Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
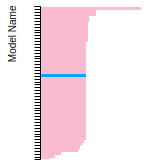
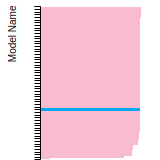
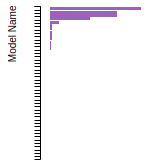
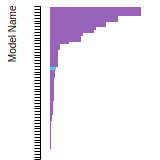
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.

437 Citations (326 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) B. Meng, D. Yuan, and S. Xu, “Atomic-Scale Characterization of Slip Deformation and Nanometric Machinability of Single-Crystal 6H-SiC,” Nanoscale Research Letters. 2019. link Times cited: 15 USED (definite) A. Javanainen et al., “Molecular Dynamics Simulations of Heavy Ion Induced Defects in SiC Schottky Diodes,” IEEE Transactions on Device and Materials Reliability. 2018. link Times cited: 8 Abstract: Heavy ion irradiation increases the leakage current in rever… read moreAbstract: Heavy ion irradiation increases the leakage current in reverse-biased SiC Schottky diodes. This letter demonstrates, via molecular dynamics simulations, that a combination of bias and ion-deposited energy is required to produce the degradation. read less USED (definite) P. G. Stubley, A. Higginbotham, and J. Wark, “Simulations of the inelastic response of silicon to shock compression,” Computational Materials Science. 2017. link Times cited: 2 USED (definite) J. Tao and Y. Sun, “The Elastic Property of Bulk Silicon Nanomaterials through an Atomic Simulation Method,” Journal of Nanomaterials. 2016. link Times cited: 3 Abstract: This paper reports a systematic study on the elastic propert… read moreAbstract: This paper reports a systematic study on the elastic property of bulk silicon nanomaterials using the atomic finite element method. The Tersoff-Brenner potential is used to describe the interaction between silicon atoms, and the atomic finite element method is constructed in a computational scheme similar to the continuum finite element method. Youngźs modulus and Poisson ratio are calculated for [100], [110], and [111] silicon nanowires that are treated as three-dimensional structures. It is found that the nanowire possesses the lowest Youngźs modulus along the [100] direction, while the [110] nanowire has the highest value with the same radius. The bending deformation of [100] silicon nanowire is also modeled, and the bending stiffness is calculated. read less USED (definite) S. Chavoshi, S. Goel, and X. Luo, “Influence of temperature on the anisotropic cutting behaviour of single crystal silicon: A molecular dynamics simulation investigation,” Journal of Manufacturing Processes. 2016. link Times cited: 79 USED (definite) S. Chavoshi, S. Xu, and X. Luo, “Dislocation-mediated plasticity in silicon during nanometric cutting : a molecular dynamics simulation study materials science in semiconductor processing,” Materials Science in Semiconductor Processing. 2016. link Times cited: 50 USED (definite) T. Susi, “Heteroatom quantum corrals and nanoplasmonics in graphene (HeQuCoG),” Research Ideas and Outcomes. 2015. link Times cited: 6 Abstract: The objective of the Heteroatom quantum corrals and nanoplas… read moreAbstract: The objective of the Heteroatom quantum corrals and nanoplasmonics in graphene (HeQuCoG) project is to create atomically precise structures made of silicon and phosphorus atoms embedded in the lattice of graphene. This will be achieved by combining proven modeling techniques with sample fabrication via carefully controlled ion implantation, and subsequent manipulation in an atomic resolution scanning transmission electron microscope (STEM). The structures will be computationally designed for interesting nanoplasmonic enhancement and quantum confinement properties, and characterized by electron energy loss spectroscopy mapping in the STEM. The expected outcome is a systematic demonstration of truly atomic-level material design and the creation of freestanding “quantum corral” structures for the first time. read less USED (definite) B. Zheng and H. Du, “A Study of Disorder Shell Effects on the Mechanical Properties of SiC Nanowires,” Strojniski Vestnik-journal of Mechanical Engineering. 2015. link Times cited: 2 Abstract: The mechanical properties of SiC nanowires were investigated… read moreAbstract: The mechanical properties of SiC nanowires were investigated using molecular dynamics simulation method. The results show that the disorder shell layer reduced the elastic modulus of SiC nanowires. This reduction mainly depends on the thickness and the atomic type of the disorder shell. Thicker C and Si disorder layers can strengthen and weaken the nanowires, respectively. Also, the core-shell wires have size-dependent strength, which can be understood by examining the variation of Young’s modulus and the volume fraction of the isolated core and isolated shell. Furthermore, the disorder coating was found to facilitate the brittle-ductile transition in the SiC core. The simulation results are expected to help the design and manufacturing of complex nanoscale architectures with desired mechanical properties. read less USED (definite) S. Goel, N. H. Faisal, X. Luo, J. Yan, and A. Agrawal, “Nanoindentation of polysilicon and single crystal silicon: Molecular dynamics simulation and experimental validation,” Journal of Physics D: Applied Physics. 2014. link Times cited: 95 Abstract: This paper presents novel advances in the deformation behavi… read moreAbstract: This paper presents novel advances in the deformation behaviour of polycrystalline and single crystal silicon using molecular dynamics (MD) simulation and validation of the same via nanoindentation experiments. In order to unravel the mechanism of deformation, four simulations were performed: indentation of a polycrystalline silicon substrate with a (i) Berkovich pyramidal and a (ii) spherical (arc) indenter, and (iii and iv) indentation of a single crystal silicon substrate with these two indenters. The simulation results reveal that high pressure phase transformation (HPPT) in silicon (Si-I to Si-II phase transformation) occurred in all cases; however, its extent and the manner in which it occurred differed significantly between polycrystalline silicon and single crystal silicon, and was the main driver of differences in the nanoindentation deformation behaviour between these two types of silicon. Interestingly, in polycrystalline silicon, the HPPT was observed to occur more preferentially along the grain boundaries than across the grain boundaries. An automated dislocation extraction algorithm (DXA) revealed no dislocations in the deformation zone, suggesting that HPPT is the primary mechanism in inducing plasticity in silicon. read less USED (definite) X. Zhang, M. Hu, K. Giapis, and D. Poulikakos, “Schemes for and Mechanisms of Reduction in Thermal Conductivity in Nanostructured Thermoelectrics,” Journal of Heat Transfer-transactions of The Asme. 2012. link Times cited: 20 Abstract: Nonequilibrium molecular dynamics (NEMD) simulations were pe… read moreAbstract: Nonequilibrium molecular dynamics (NEMD) simulations were performed to investigate schemes for enhancing the energy conversion efficiency of thermoelectric nanowires (NWs), including (1) roughening of the nanowire surface, (2) creating nanoparticle inclusions in the nanowires, and (3) coating the nanowire surface with other materials. The enhancement in energy conversion efficiency was inferred from the reduction in thermal conductivity of the nanowire, which was calculated by imposing a temperature gradient in the longitudinal direction. Compared to pristine nanowires, our simulation results show that the schemes proposed above lead to nanocomposite structures with considerably lower thermal conductivity (up to 82% reduction), implying ~5X enhancement in the ZT coefficient. This significant effect appears to have two origins: (1) increase in phonon-boundary scattering and (2) onset of interfacial interference. The results suggest new fundamental–yet realizable ways to improve markedly the energy conversion efficiency of nanostructured thermoelectrics. read less USED (definite) S. Goel, X. Luo, R. Reuben, and H. Pen, “Influence of temperature and crystal orientation on tool wear during single point diamond turning of silicon,” Wear. 2012. link Times cited: 96 USED (definite) S. Chavoshi, S. Goel, and X. Luo, “Molecular dynamics simulation investigation on the plastic flow behaviour of silicon during nanometric cutting,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 44 Abstract: Molecular dynamics (MD) simulation was carried out to acquir… read moreAbstract: Molecular dynamics (MD) simulation was carried out to acquire an in-depth understanding of the flow behaviour of single crystal silicon during nanometric cutting on three principal crystallographic planes and at different cutting temperatures. The key findings were that (i) the substrate material underneath the cutting tool was observed for the first time to experience a rotational flow akin to fluids at all the tested temperatures up to 1200 K. (ii) The degree of flow in terms of vorticity was found higher on the (1 1 1) crystal plane signifying better machinability on this orientation in accord with the current pool of knowledge (iii) an increase in the machining temperature reduces the spring-back effect and thereby the elastic recovery and (iv) the cutting orientation and the cutting temperature showed significant dependence on the location of the stagnation region in the cutting zone of the substrate. read less USED (high confidence) J. Zhou, J. Li, and J. Zhang, “Intrinsic auxeticity and mechanical anisotropy of Si9C15 siligraphene.,” Nanoscale. 2023. link Times cited: 0 Abstract: Graphene-like two-dimensional (2D) silicon carbide or siligr… read moreAbstract: Graphene-like two-dimensional (2D) silicon carbide or siligraphene has attracted remarkable attention, owing to its fascinating physical properties. Nevertheless, the first high-quality siligraphene, i.e. monolayer Si9C15, was synthesised very recently, which exhibits an excellent semiconducting behaviour. In this work, we investigate the mechanical properties of Si9C15 siligraphene by using atomistic simulations including density functional theory (DFT) calculations and molecular dynamics (MD) simulations. Both methods confirm the existence of intrinsic negative Poisson's ratios in Si9C15 siligraphene, which, as illustrated by MD simulations, is attributed to the tension-induced de-wrinkling behaviours of its intrinsic rippled configuration. Different de-wrinkling behaviours are observed in different directions of Si9C15 siligraphene, which result in the anisotropy of its auxetic properties. The fracture properties of Si9C15 siligraphene are similarly anisotropic, but relatively large fracture strains are observed in different orientations, indicating the stretchability of Si9C15 siligraphene. The stretchability together with the strain-sensitive bandgap of Si9C15 siligraphene observed in DFT calculations indicates the effectiveness of strain engineering in modulating its electronic properties. The combination of unique auxetic properties, excellent mechanical properties and tunable electronic properties may make Si9C15 siligraphene a novel 2D material with multifunctional applications. read less USED (high confidence) Z. Li et al., “A nanodispersion-in-nanograins strategy for ultra-strong, ductile and stable metal nanocomposites,” Nature Communications. 2022. link Times cited: 17 USED (high confidence) K. Lin et al., “Dynamic Strength, Reinforcing Mechanism and Damage of Ceramic Metal Composites,” International Journal of Mechanical Sciences. 2022. link Times cited: 7 USED (high confidence) J. Wang, F. Fang, and L. Li, “Cutting of Graphite at Atomic and Close-to-Atomic Scale Using Flexible Enhanced Molecular Dynamics,” Nanomanufacturing and Metrology. 2022. link Times cited: 6 USED (high confidence) M. S. Islam, I. Mia, A. Islam, C. Stampfl, and J. Park, “Temperature and interlayer coupling induced thermal transport across graphene/2D-SiC van der Waals heterostructure,” Scientific Reports. 2022. link Times cited: 4 USED (high confidence) A. Ravichandran, M. Mehta, A. Woodworth, and J. Lawson, “Molecular dynamics simulations of ultrafast radiation induced melting at metal–semiconductor interfaces,” Journal of Applied Physics. 2021. link Times cited: 3 Abstract: Metal–semiconductor contacts in silicon carbide (SiC) diodes… read moreAbstract: Metal–semiconductor contacts in silicon carbide (SiC) diodes endure damages at the interface when exposed to harsh radiation environments. Due to the rapid rise in temperature and ultrafast cooling that follows the radiation impact, the structural properties of the materials can be altered through melting, recrystallization, and amorphization. A detailed understanding of the material failure modes at the interface is lacking, specifically at the nanoscale. We use molecular simulations to investigate the ultrafast melting at tungsten (W)–SiC interfaces following radiation damage and apply deep learning techniques to track the transient evolution of the local molecular structures. We show that W near the radiation track undergoes melting and, eventually, most of it recrystallizes with a noticeable degree of undercooling, while SiC is rendered permanently amorphous. The observation of local undercooling in W films is important as it can affect the device performance even before the bulk melting temperature of the material is reached. We also show that at high temperatures, the interface undergoes a fracture-like failure. The results presented here are significant in understating the different failure modes of SiC diode materials. read less USED (high confidence) J. Clayton, M. Guziewski, J. Ligda, R. B. Leavy, and J. Knap, “A Multi-Scale Approach for Phase Field Modeling of Ultra-Hard Ceramic Composites,” Materials. 2021. link Times cited: 7 Abstract: Diamond-silicon carbide (SiC) polycrystalline composite blen… read moreAbstract: Diamond-silicon carbide (SiC) polycrystalline composite blends are studied using a computational approach combining molecular dynamics (MD) simulations for obtaining grain boundary (GB) fracture properties and phase field mechanics for capturing polycrystalline deformation and failure. An authentic microstructure, reconstructed from experimental lattice diffraction data with locally refined discretization in GB regions, is used to probe effects of local heterogeneities on material response in phase field simulations. The nominal microstructure consists of larger diamond and SiC (cubic polytype) grains, a matrix of smaller diamond grains and nanocrystalline SiC, and GB layers encasing the larger grains. These layers may consist of nanocrystalline SiC, diamond, or graphite, where volume fractions of each phase are varied within physically reasonable limits in parametric studies. Distributions of fracture energies from MD tension simulations are used in the phase field energy functional for SiC-SiC and SiC-diamond interfaces, where grain boundary geometries are obtained from statistical analysis of lattice orientation data on the real microstructure. An elastic homogenization method is used to account for distributions of second-phase graphitic inclusions as well as initial voids too small to be resolved individually in the continuum field discretization. In phase field simulations, SiC single crystals may twin, and all phases may fracture. The results of MD calculations show mean strengths of diamond-SiC interfaces are much lower than those of SiC-SiC GBs. In phase field simulations, effects on peak aggregate stress and ductility from different GB fracture energy realizations with the same mean fracture energy and from different random microstructure orientations are modest. Results of phase field simulations show unconfined compressive strength is compromised by diamond-SiC GBs, graphitic layers, graphitic inclusions, and initial porosity. Explored ranges of porosity and graphite fraction are informed by physical observations and constrained by accuracy limits of elastic homogenization. Modest reductions in strength and energy absorption are witnessed for microstructures with 4% porosity or 4% graphite distributed uniformly among intergranular matrix regions. Further reductions are much more severe when porosity is increased to 8% relative to when graphite is increased to 8%. read less USED (high confidence) E. Mareev, B. Rumiantsev, and F. Potemkin, “Study of the Parameters of Laser-Induced Shock Waves for Laser Shock Peening of Silicon,” JETP Letters. 2020. link Times cited: 6 USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, P. B. Odedeyi, M. Liman, and A. I. Jumare, “RSM and MD—a roughness predictive model and simulation comparison of monocrystalline optical grade silicon,” The International Journal of Advanced Manufacturing Technology. 2020. link Times cited: 1 USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, P. B. Odedeyi, M. Liman, and A. I. Jumare, “RSM and MD—a roughness predictive model and simulation comparison of monocrystalline optical grade silicon,” The International Journal of Advanced Manufacturing Technology. 2020. link Times cited: 0 USED (high confidence) Z. Xu et al., “Nanocutting mechanism of 6H-SiC investigated by scanning electron microscope online observation and stress-assisted and ion implant-assisted approaches,” The International Journal of Advanced Manufacturing Technology. 2020. link Times cited: 14 USED (high confidence) Z. Xu et al., “Nanocutting mechanism of 6H-SiC investigated by scanning electron microscope online observation and stress-assisted and ion implant-assisted approaches,” The International Journal of Advanced Manufacturing Technology. 2020. link Times cited: 0 USED (high confidence) X. Ge et al., “Distinguishing contributions of ceramic matrix and binder metal to the plasticity of nanocrystalline cermets,” IUCrJ. 2020. link Times cited: 4 Abstract: Contributions to plasticity from hard matrix and binder meta… read moreAbstract: Contributions to plasticity from hard matrix and binder metal in nanocrystalline cermets were studied by molecular dynamics simulations. read less USED (high confidence) B. Wen et al., “Continuous strengthening in nanotwinned diamond,” npj Computational Materials. 2019. link Times cited: 31 USED (high confidence) M. Liman, K. Abou-El-Hossein, and L. N. Abdulkadir, “Analysis of diamond nanomachining of contact lens polymers using molecular dynamics,” The International Journal of Advanced Manufacturing Technology. 2019. link Times cited: 3 USED (high confidence) J. Han et al., “Reveal the Deformation Mechanism of (110) Silicon from Cryogenic Temperature to Elevated Temperature by Molecular Dynamics Simulation,” Nanomaterials. 2019. link Times cited: 10 Abstract: Silicon undergoes a brittle-to-ductile transition as its cha… read moreAbstract: Silicon undergoes a brittle-to-ductile transition as its characteristic dimension reduces from macroscale to nanoscale. The thorough understanding of the plastic deformation mechanism of silicon at the nanoscale is still challenging, although it is essential for developing Si-based micro/nanoelectromechanical systems (MEMS/NEMS). Given the wide application of silicon in extreme conditions, it is, therefore, highly desirable to reveal the nanomechanical behavior of silicon from cryogenic temperature to elevated temperature. In this paper, large-scale molecular dynamics (MD) simulations were performed to reveal the spherical nanoindentation response and plastic deformation mechanism of (110)Si at the temperature range of 0.5 K to 573 K. Special attention was paid to the effect of temperature. Multiple pop-ins detected in load/pressure-indentation strain curves are impacted by temperature. Four featured structures induced by nanoindentation, including high-pressure phases, extrusion of α-Si, dislocations, and crack, are observed at all temperatures, consistent with experiment results. The detailed structure evolution of silicon was revealed at the atomic scale and its dependence on temperature was analyzed. Furthermore, structure changes were correlated with pop-ins in load/pressure-indentation strain curves. These results may advance our understanding of the mechanical properties of silicon. read less USED (high confidence) L. Liu et al., “MD simulation of stress-assisted nanometric cutting mechanism of 3C silicon carbide,” Industrial Lubrication and Tribology. 2019. link Times cited: 9 Abstract:
Purpose
This paper aims to reveal the mechanism for improvi… read moreAbstract:
Purpose
This paper aims to reveal the mechanism for improving ductile machinability of 3C-silicon carbide (SiC) and associated cutting mechanism in stress-assisted nanometric cutting.
Design/methodology/approach
Molecular dynamics simulation of nano-cutting 3C-SiC is carried out in this paper. The following two scenarios are considered: normal nanometric cutting of 3C-SiC; and stress-assisted nanometric cutting of 3C-SiC for comparison. Chip formation, phase transformation, dislocation activities and shear strain during nanometric cutting are analyzed.
Findings
Negative rake angle can produce necessary hydrostatic stress to achieve ductile removal by the extrusion in ductile regime machining. In ductile-brittle transition, deformation mechanism of 3C-SiC is combination of plastic deformation dominated by dislocation activities and localization of shear deformation. When cutting depth is greater than 10 nm, material removal is mainly achieved by shear. Stress-assisted machining can lead to better quality of machined surface. However, there is a threshold for the applied stress to fully gain advantages offered by stress-assisted machining. Stress-assisted machining further enhances plastic deformation ability through the active dislocations’ movements.
Originality/value
This work describes a stress-assisted machining method for improving the surface quality, which could improve 3C-SiC ductile machining ability.
read less USED (high confidence) G. Opletal, B. Sun, T. Petersen, S. Russo, and A. Barnard, “Vacancy induced formation of nanoporous silicon, carbon and silicon carbide.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 6 Abstract: Nanoporous semiconductors are used in a range of application… read moreAbstract: Nanoporous semiconductors are used in a range of applications from sensing and gas separation, to photovoltaics, rechargeable batteries, energetic materials and micro electro mechanical systems. In most cases porosity occurs in conjunction with the competing process of amorphisation, creating a complicated material that responds differently to strain and density changes, depending on the composition. In this paper we use simple computational workflow involving Monte Carlo simulation, numerical characterisation and statistical analysis to explore the development of amorphous and nanoporous carbon, silicon and silicon carbide. We show that amorphous regions in Si and SiC form in advance of nanopores, and are essential in stabilising the nanopores once developed. Carbon prefers a porous structure at lower strains than amorphisation and exhibits a bimodal change in the structure which correlates with the change in C-C bond angles from tetrahedral sp3-like bonds to hexagonal sp2-like bonds as the strain increases. These results highlight how both of these processes can be analysed simultaneously using reliable interatomic forcefields or density functionals, provided sufficient samples are included to support the statistics. read less USED (high confidence) S. Chavoshi and S. Xu, “Nanoindentation/scratching at finite temperatures: Insights from atomistic-based modeling,” Progress in Materials Science. 2019. link Times cited: 37 USED (high confidence) Y. Fan, Y. Xiang, and H. S. Shen, “Temperature-dependent negative Poisson’s ratio of monolayer graphene: Prediction from molecular dynamics simulations,” Nanotechnology Reviews. 2019. link Times cited: 25 Abstract: A temperature-dependent intrinsic property of monolayer grap… read moreAbstract: A temperature-dependent intrinsic property of monolayer graphene, the negative Poisson’s ratio (NPR), is investigated in the present study. The classical molecular dynamics (MD) method is employed and the Erhart-Albe hybrid potential, i.e. the combination of the reactive empirical bond order (REBO) and the Tersoff potentials, is used for the graphene sheet in the numerical simulation. In the simulation process, the graphene sheet is assumed to be free standing with in-plane periodical boundary condition and under an ambient temperature up to 1000 K. Our study shows that the graphene NPR is decreased with the increase of temperature. Besides, we also perform the simulation of the graphene negative temperature expansion coefficient (NTEC) as an indirect validation of the present MD model. The characteristics of the nonlinear variations for both the NPR and the NTEC of a pristine graphene sheet are investigated. Our MD results at low temperature (0.1 K) further prove the intrinsic and anisotropic property of NPR for graphene. read less USED (high confidence) L. N. Abdulkadir and K. Abou-El-Hossein, “Observed edge radius behavior during MD nanomachining of silicon at a high uncut chip thickness,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 12 USED (high confidence) L. N. Abdulkadir and K. Abou-El-Hossein, “Observed edge radius behavior during MD nanomachining of silicon at a high uncut chip thickness,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 0 USED (high confidence) B. Reischl, A. Rohl, A. Kuronen, and K. Nordlund, “Atomistic simulation of the measurement of mechanical properties of gold nanorods by AFM,” Scientific Reports. 2017. link Times cited: 6 USED (high confidence) L. Wang, H. Ke, J. Ma, and J. Liu, “Investigation of the ‘double cross’ splitting mechanism of single-crystal diamond under nanoindentation via molecular dynamics simulation,” Journal of Molecular Modeling. 2017. link Times cited: 3 USED (high confidence) D. N. Alamdary, J. Kotakoski, and T. Susi, “Structure and Energetics of Embedded Si Patterns in Graphene,” physica status solidi (b). 2017. link Times cited: 4 Abstract: Recent experiments have revealed the possibility of precise … read moreAbstract: Recent experiments have revealed the possibility of precise electron beam manipulation of silicon impurities in graphene. Motivated by these findings and studies on metal surface quantum corrals, the question arises what kind of embedded Si structures are possible within the hexagonal lattice, and how these are limited by the distortion caused by the preference of Si for sp3 hybridization. In this work, we study the geometry and stability of elementary Si patterns in graphene, including lines, hexagons, triangles, circles, and squares. Due to the size of the required unit cells, to obtain the relaxed geometries we use an empirical bond‐order potential as a starting point for density functional theory. Despite some interesting discrepancies, the classical geometries provide an effective route for the simulation of large structures. read less USED (high confidence) S. Goel, S. Chavoshi, and A. Murphy, “Molecular dynamics simulation (MDS) to study nanoscale machining processes.” 2017. link Times cited: 2 Abstract: 1 Molecular dynamics simulation (MDS) to study nanoscale cut… read moreAbstract: 1 Molecular dynamics simulation (MDS) to study nanoscale cutting processes Saurav Goel1*, Saeed Zare Chavoshi2 and Adrian Murphy3 1Precision Engineering Institute, School of Aerospace, Transport and Manufacturing, Cranfield University, Cranfield, Bedfordshire, MK430AL, UK 2Mechanical Engineering Department, Imperial College London, London, SW7 2AZ, UK 3School of Mechanical and Aerospace Engineering, Queen’s University, Belfast, BT9 5AH, UK *Corresponding author Tel.: +44 1234754132, Email address: sgoel.diamond@gmail.com read less USED (high confidence) E. Lee, T. Zhang, M. Hu, and T. Luo, “Thermal boundary conductance enhancement using experimentally achievable nanostructured interfaces - analytical study combined with molecular dynamics simulation.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 20 Abstract: Interfacial thermal resistance presents great challenges to … read moreAbstract: Interfacial thermal resistance presents great challenges to the thermal management of modern electronics. In this work, we perform an analytical study to enhance the thermal boundary conductance (TBC) of nanostructured interfaces with square-shape pillar arrays, extendable to the characteristic lengths that can be fabricated in practice. As a representative system, we investigate a SiC substrate with the square-shape pillar array combined with epitaxial GaN as the nanostructured interface. By applying a first-order ray tracing method and molecular dynamics simulations to analyze phonon incidence and transmission at the nanostructured interface, we systematically study the impact of the characteristic dimensions of the pillar array on the TBC. Based on the multi-scale analysis we provide a general guideline to optimize the nanostructured interfaces to achieve higher TBC, demonstrating that the optimized TBC value of the nanostructured SiC/GaN interfaces can be 42% higher than that of the planar SiC/GaN interfaces without nanostructures. The model used and results obtained in this study will guide the further experimental realization of nanostructured interfaces for better thermal management in microelectronics. read less USED (high confidence) C. Y. Chuang, A. Sattler, and T. Sinno, “Thermodynamic and morphological analysis of large silicon self-interstitial clusters using atomistic simulations,” Journal of Applied Physics. 2015. link Times cited: 5 Abstract: We study computationally the formation of thermodynamics and… read moreAbstract: We study computationally the formation of thermodynamics and morphology of silicon self-interstitial clusters using a suite of methods driven by a recent parameterization of the Tersoff empirical potential. Formation free energies and cluster capture zones are computed across a wide range of cluster sizes (2 < Ni < 150) and temperatures (0.65 < T/Tm < 1). Self-interstitial clusters above a critical size (Ni ∼ 25) are found to exhibit complex morphological behavior in which clusters can assume either a variety of disordered, three-dimensional configurations, or one of two macroscopically distinct planar configurations. The latter correspond to the well-known Frank and perfect dislocation loops observed experimentally in ion-implanted silicon. The relative importance of the different cluster morphologies is a function of cluster size and temperature and is dictated by a balance between energetic and entropic forces. The competition between these thermodynamic forces produces a sharp transition between the t... read less USED (high confidence) M.-Q. Le, “Prediction of Young’s modulus of hexagonal monolayer sheets based on molecular mechanics,” International Journal of Mechanics and Materials in Design. 2015. link Times cited: 31 USED (high confidence) M.-Q. Le, “Prediction of Young’s modulus of hexagonal monolayer sheets based on molecular mechanics,” International Journal of Mechanics and Materials in Design. 2014. link Times cited: 0 USED (high confidence) M.-Q. Le, “Young’s modulus prediction of hexagonal nanosheets and nanotubes based on dimensional analysis and atomistic simulations,” Meccanica. 2014. link Times cited: 17 USED (high confidence) M.-Q. Le, “Young’s modulus prediction of hexagonal nanosheets and nanotubes based on dimensional analysis and atomistic simulations,” Meccanica. 2014. link Times cited: 0 USED (high confidence) H. Chen and S. Chen, “The fracture behaviors of carbon nanotube and nanoscroll reinforced silicon matrix composites,” Carbon. 2014. link Times cited: 10 USED (high confidence) B. Kappes and C. Ciobanu, “Bandgap Opening in Metallic Carbon Nanotubes Due toSilicon Adatoms,” Cmc-computers Materials & Continua. 2013. link Times cited: 1 Abstract: Controlling the bandgap of carbon nanostructures is a key fa… read moreAbstract: Controlling the bandgap of carbon nanostructures is a key factor in the development of mainstream applications of carbon-based nanoelectronic devices. This is particularly important in the cases where it is desired that the carbon nanos- tructures are the active elements, as opposed to being conductive leads between other elements of the device. Here, we report density functional theory calculations of the effect of silicon impurities on the electronic properties of carbon nanotubes (CNTs). We have found that Si adatoms can open up a bandgap in intrinsically metallic CNTs, even when the linear density of Si atoms is low enough that they do not create an adatom chain along the tube. The bandgap opened in metallic CNTs can range up to approximately 0.47 eV, depending on adsorption site, on the linear density of Si adatoms, and on the chirality of the nanotube. We have found that a lower spatial symmetry of the charge transfer between adatom and CNT leads to a higher value of the bandgap opened, which indicates that the physical origin of the bandgap lies in the reduced spatial symmetry of the charge transferred. read less USED (high confidence) Y. Jing, Y. Sun, H. Niu, and J. Shen, “Atomistic simulations on the mechanical properties of silicene nanoribbons under uniaxial tension,” physica status solidi (b). 2013. link Times cited: 37 Abstract: The mechanical properties of silicene are investigated using… read moreAbstract: The mechanical properties of silicene are investigated using ab initio calculation and molecular dynamics simulations with different empirical potentials. The simulation results show that the calculated Young's modulus of bulk silicene with EDIP model is consistent with the ab initio calculations. The chirality has a significant effect on the critical strain and stress of bulk silicene under uniaxial tension. In addition, the Young's modulus depends strongly on the chirality and size of the silicene nanoribbon due to the edge effects. The fracture process of a silicene nanoribbon is also studied. read less USED (high confidence) S. Goel, A. Stukowski, X. Luo, A. Agrawal, and R. Reuben, “Anisotropy of single-crystal 3C–SiC during nanometric cutting,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 94 Abstract: 3C–SiC (the only polytype of SiC that resides in a diamond c… read moreAbstract: 3C–SiC (the only polytype of SiC that resides in a diamond cubic lattice structure) is a relatively new material that exhibits most of the desirable engineering properties required for advanced electronic applications. The anisotropy exhibited by 3C–SiC during its nanometric cutting is significant, and the potential for its exploitation has yet to be fully investigated. This paper aims to understand the influence of crystal anisotropy of 3C–SiC on its cutting behaviour. A molecular dynamics simulation model was developed to simulate the nanometric cutting of single-crystal 3C–SiC in nine (9) distinct combinations of crystal orientations and cutting directions, i.e. (1 1 1) 〈−1 1 0〉, (1 1 1) 〈−2 1 1〉, (1 1 0) 〈−1 1 0〉, (1 1 0) 〈0 0 1〉, (1 1 0) 〈1 1 −2〉, (0 0 1) 〈−1 1 0〉, (0 0 1) 〈1 0 0〉, (1 1 −2) 〈1 −1 0〉 and (1 −2 0) 〈2 1 0〉. In order to ensure the reliability of the simulation results, two separate simulation trials were carried out with different machining parameters. In the first trial, a cutting tool rake angle of −25°, d/r (uncut chip thickness/cutting edge radius) ratio of 0.57 and cutting velocity of 10 m s−1 were used whereas a second trial was done using a cutting tool rake angle of −30°, d/r ratio of 1 and cutting velocity of 4 m s−1. Both the trials showed similar anisotropic variation. The simulated orthogonal components of thrust force in 3C–SiC showed a variation of up to 45%, while the resultant cutting forces showed a variation of 37%. This suggests that 3C–SiC is highly anisotropic in its ease of deformation. These results corroborate with the experimentally observed anisotropic variation of 43.6% in Young's modulus of 3C–SiC. The recently developed dislocation extraction algorithm (DXA) [, ] was employed to detect the nucleation of dislocations in the MD simulations of varying cutting orientations and cutting directions. Based on the overall analysis, it was found that 3C–SiC offers ease of deformation on either (1 1 1) 〈−1 1 0〉, (1 1 0) 〈0 0 1〉, or (1 0 0) 〈1 0 0〉 setups. read less USED (high confidence) J. Schall and J. Harrison, “Reactive Bond-Order Potential for Si-, C-, and H-Containing Materials,” Journal of Physical Chemistry C. 2013. link Times cited: 8 Abstract: A new bond-order potential for modeling systems containing s… read moreAbstract: A new bond-order potential for modeling systems containing silicon, carbon, and hydrogen, such as organosilicon molecules (CxSiyHz), solid silicon, solid carbon, and alloys of silicon and carbon, is presented. This reactive potential utilizes the formalism of the second-generation reactive empirical bond-order potential (REBO) [Brenner et al. J. Phys.: Condens. Matter 2002, 14, 783] for hydrocarbons and the REBO parameters for silicon [Schall, Gao, Harrison. Phys. Rev. B 2008, 77, 115209]. Modifications to the hydrocarbon REBO potential were made to improve the description of three-atom type systems. The widespread use of Brenner’s REBO potential, its ability to model a wide range of hydrocarbon materials, and the existence of parameters for several atom types are some of the motivating factors for obtaining this Si–C–H (2B-SiCH) parametrization. The usefulness and flexibility of this potential is demonstrated by examining the properties of organosilicon molecules, the bulk, surface, and defect properties... read less USED (high confidence) Y. Jing, K. Yu, X. Qin, and J. Shen, “Composition-dependent mechanical and thermal transport properties of carbon/silicon core/shell nanowires,” Journal of Shanghai Jiaotong University (Science). 2012. link Times cited: 0 USED (high confidence) Y. Jing, K. Yu, X. Qin, and J. Shen, “Composition-dependent mechanical and thermal transport properties of carbon/silicon core/shell nanowires,” Journal of Shanghai Jiaotong University (Science). 2012. link Times cited: 0 USED (high confidence) B.-H. Kim, K.-R. Lee, Y.-C. Chung, and J.-G. Lee, “Effects of interfacial bonding in the Si-carbon nanotube nanocomposite: A molecular dynamics approach,” Journal of Applied Physics. 2012. link Times cited: 11 Abstract: We investigated the effects of interfacial bonding on the me… read moreAbstract: We investigated the effects of interfacial bonding on the mechanical properties in the Si-carbon nanotube (CNT) nanocomposite by a molecular dynamics approach. To describe the system appropriately, we used a hybrid potential that includes Tersoff, AIREBO (adaptive intermolecular reactive empirical bond order), and Lennard–Jones potentials. With increasing bonding strength at the interface of Si matrix and CNT, toughness as well as Young’s modulus and maximum strength increased steadily. CNT pull-out and load transfer on the strong CNT were identified as the main mechanisms for the enhanced properties. At optimum bonding, crack tip was deflected around CNT and the fracture proceeded in plastic mode through Si matrix owing to the strong reinforcement of CNT, and resulted in a further enhancement of toughness. At maximum bonding, however, only load transfer is operative and the fracture returned to brittle mode. We concluded that a strong interface as long as the CNT maintains its structural integrity is des... read less USED (high confidence) T. Nakajima and K. Shintani, “Controlling out‐of‐plane deformations of graphene nanobridges,” physica status solidi (b). 2011. link Times cited: 3 Abstract: Graphene nanoribbons (GNRs) can be applied to transistors, m… read moreAbstract: Graphene nanoribbons (GNRs) can be applied to transistors, mass sensors, and dust detectors. Suspended GNRs which connect terminals in electronic devices like bridges can be treated as edge‐constrained GNRs. In this paper, edge‐constrained GNRs of various sizes and initial strains are studied using molecular dynamics (MD) simulations. To induce strain in GNRs, bond lengths between carbon atoms of the initial configurations of GNR models are varied. The bond length of the energetically stable GNRs is estimated at 1.47 Å. At this bond length, GNRs obviously change the tendencies of their energies, amplitudes, and deformations. The relationships between the out‐of‐plane deformations and the sizes of GNRs, and between the out‐of‐plane deformations and strains of GNRs are studied. Under compressive strain, the out‐of‐plane deformation of GNRs is dominantly caused by buckling. The amplitude of the buckling decreases as GNRs elongate. On the other hand, under tensile strain, the out‐of‐plane deformation of GNRs is caused by ripples and thermal vibrations. The ripples show regular patterns. It is suggested we can control the amplitudes of the out‐of‐plane deformations and ripple patterns of GNRs by adjusting their strain. read less USED (high confidence) F. Zirkelbach, B. Stritzker, K. Nordlund, J. Lindner, W. Schmidt, and E. Rauls, “Combined ab initio and classical potential simulation study on silicon carbide precipitation in silicon,” Physical Review B. 2011. link Times cited: 22 Abstract: Atomistic simulations on the silicon carbide precipitation i… read moreAbstract: Atomistic simulations on the silicon carbide precipitation in bulk silicon employing both, classical potential and first-principles methods are presented. The calculations aim at a comprehensive, microscopic understanding of the precipitation mechanism in the context of controversial discussions in the literature. For the quantum-mechanical treatment, basic processes assumed in the precipitation process are calculated in feasible systems of small size. The migration mechanism of a carbon 〈1 0 0〉 interstitial and silicon 〈11 0〉 self-interstitial in otherwise defect-free silicon are investigated using density functional theory calculations. The influence of a nearby vacancy, another carbon interstitial and a substitutional defect as well as a silicon self-interstitial has been investigated systematically. Interactions of various combinations of defects have been characterized including a couple of selected migration pathways within these configurations. Most of the investigated pairs of defects tend to agglomerate allowing for a reduction in strain. The formation of structures involving strong carbon–carbon bonds turns out to be very unlikely. In contrast, substitutional carbon occurs in all probability. A long range capture radius has been observed for pairs of interstitial carbon as well as interstitial carbon and vacancies. A rather small capture radius is predicted for substitutional carbon and silicon self-interstitials. Initial assumptions regarding the precipitation mechanism of silicon carbide in bulk silicon are established and conformability to experimental findings is discussed. Furthermore, results of the accurate first-principles calculations on defects and carbon diffusion in silicon are compared to results of classical potential simulations revealing significant limitations of the latter method. An approach to work around this problem is proposed. Finally, results of the classical potential molecular dynamics simulations of large systems are examined, which reinforce previous assumptions and give further insight into basic processes involved in the silicon carbide transition. read less USED (high confidence) Y. Jing and N. Aluru, “Atomistic simulations on the mechanical properties of a silicon nanofilm covered with graphene,” Computational Materials Science. 2011. link Times cited: 19 USED (high confidence) P. Süle, “Ion-erosion induced surface nanoporosity and nanotopography on Si.,” The Journal of chemical physics. 2011. link Times cited: 4 Abstract: The low-energy ion-bombardment induced surface nanotopograph… read moreAbstract: The low-energy ion-bombardment induced surface nanotopography and the nanopatterning of Si has been simulated by atomistic simulations using an approach based on molecular dynamics (MD). In order to speed up simulations a reasonable cutoff in simulation time and increased cooling rates for keeping in hand the system temperature have been used. We get an unexpectedly rich variety of disordered nanopatterns formed by the self-organization of the crater rims and adatoms islands generated by the individual ion impacts. Our results reveal that the low-energy (0.5 keV impact energy) ion-sputtered Si surface is not smooth at the sub-20 nm length scale and deep nanoholes rule the landscape. Moreover substantial nanoporosity is found beneath the surface with the size range of a few nanometer. Scanning tunneling microscopy (STM) images are also shown obtained for low-fluence ion-sputtering of Si at 2 keV impact energy at 30° angle of incidence. STM images reveal similar features obtained by computer simulations: nanoholes can be seen with a few nanometer diameter. The overall topography landscape as well as the rms surface roughness also show similar features for the images obtained by STM or MD at 2 keV impact energy. The applied approach could make it possible the simulation of nanotopographic images at the molecular dynamics level of theory and could help resolve scanning probe microscopy images in the sub-20 nm length scale regime. read less USED (high confidence) S. Wethekam, M. Busch, C. Linsmeier, and H. Winter, “Effect of target surface on the elastic properties of fast fullerenes,” Physical Review B. 2011. link Times cited: 3 Abstract: S. Wethekam, M. Busch, ∗ Ch. Linsmeier, and H. Winter Instit… read moreAbstract: S. Wethekam, M. Busch, ∗ Ch. Linsmeier, and H. Winter Institut fur Physik, Humboldt-Universitat zu Berlin, Newtonstrasse 15, D-12489 Berlin, Germany. Max-Planck-Institut fur Plasmaphysik, EURATOM Association, Boltzmannstr. 2, D-85748 Garching, Germany. C+60 fullerenes with keV energies are scattered at grazing angles of incidence from atomically clean and flat LiF(001), KCl(001), Al(001), Be(0001), Ni(110) surfaces as well as p(2×1) and p(3×1) oxygen superstructures on Ni(110). The elastic properties of C60 are derived from a comparison of experimental data with 3D molecular dynamics simulations for different interaction potentials. In terms of a simple model for the hybridization of C60 with the surface, we find evidence for a close relation between electronic structure of the surface and elasticity of C60. read less USED (high confidence) P. Erhart and K. Albe, “Molecular Dynamics Simulations of Gas Phase Condensation of Silicon Carbide Nanoparticles,” Advanced Engineering Materials. 2005. link Times cited: 12 Abstract: Gas phase condensation of silicon and silicon carbide nanopa… read moreAbstract: Gas phase condensation of silicon and silicon carbide nanoparticles is studied by molecular-dynamics simulations. By using a recently developed bond-order potential for Si, C and SiC we investigate the fundamental processes governing nucleation and growth of SiC nanoparticles. For the case of elemental silicon particles we show that variations in the binding energy of dimers, which represent stable nuclei for the condensation process, significantly affect the long time evolution of the cluster formation process. A detailed analysis of the molecular reactions during the early stages of SiC particle growth is presented. Reactions, in which silicon monomers are formed, are dominant in case of stoichiometric composition of the precursor gas. Moreover, we find the formation of carbon-dominated species to be preferred and a sensitive dependence of the particle composition and morphology on the processing conditions, especially the cooling and precursor gas composition. read less USED (high confidence) J. Kotakoski and K. Nordlund, “Ion irradiation-induced welding of a carbon nanotube to a Si (100) surface,” MRS Proceedings. 2005. link Times cited: 1 Abstract: Carbon nanotubes (CNTs) are one of the possible building blo… read moreAbstract: Carbon nanotubes (CNTs) are one of the possible building blocks for electronic devices in the transition phase from traditional silicon-based microelectronics towards the fewnanometer regime. Remaining problems in integrating CNTs to the existing technology is the low reactivity of the CNT walls which leads to low conductance between CNTs and the other components. Because recent studies have shown that ion irradiation can be used to modify both the electrical and structural properties of CNTs, we propose that it could also be possible to use ion irradiation with low energies to enhance the conductance of these connections. We have used classical molecular dynamics simulations with empirically tted potentials to examine this possibility by irradiating a single-walled carbon nanotube (SWCNT) on a silicon substrate at room temperature. The nanotube was deposited over a trench created to the silicon substrate so that the nanotube was partly suspended. Low irradiation doses and low energies (0.2 keV–1.2 keV) were used to ensure that the irradiated CNT will not be destroyed. The simulations were carried out for silicon, carbon and neon ions. Our simulations indicate that ion irradiation will increase the number of covalent bonds between the CNT and the Si substrate. When the irradiation dose and energies are low, the damage caused to the SWCNT atomic network can be tolerable when compared to the improvement in the conductance of the contact regions. Furthermore, as the CNTs have high ability to heal the irradiation-induced damage, it is possible that the irradiation will not have a signi cant negative effect to the conductivity of the CNT in a system of this type. read less USED (low confidence) P. Yu, M. Zhong, L. Wu, Z. Chen, and S. Lu, “Multi scale simulation of crack propagation in polycrystalline SiC,” Theoretical and Applied Fracture Mechanics. 2023. link Times cited: 0 USED (low confidence) B. Zhu et al., “Atomic study on deformation behavior and anisotropy effect of 3C-SiC under nanoindentation,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) Y. Zhou, Y. Huang, J. Li, W. Lv, and F. Zhu, “Atomic scale investigation of notch evolution on 4H-SiC under different cutting surfaces and environments,” Journal of Manufacturing Processes. 2023. link Times cited: 0 USED (low confidence) V. V. Hoang, T. M. L. Nguyen, and H. T. T. Nguyen, “Formation of 2D silicon-carbide nanoribbons by cooling from the melt and out-of-plane displacements of atoms,” Journal of Nanoparticle Research. 2023. link Times cited: 0 USED (low confidence) J. Zhang, X. Shang, B. B. He, and B. Zhang, “Towards understanding the crack suppression mechanism in brittle materials with high grinding speed at different temperatures,” International Journal of Machine Tools and Manufacture. 2023. link Times cited: 0 USED (low confidence) Y. Chen, H. Liu, C. Yan, and H. Wei, “Influence of Temperature and Incidence Angle on the Irradiation Cascade Effect of 6H-SiC: Molecular Dynamics Simulations,” Micromachines. 2023. link Times cited: 0 Abstract: SiC devices have been typically subjected to extreme environ… read moreAbstract: SiC devices have been typically subjected to extreme environments and complex stresses during operation, such as intense radiation and large diurnal amplitude differences on the lunar surface. Radiation displacement damage may lead to degradation or failure of the performance of semiconductor devices. In this paper, the effects of temperature and incidence angle on the irradiation cascade effect of 6H-SiC were investigated separately using the principles of molecular dynamics. Temperatures were set to 100 K, 150 K, 200 K, 250 K, 300 K, 350 K, 400 K and 450 K. The incidence direction was parallel to the specified crystal plane, with angles of 8°, 15°, 30°, 45°, 60° and 75° to the negative direction of the Z-axis. In this paper, the six types of defects were counted, and the microscopic distribution images and trajectories of each type of defect were extracted. The results show a linear relationship between the peak of the Frenkel pair and temperature. The recombination rate of Frenkel pairs depends on the local temperature and degree of aggregation at the center of the cascade collision. Increasing the angle of incidence first inhibits and then promotes the production of total defects and Frenkel pairs. The lowest number of total defects, Frenkel pairs and antisite defects are produced at a 45° incident angle. At an incidence angle of 75°, larger size hollow clusters and anti-clusters are more likely to appear in the 6H-SiC. read less USED (low confidence) G. Hu and H. Dai, “Influence of the metal coating on nano-cutting process of cubic silicon carbide,” Proceedings of the Institution of Mechanical Engineers, Part J: Journal of Engineering Tribology. 2023. link Times cited: 0 Abstract: The effect of the metal coating on the machinability of cubi… read moreAbstract: The effect of the metal coating on the machinability of cubic silicon carbide was investigated by molecular dynamics simulation. The effect of the metal coating on the surface of the workpiece was explained using cutting force, friction coefficient, surface morphology, stress, temperature, and tool wear. The results show that the influence of metal type on cutting force, surface morphology, and stress is insignificant for coating thickness. However, the model with Cu coating has a tool suspension key number of 400 at the maximum cutting distance. The number of tool suspension keys for the Ni-Ti coating model is around 1700, indicating that the type of coating has a significant impact on tool wear. Furthermore, the results also show that in the three metals of Cu, Ni and Ni -Ti, Cu coating has the greatest impact on improving cutting performance. Among them, the average cutting force of 1.5 nm Cu coating is about 33.3% lower than that of without coating, and the tool wear is about 26.7% lower. These results demonstrate the effects of the metal coating on the workpiece surface from a theoretical point of view. read less USED (low confidence) H. Pourmirzaagha and S. Rouhi, “Molecular dynamic simulations of the heat transfer in double-layered graphene/silicene nanosheets,” Physica B: Condensed Matter. 2023. link Times cited: 0 USED (low confidence) T. Do and T. Fang, “Atomistic analysis of the phase transformation and wear regimes of textured Wurtzite-SiC hexagonality using molecular dynamics simulation,” Tribology International. 2023. link Times cited: 0 USED (low confidence) Y. Yu, X. Zhang, and L. Bai, “Nanoindentation and scratching behaviors of diamond-like carbon films by coarse-grained molecular dynamics,” Diamond and Related Materials. 2023. link Times cited: 0 USED (low confidence) M. Tahani, E. Postek, and T. Sadowski, “Investigating the Influence of Diffusion on the Cohesive Zone Model of the SiC/Al Composite Interface,” Molecules. 2023. link Times cited: 1 Abstract: Modeling metal matrix composites in finite element software … read moreAbstract: Modeling metal matrix composites in finite element software requires incorporating a cohesive zone model (CZM) to represent the interface between the constituent materials. The CZM determines the behavior of traction–separation (T–S) in this region. Specifically, when a diffusion zone is formed due to heat treatment, it becomes challenging to determine experimentally the equivalent mechanical properties of the interface. Additionally, understanding the influence of heat treatment and the creation of a diffusion zone on the T–S law is crucial. In this study, the molecular dynamics approach was employed to investigate the effect of the diffusion region formation, resulting from heat treatment, on the T–S law at the interface of a SiC/Al composite in tensile, shear, and mixed-mode loadings. It was found that the formation of a diffusion layer led to an increase in tensile and shear strengths and work of separation compared with the interfaces without heat treatment. However, the elastic and shear moduli were not significantly affected by the creation of the diffusion layer. Moreover, the numerical findings indicated that the shear strength in the diffusion region was higher when compared with the shear strength of the {111} slip plane within the fcc aluminum component of the composite material. Therefore, in the diffusion region, crack propagation did not occur in the pure shear loading case; however, shear sliding was observed at the aluminum atomic layers. read less USED (low confidence) Y. Zhou, Y. Huang, J. Li, W. Lv, and F. Zhu, “The effects of abrasive moving speed and motion mode on the thinning mechanism of SiC in three-body contact,” Physica Scripta. 2023. link Times cited: 0 Abstract: Three-body contact is the main contact type in polishing pro… read moreAbstract: Three-body contact is the main contact type in polishing process and leads to a different thinning mechanism than the two-body contact. Molecular dynamics simulation is employed to investigate the thinning mechanism of 3C-SiC substrate in three-body contact. The thinning mechanisms of 3C-SiC under different moving speeds and motion modes of diamond abrasive are compared. Through the analysis of force, temperature, potential energy, stress distribution and atomic flow field, the causes of different thinning mechanisms are explained. It is found that the influence of moving speed is mainly reflected in the temperature rise of substrate when the motion mode of abrasives is the same. The changes of motion mode will significantly alter the stress distribution, which is closely related to the damage depth and atomic flow field. When the feed speed is the same, increasing the self-rotation speed of abrasives can reduce concentration zone of hydrostatic stress and then reduce the damage depth. The self-rotation of abrasives will also change the distribution of von Mises stress, resulting in the different displacement directions of 3C-SiC atoms. Dislocations are more easily generated when the displacement direction of SiC atoms is the same. The moving speed of abrasive is found to have little effect on the thinning mechanism, while the motion mode of abrasive will significantly change the thinning mechanism. read less USED (low confidence) M. Li, X. Guo, R. Kang, D. Guo, and P. Zhou, “Study on the transformation and control mechanism of amorphous damage during the grinding process of monocrystalline silicon considering grain shapes by MD method,” Tribology International. 2023. link Times cited: 4 USED (low confidence) Y. Qu, J. Yuan, N. Deng, W. Hu, S. Wu, and H. Wang, “Effect of the thickness of amorphous silicon intermediate layer on the thermal transport of silicon/diamond interface,” Results in Physics. 2023. link Times cited: 0 USED (low confidence) D. A. Da’na, R. Shoshaa, M. Ashfaq, and M. Al‐Ghouti, “Remediation of boron, lithium, and molybdenum by date pits modified with graphene oxide and cellulose nanocrystals: Mechanistic studies,” Groundwater for Sustainable Development. 2023. link Times cited: 0 USED (low confidence) K. W. Kayang and A. N. Volkov, “Turning nanopowder into nanomaterial: Effect of continuous SiC coating on mechanical properties of Si nanoparticle arrays,” Materialia. 2023. link Times cited: 0 USED (low confidence) Z. Ou, W. Wu, and H. Dai, “Molecular dynamics simulation-based study of single-crystal 3C-SiC nano-indentation with water film,” Applied Physics A. 2023. link Times cited: 0 USED (low confidence) B. Zhu, D. Zhao, Y.-hong Niu, and H. Zhao, “Atomic study on deformation behavior and anisotropy effect of 4C–SiC during nanoindentation,” Materials Science in Semiconductor Processing. 2023. link Times cited: 1 USED (low confidence) Y. Huang, Y. Zhou, J. Li, and F. Zhu, “Understanding the role of surface mechanical properties in SiC surface machining,” Materials Science in Semiconductor Processing. 2023. link Times cited: 0 USED (low confidence) T. Do and T. Fang, “Deep insights into interaction behaviour and material removal of β-SiC wafer in nanoscale polishing,” Tribology International. 2023. link Times cited: 1 USED (low confidence) K. Nishimura and K. Saitoh, “Temperature dependence of mechanical properties and defect formation mechanisms in 3C-SiC: A molecular dynamics study,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) Y. Huang, Y. Zhou, J. Li, and F. Zhu, “Understanding of the effect of wear particles removal from the surface on grinding silicon carbide by molecular dynamics simulations,” Diamond and Related Materials. 2023. link Times cited: 0 USED (low confidence) G.-H. Liu, Z.-X. Xie, P.-Z. Jia, X.-J. Wu, and X.-K. Chen, “Effect of four-phonon scattering on the intrinsic thermal conductivity of penta-graphene,” Diamond and Related Materials. 2023. link Times cited: 0 USED (low confidence) C. Baruffi and C. Brandl, “Vacancy segregation and intrinsic coordination defects at (1 1 1) twist grain boundaries in diamond,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) P. Zhou, T. Sun, C. Wang, H. Deng, and Y. Zhu, “Two-dimensional structure evolution and formation of silicon carbides and diamonds in a nano-abrasion process,” Tribology International. 2023. link Times cited: 0 USED (low confidence) K. Yin, L. Shi, X.-N. Ma, Y. Zhong, M. Li, and X. He, “Thermal Conductivity of 3C/4H-SiC Nanowires by Molecular Dynamics Simulation,” Nanomaterials. 2023. link Times cited: 0 Abstract: Silicon carbide (SiC) is a promising material for thermoelec… read moreAbstract: Silicon carbide (SiC) is a promising material for thermoelectric power generation. The characterization of thermal transport properties is essential to understanding their applications in thermoelectric devices. The existence of stacking faults, which originate from the “wrong” stacking sequences of Si–C bilayers, is a general feature of SiC. However, the effects of stacking faults on the thermal properties of SiC are not well understood. In this study, we evaluated the accuracy of Tersoff, MEAM, and GW potentials in describing the thermal transport of SiC. Additionally, the thermal conductivity of 3C/4H-SiC nanowires was investigated using non-equilibrium molecular dynamics simulations (NEMD). Our results show that thermal conductivity exhibits an increase and then saturation as the total lengths of the 3C/4H-SiC nanowires vary from 23.9 nm to 95.6 nm, showing the size effect of molecular dynamics simulations of the thermal conductivity. There is a minimum thermal conductivity, as a function of uniform period length, of the 3C/4H-SiC nanowires. However, the thermal conductivities of nanowires weakly depend on the gradient period lengths and the ratio of 3C/4H. Additionally, the thermal conductivity of 3C/4H-SiC nanowires decreases continuously from compressive strain to tensile strain. The reduction in thermal conductivity suggests that 3C/4H-SiC nanowires have potential applications in advanced thermoelectric devices. Our study provides insights into the thermal transport properties of SiC nanowires and can guide the development of SiC-based thermoelectric materials. read less USED (low confidence) H. Dai, W. Wu, and P. Li, “Atomistic simulation on the removal mechanism of monocrystal silicon carbide with textured surface nano-machining in water lubrication,” Journal of Manufacturing Processes. 2023. link Times cited: 3 USED (low confidence) P. Wang, D. Zhou, H. Zhao, Y. Lin, A. Nie, and H. Wang, “Dislocation-mediated brittle-ductile transition of diamond under high pressure,” Diamond and Related Materials. 2023. link Times cited: 0 USED (low confidence) H. Nguyen, “Structural evolution of in-plane hybrid graphene/hexagonal boron nitride heterostructure upon heating.,” Journal of molecular graphics & modelling. 2023. link Times cited: 0 USED (low confidence) L. Liang, S. Li, P. Chai, K. Lan, and R. Yu, “Molecular Dynamics Simulation of Single-Crystal 4H-SiC Nano Scratching with Different Scratching Directions of the Tool,” Crystals. 2023. link Times cited: 2 Abstract: 4H-SiC (silicon carbide) is widely used in semiconductor dev… read moreAbstract: 4H-SiC (silicon carbide) is widely used in semiconductor devices due to its superior characteristics. However, processing techniques such as cutting, grinding, and polishing generally have problems such as low processing efficiency, high cost, difficulties guaranteeing processing quality, and serious material waste. The in-depth research on the mechanical behavior, material removal, and damage mechanism of SiC single crystals at the micro/nano scale is the foundation for solving these problems. This paper establishes a molecular dynamics simulation model for 4H-SiC single-crystal nano scratches, using three different directions of a Berkovich indenter to scratch the surface of the workpiece, studying the surface morphology, scratching force, and material removal during the scratching process. The results indicate that scratching directions of the tool varies, and the surface morphology also varies. After the scratching depth exceeds 1.6 nm, complete dislocations with a Burges vector of 1/3<12¯10> appear on the crystal subsurface, leading to the plastic removal of the material. During the process of material removal, a smaller tool rake angle removes a larger amount of material chips. By analyzing the damage layer of the workpiece, the difference in the damage layer is smaller when the scratching direction is different, but the damage layer generated by the smaller rake angle of the scratching tool is thinner. It shows that the scratching force and workpiece temperature are relatively small when the rake angle of the scratching tool is small. Therefore, when scratching 4H-SiC single crystals, choosing a tool with a smaller rake angle is more beneficial for the process. read less USED (low confidence) B. Meng, T. Chen, L. Zhang, and C. Fan, “Feasibility study on the use of single crystal silicon carbide as a tool material,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) B. Brito, G. Hai, and L. Cândido, “Isotopic effect on thermal physical properties of cubic SiC,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) X. Ning, N. Wu, Y. Wen, Q.-M. Zheng, C. Fang, and T. Chen, “Microcrack initiation and propagation in 3 C-SiC ceramic based on molecular dynamics nano-drilling,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) Y. Liu et al., “Deep learning inter-atomic potential for irradiation damage in 3C-SiC,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) M. Yimer, D. A. Wubeshet, and X. Qin, “Twin thickness-dependent tensile deformation mechanism on strengthening-softening of Si nanowires,” Heliyon. 2023. link Times cited: 0 USED (low confidence) H. Ramézani, I. Ellien, Z. E. Oufir, N. Mathieu, S. Delpeux, and S. Bhatia, “Clustering of caffeine in water and its adsorption in activated carbon: Molecular simulations and experiments,” Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2023. link Times cited: 0 USED (low confidence) Y. Huang, Y. Zhou, J. Li, and F. Zhu, “Femtosecond laser surface modification of 4H-SiC improves machinability,” Applied Surface Science. 2023. link Times cited: 2 USED (low confidence) J. Wang and F. Fang, “Controllable removal of silicon carbide at nano scale by ion-implantation assisted laser machining,” CIRP Annals. 2023. link Times cited: 0 USED (low confidence) E. Mørtsell, D. Zhao, A. Autruffe, Y. Chen, M. Sabatino, and Y. Li, “The Nature of a Low Angle Grain-Boundary in a Si Bi-Crystal with Added Fe Impurities,” SSRN Electronic Journal. 2023. link Times cited: 0 USED (low confidence) T. Han, R.-Z. Li, X. Zhang, and F. Scarpa, “Free standing nanoindentation of penta-graphene via molecular dynamics: Mechanics and deformation mechanisms,” Mechanics of Materials. 2023. link Times cited: 0 USED (low confidence) Z. Wu, L. Zhang, S. Yang, C. Wu, K. Xu, and D. Zheng, “Effects of temperature on the deformation of 6H–SiC during nanoscratching,” Wear. 2023. link Times cited: 1 USED (low confidence) N. Mitra and K. Ramesh, “Physics of molecular deformation mechanism in 6H-SiC,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 2 Abstract: Even though there have been several studies in literature of… read moreAbstract: Even though there have been several studies in literature of 6H SiC, a proper physics based understanding of the molecular deformation mechanisms of the material under different loading conditions is still lacking. Experimentally, the brittle nature of the material leads to difficulties associated with in-situ determination of molecular deformation mechanisms of the material under an applied load; whereas, the complex material structure along with the bonding environment prevents proper computational identification of different types of inelasticity mechanisms within the material. Molecular dynamics study (on successful verification of the interatomic potential with experimental results) of pristine single crystals of 6H SiC have been used to probe the physics of molecular deformation mechanisms of the material along with its inherent orientational anisotropy. The study elucidates the experimentally observed mechanisms of defect nucleation and evolution through a detailed analysis of radial distribution functions, x-ray diffraction as well as phonon vibrational studies of the single crystal. Studies have been presented at room temperature, initial high temperature and different types of confinement effects of the material (including hydrostatic and different biaxial loading cases). The confinement resulted in an increase in stress and stiffness whereas increase in initial temperature resulted in a decrease compared to uniaxial stress loading conditions at room temperature. read less USED (low confidence) Y. Wang and B. Fu, “Effect of hydrostatic strain on the thermal conductivity of β-SiC: A combined molecular dynamics and lattice dynamics investigation,” Journal of Materials Research. 2023. link Times cited: 0 Abstract: SiC is quite often used in a variety of industrial scenarios… read moreAbstract: SiC is quite often used in a variety of industrial scenarios because of its superior mechanical and thermal properties. Strain in SiC materials is ubiquitous in industrial settings, and it affects the material’s thermal conductivity. Non-equilibrium molecular dynamics and lattice dynamics from phonon perspectives were used to study the thermal conduction process of SiC under various strains (− 7% ~ + 7%). Our research demonstrates how strain affects β-SiC’s thermal conductivity. The thermal conductivity of SiC between − 7% and + 7% strain was determined using the spectral heat current method, and the effect of phonon frequencies on thermal conductivity was investigated. Lattice dynamics was also used to calculate the phonon dispersion relation, phonon group velocity, and phonon lifetime. These phonon properties were used to study thermal conductivity from the standpoint of phonon transport. Phonon lifetime first rises and reaches its maximum with an increase in compressive strain before falling off. Thermal conductivity decreases as a result of the phonon lifetime decreasing. Group velocity and phonon lifetime gradually decrease as tensile strain increases. The thermal conductivity falls as a result of both. Our findings enhance our knowledge of the thermal conduction mechanism in β-SiC. These variations in thermal conductivity are brought on by phonon properties that change as a result of strain. This conclusion could be applied to a wide variety of other materials. Graphical abstract read less USED (low confidence) Y. Chen, H. Liu, T. Gao, and H. Wei, “Simulation of the Irradiation Cascade Effect of 6H-SiC Based on Molecular Dynamics Principles,” Micromachines. 2023. link Times cited: 1 Abstract: When semiconductor materials are exposed to radiation fields… read moreAbstract: When semiconductor materials are exposed to radiation fields, cascade collision effects may form between the radiation particles in the radiation field and the lattice atoms in the target material, creating irradiation defects that can lead to degradation or failure of the performance of the device. In fact, 6H-SiC is one of the typical materials for third-generation broadband semiconductors and has been widely used in many areas of intense radiation, such as deep space exploration. In this paper, the irradiation cascade effect between irradiated particles of different energies in the radiation and lattice atoms in 6H-SiC target materials is simulated based on the molecular dynamics analysis method, and images of the microscopic trajectory evolution of PKA and SKA are obtained. The recombination rates of the Frenkel pairs were calculated at PKA energies of 1 keV, 2 keV, 5 keV, and 10 keV. The relationship between the number of defects, the spatial distribution pattern of defects, and the clustering of defects in the irradiation cascade effect of 6H-SiC materials with time and the energy of PKA are investigated. The results show that the clusters are dominated by vacant clusters and are mainly distributed near the trajectories of the SKA. The number and size of vacant clusters, the number of Frenkel pairs, and the intensity of cascade collisions of SKAs are positively correlated with the magnitude of the energy of the PKA. The recombination rate of Frenkel pairs is negatively correlated with the magnitude of the energy of PKA. read less USED (low confidence) B. Meng and C. Li, “Nano and Sub-nano Scale Friction Behavior in Rotary Processing of 6H-SiC with Different Off-Axis Angles,” Tribology Letters. 2023. link Times cited: 1 USED (low confidence) B. Yang et al., “Thermal transport mechanism of AlN/SiG/3C–SiC typical heterostructures,” Materials Today Physics. 2023. link Times cited: 1 USED (low confidence) S. Gao, H. Wang, H. Huang, and R. Kang, “Molecular simulation of the plastic deformation and crack formation in single grit grinding of 4H-SiC single crystal,” International Journal of Mechanical Sciences. 2023. link Times cited: 27 USED (low confidence) A. Zhou et al., “Investigation of nano-tribological behaviors and deformation mechanisms of Cu-Ni alloy by molecular dynamics simulation,” Tribology International. 2023. link Times cited: 7 USED (low confidence) M. Tahani, E. Postek, L. Motevalizadeh, and T. Sadowski, “Effect of Vacancy Defect Content on the Interdiffusion of Cubic and Hexagonal SiC/Al Interfaces: A Molecular Dynamics Study,” Molecules. 2023. link Times cited: 5 Abstract: The mechanical properties of ceramic–metal nanocomposites ar… read moreAbstract: The mechanical properties of ceramic–metal nanocomposites are greatly affected by the equivalent properties of the interface of materials. In this study, the effect of vacancy in SiC on the interdiffusion of SiC/Al interfaces is investigated using the molecular dynamics method. The SiC reinforcements exist in the whisker and particulate forms. To this end, cubic and hexagonal SiC lattice polytypes with the Si- and C-terminated interfaces with Al are considered as two samples of metal matrix nanocomposites. The average main and cross-interdiffusion coefficients are determined using a single diffusion couple for each system. The interdiffusion coefficients of the defective SiC/Al are compared with the defect-free SiC/Al system. The effects of temperature, annealing time, and vacancy on the self- and interdiffusion coefficients are investigated. It is found that the interdiffusion of Al in SiC increases with the increase in temperature, annealing time, and vacancy. read less USED (low confidence) Y. Zhou, Y. Huang, J. Li, W. Lv, and F. Zhu, “Polishing process of 4H-SiC under different pressures in a water environment,” Diamond and Related Materials. 2023. link Times cited: 5 USED (low confidence) Z. Bian et al., “Effects of different incidence rates of carbon and silicon clusters on the surface properties of SiC films,” Surfaces and Interfaces. 2023. link Times cited: 2 USED (low confidence) Z. Qi et al., “Aln/Diamond Interface Nanoengineering for Reducing Thermal Boundary Resistance by Molecular Dynamics Simulations,” SSRN Electronic Journal. 2023. link Times cited: 5 USED (low confidence) S. Stelmakh, S. Gierlotka, K. Skrobas, K. Stefanska-Skrobas, and B. Palosz, “Effect of surface on internal structure and apparent lattice parameter of nanocrystalline SiC grains modelled by molecular dynamics simulations,” Surface Science. 2023. link Times cited: 0 USED (low confidence) M. Tahani, E. Postek, and T. Sadowski, “Molecular Dynamics Study of Interdiffusion for Cubic and Hexagonal SiC/Al Interfaces,” Crystals. 2022. link Times cited: 5 Abstract: The mechanical properties of the SiC/Al interface are crucia… read moreAbstract: The mechanical properties of the SiC/Al interface are crucial in estimating the overall strength of this ceramic-metal composite. The present work investigates the interdiffusion at the SiC/Al interface using molecular dynamics simulations. One cubic and one hexagonal SiC with a higher probability of orientations in contact with Al are examined as two samples of metal-matrix nanocomposites with whisker and particulate reinforcements. These reinforcements with the Si- and C-terminated surfaces of the SiC/Al interfaces are also studied. The average main and cross-interdiffusion coefficients are evaluated using a single diffusion couple for each system. The effect of temperature and annealing time are analysed on the self- and interdiffusion coefficients. It is found that the diffusion of Al in SiC is similar in cubic and hexagonal SiC and as expected, the interdiffusion coefficient increases as the temperature and annealing time increase. The model after diffusion can be used to evaluate the overall mechanical properties of the interface region in future studies. read less USED (low confidence) Y. Zhou, Y. Huang, J. Li, and F. Zhu, “Effect of water film on 4H-SiC nano-indentation process,” 2022 IEEE 24th Electronics Packaging Technology Conference (EPTC). 2022. link Times cited: 1 Abstract: 4H-SiC is one of the most commonly used third-generation sem… read moreAbstract: 4H-SiC is one of the most commonly used third-generation semiconductor materials, and the machining technology of it has always been a big problem. Considering that the actual machining process is often inseparable from water, it is necessary to study the mechanical properties and deformation behavior of 4H-SiC under wet conditions. Molecular dynamics simulation is used to study the influence of water film on the indentation process of 4H-SiC. It is found that the pop-in event during loading is related to the deformation mode of 4H-SiC. The plastic deformation of 4H-SiC is accompanied by phase transformation, which can partially release the stress. Water film can advance the pop-in event because the phase transformation will be intensified after water molecules are pressed in. Water film can also narrow the gap between the maximum and minimum values of pop-in events, but it will lead to more damage and greater damage depth after indentation. Besides, the unloading process is often accompanied by the recovery of damaged structures and the water film will reduce the SiC atoms adhered to the indenter during unloading. read less USED (low confidence) Y. Huang, Y. Zhou, J. Li, and F. Zhu, “Materials removal mechanism and multi modes feature for silicon carbide during scratching,” International Journal of Mechanical Sciences. 2022. link Times cited: 11 USED (low confidence) B. Liu, H. Yang, R. Kong, X. Wang, J. Liu, and K. Pang, “Simulation and experimental study on limited cutting and heat effect of silicon carbide,” Materials Today Communications. 2022. link Times cited: 1 USED (low confidence) B. Zhao, P. Zhao, H. Liu, J. Pan, and J. Wu, “Investigation on Surface Generation Mechansim of Single-Crystal Silicon in Grinding: Surface Crystal Orientation Effect,” SSRN Electronic Journal. 2022. link Times cited: 2 USED (low confidence) J. Zhang, P. Zhang, and G. Ye, “Sintering Mechanical Properties of SiC/nano-Ag/Cu: Molecular Dynamics Simulation,” Journal of Physics: Conference Series. 2022. link Times cited: 0 Abstract: Nano-sintered silver has broad development prospects in the … read moreAbstract: Nano-sintered silver has broad development prospects in the field of electronic packaging due to its excellent thermal conductivity and feasibility at low sintering temperatures. In this paper, by using molecular dynamics simulations, a single nano-silver particle is sintered, and its melting temperature range is obtained. Then, a SiC/nano-Ag/Cu sintered sample is prepared. By observing the formation process of the sintering neck, it is found that the sintering quality can be improved by prolonging the constant temperature time. Tensile simulations are performed on the sintered samples, and the traction-separation law curve is obtained. The initial yield strength of the sintered sample is 3.03 GPa, the elastic modulus is 28.99 GPa, and the fracture energy is 11.74 N/m according to the traction-separation law curve. read less USED (low confidence) C. Liu, S. To, X. Sheng, and J. Xu, “Molecular dynamics simulation on crystal defects of single-crystal silicon during elliptical vibration cutting,” International Journal of Mechanical Sciences. 2022. link Times cited: 9 USED (low confidence) H. Huang, W. Yang, W. Ming, G. Zhang, Y. Xu, and Z. Zhang, “Mechanism of springback behavior in ultra-thin glass molding process: A molecular dynamics study,” Journal of Non-Crystalline Solids. 2022. link Times cited: 2 USED (low confidence) P. Wang et al., “Equation of state of tungsten-doped carbon based on QEOS model for laser fusion,” AIP Advances. 2022. link Times cited: 0 Abstract: Tungsten-doped diamond is employed as a promising ablator ma… read moreAbstract: Tungsten-doped diamond is employed as a promising ablator material in high-gain laser fusion target design. Unlike for pure carbon, reports on the equation of state (EOS) of tungsten-doped carbon are limited, particularly in the high-pressure range over Mbar, which is relevant to laser fusion. To complement the radiation-hydrodynamic simulations of laser fusion, we developed the EOS of tungsten-doped carbon by combining the quotidian EOS model and large-scale atomistic simulations, which provide fundamental material parameters. In this manner, the EOS of doped carbon can be efficiently constructed. The influence of tungsten doping on the diamond material parameters and EOS was observed. The application of the developed EOS was shown via typical radiation-hydrodynamic simulations of laser fusion. read less USED (low confidence) S. Sun, G. Ru, W. Qi, and W. Liu, “Molecular Dynamics Study of the Robust Superlubricity in Penta-Graphene van der Waals layered structures,” Tribology International. 2022. link Times cited: 9 USED (low confidence) B.-G. Jeong, S. Lahkar, Q. An, and K. Reddy, “Mechanical Properties and Deformation Behavior of Superhard Lightweight Nanocrystalline Ceramics,” Nanomaterials. 2022. link Times cited: 4 Abstract: Lightweight polycrystalline ceramics possess promising physi… read moreAbstract: Lightweight polycrystalline ceramics possess promising physical, chemical, and mechanical properties, which can be used in a variety of important structural applications. However, these ceramics with coarse-grained structures are brittle and have low fracture toughness due to their rigid covalent bonding (more often consisting of high-angle grain boundaries) that can cause catastrophic failures. Nanocrystalline ceramics with soft interface phases or disordered structures at grain boundaries have been demonstrated to enhance their mechanical properties, such as strength, toughness, and ductility, significantly. In this review, the underlying deformation mechanisms that are contributing to the enhanced mechanical properties of superhard nanocrystalline ceramics, particularly in boron carbide and silicon carbide, are elucidated using state-of-the-art transmission electron microscopy and first-principles simulations. The observations on these superhard ceramics revealed that grain boundary sliding induced amorphization can effectively accommodate local deformation, leading to an outstanding combination of mechanical properties. read less USED (low confidence) S. Stelmakh, K. Skrobas, K. Stefanska-Skrobas, S. Gierlotka, and B. Palosz, “Distortion of SiC lattice induced by carbon-coating on (100) and (111) surfaces - ab-initio and molecular dynamics study,” Surface Science. 2022. link Times cited: 1 USED (low confidence) Z. Dong, H. Wang, Y. Qi, X. Guo, R. Kang, and Y. Bao, “Effects of minimum uncut chip thickness on tungsten nano-cutting mechanism,” International Journal of Mechanical Sciences. 2022. link Times cited: 8 USED (low confidence) H. Dai, W. Wu, W. Fan, and H. Du, “Investigation on mechanism of ultraprecision three-body polishing of single-crystal silicon carbide with voids by molecular dynamics simulation,” Applied Physics A. 2022. link Times cited: 2 USED (low confidence) Y. Huang, Y. Zhou, J. Li, and F. Zhu, “Effect of impurity doping on 4H-SiC planarization,” 2022 23rd International Conference on Electronic Packaging Technology (ICEPT). 2022. link Times cited: 1 Abstract: Silicon carbide (SiC) is a next-generation wide-gap semicond… read moreAbstract: Silicon carbide (SiC) is a next-generation wide-gap semiconductor material, but its high hardness and brittleness limit cost reduction and quality promotion. The planarization at the packaging stage faces the doped SiC wafer, thus understanding the effect and behaviors of impurity during planarization is necessary for the final packaging performance. In this study, a series of models with various doping types (Al, B, N, P) and concentrations (1018 cm-3 – 1020 cm-3) were constructed to simulate based on molecular dynamics. The machinability effect and dynamic features of impurity were focused on in the analysis of the results. The results revealed that heavy doping affects the machinability on the nano-scale and N doping has better machinability than the other types of impurity. In addition, a potential competition mechanism between the solution strengthening and cohesive weakening maybe exist, resulting in the differences among various impurity types. read less USED (low confidence) B. Yao, Z. R. Liu, and R. F. Zhang, “EAPOTc: An integrated empirical interatomic potential optimization platform for compound solids,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) M. Xia, S.-L. Liu, S.-han Liu, J. Wu, X. Gan, and N. Zhou, “The Effect of Temperature on Silicon Nucleation from Melt in Seed-assisted Growth — a Molecular Dynamics Study,” Silicon. 2022. link Times cited: 0 USED (low confidence) X. Meng, H. Yue, W. Wu, and H. Dai, “Simulation of abrasive polishing process of single crystal silicon based on molecular dynamics,” The International Journal of Advanced Manufacturing Technology. 2022. link Times cited: 2 USED (low confidence) R. Abram, D. Chrobak, J. Byggmästar, K. Nordlund, and R. Nowak, “Comprehensive structural changes in nanoscale-deformed silicon modelled with an integrated atomic potential,” Materialia. 2022. link Times cited: 2 USED (low confidence) Z. Wu, L. Zhang, S. Yang, and C. Wu, “Effects of grain size and protrusion height on the surface integrity generation in the nanogrinding of 6H-SiC,” Tribology International. 2022. link Times cited: 13 USED (low confidence) W. Wu, Y. Hu, X. Meng, J. Dai, and H. Dai, “Molecular dynamics simulation of ion-implanted single-crystal 3C-SiC nano-indentation,” Journal of Manufacturing Processes. 2022. link Times cited: 12 USED (low confidence) Y. Fan and H. S. Shen, “Non-symmetric stiffness of origami-graphene metamaterial plates,” Composite Structures. 2022. link Times cited: 10 USED (low confidence) Y. Zhou, Y. Huang, J. Li, and F. Zhu, “Effect of water film on the nano-scratching process of 4H-SiC under the constant load,” Tribology International. 2022. link Times cited: 13 USED (low confidence) H. Wang, S. Gao, R. Kang, X. Guo, and H. Li, “Mechanical Load-Induced Atomic-Scale Deformation Evolution and Mechanism of SiC Polytypes Using Molecular Dynamics Simulation,” Nanomaterials. 2022. link Times cited: 4 Abstract: Silicon carbide (SiC) is a promising semiconductor material … read moreAbstract: Silicon carbide (SiC) is a promising semiconductor material for making high-performance power electronics with higher withstand voltage and lower loss. The development of cost-effective machining technology for fabricating SiC wafers requires a complete understanding of the deformation and removal mechanism. In this study, molecular dynamics (MD) simulations were carried out to investigate the origins of the differences in elastic–plastic deformation characteristics of the SiC polytypes, including 3C-SiC, 4H-SiC and 6H-SiC, during nanoindentation. The atomic structures, pair correlation function and dislocation distribution during nanoindentation were extracted and analyzed. The main factors that cause elastic–plastic deformation have been revealed. The simulation results show that the deformation mechanisms of SiC polytypes are all dominated by amorphous phase transformation and dislocation behaviors. Most of the amorphous atoms recovered after completed unload. Dislocation analysis shows that the dislocations of 3C-SiC are mainly perfect dislocations during loading, while the perfect dislocations in 4H-SiC and 6H-SiC are relatively few. In addition, 4H-SiC also formed two types of stacking faults. read less USED (low confidence) D. Chrobak, A. Majtyka-Piłat, G. Ziółkowski, and A. Chrobak, “Interatomic Potential for InP,” Materials. 2022. link Times cited: 0 Abstract: Classical modeling of structural phenomena occurring in InP … read moreAbstract: Classical modeling of structural phenomena occurring in InP crystal, for example plastic deformation caused by contact force, requires an interatomic interaction potential that correctly describes not only the elastic properties of indium phosphide but also the pressure-induced reversible phase transition B3↔B1. In this article, a new parametrization of the analytical bond-order potential has been developed for InP. The potential reproduces fundamental physical properties (lattice parameters, cohesive energy, stiffness coefficients) of the B3 and B1 phases in good agreement with first-principles calculations. The proposed interaction model describes the reversibility of the pressure-induced B3↔B1 phase transition as well as the formation of native point defects in the B3 phase. read less USED (low confidence) S. Sassi et al., “Energy loss in low energy nuclear recoils in dark matter detector materials,” Physical Review D. 2022. link Times cited: 5 Abstract: Recent progress in phonon-mediated detectors with eV-scale n… read moreAbstract: Recent progress in phonon-mediated detectors with eV-scale nuclear recoil energy sensitivity requires an understanding of the effect of the crystalline defects on the energy spectrum expected from dark matter or neutrino coherent scattering. We have performed molecular dynamics simulations to determine the amount of energy stored in the lattice defects as a function of the recoil direction and energy. This energy can not be observed in the phonon measurement, thus affecting the observed energy spectrum compared to the underlying true recoil energy spectrum. We describe this effect for multiple commonly used detector materials and demonstrate how the predicted energy spectrum from dark matter scattering is modified. read less USED (low confidence) S. Zhang, X. Cheng, and J. Chen, “Surface deformation, phase transition and dislocation mechanisms of single crystalline 6H-SiC in oblique nano-cutting,” Applied Surface Science. 2022. link Times cited: 16 USED (low confidence) X. Feng, K. Cao, X. Huang, G. Li, and Y. Lu, “Nanolayered CoCrFeNi/Graphene Composites with High Strength and Crack Resistance,” Nanomaterials. 2022. link Times cited: 4 Abstract: Emerging high-entropy alloy (HEA) films achieve high strengt… read moreAbstract: Emerging high-entropy alloy (HEA) films achieve high strength but generally show ineludible brittle fractures, strongly restricting their micro/nano-mechanical and functional applications. Nanolayered (NL) CoCrFeNi/graphene composites are elaborately fabricated via magnetron sputtering and the transfer process. It is uncovered that NL CoCrFeNi/graphene composite pillars exhibit a simultaneous ultra-high strength of 4.73 GPa and considerable compressive plasticity of over 20%. Detailed electron microscope observations and simulations reveal that the monolayer graphene interface can effectively block the crack propagation and stimulate dislocations to accommodate further deformation. Our findings open avenues for the fabrication of high-performance, HEA-based composites, thereby addressing the challenges and unmet needs in flexible electronics and mechanical metamaterials. read less USED (low confidence) L. Xue, G. Feng, and S. Liu, “Molecular dynamics study of temperature effect on deformation behavior of m-plane 4H–SiC film by nanoindentation,” Vacuum. 2022. link Times cited: 5 USED (low confidence) H. Wang, Z. Dong, S. Yuan, X. Guo, R. Kang, and Y. Bao, “Effects of tool geometry on tungsten removal behavior during nano-cutting,” International Journal of Mechanical Sciences. 2022. link Times cited: 19 USED (low confidence) Y. Xu, P. Zhu, R. Li, and Z. Yin, “Molecular dynamics simulations of friction behaviours on nano-textured silicon surfaces,” Molecular Simulation. 2022. link Times cited: 2 Abstract: ABSTRACT Molecular dynamics simulations have been applied to… read moreAbstract: ABSTRACT Molecular dynamics simulations have been applied to study the friction behaviours of nano-textured silicon surfaces. The effects of texture shape, texture pitch and indenter size on forces, temperature, stress and plastic deformation are investigated. It is found that the presence of the texture facilitates the reduction of friction due to the decrease the contact area. The texture shape significantly influences the tribological properties of the textured surface. The hemispherical texture has the optimum friction reduction effect, followed by cylindrical texture and lastly by cubic texture. The number of atoms that undergo phase transformation in the scratching is the maximal for the cubic texture while the smallest for the hemispherical texture. However, the texture pitch has little effect on the tribological properties of the textured surface. In addition, it is interesting to observe the indenter size effect that a larger indenter causes a smaller force and wear volume at the initial stage of scratching. The indenter size effect on tribological properties results from the variation of contact area in the scratching. The insights gained can shed light on the friction mechanism of nanoscale textured silicon surface and are beneficial to the design of micro/nanoscale devices such as micro/nanoelectromechanical systems with surface textures. read less USED (low confidence) C. Yin et al., “A Multi-Scale Simulation Study of Irradiation Swelling of Silicon Carbide,” Materials. 2022. link Times cited: 0 Abstract: Silicon carbide (SiC) is a promising structural and cladding… read moreAbstract: Silicon carbide (SiC) is a promising structural and cladding material for accident tolerant fuel cladding of nuclear reactor due to its excellent properties. However, when exposed to severe environments (e.g., during neutron irradiation), lattice defects are created in amounts significantly greater than normal concentrations. Then, a series of radiation damage behaviors (e.g., radiation swelling) appear. Accurate understanding of radiation damage of nuclear materials is the key to the design of new fuel cladding materials. Multi-scale computational simulations are often required to understand the physical mechanism of radiation damage. In this work, the effect of neutron irradiation on the volume swelling of cubic-SiC film with 0.3 mm was studied by using the combination of molecular dynamics (MD) and rate theory (RT). It was found that for C-vacancy (CV), C-interstitial (CI), Si-vacancy (SiV), Si-interstitial (SiI), and Si-antisite (SiC), the volume of supercell increases linearly with the increase of concentration of these defects, while the volume of supercell decreases linearly with the increase of defect concentration for C-antisite (CSi). Furthermore, according to the neutron spectrum of a certain reactor, one RT model was constructed to simulate the evolution of point defect under neutron irradiation. Then, the relationship between the volume swelling and the dose of neutrons can be obtained through the results of MD and RT. It was found that swelling typically increases logarithmically with radiation dose and saturates at relatively low doses, and that the critical dose for abrupt transition of volume is consistent with the available experimental data, which indicates that the rate theory model can effectively describe the radiation damage evolution process of SiC. This work not only presents a systematic study on the relationship between various point defect and excess volume, but also gives a good example of multi-scale modelling through coupling the results of binary collision, MD and RT methods, etc., regardless of the multi-scale modelling only focus on the evolution of primary point defects. read less USED (low confidence) C. Liu, W. Xu, J. Zhang, J. Xiao, X. Chen, and J. Xu, “Numerical investigation on the temperature effect in nanometric cutting of polycrystalline silicon,” International Journal of Mechanical Sciences. 2022. link Times cited: 9 USED (low confidence) Y. Liu, W. Wan, Q. Li, Z. Xiong, C. Tang, and L. Zhou, “Revisiting the Rate-Dependent Mechanical Response of Typical Silicon Structures via Molecular Dynamics,” Nanomaterials. 2022. link Times cited: 1 Abstract: Strain rate is a critical parameter in the mechanical applic… read moreAbstract: Strain rate is a critical parameter in the mechanical application of nano-devices. A comparative atomistic study on both perfect monocrystalline silicon crystal and silicon nanowire was performed to investigate how the strain rate affects the mechanical response of these silicon structures. Using a rate response model, the strain rate sensitivity and the critical strain rate of two structures were given. The rate-dependent dislocation activities in the fracture process were also discussed, from which the dislocation nucleation and motion were found to play an important role in the low strain rate deformations. Finally, through the comparison of five equivalent stresses, the von Mises stress was verified as a robust yield criterion of the two silicon structures under the strain rate effects. read less USED (low confidence) Z. Zhang et al., “Origin and evolution of a crack in silicon induced by a single grain grinding,” Journal of Manufacturing Processes. 2022. link Times cited: 34 USED (low confidence) S. Ajori and A. R. Eftekharfar, “Mechanical properties and fracture analysis of defective penta-graphene under temperature variation: Insight from molecular dynamics,” Diamond and Related Materials. 2022. link Times cited: 5 USED (low confidence) Q. Kang et al., “Mechanical properties and indentation-induced phase transformation in 4H–SiC implanted by hydrogen ions,” Ceramics International. 2022. link Times cited: 4 USED (low confidence) E. Mareev and F. Potemkin, “Dynamics of Ultrafast Phase Transitions in (001) Si on the Shock-Wave Front,” International Journal of Molecular Sciences. 2022. link Times cited: 3 Abstract: We demonstrate an ultrafast (<0.1 ps) reversible phase trans… read moreAbstract: We demonstrate an ultrafast (<0.1 ps) reversible phase transition in silicon (Si) under ultrafast pressure loading using molecular dynamics. Si changes its structure from cubic diamond to β-Sn on the shock-wave front. The phase transition occurs when the shock-wave pressure exceeds 11 GPa. Atomic volume, centrosymmetry, and the X-ray-diffraction spectrum were revealed as effective indicators of phase-transition dynamics. The latter, being registered in actual experimental conditions, constitutes a breakthrough in the path towards simple X-ray optical cross-correlation and pump-probe experiments. read less USED (low confidence) Z. Yin, H. Wu, G. Zhang, C. Mu, and L. Bai, “Wear Estimation of DLC Films Based on Energy-Dissipation Analysis: A Molecular Dynamics Study,” Materials. 2022. link Times cited: 2 Abstract: This study employs the energy-dissipation method to analyze … read moreAbstract: This study employs the energy-dissipation method to analyze the tribological behaviors of diamond-like carbon (DLC) films through molecular dynamics simulation. It is found that at small load and sliding velocity, the variation trend of average friction force is only dependent on the number of interface bonds (or contact area). However, at large load and sliding velocity, the friction mechanism is not only related to the number of interface bonds but also related to the presence of the transfer layer. The elastic–plastic deformation mainly occurs in the early sliding stage, and a part of the stored elastic potential energy is dissipated by plastic potential energy or internal frictional heat. After the sliding stabilization, over 95% of the total frictional energy is dissipated by thermal conduction, and the rest is mostly dissipated by wear. The increase in load, velocity, and temperature cause more frictional energy dissipated by elastic–plastic deformation, atomic motion, and elastic deformation instead of thermal conduction, respectively. Finally, the wear rate obtained in this work is the same order of magnitude as the experiment. Generally, this work provides an effective atomic-scale method to comprehensively analyze the microscopic wear mechanism of materials. read less USED (low confidence) M.-Q. Le, “Fracture and strength of single-atom-thick hexagonal materials,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) D. Yu, H. Zhang, J. Yi, Y. Fang, and N. Wu, “Dislocation Analysis of 3C-SiC Nanoindentation with Different Crystal Plane Groups Based on Molecular Dynamics Simulation,” Journal of Nanomaterials. 2021. link Times cited: 4 Abstract: To explore the deformation law of nanoindentation dislocatio… read moreAbstract: To explore the deformation law of nanoindentation dislocations of different crystal plane groups of 3C-SiC by cube indenter. The molecular dynamics simulation method is used to construct the different crystal plane family models of 3C-SiC, select the ensemble, set the potential function, optimize the crystal structure, and relax the indentation process. The radial distribution function, shear strain, and dislocation deformation of nanoindentation on (001), (110), and (111) planes were analyzed, respectively. In the radial distribution function, the change in
g
r
in the (110) crystal plane is the most obvious. Shear strain and dislocation occur easily at the boundary of square indentation defects. During the indentation process, the shear strain is enhanced along the atomic bond arrangement structure, (001) crystal plane shear strain is mainly concentrated around and below the indentation defects and produce a large number of cross dislocations, (110) the crystal plane shear strain is mainly concentrated in the shear strain chain extending around and below the indentation defect, which mainly produces horizontal dislocations, and (111) the crystal plane shear strain is mainly concentrated in four weeks extending on the left and right sides in the direction below the indentation defect and produces horizontal and vertical dislocations. The direction of shear stress release is related to the crystal structure. The crystal structure affects the direction of atomic slip, resulting in the results of sliding in different directions. The final dislocation rings are different, resulting in different indentation results. read less USED (low confidence) Y. Liu, Y. Ji, L. Dong, H. Xie, J. Song, and J. Li, “Effect of grinding depths on SiC nanogrinding behavior based on molecular dynamics,” Applied Physics A. 2021. link Times cited: 7 USED (low confidence) Z. Wu, L. Zhang, and S. Yang, “Effect of abrasive grain position patterns on the deformation of 6H-silicon carbide subjected to nano-grinding,” International Journal of Mechanical Sciences. 2021. link Times cited: 21 USED (low confidence) Y. Fan et al., “Nano material removal mechanism of 4H-SiC in ion implantation-assisted machining,” Computational Materials Science. 2021. link Times cited: 8 USED (low confidence) Q. Kang et al., “Modification mechanism of collaborative ions implanted into 4H-SiC by atomic simulation and experiment,” International Journal of Mechanical Sciences. 2021. link Times cited: 9 USED (low confidence) L. Xue et al., “Study of the deposition of nanopillar-patterned 4H-SiC by molecular dynamics simulation,” Applied Surface Science. 2021. link Times cited: 8 USED (low confidence) C. Liu et al., “Numerical investigation on material removal mechanism in elliptical vibration cutting of single-crystal silicon,” Materials Science in Semiconductor Processing. 2021. link Times cited: 6 USED (low confidence) Z. E. Oufir, H. Ramézani, N. Mathieu, and S. Delpeux, “Impact of adsorbent carbons and carbon surface conductivity on adsorption capacity of CO2, CH4, N2 and gas separation,” Computational Materials Science. 2021. link Times cited: 8 USED (low confidence) S. Yang, B. Cheng, J. McGeough, Y. T. Woldu, and Y. Xiaokai, “Multi-scale numerical analysis and experimental verification for nano-cutting,” Journal of Manufacturing Processes. 2021. link Times cited: 6 USED (low confidence) J. Ke, X. Chen, C. Liu, J. Zhang, H. Yang, and J. Xu, “Enhancing the ductile machinability of single-crystal silicon by laser-assisted diamond cutting,” The International Journal of Advanced Manufacturing Technology. 2021. link Times cited: 11 USED (low confidence) Z. Yin, P. Zhu, B. Li, Y. Xu, and R. Li, “Atomic Simulations of Deformation Mechanism of 3C-SiC Polishing Process with a Rolling Abrasive,” Tribology Letters. 2021. link Times cited: 6 USED (low confidence) Z. E. Oufir, H. Ramézani, N. Mathieu, S. Delpeux, and S. Bhatia, “Influence of force field used in carbon nanostructure reconstruction on simulated phenol adsorption isotherms in aqueous medium,” Journal of Molecular Liquids. 2021. link Times cited: 2 USED (low confidence) E. M. Y. Lee, A. Yu, J. D. de Pablo, and G. Galli, “Stability and molecular pathways to the formation of spin defects in silicon carbide,” Nature Communications. 2021. link Times cited: 9 USED (low confidence) B. Yao, Z. Liu, and R. Zhang, “EAPOTs: An integrated empirical interatomic potential optimization platform for single elemental solids,” Computational Materials Science. 2021. link Times cited: 3 USED (low confidence) H. Dai et al., “Molecular dynamics simulation of ultra-precision machining 3C-SiC assisted by ion implantation,” Journal of Manufacturing Processes. 2021. link Times cited: 19 USED (low confidence) A. Nayad et al., “New two-dimensional functionalised silicon nanosheets prepared by direct exfoliation of calcium disilicide with tosyl chloride,” Advances in Materials and Processing Technologies. 2021. link Times cited: 0 Abstract: ABSTRACT Among the variety of two-dimensional (2D) materials… read moreAbstract: ABSTRACT Among the variety of two-dimensional (2D) materials, silicon nanosheets (SiNSs) having nanometres thicknesses and lateral dimensions ranging from the submicrometer to the micrometre scale are highly desired for practical applications in physic area. Chemical exfoliation of calcium disilicide (CaSi2) Zintl phase is the most common route to prepare crystalline functionalised silicon nanosheets. However, the chemical functionalization of such layered materials suffers some disadvantages and needs to be improved. Herein, the synthesis of a new-layered organosilicon material is described, namely tosyl-modified silicane (Si_Ts), by the direct exfoliation of CaSi2 with p-Toluenesulfonyl chloride (tosyl chloride: TsCl), in nitrogen gas at 110°C in a sand bath. The new crystalline silicon-based functionalised material was successfully characterised by different spectroscopic techniques, which confirmed the impregnation of the organic compound onto the surface of layered nanosheets. This method opens the access of a large-scale synthesis of new hybrid organosilicon 2D materials, which are suitable for optical, electric, or photovoltaic applications. read less USED (low confidence) M. Barhoumi, N. Sfina, M. Said, and S. Znaidia, “Elastic and mechanical properties of aluminium and silicon carbide using density functional theory and beyond,” Solid State Communications. 2021. link Times cited: 3 USED (low confidence) Y. Huang, M. Wang, J. Li, and F. Zhu, “Removal behavior of micropipe in 4H-SiC during micromachining,” Journal of Manufacturing Processes. 2021. link Times cited: 11 USED (low confidence) W. Wan, C. Tang, J. Zhang, and L. Zhou, “General Molecular Dynamics Approach to Understand the Mechanical Anisotropy of Monocrystalline Silicon under the Nanoscale Effects of Point Defect,” Nanomaterials. 2021. link Times cited: 7 Abstract: Mechanical anisotropy and point defects would greatly affect… read moreAbstract: Mechanical anisotropy and point defects would greatly affect the product quality while producing silicon wafers via diamond-wire cutting. For three major orientations concerned in wafer production, their mechanical performances under the nanoscale effects of a point defect were systematically investigated through molecular dynamics methods. The results indicated anisotropic mechanical performance with fracture phenomena in the uniaxial deformation process of monocrystalline silicon. Exponential reduction caused by the point defect has been demonstrated for some properties like yield strength and elastic strain energy release. Dislocation analysis suggested that the slip of dislocations appeared and created hexagonal diamond structures with stacking faults in the [100] orientation. Meanwhile, no dislocation was observed in [110] and [111] orientations. Visualization of atomic stress proved that the extreme stress regions of the simulation models exhibited different geometric and numerical characteristics due to the mechanical anisotropy. Moreover, the regional evolution of stress concentration and crystal fracture were interrelated and mutually promoted. This article contributes to the research towards the mechanical and fracture anisotropy of monocrystalline silicon. read less USED (low confidence) Y. Huang, M. Wang, J. Li, and F. Zhu, “Effect of inclusion on 4H-SiC during nano-scratching from an atomistic perspective,” Journal of Physics: Condensed Matter. 2021. link Times cited: 11 Abstract: Inclusion, a common three-dimension defect, can be introduce… read moreAbstract: Inclusion, a common three-dimension defect, can be introduced during SiC epitaxy. In this study, we constructed nano-scratching molecular dynamics models embedded in two common types of inclusion—C-inclusion and Si-inclusion—to explore the effect of inclusion during scratching. Furthermore, the microstructure and atomistic behavior, surface morphology, scratching force, stress, and temperature were analyzed to bridge the simulation and processing parameters. The results showed that inclusion could affect the microstructure and atomistic behavior, and machinability. To eliminate inclusion completely, high penetration depth was required, but it would promote the process parameter sensitivity of inclusion. In summary, the behavior of C-inclusion embedded in SiC more likes a hard particle, while the behavior of Si-inclusion embedded in SiC more likes a soft particle. read less USED (low confidence) A. H. Howlader, M. S. Islam, and N. Ferdous, “Phonon transmission of vacancy disordered armchair silicene nanoribbon,” Optoelectronics Letters. 2021. link Times cited: 4 USED (low confidence) B. Liu, H. Yang, Z. Xu, D. Wang, and H. Ji, “Molecular Dynamics Simulation of Nanomachining Mechanism between Monocrystalline and Polycrystalline Silicon Carbide,” Advanced Theory and Simulations. 2021. link Times cited: 9 Abstract: As an advanced ceramics material, silicon carbide (SiC) is e… read moreAbstract: As an advanced ceramics material, silicon carbide (SiC) is extensively applied in numerous industries. In this study, molecular dynamics method is used to comparatively investigate the nanomachining mechanism between monocrystalline SiC (mono‐SiC) and polycrystalline SiC (poly‐SiC) ceramics. Four simulations are performed for the two materials with and without ultrasonic vibration‐assisted machining (UVAM). The diamond tool is set as a non‐rigid body and vibrated along the depth direction with 100 GHz in frequency and 0.5 nm in amplitude. The effects of material and ultrasonic vibration on the nanomachining mechanism of SiC are analyzed in depth, including the surface generation, subsurface damage, and tool wear. It is determined that the machinability of SiC ceramics can be effectively improved by UVAM. The machining‐induced damage extent of poly‐SiC is more serious than that of mono‐SiC. It is also found that UVAM can effectively reduce the machining‐induced damage, decrease the machining resistance, and increase the possibility of ductile removal, but bring about a slightly larger tool wear. read less USED (low confidence) M. Zimbone et al., “Extended defects in 3C-SiC: Stacking faults, threading partial dislocations, and inverted domain boundaries,” Acta Materialia. 2021. link Times cited: 21 USED (low confidence) C. Baruffi and C. Brandl, “On the structure of (111) twist grain boundaries in diamond: atomistic simulations with Tersoff-type interatomic potentials,” Acta Materialia. 2021. link Times cited: 9 USED (low confidence) J. Wang and F. Fang, “Nanometric cutting mechanism of silicon carbide,” CIRP Annals. 2021. link Times cited: 14 USED (low confidence) P. Fan, S. Goel, X. Luo, Y. Yan, Y. Geng, and Y. He, “Origins of ductile plasticity in a polycrystalline gallium arsenide during scratching: MD simulation study,” Applied Surface Science. 2021. link Times cited: 18 USED (low confidence) Y. Huang, M. Wang, J. Li, and F. Zhu, “Effect of abrasive particle shape on the development of silicon substrate during nano-grinding,” Computational Materials Science. 2021. link Times cited: 14 USED (low confidence) J. Kang, M. Li, H. Wu, H. Nguyen, T. Aoki, and Y. Hu, “Integration of boron arsenide cooling substrates into gallium nitride devices,” Nature Electronics. 2021. link Times cited: 33 USED (low confidence) Y. Zhou, H. Dai, and P. Li, “Mechanism of crack evolution in nano-indentation of single crystal silicon by atomistic simulations and theoretical analysis,” Proceedings of the Institution of Mechanical Engineers, Part C: Journal of Mechanical Engineering Science. 2021. link Times cited: 7 Abstract: The molecular dynamics (MD) model of nano-indentation proces… read moreAbstract: The molecular dynamics (MD) model of nano-indentation process was established to study the crack evolution in single crystal during nano-indentation. Two workpieces with different cracks and one workpiece with no crack were selected for indentation simulation in this study. The parameters of atom displacement, coordination number (CN), temperature, potential energy and loading force in the indentation process are analyzed in detail. Cracks were found to close during nano-indentation. Two modes of crack closure are observed: cooperative displacement and indentation failure. The existence of cracks will affect the size of transformation zone and the coordination number of atoms after indentation. Besides, the existence of cracks will reduce the increase of temperature and potential energy, and the closing mode of cracks is found to affect the value of indentation load. In addition, the change of stress with indentation depth at crack tip is calculated by theoretical model. The calculated stress curves reveal the evolution trend of cracks during indentation. These results provide guidance for the production of silicon wafer with higher surface quality. read less USED (low confidence) P. Zhou, N. Zhu, C. Xu, F. Niu, J. Li, and Y. Zhu, “Mechanical removal of SiC by multi-abrasive particles in fixed abrasive polishing using molecular dynamics simulation,” Computational Materials Science. 2021. link Times cited: 24 USED (low confidence) J. Clayton, J. Zorn, R. B. Leavy, M. Guziewski, and J. Knap, “Phase field modeling of diamond-silicon carbide ceramic composites with tertiary grain boundary phases,” International Journal of Fracture. 2021. link Times cited: 7 USED (low confidence) S. Singh, “Comparing different multibody reactive potentials for the elastic properties and nonlinear mechanics of the carbon nanostructures,” Mechanics of Materials. 2021. link Times cited: 0 USED (low confidence) W. Li et al., “Rate dependence and anisotropy of SiC response to ramp and wave-free quasi-isentropic compression,” International Journal of Plasticity. 2021. link Times cited: 13 USED (low confidence) Q. Ran et al., “Molecular dynamics simulation of displacement cascades in cubic silicon carbide,” Nuclear materials and energy. 2021. link Times cited: 10 USED (low confidence) J. Chen, W. Liu, and B. Wang, “Prediction of theoretical strength of diamond under complex loadings,” Extreme Mechanics Letters. 2021. link Times cited: 11 USED (low confidence) D. T. N. Tranh, V. V. Hoang, and T. T. Hanh, “Modeling glassy SiC nanoribbon by rapidly cooling from the liquid: An affirmation of appropriate potentials,” Physica B-condensed Matter. 2021. link Times cited: 6 USED (low confidence) B. Yang, H. Yang, T. Li, J. Yang, and P. Yang, “Thermal transport at 6H-SiC/graphene buffer layer/GaN heterogeneous interface,” Applied Surface Science. 2021. link Times cited: 16 USED (low confidence) V.-T. Nguyen and T. Fang, “Abrasive mechanisms and interfacial mechanics of amorphous silicon carbide thin films in chemical-mechanical planarization,” Journal of Alloys and Compounds. 2020. link Times cited: 32 USED (low confidence) M. S. Islam, I. Mia, S. Ahammed, C. Stampfl, and J. Park, “Exceptional in-plane and interfacial thermal transport in graphene/2D-SiC van der Waals heterostructures,” Scientific Reports. 2020. link Times cited: 18 USED (low confidence) P. Zhou, J. Li, Z. Wang, J. Chen, X. Li, and Y. Zhu, “Molecular dynamics study of the removal mechanism of SiC in a fixed abrasive polishing in water lubrication,” Ceramics International. 2020. link Times cited: 22 USED (low confidence) Q. Liu, Q. Liu, W. Yu, H. Luo, X. Ren, and S. Shen, “Tuning thermal resistance of SiC crystal/amorphous layered nanostructures via changing layer thickness,” Computational Materials Science. 2020. link Times cited: 2 USED (low confidence) Q. Liu, L. Li, Y. Jeng, G. Zhang, C. Shuai, and X. Zhu, “Effect of interatomic potentials on modeling the nanostructure of amorphous carbon by liquid quenching method,” Computational Materials Science. 2020. link Times cited: 9 USED (low confidence) W. Li, E. Hahn, X. Yao, T. Germann, B. Feng, and X. Zhang, “On the grain size dependence of shock responses in nanocrystalline sic ceramics at high strain rates,” Acta Materialia. 2020. link Times cited: 26 USED (low confidence) I. Chepkasov, V. S. Baidyshev, E. Sukhanova, M. Visotin, P. Süle, and Z. Popov, “Iron silicides formation on Si (100) and (111) surfaces through theoretical modeling of sputtering and annealing,” Applied Surface Science. 2020. link Times cited: 3 USED (low confidence) L. N. Abdulkadir, A. Bello, M. Bawa, and A. M. Abioye, “Nanometric behaviour of monocrystalline silicon when single point diamond turned—a molecular dynamics and response surface methodology analysis,” Engineering Research Express. 2020. link Times cited: 3 Abstract: Hard and brittle materials such as silicon and silicon carbi… read moreAbstract: Hard and brittle materials such as silicon and silicon carbide are widely used in aerospace and integrated circuit. They are often poorly machined owing to non-linearity in machining process and complexities in selecting suitable machining parameters and tool geometry. The experimental difficulty involved in observing nanoscale physical phenomena (i.e. in-process measurement problems, inaccessible contact area of tool and workpiece, and the difficulty of surface analysis) has led to the use of molecular dynamics (MD) and response surface methodology (RSM) to investigate effect of tool edge radius, rake and clearance angles on monocrystalline silicon in this research. The response of subsurface deformation depth (SSD), tool temperature, kinetic friction cutting and thrust forces to tool edge radius, rake and clearance angles showed that SSD increased as the rake angle, edge radius and clearance angle increased while kinetic friction reduced as they increased. The increase in SSD as the clearance angle increased as observed in this study can be associated to the interactive/combined influence of the effects of both edge radius and rake angle. read less USED (low confidence) J. Luo, C. Zhou, Y. Cheng, and L. Liu, “Assessing the EDIP potential for atomic simulation of carbon diffusion, segregation and solubility in silicon melt,” Journal of Crystal Growth. 2020. link Times cited: 2 USED (low confidence) A. Sharma, P. Ranjan, and R. Balasubramaniam, “Investigation of effect of uncut chip thickness to edge radius ratio on nanoscale cutting behavior of single crystal copper: MD simulation approach,” Journal of Micromanufacturing. 2020. link Times cited: 5 Abstract: Extremely small cutting depths in nanoscale cutting makes it… read moreAbstract: Extremely small cutting depths in nanoscale cutting makes it very difficult to measure the thermodynamic properties and understand the underlying mechanism and behavior of workpiece material. Highly precise single-crystal Cu is popularly employed in optical and electronics industries. This study, therefore, implements the molecular dynamics technique to analyze the cutting behavior and surface and subsurface phenomenon in the nanoscale cutting of copper workpieces with a diamond tool. Molecular dynamics simulation is carried out for different ratios of uncut chip thickness (a) to cutting edge radius (r) to investigate material removal mechanism, cutting forces, surface and subsurface defects, material removal rate (MRR), and stresses involved during the nanoscale cutting process. Calculation of forces and amount of plowing indicate that a/r = 0.5 is the critical ratio for which the average values of both increase to maximum. Material deformation mechanism changes from shear slip to shear zone deformation and then to plowing and elastic rubbing as the cutting depth/uncut chip thickness is reduced. The deformation during nano-cutting in terms of dislocation density changes with respect to cutting time. During the cutting process, it is observed that various subsurface defects like point defects, dislocations and dislocation loops, stacking faults, and stair-rod dislocation take place. read less USED (low confidence) V.-T. Nguyen and T. Fang, “Material removal and wear mechanism in abrasive polishing of SiO2/SiC using molecular dynamics,” Ceramics International. 2020. link Times cited: 26 USED (low confidence) J. Sun et al., “Investigation of Indenter-Size-Dependent Nanoplasticity of Silicon by Molecular Dynamics Simulation.” 2020. link Times cited: 9 Abstract: Silicon (Si) is commonly used in microelectronic devices and… read moreAbstract: Silicon (Si) is commonly used in microelectronic devices and micro/nanoelectromechanical systems (MEMS/NEMS) and the mechanical behavior of nanoscale silicon is very important. However, the origin ... read less USED (low confidence) Q. Zhang et al., “Designing ultrahard nanostructured diamond through internal defects and interface engineering at different length scales,” Carbon. 2020. link Times cited: 10 USED (low confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “Prediction of tool wear constants for diamond turn machining of CuBe,” Journal of Micromanufacturing. 2020. link Times cited: 3 Abstract: While several studies in diamond turning of homogeneous mate… read moreAbstract: While several studies in diamond turning of homogeneous materials like Cu, Al, and Si are well published, there is a lack of understanding about tool wear in case of heterogeneous materials like CuBe. Severity of the tool wear can be understood from the magnitude of the wear coefficients, and the magnitude of these coefficients is influenced by the wear mechanism. Hence, this study is aimed to calculate the wear coefficients from a known tool wear model in diamond turn machining of CuBe. Molecular dynamics simulation (MDS) results show that stress and temperature are responsible for increasing rate of tool wear. From the experimental results, change in the tool cutting edge radius due to wear was obtained and the temperature and stress for various a/r were found out using MDS. With these data, the wear coefficients, A & B, from a wear model for diamond turning were calculated. This methodology of using MDS to obtain stress and temperature for various a/r wherein, values of r are obtained from a single machining trial on actual material, will be useful for calculating the wear coefficients for the combination of single crystal diamond tool with various work piece materials and their activation energies. read less USED (low confidence) G. Bonny, L. Buongiorno, A. Bakaev, and N. Castin, “Models and regressions to describe primary damage in silicon carbide,” Scientific Reports. 2020. link Times cited: 2 USED (low confidence) X. Chen, C. Liu, J. Ke, J. Zhang, X. Shu, and J. Xu, “Subsurface damage and phase transformation in laser-assisted nanometric cutting of single crystal silicon,” Materials & Design. 2020. link Times cited: 51 USED (low confidence) P. Zhou, T. Sun, X. Shi, J. Li, Y. Zhu, and Z. Wang, “Atomic-scale study of vacancy defects in SiC affecting on removal mechanisms during nano-abrasion process,” Tribology International. 2020. link Times cited: 18 USED (low confidence) L. Pizzagalli and M. David, “Atomistic simulations of a helium bubble in silicon carbide,” Journal of Nuclear Materials. 2020. link Times cited: 8 USED (low confidence) V.-T. Nguyen and T. Fang, “Material removal and interactions between an abrasive and a SiC substrate: A molecular dynamics simulation study,” Ceramics International. 2020. link Times cited: 40 USED (low confidence) B. Liu, Z. Xu, Y. Wang, X. Gao, and R.-H. Kong, “Effect of ion implantation on material removal mechanism of 6H-SiC in nano-cutting: A molecular dynamics study,” Computational Materials Science. 2020. link Times cited: 29 USED (low confidence) J. Yan and S. Y. Chen, “Mechanical properties of monolayer antimony carbide: A molecular dynamics simulation,” Materials today communications. 2020. link Times cited: 0 USED (low confidence) L. Zhao, M. Alam, J. Zhang, R. Janisch, and A. Hartmaier, “Amorphization-governed elasto-plastic deformation under nanoindentation in cubic (3C) silicon carbide,” Ceramics International. 2020. link Times cited: 43 USED (low confidence) F. Saiz, “An ab initio study on liquid silicon carbide,” Journal of Physics and Chemistry of Solids. 2020. link Times cited: 6 USED (low confidence) B. Meng, D. Yuan, J. Zheng, P. Qiu, and S. Xu, “Tip-based nanomanufacturing process of single crystal SiC: Ductile deformation mechanism and process optimization,” Applied Surface Science. 2020. link Times cited: 26 USED (low confidence) G. Plummer and G. Tucker, “Bond-order potentials for theTi3AlC2andTi3SiC2MAX phases,” Physical Review B. 2019. link Times cited: 12 USED (low confidence) A. Islam, M. S. Islam, N. Ferdous, J. Park, A. G. Bhuiyan, and A. Hashimoto, “Anisotropic mechanical behavior of two dimensional silicon carbide: effect of temperature and vacancy defects,” Materials Research Express. 2019. link Times cited: 27 Abstract: Mechanical stability, which is featured by high tensile stre… read moreAbstract: Mechanical stability, which is featured by high tensile strength, is one of the most critical concerns for the reliability of next-generation nanoelectromechanical systems (NEMS). Presently, sp2 hybridized two-dimensional silicon carbide (2D-SiC) is supposed to be a novel nanomaterial to apply in nanocomposites, NEMS, and nano-energy harvesting applications because of its amazing electronic, mechanical and thermal properties. This paper explores the mechanical behavior, including fracture stress, fracture strain, and elastic modulus of both pristine and vacancy defected 2D-SiC at temperatures 300–700 K using molecular dynamics simulation. Two types of vacancy defects such as point and bi-vacancies with concentration 0.1%–1.0% are considered. Moreover, the effect of system size and strain rate on the mechanical behavior of 2D-SiC is also analyzed. A highly anisotropic mechanical behavior is found at all temperature and defect conditions. At 300 K, a fracture stress and an elastic modulus of 71.02 GPa and 637.26 GPa, respectively is obtained along the armchair direction, which is ∼24.42% and ∼14.38% higher compared to the zigzag directed fracture stress and elastic modulus. A reduction of fracture stress, fracture strain, and elastic modulus with the increase of temperature and defect concentration is also perceived in both armchair and zigzag directions. Moreover, due to the large symmetry breakdown by the point vacancy, a comparatively larger drastic reduction is noticed in the fracture behavior than the bi-vacancy at all temperatures and loading directions. These results would provide a new insight for solving the mechanical instability problem of SiC-based NEMS and nanodevices in the near future. read less USED (low confidence) C. de Tomas et al., “Transferability in interatomic potentials for carbon,” Carbon. 2019. link Times cited: 48 USED (low confidence) W. Tu, K. Wang, L. Qin, Z. Sun, and J. Chen, “Intrinsic mechanical properties and fracture mechanism of monolayer penta-graphene investigated by nanoindentation: A molecular dynamics study,” Computational Materials Science. 2019. link Times cited: 11 USED (low confidence) X. Li, H. Mizuseki, S. J. Pai, and K.-R. Lee, “Reactive molecular dynamics simulation of the amorphous carbon growth: Effect of the carbon triple bonds,” Computational Materials Science. 2019. link Times cited: 5 USED (low confidence) B. Meng, D. Yuan, J. Zheng, and S. Xu, “Molecular dynamics study on femtosecond laser aided machining of monocrystalline silicon carbide,” Materials Science in Semiconductor Processing. 2019. link Times cited: 19 USED (low confidence) Z. J. Choong, D. Huo, P. Degenaar, and A. O’Neill, “Edge chipping minimisation strategy for milling of monocrystalline silicon: A molecular dynamics study,” Applied Surface Science. 2019. link Times cited: 8 USED (low confidence) P. Zhou et al., “Molecular dynamics simulation of SiC removal mechanism in a fixed abrasive polishing process,” Ceramics International. 2019. link Times cited: 44 USED (low confidence) A. Mokhalingam, R. Ghaffari, R. Sauer, and S. S. Gupta, “Comparing quantum, molecular and continuum models for graphene at large deformations,” Carbon. 2019. link Times cited: 11 USED (low confidence) Z. Wu, W. Liu, and L. Zhang, “Effect of structural anisotropy on the dislocation nucleation and evolution in 6H SiC under nanoindentation,” Ceramics International. 2019. link Times cited: 23 USED (low confidence) J. D. Clayton, R. B. Leavy, and J. Knap, “Phase field modeling of heterogeneous microcrystalline ceramics,” International Journal of Solids and Structures. 2019. link Times cited: 14 USED (low confidence) H. Dai, F. Zhang, and J. Chen, “A study of ultraprecision mechanical polishing of single-crystal silicon with laser nano-structured diamond abrasive by molecular dynamics simulation,” International Journal of Mechanical Sciences. 2019. link Times cited: 35 USED (low confidence) Y. Li, Y. Li, and W. Xiao, “Point defects and grain boundary effects on tensile strength of 3C-SiC studied by molecular dynamics simulations,” Nuclear Engineering and Technology. 2019. link Times cited: 4 USED (low confidence) Q. Liu, Y. Li, and W. Xiao, “Oxygen impurity effects on the mechanical properties of SiC studied by first principles calculations,” Materials Today Communications. 2019. link Times cited: 5 USED (low confidence) M. Popov et al., “Ultrasmall diamond nanoparticles with unusual incompressibility,” Diamond and Related Materials. 2019. link Times cited: 12 USED (low confidence) Z. Wu, W. Liu, L. Zhang, and S. Lim, “Amorphization and Dislocation Evolution Mechanisms of Single Crystalline 6H-SiC,” Materials Engineering eJournal. 2019. link Times cited: 67 Abstract: The amorphization and dislocation evolution mechanisms of a … read moreAbstract: The amorphization and dislocation evolution mechanisms of a single crystal 6H-SiC were systematically investigated by using nano-indentation, high-resolution transmitted electron microscope (HRTEM), molecular dynamics (MD) simulations and the generalized stacking fault (GSF) energy surface analysis. Two major plastic deformation mechanisms of 6H-SiC under nano-indentation were revealed by HRTEM, i.e., (1) an amorphization region near the residual indentation mark, and (2) dislocations below the amorphization region in both the basal and prismatic planes. MD results showed that the amorphization process corresponds to the first “pop-in” event of the indentation load-displacement curve, while the dislocation nucleation and propagation are related to the consequent “pop-in” events. The amorphization is confirmed to achieve via an initial transformation from wurtzite structure to an intermediate structure, and then a further amorphization process. read less USED (low confidence) L. N. Abdulkadir and K. Abou-El-Hossein, “Diamond tool wear mode, path and tip temperature distribution considering effect of varying rake angle and duncut/Redge ratio,” Surface Topography: Metrology and Properties. 2019. link Times cited: 8 Abstract: Diamond tool wear has been proven to strongly depend on the … read moreAbstract: Diamond tool wear has been proven to strongly depend on the temperature at the cutting zone. This is because the elevated temperature so attained during machining not only lowers carbon cohesion energy thereby reducing the fracture toughness of diamond tool due to C–C bond weakening but also facilitates high rate of carbon diffusion from diamond tool into silicon workpiece leading to high tool wear. This research therefore studied the response of diamond tool wear path and mode to changing rake angle and duncutRedge ratio during ultraprecision single point diamond turning of a monocrystalline silicon. It was observed from the study that the tool with larger rake angle and high duncutRedge ratio experienced stronger cutting resistance from the workpiece thereby causing the kinetic friction to be high. Additionally, silicon carbide (Tribochemistry) formation was observed to be through both solid-state single phase and multiphase reaction which are in tandem with sp 3 –sp 2 disorder (graphitization) of diamond with bulk of the observed tool wear happening in the flank face which could be due to increasing rake angle influence on the changing duncutRedge ratio. read less USED (low confidence) S. Bringuier et al., “Atomic insight into concurrent He, D, and T sputtering and near-surface implantation of 3C-SiC crystallographic surfaces,” Nuclear Materials and Energy. 2019. link Times cited: 13 USED (low confidence) S. Winczewski and J. Rybicki, “Anisotropic mechanical behavior and auxeticity of penta-graphene: Molecular statics/molecular dynamics studies,” Carbon. 2019. link Times cited: 16 USED (low confidence) X. Li, A. Wang, and K.-R. Lee, “Role of unsaturated hydrocarbon lubricant on the friction behavior of amorphous carbon films from reactive molecular dynamics study,” Computational Materials Science. 2019. link Times cited: 12 USED (low confidence) J. Sun, Y. Guo, Q. Wang, and Y. Kawazoe, “Thermal transport properties of penta-graphene with grain boundaries,” Carbon. 2019. link Times cited: 21 USED (low confidence) Q. Liu, Z. Jia, Z. Yaowu, L. Xiong, T. Shi, and Y. Long, “On the dynamic behaviors of silicon single crystal under nanosecond laser irradiation,” Computational Materials Science. 2019. link Times cited: 3 USED (low confidence) Q. Liu et al., “A semi-empirical fracture model for silicon cleavage fracture and its molecular dynamics study,” Theoretical and Applied Fracture Mechanics. 2019. link Times cited: 8 USED (low confidence) J. Chen and B. Wang, “Existence criteria and validity of plate models for graphene-like materials,” Science China Physics, Mechanics & Astronomy. 2019. link Times cited: 5 USED (low confidence) H. Dai, J. Chen, and G. Liu, “A numerical study on subsurface quality and material removal during ultrasonic vibration assisted cutting of monocrystalline silicon by molecular dynamics simulation,” Materials Research Express. 2019. link Times cited: 30 Abstract: Molecular dynamics (MD) simulation is used to study the subs… read moreAbstract: Molecular dynamics (MD) simulation is used to study the subsurface quality and material removal of single crystal silicon with a diamond tool during ultrasonic elliptical vibration assisted cutting (UEVAC), 1D ultrasonic vibration assisted cutting (1D UVAC) and traditional cutting (TC) process. In the simulations, a long-range analytical bond order potential is used to describe the interaction inside the silicon specimen, providing a more accurate depiction of the atomic scale mechanisms of ductile plasticity, brittle fracture, and structural changes in silicon. The results show that UEVAC and 1D UVAC in cutting brittle material silicon causes a much smaller cutting force, much lower von Mises stress at the subsurface, larger material remove rate, lower compressive normal stress σ x x and σ y y , and smaller shear stress τ x y . In addition, the hydrostatic stress of subsurface for TC and 1D UVAC is much higher than that for UEVAC, which results in fewer Si-II and bct5-Si formed from the original Si-I in UEVAC. Moreover, the number of other atoms for UEVAC is overall smaller than those of using TC and 1D UVAC, which confirms that UEVAC produces a better subsurface. And atomic flow field analysis shows that the UEVAC tends to cut silicon in a more ductile mode. Besides, the temperature in front of tool edge and below the tool flank face of TC is much higher. This means that 1D UVAC and UEVAC have a positive effect on the tool life. However, the temperature in subsurface zone is overall larger, which reveals that 1D UVAC and UEVAC have a negative effect on the subsurface temperature. read less USED (low confidence) J. Fang, X. Liu, H. Lu, X. Liu, and X. Song, “Crystal defects responsible for mechanical behaviors of a WC-Co composite at room and high temperatures - a simulation study.,” Acta crystallographica Section B, Structural science, crystal engineering and materials. 2019. link Times cited: 11 Abstract: The microstructure evolution and changes in the structures o… read moreAbstract: The microstructure evolution and changes in the structures of crystal defects of the nanocrystalline WC-Co composite in the process of uniaxial compression were studied by simulations at both room and high temperatures. The deformation processes were demonstrated as a function of stress and temperature for the stages prior to and after yielding of the composite. The Peierls stresses were evaluated for Co and WC dislocations with increasing temperature. The deformation mechanisms for each stage of the stress-strain curve were disclosed, in which the effect of temperature was clarified. It was found that with the increase of stress, from elastic deformation to plastic deformation then to yielding of the composite, the dominant mechanisms are grain boundary migration, formation and motion of dislocations in Co, concurrent motion and reaction of dislocations in Co and WC, and then rotation of WC grains in combination with motion of Co and WC dislocations. At the yielding stage, sliding of WC grain boundaries plays an increasingly important role in the contribution to plastic deformation at high temperatures. With strain the proportion of mobile dislocations decreases, and dislocations pile up at triple junctions of WC grains, WC/WC grain boundaries and WC/Co phase boundaries in priority order, leading to the nucleation and propagation of microcracks in these regions. read less USED (low confidence) X. Li, A. Wang, and K.-R. Lee, “Insights on low-friction mechanism of amorphous carbon films from reactive molecular dynamics study,” Tribology International. 2019. link Times cited: 37 USED (low confidence) B. Zhu, D. Zhao, and H. Zhao, “A study of deformation behavior and phase transformation in 4H-SiC during nanoindentation process via molecular dynamics simulation,” Ceramics International. 2019. link Times cited: 54 USED (low confidence) Y. Wang, C. Wang, Y. Zhang, and H. Tan, “Graphene kirigami as reinforcement and interfacial bonding effect for toughness and strength of silicon-based nanocomposites,” Computational Materials Science. 2019. link Times cited: 3 USED (low confidence) J. Sanshan, Q. Huang, W. Tu, J. Chen, and Z. Sun, “Investigation on the phase transformation of monocrystalline silicon during nanoindentation at cryogenic temperature by molecular dynamics simulation,” Physica B: Condensed Matter. 2019. link Times cited: 7 USED (low confidence) B. Zhu, D. Zhao, Y. Tian, S. Wang, H. Zhao, and J. Zhang, “Study on the deformation mechanism of spherical diamond indenter and its influence on 3C-SiC sample during nanoindentation process via molecular dynamics simulation,” Materials Science in Semiconductor Processing. 2019. link Times cited: 28 USED (low confidence) K. Skrobas, S. Stelmakh, S. Gierlotka, and B. Palosz, “A model of density waves in atomic structure of nanodiamond by molecular dynamics simulations,” Diamond and Related Materials. 2019. link Times cited: 10 USED (low confidence) R. Chen, J. Wang, F. Fang, and X. Zhang, “Influence of buried modified layer on crack propagation and diamond turning of silicon,” Precision Engineering. 2019. link Times cited: 5 USED (low confidence) E. Eroğlu, S. Aydin, and M. Şimşek, “Stability of intrinsic and extrinsic co-decorated boron sheets with Li and Mg,” Computational Condensed Matter. 2018. link Times cited: 0 USED (low confidence) M. Mock and K. Albe, “Modelling of dislocation-solute interaction in ODS steels: Analytic bond-order potential for the iron-yttrium system,” Journal of Nuclear Materials. 2018. link Times cited: 6 USED (low confidence) T. Sipkens and K. Daun, “Effect of Surface Interatomic Potential on Thermal Accommodation Coefficients Derived from Molecular Dynamics,” The Journal of Physical Chemistry C. 2018. link Times cited: 14 Abstract: This work investigates how the interatomic surface potential… read moreAbstract: This work investigates how the interatomic surface potential influences molecular dynamics (MD)-derived thermal accommodation coefficients (TACs). Iron, copper, and silicon surfaces are considered over a range of temperatures that include their melting points. Several classes of potentials are reviewed, including two-body, three-body, and bond-order force fields. MD-derived densities and visualization of the surfaces are used to explain the differences in the parameterizations of these potentials within the context of gas–surface scattering. Finally, TACs are predicted for a range of gas–surface combinations, and recommended values of the TAC are selected that take into account the robustness and uncertainties of each of the considered parameterizations. Further, it is observed that there is a significant change in the TAC about phase changes that must be taken into account for applications with a large range of surface temperatures. read less USED (low confidence) X. Li, A. Wang, and K.-R. Lee, “Comparison of empirical potentials for calculating structural properties of amorphous carbon films by molecular dynamics simulation,” Computational Materials Science. 2018. link Times cited: 30 USED (low confidence) T. Nguyen, K. Sato, and Y. Shibutani, “Development of Fe-C interatomic potential for carbon impurities in α-iron,” Computational Materials Science. 2018. link Times cited: 10 USED (low confidence) A. Gangele and A. Pandey, “Elastic and fracture characteristics of graphene-silicon nanosheet composites using nonlinear finite element method,” International Journal of Mechanical Sciences. 2018. link Times cited: 18 USED (low confidence) T. Narumi, Y. Shibuta, and T. Yoshikawa, “Molecular dynamics simulation of interfacial growth of SiC from Si–C solution on different growth planes,” Journal of Crystal Growth. 2018. link Times cited: 3 USED (low confidence) H.-T. Nguyen, M.-Q. Le, and V. Nguyen, “Mode-I stress intensity factors of silicene, AlN, and SiC hexagonal sheets,” Materials Research Express. 2018. link Times cited: 11 Abstract: The crack-tip displacement field and molecular dynamics fini… read moreAbstract: The crack-tip displacement field and molecular dynamics finite element method with Tersoff potentials were used to find the mode-I stress intensity factors (SIF) of silicene, aluminum nitride (AlN), and silicon carbide (SiC) hexagonal sheets. Fracture properties of graphene and boronitrene are also included for comparison. It is found that KIct (KIc is mode-I critical SIF and t is the sheet’s thickness) of silicene, AlN, and SiC sheets are approximately 80, 66, and 47%; and 73, 64, and 45% smaller values of those of graphene for crack along the armchair and zigzag directions, respectively. The estimated fracture toughness of silicene is close to the experimental data of single-crystal silicon. read less USED (low confidence) J. Han et al., “Deformation mechanisms at multiple pop-ins under spherical nanoindentation of (1 1 1) Si,” Computational Materials Science. 2018. link Times cited: 14 USED (low confidence) A. Kubo, S. Nagao, and Y. Umeno, “Molecular dynamics study of deformation and fracture in SiC with angular dependent potential model,” Computational Materials Science. 2017. link Times cited: 7 USED (low confidence) S. Jiapeng, L. Cheng, J. Han, A. Ma, and L. Fang, “Nanoindentation Induced Deformation and Pop-in Events in a Silicon Crystal: Molecular Dynamics Simulation and Experiment,” Scientific Reports. 2017. link Times cited: 64 USED (low confidence) M. R. Z. Kouhpanji and U. Jafaraghaei, “A semianalytical approach for determining the nonclassical mechanical properties of materials,” Journal of the Mechanical Behavior of Materials. 2017. link Times cited: 7 Abstract: In this article, a semianalytical approach for demonstrating… read moreAbstract: In this article, a semianalytical approach for demonstrating elastic waves’ propagation in nanostructures has been presented based on the modified couple-stress theory including acceleration gradients (MCST-AG). Using the experimental results and atomic simulations, the static and dynamic length scales were calculated for several materials, zinc oxide (ZnO), silicon (Si), silicon carbide (SiC), indium antimonide (InSb), and diamond. To evaluate the predicted static and dynamic length scales as well as the presented model, the natural frequencies of a beam in addition to the phase velocity and group velocity of Si were studied and compared with the available static length scales, estimated using strain-gradient theory without considering acceleration gradients (SGT). These three criteria, natural frequency, phase velocity, and group velocity, show that the presented model is dynamically stable even for larger wavevector values. Furthermore, it is explained why the previous works, which all are based on the SGT, predicted very small values for the static length scale in the longitudinal direction comparing the static length scale in the transverse directions. read less USED (low confidence) K. Fung, C. Tang, and C. Cheung, “Molecular dynamics analysis of the effect of surface flaws of diamond tools on tool wear in nanometric cutting,” Computational Materials Science. 2017. link Times cited: 41 USED (low confidence) Q. Feng, X. Song, H. Xie, H. Wang, X. Liu, and F. Yin, “Deformation and plastic coordination in WC-Co composite — Molecular dynamics simulation of nanoindentation,” Materials & Design. 2017. link Times cited: 53 USED (low confidence) V. Levitas, H. Chen, and L. Xiong, “Triaxial-Stress-Induced Homogeneous Hysteresis-Free First-Order Phase Transformations with Stable Intermediate Phases.,” Physical review letters. 2017. link Times cited: 43 Abstract: Starting with thermodynamic predictions and following with m… read moreAbstract: Starting with thermodynamic predictions and following with molecular dynamics simulations, special triaxial compression-tension states were found for which the stresses for the instability of the crystal lattice of silicon (Si) are the same for direct and reverse phase transformations (PTs) between semiconducting Si I and metallic Si II phases. This leads to unique homogeneous and hysteresis-free first-order PTs, for which each intermediate crystal lattice along the transformation path is in indifferent thermodynamic equilibrium and can be arrested and studied by fixing the strain in one direction. By approaching these stress states, a traditional two-phase system continuously transforms to homogenous intermediate phases. Zero hysteresis and homogeneous transformations are the optimal property for various PT applications, which drastically reduce damage and energy dissipation. read less USED (low confidence) C. Tomas, I. Suarez-Martinez, and N. Marks, “Graphitization of amorphous carbons: A comparative study of interatomic potentials,” Carbon. 2016. link Times cited: 160 USED (low confidence) M. Budnitzki and M. Kuna, “Stress induced phase transitions in silicon,” Journal of The Mechanics and Physics of Solids. 2016. link Times cited: 25 USED (low confidence) H. N. Pishkenari and P. G. Ghanbari, “Vibrational properties of C60: A comparison among different inter-atomic potentials,” Computational Materials Science. 2016. link Times cited: 11 USED (low confidence) N. Zhou, C. Zhang, X. Zeng, J. Yuan, and L. Zhou, “A molecular dynamics study of the growth rate of SiC crystal and its dependence on the temperature,” International Journal of Modern Physics B. 2016. link Times cited: 1 Abstract: Molecular dynamics simulations of crystal growth of SiC in t… read moreAbstract: Molecular dynamics simulations of crystal growth of SiC in the reduced temperature range of 0.51–1.02 have been carried out. In particular, the relationship between the growth rate and the reduced temperature has been investigated by the simulations. The results show that the growth rate increases first with the temperature and then decreases dramatically after passing through a maximum. Calculations of the growth rate according to the Wilson–Frenkel model have been applied to the present system, with the required parameters of the activation energy for atomic diffusion and the free energy changes calculated by molecular dynamics simulations. The temperature dependence of the growth rate, calculated by molecular dynamics, agrees with the prediction of Wilson–Frenkel model, indicating that the crystal growth of SiC is a kind of diffusion limited growth. read less USED (low confidence) W. Su, Y. Li, C. Nie, W. Xiao, and L. Yan, “First principles study of the C/Si ratio effect on the ideal shear strength of β–SiC,” Materials Research Express. 2016. link Times cited: 3 Abstract: The effect of the C/Si atomic ratio on the ideal shear stren… read moreAbstract: The effect of the C/Si atomic ratio on the ideal shear strength of β-SiC is investigated with first principles calculations. β − SiC samples with different C/Si ratios are generated by Monte Carlo (MC) simulations with empirical inter-atomic SiC potential. Each SiC sample is sheared along the 〈 100 〉 direction and the stress-strain curve is calculated from first principles. The results show that the ideal shear strength of SiC decreases with the increase of C/Si ratio. For a non-stoichiometric SiC sample, a C–C bond inside a large carbon cluster breaks first under shear strain condition due to the internal strain around the carbon clusters. Because the band gap is narrowed under shear strain conditions, a local maximum stress appears in the elastic region of the stress-strain curve for each SiC sample at certain strain condition. The yield strength may increase with the increase of C/Si ratio. read less USED (low confidence) O. Strickson, “Numerical constitutive modelling for continuum mechanics simulation.” 2016. link Times cited: 0 USED (low confidence) A. Higginbotham et al., “Inelastic response of silicon to shock compression,” Scientific Reports. 2016. link Times cited: 20 USED (low confidence) Y. Li and W. Xiao, “First principles study of the C/Si ratio effect on the ideal tensile strength of β-SiC,” Computational Materials Science. 2015. link Times cited: 11 USED (low confidence) E. Hahn and M. Meyers, “Grain-size dependent mechanical behavior of nanocrystalline metals,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 162 USED (low confidence) N. M. Umran, N. Kaur, K. Seema, and R. Kumar, “Study of endohedral doped C60 fullerene using model potentials,” Materials Research Express. 2015. link Times cited: 4 Abstract: In this paper, we present a study of Si, Ge, Au and Tl doped… read moreAbstract: In this paper, we present a study of Si, Ge, Au and Tl doped endohedral fullerenes using model potentials. We have used Brenner potential, Lennard-Jones potential and Gupta potential to describe the C–C (Si, Ge), C–metal and metal–metal interaction, respectively. We have done total energy calculations to find out the minimum energy structures. In the case of Sin@C60 complex, a maximum of nine atoms can be doped inside the fullerene cage. The addition of a tenth Si atom leads to the breaking of the fullerene cage. Similarly for Ge, Au and Tl, a maximum of four, two and two atoms, respectively, can be doped inside the fullerene cage. It has been observed that the stability of fullerene decreases with an increase in the number of dopant atoms. These calculations prove the change of characteristics of fullerenes upon doping. read less USED (low confidence) P. Süle and M. Szendrő, “Time-lapsed graphene moiré superlattices on Cu(1 1 1),” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 7 Abstract: We report classical molecular dynamics simulations (CMD) of … read moreAbstract: We report classical molecular dynamics simulations (CMD) of the moiré superlattice of graphene on Cu(1 1 1) using a new parameterized Abell–Tersoff potential for the graphene/Cu(1 1 1) interface fitted in this paper to nonlocal van der Waals density functional theory calculations. The interfacial force field with time-lapsed CMD provides superlattices in good quantitative agreement with the available experimental results. The long range coincidence supercells with nonequivalent moiré hills have also been identified and analyzed. Spot profile analysis reveals that the moiré spots are inequivalent over large areas, and their heights are randomly distributed. This result is in accordance with recent atomic force microscopy studies. Our simulations also shed light on the transient dynamics of the moiré superlattice in atomic detail. The moiré superlattice exhibits a pattern which is dynamical rather than statically pinned to the support, and can be observed mostly via time-lapsing. The instantaneous snapshots of the periodic moiré pattern at low temperature are already weakly disordered, lacking the apparent sharpness of the time-averaged pattern and of the scanning tunneling microscopy images. This suggests the existence of competition of orders—between a static (first-order) moiré superstructure and a dynamical (second-order) moiré superstructure. read less USED (low confidence) B. Reischl, A. Kuronen, and K. Nordlund, “Nanoindentation of gold nanorods with an atomic force microscope,” Materials Research Express. 2014. link Times cited: 6 Abstract: The atomic force microscope (AFM) can be used to measure mec… read moreAbstract: The atomic force microscope (AFM) can be used to measure mechanical properties of nanoscale objects, which are too small to be studied using a conventional nanoindenter. The contact mechanics at such small scales, in proximity of free surfaces, deviate substantially from simple continuum models. We present results from atomistic computer simulations of the indentation of gold nanorods using a diamond AFM tip and give insight in the atomic scale processes, involving creation and migration of dislocations, leading to the plastic deformation of the sample under load, and explain the force–distance curves observed for different tip apex radii of curvature, as well as different crystallographic structure and orientation of the gold nanorod samples. read less USED (low confidence) M.-Q. Le and D.-T. Nguyen, “Atomistic simulations of pristine and defective hexagonal BN and SiC sheets under uniaxial tension,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2014. link Times cited: 57 USED (low confidence) M. Kozłowski, P. Sowa, A. Biborski, and R. Kozubski, “SiC (0001) and (0001¯) surfaces diffusion parameters estimated by means of atomistic Kinetic Monte Carlo simulations,” Materials Letters. 2014. link Times cited: 5 USED (low confidence) M. Joe, Y. K. Han, K.-R. Lee, H. Mizuseki, and S. Kim, “An ideal polymeric C60 coating on a Si electrode for durable Li-ion batteries,” Carbon. 2014. link Times cited: 17 USED (low confidence) K. Leung, Z. Pan, and D. Warner, “Atomistic-based predictions of crack tip behavior in silicon carbide across a range of temperatures and strain rates,” Acta Materialia. 2014. link Times cited: 25 USED (low confidence) K. Fung, C. Y. Tang, C. Cheung, and W. C. Law, “Molecular Dynamics Simulation of Plastic Deformation of Diamond at an Elevated Temperature,” Key Engineering Materials. 2014. link Times cited: 2 Abstract: Single point diamond tools are commonly used for ultraprecis… read moreAbstract: Single point diamond tools are commonly used for ultraprecision machining. At high cutting speeds, frictional contact and local heat may cause material damage to the diamond tool. The diamond crystal is softened and its mechanical strength decreases with the increase in temperature. Plastic deformation of diamonds was recently reported in some experimental studies. In this work, a molecular dynamics (MD) simulation was implemented to predict the deformation of single crystal diamond at various temperatures. Diamond is brittle at room temperature, however, it starts to exhibit plastic dislocation at a temperature above 1200 K under a confining pressure. The condition in ultraprecision machining is indeed a temperature gradient distribution at the tool tip, between the maximum temperature at the tool-workpiece interface and the average temperature at the core. The simulation results predicted that diamond deformed plastically under the gradient between 1500K and 860K. It is surprising that secondary cracks were resulted from the gradient, as comparing to a single slip obtained in an evenly distributed temperature. Bond dissociation nucleated the fractures along the (111) shuffle planes, perfect dislocation merely occurred in the hot zone and sp3-to-sp2 disorder at the cool zone. The temperature gradient created a lattice mismatch and nucleated the secondary cracks. The results give an insight that a catastrophic fracture and local material damage can occur at a diamond tool tip at the cutting temperature above 1200 K, due to softening and graphitization. read less USED (low confidence) J. Behler, “Representing potential energy surfaces by high-dimensional neural network potentials,” Journal of Physics: Condensed Matter. 2014. link Times cited: 293 Abstract: The development of interatomic potentials employing artifici… read moreAbstract: The development of interatomic potentials employing artificial neural networks has seen tremendous progress in recent years. While until recently the applicability of neural network potentials (NNPs) has been restricted to low-dimensional systems, this limitation has now been overcome and high-dimensional NNPs can be used in large-scale molecular dynamics simulations of thousands of atoms. NNPs are constructed by adjusting a set of parameters using data from electronic structure calculations, and in many cases energies and forces can be obtained with very high accuracy. Therefore, NNP-based simulation results are often very close to those gained by a direct application of first-principles methods. In this review, the basic methodology of high-dimensional NNPs will be presented with a special focus on the scope and the remaining limitations of this approach. The development of NNPs requires substantial computational effort as typically thousands of reference calculations are required. Still, if the problem to be studied involves very large systems or long simulation times this overhead is regained quickly. Further, the method is still limited to systems containing about three or four chemical elements due to the rapidly increasing complexity of the configuration space, although many atoms of each species can be present. Due to the ability of NNPs to describe even extremely complex atomic configurations with excellent accuracy irrespective of the nature of the atomic interactions, they represent a general and therefore widely applicable technique, e.g. for addressing problems in materials science, for investigating properties of interfaces, and for studying solvation processes. read less USED (low confidence) M. Kozłowski, R. Abdank-Kozubski, and C. Goyhenex, “Superstructure Transformations in High-Temperature Intermetallic Nanolayers: Atomistic Simulation,” Diffusion Foundations. 2014. link Times cited: 1 Abstract: Superstructure transformation processes in intermetallics ha… read moreAbstract: Superstructure transformation processes in intermetallics have beenstudied at the atomistic scale using Monte Carlo algorithms within two dis-tinct models: two-body interactions Ising-like system and Analytic Bond-Order Potentials. The transformation from “in-plane” to “off-plane” L10 vari-ant in [001]-oriented FePt nano-layers was observed and analysed by analyt-ical calculations providing clear explanation of the origin of the process, aswell as by “rigid-lattice” and “off-lattice” Monte Carlo simulations showingthe kinetics of the superstructure transformation. read less USED (low confidence) S. Goel, W. B. Rashid, X. Luo, A. Agrawal, and V. Jain, “A Theoretical Assessment of Surface Defect Machining and Hot Machining of Nanocrystalline Silicon Carbide,” Journal of Manufacturing Science and Engineering-transactions of The Asme. 2014. link Times cited: 49 Abstract: In this paper, a newly proposed machining method named "… read moreAbstract: In this paper, a newly proposed machining method named "surface defect machining" (SDM) was explored for machining of nanocrystalline beta silicon carbide (3C-SiC) at 300 K using MD simulation. The results were compared with isothermal high temperature machining at 1200 K under the same machining parameters, emulating ductile mode micro laser assisted machining (μ-LAM) and with conventional cutting at 300 K. In the SDM simulation, surface defects were generated on the top of the (010) surface of the 3C-SiC work piece prior to cutting, and the workpiece was then cut along the ⟨100⟩ direction using a single point diamond cutting tool at a cutting speed of 10 m/s. Cutting forces, subsurface deformation layer depth, temperature in the shear zone, shear plane angle and friction coefficient were used to characterize the response of the workpiece. Simulation results showed that SDM provides a unique advantage of decreased shear plane angle which eases the shearing action. This in turn causes an increased value of average coefficient of friction in contrast to the isothermal cutting (carried at 1200 K) and normal cutting (carried at 300 K). The increase of friction coefficient, however, was found to aid the cutting action of the tool due to an intermittent dropping in the cutting forces, lowering stresses on the cutting tool and reduced operational temperature. Analysis shows that the introduction of surface defects prior to conventional machining can be a viable choice for machining a wide range of ceramics, hard steels and composites compared to hot machining. read less USED (low confidence) K. Kang, T. Eun, M.-C. Jun, and B.-J. Lee, “Governing factors for the formation of 4H or 6H-SiC polytype during SiC crystal growth: An atomistic computational approach,” Journal of Crystal Growth. 2014. link Times cited: 30 USED (low confidence) H. Xie, F. Yin, and T. Yu, “Strain rate induced graphitization of cubic diamond film,” Applied Physics Letters. 2014. link Times cited: 5 Abstract: Using molecular dynamics simulations with a Tersoff-type for… read moreAbstract: Using molecular dynamics simulations with a Tersoff-type force field, we studied the deformation of cubic diamond film subjected to uniform strain rates at 30 K. The results show that at high strain rates, the diamond cubic phase transforms continuously to a multilayer graphene phase; and the graphitization begins from the (011) free surface and goes rapidly into the inner of the film. In this paper, we discuss the mechanism of graphitization and calculate the energy barrier of the graphitization. read less USED (low confidence) K. Fan et al., “Analytical Bond-order Potential for hcp‐Y,” Chinese Journal of Chemical Physics. 2013. link Times cited: 6 Abstract: The lattice parameters, elastic constants, cohesive energy, … read moreAbstract: The lattice parameters, elastic constants, cohesive energy, structural energy differences, as well as the properties of point defects and planar defects of hexagonal close‐packed yttrium (hcp‐Y) have been studied with ab initio density functional theory for constructing an extensive database. Based on an analytical bond-order potential scheme, empirical many‐body interatomic potential for hcp‐Y has been developed. The model is fitted to some properties of Y, e.g., the lattice parameters, elastic constants, bulk modulus, cohesive energy, vacancy formation energy, and the structural energy differences. The present potential has ability to reproduce defect properties including the self‐interstitial atoms formation energies, vacancy formation energy, divacancy binding energy, as well as the bulk properties and the thermal dynamic properties. read less USED (low confidence) J. Li, J. Li, X. Xu, Y. Zhou, M. Zhang, and X. Luo, “First-principles investigation on the electronic and magnetic properties of cubic Be0.75Mn0.25X (X = S, Se, Te),” Journal of Alloys and Compounds. 2013. link Times cited: 13 USED (low confidence) J. Zhang et al., “Superplastic nanocrystalline ceramics at room temperature and high strain rates,” Scripta Materialia. 2013. link Times cited: 19 USED (low confidence) M. Hu and D. Poulikakos, “Graphene mediated thermal resistance reduction at strongly coupled interfaces,” International Journal of Heat and Mass Transfer. 2013. link Times cited: 56 USED (low confidence) Y. Jing, L. Guo, Y. Sun, J. Shen, and N. Aluru, “Mechanical properties of a silicon nanofilm covered with defective graphene,” Surface Science. 2013. link Times cited: 9 USED (low confidence) M. Kalbáč, O. Lehtinen, A. Krasheninnikov, and J. Keinonen, “Ion‐Irradiation‐Induced Defects in Isotopically‐Labeled Two Layered Graphene: Enhanced In‐Situ Annealing of the Damage,” Advanced Materials. 2013. link Times cited: 70 Abstract: Contrary to theoretical estimates based on the conventional … read moreAbstract: Contrary to theoretical estimates based on the conventional binary collision model, experimental results indicate that the number of defects in the lower layer of the bi-layer graphene sample is smaller than in the upper layer. This observation is explained by in situ self-annealing of the defects. read less USED (low confidence) L. Pastewka, A. Klemenz, P. Gumbsch, and M. Moseler, “Screened empirical bond-order potentials for Si-C,” Physical Review B. 2013. link Times cited: 110 Abstract: Typical empirical bond-order potentials are short ranged and… read moreAbstract: Typical empirical bond-order potentials are short ranged and give ductile instead of brittle behavior for materials such as crystalline silicon or diamond. Screening functions can be used to increase the range of these potentials. We outline a general procedure to combine screening functions with bond-order potentials that does not require to refit any of the potential's properties. We use this approach to modify Tersoff's [Phys. Rev. B 39, 5566 (1989)], Erhart & Albe's [Phys. Rev. B 71, 35211 (2005)] and Kumagai et al.'s [Comp. Mater. Sci. 39, 457 (2007)] Si, C and Si-C potentials. The resulting potential formulations correctly reproduce brittle materials response, and give an improved description of amorphous phases. read less USED (low confidence) L. Briquet et al., “Reactive force field potential for carbon deposition on silicon surfaces,” Journal of Physics: Condensed Matter. 2012. link Times cited: 16 Abstract: In this paper a new interatomic potential based on the Kieff… read moreAbstract: In this paper a new interatomic potential based on the Kieffer force field and designed to perform molecular dynamics (MD) simulations of carbon deposition on silicon surfaces is implemented. This potential is a third-order reactive force field that includes a dynamic charge transfer and allows for the formation and breaking of bonds. The parameters for Si–C and C–C interactions are optimized using a genetic algorithm. The quality of the potential is tested on its ability to model silicon carbide and diamond physical properties as well as the formation energies of point defects. Furthermore, MD simulations of carbon deposition on reconstructed (100) silicon surfaces are carried out and compared to similar simulations using a Tersoff-like bond order potential. Simulations with both potentials produce similar results showing the ability to extend the use of the Kieffer potential to deposition studies. The investigation reveals the presence of a channelling effect when depositing the carbon at 45° incidence angle. This effect is due to channels running in directions symmetrically equivalent to the (110) direction. The channelling is observed to a lesser extent for carbon atoms with 30° and 60° incidence angles relative to the surface normal. On a pristine silicon surface, sticking coefficients were found to vary between 100 and 73%, depending on deposition conditions. read less USED (low confidence) L. Pastewka, M. Mrovec, M. Moseler, and P. Gumbsch, “Bond order potentials for fracture, wear, and plasticity,” MRS Bulletin. 2012. link Times cited: 55 Abstract: Coulson’s bond order is a chemically intuitive quantity that… read moreAbstract: Coulson’s bond order is a chemically intuitive quantity that measures the difference in the occupation of bonding and anti-bonding orbitals. Both empirical and rigorously derived bond order expressions have evolved in the course of time and proven very useful for atomistic modeling of materials. The latest generation of empirical formulations has recently been augmented by screening-function approaches. Using friction and wear of diamond and diamond-like carbon as examples, we demonstrate that such a screened bond order scheme allows for a faithful description of dynamical bond-breaking processes in materials far from equilibrium. The rigorous bond order expansions are obtained by systematic coarse-graining of the tight binding approximation and form a bridge between the electronic structure and the atomistic modeling hierarchies. They have enabled bottom-up derivations of bond order potentials for covalently bonded semiconductors, transition metals, and multicomponent intermetallics. The recently developed magnetic bond order potential gives a correct description of both directional covalent bonds and magnetic interactions in iron and is able to correctly predict the stability of bulk Fe polymorphs as well as the intricate properties of dislocation cores. The bond order schemes hence represent a family of reliable and powerful models that can be applied in large-scale simulations of complex processes involving fracture, wear, and plasticity. read less USED (low confidence) M. Hu, X. Zhang, D. Poulikakos, and C. Grigoropoulos, “Large ‘near junction’ thermal resistance reduction in electronics by interface nanoengineering,” International Journal of Heat and Mass Transfer. 2011. link Times cited: 54 USED (low confidence) J. Pohl and K. Albe, “Void formation in melt-grown silicon studied by molecular dynamics simulations: From grown-in faulted dislocation loops to vacancy clusters,” Applied Physics Letters. 2011. link Times cited: 5 Abstract: Molecular dynamics simulations of a dislocation based mechan… read moreAbstract: Molecular dynamics simulations of a dislocation based mechanism for void formation in silicon are presented. By studying a moving solid-liquid interface in Si, we observe the formation of dislocation loops on (111) facets consisting of coherency and anticoherency dislocations, which disband within nanoseconds into vacancy clusters of 10 or more vacancies. These vacancy clusters can act as nucleation seeds for the experimentally observed octahedral single and double voids. read less USED (low confidence) B. Jelinek et al., “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Physical Review B. 2011. link Times cited: 218 Abstract: A set of modified embedded-atom method (MEAM) potentials for… read moreAbstract: A set of modified embedded-atom method (MEAM) potentials for the interactions between Al, Si, Mg, Cu, and Fe was developed from a combination of each element's MEAM potential in order to study metal alloying. Previously published MEAM parameters of single elements have been improved for better agreement to the generalized stacking fault energy (GSFE) curves when compared with ab initio generated GSFE curves. The MEAM parameters for element pairs were constructed based on the structural and elastic properties of element pairs in the NaCl reference structure garnered from ab initio calculations, with adjustment to reproduce the ab initio heat of formation of the most stable binary compounds. The new MEAM potentials were validated by comparing the formation energies of defects, equilibrium volumes, elastic moduli, and heat of formation for several binary compounds with ab initio simulations and experiments. Single elements in their ground-state crystal structure were subjected to heating to test the potentials at elevated temperatures. An Al potential was modified to avoid formation of an unphysical solid structure at high temperatures. The thermal expansion coefficient of a compound with the composition of AA 6061 alloy was evaluated and compared with experimental values. MEAM potential tests performed in this work, utilizing the universal atomistic simulation environment (ASE), are distributed to facilitate reproducibility of the results. read less USED (low confidence) K. Fan, L. Yang, S. Peng, X. Long, Z. Wu, and X. Zu, “Analytical Interatomic Potential for HCP-Scandium,” Materials Science Forum. 2011. link Times cited: 0 Abstract: A reactive interatomic potential based on an analytical bond… read moreAbstract: A reactive interatomic potential based on an analytical bond-order scheme is developed for hexagonal close-packed (hcp) scandium, and the model is fitted to the lattice parameters, elastic constants, cohesive energy and vacancy formation energy of scandium. The potential was used to calculate the structural energy differences of bcc-hcp, fcc-hcp, sc-hcp and diamond-hcp, as well as self-interstitial atom (SIA) formation energy, vacancy migration energy, divacancy binding energy, surface energy and stacking fault energy. The developed potential is shown to be able to reproduce energetics and structural properties of hcp-scandium. read less USED (low confidence) B. Zheng and J. Lowther, “Numerical investigations into mechanical properties of hexagonal silicon carbon nanowires and nanotubes.,” Nanoscale. 2010. link Times cited: 9 Abstract: Single-crystalline hexagonal faceted silicon carbon nanowire… read moreAbstract: Single-crystalline hexagonal faceted silicon carbon nanowires and nanotubes possess simultaneous high strength and failure strain. As long as SiC nanowires or nanotubes are large or thick enough to sustain a single atomic configuration under loading, their mechanical properties are size independent. Surface atoms are firstly forced to move by stretching and then destroy the equilibrium of subsurface atoms. Then, the force in carbon-silicon bonds along the tensile directions becomes larger than that in other bonds and results in elongation by three-times of the former than that of the latter. However, the latter bonds connecting the surface to the subsurface are broken and the wires or tubes are ruptured. For thinner nanowires and nanotubes, the broken bonds don't propagate instantly, but initiate transformation from a wurtzite to a graphitic structure. This structure transformation can strengthen and plasticize SiC nanowires and nanotubes. read less USED (low confidence) E. Lampin, C. Priester, C. Krzeminski, and L. Magaud, “Graphene buffer layer on Si-terminated SiC studied with an empirical interatomic potential,” Journal of Applied Physics. 2010. link Times cited: 23 Abstract: The atomistic structure of the graphenebuffer layer on Si-te… read moreAbstract: The atomistic structure of the graphenebuffer layer on Si-terminated SiC is investigated using a modified version of the environment-dependent interatomic potential. The determination of the equilibrium state by the conjuguate gradients method suffers from a complex multiple-minima energy surface. The initial configuration is therefore modified to set the system in specific valleys of the energy surface. The solution of minimal energy forms a hexagonal pattern composed of stuck regions separated by unbonded rods that release the misfit with the SiC surface. The structure presents the experimental symmetries and a global agreement with an ab initio calculation. It is therefore expected that the interatomic potential could be used in classical molecular dynamics calculations to study the graphene growth. read less USED (low confidence) J. Pohl, M. Müller, A. Seidl, and K. Albe, “Formation of parallel (111) twin boundaries in silicon growth from the melt explained by molecular dynamics simulations,” Journal of Crystal Growth. 2010. link Times cited: 44 USED (low confidence) S. E. Wethekam, “Ladungsaustausch schneller Edelgasatome und Fullerene mit Festkörperoberflächen.” 2009. link Times cited: 3 Abstract: This work is devoted to the study of model systems for the i… read moreAbstract: This work is devoted to the study of model systems for the interaction of atoms, molecules, and their ions with solid surfaces. The thesis consists of three parts. In the first part, He atoms and ions with keV energies are scattered under grazing angles of incidence from Al(111), Al(100), and Al(110) surfaces. Fractions of surviving ions and normal energy gains of He+ ions prior to neutralization, derived from shifts of angular distributions for incident atoms and ions, are compared to results from three-dimensional Monte Carlo simulations based on theoretically calculated Auger neutralization rates and He ground-state energy shifts. From the good agreement of experimental data with simulations, a detailed microscopic understanding for a model system of ion-surface interactions is concluded. The studies are extended to noble gas atoms and surfaces with a more complex electronic structure as well as the Auger ionization process, for which a comparison to simulations based on first ab-initio calculations is presented. In the second set of experiments, the formation of doubly excited states of He atoms during collisions of He2+ ions with energies between 60 eV and 1 keV with Ni(110) and Fe(110) surfaces is studied via Auger electron spectroscopy. The electron spectra from autoionization of doubly excited states of 2`2`′ configurations show a pronounced dependence on the coverage of the target surface with adsorbates. Thermal desorption and dissolution of surface contaminations into the bulk at elevated temperatures provide an alternative interpretation of recent work where the local electron spin polarization of Ni(110) and Fe(110) surfaces was deduced from changes in the electron spectra as function of target temperature. In the third part, angular distributions, fragmentation, and charge fractions are studied for grazing scattering of C60 fullerene ions with keV energies from Al(100), Be(0001), and LiF(100) surfaces. At low energies for the motion along the surface normal, the fullerenes are scattered nearly elastically, whereas for larger normal energies, the energy loss is substantial with pronounced differences for metal and insulator surfaces. From a comparison with classical trajectory simulations, a strong perturbation of the elastic properties of the fullerene by a nearby metal surface is concluded. Shifts of angular distributions for incident C60 and C 2+ 60 projectiles for the metal surfaces are in quantitative accord with a classical over-the-barrier model and provide the first information on distances of electron transfer for positively charged fullerenes in front of metal surfaces. For the LiF(100) surface, pronounced kinematically induced internal excitations due to interactions with the periodic electric field at the surface are observed. read less USED (low confidence) F. Zirkelbach, J. Lindner, K. Nordlund, and B. Stritzker, “Molecular dynamics simulation of defect formation and precipitation in heavily carbon doped silicon,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 2009. link Times cited: 4 USED (low confidence) M. Posselt, F. Gao, and H. Bracht, “Correlation between self-diffusion in Si and the migration mechanisms of vacancies and self-interstitials: An atomistic study,” Physical Review B. 2008. link Times cited: 28 Abstract: The migration of point defects in silicon and the correspond… read moreAbstract: The migration of point defects in silicon and the corresponding atomic mobility are investigated by classical molecular dynamics simulations using the Stillinger-Weber potential and the Tersoff potential. In contrast to most of the previous studies both the point defect diffusivity and the self-diffusion coefficient per defect are calculated separately so that the diffusion-correlation factor can be determined. Simulations with both the Stillinger-Weber and the Tersoff potential show that vacancy migration is characterized by the transformation of the tetrahedral vacancy to the split vacancy and vice versa and the diffusion-correlation factor is about 0.5. This value was also derived by the statistical diffusion theory under the assumption of the same migration mechanism. The mechanisms of self-interstitial migration are more complex. The detailed study, including a visual analysis and investigations with the nudged elastic band method, reveals a variety of transformations between different self-interstitial configurations. Molecular dynamics simulations using the Stillinger-Weber potential show, that the self-interstitial migration is dominated by a dumbbell mechanism, whereas the interstitialcy mechanism prevails with the Tersoff potental. The corresponding values of the correlation factor are different, namely 0.59 and 0.69 for the dumbbell and the interstitialcy mechanism, respectively. The latter value is nearly equal to that obtainedmore » by the statistical theory which assumes the interstitialcy mechanism. Recent analysis of experimental results demonstrated, that in the framework of state-of-the-art diffusion and reaction models the best interpretation of point defect data can be given by assuming . The comparison with the present atomistic study leads to the conclusion that a dumbbell mechanism governs the self-interstitial migration in Si. Simulations using the Stillinger-Weber potential reveal two dominating migration paths which are characterized by transformation between the extended dumbbell and the dumbbell and vice versa. This process occurs either in a single {110} plane or includes a change into an equivalent {110} plane.« less read less USED (low confidence) M. Posselt, F. Gao, and W. J. Weber, “Atomistic simulations on the thermal stability of the antisite pair in 3C- and 4H-SiC,” Physical Review B. 2006. link Times cited: 16 Abstract: The thermal stability of the first-neighbor antisite pair co… read moreAbstract: The thermal stability of the first-neighbor antisite pair configurations in 3C- and 4H-SiC is investigated by a comprehensive atomistic study. At first the structure and energetics of these defects is determined in order to check the accuracy of the Gao-Weber interatomic potential used. The results are comparable with literature data obtained by the density-functional theory. Then, the lifetime of the antisite pair configurations is calculated for temperatures between 800 and 2500 K. Both in 3C- and 4H-SiC the thermal stability of the antisite pairs is rather low. In contrast to previous theoretical interpretations, the antisite pair can be therefore not correlated with the DI photoluminescence center that is stable to above 2000 K. The atomic mechanisms during the recombination of the antisite pair in 3C-SiC and of three antisite pair configurations in 4H-SiC is a modified concerted exchange. Due to the different sizes of the silicon and the carbon atoms, this process is not identical with the concerted exchange in Si. Two intermediate metastable configurations found during the recombination are similar to the bond defect in Si. Since the SiC lattice contains two types of atoms, there are also two different types of bond defects. The two bond defects canmore » be considered as the result of the incomplete recombination of a carbon vacancy and a neighboring mixed dumbbell interstitial. For selected temperatures the thermal stability of the antisite pair in 3C-SiC is investigated by molecular dynamics simulations that are based on the density-functional theory. Their results are very similar to those of the atomistic study, i.e. the Gao-Weber potential describes the antisite pair and its recombination reasonably well. The antisite pair in 4H-SiC with the two atoms on hexagonal sites has a slightly different formation energy than the other three antisite pair configurations in 4H-SiC. Its lifetime shows another dependence on the temperature, and its recombination is characterized by a separate motion of atoms.« less read less USED (low confidence) L. Feng, X. Zhang, W. Li, M. Liu, and X. Yao, “Multiple structural phase transitions in single crystal silicon subjected to dynamic loading,” Scripta Materialia. 2024. link Times cited: 0 USED (low confidence) B.-G. Jeong, Y. Shen, X. Wang, Q. An, and K. Reddy, “Evolution of shear amorphization in superhard cubic silicon carbide,” Scripta Materialia. 2023. link Times cited: 0 USED (low confidence) Y. Xu, G. Wang, and Y. Zhou, “Broadly manipulating the interfacial thermal energy transport across the Si/4H-SiC interfaces via nanopatterns,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 12 USED (low confidence) L. Xue et al., “Study of deformation mechanism of structural anisotropy in 4H–SiC film by nanoindentation,” Materials Science in Semiconductor Processing. 2022. link Times cited: 5 USED (low confidence) P. Zhao, B. Zhao, J. Pan, and J. Wu, “Superimpose mechanism of surface generation process in grinding of monocrystalline silicon using molecular dynamics simulation,” Materials Science in Semiconductor Processing. 2022. link Times cited: 10 USED (low confidence) Y. Zhou, Y. Huang, J. Li, and F. Zhu, “The effect of contact types on SiC polishing process,” Materials Science in Semiconductor Processing. 2022. link Times cited: 9 USED (low confidence) X. Huang, J. Guo, and Y. Yue, “Graphene coated 3C-SiC with improved irradiation resistance and enhanced heat conduction property after collision cascade,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 2 USED (low confidence) K. Liu, H. Wang, and X. Zhang, “Molecular Dynamics Simulation of Ductile Mode Cutting,” Springer Series in Advanced Manufacturing. 2019. link Times cited: 1 USED (low confidence) A. Bisht, A. Roy, U. S. Dixit, S. Suwas, and V. Silberschmidt, “Small-Scale Machining Simulations,” Lecture Notes on Multidisciplinary Industrial Engineering. 2019. link Times cited: 0 USED (low confidence) S. Pan, “Molecular Dynamics Simulation for Continuous Dry Friction on Fretting Interfaces,” Journal of Mechanical Engineering. 2018. link Times cited: 3 Abstract: : Aimed at exploring the continuous dry friction behavior at… read moreAbstract: : Aimed at exploring the continuous dry friction behavior at fretting interface, targeted at crystalline silicon-diamond coupled interface, the fretting interface model with single asperity for dry friction analysis is set up using LAMMPS (i.e., molecular dynamics simulation tool, MD). The micro-motion process is shown with clear images and symbolized with friction force response and the normal force. It is should be noted that during the whole process, including the intervals between direct friction contacts, there exists continuously turbulent force response. The results indicate that during the continuous single asperity contacts, influenced by the factors of stick-slip effects, deformation-recovery process and stress release of wore atoms, the friction contact intervals show friction response, possibly stronger than that of the direct friction contacts to the extent that the frictional characteristics of the coupled solid fretting interfaces can be affected. read less USED (low confidence) S. Winczewski, M. Y. Shaheen, and J. Rybicki, “Interatomic potential suitable for the modeling of penta-graphene: Molecular statics/molecular dynamics studies,” Carbon. 2018. link Times cited: 34 USED (low confidence) Y. Tang, B. Yang, H. Yang, P. Yang, J. Yang, and Y. Hu, “Numerical investigation on mechanical properties of graphene covering silicon nanofilms,” Computational Materials Science. 2017. link Times cited: 5 USED (low confidence) H. N. Pishkenari and P. G. Ghanbari, “Vibrational analysis of the fullerene family using Tersoff potential,” Current Applied Physics. 2017. link Times cited: 11 USED (low confidence) R. Jones, C. Weinberger, S. Coleman, and G. Tucker, “Introduction to Atomistic Simulation Methods.” 2016. link Times cited: 1 USED (low confidence) L. Sang, “Affect of the graphene layers on the melting temperature of silicon by molecular dynamics simulations,” Computational Materials Science. 2016. link Times cited: 8 USED (low confidence) S. Goel, X. Luo, A. Agrawal, and R. Reuben, “Diamond machining of silicon: A review of advances in molecular dynamics simulation,” International Journal of Machine Tools & Manufacture. 2015. link Times cited: 314 USED (low confidence) T. Sinno, “Atomistic Calculation of Defect Thermodynamics in Crystalline Silicon.” 2015. link Times cited: 0 USED (low confidence) J. Yeo, “Modeling and simulation of the structural evolution and thermal properties of ultralight aerogel and 2D materials.” 2014. link Times cited: 1 USED (low confidence) J. Cholewiński, M. Maździarz, G. Jurczak, and P. Dłużewski, “DISLOCATION CORE RECONSTRUCTION BASED ON FINITE DEFORMATION APPROACH AND ITS APPLICATION TO 4H-SiC CRYSTAL,” International Journal for Multiscale Computational Engineering. 2014. link Times cited: 4 Abstract: A proper reconstruction of discrete crystal structure with d… read moreAbstract: A proper reconstruction of discrete crystal structure with defects is an important problem in dislocation theory. Currently, procedures for dislocation core reconstruction presented in the literature usually neglect configuration changes. The present paper discusses a new approach, which uses an iterative algorithm to determine an atomistic configuration of the dislocation core. The mathematical background is based on finite deformation theory, in which an iterative algorithm searches for the new atomic configuration corresponding to the actual atomic configuration of the deformed crystal. Its application to the reconstruction of 4H-SiC crystal affected by the system of four threading dislocations is presented as an example. Molecular statics calculations suggest a lower potential energy, as well as dislocation core energy, per-atom energy, and per-atom stresses for the structure reconstructed by use of the iterative algorithm against the classical solution based on the Love’s equations. read less USED (low confidence) G. Ferguson and L. Curtiss, “Atomic-level modeling of organic electrolytes in lithium-ion batteries,” ChemInform. 2013. link Times cited: 5 USED (low confidence) S. Goel, X. Luo, and R. Reuben, “Molecular dynamics simulation model for the quantitative assessment of tool wear during single point diamond turning of cubic silicon carbide,” Computational Materials Science. 2012. link Times cited: 129 USED (low confidence) M. Hu, X. Zhang, K. Giapis, and D. Poulikakos, “Atomistic Mechanisms of Enhancing Energy Conversion Efficiency of Nanostructured Thermoelectrics.” 2011. link Times cited: 0 USED (low confidence) X.-C. Li, X. Shu, Y. Liu, F. Gao, and G. Lu, “Modified analytical interatomic potential for a W–H system with defects,” Journal of Nuclear Materials. 2011. link Times cited: 100 NOT USED (low confidence) Z. J. Choong, D. Huo, N. Ponon, R. Savidis, P. Degenaar, and A. O’Neill, “A novel hybrid technique to fabricate silicon-based micro-implants with near defect-free quality for neuroprosthetics application.,” Materials science & engineering. C, Materials for biological applications. 2020. link Times cited: 1 NOT USED (high confidence) J.-C. Griesser, L. Frérot, J. A. Oldenstaedt, M. Müser, and L. Pastewka, “Analytic elastic coefficients in molecular calculations: Finite strain, nonaffine displacements, and many-body interatomic potentials,” Physical Review Materials. 2023. link Times cited: 1 Abstract: Elastic constants are among the most fundamental and importa… read moreAbstract: Elastic constants are among the most fundamental and important properties of solid materials, which is why they are routinely characterized in both experiments and simulations. While conceptually simple, the treatment of elastic constants is complicated by two factors not yet having been concurrently discussed: finite-strain and non-affine, internal displacements. Here, we revisit the theory behind zero-temperature, finite-strain elastic constants and extend it to explicitly consider non-affine displacements. We further present analytical expressions for second-order derivatives of the potential energy for two-body and generic many-body interatomic potentials, such as cluster and empirical bond-order potentials. Specifically, we revisit the elastic constants of silicon, silicon carbide and silicon dioxide under hydrostatic compression and dilatation. Based on existing and new results, we outline the effect of multiaxial stress states as opposed to volumetric deformation on the limits of stability of their crystalline lattices. read less NOT USED (high confidence) F. Fang, M. Lai, J. Wang, X. Luo, J. Yan, and Y. Yan, “Nanometric cutting: Mechanisms, practices and future perspectives,” International Journal of Machine Tools and Manufacture. 2022. link Times cited: 37 NOT USED (high confidence) Y. Xie, J. Vandermause, S. Ramakers, N. Protik, A. Johansson, and B. Kozinsky, “Uncertainty-aware molecular dynamics from Bayesian active learning for phase transformations and thermal transport in SiC,” npj Computational Materials. 2022. link Times cited: 14 NOT USED (high confidence) J. M. Ortiz-Roldán, F. Montero-Chacón, E. Garcia-Perez, S. Calero, A. R. Ruiz-Salvador, and S. Hamad, “Thermostructural Characterization of Silicon Carbide Nanocomposite Materials via Molecular Dynamics Simulations,” Advanced Composite Materials. 2021. link Times cited: 1 Abstract: In this paper, we investigate the thermostructural propertie… read moreAbstract: In this paper, we investigate the thermostructural properties of a type of silicon-based nanomaterials, which we refer to as SiC@Si nanocomposites, formed by SiC crystalline nanoparticles (with the cubic phase), embedded within an amorphous Si matrix. We have followed an in silico approach to characterize the mechanical and thermal behaviour of these materials, by calculating the elastic constants, uniaxial stress-strain curves, coefficients of thermal expansion, and specific heats, at different temperatures, using interatomic potential calculations. The results obtained from our simulations suggest that this type of material presents enhanced thermal resistance features, making it suitable to be used in devices subjected to big temperature changes, such as heat sinks in micro and nanoelectronics, solar energy harvesters at high temperatures, power electronics, or in other applications in which good thermomechanical properties are required. read less NOT USED (high confidence) C. Liu et al., “Effect of tool rake angle on the material removal mechanism transition of single-crystal silicon: a molecular dynamics study,” The International Journal of Advanced Manufacturing Technology. 2021. link Times cited: 8 NOT USED (high confidence) C. Liu et al., “Molecular Dynamics Simulation on Cutting Mechanism in the Hybrid Machining Process of Single-Crystal Silicon,” Nanoscale Research Letters. 2021. link Times cited: 14 NOT USED (high confidence) A. Kubo and Y. Umeno, “Machine-Learning-Based Atomistic Model Analysis on High-Temperature Compressive Creep Properties of Amorphous Silicon Carbide,” Materials. 2021. link Times cited: 5 Abstract: Ceramic matrix composites (CMCs) based on silicon carbide (S… read moreAbstract: Ceramic matrix composites (CMCs) based on silicon carbide (SiC) are used for high-temperature applications such as the hot section in turbines. For such applications, the mechanical properties at a high temperature are essential for lifetime prediction and reliability design of SiC-based CMC components. We developed an interatomic potential function based on the artificial neural network (ANN) model for silicon-carbon systems aiming at investigation of high-temperature mechanical properties of SiC materials. We confirmed that the developed ANN potential function reproduces typical material properties of the single crystals of SiC, Si, and C consistent with first-principles calculations. We also validated applicability of the developed ANN potential to a simulation of an amorphous SiC through the analysis of the radial distribution function. The developed ANN potential was applied to a series of creep test for an amorphous SiC model, focusing on the amorphous phase, which is expected to be formed in the SiC-based composites. As a result, we observed two types of creep behavior due to different atomistic mechanisms depending on the strain rate. The evaluated activation energies are lower than the experimental values in literature. This result indicates that an amorphous region can play an important role in the creep process in SiC composites. read less NOT USED (high confidence) V. Khaki, M. Ahmadi, M. Hassanzadazar, and T. Nguyen, “Silicon Doping Effect on the Electronic Behavior of Graphene Nanoscrolls,” Journal of Electronic Materials. 2021. link Times cited: 2 NOT USED (high confidence) P. T. Jochym and J. Łażewski, “High Efficiency Configuration Space Sampling – probing the distribution of available states.” 2021. link Times cited: 5 Abstract: 1 Substantial acceleration of research and more efficient ut… read moreAbstract: 1 Substantial acceleration of research and more efficient utilization of resources can be achieved in modeling investigated phenomena by identifying the limits of system’s accessible states instead of tracing the trajectory of its evolution. The proposed strategy uses the Metropolis-Hastings Monte-Carlo sampling of the configuration space probability distribution coupled with physically-motivated prior probability distribution. We demonstrate this general idea by presenting a high performance method of generating configurations for lattice dynamics and other computational solid state physics calculations corresponding to nonzero temperatures. In contrast to the methods based on molecular dynamics, where only a small fraction of obtained data is consumed, the proposed scheme is distinguished by a considerably higher, reaching even 80%, acceptance ratio. read less NOT USED (high confidence) S. Ahammed, M. S. Islam, I. Mia, and J. Park, “Lateral and flexural thermal transport in stanene/2D-SiC van der Waals heterostructure,” Nanotechnology. 2020. link Times cited: 20 Abstract: Thermal management is one of the key challenges in nanoelect… read moreAbstract: Thermal management is one of the key challenges in nanoelectronic and optoelectronic devices. The development of a van der Waals heterostructure (vdWH) using the vertical positioning of different two-dimensional (2D) materials has recently appeared as a promising way of attaining desirable electrical, optical, and thermal properties. Here, we explore the lateral and flexural thermal conductivity of stanene/2D-SiC vdWH utilizing the reverse non-equilibrium molecular dynamics simulation and transient pump-probe technique. The effects of length, area, coupling strength and temperature on the thermal transport are studied systematically. The projected lateral thermal conductivity of a stanene/2D-SiC hetero-bilayer is found to be 66.67 W m−1K−1 , which is greater than stanene, silicene, germanene, MoSe2 and even higher than some hetero-bilayers, including MoS2/MoSe2 and stanene/silicene. The lateral thermal conductivity increases as the length increases, while it tends to decrease with increasing temperature. The computed flexural interfacial thermal resistance between stanene and 2D-SiC is 3.0622 × 10−7 K.m2 W−1, which is close to other 2D hetero-bilayers. The interfacial resistance between stanene and 2D-SiC is reduced by 70.49% and 50.118% as the temperature increases from 100 K to 600 K and the coupling factor increases from χ=0.5 to χ=5 , respectively. In addition, various phonon modes are evaluated to disclose the fundamental mechanisms of thermal transport in the lateral and flexural direction of the hetero-bilayer. These results are important in order to understand the heat transport phenomena of stanene/2D-SiC vdWH, which could be useful for enhancing their promising applications. read less NOT USED (high confidence) B. Sun, W. Ouyang, J. Gu, C. Wang, J. Wang, and L. Mi, “Formation of Moiré superstructure of epitaxial graphene on Pt(111): A molecular dynamic simulation investigation,” Materials Chemistry and Physics. 2020. link Times cited: 5 NOT USED (high confidence) M. Alam, L. Lymperakis, and J. Neugebauer, “Phase diagram of grain boundary facet and line junctions in silicon,” Physical Review Materials. 2020. link Times cited: 1 Abstract: The presence of facets and line junctions connecting facets … read moreAbstract: The presence of facets and line junctions connecting facets on grain boundaries (GBs) has a strong impact on the properties of structural, functional, and optoelectronic materials: They govern the mobility of interfaces, the segregation of impurities, as well the electronic properties. In the present paper, we employ density-functional theory and modified embedded atom method calculations to systematically investigate the energetics and thermodynamic stability of these defects. As a prototype system, we consider (cid:2) 3 tilt GBs in Si. By analyzing the energetics of different faceted GBs, we derive a diagram that describes and predicts the reconstruction of these extended defects as a function of facet length and boundary inclination angle. The phase diagram sheds light upon the fundamental mechanisms causing GB faceting phenomena. It demonstrates that the properties of faceting are not determined solely by anisotropic GB energies but by a complex interplay between geometry and microstructure, boundary energies as well as long-range strain interactions. read less NOT USED (high confidence) M. A. Caro, G. Csányi, T. Laurila, and V. L. Deringer, “Machine learning driven simulated deposition of carbon films: From low-density to diamondlike amorphous carbon,” Physical Review B. 2020. link Times cited: 29 Abstract: © 2020 American Physical Society. Amorphous carbon (a-C) mat… read moreAbstract: © 2020 American Physical Society. Amorphous carbon (a-C) materials have diverse interesting and useful properties, but the understanding of their atomic-scale structures is still incomplete. Here, we report on extensive atomistic simulations of the deposition and growth of a-C films, describing interatomic interactions using a machine learning (ML) based Gaussian approximation potential model. We expand widely on our initial work [M. A. Caro, Phys. Rev. Lett. 120, 166101 (2018)PRLTAO0031-900710.1103/PhysRevLett.120.166101] by now considering a broad range of incident ion energies, thus modeling samples that span the entire range from low-density (sp2-rich) to high-density (sp3-rich, "diamondlike") amorphous forms of carbon. Two different mechanisms are observed in these simulations, depending on the impact energy: low-energy impacts induce sp- and sp2-dominated growth directly around the impact site, whereas high-energy impacts induce peening. Furthermore, we propose and apply a scheme for computing the anisotropic elastic properties of the a-C films. Our work provides fundamental insight into this intriguing class of disordered solids, as well as a conceptual and methodological blueprint for simulating the atomic-scale deposition of other materials with ML driven molecular dynamics. read less NOT USED (high confidence) A. Islam, M. S. Islam, N. Ferdous, J. Park, and A. Hashimoto, “Vacancy-induced thermal transport in two-dimensional silicon carbide: a reverse non-equilibrium molecular dynamics study.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 23 Abstract: Because of its impressive electrical, thermal, and mechanica… read moreAbstract: Because of its impressive electrical, thermal, and mechanical properties, two-dimensional silicon carbide (2D-SiC) has recently gained tremendous attention in the field of nanoelectronics and optoelectronics. Here, we investigated the effects of various types of defects such as bi-, point-, and mixed-vacancies on the thermal conductivity of 2D-SiC using reverse non-equilibrium molecular dynamics simulation. The effects of temperature variation on the thermal conductivity of vacancy-defected 2D-SiC were also studied. A significant reduction of the thermal conductivity was observed when the concentrations of the vacancies were increased. The point vacancy resulted in the thermal conductivity decreasing more quickly as compared to bi vacancy and mixed vacancy defects. Moreover, increasing the temperature of vacancy-defected 2D-SiC further reduced the thermal conductivity due to a strong phonon-vacancy scattering effect. Because of the introduction of vacancy defects in the acoustic phonon density of states (PDOS), a softening behavior in the intensity of the characteristic peaks is perceived, and with increasing temperature, a frequency shrinking is noted in the PDOS curves, both of which contribute to the reduction of the thermal conductivity. Additionally, rapid softening of the phonon transmission spectrum and increase in entropy were obtained for the point vacancy-defected structure, which clearly confirms our findings at different vacancy concentrations as well as for types of vacancies. These findings are very much imperative for realizing heat dissipation in nano- and optoelectronic devices based on 2D-SiC as well as for demonstrating an effective method for modulating 2D-SiC thermal conductivity through defect engineering. read less NOT USED (high confidence) C. Liu, J. Zhang, J. Zhang, X. Chen, J. Xiao, and J. Xu, “A simulation investigation on elliptical vibration cutting of single-crystal silicon,” The International Journal of Advanced Manufacturing Technology. 2020. link Times cited: 0 NOT USED (high confidence) C. Liu, J. Zhang, J. Zhang, X. Chen, J. Xiao, and J. Xu, “A simulation investigation on elliptical vibration cutting of single-crystal silicon,” The International Journal of Advanced Manufacturing Technology. 2020. link Times cited: 8 NOT USED (high confidence) Y. Fan, Y. Xiang, and H. S. Shen, “Temperature-Dependent Mechanical Properties of Graphene/Cu Nanocomposites with In-Plane Negative Poisson’s Ratios,” Research. 2020. link Times cited: 35 Abstract: Negative Poisson's ratio (NPR), also known as “auxetic”… read moreAbstract: Negative Poisson's ratio (NPR), also known as “auxetic”, is a highly desired property in a wide range of future industry applications. By employing molecular dynamics (MD) simulation, metal matrix nanocomposites reinforced by graphene sheets are studied in this paper. In the simulation, single crystal copper with crystal orientation [1 1 0] is selected as the matrix and an embedded-atom method (EAM) potential is used to describe the interaction of copper atoms. An aligned graphene sheet is selected as reinforcement, and a hybrid potential, namely, the Erhart-Albe potential, is used for the interaction between a pair of carbon atoms. The interaction between the carbon atom and copper atom is approximated by the Lennard-Jones (L-J) potential. The simulation results showed that both graphene and copper matrix possess in-plane NPRs. The temperature-dependent mechanical properties of graphene/copper nanocomposites with in-plane NPRs are obtained for the first time. read less NOT USED (high confidence) C. Liu et al., “Influence of micro grooves of diamond tool on silicon cutting: a molecular dynamic study,” Molecular Simulation. 2020. link Times cited: 2 Abstract: ABSTRACT During single-point diamond turning of hard and bri… read moreAbstract: ABSTRACT During single-point diamond turning of hard and brittle materials, tool wear is a dominant factor that influences machinability. In the wear process, micro grooves on the flank face is an important character of tool wear, which leads to the formation of subcutting edges and the ductile to brittle transition. In this paper, classical molecular dynamic simulations of nanometric cutting of silicon by a diamond tool with V-shape grooves were carried out to explore the effect of groove geometry on the workpiece and tool integrity. The evolution of tool wear and machined surface integrity was discussed. Simulation result shows that grooves have a significant influence on the stress and temperature distributions of the tool, which has a great influence on tool deterioration. Grooves with sharp edges will lead to severe tool wear and bring deep subsurface damage of the machined surface. However, the subsurface damage of the machined surface can be restrained with blunt grooves since the pressure is reduced. With a comprehensive understanding and controlling of groove, tool wear can be suppressed and high-quality surface can be achieved. read less NOT USED (high confidence) C. Liu et al., “Molecular dynamic simulation of tool groove wear in nanoscale cutting of silicon,” AIP Advances. 2020. link Times cited: 5 Abstract: Tool wear is one of the bottlenecks that decrease the machin… read moreAbstract: Tool wear is one of the bottlenecks that decrease the machinability of hard and brittle materials in single point diamond turning (SPDT). Specifically, a microgroove generated on the cutting edge is an important character of tool wear, which leads to the formation of subcutting edges and facilitates the ductile to brittle transition in machining. However, the mechanism of the groove wear influence on the machined workpiece, especially the subsurface damage, is not clear just by the experimental investigations. In this paper, molecular dynamic simulations were carried out to explore the influence of groove wear on workpiece subsurface damage in SPDT of single crystal silicon. The propagation of grooves was also investigated by discussion of the stress and temperature distribution on the cutting edge. The Weierstrass-Mandelbrot function was adopted to set up groove wear on the tool flank face. It is concluded that grooves improve the atomic flowing ability and the plastic deformation in the workpiece. Moreover, the grooves can also cause polycrystal transition in the workpiece subsurface. The thickness of the subsurface damaged region is increased when groove wear becomes severe. This study contributes to the understanding of the details involved in the interaction between tool groove wear and workpiece, which is advantageous to improve the machined surface quality.Tool wear is one of the bottlenecks that decrease the machinability of hard and brittle materials in single point diamond turning (SPDT). Specifically, a microgroove generated on the cutting edge is an important character of tool wear, which leads to the formation of subcutting edges and facilitates the ductile to brittle transition in machining. However, the mechanism of the groove wear influence on the machined workpiece, especially the subsurface damage, is not clear just by the experimental investigations. In this paper, molecular dynamic simulations were carried out to explore the influence of groove wear on workpiece subsurface damage in SPDT of single crystal silicon. The propagation of grooves was also investigated by discussion of the stress and temperature distribution on the cutting edge. The Weierstrass-Mandelbrot function was adopted to set up groove wear on the tool flank face. It is concluded that grooves improve the atomic flowing ability and the plastic deformation in the workpiece. Moreo... read less NOT USED (high confidence) X.-K. Chen and K. Chen, “Thermal transport of carbon nanomaterials,” Journal of Physics: Condensed Matter. 2019. link Times cited: 84 Abstract: The diversity of thermal transport properties in carbon nano… read moreAbstract: The diversity of thermal transport properties in carbon nanomaterials enables them to be used in different thermal fields such as heat dissipation, thermal management, and thermoelectric conversion. In the past two decades, much effort has been devoted to study the thermal conductivities of different carbon nanomaterials. In this review, different theoretical methods and experimental techniques for investigating thermal transport in nanosystems are first summarized. Then, the thermal transport properties of various pure carbon nanomaterials including 1D carbon nanotubes, 2D graphene, 3D carbon foam, are reviewed in details and the associated underlying physical mechanisms are presented. Meanwhile, we discuss several important influences on the thermal conductivities of carbon nanomaterials, including size, structural defects, chemisorption and strain. Moreover, we introduce different nanostructuring pathways to manipulate the thermal conductivities of carbon-based nanocomposites and focus on the wave nature of phonons for controlling thermal transport. At last, we briefly review the potential applications of carbon nanomaterials in the fields of thermal devices and thermoelectric conversion. read less NOT USED (high confidence) N. Chen, Q. Peng, Z. Jiao, I. van Rooyen, W. Skerjanc, and F. Gao, “Analytical bond-order potential for silver, palladium, ruthenium and iodine bulk diffusion in silicon carbide,” Journal of Physics: Condensed Matter. 2019. link Times cited: 6 Abstract: The analytical bond-order potential has been developed for s… read moreAbstract: The analytical bond-order potential has been developed for simulating fission product (Ag, Pd, Ru, and I) behavior in SiC, especially for their diffusion. We have proposed adding experimentally available elastic constants and physical properties of the elements as well as important defect formation energies calculated from density functional theory simulation to the list of typical properties as the extensive fitting database. The results from molecular dynamics simulations are in a reasonable agreement with defect properties and energy barriers of their experimental/computational counterparts. The successful validation of the new potential has established a good reliability and transferability of the potentials, which enables the ability of simulation in extended scale. The kinetic behavior such as diffusion of different interstitials is then realized by applying the new interatomic potentials. The bulk diffusion is less likely to dominate the transport of the four fission products under pure thermal condition, when we refer to their extremely small values of the effective diffusion coefficients. The interstitial mechanism is hard for Pd, Ru, and I to access due to the high formation energy and high migration energy. However, it is found that the migration energy of silver is relatively low, which indicates Ag diffusion via an interstitial mechanism being feasible, especially under irradiation condition, where massive interstitials can be formed in high-temperature nuclear reactors. read less NOT USED (high confidence) A. Sarikov, A. Marzegalli, L. Barbisan, E. Scalise, F. Montalenti, and L. Miglio, “Molecular dynamics simulations of extended defects and their evolution in 3C–SiC by different potentials,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 11 Abstract: An important issue in the technology of cubic SiC (3C–SiC) m… read moreAbstract: An important issue in the technology of cubic SiC (3C–SiC) material for electronic device applications is to understand the behavior of extended defects such as partial dislocation complexes and stacking faults (SFs). Atomistic simulations using molecular dynamics (MD) are an efficient tool to tackle this issue for large systems at comparatively low computation cost. At this, proper choice of MD potential is imperative to ensure the reliability of the simulation predictions. In this work, we compare the evolution of extended defects in 3C–SiC obtained by MD simulations with Tersoff, analytical bond order, and Vashishta potentials. Key aspects of this evolution are considered including the dissociation of 60° perfect dislocations in pairs of 30° and 90° partials as well as the dependence of the partial dislocation velocity on the Burgers vector and the atomic composition of core. Tersoff potential has been found to be less appropriate in describing the dislocation behavior in 3C–SiC as compared to two other potentials, which in their turn provide qualitatively equivalent predictions. The Vashishta potential predicts much faster defect dynamics than the analytical bond order potential (ABOP). It can be applied therefore to describe the large-scale evolution of the dislocation systems and SFs. On the other hand, ABOP is more precise in predicting local atom arrangements and reconstructions of the dislocation core structures. In this respect, synergetic use of ABOP and Vashishta potential is suggested for the MD simulation study of the properties and evolution of extended defects in the 3C–SiC. read less NOT USED (high confidence) Z. Fan, Y. Wang, X. Gu, P. Qian, Y. Su, and T. Ala‐Nissila, “A minimal Tersoff potential for diamond silicon with improved descriptions of elastic and phonon transport properties,” Journal of Physics: Condensed Matter. 2019. link Times cited: 10 Abstract: Silicon is an important material and many empirical interato… read moreAbstract: Silicon is an important material and many empirical interatomic potentials have been developed for atomistic simulations of it. Among them, the Tersoff potential and its variants are the most popular ones. However, all the existing Tersoff-like potentials fail to reproduce the experimentally measured thermal conductivity of diamond silicon. Here we propose a modified Tersoff potential and develop an efficient open source code called GPUGA (graphics processing units genetic algorithm) based on the genetic algorithm and use it to fit the potential parameters against energy, virial and force data from quantum density functional theory calculations. This potential, which is implemented in the efficient open source GPUMD (graphics processing units molecular dynamics) code, gives significantly improved descriptions of the thermal conductivity and phonon dispersion of diamond silicon as compared to previous Tersoff potentials and at the same time well reproduces the elastic constants. Furthermore, we find that quantum effects on the thermal conductivity of diamond silicon at room temperature are non-negligible but small: using classical statistics underestimates the thermal conductivity by about 10% as compared to using quantum statistics. read less NOT USED (high confidence) A. Sarikov, A. Marzegalli, L. Barbisan, F. Montalenti, and L. Miglio, “Structure and Stability of Partial Dislocation Complexes in 3C-SiC by Molecular Dynamics Simulations,” Materials. 2019. link Times cited: 6 Abstract: In this work, the structure and stability of partial disloca… read moreAbstract: In this work, the structure and stability of partial dislocation (PD) complexes terminating double and triple stacking faults in 3C-SiC are studied by molecular dynamics simulations. The stability of PD complexes is demonstrated to depend primarily on the mutual orientations of the Burgers vectors of constituent partial dislocations. The existence of stable complexes consisting of two and three partial dislocations is established. In particular, two types of stable double (or extrinsic) dislocation complexes are revealed formed by two 30° partial dislocations with different orientations of Burgers vectors, or 30° and 90° partial dislocations. Stable triple PD complexes consist of two 30° partial dislocations with different orientations of their Burgers vectors and one 90° partial dislocation, and have a total Burgers vector that is equal to zero. Results of the simulations agree with experimental observations of the stable PD complexes forming incoherent boundaries of twin regions and polytype inclusions in 3C-SiC films. read less NOT USED (high confidence) A. Islam et al., “Anomalous temperature dependent thermal conductivity of two-dimensional silicon carbide,” Nanotechnology. 2019. link Times cited: 46 Abstract: Recently, two-dimensional silicon carbide (2D-SiC) has attra… read moreAbstract: Recently, two-dimensional silicon carbide (2D-SiC) has attracted considerable interest due to its exotic electronic and optical properties. Here, we explore the thermal properties of 2D-SiC using reverse non-equilibrium molecular dynamics simulation. At room temperature, a thermal conductivity of ∼313 W mK−1 is obtained for 2D-SiC which is one order higher than that of silicene. Above room temperature, the thermal conductivity deviates the normal 1/T law and shows an anomalous slowly decreasing behavior. To elucidate the variation of thermal conductivity, the phonon modes at different length and temperature are quantified using Fourier transform of the velocity auto-correlation of atoms. The calculated phonon density of states at high temperature shows a shrinking and softening of the peaks, which induces the anomaly in the thermal conductivity. On the other hand, quantum corrections are applied to avoid the freezing effects of phonon modes on the thermal conductivity at low temperature. In addition, the effect of potential on the thermal conductivity calculation is also studied by employing original and optimized Tersoff potentials. These findings provide a means for better understating as well as designing the efficient thermal management of 2D-SiC based electronics and optoelectronics in near future. read less NOT USED (high confidence) R. Li, K. Gordiz, A. Henry, P. Hopkins, E. Lee, and T. Luo, “Effect of light atoms on thermal transport across solid-solid interfaces.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 12 Abstract: Thermal transport across solid interfaces is of great import… read moreAbstract: Thermal transport across solid interfaces is of great importance for applications like power electronics. In this work, we perform non-equilibrium molecular dynamics simulations to study the effect of light atoms on the thermal transport across SiC/GaN interfaces, where light atoms refer to substitutional or interstitial defect atoms lighter than those in the pristine lattice. Various light atom doping features, such as the light atom concentration, mass of the light atom, and skin depth of the doped region, have been investigated. It is found that substituting Ga atoms in the GaN lattice with lighter atoms (e.g. boron atoms) with 50% concentration near the interface can increase the thermal boundary conductance (TBC) by up to 50%. If light atoms are introduced interstitially, a similar increase in TBC is observed. Spectral analysis of interfacial heat transfer reveals that the enhanced TBC can be attributed to the stronger coupling of mid- and high-frequency phonons after introducing light atoms. We have also further included quantum correction, which reduces the amount of enhancement, but it still exists. These results may provide a route to improve TBC across solid interfaces as light atoms can be introduced during material growth. read less NOT USED (high confidence) R. Jana, D. Savio, V. L. Deringer, and L. Pastewka, “Structural and elastic properties of amorphous carbon from simulated quenching at low rates,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 22 Abstract: We generate representative structural models of amorphous ca… read moreAbstract: We generate representative structural models of amorphous carbon (a-C) from constant-volume quenching from the liquid with subsequent relaxation of internal stresses in molecular dynamics simulations using empirical and machine-learning interatomic potentials. By varying volume and quench rate we generate structures with a range of density and amorphous morphologies. We find that all a-C samples show a universal relationship between hybridization, bulk modulus and density despite having distinctly different cohesive energies. Differences in cohesive energy are traced back to slight changes in the distribution of bond-angles that is likely linked to thermal stability of these structures. read less NOT USED (high confidence) D. Datta, R. Balasubramaniam, P. Ranjan, A. Sharma, and T. K. Roy, “Investigation of tool-workpiece interaction in nanoscale cutting: a molecular dynamics study,” International Journal of Precision Technology. 2019. link Times cited: 1 Abstract: Ductile and brittle materials differ in their physical and m… read moreAbstract: Ductile and brittle materials differ in their physical and mechanical properties and pose distinct interaction with the cutting tool while nano-machining. It is thus imperative to analyse the mechanism of material removal and tool-workpiece interaction. Towards this, molecular dynamics simulation (MDS) is carried out to study the diamond tool and workpiece interaction in the nanoscale cutting of Cu (ductile material) and Si (brittle material). Results show that material removal in Cu takes place through shear deformation by dislocations formation and their propagation while in case of Si, it takes place through phase transformation of the material in cutting zone. Force analysis of both the materials shows that machinability of Cu in nanoscale cutting is better compared to Si. Furthermore, tool wear while machining of Si with sharp edge tool is due to chipping whereas radial distribution function reveals that graphitisation of the round edge tool occurs during machining of Si. read less NOT USED (high confidence) H. Dai, H. Du, J. Chen, and G. Chen, “Influence of elliptical vibration on the behavior of silicon during nanocutting,” The International Journal of Advanced Manufacturing Technology. 2019. link Times cited: 25 NOT USED (high confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “A molecular dynamics simulation of wear mechanism of diamond tool in nanoscale cutting of copper beryllium,” The International Journal of Advanced Manufacturing Technology. 2019. link Times cited: 33 NOT USED (high confidence) K. Biswas, J. Bandyopadhyay, and D. De, “A computational study on the quantum transport properties of silicene–graphene nano-composites,” Microsystem Technologies. 2019. link Times cited: 4 NOT USED (high confidence) J. Luo, A. Alateeqi, L. Liu, and T. Sinno, “Carbon solubility in liquid silicon: A computational analysis across empirical potentials.,” The Journal of chemical physics. 2019. link Times cited: 4 Abstract: The nucleation and growth of SiC precipitates in liquid sili… read moreAbstract: The nucleation and growth of SiC precipitates in liquid silicon is important in the crystallization of silicon used for the photovoltaic industry. These processes depend strongly on the carbon concentration as well as the equilibrium solubility relative to the precipitate phase. Here, using a suite of statistical thermodynamic techniques, we calculate the solubility of carbon atoms in liquid silicon relative to the β-SiC phase. We employ several available empirical potentials to assess whether these potentials may reasonably be used to computationally analyze SiC precipitation. We find that some of the Tersoff-type potentials provide an excellent picture for carbon solubility in liquid silicon but, because of their severe silicon melting point overestimation, are limited to high temperatures where the carbon solubility is several percent, a value that is irrelevant for typical solidification conditions. Based on chemical potential calculations for pure silicon, we suggest that this well-known issue is confined to the description of the liquid phase and demonstrate that some recent potential models for silicon might address this weakness while preserving the excellent description of the carbon-silicon interaction found in the existing models. read less NOT USED (high confidence) A. Genoese, A. Genoese, and G. Salerno, “On the nanoscale behaviour of single-wall C, BN and SiC nanotubes,” Acta Mechanica. 2019. link Times cited: 21 NOT USED (high confidence) H. Dai, H. Du, J. Chen, and G. Chen, “Influence of elliptical vibration on the behavior of silicon during nanocutting,” The International Journal of Advanced Manufacturing Technology. 2019. link Times cited: 0 NOT USED (high confidence) D. Kilymis, C. Gérard, and L. Pizzagalli, “Ductile deformation of core-shell Si-SiC nanoparticles controlled by shell thickness,” Acta Materialia. 2019. link Times cited: 12 NOT USED (high confidence) Y. Lysogorskiy, T. Hammerschmidt, J. Janssen, J. Neugebauer, and R. Drautz, “Transferability of interatomic potentials for molybdenum and silicon,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 14 Abstract: Interatomic potentials are widely used in computational mate… read moreAbstract: Interatomic potentials are widely used in computational materials science, in particular for simulations that are too computationally expensive for density functional theory (DFT). Most interatomic potentials have a limited application range and often there is very limited information available regarding their performance for specific simulations. We carried out high-throughput calculations for molybdenum and silicon with DFT and a number of interatomic potentials. We compare the DFT reference calculations and experimental data to the predictions of the interatomic potentials. We focus on a large number of basic materials properties, including the cohesive energy, atomic volume, elastic coefficients, vibrational properties, thermodynamic properties, surface energies and vacancy formation energies, which enables a detailed discussion of the performance of the different potentials. We further analyze correlations between properties as obtained from DFT calculations and how interatomic potentials reproduce these correlations, and suggest a general measure for quantifying the accuracy and transferability of an interatomic potential. From our analysis we do not establish a clearcut ranking of the potentials as each potential has its strengths and weaknesses. It is therefore essential to assess the properties of a potential carefully before application of the potential in a specific simulation. The data presented here will be useful for selecting a potential for simulations of Mo or Si. read less NOT USED (high confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “A molecular dynamics simulation of wear mechanism of diamond tool in nanoscale cutting of copper beryllium,” The International Journal of Advanced Manufacturing Technology. 2019. link Times cited: 0 NOT USED (high confidence) A. Genoese, A. Genoese, and G. Salerno, “On the nanoscale behaviour of single-wall C, BN and SiC nanotubes,” Acta Mechanica. 2019. link Times cited: 0 NOT USED (high confidence) D. Prasad and N. Mitra, “An atomistic study of phase transition in cubic diamond Si single crystal subjected to static compression,” Computational Materials Science. 2019. link Times cited: 7 NOT USED (high confidence) A. H. Howlader, M. S. Islam, and A. Islam, “A Study on Phonon Transmission of (10,0) Silicon Nanotube with Atomic Vacancies,” 2018 21st International Conference of Computer and Information Technology (ICCIT). 2018. link Times cited: 2 Abstract: A systematic computer simulation has been carried out to fin… read moreAbstract: A systematic computer simulation has been carried out to find out the exclusive phonon properties of both pristine and vacancy defected (10,0) semiconductor zigzag silicon nanotube for the first time. It is found that phonons are scattered into other phonon states due to vacancy. Vacancy generates degenerate phonon branches. The simulated phonon density of states shows softening of high-frequency phonons. Quite significant reduction in the phonon transmission is observed over the whole frequency spectrum with the introduction of vacancy. Quasi ballistic phonon conduction is noticed instead of the presence of vacancy for low-frequency region. Again, high-frequency phonon localization is found in vacancy defected nanotube. The thermal conductivity decreases in a large amount with only 1 % vacancy. Moreover, entropy of the vacancy defected system is examined. read less NOT USED (high confidence) L. Pizzagalli, “Atomistic modeling of point defect contributions to swelling in Xe-implanted silicon carbide,” Journal of Nuclear Materials. 2018. link Times cited: 6 NOT USED (high confidence) D. Gobrecht, S. Bromley, J. Plane, L. Decin, and S. Cristallo, “On the onset of dust formation in AGB stars,” Proceedings of the International Astronomical Union. 2018. link Times cited: 0 Abstract: A promising candidate to initiate dust formation in oxygen-r… read moreAbstract: A promising candidate to initiate dust formation in oxygen-rich AGB stars is alumina (Al2O3) showing an emission feature around ∼13μm attributed to Al−O stretching and bending modes (Posch+99,Sloan+03). The counterpart to alumina in carbon-rich AGB atmospheres is the highly refractory silicon carbide (SiC) showing a characteristic feature around 11.3μm (Treffers74). Alumina and SiC grains are thought to represent the first condensates to emerge in AGB stellar atmospheres. We follow a bottom-up approach, starting with the smallest stoichiometric clusters (i.e. Al4O6, Si2C2), successively building up larger-sized clusters. We present new results of quantum-mechanical structure calculations of (Al2O3)n, n = 1−10 and (SiC)n clusters with n = 1−16, including potential energies, rotational constants, and structure-specific vibrational spectra. We demonstrate the energetic viability of homogeneous nucleation scenarios where monomers (Al2O3 and SiC) or dimers (Al4O6 and Si2C2) are successively added. We find significant differences between our quantum theory based results and nanoparticle properties derived from (classical) nucleation theory. read less NOT USED (high confidence) A. Alsayoud et al., “Atomistic insights into the effect of polymerization on the thermophysical properties of 2-D C60 molecular solids,” Carbon. 2018. link Times cited: 7 NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 0 NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 38 NOT USED (high confidence) S. Balagan, V. U. Nazarov, A. Shevlyagin, D. Goroshko, and N. Galkin, “Theoretical approach to embed nanocrystallites into a bulk crystalline matrix and the embedding influence on the electronic band structure and optical properties of the resulting heterostructures,” Journal of Physics: Condensed Matter. 2018. link Times cited: 0 Abstract: We develop an approach and present results of the combined m… read moreAbstract: We develop an approach and present results of the combined molecular dynamics and density functional theory calculations of the structural and optical properties of the nanometer-sized crystallites embedded in a bulk crystalline matrix. The method is designed and implemented for both compatible and incompatible lattices of the nanocrystallite (NC) and the host matrix, when determining the NC optimal orientation relative to the matrix constitutes a challenging problem. We suggest and substantiate an expression for the cost function of the search algorithm, which is the energy per supercell generalized for varying number of atoms in the latter. The epitaxial relationships at the Si/NC interfaces and the optical properties are obtained and found to be in a reasonable agreement with experimental data. Dielectric functions show significant sensitivity to the NC’s orientation relative to the matrix at energies below 0.5 eV. read less NOT USED (high confidence) F. Fang and F. Xu, “Recent Advances in Micro/Nano-cutting: Effect of Tool Edge and Material Properties,” Nanomanufacturing and Metrology. 2018. link Times cited: 97 NOT USED (high confidence) W. Li, X. Yao, and X. Zhang, “Planar impacts on nanocrystalline SiC: a comparison of different potentials,” Journal of Materials Science. 2018. link Times cited: 14 NOT USED (high confidence) K. Biswas, J. Bandyopadhyay, and D. De, “A computational study on the quantum transport properties of silicene–graphene nano-composites,” Microsystem Technologies. 2018. link Times cited: 2 NOT USED (high confidence) S. Goel and A. Stukowski, “Comment on ‘Incipient plasticity of diamond during nanoindentation’ by C. Xu, C. Liu and H. Wang, RSC Advances, 2017, 7, 36093,” RSC Advances. 2018. link Times cited: 4 Abstract: A recent molecular dynamics simulation study on nanoindentat… read moreAbstract: A recent molecular dynamics simulation study on nanoindentation of diamond carried out by Xu et al.1 has reported observation of the presence of a controversial hexagonal lonsdaleite phase of carbon in the indentation area. In this comment, we question the reported observation and attribute this anomaly to shortcomings of the long range bond order potential (LCBOP) employed in the nanoindentation study. read less NOT USED (high confidence) E. Lee and T. Luo, “Thermal transport across solid-solid interfaces enhanced by pre-interface isotope-phonon scattering,” Applied Physics Letters. 2018. link Times cited: 26 Abstract: Thermal transport across solid interfaces can play critical … read moreAbstract: Thermal transport across solid interfaces can play critical roles in the thermal management of electronics. In this letter, we use non-equilibrium molecular dynamics simulations to investigate the isotope effect on the thermal transport across SiC/GaN interfaces. It is found that engineered isotopes (e.g., 10% 15N or 71Ga) in the GaN layer can increase the interfacial thermal conductance compared to the isotopically pure case by as much as 23%. Different isotope doping features, such as the isotope concentration, skin depth of the isotope region, and its distance from the interface, are investigated, and all of them lead to increases in thermal conductance. Studies of spectral temperatures of phonon modes indicate that interfacial thermal transport due to low-frequency phonons (< 20 THz) is enhanced after isotopes are introduced. These results suggest that the enhanced thermal conductance is related to the isotope-phonon scattering, which facilitates the redistribution of phonon energy among different mod... read less NOT USED (high confidence) J. Luo, A. Alateeqi, L. Liu, and T. Sinno, “Atomistic simulations of carbon diffusion and segregation in liquid silicon,” Journal of Applied Physics. 2017. link Times cited: 9 Abstract: The diffusivity of carbon atoms in liquid silicon and their … read moreAbstract: The diffusivity of carbon atoms in liquid silicon and their equilibrium distribution between the silicon melt and crystal phases are key, but unfortunately not precisely known parameters for the global models of silicon solidification processes. In this study, we apply a suite of molecular simulation tools, driven by multiple empirical potential models, to compute diffusion and segregation coefficients of carbon at the silicon melting temperature. We generally find good consistency across the potential model predictions, although some exceptions are identified and discussed. We also find good agreement with the range of available experimental measurements of segregation coefficients. However, the carbon diffusion coefficients we compute are significantly lower than the values typically assumed in continuum models of impurity distribution. Overall, we show that currently available empirical potential models may be useful, at least semi-quantitatively, for studying carbon (and possibly other impurity) trans... read less NOT USED (high confidence) S. Bergmann, K. Albe, E. Flegel, D. Barragan-Yani, and B. Wagner, “Anisotropic solid–liquid interface kinetics in silicon: an atomistically informed phase-field model,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 10 Abstract: We present an atomistically informed parametrization of a ph… read moreAbstract: We present an atomistically informed parametrization of a phase-field model for describing the anisotropic mobility of liquid–solid interfaces in silicon. The model is derived from a consistent set of atomistic data and thus allows to directly link molecular dynamics and phase field simulations. Expressions for the free energy density, the interfacial energy and the temperature and orientation dependent interface mobility are systematically fitted to data from molecular dynamics simulations based on the Stillinger–Weber interatomic potential. The temperature-dependent interface velocity follows a Vogel–Fulcher type behavior and allows to properly account for the dynamics in the undercooled melt. read less NOT USED (high confidence) K. Biswas, “A thermally driven differential mutation approach for the structural optimization of large atomic systems.,” The Journal of chemical physics. 2017. link Times cited: 1 Abstract: A computational method is presented which is capable to obta… read moreAbstract: A computational method is presented which is capable to obtain low lying energy structures of topological amorphous systems. The method merges a differential mutation genetic algorithm with simulated annealing. This is done by incorporating a thermal selection criterion, which makes it possible to reliably obtain low lying minima with just a small population size and is suitable for multimodal structural optimization. The method is tested on the structural optimization of amorphous graphene from unbiased atomic starting configurations. With just a population size of six systems, energetically very low structures are obtained. While each of the structures represents a distinctly different arrangement of the atoms, their properties, such as energy, distribution of rings, radial distribution function, coordination number, and distribution of bond angles, are very similar. read less NOT USED (high confidence) F. Gayk, J. Ehrens, T. Heitmann, P. Vorndamme, A. Mrugalla, and J. Schnack, “Young’s moduli of carbon materials investigated by various classical molecular dynamics schemes,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 16 NOT USED (high confidence) D. Gobrecht, S. Cristallo, L. Piersanti, and S. Bromley, “Nucleation of Small Silicon Carbide Dust Clusters in AGB Stars,” The Astrophysical Journal. 2017. link Times cited: 23 Abstract: Silicon carbide (SiC) grains are a major dust component in c… read moreAbstract: Silicon carbide (SiC) grains are a major dust component in carbon-rich asymptotic giant branch stars. However, the formation pathways of these grains are not fully understood. We calculate ground states and energetically low-lying structures of (SiC)n, n = 1, 16 clusters by means of simulated annealing and Monte Carlo simulations of seed structures and subsequent quantum-mechanical calculations on the density functional level of theory. We derive the infrared (IR) spectra of these clusters and compare the IR signatures to observational and laboratory data. According to energetic considerations, we evaluate the viability of SiC cluster growth at several densities and temperatures, characterizing various locations and evolutionary states in circumstellar envelopes. We discover new, energetically low-lying structures for Si4C4, Si5C5, Si15C15, and Si16C16 and new ground states for Si10C10 and Si15C15. The clusters with carbon-segregated substructures tend to be more stable by 4–9 eV than their bulk-like isomers with alternating Si–C bonds. However, we find ground states with cage geometries resembling buckminsterfullerens (“bucky-like”) for Si12C12 and Si16C16 and low-lying stable cage structures for n ≥ 12. The latter findings thus indicate a regime of cluster sizes that differ from small clusters as well as from large-scale crystals. Thus—and owing to their stability and geometry—the latter clusters may mark a transition from a quantum-confined cluster regime to a crystalline, solid bulk-material. The calculated vibrational IR spectra of the ground-state SiC clusters show significant emission. They include the 10–13 μm wavelength range and the 11.3 μm feature inferred from laboratory measurements and observations, respectively, although the overall intensities are rather low. read less NOT USED (high confidence) A. Stukowski, E. Fransson, M. Mock, and P. Erhart, “Atomicrex—a general purpose tool for the construction of atomic interaction models,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 17 Abstract: We introduce atomicrex, an open-source code for constructing… read moreAbstract: We introduce atomicrex, an open-source code for constructing interatomic potentials as well as more general types of atomic-scale models. Such effective models are required to simulate extended materials structures comprising many thousands of atoms or more, because electronic structure methods become computationally too expensive at this scale. atomicrex covers a wide range of interatomic potential types and fulfills many needs in atomistic model development. As inputs, it supports experimental property values as well as ab initio energies and forces, to which models can be fitted using various optimization algorithms. The open architecture of atomicrex allows it to be used in custom model development scenarios beyond classical interatomic potentials while thanks to its Python interface it can be readily integrated e.g., with electronic structure calculations or machine learning algorithms. read less NOT USED (high confidence) I. Choi et al., “Laser-induced phase separation of silicon carbide,” Nature Communications. 2016. link Times cited: 71 NOT USED (high confidence) J. Chen, B. Wang, and Y. Hu, “Existence Criterion of Low-Dimensional Materials,” arXiv: Materials Science. 2016. link Times cited: 12 NOT USED (high confidence) A. Galashev, O. Rakhmanova, and Y. Zaikov, “Defect silicene and graphene as applied to the anode of lithium-ion batteries: Numerical experiment,” Physics of the Solid State. 2016. link Times cited: 15 NOT USED (high confidence) S. Chavoshi and X. Luo, “Atomic-scale characterization of occurring phenomena during hot nanometric cutting of single crystal 3C-SiC,” RSC Advances. 2016. link Times cited: 41 Abstract: Nanometric cutting of single crystal 3C–SiC on the three pri… read moreAbstract: Nanometric cutting of single crystal 3C–SiC on the three principal crystal orientations at various cutting temperatures spanning from 300 K to 3000 K was investigated by the use of molecular dynamics (MD) simulation. The dominance of the (111) cleavage was observed for all the tested temperatures. An observation of particular interest was the shift to the (110) cleavage at cutting temperatures higher than 2000 K. Another key finding was the increase of anisotropy in specific cutting energy from ∼30% at 300 K to ∼44% at 1400 K, followed by a drop to ∼36% and ∼24% at 1700 K and 2000 K, respectively. The obtained results also indicated that the specific cutting energies required for cutting surfaces of different orientations decrease by 33–43% at 2000 K compared to what are required at 300 K. Moreover, the position of the stagnation region was observed to vary with changes in temperature and crystallographic orientation. Further analysis revealed that the subsurface deformation was maximum on the (111) surface whereas it was minimum on the (110) plane. This is attributable to the occurrence of cleavage and the location of the stagnation region. In addition, the amount of subsurface damage scaled linearly with the increase of cutting temperature. A vortex flow of atoms beneath the cutting tool was also observed, which is qualitatively analogous to the plastic flow of silicon. The simulations also predicted that the atom-by-atom attrition wear and plastic deformation of the diamond cutting tool could be alleviated while cutting at high temperatures. Nevertheless, chemical wear i.e. dissolution–diffusion and adhesion wear is plausible to be accelerated at high temperatures. read less NOT USED (high confidence) D. Kaiser, S. Ghosh, S. Han, and T. Sinno, “Modeling and simulation of compositional engineering in SiGe films using patterned stress fields.” 2016. link Times cited: 2 Abstract: Semiconductor alloys such as silicon–germanium (SiGe) offer … read moreAbstract: Semiconductor alloys such as silicon–germanium (SiGe) offer attractive environments for engineering quantum-confined structures that are the basis for a host of current and future optoelectronic devices. Although vertical stacking of such structures is routinely achieved via heteroepitaxy, lateral manipulation has proven much more challenging. We have recently demonstrated that a patterned elastic stress field applied, with an array of nanoscale indenters, to an initially compositionally uniform SiGe substrate will drive atomic interdiffusion leading to compositional patterns in the near-surface region of the substrate. While this approach may offer a potentially efficient and robust pathway to producing laterally ordered arrays of quantum-confined structures, optimizing it with respect to the various process parameters, such as indenter array geometry, annealing history, and SiGe substrate thickness and composition, is highly challenging. Here, a mesoscopic model based on coarse-grained lattice kinetic Monte Carlo simulation is presented that describes quantitatively the atomic interdiffusion processes in SiGe alloy films subjected to applied stress. We first show that the model provides predictions that are quantitatively consistent with experimental measurements. Then, the model is used to investigate the impact of several process parameters such as indenter shape and pitch. We find that certain indenter configurations produce compositional patterns that are favorable for engineering lateral arrays of quantum-confined structures. read less NOT USED (high confidence) T. Hanashiro, K. Saitoh, T. Sato, K. Nishimura, M. Takuma, and Y. Takahashi, “Molecular Dynamics Study on Ductile Behavior of SiC during Nanoindentation,” Tribology Online. 2016. link Times cited: 6 Abstract: In order to clarify the plastic deformation mechanism of sil… read moreAbstract: In order to clarify the plastic deformation mechanism of silicon carbide in cubic phase (3C-SiC), molecular dynamics (MD) simulations are performed on the nanoindentation using a spherical indenter. Transition from elastic deformation to plastic deformation has been confirmed by the phenomenon called pop-in in the load-displacement curves during nanoindentation. Dislocations on {1 1 1} slip planes are found during indentation. In order to analyze internal defects, common neighbor analysis (CNA) is slightly modified so that it is suitable for the analysis of slips of zinc-blend structure. In our method, the CNA is applied separately to sub-lattice of Si or C in the same SiC. By this method, structural changes are confirmed in a region with the shape of square pyramid when the pop-in behavior occurs. By measuring the atomic distances along the region of misalignment, it was confirmed that there is certainly atomic sliding by crystalline slip. Furthermore, it is found that, with increase of loading, dislocation loops spread along {1 1 1} slip planes. read less NOT USED (high confidence) S. Zhao, E. Hahn, B. Kad, B. Remington, E. Bringa, and M. Meyers, “Shock compression of [001] single crystal silicon,” The European Physical Journal Special Topics. 2016. link Times cited: 4 NOT USED (high confidence) S. Chavoshi and X. Luo, “Molecular dynamics simulation study of deformation mechanisms in 3C-SiC during nanometric cutting at elevated temperatures,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2016. link Times cited: 102 NOT USED (high confidence) X. W. Zhou, D. Ward, and M. E. Foster, “An analytical bond‐order potential for carbon,” Journal of Computational Chemistry. 2015. link Times cited: 38 Abstract: Carbon is the most widely studied material today because it … read moreAbstract: Carbon is the most widely studied material today because it exhibits special properties not seen in any other materials when in nano dimensions such as nanotube and graphene. Reduction of material defects created during synthesis has become critical to realize the full potential of carbon structures. Molecular dynamics (MD) simulations, in principle, allow defect formation mechanisms to be studied with high fidelity, and can, therefore, help guide experiments for defect reduction. Such MD simulations must satisfy a set of stringent requirements. First, they must employ an interatomic potential formalism that is transferable to a variety of carbon structures. Second, the potential needs to be appropriately parameterized to capture the property trends of important carbon structures, in particular, diamond, graphite, graphene, and nanotubes. Most importantly, the potential must predict the crystalline growth of the correct phases during direct MD simulations of synthesis to achieve a predictive simulation of defect formation. Because an unlimited number of structures not included in the potential parameterization are encountered, the literature carbon potentials are often not sufficient for growth simulations. We have developed an analytical bond order potential for carbon, and have made it available through the public MD simulation package LAMMPS. We demonstrate that our potential reasonably captures the property trends of important carbon phases. Stringent MD simulations convincingly show that our potential accounts not only for the crystalline growth of graphene, graphite, and carbon nanotubes but also for the transformation of graphite to diamond at high pressure. © 2015 Wiley Periodicals, Inc. read less NOT USED (high confidence) P. Käshammer and T. Sinno, “A mechanistic study of impurity segregation at silicon grain boundaries,” Journal of Applied Physics. 2015. link Times cited: 28 Abstract: The segregation behavior of carbon and oxygen atoms at vario… read moreAbstract: The segregation behavior of carbon and oxygen atoms at various silicon grain boundaries was studied using a combination of atomistic simulation and analytical modeling. First, quasi-lattice Grand Canonical Monte Carlo simulations were used to compute segregation isotherms as a function of grain boundary type, impurity atom loading level, and temperature. Next, the atomistic results were employed to regress different analytical segregation models and extract thermodynamic and structural properties. The multilayer Brunauer–Emmett–Teller (BET) isotherm was found to quantitatively capture all the simulation conditions probed in this work, while simpler, single layer models such as the Langmuir-McLean model did not. Some of the BET parameters, namely, the binding free energy of the first adsorption layer and the impurity holding capacity of each layer, were tested for correlation with various measures of grain boundary structure and/or mechanical properties. It was found that certain measures of the atomistic stress distribution correlate strongly with the first-layer binding free energy for substitutional carbon atoms, while common grain boundary identifiers such as sigma value and energy density are not useful in this regard. Preliminary analysis of the more complex case of interstitial oxygen segregation showed that similar measures based on atomistic stress also may be useful here, but more systematic correlative studies are needed to develop a comprehensive picture. read less NOT USED (high confidence) P. Philipp, A. Jana, L. Briquet, T. Wirtz, and G. Henrion, “Molecular dynamics simulation on the initial stage of 1 eV carbon deposition on silicon,” Journal of Physics D: Applied Physics. 2015. link Times cited: 3 Abstract: The deposition process of 1 eV carbon on silicon has been in… read moreAbstract: The deposition process of 1 eV carbon on silicon has been investigated by molecular dynamics (MD) simulations up to a fluence of 5.3 × 1014 atoms cm−2 which corresponds more or less to monolayer coverage. At such low impact energies, atoms are expected to stay on the sample surface, which is also observed up to a fluence of 2 × 1014 atoms cm−2. For higher fluence, carbon atoms start mixing into the silicon substrate. This process seems to get initiated by the increasing strain caused by the carbon atoms deposited on the silicon surface, and which leads to some gradual distortions. The latter are important for the migration of carbon atoms into the silicon lattice. During the whole process the top part of the silicon sample gets amorphized and the coordination of the carbon atoms increases from 1 or 2 to mostly 4-fold coordinated carbon atoms. The process can be considered as the starting point of silicon carbide formation and allows to explain how nm thick films can be formed from 1 eV deposition energies. The low carbon concentration of about 7% in the modified layer is, however, too low to observe a transition towards the latter. read less NOT USED (high confidence) C. E. Rowland et al., “Silicon nanocrystals at elevated temperatures: retention of photoluminescence and diamond silicon to β-silicon carbide phase transition.,” ACS nano. 2014. link Times cited: 14 Abstract: We report the photoluminescence (PL) properties of colloidal… read moreAbstract: We report the photoluminescence (PL) properties of colloidal Si nanocrystals (NCs) up to 800 K and observe PL retention on par with core/shell structures of other compositions. These alkane-terminated Si NCs even emit at temperatures well above previously reported melting points for oxide-embedded particles. Using selected area electron diffraction (SAED), powder X-ray diffraction (XRD), liquid drop theory, and molecular dynamics (MD) simulations, we show that melting does not play a role at the temperatures explored experimentally in PL, and we observe a phase change to β-SiC in the presence of an electron beam. Loss of diffraction peaks (melting) with recovery of diamond-phase silicon upon cooling is observed under inert atmosphere by XRD. We further show that surface passivation by covalently bound ligands endures the experimental temperatures. These findings point to covalently bound organic ligands as a route to the development of NCs for use in high temperature applications, including concentrated solar cells and electrical lighting. read less NOT USED (high confidence) S. Goel, “The current understanding on the diamond machining of silicon carbide,” Journal of Physics D: Applied Physics. 2014. link Times cited: 139 Abstract: The Glenn Research Centre of NASA, USA (www.grc.nasa.gov/WWW… read moreAbstract: The Glenn Research Centre of NASA, USA (www.grc.nasa.gov/WWW/SiC/, silicon carbide electronics) is in pursuit of realizing bulk manufacturing of silicon carbide (SiC), specifically by mechanical means. Single point diamond turning (SPDT) technology which employs diamond (the hardest naturally-occurring material realized to date) as a cutting tool to cut a workpiece is a highly productive manufacturing process. However, machining SiC using SPDT is a complex process and, while several experimental and analytical studies presented to date aid in the understanding of several critical processes of machining SiC, the current knowledge on the ductile behaviour of SiC is still sparse. This is due to a number of simultaneously occurring physical phenomena that may take place on multiple length and time scales. For example, nucleation of dislocation can take place at small inclusions that are of a few atoms in size and once nucleated, the interaction of these nucleations can manifest stresses on the micrometre length scales. The understanding of how these stresses manifest during fracture in the brittle range, or dislocations/phase transformations in the ductile range, is crucial to understanding the brittle–ductile transition in SiC. Furthermore, there is a need to incorporate an appropriate simulation-based approach in the manufacturing research on SiC, owing primarily to the number of uncertainties in the current experimental research that includes wear of the cutting tool, poor controllability of the nano-regime machining scale (effective thickness of cut), and coolant effects (interfacial phenomena between the tool, workpiece/chip and coolant), etc. In this review, these two problems are combined together to posit an improved understanding on the current theoretical knowledge on the SPDT of SiC obtained from molecular dynamics simulation. read less NOT USED (high confidence) N. Faisal, R. Ahmed, S. Goel, and Y. Fu, “Influence of test methodology and probe geometry on nanoscale fatigue failure of diamond-like carbon film,” Surface & Coatings Technology. 2014. link Times cited: 29 NOT USED (high confidence) T. Yoon, T. Lim, T. Min, S. Hung, N. Jakse, and S. Lai, “Epitaxial growth of graphene on 6H-silicon carbide substrate by simulated annealing method.,” The Journal of chemical physics. 2013. link Times cited: 15 Abstract: We grew graphene epitaxially on 6H-SiC(0001) substrate by th… read moreAbstract: We grew graphene epitaxially on 6H-SiC(0001) substrate by the simulated annealing method. The mechanisms that govern the growth process were investigated by testing two empirical potentials, namely, the widely used Tersoff potential [J. Tersoff, Phys. Rev. B 39, 5566 (1989)] and its more refined version published years later by Erhart and Albe [Phys. Rev. B 71, 035211 (2005)]. Upon contrasting the results obtained by these two potentials, we found that the potential proposed by Erhart and Albe is generally more physical and realistic, since the annealing temperature at which the graphene structure just coming into view at approximately 1200 K is unambiguously predicted and close to the experimentally observed pit formation at 1298 K within which the graphene nucleates. We evaluated the reasonableness of our layers of graphene by calculating carbon-carbon (i) average bond-length, (ii) binding energy, and (iii) pair correlation function. Also, we compared with related experiments the various distance of separation parameters between the overlaid layers of graphene and substrate surface. read less NOT USED (high confidence) U. Monteverde, M. Migliorato, J. Pal, and D. Powell, “Elastic and vibrational properties of group IV semiconductors in empirical potential modelling,” Journal of Physics: Condensed Matter. 2013. link Times cited: 8 Abstract: We have developed an interatomic potential that with a singl… read moreAbstract: We have developed an interatomic potential that with a single set of parameters is able to accurately describe at the same time the elastic, vibrational and thermodynamics properties of semiconductors. The simultaneous inclusion of radial and angular forces of the interacting atom pairs (short range) together with the influence of the broken crystal symmetry when the atomic arrangement is out of equilibrium (long range) results in correct predictions of all of the phonon dispersion spectrum and mode-Grüneisen parameters of silicon and germanium. The long range interactions are taken into account up to the second nearest neighbours, to correctly influence the elastic and vibrational properties, and therefore represent only a marginal computational cost compared to the full treatment of other proposed potentials. Results of molecular dynamics simulations are compared with those of ab initio calculations, showing that when our proposed potential is used to perform the initial stages of the structural relaxation, a significant reduction of the computational time needed during the geometry optimization of density functional theory simulations is observed. read less NOT USED (high confidence) Y. Jing, M. Hu, and L. Guo, “Thermal conductivity of hybrid graphene/silicon heterostructures,” Journal of Applied Physics. 2013. link Times cited: 23 Abstract: The success of fabricating single layer graphene and silicon… read moreAbstract: The success of fabricating single layer graphene and silicon nanofilm (could be as thin as single layer so far) has triggered enormous interest in exploring their unique physics and novel applications. An intuitive idea is to investigate what happens if we construct a heterostructure composed of these two sheets. In this paper, we perform nonequilibrium molecular dynamics simulations to systematically investigate the in-plane thermal transport in graphene/silicon/graphene (Gr/Si/Gr) heterostructures. The effects of Si film thickness, interfacial interaction strength, and length on the thermal conductivity of the Gr/Si/Gr heterostructures are explicitly considered. Our simulations identify a unified scaling law for thickness dependence of thermal conductivity of the Gr/Si/Gr heterostructures, despite different interfacial interaction forms are used (weak van der Waals interaction and strong covalent bonding). By quantifying relative contribution from phonon polarizations and defining heat flux onto single ... read less NOT USED (high confidence) K. Henriksson, C. Björkas, and K. Nordlund, “Atomistic simulations of stainless steels: a many-body potential for the Fe–Cr–C system,” Journal of Physics: Condensed Matter. 2013. link Times cited: 65 Abstract: Stainless steels found in real-world applications usually ha… read moreAbstract: Stainless steels found in real-world applications usually have some C content in the base Fe–Cr alloy, resulting in hard and dislocation-pinning carbides—Fe3C (cementite) and Cr23C6—being present in the finished steel product. The higher complexity of the steel microstructure has implications, for example, for the elastic properties and the evolution of defects such as Frenkel pairs and dislocations. This makes it necessary to re-evaluate the effects of basic radiation phenomena and not simply to rely on results obtained from purely metallic Fe–Cr alloys. In this report, an analytical interatomic potential parameterization in the Abell–Brenner–Tersoff form for the entire Fe–Cr–C system is presented to enable such calculations. The potential reproduces, for example, the lattice parameter(s), formation energies and elastic properties of the principal Fe and Cr carbides (Fe3C, Fe5C2, Fe7C3, Cr3C2, Cr7C3, Cr23C6), the Fe–Cr mixing energy curve, formation energies of simple C point defects in Fe and Cr, and the martensite lattice anisotropy, with fair to excellent agreement with empirical results. Tests of the predictive power of the potential show, for example, that Fe–Cr nanowires and bulk samples become elastically stiffer with increasing Cr and C concentrations. High-concentration nanowires also fracture at shorter relative elongations than wires made of pure Fe. Also, tests with Fe3C inclusions show that these act as obstacles for edge dislocations moving through otherwise pure Fe. read less NOT USED (high confidence) B. Y. Thakore, S. G. Khambholja, A. Vahora, N. K. Bhatt, and A. R. Jani, “Thermodynamic properties of 3C—SiC,” Chinese Physics B. 2013. link Times cited: 13 Abstract: In the present paper, we report on the results of various th… read moreAbstract: In the present paper, we report on the results of various thermodynamic properties of 3C—SiC at high pressure and temperature using first principles calculations. We use the plane-wave pseudopotential density functional theory as implemented in Quantum ESPRESSO code for calculating various cohesive properties in ambient condition. Further, ionic motion at a finite temperature is taken into account using the quasiharmonic Debye model. The calculated thermodynamic properties, phonon dispersion curves, and phonon densities of states at different temperatures and structural phase transitions at high pressures are found to be in good agreement with experimental and other theoretical results. read less NOT USED (high confidence) P. Käshammer and T. Sinno, “Interactions of twin boundaries with intrinsic point defects and carbon in silicon,” Journal of Applied Physics. 2013. link Times cited: 22 Abstract: Although multicrystalline silicon (mc-Si) is currently the m… read moreAbstract: Although multicrystalline silicon (mc-Si) is currently the most widely used material for fabricating photovoltaic cells, its electrical properties remain limited by several types of defects, which interact in complex ways that are not yet fully understood. A particularly important phenomenon is the interaction between grain boundaries and intrinsic point defects or impurity atoms, such as carbon, oxygen, nitrogen, and various types of metals. Here, we use empirical molecular dynamics to study the interactions of Σ3{111}, Σ9{221}, and Σ27{552} twin boundaries, which account for over 50% of all grain boundaries in mc-Si, with self-interstitials, vacancies, and substitutional carbon atoms. It is shown that twin boundary-point defect interaction energies increase with twinning order and that they are predominantly attractive. We also find that twin boundary interactions with substitutional carbon are highly spatially heterogeneous, exhibiting alternating repulsive-attractive regions that correlate strongly wi... read less NOT USED (high confidence) P. Howell, “Comparison of molecular dynamics methods and interatomic potentials for calculating the thermal conductivity of silicon.,” The Journal of chemical physics. 2012. link Times cited: 74 Abstract: We compare the molecular dynamics Green-Kubo and direct meth… read moreAbstract: We compare the molecular dynamics Green-Kubo and direct methods for calculating thermal conductivity κ, using as a test case crystalline silicon at temperatures T in the range 500-1000 K (classical regime). We pay careful attention to the convergence with respect to simulation size and duration and to the procedures used to fit the simulation data. We show that in the Green-Kubo method the heat current autocorrelation function is characterized by three decay processes, of which the slowest lasts several tens of picoseconds so that convergence requires several tens of nanoseconds of data. Using the Stillinger-Weber potential we find excellent agreement between the two methods. We also use the direct method to calculate κ(T) for the Tersoff potential and find that the magnitude and the temperature-dependence are different for the two potentials and that neither potential agrees with experimental data. We argue that this implies that using the Stillinger-Weber or Tersoff potentials to predict trends in kappa as some system parameter is varied may yield results which are specific to the potential but not intrinsic to Si. read less NOT USED (high confidence) Y. Gao, Y. Jing, Q. Meng, L. Zhang, J. Liu, and X. Qin, “Investigation of the thermal‐transport properties for silicon nanofilm covered with graphene via nonequilibrium molecular dynamics,” physica status solidi (b). 2012. link Times cited: 7 Abstract: Nonequilibrium molecular dynamics (NEMD) is used to investig… read moreAbstract: Nonequilibrium molecular dynamics (NEMD) is used to investigate the thermal‐transport properties of a silicon nanofilm covered with graphene (Gr/Si/Gr nanofilm). The investigation results demonstrate that graphene can enhance the thermal‐transport properties and weaken the ballistic characteristics of silicon nanofilm. Under the action of a small strain, the thermal conductivity decreases with the growth of tensile and compressive strain, respectively. In addition, the higher‐frequency phonons in graphene give more contributions to the variation of thermal conductivity of Gr/Si/Gr nanofilm under strain. The thermal conductivity of Gr/Si/Gr nanofilm increases linearly with the increase of temperature in the lower‐temperature regime due to the quantum effect, and begins to clearly decrease when the temperature exceeds a definite value. read less NOT USED (high confidence) V. Dozhdikov, A. Basharin, and P. Levashov, “Two-phase simulation of the crystalline silicon melting line at pressures from -1 to 3 GPa.,” The Journal of chemical physics. 2012. link Times cited: 32 Abstract: Results of a numerical investigation of crystalline silicon … read moreAbstract: Results of a numerical investigation of crystalline silicon melting line within the range of pressures from -1 to 3 GPa are presented. A two-phase molecular dynamics method is applied to obtain temperature, pressure, and densities of solid and liquid phases on the melting line. Using a special procedure we ensure the strict control of the two-phase equilibrium in the simulation cell. To describe the interaction between the atoms four classic potentials have been chosen: the Stillinger-Weber one and three modified variants of the Tersoff potential. For the Stillinger-Weber and Tersoff potentials in the modification by Kumagai-Izumi-Hara-Sakai a good coincidence with experimental data on crystalline Si melting temperature is obtained within the range of pressure from 0 to 3 GPa. Calculations of the solid and liquid phase densities on the silicon melting line for the Stillinger-Weber potential are also in close agreement with experiments. read less NOT USED (high confidence) F. Zirkelbach, B. Stritzker, K. Nordlund, J. Lindner, W. Schmidt, and E. Rauls, “Defects in carbon implanted silicon calculated by classical potentials and first-principles methods,” Physical Review B. 2010. link Times cited: 7 Abstract: A comparative theoretical investigation of carbon interstiti… read moreAbstract: A comparative theoretical investigation of carbon interstitials in silicon is presented. Calculations using classical potentials are compared to first-principles density-functional theory calculations of the geometries, formation, and activation energies of the carbon dumbbell interstitial, showing the importance of a quantummechanical description of this system. In contrast to previous studies, the present first-principles calculations of the interstitial carbon migration path yield an activation energy that excellently matches the experiment. The bond-centered interstitial configuration shows a net magnetization of two electrons, illustrating the need for spin-polarized calculations. read less NOT USED (high confidence) G. Lucas, M. Bertolus, and L. Pizzagalli, “An environment-dependent interatomic potential for silicon carbide: calculation of bulk properties, high-pressure phases, point and extended defects, and amorphous structures,” Journal of Physics: Condensed Matter. 2010. link Times cited: 41 Abstract: An interatomic potential has been developed to describe inte… read moreAbstract: An interatomic potential has been developed to describe interactions in silicon, carbon and silicon carbide, based on the environment-dependent interatomic potential (EDIP) (Bazant et al 1997 Phys. Rev. B 56 8542). The functional form of the original EDIP has been generalized and two sets of parameters have been proposed. Tests with these two potentials have been performed for many properties of SiC, including bulk properties, high-pressure phases, point and extended defects, and amorphous structures. One parameter set allows us to keep the original EDIP formulation for silicon, and is shown to be well suited for modelling irradiation-induced effects in silicon carbide, with a very good description of point defects and of the disordered phase. The other set, including a new parametrization for silicon, has been shown to be efficient for modelling point and extended defects, as well as high-pressure phases. read less NOT USED (high confidence) K. Albe, J. Nord, and K. Nordlund, “Dynamic charge-transfer bond-order potential for gallium nitride,” Philosophical Magazine. 2009. link Times cited: 10 Abstract: We present an analytical interatomic potential for gallium n… read moreAbstract: We present an analytical interatomic potential for gallium nitride which is based on a new environment-dependent dynamic charge-transfer model. The model consists of a short-ranged bond-order potential that accounts for covalent/metallic interactions and an ionic Coulomb potential with effective point charges that are dynamically adjusted. In contrast to established models, these point charges are distance-dependent and vary with the number and type of nearest neighbour atoms. The basic concepts stem from the idea of bond charges. We assume pairwise symmetric charge transfer between atoms of different type forming a bond. Charge contributions of all bonds to an atomic site are weighted and added, yielding the effective charge per atom. Mulliken charges, as obtained from density-functional theory calculations within the local-density approximation, are used for adjusting the parameters and functional form of the potential. The short-range contributions are chosen as angular-dependent many-body bond-order potentials, which can be understood as an extension of a Finnis–Sinclair type potential. read less NOT USED (high confidence) T. Nakajima and K. Shintani, “Molecular dynamics study of energetics of graphene flakes,” Journal of Applied Physics. 2009. link Times cited: 18 Abstract: Molecular dynamics simulations for graphene flakes of variou… read moreAbstract: Molecular dynamics simulations for graphene flakes of various shapes are performed. The equilibrium structures of graphene flakes are obtained. Round, hexagonal, and rectangular graphene flakes are dealt with, and their sizes are varied from a few angstroms to 200 A. It is shown that for round and hexagonal graphene flakes of small size, the edge configuration influences their energy in equilibrium. Graphene nanoribbons (GNRs) of various aspect ratios are equilibrated at low temperature. The energies of the equilibrated graphene flakes with zigzag (ZZ) edges are lower than the energies of the equilibrated graphene flakes with armchair (AC) edges. This result corresponds to the scanning tunneling microscopy observations in the literature. The atomic bonds on the edges of graphene flakes with both edge configurations are reconstructed. The bond lengths of such reconstructed edges are smaller than the lengths of the atomic bonds inside them. Therefore, free graphene flakes undergo compressive edge stress and... read less NOT USED (high confidence) C. Björkas et al., “Interatomic potentials for the Be–C–H system,” Journal of Physics: Condensed Matter. 2009. link Times cited: 65 Abstract: Analytical bond-order potentials for beryllium, beryllium ca… read moreAbstract: Analytical bond-order potentials for beryllium, beryllium carbide and beryllium hydride are presented. The reactive nature of the formalism makes the potentials suitable for simulations of non-equilibrium processes such as plasma–wall interactions in fusion reactors. The Be and Be–C potentials were fitted to ab initio calculations as well as to experimental data of several different atomic configurations and Be–H molecule and defect data were used in determining the Be–H parameter set. Among other tests, sputtering, melting and quenching simulations were performed in order to check the transferability of the potentials. The antifluorite Be2C structure is well described by the Be–C potential and the hydrocarbon interactions are modelled by the established Brenner potentials. read less NOT USED (high confidence) A. D. Patrick, X. Dong, T. C. Allison, and E. Blaisten-Barojas, “Silicon carbide nanostructures: a tight binding approach.,” The Journal of chemical physics. 2009. link Times cited: 17 Abstract: A tight-binding model Hamiltonian is newly parametrized for … read moreAbstract: A tight-binding model Hamiltonian is newly parametrized for silicon carbide based on fits to a database of energy points calculated within the density functional theory approach of the electronic energy surfaces of nanoclusters and the total energy of bulk 3C and 2H polytypes at different densities. This TB model includes s and p angular momentum symmetries with nonorthogonal atomic basis functions. With the aid of the new TB model, minima of silicon carbide cagelike clusters, nanotubes, ring-shaped ribbons, and nanowires are predicted. Energetics, structure, growth sequences, and stability patterns are reported for the nanoclusters and nanotubes. The band structure of SiC nanotubes and nanowires indicates that the band gap of the nanotubes ranges from 0.57 to 2.38 eV depending on the chirality, demonstrating that these nanotubes are semiconductors or insulators. One type of nanowire is metallic, another type is semiconductor, and the rest are insulators. read less NOT USED (high confidence) K. Henriksson and K. Nordlund, “Simulations of cementite: An analytical potential for the Fe-C system,” Physical Review B. 2009. link Times cited: 72 Abstract: An analytical bond-order interatomic potential has been deve… read moreAbstract: An analytical bond-order interatomic potential has been developed for the iron-carbon system for use in molecular-dynamics and Monte Carlo simulations. The potential has been successfully fitted to cementite and Hagg carbide, which are most important crystalline polytypes among the many known metastable iron carbide phases. Predicted properties of other carbides and the simplest point defects are in good to reasonable agreement with available data from experiments and density-functional theory calculations. The potential correctly describes melting and recrystallization of cementite, making it useful for simulation of steels. We show that they correctly describe the metastability of cementite and can be used to model carbide growth and dissolution. read less NOT USED (high confidence) T. Kumagai, S. Hara, J. Choi, S. Izumi, and T. Kato, “Development of empirical bond-order-type interatomic potential for amorphous carbon structures,” Journal of Applied Physics. 2009. link Times cited: 21 Abstract: A bond-order-type interatomic potential has been developed f… read moreAbstract: A bond-order-type interatomic potential has been developed for reproducing amorphous carbon (a-C) structures. Several improvements have been incorporated into the conventional Brenner potential so that the material properties of carbon crystals remain unchanged. The main characteristics of the potential function developed in the present research are the use of a screening function instead of a cutoff function and the introduction of a dihedral angle potential around the bond between two threefold coordinated atoms. By using the developed interatomic potential, we can reproduce the material properties of a-C structures, such as the fraction of sp3-bonded atoms, radial distribution function, and ring statistics. It is found that the correction term enhances the formation of cluster structures in a-C, which is confirmed in the first-principles calculation. read less NOT USED (high confidence) K. Li, H. He, B. Xu, and B. Pan, “The stabilities of gallium nanowires with different phases encapsulated in a carbon nanotube,” Journal of Applied Physics. 2009. link Times cited: 10 Abstract: For C–Ga systems, a classical potential is developed to desc… read moreAbstract: For C–Ga systems, a classical potential is developed to describe the interaction between C and Ga atoms. By using this potential, we study the stabilities of the Ga nanowires with different phases encapsulated in a carbon nanotube (CNT). Simulations show that the encapsulated β-Ga and γ-Ga nanowires are more stable than the α-Ga nanowire in the CNT. Moreover, we find that such relative stabilities are mainly originated from the size effect of the Ga nanowires and the influence of the CNT. With performing molecular dynamics simulation at finite temperatures, the linear thermal expansion coefficient of an encapsulated Ga nanowire is predicted to be 1.38×10−4 K−1, being very close to the bulk value. The obtained stabilities as well as the thermal expansion feature of the concerned Ga nanowires are all consistent with experimental observations. read less NOT USED (high confidence) P. S. Branicio, J. Rino, C. Gan, and H. Tsuzuki, “Interaction potential for indium phosphide: a molecular dynamics and first-principles study of the elastic constants, generalized stacking fault and surface energies,” Journal of Physics: Condensed Matter. 2009. link Times cited: 31 Abstract: Indium phosphide is investigated using molecular dynamics (M… read moreAbstract: Indium phosphide is investigated using molecular dynamics (MD) simulations and density-functional theory calculations. MD simulations use a proposed effective interaction potential for InP fitted to a selected experimental dataset of properties. The potential consists of two- and three-body terms that represent atomic-size effects, charge–charge, charge–dipole and dipole–dipole interactions as well as covalent bond bending and stretching. Predictions are made for the elastic constants as a function of density and temperature, the generalized stacking fault energy and the low-index surface energies. read less NOT USED (high confidence) H. Amara, J. Roussel, C. Bichara, J. Gaspard, and F. Ducastelle, “Tight-binding potential for atomistic simulations of carbon interacting with transition metals: Application to the Ni-C system,” Physical Review B. 2008. link Times cited: 95 Abstract: We present a tight-binding potential for transition metals, … read moreAbstract: We present a tight-binding potential for transition metals, carbon, and transition-metal carbides, which has been optimized through a systematic fitting procedure. A minimal basis, including the s and p electrons of carbon and the d electrons of the transition metal, is used to obtain a transferable tight-binding model of the carbon-carbon, metal-metal, and metal-carbon interactions applicable to binary systems. The Ni-C system is more specifically discussed. The successful validation of the potential for different atomic configurations indicates a good transferability of the model and makes it a good choice for atomistic simulations sampling a large configuration space. This approach appears to be very efficient to describe interactions in systems containing carbon and transition-metal elements. By way of example, we present results concerning the epitaxial growth of graphene sheets on (111) Ni surfaces, as well as the catalytic nucleation of carbon nanotubes. read less NOT USED (high confidence) C. Sanz-Navarro et al., “Molecular Dynamics Simulations of Carbon-Supported Ni Clusters Using the Reax Reactive Force Field,” Journal of Physical Chemistry C. 2008. link Times cited: 30 Abstract: Molecular dynamics simulations have been performed using a R… read moreAbstract: Molecular dynamics simulations have been performed using a Reax force field for C/H/Ni systems to study the structural changes of an Ni_(100) cluster adsorbed on a carbon platelet. Three different edges of a carbon platelet are considered. We find a complete restructuring of the initial structure of the Ni_(100) clusters adsorbed on the armchair and zigzag edges. Nonetheless, the mean Ni−Ni bond length hardly changes. Several preferential sites on each of the graphite edges are identified. Diffusion of the entire cluster is found both for adsorption on the basal plane and for binding to a hydrogen terminated graphite edge. read less NOT USED (high confidence) E. Moore, “Computational modelling of inorganic solids.” 2008. link Times cited: 10 Abstract: This report covers papers published in 2011 dealing with the… read moreAbstract: This report covers papers published in 2011 dealing with the application of computational techniques to inorganic solids. It deals mainly with continuous solids that are ionic in nature; work on metals and MOFs is excluded. Special attention is given to solids used in solid oxide fuel cells, iron-based superconductors, zeolites and systems of interest to the life and earth sciences. Relevant advances in computational methods are also covered. read less NOT USED (high confidence) R. V. Pulikollu and S. Mukhopadhyay, “Nanoscale coatings for control of interfacial bonds and nanotube growth,” Applied Surface Science. 2007. link Times cited: 27 NOT USED (high confidence) P. Vashishta, R. Kalia, A. Nakano, and J. Rino, “Interaction potential for silicon carbide: A molecular dynamics study of elastic constants and vibrational density of states for crystalline and amorphous silicon carbide,” Journal of Applied Physics. 2007. link Times cited: 279 Abstract: An effective interatomic interaction potential for SiC is pr… read moreAbstract: An effective interatomic interaction potential for SiC is proposed. The potential consists of two-body and three-body covalent interactions. The two-body potential includes steric repulsions due to atomic sizes, Coulomb interactions resulting from charge transfer between atoms, charge-induced dipole-interactions due to the electronic polarizability of ions, and induced dipole-dipole (van der Waals) interactions. The covalent characters of the Si–C–Si and C–Si–C bonds are described by the three-body potential. The proposed three-body interaction potential is a modification of the Stillinger-Weber form proposed to describe Si. Using the molecular dynamics method, the interaction potential is used to study structural, elastic, and dynamical properties of crystalline (3C), amorphous, and liquid states of SiC for several densities and temperatures. The structural energy for cubic (3C) structure has the lowest energy, followed by the wurtzite (2H) and rock-salt (RS) structures. The pressure for the structural t... read less NOT USED (high confidence) J. Kotakoski and K. Nordlund, “Binding a carbon nanotube to the Si(100) surface using ion irradiation—an atomistic simulation study,” New Journal of Physics. 2006. link Times cited: 7 Abstract: Using carbon nanotubes (CNTs) as building blocks in silicon-… read moreAbstract: Using carbon nanotubes (CNTs) as building blocks in silicon-based electronics requires good electric contacts between the tubes and other devices. Recent experimental and theoretical works have shown that irradiation can be used to modify both the structure and the electrical properties of nanotubes, and also to create new covalent bonds to different nanotube structures. In this study, we have used atomistic computer simulations with analytical, empirically fitted interaction models, to examine the possibility to enhance binding between a CNT and a silicon substrate with C, Si and Ne ion irradiation. Low irradiation doses (<2.8×1014 ions/cm2) and energies (0.2–2.0 keV) were used, to ensure that the irradiated nanotube will not be destroyed. Our results indicate, that ion irradiation can be used to create new covalent bonds, and also to increase the binding energy between these structures, when the irradiation doses and energies are carefully chosen. We found that a typical number of created new covalent C–Si bonds is 0.5–0.9 (1014 ions/cm2)−1, and a typical increase in the binding energy between the structures is 100–400% for moderate irradiation doses. read less NOT USED (high confidence) P. Erhart, N. Juslin, O. Goy, K. Nordlund, R. Müller, and K. Albe, “Analytic bond-order potential for atomistic simulations of zinc oxide,” Journal of Physics: Condensed Matter. 2006. link Times cited: 75 Abstract: An interatomic potential for zinc oxide and its elemental co… read moreAbstract: An interatomic potential for zinc oxide and its elemental constituents is derived based on an analytical bond-order formalism. The model potential provides a good description of the bulk properties of various solid structures of zinc oxide including cohesive energies, lattice parameters, and elastic constants. For the pure elements zinc and oxygen the energetics and structural parameters of a variety of bulk phases and in the case of oxygen also molecular structures are reproduced. The dependence of thermal and point defect properties on the cutoff parameters is discussed. As exemplary applications the irradiation of bulk zinc oxide and the elastic response of individual nanorods are studied. read less NOT USED (high confidence) N. Juslin et al., “Analytical interatomic potential for modeling nonequilibrium processes in the W–C–H system,” Journal of Applied Physics. 2005. link Times cited: 264 Abstract: A reactive interatomic potential based on an analytical bond… read moreAbstract: A reactive interatomic potential based on an analytical bond-order scheme is developed for the ternary system W–C–H. The model combines Brenner’s hydrocarbon potential with parameter sets for W–W, W–C, and W–H interactions and is adjusted to materials properties of reference structures with different local atomic coordinations including tungsten carbide, W–H molecules, as well as H dissolved in bulk W. The potential has been tested in various scenarios, such as surface, defect, and melting properties, none of which were considered in the fitting. The intended area of application is simulations of hydrogen and hydrocarbon interactions with tungsten, which have a crucial role in fusion reactor plasma-wall interactions. Furthermore, this study shows that the angular-dependent bond-order scheme can be extended to second nearest-neighbor interactions, which are relevant in body-centered-cubic metals. Moreover, it provides a possibly general route for modeling metal carbides. © 2005 American Institute of Physics. DOI: 10.1063/1.2149492 read less NOT USED (high confidence) S. Lai, I. Setiyawati, T. Yen, and Y. H. Tang, “Studying lowest energy structures of carbon clusters by bond-order empirical potentials,” Theoretical Chemistry Accounts. 2016. link Times cited: 10 NOT USED (high confidence) R. Alsayegh, “Vision-augmented molecular dynamics simulation of nanoindentation,” Journal of Nanomaterials. 2015. link Times cited: 7 Abstract: We present a user-friendly vision-augmented technique to car… read moreAbstract: We present a user-friendly vision-augmented technique to carry out atomic simulation using hand gestures. The system is novel in its concept as it enables the user to directly manipulate the atomic structures on the screen, in 3D space using hand gestures, allowing the exploration and visualisation of molecular interactions at different relative conformations. The hand gestures are used to pick and place atoms on the screen allowing thereby the ease of carrying out molecular dynamics simulation in a more efficient way. The end result is that users with limited expertise in developing molecular structures can now do so easily and intuitively by the use of body gestures to interact with the simulator to study the system in question. The proposed system was tested by simulating the crystal anisotropy of crystalline silicon during nanoindentation. A long-range (Screened bond order) Tersoff potential energy function was used during the simulation which revealed the value of hardness and elastic modulus being similar to what has been found previously from the experiments. We anticipate that our proposed system will open up new horizons to the current methods on how an MD simulation is designed and executed. read less NOT USED (high confidence) Z. Sarvi, M. Shariyat, and M. Asgari, “Closed-form Molecular Mechanics Formulations for the 3D Local Buckling and 2D Effective Young’s Modulus of the Nanosheets,” Applied and Computational Mechanics. 2015. link Times cited: 1 Abstract: A closed form three-dimensional solution is presented for de… read moreAbstract: A closed form three-dimensional solution is presented for determination of the local buckling (cell buckling) load of the nanosheets. Moreover, an expression is proposed for the effective 2D Young’s modulus of the unit cell of the nanosheet. In this regard, a three-dimensional efficient space-frame-like geometrical model with angular and extensional compliances is considered to investigate stability and effective Young’s modulus of the nanosheet in terms of the generally possible relative movements of the atoms of the unit cell, in the Cartesian coordinates. The molecular dynamics approach is employed in development of the formulation, taking into account the force constants and bond characteristics. The governing equations are derived based on the principle of minimum total potential energy. Results of the special cases of each of the proposed expressions are verified by the results available in literature or results of the traditional approaches. Comparisons are made with various buckling results reported for different nanosheets, based on different approaches of determination of the stiffness parameters, and a good agreement is noticed. read less NOT USED (high confidence) C. Jiang, D. Morgan, and I. Szlufarska, “Structures and stabilities of small carbon interstitial clusters in cubic silicon carbide,” Acta Materialia. 2014. link Times cited: 19 NOT USED (definite) Q. Zhang, X. Ma, and Y. Zhao, “Adhesion Behavior between Multilayer Graphene and Semiconductor Substrates,” Applied Sciences. 2018. link Times cited: 2 Abstract: A high bonding strength between graphene and a semiconductor… read moreAbstract: A high bonding strength between graphene and a semiconductor surface is significant to the performance of graphene-based Micro-Electro Mechanical Systems/Nano-Electro Mechanical Systems (MEMS/NEMS) devices. In this paper, by applying a series of constant vertical upward velocities (Vup) to the topmost layer of graphene, the exfoliation processes of multilayer graphene (one to ten layers) from an Si semiconductor substrate were simulated using the molecular dynamics method, and the bonding strength was calculated. The critical exfoliation velocities, adhesion forces, and adhesion energies to exfoliate graphene were obtained. In a system where the number of graphene layers is two or three, there are two critical exfoliation velocities. Graphene cannot be exfoliated when the Vup is lower than the first critical velocity, although the total number of graphene layers can be exfoliated when the Vup is in the range between the first critical velocity and second critical velocity. Only the topmost layer can be exfoliated to be free from the Si surface if the applied Vup is greater than the second critical velocity. In systems where the number of graphene layers is four to ten, only the topmost layer can be free and exfoliated if the exfoliation velocity is greater than the critical velocity. It was found that a relatively low applied Vup resulted in entire graphene layers peeling off from the substrate. The adhesion forces of one-layer to ten-layer graphene systems were in the range of 25.04 nN–74.75 nN, and the adhesion energy levels were in the range of 73.5 mJ/m2–188.45 mJ/m2. These values are consistent with previous experimental results, indicating a reliable bond strength between graphene and Si semiconductor surfaces. read less NOT USED (definite) C. Ruestes, I. A. Alhafez, and H. Urbassek, “Atomistic Studies of Nanoindentation—A Review of Recent Advances.” 2017. link Times cited: 45 Abstract: This review covers areas where our understanding of the mech… read moreAbstract: This review covers areas where our understanding of the mechanisms underlying nanoindentation has been increased by atomistic studies of the nanoindentation process. While such studies have been performed now for more than 20 years, recent investigations have demonstrated that the peculiar features of nanoplasticity generated during indentation can be analyzed in considerable detail by this technique. Topics covered include: nucleation of dislocations in ideal crystals, effect of surface orientation, effect of crystallography (fcc, bcc, hcp), effect of surface and bulk damage on plasticity, nanocrystalline samples, and multiple (sequential) indentation. In addition we discuss related features, such as the influence of tip geometry on the indentation and the role of adhesive forces, and how pre-existing plasticity affects nanoindentation. read less NOT USED (definite) H. Xie, X. Song, F. Yin, and Y. Zhang, “Effect of WC/Co coherency phase boundaries on Fracture toughness of the nanocrystalline cemented carbides,” Scientific Reports. 2016. link Times cited: 30 NOT USED (definite) S. Goel, A. Kovalchenko, A. Stukowski, and G. Cross, “Influence of microstructure on the cutting behaviour of silicon,” Acta Materialia. 2016. link Times cited: 143 NOT USED (definite) S. Chavoshi and X. Luo, “An atomistic simulation investigation on chip related phenomena in nanometric cutting of single crystal silicon at elevated temperatures,” Computational Materials Science. 2016. link Times cited: 50 NOT USED (definite) H. Tetlow, J. Boer, I. Ford, D. Vvedensky, J. Coraux, and L. Kantorovich, “Growth of Epitaxial Graphene: Theory and Experiment,” arXiv: Materials Science. 2014. link Times cited: 222 NOT USED (definite) Y. J. Lv and M. Chen, “Thermophysical Properties of Undercooled Alloys: An Overview of the Molecular Simulation Approaches,” International Journal of Molecular Sciences. 2011. link Times cited: 20 Abstract: We review the studies on the thermophysical properties of un… read moreAbstract: We review the studies on the thermophysical properties of undercooled metals and alloys by molecular simulations in recent years. The simulation methods of melting temperature, enthalpy, specific heat, surface tension, diffusion coefficient and viscosity are introduced and the simulated results are summarized. By comparing the experimental results and various theoretical models, the temperature and the composition dependences of the thermophysical properties in undercooled regime are discussed. read less NOT USED (definite) F. Fang, X. Zhang, W. Gao, Y. B. Guo, G. Byrne, and H. N. Hansen, “Nanomanufacturing—Perspective and applications,” Cirp Annals-manufacturing Technology. 2017. link Times cited: 113 |
 Tersoff_LAMMPS_ErhartAlbe_2005_SiC__MO_903987585848_005
Tersoff_LAMMPS_ErhartAlbe_2005_SiC__MO_903987585848_005