Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
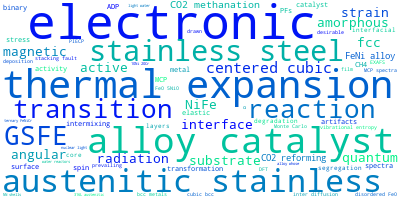
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
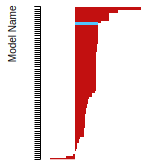
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
243 Citations (166 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) J. J. Moller et al., “110
planar faults in strained bcc metals: Origins and implications of a commonly observed artifact of classical potentials,” Physical Review Materials. 2018. link Times cited: 18 Abstract: Large-scale atomistic simulations with classical potentials … read moreAbstract: Large-scale atomistic simulations with classical potentials can provide valuable insights into microscopic deformation mechanisms and defect-defect interactions in materials. Unfortunately, these assets often come with the uncertainty of whether the observed mechanisms are based on realistic physical phenomena or whether they are artifacts of the employed material models. One such example is the often reported occurrence of stable planar faults (PFs) in body-centered cubic (bcc) metals subjected to high strains, e.g., at crack tips or in strained nano-objects. In this paper, we study the strain dependence of the generalized stacking fault energy (GSFE) of {110} planes in various bcc metals with material models of increasing sophistication, i.e., (modified) embedded atom method, angular-dependent, Tersoff, and bond-order potentials as well as density functional theory. We show that under applied tensile strains the GSFE curves of many classical potentials exhibit a local minimum which gives rise to the formation of stable PFs. These PFs do not appear when more sophisticated material models are used and have thus to be regarded as artifacts of the potentials. We demonstrate that the local GSFE minimum is not formed for reasons of symmetry and we recommend including the determination of the strain-dependent (110) GSFE as a benchmark for newly developed potentials. read less USED (high confidence) S. S. Kliavinek and L. Kolotova, “Molecular Dynamics Simulation of Glass Transition of the Supercooled Zr–Nb Melt,” Journal of Experimental and Theoretical Physics. 2020. link Times cited: 4 USED (high confidence) K. Li and C. Fu, “Ground-state properties and lattice-vibration effects of disordered Fe-Ni systems for phase stability predictions,” Physical Review Materials. 2020. link Times cited: 6 Abstract: By means of density functional theory, we perform a focused … read moreAbstract: By means of density functional theory, we perform a focused study of both body-centered-cubic (bcc) and face-centered-cubic (fcc) Fe-Ni random solid solutions, represented by special quasirandom structures. The whole concentration range and various magnetic configurations are considered. Excellent agreement on the concentration dependence of magnetization is found between our results and experimental data, except in the Invar region. Some locally antiferromagnetic fcc structures are proposed to approach experimental values of magnetization. Vibrational entropies of ordered and disordered systems are calculated for various concentrations, showing an overall good agreement with available experimental data. The vibrational entropy systematically contributes to stabilize disordered rather than ordered structures and is not negligible compared to the configurational entropy. Free energy of mixing is estimated by including the vibrational and ideal configurational entropies. From them, low- and intermediate-temperature Fe-Ni phase diagrams are constructed, showing a better agreement with experimental data than the one from a recent thermodynamic assessment for some phase boundaries below 700 K. The determined order-disorder transition temperatures for the $\mathrm{L}{1}_{0}$ and $\mathrm{L}{1}_{2}$ phases are in good agreement with the experimental values, suggesting an important contribution of vibrational entropy. read less USED (high confidence) S. Mahmoud and N. Mousseau, “Long-time point defect diffusion in ordered nickel-based binary alloys: How small kinetic differences can lead to completely long-time structural evolution,” Materialia. 2018. link Times cited: 15 USED (high confidence) K. Ray, R. Bhardwaj, B. Singh, and G. Deo, “Developing descriptors for CO2 methanation and CO2 reforming of CH4 over Al2O3 supported Ni and low-cost Ni based alloy catalysts.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 14 Abstract: The catalytic performance of Ni can be modified by alloying … read moreAbstract: The catalytic performance of Ni can be modified by alloying with a suitable amount (25% of total metal loading) of another low-cost metal such as Fe, Co or Cu. These alumina supported Ni and Ni based alloy catalysts are gaining attention for certain important reactions due to their promising activity, stability, selectivity and low-cost. The reduced form of the supported Ni-M (M = Fe, Co or Cu) catalysts formed different Ni-M alloys. To understand the reactivity trends for CO2 methanation and CO2 reforming of CH4 (DRM), we analyzed the correlations between turnover frequencies and the d-density of states (d-DOS) based electronic properties of surface Ni in Ni and Ni-M model catalysts. The composition and components of the most active catalysts for each reaction were different. The dissimilar trend in activity of the Ni and Ni-M alloy catalysts resulted in different descriptors for the two reactions. The Ni-Fe alloy catalyst (with a Ni to Fe ratio of 3 : 1) was the most active in CO2 methanation due to the minimum number of Ni d-density of states at the EF. In contrast, the Ni-Co alloy catalyst (with a Ni to Co ratio of 3 : 1) was the most active in the CO2 reforming of CH4 due to the lowest d-band center (with respect to Ni d-density of states), and the Ni-Cu alloy catalyst (with a Ni to Cu ratio of 3 : 1) was the least active for both reactions. Moreover, step sites were better correlated for CO2 methanation, whereas terrace sites were better correlated for the CO2 reforming of CH4. read less USED (high confidence) R. Masrour et al., “Spin and Orbital Magnetisms of NiFe Compound: Density Functional Theory Study and Monte Carlo Simulation,” Chinese Physics Letters. 2018. link Times cited: 2 Abstract: The self-consistent ab initio calculations based on the dens… read moreAbstract: The self-consistent ab initio calculations based on the density functional theory approach using the full potential linear augmented plane wave method are performed to investigate both the electronic and magnetic properties of the NiFe compound. Polarized spin within the framework of the ferromagnetic state between magnetic ions is considered. Also, magnetic moments considered to lie along (001) axes are computed. The Monte Carlo simulation is used to study the magnetic properties of NiFe. The transition temperature TC, hysteresis loop, coercive field and remanent magnetization of the NiFe compound are obtained using the Monte Carlo simulation. read less USED (high confidence) D. Benea, J. Min’ar, H. Ebert, and L. Chioncel, “Magnetic Compton profiles of disordered Fe0.5Ni0.5 and ordered FeNi alloys,” Physical Review B. 2017. link Times cited: 5 Abstract: We study the magnetic Compton profile (MCP) of the disordere… read moreAbstract: We study the magnetic Compton profile (MCP) of the disordered Fe0.5Ni0.5 and of the ordered FeNi alloys and discuss the interplay between structural disorder and electronic correlations. The coherent potential approximation is employed to model the substitutional disorder within the single-site approximation, while local electronic correlations are captured with the dynamical mean field theory. Comparison with the experimental data reveals the limitation of local spin-density approximation in the low momentum region, where we show that including local but dynamic correlations the experimental spectra is excellently described. We further show that using local spin-density approximation no significant difference is seen between the MCP spectra of the disordered Fe0.5Ni0.5 and a hypothetical FeNi alloy having the ordered CuAu L1(0) structure. Only by including the electronic correlations, the spectra significantly separate, from the second Brillouin zone boundary down to zero momenta. The difference between the MCP spectra of ordered and disordered alloys is discussed also in terms of the atomic-type decompositions. Finally based on the presented calculations we predict the shape of the MCP profile for the ordered FeNi alloy along the [111] direction. read less USED (high confidence) A. Takeuchi, K. Takenaka, Y. Zhang, Y. C. Wang, and A. Makino, “Stress-Enhanced Transformations from Hypothetical B2 to Stable L10 and Amorphous to fcc Phases in Fe50Ni50 Binary Alloy by Molecular Dynamic Simulations,” Materials Transactions. 2017. link Times cited: 1 Abstract: Molecular dynamics (MD) simulations were performed for an Fe… read moreAbstract: Molecular dynamics (MD) simulations were performed for an Fe50Ni50 (at.%) alloy with NTp ensemble to keep the number of atoms (N), temperature (T = 673 K), and pressure (p ∼ 101.325 kPa) constant under a GrujicicZhou-type MD potential from an Embedded Atom Method scheme with a cut-off distance of 1 nm. An Fe50Ni50 alloy was initially created as a hypothetical chemically-ordered B2 structure with a 12 × 12 × 12 supercell comprising 3456 atoms. Subsequently, it was annealed at 673 K, without the application of stress, and then under a uniaxial tension of ∼290 MPa, and shear stresses of ∼570 and ∼2940 MPa. The results revealed that stress contributed to a change in the transformation scheme to the L10 phase from partially to fully of the system with a reduction of time. On the other hand, an as-quenched amorphous phase under a shear stress of ∼680 MPa, transformed to a disordered fcc-derivative phase. Therefore it is clear that stresses in MD simulations play a crucial role in enhancing the atomic motion during a transformation. [doi:10.2320/matertrans.M2016162] read less USED (high confidence) K. Tong, F. Ye, M. Gao, M. Lei, and C. Zhang, “Interatomic potential for Fe–Cr–Ni–N system based on the second nearest-neighbor modified embedded-atom method,” Molecular Simulation. 2016. link Times cited: 7 Abstract: The interatomic potential for Fe–Cr–Ni–N system based on the… read moreAbstract: The interatomic potential for Fe–Cr–Ni–N system based on the second nearest-neighbour modified embedded-atom method has been developed in this work. The potential is based on those for the corresponding lower order systems. The potential parameters for the binary systems, Cr–N, Ni–N, Ni–Fe and Ni–Cr, were determined by fitting the lattice constants, elastic properties, heat of solution and defect binding energies. The potential parameters for the ternary systems were calculated based on the corresponding binary systems. Then, all of them were applied to the quaternary system Fe–Cr–Ni–N to confirm their validity by a simulation of the lattice constants of AISI 316 austenitic stainless steel with a range of nitrogen content. The results were in good agreement with the previous observations and calculations. read less USED (high confidence) E. Sak-Saracino and H. Urbassek, “Temperature-induced phase transformation of Fe1-xNix alloys: molecular-dynamics approach,” The European Physical Journal B. 2015. link Times cited: 17 USED (high confidence) G. Yang, X. Gao, J. Li, and L. Kong, “Orientation dependences of atomic structures in chemically heterogeneous Cu50Ta50/Ta glass-crystal interfaces,” Journal of Applied Physics. 2015. link Times cited: 4 Abstract: Molecular dynamics simulations based on an angular-dependent… read moreAbstract: Molecular dynamics simulations based on an angular-dependent potential were performed to examine the structural properties of chemically heterogeneous interfaces between amorphous Cu 50 Ta 50 and crystalline Ta. Several phenomena, namely, layering, crystallization, intermixing, and composition segregation, were observed in the Cu 50 Ta 50 region adjacent to the Ta layers. These interfacial behaviors are found to depend on the orientation of the underlying Ta substrate: Layering induced by Ta(110) extends the farthest into Cu 50 Ta 50, crystallization in the Cu 50 Ta 50 region is most significant for interface against Ta(100), while inter-diffusion is most pronounced for Ta(111). It turns out that the induced layering behavior is dominated by the interlayer distances of the underlying Ta layers, while the degree of inter-diffusion is governed by the openness of the Ta crystalline layers. In addition, composition segregations are observed in all interface models, corresponding to the immiscible nature of the Cu-Ta system. Furthermore, Voronoi polyhedra ⟨0,5,2,6⟩ and ⟨0,4,4,6⟩ are found to be abundant in the vicinity of the interfaces for all models, whose presence is believed to facilitate the structural transition between amorphous and body centered cubic. read less USED (high confidence) C. Healy and G. Ackland, “Molecular dynamics simulations of compression–tension asymmetry in plasticity of Fe nanopillars,” Acta Materialia. 2014. link Times cited: 79 USED (high confidence) G. Bonny, N. Castin, and D. Terentyev, “Interatomic potential for studying ageing under irradiation in stainless steels: the FeNiCr model alloy,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 215 Abstract: The degradation of austenitic stainless steels in a radiatio… read moreAbstract: The degradation of austenitic stainless steels in a radiation environment is a known problem for the in-core components of nuclear light water reactors. For a better understanding of the prevailing mechanisms responsible for the materials' degradation, large-scale atomistic simulations are desirable. In this framework and as a follow-up on Bonny et al (2011 Modelling Simul. Mater. Sci. Eng. 19 085008), we developed an embedded atom method type interatomic potential for the ternary FeNiCr system to model the production and evolution of radiation defects. Special attention has been drawn to the Fe10Ni20Cr alloy, whose properties were ensured to be close to those of 316L austenitic stainless steels. The potential is extensively benchmarked against density functional theory calculations and the potential developed in our earlier work. As a first validation, the potential is used in AKMC simulations to simulate thermal annealing experiments in order to determine the self-diffusion coefficients of the components in FeNiCr alloys around the Fe10Ni20Cr composition. The results from these simulations are consistent with experiments, i.e., DCr > DNi > DFe. read less USED (high confidence) C. V. Singh and D. Warner, “An Atomistic-Based Hierarchical Multiscale Examination of Age Hardening in an Al-Cu Alloy,” Metallurgical and Materials Transactions A. 2013. link Times cited: 33 USED (high confidence) N. Medvedeva, A. Murthy, V. Richards, D. Aken, and J. Medvedeva, “First principle study of cobalt impurity in bcc Fe with Cu precipitates,” Journal of Materials Science. 2013. link Times cited: 19 USED (high confidence) T. Yokoyama, “Path Integral Effective Classical Potential Method Applied to Anharmonicity and Quantum Effects in Thermal Expansion of Invar Alloy,” E-journal of Surface Science and Nanotechnology. 2012. link Times cited: 2 Abstract: Extended X-ray Absorption Fine Structure (EXAFS) spectroscop… read moreAbstract: Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy is a powerful experimental technique to investigate anharmonic vibrational properties of solids especially when one combines experimental EXAFS with quantum mechanical theoretical evaluations. In this article, the path-integral effective-classical-potential (PIECP) theory is applied to temperature dependence of EXAFS in a real system. The anharmonicity and quantum effects in the Invar alloy Fe64.6Ni35.4 that shows anomalously small thermal expansion are investigated. Experimental Fe and Ni K-edge EXAFS measurements and the computational PIECP simulations have been performed. It is experimentally revealed that the first nearest-neighbor (NN) shells around Fe show almost no thermal expansion, while those around Ni exhibit meaningful but smaller expansion than that of fcc Ni. At low temperature (<100 K), the vibrational quantum effect is found to play an essentially important role, which is confirmed by comparing the quantum mechanical simulations to the classical ones, the latter of which exhibit large (normal) thermal expansion at low temperature. It is also clarified that thermal expansion for the Ni-Ni and Ni-Fe pairs is noticeably suppressed, even though the Ni electronic state may not vary depending on the temperature. On the other hand, the anharmonicity (asymmetric distribution) clearly exist for all the first-NN shells as in the case of the normal thermal expansion system, where thermal expansion originates almost exclusively from the anharmonic interatomic potential. [DOI: 10.1380/ejssnt.2012.486] read less USED (high confidence) G. Bonny, D. Terentyev, R. Pasianot, S. Poncé, and A. Bakaev, “Interatomic potential to study plasticity in stainless steels: the FeNiCr model alloy,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 180 Abstract: Austenitic stainless steels are commonly used materials for … read moreAbstract: Austenitic stainless steels are commonly used materials for in-core components of nuclear light water reactors. In service, such components are exposed to harsh conditions: intense neutron irradiation, mechanical and thermal stresses, and aggressive corrosion environment which all contribute to the components' degradation. For a better understanding of the prevailing mechanisms responsible for the materials degradation, large-scale atomistic simulations are desirable. In this framework we developed an embedded atom method type interatomic potential for the ternary FeNiCr system to model movement of dislocations and their interaction with radiation defects. Special attention has been drawn to the Fe–10Ni–20Cr alloy, whose properties were ensured to be close to those of 316L austenitic stainless steel. In particular, the stacking fault energy and elastic constants are well reproduced. The fcc phase for the Fe–10Ni–20Cr random alloy was proven to be stable in the temperature range 0–900 K and under shear strain up to 5%. For the same alloy the stable glide of screw dislocations and stability of Frank loops was confirmed. read less USED (high confidence) Shang-Da 尚达 Chen 陈, T. Wang 王, De-Li 德立 Zheng 郑, and Yi-Chun 益春 Zhou 周, “The effect of deposition temperature on the intermixing and microstructure of Fe/Ni thin film,” Chinese Physics B. 2010. link Times cited: 1 Abstract: The physical vapour deposition of Ni atoms on α-Fe(001) surf… read moreAbstract: The physical vapour deposition of Ni atoms on α-Fe(001) surface under different deposition temperatures were simulated by molecular dynamics to study the intermixing and microstructure of the interfacial region. The results indicate that Ni atoms hardly penetrate into Fe substrate while Fe atoms easily diffuse into Ni deposition layers. The thickness of the intermixing region is temperature-dependent, with high temperatures yielding larger thicknesses. The deposited layers are mainly composed of amorphous phase due to the abnormal deposition behaviour of Ni and Fe. In the deposited Ni-rich phase, the relatively stable metallic compound B2 structured FeNi is found under high deposition temperature conditions. read less USED (high confidence) A. Hashibon, P. Schravendijk, C. Elsässer, and P. Gumbsch, “Atomistic study of structure and stability of thin Ni films on Fe surfaces,” Philosophical Magazine. 2009. link Times cited: 12 Abstract: The adhesion and residual strain of Ni thin-film coatings on… read moreAbstract: The adhesion and residual strain of Ni thin-film coatings on γ-Fe and α-Fe substrates are investigated by ab initio calculations using density functional theory (DFT) in the local density approximation (LDA), and by an empirical Finnis–Sinclair type interatomic potential utilizing angle dependent terms. The results from the DFT and empirical potentials agree for strained coherent interfaces. The phase stability and structural transitions are studied for incoherent interfaces via molecular dynamics and static relaxation methods. It is found that the transition in thin Ni films from bcc to fcc structure occurs for three or four monolayers and is accompanied by a reorientation of the Ni film with respect to the Fe substrate. read less USED (low confidence) T. Yang, X. Han, W. Li, X. Chen, and P. Liu, “Angular dependent potential for Al-Zr binary system to study the initial heterogeneous nucleation behavior of liquid Al on L12-Al3Zr,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) H. Mei et al., “Development of Machine Learning and Empirical Interatomic Potentials for the Binary Zr-Sn System,” Journal of Nuclear Materials. 2023. link Times cited: 0 USED (low confidence) J. J. Li, N. Zagni, W. D. Neilson, R. L. Gray, and S. Murphy, “The incorporation of xenon at point defects and bubbles in uranium mononitride,” Journal of Nuclear Materials. 2023. link Times cited: 0 USED (low confidence) S. Starikov, V. Jamebozorgi, D. Smirnova, R. Drautz, and M. Mrovec, “Atomistic simulations of pipe diffusion in bcc transition metals,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) B. Yao, Z. R. Liu, D. Legut, and R. F. Zhang, “Hybrid potential model with high feasibility and flexibility for metallic and covalent solids,” Physical Review B. 2023. link Times cited: 0 USED (low confidence) B. Beeler and Y. Zhang, “The reconciliation and validation of a combined interatomic potential for the description of Xe in γU-Mo,” Frontiers in Nuclear Engineering. 2023. link Times cited: 0 Abstract: A U-Mo alloy has been selected as the fuel design for the co… read moreAbstract: A U-Mo alloy has been selected as the fuel design for the conversion of high-performance research reactors in the United States. Efforts are ongoing to describe the fuel evolution as a function of time, for a variety of different reactor conditions. The accurate prediction of fuel evolution under irradiation requires the implementation of correct thermodynamic properties into mesoscale and continuum-level fuel performance modeling codes. Molecular dynamics has proven to be a valuable tool to parameterize or inform these higher-length scale models. However, there are currently inaccuracies in the only available U-Mo-Xe potential, which limits the predictive capabilities of molecular dynamics to inform critical phenomena in these fuel systems such as fission gas swelling. This work provides an updated U-Mo-Xe ternary interatomic potential which combines existing potentials in a reconciled format. The validation of the interatomic potential is performed by analyzing the phase stability and vacancy formation energies. Subsequently, Xe solution energies and an equation of state to describe Xe bubbles in U-Mo are calculated, providing 1) evidence of the significant differences between the prior ternary potential and the currently presented potential, and 2) updated data/tools for implementation into mesoscale simulation methodologies to study fission gas bubble evolution. read less USED (low confidence) H. Yuan, Q. Wei, X. Jia, M. Zhang, Z. Wu, and X. Zhu, “The stability, mechanical, and electronic properties of the Cr–Mo–B system: First-principles predictions,” Ceramics International. 2023. link Times cited: 0 USED (low confidence) I. A. Alhafez, O. Deluigi, D. Tramontina, C. Ruestes, E. Bringa, and H. Urbassek, “Simulated nanoindentation into single-phase fcc Fe\documentclass[12pt]minimal \usepackageamsmath \usepackagewasysym \usepackageamsfonts \usepackageamssymb \usepackageamsbsy \usepackagemathrsfs \usepackageupgreek \setlength\oddsidemargin-69pt \begindocument$_x$\enddocument,” Scientific Reports. 2023. link Times cited: 1 USED (low confidence) Y. Xu, Y. Xiao, Y. Wang, and W. Mi, “Spin Control and Multifunctional Photocurrent Switch in Chiral Hybrid Organic–Inorganic Perovskites,” Chemistry of Materials. 2023. link Times cited: 0 USED (low confidence) A. Zelenina, I. Gordeev, and L. Kolotova, “Atomistic simulation of Si-Al nanosponge structure features produced by laser printing method,” Journal of Non-Crystalline Solids. 2023. link Times cited: 0 USED (low confidence) S. Starikov and D. Smirnova, “Details of structure transformations in pure uranium and U-Mo alloys: insights from classical atomistic simulation,” Journal of Nuclear Materials. 2023. link Times cited: 1 USED (low confidence) Y. Xu, G. Wang, J. Shen, P. Qian, and Y. Su, “Structural features, thermal stability and catalytic implication of Fe–Ni nanoparticles,” Journal of Solid State Chemistry. 2023. link Times cited: 0 USED (low confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (low confidence) A. Sharma et al., “Solid-solution and precipitation softening effects in defect-free faceted Nickel-Iron nanoparticles,” Acta Materialia. 2022. link Times cited: 5 USED (low confidence) A. Hernandez and T. Mueller, “Generalizability of Functional Forms for Interatomic Potential Models Discovered by Symbolic Regression,” ArXiv. 2022. link Times cited: 0 Abstract: In recent years there has been great progress in the use of … read moreAbstract: In recent years there has been great progress in the use of machine learning algorithms to develop interatomic potential models. Machine-learned potential models are typically orders of magnitude faster than density functional theory but also orders of magnitude slower than physics-derived models such as the embedded atom method. In our previous work, we used symbolic regression to develop fast, accurate and transferrable interatomic potential models for copper with novel functional forms that resemble those of the embedded atom method. To determine the extent to which the success of these forms was specific to copper, here we explore the generalizability of these models to other face-centered cubic transition metals and analyze their out-of-sample performance on several material properties. We found that these forms work particularly well on elements that are chemically similar to copper. When compared to optimized Sutton-Chen models, which have similar complexity, the functional forms discovered using symbolic regression perform better across all elements considered except gold where they have a similar performance. They perform similarly to a moderately more complex embedded atom form on properties on which they were trained, and they are more accurate on average on other properties. We attribute this improved generalized accuracy to the relative simplicity of the models discovered using symbolic regression. The genetic programming models are found to outperform other models from the literature about 50% of the time in a variety of property predictions, with about 1/10th the model complexity on average. We discuss the implications of these results to the broader application of symbolic regression to the development of new potentials and highlight how models discovered for one element can be used to seed new searches for different elements. read less USED (low confidence) Y. Yu et al., “Mechanical properties and chemical bonding transitions of Nb/NbC and α-Fe/NbC interfaces in Fe-Nb-C composites,” Materials Today Communications. 2022. link Times cited: 3 USED (low confidence) A. Mangla, G. Deo, and P. A. Apte, “Cooperative freezing of the L12 ordered domains at the critical cooling temperature of Ni3Fe alloy,” Journal of Statistical Mechanics: Theory and Experiment. 2022. link Times cited: 0 Abstract: It is well known that Ni3Fe transforms from a disordered sol… read moreAbstract: It is well known that Ni3Fe transforms from a disordered solid solution to an ordered intermetallic with L12 superstructure when the alloy is cooled slowly. Here we elucidate the underlying cooperative phenomenon and the atomistic mechanism of this ordering process based on simulations using embedded atom potentials. As the simulated alloy is cooled from the disordered state to the critical cooling temperature (T c), Ni atoms with L12 order [denoted as Ni(L12 ⩾ 1) atoms] increase significantly along with Ni atoms having the least deviation from L12 local order (denoted as Ni([IP]3) atoms). The ordering (up to T c) occurs predominantly through random increase in Ni(L12 ⩾ 1) atoms throughout the system, as indicated by absence of long-range order. At T c, L12 ordered domains formed by Ni(L12 ⩾ 1) atoms ‘freeze’, i.e. these domains, collectively, achieve a threshold strength against thermal fluctuations. This is indicated by (i) dissipation of large-scale fluctuations of Ni(L12 ⩾ 1) atoms at T c and (ii) the growth of the L12 domains through propagation (at the expense of atoms with non-L12 local environment) as the alloy is cooled below T c. The stability threshold of the L12 ordered domains at T c is qualitatively consistent with (i) the critical slowing down, i.e. a significant increase in annealing time (to about 41 days) at 497 °C close to T c (∼500 °C) and (ii) sharp changes in bulk properties (due to loss of stability of the domains) when the alloy is heated across T c to about 550 °C. Further, the experimental long-range order parameter values as a function of reduced temperature are in reasonable agreement with the corresponding values of the simulated alloys. The contribution of Ni([IP]3) atoms to ordering in the actual alloy is potentially significant since such atoms together with nearest neighbours constitute about 75% of the total atoms in the simulated alloys at T c. read less USED (low confidence) S. Mandal, M. Debata, P. Sengupta, and S. Basu, “L10 FeNi: a promising material for next generation permanent magnets,” Critical Reviews in Solid State and Materials Sciences. 2022. link Times cited: 5 Abstract: Permanent magnets (PM) find widespread application in energy… read moreAbstract: Permanent magnets (PM) find widespread application in energy conversion, telecommunication, data storage, sensors, electronic gadgets, etc. Even though the market for PM is dominated by rare earth (RE) based magnets like Nd-Fe-B and Sm-Co, the recent crisis of RE elements and supply constraints have evoked the necessity of new PM materials for sustainable development. Owing to the predicted high value of (BH)max , the abundant availability of constituent elements (Fe, Ni), and presence in natural meteorites, L10 FeNi has drawn the attraction of the scientific community. Therefore, in this article, L10 FeNi (tetrataenite) is extensively reviewed as one of the most suitable candidates for future permanent magnetic material. Although L10 FeNi has shown immense potential for PM application due to its high magnetocrystalline anisotropy and magnetic saturation, the bulk synthesis of this material is not yet achieved. The problems in laboratory synthesis of L10 FeNi and the technological limitations for practical use are dominated by the slow diffusion of Ni in the FeNi lattice around the low order-disorder temperature (∼593 K). Artificial techniques with a low-temperature synthesis of ordered L10 FeNi are highlighted and the properties of L10 FeNi thin films are also presented coherently. read less USED (low confidence) B. Yao, Z. R. Liu, and R. F. Zhang, “EAPOTc: An integrated empirical interatomic potential optimization platform for compound solids,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) M. Zhou, B. Fu, Q. Hou, L. Wu, and R. Pan, “Determining the diffusion behavior of point defects in zirconium by a multiscale modelling approach,” Journal of Nuclear Materials. 2022. link Times cited: 3 USED (low confidence) Y. Xu, G. Wang, P. Qian, and Y. Su, “Element segregation and thermal stability of Ni–Pd nanoparticles,” Journal of Materials Science. 2022. link Times cited: 6 USED (low confidence) Y. Xu, G. Wang, P. Qian, and Y. Su, “Element segregation and thermal stability of Ni–Rh nanoparticles,” Journal of Solid State Chemistry. 2022. link Times cited: 6 USED (low confidence) C. Xiong, M. Ma, and C. Wu, “Explorations of stability, mechanical and thermal properties of cubic TiM2Al (M = Fe, Co, Ni, and Cu) compounds,” Solid State Communications. 2021. link Times cited: 1 USED (low confidence) S. S. Kliavinek and L. Kolotova, “Modeling of glass transition process and elastic properties of Zr-Nb amorphous alloys,” Journal of Non-crystalline Solids. 2021. link Times cited: 1 USED (low confidence) V. Maksimenko, A. Lipnitskii, V. Saveliev, I. Nelasov, and A. Kartamyshev, “Prediction of the diffusion characteristics of the V-Cr system by molecular dynamics based on N-body interatomic potentials,” Computational Materials Science. 2021. link Times cited: 5 USED (low confidence) S. Starikov and D. Smirnova, “Optimized interatomic potential for atomistic simulation of Zr-Nb alloy,” Computational Materials Science. 2021. link Times cited: 15 USED (low confidence) X. Chen et al., “Machine learning enhanced empirical potentials for metals and alloys,” Comput. Phys. Commun. 2021. link Times cited: 5 USED (low confidence) C.-yang Ran et al., “Study on Si-like and topologically close-packed structures during rapid solidification of Au-Si alloys,” Journal of Non-Crystalline Solids. 2021. link Times cited: 3 USED (low confidence) S. Starikov et al., “Angular-dependent interatomic potential for large-scale atomistic simulation of iron: Development and comprehensive comparison with existing interatomic models,” Physical Review Materials. 2021. link Times cited: 16 Abstract: The development of classical interatomic potential for iron … read moreAbstract: The development of classical interatomic potential for iron is a quite demanding task with a long history background. A new interatomic potential for simulation of iron was created with a focus on description of crystal defects properties. In contrast with previous studies, here the potential development was based on force-matching method that requires only ab initio data as reference values. To verify our model, we studied various features of body-centered-cubic iron including the properties of point defects (vacancy and self-interstitial atom), the Peierls energy barrier for dislocations (screw and mix types), and the formation energies of planar defects (surfaces, grain boundaries, and stacking fault). The verification also implies thorough comparison of a potential with 11 other interatomic potentials reported in literature. This potential correctly reproduces the largest number of iron characteristics which ensures its advantage and wider applicability range compared to the other considered classical potentials. Here application of the model is illustrated by estimation of self-diffusion coefficients and the calculation of fcc lattice properties at high temperature. read less USED (low confidence) H. Tang et al., “Synthesis of paracrystalline diamond,” Nature. 2021. link Times cited: 52 USED (low confidence) D. S. Oliveira and M. Cotta, “Role of Group V Atoms during GaAs Nanowire Growth Revealed by Molecular Dynamics Simulations: Implications in the Formation of Sharp Interfaces.” 2021. link Times cited: 3 Abstract: Understanding atomistic mechanisms for catalyst-assisted nan… read moreAbstract: Understanding atomistic mechanisms for catalyst-assisted nanowire growth is an essential step to improve control over the properties of these versatile nanomaterials. However, in silico approaches ... read less USED (low confidence) N. Pandya, A. Mevada, and P. Gajjar, “Structural, vibrational and electronic properties of D03 FeNi3,” Materials Today: Proceedings. 2020. link Times cited: 0 USED (low confidence) C. Lu et al., “Influence of applied electric field on atom diffusion behavior and mechanism for W/NiFe interface in diffusion bonding of Steel/NiFe interlayer/W by spark plasma sintering,” Applied Surface Science. 2020. link Times cited: 5 USED (low confidence) S. Zhao, D. Chen, G. Yeli, and J. Kai, “Atomistic insight into the effects of order, disorder and their interface on defect evolution,” Journal of Alloys and Compounds. 2020. link Times cited: 8 USED (low confidence) S. Starikov, I. Gordeev, Y. Lysogorskiy, L. Kolotova, and S. Makarov, “Optimized interatomic potential for study of structure and phase transitions in Si-Au and Si-Al systems,” Computational Materials Science. 2020. link Times cited: 19 USED (low confidence) A. Morell-Pacheco et al., “Ni coating on 316L stainless steel using cage plasma treatment: Feasibility and swelling studies,” Journal of Nuclear Materials. 2020. link Times cited: 12 USED (low confidence) D. S. Oliveira, M. A. Cotta, and J. E. Padilha, “Interatomic potential for atomistic simulation of self-catalyzed GaAs nanowires growth,” Computational Materials Science. 2020. link Times cited: 5 USED (low confidence) M. I. Pascuet, G. Bonny, G. Monnet, and L. Malerba, “The effect on the mechanical response of Cr and Ni segregation on dislocation lines in bcc Fe,” Journal of Nuclear Materials. 2020. link Times cited: 3 USED (low confidence) Z. Wen, Z. Zou, S.-chao Zhang, and Y.-hong Zhao, “First-principles study of phase stability, elastic and thermodynamic properties of AlCrFeNi medium-entropy alloys,” International Journal of Modern Physics B. 2020. link Times cited: 4 Abstract: We have applied the first-principles method to predict the p… read moreAbstract: We have applied the first-principles method to predict the phase stability, elastic and thermodynamic properties of ternary (AlCrFe, AlCrNi, CrFeNi and AlFeNi) and quaternary (AlCrFeNi) medium-entr... read less USED (low confidence) Y. Xiao, T. Hu, X. Zhao, F. Hu, H. Yang, and C. Li, “Thermo-selenizing to rationally tune surface composition and evolve structure of stainless steel to electrocatalytically boost oxygen evolution reaction,” Nano Energy. 2020. link Times cited: 34 USED (low confidence) E. Fomin and A. Mayer, “Slip of low-angle tilt grain boundary (110) in FCC metals at perpendicular shear,” International Journal of Plasticity. 2020. link Times cited: 13 USED (low confidence) L. Tuan, T. V. Hoan, and N. Duy, “Zero-magnetization FeNi2Mo alloy: An ab initio simulation result,” Computational Materials Science. 2020. link Times cited: 0 USED (low confidence) S. Starikov and V. Tseplyaev, “Two-scale simulation of plasticity in molybdenum: Combination of atomistic simulation and dislocation dynamics with non-linear mobility function,” Computational Materials Science. 2020. link Times cited: 9 USED (low confidence) X. Chen, X. Gao, Y. Zhao, D. Lin, W. Chu, and H. Song, “TensorAlloy: An automatic atomistic neural network program for alloys,” Comput. Phys. Commun. 2020. link Times cited: 10 USED (low confidence) M. Wang, G. W. Zhang, and H. Xu, “Investigation on properties of FeNi intermetallics under pressure by First-principles,” Journal of Physics: Conference Series. 2020. link Times cited: 3 Abstract: In this paper, the structural stabilities, elastic, electron… read moreAbstract: In this paper, the structural stabilities, elastic, electronic and magnetic properties of three binary Fe-Ni intermetallics with different structure under pressure have been systematically investigated by first-principle method based on density functional theory. The results indicated that the lattice parameters and bulk modulus of Fe-Ni compounds at zero pressure match well with other experimental data and available theoretical calculated values. The calculated energy-volume and pressure-volume expressed that all the compounds are mechanical stability under pressure, and the volume variation increase with the increasing iron content under pressure. The shear deformation resistant and volume deformation resistant are estimated by elastic constant Cij and bulk modulus B. Meanwhile, applied pressure improved the ductility of the Fe-Ni compounds, elastic anisotropy of three compounds under pressure are arranged in the following order: Fe3Ni>FeNi>FeNi3. Debye temperature ΘD of compounds gradually increase with the increase of iron contents and pressure, the temperature dependence of the linear thermal expansion coefficient α and heat capacity (CV and CP) are also calculated based on quasi-harmonic Debye model under pressure from 0 to 50GPa and various temperature. The results gain a better understanding of iron-nickel alloys. read less USED (low confidence) P. van Helden et al., “Cobalt-nickel bimetallic Fischer-Tropsch catalysts: A combined theoretical and experimental approach,” Catalysis Today. 2020. link Times cited: 25 USED (low confidence) C. Fu et al., “Structure and thermal expansion of coordination shells in solid and liquid Invar alloys by molecular dynamics study,” Journal of Applied Physics. 2020. link Times cited: 3 Abstract: Classical molecular dynamics simulations have been performed… read moreAbstract: Classical molecular dynamics simulations have been performed to study the atomic structures and thermal expansion of coordination shells in solid and liquid Invar alloys. Analysis of atomic structures reveals that there is an attraction between Fe-Ni nearest pairs, and that structural order still exists in the liquid Invar alloy. Fe—Ni bonds are found to have the smallest thermal expansion in the solid Invar alloy among three types of bonds, which plays an important role in the Invar effect. We also discover that the thermal expansion coefficient will gradually get close to the macroscopic level as the coordination shell number increases in Invar alloys. It is until the 5th coordination shell in the solid state and the 4th coordination shell in the liquid state that the thermal expansions of the coordination shells can reach the macroscale value. This study further promotes the understanding of the thermal expansions from the atomic scale.Classical molecular dynamics simulations have been performed to study the atomic structures and thermal expansion of coordination shells in solid and liquid Invar alloys. Analysis of atomic structures reveals that there is an attraction between Fe-Ni nearest pairs, and that structural order still exists in the liquid Invar alloy. Fe—Ni bonds are found to have the smallest thermal expansion in the solid Invar alloy among three types of bonds, which plays an important role in the Invar effect. We also discover that the thermal expansion coefficient will gradually get close to the macroscopic level as the coordination shell number increases in Invar alloys. It is until the 5th coordination shell in the solid state and the 4th coordination shell in the liquid state that the thermal expansions of the coordination shells can reach the macroscale value. This study further promotes the understanding of the thermal expansions from the atomic scale. read less USED (low confidence) D. Smirnova et al., “Atomistic description of self-diffusion in molybdenum: A comparative theoretical study of non-Arrhenius behavior,” Physical Review Materials. 2020. link Times cited: 16 Abstract: According to experimental observations, the temperature depe… read moreAbstract: According to experimental observations, the temperature dependence of self-diffusion coefficient in most body-centered cubic metals (bcc) exhibits non-Arrhenius behavior. The origin of this behavio ... read less USED (low confidence) E. Kirova and V. Pisarev, “System size effect on crystal nuclei morphology in supercooled metallic melt,” Journal of Crystal Growth. 2019. link Times cited: 8 USED (low confidence) S. Starikov, M. Mrovec, and R. Drautz, “Study of Grain Boundary Self-Diffusion in Iron with Different Atomistic Models,” MatSciRN: Computational Studies of Inorganic & Organic Materials (Topic). 2019. link Times cited: 22 USED (low confidence) T. Todorova, M. Gaier, J. Zwanziger, and K. Plucknett, “Understanding the elastic and thermal response in TiC-based ceramic-metal composite systems: First-principles and mechanical studies,” Journal of Alloys and Compounds. 2019. link Times cited: 14 USED (low confidence) J. Fürnkranz, “Publication list,” Journal of Physics: Conference Series. 2019. link Times cited: 10 Abstract: PUBLICATION LIST of VICTOR MANUEL VILLANUEVA SANDOVAL availa… read moreAbstract: PUBLICATION LIST of VICTOR MANUEL VILLANUEVA SANDOVAL available in this PDF. read less USED (low confidence) C. Yang and L. Qi, “Modified embedded-atom method potential of niobium for studies on mechanical properties,” Computational Materials Science. 2019. link Times cited: 17 USED (low confidence) H. Bhattarai, K. E. Newman, and J. Gezelter, “Polarizable potentials for metals: The density readjusting embedded atom method (DR-EAM),” Physical Review B. 2019. link Times cited: 6 Abstract: In simulations of metallic interfaces, a critical aspect of … read moreAbstract: In simulations of metallic interfaces, a critical aspect of metallic behavior is missing from the some of the most widely used classical molecular dynamics force fields. We present a modification of the embedded atom method (EAM) which allows for electronic polarization of the metal by treating the valence density around each atom as a fluctuating dynamical quantity. The densities are represented by a set of additional fluctuating variables (and their conjugate momenta) which are propagated along with the nuclear coordinates. This ``density readjusting EAM'' (DR-EAM) preserves nearly all of the useful qualities of traditional EAM, including bulk elastic properties and surface energies. However, it also allows valence electron density to migrate through the metal in response to external perturbations. We show that DR-EAM can successfully model polarization in response to external charges, capturing the image charge effect in atomistic simulations. DR-EAM also captures some of the behavior of metals in the presence of uniform electric fields, predicting surface charging and shielding internal to the metal. We further show that it predicts charge transfer between the constituent atoms in alloys, leading to novel predictions about unit cell geometries in layered $\mathrm{L}{1}_{0}$ structures. read less USED (low confidence) L. Belkacemi, E. Meslin, B. Décamps, B. Radiguet, and J. Henry, “Radiation-induced bcc-fcc phase transformation in a Fe 3%Ni alloy,” Acta Materialia. 2018. link Times cited: 28 USED (low confidence) D. Smirnova, S. Starikov, and A. Vlasova, “New interatomic potential for simulation of pure magnesium and magnesium hydrides,” Computational Materials Science. 2018. link Times cited: 17 USED (low confidence) G. Giannopoulos et al., “L10-FeNi films on Au-Cu-Ni buffer-layer: a high-throughput combinatorial study,” Scientific Reports. 2018. link Times cited: 13 USED (low confidence) A. Mangla, G. Deo, and P. A. Apte, “NiFe local ordering in segregated Ni3Fe alloys: A simulation study using angular dependent potential,” Computational Materials Science. 2018. link Times cited: 8 USED (low confidence) B. Beeler, Y. Zhang, M. Okuniewski, and C. Deo, “Calculation of the displacement energy of α and γ uranium,” Journal of Nuclear Materials. 2018. link Times cited: 18 USED (low confidence) D. Smirnova, S. Starikov, and I. Gordeev, “Evaluation of the structure and properties for the high-temperature phase of zirconium from the atomistic simulations,” Computational Materials Science. 2018. link Times cited: 13 USED (low confidence) E. Schmidt, A. T. Fowler, J. Elliott, and P. Bristowe, “Learning models for electron densities with Bayesian regression,” Computational Materials Science. 2018. link Times cited: 12 USED (low confidence) H. Jiang, “Theoretical Models for Bimetallic Surfaces and Nanoalloys.” 2018. link Times cited: 0 USED (low confidence) D. Pandey, K. Ray, R. Bhardwaj, S. Bojja, K. Chary, and G. Deo, “Promotion of unsupported nickel catalyst using iron for CO2 methanation,” International Journal of Hydrogen Energy. 2018. link Times cited: 56 USED (low confidence) S. Starikov, N. Lopanitsyna, D. Smirnova, and S. Makarov, “Atomistic simulation of Si-Au melt crystallization with novel interatomic potential,” Computational Materials Science. 2018. link Times cited: 20 USED (low confidence) S. Starikov, L. Kolotova, A. Kuksin, D. Smirnova, and V. Tseplyaev, “Atomistic simulation of cubic and tetragonal phases of U-Mo alloy: Structure and thermodynamic properties,” Journal of Nuclear Materials. 2018. link Times cited: 46 USED (low confidence) K. Ray and G. Deo, “A potential descriptor for the CO2 hydrogenation to CH4 over Al2O3 supported Ni and Ni-based alloy catalysts,” Applied Catalysis B-environmental. 2017. link Times cited: 72 USED (low confidence) A. Kubo, S. Nagao, and Y. Umeno, “Molecular dynamics study of deformation and fracture in SiC with angular dependent potential model,” Computational Materials Science. 2017. link Times cited: 7 USED (low confidence) Y. Wang, J. Yin, X. Liu, R. Wang, H. Hou, and J. Wang, “Precipitation kinetics in binary Fe–Cu and ternary Fe–Cu–Ni alloys via kMC method,” Progress in Natural Science: Materials International. 2017. link Times cited: 15 USED (low confidence) C.-jun Wu, B.-J. Lee, and X. Su, “Modified embedded-atom interatomic potential for Fe-Ni, Cr-Ni and Fe-Cr-Ni systems,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2017. link Times cited: 60 USED (low confidence) S. Makarov et al., “Efficient Second-Harmonic Generation in Nanocrystalline Silicon Nanoparticles.,” Nano letters. 2017. link Times cited: 139 Abstract: Recent trends to employ high-index dielectric particles in n… read moreAbstract: Recent trends to employ high-index dielectric particles in nanophotonics are motivated by their reduced dissipative losses and large resonant enhancement of nonlinear effects at the nanoscale. Because silicon is a centrosymmetric material, the studies of nonlinear optical properties of silicon nanoparticles have been targeting primarily the third-harmonic generation effects. Here we demonstrate, both experimentally and theoretically, that resonantly excited nanocrystalline silicon nanoparticles fabricated by an optimized laser printing technique can exhibit strong second-harmonic generation (SHG) effects. We attribute an unexpectedly high yield of the nonlinear conversion to a nanocrystalline structure of nanoparticles supporting the Mie resonances. The demonstrated efficient SHG at green light from a single silicon nanoparticle is 2 orders of magnitude higher than that from unstructured silicon films. This efficiency is significantly higher than that of many plasmonic nanostructures and small silicon nanoparticles in the visible range, and it can be useful for a design of nonlinear nanoantennas and silicon-based integrated light sources. read less USED (low confidence) D. Smirnova and S. Starikov, “An interatomic potential for simulation of Zr-Nb system,” Computational Materials Science. 2017. link Times cited: 37 USED (low confidence) P. Pokatashkin, P. Korotaev, and A. Yanilkin, “Amorphization in .ALPHA.-boron: A molecular dynamics study,” Physical Review B. 2017. link Times cited: 3 USED (low confidence) G. Bonny, A. Bakaev, P. Olsson, C. Domain, E. Zhurkin, and M. Posselt, “Interatomic potential to study the formation of NiCr clusters in high Cr ferritic steels,” Journal of Nuclear Materials. 2017. link Times cited: 17 USED (low confidence) P. Korotaev, A. Kuksin, P. Pokatashkin, and A. Yanilkin, “Quantum and classical molecular dynamics simulation of boron carbide behavior under pressure.” 2017. link Times cited: 3 Abstract: We present the study of boron carbide behavior under pressur… read moreAbstract: We present the study of boron carbide behavior under pressure using a multiscale approach. Both quantum and classical molecular dynamics simulations are implemented at this work. Specific phase transitions of boron carbide: chain bending and disordering are discussed and stress-phase diagram is constructed. Interatomic angular dependent potential is obtained. We present a study of grain slipping along amorphous zones, as this phenomenon is to be investigated for the construction of the microscopic model of deformation under shock wave loading. read less USED (low confidence) J. Teeriniemi, M. M. Melander, S. Lipasti, R. Hatz, and K. Laasonen, “Fe-Ni Nanoparticles: A Multiscale First-Principles Study to Predict Geometry, Structure, and Catalytic Activity,” Journal of Physical Chemistry C. 2017. link Times cited: 21 Abstract: Nanoparticles of iron and nickel are promising candidates as… read moreAbstract: Nanoparticles of iron and nickel are promising candidates as nanosized soft magnetic materials and as catalysts for carbon nanotube synthesis and CO methanation, among others. To understand geometry- and size-dependent properties of these nanoparticles, phase diagram of Fe/Ni alloy nanoparticles was calculated by density functional theory and cluster expansion method. Ground state convex is presented for face-centered cubic (FCC), body-centered cubic (BCC), and icosahedral (ICO) particles. Previous experimental observations were explained by using multiscale model for particles with realistic size (diameter ≥2 nm). At size 1.5 nm, geometry changes from BCC at low X(Ni) to icosahedral at high X(Ni). FCC is stabilized over icosahedral geometry by increasing number of atoms from 561 to 923. In large FCC particles, there is enrichment of Fe atoms from core to shell beneath surface, while surface and core are enriched by Ni atoms. Catalytic enhancement effect in CO methanation was found to be due to Ni incorpo... read less USED (low confidence) H. Fang et al., “Product tunable behavior of carbon nanotubes-supported Ni?Fe catalysts for guaiacol hydrodeoxygenation,” Applied Catalysis A-general. 2017. link Times cited: 135 USED (low confidence) V. Kuznetsov, “Fe-Ni Binary Phase Diagram Evaluation,” MSI Eureka. 2016. link Times cited: 0 USED (low confidence) Z. Niu and L. Cai, “Ab initio phase stability of some cubic phases of ordered Ni-Fe alloys at high temperatures and pressures,” Computational Materials Science. 2016. link Times cited: 6 USED (low confidence) N. Pandya, A. Mevada, and P. Gajjar, “Lattice dynamical and thermodynamic properties of FeNi3, FeNi and Fe3Ni invar materials,” Computational Materials Science. 2016. link Times cited: 29 USED (low confidence) Y. Duan and H. Guan, “Chapter 3 Fe-Based Composite Absorbers.” 2016. link Times cited: 0 USED (low confidence) L. Ding et al., “Crystal Structures, Stabilities, Electronic Properties, and Hardness of MoB2: First-Principles Calculations.,” Inorganic chemistry. 2016. link Times cited: 37 Abstract: On the basis of the first-principles techniques, we perform … read moreAbstract: On the basis of the first-principles techniques, we perform the structure prediction for MoB2. Accordingly, a new ground-state crystal structure WB2 (P63/mmc, 2 fu/cell) is uncovered. The experimental synthesized rhombohedral R3̅m and hexagonal AlB2, as well as theoretical predicted RuB2 structures, are no longer the most favorite structures. By analyzing the elastic constants, formation enthalpies, and phonon dispersion, we find that the WB2 phase is thermodynamically and mechanically stable. The high bulk modulus B, shear modulus G, low Poisson's ratio ν, and small B/G ratio are benefit to its low compressibility. When the pressure is 10 GPa, a phase transition is observed between the WB2-MoB2 and the rhombohedral R3̅m MoB2 phases. By analyzing the density of states and electron density, we find that the strong covalent is formed in MoB2 compounds, which contributes a great deal to its low compressibility. Furthermore, the low compressibility is also correlated with the local buckled structure. read less USED (low confidence) S. Hocker, M. Hummel, P. Binkele, H. Lipp, and S. Schmauder, “Molecular dynamics simulations of tensile tests of Ni-, Cu-, Mg- and Ti-alloyed aluminium nanopolycrystals,” Computational Materials Science. 2016. link Times cited: 12 USED (low confidence) D. Pandey and G. Deo, “Determining the Best Composition of a Ni–Fe/Al2O3 Catalyst used for the CO2 Hydrogenation Reaction by Applying Response Surface Methodology,” Chemical Engineering Communications. 2016. link Times cited: 13 Abstract: Response surface methodology (RSM) with central composite de… read moreAbstract: Response surface methodology (RSM) with central composite design (CCD) was applied to determine the composition of an alumina-supported nickel-iron (Ni–Fe) catalyst that provided the highest CH4 yield for the CO2 hydrogenation reaction. This involved synthesis of alumina-supported Ni–Fe catalysts of compositions that were specified by CCD. The catalysts were then tested for the CO2 hydrogenation reaction, and a model equation was developed that related the catalyst composition to the CH4 yield. The model equation was validated by analysis of variance, and it was found to adequately represent the experimental data. The model equation predicted that the alumina-supported Ni–Fe catalyst containing 32.8% Ni and 7.7% Fe would provide the highest CH4 yield. A catalyst with this specific composition and the same metal deposition method and two other catalysts of the same composition but different metal deposition metal were also synthesized, characterized, and tested for the CO2 hydrogenation reaction. The three catalysts did show activities similar to those predicted by the model equation. Furthermore, characterization and reaction studies revealed that the three catalysts were similar, suggesting that the metal deposition methods do not have any effect on the catalytic activity. read less USED (low confidence) A. Kuksin, S. Starikov, D. Smirnova, and V. Tseplyaev, “The diffusion of point defects in uranium mononitride: Combination of DFT and atomistic simulation with novel potential,” Journal of Alloys and Compounds. 2016. link Times cited: 24 USED (low confidence) D. Pandey and G. Deo, “Effect of support on the catalytic activity of supported Ni–Fe catalysts for the CO2 methanation reaction,” Journal of Industrial and Engineering Chemistry. 2016. link Times cited: 101 USED (low confidence) V. Tseplyaev and S. Starikov, “The atomistic simulation of pressure-induced phase transition in uranium mononitride,” Journal of Physics: Conference Series. 2015. link Times cited: 12 Abstract: Phase transition in uranium mononitride (UN) at high pressur… read moreAbstract: Phase transition in uranium mononitride (UN) at high pressure has been studied using molecular dynamics. At low pressure, UN has the cubic structure like NaCl (with the space group Fm3̅m). The research based on Gibbs energy calculation shows that cubic UN turns into rhombohedral face-centered structure (with the space group R3̅m) at pressure about 32 GPa. It is shown that parameters of R3̅m-structure change at increasing of the pressure. At various pressures, the parameters of structures with isotropic stress tensor are different. read less USED (low confidence) N. S. Mikhaleva, M. Visotin, Z. Popov, A. Kuzubov, and A. Fedorov, “Ab initio and empirical modeling of lithium atoms penetration into silicon,” Computational Materials Science. 2015. link Times cited: 4 USED (low confidence) D. Smirnova, A. Kuksin, and S. Starikov, “Investigation of point defects diffusion in bcc uranium and U–Mo alloys,” Journal of Nuclear Materials. 2015. link Times cited: 47 USED (low confidence) Y.-xia Liu, H. Wang, H. Wu, D. Xu, and R. Yang, “A mean-field interatomic potential for a multi-component β-type titanium alloy,” Computational Materials Science. 2014. link Times cited: 2 USED (low confidence) P. Brommer, A. Kiselev, D. Schopf, P. Beck, J. Roth, and H. Trebin, “Classical interaction potentials for diverse materials from ab initio data: a review of potfit,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 76 Abstract: Force matching is an established technique to generate effec… read moreAbstract: Force matching is an established technique to generate effective potentials for molecular dynamics simulations from first-principles data. This method has been implemented in the open source code potfit. Here, we present a review of the method and describe the main features of the code. Particular emphasis is placed on the features added since the initial release: interactions represented by analytical functions, differential evolution as optimization method, and a greatly extended set of interaction models. Beyond the initially present pair and embedded-atom method potentials, potfit can now also optimize angular dependent potentials, charge and dipolar interactions, and electron-temperature-dependent potentials. We demonstrate the functionality of these interaction models using three example systems: phonons in type I clathrates, fracture of α-alumina, and laser-irradiated silicon. read less USED (low confidence) R. Matsumoto, S. Seki, S. Taketomi, and N. Miyazaki, “Hydrogen-related phenomena due to decreases in lattice defect energies—Molecular dynamics simulations using the embedded atom method potential with pseudo-hydrogen effects,” Computational Materials Science. 2014. link Times cited: 18 USED (low confidence) G. Bonny, D. Terentyev, E. Zhurkin, and L. Malerba, “Monte Carlo study of decorated dislocation loops in FeNiMnCu model alloys,” Journal of Nuclear Materials. 2014. link Times cited: 45 USED (low confidence) D. Schopf, H. Euchner, and H. Trebin, “Effective potentials for simulations of the thermal conductivity of type-I semiconductor clathrate systems,” Physical Review B. 2014. link Times cited: 13 USED (low confidence) G. Yang, J. Li, Q. Shi, and L. Kong, “Structural and dynamical properties of heterogeneous solid–liquid Ta–Cu interfaces: A molecular dynamics study,” Computational Materials Science. 2014. link Times cited: 16 USED (low confidence) A. Fedorov, A. A. Kuzubov, N. Eliseeva, Z. Popov, M. Visotin, and N. Galkin, “Theoretical Study of the Lithium Diffusion in the Crystalline and Amorphous Silicon as well as on its Surface,” Solid State Phenomena. 2014. link Times cited: 2 Abstract: Using the PAW DFT-GGA method and numerical solving of master… read moreAbstract: Using the PAW DFT-GGA method and numerical solving of master equation the diffusion rates of lithium atoms inside both crystal and amorphous silicon of LixSi (x= 0..0.5) composition have been calculated for different temperatures. It is shown the diffusion rate for amorphous silicon is ~10 times greater than that for the crystal silicon. For both structures the rate is increased by 1.5-2 orders of magnitude while the lithium concentration is increased up to 0.5 value. This should result in that the LixSi/Si interface will be sharp. This fact has been further confirmed using molecular dynamic calculations based on Angular Dependent Potential (ADP) model. Also binding energies of Li atoms lying on different sites of Si (001) surface as well as the potential barriers for the atom jumps both along the surface and in the subsurface layers have been calculated. The data show the Li atoms move along the surface very easily but their jumps into subsurface layers are very difficult due to the high potential barrier values. read less USED (low confidence) D. Pandey and G. Deo, “Promotional effects in alumina and silica supported bimetallic Ni–Fe catalysts during CO2 hydrogenation,” Journal of Molecular Catalysis A-chemical. 2014. link Times cited: 59 USED (low confidence) J. Wu and G. Yang, “Phase stability and physical properties of technetium borides: A first-principles study,” Computational Materials Science. 2014. link Times cited: 10 USED (low confidence) S. Petegem, J. Zimmermann, S. Brandstetter, X. Sauvage, M. Legros, and H. Swygenhoven, “Microstructure and deformation mechanisms in nanocrystalline Ni–Fe. Part I. Microstructure,” Acta Materialia. 2013. link Times cited: 22 USED (low confidence) J. Michalka, P. McIntyre, and J. Gezelter, “Molecular Dynamics Simulations of the Surface Reconstructions of Pt(557) and Au(557) under Exposure to CO,” Journal of Physical Chemistry C. 2013. link Times cited: 5 Abstract: The mechanism and dynamics of surface reconstructions of Pt(… read moreAbstract: The mechanism and dynamics of surface reconstructions of Pt(557) and Au(557) exposed to various coverages of carbon monoxide (CO) were investigated using molecular dynamics simulations. Metal–CO interactions were parametrized from experimental data and plane-wave density functional theory (DFT) calculations. The large difference in binding strengths of the Pt–CO and Au–CO interactions was found to play a significant role in step-edge stability and adatom diffusion constants. Various mechanisms for CO-mediated step wandering and step doubling were investigated on the Pt(557) surface. We find that the energetics of CO adsorbed to the surface can explain the step-doubling reconstruction observed on Pt(557) and the lack of such a reconstruction on the Au(557) surface. However, more complicated reconstructions into triangular clusters that have been seen in recent experiments were not observed in these simulations. read less USED (low confidence) J. Liu, R. Davidchack, and H. Dong, “Molecular dynamics calculation of solid–liquid interfacial free energy and its anisotropy during iron solidification,” Computational Materials Science. 2013. link Times cited: 41 USED (low confidence) S. Cazottes, A. Fnidiki, M. Coïsson, D. Lemarchand, and F. Danoix, “Correlation between microstructure at fine scale and magnetic properties of magnetoresistive Cu80Fe10Ni10 ribbons: Modeling of magnetization,” Journal of Magnetism and Magnetic Materials. 2013. link Times cited: 1 USED (low confidence) H. Kotan, K. Darling, M. Saber, C. Koch, and R. Scattergood, “Effect of zirconium on grain growth and mechanical properties of a ball-milled nanocrystalline FeNi alloy,” Journal of Alloys and Compounds. 2013. link Times cited: 39 USED (low confidence) G. Zhu, “Recycling of Glass Fibre Reinforced Aluminium Laminates and Silicon Removal from Aerospace Al Alloy.” 2012. link Times cited: 0 Abstract: Aerospace aluminium alloys (7xxx and 2xxx series Al alloy) i… read moreAbstract: Aerospace aluminium alloys (7xxx and 2xxx series Al alloy) is one of the important Al alloys in our life. The recycling of aerospace Al alloy plays a significant role in sustainable development of Al industry. The fibre reinforced metal laminates GLARE including 67 wt.% 2024 Al alloy was used as upper fuselage in Airbus A380, but the solution for GLARE recycling is not available. Thermal recycling which uses high temperature to decompose the resin and separate the reinforcement fibres and fillers, has been used in the thermal delamination of Lacomet (one member of fibre metal laminates family). Similar to the thermal recycling of Lacomet, a practical solution for GLARE recycling in laboratory-scale was developed in this research.The recycling of GLARE consists of two steps. The first step is the separation of S2-glass fibre and 2024 Al sheets after the decomposition of resins in GLARE under thermal condition, the decomposition behaviour as well as the decomposition kinetics of resins in GLARE were studied. The second step is the re-melting and refining of the separated 2024 Al sheets, the critical influence factors such as the salt flux composition, refining temperature, the size of 2024 Al scrap and the ratio of salt flux to scarp were discussed in this research. Si is a harmful impurity for aerospace Al alloys, and Si concentration is strictly controlled in aerospace Al alloys. But a small amount of Al alloy scrap with high Si concentration is usually mixed together with aerospace Al alloy scrap during the recycling of aerospace Al alloys, thus Si concentration in final secondary aerospace alloys exceeds the upper limit of nominal concentration. Besides the improvement of the efficiency of scrap classification, the removal of impurity Si from recycled aerospace Al alloys is also an option to improve the quality of secondary aerospace Al alloys. In this research, the first attempt on Si removal from Al in laboratory-scale was conducted by using Ti addition method. read less USED (low confidence) D. Wang, B. Wang, and Y. Wang, “New Crystal Structures of IrB and IrB2: First-Principles Calculations,” Journal of Physical Chemistry C. 2012. link Times cited: 17 Abstract: Superhard IrB1.35 [Chem. Mater.2009, 21, 1407] and IrB1.1 fi… read moreAbstract: Superhard IrB1.35 [Chem. Mater.2009, 21, 1407] and IrB1.1 film [ACS Appl. Mater. Interfaces2010, 2, 581] have been synthesized in experiment, but the structural formulas of iridium borides with integral ratio between Ir and B atoms are still undefined up to now. Here, we use a combination of particle-swarm optimization technique and first-principles calculations to explore the crystal structures of IrB and IrB2. We demonstrate that the new phase P1–IrB belongs to the orthorhombic Pnma space group, while P5–IrB2 (space group Pmmn) has a same structure type with OsB2. At the pressure of about 5 GPa, a phase transition occurs between the Pnma and anti-NiAs phases for P1–IrB. Further phonon and elastic constants calculations imply that both P1–IrB and P5–IrB2 are dynamically and mechanically stable and are potential low compressible materials because of their high bulk moduli. The analysis of density of states and chemical bonding indicates that the formation of strong covalent bonding in these compounds cont... read less USED (low confidence) H. Kotan, M. Saber, C. Koch, and R. Scattergood, “Effect of annealing on microstructure, grain growth, and hardness of nanocrystalline Fe–Ni alloys prepared by mechanical alloying,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2012. link Times cited: 46 USED (low confidence) Y. Zhang, D. Yu, and K. Wang, “Atomistic Simulation of the Orientation-dependent Plastic Deformation Mechanisms of Iron Nanopillars,” Journal of Materials Science & Technology. 2012. link Times cited: 9 USED (low confidence) R. Subbaraman and S. Sankaranarayanan, “Momentum induced coalescence and alloying of Fe-Ni nanoclusters: A molecular dynamics simulation study,” Chemical Physics Letters. 2012. link Times cited: 4 USED (low confidence) D. Connétable, M. Mathon, and J. Lacaze, “First principle energies of binary and ternary phases of the Fe–Nb–Ni–Cr system,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2011. link Times cited: 41 USED (low confidence) B. Wang, X. Li, Y. Wang, and Y. Tu, “Phase Stability and Physical Properties of Manganese Borides: A First-Principles Study,” Journal of Physical Chemistry C. 2011. link Times cited: 50 Abstract: The thermodynamic and mechanical stabilities for the Mn–B sy… read moreAbstract: The thermodynamic and mechanical stabilities for the Mn–B system are investigated using the first-principles calculations method with density functional theory. The negative formation enthalpies of Mn2B–Al2Cu (Mn2B–Al2Cu represents Mn2B in the Al2Cu structure type, the same hereinafter), MnB–CrB, MnB–FeB, MnB2–ReB2, MnB2–AlB2, MnB3–TcP3, and MnB4 indicate that they are thermodynamically stable at zero pressure. It is found that MnB2–ReB2 is more energetically favorable than synthetic MnB2–AlB2, which indicates that experimental synthesized MnB2 is a metastable phase. Among these studied compounds, monoclinic MnB4 has the largest shear modulus, the largest Young’s modulus, and the smallest Poisson’s ratio. The results of density of states and Mulliken overlap population reveal the strong covalent bonding, which results in the high bulk and shear moduli as well as small Poisson’s ratio of MnB2–ReB2 and MnB4. An analysis of the elastic constants, elastic moduli, formation enthalpy, electronic structure, and ... read less USED (low confidence) S. Petegem, J. Zimmermann, and H. Swygenhoven, “Yield point phenomenon during strain rate change in nanocrystalline Ni–Fe,” Scripta Materialia. 2011. link Times cited: 15 USED (low confidence) B. Jelinek et al., “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Physical Review B. 2011. link Times cited: 218 Abstract: A set of modified embedded-atom method (MEAM) potentials for… read moreAbstract: A set of modified embedded-atom method (MEAM) potentials for the interactions between Al, Si, Mg, Cu, and Fe was developed from a combination of each element's MEAM potential in order to study metal alloying. Previously published MEAM parameters of single elements have been improved for better agreement to the generalized stacking fault energy (GSFE) curves when compared with ab initio generated GSFE curves. The MEAM parameters for element pairs were constructed based on the structural and elastic properties of element pairs in the NaCl reference structure garnered from ab initio calculations, with adjustment to reproduce the ab initio heat of formation of the most stable binary compounds. The new MEAM potentials were validated by comparing the formation energies of defects, equilibrium volumes, elastic moduli, and heat of formation for several binary compounds with ab initio simulations and experiments. Single elements in their ground-state crystal structure were subjected to heating to test the potentials at elevated temperatures. An Al potential was modified to avoid formation of an unphysical solid structure at high temperatures. The thermal expansion coefficient of a compound with the composition of AA 6061 alloy was evaluated and compared with experimental values. MEAM potential tests performed in this work, utilizing the universal atomistic simulation environment (ASE), are distributed to facilitate reproducibility of the results. read less USED (low confidence) V. Tatarenko, O. Sobol, D. Leonov, Y. A. Kunyts’kyy, and S. Bokoch, “Statistical Thermodynamics and Physical Kinetics of Structural Changes of Quasi-Binary Solid Solutions Based on the Close-Packed Simple Lattices (According to the Data About Evolution of a Pattern of Scattering of Waves of Various Kinds).” 2011. link Times cited: 2 USED (low confidence) J. Fan, “Applications of Atomistic Simulation in Ceramics and Metals.” 2010. link Times cited: 0 USED (low confidence) J. Fan, “Quantum Mechanics and Its Energy Linkage with Atomistic Analysis.” 2010. link Times cited: 0 USED (low confidence) E. Zhao, J. Meng, Y. Ma, and Z. Wu, “Phase stability and mechanical properties of tungsten borides from first principles calculations.,” Physical chemistry chemical physics : PCCP. 2010. link Times cited: 117 Abstract: The phase stability and mechanical properties of tungsten bo… read moreAbstract: The phase stability and mechanical properties of tungsten borides W(2)B, WB, WB(2), W(2)B(5) and WB(4) were extensively studied by first-principles calculations within density functional theory. The thermodynamic and mechanical stabilities were examined. Our calculations on the enthalpy-pressure relationship and convex hulls have demonstrated that at zero pressure, the experimentally observed W(2)B-W(2)B (W(2)B-W(2)B represents W(2)B in W(2)B structure type, the same hereinafter) and WB-WB, and assumed WB(2)-ReB(2) phases are stable against decomposition into other components. The estimated hardness of WB(2)-ReB(2) is 39.4 GPa, suggesting that it is a potentially hard compound. At 60 GPa, the most stable phases are WB-WB and WB(2)-WB(2). WB-WB, WB(2)-AlB(2) and WB(4) are the ground state phases at 100 GPa. The phase transition mechanism for WB(2) was discussed. The synthesis of WB(2)-AlB(2) could be conducted at high pressures. read less USED (low confidence) S. Simonetti, G. Brizuela, and A. Juan, “Study of the adsorption, electronic structure and bonding of C2H4 on the FeNi(111) surface,” Applied Surface Science. 2010. link Times cited: 4 USED (low confidence) G. Cacciamani, A. Dinsdale, M. Palumbo, and A. Pasturel, “The Fe–Ni system: Thermodynamic modelling assisted by atomistic calculations,” Intermetallics. 2010. link Times cited: 145 USED (low confidence) F. Popa, O. Isnard, I. Chicinaș, and V. Pop, “Synthesis of nanocrystalline Supermalloy powders by mechanical alloying: A thermomagnetic analysis,” Journal of Magnetism and Magnetic Materials. 2010. link Times cited: 28 USED (low confidence) S. Barabash, R. Chepulskii, V. Blum, and A. Zunger, “First-principles determination of low-temperature order and ground states of Fe-Ni, Fe-Pd, and Fe-Pt,” Physical Review B. 2009. link Times cited: 43 USED (low confidence) L. Sandoval, H. Urbassek, and P. Entel, “Solid-solid phase transitions and phonon softening in an embedded-atom method model for iron,” Physical Review B. 2009. link Times cited: 36 USED (low confidence) P. Olsson, “Semi-empirical atomistic study of point defect properties in BCC transition metals,” Computational Materials Science. 2009. link Times cited: 61 USED (low confidence) S. Vitta, V. Sinha, and D. Bahadur, “Magnetic properties of (Fe)1−x–(Al2O3)x and (Fe50Ni50)1−x–(Al2O3)x nanocomposite magnetic media synthesized using gel like Al2O3 matrix,” Journal of Alloys and Compounds. 2009. link Times cited: 1 USED (low confidence) S. Cazottes, F. Danoix, A. Fnidiki, D. Lemarchand, and M. Baricco, “Influence of structural parameters on magnetoresistive properties of CuFeNi melt spun ribbons.,” Ultramicroscopy. 2009. link Times cited: 11 USED (low confidence) M. Mathon, D. Connétable, B. Sundman, and J. Lacaze, “Calphad-type assessment of the Fe–Nb–Ni ternary system,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2009. link Times cited: 40 USED (low confidence) J. Keyzer, G. Cacciamani, N. Dupin, and P. Wollants, “Thermodynamic modeling and optimization of the Fe–Ni–Ti system,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2009. link Times cited: 102 USED (low confidence) G. Rahman and I. Kim, “A First-principles Study on Magnetic and Electronic Properties of Ni Impurity in bcc Fe,” Journal of Magnetics. 2008. link Times cited: 9 Abstract: The magnetic and electronic properties of Ni impurity in bcc… read moreAbstract: The magnetic and electronic properties of Ni impurity in bcc Fe (Ni₁Fe 26 ) are investigated using the full potential linearized augmented plane wave (FLAPW) method based the generalized gradient approximation (GGA). We found that the Ni impurity in bcc Fe increases both the lattice constant and the magnetic moment of bcc Fe. The calculated equilibrium lattice constant of Ni₁Fe 26 in the ferromagnetic state was 2.84 A, which is slightly larger than that of bcc Fe (2.83 A). The averaged magnetic moment per atom of Ni₁Fe 26 unit cell was calculated to be 2.24 μ B , which is greater than that of bcc Fe (2.17 μ B ). The enhancement of magnetic moment of Ni₁Fe 26 is mainly contributed by the nearest neighbor Fe atom of Ni, i.e., Fe1, and this can be explained by the spin flip of Fe1 d states. The density of states shows that Ni impurity forms a virtual bound state (VBS), which is contributed by Ni e g↓ states. We suggest that the VBS caused by the Ni impurity is responsible for the spin flip of Fe1 d states. read less USED (low confidence) J. Li, Y. Dai, X. Dai, T. Wang, and B. Liu, “Development of n-body potentials for hcp–bcc and fcc–bcc binary transition metal systems,” Computational Materials Science. 2008. link Times cited: 20 USED (low confidence) S. Olivier, R. Conte, and A. Fortunelli, “Derivation of an empirical potential for gold with angular corrections,” Physical Review B. 2008. link Times cited: 13 Abstract: From a detailed analysis of density-functional calculations … read moreAbstract: From a detailed analysis of density-functional calculations on gold model clusters and surfaces, an empirical potential for gold, which includes angular corrections, is derived. This potential introduces higher-order nonlinear terms (specifically, the product dipole-quadrupole) that do not seem to have been previously used, but that are necessary to describe directionality effects in the gold-gold interaction. Preliminary tests show that the proposed empirical potential possesses novel features with respect to the existing ones, such as a strong tendency of small Au clusters toward cage configurations, and represents a good starting point for future investigations. read less USED (low confidence) S. Vitta, A. Khuntia, G. Ravikumar, and D. Bahadur, “Electrical and magnetic properties of nanocrystalline Fe100-xNix alloys,” Journal of Magnetism and Magnetic Materials. 2008. link Times cited: 42 USED (low confidence) A. Ruban, S. Khmelevskyi, P. Mohn, and B. Johansson, “Magnetic state, magnetovolume effects, and atomic order in Fe65Ni35 Invar alloy : A first principles study,” Physical Review B. 2007. link Times cited: 51 Abstract: We employ the locally self-consistent Green's function … read moreAbstract: We employ the locally self-consistent Green's function technique and exact muffin-tin orbital method to investigate magnetic state and ground state properties of Invar Fe65Ni35 alloy. We show that ... read less USED (low confidence) G. Spanos et al., “A methodology to aid in the design of naval steels: Linking first principles calculations to mesoscale modeling,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2007. link Times cited: 17 USED (low confidence) X. Zhong, J. Zhu, A. Zhang, and S. Mou, “Investigation of electronic structures of ordered and disordered Ni3Fe by electron energy loss spectroscopy,” Applied Physics Letters. 2006. link Times cited: 5 Abstract: The electronic structures of ordered and disordered Ni3Fe ha… read moreAbstract: The electronic structures of ordered and disordered Ni3Fe have been investigated by electron energy loss spectroscopy. The threshold energies of the L2,3 edges of Fe, and then Ni, are different in the two phases and the white-line ratios L3∕L2 of Ni and Fe from ordered Ni3Fe are lower than those from disordered Ni3Fe, while the 3d occupancies of Ni and Fe atoms in ordered Ni3Fe are less than those in disordered Ni3Fe. The s-p-d rehybridization and the intraband redistribution of the d-band electrons of Ni and Fe atoms are suggested to explain the authors’ experimental results. read less USED (low confidence) G. Cacciamani, J. Keyzer, R. Ferro, U. E. Klotz, J. Lacaze, and P. Wollants, “Critical evaluation of the Fe-Ni, Fe-Ti and Fe-Ni-Ti alloy systems,” Intermetallics. 2006. link Times cited: 166 USED (low confidence) H. Chamati, N. Papanicolaou, Y. Mishin, and D. Papaconstantopoulos, “Embedded-atom potential for Fe and its application to self-diffusion on Fe(1 0 0),” Surface Science. 2006. link Times cited: 178 USED (low confidence) A. Lewis et al., “Two- and three-dimensional microstructural characterization of a super-austenitic stainless steel,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2006. link Times cited: 98 USED (low confidence) F. Wang et al., “Atomic-scale simulations in multi-component alloys and compounds: A review on advances in interatomic potential,” Journal of Materials Science & Technology. 2023. link Times cited: 9 USED (low confidence) W. Wang et al., “Big data-assisted digital twins for the smart design and manufacturing of advanced materials: from atoms to products,” Journal of Materials Informatics. 2022. link Times cited: 6 Abstract: Motivated by the ever-increasing wealth of data boosted by n… read moreAbstract: Motivated by the ever-increasing wealth of data boosted by national strategies in terms of data-driven Integrated Computational Materials Engineering (ICME), Materials Genome Engineering, Materials Genome Infrastructures, Industry 4.0, Materials 4.0 and so on, materials informatics represents a unique strategy in revealing the fundamental relationships in the development and manufacturing of advanced materials. Materials developments are becoming ever more integrated with robust data-driven and data-intensive technologies. In the present review, big data-assisted digital twins (DTs) for the smart design and manufacturing of advanced materials are presented from the perspective of the digital thread. In the introduction of the DT design paradigm in the ICME era, the simulation aspects of DT and the data and design infrastructures are discussed. Referring to the simulation and theoretical factors of DTs, high-throughput simulation and automation and artificial intelligence-assisted multiscale atomistic modeling are detailed through several cases studies. With respect to data and data mining technologies, entropy and its application for attribute selection in decision trees are discussed to emphasize knowledge-based modeling, simulation and data analysis in machine learning coherently. Guided by the perspectives and case studies of the digital thread, we present our recent work on the design, manufacturing and product service via big data-assisted DTs for smart design and manufacturing by integrating some of these advanced concepts and technologies. It is believed that big data-assisted DTs for smart design and manufacturing effectively support better products with the application of novel materials by reducing the time and cost of materials design and deployment. read less USED (low confidence) J. F. Troncoso and V. Turlo, “Evaluating the applicability of classical and neural network interatomic potentials for modeling body centered cubic polymorph of magnesium,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 2 Abstract: Magnesium (Mg) is one of the most abundant metallic elements… read moreAbstract: Magnesium (Mg) is one of the most abundant metallic elements in nature and presents attractive mechanical properties in the industry. Particularly, it has a low density and relatively high strength/weight and stiffness/weight ratios, which make it one of the most attractive lightweight metals. However, the huge potential of Mg is restricted by its low ductility, associated with its hexagonal close packed (hcp) structure. This problem can be solved if Mg adopts the body centered cubic (bcc) structure, which is stable at high pressure or in confinement with stiff bcc metals like Nb. Molecular dynamics method is a magnificent tool to study material’s structure and deformation mechanisms at the atomic level, however, requiring accurate interatomic potentials. The majority of the interatomic potentials available in the literature for Mg have only been fitted to the properties of its stable hcp phase. In the present work, we perform systematic study of applicability of currently available Mg potentials to modeling the properties of metastable bcc polymorph of Mg, taking into account cohesive energy curves, elastic constants, stacking fault energies, and phonon dispersion curves. We conclude that the modified embedded atom method (MEAM) potentials are the most suitable for investigating bcc Mg in Mg/Nb nano-composites, while the properties of high-pressure bcc Mg would be better modeled by neural network interatomic potentials after different local atomic environments corresponding to bcc Mg being included into the fitting database. read less USED (low confidence) G. Wang, Y. Xu, P. Qian, and Y. Su, “ADP potential for the Au-Rh system and its application in element segregation of nanoparticles,” Computational Materials Science. 2021. link Times cited: 6 USED (low confidence) С. Волегов, Р. М. Герасимов, and Р. П. Давлятшин, “MODELS OF MOLECULAR DYNAMICS: A REVIEW OF EAM-POTENTIALS. PART 2. POTENTIALS FOR MULTI-COMPONENT SYSTEMS.” 2018. link Times cited: 1 Abstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликова… read moreAbstract: Получена: 18 мая 2018 г. Принята: 25 июня 2018 г. Опубликована: 29 июня 2018 г. В статье представлена вторая часть обзора современных подходов и работ, посвященных построению потенциалов межатомного взаимодействия с использованием методологии погруженного атома (EAM-потенциалы). Эта часть обзора посвящена одной из наиболее остро стоящих проблем в молекулярной динамике – вопросам построения потенциалов, которые были бы пригодны для описания структуры и физико-механических свойств многокомпонентных (в первую очередь – бинарных и тернарных) материалов. Отмечены первые работы, в которых предлагались подходы к построению функций перекрестного взаимодействия для сплавов никеля и меди – как с использованием методологии EAM, так и несколько отличающийся по процедуре построения потенциал типа Финисса-Синклера. Рассматриваются работы, в которых производится сопоставление различных подходов к построению потенциалов, а также к процедуре идентификации их параметров на примере одних и тех же многокомпонентных систем (типа Al-Ni или Cu-Au). Кроме того, особый интерес представляют некоторые тернарные системы, например Fe–Ni–Cr, W–H– He или U–Mo–Xe, которые являются ключевыми для материалов атомной энергетики и которые в последние годы активно изучаются как возможные материалы для использования в термоядерных ректорах. Приведены примеры работ, в которых предлагаются и исследуются потенциалы для описания многокомпонентных систем, пригодных для использования в аэрокосмической промышленности и изготовленных прежде всего на основе никеля. Рассмотрены результаты исследований различных интерметаллических соединений, отмечены работы, в которых при помощи построенного EAM потенциала удалось количественно точно описать фазовые диаграммы соединений и вычислить характеристики фазовых переходов. read less USED (low confidence) S. Chentouf and P. Maugis, “Structural, energetic and dynamical properties of ordered and disordered bcc Fe25at.%Ni alloys: A first-principles study,” Computational Materials Science. 2017. link Times cited: 4 USED (low confidence) N. Pandya, A. Mevada, and P. Gajjar, “Structural, Vibrational and Electronic properties of FeNi,” Materials Today: Proceedings. 2016. link Times cited: 0 USED (low confidence) R. Jones, C. Weinberger, S. Coleman, and G. Tucker, “Introduction to Atomistic Simulation Methods.” 2016. link Times cited: 1 USED (low confidence) C. Müller, S. Parviainen, F. Djurabekova, K. Nordlund, and R. Spolenak, “The as-deposited structure of co-sputtered Cu–Ta alloys, studied by X-ray diffraction and molecular dynamics simulations,” Acta Materialia. 2015. link Times cited: 34 USED (low confidence) C. Müller, “Phase separation in co-sputtered immiscible Cu-Ta alloy films.” 2014. link Times cited: 2 USED (low confidence) S. Seki, R. Matsumoto, Y. Inoue, S. Taketomi, and N. Miyazaki, “Development of EAM Potential for Fe with Pseudo-Hydrogen Effects and Molecular Dynamics Simulation of Hydrogen Embrittlement,” Journal of The Society of Materials Science, Japan. 2012. link Times cited: 6 Abstract: Numerous studies have reported that solute hydrogen atoms an… read moreAbstract: Numerous studies have reported that solute hydrogen atoms and lattice defects have strong interactions, and that hydrogen atoms significantly change the stability and/or mobility of lattice defects. Although molecular dynamics (MD) simulations can treat complicated interactions of various lattice defects, the time scale is insufficient to treat hydrogen diffusion so as to influence the lattice-defect generation and cooperative motion of hydrogen atoms and lattice defects. Here we developed an interatomic potential for Fe with pseudo-hydrogen effects on lattice-defect energies and performed MD simulations of tensile loading. First, we estimated the lattice-defect energies of Fe and hydrogen-trap energies of lattice defects by using first-principle calculations and evaluated the lattice-defect energies under a practical gaseous hydrogen environment. Second, we refitted the existing embedded-atom-method potential for Fe to represent the lattice-defect energies amended by hydrogen effects. Finally, we confirmed that our potential is applicable for various phenomena by estimating the reproducibility of grain-boundary energies that are not employed for potential fitting. Our tensile-loading simulations of a nano specimen show that hydrogen reduces elongation at rupture. read less USED (low confidence) K. Benkert and F. Gähler, “Molecular Dynamics on NEC Vector Systems.” 2007. link Times cited: 2 NOT USED (high confidence) M. Trochet, F. Berthier, and P. Pernot, “Sensitivity analysis and uncertainty propagation for SMA-TB potentials,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (high confidence) K. Li, C. Fu, M. Nastar, F. Soisson, and M. Lavrentiev, “Magnetochemical effects on phase stability and vacancy formation in fcc Fe-Ni alloys,” Physical Review B. 2022. link Times cited: 6 Abstract: We investigate phase stability and vacancy formation in fcc … read moreAbstract: We investigate phase stability and vacancy formation in fcc Fe-Ni alloys over a broad composition-temperature range, via a density functional theory parametrized effective interaction model, which includes explicitly spin and chemical variables. On-lattice Monte Carlo simulations based on this model are used to predict the temperature evolution of the magnetochemical phase. The experimental composition-dependent Curie and chemical order-disorder transition temperatures are successfully predicted. We point out a significant effect of chemical and magnetic orders on the magnetic and chemical transitions, respectively. The resulting phase diagram shows a magnetically driven phase separation around 10-40% Ni and 570-700 K, between ferromagnetic and paramagnetic solid solutions, in agreement with experimental observations. We compute vacancy formation magnetic free energy as a function of temperature and alloy composition. We identify opposite magnetic and chemical disordering effects on vacancy formation in the alloys with 50% and 75% Ni. We find that thermal magnetic effects on vacancy formation are much larger in concentrated Fe-Ni alloys than in fcc Fe and Ni due to a stronger magnetic interaction. read less NOT USED (high confidence) A. Front et al., “Simulation of thermodynamic properties of magnetic transition metals from an efficient tight-binding model: The case of cobalt and beyond,” Physical Review B. 2021. link Times cited: 0 Abstract: Alexis Front, ∗ Georg Daniel Förster, 2, † Van-Truong Tran, … read moreAbstract: Alexis Front, ∗ Georg Daniel Förster, 2, † Van-Truong Tran, Chu-Chun Fu, Cyrille Barreteau, François Ducastelle, and Hakim Amara 5, ‡ Laboratoire d’Etude des Microstructures, ONERA-CNRS, UMR104, Université Paris-Saclay, BP 72, Châtillon Cedex, 92322, France Interfaces, Confinement, Matériaux et Nanostructures (ICMN), CNRS, Université d’Orléans, Orléans, France Université Paris-Saclay, CEA, Service de Recherches de Métallurgie Physique, 91191 Gif-sur-Yvette, France DRF-Service de Physique de l’Etat Condensé, CEA-CNRS, Université Paris-Saclay, F-91191 Gif-sur-Yvette, France Université de Paris, Laboratoire Matériaux et Phénomènes Quantiques (MPQ), CNRS-UMR7162, 75013 Paris, France read less NOT USED (high confidence) Y. Mishin, “Machine-Learning Interatomic Potentials for Materials Science,” Electrical Engineering eJournal. 2021. link Times cited: 103 NOT USED (high confidence) Y.-S. Lin, G. P. P. Pun, and Y. Mishin, “Development of a physically-informed neural network interatomic potential for tantalum,” Computational Materials Science. 2021. link Times cited: 9 NOT USED (high confidence) H. Bhattarai, K. E. Newman, and J. Gezelter, “The role of polarizability in the interfacial thermal conductance at the gold-water interface.,” The Journal of chemical physics. 2020. link Times cited: 3 Abstract: We have studied the interfacial thermal conductance, G, of t… read moreAbstract: We have studied the interfacial thermal conductance, G, of the flat Au(111)-water interface using non-equilibrium molecular dynamics simulations. We utilized two metal models, one based on the embedded atom method (EAM) and the other including metallic polarizability via a density readjusting EAM. These were combined with three popular water models, SPC/E, TIP4P, and TIP4P-FQ, to understand the role of polarizability in the thermal transport process. A thermal flux was introduced using velocity shearing and scaling reverse non-equilibrium molecular dynamics, and transport coefficients were measured by calculating the resulting thermal gradients and temperature differences at the interface. Our primary finding is that the computed interfacial thermal conductance between a bare metal interface and water increases when polarizability is taken into account in the metal model. Additional work to understand the origin of the conductance difference points to changes in the local ordering of the water molecules in the first two layers of water above the metal surface. Vibrational densities of states on both sides of the interface exhibit interesting frequency modulation close to the surface but no obvious differences due to metal polarizability. read less NOT USED (high confidence) Y. Wang, K. Li, F. Soisson, and C. Becquart, “Combining DFT and CALPHAD for the development of on-lattice interaction models: The case of Fe-Ni system,” Physical Review Materials. 2020. link Times cited: 6 Abstract: We present a model of pair interactions on rigid lattice to … read moreAbstract: We present a model of pair interactions on rigid lattice to study the thermodynamic properties of iron-nickel alloys. The pair interactions are fitted at 0 K on ab initio calculations of formation enthalpies of ordered and disordered (special quasi-random) structures. They are also systematically fitted on the Gibbs free energy of the γ Fe-Ni solid solution as described in a CALPHAD (CALculation of PHAse Diagrams) study by Cacciamani et al. This allows the effects of finite temperature, especially those of magnetic transitions, to be accurately described. We show that the ab initio and CALPHAD data for the γ solid solution and for the FeNi 3 -L1 2 ordered phase can be well reproduced, in a large domain of composition and temperature, using first and second neighbor pair interactions which depend on temperature and local alloy composition. The procedure makes it possible to distinguish and separately compare magnetic, chemical and configuration enthalpies and entropies. We discuss the remaining differences between the pair interaction model and CAL-PHAD, which are mainly due to the treatment of the short-range order and configurational entropy of the solid solution. The FCC phase diagram of the Fe-Ni system is determined by Monte Carlo simulations in the semi-grand canonical ensemble and is compared with experimental studies and other models. We especially discuss the stability of the FeNi-L1 0 phase at low temperature. read less NOT USED (high confidence) Y. Wang et al., “A Thermochemical Database from High-throughput First-Principles Calculations and Its Application to Analyzing Phase Evolution in AM-fabricated IN718,” Acta Materialia. 2020. link Times cited: 3 NOT USED (high confidence) G. P. P. Pun, V. Yamakov, J. Hickman, E. Glaessgen, and Y. Mishin, “Development of a general-purpose machine-learning interatomic potential for aluminum by the physically informed neural network method,” Physical Review Materials. 2020. link Times cited: 13 Abstract: Interatomic potentials constitute the key component of large… read moreAbstract: Interatomic potentials constitute the key component of large-scale atomistic simulations of materials. The recently proposed physically-informed neural network (PINN) method combines a high-dimensional regression implemented by an artificial neural network with a physics-based bond-order interatomic potential applicable to both metals and nonmetals. In this paper, we present a modified version of the PINN method that accelerates the potential training process and further improves the transferability of PINN potentials to unknown atomic environments. As an application, a modified PINN potential for Al has been developed by training on a large database of electronic structure calculations. The potential reproduces the reference first-principles energies within 2.6 meV per atom and accurately predicts a wide spectrum of physical properties of Al. Such properties include, but are not limited to, lattice dynamics, thermal expansion, energies of point and extended defects, the melting temperature, the structure and dynamic properties of liquid Al, the surface tensions of the liquid surface and the solid-liquid interface, and the nucleation and growth of a grain boundary crack. Computational efficiency of PINN potentials is also discussed. read less NOT USED (high confidence) P. Yu, G. Zhu, and M. Wen, “Application of grand-canonical ensemble Monte Carlo simulation in metals using cavity-biased method,” Molecular Simulation. 2020. link Times cited: 1 Abstract: ABSTRACT A critic issue of the application of the convention… read moreAbstract: ABSTRACT A critic issue of the application of the conventional grand-canonical Monte Carlo (GCMC) method in high-density systems is the low acceptance ratio of insertion. Previous studies have revealed that this can be overcome by the cavity-biased (CB) insertion method in simulations of vapours, fluids and liquids. Here, we demonstrate that the method is also highly efficient in metals. Using the Fe–H system as an example, we find that the acceptance ratio of inserting H into Fe lattice is increased by several times using the CB GCMC method. The method is more valid than the conventional one at bulk H concentration over 5‰, implying that the CB GCMC method is highly efficient when there are deep traps for H in simulation systems, i.e. dislocations and interfaces. Application of the method in nanocrystalline Fe shows that the CPU time required for obtaining an equilibrium distribution of H is reduced by 60%. read less NOT USED (high confidence) J. A. Flores-Livas, “Crystal structure prediction of magnetic materials,” Journal of Physics: Condensed Matter. 2020. link Times cited: 11 Abstract: We present a methodology to predict magnetic systems using a… read moreAbstract: We present a methodology to predict magnetic systems using ab initio methods. By employing crystal structure method and spin-polarized calculations, we explore the relation between crystalline structures and their magnetic properties. In this work, testbed cases of transition metal alloys (FeCr, FeMn, FeCo and FeNi) are study in the ferromagnetic case. We find soft-magnetic properties for FeCr, FeMn while for FeCo and FeNi hard-magnetic are predicted. In particular, for the family of FeNi, a candidate structure with energy lower than the tetrataenite was found. The structure has a saturation magnetization (Ms) of 1.2 MA m−1, magnetic anisotropy energy (MAE) above 1200 kJ m−3 and hardness value close to 1. Theoretically, this system made of abundant elements could be the right candidate for permanent magnet applications. Comparing with the state-of-the-art (Nd2Fe14B) hard-magnet, (Ms of 1.28 MA m−1 and MAE of 4900 kJ m−3) is appealing to explore this low energy polymorph of FeNi further. Considering the relatively limited number of magnets, predicting a new system may open routes for free rare-earth magnets. Furthermore, the use of the computational algorithm as the one presented in this work, hold promises in this field for which in near future improvements will allow to study numerous complex systems, larger simulations cells and tackled long-range antiferromagnetic cases. read less NOT USED (high confidence) Y. Ikeda, I. Tanaka, J. Neugebauer, and F. Körmann, “Impact of interstitial C on phase stability and stacking-fault energy of the CrMnFeCoNi high-entropy alloy,” Physical Review Materials. 2019. link Times cited: 44 Abstract: Interstitial alloying in CrMnFeCoNi-based high-entropy alloy… read moreAbstract: Interstitial alloying in CrMnFeCoNi-based high-entropy alloys is known to modify their mechanical properties. Specifically, strength can be increased due to interstitial solid-solution hardening, while simultaneously affecting ductility. In this paper, first-principles calculations are carried out to analyze the impact of interstitial C atoms on CrMnFeCoNi in the fcc and the hcp phases. Our results show that C solution energies are widely spread and sensitively depend on the specific local environments. Using the computed solution-energy distributions together with statistical mechanics concepts, we determine the impact of C on the phase stability. C atoms are found to stabilize the fcc phase as compared to the hcp phase, indicating that the stacking-fault energy of CrMnFeCoNi increases due to C alloying. Using our extensive set of first-principles computed solution energies, correlations between them and local environments around the C atoms are investigated. This analysis reveals, e.g., that the local valence-electron concentration around a C atom is well correlated with its solution energy. read less NOT USED (high confidence) E. Schmidt, “Atomistic modelling of precipitation in Ni-base superalloys.” 2019. link Times cited: 0 Abstract: The presence of the ordered γ ′ phase (Ni3Al) in Ni-base sup… read moreAbstract: The presence of the ordered γ ′ phase (Ni3Al) in Ni-base superalloys is fundamental to the performance of engineering components such as turbine disks and blades which operate at high temperatures and loads. Hence for these alloys it is important to optimize their microstructure and phase composition. This is typically done by varying their chemistry and heat treatment to achieve an appropriate balance between γ ′ content and other constituents such as carbides, borides, oxides and topologically close packed phases. In this work we have set out to investigate the onset of γ ′ ordering in Ni-Al single crystals and in Ni-Al bicrystals containing coincidence site lattice grain boundaries (GBs) and we do this at high temperatures, which are representative of typical heat treatment schedules including quenching and annealing. For this we use the atomistic simulation methods of molecular dynamics (MD) and density functional theory (DFT). In the first part of this work we develop robust Bayesian classifiers to identify the γ ′ phase in large scale simulation boxes at high temperatures around 1500 K. We observe significant γ ′ ordering in the simulations in the form of clusters of γ ′-like ordered atoms embedded in a γ host solid solution and this happens within 100 ns. Single crystals are found to exhibit the expected homogeneous ordering with slight indications of chemical composition change and a positive correlation between the Al concentration and the concentration of γ ′ phase. In general, the ordering is found to take place faster in systems with GBs and preferentially adjacent to the GBs. The sole exception to this is the Σ3 (111) tilt GB, which is a coherent twin. An analysis of the ensemble and time lag average displacements of the GBs reveals mostly ‘anomalous diffusion’ behaviour. Increasing the Al content from pure Ni to Ni 20 at.% Al was found to either consistently increase or decrease the mobility of the GB as seen from the changing slope of the time lag displacement average. The movement of the GB can then be characterized as either ‘super’ or ‘sub-diffusive’ and is interpreted in terms of diffusion induced grain boundary migration, which is posited as a possible precursor to the appearance of serrated edge grain boundaries. In the second part of this work we develop a method for the training of empirical interatomic read less NOT USED (high confidence) J. Einsle et al., “Nanomagnetic properties of the meteorite cloudy zone,” Proceedings of the National Academy of Sciences. 2018. link Times cited: 30 Abstract: Significance The cloudy zone is naturally occurring nanocomp… read moreAbstract: Significance The cloudy zone is naturally occurring nanocomposite found in Fe–Ni metal-bearing meteorites. It not only is a potent carrier of paleomagnetic information from the early solar system but also shows promise as a sustainable alternative to rare earth-based permanent magnets. Here we explain how the remarkable magnetic properties of the cloudy zone are linked to its 3D chemical, crystallographic, and magnetic architecture, using a state-of-the-art combination of nanometer to subnanometer resolution tomography and micromagnetic simulations. We discover the mechanism by which paleomagnetic information becomes encoded into the cloudy zone and, inspired by our findings, point toward potential pathways to optimize synthetic analogues of the cloudy zone for industrial applications. Meteorites contain a record of their thermal and magnetic history, written in the intergrowths of iron-rich and nickel-rich phases that formed during slow cooling. Of intense interest from a magnetic perspective is the “cloudy zone,” a nanoscale intergrowth containing tetrataenite—a naturally occurring hard ferromagnetic mineral that has potential applications as a sustainable alternative to rare-earth permanent magnets. Here we use a combination of high-resolution electron diffraction, electron tomography, atom probe tomography (APT), and micromagnetic simulations to reveal the 3D architecture of the cloudy zone with subnanometer spatial resolution and model the mechanism of remanence acquisition during slow cooling on the meteorite parent body. Isolated islands of tetrataenite are embedded in a matrix of an ordered superstructure. The islands are arranged in clusters of three crystallographic variants, which control how magnetic information is encoded into the nanostructure. The cloudy zone acquires paleomagnetic remanence via a sequence of magnetic domain state transformations (vortex to two domain to single domain), driven by Fe–Ni ordering at 320 °C. Rather than remanence being recorded at different times at different positions throughout the cloudy zone, each subregion of the cloudy zone records a coherent snapshot of the magnetic field that was present at 320 °C. Only the coarse and intermediate regions of the cloudy zone are found to be suitable for paleomagnetic applications. The fine regions, on the other hand, have properties similar to those of rare-earth permanent magnets, providing potential routes to synthetic tetrataenite-based magnetic materials. read less NOT USED (high confidence) F. Meneses, A. Pedernera, C. Blanco, N. Bajales, S. Urreta, and P. G. Bercoff, “L10-FeNi ordered phase in AC electrodeposited iron-nickel biphasic nanowires,” Journal of Alloys and Compounds. 2018. link Times cited: 3 NOT USED (high confidence) C. Howells and Y. Mishin, “Angular-dependent interatomic potential for the binary Ni–Cr system,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 26 Abstract: A new interatomic potential has been developed for the Ni–Cr… read moreAbstract: A new interatomic potential has been developed for the Ni–Cr system in the angular-dependent potential (ADP) format by fitting the potential parameters to a set of experimental and first-principles data. The ADP potential reproduces a wide range of properties of both elements as well as binary alloys with reasonable accuracy, including thermal and mechanical properties, defects, melting points of Ni and Cr, and the Ni–Cr phase diagram. The potential can be used for atomistic simulations of solidification, mechanical behavior and microstructure of the Ni-based and Cr-based phases as well as two-phase alloys. read less NOT USED (high confidence) T. Hamaoka and M. Takeguchi, “Displacement fields around Guinier–Preston 1 zones in Al–Cu alloys: investigations using MD and ADF-STEM image simulations,” Philosophical Magazine. 2018. link Times cited: 2 Abstract: ABSTRACT The displacements of atoms around Guinier–Preston 1… read moreAbstract: ABSTRACT The displacements of atoms around Guinier–Preston 1 (GP1) zones in Al–Cu alloys at a temperature of 295 K have been investigated using molecular dynamics (MD) simulations. The magnitude of the displacements at the first Al layer adjacent to a GP1 zone reached 24% strain and this value decreased with distance from the zone to zero at about 40th Al layer. Strain fields near a GP1 zone were evaluated by calculating strain tensors around individual atoms. The obtained strain maps suggested that the equilibrium Cu content in a GP1 zone should be 100%. The atomic configurations determined by MD simulations were utilised for multislice image simulations of annular dark-field scanning transmission electron microscopy (ADF-STEM) to investigate the effect of foil thickness and the depth position of a GP1 zone in a foil on ADF-STEM images. The atomic displacements in ADF-STEM images were smaller than the MD result due to foil thickness and electron channelling. read less NOT USED (high confidence) S. Hocker, H. Lipp, E. Eisfeld, S. Schmauder, and J. Roth, “Precipitation strengthening in Cu-Ni-Si alloys modeled with ab initio based interatomic potentials.,” The Journal of chemical physics. 2018. link Times cited: 3 Abstract: Effective interaction potentials suitable for Cu/δ-Ni2Si and… read moreAbstract: Effective interaction potentials suitable for Cu/δ-Ni2Si and Cu/β-Ni3Si are developed. We optimise the potential parameters of an embedded atom method potential to reproduce forces, energies, and stresses obtained from ab initio calculations. Details of the potential generation are given, and its validation is demonstrated. The potentials are used in molecular dynamics simulations of shear tests to study the interactions of edge dislocations with coherent δ-Ni2Si and β-Ni3Si precipitates embedded in a copper matrix. In spite of significantly different crystallographic structures of copper and δ-Ni2Si which usually result in circumvention of dislocations, we also observed cutting processes in our simulations. Dislocations cut for a specific orientation of the δ-Ni2Si precipitate and in some cases where dislocation loops originating from previous circumvention processes are present in the glide plane. It is found that β-Ni3Si precipitates have a similar effect on precipitation strengthening as δ-Ni2Si. Dislocations usually cut β-Ni3Si but increased coherency strain can lead to circumvention processes. read less NOT USED (high confidence) J. Hwang, H. Lee, and S. Yi, “Formation and Magnetic Properties of Nanocomposites in Rapidly Solidified Fe42Ni41.7C7Si4.5B3.9P0.9 (at%) Ribbons,” Metals and Materials International. 2018. link Times cited: 13 NOT USED (high confidence) L. Hale and C. Becker, “Vacancy dissociation in body-centered cubic screw dislocation cores,” Computational Materials Science. 2017. link Times cited: 9 NOT USED (high confidence) A. Rokhmanenkov, A. Kuksin, and A. Yanilkin, “Simulation of hydrogen diffusion in TiHx structures,” Physics of Metals and Metallography. 2017. link Times cited: 9 NOT USED (high confidence) A. Stukowski, E. Fransson, M. Mock, and P. Erhart, “Atomicrex—a general purpose tool for the construction of atomic interaction models,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 17 Abstract: We introduce atomicrex, an open-source code for constructing… read moreAbstract: We introduce atomicrex, an open-source code for constructing interatomic potentials as well as more general types of atomic-scale models. Such effective models are required to simulate extended materials structures comprising many thousands of atoms or more, because electronic structure methods become computationally too expensive at this scale. atomicrex covers a wide range of interatomic potential types and fulfills many needs in atomistic model development. As inputs, it supports experimental property values as well as ab initio energies and forces, to which models can be fitted using various optimization algorithms. The open architecture of atomicrex allows it to be used in custom model development scenarios beyond classical interatomic potentials while thanks to its Python interface it can be readily integrated e.g., with electronic structure calculations or machine learning algorithms. read less NOT USED (high confidence) K. L. Joshi and S. Chaudhuri, “Empirical force field-based kinetic Monte Carlo simulation of precipitate evolution and growth in Al–Cu alloys,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 12 Abstract: Ability to accelerate the morphological evolution of nanosca… read moreAbstract: Ability to accelerate the morphological evolution of nanoscale precipitates is a fundamental challenge for atomistic simulations. Kinetic Monte Carlo (KMC) methodology is an effective approach for accelerating the evolution of nanoscale systems that are dominated by so-called rare events. The quality and accuracy of energy landscape used in KMC calculations can be significantly improved using DFT-informed interatomic potentials. Using newly developed computational framework that uses molecular simulator LAMMPS as a library function inside KMC solver SPPARKS, we investigated formation and growth of Guiner–Preston (GP) zones in dilute Al–Cu alloys at different temperature and copper concentrations. The KMC simulations with angular dependent potential (ADP) predict formation of coherent disc-shaped monolayers of copper atoms (GPI zones) in early stage. Such monolayers are then gradually transformed into energetically favored GPII phase that has two aluminum layers sandwiched between copper layers. We analyzed the growth kinetics of KMC trajectory using Johnson–Mehl–Avrami (JMA) theory and obtained a phase transformation index close to 1.0. In the presence of grain boundaries, the KMC calculations predict the segregation of copper atoms near the grain boundaries instead of formation of GP zones. The computational framework presented in this work is based on open source potentials and MD simulator and can predict morphological changes during the evolution of the alloys in the bulk and around grain boundaries. read less NOT USED (high confidence) J. Michalka, A. P. Latham, and J. Gezelter, “CO-Induced Restructuring on Stepped Pt Surfaces: A Molecular Dynamics Study,” Journal of Physical Chemistry C. 2016. link Times cited: 7 Abstract: The effects of plateau width and step-edge kinking on carbon… read moreAbstract: The effects of plateau width and step-edge kinking on carbon monoxide (CO)-induced restructuring of platinum surfaces were explored using molecular dynamics (MD) simulations. Platinum crystals displaying four different vicinal surfaces [(321), (765), (112), and (557)] were constructed and exposed to partial coverages of carbon monoxide. Platinum–CO interactions were fit to recent experimental data and density functional theory (DFT) calculations, providing a classical interaction model that captures the atop binding preference on Pt. The differences in Pt–Pt binding strength between edge atoms on the various facets were found to play a significant role in step-edge wandering and reconstruction events. Because the mechanism for step doubling relies on a stochastic meeting of two wandering edges, the widths of the plateaus on the original surfaces were also found to play a role in these reconstructions. On the Pt(321) surfaces, the CO adsorbate was found to assist in reordering the kinked step edges into st... read less NOT USED (high confidence) H. Lu, Z. Liu, X. Yan, D. Li, L. Parent, and H. Tian, “Electron work function–a promising guiding parameter for material design,” Scientific Reports. 2016. link Times cited: 69 NOT USED (high confidence) M. Sansa, F. Ribeiro, A. Dhouib, and G. Tréglia, “Effect of magnetism on surface segregation in FeNi alloys,” Journal of Physics: Condensed Matter. 2016. link Times cited: 1 Abstract: Modelling the segregation of the various chemical species in… read moreAbstract: Modelling the segregation of the various chemical species in the vicinity of crystallographic defects in FeNi alloys is essential because it affects the macroscopic properties of these materials, which are widely used in technological applications. We present here a theoretical study of surface segregation, within a mean-field approach based on the tight-binding Ising model grounded on density functional theory calculations. The most important result is that, although FeNi presents none of the driving forces (i.e. surface energy, size mismatch) which generally favour surface enrichment in the same element in the whole range of concentrations, there exists a wide temperature range in which Ni is found to segregate at the surface irrespective of the concentration. This is due to a complex interplay between magnetic and ordering/phase separation effects. read less NOT USED (high confidence) G. P. P. Pun, K. Darling, L. Kecskes, and Y. Mishin, “Angular-dependent interatomic potential for the Cu–Ta system and its application to structural stability of nano-crystalline alloys,” Acta Materialia. 2015. link Times cited: 92 NOT USED (high confidence) M. Wen, S. Whalen, R. Elliott, and E. Tadmor, “Interpolation effects in tabulated interatomic potentials,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 23 Abstract: Empirical interatomic potentials are widely used in atomisti… read moreAbstract: Empirical interatomic potentials are widely used in atomistic simulations due to their ability to compute the total energy and interatomic forces quickly relative to more accurate quantum calculations. The functional forms in these potentials are sometimes stored in a tabulated format, as a collection of data points (argument–value pairs), and a suitable interpolation (often spline-based) is used to obtain the function value at an arbitrary point. We explore the effect of these interpolations on the potential predictions by calculating the quasi-harmonic thermal expansion and finite-temperature elastic constant of a one-dimensional chain compared with molecular dynamics simulations. Our results show that some predictions are affected by the choice of interpolation regardless of the number of tabulated data points. Our results clearly indicate that the interpolation must be considered part of the potential definition, especially for lattice dynamics properties that depend on higher-order derivatives of the potential. This is facilitated by the Knowledgebase of Interatomic Models (KIM) project, in which both the tabulated data (‘parameterized model’) and the code that interpolates them to compute energy and forces (‘model driver’) are stored and given unique citeable identifiers. We have developed cubic and quintic spline model drivers for pair functional type models (EAM, FS, EMT) and uploaded them to the OpenKIM repository (https://openkim.org). read less NOT USED (high confidence) J. Jalkanen and M. Müser, “Systematic analysis and modification of embedded-atom potentials: case study of copper,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 10 Abstract: In this study, we evaluate the functionals of different embe… read moreAbstract: In this study, we evaluate the functionals of different embedded-atom methods (EAM) by fitting their free parameters to ab-initio results for copper. Our emphasis lies on testing the transferability of the potentials between systems which vary in their spatial dimension and geometry. The model structures encompass zero-dimensional clusters, one-dimensional chains, two-dimensional tilings, and three-dimensional bulk systems. To avoid having to mimic charge transfer, which is outside the scope of conventional EAM potentials, we focus on structures, in which all atoms are symmetrically equivalent. We find that the simple, four-parameter Gupta EAM potential is overall satisfactory. Adding complexity to it decreases the errors on our set of structures only by marginal amounts, unless EAM is modified to depend also on density gradients, higher-order derivatives, or related terms. All tested conventional EAM functions reveal similar problems: the binding energy of closed-packed systems is overestimated in comparison to open or planar geometries, and structures with small coordination tend to be too rigid. These deficiencies can be fixed in terms of a systematically modified embedded-atom method (SMEAM), which reproduces DFT results on bond lengths, binding energies, and stiffnesses or bulk moduli by typically O(1%), O(5%), and O(15%) accuracy, respectively. SMEAM also predicts the radial distribution function of liquid copper quite accurately. Yet, it does not overcome the difficulty to reproduce the elastic tensor elements of a hypothetical diamond lattice. read less NOT USED (high confidence) L. Ding, P. Shao, F. Zhang, X.-F. Huang, and T. Yuan, “Structure, Relative Stabilities, Physical Properties, and Hardness of Osmium Carbides with Various Stoichiometries: First-Principle Investigations,” Journal of Physical Chemistry C. 2015. link Times cited: 14 Abstract: Using the first-principles calculations, the structural feat… read moreAbstract: Using the first-principles calculations, the structural features, mechanical properties, formation enthalpies, electronic properties and hardness of osmium carbides with various stoichometries have been investigated systematically. The structural stability, thermodynamic stability together with mechanical stability show that Re2N–Os2C, OsB2–OsC2, trigonal P3m1 OsC2, trigonal P3m1 OsC3, orthorhombic Cmmm OsC4 and Ru2Ge3–Os2C3 are the most stable structure for each kind of compounds. But, OsB2–OsC2 and Ru2Ge3–Os2C3 are dynamically unstable based on the calculation of phonon dispersion. The formation enthalpies under high pressure indicate that the Re2N–Os2C, P3m1 OsC3, Cmmm OsC4 and Os2Si3–Os2C3 (P4c2) have structural stabilities in the entire range of pressure. While for OsC2, there is a high pressure phase transition exists above 40 GPa. In addition, all the studied osmium carbides exhibit metallic behavior and strong covalent bonding. According to the calculated Vicker hardness based on a semiempirica... read less NOT USED (high confidence) T. Mohri, “First-principles calculations of stability and phase equilibria in the Fe–Ni system,” Journal of Materials Science. 2015. link Times cited: 16 NOT USED (high confidence) D. Smirnova et al., “Atomistic modeling of the self-diffusion in γ-U and γ-U-Mo,” The Physics of Metals and Metallography. 2015. link Times cited: 31 NOT USED (high confidence) P. Pokatashkin, A. Kuksin, and A. Yanilkin, “Angular dependent potential for α-boron and large-scale molecular dynamics simulations,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 9 Abstract: Both quantum mechanical and molecular-dynamics (MD) simulati… read moreAbstract: Both quantum mechanical and molecular-dynamics (MD) simulations of α-boron are done at this work. Angular dependent interatomic potential (ADP) for boron is obtained using force-matching technique. Fitting data are based on ab initio results within −20..100 GPa pressure range and temperatures up to 2000 K. Characteristics of α-boron, obtained using ADP potential such as bond lengths at equilibrium condition, bulk modulus, pressure-volume relations, Gruneisen coefficient, thermal expansion coefficient are in good agreement with both ab initio data, obtained in this work and known experimental data. As an example of application, the propagation of shock waves through a single crystal of α-boron is also explored by large-scale MD simulations. read less NOT USED (high confidence) M. Basire, J. Soudan, and C. Angelie, “Nanothermodynamics of large iron clusters by means of a flat histogram Monte Carlo method.,” The Journal of chemical physics. 2014. link Times cited: 2 Abstract: The thermodynamics of iron clusters of various sizes, from 7… read moreAbstract: The thermodynamics of iron clusters of various sizes, from 76 to 2452 atoms, typical of the catalyst particles used for carbon nanotubes growth, has been explored by a flat histogram Monte Carlo (MC) algorithm (called the σ-mapping), developed by Soudan et al. [J. Chem. Phys. 135, 144109 (2011), Paper I]. This method provides the classical density of states, gp(Ep) in the configurational space, in terms of the potential energy of the system, with good and well controlled convergence properties, particularly in the melting phase transition zone which is of interest in this work. To describe the system, an iron potential has been implemented, called "corrected EAM" (cEAM), which approximates the MEAM potential of Lee et al. [Phys. Rev. B 64, 184102 (2001)] with an accuracy better than 3 meV/at, and a five times larger computational speed. The main simplification concerns the angular dependence of the potential, with a small impact on accuracy, while the screening coefficients S(ij) are exactly computed with a fast algorithm. With this potential, ergodic explorations of the clusters can be performed efficiently in a reasonable computing time, at least in the upper half of the solid zone and above. Problems of ergodicity exist in the lower half of the solid zone but routes to overcome them are discussed. The solid-liquid (melting) phase transition temperature T(m) is plotted in terms of the cluster atom number N(at). The standard N(at)(-1/3) linear dependence (Pawlow law) is observed for N(at) >300, allowing an extrapolation up to the bulk metal at 1940 ±50 K. For N(at) <150, a strong divergence is observed compared to the Pawlow law. The melting transition, which begins at the surface, is stated by a Lindemann-Berry index and an atomic density analysis. Several new features are obtained for the thermodynamics of cEAM clusters, compared to the Rydberg pair potential clusters studied in Paper I. read less NOT USED (high confidence) A. Kubo, J. Wang, and Y. Umeno, “Development of interatomic potential for Nd–Fe–B permanent magnet and evaluation of magnetic anisotropy near the interface and grain boundary,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 9 Abstract: An interatomic potential based on the angular-dependent pote… read moreAbstract: An interatomic potential based on the angular-dependent potential model was developed for the neodymium magnet (Nd2Fe14B) and Nd crystal system. The developed potential very accurately reproduces the complex crystal structure of Nd2Fe14B together with other important crystal properties, including lattice parameters and elastic coefficients. Molecular dynamics simulations using the potential were performed for the Nd/Nd2Fe14B interfaces and a Nd2Fe14B grain boundary to determine their structures at the atomic level. The magnetoelastic anisotropy in the vicinity of the interfaces and the grain boundary was also evaluated. We find that the disorderliness of atomic structures and the magnetoelastic anisotropy strongly depend on the interface and grain boundary orientations. read less NOT USED (high confidence) M. Lavrentiev, J. Wróbel, D. Nguyen-Manh, and S. Dudarev, “Magnetic and thermodynamic properties of face-centered cubic Fe-Ni alloys.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 48 Abstract: A model lattice ab initio parameterized Heisenberg-Landau ma… read moreAbstract: A model lattice ab initio parameterized Heisenberg-Landau magnetic cluster expansion Hamiltonian spanning a broad range of alloy compositions and a large variety of chemical and magnetic configurations has been developed for face-centered cubic Fe-Ni alloys. The thermodynamic and magnetic properties of the alloys are explored using configuration and magnetic Monte Carlo simulations over a temperature range extending well over 1000 K. The predicted face-centered cubic-body-centered cubic coexistence curve, the phase stability of ordered Fe3Ni, FeNi, and FeNi3 intermetallic compounds, and the predicted temperatures of magnetic transitions simulated as functions of alloy composition agree well with experimental observations. Simulations show that magnetic interactions stabilize the face-centered cubic phase of Fe-Ni alloys. Both the model Hamiltonian simulations and ab initio data exhibit a particularly large number of magnetic configurations in a relatively narrow range of alloy compositions corresponding to the occurrence of the Invar effect. read less NOT USED (high confidence) Y. Duan, S. Gu, M. Wen, and Z. Liu, “The evolution of electronic configuration and magnetic characterization of Fe9Ni1, Fe8Ni2 alloy in theoretical calculation,” The European Physical Journal B. 2013. link Times cited: 4 NOT USED (high confidence) B. Wang, D. Wang, Z. Cheng, X. Wang, and Y. Wang, “Phase stability and elastic properties of chromium borides with various stoichiometries.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2013. link Times cited: 22 Abstract: Phase stability is important to the application of materials… read moreAbstract: Phase stability is important to the application of materials. By first-principles calculations, we establish the phase stability of chromium borides with various stoichiometries. Moreover, the phases of CrB3 and CrB4 have been predicted by using a newly developed particle swarm optimization (PSO) algorithm. Formation enthalpy-pressure diagrams reveal that the MoB-type structure is more energetically favorable than the TiI-type structure for CrB. For CrB2, the WB2-type structure is preferred at zero pressure. The predicted new phase of CrB3 belongs to the hexagonal P-6m2 space group and it transforms into an orthorhombic phase as the pressure exceeds 93 GPa. The predicted CrB4 (space group: Pnnm) phase is more energetically favorable than the previously proposed Immm structure. The mechanical and thermodynamic stabilities of predicted CrB3 and CrB4 are verified by the calculated elastic constants and formation enthalpies. The full phonon dispersion calculations confirm the dynamic stability of WB2 -type CrB2 and predicted CrB3. The large shear moduli, large Young's moduli, low Poisson ratios, and low bulk and shear modulus ratios of CrB4-PSC and CrB4-PSD indicate that they are potential hard materials. Analyses of Debye temperature, electronic localization function, and electronic structure provide further understanding of the chemical and physical properties of these borides. read less NOT USED (high confidence) D. Lin, S. S. Wang, D. Peng, M. Li, and X. D. Hui, “An n-body potential for a Zr–Nb system based on the embedded-atom method,” Journal of Physics: Condensed Matter. 2013. link Times cited: 49 Abstract: A novel n-body potential for an Zr–Nb system was developed i… read moreAbstract: A novel n-body potential for an Zr–Nb system was developed in the framework of the embedded-atom method. All the parameters of the constructed potential have been systematically evaluated by fitting to the ground state properties obtained from experimental measurements and first-principles calculations for pure elements and some alloys. It is shown that most of the static thermodynamics properties for Zr and Nb can be well reproduced by using the present potential. Some calculation results based on the present model are even closer to the experimental data than those based on previous potential models. The ground state properties of hypothetical Zr–Nb alloys were also calculated and found to be in agreement with first-principles calculations. Furthermore, the formation energies of random solid solutions of Zr–Nb with lattices of body centered cubic (bcc) and hexagonal close packed (hcp) type were calculated by fitting the energy–volume relations to Rose’s equation of state. These values were compared with those obtained by first-principles calculations based on special quasirandom structure models and the Miedema-ZSL-07 model (the improved Miedema model proposed by Zhang, Sheng and Liu in 2007). It is indicated that our n-body constructed potential for a Zr–Nb alloy provides an effective description for the interaction between the dissimilar ion interactions for hcp–bcc systems. read less NOT USED (high confidence) A. Mirzoev, M. M. Yalalov, D. Mirzaev, and K. Okishev, “Calculations of energies of mixing of atoms in α- and γ-phases of Fe-Ni alloys by ab initio modeling method,” The Physics of Metals and Metallography. 2013. link Times cited: 2 NOT USED (high confidence) S. Cazottes, F. Vurpillot, A. Fnidiki, D. Lemarchand, M. Baricco, and F. Danoix, “Nanometer Scale Tomographic Investigation of Fine Scale Precipitates in a CuFeNi Granular System by Three-Dimensional Field Ion Microscopy,” Microscopy and Microanalysis. 2012. link Times cited: 7 Abstract: The microstructure of Cu80Fe10Ni10 (at. %) granular ribbons … read moreAbstract: The microstructure of Cu80Fe10Ni10 (at. %) granular ribbons was investigated by means of three-dimensional field ion microscopy (3D FIM). This ribbon is composed of magnetic precipitates embedded in a nonmagnetic matrix. The magnetic precipitates have a diameter smaller than 5 nm in the as-spun state and are coherent with the matrix. No accurate characterization of such a microstructure has been performed so far. A tomographic characterization of the microstructure of melt spun and annealed Cu80Fe10Ni10 ribbon was achieved with 3D FIM at the atomic scale. A precise determination of the size distribution, number density, and distance between the precipitates was carried out. The mean diameter for the precipitates is 4 nm in the as-spun state. After 2 h at 350°C, there is an increase of the size of the precipitates, while after 2 h at 400°C the mean diameter of the precipitates decreases. Those data were used as inputs in models that describe the magnetic and magnetoresistive properties of this alloy. read less NOT USED (high confidence) T. Lee, M. Baskes, S. Valone, and J. Doll, “Atomistic modeling of thermodynamic equilibrium and polymorphism of iron,” Journal of Physics: Condensed Matter. 2012. link Times cited: 52 Abstract: We develop two new modified embedded-atom method (MEAM) pote… read moreAbstract: We develop two new modified embedded-atom method (MEAM) potentials for elemental iron, intended to reproduce the experimental phase stability with respect to both temperature and pressure. These simple interatomic potentials are fitted to a wide variety of material properties of bcc iron in close agreement with experiments. Numerous defect properties of bcc iron and bulk properties of the two close-packed structures calculated with these models are in reasonable agreement with the available first-principles calculations and experiments. Performance at finite temperatures of these models has also been examined using Monte Carlo simulations. We attempt to reproduce the experimental iron polymorphism at finite temperature by means of free energy computations, similar to the procedure previously pursued by Müller et al (2007 J. Phys.: Condens. Matter 19 326220), and re-examine the adequacy of the conclusion drawn in the study by addressing two critical aspects missing in their analysis: (i) the stability of the hcp structure relative to the bcc and fcc structures and (ii) the compatibility between the temperature and pressure dependences of the phase stability. Using two MEAM potentials, we are able to represent all of the observed structural phase transitions in iron. We discuss that the correct reproductions of the phase stability among three crystal structures of iron with respect to both temperature and pressure are incompatible with each other due to the lack of magnetic effects in this class of empirical interatomic potential models. The MEAM potentials developed in this study correctly predict, in the bcc structure, the self-interstitial in the 〈110〉 orientation to be the most stable configuration, and the screw dislocation to have a non-degenerate core structure, in contrast to many embedded-atom method potentials for bcc iron in the literature. read less NOT USED (high confidence) W. Zhu, R. Wang, G. Shu, P. Wu, and H. Xiao, “First-principles study of the structure, mechanical properties, and phase stability of crystalline zirconia under high pressure,” Structural Chemistry. 2012. link Times cited: 12 NOT USED (high confidence) H. Hou, R. Wang, J. Wang, X. Liu, G. Chen, and P. Huang, “An analytic bond-order potential for the Fe–Cu system,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 5 Abstract: An angular-dependent analytic bond-order potential (ABOP) fo… read moreAbstract: An angular-dependent analytic bond-order potential (ABOP) for copper and Fe–Cu system was developed, based on the ABOP of pure iron introduced by Müller et al (2007 J. Phys.: Condens. Matter 19 326220). The potential parameters for the present ABOP model of copper were determined by fitting to the experimental data of the basic properties of fcc Cu and ab initio calculated properties of bcc Cu. The model predicts the vacancy formation energy in good agreement with the experimental result, although no vacancy formation information was used in the fitting of the model parameters. The melting point of Cu is also properly reproduced. The Fe–Cu binary system was described by adding two independent cross parameters in the potential model. The cross parameters were fitted using the ab initio data of the formation energies and lattice parameters of fictitious Fe–Cu alloys. The potential was applied to investigate the point defects and small defect clusters in dilute Fe–Cu alloys. The results were compared with the ab initio data and the values obtained with other potentials. read less NOT USED (high confidence) M. Byshkin and M. Hou, “Phase transformations and segregation in Fe–Ni alloys and nanoalloys,” Journal of Materials Science. 2012. link Times cited: 16 NOT USED (high confidence) G. Rahman, I. Kim, and H. Bhadeshia, “A first-principles investigation on the effects of magnetism on the Bain transformation of α-phase FeNi systems,” Journal of Applied Physics. 2012. link Times cited: 9 Abstract: The effects of magnetism on the Bain transformation of α-pha… read moreAbstract: The effects of magnetism on the Bain transformation of α-phase FeNi systems are investigated by using the full potential linearized augmented plane wave method based on the generalized gradient approximation. We found that Ni impurity in bcc Fe increases the lattice constant in the ferromagnetic (FM) states, but not in the nonmagnetic (NM) states. The shear modulus, G, and Young’s modulus, E, of bcc Fe are also increased by raising the concentration of nickel. All the compositions considered show high shear anisotropy, and the ratio of the bulk to shear modulus is greater than 1.75, implying ductility. The mean sound velocities in the [100] directions are greater than in the [110] directions. The Bain transformation, which is a component of martensitic transformation, has also been studied to reveal that NixFe1−x alloys are elastically unstable in the NM states, but not so in the FM states. The electronic structures explain these results in terms of the density of states at the Fermi level. It is evident ... read less NOT USED (high confidence) H. Sheng, M. Kramer, A. Cadien, T. Fujita, and M. Chen, “Highly optimized embedded-atom-method potentials for fourteen fcc metals,” Physical Review B. 2011. link Times cited: 387 Abstract: Highly optimized embedded-atom-method (EAM) potentials have … read moreAbstract: Highly optimized embedded-atom-method (EAM) potentials have been developed for 14 face-centered-cubic (fcc) elements across the periodic table. The potentials were developed by fitting the potential-energy surface (PES) of each element derived from high-precision first-principles calculations. The as-derived potential-energy surfaces were shifted and scaled to match experimental reference data. In constructing the PES, a variety of properties of the elements were considered, including lattice dynamics, mechanical properties, thermal behavior, energetics of competing crystal structures, defects, deformation paths, liquid structures, and so forth. For each element, the constructed EAM potentials were tested against the experiment data pertaining to thermal expansion, melting, and liquid dynamics via molecular dynamics computer simulation. The as-developed potentials demonstrate high fidelity and robustness. Owing to their improved accuracy and wide applicability, the potentials are suitable for high-quality atomistic computer simulation of practical applications. read less NOT USED (high confidence) C. V. Singh, A. J. Mateos, and D. Warner, “Atomistic simulations of dislocation–precipitate interactions emphasize importance of cross-slip,” Scripta Materialia. 2011. link Times cited: 61 NOT USED (high confidence) S. Bokoch and V. Tatarenko, “Interatomic Interactions in F.C.C.-Ni–Fe Alloys.” 2010. link Times cited: 5 Abstract: The authors express their appreciation to Dr. H. M. Zapolsky… read moreAbstract: The authors express their appreciation to Dr. H. M. Zapolsky, Prof. D. Blavette and Prof. D. Ledue (GPM, UMR 6634 CNRS, Universite de Rouen, France) for very constructive discussion of the results obtained in the course of a given work. One of the authors (S. M. B.) acknowledges the partial financial assistance he has received from GPM (Rouen, France) as well as Nanoscience Foundation (Grenoble, France). We thank Prof. M. S. Blanter (Moscow State Academy of Instrumental Engineering and Information Science, Russia), Dr. R. V. Chepulskii (Duke University, U.S.A.), Prof. B. Schonfeld (ETH, Laboratory of Metal Physics and Technology, Switzerland), Dr. G. E. Ice (Oak Ridge National Laboratory, U.S.A.) and Mr. D. Pavlyuchkov (Forschungszentrum Jьlich GmbH, Germany) for kindly providing their publications and communicating important references. We apologize to other researchers, which have actively worked on the various problems related with our work, whose relevant publications are not referenced and discussed in a given paper because of volume limitations. This work has been inspired by discussions at the several congresses and conferences, primarily, European Congress on Advanced Materials and Processes ‘EUROMAT2007’ (10—13 Sept. 2007, Nurnberg, FRG), ‘Contemporary Problems of Metal Physics’ (7—9 Oct. 2008, Kyyiv, Ukraine), etc. read less NOT USED (high confidence) C. V. Singh and D. Warner, “Mechanisms of Guinier–Preston zone hardening in the athermal limit,” Acta Materialia. 2010. link Times cited: 78 NOT USED (high confidence) I. Vernyhora, S. Bokoch, and V. Tatarenko, “Interplay of Magnetic and Structural Properties of F.C.C.-Ni–Fe Alloys: Study of Statistical Thermodynamics and Kinetics by Means of Methods of Computer Simulation.” 2010. link Times cited: 4 NOT USED (high confidence) E. Zhao, J. Wang, J. Meng, and Z. Wu, “Phase stability and mechanical properties of rhenium borides by first‐principles calculations,” Journal of Computational Chemistry. 2010. link Times cited: 33 Abstract: The phase stability and elastic properties of ReB system we… read moreAbstract: The phase stability and elastic properties of ReB system were systematically investigated by use of the density functional theory. The formation enthalpies are negative for Re3B, Re7B3, Re2B, ReB, Re2B3, and ReB2, indicating that they are thermodynamically stable. Re7B3, Re2B, ReB, Re2B3, and ReB2 are mechanically stable. Combining the study of enthalpy and pressure relationship with the convex hull, it was found that the ground state phases are Re3B, Re7B3, and ReB2 at zero pressure, in agreement with the experimental observations. At the pressure of 90 GPa, Re3B, and ReB2 are the most stable phases. © 2010 Wiley Periodicals, Inc. J Comput Chem 2010 read less NOT USED (high confidence) J.-min Zhang, Z.-L. Lin, Y. Zhang, and V. Ji, “Structural stability and theoretical strength of Cu crystal under equal biaxial loading,” Pramana. 2010. link Times cited: 0 NOT USED (high confidence) Y. Mishin, M. Asta, and J. Li, “Atomistic modeling of interfaces and their impact on microstructure and properties,” Acta Materialia. 2010. link Times cited: 418 NOT USED (high confidence) G. Bonny, R. Pasianot, and L. Malerba, “Fitting interatomic potentials consistent with thermodynamics: Fe, Cu, Ni and their alloys,” Philosophical Magazine. 2009. link Times cited: 24 Abstract: In computational materials science, many atomistic methods h… read moreAbstract: In computational materials science, many atomistic methods hinge on an interatomic potential to describe material properties. In alloys, besides a proper description of problem-specific properties, a reasonable reproduction of the experimental phase diagram by the potential is essential. In this framework, two complementary methods were developed to fit interatomic potentials to the thermodynamic properties of an alloy. The first method involves the zero Kelvin phase diagram and makes use of the concept of the configuration polyhedron. The second method involves phase boundaries at finite temperature and is based on the cluster variation method. As an example for both techniques, they are applied to the Fe–Cu, Fe–Ni and Cu–Ni systems. The resulting potentials are compared to those found in the literature and are found to reproduce the experimental phase diagram more consistently than the latter. read less NOT USED (high confidence) G. P. P. Pun and Y. Mishin, “Development of an interatomic potential for the Ni-Al system,” Philosophical Magazine. 2009. link Times cited: 341 Abstract: We construct an interatomic potential for the Ni-Al system w… read moreAbstract: We construct an interatomic potential for the Ni-Al system within the embedded-atom method formalism. The potential is based on previously developed accurate potentials for pure Ni and Al. The cross-interactions are fitted to experimental cohesive energy, lattice parameter and elastic constants of B2-NiAl, as well as to ab initio formation energies of several real or imaginary intermetallic compounds with different crystal structures and chemical compositions. The potential accurately reproduces a variety of physical properties of the NiAl and Ni3Al phases, and shows reasonable agreement with experimental and ab initio data for phase stability across the Ni-Al phase diagram. Most of the properties reproduced by the new potential were not involved in the fitting process, which demonstrates its excellent transferability. Advantages and certain weaknesses of the new potential in comparison with other existing potentials are discussed in detail. The potential is expected to be especially suitable for simulations of heterophase interfaces and mechanical behavior of Ni-Al alloys. read less NOT USED (high confidence) G. Bonny, R. Pasianot, and L. Malerba, “Fe–Ni many-body potential for metallurgical applications,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 119 Abstract: A many-body interatomic potential for the Fe–Ni system is fi… read moreAbstract: A many-body interatomic potential for the Fe–Ni system is fitted, capable of describing both the ferritic and austenitic phase. The Fe–Ni system exhibits two stable ordered intermetallic phases, namely, L10 FeNi and L12 FeNi3, that are key issues to be tackled when creating a Fe–Ni potential consistent with thermodynamics. A procedure, based on a rigid lattice Ising model and the theory of correlation functions space, is developed to address all the intermetallics that are possible ground states of the system. While controlling the ground states of the system, the mixing enthalpy and defect properties were fitted. Both bcc and fcc defect properties are compared with density functional theory calculations and other potentials found in the literature. Finally, the potential is thermodynamically validated by constructing the alloy phase diagram. It is shown that the experimental phase diagram is reproduced reasonably well and that our potential gives a globally improved description of the Fe–Ni system in the whole concentration range with respect to the potentials found in the literature. read less NOT USED (high confidence) P. Jasen, E. González, G. González, L. Moro, and A. Juan, “The effect of carbon on the electronic structure of FeNi alloys with a stacking fault,” physica status solidi (b). 2008. link Times cited: 0 Abstract: The bonding of C to Fe and Ni in Fe50Ni50(L10) alloy with a … read moreAbstract: The bonding of C to Fe and Ni in Fe50Ni50(L10) alloy with a stacking fault (SF) is analyzed using density functional calculations (DFT). The changes in the electronic structure of the bulk alloy upon SF introduction and after C absorption is addressed. C locates in an octahedral site with Ni atoms in its base and capped with Fe atoms. The Fe–Ni and Ni–Ni bonding decrease while the Fe–Fe bonding increases when the SF is introduced. The effect of C is to reduce the Fe–Ni and Ni–Ni overlap population (OP) up to 75% of its original value, while the Fe–Fe OP only changes by 3.6%. Both Fe and Ni bond to C atom at a distance of 1.80 Å, which is close to that in Fe and Ni carbides metallic, clusters or C as a impurity at grain boundaries or dislocations. (© 2008 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) J.-min Zhang, J. Z. Wang, and K. Xu, “MAEAM investigation of the structural stability and theoretical strength of Fe crystals under uniaxial loading,” Crystal Research and Technology. 2008. link Times cited: 1 Abstract: The structural stability and theoretical strength of BCC cry… read moreAbstract: The structural stability and theoretical strength of BCC crystal Fe under uniaxial loading have been investigated with the modified analytic embedded‐atom method (MAEAM). Even if an orthorhombic path is applied, the deformation is spontaneous along the tetragonal path till Milstein modified Born criterion B22‐B23>0 is violated at λ1=0.9064 in the compressive region. The branched orthogonal path with lower compressive stress σ1 and energy E is preferred over the conventional tetragonal Bain path. A stress‐free FCC phase with the local maximum energy of ‐4.2186eV appearing either in compressive region (orthorhombic path) at λ1=0.8923 or in tensile region (tetragonal path) at λ1=1.2619 is unstable and would slip spontaneously into its near neighbor stress‐free mBCT phase with the local minimum energy of ‐4.2270eV. The initial BCC phase with the lowest energy of ‐4.280eV is the most stable in correspondence with the actual behavior of Fe. Furthermore, the stable region ranges from ‐79.7eV/nm3 to 30.6eV/nm3 in the theoretical strength or from 0.9064 to 1.1788 in the stretch λ1 correspondingly. (© 2008 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) S. Cazottes, G. Wang, A. Fnidiki, D. Lemarchand, P. Renault, and F. Danoix, “Transmission electron microscopy and X-ray diffraction study of microstructural evolution in magnetoresistive Cu–Fe–Ni ribbons,” Philosophical Magazine. 2008. link Times cited: 6 Abstract: The evolution of the microstructure of a granular Cu80Fe10Ni… read moreAbstract: The evolution of the microstructure of a granular Cu80Fe10Ni10 (at%) melt-spun ribbon is studied by transmission electron microscopy (TEM), energy-filtered transmission electron microscopy (EFTEM) and X-ray diffraction. This system is interesting as large giant magnetoresistance (GMR) values have been measured for this composition. We have shown the presence of two face-centred cubic phases, an (Fe,Ni)-rich phase and a Cu-rich phase. The lattice parameters of these two phases are close and no diffraction or elastic contrast is involved in displaying the two phases in TEM bright-field mode. With EFTEM imaging, we have shown the presence of a fine-scale (Fe,Ni)-rich precipitation inside the Cu-rich fcc matrix. The precipitates are 2–4 nm in the as-spun state and 4–6 nm after annealing for 2 h at 400°C. The lattice parameter of the Cu-rich phase in the as-spun sample is 0.3608 nm and 0.3610 nm for the (Fe,Ni)-rich phase. After a 24-h annealing treatment at 600°C, the mean diameter of the particle is 20 nm and the lattice parameter of the (Fe,Ni)-rich phase has decreased to 0.3600 nm, while that of the Cu-rich phase has increased to 0.3613 nm, which is consistent with a segregation of Fe and Ni in the precipitates. The composition and volume fraction of the two phases measured for this annealed sample are in good agreement with the Thermocalc® predictions. read less NOT USED (high confidence) T. Radchenko and V. Tatarenko, “Fe–Ni Alloys at High Pressures and Temperatures: Statistical Thermodynamics and Kinetics of the L1_2 or D0_19 Atomic Order.” 2008. link Times cited: 19 NOT USED (high confidence) M. Müller, P. Erhart, and K. Albe, “Analytic bond-order potential for bcc and fcc iron—comparison with established embedded-atom method potentials,” Journal of Physics: Condensed Matter. 2007. link Times cited: 177 Abstract: A new analytic bond-order potential for iron is presented th… read moreAbstract: A new analytic bond-order potential for iron is presented that has been fitted to experimental data and results from first-principles calculations. The angular-dependent functional form allows a proper description of a large variety of bulk, surface and defect properties, including the Bain path, phonon dispersions, defect diffusivities and defect formation energies. By calculating Gibbs free energies of body-centred cubic (bcc) and face-centred cubic (fcc) iron as a function of temperature, we show that this potential is able to reproduce the transitions from α-iron to γ-iron and δ-iron before the melting point. The results are compared to four widely used embedded-atom-method potentials for iron. read less NOT USED (high confidence) Y. Mishin and A. Lozovoi, “Angular-dependent interatomic potential for tantalum,” Acta Materialia. 2006. link Times cited: 70 NOT USED (high confidence) A. Hadj-Larbi, A. Ziane, S. Bouarab, and C. Demangeat, “Effect on alloying at the Fe/Ni(001) interfaces on the interlayer exchange coupling,” The European Physical Journal B - Condensed Matter and Complex Systems. 2006. link Times cited: 3 NOT USED (high confidence) T. Tsuru, “Descriptions of Dislocation via First Principles Calculations,” The Plaston Concept. 2022. link Times cited: 0 NOT USED (high confidence) S. Koch, “Development of RF-MEAM interaction potentials for Fe-Y.” 2019. link Times cited: 0 Abstract: Der Fokus dieser Arbeit lag zunachst auf einer simulationsge… read moreAbstract: Der Fokus dieser Arbeit lag zunachst auf einer simulationsgestutzen Untersuchung uber die Entsteh- ungsmechanismen von Oxidteilchen in ODS-Stahlen. Hierbei bilden empirische Wechselwirkungs- potenziale von Eisen-Yttrium-Sauerstoff (Fe-Y-O) die Grundlage fur eine Beschreibung dieser Oxid- teilchen-Bildungs-Prozesse in Molekulardynamik (MD) Simulationen, die auch Eigenschaften von Versetzungen und anderen Bestrahlungs-Panomenen detailiert zur weiteren Aufklarung behandeln konnen.
Zu diesem Zweck ist das speziell auf die Simulation zugeschnittene Anfitten der o.g. MD Potenziale (hier fur Fe-Y-O) notwendig. Hierzu dienen die zuvor durchgefuhrten ab-initio (DFT) Rechnungen als Daten- referenzgrundlage (z.B. von Phasen oder Defekten) zur Optimierung der Potenzialparameter wahrend des Anfittens, um ein moglichst exaktes MD Potenzial zu erzeugen, dass die ab-initio Daten auf groseren MD Skalen detailgetreu abbildet. Im ersten Drittel dieses Projektes wurden mehrere Potenziale fur die einzelnen Metall-Komponenten, Fe-Fe und Y-Y, erzeugt. Dabei stellte sich heraus, dass etablierte Standardmethoden nicht in der Lage sind genaue Fe-Y Potenziale als Teillosung fur das Fe-Y-O Problem zu erzeugen. Dabei wurde eine Kombination aus dem (M)EAM Modell und zur Optimierung eine LSM gestutzte Software (POTFIT) genutzt. Die Komplexitat des Problems liegt in den richtungsabhangigen Atombindungen, die die hier entwickelten fortgeschrittenen Simulations- und Fitmethoden benotigen.
Im ersten Schritt von drei Schritten (chapter 3) wurden zunachst einmal die Defizite der Standard-Fittechniken evaluiert, indem die wahrend des Fitting-Prozesses gefundenen Parametersets im EAM Formalismus mit der flexiblen Software POTFIT auf ihre Eignung hin grundlich untersucht worden sind. Die hierfur genutzten Fitfunktionen wurden ursprunglich Anfang 2000 von Zhou und Wadley entwickelt. Hierbei liegt die Ursache fur die dann entdeckte Parameterset-Problematik darin, dass zur Beschreibung des Fe-Y Systems das Model aus drei Potentialkomponenten besteht: Fe-Fe, Y-Y und Fe-Y. Fur diese einzelnen Komponenten sind die Potentialparameter erfolgreich angefittet worden mit Bezug zur Gitterkonstante und Bindungs- bzw. Kohasionsenergie (beides mit 1% Genauigkeit bezgl. DFT Rechnungen) sowie zu allen elastischen Konstanten (5% Genauigkeit bezgl. Experimente). All dies unter Zuhilfenahme von Parametersuchraum-beschrankenden Techniken, die zur Einhaltung der oben genannten Eigenschaften dienen und urspurnglich von Johnson & Oh sind. Selbst kompliziertere Defekteigenschaften, wie Zwischengitter- und Leerstellenbildungsenergien wurden erfolgreich angefittet. Das hier entwickelte EAM Potenzial fur Y-Y ist z.B. in der Lage bei Eigenzwischengitteratomen die basal oktaedrische Position von Zwischengitteratomen (ZA) im Yttrium hcp-Gitter als Grundzustand und die Transition eines jeden ZAs aus einer anderen Position, wie zuvor in DFT berechnet, zu reproduzieren.
Zur Bildung des angestrebten Fe-Y Potenzials wurden diese beiden Komponenten, Fe-Fe und Y-Y, zum weiteren Fitten in dem weitgefacherten und komplexen Fe-Y Potzenzialsuchraum genutzt. Die Parametersets wurden mit sogenannten hier entwickelten Hauptparameter (Key Driver) systematisch untersucht. Ein flexibleres Konzept statt der starreren Universal Binding Relations in Abhangigkeit von der Rose Gleichung. Dieser Hauptparameter zeigte eindeutig, dass die Nutzung der Rose Gleichung zur Parametersuchraum-Minimierung den Suchraum dahingehend einschrankt, sodass ein akkurates Anfitten der hier genutzten 900 DFT Datensets nicht mehr moglich ist. Allerdings ist die Orientierung im Parametersuchraum mit dieser Rose Gleichung bei standardmasigen Optimierungsmethoden (wie LSM) unabdingbar, da ohne diese die benotigten globalen Optima fur die Parameter nicht auffindbar sind.
Als aufklarendes Testverfahren zur weiteren Ergrundung dieser Problematik und Prufung zur Eignung fur Fe-Y Potenziale und den anschliesenden Simulationen diente der Versuch, 9 verschiedene Bindungs-energien von Yttrium-Leerstellenclustern mit ansteigender Leerstellenzahl zu reproduzieren. Dieser Test konnte von diesen Potenzialen nur teilweise erfullt werden und wurde auf die fehlende Beschreibung der Bindungswinkelabhangigkeit im Modell zuruckgefuhrt. Die Erweiterung von EAM durch MEAM mit Winkelabhangigkeit ist jedoch keineswegs eine zufriedenstellende Losung, da MEAM alternativlos auf der irrefuhrenden Rose Gleichung beruht. Daher war die Benutzung des ubersichtlicheren EAM Typs aus zwei Grunden nutzlich: 1. MEAM braucht die Rose Gleichung um diesen komplexen Formalismus zu beherrschen mit denselben Problemen wie in EAM, aber dieses grundlegende Problem ist in MEAM deutlich schwerer zu identifizieren als in EAM. 2. Die mit EAM gefundenen, angefitteten Parameter sind eine hervorragende Startparameter-Grundlage fur den verbesserten darauffolgenden RF-MEAM Typ.
Im zweiten Schritt wurde das Problem aus dem ersten Schritt gelost, indem ein modifizierter MEAM Spezialtyp im referenzlosen Format (RF-MEAM) angewandt worden ist. Im Gegensatz zum herkommlichen MEAM wird hier die Rose Gleichung durch mehr DFT Daten und insbesondere einer intelligenteren Machine Learning ahnlichen Genetic Algorithmus (GA) Optimiertechnik ersetzt, die allerdings eine bedachte Startparameterwahl vorraussetzt, womit Schritt 1 wieder ins Spiel kommt. Die genutzte fortgeschrittene MEAMfit Software, die per GA funktioniert, wurde zwischen 2016 und 2017 funktionierend eigens dafur implementiert. Mit den in Schritt 1 gefitteten Parametern und Set-Auswahltechniken konnten die weiterfuhrenden Fits mit optimalen Startparametern durchgefuhrt werden.
Auf dieser Stufe waren diese Fits mit der speziell verbesserten Technik in der Lage ein detailgetreues Fe-Y Potenzial zu generieren, das sowohl alle Phasen (Fe2Y, Fe3Y, Fe5Y, Fe23Y6 und Fe17Y2 sowohl als auch reines Fe und Y) als auch die gesamte Defektdatenbasis mit einer durchschnittlichen Abweichung von ≈11% erfolgreich abbildet. Zusatzlich bestatigend zu dieser allgemeinen Ubereinstimmung wurde konsequenterweise der in Schritt 1 entwickelte Test hervorragend mit einmaliger Genauigkeit bestanden, mit max. 5% Abweichung von den komlexen o.g. Y-Leerstellen Bildungsenergien. Allerdings konnte ein systematischer Fehlertrend aufgespurt werden, der Schwachen in der Fe-Fe Komponente offenbarte. Als Folge dessen wurde umgehend diese Komponente durch ein anderes etabliertes Fe-Fe Potenzial von G. Ackland mit einer extrem genauen Schmelztemperatur (nur 3% Abweichung vom Exp.) ausgetauscht. Mit diesem genauen Potenzial konnte zum ersten Mal die Clusterbildung von gelosten Yttrium Atomen in einer Eisenschmelze erfolgreich per MD Simulation auf atomarer Ebene nachgestellt werden oberhalb von 1750 K. Temperaturen darunter hatten eine Ausscheidungsbildung von Y mit sehr geringer Y-Loslichkeit (<0.1%) in Ubereinstimmung mit den Experimenten zur Folge. Dies wurde durch den Pot. Typ A ermoglicht, der aber die energetische Reihenfolge bei den Fe-Y Phasen nicht ganz genau einhalt. Typ B hingegen halt diese ein, dort fehlt aber die Y-Clusterbildungsneigung. Durch den gebotenen Praxisbezug zur Metallurgie mussen die Loslichkeit und Clusterbildung gleichzeitig in der Simulation genau reproduzierbar sein, was aber weder Typ A noch B kann, was zum Typ A/B Dilemma fuhrt.
Dieses Typ A/B Dilemma (Phasen oder Defekt Genauigkeit) fuhrt zum letzten dritten Schritt (chapter 5). Darin ist zusatzlich die Strukturaufklarung von der Fe17Y2 Phase mit Vergleichen zu exp. EXAFS Spektren unserer Kollaborationspartner vom ISSP (Riga) enthalten. Diese Aufklarung dient auch dazu die fehlenden magnetischen Abhangigkeiten im Potenzial zu kompensieren, da die Phasenreihenfolge mit sehr feinen Energieunterschieden wohl stark von magnetischen Wechselwirkungen gepragt ist. Obwohl Potenzial Typ B diesen (Magnetismus) nicht direkt beachtet, ist es in der Lage das tatsachlich gemessene EXAFS Spektrum grostenteils genau wiederzugeben. Allerdings offenbart eine einzige ausgepragte Phasenverschiebung, dass die angenommene hcp Struktur durch eine unterschwellige rhombohedrale Komponente, die sporadisch in der c-Gitterrichtung auftritt, korrigiert werden muss. AIMD (DFT) Berechnungen in Kooperation mit der University of Edinburgh bestatigen dies und zeigen sogar, dass magnetische Wechselwirkungen diese Strukturmischung stabilisieren. Endgultig bestatigt werden konnte dies mit der genauen EXAFS Spektren Reproduktion mit dem durch AIMD verbesserten nochmals gefitteten Potenzialtyp B, der als neuer Typ C durch AIMD indirekt den Einfluss der magnetischen Wechselwirkungen mit einschliest. Diese erstmalige nahezu deckungsgleiche MD Simulation eines EXAFS Spektrums von einem komplexen metallischen Alloy, hier Fe-Y, stellt eine bisher unerreichte Verbesserung dar. Schlieslich lost Typ C das Typ A/B Dilemma und ernoglicht eine genaue gleichzeitige MD Modellierung von Phasen- und Defekten in Fe-Y – ein Durchbruch in der MD-Potenzialentwicklung. read less NOT USED (high confidence) B. Nayak and F. Meyer, “Tetrataenite in terrestrial rock,” American Mineralogist. 2015. link Times cited: 15 Abstract: Tetrataenite is an equiatomic and highly ordered, non-cubic … read moreAbstract: Tetrataenite is an equiatomic and highly ordered, non-cubic Fe-Ni alloy mineral that forms in meteorites from the distortion of fcc taenite due to extremely slow cooling. The mineral has drawn much attention of the scientific community because of its superb magnetic properties, which may make the phase an alternative to the REE-based permanent magnets. Barring only a few passing mentions, the mineral has never been described from any terrestrial rock. Here we report the characteristics of terrestrial tetrataenite from an ophiolite-hosted Ni-bearing magnetite body from the Indo-Myanmar ranges, northeast India. Although the mineral assemblage surrounding it is very similar to that found in the meteorites, the postulated cooling regimes cannot be similar. The mineral is formed as a consequence of hydrothermal alteration of ferromagnesian minerals of the olivine and pyroxene groups. Iron and nickel were released from the silicates and precipitated in the form of Fe-Ni alloy at low temperature in extremely reducing conditions with a lack of sulfur. Our findings suggest a low-temperature hydrothermal origin of tetrataenite warrants a re-examination of the Fe-Ni phase diagram at low temperatures and puts a question mark on the age-old concept of tetrataenite formation as due solely to extremely slow cooling of fcc taenite in meteorites. It also opens up a new vista for adoption of a hydrothermal route to synthesize this rare material. read less NOT USED (high confidence) D. Schopf, “Effective potentials for numerical investigations of complex intermetallic phases.” 2013. link Times cited: 0 Abstract: The class of Complex Metallic Alloys (CMAs) is interesting f… read moreAbstract: The class of Complex Metallic Alloys (CMAs) is interesting for its wide range of physical properties. There are materials that exhibit high hardness at low density or good corrosion resistance, which is important for technological applications. Other compounds are superconductors, have strong anisotropic transport coefficients or exhibit a novel magnetic memory effect. The theoretical investigation of CMAs is often very challenging because of their inherent complexity and large unit cells with up to several thousand atoms. Molecular dynamics simulations with classical interaction potentials are well suited for this task – they can handle hundreds of thousands of atoms in reasonable time. Such simulations can provide insight into static and dynamic processes at finite temperatures on an atomistic level.
The accuracy of these simulations depends strongly on the quality of the employed interaction potentials. To generate physically relevant potentials the force-matching method can be applied. A computer code called potfit has been developed at the Institute for Theoretical and Applied Physics (ITAP) especially for this task. It uses a large database of quantum-mechanically calculated reference data, forces on individual atoms and cohesive energies, to generate effective potentials. The parameters of the potential are optimized in such a way that the reference data are reproduced as accurately as possible.
The potfit program has been greatly enhanced as part of this thesis. The optimization of analytic potentials, new interaction models as well as a new optimization algorithm were implemented. Potentials for two different complex metallic alloy systems have been generated and used to study their properties with molecular dynamics simulations.
The first system is an approximant to the decagonal Al-Pd-Mn quasicrystal. A potential which can reproduce the cohesive energy with high accuracy was generated. With the help of this potential a refinement of the experimentally poorly determined structure model could be performed.
The second class of potentials was fitted for intermetallic clathrate systems. They have interesting thermoelectric properties which originate from their special structure. Silicon- and germanium-based clathrate potentials were derived and the influence of the complex structure on the thermal conductivity has been studied.
Komplexe Intermetallische Verbindungen (CMAs) sind aufgrund ihrer vielfaltigen physikalischen Eigenschaften sehr interessant fur technologische Anwendungen. Dabei ist z.B. hohe Harte bei geringer Dichte und Korrosionsbestandigkeit wichtig. Neben Supraleitern gibt es Materialien mit anisotropen Transporteigenschaften oder einem neuartigen magnetischen Memory Effekt. Theoretische Untersuchungen von CMAs stellen durch ihre inharente Komplexitat und die riesigen Einheitszellen mit mehreren tausend Atomen oft eine grose Herausforderung dar. Molekulardynamiksimulationen mit effektiven Potenzialen konnen dazu eingesetzt werden; sie ermoglichen die Berechnung von hunderttausenden von Atome in annehmbarer Zeit. Damit kann ein Einblick in sowohl statische als auch dynamische Prozesse auf atomarer Ebene gewonnen werden.
Die Ergebenisse solcher Simulationen hangen jedoch sehr stark von der Qualitat der eingesetzten Wechselwirkung ab. Um physikalisch gerechtfertigte Potenziale zu erzeugen, kann die Force-Matching-Methode angewandt werden. Dazu wurde am Institut fur Theoretische und Angewandte Physik (ITAP) ein Programm mit dem Namen potfit entwickelt. Es verwendet eine grose Datenbank von quantenmechanisch berechneten Referenzgrosen wie z.B. Krafte auf die einzelnen Atome und die Kohasionsenergie, um effektive Potenziale zu generieren. Die freien Parameter des Potenzials werden optimiert, um die Referenzdaten so gut wie moglich zu reproduzieren.
Fur diese Arbeit wurde potfit deutlich erweitert. Es konnen nun analytisch definierte Potenziale optimiert werden, neue Wechselwirkungen wurden implementiert und ein neuer Optimierungsalgorithmus wurde hinzugefugt. Damit wurden effektive Potenziale fur zwei verschiedene CMA Systeme gefittet und deren Eigenschaften mit Molekulardynamik untersucht.
Fur die Approximanten eines decagonalen Al-Pd-Mn Quasikristalls, den Xi-Phasen, wurde ein Potenzial fur die Strukturbestimmung erzeugt. Es kann die Kohasionsenergien verschiedener Strukturen mit groser Genauigkeit wiedergeben. Ein aus experimentellen Daten ungenau bestimmtes Strukturmodell konnte damit erheblich verbessert werden.
Auserdem wurden Potenziale fur Intermetallische Klathrate erzeugt. Diese Systeme besitzen interessante thermoelektrische Eigenschaften aufgrund ihrer speziellen Kafigstruktur. Effektive Wechselwirkungen fur silizium- und germaniumbasierte Klathrate wurden erzeugt. Damit wurde der Einfluss der komplexen Struktur auf die thermische Leitfahigkeit des Gitters untersucht. read less NOT USED (high confidence) S. Sonntag, “Computer simulations of laser ablation from simple metals to complex metallic alloys.” 2011. link Times cited: 10 Abstract: In this work, a method for computer simulations of laser abl… read moreAbstract: In this work, a method for computer simulations of laser ablation in metals is presented. The ambitious task to model the physical processes, that occur on different time and length scales, is overcome to some extent by the combination of two techniques: Molecular dynamics and finite differences. The former is needed to achieve atomistic resolution of the processes involved. Material failure like melting, vaporization or spallation occur on the atomic scale. Light absorption and electronic heat conduction, which plays the major role in metals, is described by a generalized heat conduction equation solved by the finite differences method. From the so-called Two-Temperature Model temperature, density and pressure evolution - both in time and space - can be derived. With this, various studies on laser heated metals were done. For reasons discussed in more detail later, aluminum was chosen as a model system for most simulations on isotropic materials. As a more complex structure, the metallic alloy Al13Co4 was used because of its special material properties. As an approximant to the decagonal phase of Al-Ni-Co, the alloy shows an anisotropy in its transport properties, e.g. an anisotropic heat conduction.
It will be shown, that the model is able to describe the physics in laser heated solids on time scales from 100 fs up to the ns-scale properly. Great insight was gained about the processes occuring during and shortly after the laser pulse. Many of the quantities interesting for experimentalists can be predicted by the theory. From the simulations relevant parameters like the electron-cooling time or the important ablation threshold were calculated. All values match their experimental counterpart very well.
Die vorliegende Arbeit beschaftigt sich mit der Laserablation in Metallen. Ziel ist es, mit Hilfe von numerischen Simulationen das Verhalten von Metallen nach der Bestrahlung mit intensiven Laserpulsen vorherzusagen. Die Arbeit ist inhaltlich in zwei Teile gegliedert. In der ersten Halfte werden theoretische Grundlagen, eine qualitative Beschreibung der Ablation und die Implementierung des Modells gegeben. Im zweiten Teil folgen Ergebnisse sowie, falls vorhanden, Vergleiche mit Experimenten. Die Arbeit schliest mit einer Zusammenfassung und einem Ausblick. read less NOT USED (high confidence) H. Euchner, “Lattice dynamics of complex metallic alloys.” 2011. link Times cited: 0 Abstract: Throughout this thesis the lattice dynamics in CMA phases wi… read moreAbstract: Throughout this thesis the lattice dynamics in CMA phases with different structural and dynamical peculiarities have been studied in experiment and simulation. While inelastic neutron and X-ray scattering enabled an experimental approach to dynamical quantities as dispersion curves, vibrational density of states or dynamical structure factors, the theoretical approach was based on ab-initio and molecular dynamics simulations. Experimental results could be analyzed and interpreted by means of computer simulations, thus yielding insight into dynamical processes on an atomistic level. Indeed, this combination of experiment and simulation proved to be a powerful tool for the investigation of different dynamical phenomena.
In the Mg-Zn system the impact of structural complexity on vibrational properties was studied. Pure hcp Zn and the MgZn2 Laves phase were used as rather simple reference structures and compared to the structurally more complex Mg2Zn11 Pauling triacontahedral phase. While MgZn2 showed the behavior of an almost perfectly harmonic solid, Mg2Zn11 turned out to exhibit quite unusual dynamical features. In the case of MgZn2 experimental results from INS could be reproduced with high accuracy. For Mg2Zn11 experimental results and DFT calculations first evidenced non-negligible discrepancies. After reinvestigating the structure of Mg2Zn11 with both, experimental and computational methods, a partially occupied Zn site could be spotted as possible source of the occurring discrepancies. Surprisingly, the partially vacant Zn1 position, at the center of the mini-Bergman cluster proved to exert a strong influence on stability and dynamics of this system. After taking vacancy disorder into account, the experimental results could be decently reproduced and differences could be understood. With this knowledge the experimental GVDOS was finally interpreted in terms of distinct atomic motions, thus connecting macroscopic properties with processes on atomistic scale.
The second Zn-based CMA phase that was explored, is the ScZn6 1/1-approximant. The structure of this phase is closely related to the Cd-based binary icosahedral quasicrystals in the Cd-Yb and Cd-Ca system, thus making it an interesting phase with respect to structure and dynamics of quasicrystals like Mg-Zn-Sc. Secondly, the ScZn6 1/1-approximant evidences an order-disorder phase transition at about 150 K. The dynamical aspects of this phase transition were investigated throughout this work, using quasielastic neutron scattering and molecular dynamics methods. Interestingly, the phase transition could be shown to be closely related to a freezing in of the tetrahedral shell in the center of the Tsai-type cluster building blocks. In fact, experiment and calculation clearly evidenced a dynamic disorder of the tetrahedral shell above the transition temperature. The tetrahedral shell is constantly reorienting between different, energetically equivalent configurations. From neutron scattering experiments the residence time between two tetrahedron jumps could be estimated to be of the order of a few ps, while it was overestimated by the conducted MD simulations. These results thus answer the controversially debated question about the nature of the disorder in ScZn6 in favor of a dynamic process. Finally the dynamic reorientations of the tetrahedron are highly interesting with respect to entropical stabilization, a possible candidate for quasicrystal stabilization.
In the last part of the thesis the clathrate system Ba-Ge-Ni, was studied with respect to its cage-like structure and the resulting effects on its dynamical properties. Inelastic neutron scattering experiments nicely evidenced a flat dispersionless optic-like phonon branch, which by means of DFT could be shown to stem from localized motions of the encaged Ba atoms - so-called rattling modes. The cage structure of the Ba-Ge-Ni clathrates furthermore made a decomposition into different subsystems possible, such that their contributions to the vibrational spectrum could be analyzed. A comparison to a hypothetical Ge46 structure could be used to elaborate the influence of the encaged Ba-atoms and the host-lattice, respectively. Interestingly, the introduction of Ba-atoms creates a localized, dispersionless phonon branch at rather low energy, which interacts with the acoustic modes of the host structure, resulting in a reduction of the velocity of sound. Thus the low lattice thermal conductivity in this phase seems to be related to both, rattling modes of Ba guest atoms and reduced velocity of sound of the host framework.
Im Rahmen dieser Arbeit wurde die Gitterdynamik verschiedener CMA-Phasen mit Neutronen- bzw. Rontgenstreuung experimentell untersucht und dann anhand von Simulationen bezuglich verschiedener dynamischer Aspekte analysiert und interpretiert.
Im Mg-Zn System konnte der konkrete Einfluss von struktureller Komplexitat auf die Vibrationseigenschaften studiert und am Beispiel der bei den Phasen MgZn2 und Mg2Zn11 dargestellt werden. Ein besonderes Augenmerk wurde hierbei auf die Analyse der auftretenden niedrigenergetischen Moden in Mg2Zn11 gelegt.
Fur den ScZn6 1/1-Approximanten gelang es, den engen Zusammenhang der inneren Tetraederschalen mit dem Ordnungs-Unordnungs-Phasenubergang herauszuarbeiten und zu zeigen, dass die Unordnung oberhalb des Phasenuberganges von dynamischer Natur ist. Somit war es moglich, die viel diskutierte Frage uber die Tetraederunordnung oberhalb des Phasenuberganges eindeutig zu beantworten.
Im Clathrat-System Ba-Ge-Ni wurde das Phononenspektrum mit Bezug auf die niedrige thermische Leitfahigkeit des Gitters im Detail untersucht.
Die bei tiefen Energien auftretenden rattling modes sowie deren Einfluss auf die die akustischen Phononmoden konnten hier als Ursachen fur die niedrige thermische Leitfahigkeit ausgemacht werden. read less NOT USED (high confidence) H. Trebin et al., “Simulating structure and physical properties of complex metallic alloys.” 2008. link Times cited: 3 Abstract: An introduction is presented to numerical methods, by which … read moreAbstract: An introduction is presented to numerical methods, by which the behavior of complex metallic alloys can be simulated. We primarily consider the molecular dynamics (MD) technique as implemented in our software package IMD, where Newton’s equations of motion are solved for all atoms in a solid. After a short discourse on integration algorithms, some possible types of interactions are addressed. Already simple model potentials, as for example the Lennard-Jones-Gauss potential, can give rise to complex structures, where the characteristic length scales of the order by far exceed the range of the pair interaction. Realistic interactions are modelled by highly parametrized effective potentials, like the EAM (Embedded Atom Method) potential. Our program potfit allows to fit the parameters such that data from experiment or from ab-initio calculations are well reproduced. Several applications of the methods are outlined, notably the simulation of aluminium diffusion in quasicrystalline d-Al-Ni-Co, the computation of the phonon dispersion via the dynamical structure factor of MgZn2, the propagation of cracks in NbCr2, and an order-disorder phase transition in CaCd6. read less NOT USED (high confidence) P. Brommer, “Development and test of interaction potentials for complex metallic alloys.” 2008. link Times cited: 8 Abstract: Complex metallic alloys and quasicrystals show extraordinary… read moreAbstract: Complex metallic alloys and quasicrystals show extraordinary physical properties relevant for technological applications, for example hardness at low density. In the study of these systems, atomistic simulation with classical interaction potentials is a very promising tool. Such simulations require classical effective potentials describing the cohesive energy as a function of the atomic coordinates. The quality of the simulation depends crucially on the accuracy with which this potential describes the real interactions. One way to generate physically relevant potentials is the force matching method, where the parameters of a potential are adjusted to optimally reproduce the forces on individual atoms determined from quantum-mechanical calculation. The programme package potfit developed as part of this thesis implements the force matching method efficiently. Potentials are generated for a number of complex metallic alloy systems. A potential for the decagonal basic Ni-rich Al-Co-Ni quasicrystal is used to simulate diffusion processes and melting. In the CaCd6 system built from multishelled clusters, the shape and orientation of the innermost cluster shell is studied. Finally, phonon dispersion in the Mg-Zn system is determined and compared to experiment. The programme potfit is shown to be an effective tool for generating physically justified effective potentials. Potentials created with potfit can greatly improve the understanding of complex metallic alloys through atomistic simulations.
Komplexe intermetallische Verbindungen und Quasikristalle zeigen ausergewohnliche physikalische Eigenschaften, wie z.B. Harte bei geringer Dichte. Bei der Untersuchung dieser Systeme sind atomistische Simulationen mit klassischen Wechselwirkungspotenzialen ein wichtiges Werkzeug. Fur solche Simulationen benotigt man klassische effektive Potenziale, die die Bindungsenergie als eine Funktion der Atomkoordinaten beschreiben. Die Qualitat der Simulation hangt entscheidend von der Genauigkeit ab, mit der diese Potenziale die echten Wechselwirkungen wiedergeben. Eine Moglichkeit, physikalisch relevante Potenziale zu erzeugen, ist die Force-Matching-Methode. Dabei werden die Parameter eines Potenzials so angepasst, dass die mit quantenmechanischen Methoden bestimmten Krafte auf die einzelnen Atome bestmoglich reproduziert werden. Das als Teil dieser Arbeit entwickelte Programmpaket potfit implementiert die Force-Matching-Methode effizient. Fur einige komplexe intermetallische Verbindungen werden Potenziale bestimmt. In dekagonalen Al-Co-Ni-Quasikristallen werden mit Hilfe eines Potenzials Diffusionsprozesse und Schmelzvorgange simuliert. In der aus mehrschaligen Clustern bestehenden CaCd6-Verbindung wird die Struktur der innersten Clusterschale untersucht. Schlieslich wird die Phononendispersion im Mg-Zn-System bestimmt und mit experimentellen Ergebnissen verglichen. Es wird gezeigt, dass das Programm potfit ein effektives Werkzeug zur Erzeugung physikalisch gerechtfertigter Wechselwirkungen ist. Potenziale, die mit potfit erzeugt werden, konnen zum Verstandnis komplexer metallischer Verbindungen durch atomistische Simulationen viel beitragen. read less NOT USED (definite) G. P. P. Pun, R. Batra, R. Ramprasad, and Y. Mishin, “Physically informed artificial neural networks for atomistic modeling of materials,” Nature Communications. 2018. link Times cited: 188 NOT USED (definite) P. Loginov, D. Sidorenko, M. Bychkova, M. Petrzhik, and E. Levashov, “Mechanical Alloying as an Effective Way to Achieve Superior Properties of Fe–Co–Ni Binder Alloy.” 2017. link Times cited: 22 Abstract: This study addresses the fabrication of nanocrystalline Fe–C… read moreAbstract: This study addresses the fabrication of nanocrystalline Fe–Co–Ni alloy using two operations: mechanical alloying (MA) of elemental powders and hot pressing (HP). The evolution of the phase composition and structure of the powder particles after MA was investigated. Ball milling with rotation speed 700 rpm for 15–20 min allows the production of a bcc Fe-based supersaturated solid solution. During the HP of this powder, this solution decomposes into a bcc (Fe) solid solution and fcc Fe3Ni precipitates, which act as a recrystallization barrier at elevated temperatures. This factor, along with the solid solution strengthening of the (α–Fe) matrix and high concentration of lattice defects (dislocations and twins), provides high mechanical properties (ultimate bending strength of 2000 MPa and hardness of 108 HRB) and wear resistance of the alloy. The developed Fe–Co–Ni alloy is promising for use as a binder in diamond tools designed for machining abrasive materials. read less NOT USED (definite) M. Wen, J. Li, P. Brommer, R. Elliott, J. Sethna, and E. Tadmor, “A KIM-compliant potfit for fitting sloppy interatomic potentials: application to the EDIP model for silicon,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 16 Abstract: Fitted interatomic potentials are widely used in atomistic s… read moreAbstract: Fitted interatomic potentials are widely used in atomistic simulations thanks to their ability to compute the energy and forces on atoms quickly. However, the simulation results crucially depend on the quality of the potential being used. Force matching is a method aimed at constructing reliable and transferable interatomic potentials by matching the forces computed by the potential as closely as possible, with those obtained from first principles calculations. The potfit program is an implementation of the force-matching method that optimizes the potential parameters using a global minimization algorithm followed by a local minimization polish. We extended potfit in two ways. First, we adapted the code to be compliant with the KIM Application Programming Interface (API) standard (part of the Knowledgebase of Interatomic Models project). This makes it possible to use potfit to fit many KIM potential models, not just those prebuilt into the potfit code. Second, we incorporated the geodesic Levenberg–Marquardt (LM) minimization algorithm into potfit as a new local minimization algorithm. The extended potfit was tested by generating a training set using the KIM environment-dependent interatomic potential (EDIP) model for silicon and using potfit to recover the potential parameters from different initial guesses. The results show that EDIP is a ‘sloppy model’ in the sense that its predictions are insensitive to some of its parameters, which makes fitting more difficult. We find that the geodesic LM algorithm is particularly efficient for this case. The extended potfit code is the first step in developing a KIM-based fitting framework for interatomic potentials for bulk and two-dimensional materials. The code is available for download via https://www.potfit.net. read less NOT USED (definite) A. Makino, P. Sharma, K. Sato, A. Takeuchi, Y. Zhang, and K. Takenaka, “Artificially produced rare-earth free cosmic magnet,” Scientific Reports. 2015. link Times cited: 61 NOT USED (definite) J. Gattacceca et al., “Metal phases in ordinary chondrites: Magnetic hysteresis properties and implications for thermal history,” Meteoritics & Planetary Science. 2014. link Times cited: 56 Abstract: Magnetic properties are sensitive proxies to characterize Fe… read moreAbstract: Magnetic properties are sensitive proxies to characterize FeNi metal phases in meteorites. We present a data set of magnetic hysteresis properties of 91 ordinary chondrite falls. We show that hysteresis properties are distinctive of individual meteorites while homogeneous among meteorite subsamples. Except for the most primitive chondrites, these properties can be explained by a mixture of multidomain kamacite that dominates the induced magnetism and tetrataenite (both in the cloudy zone as single‐domain grains, and as larger multidomain grains in plessite and in the rim of zoned taenite) dominates the remanent magnetism, in agreement with previous microscopic magnetic observations. The bulk metal contents derived from magnetic measurements are in agreement with those estimated previously from chemical analyses. We evidence a decreasing metal content with increasing petrologic type in ordinary chondrites, compatible with oxidation of metal during thermal metamorphism. Types 5 and 6 ordinary chondrites have higher tetrataenite content than type 4 chondrites. This is compatible with lower cooling rates in the 650–450 °C interval for higher petrographic types (consistent with an onion‐shell model), but is more likely the result of the oxidation of ordinary chondrites with increasing metamorphism. In equilibrated chondrites, shock‐related transient heating events above approximately 500 °C result in the disordering of tetrataenite and associated drastic change in magnetic properties. As a good indicator of the amount of tetrataenite, hysteresis properties are a very sensitive proxy of the thermal history of ordinary chondrites, revealing low cooling rates during thermal metamorphism and high cooling rates (e.g., following shock reheating or excavation after thermal metamorphism). Our data strengthen the view that the poor magnetic recording properties of multidomain kamacite and the secondary origin of tetrataenite make equilibrated ordinary chondrites challenging targets for paleomagnetic study. read less |