Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
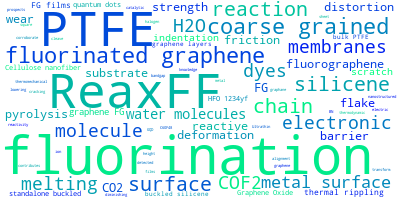
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
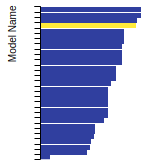
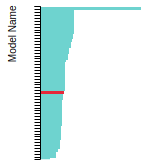
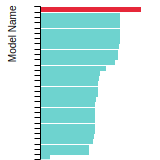
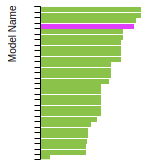
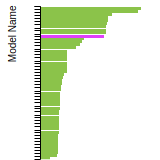
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
79 Citations (32 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) B. Koo, N. Subramanian, and A. Chattopadhyay, “Molecular dynamics study of brittle fracture in epoxy-based thermoset polymer,” Composites Part B-engineering. 2016. link Times cited: 52 USED (high confidence) R. Palacios-Rivera et al., “Surface specificity and mechanistic pathway of de-fluorination of C60F48 on coinage metals,” Nanoscale Advances. 2020. link Times cited: 1 Abstract: We provide experimental and theoretical understanding on fun… read moreAbstract: We provide experimental and theoretical understanding on fundamental processes taking place at room temperature when a fluorinated fullerene dopant gets close to a metal surface. By employing scanning tunneling microscopy and photoelectron spectroscopies, we demonstrate that the on-surface integrity of C60F48 depends on the interaction with the particular metal it approaches. Whereas on Au(111) the molecule preserves its chemical structure, on more reactive surfaces such as Cu(111) and Ni(111), molecules interacting with the bare metal surface lose the halogen atoms and transform to C60. Though fluorine–metal bonding can be detected depending on the molecular surface density, no ordered fluorine structures are observed. We show the implications of the metal-dependent de-fluorination in the electronic structure of the molecules and the energy alignment at the molecule–metal interface. Molecular dynamics simulations with ReaxFF reactive force field corroborate the experimental facts and provide a detailed mechanistic picture of the surface-induced de-fluorination, which involves the rotation of the molecule on the surface. Outstandingly, a thermodynamic analysis indicates that the effect of the metal surface is lowering and diminishing the energy barrier for C–F cleave, demonstrating the catalytic role of the surface. The present study contributes to in-depth knowledge of the mechanisms that affect the degree of stability of chemical species on surfaces, which is essential to advance our understanding of the chemical reactivity of metals and their role in on-surface chemical reactions. read less USED (high confidence) N. Nebogatikova et al., “Fluorinated graphene nanoparticles with 1–3 nm electrically active graphene quantum dots,” Nanotechnology. 2020. link Times cited: 8 Abstract: A new approach to creating a new and locally nanostructured … read moreAbstract: A new approach to creating a new and locally nanostructured graphene-based material is reported. We studied the electric and structural properties of partially fluorinated graphene (FG) films obtained from an FG-suspension and nanostructured by high-energy Xe ions. Local shock heating in ion tracks is suggested to be the main force driving the changes. It was found that ion irradiation leads to the formation of locally thermally expanded FG and its cracking into nanoparticles with small (∼1.5–3 nm) graphene quantum dots (GQD), embedded in them. The bandgap of GQD was estimated as 1 -1.5 eV. A further developed approach was applied to correct the functional properties of printed FG-based crossbar memristors. Dielectric FG films with small quantum dots may offer prospects in graphene-based electronics due to their stability and promising properties. read less USED (high confidence) P. Liu, C. Milletto, S. Monti, C. Zhu, and A. Mathew, “Design of ultrathin hybrid membranes with improved retention efficiency of molecular dyes,” RSC Advances. 2019. link Times cited: 11 Abstract: Ultrathin layers of 2,2,6,6-Tetramethyl-1-piperidinyloxy (TE… read moreAbstract: Ultrathin layers of 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO) Oxidized Cellulose Nanofibers (TOCNF) embedded with Graphene Oxide nanosheets (GOs) in different ratios were built, via the blade coating technique, on a polyvinylidene difluoride (PVDF) substrate to obtain superior membranes for separating water pollutants from aqueous media. Cellulose nanofiber–graphene oxide hybrid materials have shown a great potential for water purification due to their active microporous structure with extended areas rich in negatively charged carboxyl functional groups capable of adsorbing positively charged contaminants efficiently. In contrast to the pristine free-standing TOCNF films, which are completely impermeable, the ultrathin (68 nm thick) hybrid coating with a 100 : 1 TOCNF : GO ratio showed a stable water permeability (816 ± 3.4 L m−2 h−1 bar−1) higher than that of common polymeric membranes, and a very efficient size selectivity during filtration of water contaminated by various types of dyes. The membranes had high retention efficiency (82–99%) for dyes with hydrated radii greater than ≈0.5 nm due to the favorable combination of electrostatic/hydrophobic interactions with the hybrid matrices and steric entrapment controlled by the pore size. This was confirmed by theoretical calculations that revealed both the structure and dynamic behavior of the dyes in the complex environment of the membranes. read less USED (high confidence) Y. Cao, C. Liu, X. Xu, E. Huo, and Y. Pu, “Influence of water on HFO-1234yf oxidation pyrolysis via ReaxFF molecular dynamics simulation,” Molecular Physics. 2019. link Times cited: 18 Abstract: ABSTRACT The effect of water molecules on HFO-1234yf oxidati… read moreAbstract: ABSTRACT The effect of water molecules on HFO-1234yf oxidation pyrolysis was investigated by ReaxFF-molecular dynamics simulation from 1900 to 4200 K. The initial pyrolysis of HFO-1234yf starts around 2500 K and the water molecules participate in chemical reactions at 2800 K when the reactants pyrolysis reached the highest reaction rate. The primary products including HF, COF2 and CO2 are observed at 2600, 2700 and 2900 K, respectively. The influence of water molecules on products is mainly reflected in the promotion activity on the conversion from COF2 to CO2 and the generation of HF molecules. Four formation pathways are observed and calculated to further elucidate the procedure of pyrolysis. The main conversion process from H2O to HF is the •F + H2O = HF+•OH reaction, and the paths from H2O to •OH radical and COF2 to •CFO radical which are promoted by •F and •H radical, respectively, have relatively low energy barriers of 10.44 and 40.29 kJ/mol, and both reaction processes released HF molecules. GRAPHICAL ABSTRACT read less USED (high confidence) D. Kim et al., “Active site localization of methane oxidation on Pt nanocrystals,” Nature Communications. 2018. link Times cited: 48 USED (high confidence) J. Johnston, B. Koo, N. Subramanian, and A. Chattopadhyay, “Modeling the molecular structure of the carbon fiber/polymer interphase for multiscale analysis of composites,” Composites Part B-engineering. 2017. link Times cited: 59 USED (high confidence) S. Singh, S. Costamagna, M. Neek‐Amal, and F. Peeters, “Melting of Partially Fluorinated Graphene: From Detachment of Fluorine Atoms to Large Defects and Random Coils,” arXiv: Materials Science. 2013. link Times cited: 15 Abstract: The melting of fluorographene is very unusual and depends st… read moreAbstract: The melting of fluorographene is very unusual and depends strongly on the degree of fluorination. For temperatures below 1000 K, fully fluorinated graphene (FFG) is thermo-mechanically more stable than graphene but at T$_m\approx$2800 K FFG transits to random coils which is almost twice lower than the melting temperature of graphene, i.e. 5300 K. For fluorinated graphene (PFG) up to 30 % ripples causes detachment of individual F-atoms around 2000 K while for 40-60 % fluorination, large defects are formed beyond 1500 K and beyond 60% of fluorination F-atoms remain bonded to graphene until melting. The results agree with recent experiments on the dependence of the reversibility of the fluorination process on the percentage of fluorination. read less USED (high confidence) M. Neek‐Amal, A. Sadeghi, G. Berdiyorov, and F. Peeters, “Realization of free-standing silicene using bilayer graphene,” Applied Physics Letters. 2013. link Times cited: 69 Abstract: The available synthesized silicene-like structures have been… read moreAbstract: The available synthesized silicene-like structures have been only realized on metallic substrates which are very different from the standalone buckled silicene, e.g., the Dirac cone of silicene is destroyed due to lattice distortion and the interaction with the substrate. Using graphene bilayer as a scaffold, a route is proposed to synthesize silicene with electronic properties decoupled from the substrate. The buckled hexagonal arrangement of silicene between the graphene layers is found to be very similar to the theoretically predicted standalone buckled silicene which is only very weakly van der Waals coupled to the graphene layers with a graphite-like interlayer distance of 3.42 A and without any lattice distortion. We found that these stacked layers are stable well above room temperature. read less USED (high confidence) M. Brownell and A. Nair, “Deformation mechanisms of polytetrafluoroethylene at the nano- and microscales.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 16 Abstract: Polytetrafluoroethylene (PTFE) has not only a low coefficien… read moreAbstract: Polytetrafluoroethylene (PTFE) has not only a low coefficient of friction but also a high wear rate. Numerous studies investigating the possibility of reducing wear and the mechanisms of deformation have been conducted using experimental and computational studies. In this paper the deformation and mechanical failure of single chains and bulk PTFE are investigated using molecular dynamics (MD). Due to the length scale limitations of the model that MD simulations can investigate, a coarse-grained model is developed and PTFE particles of varying densities are generated to investigate their mechanical properties at the microscale. The coarse-grained potential parameters and coarse-grained model of PTFE are derived from first principles based ReaxFF simulations and are then used to investigate the microscale mechanical properties of PTFE. The MD study indicates that temperature has a pronounced effect on the maximum strength of a single chain, the elastic modulus is dependent on chain length, and chains shorter than 100 Å have an increase in maximum strength. During indentation and scratch the frictional coefficient of bulk PTFE is dependent on the direction of the scratch on the PTFE substrate. The coarse-grained PTFE simulations show that indentation force is dependent on the density of PTFE, and the smoother the surface of the particle the lower the coefficient of friction. The coarse-grained model that connects the atomic and macroscales of PTFE will lead to a direct comparison with experimental PTFE thin films. read less USED (low confidence) C. M. Miliante, J. P. D. de Matos, and A. Muniz, “Structural, electronic and mechanical properties of double core carbon nanothreads,” Carbon. 2023. link Times cited: 0 USED (low confidence) U. Nwankwo, Y.-D. Wang, C. Lam, and N. Onofrio, “Charge equilibration model with shielded long-range Coulomb for reactive molecular dynamics simulations.,” The Journal of chemical physics. 2023. link Times cited: 1 Abstract: Atomic description of electrochemical systems requires react… read moreAbstract: Atomic description of electrochemical systems requires reactive interaction potential to explicitly describe the chemistry between atoms and molecules and the evolving charge distribution and polarization effects. Calculating Coulomb electrostatic interactions and polarization effects requires a better estimate of the partial charge distribution in molecular systems. However, models such as reactive force fields and charge equilibration (QEq) include Coulomb interactions up to a short-distance cutoff for better computational speeds. Ignoring long-distance electrostatic interaction affects the ability to describe electrochemistry in large systems. We studied the long-range Coulomb effects among charged particles and extended the QEq method to include long-range effects. By this extension, we anticipate a proper account of Coulomb interactions in reactive molecular dynamics simulations. We validate the approach by computing charges on a series of metal-organic frameworks and some simple systems. Results are compared to regular QEq and quantum mechanics calculations. The study shows slightly overestimated charge values in the regular QEq approach. Moreover, our method was combined with Ewald summation to compute forces and evaluate the long-range effects of simple capacitor configurations. There were noticeable differences between the calculated charges with/without long-range Coulomb interactions. The difference, which may have originated from the long-range influence on the capacitor ions, makes the Ewald method a better descriptor of Coulomb electrostatics for charged electrodes. The approach explored in this study enabled the atomic description of electrochemical systems with realistic electrolyte thickness while accounting for the electrostatic effects of charged electrodes throughout the dielectric layer in devices like batteries and emerging solid-state memory. read less USED (low confidence) Q. Liu, K. Huang, X. Zhu, and G. Wang, “Study on the Thermal Decomposition of D-4OH PFPE Lubricant by Reactive Molecular Dynamic Simulation for HAMR,” IEEE Transactions on Magnetics. 2023. link Times cited: 2 Abstract: Heat-assisted magnetic recording (HAMR) is one of the ways o… read moreAbstract: Heat-assisted magnetic recording (HAMR) is one of the ways of ultrahigh density storage technology. The pulsed laser in the HAMR head heats the nano-region of the disk to Curie temperature. The laser heating causes the decomposition of the PFPE lubricant, which indirectly leads to the failure of head-disk interface (HDI). The reactive molecular dynamics (MDs) simulations are performed to research the thermal decomposition of single and multiple D-4OH lubricants. The effect of three different heating rates on the thermal decomposition process of a single D-4OH lubricant is investigated. The decomposition processes of single and multi-D-4OH molecular chains are the same: the functional end groups degrade first and then the main chain. These results are useful for the optimization of PFPE lubricants for HAMR disks. read less USED (low confidence) Y. Zhang, T. Hu, R. Hu, S. Jiang, C. Zhang, and H. Hou, “Thermal, Mechanical and Dielectric Properties of Polyimide Composite Films by In-Situ Reduction of Fluorinated Graphene,” Molecules. 2022. link Times cited: 8 Abstract: Materials with outstanding mechanical properties and excelle… read moreAbstract: Materials with outstanding mechanical properties and excellent dielectric properties are increasingly favored in the microelectronics industry. The application of polyimide (PI) in the field of microelectronics is limited because of the fact that PI with excellent mechanical properties does not have special features in the dielectric properties. In this work, PI composite films with high dielectric properties and excellent mechanical properties are fabricated by in-situ reduction of fluorinated graphene (FG) in polyamide acid (PAA) composites. The dielectric permittivity of pure PI is 3.47 and the maximum energy storage density is 0.664 J/cm3 at 100 Hz, while the dielectric permittivity of the PI composite films reaches 235.74 under the same conditions, a 68-times increase compared to the pure PI, and the maximum energy storage density is 5.651, a 9-times increase compared to the pure PI films. This method not only solves the problem of the aggregation of the filler particles in the PI matrix and maintains the intrinsic excellent mechanical properties of the PI, but also significantly improves the dielectric properties of the PI. read less USED (low confidence) S. Li et al., “Non-clustering of sp 3 fluorine adatoms on pristine graphene surface,” Journal of Physics: Condensed Matter. 2022. link Times cited: 0 Abstract: Fluorination can change graphene’s properties, and which is … read moreAbstract: Fluorination can change graphene’s properties, and which is theoretically relative to fluorination pattern of sp 3 fluorine adatoms on graphene surface. The common view for the pattern is that it can easily form as a large cluster for the low migration barrier of fluorine adatoms on pristine graphene surface. Here, we report that sp 3 fluorine adatoms are well-dispersed rather than clustered due to that the intensity ratio of 1.8 for C–CF/CF peaks (R) of fluorinated graphene is much higher than R ≈ 0 for clustered pattern. The low magnetic inducing efficiency of 1 µ B/1000F adatoms indicates that the ‘nonmagnetic’ fluorine pairs rather than ‘magnetic’ fluorine ‘points’ dominate the well-dispersed sp 3 pattern. Our findings introduce a new insight into the fluorination structure properties of fluorinated and other sp 3 functionalized such as hydrogenated, chlorinated, or hydroxylated graphene and other carbon materials. read less USED (low confidence) T. Wang, X. Liu, S. Huang, A. Waheed, and M. He, “Modelling co-gasification of plastic waste and lignin in supercritical water using reactive molecular dynamics simulations,” International Journal of Hydrogen Energy. 2022. link Times cited: 10 USED (low confidence) Y. Pedram, F. Marsusi, and S. Yousefbeigi, “Melting process of fluorinated graphene: A molecular dynamics study,” Chemical Physics Letters. 2021. link Times cited: 1 USED (low confidence) T. Wang, X. Liu, H. Liu, and M. He, “Synergistic effect of supercritical water and nano-catalyst on lignin gasification,” International Journal of Hydrogen Energy. 2021. link Times cited: 12 USED (low confidence) Y.-E. Liu, J. Hu, H. Hou, and B. Wang, “ReaxFF reactive force field development and application for molecular dynamics simulations of heptafluoroisobutyronitrile thermal decomposition,” Chemical Physics Letters. 2020. link Times cited: 4 USED (low confidence) J. J. Schichtel and A. Chattopadhyay, “An atomistic study of damage and localized anisotropic mechanical property evolution in thermoset polymers,” International Journal of Mechanical Sciences. 2020. link Times cited: 1 USED (low confidence) K. Sadki, F. Z. Zanane, M. Ouahman, and L. B. Drissi, “Molecular dynamics study of pristine and defective hexagonal BN, SiC and SiGe monolayers,” Materials Chemistry and Physics. 2020. link Times cited: 13 USED (low confidence) Y. Jiang, D. Choudhury, M. Brownell, A. Nair, J. A. Goss, and M. Zou, “The effects of annealing conditions on the wear of PDA/PTFE coatings,” Applied Surface Science. 2019. link Times cited: 26 USED (low confidence) K. Venkatesan, N. Subramanian, A. Rai, and A. Chattopadhyay, “Atomistically informed multiscale modeling of radially grown nanocomposite using a continuum damage mechanics approach,” Carbon. 2019. link Times cited: 10 USED (low confidence) J. Silveira and A. Muniz, “Diamond nanothread-based 2D and 3D materials: Diamond nanomeshes and nanofoams,” Carbon. 2018. link Times cited: 17 USED (low confidence) N. Subramanian, B. Koo, K. Venkatesan, and A. Chattopadhyay, “Interface mechanics of carbon fibers with radially-grown carbon nanotubes,” Carbon. 2018. link Times cited: 22 USED (low confidence) B. Saha and A. Datta, “Reactive Molecular Dynamics Simulations of Self-Assembly of Polytwistane and Its Application for Nanofibers,” The Journal of Physical Chemistry C. 2018. link Times cited: 10 Abstract: We investigated the self-assembly and mechanical properties … read moreAbstract: We investigated the self-assembly and mechanical properties of polytwistane (PT), particularly a seven-strand PT rope, using reactive force field-based molecular dynamics simulations at different temperatures. We show that upon self-assembly due to strong van der Waals interaction among PT units, PTs form a twisted structure (ropelike) with a twisting angle of ∼0.16 rad/nm at 300 K, which makes them mechanically stronger. The PT rope has high Young’s modulus (∼0.45 TPa) at 300 K. Interestingly, Young’s modulus increases with temperature for the seven-strand PT rope, whereas it decreases with temperature for a single-strand PT. This is because in the case of the seven-strand PT, the twisting angle also contributes to the elastic property of the PT rope and twisting depends on the temperature. We estimate a maximum load transfer of ∼1.1 and ∼3.3 nN to the central unit at 100 and 300 K, respectively. Hence, the amount of load transfer critically depends on the twisting in the rope. The fracture behavior of t... read less USED (low confidence) Y. Cao, C. Liu, H. Zhang, X. Xu, and Q. Li, “Thermal decomposition of HFO-1234yf through ReaxFF molecular dynamics simulation,” Applied Thermal Engineering. 2017. link Times cited: 51 USED (low confidence) B. Koo, E. M. Nofen, A. Chattopadhyay, and L. L. Dai, “Dimeric anthracene-based mechanophore for damage precursor detection in epoxy-based thermoset polymer matrix: Characterization and atomistic modeling,” Computational Materials Science. 2017. link Times cited: 10 USED (low confidence) R. Li, Y. Tao, Z.-li Liu, and Y. Tan, “Edge-functionalization of blue phosphorene nanoribbons: Studied from first-principle methods,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 6 USED (low confidence) R. Li, Z.-li Liu, Y. Gu, W. Zhang, and Y. Tan, “Oxygen- and hydroxyl-edge termination of silicene nanoribbons studied by first-principles calculations,” Physica E-low-dimensional Systems & Nanostructures. 2016. link Times cited: 9 USED (low confidence) S. Winczewski, M. Y. Shaheen, and J. Rybicki, “Interatomic potential suitable for the modeling of penta-graphene: Molecular statics/molecular dynamics studies,” Carbon. 2018. link Times cited: 34 USED (low confidence) N. Subramanian and A. Chattopadhyay, “Molecular Dynamics Simulations for the Analysis of Nano-engineered Fuzzy Fiber Composites.” 2018. link Times cited: 1 NOT USED (low confidence) S. Y. Misyura, V. S. Morozov, and V. A. Andryushchenko, “The influence of various factors on the transition of the fluorinated graphene wettability from superhydrophilicity to high hydrophobicity,” Surfaces and Interfaces. 2024. link Times cited: 0 NOT USED (low confidence) X. Wang, S. Liu, H. Han, X. Liu, and X. Wang, “Research progress in insulating and thermal conductivity of fluorinated graphene and its polyimide composites,” IET Nanodielectrics. 2023. link Times cited: 0 Abstract: The demand for innovative thermal management materials with … read moreAbstract: The demand for innovative thermal management materials with superior thermal conductivity and electrical insulating properties has significantly increased with the development of portable and flexible electronic gadgets. Fluorinated graphene (FG) has recently attracted the attention of the scientific community because of its exceptional thermal conductivity and electrical insulating qualities. This work aims to provide a detailed analysis of the structure‐property relationships inherent in FG, including both chemical and physical properties, and to explain the FG manufacturing process. Special attention should be paid to a thorough analysis of the thermodynamic conduction mechanism exhibited by FG, including the effects of corrugation size, fluorine coverage, and fluorine atom distribution on its thermal conductivity. The essay also examines in‐depth the most current and cutting‐edge developments addressing the utilisation of FG as a functional filler in composite‐modified polyimide (PI) materials. Furthermore, it has been noted as a crucial component in answering the needs for possible applications by maximising thermal conductivity and mechanical qualities in FG/PI composites through particular FG structural engineering and increased FG‐PI interaction. As a result, these elements serve as the main focus of ongoing research projects, highlighting important directions for development and investigation. read less NOT USED (low confidence) B. B. de Souza, S. A. Hewage, J. A. Kewalramani, A. C. van Duin, and J. N. Meegoda, “A ReaxFF-based molecular dynamics study of the destruction of PFAS due to ultrasound.,” Environmental pollution. 2023. link Times cited: 2 NOT USED (low confidence) J. Chen and W. Ge, “Computational study on the order-of-magnitude difference in thermal conductivity between graphene and graphane nanoribbons,” Diamond and Related Materials. 2022. link Times cited: 1 NOT USED (low confidence) M. Singla and N. Jaggi, “Theoretical investigations of hydrogen gas sensing and storage capacity of graphene-based materials: A review,” Sensors and Actuators A: Physical. 2021. link Times cited: 16 NOT USED (low confidence) Y.-E. Liu, J. Hu, H. Hou, and B. Wang, “Development and application of a ReaxFF reactive force field for molecular dynamics of perfluorinatedketones thermal decomposition,” Chemical Physics. 2020. link Times cited: 12 NOT USED (low confidence) J. Biskupek et al., “Bond Dissociation and Reactivity of HF and H2O in a Nano Test Tube.,” ACS nano. 2020. link Times cited: 12 Abstract: Molecular motion and bond dissociation are two of the most f… read moreAbstract: Molecular motion and bond dissociation are two of the most fundamental phenomena underpinning properties of molecular materials. We have entrapped HF and H2O molecules within the fullerene C60 cage, encapsulated within a single-walled carbon nanotube (X@C60)@SWNT where X = HF or H2O. (X@C60)@SWNT represents a class of molecular nanomaterial composed of a guest within a molecular host within a nanoscale host, enabling investigations of the interactions of isolated single di- or tri-atomic molecules with the electron beam. The use of the electron beam simultaneously as a stimulus of chemical reactions in molecules and as a sub-Å resolution imaging probe allows investigations of the molecular dynamics and reactivity in real time and at the atomic scale, which are probed directly by chromatic and spherical aberration corrected high resolution transmission electron microscopy (Cc/Cs-corrected HRTEM) imaging, or indirectly by vibrational electron energy loss spectroscopy (EELS) in situ during scanning transmission electron microscopy (STEM) experiments. Experimental measurements indicate that the electron beam triggers homolytic dissociation of the H-F or H-O bonds, respectively, causing the expulsion of the hydrogen atoms from the fullerene cage, leaving fluorine or oxygen behind. Due to a difference in the mechanisms of penetration through the carbon lattice available for F or O atoms, atomic fluorine inside the fullerene cage appears to be more stable than the atomic oxygen under the same conditions. The use of (X@C60)@SWNT, where each molecule X is 'packaged' separately from each other, in combination with the electron microscopy methods and density functional theory (DFT) modelling in this work, enable bond dynamics and reactivity of individual atoms to be probed directly at the single-molecule level. read less NOT USED (low confidence) J. Wang, J. Song, X. Mu, and M. Sun, “Optoelectronic and photoelectric properties and applications of graphene-based nanostructures,” Materials Today Physics. 2020. link Times cited: 44 NOT USED (low confidence) Z. Zheng, H. Zhan, Y. Nie, X. Xu, and Y. T. Gu, “Role of Nitrogen on the Mechanical Properties of the Novel Carbon Nitride Nanothreads,” The Journal of Physical Chemistry C. 2019. link Times cited: 7 Abstract: Carbon nanothread (C-NTH) is a new ultrathin one-dimensional… read moreAbstract: Carbon nanothread (C-NTH) is a new ultrathin one-dimensional sp3 carbon nanostructure, which exhibits promising applications in novel carbon nanofibers and nanocomposites. Recently, researchers have successfully developed a new alternative structure - ultrathin carbon nitride nanothread (CN-NTH). In this work, we investigate the mechanical properties of CN-NTHs through large-scale molecular dynamics simulations. Comparing with their C-NTH counterparts, CN-NTHs are found to exhibit a higher tensile and bending stiffness. In particular, because of the bond redistribution, the CN-NTHs in the polymer I group and tube (3,0) group are found to possess a higher failure strain than their C-NTH counterparts. However, the CN-NTH in the polytwistane group has a smaller failure strain compared with the pristine C-NTH. According to the atomic configurations, the presence of nitrogen atoms always leads to stress/strain concentrations for the nanothreads under tensile deformation. This study provides a comprehensive understanding of the mechanical properties of CN-NTHs, which should shed light on their potential applications such as fibers or reinforcements for nanocomposites. read less NOT USED (low confidence) P. Demingos and A. Muniz, “Carbon nanothreads from polycyclic aromatic hydrocarbon molecules,” Carbon. 2018. link Times cited: 21 NOT USED (low confidence) L. B. Drissi, N. Kanga, and F. Ramadan, “Excitonic and fluorination effects on optoelectronic response of GeC hybrid,” Computational Condensed Matter. 2018. link Times cited: 8 NOT USED (low confidence) D. D. Chronopoulos, A. Bakandritsos, M. Pykal, R. Zbořil, and M. Otyepka, “Chemistry, properties, and applications of fluorographene,” Applied Materials Today. 2017. link Times cited: 194 NOT USED (low confidence) B. Koo, R. Gunckel, A. Chattopadhyay, and L. L. Dai, “Parametric Study for Dimeric Anthracene-Based Mechanophore-Embedded Thermoset Polymer Matrix Using Molecular Dynamics,” MRS Advances. 2017. link Times cited: 0 Abstract: This paper presents a parametric study to investigate the ef… read moreAbstract: This paper presents a parametric study to investigate the effect of relevant design variables on mechanochemical reaction and mechanical properties of a mechanophore-embedded thermoset polymer matrix. Mechanophores emit fluorescence when a specific covalent bond breaks due to external stress, and thus have attracted immense research interest as a damage sensor. Recently, a mechanophore named dimeric 9-anthracene carboxylic acid (Di-AC) was synthesized successfully and incorporated into epoxy-based thermoset polymer matrix to detect damage precursor. However, there is significant potential in modeling the complex mechanochemistry associated with the Di-AC to obtain a better understanding of this mechanophore and its interaction with the host thermoset material. In this study, a hybrid MD simulation methodology is employed to explore this complex mechanochemistry along with the investigation of the effect of design parameters on the mechanophore performance. The hybrid MD simulation method enables the simulation of Di-AC synthesis, epoxy curing, and mechanical loading test; therefore, the experimental process performed can be emulated accurately. Previously, the hybrid MD method showed the capability of capturing experimentally observed phenomena such as early signal detection and yield strength variation between neat epoxy system and epoxy with 5 wt% Di-AC thermoset polymer. In this paper, the effect of curing temperature on mechanophore activation and mechanical properties is investigated. A series of temperatures are used in the curing simulation, which are experimentally achievable. Results show that curing temperature below glass transition temperature maintains early signal detection and yield strength decreases when the curing temperature increases above the glass transition temperature. Good correlation is observed with experimental results. read less NOT USED (low confidence) B. Saha, S. M. Pratik, and A. Datta, “Coexistence of Normal and Auxetic Behavior in a Thermally and Chemically Stable sp3 Nanothread: Poly[5]asterane.,” Chemistry. 2017. link Times cited: 8 Abstract: A one-dimensional nanostructure with sp3 -hybridized carbon … read moreAbstract: A one-dimensional nanostructure with sp3 -hybridized carbon atoms, namely, poly[5]asterane (PA), is predicted by means of electronic structure calculations and reactive molecular dynamics simulations. Thermochemical analysis based on homodesmotic reactions showed that the formation of poly[5]asterane is more favorable than that of polytriangulane and comparable to that of polytwistane. A plane-wave DFT approach gave a computed Young's modulus of about 0.84 TPa, which is quite promising and comparable to those of other sp3 -hybridized nanothreads. Simulations of the desorption of hydrogen atoms from PA showed a high activation energy (Ea ≈52 kcal mol-1 ), which again indicates substantial chemical stability. Interestingly, PA was shown to exhibit auxetic behavior (negative Poisson's ratio). Thus, PA is advocated as a new mechanically and chemically stable nanothread with exotic auxetic behavior. read less NOT USED (low confidence) J. Silveira and A. Muniz, “First-principles calculation of the mechanical properties of diamond nanothreads,” Carbon. 2017. link Times cited: 38 NOT USED (low confidence) L. B. Drissi, F. Ramadan, and N. Kanga, “Fluorination-control of electronic and magnetic properties in GeC-hybrid,” Chemical Physics Letters. 2016. link Times cited: 17 NOT USED (low confidence) J. Müller and B. Hartke, “reaxFF Reactive Force Field for Disulfide Mechanochemistry, Fitted to Multireference ab Initio Data.,” Journal of chemical theory and computation. 2016. link Times cited: 28 Abstract: Mechanochemistry, in particular in the form of single-molecu… read moreAbstract: Mechanochemistry, in particular in the form of single-molecule atomic force microscopy experiments, is difficult to model theoretically, for two reasons: Covalent bond breaking is not captured accurately by single-determinant, single-reference quantum chemistry methods, and experimental times of milliseconds or longer are hard to simulate with any approach. Reactive force fields have the potential to alleviate both problems, as demonstrated in this work: Using nondeterministic global parameter optimization by evolutionary algorithms, we have fitted a reaxFF force field to high-level multireference ab initio data for disulfides. The resulting force field can be used to reliably model large, multifunctional mechanochemistry units with disulfide bonds as designed breaking points. Explorative calculations show that a significant part of the time scale gap between AFM experiments and dynamical simulations can be bridged with this approach. read less NOT USED (low confidence) D. Kirilenko and P. Brunkov, “Measuring the height-to-height correlation function of corrugation in suspended graphene.,” Ultramicroscopy. 2016. link Times cited: 5 NOT USED (low confidence) I. Nikiforov, B. Hourahine, T. Frauenheim, and T. Dumitricǎ, “Formation of Helices in Graphene Nanoribbons under Torsion.,” The journal of physical chemistry letters. 2014. link Times cited: 16 Abstract: We use objective boundary conditions and self-consistent cha… read moreAbstract: We use objective boundary conditions and self-consistent charge density-functional-based tight-binding to simulate at the atomistic scale the formation of helices in narrow graphene nanoribbons with armchair edges terminated with fluorine and hydrogen. We interpret the microscopic data using an inextensible, unshearable elastic rod model, which considers both bending and torsional strains. When fitted to the atomistic data, the simple rod model uses closed-form solutions for a cubic equation to predict the strain energy and morphology at a given twist angle and the crossover point between pure torsion and a helix. Our modeling and simulation bring key insights into the origin of the helical graphene morphologies stored inside of carbon nanotubes. They can be useful for designing chiral nanoribbons with tailored properties. read less NOT USED (low confidence) P. Scuracchio, A. Dobry, S. Costamagna, and F. M. Peeters, “Tuning the polarized quantum phonon transmission in graphene nanoribbons,” Nanotechnology. 2014. link Times cited: 5 Abstract: We propose systems that allow a tuning of the phonon transmi… read moreAbstract: We propose systems that allow a tuning of the phonon transmission function T(ω) in graphene nanoribbons by using C13 isotope barriers, antidot structures, and distinct boundary conditions. Phonon modes are obtained by an interatomic fifth-nearest neighbor force-constant model (5NNFCM) and T(ω) is calculated using the non-equilibrium Green’s function formalism. We show that by imposing partial fixed boundary conditions it is possible to restrict contributions of the in-plane phonon modes to T(ω) at low energy. On the contrary, the transmission functions of out-of-plane phonon modes can be diminished by proper antidot or isotope arrangements. In particular, we show that a periodic array of them leads to sharp dips in the transmission function at certain frequencies &ohgr; &ngr; ?> which can be pre-defined as desired by controlling their relative distance and size. With this, we demonstrated that by adequate engineering it is possible to govern the magnitude of the ballistic transmission functions T ( &ohgr; ) ?> in graphene nanoribbons. We discuss the implications of these results in the design of controlled thermal transport at the nanoscale as well as in the enhancement of thermo-electric features of graphene-based materials. read less NOT USED (low confidence) T. Stauch, “Development and Application of Quantum Chemical Methods for the Description of Molecules Under Mechanical Stress.” 2016. link Times cited: 0 Abstract: In mechanochemistry, forces are used to initiate chemical re… read moreAbstract: In mechanochemistry, forces are used to initiate chemical reactions. Although mechanochemical reactions have been conducted for millennia, a fundamental understanding of the relevant processes at the molecular level is still unavailable. Nevertheless, mechanochemistry is a lively field with an extraordinarily wide range of applications. Several approaches to apply forces to single molecules are commonly used today. One such approach is Single-Molecule Force Spectroscopy, where forces are transmitted from the cantilever of an Atomic Force Microscope to a macromolecule that is anchored to a glass surface. Moving the cantilever away from the surface exerts a pulling force on the molecule. In another class of mechanochemical experiments, ultrasound baths are used to rupture polymers mechanically or to transduce forces to a mechanically susceptible moiety. This capability of ultrasound baths is due to the collapse of cavitational bubbles in the liquids, which generates tensile forces. Furthermore, ball milling or grinding techniques can be used to crush solids, thereby applying forces to molecules. This procedure can be applied to make and break covalent bonds. Due to the lack of solvent, mechanochemical synthesis in a ball mill shows enormous potential as a sustainable and environment-friendly alternative to thermochemistry.
Despite this rich body and long history of experimental mechanochemical procedures, the underlying processes are not well understood at the molecular level. However, such a comprehension is desperately needed for the optimization of mechanochemical syntheses. The use of quantum chemical methods to describe mechanochemical processes, which is called quantum mechanochemistry, has proven to be of tremendous value in understanding mechanochemistry at its most fundamental level. Quantum chemical methods for the description of molecules under external forces afford predictions on force-induced changes in molecular geometry, reactivity and spectroscopic properties. Moreover, force analysis tools are available that can be used to identify the mechanically relevant degrees of freedom in a molecule or its force-bearing scaffold, thereby rationalizing mechanochemical reactivity.
During my PhD work, I have developed the JEDI (Judgement of Energy DIstribution) analysis, which is a quantum chemical force analysis tool for the distribution of stress energy in a mechanically deformed molecule. Based on the harmonic approximation, an energy is calculated for each bond, bending and torsion in a molecule, thus allowing the identification of the mechanically most strained regions in a molecule as well as the rationalization of mechanochemical processes. When a molecule is stretched, some internal modes store more energy than others. This leads to particularly large displacements of certain modes and to the preconditioning of selected bonds for rupture. Using the JEDI analysis I investigated the mechanochemical properties of polymer strands that are tangled into knots. In analogy to ropes, polymer strands are weakened by the ubiquitous overhand knot by approximately 50% and the point of bond rupture is located at the “entry” or “exit” of the knot. The JEDI analysis revealed the reason for this behavior. Upon stretching, most stress energy is stored in the torsions of the curved part of the knot and only a remarkably small amount of energy is used to stretch the bonds that ultimately break. This observation leads to the physical picture that the knot “chokes off” the chain in its immediate vicinity. In this process, the torsions act as work funnels that effectively localize the mechanical energy in the knot, thus preconditioning the covalent bonds at its entry and exit for bond rupture.
Besides the description of mechanical deformation in the ground state, the JEDI analysis can be used in the electronically excited state to quantify the energy gained by relaxation on the excited state potential energy surface (PES). For this, the harmonic approximation needs to be applicable on the excited state PES of interest. The physical process that is described by the excited state JEDI analysis is fundamentally different from the ground state variant. While in the ground state JEDI analysis the distribution of stress energy in a mechanically deformed molecule is analyzed, i.e. energy is expended for deformation, the excited state JEDI analysis quantifies the energy gained by the relaxation of each internal mode upon relaxation on the excited state PES, i.e. energy becomes available. With the excited state JEDI analysis, the mechanical efficiency of molecular photoswitches can be calculated. The spatial extension of a photoswitch that undergoes, e.g., cis-trans-photoisomerization, changes significantly during this process and forces are exerted on the environment. However, other internal modes of the photoswitch that do not contribute to the change in spatial extension change as a side effect of the relaxation on the excited state PES and a certain amount of energy is wasted on them. This effect limits the mechanical efficiency of photoswitches. Using the excited state JEDI analysis, I investigated the mechanical efficiency of the stiff-stilbene photoswitch, which had been used in an experiment in literature to accelerate the electrocyclic ring opening of cyclobutene by photoisomerization. I found that the mechanical efficiency of stiff-stilbene is much too low to account for the observed enhancement of the reaction. A much more reasonable physical explanation is that excess energy from absorption of a photon is dissipated as heat, which accelerates the rupture of the thermally labile bond in cyclobutene.
Furthermore, I used the JEDI analysis to investigate methods for the stabilization of strained hydrocarbons. Angle-strained cycloalkynes with a ring size smaller than eight carbon atoms are highly unstable under normal laboratory conditions, since the C≡C−C bond angles deviate substantially from linearity. Applying an external force in an appropriate direction partially linearizes these bond angles, thus leading to a stabilization of the cycloalkyne. Incorporating cycloheptyne into a macrocycle with stiff-stilbene, however, does not lead to a significant stabilization, since the mechanical efficiency of stiff-stilbene is low. Coupling cycloheptyne to another strained hydrocarbon, on the other hand, stabilizes the molecule tremendously. In particular, cycloheptyne was coupled to a strained cyclophane in a condensed macrocycle and I found that appropriate linking leads to a loss of strain in both hydrocarbons.
In addition to the development of the JEDI force analysis tool, I used existing quantum mechanochemical methods to develop molecular force probes. This class of molecules can be incorporated into larger systems like polymers and proteins and can be used to monitor forces acting in different regions of the macromolecules in real-time via force-induced changes in the spectroscopic signals of the force probes. I found that the reduction of point group symmetry upon mechanical deformation of the molecular force probe is a profitable feature, since electronically excited states that are degenerate in the unperturbed state can split up upon application of an external force. This effect can lead to the generation of new peaks in the spectrum, thus allowing the precise identification of the force probe signal. Additionally, I incorporated molecular force probes into the backbone of proteins without disturbing their natural fold. The formation of hydrogen bonds between the force probe and neighboring strands in a β-sheet preserves the secondary and tertiary structure of the protein and allows the identification of the pulling direction. The application of forces along and perpendicular to the backbone yields pronounced and clearly distinguishable signals of the force probes in the infrared and Raman spectra. Advantageously, the intensities of these signals are proportional to the external force at selected points of the spectrum, which makes the molecules “force rulers”. The signals of the force probes can be intensified and shifted to a transparent window in the protein spectrum by isotopic substitution. read less NOT USED (low confidence) L. Liu, M. Zhang, Z. Xiong, D. Hu, G. Wu, and P. Chen, “Ammonia borane assisted solid exfoliation of graphite fluoride for facile preparation of fluorinated graphene nanosheets,” Carbon. 2015. link Times cited: 22 NOT USED (high confidence) T. Wang, X. Liu, J. Xu, W. Afzal, and M. He, “Simulation of Softwood Lignin Gasification in Supercritical Carbon Dioxide,” SSRN Electronic Journal. 2022. link Times cited: 6 Abstract: : The abundant and renewable lignin resource has attracted i… read moreAbstract: : The abundant and renewable lignin resource has attracted increasingly attention. Supercritical carbon dioxide has excellent properties in catalytic gasification. In this paper, a molecular model of softwood-lignin was established to as a reactant. ReaxFF molecular dynamics simulation method was used to simulate the gasification of softwood in supercritical carbon dioxide environment with nano-platinum particles as catalysts. The method of slow temperature rise (80K/ps) was adopted, and the system was studied at a constant temperature 1700K. The cleavage of different bond at the initial stage of reaction were studied, and the different gasification production under the action of catalysts in supercritical carbon dioxide were analyzed and compared with the non-catalyst condition. The result shows with the addition of catalyst, the amount of CO 2 cracking increases, the number of H 2 and CO molecules produced was increased, and the reaction rate increases. read less NOT USED (high confidence) D. Veeman, M. Sai, V. Rajkumar, M. Ravichandran, and S. Manivannan, “Graphene for Thermal Storage Applications: Characterization, Simulation and Modelling,” Journal of Electronic Materials. 2021. link Times cited: 2 NOT USED (high confidence) J. Hu, Z. Wilde, P. Peralta, C. Muhich, and J. Oswald, “Predicting Hugoniot equation of state in erythritol with ab initio and reactive molecular dynamics,” Journal of Applied Physics. 2021. link Times cited: 0 Abstract: Erythritol has been proposed as an inert surrogate for devel… read moreAbstract: Erythritol has been proposed as an inert surrogate for developing theoretical and computational models to study aging in energetic materials. In this work, we present a comparison of mechanical and shock properties of erythritol computed using the ReaxFF reactive force field and from ab initio calculations employing density functional theory (DFT). We screened eight different ReaxFF parameterizations, of which the CHO parameters developed for hydrocarbon oxidation provide the most accurate predictions of mechanical properties and the crystal structure of erythritol. Further validation of the applicability of this ReaxFF parameterization for modeling erythritol is demonstrated by comparing predictions of the elastic constants, crystal structure, vibrational density of states, and Hugoniot curves against DFT calculations. The ReaxFF predictions are in close agreement with the DFT simulations for the elastic constants and shock Hugoniot when the crystal is loaded along its c axis but show as much as 30% disagreement in the elastic constants in the a b plane and 12% difference in shock pressures when shocked along the a or b crystal axes. Last, we compare thermomechanical properties predicted from classical molecular dynamics with those calculated using the quasi-harmonic approximation and show that quantum mechanical effects produce large discrepancies in the computed values of heat capacity and thermal expansion coefficients compared with classical assumptions. Combining classical molecular dynamics predictions of mechanical behavior with phonon-based calculations of thermal behaviors, we show that predicted shock-induced temperatures for pressures up to 6.5 GPa do not exceed the pressure-dependent melting point of erythritol. read less NOT USED (high confidence) S. Zhao et al., “Influence of fluorinated graphene-modified epoxy coatings on the corrosion behaviour of 2024 aluminium alloy,” RSC Advances. 2021. link Times cited: 8 Abstract: This study provides an enhanced corrosion resistance of epox… read moreAbstract: This study provides an enhanced corrosion resistance of epoxy resin (EP) by embedding fluorinated graphene (FG) into the epoxy matrix. FG with different fluorine contents was obtained by reacting nitrogen trifluoride (NF3) gas with GO and then incorporated into the EP matrix to fabricate the different composites. Through a series of characterization methods, the chemical composition and microstructures of FG were systematically analyzed, and its corrosion resistance was also studied. Results revealed that F atoms were bonded to the GO surface to form C–F covalent bonds, and an FG lamellar thickness less than 2 nm. The contact angle of the coatings increased with the incorporation of FG, and the coating resistance of FG2/EP coating was 3 orders of magnitude more than that of the EP coating after immersion for 4080 h. Thus, the incorporation of FG into epoxy matrix significantly enhanced its hydrophobic properties and barrier performance, which was beneficial to improving the long-term corrosion resistance of the coating. read less NOT USED (high confidence) J. Xu, T. Hao, and Z. Hossain, “Electronic structure basis of strength and toughness in fluoropolymers,” Journal of Applied Physics. 2020. link Times cited: 0 Abstract: Using density functional theory calculations, we investigate… read moreAbstract: Using density functional theory calculations, we investigate and report the electronic structure basis of symmetry-breaking in atomic structures and variations in extreme mechanical properties of a number of widely used polymers, namely, polyvinylidene fluoride (PVDF), polytetrafluoroethylene, polyethylene, and three structural variants of PVDF. All of these polymers consist of C–C bonds in the backbone and a combination of C–H and C–F bonds attached to the backbone. Our results show that the relative proportion and arrangement of the C–H and C–F bonds, hereafter called the chemical heterogeneity, lead to two distinct sets of structural symmetries and mechanical properties. The set with the lower chemical heterogeneity possesses higher structural symmetry and exhibits higher strength and toughness. Electronic population analysis shows that chemical heterogeneity breaks the structural symmetry and alters deformation induced “electron redistribution patterns” in the C–C backbone. Strong variations in electron redistribution serve as the basis for distinctive mechanical behavior among the polymers: the higher the structural variation and electron redistribution, the lower the bond rupture force and toughness of the polymer. The bond rupture process is initiated by a decrease in the electronic population in the backbone, while the electronic interactions at the C–H and C–F bonds dictate where, how, and which bonds rupture. Furthermore, in the linear regime of mechanical deformation, the force–strain behavior is insensitive to the chemical heterogeneity and controlled by the strong covalent interactions of the C–C bonds. In the nonlinear regime, the chemical heterogeneity dramatically reduces the strength and toughness of the polymer chain. read less NOT USED (high confidence) M. Khatami, G. Gaddemane, M. L. V. de Put, M. Moravvej-Farshi, and W. Vandenberghe, “Electronic transport properties of hydrogenated and fluorinated graphene: a computational study,” Journal of Physics: Condensed Matter. 2020. link Times cited: 3 Abstract: Hydrogenation and fluorination have been presented as two po… read moreAbstract: Hydrogenation and fluorination have been presented as two possible methods to open a bandgap in graphene, required for field-effect transistor applications. In this work, we present a detailed study of the phonon-limited mobility of electrons and holes in hydrogenated graphene (graphane) and fluorinated graphene (graphene fluoride). We pay special attention to the out-of-plane acoustic (ZA) phonons, responsible for the highest scattering rates in graphane and graphene fluoride. Considering the most adverse cut-off for long-wavelength ZA phonons, we have obtained electron (hole) mobilities of 28 (41) cm2 V−1 s−1 for graphane and 96 (30) cm2 V−1 s−1 for graphene fluoride. Nonetheless, for a more favorable cut-off wavelength of ∼2.6 nm, significantly higher electron (hole) mobilities of 233 (389) cm2 V−1 s−1 for graphane and 460 (105) cm2 V−1 s−1 for graphene fluoride are achieved. Moreover, while complete suppression of ZA phonons can increase the electron (hole) mobility in graphane up to 278 (391) cm2 V−1 s−1, it does not affect the carrier mobilities in graphene fluoride. Velocity–field characteristics reveal that the electron velocity in graphane saturates at an electric field of ∼4 × 105 V cm−1. Comparing the mobilities with other two-dimensional (2D) semiconductors, we find that hydrogenation and fluorination are two promising avenues to realize a 2D semiconductor while providing good carrier mobilities. read less NOT USED (high confidence) R. Yamaletdinov, Y. A. Nikiforov, L. G. Bulusheva, and A. Okotrub, “Fluorine patterning of graphene: effects of fluorine content and temperature.,” Nanoscale. 2020. link Times cited: 8 Abstract: In this paper we present a successful approach for the gener… read moreAbstract: In this paper we present a successful approach for the generation of partially fluorinated graphene structures. A computationally simple model optimized on a large density functional theory dataset quickly and precisely predicts experimentally observed structures. From the analysis of the structural diversity of fluorinated graphene in a wide range of synthesis temperatures, the general structural patterns are identified and the conditions for their achievement are determined. In addition, to facilitate further studies of fluorinated graphene, we present a ready-to-use GenCF code that implements the described structure generator. read less NOT USED (high confidence) R. Yamaletdinov, V. L. Katkov, Y. A. Nikiforov, A. Okotrub, and V. Osipov, “Effect of Fluorine Patterns on Electronic Transport in Fluorinated Graphene,” Advanced Theory and Simulations. 2019. link Times cited: 9 Abstract: Within the framework of stochastic reactive molecular dynami… read moreAbstract: Within the framework of stochastic reactive molecular dynamics simulations a statistical method for generating fluorinated graphene structures with desirable fluorine distribution is developed. Electronic transport properties of fluorinated graphene in a wide range of functionalization degrees and system ordering are investigated. A strong correlation between irregularities in fluorine distribution and electronic properties is found. In particular, proposed consideration allows for the reproduction of both the experimentally observed electron–hole asymmetry in transport properties of fluorinated graphene and a recently revealed conductivity peak at 10% fluoride content. read less NOT USED (high confidence) J. M. Sousa et al., “Elastic properties of graphyne-based nanotubes,” Computational Materials Science. 2019. link Times cited: 30 NOT USED (high confidence) F. Marsusi, N. Drummond, and M. Verstraete, “The physics of single-side fluorination of graphene: DFT and DFT + U studies,” Carbon. 2019. link Times cited: 23 NOT USED (high confidence) S. Dabaghmanesh, M. Neek‐Amal, B. Partoens, and E. Neyts, “The formation of Cr 2 O 3 nanoclusters over graphene sheet and carbon nanotubes,” Chemical Physics Letters. 2017. link Times cited: 4 NOT USED (high confidence) N. Subramanian, A. Rai, and A. Chattopadhyay, “Atomistically derived cohesive behavior of interphases in carbon fiber reinforced CNT nanocomposites,” Carbon. 2017. link Times cited: 24 NOT USED (high confidence) H. Xiao, X. Shi, F. Hao, X. Liao, Y. Zhang, and X. Chen, “Development of a Transferable Reactive Force Field of P/H Systems: Application to the Chemical and Mechanical Properties of Phosphorene.,” The journal of physical chemistry. A. 2017. link Times cited: 34 Abstract: We developed ReaxFF parameters for phosphorus and hydrogen t… read moreAbstract: We developed ReaxFF parameters for phosphorus and hydrogen to give a good description of the chemical and mechanical properties of pristine and defected black phosphorene. ReaxFF for P/H is transferable to a wide range of phosphorus- and hydrogen-containing systems including bulk black phosphorus, blue phosphorene, edge-hydrogenated phosphorene, phosphorus clusters, and phosphorus hydride molecules. The potential parameters were obtained by conducting global optimization with respect to a set of reference data generated by extensive ab initio calculations. We extended ReaxFF by adding a 60° correction term, which significantly improved the description of phosphorus clusters. Emphasis was placed on the mechanical response of black phosphorene with different types of defects. Compared to the nonreactive SW potential ( Jiang , J.-W. Nanotechnology 2015 , 26 , 315706 ), ReaxFF for P/H systems provides a significant improvement in describing the mechanical properties of the pristine and defected black phosphorene, as well as the thermal stability of phosphorene nanotubes. A counterintuitive phenomenon is observed that single vacancies weaken the black phosphorene more than double vacancies with higher formation energy. Our results also showed that the mechanical response of black phosphorene is more sensitive to defects in the zigzag direction than that in the armchair direction. In addition, we developed a preliminary set of ReaxFF parameters for P/H/O/C to demonstrate that the ReaxFF parameters developed in this work could be generalized to oxidized phosphorene and P-containing 2D van der Waals heterostructures. That is, the proposed ReaxFF parameters for P/H systems establish a solid foundation for modeling of a wide range of P-containing materials. read less NOT USED (high confidence) T. Stauch and A. Dreuw, “Advances in Quantum Mechanochemistry: Electronic Structure Methods and Force Analysis.,” Chemical reviews. 2016. link Times cited: 113 Abstract: In quantum mechanochemistry, quantum chemical methods are us… read moreAbstract: In quantum mechanochemistry, quantum chemical methods are used to describe molecules under the influence of an external force. The calculation of geometries, energies, transition states, reaction rates, and spectroscopic properties of molecules on the force-modified potential energy surfaces is the key to gain an in-depth understanding of mechanochemical processes at the molecular level. In this review, we present recent advances in the field of quantum mechanochemistry and introduce the quantum chemical methods used to calculate the properties of molecules under an external force. We place special emphasis on quantum chemical force analysis tools, which can be used to identify the mechanochemically relevant degrees of freedom in a deformed molecule, and spotlight selected applications of quantum mechanochemical methods to point out their synergistic relationship with experiments. read less NOT USED (high confidence) J. Szala-Bilnik, M. Gomes, and A. Pádua, “Solvation of C60 fullerene and C60F48 fluorinated fullerene in molecular and ionic liquids.,” Journal of Physical Chemistry C. 2016. link Times cited: 9 Abstract: The association and dispersion of C60 fullerene and C60F48 f… read moreAbstract: The association and dispersion of C60 fullerene and C60F48 fluorinated fullerene in molecular and ionic solvents are studied using molecular dynamics simulations and dissolution experiments, with the aim of improving our understanding of nanocarbon materials interacting with liquid media. Fullerenes have uniform sizes and are devoid of edges and were chosen as models of wider classes of carbon nanomaterials. We parametrized a new atomistic force field for fluorinated nanocarbon materials and then used molecular dynamics to calculate structural quantities and potential of mean force. The aggregation in different solvents was assessed by UV/vis spectroscopy. The solvents considered include water, organic molecules, and ionic liquids, and we studied the effects of functional groups on the cation and of changing anions. Ionic liquids with a long alkyl chain on the cation are better solvents for C60F48 than for C60, revealing the stronger hydrophobic nature of C60F48. Surprisingly, an ionic liquid with a benzy... read less NOT USED (high confidence) B. Koo, A. Chattopadhyay, and L. L. Dai, “Atomistic modeling framework for a cyclobutane-based mechanophore-embedded nanocomposite for damage precursor detection,” Computational Materials Science. 2016. link Times cited: 11 NOT USED (high confidence) H. Zhan, G. Zhang, J. Bell, and Y. T. Gu, “Graphene with Patterned Fluorination: Morphology Modulation and Implications,” Journal of Physical Chemistry C. 2015. link Times cited: 11 Abstract: Recent years have witnessed a large volume of works on the m… read moreAbstract: Recent years have witnessed a large volume of works on the modification of graphene; however, an understanding of the associated morphology or mechanical properties changes is still lacking, which is vital for its engineering implementation. By taking the C4F fluorination as an example, we find that the morphology of both graphene sheet (GS) and graphene nanoribbon (GNR) can be effectively tailored by fluorination patterning via molecular dynamics simulations. The fluorine atom produces out-of-plane forces which trigger several intriguing morphology changes to monolayer graphene, including zigzag, folded, ruffle, nanoscroll, and chain structures. Notably, for multilayer GNR, the delamination and climbing phenomena of the surface layer are observed. Further studies show that the fluorination pattern can also be utilized to modulate the mechanical properties of graphene, e.g., about 40% increase of the effective yield strain is observed for the examined GNR with fluorination patterns. This study not only de... read less NOT USED (high confidence) N. Subramanian, A. Rai, and A. Chattopadhyay, “Atomistically informed stochastic multiscale model to predict the behavior of carbon nanotube-enhanced nanocomposites,” Carbon. 2015. link Times cited: 47 NOT USED (high confidence) R. Li, J. Zhou, Y. Han, J. Dong, and Y. Kawazoe, “A new (2 × 1) reconstructed edge structure of zigzag Si nanoribbon: first principles study.,” The Journal of chemical physics. 2013. link Times cited: 16 Abstract: Based upon the first principles calculations, a new (2 × 1) … read moreAbstract: Based upon the first principles calculations, a new (2 × 1) reconstructed edge structure (edge-4) with a triangle-pentagon pair topological defect at its edges is found for the zigzag Si nanoribbon, which is different from the previously found ones (edge-2 and edge-3) and more stable in energy than them. More interestingly, it is found that the edge-2 and edge-3 can transform into the new edge-4 under a little bit compression force along the ribbon edge, and the edge-4 could also be transformed into the edge-3 by a tensile strain larger than 9%. The calculated vibrational modes of the edge-4 show that two new characteristic vibrational edge defect modes appear at 434 cm(-1) and 515 cm(-1), which could be used in experiment to distinguish easily the new edge-4 from the edge-2 and edge-3. Finally, a sharp peak near the Fermi level is found to exist in the projected density of states from the edge's pz orbital of edge-4, making its energy bands spin-split and the antiferromagnetic state to be its ground state. read less NOT USED (high confidence) F. Karlický, K. K. R. Datta, M. Otyepka, and R. Zbořil, “Halogenated graphenes: rapidly growing family of graphene derivatives.,” ACS nano. 2013. link Times cited: 320 Abstract: Graphene derivatives containing covalently bound halogens (g… read moreAbstract: Graphene derivatives containing covalently bound halogens (graphene halides) represent promising two-dimensional systems having interesting physical and chemical properties. The attachment of halogen atoms to sp(2) carbons changes the hybridization state to sp(3), which has a principal impact on electronic properties and local structure of the material. The fully fluorinated graphene derivative, fluorographene (graphene fluoride, C1F1), is the thinnest insulator and the only stable stoichiometric graphene halide (C1X1). In this review, we discuss structural properties, syntheses, chemistry, stabilities, and electronic properties of fluorographene and other partially fluorinated, chlorinated, and brominated graphenes. Remarkable optical, mechanical, vibrational, thermodynamic, and conductivity properties of graphene halides are also explored as well as the properties of rare structures including multilayered fluorinated graphenes, iodine-doped graphene, and mixed graphene halides. Finally, patterned halogenation is presented as an interesting approach for generating materials with applications in the field of graphene-based electronic devices. read less NOT USED (high confidence) M. Radue, “Molecular Modeling of Aerospace Polymer Matrices Including Carbon Nanotube-Enhanced Epoxy.” 2017. link Times cited: 2 NOT USED (high confidence) H. Xiao, “Low-dimensional Material: Structure-property Relationship and Applications in Energy and Environmental Engineering.” 2017. link Times cited: 0 Abstract: Low-dimensional Material: Structure-property Relationship an… read moreAbstract: Low-dimensional Material: Structure-property Relationship and Applications in Energy and Environmental Engineering Hang Xiao In the past several decades, low-dimensional materials (0D materials, 1D materials and 2D materials) have attracted much interest from both the experimental and theoretical points of view. Because of the quantum confinement effect, low-dimensional materials have exhibited a kaleidoscope of fascinating phenomena and unusual physical and chemical properties, shedding light on many novel applications. Despite the enormous success has been achieved in the research of low-dimensional materials, there are three fundamental challenges of research in lowdimensional materials: 1) Develop new computational tools to accurately describe the properties of lowdimensional materials with low computational cost. 2) Predict and synthesize new low-dimensional materials with novel properties. 3) Reveal new phenomenon induced by the interaction between low-dimensional materials and the surrounding environment. In this thesis, atomistic modelling tools have been applied to address these challenges. We first developed ReaxFF parameters for phosphorus and hydrogen to give an accurate description of the chemical and mechanical properties of pristine and defected black phosphorene. ReaxFF for P/H is transferable to a wide range of phosphorus and hydrogen containing systems including bulk black phosphorus, blue phosphorene, edge-hydrogenated phosphorene, phosphorus clusters and phosphorus hydride molecules. The potential parameters were obtained by conducting global optimization with respect to a set of reference data generated by extensive ab initio calculations. We extended ReaxFF by adding a 60° correction term which significantly improved the description of phosphorus clusters. Emphasis was placed on the mechanical response of black phosphorene with different types of defects. Compared to the nonreactive SW potential of phosphorene, ReaxFF for P/H systems provides a significant improvement in describing the mechanical properties of the pristine and defected black phosphorene, as well as the thermal stability of phosphorene nanotubes. A counterintuitive phenomenon was observed that single vacancies weaken the black phosphorene more than double vacancies with higher formation energy. Our results also showed that the mechanical response of black phosphorene is more sensitive to defects in the zigzag direction than that in the armchair direction. Since ReaxFF allows straightforward extensions to the heterogeneous systems, such as oxides, nitrides, the proposed ReaxFF parameters for P/H systems also underpinned the reactive force field description of heterogeneous P systems, including Pcontaining 2D van der Waals heterostructures, oxides, etc. Based on the evolutionary algorithm driven structural search, we proposed a new stable trisulfur dinitride (S3N2) 2D crystal that is a covalent network composed solely of S-N σ bonds. S3N2 crystal is dynamically, thermally and chemically stable as confirmed by the computed phonon spectrum and ab initio molecular dynamics simulations. GW calculations showed that the 2D S3N2 crystal is a wide, direct band-gap (3.92 eV) semiconductor with a small hole effective mass. The anisotropic optical response of 2D S3N2 crystal was revealed by GW-BSE calculations. Our result not only marked the prediction of the first 2D crystal composed of nitrogen and sulfur, but also underpinned potential innovations in 2D electronics, optoelectronics, etc. Inspired by the discovery of S3N2 2D crystal, we proposed a new 2D crystal, diphosphorus trisulfide (P2S3), based on the extensive evolutionary algorithm driven structural search. The 2D P2S3 crystal was confirmed to be dynamically, thermally and chemically stable by the computed phonon spectrum and ab initio molecular dynamics simulations. This 2D crystalline phase of P2S3 corresponds to the global minimum in the Born-Oppenheimer surface of the phosphorus sulfide monolayers with 2:3 stoichiometry. It is a wide band gap (4.55 eV) semiconductor with P-S σ bonds. The electronic properties of P2S3 structure can be tuned by stacking into multilayer P2S3 structures, forming P2S3 nanoribbons or rolling into P2S3 nanotubes, expanding its potential applications for the emerging field of 2D electronics. Then we showed that the hydrolysis reaction is strongly affected by relative humidity. The hydrolysis of CO3 with n = 1-8 water molecules was investigated by ab initio method. For n = 15 water molecules, all the reactants follow a stepwise pathway to the transition state. For n = 6-8 water molecules, all the reactants undergo a direct proton transfer to the transition state with overall lower activation free energy. The activation free energy of the reaction is dramatically reduced from 10.4 to 2.4 kcal/mol as the number of water molecules increases from 1 to 6. Meanwhile, the degree of the hydrolysis of CO3 is significantly increased compared to the bulk water solution scenario. The incomplete hydration shells facilitate the hydrolysis of CO3 with few water molecules to be not only thermodynamically favorable but also kinetically favorable. We showed that the chemical kinetics is not likely to constrain the speed of CO2 air capture driven by the humidity-swing. Instead, the pore-diffusion of ions is expected to be the time-limiting step in the humidity driven CO2 air capture. The effect of humidity on the speed of CO2 air capture was studied by conducting CO2 absorption experiment using IER with a high ratio of CO3 to H2O molecules. Our result is able to provide valuable insights to designing efficient CO2 air-capture sorbents. Lastly, the self-assembly mechanism of one-end-open carbon nanotubes (CNTs) suspended in an aqueous solution was studied by molecular dynamics simulations. It was shown that two one-end-open CNTs with different diameters can coaxially self-assemble into a nanocapsule. The nanocapsules formed were stable in aqueous solution under ambient conditions, and the pressure inside the nanocapsule was much higher than the ambient pressure due to the van der Waals interactions between two parts of the nanocapsule. The effects of the normalized radius difference, normalized inter-tube distance and aspect ratio of the CNT pairs were systematically explored. The electric field response of nanocapsules was studied with ab initio molecular dynamics simulations, which showed that nanocapsules can be opened by applying an external electric field, due to the polarization of carbon atoms. This discovery not only shed light on a simple yet robust nanocapsule self-assembly mechanism, but also underpinned potential innovations in drug delivery, nano-reactors, etc. read less NOT USED (definite) J. Wang, X. Mu, and M. Sun, “The Thermal, Electrical and Thermoelectric Properties of Graphene Nanomaterials,” Nanomaterials. 2019. link Times cited: 51 Abstract: Graphene, as a typical two-dimensional nanometer material, h… read moreAbstract: Graphene, as a typical two-dimensional nanometer material, has shown its unique application potential in electrical characteristics, thermal properties, and thermoelectric properties by virtue of its novel electronic structure. The field of traditional material modification mainly changes or enhances certain properties of materials by mixing a variety of materials (to form a heterostructure) and doping. For graphene as well, this paper specifically discusses the use of traditional modification methods to improve graphene’s electrical and thermoelectrical properties. More deeply, since graphene is an atomic-level thin film material, its shape and edge conformation (zigzag boundary and armchair boundary) have a great impact on performance. Therefore, this paper reviews the graphene modification field in recent years. Through the change in the shape of graphene, the change in the boundary structure configuration, the doping of other atoms, and the formation of a heterostructure, the electrical, thermal, and thermoelectric properties of graphene change, resulting in broader applications in more fields. Through studies of graphene’s electrical, thermal, and thermoelectric properties in recent years, progress has been made not only in experimental testing, but also in theoretical calculation. These aspects of graphene are reviewed in this paper. read less NOT USED (definite) D. Kirilenko, A. Gorodetsky, and M. Baidakova, “Influence of charge carriers on corrugation of suspended graphene,” Solid State Communications. 2018. link Times cited: 1 NOT USED (definite) N. Subramanian, B. Koo, A. Rai, and A. Chattopadhyay, “Molecular dynamics-based multiscale damage initiation model for CNT/epoxy nanopolymers,” Journal of Materials Science. 2018. link Times cited: 15 |