Y. Mishin, “Development of a physically-informed neural network interatomic potential for tantalum,” Computational Materials Science. 2021. link Times cited: 9 NOT USED (high confidence) F. Musil, A. Grisafi, A. P. Bart’ok, C. Ortner, G. Csányi, and M. Ceriotti, “Physics-Inspired Structural Representations for Molecules and Materials.,” Chemical reviews. 2021. link Times cited: 210 Abstract: The first step in the construction of a regression model or … read moreAbstract: The first step in the construction of a regression model or a data-driven analysis, aiming to predict or elucidate the relationship between the atomic-scale structure of matter and its properties, involves transforming the Cartesian coordinates of the atoms into a suitable representation. The development of atomic-scale representations has played, and continues to play, a central role in the success of machine-learning methods for chemistry and materials science. This review summarizes the current understanding of the nature and characteristics of the most commonly used structural and chemical descriptions of atomistic structures, highlighting the deep underlying connections between different frameworks and the ideas that lead to computationally efficient and universally applicable models. It emphasizes the link between properties, structures, their physical chemistry, and their mathematical description, provides examples of recent applications to a diverse set of chemical and materials science problems, and outlines the open questions and the most promising research directions in the field. read less NOT USED (high confidence) S. Atlas, “Embedding Quantum Statistical Excitations in a Classical Force Field.,” The journal of physical chemistry. A. 2021. link Times cited: 3 Abstract: Quantum-mechanically driven charge polarization and charge t… read moreAbstract: Quantum-mechanically driven charge polarization and charge transfer are ubiquitous in biomolecular systems, controlling reaction rates, allosteric interactions, ligand-protein binding, membrane transport, and dynamically driven structural transformations. Molecular dynamics (MD) simulations of these processes require quantum mechanical (QM) information in order to accurately describe their reactive dynamics. However, current techniques-empirical force fields, subsystem approaches, ab initio MD, and machine learning-vary in their ability to achieve a consistent chemical description across multiple atom types, and at scale. Here we present a physics-based, atomistic force field, the ensemble DFT charge-transfer embedded-atom method, in which QM forces are described at a uniform level of theory across all atoms, avoiding the need for explicit solution of the Schrödinger equation or large, precomputed training data sets. Coupling between the electronic and atomistic length scales is effected through an ensemble density functional theory formulation of the embedded-atom method originally developed for elemental materials. Charge transfer is expressed in terms of ensembles of ionic state basis densities of individual atoms, and charge polarization, in terms of atomic excited-state basis densities. This provides a highly compact yet general representation of the force field, encompassing both local and system-wide effects. Charge rearrangement is realized through the evolution of ensemble weights, adjusted at each dynamical time step via chemical potential equalization. read less NOT USED (high confidence) S. L. Batzner et al., “E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials,” Nature Communications. 2021. link Times cited: 523 NOT USED (high confidence) B. Huang and O. A. von Lilienfeld, “Ab Initio Machine Learning in Chemical Compound Space,” Chemical Reviews. 2020. link Times cited: 67 Abstract: Chemical compound space (CCS), the set of all theoretically … read moreAbstract: Chemical compound space (CCS), the set of all theoretically conceivable combinations of chemical elements and (meta-)stable geometries that make up matter, is colossal. The first-principles based virtual sampling of this space, for example, in search of novel molecules or materials which exhibit desirable properties, is therefore prohibitive for all but the smallest subsets and simplest properties. We review studies aimed at tackling this challenge using modern machine learning techniques based on (i) synthetic data, typically generated using quantum mechanics based methods, and (ii) model architectures inspired by quantum mechanics. Such Quantum mechanics based Machine Learning (QML) approaches combine the numerical efficiency of statistical surrogate models with an ab initio view on matter. They rigorously reflect the underlying physics in order to reach universality and transferability across CCS. While state-of-the-art approximations to quantum problems impose severe computational bottlenecks, recent QML based developments indicate the possibility of substantial acceleration without sacrificing the predictive power of quantum mechanics. read less NOT USED (high confidence) Y. Ouyang, Z. Zhang, C. Yu, J. He, G. Yan, and J. C. hyperlinks, “Accuracy of Machine Learning Potential for Predictions of Multiple-Target Physical Properties,” Chinese Physics Letters. 2020. link Times cited: 9 NOT USED (high confidence) C. Sutton and S. Levchenko, “First-Principles Atomistic Thermodynamics and Configurational Entropy,” Frontiers in Chemistry. 2020. link Times cited: 28 Abstract: In most applications, functional materials operate at finite… read moreAbstract: In most applications, functional materials operate at finite temperatures and are in contact with a reservoir of atoms or molecules (gas, liquid, or solid). In order to understand the properties of materials at realistic conditions, statistical effects associated with configurational sampling and particle exchange at finite temperatures must consequently be taken into account. In this contribution, we discuss the main concepts behind equilibrium statistical mechanics. We demonstrate how these concepts can be used to predict the behavior of materials at realistic temperatures and pressures within the framework of atomistic thermodynamics. We also introduce and discuss methods for calculating phase diagrams of bulk materials and surfaces as well as point defect concentrations. In particular, we describe approaches for calculating the configurational density of states, which requires the evaluation of the energies of a large number of configurations. The cluster expansion method is therefore also discussed as a numerically efficient approach for evaluating these energies. read less NOT USED (high confidence) L. Messina et al., “A DFT-driven multifidelity framework for constructing efficient energy models for atomic-scale simulations,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2020. link Times cited: 3 NOT USED (high confidence) E. Vivas, P. Lara, and M. Alvaro, “Structural and transport properties for the superionic conductors AgI and RbAg4I5 through molecular dynamic simulation,” Ionics. 2020. link Times cited: 4 NOT USED (high confidence) V. L. Deringer, M. A. Caro, and G. Csányi, “A general-purpose machine-learning force field for bulk and nanostructured phosphorus,” Nature Communications. 2020. link Times cited: 75 NOT USED (high confidence) M. P. Bircher, A. Singraber, and C. Dellago, “Improved description of atomic environments using low-cost polynomial functions with compact support,” Machine Learning: Science and Technology. 2020. link Times cited: 9 Abstract: The prediction of chemical properties using machine learning… read moreAbstract: The prediction of chemical properties using machine learning techniques calls for a set of appropriate descriptors that accurately describe atomic and, on a larger scale, molecular environments. A mapping of conformational information on a space spanned by atom-centred symmetry functions (SF) has become a standard technique for energy and force predictions using high-dimensional neural network potentials (HDNNP). An appropriate choice of SFs is particularly crucial for accurate force predictions. Established atom-centred SFs, however, are limited in their flexibility, since their functional form restricts the angular domain that can be sampled without introducing problematic derivative discontinuities. Here, we introduce a class of atom-centred SFs based on polynomials with compact support called polynomial symmetry functions (PSF), which enable a free choice of both, the angular and the radial domain covered. We demonstrate that the accuracy of PSFs is either on par or considerably better than that of conventional, atom-centred SFs. In particular, a generic set of PSFs with an intuitive choice of the angular domain inspired by organic chemistry considerably improves prediction accuracy for organic molecules in the gaseous and liquid phase, with reductions in force prediction errors over a test set approaching 50% for certain systems. Contrary to established atom-centred SFs, computation of PSF does not involve any exponentials, and their intrinsic compact support supersedes use of separate cutoff functions, facilitating the choice of their free parameters. Most importantly, the number of floating point operations required to compute polynomial SFs introduced here is considerably lower than that of other state-of-the-art SFs, enabling their efficient implementation without the need of highly optimised code structures or caching, with speedups with respect to other state-of-the-art SFs reaching a factor of 4.5 to 5. This low-effort performance benefit substantially simplifies their use in new programs and emerging platforms such as graphical processing units. Overall, polynomial SFs with compact support improve accuracy of both, energy and force predictions with HDNNPs while enabling significant speedups compared to their well-established counterparts. read less NOT USED (high confidence) A. Allen, G. Dusson, C. Ortner, and G. Csányi, “Atomic permutationally invariant polynomials for fitting molecular force fields,” Machine Learning: Science and Technology. 2020. link Times cited: 26 Abstract: We introduce and explore an approach for constructing force … read moreAbstract: We introduce and explore an approach for constructing force fields for small molecules, which combines intuitive low body order empirical force field terms with the concepts of data driven statistical fits of recent machine learned potentials. We bring these two key ideas together to bridge the gap between established empirical force fields that have a high degree of transferability on the one hand, and the machine learned potentials that are systematically improvable and can converge to very high accuracy, on the other. Our framework extends the atomic permutationally invariant polynomials (aPIP) developed for elemental materials in (2019 Mach. Learn.: Sci. Technol. 1 015004) to molecular systems. The body order decomposition allows us to keep the dimensionality of each term low, while the use of an iterative fitting scheme as well as regularisation procedures improve the extrapolation outside the training set. We investigate aPIP force fields with up to generalised 4-body terms, and examine the performance on a set of small organic molecules. We achieve a high level of accuracy when fitting individual molecules, comparable to those of the many-body machine learned force fields. Fitted to a combined training set of short linear alkanes, the accuracy of the aPIP force field still significantly exceeds what can be expected from classical empirical force fields, while retaining reasonable transferability to both configurations far from the training set and to new molecules. read less NOT USED (high confidence) M. Wagih, P. M. Larsen, and C. Schuh, “Learning grain boundary segregation energy spectra in polycrystals,” Nature Communications. 2020. link Times cited: 62 NOT USED (high confidence) A. Ferrari et al., “Frontiers in atomistic simulations of high entropy alloys,” Journal of Applied Physics. 2020. link Times cited: 41 Abstract: The field of atomistic simulations of multicomponent materia… read moreAbstract: The field of atomistic simulations of multicomponent materials and high entropy alloys is progressing rapidly, with challenging problems stimulating new creative solutions. In this Perspective, we present three topics that emerged very recently and that we anticipate will determine the future direction of research of high entropy alloys: the usage of machine-learning potentials for very accurate thermodynamics, the exploration of short-range order and its impact on macroscopic properties, and the more extensive exploitation of interstitial alloying and high entropy alloy surfaces for new technological applications. For each of these topics, we briefly summarize the key achievements, point out the aspects that still need to be addressed, and discuss possible future improvements and promising directions. read less NOT USED (high confidence) Y. Liu, J.-yue Yang, G. Xin, L. Liu, G. Csányi, and B. Cao, “Machine learning interatomic potential developed for molecular simulations on thermal properties of β-Ga2O3.,” The Journal of chemical physics. 2020. link Times cited: 46 Abstract: The thermal properties of β-Ga2O3 can significantly affect t… read moreAbstract: The thermal properties of β-Ga2O3 can significantly affect the performance and reliability of high-power electronic devices. To date, due to the absence of a reliable interatomic potential, first-principles calculations based on density functional theory (DFT) have been routinely used to probe the thermal properties of β-Ga2O3. DFT calculations can only tackle small-scale systems due to the huge computational cost, while the thermal transport processes are usually associated with large time and length scales. In this work, we develop a machine learning based Gaussian approximation potential (GAP) for accurately describing the lattice dynamics of perfect crystalline β-Ga2O3 and accelerating atomic-scale simulations. The GAP model shows excellent convergence, which can faithfully reproduce the DFT potential energy surface at a training data size of 32 000 local atomic environments. The GAP model is then used to predict ground-state lattice parameters, coefficients of thermal expansion, heat capacity, phonon dispersions at 0 K, and anisotropic thermal conductivity of β-Ga2O3, which are all in excellent agreement with either the DFT results or experiments. The accurate predictions of phonon dispersions and thermal conductivities demonstrate that the GAP model can well describe the harmonic and anharmonic interactions of phonons. Additionally, the successful application of our GAP model to the phonon density of states of a 2500-atom β-Ga2O3 structure at elevated temperature indicates the strength of machine learning potentials to tackle large-scale atomic systems in long molecular simulations, which would be almost impossible to generate with DFT-based molecular simulations at present. read less NOT USED (high confidence) J. Ellis et al., “Accelerating Finite-temperature Kohn-Sham Density Functional Theory with Deep Neural Networks,” ArXiv. 2020. link Times cited: 25 Abstract: We present a numerical modeling workflow based on machine le… read moreAbstract: We present a numerical modeling workflow based on machine learning (ML) which reproduces the the total energies produced by Kohn-Sham density functional theory (DFT) at finite electronic temperature to within chemical accuracy at negligible computational cost. Based on deep neural networks, our workflow yields the local density of states (LDOS) for a given atomic configuration. From the LDOS, spatially-resolved, energy-resolved, and integrated quantities can be calculated, including the DFT total free energy, which serves as the Born-Oppenheimer potential energy surface for the atoms. We demonstrate the efficacy of this approach for both solid and liquid metals and compare results between independent and unified machine-learning models for solid and liquid aluminum. Our machine-learning density functional theory framework opens up the path towards multiscale materials modeling for matter under ambient and extreme conditions at a computational scale and cost that is unattainable with current algorithms. read less NOT USED (high confidence) S. Lee et al., “Applying Machine Learning Algorithms to Predict Potential Energies and Atomic Forces during C-H Activation,” Journal of the Korean Physical Society. 2020. link Times cited: 2 Abstract: Molecular dynamics (MD) simulations are useful in understand… read moreAbstract: Molecular dynamics (MD) simulations are useful in understanding the interaction between solid materials and molecules. However, performing MD simulations is possible only when interatomic potentials are available and constructing such interatomic potentials usually requires additional computational work. Recently, generating interatomic potentials was shown to be much easier when machine learning (ML) algorithms were used. In addition, ML algorithms require new descriptors for improved performance. Here, we present an ML approach with several categories of atomic descriptors to predict the parameters necessary for MD simulations, such as the potential energies and the atomic forces. We propose several atomic descriptors based on structural information and find that better descriptors can be generated from eXtreme gradient boosting (XGBoost). Moreover, we observe fewer descriptors that perform better in predicting the potential energies and the forces during methane activation processes on a catalytic Pt(111) surface. These results were consistently observed in two different ML algorithms: fully-connected neural network (FNN) and XGBoost. Taking into account the advantages of FNN and XGBoost, we propose an efficient ML model for estimating potential energies. Our findings will be helpful in developing new ML potentials for long-time MD simulations. read less NOT USED (high confidence) A. Goryaeva, C. Lapointe, C. Dai, J. Dérès, J. Maillet, and M. Marinica, “Reinforcing materials modelling by encoding the structures of defects in crystalline solids into distortion scores,” Nature Communications. 2020. link Times cited: 27 NOT USED (high confidence) T. W. Ko, J. A. Finkler, S. Goedecker, and J. Behler, “A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer,” Nature Communications. 2020. link Times cited: 177 NOT USED (high confidence) G. P. P. Pun, V. Yamakov, J. Hickman, E. Glaessgen, and Y. Mishin, “Development of a general-purpose machine-learning interatomic potential for aluminum by the physically informed neural network method,” Physical Review Materials. 2020. link Times cited: 13 Abstract: Interatomic potentials constitute the key component of large… read moreAbstract: Interatomic potentials constitute the key component of large-scale atomistic simulations of materials. The recently proposed physically-informed neural network (PINN) method combines a high-dimensional regression implemented by an artificial neural network with a physics-based bond-order interatomic potential applicable to both metals and nonmetals. In this paper, we present a modified version of the PINN method that accelerates the potential training process and further improves the transferability of PINN potentials to unknown atomic environments. As an application, a modified PINN potential for Al has been developed by training on a large database of electronic structure calculations. The potential reproduces the reference first-principles energies within 2.6 meV per atom and accurately predicts a wide spectrum of physical properties of Al. Such properties include, but are not limited to, lattice dynamics, thermal expansion, energies of point and extended defects, the melting temperature, the structure and dynamic properties of liquid Al, the surface tensions of the liquid surface and the solid-liquid interface, and the nucleation and growth of a grain boundary crack. Computational efficiency of PINN potentials is also discussed. read less NOT USED (high confidence) A. Goscinski, G. Fraux, G. Imbalzano, and M. Ceriotti, “The role of feature space in atomistic learning,” Machine Learning: Science and Technology. 2020. link Times cited: 23 Abstract: Efficient, physically-inspired descriptors of the structure … read moreAbstract: Efficient, physically-inspired descriptors of the structure and composition of molecules and materials play a key role in the application of machine-learning techniques to atomistic simulations. The proliferation of approaches, as well as the fact that each choice of features can lead to very different behavior depending on how they are used, e.g. by introducing non-linear kernels and non-Euclidean metrics to manipulate them, makes it difficult to objectively compare different methods, and to address fundamental questions on how one feature space is related to another. In this work we introduce a framework to compare different sets of descriptors, and different ways of transforming them by means of metrics and kernels, in terms of the structure of the feature space that they induce. We define diagnostic tools to determine whether alternative feature spaces contain equivalent amounts of information, and whether the common information is substantially distorted when going from one feature space to another. We compare, in particular, representations that are built in terms of n-body correlations of the atom density, quantitatively assessing the information loss associated with the use of low-order features. We also investigate the impact of different choices of basis functions and hyperparameters of the widely used SOAP and Behler–Parrinello features, and investigate how the use of non-linear kernels, and of a Wasserstein-type metric, change the structure of the feature space in comparison to a simpler linear feature space. read less NOT USED (high confidence) A. Lunghi, “Insights into the Spin-Lattice Dynamics of Organic Radicals Beyond Molecular Tumbling: A Combined Molecular Dynamics and Machine-Learning Approach,” Applied Magnetic Resonance. 2020. link Times cited: 7 NOT USED (high confidence) A. Grisafi, J. Nigam, and M. Ceriotti, “Multi-scale approach for the prediction of atomic scale properties,” Chemical Science. 2020. link Times cited: 23 Abstract: Electronic nearsightedness is one of the fundamental princip… read moreAbstract: Electronic nearsightedness is one of the fundamental principles that governs the behavior of condensed matter and supports its description in terms of local entities such as chemical bonds. Locality also underlies the tremendous success of machine-learning schemes that predict quantum mechanical observables – such as the cohesive energy, the electron density, or a variety of response properties – as a sum of atom-centred contributions, based on a short-range representation of atomic environments. One of the main shortcomings of these approaches is their inability to capture physical effects ranging from electrostatic interactions to quantum delocalization, which have a long-range nature. Here we show how to build a multi-scale scheme that combines in the same framework local and non-local information, overcoming such limitations. We show that the simplest version of such features can be put in formal correspondence with a multipole expansion of permanent electrostatics. The data-driven nature of the model construction, however, makes this simple form suitable to tackle also different types of delocalized and collective effects. We present several examples that range from molecular physics to surface science and biophysics, demonstrating the ability of this multi-scale approach to model interactions driven by electrostatics, polarization and dispersion, as well as the cooperative behavior of dielectric response functions. read less NOT USED (high confidence) A. Seko, “Machine learning potentials for multicomponent systems: The Ti-Al binary system,” Physical Review B. 2020. link Times cited: 13 Abstract: Machine learning potentials (MLPs) are becoming powerful too… read moreAbstract: Machine learning potentials (MLPs) are becoming powerful tools for performing accurate atomistic simulations and crystal structure optimizations. An approach to developing MLPs employs a systematic set of polynomial invariants including high-order ones to represent the neighboring atomic density. In this study, a formulation of the polynomial invariants is extended to the case of multicomponent systems. The extended formulation is more complex than the formulation for elemental systems. This study also shows its application to the Ti-Al binary system. As a result, an MLP with the lowest error and MLPs with high computational cost performance are selected from the many MLPs developed systematically. The predictive powers of the developed MLPs for many properties, such as the formation energy, elastic constants, thermodynamic properties, and mechanical properties, are examined. The MLPs exhibit high predictive power for the properties in a wide variety of ordered structures. The present scheme should be systematically applicable to other multicomponent systems. read less NOT USED (high confidence) L. Miao and L.-wang Wang, “Liquid to crystal Si growth simulation using machine learning force field.,” The Journal of chemical physics. 2020. link Times cited: 4 Abstract: Machine learning force field (ML-FF) has emerged as a potent… read moreAbstract: Machine learning force field (ML-FF) has emerged as a potential promising approach to simulate various material phenomena for large systems with ab initio accuracy. However, most ML-FFs have been used to study the phenomena relatively close to the equilibrium ground states. In this work, we have studied a far from equilibrium system of liquid to crystal Si growth using ML-FF. We found that our ML-FF based on ab initio decomposed atomic energy can reproduce all the aspects of ab initio simulated growth, from local energy fluctuations to transition temperatures, to diffusion constant, and growth rates. We have also compared the growth simulation with the Stillinger-Weber classical force field and found significant differences. A procedure is also provided to correct a systematic fitting bias in the ML-FF training process, which exists in all training models, otherwise critical results like transition temperature will be wrong. read less NOT USED (high confidence) K. Shimizu, E. Arguelles, W. Li, Y. Ando, E. Minamitani, and S. Watanabe, “Phase stability of Au-Li binary systems studied using neural network potential,” Physical Review B. 2020. link Times cited: 7 Abstract: The miscibility of Au and Li exhibits a potential applicatio… read moreAbstract: The miscibility of Au and Li exhibits a potential application as an adhesion layer and electrode material in secondary batteries. Here, to explore alloying properties, we constructed a neural network potential (NNP) of Au-Li binary systems based on density functional theory (DFT) calculations. To accelerate construction of NNPs, we proposed an efficient and inexpensive method of structural dataset generation. The predictions by the constructed NNP on lattice parameters and phonon properties agree well with those obtained by DFT calculations. We also investigated the mixing energy of Au$_{1-x}$Li$_{x}$ with fine composition grids, showing excellent agreement with DFT verifications. We found the existence of various compositions with structures on and slightly above the convex hull, which can explain the lack of consensus on the Au-Li stable phases in previous studies. Moreover, we newly found Au$_{0.469}$Li$_{0.531}$ as a stable phase, which has never been reported elsewhere. Finally, we examined the alloying process starting from the phase separated structure to the complete mixing phase. We found that when multiple adjacent Au atoms dissolved into Li, the alloying of the entire Au/Li interface started from the dissolved region. This paper demonstrates the applicability of NNPs toward miscible phases and provides the understanding of the alloying mechanism. read less NOT USED (high confidence) B. Parsaeifard et al., “An assessment of the structural resolution of various fingerprints commonly used in machine learning,” Machine Learning: Science and Technology. 2020. link Times cited: 40 Abstract: Atomic environment fingerprints are widely used in computati… read moreAbstract: Atomic environment fingerprints are widely used in computational materials science, from machine learning potentials to the quantification of similarities between atomic configurations. Many approaches to the construction of such fingerprints, also called structural descriptors, have been proposed. In this work, we compare the performance of fingerprints based on the overlap matrix, the smooth overlap of atomic positions, Behler–Parrinello atom-centered symmetry functions, modified Behler–Parrinello symmetry functions used in the ANI-1ccx potential and the Faber–Christensen–Huang–Lilienfeld fingerprint under various aspects. We study their ability to resolve differences in local environments and in particular examine whether there are certain atomic movements that leave the fingerprints exactly or nearly invariant. For this purpose, we introduce a sensitivity matrix whose eigenvalues quantify the effect of atomic displacement modes on the fingerprint. Further, we check whether these displacements correlate with the variation of localized physical quantities such as forces. Finally, we extend our examination to the correlation between molecular fingerprints obtained from the atomic fingerprints and global quantities of entire molecules. read less NOT USED (high confidence) G. Sivaraman et al., “Machine-learned interatomic potentials by active learning: amorphous and liquid hafnium dioxide,” npj Computational Materials. 2020. link Times cited: 97 NOT USED (high confidence) A. S. Christensen and A. V. Lilienfeld, “On the role of gradients for machine learning of molecular energies and forces,” Machine Learning: Science and Technology. 2020. link Times cited: 78 Abstract: The accuracy of any machine learning potential can only be a… read moreAbstract: The accuracy of any machine learning potential can only be as good as the data used in the fitting process. The most efficient model therefore selects the training data that will yield the highest accuracy compared to the cost of obtaining the training data. We investigate the convergence of prediction errors of quantum machine learning models for organic molecules trained on energy and force labels, two common data types in molecular simulations. When training models for the potential energy surface of a single molecule, we find that the inclusion of atomic forces in the training data increases the accuracy of the predicted energies and forces 7-fold, compared to models trained on energy only. Surprisingly, for models trained on sets of organic molecules of varying size and composition in non-equilibrium conformations, inclusion of forces in the training does not improve the predicted energies of unseen molecules in new conformations. Predicted forces, however, improve about 7-fold. For the systems studied, we find that force labels and energy labels contribute equally per label to the convergence of the prediction errors. The optimal choice of what type of training data to include depends on several factors: the computational cost of acquiring the force and energy labels for training, the application domain, the property of interest and the complexity of the machine learning model. Based on our observations we describe key considerations for the creation of new datasets for potential energy surfaces of molecules which maximize the efficiency of the resulting machine learning models. read less NOT USED (high confidence) I. Novikov, K. Gubaev, E. Podryabinkin, and A. Shapeev, “The MLIP package: moment tensor potentials with MPI and active learning,” Machine Learning: Science and Technology. 2020. link Times cited: 220 Abstract: The subject of this paper is the technology (the ‘how’) of c… read moreAbstract: The subject of this paper is the technology (the ‘how’) of constructing machine-learning interatomic potentials, rather than science (the ‘what’ and ‘why’) of atomistic simulations using machine-learning potentials. Namely, we illustrate how to construct moment tensor potentials using active learning as implemented in the MLIP package, focusing on the efficient ways to automatically sample configurations for the training set, how expanding the training set changes the error of predictions, how to set up ab initio calculations in a cost-effective manner, etc. The MLIP package (short for Machine-Learning Interatomic Potentials) is available at https://mlip.skoltech.ru/download/. read less NOT USED (high confidence) M. Paleico and J. Behler, “Global optimization of copper clusters at the ZnO(101¯0) surface using a DFT-based neural network potential and genetic algorithms.,” The Journal of chemical physics. 2020. link Times cited: 28 Abstract: The determination of the most stable structures of metal clu… read moreAbstract: The determination of the most stable structures of metal clusters supported at solid surfaces by computer simulations represents a formidable challenge due to the complexity of the potential-energy surface. Here, we combine a high-dimensional neural network potential, which allows us to predict the energies and forces of a large number of structures with first-principles accuracy, with a global optimization scheme employing genetic algorithms. This very efficient setup is used to identify the global minima and low-energy local minima for a series of copper clusters containing between four and ten atoms adsorbed at the ZnO(101¯0) surface. A series of structures with common structural features resembling the Cu(111) and Cu(110) surfaces at the metal-oxide interface has been identified, and the geometries of the emerging clusters are characterized in detail. We demonstrate that the frequently employed approximation of a frozen substrate surface in global optimization can result in missing the most relevant structures. read less NOT USED (high confidence) J. Westermayr and P. Marquetand, “Machine Learning for Electronically Excited States of Molecules,” Chemical Reviews. 2020. link Times cited: 166 Abstract: Electronically excited states of molecules are at the heart … read moreAbstract: Electronically excited states of molecules are at the heart of photochemistry, photophysics, as well as photobiology and also play a role in material science. Their theoretical description requires highly accurate quantum chemical calculations, which are computationally expensive. In this review, we focus on not only how machine learning is employed to speed up such excited-state simulations but also how this branch of artificial intelligence can be used to advance this exciting research field in all its aspects. Discussed applications of machine learning for excited states include excited-state dynamics simulations, static calculations of absorption spectra, as well as many others. In order to put these studies into context, we discuss the promises and pitfalls of the involved machine learning techniques. Since the latter are mostly based on quantum chemistry calculations, we also provide a short introduction into excited-state electronic structure methods and approaches for nonadiabatic dynamics simulations and describe tricks and problems when using them in machine learning for excited states of molecules. read less NOT USED (high confidence) J. Nigam, S. Pozdnyakov, and M. Ceriotti, “Recursive evaluation and iterative contraction of N-body equivariant features.,” The Journal of chemical physics. 2020. link Times cited: 45 Abstract: Mapping an atomistic configuration to a symmetrized N-point … read moreAbstract: Mapping an atomistic configuration to a symmetrized N-point correlation of a field associated with the atomic positions (e.g., an atomic density) has emerged as an elegant and effective solution to represent structures as the input of machine-learning algorithms. While it has become clear that low-order density correlations do not provide a complete representation of an atomic environment, the exponential increase in the number of possible N-body invariants makes it difficult to design a concise and effective representation. We discuss how to exploit recursion relations between equivariant features of different order (generalizations of N-body invariants that provide a complete representation of the symmetries of improper rotations) to compute high-order terms efficiently. In combination with the automatic selection of the most expressive combination of features at each order, this approach provides a conceptual and practical framework to generate systematically improvable, symmetry adapted representations for atomistic machine learning. read less NOT USED (high confidence) M. Eckhoff, F. Schönewald, M. Risch, C. Volkert, P. Blöchl, and J. Behler, “Closing the gap between theory and experiment for lithium manganese oxide spinels using a high-dimensional neural network potential,” Physical Review B. 2020. link Times cited: 17 Abstract: Many positive electrode materials in lithium ion batteries i… read moreAbstract: Many positive electrode materials in lithium ion batteries include transition metals, which are difficult to describe by electronic structure methods like density functional theory (DFT) due to the presence of multiple oxidation states. A prominent example is the lithium manganese oxide spinel ${\mathrm{Li}}_{x}{\mathrm{Mn}}_{2}{\mathrm{O}}_{4}$ with $0\ensuremath{\le}x\ensuremath{\le}2$. While DFT, employing the local hybrid functional PBE0r, provides a reliable description, the need for extended computer simulations of large structural models remains a significant challenge. Here, we close this gap by constructing a DFT-based high-dimensional neural network potential (HDNNP) providing accurate energies and forces at a fraction of the computational costs. As different oxidation states and the resulting Jahn-Teller distortions represent a new level of complexity for HDNNPs, the potential is carefully validated by performing x-ray diffraction experiments. We demonstrate that the HDNNP provides atomic level details and is able to predict a series of properties like the lattice parameters and expansion with increasing Li content or temperature, the orthorhombic to cubic transition, the lithium diffusion barrier, and the phonon frequencies. We show that for understanding these properties access to large time and length scales as enabled by the HDNNP is essential to close the gap between theory and experiment. read less NOT USED (high confidence) Y. Zhang, C. Hu, and B. Jiang, “Accelerating atomistic simulations with piecewise machine-learned ab Initio potentials at a classical force field-like cost.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 12 Abstract: Recently, machine learning methods have become easy-to-use t… read moreAbstract: Recently, machine learning methods have become easy-to-use tools for constructing high-dimensional interatomic potentials with ab initio accuracy. Although machine-learned interatomic potentials are generally orders of magnitude faster than first-principles calculations, they remain much slower than classical force fields, at the price of using more complex structural descriptors. To bridge this efficiency gap, we propose an embedded atom neural network approach with simple piecewise switching function-based descriptors, resulting in a favorable linear scaling with the number of neighbor atoms. Numerical examples validate that this piecewise machine-learning model can be over an order of magnitude faster than various popular machine-learned potentials with comparable accuracy for both metallic and covalent materials, approaching the speed of the fastest embedded atom method (i.e. several μs per atom per CPU core). The extreme efficiency of this approach promises its potential in first-principles atomistic simulations of very large systems and/or in a long timescale. read less NOT USED (high confidence) J. Byggmastar, K. Nordlund, and F. Djurabekova, “Gaussian approximation potentials for body-centered-cubic transition metals,” Physical Review Materials. 2020. link Times cited: 22 Abstract: We develop a set of machine-learning interatomic potentials … read moreAbstract: We develop a set of machine-learning interatomic potentials for elemental V, Nb, Mo, Ta, and W using the Gaussian approximation potential framework. The potentials show good accuracy and transferability for elastic, thermal, liquid, defect, and surface properties. All potentials are augmented with accurate repulsive potentials, making them applicable to radiation damage simulations involving high-energy collisions. We study melting and liquid properties in detail and use the potentials to provide melting curves up to 400 GPa for all five elements. read less NOT USED (high confidence) Q. Wang, J. Ding, L. Zhang, E. Podryabinkin, A. Shapeev, and E. Ma, “Predicting the propensity for thermally activated β events in metallic glasses via interpretable machine learning,” npj Computational Materials. 2020. link Times cited: 34 NOT USED (high confidence) E. R. M. Davidson, T. Daff, G. Csányi, and M. Finnis, “Grand canonical approach to modeling hydrogen trapping at vacancies in
α−Fe,” Physical Review Materials. 2020. link Times cited: 5 NOT USED (high confidence) C. Lapointe et al., “Machine learning surrogate models for prediction of point defect vibrational entropy,” Physical Review Materials. 2020. link Times cited: 5 Abstract: The temperature variation of the defect densities in a cryst… read moreAbstract: The temperature variation of the defect densities in a crystal depends on vibrational entropy. This contribution to the system thermodynamics remains computationally challenging as it requires a diagonalization of the system's Hessian which scales as $O({N}^{3})$ for a crystal made of $N$ atoms. Here, to circumvent such a heavy computational task and make it feasible even for systems containing millions of atoms, the harmonic vibrational entropy of point defects is estimated directly from the relaxed atomic positions through a linear-in-descriptor machine learning approach of order $O(N)$. With a size-independent descriptor dimension and fixed model parameters, an excellent predictive power is demonstrated on a wide range of defect configurations, supercell sizes, and external deformations well outside the training database. In particular, formation entropies in a range of $250{k}_{B}$ are predicted with less than $1.6{k}_{B}$ error from a training database whose formation entropies span only $25{k}_{B}$ (training error less than $1.0{k}_{B}$). This exceptional transferability is found to hold even when the training is limited to a low-energy superbasin in the phase space while the tests are performed for a different liquid-like superbasin at higher energies. read less NOT USED (high confidence) C. Zhai, T. Li, H. Shi, and J. Yeo, “Discovery and design of soft polymeric bio-inspired materials with multiscale simulations and artificial intelligence.,” Journal of materials chemistry. B. 2020. link Times cited: 28 Abstract: Materials chemistry is at the forefront of the global "… read moreAbstract: Materials chemistry is at the forefront of the global "Fourth Industrial Revolution", in part by establishing a "Materials 4.0" paradigm. A key aspect of this paradigm is developing methods to effectively integrate hardware, software, and biological systems. Towards this end, we must have intimate knowledge of the virtual space in materials design: materials omics (materiomics), materials informatics, computational modelling and simulations, artificial intelligence (AI), and big data. We focus on the discovery and design of next-generation bio-inspired materials because the design space is so huge as to be almost intractable. With nature providing researchers with specific guiding principles, this material design space may be probed most efficiently through digital, high-throughput methods. Therefore, to enhance awareness and adoption of digital approaches in soft polymeric bio-inspired materials discovery and design, we detail multiscale simulation techniques in soft matter from the molecular level to the macroscale. We also highlight the unique role that artificial intelligence and materials databases will play in molecular simulations as well as soft materials discovery. Finally, we showcase several case studies that concretely apply computational modelling and simulations for integrative soft bio-inspired materials design with experiments. read less NOT USED (high confidence) F. Brieuc, C. Schran, F. Uhl, H. Forbert, and D. Marx, “Converged quantum simulations of reactive solutes in superfluid helium: The Bochum perspective.,” The Journal of chemical physics. 2020. link Times cited: 18 Abstract: Superfluid helium has not only fascinated scientists for cen… read moreAbstract: Superfluid helium has not only fascinated scientists for centuries but is also the ideal matrix for the investigation of chemical systems under ultra-cold conditions in helium nanodroplet isolation experiments. Together with related experimental techniques such as helium tagging photodissociation spectroscopy, these methods have provided unique insights into many interesting systems. Complemented by theoretical work, they were additionally able to greatly expand our general understanding of manifestations of superfluid behavior in finite sized clusters and their response to molecular impurities. However, most theoretical studies up to now have not included the reactivity and flexibility of molecular systems embedded in helium. In this perspective, the theoretical foundation of simulating fluxional molecules and reactive complexes in superfluid helium is presented in detail. Special emphasis is put on recent developments for the converged description of both the molecular interactions and the quantum nature of the nuclei at ultra-low temperatures. As a first step, our hybrid path integral molecular dynamics/bosonic path integral Monte Carlo method is reviewed. Subsequently, methods for efficient path integral sampling tailored for this hybrid coupling scheme are discussed while also introducing new developments to enhance the accurate incorporation of the solute⋯solvent coupling. Finally, highly accurate descriptions of the interactions in solute⋯helium systems using machine learning techniques are addressed. Our current automated and adaptive fitting procedures to parameterize high-dimensional neural network potentials for both the full-dimensional potential energy surface of solutes and the solute⋯solvent interaction potentials are concisely presented. They are demonstrated to faithfully represent many-body potential functions able to describe chemically complex and reactive solutes in helium environments seamlessly from one He atom up to bulk helium at the accuracy level of coupled cluster electronic structure calculations. Together, these advances allow for converged quantum simulations of fluxional and reactive solutes in superfluid helium under cryogenic conditions. read less NOT USED (high confidence) C. Schran, K. Brezina, and O. Marsalek, “Committee neural network potentials control generalization errors and enable active learning,” The Journal of chemical physics. 2020. link Times cited: 71 Abstract: It is well known in the field of machine learning that commi… read moreAbstract: It is well known in the field of machine learning that committee models improve accuracy, provide generalization error estimates, and enable active learning strategies. In this work, we adapt these concepts to interatomic potentials based on artificial neural networks. Instead of a single model, multiple models that share the same atomic environment descriptors yield an average that outperforms its individual members as well as a measure of the generalization error in the form of the committee disagreement. We not only use this disagreement to identify the most relevant configurations to build up the model's training set in an active learning procedure but also monitor and bias it during simulations to control the generalization error. This facilitates the adaptive development of committee neural network potentials and their training sets while keeping the number of ab initio calculations to a minimum. To illustrate the benefits of this methodology, we apply it to the development of a committee model for water in the condensed phase. Starting from a single reference ab initio simulation, we use active learning to expand into new state points and to describe the quantum nature of the nuclei. The final model, trained on 814 reference calculations, yields excellent results under a range of conditions, from liquid water at ambient and elevated temperatures and pressures to different phases of ice, and the air-water interface-all including nuclear quantum effects. This approach to committee models will enable the systematic development of robust machine learning models for a broad range of systems. read less NOT USED (high confidence) B. Onat, C. Ortner, and J. Kermode, “Sensitivity and dimensionality of atomic environment representations used for machine learning interatomic potentials.,” The Journal of chemical physics. 2020. link Times cited: 27 Abstract: Faithfully representing chemical environments is essential f… read moreAbstract: Faithfully representing chemical environments is essential for describing materials and molecules with machine learning approaches. Here, we present a systematic classification of these representations and then investigate (i) the sensitivity to perturbations and (ii) the effective dimensionality of a variety of atomic environment representations and over a range of material datasets. Representations investigated include atom centered symmetry functions, Chebyshev Polynomial Symmetry Functions (CHSF), smooth overlap of atomic positions, many-body tensor representation, and atomic cluster expansion. In area (i), we show that none of the atomic environment representations are linearly stable under tangential perturbations and that for CHSF, there are instabilities for particular choices of perturbation, which we show can be removed with a slight redefinition of the representation. In area (ii), we find that most representations can be compressed significantly without loss of precision and, further, that selecting optimal subsets of a representation method improves the accuracy of regression models built for a given dataset. read less NOT USED (high confidence) A. Tran, J. Tranchida, T. Wildey, and A. Thompson, “Multi-fidelity machine-learning with uncertainty quantification and Bayesian optimization for materials design: Application to ternary random alloys,” The Journal of chemical physics. 2020. link Times cited: 42 Abstract: We present a scale-bridging approach based on a multi-fideli… read moreAbstract: We present a scale-bridging approach based on a multi-fidelity (MF) machine-learning (ML) framework leveraging Gaussian processes (GP) to fuse atomistic computational model predictions across multiple levels of fidelity. Through the posterior variance of the MFGP, our framework naturally enables uncertainty quantification, providing estimates of confidence in the predictions. We used density functional theory as high-fidelity prediction, while a ML interatomic potential is used as low-fidelity prediction. Practical materials' design efficiency is demonstrated by reproducing the ternary composition dependence of a quantity of interest (bulk modulus) across the full aluminum-niobium-titanium ternary random alloy composition space. The MFGP is then coupled to a Bayesian optimization procedure, and the computational efficiency of this approach is demonstrated by performing an on-the-fly search for the global optimum of bulk modulus in the ternary composition space. The framework presented in this manuscript is the first application of MFGP to atomistic materials simulations fusing predictions between density functional theory and classical interatomic potential calculations. read less NOT USED (high confidence) E. Bedolla, L. C. Padierna, and R. Castañeda-Priego, “Machine learning for condensed matter physics,” Journal of Physics: Condensed Matter. 2020. link Times cited: 47 Abstract: Condensed matter physics (CMP) seeks to understand the micro… read moreAbstract: Condensed matter physics (CMP) seeks to understand the microscopic interactions of matter at the quantum and atomistic levels, and describes how these interactions result in both mesoscopic and macroscopic properties. CMP overlaps with many other important branches of science, such as chemistry, materials science, statistical physics, and high-performance computing. With the advancements in modern machine learning (ML) technology, a keen interest in applying these algorithms to further CMP research has created a compelling new area of research at the intersection of both fields. In this review, we aim to explore the main areas within CMP, which have successfully applied ML techniques to further research, such as the description and use of ML schemes for potential energy surfaces, the characterization of topological phases of matter in lattice systems, the prediction of phase transitions in off-lattice and atomistic simulations, the interpretation of ML theories with physics-inspired frameworks and the enhancement of simulation methods with ML algorithms. We also discuss in detail the main challenges and drawbacks of using ML methods on CMP problems, as well as some perspectives for future developments. read less NOT USED (high confidence) C. Mangold et al., “Transferability of neural network potentials for varying stoichiometry: Phonons and thermal conductivity of MnxGey compounds,” Journal of Applied Physics. 2020. link Times cited: 24 Abstract: Germanium manganese compounds exhibit a variety of stable an… read moreAbstract: Germanium manganese compounds exhibit a variety of stable and metastable phases with different stoichiometry. These materials entail interesting electronic, magnetic and thermal properties both in their bulk form and as heterostructures. Here we develop and validate a transferable machine learning potential, based on the high-dimensional neural network formalism, to enable the study of Mn$_x$Ge$_y$ materials over a wide range of compositions. We show that a neural network potential fitted on a minimal training set reproduces successfully the structural and vibrational properties and the thermal conductivity of systems with different local chemical environments, and it can be used to predict phononic effects in nanoscale heterostructures. read less NOT USED (high confidence) J. George, G. Hautier, A. Bartók, G. Csányi, and V. L. Deringer, “Combining phonon accuracy with high transferability in Gaussian approximation potential models.,” The Journal of chemical physics. 2020. link Times cited: 26 Abstract: Machine learning driven interatomic potentials, including Ga… read moreAbstract: Machine learning driven interatomic potentials, including Gaussian approximation potential (GAP) models, are emerging tools for atomistic simulations. Here, we address the methodological question of how one can fit GAP models that accurately predict vibrational properties in specific regions of configuration space while retaining flexibility and transferability to others. We use an adaptive regularization of the GAP fit that scales with the absolute force magnitude on any given atom, thereby exploring the Bayesian interpretation of GAP regularization as an "expected error" and its impact on the prediction of physical properties for a material of interest. The approach enables excellent predictions of phonon modes (to within 0.1 THz-0.2 THz) for structurally diverse silicon allotropes, and it can be coupled with existing fitting databases for high transferability across different regions of configuration space, which we demonstrate for liquid and amorphous silicon. These findings and workflows are expected to be useful for GAP-driven materials modeling more generally. read less NOT USED (high confidence) J. S. Smith et al., “The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules,” Scientific Data. 2020. link Times cited: 1 NOT USED (high confidence) S. Jindal and S. Bulusu, “Structural evolution in gold nanoparticles using artificial neural network based interatomic potentials.,” The Journal of chemical physics. 2020. link Times cited: 5 Abstract: Relativistic effects of gold make its behavior different fro… read moreAbstract: Relativistic effects of gold make its behavior different from other metals. Unlike silver and copper, gold does not require symmetrical structures as the stable entities. We present the evolution of gold from a cluster to a nanoparticle by considering a majority of stable structural possibilities. Here, an interatomic potential (artificial neural network), trained on quantum mechanical data comprising small to medium sized clusters, gives exceptional results for larger size clusters. We have explored the potential energy surface for "magic" number clusters 309, 561, and 923. This study reveals that these clusters are not completely symmetric, but they require a distorted symmetric core with amorphous layers of atoms over it. The amorphous geometries tend to be more stable in comparison to completely symmetric structures. The first ever gold cluster to hold an icosahedron-Au13 was identified at Au60 [S. Pande et al., J. Phys. Chem. Lett. 10, 1820 (2019)]. Through our study, we have found a plausible evolution of a symmetric core as the size of the nanoparticle increases. The stable cores were found at Au160, Au327, and Au571, which can be recognized as new magic numbers. Au923 is found to have a stable symmetric core of 147 atoms covered with layers of atoms that are not completely amorphous. This shows the preference of symmetric structures as the size of the nanoparticle increases (<3.3 nm). read less NOT USED (high confidence) R. Batra and S. Sankaranarayanan, “Machine learning for multi-fidelity scale bridging and dynamical simulations of materials,” Journal of Physics: Materials. 2020. link Times cited: 12 Abstract: Molecular dynamics (MD) is a powerful and popular tool for u… read moreAbstract: Molecular dynamics (MD) is a powerful and popular tool for understanding the dynamical evolution of materials at the nano and mesoscopic scales. There are various flavors of MD ranging from the high fidelity albeit computationally expensive ab initio MD to relatively lower fidelity but much more efficient classical MD such as atomistic and coarse-grained models. Each of these different flavors of MD have been independently used by materials scientists to bring about breakthroughs in materials discovery and design. A significant gulf exists between the various MD flavors, each having varying levels of fidelity. The accuracy of DFT or ab initio MD is generally much higher than that of classical atomistic simulations which is higher than that of coarse-grained models. Multi-fidelity scale bridging to combine the accuracy and flexibility of ab initio MD with efficiency classical MD has been a longstanding goal. The advent of big-data analytics has brought to the forefront powerful machine learning methods that can be deployed to achieve this goal. Here, we provide our perspective on the challenges in multi-fidelity scale bridging and trace the developments leading up to the use of machine learning algorithms and data-science towards addressing this grand challenge. read less NOT USED (high confidence) M. Cusentino, M. Wood, and A. Thompson, “Explicit Multi-element Extension of the Spectral Neighbor Analysis Potential for Chemically Complex Systems.,” The journal of physical chemistry. A. 2020. link Times cited: 35 Abstract: A natural extension of the descriptors used in the Spectral … read moreAbstract: A natural extension of the descriptors used in the Spectral Neighbor Analysis Potential (SNAP) method is derived to treat atomic interactions in chemically complex systems. Atomic environment descriptors within SNAP are obtained from a basis function expansion of the weighted density of neighboring atoms. This new formulation instead partitions the neighbor density into partial densities for each chemical element, thus leading to explicit multi-element descriptors. For Nelem chemical elements, the number of descriptors increases as Ο(Nelem3), while the computational cost of the force calculation as implemented in LAMMPS is limited to Ο(Nelem2) and the favorable linear scaling in the number of atoms is retained. We demonstrate these chemically aware descriptors by producing an interatomic potential for indium phosphide capable of capturing high-energy defects that result from radiation damage cascades. This new explicit multi-element SNAP method reproduces the relaxed defect formation energies with substantially greater accuracy than weighted-density SNAP, while retaining accurate representation of the bulk indium phosphide properties. read less NOT USED (high confidence) M. Wen and E. Tadmor, “Uncertainty quantification in molecular simulations with dropout neural network potentials,” npj Computational Materials. 2020. link Times cited: 46 NOT USED (high confidence) A. M. Cooper, J. Kästner, A. Urban, and N. Artrith, “Efficient training of ANN potentials by including atomic forces via Taylor expansion and application to water and a transition-metal oxide,” npj Computational Materials. 2020. link Times cited: 39 NOT USED (high confidence) T. Mueller, A. Hernandez, and C. Wang, “Machine learning for interatomic potential models.,” The Journal of chemical physics. 2020. link Times cited: 189 Abstract: The use of supervised machine learning to develop fast and a… read moreAbstract: The use of supervised machine learning to develop fast and accurate interatomic potential models is transforming molecular and materials research by greatly accelerating atomic-scale simulations with little loss of accuracy. Three years ago, Jörg Behler published a perspective in this journal providing an overview of some of the leading methods in this field. In this perspective, we provide an updated discussion of recent developments, emerging trends, and promising areas for future research in this field. We include in this discussion an overview of three emerging approaches to developing machine-learned interatomic potential models that have not been extensively discussed in existing reviews: moment tensor potentials, message-passing networks, and symbolic regression. read less NOT USED (high confidence) A. V. der Ven, Z. Deng, S. Banerjee, and S. Ong, “Rechargeable Alkali-Ion Battery Materials: Theory and Computation.,” Chemical reviews. 2020. link Times cited: 116 Abstract: Since its development in the 1970s, the rechargeable alkali-… read moreAbstract: Since its development in the 1970s, the rechargeable alkali-ion battery has proven to be a truly transformative technology, providing portable energy storage for devices ranging from small portable electronics to sizable electric vehicles. Here, we present a review of modern theoretical and computational approaches to the study and design of rechargeable alkali-ion battery materials. Starting from fundamental thermodynamics and kinetics phenomenological equations, we rigorously derive the theoretical relationships for key battery properties, such as voltage, capacity, alkali diffusivity, and other electrochemically relevant computable quantities. We then present an overview of computational techniques for the study of rechargeable alkali-ion battery materials, followed by a critical review of the literature applying these techniques to yield crucial insights into battery operation and performance. Finally, we provide perspectives on outstanding challenges and opportunities in the theory and computation of rechargeable alkali-ion battery materials. read less NOT USED (high confidence) S. Desai, S. Reeve, and J. Belak, “Implementing a neural network interatomic model with performance portability for emerging exascale architectures,” Comput. Phys. Commun. 2020. link Times cited: 9 NOT USED (high confidence) C. Chen, Y. Zuo, W. Ye, X.-G. Li, Z. Deng, and S. Ong, “A Critical Review of Machine Learning of Energy Materials,” Advanced Energy Materials. 2020. link Times cited: 268 Abstract: Machine learning (ML) is rapidly revolutionizing many fields… read moreAbstract: Machine learning (ML) is rapidly revolutionizing many fields and is starting to change landscapes for physics and chemistry. With its ability to solve complex tasks autonomously, ML is being exploited as a radically new way to help find material correlations, understand materials chemistry, and accelerate the discovery of materials. Here, an in‐depth review of the application of ML to energy materials, including rechargeable alkali‐ion batteries, photovoltaics, catalysts, thermoelectrics, piezoelectrics, and superconductors, is presented. A conceptual framework is first provided for ML in materials science, with a broad overview of different ML techniques as well as best practices. This is followed by a critical discussion of how ML is applied in energy materials. This review is concluded with the perspectives on major challenges and opportunities in this exciting field. read less NOT USED (high confidence) J. Weinreich, A. Romer, M. Paleico, and J. Behler, “Properties of α-Brass Nanoparticles I: Neural Network Potential Energy Surface.” 2020. link Times cited: 22 Abstract: Binary metal clusters are of high interest for applications … read moreAbstract: Binary metal clusters are of high interest for applications in heterogeneous catalysis and have received much attention in recent years. To gain insights into their structure and composition at the atomic scale, computer simulations can provide valuable information if reliable interatomic potentials are available. In this paper we describe the construction of a high-dimensional neural network potential (HDNNP) intended for simulations of large brass nanoparticles with thousands of atoms, which is also applicable to bulk $\alpha$-brass and its surfaces. The HDNNP, which is based on reference data obtained from density-functional theory calculations, is very accurate with a root mean square error of 1.7 meV/atom for total energies and 39 meV/{\AA} for the forces of structures not included in the training set. The potential has been thoroughly validated for a wide range of energetic and structural properties of bulk $\alpha$-brass, its surfaces as well as clusters of different size and composition demonstrating its suitability for large-scale molecular dynamics and Monte Carlo simulations with first principles accuracy. read less NOT USED (high confidence) H. Yanxon, D. Zagaceta, B. Wood, and Q. Zhu, “Neural network potential from bispectrum components: A case study on crystalline silicon.,” The Journal of chemical physics. 2020. link Times cited: 13 Abstract: In this article, we present a systematic study on developing… read moreAbstract: In this article, we present a systematic study on developing machine learning force fields (MLFFs) for crystalline silicon. While the main-stream approach of fitting a MLFF is to use a small and localized training set from molecular dynamics simulations, it is unlikely to cover the global features of the potential energy surface. To remedy this issue, we used randomly generated symmetrical crystal structures to train a more general Si-MLFF. Furthermore, we performed substantial benchmarks among different choices of material descriptors and regression techniques on two different sets of silicon data. Our results show that neural network potential fitting with bispectrum coefficients as descriptors is a feasible method for obtaining accurate and transferable MLFFs. read less NOT USED (high confidence) S. Yin, J. Ding, M. Asta, and R. Ritchie, “Ab initio modeling of the energy landscape for screw dislocations in body-centered cubic high-entropy alloys,” npj Computational Materials. 2019. link Times cited: 55 NOT USED (high confidence) X.-G. Li, C. Chen, H. Zheng, Y. Zuo, and S. Ong, “Complex strengthening mechanisms in the NbMoTaW multi-principal element alloy,” npj Computational Materials. 2019. link Times cited: 114 NOT USED (high confidence) M. Amsler et al., “FLAME: A library of atomistic modeling environments,” Comput. Phys. Commun. 2019. link Times cited: 19 NOT USED (high confidence) Y. Hong, B. Hou, H. Jiang, and J. Zhang, “Machine learning and artificial neural network accelerated computational discoveries in materials science,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2019. link Times cited: 55 Abstract: Artificial intelligence (AI) has been referred to as the “fo… read moreAbstract: Artificial intelligence (AI) has been referred to as the “fourth paradigm of science,” and as part of a coherent toolbox of data‐driven approaches, machine learning (ML) dramatically accelerates the computational discoveries. As the machinery for ML algorithms matures, significant advances have been made not only by the mainstream AI researchers, but also those work in computational materials science. The number of ML and artificial neural network (ANN) applications in the computational materials science is growing at an astounding rate. This perspective briefly reviews the state‐of‐the‐art progress in some supervised and unsupervised methods with their respective applications. The characteristics of primary ML and ANN algorithms are first described. Then, the most critical applications of AI in computational materials science such as empirical interatomic potential development, ML‐based potential, property predictions, and molecular discoveries using generative adversarial networks (GAN) are comprehensively reviewed. The central ideas underlying these ML applications are discussed, and future directions for integrating ML with computational materials science are given. Finally, a discussion on the applicability and limitations of current ML techniques and the remaining challenges are summarized. read less NOT USED (high confidence) G. Dusson et al., “Atomic cluster expansion: Completeness, efficiency and stability,” J. Comput. Phys. 2019. link Times cited: 76 NOT USED (high confidence) Y. Zhang, A. Lunghi, and S. Sanvito, “Pushing the limits of atomistic simulations towards ultra-high temperature: A machine-learning force field for ZrB2,” Acta Materialia. 2019. link Times cited: 12 NOT USED (high confidence) J. S. Smith et al., “The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules,” Scientific Data. 2019. link Times cited: 113 NOT USED (high confidence) C. van der Oord, G. Dusson, G. Csányi, and C. Ortner, “Regularised atomic body-ordered permutation-invariant polynomials for the construction of interatomic potentials,” Machine Learning: Science and Technology. 2019. link Times cited: 54 Abstract: We investigate the use of invariant polynomials in the const… read moreAbstract: We investigate the use of invariant polynomials in the construction of data-driven interatomic potentials for material systems. The ‘atomic body-ordered permutation-invariant polynomials’ comprise a systematic basis and are constructed to preserve the symmetry of the potential energy function with respect to rotations and permutations. In contrast to kernel based and artificial neural network models, the explicit decomposition of the total energy as a sum of atomic body-ordered terms allows to keep the dimensionality of the fit reasonably low, up to just 10 for the 5-body terms. The explainability of the potential is aided by this decomposition, as the low body-order components can be studied and interpreted independently. Moreover, although polynomial basis functions are thought to extrapolate poorly, we show that the low dimensionality combined with careful regularisation actually leads to better transferability than the high dimensional, kernel based Gaussian Approximation Potential. read less NOT USED (high confidence) M. Wen and E. Tadmor, “Hybrid neural network potential for multilayer graphene,” Physical Review B. 2019. link Times cited: 40 Abstract: Monolayer and multilayer graphene are promising materials fo… read moreAbstract: Monolayer and multilayer graphene are promising materials for applications such as electronic devices, sensors, energy generation and storage, and medicine. In order to perform large-scale atomistic simulations of the mechanical and thermal behavior of graphene-based devices, accurate interatomic potentials are required. Here, we present a new interatomic potential for multilayer graphene structures referred to as "hNN--Gr$_x$." This hybrid potential employs a neural network to describe short-range interactions and a theoretically-motivated analytical term to model long-range dispersion. The potential is trained against a large dataset of monolayer graphene, bilayer graphene, and graphite configurations obtained from ab initio total-energy calculations based on density functional theory (DFT). The potential provides accurate energy and forces for both intralayer and interlayer interactions, correctly reproducing DFT results for structural, energetic, and elastic properties such as the equilibrium layer spacing, interlayer binding energy, elastic moduli, and phonon dispersions to which it was not fit. The potential is used to study the effect of vacancies on thermal conductivity in monolayer graphene and interlayer friction in bilayer graphene. The potential is available through the OpenKIM interatomic potential repository at \url{this https URL}. read less NOT USED (high confidence) V. L. Deringer, M. A. Caro, and G. Csányi, “Machine Learning Interatomic Potentials as Emerging Tools for Materials Science,” Advanced Materials. 2019. link Times cited: 245 Abstract: Atomic‐scale modeling and understanding of materials have ma… read moreAbstract: Atomic‐scale modeling and understanding of materials have made remarkable progress, but they are still fundamentally limited by the large computational cost of explicit electronic‐structure methods such as density‐functional theory. This Progress Report shows how machine learning (ML) is currently enabling a new degree of realism in materials modeling: by “learning” electronic‐structure data, ML‐based interatomic potentials give access to atomistic simulations that reach similar accuracy levels but are orders of magnitude faster. A brief introduction to the new tools is given, and then, applications to some select problems in materials science are highlighted: phase‐change materials for memory devices; nanoparticle catalysts; and carbon‐based electrodes for chemical sensing, supercapacitors, and batteries. It is hoped that the present work will inspire the development and wider use of ML‐based interatomic potentials in diverse areas of materials research. read less NOT USED (high confidence) R. Vasudevan et al., “Materials science in the artificial intelligence age: high-throughput library generation, machine learning, and a pathway from correlations to the underpinning physics,” MRS Communications. 2019. link Times cited: 14 Abstract: The use of statistical/machine learning (ML) approaches to m… read moreAbstract: The use of statistical/machine learning (ML) approaches to materials science is experiencing explosive growth. Here, we review recent work focusing on the generation and application of libraries from both experiment and theoretical tools. The library data enables classical correlative ML and also opens the pathway for exploration of underlying causative physical behaviors. We highlight key advances facilitated by this approach and illustrate how modeling, macroscopic experiments, and imaging can be combined to accelerate the understanding and development of new materials systems. These developments point toward a data-driven future wherein knowledge can be aggregated and synthesized, accelerating the advancement of materials science. read less NOT USED (high confidence) D. Tanner, M. A. Caro, S. Schulz, and E. O’Reilly, “Fully analytic valence force field model for the elastic and inner elastic properties of diamond and zincblende crystals,” Physical Review B. 2019. link Times cited: 6 Abstract: Using a valence force field model based on that introduced b… read moreAbstract: Using a valence force field model based on that introduced by Martin, we present three related methods through which we analytically determine valence force field parameters. The methods introduced allow easy derivation of valence force field parameters in terms of the Kleinman parameter $\ensuremath{\zeta}$ and bulk properties of zincblende and diamond crystals. We start with a model suited for covalent and weakly ionic materials, where the valence force field parameters are derived in terms of $\ensuremath{\zeta}$ and the bulk elastic constants ${C}_{11}, {C}_{12}$, and ${C}_{44}$. We show that this model breaks down as the material becomes more ionic and specifically when the elastic anisotropy factor $A=2{C}_{44}/({C}_{11}\ensuremath{-}{C}_{12})g2$. The analytic model can be stabilized for ionic materials by including Martin's electrostatic terms with effective cation and anion charges in the valence force field model. Inclusion of effective charges determined via the optical phonon mode splitting provides a stable model for all but two of the materials considered (zincblende GaN and AlN). A stable model is obtained for all materials considered by also utilizing the inner elastic constant ${E}_{11}$ to determine the magnitude of the effective charges used in the Coulomb interaction. Test calculations show that the models describe well structural relaxation in superlattices and alloys and reproduce key phonon band structure features. read less NOT USED (high confidence) J. Byggmastar, A. Hamedani, K. Nordlund, and F. Djurabekova, “Machine-learning interatomic potential for radiation damage and defects in tungsten,” Physical Review B. 2019. link Times cited: 58 Abstract: We introduce a machine-learning interatomic potential for tu… read moreAbstract: We introduce a machine-learning interatomic potential for tungsten using the Gaussian Approximation Potential framework. We specifically focus on properties relevant for simulations of radiation-induced collision cascades and the damage they produce, including a realistic repulsive potential for the short-range many-body cascade dynamics and a good description of the liquid phase. Furthermore, the potential accurately reproduces surface properties and the energetics of vacancy and self-interstitial clusters, which have been long-standing deficiencies of existing potentials. The potential enables molecular dynamics simulations of radiation damage in tungsten with unprecedented accuracy. read less NOT USED (high confidence) E. J. Ragasa, C. J. O’Brien, R. G. Hennig, S. Foiles, and S. Phillpot, “Multi-objective optimization of interatomic potentials with application to MgO,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 6 Abstract: The parameterization of a functional form for an interatomic… read moreAbstract: The parameterization of a functional form for an interatomic potential is treated as a problem in multi-objective optimization. An autonomous, machine-learning approach based on the identification of the Pareto hypersurface of errors in predicted properties allows the development of an ensemble of parameterizations with high materials fidelity and robustness. The efficacy of this approach is illustrated for the simple example of a Buckingham potential for MgO. This approach also provides a strong foundation for uncertainty quantification of potential parameterizations. read less NOT USED (high confidence) J. Schmidt, M. R. G. Marques, S. Botti, and M. A. L. Marques, “Recent advances and applications of machine learning in solid-state materials science,” npj Computational Materials. 2019. link Times cited: 1226 NOT USED (high confidence) S. Desai, M. Hunt, and A. Strachan, “Online Tools for Uncertainty Quantification in nanoHUB,” JOM. 2019. link Times cited: 3 NOT USED (high confidence) X. Qian, S. Peng, X. Li, Y. Wei, and R. Yang, “Thermal conductivity modeling using machine learning potentials: application to crystalline and amorphous silicon,” Materials Today Physics. 2019. link Times cited: 53 NOT USED (high confidence) H. Gao, J. Wang, and J. Sun, “Improve the performance of machine-learning potentials by optimizing descriptors.,” The Journal of chemical physics. 2019. link Times cited: 13 Abstract: Machine-learning (ML) potentials are promising in atomic sim… read moreAbstract: Machine-learning (ML) potentials are promising in atomic simulations due to their comparable accuracy to density functional theory but much lower computational cost. The descriptors to represent atomic environments are of high importance to the performance of ML potentials. Here, we implemented the descriptor in a differentiable way and found that ML potentials with optimized descriptors have some advantages compared with the ones without descriptor optimization, especially when the training dataset is small. Taking aluminum as an example, the trained potentials with proper descriptors can not only predict energies and forces with high accuracy of the first-principles calculations but also reproduce the statistical results of dynamical simulations. These predictions validate the efficiency of our method, which can be applied to improving the performance of machine learning interatomic potentials and will also strongly expand its applications. read less NOT USED (high confidence) M. Höhnerbach and P. Bientinesi, “Accelerating AIREBO: Navigating the Journey from Legacy to High‐Performance Code,” Journal of Computational Chemistry. 2019. link Times cited: 6 Abstract: Despite initiatives to improve the quality of scientific sof… read moreAbstract: Despite initiatives to improve the quality of scientific software, there still is a large presence of legacy code. The focus of such code is usually on domain‐science features, rather than maintainability or highest performance. Additionally, architecture specific optimizations often result in less maintainable code. In this article, we focus on the AIREBO potential from LAMMPS, which exhibits large and complex computational kernels, hindering any systematic optimization. We suggest an approach based on complexity‐reducing refactoring and hardware abstraction and present the journey from the C++ port of a previous Fortran code to performance‐portable, KNC‐hybrid, vectorized, scalable, and optimized code supporting full and reduced precision. The journey includes extensive testing that fixed bugs in the original code. Large‐scale, full‐precision runs sustain speedups of more than 4× (KNL) and 3× (Skylake). © 2019 Wiley Periodicals, Inc. read less NOT USED (high confidence) H. Babaei, R. Guo, A. Hashemi, and S. Lee, “Machine-learning-based interatomic potential for phonon transport in perfect crystalline Si and crystalline Si with vacancies,” Physical Review Materials. 2019. link Times cited: 30 Abstract: We report that single interatomic potential, developed using… read moreAbstract: We report that single interatomic potential, developed using Gaussian regression of density functional theory calculation data, has high accuracy and flexibility to describe phonon transport with ab initio accuracy in two different atomistic configurations: perfect crystalline Si and crystalline Si with vacancies. The high accuracy of second- and third-order force constants from the Gaussian approximation potential (GAP) are demonstrated with phonon dispersion, Gruneisen parameter, three-phonon scattering rate, phonon-vacancy scattering rate, and thermal conductivity, all of which are very close to the results from density functional theory calculation. We also show that the widely used empirical potentials (Stillinger-Weber and Tersoff) produce much larger errors compared to the GAP. The computational cost of GAP is higher than the two empirical potentials, but five orders of magnitude lower than the density functional theory calculation. Our work shows that GAP can provide a new opportunity for studying phonon transport in partially disordered crystalline phases with the high predictive power of ab initio calculation but at a feasible computational cost. read less NOT USED (high confidence) A. Glielmo, C. Zeni, ’A. Fekete, and A. Vita, “Building Nonparametric n-Body Force Fields Using Gaussian Process Regression,” Machine Learning Meets Quantum Physics. 2019. link Times cited: 8 NOT USED (high confidence) J. Vandermause et al., “On-the-fly active learning of interpretable Bayesian force fields for atomistic rare events,” npj Computational Materials. 2019. link Times cited: 189 NOT USED (high confidence) T. Pollock and A. V. der Ven, “The evolving landscape for alloy design,” MRS Bulletin. 2019. link Times cited: 19 Abstract: The discovery, design, and development of new alloys have lo… read moreAbstract: The discovery, design, and development of new alloys have long been critical elements of advanced engineering systems. Challenged by their chemical and structural complexity, this design process is, however, often too slow. This article highlights progress in theory, computation, data, and advanced experimental techniques that are advancing our capabilities for rapid discovery and design of new multicomponent alloys. Applied across the length scales, these new capabilities support exploration across broad composition spaces; examples of new materials and associated advances in the understanding of underlying thermochemical and thermomechanical phenomena are presented. We highlight current challenges, gaps, and specific areas that, if further developed, could have future high payoff. read less NOT USED (high confidence) M. R. G. Marques, J. Wolff, C. Steigemann, and M. Marques, “Neural network force fields for simple metals and semiconductors: construction and application to the calculation of phonons and melting temperatures.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 17 Abstract: We present a practical procedure to obtain reliable and unbi… read moreAbstract: We present a practical procedure to obtain reliable and unbiased neural network based force fields for solids. Training and test sets are efficiently generated from global structural prediction runs, at the same time assuring the structural variety and importance of sampling the relevant regions of phase space. The neural networks are trained to yield not only good formation energies, but also accurate forces and stresses, which are the quantities of interest for molecular dynamics simulations. Finally, we construct, as an example, several force fields for both semiconducting and metallic elements, and prove their accuracy for a variety of structural and dynamical properties. These are then used to study the melting of bulk copper and gold. read less NOT USED (high confidence) Z. Aitken, V. Sorkin, and Y.-W. Zhang, “Atomistic modeling of nanoscale plasticity in high-entropy alloys,” Journal of Materials Research. 2019. link Times cited: 32 Abstract: Lattice structures, defect structures, and deformation mecha… read moreAbstract: Lattice structures, defect structures, and deformation mechanisms of high-entropy alloys (HEAs) have been studied using atomistic simulations to explain their remarkable mechanical properties. These atomistic simulation techniques, such as first-principles calculations and molecular dynamics allow atomistic-level resolution of structure, defect configuration, and energetics. Following the structure–property paradigm, such understandings can be useful for guiding the design of high-performance HEAs. Although there have been a number of atomistic studies on HEAs, there is no comprehensive review on the state-of-the-art techniques and results of atomistic simulations of HEAs. This article is intended to fill the gap, providing an overview of the state-of-the-art atomistic simulations on HEAs. In particular, we discuss how atomistic simulations can elucidate the nanoscale mechanisms of plasticity underlying the outstanding properties of HEAs, and further present a list of interesting problems for forthcoming atomistic simulations of HEAs. read less NOT USED (high confidence) M. Wood, M. Cusentino, B. Wirth, and A. Thompson, “Data-driven material models for atomistic simulation,” Physical Review B. 2019. link Times cited: 37 Abstract: The central approximation made in classical molecular dynami… read moreAbstract: The central approximation made in classical molecular dynamics simulation of materials is the interatomic potential used to calculate the forces on the atoms. Great effort and ingenuity is required to construct viable functional forms and find accurate parametrizations for potentials using traditional approaches. Machine learning has emerged as an effective alternative approach to develop accurate and robust interatomic potentials. Starting with a very general model form, the potential is learned directly from a database of electronic structure calculations and therefore can be viewed as a multiscale link between quantum and classical atomistic simulations. Risk of inaccurate extrapolation exists outside the narrow range of time and length scales where the two methods can be directly compared. In this work, we use the spectral neighbor analysis potential (SNAP) and show how a fit can be produced with minimal interpolation errors which is also robust in extrapolating beyond training. To demonstrate the method, we have developed a tungsten-beryllium potential suitable for the full range of binary compositions. Subsequently, large-scale molecular dynamics simulations were performed of high energy Be atom implantation onto the (001) surface of solid tungsten. The machine learned W-Be potential generates a population of implantation structures consistent with quantum calculations of defect formation energies. A very shallow ($l2\phantom{\rule{0.16em}{0ex}}\mathrm{nm}$) average Be implantation depth is predicted which may explain ITER diverter degradation in the presence of beryllium. read less NOT USED (high confidence) M. Ceriotti, M. J. Willatt, and G. Csányi, “Machine Learning of Atomic-Scale Properties Based on Physical Principles,” Handbook of Materials Modeling. 2019. link Times cited: 30 NOT USED (high confidence) Z. Deng, C. Chen, X.-G. Li, and S. Ong, “An electrostatic spectral neighbor analysis potential for lithium nitride,” npj Computational Materials. 2019. link Times cited: 62 NOT USED (high confidence) A. Seko, A. Togo, and I. Tanaka, “Group-theoretical high-order rotational invariants for structural representations: Application to linearized machine learning interatomic potential,” Physical Review B. 2019. link Times cited: 22 Abstract: Many rotational invariants for crystal structure representat… read moreAbstract: Many rotational invariants for crystal structure representations have been used to describe the structure-property relationship by machine learning. The machine learning interatomic potential (MLIP) is one of the applications of rotational invariants, which provides the relationship between the energy and the crystal structure. Since the MLIP requires the highest accuracy among machine learning estimations of the structure-property relationship, the enumeration of rotational invariants is useful for constructing MLIPs with the desired accuracy. In this study, we introduce high-order linearly independent rotational invariants up to the sixth order based on spherical harmonics and apply them to linearized MLIPs for elemental aluminum. A set of rotational invariants is derived by the general process of reducing the Kronecker products of irreducible representations (Irreps) for the SO(3) group using a group-theoretical projector method. A high predictive power for a wide range of structures is accomplished by using high-order invariants with low-order invariants equivalent to pair and angular structural features. read less NOT USED (high confidence) C. Zeni, K. Rossi, A. Glielmo, and F. Baletto, “On machine learning force fields for metallic nanoparticles,” Advances in Physics: X. 2019. link Times cited: 25 Abstract: ABSTRACT Machine learning algorithms have recently emerged a… read moreAbstract: ABSTRACT Machine learning algorithms have recently emerged as a tool to generate force fields which display accuracies approaching the ones of the ab-initio calculations they are trained on, but are much faster to compute. The enhanced computational speed of machine learning force fields results key for modelling metallic nanoparticles, as their fluxionality and multi-funneled energy landscape needs to be sampled over long time scales. In this review, we first formally introduce the most commonly used machine learning algorithms for force field generation, briefly outlining their structure and properties. We then address the core issue of training database selection, reporting methodologies both already used and yet unused in literature. We finally report and discuss the recent literature regarding machine learning force fields to sample the energy landscape and study the catalytic activity of metallic nanoparticles. Graphical abstract read less NOT USED (high confidence) A. Samanta, “Representing local atomic environment using descriptors based on local correlations.,” The Journal of chemical physics. 2018. link Times cited: 4 Abstract: Statistical learning of material properties is an emerging t… read moreAbstract: Statistical learning of material properties is an emerging topic of research and has been tremendously successful in areas such as representing complex energy landscapes as well as in technologically relevant areas, like identification of better catalysts and electronic materials. However, analysis of large data sets to efficiently learn characteristic features of a complex energy landscape, for example, depends on the ability of descriptors to effectively screen different local atomic environments. Thus, discovering appropriate descriptors of bulk or defect properties and the functional dependence of such properties on these descriptors remains a difficult and tedious process. To this end, we develop a framework to generate descriptors based on many-body correlations that can effectively capture intrinsic geometric features of the local environment of an atom. These descriptors are based on the spectrum of two-body, three-body, four-body, and higher order correlations between an atom and its neighbors and are evaluated by calculating the corresponding two-body, three-body, and four-body overlap integrals. They are invariant to global translation, global rotation, reflection, and permutations of atomic indices. By systematically testing the ability to capture the local atomic environment, it is shown that the local correlation descriptors are able to successfully reconstruct structures containing 10-25 atoms which was previously not possible. read less NOT USED (high confidence) S. Jindal and S. Bulusu, “A transferable artificial neural network model for atomic forces in nanoparticles.,” The Journal of chemical physics. 2018. link Times cited: 10 Abstract: We have designed a new method to fit the energy and atomic f… read moreAbstract: We have designed a new method to fit the energy and atomic forces using a single artificial neural network (SANN) for any number of chemical species present in a molecular system. The traditional approach for fitting the potential energy surface for a multicomponent system using artificial neural network (ANN) is to consider n number of networks for n number of chemical species in the system. This shoots the computational cost and makes it difficult to apply to a system containing more number of species. We present a new strategy of using a SANN to compute energy and forces of a chemical system. Since atomic forces are significant for geometry optimizations and molecular dynamics simulations for any chemical system, their accurate prediction is of utmost importance. So, to predict the atomic forces, we have modified the traditional way of fitting forces from underlying energy expression. We have applied our strategy to study geometry optimizations and dynamics in gold-silver nanoalloys and thiol protected gold nanoclusters. Also, force fitting has made it possible to train smaller sized systems and extrapolate the parameters to make accurate predictions for larger systems. This proposed strategy has definitely made the mapping and fitting of atomic forces easier and can be applied to a wide variety of molecular systems. read less NOT USED (high confidence) I. Novikov and A. Shapeev, “Improving accuracy of interatomic potentials: more physics or more data? A case study of silica,” Materials Today Communications. 2018. link Times cited: 35 NOT USED (high confidence) M. J. Willatt, F. Musil, and M. Ceriotti, “Atom-density representations for machine learning.,” The Journal of chemical physics. 2018. link Times cited: 125 Abstract: The applications of machine learning techniques to chemistry… read moreAbstract: The applications of machine learning techniques to chemistry and materials science become more numerous by the day. The main challenge is to devise representations of atomic systems that are at the same time complete and concise, so as to reduce the number of reference calculations that are needed to predict the properties of different types of materials reliably. This has led to a proliferation of alternative ways to convert an atomic structure into an input for a machine-learning model. We introduce an abstract definition of chemical environments that is based on a smoothed atomic density, using a bra-ket notation to emphasize basis set independence and to highlight the connections with some popular choices of representations for describing atomic systems. The correlations between the spatial distribution of atoms and their chemical identities are computed as inner products between these feature kets, which can be given an explicit representation in terms of the expansion of the atom density on orthogonal basis functions, that is equivalent to the smooth overlap of atomic positions power spectrum, but also in real space, corresponding to n-body correlations of the atom density. This formalism lays the foundations for a more systematic tuning of the behavior of the representations, by introducing operators that represent the correlations between structure, composition, and the target properties. It provides a unifying picture of recent developments in the field and indicates a way forward toward more effective and computationally affordable machine-learning schemes for molecules and materials. read less NOT USED (high confidence) X.-G. Li, C. Hu, C. Chen, Z. Deng, J. Luo, and S. Ong, “Quantum-accurate spectral neighbor analysis potential models for Ni-Mo binary alloys and fcc metals,” Physical Review B. 2018. link Times cited: 61 Abstract: In recent years, efficient interatomic potentials approachin… read moreAbstract: In recent years, efficient interatomic potentials approaching the accuracy of density functional theory (DFT) calculations have been developed using rigorous atomic descriptors satisfying strict invariances, for example, for translation, rotation, permutation of homonuclear atoms, among others. In this paper, we generalize the spectral neighbor analysis potential (SNAP) model to bcc-fcc binary alloy systems. We demonstrate that machine-learned SNAP models can yield significant improvements even over the well-established high-performing embedded atom method (EAM) and modified EAM potentials for fcc Cu and Ni. We also report on the development of a SNAP model for the fcc Ni-bcc Mo binary system by machine learning a carefully constructed large computed data set of elemental and intermetallic compounds. We demonstrate that this binary Ni-Mo SNAP model can achieve excellent agreement with experiments in the prediction of a Ni-Mo phase diagram as well as near-DFT accuracy in the prediction of many key properties, such as elastic constants, formation energies, melting points, etc., across the entire binary composition range. In contrast, the existing Ni-Mo EAM has significant errors in the prediction of the phase diagram and completely fails in binary compounds. This paper provides a systematic model development process for multicomponent alloy systems, including an efficient procedure to optimize the hyperparameters in the model fitting, and paves the way for long-time large-scale simulations of such systems. read less NOT USED (high confidence) I. Novikov, Y. V. Suleimanov, and A. Shapeev, “Automated calculation of thermal rate coefficients using ring polymer molecular dynamics and machine-learning interatomic potentials with active learning.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 25 Abstract: We propose a methodology for the fully automated calculation… read moreAbstract: We propose a methodology for the fully automated calculation of thermal rate coefficients of gas phase chemical reactions, which is based on combining ring polymer molecular dynamics (RPMD) and machine-learning interatomic potentials actively learning on-the-fly. Based on the original computational procedure implemented in the RPMDrate code, our methodology gradually and automatically constructs the potential energy surfaces (PESs) from scratch with the data set points being selected and accumulated during the RPMDrate simulation. Such an approach ensures that our final machine-learning model provides a reliable description of the PES that avoids artifacts during exploration of the phase space by RPMD trajectories. We tested our methodology on two representative thermally activated chemical reactions studied recently by RPMDrate at temperatures within the interval of 300-1000 K. The corresponding PESs were generated by fitting to only a few thousand automatically generated structures (less than 5000) while the RPMD rate coefficients showed deviation from the reference values within the typical convergence error of RPMDrate. In future, we plan to apply our methodology to chemical reactions that proceed via complex-formation thus providing a completely general tool for calculating RPMD thermal rate coefficients for any polyatomic gas phase chemical reaction. read less NOT USED (high confidence) P. C. Myint and J. Belof, “Rapid freezing of water under dynamic compression,” Journal of Physics: Condensed Matter. 2018. link Times cited: 14 Abstract: Understanding the behavior of materials at extreme pressures… read moreAbstract: Understanding the behavior of materials at extreme pressures is a central issue in fields like aerodynamics, astronomy, and geology, as well as for advancing technological grand challenges such as inertial confinement fusion. Dynamic compression experiments to probe high-pressure states often encounter rapid phase transitions that may cause the materials to behave in unexpected ways, and understanding the kinetics of these phase transitions remains an area of great interest. In this review, we examine experimental and theoretical/computational efforts to study the freezing kinetics of water to a high-pressure solid phase known as ice VII. We first present a detailed analysis of dynamic compression experiments in which water has been observed to freeze on sub-microsecond time scales to ice VII. This is followed by a discussion of the limitations of currently available molecular and continuum simulation methods in modeling these experiments. We then describe how our phase transition kinetics models, which are based on classical nucleation theory, provide a more physics-based framework that overcomes some of these limitations. Finally, we give suggestions on future experimental and modeling work on the liquid–ice VII transition, including an outline of the development of a predictive multiscale model in which molecular and continuum simulations are intimately coupled. read less NOT USED (high confidence) A. Bartók, J. Kermode, N. Bernstein, and G. Csányi, “Machine Learning a General-Purpose Interatomic Potential for Silicon,” Physical Review X. 2018. link Times cited: 291 Abstract: The success of first principles electronic structure calcula… read moreAbstract: The success of first principles electronic structure calculation for predictive modeling in chemistry, solid state physics, and materials science is constrained by the limitations on simulated length and time scales due to computational cost and its scaling. Techniques based on machine learning ideas for interpolating the Born-Oppenheimer potential energy surface without explicitly describing electrons have recently shown great promise, but accurately and efficiently fitting the physically relevant space of configurations has remained a challenging goal. Here we present a Gaussian Approximation Potential for silicon that achieves this milestone, accurately reproducing density functional theory reference results for a wide range of observable properties, including crystal, liquid, and amorphous bulk phases, as well as point, line, and plane defects. We demonstrate that this new potential enables calculations that would be extremely expensive with a first principles electronic structure method, such as finite temperature phase boundary lines, self-diffusivity in the liquid, formation of the amorphous by slow quench, and dynamic brittle fracture. We show that the uncertainty quantification inherent to the Gaussian process regression framework gives a qualitative estimate of the potential's accuracy for a given atomic configuration. The success of this model shows that it is indeed possible to create a useful machine-learning-based interatomic potential that comprehensively describes a material, and serves as a template for the development of such models in the future. read less NOT USED (high confidence) O. A. von Lilienfeld, “Quantum Machine Learning im chemischen Raum,” Angewandte Chemie. 2018. link Times cited: 2 NOT USED (high confidence) B. Huang and O. A. V. Lilienfeld, “Quantum Machine Learning in Chemical Compound Space,” Angewandte Chemie. 2018. link Times cited: 103 Abstract: Rather than numerically solving the computationally demandin… read moreAbstract: Rather than numerically solving the computationally demanding equations of quantum or statistical mechanics, machine learning methods can infer approximate solutions, interpolating previously acquired property data sets of molecules and materials. The case is made for quantum machine learning: An inductive molecular modeling approach which can be applied to quantum chemistry problems. read less NOT USED (high confidence) G. Sosso, V. L. Deringer, S. Elliott, and G. Csányi, “Understanding the thermal properties of amorphous solids using machine-learning-based interatomic potentials,” Molecular Simulation. 2018. link Times cited: 59 Abstract: Understanding the thermal properties of disordered systems i… read moreAbstract: Understanding the thermal properties of disordered systems is of fundamental importance for condensed matter physics - and for practical applications as well. While quantities such as the thermal conductivity are usually well characterised experimentally, their microscopic origin is often largely unknown - hence the pressing need for molecular simulations. However, the time and length scales involved with thermal transport phenomena are typically well beyond the reach of ab initio calculations. On the other hand, many amorphous materials are characterised by a complex structure, which prevents the construction of classical interatomic potentials. One way to get past this deadlock is to harness machine-learning (ML) algorithms to build interatomic potentials: these can be nearly as computationally efficient as classical force fields while retaining much of the accuracy of first-principles calculations. Here, we discuss neural network potentials (NNPs) and Gaussian approximation potentials (GAPs), two popular ML frameworks. We review the work that has been devoted to investigate, via NNPs, the thermal properties of phase-change materials, systems widely used in non-volatile memories. In addition, we present recent results on the vibrational properties of amorphous carbon, studied via GAPs. In light of these results, we argue that ML-based potentials are among the best options available to further our understanding of the vibrational and thermal properties of complex amorphous solids. read less NOT USED (high confidence) E. Podryabinkin, E. Tikhonov, A. Shapeev, and A. Oganov, “Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning,” Physical Review B. 2018. link Times cited: 177 Abstract: We propose a methodology for crystal structure prediction th… read moreAbstract: We propose a methodology for crystal structure prediction that is based on the evolutionary algorithm USPEX and the machine-learning interatomic potentials actively learning on-the-fly. Our methodology allows for an automated construction of an interatomic interaction model from scratch, replacing the expensive density functional theory (DFT) and giving a speedup of several orders of magnitude. Predicted low-energy structures are then tested on DFT, ensuring that our machine-learning model does not introduce any prediction error. We tested our methodology on prediction of crystal structures of carbon, high-pressure phases of sodium, and boron allotropes, including those that have more than 100 atoms in the primitive cell. All the the main allotropes have been reproduced, and a hitherto unknown 54-atom structure of boron has been predicted with very modest computational effort. read less NOT USED (high confidence) A. Glielmo, C. Zeni, and A. Vita, “Efficient nonparametric n -body force fields from machine learning,” Physical Review B. 2018. link Times cited: 92 Abstract: The authors present a scheme to construct classical $n$-body… read moreAbstract: The authors present a scheme to construct classical $n$-body force fields using Gaussian Process (GP) Regression, appropriately mapped over explicit n-body functions (M-FFs). The procedure is possible, and will yield accurate forces, whenever prior knowledge allows to restrict the interactions to a finite order $n$, so that the ``universal approximator'' resolving power of standard GPs or Neural Networks is not needed. Under these conditions, the proposed construction preserves flexibility of training, systematically improvable accuracy, and a clear framework for validation of the underlying machine learning technique. Moreover, the M-FFs are as fast as classical parametrized potentials, since they avoid lengthy summations over database entries or weight parameters. read less NOT USED (high confidence) R. Hafizi, S. Ghasemi, S. J. Hashemifar, and H. Akbarzadeh, “A neural-network potential through charge equilibration for WS2: From clusters to sheets.,” The Journal of chemical physics. 2017. link Times cited: 9 Abstract: In the present work, we use a machine learning method to con… read moreAbstract: In the present work, we use a machine learning method to construct a high-dimensional potential for tungsten disulfide using a charge equilibration neural-network technique. A training set of stoichiometric WS2 clusters is prepared in the framework of density functional theory. After training the neural-network potential, the reliability and transferability of the potential are verified by performing a crystal structure search on bulk phases of WS2 and by plotting energy-area curves of two different monolayers. Then, we use the potential to investigate various triangular nano-clusters and nanotubes of WS2. In the case of nano-structures, we argue that 2H atomic configurations with sulfur rich edges are thermodynamically more stable than the other investigated configurations. We also studied a number of WS2 nanotubes which revealed that 1T tubes with armchair chirality exhibit lower bending stiffness. read less NOT USED (high confidence) H. Wang, L. Zhang, J. Han, and E. Weinan, “DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics,” ArXiv. 2017. link Times cited: 653 NOT USED (high confidence) A. Takahashi, A. Seko, and I. Tanaka, “Linearized machine-learning interatomic potentials for non-magnetic elemental metals: Limitation of pairwise descriptors and trend of predictive power.,” The Journal of chemical physics. 2017. link Times cited: 20 Abstract: Machine-learning interatomic potential (MLIP) has been of gr… read moreAbstract: Machine-learning interatomic potential (MLIP) has been of growing interest as a useful method to describe the energetics of systems of interest. In the present study, we examine the accuracy of linearized pairwise MLIPs and angular-dependent MLIPs for 31 elemental metals. Using all of the optimal MLIPs for 31 elemental metals, we show the robustness of the linearized frameworks, the general trend of the predictive power of MLIPs, and the limitation of pairwise MLIPs. As a result, we obtain accurate MLIPs for all 31 elements using the same linearized framework. This indicates that the use of numerous descriptors is the most important practical feature for constructing MLIPs with high accuracy. An accurate MLIP can be constructed using only pairwise descriptors for most non-transition metals, whereas it is very important to consider angular-dependent descriptors when expressing interatomic interactions of transition metals. read less NOT USED (high confidence) T. D. Huan, R. Batra, J. Chapman, S. Krishnan, L. Chen, and R. Ramprasad, “A universal strategy for the creation of machine learning-based atomistic force fields,” npj Computational Materials. 2017. link Times cited: 201 NOT USED (high confidence) A. Rohskopf, H. Seyf, K. Gordiz, T. Tadano, and A. Henry, “Empirical interatomic potentials optimized for phonon properties,” npj Computational Materials. 2017. link Times cited: 35 NOT USED (high confidence) C. Chen, Z. Deng, R. Tran, H. Tang, I. Chu, and S. Ong, “Accurate Force Field for Molybdenum by Machine Learning Large Materials Data,” arXiv: Computational Physics. 2017. link Times cited: 94 Abstract: In this work, we present a highly accurate spectral neighbor… read moreAbstract: In this work, we present a highly accurate spectral neighbor analysis potential (SNAP) model for molybdenum (Mo) developed through the rigorous application of machine learning techniques on large materials data sets. Despite Mo's importance as a structural metal, existing force fields for Mo based on the embedded atom and modified embedded atom methods still do not provide satisfactory accuracy on many properties. We will show that by fitting to the energies, forces and stress tensors of a large density functional theory (DFT)-computed dataset on a diverse set of Mo structures, a Mo SNAP model can be developed that achieves close to DFT accuracy in the prediction of a broad range of properties, including energies, forces, stresses, elastic constants, melting point, phonon spectra, surface energies, grain boundary energies, etc. We will outline a systematic model development process, which includes a rigorous approach to structural selection based on principal component analysis, as well as a differential evolution algorithm for optimizing the hyperparameters in the model fitting so that both the model error and the property prediction error can be simultaneously lowered. We expect that this newly developed Mo SNAP model will find broad applications in large-scale, long-time scale simulations. read less NOT USED (high confidence) K. Faber et al., “The role of ceramic and glass science research in meeting societal challenges: Report from an NSF-sponsored workshop.” 2017. link Times cited: 21 Abstract: Under the sponsorship of the U.S. National Science Foundatio… read moreAbstract: Under the sponsorship of the U.S. National Science Foundation, a workshop on emerging research opportunities in ceramic and glass science was held in September 2016. Reported here are proceedings of the workshop. The report details eight challenges identified through workshop discussions: Ceramic processing: Programmable design and assembly; The defect genome: Understanding, characterizing, and predicting defects across time and length scales; Functionalizing defects for unprecedented properties; Ceramic flatlands: Defining structure-property relations in free-standing, supported, and confined two-dimensional ceramics; Ceramics in the extreme: Discovery and design strategies; Ceramics in the extreme: Behavior of multimaterial systems; Understanding and exploiting glasses and melts under extreme conditions; and Rational design of functional glasses guided by predictive modeling. It is anticipated that these challenges, once met, will promote basic understanding and ultimately enable advancements within multiple sectors, including energy, environment, manufacturing, security, and health care. read less NOT USED (high confidence) A. Shapeev, “Accurate representation of formation energies of crystalline alloys with many components,” Computational Materials Science. 2016. link Times cited: 44 NOT USED (high confidence) E. Podryabinkin and A. Shapeev, “Active learning of linearly parametrized interatomic potentials,” Computational Materials Science. 2016. link Times cited: 351 NOT USED (high confidence) J. Behler, “Perspective: Machine learning potentials for atomistic simulations.,” The Journal of chemical physics. 2016. link Times cited: 874 Abstract: Nowadays, computer simulations have become a standard tool i… read moreAbstract: Nowadays, computer simulations have become a standard tool in essentially all fields of chemistry, condensed matter physics, and materials science. In order to keep up with state-of-the-art experiments and the ever growing complexity of the investigated problems, there is a constantly increasing need for simulations of more realistic, i.e., larger, model systems with improved accuracy. In many cases, the availability of sufficiently efficient interatomic potentials providing reliable energies and forces has become a serious bottleneck for performing these simulations. To address this problem, currently a paradigm change is taking place in the development of interatomic potentials. Since the early days of computer simulations simplified potentials have been derived using physical approximations whenever the direct application of electronic structure methods has been too demanding. Recent advances in machine learning (ML) now offer an alternative approach for the representation of potential-energy surfaces by fitting large data sets from electronic structure calculations. In this perspective, the central ideas underlying these ML potentials, solved problems and remaining challenges are reviewed along with a discussion of their current applicability and limitations. read less NOT USED (high confidence) G. Hegde and R. Bowen, “Machine-learned approximations to Density Functional Theory Hamiltonians,” Scientific Reports. 2016. link Times cited: 66 NOT USED (high confidence) P. Dolgirev, I. Kruglov, and A. Oganov, “Machine learning scheme for fast extraction of chemically interpretable interatomic potentials,” AIP Advances. 2016. link Times cited: 47 Abstract: We present a new method for a fast, unbiased and accurate re… read moreAbstract: We present a new method for a fast, unbiased and accurate representation of interatomic interactions. It is a combination of an artificial neural network and our new approach for pair potential reconstruction. The potential reconstruction method is simple and computationally cheap and gives rich information about interactions in crystals. This method can be combined with structure prediction and molecular dynamics simulations, providing accuracy similar to ab initio methods, but at a small fraction of the cost. We present applications to real systems and discuss the insight provided by our method. read less NOT USED (high confidence) H. Wilson and A. Barnard, “Water bilayers on ZnO(100) surfaces: data-driven structural search,” RSC Advances. 2016. link Times cited: 2 Abstract: Data science methods hold enormous promise for enhancing the… read moreAbstract: Data science methods hold enormous promise for enhancing the efficiency of multiple aspects of theoretical materials science. Here we demonstrate an approach for the use of data science methods for a structural search for high-stability atomic structures in ab initio simulations, via the analysis of a large set of candidate structures. By partitioning a large data set of structures over an appropriate set of variables, we are able to identify a small fraction of structural space into which all low-energy structures are concentrated. Structural search methods may then be applied in the identified area. The method is demonstrated on the problem of finding stable structures of the water bilayer on the ZnO(100) surface. read less NOT USED (high confidence) A. Shapeev, “Moment Tensor Potentials: A Class of Systematically Improvable Interatomic Potentials,” Multiscale Model. Simul. 2015. link Times cited: 533 Abstract: Density functional theory offers a very accurate way of comp… read moreAbstract: Density functional theory offers a very accurate way of computing materials properties from first principles. However, it is too expensive for modelling large-scale molecular systems whose properties are, in contrast, computed using interatomic potentials.
The present paper considers, from a mathematical point of view, the problem of constructing interatomic potentials that approximate a given quantum-mechanical interaction model. In particular, a new class of systematically improvable potentials is proposed, analyzed, and tested on an existing quantum-mechanical database. read less NOT USED (high confidence) T. B. Costa, S. D. Bond, D. Littlewood, and S. Moore, “Peridynamic Multiscale Finite Element Methods.” 2015. link Times cited: 4 Abstract: The problem of computing quantum-accurate design-scale solut… read moreAbstract: The problem of computing quantum-accurate design-scale solutions to mechanics problems is rich with applications and serves as the background to modern multiscale science research. The prob- lem can be broken into component problems comprised of communicating across adjacent scales, which when strung together create a pipeline for information to travel from quantum scales to design scales. Traditionally, this involves connections between a) quantum electronic structure calculations and molecular dynamics and between b) molecular dynamics and local partial differ- ential equation models at the design scale. The second step, b), is particularly challenging since the appropriate scales of molecular dynamic and local partial differential equation models do not overlap. The peridynamic model for continuum mechanics provides an advantage in this endeavor, as the basic equations of peridynamics are valid at a wide range of scales limiting from the classical partial differential equation models valid at the design scale to the scale of molecular dynamics. In this work we focus on the development of multiscale finite element methods for the peridynamic model, in an effort to create a mathematically consistent channel for microscale information to travel from the upper limits of the molecular dynamics scale to the design scale. In particular, wemore » first develop a Nonlocal Multiscale Finite Element Method which solves the peridynamic model at multiple scales to include microscale information at the coarse-scale. We then consider a method that solves a fine-scale peridynamic model to build element-support basis functions for a coarse- scale local partial differential equation model, called the Mixed Locality Multiscale Finite Element Method. Given decades of research and development into finite element codes for the local partial differential equation models of continuum mechanics there is a strong desire to couple local and nonlocal models to leverage the speed and state of the art of local models with the flexibility and accuracy of the nonlocal peridynamic model. In the mixed locality method this coupling occurs across scales, so that the nonlocal model can be used to communicate material heterogeneity at scales inappropriate to local partial differential equation models. Additionally, the computational burden of the weak form of the peridynamic model is reduced dramatically by only requiring that the model be solved on local patches of the simulation domain which may be computed in parallel, taking advantage of the heterogeneous nature of next generation computing platforms. Addition- ally, we present a novel Galerkin framework, the 'Ambulant Galerkin Method', which represents a first step towards a unified mathematical analysis of local and nonlocal multiscale finite element methods, and whose future extension will allow the analysis of multiscale finite element methods that mix models across scales under certain assumptions of the consistency of those models.« less read less NOT USED (high confidence) R. Drautz, T. Hammerschmidt, M. Cak, and D. Pettifor, “Bond-order potentials: derivation and parameterization for refractory elements,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 30 Abstract: The bond-order potentials are derived from density functiona… read moreAbstract: The bond-order potentials are derived from density functional theory by a systematic coarse graining of the electronic structure. Within their functional form the bond-order potentials comprise covalent bond formation, charge transfer and magnetism. We review the derivation of the bond-order potentials from density functional theory and discuss their application to the simulation of refractory transition metals. We show that the derived functional form of the bond-order potentials ensures the transferability of the potentials to atomic environments that have not been taken into account in the parameterization. read less NOT USED (high confidence) S. Levy, K. B. Ferreira, P. Bridges, A. Thompson, and C. Trott, “A study of the viability of exploiting memory content similarity to improve resilience to memory errors,” The International Journal of High Performance Computing Applications. 2015. link Times cited: 2 Abstract: Building the next-generation of extreme-scale distributed sy… read moreAbstract: Building the next-generation of extreme-scale distributed systems will require overcoming several challenges related to system resilience. As the number of processors in these systems grow, the failure rate increases proportionally. One of the most common sources of failure in large-scale systems is memory. In this paper, we propose a novel runtime for transparently exploiting memory content similarity to improve system resilience by reducing the rate at which memory errors lead to node failure. We evaluate the viability of this approach by examining memory snapshots collected from eight high-performance computing (HPC) applications and two important HPC operating systems. Based on the characteristics of the similarity uncovered, we conclude that our proposed approach shows promise for addressing system resilience in large-scale systems. read less NOT USED (high confidence) Q. Zeng, B. Chen, D. Kang, and J. Dai, “Large scale and quantum accurate molecular dynamics simulation: Liquid iron under extreme condition,” Acta Physica Sinica. 2023. link Times cited: 0 Abstract: Liquid iron is the major component of planetary cores. Its s… read moreAbstract: Liquid iron is the major component of planetary cores. Its structure and dynamics under high pressure and temperature is of great significance in studying geophysics and planetary science. However, for experimental techniques, it is still difficult to generate and probe such a state of matter under extreme conditions, while for theoretical method like molecular dynamics simulation, the reliable estimation of dynamic properties requires both large simulation size and ab initio accuracy, resulting in unaffordable computational costs for traditional method. Owing to the technical limitation, the understanding of such matters remains limited. In this work, combining molecular dynamics simulation, we establish a neural network potential energy surface model to study the static and dynamic properties of liquid iron at its extreme thermodynamic state close to core-mantle boundary. The implementation of deep neural network extends the simulation scales from one hundred atoms to millions of atoms within quantum accuracy. The estimated static and dynamic structure factor show good consistency with all available X-ray diffraction and inelastic X-ray scattering experimental observations, while the empirical potential based on embedding-atom-method fails to give a unified description of liquid iron across a wide range of thermodynamic conditions. We also demonstrate that the transport property like diffusion coefficient exhibits a strong size effect, which requires more than at least ten thousands of atoms to give a converged value. Our results show that the combination of deep learning technology and molecular modelling provides a way to describe matter realistically under extreme conditions. read less NOT USED (high confidence) Y. Yang et al., “Taking materials dynamics to new extremes using machine learning interatomic potentials,” Journal of Materials Informatics. 2021. link Times cited: 5 Abstract: Understanding materials dynamics under extreme conditions of… read moreAbstract: Understanding materials dynamics under extreme conditions of pressure, temperature, and strain rate is a scientific quest that spans nearly a century. Atomic simulations have had a considerable impact on this endeavor because of their ability to uncover materials’ microstructure evolution and properties at the scale of the relevant physical phenomena. However, this is still a challenge for most materials as it requires modeling large atomic systems (up to millions of particles) with improved accuracy. In many cases, the availability of sufficiently accurate but efficient interatomic potentials has become a serious bottleneck for performing these simulations as traditional potentials fail to represent the multitude of bonding. A new class of potentials has emerged recently, based on a different paradigm from the traditional approach. The new potentials are constructed by machinelearning with a high degree of fidelity from quantum-mechanical calculations. In this review, a brief introduction to the central ideas underlying machine learning interatomic potentials is given. In particular, the coupling of machine learning models with domain knowledge to improve accuracy, computational efficiency, and interpretability is highlighted. Subsequently, we demonstrate the effectiveness of the domain knowledge-based approach in certain select problems related to the kinetic response of warm dense materials. It is hoped that this review will inspire further advances in the understanding of matter under extreme conditions. read less NOT USED (high confidence) A. P. Moore, C. Deo, M. Baskes, M. Okuniewski, and D. McDowell, “Understanding the uncertainty of interatomic potentials’ parameters and formalism,” Computational Materials Science. 2017. link Times cited: 17 |
 SNAP_ThompsonSwilerTrott_2015_Ta__MO_359768485367_000
SNAP_ThompsonSwilerTrott_2015_Ta__MO_359768485367_000