Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
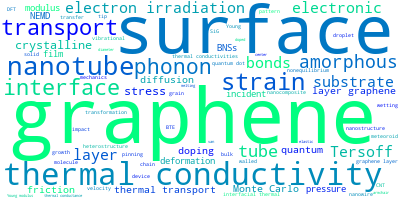
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
2453 Citations (222 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) Q. Zhang, X. Ma, and Y. Zhao, “Adhesion Behavior between Multilayer Graphene and Semiconductor Substrates,” Applied Sciences. 2018. link Times cited: 2 Abstract: A high bonding strength between graphene and a semiconductor… read moreAbstract: A high bonding strength between graphene and a semiconductor surface is significant to the performance of graphene-based Micro-Electro Mechanical Systems/Nano-Electro Mechanical Systems (MEMS/NEMS) devices. In this paper, by applying a series of constant vertical upward velocities (Vup) to the topmost layer of graphene, the exfoliation processes of multilayer graphene (one to ten layers) from an Si semiconductor substrate were simulated using the molecular dynamics method, and the bonding strength was calculated. The critical exfoliation velocities, adhesion forces, and adhesion energies to exfoliate graphene were obtained. In a system where the number of graphene layers is two or three, there are two critical exfoliation velocities. Graphene cannot be exfoliated when the Vup is lower than the first critical velocity, although the total number of graphene layers can be exfoliated when the Vup is in the range between the first critical velocity and second critical velocity. Only the topmost layer can be exfoliated to be free from the Si surface if the applied Vup is greater than the second critical velocity. In systems where the number of graphene layers is four to ten, only the topmost layer can be free and exfoliated if the exfoliation velocity is greater than the critical velocity. It was found that a relatively low applied Vup resulted in entire graphene layers peeling off from the substrate. The adhesion forces of one-layer to ten-layer graphene systems were in the range of 25.04 nN–74.75 nN, and the adhesion energy levels were in the range of 73.5 mJ/m2–188.45 mJ/m2. These values are consistent with previous experimental results, indicating a reliable bond strength between graphene and Si semiconductor surfaces. read less USED (definite) R. Dettori and L. Colombo, “THERMAL PROPERTIES OF TPD-BASED ORGANIC GLASSES,” Istituto Lombardo - Accademia di Scienze e Lettere - Incontri di Studio. 2018. link Times cited: 1 Abstract: Glassy materials are condensed matter systems showing physic… read moreAbstract: Glassy materials are condensed matter systems showing physical properties in between solids and liquids and retaining information about the thermal history they have been subjected to and the way they have been prepared. Formally, this implies that their configurational energy landscape is a complex multi-dimensional surface, showing quite a few basins with different depths, widths, and shapes: the system can be trapped in any of them, assuming very unlike physical properties. Recently, it has been demonstrated experimentally that a glassy system can be grown by physical vapor deposition of organic molecules on a substrate. The physics of such organic glasses is enriched by a new feature, namely: the anisotropic molecular structure of the basic building blocks used to assemble the film. TPD-based organic glasses have been generated by atomistic simulations that mimic vapor deposition and their thermal properties have been accordingly calculated. Simulations generate a rational phenomenology, providing robust evidence that heat transfer is not isotropic but, rather, correlated to an inherent molecular property, namely the axial structure of the TPD molecule. Furthermore, we present the first theoretical prediction of the specific heat trend versus temperature, showing in the quantum regime an intriguing anomaly with respect to crystalline systems. read less USED (definite) R. Li, S. Wang, and Q. Peng, “Tuning the Slide-Roll Motion Mode of Carbon Nanotubes via Hydroxyl Groups,” Nanoscale Research Letters. 2018. link Times cited: 11 USED (definite) S. Zhang et al., “Response to Extreme Temperatures of Mesoporous Silica MCM-41: Porous Structure Transformation Simulation and Modification of Gas Adsorption Properties.,” Langmuir : the ACS journal of surfaces and colloids. 2016. link Times cited: 12 Abstract: Molecular dynamics (MD) and Monte Carlo (MC) simulations wer… read moreAbstract: Molecular dynamics (MD) and Monte Carlo (MC) simulations were applied together for the first time to reveal the porous structure transformation mechanisms of mesoporous silica MCM-41 subjected to temperatures up to 2885 K. Silica was experimentally characterized to inform the models and enable prediction of changes in gas adsorption/separation properties. MD simulations suggest that the pore closure process is activated by a collective diffusion of matrix atoms into the porous region, accompanied by bond reformation at the surface. Degradation is kinetically limited, such that complete pore closure is postponed at high heating rates. We experimentally observe decreased gas adsorption with increasing temperature in mesoporous silica heated at fixed rates, due to pore closure and structural degradation consistent with simulation predictions. Applying the Kissinger equation, we find a strong correlation between the simulated pore collapse temperatures and the experimental values which implies an activation energy of 416 ± 17 kJ/mol for pore closure. MC simulations give the adsorption and selectivity for thermally treated MCM-41, for N2, Ar, Kr, and Xe at room temperature within the 1-10 000 kPa pressure range. Relative to pristine MCM-41, we observe that increased surface roughness due to decreasing pore size amplifies the difference of the absolute adsorption amount differently for different adsorbate molecules. In particular, we find that adsorption of strongly interacting molecules can be enhanced in the low-pressure region while adsorption of weakly interacting molecules is inhibited. This then results in higher selectivity in binary mixture adsorption in mesoporous silica. read less USED (definite) M. Höhnerbach, A. Ismail, and P. Bientinesi, “The Vectorization of the Tersoff Multi-body Potential: An Exercise in Performance Portability,” SC16: International Conference for High Performance Computing, Networking, Storage and Analysis. 2016. link Times cited: 33 Abstract: Molecular dynamics simulations, an indispensable research to… read moreAbstract: Molecular dynamics simulations, an indispensable research tool in computational chemistry and materials science, consume a significant portion of the supercomputing cycles around the world. We focus on multi-body potentials and aim at achieving performance portability. Compared with well-studied pair potentials, multibody potentials deliver increased simulation accuracy but are too complex for effective compiler optimization. Because of this, achieving cross-platform performance remains an open question. By abstracting from target architecture and computing precision, we develop a vectorization scheme applicable to both CPUs and accelerators. We present results for the Tersoff potential within the molecular dynamics code LAMMPS on several architectures, demonstrating efficiency gains not only for computational kernels, but also for large-scale simulations. On a cluster of Intel Xeon Phi's, our optimized solver is between 3 and 5 times faster than the pure MPI reference. read less USED (definite) X. Zhang and G. Wu, “Effect of Strain on Thermal Conductivity of Si Thin Films,” Journal of Nanomaterials. 2016. link Times cited: 5 Abstract: Nonequilibrium molecular dynamics (NEMD) simulations are emp… read moreAbstract: Nonequilibrium molecular dynamics (NEMD) simulations are employed to gain an understanding of the effect of strain on the thermal conductivity of Si thin films. The analysis shows that the strain has an appreciable influence on the thermal conductivity of Si thin films. The thermal conductivity decreases as the tensile strain increases and increases as the compressive strain increases. The decrease of the phonon velocities and surface reconstructions generated under strain could explain well the effects of strain on the thermal conductivity of Si thin films. read less USED (definite) É. Maras, O. Trushin, A. Stukowski, T. Ala‐Nissila, and H. Jónsson, “Global transition path search for dislocation formation in Ge on Si(001),” Comput. Phys. Commun. 2016. link Times cited: 281 USED (definite) Z. Tong, Y. Liang, X. Yang, and X. Luo, “Investigation on the thermal effects during nanometric cutting process while using nanoscale diamond tools,” The International Journal of Advanced Manufacturing Technology. 2014. link Times cited: 0 USED (definite) L. Xie, P. Brault, J. Bauchire, A. Thomann, and L. Bedra, “Molecular dynamics simulations of clusters and thin film growth in the context of plasma sputtering deposition,” Journal of Physics D: Applied Physics. 2014. link Times cited: 40 Abstract: Carrying out molecular dynamics (MD) simulations is a releva… read moreAbstract: Carrying out molecular dynamics (MD) simulations is a relevant way to understand growth phenomena at the atomic scale. Initial conditions are defined for reproducing deposition conditions of plasma sputtering experiments. Two case studies are developed to highlight the implementation of MD simulations in the context of plasma sputtering deposition: ZrxCu1−x metallic glass and AlCoCrCuFeNi high entropy alloy thin films deposited onto silicon. Effects of depositing atom kinetic energies and atomic composition are studied in order to predict the evolution of morphologies and atomic structure of MD grown thin films. Experimental and simulated x-ray diffraction patterns are compared. read less USED (definite) H. Zhao et al., “Molecular Dynamics Simulation of the Crystal Orientation and Temperature Influences in the Hardness on Monocrystalline Silicon,” Journal of Nanomaterials. 2014. link Times cited: 17 Abstract: A nanoindentation simulation using molecular dynamic (MD) me… read moreAbstract: A nanoindentation simulation using molecular dynamic (MD) method was carried out to investigate the hardness behavior of monocrystalline silicon with a spherical diamond indenter. In this study, Tersoff potential was used to model the interaction of silicon atoms in the specimen, and Morse potential was used to model the interaction between silicon atoms in the specimen and carbon atoms in the indenter. Simulation results indicate that the silicon in the indentation zone undergoes phase transformation from diamond cubic structure to body-centred tetragonal and amorphous structure upon loading of the diamond indenter. After the unloading of the indenter, the crystal lattice reconstructs, and the indented surface with a residual dimple forms due to unrecoverable plastic deformation. Comparison of the hardness of three different crystal surfaces of monocrystalline silicon shows that the (0 0 1) surface behaves the hardest, and the (1 1 1) surface behaves the softest. As for the influence of the indentation temperature, simulation results show that the silicon material softens and adhesiveness of silicon increases at higher indentation temperatures. read less USED (definite) Y. H. Lin, S. Jian, Y. Lai, and P.-F. Yang, “Molecular Dynamics Simulation of Nanoindentation-induced Mechanical Deformation and Phase Transformation in Monocrystalline Silicon,” Nanoscale Research Letters. 2008. link Times cited: 42 USED (definite) “The Tersoff many-body potential: Sustainable performance through vectorization,” ArXiv. 2017. link Times cited: 0 Abstract: Molecular dynamics models materials by simulating each indiv… read moreAbstract: Molecular dynamics models materials by simulating each individual particle's trajectory. Many-body potentials lead to a more accurate trajectory simulation, and are used in materials science and computational chemistry. We present optimization results for one multi-body potential on a range of vector instruction sets, targeting both CPUs and accelerators like the Intel Xeon Phi. Parallelization of MD simulations is well-studied; by contrast, vectorization is relatively unexplored. Given the prevalence and power of modern vector units, exploiting them is imperative for high performance software. When running on a highly parallel machine, any improvement to the scalar performance is paid back in hundreds or thousands of saved core hours. Vectorization is already commonly used in the optimization or pair potentials; multi-body potentials pose new, unique challenges. Indeed, their optimization pushes the boundaries of current compilers, forcing us to use explicit vectorization techniques for now. In this study, we add an optimized implementation of Tersoff potential to the LAMMPS molecular dynamics simulation package. To reduce the burden of explicit vectorization, we abstract from the specific vector instruction set and desired precision: From one algorithm, we get optimized implementations for many platforms, from SSE4.2 to AVX512, and the Intel Xeon Phi. We compare the kernels across different architectures, and determine suitable architecture-dependent parameters. Our optimizations benefit any architecture, but have a disproportionate effect on the Intel Xeon Phi, which beats the CPU (2xE5-2650) after optimization. read less USED (high confidence) W. Xiong, W. Zhou, P. Sun, and S. Yuan, “Enhanced hydrogen-gas permeation through rippled graphene,” Physical Review B. 2023. link Times cited: 1 Abstract: The penetration of atomic hydrogen through defect-free graph… read moreAbstract: The penetration of atomic hydrogen through defect-free graphene was generally predicted to have a barrier of at least several eV, which is much higher than the 1 eV barrier measured for hydrogen-gas permeation through pristine graphene membranes. Herein, our density functional theory calculations show that ripples, which are ubiquitous in atomically thin crystals and mostly overlooked in the previous simulations, can significantly reduce the barriers for all steps constituting the mechanism of hydrogen-gas permeation through graphene membranes, including dissociation of hydrogen molecules, reconstruction of the dissociated hydrogen atoms and their flipping across graphene. Especially, the flipping barrier of hydrogen atoms from a cluster configuration is found to decrease rapidly down to<1 eV with increasing ripples' curvature. The estimated hydrogen permeation rates by fully considering the distribution of ripples with all realistic curvatures and the major reaction steps that occurred on them are quite close to the experimental measurements. Our work provides insights into the fundamental understanding of hydrogen-gas permeation through graphene membranes and emphasizes the importance of nanoscale non-flatness (ripples) in explaining many surface and transport phenomena (for example, functionalization, corrosion and separation) in graphene and other two-dimensional materials. read less USED (high confidence) R. Smeyers, M. Milošević, and L. Covaci, “Strong gate-tunability of flat bands in bilayer graphene due to moiré encapsulation between hBN monolayers.,” Nanoscale. 2022. link Times cited: 3 Abstract: When using hexagonal boron-nitride (hBN) as a substrate for … read moreAbstract: When using hexagonal boron-nitride (hBN) as a substrate for graphene, the resulting moiré pattern creates secondary Dirac points. By encapsulating a multilayer graphene within aligned hBN sheets the controlled moiré stacking may offer even richer benefits. Using advanced tight-binding simulations on atomistically-relaxed heterostructures, here we show that the gap at the secondary Dirac point can be opened in selected moiré-stacking configurations, and is independent of any additional vertical gating of the heterostructure. On the other hand, gating can broadly tune the gap at the principal Dirac point, and may thereby strongly compress the first moiré mini-band in width against the moiré-induced gap at the secondary Dirac point. We reveal that in hBN-encapsulated bilayer graphene this novel mechanism can lead to isolated bands flatter than 10 meV under moderate gating, hence presenting a convenient pathway towards electronically-controlled strongly-correlated states on demand. read less USED (high confidence) P. Liu, Q. Pei, and Y.-W. Zhang, “Failure modes and mechanisms of layered h-BN under local energy injection,” Scientific Reports. 2022. link Times cited: 1 USED (high confidence) Y. Zhan et al., “Boron-modified perhydropolysilazane towards facile synthesis of amorphous SiBN ceramic with excellent thermal stability,” Journal of Advanced Ceramics. 2022. link Times cited: 7 USED (high confidence) A. Giri, C. Dionne, and P. Hopkins, “Atomic coordination dictates vibrational characteristics and thermal conductivity in amorphous carbon,” npj Computational Materials. 2022. link Times cited: 9 USED (high confidence) K. Zongo, Béland, and O. ‐ Plamondon, “First-principles database for fitting a machine-learning silicon interatomic force field,” MRS Advances. 2022. link Times cited: 0 Abstract: Data-driven machine learning has emerged to address the limi… read moreAbstract: Data-driven machine learning has emerged to address the limitations of traditional methods when modeling interatomic interactions in materials, such as electronic density functional theory (DFT) and semi-empirical potentials. These machine-learning frameworks involve mathematical models coupled to quantum mechanical data. In the present article, we focus on the moment tensor potential (MTP) machine-learning framework. More specifically, we provide an account of the development of a preliminary MTP for silicon, including details pertaining to the construction of a DFT database. read less USED (high confidence) M. Z. Dehaghani et al., “Dynamics of Antimicrobial Peptide Encapsulation in Carbon Nanotubes: The Role of Hydroxylation,” International Journal of Nanomedicine. 2022. link Times cited: 10 Abstract: Introduction Carbon nanotubes (CNTs) have been widely employ… read moreAbstract: Introduction Carbon nanotubes (CNTs) have been widely employed as biomolecule carriers, but there is a need for further functionalization to broaden their therapeutic application in aqueous environments. A few reports have unraveled biomolecule–CNT interactions as a measure of response of the nanocarrier to drug-encapsulation dynamics. Methods Herein, the dynamics of encapsulation of the antimicrobial peptide HA-FD-13 (accession code 2L24) into CNTs and hydroxylated CNTs (HCNTs) is discussed. Results The van der Waals (vdW) interaction energy of CNT–peptide and HCNT–peptide complexes decreased, reaching −110.6 and −176.8 kcal.Mol−1, respectively, once encapsulation of the peptide inside the CNTs had been completed within 15 ns. The free energy of the two systems decreased to −43.91 and −69.2 kcal.Mol−1 in the same order. Discussion The peptide was encased in the HCNTs comparatively more rapidly, due to the presence of both electrostatic and vdW interactions between the peptide and HCNTs. However, the peptide remained encapsulated throughout the vdW interaction in both systems. The negative values of the free energy of the two systems showed that the encapsulation process had occurred spontaneously. Of note, the lower free energy in the HCNT system suggested more stable peptide encapsulation. read less USED (high confidence) M. Gatchell et al., “Survival of polycyclic aromatic hydrocarbon knockout fragments in the interstellar medium,” Nature Communications. 2021. link Times cited: 10 USED (high confidence) S. Kohara et al., “Relationship between diffraction peak, network topology, and amorphous-forming ability in silicon and silica,” Scientific Reports. 2021. link Times cited: 7 USED (high confidence) A. Albooyeh, A. Dadrasi, A. H. Mashhadzadeh, and M. Saeb, “Theory for designing mechanically stable single- and double-walled SiGe nanopeapods,” Journal of Molecular Modeling. 2021. link Times cited: 1 USED (high confidence) B. Chava, Y. Wang, and S. Das, “Boron Nitride Nanotube–Salt–Water Hybrid: Toward Zero-Dimensional Liquid Water and Highly Trapped Immobile Single Anions Inside One-Dimensional Nanostructures,” Journal of Physical Chemistry C. 2021. link Times cited: 3 USED (high confidence) M. Tong, Y. Jiang, L.-Y. Wang, C. Wang, and C. Tang, “Frictional characteristics of graphene layers with embedded nanopores,” Nanotechnology. 2021. link Times cited: 3 Abstract: Graphite possessing extraordinary frictional properties has … read moreAbstract: Graphite possessing extraordinary frictional properties has been widely used as solid lubricants. Interesting frictional characteristics have been observed for pristine graphene layers, for defective graphene, the frictional signal shows richer behaviors such as those found in topological defective graphene and graphene step edges. Recently discovered nanoporous graphene represents a new category of defect in graphene and its impact on graphene frictional properties has not yet been explored. In this work, we perform molecular dynamics simulations on the frictional responses of nanoporous graphene layers when slid using a silicon tip. We show that the buried nanopore raises maximum friction signal amplitude while preserving the stick-slip character, the size of the nanopore plays a key role in determining the maximum frictional force. Negative friction is observed when the silicon tip scanned towards the center of the nanopore, this phenomenon originates from the asymmetrical variation of the in-plane strain and the out-of-plane deformation when indented by the silicon tip. Moreover, the layer dependent frictional character is examined for the buried graphene nanopores, showing that increasing graphene layers weakens the effect of nanopore on the frictional signal. read less USED (high confidence) S. Guthrie, Y. Gao, K. Stone, B. Xu, and G. Giri, “Probing Molecular Assembly of Small Organic Molecules during Meniscus-Guided Coating Using Experimental and Molecular Dynamics Approaches,” Journal of Physical Chemistry C. 2021. link Times cited: 3 Abstract: Evaporation-based thin-film coating processes are ubiquitous… read moreAbstract: Evaporation-based thin-film coating processes are ubiquitous in fields such as organic electronics, dye coatings, and the pharmaceutical industry. Based on experimental observations of enhanced thi... read less USED (high confidence) N. B. C. Mansour, M.-L. Ouiten, A. Soldera, A. Szymczyk, and A. Ghoufi, “Static dielectric permittivity of ionic liquids ultraconfined in carbon nanotubes,” Nano Express. 2021. link Times cited: 1 Abstract: In this work the parallel component of the static dielectric… read moreAbstract: In this work the parallel component of the static dielectric permittivity, ϵII of ionic liquids ultraconfined into flexible carbon nanotubes of radius of 1.2 nm and 2.4 nm is evaluated from molecular dynamics simulations. We show an enhancement of ϵII with respect to bulk value and a counter-intuitive temperature dependence. Indeed an increase of ϵII as a function of the temperature opposed to a bulk behavior is evidenced. Increase in ϵII is the result of the strong orientation of ionic liquid close to the pore wall. The temperature dependence is the consequence of the thermal fluctuations increasing the dipolar fluctuations such that the strong orientation is conserved. Eventually, we show a molecular stacking between [C4mim+][Tf2N−] and CNT decreasing dipolar fluctuations close to the CNT surface reducing ϵII . read less USED (high confidence) I. Syuhada, N. Hauwali, A. Rosikhin, E. Sustini, F. A. Noor, and T. Winata, “Bond order redefinition needed to reduce inherent noise in molecular dynamics simulations,” Scientific Reports. 2021. link Times cited: 1 USED (high confidence) C. Woods et al., “Charge-polarized interfacial superlattices in marginally twisted hexagonal boron nitride,” Nature Communications. 2021. link Times cited: 0 USED (high confidence) M. Z. Dehaghani et al., “Insight into the Self-Insertion of a Protein Inside the Boron Nitride Nanotube,” ACS Omega. 2020. link Times cited: 20 Abstract: Nanotubes have been considered as promising candidates for p… read moreAbstract: Nanotubes have been considered as promising candidates for protein delivery purposes due to distinct features such as their large enough volume of cavity to encapsulate the protein, providing the sustain and target release. Moreover, possessing the properties of suitable cell viabilities, and biocompatibility on the wide range of cell lines as a result of structural stability, chemical inertness, and noncovalent wrapping ability, boron nitride nanotubes (BNNTs) have caught further attention as protein nanocarriers. However, to assess the encapsulation process of the protein into the BNNT, it is vital to comprehend the protein–BNNT interaction. In the present work, the self-insertion process of the protein SmtA, metallothionein, into the BNNT has been verified by means of the molecular dynamics (MD) simulation under NPT ensemble. It was revealed that the protein was self-inserted into the BNNT through the protein–BNNT van der Waals (vdW) interaction, which descended and reached the average value of −189.63 kcal·mol–1 at 15 ns of the simulation time. The potential mean force (PMF) profile of the encapsulated protein with increasing trend, which was obtained via the pulling process unraveled that the encapsulation of the protein into the BNNT cavity proceeded spontaneously and the self-inserted protein had reasonable stability. Moreover, due to the strong hydrogen interactions between the nitrogen atoms of BNNT and hydrogen atoms of SmtA, there was no evidence of an energy barrier in the vicinity of the BNNT entrance, which resulted in the rapid adsorption of this protein into the BNNT. read less USED (high confidence) E. Mareev, B. Rumiantsev, and F. Potemkin, “Study of the Parameters of Laser-Induced Shock Waves for Laser Shock Peening of Silicon,” JETP Letters. 2020. link Times cited: 6 USED (high confidence) H. G. Ozcelik, E. Satiroglu, and M. Barisik, “Size dependent influence of contact line pinning on wetting of nano-textured/patterned silica surfaces.,” Nanoscale. 2020. link Times cited: 8 Abstract: Wetting behavior on a heterogeneous surface undergoes contac… read moreAbstract: Wetting behavior on a heterogeneous surface undergoes contact angle hysteresis as the droplet stabilized at a metastable state with a contact angle significantly different from its equilibrium value due to contact line pinning. However, there is a lack of consensus on how to calculate the influence of pinning forces. In general, the pinning effect can be characterized as (i) microscopic behavior when a droplet is pinned and the contact angle increases/decreases as the droplet volume increases/decreases and (ii) macroscopic behavior as the pinning effects decrease and ultimately, disappear with the increase of the droplet size. The current work studied both behaviors using molecular dynamics (MD) simulation with more than 300 different size water droplets on silica surfaces with three different patterns across two different wetting conditions. Results showed that the contact angle increases linearly with increasing droplet volume through the microscopic behavior, while the droplet is pinned on top of a certain number of patterns. When we normalized the droplet size with the corresponding pattern size, we observed a "wetting similarity" that linear microscopic contact angle variations over different size heterogeneities continuously line up. This shows that the pinning force remains constant and the resulting pinning effects are scalable by the size ratio between the droplet and pattern, independent of the size-scale. The slope of these microscopic linear variations decreases with an increase in the droplet size as observed through the macroscopic behavior. We further found a universal behavior in the variation of the corresponding pinning forces, independent of the wetting condition. In macroscopic behavior, pinning effects become negligible and the contact angle reaches the equilibrium value of the corresponding surface when the diameter of the free-standing droplet is approximately equal to 24 times the size of the surface structure. We found that the pinning effect is scalable with the droplet volume, not the size of the droplet base. read less USED (high confidence) C. Woods et al., “Charge-polarized interfacial superlattices in marginally twisted hexagonal boron nitride,” Nature Communications. 2020. link Times cited: 113 USED (high confidence) M. Rahman, S. Mitra, M. Motalab, and P. Bose, “Investigation on the mechanical properties and fracture phenomenon of silicon doped graphene by molecular dynamics simulation,” RSC Advances. 2020. link Times cited: 17 Abstract: Silicon doping is an effective way to modulate the bandgap o… read moreAbstract: Silicon doping is an effective way to modulate the bandgap of graphene that might open the door for graphene to the semiconductor industries. However, the mechanical properties of silicon doped graphene (SiG) also plays an important role to realize its full potential application in the electronics industry. Electronic and optical properties of silicon doped graphene are well studied, but, our understanding of mechanical and fracture properties of the doped structure is still in its infancy. In this study, molecular dynamics (MD) simulations are conducted to investigate the tensile properties of SiG by varying the concentration of silicon. It is found that as the concentration of silicon increases, both fracture stress and strain of graphene reduces substantially. Our MD results also suggest that only 5% of silicon doping can reduce the Young's modulus of graphene by ∼15.5% along the armchair direction and ∼13.5% along the zigzag direction. Tensile properties of silicon doped graphene have been compared with boron and nitrogen doped graphene. The effect of temperature, defects and crack length on the stress–strain behavior of SiG has also been investigated. Temperature studies reveal that SiG is less sensitive to temperature compared to free stranding graphene, additionally, increasing temperature causes deterioration of both fracture stress and strain of SiG. Both defects and cracks reduce the fracture stress and fracture strain of SiG remarkably, but the sensitivity to defects and cracks for SiG is larger compared to graphene. Fracture toughness of pre-cracked SiG has been investigated and results from MD simulations are compared with Griffith's theory. It has been found that for nano-cracks, SiG with larger crack length deviates more from Griffith's criterion and the degree of deviation is larger compared to graphene. Fracture phenomenon of pre-cracked SiG and the effect of strain rate on the tensile properties of SiG have been reported as well. These results will aid the design of SiG based semiconducting nanodevices. read less USED (high confidence) S. I. Kundalwal, V. Choyal, N. Luhadiya, and V. Choyal, “Effect of carbon doping on electromechanical response of boron nitride nanosheets,” Nanotechnology. 2020. link Times cited: 17 Abstract: The electromechanical response of hexagonal-boron nitride na… read moreAbstract: The electromechanical response of hexagonal-boron nitride nanosheets (h-BNSs) was studied via molecular dynamics simulations (MDS) with a three-body Tersoff potential force field using a charge-dipole (C-D) potential model. Carbon (C)-doped h-BNSs with triangular, trapezoidal and circular pores were considered. The elastic and piezoelectric coefficients of h-BNSs under tension and shear loading conditions were determined. The induced polarization in h-BNSs was found to depend on the local arrangement of C atoms around B and N atoms, and the polarization increases if C atoms are surrounded by N atoms. This is attributed to the generation of higher dipole moments due to C–N bonds compared with C–B bonds. At ∼5.5% C-doping concentration, the axial piezoelectric coefficient of doped h-BNSs with triangular and trapezoidal pores increased by 18.5% and 3.5%, respectively, while it reduced by 22.5% in the case of circular pores compared to pristine h-BNS. The shear piezoelectric coefficient of C-doped h-BNSs with triangular and trapezoidal pores increased by 20.5% and 1%, respectively, while it reduced by 7% in case of circular pores. Young’s moduli of C-doped h-BNSs with triangular, trapezoidal and circular pores increased by 9%, 7.5% and 5.5%, respectively, due to the C–C bonds being stronger than all other bonds. The respective improvements in shear moduli are 8.5%, 5% and 5%. The elastic and piezoelectric properties of armchair h-BNSs were found to be higher than zigzag h-BNSs. The results also reveal that the piezoelectric coefficient increases as doping increases; it reaches its maximum value around 0.41 C m−2 at 12.6% C-doping concentration and then starts decreasing. The present work shows that we can engineer the electromechanical response of h-BNSs via novel pathways such as different types and size of pores as well as C-doping concentration to suit a particular nanoelectromechanical systems (NEMS) application. read less USED (high confidence) G. Guttormsen, A. Fletcher, and M. Oppenheim, “Atomic‐Scale Simulations of Meteor Ablation,” Journal of Geophysical Research: Space Physics. 2020. link Times cited: 2 Abstract: Meteoroids smaller than a microgram constantly bombard the E… read moreAbstract: Meteoroids smaller than a microgram constantly bombard the Earth, depositing material in the mesosphere and lower thermosphere. Meteoroid ablation, the explosive evaporation of meteoroids due to erosive impacts of atmospheric particles, consists of sputtering and thermal ablation. This paper presents the first atomic‐scale modeling of sputtering, the initial stage of ablation where hypersonic collisions between the meteoroid and atmospheric particles cause the direct ejection of atoms from the meteoroid surface. Because meteoroids gain thermal energy from these particle impacts, these interactions are important for thermal ablation as well. In this study, a molecular dynamics simulator calculates the energy distribution of the sputtered particles as a function of the species, velocity, and angle of the incoming atmospheric particles. The sputtering yield generally agrees with semi‐empirical equations at normal incidence but disagrees with the generally accepted angular dependence. Λ, the fraction of energy from a single atmospheric particle impact incorporated into the meteoroid, was found to be less than 1 and dependent on the velocity, angle, atmospheric species, and meteoroid material. Applying this new Λ to an ablation model results in a slower meteoroid temperature increase and mass loss rate as a function of altitude. This alteration results in changes in the expected electron line densities and visual magnitudes of meteoroids. Notably, this analysis leads to the prediction that meteoroids will generally ablate 1–4 km lower than previously predicted. This affects analysis of radar and visual measurements, as well as determination of meteoroid mass. read less USED (high confidence) A. Zare, P. Sedigh, and A. Montazeri, “Enhancing multi-functional capabilities of boron nitride nanosheets through defect engineering,” Journal of Materials Science. 2020. link Times cited: 2 USED (high confidence) R. Mukuno and M. Ishimaru, “Effects of temperatures on pressure-induced structural changes in amorphous Si: A molecular-dynamics study.” 2020. link Times cited: 0 Abstract: The structural changes of amorphous silicon (a-Si) under com… read moreAbstract: The structural changes of amorphous silicon (a-Si) under compressive pressure were examined by molecular-dynamics simulations using the Tersoff interatomic potential. aSi prepared by melt-quenching methods was pressurized up to 30 GPa under different temperatures (300K and 500K). The density of a-Si increased from 2.26 to 3.24 g/cm with pressure, suggesting the occurrence of the low-density to high-density amorphous phase transformation. This phase transformation occurred at the lower pressure with increasing the temperature, because the activation barrier for amorphous-to-amorphous phase transformation could be exceeded by thermal energy. The coordination number increased with pressure and time, and it was saturated at different values depending on the pressure. This suggested the existence of different metastable atomic configurations in a-Si. Atomic pair-distribution functions and bond-angle distribution functions suggested that the short-range ordered structure of high-density a-Si is similar to the structure of the high-pressure phase of crystalline Si (β-tin and Imma structures). read less USED (high confidence) C. Nguyen and A. Beskok, “Water desalination performance of h-BN and optimized charged graphene membranes,” Microfluidics and Nanofluidics. 2020. link Times cited: 12 USED (high confidence) C. Nguyen and A. Beskok, “Water desalination performance of h-BN and optimized charged graphene membranes,” Microfluidics and Nanofluidics. 2020. link Times cited: 0 USED (high confidence) H. Tafrishi, S. Sadeghzadeh, F. Molaei, and H. Siavoshi, “Investigating the effects of adding hybrid nanoparticles, graphene and boron nitride nanosheets, to octadecane on its thermal properties,” RSC Advances. 2020. link Times cited: 20 Abstract: Octadecane is an alkane that is used to store thermal energy… read moreAbstract: Octadecane is an alkane that is used to store thermal energy at ambient temperature as a phase change material. A molecular dynamics study was conducted to investigate the effects of adding graphene and a boron nitride nanosheet on the thermal and structural properties of octadecane paraffin. The PCFF force field for paraffin, AIREBO potential for graphene, Tersoff potential for the boron nitride nanosheet, and Lennard-Jones potential for the van der Waals interaction between the nanoparticles and n-alkanes were used. Equilibrium and nonequilibrium molecular dynamics simulations were used to study the nano-enhanced phase change material properties. Results showed that the nanocomposite had a lower density change, more heat capacity (except at 300 K), more thermal conductivity, and a lower diffusion coefficient in comparison with pure paraffin. Additionally, the nanocomposite had a higher melting point, higher phonon density of state and radial distribution function peaks. read less USED (high confidence) Y. Dong, Y. Tao, R. Feng, Y. Zhang, Z. Duan, and H. Cao, “Phonon dissipation in friction with commensurate–incommensurate transition between graphene membranes,” Nanotechnology. 2020. link Times cited: 12 Abstract: To examine phonon transport during the friction process of c… read moreAbstract: To examine phonon transport during the friction process of commensurate–incommensurate transition, the vibrational density of states of contact surfaces is calculated based on molecular dynamics simulations. The results indicate that, compared with the static state, the relative sliding of the contact surfaces causes a blue shift in the interfacial phonon spectrum in or close to commensurate contact, whereas the contrast of the phonon spectrum in incommensurate contact is almost indiscernible. Further findings suggest that the cause of friction can be attributed to the excitation of new in-plane acoustic modes, which provide the most efficient energy dissipation channels in the friction process. In addition, when the tip and the substrate are subjected to a same biaxial compressive/tensile strain, fewer new acoustic modes are excited than in the no strain case. Thus, the friction can be controlled by applying in-plane strain even in commensurate contact. The contribution of the excited acoustic modes to friction at various frequency bands is also calculated, which provides theoretical guidance for controlling friction by adjusting excitation phonon modes. read less USED (high confidence) N. Takahashi, Y. Liu, and C. Kaneta, “Materials informatics approach for design of Si/Ge layered nanostructures with low thermal conductivity,” Japanese Journal of Applied Physics. 2020. link Times cited: 0 Abstract: We report an efficient method based on the materials informa… read moreAbstract: We report an efficient method based on the materials informatics approach to predict low thermal conductivity structures using a descriptor obtained by phonon mode calculations. For a small number of Si/Ge layered structures, we perform normal mode analysis to calculate the participation ratio for each phonon mode and calculations of thermal conductivity in the stacking direction using the perturbed molecular dynamics (MD) method. The descriptor for the thermal conductivity is defined using the participation ratios averaged in the acoustic phonon frequency ranges with their lower and upper limits independently optimized. By employing the descriptor and genetic algorithm, low thermal conductivity structures are recommended among a huge number of possible structures. The recommended structures are confirmed to have very small thermal conductivities from the results of the perturbed MD calculations. By employing the method, we can design Si/Ge layered structures with low thermal conductivity at very low computational cost. read less USED (high confidence) H. Ozcelik, A. Ozdemir, B. H. Kim, and M. Barisik, “Wetting of single crystalline and amorphous silicon surfaces: effective range of intermolecular forces for wetting,” Molecular Simulation. 2020. link Times cited: 11 Abstract: ABSTRACT Wetting at nanoscale is a property of a three-dimen… read moreAbstract: ABSTRACT Wetting at nanoscale is a property of a three-dimensional region with a finite length into the solid domain from the surface. Understanding the extent of the solid region effective on wetting is important for recent coating applications as well as for both crystalline and amorphous solids of different atomic ordering. For such a case, we studied the wetting behaviour of silicon surfaces at various crystalline and amorphous states. Molecular distributions of amorphous systems were varied by changing the amorphisation conditions of silicon. Semi-cylindrical water droplets were formed on the surfaces to be large enough to remain independent of line tension and Tolman length effects. Contact angles showed up to 38% variation by the change in the atomic orientation of silicon. Instead of a homogeneous solid density definition, we calculated different solid densities for a given surface measured inside different extents from the interface. We correlated the observed wetting variation with each of these different solid densities to determine which extent governs the wetting variation. We observed that the variation of solid density measured inside a 0.13 nm extent from the surface reflected the variation of wetting angle better for both single crystalline and amorphous silicon surfaces. read less USED (high confidence) M. Li, Z. Guo, and T. Chang, “Adhesion and stress-enhanced elastocaloric effect in graphene,” Science China Technological Sciences. 2020. link Times cited: 5 USED (high confidence) E. Hodille, J. Byggmästar, E. Safi, and K. Nordlund, “Sputtering of beryllium oxide by deuterium at various temperatures simulated with molecular dynamics,” Physica Scripta. 2020. link Times cited: 12 Abstract: The sputtering yield of beryllium oxide (BeO) by incident de… read moreAbstract: The sputtering yield of beryllium oxide (BeO) by incident deuterium (D) ions, for energies from 10 eV to 200 eV, has been calculated for temperatures between 300 K and 800 K using classical molecular dynamics. First, cumulative irradiations are carried out to build up a concentration of D in the material, equal to the experimentally measured concentration, that varies from an atomic fraction of 0.12 (300 K–500 K) to 0.02 (800 K). After building up the concentration of D, non-cumulative irradiations are carried out to estimate the sputtering yields of BeO. For all incident energies, the sputtering yield peaks at 500 K, being closely related to the decrease of the concentration of D above this temperature. At 10 eV, the concentration of D on the surface drives the temperature dependence, while above 30 eV, it is the amount of surface damage created during the cumulative irradiation. read less USED (high confidence) D. Bruns, A. Nojeh, A. Phani, and J. Rottler, “Heat transport in carbon nanotubes: Length dependence of phononic conductivity from the Boltzmann transport equation and molecular dynamics,” Physical Review B. 2019. link Times cited: 10 Abstract: In this article, we address lattice heat transport in single… read moreAbstract: In this article, we address lattice heat transport in single-walled carbon nanotubes (CNTs) by a quantum mechanical calculation of three-phonon scattering rates in the framework of the Boltzmann transport equation (BTE) and classical molecular dynamics (MD) simulation. Under a consistent choice of an empirical, realistic atomic interaction potential, we compare the tube length dependence of the lattice thermal conductivity (TC) at room temperature determined from an iterative solution of the BTE and from a nonequilibrium MD (NEMD) approach. Qualitatively similar trends are found in the limit of short tubes, where an extensive regime of ballistic heat transport prevailing in CNTs of lengths $L\lesssim 1\,\rm{\mu m}$ is independently confirmed. In the limit of long tubes, the BTE approach suggests a saturation of TC with tube length, whereas direct NEMD simulations of tubes extending up to $L=10\,\rm{\mu m}$ are demonstrated to be insufficient to settle the question of whether a fully diffusive heat transport regime and an intrinsic value of TC exist for CNTs. Noting that acoustic phonon lifetimes lie at the heart of a saturation of TC with tube length as per the BTE framework, we complement the quantum mechanical prediction of acoustic phonon lifetimes with an analysis of phonon modes in the framework of equilibrium MD (EMD). A normal mode analysis (NMA) with an emphasis on long wavelength acoustic modes corroborates the BTE prediction that heat transport in CNTs in the long tube limit is governed by the low attenuation rates of longitudinal and twisting phonons. read less USED (high confidence) A. Tlili, V. Giordano, Y. Beltukov, P. Desmarchelier, S. Merabia, and A. Tanguy, “Enhancement and anticipation of the Ioffe-Regel crossover in amorphous/nanocrystalline composites.,” Nanoscale. 2019. link Times cited: 13 Abstract: Nanocomposites made of crystalline nanoinclusions embedded i… read moreAbstract: Nanocomposites made of crystalline nanoinclusions embedded in an amorphous matrix are at the forefront of current research for energy harvesting applications. However, the microscopic mechanisms leading alternatively to an effectively reduced or enhanced thermal transport still escape understanding. In this work, we present a molecular dynamics simulation study of model systems, where for the first time we combine a microscopic investigation of phonon dynamics with the macroscopic thermal conductivity calculation, to shed light on thermal transport in these materials. We clearly show that crystalline nanoinclusions represent a novel scattering source for vibrational waves, modifying the nature of low energy vibrations and significantly anticipating the propagative-to-diffusive crossover (Ioffe-Regel), usually located at energies of few THz in amorphous materials. Moreover, this crossover position can be tuned by changing the elastic contrast between nanoinclusions and the matrix, and anticipated by a factor as large as 10 for a harder inclusion. While the propagative contribution to thermal transport is drastically reduced, the calculated thermal conductivity is not significantly affected in the chosen system, as the diffusive contribution dominates heat transport when all phonons are thermally populated. These findings allow finally to understand the panoply of contradictory results reported on thermal transport in nanocomposites and give clear indications to the characteristics that the parent phases should have for efficiently reducing heat transport in a nanocomposite. read less USED (high confidence) P. Novikov, A. Dvurechenskii, and K. Pavsky, “Energy Surface of Pit-Patterned Templates for Growth of Space-Arranged Arrays of Quantum Dots – Molecular Dynamics Calculations Using High-Efficiency Algorithms,” 2019 International Multi-Conference on Industrial Engineering and Modern Technologies (FarEastCon). 2019. link Times cited: 0 Abstract: Using MD simulation the energy surface of pit-patterned Si s… read moreAbstract: Using MD simulation the energy surface of pit-patterned Si substrate is obtained. The physical model is based on empirical Tersoff potential, which governs the dynamics of an atomistic crystal system including Si and Ge atoms. The pits at the surface of Si substrate assume the shape of inverted truncated pyramid. The analysis of the energy surface mapped by MD calculations shows that the dense space-arranged array of nanoislands with up to 4 nanoislands per pit may be grown by the deposition of Ge on Si pit-patterned substrate. The calculations were carried using algorithm of parallel programming. The scheme of the parallel algorithm for Verlet neighbor list formation is suggested. The results of calculations using parallel algorithm are presented in form of the speedup depending on the cores number. read less USED (high confidence) X. Zhou et al., “Kinking effects and transport properties of coaxial BN-C nanotubes as revealed by in situ transmission electron microscopy and theoretical analysis,” APL Materials. 2019. link Times cited: 0 Abstract: The insights into transport behavior and the effects of bend… read moreAbstract: The insights into transport behavior and the effects of bending on heterostructures constructed from boron nitride (BN) and carbon (C) nanotubes are important for their flexible device applications because the two systems have equally excellent mechanical but completely different electrical properties. In this work, coaxial BN–C nanotubes have been fabricated and their intrinsic transport properties, as well as structural and electrical response to bending deformation, are studied inside a high-resolution transmission electron microscope. Ballistic, diffusive, and hopping transports within different tube length ranges have been observed. When bending deformation was applied to the tubes, although severe kinking becomes apparent, their transport properties are not notably affected. Meanwhile, both theoretical and experimental analyses confirm that the kink positions depend on the ratio of tube diameter to its length. Possible formation of quantum dots, directly within the kink areas, was predicted through calculations of electron density redistribution between nanotube walls at bending.The insights into transport behavior and the effects of bending on heterostructures constructed from boron nitride (BN) and carbon (C) nanotubes are important for their flexible device applications because the two systems have equally excellent mechanical but completely different electrical properties. In this work, coaxial BN–C nanotubes have been fabricated and their intrinsic transport properties, as well as structural and electrical response to bending deformation, are studied inside a high-resolution transmission electron microscope. Ballistic, diffusive, and hopping transports within different tube length ranges have been observed. When bending deformation was applied to the tubes, although severe kinking becomes apparent, their transport properties are not notably affected. Meanwhile, both theoretical and experimental analyses confirm that the kink positions depend on the ratio of tube diameter to its length. Possible formation of quantum dots, directly within the kink areas, was predicted through ... read less USED (high confidence) T. Riedl, V. Kunnathully, A. Trapp, T. Langer, D. Reuter, and J. Lindner, “Strain-driven InAs island growth on top of GaAs(111) nanopillars,” Physical Review Materials. 2019. link Times cited: 2 Abstract: We analyze the shape and position of heteroepitaxial InAs is… read moreAbstract: We analyze the shape and position of heteroepitaxial InAs islands on the top face of cylindrical GaAs(111)A nanopillars experimentally and theoretically. Catalyst-free molecular beam epitaxial growth of InAs at low temperatures on GaAs nanopillars results in InAs islands with diameters < 30 nm exhibiting predominantly rounded triangular in-plane shapes. The islands show a tendency to grow at positions displaced from the center towards the pillar edge. Atomistic molecular statics simulations evidence that triangular-prismatic islands centered to the pillar axis with diameters smaller than that of the nanopillars are energetically favored. Moreover, we reveal the existence of minimum-energy states for off-axis island positions, in agreement with the experiment. These findings are interpreted by evaluating the spatial strain distributions and the number of broken bonds of surface atoms as a measure for the surface energy. The preferred off-axis island positions can be understood in terms of an increased compliancy of the GaAs nanopillar beneath the island because of the vicinity of free surfaces, leading to a reduction of strain energy. The influence of surface steps on the energy of the system is addressed as well. read less USED (high confidence) K. Toprak and G. Ersavaş, “Molecular Dynamics Study of the Thermal Conductivity of Graphene Coated Copper,” Proceedings of the 5th World Congress on Mechanical, Chemical, and Material Engineering. 2019. link Times cited: 0 Abstract: In this study, the thermal conductivity of various size of p… read moreAbstract: In this study, the thermal conductivity of various size of pure copper, pure graphene and, different number of layer graphene coated copper models are studied using non-equilibrium molecular dynamics (NEMD) simulations. Our findings show that the thermal conductivity of graphene coated copper is higher than the uncoated ones. Furthermore, results also indicate that single layer graphene (SLG) model has the highest thermal conductivity as compared to the other model. Even though multiple layer graphene (MLG) has lower thermal conductivity value compare to SLG, this study shows that the thermal conductivity of MLG coated copper has higher thermal conductivity than SLG coated one. The most important finding in this study suggests that the thermal conductivity of copper can be improved using high thermal conductivity materials like graphene. read less USED (high confidence) M. Li, Z. Guo, and T. Chang, “Adhesion and stress-enhanced elastocaloric effect in graphene,” Science China Technological Sciences. 2019. link Times cited: 0 USED (high confidence) F. Liu et al., “Enhancement of thermal energy transport across the graphene/h-BN heterostructure interface.,” Nanoscale. 2019. link Times cited: 17 Abstract: Enhancing thermal energy transport is critical for the appli… read moreAbstract: Enhancing thermal energy transport is critical for the applications of 2-dimensional materials. Here, we explored the methods of enhancing the interfacial thermal energy transport across the graphene (GR)/hexagonal boron nitride (h-BN) heterostructure interface, and revealed the enhancement mechanisms of interfacial thermal energy transport by applying non-equilibrium molecular dynamics (NEMD) simulations. The computational results indicated that both doping and interface topography optimization could effectively improve the interfacial thermal conductance (ITC) of the GR/h-BN heterostructure. In particular, the enhancement of the zigzag interface topography led to a much better result than the other methods. Doping and interface topography optimization increased the overlap of the phonon density of states (PDOS). Temperature had a negligible effect on the ITC of the GR/h-BN heterostructure when the temperature exceeded 600 K. read less USED (high confidence) A. Senturk, A. Oktem, and A. E. S. Konukman, “An investigation on the thermo-mechanical properties of boron-doped g-C3N4,” Applied Physics A. 2019. link Times cited: 11 USED (high confidence) T. Jiang, Z. Wang, X. Ruan, and Y. Zhu, “Equi-biaxial compressive strain in graphene: Grüneisen parameter and buckling ridges,” 2D Materials. 2018. link Times cited: 19 Abstract: Strain and defects in graphene have critical impact on morph… read moreAbstract: Strain and defects in graphene have critical impact on morphology and properties of graphene. Here we report equi-biaxial compressive strain in monolayer graphene on SiO2 and Si3N4 substrates induced by thermal cycling in vacuum. The equi-biaxial strain is attributed to the mismatch in coefficient of thermal expansion between graphene and the substrate and sliding of graphene on the substrate. The sliding occurs during heating at the temperatures of 390 and 360 K for graphene on SiO2 and Si3N4 substrates, respectively. The biaxial Grüneisen parameter is determined to be 1.95 and 3.15 for G and 2D Raman bands of graphene, respectively. As the heating temperature exceeds a threshold temperature (1040 K for graphene/SiO2 and 640 K for graphene/Si3N4), buckling ridges are observed in graphene after the thermal cycle, from which the biaxial buckling strain of graphene on SiO2 and Si3N4 substrates are obtained as 0.21% and 0.22%, respectively. Importantly, the induced buckling ridges in graphene exhibit a pattern representing the symmetry of graphene crystal structure, which indicates that graphene relieves the compressive stress mainly along its lattice symmetry directions. These thermally induced graphene ridges are also found reminiscent of those in the synthesized graphene, suggesting the same origin of formation of the buckling ridges under biaxial compression. read less USED (high confidence) G. C. Loh, “Fast water desalination by carbon-doped boron nitride monolayer: transport assisted by water clustering at pores,” Nanotechnology. 2018. link Times cited: 6 Abstract: The well-being of the ever-escalating world population hinge… read moreAbstract: The well-being of the ever-escalating world population hinges largely upon the adequacy of clean, fresh water. Desalination is one of the most promising approaches in such an endeavor. Using molecular dynamics simulations, we take a close look at nanoporous hexagonal boron nitride nanosheets as desalination membranes, and study how C dopants affect their performance. The calculations predict that the desalination performance of C-doped BN membranes compares favorably to that of MoS2 membranes: the water flux through the 0% (0CB–0CN), 25% (3CB–0CN), 75% (3CB–6CN), and 100% C terminated BN membrane (6CB–6CN) is 29.9, 47.5, 95.3, and 81.5 molecules ns−1 per pore, respectively, and there is a strong correlation between the water flux and the axial diffusion coefficient. Through our study of the effect of C content on the desalination performance, it is found that more clustering of water molecules at membrane pores due to a smaller hydration free energy and pore energy barrier assists water transport through the pores, and allows a greater water flux. read less USED (high confidence) M. Dewapriya and R. Rajapakse, “MD Simulation of Elastic Field at an Inhomogeneity in Graphene,” 2018 4th IEEE International Conference on Emerging Electronics (ICEE). 2018. link Times cited: 1 Abstract: A comprehensive molecular dynamics study is conducted to inv… read moreAbstract: A comprehensive molecular dynamics study is conducted to investigate the elastic field at an atomic inhomogeneity in graphene in the form of a circular hole or a circular boron-nitride inclusion. In addition, the effect on the stress field due to the interaction between an inhomogeneity and a crack is investigated. The results confirm that consideration must be given to the mechanical properties of the resulting system when atomic defects and inclusions are introduced to graphene to tailor optical and electronic properties. read less USED (high confidence) X. Zhang, C. Gong, and G. Wu, “Effects of Strains on Thermal Conductivity of Si/Ge Superlattices,” Journal of Wuhan University of Technology-Mater. Sci. Ed. 2018. link Times cited: 1 USED (high confidence) G. Mallick and R. Elder, “Graphene/hexagonal boron nitride heterostructures: Mechanical properties and fracture behavior from nanoindentation simulations,” Applied Physics Letters. 2018. link Times cited: 16 Abstract: In-plane or vertically stacked heterostructures containing m… read moreAbstract: In-plane or vertically stacked heterostructures containing multiple 2D materials are promising for emerging applications, such as flexible electronics, piezoelectric sensors, and molecular separations. However, utilizing heterostructures requires a fundamental understanding of their mechanics, which is currently lacking. Here, we use reactive molecular dynamics to simulate nanoindentation of stacked hexagonal boron nitride (h-BN) and graphene structures, 2D materials with similar structures but differing electronic properties. We calculate the Young's modulus, bending rigidity, ultimate strength, and the fracture strain of monolayers, homogeneous and heterogeneous bilayers, and alternating trilayers. Their mechanics are broadly similar, although graphene provides mild reinforcement to heterostructures. Further, we characterize the puncture created by nanoindentation, where we find that graphene allows smaller pores with a rougher fracture surface and more cleaved bonds than h-BN, which we attribute to differences in toughness. Our results demonstrate that these layered heterostructures maintain their mechanical robustness regardless of stacking order and provide insight into the influence of layer ordering in separation or passivation applications.In-plane or vertically stacked heterostructures containing multiple 2D materials are promising for emerging applications, such as flexible electronics, piezoelectric sensors, and molecular separations. However, utilizing heterostructures requires a fundamental understanding of their mechanics, which is currently lacking. Here, we use reactive molecular dynamics to simulate nanoindentation of stacked hexagonal boron nitride (h-BN) and graphene structures, 2D materials with similar structures but differing electronic properties. We calculate the Young's modulus, bending rigidity, ultimate strength, and the fracture strain of monolayers, homogeneous and heterogeneous bilayers, and alternating trilayers. Their mechanics are broadly similar, although graphene provides mild reinforcement to heterostructures. Further, we characterize the puncture created by nanoindentation, where we find that graphene allows smaller pores with a rougher fracture surface and more cleaved bonds than h-BN, which we attribute to dif... read less USED (high confidence) R. K. Defo, X. Zhang, D. O. Bracher, G. Kim, E. Hu, and E. Kaxiras, “Energetics and kinetics of vacancy defects in
4H
-SiC,” Physical Review B. 2018. link Times cited: 21 Abstract: Defect engineering in wide-gap semiconductors is important i… read moreAbstract: Defect engineering in wide-gap semiconductors is important in controlling the performance of single-photon emitter devices. The effective incorporation of defects depends strongly on the ability to control their formation and location, as well as to mitigate attendant damage to the material. In this study, we combine density functional theory (DFT), molecular dyamics (MD), and kinetic Monte Carlo (kMC) simulations to study the energetics and kinetics of the silicon monovacancy (VSi) and related defects in 4H-silicon carbide (SiC). We obtain the defect formation energy for VSi in various charge states and use MD simulations to model the ion implantation process for creating defects. We also study the effects of high-temperature annealing on defect position and stability using kMC and analytical models. Using a larger (480-atom) supercell than previous studies, we obtain the temperature-dependent diffusivity of VSi in various charge states and find significantly lower barriers to diffusion than previous estimates. In addition, we examine the recombination with interstitial Si and conversion of VSi into CSiVC during annealing, and propose methods for using strain to reduce changes in defect concentrations. Our results provide guidance for experimental efforts to control the position and density of VSi defects within devices, helping realize their potential as solid-state qubits. read less USED (high confidence) K. Garrity, “Combined cluster and atomic displacement expansion for solid solutions and magnetism,” Physical Review B. 2018. link Times cited: 4 Abstract: Finite temperature disordered solid solutions and magnetic m… read moreAbstract: Finite temperature disordered solid solutions and magnetic materials are difficult to study directly using first principles calculations, due to the large unit cells and many independent samples that are required. In this work, we develop a combined cluster expansion and atomic displacement expansion, which we fit to first principles energies, forces, and stresses. We then use the expansion to calculate thermodynamic quantities at nearly first principles levels of accuracy. We demonstrate that by treating all the relevant degrees of freedom explicitly, we can achieve improved convergence properties as compared to a simple cluster expansion, and our model naturally includes both configurational and vibrational entropy. In addition, we can treat coupling between structural and chemical or magnetic degrees of freedom. As examples, we use our expansion to calculate properties of Si$_{1-x}$Ge$_x$, magnetic MnO, Al with vacancies, and Ba$_x$Sr$_{1-x}$TiO$_3$. read less USED (high confidence) A. Giri and P. Hopkins, “Giant reduction and tunability of the thermal conductivity of carbon nanotubes through low-frequency resonant modes,” Physical Review B. 2018. link Times cited: 13 Abstract: Manipulating thermal transport by designing materials with c… read moreAbstract: Manipulating thermal transport by designing materials with control of their properties over the entire spectral range of vibrational frequencies would provide a unique path to create solids with designer thermal conductivities. Traditional routes of nanostructuring to reduce the vibrational thermal conductivity of solids typically target narrow bands of the vibrational energy spectrum, which is often based on the characteristic dimensions of the nanostructure. In this work, we demonstrate the ability to simultaneously impact the phonon transport of both highand low-frequency modes by creating defects that act as both high-frequency phonon scattering sites while coherently manipulating low-frequency waves via resonance with the long wavelength phonons. We use atomistic simulations to identify fullerenes functionalized on the sidewalls of carbon nanotubes (CNT) as efficient phonon blocks realized through localized resonances that appear due to hybridization between the modes of the fullerene and the underlying CNT. We show that with a large surface coverage and high periodicity in the inclusion of the covalently bonded fullerenes, the thermal conductivity of individual CNTs can be lowered by more than an order of magnitude, thus providing a large tunability in the thermal transport across these materials. We prescribe the large reduction in thermal conductivity to a combination of resonant phonon localization effects leading to phonon band anticrossings and vibrational scattering effects due to the inclusion of the strongly bonded fullerene molecules. read less USED (high confidence) J. A. Stewart et al., “Characterizing single isolated radiation-damage events from molecular dynamics via virtual diffraction methods,” Journal of Applied Physics. 2018. link Times cited: 18 Abstract: The evolution and characterization of single-isolated-ion-st… read moreAbstract: The evolution and characterization of single-isolated-ion-strikes are investigated by combining atomistic simulations with selected-area electron diffraction (SAED) patterns generated from these simulations. Five molecular dynamics simulations are performed for a single 20 keV primary knock-on atom in bulk crystalline Si. The resulting cascade damage is characterized in two complementary ways. First, the individual cascade events are conventionally quantified through the evolution of the number of defects and the atomic (volumetric) strain associated with these defect structures. These results show that (i) the radiation damage produced is consistent with the Norgett, Robinson, and Torrens model of damage production and (ii) there is a net positive volumetric strain associated with the cascade structures. Second, virtual SAED patterns are generated for the resulting cascade-damaged structures along several zone axes. The analysis of the corresponding diffraction patterns shows the SAED spots approximately doubling in size, on average, due to broadening induced by the defect structures. Furthermore, the SAED spots are observed to exhibit an average radial outward shift between 0.33% and 0.87% depending on the zone axis. This characterization approach, as utilized here, is a preliminary investigation in developing methodologies and opportunities to link experimental observations with atomistic simulations to elucidate microstructural damage states.The evolution and characterization of single-isolated-ion-strikes are investigated by combining atomistic simulations with selected-area electron diffraction (SAED) patterns generated from these simulations. Five molecular dynamics simulations are performed for a single 20 keV primary knock-on atom in bulk crystalline Si. The resulting cascade damage is characterized in two complementary ways. First, the individual cascade events are conventionally quantified through the evolution of the number of defects and the atomic (volumetric) strain associated with these defect structures. These results show that (i) the radiation damage produced is consistent with the Norgett, Robinson, and Torrens model of damage production and (ii) there is a net positive volumetric strain associated with the cascade structures. Second, virtual SAED patterns are generated for the resulting cascade-damaged structures along several zone axes. The analysis of the corresponding diffraction patterns shows the SAED spots approximately... read less USED (high confidence) A. Abramyan, N. Bessonov, L. V. Mirantsev, and A. Chevrychkina, “Equilibrium structures and flows of polar and nonpolar liquids in different carbon nanotubes,” The European Physical Journal B. 2018. link Times cited: 5 USED (high confidence) B. Cao, J.-H. Zou, G. Hu, and G. Cao, “Enhanced thermal transport across multilayer graphene and water by interlayer functionalization,” Applied Physics Letters. 2018. link Times cited: 61 Abstract: Graphene has attracted enormous attention due to its extraor… read moreAbstract: Graphene has attracted enormous attention due to its extraordinary physical properties, which have potential for increasing the thermal conductivity of nanocomposites or nanofluids, and the thermal resistance between graphene and the surrounding matrices arises as an important issue. In this paper, the thermal transport at the graphene-water interface is investigated by molecular dynamics simulations. The interfacial thermal resistance decreases with the graphene layer number. Interlayer functionalization by oxygen atoms is applied to tune the interfacial thermal resistance. A peak thermal resistance reduction of nearly 50% is generated with the oxygen ratio of only 0.5% for two-layer graphene. Based on the analyses of vibrational density of states, it is found that lower thermal resistance is consistent with more vibrational density of states overlaps at the interface. Our results are instructive for improving the interfacial thermal transport in graphene-based nanocomposites and nanofluids. read less USED (high confidence) A. Vahedi and M. H. S. Lahidjani, “Tunable thermal conductivity along graphene/hexagonal boron-nitride polycrystalline heterostructures,” The European Physical Journal Plus. 2017. link Times cited: 10 USED (high confidence) L. Wang, H. Ke, J. Ma, and J. Liu, “Investigation of the ‘double cross’ splitting mechanism of single-crystal diamond under nanoindentation via molecular dynamics simulation,” Journal of Molecular Modeling. 2017. link Times cited: 3 USED (high confidence) H. Ko, I. Szlufarska, and D. Morgan, “Cs diffusion in SiC high-energy grain boundaries,” arXiv: Materials Science. 2017. link Times cited: 3 Abstract: Cesium (Cs) is a radioactive fission product whose release i… read moreAbstract: Cesium (Cs) is a radioactive fission product whose release is of concern for Tristructural-Isotropic (TRISO) fuel particles. In this work, Cs diffusion through high energy grain boundaries (HEGBs) of cubic-SiC is studied using an ab-initio based kinetic Monte Carlo (kMC) model. The HEGB environment was modeled as an amorphous SiC (a-SiC), and Cs defect energies were calculated using density functional theory (DFT). From defect energies, it was suggested that the fastest diffusion mechanism as Cs interstitial in an amorphous SiC. The diffusion of Cs interstitial was simulated using a kMC, based on the site and transition state energies sampled from the DFT. The Cs HEGB diffusion exhibited an Arrhenius type diffusion in the range of 1200-1600{\deg}C. The comparison between HEGB results and the other studies suggests not only that the GB diffusion dominates the bulk diffusion, but also that the HEGB is one of the fastest grain boundary paths for the Cs diffusion. The diffusion coefficients in HEGB are clearly a few orders of magnitude lower than the reported diffusion coefficients from in- and out-of- pile samples, suggesting that other contributions are responsible, such as a radiation enhanced diffusion. read less USED (high confidence) D. K. Das and M. Ghosh, “On Mechanical Properties of Graphene Sheet Estimated Using Molecular Dynamics Simulations,” Journal of Materials Engineering and Performance. 2017. link Times cited: 2 USED (high confidence) L. Giacomozzi et al., “Knockout driven fragmentation of porphyrins.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 2 Abstract: We have studied collisions between tetraphenylporphyrin cati… read moreAbstract: We have studied collisions between tetraphenylporphyrin cations and He or Ne at center-of-mass energies in the range 50-110 eV. The experimental results were interpreted in view of density functional theory calculations of dissociation energies and classical molecular dynamics simulations of how the molecules respond to the He/Ne impact. We demonstrate that prompt atom knockout strongly contributes to the total destruction cross sections. Such impulse driven processes typically yield highly reactive fragments and are expected to be important for collisions with any molecular system in this collision energy range, but have earlier been very difficult to isolate for biomolecules. read less USED (high confidence) P. Hirvonen, Z. Fan, M. Ervasti, A. Harju, K. Elder, and T. Ala‐Nissila, “Energetics and structure of grain boundary triple junctions in graphene,” Scientific Reports. 2017. link Times cited: 19 USED (high confidence) U. Khalilov et al., “Nanoscale mechanisms of CNT growth and etching in plasma environment,” Journal of Physics D: Applied Physics. 2017. link Times cited: 14 Abstract: Plasma-enhanced chemical deposition (PECVD) of carbon nanotu… read moreAbstract: Plasma-enhanced chemical deposition (PECVD) of carbon nanotubes has already been shown to allow chirality control to some extent. In PECVD, however, etching may occur simultaneously with the growth, and the occurrence of intermediate processes further significantly complicates the growth process. We here employ a computational approach with experimental support to study the plasma-based formation of Ni nanoclusters, Ni-catalyzed CNT growth and subsequent etching processes, in order to understand the underpinning nanoscale mechanisms. We find that hydrogen is the dominant factor in both the re-structuring of a Ni film and the subsequent appearance of Ni nanoclusters, as well as in the CNT nucleation and etching processes. The obtained results are compared with available theoretical and experimental studies and provide a deeper understanding of the occurring nanoscale mechanisms in plasma-assisted CNT nucleation and growth. read less USED (high confidence) S. Tan, T. Xia, Y. Shi, J. Pfaendtner, S. Zhao, and Y. He, “Enhancing the Oxidation of Toluene with External Electric Fields: a Reactive Molecular Dynamics Study,” Scientific Reports. 2017. link Times cited: 23 USED (high confidence) Y. Furukawa and Y. Matsushita, “First-Principles Prediction of Densities of Amorphous Materials: The Case of Amorphous Silicon,” Journal of the Physical Society of Japan. 2017. link Times cited: 2 Abstract: A novel approach to predict the atomic densities of amorphou… read moreAbstract: A novel approach to predict the atomic densities of amorphous materials is explored on the basis of Car–Parrinello molecular dynamics (CPMD) in density functional theory. Despite the determination of the atomic density of matter being crucial in understanding its physical properties, no first-principles method has ever been proposed for amorphous materials until now. We have extended the conventional method for crystalline materials in a natural manner and pointed out the importance of the canonical ensemble of the total energy in the determination of the atomic densities of amorphous materials. To take into account the canonical distribution of the total energy, we generate multiple amorphous structures with several different volumes by CPMD simulations and average the total energies at each volume. The density is then determined as the one that minimizes the averaged total energy. In this study, this approach is implemented for amorphous silicon (a-Si) to demonstrate its validity, and we have determined... read less USED (high confidence) M. Desanker et al., “Alkyl-Cyclens as Effective Sulfur- and Phosphorus-Free Friction Modifiers for Boundary Lubrication.,” ACS applied materials & interfaces. 2017. link Times cited: 41 Abstract: Modern automotive engines operate at higher power densities … read moreAbstract: Modern automotive engines operate at higher power densities than ever before, driving a need for new lubricant additives capable of reducing friction and wear further than ever before while not poisoning the catalytic converter. Reported in this paper is a new class of molecular friction modifier (FM), represented by 1,4,7,10-tetradodecyl-1,4,7,10-tetraazacyclododecane (1a), designed to employ thermally stable, sulfur- and phosphorus-free alkyl-substituted nitrogen heterocycles with multiple nitrogen centers per molecule. The multiple nitrogen centers enable cooperative binding to a surface which provides strong surface adsorption and lubricant film durability in the boundary lubrication (BL) regime. A 1 wt % loading of the cyclen FM 1a in Group III base oil exhibits strong surface adsorption, leading to excellent reductions in friction (70%) and wear (95%) versus the pure Group III oil across a wide temperature range. The lubricant with the new FM additive also outperforms two commercially available noncyclic amine-based FMs and a fully formulated commercial 5W30 motor oil. read less USED (high confidence) N. Winter, M. Becton, L. Zhang, and X. Wang, “Effects of pore design on mechanical properties of nanoporous silicon,” Acta Materialia. 2017. link Times cited: 23 USED (high confidence) X. Zhang, C. Gong, and G. Wu, “Strain Effect Analysis on Thermal Conductivity of Ge Thin Films,” Rare Metal Materials and Engineering. 2017. link Times cited: 1 USED (high confidence) G. Argentero et al., “Unraveling the 3D Atomic Structure of a Suspended Graphene/hBN van der Waals Heterostructure,” Nano Letters. 2017. link Times cited: 79 Abstract: In this work we demonstrate that a free-standing van der Waa… read moreAbstract: In this work we demonstrate that a free-standing van der Waals heterostructure, usually regarded as a flat object, can exhibit an intrinsic buckled atomic structure resulting from the interaction between two layers with a small lattice mismatch. We studied a freely suspended membrane of well-aligned graphene on a hexagonal boron nitride (hBN) monolayer by transmission electron microscopy (TEM) and scanning TEM (STEM). We developed a detection method in the STEM that is capable of recording the direction of the scattered electron beam and that is extremely sensitive to the local stacking of atoms. A comparison between experimental data and simulated models shows that the heterostructure effectively bends in the out-of-plane direction, producing an undulated structure having a periodicity that matches the moiré wavelength. We attribute this rippling to the interlayer interaction and also show how this affects the intralayer strain in each layer. read less USED (high confidence) Z. Wang and X. Ruan, “On the domain size effect of thermal conductivities from equilibrium and nonequilibrium molecular dynamics simulations,” Journal of Applied Physics. 2017. link Times cited: 31 Abstract: Equilibrium molecular dynamics (EMD) simulations with the Gr… read moreAbstract: Equilibrium molecular dynamics (EMD) simulations with the Green-Kubo formula and nonequilibrium molecular dynamics (NEMD) simulations with the Fourier's Law are two widely used methods for calculating thermal conductivities of materials. It is well known that both methods suffer from domain size effects, especially for NEMD. But the underlying mechanisms and their comparison have not been much quantitatively studied before. In this paper, we investigate their domain size effects by using crystalline silicon at 1000 K, graphene at 300 K, and silicene at 300 K as model material systems. The thermal conductivity of silicon from EMD simulations increases normally with the increasing domain size and converges at a size of around 4×4×4 nm3. The converging trend agrees well with the wavelength-accumulated thermal conductivity. The thermal conductivities of graphene and silicene from EMD simulations decrease abnormally with the increasing domain size and converge at a size of around 10×10 nm2. We ascribe the anom... read less USED (high confidence) Z. Wang, S. Safarkhani, G. Lin, and X. Ruan, “Uncertainty quantification of thermal conductivities from equilibrium molecular dynamics simulations,” International Journal of Heat and Mass Transfer. 2016. link Times cited: 30 USED (high confidence) L. Bai, N. Srikanth, G. Kang, and K. Zhou, “Influence of Third Particle on the Tribological Behaviors of Diamond-like Carbon Films,” Scientific Reports. 2016. link Times cited: 25 USED (high confidence) E. Safi, J. Polvi, A. Lasa, and K. Nordlund, “Atomistic simulations of deuterium irradiation on iron-based alloys in future fusion reactors,” Nuclear materials and energy. 2016. link Times cited: 2 USED (high confidence) T. Schmidt, R. Albuquerque, R. Kempe, and S. Kümmel, “Investigating the electronic structure of a supported metal nanoparticle: Pd in SiCN.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 4 Abstract: We investigate the electronic structure of a Palladium nanop… read moreAbstract: We investigate the electronic structure of a Palladium nanoparticle that is partially embedded in a matrix of silicon carbonitride. From classical molecular dynamics simulations we first obtain a representative atomic structure. This geometry then serves as input to density-functional theory calculations that allow us to access the electronic structure of the combined system of particle and matrix. In order to make the computations feasible, we devise a subsystem strategy for calculating the relevant electronic properties. We analyze the Kohn-Sham density of states and pay particular attention to d-states which are prone to be affected by electronic self-interaction. We find that the density of states close to the Fermi level is dominated by states that originate from the Palladium nanoparticle. The matrix has little direct effect on the electronic structure of the metal. Our results contribute to explaining why silicon carbonitride does not have detrimental effects on the catalytic properties of palladium particles and can serve positively as a stabilizing mechanical support. read less USED (high confidence) B. Zhao 赵, Y. Wang 王, C. Liu 刘, and X. Wang 王, “Molecular dynamics simulation of structural change at metal/semiconductor interface induced by nanoindenter,” Chinese Physics B. 2016. link Times cited: 0 Abstract: The structures of the Si/Cu heterogenous interface impacted … read moreAbstract: The structures of the Si/Cu heterogenous interface impacted by a nanoindenter with different incident angles and depths are investigated in detail using molecular dynamics simulation. The simulation results suggest that for certain incident angles, the nanoindenter with increasing depth can firstly increase the stress of each atom at the interface and it then introduces more serious structural deformation of the Si/Cu heterogenous interface. A nanoindenter with increasing incident angle (absolute value) can increase the length of the Si or Cu extended atom layer. It is worth mentioning that when the incident angle of the nanoindenter is between −45° and 45°, these Si or Cu atoms near the nanoindenter reach a stable state, which has a lower stress and a shorter length of the Si or Cu extended atom layer than those of the other incident angles. This may give a direction to the planarizing process of very large scale integration circuits manufacture. read less USED (high confidence) Z. Bai, L. Zhang, H. Li, and L. Liu, “Nanopore Creation in Graphene by Ion Beam Irradiation: Geometry, Quality, and Efficiency.,” ACS applied materials & interfaces. 2016. link Times cited: 56 Abstract: Ion beam irradiation is a promising approach to fabricate na… read moreAbstract: Ion beam irradiation is a promising approach to fabricate nanoporous graphene for various applications, including DNA sequencing, water desalination, and phase separation. Further advancement of this approach and rational design of experiments all require improved mechanistic understanding of the physical drilling process. Here, we demonstrate that, by using oblique ion beam irradiation, the nanopore family is significantly expanded to include more types of nanopores of tunable geometries. With the hopping, sweeping, and shoving mechanisms, ions sputter carbon atoms even outside the ion impact zone, leading to extended damage particularly at smaller incident angles. Moreover, with lower energies, ions may be absorbed to form complex ion-carbon structures, making the graphene warped or curly at pore edges. Considering both efficiency and quality, the optimal ion energy is identified to be 1000 eV at an incident angle of 30° with respect to the graphene sheet and 400-500 eV at higher incident angles. All of these results suggest the use of oblique ion beam and moderate energy levels to efficiently fabricate high-quality nanopores of tunable geometries in graphene for a wide range of applications. read less USED (high confidence) X.-Y. Sun, Y. Qi, W. Ouyang, X.-Q. Feng, and Q. Li, “Energy corrugation in atomic-scale friction on graphite revisited by molecular dynamics simulations,” Acta Mechanica Sinica. 2016. link Times cited: 21 USED (high confidence) H. Seyf and A. Henry, “A method for distinguishing between propagons, diffusions, and locons,” Journal of Applied Physics. 2016. link Times cited: 81 Abstract: The majority of intuition on phonon transport has been deriv… read moreAbstract: The majority of intuition on phonon transport has been derived from studies of homogenous crystalline solids, where the atomic composition and structure are periodic. For this specific class of materials, the solutions to the equations of motions for the atoms (in the harmonic limit) result in plane wave modulated velocity fields for the normal modes of vibration. However, it has been known for several decades that whenever a system lacks periodicity, either compositional or structural, the normal modes of vibration can still be determined (in the harmonic limit), but the solutions take on different characteristics and many modes may not be plane wave modulated. Previous work has classified the types of vibrations into three primary categories, namely, propagons, diffusions, and locons. One can use the participation ratio to distinguish locons, from propagons and diffusons, which measures the extent to which a mode is localized. However, distinguishing between propagons and diffusons has remained a challe... read less USED (high confidence) A. Galashev and V. Polukhin, “Computer simulation of heating of nickel and mercury on graphene,” Russian Metallurgy (Metally). 2016. link Times cited: 0 USED (high confidence) A. Giri, J. L. Braun, and P. Hopkins, “Effect of crystalline/amorphous interfaces on thermal transport across confined thin films and superlattices,” Journal of Applied Physics. 2016. link Times cited: 36 Abstract: We report on the thermal boundary resistances across crystal… read moreAbstract: We report on the thermal boundary resistances across crystalline and amorphous confined thin films and the thermal conductivities of amorphous/crystalline superlattices for Si/Ge systems as determined via non-equilibrium molecular dynamics simulations. Thermal resistances across disordered Si or Ge thin films increase with increasing length of the interfacial thin films and in general demonstrate higher thermal boundary resistances in comparison to ordered films. However, for films ≲3 nm, the resistances are highly dependent on the spectral overlap of the density of states between the film and leads. Furthermore, the resistances at a single amorphous/crystalline interface in these structures are much lower than those at interfaces between the corresponding crystalline materials, suggesting that diffusive scattering at an interface could result in higher energy transmissions in these systems. We use these findings, together with the fact that high mass ratios between amorphous and crystalline materials can... read less USED (high confidence) T. Fang, W.-J. Chang, Y.-L. Feng, and C. Weng, “Tensile fracture of graphene nanoribbons encapsulated in single-walled carbon nanotubes,” Acta Mechanica. 2016. link Times cited: 4 USED (high confidence) T. Fang, W.-J. Chang, Y.-L. Feng, and C. Weng, “Tensile fracture of graphene nanoribbons encapsulated in single-walled carbon nanotubes,” Acta Mechanica. 2016. link Times cited: 0 USED (high confidence) D. Kaiser, S. Ghosh, S. Han, and T. Sinno, “Modeling and simulation of compositional engineering in SiGe films using patterned stress fields.” 2016. link Times cited: 2 Abstract: Semiconductor alloys such as silicon–germanium (SiGe) offer … read moreAbstract: Semiconductor alloys such as silicon–germanium (SiGe) offer attractive environments for engineering quantum-confined structures that are the basis for a host of current and future optoelectronic devices. Although vertical stacking of such structures is routinely achieved via heteroepitaxy, lateral manipulation has proven much more challenging. We have recently demonstrated that a patterned elastic stress field applied, with an array of nanoscale indenters, to an initially compositionally uniform SiGe substrate will drive atomic interdiffusion leading to compositional patterns in the near-surface region of the substrate. While this approach may offer a potentially efficient and robust pathway to producing laterally ordered arrays of quantum-confined structures, optimizing it with respect to the various process parameters, such as indenter array geometry, annealing history, and SiGe substrate thickness and composition, is highly challenging. Here, a mesoscopic model based on coarse-grained lattice kinetic Monte Carlo simulation is presented that describes quantitatively the atomic interdiffusion processes in SiGe alloy films subjected to applied stress. We first show that the model provides predictions that are quantitatively consistent with experimental measurements. Then, the model is used to investigate the impact of several process parameters such as indenter shape and pitch. We find that certain indenter configurations produce compositional patterns that are favorable for engineering lateral arrays of quantum-confined structures. read less USED (high confidence) A. V. Sidorenkov, S. Kolesnikov, and A. Saletsky, “Molecular dynamics simulation of graphene on Cu (111) with different Lennard-Jones parameters,” The European Physical Journal B. 2016. link Times cited: 14 USED (high confidence) M. Wolf et al., “Hydrogenated pyrene: Statistical single-carbon loss below the knockout threshold,” The European Physical Journal D. 2016. link Times cited: 14 USED (high confidence) C. da Silva, F. Saiz, D. A. Romero, and C. Amon, “Coherent phonon transport in short-period two-dimensional superlattices of graphene and boron nitride,” Physical Review B. 2016. link Times cited: 28 Abstract: Promoting coherent transport of phonons at material interfac… read moreAbstract: Promoting coherent transport of phonons at material interfaces is a promising strategy for controlling thermal transport in nanostructures and an alternative to traditional methods based on structural defects. Coherent transport is particularly relevant in short-period heterostructures with smooth interfaces and long-wavelength heat-carrying phonons, such as two-dimensional superlattices of graphene and boron nitride. In this work, we predict phonon properties and thermal conductivities in these superlattices using a normal mode decomposition approach. We study the variation of the frequency dependence of these properties with the periodicity and interface configuration (zigzag and armchair) for superlattices with period lengths within the coherent regime. Our results showed that the thermal conductivity decreases significantly from the first period length (0.44 nm) to the second period length (0.87 nm), 13% across the interfaces and 16% along the interfaces. For greater periods, the conductivity across the interfaces continues decreasing at a smaller rate of 11 W/mK per period length increase (0.43 nm), driven by changes in the phonon group velocities (coherent effects). In contrast, the conductivity along the interfaces slightly recovers at a rate of 2 W/mK per period, driven by changes in the phonon relaxation times (diffusive effects). By changing the interface configuration from armchair to zigzag, the conductivities for all period lengths increase by approximately 7% across the interfaces and 19% along the interfaces. read less USED (high confidence) K. Kunal and N. Aluru, “Characterizing phonon dynamics using stochastic sampling,” Journal of Applied Physics. 2016. link Times cited: 0 Abstract: Predicting phonon relaxation time from molecular dynamics (M… read moreAbstract: Predicting phonon relaxation time from molecular dynamics (MD) requires a long simulation time to compute the mode energy auto-correlation function. Here, we present an alternative approach to infer the phonon life-time from an approximate form of the energy auto-correlation function. The method requires as an input a set of sampled equilibrium configurations. A stochastic sampling method is used to generate the equilibrium configurations. We consider a truncated Taylor series expansion of the phonon energy auto-correlation function. The different terms in the truncated correlation function are obtained using the stochastic sampling approach. The expansion terms, thus, obtained are in good agreement with the corresponding values obtained using MD. We then use the approximate function to compute the phonon relaxation time. The relaxation time computed using this method is compared with that obtained from the exact correlation function. The two values are in agreement with each other. read less USED (high confidence) A. Özden, A. Kandemir, F. Ay, N. K. Perkgöz, and C. Sevik, “Thermal Conductivity Suppression in Nanostructured Silicon and Germanium Nanowires,” Journal of Electronic Materials. 2016. link Times cited: 8 USED (high confidence) D. Spiteri, J. Anaya, and M. Kuball, “The effects of grain size and grain boundary characteristics on the thermal conductivity of nanocrystalline diamond,” Journal of Applied Physics. 2016. link Times cited: 25 Abstract: Molecular dynamics simulation was used to study the effects … read moreAbstract: Molecular dynamics simulation was used to study the effects of each grain dimension and of grain boundary characteristics on the inter-grain thermal boundary resistance (TBR) and intragrain thermal conductivity of nanocrystalline diamond. The effect of the grain boundaries perpendicular to the heat flow was studied using a multiple slab configuration, which greatly reduced the artifacts associated with the heat source/sink. The TBR between the slabs was found to be more sensitive to the atomic arrangement at the boundary than to the tilt angle between the slabs. When the atomic arrangement at the interface was altered from the minimum energy configuration, the TBR increased by a factor of three, suggesting that a sub-optimal interface quality between the grains could play a large role in reducing the thermal conductivity of nanocrystalline diamond. The thermal conductivity between the boundaries was found to be similar to the bulk value, even when the boundaries were only 25 nm apart. The effect of grain ... read less USED (high confidence) S. Hyun, Y. Park, and H.-tae Kim, “Computational characterizations on the grain-size-dependent properties of polycrystalline nanomaterials,” Journal of the Korean Physical Society. 2015. link Times cited: 6 Abstract: The microstructures of real nanomaterials can be quite compl… read moreAbstract: The microstructures of real nanomaterials can be quite complex with variety of grain sizes aligned in different crystal orientations and structural defects possibly created in a fabrication process. Material properties of these polycrystalline materials are generally known strongly dependent on the nanoscale morphology. First principle calculations based on the density functional theory need to be employed in these atomic characterizations; however, it may not be suitable for the polycrystalline nanomaterials for which large number of atoms is required in the simulation model. Instead, a mesoscale computer simulation scheme is employed to investigate these morphology-dependent mechanical properties of polycrystalline materials. We demonstrated the Voronoi construction of various polycrystalline atomic models such as two-dimensional graphene and three-dimensional silicon carbide. General behavior of the mechanical characteristics of the bulk nanostructured silicon carbide (SiC) was addressed, particularly the contribution of grain sizes. From this study, the optimal grain size was determined near 10 nm under tensile and compressive deformations. read less USED (high confidence) X.-Y. Sun, Y. Qi, W. Ouyang, X.-Q. Feng, and Q. Li, “Energy corrugation in atomic-scale friction on graphite revisited by molecular dynamics simulations,” Acta Mechanica Sinica. 2015. link Times cited: 0 USED (high confidence) B. Mortazavi, L. Pereira, J.-W. Jiang, and T. Rabczuk, “Modelling heat conduction in polycrystalline hexagonal boron-nitride films,” Scientific Reports. 2015. link Times cited: 107 USED (high confidence) S. Kida, M. Yamamoto, K. Tada, H. Kawata, Y. Hirai, and M. Yasuda, “Correlation between electron-irradiation defects and applied stress in graphene: A molecular dynamics study,” Journal of Vacuum Science and Technology. 2015. link Times cited: 5 Abstract: Molecular dynamics (MD) simulations are performed to study t… read moreAbstract: Molecular dynamics (MD) simulations are performed to study the correlation between electron irradiation defects and applied stress in graphene. The electron irradiation effect is introduced by the binary collision model in the MD simulation. By applying a tensile stress to graphene, the number of adatom-vacancy (AV) and Stone–Wales (SW) defects increase under electron irradiation, while the number of single-vacancy defects is not noticeably affected by the applied stress. Both the activation and formation energies of an AV defect and the activation energy of an SW defect decrease when a tensile stress is applied to graphene. Applying tensile stress also relaxes the compression stress associated with SW defect formation. These effects induced by the applied stress cause the increase in AV and SW defect formation under electron irradiation. read less USED (high confidence) M. Cusentino, K. Hammond, F. Sefta, N. Juslin, and B. Wirth, “A comparison of interatomic potentials for modeling tungsten–hydrogen–helium plasma–surface interactions,” Journal of Nuclear Materials. 2015. link Times cited: 25 USED (high confidence) P. Novikov, A. Nenashev, S. Rudin, A. Polyakov, and A. Dvurechenskii, “Simulating the nucleation and growth of Ge quantum dots on Si using high-efficiency algorithms,” Nanotechnologies in Russia. 2015. link Times cited: 4 USED (high confidence) A. Sgouros, G. Kalosakas, M. Sigalas, and K. Papagelis, “Exotic carbon nanostructures obtained through controllable defect engineering,” RSC Advances. 2015. link Times cited: 8 Abstract: We numerically demonstrate the spontaneous formation of vari… read moreAbstract: We numerically demonstrate the spontaneous formation of various 3D carbon nanostructures, like multi-tube carbon nanotubes, nanopyramids, nanocubes, artificially rippled graphene, and other exotic nanomaterials, starting from graphene nanoribbons and inducing controllably engineered defects consisting of carbon adatoms or inverse Stone–Wales defects. The evolution of the initial defected planar structures towards the final 3D nanoarchitectures is obtained through molecular dynamics simulations, using different force fields to ensure the reproducibility of the derived results. The presented carbon nanostructures of different shapes, sizes, and morphologies, can be used in applications ranging from storage of hydrogen or other molecules, enhanced chemical reactions or catalysis in confined compartments, to drug delivery nanodevices and biosensors. read less USED (high confidence) C. K. Oliveira et al., “Crystal-oriented wrinkles with origami-type junctions in few-layer hexagonal boron nitride,” Nano Research. 2015. link Times cited: 32 USED (high confidence) Y. S. Buranova, B. Kulnitskiy, I. Perezhogin, and V. Blank, “Structure of boron nitride nanotubes,” Crystallography Reports. 2015. link Times cited: 3 USED (high confidence) M. R. Price, A. Ovcharenko, R. Thangaraj, and B. Raeymaekers, “Deformation of Ultra-Thin Diamond-Like Carbon Coatings Under Combined Loading on a Magnetic Recording Head,” Tribology Letters. 2015. link Times cited: 8 USED (high confidence) I. Ostanin, R. Ballarini, and T. Dumitricǎ, “Distinct element method for multiscale modeling of cross-linked carbon nanotube bundles: From soft to strong nanomaterials,” Journal of Materials Research. 2015. link Times cited: 17 Abstract: Predicting the impact of cross-links on the mechanics of car… read moreAbstract: Predicting the impact of cross-links on the mechanics of carbon nanotube-based materials is a challenging endeavor, as the micro- and nanostructure is composed of continuous nanofibers, discontinuous interfaces, and covalent bridges. Here we demonstrate a new modeling solution in the context of the distinct element method (DEM). By representing nanotubes as bonded cylinder segments undergoing van der Waals adhesion, viscous friction, and contact bonding, we are able to simulate how cross-linking transforms a soft bundle into a strong one. We predict that the sp ^3- sp cross-links formed by interstitial carbon atoms can improve the tensile strength by an order of magnitude, in agreement with experiment and molecular dynamics simulations. The DEM methodology allows performing the multiscale simulation needed for developing strategies to further enhance the mechanical performance. read less USED (high confidence) N. Rajput, Z. Tong, and X. Luo, “Investigation of ion induced bending mechanism for nanostructures,” Materials Research Express. 2014. link Times cited: 16 Abstract: Ion induced bending is a promising controlled technique for … read moreAbstract: Ion induced bending is a promising controlled technique for manipulating nanoscale structures. However, the underlying mechanism of the process is not well understood. In this letter, we report a detailed study of the bending mechanism of Si nanowires (NWs) under Ga+ irradiation. The microstructural changes in the NW due to ion beam irradiation are studied and molecular dynamics simulations are used to explore the ion–NW interaction processes. The simulation results are compared with the microstructural studies of the NW. The investigations inform a generic understanding of the bending process in crystalline materials, which we suggest to be feasible as a versatile manipulation and integration technique in nanotechnology. read less USED (high confidence) A. Galashev, “Computer simulation of the thermal stability of nickel films on two-layer graphene,” High Temperature. 2014. link Times cited: 14 USED (high confidence) A. Galashev and V. Polukhin, “Compaction of a copper film on graphene by argon-beam bombardment: Computer experiment,” Journal of Surface Investigation. X-ray, Synchrotron and Neutron Techniques. 2014. link Times cited: 15 USED (high confidence) A. Sgouros, M. Neupane, M. Sigalas, N. Aravantinos-Zafiris, and R. Lake, “Nanoscale phononic interconnects in THz frequencies.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 8 Abstract: Phononic computing is emerging as an alternative computing p… read moreAbstract: Phononic computing is emerging as an alternative computing paradigm to the conventional electronic and optical computing. In this study, we propose and analyze various phononic interconnects, such as nano-scaled phononic resonators, waveguides and switches, on the 〈111〉 surface of 3C-SiC and 3C-GeSi with substitutional and vacancy defects. This is achieved by simultaneously introducing defects of various types, and by varying their specific locations on the surface. To calculate the intrinsic and the defect-induced vibrational properties, such as the phononic bandgap and the variation in the phonon spectra, the total phonon density of states (TPDOS) and the partial phonon density of states (PPDOS) were calculated using molecular dynamics simulations with semi-empirical potentials. The proposed phononic interconnects, in conjunction with electronic and/or photonic interconnects, can be used in the current and future devices. read less USED (high confidence) M. Yamamoto, Y. Asayama, M. Yasuda, H. Kawata, and Y. Hirai, “Defect formation and transformation in graphene under electron irradiation: A molecular dynamics study,” Journal of Vacuum Science & Technology. B. Nanotechnology and Microelectronics: Materials, Processing, Measurement, and Phenomena. 2014. link Times cited: 5 Abstract: Molecular dynamics simulations were performed to study defec… read moreAbstract: Molecular dynamics simulations were performed to study defect formation and transformation in graphene under electron irradiation. The single-vacancy was the most frequently formed defect and the number of defects did not depend on the defect formation energy for normal incidence. The single-vacancy transformed to other types of defects and migrated in graphene by heating. The recovery energies of adatom-vacancy and pentagon–heptagon defects were relatively small. The Stone–Wales defect was the most stable, and did not easily recover. In the single atomic chain formation process from graphene by electron irradiation, competition between defect formation by electron collision and the recovery by heating was observed. read less USED (high confidence) A. Galashev and O. Rakhmanova, “Mechanical and thermal stability of graphene and graphene-based materials,” Physics—Uspekhi. 2014. link Times cited: 113 Abstract: Graphene has rapidly become one of the most popular material… read moreAbstract: Graphene has rapidly become one of the most popular materials for technological applications and a test material for new condensed matter ideas. This paper reviews the mechanical properties of graphene and effects related to them that have recently been discovered experimentally or predicted theoretically or by simulation. The topics discussed are of key importance for graphene's use in integrated electronics, thermal materials, and electromechanical devices and include the following: graphene transformation into other hybridization forms; stability to stretching and compression; ion-beam-induced structural modifications; how defects and graphene edges affect the electronic properties and thermal stability of graphene and related composites. read less USED (high confidence) M. Stockett et al., “Fragmentation of anthracene C₁₄H₁₀, acridine C₁₃H₉N and phenazine C₁₂H₈N₂ ions in collisions with atoms.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 21 Abstract: We report experimental total, absolute, fragmentation cross … read moreAbstract: We report experimental total, absolute, fragmentation cross sections for anthracene C14H10, acridine C13H9N, and phenazine C12H8N2 ions colliding with He at center-of-mass energies close to 100 eV. In addition, we report results for the same ions colliding with Ne, Ar, and Xe at higher energies. The total fragmentation cross sections for these three ions are the same within error bars for a given target. The measured fragment mass distributions reveal significant contributions from both delayed (≫10(-12) s) statistical fragmentation processes as well as non-statistical, prompt (∼10(-15) s), single atom knockout processes. The latter dominate and are often followed by secondary statistical fragmentation. Classical Molecular Dynamics (MD) simulations yield separate cross sections for prompt and delayed fragmentation which are consistent with the experimental results. The intensity of the single C/N-loss peak, the signature of non-statistical fragmentation, decreases with the number of N atoms in the parent ion. The fragment intensity distributions for losses of more than one C or N atom are rather similar for C14H10 and C13H9N but differ strongly for C12H8N2 where weak C-N bonds often remain in the fragments after the first fragmentation step. This greatly increases their probability to fragment further. Distributions of internal energy remaining in the fragments after knockout are obtained from the MD simulations. read less USED (high confidence) M. Park, I. Lee, and Y.-S. Kim, “Lattice thermal conductivity of crystalline and amorphous silicon with and without isotopic effects from the ballistic to diffusive thermal transport regime,” Journal of Applied Physics. 2014. link Times cited: 23 Abstract: Thermal conductivity of a material is an important physical … read moreAbstract: Thermal conductivity of a material is an important physical parameter in electronic and thermal devices, and as the device size shrinks down, its length-dependence becomes unable to be neglected. Even in micrometer scale devices, materials having a long mean free path of phonons, such as crystalline silicon (Si), exhibit a strong length dependence of the thermal conductivities that spans from the ballistic to diffusive thermal transport regime. In this work, through non-equilibrium molecular-dynamics (NEMD) simulations up to 17 μm in length, the lattice thermal conductivities are explicitly calculated for crystalline Si and up to 2 μm for amorphous Si. The Boltzmann transport equation (BTE) is solved within a frequency-dependent relaxation time approximation, and the calculated lattice thermal conductivities in the BTE are found to be in good agreement with the values obtained in the NEMD. The isotopic effects on the length-dependent lattice thermal conductivities are also investigated both in the crystalline and amorphous Si. read less USED (high confidence) F. Al-Dirini, F. Hossain, A. Nirmalathas, and E. Skafidas, “All-Graphene Planar Double Barrier Resonant Tunneling Diodes,” IEEE Journal of the Electron Devices Society. 2014. link Times cited: 16 Abstract: In this work, we propose an atomically-thin all-graphene pla… read moreAbstract: In this work, we propose an atomically-thin all-graphene planar double barrier resonant tunneling diode that can be realized within a single graphene nanoribbon. The proposed device does not require any doping or external gating and can be fabricated using minimal process steps. The planar architecture of the device allows a simple in-plane connection of multiple devices in parallel without any extra processing steps during fabrication, enhancing the current driving capabilities of the device. Quantum mechanical simulation results, based on non-equilibrium Green's function formalism and the extended Huckel method, show promising device performance with a high reverse-to-forward current rectification ratio exceeding 50 000, and confirm the presence of negative differential resistance within the device's current-voltage characteristics. read less USED (high confidence) Y. Wei, H. Zhan, K. Xia, W. Zhang, S. Sang, and Y. T. Gu, “Resonance of graphene nanoribbons doped with nitrogen and boron: a molecular dynamics study,” Beilstein Journal of Nanotechnology. 2014. link Times cited: 6 Abstract: Summary Based on its enticing properties, graphene has been … read moreAbstract: Summary Based on its enticing properties, graphene has been envisioned with applications in the area of electronics, photonics, sensors, bio-applications and others. To facilitate various applications, doping has been frequently used to manipulate the properties of graphene. Despite a number of studies conducted on doped graphene regarding its electrical and chemical properties, the impact of doping on the mechanical properties of graphene has been rarely discussed. A systematic study of the vibrational properties of graphene doped with nitrogen and boron is performed by means of a molecular dynamics simulation. The influence from different density or species of dopants has been assessed. It is found that the impacts on the quality factor, Q, resulting from different densities of dopants vary greatly, while the influence on the resonance frequency is insignificant. The reduction of the resonance frequency caused by doping with boron only is larger than the reduction caused by doping with both boron and nitrogen. This study gives a fundamental understanding of the resonance of graphene with different dopants, which may benefit their application as resonators. read less USED (high confidence) A. Galashev, “Computer simulation of heating of nickel films on two-layer graphene,” Physics of the Solid State. 2014. link Times cited: 5 USED (high confidence) B. Mortazavi and G. Cuniberti, “Mechanical properties of polycrystalline boron-nitride nanosheets,” RSC Advances. 2014. link Times cited: 90 Abstract: The first molecular dynamics (MD) study was conducted to exp… read moreAbstract: The first molecular dynamics (MD) study was conducted to explore mechanical-failure response of ultra-fine grained single-layer boron-nitride films. We used MD simulations to construct relatively large molecular models of polycrystalline structures with random grain configurations. By applying uniaxial tensile loading, we then studied the grain size effect on the mechanical response of polycrystalline boron-nitride nanosheets. Our results reveal that by decreasing the grain size, the elastic modulus of polycrystalline films decreases gradually. Interestingly, our MD results reveal that ultra-fine grained samples could present a tensile strength of around half that of the pristine films. Our investigation suggests that experimentally fabricated polycrystalline boron nitride nanosheets can exhibit remarkably high mechanical properties. read less USED (high confidence) B. Javvaji, D. R. Mahapatra, and S. Raha, “Molecular dynamics study of phonon screening in graphene,” Smart Structures. 2014. link Times cited: 0 Abstract: Phonon interaction with electrons or phonons or with structu… read moreAbstract: Phonon interaction with electrons or phonons or with structural defects result in a phonon mode conversion. The mode conversion is governed by the frequency wave-vector dispersion relation. The control over phonon mode or the screening of phonon in graphene is studied using the propagation of amplitude modulated phonon wave-packet. Control over phonon properties like frequency and velocity opens up several wave guiding, energy transport and thermo-electric applications of graphene. One way to achieve this control is with the introduction of nano-structured scattering in the phonon path. Atomistic model of thermal energy transport is developed which is applicable to devices consisting of source, channel and drain parts. Longitudinal acoustic phononmode is excited fromone end of the device. Molecular dynamics based time integration is adopted for the propagation of excited phonon to the other end of the device. The amount of energy transfer is estimated from the relative change of kinetic energy. Increase in the phonon frequency decreases the kinetic energy transmission linearly in the frequency band of interest. Further reduction in transmission is observed with the tuning of channel height of the device by increasing the boundary scattering. Phonon mode selective transmission control have potential application in thermal insulation or thermo-electric application or photo-thermal amplification. read less USED (high confidence) M. Shen and P. Keblinski, “Ballistic vs. diffusive heat transfer across nanoscopic films of layered crystals,” Journal of Applied Physics. 2014. link Times cited: 15 Abstract: We use non-equilibrium molecular dynamics to study the heat … read moreAbstract: We use non-equilibrium molecular dynamics to study the heat transfer mechanism across sandwich interfacial structures of Si/n-atomic-layers/Si, with 1 ≤ n ≤ 20 and atomic layers composed of WSe2 and/or graphene. In the case of WSe2 sheets, we observe that the thermal resistance of the sandwich structure is increasing almost linearly with the number of WSe2 sheets, n, indicating a diffusive phonon transport mechanism. By contrast in the case of n graphene layers, the interfacial thermal resistance is more or less independent on the number of layers for 1 ≤ n ≤ 10, and is associated with ballistic phonon transport mechanism. We attribute the diffusive heat transfer mechanism across WSe2 sheets to abundant low frequency and low group velocity optical modes that carry most of the heat across the interface. By contrast, in graphene, acoustic modes dominate the thermal transport across the interface and render a ballistic heat flow mechanism. read less USED (high confidence) B. J. Yang, H. Shin, H.-W. Kim, and H. Lee, “Strain rate and adhesive energy dependent viscoplastic damage modeling for nanoparticulate composites: Molecular dynamics and micromechanical simulations,” Applied Physics Letters. 2014. link Times cited: 13 Abstract: A viscoplastic damage model based on molecular dynamics (MD)… read moreAbstract: A viscoplastic damage model based on molecular dynamics (MD) and micromechanics is proposed to predict the rate-dependent inelastic behavior of nanoparticle-reinforced polymer composites. The constitutive equation is developed by combining the solution of the elastic problem and Laplace-transformed superposition principle. The MD simulation is then conducted to derive the interfacial adhesive energy of nanocomposites (silica/nylon-6), and the MD results are applied to the viscoplastic damage model. Influences of the strain rate sensitivity and the interfacial debonding damage on nanocomposites are discussed, and predictions from the proposed approach are compared with experimental measurements to elucidate the potential of the formulation. read less USED (high confidence) S. Cea et al., “Process modeling for advanced device technologies,” Journal of Computational Electronics. 2014. link Times cited: 8 USED (high confidence) Y.-Y. Liu, W.-X. Zhou, L.-M. Tang, and K. Chen, “Core-shell nanowire serves as heat cable,” Applied Physics Letters. 2013. link Times cited: 24 Abstract: To analyze the thermal transport properties in core-shell na… read moreAbstract: To analyze the thermal transport properties in core-shell nanowires, we calculate systematically the distributions of heat flux in InAs/GaAs and GaAs/InAs core-shell nanowires by using nonequilibrium molecular dynamics simulations. The results show that for InAs/GaAs core-shell nanowires, the heat current tends to transport in the shell, while for GaAs/InAs core-shell nanowires the heat current tends to transport through the core. Moreover, a simple equation is presented to describe the relationship of the thermal conductance among the core, the tubular shell, and core-shell nanowire. It is suggested that the core-shell nanowires can be served as heat cable. read less USED (high confidence) S. Hida, T. Hori, T. Shiga, J. Elliott, and J. Shiomi, “Thermal resistance and phonon scattering at the interface between carbon nanotube and amorphous polyethylene,” International Journal of Heat and Mass Transfer. 2013. link Times cited: 75 USED (high confidence) H. Muramatsu et al., “Boron-assisted coalescence of parallel multi-walled carbon nanotubes,” RSC Advances. 2013. link Times cited: 2 Abstract: Coalescing carbon nanotubes is a major challenge for designi… read moreAbstract: Coalescing carbon nanotubes is a major challenge for designing structures with novel physical and chemical properties and for creating three-dimensional carbon networks with improved mechanical and transport properties. We have coalesced adjacent triple walled carbon nanotubes (TWNTs) covalently, using catalytic boron atoms at high temperatures. The two outermost and then the two inner nanotubes of adjacent TWNTs merged in order to create an enlarged flattened double-walled carbon nanotube which encapsulated the two innermost single-walled carbon nanotubes. read less USED (high confidence) C. Cassidy, D. V. Singh, P. Grammatikopoulos, F. Djurabekova, K. Nordlund, and M. Sowwan, “Inoculation of silicon nanoparticles with silver atoms,” Scientific Reports. 2013. link Times cited: 37 USED (high confidence) M. Yasuda, Y. Chihara, K. Tada, H. Kawata, and Y. Hirai, “Correlation between electron irradiation defects and applied stress in carbon nanotubes: A molecular dynamics study,” Journal of Vacuum Science & Technology. B. Nanotechnology and Microelectronics: Materials, Processing, Measurement, and Phenomena. 2013. link Times cited: 3 Abstract: Molecular dynamics simulations have been performed to study … read moreAbstract: Molecular dynamics simulations have been performed to study the correlation between electron irradiation defects and applied stress in single-walled carbon nanotubes. The electron irradiation effect is modeled based on the Monte Carlo method using binary collision theory. The simulations show that the number of knock-on defects is not significantly affected by applied stress. Electron collision causes early stage defects such as bond breaking and punched-out atoms. The applied stresses become nonequilibrium and concentrates around the defect site. The biased stresses become driving forces and cause bond shifts and rotations, which results in the formation of pentagon–heptagon pairs and Stone–Wales defects. read less USED (high confidence) Q. Zhang and D.-feng Diao, “Potential of graphene layer controlling nano-wear during C60 intrusion by molecular dynamics simulation,” Wear. 2013. link Times cited: 11 USED (high confidence) S. Cea et al., “Process modeling for advanced device technologies,” Journal of Computational Electronics. 2013. link Times cited: 0 USED (high confidence) H. Bao, C. Shao, S. Luo, and M. Hu, “Enhancement of interfacial thermal transport by carbon nanotube-graphene junction,” Journal of Applied Physics. 2013. link Times cited: 40 Abstract: Due to the high intrinsic thermal conductivity, carbon nanot… read moreAbstract: Due to the high intrinsic thermal conductivity, carbon nanotubes are very promising to serve as effective thermal interface materials for microelectronics or other cooling applications. However, the performance of carbon nanotube based thermal interface material is strongly limited by the small effective contact area and weak bonding at carbon nanotube and material interface. Here, we propose a junction structure that the carbon nanotube is bonded with a monolayer graphene, which could potentially enhance the interface thermal conductance. Molecular dynamics simulations show that the interface thermal conductance can be enhanced by at least 40% compared to direct carbon nanotube and silicon interface with strong covalent bonding, while for weak van der Waals bonding the conductance can be enhanced by almost one order of magnitude. The enhancement of thermal conductance is attributed to the efficient thermal transport between carbon nanotube and graphene, as well as the good contact between graphene and si... read less USED (high confidence) D. R. Robinson and M. Wilson, “The liquid⟷amorphous transition and the high pressure phase diagram of carbon,” Journal of Physics: Condensed Matter. 2013. link Times cited: 4 Abstract: The phase diagram of carbon is mapped to high pressure using… read moreAbstract: The phase diagram of carbon is mapped to high pressure using a computationally-tractable potential model. The use of a relatively simple (Tersoff-II) potential model allows a large range of phase space to be explored. The coexistence (melting) curve for the diamond crystal/liquid dyad is mapped directly by modelling the solid/liquid interfaces. The melting curve is found to be re-entrant and belongs to a conformal class of diamond/liquid coexistence curves. On supercooling the liquid a phase transition to a tetrahedral amorphous form (ta-C) is observed. The liquid ⟷ amorphous coexistence curve is mapped onto the pT plane and is found to also be re-entrant. The entropy changes for both melting and the amorphous ⟶ liquid transitions are obtained from the respective coexistence curves and the associated changes in molar volume. The structural change on amorphization is analysed at different points on the coexistence curve including for transitions that are both isochoric and isocoordinate (no change in nearest-neighbour coordination number). The conformal nature of the melting curve is highlighted with respect to the known behaviour of Si. The relationship of the observed liquid/amorphous coexistence curve to the Si low- and high-density amorphous (LDA/HDA) transition is discussed. read less USED (high confidence) Y. Matsuda, S. King, M. Oliver, and R. Dauskardt, “Moisture-assisted cracking and atomistic crack path meandering in oxidized hydrogenated amorphous silicon carbide films,” Journal of Applied Physics. 2013. link Times cited: 8 Abstract: Moisture-assisted cracking of silica-derived materials resul… read moreAbstract: Moisture-assisted cracking of silica-derived materials results from a stress-enhanced reaction between water molecules and moisture-sensitive SiOSi bonds at the crack tip. We report the moisture-assisted cracking of oxidized hydrogenated amorphous silicon carbide films (a-SiCO:H) consisting of both moisture-sensitive SiOSi bonds and moisture-insensitive bonds. The sensitivity of the films to moisture-assisted cracking was observed to increase with the SiOSi bond density, ρSiOSi. This sensitivity was correlated with the number of SiOSi bonds ruptured, NSiOSi, through an atomistic kinetic fracture model. By comparing these correlated NSiOSi values with those estimated by a planar crack model, we demonstrated that at the atomistic scale the crack path meanders three-dimensionally so as to intercept the most SiOSi bonds. This atomistic crack path meandering was verified by a computational method based on graph theory and molecular dynamics. Our findings could provide a basis for better underst... read less USED (high confidence) E. H. Cook, M. Buehler, and Z. Spakovszky, “Mechanism of friction in rotating carbon nanotube bearings,” Journal of The Mechanics and Physics of Solids. 2013. link Times cited: 87 USED (high confidence) J. Zang and Y.-pu Zhao, “A diffusion and curvature dependent surface elastic model with application to stress analysis of anode in lithium ion battery,” International Journal of Engineering Science. 2012. link Times cited: 47 USED (high confidence) J. Feng, X. Qian, C. Huang, and J. Li, “Strain-engineered artificial atom as a broad-spectrum solar energy funnel,” Nature Photonics. 2012. link Times cited: 896 USED (high confidence) Y. Asayama, M. Yasuda, K. Tada, H. Kawata, and Y. Hirai, “Molecular dynamics study of the structural modification of graphene by electron irradiation,” Journal of Vacuum Science & Technology. B. Nanotechnology and Microelectronics: Materials, Processing, Measurement, and Phenomena. 2012. link Times cited: 10 Abstract: Molecular dynamics simulations have been used to study the s… read moreAbstract: Molecular dynamics simulations have been used to study the structural modification of graphene by electron irradiation. The authors used the Monte Carlo method to introduce the interaction between incident electrons and carbon atoms in graphene. Then, the effects of electron energy and incident angle on irradiation defects in single-layer graphene were studied, and the cutting of single-layer graphene using different methods of electron irradiation was compared. Following this, the authors simulated the process of single atom chain formation from single-layer graphene using electron irradiation. They also demonstrated the formation of three-dimensional structures, such as tubular structures and nanotube junctions, in bilayer graphene by electron irradiation. The simulations show the capability of structural modification of graphene to a variety of nanostructures by electron irradiation.Molecular dynamics simulations have been used to study the structural modification of graphene by electron irradiation. The authors used the Monte Carlo method to introduce the interaction between incident electrons and carbon atoms in graphene. Then, the effects of electron energy and incident angle on irradiation defects in single-layer graphene were studied, and the cutting of single-layer graphene using different methods of electron irradiation was compared. Following this, the authors simulated the process of single atom chain formation from single-layer graphene using electron irradiation. They also demonstrated the formation of three-dimensional structures, such as tubular structures and nanotube junctions, in bilayer graphene by electron irradiation. The simulations show the capability of structural modification of graphene to a variety of nanostructures by electron irradiation. read less USED (high confidence) A. Sgouros, M. Sigalas, G. Kalosakas, K. Papagelis, and N. Papanicolaou, “Phononic band gap engineering in graphene,” Journal of Applied Physics. 2012. link Times cited: 15 Abstract: Using ab initio and molecular dynamics simulations with semi… read moreAbstract: Using ab initio and molecular dynamics simulations with semi-empirical potentials, the phonon density of states (PnDOS) of graphene with different types of defects such as substitution atoms (Si), carbon isotopes (12C and 14C), and vacancies was calculated. The main interest was to investigate the possibility to generate phononic band gaps (PBGs) in the PnDOS of graphene, since the derived structures may have sufficiently low thermal conductivity and find applications in improved thermoelectric materials. From all the studied defect types, the silicon substitution is the only one that creates PBGs. read less USED (high confidence) I. Chang and J. W. Chou, “A molecular analysis of carbon nanotori formation,” Journal of Applied Physics. 2012. link Times cited: 11 Abstract: This study uses molecular dynamics simulation to examine the… read moreAbstract: This study uses molecular dynamics simulation to examine the geometric criteria and stability of forming a perfect carbon nanotorus without pentagon-heptagon defects or surface buckles. Various nanotube diameters and nanoring diameters of both armchair and zigzag nanotori were relaxed at room temperature, and the equilibrated atomic configurations were inspected. This study uses the coordinate parameter, which illustrates the atomic arrangement around each atom, as an indicator of buckles to avoid misjudgment caused by transient or thermal disturbance. For each nanotube diameter, there exists a critical nanoring diameter beyond which the perfect carbon nanotori can form. This study examines the binding potential energy and deformation energy of the relaxed nanotorus model, showing that the critical nanoring diameter cannot be easily predicted through critical energy consideration because buckling is a form of structural instability. Results show that the structural stability of a perfect nanoring primarily depends on the nanotube diameter and nanoring diameter, whereas its chirality has little effect, and one empirical relation is fitted to determine the critical nanoring diameters. read less USED (high confidence) M. Wilson, “Model investigations of network-forming materials.,” Physical chemistry chemical physics : PCCP. 2012. link Times cited: 16 Abstract: Recent advances in the study of network-forming materials ar… read moreAbstract: Recent advances in the study of network-forming materials are described for systems dominated both by ionic and covalent interatomic interactions. Modelling strategies are described which focus both on describing specific systems of interest and on modelling the systematic evolution of network topology. The effect of network topology on the presence of ordering both on intermediate- and extended-length-scales is discussed. The effect of the topology on the mechanical rigidity is also described and analysed in terms of a mean coordination model. In addition, the isomorphology between amorphous silicon and the silicon sub-lattice in SiO(2) is described. Polyamorphism in Si and ZnCl(2) is analysed and discussed. Finally, the study of reduced (two) dimensional systems is discussed for carbon, silicon and germanium. read less USED (high confidence) V. Popok, J. Samela, K. Nordlund, and V. Popov, “Impact of keV-energy argon clusters on diamond and graphite,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2012. link Times cited: 7 USED (high confidence) T. Shiga, S. Konabe, J. Shiomi, T. Yamamoto, S. Maruyama, and S. Okada, “Graphene-diamond hybrid structure as spin-polarized conducting wire with thermally efficient heat sinks,” Applied Physics Letters. 2012. link Times cited: 20 Abstract: We have theoretically investigated electronic, magnetic, and… read moreAbstract: We have theoretically investigated electronic, magnetic, and thermal properties of a graphene-diamond hybrid structure consisting of a graphene nanoribbon with zigzag edges connected to diamond surfaces. From the first-principles calculation, we found that the hybrid structure is stable and that the ferro-magnetically ordered edge state appears around the graphene-diamond. On the other hand, from the non-equilibrium molecular dynamics simulations, we found that the thermal conductance at the interface between the graphene and diamond is 7.01±0.05GWm-2K-1 at the room temperature, which is much larger than that for covalently bonded interface between carbon nanotube and silicon. Thus, we propose that the hybrid structure is a potential candidate for spin-polarized conducting wires with thermally efficient heat sinks. read less USED (high confidence) K. Yashiro and M. Fujihara, “Molecular dynamics study on atomic elastic stiffness in Si under tension: homogenization by external loading and its limit,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 9 Abstract: As a series of studies that discuss the onset of inelastic d… read moreAbstract: As a series of studies that discuss the onset of inelastic deformation based on atomic elastic stiffness (AES), we investigated the AES in silicon by the Tersoff interatomic potential. For a comprehensive discussion including the effect of structural inhomogeneity by surface and grain boundaries, we performed tensile simulations on bulk/nanowire of Si single crystal, laminate bulk/bamboo nanowire with Σ5 twist grain boundary under a very low temperature (T = 1 K). Not only the stress–strain response, but also the AESs at each atom point, , were evaluated numerically by (Voigt notation) against local strain perturbation. The deviation of vanishes/diminishes by tension both in the homogeneous case of bulk perfect lattice and inhomogeneous ones with surface and grain boundaries; however, there is a certain limit for the homogenization. That is, the subtle deviation of AES in the perfect bulk vanishes by tension but it increases again like an onset of resonance, showing precursor stress decrease just before the unstable stress drop. In the inhomogeneous cases, we demonstrated that the near-zero AESs at the initial structural defects, e.g. surface or grain boundary, do not change but the positive AESs of other stable atoms approach zero by tension. When these distributions overlap each other, the standard deviation of AES increases again as if it were the first homogenization limit. However, the real homogenization starts at that point; that is, the AES distribution changes its shape to have a single peak at the border, suggesting that the difference of initial defects and other stable part vanishes before the system instability. read less USED (high confidence) P. Li and D.-feng Diao, “Molecular dynamics simulation of intrusion of a C60 molecule ball into sliding contact space,” Lubrication Science. 2012. link Times cited: 2 Abstract: The intrusion process of a C60 ball into a sliding contact s… read moreAbstract: The intrusion process of a C60 ball into a sliding contact space with an included angle made up by two silicon substrates (100) was simulated using the molecular dynamics approach. The simulation was carried out using Tersoff potential of C and Si atoms at room temperature of 300 K. The included angle was defined as initial entry angle changing from 20° to 90° in the simulation for studying the effect of the initial entry angle during the intrusion process. The dependence of the initial entry angle on the number of sticking Si atoms of upper substrate was calculated. The results showed that the number of sticking atoms increased with the increasing of initial entry angle, and the number of sticking atoms was divided into three regions with different slopes, which could be used to evaluate the intrusion performance of a C60 ball into the sliding contact space. Copyright © 2011 John Wiley & Sons, Ltd. read less USED (high confidence) C. Reinke et al., “Thermal conductivity prediction of nanoscale phononic crystal slabs using a hybrid lattice dynamics-continuum mechanics technique,” AIP Advances. 2011. link Times cited: 26 Abstract: Recent work has demonstrated that nanostructuring of a semic… read moreAbstract: Recent work has demonstrated that nanostructuring of a semiconductor material to form a phononic crystal (PnC) can significantly reduce its thermal conductivity. In this paper, we present a classical method that combines atomic-level information with the application of Bloch theory at the continuum level for the prediction of the thermal conductivity of finite-thickness PnCs with unit cells sized in the micron scale. Lattice dynamics calculations are done at the bulk material level, and the plane-wave expansion method is implemented at the macrosale PnC unit cell level. The combination of the lattice dynamics-based and continuum mechanics-based dispersion information is then used in the Callaway-Holland model to calculate the thermal transport properties of the PnC. We demonstrate that this hybrid approach provides both accurate and efficient predictions of the thermal conductivity. read less USED (high confidence) Y. Chihara, M. Yasuda, S. Wakuda, H. Kawata, and Y. Hirai, “Computational study of electron-irradiation effects in carbon nanomaterials on substrates,” Journal of Vacuum Science & Technology. B. Nanotechnology and Microelectronics: Materials, Processing, Measurement, and Phenomena. 2011. link Times cited: 7 Abstract: Molecular dynamics simulation is performed to study electron… read moreAbstract: Molecular dynamics simulation is performed to study electron-irradiation effects in carbon nanomaterials on substrates. The interaction between an incident electron and a carbon atom in target nanomaterials is introduced by the Monte Carlo method. Collisions of the backscattered electrons from the substrate are also introduced. The distributions of energy and the exit angle of backscattered electrons are calculated using Monte Carlo simulation of electron scattering in the substrate. Structural changes become more remarkable when the carbon nanomaterials are on the substrates. The threshold energy and the characteristics of structural changes by backscattered electrons are also discussed. read less USED (high confidence) F. Schäffel, M. W. B. Wilson, and J. Warner, “Motion of light adatoms and molecules on the surface of few-layer graphene.,” ACS nano. 2011. link Times cited: 28 Abstract: Low-voltage aberration-corrected transmission electron micro… read moreAbstract: Low-voltage aberration-corrected transmission electron microscopy (TEM) is applied to investigate the feasibility of continuous electron beam cleaning of graphene and monitor the removal of residual species as present on few-layer graphene (FLG) surfaces. This combined approach allows us to detect light adatoms and evaluate their discontinuous sporadic motional behavior. Furthermore, the formation and dynamic behavior of isolated molecules on the FLG surface can be captured. The preferential source of adatoms and adsorbed molecules appeared to be carbonaceous clusters accumulated from residual solvents on the graphene surface. TEM image simulations provide potential detail on the observed molecular structures. Molecular dynamics simulations confirm the experimentally observed dynamics occurring on the energy scale imposed by the presence of the 80 kV electron beam and help elucidate the underlying mechanisms. read less USED (high confidence) D. Tang et al., “Mechanical properties of bamboo-like boron nitride nanotubes by in situ TEM and MD simulations: strengthening effect of interlocked joint interfaces.,” ACS nano. 2011. link Times cited: 63 Abstract: Understanding the influence of interfacial structures on the… read moreAbstract: Understanding the influence of interfacial structures on the nanoarchitecture mechanical properties is of particular importance for its mechanical applications. Due to a small size of constituting nanostructural units and a consequently high volume ratio of such interfacial regions, this question becomes crucial for the overall mechanical performance. Boron nitride bamboo-like nanotubes, called hereafter boron nitride nanobamboos (BNNBs), are composed of short BN nanotubular segments with specific interfaces at the bamboo-shaped joints. In this work, the mechanical properties of such structures are investigated by using direct in situ transmission electron microscopy tensile tests and molecular dynamics simulations. The mechanical properties and deformation behaviors are correlated with the interfacial structure under atomic resolution, and a geometry strengthening effect is clearly demonstrated. Due to the interlocked joint interfacial structures and compressive interfacial stresses, the deformation mechanism is switched from an interplanar sliding mode to an in-plane tensile elongation mode. As a result of such a specific geometry strengthening effect, the BNNBs show high tensile fracture strength and Young's modulus up to 8.0 and 225 GPa, respectively. read less USED (high confidence) X. Zhang and Z.-wei Sun, “Influences of vacancy defects on thermal conductivities of Ge thin films,” Rare Metals. 2011. link Times cited: 3 USED (high confidence) H. Lan, C. Liu, Y. Cui, and T. Kato, “Molecular simulations of sliding process between Fe and DLC films on various boundary conditions,” 2011 Seventh International Conference on Natural Computation. 2011. link Times cited: 0 Abstract: Diamond-like carbon (DLC) films have been extensively studie… read moreAbstract: Diamond-like carbon (DLC) films have been extensively studied over the past decades due to their unique combination of properties, such as a low friction coefficient, high hardness, high wear resistance and chemical inertness. But many results of DLC films exhibit a wide range of sometimes contradictory tribological properties. In the experiment, isolating the influences of factors, such as film structure, testing conditions and environments effects has proven different. In the paper, molecular dynamics (MD) simulations were used to study a sliding friction process between Fe and DLC films on various boundary conditions (such as: water, oil and no lubrication). The results have been shown that for all the boundary conditions, boundary lubrication occurs, even where there is no lubricant between the Fe and DLC films. The Lubrication boundaries have little effect on the tribological properties between Fe and DLC films. The friction forces for all the cases are almost the same. read less USED (high confidence) J. A. Driscoll, S. Bubin, W. R. French, and K. Varga, “Time-dependent density functional study of field emission from nanotubes composed of C, BN, SiC, Si, and GaN,” Nanotechnology. 2011. link Times cited: 7 Abstract: Field emission from various types of nanotubes is studied by… read moreAbstract: Field emission from various types of nanotubes is studied by propagating the electronic density in real space and time using time-dependent density functional theory. Capped (5, 5) C, BN, SiC, Si, and GaN nanotubes are considered. The GaN, SiC, and Si nanotubes were found to be significantly better field emitters than C and BN nanotubes, both in terms of current magnitude and sharpness of peaks in the energy spectra. By analyzing the electronic structure of the various systems it is seen that the nanotubes with the highest currents have electron densities that extend significantly from the nanotube in the emission direction. read less USED (high confidence) G. Balasubramanian and I. Puri, “Heat conduction across a solid-solid interface: Understanding nanoscale interfacial effects on thermal resistance,” Applied Physics Letters. 2011. link Times cited: 76 Abstract: Phonons scatter and travel ballistically in systems smaller … read moreAbstract: Phonons scatter and travel ballistically in systems smaller than the phonon mean free path. At larger lengths, the transport is instead predominantly diffusive. We employ molecular dynamics simulations to describe the length dependence of the thermal conductivity. The simulations show that the interfacial thermal resistance Rk for a Si-Ge superlattice is inversely proportional to its length, but reaches a constant value as the system dimension becomes larger than the phonon mean free path. This nanoscale effect is incorporated into an accurate continuum model by treating the interface as a distinct material with an effective thermal resistance equal to Rk. read less USED (high confidence) C. Wu, T. Fang, and C. Chan, “A molecular dynamics simulation of the mechanical characteristics of a C60-filled carbon nanotube under nanoindentation using various carbon nanotube tips,” Carbon. 2011. link Times cited: 43 USED (high confidence) T. Luo and J. Lloyd, “Molecular dynamics study of thermal transport in GaAs-self-assembly monolayer-GaAs junctions with ab initio characterization of thiol-GaAs bonds,” Journal of Applied Physics. 2011. link Times cited: 33 Abstract: Thermal dissipation in molecular electronic devices is a cri… read moreAbstract: Thermal dissipation in molecular electronic devices is a critical issue for the proper functioning of such devices. In this work, molecular dynamics (MD) simulations were carried out to study the thermal energy transport in GaAs-SAM (self-assembly monolayer)-GaAs junctions, with alkanedithiols being the SAM molecules. In order to characterize the molecule-GaAs interface, ab initio density functional theory (DFT) was used to study the structural and binding properties of alkanethiolates on GaAs(001) surfaces. Parameters of classical potentials, which were used to model the molecule-GaAs interactions, were obtained by fitting to the results from the DFT calculations. Then, nonequilibrium MD (NEMD) simulations were performed to reveal the GaAs-SAM interfacial thermal conductance at different temperatures. The results from this work showed that the GaAs-SAM interfaces are the major sources of thermal resistance in the GaAs-SAM-GaAs junctions. The delocalized phonon modes carry thermal energy efficiently insid... read less USED (high confidence) D. Konatham, K. Bui, D. Papavassiliou, and A. Striolo, “Simulation insights into thermally conductive graphene-based nanocomposites,” Molecular Physics. 2011. link Times cited: 54 Abstract: Dispersing nanoparticles in a polymer can enhance both mecha… read moreAbstract: Dispersing nanoparticles in a polymer can enhance both mechanical and transport properties. Nanocomposites with high thermal conductivity could be obtained by using thermally conductive nanoparticles. Carbon-based nanoparticles are extremely promising, although high resistances to heat transfer from the nanoparticles to the polymer matrix could cause significant limitations. This work focuses on graphene sheets (GS) dispersed within n-octane. Although pristine GS agglomerate, equilibrium molecular dynamic simulations suggest that when the GS are functionalized with short branched hydrocarbons along the GS edges, they remain well dispersed. Results are reported from equilibrium and non-equilibrium molecular dynamics simulations to assess the effective interactions between dispersed GS, the self-assembly of GS, and the heat transfer through the GS–octane nanocomposite. Tools are designed to understand the effect of GS size, solvent molecular weight and molecular architecture on GS dispersability and GS–octane thermal conductivity. Evidence is provided for the formation of nematic phases when the GS volume fraction increases within octane. The atomic-level results are input for a coarse-grained Monte Carlo simulation that predicts anisotropic thermal conductivity for GS-based composites when the GS show nematic phases. read less USED (high confidence) X. Chang, Y. Ge, and J. Dong, “Ripples of AA and AB stacking bilayer graphenes,” The European Physical Journal B. 2010. link Times cited: 5 USED (high confidence) S.-heon Lee, K. Shin, and W. Lee, “Modeling of CNTs and CNT-Matrix Interfaces in Continuum-Based Simulations for Composite Design,” Korean Journal of Materials Research. 2010. link Times cited: 2 Abstract: A series of molecular dynamic (MD), finite element (FE) and … read moreAbstract: A series of molecular dynamic (MD), finite element (FE) and ab initio simulations are carried out to establishsuitable modeling schemes for the continuum-based analysis of aluminum matrix nanocomposites reinforced with carbonnanotubes (CNTs). From a comparison of the MD with FE models and inferences based on bond structures and electrondistributions, we propose that the effective thickness of a CNT wall for its continuum representation should be related to thegraphitic inter-planar spacing of 3.4 A. We also show that shell element representation of a CNT structure in the FE modelsproperly simulated the carbon-carbon covalent bonding and long-range interactions in terms of the load-displacement behaviors.Estimation of the effective interfacial elastic properties by ab initio simulations showed that the in-plane interfacial bond strengthis negligibly weaker than the normal counterpart due to the nature of the weak secondary bonding at the CNT-Al interface.Therefore, we suggest that a third-phase solid element representation of the CNT-Al interface in nanocomposites is not physicallymeaningful and that spring or bar element representation of the weak interfacial bonding would be more appropriate as in the casesof polymer matrix counterparts. The possibility of treating the interface as a simply contacted phase boundary is also discussed.Key wordscarbon nanotube, molecular dynamic simulation; ab initio simulation; nanocomposite. read less USED (high confidence) C. Lin, H. Wang, and W. Yang, “The thermomutability of single-walled carbon nanotubes by constrained mechanical folding,” Nanotechnology. 2010. link Times cited: 8 Abstract: The thermomutability of single-walled carbon nanotubes (SWCN… read moreAbstract: The thermomutability of single-walled carbon nanotubes (SWCNT) with an ultrahigh aspect ratio has been systematically investigated by molecular dynamics simulations. A constraining matrix is necessary to transform the tube-long Euler instability to a localized cross-section flattening which can effectively hinder phonon transportation at the throttling kinks. A thermal conductivity reduction of ∼ 50% was obtained for a 100 nm (5, 5) SWCNT when embedded in a compliant matrix (E ∼ 0.4 GPa). An even larger reduction (∼80%) can be achieved for a stiffer matrix (40 GPa). The thermal conductivity decreases abruptly at the buckling portion which further develops into folding and connected spirals. The constrained mechanical folding is highly desirable for realizing ultra-sensitive thermal sensors or switches. read less USED (high confidence) R. Jones, J. Templeton, G. Wagner, D. Olmsted, and N. A. Modine, “Electron transport enhanced molecular dynamics for metals and semi‐metals,” International Journal for Numerical Methods in Engineering. 2010. link Times cited: 21 Abstract: In this work we extend classical molecular dynamics by coupl… read moreAbstract: In this work we extend classical molecular dynamics by coupling it with an electron transport model known as the two temperature model. This energy balance between free electrons and phonons was first proposed in 1956 by Kaganov et al. but has recently been utilized as a framework for coupling molecular dynamics to a continuum description of electron transport. Using finite element domain decomposition techniques from our previous work as a basis, we develop a coupling scheme that preserves energy and has local control of temperature and energy flux via a Gaussian isokinetic thermostat. Unlike the previous work on this subject, we employ an efficient, implicit time integrator for the fast electron transport which enables larger stable time steps than the explicit schemes commonly used. A number of example simulations are given that validate the method, including Joule heating of a copper nanowire and laser excitation of a suspended carbon nanotube with its ends embedded in a conducting substrate. Published in 2010 by John Wiley & Sons, Ltd. read less USED (high confidence) J. Warner and M. W. B. Wilson, “Elastic distortions of carbon nanotubes induced by chiral fullerene chains.,” ACS nano. 2010. link Times cited: 15 Abstract: We show that when Y@C82 metallofullerenes are inserted into … read moreAbstract: We show that when Y@C82 metallofullerenes are inserted into single-walled carbon nanotubes (SWNTs) with large diameters of 2 nm, the minimum energy configuration is a double-helix chiral structure extending over hundreds of nanometers. We demonstrate rotation of the double-helix fullerene chain within the nanotube host that induces real time elastic distortions of the nanotube in a crank-shaft manner. Molecular dynamics simulations, employing an atomic description of the confining SWNT and a reduced description of Y@C82, reproduce the key experimental observations. read less USED (high confidence) S. Lu and C. Cho, “A New Extension of Cauchy–Born Rule for Monolayer Crystal Films,” Nanoscale Research Letters. 2010. link Times cited: 0 USED (high confidence) M. Yasuda, K. Tada, and Y. Hirai, “Molecular Dynamics Study on Mold and Pattern Breakages in Nanoimprint Lithography.” 2010. link Times cited: 7 Abstract: Nanoimprint lithography (NIL) is one of the promising techno… read moreAbstract: Nanoimprint lithography (NIL) is one of the promising technologies for the fabrication of nanostructures at low cost (Chou et al., 1995) (Chou et al., 1996). In NIL, understanding the deformation behaviour of polymer during imprinting processes is an essential issue for high-speed and uniformed fabrication. Since numerical simulations can be efficient approaches for this issue, several studies using continuum mechanics are performed (Hirai et al., 2001) (Hirai et al., 2004) (Song et al., 2008). Continuum mechanics successfully predict the material deformation in submicron scale. However, as the pattern size becomes smaller than several tens of nanometers, continuum mechanics fails to analyze the material behaviour. Single-nanometre resolution has experimentally been demonstrated in NIL (Hua et al., 2004) (Hua et al., 2006). For the exact analysis of the material deformation in nanoscale system, the behaviour of atoms or molecules should be considered. Molecular dynamics (MD) simulation is a useful tool to study the deformation mechanism of the materials in atomic scale. Several MD studies on NIL process are reported. Kang et al. propose a MD simulation model of a NIL process imprinting an α-quartz stamp into an amorphous poly-(methylmethacrylate) film (Kang et al., 2007). In their study, the distributions of density and stress in the polymer film are calculated for the detail analysis of deformation behaviour. The qualitative agreement between the MD simulation and the experimental data for the density variation of patterned polymer is reported (Woo et al., 2007). Mold geometry effect on springback phenomenon in NIL process is also studied with the MD simulation (Yang et al., 2009). For metal direct imprinting, more MD studies are performed. Process parameters such as stamp taper angle, imprint depth, temperature and punch velocity are investigated for copper imprinting (Hsu et al., 2004) (Hsu et al., 2005). The mechanism of the atomic-scale friction is studied for aluminium imprinting (Hsieh & Sung, 2007). The metal film thickness effect on pattern formation is also studied (Cheng et al., 2007). Agreement between MD simulation and experimental results is reported for temperature effects on gold imprinting (Hsiung et al., 2009). MD simulation of nanoimprint for alloys is demonstrated (Fang et al., 2007). In order to save computational time, a multi-scale simulation for nanoimprint process that mixes the atomistic and continuum approaches is proposed (Wu & Lin, 2008). Recently, MD simulation of roller nanoimprint process is performed (Wu et al., 2009). read less USED (high confidence) G.-Q. Li, J.-K. Deng, and C. Jun, “Dependence of Conductance of Corrugated Graphene Quantum Dot on Geometrical Features,” Communications in Theoretical Physics. 2009. link Times cited: 1 Abstract: Dependence of conductance of corrugated graphene quantum dot… read moreAbstract: Dependence of conductance of corrugated graphene quantum dot (CGQD) on geometrical features including length, width, connection and edge is investigated by the first principles calculations. The results demonstrate that the conductance of CGQD with different geometrical features is different from each other. The positions and amplitudes of discrete levels in densities of states and transmission coefficients are sensitive to geometrical features. The I-V characteristics of graphene are modified by size and edge, it is surprise the current does not change monotonously but oscillatory with length. And they are slight change for different connections. read less USED (high confidence) P. Palla, S. Giordano, and L. Colombo, “Interface elasticity in nanostructured silicon,” Physical Review B. 2009. link Times cited: 7 Abstract: We investigate through atomistic simulations the mechanical … read moreAbstract: We investigate through atomistic simulations the mechanical behavior of a c-Si nanowire embedded in an elastically different c-Si. The results are compared with the continuum predictions based on the elasticity theory. The observed deviations between the two approaches are due to the presence of the disordered interface in the atom-resolved system which, for small wires, induces sizable prestrain into the sample, also in absence of any external loads. Finally, we develop a continuum model fully exploiting such interface effects provided by the atomistic simulations. read less USED (high confidence) X. Liu, D. Cheng, and D. Cao, “The structure, energetics and thermal evolution of SiGe nanotubes,” Nanotechnology. 2009. link Times cited: 20 Abstract: The structure, energetics and thermal behavior of all the Si… read moreAbstract: The structure, energetics and thermal behavior of all the SiGe nanotubes in armchair and zigzag structures (n = 4–10) and two atomic arrangement types are investigated using the ab initio method and classical molecular dynamics simulations. Gearlike and puckering configurations of SiGe nanotubes are obtained. The simulation results indicate that large-diameter nanotubes are more stable than small-diameter ones. Moreover, the type 1 (alternating atom arrangement type) zigzag nanotubes are always more energetically favorable than the type 2 (layered atom arrangement type) zigzag nanotubes. During the melting process, the melting-like structural transformations from the initial nanotube to the compact nanowire take place first, and then the compact nanowires are changed into agglomerate structures at higher temperature. It is also found that the melting-like temperatures of Ge-substituted silicon nanotubes decrease with increase of the Ge concentration. read less USED (high confidence) M. Griebel, J. Hamaekers, and F. Heber, “A molecular dynamics study on the impact of defects and functionalization on the Young modulus of boron–nitride nanotubes,” Computational Materials Science. 2009. link Times cited: 58 USED (high confidence) H.-J. Kim, T. Oh, and D.-E. Kim, “Comparison of Indentation and Scribing Behaviors of Crystalline and Initially Deformed Silicon Tips by Molecular Dynamics Simulation,” IEEE Transactions on Magnetics. 2009. link Times cited: 4 Abstract: Silicon probe tips are used widely in micro and nano-systems… read moreAbstract: Silicon probe tips are used widely in micro and nano-systems such as AFM, MEMS, and probe recording. The mechanical integrity of the tip is important to assure reliable performance of the tip during contact as well as sliding. Crystalline silicon normally forms a tetrahedral structure, however, under high pressure it is known that the structure transforms to a different phase. This can cause a change in the contact phenomena. In this work, the silicon probe tip deformation process during nano-indentation was investigated by using molecular dynamics simulation. In addition, scribing simulation was carried out to observe the frictional characteristics of crystalline and amorphous silicon structures. The simulation results showed that the structure of silicon near the surface was permanently deformed at a contact stress of approximately 17 GN/m2 and the deformation process could be monitored by observing the bond-angle distribution graph. It was also found that the atomic structure of the silicon tip in the contact region affected the frictional behavior of the tip with respect to fluctuation periodicity and magnitude. read less USED (high confidence) S. Y. Kim and H. S. Park, “Multilayer friction and attachment effects on energy dissipation in graphene nanoresonators,” Applied Physics Letters. 2009. link Times cited: 46 Abstract: We utilize classical molecular dynamics to study the effects… read moreAbstract: We utilize classical molecular dynamics to study the effects of intrinsic, interlayer friction between graphene monolayers, as well as extrinsic attachment or clamping strength between graphene and a model silicon substrate on the energy dissipation (Q-factors) of oscillating graphene nanoresonators. Both interlayer friction and attachment effects are found to significantly degrade the graphene Q-factors, with an increase in energy dissipation with increasing temperature, while both effects are found to be strongly dependent on the strength of the van der Waals interactions, either between adjacent layers of graphene or between graphene and the underlying substrate. read less USED (high confidence) S.-M. Jeong and T. Kitamura, “Phase Transformation of Silicon under Nonhydrostatic Stress State: Formation of Si II Phase under a Round Indenter,” Japanese Journal of Applied Physics. 2009. link Times cited: 1 Abstract: Silicon transforms from a diamond cubic structure to a β-Sn … read moreAbstract: Silicon transforms from a diamond cubic structure to a β-Sn structure as induced by mechanical stress, the magnitude of which changes depending on the stress state. In this study, the origin of the transformed silicon is explored with a stress criterion independent of the stress state. As an illustration of a nonhydrostatic stress state, the stress distribution of silicon under a round indenter is analyzed by finite element analysis. As a result, the initial formation of β-Sn structured silicon is successfully explained using the new stress criterion. read less USED (high confidence) L. Bagolini, A. Mattoni, and L. Colombo, “Electronic localization and optical absorption in embedded silicon nanograins,” Applied Physics Letters. 2009. link Times cited: 14 Abstract: We study the spatial distribution of electron states in crys… read moreAbstract: We study the spatial distribution of electron states in crystalline Si nanograins embedded into amorphous silicon. We prove that it is not possible to tune the absorption gap by only controlling the size of the grain, since no quantum confinement there occurs. The absorption properties of such a two-phase system are rather controlled by the population of localized electron states generated by large angular distortions of Si–Si bonds. read less USED (high confidence) R. Gatti, A. Marzegalli, V. Zinovyev, F. Montalenti, and L. Miglio, “Modeling the plastic relaxation onset in realistic SiGe islands on Si(001),” Physical Review B. 2008. link Times cited: 55 Abstract: A detailed investigation of plastic relaxation onset in hete… read moreAbstract: A detailed investigation of plastic relaxation onset in heteroepitaxial SiGe islands on Si(001) is presented. The strain field induced by a straight misfit-dislocation segment is modeled by finite-element-method (FEM) calculations in three dimensions, fully taking into account the interaction with the multifaceted free surfaces of realistic islands. The total elastic energies before and after the placement of a $60\ifmmode^\circ\else\textdegree\fi{}$ dislocation segment in the most favorable position are therefore evaluated by a full FEM approach, for different island sizes and compositions. The critical volumes with composition for inserting the dislocation are finally obtained and successfully compared with the data in a report by Marzegalli et al. [Phys. Rev. Lett. 99, 235505 (2007)], where experimental values are compared to a simpler approach. read less USED (high confidence) M. Suri and T. Dumitricǎ, “Efficient sticking of surface-passivated Si nanospheres via phase-transition plasticity,” Physical Review B. 2008. link Times cited: 17 Abstract: The fundamental understanding of the nanoparticlesurface col… read moreAbstract: The fundamental understanding of the nanoparticlesurface collision modes in the low-energy regime of less than 1e V/atom is of considerable importance because achieving efficient sticking to surfaces with preservation of a grainy structure are key issues for creating new materials. Nanoparticle production in the gas phase, coupled with the application of chemical coatings and the deposition onto substrates, is a combination of aerosol technologies used for manufacturing novel nanostructured surfaces and thin films. 1‐6 Chemical passivation in the gas phase is essential for preventing coalescence and large particle growth but makes the deposition step challenging. Deposition strategies have been developed, some necessitating the creation of defects in the substrate in order to pin the impinging nanoparticles, 1 which otherwise would be reflected. Remarkably, read less USED (high confidence) K. Tada, M. Yasuda, Y. Kimoto, H. Kawata, and Y. Hirai, “Molecular Dynamics Study of Yield Stress of Si Mold for Nanoimprint Lithography,” Japanese Journal of Applied Physics. 2008. link Times cited: 11 Abstract: Molecular dynamics studies are carried out to investigate th… read moreAbstract: Molecular dynamics studies are carried out to investigate the fracture mechanism of a crystalline Si mold for nanoimprint lithography (NIL). The stress–strain characteristics are evaluated for mold models with various crystalline orientations, and temperature effects on strength are calculated. It is found that the behavior of the fracture and the yield stress of the mold are strongly associated with the configurations of the {111} planes in the mold. The simulation results indicate that crystals with a {100} top surface are more suitable for a mold with arbitrary patterns than those with a {110} top surface because of the weaker dependence of yield stress on crystalline orientation. read less USED (high confidence) D. Grimm et al., “Synthesis of SWCNT rings made by two Y junctions and possible applications in electron interferometry.,” Small. 2007. link Times cited: 14 USED (high confidence) P. Valentini and T. Dumitricǎ, “Molecular Dynamics Simulations of Nanoparticle-Surface Collisions in Crystalline Silicon,” Journal of Nano Research. 2007. link Times cited: 7 Abstract: We present a microscopic description for the impacting proce… read moreAbstract: We present a microscopic description for the impacting process of silicon nanospheres onto a silicon substrate. In spite of the relatively low energy regime considered (up to 1 eV/atom), the impacting process exhibits a rich behavior: A rigid Hertzian model is valid for speeds below 500 m/s, while a quasi-ellipsoidal deformation regime emerges at larger speeds. Furthermore, for speeds up to 1000 m/s the particle undergoes a soft landing and creates a long-lived coherent surface phonon. Higher speeds lead to a rapid attenuation of the coherent phonon due to a partial diamond cubic to-tin phase transformation occurring in the particle. read less USED (high confidence) D. Grimm, A. Latgé, and P. Venezuela, “Standing‐wave observations in single‐wall carbon nanotube quantum dots and Y‐junction rings,” physica status solidi (b). 2007. link Times cited: 0 Abstract: Standing electronic wave formation inside single walled carb… read moreAbstract: Standing electronic wave formation inside single walled carbon nanotube Y‐junction rings and quantum dots with a quantum‐dot‐like quadratic dispersion is reported. Remarkably, the detailed structural defect configurations play only a minor role and the standing wave oscillation dispersions are mainly triggered by the electronic properties of the pristine constituent tubes. This raises the possibility of using both structures as nanoscale tuneable electronic switching devices. Near the Fermi level, quantum dots show smooth dispersing oscillations reminiscent of the finite pristine tube eigenvalues. However, localized density of states peaks are found in YJRs, which are identified as interference effects of the double‐slit interferometer. (© 2007 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less USED (high confidence) J. Adhikari and A. Kumar, “Study of structural and thermodynamic properties of GaAs and InAs using Monte Carlo simulations,” Molecular Simulation. 2007. link Times cited: 6 Abstract: Binary compound semiconductor alloys such as GaAs and InAs f… read moreAbstract: Binary compound semiconductor alloys such as GaAs and InAs find extensive use in our daily lives. This study predicts the structural and thermodynamic properties such as the lattice constant, linear thermal expansion coefficient, nearest neighbour distances and molar heat capacities at constant volume, and their variations with temperature using Monte Carlo simulations. The Tersoff potential model is used to describe the interatomic interactions and the model is validated by comparing the predicted properties against experimental data for GaAs. The simulation results for the GaAs alloy show good agreement with literature data for lattice constant and bond length measurements. Linear thermal expansion coefficients are overestimated consistently as compared to experimental data for all temperatures. Low temperature range thermal expansion coefficient data capture qualitative behaviour but is unable to accurately predict quantitative data. The specific heat at constant volume measured at high temperatures follows the Dulong–Petit law. Having established the validity of the Tersoff potential in modelling III–V binary alloys, the same properties and their variance with temperature are determined for the InAs alloy. read less USED (high confidence) W. Liu et al., “Surface reconstruction and core distortion of silicon and germanium nanowires,” Nanotechnology. 2007. link Times cited: 9 Abstract: We report the results of molecular dynamics simulations for … read moreAbstract: We report the results of molecular dynamics simulations for structures of pristine silicon nanowires and germanium nanowires with bulk cores oriented along the [110] direction and bounded by the (100) and (110) surfaces in the lateral direction. We found that the (100) surfaces for both silicon and germanium nanowires undergo 2 × 1 dimerization while their (110) surfaces do not show reconstruction. The direction of the dimer rows is either parallel or perpendicular to the wire axis depending on the orientation of the surface dangling bonds. The dimer length for Si is in good agreement with the result obtained by first-principles calculations. However, the geometry of Si dimers belongs to the symmetrical 2 × 1 reconstruction rather than the asymmetrical buckled dimers. We also show that surface reconstruction of a small nanowire induces significant change in the lattice spacing for the atoms not on the (100) surface, resulting in severe structural distortion of the core of the nanowire. read less USED (high confidence) J. Hsieh, J.-M. Lu, M. Huang, and C. Hwang, “Theoretical variations in the Young’s modulus of single-walled carbon nanotubes with tube radius and temperature: a molecular dynamics study,” Nanotechnology. 2006. link Times cited: 75 Abstract: This study uses molecular dynamics simulations to investigat… read moreAbstract: This study uses molecular dynamics simulations to investigate the intrinsic thermal vibrations of a single-walled carbon nanotube (SWNT) modelled as a clamped cantilever. Using an elastic model defined in terms of the tube length, the tube radius and the tube temperature, the standard deviation of the vibrational amplitude of the tube’s free end is calculated and the Young’s modulus of the SWNT evaluated. The numerical results reveal that the value of the Young’s modulus is independent of the tube length, but decreases with increasing tube radius. At large tube radii, the Young’s modulus value approaches the in-plane modulus of graphene, which can be regarded as an SWNT of infinitely large radius. The results also indicate that the Young’s modulus is insensitive to changes in the tube temperature at temperatures of less than approximately 1100 K, but decreases significantly at higher temperatures. read less USED (high confidence) M. Endo et al., “Nanotube coalescence-inducing mode: a novel vibrational mode in carbon systems.,” Small. 2006. link Times cited: 70 USED (high confidence) S. Pizzini, S. Binetti, A. Donne, A. Marzegalli, and J. Rabier, “Optical properties of shuffle dislocations in silicon,” Applied Physics Letters. 2006. link Times cited: 12 Abstract: The radiative recombination processes in dislocated float zo… read moreAbstract: The radiative recombination processes in dislocated float zone silicon samples deformed under gigapascal stresses were studied by photoluminescence (PL) spectroscopy. The observed shuffle dislocations present a reconstructed core and their generation is accompanied by the introduction of point defects and point defect clusters, whose signature is evident in the PL spectra. A broad band around 1eV is the only PL feature which could be directly related to shuffle dislocations and it is explained conjecturing strain field induced gap changes, as confirmed by molecular dynamics simulations. read less USED (high confidence) D. Mulliah, S. Kenny, E. Mcgee, R. Smith, A. Richter, and B. Wolf, “Atomistic modelling of ploughing friction in silver, iron and silicon,” Nanotechnology. 2006. link Times cited: 44 Abstract: Molecular dynamics (MD) simulations of atomic-scale stick–sl… read moreAbstract: Molecular dynamics (MD) simulations of atomic-scale stick–slip have been performed for a diamond tip in contact with the (100) surface of fcc Ag, bcc Fe, Si and H-terminated Si, at a temperature of 300 K. Simulations were carried out at different support displacements between 5 and 15 Å. The simulations illustrate the important mechanisms that take place during stick–slip. In particular, for the case of the metals they show a direct link between tip slip events and the emission of dislocations from the point of contact of the tip with the substrate. This occurs both during indentation and scratching. For the case of silicon, no slip events were observed and no subsurface dislocations were generated underneath the scratch groove. At the deeper support displacement of 15 Å the silicon atoms undergo some local phase transformations and the atom coordination number varies between 5 and 8, with the majority being five-fold or six-fold coordinated. Both the dynamic and the static friction coefficients were found to be higher for Si compared to the corresponding values for H-terminated Si. Comparisons were made between the MD simulations and experimental measurements for indentation on the (100) surface of Si and Al. A good qualitative agreement was observed between the experimental and theoretical results. However, in both the cases of Si and metals the MD simulations give a contact pressure under load that is depth dependent and values that are higher than experimental nanohardness values. read less USED (high confidence) S. Hai-yang, S. He-ming, and Z. Guo-xiang, “Molecular Dynamics Study of Effects of Si-Doping Upon Structure and Mechanical Properties of Carbon Nanotube,” Communications in Theoretical Physics. 2006. link Times cited: 24 Abstract: In this paper, a Si-doped single-walled carbon nanotube (SWC… read moreAbstract: In this paper, a Si-doped single-walled carbon nanotube (SWCNT) (7,7) and several perfect armchair SWCNTs are investigated using the classical molecular dynamics simulations method. The inter-atomic short-range interaction is represented by empirical Tersoff bond order potential. The computational results show that the axial Young's modulus of the perfect SWCNTs are in the range of 1.099±0.005 TPa, which is in good agreement with the existing experimental results. From our simulation, the Si-doping decreases the Young's modulus of SWCNT, and with the increased strain levels, the effect of Si-doped layer in enhancing the local stress level increases. The Young's modulus of armchair SWCNTs are weakly affected by tube radius. read less USED (high confidence) C. Wei, “Adhesion and reinforcement in carbon nanotube polymer composite,” Applied Physics Letters. 2006. link Times cited: 75 Abstract: Temperature dependent adhesion behavior and reinforcement in… read moreAbstract: Temperature dependent adhesion behavior and reinforcement in carbon nanotube (CNT)-polymer (polyethylene) composite is studied through molecular dynamics simulations. The interfacial shear stress through van der Waals interactions is found to increase linearly with applied tensile strains along the nanotube axis direction, until the noncovalent bonds between CNTs and molecules break successively. A lower bound value about 46MPa is found for the shear strength at low temperatures. Direct stress-strain calculations show significant reinforcements in the composite in a wide temperature range, with ∼200% increase in the Young’s modulus when adding 6.5% volume ratio of short CNTs, and comparisons with the Halpin–Tsai formula are discussed. read less USED (high confidence) C.-Y. Chen and D. Kopelevich, “Quasi-one-dimensional nanostructures and efficient heat transfer in nanoscale devices,” SPIE Optics East. 2005. link Times cited: 0 Abstract: The steady decrease of the feature size of integrated circui… read moreAbstract: The steady decrease of the feature size of integrated circuits towards the nanometer scale leads to an increase in generated heat per unit area. Hence, efficient transfer of heat away from hotspots of integrated circuits becomes a crucial issue in the design of new generations of electronic devices. The importance of efficient thermal transport is even more pronounced in moving parts of nanoelectromechanical systems (NEMS). Recent research has shown that low-dimensional nanomaterials possess high thermal conductivity and hence are promising candidates for efficient heat reduction in nanodevices. In this talk, we present results of theoretical modeling of heat transport in one-dimensional (e.g. long chain molecules) and quasi-one-dimensional (e.g. carbon nanotubes) nanostructures. The study is performed under the assumption that the contribution of electrons to thermal conductivity is negligible and therefore the heat transfer is solely due to nonlinear interactions between vibrations of atoms in a nanostructure. We investigate the role of various lattice vibration modes in the heat transport with a particular focus on nonlinear localized vibration modes (breathers). These modes are highly localized and have properties qualitatively different from the linear phonon vibration modes. In particular, breathers are very stable and, at certain conditions, they move at a constant velocity which exceeds the speed of sound. This property of breathers suggests their potential use in efficient transfer of heat away from hotspots in a nanoscopic device. read less USED (high confidence) P. Erhart and K. Albe, “Molecular Dynamics Simulations of Gas Phase Condensation of Silicon Carbide Nanoparticles,” Advanced Engineering Materials. 2005. link Times cited: 12 Abstract: Gas phase condensation of silicon and silicon carbide nanopa… read moreAbstract: Gas phase condensation of silicon and silicon carbide nanoparticles is studied by molecular-dynamics simulations. By using a recently developed bond-order potential for Si, C and SiC we investigate the fundamental processes governing nucleation and growth of SiC nanoparticles. For the case of elemental silicon particles we show that variations in the binding energy of dimers, which represent stable nuclei for the condensation process, significantly affect the long time evolution of the cluster formation process. A detailed analysis of the molecular reactions during the early stages of SiC particle growth is presented. Reactions, in which silicon monomers are formed, are dominant in case of stoichiometric composition of the precursor gas. Moreover, we find the formation of carbon-dominated species to be preferred and a sensitive dependence of the particle composition and morphology on the processing conditions, especially the cooling and precursor gas composition. read less USED (high confidence) H. Shim, L. Zhou, H.-C. Huang, and T. Cale, “Nanoplate elasticity under surface reconstruction,” Applied Physics Letters. 2005. link Times cited: 71 Abstract: Using classical molecular statics simulations, we show that … read moreAbstract: Using classical molecular statics simulations, we show that nanoplate elasticity strongly depends on surface reconstruction and alignment of bond chains. Because of its well-established surface reconstructions and the readily available interatomic potential, diamond-cubic silicon is the prototype of this study. We focus on silicon nanoplates of high-symmetry surfaces, {111} and {100}; with 7×7 and 2×1 reconstructions. Nanoplates with unreconstructed {111} surfaces are elastically stiffer than bulk. In contrast, the same nanoplates with 7×7 reconstructed {111} surfaces are elastically softer than bulk. On {100} surfaces, the 2×1 surface reconstruction has little impact. The bond chains are along one of the two ⟨110⟩ directions, making the two ⟨110⟩ directions nonequivalent. The alignment of the bond chains on the opposite surfaces of a nanoplate dictates its elastic anisotropy. The sensitivity of nanoplate elasticity on details of surface atomic arrangements may impact the application of nanoplates (or nan... read less USED (high confidence) N. Moloi and M. Ali, “An Iterative Global Optimization Algorithm for Potential Energy Minimization,” Computational Optimization and Applications. 2005. link Times cited: 40 USED (high confidence) A. Marzegalli, F. Montalenti, and L. Miglio, “Stability of shuffle and glide dislocation segments with increasing misfit in Ge∕Si1−xGex(001) epitaxial layers,” Applied Physics Letters. 2005. link Times cited: 15 Abstract: Using molecular dynamics simulations, based on Tersoff poten… read moreAbstract: Using molecular dynamics simulations, based on Tersoff potentials, we show that at typical experimental temperatures high compressive strain regimes suppress the formation of partial glide dislocations, while enhancing the gliding of the shuffle segments. Despite being qualitative in nature, these results suggest that strain relaxation in thin virtual substrates at high misfit may occur with a different modality than in thick graded layers, as indicated by preliminary experimental results by low-energy plasma enhanced chemical vapor deposition. read less USED (high confidence) Y. Jeng, P. Tsai, and T. Fang, “Molecular dynamics investigation of the mechanical properties of gallium nitride nanotubes under tension and fatigue,” Nanotechnology. 2004. link Times cited: 47 Abstract: This study adopts a classical molecular dynamics (MD) simula… read moreAbstract: This study adopts a classical molecular dynamics (MD) simulation with the realistic Tersoff many-body potential model to investigate the mechanical properties of gallium nitride (GaN) nanotubes. The investigation focuses primarily on the mechanical properties of (n,0) and (n,n) GaN nanotubes since these particular nanotubes represent two extreme cases. The present results indicate that under small strain conditions, mechanical properties such as Young’s modulus are insensitive to the wrapping angle. Conversely, the wrapping angle has a significant influence upon these mechanical properties under large strain conditions. It is demonstrated that (9,0) GaN nanotubes are far less resistant to bond rotation. Under large tensile strain conditions, due to the unfavourable bond orientations induced by Stone–Wales (SW) transformation, the bonds in (n,0) GaN tubes quickly degenerate. Moreover, the present results suggest that the tensile strength of a nanotube is strongly sensitive to the temperature and strain rate. Regarding the fatigue test, this study uses a standard theoretical model to derive curves of amplitude stress versus number of cycles for the current nanotubes. The results demonstrate that the fatigue limit of GaN nanotubes increases with increasing temperature. read less USED (high confidence) Y. Umeno and T. Kitamura, “Ab initio molecular dynamics study on the formation process of Al layers on Si(001) surface,” Modelling and Simulation in Materials Science and Engineering. 2004. link Times cited: 2 Abstract: To clarify the early stage of the Al formation process on th… read moreAbstract: To clarify the early stage of the Al formation process on the Si(001) surface, we conduct ab initio molecular dynamics calculations of the precipitation of Al atoms on to the Si surface based on the pseudopotential plane wave basis method. We examine two candidate structures of the first Al layer, where the Al dimers are between Si dimer rows: Al dimers perpendicular (case A) and parallel (case B) to Si dimers. In case A, the distance between the first and the second layers is shorter than that between the (111) planes in an Al crystal. Results show that the charge density of the Al–Al bonds is 0.03–0.04 au−3 and that of the Al–Si bonds is 0.06 au−3. The bond lengths are 0.27 nm, which are almost the same as those in the Al single crystal. Al atoms precipitated after the formation of the two layers can move onto the surface. In case B, the first layer of Al atoms are arranged on a groove between the Si dimer rows and form bonds with the Si dimers, where the charge population of the bonds is similar to that in case A. These results suggest that dense layers of Al can be formed on the surface. read less USED (high confidence) D. Fischer, A. Curioni, S. Billeter, and W. Andreoni, “Effects of nitridation on the characteristics of silicon dioxide: dielectric and structural properties from ab initio calculations.,” Physical review letters. 2004. link Times cited: 27 Abstract: By combining ab initio calculations and classical molecular … read moreAbstract: By combining ab initio calculations and classical molecular dynamics, we determine how the inclusion of nitrogen in a silica matrix changes its dielectric constant, and elucidate the underlying mechanisms. We find that there is an entire range of nitrogen concentrations (up to approximately 25%) for which the structural pattern of the oxide is preserved in bulk SiON, and the dielectric constant increases mainly because of the variation of the ionic polarizability. This behavior is not sensitive to hydrogen passivation of nitrogen. The few defects, which are associated with electron states near the gap, are mainly centered on undercoordinated nitrogen and undercoordinated silicon, and tend to be removed by hydrogen. read less USED (high confidence) T. Y. Kim, S. Han, and H.-M. Lee, “Nanomechanical Behavior of β-SiC Nanowire in Tension: Molecular Dynamics Simulations,” Materials Transactions. 2004. link Times cited: 35 Abstract: The molecular dynamics (MD) simulation employing a Tersoff p… read moreAbstract: The molecular dynamics (MD) simulation employing a Tersoff potential was performed to examine the nanomechanical behavior of the � SiC nanowire in tension. The elongation was much larger than that of the bulk � -SiC. We observed non-homogeneous deformation, and the fracture behavior was found to depend on size, orientation and temperature of the specimen. The Young’s modulus calculated in this study generally decreased with temperatures and increased with the radius, namely, the diameter of the � -SiC nanowire as long as the length scale remained the same. The initial orientation was found to have a more serious effect on the Young’s modulus than size and temperature. The [111] Young’s modulus is much higher than that of the [001] orientation. The fracture of the � -SiC nanowire in the [001] orientation showed two different modes, which is brittle at 100 K and ductile at 300 and 500 K. The ductile fracture was accompanied by formation of an atomic chain. In the [111] orientation, it was always fractured in the ductile mode and thus an atomic chain was formed before rupture. read less USED (high confidence) L. Martinelli et al., “Formation of strain-induced Si-rich and Ge-rich nanowires at misfit dislocations in SiGe: A model supported by photoluminescence data,” Applied Physics Letters. 2004. link Times cited: 15 Abstract: Molecular dynamics simulations with the Tersoff potential of… read moreAbstract: Molecular dynamics simulations with the Tersoff potential of the strain distribution around 60° misfit dislocation in a heteroepitaxial SiGe film confirm that highly compressed and expanded, cylindrical nanometer-sized regions appear on opposite sides of the (111) glide plane. Such a configuration is suggested to generate opposite chemical potential gradients for Si and Ge diffusion and, as verified by a Monte Carlo simulation, in the formation of Si-rich and Ge-rich nanowires along the dislocation core. This model is supported by photoluminescence measurements as a function of annealing temperature and time. read less USED (high confidence) L. Zhou and H.-C. Huang, “Are surfaces elastically softer or stiffer,” Applied Physics Letters. 2004. link Times cited: 351 Abstract: This letter addresses the issue of surface softening versus … read moreAbstract: This letter addresses the issue of surface softening versus stiffening during elastic deformation. Using a combination of molecular statics and ab initio calculations, we show that a solid surface can be either softer or stiffer elastically than the corresponding bulk. Whether a particular surface is softer or stiffer depends on the competition between atomic coordination and electron redistribution (which sometimes is referred as bond saturation) on the surface. Taking Cu as an example, we demonstrate that the Young’s modulus along 〈110〉 direction on {100} surface is larger than its bulk counterpart; meanwhile, it is smaller along 〈100〉 direction on {100} surface. read less USED (high confidence) C. Wei, K. Cho, and D. Srivastava, “Tensile yielding of multiwall carbon nanotubes,” Applied Physics Letters. 2003. link Times cited: 33 Abstract: The tensile yielding of multiwall carbon nanotubes (MWCNTs) … read moreAbstract: The tensile yielding of multiwall carbon nanotubes (MWCNTs) has been studied using molecular-dynamics simulations and a transition state theory based model. We find a strong dependence of the yielding on the strain rate. A critical strain rate has been predicted above/below which yielding strain of a MWCNT is larger/smaller than that of the corresponding single-wall carbon nanotubes (CNTs). At an experimentally feasible strain rate of 1%/h and T=300 K, the yield strain of a MWCNT is estimated to be about 3%–4% higher than that of an equivalent single-wall CNT. This is in good agreement with recent experimental observations. read less USED (high confidence) D. A. Alman and D. Ruzic, “Molecular dynamics calculation of carbon/hydrocarbon reflection coefficients on a hydrogenated graphite surface,” Journal of Nuclear Materials. 2003. link Times cited: 43 USED (high confidence) Y. Zhao, R. Smalley, and B. Yakobson, “Coalescence of fullerene cages: Topology, energetics, and molecular dynamics simulation,” Physical Review B. 2002. link Times cited: 50 Abstract: Center for Nanoscale Science and Technology and Department o… read moreAbstract: Center for Nanoscale Science and Technology and Department of Mechanical Engineering and Materials Science, Rice University,Houston, Texas 77005~Received 21 June 2002; published 18 November 2002!Sequential atomic rearrangements leading to the coalescence of fullerene cages or tubes are derived bytopological analysis. Qualitative reasoning assists the search for the minimum-energy path, which consists ofa jump-to-contact formation of covalent bonds between the separate cages and the following ‘‘plastic flow’’ byexclusively Stone-Wales bond rotations. A connecting neck forms and grows gradually until the separateclusters are completely fused into a coherent unit. The most favorable path is determined by comparison of thecalculated energies and is further supported by molecular dynamics simulations. Results are presented forC read less USED (high confidence) S. Nakhmanson and N. Mousseau, “Crystallization study of model tetrahedral semiconductors,” Journal of Physics: Condensed Matter. 2002. link Times cited: 13 Abstract: The microscopic mechanisms leading to crystallization are no… read moreAbstract: The microscopic mechanisms leading to crystallization are not yet fully understood. This is due, in part, to the lack of atomistic as well as interatomic interaction models for a wide range of materials that can lead to crystallization on a computer-simulation timescale, i.e. < 100 ns. While the nucleation in close-packed systems has been extensively studied, there are almost no numerical results for covalent tetrahedral semiconductors. We present here the simulation results of crystallization from the liquid and amorphous states of a 1000-atom model of silicon, described with a modified Stillinger?Weber potential. With this potential, it is possible to crystallize the model in as little as a few nanoseconds, which opens a door to detailed studies of the nucleation processes in covalent systems. Using topological analysis, we also present a first characterization of the structural fluctuations of the nucleation centres in this system and give a rough estimate for the critical size of these centres. read less USED (high confidence) G. Samsonidze, G. G. Samsonidze, and B. Yakobson, “Energetics of Stone-Wales defects in deformations of monoatomic hexagonal layers,” Computational Materials Science. 2002. link Times cited: 70 USED (high confidence) C. Herrero, “The isotopic mass and lattice parameter of diamond; a path-integral simulation,” Journal of Physics: Condensed Matter. 2001. link Times cited: 5 Abstract: The dependence of the lattice parameter of diamond upon the … read moreAbstract: The dependence of the lattice parameter of diamond upon the isotopic mass has been studied by path-integral Monte Carlo simulations in the isothermal-isobaric ensemble. This computational method provides us with a quantitative and nonperturbative procedure for analysing such anharmonic effects. Atomic nuclei were treated as quantum particles interacting via a Tersoff-type potential. At 300 K, the difference Δa between the lattice parameter of isotopically pure crystals of 12C and 13C is found to be 6.1×10-4 A, in good agreement with experimental results. This difference decreases under an applied external pressure, and for 4000 kbar we obtain Δa = 2.4×10-4 A. read less USED (high confidence) W. Sekkal, A. Zaoui, A. Laref, M. Certier, and H. Aourag, “Molecular dynamics simulation of CuI using a three-body potential,” Journal of Physics: Condensed Matter. 2000. link Times cited: 27 Abstract: A three-body potential coupled with a molecular dynamics met… read moreAbstract: A three-body potential coupled with a molecular dynamics method have been used to simulate structural properties of CuI in the zincblende and tetragonal phases. It is found that the diffusion constant is well reproduced for the α-phase of CuI using this model rather than a two-body potential. This study predicts also the presence of cation disorder at elevated temperature within the tetragonal phase of CuI. read less USED (high confidence) A. Ivanovskii, “Simulation of nanotubular forms of matter,” Russian Chemical Reviews. 1999. link Times cited: 22 Abstract: Data on the electronic and chemical structure of a new quasi… read moreAbstract: Data on the electronic and chemical structure of a new quasi-one-dimensional form of matter, viz., nanotubulenes, are generalised and systematised. Methods and approaches used in modern quantum chemistry for the simulation of the composition, structure, and properties of isolated tubulenes based on layered phases (graphite, boron nitride, boron carbide and boron carbonitride), nanotubular composites and nanotube crystals are described. The role of quantum theory in the development of the concepts of fundamental properties of substances in the nanotubular form and methods of their targeted modification is discussed. Prognostic potentials of theoretical models in solving material science problems are considered. The bibliography includes 197 references read less USED (high confidence) B. Yakobson, C. Brabec, and J. Bernholc, “Structural mechanics of carbon nanotubes: From continuum elasticity to atomistic fracture,” Journal of Computer-Aided Materials Design. 1996. link Times cited: 51 USED (high confidence) L. Alonso, J. A. Alonso, and M. J. López, “Computer simulations of the structure of nanoporous carbons and higher density phases of carbon.” 2018. link Times cited: 1 USED (high confidence) F. F. D. Oliveira, “Forefront engineering of nitrogen-vacancy centers in diamond for quantum technologies.” 2017. link Times cited: 0 Abstract: The revolution being led by the next generation of quantum t… read moreAbstract: The revolution being led by the next generation of quantum technologies. Since the beginning of the 20th century, the rise of quantum physics has revolutionized the human comprehension of the universe. At that time, several experimental observations pushed physicists to think outside the classical Newtonian mechanics and electromagnetism theories. For instance, the pioneer study of the electromagnetic radiation of a blackbody by several scientists such as Max Planck and Lord Rayleigh is considered by many the first gearing event that challenged the so-called classical physical concepts of light and matter. The following breakthrough works involving the wave-particle duality concept to explain the particle-like behavior of electromagnetic waves and the photoelectric effect have then led to the foundations of quantum mechanics. Although very controversial at that time, quantum mechanics began to expand and gain further grounds after the mathematical formulation developed by Ervin Schrödinger in 1926 [1] and subsequent studies. Yet, since that time, a question has intrigued scientists from many different research fields: can the concepts of quantum mechanics be somehow implemented in something feasible (i.e. a device) for the long benefit of the society? The answer came quickly by the development of a ground-breaking first-generation of quantum technologies such as the laser and the global positioning system (GPS), which are devices based primarily on the quantum principle of coherence. These events resulted in quantum physics to be evolved from essentially a conceptual framework, to provide new inspirations for realistic technological applications. A particular field that has always been of broad interest is the capability of store, transmit and process information. With the rise of the industrial applications of semiconductor technology, especially the development of silicon-based micro-electronic devices in the late 1950s [2], the digitally-encoded type of information became popular and widely-spread within many different areas. Recent advances in microand nano-structuring, and a rapid progress in the material synthesis and development of new platforms led to a continuous increase of transmission speed and storage capacity of information in modern devices. Since the invention of integrated circuits, Moore’s law [3] has reasonably predicted the time evolution related to the density of electronic components that can be packed on a read less USED (high confidence) F. Al-Dirini, M. A. Mohammed, F. Hossain, T. Nirmalathas, and E. Skafidas, “All-Graphene Planar Double-Quantum-Dot Resonant Tunneling Diodes,” IEEE Journal of the Electron Devices Society. 2016. link Times cited: 15 Abstract: This paper proposes a new class of resonant tunneling diodes… read moreAbstract: This paper proposes a new class of resonant tunneling diodes (RTDs) that are planar and realizable with a single graphene nanoribbon. Unlike conventional RTDs, which incorporate vertical quantum well regions, the proposed devices incorporate two confined planar quantum dots within the single graphene nanoribbon, giving rise to a pronounced negative differential resistance (NDR) effect. The proposed devices, termed here as planar double-quantum-dot RTDs, and their transport properties are investigated using quantum simulations based on nonequilibrium Green's function formalism and the extended Huckel method. The proposed devices exhibit a unique current-voltage waveform consisting of a single pronounced current peak with an extremely high, in the order of 104, peak-to-valley ratio. The position of the current peak can be tuned between discrete voltage levels, allowing digitized tunability, which is exploited to realize multi-peak NDR devices. read less USED (high confidence) A. R. Setoodeh, H. Badjian, and H. S. Jahromi, “Atomistic study of mono/multi-atomic vacancy defects on the mechanical characterization of boron-doped graphene sheets,” Journal of Molecular Modeling. 2016. link Times cited: 16 USED (high confidence) A. Adnan and S. F. Ferdous, “Computational design of novel carbon enriched Si1 xCx ceramics: A molecular dynamics simulation study,” Computational Materials Science. 2015. link Times cited: 2 USED (high confidence) Z. Hui and X. Liu, “Molecular Dynamics Simulation of Coarse-Grain Model of Silicon Functionalized Graphene.” 2015. link Times cited: 0 Abstract: The electronic transport, the storage capacity and the servi… read moreAbstract: The electronic transport, the storage capacity and the service life of the anode material for lithium ion batteries will be reduced seriously in the event of the material layering or cracking, so the anode material must have strong mechanical reliability. Firstly, in view of the traditional molecular dynamics (MD) limited by the geometric scales of the model of Silicon functionalized graphenen (SFG) as lithium ion batteries anode material, some full atomic models of SFG were established using Tersoff potential and Lennard-Jones potential, and used to calculate the modulus and the adhesion properties. What’s more, the assertion of mechanical equilibrium condition and energy conservation between full atomic and coarse-grain models through elastic strain energy were enforced to arrive at model parameters. The model of SFG coarse-grain bead-spring elements and its system energy function were obtained via full atomic simulations. Finally, the validity of the SFG coarse-grain model was verified by comparing the tensile property of coarse-grain model with full atoms model. read less USED (high confidence) P. Sengupta, S. Steiger, S. Lee, H. Ryu, and G. Klimeck, “Multiscale Modeling of Quantum Dot Heterostructures.” 2011. link Times cited: 4 Abstract: A multiscale approach was adopted for the calculation of con… read moreAbstract: A multiscale approach was adopted for the calculation of confined states in self-assembled semiconductor quantum dots (QDs). While results close to experimental data have been obtained with a combination of atomistic strain and tight-binding (TB) electronic structure description for the confined quantum states in the QD, the TB calculation requires substantial computational resources. To alleviate this problem an integrated approach was adopted to compute the energy states from a continuum 8-band k.p Hamiltonian under the influence of an atomistic strain field. Such multiscale simulations yield a roughly six-fold faster simulation. Atomic-resolution strain is added to the k.p Hamiltonian through interpolation onto a coarser continuum grid. Sufficient numerical accuracy is obtained by the multiscale approach. Optical transition wavelengths are within 7% of the corresponding TB results with a proper splitting of p-type sub-bands. The systematically lower emission wavelengths in k.p are attributable to an underestimation of the coupling between the conduction and valence bands. read less USED (high confidence) W. K. Liu and C. McVeigh, “Predictive multiscale theory for design of heterogeneous materials,” Computational Mechanics. 2008. link Times cited: 57 USED (high confidence) T. Hawa and M. Zachariah, “Coalescence kinetics of unequal sized nanoparticles,” Journal of Aerosol Science. 2006. link Times cited: 128 USED (high confidence) G. Benedek, E. Galvani, and S. Sanuinetti, “A hypothetical new class of superhard materials,” Il Nuovo Cimento D. 1995. link Times cited: 6 USED (low confidence) Z. Hao, H. Zhang, Y. Fan, and G. Cui, “Wear mechanism of nanotwinned cBN tools in nano-cutting Ni-Cr-Fe alloy by molecular dynamics simulation,” Journal of Manufacturing Processes. 2022. link Times cited: 5 USED (low confidence) Y. Zhao et al., “Molecular Dynamics Simulations of the Thermally and Stress-Activated Glide of a ⟨0001⟩11̅00 Screw Dislocation in AlN,” Crystal Growth & Design. 2021. link Times cited: 1 USED (low confidence) A. Mosavi et al., “Atomic interactions between rock substrate and water-sand mixture with and without graphene nanosheets via molecular dynamics simulation,” Journal of Molecular Liquids. 2020. link Times cited: 6 USED (low confidence) J. Rouviere, F. Lançon, and O. H. H. Duparc, “Atomic structures of Si and Ge Σ = 13 [0 0 1] tilt grain boundaries studied by high-resolution electron microscopy and atomistic simulations,” Philosophical Magazine. 2013. link Times cited: 5 Abstract: By combining high-resolution electron microscopy and atomist… read moreAbstract: By combining high-resolution electron microscopy and atomistic simulations, the atomic structures of several interfaces, {5 1 0}, {2 3 0} and {8 1 0}/{7 4 0}, in germanium and in silicon Σ = 13 [0 0 1] tilt grain boundaries (TGBs) are studied using bicrystals prepared in two different ways from the melt. The interfaces are characterized by either transmission electron microscopy or scanning transmission electron microscopy (STEM). The Si TGB shows only one interface, {1 5 0} with one interfacial structure. The Ge TGB contains many facets. In Ge, observations performed in two perpendicular directions, [0 0 1] and [ 5 0], confirm that the {5 1 0} interface has two different structures. One structure, called M-structure, is periodic along [0 0 1] and has tetracoordinated atoms. The other structure, called U-structure, is more peculiar as it contains a fixed part surrounding a variable complex core. High-resolution STEM, realised in modern microscopes equipped with a probe Cs-corrector, is a very effective technique for structure determination of grain boundaries (GBs). However, current limitations for high-resolution study of GBs are the structural changes under the electron beam and the limited number of crystallographic axes suitable for atomic-resolution imaging. The structures of GB atomistic models can be ordered according to their calculated energies. It appears that energies calculated using empirical potentials, like Tersoff or Stillinger-Weber potentials, do not give the same classification as ab initio calculations and cannot be used to determine the structure of lowest energy. This structure is the M-structure, the structure observed in the Si bicrystal. read less USED (low confidence) X. W. Zhou, F. Doty, and P. Yang, “Atomistic simulation study of atomic size effects on B1 (NaCl), B2 (CsCl), and B3 (zinc-blende) crystal stability of binary ionic compounds,” Computational Materials Science. 2011. link Times cited: 15 USED (low confidence) H. Dai, F. Zhang, and Y. Zhou, “Numerical study of three-body diamond abrasive polishing single crystal Si under graphene lubrication by molecular dynamics simulation,” Computational Materials Science. 2020. link Times cited: 21 NOT USED (low confidence) M. Yu, R. Lou, H. Li, F. Wang, J. Wang, and K. Wang, “Reactive force field molecular dynamics (ReaxFF-MD) simulation of lignite combustion under an external electric field,” Fuel. 2024. link Times cited: 0 NOT USED (low confidence) H. Sakakima et al., “Development of charge-transfer interatomic potential for O-Fe-P-Zn systems and its application to tribochemical reactions between ZnDTP-derived tribofilm and iron oxide,” Computational Materials Science. 2024. link Times cited: 0 NOT USED (low confidence) A. Hirano, H. Sakakima, A. Hatano, and S. Izumi, “Long-range Tersoff potential for silicon to reproduce 30° partial dislocation migration,” Computational Materials Science. 2024. link Times cited: 0 NOT USED (low confidence) M. H. Pebdani, R. Sabetvand, and I. Pishkar, “Molecular dynamics study of vacancy effect on mechanical properties of polyurethane–graphene nanocomposite,” Journal of the Brazilian Society of Mechanical Sciences and Engineering. 2023. link Times cited: 0 NOT USED (low confidence) P. Gao et al., “Redesign and Accelerate the AIREBO Bond-Order Potential on the New Sunway Supercomputer,” IEEE Transactions on Parallel and Distributed Systems. 2023. link Times cited: 0 Abstract: Molecular dynamics (MD) is one of the most crucial computer … read moreAbstract: Molecular dynamics (MD) is one of the most crucial computer simulation methods for understanding real-world processes at the atomic level. Reactive potentials based on the bond order concept have the ability to model dynamic bond breaking and formation with close to quantum mechanical (QM) precision without actually requiring expensive QM calculations. In this article, we focus on the adaptive intermolecular reactive empirical bond-order (AIREBO) potential in LAMMPS for the simulation of carbon and hydrocarbon systems on the new Sunway supercomputer. To achieve scalable performance, we propose a parallel two-level building scheme and periodic buffering strategy for the tailored data design to explore data locality and data reuse. Furthermore, we design two optimized nearest-neighbor access algorithms: the redistribution of accumulated coefficients algorithm and the double-end search connectivity algorithm. Finally, we implement parallel force computation with an AoS data layout and hardware/software co-cache. In addition, we have designed a low-overhead atomic operation-based load balancing method and vectorization. The overall performance of AIREBO achieves a speedup of nearly $20\times$20× on a single core group (CG), and more than $5\times$5× and $4\times$4× over an Intel Xeon E5 2680 v3 core and an Intel Xeon Gold 6138 core, respectively. Compared with the Intel accelerator package in LAMMPS, our performance further achieves $3.0\times$3.0× of an Intel Xeon E5 2680 v3 core and is better than that of an Intel Xeon Gold 6138 core. We complete the validation of the results in no more than 20.5 hours on a single node with 2,000,000 running steps (i.e., 1 ns). Our experiments show that the simulation of 2,139,095,040 atoms on 798,720 ((1MPE+64CPEs) × 12,288 processes) cores exhibits a parallel efficiency of 88% under weak scaling. read less NOT USED (low confidence) H. Niu et al., “The nano-pumping process of C20 molecules from carbon nanotube at the different external electric fields and atomic defects: A molecular dynamics approach,” Diamond and Related Materials. 2023. link Times cited: 0 NOT USED (low confidence) E. Strand, F. Tourlomousis, and N. Gershenfeld, “Mesoscale material modeling with memoryless isotropic point particles,” Journal of Computational Science. 2023. link Times cited: 0 NOT USED (low confidence) B. Mei et al., “The effect of the initial temperature, pressure, and shape of carbon nanopores on the separation process of SiO2 molecules from water vapor by molecular dynamics simulation.,” Chemosphere. 2023. link Times cited: 0 NOT USED (low confidence) M. Shi, S. Zhang, and G. Chern, “Machine learning force-field models for metallic spin glass,” ArXiv. 2023. link Times cited: 0 Abstract: Metallic spin glass systems, such as dilute magnetic alloys,… read moreAbstract: Metallic spin glass systems, such as dilute magnetic alloys, are characterized by randomly distributed local moments coupled to each other through a long-range electron-mediated effective interaction. We present a scalable machine learning (ML) framework for dynamical simulations of metallic spin glasses. A Behler-Parrinello type neural-network model, based on the principle of locality, is developed to accurately and efficiently predict electron-induced local magnetic fields that drive the spin dynamics. A crucial component of the ML model is a proper symmetry-invariant representation of local magnetic environment which is direct input to the neural net. We develop such a magnetic descriptor by incorporating the spin degrees of freedom into the atom-centered symmetry function methods which are widely used in ML force-field models for quantum molecular dynamics. We apply our approach to study the relaxation dynamics of an amorphous generalization of the s-d model. Our work highlights the promising potential of ML models for large-scale dynamical modeling of itinerant magnets with quenched disorder. read less NOT USED (low confidence) J. Li et al., “Enhance the Strong Scaling of LAMMPS on Fugaku,” Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. 2023. link Times cited: 0 Abstract: Physical phenomenon such as protein folding requires simulat… read moreAbstract: Physical phenomenon such as protein folding requires simulation up to microseconds of physical time, which directly corresponds to the strong scaling of molecular dynamics(MD) on modern supercomputers. In this paper, we present a highly scalable implementation of the state-of-the-art MD code LAMMPS on Fugaku by exploiting the 6D mesh/torus topology of the TofuD network. Based on our detailed analysis of the MD communication pattern, we first adapt coarse-grained peer-to-peer ghost-region communication with uTofu interface, then further improve the scalability via fine-grained thread pool. Finally, Remote direct memory access (RDMA) primitives are utilized to avoid buffer overhead. Numerical results show that our optimized code can reduce 77% of the communication time, improving the performance of baseline LAMMPS by a factor of 2.9x and 2.2x for Lennard-Jones and embedded-atom method potentials when scaling to 36, 846 computing nodes. Our optimization techniques can also benefit other applications with stencil or domain decomposition methods. read less NOT USED (low confidence) Y. Wang, J. Zhao, and Z. Tang, “Molecular dynamics simulation of the effect of carbon nanotubes on liquid argon phase transition behavior on the platinum plate,” Journal of Molecular Liquids. 2023. link Times cited: 0 NOT USED (low confidence) X. Guo et al., “The computational study of external heat flux and silicon doping effect on displacement of C20 molecule in a carbon nanotube (CNT): A molecular dynamics method,” Results in Physics. 2023. link Times cited: 0 NOT USED (low confidence) A. Kanani, M. Mahnama, and E. Ghavaminezahd, “Investigation of vibration of carbon nanotube and quality factor with confined and submerged fluid under hammer impact Test: A molecular dynamics study,” Journal of Molecular Liquids. 2023. link Times cited: 0 NOT USED (low confidence) M. Shahryari, A. Nazari-Golshan, S. Nourazar, and M. Abedi, “Investigating the atomic behavior of carbon nanotubes as nanopumps,” Scientific Reports. 2023. link Times cited: 1 NOT USED (low confidence) K. Kirill, S. Denis, and K. Platon, “The Way to Analyse MD Simulation Results of Cluster Ion Bombardment,” 2023 International Conference on Electrical Engineering and Photonics (EExPolytech). 2023. link Times cited: 0 Abstract: Molecular Dynamics (MD) is a simulation method commonly empl… read moreAbstract: Molecular Dynamics (MD) is a simulation method commonly employed in scientific research, specifically in the field of solid state physics. In particular, MD is a valuable tool for analysing rapid processes that take place on the surface, such as the stopping and fragmentation of accelerated cluster ions, sputtering of the target material and the formation of crater and rim. These phenomena necessitate the analysis of substantial amounts of data, encompassing the velocity, coordinates, and energies of each atom in the simulation. In this contribution, we propose scalable and automated methods to analyse size of impact crater, rim characteristics, surface deformation and the energy and directions of sputtered particles. To illustrate the efficacy of these methods, we present results obtained during MD simulation of (100) Si crystal surface bombardment with accelerated C60 molecules. read less NOT USED (low confidence) Y. Huang and H. Chen, “A detailed reaction mechanism for hexamethyldisiloxane combustion via experiments and ReaxFF molecular dynamics simulations,” International Journal of Chemical Kinetics. 2023. link Times cited: 0 Abstract: Hexamethyldisiloxane (HMDSO) is one of the main impurities i… read moreAbstract: Hexamethyldisiloxane (HMDSO) is one of the main impurities in the syngas produced from sewage and landfill plants. In order to utilize this syngas or control the characteristics of the generated silica particles, it is crucial to understand the chemical kinetics of HMDSO combustion. This study investigated the process of HMDSO combustion using synchrotron radiation mass spectrometry (SRMS), gas chromatography (GC), and ReaxFF molecular dynamics simulations. First, the force field used for ReaxFF simulation was validated by comparing the energies of different bond lengths, bond angles, and dihedral angles with the ones from DFT calculations. Good agreements were found. Then, ReaxFF simulations of HMDSO combustion with this force field were conducted under various conditions, which include different equivalence ratios (0.67, 1.0, and 1.5) and temperatures ranging from 2000 to 3500 K. The oxidation characteristics of HMDSO were analyzed, including the evolution of gas products and particle formation. Finally, based on the results from experiments and ReaxFF simulations, the reaction pathways, reaction lists, and reaction kinetics data during HMDSO combustion were obtained. A detailed reaction mechanism was proposed and validated by applying it in modeling the H2/HMDSO/O2 combustion systems. The temperature and part of the gas products such as CO and CO2 as well as SiO could be well predicted. read less NOT USED (low confidence) M. R. P. M. Tavares, D. Rolón, J. C. B. Kober, S. Kühne, R. B. Schroeter, and D. Oberschmidt, “Simulation of gallium phosphide cutting mechanism in ductile regime using molecular dynamics,” NanoScience + Engineering. 2023. link Times cited: 0 Abstract: Gallium Phosphide (GaP) is a semiconductor with advantageous… read moreAbstract: Gallium Phosphide (GaP) is a semiconductor with advantageous optical properties for near- and middle infrared optical systems. However, optical applications of GaP are limited by its current low machinability. To cut brittle semiconductors such as GaP and generate optical quality surfaces, it is necessary to induce a High-Pressure Phase Transformation (HPPT) so that a phase is formed that behaves ductile when machined. Along the cutting process this can be achieved by applying a negative rake angle. Otherwise, cracks will appear on the machined surface, worsening its optical capabilities. A HPPT of GaP happens at an atomic scale when a zincblende structure changes into a β-tin one. The β-tin structure behaves ductile and is metastable. Hence, the metastable β-tin phase cannot be observed during the cutting process. Therefore, atomistic simulation, such as Classic Molecular Dynamics Simulation (CMDS), is required to study the machinability under HPPT. In this work, CMDS were used to analyze GaP cutting mechanisms. A diamond tool was modelled with a cutting edge radius rβ = 10 nm, rake angle γ = -20 º, and clearance angle α = 10 º. The cut was performed with a depth of cut ap = 12 nm along the [100]-direction in a zincblende GaP workpiece. Stacking faults were found on the shear zone, {111}-planes, by two different post processes approaches. HPPT was found in the deformation zone only. A stagnation zone was found in front of the cutting edge proceeding a crack nucleation. read less NOT USED (low confidence) H. Pourmirzaagha and S. Rouhi, “Molecular dynamic simulations of the heat transfer in double-layered graphene/silicene nanosheets,” Physica B: Condensed Matter. 2023. link Times cited: 0 NOT USED (low confidence) T. Panczyk and K. Nieszporek, “Formation of degraded LDPE surfaces using mechanical cleavage and shock compression analyzed by means of molecular dynamics simulations,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) P. A. Taylor and M. J. Stevens, “Explicit solvent machine-learned coarse-grained model of sodium polystyrene sulfonate to capture polymer structure and dynamics,” The European Physical Journal E. 2023. link Times cited: 0 NOT USED (low confidence) Z. Yan, J. Zhao, R. Liu, B. Liu, Y.-lin Shao, and M. Liu, “An insight into sintering mechanisms of silicon carbide nanoparticles with additives using MD simulation,” Powder Technology. 2023. link Times cited: 0 NOT USED (low confidence) Y. Yu, X. Zhang, and L. Bai, “Nanoindentation and scratching behaviors of diamond-like carbon films by coarse-grained molecular dynamics,” Diamond and Related Materials. 2023. link Times cited: 0 NOT USED (low confidence) D. Hu, Z. Li, Y. Liu, C. Ding, C.-L. Miao, and H. Wang, “Coal Pyrolysis law and mechanism of index gas generation in Linsheng Mine,” Journal of Molecular Structure. 2023. link Times cited: 0 NOT USED (low confidence) M. Grigoryeva et al., “Modeling of Short-Pulse Laser Interactions with Monolithic and Porous Silicon Targets with an Atomistic–Continuum Approach,” Nanomaterials. 2023. link Times cited: 0 Abstract: The acquisition of reliable knowledge about the mechanism of… read moreAbstract: The acquisition of reliable knowledge about the mechanism of short laser pulse interactions with semiconductor materials is an important step for high-tech technologies towards the development of new electronic devices, the functionalization of material surfaces with predesigned optical properties, and the manufacturing of nanorobots (such as nanoparticles) for bio-medical applications. The laser-induced nanostructuring of semiconductors, however, is a complex phenomenon with several interplaying processes occurring on a wide spatial and temporal scale. In this work, we apply the atomistic–continuum approach for modeling the interaction of an fs-laser pulse with a semiconductor target, using monolithic crystalline silicon (c-Si) and porous silicon (Si). This model addresses the kinetics of non-equilibrium laser-induced phase transitions with atomic resolution via molecular dynamics, whereas the effect of the laser-generated free carriers (electron–hole pairs) is accounted for via the dynamics of their density and temperature. The combined model was applied to study the microscopic mechanism of phase transitions during the laser-induced melting and ablation of monolithic crystalline (c-Si) and porous Si targets in a vacuum. The melting thresholds for the monolithic and porous targets were found to be 0.32 J/cm2 and 0.29 J/cm2, respectively. The limited heat conduction mechanism and the absence of internal stress accumulation were found to be involved in the processes responsible for the lowering of the melting threshold in the porous target. The results of this modeling were validated by comparing the melting thresholds obtained in the simulations to the experimental values. A difference in the mechanisms of ablation of the c-Si and porous Si targets was considered. Based on the simulation results, a prediction regarding the mechanism of the laser-assisted production of Si nanoparticles with the desired properties is drawn. read less NOT USED (low confidence) P. Belardinelli, S. Lenci, and F. Alijani, “Imperfection-induced internal resonance in nanotube resonators,” Journal of Sound and Vibration. 2023. link Times cited: 0 NOT USED (low confidence) J. F. Wang, P. H. Li, X. B. Tian, S. Shi, and L.-ho Tam, “Molecular investigation on temperature-dependent mechanical properties of PMMA/CNT nanocomposite,” Engineering Fracture Mechanics. 2023. link Times cited: 0 NOT USED (low confidence) M. Z. Dehaghani et al., “Evaluation of water desalination performances of functionalized nanoporous graphene membranes by molecular dynamics simulation,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) S. A. Mousavi and A. Montazeri, “Predicting mechanical properties of defective h-BN nanosheets using Data-Driven models,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) D. Feng, Z. Zhao, X. Zhang, and Y. Feng, “Carbon-based nanoadditives induced enhancement of phase change thermal properties of sugar alcohol and interfacial heat transport mechanisms,” Composites Science and Technology. 2023. link Times cited: 0 NOT USED (low confidence) C. Yang, B. Wu, W. Deng, S. Li, J. Jin, and Q. Peng, “Assessment of the Interatomic Potentials of Beryllium for Mechanical Properties,” Crystals. 2023. link Times cited: 0 Abstract: Beryllium finds widespread applications in nuclear energy, w… read moreAbstract: Beryllium finds widespread applications in nuclear energy, where it is required to service under extreme conditions, including high-dose and high-dose rate radiation with constant bombardments of energetic particles leading to various kinds of defects. Though it is generally known that defects give rise to mechanical degradation, the quantitative relationship between the microstructure and the corresponding mechanical properties remains elusive. Here we have investigated the mechanical properties of imperfect hexagonal close-packed (HCP) beryllium via means of molecular dynamics simulations. We have examined the beryllium crystals with void, a common defect under in-service conditions. We have assessed three types of potentials, including MEAM, Finnis–Sinclair, and Tersoff. The volumetric change with pressure based on MEAM and Tersoff and the volumetric change with temperature based on MEAM are consistent with the experiment. Through cross-comparison on the results from performing hydrostatic compression, heating, and uniaxial tension, the MEAM type potential is found to deliver the most reasonable predictions on the targeted properties. Our atomistic insights might be helpful in atomistic modeling and materials design of beryllium for nuclear energy. read less NOT USED (low confidence) M. Hou, X. Zhou, Z. Yan, M. Liu, and B. Liu, “Molecular dynamics study of the irradiation damage accumulation in beryllium oxide at different temperatures,” Materialia. 2023. link Times cited: 0 NOT USED (low confidence) Y.-F. Wu, W. Yu, and S. Shen, “Developing an analytical bond-order potential for Hf/Nb/Ta/Zr/C system using machine learning global optimization,” Ceramics International. 2023. link Times cited: 0 NOT USED (low confidence) F. Liu et al., “Interfacial mechanical properties of periodic wrinkled graphene/polyethylene nanocomposite,” Physica Scripta. 2023. link Times cited: 0 Abstract: Molecular dynamics simulations have been performed to invest… read moreAbstract: Molecular dynamics simulations have been performed to investigate the interfacial mechanical properties of periodic wrinkled graphene (GR) with polyethylene (PE) matrix. The influences of amplitude (H), wavelength (λ), and vacancy defect for the periodic wrinkled GR on the interfacial mechanical properties were considered and the potential mechanisms were analyzed. The results indicate that the interfacial mechanical properties of GR with periodic wrinkles are superior to that of flat GR, especially when the H/λ=0.51 the interfacial strength enhances ∼29.3%. Through the radial distribution function (RDF) analysis we found that the stronger interfacial mechanical properties are, the more PE molecular chains are attached to the GR when the GR is separated from the PE matrix. In addition, we found that vacancy defect in periodic wrinkled GR does not always degrade the interfacial mechanical properties, and when the vacancy defect content is 20%, the interfacial mechanical properties can be improved, as the vacancy defect reduces the interfacial distance and increases the roughness of the interface. read less NOT USED (low confidence) X. Jiang, H. Sun, K. Choudhary, H. Zhuang, and Q. Nian, “Interpretable Ensemble Learning for Materials Property Prediction with Classical Interatomic Potentials: Carbon as an Example,” ArXiv. 2023. link Times cited: 0 Abstract: Machine learning (ML) is widely used to explore crystal mate… read moreAbstract: Machine learning (ML) is widely used to explore crystal materials and predict their properties. However, the training is time-consuming for deep-learning models, and the regression process is a black box that is hard to interpret. Also, the preprocess to transfer a crystal structure into the input of ML, called descriptor, needs to be designed carefully. To efficiently predict important properties of materials, we propose an approach based on ensemble learning consisting of regression trees to predict formation energy and elastic constants based on small-size datasets of carbon allotropes as an example. Without using any descriptor, the inputs are the properties calculated by molecular dynamics with 9 different classical interatomic potentials. Overall, the results from ensemble learning are more accurate than those from classical interatomic potentials, and ensemble learning can capture the relatively accurate properties from the 9 classical potentials as criteria for predicting the final properties. read less NOT USED (low confidence) A. Chandra et al., “Reinforcement learning based hybrid bond-order coarse-grained interatomic potentials for exploring mesoscale aggregation in liquid-liquid mixtures.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: Exploring mesoscopic physical phenomena has always been a ch… read moreAbstract: Exploring mesoscopic physical phenomena has always been a challenge for brute-force all-atom molecular dynamics simulations. Although recent advances in computing hardware have improved the accessible length scales, reaching mesoscopic timescales is still a significant bottleneck. Coarse-graining of all-atom models allows robust investigation of mesoscale physics with a reduced spatial and temporal resolution but preserves desired structural features of molecules, unlike continuum-based methods. Here, we present a hybrid bond-order coarse-grained forcefield (HyCG) for modeling mesoscale aggregation phenomena in liquid-liquid mixtures. The intuitive hybrid functional form of the potential offers interpretability to our model, unlike many machine learning based interatomic potentials. We parameterize the potential with the continuous action Monte Carlo Tree Search (cMCTS) algorithm, a reinforcement learning (RL) based global optimizing scheme, using training data from all-atom simulations. The resulting RL-HyCG correctly describes mesoscale critical fluctuations in binary liquid-liquid extraction systems. cMCTS, the RL algorithm, accurately captures the mean behavior of various geometrical properties of the molecule of interest, which were excluded from the training set. The developed potential model along with the RL-based training workflow could be applied to explore a variety of other mesoscale physical phenomena that are typically inaccessible to all-atom molecular dynamics simulations. read less NOT USED (low confidence) Z. Xiong, Y. Yu, H.-C. Chen, and L. Bai, “A coarse-grained study on mechanical behaviors of diamond-like carbon based on machine learning,” Nanotechnology. 2023. link Times cited: 2 Abstract: Diamond-like carbon (DLC) films have broad application poten… read moreAbstract: Diamond-like carbon (DLC) films have broad application potential due to their high hardness, high wear resistance, and self-lubricating properties. However, considering that DLC films are micron-scale, neither finite element methods nor macroscopic experiments can reveal their deformation and failure mechanisms. Here we propose a coarse-grained molecular dynamics (CGMD) approach which expands the capabilities of molecular dynamics simulations to uniaxial tensile behavior of DLC films at a higher scale. The Tersoff potential is modified by high-throughput screening calculations for CGMD. Given this circumstance, machine learning (ML) models are employed to reduce the high-throughput computational cost by 86%, greatly improving the efficiency of parameter optimization in second- and fourth-order CGMD. The final obtained coarse-grained tensile curves fit well with that of the all-atom curves, showing that the ML-based CGMD method can investigate DLC films at higher scales while saving a large number of computational resources, which is important for promoting the research and production of high-performance DLC films. read less NOT USED (low confidence) Q. Mao, M. Feng, X. Jiang, Y. Ren, K. Luo, and A. V. van Duin, “Classical and reactive molecular dynamics: Principles and applications in combustion and energy systems,” Progress in Energy and Combustion Science. 2023. link Times cited: 10 NOT USED (low confidence) Y. Xie et al., “A computational study of the effect of external heat flux and electric field on the nano-pumping of C20 molecules in carbon nanotubes by molecular dynamics simulation,” Journal of Materials Research and Technology. 2023. link Times cited: 1 NOT USED (low confidence) H. Han, W. Ye, F. Zhang, D.-sheng Zhu, Y. Shen, and X. Xiong, “Molecular dynamics simulation study of nano-cutting interaction mechanisms in grain boundary affect zone segregated Cu alloys,” Journal of Nanoparticle Research. 2023. link Times cited: 0 NOT USED (low confidence) Y. Zhang et al., “Amorphous Carbon Interlayer Modulated Interfacial Thermal Conductance between Cu and Diamond,” Applied Surface Science. 2023. link Times cited: 0 NOT USED (low confidence) W. Zhu et al., “Effect of fracture behavior variables on hydraulic fracturing optimization by adding graphene nanosheets to sand/water mixtures: A molecular dynamics approach,” Journal of Molecular Liquids. 2023. link Times cited: 0 NOT USED (low confidence) A. H. Banna and S. Roy, “Investigating surface effect on stress concentration in amorphous carbon materials with nano-scale pores: A molecular dynamics study,” Mechanics of Materials. 2023. link Times cited: 0 NOT USED (low confidence) L. Weidong, M. Al‐Bahrani, A. Alizadeh, N. Nasajpour-Esfahani, M. Shamsborhan, and D. Toghraie, “Variable electric field and atomic percentage of doping on the displacement process of C20 molecules in a nanotube with molecular dynamics simulation,” TrAC Trends in Analytical Chemistry. 2023. link Times cited: 1 NOT USED (low confidence) W.-L. Tang et al., “Tribological performance and lubrication mechanism of carbon nitride nanosheets as novel and high-efficiency additives for water lubrication,” Journal of Molecular Liquids. 2023. link Times cited: 0 NOT USED (low confidence) J. Trieschmann, L. Vialetto, and T. Gergs, “Machine learning for advancing low-temperature plasma modeling and simulation,” ArXiv. 2023. link Times cited: 0 Abstract: Machine learning has had an enormous impact in many scientif… read moreAbstract: Machine learning has had an enormous impact in many scientific disciplines. Also in the field of low-temperature plasma modeling and simulation it has attracted significant interest within the past years. Whereas its application should be carefully assessed in general, many aspects of plasma modeling and simulation have benefited substantially from recent developments within the field of machine learning and data-driven modeling. In this survey, we approach two main objectives: (a) We review the state-of-the-art focusing on approaches to low-temperature plasma modeling and simulation. By dividing our survey into plasma physics, plasma chemistry, plasma-surface interactions, and plasma process control, we aim to extensively discuss relevant examples from literature. (b) We provide a perspective of potential advances to plasma science and technology. We specifically elaborate on advances possibly enabled by adaptation from other scientific disciplines. We argue that not only the known unknowns, but also unknown unknowns may be discovered due to the inherent propensity of data-driven methods to spotlight hidden patterns in data. read less NOT USED (low confidence) R. Cappabianca, P. D. Angelis, M. Fasano, E. Chiavazzo, and P. Asinari, “An Overview on Transport Phenomena within Solid Electrolyte Interphase and Their Impact on the Performance and Durability of Lithium-Ion Batteries,” Energies. 2023. link Times cited: 0 Abstract: The nature of the electrode–electrolyte interface has an imp… read moreAbstract: The nature of the electrode–electrolyte interface has an impact on the performance and durability of lithium-ion batteries (LIBs). The initial electrolyte’s thermodynamic instability at the anode–electrolyte interface in LIBs results in the formation of a passivation layer, called solid electrolyte interphase (SEI). The initial dense and intact layer allows Li+ transport and restricts electron tunneling, thus preventing electrolyte decomposition and ensuring the electrochemical stability of a battery. However, the growth of this layer can reduce the availability of active lithium and electrolyte, and ultimately lead to an irreversible battery capacity fade. Investigating the transport phenomena of lithium ions within SEI is crucial for understanding its formation and growth. Nonetheless, accurately describing all relevant mechanisms is challenging due to its complex and multiscale nature. An overview of current computational efforts to study Li+ transport within SEI is given in this article, ranging from electronic/atomistic scale simulations to macroscopic models. The drawbacks and advantages of the proposed numerical approaches are summarized along with the obstacles that need to be overcome to obtain accurate experimental data, identified on the basis of the most recent literature evidence. We highlight collaboration gaps between modeling and experimental approaches, as well as the urgent need for new multiscale models, to gain a better understanding of such a crucial transport phenomenon. read less NOT USED (low confidence) M. Hou, X. Zhou, M. Liu, and B. Liu, “Molecular Dynamics Simulation of High Temperature Mechanical Properties of Nano-Polycrystalline Beryllium Oxide and Relevant Experimental Verification,” Energies. 2023. link Times cited: 1 Abstract: This article investigated the deformation behavior of nano-p… read moreAbstract: This article investigated the deformation behavior of nano-polycrystalline beryllium oxide under tensile and compressive stress using the molecular dynamics simulation method. Both the tensile and compressive test results indicate that beryllium oxide breaks mainly along grain boundaries. At low temperature, there is little internal deformation of beryllium oxide grains. When the temperature is above 1473 K, the internal deformation of beryllium oxide grains also occurs, and the phenomenon becomes more obvious with the increase in temperature. This deformation within the grain should be plastic. The flexural strength fracture morphology of beryllium oxide also shows that the fracture mode of beryllium oxide is a brittle fracture at low temperature, while the slip bands appear at 1773 K. This indicates that beryllium oxide, as a ceramic material, can also undergo plastic deformation under high temperature and stress. read less NOT USED (low confidence) A. Sharma, S. Sharma, and S. Ajori, “Molecular dynamics simulation of the mechanical and thermal properties of phagraphene nanosheets and nanotubes: a review,” Journal of Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) X. Zhou et al., “The molecular dynamics description of electric field effect on nano-pumping performance of boron-nitride nanotube (BNNT) in the presence of vacancy defect,” Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2023. link Times cited: 2 NOT USED (low confidence) S. S. Taheri and M. M. S. Fakhrabadi, “Elastic and inelastic behavior of boron nitride nanocones at finite strains,” Physics Letters A. 2023. link Times cited: 0 NOT USED (low confidence) M. Siron, N. Chandrasekhar, and K. Persson, “Enabling automated high-throughput Density Functional Theory studies of amorphous material surface reactions,” Computational Materials Science. 2023. link Times cited: 2 NOT USED (low confidence) M. Hou, X. Zhou, and B. Liu, “Stress corrosion phenomenon of BeO at room temperature and its mechanism: Experimental and molecular dynamics study,” Materials Today Communications. 2023. link Times cited: 0 NOT USED (low confidence) S. González-Tortuero, M. A. Garrido, and J. Rodríguez, “An adhesion study in Ni and Cu nanocontacts from a molecular dynamics perspective,” European Journal of Mechanics - A/Solids. 2023. link Times cited: 3 NOT USED (low confidence) B. P. Asadollahi, M. P. Panah, and R. Sabetvand, “Molecular Dynamics Study of SiC Nanoparticle Effect on Crack Growth in Ti-6Al-4V Alloy,” Journal of Materials Engineering and Performance. 2023. link Times cited: 0 NOT USED (low confidence) S. Paul, D. Schwen, M. Short, and K. Momeni, “A Modified Embedded-Atom Method Potential for a Quaternary Fe-Cr-Si-Mo Solid Solution Alloy,” Materials. 2023. link Times cited: 0 Abstract: Ferritic-martensitic steels, such as T91, are candidate mate… read moreAbstract: Ferritic-martensitic steels, such as T91, are candidate materials for high-temperature applications, including superheaters, heat exchangers, and advanced nuclear reactors. Considering these alloys’ wide applications, an atomistic understanding of the underlying mechanisms responsible for their excellent mechano-chemical properties is crucial. Here, we developed a modified embedded-atom method (MEAM) potential for the Fe-Cr-Si-Mo quaternary alloy system—i.e., four major elements of T91—using a multi-objective optimization approach to fit thermomechanical properties reported using density functional theory (DFT) calculations and experimental measurements. Elastic constants calculated using the proposed potential for binary interactions agreed well with ab initio calculations. Furthermore, the computed thermal expansion and self-diffusion coefficients employing this potential are in good agreement with other studies. This potential will offer insightful atomistic knowledge to design alloys for use in harsh environments. read less NOT USED (low confidence) Y. Wang, W. Nie, L.-zhi Wang, D. Zhang, K. Niu, and W. Xia, “Understanding the graphene-polymer interfacial mechanical behavior via coarse-grained modeling,” Computational Materials Science. 2023. link Times cited: 2 NOT USED (low confidence) Y. Dong, W. Hui, F. Lian, and Y. Ding, “Phononic origin of strain-controlled friction force,” Tribology International. 2023. link Times cited: 1 NOT USED (low confidence) H. Xu, Z. Li, Z. Zhang, S. Liu, S. Shen, and Y. Guo, “High-Accuracy Neural Network Interatomic Potential for Silicon Nitride,” Nanomaterials. 2023. link Times cited: 1 Abstract: In the field of machine learning (ML) and data science, it i… read moreAbstract: In the field of machine learning (ML) and data science, it is meaningful to use the advantages of ML to create reliable interatomic potentials. Deep potential molecular dynamics (DEEPMD) are one of the most useful methods to create interatomic potentials. Among ceramic materials, amorphous silicon nitride (SiNx) features good electrical insulation, abrasion resistance, and mechanical strength, which is widely applied in industries. In our work, a neural network potential (NNP) for SiNx was created based on DEEPMD, and the NNP is confirmed to be applicable to the SiNx model. The tensile tests were simulated to compare the mechanical properties of SiNx with different compositions based on the molecular dynamic method coupled with NNP. Among these SiNx, Si3N4 has the largest elastic modulus (E) and yield stress (σs), showing the desired mechanical strength owing to the largest coordination numbers (CN) and radial distribution function (RDF). The RDFs and CNs decrease with the increase of x; meanwhile, E and σs of SiNx decrease when the proportion of Si increases. It can be concluded that the ratio of nitrogen to silicon can reflect the RDFs and CNs in micro level and macro mechanical properties of SiNx to a large extent. read less NOT USED (low confidence) Y. Lei and Y. Yan, “BORON NITRIDE NANOSHEET SURFACE REINFORCEMENT OF ALUMINUM,” Journal of Applied Mechanics and Technical Physics. 2023. link Times cited: 0 NOT USED (low confidence) E. Ivanova and V. D. Tur, “The body‐point model and its application to describe the motion of an electron near the nucleus of a hydrogen atom,” ZAMM ‐ Journal of Applied Mathematics and Mechanics / Zeitschrift für Angewandte Mathematik und Mechanik. 2023. link Times cited: 1 Abstract: We consider the motion of a body‐point near an attracting ce… read moreAbstract: We consider the motion of a body‐point near an attracting center. The body‐point is defined as a particle with the following properties. (a) The body‐point occupies zero volume in space like a point mass. (b) The body‐point has both translational and rotational degrees of freedom like a rigid body. (c) The body‐point is characterized by a larger number of moments of inertia than an ordinary rigid body. Due to the additional moments of inertia, the body‐point acquires dynamic properties that are fundamentally different from the dynamic properties of an ordinary rigid body. We show that the trajectory of the body‐point moving near the attracting center is a spatial curve, and not a flat one, as it would be in the case of a point mass or a rigid body. We study in detail a special case of the body‐point motion, in which the magnitude of the momentum vector remains constant. We show that, in this special case, the region of space where the trajectory of the body‐point is located can be interpreted as the orbital of an electron in a hydrogen atom. Assuming that the body‐point models the electron in the ground energy state, we determine the parameters of our model. We emphasize that the problem of the body‐point motion near an attracting center is considered for the first time, and therefore all theoretical results presented in this paper are novel. Also, for the first time, the model of the body‐point in a central potential field is used to describe the behavior of an electron in a hydrogen atom. read less NOT USED (low confidence) M. Eghbalian, R. Ansari, and S. Haghighi, “Elastic Properties of Randomly Dispersed Functionalized Silicon Carbide Nanotubes/Polymer Nanocomposites: Combined Multiscale Molecular Dynamics and Finite Element Modeling,” Silicon. 2023. link Times cited: 1 NOT USED (low confidence) M. Maździarz, “Transferability of interatomic potentials for silicene,” Beilstein Journal of Nanotechnology. 2023. link Times cited: 1 Abstract: The ability of various interatomic potentials to reproduce t… read moreAbstract: The ability of various interatomic potentials to reproduce the properties of silicene, that is, 2D single-layer silicon, polymorphs was examined. Structural and mechanical properties of flat, low-buckled, trigonal dumbbell, honeycomb dumbbell, and large honeycomb dumbbell silicene phases, were obtained using density functional theory and molecular statics calculations with Tersoff, MEAM, Stillinger–Weber, EDIP, ReaxFF, COMB, and machine-learning-based interatomic potentials. A quantitative systematic comparison and a discussion of the results obtained are reported. read less NOT USED (low confidence) S. Ghorbanali, E. Zaminpayma, and H. Mobarakinia, “Stability, mechanical and electronic properties of Occ carbon allotropes: Four new tetragonal 3D superhard carbon crystals,” Diamond and Related Materials. 2023. link Times cited: 1 NOT USED (low confidence) Y. Dong et al., “Decoding the phonon transport of structural lubrication at silicon/silicon interface,” Nanotechnology. 2023. link Times cited: 2 Abstract: Although the friction characteristics under different contac… read moreAbstract: Although the friction characteristics under different contact conditions have been extensively studied, the mechanism of phonon transport at the structural lubrication interface is not extremely clear. In this paper, we firstly promulgate that there is a 90°-symmetry of friction force depending on rotation angle at Si/Si interface, which is independent of normal load and temperature. It is further found that the interfacial temperature difference under incommensurate contacts is much larger than that in commensurate cases, which can be attributed to the larger interfacial thermal resistance (ITR). The lower ITR brings greater energy dissipation in commensurate sliding, and the reason for that is more effective energy dissipation channels between the friction surfaces, making it easier for the excited phonons at the washboard frequency and its harmonics to transfer through the interface. Nevertheless, the vibrational frequencies of the interfacial atoms between the tip and substrate during the friction process do not match in incommensurate cases, and there is no effective energy transfer channel, thus presenting the higher ITR and lower friction. Eventually, the number of excited phonons on contact surfaces reveals the amount of frictional energy dissipation in different contact states. read less NOT USED (low confidence) H. Niu et al., “A machine-learning interatomic potential to understand primary radiation damage of silicon,” Computational Materials Science. 2023. link Times cited: 3 NOT USED (low confidence) Y. Wang, Q. Mao, Z. Wang, K. Luo, L. Zhou, and H. Wei, “A ReaxFF molecular dynamics study of polycyclic aromatic hydrocarbon oxidation assisted by nitrogen oxides,” Combustion and Flame. 2023. link Times cited: 2 NOT USED (low confidence) G. Cao et al., “Molecular dynamics simulation of the mechanical characteristics of brick structure reinforced with graphene nanosheet,” Solid State Communications. 2023. link Times cited: 0 NOT USED (low confidence) S. Yan, D. Xia, N.-C. Lai, B. Jiang, and X. Liu, “New insight into the synergistic reactions involved in the hydrothermal co-liquefaction of synthetic polymer wastes by molecular dynamics and DFT methods.,” Journal of hazardous materials. 2023. link Times cited: 0 NOT USED (low confidence) M. Fan, C. Zhou, J. Duan, J. Gong, and Z.-yi Jiang, “Effect of indentation rate on deformation behavior and mechanical properties of FeO/Fe is simulated based on molecular dynamics,” Applied Physics A. 2023. link Times cited: 0 NOT USED (low confidence) A. Pedrielli, M. Dapor, K. Gkagkas, S. Taioli, and N. Pugno, “Mechanical Properties of Twisted Carbon Nanotube Bundles with Carbon Linkers from Molecular Dynamics Simulations,” International Journal of Molecular Sciences. 2023. link Times cited: 3 Abstract: The manufacturing of high-modulus, high-strength fibers is o… read moreAbstract: The manufacturing of high-modulus, high-strength fibers is of paramount importance for real-world, high-end applications. In this respect, carbon nanotubes represent the ideal candidates for realizing such fibers. However, their remarkable mechanical performance is difficult to bring up to the macroscale, due to the low load transfer within the fiber. A strategy to increase such load transfer is the introduction of chemical linkers connecting the units, which can be obtained, for example, using carbon ion-beam irradiation. In this work, we investigate, via molecular dynamics simulations, the mechanical properties of twisted nanotube bundles in which the linkers are composed of interstitial single carbon atoms. We find a significant interplay between the twist and the percentage of linkers. Finally, we evaluate the suitability of two different force fields for the description of these systems: the dihedral-angle-corrected registry-dependent potential, which we couple for non-bonded interaction with either the AIREBO potential or the screened potential ReboScr2. We show that both of these potentials show some shortcomings in the investigation of the mechanical properties of bundles with carbon linkers. read less NOT USED (low confidence) M. Eghbalian, R. Ansari, and S. Haghighi, “Molecular dynamics investigation of the mechanical properties and fracture behaviour of hydroxyl-functionalised carbon and silicon carbide nanotubes-reinforced polymer nanocomposites,” Molecular Simulation. 2023. link Times cited: 2 Abstract: ABSTRACT The tensile properties and fracture mechanism of hy… read moreAbstract: ABSTRACT The tensile properties and fracture mechanism of hydroxyl-functionalized silicon carbide nanotubes (O-fSiCNTs) inserted into polymer matrices are explored and the outcomes are compared to results for the hydroxyl-functionalized carbon nanotubes (O-fCNTs) incorporated in similar matrices. The molecular dynamics (MD) method is used and the simulations are based on the notion of representative volume elements (RVEs). The incorporation of chemisorbed nanotubes in polymers has a profound effect on the enhancement of their mechanical properties. The O-fSiCNTs inside the polyethylene (PE) and polypropylene (PP) (O-fSiCNTs/PE and O-fSiCNTs/PP) possess lower Young’s modulus, maximum stress, and strain energy as compared to the O-fCNTs/PE and O-fCNTs/PP. The zigzag O-fSiCNTs/polymer experiences lower bearable maximum strains in response to imposed loads in comparison with the O-fCNTs/polymer which is opposite to what occurs in the armchair O-fSiCNTs and O-fCNTs/polymer. The more the functionalization degree is, the weaker the structure is and its stiffness, tensile strength, tolerable strain before fracture, and ability to absorption of internal energy decline. Not only are the zigzag O-fSiCNTs/polymer stiffer than the armchair O-fSiCNTs/polymer in every percent of functionalization, but also as compared to the armchair ones, they show a lower decrease in the variation of Young’s modulus with increasing the functionalization percentage. read less NOT USED (low confidence) E. Ghavanloo, H. Rafii-Tabar, A. Kausar, G. Giannopoulos, and S. A. Fazelzadeh, “Experimental and computational physics of fullerenes and their nanocomposites: Synthesis, thermo-mechanical characteristics and nanomedicine applications,” Physics Reports. 2023. link Times cited: 6 NOT USED (low confidence) A. Khaitan et al., “Characterization of quenched MD simulated porous carbon electrodes for supercapacitors,” Materials Today: Proceedings. 2023. link Times cited: 0 NOT USED (low confidence) G. Wang et al., “Theoretical and experimental studies of nucleation and interface structure between carbon nanotubes and metals,” Journal of Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) Z. Bian et al., “Effects of different incidence rates of carbon and silicon clusters on the surface properties of SiC films,” Surfaces and Interfaces. 2023. link Times cited: 2 NOT USED (low confidence) A. Akhunova, L. Galiakhmetova, and J. Baimova, “The Effects of Dislocation Dipoles on the Failure Strength of Wrinkled Graphene from Atomistic Simulation,” Applied Sciences. 2022. link Times cited: 3 Abstract: This research paper studies the fracture and mechanical prop… read moreAbstract: This research paper studies the fracture and mechanical properties of rippled graphene containing dislocation dipoles. The atomistic simulation is performed to study the deformation behavior of pristine and defective wrinkled graphene. Graphene wrinkling considerably decreases the ultimate tensile strength of graphene with and without defects but increases the fracture strain. For graphene with the dislocation dipoles, temperature increase slightly affects mechanical properties, in contrast to graphene and graphene with Stone–Wales defect. The extremely similar slopes of the stress-strain curves for graphene with the dislocation dipoles with different arms imply that the distance between dislocations in the dipole does not have noticeable effects on the elastic modulus and strength of graphene. Defects in graphene can also affect its wrinkling; for example, preventing wrinkle formation. read less NOT USED (low confidence) J. Li, Y. Huang, Y. Zhou, and F. Zhu, “The effect of BNNS distribution on the plastic deformation of BNNS/ Al composites during the nanoindentation,” 2022 IEEE 24th Electronics Packaging Technology Conference (EPTC). 2022. link Times cited: 0 Abstract: Although it has been demonstrated that BNNS/Al composites ha… read moreAbstract: Although it has been demonstrated that BNNS/Al composites have excellent tensile and compressive properties, the effect of BNNS's distribution on the hardness of BNNS/Al composites has been rarely studied. In this work, molecular dynamics (MD) simulations are performed to obtain the nanoindentation process of the BNNS/Al models with different distribution angles of BNNS. The results show that the distribution angle of BNNS in the Al matrix has little effect on the hardness and dislocation density. However, different angles have a great influence on the deformation of the Al matrix. When the plane of BNNS is in the same direction as the slippage, the atomic strain of the Al matrix can extend to the bottom of the material. While other distributions have a hindering effect on the atomic strain of the Al matrix. In addition, the distribution of BNNS also changes the extension direction of the phase transition of the Al matrix. read less NOT USED (low confidence) A. Bahadoran et al., “Separation of SiO2 nanoparticles from H2O vapour using graphene nano-pores in the presence of an external electric field: A molecular dynamics approach,” Carbon. 2022. link Times cited: 0 NOT USED (low confidence) A. S. Grossek, A. Niggas, R. Wilhelm, F. Aumayr, and C. Lemell, “Model for Nanopore Formation in Two-Dimensional Materials by Impact of Highly Charged Ions,” Nano Letters. 2022. link Times cited: 5 Abstract: We present a first qualitative description of the atomic dyn… read moreAbstract: We present a first qualitative description of the atomic dynamics in two-dimensional (2D) materials induced by the impact of slow, highly charged ions. We employ a classical molecular dynamics simulation for the motion of the target atoms combined with a Monte Carlo model for the diffusive charge transport within the layer. Depending on the velocity of charge transfer (hopping time or hole mobility) and the number of extracted electrons which, in turn, depends on the charge state of the impinging ions, we find regions of stability of the 2D structure as well as parameter combinations for which nanopore formation due to Coulomb repulsion is predicted. read less NOT USED (low confidence) Y. Zhang, Q. Lin, and B. Jiang, “Atomistic neural network representations for chemical dynamics simulations of molecular, condensed phase, and interfacial systems: Efficiency, representability, and generalization,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2022. link Times cited: 10 Abstract: Machine learning techniques have been widely applied in many… read moreAbstract: Machine learning techniques have been widely applied in many fields of chemistry, physics, biology, and materials science. One of the most fruitful applications is machine learning of the complicated multidimensional function of potential energy or related electronic properties from discrete quantum chemical data. In particular, substantial efforts have been dedicated to developing various atomistic neural network (AtNN) representations, which refer to a family of methods expressing the targeted physical quantity as a sum of atomic components represented by atomic NNs. This class of approaches not only fully preserves the physical symmetry of the system but also scales linearly with respect to the size of a system, enabling accurate and efficient chemical dynamics and spectroscopic simulations in complicated systems and even a number of variably sized systems across the phases. In this review, we discuss different strategies in developing highly efficient and representable AtNN potentials, and in generalizing these scalar AtNN models to learn vectorial and tensorial quantities with the correct rotational equivariance. We also review active learning algorithms to generate practical AtNN models and present selected examples of AtNN applications in gas‐surface systems to demonstrate their capabilities of accurately representing both molecular systems and condensed phase systems. We conclude this review by pointing out remaining challenges for the further development of more reliable, transferable, and scalable AtNN representations in more application scenarios. read less NOT USED (low confidence) X. Zhang, H. Nguyen, X. Zhang, P. Ajayan, J. Wen, and H. Espinosa, “Atomistic measurement and modeling of intrinsic fracture toughness of two-dimensional materials,” Proceedings of the National Academy of Sciences of the United States of America. 2022. link Times cited: 6 Abstract: Significance The promise of computational material design is… read moreAbstract: Significance The promise of computational material design is based on atomistic models with predictive capabilities. While quantum chemistry simulations can be used during unit cell property screening of two-dimensional (2D) materials, device-level analysis requires large-scale atomistic simulations employing interatomic potentials. We present advances in experimentation and machine learning–inspired algorithms for accurate interatomic potential selection, parameterization, and validation. Through in-situ high-resolution transmission electron microscopy atomistic measurements, we directly compare atomic structures and material fracture toughness with those predicted by molecular dynamics simulations employing a potential informed by ab initio data in the nonequilibrium regime. The integrated framework provides a robust approach to obtaining intrinsic mechanical properties of 2D materials (in their pristine and defective states) and informs the analysis of device reliability with unprecedented accuracy. read less NOT USED (low confidence) X. Liu et al., “Phase change process in a porous Carbon-Paraffin matrix with different volume fractions of copper oxide Nanoparticles: A molecular dynamics study,” Journal of Molecular Liquids. 2022. link Times cited: 19 NOT USED (low confidence) S. Haseen and P. Kroll, “Analyzing the Effect of Composition, Density, and the Morphology of the ‘free’ Carbon Phase on Elastic Moduli in Silicon Oxycarbide Ceramics,” Journal of the European Ceramic Society. 2022. link Times cited: 1 NOT USED (low confidence) R. Tong, Y. Wang, T. Zhang, J. Du, and G. Liu, “Friction properties of the single-crystal Si in collision sliding contacts under different lubrication conditions,” Surface and Coatings Technology. 2022. link Times cited: 0 NOT USED (low confidence) A. Hernandez and T. Mueller, “Generalizability of Functional Forms for Interatomic Potential Models Discovered by Symbolic Regression,” ArXiv. 2022. link Times cited: 0 Abstract: In recent years there has been great progress in the use of … read moreAbstract: In recent years there has been great progress in the use of machine learning algorithms to develop interatomic potential models. Machine-learned potential models are typically orders of magnitude faster than density functional theory but also orders of magnitude slower than physics-derived models such as the embedded atom method. In our previous work, we used symbolic regression to develop fast, accurate and transferrable interatomic potential models for copper with novel functional forms that resemble those of the embedded atom method. To determine the extent to which the success of these forms was specific to copper, here we explore the generalizability of these models to other face-centered cubic transition metals and analyze their out-of-sample performance on several material properties. We found that these forms work particularly well on elements that are chemically similar to copper. When compared to optimized Sutton-Chen models, which have similar complexity, the functional forms discovered using symbolic regression perform better across all elements considered except gold where they have a similar performance. They perform similarly to a moderately more complex embedded atom form on properties on which they were trained, and they are more accurate on average on other properties. We attribute this improved generalized accuracy to the relative simplicity of the models discovered using symbolic regression. The genetic programming models are found to outperform other models from the literature about 50% of the time in a variety of property predictions, with about 1/10th the model complexity on average. We discuss the implications of these results to the broader application of symbolic regression to the development of new potentials and highlight how models discovered for one element can be used to seed new searches for different elements. read less NOT USED (low confidence) K. Karasev, D. Strizhkin, and P. Karaseov, “Convenient Way to Create an MD Model of a Hot Crystal with an Open Surface,” 2022 International Conference on Electrical Engineering and Photonics (EExPolytech). 2022. link Times cited: 2 Abstract: Molecular Dynamics simulation is a widely used approach in r… read moreAbstract: Molecular Dynamics simulation is a widely used approach in research, particularly in solid state physics. Oscillations of an open surface with a large amplitude could arise during thermalisation of crystal model at some non-zero temperature. This artefact prevents correct determination of various system parameters during further simulations. As an example, number of point defects generated by a stopping ion could exhibit unphysical drastic increase. Several methods for system heating and relaxation exist, but they either take a long computation time or do not result in a low enough open surface oscillation amplitude. In this contribution, we propose fast and convenient technique to create a model of a large open-surface crystal at an elevated temperature. Efficiency and scalability of this method is demonstrated by construction of $40\times 40\times 31$ unit cell sized Si crystal from the small $8\times 8\times 16$ unit cell equilibrated seed cells. Amplitude of the (100) open surface oscillations of less than 0.1 A is demonstrated. This technique would be useful for those who is intended to perform MD simulations of large systems using accessible desktop computer systems. read less NOT USED (low confidence) Z. Liu, X. Sun, J. Xie, X. Zhang, and J. Li, “Interfacial thermal transport properties and its effect on thermal conductivity of functionalized BNNS/epoxy composites,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 3 NOT USED (low confidence) C. Wang, Z. Wang, Z. Sun, L. Zhu, Y. Li, and T. Li, “Molecular dynamics simulation of single droplet behavior on the windward side of a fiber filter during coalescence,” Chemical Engineering Science. 2022. link Times cited: 2 NOT USED (low confidence) J. Li, Y. Huang, Y. Zhou, and F. Zhu, “Role of boron nitride nanosheets coating on aluminum substrates during the nanoindentation from the atomic perspective,” Applied Surface Science. 2022. link Times cited: 2 NOT USED (low confidence) Z. Ge, H. Li, and X. Cheng, “Research on phase transition induced plastic deformation in nanoindentation of single crystal diamond,” Diamond and Related Materials. 2022. link Times cited: 0 NOT USED (low confidence) I. Jeon, T. Yun, and S. Yang, “Classical, Coarse-Grained, and Reactive Molecular Dynamics Simulations on Polymer Nanocomposites,” Multiscale Science and Engineering. 2022. link Times cited: 5 NOT USED (low confidence) X. Han, “Investigate the constrained-microplasticity of nano-polycrystal silicon in nanomachining using atomic simulation method,” Applied Physics A. 2022. link Times cited: 1 NOT USED (low confidence) Y. Rostamiyan, N. Shahab, C. Spitas, and A. H. Mashhadzadeh, “Mechanical properties of multi-walled beryllium-oxide nanotubes: a molecular dynamics simulation study,” Journal of Molecular Modeling. 2022. link Times cited: 2 NOT USED (low confidence) A. Tanhadoust, M. Jahanshahi, and A. Khoei, “Temperature-dependent multiscale modeling of graphene sheet under finite deformation,” Diamond and Related Materials. 2022. link Times cited: 1 NOT USED (low confidence) Y. Lin et al., “Interfacial mechanical properties of tetrahydrofuran hydrate-solid surfaces: Implications for hydrate management.,” Journal of colloid and interface science. 2022. link Times cited: 9 NOT USED (low confidence) S. Im, H. Kim, W. Kim, H. Chung, and M. Cho, “Discovering constitutive equations of crystal structures by sparse identification,” International Journal of Mechanical Sciences. 2022. link Times cited: 0 NOT USED (low confidence) Y. Wang and X. Zhou, “Molecular Dynamics Simulation of Fe-Based Metal Powder Oxidation during Laser Powder Bed Fusion,” Materials. 2022. link Times cited: 4 Abstract: Because the laser powder bed fusion process is generally com… read moreAbstract: Because the laser powder bed fusion process is generally completed in a confined space and in a very short time, it is difficult to study material oxidation during this process using traditional methods. To address this knowledge gap, in this work, we used molecular dynamics (MDs) based on a reaction force field (ReaxFF) to clarify the atomic-level interaction mechanism between metal atoms and oxygen molecules during laser powder bed fusion. The ReaxFF potential energy model has variable charges that can dynamically handle charge changes between atoms and the breaking and formation of chemical bonds that occur during oxidation reactions. We investigated the effects of laser power, scanning speed, region position, and oxygen concentration on powder oxidation. The results show that the laser power and scanning speed affected the oxidation degree by changing the energy input density, and the oxidation degree increased with the energy input density. Different forms of oxidation occurred near the melt channel due to the existence of a temperature gradient, and the degree of oxidation increased with the temperature. Atoms in the metal powder model underwent selective oxidation, which was related to the potential energy of their atomic position. A larger potential energy made it easier for iron atoms to overcome the energy barrier during the initial stage of oxidation, making them easier to oxidize. read less NOT USED (low confidence) Y. Liu et al., “Investigations on microstructure and mechanical properties of boron nitride fiber using experimental and numerical methods,” Materials Today Communications. 2022. link Times cited: 0 NOT USED (low confidence) M. Diego et al., “Ultrafast nano generation of acoustic waves in water via a single carbon nanotube,” Photoacoustics. 2022. link Times cited: 7 NOT USED (low confidence) M. Eghbalian, R. Ansari, and S. Haghighi, “A combined molecular dynamics-finite element multiscale modeling to analyze the mechanical properties of randomly dispersed, chemisorbed carbon nanotubes/polymer nanocomposites,” Mechanics of Advanced Materials and Structures. 2022. link Times cited: 5 Abstract: A two-stage molecular dynamics (MD)-finite element (FE) mode… read moreAbstract: A two-stage molecular dynamics (MD)-finite element (FE) modeling method is developed based on the concepts of representative volume element (RVE) and equivalent solid fibers (ESFs) containing functionalized carbon nanotubes (ESFs-fCNTs). First, the influences of nanotubes’ chirality, different percent of functionalization ( ), various functional atoms, and polymers on the tensile and shear properties of the fCNTs inserted into the polymer matrix (fCNTs/polymer) are discovered using MD simulations. Then, using MD information as input data, the effective Young’s modulus of polymeric unit cell strengthened by ESFs-fCNTs (ESFs-fCNTs/polymer) is explored through FE modeling. The ratio of effective Young’s modulus of the unit cell ( ) to Young’s modulus of the polymeric cube ( ) is reported and all findings ( ) are compared to the ESFs-pure CNTs/polymer results as well. It is found that longitudinal Young’s modulus ( ) of nanofillers/polymer RVEs affects remarkably the of the ESFs-nanofillers/polymer nanocomposites. The decreases by increasing the Generally, the reinforcing impact of zigzag nanotubes compared to armchair ones on the of polymer RVEs is more considerable. Additionally, FE-based results illustrate that as the volume fraction of ESFs ( ) increases, the is enhanced. At a specific the reinforcing effect of the ESFs-armchair and zigzag fCNTs is more in favor of polyethylene nanocomposites than that of the polypropylene systems. GRAPHICAL ABSTRACT read less NOT USED (low confidence) I. Kosarev, S. Dmitriev, A. Semenov, and E. Korznikova, “Stability of Strained Stanene Compared to That of Graphene,” Materials. 2022. link Times cited: 1 Abstract: Stanene, composed of tin atoms, is a member of 2D-Xenes, two… read moreAbstract: Stanene, composed of tin atoms, is a member of 2D-Xenes, two-dimensional single element materials. The properties of the stanene can be changed and improved by applying deformation, and it is important to know the range of in-plane deformation that the stanene can withstand. Using the Tersoff interatomic potential for calculation of phonon frequencies, the range of stability of planar stanene under uniform in-plane deformation is analyzed and compared with the known data for graphene. Unlike atomically flat graphene, stanene has a certain thickness (buckling height). It is shown that as the tensile strain increases, the thickness of the buckled stanene decreases, and when a certain tensile strain is reached, the stanene becomes absolutely flat, like graphene. Postcritical behaviour of stanene depends on the type of applied strain: critical tensile strain leads to breaking of interatomic bonds and critical in-plane compressive strain leads to rippling of stanene. It is demonstrated that application of shear strain reduces the range of stability of stanene. The existence of two energetically equivalent states of stanene is shown, and consequently, the possibility of the formation of domains separated by domain walls in the stanene is predicted. read less NOT USED (low confidence) J. Han, Y. Chen, J. Wang, G. Zhang, and H. Wang, “A review of molecular dynamics simulation in studying surface generation mechanism in ultra-precision cutting,” The International Journal of Advanced Manufacturing Technology. 2022. link Times cited: 2 NOT USED (low confidence) W. He, D. Wang, W. Ming, J. Ma, K. Liu, and J. Du, “Research progress on cutting machining simulation technology of metallic glasses,” The International Journal of Advanced Manufacturing Technology. 2022. link Times cited: 2 NOT USED (low confidence) G. Miloshevsky, “Ultrafast laser matter interactions: modeling approaches, challenges, and prospects,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 4 Abstract: The irradiation of the target surface by an ultrafast femtos… read moreAbstract: The irradiation of the target surface by an ultrafast femtosecond (fs) laser pulse produces the extreme non-equilibrium states of matter and subsequent phase transformations. Computational modeling and simulation is a very important tool for gaining insight into the physics processes that govern the laser–matter interactions, and, specifically, for quantitative understanding the laser light absorption, electron–ion energy exchange, spallation, melting, warm dense matter regime, vaporization, and expansion of plasma plume. High-fidelity predictive modeling of a variety of these multi-physics processes that take place at various time and length scales is extremely difficult, requiring the coupled multi-physics and multi-scale models. This topical review covers progress and advances in developing the modeling approaches and performing the state-of-the-art simulations of fs laser-pulse interactions with solids and plasmas. A complete kinetic description of a plasma based on the most accurate Vlasov–Maxwell set of equations is first presented and discussed in detail. After that an exact kinetic model that encompasses the microscopic motions of all the individual particles, their charge and current densities, generated electric and magnetic fields, and the effects of these fields on the motion of charged particles in a plasma is briefly reviewed. The methodology of kinetic particle-in-cell (PIC) approach that is well suitable for computational studies of the non-linear processes in laser–plasma interactions is then presented. The hydrodynamic models used for the description of plasmas under the assumption of a local thermodynamic equilibrium include the two-fluid and two-temperature model and its simplifications. The two-temperature model coupled with molecular dynamics (MD) method is finally discussed. Examples are illustrated from research areas such as applications of the fully kinetic, PIC, hydrodynamic, and MD models to studies of ultrafast laser–matter interactions. Challenges and prospects in the development of computational models and their applications to the modeling of ultrafast intense laser–solid and laser–plasma interactions are overviewed. read less NOT USED (low confidence) C. Hou et al., “Atomistic simulation of low-dimensional nanostructures toward extreme-scale supercomputing,” CCF Transactions on High Performance Computing. 2022. link Times cited: 0 NOT USED (low confidence) B. Luo et al., “Novel atomic-scale graphene metamaterials with broadband electromagnetic wave absorption and ultra-high elastic modulus,” Carbon. 2022. link Times cited: 8 NOT USED (low confidence) B. Yao, Z. R. Liu, and R. F. Zhang, “EAPOTc: An integrated empirical interatomic potential optimization platform for compound solids,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) Y. Qiu, W. Zhong, and A. Yu, “The molecular dynamics simulation of lignite combustion process in O2/CO2 atmosphere with ReaxFF force field,” Powder Technology. 2022. link Times cited: 4 NOT USED (low confidence) G. Mora-Barzaga et al., “Enhancing the Thermal Conductivity of Amorphous Carbon with Nanowires and Nanotubes,” Nanomaterials. 2022. link Times cited: 1 Abstract: The thermal conductivity of nanostructures can be obtained u… read moreAbstract: The thermal conductivity of nanostructures can be obtained using atomistic classical Molecular Dynamics (MD) simulations, particularly for semiconductors where there is no significant contribution from electrons to thermal conduction. In this work, we obtain and analyze the thermal conductivity of amorphous carbon (aC) nanowires (NW) with a 2 nm radius and aC nanotubes (NT) with 0.5, 1 and 1.3 nm internal radii and a 2 nm external radius. The behavior of thermal conductivity with internal radii, temperature and density (related to different levels of sp3 hybridization), is compared with experimental results from the literature. Reasonable agreement is found between our modeling results and the experiments for aC films. In addition, in our simulations, the bulk conductivity is lower than the NW conductivity, which in turn is lower than the NT conductivity. NTs thermal conductivity can be tailored as a function of the wall thickness, which surprisingly increases when the wall thickness decreases. While the vibrational density of states (VDOS) is similar for bulk, NW and NT, the elastic modulus is sensitive to the geometrical parameters, which can explain the enhanced thermal conductivity observed for the simulated nanostructures. read less NOT USED (low confidence) R. Badal, M. Friedrich, and J. Seutter, “Existence of quasi-static crack evolution for atomistic systems,” Forces in Mechanics. 2022. link Times cited: 0 NOT USED (low confidence) E. Hodille, J. Byggmästar, Y. Ferro, and K. Nordlund, “Molecular dynamics study of hydrogen isotopes at the Be/BeO interface,” Journal of Physics: Condensed Matter. 2022. link Times cited: 3 Abstract: Molecular dynamics simulations are used to investigate the b… read moreAbstract: Molecular dynamics simulations are used to investigate the behaviour of D atoms at two interfaces between beryllium (Be) and beryllium oxide (BeO). After relaxation of the simulation cell, there are (a) localised defects at the interface and (b) a hexagonal misfit dislocation network creating a succession of compressed and expanded area from each side of the interface. The simulations between 750 K and 1500 K for tens to hundreds of nanoseconds show that both interfaces act as trapping sites for D atoms. The simulations also show that D atoms tend to migrate in the material where the hydrogen isotope solubility is the highest as predicted by thermodynamics. However, the simulations also shows that there are additional kinetic barriers (D trapping sites, D2 formation/dissociation in BeO) that slow down the path to equilibrium. These additional kinetic barriers may influence the fuel retention and permeation in Be materials. read less NOT USED (low confidence) R. Abram, D. Chrobak, J. Byggmästar, K. Nordlund, and R. Nowak, “Comprehensive structural changes in nanoscale-deformed silicon modelled with an integrated atomic potential,” Materialia. 2022. link Times cited: 2 NOT USED (low confidence) A. M. Bell and P. Mulheran, “Modelling multi-scale material growth and erosion under energetic atomic deposition,” Molecular Simulation. 2022. link Times cited: 0 Abstract: ABSTRACT We study crystalline surface evolution in extreme e… read moreAbstract: ABSTRACT We study crystalline surface evolution in extreme environments, where high-energy atoms impinge on a crystalline surface to cause sputtering, growth and surface roughening. For our model system, we study the evolution of the Ni(111) surface under Ni atom bombardment, using Molecular Dynamics (MD) simulation and a Sutton-Chen force field, where the uppermost surface layers are free to move, supported by thermostatically controlled layers above a rigid template. The MD statistics of sputtering and sticking are used to aid the development of a computationally efficient kinetic Monte Carlo (KMC) code. Comparisons between the simulation surface morphologies are used to tune the KMC growth rules so that the key statistical features of the MD structures are captured by the KMC model. This model is then employed to explore the predicted behaviour over length and times scales much larger than those accessible to MD. While the MD-KMC approach is well known, this application to the complex surface growth encountered in energetic atomic bombardment illuminates the complexities of relating atomistic events to morphological evolution. The work shows how simulation methodology can be extended to provide predictive capabilities, paving the way for design tools for engineering processes such as plasma deposition film growth. read less NOT USED (low confidence) M. A. Torkaman-Asadi and M. A. Kouchakzadeh, “Atomistic simulations of mechanical properties and fracture of graphene: A review,” Computational Materials Science. 2022. link Times cited: 14 NOT USED (low confidence) W. Chen et al., “Engineering Electronic Platinum–Carbon Support Interaction to Tame Carbon Monoxide Activation,” Fundamental Research. 2022. link Times cited: 3 NOT USED (low confidence) D. P. Ranjan, M. A. Owhal, D. Chakrabarti, D. S. Belgamwar, T. Roy, and D. R. Balasubramaniam, “Fundamental Insights of Mechanical Polishing on Polycrystalline Cu Through Molecular Dynamics Simulations,” SSRN Electronic Journal. 2022. link Times cited: 9 NOT USED (low confidence) B. Luo, L. Wu, Z. Zhang, G. Li, and E. Tian, “A triatomic carbon and derived pentacarbides with superstrong mechanical properties,” iScience. 2022. link Times cited: 7 NOT USED (low confidence) N. Kametani, M. Nakamura, K. Yashiro, and T. Takaki, “Investigating residual stress evolution in the deposition process of diamond-like carbon film through molecular dynamics,” Computational Materials Science. 2022. link Times cited: 8 NOT USED (low confidence) L. Patra and R. Pandey, “Mechanical properties of 2D materials: A review on molecular dynamics based nanoindentation simulations,” Materials Today Communications. 2022. link Times cited: 8 NOT USED (low confidence) W. Wan, C. Tang, and W. Zou, “Exploring Silicon [001] Small Angle Symmetric Tilt Grain Boundaries: Structures, Energies and Stress fields,” Applied Surface Science. 2022. link Times cited: 4 NOT USED (low confidence) V. Ivashchenko, P. Turchi, R. V. Shevchenko, L. Gorb, J. Leszczynski, and A. Kozak, “An effect of nitrogen incorporation on the structure and properties of amorphous SiC: first-principles molecular dynamics simulations,” Thin Solid Films. 2022. link Times cited: 0 NOT USED (low confidence) F. Molaei, “Understanding the Anisotropic Mechanical Behavior of Single‐Crystalline Alpha Quartz From the Insight of Molecular Dynamics,” Journal of Geophysical Research: Solid Earth. 2022. link Times cited: 1 Abstract: Quartz is among Earth's most abundant minerals, which h… read moreAbstract: Quartz is among Earth's most abundant minerals, which has several stable polymorphs in nature. In this study, molecular dynamics simulations are used to investigate the mechanical properties of crystalline alpha quartz. The obtained results specify that tensile stress under uniaxial tension is greater in z (c‐axis) than in other directions (290 vs. 115 GPa and 190 GPa). This outcome confirms that crystalline quartz has an anisotropic behavior under applied load, and as a result, Young's modulus varies in different directions. Furthermore, the effect of existing central cracks with different lengths are considered and the results analyzed. According to the results, central cracks decrease average stress and strain, and this reduction is higher in the z‐direction([0 0 0 1]). Additionally, it was found that the strain rate affects the stress‐strain behavior of the models; however, the strain rate plays a negligible role when the central crack length extends. In terms of potential energy, simulation results indicate that potential energy is the highest in the model without any crack and reduces by growing the central crack size. read less NOT USED (low confidence) Y. Dong, W. Hui, F. Lian, Y. Ding, and Z. Rui, “Phononic Friction in Monolayer/Bilayer Graphene,” Tribology Letters. 2022. link Times cited: 5 NOT USED (low confidence) E. Mareev, A. Pushkin, E. Migal, K. Lvov, S. Stremoukhov, and F. Potemkin, “Single-shot femtosecond bulk micromachining of silicon with mid-IR tightly focused beams,” Scientific Reports. 2022. link Times cited: 8 NOT USED (low confidence) M. H. Pebdani and R. Sabetvand, “Mechanical properties of carbon nanotube reinforced polyurethane matrix using computational method: a molecular dynamics study,” Physica Scripta. 2022. link Times cited: 1 Abstract: The reinforcing nanostructures can be made up of nanoparticl… read moreAbstract: The reinforcing nanostructures can be made up of nanoparticles, nanosheets or nanofibres such as carbon nanotubes (CNTs) and graphene nanosheets. To investigate the reinforce mechanism, the changes in mechanical behavior of CNT reinforced Polyurethane (PU) matrix with various chirality was studied using molecular dynamics (MD) method in current work. We used the DREIDING and Tersoff force-fields for simulation of the PU and CNT samples, respectively. To report the mechanical properties of pristine PU matrix and reinforced PU/CNT structure, some physical parameters such as interaction energy between polymer chains and nanotube atoms, ultimate strength, and Young’s modulus are calculated. MD outputs indicated inserting CNT with zigzag edge into pristine matrix enlarged the Young’s modulus by 17.10% and the ultimate strength by 25.69%. These results indicated the promising effect of CNT-based nanostructures on the mechanical properties of PU matrix. read less NOT USED (low confidence) Y. Xu, Q. Cao, L. Li, X. Zhang, H. Li, and F. Huang, “Mechanical Response of Graphene with Nanopore under Nanoindentation via Molecular Dynamics Simulations,” Surfaces and Interfaces. 2022. link Times cited: 5 NOT USED (low confidence) G. G. Vidable, R. González, F. Valencia, N. Amigo, D. Tramontina, and E. Bringa, “Simulations of plasticity in diamond nanoparticles showing ultrahigh strength,” Diamond and Related Materials. 2022. link Times cited: 8 NOT USED (low confidence) C. C. Sluss, J. Pittman, D. Nicholson, and D. Keffer, “Exploration of Entropy Pair Functional Theory,” Entropy. 2022. link Times cited: 1 Abstract: Evaluation of the entropy from molecular dynamics (MD) simul… read moreAbstract: Evaluation of the entropy from molecular dynamics (MD) simulation remains an outstanding challenge. The standard approach requires thermodynamic integration across a series of simulations. Recent work Nicholson et al. demonstrated the ability to construct a functional that returns excess entropy, based on the pair correlation function (PCF); it was capable of providing, with acceptable accuracy, the absolute excess entropy of iron simulated with a pair potential in both fluid and crystalline states. In this work, the general applicability of the Entropy Pair Functional Theory (EPFT) approach is explored by applying it to three many-body interaction potentials. These potentials are state of the art for large scale models for the three materials in this study: Fe modelled with a modified embedded atom method (MEAM) potential, Cu modelled with an MEAM and Si modelled with a Tersoff potential. We demonstrate the robust nature of EPFT in determining excess entropy for diverse systems with many-body interactions. These are steps toward a universal Entropy Pair Functional, EPF, that can be applied with confidence to determine the entropy associated with sophisticated optimized potentials and first principles simulations of liquids, crystals, engineered structures, and defects. read less NOT USED (low confidence) A. Eldridge, A. Rodriguez, M. Hu, and J. Hu, “Genetic programming-based learning of carbon interatomic potential for materials discovery,” ArXiv. 2022. link Times cited: 1 Abstract: Efficient and accurate interatomic potential functions are cr… read moreAbstract: Efficient and accurate interatomic potential functions are critical to computational study of materials while searching for structures with desired properties. Traditionally, potential functions or energy landscapes are designed by experts based on theoretical or heuristic knowledge. Here, we propose a new approach to leverage strongly typed parallel genetic programming (GP) for potential function discovery. We use a multi-objective evolutionary algorithm with NSGA-III selection to optimize individual age, fitness, and complexity through symbolic regression. With a DFT dataset of 863 unique carbon allotrope configurations drawn from 858 carbon structures, the generated potentials are able to predict total energies within ± 7 . 70 eV at low computational cost while generalizing well across multiple carbon structures. Our code is open source and available at http://www.github. com/usccolumbia/mlpotential . read less NOT USED (low confidence) D. Jacobson and G. Thompson, “Revisting Lennard Jones, Morse, and N-M potentials for metals,” Computational Materials Science. 2022. link Times cited: 9 NOT USED (low confidence) K. Yeo, K. Park, and S. Jeong, “Neural network approach to diffusion of B and N adatoms on the Pt(111) surface,” Current Applied Physics. 2022. link Times cited: 2 NOT USED (low confidence) J. Zhang, J. Wang, Z. Li, J. Zhu, and B. Lu, “Molecular Dynamics Simulation and Gas Generation Tracking of Pyrolysis of Bituminous Coal,” ACS Omega. 2022. link Times cited: 2 Abstract: To study the generation rules of organic molecules or fragme… read moreAbstract: To study the generation rules of organic molecules or fragments and the generation paths of some hydrocarbon gases (C2H2/C2H4) and inorganic gases (CO2/H2O/H2/H2S) in the pyrolysis process of bituminous coal at 1000–5000 K, the ReaxFF molecular dynamics module in AMS software was used to simulate the pyrolysis behavior of the Hongqingliang model, Gaojialiang model, and Wiser model. With the increase of temperature, the system potential energy decreases, the endothermic efficiency increases first and then decreases, the fragments of C1–C4 fragments increase, and the gas molecules generated increase. In the pyrolysis process, the oxygen-containing functional groups and hydrogen groups formed H2O and H2 with the increase of temperature. H2S as an intermediate product is always maintained in dynamic equilibrium. CO2 comes from the decarboxylation of the carboxyl groups. When the temperature is lower than 3000 K, C2H4 and C2H2 are mainly formed by the adjacent carbon structure in coal molecules, and C2H4 is formed from the ethyl side chain, the naphthenic structure, and the unstable aromatic rings. C2H2 is formed from naphthene structures and aromatic rings with multiple side chains. When the temperature is higher than 3000 K, C2H4 and C2H2 are mainly formed by the random combination of free radicals generated by the crushing of coal molecules. The research results are of great significance to coal pyrolysis and clean utilization of coal. read less NOT USED (low confidence) N. Zhang et al., “Near-Interface Defects in Graphene/H-BN In-Plane Heterostructures: Insights into the Interfacial Thermal Transport,” Nanomaterials. 2022. link Times cited: 3 Abstract: Based on nonequilibrium molecular dynamics (NEMD) and nonequ… read moreAbstract: Based on nonequilibrium molecular dynamics (NEMD) and nonequilibrium Green’s function simulations, the interfacial thermal conductance (ITC) of graphene/h-BN in-plane heterostructures with near-interface defects (monovacancy defects, 585 and f5f7 double-vacancy defects) is studied. Compared to pristine graphene/h-BN, all near-interface defects reduce the ITC of graphene/h-BN. However, differences in defective structures and the wrinkles induced by the defects cause significant discrepancies in heat transfer for defective graphene/h-BN. The stronger phonon scattering and phonon localization caused by the wider cross-section in defects and the larger wrinkles result in the double-vacancy defects having stronger energy hindrance effects than the monovacancy defects. In addition, the approximate cross-sections and wrinkles induced by the 585 and f5f7 double-vacancy defects provide approximate heat hindrance capability. The phonon transmission and vibrational density of states (VDOS) further confirm the above results. The double-vacancy defects in the near-interface region have lower low-frequency phonon transmission and VDOS values than the monovacancy defects, while the 585 and f5f7 double-vacancy defects have similar low-frequency phonon transmission and VDOS values at the near-interface region. This study provides physical insight into the thermal transport mechanisms in graphene/h-BN in-plane heterostructures with near-interface defects and provides design guidelines for related devices. read less NOT USED (low confidence) J. Byggmästar, K. Nordlund, and F. Djurabekova, “Simple machine-learned interatomic potentials for complex alloys,” Physical Review Materials. 2022. link Times cited: 5 Abstract: Developing data-driven machine-learning interatomic potentia… read moreAbstract: Developing data-driven machine-learning interatomic potentials for materials containing many elements becomes increasingly challenging due to the vast configuration space that must be sampled by the training data. We study the learning rates and achievable accuracy of machine-learning interatomic potentials for many-element alloys with different combinations of descriptors for the local atomic environments. We show that for a five-element alloy system, potentials using simple low-dimensional descriptors can reach meV/atom-accuracy with modestly sized training datasets, significantly outperforming the high-dimensional SOAP descriptor in data efficiency, accuracy, and speed. In particular, we develop a computationally fast machine-learned and tabulated Gaussian approximation potential (tabGAP) for Mo–Nb–Ta–V–W alloys with a combination of two-body, three-body, and a new simple scalar many-body density descriptor based on the embedded atom method. read less NOT USED (low confidence) X. Meng, W. Wu, B. Liao, and H. Dai, “Atomic simulation of textured silicon carbide surface ultra-precision polishing,” Ceramics International. 2022. link Times cited: 17 NOT USED (low confidence) H. Zhang, M. Shukla, S. Larson, A. Rajendran, and S. Jiang, “Molecular dynamics study of anisotropic shock responses in oriented α-quartz single crystal,” Journal of Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) Z. Hao, H. Zhang, and Y. Fan, “Mechanical response of nanoindentation and material strengthening mechanism of nt-cBN superhard materials based on molecular dynamics,” International Journal of Refractory Metals and Hard Materials. 2022. link Times cited: 3 NOT USED (low confidence) S. Haghighi, R. Ansari, and Y. Keramati, “A molecular dynamics study on the vibrational behavior of perfect and defective hybrid carbon boron-nitride heteronanotubes,” Diamond and Related Materials. 2022. link Times cited: 3 NOT USED (low confidence) M. Shahryari, A. Nazari-Golshan, and S. Nourazar, “The study of heat flux and external electric field effects on carbon nanotube behavior as an atomic nano-pump,” Applied Physics A. 2022. link Times cited: 4 NOT USED (low confidence) F. Uesugi and M. Ishii, “Classification for transmission electron microscope images from different amorphous states using persistent homology,” Microscopy. 2022. link Times cited: 1 Abstract: It is difficult to discriminate the amorphous state using a … read moreAbstract: It is difficult to discriminate the amorphous state using a transmission electron microscope (TEM). We discriminated different amorphous states on TEM images using persistent homology, which is a mathematical analysis technique that employs the homology concept and focuses on ‘holes’. The structural models of the different amorphous states, that is, amorphous and liquid states, were created using classical molecular dynamic simulation. TEM images in several defocus conditions were simulated by the multi-slice method using the created amorphous and liquid states, and their persistent diagrams were calculated. Finally, logistic regression and support vector classification machine learning algorithms were applied for discrimination. Consequently, we found that the amorphous and liquid phases can be discriminated by more than 85%. Because the contrast of TEM images depends on sample thickness, focus, lens aberration, etc., radial distribution function cannot be classified; however, the persistent homology can discriminate different amorphous states in a wide focus range. read less NOT USED (low confidence) A. Chaurasia and A. Parashar, “Molecular Dynamics Study of Anisotropic Shock Response in Mono- and Bicrystalline Boron Nitride Nanosheets: Implications for Shock-Resistant Solid-State Devices,” ACS Applied Nano Materials. 2022. link Times cited: 5 NOT USED (low confidence) A. V. Lun-Fu, A. Bubenchikov, M. Bubenchikov, and V. Ovchinnikov, “Computational Analysis of Strain-Induced Effects on the Dynamic Properties of C60 in Fullerite,” Crystals. 2022. link Times cited: 7 Abstract: A hybrid discrete-continuous physical and mathematical model… read moreAbstract: A hybrid discrete-continuous physical and mathematical model is used to study what deformation characteristics cause the rolling effect of C60 fullerene in a fullerite crystal. The interaction of fullerene atoms with surrounding molecules is described using a centrally symmetric interaction potential, in which the surrounding molecules are considered as a spherical surface of uniformly distributed carbon atoms. The rotational motion of fullerene is described by the Euler dynamic equations. The results of a numerical study of the influence of the rate, magnitude, and direction of strain on the dynamic characteristics of the rotational and translational motion of C60 fullerene in a crystalline fragment are presented. read less NOT USED (low confidence) Y. Lin and C.-Y. Wu, “Amorphous silica glass nano-grooving behavior investigated using molecular dynamics method,” Proceedings of the Institution of Mechanical Engineers, Part B: Journal of Engineering Manufacture. 2022. link Times cited: 3 Abstract: This work uses molecular dynamics to simulate the grooving b… read moreAbstract: This work uses molecular dynamics to simulate the grooving behavior of optical silica glass. The amorphous SiO2 (silica glass) was fabricated using a melting-quenching process, and the critical cut depth, and mechanism of the chip formation, were explored using a molecular dynamics simulation. The analytical results indicated that the tool edge radius affected the critical cut depth and roughness of the machined surface. A larger tool edge radius had a larger equivalent negative rake angle at the same depth of cut, causing a larger critical cut depth, while producing a smoother machined surface. In addition, the uncut chip thickness affected the tangential force and thrust force distribution weighting of the tool. The temperature field analysis revealed that a groove formed by the chip formation mechanism resulted in a higher workpiece temperature, causing the brittle material to exhibit ductile cutting behavior during the nanogrooving process. However, grooves formed by the scratching-indenting mechanism had a lower workpiece temperature. read less NOT USED (low confidence) C. Chen and S. Ong, “A universal graph deep learning interatomic potential for the periodic table,” Nature Computational Science. 2022. link Times cited: 98 NOT USED (low confidence) A. Jam, N. N. Jam, M. Izadifar, and T. Rabczuk, “Molecular dynamics study on the crack propagation in carbon doped polycrystalline boron-nitride nanosheets,” Computational Materials Science. 2022. link Times cited: 5 NOT USED (low confidence) B. Brito, L. Cândido, J. Rabelo, and G. Hai, “Path-integral Monte Carlo simulations on the thermodynamic properties of single-layer hexagonal boron nitride,” Computational Condensed Matter. 2022. link Times cited: 2 NOT USED (low confidence) E. Mareev and F. Potemkin, “Dynamics of Ultrafast Phase Transitions in (001) Si on the Shock-Wave Front,” International Journal of Molecular Sciences. 2022. link Times cited: 3 Abstract: We demonstrate an ultrafast (<0.1 ps) reversible phase trans… read moreAbstract: We demonstrate an ultrafast (<0.1 ps) reversible phase transition in silicon (Si) under ultrafast pressure loading using molecular dynamics. Si changes its structure from cubic diamond to β-Sn on the shock-wave front. The phase transition occurs when the shock-wave pressure exceeds 11 GPa. Atomic volume, centrosymmetry, and the X-ray-diffraction spectrum were revealed as effective indicators of phase-transition dynamics. The latter, being registered in actual experimental conditions, constitutes a breakthrough in the path towards simple X-ray optical cross-correlation and pump-probe experiments. read less NOT USED (low confidence) J. Yan, M. Xiong, L. Tong, H. Ding, and Z. Lei, “Spontaneous Arched Graphene Under Uniaxial Compression and Bistable Interswitch Behaviors of Single-Layer Graphene,” Journal of Vibration Engineering & Technologies. 2022. link Times cited: 2 NOT USED (low confidence) A. James, C. John, A. Melekamburath, M. Rajeevan, and R. Swathi, “A journey toward the heaven of chemical fidelity of intermolecular force fields,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2022. link Times cited: 1 Abstract: Alongside the evolution of density functional theory into a … read moreAbstract: Alongside the evolution of density functional theory into a new era led by the dispersion‐corrected hybrid density functional theory approaches, formulation of a new generation of intermolecular potentials has also taken the center stage. An ideal potential formulation should desirably possess simplicity of functional forms, physically meaningful parameters, separability of various terms into atomic‐level contributions, computational tractability, ability to capture non‐additivity of interactions, transferability across different chemical species, and crucially, chemical fidelity in terms of reproducing the benchmark data. The Lennard‐Jones potential, one of the popular intermolecular pair potentials for performing large‐scale simulations fails to capture the intricate features of molecular interactions. Woven around the central theme of anisotropy in the nature of intermolecular interactions, herein, we describe various landmark contributions in the quest for chemical fidelity of empirical potential formulations that include (i) incorporation of the anisotropic nature of exchange‐repulsion and dispersion contributions, (ii) multipolar description of the dispersion terms, (iii) damping functions to provide an accurate description of the asymptotes, and (iv) transferability of intermolecular interaction terms. We illustrate the nuances of intermolecular force field development in the context of modeling the non‐covalent interactions governing the (i) binding of atoms and molecules with carbon nanostructures, (ii) molecular aggregates of polycyclic aromatic hydrocarbons, and (iii) interlayer interactions in layered nanostructures. We exemplify the hierarchy of empirical potentials by depicting them on the various rungs of the Jacob's ladder equivalent of density functional theory for the intermolecular force fields. Finally, we discuss some possible futuristic directions in intermolecular force field development. read less NOT USED (low confidence) S. Ababtin et al., “Single-wall carbon nanotube mechanical behavior using the modified embedded atom method with bond order (MEAM-BO),” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 1 Abstract: We report the capability of the modified embedded atom metho… read moreAbstract: We report the capability of the modified embedded atom method with bond order (MEAM-BO) potential to capture single-wall carbon nanotube (SWCNT) mechanical properties accurately by calculating the wavenumber of the radial breathing mode (RBM), elastic properties, and folding energy (ΔE), which are all associated with bond curvature. We find that the existing MEAM-BO potential gives results that correlate well with experimental and density functional theory (DFT) results with the exception of the folding energy. The MEAM-BO potential parameters are updated to produce a MEAM-BO* potential for the SWCNT system. Interestingly, including the SWCNT data base, improved the previous fit of the C-H binary system. Previous studies in the literature concluded that the CNTs diameter, d, was inversely proportional to the RBM wavenumber and folding energy. When comparing MEAM-BO*, MEAM-BO, REBO, and ReaxFF with DFT results, we found that MEAM-BO* gave the closest results to DFT for the RBM wavenumbers, folding energy, and SWCNT Young’s modulus, especially for small diameter SWCNTs. We conclude that MEAM-BO* captures SWCNT curvature effects and unsaturated hydrocarbons bond behavior. read less NOT USED (low confidence) S. Manna et al., “Learning in continuous action space for developing high dimensional potential energy models,” Nature Communications. 2022. link Times cited: 16 NOT USED (low confidence) B. Sharma and A. G. Rajan, “How Grain Boundaries and Interfacial Electrostatic Interactions Modulate Water Desalination via Nanoporous Hexagonal Boron Nitride.,” The journal of physical chemistry. B. 2022. link Times cited: 5 Abstract: To fulfill the increasing demand for drinking water, researc… read moreAbstract: To fulfill the increasing demand for drinking water, researchers are currently exploring nanoporous two-dimensional materials, such as hexagonal boron nitride (hBN), as potential desalination membranes. A prominent, yet unsolved challenge is to understand how such membranes will perform in the presence of defects or surface charge in the membrane material. In this work, we study the effect of grain boundaries (GBs) and interfacial electrostatic interactions on the desalination performance of bicrystalline nanoporous hBN using classical molecular dynamics simulations supported by quantum-mechanical density functional theory (DFT) calculations. We investigate three different nanoporous bicrystalline hBN configurations, with symmetric tilt GBs having misorientation angles of 13.2, 21.8, and 32.2°. Using lattice dynamics calculations, we find that grain boundaries alter the areas and shapes of nanopores in bicrystalline hBN, as compared to the nanopores in monocrystalline hBN. We observe that, although bicrystalline nanoporous hBN with a misorientation angle of 13.2° shows an improved water flow rate by ∼30%, it demonstrates reduced Na+ ion rejection by ∼6%, as compared to monocrystalline hBN. We also uncover the role of the nanopore shape in water desalination, finding that more elongated pores with smaller sizes (in 21.8- and 32.2°-misoriented bicrystalline hBN) can match water permeation through less elongated pores of slightly larger sizes, with a concomitant ∼3-4% decrease in Na+ rejection. Simulations also predict that the water flow rate is significantly affected by interfacial electrostatic interactions. Indeed, the water flow rate is the highest when altered partial charges on B and N atoms were determined using DFT calculations, as compared to when no partial charges or bulk partial charges (i.e., charged hBN) were considered. Overall, our work on water/ion transport through nanopores in bicrystalline hBN indicates that the presence of GBs and surface charge can lead, respectively, to a decrease in the ion rejection and water permeation performance of hBN membranes. read less NOT USED (low confidence) J. F. Wang, J. P. Yang, L.-ho Tam, and W. Zhang, “Effect of CNT volume fractions on nonlinear vibrations of PMMA/CNT composite plates: A multiscale simulation,” Thin-Walled Structures. 2022. link Times cited: 20 NOT USED (low confidence) J. Wang et al., “A deep learning interatomic potential developed for atomistic simulation of carbon materials,” Carbon. 2022. link Times cited: 18 NOT USED (low confidence) C. Hou et al., “Atomistic Simulation toward Real-scale Microprocessor Circuits,” Chemical Physics Letters. 2022. link Times cited: 1 NOT USED (low confidence) L. Chen, Z. Li, S.-S. Xu, and A. Sha, “Interaction Between Edge Dislocation and Single-Layered Graphene in Aluminum Matrix Investigated by Molecular Dynamics Simulation,” Journal of Physics: Conference Series. 2022. link Times cited: 0 Abstract: The influence of graphene on dislocation movement and subseq… read moreAbstract: The influence of graphene on dislocation movement and subsequent mechanical response of aluminum is investigated by the computational method of molecular dynamics simulation. A Lennard–Jones potential describing Al-C interaction was obtained through ab initio calculation. It was observed that the 2D graphene could reinforce Al matrix similar to the traditional Orowan mechanism. The Al/graphene interface first attract the gliding dislocation to reduce the system energy, which is unlike the grain boundary to repel gliding dislocations through pile-up mechanism. With the increase of stress, dislocation attracted and trapped at the front of graphene could glide along the interface and finally bypass it through climbing when graphene is orientated out of the shear plane. In addition, the strengthening ability of graphene is size dependent, showing a linear relationship between strength increment and graphene size. read less NOT USED (low confidence) Y. Zhao et al., “Glide Mobility of a-Type Edge Dislocations in Aluminum Nitride by Molecular Dynamics Simulation,” ACS Omega. 2021. link Times cited: 0 Abstract: Classical molecular dynamics simulations are performed to in… read moreAbstract: Classical molecular dynamics simulations are performed to investigate the motion of a-type edge dislocations in wurtzite aluminum nitride (AlN). The nucleation and propagation of kinks are observed via the dislocation extraction algorithm. Our simulation results show that the nucleation energy of the kink pair in AlN is 1.2 eV and that the migration energy is 2.8 eV. The Peierls stress of the 1/3⟨112̅0⟩{101̅0} edge dislocation at 0 K is 15.9 GPa. The viscous motion of dislocations occurs when τ > τp, and the dislocation velocity is inversely proportional to the temperature and directly proportional to the applied stress. Below room temperature, the value of the critical resolved shear stress (CRSS) on the prismatic plane is the lowest, which suggests that the dislocation mobility on the prismatic plane is the easiest. The CRSS on the pyramidal plane is always the highest at all temperatures, which suggests that pyramidal slip is the hardest among these three slip systems. read less NOT USED (low confidence) S. Ajori, A. Ameri, and R. Ansari, “High velocity impact analysis of free-free carbon nanotubes.,” Journal of molecular graphics & modelling. 2021. link Times cited: 0 NOT USED (low confidence) Q. Wang, N. Gui, X. Huang, X. Yang, J. Tu, and S. Jiang, “The effect of temperature and cascade collision on thermal conductivity of 3C-SiC: A molecular dynamics study,” International Journal of Heat and Mass Transfer. 2021. link Times cited: 10 NOT USED (low confidence) J. Wang, S. Wu, H. Xie, and L. Xiong, “Theoretical study on thermal properties of molybdenum disulfide/silicon heterostructures,” Computational Materials Science. 2021. link Times cited: 0 NOT USED (low confidence) F. Hasheminia, Y. Bahari, A. Rajabpour, and S. Arabha, “Elucidation of thermo-mechanical properties of silicon nanowires from a molecular dynamics perspective,” Computational Materials Science. 2021. link Times cited: 8 NOT USED (low confidence) S. Wyant, A. Rohskopf, and A. Henry, “Machine learned interatomic potentials for modeling interfacial heat transport in Ge/GaAs,” Computational Materials Science. 2021. link Times cited: 4 NOT USED (low confidence) M. Eghbalian, R. Ansari, and S. Haghighi, “Molecular dynamics study of mechanical properties and fracture behavior of carbon and silicon carbide nanotubes under chemical adsorption of atoms,” Diamond and Related Materials. 2021. link Times cited: 10 NOT USED (low confidence) A. Yarahmadi, M. Hashemian, D. Toghraie, R. Abedinzadeh, and S. A. Eftekhari, “Investigation of mechanical properties of epoxy-containing Detda and Degba and graphene oxide nanosheet using molecular dynamics simulation,” Journal of Molecular Liquids. 2021. link Times cited: 9 NOT USED (low confidence) S. B. O., A.-S. Smith, and P. Steinmann, “Configurational Forces in Bond Order Potentials,” PAMM. 2021. link Times cited: 0 Abstract: In this contribution, a configurational mechanics framework … read moreAbstract: In this contribution, a configurational mechanics framework is elaborated to assess the applicability of atomistic configurational forces in fracture of crystalline lattices. To this end, an analytical interatomic potential is reformulated in terms of the material positions occupied by the atoms participating in two‐ and three‐body interactions. It is demonstrated that such a potential satisfies the requirements of invariances i.e., translational, rotational and parity. The focus of this work is developing the configurational setting for the bond order Tersoff potential. Two‐dimensional pre‐cracked mono‐layer graphene modelled with the Tersoff potential is chosen to study the configurational force approach in determining energy release during crack propagation into the lattice. read less NOT USED (low confidence) J. F. Wang, S. Shi, Y. Z. Liu, J. P. Yang, and L.-ho Tam, “Multiscale simulation of temperature- and pressure-dependent nonlinear dynamics of PMMA/CNT composite plates,” Nonlinear Dynamics. 2021. link Times cited: 6 NOT USED (low confidence) D. Gara, S. potnuru, G. Raghavendra, and P. S. Prasad, “Determination of third order elastic constants from Morse potential for a Nitinol shape memory alloy,” Proceedings of the Institution of Mechanical Engineers, Part L: Journal of Materials: Design and Applications. 2021. link Times cited: 0 Abstract: The current paper determines the third order elastic constan… read moreAbstract: The current paper determines the third order elastic constants of the Ni-Ti based shape memory alloy from Morse potential using second nearest neighbour embedded atomic model (2NNEAM) in LAMMPS Molecular Dynamics (MD) simulation package. The complexities involved in the current work are lack of experimental data, such as amount of stress induced for the first order martensitic phase transition and plane about which the phase transformation takes place, which in our current work is considered as the amount of vibrations that the inherent anharmonicity possessed by the Shape memory alloy (SMA) for the first order phase transition. It is further attempted to justify the third elastic constants that are evaluated using VRH approximation, but however VRH approximation lacked stability and hence it is concluded that Morse potential is suitable. read less NOT USED (low confidence) M. Eghbalian, R. Ansari, and S. Haghighi, “On the mechanical properties and fracture analysis of polymer nanocomposites reinforced by functionalized silicon carbide nanotubes: A molecular dynamics investigation.,” Journal of molecular graphics & modelling. 2021. link Times cited: 8 NOT USED (low confidence) S. Zhang et al., “Atomistic observations on the structure evolution of glass-ceramics induced by the cascade collisions,” Applied Surface Science. 2021. link Times cited: 1 NOT USED (low confidence) Y. Wang, K. Niu, and Y. Wu, “Multiscale modelling of graphene sheet and its application in laminated composites,” Composite Structures. 2021. link Times cited: 8 NOT USED (low confidence) P. Gao et al., “lMFF: Efficient and Scalable layered Materials Force Field on Heterogeneous Many-Core Processors,” SC21: International Conference for High Performance Computing, Networking, Storage and Analysis. 2021. link Times cited: 7 Abstract: LAMMPS is one of the most popular Molecular Dynamic (MD) pac… read moreAbstract: LAMMPS is one of the most popular Molecular Dynamic (MD) packages and is widely used in the field of physics, chemistry and materials simulation. Layered Materials Force Field (LMFF) is our expansion of the LAMMPS potential function based on the Tersoff potential and inter-layer potential (ILP) in LAMMPS. LMFF is designed to study layered materials such as graphene and boron hexanitride. It is universal and does not depend on any platform. We have also carried out a series of optimizations on LMFF and the optimization work is carried out on the new generation of Sunway supercomputer, called SWLMFF. Experiments show that our implementation is efficient, scalable and portable. When generic LMFF is ported to Intel Xeon Gold 6278C, $2\times$ performance improvement is achieved. For the optimized SWLMFF, the overall performance improvement is nearly $200-330\times$ compared to the original ILP and Tersoff potentials. And SWLMFF has good parallel efficiency of 95%-100% under weak scaling with 2.7 million atoms on a single process. The maximum atomic system simulated by SWLMFF is close to $2^{31}$ atoms. And nanosecond simulations in one day can be realized. read less NOT USED (low confidence) M. Zojaji, A. Hydarinasab, S. Hashemabadi, and M. Mehranpour, “Rheological study of the effects of size/shape of graphene oxide and SiO2 nanoparticles on shear thickening behaviour of polyethylene glycol 400-based fluid: molecular dynamics simulation,” Molecular Simulation. 2021. link Times cited: 3 Abstract: ABSTRACT In this computational study, the effect of the shap… read moreAbstract: ABSTRACT In this computational study, the effect of the shape and size of graphene oxide (GO) and SiO2 nanoparticles on the shear thickening Behaviour of the fluids was reported with the molecular dynamics (MD) approach. For this purpose, the viscosity of fluids with C, Si, O, and H atomic arrangements was determined by Tersoff and Lenard-Jones (LJ) interatomic force fields. Atomic stability of the simulated structures was detected after 1.000.000 time-steps, demonstrating the validity of the PEG-400-based STF. Additionally, MD simulation results indicated that addition of zigzag GO and cubic SiO2 nanoparticles to the pristine fluid would maximise the viscosity of this atomic structure. Numerically, by adding these nanostructures, the viscosity of the simulated fluid was converged to 88 Pa. s and 94 Pa. s, respectively. The jamming viscosity (discontinuous shear-thickening) changes occurred in 70 and 80 s−1 shear rates by adding GO and SiO2 nanoparticles to the pristine fluid. read less NOT USED (low confidence) S. Zhang et al., “Insight into the roles of the glassy phase in glass-ceramics during the cascade collisions,” Computational Materials Science. 2021. link Times cited: 0 NOT USED (low confidence) S. Zhang, X. Guo, C. Zhang, Z. Jin, R. Kang, and D. Guo, “Modeling the heterogeneity response induced by the cascade collisions of glass-ceramics,” Computational Materials Science. 2021. link Times cited: 0 NOT USED (low confidence) Z. E. Oufir, H. Ramézani, N. Mathieu, and S. Delpeux, “Impact of adsorbent carbons and carbon surface conductivity on adsorption capacity of CO2, CH4, N2 and gas separation,” Computational Materials Science. 2021. link Times cited: 8 NOT USED (low confidence) R. A. Moghadam et al., “Water molecules adsorption by a porous carbon matrix in the presence of NaCl impurities using molecular dynamic simulation,” Journal of Molecular Liquids. 2021. link Times cited: 2 NOT USED (low confidence) C. Ji, X. Cai, Z. Zhou, F. Dong, S. Liu, and B. Gao, “Effects of intermetallic compound layer thickness on the mechanical properties of silicon-copper interface,” Materials & Design. 2021. link Times cited: 6 NOT USED (low confidence) A. Lahti, R. Östermark, and K. Kokko, “Optimization of SiO2 with GHA and basin hopping,” Computational Materials Science. 2021. link Times cited: 1 NOT USED (low confidence) F. Molaei, “Molecular dynamics simulation of edge crack propagation in single crystalline alpha quartz.,” Journal of molecular graphics & modelling. 2021. link Times cited: 6 NOT USED (low confidence) H. Dai, W. Wu, and Y. Hu, “Lubricating effect of graphene during ultra-precision mechanical polishing by atomic scale simulation,” Proceedings of the Institution of Mechanical Engineers, Part B: Journal of Engineering Manufacture. 2021. link Times cited: 1 Abstract: As a new two-dimensional material with unique friction and w… read moreAbstract: As a new two-dimensional material with unique friction and wear properties, graphene often serves as a solid lubricant. In order to better understand the lubrication effect of graphene in the process of three-body polishing of single crystal silicon with diamond abrasive, a molecular dynamics model of this process was established in this study. Further, the changes of coordination number, friction coefficient, temperature, potential energy, stress, and surface/subsurface damage in the process of three-body polishing were analyzed in detail. The results showed that graphene lubrication could enhance the heat dissipation and reduce the number of defect atoms, friction coefficient, potential energy, stress, and chips. Therefore, less subsurface damage and material resistance were observed in the workpiece with graphene lubrication during machining. In general, graphene can be used as a high-quality solid lubricant in the three-body polishing of single crystal silicon using diamond abrasive because of its excellent lubricating effect. read less NOT USED (low confidence) A. Priyadarsini and B. Mallik, “Amphiphilicity of Intricate Layered Graphene/g-C3N4 Nanosheets.,” The journal of physical chemistry. B. 2021. link Times cited: 5 Abstract: The hybrid heterostructure of the tri-s-triazine form of gra… read moreAbstract: The hybrid heterostructure of the tri-s-triazine form of graphitic carbon nitride (g-C3N4), a stable two-dimensional material, results from intricate layer formation with graphene. In this material, g-C3N4, an amphiphilic material, stabilizes Pickering emulsions as an emulsifier and can effectively disperse graphene. Due to the various technological applications of the hybrid nanosheets in an aqueous environment, it is essential to study the interaction of water molecules with graphene and g-C3N4 (Gr/g-C3N4)-combined heterostructure. Although few studies have been performed signifying the water orientation in the interfacial layer, we find that there is a lack of detailed studies using various dynamical and structural properties of the interfacial water molecules. The interface of the Gr/g-C3N4 hybrid structure, one of the rarely found amphiphilic interfaces (on the g-C3N3 side), is appropriate for exploring the water affinity due to the availability of heterogeneous interfacial aqueous interactions. We adopted classical molecular dynamics simulations using two models for water molecules to study the structure and dynamics of an aqueous interface. We have correlated the structural properties to dynamics and spectral properties to understand the overall behavior of the amphiphilic interface. Our results branch into two significant hydrogen bond (HB) properties in HB count and HB strength among the water molecules in the different layers. The HB counts in the different layers of water are correlated using the average distance distribution (PrO4), tetrahedral order parameters, HB donor/acceptor count, and total HBs per water molecule. A conspicuous difference is found in the HB count and related dynamics of the system. The HB lifetime and diffusion coefficient hint at the equivalent strength of HBs in the different layers. All the findings conclude that the amphiphilicity of the Gr/g-C3N4 interface can help in understanding various interfacial physical and chemical processes. read less NOT USED (low confidence) X. Yan, Y. Feng, L. Qiu, and X. Zhang, “Thermal conductivity and phase change characteristics of hierarchical porous diamond/erythritol composite phase change materials,” Energy. 2021. link Times cited: 25 NOT USED (low confidence) L. Zhao et al., “The improvement of mechanical properties of conventional concretes using carbon nanoparticles using molecular dynamics simulation,” Scientific Reports. 2021. link Times cited: 9 NOT USED (low confidence) M. Lbadaoui-Darvas, G. Garberoglio, K. Karadima, M. Cordeiro, A. Nenes, and S. Takahama, “Molecular simulations of interfacial systems: challenges, applications and future perspectives,” Molecular Simulation. 2021. link Times cited: 12 Abstract: ABSTRACT We present a comprehensive review of methods and ap… read moreAbstract: ABSTRACT We present a comprehensive review of methods and applications of molecular simulations of interfacial systems. We give a detailed overview of the main techniques and major challenges in the following aspects of solid and fluid surfaces: adsorption at solid surfaces, interfacial transport and surface-to-bulk partitioning. We summarise methods to estimate macroscopic properties interfaces (adsorption isotherms, surface tension and contact angle) and ways to extract quantitative information about fluctuating liquid surfaces. We demonstrate the usage of these methods on recent applications from the fields of atmospheric science, material science and biophysics. The two main goals of this review are: (i) to provide guidance in practical questions, such as choosing software, force field, level of theory, system geometry, and finding the appropriate selective surface analysis methods based on the type of the interface and the nature of the physical problem to be studied; and (ii) to highlight domains where molecular simulations can bring about substantial advances in our understanding in important questions of applied science as a function of future method development and adaptation for applied fields. read less NOT USED (low confidence) A. Hosseini and M. N. Nasrabadi, “Investigation of vacancy defects and temperature effects on the GaN bombarding with argon atoms: Molecular dynamics simulation,” Materials Chemistry and Physics. 2021. link Times cited: 3 NOT USED (low confidence) S. Yuan et al., “Gradient nanotwinned CrCoNi medium-entropy alloy with strength-ductility synergy,” Scripta Materialia. 2021. link Times cited: 31 NOT USED (low confidence) M. Parvaiz et al., “Modeling and simulation of carbon nanotube amino-acid sensor: A first-principles study,” Computational and Theoretical Chemistry. 2021. link Times cited: 4 NOT USED (low confidence) G. Wang, G. Zhao, J. Song, and Q. Ding, “Study on the tribological properties of copper coated by graphene and h-BN from the atomic scale,” Applied Surface Science. 2021. link Times cited: 16 NOT USED (low confidence) X. Yan et al., “Excellent heat transfer and phase transformation performance of erythritol/graphene composite phase change materials,” Composites Part B: Engineering. 2021. link Times cited: 51 NOT USED (low confidence) B. Zhang, H. Yang, H. Liu, J. Hao, and L. Xinhua, “Crystallographic features and microstructure evolution of sandwich warm laser polishing: The case of aluminum foil,” Applied Surface Science. 2021. link Times cited: 8 NOT USED (low confidence) A. V. Lun-Fu, A. Bubenchikov, M. Bubenchikov, and V. Ovchinnikov, “Molecular Dynamics Study of Collective Behavior of Carbon Nanotori in Columnar Phase,” Crystals. 2021. link Times cited: 8 Abstract: Supramolecular interaction of carbon nanotori in a columnar … read moreAbstract: Supramolecular interaction of carbon nanotori in a columnar phase is described using the methods of classical molecular dynamics. The collective behavior and dynamic properties of toroidal molecules arising under the action of the van der Waals forces are studied. The conditions under which columnar structures based on molecular tori become unstable and rearrange into another structure are investigated. The reasons for the appearance of two types of directed rotational motion from the chaotic motion of molecules are discussed. read less NOT USED (low confidence) R. Qiu, X. Yu, D. Wang, S. Zhang, D. Kang, and J. Dai, “Nanoscale Topological Morphology Transition and Controllable Thermal Conductivity of Wrinkled Hexagonal Boron Nitride: Implications for Thermal Manipulation and Management,” ACS Applied Nano Materials. 2021. link Times cited: 1 NOT USED (low confidence) J. Li, Y. Huang, B. Zeng, C. Feng, and F. Zhu, “Mechanical Response of BNNS-reinforced Aluminum Composites under Uniaxial Compression,” 2021 22nd International Conference on Electronic Packaging Technology (ICEPT). 2021. link Times cited: 0 Abstract: Boron nitride nanosheet (BNNS) has been widely used as the r… read moreAbstract: Boron nitride nanosheet (BNNS) has been widely used as the reinforcing material in metal matrix composites because of excellent thermal, electrical and mechanical properties. Chirality directions of boron nitride nanosheet include armchair and zigzag. In this paper, the influence of temperature on the mechanical reponses of aluminum (Al) and BNNS/Al composite under uniaxial compression loads haa been investigated by molecular dynamics (MD) simulations. MD simulation results show that there is a negative relationship between temperature and ultimate stress of the above material, and ultimate stress decreases as the temperatue increases. The relationship between critical strain and temperature is the same response. In addition, BNNS plays a significant role in improving ultimate stress and Young's modulus of the nanocomposite, despite its small volume fraction. However, compression loads along different chiral directions can also result in some diversities of mechanical response. For different temperature conditions, the ultimate stress of zigzag BNNS/Al composite is bigger than others in general, and armchair BNNS/Al composite shows the better temperature stability. read less NOT USED (low confidence) B. Yao, Z. Liu, and R. Zhang, “EAPOTs: An integrated empirical interatomic potential optimization platform for single elemental solids,” Computational Materials Science. 2021. link Times cited: 3 NOT USED (low confidence) F. Afsharirad, S. Mousanezhad, H. Biglari, and O. Rahmani, “Molecular dynamics of axial interwall van der Waals force and mechanical vibration of double-walled carbon nanotubes,” Materials today communications. 2021. link Times cited: 3 NOT USED (low confidence) A. Thompson et al., “LAMMPS - A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales,” Computer Physics Communications. 2021. link Times cited: 2377 NOT USED (low confidence) X. Qian and R. Yang, “Machine learning for predicting thermal transport properties of solids,” Materials Science and Engineering: R: Reports. 2021. link Times cited: 34 NOT USED (low confidence) Y. Singh, P. K. Patra, K. O. Obodo, and D. P. Rai, “Electronic and mechanical properties of (6,1) single-walled carbon nanotubes with different tube diameters: a theoretical study,” Carbon Letters. 2021. link Times cited: 5 NOT USED (low confidence) Z. Lu, N. P. Smith, M. Prange, R. A. Bunker, J. Orrell, and A. Chaka, “Effect of interfacial structures on phonon transport across atomically precise Si/Al heterojunctions,” Physical Review Materials. 2021. link Times cited: 3 Abstract: Phonons are important carriers of energy and information in … read moreAbstract: Phonons are important carriers of energy and information in many cryogenic devices used for quantum information science and in fundamental physics experiments such as dark matter detectors. In these systems phonon behaviors can be dominated by interfaces and their atomic structures; hence, there is increasing demand for a more detailed understanding of interfacial phonon transport in relevant material systems. Previous studies have focused on understanding thermal transport over the entire phonon spectrum at and above room temperature. At ultralow temperatures, however, knowledge is missing regarding athermal phonon behavior due to the challenge in modeling the extreme conditions in microscale, heterogeneous cryogenic systems, as well as extracting single-phonon information from a large ensemble. In this paper, we delineate the effects of interfacial atomic structures on phonon transport using a combination of classical molecular dynamics (MD) and phonon wave-packet simulations, to illustrate the consistency and differences between the ensemble- and single-phonon dynamics. We consider three single-crystal Si surface reconstructions---$(1\ifmmode\times\else\texttimes\fi{}1), (\sqrt{3}\ifmmode\times\else\texttimes\fi{}\sqrt{3})$ and $(7\ifmmode\times\else\texttimes\fi{}7)$---and model both experimentally observed $\mathrm{Si}(1\ifmmode\times\else\texttimes\fi{}1)/\mathrm{Al}$ interfaces and hypothesized $\mathrm{Si}(\sqrt{3}\ifmmode\times\else\texttimes\fi{}\sqrt{3})$/Al and $\mathrm{Si}(7\ifmmode\times\else\texttimes\fi{}7)/\mathrm{Al}$ interfaces. The overall interfacial thermal conductance calculated from non-equilibrium MD shows that for the $\mathrm{Si}(1\ifmmode\times\else\texttimes\fi{}1)/\mathrm{Al}$ system, the presence of Al twin boundaries can hinder phonon transport and reduce thermal conductance by 2--12% relative to single-crystal Al; whereas the Si $(\sqrt{3}\ifmmode\times\else\texttimes\fi{}\sqrt{3})$ and $(7\ifmmode\times\else\texttimes\fi{}7)$ reconstructions can enhance it by 6--19%. Normal mode decomposition reveals that both the increase and decrease in conductance are related to inelastic phonon scattering. Single-phonon wave-packet simulations predict phonon transport properties consistent with non-equilibrium MD, while further suggesting that phonon polarization conversion is significant even when elastic transmission dominates, and that the interfacial structures have anisotropic impacts on atomic vibrations along different lattice directions. Our findings suggest avenues for achieving selective phonon transport via controlling interfacial structures of materials using atomically precise fabrication techniques, and that the phonon wave-packet formalism is a potentially powerful method for developing a detailed understanding of non-equilibrium phenomena in the low-temperature limit. read less NOT USED (low confidence) J. Wang, Q. Hou, F. Zeng, and G.-J. Guo, “Gas generation mechanisms of bituminous coal under shear stress based on ReaxFF molecular dynamics simulation,” Fuel. 2021. link Times cited: 12 NOT USED (low confidence) F. Liu et al., “Strain effects on the interfacial thermal conductance of graphene/h-BN heterostructure,” Nano Materials Science. 2021. link Times cited: 3 NOT USED (low confidence) L. Nan, N. Ding, T. Jiang, L. Liu, and F. Zaïri, “Mechanical properties of nanocracks in hybrid graphene/hexagonal boron nitride sheets,” Journal of Mechanics of Materials and Structures. 2021. link Times cited: 0 NOT USED (low confidence) L. Pereira, “Investigating mechanical properties and thermal conductivity of 2D carbon-based materials by computational experiments,” Computational Materials Science. 2021. link Times cited: 9 NOT USED (low confidence) C.-C. Chiang, J. Breslin, S. Weeks, and Z. Meng, “Dynamic Mechanical Behaviors of Nacre-Inspired Graphene-Polymer Nanocomposites Depending on Internal Nanostructures.,” Extreme Mechanics Letters. 2021. link Times cited: 2 NOT USED (low confidence) S. S. M. Khadem et al., “MEL zeolite nanosheet membranes for water purification: insights from molecular dynamics simulations,” Journal of Nanostructure in Chemistry. 2021. link Times cited: 9 NOT USED (low confidence) C. Huang, X. Peng, and B. Yang, “Effect of heterointerface on the indentation behavior of nano-laminated c-BN/diamond composites,” Ceramics International. 2021. link Times cited: 6 NOT USED (low confidence) N. Goga et al., “A Review of Recent Developments in Molecular Dynamics Simulations of the Photoelectrochemical Water Splitting Process,” Catalysts. 2021. link Times cited: 8 Abstract: In this review, we provide a short overview of the Molecular… read moreAbstract: In this review, we provide a short overview of the Molecular Dynamics (MD) method and how it can be used to model the water splitting process in photoelectrochemical hydrogen production. We cover classical non-reactive and reactive MD techniques as well as multiscale extensions combining classical MD with quantum chemical and continuum methods. Selected examples of MD investigations of various aqueous semiconductor interfaces with a special focus on TiO2 are discussed. Finally, we identify gaps in the current state-of-the-art where further developments will be needed for better utilization of MD techniques in the field of water splitting. read less NOT USED (low confidence) M. Naderi and A. Karimipour, “Two-phase solid/liquid mixture of water/carbon nanotubes at the equilibration phase of atomic structures: Atomic value effects in a microchannel using the molecular dynamics method,” Journal of Molecular Liquids. 2021. link Times cited: 6 NOT USED (low confidence) P. Steinmann, A. Smith, E. Birang, A. McBride, and A. Javili, “Atomistic two-, three- and four-body potentials. Spatial and material settings,” Journal of The Mechanics and Physics of Solids. 2021. link Times cited: 5 NOT USED (low confidence) R. Sabetvand and H. Jami, “The study of boron-nitride nanotube behavior as an atomic nano-pump for biomedicine applications,” Journal of Molecular Modeling. 2021. link Times cited: 4 NOT USED (low confidence) Y. Karaaslan, J. Haskins, H. Yapicioglu, and C. Sevik, “Influence of randomly distributed vacancy defects on thermal transport in two-dimensional group-III nitrides,” Journal of Applied Physics. 2021. link Times cited: 3 Abstract: Efficient thermal transport control is a fundamental issue f… read moreAbstract: Efficient thermal transport control is a fundamental issue for electronic device applications such as information, communication, and energy storage technologies in modern electronics in order to achieve desired thermal conditions. Structural defects in materials provide a mechanism to adjust the thermal transport properties of these materials on demand. In this context, the effect of structural defects on lattice thermal conductivities of two-dimensional hexagonal binary group-III nitride (XN, X = B, Al, and Ga) semiconductors is systematically investigated by means of classical molecular dynamics simulations performed with recently developed transferable inter-atomic potentials accurately describing defect energies. Here, two different Green–Kubo based approaches and another approach based on non-equilibrium molecular dynamics are compared in order to get an overall understanding. Our investigation clearly shows that defect concentrations of 3 % decrease the thermal conductivity of systems containing these nitrites up to 95 %. Results hint that structural defects can be used as effective adjustment parameters in controlling thermal transport properties in device applications associated with these materials. read less NOT USED (low confidence) S. Fujii and A. Seko, “Structure and lattice thermal conductivity of grain boundaries in silicon by using machine learning potential and molecular dynamics,” Computational Materials Science. 2021. link Times cited: 8 NOT USED (low confidence) B. M. R. Raj, S. Singh, and K. Mali, “Multiscale based finite element modeling for the nonlinear bending and postbuckling analyses of some noncarbon nanomaterials,” International Journal of Non-linear Mechanics. 2021. link Times cited: 0 NOT USED (low confidence) J. F. Wang, J. P. Yang, L.-ho Tam, and W. Zhang, “Molecular dynamics-based multiscale nonlinear vibrations of PMMA/CNT composite plates,” Mechanical Systems and Signal Processing. 2021. link Times cited: 20 NOT USED (low confidence) K. Skrobas, K. Stefanska-Skrobas, S. Stelmakh, S. Gierlotka, and B. Palosz, “Surface free energy of diamond nanocrystals - a molecular dynamics study of its size dependence.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 4 Abstract: The dependency of the surface free energy (SFE) of diamond n… read moreAbstract: The dependency of the surface free energy (SFE) of diamond nanocrystals on particle size was studied by means of molecular dynamics (MD) and DFT simulations. It was demonstrated how to avoid the ambiguities in calculating the surface area of very small crystallites by expressing the particle size in terms of the number of atoms which we called the number of atoms convention (NAC) rather than in units of length. The NAC method was applied to a set of models terminated with either (100) or (111) crystal faces. The MD simulations were done for two widely used potentials, i.e. Tersoff and AIREBO. Both potentials show appreciable changes in surface free energy with decreasing crystal size but in opposite directions. In the limit of an infinite crystal both tested potentials give the energy of the (100) surface to be more than two times higher than that of the (111) surface. Also the absolute figures calculated from the AIREBO potential are twice larger than those from the Tersoff potential. DFT simulations of the selected small particles confirmed the MD calculations based on the AIREBO results for the (111) surface but for the (100) surface the values were considerably smaller. read less NOT USED (low confidence) B. Yang et al., “Strengthening effects of penetrating twin boundary and phase boundary in polycrystalline diamond,” Diamond and Related Materials. 2021. link Times cited: 12 NOT USED (low confidence) M. J. Tolladay, F. Scarpa, and N. Allan, “Interatomic forces breaking carbon-carbon bonds,” Carbon. 2021. link Times cited: 5 NOT USED (low confidence) S. A. Eftekhari, D. Toghraie, M. Hekmatifar, and R. Sabetvand, “Mechanical and thermal stability of armchair and zig-zag carbon sheets using classical MD simulation with Tersoff potential,” Physica E-low-dimensional Systems & Nanostructures. 2021. link Times cited: 7 NOT USED (low confidence) E. A. Bea, A. M. Viotti, M. F. Carusela, A. Monastra, and A. Soba, “Assessment, improvement, and comparison of different computational tools used for the simulation of heat transport in nanostructures,” SIMULATION. 2021. link Times cited: 1 Abstract: In this work we compare different implementations of two int… read moreAbstract: In this work we compare different implementations of two interatomic potential models, one the empirical Tersoff–Brenner and the other the semi-empirical tight-binding, to be used in the thermal transport study of silicon nanosystems. The calculations are based on molecular dynamics simulations. In the case of Tersoff–Brenner potential, two free software packages were used, while for tight-binding potential, an in-house code was developed. Both approaches require an enormous amount of computing effort, so the use of acceleration tools for adequate performance is crucial. We present a detailed study of each computational tool used: efficiency, advantages and disadvantages, and the results of application to the calculation of thermal conductance of structured silicon nanocrystals subjected to a temperature gradient. read less NOT USED (low confidence) D. K. Das, A. Mallick, and S. Singh, “Estimating thermal properties of plumbene by multiscale modeling using molecular dynamics simulation technique,” Mechanics of Advanced Materials and Structures. 2021. link Times cited: 3 Abstract: To estimate the thermal properties of plumbene under differe… read moreAbstract: To estimate the thermal properties of plumbene under different temperatures, a plumbene sheet is developed by multi-scale modeling. Plumbene sheets of variable sample sizes are also investigated to obtain a broader view of thermal properties of the material for useful application in several engineering fields. Melting point, specific heat at constant volume and pressure, heat of fusion and the coefficient of linear and surface expansion within a temperature range of 318–398 K are also determined. Present applied techniques will guide experimental approaches for designing of plumbene sheets with specific thermophysical properties for targeted applications. read less NOT USED (low confidence) K. K. Gupta, T. Mukhopadhyay, A. Roy, L. Roy, and S. Dey, “Sparse machine learning assisted deep computational insights on the mechanical properties of graphene with intrinsic defects and doping,” Journal of Physics and Chemistry of Solids. 2021. link Times cited: 23 NOT USED (low confidence) T. P. Sheerin, D. Tanner, and S. Schulz, “Atomistic analysis of piezoelectric potential fluctuations in zinc-blende InGaN/GaN quantum wells: A Stillinger-Weber potential based analysis,” Physical Review B. 2021. link Times cited: 4 Abstract: Title Atomistic analysis of piezoelectric potential fluctuat… read moreAbstract: Title Atomistic analysis of piezoelectric potential fluctuations in zinc-blende InGaN/GaN quantum wells: A Stillinger-Weber potential based analysis Author(s) Sheerin, Thomas P.; Tanner, Daniel S. P.; Schulz, Stefan Publication date 2021-04-19 Original citation Sheerin, T. P., Tanner, D. S. P. and Schulz, S. (2021) 'Atomistic analysis of piezoelectric potential fluctuations in zinc-blende InGaN/GaN quantum wells: A Stillinger-Weber potential based analysis', Physical Review B, 103(16), 165201 (13pp). doi: 10.1103/PhysRevB.103.165201 Type of publication Article (peer-reviewed) Link to publisher's version http://dx.doi.org/10.1103/PhysRevB.103.165201 Access to the full text of the published version may require a subscription. Rights © 2021, American Physical Society. All rights reserved. Item downloaded from http://hdl.handle.net/10468/11250 read less NOT USED (low confidence) A. Allouch et al., “Statistical abundance and stability of carbon nanostructures by combined condensation-annealing molecular dynamics simulations,” Computational and Theoretical Chemistry. 2021. link Times cited: 3 NOT USED (low confidence) Y. Liu et al., “Large-scale generation and characterization of amorphous boron nitride and its mechanical properties in atomistic simulations,” Journal of Non-crystalline Solids. 2021. link Times cited: 6 NOT USED (low confidence) D. Ni, W. Wu, Y. Guo, S. Gong, and Q. Wang, “Identifying key parameters for predicting materials with low defect generation efficiency by machine learning,” Computational Materials Science. 2021. link Times cited: 2 NOT USED (low confidence) S. S. M. Khadem, A. H. Mashhadzadeh, S. Habibzadeh, M. Munir, E. Lima, and M. Saeb, “A theoretical probe into the effects of material and operational variables on water purification with zeolite membranes,” Microporous and Mesoporous Materials. 2021. link Times cited: 12 NOT USED (low confidence) S. Singh, “Comparing different multibody reactive potentials for the elastic properties and nonlinear mechanics of the carbon nanostructures,” Mechanics of Materials. 2021. link Times cited: 0 NOT USED (low confidence) S. Chen, B. Shen, Z. Huang, Z. Ji, Q. Lin, and Z. Zhang, “Controlled friction on graphene via substrate deformation induced atomic pinning effect,” Computational Materials Science. 2021. link Times cited: 3 NOT USED (low confidence) W. Sekkal and A. Zaoui, “Novel properties of nano-engineered cementitious materials with fullerene buckyballs,” Cement & Concrete Composites. 2021. link Times cited: 3 NOT USED (low confidence) S. Singh, B. M. R. Raj, K. Mali, and G. Watts, “Elastic Properties and Nonlinear Elasticity of the Noncarbon Hexagonal Lattice Nanomaterials Based on the Multiscale Modeling,” Journal of Engineering Materials and Technology-transactions of The Asme. 2021. link Times cited: 3 Abstract:
This study presents the elastic properties and nonlinear e… read moreAbstract:
This study presents the elastic properties and nonlinear elasticity of the two-dimensional noncarbon nanomaterials of hexagonal lattice structures having molecular structure XY. Four nitride-based and two phosphide-based two-dimensional nanomaterials, having graphene-like hexagonal lattice structure, are considered in the present study. The four empirical parameters associated with the attractive and repulsive terms of the Tersoff–Brenner potential are calibrated for noncarbon nanomaterials and tested for elastic properties, nonlinear constitutive behavior, bending modulus, bending and torsional energy. The mathematical identities for the tangent constitutive matrix in terms of the interatomic potential function are derived through an atomistic–continuum coupled multiscale framework of the extended version of Cauchy–Born rule. The results obtained using newly calibrated empirical parameters for cohesive energy, bond length, elastic properties, and bending rigidity are compared with those reported in the literature through experimental investigations and quantum mechanical calculations. The continuum approximation is attained through the finite element method. Multiscale evaluations for elastic properties and nonlinear stretching of the nanosheets under in-plane loads are also compared with those obtained from atomistic simulations. read less NOT USED (low confidence) J. Feng, S. Ma, K. Zhang, S. Hao, and B. Li, “Static and dynamic buckling behavior of CNTS with S-W defects,” International Journal of Modern Physics B. 2021. link Times cited: 2 Abstract: The excellent performance of carbon nanotubes (CNTs) allows … read moreAbstract: The excellent performance of carbon nanotubes (CNTs) allows them to be widely employed in various micro- and nano-electromechanical devices. However, different imperfections such as Stone–Wales (S-W) defects often arise in these structures during the preparation process. In this paper, special attention is paid to the effects of the number and location of defects as well as the diameter and chiral angle of CNTs on the static and dynamic buckling of CNTs with S-W defects. First, LAMMPS software is used to simulate the molecular dynamics (MDs) of CNTs with S-W defects, and their static buckling performances are discussed. Then based on the static buckling data, the dynamic buckling vibration performance of CNTs with S-W defects is analyzed in the context of the nonlocal elastic theory. Finally, the effective range of nonlocal parameters is established via the MD modeling. The results show that the existence of S-W defects will reduce the buckling performance and vibration characteristics of CNTs, and an increase in the number of defects will aggravate the influence of diameter and chiral angle on the buckling performance as well as the natural frequency and amplitude of the nanotube’s axial vibration. read less NOT USED (low confidence) S. Yoo, B. Lee, and K. Kang, “Density functional theory study of the mechanical behavior of silicene and development of a Tersoff interatomic potential model tailored for elastic behavior,” Nanotechnology. 2021. link Times cited: 8 Abstract: Silicene, a graphene-like 2D material made from Si atoms, ha… read moreAbstract: Silicene, a graphene-like 2D material made from Si atoms, has been fabricated and studied for its promising applications in micro/nanoelectronics. For the reliable function of silicene devices, it is important to investigate silicene’s mechanical properties. In this study, the authors conducted density functional theory (DFT) simulations of mechanical tests of silicene and investigated the elastic modulus and mechanical response such as structural transformation. In addition, the authors optimized the Tersoff potential parameters using a gradient-based minimization with a grid search method in hyperdimensional parameter space, to match the DFT calculation results in the elastic regime. With the new parameter set, the elastic moduli of silicene in the zigzag (ZZ) and armchair (AC) directions were computed with molecular statics (MS) simulations and compared with those of other Si interatomic potential models and DFT results. In addition, uniaxial tensile tests along the ZZ and AC directions were performed to examine how far the Tersoff model is transferable with our new parameter set to describe the nonlinear mechanical behavior of silicene. The results of uniaxial tensile tests suggest that the angle penalty function in the Tersoff model needs to be modified and that the stress–strain curve predicted with this modification shows improvement compared to the original function. read less NOT USED (low confidence) S. Sankaranarayanan et al., “Learning in Continuous Action Space for Determination of High Dimensional Potential Energy Surfaces.” 2021. link Times cited: 0 Abstract:
Reinforcement learning (RL) approaches that combine a tree… read moreAbstract:
Reinforcement learning (RL) approaches that combine a tree search with deep learning have found remarkable success in searching exorbitantly large, albeit discrete action spaces, as demonstrated recently in board games like chess, Shogi, and Go. Many real-world materials discovery and design applications, however, involve multi-dimensional search problems and learning domains that have continuous action spaces. Exploring high-dimensional potential energy surfaces (PES) of materials to represent inter- and intra-molecular interactions, for example, involves a continuous action search to find optimal potential parameters or coefficients. Traditionally, these searches are time consuming (often several years for a single system) and have been driven by human intuition and/or expertise and more recently by global/local optimization searches that have issues with convergence and/or do not scale well with the search dimensionality. Here, in a departure from discrete action and other gradient-based approaches, we introduce a RL strategy based on decision trees that incorporates modified rewards for improved exploration, efficient sampling during playouts, and a “window scaling scheme” for enhanced exploitation, to enable efficient and scalable search for continuous action space problems. Using high-dimensional artificial landscapes and control RL problems, we successfully benchmark our approach against popular global optimization schemes and state-of-the-art policy gradient methods, respectively. We further demonstrate its efficacy to perform high-throughput PES search for 54 different elemental systems across the Periodic table, in- including alkali, alkaline-earth, transition metals, metalloids, as well as non-metals. Using a well-sampled (∼165,000 configurations) first-principles derived training and test dataset, we demonstrate that the new class of RL trained bond-order potentials capture the size-dependent energetic landscape from few atom clusters to bulk (energy errors << 200 meV/atom over a 3-6 eV sampled range) as well as their dynamics (force errors << 0.5 eV/A over a 50-100 eV/A range). We analyze the error trends across different
elements in the latent space and trace their origin to elemental structural diversity and the smoothness of the element energy surface. Finally, we run molecular dynamics using these RL trained potentials and perform a comprehensive test of dynamic stability of more than 40,000 clusters sampled for different elements across the Periodic table. Our newly developed high-quality potentials will enable accelerated nanoscale materials design and discovery. Broadly, our RL strategy will be applicable to many other physical science problems involving search over continuous action spaces. read less NOT USED (low confidence) S. I. Kundalwal, V. Choyal, V. Choyal, S. Nevhal, and N. Luhadiya, “Enhancement of piezoelectric and flexoelectric response of boron nitride sheet superlattices via interface and defect engineering,” Physica E-low-dimensional Systems & Nanostructures. 2021. link Times cited: 9 NOT USED (low confidence) H. Mes-adi, K. Saadouni, and M. Mazroui, “Effect of incident angle on the microstructure proprieties of Cu thin film deposited on Si (001) substrate,” Thin Solid Films. 2021. link Times cited: 8 NOT USED (low confidence) X. Zhang, Z. Chen, H. Chen, and L. Xu, “Comparative studies of thermal conductivity for bilayer graphene with different potential functions in molecular dynamic simulations,” Results in physics. 2021. link Times cited: 11 NOT USED (low confidence) S. Stelmakh, K. Skrobas, S. Gierlotka, and B. Palosz, “The shape and surface structure of detonation nanodiamond purified in oxidizing chemical environment,” Diamond and Related Materials. 2021. link Times cited: 3 NOT USED (low confidence) Y. Lysogorskiy et al., “Performant implementation of the atomic cluster expansion (PACE) and application to copper and silicon,” npj Computational Materials. 2021. link Times cited: 84 NOT USED (low confidence) H. Yu and A. Banerjee, “Density functional theory method for twisted geometries with application to torsional deformations in group-IV nanotubes,” J. Comput. Phys. 2021. link Times cited: 7 NOT USED (low confidence) X.-Y. Zhou, H. Fu, J.-hua Zhu, and X. Yang, “Atomistic simulations of the surface severe plastic deformation-induced grain refinement in polycrystalline magnesium: The effect of processing parameters,” Journal of Magnesium and Alloys. 2021. link Times cited: 7 NOT USED (low confidence) R. Sabetvand, D. Toghraie, and M. Hekmatifar, “The molecular dynamics study of boron-nitride nanosheet roughness after atomic bombardment process,” Journal of Molecular Liquids. 2021. link Times cited: 4 NOT USED (low confidence) X. Luo et al., “Atomistic simulation of amorphization during AlN nanoindentation,” Ceramics International. 2021. link Times cited: 3 NOT USED (low confidence) A. Priyadarsini and B. Mallik, “Effects of Doped N, B, P, and S Atoms on Graphene toward Oxygen Evolution Reactions,” ACS Omega. 2021. link Times cited: 11 Abstract: Molecular oxygen and hydrogen can be obtained from the water… read moreAbstract: Molecular oxygen and hydrogen can be obtained from the water-splitting process through the electrolysis technique. However, harnessing energy is very challenging in this way due to the involvement of the 4e– reaction pathway, which is associated with a substantial amount of reaction barrier. After the report of the first N-doped graphene acting as an oxygen reduction reaction catalyst, the scientific community set out on exploring more reliable doping materials, better material engineering techniques, and developing computational models to explain the interfacial reactions. In this study, we modeled the graphene surface with four different nonmetal doping atoms N, B, P, and S individually by replacing a carbon atom from one of the graphitic positions. We report the mechanism of the complete catalytic cycle for each of the doped surfaces by the doping atom. The energy barriers for individual steps were explored using the biased first-principles molecular dynamics simulations to overcome the high reaction barrier. We explain the active sites and provide a comparison between the activation energy obtained by the application of two computational methods. Observing the rate-determining step, that is, oxo–oxo bond formation, S-doped graphene is the most effective. In contrast, N-doped graphene seems to be the least useful for oxygen evolution catalysis compared to the undoped graphene surface. B-doped graphene and P-doped graphene have an equivalent impact on the catalytic cycle. read less NOT USED (low confidence) A. Batuer, D. Chen, X. He, and Z. Huang, “Simulation methods of cotton pyrolysis based on ReaxFF and the influence of volatile removal ratio on volatile evolution and char formation,” Chemical Engineering Journal. 2021. link Times cited: 29 NOT USED (low confidence) Z.-H. Yang, “Speed-dependent adaptive partitioning in QM/MM MD simulations of displacement damage in solid-state systems.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 1 Abstract: Solids undergo displacement damage (DD) when interacting wit… read moreAbstract: Solids undergo displacement damage (DD) when interacting with energetic particles, which may happen during the fabrication of semiconductor devices, in harsh environments and in certain analysis techniques. Simulations of DD generation are usually carried out using classical molecular dynamics (MD), but classical MD does not account for all the effects in DD, as demonstrated by ab initio calculations of model systems in the literature. A complete ab initio simulation of DD generation is impractical due to the large number of atoms involved. In my previous paper [Yang, Phys. Chem. Chem. Phys., 2020, 22, 19307], I developed an adaptive-center (AC) method for the adaptive-partitioning (AP) of quantum mechanics/molecular mechanics (QM/MM) simulations, allowing the active region centers and the QM/MM partition to be determined on-the-fly for energy-conserving AP-QM/MM methods. I demonstrated that the AC-AP-QM/MM is applicable to the simulation of DD generation, so that the active regions can be treated using an ab initio method. The AC method could not be used to identify the fast-moving recoil ions in DD generation as active region centers, however, and the accuracy is negatively affected by the rapid change in the QM/MM partition of the system. In this paper, I extend the AC method and develop a speed-dependent adaptive-center (SDAC) method for accurate AP-QM/MM simulations of DD. The SDAC method is applicable to general problems with speed-dependent active regions, and is compatible with all existing energy-conserving partitioning-by-distance AP-QM/MM methods. The artifact due to the speed-dependent potential energy surface can be made small by choosing suitable criteria. I demonstrate the SDAC method by simulations of DD generation in bulk silicon. read less NOT USED (low confidence) A. A. Zhuravlev, K. Abgaryan, and D. Reviznikov, “Multiscale Discrete Element Modeling,” Symmetry. 2021. link Times cited: 1 Abstract: A multiscale approach to discrete element modeling is presen… read moreAbstract: A multiscale approach to discrete element modeling is presented. A distinctive feature of the method is that each macroscopic discrete element has an associated atomic sample representing the material’s atomic structure. The dynamics of the elements on macro and micro levels are described by systems of ordinary differential equations, which are solved in a self-consistent manner. A full cycle of multiscale simulations is applied to polycrystalline silicon. Macroscale elastic properties of silicon were obtained only using data extracted from the quantum mechanical properties. The results of computational experiments correspond well to the reference data. read less NOT USED (low confidence) I. A. Balyakin and S. Sadovnikov, “Simulations of ZnS deposition on Ag2S surface and formation of Ag2S/ZnS heteronanostructure,” IOP Conference Series: Materials Science and Engineering. 2021. link Times cited: 1 Abstract: Simulation of ZnS deposition from aqueous solution on a surf… read moreAbstract: Simulation of ZnS deposition from aqueous solution on a surface of crystalline Ag2S has been performed to determine the features of formation of Ag2S/ZnS heteronanostructures. The classical molecular dynamics and density functional theory have been used to study the features of the initial stages of ZnS growth on Ag2S [001] surface. By classical molecular dynamics it was established that sulfur atoms initially are adsorbed on the Ag2S. Zinc atoms were shown to be adsorbed after sulfur atoms. However, the final location of the first adsorbed Zn layer was closer to the Ag2S surface than location of the first adsorbed S layer. Density functional theory calculation showed that Zn atoms were indeed closer to Ag2S surface and confirmed classical molecular dynamics results. However, density functional theory results are more precise since they made possible to account for Ag2S surface reconstruction. read less NOT USED (low confidence) S. Ajori, F. Sadeghi, and R. Ansari, “Dynamic behavior of chloride ion-electrically charged open carbon nanocone oscillators: A molecular dynamics study,” Proceedings of the Institution of Mechanical Engineers, Part C: Journal of Mechanical Engineering Science. 2021. link Times cited: 2 Abstract: This paper is intended to study the dynamic oscillatory beha… read moreAbstract: This paper is intended to study the dynamic oscillatory behavior of chloride ion inside electrically charged open carbon nanocones (CNCs) using the molecular dynamics (MD) simulations. The small and wide ends of nanocone are assumed to be identically and uniformly charged with positive electric charges. In the simulation, the Tersoff-Brenner (TB) and the Lennard-Jones (LJ) potential functions are employed to evaluate the interatomic interactions between carbon atoms and the van der Waals (vdW) interactions between the ion and the nanocone, respectively. The Coulomb potential is also adopted to evaluate the electrostatic interactions between the ion and the electric charges distributed at both ends of nanocone. Numerical results are presented to examine the effects of magnitude of electric charges, initial separation distance and initial velocity on the mechanical oscillatory behavior of system and the obtained results are also compared with the ones related to an uncharged nanocone. It is found that operating frequency as well as escape velocity enhance considerably as a result of electrostatic interactions. It is further found that regardless of the value of electric charges, optimal oscillation frequency is achievable when no initial velocity is imposed on the ion initially located inside of nanocone with an offset of 2 Å from its small end. read less NOT USED (low confidence) H. J. Kim and K. Chung, “Atomistic investigation of the effect of contact condition on frictional properties of nanowire,” Applied Surface Science. 2020. link Times cited: 4 NOT USED (low confidence) W. Shao et al., “High-Temperature Sliding Friction Behavior of Amorphous Carbon Films: Molecular Dynamics Simulation.,” Langmuir : the ACS journal of surfaces and colloids. 2020. link Times cited: 7 Abstract: With the development of the aerospace industry, the requirem… read moreAbstract: With the development of the aerospace industry, the requirement for mechanical parts, which are serviced under extreme conditions such as high temperature, is more and more severe. Amorphous carbon (a-C) films are widely used in the aviation field as a protective coating because of their excellent antiwear and friction-reduction properties. However, a-C films are vulnerable to failure in a high-temperature environment, and a series of complex changes in the friction process make it a challenge to put forward the friction mechanism. Here, the sliding friction behaviors of amorphous carbon (a-C) films at different simulated temperatures (STs) (300-1300 K) were analyzed by molecular dynamics. The density, average coordination number, and local residual stress as well as the hybridization of sp, sp2, and sp3 of a-C films were analyzed to reveal the high-temperature sliding friction mechanism of a-C films. The results show that the friction coefficient (μ) of a-C films increased with increase in ST. Meanwhile, the friction mechanisms of a-C films are different at an ST lower than 800 K and higher than 1100 K. Compared with those before sliding, the local residual stress of all a-C films is relaxed, which causes transformation of sp3 into sp2. Moreover, when ST is lower than 800 K, the μ increased with increase in sp3%. When ST is higher than 1100 K, the stability of a-C films is broken, which results in the rapid increase in μ. read less NOT USED (low confidence) P. Gao et al., “Millimeter-Scale and Billion-Atom Reactive Force Field Simulation on Sunway Taihulight,” IEEE Transactions on Parallel and Distributed Systems. 2020. link Times cited: 13 Abstract: Large-scale molecular dynamics (MD) simulations on supercomp… read moreAbstract: Large-scale molecular dynamics (MD) simulations on supercomputers play an increasingly important role in many research areas. With the capability of simulating charge equilibration (QEq), bonds and so on, Reactive force field (ReaxFF) enables the precise simulation of chemical reactions. Compared to the first principle molecular dynamics (FPMD), ReaxFF has far lower requirements on computational resources so that it can achieve higher efficiencies for large-scale simulations. In this article, we present our efforts on scaling ReaxFF on the Sunway TaihuLight Supercomputer (TaihuLight). We have carefully redesigned the force analysis and neighbor list building steps. By applying fine-grained optimizations we gain better single process performance. For the many-body interactions, we propose an isolated computation and update strategy and implement inverse trigonometric functions. For QEq, we implement a pipelined conjugate gradient (CG) approach to achieving better scalability. Furthermore, we reorganize the data layout and implement the update operation based on data locality in ReaxFF. Our experiments show that this approach can simulate chemical reactions with 1,358,954,496 atoms using 4,259,840 cores with a performance of 0.015 ns/day. To our best knowledge, this is the first realization of chemical reaction simulation with a millimeter-scale force field. read less NOT USED (low confidence) S. D. D. Nath, N. K. Peyada, and S.-G. Kim, “On the elastic modulus, and ultimate strength of Ge, Ge-Si nanowires,” Computational Materials Science. 2020. link Times cited: 2 NOT USED (low confidence) W. Sun, J. Jiang, and P. Chen, “Microstructural evolution, shocking sintering mechanism and dynamic mechanical behaviours of silica nanoparticles acting as catalyst carrier in energetic nanomaterials during shock-wave impact.” 2020. link Times cited: 1 NOT USED (low confidence) Y. Yu, W. Tang, Z. Liu, and L. Bai, “Deformation mechanisms of Si-doped diamond-like carbon films under uniaxial tension conditions,” Diamond and Related Materials. 2020. link Times cited: 4 NOT USED (low confidence) P. Novikov, K. Pavsky, and A. Baranov, “Quick search of neighbour particles in molecular dynamics simulations,” Journal of Physics: Conference Series. 2020. link Times cited: 1 Abstract: Quicksort algorithm as an assistant preliminary tool is prop… read moreAbstract: Quicksort algorithm as an assistant preliminary tool is proposed for acceleration of neighbour search procedure in the frame of molecular dynamics simulations. Simple estimations are made showing that the number of operations required to determine all the neighbours within a system of particles can be reduced by 2-3 orders of magnitude due to the preliminary sorting. Test molecular dynamics simulations carried out for virtual crystal structure containing atoms manifest 9-fold acceleration achieved by the modified algorithm. read less NOT USED (low confidence) R. Yamaletdinov and Y. V. Pershin, “Kinks in buckled graphene uncompressed and compressed in the longitudinal direction,” arXiv: Mesoscale and Nanoscale Physics. 2020. link Times cited: 1 NOT USED (low confidence) S. Stelmakh, K. Skrobas, S. Gierlotka, S. Vogel, and B. Palosz, “Atomic structure and grain shape evolution of nanodiamond during annealing in oxidizing atmosphere from neutron diffraction and MD simulations,” Diamond and Related Materials. 2020. link Times cited: 5 NOT USED (low confidence) B. Chava, Y. Wang, V. Sivasankar, and S. Das, “Water-free Localization of Anion at Anode for Small-Concentration Water-in-Salt Electrolytes Confined in Boron-Nitride Nanotube.” 2020. link Times cited: 3 NOT USED (low confidence) A. Galashev and O. Rakhmanova, “Computational study of the formation of aluminum-graphene nanocrystallites,” Physics Letters A. 2020. link Times cited: 2 NOT USED (low confidence) J. Christenson, M. Kroonblawd, R. Austin, L. Fried, and R. Phillips, “Simulating transient heat transfer in graphene at finite Knudsen number via the Boltzmann transport equation and molecular dynamics,” Physical Review B. 2020. link Times cited: 2 NOT USED (low confidence) H. Shabbir and M. Hartmann, “A high coordination of cross-links in fiber bundles prevents local strain concentrations,” Computational Materials Science. 2020. link Times cited: 0 NOT USED (low confidence) K. Talaat, M. El-Genk, and B. Cowen, “Extrapolation of thermal conductivity in non-equilibrium molecular dynamics simulations to bulk scale,” International Communications in Heat and Mass Transfer. 2020. link Times cited: 2 NOT USED (low confidence) T. Fukuya and Y. Shibuta, “Machine learning approach to automated analysis of atomic configuration of molecular dynamics simulation,” Computational Materials Science. 2020. link Times cited: 12 NOT USED (low confidence) S. Sadovnikov and I. A. Balyakin, “Molecular dynamics simulations of zinc sulfide deposition on silver sulfide from aqueous solution,” Computational Materials Science. 2020. link Times cited: 6 NOT USED (low confidence) J. Hao, S. Jin, G. Lu, and H. Xu, “Migration energy barriers and diffusion anisotropy of point defects on tungsten surfaces,” Computational Materials Science. 2020. link Times cited: 11 NOT USED (low confidence) X. Duan et al., “Cell-List based Molecular Dynamics on Many-Core Processors: A Case Study on Sunway TaihuLight Supercomputer,” SC20: International Conference for High Performance Computing, Networking, Storage and Analysis. 2020. link Times cited: 1 Abstract: Molecular dynamics (MD) simulations are playing an increasin… read moreAbstract: Molecular dynamics (MD) simulations are playing an increasingly important role in several research areas. The most frequently used potentials in MD simulations are pair-wise potentials. Due to the memory wall, computing pair-wise potentials on many-core processors are usually memory bounded. In this paper, we take the SW26010 processor as an exemplary platform to explore the possibility to break the memory bottleneck by improving data reusage via cell-list-based methods. We use cell-lists instead of neighbor-lists in the potential computation, and apply a number of novel optimization methods. Theses methods include: an adaptive replica arrangement strategy, a parameter profile data structure, and a particle-cell cutoff checking filter. An incremental cell-list building method is also realized to accelerate the construction of cell-lists. Furthermore, we have established an open source standalone framework, ESMD, featuring the techniques above. Experiments show that ESMD is 50$\sim$170% faster than previous ports on a single node, and can scale to 1,024 nodes with a weak scalibility of 95%. read less NOT USED (low confidence) D. Gobbo, P. Ballone, and B. Garabato, “Coarse-Grained Model of Entropy-Driven Demixing.,” The journal of physical chemistry. B. 2020. link Times cited: 2 Abstract: Entropy-driven demixing transitions play an important role i… read moreAbstract: Entropy-driven demixing transitions play an important role in a variety of phenomena in solution chemistry, in mixtures of ionic liquids, in polymers, and in biosystems. A simple coarse-grained model of a binary (A and B) fluid mixture of Lennard-Jones particles carrying classical harmonic oscillators whose frequency decreases with increasing homo-coordination separates into two nearly pure phases with increasing T, as the entropy gain in lowering the oscillators' frequency overcomes the potential energy and ideal entropy advantage of the homogeneous phase. We characterize features of the demixing transition and outline physical questions that can be addressed by this simple and inexpensive model. Besides and beyond these conceptual points, we provide examples of how the model could be adapted to real systems, aiming at their quantitative description by a coarse-grained model made of particles carrying momentum, energy, and entropy. read less NOT USED (low confidence) B. Yang, D. Li, H. Yang, J. Wang, and P. Yang, “Thermal conductivity enhancement of defective graphene nanoribbons,” International Communications in Heat and Mass Transfer. 2020. link Times cited: 8 NOT USED (low confidence) J. Tan, Y. Wang, and Y. Guo, “Humidity effect on peeling of monolayer graphene and hexagonal boron nitride,” Nanotechnology. 2020. link Times cited: 3 Abstract: Ambient humidity introduces water adsorption and intercalati… read moreAbstract: Ambient humidity introduces water adsorption and intercalation at the surfaces and interfaces of low-dimensional materials. Our extensive molecular dynamics (MD) simulations reveal the completely opposite contributions of interfacial water to the peeling of monolayer graphene and hexagonal boron nitride (h-BN) sheets from graphite and BN substrates. For graphene, interfacial water decreases the peeling force, due to lower adhesion at the graphene/water interface. The peeling force of h-BN increases with an increase in the thickness of interfacial water, owing to stronger adhesion at the h-BN/water interface and the detachment of the water layer from the substrates. In this work, a theoretical model considering graphene/water and water/substrate interfacial adhesion energies is established, to predict the peeling forces of graphene and h-BN, which coincides well with the peeling forces predicted by the MD simulations. Our results should provide a deeper insight into the effect of interfacial water, induced by ambient humidity, on mechanical exfoliation and the transfer of two-dimensional van der Waals crystals. read less NOT USED (low confidence) R. Tromer et al., “Electronic, optical and thermoelectric properties of boron-doped nitrogenated holey graphene.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 8 Abstract: We employ first principles calculations to investigate the e… read moreAbstract: We employ first principles calculations to investigate the electronic, optical, and thermoelectric properties of ten boron-doped nitrogenated holey graphene (NHG) monolayers. We find that most of the proposed structures remain stable during ab initio molecular dynamics simulations, in spite of their increased formation energies. Density functional theory calculations employing a hybrid functional predict band gaps ranging from 0.73 eV to 2.30 eV. In general, we find that boron doping shifts optical absorption towards the visible spectrum, and also reduces light reflection in this region. On the other hand, the magnitude of optical absorption coefficients are reduced. Regarding the thermoelectric properties, we predict that boron doping can enhance the figure of merit ZT of NHG by up to 55%. Our results indicate that boron-doped NHG monolayers may find application in solar cells and thermoelectric devices. read less NOT USED (low confidence) J. Chapman and R. Ramprasad, “Nanoscale Modeling of Surface Phenomena in Aluminum Using Machine Learning Force Fields,” The Journal of Physical Chemistry C. 2020. link Times cited: 7 Abstract: The study of nano-scale surface phenomena is essential in un… read moreAbstract: The study of nano-scale surface phenomena is essential in understanding the physical processes that aid in technologically relevant applications, such as catalysis, material growth, and failure nuc... read less NOT USED (low confidence) C. Pham, R. Lindsey, L. Fried, and N. Goldman, “Calculation of the detonation state of HN3 with quantum accuracy.,” The Journal of chemical physics. 2020. link Times cited: 14 Abstract: HN3 is a unique liquid energetic material that exhibits ultr… read moreAbstract: HN3 is a unique liquid energetic material that exhibits ultrafast detonation chemistry and a transition to metallic states during detonation. We combine the Chebyshev interaction model for efficient simulation (ChIMES) many-body reactive force field and the extended-Lagrangian multiscale shock technique molecular dynamics method to calculate the detonation properties of HN3 with the accuracy of Kohn-Sham density-functional theory. ChIMES is based on a Chebyshev polynomial expansion and can accurately reproduce density-functional theory molecular dynamics (DFT-MD) simulations for a wide range of unreactive and decomposition conditions of liquid HN3. We show that addition of random displacement configurations and the energies of gas-phase equilibrium products in the training set allows ChIMES to efficiently explore the complex potential energy surface. Schemes for selecting force field parameters and the inclusion of stress tensor and energy data in the training set are examined. Structural and dynamical properties and chemistry predictions for the resulting models are benchmarked against DFT-MD. We demonstrate that the inclusion of explicit four-body energy terms is necessary to capture the potential energy surface across a wide range of conditions. Our results generally retain the accuracy of DFT-MD while yielding a high degree of computational efficiency, allowing simulations to approach orders of magnitude larger time and spatial scales. The techniques and recipes for MD model creation we present allow for direct simulation of nanosecond shock compression experiments and calculation of the detonation properties of materials with the accuracy of Kohn-Sham density-functional theory. read less NOT USED (low confidence) Z. Lu, A. Chaka, and P. Sushko, “Thermal conductance enhanced via inelastic phonon transport by atomic vacancies at Cu/Si interfaces,” Physical Review B. 2020. link Times cited: 10 NOT USED (low confidence) Q. Zhang et al., “Designing ultrahard nanostructured diamond through internal defects and interface engineering at different length scales,” Carbon. 2020. link Times cited: 10 NOT USED (low confidence) Y. Jiao and J. Fish, “Coupled thermodynamically consistent thermo-mechanical model of silica glass subjected to hypervelocity impact,” Computer Methods in Applied Mechanics and Engineering. 2020. link Times cited: 3 NOT USED (low confidence) B. Rice, W. Mattson, J. Larentzos, and E. Byrd, “Heuristics for chemical species identification in dense systems.,” The Journal of chemical physics. 2020. link Times cited: 6 Abstract: A new approach to identify chemical species from molecular d… read moreAbstract: A new approach to identify chemical species from molecular dynamics (MD) simulations of reacting materials under extreme temperatures and pressures is presented. The approach is based on bond-distance and vibrational criteria, derived from the examination of atomic behavior during a density functional theory MD simulation of an overdriven shock of the explosive pentaerythritol tetranitrate. For comparison, the trajectory was analyzed using popular bonding criteria commonly used in analysis of reactive MD simulations, including distance, distance-time, and bond-order criteria. Cluster analyses using the new time-dependent bond definition approach presented here and a bond-order approach revealed that species and their corresponding lifetimes were strongly dependent on the chosen approach, indicating significant implications for the development of chemical mechanisms and chemical kinetics models using the results of reactive MD simulations. read less NOT USED (low confidence) K. K. Gupta, T. Mukhopadhyay, A. Roy, and S. Dey, “Probing the compound effect of spatially varying intrinsic defects and doping on mechanical properties of hybrid graphene monolayers,” Journal of Materials Science & Technology. 2020. link Times cited: 22 NOT USED (low confidence) A. Batuer, D. Chen, Q. Xin, X. He, J. Zhang, and Z. Huang, “Mechanical properties of waste cotton and their changes during early pyrolysis,” Journal of Analytical and Applied Pyrolysis. 2020. link Times cited: 1 NOT USED (low confidence) H. Ghasemi, J. Rutledge, and H. Yazdani, “Mechanical properties of defective cyanoethynyl (2D polyaniline – C3N): A comparative molecular dynamics study versus graphene and hexagonal boron nitride,” Physica E-low-dimensional Systems & Nanostructures. 2020. link Times cited: 4 NOT USED (low confidence) P. Marepalli, S. Mathur, and J. Murthy, “Automatic differentiation approach for property computations in nanoscale thermal transport,” Comput. Phys. Commun. 2020. link Times cited: 1 NOT USED (low confidence) R. B. Dehkordi, D. Toghraie, M. Hashemian, F. Aghadavoudi, and M. Akbari, “Molecular dynamics simulation of ferro-nanofluid flow in a microchannel in the presence of external electric field: Effects of Fe3O4 nanoparticles,” International Communications in Heat and Mass Transfer. 2020. link Times cited: 20 NOT USED (low confidence) Y. Zhang, C. Hu, and B. Jiang, “Accelerating atomistic simulations with piecewise machine-learned ab Initio potentials at a classical force field-like cost.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 12 Abstract: Recently, machine learning methods have become easy-to-use t… read moreAbstract: Recently, machine learning methods have become easy-to-use tools for constructing high-dimensional interatomic potentials with ab initio accuracy. Although machine-learned interatomic potentials are generally orders of magnitude faster than first-principles calculations, they remain much slower than classical force fields, at the price of using more complex structural descriptors. To bridge this efficiency gap, we propose an embedded atom neural network approach with simple piecewise switching function-based descriptors, resulting in a favorable linear scaling with the number of neighbor atoms. Numerical examples validate that this piecewise machine-learning model can be over an order of magnitude faster than various popular machine-learned potentials with comparable accuracy for both metallic and covalent materials, approaching the speed of the fastest embedded atom method (i.e. several μs per atom per CPU core). The extreme efficiency of this approach promises its potential in first-principles atomistic simulations of very large systems and/or in a long timescale. read less NOT USED (low confidence) Z.-H. Yang, “On-the-fly determination of active region centers in adaptive-partitioning QM/MM.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 4 Abstract: Quantum mechanics/molecular mechanics (QM/MM) methods are wi… read moreAbstract: Quantum mechanics/molecular mechanics (QM/MM) methods are widely used in molecular dynamics (MD) simulations of large systems. By partitioning the system into active and environmental regions and treating them with different levels of theory, QM/MM methods achieve accuracy and efficiency at the same time. Adaptive-partitioning (AP) QM/MM allows the partition of the system to change during the MD simulation, making it possible to simulate processes in which the active and environmental regions exchange atoms or molecules, such as processes in solutions or solids. AP-QM/MM methods usually partition the system according to distances to centers of active regions. For energy-conserving AP-QM/MM methods, these centers are chosen beforehand and remain fixed during the MD simulation, making it difficult to simulate processes in which active regions may occur or vanish. In this paper, I develop an adaptive-center (AC) method that allows on-the-fly determination of the centers of active regions according to any geometrical criterion or any criterion dependent on the potential energy. The AC method is compatible with all existing energy-conserving AP-QM/MM methods, and the resulting potential energy surface is smooth. The application of the AC method is demonstrated with two examples in solid systems. read less NOT USED (low confidence) M. S. Mrudul, S. Thomas, and K. Ajith, “Anharmonicities in the temperature-dependent bending rigidity of BC3 monolayer,” Journal of Physics and Chemistry of Solids. 2020. link Times cited: 1 NOT USED (low confidence) M. Motlag et al., “Molecular-Scale Nanodiamond with High-Density Color Centers Fabricated from Graphite by Laser Shocking.” 2020. link Times cited: 4 NOT USED (low confidence) M. Sengul et al., “An Initial Design-enhanced Deep Learning-based Optimization Framework to Parameterize Multicomponent ReaxFF Force Fields,” ChemRxiv. 2020. link Times cited: 5 Abstract: ReaxFF is an empirical interatomic potential capable of simu… read moreAbstract: ReaxFF is an empirical interatomic potential capable of simulating reactions in complex chemical processes and thus determine the dynamical evolution of the molecular systems. A drawback of this method is the necessity of a significant preprocessing to adapt it to a chemical system of nterest. One of the preprocessing steps is the optimization of force field parameters that are used to tune interatomic interactions to mimic ones obtained by quantum chemistry-based methods. The optimization of these parameters is a very complex high dimensional problem. Here, we propose an INitial-DEsign Enhanced Deep learning-based OPTimization (INDEEDopt) framework to be used in ReaxFF parameterization. The procedure starts with a Latin Hypercube Design (LHD) algorithm that is used to explore the parameter landscape extensively. The LHD passes the information about explored regions to a deep learning model for training. The deep learning model finds the minimum discrepancy regions and eliminates unfeasible regions, which originate from the unphysical atomistic interactions, and constructs a more comprehensive understanding of a physically meaningful parameter space. The procedure was successfully used to parametrize a nickel-chromium binary force field and a tungsten-sulfide-carbon-hydrogen quaternary force field and produced improved accuracies in shorter periods time compared to conventional optimization method. read less NOT USED (low confidence) M. Zheng, P. Yang, Z. Wang, X. Li, and L. Guo, “Capturing the dynamic profiles of products in Hailaer brown coal pyrolysis with reactive molecular simulations and experiments,” Fuel. 2020. link Times cited: 34 NOT USED (low confidence) S. Chen et al., “Giant low-temperature anharmonicity in silicon nanocrystals,” Physical Review Materials. 2020. link Times cited: 1 NOT USED (low confidence) Z. J. Choong, D. Huo, N. Ponon, R. Savidis, P. Degenaar, and A. O’Neill, “A novel hybrid technique to fabricate silicon-based micro-implants with near defect-free quality for neuroprosthetics application.,” Materials science & engineering. C, Materials for biological applications. 2020. link Times cited: 1 NOT USED (low confidence) A. Khokonov and Z. A. Akhmatov, “Bending vibrations of free and microdroplet-loaded graphene in the framework of the molecular dynamics method,” Journal of Physics: Conference Series. 2020. link Times cited: 1 Abstract: Molecular dynamic (MD) modeling revealed that takes place sw… read moreAbstract: Molecular dynamic (MD) modeling revealed that takes place swing up of transverse vibrations of graphene atoms with their transition to bending vibrations of membrane type. The amplitudes of such oscillations can reach large values, which considerably exceed the interatomic distances already for samples of micron sizes and amount to 10−2 from the length of the sample. The results of MD simulation were compared with the characteristic vibrational frequencies of graphene in the approximation of the tensioned membrane. The behavior of the graphene membrane during it loading with a liquid metal microdroplet has been considered also. Contact angles and relative stretching of the membrane outside and under the drop allow us to calculate the surface tension of the drop. read less NOT USED (low confidence) A. Volkov and A. H. Banna, “Mesoscopic computational model of covalent cross-links and mechanisms of load transfer in cross-linked carbon nanotube films with continuous networks of bundles,” Computational Materials Science. 2020. link Times cited: 4 NOT USED (low confidence) S. Ajori, H. Parsapour, and R. Ansari, “A comprehensive analysis of the mechanical properties and fracture analysis of metallic glass nanocomposites reinforced by carbon nanotubes and Cu nanowires: A molecular dynamics study,” Mechanics of Advanced Materials and Structures. 2020. link Times cited: 9 Abstract: Reinforced by nanowires (NWs), carbon nanotubes (CNTs) and N… read moreAbstract: Reinforced by nanowires (NWs), carbon nanotubes (CNTs) and NW encapsulated CNT (NW@CNT), tensile behavior of various types of Cu-Zr based metallic glass (MG) nanocomposites are studied using molecular dynamics (MD) simulations. It is observed that pure two-toms alloy MG and the one reinforced with bigger CNT demonstrates higher tensile properties than other types of MGs. Further, it is observed that the ultimate strength of reinforced MGs with individual CNTs is slightly higher than that of NW@CNT reinforced analogous. In this case, it is noticed that reinforced three-atoms Cu-Zr MG nanocomposites including Ti atoms demonstrate the highest ultimate strength and strain. read less NOT USED (low confidence) H. Mes-adi, Y. Lachtioui, K. Saadouni, and M. Mazroui, “Morphology and surface properties of Cu thin film on Si (001),” Thin Solid Films. 2020. link Times cited: 12 NOT USED (low confidence) J. Hickman and Y. Mishin, “Thermal conductivity and its relation to atomic structure for symmetrical tilt grain boundaries in silicon.,” Physical review materials. 2020. link Times cited: 9 Abstract: We perform a systematic study of thermal resistance/conducta… read moreAbstract: We perform a systematic study of thermal resistance/conductance of tilt grain boundaries (GBs) in Si using classical molecular dynamics. The GBs studied are naturally divided into three groups according to the structural units forming the GB core. We find that, within each group, the GB thermal conductivity strongly correlates with the excess GB energy. All three groups predict nearly the same GB conductivity extrapolated to the high-energy limit. This limiting value is close to the thermal conductivity of amorphous Si, suggesting similar heat transport mechanisms. While the lattice thermal conductivity decreases with temperature, the GB conductivity slightly increases. However, at high temperatures it turns over and starts decreasing if the GB structure undergoes a premelting transformation. Analysis of vibrational spectra of GBs resolved along different directions sheds light on the mechanisms of their thermal resistance. The existence of alternating tensile and compressive atomic environments in the GB core gives rise to localized vibrational modes, frequency gaps creating acoustic mismatch with lattice phonons, and anharmonic vibrations of loosely-bound atoms residing in open atomic environments. read less NOT USED (low confidence) M. Cusentino, M. Wood, and A. Thompson, “Explicit Multi-element Extension of the Spectral Neighbor Analysis Potential for Chemically Complex Systems.,” The journal of physical chemistry. A. 2020. link Times cited: 35 Abstract: A natural extension of the descriptors used in the Spectral … read moreAbstract: A natural extension of the descriptors used in the Spectral Neighbor Analysis Potential (SNAP) method is derived to treat atomic interactions in chemically complex systems. Atomic environment descriptors within SNAP are obtained from a basis function expansion of the weighted density of neighboring atoms. This new formulation instead partitions the neighbor density into partial densities for each chemical element, thus leading to explicit multi-element descriptors. For Nelem chemical elements, the number of descriptors increases as Ο(Nelem3), while the computational cost of the force calculation as implemented in LAMMPS is limited to Ο(Nelem2) and the favorable linear scaling in the number of atoms is retained. We demonstrate these chemically aware descriptors by producing an interatomic potential for indium phosphide capable of capturing high-energy defects that result from radiation damage cascades. This new explicit multi-element SNAP method reproduces the relaxed defect formation energies with substantially greater accuracy than weighted-density SNAP, while retaining accurate representation of the bulk indium phosphide properties. read less NOT USED (low confidence) L. V. Mirantsev and A. Abramyan, “Couette flows between various bounding substrates,” Physics Letters A. 2020. link Times cited: 2 NOT USED (low confidence) S. A. A. Kalkhoran, M. Vahdati, and J. Yan, “Effect of relative tool sharpness on subsurface damage and material recovery in nanometric cutting of mono-crystalline silicon: A molecular dynamics approach,” Materials Science in Semiconductor Processing. 2020. link Times cited: 19 NOT USED (low confidence) J. Chapman, R. Batra, and R. Ramprasad, “Machine learning models for the prediction of energy, forces, and stresses for Platinum,” Computational Materials Science. 2020. link Times cited: 18 NOT USED (low confidence) M. Pelliciari and A. Tarantino, “Equilibrium paths of a three-bar truss in finite elasticity with an application to graphene,” Mathematics and Mechanics of Solids. 2020. link Times cited: 18 Abstract: This paper presents the formulation of the equilibrium probl… read moreAbstract: This paper presents the formulation of the equilibrium problem of a three-bar truss in the nonlinear context of finite elasticity. The bars are composed of a homogeneous, isotropic, and compressible hyperelastic material. The equilibrium equations in the deformed configuration are derived under the assumption of homogeneous deformations and the stability of the solutions is assessed through the energy criterion. The general formulation is then specialized for a compressible Mooney–Rivlin material. The results for both vertical and horizontal load cases show unexpected post-critical behaviors involving several branches, stable asymmetrical configurations, bifurcation, and snap-through. The three-bar truss studied here is not only a benchmark test for the numerical analysis of nonlinear truss structures, but also a representative system for the unit cell of the graphene hexagonal lattice. Therefore, an application to graphene is performed by simulating the covalent bonds between carbon atoms as the bars of the truss, characterized by the modified Morse potential. The results provide insights on the internal mechanisms that take place when graphene undergoes large in-plane deformations, whose influence should be considered when developing molecular mechanics and continuum models in nonlinear elasticity. read less NOT USED (low confidence) B. Shen, S. Chen, Z. Huang, Z. Ji, Q. Lin, and Z. Zhang, “Elucidating the atomic mechanism of the lubricity of graphene on the diamond substrate,” Applied Surface Science. 2020. link Times cited: 17 NOT USED (low confidence) X. Li and Z. Huang, “A Non-Ordinary State-Based Peridynamic Constitutive Model Based on Three-Body Interaction Potential and Its Implementation on Algorithm,” Journal of Peridynamics and Nonlocal Modeling. 2020. link Times cited: 5 NOT USED (low confidence) X. Li and Z. Huang, “A Non-Ordinary State-Based Peridynamic Constitutive Model Based on Three-Body Interaction Potential and Its Implementation on Algorithm,” Journal of Peridynamics and Nonlocal Modeling. 2020. link Times cited: 0 NOT USED (low confidence) F. Guo et al., “Intelligent-ReaxFF: Evaluating the reactive force field parameters with machine learning,” Computational Materials Science. 2020. link Times cited: 29 NOT USED (low confidence) R. Tromer, I. M. Felix, A. Freitas, S. Azevedo, and L. Pereira, “Diboron-porphyrin monolayer: A new 2D semiconductor,” Computational Materials Science. 2020. link Times cited: 16 NOT USED (low confidence) S. Im, W. Kim, H. Kim, and M. Cho, “Artificial neural network modeling of anisotropic hyperelastic materials based on computational crystal structure data.” 2020. link Times cited: 4 NOT USED (low confidence) A. Dadrasi, A. Albooyeh, and A. H. Mashhadzadeh, “Mechanical properties of silicon-germanium nanotubes: A molecular dynamics study,” Applied Surface Science. 2019. link Times cited: 32 NOT USED (low confidence) G. Plummer and G. Tucker, “Bond-order potentials for theTi3AlC2andTi3SiC2MAX phases,” Physical Review B. 2019. link Times cited: 12 NOT USED (low confidence) A. Braides, B. Schmidt, U. Stefanelli, and F. Theil, “Emergence of Structures in Particle Systems: Mechanics, Analysis and Computation,” Oberwolfach Reports. 2019. link Times cited: 0 Abstract: . The meeting focused on the last advances in particle syste… read moreAbstract: . The meeting focused on the last advances in particle systems. The talks covered a broad range of topics, ranging from questions in crystallization and atomistic systems to mesoscopic models of defects to machine learning approaches and computational aspects. study non-convex perturbations of the massless free Gaussian field (GFF) in dimensions d = 2 , 3. We first the strict convexity of the surface tension for large enough β (low temperatures) and sufficiently small tilt, using multi-scale (renormalisation group theory) techniques based on a finite range decomposition of the underlying background Gaussian in read less NOT USED (low confidence) Y. Nan, D. Tan, J. Zhao, M. Willatzen, and Z. L. Wang, “Shape- and size dependent piezoelectric properties of monolayer hexagonal boron nitride nanosheets,” Nanoscale Advances. 2019. link Times cited: 8 Abstract: We use molecular dynamics simulations (MD) to study piezoele… read moreAbstract: We use molecular dynamics simulations (MD) to study piezoelectric properties of hexagonal boron nitride nanosheets (BNNS) and reveal how piezoelectric properties depend on size and shape. We first analyze how the macroscopic shape affects the full 2D structure symmetry and its piezoelectric tensor. In particular, we demonstrate that a hexagonal (rectangular)-shaped BNNS belongs to the hexagonal 6̄m2 (monoclinic m) point group. Our simulation results show that the piezoelectric constants of BNNS depend strongly on the macroscopic shape, in agreement with the symmetry of the structure, but are nearly independent of the macroscopic size. The present study provides a detailed understanding of the piezoelectric properties of finite size BNNS and guidance to future experiments and optimization of 2D piezoelectric materials in general. read less NOT USED (low confidence) B. Wang, Y. Chen, and C. Hou, “A Communication-Avoiding Algorithm for Molecular Dynamics Simulation,” International Conference on Algorithms and Architectures for Parallel Processing. 2019. link Times cited: 0 NOT USED (low confidence) S. Takamoto, S. Izumi, and J. Li, “TeaNet: universal neural network interatomic potential inspired by iterative electronic relaxations,” ArXiv. 2019. link Times cited: 29 NOT USED (low confidence) A. Senturk, A. Oktem, and A. E. S. Konukman, “Investigation of interfacial thermal resistance of hybrid graphene/hexagonal boron nitride,” International Journal of Mechanics and Materials in Design. 2019. link Times cited: 9 NOT USED (low confidence) J. Yan, L. Tong, R. Luo, and D. Gao, “Thickness of monolayer h-BN nanosheet and edge effect on free vibration behaviors,” International Journal of Mechanical Sciences. 2019. link Times cited: 15 NOT USED (low confidence) Y. Katoh and L. Snead, “Silicon carbide and its composites for nuclear applications – Historical overview,” Journal of Nuclear Materials. 2019. link Times cited: 106 NOT USED (low confidence) T. Jiang et al., “Mechanical properties of hydrogenated amorphous silicon (a-Si:H) particles,” Journal of Applied Physics. 2019. link Times cited: 6 Abstract: A nanoindenter was used to compress individual particles of … read moreAbstract: A nanoindenter was used to compress individual particles of hydrogenated amorphous silicon (a-Si:H) ranging in diameter from 290 nm to 780 nm. The colloidal synthesis used to produce the particles enables the hydrogen content to be manipulated over a wide range, from about 5 at. % to 50 at. %, making these a-Si:H particles promising for applications in lithium ion batteries, hydrogen storage, and optical metamaterials. Force-displacement curves generated using a tungsten probe flattened with focused ion beam exhibited elastic and then plastic deformations, followed by fracture and crushing of the particles. For particles with 5% and 50% H, Young's moduli, yield strengths, and compressive strengths were 73.5(±19.5) GPa, 5.8 GPa, and 3.2(±0.1)–9.3(±0.6) GPa and 31.2(±9.0) GPa, 2.5 GPa, and 1.8 (±0.3)–5.3(±0.8) GPa, respectively. Particles with more hydrogen were significantly more compliant and weaker. This is consistent with atomistically detailed molecular dynamics simulations, which revealed compression forms of an interphase of H atom clusters that weakens the material.A nanoindenter was used to compress individual particles of hydrogenated amorphous silicon (a-Si:H) ranging in diameter from 290 nm to 780 nm. The colloidal synthesis used to produce the particles enables the hydrogen content to be manipulated over a wide range, from about 5 at. % to 50 at. %, making these a-Si:H particles promising for applications in lithium ion batteries, hydrogen storage, and optical metamaterials. Force-displacement curves generated using a tungsten probe flattened with focused ion beam exhibited elastic and then plastic deformations, followed by fracture and crushing of the particles. For particles with 5% and 50% H, Young's moduli, yield strengths, and compressive strengths were 73.5(±19.5) GPa, 5.8 GPa, and 3.2(±0.1)–9.3(±0.6) GPa and 31.2(±9.0) GPa, 2.5 GPa, and 1.8 (±0.3)–5.3(±0.8) GPa, respectively. Particles with more hydrogen were significantly more compliant and weaker. This is consistent with atomistically detailed molecular dynamics simulations, which revealed compression ... read less NOT USED (low confidence) R. Ravinder, P. Garg, and N. Krishnan, “Glass Transition and Crystallization in Hexagonal Boron Nitride: Crucial Role of Orientational Order,” Advanced Theory and Simulations. 2019. link Times cited: 1 Abstract: While extensive studies have been carried out on 2D crystals… read moreAbstract: While extensive studies have been carried out on 2D crystals, their disordered counterpart, namely 2D glasses, remain poorly explored. Using molecular dynamics simulation, it is demonstrated that hexagonal boron nitride (h‐BN) can exhibit crystallization and glass transition. Similar to archetypical glasses, it is observed that the glassy structures are disordered, marked by the absence of any long‐range order while exhibiting some short‐range order. Further, the structures obtained are stable in three dimensions, confirming the realistic nature of the 2D glasses. Interestingly, it is observed that the orientational order of the h‐BN structure with respect to density as well as ground state enthalpy follows a master curve for both crystallization and glass transition, revealing thermally accessible regions for the structure. Further, the standard deviation of orientational order provides information about the spatial heterogeneity in the structure, the rearrangement of which results in the formation of crystal/glass. This suggests that the orientational order may play a crucial role in controlling the propensity for crystallization/glass transition in systems having directional bonds. read less NOT USED (low confidence) R. Marimpul, T. Winata, F. A. Noor, I. Syuhada, and A. Rosikhin, “Molecular dynamics simulation of platinum film growth based on thermal evaporation method,” IOP Conference Series: Materials Science and Engineering. 2019. link Times cited: 0 Abstract: Platinum film growth using thermal evaporation method was st… read moreAbstract: Platinum film growth using thermal evaporation method was studied using molecular dynamics simulation. This platinum film was intended as catalyst film for graphene growth. Tersoff, Eam and Lennard-Jones potential were used to describe interaction of Si-Si, Pt-Pt and Pt-Si respectively. Deposition process was performed with low incident energy to represent thermal evaporation method. Our simulation found that heating temperature at 400 K produced platinum film with higher percentage of crystal structure than other heating condition 300K, 500K & 600K. We also found transition phase from fcc to bcc at 600K. read less NOT USED (low confidence) X. Li, H. Mizuseki, S. J. Pai, and K.-R. Lee, “Reactive molecular dynamics simulation of the amorphous carbon growth: Effect of the carbon triple bonds,” Computational Materials Science. 2019. link Times cited: 5 NOT USED (low confidence) Y. Wang and J. Shi, “Phase transformation of monocrystalline silicon by nanoindentation – Effect of processing temperature,” Materials Science in Semiconductor Processing. 2019. link Times cited: 5 NOT USED (low confidence) H. Calderón et al., “Enhanced elastic behavior of all-carbon composites reinforced by in-situ synthesized morphed graphene,” Carbon. 2019. link Times cited: 11 NOT USED (low confidence) J.-H. Zou, X.-T. Xu, and B. Cao, “Size-dependent mode contributions to the thermal transport of suspended and supported graphene,” Applied Physics Letters. 2019. link Times cited: 15 Abstract: Graphene is promising for thermal management applications du… read moreAbstract: Graphene is promising for thermal management applications due to its superior thermal conductivity, but the inherent thermal transport mechanism is not fully understood. In this work, we directly extract the mode contributions to the thermal conductivity of free-standing and supported graphene nanoribbons using nonequilibrium molecular dynamics simulations. With the aid of the mode analyses, it is uncovered that the acoustic contribution increases with respect to the characteristic length in suspended graphene, and such a size-dependency is compromised in supported graphene. In addition, the contribution of optical modes could be more than 25% for heat conduction in narrow graphene nanoribbons because of strong boundary scatterings. Furthermore, the LA and TA modes rather than the ZA modes are dominant heat carriers in suspended and supported graphene owing to their large group velocities and long mean free path. Our results are instructive for understanding the mode thermal transport in free and supported graphene. read less NOT USED (low confidence) F. Cornacchia, N. Fantuzzi, R. Luciano, and R. Penna, “Solution for cross- and angle-ply laminated Kirchhoff nano plates in bending using strain gradient theory,” Composites Part B: Engineering. 2019. link Times cited: 34 NOT USED (low confidence) L. Fan and W. Yao, “Regulation and coupling effects on mechanical properties of copper-graphene/h-BN layered heterostructure via ion irradiation, interlayer sp3 bonds and temperature,” Materials Research Express. 2019. link Times cited: 4 Abstract: In this paper, by using the molecular dynamics method, vario… read moreAbstract: In this paper, by using the molecular dynamics method, various defects have been created in graphene/h-BN with interlayer sp3 bonds (G/BN-sp3) by using ion irradiation. The copper-graphene/h-BN layered heterostructure (Cu-G/BN-sp3) were prepared by inserting G/BN-sp3 after irradiation damage into the single crystal copper matrix. The coupling effects of ion irradiation, interlayer sp3 bonds and temperature on defective copper-graphene/h-BN layered heterostructure were studied. The results show with the increase of ion irradiation doses, the mechanical properties of copper layered heterostructure decrease. When defects (caused by ion irradiation) and sp3 bonds coexist, if the defects are created around the sp3 bonds, the composite system is more vulnerable to damage. The location of the defects is an important factor to determine the crack position and fracture direction of copper layered heterostructure. Although ion irradiation and sp3 defects have adverse effects on mechanical properties, the mechanical properties of pure copper are still improved by introducing graphene/h-BN heterostructure. It provides a new way to enhance the radiation resistance and corrosion resistance of copper matrix composites. Meanwhile, controlling and strengthening the properties of copper matrix composites from a new perspective. read less NOT USED (low confidence) F. Cornacchia, F. Fabbrocino, N. Fantuzzi, R. Luciano, and R. Penna, “Analytical solution of cross- and angle-ply nano plates with strain gradient theory for linear vibrations and buckling,” Mechanics of Advanced Materials and Structures. 2019. link Times cited: 40 Abstract: Vibrations and buckling of Kirchhoff nano plates are investi… read moreAbstract: Vibrations and buckling of Kirchhoff nano plates are investigated using second-order strain gradient theory. The Navier displacement field has been considered for two different sets of boundary conditions and stacking sequences. Different geometries and material properties for isotropic, orthotropic cross- and angle-ply laminates are considered, and numerical simulations are discussed in terms of plate aspect ratio and non local ratio. A comparison with the classical analytical solution is provided whenever possible for buckling loads and fundamental frequencies. read less NOT USED (low confidence) S. H. Boroushak, S. Ajori, and R. Ansari, “Characterization of the structural instability of BxCyNz heteronanotubes via molecular dynamics simulations,” Materials Research Express. 2019. link Times cited: 6 Abstract: Carbon nanotubes (CNTs) possess unique structural properties… read moreAbstract: Carbon nanotubes (CNTs) possess unique structural properties which can be modified by several methods such as partial and full atom substitution. In this method also known as doping, novel hybrid tubular structures with desired and new characteristics can be synthesized. To this end, Boron (B) and Nitrogen (N) are selected as dopants and then by using molecular dynamics (MD) simulations the structural behavior of the new heteronanotubes are investigated. Moreover, the buckling behavior of these novel nanotube alongside the pure CNT and BN nanotube (BNNT) are studied. Apparently, the critical forces of the newly formed structures are computed between those of pure CNT and BNNT. Further, a combination of the doped structures and pure ones are used to simulate hybrid double-wall nanotubes and then their buckling response under axial compressive load is studied. Attained results demonstrated that double-walled hybrid structures possess mechanical stabilities lower and higher than pure double-walled CNT and BNNT, respectively. read less NOT USED (low confidence) K. Lee, D. Yoo, W. Jeong, and S. Han, “SIMPLE-NN: An efficient package for training and executing neural-network interatomic potentials,” Comput. Phys. Commun. 2019. link Times cited: 75 NOT USED (low confidence) E. Mahdavi, R. Khaledialidusti, and A. Barnoush, “Rheological properties of super critical CO2 with Al2O3: Material type, size and temperature effect,” Journal of Molecular Liquids. 2019. link Times cited: 10 NOT USED (low confidence) Y. He, “Modeling of Graphene‐Based Electronics: From Material Properties to Circuit Simulations,” Handbook of Graphene. 2019. link Times cited: 0 NOT USED (low confidence) T. D. Huan, R. Batra, J. Chapman, C. Kim, A. Chandrasekaran, and R. Ramprasad, “Iterative-Learning Strategy for the Development of Application-Specific Atomistic Force Fields,” The Journal of Physical Chemistry C. 2019. link Times cited: 18 Abstract: Emerging data-driven approaches in materials science have tr… read moreAbstract: Emerging data-driven approaches in materials science have triggered the development of numerous machine-learning force fields. In practice, they are constructed by training a statistical model on a reference database to predict potential energy and/or atomic forces. Although most of the force fields can accurately recover the properties of the training set, some of them are becoming useful for actual molecular dynamics simulations. In this work, we employ a simple iterative-learning strategy for the development of machine-learning force fields targeted at specific simulations (applications). The strategy involves (1) preparing and fingerprinting a diverse reference database of atomic configurations and forces, (2) generating a pool of machine-learning force fields by learning the reference data, (3) validating the force fields against a series of targeted applications, and (4) selectively and recursively improving the force fields that are unsuitable for a given application while keeping their performance... read less NOT USED (low confidence) L. V. Mirantsev, “Superfluidity inside carbon nanotubes.,” Physical review. E. 2019. link Times cited: 1 Abstract: Molecular dynamics simulations of equilibrium structures and… read moreAbstract: Molecular dynamics simulations of equilibrium structures and flows of nonpolar argon atoms confined by single-walled carbon nanotubes (SWCNTs) with circular cross section and rectangular cross section having the same area and the ratio between its sides 1:4 have been performed. It has been shown that, inside these SWCNTs, argon atoms form the spatially ordered structures and, under action of external driving forces they move collectively along SWCNT's axes. It has been also obtained that there are two regimes of such collective movement. In the first regime, when the driving external force f_{x0} is lower than a certain critical value f_{xc}, argon atoms flow through these SWCNTs with the finite average flow velocity. In the second regime, when the driving external force f_{x0} exceeds f_{xc}, the retarding friction force acting on argon atoms from bounding wall carbon atoms gradually drops to zero and the average flow velocity exhibits an unlimited growth. Moreover, when the retarding friction force becomes close to zero, the fluid will continue to flow with the same constant velocity at switched off external driving force. Hence, in the second regime, argon atoms inside the above-mentioned SWCNTs demonstrate the ballistic frictionless flows which resemble the superfluidic liquid flow. It has been shown that collective frictionless ballistic flows of argon atoms through SWCNTs are caused by the crystalline structure of SWCNT's bounding walls and, for the same SWCNTs with random distribution of carbon atoms on the bounding walls, one can observe only the first regime of the argon atom flows. read less NOT USED (low confidence) B. Lee and S. Park, “Applying Tersoff-potential and bond-softening models in a molecular dynamics study of femtosecond laser processing,” Journal of Applied Physics. 2019. link Times cited: 3 Abstract: In the molecular dynamics study of short-pulsed laser proces… read moreAbstract: In the molecular dynamics study of short-pulsed laser processing of semiconductors, potential models capable of describing the atomistic behavior during high electronic excitations is the most critical issue at the current stage. This study of the molecular dynamics adopts the Tersoff-potential model to analyze the ultrafast laser processing of silicon. The model was modified to include electronic excitation effects by reducing the attraction of the antibonding state by half. It offers an excellent description of the experimental behavior during nonthermal melting. Subpicosecond melting is achieved above certain threshold levels of superheating and carrier density as required in experiments. Energy conservation is demonstrated with a bandgap energy of the order obtained in experiments. The modification of the potential mimics an absorption of bandgap energy and a subsequent lattice heating on a time scale within 0.3 ps. The melting kinetics establishes a correlation between nonthermal melting and thermal bulk melting. For superheating of less than two, the electronic melting of bond softening proceeds via homogeneous nucleation. The associated thermal theory, corrected with a limit on the nucleus radius to bond length, is still valid for the higher superheating regime. The original Tersoff model shows that this superheating by a factor of two is isothermal for spallation—the lowest-energy ablative mechanism. Its proximity to the evaporating point suggests the role of thermal boiling during spallation.In the molecular dynamics study of short-pulsed laser processing of semiconductors, potential models capable of describing the atomistic behavior during high electronic excitations is the most critical issue at the current stage. This study of the molecular dynamics adopts the Tersoff-potential model to analyze the ultrafast laser processing of silicon. The model was modified to include electronic excitation effects by reducing the attraction of the antibonding state by half. It offers an excellent description of the experimental behavior during nonthermal melting. Subpicosecond melting is achieved above certain threshold levels of superheating and carrier density as required in experiments. Energy conservation is demonstrated with a bandgap energy of the order obtained in experiments. The modification of the potential mimics an absorption of bandgap energy and a subsequent lattice heating on a time scale within 0.3 ps. The melting kinetics establishes a correlation between nonthermal melting and thermal ... read less NOT USED (low confidence) L. Giacomozzi et al., “Decay pathways for protonated and deprotonated adenine molecules.,” The Journal of chemical physics. 2019. link Times cited: 0 Abstract: We have measured fragment mass spectra and total destruction… read moreAbstract: We have measured fragment mass spectra and total destruction cross sections for protonated and deprotonated adenine following collisions with He at center-of-mass energies in the 20-240 eV range. Classical and ab initio molecular dynamics simulations are used to provide detailed information on the fragmentation pathways and suggest a range of alternative routes compared to those reported in earlier studies. These new pathways involve, for instance, losses of HNC molecules from protonated adenine and losses of NH2 or C3H2N2 from deprotonated adenine. The present results may be important to advance the understanding of how biomolecules may be formed and processed in various astrophysical environments. read less NOT USED (low confidence) Z. Chen, Y. Cao, W. Tian, and Y. Wang, “Surface roughness analysis of Cu films deposited on Si substrates: A molecular dynamic analysis,” Journal of Applied Physics. 2019. link Times cited: 4 Abstract: Cu is a promising material to replace Al and Au in integrate… read moreAbstract: Cu is a promising material to replace Al and Au in integrated circuits and microscale devices because of its low electrical resistivity, high electromigration resistance, and low cost. However, surface roughness affects the contact resistance of these devices, especially when the device is on a microscale or nanoscale. This paper focuses on surface roughness analysis of Cu films deposited on Si substrates by molecular dynamic simulation based on the mechanism of physical vapor deposition. The effects of film thickness, deposition temperature, deposition interval, and reflow temperature on the surface roughness of Cu films are studied in detail. The simulation results show that the surface roughness can be improved by appropriate adjustments of these parameters. They also provide a foundation for further work on the deposition of Cu films on Si substrates.Cu is a promising material to replace Al and Au in integrated circuits and microscale devices because of its low electrical resistivity, high electromigration resistance, and low cost. However, surface roughness affects the contact resistance of these devices, especially when the device is on a microscale or nanoscale. This paper focuses on surface roughness analysis of Cu films deposited on Si substrates by molecular dynamic simulation based on the mechanism of physical vapor deposition. The effects of film thickness, deposition temperature, deposition interval, and reflow temperature on the surface roughness of Cu films are studied in detail. The simulation results show that the surface roughness can be improved by appropriate adjustments of these parameters. They also provide a foundation for further work on the deposition of Cu films on Si substrates. read less NOT USED (low confidence) S. Ajori, H. Parsapour, R. Ansari, and A. Ameri, “Buckling behavior of various metallic glass nanocomposites reinforced by carbon nanotube and Cu nanowire: A molecular dynamics simulation study,” Materials Research Express. 2019. link Times cited: 19 Abstract: The reinforcement of various materials by nanofillers as nan… read moreAbstract: The reinforcement of various materials by nanofillers as nanocomposites has recently received the attention of many researchers. In the present research, molecular dynamics simulations are used to investigate the influence of nanowire (NW)/carbon nanotube (CNT) reinforcement on the buckling behavior of metallic glass matrix nanocomposites (MGMNCs). The buckling characteristics of nanocomposites made by adding Cu NWs, CNTs and Cu NW-encapsulated CNTs to metallic glass matrices are studied. The results demonstrate that MG alloys comprising just two elements (Cu and Zr) with higher Cu percentage have higher mechanical stability. Also, it is observed that adding NW leads to a negative effect on the buckling behavior, while adding CNT and NW-encapsulated CNT considerably increases the buckling force and strain of the metallic glass models. Moreover, it is found that the filled CNT is the most effective nanofiller for amending the buckling behavior of metallic glasses. Furthermore, as the size of nanofillers gets larger, the critical force increases and the critical strain decreases. read less NOT USED (low confidence) J. V. Michelin, L. G. Gonçalves, and J. Rino, “On the transferability of interaction potentials for condensed phases of silicon,” Journal of Molecular Liquids. 2019. link Times cited: 6 NOT USED (low confidence) B. Babu and B. P. Patel, “A new computationally efficient finite element formulation for nanoplates using second-order strain gradient Kirchhoff’s plate theory,” Composites Part B: Engineering. 2019. link Times cited: 49 NOT USED (low confidence) M. Izadifar et al., “Fracture toughness of various percentage of doping of boron atoms on the mechanical properties of polycrystalline graphene: A molecular dynamics study,” Physica E: Low-dimensional Systems and Nanostructures. 2019. link Times cited: 15 NOT USED (low confidence) G. He, C. Xu, C. Liu, H. Liu, and H. Wang, “Grain size and temperature effects on the indentation induced plastic deformations of nano polycrystalline diamond,” Applied Surface Science. 2019. link Times cited: 12 NOT USED (low confidence) X. Wei, H. Zhong, Q. Yang, E. Yao, Y. Zhang, and H. Zou, “Studying the mechanisms of natural rubber pyrolysis gas generation using RMD simulations and TG-FTIR experiments,” Energy Conversion and Management. 2019. link Times cited: 56 NOT USED (low confidence) H. Nguyen, “Graphene layer of hybrid graphene/hexagonal boron nitride model upon heating,” Carbon Letters. 2019. link Times cited: 9 NOT USED (low confidence) J. Yan, S. Lai, and L. He, “Nonlinear dynamic behavior of single-layer graphene under uniformly distributed loads,” Composites Part B: Engineering. 2019. link Times cited: 18 NOT USED (low confidence) S. Smidstrup et al., “QuantumATK: an integrated platform of electronic and atomic-scale modelling tools,” Journal of Physics: Condensed Matter. 2019. link Times cited: 638 Abstract: QuantumATK is an integrated set of atomic-scale modelling to… read moreAbstract: QuantumATK is an integrated set of atomic-scale modelling tools developed since 2003 by professional software engineers in collaboration with academic researchers. While different aspects and individual modules of the platform have been previously presented, the purpose of this paper is to give a general overview of the platform. The QuantumATK simulation engines enable electronic-structure calculations using density functional theory or tight-binding model Hamiltonians, and also offers bonded or reactive empirical force fields in many different parametrizations. Density functional theory is implemented using either a plane-wave basis or expansion of electronic states in a linear combination of atomic orbitals. The platform includes a long list of advanced modules, including Green’s-function methods for electron transport simulations and surface calculations, first-principles electron-phonon and electron-photon couplings, simulation of atomic-scale heat transport, ion dynamics, spintronics, optical properties of materials, static polarization, and more. Seamless integration of the different simulation engines into a common platform allows for easy combination of different simulation methods into complex workflows. Besides giving a general overview and presenting a number of implementation details not previously published, we also present four different application examples. These are calculations of the phonon-limited mobility of Cu, Ag and Au, electron transport in a gated 2D device, multi-model simulation of lithium ion drift through a battery cathode in an external electric field, and electronic-structure calculations of the composition-dependent band gap of SiGe alloys. read less NOT USED (low confidence) T. Fang, C.-guo Shen, Y. Fan, and W.-J. Chang, “Fracture characteristics of silicene nanosheet with a crack under tension estimated using molecular dynamics simulation,” Superlattices and Microstructures. 2019. link Times cited: 5 NOT USED (low confidence) A. Raj and J. Eapen, “Phonon dispersion using the ratio of zero-time correlations among conjugate variables: Computing full phonon dispersion surface of graphene,” Comput. Phys. Commun. 2019. link Times cited: 4 NOT USED (low confidence) S. Bringuier et al., “Atomic insight into concurrent He, D, and T sputtering and near-surface implantation of 3C-SiC crystallographic surfaces,” Nuclear Materials and Energy. 2019. link Times cited: 13 NOT USED (low confidence) V. Kuryliuk, O. Nepochatyi, P. Chantrenne, D. Lacroix, and M. Isaiev, “Thermal conductivity of strained silicon: Molecular dynamics insight and kinetic theory approach,” Journal of Applied Physics. 2019. link Times cited: 15 Abstract: In this work, we investigated tensile and compression forces… read moreAbstract: In this work, we investigated tensile and compression forces effect on the thermal conductivity of silicon. We used equilibrium molecular dynamics approach for the evaluation of thermal conductivity considering different interatomic potentials. More specifically, we tested Stillinger-Weber, Tersoff, Environment-Dependent Interatomic Potential and Modified Embedded Atom Method potentials for the description of silicon atom motion under different strain and temperature conditions. Additionally, we extracted phonon density of states and dispersion curves from molecular dynamics simulations. These data were used for direct calculations of thermal conductivity considering the kinetic theory approach. Comparison of molecular dynamics and kinetic theory simulations results as a function of strain and temperature allowed us to investigate the different factors affecting the thermal conductivity of strained silicon. read less NOT USED (low confidence) X. Li, A. Wang, and K.-R. Lee, “Role of unsaturated hydrocarbon lubricant on the friction behavior of amorphous carbon films from reactive molecular dynamics study,” Computational Materials Science. 2019. link Times cited: 12 NOT USED (low confidence) Q. Cai et al., “High thermal conductivity of high-quality monolayer boron nitride and its thermal expansion,” Science Advances. 2019. link Times cited: 271 Abstract: Atomically thin boron nitride is one of the best thermal con… read moreAbstract: Atomically thin boron nitride is one of the best thermal conductors among semiconductors and insulators. Heat management has become more and more critical, especially in miniaturized modern devices, so the exploration of highly thermally conductive materials with electrical insulation is of great importance. Here, we report that high-quality one-atom-thin hexagonal boron nitride (BN) has a thermal conductivity (κ) of 751 W/mK at room temperature, the second largest κ per unit weight among all semiconductors and insulators. The κ of atomically thin BN decreases with increased thickness. Our molecular dynamic simulations accurately reproduce this trend, and the density functional theory (DFT) calculations reveal the main scattering mechanism. The thermal expansion coefficients of monolayer to trilayer BN at 300 to 400 K are also experimentally measured for the first time. Owing to its wide bandgap, high thermal conductivity, outstanding strength, good flexibility, and excellent thermal and chemical stability, atomically thin BN is a strong candidate for heat dissipation applications, especially in the next generation of flexible electronic devices. read less NOT USED (low confidence) J. Byggmästar, M. J. Nagel, K. Albe, K. Henriksson, and K. Nordlund, “Analytical interatomic bond-order potential for simulations of oxygen defects in iron,” Journal of Physics: Condensed Matter. 2019. link Times cited: 11 Abstract: We present an analytical bond-order potential for the Fe–O s… read moreAbstract: We present an analytical bond-order potential for the Fe–O system, capable of reproducing the basic properties of wüstite as well as the energetics of oxygen impurities in -iron. The potential predicts binding energies of various small oxygen-vacancy clusters in -iron in good agreement with density functional theory results, and is therefore suitable for simulations of oxygen-based defects in iron. We apply the potential in simulations of the stability and structure of Fe/FeO interfaces and FeO precipitates in iron, and observe that the shape of FeO precipitates can change due to formation of well-defined Fe/FeO interfaces. The interface with crystalline Fe also ensures that the precipitates never become fully amorphous, no matter how small they are. read less NOT USED (low confidence) E. Hodille, J. Byggmästar, E. Safi, and K. Nordlund, “Molecular dynamics simulation of beryllium oxide irradiated by deuterium ions: sputtering and reflection,” Journal of Physics: Condensed Matter. 2019. link Times cited: 10 Abstract: The sputtering and reflection properties of wurtzite berylli… read moreAbstract: The sputtering and reflection properties of wurtzite beryllium oxide (BeO) subjected to deuterium (D) ions bombardment at 300 K with ion energy between 10 eV and 200 eV is studied by classical molecular dynamics. Cumulative irradiations of wurtzite BeO show a D concentration threshold above which an ‘unphysical dramatic’ sputtering is observed. From the cumulative irradiations, simulation cells with different D concentrations are used to run non-cumulative irradiations at different concentrations. Using a D concentration close to the experimentally determined saturation concentration (0.12 atomic fraction), the simulations are able to reproduce accurately the experimental sputtering yield of BeO materials. The processes driving the sputtering of beryllium (Be) and oxygen (O) atoms as molecules are subsequently determined. At low irradiation energy, between 10 eV and 80 eV, swift chemical sputtering (SCS) is dominant and produces mostly ODz molecules. At high energy, the sputtered molecules are mostly BexOy molecules (mainly BeO dimer). Four different processes are associated to the formation of such molecules: the physical sputtering of BeO dimer, the delayed SCS not involving D ions and the detachment-induced sputtering. The physical sputtering of BeO dimer can be delayed if the sputtering event implies two interactions with the incoming ion (first interaction in its way in the material, the other in its way out if it is backscattered). The detachment-induced sputtering is a characteristic feature of the ‘dramatic’ sputtering and is mainly observed when the concentration of D is close to the threshold leading to this sputtering regime. read less NOT USED (low confidence) X. Li, A. Wang, and K.-R. Lee, “Insights on low-friction mechanism of amorphous carbon films from reactive molecular dynamics study,” Tribology International. 2019. link Times cited: 37 NOT USED (low confidence) Q. Qiao, C. Liu, W. Gao, and L. Huang, “Graphene oxide model with desirable structural and chemical properties,” Carbon. 2019. link Times cited: 22 NOT USED (low confidence) S. Stelmakh, K. Skrobas, S. Gierlotka, and B. Palosz, “Atomic structure of nanodiamond and its evolution upon annealing up to 1200 °C: Real space neutron diffraction analysis supported by MD simulations,” Diamond and Related Materials. 2019. link Times cited: 7 NOT USED (low confidence) Z. Hui and Y. Chang, “Phonon heat transport properties of graphene based on molecular dynamics simulations and lattice dynamics,” International Journal of Modern Physics B. 2019. link Times cited: 1 Abstract: To choose an ideal method to study the phonon properties of … read moreAbstract: To choose an ideal method to study the phonon properties of graphene, the results of thermal conductivity (TC) of graphene computed using the equilibrium molecular dynamics (EMD), reverse nonequilibrium molecular dynamics (RNEMD) and direct nonequilibrium molecular dynamics (DNEMD) with Tersoff potential are compared, and we find that all of them are very close to each other. While two of them have been compared in the past, there is a lack of comparison of the three methods. Eventually, we choose the Green–Kubo method to study the temperature dependence of TC in graphene and find that the [Formula: see text] diverges with the system temperature T as [Formula: see text]T[Formula: see text] with [Formula: see text] and [Formula: see text] for the direction of armchair and zigzag, respectively, which is in reasonable agreement with the one in recent theoretical and experimental researches. To gain further insight into the TC, the phonon dispersion and the phonon density of states (PDOS), which depend on evaluating the eigenvalues and the eigenvectors of dynamical matrix, are calculated for graphene with dimensions of 30 × 30 unit cell by a combination of EMD simulations and lattice dynamics calculations. read less NOT USED (low confidence) H. Loulijat, A. Koumina, and H. Zerradi, “The effect of the thermal vibration of graphene nanosheets on viscosity of nanofluid liquid argon containing graphene nanosheets,” Journal of Molecular Liquids. 2019. link Times cited: 10 NOT USED (low confidence) C. Huang, X. Peng, B. Yang, S. Weng, Y. Zhao, and T. Fu, “Grain size dependence of tensile properties in nanocrystalline diamond,” Computational Materials Science. 2019. link Times cited: 17 NOT USED (low confidence) Z. Wang et al., “Stress-driven grain re-orientation and merging behaviour found in oxidation of zirconium alloy using in-situ method and MD simulation,” Corrosion Science. 2019. link Times cited: 3 NOT USED (low confidence) J. Wang, X. Zhang, F. Fang, and R. Chen, “Diamond cutting of micro-structure array on brittle material assisted by multi-ion implantation,” International Journal of Machine Tools and Manufacture. 2019. link Times cited: 29 NOT USED (low confidence) N. Nayir, A. V. van Duin, and S. Erkoç, “Development of a ReaxFF Reactive Force Field for Interstitial Oxygen in Germanium and Its Application to GeO2/Ge Interfaces,” The Journal of Physical Chemistry C. 2019. link Times cited: 10 Abstract: We developed the ReaxFF force field parameters for Ge/O/H in… read moreAbstract: We developed the ReaxFF force field parameters for Ge/O/H interactions, specifically targeted for the applications of Ge/GeO2 interfaces and O-diffusion in bulk Ge. The original training set, taken... read less NOT USED (low confidence) A. Senturk, A. Oktem, and A. E. S. Konukman, “Investigation of interfacial thermal resistance of hybrid graphene/hexagonal boron nitride,” International Journal of Mechanics and Materials in Design. 2019. link Times cited: 0 NOT USED (low confidence) M. Nowruzpour, S. Sarkar, J. Reddy, and D. Roy, “A derivative-free upscaled theory for analysis of defects,” Journal of the Mechanics and Physics of Solids. 2019. link Times cited: 9 NOT USED (low confidence) K. Skrobas, S. Stelmakh, S. Gierlotka, and B. Palosz, “A model of density waves in atomic structure of nanodiamond by molecular dynamics simulations,” Diamond and Related Materials. 2019. link Times cited: 10 NOT USED (low confidence) X. Wang and D. Jing, “Determination of thermal conductivity of interfacial layer in nanofluids by equilibrium molecular dynamics simulation,” International Journal of Heat and Mass Transfer. 2019. link Times cited: 43 NOT USED (low confidence) P. Tsai and Y. Jeng, “Coalescence and epitaxial self-assembly of Cu nanoparticles on graphene surface: A molecular dynamics study,” Computational Materials Science. 2019. link Times cited: 17 NOT USED (low confidence) M. Dewapriya and R. Rajapakse, “Atomistic and continuum modelling of stress field at an inhomogeneity in graphene,” Materials & Design. 2018. link Times cited: 7 NOT USED (low confidence) M. Zheng, X. Li, and L. Guo, “Investigation of N behavior during coal pyrolysis and oxidation using ReaxFF molecular dynamics,” Fuel. 2018. link Times cited: 52 NOT USED (low confidence) Q. Ding, N. Ding, L. Liu, N. Li, and C. M. L. Wu, “Investigation on mechanical performances of grain boundaries in hexagonal boron nitride sheets,” International Journal of Mechanical Sciences. 2018. link Times cited: 17 NOT USED (low confidence) J. M. Wynn, “First-principles structure prediction of extreme nanowires.” 2018. link Times cited: 0 Abstract: Low-dimensional systems are an important and intensely studi… read moreAbstract: Low-dimensional systems are an important and intensely studied area of condensed matter physics. When a material is forced to adopt a low-dimensional structure, its behaviour is often dramatically different to that of the bulk phase. It is vital to predict the structures of low-dimensional systems in order to reliably predict their properties. To this end, the ab initio random structure searching (AIRSS) method, which has previously been used to identify the structures of bulk materials, has been extended to deal with the case of nanowires encapsulated inside carbon nanotubes. Such systems are a rapidly developing area of research with important nanotechnological applications, including information storage, energy storage and chemical sensing. The extended AIRSS method for encapsulated nanowires (ENWs) was implemented and used to identify the structures formed by germanium telluride, silver chloride, and molybdenum diselenide ENWs. In each of these cases, a number of novel nanowire structures were identified, and a phase diagram predicting the ground state nanowire structure as a function of the radius of the encapsulating nanotube was calculated. In the case of germanium telluride, which is a technologically important phase-change material, the potential use of GeTe ENWs as switchable nanoscale memory devices was investigated. The vibrational properties of silver chloride ENWs were also considered, and a novel scheme was developed to predict the Raman spectra of systems which can be decomposed into multiple weakly interacting subsystems. This scheme was used to obtain a close approximation to the Raman spectra of AgCl ENWs at a fraction of the computational cost that would otherwise be necessary. The encapsulation of AgCl was shown to produce substantial shifts in the Raman spectra of nanotubes, providing an important link with experiment. read less NOT USED (low confidence) Y. Chen and A. Diaz, “Physical foundation and consistent formulation of atomic-level fluxes in transport processes,” Physical Review E. 2018. link Times cited: 26 Abstract: Irving and Kirkwood derived the transport equations from the… read moreAbstract: Irving and Kirkwood derived the transport equations from the principles of classical statistical mechanics using the Dirac delta to define local densities. Thereby, formulas for fluxes were obtained in terms of molecular variables. The Irving and Kirkwood formalism has inspired numerous formulations. Many of the later developments, however, considered it more rigorous to replace the Dirac delta with a continuous volume-weighted averaging function and subsequently defined fluxes as a volume density. Although these volume-averaged flux formulas have dominated the literature for decades and are widely implemented in popular molecular dynamics (MD) software, they are a departure from the well-established physical concept of fluxes. In this work, we review the historical developments that led to the unified physical concept of fluxes for transport phenomena. We then use MD simulations to show that these popular flux formulas conserve neither momentum nor energy; nor do they produce fluxes that are consistent with their physical definitions. We also use two different approaches to derive fluxes for general many-body potentials. The results of the formulation show that atomistic formulas for fluxes can be fully consistent with the physical definitions of fluxes and conservation laws. read less NOT USED (low confidence) C. Hu, V. Michaud-Rioux, W. Yao, and H. Guo, “Moiré Valleytronics: Realizing Dense Arrays of Topological Helical Channels.,” Physical review letters. 2018. link Times cited: 13 Abstract: We propose a general and robust platform, the moiré valleytr… read moreAbstract: We propose a general and robust platform, the moiré valleytronics, to realize high-density arrays of 1D topological helical channels in real materials at room temperature. We demonstrate the idea using a long-period 1D moiré pattern of graphene on hBN by first-principles calculation. Through calculating the Berry curvature and topological charge of the electronic structure associated with various local graphene/hBN stackings in the moiré pattern, it is revealed that the helical channel arrays originate intrinsically from the periodic modulation of the local topological orders by the moiré pattern. For a freestanding wavelike moiré pattern, two groups of helical channel arrays are spatially separated out of plane, validating the structural robustness of the moiré topology. The generality and experimental feasibility of moiré valleytronics are demonstrated by investigating a broad range of moiré systems. read less NOT USED (low confidence) Z. Liu, J. Li, C. Zhou, and W. Zhu, “A molecular dynamics study on thermal and rheological properties of BNNS-epoxy nanocomposites,” International Journal of Heat and Mass Transfer. 2018. link Times cited: 42 NOT USED (low confidence) X. Nie, L. Zhao, S. Deng, and Y. Zhang, “Molecular dynamic study on crossover of equilibrium time of conduction for silicon/silicon and silicon/silicon carbide pairs on nanoscale,” International Communications in Heat and Mass Transfer. 2018. link Times cited: 3 NOT USED (low confidence) V. Singla, A. Verma, and A. Parashar, “A molecular dynamics based study to estimate the point defects formation energies in graphene containing STW defects,” Materials Research Express. 2018. link Times cited: 21 Abstract: In the present article, molecular dynamics based simulations… read moreAbstract: In the present article, molecular dynamics based simulations have been performed to estimate the vacancy formation and displacement threshold energies in a defective graphene nanosheet. Pristine graphene is a hypothetical concept, as its synthesis often results in a nanosheet containing various geometrical and atomic defects such as grain boundaries and dislocations. Stone Thrower Wales, a type of defect that are either present in grain boundaries or generated through experimental means such as ion beam and electron beam irradiation techniques. The simulations performed in this investigation shall help in the characterization and determining suitability of defective graphene with STW defects for radiation shielding purposes and future space research. Moreover, this study will be valuable in bringing new insights for guiding and modifying the design of graphene-based nanomaterials exposed to radiation environments. read less NOT USED (low confidence) A. Jam, R. Abadi, M. Izadifar, and T. Rabczuk, “Molecular dynamics study on the mechanical properties of carbon doped single-layer polycrystalline boron-nitride nanosheets,” Computational Materials Science. 2018. link Times cited: 20 NOT USED (low confidence) A. Sharma, D. Datta, and R. Balasubramaniam, “Molecular dynamics simulation to investigate the orientation effects on nanoscale cutting of single crystal copper,” Computational Materials Science. 2018. link Times cited: 66 NOT USED (low confidence) J. Rabelo and L. Cândido, “Strong anharmonicity in pristine graphene,” Journal of Physics Communications. 2018. link Times cited: 1 Abstract: The thermodynamic coefficients of a free standing infinite g… read moreAbstract: The thermodynamic coefficients of a free standing infinite graphene monolayer are calculated using the quasi-classical unsymmetrized self-consistent field method (USF). The basic nonlinear integral equations of this theory are solved numerically in the strong anharmonic approximation. The isothermal and adiabatic elastic bulk moduli, the isochoric and isobaric heat capacities, the thermal expansion, thermal pressure coefficient, and the macroscopic Gruneisen parameter are calculated in terms of the derivatives of a specifically chosen interatomic potential function for different values of stress and for temperatures ranging from below room temperature up to the point of loss of thermodynamic stability. The nearest-neighbor distances vary from ≳1.4 Å to ≲1.8 Å for zero stress. Under stress, these distances decrease. At room temperature the molar heat capacities are ∼5.0 Jmol−1K−1. The elasticity moduli vary from 15.0 eV Å − 2 up to zero at the temperature of loss of stability and are increased by stress. The thermal expansion coefficient has a strong dependence on the temperature and is negative for temperatures lower than ∼340 K. For high temperatures it monotonically increases and decreases with stress. The macroscopic Gruneisen parameter has a strong nonlinear dependence with temperature and is estimated in about 3.0 ÷ 3.7 ; at ∼340 K its value decreases to ∼1.0 K and for even lower temperature it shows a peak and deep structure similar to what has been earlier reported for fullerene C60. read less NOT USED (low confidence) J. Amraei, J. E. Jam, B. Arab, and R. Firouz-Abadi, “Effect of interphase zone on the overall elastic properties of nanoparticle-reinforced polymer nanocomposites,” Journal of Composite Materials. 2018. link Times cited: 24 Abstract: In the current work, the effect of interphase region on the … read moreAbstract: In the current work, the effect of interphase region on the mechanical properties of polymer nanocomposites reinforced with nanoparticles is studied. For this purpose, a closed-form interphase model as a function of radial distance based on finite-size representative volume element is suggested to estimate the mechanical properties of particle-reinforced nanocomposites. The effective Young’s and shear moduli of thermoplastic polycarbonate-based nanocomposites for a wide range of sizes and volume fractions of silicon carbide nanoparticles are investigated using the proposed interphase model and molecular dynamics simulations. In order to investigate the effect of particle size, several unit cells of the same volume fraction, but with different particle radii have been considered. The micromechanics-based homogenization results are in good agreement with the results of molecular dynamics simulations for all models. This study demonstrates that the suggested micromechanical interphase model has the capacity to estimate effective mechanical properties of polymer-based nanocomposites reinforced with spherical inclusions. read less NOT USED (low confidence) A. Vakis et al., “Modeling and simulation in tribology across scales: An overview,” Tribology International. 2018. link Times cited: 351 NOT USED (low confidence) D. Hong, L. Liu, S. Zhang, and X. Guo, “Effect of cooling rate on the reaction of volatiles from low-rank coal pyrolysis: Molecular dynamics simulations using ReaxFF,” Fuel Processing Technology. 2018. link Times cited: 22 NOT USED (low confidence) H. Tian, X. Yang, G. Yang, and B. Zhang, “Instability of rapidly accelerating rupture fronts in nanostrips of monolayer hexagonal boron nitride,” Engineering Fracture Mechanics. 2018. link Times cited: 4 NOT USED (low confidence) C. Huang et al., “Effects of strain rate and annealing temperature on tensile properties of nanocrystalline diamond,” Carbon. 2018. link Times cited: 33 NOT USED (low confidence) Y. Luo, M. Lin, N. Zhou, H. Huang, C.-T. Tsai, and L. Zhou, “Molecular dynamics simulation study of the microstructure of a-Si:H thin film grown by oblique-angle deposition,” Physica B: Condensed Matter. 2018. link Times cited: 7 NOT USED (low confidence) H. Sadouki et al., “Prediction of half-metallicity in the NaS, NaSe and NaTe alkali-metal chalcogenides using first principles,” International Journal of Computational Materials Science and Engineering. 2018. link Times cited: 4 Abstract: First-principles full-potential linearized augmented plane-w… read moreAbstract: First-principles full-potential linearized augmented plane-wave method based on density functional theory is used to investigate the structural, electronic and magnetic properties of NaS, NaSe and NaTe alkali-metal chalcogenides binary compounds. These compounds in different crystalline phases: NaCl (B1), CsCl (B2), ZB (B3), NiAs (B81), WZ (B4) and Pnma were calculated within the generalized gradient approximation (GGA-PBE) and the modified Becke–Johnson approach (mBJ-GGA-PBE) for the exchange-correlation energy and potential. We found that the most stable phase for the NaX binary compounds is the nonmagnetic Pnma phase. The calculated lattice parameters, bulk moduli, their first-pressure derivatives and internal parameters are in good agreement with the other theoretical data. The electronic band structure and density of states show that half-metallic and magnetic character arises, which can be attributed to the presence of spin-polarized [Formula: see text] orbitals in the group VI elements. The NaS, NaSe and NaTe binary compounds show half-metallic character in ZB and WZ phases, with an integer magnetic moment of 1 [Formula: see text] per formula unit and half-metallic gaps. read less NOT USED (low confidence) S. Liu, M. Hao, and B.-S. Lee, “Student Cluster Competition 2017, team Nanyang Technological University: Reproducing vectorization of the Tersoff multi-body potential on the Intel Broadwell architecture,” Parallel Comput. 2018. link Times cited: 0 NOT USED (low confidence) R. Abadi, M. Izadifar, M. Sepahi, N. Alajlan, and T. Rabczuk, “Computational modeling of graphene nanopore for using in DNA sequencing devices,” Physica E: Low-dimensional Systems and Nanostructures. 2018. link Times cited: 8 NOT USED (low confidence) J. Harrison, J. Schall, S. Maskey, P. Mikulski, M. T. Knippenberg, and B. Morrow, “Review of force fields and intermolecular potentials used in atomistic computational materials research,” Applied Physics Reviews. 2018. link Times cited: 99 Abstract: Molecular simulation is a powerful computational tool for a … read moreAbstract: Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferability. When selecting a potential energy function for use in molecular simulations, the accuracy, transferability, and computational speed must all be considered. In this focused review, some of the more commonly used potential energy functions for molecular simulations are reviewed with an eye toward presenting their general forms, strengths, and weaknesses.Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferabilit... read less NOT USED (low confidence) Q. Zhang, Y. Zhao, X. Ma, Y. Zhao, and X. Pang, “Study on bonding mechanism of graphene on silicon substrate,” Modern Physics Letters B. 2018. link Times cited: 1 Abstract: The study of bonding mechanism is important for the applicat… read moreAbstract: The study of bonding mechanism is important for the application of graphene in micro-electro-mechanical system (MEMS). Using molecular dynamics (MD) simulation, the adhesion behavior of graphene on silicon substrate was observed, by applying a constant vertical upward exfoliation velocity to graphene. The effects of silicon substrate size, graphene layer number and exfoliation velocity on adhesion properties of graphene were studied. The minimum velocity to exfoliate monolayer graphene was 4.3 Å/ps, and the maximum adhesive force was 25.04 nN. For two-layer graphene, velocity was applied on the top layer, 5.2 Å/ps and 12 Å/ps were the critical velocities: when the velocity was no more than 5.2 Å/ps, the top layer cannot be exfoliated; as the velocity was in the range of 5.2–12 Å/ps, the second layer was driven upward together with the top layer, because of interlayer interaction between graphene layers; when the velocity increased greater than 12 Å/ps, the top layer graphene was broken through the bonding forces of substrate and the second layer, and exfoliated alone. It can be concluded that the velocity to exfoliate graphene was extremely high, and the adhesion energy was 299.81 mJ/m2 and 323.41 mJ/m2 for exfoliating monolayer and two-layer graphene respectively, thus the adhesive strength between graphene and silicon was very strong. read less NOT USED (low confidence) J. Yan and S. Lai, “Superelasticity and wrinkles controlled by twisting circular graphene,” Computer Methods in Applied Mechanics and Engineering. 2018. link Times cited: 21 NOT USED (low confidence) T. Sipkens and K. Daun, “Effect of Surface Interatomic Potential on Thermal Accommodation Coefficients Derived from Molecular Dynamics,” The Journal of Physical Chemistry C. 2018. link Times cited: 14 Abstract: This work investigates how the interatomic surface potential… read moreAbstract: This work investigates how the interatomic surface potential influences molecular dynamics (MD)-derived thermal accommodation coefficients (TACs). Iron, copper, and silicon surfaces are considered over a range of temperatures that include their melting points. Several classes of potentials are reviewed, including two-body, three-body, and bond-order force fields. MD-derived densities and visualization of the surfaces are used to explain the differences in the parameterizations of these potentials within the context of gas–surface scattering. Finally, TACs are predicted for a range of gas–surface combinations, and recommended values of the TAC are selected that take into account the robustness and uncertainties of each of the considered parameterizations. Further, it is observed that there is a significant change in the TAC about phase changes that must be taken into account for applications with a large range of surface temperatures. read less NOT USED (low confidence) L. Chen, H. Wang, F. Wang, D. Geng, J. Wu, and J. Lu, “Thermal Decomposition Mechanism of 2,2′,4,4′,6,6′-Hexanitrostilbene by ReaxFF Reactive Molecular Dynamics Simulations,” The Journal of Physical Chemistry C. 2018. link Times cited: 37 Abstract: 2,2′,4,4′,6,6′-Hexanitrostilbene (HNS) is an explosive with … read moreAbstract: 2,2′,4,4′,6,6′-Hexanitrostilbene (HNS) is an explosive with increased heat resistance, and its mechanism of thermal decomposition is of interest. In this paper, the decomposition processes of HNS at various temperatures (2500, 2750, 3000, 3250, and 3500 K) are calculated by large-scale reactive molecular dynamics simulations. The initial reactions and the evolution of clusters (whose molecular weight is larger than HNS) are analyzed. The reaction kinetics parameters are fitted. The results show that the main initial decomposition mechanisms of HNS are C–NO2 bond dissociation and nitro-nitrite (NO2–ONO) isomerization. During decomposition, O atoms are less likely to be released from the cluster than H and N atoms. Low temperatures tend to produce larger clusters, and clusters at higher temperatures tend to decompose. The thermal decomposition of HNS is a combination of single-molecule and bimolecular decomposition mechanisms. The dimerization reaction is clearly weakened, and the C–N bond cleavage is still... read less NOT USED (low confidence) X. Qin, W. Yan, X. Guo, T. Gao, and Q. Xie, “Molecular Dynamics Simulations of Si ion Substituted Graphene by Bombardment,” IOP Conference Series: Materials Science and Engineering. 2018. link Times cited: 1 Abstract: Molecular dynamics simulations with Tersoff-Ziegler-Biersack… read moreAbstract: Molecular dynamics simulations with Tersoff-Ziegler-Biersack-Littmark (Tersoff-ZBL) potential and adaptive intermolecular reactive empirical bond order (AIREBO) potential are performed to study the substitutional process of silicon ions by bombardment. The silicon ions bombardment of graphene is simulated at energies 100 eV, 100 eV, 68 eV and 67 eV, respectively. All silicon atoms are substitute for the relevant carbon atoms at these energies. And a perfect region of SiC structure in graphene sheet is observed, this approach can viewed as a new preparation of graphene-based SiC electronics in theory. read less NOT USED (low confidence) X. Li, A. Wang, and K.-R. Lee, “Comparison of empirical potentials for calculating structural properties of amorphous carbon films by molecular dynamics simulation,” Computational Materials Science. 2018. link Times cited: 30 NOT USED (low confidence) G. Lazzaroni and U. Stefanelli, “Chain-like ground states in three dimensions,” Transactions of Mathematics and Its Applications. 2018. link Times cited: 5 Abstract:
We investigate the minimization of configurational energie… read moreAbstract:
We investigate the minimization of configurational energies of Brenner type. These include two- and three-body interaction terms, which favor the alignment of first neighbors. In particular, such configurational energies arise in connection with the molecular-mechanical modeling of covalent$sp$-bonding in carbon. Ground states in three dimensions are characterized and the stability of chains and rings is discussed. The interaction energy is then augmented with terms corresponding to weaker interactions favoring the stratification of configurations. This gives rise to stratified structures, which are reminiscent of nanoscrolls and multi-wall nanotubes. Optimal stratified configurations are identified and their geometry is discussed. read less NOT USED (low confidence) S. Bazrafshan and A. Rajabpour, “Engineering of thermal transport in graphene using grain size, strain, nitrogen and boron doping; a multiscale modeling,” International Journal of Heat and Mass Transfer. 2018. link Times cited: 31 NOT USED (low confidence) K. Yin et al., “Generating Sub-nanometer Pores in Single-Layer MoS2 by Heavy-Ion Bombardment for Gas Separation: A Theoretical Perspective.,” ACS applied materials & interfaces. 2018. link Times cited: 30 Abstract: Single-layer molybdenum disulfide (MoS2) filters with nanome… read moreAbstract: Single-layer molybdenum disulfide (MoS2) filters with nanometer-size pores have attracted great attention recently due to their promising performance for membrane separation. Generating nanopores in MoS2 controllably, however, is still a challenging task, which greatly limits the real application of MoS2 filters. In this work, the pore forming process in single-layer MoS2 by heavy-ion bombardment was investigated in detail using molecular dynamics simulations. We found that pores with sub-nanometer size (0.6-1.2 nm) can be created in the MoS2 sheet by single-ion bombardment, with a probability as high as 0.8 pores per incident ion. The size and shape of the nanopore can be tuned controllably by adjusting bombardment parameters. Furthermore, the performance of the MoS2 filter with these sub-nanometer-size pores for separation of He, Ne, H2, Ar, and Kr gases was evaluated by density functional theory-based first-principles calculations. The MoS2 filter was found to show much higher selectivity for separating H2/He and He/Ne than that reported for graphene and other membranes. Such high selectivity was attributed to the interaction between gases and the charged edge of pores in MoS2. Our results suggest the potential application of ion beam technology in single-layer MoS2 for membrane separation. read less NOT USED (low confidence) M. Sadat, K. Muralidharan, and L. Zhang, “Reactive molecular dynamics simulation of the mechanical behavior of sodium aluminosilicate geopolymer and calcium silicate hydrate composites,” Computational Materials Science. 2018. link Times cited: 31 NOT USED (low confidence) T. Nguyen, K. Sato, and Y. Shibutani, “Development of Fe-C interatomic potential for carbon impurities in α-iron,” Computational Materials Science. 2018. link Times cited: 10 NOT USED (low confidence) X. Yuan and Y. Wang, “Radial deformation of single-walled carbon nanotubes adhered to solid substrates and variations of energy: Atomistic simulations and continuum analysis,” International Journal of Solids and Structures. 2018. link Times cited: 8 NOT USED (low confidence) X. Duan, B. He, M. Guo, Z. Liu, Y. Wen, and B. Shan, “Lattice inversion modified embedded atom method for FCC metals,” Computational Materials Science. 2018. link Times cited: 8 NOT USED (low confidence) M. A. Lively, S. X. Bennett, and J. Allain, “Molecular dynamics studies of ion beam implantation and patterning of silicon: Effect of noble gas cluster formation,” Physical Review B. 2018. link Times cited: 7 NOT USED (low confidence) A. Sgouros, G. Kalosakas, G. Kalosakas, K. Papagelis, and C. Galiotis, “Compressive response and buckling of graphene nanoribbons,” Scientific Reports. 2018. link Times cited: 22 NOT USED (low confidence) G. Shchygol, A. Yakovlev, T. Trnka, A. C. T. van Duin, and T. Verstraelen, “Systematic Comparison of Monte Carlo Annealing and Covariance Matrix Adaptation for the Optimization of ReaxFF Parameters.” 2018. link Times cited: 3 Abstract: ReaxFF is a computationally efficient force field to simulat… read moreAbstract: ReaxFF is a computationally efficient force field to simulate complex reactive dynamics in extended molecular models with diverse chemistries, if reliable force-field parameters are available for the chemistry of interest. If not, they must be calibrated by minimizing the error ReaxFF makes on a relevant training set. Because this optimization is far from trivial, many methods, in particular genetic algorithms (GAs), have been developed to search for the global optimum in parameter space. Recently, two alternative parameter calibration techniques were proposed, i.e.\ Monte-Carlo Force Field optimizer (MCFF) and Covariance Matrix Adaptation Evolutionary Strategy (CMA-ES), which have the potential to find good parameters at a relatively low computational cost. In this work, these two methods are tested, as implemented in ADF2018, using three ReaxFF training sets, which have previously been used to benchmark GAs. Even though MCFF and CMA-ES should not be considered as exhaustive global optimizers, they can find parameters that are comparable in quality to those obtained with GAs. We observe that CMA-ES leads to slightly better results and is less sensitive to the initial guess of the parameters. Concrete recipes are provided for obtaining similar results with new training sets.Besides optimization recipes, a successful ReaxFF parameterization requires the design of a good training set. At every trial set of parameters, ReaxFF is used to optimize molecular geometries in the training set. When the optimization of some geometries fails easily, it becomes increasingly difficult to find the optimal parameters. We have addressed this issue by fixing several bugs in the ReaxFF forces and by improving the robustness of the geometry optimization. These improvements cannot eliminate all geometry convergence issues and we recommend to avoid very flexible geometries in the training set.Both MCFF and CMA-ES are still liable to converge to sub- or near-optimal parameters, which we detected by repeating the calibration with different random seeds. The existence of distinct near-optimal parameter vectors is a general pattern throughout our study and provides opportunities to improve the training set or to detect overfitting artifacts. read less NOT USED (low confidence) S. Singh and B. P. Patel, “Effect of initial strain and material nonlinearity on the nonlinear static and dynamic response of graphene sheets,” Journal of Sound and Vibration. 2018. link Times cited: 8 NOT USED (low confidence) G. Zhu, J. Sun, L. Zhang, and Z. Gan, “Molecular dynamics simulation of temperature effects on deposition of Cu film on Si by magnetron sputtering,” Journal of Crystal Growth. 2018. link Times cited: 20 NOT USED (low confidence) Z. Cong and S. Lee, “Study of mechanical behavior of BNNT-reinforced aluminum composites using molecular dynamics simulations,” Composite Structures. 2018. link Times cited: 43 NOT USED (low confidence) F. Dias and W. S. Machado, “The effects of computational time parameter in the thermal conductivity of single-walled carbon nanotubes by molecular dynamics simulation,” Computational Condensed Matter. 2018. link Times cited: 4 NOT USED (low confidence) A. Dasmahapatra and P. Kroll, “Modeling amorphous silicon nitride: A comparative study of empirical potentials,” Computational Materials Science. 2018. link Times cited: 12 NOT USED (low confidence) F. Krause, D. Bredemeier, M. Schowalter, T. Mehrtens, T. Grieb, and A. Rosenauer, “Using molecular dynamics for multislice TEM simulation of thermal diffuse scattering in AlGaN.,” Ultramicroscopy. 2018. link Times cited: 13 NOT USED (low confidence) S. Singh and B. P. Patel, “Mathematical Treatise to Model Dihedral Energy in the Multiscale Modeling of Two-Dimensional Nanomaterials,” Journal of Applied Mechanics. 2018. link Times cited: 3 NOT USED (low confidence) P. Gong, Z. Ye, L. Yuan, and P. Egberts, “Evaluation of wetting transparency and surface energy of pristine and aged graphene through nanoscale friction,” Carbon. 2018. link Times cited: 31 NOT USED (low confidence) C. Huang et al., “Anisotropy effects in diamond under nanoindentation,” Carbon. 2018. link Times cited: 46 NOT USED (low confidence) A. Kucherik, A. Osipov, S. Arakelian, S. V. Garnov, A. V. Povolotckaya, and S. Kutrovskaya, “The laser-assisted synthesis of linear carbon chains stabilized by noble metal particle,” Journal of Physics: Conference Series. 2018. link Times cited: 7 Abstract: The problem to induce the Long Linear Carbon Chain (LLCC)/ca… read moreAbstract: The problem to induce the Long Linear Carbon Chain (LLCC)/carbyne is very principal in both physics and applications, especially to control the electrophysical properties due to high polarizability. General approach to fabricate the LLCC is based on the concept to compensate the process of instability for linear chain (due to form of globules and bundles) by some external interaction. In this paper we considered features of laser experiments for the problem. Laser-induced LLCC in colloidal system with carbon nanoparticles has been obtained by us in two cases: with shungite target in liquid and laser synthesis of metal complexes with carbyne. Some possible applications in nanophotonics are discussed in respect of high/hopping electroconductivity. read less NOT USED (low confidence) M. Dewapriya, S. Meguid, and R. Rajapakse, “Atomistic modelling of crack-inclusion interaction in graphene,” Engineering Fracture Mechanics. 2018. link Times cited: 13 NOT USED (low confidence) K. Yashiro, “Local lattice instability analysis on mode I crack tip in β-SiC: Characteristics in binary covalent crystal,” Computational Materials Science. 2018. link Times cited: 6 NOT USED (low confidence) J. Sarkar and D. K. Das, “Nanoindentation study of mechanical behavior and response of a single layer pristine silicene sheet using molecular dynamics simulations,” Computational Materials Science. 2018. link Times cited: 14 NOT USED (low confidence) R. Dongol, L. Wang, A. Cormack, and S. Sundaram, “Molecular dynamics simulation of sodium aluminosilicate glass structures and glass surface-water reactions using the reactive force field (ReaxFF),” Applied Surface Science. 2018. link Times cited: 36 NOT USED (low confidence) S. Ajori, R. Ansari, and F. Sadeghi, “Molecular dynamics study of gigahertz nanomechanical oscillators based on an ion inside a series of electrically charged carbon nanotubes,” European Journal of Mechanics A-solids. 2018. link Times cited: 18 NOT USED (low confidence) M. Izadifar, R. Abadi, A. N. Shirazi, N. Alajlan, and T. Rabczuk, “Nanopores creation in boron and nitrogen doped polycrystalline graphene: A molecular dynamics study,” Physica E-low-dimensional Systems & Nanostructures. 2018. link Times cited: 13 NOT USED (low confidence) Y. Guo, H.-F. Liu, H.-L. Zeng, X. Yan, and X.-C. Song, “Edge defect switched dual spin filter in zigzag hexagonal boron nitride nanoribbons.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 10 Abstract: Unlike graphene nanoribbons, zigzag monolayer hexagonal boro… read moreAbstract: Unlike graphene nanoribbons, zigzag monolayer hexagonal boron nitride nanoribbons (ZBNNRs) possess two distinct edges (B and N edges). Using first-principles calculations, we investigate the spin-dependent electronic transport of ZBNNRs with edge defects. It is found that the defects could make the system operate as a dual spin filter, where the direction of spin polarization is switched by the defect. Further analysis shows that the transmission eigenchannels for the opposite spins reside spatially separated on opposite edges. The defect on one edge could suppress the transmission for only one spin component, but preserve that for the other spin, resulting in a dual spin filter effect. This effect is found to be unaffected by the width of the ribbon and the length of the defect. Moreover, by constructing defects on both edges, the system exhibits two transmission peaks with opposite spins residing discretely on both sides of the Fermi level, suggesting that an electrically controlled dual spin filter based on ZBNNRs is also realizable. As controllable defects have been experimentally fabricated on monolayer boron nitride [T. Pham, A. L. Gibb, Z. Li, S. M. Gilbert, C. Song, S. G. Louie and A. Zettl, Nano Lett., 2016, 16, 7142-7147], our results may shed light on the development of B/N-based spintronic devices. read less NOT USED (low confidence) J. Yeo et al., “Multiscale modeling of keratin, collagen, elastin and related human diseases: Perspectives from atomistic to coarse-grained molecular dynamics simulations.,” Extreme Mechanics Letters. 2018. link Times cited: 30 NOT USED (low confidence) Y. Tuo et al., “Insight into the support effect on the particle size effect of Pt/C catalysts in dehydrogenation,” Journal of Catalysis. 2018. link Times cited: 63 NOT USED (low confidence) R. Abadi, A. N. Shirazi, M. Izadifar, M. Sepahi, and T. Rabczuk, “Fabrication of nanopores in polycrystalline boron-nitride nanosheet by using Si, SiC and diamond clusters bombardment,” Computational Materials Science. 2018. link Times cited: 16 NOT USED (low confidence) S. Singh and B. P. Patel, “Nonlinear elastic properties of graphene sheet using MM3 potential under finite deformation,” Composites Part B-engineering. 2018. link Times cited: 16 NOT USED (low confidence) Z. Rashid, L. Zhu, and W. Li, “Effect of confinement on anharmonic phonon scattering and thermal conductivity in pristine silicon nanowires,” Physical Review B. 2018. link Times cited: 7 NOT USED (low confidence) S. Riedel and S. G. Mayr, “Reagent-Free Programming of Shape-Memory Behavior in Gelatin by Electron Beams: Experiments and Modeling,” Physical review applied. 2018. link Times cited: 7 NOT USED (low confidence) A. Shahabodini, R. Ansari, and M. Darvizeh, “Atomistic-continuum modeling of vibrational behavior of carbon nanotubes using the variational differential quadrature method,” Composite Structures. 2018. link Times cited: 16 NOT USED (low confidence) A. Raj and J. Eapen, “Computing Phonon Dispersion using Fast Zero-Point Correlations of Conjugate Variables,” MRS Advances. 2018. link Times cited: 2 Abstract: Time correlations of dynamic variables in the reciprocal spa… read moreAbstract: Time correlations of dynamic variables in the reciprocal space offer a rich theoretical setting for computing the phonon dispersion curves, particularly for systems with marked anharmonic interactions. Present techniques primarily rely either on the equipartition of energy between the phonon modes or on the oscillation of the time correlation of the normal mode projections. The former can lead to numerical errors due to deviation from equipartition while the latter usually requires long simulations for computing the time correlations. We investigate a different approach using the ratio of the normal mode expectation value of two conjugate variables – velocity and acceleration. Since only the correlations at the initial time (t=0) are needed, this approach is computationally attractive. In this work, we employ this method to extract the full Brillouin zone phonon dispersion for graphene. read less NOT USED (low confidence) O. Matsiaka, C. J. Penington, R. Baker, and M. Simpson, “Discrete and Continuum Approximations for Collective Cell Migration in a Scratch Assay with Cell Size Dynamics,” Bulletin of Mathematical Biology. 2018. link Times cited: 8 NOT USED (low confidence) S. Singh and B. P. Patel, “A computationally efficient multiscale finite element formulation for dynamic and postbuckling analyses of carbon nanotubes,” Computers & Structures. 2018. link Times cited: 16 NOT USED (low confidence) C. Huang et al., “Molecular dynamics simulations for responses of nanotwinned diamond films under nanoindentation,” Ceramics International. 2017. link Times cited: 40 NOT USED (low confidence) R. S. Okatiev and I. Zubko, “Exact expressions for the bending rigidity and elastic moduli of a graphene sheet derived with the geometric potential of carbon.” 2017. link Times cited: 0 Abstract: In some cases, the classical interatomic interaction potenti… read moreAbstract: In some cases, the classical interatomic interaction potentials allow quite an accurate estimation of the elastic moduli of a graphene sheet in its plane. However, they often provide lowered values of the bending rigidity. For a correct prediction of the latter, it is necessary to take into account the response of the covalent bond of carbon atoms in graphene to lattice bending. A new potential considering for the geometric structure of the sp2-hybridized carbon electron shell and containing an independent energy parameter (responsible only for bond bending) is built using invariants of vector triads which set up directions of carbon atom covalent bonds in graphene. The obtained potential gives realistic values of the graphene bending rigidity. Exact expressions have been derived in the form of finite sums for the computation of the bending rigidity and elastic moduli of the graphene sheet. The equations contain parameters of the suggested potential and coordinates of carbon atoms in a reference equilibri... read less NOT USED (low confidence) M. Dewapriya and S. Meguid, “Atomistic simulations of nanoscale crack-vacancy interaction in graphene,” Carbon. 2017. link Times cited: 24 NOT USED (low confidence) N. H. Giang, T. T. Hanh, L. N. Ngoc, N. T. Nga, and V. V. Hoang, “Formation of graphene on BN substrate by vapor deposition method and size effects on its structure,” Physica B-condensed Matter. 2017. link Times cited: 4 NOT USED (low confidence) X. Liu et al., “Toward the multiscale nature of stress corrosion cracking,” Nuclear Engineering and Technology. 2017. link Times cited: 18 NOT USED (low confidence) H. Xiang, H. Li, and X. Peng, “Comparison of different interatomic potentials for MD simulations of AlN,” Computational Materials Science. 2017. link Times cited: 21 NOT USED (low confidence) L. Nan, N. Ding, S. Qu, L. Liu, W. Guo, and C. M. L. Wu, “Mechanical properties and failure behavior of hexagonal boron nitride sheets with nano-cracks,” Computational Materials Science. 2017. link Times cited: 32 NOT USED (low confidence) J. Yan, L. Tong, and P. Xiang, “Free vibration analysis of single-walled boron nitride nanotubes based on a computational mechanics framework,” Superlattices and Microstructures. 2017. link Times cited: 6 NOT USED (low confidence) W. Ge et al., “Discrete simulation of granular and particle-fluid flows: from fundamental study to engineering application,” Reviews in Chemical Engineering. 2017. link Times cited: 82 Abstract: Multiphase chemical reactors with characteristic multiscale … read moreAbstract: Multiphase chemical reactors with characteristic multiscale structures are intrinsically discrete at the elemental scale. However, due to the lack of multiscale models and the limitation of computational capability, such reactors are traditionally treated as continua through straightforward averaging in engineering simulations or as completely discrete systems in theoretical studies. The continuum approach is advantageous in terms of the scale and speed of computation but does not always give good predictions, which is, in many cases, the strength of the discrete approach. On the other hand, however, the discrete approach is too computationally expensive for engineering applications. Developments in computing science and technologies and encouraging progress in multiscale modeling have enabled discrete simulations to be extended to engineering systems and represent a promising approach to virtual process engineering (VPE, or virtual reality in process engineering). In this review, we analyze this emerging trend and emphasize that multiscale discrete simulations (MSDS), that is, considering multiscale structures in discrete simulation through rational coarse-graining and coupling between discrete and continuum methods with the effect of mesoscale structures accounted in both cases, is an effective way forward, which can be complementary to the continuum approach that is being improved by multiscale modeling also. For this purpose, our review is not meant to be a complete summary to the literature on discrete simulation, but rather a demonstration of its feasibility for engineering applications. We therefore discuss the enabling methods and technologies for MSDS, taking granular and particle-fluid flows as typical examples in chemical engineering. We cover the spectrum of modeling, numerical methods, algorithms, software implementation and even hardware-software codesign. The structural consistency among these aspects is shown to be the pivot for the success of MSDS. We conclude that with these developments, MSDS could soon become, among others, a mainstream simulation approach in chemical engineering which enables VPE. read less NOT USED (low confidence) O. Matsiaka, C. J. Penington, R. Baker, and M. Simpson, “Discrete and Continuum Approximations for Collective Cell Migration in a Scratch Assay with Cell Size Dynamics,” Bulletin of Mathematical Biology. 2017. link Times cited: 17 Abstract: Scratch assays are routinely used to study the collective sp… read moreAbstract: Scratch assays are routinely used to study the collective spreading of cell populations. In general, the rate at which a population of cells spreads is driven by the combined effects of cell migration and proliferation. To examine the effects of cell migration separately from the effects of cell proliferation, scratch assays are often performed after treating the cells with a drug that inhibits proliferation. Mitomycin-C is a drug that is commonly used to suppress cell proliferation in this context. However, in addition to suppressing cell proliferation, mitomycin-C also causes cells to change size during the experiment, as each cell in the population approximately doubles in size as a result of treatment. Therefore, to describe a scratch assay that incorporates the effects of cell-to-cell crowding, cell-to-cell adhesion, and dynamic changes in cell size, we present a new stochastic model that incorporates these mechanisms. Our agent-based stochastic model takes the form of a system of Langevin equations that is the system of stochastic differential equations governing the evolution of the population of agents. We incorporate a time-dependent interaction force that is used to mimic the dynamic increase in size of the agents. To provide a mathematical description of the average behaviour of the stochastic model we present continuum limit descriptions using both a standard mean-field approximation and a more sophisticated moment dynamics approximation that accounts for the density of agents and density of pairs of agents in the stochastic model. Comparing the accuracy of the two continuum descriptions for a typical scratch assay geometry shows that the incorporation of agent growth in the system is associated with a decrease in accuracy of the standard mean-field description. In contrast, the moment dynamics description provides a more accurate prediction of the evolution of the scratch assay when the increase in size of individual agents is included in the model. read less NOT USED (low confidence) S. Thomas, K. Ajith, and M. C. Valsakumar, “Effect of ripples on the finite temperature elastic properties of hexagonal boron nitride using strain-fluctuation method,” Superlattices and Microstructures. 2017. link Times cited: 10 NOT USED (low confidence) P. Zhang, R. Zhu, M. Jiang, Y. Song, D. Zhang, and Y. Cui, “Size effect caused significant reduction of thermal conductivity of GaAs/AlAs distributed Bragg reflector used in semiconductor disk laser,” Optics and Laser Technology. 2017. link Times cited: 4 NOT USED (low confidence) M. A. Lively, B. Holybee, M. Y. Toriyama, and J. Allain, “Massive-scale molecular dynamics of ion-irradiated III–V compound semiconductors at the onset of nanopatterning,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 1 NOT USED (low confidence) M. Izadifar, R. Abadi, A. Jam, and T. Rabczuk, “Investigation into the effect of doping of boron and nitrogen atoms in the mechanical properties of single-layer polycrystalline graphene,” Computational Materials Science. 2017. link Times cited: 25 NOT USED (low confidence) A. Giri and P. Hopkins, “Role of interfacial mode coupling of optical phonons on thermal boundary conductance,” Scientific Reports. 2017. link Times cited: 15 NOT USED (low confidence) Y. Luo, X. Sui, Y. He, H. Huang, N. Zhou, and L. Zhou, “The influence of annealing temperature upon the structure of a-Si:H/c-Si thin films,” Journal of Non-crystalline Solids. 2017. link Times cited: 10 NOT USED (low confidence) S. Bazrafshan and A. Rajabpour, “Thermal transport engineering in amorphous graphene: Non-equilibrium molecular dynamics study,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 44 NOT USED (low confidence) S. F. Ferdous and A. Adnan, “Mode-I Fracture Toughness Prediction of Diamond at the Nanoscale,” Journal of Nanomechanics and Micromechanics. 2017. link Times cited: 10 Abstract: AbstractIn this paper, the fracture process of nanoscale dia… read moreAbstract: AbstractIn this paper, the fracture process of nanoscale diamond is analyzed using atomistic simulations and fracture toughness obtained using four different continuum fracture-mechanics theories. ... read less NOT USED (low confidence) S. Zhang et al., “Computational and Experimental Studies on Novel Materials for Fission Gas Capture.” 2017. link Times cited: 1 NOT USED (low confidence) C. Lin, X. Zhang, and Z. Rao, “Theoretical prediction of thermal transport in BC2N monolayer,” Nano Energy. 2017. link Times cited: 39 NOT USED (low confidence) M. Faruq, A. Villesuzanne, M. Guo, and G. Shao, “Structure, Melting and Transport Properties of Binary Liquid Pd-Si Metal Alloys: Molecular Dynamics Simulations.” 2017. link Times cited: 4 Abstract: Quantum Sutton-Chen (Q-SC) potentials for molecular dynamic … read moreAbstract: Quantum Sutton-Chen (Q-SC) potentials for molecular dynamic (MD) simulation were derived for the Pd-Si system, which were then used to obtain an atomistic description of melting and transport properties for palladium metal, metallic silicon and their alloys. Melting and structural properties were investigated by analysing the radial distribution function, enthalpy, density, and diffusion coefficient as a function of temperature. The agreement between the MD/Q-SC results and experimental values for the estimated melting points and structural properties was excellent for both pure elements: Pd and metallic Si, while melting of alloys was shown to be complicated by chemical association between the interacting constituents, which led to difficulty in the dissociation of long-range order and thus significant overshoot in calculated melting points owing to high heating rate for achievable MD execution. read less NOT USED (low confidence) X. Wang, X. Wang, H. Qi, X. Song, and R. Li, “A study on elastic properties of carbon nanocones based on a temperature-related quasi-continuum model,” Fullerenes, Nanotubes and Carbon Nanostructures. 2017. link Times cited: 1 Abstract: ABSTRACT In the present paper, general analytical formulas f… read moreAbstract: ABSTRACT In the present paper, general analytical formulas for calculating the Young's modulus and Poisson's ratio of single-walled carbon nanocones (SWCNCs) at finite temperatures are derived based on the proposed temperature-related multiscale quasi-continuum (QC) model. To this end, a temperature-related higher-order Cauchy-Born (THCB) rule is employed to establish the hyper-elastic constitutive model of SWCNCs. With use of the proposed approach, the influences of the temperature, apex angle, rotation angle of cutting lines and top end radius on the Young's moduli and Poisson's ratios of SWCNCs are investigated systematically. read less NOT USED (low confidence) J. Liu and X. Guo, “ReaxFF molecular dynamics simulation of pyrolysis and combustion of pyridine,” Fuel Processing Technology. 2017. link Times cited: 65 NOT USED (low confidence) S. Thomas, K. Ajith, and M. C. Valsakumar, “Empirical potential influence and effect of temperature on the mechanical properties of pristine and defective hexagonal boron nitride,” Materials Research Express. 2017. link Times cited: 11 Abstract: The major objective of this work is to present results of a … read moreAbstract: The major objective of this work is to present results of a classical molecular dynamics study to investigate the effect of changing the cut-off distance in the empirical potential on the stress–strain relation and also the temperature dependent Young’s modulus of pristine and defective hexagonal boron nitride. As the temperature increases, the computed Young’s modulus shows a significant decrease along both the armchair and zigzag directions. The computed Young’s modulus shows a trend in keeping with the structural anisotropy of h-BN. The variation of Young’s modulus with system size is elucidated. The observed mechanical strength of h-BN is significantly affected by the vacancy and Stone–Wales type defects. The computed room temperature Young’s modulus of pristine h-BN is 755 GPa and 769 GPa respectively along the armchair and zigzag directions. The decrease of Young’s modulus with increase in temperature has been analyzed and the results show that the system with zigzag edge shows a higher value of Young’s modulus in comparison to that with armchair edge. As the temperature increases, the computed stiffness decreases and the system with zigzag edge possesses a higher value of stiffness as compared to the armchair counterpart and this behaviour is consistent with the variation of Young’s modulus. The defect analysis shows that presence of vacancy type defects leads to a higher Young’s modulus, in the studied range with different percentage of defect concentration, in comparison with Stone–Wales defect. The variations in the peak position of the computed radial distribution function reveals the changes in the structural features of systems with zigzag and armchair edges in the presence of applied stress. read less NOT USED (low confidence) E. Lotfi and M. Neek‐Amal, “Temperature distribution in graphene doped with nitrogen and graphene with grain boundary.,” Journal of molecular graphics & modelling. 2017. link Times cited: 12 NOT USED (low confidence) Z. Bažant and J. Le, “Probabilistic Mechanics of Quasibrittle Structures: Strength, Lifetime, and Size Effect.” 2017. link Times cited: 76 NOT USED (low confidence) P. Zhang, M. Jiang, R. Zhue, D. Zhang, and Y. Song, “Thermal conductivity of GaAs/AlAs distributed Bragg reflectors in semiconductor disk laser: comparison of molecular dynamics simulation and analytic methods.,” Applied optics. 2017. link Times cited: 5 Abstract: GaAs/AlAs distributed Bragg reflectors (DBRs) are widely use… read moreAbstract: GaAs/AlAs distributed Bragg reflectors (DBRs) are widely used in the gain chips of 1 μm wave band semiconductor disk lasers (SDLs) as an end/folded cavity mirror. Because the generated redundant heat in the active region of a gain chip mainly dissipates through the DBR, thermal conductivities of DBRs are crucial for the output performance of SDLs. For the purpose of more reasonable semiconductor wafer design, to improve the thermal management of SDLs, accurate thermal conductivities of DBRs with various layer thicknesses are under considerable requirement. By the use of the equilibrium molecular dynamics (EMD) simulation and the Tersoff potential, thermal conductivities of GaAs/AlAs superlattices with different layer thickness are calculated, and computed results are compared with reported data to verify the validity of the EMD simulation. The computed thermal conductivities of GaAs/AlAs DBRs using the EMD method show significant reduction in contrast to the bulk value. Compared to EMD simulation, analytic methods result in smaller values of thermal conductivities and get close to the bulk value much more slowly with increasing layer thickness. In the layer thickness of interest (60-100 nm), the Matthiessen rule with α=1 for GaAs and α=0.5 for AlAs is a practicable tool for thermal conductivity estimation. read less NOT USED (low confidence) F. Memarian, A. Fereidoon, S. Khodaei, A. H. Mashhadzadeh, and M. Ganji, “Molecular dynamic study of mechanical properties of single/double wall SiCNTs: Consideration temperature, diameter and interlayer distance,” Vacuum. 2017. link Times cited: 27 NOT USED (low confidence) J. Zhang, J. Zhang, Z. Wang, A. Hartmaier, Y. Yan, and T. Sun, “Interaction between phase transformations and dislocations at incipient plasticity of monocrystalline silicon under nanoindentation,” Computational Materials Science. 2017. link Times cited: 37 NOT USED (low confidence) B. Mortazavi, A. Lherbier, Z. Fan, A. Harju, T. Rabczuk, and J. Charlier, “Thermal and electronic transport characteristics of highly stretchable graphene kirigami.,” Nanoscale. 2017. link Times cited: 23 Abstract: For centuries, cutting and folding papers with special patte… read moreAbstract: For centuries, cutting and folding papers with special patterns have been used to build beautiful, flexible and complex three-dimensional structures. Inspired by the old idea of kirigami (paper cutting), and the outstanding properties of graphene, recently graphene kirigami structures were fabricated to enhance the stretchability of graphene. However, the possibility of further tuning the electronic and thermal transport along the 2D kirigami structures has remained original to investigate. We therefore performed extensive atomistic simulations to explore the electronic, heat and load transfer along various graphene kirigami structures. The mechanical response and thermal transport were explored using classical molecular dynamics simulations. We then used a real-space Kubo-Greenwood formalism to investigate the charge transport characteristics in graphene kirigami. Our results reveal that graphene kirigami structures present highly anisotropic thermal and electrical transport. Interestingly, we show the possibility of tuning the thermal conductivity of graphene by four orders of magnitude. Moreover, we discuss the engineering of kirigami patterns to further enhance their stretchability by more than 10 times as compared with pristine graphene. Our study not only provides a general understanding concerning the engineering of electronic, thermal and mechanical response of graphene, but more importantly can also be useful to guide future studies with respect to the synthesis of other 2D material kirigami structures, to reach highly flexible and stretchable nanostructures with finely tunable electronic and thermal properties. read less NOT USED (low confidence) M. Wood, M. Cherukara, E. Antillon, and A. Strachan, “Molecular Dynamics Simulations of Shock Loading of Materials: A Review and Tutorial.” 2017. link Times cited: 14 NOT USED (low confidence) V. Kuryliuk and O. Korotchenkov, “Atomistic simulation of the thermal conductivity in amorphous SiO 2 matrix/Ge nanocrystal composites,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 5 NOT USED (low confidence) A. Shahabodini, R. Ansari, and M. Darvizeh, “Multiscale modeling of embedded graphene sheets based on the higher-order Cauchy-Born rule: Nonlinear static analysis,” Composite Structures. 2017. link Times cited: 23 NOT USED (low confidence) Z.-L. Liu, R. Li, X.-L. Zhang, N. Qu, and L. Cai, “Direct anharmonic correction method by molecular dynamics,” Comput. Phys. Commun. 2017. link Times cited: 3 NOT USED (low confidence) O. Matsiaka, C. J. Penington, R. Baker, and M. Simpson, “Continuum approximations for lattice-free multi-species models of collective cell migration,” bioRxiv. 2017. link Times cited: 0 Abstract: Cell migration within tissues involves the interaction of ma… read moreAbstract: Cell migration within tissues involves the interaction of many cells from distinct subpopulations. In this work, we present a discrete model of collective cell migration where the motion of individual cells is driven by random forces, short range repulsion forces to mimic crowding, and longer range attraction forces to mimic adhesion. This discrete model can be used to simulate a population of cells that is composed of K ≥ 1 distinct subpopulations. To analyse the discrete model we formulate a hierarchy of moment equations that describe the spatial evolution of the density of agents, pairs of agents, triplets of agents, and so forth. To solve the hierarchy of moment equations we introduce two forms of closure: (i) the mean field approximation, which effectively assumes that the distributions of individual agents are independent; and (ii) a moment dynamics description that is based on the Kirkwood superposition approximation. The moment dynamics description provides an approximate way of incorporating spatial patterns, such as agent clustering, into the continuum description. Comparing the performance of the two continuum descriptions confirms that both perform well when adhesive forces are sufficiently weak. In contrast, the moment dynamics description outperforms the mean field model when adhesive forces are sufficiently large. This is a first attempt to provide an accurate continuum description of a lattice-free, multi-species model of collective cell migration. read less NOT USED (low confidence) A. Lazutin, A. A. Glagoleva, V. Vasilevskaya, and A. Khokhlov, “Computer synthesis of hypercrosslinked polystyrene: All-atom simulations,” Low Temperature Physics. 2017. link Times cited: 1 Abstract: For the first time a special force field ReaxFF is used to d… read moreAbstract: For the first time a special force field ReaxFF is used to describe the synthesis of polymer networks and for all-atom simulations of intermolecular cross linking in polystyrene. The density, specific surface, and coefficient of thermal expansion for sample networks with different degrees of crosslinking are calculated in the all-atom model. The results are in agreement with experimental data. read less NOT USED (low confidence) X. Wu, H. Zhao, D. Yan, and J. Pei, “Doping of graphene using ion beam irradiation and the atomic mechanism,” Computational Materials Science. 2017. link Times cited: 18 NOT USED (low confidence) R. Ansari, F. Sadeghi, and S. Ajori, “Oscillation characteristics of carbon nanotori molecules along carbon nanotubes under various system parameters,” European Journal of Mechanics A-solids. 2017. link Times cited: 14 NOT USED (low confidence) S. J. Mahdizadeh and G. Akhlamadi, “Optimized Tersoff empirical potential for germanene.,” Journal of molecular graphics & modelling. 2017. link Times cited: 21 NOT USED (low confidence) S. Goel, S. Chavoshi, and A. Murphy, “Molecular dynamics simulation (MDS) to study nanoscale machining processes.” 2017. link Times cited: 2 Abstract: 1 Molecular dynamics simulation (MDS) to study nanoscale cut… read moreAbstract: 1 Molecular dynamics simulation (MDS) to study nanoscale cutting processes Saurav Goel1*, Saeed Zare Chavoshi2 and Adrian Murphy3 1Precision Engineering Institute, School of Aerospace, Transport and Manufacturing, Cranfield University, Cranfield, Bedfordshire, MK430AL, UK 2Mechanical Engineering Department, Imperial College London, London, SW7 2AZ, UK 3School of Mechanical and Aerospace Engineering, Queen’s University, Belfast, BT9 5AH, UK *Corresponding author Tel.: +44 1234754132, Email address: sgoel.diamond@gmail.com read less NOT USED (low confidence) T. Rieger, T. Riedl, E. Neumann, D. Grützmacher, J. Lindner, and A. Pawlis, “Strain Compensation in Single ZnSe/CdSe Quantum Wells: Analytical Model and Experimental Evidence.,” ACS applied materials & interfaces. 2017. link Times cited: 3 Abstract: The lattice mismatch between CdSe and ZnSe is known to limit… read moreAbstract: The lattice mismatch between CdSe and ZnSe is known to limit the thickness of ZnSe/CdSe quantum wells on GaAs (001) substrates to about 2-3 monolayers. We demonstrate that this thickness can be enhanced significantly by using In0.12Ga0.88As pseudo substrates, which generate alternating tensile and compressive strains in the ZnSe/CdSe/ZnSe layers resulting in an efficient strain compensation. This method enables to design CdSe/ZnSe quantum wells with CdSe thicknesses ranging from 1 to 6 monolayers, covering the whole visible spectrum. The strain compensation effect is investigated by high resolution transmission electron microscopy and supported by molecular statics simulations. The model approach with the supporting experimental measurements is sufficiently general to be also applied to other highly mismatched material combinations for the design of advanced strained heterostructures. read less NOT USED (low confidence) J. Hao, X. Shu, S. Jin, X. Zhang, Y. Zhang, and G. Lu, “A comparison of interatomic potentials for modeling tungsten nanocluster structures,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 4 NOT USED (low confidence) C. Huang et al., “Nanoindentation of ultra-hard cBN films: A molecular dynamics study,” Applied Surface Science. 2017. link Times cited: 29 NOT USED (low confidence) C. Lin and Z. Rao, “Thermal conductivity enhancement of paraffin by adding boron nitride nanostructures: A molecular dynamics study,” Applied Thermal Engineering. 2017. link Times cited: 58 NOT USED (low confidence) Z. Bergstrom, M. Cusentino, and B. Wirth, “A Molecular Dynamics Study of Subsurface Hydrogen-Helium Bubbles in Tungsten,” Fusion Science and Technology. 2017. link Times cited: 36 Abstract: Fusion reactor materials experience high ion fluxes and oper… read moreAbstract: Fusion reactor materials experience high ion fluxes and operating temperatures, which will ultimately produce subsurface helium and hydrogen bubbles in the tungsten divertor that can cause surface degradation and impact core plasma performance. Molecular dynamics simulations have been used to evaluate the behavior of hydrogen and helium near a 2-nm bubble or void below a tungsten surface as a function of surface orientation, temperature, gas atom concentration, initial hydrogen distribution, and depth below the surface. A clear tendency for hydrogen to segregate to the bubble-matrix interface is observed in these simulations, regardless of the initial spatial distribution of the hydrogen or simulation parameters. This segregation is due in part to a local minimum in the hydrogen energy at the periphery of the bubble. Further work is required to fully characterize the mechanism of this behavior and to assess the quantities of hydrogen in the bubble and at the bubble periphery. read less NOT USED (low confidence) J. Rabe, N. Severin, and M. F. Gholami, “Folding of Graphene and Other Two-dimensional Materials,” On Folding. 2016. link Times cited: 2 NOT USED (low confidence) N. Karkalos and A. Markopoulos, “Modeling Nano-Metric Manufacturing Processes with Molecular Dynamics Method: A Review,” Current Nanoscience. 2016. link Times cited: 9 NOT USED (low confidence) W.-L. Lv and A. Henry, “Examining the Validity of the Phonon Gas Model in Amorphous Materials,” Scientific Reports. 2016. link Times cited: 50 NOT USED (low confidence) W.-L. Lv, M. Winters, G. Weinberg, and A. Henry, “Understanding Divergent Thermal Conductivity in Single Polythiophene Chains Using Green-Kubo Modal Analysis and Sonification.,” The journal of physical chemistry. A. 2016. link Times cited: 8 Abstract: We used molecular dynamics simulations and the Green-Kubo mo… read moreAbstract: We used molecular dynamics simulations and the Green-Kubo modal analysis (GKMA) method as well as sonification to study the modal contributions to thermal conductivity in individual polythiophene chains. The simulations suggest that it is possible to achieve divergent thermal conductivity in individual polythiophene chains of certain lengths, with periodic boundary conditions. Application of the GKMA method further allowed for exact pinpointing of the modes responsible for the anomalous behavior. The analysis showed that transverse vibrations in the plane of the aromatic rings at low frequencies ∼0.05 THz are primarily responsible for the divergence. Within the integration time, one mode in particular exhibits a thermal conductivity contribution greater than 100 W m-1 K-1. Further investigation showed that the divergence arises from persistent correlation between the three lowest frequency modes on chains that have exact multiples of 30 unit cells in length. Sonification of the mode heat fluxes revealed regions where the heat flux amplitude yields a somewhat sinusoidal envelope with a long period similar to the relaxation time. This characteristic in the divergent mode heat fluxes gives rise to the overall thermal conductivity divergence, which strongly supports earlier hypotheses that attribute the divergence to correlated phonon-phonon scattering/interactions as opposed to a lack of scattering/interaction among modes (e.g., infinite relaxation time/ballistic transport). read less NOT USED (low confidence) I. Tsoulos, A. Tzallas, and D. Tsalikakis, “PDoublePop: An implementation of parallel genetic algorithm for function optimization,” Comput. Phys. Commun. 2016. link Times cited: 18 NOT USED (low confidence) S. Volz, “Relevant Semiempirical Potentials for Phonon Properties.” 2016. link Times cited: 1 NOT USED (low confidence) N. Aktaev and G. Remnev, “Modeling of carbon penetration into silicon structure under the action of pulsed high-intensity ion beam,” Surface & Coatings Technology. 2016. link Times cited: 3 NOT USED (low confidence) K. Müller-Caspary et al., “Materials characterisation by angle-resolved scanning transmission electron microscopy,” Scientific Reports. 2016. link Times cited: 32 NOT USED (low confidence) A. Dmitriev and A. Nikonov, “Molecular dynamics study of the frictional properties of silica nanoparticles in an amorphous state.” 2016. link Times cited: 0 Abstract: In the paper, simulation of the treatment of two silica crys… read moreAbstract: In the paper, simulation of the treatment of two silica crystals with an amorphous interlayer was carried out using the method of molecular dynamics. The three-body interatomic interaction suggested by Tersoff was used. We studied sliding behavior under two different thermal conditions: ambient and elevated temperature. The simulation results have revealed several processes realized in the contact area caused by a shear loading. Depending on temperature and value of external compression, we observed smooth sliding or stick-slip motion of silicon and oxygen atoms within amorphous interlayer. We compare the time dependencies of resistance forces for the studied specimens. In spite of loading conditions even in case of stick-slip sliding the mean value of resistance force for simulated specimens is very low. The last can explain the experimentally observed low friction properties of polymer nano-composite materials with silica nanoparticles inclusions. read less NOT USED (low confidence) S. Armaković and S. Armaković, “Investigation of boron modified graphene nanostructures; optoelectronic properties of graphene nanoparticles and transport properties of graphene nanosheets,” Journal of Physics and Chemistry of Solids. 2016. link Times cited: 13 NOT USED (low confidence) A. Bharti and T. Banerjee, “Reactive force field simulation studies on the combustion behavior of n-octanol,” Fuel Processing Technology. 2016. link Times cited: 36 NOT USED (low confidence) Y. G. Evtushenko, S. Lurie, and M. Posypkin, “New optimization problems arising in modelling of 2D-crystal lattices.” 2016. link Times cited: 3 Abstract: The paper considers the problem of finding the structure of … read moreAbstract: The paper considers the problem of finding the structure of a fragment of two-dimensional crystal lattice with the minimal energy. Atoms in a lattice reside on parallel lines (layers). The interatomic distances are the same within one layer but can differ for distinct layers. The energy of the piece of material is computed using so-called potential functions. We used Lennard-Jones, Morse and Tersoff potentials. The proposed formulation can serve as a scalable complex non-smooth optimization test. The paper evaluates various optimization techniques for the problem under consideration, compares their performances and draws the conclusion about the best choice of optimization methods for the problem under test. As a result we were able to locate minima meaningful from the physical point of view, e.g. reproducing graphene lattice. read less NOT USED (low confidence) B. Vasić, A. Matković, R. Gajić, and I. Stanković, “Wear properties of graphene edges probed by atomic force microscopy based lateral manipulation,” Carbon. 2016. link Times cited: 42 NOT USED (low confidence) J. D. Lee and K. P. Robert, “Multiscale atomistic modeling of fracture subjected to cyclic loading.” 2016. link Times cited: 2 Abstract: It is an established fact that multiscale modeling is an eff… read moreAbstract: It is an established fact that multiscale modeling is an effective way of studying materials over a realistic length scale. In this work, we demonstrate the use of sequential and concurrent multiscale modeling to study the effect of cyclic loading on both the atomic and continuum regions, of graphene, a material which comes with its own set of unique properties. Moreover, to further strengthen this work, we have studied the temperature effects during the cyclic loading, by analyzing the effect of loading and varying temperature gradients. read less NOT USED (low confidence) X. Zhang and X. Wu, “Influence of surface roughness on thermal properties of single crystalline Ge thin films,” Computational Materials Science. 2016. link Times cited: 8 NOT USED (low confidence) J. Yan, L. Zhang, and K. Liew, “A multiscale computational framework for the analysis of graphene involving geometrical and material nonlinearities,” Computer Methods in Applied Mechanics and Engineering. 2016. link Times cited: 15 NOT USED (low confidence) Y. Zhao et al., “Molecular dynamics simulation of nano-indentation of (111) cubic boron nitride with optimized Tersoff potential,” Applied Surface Science. 2016. link Times cited: 25 NOT USED (low confidence) P. Tredak, W. Rudnicki, and J. Majewski, “Efficient implementation of the many-body Reactive Bond Order (REBO) potential on GPU,” J. Comput. Phys. 2016. link Times cited: 4 NOT USED (low confidence) F. Cipcigan, V. Sokhan, J. Crain, and G. Martyna, “Electronic coarse graining enhances the predictive power of molecular simulation allowing challenges in water physics to be addressed,” J. Comput. Phys. 2016. link Times cited: 13 NOT USED (low confidence) D. Hong, L. Liu, Y. Huang, C. Zheng, and X. Guo, “Chemical Effect of H2O on CH4 Oxidation during Combustion in O2/H2O Environments,” Energy & Fuels. 2016. link Times cited: 43 Abstract: The effect of H2O reactivity on CH4 oxidation in O2/H2O comb… read moreAbstract: The effect of H2O reactivity on CH4 oxidation in O2/H2O combustion was studied using the reactive molecular dynamics (ReaxFF-MD) method. Simulations were performed under fuel-rich, stoichiometric, and fuel-lean conditions at the temperature 2400–3600 K with a high concentration of H2O. The results obtained under fuel-rich conditions showed that replacing N2 gas with H2O inhibited the oxidation rate of CH4 at low temperatures due to equilibrium reasons and the third body efficiency of H2O. However, the presence of H2O advanced the oxidation of CH4 because of H2O reactivity at high temperatures. The amount of OH radicals and H2 molecules in fuel-rich O2/H2O combustion was obviously greater than that in O2/N2 combustion, which proved that H2O molecules mainly take part in the reactions through H2O + H → H2 + OH and H2O + O → OH + OH. The activity of OH radicals is higher than that of H radicals in CH4 oxidation. Therefore, a high concentration of H2O promoted the reaction rate of CH4 at high temperatures. Me... read less NOT USED (low confidence) S. Singh and B. P. Patel, “Nonlinear dynamic response of single layer graphene sheets using multiscale modelling,” European Journal of Mechanics A-solids. 2016. link Times cited: 16 NOT USED (low confidence) B. Liu, J. Tao, X. Chen, Y. Zhang, Y. Jiang, and Y. Qian, “Numerical investigation of the effects of phosphorus on the mechanical responses of [1 1 0]-oriented silicon nano-wires,” Microelectron. Reliab. 2016. link Times cited: 1 NOT USED (low confidence) T. Fang, W.-J. Chang, Y.-L. Feng, and D. Lu, “Torsional characteristics of graphene nanoribbons encapsulated in single-walled carbon nanotubes,” Physica E-low-dimensional Systems & Nanostructures. 2016. link Times cited: 5 NOT USED (low confidence) A. Mourad and I. Zalzali, “Discrete and homogenized mechanical models for single walled carbon nanotubes,” Mathematics and Mechanics of Solids. 2016. link Times cited: 1 Abstract: In this paper, we propose two mechanical models for the sing… read moreAbstract: In this paper, we propose two mechanical models for the single walled carbon nanotubes: (a) a discrete mechanical model at the nanoscale level based on replacing the atomic structure by a discrete lattice of elastic bars and (b) a homogenized continuous mechanical model based on atomistic experimental results on the stretching and angular variations of carbon atomic bonds. Two different interatomic mechanical models (linear and exponential) for the stretching–compression behavior of the carbon–carbon atomic bond have been used. The numerical results show that the homogenized asymptotic model is a good approximation of the full discrete model. The former has the advantage of requiring very little computational resources whereas the latter requires a huge computational cost hence limiting its application to structures with a small number of atoms and atomic bonds. Moreover, the obtained results show that the axial Young’s modulus is almost insensitive to the radius and the chirality but it is very sensitive to the choice of the interatomic mechanical models and their corresponding elastic constants. The linear interatomic model seems to be invalid when used for large deformation of the nanotubes, i.e. it is valid only for small displacements. On the other hand, and most importantly, the stress–strain curves obtained from the homogenized model using the exponential interatomic potential show that the nanotube type determines completely its mechanical characteristics. For instance, the tensile strength and elongation at break are the largest in the case of the armchair nanotubes and the smallest in the case of the zigzag nanotubes. read less NOT USED (low confidence) S. Debroy, V. P. K. Miriyala, K. V. Sekhar, S. G. Acharyya, and A. Acharyya, “Self healing nature of bilayer graphene,” Superlattices and Microstructures. 2016. link Times cited: 7 NOT USED (low confidence) M. Dewapriya and R. Rajapakse, “Development of a homogenous nonlinear spring model characterizing the interfacial adhesion properties of graphene with surface defects,” Composites Part B-engineering. 2016. link Times cited: 16 NOT USED (low confidence) M. M. Islam, G. Kolesov, T. Verstraelen, E. Kaxiras, and A. V. van Duin, “eReaxFF: A Pseudoclassical Treatment of Explicit Electrons within Reactive Force Field Simulations.,” Journal of chemical theory and computation. 2016. link Times cited: 70 Abstract: We present a computational tool, eReaxFF, for simulating exp… read moreAbstract: We present a computational tool, eReaxFF, for simulating explicit electrons within the framework of the standard ReaxFF reactive force field method. We treat electrons explicitly in a pseudoclassical manner that enables simulation several orders of magnitude faster than quantum chemistry (QC) methods, while retaining the ReaxFF transferability. We delineate here the fundamental concepts of the eReaxFF method and the integration of the Atom-condensed Kohn-Sham DFT approximated to second order (ACKS2) charge calculation scheme into the eReaxFF. We trained our force field to capture electron affinities (EA) of various species. As a proof-of-principle, we performed a set of molecular dynamics (MD) simulations with an explicit electron model for representative hydrocarbon radicals. We establish a good qualitative agreement of EAs of various species with experimental data, and MD simulations with eReaxFF agree well with the corresponding Ehrenfest dynamics simulations. The standard ReaxFF parameters available in the literature are transferrable to the eReaxFF method. The computationally economic eReaxFF method will be a useful tool for studying large-scale chemical and physical systems with explicit electrons as an alternative to computationally demanding QC methods. read less NOT USED (low confidence) A. Khoei and M. Khorrami, “Mechanical properties of graphene oxide: A molecular dynamics study,” Fullerenes, Nanotubes and Carbon Nanostructures. 2016. link Times cited: 43 Abstract: ABSTRACT In this paper, the mechanical properties of graphen… read moreAbstract: ABSTRACT In this paper, the mechanical properties of graphene oxide are obtained using the molecular dynamics analysis, including the ultimate stress, Young modulus, shear modulus and elastic constants, and the results are compared with those of pristine graphene. It is observed that the increase of oxide agents (–O) and (–OH) leads to the increase of C–C bond length at each hexagonal lattice and as a result, alter the mechanical properties of the graphene sheet. It is shown that the elasticity modulus and ultimate tensile strength of graphene oxides (–O) and (–OH) decrease significantly causing the failure behavior of graphene sheet changes from the brittle to ductile. The results of shear loading tests illustrate that the increase of oxide agents (–O/–OH) results in the decrease of ultimate shear stress and shear module of the graphene sheet. It is shown that the increase of oxide agents in the graphene sheet leads to decrease of the elastic constants, in which the reduction of elastic properties in the armchair direction is more significant than the zigzag direction. Moreover, the graphene sheet with oxide agents (–O) and (–O/–OH) presents an anisotropic behavior. read less NOT USED (low confidence) X. Liu, G. Zhang, and Y.-W. Zhang, “Topological Defects at the Graphene/h-BN interface Abnormally Enhance Its Thermal Conductance.,” Nano letters. 2016. link Times cited: 110 Abstract: Low thermal conductance across interface is often the limiti… read moreAbstract: Low thermal conductance across interface is often the limiting factor in managing heat in many advanced device applications. The most commonly used approach to enhance the thermal conductance is to reduce/eliminate the interfacial structural defects. Using a graphene/h-BN (Gr/h-BN) interface, we show surprisingly that topological defects are able to enhance the thermal conductance across the interface. It is found that the phonon transmission across the Gr/h-BN interface with 5|7 defects is higher than that of the pristine interface, which is in strong contrast to the common notion that interface defects promote phonon scattering. By analyzing the strain distribution and phonon vibrational spectra, we find that this abnormal enhancement in interfacial thermal conductance originates from the localization of the stress fields arising from misfit dislocations and their out-of-plane deformations at the interface. In the presence of the defects, the overall mismatch strain is reduced. In addition, the out-of-plane deformations screen the long-ranged dislocation strain fields, resulting in the stress fields to be localized only at the cores of the defects. This abnormal mechanism provides a new dimension to enhance the interfacial thermal conductance in two-dimensional heterostructures. read less NOT USED (low confidence) H. Li, X. Tang, F. Chen, H. Huang, J. Liu, and D. Chen, “Molecular dynamics study of radiation damage and microstructure evolution of zigzag single-walled carbon nanotubes under carbon ion incidence,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2016. link Times cited: 5 NOT USED (low confidence) Z. Zhang, A. Stukowski, and H. Urbassek, “Interplay of dislocation-based plasticity and phase transformation during Si nanoindentation,” Computational Materials Science. 2016. link Times cited: 28 NOT USED (low confidence) M. Malakouti and A. Montazeri, “Nanomechanics analysis of perfect and defected graphene sheets via a novel atomic-scale finite element method,” Superlattices and Microstructures. 2016. link Times cited: 20 NOT USED (low confidence) D. Pan, T.-C. Wang, C. Wang, W. Guo, and Y. Yao, “Self-assembled chiral phosphorus nanotubes from phosphorene: a molecular dynamics study,” RSC Advances. 2016. link Times cited: 15 Abstract: Controlled syntheses in nanoscale structures should be expec… read moreAbstract: Controlled syntheses in nanoscale structures should be expected and phosphorous nanotubes with predefined chiralities are important in electronic devices with tunable bandgap. Here, incorporating molecular dynamics simulations with theoretical analyses, we show that a zigzag phosphorene nanoribbon can self-assemble and form a corresponding chiral phosphorous nanotube surrounding a template armchair phosphorous nanotube. The van der Waals potential between the nanoribbon and the nanotube is transformed to the intrinsic deformed and chemical bonding energies of the synthesized tube together with partial kinetic energy. The self-assembly process has an apparent temperature dependence and size effect and the formed chiral tube is thermodynamically stable. Also, the chirality and measurement can be tuned by the radius of template tube and the aspect ratio of raw ribbon. The study suggests a novel and feasible approach for controlled synthesis of phosphorous nanotubes and thus is of great interest for semiconductor device applications. read less NOT USED (low confidence) K. Firestein et al., “Structural analysis and atomic simulation of Ag/BN nanoparticle hybrids obtained by Ag ion implantation.” 2016. link Times cited: 19 NOT USED (low confidence) S. Bhattacharyya, S. Otake, S. Ono, R. Kuwahara, and K. Ohno, “Growth of Graphene Nanocoil in a SiC Container: A Molecular Dynamics Study,” Advances in Materials Physics and Chemistry. 2016. link Times cited: 0 Abstract: Graphene nano structures find their application in modern na… read moreAbstract: Graphene nano structures find their application in modern nano electronics because of their excellent mechanical, unique electronics and electrical properties owing to the ballistic transport, where the charge carriers can move freely without getting scattered. In this work, we show a possibility to grow a miniaturised coil from a pre-existing graphene coil layer using molecular dynamics simulation. From our calculations it was observed that isolated carbon atoms get attached to the edge of the initial graphene coil and form an extension of the coil structure. The growth process depends strongly on the chirality of the growth front as well as the growth temperature. An optimal temperature between 2000 - 2300 K was proposed for all the edge structures except armchair (2500 K) type for the maximum number of the new hexagonal rings. Our results predict a technique that can be adopted experimentally to grow graphene nanocoil. read less NOT USED (low confidence) W.-L. Lv and A. Henry, “Phonon transport in amorphous carbon using Green-Kubo modal analysis,” Applied Physics Letters. 2016. link Times cited: 31 Abstract: Amorphous carbon (a-C) is an important material often used i… read moreAbstract: Amorphous carbon (a-C) is an important material often used in microelectronics. Using a recently developed approach, termed Green–Kubo modal analysis, we were able to calculate the thermal conductivity of a-C, which yielded excellent agreement with experiments, by employing a simple correction to the specific heat. The results show that the heat capacity substantially limits the thermal conductivity of a-C at room temperature and it is dominated by contributions from diffusons between 10 and 40 THz. Furthermore, the phonon relaxation times in a-C do not vary significantly with increasing temperature, which is quite unusual by comparison with the behavior observed for other materials. read less NOT USED (low confidence) A. Tabarraei and X. Wang, “Anomalous thermal conductivity of monolayer boron nitride,” Applied Physics Letters. 2016. link Times cited: 31 Abstract: In this paper, we use nonequilibrium molecular dynamics mode… read moreAbstract: In this paper, we use nonequilibrium molecular dynamics modeling to investigate the thermal properties of monolayer hexagonal boron nitride nanoribbons under uniaxial strain along their longitudinal axis. Our simulations predict that hexagonal boron nitride shows an anomalous thermal response to the applied uniaxial strain. Contrary to three dimensional materials, under uniaxial stretching, the thermal conductivity of boron nitride nanoribbons first increases rather than decreasing until it reaches its peak value and then starts decreasing. Under compressive strain, the thermal conductivity of monolayer boron nitride ribbons monolithically reduces rather than increasing. We use phonon spectrum and dispersion curves to investigate the mechanism responsible for the unexpected behavior. Our molecular dynamics modeling and density functional theory results show that application of longitudinal tensile strain leads to the reduction of the group velocities of longitudinal and transverse acoustic modes. Such a phonon softening mechanism acts to reduce the thermal conductivity of the nanoribbons. On the other hand, a significant increase in the group velocity (stiffening) of the flexural acoustic modes is observed, which counteracts the phonon softening effects of the longitudinal and transverse modes. The total thermal conductivity of the ribbons is a result of competition between these two mechanisms. At low tensile strain, the stiffening mechanism overcomes the softening mechanism which leads to an increase in the thermal conductivity. At higher tensile strain, the softening mechanism supersedes the stiffening and the thermal conductivity slightly reduces. Our simulations show that the decrease in the thermal conductivity under compressive strain is attributed to the formation of buckling defects which reduces the phonon mean free path. read less NOT USED (low confidence) D. Papavassiliou, K. Bui, and H. Nguyen, “11. Thermal Boundary Resistance Effects in Carbon Nanotube Composites.” 2016. link Times cited: 1 NOT USED (low confidence) G. Ghadyani and M. Rahmandoust, “Computational Nanomechanics Investigation Techniques.” 2016. link Times cited: 1 Abstract: Today, many fields of rapidly growing research about nanomat… read moreAbstract: Today, many fields of rapidly growing research about nanomaterials and nanodevices are dependent on a combined detailed investigation between nanoscience and engineering. Hence, current nanotechnological engineering requires a vital linkage between fundamental research about the nature of the materials, which should be sought in nanoscience, and engineering investigation tools through simulations and modeling in computational nanomechanics. This linkage is necessary to obtain a comprehensive picture of the properties and characteristics of the studied nanomaterials and nanodevices under various conditions. In this chapter, a review of the fundamental concepts of the Newtonian mechanics, including Lagrangian and Hamiltonian functions is provided first. The developed equations of motion of a system with interacting material points are introduced then. After that, based on the physics of nanosystems, which can be applicable in any material phases, basic concepts of molecular dynamic simulations are introduced. Some interatomic potentials from which Morse function is recognized as an accurate definition are discussed in the next step for defining the natural bond length. With the purpose of decreasing computational effort, the cut-off radius is considered to limit atomic interactions to immediate neighbors only. The link between molecular dynamics and quantum mechanics is then explained using a simple classical example of two interacting hydrogen atoms, and the major limitations of the simulation method are discussed. Length and timescale limitation of molecular dynamics simulation technique are the major reasons behind opting multiscale simulations rather than molecular dynamics, which are explained briefly at the final sections of this chapter. read less NOT USED (low confidence) E. Hernández, Y. Takada, and T. Yamamoto, “Mechanical Properties of Inorganic Nanostructures.” 2016. link Times cited: 0 NOT USED (low confidence) T. Ito, T. Akiyama, and K. Nakamura, “Systematic approach to developing empirical interatomic potentials for III–N semiconductors,” Japanese Journal of Applied Physics. 2016. link Times cited: 5 Abstract: A systematic approach to the derivation of empirical interat… read moreAbstract: A systematic approach to the derivation of empirical interatomic potentials is developed for III–N semiconductors with the aid of ab initio calculations. The parameter values of empirical potential based on bond order potential are determined by reproducing the cohesive energy differences among 3-fold coordinated hexagonal, 4-fold coordinated zinc blende, wurtzite, and 6-fold coordinated rocksalt structures in BN, AlN, GaN, and InN. The bond order p is successfully introduced as a function of the coordination number Z in the form of p = a exp(−bZn) if Z ≤ 4 and p = (4/Z)α if Z ≥ 4 in empirical interatomic potential. Moreover, the energy difference between wurtzite and zinc blende structures can be successfully evaluated by considering interaction beyond the second-nearest neighbors as a function of ionicity. This approach is feasible for developing empirical interatomic potentials applicable to a system consisting of poorly coordinated atoms at surfaces and interfaces including nanostructures. read less NOT USED (low confidence) G. Rajasekaran, R. Kumar, and A. Parashar, “Tersoff potential with improved accuracy for simulating graphene in molecular dynamics environment,” Materials Research Express. 2016. link Times cited: 71 Abstract: Graphene is an elementary unit for various carbon based nano… read moreAbstract: Graphene is an elementary unit for various carbon based nanostructures. The recent technological developments have made it possible to manufacture hybrid and sandwich structures with graphene. In order to model these nanostructures in atomistic scale, a compatible interatomic potential is required to successfully model these nanostructures. In this article, an interatomic potential with modified cut-off function for Tersoff potential was proposed to avoid overestimation and also to predict the realistic mechanical behavior of single sheet of graphene. In order to validate the modified form of cut-off function for Tersoff potential, simulations were performed with different set of temperatures and strain rates, and results were made to compare with available experimental data and molecular dynamics simulation results obtained with the help of other empirical interatomic potentials. read less NOT USED (low confidence) S. De, K. Kunal, and N. Aluru, “Mixed role of surface on intrinsic losses in silicon nanostructures,” Journal of Applied Physics. 2016. link Times cited: 9 Abstract: We utilize molecular dynamics simulations and show opposing … read moreAbstract: We utilize molecular dynamics simulations and show opposing roles of surface on dissipation in nanostructures. While the surface defects always aid in the entropy generation process, the scattering of phonons from rough surfaces can suppress Akhiezer damping. For the case of a silicon (2 × 1) reconstructed surface, the former dominates and Q−1 (Q is the quality factor) is found to increase with the decrease in size. However, different scaling trends are observed in the case of a hydrogen (H) terminated silicon surface with no defects and dimers. Particularly, in the case of a H-terminated silicon, if the resonator is operated with a frequency Ω such that Ωτph 1. A simplified model, based on two phonon groups (with positive and negative Gruneisen parameters), is considered to explain the observed trend. We show that the equilibration time between the two mode groups d... read less NOT USED (low confidence) R. Ansari, S. Ajori, and A. Ameri, “Stability characteristics and structural properties of single- and double-walled boron-nitride nanotubes under physical adsorption of Flavin mononucleotide (FMN) in aqueous environment using molecular dynamics simulations,” Applied Surface Science. 2016. link Times cited: 22 NOT USED (low confidence) M. Maslov, A. I. Podlivaev, and K. Katin, “Nonorthogonal tight-binding model with H–C–N–O parameterisation,” Molecular Simulation. 2016. link Times cited: 49 Abstract: A parametric nonorthogonal tight-binding model (NTBM1) with … read moreAbstract: A parametric nonorthogonal tight-binding model (NTBM1) with the set of parameters for H–C–N–O systems is presented. This model compares well with widely used semi-empirical AM1 and PM3/PM7 models but contains less fitting parameters per atom. All NTBM1 parameters are derived based on a criterion of the best agreement between the calculated and experimental values of bond lengths, valence angles and binding energies for various H–C–N–O molecules. Results for more than 200 chemical compounds are reported. Parameters are currently available for hydrogen, carbon, nitrogen, oxygen atoms and corresponding interatomic interactions. The model has a good transferability and can be used for both relaxation of large molecular systems (e.g., high-molecular compounds or covalent cluster complexes) and long-timescale molecular dynamics simulation (e.g., modelling of thermal decomposition processes). The program package based on this model is available for download at no cost from http://ntbm.info. read less NOT USED (low confidence) J. Zhen et al., “Molecular dynamics study of structural damage in amorphous silica induced by swift heavy-ion radiation,” Radiation Effects and Defects in Solids. 2016. link Times cited: 16 Abstract: ABSTRACT In this paper, the radiation defects induced by the… read moreAbstract: ABSTRACT In this paper, the radiation defects induced by the swift heavy ions and the recoil atoms in amorphous SiO2 were studied. The energy of recoil atoms induced by the incident Au ions in SiO2 was calculated by using Monte Carlo method. Results show that the average energies of recoils reach the maximum (200 eV for Si and 130 eV for O, respectively) when the incident energy of Au ion is 100 MeV. Using Tersoff/zbl potential with the newly built parameters, the defects formation processes in SiO2 induced by the recoils were studied by using molecular dynamics method. The displacement threshold energies (Ed) for Si and O atoms are found to be 33.5 and 16.3 eV, respectively. Several types of under- and over-coordinated Si and O defects were analyzed. The results demonstrate that Si3, Si5, and O1 are the mainly defects in SiO2 after radiation. Besides, the size of cylindrical damage region produced by a single recoil atom was calculated. The calculation shows that the depth and the radius are up to 2.0 and 1.4 nm when the energy of recoils is 200 eV. Finally, it is estimated that the Au ion would induce a defected track with a diameter of 4 nm in SiO2. read less NOT USED (low confidence) N. Artrith and A. Urban, “An implementation of artificial neural-network potentials for atomistic materials simulations: Performance for TiO2,” Computational Materials Science. 2016. link Times cited: 350 NOT USED (low confidence) A. F. Hidayat, A. Rosikhin, I. Syuhada, and T. Winata, “Effects of temperature on Cu structure deposited on Si substrate: A molecular dynamics study.” 2016. link Times cited: 2 Abstract: The deposition process of copper onto silicon substrate was … read moreAbstract: The deposition process of copper onto silicon substrate was studied by the molecular dynamics method. Tersoff, MEAM, and Morse potentials were used to describe the interaction of Si-Si Cu-Cu, and Cu-Si, respectively. Deposition process was performed using NVE ensemble and applying Berendsen thermostat with 0,2 fs timestep for 100 ps. The effect of substrate temperature on the percentage of amorphous structure, radial distribution function (RDF), and coordination number was investigated. The result was indicated that at 300 K, the percentage of amorphous structure was relatively lower compared to another temperature. First peaks of RDF at each temperature were found at radius 3,05 A and were still relatively wide, indicating short-range order structure. read less NOT USED (low confidence) K. Yashiro, “Molecular dynamics study on atomic elastic stiffness at mode I crack tip in Si: Precursor instability in their eigenvalue before crack propagation,” Computational Materials Science. 2016. link Times cited: 8 NOT USED (low confidence) J. Han, V. Vítek, and D. Srolovitz, “Grain-boundary metastability and its statistical properties,” Acta Materialia. 2016. link Times cited: 109 NOT USED (low confidence) R. Kumar and A. Parashar, “Atomistic modeling of BN nanofillers for mechanical and thermal properties: a review.,” Nanoscale. 2016. link Times cited: 69 Abstract: Due to their exceptional mechanical properties, thermal cond… read moreAbstract: Due to their exceptional mechanical properties, thermal conductivity and a wide band gap (5-6 eV), boron nitride nanotubes and nanosheets have promising applications in the field of engineering and biomedical science. Accurate modeling of failure or fracture in a nanomaterial inherently involves coupling of atomic domains of cracks and voids as well as a deformation mechanism originating from grain boundaries. This review highlights the recent progress made in the atomistic modeling of boron nitride nanofillers. Continuous improvements in computational power have made it possible to study the structural properties of these nanofillers at the atomistic scale. read less NOT USED (low confidence) L. Madeira and S. Vitiello, “Properties of heavy rare-gases adlayers on graphene substrates,” Surface Science. 2015. link Times cited: 6 NOT USED (low confidence) A. Adnan and S. F. Ferdous, “Mechanical properties of computationally designed novel carbon enriched Si1−xCx ceramics: A molecular dynamics simulation study,” Computational Materials Science. 2015. link Times cited: 9 NOT USED (low confidence) Y. Wu, P. Krstic, F. Zhou, and F. Meyer, “Damage at a tungsten surface induced by impacts of self-atoms,” Journal of Nuclear Materials. 2015. link Times cited: 3 NOT USED (low confidence) P. Molian, “Three-Dimensional Printing of Nanoscale Powders Using Laser Shockwaves,” Journal of Micro and Nano-Manufacturing. 2015. link Times cited: 1 Abstract: A new three-dimensional (3D) printing process designated as … read moreAbstract: A new three-dimensional (3D) printing process designated as shockwave-induced freeform technique (SWIFT) is explored for fabricating microparts from nanopowders. SWIFT consists of generating shockwaves using a laser beam, applying these shocks to pressure sinter nanoparticles at room temperature, and creating structures and devices by the traditional layer-by-layer formation. Shockwave cold compaction of nanoscale powders has the capability to overcome limitations, such as shrinkage, porosity, rough surface, and wide tolerance, normally encountered in hot sintering processes, such as selective laser sintering. In this study, the window of operating parameters and the underlying physics of SWIFT were investigated using a high-energy Q-switched Nd: YAG laser and nanodiamond (ND) powders. Results indicate the potential of SWIFT for fabricating high-performance diamond microtools with high aspect ratios, smooth surfaces, and sharp edges. The drawback is that the SWIFT process does not work for micro-sized powders. read less NOT USED (low confidence) J. Boer, “Theoretical studies of epitaxial graphene formation on metal surfaces.” 2015. link Times cited: 0 Abstract: In this thesis we develop a set of pheneomenological models … read moreAbstract: In this thesis we develop a set of pheneomenological models that we apply to the problem of epitaxial growth of graphene on metal substrates. The high temperature and typically low flux conditions under which graphene growth occurs are such that state of the art techniques such as kinetic Monte Carlo (kMC) are extremely difficult to apply to study the growth processes. Rather we utilise simpler theories based on rate equations and also develop a technique based on the phase-field method of island front tracking. The latter method may be considered to be an addition to the class of techniques known as "island dynamics" models [1]. We use rate equations to study the nucleation and growth of graphene [2] and to explore the dehydrogenation sequence of ethylene CH read less NOT USED (low confidence) T. Fang, W.-J. Chang, and Y.-L. Feng, “Mechanical characteristics of graphene nanoribbons encapsulated in single-walled carbon nanotubes using molecular dynamics simulations,” Applied Surface Science. 2015. link Times cited: 28 NOT USED (low confidence) E. Hernández, C. Herrero, and J. Soler, “A chain-of-states acceleration method for the efficient location of minimum energy paths.,” The Journal of chemical physics. 2015. link Times cited: 3 Abstract: We describe a robust and efficient chain-of-states method fo… read moreAbstract: We describe a robust and efficient chain-of-states method for computing Minimum Energy Paths (MEPs) associated to barrier-crossing events in poly-atomic systems, which we call the acceleration method. The path is parametrized in terms of a continuous variable t ∈ [0, 1] that plays the role of time. In contrast to previous chain-of-states algorithms such as the nudged elastic band or string methods, where the positions of the states in the chain are taken as variational parameters in the search for the MEP, our strategy is to formulate the problem in terms of the second derivatives of the coordinates with respect to t, i.e., the state accelerations. We show this to result in a very simple and efficient method for determining the MEP. We describe the application of the method to a series of test cases, including two low-dimensional problems and the Stone-Wales transformation in C60. read less NOT USED (low confidence) N. S. Mikhaleva, M. Visotin, Z. Popov, A. Kuzubov, and A. Fedorov, “Ab initio and empirical modeling of lithium atoms penetration into silicon,” Computational Materials Science. 2015. link Times cited: 4 NOT USED (low confidence) S. Singh and B. P. Patel, “Atomistic–continuum coupled model for nonlinear analysis of single layer graphene sheets,” International Journal of Non-linear Mechanics. 2015. link Times cited: 17 NOT USED (low confidence) S. Debroy, V. P. K. Miriyala, K. V. Sekhar, S. G. Acharyya, and A. Acharyya, “Graphene heals thy cracks,” Computational Materials Science. 2015. link Times cited: 12 NOT USED (low confidence) B. Liu, H. Zhang, J. Tao, X. Chen, and Y.-A. Zhang, “Comparative investigation of a newly optimized modified embedded atom method potential with other potentials for silicon,” Computational Materials Science. 2015. link Times cited: 7 NOT USED (low confidence) J. Zhang, F. Xu, Y. Hong, Q. Xiong, and J. Pan, “A comprehensive review on the molecular dynamics simulation of the novel thermal properties of graphene,” RSC Advances. 2015. link Times cited: 65 Abstract: This review summarizes state-of-the-art progress in the mole… read moreAbstract: This review summarizes state-of-the-art progress in the molecular dynamics (MD) simulation of the novel thermal properties of graphene. The novel thermal properties of graphene, which include anisotropic thermal conductivity, decoupled phonon thermal transport, thermal rectification and tunable interfacial thermal conductance, have attracted enormous interest in the development of next-generation nano-devices. Molecular dynamics simulation is one of the main approaches in numerical simulation of the novel thermal properties of graphene. In this paper, the widely used potentials of MD for modeling the novel thermal properties of graphene are described first. Then MD simulations of anisotropic thermal conductivity, decoupled phonon thermal transport, thermal rectification and tunable interfacial thermal conductance are discussed. Finally, the paper concludes with highlights on both the current status and future directions of the MD simulation of the novel thermal properties of graphene. read less NOT USED (low confidence) A. Fereidoon, M. Mostafaei, M. Ganji, and F. Memarian, “Atomistic simulations on the influence of diameter, number of walls, interlayer distance and temperature on the mechanical properties of BNNTs,” Superlattices and Microstructures. 2015. link Times cited: 20 NOT USED (low confidence) R. Ansari, S. Ajori, and F. Sadeghi, “Molecular dynamics investigation into the electric charge effect on the operation of ion-based carbon nanotube oscillators,” Journal of Physics and Chemistry of Solids. 2015. link Times cited: 29 NOT USED (low confidence) J. Zhang, Y. Hong, Z. Tong, Z. Xiao, H. Bao, and Y. Yue, “Molecular dynamics study of interfacial thermal transport between silicene and substrates.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 41 Abstract: In this work, the interfacial thermal transport across silic… read moreAbstract: In this work, the interfacial thermal transport across silicene and various substrates, i.e., crystalline silicon (c-Si), amorphous silicon (a-Si), crystalline silica (c-SiO2) and amorphous silica (a-SiO2) are explored by classical molecular dynamics (MD) simulations. A transient pulsed heating technique is applied in this work to characterize the interfacial thermal resistance in all hybrid systems. It is reported that the interfacial thermal resistances between silicene and all substrates decrease nearly 40% with temperature from 100 K to 400 K, which is due to the enhanced phonon couplings from the anharmonicity effect. Analysis of phonon power spectra of all systems is performed to interpret simulation results. Contradictory to the traditional thought that amorphous structures tend to have poor thermal transport capabilities due to the disordered atomic configurations, it is calculated that amorphous silicon and silica substrates facilitate the interfacial thermal transport compared with their crystalline structures. Besides, the coupling effect from substrates can improve the interface thermal transport up to 43.5% for coupling strengths χ from 1.0 to 2.0. Our results provide fundamental knowledge and rational guidelines for the design and development of the next-generation silicene-based nanoelectronics and thermal interface materials. read less NOT USED (low confidence) J. Wang and H. Xie, “Molecular dynamic investigation on the structures and thermal properties of carbon nanotube interfaces,” Applied Thermal Engineering. 2015. link Times cited: 8 NOT USED (low confidence) A. Fereidoon, M. Khorasani, M. Ganji, and F. Memarian, “Atomistic simulation study of mechanical properties of periodic graphene nanobuds,” Computational Materials Science. 2015. link Times cited: 7 NOT USED (low confidence) B. Cowen and M. El-Genk, “On force fields for molecular dynamics simulations of crystalline silica,” Computational Materials Science. 2015. link Times cited: 31 NOT USED (low confidence) F. Memarian, A. Fereidoon, and M. Ganji, “Graphene Young’s modulus: Molecular mechanics and DFT treatments,” Superlattices and Microstructures. 2015. link Times cited: 90 NOT USED (low confidence) R. Ansari, A. Shahabodini, and H. Rouhi, “A nonlocal plate model incorporating interatomic potentials for vibrations of graphene with arbitrary edge conditions,” Current Applied Physics. 2015. link Times cited: 37 NOT USED (low confidence) M. P. Ariza and J. P. Mendez, “Stability of discrete topological defects in graphene,” Journal of Mechanics of Materials and Structures. 2015. link Times cited: 3 NOT USED (low confidence) A. Tabarraei and X. Wang, “A molecular dynamics study of nanofracture in monolayer boron nitride,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 35 NOT USED (low confidence) J. Han, V. Vitek, and D. Srolovitz, “The interplay between grain boundary structure and defect sink/annealing behavior,” IOP Conference Series: Materials Science and Engineering. 2015. link Times cited: 7 Abstract: We present a series of results from atomistic simulations in… read moreAbstract: We present a series of results from atomistic simulations in three different materials (3 crystal structures) that demonstrate that the multiplicity of grain boundary (GB) structures at fixed macroscopic GB degrees of freedom is both extremely large and ubiquitous. The GB energy vs. misorientation curve that is commonly discussed is in fact a wide band, with many GB states very close in energy. The existence of so many GB states suggests that GB configurational entropy Sc is important for GB properties. We demonstrate that the GB Sc consists of two major contributions, one of which is geometric in nature and one that depends on bonding. We then show how this concept can be employed to predict GB relaxation dynamics by analogy with Adam-Gibbs theory, originally derived to predict the properties of glass forming liquids. Finally, we apply these predictions to understand GB denuded zone size during irradiation. read less NOT USED (low confidence) W. Kim, H. Chung, and M. Cho, “Anisotropic hyperelastic modeling for face-centered cubic and diamond cubic structures,” Computer Methods in Applied Mechanics and Engineering. 2015. link Times cited: 10 NOT USED (low confidence) M. A. Balatero, G. J. Paylaga, N. T. Paylaga, and R. Bantaculo, “Molecular Dynamics Simulations of Thermal Conductivity of Germanene Nanoribbons (GeNR) with Armchair and Zigzag Chirality,” Applied Mechanics and Materials. 2015. link Times cited: 9 Abstract: Germanene, an allotrope of germanium which is a two dimensio… read moreAbstract: Germanene, an allotrope of germanium which is a two dimensional material with sp2 hybridization, has almost the same properties with graphene except for its buckled structure. In this study, germanium nanoribbon (GeNR) is use for it is still a new material for nanoscale level of research. In this paper, we investigate the effect of chirality on the thermal conductivity of zigzag GeNR (ZGeNR) and armchair GeNR (AGeNR) chiralities using equilibrium molecular dynamics with varied lengths at fixed temperature and varied temperatures at fixed length. The simulations were carried out in Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) using Tersoff potential for the Ge-Ge interactions. The thermal conductivity is calculated using Green-Kubo method. It is found that the chirality can affect the thermal conductivity of GeNR. Our results show that thermal conductivity of AGeNR is higher than ZGeNR in both increasing temperatures and lengths similar to the thermal conductivity behavior obtained in silicene nanoribbons [Int. J. Mech. Mater. Des. 9 (2013) 105]. read less NOT USED (low confidence) J. Yan and K. Liew, “Predicting elastic properties of single-walled boron nitride nanotubes and nanocones using an atomistic-continuum approach,” Composite Structures. 2015. link Times cited: 30 NOT USED (low confidence) A. Abramyan, N. Bessonov, L. V. Mirantsev, and N. Reinberg, “Influence of liquid environment and bounding wall structure on fluid flow through carbon nanotubes,” Physics Letters A. 2015. link Times cited: 6 NOT USED (low confidence) T. Shen et al., “Formation of multiple dislocations in Si solid-phase epitaxy regrowth process using stress memorization technique,” Computational Materials Science. 2015. link Times cited: 2 NOT USED (low confidence) R. Tanaka, K. Takeuchi, and K. Yuge, “Application of Grid Increment Cluster Expansion to Modeling Potential Energy Surface of Cu-Based Alloys,” Materials Transactions. 2015. link Times cited: 3 Abstract: We demonstrate the applicability of extended cluster expansi… read moreAbstract: We demonstrate the applicability of extended cluster expansion technique, GICE, to calculation of a potential energy surface (PES) at discrete position in terms of atomic arrangement with an example of Cu and Cu-Ti binary system on fcc lattice. We find that the proposed CE successfully predicts total energy within error of 0.5meV/atom for Cu and 1.2meV/atom for Cu-Ti with respect to DFT calculation, which indicates that this method can model the PES and possesses potential to formulate physical properties in terms of atomic arrangement. [doi:10.2320/matertrans.M2015024] read less NOT USED (low confidence) P. Gautreau, Y. Chu, T. Ragab, and C. Basaran, “Phonon–phonon scattering rates in single walled carbon nanotubes,” Computational Materials Science. 2015. link Times cited: 9 NOT USED (low confidence) S. Kerdsongpanya, “Design of Transition-Metal Nitride Thin Films for Thermoelectrics.” 2015. link Times cited: 0 Abstract: Thermoelectric devices are one of the promising energy harve… read moreAbstract: Thermoelectric devices are one of the promising energy harvesting technologies, because of their ability to convert heat (temperature gradient) to electricity by the Seebeck effect. Furthermore, th ... read less NOT USED (low confidence) E. D. Monterola, N. T. Paylaga, G. J. Paylaga, and R. Bantaculo, “Anomalous Effect on the Phononic Thermal Conductivity of Silicene Nanoribbon by Hydrogenation,” Advanced Materials Research. 2015. link Times cited: 3 Abstract: Silicene is a two-dimensional (2D) allotrope of silicon know… read moreAbstract: Silicene is a two-dimensional (2D) allotrope of silicon known to have a lower thermal conductivity than graphene; thus, more suitable for thermoelectric applications. This paper investigates the effect of hydrogenation on the thermal conductivity of silicene nanoribbon (SiNR) using equilibrium molecular dynamics (EMD) simulations. The simulations were carried out in Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) using a modified Tersoff potential that considers both Si-Si and Si-H interactions. The thermal conductivity of fully hydrogenated silicene nanoribbon (H-SiNR), also known as silicane nanoribbon, was found to be higher than that of pristine SiNR in all the temperatures and dimensions considered here. This anomalous enhancement in the thermal conductivity is similar to that found in hydrogenated silicon nanowires (H-SiNWs). A mechanism for this anomalous effect has been proposed relating the hydrogenation of SiNR with the stiffening and increase of the acoustic out-of-plane flexural (ZA) phonon modes. Also, for both SiNR and H-SiNR, the thermal conductivities generally increase as the dimensions are increased while they generally decrease as the temperatures are increased, in agreement to other reports. read less NOT USED (low confidence) C. G. G. Alipala, G. J. Paylaga, N. T. Paylaga, and R. Bantaculo, “Thermal Conductivity of Silicon-Graphene Nanoribbon (SiGNR): An Equilibrium Molecular Dynamics (EMD) Simulation,” Advanced Materials Research. 2015. link Times cited: 2 Abstract: Silicon-graphene nanoribbon (SiGNR), an allotrope of silicon… read moreAbstract: Silicon-graphene nanoribbon (SiGNR), an allotrope of silicon carbide with sp2 hybridization, gains interest nowadays in the world of two-dimensional materials. In this study, the thermal conductivity of SiGNR is investigated and compared to that of graphene nanoribbon (GNR) and silicene nanoribbon (SiNR). Molecular Dynamics using Tersoff potential through Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) using the Green-Kubo method is employed to predict the thermal conductivity of silicon-graphene materials with armchair chirality. The temperature is varied from 50 K, 77 K, 150 K, 300 K, 500 K, 700 K, 1000 K, 1200 K, and 1500 K with a fixed width of 10 nm and length of 50 nm. The length of the materials is also varied from 10 nm, 20 nm, 30 nm, 40 nm and 50 nm with a fixed temperature of 300 K. Our results show that the thermal conductivity of SiGNR is higher than that of GNR and is approximately 50% larger at room temperature, which may be attributed to the presence of Si atoms inducing larger flexural phonon density of states than in GNR and SiNR. Also, the thermal conductivity of SiGNR follows the same length-dependent behavior of GNR due to its long mean free path. This study presents new insights into the thermal properties of silicon-graphene which will be significant for nanoelectronic applications. read less NOT USED (low confidence) J.-W. Jiang, “Parametrization of Stillinger–Weber potential based on valence force field model: application to single-layer MoS2 and black phosphorus,” Nanotechnology. 2015. link Times cited: 223 Abstract: We propose parametrizing the Stillinger–Weber potential for … read moreAbstract: We propose parametrizing the Stillinger–Weber potential for covalent materials starting from the valence force-field model. All geometrical parameters in the Stillinger–Weber potential are determined analytically according to the equilibrium condition for each individual potential term, while the energy parameters are derived from the valence force-field model. This parametrization approach transfers the accuracy of the valence force field model to the Stillinger–Weber potential. Furthermore, the resulting Stilliinger–Weber potential supports stable molecular dynamics simulations, as each potential term is at an energy-minimum state separately at the equilibrium configuration. We employ this procedure to parametrize Stillinger–Weber potentials for single-layer MoS2 and black phosphorous. The obtained Stillinger–Weber potentials predict an accurate phonon spectrum and mechanical behaviors. We also provide input scripts of these Stillinger–Weber potentials used by publicly available simulation packages including GULP and LAMMPS. read less NOT USED (low confidence) X. Zhao et al., “Exploration of tetrahedral structures in silicate cathodes using a motif-network scheme,” Scientific Reports. 2015. link Times cited: 26 NOT USED (low confidence) A. Akimov and O. Prezhdo, “Large-Scale Computations in Chemistry: A Bird’s Eye View of a Vibrant Field.,” Chemical reviews. 2015. link Times cited: 171 NOT USED (low confidence) J. F. Thekkethala and S. P. Sathian, “The effect of graphene layers on interfacial thermal resistance in composite nanochannels with flow,” Microfluidics and Nanofluidics. 2015. link Times cited: 12 NOT USED (low confidence) A. Garg, V. Vijayaraghavan, J. Lam, P. Singru, and L. Gao, “A molecular simulation based computational intelligence study of a nano-machining process with implications on its environmental performance,” Swarm Evol. Comput. 2015. link Times cited: 30 NOT USED (low confidence) R. Bari et al., “Liquid phase exfoliation and crumpling of inorganic nanosheets.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 66 Abstract: Here we demonstrate through experiment and simulation the po… read moreAbstract: Here we demonstrate through experiment and simulation the polymer-assisted dispersion of inorganic 2D layered nanomaterials such as boron nitride nanosheets (BNNSs), molybdenum disulfide nanosheets (MoS2), and tungsten disulfide nanosheets (WS2), and we show that spray drying can be used to alter such nanosheets into a crumpled morphology. Our data indicate that polyvinylpyrrolidone (PVP) can act as a dispersant for the inorganic 2D layered nanomaterials in water and a range of organic solvents; the effectiveness of our dispersion process was characterized by UV-vis spectroscopy, microscopy and dynamic light scattering. Molecular dynamics simulations confirm that PVP readily physisorbs to BNNS surfaces. Collectively, these results indicate that PVP acts as a general dispersant for nanosheets. Finally, a rapid spray drying technique was utilized to convert these 2D dispersed nanosheets into 3D crumpled nanosheets; this is the first report of 3D crumpled inorganic nanosheets of any kind. Electron microscopy images confirm that the crumpled nanosheets (1-2 μm in diameter) show a distinctive morphology with dimples on the surface as opposed to a wrinkled, compressed surface, which matches earlier simulation results. These results demonstrate the possibility of scalable production of inorganic nanosheets with tailored morphology. read less NOT USED (low confidence) Z. Hui, P. He, Y. Dai, and A. Wu, “The optimal initial configuration of silicon-functionalized graphene for lithium-ion battery anode,” Proceedings of the Institution of Mechanical Engineers, Part N: Journal of Nanoengineering and Nanosystems. 2015. link Times cited: 0 Abstract: Silicon-functionalized graphene in its initial configuration… read moreAbstract: Silicon-functionalized graphene in its initial configuration, as anode materials for lithium-ion battery, will directly affect the battery’s reversible capacity, charge and discharge rate and service life. To present its optimal initial configuration, the relaxation and stretching properties of silicon-functionalized graphene were studied using molecular dynamics simulation with the Tersoff potential, the Lennard–Jones potential and the velocity Verlet time-stepping algorithm. In this study, many models of silicon-functionalized graphene with different arrangement of silicon atoms, different Si/C ratios, different vacancy defect ratios, different tension rates and different temperatures were primarily developed respectively to simulate the influence of different configurations on the volume, potential energy, elastic modulus, tensile strain, strength and other properties of the model, and we found that (1) the model with random arrangement of silicon atoms possessed biggest system potential energy, biggest volume and highest mechanics property among all models. (2) With the increasing amount of silicon atoms, the wavy corrugations and the peak became clearer, the potential energy decreased and volume increased. The model with Si/C ratio of 3.28% possessed highest mechanics property. (3) With the increasing vacancy, the system’s potential energy increased and volume decreased. The model with a vacancy defect ratio of 1% possessed highest mechanics property parameters. (4) Mechanics properties were the highest at the temperature of 300 K. read less NOT USED (low confidence) P. Süle and M. Szendrő, “Time-lapsed graphene moiré superlattices on Cu(1 1 1),” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 7 Abstract: We report classical molecular dynamics simulations (CMD) of … read moreAbstract: We report classical molecular dynamics simulations (CMD) of the moiré superlattice of graphene on Cu(1 1 1) using a new parameterized Abell–Tersoff potential for the graphene/Cu(1 1 1) interface fitted in this paper to nonlocal van der Waals density functional theory calculations. The interfacial force field with time-lapsed CMD provides superlattices in good quantitative agreement with the available experimental results. The long range coincidence supercells with nonequivalent moiré hills have also been identified and analyzed. Spot profile analysis reveals that the moiré spots are inequivalent over large areas, and their heights are randomly distributed. This result is in accordance with recent atomic force microscopy studies. Our simulations also shed light on the transient dynamics of the moiré superlattice in atomic detail. The moiré superlattice exhibits a pattern which is dynamical rather than statically pinned to the support, and can be observed mostly via time-lapsing. The instantaneous snapshots of the periodic moiré pattern at low temperature are already weakly disordered, lacking the apparent sharpness of the time-averaged pattern and of the scanning tunneling microscopy images. This suggests the existence of competition of orders—between a static (first-order) moiré superstructure and a dynamical (second-order) moiré superstructure. read less NOT USED (low confidence) A. Galashev, “Computer study of the removal of Cu from the graphene surface using Ar clusters,” Computational Materials Science. 2015. link Times cited: 22 NOT USED (low confidence) X. Duan, B. Zhou, Y. Wen, R. Chen, H. Zhou, and B. Shan, “Lattice inversion modified embedded atom method for bcc transition metals,” Computational Materials Science. 2015. link Times cited: 14 NOT USED (low confidence) J. Chen, G. Zhang, and B. Li, “Molecular Dynamics Simulations for Computing Thermal Conductivity of Nanomaterials: Molecular Dynamics Simulations for Computing Thermal Conductivity of Nanomaterials.” 2015. link Times cited: 2 NOT USED (low confidence) R. Rozada et al., “From graphene oxide to pristine graphene: revealing the inner workings of the full structural restoration.,” Nanoscale. 2015. link Times cited: 80 Abstract: High temperature annealing is the only method known to date … read moreAbstract: High temperature annealing is the only method known to date that allows the complete repair of a defective lattice of graphenes derived from graphite oxide, but most of the relevant aspects of such restoration processes are poorly understood. Here, we investigate both experimentally (scanning probe microscopy) and theoretically (molecular dynamics simulations) the thermal evolution of individual graphene oxide sheets, which is rationalized on the basis of the generation and the dynamics of atomic vacancies in the carbon lattice. For unreduced and mildly reduced graphene oxide sheets, the amount of generated vacancies was so large that they disintegrated at 1773-2073 K. By contrast, highly reduced sheets survived annealing and their structure could be completely restored at 2073 K. For the latter, a minor atomic-sized defect with six-fold symmetry was observed and ascribed to a stable cluster of nitrogen dopants. The thermal behavior of the sheets was significantly altered when they were supported on a vacancy-decorated graphite substrate, as well as for the overlapped/stacked sheets. In these cases, a net transfer of carbon atoms between neighboring sheets via atomic vacancies takes place, affording an additional healing process. Direct evidence of sheet coalescence with the step edge of the graphite substrate was also gathered from experiments and theory. read less NOT USED (low confidence) C. Zou, Y. Shin, A. V. van Duin, H. Fang, and Z.-kui Liu, “Molecular dynamics simulations of the effects of vacancies on nickel self-diffusion, oxygen diffusion and oxidation initiation in nickel, using the ReaxFF reactive force field,” Acta Materialia. 2015. link Times cited: 76 NOT USED (low confidence) J. Xu et al., “Engineering molecular dynamics simulation in chemical engineering,” Chemical Engineering Science. 2015. link Times cited: 16 NOT USED (low confidence) J. Joseph and Y. Lu, “Uniform shell modeling of vertically aligned multi-walled carbon nanotube arrays,” International Journal of Computational Materials Science and Engineering. 2014. link Times cited: 1 Abstract: Vertically aligned multi-walled carbon nanotube arrays (VA-M… read moreAbstract: Vertically aligned multi-walled carbon nanotube arrays (VA-MWCNTs) are novel carbon nanomaterials that have broader applications than individual carbon nanotubes (CNTs). This paper presents the uniform shell modeling of VA-MWCNTs which allow effectively design and characterize these complex materials. Multi-walled nanotubes in the arrays are approximated as equivalent single-walled CNTs and modeled with shell elements. The stiffness and Young's modulus obtained from the equivalent thickness method are comparable to those obtained from the modeling of actual multi-wall structures, but at much efficient computational efforts. VA-MWCNTs have also been designed at various architectures (square, FCC) and areal densities, and the resultant mechanical properties are analyzed. read less NOT USED (low confidence) M. Yaghoobi and G. Voyiadjis, “Effect of boundary conditions on the MD simulation of nanoindentation,” Computational Materials Science. 2014. link Times cited: 71 NOT USED (low confidence) X. Duan, B. Zhou, R. Chen, H. Zhou, Y. Wen, and B. Shan, “Development of lattice inversion modified embedded atom method and its applications,” Current Applied Physics. 2014. link Times cited: 11 NOT USED (low confidence) G. Ciccotti, M. Ferrario, and C. Schuette, “Molecular Dynamics Simulation,” Mechanics of Particle‐ and Fiber‐Reinforced Polymer Nanocomposites. 2014. link Times cited: 571 NOT USED (low confidence) Y.-Y. Liu, W.-X. Zhou, L.-M. Tang, and K. Chen, “An important mechanism for thermal rectification in graded nanowires,” Applied Physics Letters. 2014. link Times cited: 53 Abstract: In the quest for the origin of the different thermal rectify… read moreAbstract: In the quest for the origin of the different thermal rectifying behavior of two graded nanowires, we reveal the important role that standing waves play in the thermal transport properties of such graded structures. Evidence for the existence of standing waves is given from two angles, and one possible scenario of the origin of the standing wave is presented. The key point is that the formation of the standing wave, which greatly hinders the propagation of phonon waves, occurs only when the narrow end of the nanowire is at a higher temperature than the wide end, making the heat current flow preferably from the wide end to the narrow end. read less NOT USED (low confidence) S. Bhoi, T. Banerjee, and K. Mohanty, “Molecular dynamic simulation of spontaneous combustion and pyrolysis of brown coal using ReaxFF,” Fuel. 2014. link Times cited: 131 NOT USED (low confidence) P. Zhu, C. Qiu, F. Fang, D. Yuan, and X. Shen, “Molecular dynamics simulations of nanometric cutting mechanisms of amorphous alloy,” Applied Surface Science. 2014. link Times cited: 84 NOT USED (low confidence) M. P. Haag and M. Reiher, “Studying chemical reactivity in a virtual environment.,” Faraday discussions. 2014. link Times cited: 34 Abstract: Chemical reactivity of a set of reactants is determined by i… read moreAbstract: Chemical reactivity of a set of reactants is determined by its potential (electronic) energy (hyper)surface. The high dimensionality of this surface renders it difficult to efficiently explore reactivity in a large reactive system. Exhaustive sampling techniques and search algorithms are not straightforward to employ as it is not clear which explored path will eventually produce the minimum energy path of a reaction passing through a transition structure. Here, the chemist's intuition would be of invaluable help, but it cannot be easily exploited because (1) no intuitive and direct tool for the scientist to manipulate molecular structures is currently available and because (2) quantum chemical calculations are inherently expensive in terms of computational effort. In this work, we elaborate on how the chemist can be reintroduced into the exploratory process within a virtual environment that provides immediate feedback and intuitive tools to manipulate a reactive system. We work out in detail how this immersion should take place. We provide an analysis of modern semi-empirical methods which already today are candidates for the interactive study of chemical reactivity. Implications of manual structure manipulations for their physical meaning and chemical relevance are carefully analysed in order to provide sound theoretical foundations for the interpretation of the interactive reactivity exploration. read less NOT USED (low confidence) Z. V. Smagina, N. Stepina, V. Zinovyev, P. Novikov, P. Kuchinskaya, and A. Dvurechenskii, “Chains of quantum dot molecules grown on Si surface pre-patterned by ion-assisted nanoimprint lithography,” Applied Physics Letters. 2014. link Times cited: 7 Abstract: An original approach based on the combination of nanoimprint… read moreAbstract: An original approach based on the combination of nanoimprint lithography and ion irradiation through mask has been developed for fabrication of large-area periodical pattern on Si(100). Using the selective etching of regions amorphized by ion irradiation ordered structures with grooves and ridges were obtained. The shape and depth of the relief were governed by ion energy and by the number of etching stages as well. Laterally ordered chains of Ge quantum dots were fabricated by molecular beam epitaxy of Ge on the pre-patterned Si substrates. For small amount of Ge deposited chains contain separate quantum dot molecules. The increase of deposition amount leads to overlapping of quantum dot molecules with formation of dense homogeneous chains of quantum dots. It was shown that the residual irradiation-induced bulk defects underneath the grooves suppress nucleation of Ge islands at the bottom of grooves. On pre-patterned substrates with whole defect regions, etched quantum dots grow at the bottom of grooves.... read less NOT USED (low confidence) D. Huang, F. Gao, and D. Cardimona, “Multi-timescale microscopic theory for radiation degradation of electronic and optoelectronic devices,” arXiv: Materials Science. 2014. link Times cited: 2 Abstract: A multi-timescale hybrid model is proposed to study microsco… read moreAbstract: A multi-timescale hybrid model is proposed to study microscopically the degraded performance of electronic devices, covering three individual stages of radiation effects studies, including ultrafast displacement cascade, intermediate defect stabilization and cluster formation, as well as slow defect reaction and migration. Realistic interatomic potentials are employed in molecular-dynamics calculations for the first two stages up to 100\,ns as well as for the system composed of layers with thickness of hundreds times of lattice constant. These quasi-steady-state results for individual layers are input into a rate-diffusion theory as initial conditions to calculate the steady-state distribution of point defects in a mesoscopic-scale layered-structure system, including planar biased dislocation loops and spherical neutral voids, on a much longer time scale. Assisted by the density-functional theory for specifying electronic properties of point defects, the resulting spatial distributions of these defects and clusters are taken into account in studying the degradation of electronic and optoelectronic devices, e.g., carrier momentum-relaxation time, defect-mediated non-radiative recombination, defected-assisted tunneling of electrons and defect or charged-defect Raman scattering as well. Such theoretical studies are expected to be crucial in fully understanding the physical mechanism for identifying defect species, performance degradations in field-effect transistors, photodetectors, light-emitting diodes and solar cells, and in the development of effective mitigation methods during their microscopic structure design stages. read less NOT USED (low confidence) D. Delgado and R. Vila, “Hydrogen species in diamond: Molecular dynamics simulation in bulk diamond for fusion applications,” Journal of Nuclear Materials. 2014. link Times cited: 1 NOT USED (low confidence) E. Cieren, L. Colombet, S. Pitoiset, and R. Namyst, “ExaStamp: A Parallel Framework for Molecular Dynamics on Heterogeneous Clusters,” Euro-Par Workshops. 2014. link Times cited: 8 NOT USED (low confidence) H. Yasuoka, T. Imae, M. Kaneda, and K. Suga, “Molecular dynamics simulation for flow characteristics in nanochannels and single walled carbon nanotubes,” Journal of Physics: Conference Series. 2014. link Times cited: 1 Abstract: Flows in graphite-, diamond- and silicon-walled nanochannels… read moreAbstract: Flows in graphite-, diamond- and silicon-walled nanochannels are discussed by performing molecular dynamics simulations. Flows in carbon nanotubes (CNTs) and graphene- walled nanochannels are also investigated. It is found that the flow rate in the graphite-walled channel tends to be the largest because of its slippery wall structure by the short bond length and the high molecular density of the CNTs. The flow rate in the single walled CNT at a very narrow diameter tends to increase although such a tendency is not seen in the graphene-walled channel. read less NOT USED (low confidence) S. Saptarshi and G. Smitha, “Methodology for Evaluating Theproperties of Carbon Nano Tubes for Engineering Applications.” 2014. link Times cited: 0 Abstract: Owing to the superior mechanical properties of Carbon Nanotu… read moreAbstract: Owing to the superior mechanical properties of Carbon Nanotubes (CNTs), research has been initiated to use this nanomaterial in construction industry. The elastic modulus of CNT is of great importance in order to use CNTs in construction industry. For judicious and effective application of CNT in construction industry where mass scale utilization is inevitable, it is important to accurately assess the properties of CNTs since these nano fiber materials are extremely cost sensitive. There is no well established tool or method available for the engineers to determine the properties of CNT, other than to accept the manufacturer’s data. Nanoscale continuum theory uses a representative volume to determine the elastic modulus which does not include the effect of radius and aspect ratio. In the present study, numerical model is developed and the validated one is used to study the effects of geometrical variability’s on the properties of CNT. Further, a methodology has been proposed to determine the mechanical properties, such as tensile stiffness, of the Carbon nanotube with a given structural arrangement (chirality and radius) if the same for the basic configuration, graphene, is known. The procedure discussed in this study will be helpful to the engineers to easily estimate the properties such as elastic modulus or tensile stiffness of CNTs before incorporating into conventional materials for developing new materials with enhanced properties. read less NOT USED (low confidence) Y. Fu and J. H. Song, “On computing stress in polymer systems involving multi-body potentials from molecular dynamics simulation.,” The Journal of chemical physics. 2014. link Times cited: 14 Abstract: Hardy stress definition has been restricted to pair potentia… read moreAbstract: Hardy stress definition has been restricted to pair potentials and embedded-atom method potentials due to the basic assumptions in the derivation of a symmetric microscopic stress tensor. Force decomposition required in the Hardy stress expression becomes obscure for multi-body potentials. In this work, we demonstrate the invariance of the Hardy stress expression for a polymer system modeled with multi-body interatomic potentials including up to four atoms interaction, by applying central force decomposition of the atomic force. The balance of momentum has been demonstrated to be valid theoretically and tested under various numerical simulation conditions. The validity of momentum conservation justifies the extension of Hardy stress expression to multi-body potential systems. Computed Hardy stress has been observed to converge to the virial stress of the system with increasing spatial averaging volume. This work provides a feasible and reliable linkage between the atomistic and continuum scales for multi-body potential systems. read less NOT USED (low confidence) J. F. Thekkethala and S. P. Sathian, “The effect of graphene layers on interfacial thermal resistance in composite nanochannels with flow,” Microfluidics and Nanofluidics. 2014. link Times cited: 2 NOT USED (low confidence) K. Fung, C. Y. Tang, C. Cheung, and W. C. Law, “Molecular Dynamics Simulation of Plastic Deformation of Diamond at an Elevated Temperature,” Key Engineering Materials. 2014. link Times cited: 2 Abstract: Single point diamond tools are commonly used for ultraprecis… read moreAbstract: Single point diamond tools are commonly used for ultraprecision machining. At high cutting speeds, frictional contact and local heat may cause material damage to the diamond tool. The diamond crystal is softened and its mechanical strength decreases with the increase in temperature. Plastic deformation of diamonds was recently reported in some experimental studies. In this work, a molecular dynamics (MD) simulation was implemented to predict the deformation of single crystal diamond at various temperatures. Diamond is brittle at room temperature, however, it starts to exhibit plastic dislocation at a temperature above 1200 K under a confining pressure. The condition in ultraprecision machining is indeed a temperature gradient distribution at the tool tip, between the maximum temperature at the tool-workpiece interface and the average temperature at the core. The simulation results predicted that diamond deformed plastically under the gradient between 1500K and 860K. It is surprising that secondary cracks were resulted from the gradient, as comparing to a single slip obtained in an evenly distributed temperature. Bond dissociation nucleated the fractures along the (111) shuffle planes, perfect dislocation merely occurred in the hot zone and sp3-to-sp2 disorder at the cool zone. The temperature gradient created a lattice mismatch and nucleated the secondary cracks. The results give an insight that a catastrophic fracture and local material damage can occur at a diamond tool tip at the cutting temperature above 1200 K, due to softening and graphitization. read less NOT USED (low confidence) S. Ajori and R. Ansari, “Torsional buckling behavior of boron-nitride nanotubes using molecular dynamics simulations,” Current Applied Physics. 2014. link Times cited: 26 NOT USED (low confidence) R. Ansari and S. Ajori, “Molecular dynamics study of the torsional vibration characteristics of boron-nitride nanotubes,” Physics Letters A. 2014. link Times cited: 31 NOT USED (low confidence) S. Lee and W. Lu, “Controlling the number of graphene sheets exfoliated from graphite by designed normal loading and frictional motion,” Journal of Applied Physics. 2014. link Times cited: 8 Abstract: We use molecular dynamics to study the exfoliation of patter… read moreAbstract: We use molecular dynamics to study the exfoliation of patterned nanometer-sized graphite under various normal loading conditions for friction-induced exfoliation. Using highly ordered pyrolytic graphite (HOPG) as well as both amorphous and crystalline SiO2 substrate as example systems, we show that the exfoliation process is attributed to the corrugation of the HOPG surface and the atomistic roughness of the substrate when they contact under normal loading. The critical normal strain, at which the exfoliation occurs, is higher on a crystalline substrate than on an amorphous substrate. This effect is related to the atomistic flatness and stiffness of the crystalline surface. We observe that an increase of the van der Waals interaction between the graphite and the substrate results in a decrease of the critical normal strain for exfoliation. We find that the magnitude of the normal strain can effectively control the number of exfoliated graphene layers. This mechanism suggests a promising approach of applyi... read less NOT USED (low confidence) C. M. Handley and J. Behler, “Next generation interatomic potentials for condensed systems,” The European Physical Journal B. 2014. link Times cited: 66 NOT USED (low confidence) A. Yankovich et al., “Picometre-precision analysis of scanning transmission electron microscopy images of platinum nanocatalysts,” Nature Communications. 2014. link Times cited: 234 NOT USED (low confidence) R. Ganeev, “Characterization of Plasma Harmonics.” 2014. link Times cited: 0 NOT USED (low confidence) Y. Chen, F. Fang, X. Zhang, and X. Hu, “Molecular dynamics of nanometric processing of ion implanted monocrystalline silicon surfaces,” Transactions of Tianjin University. 2014. link Times cited: 2 NOT USED (low confidence) J. Eapen, K. Murty, and T. Burchell, “Understanding Creep Mechanisms in Graphite with Experiments, Multiscale Simulations, and Modeling.” 2014. link Times cited: 0 Abstract: Disordering mechanisms in graphite have a long history with … read moreAbstract: Disordering mechanisms in graphite have a long history with conflicting viewpoints. Using Raman and x-ray photon spectroscopy, electron microscopy, x-ray diffraction experiments and atomistic modeling and simulations, the current project has developed a fundamental understanding of early-to-late state radiation damage mechanisms in nuclear reactor grade graphite (NBG-18 and PCEA). We show that the topological defects in graphite play an important role under neutron and ion irradiation. read less NOT USED (low confidence) Y. Wang, J. Shi, and C. Ji, “A numerical study of residual stress induced in machined silicon surfaces by molecular dynamics simulation,” Applied Physics A. 2014. link Times cited: 58 NOT USED (low confidence) P. Tsai and Y. Jeng, “Theoretical investigation of thermally induced coalescence mechanism of single-wall carbon nanohorns and their mechanical properties,” Computational Materials Science. 2014. link Times cited: 5 NOT USED (low confidence) J. Yan, L.-W. Zhang, K. Liew, and L. He, “A higher-order gradient theory for modeling of the vibration behavior of single-wall carbon nanocones,” Applied Mathematical Modelling. 2014. link Times cited: 34 NOT USED (low confidence) B. Yakobson and T. Dumitricǎ, “Retracted: Nanomechanics: Physics Between Engineering and Chemistry.” 2014. link Times cited: 0 NOT USED (low confidence) Y. Chen, F. Fang, X. Zhang, and X. Hu, “Molecular dynamics of nanometric processing of ion implanted monocrystalline silicon surfaces,” Transactions of Tianjin University. 2014. link Times cited: 0 NOT USED (low confidence) M. C. Nguyen, X. Zhao, C. Wang, and K. Ho, “sp3-hybridized framework structure of group-14 elements discovered by genetic algorithm,” Physical Review B. 2014. link Times cited: 21 Abstract: Group-14 elements, including C, Si, Ge, and Sn, can form var… read moreAbstract: Group-14 elements, including C, Si, Ge, and Sn, can form various stable and metastable structures. Finding new metastable structures of group-14 elements with desirable physical properties for new technological applications has attracted a lot of interest. Using a genetic algorithm, we discovered a new low-energy metastable distorted sp3-hybridized framework structure of the group-14 elements. It has P42/mnm symmetry with 12 atoms per unit cell. The void volume of this structure is as large as 139.7A3 for Si P42/mnm, and it can be used for gas or metal-atom encapsulation. Band-structure calculations show that P42/mnm structures of Si and Ge are semiconducting with energy band gaps close to the optimal values for optoelectronic or photovoltaic applications. With metal-atom encapsulation, the P42/mnm structure would also be a candidate for rattling-mediated superconducting or used as thermoelectric materials. read less NOT USED (low confidence) S. Goel, “The current understanding on the diamond machining of silicon carbide,” Journal of Physics D: Applied Physics. 2014. link Times cited: 139 Abstract: The Glenn Research Centre of NASA, USA (www.grc.nasa.gov/WWW… read moreAbstract: The Glenn Research Centre of NASA, USA (www.grc.nasa.gov/WWW/SiC/, silicon carbide electronics) is in pursuit of realizing bulk manufacturing of silicon carbide (SiC), specifically by mechanical means. Single point diamond turning (SPDT) technology which employs diamond (the hardest naturally-occurring material realized to date) as a cutting tool to cut a workpiece is a highly productive manufacturing process. However, machining SiC using SPDT is a complex process and, while several experimental and analytical studies presented to date aid in the understanding of several critical processes of machining SiC, the current knowledge on the ductile behaviour of SiC is still sparse. This is due to a number of simultaneously occurring physical phenomena that may take place on multiple length and time scales. For example, nucleation of dislocation can take place at small inclusions that are of a few atoms in size and once nucleated, the interaction of these nucleations can manifest stresses on the micrometre length scales. The understanding of how these stresses manifest during fracture in the brittle range, or dislocations/phase transformations in the ductile range, is crucial to understanding the brittle–ductile transition in SiC. Furthermore, there is a need to incorporate an appropriate simulation-based approach in the manufacturing research on SiC, owing primarily to the number of uncertainties in the current experimental research that includes wear of the cutting tool, poor controllability of the nano-regime machining scale (effective thickness of cut), and coolant effects (interfacial phenomena between the tool, workpiece/chip and coolant), etc. In this review, these two problems are combined together to posit an improved understanding on the current theoretical knowledge on the SPDT of SiC obtained from molecular dynamics simulation. read less NOT USED (low confidence) M. Gatchell et al., “Non-statistical fragmentation of PAHs and fullerenes in collisions with atoms,” International Journal of Mass Spectrometry. 2014. link Times cited: 34 NOT USED (low confidence) A. Galashev, “Computer stability test for aluminum films heated on a graphene sheet,” Technical Physics. 2014. link Times cited: 20 NOT USED (low confidence) H. Zettergren et al., “Bond formation in C+59 – C60 collisions,” Journal of Physics: Conference Series. 2014. link Times cited: 1 Abstract: In this work, we show that keV-ions are able to remove singl… read moreAbstract: In this work, we show that keV-ions are able to remove single carbon atoms from individual fullerenes in clusters of C60 molecules. This very efficiently leads to the formation of exotic C+119 dumbbell molecules through secondary C+59 + C60 collisions within the fragmenting cluster. Such molecular fusion processes are inherently different from those induced by photons where only products with even numbers of carbon atoms are observed. Thus, ion collisions ignite unique and hitherto overlooked secondary reactions in small aggregates of matter. This relates to the question on how complex molecules may form in e.g. space. read less NOT USED (low confidence) J. N. Sarma, R. Chowdhury, R. Jayaganthan, and F. Scarpa, “Atomistic Studies on Tensile Mechanics of BN Nanotubes in the Presence of Defects,” International Journal of Nanoscience. 2014. link Times cited: 8 Abstract: Boron nitride nanotubes (BNNTs) are of immense importance du… read moreAbstract: Boron nitride nanotubes (BNNTs) are of immense importance due to their many interesting functional features, notably biocompatibility and piezoelectricity and dominant mechanical strength as compared to carbon nanotubes (CNTs). The reliable implementation of these structures in an application is inherently related to its mechanical characteristics under external loads. The presence of defects in these structures severely affects the tensile properties. The effect of presence of point, line and Stone–Wales (SW) defects on the tensile behavior of BNNTs is systematically investigated by applying reactive force fields in molecular dynamics (MD) framework. Reactive force fields effectively describe the bond breaking and bond forming mechanism for BNNTs that are important for a practical situation. The Young's modulus of single-walled (10,0) BNNTs of length 100 nm has been found to be nearly 1.098 TPa, in good agreement with the available reports. The presence of defects has been shown to significantly reduce the tensile strength of the tube, while the number and separation of the defects effectively contribute to the percentage reduction. In addition, the effect of tube diameter and also the initial temperature are observed to strongly influence the tensile characteristics of BNNTs, indicating increased auxetic behavior than CNTs. read less NOT USED (low confidence) H. Xie, M. Hu, and H. Bao, “Thermal conductivity of silicene from first-principles,” Applied Physics Letters. 2014. link Times cited: 148 Abstract: Silicene, as a graphene-like two-dimensional material, now r… read moreAbstract: Silicene, as a graphene-like two-dimensional material, now receives exceptional attention of a wide community of scientists and engineers beyond graphene. Despite extensive study on its electric property, little research has been done to accurately calculate the phonon transport of silicene so far. In this paper, thermal conductivity of monolayer silicene is predicted from first-principles method. At 300 K, the thermal conductivity of monolayer silicene is found to be 9.4 W/mK and much smaller than bulk silicon. The contributions from in-plane and out-of-plane vibrations to thermal conductivity are quantified, and the out-of-plane vibration contributes less than 10% of the overall thermal conductivity, which is different from the results of the similar studies on graphene. The difference is explained by the presence of small buckling, which breaks the reflectional symmetry of the structure. The flexural modes are thus not purely out-of-plane vibration and have strong scattering with other modes. read less NOT USED (low confidence) M. Kozłowski, R. Abdank-Kozubski, and C. Goyhenex, “Superstructure Transformations in High-Temperature Intermetallic Nanolayers: Atomistic Simulation,” Diffusion Foundations. 2014. link Times cited: 1 Abstract: Superstructure transformation processes in intermetallics ha… read moreAbstract: Superstructure transformation processes in intermetallics have beenstudied at the atomistic scale using Monte Carlo algorithms within two dis-tinct models: two-body interactions Ising-like system and Analytic Bond-Order Potentials. The transformation from “in-plane” to “off-plane” L10 vari-ant in [001]-oriented FePt nano-layers was observed and analysed by analyt-ical calculations providing clear explanation of the origin of the process, aswell as by “rigid-lattice” and “off-lattice” Monte Carlo simulations showingthe kinetics of the superstructure transformation. read less NOT USED (low confidence) Z. Zhang, Y. Chen, and H. Zheng, “A modified Stillinger–Weber potential-based hyperelastic constitutive model for nonlinear elasticity,” International Journal of Solids and Structures. 2014. link Times cited: 25 NOT USED (low confidence) A. Sgouros, M. Sigalas, K. Papagelis, G. Kalosakas, and G. Kalosakas, “Transforming graphene nanoribbons into nanotubes by use of point defects,” Journal of Physics: Condensed Matter. 2014. link Times cited: 11 Abstract: Using molecular dynamics simulations with semi-empirical pot… read moreAbstract: Using molecular dynamics simulations with semi-empirical potentials, we demonstrate a method to fabricate carbon nanotubes (CNTs) from graphene nanoribbons (GNRs), by periodically inserting appropriate structural defects into the GNR crystal structure. We have found that various defect types initiate the bending of GNRs and eventually lead to the formation of CNTs. All kinds of carbon nanotubes (armchair, zigzag, chiral) can be produced with this method. The structural characteristics of the resulting CNTs, and the dependence on the different type and distribution of the defects, were examined. The smallest (largest) CNT obtained had a diameter of ∼5 Å (∼39 Å). Proper manipulation of ribbon edges controls the chirality of the CNTs formed. Finally, the effect of randomly distributed defects on the ability of GNRs to transform into CNTs is considered. read less NOT USED (low confidence) G. Opletal and S. Russo, “Atomistic Modeling and Simulations of Chalcogenide Glasses.” 2014. link Times cited: 1 NOT USED (low confidence) X. Qin, T. Gao, W. Yan, X. Guo, and Q. Xie, “Molecular dynamics simulation of graphene bombardment with Si ion,” Journal of Molecular Structure. 2014. link Times cited: 14 NOT USED (low confidence) S. Hollerer, “Numerical validation of a concurrent atomistic-continuum multiscale method and its application to the buckling analysis of carbon nanotubes,” Computer Methods in Applied Mechanics and Engineering. 2014. link Times cited: 17 NOT USED (low confidence) M. C. Nguyen, X. Zhao, Y. Wang, C. Wang, and K. Ho, “Genetic algorithm prediction of crystal structure of metastable Si-IX phase,” Solid State Communications. 2014. link Times cited: 7 NOT USED (low confidence) Y. Sun and K. Liew, “Effect of higher-order deformation gradients on buckling of single-walled carbon nanotubes,” Composite Structures. 2014. link Times cited: 18 NOT USED (low confidence) B. Mortazavi, M. Pötschke, and G. Cuniberti, “Multiscale modeling of thermal conductivity of polycrystalline graphene sheets.,” Nanoscale. 2014. link Times cited: 94 Abstract: We developed a multiscale approach to explore the effective … read moreAbstract: We developed a multiscale approach to explore the effective thermal conductivity of polycrystalline graphene sheets. By performing equilibrium molecular dynamics (EMD) simulations, the grain size effect on the thermal conductivity of ultra-fine grained polycrystalline graphene sheets is investigated. Our results reveal that the ultra-fine grained graphene structures have thermal conductivity one order of magnitude smaller than that of pristine graphene. Based on the information provided by the EMD simulations, we constructed finite element models of polycrystalline graphene sheets to probe the thermal conductivity of samples with larger grain sizes. Using the developed multiscale approach, we also investigated the effects of grain size distribution and thermal conductivity of grains on the effective thermal conductivity of polycrystalline graphene. The proposed multiscale approach on the basis of molecular dynamics and finite element methods could be used to evaluate the effective thermal conductivity of polycrystalline graphene and other 2D structures. read less NOT USED (low confidence) Y. Liu, G. F. Zhou, L. He, and H. Ye, “Studying the rotation induced super-lattices on graphite using a type-criterion potential based molecular dynamics method,” Computational Materials Science. 2014. link Times cited: 0 NOT USED (low confidence) R. E. Roman and S. W. Cranford, “Mechanical properties of silicene,” Computational Materials Science. 2014. link Times cited: 89 NOT USED (low confidence) J. M. Rodríguez-Aguirre and M. M. Jakas, “Classical-trajectory calculations of the electronic stopping cross-section for low-energy H and H+ projectiles by H2-molecules,” Radiation Effects and Defects in Solids. 2014. link Times cited: 0 Abstract: A model that enables the classical-trajectory simulation of … read moreAbstract: A model that enables the classical-trajectory simulation of the interaction between an atomic particle and a target containing one or more electrons is devised. It makes use of the so-called Gaussian kernel approximation and ad-hoc potentials. In this way, the most relevant quantum properties of the electron can be preserved and, at the same time, still using classical mechanics to solve the response of the electronic system to the presence of a moving, heavy charge. As a first step to assessing the proposed model we calculate the electronic stopping cross-section for 1–20 keV Protons and Hydrogen impinging upon atomic and molecular Hydrogen targets. The results show a fairly good agreement between experiments and previous theoretical calculations over the entire bombarding energy studied in this paper. read less NOT USED (low confidence) A. Khoei, H. DorMohammadi, and A. Aramoon, “A temperature-related boundary Cauchy–Born method for multi-scale modeling of silicon nano-structures,” Physics Letters A. 2014. link Times cited: 8 NOT USED (low confidence) W. M. Brown and M. Yamada, “Implementing molecular dynamics on hybrid high performance computers - Three-body potentials,” Comput. Phys. Commun. 2013. link Times cited: 106 NOT USED (low confidence) X.-Y. Sun, C. Li, Y. T. Gu, and X.-Q. Feng, “Mechanical properties of bioinspired bicontinuous nanocomposites,” Science & Engineering Faculty. 2013. link Times cited: 14 NOT USED (low confidence) J. Yan, K. Liew, and L. He, “Buckling and post-buckling of single-wall carbon nanocones upon bending,” Composite Structures. 2013. link Times cited: 24 NOT USED (low confidence) Z. Q. Li, J. Wang, and T. Sun, “Atomistic Simulations of Ultrashort Pulsed Laser Ablation of Polycrystalline Diamond,” Current Nanoscience. 2013. link Times cited: 10 NOT USED (low confidence) M. Tewes et al., “Quantitative Composition Evaluation from HAADF-STEM in GeSi/Si Heterostructures,” Journal of Physics: Conference Series. 2013. link Times cited: 8 Abstract: High-angle annular dark field scanning transmission electron… read moreAbstract: High-angle annular dark field scanning transmission electron microscopy has been successfully used for composition evaluation in various material systems. In this work, the quantitative applicability of this method to GeSi/Si heterostructures was studied. Reference images were simulated by frozen lattice multislice simulations for different Ge concentrations accounting for static atomic displacements and biaxial strain due to pseudomorphic growth. Specimen thickness and composition are obtained by comparison of simulated and normalised experimental intensities. The measured thickness of a pure Si wedge specimen is compared to thickness determined from Pendellösung fringes in dark field micrographs. The deviation is below 10 nm coinciding with the accuracy of prior works. The composition of a GeSi-layer structure was measured in a calibration sample of known concentration and good agreement is found. Two-dimensional concentration maps of a GeSi/Si transistor structure were created. Measured concentrations agree with nominal values. However, strain fields in the Si lead to a variation of the image intensity causing an artificial fluctuation of the measured concentrations of ±4%. read less NOT USED (low confidence) F. Meyer et al., “Surface-morphology changes and damage in hot tungsten by impact of 80 eV – 12 keV He-ions and keV-energy self-atoms,” Journal of Physics: Conference Series. 2013. link Times cited: 7 Abstract: We report results of measurements on the evolution of the su… read moreAbstract: We report results of measurements on the evolution of the surface morphology of a hot tungsten surface due to impacting low-energy (80 – 12,000 eV) He ions, performed at the ORNL Multicharged Ion Research Facility (MIRF). Surface-morphology changes were investigated over a broad range of fluences, energies and temperatures for both virgin and pre-damaged W-targets. At low fluences, ordered coral-like and ridge-like surface structures are observed, with great grain-to-grain variability. At the largest fluences, individual grain characteristics disappear in FIB/SEM scans, and the entire surface is covered by a multitude of near-surface bubbles with a broad range of sizes, and disordered whisker growth, while in top-down SEM imaging the surface is virtually indistinguishable from the nanofuzz produced on linear plasma devices. These features are evident at progressively lower fluences as the He-ion energy is increased. In addition, simulations were carried out of damage caused by cumulative bombardment of 1 keV W self-atoms, using LAMMPS at the Kraken supercomputing facility of the University of Tennessee. The simulations show strong defect-recombination effects that lead to a saturation of the total defect number after a few hundred impacts, while sputtering and implantation lead to an imbalance of the vacancy and interstitial numbers. read less NOT USED (low confidence) B. Motevalli, A. Montazeri, J. Z. Liu, and H. Rafii-Tabar, “Comparison of continuum-based and atomistic-based modeling of axial buckling of carbon nanotubes subject to hydrostatic pressure,” Computational Materials Science. 2013. link Times cited: 11 NOT USED (low confidence) Y. Sasajima, T. Osada, N. Ishikawa, and A. Iwase, “Computer simulation of structural modifications induced by highly energetic ions in uranium dioxide,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2013. link Times cited: 3 NOT USED (low confidence) B. Mortazavi and S. Ahzi, “Thermal conductivity and tensile response of defective graphene: A molecular dynamics study,” Carbon. 2013. link Times cited: 231 NOT USED (low confidence) U. Monteverde, M. Migliorato, J. Pal, and D. Powell, “Elastic and vibrational properties of group IV semiconductors in empirical potential modelling,” Journal of Physics: Condensed Matter. 2013. link Times cited: 8 Abstract: We have developed an interatomic potential that with a singl… read moreAbstract: We have developed an interatomic potential that with a single set of parameters is able to accurately describe at the same time the elastic, vibrational and thermodynamics properties of semiconductors. The simultaneous inclusion of radial and angular forces of the interacting atom pairs (short range) together with the influence of the broken crystal symmetry when the atomic arrangement is out of equilibrium (long range) results in correct predictions of all of the phonon dispersion spectrum and mode-Grüneisen parameters of silicon and germanium. The long range interactions are taken into account up to the second nearest neighbours, to correctly influence the elastic and vibrational properties, and therefore represent only a marginal computational cost compared to the full treatment of other proposed potentials. Results of molecular dynamics simulations are compared with those of ab initio calculations, showing that when our proposed potential is used to perform the initial stages of the structural relaxation, a significant reduction of the computational time needed during the geometry optimization of density functional theory simulations is observed. read less NOT USED (low confidence) Y. Wang, J. Shi, and C. Ji, “A numerical study of residual stress induced in machined silicon surfaces by molecular dynamics simulation,” Applied Physics A. 2013. link Times cited: 0 NOT USED (low confidence) R. Ansari, R. Gholami, and S. Ajori, “Torsional Vibration Analysis of Carbon Nanotubes Based on the Strain Gradient Theory and Molecular Dynamic Simulations,” Journal of Vibration and Acoustics. 2013. link Times cited: 37 Abstract: In the current study, the torsional vibration of carbon nano… read moreAbstract: In the current study, the torsional vibration of carbon nanotubes is examined using the strain gradient theory and molecular dynamic simulations. The model developed based on this gradient theory enables us to interpret size effect through introducing material length scale parameters. The model accommodates the modified couple stress and classical models when two or all material length scale parameters are set to zero, respectively. Using Hamilton's principle, the governing equation and higher-order boundary conditions of carbon nanotubes are obtained. The generalized differential quadrature method is utilized to discretize the governing differential equation of the present model along with two boundary conditions. Then, molecular dynamic simulations are performed for a series of carbon nanotubes with different aspect ratios and boundary conditions, the results of which are matched with those of the present strain gradient model to extract the appropriate value of the length scale parameter. It is found that the present model with properly calibrated value of length scale parameter has a good capability to predict the torsional vibration behavior of carbon nanotubes. read less NOT USED (low confidence) I. Chang and C.-M. Huang, “Vibrational Behavior of Single-Walled Carbon Nanotubes: Atomistic Simulations,” Japanese Journal of Applied Physics. 2013. link Times cited: 7 Abstract: This study examines the vibrational behaviors of both armcha… read moreAbstract: This study examines the vibrational behaviors of both armchair and zigzag carbon nanotubes (CNTs). The natural longitudinal/flexural/torsional/radial frequencies of CNTs were extracted and analyzed simultaneously from an equilibrium molecular dynamics (MD) simulation without imposing any initial modal displacement or force. Initial random atomic velocities, which were assigned to fit the simulated temperature, could be considered as an excitation on CNTs composing of wide range of spatial frequencies. The position and velocity of each atom at every time step was calculated using finite difference algorithm, and fast Fourier transform (FFT) was used to perform time-to-frequency domain transform. The effects of CNT length, radius, chirality, and boundary condition on the vibrational behaviors of CNTs were systematically examined. Moreover, the simulated natural frequencies and mode shapes were compared with the predictions based on continuum theories, i.e., rod, Euler–Bernoulli beam and nonlocal Timoshenko beam, to examine their applicability in nanostructures. read less NOT USED (low confidence) S. D. Nath, “Study of the effect of sizes on the structural properties of SiO2 glass by molecular dynamics simulations,” Journal of Non-crystalline Solids. 2013. link Times cited: 8 NOT USED (low confidence) L. Chen, Y. Wang, H. Bu, and Y. Chen, “Simulations of the anisotropy of friction force between a silicon tip and a substrate at nanoscale,” Proceedings of the Institution of Mechanical Engineers, Part N: Journal of Nanoengineering and Nanosystems. 2013. link Times cited: 3 Abstract: In this article, the anisotropy of the friction force with a… read moreAbstract: In this article, the anisotropy of the friction force with a variety of rotation angles between a silicon tip and a substrate was investigated using molecular dynamics simulations. When the silicon tip slides over the silicon substrate under incommensurate contacting surfaces, the results of the simulations illustrate that the sliding friction forces decrease with the increase of the rotation angle from 0° to 45° and increase with the increase of the rotation angle from 45° to 90°. Furthermore, an approximate symmetrical curve of the friction force for the 45° angle can be obtained, and the friction force is minimum and close to superlubricity at the 45° angle. However, at the same angle, the friction force stops decreasing with the increase of the temperature and its magnitude depends mainly on the degree of the commensurability of the contacting surfaces rather than the temperature. read less NOT USED (low confidence) T. Liang et al., “Classical atomistic simulations of surfaces and heterogeneous interfaces with the charge-optimized many body (COMB) potentials,” Materials Science & Engineering R-reports. 2013. link Times cited: 207 NOT USED (low confidence) N. Podolska and A. I. Zhmakin, “Semiconductor nanostructure properties. Molecular Dynamic Simulations,” Journal of Physics: Conference Series. 2013. link Times cited: 0 Abstract: The need for research is based on the fact that development … read moreAbstract: The need for research is based on the fact that development of non-planar semiconductor nanosystems and nanomaterials with controlled properties is an important scientific and industrial problem. So, final scientific and technological problem is the creation of adequate modern methods and software for growth and properties simulation and optimization of various III-V (GaAs, InAs, InP, InGaAs etc.) nanostructures (e.g. nanowires) with controlled surface morphology, crystal structure, optical, transport properties etc. Accordingly, now we are developing a specialized computer code for atomistic simulation of structural (distribution of atoms and impurities, elastic and force constants, strain distribution etc.) and thermodynamic (mixing energy, interaction energy, surface energy etc.) properties of the nanostructures. Some simulation results are shown too. read less NOT USED (low confidence) X. W. Zhou, D. Ward, J. E. Martin, F. Swol, J. Cruz-Campa, and D. Zubia, “Stillinger-Weber potential for the II-VI elements Zn-Cd-Hg-S-Se-Te,” Physical Review B. 2013. link Times cited: 86 Abstract: X. W. Zhou,1,* D. K. Ward,2 J. E. Martin,3 F. B. van Swol,4 … read moreAbstract: X. W. Zhou,1,* D. K. Ward,2 J. E. Martin,3 F. B. van Swol,4 J. L. Cruz-Campa,5 and D. Zubia6 1Mechanics of Materials Department, Sandia National Laboratories, Livermore, California 94550, USA 2Radiation and Nuclear Detection Materials and Analysis Department, Sandia National Laboratories, Livermore, California 94550, USA 3Nanoscale Sciences Department, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA 4Computational Materials and Data Science Department, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA 5MEMS Technologies Department, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA 6Department of Electrical Engineering, University of Texas at El Paso, El Paso, Texas 79968, USA (Received 30 May 2013; published 9 August 2013; corrected 13 November 2013) read less NOT USED (low confidence) C. Hou et al., “Petascale molecular dynamics simulation of crystalline silicon on Tianhe-1A,” The International Journal of High Performance Computing Applications. 2013. link Times cited: 24 Abstract: An efficient and highly scalable bond-order potential code h… read moreAbstract: An efficient and highly scalable bond-order potential code has been developed for the molecular dynamics simulation of bulk silicon, reaching 1.87 Pflops (floating point operations per second) in single precision on 7168 graphic processing units (GPUs) of the Tianhe-1A system. Furthermore, by coupling GPUs and central processing units, we also simulated surface reconstruction of crystalline silicon at the sub-millimeter scale with more than 110 billion atoms, reaching 1.17 Pflops in single precision plus 92.1 Tflops in double precision on the entire Tianhe-1A system. Such simulations can provide unprecedented insight into a variety of microscopic behaviors or structures, such as doping, defects, grain boundaries, and surface reactions. read less NOT USED (low confidence) C. Björkas and K. Nordlund, “Variables affecting simulated Be sputtering yields,” Journal of Nuclear Materials. 2013. link Times cited: 12 NOT USED (low confidence) J.-W. Jiang, H. S. Park, and T. Rabczuk, “Molecular dynamics simulations of single-layer molybdenum disulphide (MoS2): Stillinger-Weber parametrization, mechanical properties, and thermal conductivity,” Journal of Applied Physics. 2013. link Times cited: 303 Abstract: We present a parameterization of the Stillinger-Weber potent… read moreAbstract: We present a parameterization of the Stillinger-Weber potential to describe the interatomic interactions within single-layer MoS2 (SLMoS2). The potential parameters are fitted to an experimentally obtained phonon spectrum, and the resulting empirical potential provides a good description for the energy gap and the crossover in the phonon spectrum. Using this potential, we perform classical molecular dynamics simulations to study chirality, size, and strain effects on the Young's modulus and the thermal conductivity of SLMoS2. We demonstrate the importance of the free edges on the mechanical and thermal properties of SLMoS2 nanoribbons. Specifically, while edge effects are found to reduce the Young's modulus of SLMoS2 nanoribbons, the free edges also reduce the thermal stability of SLMoS2 nanoribbons, which may induce melting well below the bulk melt temperature. Finally, uniaxial strain is found to efficiently manipulate the thermal conductivity of infinite, periodic SLMoS2. read less NOT USED (low confidence) J. Yan, J. Yan, K. Liew, and L. He, “Free vibration analysis of single-walled carbon nanotubes using a higher-order gradient theory,” Journal of Sound and Vibration. 2013. link Times cited: 45 NOT USED (low confidence) I. Szlufarska, K. Ramesh, and D. Warner, “Simulating Mechanical Behavior of Ceramics Under Extreme Conditions,” Annual Review of Materials Research. 2013. link Times cited: 14 Abstract: The mechanical behavior of ceramics in extreme environments … read moreAbstract: The mechanical behavior of ceramics in extreme environments can be qualitatively different from that observed at ambient conditions and at typical loading rates. For instance, during shock loading the fracture of ceramics is not controlled by the largest flaw. Computer simulations play an increasingly important role in understanding and predicting material behavior, in particular under conditions in which experiments might be challenging or expensive. Here, we review the strengths and limitations of simulation techniques that are most commonly used to model the mechanical behavior of ceramics. We discuss specific application areas of simulations, focusing on the effects of high strain rate, confined deformation volume, altered material chemistry, and high temperature. We conclude by providing examples of future opportunities for modeling studies in this field. read less NOT USED (low confidence) K. Tada, M. Yasuda, T. Mitsueda, R. Honda, H. Kawata, and Y. Hirai, “Molecular dynamics study of electron irradiation effects on mechanical properties of carbon nanotubes,” Microelectronic Engineering. 2013. link Times cited: 9 NOT USED (low confidence) G. Norman and V. Stegailov, “Stochastic theory of the classical molecular dynamics method,” Mathematical Models and Computer Simulations. 2013. link Times cited: 0 NOT USED (low confidence) R. Promyoo, H. El-Mounayri, V. K. Karingula, and K. Varahramyan, “AFM-Based Nanofabrication: Modeling, Simulation, and Experimental Verification.” 2013. link Times cited: 2 Abstract: Recent developments in science and engineering have advanced… read moreAbstract: Recent developments in science and engineering have advanced the fabrication techniques for micro/nanodevices. Among them, atomic force microscope (AFM) has already been used for nanomachining and fabrication of micro/nanodevices. In this paper, a computational model for AFM-based nanofabrication processes is being developed. Molecular Dynamics (MD) technique is used to model and simulate mechanical indentation and scratching at the nanoscale. The effects of AFM-tip radius and crystal orientation are investigated. The simulation is also used to study the effect of the AFM tip speed on the indentation force at the interface between the tip and the substrate/workpiece The material deformation and indentation geometry are extracted from the final locations of atoms, which are displaced by the rigid indenter. Material properties including modulus of elasticity and hardness are estimated. It is found that properties vary significantly at the nanoscale. AFM is used to conduct actual nanoindentation and scratching, to validate the MD simulation. Qualitative agreement is found. Finally, AFM-based fabrication of nanochannels/nanofluidic devices is conducted using different applied forces, scratching length, and feed rate.Copyright © 2013 by ASME read less NOT USED (low confidence) B. Li, “Formation of helicity in an armchair single-walled carbon nanotube during tensile loading,” Computational Materials Science. 2013. link Times cited: 4 NOT USED (low confidence) R. Ansari, A. Shahabodini, and H. Rouhi, “A thickness-independent nonlocal shell model for describing the stability behavior of carbon nanotubes under compression,” Composite Structures. 2013. link Times cited: 35 NOT USED (low confidence) I. Chang, “Molecular dynamics investigation of carbon nanotube resonance,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 18 Abstract: In this work, a methodology to directly extract resonant inf… read moreAbstract: In this work, a methodology to directly extract resonant information from an equilibrium molecular dynamics simulation is proposed and demonstrated by analyzing the vibrational behavior of carbon nanotubes (CNTs). Different vibrational motions, i.e. longitudinal, transverse, rotational and radial, could be easily distinguished and computed through the time sequence of the velocity components of atoms at the equilibrating process. Fast Fourier transform is adopted to perform the transformation of vibration information from time to frequency domain. The effects of CNT length, radius and boundary condition on the resonant behaviors of CNTs are systematically investigated. Moreover, the simulation results are compared with those predicted based on the Euler–Bernoulli beam theory. Note that the simulated longitudinal and rotational resonant behaviors agree quite well with the theoretical prediction and a slight deviation is observed in the transverse prediction. read less NOT USED (low confidence) S. Di, Z. Yao, M. Daymond, and F. Gao, “Molecular dynamics simulations of irradiation cascades in alpha-zirconium under macroscopic strain,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2013. link Times cited: 36 NOT USED (low confidence) A. Meinander, C. Björkas, and K. Nordlund, “The effect of hydrocarbon chemistry on sputtering in mixed Be–C–H materials,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2013. link Times cited: 2 NOT USED (low confidence) T. Vazhappilly and D. Micha, “Atomic modeling of structural and optical properties of amorphous silicon,” Chemical Physics Letters. 2013. link Times cited: 7 NOT USED (low confidence) C. Hou, J. Xu, P. Wang, W. Huang, and X. Wang, “Efficient GPU-accelerated molecular dynamics simulation of solid covalent crystals,” Comput. Phys. Commun. 2013. link Times cited: 28 NOT USED (low confidence) A. Oluwajobi and X. Chen, “Effects of interatomic potentials on the determination of the minimum depth of cut in nanomachining,” International Journal of Abrasive Technology. 2013. link Times cited: 4 Abstract: The minimum depth of cut (MDC) is a major limiting factor on… read moreAbstract: The minimum depth of cut (MDC) is a major limiting factor on achievable accuracy in nanomachining, because the generated surface roughness is primarily attributed to the ploughing process when the uncut chip thickness is less than the MDC. This paper presents the material removal in a nanomachining process, where a sharp diamond tool with an edge radius of few atoms acts on a crystalline copper workpiece. The molecular dynamics (MD) simulation results show the phenomena of rubbing, ploughing and cutting. The formation of chip occurred from the depth of cut thickness of 1~1.5 nm. Also, the effects of the interatomic potentials on the MDC have been presented. read less NOT USED (low confidence) V. Tomar, “Timescaling in Multiscale Mechanics of Nanowires and Nanocrystalline Materials.” 2013. link Times cited: 0 NOT USED (low confidence) V. Brázdová and D. Bowler, “Calculating Energies and Forces.” 2013. link Times cited: 0 NOT USED (low confidence) C. Ciobanu, C. Wang, and K. Ho, “Other Methodologies for Investigating Atomic Structure.” 2013. link Times cited: 0 NOT USED (low confidence) D. Antonov et al., “Statistical investigations on nitrogen-vacancy center creation,” Applied Physics Letters. 2013. link Times cited: 34 Abstract: Quantum information technologies require networks of interac… read moreAbstract: Quantum information technologies require networks of interacting defect bits. Color centers, especially the nitrogen vacancy (NV−) center in diamond, represent one promising avenue, toward the realisation of such devices. The most successful technique for creating NV− in diamond is ion implantation followed by annealing. Previous experiments have shown that shallow nitrogen implantation (<10 keV) results in NV− centers with a yield of 0.01%–0.1%. We investigate the influence of channeling effects during shallow implantation and statistical diffusion of vacancies using molecular dynamics and Monte Carlo simulation techniques. Energy barriers for the diffusion process were calculated using density functional theory. Our simulations show that 25% of the implanted nitrogens form a NV center, which is in good agreement with our experimental findings. read less NOT USED (low confidence) Y. Dong, Q. Li, and A. Martini, “Molecular dynamics simulation of atomic friction: A review and guide,” Journal of Vacuum Science and Technology. 2013. link Times cited: 158 Abstract: This paper reviews recent progress in molecular dynamics sim… read moreAbstract: This paper reviews recent progress in molecular dynamics simulation of atomic-scale friction measured by an atomic force microscopy. Each section of the review focuses on an individual condition or parameter that affects atomic friction including materials, surfaces, compliance, contact area, normal load, temperature, and velocity. The role each parameter plays is described in the context of both experimental measurements and simulation predictions. In addition, the discussion includes an overview of the research community's current understanding of observed effects, guidelines for implementation of those effects in an atomistic simulation, and suggestions for future research to address open questions. Taken together, this review conveys the message that friction at the atomic scale is affected by many interrelated parameters and that the use of molecular dynamics simulation as a predictive tool can be accomplished only through careful model design. read less NOT USED (low confidence) M. H. Khadem and A. Wemhoff, “Comparison of Green–Kubo and NEMD heat flux formulations for thermal conductivity prediction using the Tersoff potential,” Computational Materials Science. 2013. link Times cited: 57 NOT USED (low confidence) J. Thibault, J. Rouviere, and A. Bourret, “Grain Boundaries in Semiconductors,” Materials Science and Technology. 2013. link Times cited: 2 Abstract: The sections in this article are
Introduction
Grain Bo… read moreAbstract: The sections in this article are
Introduction
Grain Boundary Structure: Concepts and Tools
Grain Boundary Definitions
Geometrical Concepts
Dislocation Model
Primary Dislocation Network
Secondary Dislocation Network
Stress Field Associated with Grain Boundaries
Structural Unit Descriptions
Stick and Ball Structural Units
Energetic Structural Units
Algebraic Structural Units
Structural Units and Dislocations/Disclinations
The Limits of the Structural Unit Descriptions
Computer Simulation Techniques
Methods
Boundary Conditions
Interaction Laws
Experimental Techniques
Grain Boundary Structure: Experience and Simulation Results
Silicon and Germanium
Tilt Grain Boundaries
Twist Grain Boundaries
Diamond
SiC
GaAs
GaN
AlN
NiO
Comments on Grain Boundary Structures
Electrical Properties of Grain Boundaries
Introduction
Electrical Effects Induced by Grain Boundaries
Electronic States Associated with a Grain Boundary
Potential Barrier and Transport Properties
Dynamic Properties and Recombination Properties
Experimental Methods for Measuring the Grain Boundary Electrical Activity
Methods Based on Transport
Transient Methods
Correlation Between Electrical Activity and Structure
Transport Experiments in Bicrystals
Transient Properties Measured on Bicrystals
Emission and Capture Properties of Silicon and Germanium Grain Boundaries
Polycrystalline Silicon
Intrinsic or Extrinsic Origin of Electrical Activity of Grain Boundaries
Impurity Segregation and Precipitation Induced by Grain Boundaries
Introduction
Dopant Elements
Oxygen and Sulfur
Transition Elements
Copper
Nickel
Iron
Conclusions
Mechanical Properties of Grain Boundaries in Semiconductors
Introduction
Interaction Between Dislocations and Grain Boundaries
Dislocation Absorption
Dislocation Transmission Across Grain Boundaries
Grain Boundaries as a Dislocation Source
Grain Boundary Dislocation Movement
Physical Consequences
Grain Boundary Migration
Recovery of the Grain Boundary Structure and Cavitation
Deformation Modelling
Conclusions read less NOT USED (low confidence) A. Shokuhfar and S. Ebrahimi-Nejad, “Effects of structural defects on the compressive buckling of boron nitride nanotubes,” Physica E-low-dimensional Systems & Nanostructures. 2013. link Times cited: 33 NOT USED (low confidence) M. Li, Z. Kang, P. Yang, X. Meng, and Y. Lu, “Molecular dynamics study on buckling of single-wall carbon nanotube-based intramolecular junctions and influence factors,” Computational Materials Science. 2013. link Times cited: 24 NOT USED (low confidence) J. Wojdel, P. Hermet, M. Ljungberg, P. Ghosez, and J. Íñiguez, “First-principles model potentials for lattice-dynamical studies: general methodology and example of application to ferroic perovskite oxides,” Journal of Physics: Condensed Matter. 2013. link Times cited: 78 Abstract: We present a scheme to construct model potentials, with para… read moreAbstract: We present a scheme to construct model potentials, with parameters computed from first principles, for large-scale lattice-dynamical simulations of materials. We mimic the traditional solid-state approach to the investigation of vibrational spectra, i.e., we start from a suitably chosen reference configuration of the compound and describe its energy as a function of arbitrary atomic distortions by means of a Taylor series. Such a form of the potential-energy surface is general, trivial to formulate for any material, and physically transparent. Further, such models involve clear-cut approximations, their precision can be improved in a systematic fashion, and their simplicity allows for convenient and practical strategies to compute/fit the potential parameters. We illustrate our scheme with two challenging cases in which the model potential is strongly anharmonic, namely, the ferroic perovskite oxides PbTiO3 and SrTiO3. Studying these compounds allows us to better describe the connection between the so-called effective-Hamiltonian method and ours (which may be seen as an extension of the former), and to show the physical insight and predictive power provided by our approach—e.g., we present new results regarding the factors controlling phase-transition temperatures, novel phase transitions under elastic constraints, an improved treatment of thermal expansion, etc. read less NOT USED (low confidence) S. Taioli et al., “Non-adiabatic ab initio molecular dynamics of supersonic beam epitaxy of silicon carbide at room temperature.,” The Journal of chemical physics. 2013. link Times cited: 12 Abstract: In this work, we investigate the processes leading to the ro… read moreAbstract: In this work, we investigate the processes leading to the room-temperature growth of silicon carbide thin films by supersonic molecular beam epitaxy technique. We present experimental data showing that the collision of fullerene on a silicon surface induces strong chemical-physical perturbations and, for sufficient velocity, disruption of molecular bonds, and cage breaking with formation of nanostructures with different stoichiometric character. We show that in these out-of-equilibrium conditions, it is necessary to go beyond the standard implementations of density functional theory, as ab initio methods based on the Born-Oppenheimer approximation fail to capture the excited-state dynamics. In particular, we analyse the Si-C(60) collision within the non-adiabatic nuclear dynamics framework, where stochastic hops occur between adiabatic surfaces calculated with time-dependent density functional theory. This theoretical description of the C(60) impact on the Si surface is in good agreement with our experimental findings. read less NOT USED (low confidence) B. Mortazavi et al., “Experimental and multiscale modeling of thermal conductivity and elastic properties of PLA/expanded graphite polymer nanocomposites,” Thermochimica Acta. 2013. link Times cited: 79 NOT USED (low confidence) S. Ajori, R. Ansari, and M. Mirnezhad, “Mechanical properties of defective γ-graphyne using molecular dynamics simulations,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2013. link Times cited: 61 NOT USED (low confidence) L. Pastewka, A. Klemenz, P. Gumbsch, and M. Moseler, “Screened empirical bond-order potentials for Si-C,” Physical Review B. 2013. link Times cited: 110 Abstract: Typical empirical bond-order potentials are short ranged and… read moreAbstract: Typical empirical bond-order potentials are short ranged and give ductile instead of brittle behavior for materials such as crystalline silicon or diamond. Screening functions can be used to increase the range of these potentials. We outline a general procedure to combine screening functions with bond-order potentials that does not require to refit any of the potential's properties. We use this approach to modify Tersoff's [Phys. Rev. B 39, 5566 (1989)], Erhart & Albe's [Phys. Rev. B 71, 35211 (2005)] and Kumagai et al.'s [Comp. Mater. Sci. 39, 457 (2007)] Si, C and Si-C potentials. The resulting potential formulations correctly reproduce brittle materials response, and give an improved description of amorphous phases. read less NOT USED (low confidence) J. Schall and J. Harrison, “Reactive Bond-Order Potential for Si-, C-, and H-Containing Materials,” Journal of Physical Chemistry C. 2013. link Times cited: 8 Abstract: A new bond-order potential for modeling systems containing s… read moreAbstract: A new bond-order potential for modeling systems containing silicon, carbon, and hydrogen, such as organosilicon molecules (CxSiyHz), solid silicon, solid carbon, and alloys of silicon and carbon, is presented. This reactive potential utilizes the formalism of the second-generation reactive empirical bond-order potential (REBO) [Brenner et al. J. Phys.: Condens. Matter 2002, 14, 783] for hydrocarbons and the REBO parameters for silicon [Schall, Gao, Harrison. Phys. Rev. B 2008, 77, 115209]. Modifications to the hydrocarbon REBO potential were made to improve the description of three-atom type systems. The widespread use of Brenner’s REBO potential, its ability to model a wide range of hydrocarbon materials, and the existence of parameters for several atom types are some of the motivating factors for obtaining this Si–C–H (2B-SiCH) parametrization. The usefulness and flexibility of this potential is demonstrated by examining the properties of organosilicon molecules, the bulk, surface, and defect properties... read less NOT USED (low confidence) R. Ansari, A. Shahabodini, H. Rouhi, and A. Alipour, “Thermal Buckling Analysis of Multi-Walled Carbon Nanotubes Through a Nonlocal Shell Theory Incorporating Interatomic Potentials,” Journal of Thermal Stresses. 2013. link Times cited: 20 Abstract: Described in the current study is the thermal buckling behav… read moreAbstract: Described in the current study is the thermal buckling behavior of multi-walled carbon nanotubes (WCNTs) via a nonlocal atomistic-based shell model. The model including the effects of small-scale length and the van der Waals (vdW) forces between adjacent nanotubes is established through the incorporation of the interatomic potential into the nonlocal Flügge shell theory. This model links the strain energy density induced in the continuum to Eringen's nonlocal constitutive relations. The set of coupled field equations are analytically solved for two types of temperature distribution. The present model is of a distinguishing feature which is its independence from the widely scattered values of Young's modulus and the effective wall thickness of carbon nanotubes. read less NOT USED (low confidence) A. Mashreghi, “Thermal expansion/contraction of boron nitride nanotubes in axial, radial and circumferential directions,” Computational Materials Science. 2012. link Times cited: 11 NOT USED (low confidence) Y. Jeng, “Development of Innovative Algorithm for Nanomechanics and its Applications to the Characterization of Materials,” Key Engineering Materials. 2012. link Times cited: 0 Abstract: Understanding major mechanisms affecting material strength s… read moreAbstract: Understanding major mechanisms affecting material strength such as grain size, grain orientation and dislocation mechanism from atomistic viewpoint can empower scientists and engineers with the capability to produce vastly strengthened materials. Computational studies can offer the possibility of carrying out simulations of material properties at both larger length scales and longer times than direct atomistic calculations. The study has conducted theoretical modeling and experimental testing to investigate nanoscale mechanisms related to material strength and interfacial performance. Various computational algorithms in nanomechanics including energy minimization, molecular dynamics and hybrid approaches that mix atomistic and continuum methods to bridge the length and time scales have been used to thoroughly study the deformation and strengthening mechanisms. Our study has also performed experiments including depth-sensing indentation technique and in-situ pico-indentation to characterize the nanomechanisms related to material strength and tribological performance. In this project, we have developed the innovative mutil-scale algorithms in the area of nanomechanics. These approaches were used to studies the defect effect on the mechanical properties of thin film, mechanical properties of nanotubes, and tribological phenomena at nanoscale interfaces. read less NOT USED (low confidence) J. Yan, K. Liew, and L. He, “A mesh-free computational framework for predicting buckling behaviors of single-walled carbon nanocones under axial compression based on the moving Kriging interpolation,” Computer Methods in Applied Mechanics and Engineering. 2012. link Times cited: 34 NOT USED (low confidence) J. Yan, K. Liew, and L. He, “Predicting mechanical properties of single-walled carbon nanocones using a higher-order gradient continuum computational framework,” Composite Structures. 2012. link Times cited: 32 NOT USED (low confidence) M. Joe, M. Moon, and K.-R. Lee, “Atomistic simulations of diamond-like carbon growth,” Thin Solid Films. 2012. link Times cited: 12 NOT USED (low confidence) H. Zhao, C. Shi, P. Zhang, L. Zhang, H. Huang, and J. Yan, “Research on the effects of machining-induced subsurface damages on mono-crystalline silicon via molecular dynamics simulation,” Applied Surface Science. 2012. link Times cited: 62 NOT USED (low confidence) M. L. Nietiadi, Y. Rosandi, M. Kopnarski, and H. Urbassek, “Sputtering of dimers off a silicon surface,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2012. link Times cited: 5 NOT USED (low confidence) A. Bouhekka, A. Kebab, J. D. Sib, Y. Bouizem, M. Benbekhti, and L. Chahed, “Monte-Carlo simulation of hydrogenated amorphous silicon growth,” Journal of the Association of Arab Universities for Basic and Applied Sciences. 2012. link Times cited: 1 NOT USED (low confidence) A. Dongare, B. Lamattina, and A. Rajendran, “Strengthening Behavior and Tension–Compression Strength–Asymmetry in Nanocrystalline Metal–Ceramic Composites,” Journal of Engineering Materials and Technology-transactions of The Asme. 2012. link Times cited: 10 NOT USED (low confidence) A. Khoei and H. DorMohammadi, “Validity and size-dependency of Cauchy―Born hypothesis with Tersoff potential in silicon nano-structures,” Computational Materials Science. 2012. link Times cited: 8 NOT USED (low confidence) X. Lu, M. Chen, D. Qiu, L. Fan, C. Wang, and H. Wang, “Dynamics behavior and defects evolution of silicon nitride nanowires under tension and compression load: A molecular dynamics study,” Computational Materials Science. 2012. link Times cited: 19 NOT USED (low confidence) M. Yasuda, Y. Chihara, R. Mimura, Y. Kimoto, H. Kawata, and Y. Hirai, “Performance evaluation of carbon nanotube-based oscillators and bearings under electron irradiation: Molecular dynamics study,” Microelectronic Engineering. 2012. link Times cited: 10 NOT USED (low confidence) C. Lan et al., “Molecular dynamics simulations of ion range profiles for heavy ions in light targets,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2012. link Times cited: 9 NOT USED (low confidence) S. Hwang, Y. Li, and Z.-H. Hong, “Molecular Dynamic Simulation for Co Cluster Deposition on Si Substrate,” Advanced Materials Research. 2012. link Times cited: 2 Abstract: Molecular dynamic simulation for Co cluster deposition on Si… read moreAbstract: Molecular dynamic simulation for Co cluster deposition on Si substrate was investigated in this work. The surface roughness and the interface mixing will be evaluated for the deposited film quality under different incident energies and substrate temperatures. The effect of thermal annealing on the ability of gap filling will be discussed by a slip vector. The results indicate that the incident energy has dominant effect on the surface roughness, and there is a minimum surface roughness value around the incident energy of 8 eV. However, the substrate temperature has little effect on the surface roughness. For interface mixing, the simulation indicates the easy diffusion of Co atoms into Si substrate. However, increasing either the incident energy or the substrate temperature could not change much the mixing conditions. As for the ability of gap filling, it is clear that the thermal annealing does improve this ability and obtains better surface roughness and interface mixing. read less NOT USED (low confidence) R. Ansari and E. Kazemi, “Detailed investigation on single water molecule entering carbon nanotubes,” Applied Mathematics and Mechanics. 2012. link Times cited: 9 NOT USED (low confidence) R. Ansari and E. Kazemi, “Detailed investigation on single water molecule entering carbon nanotubes,” Applied Mathematics and Mechanics. 2012. link Times cited: 0 NOT USED (low confidence) D. C. Hannah et al., “On the origin of photoluminescence in silicon nanocrystals: pressure-dependent structural and optical studies.,” Nano letters. 2012. link Times cited: 127 Abstract: A lack of consensus persists regarding the origin of photolu… read moreAbstract: A lack of consensus persists regarding the origin of photoluminescence in silicon nanocrystals. Here we report pressure-dependences of X-ray diffraction and photoluminescence from alkane-terminated colloidal particles. We determine the diamond-phase bulk modulus, observe multiple phase transitions, and importantly find a systematic photoluminescence red shift that matches the X(conduction)-to-Γ(valence) transition of bulk crystalline silicon. These results, reinforced by calculations, suggest that the efficient photoluminescence, frequently attributed to defects, arises instead from core-states that remain highly indirect despite quantum confinement. read less NOT USED (low confidence) B. D. Jensen, A. Bandyopadhyay, K. Wise, and G. Odegard, “Parametric Study of ReaxFF Simulation Parameters for Molecular Dynamics Modeling of Reactive Carbon Gases.,” Journal of chemical theory and computation. 2012. link Times cited: 35 Abstract: The development of innovative carbon-based materials can be … read moreAbstract: The development of innovative carbon-based materials can be greatly facilitated by molecular modeling techniques. Although the Reax Force Field (ReaxFF) can be used to simulate the chemical behavior of carbon-based systems, the simulation settings required for accurate predictions have not been fully explored. Using the ReaxFF, molecular dynamics (MD) simulations are used to simulate the chemical behavior of pure carbon and hydrocarbon reactive gases that are involved in the formation of carbon structures such as graphite, buckyballs, amorphous carbon, and carbon nanotubes. It is determined that the maximum simulation time step that can be used in MD simulations with the ReaxFF is dependent on the simulated temperature and selected parameter set, as are the predicted reaction rates. It is also determined that different carbon-based reactive gases react at different rates, and that the predicted equilibrium structures are generally the same for the different ReaxFF parameter sets, except in the case of the predicted formation of large graphitic structures with the Chenoweth parameter set under specific conditions. read less NOT USED (low confidence) B. Mortazavi and S. Ahzi, “Molecular dynamics study on the thermal conductivity and mechanical properties of boron doped graphene,” Solid State Communications. 2012. link Times cited: 85 NOT USED (low confidence) Y.-T. Cheng et al., “Atomistic simulations of the adsorption and migration barriers of Cu adatoms on ZnO surfaces using COMB potentials,” Surface Science. 2012. link Times cited: 25 NOT USED (low confidence) V. Dozhdikov, A. Basharin, and P. Levashov, “Two-phase simulation of the crystalline silicon melting line at pressures from -1 to 3 GPa.,” The Journal of chemical physics. 2012. link Times cited: 32 Abstract: Results of a numerical investigation of crystalline silicon … read moreAbstract: Results of a numerical investigation of crystalline silicon melting line within the range of pressures from -1 to 3 GPa are presented. A two-phase molecular dynamics method is applied to obtain temperature, pressure, and densities of solid and liquid phases on the melting line. Using a special procedure we ensure the strict control of the two-phase equilibrium in the simulation cell. To describe the interaction between the atoms four classic potentials have been chosen: the Stillinger-Weber one and three modified variants of the Tersoff potential. For the Stillinger-Weber and Tersoff potentials in the modification by Kumagai-Izumi-Hara-Sakai a good coincidence with experimental data on crystalline Si melting temperature is obtained within the range of pressure from 0 to 3 GPa. Calculations of the solid and liquid phase densities on the silicon melting line for the Stillinger-Weber potential are also in close agreement with experiments. read less NOT USED (low confidence) T. Liang, B. Devine, S. Phillpot, and S. Sinnott, “Variable charge reactive potential for hydrocarbons to simulate organic-copper interactions.,” The journal of physical chemistry. A. 2012. link Times cited: 89 Abstract: A variable charge reactive empirical potential for carbon-ba… read moreAbstract: A variable charge reactive empirical potential for carbon-based materials, hydrocarbons, organometallics, and their interfaces is developed within the framework of charge optimized many-body (COMB) potentials. The resulting potential contains improved expressions for the bond order and self-energy, which gives a flexible, robust, and integrated treatment of different bond types in multicomponent and multifunctional systems. It furthermore captures the dissociation and formation of the chemical bonds and appropriately and dynamically determines the associated charge transfer, thus providing a powerful method to simulate the complex chemistry of many-atom systems in changing environments. The resulting COMB potential is used in a classical molecular dynamics simulation of the room temperature, low energy deposition of ethyl radicals on the Cu (111) surface (a system with ∼5000 atoms) to demonstrate its capabilities at describing organic-metal interactions in a dynamically changing environment. read less NOT USED (low confidence) Y. Wang, B. Qiu, and X. Ruan, “Edge effect on thermal transport in graphene nanoribbons: A phonon localization mechanism beyond edge roughness scattering,” Applied Physics Letters. 2012. link Times cited: 93 Abstract: Equilibrium molecular dynamics simulations show that graphen… read moreAbstract: Equilibrium molecular dynamics simulations show that graphene nanoribbons (GNRs) with zigzag edges have higher thermal conductivity (κ) than armchair-edged ones, and the difference diminishes with increasing temperature or ribbon width. The dominant phonon wavelength for thermal transport can be much longer (by orders of magnitude) than the difference between the “roughness” of smooth zigzag and armchair edges. Therefore, the roughness scattering theory is not sufficient to explain the largely different κ of GNRs with different edge chiralities. Cross-sectional decomposition of the steady-state heat flux shows significant suppression of thermal transport at edges, especially in armchair ones. This behavior is explored by phonon spectra analysis. Considerable phonon localization at edges is concluded to underlie the edge-chirality dependent κ of GNRs. read less NOT USED (low confidence) J. Yan, K. Liew, and L. He, “Analysis of single-walled carbon nanotubes using the moving Kriging interpolation,” Computer Methods in Applied Mechanics and Engineering. 2012. link Times cited: 32 NOT USED (low confidence) X. Shen, X. Shen, Y. Xiao, W. Dong, X. Yan, and H. F. Busnengo, “Molecular dynamics simulations based on reactive force-fields for surface chemical reactions,” Computational and Theoretical Chemistry. 2012. link Times cited: 5 NOT USED (low confidence) R. Promyoo, H. El-Mounayri, and K. Varahramyan, “AFM-Based Nanoindentation Process: A Comparative Study.” 2012. link Times cited: 4 Abstract: Atomic force microscopy (AFM) has been widely used for nanom… read moreAbstract: Atomic force microscopy (AFM) has been widely used for nanomachining and fabrication of micro/nanodevices. This paper describes the development and validation of computational models for AFM-based nanomachining. Molecular Dynamics (MD) technique is used to model and simulate mechanical indentation at the nanoscale for different types of materials, including gold, copper, aluminum, and silicon. The simulation allows for the prediction of indentation forces at the interface between an indenter and a substrate. The effects of tip materials on machined surface are investigated. The material deformation and indentation geometry are extracted based on the final locations of the atoms, which have been displaced by the rigid tool. In addition to the modeling, an AFM was used to conduct actual indentation at the nanoscale, and provide measurements to which the MD simulation predictions can be compared. The MD simulation results show that surface and subsurface deformation found in the case of gold, copper and aluminum have the same pattern. However, aluminum has more surface deformation than other materials. Two different types of indenter tips including diamond and silicon tips were used in the model. More surface and subsurface deformation can be observed for the case of nanoindentation with diamond tip. The indentation forces at various depths of indentation were obtained. It can be concluded that indentation force increases as depth of indentation increases. Due to limitations on computational time, the quantitative values of the indentation force obtained from MD simulation are not comparable to the experimental results. However, the increasing trends of indentation force are the same for both simulation and experimental results.Copyright © 2012 by ASME read less NOT USED (low confidence) R. Ansari, A. Shahabodini, A. Alipour, and H. Rouhi, “Stability of a single-layer graphene sheet with various edge conditions: a non-local plate model including interatomic potentials,” Proceedings of the Institution of Mechanical Engineers, Part N: Journal of Nanoengineering and Nanosystems. 2012. link Times cited: 12 Abstract: In this article, the biaxial buckling of a single layer grap… read moreAbstract: In this article, the biaxial buckling of a single layer graphene sheet is investigated. The model is established through the incorporation of an interatomic potential into non-local elastic plate theory to take into account the size effects and to circumvent the use of the Young’s modulus of a single layer graphene sheet since there is no accurate value of this property available in the literature. The model links the strain energy density induced in the continuum to Eringen’s non-local constitutive relations. By using the Galerkin method, explicit formulas for the critical buckling stresses of a single layer graphene sheet with arbitrary edge supports are derived from its static deflection due to a uniformly distributed load. The influences of the small size of the system and boundary conditions on the critical buckling load of the single layer graphene sheet are studied. It is found that the critical buckling load at large side lengths is almost independent of the type of boundary condition and compressive loading and is nearly immune to size effects. The analytical expressions provide a simple and quick way to evaluate accurate values of the critical buckling load. Indeed, the lack of complexity in the formulas allows a simple prediction of the value of the scale parameter that closely matches the one obtained using the complex molecular dynamics simulation technique. read less NOT USED (low confidence) B. Mortazavi and Y. Rémond, “Investigation of tensile response and thermal conductivity of boron-nitride nanosheets using molecular dynamics simulations,” Physica E-low-dimensional Systems & Nanostructures. 2012. link Times cited: 121 NOT USED (low confidence) R. Ansari, F. Alisafaei, A. Alipour, and E. Mahmoudinezhad, “On the van der Waals interaction of carbon nanocones,” Journal of Physics and Chemistry of Solids. 2012. link Times cited: 18 NOT USED (low confidence) J. Palmer and K. Gubbins, “Atomistic models for disordered nanoporous carbons using reactive force fields,” Microporous and Mesoporous Materials. 2012. link Times cited: 75 NOT USED (low confidence) S. Plimpton and A. Thompson, “Computational aspects of many-body potentials,” MRS Bulletin. 2012. link Times cited: 279 Abstract: We discuss the relative complexity and computational cost of… read moreAbstract: We discuss the relative complexity and computational cost of several popular many-body empirical potentials, developed by the materials science community over the past 30 years. The inclusion of more detailed many-body effects has come at a computational cost, but the cost still scales linearly with the number of atoms modeled. This is enabling very large molecular dynamics simulations with unprecedented atomic-scale fidelity to physical and chemical phenomena. The cost and scalability of the potentials, run in serial and parallel, are benchmarked in the LAMMPS molecular dynamics code. Several recent large calculations performed with these potentials are highlighted to illustrate what is now possible on current supercomputers. We conclude with a brief mention of high-performance computing architecture trends and the research issues they raise for continued potential development and use. read less NOT USED (low confidence) B. Uberuaga, S. Stuart, W. Windl, M. Masquelier, and A. Voter, “Fullerene and graphene formation from carbon nanotube fragments,” Computational and Theoretical Chemistry. 2012. link Times cited: 13 NOT USED (low confidence) R. Ansari, S. Ajori, and B. Arash, “Vibrations of single- and double-walled carbon nanotubes with layerwise boundary conditions: A molecular dynamics study,” Current Applied Physics. 2012. link Times cited: 78 NOT USED (low confidence) J. C. Slattery, K. Fu, and E. Oh, “The mechanics and thermodynamics of edge fracture: the critical energy release rate, the compatibility constraint, and the bond potential,” Philosophical Magazine. 2012. link Times cited: 3 Abstract: Following Gurtin and many others, the critical energy releas… read moreAbstract: Following Gurtin and many others, the critical energy release rate is commonly identified as an ill-defined surface energy. The primary objectives of this paper are to clarify the definition of this surface energy and the role of the entropy inequality in the discussion of critical conditions. In view of an increasing emphasis on ab initio computations, a secondary objective is to show how the critical energy release rate and the compatibility constraint 1 can be used to solve a problem for which we have experimental data, using only ab initio estimates of surface tension and bond potential, both of which are increasingly available. read less NOT USED (low confidence) Y. Wang, S. Chen, and X. Ruan, “Tunable thermal rectification in graphene nanoribbons through defect engineering: A molecular dynamics study,” Applied Physics Letters. 2012. link Times cited: 96 Abstract: Using non-equilibrium molecular dynamics, we show that asymm… read moreAbstract: Using non-equilibrium molecular dynamics, we show that asymmetrically defected graphene nanoribbons (GNR) are promising thermal rectifiers. The optimum conditions for thermal rectification (TR) include low temperature, high temperature bias, ∼1% concentration of single-vacancy or substitutional silicon defects, and a moderate partition of the pristine and defected regions. TR ratio of ∼80% is found in a 14-nm long and 4-nm wide GNR at a temperature of 200 K and bias of 90 K, where heat conduction is in the ballistic regime since the bulk effective phonon mean-free-path is around 775 nm. As the GNR length increases towards the diffusive regime, the TR ratio decreases and eventually stabilizes at a length-independent value of about 3%–5%. This work extends defect engineering to 2D materials for achieving TR. read less NOT USED (low confidence) E. Bellido and J. Seminario, “Graphene-Based Vibronic Devices,” Journal of Physical Chemistry C. 2012. link Times cited: 14 Abstract: Molecular dynamic simulations are used to model the vibratio… read moreAbstract: Molecular dynamic simulations are used to model the vibrational bending modes of graphene ribbons of several sizes to calculate frequencies of the ribbons and determine the relationship between the size of the ribbon and their corresponding resonance frequencies. These ribbons can be utilized to fabricate several types of vibronic devices such as NEMS, sensors, terahertz generators, and devices for encoding, transferring, and processing information. The interaction of a graphene vibronic device with water and isopropyl alcohol molecules demonstrates that this device can be used as a very sensitive vibronic molecular sensor that is able to distinguish the chemical nature of the sensed molecule. The electrical properties of the graphene vibronic devices are also calculated for two cases, armchair and zigzag border. The zigzag border demonstrated in this work has the potential to generate THz electrical signals. read less NOT USED (low confidence) S. Hwang, Y. Li, and Z.-H. Hong, “Molecular dynamic simulation for Cu cluster deposition on Si substrate,” Computational Materials Science. 2012. link Times cited: 43 NOT USED (low confidence) X. Wang and X. Guo, “Numerical simulation for finite deformation of single-walled carbon nanotubes at finite temperature using temperature-related higher order Cauchy-Born rule based quasi-continuum model,” Computational Materials Science. 2012. link Times cited: 27 NOT USED (low confidence) A. Danescu, “Hyper-pre-stress vs. strain-gradient for surface relaxation in diamond-like structures,” Journal of The Mechanics and Physics of Solids. 2012. link Times cited: 8 NOT USED (low confidence) Y. Lee and G. Hwang, “Force-matching-based parameterization of the Stillinger-Weber potential for thermal conduction in silicon,” Physical Review B. 2012. link Times cited: 38 NOT USED (low confidence) L. Chen and S. Kumar, “Thermal transport in graphene supported on copper,” Journal of Applied Physics. 2012. link Times cited: 91 Abstract: We investigate the thermal transport in isolated single laye… read moreAbstract: We investigate the thermal transport in isolated single layer graphene (SLG) and SLG supported on Cu substrate using equilibrium molecular dynamics simulations and relaxation time approximation (RTA) method. We observe significant changes in the SLG dispersion curve in low frequency and low wave-vector region due to the interaction with Cu substrate. Several new phonon modes related to out-of-plane vibrations appear at the low frequency and small wave vector regions, but their contribution to graphene thermal conductivity is negligible. The thermal conductivity of graphene decreases by 44% due to the interactions with Cu substrate for high interaction strength parameter in Lennard-Jones potential formulation for graphene-Cu interaction. The phonon mode analysis through the RTA approach shows that the acoustic phonons dominate the thermal transport for both isolated and supported graphenes. The longitudinal acoustic (LA), transverse acoustic (TA), and out-of-plane acoustic (ZA) phonons contribute 654, 330,... read less NOT USED (low confidence) F. Graziani et al., “Large-scale molecular dynamics simulations of dense plasmas: The Cimarron Project,” High Energy Density Physics. 2012. link Times cited: 82 NOT USED (low confidence) A. Volkov, T. Shiga, D. Nicholson, J. Shiomi, and L. Zhigilei, “Effect of bending buckling of carbon nanotubes on thermal conductivity of carbon nanotube materials,” Journal of Applied Physics. 2012. link Times cited: 40 Abstract: The effect of bending buckling of carbon nanotubes (CNTs) on… read moreAbstract: The effect of bending buckling of carbon nanotubes (CNTs) on thermal conductivity of CNT materials is investigated in atomistic and mesoscopic simulations. Nonequilibrium molecular dynamics simulations of the thermal conductance through an individual buckling kink in a (10,10) single-walled CNT reveal a strong dependence (close to inverse proportionality) of the thermal conductance of the buckling kink on the buckling angle. The value of the buckling kink conductance divided by the cross-sectional area of the CNT ranges from 40 to 10 GWm−2 K−1 as the buckling angle changes from 20 to 110°. The predictions of the atomistic simulations are used for parameterization of a mesoscopic model that enables calculations of thermal conductivity of films composed of thousands of CNTs arranged into continuous networks of bundles. The results of mesoscopic simulations demonstrate that the conductivity of CNT films is sensitive to the angular dependence of the buckling kink conductance and the length of the individual C... read less NOT USED (low confidence) F. Castro-Marcano, A. M. Kamat, M. F. Russo, A. Duin, and J. Mathews, “Combustion of an Illinois No. 6 coal char simulated using an atomistic char representation and the ReaxFF reactive force field,” Combustion and Flame. 2012. link Times cited: 297 NOT USED (low confidence) L. Peng and J. R. Morris, “Structure and hydrogen adsorption properties of low density nanoporous carbons from simulations,” Carbon. 2012. link Times cited: 41 NOT USED (low confidence) B. Mortazavi, S. Ahzi, V. Toniazzo, and Y. Rémond, “Nitrogen doping and vacancy effects on the mechanical properties of graphene: A molecular dynamics study,” Physics Letters A. 2012. link Times cited: 78 NOT USED (low confidence) D. Konatham, D. Papavassiliou, and A. Striolo, “Thermal boundary resistance at the graphene–graphene interface estimated by molecular dynamics simulations,” Chemical Physics Letters. 2012. link Times cited: 58 NOT USED (low confidence) K. Bui, H. Nguyen, C. Cousin, A. Striolo, and D. Papavassiliou, “Thermal Behavior of Double-Walled Carbon Nanotubes and Evidence of Thermal Rectification,” Journal of Physical Chemistry C. 2012. link Times cited: 21 Abstract: The thermal boundary resistance (TBR, also known as Kapitza … read moreAbstract: The thermal boundary resistance (TBR, also known as Kapitza resistance) between carbon nanotubes and a surrounding polymeric matrix (e.g., epoxy, polystyrene) is known to hinder the development of thermally conductive, nanotube-based composite materials. Because the carbon nanotube to carbon nanotube TBR can be even higher than that between a nanotube and the polymer matrix, a fundamental question is whether thermal transfer along multiwalled carbon nanotubes occurs solely via the outmost nanotube. To address this question, molecular dynamics simulations were conducted in double-walled carbon nanotubes. It is found that the resistance to heat transfer between the two nanotubes is higher than the TBR typically reported between nanotubes and polymeric matrix materials, although heat transfer occurs more easily when the two nanotubes have different chirality. Further, and probably more importantly in terms of fundamental knowledge, the thermal boundary resistance is found to be up to 120% lower when thermal ... read less NOT USED (low confidence) E. Bellido and J. Seminario, “Molecular Dynamics Simulations of Ion-Bombarded Graphene,” Journal of Physical Chemistry C. 2012. link Times cited: 50 Abstract: Using molecular dynamics simulations and a hybrid Tersoff-ZB… read moreAbstract: Using molecular dynamics simulations and a hybrid Tersoff-ZBL potential, the effects of irradiating graphene with a carbon ion at several positions and several energies from 0.1 eV to 100 keV are studied. The simulations show four types of processes: absorption, reflection, transmission, and vacancy formation. At energies below 10 eV, the dominant process is reflection; between 10 and 100 eV, it is absorption; and between 100 eV and 100 keV, the dominant process is transmission. Vacancy formation is a low-probability process that takes place at energies above 30 eV. Three types of defects are found: adatom, single vacancy, and 5–8–5 defect formed from a double-vacancy defect. The simulations provide a fundamental understanding of the graphene carbon bombardment and the parameters to develop graphene devices by controlling defect formation. read less NOT USED (low confidence) R. Ansari, E. Kazemi, E. Mahmoudinezhad, and F. Sadeghi, “Preferred Position and Orientation of Anticancer Drug Cisplatin During Encapsulation Into Single-Walled Carbon Nanotubes,” Journal of Nanotechnology in Engineering and Medicine. 2012. link Times cited: 4 NOT USED (low confidence) B. Mortazavi, A. Rajabpour, S. Ahzi, Y. Rémond, and S. M. V. Allaei, “Nitrogen doping and curvature effects on thermal conductivity of graphene: A non-equilibrium molecular dynamics study,” Solid State Communications. 2012. link Times cited: 98 NOT USED (low confidence) C. Wong and V. Vijayaraghavan, “Nanomechanics of imperfectly straight single walled carbon nanotubes under axial compression by using molecular dynamics simulation,” Computational Materials Science. 2012. link Times cited: 34 NOT USED (low confidence) H. Whitlow and S. Nakagawa, “Ordering effects in extreme high-resolution depth profiling with MeV ion beams,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2012. link Times cited: 0 NOT USED (low confidence) B. Mortazavi, Y. Rémond, S. Ahzi, and V. Toniazzo, “Thickness and chirality effects on tensile behavior of few-layer graphene by molecular dynamics simulations,” Computational Materials Science. 2012. link Times cited: 67 NOT USED (low confidence) C. Hou and W. Ge, “A NOVEL MODE AND ITS VERIFICATION OF PARALLEL MOLECULAR DYNAMICS SIMULATION WITH THE COUPLING OF GPU AND CPU,” International Journal of Modern Physics C. 2012. link Times cited: 4 Abstract: Graphics processing unit (GPU) is becoming a powerful comput… read moreAbstract: Graphics processing unit (GPU) is becoming a powerful computational tool in scientific and engineering fields. In this paper, for the purpose of the full employment of computing capability, a novel mode for parallel molecular dynamics (MD) simulation is presented and implemented on basis of multiple GPUs and hybrids with central processing units (CPUs). Taking into account the interactions between CPUs, GPUs, and the threads on GPU in a multi-scale and multilevel computational architecture, several cases, such as polycrystalline silicon and heat transfer on the surface of silicon crystals, are provided and taken as model systems to verify the feasibility and validity of the mode. Furthermore, the mode can be extended to MD simulation of other areas such as biology, chemistry and so forth. read less NOT USED (low confidence) M. Treacy and J. Gibson, “Examination of a Polycrystalline Thin-Film Model to Explore the Relation between Probe Size and Structural Correlation Length in Fluctuation Electron Microscopy,” Microscopy and Microanalysis. 2012. link Times cited: 13 Abstract: We examine simulated electron microdiffraction patterns from… read moreAbstract: We examine simulated electron microdiffraction patterns from models of thin polycrystalline silicon. The models are made by a Voronoi tessellation of random points in a box. The Voronoi domains are randomly selected to contain either a randomly-oriented cubic crystalline grain or a region of continuous random network material. The microdiffraction simulations from coherent probes of different widths are computed at the ideal kinematical limit, ignoring inelastic and multiple scattering. By examining the normalized intensity variance that is obtained in fluctuation electron microscopy experiments, we confirm that intensity fluctuations increase monotonically with the percentage of crystalline grains in the material. However, anomalously high variance is observed for models that have 100% crystalline grains with no imperfections. We confirm that the reduced normalized variance, V(k,R) − 1, that is associated with four-body correlations at scattering vector k, varies inversely with specimen thickness. Further, for probe sizes R larger than the mean grain size, we confirm that the reduced normalized variance obeys the predicted form given by Gibson et al. [Ultramicroscopy, 83, 169–178 (2000)] for the kinematical coherent scattering limit. read less NOT USED (low confidence) F. Nishimura, T. Shiga, S. Maruyama, K. Watanabe, and J. Shiomi, “Thermal Conductance of Buckled Carbon Nanotubes,” Japanese Journal of Applied Physics. 2012. link Times cited: 11 Abstract: Knowledge of thermal conductance of carbon nanotubes under m… read moreAbstract: Knowledge of thermal conductance of carbon nanotubes under mechanical deformation is important to characterize the robustness of carbon nanotube heat conduction. In this study, using molecular dynamics simulations, we have calculated thermal conductance of an elastically buckled single-walled carbon nanotube. A local buckle was formed by mechanically bending a carbon nanotube at an angle of 60°, and thermal conductance through the buckle was calculated by a nonequilibrium molecular dynamics approach. The thermal conductance exhibits strong diameter dependence, correlated with the strain energy generated in the buckle. Despite the highly strained deformation, the thermal resistance across a buckle is similar to that of a point defect and heterotube junction, revealing a robust nature of carbon nanotube heat conduction to buckling deformation. read less NOT USED (low confidence) Y. Umeno and J. Negami, “Atomistic Simulation of Stress-Induced Grain Boundary Diffusion: For Tin-Whisker Problem,” Materials Science Forum. 2012. link Times cited: 2 Abstract: The problem of whisker formation in tin (Sn) wiring in small… read moreAbstract: The problem of whisker formation in tin (Sn) wiring in small electronic devices has become an important issue with the requirement of lead-free wiring, because doping of Pb to reduce whisker formation cannot be applied. It is therefore urged to better understand stress migration in tin, which is suspected to play a key role in whisker growth. We aim to study grain boundary diffusion in tin by atomistic simulation. After constructing an efficient interatomic potential suitable for diffusion of atoms using the genetic algorithm (GA), we perform molecular dynamics (MD) simulation of grain boundary diffusion in Sn under stress. We find that the magnitude of stress effect on diffusion depends on the boundary structure. Moreover, we examine the effect of impurities on vacancy migration by ab initio calculation to find atom doping that has potential to suppress diffusion. read less NOT USED (low confidence) E. Tadmor and R. E. Miller, “Modeling Materials: Continuum, Atomistic and Multiscale Techniques.” 2011. link Times cited: 395 Abstract: 1. Introduction Part I. Continuum Mechanics and Thermodynami… read moreAbstract: 1. Introduction Part I. Continuum Mechanics and Thermodynamics: 2. Essential continuum mechanics and thermodynamics Part II. Atomistics: 3. Lattices and crystal structures 4. Quantum mechanics of materials 5. Empirical atomistic models of materials 6. Molecular statics Part III. Atomistic Foundations of Continuum Concepts: 7. Classical equilibrium statistical mechanics 8. Microscopic expressions for continuum fields 9. Molecular dynamics Part IV. Multiscale Methods: 10. What is multiscale modeling? 11. Atomistic constitutive relations for multilattice crystals 12. Atomistic/continuum coupling: static methods 13. Atomistic/continuum coupling: finite temperature and dynamics Appendix References Index. read less NOT USED (low confidence) L. Priester, “Grain Boundary Structures and Defects.” 2011. link Times cited: 0 NOT USED (low confidence) H. Kim and V. Tomar, “Nanometer to Micron Scale Atomistic Mechanics of Silicon Using Atomistic Simulations at Accelerated Time Steps,” Journal of Nanomechanics and Micromechanics. 2011. link Times cited: 2 Abstract: Atomistic simulations have a unique capability to reveal the… read moreAbstract: Atomistic simulations have a unique capability to reveal the material deformation mechanisms and the corresponding deformation-based constitutive behavior. However, atomistic simulations are limited by the accessible length and time scales. In the present work, an equivalent crystal lattice method is used to analyze atomistic mechanical deformation of nanometer- to micrometer-sized polycrystalline silicon (Si) samples at accelerated time steps. The equivalent crystal lattice method’s validity is verified by the results of classical molecular dynamics (MD) simulations at MD strain rates. The method is then used to predict material behavior at subcontinuum length scales. An extrapolation of the thin film polycrystalline silicon stress-strain relationships to lower strain-rate values indicates that the thin film peak stress values at the experimental strain rates are in agreement with experimental values. Analyses reveal that the peak stress values in the case of polycrystalline Si follow inverse Hall-Petch ... read less NOT USED (low confidence) C. Bulutay, “Quadrupolar spectra of nuclear spins in strained InGaAs quantum dots,” Bulletin of the American Physical Society. 2011. link Times cited: 50 Abstract: Self-assembled quantum dots (QDs) are born out of lattice mi… read moreAbstract: Self-assembled quantum dots (QDs) are born out of lattice mismatched ingredients where strain plays an indispensable role. Through the electric quadrupolar coupling, strain affects the magnetic environment as seen by the nuclear spins. To guide prospective single-QD nuclear magnetic resonance (NMR) as well as dynamic nuclear spin polarization experiments, an atomistic insight to the strain and quadrupolar field distributions is presented. A number of implications of the structural and compositional profile of the QD have been identified. A high aspect ratio of the QD geometry enhances the quadrupolar interaction. The inclined interfaces introduce biaxiality and the tilting of the major quadrupolar principal axis away from the growth axis; the alloy mixing of gallium into the QD enhances both of these features while reducing the quadrupolar energy. Regarding the NMR spectra, both Faraday and Voigt geometries are investigated, unraveling in the first place the extend of inhomogeneous broadening and the appearance of the normally-forbidden transitions. Moreover, it is shown that from the main extend of the NMR spectra the alloy mole fraction of a single QD can be inferred. By means of the element-resolved NMR intensities it is found that In nuclei has a factor of five dominance over those of As. In the presence of an external magnetic field, the borderlines between the quadrupolar and Zeeman regimes are extracted as 1.5 T for In and 1.1 T for As nuclei. At these values the nuclear spin depolarization rates of the respective nuclei get maximized due to the noncollinear secular hyperfine interaction with a resident electron in the QD. read less NOT USED (low confidence) M. Byshkin, “Bond-coordination lattice model for phase transformations in carbon,” Diamond and Related Materials. 2011. link Times cited: 2 NOT USED (low confidence) J. Liu, J. Kong, D. Lei, Y. L. Zhang, H. F. Li, and X. Zhao, “Molecular Dynamics Simulation of Nanoindentation on Diamond Crystal [100] Surface,” Advanced Materials Research. 2011. link Times cited: 0 Abstract: The nanoindentation of diamond crystal [100] surface is stud… read moreAbstract: The nanoindentation of diamond crystal [100] surface is studied in this paper, by using molecular dynamics simulation method and Tersoff potential. The total number of atoms in the model is exceed to 2,000,000. The crystal structure changes and the bond formations of C atoms under pressure load are analyzed. A light load causes lattice distortion but cannot cause bond breaking or hybridization transition from sp3 to sp2. When the load is enough heavy, the energy be imposed on the workpiece will beyond the range of lattice distortion, which can cause bond break and hybridization transition from sp3 to sp2. read less NOT USED (low confidence) W. Ge et al., “Meso-scale oriented simulation towards virtual process engineering (VPE)-The EMMS Paradigm,” Chemical Engineering Science. 2011. link Times cited: 126 NOT USED (low confidence) B. Devine et al., “Atomistic simulations of copper oxidation and Cu/Cu2O interfaces using charge-optimized many-body potentials,” Physical Review B. 2011. link Times cited: 64 Abstract: Bryce Devine,1 Tzu-Ray Shan( ),1 Yu-Ting Cheng( ),1 Alan J. … read moreAbstract: Bryce Devine,1 Tzu-Ray Shan( ),1 Yu-Ting Cheng( ),1 Alan J. H. McGaughey,1,2 Minyoung Lee,2 Simon R. Phillpot,1 and Susan B. Sinnott1,* 1Department of Materials Science and Engineering, University of Florida, Gainesville, FL 32611-6400, USA 2Department of Mechanical Engineering, Carnegie-Mellon University, Pittsburgh, PA, 15213, USA (Received 14 February 2011; revised manuscript received 21 July 2011; published 12 September 2011) read less NOT USED (low confidence) R. Ansari, B. Motevalli, A. Montazeri, and S. Ajori, “Fracture analysis of monolayer graphene sheets with double vacancy defects via MD simulation,” Solid State Communications. 2011. link Times cited: 73 NOT USED (low confidence) D. Chrobak, N. Tymiak, A. Beaber, O. Ugurlu, W. Gerberich, and R. Nowak, “Deconfinement leads to changes in the nanoscale plasticity of silicon.,” Nature nanotechnology. 2011. link Times cited: 136 NOT USED (low confidence) R. Li, Y.-zhong Hu, and H. Wang, “A Study on Mechanism of Tribological Behavior of Carbon Nanotubes,” Advanced Materials Research. 2011. link Times cited: 1 Abstract: This paper investigates mechanism of tribological behavior o… read moreAbstract: This paper investigates mechanism of tribological behavior of carbon nanotubes by using universal tribometer-II and molecular dynamics simulations. The experiment results indicate that multi-walled carbon nanotubes film with mixed acid treatment has better surface quality and less impurities but higher friction than pristine carbon nanotubes film. The reason is that mixed acid treatment introduces carboxyl group and more defects which increases dangling bonds of carbon nanotubes. Breaking of dangling bonds increases friction force when sliding and shearing occurs. Molecular dynamics simulation of shearing between silicon surfaces and single-walled carbon nanotube bundles without defect shows low lateral forces because only van der walls force exists between silicon surfaces and carbon nanotubes owing to no dangling bonds. The result is consistent to the conclusion inferred from experiment. Therefore excellent performance is expected when carbon nanotubes treated with little defects are used as lubricant or addictives. read less NOT USED (low confidence) P. Czekala, H. Lin, W. Hofer, and A. Gulans, “Acetylene adsorption on silicon (100)-(4×2) revisited,” Surface Science. 2011. link Times cited: 15 NOT USED (low confidence) K. Liew, J. Yan, Y. Sun, and L. He, “Investigation of temperature effect on the mechanical properties of single-walled carbon nanotubes,” Composite Structures. 2011. link Times cited: 35 NOT USED (low confidence) A. Dongare and B. Lamattina, “Deformation and Failure Mechanisms in Ceramic-Reinforced Metal-Matrix Composites at Atomic Scales.” 2011. link Times cited: 1 NOT USED (low confidence) Z. Wei, Z. Ni, K. Bi, M. Chen, and Y. Chen, “In-plane lattice thermal conductivities of multilayer graphene films,” Carbon. 2011. link Times cited: 158 NOT USED (low confidence) S. Arghavan and A. V. Singh, “On the vibrations of single-walled carbon nanotubes,” Journal of Sound and Vibration. 2011. link Times cited: 61 NOT USED (low confidence) H. Qian, A. V. van Duin, K. Morokuma, and S. Irle, “Reactive Molecular Dynamics Simulation of Fullerene Combustion Synthesis: ReaxFF vs DFTB Potentials.,” Journal of chemical theory and computation. 2011. link Times cited: 74 Abstract: The dynamic fullerene self-assembly process during benzene c… read moreAbstract: The dynamic fullerene self-assembly process during benzene combustion was studied using classical Reactive Force Field (ReaxFF) nonequilibrium molecular dynamics (MD) simulations. In order to drive the combustion process, the hydrogen to carbon (H/C) ratio was gradually reduced during the course of the MD simulations. Target temperatures of 2500 and 3000 K were maintained by using a Berendsen thermostat. Simulation conditions and hydrogen removal strategies were chosen to match closely a previous quantum chemical MD (QM/MD) study based on the density-functional tight-binding (DFTB) potential ( Saha et al. ACS Nano 2009 , 3 , 2241 ) to allow a comparison between the two different potentials. Twenty trajectories were computed at each target temperature, and hydrocarbon cluster size, CxHy composition, average carbon cluster curvature, carbon hybridization type, and ring count statistics were recorded as a function of time. Similarly as in the QM/MD simulations, only giant fullerene cages in the range from 155 to 212 carbon atoms self-assembled, and no C60 cages were observed. The most notable difference concerned the time required for completing cage self-assembly: Depending on temperature, it takes between 50 and 150 ps in DFTB/MD simulations but never less than 100 ps and frequently several 100s ps in ReaxFF/MD simulations. In the present system, the computational cost of ReaxFF/MD is about 1 order of magnitude lower than that of the corresponding DFTB/MD. Overall, the ReaxFF/MD simulations method paints a qualitatively similar picture of fullerene formation in benzene combustion when compared to direct MD simulations based on the DFTB potential. read less NOT USED (low confidence) F. Delogu, “Melting of Pb clusters encapsulated in large fullerenes,” Chemical Physics. 2011. link Times cited: 0 NOT USED (low confidence) P. Novikov, J. V. Smagina, D. Vlasov, A. Deryabin, A. Kozhukhov, and A. Dvurechenskii, “Space arrangement of Ge nanoislands formed by growth of Ge on pit-patterned Si substrates,” Journal of Crystal Growth. 2011. link Times cited: 5 NOT USED (low confidence) Z. V. Smagina, P. Novikov, V. Zinovyev, V. Armbrister, S. Teys, and A. Dvurechenskii, “Molecular-beam epitaxial growth of Ge/Si nanostructures under low-energy ion irradiation,” Journal of Crystal Growth. 2011. link Times cited: 2 NOT USED (low confidence) E. H. Feng and R. Jones, “Carbon nanotube cantilevers for next-generation sensors,” Physical Review B. 2011. link Times cited: 23 NOT USED (low confidence) Z. Xing-li and S. Zhaowei, “Effects of vacancy structural defects on the thermal conductivity of silicon thin films,” Journal of Semiconductors. 2011. link Times cited: 9 Abstract: Vacancy structural defect effects on the lattice thermal con… read moreAbstract: Vacancy structural defect effects on the lattice thermal conductivity of silicon thin films have been investigated with non-equilibrium molecular dynamics simulation. The lattice thermal conductivities decrease with increasing vacancy concentration at all temperatures from 300 to 700 K. Vacancy defects decrease the sample thermal conductivity, and the temperature dependence of thermal conductivity becomes less significant as the temperature increases. The molecular dynamics result is in good agreement with the theoretical analysis values obtained based on the Boltzmann equation. In addition, theoretical analysis indicates that the reduction in the lattice thermal conductivity with vacancy defects can be explained by the enhanced point-defect scattering due to lattice strain. read less NOT USED (low confidence) X. Zhang and Z.-wei Sun, “Thermal Conductivity of Silicon Thin Films Predicted by Molecular Dynamics Simulations and Theoretical Calculation,” Applied Mechanics and Materials. 2011. link Times cited: 3 Abstract: Molecular, dynamics simulation and the Boltzmann transport e… read moreAbstract: Molecular, dynamics simulation and the Boltzmann transport equation are used respectively to analyze the phonon transport in Si thin film. The MD result is in good agreement with the theoretical analysis values. The results show that the calculated thermal conductivity decreases almost linearly as the film thickness reduced and is almost independent of the temperature at the nanoscale. It was observed from the simulation results that there exists the obvious size effect on the thermal conductivity. read less NOT USED (low confidence) L. Trandinh, Y.-M. Ryu, W. Kang, and S. Cheon, “A molecular dynamics simulation on the defect structure in silicon under indentation.” 2011. link Times cited: 0 Abstract: ,In this paper, the symmetric axis parameter method, which w… read moreAbstract: ,In this paper, the symmetric axis parameter method, which was proposed to identify defects, dislocations and stacking fault, with perfect structures in the zinc-blende materials, was introduced as a way to distinguish between elastic and plastic deformation. LAMMPS, a molecular dynamics programme of Sandia National Laboratories, was used to perform nanoindentation simulation on silicon, a zinc-blende material. Defects in silicon (111) under spherical indentation showed the threefold pattern and the slip system in the form of ring crack. Also simulation results show good agreement with experimental results and existing theoretical analyses. read less NOT USED (low confidence) J. Bhattacharjee and J. Neaton, “Interfacing carbon nanotubes of arbitrary chiralities into linear heterojunctions,” Physical Review B. 2011. link Times cited: 2 Abstract: Motivated by recent advances in synthesis and characterizati… read moreAbstract: Motivated by recent advances in synthesis and characterization of carbon nanotube (CNT) heterojunctions, we introduce a systematic approach for obtaining atomic geometries that connect two carbon nanotubes of different chiralities. Using our approach, it is straightforward to construct atomic interface geometries between two single-walled CNT's of arbitrary chiralities arranged at different orientations and angles. Our method generalizes existing approaches and is readily applicable to joining domains of graphene nanoribbons as well. As an example, we focus on linear heterojunctions, and we postulate the minimum number of simple topological defects required at the interface, and the preferred spatial arrangements, to obtain maximally linear heterojunctions given any two arbitrary chiralities. We also provide a physical picture of the defect structure of the resultant interface geometries using the results of classical force-field simulations. read less NOT USED (low confidence) A. Montazeri, M. Sadeghi, R. Naghdabadi, and H. Rafii-Tabar, “Multiscale modeling of the effect of carbon nanotube orientation on the shear deformation properties of reinforced polymer-based composites,” Physics Letters A. 2011. link Times cited: 23 NOT USED (low confidence) D. K. Samarakoon and X.-Q. Wang, “Structural and Electronic Properties of Hydrogenated Graphene.” 2011. link Times cited: 2 Abstract: DEPARTMENT OF PHYSICS SAMARAKOON, DUMINDA K. B.SC. UNIVERSIT… read moreAbstract: DEPARTMENT OF PHYSICS SAMARAKOON, DUMINDA K. B.SC. UNIVERSITY OF PERADENIYA, 2004 M.SC. UNIVERSITY OF PERADENIYA, 2007 STRUCTURAL AND ELECTRONIC PROPERTIES OF HYDROGENATED GRAPHENE Committee Chair: Xiao-Qian Wang, Ph.D. Thesis dated May 2011 Graphane is a two-dimensional system consisting of a single planar layer of fully saturated carbon atoms, which has recently been realized experimentally through hydrogenation of graphene membranes. We have studied the stability of chair, boat, and twist-boat graphane structures using first-principles density functional calculations. Our results indicate that locally stable twist-boat membranes significantly contribute to the experimentally observed lattice contraction. The band gaps of graphane nanoribbons decrease monotonically with the increase of the ribbon width and are insensitive to the edge structure. We also have studied the electronic structural characteristics in a hydrogenated bilayer graphene under a perpendicular electric bias. The bias voltage applied between the two hydrogenated graphene layers allows continuously tuning the band gap and leads a transition from semiconducting to metallic state. Desorption of hydrogen from one layer in the chair conformation yields a ferromagnetic semiconductor with tunable band gap. STRUCTURAL AND ELECTRONIC PROPERTIES OF HYDROGENATED GRAPHENE A THESIS SUBMITTED TO THE FACULTY OF CLARK ATLANTA UNIVERSITY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE MASTER OF SCIENCE BY DUMINDA K. SAMARAKOON DEPARTMENT OF PHYSICS ATLANTA, GEORGIA MAY 2011 read less NOT USED (low confidence) D. Bai, “Size, Morphology and Temperature Dependence of the Thermal Conductivity of Single-Walled Silicon Carbide Nanotubes,” Fullerenes, Nanotubes and Carbon Nanostructures. 2011. link Times cited: 11 Abstract: The thermal conductivity of single-walled silicon carbide na… read moreAbstract: The thermal conductivity of single-walled silicon carbide nanotubes (SW-SiCNTs) has been investigated by molecular dynamics (MD) simulation using the many-body Tersoff potential. To validate the reliability of the simulations code, the following measures have been taken: The calculated potential energies of SW-SiCNTs and the calculated thermal conductivities of single-walled carbon nanotubes (SWCNTs) are, respectively, compared with available data, and both comparisons are in good agreement. To investigate the size (tube length and diameter), morphology (chirality and the atom arrangement) and temperature dependence of the thermal conductivity of SW-SiCNTs, the thermal conductivities of SW-SiCNTs with different sizes, morphologies and temperatures, are calculated and compared with each other. It is found that (1) as the temperature increases, the thermal conductivity decreases at different rate, which depends on the tube morphology; (2) as long as the length increases, the thermal conductivity increases correspondingly; (3) the thermal conductivity depends on the tube diameter and exhibits a peaking behavior as a function of diameter; (4) atom arrangement strongly affects the thermal conductivity not only in quantity but also in the extent of dependence on chirality; and (5) the thermal conductivity is dependent on the chirality of nanotube with different extent. read less NOT USED (low confidence) Z. Wei, Z. Ni, K. Bi, M. Chen, and Y. Chen, “Interfacial thermal resistance in multilayer graphene structures,” Physics Letters A. 2011. link Times cited: 84 NOT USED (low confidence) Q.-D. Wang, J. Wang, J.-Q. Li, N. Tan, and X. Li, “Reactive molecular dynamics simulation and chemical kinetic modeling of pyrolysis and combustion of n-dodecane,” Combustion and Flame. 2011. link Times cited: 173 NOT USED (low confidence) Y. Wang, H. Lan, and C. Liu, “Molecular Dynamics Simulations on Sliding Friction between Amorphous Si-DLC Films,” Materials Science Forum. 2011. link Times cited: 0 Abstract: Diamond-like carbon films have been extensively studied over… read moreAbstract: Diamond-like carbon films have been extensively studied over the past decades due to their unique combination of properties, in particular, Si-DLC films are of significant interest for tribological effects. They possess the potential to improve wear performance in humid atmospheres and at higher temperatures. MD simulations were carried out to generate Si-DLC films at different silicon contents from 0 to 50%, in order to theoretically investigate the influence of silicon contents on microstructures and tribological properties between Si-DLC films. The results show that the sp3/sp2 ratio in Si-DLC films increases with the increasing silicon content. The MD simulation results suggest that the friction force increases with addition of silicon to DLC films. The bond numbers of interfilms have showed that the silicon addition promotes the bonding of interfilms forming, which results in the friction force increased. read less NOT USED (low confidence) T. Nakayama, Y. Kangawa, and K. Shiraishi, “Atomic Structures and Electronic Properties of Semiconductor Interfaces.” 2011. link Times cited: 14 NOT USED (low confidence) P. Valentini, T. Schwartzentruber, and I. Cozmuta, “Simulation of Gas-Surface Interactions using ReaxFF Reactive Molecular Dynamics: Oxygen Adsorption on Platinum.” 2010. link Times cited: 3 Abstract: Atomistic simulations equipped with the ab initio based clas… read moreAbstract: Atomistic simulations equipped with the ab initio based classical reactive force fleld ReaxFF are used to study adsorption of oxygen on a Pt(111) surface. Molecular Dynamics (MD) simulations are used to study the adsorption dynamics of O2 on Pt(111) for both normal and oblique impacts, whereas Grand Canonical Monte Carlo (GCMC) calculations are employed to study the surface coverage of atomic oxygen on the same platinum surface. Overall, good quantitative agreement with the experimental data is found. Our MD simulations reproduce the characteristic minimum of the trapping probability at kinetic incident energies around 0.1 eV. This feature is determined by the presence of a physisorption well in the ReaxFF Potential Energy Surface (PES) and the progressive suppression of a steering mechanism when increasing the translational kinetic energy (or the molecule’s rotational energy) because of steric hindrance. In the energy range between 0.1 eV and 0.4 eV, the sticking probability increases, similarly to molecular beam sticking data. For very energetic impacts (above 0.4 eV), ReaxFF predicts sticking probabilities lower than experimental sticking data by almost a factor of 3, due to an overall less attractive ReaxFF PES compared to experiments and DFT. For oblique impacts, the trapping probability does not scale with the total incident kinetic energy, but is reduced by the non-zero parallel momentum because of the PES corrugation. Furthermore, our simulations predict quasi-specular (slightly supraspecular) distributions of angles of re∞ection, in accordance with molecular beam experiments. With GCMC simulations, a coverage of about 0.25 is determined at ultra-vacuum conditions (» 10 i10 atm), reproducing the experimental observations. Further reflning of the potential parameters will be aimed to improve the agreement of sticking results at high Ei (by including direct dissociation pathways in the training set) and to reproduce the p(2£ 2) adsorbates surface structure at 0.25 coverage (by strengthening the lateral repulsion between adsorbed O atoms). read less NOT USED (low confidence) B.-J. Lee, W. Ko, H.-K. Kim, and E.-H. Kim, “The modified embedded-atom method interatomic potentials and recent progress in atomistic simulations,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2010. link Times cited: 137 NOT USED (low confidence) A. Oluwajobi and X. Chen, “The fundamentals of modelling abrasive machining using molecular dynamics,” International Journal of Abrasive Technology. 2010. link Times cited: 17 Abstract: The development of ultra-precision processes which can achie… read moreAbstract: The development of ultra-precision processes which can achieve excellent surface finish and tolerance at the nanometre level is now a critical requirement for many industrial applications. At present, it is very difficult to observe the diverse microscopic physical phenomena occurring in nanometric machining through experiments. The use of molecular dynamics (MD) simulation has proved to be an effective tool for the prediction and the analysis of these processes at the nanometre scale. The crucial task in a MD simulation is the selection of the potential function. The lack of clear understanding about the scope and the limitations of a given potential function may lead to nonsensical results. This article presents the backgrounds of popular potentials used in the modelling of materials processes and the algorithms for the solution of the equations encountered in the simulation. Current applications of MD in abrasive machining are reviewed. read less NOT USED (low confidence) S. Hamaguchi, “Plasma-surface Interactions in Material Processing,” Journal of Physics: Conference Series. 2010. link Times cited: 5 Abstract: In plasma processes such as reactive ion etching and thin fi… read moreAbstract: In plasma processes such as reactive ion etching and thin film deposition for microelectronics device fabrication, atomic-level control of surface morphologies and compositions of processed materials has become increasingly important as the device sizes diminish to the nano-meter range. While various species such as ions, neutral radicals, electrons and photons simultaneously hit the material surface in a plasma, the plasma-surface interactions can be best understood if individual elementary processes such as interaction of specific species with the surface at specific incident energy are studied separately under well controlled conditions. In this article, a molecular dynamics (MD) simulation technique is reviewed as a means to analyse plasma-surface interactions in such a manner and some sample simulation results for polymer etching and diamond-like carbon (DLC) deposition are presented. read less NOT USED (low confidence) Z. Kang, M. Li, and Q. Tang, “Buckling behavior of carbon nanotube-based intramolecular junctions under compression: Molecular dynamics simulation and finite element analysis,” Computational Materials Science. 2010. link Times cited: 51 NOT USED (low confidence) E. S. Zijlstra and M. E. Garcia, “Laser‐Induced Softening of Lattice Vibrations.” 2010. link Times cited: 5 NOT USED (low confidence) K. Tada, S. Horimoto, Y. Kimoto, M. Yasuda, H. Kawata, and Y. Hirai, “Molecular dynamics study on compressive strength of monocrystalline, nanocrystalline and amorphous Si mold for nanoimprint lithography,” Microelectronic Engineering. 2010. link Times cited: 7 NOT USED (low confidence) S. Huh and S. Hyun, “Energetic impact deposition of carbon nanotubes: Mechanism, nanopatterning, and field emission property,” Carbon. 2010. link Times cited: 2 NOT USED (low confidence) R. Rurali and E. Hernández, “Thermally induced directed motion of fullerene clusters encapsulated in carbon nanotubes,” Chemical Physics Letters. 2010. link Times cited: 36 NOT USED (low confidence) A. Montazeri, M. Sadeghi, R. Naghdabadi, and H. Rafii-Tabar, “Computational modeling of the transverse-isotropic elastic properties of single-walled carbon nanotubes,” Computational Materials Science. 2010. link Times cited: 20 NOT USED (low confidence) V. Haxha et al., “Control of strain in GaSbAs/InAs/GaAs quantum dots.” 2010. link Times cited: 5 Abstract: We discuss strain simulations of quantum dot structures cove… read moreAbstract: We discuss strain simulations of quantum dot structures covered with a GaSbAs strain reducing capping layer in the presence of Sb segregation. Cross Sectional Scanning Tunneling Microscopy shows strong Sb and In segregation in the material surrounding the quantum dot. Using the three layer model originally proposed for the SiGe system by D. J. Godbey, M. G. Ancona, J. Vac. Sci. Technol. A 15, 976 (1997) we accurately calculate the segregation profile and include a non uniform composition to our models. Using atomistic modeling, we present strain maps of the quantum dot structures that show the propagation of the strain into the GaAs region is strongly affected by the shape and composition of the strain reduction layer. read less NOT USED (low confidence) K. Bi, Y. Chen, M. Chen, and Y. Wang, “The influence of structure on the thermal conductivities of low-dimensional carbon materials,” Solid State Communications. 2010. link Times cited: 7 NOT USED (low confidence) S. Lu and C. Cho, “THE STUDY OF INSCRIBED SURFACE OF ARMCHAIR CARBON NANOTUBES,” Modern Physics Letters B. 2010. link Times cited: 0 Abstract: It is natural to treat the monolayer crystalline film as the… read moreAbstract: It is natural to treat the monolayer crystalline film as the surface fixed by the positions of atoms. However, this type surface is not well-suited to the monolayer crystalline film. It has been discovered that the mechanical properties of a monolayer crystalline film can be described exquisitely by its inscribed surface. Depending on this inscribed surface of armchair carbon nanotubes, the present study shows the reason why CNTs cannot be simply considered as a thin shell with constant thickness. Application of the present model to armchair CNTs predicts that the ranges of Young's modulus and effective thickness are respectively from 2.407 to 3.209 TPa and from 0.082 to 0.073 nm, which are in good agreement with previous theoretical studies and experimental observations. read less NOT USED (low confidence) Y. Ge, W. Fa, J. Zhou, and J. Dong, “Single-walled gold nanotubes grown in carbon nanotubes: Molecular dynamics simulations,” Physics Letters A. 2010. link Times cited: 6 NOT USED (low confidence) V. Haxha and M. Migliorato, “Calculating strain using atomistic simulations: A review.” 2010. link Times cited: 0 Abstract: We present a short review of methods of evaluating of strain… read moreAbstract: We present a short review of methods of evaluating of strain from atomistic models in the context of linear elasticity. read less NOT USED (low confidence) B. Venkatachari and I. Cozmuta, “Atomistic modeling of the decomposition of charring ablators.” 2010. link Times cited: 3 Abstract: The composition of the pyrolysis gases that are injected int… read moreAbstract: The composition of the pyrolysis gases that are injected into the boundary layer gas from an ablative thermal protection system (TPS) has a significant impact on the aerothermodynamics near the TPS surface. Recent analysis revealed that predicted surface response (surface temperature) of the TPS is very sensitive to the initial composition of the pyrolysis gases in the pyrolysis zone and leads to large uncertainties. This sensitivity study used a recently developed high-fidelity numerical model ‐ one that accounts for the transport and reaction of the pyrolysis gases through the char in the surface ablation process, using both equilibrium and detailed finite-rate chemistry. For most carbon (PICA) or silicon (AVCOAT) based ablators the decomposing phase is a phenolic resin for which the underlying mechanism of thermal decomposition is unknown; the by-products of the thermal decomposition have also not been experimentally determined. The current study represents an attempt to obtain an improved fundamental understanding of the thermal decomposition process of phenolic resins, using reactive force field (ReaxFF/LAMMPS) atomistic simulations and characterize the composition of the resulting pyrolysis gas at various temperatures. In this study, decomposition pathways along with the composition of the resulting pyrolysis gases at different temperatures are determined for phenol and one other sub-structure of the PICA monomer, as a precursor for future studies on the thermal decomposition of more complex substances like PICA and other phenolic-resin based ablators. Results from these simulations are outlined in terms of detailing the decomposition pathway and by comparing the resulting composition of the pyrolysis gases against available experimental data. Potential use of these simulation tools in constructing simplified chemical kinetic models, which has wide implications for TPS modeling, is also discussed. read less NOT USED (low confidence) D. Holec, M. Hartmann, F. Fischer, F. Rammerstorfer, P. Mayrhofer, and O. Paris, “Curvature-induced excess surface energy of fullerenes: Density functional theory and Monte Carlo simulations,” Physical Review B. 2010. link Times cited: 29 Abstract: Carbon nanostructures are investigated using a multiscale ap… read moreAbstract: Carbon nanostructures are investigated using a multiscale approach based on density functional theory DFT and Monte Carlo MC simulations. The structure of small fullerenes is calculated using DFT, and simple models are employed to determine classical potential functions which are then used in MC simulations to investigate larger structures. The structural parameters as obtained by DFT and by MC simulations are cross validated for small fullerenes, allowing to understand the effect of the approximations made in MC simulations. It is found that MC overestimates the numerical value of the excess surface energy of carbon nanostructures but the functional dependence, i.e., the decay exponent as a function of the fullerene size, is accurately described. The MC results reveal that bond torsion is the dominant term of the total curvature energy. The combination of DFT and MC allows to get reliable estimates for the excess surface energy of fullerenes as a function of radius for a wide range of fullerene sizes, which may serve as an important input for large-scale finite-element modeling of more complex systems. read less NOT USED (low confidence) Y.-C. Wang, Q. Kuo, and C. Chen, “Interactions between a buckled carbon nanotube and fullerene via molecular-dynamics simulations.” 2010. link Times cited: 0 Abstract: Mechanically buckled components may exhibit negative stiffne… read moreAbstract: Mechanically buckled components may exhibit negative stiffness, namely negative slopes of load‐displacement curves. Negative‐stiffness components are unstable under load control, but may be stabilized when embedded with positive‐stiffness matrix in a form of composite. Negative‐stiffness composites have been shown to exhibit stiffness greater than diamond, and their viscoelastic damping can be largely increased due to the negative‐stiffness inclusions. Combining the negative‐stiffness effects and nano‐scale systems, we studied the mechanical system of a buckled carbon nanotube being compressed laterally with a carbon fullerene with molecular‐dynamics (MD) simulations to explore its high effective stiffness. The buckled nanotube was laterally compressed with a C20 fullerene. The fullerenes are attached to the buckled nanotube via the carbon‐carbon bonding with the Tersoff‐Brenner interatomic potential. The force exerting on the fullerene and the corresponding displacement are monitored and recorded at each... read less NOT USED (low confidence) B. Kan, J. Ding, G. Cheng, X. Q. Wang, Z. Fan, and Z. Ling, “Frequency shift of Single-Walled Carbon Nanotube under axial load,” International Journal of Surface Science and Engineering. 2010. link Times cited: 0 NOT USED (low confidence) J.-Y. Wu, H.-C. Wang, J.-S. Chen, K. Chen, and K. Ting, “Using Molecular Dynamics Simulation and Parallel Computing Technique of the Deposition of Diamond-Like Carbon Thin Films,” Methods and Tools of Parallel Programming Multicomputers. 2010. link Times cited: 0 NOT USED (low confidence) M. Timonova and B. Thijsse, “Thermodynamic properties and phase transitions of silicon using a new MEAM potential,” Computational Materials Science. 2010. link Times cited: 13 NOT USED (low confidence) E. Oh, “Elastic properties of boron-nitride nanotubes through the continuum lattice approach,” Materials Letters. 2010. link Times cited: 105 NOT USED (low confidence) S. Habibi, M. Farid, and M. Kadivar, “Continuum mechanics ability to predict the material response at atomic scale,” Computational Materials Science. 2010. link Times cited: 5 NOT USED (low confidence) E. H. Feng and R. Jones, “Equilibrium thermal vibrations of carbon nanotubes,” Physical Review B. 2010. link Times cited: 24 NOT USED (low confidence) I. Nikiforov, D.-B. Zhang, R. James, and T. Dumitricǎ, “Wavelike rippling in multiwalled carbon nanotubes under pure bending,” Applied Physics Letters. 2010. link Times cited: 59 Abstract: Objective molecular dynamics is used to systematically inves… read moreAbstract: Objective molecular dynamics is used to systematically investigate elastic bending in carbon nanotubes up to 4.2 nm in diameter. A contrasting behavior is revealed: While single-wall tubes buckle in a gradual way, with a clear intermediate regime before they fully buckle, multiwalled tubes with closed cores exhibit a rate- and size-independent direct transition to an unusual wavelike mode with a 1 nm characteristic length. This rippling mode has a nearly-linear bending response and causes a ∼35% reduction in the stiffness of the thickest multiwalled tubes. read less NOT USED (low confidence) K. Katin and A. Podlivaev, “Dynamic characteristics of the low-temperature decomposition of the C20 fullerene,” Physics of the Solid State. 2010. link Times cited: 16 NOT USED (low confidence) J. W. Lee, A. Meade, E. Barrera, and J. Templeton, “Dependencies of the thermal conductivity of individual single-walled carbon nanotubes,” Proceedings of the Institution of Mechanical Engineers, Part N: Journal of Nanoengineering and Nanosystems. 2010. link Times cited: 7 Abstract: This work is aimed at assessing the sensitivity of carbon na… read moreAbstract: This work is aimed at assessing the sensitivity of carbon nanotube (CNT) thermal conductivity to physical and numerical parameters owing to its wide variation in the literature. CNTs of various lengths, chiralities, and temperatures are simulated with molecular dynamics. The Tersoff and AIREBO potentials are also compared in this study. Thermal conductivity is computed with two different non-equilibrium molecular dynamics (NEMD) methods, which show interestingly divergent results; exploring the CNT phonon density of states reveals a proximate cause for the differences. read less NOT USED (low confidence) X. Guo and T. Zhang, “A study on the bending stiffness of single-walled carbon nanotubes and related issues,” Journal of The Mechanics and Physics of Solids. 2010. link Times cited: 23 NOT USED (low confidence) D. Camacho and Y. Niquet, “Application of Keating’s valence force field model to non-ideal wurtzite materials,” Physica E-low-dimensional Systems & Nanostructures. 2010. link Times cited: 52 NOT USED (low confidence) Z. Ni, H. Bu, M. Zou, H. Yi, K. Bi, and Y. Chen, “Anisotropic mechanical properties of graphene sheets from molecular dynamics,” Physica B-condensed Matter. 2010. link Times cited: 266 NOT USED (low confidence) N. Kaur, S. Gupta, V. Jindal, and K. Dharamvir, “Pressure induced transformations in condensed and molecular phases of C60,” Carbon. 2010. link Times cited: 11 NOT USED (low confidence) W. M. Brown, A. Thompson, and P. Schultz, “Efficient hybrid evolutionary optimization of interatomic potential models.,” The Journal of chemical physics. 2010. link Times cited: 19 Abstract: The lack of adequately predictive atomistic empirical models… read moreAbstract: The lack of adequately predictive atomistic empirical models precludes meaningful simulations for many materials systems. We describe advances in the development of a hybrid, population based optimization strategy intended for the automated development of material specific interatomic potentials. We compare two strategies for parallel genetic programming and show that the Hierarchical Fair Competition algorithm produces better results in terms of transferability, despite a lower training set accuracy. We evaluate the use of hybrid local search and several fitness models using system energies and/or particle forces. We demonstrate a drastic reduction in the computation time with the use of a correlation-based fitness statistic. We show that the problem difficulty increases with the number of atoms present in the systems used for model development and demonstrate that vectorization can help to address this issue. Finally, we show that with the use of this method, we are able to "rediscover" the exact model for simple known two- and three-body interatomic potentials using only the system energies and particle forces from the supplied atomic configurations. read less NOT USED (low confidence) P. Mikulski, M. T. Knippenberg, and J. A. Harrison, “Merging bond-order potentials with charge equilibration.,” The Journal of chemical physics. 2009. link Times cited: 23 Abstract: A method is presented for extending any bond-order potential… read moreAbstract: A method is presented for extending any bond-order potential (BOP) to include charge transfer between atoms through a modification of the split-charge equilibration (SQE) formalism. Variable limits on the maximum allowed charge transfer between atomic pairs are defined by mapping bond order to an amount of shared charge in each bond. Charge transfer is interpreted as an asymmetry in how the shared charge is distributed between the atoms of the bond. Charge equilibration (QE) assesses the asymmetry of the shared charge, while the BOP converts this asymmetry to the actual amount of charge transferred. When applied to large molecules, this BOP/SQE method does not exhibit the unrealistic growth of charges that is often associated with QE models. read less NOT USED (low confidence) E. Hernández et al., “Using Thermal Gradients for Actuation in the Nanoscale.” 2009. link Times cited: 0 NOT USED (low confidence) O. Okeke and J. Lowther, “Molecular dynamics of binary metal nitrides and ternary oxynitrides,” Physica B-condensed Matter. 2009. link Times cited: 5 NOT USED (low confidence) M. Ali, “A recursive topographical differential evolution algorithm for potential energy minimization,” Journal of Industrial and Management Optimization. 2009. link Times cited: 2 Abstract: The problem of the determination of the minimum energy confi… read moreAbstract: The problem of the determination of the minimum energy configuration of an
arrangement of $N$ point particles under the interaction of their
interatomic forces is discussed. The interatomic force is described by
a classical many body potential, namely the Tersoff potential for silicon.
We propose a global optimization algorithm for minimization of
energy of clusters of particles using Tersoff potential. The algorithm
combines the topographical differential evolution (TDE) with
the modified recursive procedure of
the recursive differential evolution (RDE) algorithm.
It also introduces an initialization procedure for the population
set. Two important features
of the new algorithm are that it makes use of the \lq graph
minima' for local search, and that the initial population set
is generated with low function values.
The global minima of clusters consisting of up to
20 particles are investigated. The new algorithm is compared with a recent
genetic algorithm. read less NOT USED (low confidence) J. Wu, Z. Zhang, B. Liu, K. Hwang, and Y. Huang, “Numerical analyses for the atomistic-based shell theory of carbon nanotubes,” International Journal of Plasticity. 2009. link Times cited: 7 NOT USED (low confidence) H. Bu, Y. Chen, M. Zou, H. Yi, K. Bi, and Z. Ni, “Atomistic simulations of mechanical properties of graphene nanoribbons,” Physics Letters A. 2009. link Times cited: 145 NOT USED (low confidence) S. E. Wethekam, “Ladungsaustausch schneller Edelgasatome und Fullerene mit Festkörperoberflächen.” 2009. link Times cited: 3 Abstract: This work is devoted to the study of model systems for the i… read moreAbstract: This work is devoted to the study of model systems for the interaction of atoms, molecules, and their ions with solid surfaces. The thesis consists of three parts. In the first part, He atoms and ions with keV energies are scattered under grazing angles of incidence from Al(111), Al(100), and Al(110) surfaces. Fractions of surviving ions and normal energy gains of He+ ions prior to neutralization, derived from shifts of angular distributions for incident atoms and ions, are compared to results from three-dimensional Monte Carlo simulations based on theoretically calculated Auger neutralization rates and He ground-state energy shifts. From the good agreement of experimental data with simulations, a detailed microscopic understanding for a model system of ion-surface interactions is concluded. The studies are extended to noble gas atoms and surfaces with a more complex electronic structure as well as the Auger ionization process, for which a comparison to simulations based on first ab-initio calculations is presented. In the second set of experiments, the formation of doubly excited states of He atoms during collisions of He2+ ions with energies between 60 eV and 1 keV with Ni(110) and Fe(110) surfaces is studied via Auger electron spectroscopy. The electron spectra from autoionization of doubly excited states of 2`2`′ configurations show a pronounced dependence on the coverage of the target surface with adsorbates. Thermal desorption and dissolution of surface contaminations into the bulk at elevated temperatures provide an alternative interpretation of recent work where the local electron spin polarization of Ni(110) and Fe(110) surfaces was deduced from changes in the electron spectra as function of target temperature. In the third part, angular distributions, fragmentation, and charge fractions are studied for grazing scattering of C60 fullerene ions with keV energies from Al(100), Be(0001), and LiF(100) surfaces. At low energies for the motion along the surface normal, the fullerenes are scattered nearly elastically, whereas for larger normal energies, the energy loss is substantial with pronounced differences for metal and insulator surfaces. From a comparison with classical trajectory simulations, a strong perturbation of the elastic properties of the fullerene by a nearby metal surface is concluded. Shifts of angular distributions for incident C60 and C 2+ 60 projectiles for the metal surfaces are in quantitative accord with a classical over-the-barrier model and provide the first information on distances of electron transfer for positively charged fullerenes in front of metal surfaces. For the LiF(100) surface, pronounced kinematically induced internal excitations due to interactions with the periodic electric field at the surface are observed. read less NOT USED (low confidence) Jos, P. Rino, G. O. Cardozo, and A. Picinin, “Atomistic Modeling of the Structural and Thermal Conductivity of the InSb,” Cmc-computers Materials & Continua. 2009. link Times cited: 6 Abstract: A new parametrization for the previous empirical interatomic… read moreAbstract: A new parametrization for the previous empirical interatomic potential for indium antimonite is presented. This alternative parametrization is designed to correct the energetic sequence of structures. The effective empirical interatomic potential proposed consists of two and three body interactions which has the same functional form of the interatomic potential proposed by Vashishta et. al. to study other semiconductors (Branicio et al., 2003; Ebbsjo et al., 2000; Shimojo et al., 2000; Vashishta et al., 2008). Molecular dynamics simulations (MD) are performed to study high pressure phases of InSb up to 70 GPa and its thermal conductivity as a function of temperature. The rock-salt to cesium chloride, expected to occur at high pressures, is observed with the proposed interatomic potential. read less NOT USED (low confidence) P. Valentini, T. Schwartzentruber, and I. Cozmuta, “A Mechanism-Based Finite-Rate Surface Catalysis Model for Simulating Reacting Flows.” 2009. link Times cited: 9 Abstract: A mechanism-based finite-rate wall boundary condition is imp… read moreAbstract: A mechanism-based finite-rate wall boundary condition is implemented in a state-ofthe-art finite volume CFD thermochemical nonequilibrium code to study a high enthalpy CO2 flow over blunt bodies. All the relevant surface processes responsible for the catalytic behavior of the wall are accounted for, including adsorption and desorption (both atomic and molecular), and Eley-Rideal and Langmuir-Hinshelwood recombinations. The model only requires the specification of the reaction rates for each of the processes considered, and the law of mass action is used to compute surface coverages and mass fluxes produced or consumed at the wall due to its catalytic activity. The kinetic rates are chosen to describe a platinum surface, with a fairly high degree of catalycity with respect to CO oxidation. As expected, the predicted heat flux is intermediate between the two extrema, namely the non-catalytic and supercatalytic wall assumptions. Because the only input of the model are the reaction rates, which are usually unavailable or affected by a large experimental uncertainty, the use of Molecular Dynamics simulations employing the Quantum Chemistry based reactive force field ReaxFF is proposed as a novel approach to both determine and characterize each of the underlying processes which collectively cause the wall catalytic activity. Because (dissociative) adsorption is a fundamental step leading to surface recombinations, the sticking of O2 on Pt(111) is studied using ReaxFF Molecular Dynamics simulations. read less NOT USED (low confidence) D. Brenner, C. T. White, M. Elert, and F. Walker, “Chemical model for intrinsic detonation velocities,” International Journal of Quantum Chemistry. 2009. link Times cited: 1 Abstract: Straightforward considerations suggest that chemical reactio… read moreAbstract: Straightforward considerations suggest that chemical reactions including fragmentation processes at or near the front of a chemically sustained shock wave can be important in regulating the shock velocity. This picture is supported by results of recent computer simulations of a model A-B energetic material. read less NOT USED (low confidence) K. Chandraseker, S. Mukherjee, J. T. Paci, and G. Schatz, “An atomistic-continuum Cosserat rod model of carbon nanotubes,” Journal of The Mechanics and Physics of Solids. 2009. link Times cited: 75 NOT USED (low confidence) V. Perebeinos and J. Tersoff, “Valence force model for phonons in graphene and carbon nanotubes,” Physical Review B. 2009. link Times cited: 46 Abstract: Many calculations require a simple classical model for the i… read moreAbstract: Many calculations require a simple classical model for the interactions between sp 2 -bonded carbon atoms, as in graphene or carbon nanotubes. Here we present a valence force model to describe these interactions. The calculated phonon spectrum of graphene and the nanotube breathing-mode energy agree well with experimental measurements and with ab initio calculations. The model does not assume an underlying lattice, so it can also be directly applied to distorted structures. The characteristics and limitations of the model are discussed. read less NOT USED (low confidence) P. Mantena, A. Al-ostaz, and A. Cheng, “Dynamic response and simulations of nanoparticle-enhanced composites,” Composites Science and Technology. 2009. link Times cited: 30 NOT USED (low confidence) H.-C. Cheng, Y.-L. Liu, Y. Hsu, and W.-H. Chen, “Atomistic-continuum modeling for mechanical properties of single-walled carbon nanotubes,” International Journal of Solids and Structures. 2009. link Times cited: 78 NOT USED (low confidence) M. Ruda, D. Farkas, and G. García, “Atomistic simulations in the Fe–C system,” Computational Materials Science. 2009. link Times cited: 79 NOT USED (low confidence) V. Haxha et al., “Empirical bond order potential calculations of the elastic properties of epitaxial InGaSbAs layers,” Microelectron. J. 2009. link Times cited: 2 NOT USED (low confidence) E. Salmon, A. Duin, F. Lorant, P. Marquaire, and W. Goddard, “Thermal decomposition process in algaenan of Botryococcus braunii race L. Part 2: Molecular dynamics simulations using the ReaxFF reactive force field,” Organic Geochemistry. 2009. link Times cited: 95 NOT USED (low confidence) S. Heo and S. Sinnott, “Computational investigation of the mechanical properties of nanomaterials,” Diamond and Related Materials. 2009. link Times cited: 8 NOT USED (low confidence) A. Page and B. Moghtaderi, “Molecular dynamics simulation of the low-temperature partial oxidation of CH4.,” The journal of physical chemistry. A. 2009. link Times cited: 44 Abstract: Low-temperature partial oxidation of methane was investigate… read moreAbstract: Low-temperature partial oxidation of methane was investigated using reactive molecular dynamics (MD) and quantum mechanical (QM) methods. In particular, the ReaxFF hydrocarbon force field [Chenoweth, K.; et al. J. Phys. Chem. A 2008, 112, 1040] was employed to simulate a [20 CH(4) + 10 O(2)] model system at 500 degrees C. The chemical mechanism of the partial oxidation of methane was proposed on the basis of analysis of the computed trajectory of this model system. The partial oxidation of methane was observed to be initiated by the abstraction of hydrogen from CH(4) by O(2) and the atomization of CH(4) itself. Subsequent radical recombination between hydrogen atoms and the dehydrogenation of CH(4) were the primary pathways by which H(2) was formed. In agreement with current models of low-temperature combustion, radicals including H(3)C-OO and H(2)C-OO were also observed during the MD simulation. The observed reaction mechanism was subsequently analyzed using QM methods. For instance, structural features of prominent radical species observed during the MD simulation were analyzed using density functional theory (DFT) and coupled-cluster (CCSD(T)) methods. Enthalpies of reaction of all observed chemical processes were calculated using DFT and the W1 composite method. Where possible, comparisons with experimental data were made. read less NOT USED (low confidence) A. Imtani and V. Jindal, “Pressure effects on bond lengths and shape of zigzag single-walled carbon nanotubes,” arXiv: Materials Science. 2008. link Times cited: 18 NOT USED (low confidence) S. Lu, C. Cho, and L. Song, “ENERGY OF ARMCHAIR NANOTUBE USING THE MODIFIED CAUCHY-BORN RULE,” International Journal of Modern Physics B. 2008. link Times cited: 2 Abstract: Due to the difference of nanotube diameters, the single-wall… read moreAbstract: Due to the difference of nanotube diameters, the single-walled carbon nanotubes (SWCNTs) show the different energy and mechanical properties. In order to take the effect of the curvature of nanotubes into account in the modeling of those structures, the present paper proposes an atomistic based continuum model with using a type of modified Cauchy-Born to link the continuum strain energy to the interatomic potential. This modified Cauchy-Born is developed by incorporating the concept of differential mean value theorem into the standard Cauchy-Born rule. The present model not only can bridge the microscopic and macroscopic length scales, but also can investigate the curvature effect of a single layer film on the continuum level. Application of the current model to armchair carbon nanotubes and graphite shows an excellent prediction of the size dependent strain energy which are compared in a good agreement with the existing experimental and theoretical results. read less NOT USED (low confidence) A. Imtani and V. Jindal, “Modeling and characterizing single-walled carbon nanotubes by pressure probe,” arXiv: Materials Science. 2008. link Times cited: 4 NOT USED (low confidence) A. Imtani and V. Jindal, “Structure of chiral single-walled carbon nanotubes under hydrostatic pressure,” Computational Materials Science. 2008. link Times cited: 19 NOT USED (low confidence) X. W. Zhou and F. Doty, “Embedded-ion method: An analytical energy-conserving charge-transfer interatomic potential and its application to the La-Br system,” Physical Review B. 2008. link Times cited: 30 NOT USED (low confidence) P. Schelling, “Phase behavior and kinetics of a new bond-order potential for silicon,” Computational Materials Science. 2008. link Times cited: 24 NOT USED (low confidence) K. Amara, B. Soudini, D. Rached, and A. Boudali, “Molecular dynamics simulations of the structural, elastic and thermodynamic properties of cubic BBi,” Computational Materials Science. 2008. link Times cited: 16 NOT USED (low confidence) I. Solov’yov, M. Mathew, A. Solov’yov, and W. Greiner, “Liquid surface model for carbon nanotube energetics.,” Physical review. E, Statistical, nonlinear, and soft matter physics. 2008. link Times cited: 19 Abstract: In the present paper we developed a model for calculating th… read moreAbstract: In the present paper we developed a model for calculating the energy of single-wall carbon nanotubes of arbitrary chirality. This model, which we call as the liquid surface model, predicts the energy of a nanotube with relative error less than 1% once its chirality and the total number of atoms are known. The parameters of the liquid surface model and its potential applications are discussed. The model has been suggested for open end and capped nanotubes. The influence of the catalytic nanoparticle, atop which nanotubes grow, on the nanotube stability is also discussed. The suggested model gives an important insight in the energetics and stability of nanotubes of different chirality and might be important for the understanding of nanotube growth process. For the computations we use empirical Brenner and Tersoff potentials and discuss their applicability to the study of carbon nanotubes. From the calculated energies we determine the elastic properties of the single-wall carbon nanotubes (Young modulus, curvature constant) and perform a comparison with available experimental measurements and earlier theoretical predictions. read less NOT USED (low confidence) I. Atsushi, N. Hiroaki, and T. Arimichi, “Molecular Dynamics Simulation of the Chemical Interaction between Hydrogen Atom and Graphene,” Journal of the Physical Society of Japan. 2008. link Times cited: 34 Abstract: We report the chemical interaction between a single hydrogen… read moreAbstract: We report the chemical interaction between a single hydrogen atom and graphene via a classical molecular dynamics simulation using a modified Brenner empirical bond order potential. Three interactions, that is, adsorption, reflection, and penetration, are observed in our simulation. The rates of the interactions depend on the incident energy of the hydrogen atom and the graphene temperature. This dependence can be explained by the following mechanisms: (1) The hydrogen atom experiences a repulsive force due to π electrons. (2) The graphene adsorbs the hydrogen atom and transforms its structure to an “overhang” configuration such as the sp 3 state. (3) The expansion of the six-membered ring causes the loss of the kinetic energy of the hydrogen atom during penetration. read less NOT USED (low confidence) V. Haxha et al., “The use of Abel-Tersoff potentials in atomistic simulations of InGaAsSb/GaAs.” 2008. link Times cited: 2 Abstract: In this paper we show the use of an optimally parameterized … read moreAbstract: In this paper we show the use of an optimally parameterized empirical potential of the Abell-Tersoff type for atomistic simulations of the elastic properties of the epitaxially grown quaternary alloy InGaAsSb. We find that the strain energy as a function of composition does not follow intuitive averages between the binary constituents. Furthermore we will provide an explanation for the often observed decomposition into ternary components. The predictions of our model appear to be substantiated by experimental evidence of growth of InAs self assembled quantum dots capped by GaSbAs. read less NOT USED (low confidence) D. Bandyopadhyay and M. Kumar, “The electronic structures and properties of transition metal-doped silicon nanoclusters: A density functional investigation,” Chemical Physics. 2008. link Times cited: 28 NOT USED (low confidence) V. Jindal and A. Imtani, “Bond lengths of armchair single-waled carbon nanotubes and their pressure dependence,” Computational Materials Science. 2008. link Times cited: 31 NOT USED (low confidence) J. Wu, K. Hwang, Y. Huang, and J. Song, “A Finite-Deformation Shell Theory for Carbon Nanotubes Based on the Interatomic Potential—Part I: Basic Theory,” Journal of Applied Mechanics. 2008. link Times cited: 7 Abstract: A finite-deformation shell theory for carbon nanotubes (CNTs… read moreAbstract: A finite-deformation shell theory for carbon nanotubes (CNTs) is established directly from the interatomic potential for carbon to account for the effect of bending and curvature. Its constitutive relation accounts for the nonlinear multibody atomistic interactions and therefore can model the important effect of CNT chirality and radius. The equilibrium equations and boundary conditions are obtained for the symmetric stresses and bending moments, which are different from many existing shell theories that involve asymmetric stress and bending moments. The theory is used in Part II of this paper to study the instability of carbon nanotubes subjected to different loadings. read less NOT USED (low confidence) V. Haxha et al., “The use of Abell–Tersoff potentials in atomistic simulations of InGaAsSb/GaAs,” Optical and Quantum Electronics. 2008. link Times cited: 2 NOT USED (low confidence) H. Ohta, A. Iwakawa, K. Eriguchi, and K. Ono, “An interatomic potential model for molecular dynamics simulation of silicon etching by Br+-containing plasmas,” Journal of Applied Physics. 2008. link Times cited: 22 Abstract: An interatomic potential model for Si–Br systems has been de… read moreAbstract: An interatomic potential model for Si–Br systems has been developed for performing classical molecular dynamics (MD) simulations. This model enables us to simulate atomic-scale reaction dynamics during Si etching processes by Br+-containing plasmas such as HBr and Br2 plasmas, which are frequently utilized in state-of-the-art techniques for the fabrication of semiconductor devices. Our potential form is based on the well-known Stillinger–Weber potential function, and the model parameters were systematically determined from a database of potential energies obtained from ab initio quantum-chemical calculations using GAUSSIAN03. For parameter fitting, we propose an improved linear scheme that does not require any complicated nonlinear fitting as that in previous studies [H. Ohta and S. Hamaguchi, J. Chem. Phys. 115, 6679 (2001)]. In this paper, we present the potential derivation and simulation results of bombardment of a Si(100) surface using a monoenergetic Br+ beam. read less NOT USED (low confidence) M. Maslov, “Theoretical modeling of the cubane-based chains, networks and bulks.” 2008. link Times cited: 2 Abstract: We have carried out quantum-mechanical calculations of the q… read moreAbstract: We have carried out quantum-mechanical calculations of the quasione-dimensional, quasitwo-dimensional and bulk cubane-based structures by means of a nonorthogonal tight- binding potential. The possible existence of these compounds is predicted. Geometries and energetic properties are calculated. We found that the density of supercubane structure is only 2.7 g/cm3 and it is not a high-density polymorph. The intermolecular bond in such a structure equals to 1.460 A. read less NOT USED (low confidence) J. Adhikari, “Molecular simulation study of the structural properties in InxGa1−xAs alloys: Comparison between Valence Force Field and Tersoff potential models,” Computational Materials Science. 2008. link Times cited: 6 NOT USED (low confidence) P. Koumoutsakos, “Multiscale Modeling and Simulation for Fluid Mechanics at the Nanoscale.” 2008. link Times cited: 2 NOT USED (low confidence) D. Shiri, Y. Kong, A. Buin, and M. Anantram, “Strain induced change of bandgap and effective mass in silicon nanowires,” Applied Physics Letters. 2008. link Times cited: 135 Abstract: This work computationally investigates the electromechanical… read moreAbstract: This work computationally investigates the electromechanical properties of hydrogen passivated silicon nanowires under uniaxial tensile strain. It has been observed that bandgap changes can be as large as 60 and 100 meV per 1% axial strain for [100] and [110] nanowires, respectively. This rate of change in the bandgap is independent of nanowire size and depends only on the growth direction. More importantly, the nature of the bandgap can reversibly change from indirect to direct as a function of strain. It is also observed that for larger diameter nanowires, the indirect-to-direct transition occurs at smaller compressive strain. read less NOT USED (low confidence) R. Khare, S. L. Mielke, J. T. Paci, G. Schatz, and T. Belytschko, “A simple energy-scaling scheme for fine-tuning empirical potentials for coupled quantum mechanical/molecular mechanical studies,” Chemical Physics Letters. 2008. link Times cited: 3 NOT USED (low confidence) L. Sun et al., “Electronic structures of SiC nanoribbons.,” The Journal of chemical physics. 2008. link Times cited: 215 Abstract: Electronic structures of SiC nanoribbons have been studied b… read moreAbstract: Electronic structures of SiC nanoribbons have been studied by spin-polarized first-principles calculations. The armchair nanoribbons are nonmagnetic semiconductors, while the zigzag nanoribbons are magnetic metals. The spin polarization in the zigzag SiC nanoribbons is originated from the unpaired electrons localized on the ribbon edges. Interestingly, the zigzag nanoribbons narrower than approximately 4 nm present half-metallic behavior. Without the aid of external field or chemical modification, the metal-free half-metallicity predicted for narrow SiC zigzag nanoribbons opens a facile way for nanomaterial-based spintronics applications. read less NOT USED (low confidence) P. Tian, “Molecular dynamics simulations of nanoparticles.” 2008. link Times cited: 37 Abstract: A review of molecular dynamics simulation studies of nanopar… read moreAbstract: A review of molecular dynamics simulation studies of nanoparticles is presented. While research on nanoparticles and their usage in industries, healthcare, and biomedical sciences has been very active, real time observation and analysis of some dynamical and thermodynamic properties and physical mechanisms underlying many of the special characteristics of various nanoparticles are not easily achieved experimentally. Due to the rapid development of the computational algorithms and available computational resources to scientific researchers and relatively small sizes of nanoparticles, molecular dynamics (MD) simulations, together with other computational methods, occupy an increasingly important niche in this rapidly developing and expanding field. As part of the Annual Reports, the focus of this review is on the research published during the last year. A brief survey of fundamentals of MD simulations is given first, followed by how various MD methodologies are utilized for the investigations of the nucleation and melting behavior of various metallic nanoparticles, for the understanding of structural and physiochemical properties of metal oxide and semiconductor nanoparticles; and for the studies of interactions of nanoparticles with their surrounding materials and among themselves. The role of multiscale modeling, involving both methods and applications, in nanoparticle research is discussed. The challenges and opportunities in the future are briefly discussed at the end. read less NOT USED (low confidence) Y. Sun and K. Liew, “The buckling of single-walled carbon nanotubes upon bending: The higher order gradient continuum and mesh-free method,” Computer Methods in Applied Mechanics and Engineering. 2008. link Times cited: 100 NOT USED (low confidence) Y. Sun and K. Liew, “Mesh-free simulation of single-walled carbon nanotubes using higher order Cauchy–Born rule,” Computational Materials Science. 2008. link Times cited: 45 NOT USED (low confidence) G. Li, J. Cai, J. Deng, A. Rocha, and S. Sanvito, “The difference of the transport properties of graphene with corrugation structure and with flat structure,” Applied Physics Letters. 2008. link Times cited: 9 Abstract: The transport properties of devices made from graphene ribbo… read moreAbstract: The transport properties of devices made from graphene ribbons with either perfectly flat or corrugated structures and sandwiched between metallic electrodes are investigated with first principles method. The relaxed geometry of the devices is obtained by using molecular dynamics based on the Tersoff’s potential, while the transport is evaluated with a combination of density functional theory and the nonequilibrium Green’s function method. In general, the transport properties of the two graphene structures differ from each other. In particular, we find that corrugation greatly enhances the conductance through the device. read less NOT USED (low confidence) J.-M. Lu, Y.-C. Wang, J.-G. Chang, M.-H. Su, and C. Hwang, “Molecular-Dynamic Investigation of Buckling of Double-Walled Carbon Nanotubes under Uniaxial Compression(Condensed matter: structure and mechanical and thermal properties),” Journal of the Physical Society of Japan. 2008. link Times cited: 7 Abstract: This paper studies the buckling phenomena and mechanical beh… read moreAbstract: This paper studies the buckling phenomena and mechanical behavior of single-walled carbon nanotubes (SWNTs) and double-walled carbon nanotubes (DWNTs) via molecular dynamics simulations. The Tersoff interatomic C–C potential is adopted. Using a dimensionless parameter, slenderness ratio (SR, the ratio of length to diameter), we investigate the mechanical behavior of long and short nanotubes under compression through their buckling modes, total strain energy and strain energy density, as well as post-buckling. The curvatures of strain energy provide a means to measure the Young’s modulus of the nanotubes. Moreover, jumps in either the strain energy or strain energy density indicate identical mechanical buckling strains, and are studied in relation to buckling modes. In our simulations, a transition time is observed for short nanotubes to reach stable vase-like buckling mode, indicating a time-dependent property of nanotubes. Furthermore, nanotubes with small SR can bear higher compressive load after their first buckling. In addition, nanotubes with same chirality exhibit roughly the same elastic modulus, regardless of their lengths, when applied compressive strains are less than 5% strain. However, long nanotubes show smaller buckling strength. Effects of temperature at 300 K on buckling strength for SWNT are also discussed in connection to our present study at 1 K. read less NOT USED (low confidence) K. Nordlund and S. Dudarev, “Interatomic potentials for simulating radiation damage effects in metals,” Comptes Rendus Physique. 2008. link Times cited: 29 NOT USED (low confidence) L. Vattuone, L. Savio, and M. Rocca, “Bridging the structure gap: Chemistry of nanostructured surfaces at well-defined defects,” Surface Science Reports. 2008. link Times cited: 103 NOT USED (low confidence) J. Leininger, C. Minot, and F. Lorant, “Two theoretical simulations of hydrocarbons thermal cracking: Reactive force field and density functional calculations,” Journal of Molecular Structure-theochem. 2008. link Times cited: 18 NOT USED (low confidence) J.-M. Lu, C. C. Hwang, Q. Kuo, and Y. Wang, “Mechanical buckling of multi-walled carbon nanotubes: The effects of slenderness ratio,” Physica E-low-dimensional Systems & Nanostructures. 2008. link Times cited: 25 NOT USED (low confidence) N. Kaur, S. Gupta, K. Dharamvir, and V. Jindal, “The formation of dimerized molecules of C60 and their solids,” Carbon. 2008. link Times cited: 6 NOT USED (low confidence) Y. Lin, T.-C. Chen, P.-F. Yang, S. Jian, and Y. Lai, “Atomic-level simulations of nanoindentation-induced phase transformation in mono-crystalline silicon,” Applied Surface Science. 2007. link Times cited: 52 NOT USED (low confidence) Y. Sasajima, T. Akabane, T. Nakazawa, and A. Iwase, “Computer simulation of high-energy-beam irradiation of single-crystalline silicon,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 8 NOT USED (low confidence) B. Jeong, J.-K. Lim, and S. Sinnott, “Multiscale-failure criteria of carbon nanotube systems under biaxial tension–torsion,” Nanotechnology. 2007. link Times cited: 12 Abstract: The failure criteria for carbon nanotube system fracture und… read moreAbstract: The failure criteria for carbon nanotube system fracture under biaxial tensile–torsional loads are developed based on a multiscale approach that adopts continuum mechanics models to describe atomistic predictions of failure from molecular dynamics simulations. The failure strength or envelope of carbon nanotube systems under this type of loading is significantly different from what occurs under uniaxial tensile loading and, importantly, is different from the predictions of failure criteria for macroscopic objects. The failure criteria developed here can be used to design carbon nanotube-based devices and materials, such as nanoelectromechanical systems and nanocomposites, which undergo biaxial tensile–torsional loading. read less NOT USED (low confidence) M. Müller, P. Erhart, and K. Albe, “Thermodynamics of L 1 0 ordering in FePt nanoparticles studied by Monte Carlo simulations based on an analytic bond-order potential,” Physical Review B. 2007. link Times cited: 64 Abstract: The size dependence of the order-disorder transition in FePt… read moreAbstract: The size dependence of the order-disorder transition in FePt nanoparticles with an $L{1}_{0}$ structure is investigated by means of Monte Carlo simulations based on an analytic bond-order potential for FePt. A cross parametrization for the Fe-Pt interaction is proposed, which complements existing potentials for the constituents Fe and Pt. This FePt potential properly describes structural properties of ordered and disordered phases, surface energies, and the $L{1}_{0}$ to $A1$ transition temperature in bulk FePt. The potential is applied for examining the ordering behavior in small particles. The observed lowering of the order-disorder transition temperature with decreasing particle size confirms previous lattice-based Monte Carlo simulations [M. M\"uller and K. Albe, Phys. Rev. B 72, 094203 (2005)]. Although a distinctly higher amount of surface induced disorder is found in comparison to previous studies based on lattice-type Hamiltonians, the presence of lattice strain caused by the tetragonal distortion of the $L{1}_{0}$ structure does not have a significant influence on the depression of the ordering temperature with decreasing particle size. read less NOT USED (low confidence) C. Wei, “Structural phase transition of alkane molecules in nanotube composites,” Physical Review B. 2007. link Times cited: 26 Abstract: The structural phase transition and crystallization process … read moreAbstract: The structural phase transition and crystallization process of alkane molecules in carbon nanotube (CNT) composites are studied through molecular dynamics simulations. An isotropic-to-nematic transition with molecules aligned with embedded nanotubes is found. Further smectic transition of the alkane molecules is found with small radius armchaired CNT (5, 5) as a nucleation site, where molecules form lamellar layers along the nanotube axis and have two-dimensional structure ordering in planes perpendicular to the tube axis, in analogy to a crystalline polymer. While the translational diffusions are limited in the smectic phase, the molecules have rotation freedom along its long axis with small energy barriers $\ensuremath{\sim}1\char21{}2{k}_{B}T$ $(T=300\phantom{\rule{0.3em}{0ex}}\mathrm{K})$ for decane molecules studied here. The molecule crystallization is found to strongly dependent on CNT chirality, where a lack of two-dimensional ordering of molecules around a similar radius zigzag CNT (10, 0) is due to diverse molecule assembly domains with tilted wrapping angles at the interface, controlled by the interactions with the substrate nanotube lattice. The influence of strength and nature of the interfacial interactions between molecules and nanotubes is also studied. The results in this study would be useful and important for the understanding of structural phases in hybrid polymer materials and for the designs of nanotube based high performance composites. read less NOT USED (low confidence) G. Yong and D. Jin-ming, “Heat Conductivity of One-Dimensional Carbon Chain in an External Potential,” Chinese Physics Letters. 2007. link Times cited: 2 Abstract: The heat transport in a one-dimensional (1D) carbon nanowire… read moreAbstract: The heat transport in a one-dimensional (1D) carbon nanowire (CNW) lying in an external potential with different amplitudes and periods is studied by the non-equilibrium molecular dynamics method. It is found that the thermal conductivity of CNW is always anomalous, increasing with the CNW length and obeying the power law κ~N, in which α decreases with the increasing external potential amplitude. The thermal conductivity could be enhanced by the external potential with rather larger amplitudes, which means that an applied external potential could be an efficient tool to improve the heat conductivity of a real 1D material. In addition, the effect of different periods of the external potential is studied, finding the external potential with an incommensurate period leads to the smaller α value. read less NOT USED (low confidence) J. Ryoo, P. Hajela, J. Suhr, and N. Koratkar, “Estimation of Young’s modulus of single-walled carbon nanotube using cellular automata,” Adv. Eng. Softw. 2007. link Times cited: 7 NOT USED (low confidence) S.-M. Jeong and T. Kitamura, “Structural Transformation of Single Crystal Silicon under Uniaxial Stress,” Key Engineering Materials. 2007. link Times cited: 2 Abstract: The diamond structure of single crystal silicon transforms t… read moreAbstract: The diamond structure of single crystal silicon transforms to other structures under mechanical stress. We investigate the structural transformation of diamond cubic structure to betatin structure in silicon under uniaxial stress using atomistic simulation on the basis of the Tersoff potential. As a result, under extensive compressive strain, the structural transformation from Si-I to Si-II is found. read less NOT USED (low confidence) H. Whitlow and S. Nakagawa, “Low-energy primary knock on atom damage distributions near MeV proton beams focused to nanometre dimensions,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 14 NOT USED (low confidence) K. Chandraseker and S. Mukherjee, “Atomistic-continuum and ab initio estimation of the elastic moduli of single-walled carbon nanotubes,” Computational Materials Science. 2007. link Times cited: 60 NOT USED (low confidence) W. Chen, H. C. Cheng, and Y. Hsu, “Mechanical Properties of Carbon Nanotubes Using Molecular Dynamics Simulations with the Inlayer van der Waals Interactions,” Cmes-computer Modeling in Engineering & Sciences. 2007. link Times cited: 34 Abstract: The evaluation of the fundamental mechanical properties of s… read moreAbstract: The evaluation of the fundamental mechanical properties of single/multi-walled carbon nanotubes(S/MWCNTs) is of great importance for their industrial applications. The present work is thus devoted to the determination of various mechanical properties of S/MWCNTs using molecular dynamics (MD) simulations. The study first focuses on the exploration of the effect of the weak inlayer van der Waals (vdW) atomistic interactions on the mechanical properties of S/MWCNTs. Secondly, in addition to the zig-zag and armchair types of CNTs, the hybrid type of MWCNTs that comprise a zig-zag outer tube and an inner armchair tube is also analyzed. Thirdly, the investigation is extended to deal with the influence of the axial orientation mismatch between the inner and outer layers of MWCNTs on the associated mechanical properties. Lastly, the behaviors of the interlayer shear force/strength of MWCNTs are discussed in detail. In the MD simulations, the force field between two carbon atoms is modeled with the Tersoff-Brenner (TB) potential while the inlayer/interlayer vdW atomistic interactions are simulated with the Lennard-Jones (L-J) potential. The effectiveness of the MD simulations is demonstrated by comparing the computed results with the theoretical/experimental data available in literature. Some interesting and essential results are pre1 Tsing Hua Chair Professor, Department of Power Mechanical Engineering, National Tsing Hua University, Hsinchu, Taiwan 30013, R.O.C., whchen@pme.nthu.edu.tw 2 Professor, Department of Aerospace and Systems Engineering, Feng Chia University, Taichung, Taiwan 40724, R.O.C., hccheng@fcu.edu.tw 3 Graduate Student, Department of Power Mechanical Engineering, National Tsing Hua University, Hsinchu, Taiwan 30013, R.O.C. sented. With different dimensions and geometries of CNTs, the inlayer vdW atomistic interactions can have up to about 9% increase of the elastic moduli, 27% decrease of the Poisson’s ratios, 12% growth of the shear moduli, and 13% enhancement of the interlayer shear strength. The mechanical properties of the hybrid MWCNTs are found to be midway between the zig-zag and armchair MWCNTs. It is also detected that the axial orientation mismatch between the inner and outer layers of a double-walled CNT has a trivial impact on the mechanical properties of CNTs. To separate the inner layer of a double-walled CNT from its outer layer, it requires a minimum external force of 0.889nN for the zig-zag type, 0.550 nN for hybrid type and 0.493nN for the armchair type. In summary, the effect of the inlayer vdW atomistic interactions can not be neglected and should receive attention in the MD simulations of the mechanical properties of CNTs. Keyword: Molecular Dynamics Simulation, Carbon Nanotubes, Inlayer van der Waals Force, Mechanical Properties. read less NOT USED (low confidence) N. Chakraborti, S. Das, R. Jayakanth, R. Peköz, and S. Erkoç, “Genetic Algorithms Applied to Li+ Ions Contained in Carbon Nanotubes: An Investigation Using Particle Swarm Optimization and Differential Evolution Along with Molecular Dynamics,” Materials and Manufacturing Processes. 2007. link Times cited: 28 Abstract: Empirical potentials based upon two and three body interacti… read moreAbstract: Empirical potentials based upon two and three body interactions were applied to the Li+–C system, assuming the Li+ ions to be distributed inside high-symmetry, single walled carbon nanotubes of different chirality. Structural optimizations for various assemblages were conducted using evolutionary and genetic algorithms, where differential evolution and particle swarm optimization techniques worked satisfactorily. The results were compared with the outcome of some rigorous molecular dynamics simulations. The potential for using the carbon nanotubes in the negative electrode of lithium ion batteries was also critically examined. read less NOT USED (low confidence) T. Sinno, “A bottom-up multiscale view of point-defect aggregation in silicon,” Journal of Crystal Growth. 2007. link Times cited: 32 NOT USED (low confidence) H. W. Zhang, L. Wang, and J. B. Wang, “Computer simulation of buckling behavior of double-walled carbon nanotubes with abnormal interlayer distances,” Computational Materials Science. 2007. link Times cited: 28 NOT USED (low confidence) T. Kumagai, S. Izumi, S. Hara, and S. Sakai, “Development of bond-order potentials that can reproduce the elastic constants and melting point of silicon for classical molecular dynamics simulation,” Computational Materials Science. 2007. link Times cited: 148 NOT USED (low confidence) P. Tsai and T. Fang, “A molecular dynamics study of the nucleation, thermal stability and nanomechanics of carbon nanocones,” Nanotechnology. 2007. link Times cited: 58 Abstract: In this study, the nucleation mechanism of carbon nanocones … read moreAbstract: In this study, the nucleation mechanism of carbon nanocones is investigated using molecular dynamics (MD) simulations and structural analyses and is compared with that of carbon nanotubes. It is shown that the structural stability of carbon nanocones is sensitive to the cone apex angle. Specifically, an increase in the conical angle results in a moderate improvement in the structural stability of the nanocone as a result of a lower strain energy in the capped mantle. The simulation results also show that the melting temperature of the nanocone increases with increasing conical angle. Furthermore, it is observed that a metastable tube-like structure is formed in carbon nanocones with a lower conical angle at temperatures ranging from 2400 to 3600 K. Finally, the numerical simulations reveal that the mechanical properties of carbon nanocones under nanoindentation are strongly dependent on the conical angle. For carbon nanocones with a large conical angle, the high deformation-promoted reactivity and reversible mechanical response have been performed due to highly symmetrical networks. read less NOT USED (low confidence) C. Wang and K. Ho, “Tight‐Binding Molecular Dynamics Studies of Covalent Systems.” 2007. link Times cited: 3 NOT USED (low confidence) D. Powell, M. Migliorato, and A. Cullis, “Optimized Tersoff potential parameters for tetrahedrally bonded III-V semiconductors,” Physical Review B. 2007. link Times cited: 64 Abstract: We address the issue of accurate parametrization for the Abe… read moreAbstract: We address the issue of accurate parametrization for the Abell-Tersoff empirical potential applied to tetrahedrally bonded semiconductor materials. Empirical potential methods for structural relaxation are widely used for group IV semiconductors while, with few notable exceptions, work on III-V materials has not been extensive. In the case of the Abell-Tersoff potential parametrizations exist only for III-As and III-N, and are designed to correctly predict only a limited number of cohesive and elastic properties. In this work we show how by fitting to a larger set of cohesive and elastic properties calculated from density functional theory, we are able to obtain parameters for III-As, III-N, III-P, and III-Sb zinc blende semiconductors, which can also correctly predict important nonlinear effects in the strain. read less NOT USED (low confidence) J. Yu, S. Sinnott, and S. Phillpot, “Charge optimized many-body potential for the Si/SiO2 system,” Physical Review B. 2007. link Times cited: 151 Abstract: A dynamic-charge, many-body potential for the Si/SiO{sub 2} … read moreAbstract: A dynamic-charge, many-body potential for the Si/SiO{sub 2} system, based on an extended Tersoff potential for semiconductors, is proposed and implemented. The validity of the potential function is tested for both pure silicon and for five polymorphs of silica, for which good agreement is found between the calculated and experimental structural parameters and energies. The dynamic charge transfer intrinsic to the potential function allows the interface properties to be captured automatically, as demonstrated for the silicon/{beta}-cristobalite interface. read less NOT USED (low confidence) P. Valavala, T. Clancy, G. Odegard, and T. Gates, “Nonlinear multiscale modeling of polymer materials,” International Journal of Solids and Structures. 2007. link Times cited: 62 NOT USED (low confidence) Z.-H. Zhang, H.-mao Liu, Y. Xiong, W.-J. Xu, and Y.-hong Tang, “UV–vis Absorption and PL Properties of Self-Assembled Silicon Nanotubes,” Chinese Physics Letters. 2007. link Times cited: 4 Abstract: Silicon nanotubes (SiNTs) are novel one-dimensional nanomate… read moreAbstract: Silicon nanotubes (SiNTs) are novel one-dimensional nanomaterials, which have potential applications in nano-photoelectric devices, sensors and field-emission devices. The self-assembled silicon nanotubes have clear structures without metal catalysts. The structures are confirmed by TEM and HRTEM, and the UV-vis absorption spectra with an absorption peak near 685 nm and PL spectra with widened strong emission near 436 nm are measured by UV–vis spectrometer and spectrofluorophotometer. read less NOT USED (low confidence) L. Brutzel and J. Crocombette, “Classical molecular dynamics study of primary damage created by collision cascade in a ZrC matrix,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 31 NOT USED (low confidence) S. Stuart, P. Krstic, T. A. Embry, and C. Reinhold, “Methane production by deuterium impact at carbon surfaces,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 22 NOT USED (low confidence) M. Mrovec, M. Moseler, C. Elsasser, and P. Gumbsch, “Atomistic modeling of hydrocarbon systems using analytic bond-order potentials,” Progress in Materials Science. 2007. link Times cited: 27 NOT USED (low confidence) S. Bukkapatnam, M. Malshe, P. Agrawal, L. Raff, and R. Komanduri, “Parametrization of interatomic potential functions using a genetic algorithm accelerated with a neural network,” Physical Review B. 2006. link Times cited: 17 NOT USED (low confidence) M. Aly, E. Ng, S. Veldhuis, and M. Elbestawi, “Prediction of cutting forces in the micro-machining of silicon using a ‘hybrid molecular dynamic-finite element analysis’ force model,” International Journal of Machine Tools & Manufacture. 2006. link Times cited: 31 NOT USED (low confidence) J. Dziedzic, E. Principi, and J. Rybicki, “Analysis of the mixing rules for the Stillinger–Weber potential: a case-study of Ge–Si interactions in the liquid phase,” Journal of Non-crystalline Solids. 2006. link Times cited: 6 NOT USED (low confidence) K. Chandraseker, S. Mukherjee, and Y. Mukherjee, “Modifications to the Cauchy–Born rule: Applications in the deformation of single-walled carbon nanotubes,” International Journal of Solids and Structures. 2006. link Times cited: 36 NOT USED (low confidence) F. S. A. Muriefah, F. Luca, and A. Togbé, “Computational Methods.” 2006. link Times cited: 171 NOT USED (low confidence) O. A. Oviedo, C. Mayer, G. Staikov, E. Leiva, and W. Lorenz, “Low-dimensional metallic nanostructures and their electrochemical relevance: Energetics and phenomenological approach,” Surface Science. 2006. link Times cited: 6 NOT USED (low confidence) N. Kaur, K. Dharamvir, and V. Jindal, “Behaviour of a bucky-ball under extreme internal and external pressures,” arXiv: Materials Science. 2006. link Times cited: 6 NOT USED (low confidence) M. Bachlechner et al., “Mechanisms of pit formation at strained crystallineSi(111)∕Si3N4(0001)interfaces: Molecular-dynamics simulations,” Physical Review B. 2006. link Times cited: 8 NOT USED (low confidence) H. Nakamura and A. Ito, “Molecular dynamics simulation of sputtering process of hydrogen and graphene sheets,” Molecular Simulation. 2006. link Times cited: 15 Abstract: To clarify the yielding mechanism of small hydrocarbon molec… read moreAbstract: To clarify the yielding mechanism of small hydrocarbon molecules in chemical sputtering between hydrogen and graphene sheets, we made a classical molecular dynamics simulation with modified Brenner's REBO potential, which we proposed to deal with chemical reaction. As the simulation model, we prepared a more realistic physical system, which is composed of 160 incident hydrogen atoms and ten graphene multilayers, than our previous model. From the present work, we found the following fact: breaking the covalent bonds between carbon atoms by hydrogen does not play an important role during the destruction process of graphene structure, but momentum transfer from incident hydrogen to graphene causes it to destroy graphene structure. Moreover, it is found that almost all fragments of graphene sheets form chain-shaped molecules, and that yielded hydrocarbon molecules are composed of carbon chain and single hydrogen-atom. read less NOT USED (low confidence) Z. Huang, Z. N. Guo, X. Chen, T. Yue, S. To, and W. Lee, “Molecular Dynamics Simulation for Ultrafine Machining,” Materials and Manufacturing Processes. 2006. link Times cited: 20 Abstract: This article surveys the advances of molecular dynamics (MD)… read moreAbstract: This article surveys the advances of molecular dynamics (MD) simulation in the research of ultrafine machining and related technologies. Modeling methods, including interatomic potentials and boundary conditions, are addressed. Algorithm strategies for MD simulations are discussed. By applying simulations with Tersoff potential, a case study of the material removal mechanism of the polishing based on coupling vibrations of liquid (PCVL) is presented. read less NOT USED (low confidence) S. Xiao and W. Hou, “Studies of Size Effects on Carbon Nanotubes’ Mechanical Properties by Using Different Potential Functions,” Fullerenes, Nanotubes and Carbon Nanostructures. 2006. link Times cited: 62 Abstract: We use molecular mechanics calculations to study size effect… read moreAbstract: We use molecular mechanics calculations to study size effects on mechanical properties of carbon nanotubes. Both single‐walled nanotubes (SWNTs) and multi‐walled nanotubes (MWNTs) are considered. The size‐dependent Young's modulus decreases with the increasing tube diameter for a reactive empirical bond order (REBO) potential function. However, we observe a contrary trend if we use other potential functions such as the modified Morse potential function and the universal force field (UFF). Such confliction is only obtained for small tubes within cutoff diameters (3 nm for REBO and 1.5 nm for others). In light of these predictions, Young's moduli of large nanotubes concur with experimental results for all the potential functions. No matter which potential function is used, the Poisson's ratio decreases with the increasing tube diameter. We also study the chirality effects on mechanical properties of SWNTs. We find that the Young's moduli are insensitive to the chirality of nanotubes. The chirality effect on the Poisson's ratio is significant for the UFF but not the REBO or modified Morse potential functions. read less NOT USED (low confidence) I. Belabbas, P. Ruterana, J. Chen, and G. Nouet, “The atomic and electronic structure of dislocations in Ga-based nitride semiconductors,” Philosophical Magazine. 2006. link Times cited: 18 Abstract: The atomic and electronic properties of dislocations in III–… read moreAbstract: The atomic and electronic properties of dislocations in III–N semiconductor layers, especially GaN, are presented. The atomic structure of the edge threading dislocation is now well established with three different cores (8 or full core, 5/7 or open core, and 4-atom ring). The use of atomistic simulations has confirmed these atomic structures and has given a good understanding of the electronic structure of the screw dislocation. Partial dislocations which are mostly confined in the area close to the substrate are now also being investigated. It is becoming clear that the electrical activity of all these defects is dependent on the layer quality, which is governed by the growth conditions. read less NOT USED (low confidence) S. Nagao, K. Nordlund, and R. Nowak, “Anisotropic elasticity of IVB transition-metal mononitrides determined by ab initio calculations,” Physical Review B. 2006. link Times cited: 43 Abstract: Elastic parameters of IVB transition-metal mononitrides, TiN… read moreAbstract: Elastic parameters of IVB transition-metal mononitrides, TiN, ZrN, and HfN in the cubic NaCl crystal structure have been calculated by means of density-functional theory with the generalized gradient approximation. The elastic constants c{sub 11}, c{sub 12}, and c{sub 44} were shown to be sufficiently converged with the density of the k-point mesh in the deformed Brillouin zone to discuss the elastic anisotropy of the systems. It was found that the anisotropy coefficient {kappa}{identical_to}(c{sub 11}-c{sub 12})/2c{sub 44} increases with the atomic number of the metal element, i.e., HfN exhibits as strong anisotropy as {kappa}=2.02. The Young's modulus of HfN along is approximately two times higher than that along . Moreover, analysis of the deformation energy by the applied strain modes shows that this elastic anisotropy originates from the strong covalent bonding between metal and nitrogen atoms along . read less NOT USED (low confidence) S. Billeter, A. Curioni, D. Fischer, and W. Andreoni, “Ab initio derived augmented Tersoff potential for silicon oxynitride compounds and their interfaces with silicon,” Physical Review B. 2006. link Times cited: 42 Abstract: Coordination-dependent interatomic potentials are proposed f… read moreAbstract: Coordination-dependent interatomic potentials are proposed for silicon oxides and oxynitrides\char22{}also hydrogenated ones\char22{}with a functional form based on the widely used Tersoff silicon potential. They are intended for an accurate sampling of the configurational space of realistic silicon oxynitride systems and their interfaces with silicon, including defects and changes of oxidation states. The parameters, which are given in the text, are obtained by simultaneously mapping forces and energies onto the results of density-functional-theory calculations performed for a set of diverse systems and configurations and a wide composition range. Application to a larger set of systems and configurations shows the transferability of these augmented Tersoff potentials and their validity in predicting bulk lattice parameters, energetics of defect relaxation, and vibrational spectra. read less NOT USED (low confidence) W. Moon and C. H. Choi, “Molecular-dynamics study of inversion domain boundary in w-GaN,” Physics Letters A. 2006. link Times cited: 7 NOT USED (low confidence) J. Lu and L. Zhang, “Analysis of localized failure of single-wall carbon nanotubes,” Computational Materials Science. 2006. link Times cited: 39 NOT USED (low confidence) J. Dai, W. Seider, and T. Sinno, “Lattice kinetic Monte Carlo simulations of defect evolution in crystals at elevated temperature,” Molecular Simulation. 2006. link Times cited: 22 Abstract: A lattice kinetic Monte Carlo (LKMC) model for vacancy diffu… read moreAbstract: A lattice kinetic Monte Carlo (LKMC) model for vacancy diffusion and aggregation in crystalline silicon at elevated temperature is developed and analyzed in detail by comparing predicted cluster aggregation, thermodynamics, structures and diffusivities with properties obtained from molecular dynamics (MD) simulations. The lattice KMC model is based on a long-range bond-counting scheme in which the bond energies are determined by regression to a single non-equilibrium MD simulation of vacancy aggregation. It is shown that the resulting KMC model is able to capture important high temperature entropic contributions by coarse-graining off-lattice relaxations around defect clusters. read less NOT USED (low confidence) K. Chandraseker and S. Mukherjee, “Coupling of Extension and Twist in Single-Walled Carbon Nanotubes,” Journal of Applied Mechanics. 2006. link Times cited: 45 Abstract: This paper presents a study of the deformation behavior of s… read moreAbstract: This paper presents a study of the deformation behavior of single-walled carbon nanotubes (SWNTs) subjected to extension and twist. The interatomic force description is provided by the Tersoff-Brenner potential for carbon. The rolling of a flat graphene sheet into a SWNT is first simulated by minimizing the energy per atom, the end result being the configuration of an undeformed SWNT The Cauchy-Born rule is then used to connect the atomistic and continuum descriptions of the deformation of SWNTs, and leads to a multilength scale mechanics framework for simulating deformation of SWNTs under applied loads. Coupled extension and twist of SWNTs is considered next. As an alternative to the Cauchy-Born rule for coupled extension-twist problems, a direct map is formulated. Analytic expressions are derived for the deformed bond lengths using the Cauchy-Born rule and the direct map for this class of deformations. Numerical results are presented for kinematic coupling, for imposed extension and imposed twist problems, using the Cauchy-Born rule as well as the direct map, for representative chiral, armchair and zig-zag SWNTs. Results from both these approaches are carefully compared. read less NOT USED (low confidence) X. Guo, J. Wang, and H. Zhang, “Mechanical properties of single-walled carbon nanotubes based on higher order Cauchy–Born rule,” International Journal of Solids and Structures. 2006. link Times cited: 139 NOT USED (low confidence) C. To, “Bending and shear moduli of single-walled carbon nanotubes,” Finite Elements in Analysis and Design. 2006. link Times cited: 111 NOT USED (low confidence) K. Bi, Y. Chen, J. Yang, Y. Wang, and M. Chen, “Molecular dynamics simulation of thermal conductivity of single-wall carbon nanotubes,” Physics Letters A. 2006. link Times cited: 66 NOT USED (low confidence) V. Hùng, K. Masuda-Jindo, and P. Hanh, “Application of the statistical moment method to thermodynamic quantities of silicon,” Journal of Physics: Condensed Matter. 2006. link Times cited: 20 Abstract: The lattice constants, thermal expansion coefficients, speci… read moreAbstract: The lattice constants, thermal expansion coefficients, specific heats at constant volume and those at constant pressure, Cv and Cp, second cumulants, and Lindemann ratio are derived analytically for diamond cubic semiconductors, using the statistical moment method. The calculated thermodynamic quantities of the Si crystal are in good agreement with the experimental results. We also find the characteristic negative thermal expansion in the Si crystal at low temperatures. read less NOT USED (low confidence) A. Heim, N. Grønbech-Jensen, T. Germann, B. Holian, E. Kober, and P. Lomdahl, “Influence of interatomic bonding potentials on detonation properties.,” Physical review. E, Statistical, nonlinear, and soft matter physics. 2006. link Times cited: 11 Abstract: The dependences of the macroscopic detonation properties of … read moreAbstract: The dependences of the macroscopic detonation properties of a two-dimensional (2D) diatomic (AB) molecular system on the fundamental molecular properties were investigated. This includes examining the detonation velocity, reaction zone thickness, and critical width as functions of the exothermicity (Q) of the gas-phase reaction [AB --> (1/2)(A(2) + B(2))] and the gas-phase dissociation energy (D(e)(AB)) for AB --> A + B . Following previous work, molecular dynamics (MD) simulations with a reactive empirical bond-order potential were used to characterize the shock-induced response of a diatomic AB molecular solid, which exothermically reacts to produce A2 and B2 gaseous products. Nonequilibrium MD simulations reveal that there is a linear dependence between the square of the detonation velocity and both of these molecular parameters. The detonation velocities were shown to be consistent with the Chapman-Jouguet (CJ) model, demonstrating that these dependences arise from how the equation of state of the products and reactants are affected. Equilibrium MD simulations of microcanonical ensembles were used to determine the CJ states for varying Q 's, and radial distribution functions characterize the atomic structure. The character of this material near the CJ conditions was found to be somewhat unusual, consisting of polyatomic clusters rather than discrete molecular species. It was also found that there was a minimum value of Q and a maximum value of (D(e)(AB)) for which a pseudo-one-dimensional detonation could not be sustained. The reaction zone of this material was characterized under both equilibrium (CJ) and transient (underdriven) conditions. The basic structure is consistent with the Zeldovich-von Neumann-Döring model, with a sharp shock rise and a reaction zone that extends to 200-300 Angstrom. The underdriven systems show a buildup process which requires an extensive time to approach equilibrium conditions. The rate stick failure diameter (critical width in 2D) was also found to depend on Q and (D(e)(AB)). The dependence on Q could be explained in terms of the reaction zone properties. read less NOT USED (low confidence) J. Los, L. Ghiringhelli, E. Meijer, and A. Fasolino, “Improved long-range reactive bond-order potential for carbon. I. Construction (Correction on vol 72, pg 214102, 2005),” Acta Crystallographica Section B-structural Science. 2005. link Times cited: 181 Abstract: We present LCBOPII, an improvement of the long-range carbon … read moreAbstract: We present LCBOPII, an improvement of the long-range carbon bond-order potential (LCBOP) by Los and Fasolino [Phys. Rev. B 68, 024107 (2003)]. LCBOPII contains a coordination dependent medium range term for bond distances between 1.7 and $4\phantom{\rule{0.3em}{0ex}}\mathrm{\AA{}}$, meant to reproduce the dissociation energy curves for single, double, and triple bonds and improve the reactive properties as well as the description of the liquid and of low coordinated phases. Other features of LCBOPII are a coordination dependent angular correlation, a correction for antibonding states, and a conjugation dependent torsional interaction based on ab initio calculations of the torsional barriers for a set of molecular configurations. We present results for the geometry and energetics of the graphite-to-diamond transformation and of the vacancy in diamond and graphite as well as the prediction of the energy barrier of the 5-77-5 defect in graphite and graphene for which ab initio results are available only for unsuitably small samples. In the accompanying paper (Ghiringhelli et al., Phys. Rev. B 72, 214103 (2005) we use LCBOPII to evaluate several properties, including the equation of state, of liquid carbon. read less NOT USED (low confidence) A. Tekin and B. Hartke, “GLOBAL GEOMETRY OPTIMIZATION OF SILICON CLUSTERS EMPLOYING EMPIRICAL POTENTIALS, DENSITY FUNCTIONALS, AND AB INITIO CALCULATIONS,” Journal of Theoretical and Computational Chemistry. 2005. link Times cited: 13 Abstract: Sin clusters in the size range n = 4–30 have been investigat… read moreAbstract: Sin clusters in the size range n = 4–30 have been investigated using a combination of global structure optimization methods with DFT and ab initio calculations. One of the central aims is to provide explanations for the structural transition from prolate to spherical outer shapes at about n = 25, as observed in ion mobility measurements. Firstly, several existing empirical potentials for silicon and a newly generated variant of one of them were better adapted to small silicon clusters, by global optimization of their parameters. The best resulting empirical potentials were then employed in global cluster structure optimizations. The most promising structures from this stage were relaxed further at the DFT level with the hybrid B3LYP functional. For the resulting structures, single point energies have been calculated at the LMP2 level with a reasonable medium-sized basis set, cc-pVTZ. These DFT and LMP2 calculations were also carried out for the best structures proposed in the literature, including the most recent ones, to obtain the currently best and most complete overall picture of the structural preferences of silicon clusters. In agreement with recent findings, results obtained at the DFT level do support the shape transition from prolate to spherical structures, beginning with Si26 (albeit not completely without problems). In stark contrast, at the LMP2 level, the dominance of spherical structures after the transition region could not be confirmed. Instead, just as below the transition region, prolate isomers are obtained as the lowest-energy structures for n ≤ 29. We conclude that higher (probably multireference) levels of theoretical treatments are needed before the puzzle of the silicon cluster shape transition at n = 25 can safely be considered as explained. read less NOT USED (low confidence) G. Wu, Z.-wei Sun, X. Kong, and D. Zhao, “Molecular dynamics simulation on the out‐of plane thermal conductivity of single‐crystal silicon thin films,” Aircraft Engineering and Aerospace Technology. 2005. link Times cited: 2 Abstract: Purpose – Combining the characteristic of satellite “minisiz… read moreAbstract: Purpose – Combining the characteristic of satellite “minisize nucleus” non‐equilibrium molecular dynamics (NEMD) method is used. We select corresponding Tersoff potential energy function to build model and, respectively, simulate thermal conductivities of silicon nanometer thin film.Design/methodology/approach – NEMD method is used, and the corresponding Tersoff potential energy function is used to build model.Findings – The thermal conductivities of silicon nanometer thin film are markedly below the corresponding thermal conductivities of their crystals under identical temperature. The thermal conductivities are rising with the increase of thickness of thin film; what's more, the conductivities have a linear approximation with thickness of the thin film.Research limitations/implications – It is difficult to do physics experiment.Practical implications – The findings have some theory guidance to analyze satellite thermal control.Originality/value – The calculation results of thermal conductivities specify... read less NOT USED (low confidence) D. Murdick, X. W. Zhou, and H. Wadley, “Assessment of interatomic potentials for molecular dynamics simulations of GaAs deposition,” Physical Review B. 2005. link Times cited: 22 Abstract: Computational studies of atomic assembly processes during Ga… read moreAbstract: Computational studies of atomic assembly processes during GaAs vapor deposition require interatomic potentials that are able to reasonably predict the structures and energies of a molecular arsenic vapor, a variety of elemental gallium and arsenic lattices, binary GaAs lattices, GaAs lattice defects, and 001 GaAs surfaces. These properties were systematically evaluated and compared to ab initio and experimental data for one Tersoff and two Stillinger-Weber SW GaAs interatomic potentials. It was observed that bulk and arsenic molecular properties calculated by the Tersoff parametrization matched density functional predictions and experimental observations significantly better than either of the SW parametrizations. These trends can be related to the bonding physics included in each potential format. Surface free energy calculations indicate that none of these potentials correctly predict the low-energy surface reconstructions of the GaAs 001 surface. Simulated As2 molecular bonding with gallium-rich GaAs 001 surfaces indicate a high sticking probability for SW potentials, which is in good agreement with experimental observations at low growth temperatures. However, the Tersoff parametrization resulted in an unphysically high desorption probability for As2 over a wide range of surface temperatures. read less NOT USED (low confidence) Y. H. Tang, L. Pei, Y. W. Chen, and C. Guo, “Self-assembled silicon nanotubes under supercritically hydrothermal conditions.,” Physical review letters. 2005. link Times cited: 111 Abstract: Self-assembled silicon nanotubes with one-dimensional struct… read moreAbstract: Self-assembled silicon nanotubes with one-dimensional structure have been synthesized from silicon monoxide powder under supercritically hydrothermal conditions with a temperature of 470 degrees C and a pressure of 6.8 MPa. The silicon nanotubes were identified by transmission electron microscopy and high-resolution transmission electron microscopy. The results show that the silicon nanotubes (SiNT) have closed caps. The structures of the silicon nanotubes are hollow inner pore, crystalline silicon wall layers with a 0.31 nm interplanar spacing and 2-3 nm amorphous silica outer layers. Pure crystalline silicon nanotubes survive after etching the silicon nanotubes with 5% HF acid for enough time to imply that the self-assembled silicon nanotubes are stable. A possible theoretical reason for the growth of SiNTs from SiO under supercritically hydrothermal conditions was also proposed. read less NOT USED (low confidence) Q. Lu and B. Bhattacharya, “The role of atomistic simulations in probing the small-scale aspects of fracture—a case study on a single-walled carbon nanotube,” Engineering Fracture Mechanics. 2005. link Times cited: 71 NOT USED (low confidence) H. Zhang, J. B. Wang, and X. Guo, “Predicting the elastic properties of single-walled carbon nanotubes,” Journal of The Mechanics and Physics of Solids. 2005. link Times cited: 89 NOT USED (low confidence) J. Ma, D. Huo, and F. Cui, “Simulation study on radius dependence in rare-gas atoms injection into single-wall carbon nanotubes,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 0 NOT USED (low confidence) C. Rocha, A. Latgé, and L. Chico, “Metallic carbon nanotube quantum dots under magnetic fields,” Physical Review B. 2005. link Times cited: 20 Abstract: Quantum dots made of individual metallic carbon nanotubes ar… read moreAbstract: Quantum dots made of individual metallic carbon nanotubes are theoretically studied under the influence of a magnetic field applied in the axial direction. After assessing the mechanical stability of the heterostructure by Monte Carlo simulations, the dependence of the electronic properties on the size of the nanotube quantum dot and applied magnetic field has been investigated within the Peierls approximation in a tight-binding model. The transport gaps induced by the magnetic field are found to be different from those of the perfect constituent tubes. Due to the presence of topological defects, some physical properties exhibit a lack of periodicity in the magnetic flux. The spin coupling to the magnetic field is also incorporated via a Zeeman term in the Hamiltonian; we have found huge differences between the up and down local densities of states which may be explored for future applications of carbon nanotube quantum dots as spintronic devices. Finally, the temperature dependence of the magnetic properties has also been addressed. We have found a diamagnetic response very similar to that of perfect tubes. read less NOT USED (low confidence) M. J. López, I. Cabria, N. H. March, and J. A. Alonso, “Structural and thermal stability of narrow and short carbon nanotubes and nanostrips,” Carbon. 2005. link Times cited: 35 NOT USED (low confidence) P. Liu, R. Kukta, and D. Kouris, “Strain-modulated adatom and surface vacancy pair interactions,” Journal of Applied Mechanics. 2005. link Times cited: 1 Abstract: Adsorbed atoms (adatoms) and vacancies have a significant ro… read moreAbstract: Adsorbed atoms (adatoms) and vacancies have a significant role to play in the physics of surfaces and the mechanisms of film growth on a substrate. This paper investigates the effect of applied or residual strain on the energetic interaction between pairs of adatoms and vacancies. The analysis is based on a continuum-level point-defect model, where adatoms and vacancies have strain-dependent properties. Atomistic calculations are used to evaluate the defect properties for Si(111). The result is used as input for the defect model in order to investigate the strength and character of the interaction versus strain, separation distance, and relative orientation of the defects. It is found that strain may cause the defects to align in certain direction and modulate their interaction between repulsion and attraction, providing a mechanism for controlled building of nanostructures. read less NOT USED (low confidence) D. Grimm, P. Venezuela, and A. Latgé, “Thermal and mechanical stability of Y-shaped carbon nanotubes,” Physical Review B. 2005. link Times cited: 14 NOT USED (low confidence) V. Ivashchenko, P. Turchi, V. Shevchenko, L. A. Ivashchenko, and O. Shramko, “Simulations of pressure-induced phase transitions in amorphousSixC1−xalloys,” Physical Review B. 2005. link Times cited: 6 NOT USED (low confidence) N. Lorente, R. Rurali, and H. Tang, “Single-molecule manipulation and chemistry with the STM,” Journal of Physics: Condensed Matter. 2005. link Times cited: 59 Abstract: We review recent theoretical work on the manipulation of sin… read moreAbstract: We review recent theoretical work on the manipulation of single molecules with scanning probes, in particular the scanning tunnelling microscope (STM). The aim of theories and simulations is to account for the processes, ideally at a quantitative level, that permit the controlled manipulation of matter at the atomic scale in adsorbed molecular systems. In order to achieve this, simulations rely on total energy and electronic structure calculations where a trade-off is made between the size of the system and the accuracy of the calculation. This first stage of the calculation yields the basic quantities used for the second stage: the evaluation of the coupled electron–nuclear dynamics. This second stage is a formidable task and many approximations are involved. In this review, we will present some of the customary approximations regarding the theoretical study of mechanical and inelastic manipulations. Mechanical manipulations use the interaction between the acting probe (usually a metallic tip) and the targeted adsorbate. We review recent results in the field of adsorbate mechanical manipulations and explain how manipulations can be effected by using the interaction between the probe’s tip and certain molecular groups of complex chemisorbed molecular systems. On the other hand, inelastic manipulations use the tunnelling current to convey energy with sub-ångström precision. This current can excite localized vibrations that can induce measurable variations of the tunnelling conductance, hence providing a means of detecting single-molecule vibrations. This current can also inject energy in a few reaction coordinates. Recently, the possibility of vibrational selective manipulations of NH3/Cu(100) has been experimentally demonstrated. The theory presented here addresses the actual pathways accessed when the molecule is excited by the tunnelling current from an STM. read less NOT USED (low confidence) A. Latgé, D. Grimm, and P. Venezuela, “Y-shaped carbon nanotubes: structural stability and transport properties,” Journal of Molecular Catalysis A-chemical. 2005. link Times cited: 6 NOT USED (low confidence) G. Wu and J. Dong, “Anomalous heat conduction in a carbon nanowire : Molecular dynamics calculations,” Physical Review B. 2005. link Times cited: 21 Abstract: Heat conduction of a real quasi-one dimensional material, th… read moreAbstract: Heat conduction of a real quasi-one dimensional material, the finite length carbon nanowire (CNW), inserted into the single-walled carbon nanotube (SWNT) has been studied by the molecular dynamical (MD) method, in which both of the longitudinal as well as transverse motions of the chain atoms in the SWNT have been permitted. It is found that the thermal conductivity $\kappa $ of the carbon nanowire is very high at room temperature, and diverges more likely with the chain length logarithmically. read less NOT USED (low confidence) M. Longhurst and N. Quirke, “The radial breathing mode of carbon nanotubes,” Molecular Simulation. 2005. link Times cited: 14 Abstract: We report an extensive set of results for the radial breathi… read moreAbstract: We report an extensive set of results for the radial breathing modes (RBM) of infinite and finite length single walled carbon nanotubes using the second generation reactive empirical bond order potential (REBO) developed by Brenner et al. As expected, the frequency, ν of the RBM is inversely proportional to the nanotube radius, R 0, We find two different linear fits to the data, one for zigzag tubes (3.25 THz nm) and one for armchair tubes For finite tubes, the RBM rapidly approaches the infinite length value for nanotubes greater than 5 nm in length. read less NOT USED (low confidence) Y. Jeng, P. Tsai, and T. Fang, “Effects of temperature, strain rate, and vacancies on tensile and fatigue behaviors of silicon-based nanotubes,” Physical Review B. 2005. link Times cited: 32 Abstract: This paper adopts the Tersoff-Brenner many-body potential fu… read moreAbstract: This paper adopts the Tersoff-Brenner many-body potential function to perform molecular dynamics simulations of the tensile and fatigue behaviors of hypothetical silicon-based tubular nanostructures at various temperatures, strain rates, and vacancy percentages. The tensile test results indicate that with a predicted Young's modulus of approximately $60\phantom{\rule{0.3em}{0ex}}\mathrm{GPa}$, silicon nanotubes $(\mathrm{SiNTs})$ are significantly less stiff than conventional carbon nanotubes. It is observed that the presence of hydrogen has a significant influence on the tensile strength of $\mathrm{SiNTs}$. Additionally, the present results indicate that the tensile strength clearly decreases with increasing temperature and with decreasing strain rate. Moreover, it is shown that the majority of the mechanical properties considered in the present study decrease with an increasing vacancy percentage. Regarding the fatigue tests, this study uses a standard theoretical model to derive curves of amplitude stress versus number of cycles for the current nanotubes. The results demonstrate that the fatigue limit of $\mathrm{SiNTs}$ increases with a decreasing vacancy percentage and with increasing temperature. read less NOT USED (low confidence) P. Erhart and K. Albe, “Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide,” Physical Review B. 2005. link Times cited: 462 Abstract: We present an analytical bond-order potential for silicon, c… read moreAbstract: We present an analytical bond-order potential for silicon, carbon, and silicon carbide that has been optimized by a systematic fitting scheme. The functional form is adopted from a preceding work {\}Phys. Rev. B 65, 195124 (2002) and is built on three independently fitted potentials for Si-Si, C-C, and Si-C interaction. For elemental silicon and carbon, the potential perfectly reproduces elastic properties and agrees very well with first-principles results for high-pressure phases. The formation enthalpies of point defects are reasonably reproduced. In the case of silicon stuctural features of the melt agree nicely with data taken from literature. For silicon carbide the dimer as well as the solid phases B1, B2, and B3 were considered. Again, elastic properties are very well reproduced including internal relaxations under shear. Comparison with first-principles data on point defect formation enthalpies shows fair agreement. The successful validation of the potentials for configurations ranging from the molecular to the bulk regime indicates the transferability of the potential model and makes it a good choice for atomistic simulations that sample a large configuration space. read less NOT USED (low confidence) M. S. Valipa, S. Sriraman, E. Aydil, and D. Maroudas, “Atomic-scale analysis of fundamental mechanisms of surface valley filling during plasma deposition of amorphous silicon thin films,” Surface Science. 2005. link Times cited: 9 NOT USED (low confidence) F. Cui, J. Ma, D. Huo, and Z. Chen, “Computer simulation of rare-gas atoms injection into single-wall carbon nanotube,” Physics Letters A. 2004. link Times cited: 3 NOT USED (low confidence) R. Avichail-Bibi, D. Fuks, A. Kiv, T. Maximova, Y. Roizin, and M. Gutman, “A model of the trapping media in microFLASH® memory cells,” Journal of Materials Processing Technology. 2004. link Times cited: 0 NOT USED (low confidence) Y. Jeng, P. Tsai, and T. Fang, “Effects of temperature and vacancy defects on tensile deformation of single-walled carbon nanotubes,” Journal of Physics and Chemistry of Solids. 2004. link Times cited: 62 NOT USED (low confidence) S. Izumi, S. Hara, T. Kumagai, and S. Sakai, “Classification of amorphous-silicon microstructures by structural parameters: molecular dynamics study,” Computational Materials Science. 2004. link Times cited: 13 NOT USED (low confidence) J. Kang and H. Hwang, “Atomistic study of III-nitride nanotubes,” Computational Materials Science. 2004. link Times cited: 45 NOT USED (low confidence) A. Marzegalli et al., “Relaxed SiGe heteroepitaxy on Si with very thin buffer layers: experimental LEPECVD indications and an interpretation based on strain-dependent dislocation nature,” Microelectronic Engineering. 2004. link Times cited: 6 NOT USED (low confidence) J. Chen, A. Béré, G. Nouet, and P. Ruterana, “Analysis of faceting of grain boundaries in GaN,” Superlattices and Microstructures. 2004. link Times cited: 2 NOT USED (low confidence) S. Goumri‐Said, M. Kanoun, A. E. Merad, G. Merad, and H. Aourag, “Empirical molecular dynamics study of structural, elastic and thermodynamic properties of zinc-blende-like SiGe compound,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 2004. link Times cited: 7 NOT USED (low confidence) S. Goumri‐Said, M. Kanoun, A. Merad, G. Merad, and H. Aourag, “Prediction of structural and thermodynamic properties of zinc-blende AlN: molecular dynamics simulation,” Chemical Physics. 2004. link Times cited: 49 NOT USED (low confidence) M. Buehler, Y. Kong, and H. Gao, “Deformation Mechanisms of Very Long Single-Wall Carbon Nanotubes Subject to Compressive Loading,” Journal of Engineering Materials and Technology-transactions of The Asme. 2004. link Times cited: 121 Abstract: We report atomistic studies of single-wall carbon nanotubes … read moreAbstract: We report atomistic studies of single-wall carbon nanotubes with very large aspect ratiossubject to compressive loading. These long tubes display significantly different mechani-cal behavior than tubes with smaller aspect ratios. We distinguish three different classesof mechanical response to compressive loading. While the deformation mechanism ischaracterized by buckling of thin shells in nanotubes with small aspect ratios, it is re-placed by a rod-like buckling mode above a critical aspect ratio, analogous to the Eulertheory in continuum mechanics. For very large aspect ratios, a nanotube is found tobehave like a flexible macromolecule which tends to fold due to vdW interactions betweendifferent parts of the carbon nanotube. This suggests a shell-rod-wire transition of themechanical behavior of carbon nanotubes with increasing aspect ratios. While continuummechanics concepts can be used to describe the first two types of deformation, statisticalmethods will be necessary to describe the dynamics of wire-like long tubes.@DOI: 10.1115/1.1751181#Keywords: Carbon Nanotubes, Mechanical Properties, Buckling Folding, Shell, Rod,Macromolecule read less NOT USED (low confidence) H. Jiang, B. Liu, Y. Huang, and K. Hwang, “Thermal Expansion of Single Wall Carbon Nanotubes,” Journal of Engineering Materials and Technology-transactions of The Asme. 2004. link Times cited: 300 Abstract: We have developed an analytical method to determine the coef… read moreAbstract: We have developed an analytical method to determine the coefficient of thermal expansion (CTE) for single wall carbon nanotubes (CNTs). We have found that all CTEs are negative at low and room temperature and become positive at high temperature. As the CNT diameter decreases, the range of negative CTE shrinks. The CTE in radial direction of the CNT is less than that in the axial direction for armchair CNTs, but the opposite holds for zigzag CNTs. The radial CTE is independent of the CNT helicity, while the axial CTE shows a strong helicity dependence. read less NOT USED (low confidence) A. V. Vershinin, A. V. Zverev, N. Shwartz, and Z. Yanovitskaja, “Adatom potential relief on Si (111) - 7/spl times/7 surface,” 2004 International Siberian Workshop on Electron Devices and Materials. 2004. link Times cited: 0 Abstract: Simulation of Adatom potential relief on Si (111) - 7/spl ti… read moreAbstract: Simulation of Adatom potential relief on Si (111) - 7/spl times/7 surface was carried out using Tersoff potential. Positions of adatom localization on reconstructed surface were determined. Three absolute energy minimums are found around each rest atom of the height H/sub 1/=2.3 /spl Aring/ over lower monolayer of upper bilayer. Energy barriers of diffusion hops have been estimated: for hops around rest atom 0.75 eV, for hops from one rest atom to neighbor rest-atom 1.25 eV and for transition from one half-cell to the other 1.75 eV. read less NOT USED (low confidence) W. Moon, M. Son, J. H. Lee, and H. Hwang, “Molecular dynamics simulation of C60 encapsulated in boron nitride nanotubes,” physica status solidi (b). 2004. link Times cited: 6 Abstract: We investigate the C60 chain encapsulated in boron nitride (… read moreAbstract: We investigate the C60 chain encapsulated in boron nitride (BN) nanotubes using molecular‐dynamics simulation. The most favorable BN nanotubes for encapsulation of C60 molecules are (10, 10) and (17, 0) with energy gains of 3.83 and 3.61 eV per C60 for (n, n) and (n, 0) BN nanotubes, respectively. For the diffusion of a C60 into the tube, the position of atoms of a C60 must be not located above the outer wall of the BN nanotube. The C60 located above the outer wall is quickly absorbed and moves on the surface of the tube. The C60 absorbed on the surface is not spontaneously encapsulated inside the tube, which is due to the energy barrier (0.48 eV) of the edge of the BN nanotube. We also calculate the energy barrier for drawing C60 outside the (10, 10) BN nanotube, which is above 3.92 eV. (© 2004 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (low confidence) C. R. S. Silva, J. F. Justo, and A. Fazzio, “On the reversibility of hydrogen effects on the properties of amorphous silicon carbide,” Journal of Non-crystalline Solids. 2004. link Times cited: 4 NOT USED (low confidence) B. Thijsse, T. Klaver, and E. Haddeman, “Molecular Dynamics simulation of silicon sputtering: sensitivity to the choice of potential,” Applied Surface Science. 2004. link Times cited: 24 NOT USED (low confidence) W. Zhang, Z. Zhu, F. Wang, T. Wang, L. Sun, and Z. Wang, “Chirality dependence of the thermal conductivity of carbon nanotubes,” Nanotechnology. 2004. link Times cited: 115 Abstract: The thermal conductivities of three types of single-wall car… read moreAbstract: The thermal conductivities of three types of single-wall carbon nanotubes are studied using the homogeneous non-equilibrium Green–Kubo method based on the Brenner potential. The thermal conductivity of a carbon nanotube is found to have dependence on its chirality. The thermal conductivities of three types of nanotube seem to have similar temperature dependence. The thermal conductivity of the chiral nanotube is lower than that of the other two types of nanotube. read less NOT USED (low confidence) K. Nordlund, “Atomistic simulation of radiation effects in carbon-based materials and nitrides,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2004. link Times cited: 13 NOT USED (low confidence) E. Duffour and P. Malfreyt, “MD simulations of the collision between a copper ion and a polyethylene surface: an application to the plasma–insulating material interaction,” Polymer. 2004. link Times cited: 8 NOT USED (low confidence) N.-X. Wei, G. Wu, and J. Dong, “Heat conduction in a carbon chain,” Physics Letters A. 2004. link Times cited: 8 NOT USED (low confidence) K. Liew, X. He, and C. Wong, “On the study of elastic and plastic properties of multi-walled carbon nanotubes under axial tension using molecular dynamics simulation,” Acta Materialia. 2004. link Times cited: 380 NOT USED (low confidence) W. Moon and H. Hwang, “Molecular-dynamics simulation of structure and thermal behaviour of boron nitride nanotubes,” Nanotechnology. 2004. link Times cited: 89 Abstract: We investigate the structure and thermal behaviour of boron … read moreAbstract: We investigate the structure and thermal behaviour of boron nitride (BN) nanotubes using molecular-dynamics simulations based on the Tersoff-like potential. The strain energy decreases with increasing diameter, which is proportional to the inverse square of the tube diameter on the basis of continuum elastic theory. The disintegration temperature of zigzag nanotubes is smaller than that of armchair nanotubes of nearly the same diameter and increases with increasing diameter due to the decrease in strain energy. Despite homoelemental bonds, the Stone–Wales (SW) defect is found in BN nanotubes during thermal treatment. The formation energy of the SW defect increases with increasing tube diameter. These results agree well with the trend for carbon nanotubes. read less NOT USED (low confidence) P. Zhang, H. Jiang, Y. Huang, P. Geubelle, and K. Hwang, “An atomistic-based continuum theory for carbon nanotubes: Analysis of fracture nucleation,” Journal of The Mechanics and Physics of Solids. 2004. link Times cited: 108 NOT USED (low confidence) Y. Jin, “DISCRETE ATOMISTIC AND CONTINUUM FRACTURE PARAMETER MODELING OF A GRAPHENE SHEET.” 2004. link Times cited: 0 Abstract: Macroscopic fracture parameters are investigated using molec… read moreAbstract: Macroscopic fracture parameters are investigated using molecular mechanics simulations for a graphene sheet containing atomic-scale cracks. In the discrete atomistic modeling the interatomic forces are described based on Tersoff-Brenner potential. Elastic energy release rates of the graphene sheet under symmetric (Mode I) and antisymmetric (Mode II) small deformation are directly calculated from global energy approach and local force approach using the principle of virtual work respectively. The energy release rates are also calculated through homogenized material properties based on linear elastic fracture mechanics. The results show good agreement between discrete atomistic and continuum mechanics modeling for fracture parameters and deformed crack surface profile. This establishes connections of fracture parameters between microscopic and macroscopic description of fracture in covalently bonded solids. read less NOT USED (low confidence) K. Liew, C. Wong, X. He, M. Tan, and S. Meguid, “Nanomechanics of single and multiwalled carbon nanotubes,” Physical Review B. 2004. link Times cited: 305 Abstract: Buckling behavior of single-walled and multiwalled carbon na… read moreAbstract: Buckling behavior of single-walled and multiwalled carbon nanotubes is studied under axial compression in this work. Brenner's "second generation" empirical potential is used to describe the many-body short-range interatomic interactions for single-walled carbon nanotubes, while the Lennard Jones model for the van der Waals potential is added for multiwalled carbon nanotubes. Single-, two-, three-, and four-walled nanotubes are considered in the simulations in order to examine the effects of the number of layers on the structural properties of the multiwalled nanotubes. Results indicate that there exists an optimum diameter for single-walled nanotubes at which the buckling load reaches its maximum value. The buckling load increases rapidly with the increase of the diameter up to the optimum diameter. A further increment beyond this diameter results in a slow decline in buckling load until a steady value is reached. The effects of layers on the buckling load of multiwalled nanotubes are also examined. read less NOT USED (low confidence) F. Aoumeur-Benkabou and B. Belgoumène, “Structural and dynamical properties of SrO in the rock-salt phase,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2004. link Times cited: 9 NOT USED (low confidence) S.-H. Park, H. Kim, D.-K. Lee, J. S. Lee, Y. Choi, and O. D. Kwon, “Heterogeneous crystallization of amorphous silicon expedited by external force fields: a molecular dynamics study,” Superlattices and Microstructures. 2004. link Times cited: 6 NOT USED (low confidence) P. Gunes, Şi̇mşek S., and S. Erkoç, “a Comparative Study of Empirical Potential Energy Functions,” International Journal of Modern Physics C. 2004. link Times cited: 2 Abstract: A comparative study has been performed for silicon microclus… read moreAbstract: A comparative study has been performed for silicon microclusters, Si3 and Si4, considering fifteen different empirical potential energy functions. It has been found that only two of the empirical potential energy functions give linear structure more stable for Si3, the remaining potential functions give triangular structure as more stable. In the case of Si4 microclusters eight potential functions give open tetrahedral structure as more stable, two functions give perfect tetrahedral as more stable, three functions give square structure as more stable, and two functions give linear structure as more stable. read less NOT USED (low confidence) H. Rafii-Tabar, “Computational modelling of thermo-mechanical and transport properties of carbon nanotubes,” Physics Reports. 2004. link Times cited: 190 NOT USED (low confidence) W. Moon and H. Hwang, “Molecular-dynamics simulation of defect formation energy in boron nitride nanotubes,” Physics Letters A. 2004. link Times cited: 27 NOT USED (low confidence) J. Kang and H. Hwang, “Molecular Dynamics Simulations of Single-wall GaN Nanotubes,” Molecular Simulation. 2004. link Times cited: 16 Abstract: We have investigated the structural properties and the therm… read moreAbstract: We have investigated the structural properties and the thermal behavior of single-wall GaN nanotubes using atomistic simulations based on the Tersoff-type potential. The Tersoff potential for GaN has effectively described the properties of GaN nanotubes. The caloric curves of single-wall GaN nanotubes were divided into three regions corresponding to nanotube, disintegrating range and vapor. Since the stability or the stiffness of the tube decreased with increasing curving strain energy of sheet-to-tube, the disintegration temperatures of GaN nanotubes were closely related to the curving strain energy of sheet-to-tube. read less NOT USED (low confidence) D. Grimm, R. Muniz, and A. Latgé, “From straight carbon nanotubes to Y-shaped junctions and rings,” Physical Review B. 2003. link Times cited: 23 Abstract: We explore quantum interference effects in the electronic tr… read moreAbstract: We explore quantum interference effects in the electronic transport properties of single-walled carbon nanotube junctions. Remarkable changes in the electrical conductance are found by varying the length of one of the junction's arms. Owing to the relatively high electron mobility in carbon nanotubes, we show that a double-slit-like electron interferometer may be fabricated by joining two Y-shaped carbon nanotube junctions. These nanostructures have very interesting electrical characteristics that may be manipulated by altering geometrical aspects of the junctions. read less NOT USED (low confidence) C. Lang, D. Nguyen-Manh, and D. Cockayne, “Nonuniform alloying in Ge(Si)/Si(001) quantum dots,” Journal of Applied Physics. 2003. link Times cited: 13 Abstract: The composition profile of pyramid shaped Ge(Si)/Si(001) qua… read moreAbstract: The composition profile of pyramid shaped Ge(Si)/Si(001) quantum dots has been modeled using a combination of atomistic total energy calculations and a Metropolis Monte Carlo process. The analysis of the non-uniform composition profile has revealed the important, separate roles of the strain energy, the surface energy, and the mixing energy as driving forces of the alloying and segregation process. The surface energy was found to drive a redistribution of Ge into the surface layer of the quantum dot, which was followed by two Si-rich layers. In the vertical direction Si is found to redistribute to the bottom resulting in a Ge-rich apex of the quantum dot. This result is compared to a phenomenological description of the composition profile by Tersoff [Phys. Rev. Lett. 81, 3183 (1998)]. The possibility of a misinterpretation of experimental measurements of the composition profile as a result of the Ge-rich surface layer is discussed. read less NOT USED (low confidence) Y. Kimoto et al., “Molecular Dynamics Study of Double-Walled Carbon Nanotubes for Nano-Mechanical Manipulation,” Japanese Journal of Applied Physics. 2003. link Times cited: 24 Abstract: Double-walled carbon nanotubes (DWNTs) are expected to be us… read moreAbstract: Double-walled carbon nanotubes (DWNTs) are expected to be useful as elements in nano-mechanical systems such as nanobearings and nanosliders. A molecular dynamics simulation is carried out to estimate the relative motion between the inner and outer tubes. The force required to pull the inner tube out of the outer tube is evaluated quantitatively by pulling the inner tube under a constant velocity for DWNTs with various inter-tube spacings and chiralities. When the inner tube is pulled under smaller constant force, the inner tube vibrates inside the outer tube without being pulled out, and an energetics is applied to explain the critical force and vibrational amplitude. The constant force induces not only vibration along the tube axis but also rotation around the tube axis, which indicates the possibility of creating a slider crank mechanism using a DWNT. read less NOT USED (low confidence) W. Moon and H. Hwang, “Atomistic study of elastic constants and thermodynamic properties of cubic boron nitride,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 2003. link Times cited: 11 NOT USED (low confidence) A. Selezenev, A. Aleynikov, N. S. Gantchuk, P. V. Yermakov, J. Labanowski, and A. Korkin, “SAGE MD: molecular-dynamic software package to study properties of materials with different models for interatomic interactions,” Computational Materials Science. 2003. link Times cited: 12 NOT USED (low confidence) R. Rurali and E. Hernández, “Trocadero: a multiple-algorithm multiple-model atomistic simulation program,” Computational Materials Science. 2003. link Times cited: 61 NOT USED (low confidence) M. Kanoun, A. E. Merad, H. Aourag, J. Cibert, and G. Merad, “Molecular-dynamics simulations of structural and thermodynamic properties of ZnTe using a three-body potential,” Solid State Sciences. 2003. link Times cited: 25 NOT USED (low confidence) F. Z. Aoumeur, K. Benkabou, and B. Belgoumène, “Structural and dynamical properties of ZnO in zinc-blende and rocksalt phases,” Physica B-condensed Matter. 2003. link Times cited: 27 NOT USED (low confidence) W. Moon and H. Hwang, “Structural and thermodynamic properties of GaN: a molecular dynamics simulation,” Physics Letters A. 2003. link Times cited: 39 NOT USED (low confidence) W. Moon, M. Son, and H. Hwang, “Molecular-dynamics simulation of structural properties of cubic boron nitride,” Physica B-condensed Matter. 2003. link Times cited: 33 NOT USED (low confidence) H. Koga, Y. Nakamura, and S. Watanabe, “Repulsion-Induced Order Formation in Graphite-Diamondlike Transition of Boron Nitride: A Molecular Dynamics Study,” Journal of the Physical Society of Japan. 2003. link Times cited: 0 Abstract: Repulsion-induced order formation is shown to be crucial to … read moreAbstract: Repulsion-induced order formation is shown to be crucial to the occurrence of the transition from rhombohedral boron nitride (rBN) to cubic boron nitride (cBN), by performing molecular dynamics simulations. Due to the repulsion among B (N), a face-centered-cubic (fcc) lattice of B (N) is formed under compression. This restores the stacking sequence of basal planes, which is disordered during the compression. The fcc lattice directly becomes the fcc lattice of cBN. These results imply the possibility of controlling the transition by controlling the repulsion. read less NOT USED (low confidence) K. Gärtner and B. Weber, “Molecular dynamics simulations of solid-phase epitaxial growth in silicon,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 19 NOT USED (low confidence) G. J. Sibona, S. Schreiber, R. Hoppe, B. Stritzker, and A. Revnic, “Numerical simulation of the production processes of layered materials,” Materials Science in Semiconductor Processing. 2003. link Times cited: 8 NOT USED (low confidence) R. Komanduri, N. Chandrasekaran, and L. Raff, “Molecular dynamic simulations of uniaxial tension at nanoscale of semiconductor materials for micro-electro-mechanical systems (MEMS) applications,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2003. link Times cited: 48 NOT USED (low confidence) M. Medvedeva, I. A. Wojciechowski, and B. Garrison, “Enhancement of cluster yield under gold dimer oblique bombardment of the silicon surface,” Applied Surface Science. 2003. link Times cited: 3 NOT USED (low confidence) C. Wei, D. Srivastava, and K. Cho, “Molecular Dynamics Study of Temperature Dependent Plastic Collapse of Carbon Nanotubes under Axial Compression,” Cmes-computer Modeling in Engineering & Sciences. 2002. link Times cited: 17 Abstract: The temperature dependence of the plastic collapse of single… read moreAbstract: The temperature dependence of the plastic collapse of single-wall carbon nanotubes under axial compression has been studied with classical molecular dynamics simulations using Tersoff-Brenner potential for C-C interactions. At zero temperature, an (8,0) singlewall carbon nanotube under axial compression collapses by forming fins-like structure which remains within the elastic limit of the system, in agreement of previous molecular dynamics study. At finite temperatures, however, we find that temperature dependent fluctuations can activate the formation of sp3 bonds, in agreement with a recently proposed plastic collapse mechanism of the same nanotube with a generalized tight-binding molecular dynamics description. Furthermore, Stone-Wales defects are also found in the plastically collapsed structures. The thermal fluctuations are shown to drive nanotubes to overcome the energy barriers leading to plastically collapsed structures which have significantly lower strain energy (0.1eV/atom) than fins-like structure. Since their discovery in 1991, [Iijima (1991), Iijima and Ichihashi (1993)] singleand multi-walled carbon nanotubes (CNTs) are shown to have exceptionally strong and stiff mechanical characteristics along the axis of the tube, and to be very flexible normal to the tube axis.[Iijima, Brabc, Maiti, Berholc (1996); Despres, Daguerre, Lafdi (1995); Chopra, Bebedict, Crespi, Cohen, Louse, Zettl (1995); Ruoff, Lorents (1995); Yakobson, Brabec, Bernholc (1996); Falvo, Clary, Taylor, Chi, Brooks, Washburn, Superfine (1997); Srivastava, Barnard (1997); Knechtel, Dusberg, Blau (1998); Clauss, Bergeron, Johnson (1998); Bernholc, Brabec, Buongioron, Maiti, Yakobson (1998)] 1 Department of Mechanical Engineering, Stanford University, California 94305-4040 2 NASA Ames Research Center, MS T27A-1, Moffett Field, California 94035-1000 For axial deformations, the Young’s modulus of singlewalled CNTs can be larger than 1 TPa, and some efforts have been made to take advantage of this strength to use CNT as a reinforcing fiber in nanotube-polymer composite matrix. [Schadler, Giannaris, Ajayan (1998); Andrews, Jacques, Rao, Rantell, Derbyshire, Y. Chen, J. Chen, Haddon (1999); Ajayan, Schadler, Giannaris, Rubio (2000); Qian, Dckey, Andrews, Rantell (2000)] To develop a fully optimized nanotube composite materials, it is important to characterize the nanomechanics of CNTs. Many experimental and theoretical efforts have been made to study CNT mechanics. [Iijima, Brabc, Maiti, Berholc (1996); Despres, Daguerre, Lafdi (1995); Chopra, Bebedict, Crespi, Cohen, Louse, Zettl (1995); Ruoff, Lorents (1995); Yakobson, Brabec, Bernholc (1996); Falvo, Clary, Taylor, Chi, Brooks, Washburn, Superfine (1997); Srivastava, Barnard (1997); Knechtel, Dusberg, Blau (1998); Clauss, Bergeron, Johnson (1998); Bernholc, Brabec, Buongioron, Maiti, Yakobson (1998)] The initial investigations, using classical molecular dynamics (MD) simulations with Tersoff-Brenner potential, have shown extreme stiffness of the tubes under axial compression, and the system is shown to remain in elastic limit even for very large deformations (up to 15%). [Yakobson, Brabec, Bernholc (1996); Srivastava, Barnard (1997); Bernholc, Brabec, Buongioron, Maiti, Yakobson (1998)] Non-linear elastic instabilities, with the appearance of fins-like structure, were observed in MD simulations, but the system remained within elastic limit and returned to the original unstrained state as soon as constraining forces were removed. However, more accurate generalized tight-binding (TB) MD simulations [Srivastava, Menon, Cho (1999)] have recently shown that an axial compression leads to a plastic collapse of the system that is driven by a graphitic (sp2) to diamondlike (sp3) bonding transition at the location of a collapse. At zero or low temperatures, classical MD simulations find that CNT remains within elastic limit upto 256 Copyright c 2002 Tech Science Press CMES, vol.3, no.2, pp.255-261, 2002 1.2 1.3 1.4 1.5 1.6 1.7 read less NOT USED (low confidence) D. Qian, G. Wagner, W. K. Liu, M.-F. Yu, and R. Ruoff, “Mechanics of carbon nanotubes,” Applied Mechanics Reviews. 2002. link Times cited: 1115 Abstract: Soon after the discovery of carbon nanotubes, it was realize… read moreAbstract: Soon after the discovery of carbon nanotubes, it was realized that the theoretically predicted mechanical properties of these interesting structures--including high strength, high stiffness, low density and
structural perfection--could make them ideal for a wealth of technological applications. The experimental
verification, and in some cases refutation, of these predictions, along with a number of computer simulation methods applied to their modeling, has led over the past decade to an improved but by no means complete understanding of the mechanics of carbon nanotubes. We review the theoretical predictions and discuss the experimental techniques that are most often used for the challenging tasks of visualizing and manipulating these tiny structures. We also outline the computational approaches that have been taken, including ab initio quantum mechanical simulations, classical molecular dynamics, and continuum models. The development of multiscale and multiphysics models and simulation tools naturally arises as a result of the link between basic scientific research and engineering application; while this issue is still under intensive study, we present here some of the approaches to this topic. Our concentration throughout is on the exploration of mechanical properties such as Young's modulus, bending stiffness, buckling criteria, and tensile and compressive strengths. Finally, we discuss several examples of exciting applications that take advantage of these properties, including nanoropes, filled nanotubes, nanoelectromechanical systems, nanosensors, and nanotube-reinforced polymers. This review article cites 349 references. read less NOT USED (low confidence) Y. Umeno, T. Kitamura, K. Date, M. Hayashi, and T. Iwasaki, “Optimization of interatomic potential for Si/SiO2 system based on force matching,” Computational Materials Science. 2002. link Times cited: 25 NOT USED (low confidence) C. Sbraccia, P. Silvestrelli, and F. Ancilotto, “Modified XB potential for simulating interactions of organic molecules with Si surfaces,” Surface Science. 2002. link Times cited: 26 NOT USED (low confidence) J. Chen, A. Hairie, E. Paumier, and G. Nouet, “Energy of the Two Variants 11A and 11B in Silicon and Germanium by the Semi-Empirical Tight-Binding Method,” Physica Status Solidi B-basic Solid State Physics. 2002. link Times cited: 2 Abstract: A semi-empirical tight-binding model was used to study the t… read moreAbstract: A semi-empirical tight-binding model was used to study the total energy of Σ = 11 tilt grain boundaries in silicon and germanium. At low temperature the structures A in silicon and B in germanium were found to be more stable in agreement with the experimental results. read less NOT USED (low confidence) A. Pérez-Garrido and A. Urbina, “Metal–semiconductor heterojunctions in T-shaped carbon nanotubes,” Carbon. 2002. link Times cited: 27 NOT USED (low confidence) P. Zhang, Y. Huang, H. Gao, and K. Hwang, “Fracture Nucleation in Single-Wall Carbon Nanotubes Under Tension: A Continuum Analysis Incorporating Interatomic Potentials,” Journal of Applied Mechanics. 2002. link Times cited: 117 Abstract: Carbon nanotubes show great promise for applications ranging… read moreAbstract: Carbon nanotubes show great promise for applications ranging from nanocomposites, nanoelectronic components, nanosensors, to nanoscale mechanical probes. These materials exhibit very attractive mechanical properties with extraordinarily high stiffness and strength, and are of great interest to researchers from both atomistic and continuum points of view. In this paper, we intend to develop a continuum theory of fracture nucleation in single-walled carbon nanotubes by incorporating interatomic potentials between carbon atoms into a continuum constitutive model for the nanotube wall. In this theory, the fracture nucleation is viewed as a bifurcation instability of a homogeneously deformed nanotube at a critical strain. An eigenvalue problem is set up to determine the onset of fracture, with results in good agreement with those from atomistic studies. ©2002 ASME read less NOT USED (low confidence) M. Huhtala, A. Kuronen, and K. Kaski, “Carbon nanotube structures: molecular dynamics simulation at realistic limit,” Computer Physics Communications. 2002. link Times cited: 49 NOT USED (low confidence) P. Zhang, Y. Huang, P. Geubelle, P. Klein, and K. Hwang, “The elastic modulus of single-wall carbon nanotubes: a continuum analysis incorporating interatomic potentials,” International Journal of Solids and Structures. 2002. link Times cited: 413 NOT USED (low confidence) F. Gao and W. J. Weber, “Empirical potential approach for defect properties in 3C-SiC,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2002. link Times cited: 92 NOT USED (low confidence) S. Tomita, P. Hvelplund, S. Nielsen, and T. Muramoto, “C 59 -ion formation in high-energy collisions between cold C - 60 and noble gases,” Physical Review A. 2002. link Times cited: 23 Abstract: A high kinetic energy beam of C 6 0 ions produced by electro… read moreAbstract: A high kinetic energy beam of C 6 0 ions produced by electrospray was collided with He and Ne targets, and the cross sections for production of positive and negative fragment ions were determined. In these experiments we observed both C 5 9 + and C 5 9 fragment ions with He and Ne target gases. Since the initial internal energy of the ions is low, it is possible to knock out one carbon atom from the fullerene cage and thereby form C 5 9 ions with lifetimes long enough to allow them to be detected (of the order of 10 μs). Comparisons with molecular dynamic simulations show that the cross sections for formation of C 5 9 - and also C 5 8 can be explained as the result of a "prompt" knock-out process. read less NOT USED (low confidence) N. Chaâbane, H. Vach, and G. H. Peslherbe, “Complex dynamics during the reactive scattering of Si+ (2P) and H2,” Journal of Non-crystalline Solids. 2002. link Times cited: 2 NOT USED (low confidence) U. Kaiser, J. Biskupek, D. Muller, K. Gärtner, and C. Schubert, “Properties of GeSi Nanocrystals Embedded in Hexagonal SiC,” Crystal Research and Technology. 2002. link Times cited: 14 Abstract: In this paper high-resolution electron microscopy investigat… read moreAbstract: In this paper high-resolution electron microscopy investigations and molecular dynamics simulations of GeSi nanocrystals buried in 4H-SiC are performed, showing that the experimentally observed shapes of the GeSi nanocrystals are strongly correlated with their orientational relationships. Measurements of the lattice spacing suggest that the nanocrystals are strained. Quantum confinement in selected nanocrystals has been detected using spatially-resolved electron energy loss spectroscopy performed in conjunction with atomic resolution annular dark-field scanning TEM. read less NOT USED (low confidence) K. Shiraishi et al., “First principles and macroscopic theories of semiconductor epitaxial growth,” Journal of Crystal Growth. 2002. link Times cited: 15 NOT USED (low confidence) K. Nordlund, “Computational materials science of ion irradiation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2002. link Times cited: 18 NOT USED (low confidence) M. Ruda, D. Farkas, and J. Abriata, “Interatomic potentials for carbon interstitials in metals and intermetallics,” Scripta Materialia. 2002. link Times cited: 53 NOT USED (low confidence) I. Jang, B. Ni, and S. Sinnott, “Study of angular influence of C3H5+ ion deposition on polystyrene surfaces using molecular dynamics simulations,” Journal of Vacuum Science and Technology. 2002. link Times cited: 10 Abstract: The influence of incident angle on the interaction of polyat… read moreAbstract: The influence of incident angle on the interaction of polyatomic hydrocarbon ions (C3H5+) with polystyrene surfaces is examined using classical molecular dynamics simulations. The forces are determined using the reactive empirical bond order method developed by Tersoff and parametrized by Brenner. The total incident energy is 50 eV and the angles considered are 0° (normal to the surface), 15°, 45°, and 75°. At each angle, the outcomes of 80 trajectories are compiled and averaged. The results show that intact ions scatter from the surface in only 2% of the trajectories and that the ions dissociate in 61% of the trajectories at normal incidence. At 75°, intact ions scatter away in 56% and they dissociate in only 30% of the trajectories. The largest total amount of carbon is deposited at normal incident angles. However, more ions or ion fragments are predicted to remain near the surface (penetrate 3.5–5.5 A) at 45°. This is because ion fragments tend to penetrate more deeply (6–7 A) into the surface at small... read less NOT USED (low confidence) C. Wei, K. Cho, and D. Srivastava, “Tensile strength of carbon nanotubes under realistic temperature and strain rate,” Physical Review B. 2002. link Times cited: 205 Abstract: Strain rate and temperature dependence of the tensile streng… read moreAbstract: Strain rate and temperature dependence of the tensile strength of single-wall carbon nanotubes has been investigated with molecular dynamics simulations. The tensile failure or yield strain is found to be strongly dependent on the temperature and strain rate. A transition state theory based predictive model is developed for the tensile failure of nanotubes. Based on the parameters fitted from high-strain rate and temperature dependent molecular dynamics simulations, the model predicts that a defect free micrometer long single-wall nanotube at 300 K, stretched with a strain rate of 1%/hour, fails at about 9 plus or minus 1% tensile strain. This is in good agreement with recent experimental findings. read less NOT USED (low confidence) N. Mousseau, G. Barkema, and S. Nakhmanson, “Recent developments in the study of continuous random networks,” Philosophical Magazine B. 2002. link Times cited: 11 Abstract: We report on recent progress in the development of new techn… read moreAbstract: We report on recent progress in the development of new techniques to generate high-quality models of continuous random networks, which are used as models for elemental and binary tetrahedral semiconductors such as amorphous Si and amorphous GaAs. The availability of such models has allowed us to look at a number of outstanding issues regarding their electronic properties, the fundamental role of defects and dynamics. We describe briefly our modifications made to the Wooten-Winer-Weaire bond-switching algorithm, allowing us to produce low-strain amorphous and paracrystalline networks of up to 10000 atoms. Then some of the structural and electronic properties of these models are presented. We also discuss briefly some recent results on the recrystallization of amorphous networks. read less NOT USED (low confidence) R. Komanduri and L. Raff, “A review on the molecular dynamics simulation of machining at the atomic scale,” Proceedings of the Institution of Mechanical Engineers, Part B: Journal of Engineering Manufacture. 2001. link Times cited: 138 Abstract: Molecular dynamics (MD) simulation, like other simulation te… read moreAbstract: Molecular dynamics (MD) simulation, like other simulation techniques, such as the finite difference method (FDM), or the finite element method (FEM) can play a significant role in addressing a number of machining problems at the atomic scale. It may be noted that atomic simulations are providing new data and exciting insights into various manufacturing processes and tribological phenomenon that cannot be obtained readily in any other way—theory, or experiment. In this paper, the principles of MD simulation, relative advantages and current limitations, and its application to a range of machining problems are reviewed. Machining problems addressed include: (a) the mechanics of nanometric cutting of non-ferrous materials, such as copper and aluminium; (b) the mechanics of nanometric cutting of semiconductor materials, such as silicon and germanium; (c) the effect of various process parameters, including rake angle, edge radius and depth of cut on cutting and thrust forces, specific force ratio, energy, and subsurface deformation of the machined surface; the objective is the development of a process that is more efficient and effective in minimizing the surface or subsurface damage; (d) modelling of the exit failures in various work materials which cause burr formation in machining; (e) simulation of work materials with known defect structure, such as voids, grain boundaries, second phase particles; shape, size and density of these defects can be varied using MD simulation as well as statistical mechanical or Monte Carlo approaches; (f) nanometric cutting of nanostructures; (g) investigation of the nanometric cutting of work materials of known crystallographic orientation; (h) relative hardness of the tool material with respect to the work material in cutting; a range of hardness values from the tool being softer than the work material to the tool being several times harder than the work material is considered; and (i) the tool wear in nanometric cutting of iron with a diamond tool. The nature of deformation in the work material ahead of the tool, subsurface deformation, nature of variation of the forces and their ratio, and specific energy with cutting conditions are investigated by this method. read less NOT USED (low confidence) A. Lamzatouar, M. E. Kajbaji, A. Charaï, M. Benaissa, O. H. Duparc, and J. Thibault, “The atomic structure of Σ=33144〈011〉 (θ=20.05°) tilt grain boundary in germanium,” Scripta Materialia. 2001. link Times cited: 5 NOT USED (low confidence) S. Walch, S. Ramalingam, S. Sriraman, E. Aydil, and D. Maroudas, “Mechanisms and energetics of SiH3 adsorption on the pristine Si(0 0 1)- (2×1) surface,” Chemical Physics Letters. 2001. link Times cited: 18 NOT USED (low confidence) K. Nordlund, J. Peltola, J. Nord, J. Keinonen, and R. Averback, “Defect clustering during ion irradiation of GaAs: Insight from molecular dynamics simulations,” Journal of Applied Physics. 2001. link Times cited: 56 Abstract: Defect formation in compound semiconductors such as GaAs und… read moreAbstract: Defect formation in compound semiconductors such as GaAs under ion irradiation is not as well understood as in Si and Ge. We show how a combination of ion range calculations and molecular dynamics computer simulations can be used to predict the atomic-level damage structures produced by MeV ions. The results show that the majority of damage produced in GaAs both by low-energy self-recoils and 6 MeV He ions is in clusters, and that a clear majority of the isolated defects are interstitials. Implications of the results for suggested applications are also discussed. read less NOT USED (low confidence) R. Sahara, H. Mizuseki, K. Ohno, H. Kubo, and Y. Kawazoe, “Lattice Monte Carlo simulation with a renormalized potential in Si,” Journal of Crystal Growth. 2001. link Times cited: 3 NOT USED (low confidence) K. Nordlund, E. Salonen, J. Keinonen, and C. Wu, “Sputtering of hydrocarbons by ion-induced breaking of chemical bonds,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2001. link Times cited: 14 NOT USED (low confidence) S. Fagan, R. Mota, R. J. Baierle, G. Paiva, A. J. D. Silva, and A. Fazzio, “Stability investigation and thermal behavior of a hypothetical silicon nanotube,” Journal of Molecular Structure-theochem. 2001. link Times cited: 88 NOT USED (low confidence) R. Neuendorf, R. Palmer, and R. Smith, “Low energy deposition of size-selected Si clusters onto graphite,” Chemical Physics Letters. 2001. link Times cited: 18 NOT USED (low confidence) E. Ziemniak and C. Jung, “A model for monolayer deposition with interacting particles,” Chaos Solitons & Fractals. 2001. link Times cited: 0 NOT USED (low confidence) D. Nicholson and K. Travis, “Molecular Simulation of Transport in a Single Micropore,” Membrane Science and Technology. 2000. link Times cited: 10 NOT USED (low confidence) W. Zhu, Z. Pan, Y. Ho, and Y. Wang, “Simulations of C28 chemisorption on diamond (001)-(2×1) surface: The comparison between cluster–cluster interaction and cluster–surface interaction,” Journal of Applied Physics. 2000. link Times cited: 10 Abstract: In this article, the dynamic behavior of C28 chemisorption o… read moreAbstract: In this article, the dynamic behavior of C28 chemisorption on diamond (001)-(2×1) surface was investigated by molecular dynamics simulation. The many-body Brenner potential was employed to describe the interaction between carbon atoms. With the incident energy ranging from 25 to 40 eV, the single C28 was found to have more than 50% of the probability to be chemisorbed on a diamond surface and to form two C–C bonds with one dimer of the surface. Then the chemisorption of two C28 clusters was simulated at the above energy range. The cluster–cluster interaction was found to hinder the next incident cluster to be chemisorbed. Besides, the juxtaposition configuration of two C28 on the surface was observed when their impact points were along the same dimer row. For multicluster impacting, when two or three clusters formed a nucleation site, the forthcoming cluster was easily to be adsorbed close to it. The growth of the C28 cluster assembled film is typically a three dimensional island mode. Our study also showed that within the energy range the C28 clusters retained their cage structure after chemisorption. This is in agreement with experimental results.In this article, the dynamic behavior of C28 chemisorption on diamond (001)-(2×1) surface was investigated by molecular dynamics simulation. The many-body Brenner potential was employed to describe the interaction between carbon atoms. With the incident energy ranging from 25 to 40 eV, the single C28 was found to have more than 50% of the probability to be chemisorbed on a diamond surface and to form two C–C bonds with one dimer of the surface. Then the chemisorption of two C28 clusters was simulated at the above energy range. The cluster–cluster interaction was found to hinder the next incident cluster to be chemisorbed. Besides, the juxtaposition configuration of two C28 on the surface was observed when their impact points were along the same dimer row. For multicluster impacting, when two or three clusters formed a nucleation site, the forthcoming cluster was easily to be adsorbed close to it. The growth of the C28 cluster assembled film is typically a three dimensional island mode. Our study also show... read less NOT USED (low confidence) T. Lenosky et al., “Highly optimized empirical potential model of silicon,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 145 Abstract: We fit an empirical potential for silicon using the modified… read moreAbstract: We fit an empirical potential for silicon using the modified embedded atom (MEAM) functional form, which contains a nonlinear function of a sum of pairwise and three-body terms. The three-body term is similar to the Stillinger-Weber form. We parametrized our model using five cubic splines, each with 10 fitting parameters, and fitted the parameters to a large database using the force-matching method. Our model provides a reasonable description of energetics for all atomic coordinations, Z, from the dimer (Z = 1) to fcc and hcp (Z = 12). It accurately reproduces phonons and elastic constants, as well as point defect energetics. It also provides a good description of reconstruction energetics for both the 30° and 90° partial dislocations. Unlike previous models, our model accurately predicts formation energies and geometries of interstitial complexes - small clusters, interstitial-chain and planar {311} defects. read less NOT USED (low confidence) S. Walch, S. Ramalingam, E. Aydil, and D. Maroudas, “Mechanism and energetics of dissociative adsorption of SiH3 on the hydrogen-terminated Si(0 0 1)-(2×1) surface,” Chemical Physics Letters. 2000. link Times cited: 29 NOT USED (low confidence) F. Benkabou, H. Aourag, and M. Certier, “Atomistic study of zinc-blende CdS, CdSe, ZnS, and ZnSe from molecular dynamics,” Materials Chemistry and Physics. 2000. link Times cited: 80 NOT USED (low confidence) B. Lebouvier, A. Hairie, G. Nouet, and E. Paumier, “Analysis of Distortions in 110 Tilt Silicon Bicrystals,” Physica Status Solidi B-basic Solid State Physics. 2000. link Times cited: 0 Abstract: The empirical potentials used for defect simulation in silic… read moreAbstract: The empirical potentials used for defect simulation in silicon are fitted on elastic or phonon properties. Usually they are not able to take into account all the distortions present in a defected material. On the basis of the potential proposed by Vanderbilt, Taole and Narasimhan a method is presented to separate the contribution of distortions linked to elastic properties from the contribution of distortions linked to phonon properties. The method is applied to grain boundaries in silicon simulated by potentials in the harmonic approximation. The phonon contribution is found slightly predominant with respect to the elastic one. read less NOT USED (low confidence) M. Kanoun, W. Sekkal, H. Aourag, and G. Merad, “Molecular-dynamics study of the structural, elastic and thermodynamic properties of cadmium telluride,” Physics Letters A. 2000. link Times cited: 50 NOT USED (low confidence) T. Sinno, E. Dornberger, W. Ammon, R. A. Brown, and F. Dupret, “Defect engineering of Czochralski single-crystal silicon,” Materials Science & Engineering R-reports. 2000. link Times cited: 112 NOT USED (low confidence) S. Sriraman, S. Ramalingam, E. Aydil, and D. Maroudas, “Abstraction of hydrogen by SiH radicals from hydrogenated amorphous silicon surfaces,” Surface Science. 2000. link Times cited: 9 NOT USED (low confidence) C. Herrero, “Path-integral Monte Carlo study of amorphous silicon,” Journal of Non-crystalline Solids. 2000. link Times cited: 8 NOT USED (low confidence) M. Mäki-Jaskari, K. Kaski, and A. Kuronen, “Simulations of crack initiation in silicon,” Computational Materials Science. 2000. link Times cited: 6 NOT USED (low confidence) K. Koivusaari, T. Rantala, and S. Leppävuori, “Calculated electronic density of states and structural properties of tetrahedral amorphous carbon,” Diamond and Related Materials. 2000. link Times cited: 14 NOT USED (low confidence) H. Rafii-Tabar, “Modelling the nano-scale phenomena in condensed matter physics via computer-based numerical simulations,” Physics Reports. 2000. link Times cited: 163 NOT USED (low confidence) Y. Saito, N. Sasaki, H. Moriya, A. Kagatsume, and S. Noro, “Parameter Optimization of Tersoff Interatomic Potentials Using a Genetic Algorithm,” Jsme International Journal Series A-solid Mechanics and Material Engineering. 2000. link Times cited: 10 Abstract: A method that gives the parameters of advanced Tersoff inter… read moreAbstract: A method that gives the parameters of advanced Tersoff interatomic potentials for describing nonequilibrium atomic structures has been developed. This method uses a genetic algorithm to optimize the Tersoff potential parameters fitted to first-principles-calculated cohesive energies of various carbon systems, including bulk systems with atomic defects and amorphous, surface, or cluster systems under stress. These optimized parameters converge towards a set of Tersoff potential parameters that well describes not only crystals but also amorphous systems. read less NOT USED (low confidence) S. Jiang and J. Belak, “Chapter 15 - Molecular Dynamics of Thin Films under Shear,” Theoretical and Computational Chemistry. 1999. link Times cited: 1 NOT USED (low confidence) K. Nordlund and A. Kuronen, “Non-equilibrium properties of GaAs interatomic potentials,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 18 NOT USED (low confidence) J. Spence, “Atomistics and mechanical properties of silicon,” Acta Materialia. 1999. link Times cited: 8 NOT USED (low confidence) H. Ichikawa, R. Sahara, H. Mizuseki, K. Ohno, and Y. Kawazoe, “Monte Carlo Simulation of Cu–Au Alloys on FCC Lattice with a Renormalized Potential,” Materials Transactions Jim. 1999. link Times cited: 4 Abstract: Monte Carlo simulation of an FCC lattice-gas model is carrie… read moreAbstract: Monte Carlo simulation of an FCC lattice-gas model is carried out to study order-disorder phase transitions. To study an actual Cu-Au alloys as quantitatively as possible, a Finnis-Sinclair-type potential, which has been used widely for molecular dynamics (MD) simulations, is mapped onto the FCC model by using the potential renormalization technique proposed by one of us. Using this renormalized potential, we find that the linear expansion coefficient of Cu and Au crystals and the transition temperatures are greatly improved when compared with the case of using the MD potential directly on the lattice. read less NOT USED (low confidence) D. Ashenford, F. Long, W. Hagston, B. Lunn, and A. Matthews, “Experimental and theoretical studies of the low-temperature growth of chromia and alumina,” Surface & Coatings Technology. 1999. link Times cited: 41 NOT USED (low confidence) W. Sekkal, A. Laref, A. Zaoui, H. Aourag, and M. Certier, “Atomistic simulation of a high-pressure phase of AgI using a three-body potential,” Solid State Communications. 1999. link Times cited: 12 NOT USED (low confidence) C. Wang, B. Pan, J. Xiang, and K. Ho, “Adatom vacancies on the Si(111)-(7 × 7) surface,” Surface Science. 1999. link Times cited: 5 NOT USED (low confidence) A. Petukhov, A. Fasolino, D. Passerone, and F. Ercolessi, “Reconstruction of Diamond (001) Surface: A Monte Carlo Study with the Tersoff Potential,” Physica Status Solidi (a). 1999. link Times cited: 6 NOT USED (low confidence) S. Hobday, R. Smith, and J. BelBruno, “Applications of genetic algorithms and neural networks to interatomic potentials,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 34 NOT USED (low confidence) W. J. Zhu, Z. Pan, Y. Ho, and Z. Man, “Impact-induced chemisorption of C2H2 on diamond(001) surfaces: a molecular dynamics simulation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 8 NOT USED (low confidence) A. Dyson and P. Smith, “Improved empirical interatomic potential for C—Si—H systems,” Molecular Physics. 1999. link Times cited: 30 Abstract: The Brenner hydrocarbon potential was extended recently to i… read moreAbstract: The Brenner hydrocarbon potential was extended recently to include interactions with silicon. This extended Brenner potential has now been improved by the fitting of bond order correction terms, and the introduction of an adjustable parameter into the angular function. The new potential gives an excellent description of small Si m H n molecules and radicals. Its treatment of the low index surfaces of silicon and β-SiC is also significantly improved, although the recently proposed non-dimerized structure for the silicon terminated (001) surface of β-SiC is not described properly. Calculations of the chemisorption of C2H2 and CH3 onto the (001) surfaces of silicon and β-SiC using this improved potential are reported. Also presented are some initial results of molecular dynamics simulations of the Si(111) 7 × 7:CH3 and hydrogenated Si(001) 2 × 1:C2H2 chemisorption systems. read less NOT USED (low confidence) M. Salmi, M. Alatalo, T. Ala‐Nissila, and R. Nieminen, “Energetics and diffusion paths of gallium and arsenic adatoms on flat and stepped GaAs(001) surfaces,” Surface Science. 1999. link Times cited: 33 NOT USED (low confidence) C. Wang, B. Pan, and K. Ho, “An environment-dependent tight-binding potential for Si,” Journal of Physics: Condensed Matter. 1999. link Times cited: 116 Abstract: We present a new generation of tight-binding model for silic… read moreAbstract: We present a new generation of tight-binding model for silicon which goes beyond the traditional two-centre approximation and allows the tight-binding parameters to scale according to the bonding environment. We show that the new model improves remarkably the accuracy and transferability of the potential for describing the structures and energies of silicon surfaces, in addition to the properties of silicon in the bulk diamond structure. read less NOT USED (low confidence) B. Weber, D. Stock, and K. Gärtner, “MD simulations of ion beam induced epitaxial crystallization at a-Si/c-Si interfaces: interface structure and elementary processes of crystallization,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 15 NOT USED (low confidence) E. Burgos, E. Halac, and H. Bonadeo, “A semi-empirical potential for the statics and dynamics of covalent carbon systems,” Chemical Physics Letters. 1998. link Times cited: 13 NOT USED (low confidence) T. Akiyama, A. Oshiyama, and O. Sugino, “Magic Numbers of Multivacancy in Crystalline Si: Tight-Binding Studies for the Stability of the Multivacancy,” Journal of the Physical Society of Japan. 1998. link Times cited: 16 Abstract: We perform microscopic total-energy calculations based on a … read moreAbstract: We perform microscopic total-energy calculations based on a transferable tight-binding model combined with conjugate-gradient minimization technique for various multivacancies V n in Si. We find that stable multivacancies (magic numbers of negative Si clusters) are V 6 , V 10 , V 14 , V 17 , V 22 , V 26 and V 35 . We also find that both topological networks of vacant sites and relaxation of surrounding atoms are crucial to determine energetics of the multivacancies. read less NOT USED (low confidence) S. Ramalingam, D. Maroudas, E. Aydil, and S. Walch, “Abstraction of hydrogen by SiH3 from hydrogen-terminated Si(001)-(2×1) surfaces,” Surface Science. 1998. link Times cited: 48 NOT USED (low confidence) J. Lukes, D. Li, X.-gang Liang, and C. Tien, “Molecular Dynamics Study of Solid Thin-Film Thermal Conductivity,” Heat Transfer: Volume 4 — Heat Transfer in Materials Processing. 1998. link Times cited: 169 Abstract:
This study explores the feasibility of using the molecular… read moreAbstract:
This study explores the feasibility of using the molecular dynamics computational technique to predict the thermal conductivity of solid thin films in the direction perpendicular to the film plane. The results show that thermal conductivity, as expected from thin-film experimental data and theoretical predictions, decreases as film thickness is reduced. In the large-size limit, this method yields thermal conductivities which asymptote to a value comparable to experimental data. The calculations modestly overpredict thermal conductivity, probably due to the use of a too-steep intermolecular potential. Most interestingly, an unusual wave effect is revealed for thin film thermal conductivity. This effect may be a manifestation of phonon wave interference analogous to the interference of light which determines the radiative properties of thin films.
It is also found that there are some temperature and computational domain size limitations on the applicability of molecular dynamics to the study of solid systems. A regime map is developed which delineates the conditions necessary for molecular dynamics to produce physically meaningful results. This work shows that molecular dynamics, applied under the correct conditions, is a viable tool for calculating the thermal conductivity of solid thin films. More generally, this work demonstrates the potential of molecular dynamics for ascertaining microscale thermophysical properties in more complex structures. read less NOT USED (low confidence) M. Nardelli, C. Roland, and J. Bernholc, “Theoretical bounds for multiwalled carbon nanotube growth,” Chemical Physics Letters. 1998. link Times cited: 15 NOT USED (low confidence) V. Rosato, J. Lascovich, A. Santoni, and L. Colombo, “On the Use of the Reverse Monte Carlo Technique to Generate Amorphous Carbon Structures,” International Journal of Modern Physics C. 1998. link Times cited: 10 Abstract: The reverse Monte Carlo (RMC) technique has been used to gen… read moreAbstract: The reverse Monte Carlo (RMC) technique has been used to generate atomic structures of amorphous carbon based on the radial distribution functions and the fraction of differently coordinated sites measured on experimental samples. The resulting structures have been subsequently relaxed via a Tight Binding Molecular Dynamics simulation (TBMD). The radial distribution function, the energy and the fraction of 2-, 3- and 4-fold coordinated sites, evaluated on the relaxed structures, have been compared to those calculated for atomic systems generated on the basis of the "conventional" numerical melt-quench technique. We thus suggest the possibility of using RMC modeling as a useful and convenient tool for generating amorphous structures to be used as initial configurations in Molecular Dynamics simulations. read less NOT USED (low confidence) T. Motooka, “The role of defects during amorphization and crystallization processes in ion implanted Si,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1998. link Times cited: 11 NOT USED (low confidence) R. Webb, Jiménez-Rodrı́guez J. J., M. Kerford, and S. Silva, “The formation of diamond-like carbon films due to molecular impacts on graphite,” Diamond and Related Materials. 1998. link Times cited: 5 NOT USED (low confidence) W. Sekkal, H. Aourag, and M. Certier, “Molecular dynamics simulation of high pressure phases of CuCl and CuBr,” Journal of Physics and Chemistry of Solids. 1998. link Times cited: 29 NOT USED (low confidence) C. R. Zacharias, M. R. Lemes, and A. D. Pino, “Combining genetic algorithm and simulated annealing: A molecular geometry optimization study,” Journal of Molecular Structure-theochem. 1998. link Times cited: 42 NOT USED (low confidence) A. Marinopoulos, V. Vítek, and J. Bassani, “Local and Effective Elastic Properties of Grain Boundaries in Silicon,” Physica Status Solidi (a). 1998. link Times cited: 13 Abstract: When considering the mechanical behaviour of materials an im… read moreAbstract: When considering the mechanical behaviour of materials an important property is the tensor of elastic moduli. Recently, local elastic moduli of interfaces have been defined and studied for metallic materials [1 to 3]. In these works grain boundaries are regarded as heterogeneous continua composed of ‘phases’ associated with individual atoms which possess elastic moduli identified with the atomic-level moduli evaluated at corresponding atomic positions. From this representation it is possible to define the ‘effective’ moduli of the grain boundary region. In this paper this concept is developed for materials with covalent character of bonding, specifically silicon. Using the Tersoff's potential [4, 5], the atomic-level and effective elastic moduli of the interfacial region have been evaluated for three alternate structures of the Σ = 3 (112-)/[11-0] tilt boundary. These calculations are then compared with the continuum bounds on the effective moduli evaluated using the classical minimum-energy principles of elasticity. The effective moduli calculated in the atomistic framework are generally within the continuum bounds and thus the present study demonstrates that the heterogeneous continuum model of the interfaces is appropriate for the description of the elastic properties of grain boundaries in silicon. An important aspect addressed in this study is the uniqueness of interfacial elastic moduli since their evaluation involves the energy associated with an atom which cannot be defined uniquely. The calculations have been made for two different partitions of the total energy into energies associated with individual atoms. These two partitions lead to almost identical results for the effective moduli and continuum bounds when the tensor of the atomic-level moduli is positive definite. When some atomic-level moduli are not positive definite the results may depend on the chosen energy partition. read less NOT USED (low confidence) M. Böhm, R. Ramírez, and J. Schulte, “Electrons and nuclei of C6H6 and C6D6; a combined Feynman path integral – ab initio approach,” Chemical Physics. 1998. link Times cited: 20 NOT USED (low confidence) A. Charaï, L. Fares, C. Alfonso, L. Roussel, and J. Rouviere, “Interfacial Modification Induced by Equilibrium Segregation in Ge(S) Bicrystal,” Surface Review and Letters. 1998. link Times cited: 4 Abstract: An original experimental procedure is set up in order to rel… read moreAbstract: An original experimental procedure is set up in order to relate unambiguously the equilibrium impurity segregation in a grain boundary to the structural modifications. This is clearly established in the case of the Ge(S) Σ=25 grain boundary. Using HREN, EDS and EELS, we show that the sulfur segregation occurs on the dislocation cores of the grain boundary and that it stabilizes the lowest energy structures of the initial one. read less NOT USED (low confidence) A. Dyson and P. V. Smith, “Empirical potential study of the chemisorption of C2H2 and CH3 on the β-SiC(001) surface,” Surface Science. 1998. link Times cited: 26 NOT USED (low confidence) C. Goringe, D. Bowler, and E. Hernández, “Tight-binding modelling of materials,” Reports on Progress in Physics. 1997. link Times cited: 453 Abstract: The tight-binding method of modelling materials lies between… read moreAbstract: The tight-binding method of modelling materials lies between the very accurate, very expensive, ab initio methods and the fast but limited empirical methods. When compared with ab initio methods, tight-binding is typically two to three orders of magnitude faster, but suffers from a reduction in transferability due to the approximations made; when compared with empirical methods, tight-binding is two to three orders of magnitude slower, but the quantum mechanical nature of bonding is retained, ensuring that the angular nature of bonding is correctly described far from equilibrium structures. Tight-binding is therefore useful for the large number of situations in which quantum mechanical effects are significant, but the system size makes ab initio calculations impractical. In this paper we review the theoretical basis of the tight-binding method, and the range of approaches used to exactly or approximately solve the tight-binding equations. We then consider a representative selection of the huge number of systems which have been studied using tight-binding, identifying the physical characteristics that favour a particular tight-binding method, with examples drawn from metallic, semiconducting and ionic systems. Looking beyond standard tight-binding methods we then review the work which has been done to improve the accuracy and transferability of tight-binding, and moving in the opposite direction we consider the relationship between tight-binding and empirical models. read less NOT USED (low confidence) C. Wang and K. Ho, “Material simulations with tight-binding molecular dynamics,” Journal of Phase Equilibria. 1997. link Times cited: 15 NOT USED (low confidence) K. Komvopoulos and W. Yan, “Molecular dynamics simulation of single and repeated indentation,” Journal of Applied Physics. 1997. link Times cited: 27 Abstract: Atomic-scale material responses of dynamic metal-like substr… read moreAbstract: Atomic-scale material responses of dynamic metal-like substrates due to single and repeated indentation by metal-like or covalent rigid tips are interpreted in light of three-dimensional molecular dynamics simulation results. Single-indentation results for a face-centered-cubic metal substrate indented by a relatively blunt tip of the same material and a sharper pyramidal tip of a covalent material are compared to elucidate the effects of interfacial atomic potential and tip shape on the deformation behavior. Force hysteresis occurs due to inelastic deformation and heating of the substrate. The abrupt decrease of the repulsive force during penetration is attributed to permanent deformation. The formation of a connective neck during unloading observed only in the indentations involving the metal tip is associated with the stronger interatomic forces and larger tip surface area in these simulations. The rapid decrease of the attractive force during retraction of the tip is associated with the reconstruction of the elongated neck. Results for different metallic substrates repeatedly indented by a rigid covalent tip up to a fixed maximum depth or maximum normal force are presented in order to reveal the evolution of deformation and heating in the substrate with indentation cycles. Repeated indentation gives rise to behaviors resembling cyclic hardening and softening observed at the macroscale. read less NOT USED (low confidence) G. Sh, P. Kodali, B. Garrison, and N. Winograd, “Angle-resolved SIMS studies of AlxGa(1 − x)As 001 (2 × 4) surface reconstruction,” Surface Science. 1997. link Times cited: 0 NOT USED (low confidence) S. A. Fedotov, A. A. Efimchik, and A. Byeli, “DLC growth by ion beam assisted deposition : a molecular simulation,” Diamond and Related Materials. 1997. link Times cited: 8 NOT USED (low confidence) Z. Zhang, F. Wu, and M. Lagally, “AN ATOMISTIC VIEW OF Si(001) HOMOEPITAXY1,” Annual Review of Materials Science. 1997. link Times cited: 25 Abstract: ▪ Abstract Growth of thin films from atoms deposited from th… read moreAbstract: ▪ Abstract Growth of thin films from atoms deposited from the gas phase is intrinsically a non-equilibrium phenomenon dictated by a competition between kinetics and thermodynamics. Precise control of the growth becomes possible only after achieving an understanding of this competition. In this review, we present an atomistic view of the various kinetic aspects in a model system, the epitaxy of Si on Si(001), as revealed by scanning tunneling microscopy and total-energy calculations. Fundamentally important issues investigated include adsorption dynamics and energetics, adatom diffusion, nucleation, sticking, and detachment. We also briefly discuss the inverse process of growth, removal by sputtering or etching. We aim our discussions to an understanding at a quantitative level whenever possible. read less NOT USED (low confidence) K. Nordlund and T. Mattila, “Hillock formation on ion-irradiated graphite surfaces,” Radiation Effects and Defects in Solids. 1997. link Times cited: 5 Abstract: Scanning probe microscopy experiments show that ion irradiat… read moreAbstract: Scanning probe microscopy experiments show that ion irradiation of (0001) graphite results in the formation of isolated defects comprising of a few tens of atoms. We use molecular dynamics simulations and density-functional theory calculations to study the formation probabilities of these defects. We identify different defect structures which correspond to experimentally observed hillocks on graphite surfaces. We find that the predominant source of defects are vacancies and interlayer interstitials, and identify a three-atom carbon ring defect on the graphite surface. read less NOT USED (low confidence) I. Jenčič, J. Peternelj, and I. Robertson, “Randomization-and-relaxation model revisited,” Radiation Effects and Defects in Solids. 1997. link Times cited: 0 Abstract: The interatomic potentials of Stillinger-Weber and Tersoff w… read moreAbstract: The interatomic potentials of Stillinger-Weber and Tersoff were incorporated into the randomization-and-relaxation model, which was originally developed for modelling amorphous silicon by using the Keating interatomic potential. The inclusion of more recent and more complicated interatomic potentials resulted in a more sophisticated set of bond switching rules which form the basis for the randomization-and-relaxation algorithm. This improved model was then used to model small isolated amorphous zones which are produced by individual heavy ions during ion implantation in silicon. The temperature evolution during zone creation was calculated by using idealized thermal spike model. The structure and stability of these amorphous zones was examined with respect to the energy of incoming ion and with respect to the interatomic potential employed. It was established that significantly lower spike energy is required to create a stable amorphous region than in the simulation where the Keating potential wa... read less NOT USED (low confidence) M. Shapiro, “Using molecular dynamics simulations to investigate sputtering processes,” Radiation Effects and Defects in Solids. 1997. link Times cited: 8 Abstract: The popularity of molecular dynamics (MD) techniques for the… read moreAbstract: The popularity of molecular dynamics (MD) techniques for the investigation of sputtering processes continues to grow, driven in large measure by the rapid increase in the performance-to-price ratio for modern workstations and high-end personal computers. The ready availability of these inexpensive, powerful computing platforms has encouraged researchers to use MD methods to better understand a variety of problems in sputtering. These include studies of: (1) sputtering induced by complex projectiles: (2) the ejection of small clusters during sputtering: (3) the role of inelastic effects during sputtering: (4) sputtering from complex target materials; and (5) chemical effects during sputtering. Increases in computing power also have made it possible to use more realistic many-body potentials in these simulations. This paper reviews some of the recent literature in these areas, and provides an overview of the progress made in the past few years. read less NOT USED (low confidence) J. Domínguez-Vázquez, E. P. Andribet, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “A hybrid MC–MD calculation study,” Radiation Effects and Defects in Solids. 1997. link Times cited: 4 Abstract: It is widely accepted that the binary collision approximatio… read moreAbstract: It is widely accepted that the binary collision approximation (BCA) is rather accurate in the high energy regime of a collision cascade while multiple interaction describes better the low energy and post collisional regimes. It will be therefore desirable to determine the grade of accuracy when both regimes are treated by their corresponding approximations in a single case study. A silicon sample self-bombarded by 2.5 keV ions has been studied. First of all, 1000 trajectories have been calculated by means of the molecular dynamic MD-TOPS code in order to have a reasonable statistic. These results will be considered as a reference when comparing with a second set of 1000 trajectories calculated within the BC approximation using the Monte Carlo MC-TOPS code in the 2.5–0.25 keV energy range and resuming the calculation in the low energy regime by MD. Input parameters required by MC calculations, such as the displacement energy, E d, bulk energy, E b, and surface binding energy, U, for silicon have b... read less NOT USED (low confidence) R. Jones, D. Steiner, H. Heinisch, G. Newsome, and H. Kerch, “OXIDATION RESISTANT CERAMIC MATRIX COMPOSITES,” Journal of Nuclear Materials. 1997. link Times cited: 43 NOT USED (low confidence) S. A. Fedotov, A. A. Efimchik, and A. Byeli, “Ion Beam Assisted Deposition: A Molecular Dynamics Simulation,” Materials and Manufacturing Processes. 1997. link Times cited: 2 Abstract: Molecular dynamics simulation of C film growth by ion assist… read moreAbstract: Molecular dynamics simulation of C film growth by ion assisted deposition is reported. The developed kinetic model of ion assistance offers a possibility to consider relatively low energies of assisting ions (lower than 1 keV) compared to traditional models. C-C atomic interactions are calculated using the Tersoff potential (1). Effects of deposition and assistance modes on the film properties are discussed. A conclusion is made that the Tersoff potential cannot provide accurate simulation of the low-density C phase formation, because it does not describe explicitly the passage between sp3-sp sp3 hybridizations. read less NOT USED (low confidence) M. López, P. A. Marcos, A. Rubio, and J. Alonso, “Thermal behaviour of carbon clusters and small fullerenes,” Zeitschrift für Physik D Atoms,Molecules and Clusters. 1997. link Times cited: 7 NOT USED (low confidence) Lyapin and Brazhkin, “Pressure-induced lattice instability and solid-state amorphization.,” Physical review. B, Condensed matter. 1996. link Times cited: 26 NOT USED (low confidence) P. Stephenson, M. Radny, and P. V. Smith, “A modified Stillinger-Weber potential for modelling silicon surfaces,” Surface Science. 1996. link Times cited: 19 NOT USED (low confidence) C. Wang and K. Ho, “Tight-binding molecular dynamics for materials simulations,” Journal of Computer-Aided Materials Design. 1996. link Times cited: 12 NOT USED (low confidence) E. Kaxiras, “Review of atomistic simulations of surface diffusion and growth on semiconductors,” Computational Materials Science. 1996. link Times cited: 17 NOT USED (low confidence) E. P. Andribet, J. Domínguez-Vázquez, Pérez-Martı́n A., E. Alonso, and Jiménez-Rodrı́guez J. J., “Empirical approach for the interatomic potential of carbon,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1996. link Times cited: 4 NOT USED (low confidence) Y. Xing, Y. Xia, and L. Mei, “Formation of Endohedral Complexes in Collisions of H 2 and D 2 with C 60,” Chinese Physics Letters. 1996. link Times cited: 1 Abstract: Formations of endohedral complexes in collisions of H2 and D… read moreAbstract: Formations of endohedral complexes in collisions of H2 and D2 with C60 molecules have been studied using a molecular dynamics simulation method. H@C60 complex is produced when a H2 molecule collides with a C60 molecule along the direction from the center of a hexagonal ring into the center of the C60 at collision energy E0 = 40 eV. At the same collision condition, a D2 molecule colliding with a C60 molecule forms a D2@C60 complex. read less NOT USED (low confidence) M. C. Yang, C. Kim, H. Lee, and H.-G. Kang, “Heavy ion-surface interaction at low energy: scattering of 3–300 eV Cs+, Xe+, and Ar+ from the Si surface,” Surface Science. 1996. link Times cited: 10 NOT USED (low confidence) Y. Xia, Y. Xing, C. Tan, L. Mei, and H. Yang, “Fusion of two C60 molecules and fragmentation of the fusion product caused by C60C60 collisions,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1996. link Times cited: 4 NOT USED (low confidence) G. Jungnickel, T. Köhler, T. Frauenheim, M. Haase, P. Blaudeck, and U. Stephan, “Structure and chemical bonding in amorphous diamond,” Diamond and Related Materials. 1996. link Times cited: 12 NOT USED (low confidence) D. Timpel, K. Scheerschmidt, and S. Ruvimov, “HREM simulations of particles and interfaces refined by molecular dynamics relaxations,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 1996. link Times cited: 2 NOT USED (low confidence) T. Motooka, “Atomistic simulations of amorphization processes in ion-implanted Si : roles of defects during amorphization, relaxation, and crystallization,” Thin Solid Films. 1996. link Times cited: 9 NOT USED (low confidence) W. Husinsky and G. Betz, “Fundamental aspects of SNMS for thin film characterization: experimental studies and computer simulations,” Thin Solid Films. 1996. link Times cited: 17 NOT USED (low confidence) T. Motooka, Y. Hiroyama, R. Suzuki, T. Ohdaira, Y. Hirano, and F. Sato, “Role of defects during amorphization and relaxation processes in Si,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1995. link Times cited: 7 NOT USED (low confidence) T. Raz and R. Levine, “On the burning of air,” Chemical Physics Letters. 1995. link Times cited: 38 NOT USED (low confidence) C.-ru Wang, R.-B. Huang, Z.-yang Liu, and L.-S. Zheng, “Statistical size distribution of laser generated clusters,” Chemical Physics. 1995. link Times cited: 10 NOT USED (low confidence) A. Marinopoulos, V. Vítek, and A. Carlsson, “Significance of non-central forces in atomistic studies of grain boundaries in bcc transition metals,” Philosophical Magazine. 1995. link Times cited: 40 Abstract: The effects of non-central forces in atomistic studies of gr… read moreAbstract: The effects of non-central forces in atomistic studies of grain boundaries in molybdenum and tungsten, the transition metals with half-filled d-band, are investigated. For this purpose we have used two different types of potential which include different number of moments of the local density of electronic states when evaluating the total energy: the central-force Finnis-Sinclair potentials which include the scalar second moment and the potentials constructed by Carlsson which include the fourth and the matrix second moments. The energy terms associated with these two moments represent non-central interactions and assure that the bcc-fcc structural energy difference is reproduced with good accuracy. For the three boundaries studied, the non-central forces have been found to be very important in determining the lowest energy structures. In particular, the energy differences between multiple structures depend on specific orientations and geometries of the atomic clusters at and near the interface. ... read less NOT USED (low confidence) G. Benedek, E. Galvani, S. Sanguinetti, and S. Serra, “Hallow diamonds: stability and elastic properties,” Chemical Physics Letters. 1995. link Times cited: 28 NOT USED (low confidence) X. Liu, “New model of potential energy functions for atomic solids. Part 2. New potentials of silicon and germanium crystals,” Journal of Molecular Structure-theochem. 1995. link Times cited: 1 NOT USED (low confidence) R. Smith, K. Beardmore, A. Gras-marti, R. Kirchner, and R. Webb, “A molecular dynamics study of energetic particle impacts on carbon and silicon,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1995. link Times cited: 20 NOT USED (low confidence) J. Crain, G. Ackland, and S. Clark, “Exotic structures of tetrahedral semiconductors,” Reports on Progress in Physics. 1995. link Times cited: 29 Abstract: Recent experimental and theoretical studies of exotic forms … read moreAbstract: Recent experimental and theoretical studies of exotic forms of tetrahedrally coordinated semiconductors are reviewed. These unusual phases are synthesized as long-lived metastable forms of the elemental semiconductors silicon and germanium by the application and subsequent removal of high pressure. Rather than being simply crystallographic oddities, the bonding arrangements in these phases show many similarities to those found in amorphous semiconductors. As a result, these dense structures have been used as so-called 'complex crystal' models for the amorphous state. Advances in experimental and computational techniques have recently allowed for detailed study of the structural, electronic and vibrational properties of these phases to be made under variable temperature and pressure conditions. In view of the considerable difficulties associated with performing theoretical studies of non-crystalline solids, the BC8 and ST12 structures are useful in that an understanding of their properties provides insight into the essential physics of amorphous tetrahedral semiconductors. read less NOT USED (low confidence) R. Ramírez and M. Bohm, “On the delocalization of the C nuclei in the C60 molecule; a Feynman path-integral Monte Carlo study,” Journal of Physics: Condensed Matter. 1995. link Times cited: 16 Abstract: Feynman path-integral Monte Carlo simulations have been perf… read moreAbstract: Feynman path-integral Monte Carlo simulations have been performed to study finite-temperature properties of the C60 molecule in a temperature range between 50 and 800 K. The interaction between the C atoms was modelled by the empirical Tersoff potential that reliably reproduces several properties of fullerenes like the binding energy and the alternation between the long and short C-C bonds. We show that the delocalization of the C nuclei is comparable to the difference between the lengths of the long and short bonds. The importance of quantum effects for the C nuclei is illustrated by comparing classical and quantum results for properties like the total energy and the radial and angular distribution functions. read less NOT USED (low confidence) P. Ashu, J. Jefferson, A. Cullis, W. Hagston, and C. Whitehouse, “Molecular dynamics simulation of (100)InGaAs/GaAs strained-layer relaxation processes,” Journal of Crystal Growth. 1995. link Times cited: 38 NOT USED (low confidence) C. Brabec, A. Maiti, C. Roland, and J. Bernholc, “Growth of carbon nanotubes: a molecular dynamics study,” Chemical Physics Letters. 1995. link Times cited: 47 NOT USED (low confidence) J. Glosli, M. R. Philpott, and J. Belak, “Molecular Dynamics Simulation of Mechanical Deformation of Ultra-Thin Amorphous Carbon Films,” MRS Proceedings. 1995. link Times cited: 8 Abstract: Amorphous carbon films approximately 20nm thick are used thr… read moreAbstract: Amorphous carbon films approximately 20nm thick are used throughout the computer industry as protective coatings on magnetic storage disks. The structure and function of this family of materials at the atomic level is poorly understood. Recently. we simulated the growth of a:C and a:CH films 1 to 5 nm thick using Brenner`s bond-order potential model with added torsional energy terms. The microstructure shows a propensity towards graphitic structures at low deposition energy ( 20eV). In this paper we present simulations of the evolution of this microstructure for the dense 20eV films during a simulated indentation by a hard diamond tip. We also simulate sliding, the tip across the surface to study dynamical processes like friction, energy transport and microstructure evolution during sliding. read less NOT USED (low confidence) J. Belak, J. Glosli, D. B. Boercker, and I. Stowers, “Molecular dynamics simulation of mechanical deformation of ultra-thin metal and ceramic films,” MRS Proceedings. 1995. link Times cited: 15 Abstract: We present an overview of the molecular dynamics computer si… read moreAbstract: We present an overview of the molecular dynamics computer simulation method as employed in the study of the mechanical properties of surfaces at the manometer scale. The embedded atom method is used to model a clean metal surface and the bond-order model is used to model ceramic surfaces. The computer experiment consists of the indentation and scraping of a hard diamond-like tool into and across the surface. Results are presented for the (111) surface of copper and silver and for the (100) surface of silicon. We explicitly demonstrate in our point indentation simulations that nanoscale plasticity in metals takes place by nondislocation mechanisms, a result suggested by recent nanoindentation experiments. We also observe the surface to accommodate nearly the entire volume of the tip and the annealing out of plastic work as the tip is removed. In our orthogonal cutting simulation, we observe an interesting phenomenon: the system dynamically reorients the gain in front of the tool tip to minimize the work performed on the shear plane (i.e. the shear plane becomes an easy slip plane). Silicon transforms into an amorphous state which then flows plastically. read less NOT USED (low confidence) K. Nordlund, “Molecular dynamics simulation of ion ranges in the 1–100 keV energy range,” Computational Materials Science. 1995. link Times cited: 382 NOT USED (low confidence) A. Andriotis, “The scaling of the tight-binding Hamiltonian,” Journal of Physics: Condensed Matter. 1995. link Times cited: 2 Abstract: We re-examine the variation of the Slater-Koster tight-bindi… read moreAbstract: We re-examine the variation of the Slater-Koster tight-binding (SKTB) parameters with the interatomic distance and the lattice structure. It is shown that when volume effects are separated from lattice (field) effects, the scaling with the volume can be described in terms of the electron density (or equivalently the parameter rs) while the scaling with the lattice structure can be described in terms of the number of nearest neighbours. The proposed scaling form appears to fit very accurately the SKTB parameters of Si obtained by the TB-LMTO method. read less NOT USED (low confidence) Y. Xia, C. Tan, Y. Xing, H. Yang, X. Sun, and B. Gong, “Molecular-Dynamics Simulation of Surface Relaxation for Tersoff-Dodson Type (100) Si,” Chinese Physics Letters. 1994. link Times cited: 1 Abstract: Surface relaxation and lattice dynamics of (100) Si have bee… read moreAbstract: Surface relaxation and lattice dynamics of (100) Si have been studied using Tersoff-Dodson type Si potential. The average temperature of the lattice is studied as well. The temperature fluctuates with a frequency of 9.5 × 1012 Hz, that is about the average frequency of the optical phonons in Si. The (100) Si surface relaxes inward by 0.86 Å, and a reduction of 19% in the first interlayer spacing is found. read less NOT USED (low confidence) N. Lehto, S. Marklund, and W. Yongliang, “ELECTRON-STATES OF A STACKING-FAULT RIBBON IN SILICON,” Solid State Communications. 1994. link Times cited: 2 NOT USED (low confidence) G. Betz and K. Wien, “Energy and angular distributions of sputtered particles,” International Journal of Mass Spectrometry and Ion Processes. 1994. link Times cited: 198 NOT USED (low confidence) Z. Jiang and R. A. Brown, “Modelling oxygen defects in silicon crystals using an empirical interatomic potential,” Chemical Engineering Science. 1994. link Times cited: 14 NOT USED (low confidence) T. Ohira, T. Inamuro, and T. Adachi, “Molecular dynamics simulation of amorphous silicon with Tersoff potential,” Solar Energy Materials and Solar Cells. 1994. link Times cited: 12 NOT USED (low confidence) D. Maroudas and S. Pantelides, “Point defects in crystalline silicon, their migration and their relation to the amorphous phase,” Chemical Engineering Science. 1994. link Times cited: 11 NOT USED (low confidence) M. Tang and S. Yip, “Lattice instability in β‐SiC and simulation of brittle fracture,” Journal of Applied Physics. 1994. link Times cited: 47 Abstract: Brittle fracture of β‐SiC (polytype 3C) under hydrostatic te… read moreAbstract: Brittle fracture of β‐SiC (polytype 3C) under hydrostatic tension has been modeled by molecular dynamics simulation using an interatomic potential function that treats the solid as fully covalent. The critical stress at which the lattice becomes structurally unstable is shown to agree quantitatively with that predicted by stability analysis based on elastic stiffness coefficients. The instability mode is the spinodal (vanishing of bulk modulus), and decohesion occurs as spontaneous nucleation of cracking on {111} shuffle planes. Atomic relaxation on the newly generated cracked surfaces appears to take place immediately following crack opening. read less NOT USED (low confidence) G. Betz, R. Kirchner, W. Husinsky, F. Rüdenauer, and H. Urbassek, “Molecular dynamics study of sputtering of Cu (111) under Ar ion bombardment,” Radiation Effects and Defects in Solids. 1994. link Times cited: 41 Abstract: We have used the molecular dynamics (MD) technique using man… read moreAbstract: We have used the molecular dynamics (MD) technique using many-body interaction potentials to analyse in detail the processes leading to sputter emission, in order to gain a microscopic understanding of low energy bombardment phenomena. Calculations were performed for a Cu (111) single crystal surface bombarded with Ar atoms in the energy range from 10–1000 eV. The results presented for low bombarding energies are mainly concerned with the near sputtering threshold behaviour, yields and depth of origin of sputtered atoms. Furthermore, it is found, that in addition to sputtered atoms, a large number of ad-atoms at the surface are generated during the evolution of the collision cascade. At higher energies the question of cluster emission and especially their energy distribution and angular distribution are addressed. It was found that the energy distributions for the dimers and monomer atoms exhibit a similar dependence on emission energy as has been observed recently also experimentally. For atoms ... read less NOT USED (low confidence) S. Marklund and W. Yongliang, “Influence of the 90° partial dislocation core structure in silicon on energy levels,” Solid State Communications. 1994. link Times cited: 2 NOT USED (low confidence) J. Bernholc, C. Brabec, A. Maiti, and J. Yi, “Structural transformations, reactions, and electronic properties of fullerenes, onions, and buckytubes,” Computational Materials Science. 1994. link Times cited: 1 NOT USED (low confidence) G. Gilmer and C. Roland, “Applications of molecular dynamics methods to low energy ion beams and film deposition processes,” Radiation Effects and Defects in Solids. 1994. link Times cited: 12 Abstract: Molecular dynamics methods are used to model the impingement… read moreAbstract: Molecular dynamics methods are used to model the impingement of low energy ions onto crystalline targets, and the effects of these beams on thin film deposition. Simulations of the deposition of silicon films show that the structure of deposits can often be improved by the use of low energy ion beams instead of the conventional thermal beam. We examine the influence of beam energy on the formation of amorphous or crystalline deposits. The influence of ion beams on surface diffusion rates and the interdiffusion between atomic layers near the surface are also considered. Cluster deposition is treated, and the results suggest that cluster beams would be effective for depositing smooth films of materials that do not wet the substrate. We discuss the use of special purpose computers and signal processing boards to extend the time scales of molecular dynamics simulations. Rapid advances in computer hardware, algorithms, and the development of accurate interatomic potentials are dramatically increasing ... read less NOT USED (low confidence) J. Keinonen, A. Kuronen, K. Nordlund, R. Nieminen, and A. Seitsonen, “First-principles simulation of collision cascades in Si to test pair-potentials for Si-Si interaction at 10 eV–5 keV,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1994. link Times cited: 22 NOT USED (low confidence) A. El-Azab and N. Ghoniem, “Molecular dynamics simulations of low energy cascades in β-SiC,” Radiation Effects and Defects in Solids. 1994. link Times cited: 0 Abstract: The dynamics of point defect production in β-SiC is studied … read moreAbstract: The dynamics of point defect production in β-SiC is studied using the Molecular Dynamics (MD) technique. A hybrid pair/three-body potential developed by E. Pearson et al. 10 is used to model interatomic forces. The bulk displacement energies are found for Si and C atoms along selected crystallographic directions within the 〈111〉 tetrahedral gaps. It is found that Si atoms have higher displacement energies than C atoms for all directions. Si displacement energy is found to be ∼52 eV, while that of C is only ∼10 eV through the 〈111〉 gap. Focused cascades along the close-packed [111] direction contribute to displacements in β-SiC but, replacement collision sequences are not likely to occur. Displaced atoms come to equilibrium in hexagonal interstitial sites between the (111) planes in most cases. Also, trivacancies tend to occur on the (111) carbon planes. The equilibrium cascade configurations are observed to be highly non-stoichiometric with the majority of displacements being of C type. read less NOT USED (low confidence) G. Jungnickel et al., “Structure and chemical bonding in high density amorphous carbon,” Diamond and Related Materials. 1994. link Times cited: 13 NOT USED (low confidence) T. Frauenheim, U. Stephan, P. Blaudeck, D. Porezag, H. Busmann, and W. Zimmermann-Edling, “Stability and reconstruction of diamond (100) and (111) surfaces,” Diamond and Related Materials. 1994. link Times cited: 17 NOT USED (low confidence) T. Frauenheim, U. Stephan, P. Blaudeck, and G. Jungnickel, “Molecular dynamic investigations of amorphous carbon: π bonding vs. electronic defect generation,” Diamond and Related Materials. 1994. link Times cited: 15 NOT USED (low confidence) G. Tóth and G. Náray‐Szabó, “Novel semiempirical method for quantum Monte Carlo simulation: Application to amorphous silicon,” Journal of Chemical Physics. 1994. link Times cited: 17 Abstract: We propose a novel bulk phase Monte Carlo simulation techniq… read moreAbstract: We propose a novel bulk phase Monte Carlo simulation technique, in which the energy is calculated by quantum mechanical methods. The semiempirical fragment self‐consistent field technique applied divides the periodic simulation cell into two parts. The first is the subsystem where the important change (the random movement of an atom or molecule) takes place and the second is the environment exerting only secondary effects on the former. Expanding the electronic wave function on the basis of strictly localized molecular orbitals and/or atomic hybrid orbitals the wave function of the environment is obtained from simple coupled 2×2 secular equations. The conventional self‐consistent field equations, with a perturbation term in the Fockian, have to be solved only for the subsystem. In this way the computational efforts are decreased drastically, as the dependence on the number of atoms in the environment reduces to quadratic instead of cubic or quartic as in conventional semiempirical or ab initio methods, re... read less NOT USED (low confidence) W. Bacsa and J. Lannin, “Intermolecular coupling of monolayer C60,” Journal of Electron Spectroscopy and Related Phenomena. 1993. link Times cited: 1 NOT USED (low confidence) L. Zeger and E. Kaxiras, “Compact carbon clusters with tetrahedral bonding and icosahedral symmetry,” Computational Materials Science. 1993. link Times cited: 5 NOT USED (low confidence) J. Spence, Y. M. Huang, and O. Sankey, “Lattice trapping and surface reconstruction for silicon cleavage on (111). Ab-initio quantum molecular dynamics calculations,” Acta Metallurgica Et Materialia. 1993. link Times cited: 71 NOT USED (low confidence) C. S. Carmer, B. Weiner, and M. Frenklach, “Molecular dynamics with combined quantum and empirical potentials: C2H2 adsorption on Si(100),” Journal of Chemical Physics. 1993. link Times cited: 71 Abstract: Classical trajectory calculations were employed to study the… read moreAbstract: Classical trajectory calculations were employed to study the reaction of acetylene with dimer sites on the Si(100) surface at 105 K. Two types of potential energy functions were combined to describe interactions for different regions of the model surface. A quantum mechanical potential based on the semiempirical AM1 Hamiltonian was used to describe interactions between C2H2 and a portion of the silicon surface, while an empirically parametrized potential was developed to extend the size of the surface and simulate the dynamics of the surrounding silicon atoms. Reactions of acetylene approaching different sites were investigated, directly above a surface dimer, and between atoms from separate dimers. In all cases, the outcome of C2H2 surface collisions was controlled by the amount of translational energy possessed by the incoming molecule. Acetylene molecules with high translational energy reacted with silicon dimers to form surface species with either one or two Si–C bonds. Those molecules with low transl... read less NOT USED (low confidence) D. Anderson, J. Kieffer, and S. Klarsfeld, “Molecular dynamic simulations of the infrared dielectric response of silica structures,” Journal of Chemical Physics. 1993. link Times cited: 22 Abstract: The molecular dynamic simulation technique was used to model… read moreAbstract: The molecular dynamic simulation technique was used to model the vibrational behavior of crystalline (α and β cristobalite) and amorphous silica structures. To this end a refined potential function was developed, which allows one to reproduce the correct structural geometries, the corresponding infrared spectra, and to observe a reversible phase transformation between α and β cristobalite. The complex dielectric constants in the infrared frequency range were calculated from the dipole moment time correlation functions. While idealized cristobalite exhibits the simplest spectrum with only two narrow bands, the increase of structural complexity and reduction of symmetry characteristic for the real cristobalites and amorphous silica, creates additional features in the infrared spectra. These structural changes predominantly affect the coordination of oxygen, and generate a broader spread in the normal modes characterizing the vibrations of this species. A unique method for the identification of atomic trajec... read less NOT USED (low confidence) J. Belak, D. B. Boercker, and I. Stowers, “Simulation of Nanometer-Scale Deformation of Metallic and Ceramic Surfaces,” MRS Bulletin. 1993. link Times cited: 79 NOT USED (low confidence) Tsumuraya, Ishibashi, and Kusunoki, “Statistics of Voronoi polyhedra in a model silicon glass.,” Physical review. B, Condensed matter. 1993. link Times cited: 14 Abstract: We clarify the local structure in a model silicon glass by u… read moreAbstract: We clarify the local structure in a model silicon glass by use of Voronoi-polyhedron analysis. The glass is produced by molecular dynamics with a Stillinger-Weber potential. The atoms in the glass are nearly distinguishable: there are about 200 types in the system with 216 atoms. The analysis clarifies that the polyhedra are formed by a small number of large-area polygons or by a large number of small-area polygons. This feature is different from those in Lennard-Jones glasses or metallic glasses and is attributed to the loose-packed structure even in the glass state, in which the atoms still have directional bonding read less NOT USED (low confidence) E. Beam, “Computer simulation of the surface topology of (001) silicon resulting from the termination of 12〈110〉 edge dislocations with Burgers vectors parallel to the surface,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 1993. link Times cited: 0 NOT USED (low confidence) H. Bergsåker, F. Lama, R. Smith, and R. Webb, “A comparison of TRIM and molecular dynamics in calculating the backscattering yield of carbon incident on graphite,” Vacuum. 1993. link Times cited: 4 NOT USED (low confidence) M. Ali and R. Smith, “The structure of small clusters ejected by ion bombardment of solids,” Vacuum. 1993. link Times cited: 11 NOT USED (low confidence) A. Parisini and A. Bourret, “Diamond hexagonal silicon phase and 113 defects Energy calculations and new defect models,” Philosophical Magazine. 1993. link Times cited: 26 Abstract: The hypothesis that the precipitation of self-interstitial S… read moreAbstract: The hypothesis that the precipitation of self-interstitial Si atoms leads to the formation of the diamond hexagonal Si phase, inside the structure of {113} stacking faults and rod-like defects, is reviewed on the basis of calculations of the total energy of the defects and high-resolution electron microscopy (HREM) image simulations. The relaxed atomic structures of several {113} defect models is obtained by the statics molecular method. The results of the calculations of the total energy of these models show that three new models present an energy lower than all other previously reported models of the {113} defects. The displacement vectors obtained from these models agree with available experimental data. These models also account for the experimentally observed transformation of the {113} defects into {111} faulted loops and perfect loops. From HREM image simulations it is shown that the major features of the experimental images are well reproduced in the simulated images. These findings allow... read less NOT USED (low confidence) W. Choi, C. Kim, and H. Kang, “Interactions of low energy (10–600 eV) noble gas ions with a graphite surface: surface penetration, trapping and self-sputtering behaviors,” Surface Science. 1993. link Times cited: 51 NOT USED (low confidence) J. R. Smith and D. Srolovitz, “Developing potentials for atomistic simulations,” Modelling and Simulation in Materials Science and Engineering. 1992. link Times cited: 14 Abstract: A small group of researchers met recently to review the new … read moreAbstract: A small group of researchers met recently to review the new and rapidly growing field of many-atom potentials for solids. The workshop was held on 25-27 September 1991, in Ann Arbor, MI, and was commissioned by the Air Force Office of Scientific Research. Some classes of materials are being treated well by many-atom potentials, while others are only now being considered. Combinations of materials including more than one type of bond seem clearly beyond our present capabilities. The systematics of many-atom potential development is in its infancy, and progress appears to be rapid. read less NOT USED (low confidence) C. T. White, D. Robertson, and D. Brenner, “Dissociative phase transitions from hypervelocity impacts,” Physica A-statistical Mechanics and Its Applications. 1992. link Times cited: 5 NOT USED (low confidence) A. El-Azab and N. Ghoniem, “Molecular dynamics study of the displacement threshold surfaces and the stability of Frenkel pairs in β-SiC,” Journal of Nuclear Materials. 1992. link Times cited: 22 NOT USED (low confidence) W. Niessen and V. G. Zakrzewski, “Complex Electron Affinity Processes in Clusters of S and Si.” 1992. link Times cited: 2 Abstract: Vertical and in some cases adiabatic electron affinities are… read moreAbstract: Vertical and in some cases adiabatic electron affinities are calculated for the clusters S4 and Sin, n = 3 – 7 with large basis sets. The effects of electron correlation are taken into account by CI and Green function techniques. The clusters show a complex behaviour upon electron attachment. The isomers of 84 show normal electron capture processes as well as electron attachment with shake-up. The Si clusters show multiple affinity states resulting from capture of an electron into different orbitals: Si3 C2v has at least three, Si4 D2h four, Si5 D3h two, Si6 D4v one, Si6 C2v three and Si7 D5h two affinity states (vertical processes: Sin + e− ± Sin + hν). For the Sin clusters in some cases shake-up affinities are calculated which are positive. The effects of electron correlation on the electron affinities are extremely large for the Si clusters in particular. In several cases the differences between the adiabatic and vertical electron affinities are very large amounting up to 1.5 eV. read less NOT USED (low confidence) L. Corrales and P. Rossky, “A novel semi-empirical potential for covalently bonded materials,” Chemical Physics Letters. 1992. link Times cited: 8 NOT USED (low confidence) S. Marklund and Y.-L. Wang, “Energy level calculations of the reconstructed 90° partial dislocation in silicon,” Solid State Communications. 1992. link Times cited: 8 NOT USED (low confidence) R. Smith, “A semi-empirical many-body interatomic potential for modelling dynamical processes in gallium arsenide,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1992. link Times cited: 55 NOT USED (low confidence) B. Garrison, E. J. Dawnkaski, D. Srivastava, and D. Brenner, “Molecular Dynamics Simulations of Dimer Opening on a Diamond 001(2x1) Surface,” Science. 1992. link Times cited: 163 Abstract: Computer simulations of hydrocarbon and related molecules us… read moreAbstract: Computer simulations of hydrocarbon and related molecules using empirical force fields have become important tools for studying a number of biological and related processes at the atomic scale. Traditional force fields, however, cannot be used to simulate dynamic chemical reactivity that involves changes in atomic hybridization. Application of a many-body potential function allows such reactivity to occur in a computer simulation. Simulations of the reaction of small hydrocarbon molecules adsorbed on a reconstructed diamond {001}(2x1) surface suggest that these hydrocarbons are highly reactive species and that initial stages of diamond growth proceed through a dimer-opening mechanism. Rates estimated from transition state theory of two interconversions between states where the dimer is open and closed are given. read less NOT USED (low confidence) U. Rothlisberger, W. Andreoni, and P. Giannozzi, “Thirteen‐atom clusters: Equilibrium geometries, structural transformations, and trends in Na, Mg, Al, and Si,” Journal of Chemical Physics. 1992. link Times cited: 147 Abstract: We report the results of an extensive structural study of Na… read moreAbstract: We report the results of an extensive structural study of Na13, Mg13, Al13, and Si13 carried out with the Car–Parrinello method. Several and mostly unforeseen noncrystalline structures are discovered to characterize the low portion of the potential energy surface. Crystalline structures are shown either to correspond to high‐energy local minima or to be highly unstable. The low‐energy structural pattern appears to change significantly from one element to the other. Specific characteristics as well as trends are discussed. read less NOT USED (low confidence) D. A. Jelski, B. Swift, T. Rantala, X. Xia, and T. George, “Structure of the Si45 cluster,” Journal of Chemical Physics. 1991. link Times cited: 46 Abstract: Six structures for the Si45 cluster are compared using a tig… read moreAbstract: Six structures for the Si45 cluster are compared using a tight‐binding model. Two new structures are proposed which appear to be the low‐energy isomers and to explain much of the existing experimental data. Cluster reactivity is distinguished from cluster stability, and several reasons are discussed which may lead to a reactive or unreactive species. These criteria are applied to the Si45 isomers, and the results are also correlated with experimental data. read less NOT USED (low confidence) R. Smith and R. Webb, “Long-range channelling in low energy ion implantation into silicon,” Philosophical Magazine Letters. 1991. link Times cited: 13 Abstract: Low energy (50 eV) implantation of boron and silicon into cr… read moreAbstract: Low energy (50 eV) implantation of boron and silicon into crystalline Si through the {110} and {100} faces is studied by a molecular dynamics simulation and the results compared with a binary-collision crystalline computer model and SIMS data. It is found that long-range channelling of the B particles takes place and that their ranges far exceed those predicted by transport theory in random media or Monte-Carlo computer models. It is found that channelling of B occurs only in the 〈110〉 direction. The Si atoms which are displaced by more than the nearest-neighbour spacing always originate from near to the surface as a result of direct knock-ons from ions which do not channel. These displacements show a distinct angular dependence because of the crystalline nature of the solid. For the Si implantation, the attractive forces between the incoming ion and the crystal atoms play an important role in limiting the ion range. read less NOT USED (low confidence) R. Virkkunen, K. Laasonen, and R. Nieminen, “Molecular dynamics using the tight-binding approximation: application to liquid silicon,” Journal of Physics: Condensed Matter. 1991. link Times cited: 32 Abstract: The authors present molecular dynamical simulations of liqui… read moreAbstract: The authors present molecular dynamical simulations of liquid silicon using the tight-binding approximation for electron-mediated interactions. Several structural and dynamical properties of liquid silicon are calculated and compared with the results of ab initio and classical molecular dynamics. The tight-binding model with parameters fitted to bulk crystalline properties is found to be very successful in characterizing the liquid state, which facilitates large-scale dynamical simulations. read less NOT USED (low confidence) A. Al-Derzi, R. Johnston, J. Murrell, and J. Rodriguez-Ruiz, “Potential energy functions for atomic solids: III. Fitting phonon frequencies and elastic constants of diamond structures,” Molecular Physics. 1991. link Times cited: 39 NOT USED (low confidence) W. Tiller, “The role of ledges in stress tensor-mediated surface processes for Si and GaAs,” Metallurgical Transactions A. 1991. link Times cited: 0 NOT USED (low confidence) M. Heggie, “Semiclassical interatomic potential for carbon and its application to the self-interstitial in graphite,” Journal of Physics: Condensed Matter. 1991. link Times cited: 33 Abstract: A semiclassical interatomic potential for carbon is discusse… read moreAbstract: A semiclassical interatomic potential for carbon is discussed which is based on the proximity cell (the Wigner-Seitz cell) around each atom. It introduces three internal degrees of freedom per atom, representing the magnitude and direction of the p orbital that is not involved in sp hybridization. Its direct interpolation between sp2 and sp3 configurations combined with good elastic properties allows its use on problematic defects, such as the interplanar interstitial in graphite, which is given as an example. read less NOT USED (low confidence) M. Kohyama, “On the transferable SETB method for Si,” Journal of Physics: Condensed Matter. 1991. link Times cited: 21 Abstract: The two types of transferable semi-empirical tight-binding (… read moreAbstract: The two types of transferable semi-empirical tight-binding (SETB) method for Si recently proposed by Goodwin et al. (1989) and by Sawada, which are intended to reproduce the binding energies and equilibrium volumes of variously coordinated structures of Si, have been examined and compared with each other. It has been found that there are some drawbacks in the method proposed by Goodwin et al, and that the method proposed by Sawada is much superior. The parameters in the Sawada method have been readjusted in order to apply this method to lattice defects or disordered systems of Si. The present results indicate the importance of incorporating the dependence on the local environment into the repulsive energy in the transferable SETB method. This can be explained by the origin of the repulsive energy. read less NOT USED (low confidence) A. Levi, D. A. Smith, and J. Wetzel, “Calculated structures for [001] symmetrical tilt grain boundaries in silicon,” Journal of Applied Physics. 1991. link Times cited: 19 Abstract: The structures of [001] tilt boundaries in silicon have been… read moreAbstract: The structures of [001] tilt boundaries in silicon have been systematically investigated by computer modeling, using the harmonic Keating potential to describe the interatomic forces. The full angular range of symmetrical tilt boundaries can be described in terms of linear combinations of characteristic groupings of atoms. More than one stable relaxed structure has been found for most grain boundaries. In all cases the relaxed bicrystal consists of localized groups of pure edge or 45° dislocation cores embedded in a tetrahedrally coordinated, stable structure. read less NOT USED (low confidence) F. Zerbetto, “Annealing graphite-like structures. A Monte Carlo-quantum chemical study.” 1991. link Times cited: 3 NOT USED (low confidence) J. H. Wilson, J. Todd, and A. Sutton, “Modelling of silicon surfaces: a comparative study,” Journal of Physics: Condensed Matter. 1990. link Times cited: 25 Abstract: A theoretical study of the Si(110)-1*1, Si(100)-2*1, Si(111)… read moreAbstract: A theoretical study of the Si(110)-1*1, Si(100)-2*1, Si(111)-2*1 and Si(113)-1*1 surfaces is presented. The authors use both the semi-empirical tight-binding bond model and the classical potential of Stillinger and Weber to describe interatomic forces. Energy minimization calculations are carried out in order to deduce the stable atomic configurations. The authors show that the semi-empirical tight-binding approach can produce results in reasonable agreement with other experimental and theoretical work and they demonstrate that charge transfer is not an important factor governing the stability of these surfaces. In a comparative study, involving not only static energy minimization but also Monte Carlo simulated annealing, the authors show why the classical potential does not perform well in describing surface atomic structure. read less NOT USED (low confidence) N. Blais and J. Stine, “A model of reactive dynamics in a detonation,” Journal of Chemical Physics. 1990. link Times cited: 7 Abstract: Classical trajectories are used to examine the importance of… read moreAbstract: Classical trajectories are used to examine the importance of many‐body interactions in the chemical reactivity of condensed phase explosives under the high density conditions that are characteristic of a detonation wave. We have constructed a model based on the explosive liquid nitric oxide, and we examine how reactions occur when the system is compressed rapidly to about double liquid density. The probability of reaction is investigated with a realistic potential energy surface that is derived for six atoms but is equally applicable to four atoms. The model was found to have the proper energy characteristics to simulate an explosive material. We find that the probability of forming detonation products is higher for six atoms than for four atoms, ranging from a factor of 4 to a factor of 25 depending on the potential; more than can be accounted for on the basis of a statistical analysis. The details of the trajectories that lead to reaction products differ considerably between the four‐ and six‐atom trial... read less NOT USED (low confidence) T. D. de la Rubia and M. Guinan, “Progress in the development of a molecular dynamics code for high-energy cascade studies,” Journal of Nuclear Materials. 1990. link Times cited: 146 NOT USED (low confidence) M. Kohyama, S. Kose, M. Kinoshita, and R. Yamamoto, “The self-consistent tight-binding method: application to silicon and silicon carbide,” Journal of Physics: Condensed Matter. 1990. link Times cited: 27 Abstract: The self-consistent tight-binding (SCTB) model proposed by M… read moreAbstract: The self-consistent tight-binding (SCTB) model proposed by Majewski and Vogl (1987) has been extended to be applicable for calculations of lattice defects in solids or disordered systems with both ionic and covalent characters that cannot be treated using other types of tight-binding theories. The precise formulation of electronic structure, total energy and atomic forces in the supercell technique has been presented. In order to apply this method to lattice defects in SiC, the parameters and functional forms have been examined so as to reproduce the basic properties of Si, SiC and C. The nature of the bonding and the phase stability in Si and SiC have been analysed by the present SCTB method. read less NOT USED (low confidence) A. F. Bakker, G. Gilmer, M. Grabow, and K. Thompson, “A special purpose computer for molecular dynamics calculations,” Journal of Computational Physics. 1990. link Times cited: 27 NOT USED (low confidence) D. Brenner and B. Garrison, “Gas‐Surface Reactions: Molecular Dynamics Simulations of Real Systems,” Advances in Chemical Physics. 1990. link Times cited: 8 NOT USED (low confidence) T. Weber and F. Stillinger, “Dynamical branching during fluorination of the dimerized Si(100) surface: A molecular dynamics study,” Journal of Chemical Physics. 1990. link Times cited: 70 Abstract: Collections of classical trajectories have been numerically … read moreAbstract: Collections of classical trajectories have been numerically generated for individual F2 molecules impinging at normal incidence on a Si(100) surface at 0 K dimerized in a p(2×1) pattern. A linear combination of two‐atom and three‐atom interaction functions represents the potential energy. Trajectories fall into four categories: (a) non‐reactive F2 rebound, (b) monofluorination at a surface dangling bond with energetic expulsion into the vacuum of the remaining F atom, (c) difluorination of a pair of dangling bonds, and (d) monofluorination with retention of the second F in a weakly bound Si–F⋅⋅⋅F surface complex. Surface patterns for difluorination, (c), indicate absence of surface diffusion during this mode of chemisorption. Increasing either the translational kinetic energy or the vibrational excitation of the incident F2 appears to enhance its surface reactivity. read less NOT USED (low confidence) A. Rockett, “The influence of surface structure on growth of Si(001)2×1 from the vapor phase,” Surface Science. 1990. link Times cited: 23 NOT USED (low confidence) R. Smith, D. E. Harrison, and B. Garrison, “Simulation of keV particle bombardment of covalent materials: An investigation of the yield dependence on incidence angle,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1990. link Times cited: 18 NOT USED (low confidence) E. Kaxiras, “Structural model for a covalently bonded Si45 cluster,” Chemical Physics Letters. 1989. link Times cited: 45 NOT USED (low confidence) S. Marklund, “Vacancy concentration in the core of the 90° reconstructred partial dislocation in silicon,” Solid State Communications. 1989. link Times cited: 2 NOT USED (low confidence) P. Ashu and C. Matthai, “Computer simulation of Si and Ge adatoms and thin layers on Si substrates,” Journal of Physics: Condensed Matter. 1989. link Times cited: 4 Abstract: The molecular dynamics method is used to determine minimum e… read moreAbstract: The molecular dynamics method is used to determine minimum energy configurations of Si and Ge adatoms on a Si surface. Results for thin layers of Ge on Si substrates and Si-Ge superlattices are also presented. read less NOT USED (low confidence) Smith, Harrison, and Garrison, “keV particle bombardment of semiconductors: A molecular-dynamics simulation.,” Physical review. B, Condensed matter. 1989. link Times cited: 126 Abstract: Molecular-dynamics simulations have been performed for the k… read moreAbstract: Molecular-dynamics simulations have been performed for the keV particlebombardment of Si/l brace/110/r brace/ and Si/l brace/100/r brace/ using a many-body potential developed byTersoff. Energy and angle distributions are presented along with an analysis ofthe important ejection mechanisms. We have developed a computer logic that onlyintegrates the equations of motion of the atoms that are struck, thusdecreasing the computer time by a factor of 3 from a completemolecular-dynamics simulation. read less NOT USED (low confidence) L. Feng, X. Zhang, W. Li, M. Liu, and X. Yao, “Multiple structural phase transitions in single crystal silicon subjected to dynamic loading,” Scripta Materialia. 2024. link Times cited: 0 NOT USED (low confidence) A. Casto et al., “Water filling in carbon nanotubes with different wettability and implications on nanotube/water heat transfer via atomistic simulations,” International Journal of Heat and Mass Transfer. 2023. link Times cited: 3 NOT USED (low confidence) H. Wang et al., “Molecular dynamics study of the effects of the porosity and initial pressure on phase transition of porous phase change materials,” Journal of Energy Storage. 2023. link Times cited: 1 NOT USED (low confidence) “Investigation of the heat transport in intersected graphene,” International Journal of Heat and Mass Transfer. 2023. link Times cited: 2 NOT USED (low confidence) F. Liu et al., “Insights into the interfacial thermal transport properties of in-plane graphene/h-BN heterostructure with grain boundary,” International Journal of Heat and Mass Transfer. 2023. link Times cited: 1 NOT USED (low confidence) I. M. Felix and L. Pereira, “Thermal conductivity of Thue–Morse and double-period quasiperiodic graphene-hBN superlattices,” International Journal of Heat and Mass Transfer. 2022. link Times cited: 5 NOT USED (low confidence) K. Zhou and B. Liu, “Potential energy functions,” Molecular Dynamics Simulation. 2022. link Times cited: 0 NOT USED (low confidence) Y. Karaaslan, “Coherent and incoherent phonon thermal transport in group-III nitride monolayer superlattices with Tersoff type interatomic potential,” Physica E: Low-dimensional Systems and Nanostructures. 2022. link Times cited: 2 NOT USED (low confidence) S. Li, S. Sajadi, K. Alharbi, M. El-Shorbagy, and I. Tlili, “The molecular dynamics study of vacancy defect influence on carbon nanotube performance as drug delivery system,” Engineering Analysis with Boundary Elements. 2022. link Times cited: 24 NOT USED (low confidence) H. Jia et al., “Dual-functional graphene/carbon nanotubes thick film: Bidirectional thermal dissipation and electromagnetic shielding,” Carbon. 2021. link Times cited: 44 NOT USED (low confidence) H. Parsapour, S. Ajori, and R. Ansari, “A molecular dynamics study on the tensile characteristics of various metallic glass nanocomposites reinforced by Weyl semimetals three-dimensional graphene network,” European Journal of Mechanics A-solids. 2021. link Times cited: 7 NOT USED (low confidence) S. Fang and Y. Hu, “Cyclo[18]carbon as an ultra-elastic molecular O-ring with unique mechanical properties,” Carbon. 2021. link Times cited: 28 NOT USED (low confidence) Y. Liu et al., “Mechanical properties and thickness-determined fracture mode of hexagonal boron nitride nanosheets under nanoindentation simulations,” Computational Materials Science. 2021. link Times cited: 12 NOT USED (low confidence) L.-G. Dai, G. Chen, and Z. Shan, “Study on ultra-high speed nano-grinding of monocrystalline copper with V-shaped diamond abrasive grains based on molecular dynamics method,” Diamond and Related Materials. 2021. link Times cited: 9 NOT USED (low confidence) M. Hummel, W. Verestek, and S. Schmauder, “Molecular Dynamics Simulations—A Time and Length Scale Investigation.” 2021. link Times cited: 0 NOT USED (low confidence) P. Singh and I. S. Chahal, “Atomistic Study on the Mechanical Behavior of Silicon-Base Nanotubes.” 2021. link Times cited: 0 Abstract: Received Feb 10, 2021 Revised Mar 16, 2021 Accepted May 14, … read moreAbstract: Received Feb 10, 2021 Revised Mar 16, 2021 Accepted May 14, 2021 Recently, silicon nanotubes (SiNTs) have been successfully synthesized and have attracted many researchers to work on the different aspects of them. In the present study, the stress-strain curve along with the Young’s modulus as a significant mechanical property of single walled silicon nanotubes at different diameters are determined. The simulation is performed by the use of molecular dynamics based on the Tersoff-Brenner many-body potential energy function. The results of the total strain energy of nanotubes as an accurate and effective methodology are used to establish appropriate expressions for evaluating Young’s modulus of the nanotubes. Keyword: read less NOT USED (low confidence) L. Marqués, M. Aboy, P. López, I. Santos, L. Pelaz, and G. Fisicaro, “Atomistic modeling of laser-related phenomena,” Laser Annealing Processes in Semiconductor Technology. 2021. link Times cited: 0 NOT USED (low confidence) A. Albooyeh, A. Dadrasi, and A. H. Mashhadzadeh, “Effect of point defects and low-density carbon-doped on mechanical properties of BNNTs: A molecular dynamics study,” Materials Chemistry and Physics. 2020. link Times cited: 27 NOT USED (low confidence) P. Sedigh, A. Zare, and A. Montazeri, “Evolution in aluminum applications by numerically-designed high strength boron-nitride/Al nanocomposites,” Computational Materials Science. 2020. link Times cited: 14 NOT USED (low confidence) K. Nordlund and F. Djurabekova, “Molecular Dynamics Simulations of Non-equilibrium Systems,” Handbook of Materials Modeling. 2020. link Times cited: 3 NOT USED (low confidence) R. Devanathan, “Interatomic Potentials for Nuclear Materials,” Handbook of Materials Modeling. 2020. link Times cited: 1 NOT USED (low confidence) J. Yeo, Z. Liu, and T. Ng, “Silica Aerogels: A Review of Molecular Dynamics Modelling and Characterization of the Structural, Thermal, and Mechanical Properties,” Handbook of Materials Modeling. 2020. link Times cited: 9 NOT USED (low confidence) A. K. Balerba et al., “Graphene nano-flakes on Cu low-index surfaces by density functional theory and molecular dynamics simulations.” 2020. link Times cited: 2 NOT USED (low confidence) A. Sircar and P. Patra, “Rolling and Sliding Resistance as Carbon Nanotubes are Driven on a Graphene Sheet.” 2020. link Times cited: 0 NOT USED (low confidence) P. Marepalli, S. Mathur, and J. Murthy, “An unintrusive approach to the computation of derivatives: Applications in nanoscale thermal transport.” 2020. link Times cited: 0 NOT USED (low confidence) A. C. Pierre, “Gelation,” Introduction to Sol-Gel Processing. 2020. link Times cited: 9 NOT USED (low confidence) K. Liu, H. Wang, and X. Zhang, “Molecular Dynamics Simulation of Ductile Mode Cutting,” Springer Series in Advanced Manufacturing. 2019. link Times cited: 1 NOT USED (low confidence) B. Babu and B. P. Patel, “Analytical solution for strain gradient elastic Kirchhoff rectangular plates under transverse static loading,” European Journal of Mechanics - A/Solids. 2019. link Times cited: 31 NOT USED (low confidence) M. Islam, M. Cherukara, E. Antillon, and A. Strachan, “Shock-Induced Chemistry: Molecular Dynamics and Coarse Grain Modeling,” Computational Approaches for Chemistry Under Extreme Conditions. 2019. link Times cited: 5 NOT USED (low confidence) G. Voyiadjis and M. Yaghoobi, “Size Effects During Nanoindentation: Molecular Dynamics Simulation,” Handbook of Nonlocal Continuum Mechanics for Materials and Structures. 2019. link Times cited: 5 NOT USED (low confidence) L. Zhang, “Mechanics of Carbon Nanotubes and Their Composites,” Handbook of Mechanics of Materials. 2019. link Times cited: 0 NOT USED (low confidence) A. Lahti, R. Östermark, and K. Kokko, “Optimizing Atomic Structures through Geno-Mathematical Programming,” Communications in Computational Physics. 2019. link Times cited: 1 Abstract: In this paper, we describe our initiative to utilize a moder… read moreAbstract: In this paper, we describe our initiative to utilize a modern well-tested numerical platform in the field of material physics: the Genetic Hybrid Algorithm (GHA). Our aim is to develop a powerful special-purpose tool for finding ground state structures. Our task is to find the diamond bulk atomic structure of a silicon supercell through optimization. We are using the semi-empirical Tersoff potential. We focus on a 2x2x1 supercell of cubic silicon unit cells; of the 32 atoms present, we have fixed 12 atoms at their correct positions, leaving 20 atoms for optimization. We have been able to find the known global minimum of the system in different 19-, 43and 60-parameter cases. We compare the results obtained with our algorithm to traditional methods of steepest descent, simulated annealing and basin hopping. The difficulties of the optimization task arise from the local minimum dense energy landscape of materials and a large amount of parameters. We need to navigate our way efficiently through these minima without being stuck in some unfavorable area of the parameter space. We employ different techniques and optimization algorithms to do this. AMS subject classifications: 82-08 read less NOT USED (low confidence) P. Szarek and A. Tachibana, “Contemporary analysis of the influence of adsorbents on the structure, stability, and reactivity of main group nanoparticles using regional density functional theory,” Harnessing Nanoscale Surface Interactions. 2019. link Times cited: 0 NOT USED (low confidence) G. Voyiadjis and M. Yaghoobi, “Molecular dynamics,” Size Effects in Plasticity. 2019. link Times cited: 0 NOT USED (low confidence) S. Shafraniuk, “Contribution of many-body effects into thermoelectricity and heat transport in graphene.” 2018. link Times cited: 0 NOT USED (low confidence) S. Singh and B. P. Patel, “Large deformation static and dynamic response of carbon nanotubes by mixed atomistic and continuum models,” International Journal of Mechanical Sciences. 2018. link Times cited: 13 NOT USED (low confidence) A. Dmitriev and A. Nikonov, “Molecular-dynamic study the influence of size parameter and temperature of the system on adhesive wear mechanisms.” 2018. link Times cited: 0 NOT USED (low confidence) R. Promyoo, H. El-Mounayri, and M. Agarwal, “An Experimental Study to Guide AFM-Based TBN of Nanochannels.” 2018. link Times cited: 1 NOT USED (low confidence) S. Shafraniuk, “Chapter 5 – Role of structural defects and imperfections.” 2018. link Times cited: 0 NOT USED (low confidence) Y. Luo, N. Zhou, H. Gong, H. Huang, and L. Zhou, “Effect of incident kinetic energy on a-Si:H structure: A molecular dynamics simulation study,” IOP Conference Series: Materials Science and Engineering. 2018. link Times cited: 0 Abstract: This paper utilized molecular dynamics simulation to investi… read moreAbstract: This paper utilized molecular dynamics simulation to investigate the influences of the incident kinetic energy on the structure of hydrogenated amorphous silicon (a-Si:H) thin films. The SiH3 radical impinged on the Si (001) surface with substrate temperature of 500 K in the incident kinetic energy range from 0.04 to 5.81 eV. The results showed that high incident kinetic energy could smooth the surface, promote the densification of internal structure, and increase the adsorption rate of SiH3 radical and the crystalline volume fraction. The SiHx and dangling bonds, which are the key factors that affecting the film quality, have been further analyzed, the results reflected that the increase of the incident kinetic energy will increase the content of SiH combination pattern and decrease the dangling bonds in the films, thus improving the quality of the thin films. There is a critical value, 0.64 eV, 1.45 eV or 4.04 eV, of this variation rate. Steep variation trends is corresponding to the incident kinetic energy less than this critical value, and variation trends is gentle for that above it. read less NOT USED (low confidence) B. Schultrich, “Structure of Amorphous Carbon.” 2018. link Times cited: 2 NOT USED (low confidence) H. Li and R. Q. Zhang, “Theoretical and Experimental Methods for Determining the Thermal Conductivity of Nanostructures.” 2018. link Times cited: 0 NOT USED (low confidence) D. Gerasimov and E. I. Yurin, “Numerical Experiments: Molecular Dynamics Simulations.” 2018. link Times cited: 0 NOT USED (low confidence) C. Zhang, M. Zhao, C. Hou, and W. Ge, “A multilevel-skin neighbor list algorithm for molecular dynamics simulation,” Comput. Phys. Commun. 2018. link Times cited: 2 NOT USED (low confidence) A. Shkrebtii and M. Rohlfing, “Determination of the total energy of a many-particle system.” 2018. link Times cited: 0 NOT USED (low confidence) X. Wu, “Experiment Approaches and Simulation Methods.” 2018. link Times cited: 0 NOT USED (low confidence) J. D. Lee, J. Li, Z. Zhang, and L. Wang, “Sequential and Concurrent Multiscale Modeling of Multiphysics: From Atoms to Continuum.” 2018. link Times cited: 8 NOT USED (low confidence) P. Wang, R. Xiang, and S. Maruyama, “Thermal Conductivity of Carbon Nanotubes and Assemblies.” 2018. link Times cited: 14 NOT USED (low confidence) M. Yaghoobi, “Modeling of Size Effects in Metallic Samples of Confined Volumes.” 2017. link Times cited: 0 NOT USED (low confidence) I. Giordanelli, “Correlated Deformations in Graphene.” 2017. link Times cited: 0 Abstract: In this thesis, we perform a numerical study on the conforma… read moreAbstract: In this thesis, we perform a numerical study on the conformal invariance, crumpling, and electrical properties of suspended graphene sheets. We use molecular dynamics simulations for describing the motion of the carbon atoms, and a lattice Boltzmann model to calculate the electronic flow within the hydrodynamics formalism. We find that graphene membranes exhibit correlated deformations which we study under the framework of rough surfaces and determine their Hurst (roughness) exponent. We show that, independent of the temperature, graphene membranes possess scale invariant properties, expressed for instance in a well-defined fractal dimension of the iso-height lines at the percolation threshold. Additionally, we provide numerical evidence that the iso-height lines are conformally invariant. Interestingly, iso-height lines of other rough surfaces are not necessarily conformally invariant even if they have the same Hurst exponent, e.g. random Gaussian surfaces. We compare our graphene sheets with random Gaussian surfaces with the same Hurst exponent and find not only a disagreement in the fractal dimension of the iso-height lines, but also a disagreement in the distribution of the modulus of the Fourier coefficients. The correlated random deformations in graphene are responsible for the stability of the two-dimensional material. An analysis of the two-dimensional crystal problem beyond the harmonic approximation, has led to the conclusion that atomically thin membranes can be stabilised through their deformation in the third dimension. In contrast to it, graphene membranes with sufficiently high vacancy densities have the tendency to crumple, deviating read less NOT USED (low confidence) S. Thamaraikannan, S. Pradhan, and M. R. Sunny, “Parametric study on Topology of carbon Nanotubes Effects on Mechanical properties,” Materials Today: Proceedings. 2017. link Times cited: 0 NOT USED (low confidence) A. Vakhrushev, A. Fedotov, A. V. Severjuhin, and R. Valeev, “Effect of Pore Size Parameters for Mechanisms of Nanofilm Coatings on Substrates of Porous Alumina,” Bulletin of the South Ural State University. Series "Mathematical Modelling, Programming and Computer Software. 2017. link Times cited: 1 NOT USED (low confidence) J. Zhang, Y. Hong, M. Liu, Y. Yue, Q. Xiong, and G. Lorenzini, “Molecular dynamics simulation of the interfacial thermal resistance between phosphorene and silicon substrate,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 81 NOT USED (low confidence) Y. Tamura et al., “Molecular Dynamics Simulation for Intrinsic Stress Caused by Surface Oxidation on Hydrogenated Amorphous Silicon,” Journal of The Society of Materials Science, Japan. 2017. link Times cited: 0 Abstract: It was reported that the lack of the structural stability of… read moreAbstract: It was reported that the lack of the structural stability of semiconductor silicon micro-pattern induced the lateral undulation buckling. Our previous report revealed that the intrinsic stress of the oxide film on the surface of amorphous silicon produced the compressive stress which induces the buckling failure. However, actual amorphous silicon contains hydrogen atoms. Therefore, in this study, we realize the surface oxide film fabrication on hydrogenated amorphous silicon and clarify the relationship between hydrogen concentration and intrinsic stress due to surface oxidation. As a result, regardless the hydrogen concentration, surface oxide layer contains no hydrogen atoms. In addition, it is found that the intrinsic stress is generated in the sub-oxide layer where oxidation process is not completed. As the hydrogen concentration increases, the integral value of the compressive stress decreases linearly. The stress decreases about 30 % when the hydrogen concentration reaches 25 at%. Decrease in the stress would be caused by the sparse silicon structure due to hydrogen atoms and resulting release of the strain due to surface oxidation. read less NOT USED (low confidence) K. Liew, J.-W. Yan, and L.-W. Zhang, “Classical Molecular Dynamics Simulations.” 2017. link Times cited: 8 NOT USED (low confidence) A. Soloviev, R. Gruzdev, A. V. Derkun, and E. Lähderanta, “Identification of Graphene Properties in the Framework of Molecular Dynamics.” 2017. link Times cited: 0 NOT USED (low confidence) M. Verdier, K. Termentzidis, and D. Lacroix, “Modeling Thermal Transport in Nano-Porous Semiconductors.” 2017. link Times cited: 2 NOT USED (low confidence) A. Vakhrushev, A. V. Severyukhin, A. Fedotov, and R. Valeev, “Investigation of deposition of nanofilms on a porous aluminium oxide substrate by mathematical modeling techniques.” 2016. link Times cited: 5 Abstract: В работе приведена постановка задачи и описание методики изу… read moreAbstract: В работе приведена постановка задачи и описание методики изучения процессов осаждения нанопленок на подложку из пористого алюминия. Рассмотрены уравнения, составляющие основу многочастичного потенциала, отвечающего модифицированному методу погруженного атома. В качестве осаждаемых материалов брались золото, серебро, железо, галлий, германий и палладий. Расчеты показали, что существуют разные механизмы заращивания пористой подложки из оксида алюминия данными материалами. Для каждого из типов осаждаемых атомов были зафиксированы свои процессы взаимодействия наноструктур и механизмы заращивания подложек и пор. Так, атомы серебра и золота равномерно закрывали поры нанопленкой без проникновения внутрь их, имело место лишь незначительное проседание нанопленки в поры. Атомы железа генерировали наноструктуры в безвоздушной среде над подложкой; заращивание подложки происходило по островному принципу; мелкие наноструктуры железа на подложке постепенно укрупнялись и группировались в бóльшие по размеру; наблюдалось образование наноструктуры железа внутри поры. При осаждении атомов галлия пора также полностью не зарастала, а нанопленка на поверхности подложки формировалась в виде отдельных областей; были заметны небольшие наночастицы галлия на поверхности подложки. Палладий порождал равномерную пленку с небольшим проседанием в области поры; при осаждении атомов палладия непосредственно над порой на протяжении всего этапа конденсации сохранялось отверстие, которое так и не заросло. У всех типов осаждаемых атомов имелись единичные экземпляры, которые достигали дна поры. Наиболее полное и плотное заращивание поры зафиксировано при эпитаксии галлия. Установлено, что пора, заполненная атомами, может рассматриваться как квантовая точка и использоваться для получения оптических и электрических эффектов. Сформулированы практические рекомендации для производства нанопленочных материалов различной структуры. Методики осаждения наноразмерных пленок могут применяться для контроля в конкретных технологических процессах, а также для прогнозирования и проектирования нанопленочных материалов. read less NOT USED (low confidence) S. Urata and S. Li, “Higher order Cauchy–Born rule based multiscale cohesive zone model and prediction of fracture toughness of silicon thin films,” International Journal of Fracture. 2016. link Times cited: 18 NOT USED (low confidence) A. D. Bobadilla and J. Seminario, “In Silico Assembly of Carbon-Based Nanodevices.” 2016. link Times cited: 0 NOT USED (low confidence) D. Hong, H. Shu, X. Guo, and C. Zheng, “Molecular Dynamics Simulations Study of Brown Coal Pyrolysis Using ReaxFF Method.” 2016. link Times cited: 4 NOT USED (low confidence) W. Xing, C.-X. Zhang, S. Fan, and Li-Cheng, “Research Progress on Resonant Characteristics of Graphene,” Journal of Inorganic Materials. 2016. link Times cited: 2 Abstract: The research on the resonant characteristics of graphene is … read moreAbstract: The research on the resonant characteristics of graphene is essential due to its excellence and vital significance for the future development and application of resonant sensors. Currently, the resonant characteristics of graphene are studied by experimental measurements and theoretical analysis, and the theoretical analysis is composed of the methods based on nano-scale mechanic and on classical mechanics. And the related theoretical researches on graphene are urgent because it is difficult to obtain the resonant characteristics of graphene accurately with experiments. In this paper, research methods, achievements and controversial problems are reviewed, including progress and consensus of the researches on the resonant characteristic of graphene e.g., the experiments of the resonant graphene sensor, the theoretical research methods and its classification, present situation, advantages, disadvantages, as well as the developing trend. read less NOT USED (low confidence) M. Yaghoobi and G. Voyiadjis, “Atomistic simulation of size effects in single-crystalline metals of confined volumes during nanoindentation,” Computational Materials Science. 2016. link Times cited: 47 NOT USED (low confidence) R. Jones, C. Weinberger, S. Coleman, and G. Tucker, “Introduction to Atomistic Simulation Methods.” 2016. link Times cited: 1 NOT USED (low confidence) S. Thamaraikannan and S. Pradhan, “Atomistic Study of Carbon Nanotubes: Effect of Cut-Off Distance.” 2016. link Times cited: 6 NOT USED (low confidence) R. Jones, J. Templeton, and J. Zimmerman, “Principles of Coarse-Graining and Coupling Using the Atom-to-Continuum Method.” 2016. link Times cited: 6 NOT USED (low confidence) L. Sang, “Affect of the graphene layers on the melting temperature of silicon by molecular dynamics simulations,” Computational Materials Science. 2016. link Times cited: 8 NOT USED (low confidence) M. Shai, T. Mosuang, and E. Rammutla, “Relative stability and thermodynamics properties of some graphene structures,” Materials Today: Proceedings. 2016. link Times cited: 0 NOT USED (low confidence) J. Houška, “Force field for realistic molecular dynamics simulations of ZrO2 growth,” Computational Materials Science. 2016. link Times cited: 12 NOT USED (low confidence) T. Ng, S. Joshi, J. Yeo, and Z. Liu, “Effects of Nanoporosity on the Mechanical Properties and Applications of Aerogels in Composite Structures.” 2016. link Times cited: 2 NOT USED (low confidence) A. Dmitriev, A. Nikonov, and W. Österle, “Multiscale modeling of low friction sliding behavior of a hybrid epoxy-matrix nanocomposite,” Procedia structural integrity. 2016. link Times cited: 3 NOT USED (low confidence) G. Pal and S. Kumar, “Modeling of carbon nanotubes and carbon nanotube-polymer composites,” Progress in Aerospace Sciences. 2016. link Times cited: 71 NOT USED (low confidence) G. V. Alberdi, “Object kinetic Monte Carlo simulations of irradiated tungsten for nuclear fusion reactors.” 2016. link Times cited: 1 NOT USED (low confidence) S. Goel, X. Luo, A. Agrawal, and R. Reuben, “Diamond machining of silicon: A review of advances in molecular dynamics simulation,” International Journal of Machine Tools & Manufacture. 2015. link Times cited: 314 NOT USED (low confidence) F. Fang and Y. Chen, “Nanometric cutting of crystal surfaces modified by ion implantation.” 2015. link Times cited: 1 NOT USED (low confidence) S. Singh and B. P. Patel, “Nonlinear elastic properties of graphene sheet under finite deformation,” Composite Structures. 2015. link Times cited: 30 NOT USED (low confidence) B. Johnson, J. McCallum, and M. Aziz, “Solid-Phase Epitaxy.” 2015. link Times cited: 11 NOT USED (low confidence) M. Ganchenkova and R. Nieminen, “Mechanical Properties of Silicon Microstructures.” 2015. link Times cited: 4 NOT USED (low confidence) Z.-J. Wu, “The mechanism governing cutting of hard materials with hybrid Laser/Waterjet system through controlled fracture.” 2015. link Times cited: 3 Abstract: ............................................................… read moreAbstract: ........................................................................................................ 79 5. read less NOT USED (low confidence) A. Rajabpour, L. Seidabadi, and M. Soltanpour, “Calculating the Bulk Modulus of Iron and Steel Using Equilibrium Molecular Dynamics Simulation,” Procedia Materials Science. 2015. link Times cited: 18 NOT USED (low confidence) V. Venkatesh, “Computer simulation studies of carbon nanotube and its interactions with water.” 2014. link Times cited: 1 Abstract: ii addition to defect concentration, the location of defects… read moreAbstract: ii addition to defect concentration, the location of defects in SWCNT will also affect the mechanical properties of water submerged SWCNT. For the case of capped SWCNTs, it was found that the concentration of water molecule encapsulated inside the SWCNT strongly affects the elastic properties of the SWCNT. Another study involved the transport characteristics of water molecules in CNTs using MD simulation. The transport properties of water molecules in a nano-scale channel such as CNTs is critical for its key role in designing the next generation CNT based nanofluidic devices. The effect of channel diameter, defects and the inter-layer spacing on the transport of water molecules is studied by subjecting the flow of water molecules through CNTs under pressure. The findings show that the efficiency of water transport can be improved by deploying bigger SWCNTs that have wide channel diameter. It was however found that defects in the nano-fluidic system will reduce the transport efficiency of water molecules. The results also show that the inter-layer spacing in a double-walled CNTs (DWCNTs) has a significant influence on the transport efficiency of water molecules. The investigations and conclusions obtained from this thesis is expected to further compliment the potential applications of CNTs in nano-fluidics and NEMS devices. read less NOT USED (low confidence) Z. Hu and X. Lu, “Mechanical Properties of Carbon Nanotubes and Graphene.” 2014. link Times cited: 11 NOT USED (low confidence) R. Khanna and V. Sahajwalla, “Atomistic Simulations of Properties and Phenomena at High Temperatures.” 2014. link Times cited: 3 NOT USED (low confidence) W.-H. Chen and H.-C. Cheng, “Molecular Modeling and Simulation of Physical Properties and Behavior of Low-Dimensional Carbon Allotropes.” 2014. link Times cited: 1 NOT USED (low confidence) M. Yasuda, S. Wakuda, Y. Asayama, H. Kawata, and Y. Hirai, “Interaction volume of electron beam in carbon nanomaterials: A molecular dynamics study,” MRS Proceedings. 2014. link Times cited: 1 NOT USED (low confidence) R. Ansari, S. Rouhi, and S. Ajori, “Elastic properties and large deformation of two-dimensional silicene nanosheets using molecular dynamics,” Superlattices and Microstructures. 2014. link Times cited: 45 NOT USED (low confidence) A. Ovrutsky, A. Prokhoda, and M. Rasshchupkyna, “Computer Modeling of Physical Phenomena and Processes.” 2014. link Times cited: 1 NOT USED (low confidence) J. L. Gomez-Ballesteros, A. Callejas-Tovar, L. Coelho, and P. Balbuena, “Molecular Dynamics Studies of Graphite Exfoliation Using Supercritical CO2.” 2014. link Times cited: 4 NOT USED (low confidence) C. Fang, A. Kumar, and S. Mukherjee, “Finite element analysis of single-walled carbon nanotubes based on a rod model including in-plane cross-sectional deformation,” International Journal of Solids and Structures. 2013. link Times cited: 10 NOT USED (low confidence) V. Vassilev, “Unduloid-Like Equilibrium Shapes of Single-Wall Carbon Nanotubes Under Pressure.” 2013. link Times cited: 2 Abstract: . In this work, a continuum model is used to determine in an… read moreAbstract: . In this work, a continuum model is used to determine in analytic form a class of unduloid-like equilibrium shapes of single-wall carbon nanotubes subjected to uniform hydrostatic pressure. The parametric equations of the profile curves of the foregoing shapes are presented in explicit form by means of elliptic functions and integrals. read less NOT USED (low confidence) F. V. Mackenzie and B. Thijsse, “Atomistic Modeling of Alumina/Epoxy Adhesion,” MRS Proceedings. 2013. link Times cited: 2 NOT USED (low confidence) I. Mladenov, M. Hadzhilazova, V. Vassilev, and P. Djondjorov, “Unduloid-like Equilibrium Shapes of Carbon Nanotubes Subjected to Hydrostatic Pressure.” 2013. link Times cited: 0 NOT USED (low confidence) R. Ansari, A. Shahabodini, and H. Rouhi, “Prediction of the biaxial buckling and vibration behavior of graphene via a nonlocal atomistic-based plate theory,” Composite Structures. 2013. link Times cited: 47 NOT USED (low confidence) R. Ansari, F. Sadeghi, and S. Ajori, “Continuum and molecular dynamics study of C60 fullerene–carbon nanotube oscillators,” Mechanics Research Communications. 2013. link Times cited: 41 NOT USED (low confidence) E. Mahmoudinezhad and R. Ansari, “Vibration analysis of circular and square single-layered graphene sheets: An accurate spring mass model,” Physica E-low-dimensional Systems & Nanostructures. 2013. link Times cited: 26 NOT USED (low confidence) S. Hug, “Classical molecular dynamics in a nutshell.,” Methods in molecular biology. 2013. link Times cited: 20 NOT USED (low confidence) J. Li et al., “Many-Core Programming.” 2013. link Times cited: 0 NOT USED (low confidence) P. Gamallo, L. Martin-Gondre, R. Sayós, C. Crespos, and P. Larrégaray, “Potential Energy Surfaces for the Dynamics of Elementary Gas-Surface Processes.” 2013. link Times cited: 8 NOT USED (low confidence) B. Szefler and B. Szefler, “On Molecular Dynamics of the Diamond D5 Substructures.” 2013. link Times cited: 1 NOT USED (low confidence) A. Bouhekka, “Monte Carlo.” 2013. link Times cited: 0 Abstract: Predicting mechanical strength of materials through theory a… read moreAbstract: Predicting mechanical strength of materials through theory and simulations of defect microstructures across atomic, mesoscopic and continuum scales. Developing new atomistic simulation methods for long time-scale processes, such as crystal growth and self-assembly. Applying machine learning techniques to materials research. Modeling and experiments on the metallurgical processes in metal 3D printing. Understanding microstructure-property relationship in materials for stretchable electronics, such as carbon nanotube networks and semiconducting elastomers. read less NOT USED (low confidence) V. Eyert et al., “Atomistic Simulations of Microelectronic Materials: Prediction of Mechanical, Thermal, and Electrical Properties.” 2012. link Times cited: 0 NOT USED (low confidence) G. Ackland, “1.10 – Interatomic Potential Development.” 2012. link Times cited: 10 NOT USED (low confidence) P. Khalatur, “Molecular Dynamics Simulations in Polymer Science: Methods and Main Results.” 2012. link Times cited: 28 NOT USED (low confidence) S. Seto, N. Arai, and K. Shintani, “Atomistic Study of the Morphology of Graphene on Si and SiC Substrates,” MRS Proceedings. 2012. link Times cited: 1 Abstract: The morphology of graphene on Si and SiC substrates is inves… read moreAbstract: The morphology of graphene on Si and SiC substrates is investigated using molecular- dynamics simulation. The effects of the size and orientation of graphene on its roughness, distance from the substrate, and periodic structure are examined. The roughness and distance show the size dependency which agrees with the size dependency of the ratio of the periphery length of graphene to its area. It is found there are some cases in which the roughness of graphene can be suppressed. read less NOT USED (low confidence) H. Lan, Y. Wang, and C. Liu, “Simulations of structures of amorphous SixC1−x films,” Applied Surface Science. 2012. link Times cited: 0 NOT USED (low confidence) A. Chroneos, “Appendix – Atomic-scale computer simulation of functional materials: methodologies and applications.” 2012. link Times cited: 0 NOT USED (low confidence) K. Bongsang et al., “Phonon manipulation with phononic crystals.” 2012. link Times cited: 20 Abstract: In this work, we demonstrated engineered modification of pro… read moreAbstract: In this work, we demonstrated engineered modification of propagation of thermal phonons, i.e. at THz frequencies, using phononic crystals. This work combined theoretical work at Sandia National Laboratories, the University of New Mexico, the University of Colorado Boulder, and Carnegie Mellon University; the MESA fabrication facilities at Sandia; and the microfabrication facilities at UNM to produce world-leading control of phonon propagation in silicon at frequencies up to 3 THz. These efforts culminated in a dramatic reduction in the thermal conductivity of silicon using phononic crystals by a factor of almost 30 as compared with the bulk value, and about 6 as compared with an unpatterned slab of the same thickness. This work represents a revolutionary advance in the engineering of thermoelectric materials for optimal, high-ZT performance. We have demonstrated the significant reduction of the thermal conductivity of silicon using phononic crystal structuring using MEMS-compatible fabrication techniques and in a planar platform that is amenable to integration with typical microelectronic systems. The measured reduction in thermal conductivity as compared to bulk silicon was about a factor of 20 in the cross-plane direction [26], and a factor of 6 in the in-plane direction. Since the electrical conductivity was only reduced by a corresponding factor of about 3 due to the removal of conductive material (i.e., porosity), and the Seebeck coefficient should remain constant as an intrinsic material property, this corresponds to an effective enhancement in ZT by a factor of 2. Given the number of papers in literature devoted to only a small, incremental change in ZT, the ability to boost the ZT of a material by a factor of 2 simply by reducing thermal conductivity is groundbreaking. The results in this work were obtained using silicon, a material that has benefitted from enormous interest in the microelectronics industry and that has a fairly large thermoelectric power factor. In addition, the techniques and scientific understanding developed in the research can be applied to a wide range of materials, with the caveat that the thermal conductivity of such a material be dominated by phonon, rather than electron, transport. In particular, this includes several thermoelectric materials with attractive properties at elevated temperatures (i.e., greater than room temperature), such as silicon germanium and silicon carbide. It is reasonable that phononic crystal patterning could be used for high-temperature thermoelectric devices using such materials, with applications in energy scavenging via waste-heat recovery and thermoelectric cooling for high-performance microelectronic circuits. The only part of the ZT picture missing in this work was the experimental measurement of the Seebeck coefficient of our phononic crystal devices. While a first-order approximation indicates that the Seebeck coefficient should not change significantly from that of bulk silicon, we were not able to actually verify this assumption within the timeframe of the project. Additionally, with regards to future high-temperature applications of this technology, we plan to measure the thermal conductivity reduction factor of our phononic crystals as elevated temperatures to confirm that it does not diminish, given that the nominal thermal conductivity of most semiconductors, including silicon, decreases with temperature above room temperature. We hope to have the opportunity to address these concerns and further advance the state-of-the-art of thermoelectric materials in future projects. read less NOT USED (low confidence) H. Hara, Y. Kato, G. Ichinose, and S. Irle, “QM/MD Simulations of High-Temperature SWCNT Self-capping.” 2012. link Times cited: 0 NOT USED (low confidence) S. Irle et al., “Atomistic Mechanism of Carbon Nanostructure Self-Assembly as Predicted by Nonequilibrium QM/MD Simulations.” 2012. link Times cited: 18 NOT USED (low confidence) C. Zhu, X. Liu, X.-lan Yu, N. Zhao, J. Liu, and J. Xu, “A small-angle X-ray scattering study and molecular dynamics simulation of microvoid evolution during the tensile deformation of carbon fibers,” Carbon. 2012. link Times cited: 65 NOT USED (low confidence) Y. Wang and X. Ruan, “Necessary conditions for thermal rectification and negative differential thermal conductance in graphene nanoribbons,” MRS Proceedings. 2011. link Times cited: 0 Abstract: We have studied negative differential thermal conductance (N… read moreAbstract: We have studied negative differential thermal conductance (NDTC) and thermal rectification (TR) in graphene nanoribbons (GNRs) using nonequilibrium molecular dynamics simulations. Strong ballistic transport regime and sufficient temperature gradient are found to be necessary conditions for the onset of both NDTC and TR in GNRs, while the latter also requires asymmetry in structure. Preferred direction of heat transport is also discussed for TR. read less NOT USED (low confidence) T. Nakajima and K. Shintani, “Atomistic study of the mechanical stability of multi-layered graphene nanobridges,” MRS Proceedings. 2011. link Times cited: 0 NOT USED (low confidence) E. Hristova, R. Janisch, R. Drautz, and A. Hartmaier, “Solubility of carbon in α-iron under volumetric strain and close to the Σ5(3 1 0)[0 0 1] grain boundary: Comparison of DFT and empirical potential methods,” Computational Materials Science. 2011. link Times cited: 46 NOT USED (low confidence) H. N. Pishkenari and A. Meghdari, “Effects of higher oscillation modes on TM-AFM measurements.,” Ultramicroscopy. 2011. link Times cited: 24 NOT USED (low confidence) C. Lorenz and N. Doltsinis, “Molecular Dynamics Simulation: From ‘Ab Initio’ to ‘Coarse Grained.’” 2011. link Times cited: 12 NOT USED (low confidence) X. Gong et al., “Growth behavior of GaN film along non-polar [11-20] directions,” Physica B-condensed Matter. 2011. link Times cited: 6 NOT USED (low confidence) X. Zhang 张 and Z. Sun 孙, “Effects of vacancy structural defects on the thermal conductivity of silicon thin films,” Journal of Semiconductors. 2011. link Times cited: 0 Abstract: Vacancy structural defect effects on the lattice thermal con… read moreAbstract: Vacancy structural defect effects on the lattice thermal conductivity of silicon thin films have been investigated with non-equilibrium molecular dynamics simulation. The lattice thermal conductivities decrease with increasing vacancy concentration at all temperatures from 300 to 700 K. Vacancy defects decrease the sample thermal conductivity, and the temperature dependence of thermal conductivity becomes less significant as the temperature increases. The molecular dynamics result is in good agreement with the theoretical analysis values obtained based on the Boltzmann equation. In addition, theoretical analysis indicates that the reduction in the lattice thermal conductivity with vacancy defects can be explained by the enhanced point-defect scattering due to lattice strain. read less NOT USED (low confidence) P. Cheng, J. Huang, and G. Zeng, “Applied Mathematics and Mechanics.” 2010. link Times cited: 37 Abstract: From the potential theorem, the fundamental boundary eigenpr… read moreAbstract: From the potential theorem, the fundamental boundary eigenproblems can be converted into boundary integral equations (BIEs) with the logarithmic singularity. In this paper, mechanical quadrature methods (MQMs) are presented to obtain the eigen- solutions that are used to solve Laplace's equations. The MQMs possess high accuracy and low computation complexity. The convergence and the stability are proved based on Anselone's collective and asymptotical compact theory. An asymptotic expansion with odd powers of the errors is presented. By the h 3 -Richardson extrapolation algorithm (EA), the accuracy order of the approximation can be greatly improved, and an a poste- riori error estimate can be obtained as the self-adaptive algorithms. The efficiency of the algorithm is illustrated by examples. read less NOT USED (low confidence) N. Marks, “Amorphous Carbon and Related Materials.” 2010. link Times cited: 7 NOT USED (low confidence) R. W. Nunes and J. F. Justo, “Silicon Nanowires: From Empirical to First Principles Modeling.” 2010. link Times cited: 0 NOT USED (low confidence) M. Ganchenkova and R. Nieminen, “Chapter Eleven – Mechanical Properties of Silicon Microstructures.” 2010. link Times cited: 3 NOT USED (low confidence) S. Sinnott, S. Heo, D. Brenner, J. Harrison, and D. Irving, “Computer Simulations of Nanometer-Scale Indentation and Friction.” 2010. link Times cited: 18 NOT USED (low confidence) X. Blase, G. Benedek, and M. Bernasconi, “Structural, Mechanical, and Superconducting Properties of Clathrates.” 2010. link Times cited: 9 NOT USED (low confidence) Y. Sun and K. Liew, “Multiscale Modeling of Carbon Nanotubes.” 2010. link Times cited: 2 NOT USED (low confidence) L. Zhang and C. Wang, “Effective Wall Thickness of Single-Walled Carbon Nanotubes for Multi-Scale Analysis: The Problem and a Possible Solution.” 2009. link Times cited: 6 NOT USED (low confidence) L. Rui, H. Yuan-zhong, and W. Hui, “Molecular Dynamics Study on Carbon Nanotubes Sandwiched between Si Surface.” 2009. link Times cited: 0 NOT USED (low confidence) R. Latour, “Molecular Simulation of Protein-Surface Interactions.” 2009. link Times cited: 12 NOT USED (low confidence) J. Wu, K. Hwang, and Y. Huang, “A shell theory for carbon nanotubes based on the interatomic potential and atomic structure,” Advances in Applied Mechanics. 2009. link Times cited: 12 NOT USED (low confidence) F. Gao, “Box 4: Interatomic Potential.” 2009. link Times cited: 0 NOT USED (low confidence) H. Magoariec and A. Danescu, “Macroscopic Elasticity of Nanoporous Silicon: Bulk and Surface Effects.” 2009. link Times cited: 1 NOT USED (low confidence) M. Luo, “Surface-induced size-dependent Young’s modulus in nanomaterials.” 2008. link Times cited: 0 Abstract: Nanowires and ultra-thin films have wide applications in the… read moreAbstract: Nanowires and ultra-thin films have wide applications in the quickly developed nanotechnology and nanoscience. However, their Young’s modulus varies with the size, which is seemingly contradictory to the conventional continuum elasticity. Investigating and understanding the underlying mechanism of the size-dependent elastic properties in nanomaterials is of both academic and practical significance. In this work, both theoretical modeling and virtual experiments have been made on this issue. A nanoelement, from the traction free bulk lattice, undergoes an initial relaxation, during which its morphology changes and energy reduces, which is an emphasis in this developed methodology and is a distinction from almost other existing models. With different definitions of surfaces and edges, two models for a nanomaterial – a nanowire or an ultra-thin film – are derived based on the same thermodynamics framework. Comparing with most of others’ treatment, Model I has an advantage to mathematically treat a surface phase to be two-dimensional and an edge phase to be one-dimensional. Under external loadings, the initial relaxed state is taken as the reference. Experimentally, relaxation and tension/compression tests in different loading directions have been conducted on SiC, Si and Cu crystalline nanowires with different cross-sectional sizes and ultra-thin films with different thicknesses by Molecular Dynamics (MD) simulations. This systematic study successfully illustrates the intrinsic mechanism of the size-dependent Young’s modulus in nanomaterials and the proposed methodology facilitate characterizing mechanical properties of nanomaterials to some extent when continuum concepts, such as, surface energy and surface elastic constants, are used. read less NOT USED (low confidence) J. C. Yoon and S. Dunham, “Atomistic Simulations of Epitaxial Regrowth of As-doped Silicon,” MRS Proceedings. 2008. link Times cited: 1 Abstract: We conducted molecular dynamics (MD) simulations of solid ph… read moreAbstract: We conducted molecular dynamics (MD) simulations of solid phase epitaxial growth of As-doped Si using a Tersoff potential characterized via comparison to DFT calculations, including energies of As n V clusters. The Si:As systems were initialized by amorphizing the surface region of crystalline silicon via Si ion implantation and/or selective melting. The remaining crystalline region provides dual function of controlling temperature in system without perturbing regrowth and providing seed for recrystallization. After recrystallization, isolated As atoms occupy substitutional sites, with the average number of nearest neighbors for As changing from about 3.3 in amorphous Si to 4 after crystallization. We observe V incorporation associated with high As concentrations. A small fraction of isolated As atoms have associated vacancies, while vacancies are incorporated in the majority of cases in which there are sites with two As neighbors. These observations are consistent with our previous model developed to explain kinetics of As shallow junction formation which assumed V incorporation at sites with 2 or more As nearest neighbors to account for experimental data. read less NOT USED (low confidence) J. Wu, K. Hwang, and Y. Huang, “An atomistic-based finite-deformation shell theory for single-wall carbon nanotubes,” Journal of The Mechanics and Physics of Solids. 2008. link Times cited: 126 NOT USED (low confidence) R. Sahara, H. Mizuseki, K. Ohno, and Y. Kawazoe, “Thermodynamic Properties of Materials Using Lattice-Gas Models with Renormalized Potentials.” 2008. link Times cited: 2 NOT USED (low confidence) H.-jun Shen, “Compressive and tensile properties of Ar filled carbon nano-peapods,” Materials Letters. 2007. link Times cited: 10 NOT USED (low confidence) A. Bagaturyants, M. A. Deminskii, A. Knizhnik, B. Potapkin, and S. Umanskii, “Chapter 9 Integrated Approach to Dielectric Film Growth Modeling: Growth Mechanisms and Kinetics.” 2007. link Times cited: 2 NOT USED (low confidence) M. Yasuda, T. Majima, Y. Kimoto, K. Tada, H. Kawata, and Y. Hirai, “Molecular Dynamics Study of Electron Irradiation Damages in Carbon Nanomaterials,” MRS Proceedings. 2007. link Times cited: 1 Abstract: Molecular dynamics (MD) studies are carried out to investiga… read moreAbstract: Molecular dynamics (MD) studies are carried out to investigate the electron irradiation damages in carbon nanomaterials. The interaction between an incident electron and a carbon atom is modeled based on the Monte Carlo method using the elastic scattering cross section. The electron irradiation damages in graphen, graphite, single-walled carbon nanotube (SWNT) and carbon nanopeapod are demonstrated. The cross-links among the nanostructures caused by the knock-on effect are observed as typical damages. The dependence of the damages on the electron primary energy is also shown for the SWNT. read less NOT USED (low confidence) F. Gou, M. Gleeson, and A. Kleyn, “CF interaction with Si(1 0 0)-(2 × 1): Molecular dynamics simulation,” Surface Science. 2007. link Times cited: 3 NOT USED (low confidence) J. Schall, P. Mikulski, G. M. Chateauneuf, G. Gao, and J. Harrison, “Molecular Dynamics Simulations of Tribology.” 2007. link Times cited: 8 NOT USED (low confidence) T. Yamamoto, K. Watanabe, and E. Hernández, “Mechanical Properties, Thermal Stability and Heat Transport in Carbon Nanotubes.” 2007. link Times cited: 42 NOT USED (low confidence) E. Ivanova, A. Krivtsov, and N. Morozov, “Derivation of macroscopic relations of the elasticity of complex crystal lattices taking into account the moment interactions at the microlevel,” Journal of Applied Mathematics and Mechanics. 2007. link Times cited: 41 NOT USED (low confidence) L. Zhang, “Stability analysis of atomic structures.” 2006. link Times cited: 2 Abstract: The stability and failure mechanism of a structure at the na… read moreAbstract: The stability and failure mechanism of a structure at the nanometer scale are important for understanding the mechanical behavior of nanoscale materials and structures. This thesis focuses on the material stability of atomic structures. First, the material stability of pristine carbon nanotubes is investigated at the continuum level by using the crystal elasticity theory. A homogenized continuum model is adopted. The strong ellipticity condition is employed to capture the localized failure of carbon nanotubes. The critical strain and strength predicted are reasonably comparable with experimental estimations. An atomic material stability theory is developed as the atomistic counterpart of the continuum material stability theory in nonlinear elasticity. A local instability indicator named ``atomic acoustic tensor'' is derived and utilized to detect material failure at the atomic scale. The stability criterion is based directly on the local energetic responses of an atomic site, and resorts to neither the continuum theory nor the pristine lattice. Thus, it is applicable to inhomogeneous atomic systems provided that the site energy can be reasonably defined. The atomic stability theory is combined with atomistic simulation to gain understanding on crack propagation and fracture as instabilities of bond structures. The atomic acoustic tensor is used as the indicator to detect the local instability at the crack tip, and then to decide bond breaking. Quasi-static crack growth till fracture is simulated by the atomistic finite element method, which is proposed according to the form of bond potential and lattice topology. An Eshelby-type approximate method is presented for calculating the formation energy of Stone-Wales defects. A formula is derived to show that the energy variation consists of the change of local atomic potential due to bond reconfiguration in the defective region and a higher order correction that represents the influence of the read less NOT USED (low confidence) G. Wu, X. Kong, Z.-wei Sun, and D. Zhao, “The Molecular Dynamics Simulation of Monocrystal Carbon, Silicon and Germanium Thermal Conductivity,” 2006 1st IEEE International Conference on Nano/Micro Engineered and Molecular Systems. 2006. link Times cited: 0 Abstract: In this paper the thermal conductivities of monocrystal carb… read moreAbstract: In this paper the thermal conductivities of monocrystal carbon, silicon, and germanium nanometer thin film are simulated respectively using non-equilibrium molecular dynamics (NEMD) method and corresponding Tersoff potential energy function. The simulation results indicate that the thermal conductivities of those nanometer thin films are obviously lower than the corresponding thermal conductivities of their bulk crystals under the same temperature. The thermal conductivities increase with the increasing of thin film thickness, and the conductivities have an approximately linear relationship with thickness of the thin films. The curve slope of carbon thermal conductivity is larger than that of silicon and germanium. The calculation results of thermal conductivities demonstrate distinct size effect. In normal direction, the thin film thermal conductivities of carbon, silicon and germanium crystals decline with the increasing of temperature, and the declining degree steps down in the sequence of carbon, silicon and germanium read less NOT USED (low confidence) T. Kumagai, S. Hara, S. Izumi, and S. Sakai, “Development of a Bond-Order Potential that can Reproduce the Elastic Constants and Melting Point of Silicon,” Journal of The Society of Materials Science, Japan. 2006. link Times cited: 1 Abstract: The Tersoff potential is one of the most widely used interat… read moreAbstract: The Tersoff potential is one of the most widely used interatomic potentials for silicon. However, its poor descrip-tion of the elastic constants and melting point of diamond silicon is well known. In this research, a new bond-order type interatomic potential has been developed that can reproduce the elastic constants and melting point of diamond silicon as well as the cohesive energies and equilibrium bond lengths of polytypes of silicon. We improved the original Tersoff potential function through the introduction of a flexible angular dependent term. In order to increase the robustness of the potential, systems that include a wide range of local atomic environments are employed for fitting. Optimized potential parameters were found using a genetic algorithm. The elastic constants and melting point of diamond silicon calculated using the developed potential turned out to be C 11 = 166.4GPa, C 12 = 65.3GPa, C 44 = 77.1GPa and T m = 1681K. It was also found that only elastic constants can be reproduced using the original Tersoff potential function, and that our proposed angular dependent term is a key to reproducing the melting point. read less NOT USED (low confidence) G. Betz, “Interaction of Ions and Electrons with Solid Surfaces.” 2006. link Times cited: 1 NOT USED (low confidence) C. Lang, D. Nguyen-Manh, and D. Cockayne, “Modelling Ge/Si quantum dots using finite element analysis and atomistic simulation.” 2006. link Times cited: 4 Abstract: Finite Element Analysis (FEA) and atomistic simulations are … read moreAbstract: Finite Element Analysis (FEA) and atomistic simulations are used to model Ge/Si quantum dots. The three dimensional non-uniform composition profile in Ge(Si)/Si(001) quantum dots is calculated using atomistic modelling. The results are compared to experimental data from the literature. FEA is used to model the contact angle dependence of the strain energy of the QD. An equation is fitted to the modelled dataset describing the strain energy of a uniformly alloyed, pyramid shaped Ge/Si quantum dot as a function of the contact angle. read less NOT USED (low confidence) W. Andreoni, A. Curioni, D. Fischer, S. Billeter, and C. Pignedoli, “STUDYING THE EFFECTS OF NITROGEN AND HAFNIUM INCORPORATION INTO THE SIO2/SI(100) INTERFACE WITH REPLICA-EXCHANGE MOLECULAR DYNAMICS AND DENSITYFUNCTIONAL- THEORY CALCULATIONS.” 2006. link Times cited: 0 NOT USED (low confidence) D. R. Chilla, “Numerical studies on elastic properties of graphene sheets and free vibration of carbon nanotubes by atomistic simulation and equivalent continuum modelling.” 2006. link Times cited: 0 Abstract: The present research deals with determination of elastic pro… read moreAbstract: The present research deals with determination of elastic properties of finite graphene sheets and CNTs by equivalent continuum modelling, and free vibration analysis of plain as well as fluid conveying CNTs by atomistic modelling of stiffness and mass properties. read less NOT USED (low confidence) M. Buehler, Y. Kong, H. Gao, and Y. Huang, “Self-folding and unfolding of carbon nanotubes,” Journal of Engineering Materials and Technology-transactions of The Asme. 2006. link Times cited: 43 Abstract: Carbon nanotubes (CNTs) constitute a prominent example of na… read moreAbstract: Carbon nanotubes (CNTs) constitute a prominent example of nanomaterials. In most studies on mechanical properties, the effort was concentrated on CNTs with relatively small aspect ratio of length to diameters. In contrast, CNTs with aspect ratios of several hundred can be produced with today's experimental techniques. We report atomistic-continuum studies of single-wall carbon nanotubes with very large aspect ratios subject to compressive loading. It was recently shown that these long tubes display significantly different mechanical behavior than tubes with smaller aspect ratios (Buehler, M. J., Kong, Y., and Guo, H., 2004, ASME J. Eng. Mater. Technol. 126, pp. 245-249). We distinguish three different classes of mechanical response to compressive loading. While the deformation mechanism is characterized by buckling of thin shells in nanotubes with small aspect ratios, it is replaced by a rodlike buckling mode above a critical aspect ratio, analogous to the Euler theory in continuum mechanics. For very large aspect ratios, a nanotube is found to behave like a wire that can be deformed in a very flexible manner to various shapes. In this paper, we focus on the properties of such wirelike CNTs. Using atomistic simulations carried out over a several-nanoseconds time span, we observe that wirelike CNTs behave similarly to flexible macromolecules. Our modeling reveals that they can form thermodynamically stable self-folded structures, where different parts of the CNTs attract each other through weak van der Waals (vdW) forces. This self-folded CNT represents a novel structure not described in the literature. There exists a critical length for self-folding of CNTs that depends on the elastic properties of the tube. We observe that CNTs fold below a critical temperature and unfold above another critical temperature. Surprisingly, we observe that self-folded CNTs with very large aspect ratios never unfold until they evaporate. The folding-unfolding transition can be explained by entropic driving forces that dominate over the elastic energy at elevated temperature. These mechanisms are reminiscent of the dynamics of biomolecules, such as proteins. The different stable states of CNTs are finally summarized in a schematic phase diagram of CNTs. read less NOT USED (low confidence) C. Anderson and K. Tamma, “Introduction to nanoscale, microscale, and macroscale heat transport: Characterization and bridging of space and time scales.” 2005. link Times cited: 15 NOT USED (low confidence) B. Holian, T. Germann, A. Strachan, and J. Maillet, “Non-Equilibrium Molecular Dynamics Studies of Shock and Detonation Processes in Energetic Materials.” 2005. link Times cited: 2 NOT USED (low confidence) M. Elert, S. Zybin, and C. T. White, “Shock-Induced Chemistry in Hydrocarbon Molecular Solids,” ChemInform. 2005. link Times cited: 1 NOT USED (low confidence) J. F. Justo, “Modeling Covalent Bond with Interatomic Potentials.” 2005. link Times cited: 1 NOT USED (low confidence) S. Irle, G. Zheng, M. Elstner, and K. Morokuma, “High-temperature quantum chemical molecular dynamics simulations of carbon nanostructure self-assembly processes.” 2005. link Times cited: 3 NOT USED (low confidence) Y. Mishin, “Atomistic Computer Simulation of Diffusion.” 2005. link Times cited: 7 NOT USED (low confidence) C. Wong, “Numerical simulation and modeling of carbon nanotubes.” 2005. link Times cited: 0 NOT USED (low confidence) B. Thijsse, “Silicon potentials under (ion) attack: towards a new MEAM model,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 24 NOT USED (low confidence) M. Biehl and F. Much, “Quantum Dots: Fundamentals, Applications, and Frontiers.” 2005. link Times cited: 52 NOT USED (low confidence) C. Anderson and K. Tamma, “An overview of advances in heat conduction models and approaches for prediction of thermal conductivity in thin dielectric films,” International Journal of Numerical Methods for Heat & Fluid Flow. 2004. link Times cited: 29 Abstract: We first provide an overview of some predominant theoretical… read moreAbstract: We first provide an overview of some predominant theoretical methods currently used for predicting thermal conductivity of thin dielectric films: the equation of radiative transfer, the temperature‐dependent thermal conductivity theories based on the Callaway model, and the molecular dynamics simulation. This overview also highlights temporal and spatial scale issues by looking at a unified theory that bridges physical issues presented in the Fourier and Cattaneo models. This newly developed unified theory is the so‐called C‐ and F‐processes constitutive model. This model introduces the notion of a new dimensionless heat conduction model number, which is the ratio of the thermal conductivity of the fast heat carrier F‐processes to the total thermal conductivity comprised of both the fast heat carriers F‐processes and the slow heat carriers C‐processes. Illustrative numerical examples for prediction of thermal conductivity in thin films are primarily presented. read less NOT USED (low confidence) T. Natsuki, K. Tantrakarn, and M. Endo, “Prediction of elastic properties for single-walled carbon nanotubes,” Carbon. 2004. link Times cited: 171 NOT USED (low confidence) U. Kaiser, “Characterization of Low-Dimensional Structures in SiC Using Advanced Transmission Electron Microscopy.” 2004. link Times cited: 0 NOT USED (low confidence) P. Kratzer, “Atomistic Simulations of Processes at Surfaces.” 2004. link Times cited: 1 NOT USED (low confidence) S. Izumi, S. Hara, T. Kumagai, and S. Sakai, “Elastic Properties of the Surfaces and Interfaces of Crystal and Amorphous Silicon.” 2004. link Times cited: 0 NOT USED (low confidence) E. Aydil, S. Agarwal, M. S. Valipa, S. Sriraman, and D. Maroudas, “Surface processes during growth of hydrogenated amorphous silicon,” MRS Proceedings. 2004. link Times cited: 1 NOT USED (low confidence) V. Harik and M. Salas, “Trends in Nanoscale Mechanics.” 2003. link Times cited: 16 NOT USED (low confidence) 변기량, 강정원, L. J. Ha, K. O. Kuen, and HoJungHwang, “Torsion of Hypothetical Single-Wall Silicon Nanotubes,” Journal of The Korean Institute of Electrical and Electronic Material Engineers. 2003. link Times cited: 0 NOT USED (low confidence) É. Cancès, M. Defranceschi, W. Kutzelnigg, C. Bris, and Y. Maday, “Computational quantum chemistry: A primer,” Handbook of Numerical Analysis. 2003. link Times cited: 159 NOT USED (low confidence) L. Zhou and H.-C. Huang, “Young’s Modulus Variation with Thickness of Thin Films,” MRS Proceedings. 2003. link Times cited: 1 Abstract: This paper describes atomistic determinations of the Young&a… read moreAbstract: This paper describes atomistic determinations of the Young's modulus of free standing thin films, or nanoplates. Using a combination of analytical formulation and molecular statics simulations, we show that the Young's modulus of a nanoplate may either increase or decrease with the thickness. It is the competition of bond saturation and bond loss on surfaces that dictates the increase or decrease. Taking Cu as an example, we demonstrate that the Young's modulus is larger than its bulk counterparts for nanoplates having some surfaces and loading directions, and smaller for others. read less NOT USED (low confidence) B. Yakobson and T. Dumitricǎ, “Nanomechanics: Physics between Engineering and Chemistry.” 2003. link Times cited: 2 NOT USED (low confidence) K. Nordlund, J. Nord, A. Krasheninnikov, and K. Albe, “Atomic-scale simulations of radiation effects in GaN and carbon nanotubes,” MRS Proceedings. 2003. link Times cited: 0 Abstract: Gallium nitride and carbon nanotubes have received wide inte… read moreAbstract: Gallium nitride and carbon nanotubes have received wide interest in the materials research community since the mid-1990's. The former material is already in use in optoelectronics applications, while the latter is considered to be extremely promising in a wide range of materials. Common to both materials is that ion irradiation may be useful for modifying their properties. In this paper we overview our recent molecular dynamics simulations results on ion irradiation of these materials. We employ such potentials to study the basic physics of how ion irradiation affects these materials. In particular we discuss the reasons for the high radiation hardness of GaN, and the surprising nature of vacancies and interstitials in carbon nanotubes read less NOT USED (low confidence) E. Blank, “Chapter 2 Structural imperfections in CVD diamond films,” Semiconductors and Semimetals. 2003. link Times cited: 8 NOT USED (low confidence) L. Nurminen, F. Tavazza, D. Landau, A. Kuronen, and K. Kaski, “Monte Carlo Simulation of the Surface Structure of Ge on Si(00l).” 2003. link Times cited: 1 NOT USED (low confidence) C. M. Gilmore and J. Sprague, “Molecular Dynamics Simulation of Thin Film Growth with Energetic Atoms.” 2002. link Times cited: 2 NOT USED (low confidence) K. Esfarjani, A. Farajian, Y. Hashi, and Y. Kawazoe, “Electronic, Transport and Mechanical Properties of Carbon Nanotubes.” 2002. link Times cited: 8 NOT USED (low confidence) S. Silva, J. D. Carey, R. Khan, E. Gerstner, and J. Anguita, “Amorphous carbon thin films.” 2002. link Times cited: 32 NOT USED (low confidence) J. Chen, P. Ruterana, and G. Nouet, “Theoretical analysis of tilt grain boundaries in GaN at the atomic scale,” MRS Proceedings. 2002. link Times cited: 0 NOT USED (low confidence) T. Inamura, N. Takezawa, and S. Shimada, “Importance of Micro/Macro Interaction in the Mechanism of Brittle Mode Cutting,” CIRP Annals. 2002. link Times cited: 8 NOT USED (low confidence) A. Kuronen, K. Henriksson, and K. Kaski, “Adatom Diffusion on Strained Si(001)-(2×1) Surface,” MRS Proceedings. 2002. link Times cited: 0 NOT USED (low confidence) Y. H. Kim, H. Sim, and K. Chang, “Electronic structure of collapsed C, BN, and BC3 nanotubes,” Current Applied Physics. 2001. link Times cited: 20 NOT USED (low confidence) E. Aydil et al., “In situ probing and atomistic simulation of a-Si:H plasma deposition,” MRS Proceedings. 2001. link Times cited: 14 Abstract: Hydrogenated amorphous silicon thin films deposited from SiH… read moreAbstract: Hydrogenated amorphous silicon thin films deposited from SiH4 containing plasmas are used in solar cells and thin film transistors for flat panel displays. Understanding the fundamental microscopic surface processes that lead to Si deposition and H incorporation is important for controlling the film properties. An in situ method based on attenuated total internal reflection Fourier transform infrared (ATR-FTIR) spectroscopy was developed and used to determine the surface coverage of silicon mono-, di-, and tri-hydrides as a function of deposition temperature and ion bombardment flux. Key reactions that take place on the surface during deposition are hypothesized based on the evolution of the surface hydride composition as a function of temperature and ion flux. In conjunction with the experiments, the growth of a-Si:H on H-terminated Si(001)-(2×1) surfaces was simulated through molecular dynamics. The simulation results were compared with experimental measurements to validate the simulations and to provide supporting evidence for radical-surface interaction mechanisms hypothesized based on the infrared spectroscopy data. Experimental measurements of the surface silicon hydride coverage and atomistic simulations are used synergistically to elucidate elementary processes occurring on the surface during a-Si:H deposition. read less NOT USED (low confidence) R. Wagner and E. Gulari, “Simulation of Mechanisms in Strained Silicon Germanium Epitaxy,” MRS Proceedings. 2001. link Times cited: 0 Abstract: Growth of strained semiconductors can lead to self-assembly … read moreAbstract: Growth of strained semiconductors can lead to self-assembly of interesting structures such as quantum dots. Simulation of this growth requires an accurate and efficient model for the interactions of lattice mismatched materials. We present a potential for calculating the bond energies in a diamond-like crystal of silicon and germanium. With this potential we predict the strain profiles in an embedded quantum dot and discuss the mechanisms of island formation. read less NOT USED (low confidence) C. Wei, K. Cho, and D. Srivastava, “Chemical Bonding of Polymer on Carbon Nanotube,” MRS Proceedings. 2001. link Times cited: 13 Abstract: : Recently, carbon nanotubes are considered as nanoscale fib… read moreAbstract: : Recently, carbon nanotubes are considered as nanoscale fibers, which can strengthen polymer composite materials. Nanotube-polymer composite materials can be used for micron scale devices with designed mechanical properties and smart polymer coating to protect materials under extreme physical conditions such as microsatellites. To explore these possibilities it is important to develop a detailed atomic scale understanding of the mechanical coupling between polymer matrix and embedded nanotubes. In this work we study the chemical bonding between polymer molecules and carbon nanotubes (CNTs) using molecular dynamics. Study shows that the bonding between polyethylene and a CNT is energetically favorable. Chemical bonds can be formed at multiple sites, which make the mechanical load transfer from the polymer chain to the tube more favorable. We will discuss about the resulting mechanical coupling between the CNTs and polymer matrix to develop efficient nano-composite materials. read less NOT USED (low confidence) T. Raz and R. Levine, “Essentials of Cluster Impact Chemistry.” 2001. link Times cited: 3 NOT USED (low confidence) D. Maroudas, “Modeling of radical-surface interactions in the plasma-enhanced chemical vapor deposition of silicon thin films,” Advances in Chemical Engineering. 2001. link Times cited: 33 NOT USED (low confidence) D. Tománek, “Thermal and Electrical Conductance of Carbon Nanostructures.” 2001. link Times cited: 0 NOT USED (low confidence) C. Wei and K. Cho, “Temperature and Strain-Rate Dependent Plastic Deformation of Carbon Nanotube,” MRS Proceedings. 2001. link Times cited: 4 Abstract: In this work we use classical molecular dynamics to study st… read moreAbstract: In this work we use classical molecular dynamics to study strain rate and temperature dependent plasticity of carbon nanotube (CNT) under compressive strain. We focus on two types of defects: sp3 bond formation and bond rotation. Our simulation shows that thermal fluctuations help the strained CNT to overcome the local energy barrier to obtain plastic deformation. The yielding strain of a compressed CNT found to be strain-rate and temperature dependent, and low strain rate limit of the yielding strain is estimated to be less that 6%. read less NOT USED (low confidence) T. Ito, “Atomistic simulation of epitaxial growth processes.” 2001. link Times cited: 0 NOT USED (low confidence) M. Machado, P. Piquini, R. Mota, and A. Fazzio, “Electronic and Structural Properties of Carbon Nanotubes Molecular Junction,” MRS Proceedings. 2000. link Times cited: 0 NOT USED (low confidence) M. Nardelli, J. Fattebert, D. Orlikowski, C. Roland, Q. Zhao, and J. Bernholc, “Mechanical properties, defects and electronic behavior of carbon nanotubes,” Carbon. 2000. link Times cited: 166 NOT USED (low confidence) N. H. March, “Electron theory related to mechanical properties of condensed phases,” International Journal of Quantum Chemistry. 2000. link Times cited: 0 Abstract: In this article, some recent work is surveyed on mechanical … read moreAbstract: In this article, some recent work is surveyed on mechanical properties of both metals and semiconducting silicon. In quantum chemical language, such properties as (1) cleavage forces and (2) tribological properties involve bond stretching, and here it is argued that electron correlation must play an important role. In the context of long-range dispersion forces, this is already evident in first-principles work of Lifshitz, which, particularly in simple metals, leads to an asymptotic form of cleavage force F(z), with z the interplanar separation beyond equilibrium spacing on cleavage, falling off as z{sup {minus}3}, the magnitude of this term being expressible in terms of the electron plasma frequency. Here, it is argued, from quantum chemistry, that in covalently binded systems, such as semiconducting Si, the elasticity of a covalent bond in the crystal is quite comparable with that of the H{sub 2} molecule bonding in free space. The transition from delocalized molecular-orbital theory to Heitler-London or valence bond regimes comes about as the bonds are stretched to breaking point. This situation with covalent bonds is compared and contrasted with that of a largely nondirectionally bonded metal such as Cu, in which there is a closed d shell, and a more complicated body-centered-cubicmore » Fe, in which the charge density can be demonstrated experimentally, through overlapping reflections in X-ray diffraction, to have marked directionally. Even for simple sp metals, it is shown that some of the main features of the heavier alkalis, and in particular K, Rb, and Cs, can be explained by viewing the dimer, say K{sub 2}, as the basic building block in representing the energy of the crystal of K as a function of lattice spacing and local coordination number. Finally, a brief discussion of liquid alkalis is given, mechanical properties now embracing viscosities.« less read less NOT USED (low confidence) M. Ali and A. Törn, “Optimization of Carbon and Silicon Cluster Geometry for Tersoff Potential using Differential Evolution.” 2000. link Times cited: 23 NOT USED (low confidence) H. Jónsson, “Theoretical studies of atomic-scale processes relevant to crystal growth.,” Annual review of physical chemistry. 2000. link Times cited: 102 Abstract: The study of adsorption, diffusion, island formation, and in… read moreAbstract: The study of adsorption, diffusion, island formation, and interlayer transport of atoms on a growing surface has been an active field in the past decade, because of both experimental and theoretical advances. Experiments can give detailed images of patterns formed on growing surfaces. An important challenge to the theoretical studies is the identification of dynamical processes controlling the pattern formation and overall surface morphology. This can be achieved by accurate modeling of the atomic interactions, a thorough search for active atomic-scale processes, and simulation of the growth on the experimental timescale to allow for detailed comparison with the experimental measurements. An overview of some of the theoretical methodology used in these studies and results obtained for one of the most extensively studied systems, Pt(111), is given here. A remarkable richness of phenomena has emerged from these studies, where apparently small effects can shift the balance between competing molecular processes and thereby change the morphology of a growing surface. The article concludes with a discussion of possible future directions in this research area. read less NOT USED (low confidence) T. Motooka, S. Munetoh, K. Nisihira, K. Moriguchi, and A. Shintani, “Molecular Dynamics Simulations of Solid Phase Epitaxy of Si:Growth Mechanism and Defect Formation,” MRS Proceedings. 1999. link Times cited: 0 NOT USED (low confidence) N. H. March, “Many-electron theory: Density functional approach generalized to treat spin eigenfunctions and relation to spinless low-order density matrices,” International Journal of Quantum Chemistry. 1999. link Times cited: 2 NOT USED (low confidence) T. Inamura, S. Shimada, N. Takezawa, and N. Ikawa, “Crack Initiation in Machining Monocrystalline Silicon,” Cirp Annals-manufacturing Technology. 1999. link Times cited: 44 NOT USED (low confidence) K. Kawamura, “Mineralogy by Means of Molecular Simulations,” Journal of the Mineralogical Society of Japan. 1999. link Times cited: 0 Abstract: We reviewed the computational methods of material sciences, … read moreAbstract: We reviewed the computational methods of material sciences, and described the details of molecular dynamics methods and Metropolis Monte Carlo methods from the view points of the fundamental aspects and the applications to mineral sciences. We emphasize the importance of interatomic and intermolecular interaction models to perform molecular simulation. For the further advanced applications of the current importantploblems in the earth and planetary sciences, we need to understand chemical bondings in minerals and related materials more quantitatively and effectively, and also devel op appropriate procedures for the earth and planetary materials. read less NOT USED (low confidence) F. B. Mota, J. F. Justo, and A. Fazzio, “Structural and electronic properties of silicon nitride materials,” International Journal of Quantum Chemistry. 1998. link Times cited: 20 Abstract: The authors developed an empirical potential for interaction… read moreAbstract: The authors developed an empirical potential for interactions between Si and N to describe silicon nitride systems using the Tersoff functional form. With this model, they explored the structural properties of amorphous silicon nitride through the Monte Carlo simulations and compared them to available experimental data. The empirical model provided a very good description of such properties for a-SiN{sub x} (0 < x < 1.5). Electronic structure of amorphous and point defects in crystalline silicon nitride were then studied using first-principles calculations. For such calculations, the configurations were created by the empirical model, with the relaxed structures used as input for the first-principles calculations. Atomic relaxation was later allowed in the first-principles calculations. read less NOT USED (low confidence) J. Vigneron, A. Benaissa, I. Derycke, A. Wiame, and R. Sporken, “ATOMIC MOTION AT GERMANIUM SURFACES : SCANNING TUNNELING MICROSCOPY AND MONTE CARLO SIMULATIONS,” International Journal of Quantum Chemistry. 1998. link Times cited: 0 Abstract: The observation of the motion of an adatom on the reconstruc… read moreAbstract: The observation of the motion of an adatom on the reconstructed Ge (111) is a rare event, which will be examined by way of simple adatom–surface interaction models. Estimations of the residence time of adatoms on energetically favorable sites indicate that a thermal excitation can account for casual adatom motion and that a strong tip–surface interaction is not obviously needed to explain the changes found in sequences of scanning tunneling microscopy (STM) images of the same surface areas. © 1998 John Wiley & Sons, Inc. Int J Quant Chem 70: 1093–1097, 1998 read less NOT USED (low confidence) A. Omeltchenko, A. Nakano, K. Tsuruta, R. Kalia, and P. Vashishta, “Structure and mechanical failure in nanophase silicon nitride.” 1998. link Times cited: 4 NOT USED (low confidence) D. Robertson, D. Brenner, and C. T. White, “Molecular Dynamics Analysis of Shock Phenomena.” 1998. link Times cited: 8 NOT USED (low confidence) J. F. Justo, M. Bazant, E. Kaxiras, V. Bulatov, and S. Yip, “Interatomic Potential for Condensed Phases and Bulk Defects in Silicon,” MRS Proceedings. 1997. link Times cited: 5 NOT USED (low confidence) H. Terrones and A. Mackay, “From C60 to negatively curved graphite,” Progress in Crystal Growth and Characterization of Materials. 1997. link Times cited: 31 NOT USED (low confidence) V. Rosato and M. Celino, “Tight Binding Simulations of Disordered Systems,” MRS Proceedings. 1997. link Times cited: 0 NOT USED (low confidence) L. Colombo, A. Bongiorno, and T. D. Rubia, “Formation and Binding Energies of Vacancy Clusters in Silicon,” MRS Proceedings. 1997. link Times cited: 3 Abstract: We critically readdress the problem of vacancy clustering in… read moreAbstract: We critically readdress the problem of vacancy clustering in silicon by perform large-scale tight-binding molecular dynamics simulations. We also compare the results of this quantum-mechanical approach to the widely used model-potential molecular dynamics scheme based on the Tersoff and Stillinger-Weber interatomic potentials. read less NOT USED (low confidence) N. Mousseau, “Computer Modelling of Glasses and Glassy Alloys.” 1997. link Times cited: 0 NOT USED (low confidence) J. Bernholc et al., “Real-space multigrid methods for large-scale electronic structure problems,” International Journal of Quantum Chemistry. 1997. link Times cited: 17 Abstract: We describe the development and applications of a new electr… read moreAbstract: We describe the development and applications of a new electronic structure method that uses a real-space grid as a basis. Multigrid techniques provide preconditioning and convergence acceleration at all length scales and therefore lead to particularly efficient algorithms. The salient points of our implementation include: (i) new compact discretization schemes in real space for systems with cubic, orthorhombic, and hexagonal symmetry and (ii) new multilevel algorithms for the iterative solution of Kohn–Sham and Poisson equations. The accuracy of the discretizations was tested by direct comparison with plane-wave calculations, when possible, and the results were in excellent agreement in all cases. These techniques are very suitable for use on massively parallel computers and in O(N) methods. Tests on the Cray-T3D have shown nearly linear scaling of the execution time up to the maximum number of processors (512). The above methodology was tested on a large number of systems, such as the C60 molecule, diamond, Si and GaN supercells, and quantum molecular dynamics simulations for Si. Large-scale applications include a simulation of surface melting of Si and investigations of electronic and structural properties of surfaces, interfaces, and biomolecules. © 1997 John Wiley & Sons, Inc. Int J Quant Chem 65: 531–543, 1997 read less NOT USED (low confidence) S. Ramalingam, D. Maroudas, and E. Aydil, “Atomic-Scale Analysis of the Reactivity of Radicals from Silane/Hydrogen Plasmas with Silicon Surfaces,” MRS Proceedings. 1997. link Times cited: 2 NOT USED (low confidence) N. H. March, “Forces between atoms and atomic planes in condensed metallic phases and in semiconducting silicon,” International Journal of Quantum Chemistry. 1997. link Times cited: 0 NOT USED (low confidence) T. Inamura, S. Shimada, N. Takezawa, and N. Nakahara, “Brittle/Ductile Transition Phenomena Observed in Computer Simulations of Machining Defect-Free Monocrystalline Silicon,” CIRP Annals. 1997. link Times cited: 71 NOT USED (low confidence) G. Krasko, “Energetics of Ideal Grain Boundary Fracture in Iron and the Thermodynamic Criterion of Impurity Embrittlement,” MRS Proceedings. 1996. link Times cited: 3 Abstract: Modeling of grain boundary (GB) relaxation during ideal frac… read moreAbstract: Modeling of grain boundary (GB) relaxation during ideal fracture, and the fracture energetics of a Σ3 (111) GB in Fe was performed using the modified Finnis-Sinclair semi-empirical method, and utilizing the so-called “environment-sensitive embedding energies” of impurity atoms, introduced earlier by the author. The calculations were done for both the clean GB, and GB with the following impurity atoms: H, B, C, N, O, P and S. The ideal fracture was modeled by separating the two halves of crystal normal to the GB, step-wise, minimizing the total crystal energy at every step. The interplanar distances were varied, while the Fe interatomic spacing within the hexagonal planes was held fixed. When the distance between the two crystal halves: one with the impurity and another without, exceeded the interatomic interaction cut-off radius (3.6 A), two different free surfaces - with and without the impurity - emerged. The GB and surface energies were found both for the pure Fe, and that with impurity atoms at the GB or free surface. Both the (111) GB energy and the (111) surface energy of pure Fe agree well with experimental data and results of previous semi-empirical modeling. In general, the correlation between the embrittling/ cohesion enhancing effect of impurities in GB and the difference between the GB and free surface energies agrees with the thermodynamic criterion of embrittlement. read less NOT USED (low confidence) V. Bakaev and W. Steele, “Chapter 2.1 Computer simulation of adsorption on amorphous oxide surfaces,” Studies in Surface Science and Catalysis. 1996. link Times cited: 2 NOT USED (low confidence) W. Neumann, H. Hofmeister, D. Conrad, K. Scheerschmidt, and S. Ruvimov, “Characterization of interface structures in nanocrystalline germanium by means of high-resolution electron microscopy and molecular dynamics simulations,” Zeitschrift Fur Kristallographie. 1996. link Times cited: 5 Abstract: The atomic structure of nanocrystalline particles formed by … read moreAbstract: The atomic structure of nanocrystalline particles formed by vapor deposition and subsequent annealing of amorphous thin films of germanium was studied by high resolution electron microscopy (HREM). The HREM images revealed a strongly varied multiply twinned structure. In some regions of adjacent twins contrast features were detected which were caused by an overlapping of twin lamellae. It will be shown by HREM contrast simulations that these interface types can be described by Σ = 3 n boundaries. The influence of lattice relaxations is taken into consideration by molecular dynamics simulations of the structure models. read less NOT USED (low confidence) T. Frauenheim, D. Porezag, T. Köhler, and F. Weich, “Molecular-Dynamic Simulations of Structure Formation in Complex Materials.” 1996. link Times cited: 1 NOT USED (low confidence) C. Fang, V. Prasad, R. Joshi, F. Jones, and J. Hsieh, “A process model for sputter deposition of thin films using molecular dynamics,” Thin Films. 1996. link Times cited: 4 NOT USED (low confidence) X. Liu, “NEW MODEL OF POTENTIAL-ENERGY FUNCTIONS FOR ATOMIC SOLIDS,” Journal of the Chemical Society, Faraday Transactions. 1995. link Times cited: 2 Abstract: A new theoretical model of potential-energy functions for at… read moreAbstract: A new theoretical model of potential-energy functions for atomic solids has been developed. An angular factor has been included in this model and its effect has been discussed. Using this new model a new preliminary potential for silicon crystal has been derived. The calculated phonon dispersion curve along the [q00] direction, using this new potential, has been given. A good agreement has been found with experiment. read less NOT USED (low confidence) S. Shimada, N. Ikawa, T. Inamura, N. Takezawa, H. Ohmori, and T. Sata, “Brittle-Ductile Transition Phenomena in Microindentation and Micromachining,” CIRP Annals. 1995. link Times cited: 138 NOT USED (low confidence) J. J. C. Barrett, D. Robertson, D. Brenner, and C. T. White, “Simulations of Ozone Detonation Using a Reactive Empirical Bond Order (REBO) Potential for the Oxygen System,” MRS Proceedings. 1995. link Times cited: 1 Abstract: The short length and time scales associated with chemical de… read moreAbstract: The short length and time scales associated with chemical detonations make these processes excellent candidates for study by molecular dynamics (MD) simulation. Potentials used in these simulations must have sufficient flexibility to describe gas-phase properties of isolated reactant and product molecules, high density material generated under shock compression, and allow smooth adjustment of bonding forces during chemical reaction. The REBO formalism has been shown to provide these characteristics and allow the treatment of a sufficient number atoms for sufficiently long times to demonstrate a chemically-sustained shock wave (CSSW). In this paper the authors present a REBO potential describing the oxygen system for use in MD simulation of detonation in an ozone molecular solid. The potential reproduces spectroscopic properties of isolated gas-phase O{sub 2} and O{sub 3}. It also describes an ozone molecular solid with density and speed of sound within physical norms. They observe detonation characteristics that depend on crystallographic orientation in simulations using a three dimensional ozone molecular crystal. read less NOT USED (low confidence) R. P. Messmer, “Computational materials science — a personal perspective of an industrial scientist,” Computational Materials Science. 1994. link Times cited: 6 NOT USED (low confidence) R. Smith and R. Webb, “Atomic collisions in semiconductors,” Radiation Effects and Defects in Solids. 1994. link Times cited: 1 Abstract: Energetic particle bombardment of semi-conductors (Si and Ga… read moreAbstract: Energetic particle bombardment of semi-conductors (Si and GaAs) is studied by means of Molecular Dynamics simulations using many-body potentials. The simulations show that the diamond lattice structures can lead to the trajectories of particles within the crystal being channelled even at low energies. Some results concerning damage production, low energy implantation profiles and angular distributions of ejected particles are presented. read less NOT USED (low confidence) G. Benedek, L. Colombo, B. Corona, E. Galvani, S. Sanguinetti, and S. Serra, “Structure, Stability and Properties of Covalent C 34 , C 20 , and C 22 Crystals,” MRS Proceedings. 1994. link Times cited: 2 NOT USED (low confidence) T. Frauenheim, “Political changes in East Germany with lasting impact on computer simulations of carbon-based materials,” Computational Materials Science. 1994. link Times cited: 1 NOT USED (low confidence) C. Wang, K. Ho, and C. Chan, “Material research with tight-binding molecular dynamics,” Computational Materials Science. 1994. link Times cited: 14 NOT USED (low confidence) M. M. Souza and G. Amaratunga, “Self Diffusion in Silicon Using the Ackland Potential.” 1993. link Times cited: 2 NOT USED (low confidence) G. Gilmer, “CHAPTER 8 – Atomic-scale Models of Crystal Growth.” 1993. link Times cited: 5 NOT USED (low confidence) N. Miskovsky, T. Tsong, and C.-M. Wei, “Atomic Manipulation Using Field Evaporation.” 1993. link Times cited: 0 NOT USED (low confidence) A. Charai and J. Rouviere, “Evidence by Hrem of Interfacial Modification Induced by Equilibrium Segregation in GE(S) Bicrystal,” MRS Proceedings. 1993. link Times cited: 5 NOT USED (low confidence) M. Aoki, P. Gumbsch, and D. Pettifor, “Angular-Dependent Many-Atom Bond-Order Potentials.” 1993. link Times cited: 5 NOT USED (low confidence) R. C. Mowrey, D. Brenner, B. Dunlap, J. Mintmire, and C. T. White, “Molecular-Dynamics Simulations of C 60 /He Collisions.” 1992. link Times cited: 0 NOT USED (low confidence) F. Ercolessi and J. B. Adams, “Interatomic Potentials From First-Principles Calculations,” MRS Proceedings. 1992. link Times cited: 22 Abstract: We propose a new scheme to extract “optimal” interatomic pot… read moreAbstract: We propose a new scheme to extract “optimal” interatomic potentials starting from a large number of atomic configurations (and their forces) obtained from first-principles calculations. The method appears to be able to overcome the difficulties encountered by traditional fitting approaches when using rich and complex analytical forms, and constitute a step forward towards large-scale simulations of condensed matter systems with a degree of accuracy comparable to that obtained by ab initio methods. A first exploratory application to aluminum is presented. read less NOT USED (low confidence) D. Robertson, D. Brenner, and C. T. White, “Delayed Initiation in a Model Energetic Material,” MRS Proceedings. 1992. link Times cited: 2 NOT USED (low confidence) T. Okada, S. Kambayashi, M. Yabuki, Y. Tsunashima, Y. Mirata, and S. Onga, “A new thin film Growth/Regrowth Process Design and Experimental Comparisons with Molecular Dynamic Analyses,” MRS Proceedings. 1992. link Times cited: 0 Abstract: A new concept of thin film growth/regrowth process design ta… read moreAbstract: A new concept of thin film growth/regrowth process design taking atomic motions into account using molecular dynamics is proposed. In the system, a modified many-body Tersoff-type interatomic potential for silicon has been adopted. The mathematical derivation of higher order derivatives was rigorously treated. Among many applications, the solid phase growth process was studied. It has been found from simulation studies that the solid phase growth of crystalline silicon proceeded along the [110] direction layer by layer. Furthermore, it has been obtained that all the atoms are activated in an extremely thin amorphous silicon film. Based on simulated results, an experiment using an extremely thin amorphous silicon film was carried out. It has been found that the perfect spherical silicon crystals with a uniform size and spacing can be grown from a thin amorphous silicon film. read less NOT USED (low confidence) M. Robinson, “Computer Simulation of Atomic Collision Processes in Solids,” MRS Proceedings. 1992. link Times cited: 2 Abstract: Computer simulation is a major tool for studying the interac… read moreAbstract: Computer simulation is a major tool for studying the interactions of swift ions with solids which underlie processes such as particle backscattering, ion implantation, radiation damage, and sputtering. Numerical models are classed as molecular dynamics or binary collision models, along with some intermediate types. Binary collision models are divided into those for crystalline targets and those for structureless ones. The foundations of such models are reviewed, including interatomic potentials, electron excitations, and relationships among the various types of codes. Some topics of current interest are summarized. read less NOT USED (low confidence) D. Robertson, D. Brenner, M. Elert, and C. T. White, “SIMULATIONS OF CHEMICALLY-SUSTAINED SHOCK FRONTS IN A MODEL ENERGETIC MATERIAL.” 1992. link Times cited: 8 NOT USED (low confidence) J. Villain, J. Rouviere, and I. Vilfan, “Phenomenology of Surface Reconstruction.” 1991. link Times cited: 0 NOT USED (low confidence) G. Gilmer and A. F. Bakker, “MOLECULAR DYNAMICS SIMULATIONS OF MOLECULAR BEAM EPITAXY,” Computer Aided Innovation of New Materials. 1991. link Times cited: 1 NOT USED (low confidence) M. Duesbery and G. Richardson, “The dislocation core in crystalline materials,” Critical Reviews in Solid State and Materials Sciences. 1991. link Times cited: 134 Abstract: The art of forming materials into technologically useful art… read moreAbstract: The art of forming materials into technologically useful artifacts by manipulation of the dislocation substructure dates back at least 8000 years to the Sumerian coppersmiths.1 Physical understanding of the mechanisms involved, on the other hand, began little more than 50 years ago; modem knowledge suggests that even now this understanding is far from complete. read less NOT USED (low confidence) Š. Pick, “Instabilities and reconstructions on solid surfaces: basic theoretical notions and examples,” Surface Science Reports. 1991. link Times cited: 3 NOT USED (low confidence) N. Winograd and B. Garrison, “Surface Structure and Reaction Studies by Ion-Solid Collisions,” ChemInform. 1991. link Times cited: 4 NOT USED (low confidence) R. Biswas, I. Kwon, and C. Soukoulis, “Molecular Dynamics Simulations of the Structural, Vibrational and Electronic Properties of Amorphous Silicon,” MRS Proceedings. 1990. link Times cited: 1 NOT USED (low confidence) K. Laasonen and R. Nieminen, “Molecular Dynamics and Tight-Binding.” 1990. link Times cited: 0 NOT USED (low confidence) D. Brenner, “Atomistic computer simulations of the reactive dynamics of diamond surfaces,” Carbon. 1990. link Times cited: 2 NOT USED (low confidence) A. Carlsson, “Beyond Pair Potentials in Elemental Transition Metals and Semiconductors,” Journal of Physics C: Solid State Physics. 1990. link Times cited: 169 NOT USED (low confidence) D. Pettifor, “From Exact to Approximate Theory: The Tight Binding Bond Model and Many-Body Potentials.” 1990. link Times cited: 28 NOT USED (low confidence) J. Rouviere and A. Bourret, “Multiple Structures of a [001] Σ = 13 Tilt Grain Boundary in Germanium.” 1989. link Times cited: 8 NOT USED (low confidence) U. Landman and W. Luedtke, “Molecular Dynamics Simulations of Materials: Beyond Pair Interactions.” 1989. link Times cited: 0 NOT USED (low confidence) A. Bourret and J. Rouviere, “Grain Boundary Structure Determination by HREM: A Comparison with Computer Relaxed Configurations for Pure Tilt in Germanium.” 1989. link Times cited: 13 NOT USED (low confidence) M. Heggie, “A New Interatomic Potential for Non-Metals.” 1989. link Times cited: 0 NOT USED (low confidence) A. Carlsson, “Angular Forces in Transition Metals and Diamond Structure Semiconductors.” 1989. link Times cited: 1 NOT USED (low confidence) H. Teichler, “Computer Modelling of Grain Boundaries by Use of Interatomic Potentials.” 1989. link Times cited: 8 NOT USED (low confidence) W. Andreoni, “On the Electronic and Structural Properties of Small Clusters.” 1989. link Times cited: 0 NOT USED (low confidence) V. Vítek, D. Srolovitz, and W. Morgan, “MOLECULAR DYNAMICS SIMULATION OF THE PHYSICS OF THIN FILM GROWTH ON SILICON: EFFECTS OF THE PROPERTIES OF INTERATOMIC POTENTIAL MODELS.” 1989. link Times cited: 0 NOT USED (low confidence) D. J. Oh and R. Johnson, “A Semi-Empirical Potential for Graphite,” MRS Proceedings. 1988. link Times cited: 4 NOT USED (high confidence) N. Nguyen, “Fast proper orthogonal descriptors for many-body interatomic potentials,” Physical Review B. 2022. link Times cited: 1 Abstract: The development of differentiable invariant descriptors for … read moreAbstract: The development of differentiable invariant descriptors for accurate representations of atomic environments plays a central role in the success of interatomic potentials for chemistry and materials science. We introduce a method to generate fast proper orthogonal descriptors for the construction of many-body interatomic potentials and discuss its relation to exising empirical and machine learning interatomic potentials. A traditional way of implementing the proper orthogonal descriptors has a computational complexity that scales exponentially with the body order in terms of the number of neighbors. We present an algorithm to compute the proper orthogonal descriptors with a computational complexity that scales linearly with the number of neighbors irrespective of the body order. We show that our method can enable a more efficient implementation for a number of existing potentials and provide a scalable systematic framework to construct new many-body potentials. The new potentials are demonstrated on a data set of density functional theory calculations for Tantalum and compared with other interatomic potentials. read less NOT USED (high confidence) H. Zhai and J. Yeo, “Multiscale mechanics of thermal gradient coupled graphene fracture: A molecular dynamics study,” International Journal of Applied Mechanics. 2022. link Times cited: 2 Abstract: The thermo-mechanical coupling mechanism of graphene fractur… read moreAbstract: The thermo-mechanical coupling mechanism of graphene fracture under thermal gradients possesses rich applications whereas is hard to study due to its coupled non-equilibrium nature. We employ non-equilibrium molecular dynamics to study the fracture of graphene by applying a fixed strain rate under different thermal gradients by employing different potential fields. It is found that for AIREBO and AIREBO-M, the fracture stresses do not strictly follow the positive correlations with the initial crack length. Strain-hardening effects are observed for"REBO-based"potential models of small initial defects, which is interpreted as blunting effect observed for porous graphene. The temperature gradients are observed to not show clear relations with the fracture stresses and crack propagation dynamics. Quantized fracture mechanics verifies our molecular dynamics calculations. We provide a unique perspective that the transverse bond forces share the loading to account for the nonlinear increase of fracture stress with shorter crack length. Anomalous kinetic energy transportation along crack tips is observed for"REBO-based"potential models, which we attribute to the high interatomic attractions in the potential models. The fractures are honored to be more"brittle-liked"carried out using machine learning interatomic potential (MLIP), yet incapable of simulating post-fracture dynamical behaviors. The mechanical responses using MLIP are observed to be not related to temperature gradients. The temperature configuration of equilibration simulation employing the dropout uncertainty neural network potential with a dropout rate of 0.1 is reported to be the most accurate compared with the rest. This work is expected to inspire further investigation of non-equilibrium dynamics in graphene with practical applications in various engineering fields. read less NOT USED (high confidence) A. Pedone, M. Bertani, L. Brugnoli, and A. Pallini, “Interatomic potentials for oxide glasses: Past, present, and future,” Journal of Non-Crystalline Solids: X. 2022. link Times cited: 2 NOT USED (high confidence) N. Nguyen and A. Rohskopf, “Proper orthogonal descriptors for efficient and accurate interatomic potentials,” J. Comput. Phys. 2022. link Times cited: 6 NOT USED (high confidence) A. Osinsky and N. Brilliantov, “Scaling laws in fragmentation kinetics,” Physica A: Statistical Mechanics and its Applications. 2022. link Times cited: 0 NOT USED (high confidence) H. R. Heris, M. Kateb, S. Erlingsson, and A. Manolescu, “Effects of transverse geometry on the thermal conductivity of Si and Ge nanowires,” Surfaces and Interfaces. 2022. link Times cited: 5 NOT USED (high confidence) J. M. Ortiz-Roldán, F. Montero-Chacón, E. Garcia-Perez, S. Calero, A. R. Ruiz-Salvador, and S. Hamad, “Thermostructural Characterization of Silicon Carbide Nanocomposite Materials via Molecular Dynamics Simulations,” Advanced Composite Materials. 2021. link Times cited: 1 Abstract: In this paper, we investigate the thermostructural propertie… read moreAbstract: In this paper, we investigate the thermostructural properties of a type of silicon-based nanomaterials, which we refer to as SiC@Si nanocomposites, formed by SiC crystalline nanoparticles (with the cubic phase), embedded within an amorphous Si matrix. We have followed an in silico approach to characterize the mechanical and thermal behaviour of these materials, by calculating the elastic constants, uniaxial stress-strain curves, coefficients of thermal expansion, and specific heats, at different temperatures, using interatomic potential calculations. The results obtained from our simulations suggest that this type of material presents enhanced thermal resistance features, making it suitable to be used in devices subjected to big temperature changes, such as heat sinks in micro and nanoelectronics, solar energy harvesters at high temperatures, power electronics, or in other applications in which good thermomechanical properties are required. read less NOT USED (high confidence) G. Dobrik et al., “Large-area nanoengineering of graphene corrugations for visible-frequency graphene plasmons,” Nature Nanotechnology. 2021. link Times cited: 16 NOT USED (high confidence) Z. Luo, S. A. Burrows, X. Fan, S. Smoukov, and E. Boek, “Virtual voids method to generate low-density microporous carbon structures using quenched molecular dynamics simulation,” Carbon. 2021. link Times cited: 1 NOT USED (high confidence) A. Osinsky and N. Brilliantov, “Collision fragmentation of aggregates. The role of the interaction potential between comprising particles.” 2021. link Times cited: 3 NOT USED (high confidence) S. Stephan, M. Dyga, I. A. Alhafez, J. Lenhard, H. Urbassek, and H. Hasse, “Reproducibility of atomistic friction computer experiments: a molecular dynamics simulation study,” Molecular Simulation. 2021. link Times cited: 2 Abstract: ABSTRACT The elementary processes of friction in contact pro… read moreAbstract: ABSTRACT The elementary processes of friction in contact processes of two solid bodies occur on the nanoscale and are difficult to study experimentally. Therefore, molecular dynamics simulations are often used for their elucidation. In these studies, usually, only a single simulation is carried out for each scenario and the resulting observables are evaluated. In the present work, the reliability and reproducibility of measured observables from such nanoscopic contact process simulations are assessed by means of their statistical uncertainties. Therefore, the computer experiment is carried out not only once, but it is repeated eight times, with individual runs that only differ in the initial thermal motion. This set of replicas enables an assessment of observations for distinguishing reproducible physical phenomena from stochastic coincidence. In this way, a dry and a lubricated nanoscale scratching process were studied, in which a cylindrical indenter carried out two sequential movements: it first penetrates a substrate vertically and then scratches laterally, which causes elastic and plastic deformation of the substrate. In the lubricated case, the indenter was fully immersed in the fluid. Substrate, indenter, and fluid were described by suitably parametrised Lennard–Jones potentials. Various mechanical and thermodynamic process properties were monitored in all simulation runs. The results are compared and evaluated statistically. read less NOT USED (high confidence) Y. Tanuma, T. Maekawa, and C. Ewels, “Methodological Investigation for Hydrogen Addition to Small Cage Carbon Fullerenes,” Crystals. 2021. link Times cited: 2 Abstract: Hydrogenated small fullerenes (Cn, n<60) are of interest as … read moreAbstract: Hydrogenated small fullerenes (Cn, n<60) are of interest as potential astrochemical species, and as intermediates in hydrogen catalysed fullerene growth. However computational identification of key stable species is difficult due to the vast combinatorial space of structures. In this study we explore routes to predict stable hydrogenated small fullerenes. We show that neither local fullerene geometry nor local electronic structure analysis are able to correctly predict subsequent low energy hydrogenation sites, and indeed sequential stable addition searches also sometimes fail to identify most stable hydrogenated fullerene isomers. Of the empirical and semi-empirical methods tested, GFN2-xTB consistently gives highly accurate energy correlation (r>0.99) to full DFT-LDA calculations at a fraction of the computational cost. This allows identification of the most stable hydrogenated fullerenes up to 4H for four fullerenes, namely two isomers of C28 and C40, via “brute force” systematic testing of all symmetry inequivalent combinations. The approach shows promise for wider systematic studies of smaller hydrogenated fullerenes. read less NOT USED (high confidence) E. M. Y. Lee, A. Yu, J. D. de Pablo, and G. Galli, “Stability and molecular pathways to the formation of spin defects in silicon carbide,” Nature Communications. 2021. link Times cited: 9 NOT USED (high confidence) J. Hur, Y. Abousleiman, K. Hull, and M. J. A. Qomi, “Reactive force fields for modeling oxidative degradation of organic matter in geological formations,” RSC Advances. 2021. link Times cited: 1 Abstract: In an attempt to better explore organic matter reaction and … read moreAbstract: In an attempt to better explore organic matter reaction and properties, at depth, to oxidative fluid additives, we have developed a new ReaxFF potential to model and describe the oxidative decompositions of aliphatic and aromatic hydrocarbons in the presence of the oxychlorine ClOn− oxidizers. By carefully adjusting the new H/C/O/Cl parameters, we show that the potential energies in both training and validation sets correlate well with calculated density functional theory (DFT) energies. Our parametrization yields a reliable empirical reactive force field with an RMS error of ∼1.57 eV, corresponding to a 1.70% average error. At this accuracy level, the reactive force field provides a reliable atomic-level picture of thermodynamically favorable reaction pathways governing oxidative degradation of H/C/O/Cl compounds. We demonstrate this capability by studying the structural degradation of small aromatic and aliphatic hydrocarbons in the presence of oxychlorine oxidizers in aqueous environments. We envision that such reactive force fields will be critical in understanding the oxidation processes of organic matter in geological reservoirs and the design of the next generation of reactive fluids for enhanced shale gas recovery and improved carbon dioxide adsorption and sequestration. read less NOT USED (high confidence) M. Wen, Y. Afshar, R. Elliott, and E. Tadmor, “KLIFF: A framework to develop physics-based and machine learning interatomic potentials,” Comput. Phys. Commun. 2021. link Times cited: 12 NOT USED (high confidence) M. Friedrich, M. Seitz, and U. Stefanelli, “Tilings with Nonflat Squares: A Characterization,” Milan Journal of Mathematics. 2021. link Times cited: 0 NOT USED (high confidence) X. Zhang, H. Nguyen, J. T. Paci, S. Sankaranarayanan, J. L. Mendoza-Cortes, and H. Espinosa, “Multi-objective parametrization of interatomic potentials for large deformation pathways and fracture of two-dimensional materials,” npj Computational Materials. 2021. link Times cited: 11 NOT USED (high confidence) G. Chu et al., “MD Simulation of Hundred-Billion-Metal-Atom Cascade Collision on Sunway Taihulight,” Comput. Phys. Commun. 2021. link Times cited: 7 NOT USED (high confidence) S. Ono, “Magic numbers for vibrational frequency of charged particles on a sphere,” Physical Review B. 2021. link Times cited: 1 Abstract: Finding minimum energy distribution of $N$ charges on a sphe… read moreAbstract: Finding minimum energy distribution of $N$ charges on a sphere is known as the Thomson problem. Here, we study the vibrational properties of the $N$ charges in the lowest energy state within the harmonic approximation for $10\le N\le 200$ and for selected sizes up to $N=372$. The maximum frequency $\omega_{\rm max}$ increases with $N^{3/4}$, which is rationalized by studying the lattice dynamics of a two-dimensional triangular lattice. The $N$-dependence of $\omega_{\rm max}$ identifies magic numbers of $N=12, 32, 72, 132, 192, 212, 272, 282$, and 372, reflecting both a strong degeneracy of one-particle energies and an icosahedral structure that the $N$ charges form. $N=122$ is not identified as a magic number for $\omega_{\rm max}$ because the former condition is not satisfied. The magic number concept can hold even when an average of high frequencies is considered. The maximum frequency mode at the magic numbers has no anomalously large oscillation amplitude (i.e., not a defect mode). read less NOT USED (high confidence) Y. Fuseya, H. Katsuno, K. Behnia, and A. Kapitulnik, “Nanoscale Turing patterns in a bismuth monolayer,” Nature Physics. 2021. link Times cited: 17 NOT USED (high confidence) C. Xing, J. Sheng, L. Wang, and W. Fei, “Research progress in molecular dynamics simulation of CNT and graphene reinforced metal matrix composites.” 2021. link Times cited: 4 Abstract:
Carbon nanomaterials are considered as one of the ideal ch… read moreAbstract:
Carbon nanomaterials are considered as one of the ideal choices for high-performance metal matrix composite reinforcements and one of the key directions of scientific research in recent years. Molecular dynamics simulation could be used conveniently to construct different composite material systems and study the properties of carbon nanomaterials reinforced metal matrix composites under different conditions. This review mainly introduces the molecular dynamic research progress of carbon nanotube (CNT) and graphene-reinforced metal (Cu, Al, Ni) composites. The potential functions of the carbon nanomaterials reinforced metal matrix composite simulation systems are briefly introduced. The dependence of the mechanical properties of metal matrix composites on the sizes, volume fraction and distribution states of CNT and graphene is detailed and discussed. Finally, we briefly summarize the future development direction of the molecular dynamic simulation with respect to carbon nanomaterials reinforced metal matrix composites. read less NOT USED (high confidence) I. M. P. Espinosa, T. Jacobs, and A. Martini, “Evaluation of Force Fields for Molecular Dynamics Simulations of Platinum in Bulk and Nanoparticle Forms.,” Journal of chemical theory and computation. 2021. link Times cited: 7 Abstract: Understanding the size- and shape-dependent properties of pl… read moreAbstract: Understanding the size- and shape-dependent properties of platinum nanoparticles is critical for enabling the design of nanoparticle-based applications with optimal and potentially tunable functionality. Toward this goal, we evaluated nine different empirical potentials with the purpose of accurately modeling faceted platinum nanoparticles using molecular dynamics simulation. First, the potentials were evaluated by computing bulk and surface properties-surface energy, lattice constant, stiffness constants, and the equation of state-and comparing these to prior experimental measurements and quantum mechanics calculations. Then, the potentials were assessed in terms of the stability of cubic and icosahedral nanoparticles with faces in the {100} and {111} planes, respectively. Although none of the force fields predicts all the evaluated properties with perfect accuracy, one potential-the embedded atom method formalism with a specific parameter set-was identified as best able to model platinum in both bulk and nanoparticle forms. read less NOT USED (high confidence) S. Kim, W. Park, and O. Kwon, “The Strength and Delamination of Graphene/Cu Composites with Different Cu Thicknesses,” Materials. 2021. link Times cited: 3 Abstract: This study analyzed the mechanical and fracture behavior of … read moreAbstract: This study analyzed the mechanical and fracture behavior of graphene/copper (Cu) composites with different Cu thicknesses by using molecular dynamics (MD) and representative volume element (RVE) analysis. Three graphene/Cu composite analytical models were classified as 4.8, 9.8, and 14.3 nm according to Cu thicknesses. Using MD analysis, zigzag-, armchair-, and z (thickness)-direction tensile analyses were performed for each model to analyze the effect of Cu thickness variation on graphene/Cu composite strength and delamination fracture. In the RVE analysis, the mechanical characteristics of the interface between graphene and Cu were evaluated by setting the volume fraction to 1.39, 2.04, and 4.16% of the graphene/Cu composite model, classified according to the Cu thickness. From their obtained results, whether the graphene bond is maintained has the greatest effect on the strength of graphene/Cu composites, regardless of the Cu thickness. Additionally, graphene/Cu composites are more vulnerable to armchair direction tensile forces with fracture strengths of 14.7, 8.9, and 8.2 GPa depending on the Cu thickness. The results of this study will contribute to the development of guidelines and performance evaluation standards for graphene/Cu composites. read less NOT USED (high confidence) C. W. Park, M. Kornbluth, J. Vandermause, C. Wolverton, B. Kozinsky, and J. Mailoa, “Accurate and scalable graph neural network force field and molecular dynamics with direct force architecture,” npj Computational Materials. 2021. link Times cited: 80 NOT USED (high confidence) M. Sengul et al., “INDEEDopt: a deep learning-based ReaxFF parameterization framework,” npj Computational Materials. 2021. link Times cited: 18 NOT USED (high confidence) M. Zhao, R. Dang, L. Jin, and W. Yu, “Structures and energies of Σ3 asymmetric tilt grain boundaries in silicon,” Journal of Materials Research. 2021. link Times cited: 2 Abstract: We optimize 23 silicon Σ3 asymmetric tilt grain boundaries (… read moreAbstract: We optimize 23 silicon Σ3 asymmetric tilt grain boundaries (ATGBs) using Stillinger Weber (SW), Tersoff and the optimized Modified Embedded Atom Method (MEAM) potentials. It is demonstrated that conventional GB optimization via rigid body translations in combination with atom deletions is totally incapable of driving an as-constructed flat Si grain boundary (GB) to its equilibrated state since it may inevitably cause lattice distortions in GB. But it can be easily achieved by initially introducing some pre-designed steps into as-constructed flat GB model. These steps are composed of coherent twin boundary (CTB) and symmetric incoherent twin boundary (SITB) facets. By doing so, energies of all 23 ATGBs are greatly reduced. Meanwhile, some ATGBs may have degenerate states with different structures but same energies. This work not only facilitates the structural characterization of Si Σ3 ATGBs, but may provide new insights into microstructure design in polycrystalline silicon. read less NOT USED (high confidence) I. Leven et al., “Recent Advances for Improving the Accuracy, Transferability, and Efficiency of Reactive Force Fields.,” Journal of chemical theory and computation. 2021. link Times cited: 27 Abstract: Reactive force fields provide an affordable model for simula… read moreAbstract: Reactive force fields provide an affordable model for simulating chemical reactions at a fraction of the cost of quantum mechanical approaches. However, classically accounting for chemical reactivity often comes at the expense of accuracy and transferability, while computational cost is still large relative to nonreactive force fields. In this Perspective, we summarize recent efforts for improving the performance of reactive force fields in these three areas with a focus on the ReaxFF theoretical model. To improve accuracy, we describe recent reformulations of charge equilibration schemes to overcome unphysical long-range charge transfer, new ReaxFF models that account for explicit electrons, and corrections for energy conservation issues of the ReaxFF model. To enhance transferability we also highlight new advances to include explicit treatment of electrons in the ReaxFF and hybrid nonreactive/reactive simulations that make it possible to model charge transfer, redox chemistry, and large systems such as reverse micelles within the framework of a reactive force field. To address the computational cost, we review recent work in extended Lagrangian schemes and matrix preconditioners for accelerating the charge equilibration method component of ReaxFF and improvements in its software performance in LAMMPS. read less NOT USED (high confidence) Q. Zhang, X. Pang, and Y. Zhao, “Effect of the External Velocity on the Exfoliation Properties of Graphene from Amorphous SiO2 Surface,” Crystals. 2021. link Times cited: 4 Abstract: External action has a significant influence on the formation… read moreAbstract: External action has a significant influence on the formation of high-quality graphene and the adhesion of graphene on the surface of the MEMS/NEMS device. The atomic-scale simulation and calculation can further study the exfoliation process of graphene by external actions. In multilayer graphene systems where graphene layers were simulated weakly contacted with SiO2 substrate, a constant vertical upward velocity (Vup) was applied to the topmost layer. Then two critical velocities were found, and three kinds of distinct exfoliation processes determined by critical upward velocities were observed in multilayer graphene systems. The first critical velocities are in the range of 0.5 Å/ps–3.18 Å/ps, and the second critical velocities are in the range of 9.5 Å/ps–12.1 Å/ps. When the Vup is less than the first critical velocity, all graphene layers will not be exfoliated. When Vup is between the first and second critical Vup, all layers can be exfoliated almost synchronously at last. When Vup is larger than the second critical Vup, the topmost layer can be exfoliated alone, transferring energy to the underlying layers, and the underlying layers are slowly exfoliated. The maximum exfoliation force to exfoliate the topmost layer of graphene is 3200 times larger than that of all graphene layers. Moreover, it is required 149.26 mJ/m2 to get monolayer graphene from multilayers, while peeling off all layers without effort. This study explains the difficulty to get monolayer graphene and why graphene falls off easily during the transfer process. read less NOT USED (high confidence) F. Saiz, Y. Karaaslan, R. Rurali, and C. Sevik, “Interatomic potential for predicting the thermal conductivity of zirconium trisulfide monolayers with molecular dynamics,” Journal of Applied Physics. 2021. link Times cited: 1 Abstract: We present here a new interatomic potential parameter set to… read moreAbstract: We present here a new interatomic potential parameter set to predict the thermal conductivity of zirconium trisulfide monolayers. The generated Tersoff-type force field is parameterized using data collected with first-principles calculations. We use non-equilibrium molecular dynamics simulations to predict the thermal conductivity. The generated parameters result in very good agreement in structural, mechanical, and dynamical parameters. The room temperature lattice thermal conductivity ( κ) of the considered crystal is predicted to be κ x x = 25.69 W m − 1 K − 1 and κ y y = 42.38 W m − 1 K − 1, which both agree well with their corresponding first-principles values with a discrepancy of less than 5%. Moreover, the calculated κ variation with temperature (200 and 400 K) are comparable within the framework of the accuracy of both first-principles and molecular dynamics simulations. read less NOT USED (high confidence) T. Abrams et al., “Evaluation of silicon carbide as a divertor armor material in DIII-D H-mode discharges,” Nuclear Fusion. 2021. link Times cited: 15 Abstract: Silicon carbide (SiC) represents a promising but largely unt… read moreAbstract: Silicon carbide (SiC) represents a promising but largely untested plasma-facing material (PFM) for next-step fusion devices. In this work, an analytic mixed-material erosion model is developed by calculating the physical (via SDTrimSP) and chemical (via empirical scalings) sputtering yield from SiC, Si, and C. The Si content in the near-surface SiC layer is predicted to increase during D plasma bombardment due to more efficient physical and chemical sputtering of C relative to Si. Silicon erosion from SiC thereby occurs primarily from sputtering of the enriched Si layer, rather than directly from the SiC itself. SiC coatings on ATJ graphite, manufactured via chemical vapor deposition, were exposed to repeated H-mode plasma discharges in the DIII-D tokamak to test this model. The qualitative trends from analytic modeling are reproduced by the experimental measurements, obtained via spectroscopic inference using the S/XB method. Quantitatively the model slightly under-predicts measured erosion rates, which is attributed to uncertainties in the ion impact angle distribution, as well as the effect of edge-localized modes. After exposure, minimal changes to the macroscopic or microscopic surface morphology of the SiC coatings were observed. Compositional analysis reveals Si enrichment of about 10%, in line with expectations from the erosion model. Extrapolating to a DEMO-type device, an order-of-magnitude decrease in impurity sourcing, and up to a factor of 2 decrease in impurity radiation, is expected with SiC walls, relative to graphite, if low C plasma impurity content can be achieved. These favorable erosion properties motivate further investigations of SiC as a low-Z, non-metallic PFM. read less NOT USED (high confidence) X. Wei, J. Yu, J.-L. Du, and L. Sun, “New Insights into the Pyrolysis Behavior of Polycarbonates: A Study Based on DFT and ReaxFF-MD Simulation under Nonisothermal and Isothermal Conditions,” Energy & Fuels. 2021. link Times cited: 14 Abstract: In this work, a combined ReaxFF-MD simulation and density fu… read moreAbstract: In this work, a combined ReaxFF-MD simulation and density functional theory (DFT) study was performed to study the pyrolysis behavior of polycarbonates under nonisothermal and isothermal conditions... read less NOT USED (high confidence) X. Liu et al., “Identifying the Activity Origin of a Cobalt Single‐Atom Catalyst for Hydrogen Evolution Using Supervised Learning,” Advanced Functional Materials. 2021. link Times cited: 79 Abstract: Single‐atom catalysts (SACs) have become the forefront of en… read moreAbstract: Single‐atom catalysts (SACs) have become the forefront of energy conversion studies, but unfortunately, the origin of their activity and the interpretation of the synchrotron spectrograms of these materials remain ambiguous. Here, systematic density functional theory computations reveal that the edge sites—zigzag and armchair—are responsible for the activity of the graphene‐based Co (cobalt) SACs toward hydrogen evolution reaction (HER). Then, edge‐rich (E)‐Co single atoms (SAs) were rationally synthesized guided by theoretical results. Supervised learning techniques are applied to interpret the measured synchrotron spectrum of E‐Co SAs. The obtained local environments of Co SAs, 65.49% of Co‐4N‐plane, 13.64% in Co‐2N‐armchair, and 20.86% in Co‐2N‐zigzag, are consistent with Athena fitting. Remarkably, E‐Co SAs show even better HER electrocatalytic performance than commercial Pt/C at high current density. Using the joint effort of theoretical modeling, thorough characterization of the catalysts aided by supervised learning, and catalytic performance evaluations, this study not only uncovers the activity origin of Co SACs for HER but also lays the cornerstone for the rational design and structural analysis of nanocatalysts. read less NOT USED (high confidence) B. Parsaeifard, D. De, J. A. Finkler, and S. Goedecker, “Fingerprint-Based Detection of Non-Local Effects in the Electronic Structure of a Simple Single Component Covalent System,” Condensed Matter. 2021. link Times cited: 5 Abstract: Using fingerprints used mainly in machine learning schemes o… read moreAbstract: Using fingerprints used mainly in machine learning schemes of the potential energy surface, we detect in a fully algorithmic way long range effects on local physical properties in a simple covalent system of carbon atoms. The fact that these long range effects exist for many configurations implies that atomistic simulation methods, such as force fields or modern machine learning schemes, that are based on locality assumptions, are limited in accuracy. We show that the basic driving mechanism for the long range effects is charge transfer. If the charge transfer is known, locality can be recovered for certain quantities such as the band structure energy. read less NOT USED (high confidence) J. Keith et al., “Combining Machine Learning and Computational Chemistry for Predictive Insights Into Chemical Systems,” Chemical Reviews. 2021. link Times cited: 224 Abstract: Machine learning models are poised to make a transformative … read moreAbstract: Machine learning models are poised to make a transformative impact on chemical sciences by dramatically accelerating computational algorithms and amplifying insights available from computational chemistry methods. However, achieving this requires a confluence and coaction of expertise in computer science and physical sciences. This Review is written for new and experienced researchers working at the intersection of both fields. We first provide concise tutorials of computational chemistry and machine learning methods, showing how insights involving both can be achieved. We follow with a critical review of noteworthy applications that demonstrate how computational chemistry and machine learning can be used together to provide insightful (and useful) predictions in molecular and materials modeling, retrosyntheses, catalysis, and drug design. read less NOT USED (high confidence) Y. Mishin, “Machine-Learning Interatomic Potentials for Materials Science,” Electrical Engineering eJournal. 2021. link Times cited: 103 NOT USED (high confidence) M. Haro et al., “Nano-vault architecture mitigates stress in silicon-based anodes for lithium-ion batteries,” Communications Materials. 2021. link Times cited: 8 NOT USED (high confidence) R.-S. Zhang, J.-D. He, B. Wang, and J.-W. Jiang, “Physical description of the monoclinic phase of zirconia based on the bond-order characteristic of the Tersoff potential,” Frontiers of Physics. 2021. link Times cited: 1 NOT USED (high confidence) A. Morozov, A. Zhuravlev, and D. Reviznikov, “Sparse Grid Adaptive Interpolation in Problems of Modeling Dynamic Systems with Interval Parameters,” Mathematics. 2021. link Times cited: 6 Abstract: The paper is concerned with the issues of modeling dynamic s… read moreAbstract: The paper is concerned with the issues of modeling dynamic systems with interval parameters. In previous works, the authors proposed an adaptive interpolation algorithm for solving interval problems; the essence of the algorithm is the dynamic construction of a piecewise polynomial function that interpolates the solution of the problem with a given accuracy. The main problem of applying the algorithm is related to the curse of dimension, i.e., exponential complexity relative to the number of interval uncertainties in parameters. The main objective of this work is to apply the previously proposed adaptive interpolation algorithm to dynamic systems with a large number of interval parameters. In order to reduce the computational complexity of the algorithm, the authors propose using adaptive sparse grids. This article introduces a novelty approach of applying sparse grids to problems with interval uncertainties. The efficiency of the proposed approach has been demonstrated on representative interval problems of nonlinear dynamics and computational materials science. read less NOT USED (high confidence) S. Stelmakh, K. Skrobas, S. Gierlotka, and B. Palosz, “Structure of plate-shape nanodiamonds synthesized from chloroadamantane—are they still diamonds?,” Journal of Physics: Condensed Matter. 2021. link Times cited: 1 Abstract: Atomic structure of plate-shaped nanodiamonds synthesized fr… read moreAbstract: Atomic structure of plate-shaped nanodiamonds synthesized from chloroadamantane was identified with application of large-Q powder diffraction data. Both reciprocal and real space methods of experimental data analysis were applied. Theoretical atomistic models of nanodiamonds were obtained with application of molecular dynamics (MD) simulations. It was found that examined nanodiamond samples with average grain size from 1.2 up to 2.5 nm are plates build from only six hexagonal carbon layers and they are terminated by (111)B surfaces with three dangling bonds. MD simulations showed that as a result of relaxation of surface stresses there appears a complex system of compressive and tensile strains across and parallel to the surface of the plate-nanodiamonds. Identification of the internal structure of nanodiamond was performed based on the analysis of differential interatomic distance diagrams derived from pair distribution functions G(r). Based on MD simulations an atomic model of plate-grains of diamond was elaborated. Usefulness of lattice parameters determined in a routine diffraction data analysis for characterization of nanodiamonds is questioned. As an alternative the application of the apparent lattice parameter is recommended. A dependence of the overall apparent lattice parameter 〈alp〉 on the size and shape of nanodiamond grains terminated by low index crystal faces is presented. read less NOT USED (high confidence) O. Koroleva, M. Demin, A. Mazhukin, and V. Mazhukin, “Modeling of electronic and phonon thermal conductivity of silicon in a wide temperature range,” Journal of Physics: Conference Series. 2021. link Times cited: 7 Abstract: In the present article, using the methods of mathematical mo… read moreAbstract: In the present article, using the methods of mathematical modeling, the thermal conductivity of silicon was obtained in a wide temperature range (0.3 ≼ T ≼ 3 kK), including the region of semiconductor-metal phase transformations. As it is known, there are two mechanisms of heat transfer in a solid: elastic lattice vibrations and free electrons, therefore, in the study of the thermal conductivity of silicon, the lattice and electronic components were taken into account. The lattice (phonon) thermal conductivity in this work was determined within the framework of the atomistic approach. The Stillinger–Weber and Kumagai–Izumi–Hara–Sakai interaction potentials were used for modeling. The results of the comparison of the phonon thermal conductivity obtained from the simulation results with the used interaction potentials are presented. The modeling of the thermal conductivity of the electronic subsystem of silicon with intrinsic conductivity in this work is based on the use of the quantum statistics of the electron gas using the Fermi–Dirac integrals. The total thermal conductivity of silicon, obtained as the sum of the electronic and phonon components, is compared with the experimental data. read less NOT USED (high confidence) R. Kobayashi, “nap: A molecular dynamics package with parameter-optimization programs for classical and machine-learning potentials,” J. Open Source Softw. 2021. link Times cited: 8 Abstract: The nap is a package for molecular dynamics (MD) simulation … read moreAbstract: The nap is a package for molecular dynamics (MD) simulation consisting of an MD program ( pmd ) that can perform large-scale simulation using a spatial-decomposition technique and two parameter-optimization programs: one for classical (CL) potentials ( fp.py ) and another for machine-learning (ML) potentials ( fitpot ). Since the numbers of parameters to be optimized are much different between CL and ML potentials, optimization approaches for them are also different; meta-heuristic global minimum-search algorithms for the CL potentials, in which the numbers of parameters are usually much less than one hundred, and gradient-based methods for the ML potentials. The parameters of CL potentials can be optimized to any target quantity that can be computed using the potentials since meta-heuristic methods do not require the derivatives of the quantity with respect to parameters. On the other hand, ML-potential parameters can be optimized to only energies, forces on atoms and stress components of reference systems, mainly because gradient-based methods require the derivatives of other quantities with respect to parameters, and the analytical derivatives and the coding of them are usually painful and sometimes impossible. Potentials can be used in combination with any other potential, such as pair and angular potentials, short-range and long-range potentials, CL and ML potentials. With using the nap package, users can perform MD simulation of solid-state materials with the choice of different levels of complexity (CL or ML) by creating interatomic potentials optimized to quantum-mechanical calculation data even if no potential is available. read less NOT USED (high confidence) Y.-S. Lin, G. P. P. Pun, and Y. Mishin, “Development of a physically-informed neural network interatomic potential for tantalum,” Computational Materials Science. 2021. link Times cited: 9 NOT USED (high confidence) X. Zhang and A. Beyer, “Mechanics of free-standing inorganic and molecular 2D materials.,” Nanoscale. 2021. link Times cited: 14 Abstract: The discovery of graphene has triggered a great interest in … read moreAbstract: The discovery of graphene has triggered a great interest in inorganic as well as molecular two-dimensional (2D) materials. In this review, we summarize recent progress in the mechanical characterization of free-standing 2D materials, such as graphene, hexagonal boron nitride (hBN), transition metal-dichalcogenides, MXenes, black phosphor, carbon nanomembranes (CNMs), 2D polymers, 2D metal organic frameworks (MOFs) and covalent organic frameworks (COFs). Elastic, fracture, bending and interfacial properties of these materials have been determined using a variety of experimental techniques including atomic force microscopy based nanoindentation, in situ tensile/fracture testing, bulge testing, Raman spectroscopy, Brillouin light scattering and buckling-based metrology. Additionally, we address recent advances of 2D materials in a variety of mechanical applications, including resonators, microphones and nanoelectromechanical sensors. With the emphasis on progress and challenges in the mechanical characterization of inorganic and molecular 2D materials, we expect a continuous growth of interest and more systematic experimental work on the mechanics of such ultrathin nanomaterials. read less NOT USED (high confidence) Y. Fuseya, H. Katsuno, K. Behnia, and A. Kapitulnik, “Nanometric Turing Patterns: Morphogenesis of a Bismuth Monolayer,” arXiv: Mesoscale and Nanoscale Physics. 2021. link Times cited: 11 NOT USED (high confidence) T. Sato et al., “Molecular Dynamics Study on SiO2 Interfaces of Nonfiring Solids,” Journal of Nanomaterials. 2020. link Times cited: 0 Abstract: As a sustainable ecosystem, the general firing process for c… read moreAbstract: As a sustainable ecosystem, the general firing process for ceramics emits large amounts of CO2 gas; thus in ceramics production, the focus is the nonfiring process; however, the solidification and strengthen mechanism of this nonfiring system, which essentially reacts between surface-activated ceramic particles and a solvent, has not been elucidated to date. The nonfiring process had three steps, i.e., particle surface activate process by grinding process, maintaining the active state until starting nonfiring solidification begins, and nonfiring solidification process. Thus, in this study, the reaction of silica and water was simulated by adapting molecular dynamics based on LAMMPS with ReaxFF potentials. Reproducing the activated silica surface state, three ended models called O model, Si model, and OH model were prepared which indicated ended molecules of each surface. These models and a water molecule as a solvent were bonded in the atomic scale, and the energetic state and mechanical properties were evaluated. A reacted or structured O-H-O bond was reproduced in the nonfiring process in the O-ended model. The bond was a hydrogen bond. A Si-O-Si bond was produced when a Si atom was ended on the interface. The bonded interface was able to tensile. However, the tensile strength was weaker than that of the solid silica model. The nonbonded OH model did not have tensile strength. read less NOT USED (high confidence) A. Lyashenko, E. Safi, J. Polvi, F. Djurabekova, and K. Nordlund, “Computational study of tungsten sputtering by nitrogen,” Journal of Nuclear Materials. 2020. link Times cited: 1 NOT USED (high confidence) A. Boscoboinik, S. Manzi, V. Pereyra, W. Mas, and J. Boscoboinik, “Structural evolution of two-dimensional silicates using a ‘bond-switching’ algorithm.,” Nanoscale. 2020. link Times cited: 0 Abstract: Silicates are the most abundant materials in the earth'… read moreAbstract: Silicates are the most abundant materials in the earth's crust. In recent years, two-dimensional (2D) versions of them grown on metal supports (known as bilayer silicates) have allowed their study in detail down to the atomic scale. These structures are self-containing. They are not covalently bound to the metal support but interact with it through van der Waals forces. Like their three-dimensional counterparts, the 2D-silicates can form both crystalline and vitreous structures. Furthermore, the interconversion between vitreous to crystalline structures has been experimentally observed at the nanoscale. While theoretical work has been carried out to try to understand these transformations, a limitation for ab initio methods, and even molecular dynamics methods, is the computational cost of studying large systems and long timescales. In this work, we present a simple and computationally inexpensive approach, that can be used to represent the evolution of bilayer silicates using a bond-switching algorithm. This approach allows reaching equilibrium ring size distributions as a function of a parameter that can be related to the ratio between temperature and the energy required for the bond-switching event. The ring size distributions are compared to experimental data available in the literature. read less NOT USED (high confidence) Y. Ouyang, Z. Zhang, C. Yu, J. He, G. Yan, and J. C. hyperlinks, “Accuracy of Machine Learning Potential for Predictions of Multiple-Target Physical Properties,” Chinese Physics Letters. 2020. link Times cited: 9 NOT USED (high confidence) D. Unruh, C. M. Hansen, R. V. Meidanshahi, S. Goodnick, and G. Zimányi, “From Femtoseconds to Gigaseconds: Performance Degradation in Silicon Heterojunction Solar Cells,” 2021 IEEE 48th Photovoltaic Specialists Conference (PVSC). 2020. link Times cited: 2 Abstract: a-Si/c-Si heterojunction solar cells hold the efficiency wor… read moreAbstract: a-Si/c-Si heterojunction solar cells hold the efficiency world record around 27%, yet their market penetration is delayed. One concern is the presence of an amorphous Si layer that some suspect may speed up the degradation of their performance. To address this concern, we developed the SolDeg structural simulation platform that is capable of capturing extremely slow degradation processes in a-Si. SolDeg integrates molecular dynamics methods that optimize the Si structure with femtosecond time steps, with the nudged elastic band method that captures the defect generation on time scales extending to gigaseconds. In this paper we report SolDeg simulations for Si-only heterojunctions. The SolDeg platform enabled us to determine the defect generation rate to be in the 15-20%/year range, translating into a 1-1.5%/year Voc degradation rate. These results establish that SolDeg can be a uniquely useful platform to describe degradation processes with an eye on finding strategies to mitigate the performance degradation of these promising heterojunction cells. read less NOT USED (high confidence) B. Wang, Y. Chen, and C. Hou, “Communication Optimization Strategy for Molecular Dynamics Simulation on Sunway TaihuLight,” 2020 IEEE 22nd International Conference on High Performance Computing and Communications; IEEE 18th International Conference on Smart City; IEEE 6th International Conference on Data Science and Systems (HPCC/SmartCity/DSS). 2020. link Times cited: 0 Abstract: Molecular dynamics simulation defined as simple state update… read moreAbstract: Molecular dynamics simulation defined as simple state updates over multiple time steps is the main approach to the description of the chemical, mechanical and electrical processes in many real-world application. Currently, most optimization methods focus on simplifying forces or computing parallelization. However, modern general-purpose supercomputers have succeeded on compute-intensive or memory/bandwidth-intensive applications, but the improvement of network bandwidth/latency is fairly limited. The communication overhead is critical, and severely degrades the overall performance and scalability. In this paper, we propose a communication optimization strategy to reduce inter-nodes communication overheads, including a ghost communication mode to reduce the total amount of message, a shift communication algorithm to reduce the total number of messages and a zero-copy RDMA (Remote Direct Memory Access) communication method to reduce inter-nodes memory copy overheads. We implement our ghost communication, shift communication and zero-copy RDMA communication on the third highest performance supercomputer Sunway TaihuLight in the world (before June 2020). We test molecular dynamics simulation of condensed covalent materials and scale the simulation up to 8,519,680 cores to simulate more than 50.4 million silicon atoms, where the parallel efficiency is over 80% on the whole machine. Results show that the communication optimization strategy reduces the communication time by nearly 75% compared with traditional inter-nodes MPI (Message Passing Interface) communication with memory copy and the dimension of the simulated system significantly exceeds the experimentally measurable range. Our simulation enables virtual experiments on real-world applications and makes the technology more accessible to the general scientific users by using general-purpose supercomputers. read less NOT USED (high confidence) M. S. Islam, I. Mia, S. Ahammed, C. Stampfl, and J. Park, “Exceptional in-plane and interfacial thermal transport in graphene/2D-SiC van der Waals heterostructures,” Scientific Reports. 2020. link Times cited: 18 NOT USED (high confidence) N. Piroozan and M. Sahimi, “Molecular origin of sliding friction and flash heating in rock and heterogeneous materials,” Scientific Reports. 2020. link Times cited: 1 NOT USED (high confidence) S. Romashin and V. Shorkin, “Variant of the Relationship between the Mechanical and Adhesive Properties of Solid Materials,” Mechanics of Solids. 2020. link Times cited: 1 NOT USED (high confidence) N. Walet and F. Guinea, “Flat bands, strains, and charge distribution in twisted bilayer
h−BN,” Physical Review B. 2020. link Times cited: 12 Abstract: We study the effect of twisting on bilayer graphene. The eff… read moreAbstract: We study the effect of twisting on bilayer graphene. The effect of lattice relaxation is included; we look at the electronic structure, piezo-electric charges and spontaneous polarisation. We show that the electronic structure without lattice relaxation shows a set of extremely flat in-gap states similar to Landau-levels, where the spacing scales with twist angle. With lattice relaxation we still have flat bands, but now the spectrum becomes independent of twist angle for sufficiently small angles. We describe in detail the nature of the bands, and study appropriate continuum models, at the same time explaining the spectrum We find that even though the spectra for both parallel an anti-parallel alignment are very similar, the spontaneous polarisation effects only occur for parallel alignment. We argue that this suggests a large interlayer hopping between boron and nitrogen. read less NOT USED (high confidence) Y. R. Than and R. Grimes, “Predicting radiation damage in beryllium,” Philosophical Magazine. 2020. link Times cited: 2 Abstract: ABSTRACT Displacement damage in beryllium was predicted as a… read moreAbstract: ABSTRACT Displacement damage in beryllium was predicted as a function of temperature and energy using molecular dynamics simulations. A key aim of this study was to determine if average results from large displacement cascades correspond to values predicted by the Kinchin–Pease (K–P) model. The number of residual defects remaining after 1 ps increased linearly with primary knock-on atom (PKA) energy from 0.5 keV to 2.5 keV, while the extent of residual damage was largely temperature independent from 300 K to 1100 K. The same simulation model was used to predict the directionally averaged probability of displacement as a function of displacement energy, , and thereby the threshold displacement energy at which the probability for displacement is 100%, eV. There is an excellent correspondence between the K–P prediction using and the number of residual defects remaining after the initial recovery phase. Also, by utilising , a modification to the K–P model is proposed that gives rise to an average model prediction when . read less NOT USED (high confidence) J. Ehrens et al., “Theoretical formation of carbon nanomembranes under realistic conditions using classical molecular dynamics,” Physical Review B. 2020. link Times cited: 2 Abstract: Carbon nanomembranes made from aromatic precursor molecules … read moreAbstract: Carbon nanomembranes made from aromatic precursor molecules are free standing nanometer thin materials of macroscopic lateral dimensions. Although produced in various versions for about two decades not much is known about their internal structure. Here we present a first systematic theoretical attempt to model the formation, structure, and mechanical properties of carbon nanomembranes using classical molecular dynamics simulations. read less NOT USED (high confidence) H. Tian, F. Pan, and B. Zhang, “Crack kinking in h-BN monolayer predicted by energy dissipation,” Journal of Applied Physics. 2020. link Times cited: 0 Abstract: Rapid crack propagation in a strip of a hexagonal boron nitr… read moreAbstract: Rapid crack propagation in a strip of a hexagonal boron nitride monolayer is studied by molecular dynamics. Crack kinking/branching takes place at high velocities under displacement loadings, accompanied by elastic waves dissipating the external supplied work besides the fresh surface energy. Cracks moving at a maximum velocity (∼74% of the Rayleigh wave velocity) do not branch immediately, which is governed by energy dissipation around the crack tip, instead, once the energy release rate (G) reaches a critical value of 19.75 J/m2 (∼5.9 times the surface energy density of 3.35 J/m2), kinking occurs, which shows that G could predict the onset of kinking/branching accurately. The dependences of G for crack initiation and branching on displacement-loading rate, strip size, and initial crack length are examined as well. read less NOT USED (high confidence) Z. Li, Y. Yan, J. Wang, and Y. Geng, “Molecular Dynamics Study on Tip-Based Nanomachining: A Review,” Nanoscale Research Letters. 2020. link Times cited: 8 NOT USED (high confidence) S. Im, H. Kim, W. Kim, and M. Cho, “Neural network constitutive model for crystal structures,” Computational Mechanics. 2020. link Times cited: 14 NOT USED (high confidence) S. Im, H. Kim, W. Kim, and M. Cho, “Neural network constitutive model for crystal structures,” Computational Mechanics. 2020. link Times cited: 0 NOT USED (high confidence) J. Chapman and R. Ramprasad, “Multiscale Modeling of Defect Phenomena in Platinum Using Machine Learning of Force Fields,” JOM. 2020. link Times cited: 5 NOT USED (high confidence) L. B’etermin, M. Friedrich, and U. Stefanelli, “Stability of Z2 configurations in 3D,” Nonlinearity. 2020. link Times cited: 0 Abstract: Inspired by the issue of stability of molecular structures, … read moreAbstract: Inspired by the issue of stability of molecular structures, we investigate the strict minimality of point sets with respect to configurational energies featuring two- and three-body contributions. Our main focus is on characterizing those configurations which cannot be deformed without changing distances between first neighbours or angles formed by pairs of first neighbours. Such configurations are called angle-rigid. We tackle this question in the class of finite configurations in Z2 , seen as planar three-dimensional point sets. A sufficient condition preventing angle-rigidity is presented. This condition is also proved to be necessary when restricted to specific subclasses of configurations. read less NOT USED (high confidence) E. A. Bea, M. F. Carusela, A. Soba, A. Monastra, and A. M. Viotti, “Thermal conductance of structured silicon nanocrystals,” Modelling and Simulation in Materials Science and Engineering. 2020. link Times cited: 1 Abstract: We calculate the thermal conductance of a structured silicon… read moreAbstract: We calculate the thermal conductance of a structured silicon nanocrystal with a hole of different sizes. The numerical study is based on non-equilibrium molecular dynamics simulations using two potential models for the interatomic interactions: (i) an empirical Tersoff–Brenner (Tersoff) potential; (ii) a semi-empirical tight binding (TB) potential. TB potential model predicts a similar thermal conductance for the nanocrystal with no hole and with a small size hole, which contrasts with the monotonic decrease predicted by Tersoff potential model. In addition, thermal conductance decreasing is higher for TB potential model when the surface-to-volume ratio increases. This points out that to study thermal properties of nanostructures with high surface-to-volume ratio is mandatory the use of potential models with high transferability to take adequately into account the relevant quantum physical effects due to boundaries and surfaces. read less NOT USED (high confidence) H. Lin, A. Croy, R. Gutierrez, and G. Cuniberti, “Surface-Phonon-Induced Rotational Dissipation for Nanoscale Solid-State Gears,” arXiv: Mesoscale and Nanoscale Physics. 2020. link Times cited: 3 Abstract: Compared to nanoscale friction of translational motion, the … read moreAbstract: Compared to nanoscale friction of translational motion, the mechanisms of rotational friction have received less attention. Such motion becomes an important issue for the miniaturization of mechanical machineries which often involve rotating gears. In this study, molecular dynamics simulations are performed to explore rotational friction for solid-state gears rotating on top of different substrates. In each case, viscous damping of the rotational motion is observed and found to be induced by the pure van-der-Waals interaction between gear and substrate. The influence of different gear sizes and various substrate materials is investigated. Furthermore, the rigidities of the gear and the substrate are found to give rise to different dissipation channels. Finally, it is shown that the dominant contribution to the dissipation is related to the excitation of low-frequency surface-phonons in the substrate. read less NOT USED (high confidence) S. Ajori, F. Sadeghi, and R. Ansari, “Nano-oscillators based on a C60 fullerene inside open carbon nanocones: a molecular dynamics study,” Journal of the Brazilian Society of Mechanical Sciences and Engineering. 2020. link Times cited: 1 NOT USED (high confidence) H. Guo, Z. Q. Zhao, D. Nan, Y. Cai, and J. Yan, “Predicting tensile properties of monolayer white graphene involving edge effect,” Journal of the Brazilian Society of Mechanical Sciences and Engineering. 2020. link Times cited: 1 NOT USED (high confidence) Y. Zheng, X. Zhang, M. T. S. Mobareke, M. Hekmatifar, A. Karimipour, and R. Sabetvand, “Potential energy and atomic stability of H2O/CuO nanoparticles flow and heat transfer in non-ideal microchannel via molecular dynamic approach: the Green–Kubo method,” Journal of Thermal Analysis and Calorimetry. 2020. link Times cited: 33 NOT USED (high confidence) D. Mora‐Fonz et al., “Real and virtual polymorphism of titanium selenide with robust interatomic potentials,” Journal of Materials Chemistry A. 2020. link Times cited: 5 Abstract: The first successful pairwise potential for a layered materi… read moreAbstract: The first successful pairwise potential for a layered material, TiSe2, has been parameterised to fit the experimental data, using a genetic algorithm as the optimisation tool for the parameters of the interatomic potential. read less NOT USED (high confidence) É. A. Velásquez, J. Mazo‐Zuluaga, E. Tangarife, and J. Mejía‐López, “Structural Relaxation and Crystalline Phase Effects on the Exchange Bias Phenomenon in FeF2/Fe Core/Shell Nanoparticles,” Advanced Materials Interfaces. 2020. link Times cited: 4 Abstract: In this study, the power of first‐principles methods along w… read moreAbstract: In this study, the power of first‐principles methods along with molecular dynamics and atomistic Monte Carlo simulations is employed to elucidate the effects of the structural relaxation on the exchange bias (EB) behavior of FeF2/Fe core/shell nanoparticles. The effects of the crystalline phase are also explored by studying the EB features on the related nanoparticles modeled through simple cubic, body centered cubic, and face centered cubic systems. The results indicate that effects of both structural relaxation and crystalline phase on the EB phenomenon are crucial. Noticeable differences are found in the quantitative and qualitative results, as well as in conclusions from studies which, for the sake of simplicity, have used simple cubic crystalline structures for modeling the sample of study instead of its own crystalline model. To compare these results with experimental systems, hysteresis behaviors under field cooling procedures and for a sample made up by a particle diameter distribution D = 4.3 ± 0.7 nm, which is easily affordable at present, are presented. In that sense, this study raises a warning about the conclusions derived from previous works, and offers a suggestion to pay close attention to both the crystalline model and the structural relaxation of the nanoparticle systems exhibiting EB effects. read less NOT USED (high confidence) H. Wang et al., “Investigation on electronic and mechanical properties of penta-graphene nanotubes,” Journal of Materials Science. 2020. link Times cited: 11 NOT USED (high confidence) V. Choyal and S. I. Kundalwal, “Effect of Stone–Wales defects on the mechanical behavior of boron nitride nanotubes,” Acta Mechanica. 2020. link Times cited: 18 NOT USED (high confidence) T. C. Sagar and V. Chinthapenta, “Effect of substitutional and vacancy defects on the electrical and mechanical properties of 2D-hexagonal boron nitride,” Journal of Molecular Modeling. 2020. link Times cited: 5 NOT USED (high confidence) A. Morozov, A. Zhuravlev, and D. Reviznikov, “Analysis and Optimization of an Adaptive Interpolation
Algorithm for the Numerical Solution of a System
of Ordinary Differential Equations
with Interval Parameters,” Differential Equations. 2020. link Times cited: 4 NOT USED (high confidence) A. Morozov, A. A. Zhuravlev, and D. Reviznikov, “Analysis and Optimization of an Adaptive Interpolation Algorithm for the Numerical Solution of a System of Ordinary Differential Equations with Interval Parameters,” Differential Equations. 2020. link Times cited: 0 NOT USED (high confidence) X. Han, “Investigate the mechanical property of nanopolycrystal silicon by means of the nanoindentation method,” AIP Advances. 2020. link Times cited: 2 Abstract: A comprehensive understanding of the basic deformation mecha… read moreAbstract: A comprehensive understanding of the basic deformation mechanisms is essential for novel nanomaterials with unique properties for engineering applications. Unfortunately, nanopolycrystal materials with smaller grains are difficult prepare, which makes the study of the deformation process difficult using experiments. The molecular dynamics (MD) method has already been proved to be an efficient tool kit for the nanoscale phenomenon and was gradually adopted by many researchers to investigate the mechanical deformation of nanocrystalline materials. This manuscript studies the mechanical response of specimens with internal grains separated by high angle boundaries without porosities and impurities using MD simulation methods. The results demonstrate that the partial dislocation activity takes over in nanocrystalline materials if the grain sizes are large enough. The distribution of the ideal crystal structure along the radial direction remains almost unchanged, which justifies that little lateral deformation is induced. The animation shows that many atoms are stripped by the feeding of the indenter. This type of atom removal (moves just like rain flow) is different from any kind of material stripping in the macroscopic scale. Therefore, the deformation of the substrate is generated by the coupling of dislocation and atom sliding. The distribution of dislocation is more suitable for characterizing materials deformation at small scale. In addition, a novel cone-shaped dislocation distribution is observed. With the feeding of tools, the amount of screw dislocation gradually increases while the amount of the edge dislocation gradually decreases. The simulation results also show that the grain boundary exhibits higher self-diffusivities than the perfect lattice, which is helpful in grain boundary sliding. read less NOT USED (high confidence) A. Guajardo-Cuéllar, D. Go, and M. Sen, “Analysis of Energy Transport Behavior and Geometric Effects in Graphene,” Frontiers of Mechanical Engineering. 2020. link Times cited: 0 Abstract: Graphene is an excellent heat conductor, with the potential … read moreAbstract: Graphene is an excellent heat conductor, with the potential to be used as a heat spreader for applications where there are fast, transient heat pulses. In this study we analyze and describe energy transport in graphene subject to an initial pulse of energy. We analyze the effects of using harmonic, anharmonic, and a non-linear (Tersoff) potentials to describe the transient energy transport and compare these to classical continuum descriptions. The energy pulse produces pure wave-like behavior and a spatial energy distribution that has geometric features similar to the graphene geometry itself. Depending on the potential used, the energy travels outward from the impulse location following a similar pattern as the hexagonal shape of graphene. This pattern is clearly identified when the transport is treated with a harmonic potential. Increasing the anharmonicity and non-linearity dampens this effect and results in thermal transport that does not follow the geometry of graphene. read less NOT USED (high confidence) J. Chapman and R. Ramprasad, “Predicting the dynamic behavior of the mechanical properties of platinum with machine learning.,” The Journal of chemical physics. 2020. link Times cited: 2 Abstract: Over the last few decades, computational tools have been ins… read moreAbstract: Over the last few decades, computational tools have been instrumental in understanding the behavior of materials at the nano-meter length scale. Until recently, these tools have been dominated by two levels of theory: quantum mechanics (QM) based methods and semi-empirical/classical methods. The former are time-intensive but accurate and versatile, while the latter methods are fast but are significantly limited in veracity, versatility, and transferability. Recently, machine learning (ML) methods have shown the potential to bridge the gap between these two chasms due to their (i) low cost, (ii) accuracy, (iii) transferability, and (iv) ability to be iteratively improved. In this work, we further extend the scope of ML for atomistic simulations by capturing the temperature dependence of the mechanical and structural properties of bulk platinum through molecular dynamics simulations. We compare our results directly with experiments, showcasing that ML methods can be used to accurately capture large-scale materials phenomena that are out of reach of QM calculations. We also compare our predictions with those of a reliable embedded atom method potential. We conclude this work by discussing how ML methods can be used to push the boundaries of nano-scale materials research by bridging the gap between QM and experimental methods. read less NOT USED (high confidence) A. Hamedani et al., “Insights into the primary radiation damage of silicon by a machine learning interatomic potential,” Materials Research Letters. 2020. link Times cited: 14 Abstract: ABSTRACT We develop a silicon Gaussian approximation machine… read moreAbstract: ABSTRACT We develop a silicon Gaussian approximation machine learning potential suitable for radiation effects, and use it for the first ab initio simulation of primary damage and evolution of collision cascades. The model reliability is confirmed by good reproduction of experimentally measured threshold displacement energies and sputtering yields. We find that clustering and recrystallization of radiation-induced defects, propagation pattern of cascades, and coordination defects in the heat spike phase show striking differences to the widely used analytical potentials. The results reveal that small defect clusters are predominant and show new defect structures such as a vacancy surrounded by three interstitials. GRAPHICAL ABSTRACT Impact statement Quantum-mechanical level of accuracy in simulation of primary damage was achieved by a silicon machine learning potential. The results show quantitative and qualitative differences from the damage predicted by any previous models. read less NOT USED (high confidence) M. Farzinpour, D. Toghraie, B. Mehmandoust, F. Aghadavoudi, and A. Karimipour, “Molecular dynamics simulation of ferronanofluid behavior in a nanochannel in the presence of constant and time-dependent magnetic fields,” Journal of Thermal Analysis and Calorimetry. 2020. link Times cited: 22 NOT USED (high confidence) S. Wang and K. Komvopoulos, “Structure evolution during deposition and thermal annealing of amorphous carbon ultrathin films investigated by molecular dynamics simulations,” Scientific Reports. 2020. link Times cited: 24 NOT USED (high confidence) M. Hossain, G. Pawar, B. Liaw, K. Gering, E. J. Dufek, and A. V. van Duin, “Lithium-electrolyte solvation and reaction in the electrolyte of a lithium ion battery: A ReaxFF reactive force field study.,” The Journal of chemical physics. 2020. link Times cited: 19 Abstract: In the electrode/electrolyte interface of a typical lithium-… read moreAbstract: In the electrode/electrolyte interface of a typical lithium-ion battery, a solid electrolyte interphase layer is formed as a result of electrolyte decomposition during the initial charge/discharge cycles. Electron leakage from the anode to the electrolyte reduces the Li+-ion and makes it more reactive, resulting in decomposition of the organic electrolyte. To study the Li-electrolyte solvation, solvent exchange, and subsequent solvent decomposition reactions at the anode/electrolyte interface, we have extended the existing ReaxFF reactive force field parameter sets to organic electrolyte species, such as ethylene carbonate, ethyl methyl carbonate, vinylene carbonate, and LiPF6 salt. Density Functional Theory (DFT) data describing Li-associated initiation reactions for the organic electrolytes and binding energies of Li-electrolyte solvation structures were generated and added to the existing ReaxFF training data, and subsequently, we trained the ReaxFF parameters with the aim of finding the optimal reproduction of the DFT data. In order to discern the characteristics of the Li neutral and cation, we have introduced a second Li parameter set to describe the Li+-ion. ReaxFF is trained for Li-neutral and Li+-cation to have similar solvation energies, but unlike the neutral Li, Li+ will not induce reactivity in the organic electrolyte. Solvent decomposition reactions are presumed to happen once Li+-ions are reduced to Li-atoms, which can be simulated using a Monte Carlo type atom modification within ReaxFF. This newly developed force field is capable of distinguishing between a Li-atom and a Li+-ion properly. Moreover, it is found that the solvent decomposition reaction barrier is a function of the number of ethylene carbonate molecules solvating the Li-atom. read less NOT USED (high confidence) W. Xu, Y. Jiao, and J. Fish, “An atomistically-informed multiplicative hyper-elasto-plasticity-damage model for high-pressure induced densification of silica glass,” Computational Mechanics. 2020. link Times cited: 0 NOT USED (high confidence) W. Xu, Y. Jiao, and J. Fish, “An atomistically-informed multiplicative hyper-elasto-plasticity-damage model for high-pressure induced densification of silica glass,” Computational Mechanics. 2020. link Times cited: 6 NOT USED (high confidence) A. Martini, S. Eder, and N. Dörr, “Tribochemistry: A Review of Reactive Molecular Dynamics Simulations,” Lubricants. 2020. link Times cited: 40 Abstract: Tribochemistry, the study of chemical reactions in tribologi… read moreAbstract: Tribochemistry, the study of chemical reactions in tribological interfaces, plays a critical role in determining friction and wear behavior. One method researchers have used to explore tribochemistry is “reactive” molecular dynamics simulation based on empirical models that capture the formation and breaking of chemical bonds. This review summarizes studies that have been performed using reactive molecular dynamics simulations of chemical reactions in sliding contacts. Topics include shear-driven reactions between and within solid surfaces, between solid surfaces and lubricating fluids, and within lubricating fluids. The review concludes with a perspective on the contributions of reactive molecular dynamics simulations to the current understanding of tribochemistry, as well as opportunities for this approach going forward. read less NOT USED (high confidence) L. Bikova, N. Jelyabovskaya, V. Shukhtin, and V. Ulasyuk, “Thermoelectric Structured Systems with Nanophonic Metamaterials,” DEStech Transactions on Environment, Energy and Earth Science. 2020. link Times cited: 0 Abstract: This work is aimed at analysis of methods for the developing… read moreAbstract: This work is aimed at analysis of methods for the developing of new thermoelectric systems including those based on the so-called locally resonant "nanophonic" metamaterials. These materials, with their experimental realization, are intended for use in thermoelectric systems, since they have a large value of the thermoelectric Q-factor ZT. The increase in Q is achieved by reducing the thermal conductivity of the material, which leads to an increase in the value of ZT, if the electrical conductivity remains the same. The reduction in thermal conductivity, without corresponding reduction in electrical conductivity, is achieved by choosing a configuration of thermoelectric materials consisting of a thin film with a periodic set of columns mounted on a free surface. Such a configuration qualitatively changes the base phonon spectrum of a thin film due to the hybridization mechanism between local column resonances and underlying lattice scattering. Numerical methods are used to study changes in the phonon spectrum and, as a consequence, a change (decrease) in the value of thermal conductivity depending on variations in the system architecture. read less NOT USED (high confidence) E. Giessen et al., “Roadmap on multiscale materials modeling,” Modelling and Simulation in Materials Science and Engineering. 2020. link Times cited: 91 Abstract: Modeling and simulation is transforming modern materials sci… read moreAbstract: Modeling and simulation is transforming modern materials science, becoming an important tool for the discovery of new materials and material phenomena, for gaining insight into the processes that govern materials behavior, and, increasingly, for quantitative predictions that can be used as part of a design tool in full partnership with experimental synthesis and characterization. Modeling and simulation is the essential bridge from good science to good engineering, spanning from fundamental understanding of materials behavior to deliberate design of new materials technologies leveraging new properties and processes. This Roadmap presents a broad overview of the extensive impact computational modeling has had in materials science in the past few decades, and offers focused perspectives on where the path forward lies as this rapidly expanding field evolves to meet the challenges of the next few decades. The Roadmap offers perspectives on advances within disciplines as diverse as phase field methods to model mesoscale behavior and molecular dynamics methods to deduce the fundamental atomic-scale dynamical processes governing materials response, to the challenges involved in the interdisciplinary research that tackles complex materials problems where the governing phenomena span different scales of materials behavior requiring multiscale approaches. The shift from understanding fundamental materials behavior to development of quantitative approaches to explain and predict experimental observations requires advances in the methods and practice in simulations for reproducibility and reliability, and interacting with a computational ecosystem that integrates new theory development, innovative applications, and an increasingly integrated software and computational infrastructure that takes advantage of the increasingly powerful computational methods and computing hardware. read less NOT USED (high confidence) K. Momeni et al., “Multiscale computational understanding and growth of 2D materials: a review,” npj Computational Materials. 2020. link Times cited: 85 NOT USED (high confidence) Q. Gao, Y. Han, P. Liang, and J. Meng, “Influence of an external electric field on the deprotonation reactions of an Fe3+-solvated molecule: a reactive molecular dynamics study.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 13 Abstract: The influence of an external electric field (EEF) on the dep… read moreAbstract: The influence of an external electric field (EEF) on the deprotonation reaction of Fe3+-solvated molecules was studied using reactive molecular dynamics (ReaxFF MD) simulations. It was examined in terms of changes in structural properties, kinetics, system energy, and reaction products under an EEF, and the results were further verified experimentally. The research results show that the presence of an EEF will affect the distribution of water molecules around Fe3+ and provide energy for the fracturing of O-H bonds. The increase in the state of reaction products represented by H+ also suggests that the EEF can promote the deprotonation reaction of Fe3+-solvated molecules. The viscosity of the system is significantly increased under an EEF. The experimental results for verification show that the pH of the FeCl3 solution is reduced under the action of an EEF, which means that the hydrolysis of Fe3+ has been promoted. The experimental results are consistent with the results of the MD simulations. read less NOT USED (high confidence) B. Sharma and A. Parashar, “A review on thermo-mechanical properties of bi-crystalline and polycrystalline 2D nanomaterials,” Critical Reviews in Solid State and Materials Sciences. 2020. link Times cited: 29 Abstract: Due to outstanding properties, graphene and h-BN nanosheets … read moreAbstract: Due to outstanding properties, graphene and h-BN nanosheets are emerging as a potential candidate for wide spectrum of applications in the field of engineering and bio-medical science. Graphene and h-BN nanosheets have comparable mechanical and thermal properties, whereas due to high band gap h-BN (∼5eV) have contrasting electrical conductivities. Large size graphene and h-BN nanosheets are synthesized by chemical vapor deposition technique, which results in polycrystalline atomic structure. These polycrystalline nanosheets are characterized either by experimental means or numerical simulations. Experimental techniques are considered as most accurate and practical, but cost and time involved in these techniques limits it application at the nanoscale level. On the other hand, atomistic modeling techniques are emerging as viable alternatives to the experimentations, and are accurate enough to predict the mechanical properties, fracture toughness, and thermal conductivities of polycrystalline graphene and h-BN nanosheets. This comprehensive review article encompasses different characterizing techniques used by the researchers for polycrystalline nanosheets. This review will help in elaborating the properties of polycrystalline graphene and h-BN, and also establishing a perspective on how the microstructure impacts its large-scale physical properties. read less NOT USED (high confidence) Á. Jász, Á. Rák, I. Ladjánszki, and G. Cserey, “Classical molecular dynamics on graphics processing unit architectures,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2020. link Times cited: 6 Abstract: Molecular dynamics (MD) has experienced a significant growth… read moreAbstract: Molecular dynamics (MD) has experienced a significant growth in the recent decades. Simulating systems consisting of hundreds of thousands of atoms is a routine task of computational chemistry researchers nowadays. Thanks to the straightforwardly parallelizable structure of the algorithms, the most promising method to speed‐up MD calculations is exploiting the large‐scale processing power offered by the parallel hardware architecture of graphics processing units or GPUs. Programming GPUs is becoming easier with general‐purpose GPU computing frameworks and higher levels of abstraction. In the recent years, implementing MD simulations on graphics processors has gained a large interest, with multiple popular software packages including some form of GPU‐acceleration support. Different approaches have been developed regarding various aspects of the algorithms, with important differences in the specific solutions. Focusing on published works in the field of classical MD, we describe the chosen implementation methods and algorithmic techniques used for porting to GPU, as well as how recent advances of GPU architectures will provide even more optimization possibilities in the future. read less NOT USED (high confidence) S. Damián-Vázquez, J. F. Alvarado, and E. O. Castrejón-González, “A combined force field for the silica/nickel system,” Molecular Simulation. 2020. link Times cited: 2 Abstract: ABSTRACT Silica and nickel are frequently used in the synthe… read moreAbstract: ABSTRACT Silica and nickel are frequently used in the synthesis of carbon nanotubes by the chemical vapour deposition (CVD) process. The molecular simulation of this process requires the knowledge of force fields to model the interactions occurring between those species at the atomic scale. This work proposes a combined force field to model the silica/nickel system when these components are in contact as substrate and catalyst, respectively, in the CVD process. The proposed combined force field includes the Lennard–Jones (n–m) potential for modelling the silicon/nickel pair interactions and the Buckingham potential for the oxygen/nickel pair interactions. The combined force field is completed by the Tersoff potential to model silica (SiO2) and the Sutton–Chen potential for the cohesive forces present in the nickel clusters. Parameters for the Lennard–Jones (n–m) and Buckingham pair potentials were fitted, by the least squares technique, to interaction energies data for the silica/nickel system. The energies were obtained from Ab-initio (DFT) calculations using the VASP code. It was found that the combined force field reproduces adequately, by molecular dynamics simulation, the adherence (adsorption) of nickel clusters on the silica surface. Keeping stable this configuration is crucial in modelling the carbon nanotubes synthesis by the CVD process. read less NOT USED (high confidence) B. Babu and B. P. Patel, “An improved quadrilateral finite element for nonlinear second-order strain gradient elastic Kirchhoff plates,” Meccanica. 2020. link Times cited: 7 NOT USED (high confidence) T. Mueller, A. Hernandez, and C. Wang, “Machine learning for interatomic potential models.,” The Journal of chemical physics. 2020. link Times cited: 189 Abstract: The use of supervised machine learning to develop fast and a… read moreAbstract: The use of supervised machine learning to develop fast and accurate interatomic potential models is transforming molecular and materials research by greatly accelerating atomic-scale simulations with little loss of accuracy. Three years ago, Jörg Behler published a perspective in this journal providing an overview of some of the leading methods in this field. In this perspective, we provide an updated discussion of recent developments, emerging trends, and promising areas for future research in this field. We include in this discussion an overview of three emerging approaches to developing machine-learned interatomic potential models that have not been extensively discussed in existing reviews: moment tensor potentials, message-passing networks, and symbolic regression. read less NOT USED (high confidence) V. Reshetniak and A. Aborkin, “Aluminum–Carbon Interaction at the Aluminum–Graphene and Aluminum–Graphite Interfaces,” Journal of Experimental and Theoretical Physics. 2020. link Times cited: 8 NOT USED (high confidence) Y. Karaaslan, H. Yapicioglu, and C. Sevik, “Assessment of Thermal Transport Properties of Group-III Nitrides: A Classical Molecular Dynamics Study with Transferable Tersoff-Type Interatomic Potentials,” Physical Review Applied. 2020. link Times cited: 14 Abstract: In this study, by means of classical molecular dynamics simu… read moreAbstract: In this study, by means of classical molecular dynamics simulations, we investigated the thermal transport properties of hexagonal single-layer, zinc-blend and wurtzite phases of BN, AlN, and GaN crystals, which are very promising for the application and design of high-quality electronic devices. With this in mind, we generated fully transferable Tersoff-type empirical inter-atomic potential parameter sets by utilizing an optimization procedure based on particle swarm optimization. The predicted thermal properties as well as the structural, mechanical and vibrational properties of all materials are in very good agreement with existing experimental and first-principles data. The impact of isotopes on thermal transport is also investigated and between $\sim$10 and 50\% reduction in phonon thermal transport with random isotope distribution is observed in BN and GaN crystals. Our investigation distinctly shows that the generated parameter sets are fully transferable and very useful in exploring the thermal properties of systems containing these nitrides. read less NOT USED (high confidence) C.-W. Lee, J. A. Stewart, R. Dingreville, S. Foiles, and A. Schleife, “Multiscale simulations of electron and ion dynamics in self-irradiated silicon,” Physical Review B. 2020. link Times cited: 22 Abstract: The interaction of energetic ions with the electronic and io… read moreAbstract: The interaction of energetic ions with the electronic and ionic system of target materials is an interesting but challenging multi-scale problem and understanding of the early stages after impact of heavy, initially charged ions is particularly poor. At the same time, energy deposition during these early stages determines later formation of damage cascades. We address the multi-scale character by combining real-time time-dependent density functional theory for electron dynamics with molecular dynamics simulations of damage cascades. Our first-principles simulations prove that core electrons affect electronic stopping and have an unexpected influence on the charge state of the projectile. We show that this effect is absent for light projectiles, but dominates the stopping physics for heavy projectiles. By parameterizing an inelastic energy loss friction term in the molecular dynamics simulations using our first-principles results, we also show a qualitative influence of electronic stopping physics on radiation-damage cascades. read less NOT USED (high confidence) A. Kolesnikova, M. Rozhkov, and A. Romanov, “On Fracture of Pseudo-Graphenes,” Mechanics of Solids. 2020. link Times cited: 1 NOT USED (high confidence) A. Kolesnikova, M. Rozhkov, and A. Romanov, “On Fracture of Pseudo-Graphenes,” Mechanics of Solids. 2020. link Times cited: 0 NOT USED (high confidence) S. Ajori, A. Ameri, and R. Ansari, “The effect of chitosan adsorption on the stability characteristics of single- and double-walled boron-nitride nanotubes under compressive force using molecular dynamics simulations,” Structural Chemistry. 2019. link Times cited: 1 NOT USED (high confidence) R. Li, E. Lee, and T. Luo, “A unified deep neural network potential capable of predicting thermal conductivity of silicon in different phases,” arXiv: Materials Science. 2019. link Times cited: 47 NOT USED (high confidence) A. Sircar and P. Patra, “A simple generalization of Prandtl–Tomlinson model to study nanoscale rolling friction,” Journal of Applied Physics. 2019. link Times cited: 7 Abstract: Prandtl-Tomlinson (PT) model has been very successful in exp… read moreAbstract: Prandtl-Tomlinson (PT) model has been very successful in explaining nanoscale friction in a variety of situations. However, the simplistic PT model, on account of having a point mass being dragged across a sinusoidal force field, cannot be used for studying rolling friction at nanoscales. In this manuscript, we generalize the PT model as a collection of point particles arranged in a circle of radius $R$. The resulting ``rigid body'' is driven in a composite force field by a moving spring (of stiffness $k$) connected to the center of mass of the rigid body in presence of damping. The force field is a product of the familiar sinusoidal function used in the PT model with a parametrically controlled ($\lambda$) exponentially varying function that is dependent on the vertical coordinates of the particles. Our generalized model degenerates to the standard PT model if $R \ll 1$ and $\lambda \to 0$. With $R \sim 1$ and $\lambda \to 0$, the model undergoes a transition from sticky dynamics to smooth dynamics as $k$ is increased to a critical value. The analytical expression agrees well with the simulation results. Similar analytical expressions have been derived for $ \lambda \neq 0$ as well. In this scenario, the sticky dynamics is experienced in both $x$ and $y$ directions, and our numerical results agree with the analytical solution for $x$ direction. The dynamics, investigated numerically for the general case of $R \sim 1$ and $\lambda \neq 0$, reveals several interesting aspects of nanoscale tribology including the regimes where energy dissipation due to friction is minimum. Further the results from our proposed model are in qualitative agreement with those from MD simulations as well. We believe that the simplicity of our model along with its similarity to the PT model may make it a popular tool for analyzing complicated nanotribological regimes. read less NOT USED (high confidence) B. Babu and B. P. Patel, “An improved quadrilateral finite element for nonlinear second-order strain gradient elastic Kirchhoff plates,” Meccanica. 2019. link Times cited: 0 NOT USED (high confidence) J. Hur, “Modified potential for atomistic simulation of the growth of carbon materials from binary alloy catalysts.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 3 Abstract: A new hybrid bond order potential has been developed and imp… read moreAbstract: A new hybrid bond order potential has been developed and implemented to describe carbon-bimetallic alloy interactions, which are involved in the catalytic growth of carbon materials such as graphene and carbon nanotubes on the surface of binary alloy catalysts. In carefully adjusting the parameters, the potential energy fitting correlated with the results calculated from the density functional theory (DFT) method leads to a high quality empirical force field with an average error of <4.5% only. With the PES accuracy, in total 16 (n,m) have been successfully obtained from the MD trajectories in this work, and the structural evolution including random chirality and diameter formation has been identified. The newly modified force field is expected to be useful for modelling the spontaneous growth of carbon materials, particularly tubes on binary alloy clusters, giving an idea of how these C-C, C-M, and M-M interactions affect the growth behavior of carbon nanotubes. In addition, the new FF is only valid for liquid alloy nanoparticles at this time, but the use of solid alloy nanocatalysts with the new FF can be further employed for 2-D material growth such as graphene layer growth. read less NOT USED (high confidence) Y. Lei, Y. Yan, and J. Lv, “Atomistic study of the strengthening mechanisms of graphene coated aluminum,” Nanotechnology. 2019. link Times cited: 7 Abstract: We have investigated the nano-indentation responses of graph… read moreAbstract: We have investigated the nano-indentation responses of graphene/aluminum systems via computational nano-indentation processes by using molecular dynamics simulations. The effects of system temperature, grain-orientation and bilayer graphene are also investigated. We demonstrate that, the graphene coating enlarges the load-carrying area by about 5.36 times and changes the deformation behaviors of aluminum substrate during nano-indentation processes. The load bearing capacity of graphene/Al system is significantly improved by about 4.7 times compared with that of bare Al system. It is revealed that higher system temperature weakens the ultimate indentation depth and corresponding load. The grain orientation of aluminum substrate hardly affect the indentation mechanical properties of graphene/Al system. The strengthening effect of bilayer graphene is about 1.5 times that of monolayer graphene. read less NOT USED (high confidence) S. Pozdnyakov, A. Oganov, E. Mazhnik, A. Mazitov, and I. Kruglov, “Fast general two- and three-body interatomic potential,” Physical Review B. 2019. link Times cited: 6 Abstract: We introduce a new class of machine learning interatomic pot… read moreAbstract: We introduce a new class of machine learning interatomic potentials - fast General Two- and Three-body Potential (GTTP) which are as fast as conventional empirical potentials and require computational time that remains constant with increasing fitting flexibility. GTTP does not contain any assumptions about functional form of two- and three-body interactions. These interactions can be modeled arbitrarily accurately potentially by thousands of parameters not affecting resulting computational cost. Time complexity is O(1) per every considered pair or triple of atoms. The fitting procedure is reduced to simple linear regression on ab initio calculated energies and forces and leads to effective two- and three-body potential which reproduces quantum many-body interactions as accurately as possible. Our potential can be made continuously differentiable any number of times at the expense of increased computational time. We made a number of performance tests on one-, two- and three-component systems. Flexibility of the introduced approach makes the potential transferable in terms of size and type of atomic systems. We show, that trained on randomly generated structures with just 8 atoms in the unit cell, it significantly outperforms common empirical interatomic potentials in the study of large systems, such as grain boundaries in polycrystalline materials. read less NOT USED (high confidence) A. Shahabodini, Y. Gholami, R. Ansari, and H. Rouhi, “Vibration analysis of graphene sheets resting on Winkler/Pasternak foundation: A multiscale approach,” The European Physical Journal Plus. 2019. link Times cited: 10 NOT USED (high confidence) L. Liu et al., “Molecular dynamics simulation of helium ion implantation into silicon and its migration,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2019. link Times cited: 16 NOT USED (high confidence) T. Riedl et al., “Applicability of molecular statics simulation to partial dislocations in GaAs,” arXiv: Materials Science. 2019. link Times cited: 0 NOT USED (high confidence) H. E. Sauceda, S. Chmiela, I. Poltavsky, K.-R. Muller, and A. Tkatchenko, “Construction of Machine Learned Force Fields with Quantum Chemical Accuracy: Applications and Chemical Insights,” Machine Learning Meets Quantum Physics. 2019. link Times cited: 10 NOT USED (high confidence) E. Tangarife, A. Romero, and J. Mejía‐López, “A charge optimized many-body potential for iron/iron-fluoride systems.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 2 Abstract: A classical interatomic potential for iron/iron-fluoride sys… read moreAbstract: A classical interatomic potential for iron/iron-fluoride systems is developed in the framework of the charge optimized many-body (COMB) potential. This interatomic potential takes into consideration the effects of charge transfer and many-body interactions depending on the chemical environment. The potential is fitted to a training set composed of both experimental and ab initio results of the cohesive energies of several Fe and FeF2 crystal phases, the two fluorine molecules F2 and the F2-1 dissociation energy curve, the Fe and FeF2 lattice parameters of the ground state crystalline phase, and the elastic constants of the body centered cubic Fe structure. The potential is tested in an NVT ensemble for different initial structural configurations as the crystal ground state phases, F2 molecules, iron clusters, and iron nanospheres. In particular, we model the FeF2/Fe bilayer and multilayer interfaces, as well as a system of square FeF2 nanowires immersed in an iron solid. It has been shown that there exists a reordering of the atomic positions for F and Fe atoms at the interface zone; this rearrangement leads to an increase in the charge transfer among the atoms that make the interface and put forward a possible mechanism of the exchange bias origin based on asymmetric electric charge transfer in the different spin channels. read less NOT USED (high confidence) M. Comin and L. J. Lewis, “Deep-learning approach to the structure of amorphous silicon,” Physical Review B. 2019. link Times cited: 6 Abstract: We present a deep-learning approach for modeling the atomic … read moreAbstract: We present a deep-learning approach for modeling the atomic structure of amorphous silicon ( a -Si). While accurate models of disordered systems require an ab initio description of the energy landscape which severely limits the attainable system size, large-scale models rely on empirical potentials, at the price of reduced reliability and a computational load that is still restricting for many purposes. In this paper, we explore an approach based on deep learning, particularly generative modeling that could reconcile both requirements of accuracy and efficiency by learning structural features from data. When trained on a set of observations, such models can generate new structures very efficiently with the desired level of accuracy, as determined by the data set. We first validate our approach by training a convolutional neural network to approximate the potential-energy surface of a -Si, as given by the Stillinger-Weber potential, which results in a root-mean-square error of 5.05 meV per atom—about 0 . 16% of the atomic energy. We then train a deep generative model, the Wasserstein autoencoder, for the generation of a -Si configurations. Our approach leads to models which exhibit some of the essential features of a -Si while possessing too much structural disorder, thus suggesting that the method is viable; we indicate avenues for improving it towards the generation of state-of-the-art structures. read less NOT USED (high confidence) J. Acharjee and R. P. Joshi, “Numerical evaluation of hydrogen outgassing from copper electrodes with mitigation based on a tungsten capping layer,” Physics of Plasmas. 2019. link Times cited: 6 Abstract: Outgassing remains a pertinent issue for high power applicat… read moreAbstract: Outgassing remains a pertinent issue for high power applications and is exacerbated by the high field driven, localized heating environments commonly encountered. Here, molecular dynamics simulations are performed for a simple model-based assessment of outgassing from electrodes. Our results of temperature dependent diffusion coefficients for hydrogen in copper agree well with experimental reports over a wide range spanning 300 K to 1330 K. Separate results are also obtained for transport of hydrogen to ascertain whether a grain-boundary would facilitate channeled transport or work to impede flow by clustering the gas atoms. Finally, the use of a tungsten overlayer on copper is evaluated as a material-based strategy for mitigating outgassing. It is demonstrated that a few monolayers of tungsten coating on the outer surface can be effective in significantly reducing outdiffusion at 700 K.Outgassing remains a pertinent issue for high power applications and is exacerbated by the high field driven, localized heating environments commonly encountered. Here, molecular dynamics simulations are performed for a simple model-based assessment of outgassing from electrodes. Our results of temperature dependent diffusion coefficients for hydrogen in copper agree well with experimental reports over a wide range spanning 300 K to 1330 K. Separate results are also obtained for transport of hydrogen to ascertain whether a grain-boundary would facilitate channeled transport or work to impede flow by clustering the gas atoms. Finally, the use of a tungsten overlayer on copper is evaluated as a material-based strategy for mitigating outgassing. It is demonstrated that a few monolayers of tungsten coating on the outer surface can be effective in significantly reducing outdiffusion at 700 K. read less NOT USED (high confidence) V. L. Deringer, M. A. Caro, and G. Csányi, “Machine Learning Interatomic Potentials as Emerging Tools for Materials Science,” Advanced Materials. 2019. link Times cited: 245 Abstract: Atomic‐scale modeling and understanding of materials have ma… read moreAbstract: Atomic‐scale modeling and understanding of materials have made remarkable progress, but they are still fundamentally limited by the large computational cost of explicit electronic‐structure methods such as density‐functional theory. This Progress Report shows how machine learning (ML) is currently enabling a new degree of realism in materials modeling: by “learning” electronic‐structure data, ML‐based interatomic potentials give access to atomistic simulations that reach similar accuracy levels but are orders of magnitude faster. A brief introduction to the new tools is given, and then, applications to some select problems in materials science are highlighted: phase‐change materials for memory devices; nanoparticle catalysts; and carbon‐based electrodes for chemical sensing, supercapacitors, and batteries. It is hoped that the present work will inspire the development and wider use of ML‐based interatomic potentials in diverse areas of materials research. read less NOT USED (high confidence) J. Byggmastar, A. Hamedani, K. Nordlund, and F. Djurabekova, “Machine-learning interatomic potential for radiation damage and defects in tungsten,” Physical Review B. 2019. link Times cited: 58 Abstract: We introduce a machine-learning interatomic potential for tu… read moreAbstract: We introduce a machine-learning interatomic potential for tungsten using the Gaussian Approximation Potential framework. We specifically focus on properties relevant for simulations of radiation-induced collision cascades and the damage they produce, including a realistic repulsive potential for the short-range many-body cascade dynamics and a good description of the liquid phase. Furthermore, the potential accurately reproduces surface properties and the energetics of vacancy and self-interstitial clusters, which have been long-standing deficiencies of existing potentials. The potential enables molecular dynamics simulations of radiation damage in tungsten with unprecedented accuracy. read less NOT USED (high confidence) E. J. Ragasa, C. J. O’Brien, R. G. Hennig, S. Foiles, and S. Phillpot, “Multi-objective optimization of interatomic potentials with application to MgO,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 6 Abstract: The parameterization of a functional form for an interatomic… read moreAbstract: The parameterization of a functional form for an interatomic potential is treated as a problem in multi-objective optimization. An autonomous, machine-learning approach based on the identification of the Pareto hypersurface of errors in predicted properties allows the development of an ensemble of parameterizations with high materials fidelity and robustness. The efficacy of this approach is illustrated for the simple example of a Buckingham potential for MgO. This approach also provides a strong foundation for uncertainty quantification of potential parameterizations. read less NOT USED (high confidence) A. Hashemi, R. Guo, K. Esfarjani, and S. Lee, “Ab initio
phonon transport across grain boundaries in graphene using machine learning based on small dataset,” Physical Review Materials. 2019. link Times cited: 1 Abstract: Establishing the structure-property relationship for grain b… read moreAbstract: Establishing the structure-property relationship for grain boundaries (GBs) is critical for developing next generation functional materials, but has been severely hampered due to its extremely large configurational space. Atomistic simulations with low computational cost and high predictive power are strongly desirable, but the conventional simulations using empirical interatomic potentials and density functional theory suffer from the lack of predictive power and high computational cost, respectively. A machine learning interatomic potential (MLIP) recently emerged but often requires an extensive size of the training dataset, making it a less feasible approach. Here we demonstrate that an MLIP trained with a rationally designed small training dataset can predict thermal transport across GBs in graphene with ab initio accuracy at an affordable computational cost. In particular, we employed a rational approach based on the structural unit model to find a small set of GBs that can represent the entire configurational space and thus can serve as a cost-effective training dataset for the MLIP. Only 5 GBs were found to be enough to represent the entire configurational space of graphene GBs. Using the atomistic Green’s function approach and the MLIP, we revealed that the structure-thermal resistance relation in graphene does not follow the common understanding that large dislocation density causes larger thermal resistance. In fact, thermal resistance is nearly independent of dislocation density at room temperature and is higher when the dislocation density is small at sub-room temperature. We explain this intriguing behavior with the buckling near a GB causing a strong scattering of flexural phonon modes. Our work shows that a machine learning technique combined with conventional wisdom (e.g., structural unit model) can extend the recent success of ab initio thermal transport simulation, which has been mostly limited to single crystals, to complex yet practically important polycrystals with GBs. read less NOT USED (high confidence) A. Giri and P. Hopkins, “A Review of Experimental and Computational Advances in Thermal Boundary Conductance and Nanoscale Thermal Transport across Solid Interfaces,” Advanced Functional Materials. 2019. link Times cited: 144 Abstract: Interfacial thermal resistance is the primary impediment to … read moreAbstract: Interfacial thermal resistance is the primary impediment to heat flow in materials and devices as characteristic lengths become comparable to the mean‐free paths of the energy carriers. This thermal boundary conductance across solid interfaces at the nanoscale can affect a plethora of applications. The recent experimental and computational advances that have led to significant atomistic insights into the nanoscopic thermal transport mechanisms at interfaces between various types of materials are summarized. The authors focus on discussions of works that have pushed the limits to interfacial heat transfer and drastically increased the understanding of thermal boundary conductance on the atomic and nanometer scales near solid/solid interfaces. Specifically, the role of localized interfacial modes on the energy conversion processes occurring at interfaces is emphasized in this review. The authors also focus on experiments and computational works that have challenged the traditionally used phonon gas models in interpreting the physical mechanisms driving interfacial energy transport. Finally, the authors discuss the future directions and avenues of research that can further the knowledge of heat transfer across systems with broken symmetries. read less NOT USED (high confidence) E. Zhang, Y.-H. Yao, T. Gao, D. Kang, J. Wu, and J. Dai, “The effect of external temperature gradients on thermal conductivity in non-equilibrium molecular dynamics simulations: From nanowires to bulk Si,” The Journal of Chemical Physics. 2019. link Times cited: 6 Abstract: Nonequilibrium molecular dynamics is widely used to calculat… read moreAbstract: Nonequilibrium molecular dynamics is widely used to calculate the thermal conductivity of various materials, but the influence of temperature gradient to thermal conductivity has received limited attention within current research studies. The purpose of this article is to explore the discrepancy between intrinsic and extrinsic thermal conductivities under different temperature gradients, which can be considered as external fields. The analyses of phonon density of states have shown that the temperature gradient plays a role in the external field, and a larger temperature gradient activates more low-frequency vibrational modes, which leads to larger thermal conductivities. Specially, the thermal conductivity increases linearly with the temperature gradient when using Stillinger-Weber (SW) potential. Moreover, a new formula was derived to satisfactorily fit the thermal conductivities of bulk Si and silicon nanowires (SiNWs) for various cell sizes, and the physical meaning of the formula was explained. It is shown that the SW potential and Tersoff potential of Si produce different thermal conductivities. By comparing the results of first principles simulations, the Tersoff potential gives rise to better description of vibrational modes. read less NOT USED (high confidence) Y. Gholami, A. Shahabodini, R. Ansari, and H. Rouhi, “Nonlinear vibration analysis of graphene sheets resting on Winkler–Pasternak elastic foundation using an atomistic-continuum multiscale model,” Acta Mechanica. 2019. link Times cited: 9 NOT USED (high confidence) S. Ajori, A. Ameri, and R. Ansari, “The mechanical properties and structural instability of single- and double-walled boron-nitride nanotubes functionalized with 2-methoxy-N,N-dimethylethanamine (MDE) using molecular dynamics simulations,” The European Physical Journal D. 2019. link Times cited: 4 NOT USED (high confidence) S. N. A. Kalkhoran, M. Vahdati, and J. Yan, “Molecular Dynamics Investigation of Nanometric Cutting of Single-Crystal Silicon Using a Blunt Tool,” JOM. 2019. link Times cited: 11 NOT USED (high confidence) G. Shchygol, A. Yakovlev, T. Trnka, A. V. van Duin, and T. Verstraelen, “ReaxFF Parameter Optimization with Monte Carlo and Evolutionary Algorithms: Guidelines and Insights.,” Journal of chemical theory and computation. 2019. link Times cited: 44 Abstract: ReaxFF is a computationally efficient force field to simulat… read moreAbstract: ReaxFF is a computationally efficient force field to simulate complex reactive dynamics in extended molecular models with diverse chemistries, if reliable force-field parameters are available for the chemistry of interest. If not, they must be optimized by minimizing the error ReaxFF makes on a relevant training set. Because this optimization is far from trivial, many methods, in particular genetic algorithms (GAs), have been developed to search for the global optimum in parameter space. Recently, two alternative parameter calibration techniques were proposed, i.e. Monte-Carlo Force Field optimizer (MCFF) and Covariance Matrix Adaptation Evolutionary Strategy (CMA-ES). In this work, CMA-ES, MCFF and a GA method (OGOLEM) are systematically compared using three training sets from the literature. By repeating optimizations with different random seeds and initial parameter guesses, it is shown that a single optimization run with any of these methods should not be trusted blindly: non-reproducible, poor or premature convergence are common deficiencies. GA shows the smallest risk of getting trapped into a local minimum, whereas CMA-ES is capable of reaching the lowest errors for two third of the cases, albeit not systematically. For each method, we provide reasonable default settings and our analysis offers useful guidelines for their usage in future work. An important side effect impairing the parameter optimization is numerical noise. A detailed analysis reveals that it can be reduced, e.g. by using exclusively unambiguous geometry optimizations in the training set. Even without this noise, many distinct near-optimal parameter vectors can be found, which opens new avenues for improving the training set and detecting overfitting artifacts. read less NOT USED (high confidence) A. N. Ladines, T. Hammerschmidt, and R. Drautz, “BOPcat software package for the construction and testing of tight-binding models and bond-order potentials,” Computational Materials Science. 2019. link Times cited: 6 NOT USED (high confidence) A. Dmitriev, A. Nikonov, W. Österle, and B. Jim, “VERIFICATION OF RABINOWICZ’ CRITERION BY DIRECT MOLECULAR DYNAMICS MODELING,” Facta Universitatis, Series: Mechanical Engineering. 2019. link Times cited: 6 Abstract: In the paper we use direct molecular dynamics modeling to va… read moreAbstract: In the paper we use direct molecular dynamics modeling to validate the criterion for formation of wear debris proposed by E. Rabinowicz in 1958. A conventional molecular dynamics using a classical Tersoff’s potential was applied to simulate the sliding behavior within a thin film corresponding to a tribofilm formed from silica nano-particles in amorphous-like state. The simulation was carried out by varying the initial temperature and the spatial size of the simulated crystallite. The results show the change in sliding behavior of silica-based tribofilm depending on the temperature and the size parameter of the system under consideration. Thus increasing the temperature provides smooth sliding while at moderate conditions wear process can occur via debris formation. Our estimations show good correlation between predicted critical size of the simulated system and calculated energetic characteristics. read less NOT USED (high confidence) Z. Wang, “Gear junctions between chiral boron nitride nanotubes,” Physical Review B. 2019. link Times cited: 3 Abstract: A gear effect is demonstrated at parallel and cross junction… read moreAbstract: A gear effect is demonstrated at parallel and cross junctions between boron nitride nanotubes (BNNTs) via atomistic simulations. The atoms of neighboring BNNTs are meshed together at the junctions like gear teeth through long-range non-covalent interaction, which are shown to be able to transmit motion and power. The sliding motion of a BNNT can be spontaneously translated to rotating motion of an adjoining one or viceversa at a well-defined speed ratio. The transmittable motion and force strongly depend on the helical lattice structure of BNNTs represented by a chiral angle. The motion transmission efficiency of the parallel junctions increases up to a maximum for certain BNNTs depending on displacement rates. It then decreases with increasing chiral angles. For cross junctions, the angular motion transmission ratio increases with decreasing chiral angles of the driven BNNTs, while the translational one exhibits the opposite trend. read less NOT USED (high confidence) O. Matsiaka, R. Baker, and M. Simpson, “Continuum descriptions of spatial spreading for heterogeneous cell populations: theory and experiment,” bioRxiv. 2019. link Times cited: 2 Abstract: Variability in cell populations is frequently observed in bo… read moreAbstract: Variability in cell populations is frequently observed in both in vitro and in vivo settings. Intrinsic differences within populations of cells, such as differences in cell sizes or differences in rates of cell motility, can be present even within a population of cells from the same cell line. We refer to this variability as cell heterogeneity. Mathematical models of cell migration, for example, in the context of tumour growth and metastatic invasion, often account for both undirected (random) migration and directed migration that is mediated by cell-to-cell contacts and cell-to-cell adhesion. A key feature of standard models is that they often assume that the population is composed of identical cells with constant properties. This leads to relatively simple single-species homogeneous models that neglect the role of heterogeneity. In this work, we use a continuum modelling approach to explore the role of heterogeneity in spatial spreading of cell populations. We employ a three-species heterogeneous model of cell motility that explicitly incorporates different types of experimentally-motivated heterogeneity in cell sizes: (i) monotonically decreasing; (ii) uniform; (iii) non-monotonic; and (iv) monotonically increasing distributions of cell size. Comparing the density profiles generated by the three-species heterogeneous model with density profiles predicted by a more standard single-species homogeneous model reveals that when we are dealing with monotonically decreasing and uniform distributions a simple and computationally efficient single-species homogeneous model can be remarkably accurate in describing the evolution of a heterogeneous cell population. In contrast, we find that the simpler single-species homogeneous model performs relatively poorly when applied to non-monotonic and monotonically in-creasing distributions of cell sizes. Additional results for heterogeneity in parameters describing both undirected and directed cell migration are also considered, and we find that similar results apply. read less NOT USED (high confidence) I. Talyzin, M. V. Samsonov, V. Samsonov, M. Y. Pushkar,’ and V. V. Dronnikov, “Size Dependence of the Melting Point of Silicon Nanoparticles: Molecular Dynamics and Thermodynamic Simulation,” Semiconductors. 2019. link Times cited: 12 NOT USED (high confidence) W. Yao and L. Fan, “The Effect of Ion Irradiation Induced Defects on Mechanical Properties of Graphene/Copper Layered Nanocomposites,” Metals. 2019. link Times cited: 11 Abstract: One of the miraculous functions of graphene is to use its de… read moreAbstract: One of the miraculous functions of graphene is to use its defects to alter the material properties of graphene composites and, thereby, expand the application of graphene in other fields. In this paper, various defects have been created in graphene by using ion irradiation. Defective graphene is sandwiched between two copper layers. A numerical model of Graphene/Copper layered composites after irradiation damage was established by the molecular dynamics method. The effects of ion irradiation and temperature coupling on defective graphene/copper composites were studied. The results show that there are a lot of empty defects in graphene after irradiation injury, which will produce more incomplete bonding. Although the bonds between carbon atoms can be weakened, defective graphene still enhances the mechanical properties of pure copper. At the same time, the location and arrangement of defects have a great influence on the mechanical stability of graphene/copper composites, and the arrangement of empty defects has different effects on deformation behavior and the stress transfer mechanism. It can be concluded that the defects formed by radiation have an effect on the physical properties of two-dimensional materials. Therefore, irradiation technology can be used to artificially control the formation of defects, and then make appropriate adjustments to their properties. This can not only optimize the radiation resistance and mechanical properties of nuclear materials, but also expand the application of graphene in electronic devices and other fields. read less NOT USED (high confidence) S. M. Handrigan, L. Morrissey, and S. Nakhla, “Investigating various many-body force fields for their ability to predict reduction in elastic modulus due to vacancies using molecular dynamics simulations,” Molecular Simulation. 2019. link Times cited: 6 Abstract: ABSTRACT Molecular dynamics simulations are more frequently … read moreAbstract: ABSTRACT Molecular dynamics simulations are more frequently being utilised to predict macroscale mechanical properties as a result of atomistic defects. However, the interatomic force field can significantly affect the resulting mechanical properties. While several studies exist which demonstrate the ability of various force fields to predict mechanical properties, the investigation into which is most accurate for the investigation of vacancies is limited. To obtain meaningful predictions of mechanical properties, a clear understanding of force field parameterisation is required. As such, the current study evaluates various many-body force fields to demonstrate the reduction in mechanical properties of iron and iron–chromium due to the presence of vacancies while undergoing room temperature atomistic uniaxial tension. Reduction was normalised in each case with the zero-vacancy elastic modulus, removing the need to predict an accurate nominal elastic modulus. Comparisons were made to experimental data and an empirical model from literature. It was demonstrated that accurate fitting to vacancy formation and migration energy allowed for accurate predictions. In addition, bond-order based force fields showed enhanced predictions regardless of fitting procedure. Overall, these findings highlight the need to understand capabilities and limitations of available force fields, as well as the need for enhanced parameterisation of force fields. read less NOT USED (high confidence) B. Sharma and A. Parashar, “Atomistic simulations to study the effect of grain boundaries and hydrogen functionalization on the fracture toughness of bi-crystalline h-BN nanosheets.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 17 Abstract: The aim of this research article was to investigate the effe… read moreAbstract: The aim of this research article was to investigate the effect of grain boundaries (GBs), and hydrogen functionalisation on the fracture toughness of bi-crystalline hexagonal boron nitride (h-BN) nanosheets. Molecular dynamics based simulations were performed in conjunction with the reactive force field to study the crack tip behaviour in single and bi-crystalline h-BN nanosheets. Atomistic simulations help in predicting a positive effect of the GB plane in the near vicinity of the crack tip. The density of 5|7 dislocation pairs significantly affects the fracture behaviour of bi-crystalline h-BN nanosheets. Additionally, the distance of the GB plane from the crack tip, and limited hydrogen functionalisation of GB atoms, further help in improving the fracture toughness of bi-crystalline h-BN nanosheets. Hydrogen functionalisation helps in inducing out of plane displacement at the GB plane, which helps in arresting or retarding the crack propagation. It can be concluded from the results that instead of deteriorating, geometrical defects such as GBs can also be used to tailor the fracture toughness of h-BN nanosheets. This study on the fracture toughness of bi-crystalline h-BN nanosheets helps in complementing the research on using porous h-BN nanosheets as nanomembranes for water desalination and ion separation. read less NOT USED (high confidence) Y. Jin et al., “Comparison of the oxidation resistance of synthetic ester oils DOA and TDTM: Experimental evaluation and theoretical calculation,” Lubrication Science. 2019. link Times cited: 7 Abstract: The oxidation behavior of the synthetic ester oils di‐isooct… read moreAbstract: The oxidation behavior of the synthetic ester oils di‐isooctyl adipate (DOA) and tri‐isodecyl trimellitate (TDTM) was investigated by analysing the oxidation induction time and oxidation onset temperature via pressure differential scanning calorimetry tests, and the variation in viscosity after oven accelerated oxidation tests. Moreover, theoretical calculations of the fractional free volumes and oxygen diffusion coefficients of DOA and TDTM were performed, and atomistic‐scale insights into the oxidative mechanisms of both oils were proposed based on reactive molecular dynamic (RMD) simulations. Better oxidation resistance for TDTM was confirmed with experimental observations. RMD results indicated that the C―O bonds of the alcohol chain and ester group of DOA were susceptible to oxidation. However, only the dissociation of the C―O bond of the alcohol chain initiated the oxidation of TDTM. Additionally, more degradation fragments for DOA were identified, along with the formation of a polymerized product, while no large molecules formed for TDTM. read less NOT USED (high confidence) H. Tian, B. Zhang, and Q. Li, “Ballistic response of hexagonal boron nitride monolayer under impact of a nano-projectile,” Mechanics of Materials. 2019. link Times cited: 8 NOT USED (high confidence) J. Yan, J. He, and L. Tong, “Longitudinal and Torsional Vibration Characteristics of Boron Nitride Nanotubes,” Journal of Vibration Engineering & Technologies. 2019. link Times cited: 17 NOT USED (high confidence) M. Höhnerbach and P. Bientinesi, “Accelerating AIREBO: Navigating the Journey from Legacy to High‐Performance Code,” Journal of Computational Chemistry. 2019. link Times cited: 6 Abstract: Despite initiatives to improve the quality of scientific sof… read moreAbstract: Despite initiatives to improve the quality of scientific software, there still is a large presence of legacy code. The focus of such code is usually on domain‐science features, rather than maintainability or highest performance. Additionally, architecture specific optimizations often result in less maintainable code. In this article, we focus on the AIREBO potential from LAMMPS, which exhibits large and complex computational kernels, hindering any systematic optimization. We suggest an approach based on complexity‐reducing refactoring and hardware abstraction and present the journey from the C++ port of a previous Fortran code to performance‐portable, KNC‐hybrid, vectorized, scalable, and optimized code supporting full and reduced precision. The journey includes extensive testing that fixed bugs in the original code. Large‐scale, full‐precision runs sustain speedups of more than 4× (KNL) and 3× (Skylake). © 2019 Wiley Periodicals, Inc. read less NOT USED (high confidence) O. Matsiaka, R. Baker, E. T. Shah, and M. Simpson, “Mechanistic and experimental models of cell migration reveal the importance of cell-to-cell pushing in cell invasion,” Biomedical Physics & Engineering Express. 2019. link Times cited: 6 Abstract: Moving fronts of cells are essential for development, repair… read moreAbstract: Moving fronts of cells are essential for development, repair and disease progression. Therefore, understanding and quantifying the details of the mechanisms that drive the movement of cell fronts is of wide interest. Quantitatively identifying the role of intercellular interactions, and in particular the role of cell pushing, remains an open question. In this work, we report a combined experimental-modelling approach showing that intercellular interactions contribute significantly to the spatial spreading of a population of cells. We use a novel experimental data set with PC-3 prostate cancer cells that have been pretreated with Mitomycin-C to suppress proliferation. This allows us to experimentally separate the effects of cell migration from cell proliferation, thereby enabling us to focus on the migration process in detail as the population of cells recolonizes an initially-vacant region in a series of two-dimensional experiments. We quantitatively model the experiments using a stochastic modelling framework, based on Langevin dynamics, which explicitly incorporates random motility and various intercellular forces including: (i) long range attraction (adhesion); and (ii) finite size effects that drive short range repulsion (pushing). Quantitatively comparing the ability of this model to describe the experimentally observed population-level behaviour provides us with quantitative insight into the roles of random motility and intercellular interactions. To quantify the mechanisms at play, we calibrate the stochastic model to match experimental cell density profiles to obtain estimates of cell diffusivity, D, and the amplitude of intercellular forces, f0. Our analysis shows that taking a standard modelling approach which ignores intercellular forces provides a poor match to the experimental data whereas incorporating intercellular forces, including short-range pushing and longer range attraction, leads to a faithful representation of the experimental observations. These results demonstrate a significant role of cell pushing during cell front movement and invasion. read less NOT USED (high confidence) H. Babaei, R. Guo, A. Hashemi, and S. Lee, “Machine-learning-based interatomic potential for phonon transport in perfect crystalline Si and crystalline Si with vacancies,” Physical Review Materials. 2019. link Times cited: 30 Abstract: We report that single interatomic potential, developed using… read moreAbstract: We report that single interatomic potential, developed using Gaussian regression of density functional theory calculation data, has high accuracy and flexibility to describe phonon transport with ab initio accuracy in two different atomistic configurations: perfect crystalline Si and crystalline Si with vacancies. The high accuracy of second- and third-order force constants from the Gaussian approximation potential (GAP) are demonstrated with phonon dispersion, Gruneisen parameter, three-phonon scattering rate, phonon-vacancy scattering rate, and thermal conductivity, all of which are very close to the results from density functional theory calculation. We also show that the widely used empirical potentials (Stillinger-Weber and Tersoff) produce much larger errors compared to the GAP. The computational cost of GAP is higher than the two empirical potentials, but five orders of magnitude lower than the density functional theory calculation. Our work shows that GAP can provide a new opportunity for studying phonon transport in partially disordered crystalline phases with the high predictive power of ab initio calculation but at a feasible computational cost. read less NOT USED (high confidence) A. Glielmo, C. Zeni, ’A. Fekete, and A. Vita, “Building Nonparametric n-Body Force Fields Using Gaussian Process Regression,” Machine Learning Meets Quantum Physics. 2019. link Times cited: 8 NOT USED (high confidence) Y. H. Lin and T.-C. Chen, “Nanoscale Mechanical and Mechanically-Induced Electrical Properties of Silicon Nanowires,” Crystals. 2019. link Times cited: 1 Abstract: Molecular dynamics (MD) simulation was employed to examine t… read moreAbstract: Molecular dynamics (MD) simulation was employed to examine the deformation and phase transformation of mono-crystalline Si nanowire (SiNW) subjected to tensile stress. The techniques of coordination number (CN) and centro-symmetry parameter (CSP) were used to monitor and elucidate the detailed mechanisms of the phase transformation throughout the loading process in which the evolution of structural phase change and the dislocation pattern were identified. Therefore, the relationship between phase transformation and dislocation pattern was established and illustrated. In addition, the electrical resistance and conductivity of SiNW were evaluated by using the concept of virtual electric source during loading and unloading similar to in situ electrical measurements. The effects of temperature on phase transformation of mono-crystalline SiNWs for three different crystallographically oriented surfaces were investigated and discussed. Simulation results show that, with the increase of applied stress, the dislocations are initiated first and then the phase transformation such that the total energy of the system tends to approach a minimum level. Moreover, the electrical resistance of (001)- rather than (011)- and (111)-oriented SiNWs was changed before failure. As the stress level of the (001) SiNW reaches 24 GPa, a significant amount of metallic Si-II and amorphous phases is produced from the semiconducting Si-I phase and leads to a pronounced decrease of electrical resistance. It was also found that as the temperature of the system is higher than 500 K, the electrical resistance of (001) SiNW is significantly reduced through the process of axial elongation. read less NOT USED (high confidence) J. Yan, J. He, and L. Tong, “Longitudinal and Torsional Vibration Characteristics of Boron Nitride Nanotubes,” Journal of Vibration Engineering & Technologies. 2019. link Times cited: 0 NOT USED (high confidence) E. Schmidt, “Atomistic modelling of precipitation in Ni-base superalloys.” 2019. link Times cited: 0 Abstract: The presence of the ordered γ ′ phase (Ni3Al) in Ni-base sup… read moreAbstract: The presence of the ordered γ ′ phase (Ni3Al) in Ni-base superalloys is fundamental to the performance of engineering components such as turbine disks and blades which operate at high temperatures and loads. Hence for these alloys it is important to optimize their microstructure and phase composition. This is typically done by varying their chemistry and heat treatment to achieve an appropriate balance between γ ′ content and other constituents such as carbides, borides, oxides and topologically close packed phases. In this work we have set out to investigate the onset of γ ′ ordering in Ni-Al single crystals and in Ni-Al bicrystals containing coincidence site lattice grain boundaries (GBs) and we do this at high temperatures, which are representative of typical heat treatment schedules including quenching and annealing. For this we use the atomistic simulation methods of molecular dynamics (MD) and density functional theory (DFT). In the first part of this work we develop robust Bayesian classifiers to identify the γ ′ phase in large scale simulation boxes at high temperatures around 1500 K. We observe significant γ ′ ordering in the simulations in the form of clusters of γ ′-like ordered atoms embedded in a γ host solid solution and this happens within 100 ns. Single crystals are found to exhibit the expected homogeneous ordering with slight indications of chemical composition change and a positive correlation between the Al concentration and the concentration of γ ′ phase. In general, the ordering is found to take place faster in systems with GBs and preferentially adjacent to the GBs. The sole exception to this is the Σ3 (111) tilt GB, which is a coherent twin. An analysis of the ensemble and time lag average displacements of the GBs reveals mostly ‘anomalous diffusion’ behaviour. Increasing the Al content from pure Ni to Ni 20 at.% Al was found to either consistently increase or decrease the mobility of the GB as seen from the changing slope of the time lag displacement average. The movement of the GB can then be characterized as either ‘super’ or ‘sub-diffusive’ and is interpreted in terms of diffusion induced grain boundary migration, which is posited as a possible precursor to the appearance of serrated edge grain boundaries. In the second part of this work we develop a method for the training of empirical interatomic read less NOT USED (high confidence) H. Jiang et al., “Imaging covalent bond formation by H atom scattering from graphene,” Science. 2019. link Times cited: 67 Abstract: Atom scattering reveals bond formation When molecules collid… read moreAbstract: Atom scattering reveals bond formation When molecules collide, they can form an addition complex in which new chemical bonds can form. However, if energy does not flow out of this complex and into the rest of the molecule, the new bond will usually simply dissociate. Jiang et al. observed the scattering of hydrogen atoms from graphene and interpreted their results with a first-principles potential energy surface and a dynamical simulation (see the Perspective by Hornekaer). At near-normal incidence, these experiments probe transient carbon-hydrogen bond formation when the hydrogen atoms collide with the centers of the six-atom carbon rings. Rapid intramolecular vibrational relaxation results from orbital rehybridization and structural deformations that occur during bond formation. Science, this issue p. 379; see also p. 331 Scattering of hydrogen atoms near normal incidence from graphene reveals details of transient carbon-hydrogen bond formation. Viewing the atomic-scale motion and energy dissipation pathways involved in forming a covalent bond is a longstanding challenge for chemistry. We performed scattering experiments of H atoms from graphene and observed a bimodal translational energy loss distribution. Using accurate first-principles dynamics simulations, we show that the quasi-elastic channel involves scattering through the physisorption well where collision sites are near the centers of the six-membered C-rings. The second channel results from transient C–H bond formation, where H atoms lose 1 to 2 electron volts of energy within a 10-femtosecond interaction time. This remarkably rapid form of intramolecular vibrational relaxation results from the C atom’s rehybridization during bond formation and is responsible for an unexpectedly high sticking probability of H on graphene. read less NOT USED (high confidence) J. Luo, A. Alateeqi, L. Liu, and T. Sinno, “Carbon solubility in liquid silicon: A computational analysis across empirical potentials.,” The Journal of chemical physics. 2019. link Times cited: 4 Abstract: The nucleation and growth of SiC precipitates in liquid sili… read moreAbstract: The nucleation and growth of SiC precipitates in liquid silicon is important in the crystallization of silicon used for the photovoltaic industry. These processes depend strongly on the carbon concentration as well as the equilibrium solubility relative to the precipitate phase. Here, using a suite of statistical thermodynamic techniques, we calculate the solubility of carbon atoms in liquid silicon relative to the β-SiC phase. We employ several available empirical potentials to assess whether these potentials may reasonably be used to computationally analyze SiC precipitation. We find that some of the Tersoff-type potentials provide an excellent picture for carbon solubility in liquid silicon but, because of their severe silicon melting point overestimation, are limited to high temperatures where the carbon solubility is several percent, a value that is irrelevant for typical solidification conditions. Based on chemical potential calculations for pure silicon, we suggest that this well-known issue is confined to the description of the liquid phase and demonstrate that some recent potential models for silicon might address this weakness while preserving the excellent description of the carbon-silicon interaction found in the existing models. read less NOT USED (high confidence) L. Morrissey, S. M. Handrigan, S. Subedi, and S. Nakhla, “Atomistic uniaxial tension tests: investigating various many-body potentials for their ability to produce accurate stress strain curves using molecular dynamics simulations,” Molecular Simulation. 2019. link Times cited: 13 Abstract: ABSTRACT Molecular dynamics simulations, which take place on… read moreAbstract: ABSTRACT Molecular dynamics simulations, which take place on the atomistic scale, are now being used to predict the influence of atomistic processes on macro-scale mechanical properties. However, there is a lack of clear understanding on which potential should be used when attempting to obtain these properties. Moreover, many MD studies that do test mechanical properties do not actually simulate the macro-scale laboratory tension tests used to obtain them. As such, the purpose of the current study was to evaluate the various types of potentials for their accuracy in predicting the mechanical properties of iron from an atomistic uniaxial tension test at room temperature. Results demonstrated that while EAM and MEAM potentials all under predicted the elastic modulus at room temperature, the Tersoff and ReaxFF potentials were significantly more accurate. Unlike EAM and MEAM, both the Tersoff and ReaxFF potentials are bond order based. Therefore, these results demonstrate the importance of considering bonding between atoms when modelling tensile tests. In addition, the ReaxFF potential also accurately predicted the Poisson's ratio, allowing for complete characterisation of the material's behaviour. Overall, these findings highlight the need to understand the capabilities and limitations of each potential before application to a problem outside of the initial intended use. read less NOT USED (high confidence) N. Piroozan, S. Naserifar, and M. Sahimi, “Sliding friction between two silicon-carbide surfaces,” Journal of Applied Physics. 2019. link Times cited: 6 Abstract: Sliding friction between two SiC surfaces is important due t… read moreAbstract: Sliding friction between two SiC surfaces is important due to its relevance to many practical applications. It is also important to study whether kinetic friction at the nanoscale follows Coulomb’s law. Since SiC exists both as an amorphous material and with a crystalline structure, the effect of surface roughness on the kinetic friction may also be significant. We report the results of an extensive molecular dynamics simulation of sliding friction between surfaces of the two types of SiC over a wide range of sliding velocities. The amorphous SiC was generated by the reactive force field ReaxFF, which was also used to represent the interaction potential for the simulation of sliding friction. As the sliding velocity increases, bond breaking occurs at the interface between the two surfaces, leading to their roughening and formation of excess free volume. They reduce the kinetic friction force, hence resulting in decreasing the difference between kinetic friction in the amorphous and crystalline surfaces. The average kinetic friction force depends nonlinearly on the sliding velocity V, implying that Coulomb’s law of friction is not satisfied by the surfaces that we study at the nanoscale. The average kinetic friction force F k depends on V as F k ∝ ln V.Sliding friction between two SiC surfaces is important due to its relevance to many practical applications. It is also important to study whether kinetic friction at the nanoscale follows Coulomb’s law. Since SiC exists both as an amorphous material and with a crystalline structure, the effect of surface roughness on the kinetic friction may also be significant. We report the results of an extensive molecular dynamics simulation of sliding friction between surfaces of the two types of SiC over a wide range of sliding velocities. The amorphous SiC was generated by the reactive force field ReaxFF, which was also used to represent the interaction potential for the simulation of sliding friction. As the sliding velocity increases, bond breaking occurs at the interface between the two surfaces, leading to their roughening and formation of excess free volume. They reduce the kinetic friction force, hence resulting in decreasing the difference between kinetic friction in the amorphous and crystalline surfaces. T... read less NOT USED (high confidence) M. R. G. Marques, J. Wolff, C. Steigemann, and M. Marques, “Neural network force fields for simple metals and semiconductors: construction and application to the calculation of phonons and melting temperatures.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 17 Abstract: We present a practical procedure to obtain reliable and unbi… read moreAbstract: We present a practical procedure to obtain reliable and unbiased neural network based force fields for solids. Training and test sets are efficiently generated from global structural prediction runs, at the same time assuring the structural variety and importance of sampling the relevant regions of phase space. The neural networks are trained to yield not only good formation energies, but also accurate forces and stresses, which are the quantities of interest for molecular dynamics simulations. Finally, we construct, as an example, several force fields for both semiconducting and metallic elements, and prove their accuracy for a variety of structural and dynamical properties. These are then used to study the melting of bulk copper and gold. read less NOT USED (high confidence) A. Jamr’oz and J. Majewski, “Morphology, Ordering, Stability, and Electronic Structure of Carbon‐Doped Hexagonal Boron Nitride,” physica status solidi (b). 2019. link Times cited: 6 Abstract: Theoretical studies of morphology, stability, and electronic… read moreAbstract: Theoretical studies of morphology, stability, and electronic structure of monolayer hexagonal CBN alloys with rich content of h‐BN and carbon concentration not exceeding 50% are presented. The studies are based on the bond order type of the valence force field to account for the interactions between atomic constituents and Monte Carlo method with Metropolis algorithm to establish equilibrium distribution of atoms over the lattice. It is found that the phase separation into graphene and h‐BN domains occurs in the majority of growth conditions. Only in N‐rich growth conditions, it is possible to obtain a quasi uniform distribution of carbon atoms over the boron sublattice. It is been predicted that the energy gap in stoichiometric Cx(BN)1−x alloys exhibits extremely strong bowing. read less NOT USED (high confidence) D. Damasceno, E. Mesquita, R. Rajapakse, and R. Pavanello, “Atomic-scale finite element modelling of mechanical behaviour of graphene nanoribbons,” International Journal of Mechanics and Materials in Design. 2019. link Times cited: 11 NOT USED (high confidence) B. Bauerhenne and M. E. Garcia, “Performance of state-of-the-art force fields for atomistic simulations of silicon at high electronic temperatures,” The European Physical Journal Special Topics. 2019. link Times cited: 5 NOT USED (high confidence) F. Yousefi and F. Khoeini, “Impact of topological line defects on wall roughness and thermal conductivity of carbon nanotubes: A molecular dynamics study,” AIP Advances. 2019. link Times cited: 6 Abstract: Understanding the influence of defects on thermal conductivi… read moreAbstract: Understanding the influence of defects on thermal conductivity of nanowires and nanomaterials is important due to its application for heat management in the nanodevices. In the present study, we investigate the influence of topological line defects on thermal conductivity of single-walled carbon nanotube (SWCNT) through molecular dynamics simulations. To model interaction between carbon atoms in the carbon nanotube, we employed the three-body Tersoff potential. Thermal conductivity was obtained in situations, which the 5-8-5 defects have been distributed with several patterns on the surface of carbon nanotube (CNT). We examined the impact of defect concentration and found that thermal conductivity decreases with increasing defect concentration. We also investigated the effects of length, temperature and the temperature difference between two ends of carbon nanotube on its thermal conductivity. The increase of length leads to an increment in thermal conductivity, while the increase of temperature causes thermal conductivity decreases. The cross-section of the nanotubes changes with the pattern of defect. Our results can be applicable in the heat management of carbon nanotube-based nanodevices.Understanding the influence of defects on thermal conductivity of nanowires and nanomaterials is important due to its application for heat management in the nanodevices. In the present study, we investigate the influence of topological line defects on thermal conductivity of single-walled carbon nanotube (SWCNT) through molecular dynamics simulations. To model interaction between carbon atoms in the carbon nanotube, we employed the three-body Tersoff potential. Thermal conductivity was obtained in situations, which the 5-8-5 defects have been distributed with several patterns on the surface of carbon nanotube (CNT). We examined the impact of defect concentration and found that thermal conductivity decreases with increasing defect concentration. We also investigated the effects of length, temperature and the temperature difference between two ends of carbon nanotube on its thermal conductivity. The increase of length leads to an increment in thermal conductivity, while the increase of temperature causes th... read less NOT USED (high confidence) H. Seyf, K. Gordiz, F. DeAngelis, and A. Henry, “Using Green-Kubo modal analysis (GKMA) and interface conductance modal analysis (ICMA) to study phonon transport with molecular dynamics,” Journal of Applied Physics. 2019. link Times cited: 26 Abstract: While current descriptions of thermal transport exist for we… read moreAbstract: While current descriptions of thermal transport exist for well-ordered solids, i.e., crystal lattices, new methods are needed to describe thermal transport in systems with lack of symmetry such as structurally/compositionally disordered solids and interfaces. In this tutorial, we discuss the formalism, implementation, and application of two recently developed methods, Green-Kubo modal analysis and interface conductance modal analysis, to predict the thermal conductivity and thermal interface conductance, respectively. Specifically, these methods enable the prediction of phonon contributions to transport in crystalline materials with any level of defects, up through fully amorphous solids, dilute to fully random alloys, molecules, nanostructures, and across interfaces involving any of these material classes—all within a single and unified perspective. This tutorial article not only describes the methods, but also provides example codes that can be used for their direct implementation. The design and functionality of the codes is also discussed in order to reduce the barrier to more extensive utilization of these approaches by others. read less NOT USED (high confidence) J. J. Varghese and S. H. Mushrif, “Origins of complex solvent effects on chemical reactivity and computational tools to investigate them: a review,” Reaction Chemistry & Engineering. 2019. link Times cited: 73 Abstract: Origins of solvent-induced enhancement in catalytic reactivi… read moreAbstract: Origins of solvent-induced enhancement in catalytic reactivity and product selectivity are discussed with computational methods to study them. read less NOT USED (high confidence) S. Chavoshi and S. Xu, “Nanoindentation/scratching at finite temperatures: Insights from atomistic-based modeling,” Progress in Materials Science. 2019. link Times cited: 37 NOT USED (high confidence) M. Kunaseth, S. Hannongbua, and A. Nakano, “Shift/collapse on neighbor list (SC-NBL): Fast evaluation of dynamic many-body potentials in molecular dynamics simulations,” Comput. Phys. Commun. 2019. link Times cited: 3 NOT USED (high confidence) Y. Lysogorskiy, T. Hammerschmidt, J. Janssen, J. Neugebauer, and R. Drautz, “Transferability of interatomic potentials for molybdenum and silicon,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 14 Abstract: Interatomic potentials are widely used in computational mate… read moreAbstract: Interatomic potentials are widely used in computational materials science, in particular for simulations that are too computationally expensive for density functional theory (DFT). Most interatomic potentials have a limited application range and often there is very limited information available regarding their performance for specific simulations. We carried out high-throughput calculations for molybdenum and silicon with DFT and a number of interatomic potentials. We compare the DFT reference calculations and experimental data to the predictions of the interatomic potentials. We focus on a large number of basic materials properties, including the cohesive energy, atomic volume, elastic coefficients, vibrational properties, thermodynamic properties, surface energies and vacancy formation energies, which enables a detailed discussion of the performance of the different potentials. We further analyze correlations between properties as obtained from DFT calculations and how interatomic potentials reproduce these correlations, and suggest a general measure for quantifying the accuracy and transferability of an interatomic potential. From our analysis we do not establish a clearcut ranking of the potentials as each potential has its strengths and weaknesses. It is therefore essential to assess the properties of a potential carefully before application of the potential in a specific simulation. The data presented here will be useful for selecting a potential for simulations of Mo or Si. read less NOT USED (high confidence) H. Chan et al., “Machine learning coarse grained models for water,” Nature Communications. 2019. link Times cited: 96 NOT USED (high confidence) H. E. Sauceda, S. Chmiela, I. Poltavsky, K. Müller, and A. Tkatchenko, “Molecular force fields with gradient-domain machine learning: Construction and application to dynamics of small molecules with coupled cluster forces.,” The Journal of chemical physics. 2019. link Times cited: 77 Abstract: We present the construction of molecular force fields for sm… read moreAbstract: We present the construction of molecular force fields for small molecules (less than 25 atoms) using the recently developed symmetrized gradient-domain machine learning (sGDML) approach [Chmiela et al., Nat. Commun. 9, 3887 (2018) and Chmiela et al., Sci. Adv. 3, e1603015 (2017)]. This approach is able to accurately reconstruct complex high-dimensional potential-energy surfaces from just a few 100s of molecular conformations extracted from ab initio molecular dynamics trajectories. The data efficiency of the sGDML approach implies that atomic forces for these conformations can be computed with high-level wavefunction-based approaches, such as the "gold standard" coupled-cluster theory with single, double and perturbative triple excitations [CCSD(T)]. We demonstrate that the flexible nature of the sGDML model recovers local and non-local electronic interactions (e.g., H-bonding, proton transfer, lone pairs, changes in hybridization states, steric repulsion, and n → π* interactions) without imposing any restriction on the nature of interatomic potentials. The analysis of sGDML molecular dynamics trajectories yields new qualitative insights into dynamics and spectroscopy of small molecules close to spectroscopic accuracy. read less NOT USED (high confidence) J. Yeo, G. Jung, F. J. Martín-Martínez, J. Beem, Z. Qin, and M. Buehler, “Multiscale Design of Graphyne‐Based Materials for High‐Performance Separation Membranes,” Advanced Materials. 2019. link Times cited: 29 Abstract: By varying the number of acetylenic linkages connecting arom… read moreAbstract: By varying the number of acetylenic linkages connecting aromatic rings, a new family of atomically thin graph‐n‐yne materials can be designed and synthesized. Generating immense scientific interest due to its structural diversity and excellent physical properties, graph‐n‐yne has opened new avenues toward numerous promising engineering applications, especially for separation membranes with precise pore sizes. Having these tunable pore sizes in combination with their excellent mechanical strength to withstand high pressures, free‐standing graph‐n‐yne is theoretically posited to be an outstanding membrane material for separating or purifying mixtures of either gases or liquids, rivaling or even dramatically exceeding the capabilities of current, state‐of‐art separation membranes. Computational modeling and simulations play an integral role in the bottom‐up design and characterization of these graph‐n‐yne materials. Thus, here, the state of the art in modeling α‐, β‐, γ‐, δ‐, and 6,6,12‐graphyne nanosheets for synthesizing graph‐2‐yne materials and 3D architectures thereof is discussed. Different synthesis methods are described and a broad overview of computational characterizations of graph‐n‐yne's electrical, chemical, and thermal properties is provided. Furthermore, a series of in‐depth computational studies that delve into the specifics of graph‐n‐yne's mechanical strength and porosity, which confer superior performance for separation and desalination membranes, are reviewed. read less NOT USED (high confidence) L. Wang, W. Yu, and S. Shen, “Revisiting the structures and energies of silicon 〈110〉 symmetric tilt grain boundaries,” Journal of Materials Research. 2019. link Times cited: 19 Abstract: Atomistic simulations of 18 silicon 〈110〉 symmetric tilt gra… read moreAbstract: Atomistic simulations of 18 silicon 〈110〉 symmetric tilt grain boundaries are performed using Stillinger Weber, Tersoff, and the optimized Modified Embedded Atom Method potentials. We define a novel structural unit classification through dislocation core analysis to characterize the relaxed GB structures. GBs with the misorientation angle θ ranging from 13.44° to 70.53° are solely composed of Lomer dislocation cores. For GBs with θ less than but close to 70.53°, GB ‘step’ appears and the equilibrated states with lowest GB energies can be attained only when such GB ‘step’ is located in the middle of each single periodic GB structure. For the misorientation angles in the range of 93.37° ≤ θ ≤ 148.41°, GB structures become complicated since they contain multiple types of dislocation cores. This work not only facilitates the structural characterization of silicon 〈110〉 STGBs, but also may provide new insights into mirco-structure design in multicrystalline silicon. read less NOT USED (high confidence) R. Wexler, T. Qiu, and A. Rappe, “Automatic Prediction of Surface Phase Diagrams Using Ab Initio Grand Canonical Monte Carlo,” The Journal of Physical Chemistry C. 2019. link Times cited: 44 Abstract: The properties of a material are often strongly influenced b… read moreAbstract: The properties of a material are often strongly influenced by its surfaces. Depending on the nature of the chemical bonding in a material, its surface can undergo a variety of stabilizing reconstructions that dramatically alter the chemical reactivity, light absorption, and electronic band offsets. For decades, ab initio thermodynamics has been the method of choice for computationally determining the surface phase diagram of a material under different conditions. The surfaces considered for these studies, however, are often human-selected and too few in number, leading both to insufficient exploration of all possible surfaces and to biases toward portions of the composition–structure phase space that often do not encompass the most stable surfaces. To overcome these limitations and automate the discovery of realistic surfaces, we combine density functional theory and grand canonical Monte Carlo (GCMC) into “ab initio GCMC.” This paper presents the successful application of ab initio GCMC to the study of o... read less NOT USED (high confidence) D. Prasad and N. Mitra, “An atomistic study of phase transition in cubic diamond Si single crystal subjected to static compression,” Computational Materials Science. 2019. link Times cited: 7 NOT USED (high confidence) I. Sukuba, L. Chen, M. Probst, and A. Kaiser, “A neural network interface for DL_POLY and its application to liquid water,” Molecular Simulation. 2018. link Times cited: 7 Abstract: ABSTRACT After a general discussion of neural networks poten… read moreAbstract: ABSTRACT After a general discussion of neural networks potential energy functions and their standing within the various approaches of representing the potential energy function of a system, we describe a new interface between the open source atomistic library aenet of Artrith and Urban and the DL_POLY 4 code. As an application example, the training of a neural network for liquid water is described and the network is used in a molecular dynamics simulation. The resulting thermodynamic properties are compared with those from a reference simulation with the same SPC/E model that has been used in the training. read less NOT USED (high confidence) L. V. Mirantsev and A. Abramyan, “Equilibrium structures and flows of polar and nonpolar liquids and their mixtures in carbon nanotubes with rectangular cross sections,” Computational Materials Science. 2018. link Times cited: 0 NOT USED (high confidence) R. Khaledialidusti, A. Mishra, and A. Barnoush, “Rheological properties of super critical CO2 with CuO: Multi-scale computational modeling.,” The Journal of chemical physics. 2018. link Times cited: 12 Abstract: A multi-scale computational methodology based on the density… read moreAbstract: A multi-scale computational methodology based on the density functional theory and molecular dynamics has been used to investigate the rheological properties of super critical CO2 with CuO nano-particle (NP). Density functional theory which treats the electron density as the central variable has been used to explore the adsorption of CO2 molecules on the two most stable CuO surfaces [i.e., (111) and (011)] at absolute zero. The results of this theory would provide valuable information to make CuO NPs with the surface where the CO2 adsorption is maximum in order to have a stronger mono-layer of adsorbed CO2 molecules on the surface of the NP which is the most crucial factor in formation of a stable nanofluid. The results show that the CO2 molecule is adsorbed more strongly on the (011) surface with an adsorption energy of -99.06 kJ/mol compared to the (111) surface. A computational methodology based on molecular dynamics has been used to evaluate the enhancement of the rheological properties of the super-critical CO2 liquid based nanofluid at different temperatures and pressures. In this scale, first, the CO2 liquid has been modeled by employing the condensed-phase optimized molecular potentials for atomistic simulation studies (COMPASS) force field potential and the fluid properties computed are in excellent agreement with the literature and experiment values. Second, the nanofluid has been modeled in order to study the enhancement of the fluid properties with the CuO NPs. The charged optimized many-body force field potential has been employed to consider the effect of the charge transferring between the NPs and liquid molecules and breaking of existing bonds and the formation of new bonds. The COMPASS force field potential is also employed for the interactions between CO2 molecules. The combination of these potentials is quite a new approach for the study of the super-critical (SC)-CO2 based nanofluid. The results show that the viscosity of the SC-CO2 is enhanced between 1.3 and 2.5 times under the temperature and pressure conditions studied. read less NOT USED (high confidence) M. Anafcheh, F. Naderi, Z. Khodadadi, F. Ektefa, and R. Ghafouri, “Exploring Adjacent Pentagons in Non-IPR and SW Defective Si60 and Si70 Silicon Fullerenes: a Computational Study,” Silicon. 2018. link Times cited: 1 NOT USED (high confidence) X. Duan et al., “Redesigning LAMMPS for Peta-Scale and Hundred-Billion-Atom Simulation on Sunway TaihuLight,” SC18: International Conference for High Performance Computing, Networking, Storage and Analysis. 2018. link Times cited: 41 Abstract: Large-scale molecular dynamics (MD) simulations on supercomp… read moreAbstract: Large-scale molecular dynamics (MD) simulations on supercomputers play an increasingly important role in many research areas. In this paper, we present our efforts on redesigning the widely used LAMMPS MD simulator for Sunway TaihuLight supercomputer and its ShenWei many-core architecture (SW26010). The memory constraints of SW26010 bring a number of new challenges for achieving efficient MD implementation on it. In order to overcome these constraints, we employ four levels of optimization: (1) a hybrid memory update strategy; (2) a software cache strategy; (3) customized transcendental math functions; and (4) a full pipeline acceleration. Furthermore, we redesign the code to enable all possible vectorization. Experiments show that our redesigned software on a single SW26010 processor can outperform over 100 E5-2650 v2 cores for running the latest stable release (11Aug17) of LAMMPS. We also achieve a performance of over 2.43 PFlops for a Tersoff simulation when using 16,384 nodes on Sunway TaihuLight. read less NOT USED (high confidence) X. W. Zhou, M. E. Foster, and R. Sills, “An Fe‐Ni‐Cr embedded atom method potential for austenitic and ferritic systems,” Journal of Computational Chemistry. 2018. link Times cited: 65 Abstract: Fe‐Ni‐Cr stainless‐steels are important structural materials… read moreAbstract: Fe‐Ni‐Cr stainless‐steels are important structural materials because of their superior strength and corrosion resistance. Atomistic studies of mechanical properties of stainless‐steels, however, have been limited by the lack of high‐fidelity interatomic potentials. Here using density functional theory as a guide, we have developed a new Fe‐Ni‐Cr embedded atom method potential. We demonstrate that our potential enables stable molecular dynamics simulations of stainless‐steel alloys at high temperatures, accurately reproduces the stacking fault energy—known to strongly influence the mode of plastic deformation (e.g., twinning vs. dislocation glide vs. cross‐slip)—of these alloys over a range of compositions, and gives reasonable elastic constants, energies, and volumes for various compositions. The latter are pertinent for determining short‐range order and solute strengthening effects. Our results suggest that our potential is suitable for studying mechanical properties of austenitic and ferritic stainless‐steels which have vast implementation in the scientific and industrial communities. Published 2018. This article is a U.S. Government work and is in the public domain in the USA. read less NOT USED (high confidence) X. Yuan and Y. Wang, “Adhesion of carbon nanotubes on elastic substrates with finite thickness,” Journal of Applied Physics. 2018. link Times cited: 5 Abstract: How carbon nanotubes (CNTs) interact with substrates is fund… read moreAbstract: How carbon nanotubes (CNTs) interact with substrates is fundamental for understanding their physical properties. In existing theoretical and modeling studies, the substrates are considered to be rigid with semi-infinite thickness. In this work, the effects of finite substrate thickness and elasticity are analyzed theoretically and numerically for free boundary conditions. Based on the energy-variational approach, considering the interfacial van der Waals interactions and bending strain energies stored in CNTs and substrates, the governing equations and boundary conditions are derived analytically. The theoretical predictions are in reasonable agreement with the results of molecular dynamics simulations. When the substrate is sufficiently thick, the results of the present theoretical model are entirely consistent with previous models for the infinite-thickness substrate. However, for relatively thin substrates, the effect of substrate thickness is significant due to the geometric large deformation. Three stable adhesive states (initial non-adhesive, partially adhesive, and fully wrapping states) can be achieved, dependent on the substrate thickness, the number of CNT walls, and the interfacial adhesion work. The stability of adhesive configurations is explored by analyzing the energy variations corresponding to the adhesive deformation. We show that there exist several modes of energy variations, depending on the adhesion work and the substrate-CNT bending stiffness ratio, which exhibit linear and nonlinear influences, respectively. Our results could serve as guidelines to design CNT-on-substrate systems.How carbon nanotubes (CNTs) interact with substrates is fundamental for understanding their physical properties. In existing theoretical and modeling studies, the substrates are considered to be rigid with semi-infinite thickness. In this work, the effects of finite substrate thickness and elasticity are analyzed theoretically and numerically for free boundary conditions. Based on the energy-variational approach, considering the interfacial van der Waals interactions and bending strain energies stored in CNTs and substrates, the governing equations and boundary conditions are derived analytically. The theoretical predictions are in reasonable agreement with the results of molecular dynamics simulations. When the substrate is sufficiently thick, the results of the present theoretical model are entirely consistent with previous models for the infinite-thickness substrate. However, for relatively thin substrates, the effect of substrate thickness is significant due to the geometric large deformation. Three s... read less NOT USED (high confidence) R. Ma, X. Wan, T. Zhang, N. Yang, and T. Luo, “Role of Molecular Polarity in Thermal Transport of Boron Nitride–Organic Molecule Composites,” ACS Omega. 2018. link Times cited: 20 Abstract: Understanding the role of fillers in the thermal transport o… read moreAbstract: Understanding the role of fillers in the thermal transport of composite materials is of great importance to engineering better materials. The filler induces material interfaces within the composite, which influence the thermal transport between the matrix and themselves. The filler can also alter the molecular arrangement of the matrix in its vicinity, which may also impact the thermal transport ability. In this paper, molecular dynamics simulations are performed to study the thermal transport across the matrix–filler interfaces in hexagonal boron nitride (h-BN)–organic molecule composites. Four different organic molecules are studied as the matrixes. They include hexane (C6H14), hexanamine (C6H13NH2), hexanol (C6H13OH), and hexanoic acid (C5H11COOH), which feature the same molecular backbone but increasingly different polar functional groups. The nominal local thermal conductivities of the hexane matrix with varying distances to the interface are calculated to demonstrate the influence of the filler on the thermal transport properties of the matrix. It is found that a more polar matrix exhibits a higher density in the near-interface region and a higher nominal local thermal conductivity, suggesting that the interfacial interaction can impact the local heat transfer ability of the matrix. In addition, the more polar matrix also leads to a larger interfacial thermal conductance with h-BN (hexane: 90.47 ± 14.49 MW/m2 K, hexanamine: 113.38 ± 17.72 MW/m2 K, hexanol: 136.16 ± 25.12 MW/m2 K, and hexanoic acid: 155.17 ± 24.89 MW/m2 K) because of the higher matrix density near the interface and thus more atoms exchanging energy with the filler. The results of this study may provide useful information for designing composite materials for heat transfer applications. read less NOT USED (high confidence) I. V. Vitkovskii, S. Romashin, V. S. Federyaeva, and V. Shorkin, “Theoretical Justification and Experimental Confirmation of the Selection Condition of Material for the Antidiffusion Layer of the Heat-Resistant Bimetallic Wire,” Technical Physics. 2018. link Times cited: 0 NOT USED (high confidence) S. Jha, V. Ponce, and J. Seminario, “Investigating the effects of vacancies on self-diffusion in silicon clusters using classical molecular dynamics,” Journal of Molecular Modeling. 2018. link Times cited: 14 NOT USED (high confidence) A. Mahata and T. Mukhopadhyay, “Probing the chirality-dependent elastic properties and crack propagation behavior of single and bilayer stanene.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 26 Abstract: Stanene, a quasi-two-dimensional honeycomb-like structure of… read moreAbstract: Stanene, a quasi-two-dimensional honeycomb-like structure of tin belonging to the family of 2D-Xenes (X = Si, Ge, Sn) has recently been reported to show promising electronic, optical and mechanical properties. This paper investigates the elastic moduli and crack propagation behaviour of single layer and bilayer stanene based on molecular dynamics simulations, which have been performed using the Tersoff bond order potential (BOP). We have parameterized the interlayer van der Waals interactions for the bilayer Lennard-Jones potential in the case of bilayer stanene. Density functional calculations are performed to fit the Lennard-Jones parameters for the properties which are not available from the scientific literature. The effect of temperature and strain rate on the mechanical properties of stanene is investigated for both single layer and bilayer stanene in the armchair and zigzag directions. The results reveal that both the fracture strength and strain of stanene decrease with increasing temperature, while at higher loading rate, the material is found to exhibit higher fracture strength and strain. The effect of chirality on the elastic moduli of stanene is explained on the basis of a physics-based analytical approach, wherein the fundamental interaction between the shear modulus and Young's modulus is elucidated. To provide a realistic perspective, we have investigated the compound effect of uncertainty on the elastic moduli of stanene based on an efficient analytical approach. Large-scale Monte Carlo simulations are carried out considering different degrees of stochasticity. The in-depth results on mechanical properties presented in this article will further aid the adoption of stanene as a potential nano-electro-optical substitute with exciting features such as 2D topological insulating properties with a large bandgap, the capability to support enhanced thermoelectric performance, topological superconductivity and a quantum anomalous Hall effect at near-room-temperature. read less NOT USED (high confidence) J. Byggmästar, F. Granberg, and K. Nordlund, “Effects of the short-range repulsive potential on cascade damage in iron,” Journal of Nuclear Materials. 2018. link Times cited: 52 NOT USED (high confidence) K. Kohno and M. Ishimaru, “Molecular-dynamics simulations of solid phase epitaxy in silicon: Effects of system size, simulation time, and ensemble,” Japanese Journal of Applied Physics. 2018. link Times cited: 3 Abstract: Solid phase epitaxial (SPE) recrystallization of amorphous S… read moreAbstract: Solid phase epitaxial (SPE) recrystallization of amorphous Si on a Si(001) substrate was examined by large-scale (6144–129024 Si atoms), long-time (up to 2000 ns) molecular-dynamics (MD) simulations using the empirical Tersoff interatomic potential. We particularly focused on the effects of the MD cell size, simulation time, and ensemble on the SPE growth rate. We found that the simulations under the isothermal–isochoric conditions (NVT ensemble) show a higher crystallization rate than those under the isothermal–isobaric conditions (NPT ensemble). The system size dealt with in the present MD simulation, i.e., >6144 Si atoms, was enough to estimate the SPE growth rate. The Arrhenius plot of the growth rate between 1300 and 1600 K exhibited a single activation energy, ∼2.4 eV, which is in agreement with the experimental value (∼2.7 eV). However, the growth rate at temperatures below 1300 K deviated from the extrapolated ones from 1300 to 1600 K, which is because recrystallization does not reach a steady state: long-time MD simulations are required to estimate the growth rate at low temperature. The structure analysis of amorphous/crystalline interfaces suggested that the braking of atomic bonds parallel to the interface becomes a rate-limiting step of the SPE growth. read less NOT USED (high confidence) O. Matsiaka, R. Baker, E. T. Shah, and M. Simpson, “Mechanistic and experimental models of cell migration reveal the importance of intercellular interactions in cell invasion,” bioRxiv. 2018. link Times cited: 2 Abstract: Moving fronts of cells are essential for development, repair… read moreAbstract: Moving fronts of cells are essential for development, repair and disease progression. Therefore, understanding and quantifying the details of the mechanisms that drive the movement of cell fronts is of wide interest. Quantitatively identifying the role of intercellular interactions, and in particular the role of cell pushing, remains an open question. Indeed, perhaps the most common continuum mathematical idealization of moving cell fronts is to treat the population of cells, either implicitly or explicitly, as a population of point particles undergoing a random walk that neglects intercellular interactions. In this work, we report a combined experimental-modelling approach showing that intercellular interactions contribute significantly to the spatial spreading of a population of cells. We use a novel experimental data set with PC-3 prostate cancer cells that have been pretreated with Mitomycin-C to suppress proliferation. This allows us to experimentally separate the effects of cell migration from cell proliferation, thereby enabling us to focus on the migration process in detail as the population of cells recolonizes an initially-vacant region in a series of two-dimensional experiments. We quantitatively model the experiments using a stochastic modelling framework, based on Langevin dynamics, which explicitly incorporates random motility and various intercellular forces including: (i) long range attraction (adhesion); and (ii) finite size effects that drive short range repulsion (pushing). Quantitatively comparing the ability of this model to describe the experimentally observed population-level behaviour provides us with quantitative insight into the roles of random motility and intercellular interactions. To quantify the mechanisms at play, we calibrate the stochastic model to match experimental cell density profiles to obtain estimates of cell diffusivity, D, and the amplitude of intercellular forces, f0. Our analysis shows that taking a standard modelling approach which ignores intercellular forces provides a poor match to the experimental data whereas incorporating intercellular forces, including short-range pushing and longer range attraction, leads to a faithful representation of the experimental observations. These results demonstrate a significant role for intercellular interactions in cell invasion. Author summary Moving cell fronts are routinely observed in various physiological processes, such as wound healing, malignant invasion and embryonic morphogenesis. We explore the effects of a previously overlooked mechanism that contributes to population-level front movement: pushing. Our framework is flexible and incorporates range of reasonable biological phenomena, such as random motility, cell-to-cell adhesion, and pushing. We find that neglecting finite size effects and intercellular forces, such as cell pushing, reduces our ability to mimic and predict our experimental observations. read less NOT USED (high confidence) J. Wu et al., “Reactive Molecular Dynamics Simulations of the Thermal Decomposition Mechanism of 1,3,3-Trinitroazetidine.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2018. link Times cited: 17 Abstract: 1,3,3-Trinitroazetidine (TNAZ) has a molecular formula of C3… read moreAbstract: 1,3,3-Trinitroazetidine (TNAZ) has a molecular formula of C3 H4 N4 O6 and the characteristics of low melting point, low impact sensitivity and good thermal stability. It is suitable for melt casting and pressed charges, and it has broad prospects for applications in low-sensitivity ammunition. In this study, the thermal decomposition of TNAZ crystals at high temperature was calculated by molecular dynamics simulation with the ReaxFF/lg reactive force field. The change in the potential energy of TNAZ, the formation of small-molecule products and clusters, and the initial reaction path of TNAZ were analysed. The kinetic parameters of different reaction stages in TNAZ thermal decomposition were obtained. The primary thermal decomposition reaction of TNAZ was found to be as follows: N-NO2 and C-NO2 bonds broke; a H atom on the quaternary ring was transferred to the nitro group; and the C-HNO2 and N-HNO2 bonds broke. The main decomposition products of TNAZ were thus NO2 , NO, N2 , H2 O, CO2 and HNO2 , as well as macromolecular clusters. The size of the cluster structure was related to the reaction temperature, and the higher the temperature was, the smaller the cluster size was. read less NOT USED (high confidence) P. Alaba, A. Abbas, J. Huang, and W. M. A. W. Daud, “Molybdenum carbide nanoparticle: Understanding the surface properties and reaction mechanism for energy production towards a sustainable future,” Renewable and Sustainable Energy Reviews. 2018. link Times cited: 27 NOT USED (high confidence) S. Thomas, K. Ajith, S. U. Lee, and M. C. Valsakumar, “Assessment of the mechanical properties of monolayer graphene using the energy and strain-fluctuation methods,” RSC Advances. 2018. link Times cited: 28 Abstract: Molecular statics and dynamics simulations were performed to… read moreAbstract: Molecular statics and dynamics simulations were performed to investigate the mechanical properties of a monolayer graphene sheet using an efficient energy method and strain-fluctuation method. Using the energy method, we observed that the mechanical properties of an infinite graphene sheet are isotropic, whereas for a finite sheet, they are anisotropic. This work is the first to report the temperature-dependent elastic constants of graphene between 100 and 1000 K using the strain-fluctuation method. We found that the out-of-plane thermal excursions in a graphene membrane lead to strong anharmonic behavior, which allows large deviations from isotropic elasticity. The computed Young's modulus and Poisson's ratio of a sheet with an infinite spatial extent are 0.939 TPa and 0.223, respectively. We also found that graphene sheets with both finite and infinite spatial extent satisfy the Born elastic stability conditions. We extracted the variation in bending modulus with the system size at zero kelvin (0.83 eV) using a formula derived from the Foppl–von Karman approach. When the temperature increases, the Young's modulus of the sample decreases, which effectively reduces the longitudinal and shear wave velocities. read less NOT USED (high confidence) S. Stelmakh, K. Skrobas, S. Gierlotka, and B. Palosz, “Application of PDF analysis assisted by MD simulations for determination of the atomic structure and crystal habit of CdSe nanocrystals,” Journal of Physics: Condensed Matter. 2018. link Times cited: 6 Abstract: A new methodology of performing structural analysis of nanoc… read moreAbstract: A new methodology of performing structural analysis of nanocrystals based on wide angle powder diffraction is proposed. It combines molecular dynamics simulations with the analysis of pair distribution function. The actual analysis was performed on CdSe quantum dots. MD simulations of nanocrystals with shapes defined by low-index atomic planes, (1 0 0), (1 1 0), and (1 1 1) introduced bulk and surface relaxation in initially perfect crystal lattice. In search for the best atomistic model of the actual CdSe nanocrystals, experimental structure functions S(Q) and interatomic distance functions G(r) were matched to those calculated with MD models. Eventually it was concluded that CdSe nanocrystals with dimensions of 2.5–3.5 nm assume the shape of octahedrons terminated by (1 1 1) surfaces. read less NOT USED (high confidence) S. Goel, G. Cross, A. Stukowski, E. Gamsjäger, B. Beake, and A. Agrawal, “Designing nanoindentation simulation studies by appropriate indenter choices: Case study on single crystal tungsten,” Computational Materials Science. 2018. link Times cited: 39 NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 38 NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 0 NOT USED (high confidence) T. Rabczuk, M. Kakavand, R. Uma, A. N. Shirazi, and M. Makaremi, “Thermal Conductance along Hexagonal Boron Nitride and Graphene Grain Boundaries,” Energies. 2018. link Times cited: 5 Abstract: : We carried out molecular dynamics simulations at various t… read moreAbstract: : We carried out molecular dynamics simulations at various temperatures to predict the thermal conductivity and the thermal conductance of graphene and hexagonal boron-nitride (h-BN) thin films. Therefore, several models with six different grain boundary configurations ranging from 33–140 nm in length were generated. We compared our predicted thermal conductivity of pristine graphene and h-BN with previously conducted experimental data and obtained good agreement. Finally, we computed the thermal conductance of graphene and h-BN sheets for six different grain boundary configurations, five sheet lengths ranging from 33 to 140 nm and three temperatures (i.e., 300 K, 500 K and 700 K). The results show that the thermal conductance remains nearly constant with varying length and temperature for each grain boundary. read less NOT USED (high confidence) E. Ivanova, “On the Use of the Continuum Mechanics Method for Describing Interactions in Discrete Systems with Rotational Degrees of Freedom,” Journal of Elasticity. 2018. link Times cited: 5 NOT USED (high confidence) M. El-Genk, K. Talaat, and B. Cowen, “Thermal conductivity of silicon using reverse non-equilibrium molecular dynamics,” Journal of Applied Physics. 2018. link Times cited: 13 Abstract: Simulations are performed using the reverse non-equilibrium … read moreAbstract: Simulations are performed using the reverse non-equilibrium molecular dynamics (rNEMD) method and the Stillinger-Weber (SW) potential to determine the input parameters for achieving ±1% convergence of the calculated thermal conductivity of silicon. These parameters are then used to investigate the effects of the interatomic potentials of SW, Tersoff II, Environment Dependent Interatomic Potential (EDIP), Second Nearest Neighbor, Modified Embedded-Atom Method (MEAM), and Highly Optimized Empirical Potential MEAM on determining the bulk thermal conductivity as a function of temperature (400–1000 K). At temperatures > 400 K, data collection and swap periods of 15 ns and 150 fs, system size ≥6 × 6 UC2 and system lengths ≥192 UC are adequate for ±1% convergence with all potentials, regardless of the time step size (0.1–0.5 fs). This is also true at 400 K, except for the SW potential, which requires a data collection period ≥30 ns. The calculated bulk thermal conductivities using the rNEMD method and the EDIP potential are close to, but lower than experimental values. The 10% difference at 400 K increases gradually to 20% at 1000 K.Simulations are performed using the reverse non-equilibrium molecular dynamics (rNEMD) method and the Stillinger-Weber (SW) potential to determine the input parameters for achieving ±1% convergence of the calculated thermal conductivity of silicon. These parameters are then used to investigate the effects of the interatomic potentials of SW, Tersoff II, Environment Dependent Interatomic Potential (EDIP), Second Nearest Neighbor, Modified Embedded-Atom Method (MEAM), and Highly Optimized Empirical Potential MEAM on determining the bulk thermal conductivity as a function of temperature (400–1000 K). At temperatures > 400 K, data collection and swap periods of 15 ns and 150 fs, system size ≥6 × 6 UC2 and system lengths ≥192 UC are adequate for ±1% convergence with all potentials, regardless of the time step size (0.1–0.5 fs). This is also true at 400 K, except for the SW potential, which requires a data collection period ≥30 ns. The calculated bulk thermal conductivities using the rNEMD method and the EDIP... read less NOT USED (high confidence) A. Gola and L. Pastewka, “Embedded atom method potential for studying mechanical properties of binary Cu–Au alloys,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 13 Abstract: We present an embedded atom method (EAM) potential for the b… read moreAbstract: We present an embedded atom method (EAM) potential for the binary Cu–Au system. The unary phases are described by two well-tested unary EAM potentials for Cu and Au. We fitted the interaction between Cu and Au to experimental properties of the binary intermetallic phases Cu3Au, CuAu and CuAu3. Particular attention has been paid to reproducing stacking fault energies in order to obtain a potential suitable for studying deformation in this binary system. The resulting energies, lattice constant, elastic properties and melting points are in good agreement with available experimental data. We use nested sampling to show that our potential reproduces the phase boundaries between intermetallic phases and the disordered face-centered cubic solid solution. We benchmark our potential against four popular Cu–Au EAM parameterizations and density-functional theory calculations. read less NOT USED (high confidence) R. E. Jones, J. Rimsza, and L. Criscenti, “An atomic-scale evaluation of the fracture toughness of silica glass,” Journal of Physics: Condensed Matter. 2018. link Times cited: 4 Abstract: Using an atomistic technique consistent with continuum balan… read moreAbstract: Using an atomistic technique consistent with continuum balance laws and drawing on classical fracture mechanics theory, we estimate the resistance to fracture propagation of amorphous silica. We discuss correspondence and deviations from classical linear elastic fracture mechanics theory including size dependence, rigid/floppy modes of deformation, and the effects of surface energy and stress. read less NOT USED (high confidence) A. Bartók, J. Kermode, N. Bernstein, and G. Csányi, “Machine Learning a General-Purpose Interatomic Potential for Silicon,” Physical Review X. 2018. link Times cited: 291 Abstract: The success of first principles electronic structure calcula… read moreAbstract: The success of first principles electronic structure calculation for predictive modeling in chemistry, solid state physics, and materials science is constrained by the limitations on simulated length and time scales due to computational cost and its scaling. Techniques based on machine learning ideas for interpolating the Born-Oppenheimer potential energy surface without explicitly describing electrons have recently shown great promise, but accurately and efficiently fitting the physically relevant space of configurations has remained a challenging goal. Here we present a Gaussian Approximation Potential for silicon that achieves this milestone, accurately reproducing density functional theory reference results for a wide range of observable properties, including crystal, liquid, and amorphous bulk phases, as well as point, line, and plane defects. We demonstrate that this new potential enables calculations that would be extremely expensive with a first principles electronic structure method, such as finite temperature phase boundary lines, self-diffusivity in the liquid, formation of the amorphous by slow quench, and dynamic brittle fracture. We show that the uncertainty quantification inherent to the Gaussian process regression framework gives a qualitative estimate of the potential's accuracy for a given atomic configuration. The success of this model shows that it is indeed possible to create a useful machine-learning-based interatomic potential that comprehensively describes a material, and serves as a template for the development of such models in the future. read less NOT USED (high confidence) S. Balagan, V. U. Nazarov, A. Shevlyagin, D. Goroshko, and N. Galkin, “Theoretical approach to embed nanocrystallites into a bulk crystalline matrix and the embedding influence on the electronic band structure and optical properties of the resulting heterostructures,” Journal of Physics: Condensed Matter. 2018. link Times cited: 0 Abstract: We develop an approach and present results of the combined m… read moreAbstract: We develop an approach and present results of the combined molecular dynamics and density functional theory calculations of the structural and optical properties of the nanometer-sized crystallites embedded in a bulk crystalline matrix. The method is designed and implemented for both compatible and incompatible lattices of the nanocrystallite (NC) and the host matrix, when determining the NC optimal orientation relative to the matrix constitutes a challenging problem. We suggest and substantiate an expression for the cost function of the search algorithm, which is the energy per supercell generalized for varying number of atoms in the latter. The epitaxial relationships at the Si/NC interfaces and the optical properties are obtained and found to be in a reasonable agreement with experimental data. Dielectric functions show significant sensitivity to the NC’s orientation relative to the matrix at energies below 0.5 eV. read less NOT USED (high confidence) B. Dongre, J. Carrete, A. Katre, N. Mingo, and G. Madsen, “Resonant phonon scattering in semiconductors,” Journal of Materials Chemistry C. 2018. link Times cited: 16 Abstract: Boron impurities have recently been shown to induce resonant… read moreAbstract: Boron impurities have recently been shown to induce resonant phonon scattering in 3C-SiC, dramatically lowering its thermal conductivity. The B-doped 3C-SiC is associated with an off-center relaxation of the B atom, inducing a local transition from Td to C3v symmetry. Similar relaxations in B and N-doped diamond, with a similarly large effect on the interatomic force constants (IFCs), fail to produce resonances. Here we develop an intuitive understanding of such dopant-induced resonant phonon scattering in semiconductors with the help of a 1D monoatomic chain model. We find that the phenomenon is connected to a slight asymmetry in the relaxed position of the defect, with its origin in two or more minima of the potential energy surface in close proximity. The large perturbation they introduce in the IFCs is the essential ingredient of a resonance. read less NOT USED (high confidence) A. Verma, A. Parashar, and M. Packirisamy, “Atomistic modeling of graphene/hexagonal boron nitride polymer nanocomposites: a review,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2018. link Times cited: 80 Abstract: Due to their exceptional properties, graphene and hexagonal … read moreAbstract: Due to their exceptional properties, graphene and hexagonal boron nitride (h‐BN) nanofillers are emerging as potential candidates for reinforcing the polymer‐based nanocomposites. Graphene and h‐BN have comparable mechanical and thermal properties, whereas due to high band gap in h‐BN (~5 eV), have contrasting electrical conductivities. Atomistic modeling techniques are viable alternatives to the costly and time‐consuming experimental techniques, and are accurate enough to predict the mechanical properties, fracture toughness, and thermal conductivities of graphene and h‐BN‐based nanocomposites. Success of any atomistic model entirely depends on the type of interatomic potential used in simulations. This review article encompasses different types of interatomic potentials that can be used for the modeling of graphene, h‐BN, and corresponding nanocomposites, and further elaborates on developments and challenges associated with the classical mechanics‐based approach along with synergic effects of these nano reinforcements on host polymer matrix. read less NOT USED (high confidence) M. Faruq, A. Villesuzanne, and G. Shao, “Molecular-dynamics simulations of binary Pd-Si metal alloys: Glass formation, crystallisation and cluster properties,” Journal of Non-crystalline Solids. 2018. link Times cited: 22 NOT USED (high confidence) J. Yeo et al., “Materials-by-design: computation, synthesis, and characterization from atoms to structures,” Physica Scripta. 2018. link Times cited: 36 Abstract: In the 50 years that succeeded Richard Feynman’s exposition … read moreAbstract: In the 50 years that succeeded Richard Feynman’s exposition of the idea that there is ‘plenty of room at the bottom’ for manipulating individual atoms for the synthesis and manufacturing processing of materials, the materials-by-design paradigm is being developed gradually through synergistic integration of experimental material synthesis and characterization with predictive computational modeling and optimization. This paper reviews how this paradigm creates the possibility to develop materials according to specific, rational designs from the molecular to the macroscopic scale. We discuss promising techniques in experimental small-scale material synthesis and large-scale fabrication methods to manipulate atomistic or macroscale structures, which can be designed by computational modeling. These include recombinant protein technology to produce peptides and proteins with tailored sequences encoded by recombinant DNA, self-assembly processes induced by conformational transition of proteins, additive manufacturing for designing complex structures, and qualitative and quantitative characterization of materials at different length scales. We describe important material characterization techniques using numerous methods of spectroscopy and microscopy. We detail numerous multi-scale computational modeling techniques that complements these experimental techniques: DFT at the atomistic scale; fully atomistic and coarse-grain molecular dynamics at the molecular to mesoscale; continuum modeling at the macroscale. Additionally, we present case studies that utilize experimental and computational approaches in an integrated manner to broaden our understanding of the properties of two-dimensional materials and materials based on silk and silk-elastin-like proteins. read less NOT USED (high confidence) J. Byggmästar, E. Hodille, Y. Ferro, and K. Nordlund, “Analytical bond order potential for simulations of BeO 1D and 2D nanostructures and plasma-surface interactions,” Journal of Physics: Condensed Matter. 2018. link Times cited: 18 Abstract: An analytical interatomic bond order potential for the Be–O … read moreAbstract: An analytical interatomic bond order potential for the Be–O system is presented. The potential is fitted and compared to a large database of bulk BeO and point defect properties obtained using density functional theory. Its main applications include simulations of plasma-surface interactions involving oxygen or oxide layers on beryllium, as well as simulations of BeO nanotubes and nanosheets. We apply the potential in a study of oxygen irradiation of Be surfaces, and observe the early stages of an oxide layer forming on the Be surface. Predicted thermal and elastic properties of BeO nanotubes and nanosheets are simulated and compared with published ab initio data. read less NOT USED (high confidence) A. Shiryaev et al., “Ion implantation in nanodiamonds: size effect and energy dependence,” Scientific Reports. 2018. link Times cited: 25 NOT USED (high confidence) Z. Ong, B. Qiu, S. Xu, X. Ruan, and E. Pop, “Flexural resonance mechanism of thermal transport across graphene-SiO2 interfaces,” Journal of Applied Physics. 2018. link Times cited: 24 Abstract: Understanding the microscopic mechanism of heat dissipation … read moreAbstract: Understanding the microscopic mechanism of heat dissipation at the dimensionally mismatched interface between a two-dimensional (2D) crystal and its substrate is crucial for the thermal management of devices based on 2D materials. Here, we study the lattice contribution to thermal (Kapitza) transport at graphene-SiO2 interfaces using molecular dynamics (MD) simulations and non-equilibrium Green's functions (NEGF). We find that 78 percent of the Kapitza conductance is due to sub-20 THz flexural acoustic modes, and that a resonance mechanism dominates the interfacial phonon transport. MD and NEGF estimate the classical Kapitza conductance to be hK ≈ 10 to 16 MW K−1 m−2 at 300 K, respectively, consistent with existing experimental observations. Taking into account quantum mechanical corrections, this value is approximately 28% lower at 300 K. Our calculations also suggest that hK scales as T2 at low temperatures (T < 100 K) due to the linear frequency dependence of phonon transmission across the graphene-SiO2 interface at low frequencies. Our study sheds light on the role of flexural acoustic phonons in heat dissipation from graphene to its substrate.Understanding the microscopic mechanism of heat dissipation at the dimensionally mismatched interface between a two-dimensional (2D) crystal and its substrate is crucial for the thermal management of devices based on 2D materials. Here, we study the lattice contribution to thermal (Kapitza) transport at graphene-SiO2 interfaces using molecular dynamics (MD) simulations and non-equilibrium Green's functions (NEGF). We find that 78 percent of the Kapitza conductance is due to sub-20 THz flexural acoustic modes, and that a resonance mechanism dominates the interfacial phonon transport. MD and NEGF estimate the classical Kapitza conductance to be hK ≈ 10 to 16 MW K−1 m−2 at 300 K, respectively, consistent with existing experimental observations. Taking into account quantum mechanical corrections, this value is approximately 28% lower at 300 K. Our calculations also suggest that hK scales as T2 at low temperatures (T < 100 K) due to the linear frequency dependence of phonon transmission across the graphene-SiO... read less NOT USED (high confidence) O. T. Unke and M. Meuwly, “A reactive, scalable, and transferable model for molecular energies from a neural network approach based on local information.,” The Journal of chemical physics. 2018. link Times cited: 75 Abstract: Despite the ever-increasing computer power, accurate ab init… read moreAbstract: Despite the ever-increasing computer power, accurate ab initio calculations for large systems (thousands to millions of atoms) remain infeasible. Instead, approximate empirical energy functions are used. Most current approaches are either transferable between different chemical systems, but not particularly accurate, or they are fine-tuned to a specific application. In this work, a data-driven method to construct a potential energy surface based on neural networks is presented. Since the total energy is decomposed into local atomic contributions, the evaluation is easily parallelizable and scales linearly with system size. With prediction errors below 0.5 kcal mol-1 for both unknown molecules and configurations, the method is accurate across chemical and configurational space, which is demonstrated by applying it to datasets from nonreactive and reactive molecular dynamics simulations and a diverse database of equilibrium structures. The possibility to use small molecules as reference data to predict larger structures is also explored. Since the descriptor only uses local information, high-level ab initio methods, which are computationally too expensive for large molecules, become feasible for generating the necessary reference data used to train the neural network. read less NOT USED (high confidence) X. Yang, H. Tian, and B. Zhang, “Fast crack propagation correlated with crack tip stress in 2D hexagonal atomic lattices,” International Journal of Fracture. 2018. link Times cited: 4 NOT USED (high confidence) Y. Yan, J. Lv, and S. Liu, “Chirality and grain boundary effects on indentation mechanical properties of graphene coated on nickel foil,” Nanotechnology. 2018. link Times cited: 14 Abstract: We investigate chirality and grain boundary (GB) effects on … read moreAbstract: We investigate chirality and grain boundary (GB) effects on indentation mechanical properties of graphene coated on nickel foil using molecular dynamics simulations. The models of graphene with different chirality angles, different numbers of layers and tilt GBs were established. It was found that the chirality angle of few-layer graphene had a significant effect on the load bearing capacity of graphene/nickel systems, and this turns out to be more significant when the number of layers is greater than one. The enhancement to the contact stiffness, elastic capacity and the load bearing capacity of graphene with tilt GBs was lower than that of pristine graphene. read less NOT USED (high confidence) S. Takamoto et al., “Atomistic mechanism of graphene growth on a SiC substrate: Large-scale molecular dynamics simulations based on a new charge-transfer bond-order type potential,” Physical Review B. 2018. link Times cited: 9 Abstract: Thermal decomposition of silicon carbide is a promising appr… read moreAbstract: Thermal decomposition of silicon carbide is a promising approach for the fabrication of graphene. However, the atomistic growth mechanism of graphene remains unclear. This paper describes the development of a new charge-transfer interatomic potential. Carbon bonds with a wide variety of characteristics can be reproduced by the proposed vectorized bond-order term. Large-scale thermal decomposition simulation enables us to observe the continuous growth process of the multi-ring carbon structure. The annealing simulation reveals the atomistic process by which the multi-ring carbon structure is transformed to flat graphene involving only 6-membered rings. Also, it is found that the surface atoms of the silicon carbide substrate enhance the homogeneous graphene formation. read less NOT USED (high confidence) M. Friedrich and U. Stefanelli, “Graphene ground states,” Zeitschrift für angewandte Mathematik und Physik. 2018. link Times cited: 7 NOT USED (high confidence) T. Iwata and K. Shintani, “Reduction of the thermal conductivity of a graphene/hBN heterobilayer via interlayer sp3 bonds.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 21 Abstract: Thermal conductivities (TCs) of graphene (g)/hexagonal boron… read moreAbstract: Thermal conductivities (TCs) of graphene (g)/hexagonal boron nitride (hBN) heterobilayers with interlayer sp3 bonds are computed using nonequilibrium molecular dynamics (NEMD) simulations. It is revealed that the TC of a g/hBN heterobilayer drastically decreases if there is even a few interlayer sp3 bonds, and continues to gradually decrease upon increasing their fraction up to 0.25, where the fraction of the interlayer sp3 bonds is defined by the atomic fraction of interlayer-sp3-bonded carbon atoms within graphene constituting a g/hBN heterobilayer. If their fraction exceeds 0.25, the TC of a g/hBN heterobilayer gradually increases, namely, the TC of a g/hBN heterobilayer takes a minimum at the fraction of 0.25 of the interlayer sp3 bonds. In order to understand such a behavior of the TC of the heterobilayer, the local phonon density of states (DOSs) in each of the two layers is calculated. By examining the local phonon DOSs, it was found that the existence of the minimum TC of the heterobilayer can be understood by considering both the phonon scattering and the characteristic change of the heterobilayer structure. In the range of the low fractions of interlayer sp3 bonds, the van der Waals (vdW) interactions are predominantly effective for binding the two layers, and the interlayer sp3 bonds act as phonon scatterers like defects to make the TC of the heterobilayer decrease. Upon increasing the fraction of interlayer sp3 bonds, the contribution of the interlayer sp3 bonds to the unification of the two layers becomes stronger, and hence the rigidity of the heterobilayer structure gradually increases. If their fraction exceeds 0.25, the heterobilayer structure approaches a quasi-three-dimensional one, so that the TC of the heterobilayer increases. These findings will be useful for tuning the TCs of g/hBN heterobilayers via interlayer sp3 bonds. read less NOT USED (high confidence) D. Damasceno, E. Mesquita, R. Rajapakse, and R. Pavanello, “Atomic-scale finite element modelling of mechanical behaviour of graphene nanoribbons,” International Journal of Mechanics and Materials in Design. 2018. link Times cited: 0 NOT USED (high confidence) D. K. Das and J. Sarkar, “Comparison of mechanical properties of silicene estimated using different testing procedures: A molecular dynamics study,” Journal of Applied Physics. 2018. link Times cited: 11 Abstract: Silicene, a two-dimensional allotrope and silicon counterpar… read moreAbstract: Silicene, a two-dimensional allotrope and silicon counterpart of graphene, has recently attracted scientists all over the world due to its superior material properties and thus can be a potential applicant as a reinforcing agent. The mechanical properties of silicene have been studied using several testings (tensile, bending, oscillation, and equilibrium) through the molecular dynamics (MD) simulation technique. Plastic flow occurs, and 46% elongation is observed in a silicene sheet with dimensions of (200 × 700) A for room temperature (298 K) tensile testing. The yield strength, ultimate tensile strength, Young's modulus (E), cohesive energy, and bulk modulus are found to be 18.28 GPa, 23.96 GPa, 5.25 TPa, 3.72 eV atom−1, and 3.62 TPa, respectively. For the same sample, a Poisson ratio of 0.75 is observed. An ultrahigh mechanical strength of silicene, even higher than the previously predicted value of 0.178 TPa, is observed in this study. read less NOT USED (high confidence) S. Hu, W.-guo Sun, J. Fu, Z. Zhang, W. Wu, and Y. Tang, “Initiation mechanisms and kinetic analysis of the isothermal decomposition of poly(α-methylstyrene): a ReaxFF molecular dynamics study,” RSC Advances. 2018. link Times cited: 11 Abstract: This study investigates the thermal decomposition initiation… read moreAbstract: This study investigates the thermal decomposition initiation mechanisms and kinetics of poly(α-methylstyrene) (PαMS) under isothermal conditions, using molecular dynamics simulations with the ReaxFF reactive force field. The unimolecular pyrolysis simulations show that the thermal decomposition of the PαMS molecule is initiated mainly by carbon–carbon backbone cleavage in two types at random points along the main chain that leads to different intermediates, and is accompanied by depolymerization reactions that lead to the formation of the final products. The time evolution of typical species in the process of PαMS thermal decomposition at various temperatures presents specific evolution profiles and shows a temperature-dependence effect. Isothermal decomposition kinetic analysis based on PαMS pyrolysis shows that the activation energy varies with the degree of conversion during the thermal decomposition processes, which infers that the decomposition process at different conversions may have different reaction mechanisms. read less NOT USED (high confidence) A. Glielmo, C. Zeni, and A. Vita, “Efficient nonparametric n -body force fields from machine learning,” Physical Review B. 2018. link Times cited: 92 Abstract: The authors present a scheme to construct classical $n$-body… read moreAbstract: The authors present a scheme to construct classical $n$-body force fields using Gaussian Process (GP) Regression, appropriately mapped over explicit n-body functions (M-FFs). The procedure is possible, and will yield accurate forces, whenever prior knowledge allows to restrict the interactions to a finite order $n$, so that the ``universal approximator'' resolving power of standard GPs or Neural Networks is not needed. Under these conditions, the proposed construction preserves flexibility of training, systematically improvable accuracy, and a clear framework for validation of the underlying machine learning technique. Moreover, the M-FFs are as fast as classical parametrized potentials, since they avoid lengthy summations over database entries or weight parameters. read less NOT USED (high confidence) X. Yuan and Y. Wang, “Collapsed adhesion of carbon nanotubes on silicon substrates: continuum mechanics and atomistic simulations,” Nanotechnology. 2018. link Times cited: 14 Abstract: Carbon nanotubes (CNTs) can undergo collapse from the ordina… read moreAbstract: Carbon nanotubes (CNTs) can undergo collapse from the ordinary cylindrical configurations to bilayer ribbons when adhered on substrates. In this study, the collapsed adhesion of CNTs on the silicon substrates is investigated using both classical molecular dynamics (MD) simulations and continuum analysis. The governing equations and transversality conditions are derived based on the minimum potential energy principle and the energy-variational method, considering both the van der Waals interactions between CNTs and substrates and those inside CNTs. Closed-form solutions for the collapsed configuration are obtained which show good agreement with the results of MD simulations. The stability of adhesive configurations is investigated by analyzing the energy states. It is found that the adhesive states of single-walled CNTs (SWCNTs) (n, n) on the silicon substrates can be categorized by two critical radii, 0.716 and 0.892 nm. For SWCNTs with radius larger than 0.892 nm, they would fully collapse on the silicon substrates. For SWCNTs with radius less than 0.716 nm, the initial cylindrical configuration is energetically favorable. For SWCNTs with radius between two critical radii, the radially deformed state is metastable. The non-contact ends of all collapsed SWCNTs are identical with the same arc length of 2.38 nm. Finally, the role of number of walls on the adhesive configuration is investigated quantitatively. For multi-walled CNTs with the number of walls exceeding a certain value, the cylindrical configuration is stable due to the increasing bending stiffness. The present study can be useful for the design of CNT-based nanodevices. read less NOT USED (high confidence) M. Wen, S. Shirodkar, P. Plecháč, E. Kaxiras, R. Elliott, and E. Tadmor, “A force-matching Stillinger-Weber potential for MoS2: Parameterization and Fisher information theory based sensitivity analysis,” Journal of Applied Physics. 2017. link Times cited: 25 Abstract: Two-dimensional molybdenum disulfide (MoS2) is a promising m… read moreAbstract: Two-dimensional molybdenum disulfide (MoS2) is a promising material for the next generation of switchable transistors and photodetectors. In order to perform large-scale molecular simulations of the mechanical and thermal behavior of MoS2-based devices, an accurate interatomic potential is required. To this end, we have developed a Stillinger-Weber potential for monolayer MoS2. The potential parameters are optimized to reproduce the geometry (bond lengths and bond angles) of MoS2 in its equilibrium state and to match as closely as possible the forces acting on the atoms along a dynamical trajectory obtained from ab initio molecular dynamics. Verification calculations indicate that the new potential accurately predicts important material properties including the strain dependence of the cohesive energy, the elastic constants, and the linear thermal expansion coefficient. The uncertainty in the potential parameters is determined using a Fisher information theory analysis. It is found that the parameters are... read less NOT USED (high confidence) J. Hasik, E. Tosatti, and R. Martoňák, “Quantum and classical ripples in graphene,” Physical Review B. 2017. link Times cited: 18 Abstract: Thermal ripples of graphene are well understood at room temp… read moreAbstract: Thermal ripples of graphene are well understood at room temperature, but their quantum counterpart at low temperatures are still in need of a realistic quantitative description. Here we present atomistic Path Integral Monte Carlo simulations of freestanding graphene, which show upon cooling a striking classical-quantum evolution of height and angular fluctuations. The crossover takes place at ever-decreasing temperatures for ever-increasing wavelengths so that a completely quantum regime is never attained. Low-temperature quantum graphene is flatter and smoother than classical at large scales, yet rougher at short scales. The angular fluctuation distribution of the normals can be quantitatively described by coexistence of two Gaussians, one classical, strongly T-dependent, and one quantum, about 2 degrees wide, of zero-point character. The quantum evolution of ripple-induced height and angular spread should be observable in electron diffraction in graphene and other 2D materials like $MoS_2$, bilayer graphene, BN, etc. read less NOT USED (high confidence) J. Luo, A. Alateeqi, L. Liu, and T. Sinno, “Atomistic simulations of carbon diffusion and segregation in liquid silicon,” Journal of Applied Physics. 2017. link Times cited: 9 Abstract: The diffusivity of carbon atoms in liquid silicon and their … read moreAbstract: The diffusivity of carbon atoms in liquid silicon and their equilibrium distribution between the silicon melt and crystal phases are key, but unfortunately not precisely known parameters for the global models of silicon solidification processes. In this study, we apply a suite of molecular simulation tools, driven by multiple empirical potential models, to compute diffusion and segregation coefficients of carbon at the silicon melting temperature. We generally find good consistency across the potential model predictions, although some exceptions are identified and discussed. We also find good agreement with the range of available experimental measurements of segregation coefficients. However, the carbon diffusion coefficients we compute are significantly lower than the values typically assumed in continuum models of impurity distribution. Overall, we show that currently available empirical potential models may be useful, at least semi-quantitatively, for studying carbon (and possibly other impurity) trans... read less NOT USED (high confidence) A. H. Howlader and M. S. Islam, “Phonon transmission of vacancy defected (10,0) carbon nanotube,” 2017 3rd International Conference on Electrical Information and Communication Technology (EICT). 2017. link Times cited: 4 Abstract: Carbon nanotubes (CNTs) are envisaged as the nano-building b… read moreAbstract: Carbon nanotubes (CNTs) are envisaged as the nano-building blocks in the next generation electronic devices due to their versatile potentialities. Here, the phonon properties of vacancy defected semiconductor (10,0) zigzag carbon nanotube (ZCNT) are explored using empirical interatomic potentials (EIPs). It is found that phonon modes become non-degenerate at the Γ point of Brillouin zone due to vacancies. The small changes in defect concentrations give rise to a large variation in the entropy and phonon transmission properties of the system. The thermal conductivity reduces about 50% with only 1% vacancy defects. We also address the issue of temperature effects on thermal conductivity. Moreover, quasi-ballistic low-frequency acoustic phonon transport is observed. read less NOT USED (high confidence) S. Stelmakh, K. Skrobas, S. Gierlotka, and B. Palosz, “Effect of interaction of external surfaces on the symmetry and lattice distortion of CdSe nanocrystals by molecular dynamics simulations,” Journal of Nanoparticle Research. 2017. link Times cited: 2 NOT USED (high confidence) B. Narayanan et al., “Machine learnt bond order potential to model metal-organic (Co-C) heterostructures.,” Nanoscale. 2017. link Times cited: 7 Abstract: A fundamental understanding of the inter-relationships betwe… read moreAbstract: A fundamental understanding of the inter-relationships between structure, morphology, atomic scale dynamics, chemistry, and physical properties of mixed metallic-covalent systems is essential to design novel functional materials for applications in flexible nano-electronics, energy storage and catalysis. To achieve such knowledge, it is imperative to develop robust and computationally efficient atomistic models that describe atomic interactions accurately within a single framework. Here, we present a unified Tersoff-Brenner type bond order potential (BOP) for a Co-C system, trained against lattice parameters, cohesive energies, equation of state, and elastic constants of different crystalline phases of cobalt as well as orthorhombic Co2C derived from density functional theory (DFT) calculations. The independent BOP parameters are determined using a combination of supervised machine learning (genetic algorithms) and local minimization via the simplex method. Our newly developed BOP accurately describes the structural, thermodynamic, mechanical, and surface properties of both the elemental components as well as the carbide phases, in excellent accordance with DFT calculations and experiments. Using our machine-learnt BOP potential, we performed large-scale molecular dynamics simulations to investigate the effect of metal/carbon concentration on the structure and mechanical properties of porous architectures obtained via self-assembly of cobalt nanoparticles and fullerene molecules. Such porous structures have implications in flexible electronics, where materials with high electrical conductivity and low elastic stiffness are desired. Using unsupervised machine learning (clustering), we identify the pore structure, pore-distribution, and metallic conduction pathways in self-assembled structures at different C/Co ratios. We find that as the C/Co ratio increases, the connectivity between the Co nanoparticles becomes limited, likely resulting in low electrical conductivity; on the other hand, such C-rich hybrid structures are highly flexible (i.e., low stiffness). The BOP model developed in this work is a valuable tool to investigate atomic scale processes, structure-property relationships, and temperature/pressure response of Co-C systems, as well as design organic-inorganic hybrid structures with a desired set of properties. read less NOT USED (high confidence) S. Mei and I. Knezevic, “Thermal conductivity of ternary III-V semiconductor alloys: The role of mass difference and long-range order,” arXiv: Materials Science. 2017. link Times cited: 6 Abstract: Thermal transport in bulk ternary III-V arsenide (III-As) se… read moreAbstract: Thermal transport in bulk ternary III-V arsenide (III-As) semiconductor alloys was investigated using equilibrium molecular dynamics with optimized Albe-Tersoff empirical interatomic potentials. Existing potentials for binary AlAs, GaAs, and InAs were optimized to obtain accurate phonon dispersions and temperature-dependent thermal conductivity. Calculations of thermal transport in ternary III-Vs commonly employ the virtual-crystal approximation (VCA), where the structure is assumed to be a random alloy and all group-III atoms (cations) are treated as if they have an effective weighted-average mass. Here, we showed that is critical to treat atomic masses explicitly, and that the thermal conductivity obtained with explicit atomic masses differs considerably from the value obtained with the average VCA cation mass. The larger the difference between the cation masses, the poorer the VCA prediction for thermal conductivity. The random-alloy assumption in the VCA is also challenged, because X-ray diffraction and transmission electron microscopy show order in InGaAs, InAlAs, and GaAlAs epi-layers. We calculated thermal conductivity for three common types of order [CuPt-B, CuAu-I, and triple-period-A (TPA)] and showed that the experimental results for In$_{0.53}$Ga$_{0.47}$As and In$_{0.52}$Al$_{0.48}$As, which are lattice matched to the InP substrate, can be reproduced in molecular dynamics simulation with 2% and 8% of random disorder, respectively. Based on our results, thermal transport in ternary III-As alloys appears to be governed by the competition between mass-difference scattering, which is much more pronounced than the VCA suggests, and the long-range order that these alloys support. read less NOT USED (high confidence) E. Mainini, H. Murakawa, P. Piovano, and U. Stefanelli, “Carbon-Nanotube Geometries as Optimal Configurations,” Multiscale Model. Simul. 2017. link Times cited: 12 Abstract: The fine geometry of carbon nanotubes is investigated from t… read moreAbstract: The fine geometry of carbon nanotubes is investigated from the viewpoint of molecular mechanics. Actual nanotube configurations are characterized as locally minimizers of a given configurational energy, including both two- and three-body contributions. By focusing on so-called zigzag and armchair topologies, we prove that the configurational energy is strictly minimized within specific, one-parameter families of periodic configurations. Such optimal configurations are checked to be stable with respect to a large class of small nonperiodic perturbations and do not coincide with classical rolled-up nor polyhedral geometries. read less NOT USED (high confidence) Y. Joko, R. Sasaki, and K. Shintani, “Dynamic encapsulation of corannulene molecules into a single-walled carbon nanotube.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 3 Abstract: The morphology of corannulene molecules encapsulated in a si… read moreAbstract: The morphology of corannulene molecules encapsulated in a single-walled carbon nanotube (SWCNT) is addressed using atomistic simulations. Firstly, dynamic simulation (DS) of encapsulation of corannulene molecules into a SWCNT is performed using a molecular dynamics (MD) method. It is revealed that corannulene molecules encapsulated in a SWCNT tend to form concave-concave (CC) dimers, and these dimers make stacks tilting against the SWCNT axis or take an arrangement such that their convex surfaces face the inner wall of the SWCNT. This tendency arises from the fact that the van der Waals interactions between the convex surfaces of the corannulene molecules and the inner wall of the SWCNT dominate in their dynamic encapsulation into the SWCNT, and CC dimers are favored based on the energetics. Next, conjugate gradient (CG) energy minimizations starting from two kinds of initial arrangement of corannulene molecules in a SWCNT, concave-convex (CV) and CC/convex-convex (VV) arrangements, are performed. The CG energy minimizations confirm the result of DS that CC dimers are the structural motif of corannulene molecules in a SWCNT. From the final configurations of both the simulations, the tilt angles and intermolecular distances of the stacked molecules are calculated. With increasing the SWCNT diameter, the tilt angles decrease while the intermolecular distances remain almost constant. The tilt angles of the stacked corannulene molecules are approximately expressed by a semi-analytical formula which is derived on the basis of a geometrical constraint condition. read less NOT USED (high confidence) L. Jaillet, S. Artemova, and S. Redon, “IM-UFF: Extending the universal force field for interactive molecular modeling.,” Journal of molecular graphics & modelling. 2017. link Times cited: 31 NOT USED (high confidence) H. Shabbir and M. Hartmann, “Influence of reversible cross-link coordination on the mechanical behavior of a linear polymer chain,” New Journal of Physics. 2017. link Times cited: 5 Abstract: In this work, we investigate the effect of the coordination … read moreAbstract: In this work, we investigate the effect of the coordination of cross-links (i.e., the number of monomers participating in one cross-link) on the mechanical performance of a single polymeric chain. The framework provided by the reactive empirical bond order potential is used to generically describe the ability of certain monomers to form cross-links of different coordination. A systematic investigation of the influence of the coordination of cross-links on the mechanical properties of single polymeric chains is presented by comparing systems that contain cross-links in the classical form between two monomers (dimer) and such where the cross-links are formed by three monomers (trimer). The results show that the mechanical performance crucially depends on the coordination of cross-links. The overall shape of the load-displacement curves as well as mechanical parameters like stiffness, strength and work-to-straighten the molecule are different for the different systems. While the load-displacement curve shows an overall more continuous shape for the system containing trimers compared to the system including dimers only, the mechanical parameters are consistently lower for the first system. On the other hand, in contrast to the dimer case a trimer remains stable upon detachment of one of the monomers and the bonds are more mobile. This will be of importance in the case of fiber bundles, where the loading situation is even more complicated than in the single chain system due to the presence of inter-chain cross-links. read less NOT USED (high confidence) D. Huang, A. Iurov, F. Gao, G. Gumbs, and D. Cardimona, “Many-Body Theory of Proton-Generated Point Defects for Losses of Electron Energy and Photons in Quantum Wells,” Physical review applied. 2017. link Times cited: 6 Abstract: The effects of point defects on the loss of either energies … read moreAbstract: The effects of point defects on the loss of either energies of ballistic electron beams or incident photons are studied by using a many-body theory in a multi-quantum-well system. This includes the defect-induced vertex correction to a bare polarization function of electrons within the ladder approximation as well as the intralayer and interlayer screening of defect-electron interactions are also taken into account in the random-phase approximation. The numerical results of defect effects on both energy-loss and optical-absorption spectra are presented and analyzed for various defect densities, number of quantum wells, and wave vectors. The diffusion-reaction equation is employed for calculating distributions of point defects in a layered structure. For completeness, the production rate for Frenkel-pair defects and their initial concentration are obtained based on atomic-level molecular-dynamics simulations. By combining defect-effect, diffusion-reaction and molecular-dynamics models proposed in this paper with a space-weather forecast model for the first time, it will be possible to enable specific designing for electronic and optoelectronic quantum devices that will be operated in space with radiation-hardening protection, and therefore, will effectively extend the lifetime of these satellite onboard electronic and optoelectronic devices. read less NOT USED (high confidence) V. D. Camiola and V. Tozzini, “Collective mode mining from molecular dynamics simulations: a comparative approach,” arXiv: Computational Physics. 2017. link Times cited: 2 Abstract: The evaluation of collective modes is fundamental in the ana… read moreAbstract: The evaluation of collective modes is fundamental in the analysis of molecular dynamics simulations. Several methods are available to extract that information, i.e normal mode analysis, principal component and spectral analysis of trajectories, basically differing by the quantity considered as the nodal one (frequency, amplitude, or pattern of displacement) and leading to the definition of different kinds of collective excitations and physical spectral observables. Different views converge in the harmonic regime and/or for homo-atomic systems. However, for anharmonic and out of equilibrium dynamics different quantities bring different information and only their comparison can give a complete view of the system behavior. To allow such a comparative analysis, we review and compare the different approaches, applying them in different combination to two examples of physical relevance: graphene and fullerene C60. read less NOT USED (high confidence) X. Yuan and Y. Wang, “Adhesion of single- and multi-walled carbon nanotubes to silicon substrate: atomistic simulations and continuum analysis,” Journal of Physics D: Applied Physics. 2017. link Times cited: 13 Abstract: The radial deformation of carbon nanotubes (CNTs) adhering t… read moreAbstract: The radial deformation of carbon nanotubes (CNTs) adhering to a substrate may prominently affect their mechanical and physical properties. In this study, both classical atomistic simulations and continuum analysis are carried out, to investigate the lateral adhesion of single-walled CNTs (SWCNTs) and multi-walled CNTs (MWCNTs) to a silicon substrate. A linear elastic model for analyzing the adhesion of 2D shells to a rigid semi-infinite substrate is constructed in the framework of continuum mechanics. Good agreement is achieved between the cross-section profiles of adhesive CNTs obtained by the continuum model and by the atomistic simulation approach. It is found that the adhesion of a CNT to the silicon substrate is significantly influenced by its initial diameter and the number of walls. CNTs with radius larger than a certain critical radius are deformed radially on the silicon substrate with flat contact regions. With increasing number of walls, the extent of radial deformation of a MWCNT on the substrate decreases dramatically, and the flat contact area reduces—and eventually vanishes—due to increasing equivalent bending stiffness. It is analytically predicted that large-diameter MWCNTs with a large number of walls are likely to ‘stand’ on the silicon substrate. The present work can be useful for understanding the radial deformation of CNTs adhering to a solid planar substrate. read less NOT USED (high confidence) U. Stefanelli, “Stable carbon configurations,” Bollettino dell’Unione Matematica Italiana. 2017. link Times cited: 8 NOT USED (high confidence) S. Huang and C. Zhou, “Modeling and Simulation of Nanoindentation,” JOM. 2017. link Times cited: 17 NOT USED (high confidence) Z. Zhang and H. Urbassek, “Comparative Study of Interatomic Interaction Potentials for Describing Indentation into Si Using Molecular Dynamics Simulation,” Applied Mechanics and Materials. 2017. link Times cited: 5 Abstract: We compare the performance of three interatomic interaction … read moreAbstract: We compare the performance of three interatomic interaction potentials for describing the evolution of plasticity and phase transformations in Si: the well established Stillinger-Weber potential, a recent modification used in the description of Al/Si composites, and a modification of the well known Tersoff potential. We show that the generation of dislocations and the evolution of plasticity are well described by the Stillinger-Weber potential and its modification, while the phase transformation to the high-pressure bct5 modification and the subsequent amorphization are better included in the modified Tersoff potential. read less NOT USED (high confidence) G. D. Chatzidakis, G. Kalosakas, Z. Fthenakis, and N. Lathiotakis, “A torsional potential for graphene derived from fitting to DFT results,” The European Physical Journal B. 2017. link Times cited: 5 NOT USED (high confidence) E. Lee and T. Luo, “The role of optical phonons in intermediate layer-mediated thermal transport across solid interfaces.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 13 Abstract: Thermal transport across solid interfaces plays important ro… read moreAbstract: Thermal transport across solid interfaces plays important roles in many applications, especially in the thermal management of modern power electronics. In this study, we use non-equilibrium MD (NEMD) simulations to systematically study a model SiC/GaN interface, which is an important interface in GaN-based power electronics, mated by different intermediate layers (ILs) with the focus on how the atomic masses of the ILs influence the overall thermal conductance. To isolate the mass effect, the Tersoff potential with the same parameters is used to approximate the interatomic interactions between all atoms, with the only differences between materials being their atomic masses. The NEMD results show that the thermal boundary conductance (TBC) of IL-mated interfaces depends not only on the total primitive cell mass of the IL but also on the relative masses of the atoms within the unit cell. By analyzing the vibrational power spectra (VPS) of SiC, IL, and GaN, it is found that the optical phonons play important roles in thermal transport across the solid/solid interfaces. There is an optimal mass ratio of the atoms in the unit cell of the IL that can maximize the overlap of IL optical phonon VPS with those of SiC and GaN. Furthermore, the atomic masses of a number of III-V semiconductor compounds are studied for the ILs. It is shown that when only considering the mass effect, in the classical limit, AlN will be the best IL to enhance thermal transport across SiC/GaN interfaces with an improvement of as much as 27% over that of a pristine SiC/GaN interface. Despite the known limitation of the model (e.g., absence of strain and quantum effects), the results from this work may still provide some useful information for the design of ILs to improve thermal transport across solid/solid interfaces. read less NOT USED (high confidence) J. Shang, Q. Yang, and X. Liu, “New Coarse-Grained Model and Its Implementation in Simulations of Graphene Assemblies.,” Journal of chemical theory and computation. 2017. link Times cited: 18 Abstract: Graphene is a one-atom thick layer of carbon atoms arranged … read moreAbstract: Graphene is a one-atom thick layer of carbon atoms arranged in a hexagonal pattern, which makes it the strongest material in the world. The Tersoff potential is a suitable potential for simulating the mechanical behavior of the complex covalently bonded system of graphene. In this paper, we describe a new coarse-grained (CG) potential, TersoffCG, which is based on the function form of the Tersoff potential. The TersoffCG applies to a CG model of graphene that uses the same hexagonal pattern as the atomistic model. The parameters of the TersoffCG potential are determined using structural feature and potential-energy fitting between the CG model and the atomic model. The modeling process of graphene is highly simplified using the present CG model as it avoids the necessity to define bonds/angles/dihedrals connectivity. What is more, the present CG model provides a new perspective of coarse-graining scheme for crystal structures of nanomaterials. The structural changes and mechanical properties of multilayer graphene were calculated using the new potential. Furthermore, a CG model of a graphene aerogel was built in a specific form of assembly. The chemical bonding in the joints of graphene-aerogel forms automatically during the energy relaxation process. The compressive and recover test of the graphene aerogel was reproduced to study its high elasticity. Our computational examples show that the TersoffCG potential can be used for simulations of graphene and its assemblies, which have many applications in areas of environmental protection, aerospace engineering, and others. read less NOT USED (high confidence) F. Gayk, J. Ehrens, T. Heitmann, P. Vorndamme, A. Mrugalla, and J. Schnack, “Young’s moduli of carbon materials investigated by various classical molecular dynamics schemes,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 16 NOT USED (high confidence) F. Al-Dirini et al., “Negative differential resistance in planar graphene quantum dot resonant tunneling diodes,” 2017 IEEE 17th International Conference on Nanotechnology (IEEE-NANO). 2017. link Times cited: 1 Abstract: Negative differential resistance (NDR), an electronic proper… read moreAbstract: Negative differential resistance (NDR), an electronic property present in resonant tunneling diodes, enables high performance terahertz frequency oscillators and multi-state logic and memory devices. An important measure of NDR is the peak-to-valley current ratio (PVCR) and this has been extremely lacking in solid-state NDR devices. Here we show how a dimensional mismatch between the quantum dot and the electrodes of a planar graphene Double Barrier Resonant Tunneling Diode (DB-RTD) greatly enhances the PVCR of the device up to a ratio of 103. Our findings suggest a promising future for the application of planar graphene quantum dot devices in next generation electronics. read less NOT USED (high confidence) R. Barreto, M. F. Carusela, and A. Monastra, “Thermal conductance of suspended nanoribbons: interplay between strain and interatomic potential nonlinearity,” Journal of Statistical Mechanics: Theory and Experiment. 2017. link Times cited: 1 Abstract: We investigate the role that nonlinearity in the interatomic… read moreAbstract: We investigate the role that nonlinearity in the interatomic potential has on the thermal conductance of a suspended nanoribbon when it is subjected to a longitudinal strain. To focus on the first cubic and quartic nonlinear terms of a general potential, we propose an atomic system based on an α–β Fermi–Pasta–Ulam nearest neighbor interaction. We perform classical molecular dynamics simulations to investigate the contribution of longitudinal, transversal and flexural modes to the thermal conductance as a function of the α–β parameters and the applied strain. We compare the cases where atoms are allowed to vibrate only in plane (2D) with the case of vibrations in and out of plane (3D). We find that the dependence of conductance on α and β relies on a crossover phenomenon between linear/nonlinear delocalized/localized flexural and transversal modes, driven by an on/off switch of the strain. read less NOT USED (high confidence) H. Li, R. Xu, Z. Bi, X. Shen, and K. Han, “Melting Properties of Medium-Sized Silicon Nanoclusters:
A Molecular Dynamics Study,” Journal of Electronic Materials. 2017. link Times cited: 7 NOT USED (high confidence) C. Davini, A. Favata, and R. Paroni, “A REBO-Potential-Based Model for Graphene Bending by $Γ$Γ-Convergence,” Archive for Rational Mechanics and Analysis. 2017. link Times cited: 4 NOT USED (high confidence) B. Mortazavi, H. Yang, F. Mohebbi, G. Cuniberti, and T. Rabczuk, “Graphene or h-BN paraffin composite structures for the thermal management of Li-ion batteries: A multiscale investigation,” Applied Energy. 2017. link Times cited: 129 NOT USED (high confidence) X. Chen, J. Ding, C. Jiang, Z. Liu, and N. Yuan, “Microstructure evolution of polycrystalline silicon by molecular dynamics simulation,” AIP Advances. 2017. link Times cited: 2 Abstract: Polycrystalline silicon is the dominant material in solar ce… read moreAbstract: Polycrystalline silicon is the dominant material in solar cells and plays an important role in photovoltaic industry. It is important for not only the conventional production of silicon ingots but also the direct growth of silicon wafers to control crystallization for obtaining the desired polycrystalline silicon. To the best of our knowledge, few studies have systematically reported about the effects of crystalline planes on the solidification behavior of liquid silicon and the analysis of the microstructural features of the polysilicon structure. In this study, molecular dynamics simulations were employed to investigate the solidification and microstructure evolution of polysilicon, with focus on the effects of the seed distribution and cooling rate on the growth of polycrystalline silicon. The (110), (111), and (112) planes were extruded by the (100) plane and formed the inclusion shape. The crystallization of silicon consisted of diamond-type structures is relatively high at a low cooling rate. The si... read less NOT USED (high confidence) M. Friedrich, E. Mainini, P. Piovano, and U. Stefanelli, “Characterization of Optimal Carbon Nanotubes Under Stretching and Validation of the Cauchy–Born Rule,” Archive for Rational Mechanics and Analysis. 2017. link Times cited: 9 NOT USED (high confidence) H. A. Toupkanloo and M. Fathollahi, “Molecular Dynamics Simulation of Al/NiO Thermite Reaction Using Reactive Force Field (ReaxFF),” Physical Chemistry Research. 2017. link Times cited: 3 Abstract: In this work, the thermal reaction of aluminum (Al) and nick… read moreAbstract: In this work, the thermal reaction of aluminum (Al) and nickel oxide (NiO) was investigated by molecular dynamics simulations. Some effective features of reaction such as reaction temperature, the reaction mechanism, and diffusion rate of oxygen into aluminum structure were studied. ReaxFF force field was performed to study the Al/NiO thermite reaction behavior at five different temperatures (500, 900, 1100, 1200, and 1400 K). The results obtained from the molecular dynamics simulation predict that the reaction temperature for aluminum metal and nickel oxide mixture would be 1141 K, which is in a good agreement with that of the experimental value (i.e. 1148.8 K). In addition, the mean square displacement analysis suggests that the movement of aluminum atoms is less than that of oxygen and nickel atoms. The estimated diffusion coefficient of oxygen in the aluminum / nickel oxide thermite mixtures was 4.53 × 10-8 m2 s-1. The results show that the diffusion coefficients significantly increase, by increasing temperature. read less NOT USED (high confidence) A. Shahabodini, R. Ansari, and M. Darvizeh, “Multiscale Evaluation of the Nonlinear Elastic Properties of Carbon Nanotubes Under Finite Deformation.” 2017. link Times cited: 5 Abstract: This paper deals with the calculation of the elastic propert… read moreAbstract: This paper deals with the calculation of the elastic properties for single-walled carbon nanotubes (SWCNTs) under axial deformation and hydrostatic pressure using the atomistic-based continuum approach and the deformation mapping technique. A hyperelastic model based on the higher-order Cauchy-Born (HCB) rule being applicable at finite strains and accounting for the chirality and material nonlinearity is presented. Mechanical properties of several carbon nanotubes (CNTs) are computed and compared with the existing theoretical results and a good agreement is observed. Moreover, by comparison with atomistic calculations, it is found that the present model can reproduce the energetics of axially deformed CNTs. The model is then adopted to study the dependence of the elastic properties on chirality, radius and strain which yields an upper bound on the stability limit of axially and circumferentially stretched nanotubes. The influence of chirality is found to be more prominent for smaller tubes and as the diameter increases, the anisotropy induced by finite deformations gets nullified. It is discerned that the constitutive properties of the CNT can vary with deformation in a nonlinear manner. It is also found that the CNT displays a martial softening behavior at finite tensile strains and a hardening behavior at slightly compressive strains. read less NOT USED (high confidence) X. Wang et al., “Quasi-continuum study of the buckling behavior of single-walled carbon nanocones subjected to bending under thermal loading,” Journal of Materials Research. 2017. link Times cited: 3 Abstract: In this study, the buckling behaviors of single-walled carbo… read moreAbstract: In this study, the buckling behaviors of single-walled carbon nanocones (SWCNCs) under bending at finite temperatures are predicted using a proposed multiscale quasi-continuum approach based on the temperature-dependent higher order Cauchy–Born (THCB) rule. The hyper-elastic constitutive model is derived exactly in the context of the higher order gradient theory that incorporates the details of the interatomic interaction. The numerical simulations for the deformation of SWCNCs are implemented using the developed meshless computational framework based on moving least-squares interpolation, which can precisely reproduce the deformation process and buckling patterns of SWCNCs under bending. The underlying correlations of the critical bending angle with respect to the geometry of SWCNCs and temperature are revealed by the numerical results. Furthermore, our simulation captures the transformation from the local to the global buckling process of SWCNCs, accompanied with an average strain energy jump. Our results correspond with previous studies. read less NOT USED (high confidence) J.-H. Jiang, W. Fu, J. Chen, and H. Zhao, “Anharmonicity induced thermal modulation in stressed graphene,” Science China Physics, Mechanics & Astronomy. 2017. link Times cited: 6 NOT USED (high confidence) H. Ko, A. Kaczmarowski, I. Szlufarska, and D. Morgan, “Data for: Optimization of self-interstitial clusters in 3C-SiC with Genetic Algorithm.” 2017. link Times cited: 9 NOT USED (high confidence) Z.-zhong Zhu et al., “An Efficient Scheme for Crystal Structure Prediction Based on Structural Motifs,” Journal of Physical Chemistry C. 2017. link Times cited: 10 Abstract: An efficient scheme based on structural motifs is proposed f… read moreAbstract: An efficient scheme based on structural motifs is proposed for the crystal structure prediction of materials. The key advantage of the present method comes in twofold: first, the degrees of freedom of the system are greatly reduced, since each structural motif, regardless of its size, can always be described by a set of parameters (R, θ, φ) with five degrees of freedom; second, the motifs could always appear in the predicted structures when the energies of the structures are relatively low. Both features make the present scheme a very efficient method for predicting desired materials. The method has been applied to the case of LiFePO4, an important cathode material for lithium-ion batteries. Numerous new structures of LiFePO4 have been found, compared to those currently available, demonstrating the reliability of the present methodology and illustrating the promise of the concept of structural motifs. read less NOT USED (high confidence) M. Dewapriya and S. Meguid, “Atomistic modeling of out-of-plane deformation of a propagating Griffith crack in graphene,” Acta Mechanica. 2017. link Times cited: 23 NOT USED (high confidence) S. Hu et al., “Reactive molecular dynamics simulations on the thermal decomposition of poly alpha-methyl styrene,” Journal of Molecular Modeling. 2017. link Times cited: 8 NOT USED (high confidence) S. Stelmakh, K. Skrobas, S. Gierlotka, and B. Palosz, “Effect of the surface on the internal structure of CdSe crystal lattice based on molecular dynamics simulations,” Journal of Nanoparticle Research. 2017. link Times cited: 10 NOT USED (high confidence) B. Mortazavi, “Ultra high stiffness and thermal conductivity of graphene like C3N,” Carbon. 2017. link Times cited: 217 NOT USED (high confidence) O. Matsiaka, C. J. Penington, R. Baker, and M. Simpson, “Continuum approximations for lattice-free multi-species models of collective cell migration.,” Journal of theoretical biology. 2017. link Times cited: 20 NOT USED (high confidence) C. Si, X.-dong Wang, Z. Fan, Z.-hai Feng, and B. Cao, “Impacts of potential models on calculating the thermal conductivity of graphene using non-equilibrium molecular dynamics simulations,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 64 NOT USED (high confidence) J. Zhang and S. Meguid, “Piezoelectricity of 2D nanomaterials: characterization, properties, and applications,” Semiconductor Science and Technology. 2017. link Times cited: 46 Abstract: The discovery of piezoelectricity in 2D nanomaterials repres… read moreAbstract: The discovery of piezoelectricity in 2D nanomaterials represents a milestone towards embedding low-dimensional materials into future technologies. This article reviews recent progress in the characterization, properties evaluation, and applications of piezoelectricity of 2D piezoelectric nanomaterials (PNs). To begin, an introduction to the existing 2D PNs, which exhibit a wide range of atomic structures and configurations, is presented. The nanoscale measurements and associated experimental techniques as well as the atomic simulations of the piezoelectric properties of 2D PNs are then summarized. Some of the pertinent parameters, which govern the piezoelectric properties of 2D PNs, are discussed. Furthermore, our article concludes with some potential applications including piezotronics, piezophototronics, and energy harvesting of 2D PNs, which can open the doors to the innovative design of next-generation nanoelectronics and nanodevices. Finally, we highlight perspectives and challenges for the future development of 2D PNs. read less NOT USED (high confidence) S. Rouhi and A. Atfi, “Molecular Dynamics Simulations of Adsorption of Polymer Chains on the Surface of BmNn Graphyne-Like Monolayers,” Brazilian Journal of Physics. 2017. link Times cited: 8 NOT USED (high confidence) S. Rouhi and A. Atfi, “Molecular Dynamics Simulations of Adsorption of Polymer Chains on the Surface of BmNn Graphyne-Like Monolayers,” Brazilian Journal of Physics. 2017. link Times cited: 0 NOT USED (high confidence) T. D. Nguyen, “GPU-accelerated Tersoff potentials for massively parallel Molecular Dynamics simulations,” Comput. Phys. Commun. 2017. link Times cited: 44 NOT USED (high confidence) A. Verkhovtsev, A. Korol, and A. Solovyov, “Classical molecular dynamics simulations of fusion and fragmentation in fullerene-fullerene collisions,” The European Physical Journal D. 2017. link Times cited: 8 NOT USED (high confidence) K. Choudhary, F. Y. Congo, T. Liang, C. Becker, R. Hennig, and F. Tavazza, “Evaluation and comparison of classical interatomic potentials through a user-friendly interactive web-interface,” Scientific Data. 2017. link Times cited: 21 NOT USED (high confidence) F. F. de Oliveira et al., “Tailoring spin defects in diamond by lattice charging,” Nature Communications. 2017. link Times cited: 88 NOT USED (high confidence) C. K. Addington, J. Mansell, and K. Gubbins, “Computer simulation of conductive linear sulfur chains confined in carbon nanotubes,” Molecular Simulation. 2017. link Times cited: 11 Abstract: Recent experimental studies have shown that when sulfur is c… read moreAbstract: Recent experimental studies have shown that when sulfur is confined within a carbon nanotube (CNT) of appropriate diameter it forms a covalently bonded linear chain, and this confined one-dimensional sulfur phase is metallic. Such one-dimensional phases of sulfur are not observed in the bulk material, but at pressures in excess of 83 GPa bulk sulfur does form a metallic monoclinic phase. This suggests that the 1-D conducting phase formed in confinement may arise from high pressures arising due to the strong confinement. Here we report atomistic molecular dynamics of sulfur within (8,0) CNTs, using a recently derived statistical method for calculations of the pressure tensor, we find axial pressure component values as high as bar (800 GPa) for the SWLJ case. We decompose the pressure into contributions from sulfur–sulfur (S–S) and sulfur–carbon (S–C) interactions, and find that S–S interactions are strongly attractive, while S–C interactions are strongly repulsive. read less NOT USED (high confidence) J. Schneider et al., “ATK-ForceField: a new generation molecular dynamics software package,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 63 Abstract: ATK-ForceField is a software package for atomistic simulatio… read moreAbstract: ATK-ForceField is a software package for atomistic simulations using classical interatomic potentials. It is implemented as a part of the Atomistix ToolKit (ATK), which is a Python programming environment that makes it easy to create and analyze both standard and highly customized simulations. This paper will focus on the atomic interaction potentials, molecular dynamics, and geometry optimization features of the software, however, many more advanced modeling features are available. The implementation details of these algorithms and their computational performance will be shown. We present three illustrative examples of the types of calculations that are possible with ATK-ForceField: modeling thermal transport properties in a silicon germanium crystal, vapor deposition of selenium molecules on a selenium surface, and a simulation of creep in a copper polycrystal. read less NOT USED (high confidence) B. Zhu et al., “A study on the surface quality and brittle–ductile transition during the elliptical vibration-assisted nanocutting process on monocrystalline silicon via molecular dynamic simulations,” RSC Advances. 2017. link Times cited: 50 Abstract: Molecular dynamic (MD) simulation method was applied to inve… read moreAbstract: Molecular dynamic (MD) simulation method was applied to investigate the surface quality and brittle–ductile transition of monocrystalline silicon with a diamond tool during the elliptical vibration-assisted nanocutting (EVANC) and traditional nanocutting process. In the simulations, the interaction between silicon atoms in the specimen was modeled by the Tersoff potential, whereas the Morse potential was for the description of the interactions between silicon atoms in the specimen and carbon atoms in the diamond tool. In this study, we discovered that EVANC not only changed the brittle-mode cutting into the ductile-mode cutting, but also made the phase transformation layer thinner than that in the traditional nanocutting, which leads to a better surface finish and a large rate of removal of materials. Herein, stress analysis showed that the stress-affected region of the workpiece processed by EVANC was smaller than that of the workpiece processed by the traditional nanocutting. The temperature also increased during the EVANC process. This may soften the silicon material and make the cutting easier. In EVANC, the tangential force and normal force decreased because of the change in the brittle–ductile transition. From the simulation results, EVANC removed the material in the ductile mode, which could increase the removal rate, improve the surface finish, and decrease the cutting force to reduce the tool wear. In conclusion, EVANC has positive effects on the machinability and surface finish of the silicon material. read less NOT USED (high confidence) T. Rakib, S. Mojumder, S. Das, S. Saha, and M. Motalab, “Graphene and its elemental analogue: A molecular dynamics view of fracture phenomenon,” Physica B-condensed Matter. 2017. link Times cited: 38 NOT USED (high confidence) P. Li and K. Merz, “Metal Ion Modeling Using Classical Mechanics,” Chemical Reviews. 2017. link Times cited: 230 Abstract: Metal ions play significant roles in numerous fields includi… read moreAbstract: Metal ions play significant roles in numerous fields including chemistry, geochemistry, biochemistry, and materials science. With computational tools increasingly becoming important in chemical research, methods have emerged to effectively face the challenge of modeling metal ions in the gas, aqueous, and solid phases. Herein, we review both quantum and classical modeling strategies for metal ion-containing systems that have been developed over the past few decades. This Review focuses on classical metal ion modeling based on unpolarized models (including the nonbonded, bonded, cationic dummy atom, and combined models), polarizable models (e.g., the fluctuating charge, Drude oscillator, and the induced dipole models), the angular overlap model, and valence bond-based models. Quantum mechanical studies of metal ion-containing systems at the semiempirical, ab initio, and density functional levels of theory are reviewed as well with a particular focus on how these methods inform classical modeling efforts. Finally, conclusions and future prospects and directions are offered that will further enhance the classical modeling of metal ion-containing systems. read less NOT USED (high confidence) Y. Evtushenko, S. A. Lurie, M. Posypkin, and Y. Solyaev, “Application of optimization methods for finding equilibrium states of two-dimensional crystals,” Computational Mathematics and Mathematical Physics. 2016. link Times cited: 8 NOT USED (high confidence) E. Mainini, H. Murakawa, P. Piovano, and U. Stefanelli, “Carbon-nanotube geometries: Analytical and numerical results,” Discrete and Continuous Dynamical Systems - Series S. 2016. link Times cited: 10 Abstract: We investigate carbon-nanotubes under the perspective ofgeom… read moreAbstract: We investigate carbon-nanotubes under the perspective ofgeometry optimization. Nanotube geometries are assumed to correspondto atomic configurations whichlocally minimize Tersoff-type interactionenergies. In the specific cases of so-called zigzag and armchairtopologies, candidate optimal configurations are analytically identifiedand their local minimality is numerically checked. Inparticular, these optimal configurations do not correspond neither tothe classical Rolled-up model [ 5 ] nor to themore recent polyhedral model [ 3 ]. Eventually, theelastic response of the structure under uniaxial testing is numericallyinvestigated and the validity of the Cauchy-Born rule is confirmed. read less NOT USED (high confidence) Y. G. Evtushenko, S. Lurie, M. Posypkin, and Y. Solyaev, “Application of optimization methods for finding equilibrium states of two-dimensional crystals,” Computational Mathematics and Mathematical Physics. 2016. link Times cited: 0 NOT USED (high confidence) P. Zhang, R. Zhu, M. Jiang, Y. Song, D. Zhang, and Y. Cui, “Molecular dynamics study on the thermal conductivity of multiple layers in semiconductor disk laser,” SPIE/COS Photonics Asia. 2016. link Times cited: 0 Abstract: Thermal properties of multiple layers including distributed … read moreAbstract: Thermal properties of multiple layers including distributed Bragg reflector (DBR) and multiple quantum wells (MQWs) used in the semiconductor gain element are crucial for the performance of a semiconductor disk laser (SDL). For the purpose of more reasonable semiconductor wafer design, so to improve the thermal management of SDLs, accurate thermal conductivity value of a DBR is under considerable requirement. By the use of equilibrium molecular dynamics (EMD) method, thermal conductivities of AlAs/GaAs DBRs, which were widely employed in 1μm wavelength SDLs, were calculated, and simulated results were compared with reported data. Influences of the Al composition, and the layer thickness on the thermal conductivities were focused and analyzed. read less NOT USED (high confidence) P. O. Hubin, D. Jacquemin, L. Leherte, and D. P. Vercauteren, “Parameterization of the ReaxFF reactive force field for a proline‐catalyzed aldol reaction,” Journal of Computational Chemistry. 2016. link Times cited: 11 Abstract: A parameterization of the ReaxFF reactive FF is performed us… read moreAbstract: A parameterization of the ReaxFF reactive FF is performed using a Monte Carlo Simulated Annealing procedure for the modeling of a proline‐catalyzed aldol reaction. Emphasis is put on the accurate reproduction of the relative stabilities of several key intermediates of the reaction, as well as, on the description of the reaction path bridging these intermediates based on quantum mechanical calculations. Our training sets include new criteria based on geometry optimizations and short Molecular Dynamics simulations to ensure that the trained ReaxFF potentials adequately predict the structures of all key intermediates. The transferability of the sets of parameters obtained is assessed for various steps of the considered aldol reaction, as well as for different substrates, catalysts, and reagents. This works indeed highlights the challenge of reaching transferable parameters for several reaction steps. © 2016 Wiley Periodicals, Inc. read less NOT USED (high confidence) T. Habib et al., “Cosolvents as Liquid Surfactants for Boron Nitride Nanosheet (BNNS) Dispersions.,” Langmuir : the ACS journal of surfaces and colloids. 2016. link Times cited: 19 Abstract: Despite a range of promising applications, liquid-phase exfo… read moreAbstract: Despite a range of promising applications, liquid-phase exfoliation of boron nitride nanosheets (BNNSs) is limited, both by low yield in common solvents as well as the disadvantages of using dissolved surfactants. One recently reported approach is the use of cosolvent systems to increase the as-obtained concentration of BNNS; the role of these solvents in aiding exfoliation and/or aiding colloidal stability of BNNSs is difficult to distinguish. In this paper, we have investigated the use of a t-butanol/water cosolvent to disperse BNNSs. We utilize solvent-exchange experiments to demonstrate that the t-butanol is in fact essential to colloidal stability; we then utilized molecular dynamics simulations to explore the mechanism of t-butanol/BNNS interactions. Taken together, the experimental and simulation results show that the key to the success of t-butanol (as compared to the other alcohols of higher or lower molecular weight) lies in its ability to act as a "liquid dispersant" which allows it to favorably interact with both water and BNNSs. Additionally, we show that the stable dispersions of BNNS in water/t-butanol systems may be freeze-dried to yield nonaggregated, redispersible BNNS powders, which would be useful in an array of industrial processes. read less NOT USED (high confidence) E. Davoli, P. Piovano, and U. Stefanelli, “Wulff shape emergence in graphene,” Mathematical Models and Methods in Applied Sciences. 2016. link Times cited: 28 Abstract: Graphene samples are identified as minimizers of configurati… read moreAbstract: Graphene samples are identified as minimizers of configurational energies featuring both two- and three-body atomic-interaction terms. This variational viewpoint allows for a detailed description of ground-state geometries as connected subsets of a regular hexagonal lattice. We investigate here how these geometries evolve as the number n of carbon atoms in the graphene sample increases. By means of an equivalent characterization of minimality via a discrete isoperimetric inequality, we prove that ground states converge to the ideal hexagonal Wulff shape as n →∞. Precisely, ground states deviate from such hexagonal Wulff shape by at most Kn3/4 + o(n3/4) atoms, where both the constant K and the rate n3/4 are sharp. read less NOT USED (high confidence) M. Wilson, “Structure and dynamics in network-forming materials,” Journal of Physics: Condensed Matter. 2016. link Times cited: 9 Abstract: The study of the structure and dynamics of network-forming m… read moreAbstract: The study of the structure and dynamics of network-forming materials is reviewed. Experimental techniques used to extract key structural information are briefly considered. Strategies for building simulation models, based on both targeting key (experimentally-accessible) materials and on systematically controlling key model parameters, are discussed. As an example of the first class of materials, a key target system, SiO2, is used to highlight how the changing structure with applied pressure can be effectively modelled (in three dimensions) and used to link to both experimental results and simple structural models. As an example of the second class the topology of networks of tetrahedra in the MX2 stoichiometry are controlled using a single model parameter linked to the M–X–M bond angles. The evolution of ordering on multiple length-scales is observed as are the links between the static structure and key dynamical properties. The isomorphous relationship between the structures of amorphous Si and SiO2 is discussed as are the similarities and differences in the phase diagrams, the latter linked to potential polyamorphic and ‘anomalous’ (e.g. density maxima) behaviour. Links to both two-dimensional structures for C, Si and Ge and near-two-dimensional bilayers of SiO2 are discussed. Emerging low-dimensional structures in low temperature molten carbonates are also uncovered. read less NOT USED (high confidence) J.-H. Zou, Z.-Q. Ye, and B. Cao, “Phonon thermal properties of graphene from molecular dynamics using different potentials.,” The Journal of chemical physics. 2016. link Times cited: 67 Abstract: Phonon thermal transport in graphene has attracted significa… read moreAbstract: Phonon thermal transport in graphene has attracted significant interest in recent years. Phonon thermal properties of graphene are investigated by molecular dynamics simulations using the Tersoff, Tersoff-2010, REBO, and AIREBO potentials. By calculating the phonon properties and thermal conductivity of graphene, the performance of the potentials is evaluated based on comparisons with experimental data. It shows that the Tersoff-2010 and REBO display better dispersion curves for graphene than the original Tersoff and AIREBO. The Tersoff-2010 correctly provides the Γ point phonon velocities of the LA and TA branches as well as the G peak frequency with a value of 46 THz. In addition, the acoustic phonon relaxation time derived from the Tersoff-2010 satisfies the ideal relation "τ-1 ∝ ν2." It is also found that the Tersoff-2010 provides the highest graphene thermal conductivity among the used potentials, and estimates about 30.0% contribution for flexural phonons to the total thermal conductivity. By comparison, the Tersoff-2010 potential is demonstrated to be the most suitable one to describe the phonon thermal properties of graphene. read less NOT USED (high confidence) U. Stefanelli, “Stable carbon configurations,” Bollettino dell’Unione Matematica Italiana. 2016. link Times cited: 0 NOT USED (high confidence) M. Cherukara et al., “Ab Initio-Based Bond Order Potential to Investigate Low Thermal Conductivity of Stanene Nanostructures.,” The journal of physical chemistry letters. 2016. link Times cited: 71 Abstract: We introduce a bond order potential (BOP) for stanene based … read moreAbstract: We introduce a bond order potential (BOP) for stanene based on an ab initio derived training data set. The potential is optimized to accurately describe the energetics, as well as thermal and mechanical properties of a free-standing sheet, and used to study diverse nanostructures of stanene, including tubes and ribbons. As a representative case study, using the potential, we perform molecular dynamics simulations to study stanene's structure and temperature-dependent thermal conductivity. We find that the structure of stanene is highly rippled, far in excess of other 2-D materials (e.g., graphene), owing to its low in-plane stiffness (stanene: ∼ 25 N/m; graphene: ∼ 480 N/m). The extent of stanene's rippling also shows stronger temperature dependence compared to that in graphene. Furthermore, we find that stanene based nanostructures have significantly lower thermal conductivity compared to graphene based structures owing to their softness (i.e., low phonon group velocities) and high anharmonic response. Our newly developed BOP will facilitate the exploration of stanene based low dimensional heterostructures for thermoelectric and thermal management applications. read less NOT USED (high confidence) S. Ahmad, “Criteria for the growth of fullerenes and single-walled carbon nanotubes in sooting environments,” Nanotechnology. 2016. link Times cited: 12 Abstract: The spherical curvature induced by pentagons in corannulenes… read moreAbstract: The spherical curvature induced by pentagons in corannulenes and hexagonal sheets is shown to be the basic constituent that controls the growth of fullerenes and single-walled carbon nanotubes (SWNTs) in soot forming and carbon vapour environments. Formation of the initial ring of five or six atoms is the essential step which with the addition of further pentagons and hexagons determines whether a spinning fullerene is to be formed or the cap that lifts up and leads to the formation of an SWNT. A continuum elastic model is developed to determine the criteria for the growth of these structures. The observed dominance of the growth of 14 Å diameter armchair SWNTs in sooting and carbonaceous environments is explained by using the nanoelastic model of C shells. read less NOT USED (high confidence) V. Tahouneh, M. M. Mashhadi, and M. H. Naei, “Finite element and micromechanical modeling for investigating effective material properties of polymer–matrix nanocomposites with microfiber, reinforced by CNT arrays,” International Journal of Advanced Structural Engineering. 2016. link Times cited: 5 NOT USED (high confidence) H. Nishizawa, Y. Nishimura, M. Kobayashi, S. Irle, and H. Nakai, “Three pillars for achieving quantum mechanical molecular dynamics simulations of huge systems: Divide‐and‐conquer, density‐functional tight‐binding, and massively parallel computation,” Journal of Computational Chemistry. 2016. link Times cited: 68 Abstract: The linear‐scaling divide‐and‐conquer (DC) quantum chemical … read moreAbstract: The linear‐scaling divide‐and‐conquer (DC) quantum chemical methodology is applied to the density‐functional tight‐binding (DFTB) theory to develop a massively parallel program that achieves on‐the‐fly molecular reaction dynamics simulations of huge systems from scratch. The functions to perform large scale geometry optimization and molecular dynamics with DC‐DFTB potential energy surface are implemented to the program called DC‐DFTB‐K. A novel interpolation‐based algorithm is developed for parallelizing the determination of the Fermi level in the DC method. The performance of the DC‐DFTB‐K program is assessed using a laboratory computer and the K computer. Numerical tests show the high efficiency of the DC‐DFTB‐K program, a single‐point energy gradient calculation of a one‐million‐atom system is completed within 60 s using 7290 nodes of the K computer. © 2016 Wiley Periodicals, Inc. read less NOT USED (high confidence) R. Chaudret, A. Bick, and X. Krokidis, “Theoretical Modeling of Thermal Decomposition of Methylnaphthalene Derivatives: Influence of Substituents,” Energy & Fuels. 2016. link Times cited: 6 Abstract: The kinetics and thermodynamics of thermal decomposition of … read moreAbstract: The kinetics and thermodynamics of thermal decomposition of naphthalene and several mono- and dimethylnaphathalene derivatives have been established through reactive molecular dynamics simulations using ReaxFF. These results were compared to previous theoretical and experimental studies and also compared to density functional theory calculation results. This work demonstrates that the kinetics and thermodynamics of the initial activation reaction is directly impacted by the position and number of methyl substituents. Subsequently, the activated molecules react to form either small organic or large char molecules. The char formation mechanism is shown to occur in three steps: activation, dimerization/trimerization, and condensation. Finally, temperature effects on the char formation reaction were also studied. read less NOT USED (high confidence) M. Wander and B. Bickmore, “A preliminary valence-multipole potential energy model: Al-Si-H-O system,” American Mineralogist. 2016. link Times cited: 2 Abstract: Here we test the concept that a potential energy model (forc… read moreAbstract: Here we test the concept that a potential energy model (force field) based on an expansion of the bond-valence model can use molecular geometry to make a reasonable prediction of the thermodynamic energy. The backbone of the model is a non-standard choice of structural descriptors for the energy decomposition, which relates the energy to particular aspects of the structure. Most force fields use a many-body decomposition to describe structures (with two-, three-, and possibly four-body terms, etc.), whereas ours employs a multipole expansion of the bond valence incident to each atom. This valence multipole model separates the energy associated with each atom into terms related to total bonding (valence monopole), bonding asymmetry (valence dipole), and ellipsoidal deformation (valence quadrupole). All of these are inherently multi-body terms that are calculated by combining two-body terms (bond valences). Provided bond valence sums are satisfied to within 0.2 v.u. of the ideal for all atoms, this model can provide accuracies of ~5 kJ/mol per unique atom in the Al-Si-H-O system, at least for the equilibrium structures tested here, comparable to most quantum mechanical calculations. More development is needed to produce a fully functional force field suitable for molecular dynamics simulations, but this work shows that the development of such a force field is likely to be feasible. read less NOT USED (high confidence) M. Gatchell and H. Zettergren, “Knockout driven reactions in complex molecules and their clusters,” Journal of Physics B: Atomic, Molecular and Optical Physics. 2016. link Times cited: 54 Abstract: Energetic ions lose some of their kinetic energy when intera… read moreAbstract: Energetic ions lose some of their kinetic energy when interacting with electrons or nuclei in matter. Here, we discuss combined experimental and theoretical studies on such impulse driven reactions in polycyclic aromatic hydrocarbons (PAHs), fullerenes, and pure or mixed clusters of these molecules. These studies show that the nature of excitation is important for how complex molecular systems respond to ion/atom impact. Rutherford-like nuclear scattering processes may lead to prompt atom knockout and formation of highly reactive fragments, while heating of the molecular electron clouds in general lead to formation of more stable and less reactive fragments. In this topical review, we focus on recent studies of knockout driven reactions, and present new calculations of the angular dependent threshold (displacement) energies for such processes in PAHs. The so-formed fragments may efficiently form covalent bonds with neighboring molecules in clusters. These unique molecular growth processes may be important in astrophysical environments such as low velocity shock waves. read less NOT USED (high confidence) T. Li, Z. Tang, Z. Huang, and J. Yu, “Interfacial thermal resistance of 2D and 1D carbon/hexagonal boron nitride van der Waals heterostructures,” Carbon. 2016. link Times cited: 29 NOT USED (high confidence) S. Chowdhury, B. Haque, and J. Gillespie, “Molecular dynamics simulations of the structure and mechanical properties of silica glass using ReaxFF,” Journal of Materials Science. 2016. link Times cited: 92 NOT USED (high confidence) T. Sanyal and S. Shella, “Coarse-grained models using local-density potentials optimized with the relative entropy: Application to implicit solvation.,” The Journal of chemical physics. 2016. link Times cited: 75 Abstract: Bottom-up multiscale techniques are frequently used to devel… read moreAbstract: Bottom-up multiscale techniques are frequently used to develop coarse-grained (CG) models for simulations at extended length and time scales but are often limited by a compromise between computational efficiency and accuracy. The conventional approach to CG nonbonded interactions uses pair potentials which, while computationally efficient, can neglect the inherently multibody contributions of the local environment of a site to its energy, due to degrees of freedom that were coarse-grained out. This effect often causes the CG potential to depend strongly on the overall system density, composition, or other properties, which limits its transferability to states other than the one at which it was parameterized. Here, we propose to incorporate multibody effects into CG potentials through additional nonbonded terms, beyond pair interactions, that depend in a mean-field manner on local densities of different atomic species. This approach is analogous to embedded atom and bond-order models that seek to capture multibody electronic effects in metallic systems. We show that the relative entropy coarse-graining framework offers a systematic route to parameterizing such local density potentials. We then characterize this approach in the development of implicit solvation strategies for interactions between model hydrophobes in an aqueous environment. read less NOT USED (high confidence) J. R. Boes, M. C. Groenenboom, J. Keith, and J. Kitchin, “Neural network and ReaxFF comparison for Au properties,” International Journal of Quantum Chemistry. 2016. link Times cited: 64 Abstract: We have studied how ReaxFF and Behler–Parrinello neural netw… read moreAbstract: We have studied how ReaxFF and Behler–Parrinello neural network (BPNN) atomistic potentials should be trained to be accurate and tractable across multiple structural regimes of Au as a representative example of a single-component material. We trained these potentials using subsets of 9,972 Kohn-Sham density functional theory calculations and then validated their predictions against the untrained data. Our best ReaxFF potential was trained from 848 data points and could reliably predict surface and bulk data; however, it was substantially less accurate for molecular clusters of 126 atoms or fewer. Training the ReaxFF potential to more data also resulted in overfitting and lower accuracy. In contrast, BPNN could be fit to 9,734 calculations, and this potential performed comparably or better than ReaxFF across all regimes. However, the BPNN potential in this implementation brings significantly higher computational cost. © 2016 Wiley Periodicals, Inc. read less NOT USED (high confidence) A. Oluwajobi and X. Chen, “Molecular Dynamics (MD) Simulation of Multi-pass Nanometric Machining – The Effect of Machining Conditions,” Current Nanoscience. 2016. link Times cited: 1 Abstract: Understanding material behaviour during nanoscale machining … read moreAbstract: Understanding material behaviour during nanoscale machining is critical for improving machining efficiency. This paper investigates the benefits of using Molecular Dynamics (MD) simulation in studying the effects of machining parameters in nanometric machining of copper workpiece with a diamond tool. The material behaviour under multi cutting pass conditions was examined. The copper-copper interactions were modelled by the EAM potential and the copper-diamond interactions were modelled by the Morse potential. The diamond tool was modelled as a deformable body and the Tersoff potential was applied for the carbon-carbon interactions. It was observed that the average tangential and normal components of the cutting forces increase with increase in depth of cut and they reduced in consecutive cutting passes for each depth of cut. The ratios of the tangential to the normal force components decreases as the depth of cut increases, but remain constant after the depth of cut 1.5nm. The magnitudes of the cutting forces decrease from pass 1 to pass 2, but they are identical for both pass 2 and pass 3. The least resistance to cutting was observed at 2.0nm, which may indicate the existence of a critical depth of cut in nanomachining, for tool wear reduction. After the first pass, the average tangential and normal components of the cutting forces increase with increase in the feed. Also, there is always an increase in friction from pass 1 to pass 2. In multipass processes, the arrangement should be effected with minimum overlap in the runs, for efficient machining. read less NOT USED (high confidence) E. Voyiatzis and M. Böhm, “Atomic and global mechanical properties of systems described by the Stillinger–Weber potential,” Journal of Physics: Condensed Matter. 2016. link Times cited: 0 Abstract: Analytical expressions for the stress and elasticity tensors… read moreAbstract: Analytical expressions for the stress and elasticity tensors of materials, in which the interactions are described by the Stillinger–Weber potential, are derived in the context of the stress fluctuation formalism. The derived formulas can be used both in Monte Carlo and molecular dynamics simulations. As an example of possible applications, they are employed to calculate the influence of the temperature and system size on the mechanical properties of crystalline cubic boron nitride. The system has been studied by molecular dynamics simulations. The computed mechanical properties are in good agreement with available experimental data and first principle calculations. In the studied crystalline cubic boron nitride system, the employed formalism is of higher accuracy than the ‘small-strain’ non-equilibrium method. The dominant contributions to the elastic constants stem from the Born and stress fluctuation terms. An increase in the system size reduces the statistical uncertainties in the computation of the mechanical properties. A rise of the temperature leads to a slight increase in the observed uncertainties. The derived expressions for the stress and elasticity tensors are further decomposed into sums of atomic level stress and atomic level elasticity tensors. The developed factorization enables us (i) to quantify the contribution of the various chemical groups, in the case under consideration of the different atoms, to the observed mechanical properties and (ii) to determine the elastic constants with reduced computational uncertainties. The reason is that the exact values of some terms of the proposed factorization can be determined theoretically beforehand. Thus, they can be substituted in the derived formulas leading to an enhanced convergence. read less NOT USED (high confidence) A. Jamr’oz and J. Majewski, “Ordering effects in 2D hexagonal systems of binary and ternary BCN alloys,” arXiv: Mesoscale and Nanoscale Physics. 2016. link Times cited: 3 NOT USED (high confidence) S. Thomas, K. Ajith, and M. C. Valsakumar, “Directional anisotropy, finite size effect and elastic properties of hexagonal boron nitride,” Journal of Physics: Condensed Matter. 2016. link Times cited: 43 Abstract: Classical molecular dynamics simulations have been performed… read moreAbstract: Classical molecular dynamics simulations have been performed to analyze the elastic and mechanical properties of two-dimensional (2D) hexagonal boron nitride (h-BN) using a Tersoff-type interatomic empirical potential. We present a systematic study of h-BN for various system sizes. Young’s modulus and Poisson’s ratio are found to be anisotropic for finite sheets whereas they are isotropic for the infinite sheet. Both of them increase with system size in accordance with a power law. It is concluded from the computed values of elastic constants that h-BN sheets, finite or infinite, satisfy Born’s criterion for mechanical stability. Due to the the strong in-plane sp2 bonds and the small mass of boron and nitrogen atoms, h-BN possesses high longitudinal and shear velocities. The variation of bending rigidity with system size is calculated using the Foppl–von Karman approach by coupling the in-plane bending and out-of-plane stretching modes of the 2D h-BN. read less NOT USED (high confidence) G. Sosso et al., “Crystal Nucleation in Liquids: Open Questions and Future Challenges in Molecular Dynamics Simulations,” Chemical Reviews. 2016. link Times cited: 553 Abstract: The nucleation of crystals in liquids is one of nature’s mos… read moreAbstract: The nucleation of crystals in liquids is one of nature’s most ubiquitous phenomena, playing an important role in areas such as climate change and the production of drugs. As the early stages of nucleation involve exceedingly small time and length scales, atomistic computer simulations can provide unique insights into the microscopic aspects of crystallization. In this review, we take stock of the numerous molecular dynamics simulations that, in the past few decades, have unraveled crucial aspects of crystal nucleation in liquids. We put into context the theoretical framework of classical nucleation theory and the state-of-the-art computational methods by reviewing simulations of such processes as ice nucleation and the crystallization of molecules in solutions. We shall see that molecular dynamics simulations have provided key insights into diverse nucleation scenarios, ranging from colloidal particles to natural gas hydrates, and that, as a result, the general applicability of classical nucleation theory has been repeatedly called into question. We have attempted to identify the most pressing open questions in the field. We believe that, by improving (i) existing interatomic potentials and (ii) currently available enhanced sampling methods, the community can move toward accurate investigations of realistic systems of practical interest, thus bringing simulations a step closer to experiments. read less NOT USED (high confidence) O. Böhm, S. Pfadenhauer, R. Leitsmann, P. Plänitz, E. Schreiner, and M. Schreiber, “ReaxFF+—A New Reactive Force Field Method for the Accurate Description of Ionic Systems and Its Application to the Hydrolyzation of Aluminosilicates,” Journal of Physical Chemistry C. 2016. link Times cited: 8 Abstract: In this paper we present a powerful extension of the reactiv… read moreAbstract: In this paper we present a powerful extension of the reactive force field method ReaxFF, which we call ReaxFF+. It combines the charge equilibrium scheme with the bond order principle. The main advantage of this procedure is the correct distinction and description of covalent and ionic bonds. It allows reactive molecular dynamic simulations in ionic gases and liquids. To demonstrate the accuracy of this new method, we study the hydrolyzation of aluminosilicates. Comparing the results with experimental and ab initio data, we can prove the high accuracy of our method. This shows that ReaxFF+ is a powerful force field simulation tool for reactions in acidic or alkaline environments. read less NOT USED (high confidence) X. Yang, Y. Huang, L. Wang, B. Cao, and A. To, “Formation of single carbon chain bridging two SWCNTs via tensile deformation of nanobud junction,” Materials & Design. 2016. link Times cited: 4 NOT USED (high confidence) A. M. Viotti, A. Monastra, M. F. Moreno, and M. F. Carusela, “Thermal expansion in nanoresonators,” Journal of Statistical Mechanics: Theory and Experiment. 2016. link Times cited: 1 Abstract: Inspired by some recent experiments and numerical works rela… read moreAbstract: Inspired by some recent experiments and numerical works related to nanoresonators, we perform classical molecular dynamics simulations to investigate the thermal expansion and the ability of the device to act as a strain sensor assisted by thermally-induced vibrations. The proposed model consists in a chain of atoms interacting anharmonically with both ends clamped to thermal reservoirs. We analyze the thermal expansion and resonant frequency shifts as a function of temperature and the applied strain. For the transversal modes the shift is approximately linear with strain. We also present analytical results from canonical calculations in the harmonic approximation showing that thermal expansion is uniform along the device. This prediction also works when the system operates in a nonlinear oscillation regime at moderate and high temperatures. read less NOT USED (high confidence) A. Favata, A. Micheletti, P. Podio-Guidugli, and N. Pugno, “How graphene flexes and stretches under concomitant bending couples and tractions,” Meccanica. 2016. link Times cited: 18 NOT USED (high confidence) I. Zubko, “Computation of elastic moduli of graphene monolayer in nonsymmetric formulation using energy-based approach,” Physical Mesomechanics. 2016. link Times cited: 6 NOT USED (high confidence) P. Kuopanportti, E. Hayward, C. Fu, A. Kuronen, and K. Nordlund, “Interatomic FeH potential for irradiation and embrittlement simulations,” Computational Materials Science. 2016. link Times cited: 18 NOT USED (high confidence) M. Friedrich, P. Piovano, and U. Stefanelli, “The Geometry of C60: A Rigorous Approach via Molecular Mechanics,” SIAM J. Appl. Math. 2016. link Times cited: 5 Abstract: Molecular Mechanics describes molecules as particle configur… read moreAbstract: Molecular Mechanics describes molecules as particle configurations interacting via classical potentials. These {\it configurational energies} usually consist of the sum of different phenomenological terms which are tailored to the description of specific bonding geometries. This approach is followed here to model the fullerene $C_{60}$, an allotrope of carbon corresponding to a specific hollow spherical structure of sixty atoms. We rigorously address different modeling options and advance a set of minimal requirements on the configurational energy able to deliver an accurate prediction of the fine three-dimensional geometry of $C_{60}$ as well as of its remarkable stability. In particular, the experimentally observed truncated-icosahedron structure with two different bond lengths is shown to be a strict local minimizer. read less NOT USED (high confidence) S. Rouhi, “Molecular dynamics simulation of the adsorption of polymer chains on CNTs, BNNTs and GaNNTs,” Fibers and Polymers. 2016. link Times cited: 19 NOT USED (high confidence) M. Biswas and B. Knigge, “Opportunities and Challenges of Atomistic Modeling to Simulate Amorphous Carbon Properties for Computer Hard-Disk Applications,” IEEE Transactions on Magnetics. 2016. link Times cited: 1 Abstract: Amorphous carbon is used as coating material for computer ha… read moreAbstract: Amorphous carbon is used as coating material for computer hard-disks magnetic media and recording heads. There has been significant improvement in understanding amorphous carbon's properties based on experimental observations. High data storage density requirement in the coming years necessitates the use of an ultrathin carbon overcoat while maintaining or enhancing its tribological, thermal, optical, and corrosion properties for better recording performance and reliability. Along with experimental techniques, atomistic simulations can be a useful tool to provide fundamental understanding, especially in the cases where experiments are not adequate. This review gives an overview of how atomistic modeling can provide insights into amorphous carbon properties and discuss challenges for such modeling. read less NOT USED (high confidence) C. Hou, J. Xu, W. Ge, and J. Li, “Molecular dynamics simulation overcoming the finite size effects of thermal conductivity of bulk silicon and silicon nanowires,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 13 Abstract: Nonequilibrium molecular dynamics simulation has been a powe… read moreAbstract: Nonequilibrium molecular dynamics simulation has been a powerful tool for studying the thermophysical properties of bulk silicon and silicon nanowires. Nevertheless, usually limited by the capacity and capability of computational resources, the traditional longitudinal and transverse simulation sizes are evidently restricted in a narrow range much less than the experimental scales, which seriously hinders the exploration of the thermal properties. In this research, based on a powerful and efficient molecular dynamics (MD) simulation method, the computation of thermal conductivity beyond the known Casimir size limits is realized. The longitudinal dimensions of the simulations significantly exceed the micrometer scale. More importantly, the lateral characteristic sizes are much larger than 10 nanometers, explicitly comparable with the silicon nanowires fabricated and measured experimentally, whereas the traditional simulation size is several nanometers. The powerful virtual experimental measurement provided in our simulations achieves the direct prediction of the thermal conductivity of bulk silicon and real-scale silicon nanowires, and delineates the complete longitudinal size dependence of their thermal conductivities, especially at the elusive mesoscopic scale. Furthermore, the presented measurement paves an exciting and promising way to explore in depth the thermophysical properties of other bulk covalent solids and their low-dimensional structures, such as nanowires and nanosheets. read less NOT USED (high confidence) S. Barr, G. Kedziora, A. M. Ecker, J. Moller, R. Berry, and T. Breitzman, “Bond breaking in epoxy systems: A combined QM/MM approach.,” The Journal of chemical physics. 2016. link Times cited: 11 Abstract: A novel method to combine quantum mechanics (QM) and molecul… read moreAbstract: A novel method to combine quantum mechanics (QM) and molecular mechanics has been developed to accurately and efficiently account for covalent bond breaking in polymer systems under high strain without the use of predetermined break locations. Use of this method will provide a better fundamental understanding of the mechano-chemical origins of fracture in thermosets. Since classical force fields cannot accurately account for bond breaking, and QM is too demanding to simulate large systems, a hybrid approach is required. In the method presented here, strain is applied to the system using a classical force field, and all bond lengths are monitored. When a bond is stretched past a threshold value, a zone surrounding the bond is used in a QM energy minimization to determine which, if any, bonds break. The QM results are then used to reconstitute the system to continue the classical simulation at progressively larger strain until another QM calculation is triggered. In this way, a QM calculation is only computed when and where needed, allowing for efficient simulations. A robust QM method for energy minimization has been determined, as well as appropriate values for the QM zone size and the threshold bond length. Compute times do not differ dramatically from classical molecular mechanical simulations. read less NOT USED (high confidence) T. Hynninen, T. Musso, and A. Foster, “Limitations of reactive atomistic potentials in describing defect structures in oxides,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 4 Abstract: It is difficult to achieve low expense and high accuracy in … read moreAbstract: It is difficult to achieve low expense and high accuracy in computational methods, yet it remains a key objective in atomistic approaches. In solid state physics, advanced atomistic potentials using reactive force fields have shown promise in delivering both. However, these methods have not been applied widely beyond their development environment and thus their strengths and weaknesses are not fully understood. In this work we present benchmark calculations on silica (SiO2) and hafnia (HfO2) structures, comparing a leading charge optimized many-body potential to a more advanced density functional calculation. We find that although the atomistic potential gives excellent results for bulk structures, it has severe shortcomings when applied to small systems with low coordinated atoms. We also establish clearly the components of the many-body potential and how these relate to predicted physical properties. read less NOT USED (high confidence) T. Senftle et al., “The ReaxFF reactive force-field: development, applications and future directions.” 2016. link Times cited: 1212 NOT USED (high confidence) A. M. Tamidi and Y. Sasajima, “The Relationship between Nanocluster Precipitation and Thermal Conductivity in Si/Ge Amorphous Multilayer Films,” Journal of Nanomaterials. 2016. link Times cited: 2 Abstract: We have used a molecular dynamics technique to simulate the … read moreAbstract: We have used a molecular dynamics technique to simulate the relationship between nanocluster precipitation and thermal conductivity in Si/Ge amorphous multilayer films, with and without Cu addition. In the study, the Green-Kubo equation was used to calculate thermal conductivity in these materials. Five specimens were prepared: Si/Ge layers, Si/Ge + Cu layers, Si + Cu/Ge + Cu layers, Si/Cu/Ge/Cu layers, and Si/Cu/Ge layers. The number of precipitated nanoclusters in these specimens, which is defined as the number of four-coordinate atoms, was counted along the lateral direction of the specimens. The observed results of precipitate formation were considered in relation to the thermal conductivity results. Enhancement of precipitation of nanoclusters by Cu addition, that is, densification of four-coordinate atoms, can prevent the increment of thermal conductivity. Cu dopant increases the thermal conductivity of these materials. Combining these two points, we concluded that Si/Cu/Ge is the best structure to improve the conversion efficiency of the Si/Ge amorphous multilayer films. read less NOT USED (high confidence) X. Wang, J. Wang, and X. Guo, “Finite deformation of single-walled carbon nanocones under axial compression using a temperature-related multiscale quasi-continuum model,” Computational Materials Science. 2016. link Times cited: 17 NOT USED (high confidence) C. Barrett and L.-wang Wang, “A systematic fitting procedure for accurate force field models to reproduce ab initio phonon spectra of nanostructures,” Comput. Phys. Commun. 2016. link Times cited: 3 NOT USED (high confidence) I. Giordanelli, N. Pose, M. Mendoza, and H. Herrmann, “Conformal Invariance of Graphene Sheets,” Scientific Reports. 2016. link Times cited: 19 NOT USED (high confidence) M. R. Price, A. Ovcharenko, and B. Raeymaekers, “Qualitative Evaluation of Ultra-thin Multi-layer Diamond-Like Carbon Coatings Using Molecular Dynamics Nanoindentation Simulations,” Tribology Letters. 2016. link Times cited: 15 NOT USED (high confidence) B. L. Mooney et al., “Elucidating the Properties of Surrogate Fuel Mixtures Using Molecular Dynamics,” Energy & Fuels. 2016. link Times cited: 14 Abstract: The wide compositional differences between conventional and … read moreAbstract: The wide compositional differences between conventional and alternative fuels have resulted in much research aimed at determining which alternative fuels can be used, and in what proportions, in conventional engines. Atomic-scale modeling is uniquely positioned to lend insight into this question without extensive large-scale tests. The predictive power such modeling affords could narrow the phase space that must be examined experimentally. This study utilizes molecular dynamics (MD) simulations to predict the properties of a set of pure hydrocarbons, as well as binary and multicomponent surrogate fuel mixtures for alternative fuels created from these pure components. The accuracy and transferability of the modified Lennard-Jones adaptive intermolecular reactive empirical bond-order potential (mod-LJ AIREBO) [Liu, A.; Stuart, S. J. J. Comput. Chem. 2008, 29, 601−611] was assessed by calculating densities, heats of vaporization, and bulk moduli of pure hydrocarbons and the mixtures of these hydrocarbons, i.... read less NOT USED (high confidence) E. Helgee and A. Isacsson, “Adsorption of metal atoms at a buckled graphene grain boundary using model potentials,” AIP Advances. 2016. link Times cited: 4 Abstract: Two model potentials have been evaluated with regard to thei… read moreAbstract: Two model potentials have been evaluated with regard to their ability to model adsorption of single metal atoms on a buckled graphene grain boundary. One of the potentials is a Lennard-Jones potential parametrized for gold and carbon, while the other is a bond-order potential parametrized for the interaction between carbon and platinum. Metals are expected to adsorb more strongly to grain boundaries than to pristine graphene due to their enhanced adsorption at point defects resembling those that constitute the grain boundary. Of the two potentials considered here, only the bond-order potential reproduces this behavior and predicts the energy of the adsorbate to be about 0.8 eV lower at the grain boundary than on pristine graphene. The Lennard-Jones potential predicts no significant difference in energy between adsorbates at the boundary and on pristine graphene. These results indicate that the Lennard-Jones potential is not suitable for studies of metal adsorption on defects in graphene, and that bond-order potentials are preferable. read less NOT USED (high confidence) I. Zubko, “Computation of elastic moduli of graphene monolayer in nonsymmetric formulation using energy-based approach,” Physical Mesomechanics. 2016. link Times cited: 0 NOT USED (high confidence) B. Narayanan et al., “Describing the Diverse Geometries of Gold from Nanoclusters to Bulk—A First-Principles-Based Hybrid Bond-Order Potential,” Journal of Physical Chemistry C. 2015. link Times cited: 27 Abstract: Molecular dynamics simulations using empirical force fields … read moreAbstract: Molecular dynamics simulations using empirical force fields (EFFs) are crucial for gaining fundamental insights into atomic structure and long time scale dynamics of Au nanoclusters with far-reaching applications in energy and devices. This approach is thwarted by the failure of currently available EFFs in describing the size-dependent dimensionality and diverse geometries exhibited by Au clusters (e.g., planar structures, hollow cages, tubes, pyramids, space-filled structures). Here, we mitigate this issue by introducing a new hybrid bond-order potential (HyBOP), which accounts for (a) short-range interactions via Tersoff-type BOP terms that accurately treat bond directionality and (b) long-range dispersion effects by a scaled Lennard–Jones term whose contribution depends on the local atomic density. We optimized the independent parameters for our HyBOP using a global optimization scheme driven by genetic algorithms. Moreover, to ensure good transferability of these parameters across different length sca... read less NOT USED (high confidence) D. Marshall and H. Sadeghpour, “Simulating the Formation of Carbon-rich Molecules on an idealised Graphitic Surface,” arXiv: Earth and Planetary Astrophysics. 2015. link Times cited: 3 Abstract: There is accumulating evidence for the presence of complex m… read moreAbstract: There is accumulating evidence for the presence of complex molecules, including carbon-bearing and organic molecules, in the interstellar medium. Much of this evidence comes to us from studies of chemical composition, photo- and mass-spectroscopy in cometary, meteoritic and asteroid samples, indicating a need to better understand the surface chemistry of astrophysical objects. There is also considerable interest in the origins of life-forming and life-sustaining molecules on Earth. Here, we perform reactive molecular dynamics simulations to probe the formation of carbon-rich molecules and clusters on carbonaceous surfaces resembling dust grains and meteoroids. Our results show that large chains form on graphitic surfaces at low temperatures (100K - 500K) and smaller fullerene-like molecules form at higher temperatures (2000K - 3000K). The formation is faster on the surface than in the gas at low temperatures but slower at high temperatures as surface interactions prevent small clusters from coagulation. We find that for efficient formation of molecular complexity, mobility about the surface is important and helps to build larger carbon chains on the surface than in the gas phase at low temperatures. Finally, we show that the temperature of the surface strongly determines what kind of structures forms and that low turbulent environments are needed for efficient formation. read less NOT USED (high confidence) Q. Xiong and X. Tian, “Torsional properties of hexagonal boron nitride nanotubes, carbon nanotubes and their hybrid structures: A molecular dynamics study,” AIP Advances. 2015. link Times cited: 15 Abstract: The torsional mechanical properties of hexagonal single-wall… read moreAbstract: The torsional mechanical properties of hexagonal single-walled boron nitride nanotubes (SWBNNTs), single-walled carbon nanotubes (SWCNTs), and their hybrid structures (SWBN-CNTs) are investigated using molecular dynamics (MD) simulation. Two approaches - force approach and energy approach, are adopted to calculate the shear moduli of SWBNNTs and SWCNTs, the discrepancy between two approaches is analyzed. The results show that the shear moduli of single-walled nanotubes (SWNTs), including SWBNNTs and SWCNTs are dependent on the diameter, especially for armchair SWNTs. The armchair SWNTs show the better ability of resistance the twisting comparable to the zigzag SWNTs. The effects of diameter and length on the critical values of torque of SWNTs are obtained by comparing the torsional behaviors of SWNTs with different diameters and different lengths. It is observed that the MD results of the effect of diameter and length on the critical values of torque agrees well with the prediction of continuum shell mode... read less NOT USED (high confidence) D. Belashchenko, “Hybrid potential of interparticle interaction and calculation of lithium melting curves using the molecular dynamics method,” High Temperature. 2015. link Times cited: 4 NOT USED (high confidence) X. Liu, G. Zhang, and Y.-W. Zhang, “Tunable Mechanical and Thermal Properties of One-Dimensional Carbyne Chain: Phase Transition and Microscopic Dynamics,” Journal of Physical Chemistry C. 2015. link Times cited: 53 Abstract: Recently, carbyne chain, the one-dimensional sp-hybridized c… read moreAbstract: Recently, carbyne chain, the one-dimensional sp-hybridized carbon allotrope in the form of either α-carbyne (polyyne), with alternating single and triple bonds, or β-carbyne (cumulene), with repeating double bonds, has attracted more and more attention. However, the mechanical and thermal properties of individual phases, phase-transition dynamics, and defect formation remain largely unknown. Our molecular dynamics simulations show that the critical temperature for phase transition from cumulene to polyyne is 499 K, and the phase transition is ultrafast and completed within 150 fs. To achieve perfect polyyne, however, refined temperature control is needed so as to avoid defective bonds. The bending stiffness and Young’s modulus of cumulene are significantly higher than those of polyyne, while both are comparable to those of the hardest natural materials. The large difference in the stress–strain behavior between cumulene and polyyne provides a novel route for storing mechanical energy. Furthermore, the the... read less NOT USED (high confidence) J. Tao, G. Xu, and Y. Sun, “Elastic Properties of Boron-Nitride Nanotubes through an Atomic Simulation Method,” Mathematical Problems in Engineering. 2015. link Times cited: 12 Abstract: The elastic properties of the boron-nitride nanotubes are st… read moreAbstract: The elastic properties of the boron-nitride nanotubes are studied based on an atomic simulation method that is called atomic-scale finite element method. The Tersoff-Brenner potential is used to describe the interaction between boron and nitrogen atoms, and the computational method is established in an atomic-scale scheme similar to the classical finite element method. Young’s modulus is evaluated for the boron-nitride nanotubes, and their buckling behavior is analyzed. It is shown that the diameter has an obvious influence on Young’s modulus of BNNTs, and the buckling is little related to the length of the nanotubes. read less NOT USED (high confidence) U. Monteverde et al., “Ripples, phonons and bandgap in strained graphene,” 2015 International Conference on Numerical Simulation of Optoelectronic Devices (NUSOD). 2015. link Times cited: 1 Abstract: Using a novel interatomic force field, called MMP, we study … read moreAbstract: Using a novel interatomic force field, called MMP, we study the morphology of Graphene layers under a variety of strain conditions. We report that strain induced ripples possess the “right” kind of elastic deformation that is necessary in order to produce appreciable bandgap opening, which we calculate using Tight Binding, even for low enough strain that can be accessed through realistic means. At the same time the vibrational properties, calculated from analytic derivatives of the MMP force field and used within the dynamics matrix method, can be easily linked to strain obtained from Molecular Dynamics, opening the way for accurate modelling of Raman data. We also show that our models have allowed us to realize in practice novel devices based on our predictions. read less NOT USED (high confidence) D. Taylor, “Shock Compression of Boron Carbide: A Quantum Mechanical Analysis,” Journal of the American Ceramic Society. 2015. link Times cited: 37 Abstract: The shock Hugoniot of boron carbide, from 0 to 80 GPa, has b… read moreAbstract: The shock Hugoniot of boron carbide, from 0 to 80 GPa, has been obtained using first principles quantum mechanics (density functional theory) and molecular dynamics simulation. The Hugoniot for six different structures which vary by structure or stoichiometry were computed and compared to experimental data. The effect of stoichiometry, and structural variation within a given stoichiometry, are shown to have marked effects on the shock properties with some compositions displaying bilinear behavior in the computed shock velocity-particle velocity profiles while others show a continuous Hugoniot curve with no evidence of a phase transition over the pressure range considered in this work. Two structures, B12(CBC) and B11Cp(CCB), have predicted phase transition pressures lying within the 40–50 GPa range suggested experimentally. It is shown that the phase transition is driven by deformation of the 3-atom chain within the boron carbide crystal structure which induces a discontinuous volume change at the critical shock pressure. The effect of defects, in the form of chain vacancies, on the shock response is presented and the ability of shear to significantly lower the phase transition pressure, in accord with experimental observation, is demonstrated. read less NOT USED (high confidence) X. Gu and R. Yang, “Phonon transport and thermal conductivity in two-dimensional materials,” arXiv: Materials Science. 2015. link Times cited: 50 Abstract: Two-dimensional materials, such as graphene, boron nitride a… read moreAbstract: Two-dimensional materials, such as graphene, boron nitride and transition metal dichalcogenides, have attracted increased interest due to their potential applications in electronics and optoelectronics. Thermal transport in two-dimensional materials could be quite different from three-dimensional bulk materials. This article reviews the progress on experimental measurements and theoretical modeling of phonon transport and thermal conductivity in two-dimensional materials. We focus our review on a few typical two-dimensional materials, including graphene, boron nitride, silicene, transition metal dichalcogenides, and black phosphorus. The effects of different physical factors, such as sample size, strain and defects, on thermal transport in Two-dimensional materials are summarized. We also discuss the environmental effect on the thermal transport of two-dimensional materials, such as substrate and when two-dimensional materials are presented in heterostructures and intercalated with inorganic components or organic molecules. read less NOT USED (high confidence) J. Wang, A. Rajendran, and A. Dongare, “Atomic scale modeling of shock response of fused silica and α-quartz,” Journal of Materials Science. 2015. link Times cited: 34 NOT USED (high confidence) J. Jalkanen and M. Müser, “Systematic analysis and modification of embedded-atom potentials: case study of copper,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 10 Abstract: In this study, we evaluate the functionals of different embe… read moreAbstract: In this study, we evaluate the functionals of different embedded-atom methods (EAM) by fitting their free parameters to ab-initio results for copper. Our emphasis lies on testing the transferability of the potentials between systems which vary in their spatial dimension and geometry. The model structures encompass zero-dimensional clusters, one-dimensional chains, two-dimensional tilings, and three-dimensional bulk systems. To avoid having to mimic charge transfer, which is outside the scope of conventional EAM potentials, we focus on structures, in which all atoms are symmetrically equivalent. We find that the simple, four-parameter Gupta EAM potential is overall satisfactory. Adding complexity to it decreases the errors on our set of structures only by marginal amounts, unless EAM is modified to depend also on density gradients, higher-order derivatives, or related terms. All tested conventional EAM functions reveal similar problems: the binding energy of closed-packed systems is overestimated in comparison to open or planar geometries, and structures with small coordination tend to be too rigid. These deficiencies can be fixed in terms of a systematically modified embedded-atom method (SMEAM), which reproduces DFT results on bond lengths, binding energies, and stiffnesses or bulk moduli by typically O(1%), O(5%), and O(15%) accuracy, respectively. SMEAM also predicts the radial distribution function of liquid copper quite accurately. Yet, it does not overcome the difficulty to reproduce the elastic tensor elements of a hypothetical diamond lattice. read less NOT USED (high confidence) M. Terrones, G. Terrones, and H. Terrones, “Structure, Chirality, and Formation of Giant Icosahedral Fullerenes and Spherical Graphitic Onions,” Structural Chemistry. 2015. link Times cited: 35 NOT USED (high confidence) K. Home, T. Antoni, and S. Volz, “Molecular dynamics thermal conductivity computation of a quantum cascade laser diode,” 2015 21st International Workshop on Thermal Investigations of ICs and Systems (THERMINIC). 2015. link Times cited: 0 Abstract: By using molecular dynamics technique, we have computed the … read moreAbstract: By using molecular dynamics technique, we have computed the effective cross-plane thermal conductivity of a single cascade of a quantum cascade laser diode. Additionally, the local phonon density of states was also computed. The Tersoff potential was used with coefficients found from the literature for inter-atomic forces, and the Green-Kubo relation was used to compute the conductivity from the integral of the system heat flux autocorrelation. The computed conductivity lies in the same range as measurements found in the literature, but the local density of states deviated from expected values based on local diode composition, suggesting that previous assumptions made about electron-phonon coupling are incorrect. read less NOT USED (high confidence) Z. Tong, X. Luo, J. Sun, Y. Liang, and X. Jiang, “Investigation of a scale-up manufacturing approach for nanostructures by using a nanoscale multi-tip diamond tool,” The International Journal of Advanced Manufacturing Technology. 2015. link Times cited: 14 NOT USED (high confidence) J. S. Babu, C. Mondal, S. Sengupta, and S. Karmakar, “Excess vibrational density of states and the brittle to ductile transition in crystalline and amorphous solids.,” Soft matter. 2015. link Times cited: 9 Abstract: The conditions which determine whether a material behaves in… read moreAbstract: The conditions which determine whether a material behaves in a brittle or ductile fashion on mechanical loading are still elusive and comprise a topic of active research among materials physicists and engineers. In this study, we present the results of in silico mechanical deformation experiments from two very different model solids in two and three dimensions. The first consists of particles interacting with isotropic potentials and the other has strongly direction dependent interactions. We show that in both cases, the excess vibrational density of states is one of the fundamental quantities which characterizes the ductility of the material. Our results can be checked using careful experiments on colloidal solids. read less NOT USED (high confidence) X. Qi-lin, L. Zhenhuan, and T. Xiaogeng, “The defect-induced fracture behaviors of hexagonal boron-nitride monolayer nanosheets under uniaxial tension,” Journal of Physics D: Applied Physics. 2015. link Times cited: 42 Abstract: Due to its excellent mechanical and electrical insulating pr… read moreAbstract: Due to its excellent mechanical and electrical insulating properties, the hexagonal boron-nitride (h-BN) monolayer nanosheet is regarded as a complementary addition to graphene. However, its mechanical strength can be significantly affected by various defects pre-existing in it, such as a Stone–Wales defect, a vacancy defect, an atomic anti-site defect, etc. In this work, the influences of various pre-existing defects on the fracture behaviors of an h-BN monolayer nanosheet are investigated carefully using molecular dynamics simulation. The results show that the nucleation and evolution of a fracture induced by defects in the h-BN monolayer nanosheet are directional, and that the crack always starts from the location which has a weak bond energy. An unexpected observation is that the defect propagates mostly in the zigzag direction but occasionally in the armchair direction. The fracture strength and the fracture strain of the h-BN monolayer nanosheet are reduced at different extents due to the various pre-existing defects. Additionally, for the defective h-BN monolayer nanosheets, the fracture strength and strain measured in the armchair direction is much higher than the strength found in the zigzag direction. However, the strengths measured in the armchair and zigzag directions for the defect-free h-BN monolayer nanosheets almost are identical which implies that the armchair direction has a stronger ability to resist various defects compared to the zigzag direction. read less NOT USED (high confidence) B. D. Jensen, K. Wise, and G. Odegard, “The effect of time step, thermostat, and strain rate on ReaxFF simulations of mechanical failure in diamond, graphene, and carbon nanotube,” Journal of Computational Chemistry. 2015. link Times cited: 83 Abstract: As the sophistication of reactive force fields for molecular… read moreAbstract: As the sophistication of reactive force fields for molecular modeling continues to increase, their use and applicability has also expanded, sometimes beyond the scope of their original development. Reax Force Field (ReaxFF), for example, was originally developed to model chemical reactions, but is a promising candidate for modeling fracture because of its ability to treat covalent bond cleavage. Performing reliable simulations of a complex process like fracture, however, requires an understanding of the effects that various modeling parameters have on the behavior of the system. This work assesses the effects of time step size, thermostat algorithm and coupling coefficient, and strain rate on the fracture behavior of three carbon‐based materials: graphene, diamond, and a carbon nanotube. It is determined that the simulated stress‐strain behavior is relatively independent of the thermostat algorithm, so long as coupling coefficients are kept above a certain threshold. Likewise, the stress‐strain response of the materials was also independent of the strain rate, if it is kept below a maximum strain rate. Finally, the mechanical properties of the materials predicted by the Chenoweth C/H/O parameterization for ReaxFF are compared with literature values. Some deficiencies in the Chenoweth C/H/O parameterization for predicting mechanical properties of carbon materials are observed. © 2015 Wiley Periodicals, Inc. read less NOT USED (high confidence) A. Kumar et al., “Charge optimized many-body (COMB) potential for dynamical simulation of Ni–Al phases,” Journal of Physics: Condensed Matter. 2015. link Times cited: 18 Abstract: An interatomic potential for the Ni–Al system is presented w… read moreAbstract: An interatomic potential for the Ni–Al system is presented within the third-generation charge optimized many-body (COMB3) formalism. The potential has been optimized for Ni3Al, or the γ′ phase in Ni-based superalloys. The formation energies predicted for other Ni–Al phases are in reasonable agreement with first-principles results. The potential further predicts good mechanical properties for Ni3Al, which includes the values of the complex stacking fault (CSF) and the anti-phase boundary (APB) energies for the (1 1 1) and (1 0 0) planes. It is also used to investigate dislocation propagation across the Ni3Al (1 1 0)–Ni (1 1 0) interface, and the results are consistent with simulation results reported in the literature. The potential is further used in combination with a recent COMB3 potential for Al2O3 to investigate the Ni3Al (1 1 1)–Al2O3 (0 0 01) interface, which has not been modeled previously at the classical atomistic level due to the lack of a reactive potential to describe both Ni3Al and Al2O3 as well as interactions between them. The calculated work of adhesion for this interface is predicted to be 1.85 J m−2, which is in agreement with available experimental data. The predicted interlayer distance is further consistent with the available first-principles results for Ni (1 1 1)–Al2O3 (0 0 0 1). read less NOT USED (high confidence) A. Favata, A. Micheletti, S. Ryu, and N. Pugno, “An analytical benchmark and a Mathematica program for MD codes: Testing LAMMPS on the 2nd generation Brenner potential,” Comput. Phys. Commun. 2015. link Times cited: 13 NOT USED (high confidence) B. Wirth, K. Hammond, S. Krasheninnikov, and D. Maroudas, “Challenges and opportunities of modeling plasma–surface interactions in tungsten using high-performance computing,” Journal of Nuclear Materials. 2015. link Times cited: 72 NOT USED (high confidence) K. Zhang and Q. Zhang, “Raman Signatures of Broken C–C Bonds in Single-Walled Carbon Nanotubes upon [2 + 1] Cycloaddition,” Journal of Physical Chemistry C. 2015. link Times cited: 7 Abstract: Intact or broken sidewall C–C bonds on covalently functional… read moreAbstract: Intact or broken sidewall C–C bonds on covalently functionalized single-walled carbon nanotubes (SWCNTs) play a fundamental role in preserving the excellent quantum conductance and electronic structure of SWCNTs. However, there has been a lack of experimental identification on possible sidewall C–C broken bond caused by covalent functionalizations. In this work, clear evidence of sidewall C–C bonds broken by [2 + 1] cycloaddition are observed with Raman scatterings on individual SWCNTs with various diameters and chiralities. The relationship between SWCNT chirality and dichlorocarbene grafting configurations is also observed for the first time. read less NOT USED (high confidence) O. Trushin et al., “Minimum energy path for the nucleation of misfit dislocations in Ge/Si(0 0 1) heteroepitaxy,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 9 Abstract: A possible mechanism for the formation of a 90° misfit dislo… read moreAbstract: A possible mechanism for the formation of a 90° misfit dislocation at the Ge/Si(0 0 1) interface through homogeneous nucleation is identified from atomic scale calculations where a minimum energy path connecting the coherent epitaxial state and a final state with a 90° misfit dislocation is found using the nudged elastic band method. The initial path is generated using a repulsive bias activation procedure in a model system including 75 000 atoms. The energy along the path exhibits two maxima in the energy. The first maximum occurs as a 60° dislocation nucleates. The intermediate minimum corresponds to an extended 60° dislocation. The subsequent energy maximum occurs as a second 60° dislocation nucleates in a complementary, mirror glide plane, simultaneously starting from the surface and from the first 60° dislocation. The activation energy of the nucleation of the second dislocation is 30% lower than that of the first one showing that the formation of the second 60° dislocation is aided by the presence of the first one. The simulations represent a step towards unraveling the formation mechanism of 90° dislocations, an important issue in the design of growth procedures for strain released Ge overlayers on Si(1 0 0) surfaces, and more generally illustrate an approach that can be used to gain insight into the mechanism of complex nucleation paths of extended defects in solids. read less NOT USED (high confidence) M. Migliorato et al., “Beyond ZnO nanowires for piezotronics and nanogenerators,” 2015 IEEE 15th International Conference on Nanotechnology (IEEE-NANO). 2015. link Times cited: 0 Abstract: In the past decade ZnO nanowires have been the key enabling … read moreAbstract: In the past decade ZnO nanowires have been the key enabling material for demonstrating novel electronics components in the field of piezotronics and in the first realization of a nanogenerator. What are the materials that will be crucial in demonstrating even more novel devices in future years? We propose the use of both core shell nanowires and graphene as key enablers of new functionalities. read less NOT USED (high confidence) S. Thomas, K. Ajith, S. Chandra, and M. C. Valsakumar, “Temperature dependent structural properties and bending rigidity of pristine and defective hexagonal boron nitride,” Journal of Physics: Condensed Matter. 2015. link Times cited: 58 Abstract: Structural and thermodynamical properties of monolayer prist… read moreAbstract: Structural and thermodynamical properties of monolayer pristine and defective boron nitride sheets (h-BN) have been investigated in a wide temperature range by carrying out atomistic simulations using a tuned Tersoff-type inter-atomic empirical potential. The temperature dependence of lattice parameter, radial distribution function, specific heat at constant volume, linear thermal expansion coefficient and the height correlation function of the thermally excited ripples on pristine as well as defective h-BN sheet have been investigated. Specific heat shows considerable increase beyond the Dulong–Petit limit at high temperatures, which is interpreted as a signature of strong anharmonicity present in h-BN. Analysis of the height fluctuations, ⟨h2⟩ ?>, shows that the bending rigidity and variance of height fluctuations are strongly temperature dependent and this is explained using the continuum theory of membranes. A detailed study of the height–height correlation function shows deviation from the prediction of harmonic theory of membranes as a consequence of the strong anharmonicity in h-BN. It is also seen that the variance of the height fluctuations increases with defect concentration. read less NOT USED (high confidence) A. Galashev and V. Polukhin, “Computer modeling of the structure and properties of mercury films on graphene,” Russian Journal of Physical Chemistry A. 2015. link Times cited: 4 NOT USED (high confidence) R. Ansari and S. Ajori, “A molecular dynamics study on the vibration of carbon and boron nitride double-walled hybrid nanotubes,” Applied Physics A. 2015. link Times cited: 28 NOT USED (high confidence) R. Ansari and S. Ajori, “A molecular dynamics study on the vibration of carbon and boron nitride double-walled hybrid nanotubes,” Applied Physics A. 2015. link Times cited: 0 NOT USED (high confidence) H. Chen and C. Ortner, “QM/MM Methods for Crystalline Defects. Part 1: Locality of the Tight Binding Model,” Multiscale Model. Simul. 2015. link Times cited: 40 Abstract: The tight binding model is a minimal electronic structure mo… read moreAbstract: The tight binding model is a minimal electronic structure model for molecular modeling and simulation. We show that for a finite temperature model, the total energy in this model can be decomposed into site energies, that is, into contributions from each atomic site whose influence on their environment decays exponentially. This result lays the foundation for a rigorous analysis of QM/MM coupling schemes. read less NOT USED (high confidence) S. Naserifar, W. Goddard, T. Tsotsis, and M. Sahimi, “First principles-based multiparadigm, multiscale strategy for simulating complex materials processes with applications to amorphous SiC films.,” The Journal of chemical physics. 2015. link Times cited: 9 Abstract: Progress has recently been made in developing reactive force… read moreAbstract: Progress has recently been made in developing reactive force fields to describe chemical reactions in systems too large for quantum mechanical (QM) methods. In particular, ReaxFF, a force field with parameters that are obtained solely from fitting QM reaction data, has been used to predict structures and properties of many materials. Important applications require, however, determination of the final structures produced by such complex processes as chemical vapor deposition, atomic layer deposition, and formation of ceramic films by pyrolysis of polymers. This requires the force field to properly describe the formation of other products of the process, in addition to yielding the final structure of the material. We describe a strategy for accomplishing this and present an example of its use for forming amorphous SiC films that have a wide variety of applications. Extensive reactive molecular dynamics (MD) simulations have been carried out to simulate the pyrolysis of hydridopolycarbosilane. The reaction products all agree with the experimental data. After removing the reaction products, the system is cooled down to room temperature at which it produces amorphous SiC film, for which the computed radial distribution function, x-ray diffraction pattern, and the equation of state describing the three main SiC polytypes agree with the data and with the QM calculations. Extensive MD simulations have also been carried out to compute other structural properties, as well the effective diffusivities of light gases in the amorphous SiC film. read less NOT USED (high confidence) L.-F. Wang, X. Shu, and G. Lu, “Comparison of two tungsten–helium interatomic potentials,” Journal of Materials Research. 2015. link Times cited: 4 Abstract: We have clarified the performance of two tungsten–helium ana… read moreAbstract: We have clarified the performance of two tungsten–helium analytical interatomic potentials, one of which, developed by Li et al., is a bond-order potential, and another, developed by Juslin et al., is a combination of embedded atom method potential and pair potential. Using these two potentials, we have simulated and made a full comparison of formation energy and migration energy of different defects including helium and vacancy, binding energies of helium and vacancy with helium-vacancy cluster, surface energy, as well as melting point, with reference to the corresponding results from the first-principles and experiments. read less NOT USED (high confidence) N. Onofrio and A. Strachan, “Voltage equilibration for reactive atomistic simulations of electrochemical processes.,” The Journal of chemical physics. 2015. link Times cited: 37 Abstract: We introduce electrochemical dynamics with implicit degrees … read moreAbstract: We introduce electrochemical dynamics with implicit degrees of freedom (EChemDID), a model to describe electrochemical driving force in reactive molecular dynamics simulations. The method describes the equilibration of external electrochemical potentials (voltage) within metallic structures and their effect on the self-consistent partial atomic charges used in reactive molecular dynamics. An additional variable assigned to each atom denotes the local potential in its vicinity and we use fictitious, but computationally convenient, dynamics to describe its equilibration within connected metallic structures on-the-fly during the molecular dynamics simulation. This local electrostatic potential is used to dynamically modify the atomic electronegativities used to compute partial atomic changes via charge equilibration. Validation tests show that the method provides an accurate description of the electric fields generated by the applied voltage and the driving force for electrochemical reactions. We demonstrate EChemDID via simulations of the operation of electrochemical metallization cells. The simulations predict the switching of the device between a high-resistance to a low-resistance state as a conductive metallic bridge is formed and resistive currents that can be compared with experimental measurements. In addition to applications in nanoelectronics, EChemDID could be useful to model electrochemical energy conversion devices. read less NOT USED (high confidence) A. Davydova, E. Despiau-Pujo, G. Cunge, and D. Graves, “Etching mechanisms of graphene nanoribbons in downstream H2 plasmas: insights from molecular dynamics simulations,” Journal of Physics D: Applied Physics. 2015. link Times cited: 20 Abstract: Lateral etching mechanisms of graphene nanoribbons (GNRs) wi… read moreAbstract: Lateral etching mechanisms of graphene nanoribbons (GNRs) with zigzag (ZZ) edges in downstream H2 plasmas are investigated using molecular dynamics simulations. A new etching mechanism is found, which occurs in three consecutive phases and requires a continuous exposure of GNRs to H atoms and high substrate temperatures (~800 K). Full hydrogenation of GNR free edges during phase 1 reduces the potential barriers to H chemisorption on near-edge C atoms from the basal plane. Subsequent hydrogenation of near-edge C–C dimers creates mechanical stress between C atoms (due to local sp2-to-sp3 rehybridizations) which leads to the rupture of C–C dimers bonds, unzipping locally the 1st and 2nd edge carbon rows. The unzipping then propagates randomly along the GNR edges and creates suspended linear carbon chains (phase 2). Weakened by their exposure to continuous H bombardment and strong thermal vibrations, the suspended carbon chains may then rupture, leading to the sputtering of their carbon atoms as single C atoms or C2 molecules (phase 3). Thus no formation of volatile hydrocarbon etching products is observed in this three-phase mechanism, which explains why the ribbon edges can be sharp-cut without generation of line-edge roughness, as also observed experimentally. Influence of substrate temperature on ZZ-GNRs etching is investigated and suggests the dominant mechanisms for understanding the temperature dependence of the etch rate observed experimentally (peaks at 800 K and decreases for lower or higher temperatures). read less NOT USED (high confidence) A. Loya and G. Ren, “Molecular dynamics simulation study of rheological properties of CuO–water nanofluid,” Journal of Materials Science. 2015. link Times cited: 27 NOT USED (high confidence) A. Page, F. Ding, S. Irle, and K. Morokuma, “Insights into carbon nanotube and graphene formation mechanisms from molecular simulations: a review,” Reports on Progress in Physics. 2015. link Times cited: 96 Abstract: The discovery of carbon nanotubes (CNTs) and graphene over t… read moreAbstract: The discovery of carbon nanotubes (CNTs) and graphene over the last two decades has heralded a new era in physics, chemistry and nanotechnology. During this time, intense efforts have been made towards understanding the atomic-scale mechanisms by which these remarkable nanostructures grow. Molecular simulations have made significant contributions in this regard; indeed, they are responsible for many of the key discoveries and advancements towards this goal. Here we review molecular simulations of CNT and graphene growth, and in doing so we highlight the many invaluable insights gained from molecular simulations into these complex nanoscale self-assembly processes. This review highlights an often-overlooked aspect of CNT and graphene formation—that the two processes, although seldom discussed in the same terms, are in fact remarkably similar. Both can be viewed as a 0D → 1D → 2D transformation, which converts carbon atoms (0D) to polyyne chains (1D) to a complete sp2-carbon network (2D). The difference in the final structure (CNT or graphene) is determined only by the curvature of the catalyst and the strength of the carbon–metal interaction. We conclude our review by summarizing the present shortcomings of CNT/graphene growth simulations, and future challenges to this important area. read less NOT USED (high confidence) W.-L. Lv and A. Henry, “Direct calculation of modal contributions to thermal conductivity via Green–Kubo modal analysis,” New Journal of Physics. 2015. link Times cited: 115 Abstract: We derived a new method for direct calculation of the modal … read moreAbstract: We derived a new method for direct calculation of the modal contributions to thermal conductivity, which is termed Green–Kubo modal analysis (GKMA). The GKMA method combines the lattice dynamics formalism with the Green–Kubo formula for thermal conductivity, such that the thermal conductivity becomes a direct summation of modal contributions, where one need not define the phonon velocity. As a result, the GKMA method can be applied to any material/group of atoms, where the atoms vibrate around stable equilibrium positions, which includes non-stoichiometric compounds, random alloys, amorphous materials and even rigid molecules. By using molecular dynamics simulations to obtain the time history of each mode’s contribution to the heat current, one naturally includes anharmonicity to full order and can obtain insight into the interactions between different modes through the cross-correlations. As an example, we applied the GMKA method to crystalline and amorphous silicon. The modal contributions at each frequency result from the analysis and thereby allow one to apply a quantum correction to the mode heat capacity to determine the temperature dependence of thermal conductivity. The predicted temperature dependent thermal conductivity for amorphous silicon shows the best agreement with experiments to date. The GKMA method provides new insight into the nature of phonon transport, as it casts the problem in terms of mode–mode correlation instead of scattering, and provides a general unified formalism that can be used to understand phonon–phonon interactions in essentially any class of materials or structures where the atoms vibrate around stable equilibrium sites. read less NOT USED (high confidence) A. Mitra, S. Ganguly, S. Sengupta, and P. Sollich, “Non-affine fluctuations and the statistics of defect precursors in the planar honeycomb lattice,” Journal of Statistical Mechanics: Theory and Experiment. 2015. link Times cited: 9 Abstract: Certain localised displacement fluctuations in the planar ho… read moreAbstract: Certain localised displacement fluctuations in the planar honeycomb lattice may be identified as precursors to topological defects. We show that these fluctuations are among the most pronounced non-affine distortions of an elemental coarse graining volume of the honeycomb structure at non zero temperatures. We obtain the statistics of these precursor modes in the canonical ensemble, evaluating exactly their single point and two-point spatio-temporal distributions, for a lattice with harmonic nearest neighbour and next near neighbour bonds. As the solid is destabilised by tuning interactions, the precursor fluctuations diverge and correlations become long-lived and long-ranged. read less NOT USED (high confidence) Z. Wang, T. Feng, and X. Ruan, “Thermal conductivity and spectral phonon properties of freestanding and supported silicene,” Journal of Applied Physics. 2015. link Times cited: 73 Abstract: We conduct molecular dynamics (MD) simulations to study the … read moreAbstract: We conduct molecular dynamics (MD) simulations to study the thermal conductivity of freestanding silicene and silicene supported on an amorphous silicon dioxide (SiO2) substrate in the temperature range from 300 to 900 K. The results show that the thermal conductivity decreases with increasing temperature and that the presence of the SiO2 substrate results in a great reduction, up to 78% at 300 K, to the thermal conductivity of silicene. With atomic trajectories from equilibrium MD simulations, we perform spectral energy density analysis to compute the thermal conductivities, spectral phonon relaxation times, and spectral phonon mean free paths (MFPs) of freestanding and supported silicene at 300 K. When silicene is put on a SiO2 substrate, the phonon relaxation times are decreased from 1–13 ps to less than 1 ps, and the phonon MFPs are reduced from 10–120 nm to 0–20 nm. We also calculate the thermal conductivity contributions from all phonon branches and find that the thermal conductivities of freestanding and supported silicene are mainly (>85%) contributed by the longitudinal and transverse acoustic phonons, while the out-of-plane acoustic phonons have a contribution less than 3%. Our study predicts the reduction of the thermal conductivity of silicene due to substrate effects and provides a fundamental understanding of the reduction in terms of the spectral phonon relaxation times and MFPs. read less NOT USED (high confidence) E. Bitzek, J. Kermode, and P. Gumbsch, “Atomistic aspects of fracture,” International Journal of Fracture. 2015. link Times cited: 130 NOT USED (high confidence) H. Gao et al., “Large-scale nanoshaping of ultrasmooth 3D crystalline metallic structures,” Science. 2014. link Times cited: 149 Abstract: We report a low-cost, high-throughput benchtop method that e… read moreAbstract: We report a low-cost, high-throughput benchtop method that enables thin layers of metal to be shaped with nanoscale precision by generating ultrahigh-strain-rate deformations. Laser shock imprinting can create three-dimensional crystalline metallic structures as small as 10 nanometers with ultrasmooth surfaces at ambient conditions. This technique enables the successful fabrications of large-area, uniform nanopatterns with aspect ratios as high as 5 for plasmonic and sensing applications, as well as mechanically strengthened nanostructures and metal-graphene hybrid nanodevices. Smooth surface, crystalline 3D metallic nanostructures are fabricated using a laser shock imprinting technique. Laser shock imprinting for patterning metals High-fidelity, small-scale patterning is often a tradeoff between full-pattern methods that may have limited resolution or flexiblity, and serial methods that can create high-resolution patterns but only by slow processes. Furthermore, metals have limited formability at very small scales. Gao et al. developed a method to create very smooth threedimensional crystalline metallic nanoscale structures using a laser to create shockwave impulses. The shockwave creates ultrahigh-strain-rate deformations that overcome the metal's normal strength and, thus, resistance to patterning. Science, this issue p. 1352 read less NOT USED (high confidence) V. D. Camiola, R. Farchioni, V. Pellegrini, and V. Tozzini, “Hydrogen transport within graphene multilayers by means of flexural phonons,” 2D Materials. 2014. link Times cited: 4 Abstract: Graphene sustains transverse out-of-plane mechanical vibrati… read moreAbstract: Graphene sustains transverse out-of-plane mechanical vibrations (flexural phonons). At the nanometer scale, these appear as traveling ripples, or cavities, if excited in counter-phase in alternate multilayers. In this work we explore by means of classical molecular dynamics simulations the possibility of using these moving nano-cavities to actively transport hydrogen. We find that the gas can be efficiently transported for hundreds of nanometers in the wave propagation direction, before the phonons damp down. Therefore, this effect could be used to move and pump gases through multilayers graphene based frameworks. read less NOT USED (high confidence) A. Shakouri, J. Yeo, T. Ng, Z. Liu, and H. Taylor, “Superlubricity-activated thinning of graphite flakes compressed by passivated crystalline silicon substrates for graphene exfoliation,” Carbon. 2014. link Times cited: 4 NOT USED (high confidence) T. L. Jensen, J. Moxnes, E. Unneberg, and O. Dullum, “Calculation of Decomposition Products from Components of Gunpowder by using ReaxFF Reactive Force Field Molecular Dynamics and Thermodynamic Calculations of Equilibrium Composition,” Propellants, Explosives, Pyrotechnics. 2014. link Times cited: 8 Abstract: Jensen, Tomas Lunde; Moxnes, John Fredrik; Unneberg, Erik; D… read moreAbstract: Jensen, Tomas Lunde; Moxnes, John Fredrik; Unneberg, Erik; Dullum, Ove.
Calculation of decomposition products from components of gunpowder by using ReaxFF reactive force field molecular dynamics and thermodynamic calculations of equilibrium composition. Propellants, explosives, pyrotechnics 2014 ;Volum 39.(6) s. 830-837 read less NOT USED (high confidence) S. Linas et al., “Interplay between Raman shift and thermal expansion in graphene: temperature-dependent measurements and analysis of substrate corrections,” Physical Review B. 2014. link Times cited: 44 Abstract: Measurements and calculations have shown significant disagre… read moreAbstract: Measurements and calculations have shown significant disagreement regarding the sign and variations of the thermal expansion coefficient (TEC) of graphene α(T). Here we report dedicated Raman scattering experiments conducted for graphene monolayers deposited on silicon nitride substrates and over the broad temperature range 150–900 K. The relation between those measurements for the G band and the graphene TEC, which involves correcting the measured signal for the mismatch contribution of the substrate, is analyzed based on various theoretical candidates for α(T). Contrary to calculations in the quasiharmonic approximation, a many-body potential reparametrized for graphene correctly reproduces experimental data. These results indicate that the TEC is more likely to be positive above room temperature. read less NOT USED (high confidence) A. Dongare, “Quasi-coarse-grained dynamics: modelling of metallic materials at mesoscales,” Philosophical Magazine. 2014. link Times cited: 26 Abstract: A computationally efficient modelling method called quasi-co… read moreAbstract: A computationally efficient modelling method called quasi-coarse-grained dynamics (QCGD) is developed to expand the capabilities of molecular dynamics (MD) simulations to model behaviour of metallic materials at the mesoscales. This mesoscale method is based on solving the equations of motion for a chosen set of representative atoms from an atomistic microstructure and using scaling relationships for the atomic-scale interatomic potentials in MD simulations to define the interactions between representative atoms. The scaling relationships retain the atomic-scale degrees of freedom and therefore energetics of the representative atoms as would be predicted in MD simulations. The total energetics of the system is retained by scaling the energetics and the atomic-scale degrees of freedom of these representative atoms to account for the missing atoms in the microstructure. This scaling of the energetics renders improved time steps for the QCGD simulations. The success of the QCGD method is demonstrated by the prediction of the structural energetics, high-temperature thermodynamics, deformation behaviour of interfaces, phase transformation behaviour, plastic deformation behaviour, heat generation during plastic deformation, as well as the wave propagation behaviour, as would be predicted using MD simulations for a reduced number of representative atoms. The reduced number of atoms and the improved time steps enables the modelling of metallic materials at the mesoscale in extreme environments. read less NOT USED (high confidence) A. D. Bobadilla and J. Seminario, “Argon-Beam-Induced Defects in a Silica-Supported Single-Walled Carbon Nanotube,” Journal of Physical Chemistry C. 2014. link Times cited: 8 Abstract: Ion beams can be used to tailor the structure and properties… read moreAbstract: Ion beams can be used to tailor the structure and properties of carbon nanostructures. Using molecular dynamics simulations, we explored the effects of irradiating silica-supported single-walled carbon nanotube (CNT) with an ion beam. We analyzed the defects produced at several energy levels when one argon atom collides with a single-walled CNT. At beam energies greater than 32 keV, the resulting defects were mainly single-vacancy defects. In addition to vacancy defects, we found chemisorption on the CNT sidewall, doping of the silica substrate, and cross-linking between the CNT and the substrate; these types of complex defects had a maximum probability of occurrence at around 100 eV and a close to null probability at around 100 keV. read less NOT USED (high confidence) X.-Y. Sun, Y. Xu, G.-K. Xu, and J.-hui Zhang, “Effects of interface cohesion on mechanical properties of interpenetrating phase nanocomposites,” Micro & Nano Letters. 2014. link Times cited: 4 Abstract: Molecular dynamics simulations and micromechanics model anal… read moreAbstract: Molecular dynamics simulations and micromechanics model analysis are performed to investigate the mechanical behaviours and interfacial effects of interpenetrating phase composites in the nanoscale. It is observed that the overall Young's modulus and ultimate strength of the nanocomposites vary nonlinearly with the cohesive energy of the interface. The cohesive properties affect the stiffness of the interface zone, and in turn, influence the effective Young's modulus of composites. The competition between interfacial failure and weak phase damage results in an optimal cohesive parameter of the interface, at which the composite possesses the maximal ultimate strength. The obtained results provide useful guidelines for the design and optimisation of advanced nanocomposites with superior mechanical properties. read less NOT USED (high confidence) A. Favata, A. Micheletti, P. Podio-Guidugli, and N. Pugno, “Geometry and Self-stress of Single-Wall Carbon Nanotubes and Graphene via a Discrete Model Based on a 2nd-Generation REBO Potential,” Journal of Elasticity. 2014. link Times cited: 33 NOT USED (high confidence) Z. Tong, Y. Liang, X. Yang, and X. Luo, “Investigation on the thermal effects during nanometric cutting process while using nanoscale diamond tools,” The International Journal of Advanced Manufacturing Technology. 2014. link Times cited: 40 NOT USED (high confidence) E. Neyts, M. Yusupov, C. Verlackt, and A. Bogaerts, “Computer simulations of plasma–biomolecule and plasma–tissue interactions for a better insight in plasma medicine,” Journal of Physics D: Applied Physics. 2014. link Times cited: 42 Abstract: Plasma medicine is a rapidly evolving multidisciplinary fiel… read moreAbstract: Plasma medicine is a rapidly evolving multidisciplinary field at the intersection of chemistry, biochemistry, physics, biology, medicine and bioengineering. It holds great potential in medical, health care, dentistry, surgical, food treatment and other applications. This multidisciplinary nature and variety of possible applications come along with an inherent and intrinsic complexity. Advancing plasma medicine to the stage that it becomes an everyday tool in its respective fields requires a fundamental understanding of the basic processes, which is lacking so far. However, some major advances have already been made through detailed experiments over the last 15 years. Complementary, computer simulations may provide insight that is difficult—if not impossible—to obtain through experiments. In this review, we aim to provide an overview of the various simulations that have been carried out in the context of plasma medicine so far, or that are relevant for plasma medicine. We focus our attention mostly on atomistic simulations dealing with plasma–biomolecule interactions. We also provide a perspective and tentative list of opportunities for future modelling studies that are likely to further advance the field. read less NOT USED (high confidence) N. Wang and K. Komvopoulos, “The effect of deposition energy of energetic atoms on the growth and structure of ultrathin amorphous carbon films studied by molecular dynamics simulations,” Journal of Physics D: Applied Physics. 2014. link Times cited: 14 Abstract: The growth and structure of ultrathin amorphous carbon films… read moreAbstract: The growth and structure of ultrathin amorphous carbon films was investigated by molecular dynamics simulations. The second-generation reactive-empirical-bond-order potential was used to model atomic interactions. Films with different structures were simulated by varying the deposition energy of carbon atoms in the range of 1–120 eV. Intrinsic film characteristics (e.g. density and internal stress) were determined after the system reached equilibrium. Short- and intermediate-range carbon atom ordering is examined in the context of atomic hybridization and ring connectivity simulation results. It is shown that relatively high deposition energy (i.e., 80 eV) yields a multilayer film structure consisting of an intermixing layer, bulk film and surface layer, consistent with the classical subplantation model. The highest film density (3.3 g cm−3), sp3 fraction (∼43%), and intermediate-range carbon atom ordering correspond to a deposition energy of ∼80 eV, which is in good agreement with experimental findings. read less NOT USED (high confidence) V. Vijayaraghavan, A. Garg, C. H. Wong, K. Tai, and P. Singru, “An integrated computational approach for determining the elastic properties of boron nitride nanotubes,” International Journal of Mechanics and Materials in Design. 2014. link Times cited: 39 NOT USED (high confidence) F. Soberon, “Surface activation of cyclo olefin polymer by oxygen plasma discharge: a molecular dynamics study,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 3 Abstract: Thermoplastic substrates made of cyclo olefin polymer (COP) … read moreAbstract: Thermoplastic substrates made of cyclo olefin polymer (COP) are treated with oxygen plasma discharges to introduce polar groups at the surface. This is the first step in the process of surface functionalization of COP substrates used in biosensor devices. A molecular dynamics model of basic COP structure is implemented using the second-generation reactive empirical bond order (REBO) potentials for hydrocarbon–oxygen interactions. The model includes covalent bond and Van der Waals interactions. The bombardment of a COP surface with mono-energetic atomic oxygen ions, energy in the range 1-35 eV, is simulated and reported here. The dynamics of the substrate modification reveals that the substrate top layer is de-hydrogenated and subsequently builds up an oxygen–carbon matrix layer, ∼10 Å thick. Analysis of the modified substrates indicates that surface yield is predominantly peroxide groups. read less NOT USED (high confidence) D. V. Singh, C. Cassidy, P. Grammatikopoulos, F. Djurabekova, K. Nordlund, and M. Sowwan, “Heterogeneous Gas-Phase Synthesis and Molecular Dynamics Modeling of Janus and Core–Satellite Si–Ag Nanoparticles,” Journal of Physical Chemistry C. 2014. link Times cited: 73 Abstract: Heterogeneous gas-phase condensation is a promising method o… read moreAbstract: Heterogeneous gas-phase condensation is a promising method of producing hybrid multifunctional nanoparticles with tailored composition and microstructure but also intrinsically introduces greater complexity to the nucleation process and growth kinetics. Herein, we report on the synthesis and growth modeling of silicon–silver (Si–Ag) hybrid nanoparticles using gas-aggregated cosputtering from elemental Si and Ag source targets. The final Si–Ag ensemble size was manipulated in the range 5–15 nm by appropriate tuning of the deposition parameters, while variations in the Si–Ag sputtering power ratio, from 1.8 to 2.25, allowed distinctive Janus and core–satellite structures, respectively, to be produced. Molecular dynamics simulations indicate that the individual species first form independent clusters of Si and Ag without significant intermixing. Collisions between unlike species are unstable in the early stages of growth (<100 ns), with large temperature differences resulting in rapid energy exchange and sep... read less NOT USED (high confidence) S. A. Deshmukh, G. Kamath, and S. Sankaranarayanan, “Effect of nanoscale confinement on freezing of modified water at room temperature and ambient pressure.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2014. link Times cited: 2 Abstract: Understanding the phase behavior of confined water is centra… read moreAbstract: Understanding the phase behavior of confined water is central to fields as diverse as heterogeneous catalysis, corrosion, nanofluidics, and to emerging energy technologies. Altering the state points (temperature, pressure, etc.) or introduction of a foreign surface can result in the phase transformation of water. At room temperature, ice nucleation is a very rare event and extremely high pressures in the GPa-TPa range are required to freeze water. Here, we perform computer experiments to artificially alter the balance between electrostatic and dispersion interactions between water molecules, and demonstrate nucleation and growth of ice at room temperature in a nanoconfined environment. Local perturbations in dispersive and electrostatic interactions near the surface are shown to provide the seed for nucleation (nucleation sites), which lead to room temperature liquid-solid phase transition of confined water. Crystallization of water occurs over several tens of nanometers and is shown to be independent of the nature of the substrate (hydrophilic oxide vs. hydrophobic graphene and crystalline oxide vs. amorphous diamond-like carbon). Our results lead us to hypothesize that the freezing transition of confined water can be controlled by tuning the relative dispersive and electrostatic interaction. read less NOT USED (high confidence) X. Wu and X. Li, “On consistent definitions of momentum and energy fluxes for molecular dynamics models with multi-body interatomic potentials,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 7 Abstract: Results from molecular dynamics simulations often need to be… read moreAbstract: Results from molecular dynamics simulations often need to be further processed to understand the physics on a larger scale. This paper considers the definitions of momentum and energy fluxes obtained from a control-volume approach. To assess the validity of these defined quantities, two consistency criteria are proposed. As examples, the embedded atom potential and the Tersoff potential are considered. The consistency is verified using analytical and numerical methods. read less NOT USED (high confidence) B. Mortazavi and G. Cuniberti, “Atomistic modeling of mechanical properties of polycrystalline graphene,” Nanotechnology. 2014. link Times cited: 86 Abstract: We performed molecular dynamics (MD) simulations to investig… read moreAbstract: We performed molecular dynamics (MD) simulations to investigate the mechanical properties of polycrystalline graphene. By constructing molecular models of ultra-fine-grained graphene structures, we studied the effect of different grain sizes of 1–10 nm on the mechanical response of graphene. We found that the elastic modulus and tensile strength of polycrystalline graphene decrease with decreasing grain size. The calculated mechanical proprieties for pristine and polycrystalline graphene sheets are found to be in agreement with experimental results in the literature. Our MD results suggest that the ultra-fine-grained graphene structures can show ultrahigh tensile strength and elastic modulus values that are very close to those of pristine graphene sheets. read less NOT USED (high confidence) P. Saidi, T. Frolov, J. Hoyt, and M. Asta, “An angular embedded atom method interatomic potential for the aluminum–silicon system,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 19 Abstract: A modified version of the Stillinger–Weber (SW) interatomic … read moreAbstract: A modified version of the Stillinger–Weber (SW) interatomic potential for pure Si has been developed. In contrast to the original SW form, the modified version allows one to grow diamond cubic crystal structures from the melt at high temperatures. Now, the modified SW potential has been combined with an embedded atom (EAM) description of pure Al developed by Mendelev et al to formulate an Al–Si binary potential of the angular EAM type. The Al–Si potential reproduces quite well the experimental enthalpy of mixing in the liquid. It also predicts an Al–Si phase diagram with a eutectic concentration for the liquid that agrees with experimental values within 4 at% and a eutectic temperature that differs from experimental values by just 13 K. read less NOT USED (high confidence) C. Zou, S. Raman, and A. V. van Duin, “Large-scale reactive molecular dynamics simulation and kinetic modeling of high-temperature pyrolysis of the Gloeocapsomorphaprisca microfossils.,” The journal of physical chemistry. B. 2014. link Times cited: 22 Abstract: The ability to predict accurately the thermal conversion of … read moreAbstract: The ability to predict accurately the thermal conversion of complex carbonaceous materials is of value in both petroleum exploration and refining operations. Modeling the thermal cracking of kerogen under basinal heating conditions improves the predrill prediction of oil and gas yields and quality, thereby ultimately lowering the exploration risk. Modeling the chemical structure and reactivity of asphaltene from petroleum vacuum residues enables prediction of coke formation and properties in refinery processes, thereby lowering operating cost. The chemical structure-chemical yield modeling (CS-CYM) developed by Freund et al. is more rigorous, time-consuming, and requires a great deal of chemical insight into reaction network and reaction kinetics. The present work explores the applicability of a more fundamental atomistic simulation using the quantum mechanically based reactive force field to predict the product yield and overall kinetics of decomposition of two biopolymers, namely, the Kukersite and Gutternberg. Reactive molecular dynamics (RMD) simulations were performed on systems consisting of 10(4) to 10(5) atoms at different densities and temperatures to derive the overall kinetic parameters and a lumped kinetic model for pyrolysis. The kinetic parameters derived from the simulated pyrolysis of an individual component and the mixture of all four components in Guttenberg reveal the role of cross-talk between the fragments and enhanced reactivity of component A by radicals from other components. The Arrhenius extrapolation of the model yields reasonable prediction for the overall barrier for cracking. Because simulations were run at very high temperature (T > 1500 K) to study cracking within the simulation time of up to 1 ns, it, however, led to the entropically favored ethylene formation as a dominant decomposition route. Future work will focus on evaluating the applicability of accelerated reactive MD approaches to study cracking. read less NOT USED (high confidence) K. Nordlund, C. Björkas, T. Ahlgren, A. Lasa, and A. Sand, “Multiscale modelling of plasma–wall interactions in fusion reactor conditions,” Journal of Physics D: Applied Physics. 2014. link Times cited: 58 Abstract: The interaction of fusion reactor plasma with the material o… read moreAbstract: The interaction of fusion reactor plasma with the material of the first wall involves a complex multitude of interlinked physical and chemical effects. Hence, modern theoretical treatment of it relies to a large extent on multiscale modelling, i.e. using different kinds of simulation approaches suitable for different length and time scales in connection with each other. In this review article, we overview briefly the physics and chemistry of plasma–wall interactions in tokamak-like fusion reactors, and present some of the most commonly used material simulation approaches relevant for the topic. We also give summaries of recent multiscale modelling studies of the effects of fusion plasma on the modification of the materials of the first wall, especially on swift chemical sputtering, mixed material formation and hydrogen isotope retention in tungsten. read less NOT USED (high confidence) A. Galashev and V. Polukhin, “Computer analysis of the stability of copper films on graphene,” Russian Journal of Physical Chemistry A. 2014. link Times cited: 15 NOT USED (high confidence) A. Verkhovtsev, S. Schramm, and A. Solov’yov, “Molecular dynamics study of the stability of a carbon nanotube atop a catalytic nanoparticle,” The European Physical Journal D. 2014. link Times cited: 23 NOT USED (high confidence) Y. Magnin, G. D. Förster, F. Rabilloud, F. Calvo, A. Zappelli, and C. Bichara, “Thermal expansion of free-standing graphene: benchmarking semi-empirical potentials,” Journal of Physics: Condensed Matter. 2014. link Times cited: 55 Abstract: The thermodynamical properties of free-standing graphene hav… read moreAbstract: The thermodynamical properties of free-standing graphene have been investigated under constant zero pressure as a function of temperature using Monte Carlo simulations. A variety of atomistic models have been used, including the simple three-body Stillinger potential and a series of bond-order many-body potentials based on the Tersoff–Brenner seminal models, with recent reparametrizations dedicated to graphene, extensions to medium-range or long-range dispersion corrections. In addition, we have also tested a tight-binding potential in the fourth-moment approximation. The simulations reveal significant discrepancies in the in-plane lattice parameter and the thermal expansion coefficient, which despite showing monotonically increasing variations with temperature, can be positive, negative or change sign at moderate temperature depending on the potential. Comparison with existing experimental and theoretical data obtained from complementary approaches indicates that empirical potentials limited to nearest-neighbour interactions give rather dispersed results, and that van der Waals corrections generally tend to flatten the variations of the in-plane lattice constant, in contradiction with experiment. Only the medium-range corrected potentials of Los and Fasolino, as well as the tight-binding model in the fourth-moment approximation, are reasonably close to the reference results near room temperature. Our results suggest that classical potentials should be used with caution for thermal properties. read less NOT USED (high confidence) D. Feng, Y. Feng, and X. Zhang, “Numerical Study of Thermal Conductivities of Carbon-Based Mesoporous Composites,” International Journal of Thermophysics. 2014. link Times cited: 4 NOT USED (high confidence) Y. Wang, A. Vallabhaneni, B. Qiu, and X. Ruan, “Two-Dimensional Thermal Transport in Graphene: A Review of Numerical Modeling Studies,” Nanoscale and Microscale Thermophysical Engineering. 2014. link Times cited: 54 Abstract: This article reviews recent numerical studies of thermal tra… read moreAbstract: This article reviews recent numerical studies of thermal transport in graphene, with a focus on molecular dynamics simulation, the atomistic Green’s function method, and the phonon Boltzmann transport equation method. The mode-wise phonon contribution to the intrinsic thermal conductivity (κ) of graphene and the effects of extrinsic mechanisms—for example, substrate, isotope, impurities, and defects—on κ are discussed. We also highlight the insights from numerical studies aimed at bridging the gaps between 1D, 2D, and 3D thermal transport in carbon nanotubes/graphene nanoribbons, graphene, and graphite. Numerical studies on thermal transport across the interface between graphene and other materials and nonlinear thermal transport phenomena such as thermal rectification and negative differential thermal resistance are also reviewed. read less NOT USED (high confidence) Y. Cong, J. Yvonnet, and H. Zahrouni, “Simulation of instabilities in thin nanostructures by a perturbation approach,” Computational Mechanics. 2014. link Times cited: 2 NOT USED (high confidence) K. Xia, H. Zhan, Y. Wei, and Y. T. Gu, “Tensile properties of a boron/nitrogen-doped carbon nanotube–graphene hybrid structure,” Beilstein Journal of Nanotechnology. 2014. link Times cited: 25 Abstract: Summary Doping is an effective approach that allows for the … read moreAbstract: Summary Doping is an effective approach that allows for the intrinsic modification of the electrical and chemical properties of nanomaterials. Recently, a graphene and carbon nanotube hybrid structure (GNHS) has been reported, which extends the excellent properties of carbon-based materials to three dimensions. In this paper, we carried out a first-time investigation on the tensile properties of the hybrid structures with different dopants. It is found that with the presence of dopants, the hybrid structures usually exhibit lower yield strength, Young’s modulus, and earlier yielding compared to that of a pristine hybrid structure. For dopant concentrations below 2.5% no significant reduction of Young’s modulus or yield strength could be observed. For all considered samples, the failure is found to initiate at the region where the nanotubes and graphene sheets are connected. After failure, monatomic chains are normally observed around the failure region. Dangling graphene layers without the separation of a residual CNT wall are found to adhere to each other after failure with a distance of about 3.4 Å. This study provides a fundamental understanding of the tensile properties of the doped graphene–nanotube hybrid structures, which will benefit the design and also the applications of graphene-based hybrid materials. read less NOT USED (high confidence) E. Mainini and U. Stefanelli, “Crystallization in Carbon Nanostructures,” Communications in Mathematical Physics. 2014. link Times cited: 57 NOT USED (high confidence) S. D. Nath and S.-G. Kim, “Study of the Nanomechanics of CNTs under Tension by Molecular Dynamics Simulation Using Different Potentials,” International Scholarly Research Notices. 2014. link Times cited: 21 Abstract: At four different strain rates, the tensile stress strain re… read moreAbstract: At four different strain rates, the tensile stress strain relationship of single-walled 12-12 CNT with aspect ratio 9.1 obtained by Rebo potential (Brenner, 1990), Airebo potential (Stuart et al., 2000), and Tersoff potential (Tersoff, 1988) is compared with that of Belytschko et al. (2002) to validate the present model. Five different empirical potentials such as Rebo potential (Brenner, 1990), Rebo potential (Brenner et al., 2002), Inclusion LJ with Rebo potential (Brenner, 1990), Airebo potential (Stuart et al., 2000), and Tersoff potential (Tersoff, 1988) are used to simulate CNT subjected to axial tension differing its geometry at high strain rate. In Rebo potential (Mashreghi and Moshksar, 2010) only bond-order term is used and in Rebo potential (Brenner et al., 2002) torsional term is included with the bond-order term. At high strain rate the obtained stress strain relationships of CNTs subjected to axial tension differing its geometries using five different potentials are compared with the published results and from the comparison of the results, the drawback of the published results and limitations of different potentials are evaluated and the appropriate potential is selected which is the best among all other potentials to study the elastic, elastic-plastic properties of different types of CNTs. The present study will help a new direction to get reliable elastic, elastic-plastic properties of CNTs at different strain rates. Effects of long range Van der Waals interaction and torsion affect the elastic, elastic-plastic properties of CNTs and why these two effects are really needed to consider in bond-order Rebo potential (Brenner, 1990) to get reliable elastic, elastic-plastic properties of CNTs is also discussed. Effects of length-to-diameter ratio, layering of CNTs, and different empirical potentials on the elastic, elastic-plastic properties of CNTs are discussed in graphical and tabular forms with published results as a comparative manner to understand the nanomechanics of CNTs under tension using molecular dynamics simulation. read less NOT USED (high confidence) R. N. Salaway and L. Zhigilei, “Molecular dynamics simulations of thermal conductivity of carbon nanotubes: Resolving the effects of computational parameters,” International Journal of Heat and Mass Transfer. 2014. link Times cited: 97 NOT USED (high confidence) K. Nordlund and F. Djurabekova, “Multiscale modelling of irradiation in nanostructures,” Journal of Computational Electronics. 2014. link Times cited: 42 NOT USED (high confidence) K. Li, W. Yang, J. Wei, S. Du, and Y. Li, “Modeling of metal–oxide semiconductor: Analytical bond-order potential for cupric oxide,” Chinese Physics B. 2014. link Times cited: 0 Abstract: Atomistic potentials for cupric element and cupric oxide are… read moreAbstract: Atomistic potentials for cupric element and cupric oxide are derived based on the analytical bond-order scheme that was presented by Brenner [Brenner D W, "Erratum: Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films", Phys. Rev. B 1992, 46 1948]. In this paper, for the pure cupric element, the energy and structural parameters for several bulk phases as well as dimmer structure are well reproduced. The reference data are taken from our density functional theory calculations and the available experiments. The model potential also provides a good description of the bulk properties of various solid structures of cupric oxide compound structures, including cohesive energies, lattice parameters, and elastic constants. read less NOT USED (high confidence) D. T. Ho, S. D. Park, S. Y. Kwon, K. Park, and S. Y. Kim, “Negative Poisson’s ratios in metal nanoplates,” Nature Communications. 2014. link Times cited: 112 NOT USED (high confidence) X. A. Deng, Y. Song, J. Li, and Y. Pu, “Parametrization of the Stillinger-Weber potential for Si/N/H system and its application to simulations of silicon nitride film deposition with SiH4/NH3,” Journal of Applied Physics. 2014. link Times cited: 1 Abstract: We determined the Stillinger-Weber interatomic potential par… read moreAbstract: We determined the Stillinger-Weber interatomic potential parameters for Si/N/H system based on first principles density functional calculations. This new potential can be used to perform classical molecular dynamics simulation for silicon nitride deposition on Si substrate. During the first principles calculations, cluster models have been carefully and systematically chosen to make sampling of the interatomic potential supersurface more thoroughly. Global optimization method was used to fit the ab initio data into Stillinger-Weber form. We used a recursive method to perform the classical molecular dynamics simulations for silicon nitride (SiN) film growth on Si substrate with SiH4/NH3 gas mixtures. During the simulation, we could clearly observe the silicon nitride film growth progress. In this paper, we present the details of potential derivation and simulation results with different SiH4:NH3 ratios. It is demonstrated that this new potential is suitable to describe the surface reactions of the Si/N/H s... read less NOT USED (high confidence) T. Han, Y. Luo, and C. Wang, “Effects of temperature and strain rate on the mechanical properties of hexagonal boron nitride nanosheets,” Journal of Physics D: Applied Physics. 2014. link Times cited: 106 Abstract: The effect of temperature and strain rate on mechanical prop… read moreAbstract: The effect of temperature and strain rate on mechanical properties remains an open topic in research of hexagonal boron nitride (h-BN) nanosheets. To examine these fundamental issues we have performed molecular dynamics simulations to record the stress–strain curves in tensile tests and measure Young's modulus, fracture strength and fracture strain in armchair and zigzag directions. Comparing the results obtained at different temperatures and strain rates we have quantified the effects of the two factors on the tensile properties of the h-BN nanosheets. The influence of crystal orientation is also examined in the present study. It is found that the h-BN nanosheets are basically an anisotropic material whose tensile properties vary substantially with temperature and strain rate. In particular, a yielding platform is observed for the h-BN nanomaterial at relatively low temperature. read less NOT USED (high confidence) Y. Li, T. Liang, S. Sinnott, and S. Phillpot, “A charge-optimized many-body potential for the U–UO2–O2 system,” Journal of Physics: Condensed Matter. 2013. link Times cited: 22 Abstract: Building on previous charge-optimized many-body (COMB) poten… read moreAbstract: Building on previous charge-optimized many-body (COMB) potentials for metallic α-U and gaseous O2, we have developed a new potential for UO2, which also allows the simulation of U–UO2–O2 systems. The UO2 lattice parameter, elastic constants and formation energies of stoichiometric and non-stoichiometric intrinsic defects are well reproduced. Moreover, this is the first rigid-ion potential that produces the correct deviation of the Cauchy relation, as well as the first classical interatomic potential that is able to determine the defect energies of non-stoichiometric intrinsic point defects in UO2 with an appropriate reference state. The oxygen molecule interstitial in the α-U structure is shown to decompose, with some U–O bonds approaching the natural bond length of perfect UO2. Finally, we demonstrate the capability of this COMB potential to simulate a complex system by performing a simulation of the α-U + O2 → UO2 phase transformation. We also identify a possible mechanism for uranium oxidation and the orientation of the resulting fluorite UO2 structure relative to the coordinate system of orthorhombic α-U. read less NOT USED (high confidence) C. Becker, F. Tavazza, Z. Trautt, and R. B. D. Macedo, “Considerations for choosing and using force fields and interatomic potentials in materials science and engineering,” Current Opinion in Solid State & Materials Science. 2013. link Times cited: 196 NOT USED (high confidence) D. Belashchenko, “Computer simulation of liquid metals,” Physics—Uspekhi. 2013. link Times cited: 84 Abstract: Methods for and the results of the computer simulation of li… read moreAbstract: Methods for and the results of the computer simulation of liquid metals are reviewed. Two basic methods, classical molecular dynamics with known interparticle potentials and the ab initio method, are considered. Most attention is given to the simulated results obtained using the embedded atom model (EAM). The thermodynamic, structural, and diffusion properties of liquid metal models under normal and extreme (shock) pressure conditions are considered. Liquid-metal simulated results for the Groups I–IV elements, a number of transition metals, and some binary systems (Fe–C, Fe–S) are examined. Possibilities for the simulation to account for the thermal contribution of delocalized electrons to energy and pressure are considered. Solidification features of supercooled metals are also discussed. read less NOT USED (high confidence) A. Mrugalla and J. Schnack, “Classical molecular dynamics investigations of biphenyl-based carbon nanomembranes,” Beilstein Journal of Nanotechnology. 2013. link Times cited: 9 Abstract: Summary Background: Free-standing carbon nanomembranes (CNM)… read moreAbstract: Summary Background: Free-standing carbon nanomembranes (CNM) with molecular thickness and macroscopic size are fascinating objects both for fundamental reasons and for applications in nanotechnology. Although being made from simple and identical precursors their internal structure is not fully known and hard to simulate due to the large system size that is necessary to draw definite conclusions. Results: We performed large-scale classical molecular dynamics investigations of biphenyl-based carbon nanomembranes. We show that one-dimensional graphene-like stripes constitute a highly symmetric quasi one-dimensional energetically favorable ground state. This state does not cross-link. Instead cross-linked structures are formed from highly excited precursors with a sufficient amount of broken phenyls. Conclusion: The internal structure of the CNM is very likely described by a disordered metastable state which is formed in the energetic initial process of electron irradiation and depends on the process of relaxation into the sheet phase. read less NOT USED (high confidence) T. Yoon, T. Lim, T. Min, S. Hung, N. Jakse, and S. Lai, “Epitaxial growth of graphene on 6H-silicon carbide substrate by simulated annealing method.,” The Journal of chemical physics. 2013. link Times cited: 15 Abstract: We grew graphene epitaxially on 6H-SiC(0001) substrate by th… read moreAbstract: We grew graphene epitaxially on 6H-SiC(0001) substrate by the simulated annealing method. The mechanisms that govern the growth process were investigated by testing two empirical potentials, namely, the widely used Tersoff potential [J. Tersoff, Phys. Rev. B 39, 5566 (1989)] and its more refined version published years later by Erhart and Albe [Phys. Rev. B 71, 035211 (2005)]. Upon contrasting the results obtained by these two potentials, we found that the potential proposed by Erhart and Albe is generally more physical and realistic, since the annealing temperature at which the graphene structure just coming into view at approximately 1200 K is unambiguously predicted and close to the experimentally observed pit formation at 1298 K within which the graphene nucleates. We evaluated the reasonableness of our layers of graphene by calculating carbon-carbon (i) average bond-length, (ii) binding energy, and (iii) pair correlation function. Also, we compared with related experiments the various distance of separation parameters between the overlaid layers of graphene and substrate surface. read less NOT USED (high confidence) Y. D. Fomin, “Molecular dynamics simulation of benzene in graphite and amorphous carbon slit pores,” Journal of Computational Chemistry. 2013. link Times cited: 7 Abstract: It is well known that confining a liquid into a pore strongl… read moreAbstract: It is well known that confining a liquid into a pore strongly alters the liquid behavior. Investigations of the effect of confinement are of great importance for many scientific and technological applications. Here, we present a study of the behavior of benzene confined in carbon slit pores. Two types of pores are considered–graphite and amorphous carbon ones. We show that the effect of different pore structure is of crucial importance for the benzene behavior. © 2013 Wiley Periodicals, Inc. read less NOT USED (high confidence) J. Purton, J. Crabtree, and S. C. Parker, “DL_MONTE: a general purpose program for parallel Monte Carlo simulation,” Molecular Simulation. 2013. link Times cited: 69 Abstract: Monte Carlo (MC) represents a powerful simulation tool that … read moreAbstract: Monte Carlo (MC) represents a powerful simulation tool that can be usefully applied to calculating thermodynamic data. However, such codes are normally bespoke for a particular problem and not widely applicable. In this paper, we report a new flexible and versatile MC code called DL_MONTE, which builds on the highly successful DL_POLY molecular dynamics code to allow the treatment of polymers, minerals, semiconductors and metals in a range of applications on both workstations and highly parallel supercomputers. In addition, to describe its features, we used a recent work to model the phase diagrams of mixed metal oxide nanoparticles using MgO/MnO as an illustration, adsorption of water at the MgO surface and, finally, the adsorption isotherms of CO2 in different microporous zeolites. The results demonstrate the flexibility of the methodology and how semi-grand and grand canonical MC can be readily applied. read less NOT USED (high confidence) A. Galashev and V. Polukhin, “Computer simulation of thin nickel films on single-layer graphene,” Physics of the Solid State. 2013. link Times cited: 14 NOT USED (high confidence) J. Yang, C. Mao, X. Li, and C. Liu, “On the Cauchy-Born approximation at finite temperature for alloys,” Discrete & Continuous Dynamical Systems - B. 2013. link Times cited: 4 NOT USED (high confidence) I. Leven, I. Azuri, L. Kronik, and O. Hod, “Inter-layer potential for hexagonal boron nitride.,” The Journal of chemical physics. 2013. link Times cited: 63 Abstract: A new interlayer force-field for layered hexagonal boron nit… read moreAbstract: A new interlayer force-field for layered hexagonal boron nitride (h-BN) based structures is presented. The force-field contains three terms representing the interlayer attraction due to dispersive interactions, repulsion due to anisotropic overlaps of electron clouds, and monopolar electrostatic interactions. With appropriate parameterization, the potential is able to simultaneously capture well the binding and lateral sliding energies of planar h-BN based dimer systems as well as the interlayer telescoping and rotation of double walled boron-nitride nanotubes of different crystallographic orientations. The new potential thus allows for the accurate and efficient modeling and simulation of large-scale h-BN based layered structures. read less NOT USED (high confidence) K. Henriksson, C. Björkas, and K. Nordlund, “Atomistic simulations of stainless steels: a many-body potential for the Fe–Cr–C system,” Journal of Physics: Condensed Matter. 2013. link Times cited: 65 Abstract: Stainless steels found in real-world applications usually ha… read moreAbstract: Stainless steels found in real-world applications usually have some C content in the base Fe–Cr alloy, resulting in hard and dislocation-pinning carbides—Fe3C (cementite) and Cr23C6—being present in the finished steel product. The higher complexity of the steel microstructure has implications, for example, for the elastic properties and the evolution of defects such as Frenkel pairs and dislocations. This makes it necessary to re-evaluate the effects of basic radiation phenomena and not simply to rely on results obtained from purely metallic Fe–Cr alloys. In this report, an analytical interatomic potential parameterization in the Abell–Brenner–Tersoff form for the entire Fe–Cr–C system is presented to enable such calculations. The potential reproduces, for example, the lattice parameter(s), formation energies and elastic properties of the principal Fe and Cr carbides (Fe3C, Fe5C2, Fe7C3, Cr3C2, Cr7C3, Cr23C6), the Fe–Cr mixing energy curve, formation energies of simple C point defects in Fe and Cr, and the martensite lattice anisotropy, with fair to excellent agreement with empirical results. Tests of the predictive power of the potential show, for example, that Fe–Cr nanowires and bulk samples become elastically stiffer with increasing Cr and C concentrations. High-concentration nanowires also fracture at shorter relative elongations than wires made of pure Fe. Also, tests with Fe3C inclusions show that these act as obstacles for edge dislocations moving through otherwise pure Fe. read less NOT USED (high confidence) Y. Sasajima, J. Murakami, and A. M. Tamidi, “Computer Simulation of Precipitation Process in Si/Ge Amorphous Multi-Layer Films: Effects of Cu Addition,” Materials Transactions. 2013. link Times cited: 1 Abstract: We have simulated the precipitation process in an amorphous … read moreAbstract: We have simulated the precipitation process in an amorphous Si/Ge multi-layer film, with and without Cu addition, by a molecular dynamics method. Four specimens were prepared for this study: Si/Ge layers, Si/(Ge + Cu) layers, (Si + Cu)/(Ge + Cu) layers and Si/Cu/Ge/ Cu layers. After the multi-layered films became amorphous, we tracked the movement of individual atoms at 1000K, the annealing temperature. When Cu was present in the Ge layer or both the Si and Ge layers, the precipitation of nano-clusters was less than that in Cu-free Si/Ge layers. We think that the Cu atoms block the precipitation and make the Si and Ge become more stable in the amorphous state. If Cu atoms are note present in a layer, however, like the Si layer in Si/(Ge + Cu) and Si/Cu/Ge/Cu specimens, the precipitation of nano-clusters in the Cu-free layer is enhanced. Therefore we conclude that precipitation of nano-clusters in Si/Ge layers can be controlled by how Cu atoms are added to the amorphous Si/Ge system, and that this will improve the thermoelectric performance. [doi:10.2320/matertrans.M2013191] read less NOT USED (high confidence) A. P. Jones, J. Crain, F. Cipcigan, V. Sokhan, M. Modani, and G. Martyna, “Electronically coarse-grained molecular dynamics using quantum Drude oscillators,” Molecular Physics. 2013. link Times cited: 14 Abstract: Standard molecular dynamics (MD) simulations generally make … read moreAbstract: Standard molecular dynamics (MD) simulations generally make use of a basic description of intermolecular forces which consists of fixed, pairwise, atom-centred Coulomb, van der Waals and short-range repulsive terms. Important interactions such as many-body polarisation and many-body dispersion which are sensitive to changes in the environment are usually neglected, and their effects treated effectively within mean-field approximations to reproduce a single thermodynamic state point or physical environment. This leads to difficulties in modelling the complex interfaces of interest today where the behaviour may be quite different from the regime of parameterisation. Here, we describe the construction and properties of a Gaussian coarse-grained electronic structure, which naturally generates many-body polarisation and dispersion interactions. The electronic structure arises from a fully quantum mechanical treatment of a set of distributed quantum Drude oscillators (QDOs), harmonic atoms which interact with each other and other moieties via electrostatic (Coulomb) interactions; this coarse-grained approach is capable of describing many-body polarisation and dispersion but not short-range interactions which must be parametrised. We describe how on-the-fly forces due to this exchange-free Gaussian model may be generated with linear scale in the number of atoms in the system using an adiabatic path integral molecular dynamics for quantum Drude oscillators technique (APIMD-QDO). We demonstrate the applicability of the QDO approach to realistic systems via a study of the liquid–vapour interface of water. read less NOT USED (high confidence) T. Aoki, “Molecular dynamics simulations of cluster impacts on solid targets: implantation, surface modification, and sputtering,” Journal of Computational Electronics. 2013. link Times cited: 31 NOT USED (high confidence) A. Galashev and S. Dubovik, “Molecular dynamics simulation of compression of single-layer graphene,” Physics of the Solid State. 2013. link Times cited: 7 NOT USED (high confidence) R. Bingham and P. Ballone, “Energy, structure and vibrational modes of small water clusters by a simple many-body potential mimicking polarisation effects,” Molecular Physics. 2013. link Times cited: 4 Abstract: An empirical many-body model potential able to mimic polaris… read moreAbstract: An empirical many-body model potential able to mimic polarisation effects is applied to compute cohesive, structural and vibrational properties of water clusters with up to 12 H2O molecules. The model introduces local coordination functions to account for the variation of charges and other intra- and inter-molecular force constants upon formation of hydrogen bonds among water molecules. The potential is tuned to fit the results of state of the art density functional computations, and it is shown to accurately reproduce cohesive energies, bond lengths and vibrational properties of clusters. Moreover, it reproduces the marked increase of the molecular dipole moment with increasing water–water coordination. At variance from traditional polarisable models, the energy is an explicit function of the atomic coordinates, and does not require the minimisation of the electrostatic energy or the equalisation of the electron chemical potential, and thus is suitable for large-scale simulations in materials science and in bio-chemistry/bio-physics. read less NOT USED (high confidence) Z. Li, N. Mathew, and R. C. Picu, “Dependence of Peierls stress on lattice strains in silicon,” Computational Materials Science. 2013. link Times cited: 7 NOT USED (high confidence) P. Käshammer and T. Sinno, “Interactions of twin boundaries with intrinsic point defects and carbon in silicon,” Journal of Applied Physics. 2013. link Times cited: 22 Abstract: Although multicrystalline silicon (mc-Si) is currently the m… read moreAbstract: Although multicrystalline silicon (mc-Si) is currently the most widely used material for fabricating photovoltaic cells, its electrical properties remain limited by several types of defects, which interact in complex ways that are not yet fully understood. A particularly important phenomenon is the interaction between grain boundaries and intrinsic point defects or impurity atoms, such as carbon, oxygen, nitrogen, and various types of metals. Here, we use empirical molecular dynamics to study the interactions of Σ3{111}, Σ9{221}, and Σ27{552} twin boundaries, which account for over 50% of all grain boundaries in mc-Si, with self-interstitials, vacancies, and substitutional carbon atoms. It is shown that twin boundary-point defect interaction energies increase with twinning order and that they are predominantly attractive. We also find that twin boundary interactions with substitutional carbon are highly spatially heterogeneous, exhibiting alternating repulsive-attractive regions that correlate strongly wi... read less NOT USED (high confidence) A. Marconnet, M. Panzer, and K. Goodson, “Thermal conduction phenomena in carbon nanotubes and related nanostructured materials,” Reviews of Modern Physics. 2013. link Times cited: 350 Abstract: The extremely high thermal conductivities of carbon nanotube… read moreAbstract: The extremely high thermal conductivities of carbon nanotubes have motivated a wealth of research. Progress includes innovative conduction metrology based on microfabricated platforms and scanning thermal probes as well as simulations exploring phonon dispersion and scattering using both transport theory and molecular dynamics. This article highlights these advancements as part of a detailed review of heat conduction research on both individual carbon nanotubes and nanostructured films consisting of arrays of nanotubes or disordered nanotube mats. Nanotube length, diameter, and chirality strongly influence the thermal conductivities of individual nanotubes and the transition from primarily diffusive to ballistic heat transport with decreasing temperature. A key experimental challenge, for both individual nanotubes and aligned films, is the separation of intrinsic and contact resistances. Molecular dynamics simulations have studied the impacts of specific types of imperfections on the nanotube conductance and its variation with length and chirality. While the properties of aligned films fall short of predictions based on individual nanotube data, improvements in surface engagement and postfabrication nanotube quality are promising for a variety of applications including mechanically compliant thermal contacts. read less NOT USED (high confidence) W. Li, L. Liang, S. Zhao, S. Zhang, and J. Xue, “Fabrication of nanopores in a graphene sheet with heavy ions: A molecular dynamics study,” Journal of Applied Physics. 2013. link Times cited: 60 Abstract: Molecular dynamics simulations were performed to study the f… read moreAbstract: Molecular dynamics simulations were performed to study the formation process of nanopores in a suspended graphene sheet irradiated by using energetic ions though a mask. By controlling the ion parameters including mass, energy, and incident angle, different kinds of topography were observed in the graphene sheet. Net-like defective structures with carbon atom chains can be formed at low ion fluences, which provide the possibility to functionalize the irradiated sample with subsequent chemical methods; finally a perfect nanopore with smooth edge appears when the ion fluence is high enough. We found that the dependence of ion damage efficiency on ion fluence, energy, and incident angle are different from that predicted by the semi-empirical model based on the binary-collision approximation, which results from the special structure of graphene. Our results demonstrate that it is feasible to fabricate controlled nanopores/nanostructures in graphene via heavy ion irradiation. read less NOT USED (high confidence) X. Li, M. Joe, A. Wang, and K.-R. Lee, “Stress reduction of diamond-like carbon by Si incorporation: A molecular dynamics study,” Surface & Coatings Technology. 2013. link Times cited: 24 NOT USED (high confidence) Y. Yang and W. Zhao, “Molecular dynamics simulation of the thermal-caused material removal process by the SPM-based electric nanofabrication,” 2013 13th IEEE International Conference on Nanotechnology (IEEE-NANO 2013). 2013. link Times cited: 0 Abstract: This paper intends to study the phenomena of thermal-caused … read moreAbstract: This paper intends to study the phenomena of thermal-caused material modifications in the principle of nanoscale electro spark during the SPM-based electric lithography. Since the direct observation of the electro spark process seems impossible in the nanoscale gap region, the molecular dynamics (MD) simulation method is applied to help investigate the influence of the thermal effect due to the Joule heating generated by the electro spark. The simplified heat source model is constructed based on the local temperature profile of the sample material beneath the tip, which is calculated through the Joule heating equation by the finite element method (FEM). The material removal process of local Cu and graphite sample subjected to the heat input is respectively simulated by the MD method to semi-quantitatively identify the thermal effect on the SPM-based electric nanofabrication results. read less NOT USED (high confidence) U. Monteverde, M. Migliorato, and D. Powell, “Atomistic modelling of elasticity and phonons in diamond and graphene,” 2013 13th International Conference on Numerical Simulation of Optoelectronic Devices (NUSOD). 2013. link Times cited: 0 Abstract: We present an atomistic interatomic potential that with a si… read moreAbstract: We present an atomistic interatomic potential that with a single set of parameters is able to accurately describe at the same time the elastic, vibrational and thermodynamics properties of semiconductors. We also show that the correct inclusion in the potential of short and long range interactions provides a model for the force field that accurately performs Static Dynamics and Molecular Dynamics. read less NOT USED (high confidence) D. Spiteri, J. Pomeroy, and M. Kuball, “Influence of microstructural defects on the thermal conductivity of GaN: A molecular dynamics study,” physica status solidi (b). 2013. link Times cited: 14 Abstract: The lattice thermal conductivity is known to depend on cryst… read moreAbstract: The lattice thermal conductivity is known to depend on crystal quality, but the reduction in thermal conductivity due to specific defects is presently unclear. Molecular dynamics simulations were used to investigate the impact of microstructural defects on the thermal conductivity of gallium nitride. The conductivity of a finite crystal was reduced to (39 ± 4)% by a screw dislocation density of 2.0 × 1013 cm−2 and to (51 ± 4)% by an edge dislocation of similar density, illustrating that the type of dislocation is important for thermal conductivity. The effect of stacking faults on thermal conductivity was also investigated. read less NOT USED (high confidence) L. Pereira, I. Savi’c, and D. Donadio, “Thermal conductivity of one-, two- and three-dimensional sp2 carbon,” New Journal of Physics. 2013. link Times cited: 27 Abstract: Carbon atoms can form structures in one, two and three dimen… read moreAbstract: Carbon atoms can form structures in one, two and three dimensions due to their unique chemical versatility. In terms of thermal conductivity, carbon polymorphs cover a wide range from very low values with amorphous carbon to very high values with diamond, carbon nanotubes and graphene. Schwarzites are a class of three-dimensional fully covalent sp2-bonded carbon polymorphs, with the same local chemical environment as graphene and carbon nanotubes, but negative Gaussian curvature. We calculate the thermal conductivity of a (10,0) carbon nanotube, graphene and two schwarzites with different curvature, by molecular dynamics simulations based on the Tersoff empirical potential. We find that schwarzites present a thermal conductivity two orders of magnitude smaller than nanotubes and graphene. The reason for such large difference is explained by anharmonic lattice dynamics calculations, which show that phonon group velocities and mean free paths are much smaller in schwarzites than in nanotubes and graphene. Their reduced thermal conductivity, in addition to tunable electronic properties, indicate that schwarzites could pave the way towards all-carbon thermoelectric technology with high conversion efficiency. read less NOT USED (high confidence) G. Norman and V. Stegailov, “Stochastic theory of the classical molecular dynamics method,” Mathematical Models and Computer Simulations. 2013. link Times cited: 137 NOT USED (high confidence) J. Zhang, C. Liu, and J. Fan, “Comparison of Cu thin films deposited on Si substrates with different surfaces and temperatures,” Applied Surface Science. 2013. link Times cited: 22 NOT USED (high confidence) J. Solomon, P. Chung, D. Srivastava, and E. F. Darve, “Method and Advantages of Genetic Algorithms in Parameterization of Interatomic Potentials: Metal-Oxides,” arXiv: Materials Science. 2013. link Times cited: 14 NOT USED (high confidence) A. Matsuda, Y. Nakakubo, Y. Takao, K. Eriguchi, and K. Ono, “Atomistic simulations of plasma process-induced Si substrate damage - Effects of substrate bias-power frequency,” Proceedings of 2013 International Conference on IC Design & Technology (ICICDT). 2013. link Times cited: 9 Abstract: Plasma-induced defect generation process in crystalline Si s… read moreAbstract: Plasma-induced defect generation process in crystalline Si structure was simulated by classical molecular dynamics simulations. Energy distribution functions of Ar and Cl ions incident on the Si surface (IEDF) were implemented to predict the impacts on the defect generation processes in present-day plasma process equipments. The damaged-layer thickness was confirmed to be a weak function of IEDF, which are consistent with a binary-collision-based range model and experimental results. In the case of “fin-gate structure”, the simulation results predict that the sidewall may be damaged not by the incident angular distribution of ions but by the straggling of high-energy ions near the reaction surface, which leads to an on-current degradation of FinFETs. read less NOT USED (high confidence) J. C. Castro-Palacio, L. Velazquez-Abad, M. Fernández, and J. Q. Cuador-Gil, “Molecular dynamics study of one dimensional nanoscale Si/SiO2 interfaces,” The European Physical Journal D. 2013. link Times cited: 1 NOT USED (high confidence) K. Tuleubekov, K. Volokh, K. Volokh, H. Stolarski, and S. Mogilevskaya, “Strength of graphene in biaxial tension,” European Journal of Mechanics A-solids. 2013. link Times cited: 7 NOT USED (high confidence) W. Song and S.-jin Zhao, “Development of the ReaxFF reactive force field for aluminum–molybdenum alloy,” Journal of Materials Research. 2013. link Times cited: 10 Abstract: We have developed a reactive force field within the ReaxFF f… read moreAbstract: We have developed a reactive force field within the ReaxFF framework to accurately describe reactions involving aluminum–molybdenum alloy, which are part parameters of Al–O–Mo ternary system metastable intermolecular composites. The parameters are optimized from a training set, whose data come from density functional theory (DFT) calculations and experimental value, such as heat of formation, geometry data, and equation of states, which are reproduced well by ReaxFF. Body-centered cubic molybdenum’s surface energy, vacancy formation, and two transformational paths, Bain and trigonal paths are calculated to validate the ReaxFF ability describing the defects and deformations. Some structures’ elastic constant and phonon are calculated by DFT and ReaxFF to predict the structures’ mechanics and kinetic stability. All those results indicate that the fitted parameters can describe the energy difference of various structures under various circumstances and generally represent the diffusion property but cannot reproduce the elasticity and phonon spectra so well. read less NOT USED (high confidence) S. Hollerer and C. Celigoj, “Buckling analysis of carbon nanotubes by a mixed atomistic and continuum model,” Computational Mechanics. 2013. link Times cited: 20 NOT USED (high confidence) V. L. Levshunova, G. Pokhil, and D. Tetelbaum, “Waveguide effect for hypersonic waves in silicon with dislocations,” Journal of Surface Investigation. X-ray, Synchrotron and Neutron Techniques. 2013. link Times cited: 2 NOT USED (high confidence) J. Ding, L. Zhang, Y. Zhang, and K. Han, “A reactive molecular dynamics study of n-heptane pyrolysis at high temperature.,” The journal of physical chemistry. A. 2013. link Times cited: 104 Abstract: n-Heptane is the most important straight chain paraffin in t… read moreAbstract: n-Heptane is the most important straight chain paraffin in the fossil-fuel industry. In this work, pyrolysis of n-heptane at high temperature is investigated by a series of ReaxFF based reactive molecular dynamic simulations. The pyrolysis correlated intermediate reactions, important product/intermediate distributions, and corresponding kinetics behaviors are systematically analyzed at atomistic level. The results indicate that the entire pyrolysis process is radical-dominated. The unimolecular dissociation is the main pathway of n-heptane decomposition. Initiation of the decomposition is mainly through C-C bond fission. Central C-C bonds would dissociate prior to the terminal ones. Besides, the Rice-Kossiakoff theory is proved for the pyrolysis of n-heptane at the atomistic level. To give a better description of the pyrolysis behavior, some alkane related intermolecular reactions should be considered in the mechanism. The apparent activation energy extracted from the present simulations is 43.02-54.49 kcal/mol in the temperature range 2400-3000 K, which is reasonably consistent with the experimental results. read less NOT USED (high confidence) J. Yan, K. Liew, and L.-. He, “Ultra-sensitive analysis of a cantilevered single-walled carbon nanocone-based mass detector,” Nanotechnology. 2013. link Times cited: 33 Abstract: The ultra-sensitivity of mass detectors using individual can… read moreAbstract: The ultra-sensitivity of mass detectors using individual cantilevered single-walled carbon nanocone (SWCNC) resonators is first investigated. A higher-order gradient theory, derived at the atomic level, is applied for modeling SWCNC resonators. Numerical simulations using a mesh-free computational framework based on moving Kriging interpolation are conducted to investigate the mass sensitivity of cantilevered SWCNC resonators with extra mass loading as well as with equivalent single-walled carbon nanotube (SWCNT) resonators. Comparison of the magnitude of resonant frequency shifts, the key criterion for mass sensitivity, of these two kinds of resonators demonstrates a far higher mass sensitivity for SWCNC resonators than for SWCNT resonators, thus suggesting a new method for ultra-sensitive mass detection via SWCNC resonators. The dependence of the mass sensitivity of SWCNC resonators on height and top radii has been examined. A reduction in the height of SWCNC resonators gives rise to a considerable increase in mass sensitivity. The mass sensitivity of a 6 nm high SWCNC resonator can even reach a level of 10−22 g. It is noteworthy that the top radii of SWCNC resonators have a slight effect on frequency shifts. Another interesting observed phenomenon is that a deviation in the height of 19.2° SWCNC resonators leads to little loss in precision of mass detection when the attached mass is smaller than 10−20 g. This superior characteristic indicates that SWCNC-based mass detectors have great potential in practical applications. read less NOT USED (high confidence) E. Despiau-Pujo, A. Davydova, G. Cunge, L. Delfour, L. Magaud, and D. Graves, “Elementary processes of H2 plasma-graphene interaction: A combined molecular dynamics and density functional theory study,” Journal of Applied Physics. 2013. link Times cited: 35 Abstract: Elementary interactions between H atoms and monolayer graphe… read moreAbstract: Elementary interactions between H atoms and monolayer graphene are investigated using classical molecular dynamics (CMD) and density functional theory (DFT). C-H interatomic potential curves and associated energy barriers are reported depending on the H impact position (top, bridge, hollow, vacancy, or edge sites of graphene nanoribbons). Chemisorption of atomic hydrogen and formation of molecular hydrogen from chemisorbed H states on graphene are examined. The influence of graphene temperature and incident species energy on adsorption, reflection, and penetration mechanisms is also presented. Except for impacts at graphene nanoribbon (GNR) edges or at defect locations, H atoms are shown to experience a repulsive force due to delocalized π-electrons which prevents any species with less than 0.4-0.6 eV to chemisorb on the graphene surface. C-H bond formation requires a local sp2-sp3 rehybridization resulting in structural changes of the graphene sample. Chemisorption sites with deep potential wells and no ... read less NOT USED (high confidence) T. Järvi, L. Mayrhofer, J. Polvi, K. Nordlund, L. Pastewka, and M. Moseler, “Adaptive molecular decomposition: large-scale quantum chemistry for liquids.,” The Journal of chemical physics. 2013. link Times cited: 1 Abstract: We present a linear-scaling method based on self-consistent … read moreAbstract: We present a linear-scaling method based on self-consistent charge non-orthogonal tight-binding. Linear scaling is achieved using a many-body expansion, which is adjusted dynamically to the instantaneous molecular configuration of a liquid. The method is capable of simulating liquids over large length and time scales, and also handles reactions correctly. Benchmarking on typical carbonate electrolytes used in Li-ion batteries displays excellent agreement with results from full tight-binding calculations. The decomposition slightly breaks the Hellmann-Feynman theorem, which is demonstrated by application to water. However, an additional correction also enables dynamical simulation in this case. read less NOT USED (high confidence) I. Ostanin, R. Ballarini, D. Potyondy, and T. Dumitricǎ, “A distinct element method for large scale simulations of carbon nanotube assemblies,” Journal of The Mechanics and Physics of Solids. 2013. link Times cited: 66 NOT USED (high confidence) M. H. Khadem and A. Wemhoff, “Thermal conductivity predictions of herringbone graphite nanofibers using molecular dynamics simulations.,” The Journal of chemical physics. 2013. link Times cited: 7 Abstract: Non-equilibrium molecular dynamics (NEMD) simulations are us… read moreAbstract: Non-equilibrium molecular dynamics (NEMD) simulations are used to investigate the thermal conductivity of herringbone graphite nanofibers (GNFs) at room temperature by breaking down the axial and transverse conductivity values into intralayer and interlayer components. The optimized Tersoff potential is used to account for intralayer carbon-carbon interactions while the Lennard-Jones potential is used to model the interlayer carbon-carbon interactions. The intralayer thermal conductivity of the graphene layers near room temperature is calculated for different crease angles and number of layers using NEMD with a constant applied heat flux. The edge effect on a layer's thermal conductivity is investigated by computing the thermal conductivity values in both zigzag and armchair directions of the heat flow. The interlayer thermal conductivity is also predicted by imposing hot and cold Nosé-Hoover thermostats on two layers. The limiting case of a 90° crease angle is used to compare the results with those of single-layer graphene and few-layer graphene. The axial and transverse thermal conductivities are then calculated using standard trigonometric conversions of the calculated intralayer and interlayer thermal conductivities, along with calculations of few-layer graphene without a crease. The results show a large influence of the crease angle on the intralayer thermal conductivity, and the saturation of thermal conductivity occurs when number of layers is more than three. The axial thermal conductivity, transverse thermal conductivity in the crease direction, and transverse thermal conductivity normal to the crease for the case of a five-layer herringbone GNF with a 45° crease angle are calculated to be 27 W∕m K, 263 W∕m K, and 1500 W∕m K, respectively, where the axial thermal conductivity is in good agreement with experimental measurements. read less NOT USED (high confidence) Z. Li and R. C. Picu, “Shuffle-glide dislocation transformation in Si,” Journal of Applied Physics. 2013. link Times cited: 23 Abstract: The transformation of dislocation cores from the shuffle to … read moreAbstract: The transformation of dislocation cores from the shuffle to the glide set of {111} glide planes in Si is examined in this work. The transformation is thermally activated and is favored by a resolved shear stress which applies no force on the original perfect shuffle dislocation. A resolved shear stress driving dislocation motion in the glide plane is not observed to promote the transition. The stress-dependent activation energy for the described shuffle-glide transformation mechanism is evaluated using a statistical analysis. It is observed that the transformation is not associated with an intermediate metastable state, as has been previously suggested in the literature. read less NOT USED (high confidence) I. Mladenov, P. Djondjorov, M. Hadzhilazova, and V. Vassilev, “Equilibrium Configurations of Lipid Bilayer Membranes and Carbon Nanostructures,” Communications in Theoretical Physics. 2013. link Times cited: 11 Abstract: The present article concerns the continuum modelling of the … read moreAbstract: The present article concerns the continuum modelling of the mechanical behaviour and equilibrium shapes of two types of nano-scale objects: fluid lipid bilayer membranes and carbon nanostructures. A unified continuum model is used to handle four different case studies. Two of them consist in representing in analytic form cylindrical and axisymmetric equilibrium configurations of single-wall carbon nanotubes and fluid lipid bilayer membranes subjected to uniform hydrostatic pressure. The third one is concerned with determination of possible shapes of junctions between a single-wall carbon nanotube and a flat graphene sheet or another single-wall carbon nanotube. The last one deals with the mechanical behaviour of closed fluid lipid bilayer membranes (vesicles) adhering onto a flat homogeneous rigid substrate subjected to micro-injection and uniform hydrostatic pressure. read less NOT USED (high confidence) C. Davini, “Homogenization of a graphene sheet,” Continuum Mechanics and Thermodynamics. 2013. link Times cited: 0 NOT USED (high confidence) M. Dürr and U. Höfer, “Hydrogen diffusion on silicon surfaces,” Progress in Surface Science. 2013. link Times cited: 40 NOT USED (high confidence) I. Lebedeva, A. Knizhnik, and B. Potapkin, “Predictive modeling of formation of carbon nanostructures,” Nanotechnologies in Russia. 2012. link Times cited: 1 NOT USED (high confidence) P. Howell, “Comparison of molecular dynamics methods and interatomic potentials for calculating the thermal conductivity of silicon.,” The Journal of chemical physics. 2012. link Times cited: 74 Abstract: We compare the molecular dynamics Green-Kubo and direct meth… read moreAbstract: We compare the molecular dynamics Green-Kubo and direct methods for calculating thermal conductivity κ, using as a test case crystalline silicon at temperatures T in the range 500-1000 K (classical regime). We pay careful attention to the convergence with respect to simulation size and duration and to the procedures used to fit the simulation data. We show that in the Green-Kubo method the heat current autocorrelation function is characterized by three decay processes, of which the slowest lasts several tens of picoseconds so that convergence requires several tens of nanoseconds of data. Using the Stillinger-Weber potential we find excellent agreement between the two methods. We also use the direct method to calculate κ(T) for the Tersoff potential and find that the magnitude and the temperature-dependence are different for the two potentials and that neither potential agrees with experimental data. We argue that this implies that using the Stillinger-Weber or Tersoff potentials to predict trends in kappa as some system parameter is varied may yield results which are specific to the potential but not intrinsic to Si. read less NOT USED (high confidence) A. Kumar, M. Wilson, M. Thorpe, and M. Thorpe, “Amorphous graphene: a realization of Zachariasen’s glass,” Journal of Physics: Condensed Matter. 2012. link Times cited: 49 Abstract: Amorphous graphene is a realization of a two-dimensional Zac… read moreAbstract: Amorphous graphene is a realization of a two-dimensional Zachariasen glass as first proposed 80 years ago. Planar continuous random networks of this archetypal two-dimensional network are generated by two complementary simulation methods. In the first, a Monte Carlo bond switching algorithm is employed to systematically amorphize a crystalline graphene sheet. In the second, molecular dynamics simulations are utilized to quench from the high temperature liquid state. The two approaches lead to similar results as detailed here, through the pair distribution function and the associated diffraction pattern. Details of the structure, including ring statistics and angular distortions, are shown to be sensitive to preparation conditions, and await experimental confirmation. read less NOT USED (high confidence) Y. Shin, H. Kwak, C. Zou, A. Vasenkov, and A. V. van Duin, “Development and validation of a ReaxFF reactive force field for Fe/Al/Ni alloys: molecular dynamics study of elastic constants, diffusion, and segregation.,” The journal of physical chemistry. A. 2012. link Times cited: 59 Abstract: We have developed a ReaxFF force field for Fe/Al/Ni binary a… read moreAbstract: We have developed a ReaxFF force field for Fe/Al/Ni binary alloys based on quantum mechanical (QM) calculations. In addition to the various bulk phases of the binary alloys, the (100), (110) and (111) surface energies and adatom binding energies were included in the training set for the force field parametrization of the Fe/Al/Ni binary alloys. To validate these optimized force fields, we studied (i) elastic constants of the binary alloys at finite temperatures, (ii) diffusivity of alloy components in Al/Ni alloy, and (iii) segregation on the binary alloy surfaces. First, we calculated linear elastic constants of FeAl, FeNi(3), and Ni(3)Al in the temperature range 300 to 1100 K. The temperature dependences of the elastic constants of these three alloys, showing a decrease in C(11), C(12), and C(44) as temperature increases, were in good agreement with the experimental results. We also performed ReaxFF molecular dynamics (MD) simulations for Al or Ni diffusion in the system modeled as Al/Ni mixed layers with the linear composition gradients. At 1000 K, Al diffusivity at the pure Al end was 2 orders of magnitude larger than that in the Al trace layers, probably explaining the nature of different diffusion behavior between molten metals and alloys. However, the diffusivity of Ni at the pure Ni end was only slightly larger than that in the Ni trace layers at the system temperature much lower than the melting temperature of Ni. Third, we investigated the surface segregation in L1(2)-Fe(3)Al, Fe(3)Ni, and Ni(3)Al clusters at high temperature (2500 K). From the analysis of composition distribution of the alloy components from the bulk to the surface layer, it was found that the degree of segregation depended on the chemical composition of the alloy. Al surface segregation occurred most strongly in Fe(3)Al, whereas it occurred most weakly in Ni(3)Al. These results may support the segregation mechanism that surface segregation results from the interplay between the energetic stability of the ordered bulk phase and the surface reconstruction. In addition, the surface segregation induced the depletion layers of segregating metal species (Al in Fe(3)Al and Ni(3)Al, and Ni in Fe(3)Ni) next to the segregation layers. These simulation results qualitatively agreed with early experimental observations of segregation in Fe/Al/Ni binary alloys. read less NOT USED (high confidence) J. Titantah and M. Karttunen, “Multiphase density functional theory parameterization of the interatomic potential for silver and gold,” The European Physical Journal B. 2012. link Times cited: 8 NOT USED (high confidence) J. Zhang, C. Liu, Y. Shu, and J. Fan, “Growth and properties of Cu thin film deposited on Si(0 0 1) substrate: A molecular dynamics simulation study,” Applied Surface Science. 2012. link Times cited: 50 NOT USED (high confidence) I. Solov’yov, A. Yakubovich, P. Nikolaev, I. Volkovets, and A. Solov’yov, “MesoBioNano explorer—A universal program for multiscale computer simulations of complex molecular structure and dynamics,” Journal of Computational Chemistry. 2012. link Times cited: 112 Abstract: We present a multipurpose computer code MesoBioNano Explorer… read moreAbstract: We present a multipurpose computer code MesoBioNano Explorer (MBN Explorer). The package allows to model molecular systems of varied level of complexity. In particular, MBN Explorer is suited to compute system's energy, to optimize molecular structure as well as to consider the molecular and random walk dynamics. MBN Explorer allows to use a broad variety of interatomic potentials, to model different molecular systems, such as atomic clusters, fullerenes, nanotubes, polypeptides, proteins, DNA, composite systems, nanofractals, and so on. A distinct feature of the program, which makes it significantly different from the existing codes, is its universality and applicability to the description of a broad range of problems involving different molecular systems. Most of the existing codes are developed for particular classes of molecular systems and do not permit multiscale approach while MBN Explorer goes beyond these drawbacks. On demand, MBN Explorer allows to group particles in the system into rigid fragments, thereby significantly reducing the number of dynamical degrees of freedom. Despite the universality, the computational efficiency of MBN Explorer is comparable (and in some cases even higher) than the computational efficiency of other software packages, making MBN Explorer a possible alternative to the available codes. © 2012 Wiley Periodicals, Inc. read less NOT USED (high confidence) C. Henager, F. Gao, S. Hu, G. Lin, E. Bylaska, and N. Zabaras, “Simulating Interface Growth and Defect Generation in CZT – Simulation State of the Art and Known Gaps.” 2012. link Times cited: 1 Abstract: This one-year, study topic project will survey and investiga… read moreAbstract: This one-year, study topic project will survey and investigate the known state-of-the-art of modeling and simulation methods suitable for performing fine-scale, fully 3-D modeling, of the growth of CZT crystals at the melt-solid interface, and correlating physical growth and post-growth conditions with generation and incorporation of defects into the solid CZT crystal. In the course of this study, this project will also identify the critical gaps in our knowledge of modeling and simulation techniques in terms of what would be needed to be developed in order to perform accurate physical simulations of defect generation in melt-grown CZT. The transformational nature of this study will be, for the first time, an investigation of modeling and simulation methods for describing microstructural evolution during crystal growth and the identification of the critical gaps in our knowledge of such methods, which is recognized as having tremendous scientific impacts for future model developments in a wide variety of materials science areas. read less NOT USED (high confidence) K. Volokh, “On the Strength of Graphene,” Journal of Applied Mechanics. 2012. link Times cited: 12 Abstract: Failure of a single-atomic-layer graphene sheet is analyzed … read moreAbstract: Failure of a single-atomic-layer graphene sheet is analyzed in plane tension under the varying biaxiality condition. The analysis is based on the combined use of continuum and molecular mechanics where the strain energy is expressed with the help of the Tersoff-Brenner atomistic potential. A critical failure surface is produced for strains in biaxial tension. It is found that the anisotropy of graphene has a pronounced effect on its strength. [DOI: 10.1115/1.4005582] read less NOT USED (high confidence) S. Gupta, K. Dharamvir, and V. Jindal, “Implicit phonon shifts and thermodynamical properties of rigid carbon nanotube bunches,” AIP Advances. 2012. link Times cited: 1 Abstract: We calculate phonon shifts of external modes of a bunch of c… read moreAbstract: We calculate phonon shifts of external modes of a bunch of carbon nanotubes. The bunches form a 2-dimensional hexagonal arrangement of lattice with long molecules of carbon nanotubes. A simple model based on atom-atom potential has been used to calculate the implicit anharmonicity in the phonons of carbon nanotube bundles (also called ropes or bunches) having rigid tubes, with the assumption that under hydrostatic pressure only the inter-tube distance in the bunch varies. Various bulk and thermodynamic properties like thermal expansion, bulk modulus and the Gruneisen constants and external phonon shifts which naturally enter into the calculation are also described and compared with the available data. The specific heat capacity has also been calculated. read less NOT USED (high confidence) M. Backman, N. Juslin, and K. Nordlund, “Bond order potential for gold,” The European Physical Journal B. 2012. link Times cited: 11 NOT USED (high confidence) J. Hur and S. Stuart, “Modified reactive empirical bond-order potential for heterogeneous bonding environments.,” The Journal of chemical physics. 2012. link Times cited: 11 Abstract: An improvement to the AIREBO potential for hydrocarbons is p… read moreAbstract: An improvement to the AIREBO potential for hydrocarbons is presented in which contributions to the bond order are determined by the local bonding environment around the bond, rather than the average of the environments around the two constituent atoms. This bond-centric approach decreases the errors by ~80% in the fullerene-type systems for which the original approach leads to the most severe errors. With the newly developed and parameterized method, energy errors are less than 0.7 eV for a collection of hydrocarbon molecules not used in the fitting. This modified AIREBO potential is expected to be more useful not only for the molecular hydrocarbons and fullerene isomers studied here, but also for the full range of carbon and hydrocarbon systems to which the AIREBO potential has been applied. read less NOT USED (high confidence) Z. Wei, Y. Chen, and C. Dames, “Wave packet simulations of phonon boundary scattering at graphene edges,” Journal of Applied Physics. 2012. link Times cited: 32 Abstract: Wave packet dynamics is used to investigate the scattering o… read moreAbstract: Wave packet dynamics is used to investigate the scattering of longitudinal (LA), transverse (TA), and bending-mode (ZA) phonons at the zigzag and armchair edges of suspended graphene. The interatomic forces are calculated using a linearized Tersoff potential. The strength of a boundary scattering event at impeding energy flow is described by a forward scattering coefficient, similar in spirit to a specularity parameter. For armchair boundaries, this scattering coefficient is found to depend strongly on the magnitude, direction, and polarization of the incident wavevector, while for zigzag boundaries, the forward scattering coefficient is found to always be unity regardless of wavevector and polarization. Wave packet splitting is observed for ZA phonons incident on armchair boundaries, while both splitting and mode conversion are observed for LA and TA phonons incident on both zigzag and armchair boundaries. These simulation results show that armchair boundaries impede the forward propagation of acoustic p... read less NOT USED (high confidence) S. Hollerer and C. Celigoj, “Buckling analysis of carbon nanotubes by a mixed atomistic and continuum model,” Computational Mechanics. 2012. link Times cited: 0 NOT USED (high confidence) S. Hollerer, “Buckling analysis of carbon nanotubes – a molecular statics investigation into the influence of non‐bonded interactions,” International Journal for Numerical Methods in Engineering. 2012. link Times cited: 4 Abstract: This paper analyses the buckling behaviour of single‐walled … read moreAbstract: This paper analyses the buckling behaviour of single‐walled and double‐walled carbon nanotubes. The total potential of the atomic structure consists of the bonded energy and the non‐bonded energy, both resulting from interatomic potentials, as well as the energy of external contributions. In particular, the influence of the in‐layer and inter‐layer non‐bonded interactions is investigated. These non‐bonded interactions are important to avoid the nanotubes from self‐intersection or penetration and govern the morphology of the buckled tubes. read less NOT USED (high confidence) J. Jakowski et al., “Optimization of density functional tight-binding and classical reactive molecular dynamics for high-throughput simulations of carbon materials,” Extreme Science and Engineering Discovery Environment. 2012. link Times cited: 2 Abstract: Carbon materials and nanostructures (fullerenes, nanotubes) … read moreAbstract: Carbon materials and nanostructures (fullerenes, nanotubes) are promising building blocks of nanotechnology. Potential applications include optical and electronic devices, sensors, and nano-scale machines. The multiscale character of processes related to fabrication and physics of such materials requires using a combination of different approaches such as (a) classical dynamics, (b) direct Born-Oppenheimer dynamics, (c) quantum dynamics for electrons and (d) quantum dynamics for selected nuclei. We describe our effort on optimization of classical reactive molecular dynamics and density-functional tight binding method, which is a core method in our direct and quantum dynamics studies. We find that optimization is critical for efficient use of high-end machines. Choosing the optimal configuration for the numerical library and compilers can result in four-fold speedup of direct dynamics as compared with default programming environment. The integration algorithm and parallelization approach must also be tailored for the computing environment. The efficacy of possible choices is discussed. read less NOT USED (high confidence) P. T. Araujo et al., “In situ atomic force microscopy tip-induced deformations and Raman spectroscopy characterization of single-wall carbon nanotubes.,” Nano letters. 2012. link Times cited: 16 Abstract: In this work, an atomic force microscope (AFM) is combined w… read moreAbstract: In this work, an atomic force microscope (AFM) is combined with a confocal Raman spectroscopy setup to follow in situ the evolution of the G-band feature of isolated single-wall carbon nanotubes (SWNTs) under transverse deformation. The SWNTs are pressed by a gold AFM tip against the substrate where they are sitting. From eight deformed SWNTs, five exhibit an overall decrease in the Raman signal intensity, while three exhibit vibrational changes related to the circumferential symmetry breaking. Our results reveal chirality dependent effects, which are averaged out in SWNT bundle measurements, including a previously elusive mode symmetry breaking that is here explored using molecular dynamics calculations. read less NOT USED (high confidence) J. Zhu, M. He, and F. Qiu, “Effect of Vacancy Defects on the Young’s Modulus and Fracture Strength of Graphene: A Molecular Dynamics Study,” Chinese Journal of Chemistry. 2012. link Times cited: 31 Abstract: The Young's modulus of graphene with various rectangula… read moreAbstract: The Young's modulus of graphene with various rectangular and circular vacancy defects is investigated by molecular dynamics simulation. By comparing with the results calculated from an effective spring model, it is demonstrated that the Young's modulus of graphene is largely correlated to the size of vacancy defects perpendicular to the stretching direction. And a linear reduction of Young's modulus with the increasing concentration of mono-atomic-vacancy defects (i.e., the slope of −0.03) is also observed. The fracture behavior of graphene, including the fracture strength, crack initiation and propagation are then studied by the molecular dynamics simulation, the effective spring model, and the quantized fracture mechanics. The blunting effect of vacancy edges is demonstrated, and the characterized crack tip radius of 4.44 A is observed. read less NOT USED (high confidence) Y. Li, T.-R. Shan, T. Liang, S. Sinnott, and S. Phillpot, “Classical interatomic potential for orthorhombic uranium,” Journal of Physics: Condensed Matter. 2012. link Times cited: 25 Abstract: A classical interatomic potential for uranium metal is deriv… read moreAbstract: A classical interatomic potential for uranium metal is derived within the framework of the charge optimized many body (COMB) formalism. The potential is fitted with a database obtained from experiment and density functional theory (DFT) calculations. The potential correctly predicts orthorhombic α-U to be the ground state. Good agreement with experimental values is obtained for the lattice parameters, nearest neighbor distances, and elastic constants. Molecular dynamics simulations also correctly show the anisotropy in the coefficient of thermal expansion and the temperature dependence of the nearest neighbor distances. read less NOT USED (high confidence) Y. Hwang, E.-K. Lee, H. Choi, K.-H. Yun, M. Lee, and Y.-C. Chung, “Atomic behavior of carbon atoms on a Si removed 3C-SiC (111) surface during the early stage of epitaxial graphene growth,” Journal of Applied Physics. 2012. link Times cited: 6 Abstract: The understanding of the formation of graphene at the atomic… read moreAbstract: The understanding of the formation of graphene at the atomic scale on Si-terminated 3C-SiC for obtaining high-quality graphene sheets remains elusive, although epitaxial graphene growth has been shown to be a well-known method for economical mass production of graphene/SiC heterojunctions. In this paper, the atomic behavior of carbon atoms on a Si removed 3C-SiC (111) surface for the formation of graphene buffer layer during the early stage of epitaxial graphene growth was investigated using a molecular dynamics simulation. Observation of the behavior of the remaining carbon atoms on the Si-terminated 3C-SiC (111) surface after removal of the silicon atoms revealed that graphene clusters, which were formed by sp2-bonded carbon atoms, start to appear at annealing temperatures higher than 1300 K. Our simulations indicated that the structural stability of the whole system increased as the number of sp2-bonded carbon atoms on the Si-terminated 3C-SiC (111) surface increased. It was also found that the diffusi... read less NOT USED (high confidence) H. Hou, R. Wang, J. Wang, X. Liu, G. Chen, and P. Huang, “An analytic bond-order potential for the Fe–Cu system,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 5 Abstract: An angular-dependent analytic bond-order potential (ABOP) fo… read moreAbstract: An angular-dependent analytic bond-order potential (ABOP) for copper and Fe–Cu system was developed, based on the ABOP of pure iron introduced by Müller et al (2007 J. Phys.: Condens. Matter 19 326220). The potential parameters for the present ABOP model of copper were determined by fitting to the experimental data of the basic properties of fcc Cu and ab initio calculated properties of bcc Cu. The model predicts the vacancy formation energy in good agreement with the experimental result, although no vacancy formation information was used in the fitting of the model parameters. The melting point of Cu is also properly reproduced. The Fe–Cu binary system was described by adding two independent cross parameters in the potential model. The cross parameters were fitted using the ab initio data of the formation energies and lattice parameters of fictitious Fe–Cu alloys. The potential was applied to investigate the point defects and small defect clusters in dilute Fe–Cu alloys. The results were compared with the ab initio data and the values obtained with other potentials. read less NOT USED (high confidence) M. P. Ariza, R. Serrano, J. P. Mendez, and M. Ortiz, “Stacking faults and partial dislocations in graphene,” Philosophical Magazine. 2012. link Times cited: 15 Abstract: We investigate two mechanisms of crystallographic slip in gr… read moreAbstract: We investigate two mechanisms of crystallographic slip in graphene, corresponding to glide and shuffle generalized stacking faults (GSF), and compute their γ-curves using Sandia National Laboratories Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS). We find evidence of metastable partial dislocations for the glide GSF only. The computed values of the stable and unstable stacking-fault energies are suggestive of a high stability of full dislocations against dissociation and of dislocation dipoles against annihilation. read less NOT USED (high confidence) X. Shi, B. Peng, N. Pugno, and H. Gao, “Stretch-induced softening of bending rigidity in graphene,” Applied Physics Letters. 2012. link Times cited: 34 Abstract: First principle calculations are performed to show that the … read moreAbstract: First principle calculations are performed to show that the bending rigidity of graphene can be softened considerably with in-plane stretching. This phenomenon can be attributed to stretch-induced loosening of atomic packing and should be of fundamental significance for graphene-based structures and devices. read less NOT USED (high confidence) S. Zhao, J. Xue, Y. Wang, and S. Yan, “Chemical bonding assisted damage production in single-walled carbon nanotubes induced by low-energy ions,” Applied Physics A. 2012. link Times cited: 8 NOT USED (high confidence) M. T. Knippenberg, P. Mikulski, K. E. Ryan, S. Stuart, G. Gao, and J. Harrison, “Bond-order potentials with split-charge equilibration: application to C-, H-, and O-containing systems.,” The Journal of chemical physics. 2012. link Times cited: 25 Abstract: A method for extending charge transfer to bond-order potenti… read moreAbstract: A method for extending charge transfer to bond-order potentials, known as the bond-order potential/split-charge equilibration (BOP/SQE) method [P. T. Mikulski, M. T. Knippenberg, and J. A. Harrison, J. Chem. Phys. 131, 241105 (2009)], is integrated into a new bond-order potential for interactions between oxygen, carbon, and hydrogen. This reactive potential utilizes the formalism of the adaptive intermolecular reactive empirical bond-order potential [S. J. Stuart, A. B. Tutein, and J. A. Harrison, J. Chem. Phys. 112, 6472 (2000)] with additional terms for oxygen and charge interactions. This implementation of the reactive potential is able to model chemical reactions where partial charges change in gas- and condensed-phase systems containing oxygen, carbon, and hydrogen. The BOP/SQE method prevents the unrestricted growth of charges, often observed in charge equilibration methods, without adding significant computational time, because it makes use of a quantity which is calculated as part of the underlying covalent portion of the potential, namely, the bond order. The implementation of this method with the qAIREBO potential is designed to provide a tool that can be used to model dynamics in a wide range of systems without significant computational cost. To demonstrate the usefulness and flexibility of this potential, heats of formation for isolated molecules, radial distribution functions of liquids, and energies of oxygenated diamond surfaces are calculated. read less NOT USED (high confidence) Z. Xi and B. Youn, “Predictive carbon nanotube models using the eigenvector dimension reduction (EDR) method,” Journal of Mechanical Science and Technology. 2012. link Times cited: 8 Abstract: It has been reported that a carbon nanotube (CNT) is one of … read moreAbstract: It has been reported that a carbon nanotube (CNT) is one of the strongest materials with its high failure stress and strain. Moreover, the nanotube has many favorable features, such as high toughness, great flexibility, low density, and so on. This discovery has opened new opportunities in various engineering applications, for example, a nanocomposite material design. However, recent studies have found a substantial discrepancy between computational and experimental material property predictions, in part due to defects in the fabricated nanotubes. It is found that the nanotubes are highly defective in many different formations (e.g., vacancy, dislocation, chemical, and topological defects). Recent parametric studies with vacancy defects have found that the vacancy defects substantially affect mechanical properties of the nanotubes. Given random existence of the nanotube defects, the material properties of the nanotubes can be better understood through statistical modeling of the defects. This paper presents predictive CNT models, which enable to estimate mechanical properties of the CNTs and the nanocomposites under various sources of uncertainties. As the first step, the density and location of vacancy defects will be randomly modeled to predict mechanical properties. It has been reported that the eigenvector dimension reduction (EDR) method performs probability analysis efficiently and accurately. In this paper, molecular dynamics (MD) simulation with a modified Morse potential model is integrated with the EDR method to predict the mechanical properties of the CNTs. To demonstrate the feasibility of the predicted model, probabilistic behavior of mechanical properties (e.g., failure stress, failure strain, and toughness) is compared with the precedent experiment results. read less NOT USED (high confidence) K. Farah, F. Müller-Plathe, and M. Böhm, “Classical reactive molecular dynamics implementations: state of the art.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2012. link Times cited: 71 Abstract: Reactive molecular dynamics (RMD) implementations equipped w… read moreAbstract: Reactive molecular dynamics (RMD) implementations equipped with force field approaches to simulate both the time evolution as well as chemical reactions of a broad class of materials are reviewed herein. We subdivide the RMD approaches developed during the last decade as well as older ones already reviewed in 1995 by Srivastava and Garrison and in 2000 by Brenner into two classes. The methods in the first RMD class rely on the use of a reaction cutoff distance and employ a sudden transition from the educts to the products. Due to their simplicity these methods are well suited to generate equilibrated atomistic or material-specific coarse-grained polymer structures. In connection with generic models they offer useful qualitative insight into polymerization reactions. The methods in the second RMD class are based on empirical reactive force fields and implement a smooth and continuous transition from the educts to the products. In this RMD class, the reactive potentials are based on many-body or bond-order force fields as well as on empirical standard force fields, such as CHARMM, AMBER or MM3 that are modified to become reactive. The aim with the more sophisticated implementations of the second RMD class is the investigation of the reaction kinetics and mechanisms as well as the evaluation of transition state geometries. Pure or hybrid ab initio, density functional, semi-empirical, molecular mechanics, and Monte Carlo methods for which no time evolution of the chemical systems is achieved are excluded from the present review. So are molecular dynamics techniques coupled with quantum chemical methods for the treatment of the reactive regions, such as Car-Parinello molecular dynamics. read less NOT USED (high confidence) A. Dongare, B. Lamattina, D. Irving, A. Rajendran, M. Zikry, and D. Brenner, “An angular-dependent embedded atom method (A-EAM) interatomic potential to model thermodynamic and mechanical behavior of Al/Si composite materials,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 23 Abstract: A new interatomic potential is developed for the Al/Si syste… read moreAbstract: A new interatomic potential is developed for the Al/Si system in the formulation of the recently developed angular-dependent embedded atom method (A-EAM). The A-EAM is formulated by combining the embedded atom method potential for Al with the Stillinger–Weber potential for Si. The parameters of the Al/Si cross-interactions are fitted to reproduce the structural energetics of Al/Si bulk alloys determined based on the results of density functional theory calculations and the experimentally observed mixing behavior of the AlSi liquid alloy at high temperatures. The ability to investigate the thermodynamic properties of the Al/Si system is demonstrated by computing the binary phase diagram of the Al–Si system as predicted by the A-EAM potential and comparing with that obtained using experiments. The ability to study the mechanical behavior of the Al/Si composite systems is demonstrated by investigating the micromechanisms related to dynamic failure of the Al/Si nanocomposites using MD simulations. read less NOT USED (high confidence) Z. Xi and B. Youn, “Predictive carbon nanotube models using the eigenvector dimension reduction (EDR) method,” Journal of Mechanical Science and Technology. 2012. link Times cited: 1 NOT USED (high confidence) B. V. Koten and C. Ortner, “Symmetries of 2-Lattices and Second Order Accuracy of the Cauchy-Born Model,” Multiscale Model. Simul. 2012. link Times cited: 11 Abstract: We show that the Cauchy--Born model of a single-species 2-la… read moreAbstract: We show that the Cauchy--Born model of a single-species 2-lattice is second order if the atomistic and continuum kinematics are connected in a novel way. Our proof uses a generalization to 2-lattices of the point symmetry of Bravais lattices. Moreover, by identifying similar symmetries in multispecies pair interaction models, we construct a new stored energy density, using shift gradients but not strain gradients, that is also second order accurate. These results can be used to develop highly accurate continuum models and atomistic/continuum coupling methods for materials such as graphene, hcp metals, and shape memory alloys. read less NOT USED (high confidence) D. Mathieu, “Formation Enthalpies Derived from Pairwise Interactions: A Step toward More Transferable Reactive Potentials for Organic Compounds.,” Journal of chemical theory and computation. 2012. link Times cited: 4 Abstract: A new approach to the development and parametrization of rea… read moreAbstract: A new approach to the development and parametrization of reactive potentials for organic compounds is put forward. As a byproduct of preliminary efforts in this direction, the performance of a simple representation of the energy of equilibrium structures in term of pairwise atom-atom and bond-bond contributions is investigated. For now, each contribution is assumed constant, given the multiplicity of covalent bonds, rather than computed on-the-fly from geometries and bond orders. In spite of this rough approximation, the approach performs remarkably well by comparison with semiempirical quantum chemical methods. Nevertheless, further refinement proves necessary for some unstable species involved in chemical reactions. As it stands, the present model appears as a promising basis in view of less empirical and more versatile alternatives to group contribution methods for the fast prediction of heats of formation, although much work remains to be done to demonstrate its value as a starting point toward better reactive potentials. read less NOT USED (high confidence) I. Lebedeva, A. Knizhnik, A. Popov, and B. Potapkin, “Ni-assisted transformation of graphene flakes to fullerenes,” arXiv: Mesoscale and Nanoscale Physics. 2012. link Times cited: 34 Abstract: Transformation of graphene flakes to fullerenes assisted by … read moreAbstract: Transformation of graphene flakes to fullerenes assisted by Ni clusters is investigated using molecular dynamics simulations. The bond-order potential for Ni-C systems is developed. The potential reproduces the experimental and first-principles data on the physical properties of pure Ni as well as on relative energies of carbon species on Ni surfaces and in Ni bulk. The potential is applied for molecular dynamics simulations of the transformation of graphene flakes consisting of 50 - 400 atoms with and without Ni clusters attached. Free fullerenes, fullerenes with Ni clusters attached from outside and fullerenes encapsulating Ni clusters (Ni endofullerenes) are observed to form in the presence of Ni clusters consisting of 5 - 80 atoms. Moreover, a new type of heterofullerenes with a patch made of a Ni cluster is found to form as an intermediate structure during the transformation. The Ni clusters are shown to reduce the activation energy for the graphene-fullerene transformation from 4.0 eV to 1.5 - 1.9 eV, providing the decrease of the minimal temperature at which such a transformation can be observed experimentally from about 1400 K for free graphene flakes to about 700 - 800 K. While the transformation of free graphene flakes is found to occur through formation of chains of two-coordinated carbon atoms at the flake edges, the mechanism of the Ni-assisted graphene-fullerene transformation is revealed to be based on the transfer of carbon atoms from the graphene flake to the Ni cluster and back. The way of controlled synthesis of endofullerenes with a transition metal cluster inside and heterofullerenes with a transition metal patch is also proposed. read less NOT USED (high confidence) Z. Zhang and H. Gao, “Simulating fracture propagation in rock and concrete by an augmented virtual internal bond method,” International Journal for Numerical and Analytical Methods in Geomechanics. 2012. link Times cited: 37 Abstract: This paper develops a practical approach to simulating fract… read moreAbstract: This paper develops a practical approach to simulating fracture propagation in rock and concrete based on an augmented virtual internal bond (VIB) method in which the cohesion of solid is modeled as material particles interconnected by a network of randomized virtual micro bonds described by the Xu–Needleman potential. The micro bond potential is used to derive macroscale constitutive relations via the Cauchy–Born rule. By incorporating different energy contributions due to stretch and shearing, as well as different energy levels under tension and compression of each micro bond, the derived macro constitutive laws are particularly useful for modeling quasi‐brittle materials such as rock and concrete which usually have different Poisson ratios and much higher compressive strength than tensile strength. The mesh‐size sensitivity associated with strain‐softening in the present constitutive model is addressed by adjusting material constants near the crack tip so that the biJ‐integral is kept equal to the intrinsic fracture energy of the material. Numerical examples demonstrate that the proposed VIB method is capable of simulating mixed mode fracture propagation in rock and concrete with results in consistency with relevant experimental observations. Copyright © 2011 John Wiley & Sons, Ltd. read less NOT USED (high confidence) L. Hale et al., “Dislocation morphology and nucleation within compressed Si nanospheres: A molecular dynamics study,” Computational Materials Science. 2012. link Times cited: 24 NOT USED (high confidence) G. Yun and H. S. Park, “Bridging the gap between experimental measurements and atomistic predictions of the elastic properties of silicon nanowires using multiscale modeling,” Finite Elements in Analysis and Design. 2012. link Times cited: 9 NOT USED (high confidence) F. Liu, Q. H. Tang, B. S. Shang, and T. C. Wang, “Simple optimized Brenner potential for thermodynamic properties of diamond,” Philosophical Magazine. 2012. link Times cited: 3 Abstract: We have examined the commonly used Brenner potentials in the… read moreAbstract: We have examined the commonly used Brenner potentials in the context of the thermodynamic properties of diamond. A simple optimized Brenner potential is proposed that provides very good predictions of the thermodynamic properties of diamond. It is shown that, compared to the experimental data, the lattice wave theory of molecular dynamics (LWT) with this optimized Brenner potential can accurately predict the temperature dependence of specific heat, lattice constant, Grüneisen parameters and coefficient of thermal expansion (CTE) of diamond. read less NOT USED (high confidence) C. Hou and W. Ge, “GPU-accelerated molecular dynamics simulation of solid covalent crystals,” Molecular Simulation. 2012. link Times cited: 17 Abstract: Graphics processing unit (GPU) is becoming a powerful comput… read moreAbstract: Graphics processing unit (GPU) is becoming a powerful computational tool in science and engineering. In this paper, different from previous molecular dynamics (MD) simulation with pair potentials and many-body potentials, two MD simulation algorithms implemented on a single GPU are presented to describe a special category of many-body potentials – bond order potentials used frequently in solid covalent materials, such as the Tersoff potentials for silicon crystals. The simulation results reveal that the performance of GPU implementations is apparently superior to their CPU counterpart. Furthermore, the proposed algorithms are generalised, transferable and scalable, and can be extended to the simulations with general many-body interactions such as Stillinger–Weber potential and so on. read less NOT USED (high confidence) B. L. Davis and M. Hussein, “Thermal characterization of nanoscale phononic crystals using supercell lattice dynamics,” AIP Advances. 2011. link Times cited: 33 Abstract: The concept of a phononic crystal can in principle be realiz… read moreAbstract: The concept of a phononic crystal can in principle be realized at the nanoscale whenever the conditions for coherent phonon transport exist. Under such conditions, the dispersion characteristics of both the constitutive material lattice (defined by a primitive cell) and the phononic crystal lattice (defined by a supercell) contribute to the value of the thermal conductivity. It is therefore necessary in this emerging class of phononic materials to treat the lattice dynamics at both periodicity levels. Here we demonstrate the utility of using supercell lattice dynamics to investigate the thermal transport behavior of three-dimensional nanoscale phononic crystals formed from silicon and cubic voids of vacuum. The periodicity of the voids follows a simple cubic arrangement with a lattice constant that is around an order of magnitude larger than that of the bulk crystalline silicon primitive cell. We consider an atomic-scale supercell which incorporates all the details of the silicon atomic locations and the void geometry. For this supercell, we compute the phonon band structure and subsequently predict the thermal conductivity following the Callaway-Holland model. Our findings dictate that for an analysis based on supercell lattice dynamics to be representative of the properties of the underlying lattice model, a minimum supercell size is needed along with a minimum wave vector sampling resolution. Below these minimum values, a thermal conductivity prediction of a bulk material based on a supercell will not adequately recover the value obtained based on a primitive cell. Furthermore, our results show that for the relatively small voids and void spacings we consider (where boundary scattering is dominant), dispersion at the phononic crystal unit cell level plays a noticeable role in determining the thermal conductivity. read less NOT USED (high confidence) F. Gao, J. Qu, and M. Yao, “Interfacial thermal resistance between metallic carbon nanotube and Cu substrate,” Journal of Applied Physics. 2011. link Times cited: 29 Abstract: A comprehensive model was developed to calculate the interfa… read moreAbstract: A comprehensive model was developed to calculate the interfacial thermal resistance between a metallic carbon nanotube (CNT) and a Cu substrate. The new model accounts for both phonon-mediated and electron-mediated thermal transfer at the interface, as well as the effect of electron-phonon coupling within CNT and Cu. The phonon-mediated thermal transfer was simulated using the non-equilibrium molecular dynamics, while the electron-mediated thermal transfer was computed by the non-equilibrium Green’s function method in conjunction with the density function theory. The effect of electron-phonon coupling within Cu and CNT was investigated by using the kinetic theory. Our results show that (1) electron-phonon coupling within Cu and CNT contributes significantly to the overall thermal transfer across the CNT/Cu interface, and (2) contributions to the overall thermal conductance at the CNT/Cu interface from the electron-mediated thermal transfer are comparable to that from the phonon-mediated thermal transfer. read less NOT USED (high confidence) G. Kosa, M. Bercu, and V. Grecu, “Elastic waves propagation in stressed SWCNT,” CAS 2011 Proceedings (2011 International Semiconductor Conference). 2011. link Times cited: 0 Abstract: This paper presents the study on the behaviour of deformed s… read moreAbstract: This paper presents the study on the behaviour of deformed singe wall carbon nanotubes associated to the conditions of mechanical treatments. We investigate the time evolution of the deformation wave, propagating along the carbon nanotube, and reflected at both clamped ends by using large atomistic models based on molecular dynamics calculation. The system is turned into a non-homogeneous one by the quick deformation applied at one extremity, determining local changes of elastic properties. Processes concerning mechanic wave propagation and elastic energy accumulation at specific moments and places, culminating with the tube fracture are analysed in relation with the stress-strain characteristic of the single wall carbon nanotube. read less NOT USED (high confidence) S. Hollerer, “Buckling Analysis of Carbon Nanotubes – a molecular mechanics approach using the finite element framework,” PAMM. 2011. link Times cited: 4 Abstract: In this work, a molecular mechanics model embedded in the fi… read moreAbstract: In this work, a molecular mechanics model embedded in the finite element framework is applied to analyze the buckling behaviour of carbon nanotubes. Within this model a specific finite element is set up for the underlaying interatomic potential describing the behaviour of the multi particle system. The model relies on the fully nonlinear description of the interatomic potential and the atomic kinematics. Stability points of the system are located by an accompanying eigenvalue analysis and the bifurcation point is detected using a bisection algorithm. To follow the nonlinear load‐deformation path in the area of postbuckling a branch switch is performed. With the help of this molecular mechanics model, the response of carbon nanotubes on different loading conditions with respect to buckling is studied. (© 2011 Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) T. Nakajima and K. Shintani, “Controlling out‐of‐plane deformations of graphene nanobridges,” physica status solidi (b). 2011. link Times cited: 3 Abstract: Graphene nanoribbons (GNRs) can be applied to transistors, m… read moreAbstract: Graphene nanoribbons (GNRs) can be applied to transistors, mass sensors, and dust detectors. Suspended GNRs which connect terminals in electronic devices like bridges can be treated as edge‐constrained GNRs. In this paper, edge‐constrained GNRs of various sizes and initial strains are studied using molecular dynamics (MD) simulations. To induce strain in GNRs, bond lengths between carbon atoms of the initial configurations of GNR models are varied. The bond length of the energetically stable GNRs is estimated at 1.47 Å. At this bond length, GNRs obviously change the tendencies of their energies, amplitudes, and deformations. The relationships between the out‐of‐plane deformations and the sizes of GNRs, and between the out‐of‐plane deformations and strains of GNRs are studied. Under compressive strain, the out‐of‐plane deformation of GNRs is dominantly caused by buckling. The amplitude of the buckling decreases as GNRs elongate. On the other hand, under tensile strain, the out‐of‐plane deformation of GNRs is caused by ripples and thermal vibrations. The ripples show regular patterns. It is suggested we can control the amplitudes of the out‐of‐plane deformations and ripple patterns of GNRs by adjusting their strain. read less NOT USED (high confidence) M. Alaghemandi, F. Müller-Plathe, and M. Böhm, “Thermal conductivity of carbon nanotube-polyamide-6,6 nanocomposites: reverse non-equilibrium molecular dynamics simulations.,” The Journal of chemical physics. 2011. link Times cited: 34 Abstract: The thermal conductivity of composites of carbon nanotubes a… read moreAbstract: The thermal conductivity of composites of carbon nanotubes and polyamide-6,6 has been investigated using reverse non-equilibrium molecular dynamics simulations in a full atomistic resolution. It is found, in line with experiments, that the composites have thermal conductivities, which are only moderately larger than that of pure polyamide. The composite conductivities are orders of magnitude less than what would be expected from naïve additivity arguments. This means that the intrinsic thermal conductivities of isolated nanotubes, which exceed the best-conducting metals, cannot be harnessed for heat transport, when the nanotubes are embedded in a polymer matrix. The main reason is the high interfacial thermal resistance between the nanotubes and the polymer, which was calculated in addition to the total composite thermal conductivity as well as that of the subsystem. It hinders heat to be transferred from the slow-conducting polymer into the fast-conducting nanotubes and back into the polymer. This interpretation is in line with the majority of recent simulation works. An alternative explanation, namely, the damping of the long-wavelength phonons in nanotubes by the polymer matrix is not supported by the present calculations. These modes provide most of the polymers heat conduction. An additional minor effect is caused by the anisotropic structure of the polymer phase induced by the nearby nanotube surfaces. The thermal conductivity of the polymer matrix increases slightly in the direction parallel to the nanotubes, whereas it decreases perpendicular to it. read less NOT USED (high confidence) J. Behler, “Neural network potential-energy surfaces in chemistry: a tool for large-scale simulations.,” Physical chemistry chemical physics : PCCP. 2011. link Times cited: 546 Abstract: The accuracy of the results obtained in molecular dynamics o… read moreAbstract: The accuracy of the results obtained in molecular dynamics or Monte Carlo simulations crucially depends on a reliable description of the atomic interactions. A large variety of efficient potentials has been proposed in the literature, but often the optimum functional form is difficult to find and strongly depends on the particular system. In recent years, artificial neural networks (NN) have become a promising new method to construct potentials for a wide range of systems. They offer a number of advantages: they are very general and applicable to systems as different as small molecules, semiconductors and metals; they are numerically very accurate and fast to evaluate; and they can be constructed using any electronic structure method. Significant progress has been made in recent years and a number of successful applications demonstrate the capabilities of neural network potentials. In this Perspective, the current status of NN potentials is reviewed, and their advantages and limitations are discussed. read less NOT USED (high confidence) M. J. López, I. Cabria, and J. A. Alonso, “Simulated porosity and electronic structure of nanoporous carbons.,” The Journal of chemical physics. 2011. link Times cited: 38 Abstract: Nanoporous carbon refers to a broad class of materials chara… read moreAbstract: Nanoporous carbon refers to a broad class of materials characterized by nanometer-size pores, densities lower than water, large specific surface areas, and high porosities. These materials find applications in nanocatalysis and gas adsorption, among others. The porosity structure, that determines the properties and functionalities of these materials, is still not characterized in detail. Here, we reveal the detail porosity structure and the electronic properties of a type of nanoporous carbons, the so called carbide derived carbons (CDCs), through a simulation scheme that combines large simulation cells and long time scales at the empirical level with first-principles density functional calculations. We show that the carbon network consists in one layer thick nanographenes interconnected among them. The presence of specific defects in the carbon layers (heptagons and octagons) yields to open pores. These defects are not completely removed through annealing at high temperatures. We also suggest that, in contrast with graphene which is a zero-gap semiconductor, these materials would have a metallic character, since they develop an electronic band around the Fermi level. This band arises from the electronic states localized at the edges of the nanographene layers. read less NOT USED (high confidence) M. Liao, Y.-C. Wang, S. Ju, T.-W. Lien, and L.-F. Huang, “Deformation behaviors of an armchair boron-nitride nanotube under axial tensile strains,” Journal of Applied Physics. 2011. link Times cited: 49 Abstract: Deformation behaviors of an (8,8) boron-nitride nanotube (BN… read moreAbstract: Deformation behaviors of an (8,8) boron-nitride nanotube (BNNT) under axial tensile strains were investigated via molecular dynamics (MD) simulations. The Tersoff potential was employed in the simulations with potential parameters determined by fitting the MD simulations results to those obtained from density functional theory calculations for BNNTs with the aid of the force-matching method. Variations in the axial stress, bond lengths, bond angles, radial buckling, and slip vectors with tensile strain were all examined. The axial, the radial, and tangential components of the slip vector were employed to monitor the local elongation, the local necking, and the local twisting deformations, respectively, near the tensile failure of the BNNT. From this study, it was noted that the BNNT started to fail at the failure strain of 26.7%. The components of the slip vector grew abruptly and rapidly after the failure strain, especially for the axial component. This implies that the local elongation dominates the ten... read less NOT USED (high confidence) C. D. Cruz, K. Termentzidis, P. Chantrenne, and X. Kleber, “Molecular dynamics simulations for the prediction of thermal conductivity of bulk silicon and silicon nanowires: Influence of interatomic potentials and boundary conditions,” Journal of Applied Physics. 2011. link Times cited: 62 Abstract: The reliability of molecular dynamics (MD) results depends s… read moreAbstract: The reliability of molecular dynamics (MD) results depends strongly on the choice of interatomic potentials and simulation conditions. Five interatomic potentials have been evaluated for heat transfer MD simulations of silicon, based on the description of the harmonic (dispersion curves) and anharmonic (linear thermal expansion) properties. The best interatomic potential is the second nearest-neighbor modified embedded atom method potential followed by the Stillinger-Weber, and then the Tersoff III. However, the prediction of the bulk silicon thermal conductivity leads to the conclusion that the Tersoff III potential gives the best results for isotopically pure silicon at high temperatures. The thermal conductivity of silicon nanowires as a function of cross-section and length is calculated, and the influence of the boundary conditions is studied for those five potentials. read less NOT USED (high confidence) M. Daw, J. Lawson, and C. Bauschlicher, “Interatomic potentials for Zirconium Diboride and Hafnium Diboride,” Computational Materials Science. 2011. link Times cited: 19 NOT USED (high confidence) A. Oluwajobi and X. Chen, “The effect of interatomic potentials on the molecular dynamics simulation of nanometric machining,” International Journal of Automation and Computing. 2011. link Times cited: 45 NOT USED (high confidence) Y. Chan and J. M. Hill, “Dynamics of benzene molecules situated in metal-organic frameworks,” Journal of Mathematical Chemistry. 2011. link Times cited: 7 NOT USED (high confidence) H. Wang and M. Upmanyu, “Saddles, twists, and curls: shape transitions in freestanding nanoribbons.,” Nanoscale. 2011. link Times cited: 14 Abstract: Efforts to modulate the electronic properties of atomically … read moreAbstract: Efforts to modulate the electronic properties of atomically thin crystalline nanoribbons requires precise control over their morphology. Here, we perform atomistic simulations on freestanding graphene nanoribbons (GNRs) to first identify the minimal shapes as a function of ribbon width, and then develop a core-edge framework based on classical plate theory to explore the effect of size and ribbon elasticity in more general systems. The elastic edge-edge interactions are central to stabilization of the flat phase in ultra-narrow ribbons, and their bifurcation to twisted and bent shapes at critical widths that vary inversely with edge stress. In the case of compressive edge stress, we uncover hitherto ignored saddle shapes that are energetically indistinguishable with twisted shapes in the vicinity of the bifurcation yet dominate the morphological space with increasing width. At much larger widths with negligible edge-edge interactions, rippling instabilities set in, i.e. edge ripples and midline dimples for compressive and tensile edge stresses, respectively. Simulations of tapering GNRs reveal the dynamics of these shape transitions. Our results capture the interplay between geometry and mechanics that sets the morphology of crystalline nanoribbons and also highlight the utility of the core-edge framework in developing a unified understanding of the interplay. read less NOT USED (high confidence) S. Mahajan, G. Subbarayan, and B. Sammakia, “Estimating Kapitza Resistance Between \rm Si\hbox-\rm SiO_2 Interface Using Molecular Dynamics Simulations,” IEEE Transactions on Components, Packaging and Manufacturing Technology. 2011. link Times cited: 20 Abstract: The interface between nano-scale films is of relevance in ma… read moreAbstract: The interface between nano-scale films is of relevance in many critical applications. Specifically, recent technological advances in semiconductor industry that utilize silicon-on-insulator devices have given importance to the understanding of thermal transport across ${\rm Si}{\hbox{-}}{\rm SiO}_{2}$ interface. Estimates of interfacial (Kapitza) resistance to the thermal transport across ${\rm Si}{\hbox{-}}{\rm SiO}_{2}$ films do not appear to exist at the present time. In this paper, we develop and carryout reverse non-equilibrium molecular dynamics simulations by imposing known heat flux to determine the Kapitza resistance between ${\rm Si}{\hbox{-}}{\rm SiO}_{2}$ thin films. For the ${\rm Si}{\hbox{-}}{\rm SiO}_{2}$ interface, the average Kapitza resistance for a ${\sim}{8}~{\rm\AA}$ thick oxide layer system was 0.503 ${\times}10^{-9}~{\rm m}^{2}{\rm K}/{\rm W}$ and for a ${\sim}{\rm 11.5}~{\rm\AA}$ thick oxide layer system was 0.518 $\,\times 10^{-9}~{\rm m}^{2}{\rm K}/{\rm W}$. These values were of the same order of magnitude as the Kapitza resistance values determined from the acoustic mismatch model and the diffuse mismatch model for the ${\rm Si}\hbox{-}{\rm SiO}_{2}$ interface. read less NOT USED (high confidence) A. Guz and J. Rushchitsky, “Establishing foundations of the mechanics of nanocomposites (Review),” International Applied Mechanics. 2011. link Times cited: 23 NOT USED (high confidence) M. Tungare, Y. Shi, N. Tripathi, P. Suvarna, and F. Shahedipour-Sandvik, “A Tersoff‐based interatomic potential for wurtzite AlN,” physica status solidi (a). 2011. link Times cited: 41 Abstract: Aluminum nitride (AlN) is a popular buffer layer and interla… read moreAbstract: Aluminum nitride (AlN) is a popular buffer layer and interlayer. The understanding of how AlN serves as a wetting and fracture‐mitigating layer relies on molecular pictures of the AlN layer and the interfaces. However, molecular dynamics (MD) simulation studies on AlN system, particularly on its wurtzite phase, have been limited. This is because most existing interatomic force fields of AlN target the less common zinc blende phase. Here, we report a new Tersoff‐based AlN force field for its wurtzite structure. This potential has been extensively tested in terms of lattice parameters, bulk modulus, cohesive energy, and heat capacity. In addition, thermal expansion coefficient (TEC) of wurtzite AlN, a key property to precisely model heterostructures, has been calculated using MD method. The value of 2.66 × 10−6 K−1 calculated at 300 K for TEC is in excellent agreement with the reported experimental value. read less NOT USED (high confidence) T. Watanabe, “Dynamic bond-order force field,” Journal of Computational Electronics. 2011. link Times cited: 8 NOT USED (high confidence) N. Legenski et al., “Force fields for metallic clusters and nanoparticles,” Journal of Computational Chemistry. 2011. link Times cited: 10 Abstract: Atomic force fields for simulating copper, silver, and gold … read moreAbstract: Atomic force fields for simulating copper, silver, and gold clusters and nanoparticles are developed. Potential energy functions are obtained for both monatomic and binary metallic systems using an embedded atom method. Many cluster configurations of varying size and shape are used to constrain the parametrization for each system. Binding energies for these training clusters were computed using density functional theory (DFT) with the Perdew‐Wang exchange‐correlation functional in the generalized gradients approximation. Extensive testing shows that the many‐body potentials are able to reproduce the DFT energies for most of the structures that were included in the training set. The force fields were used to calculate surface energies, buk structures, and thermodynamic properties. The results are in good agreement with the DFT values and consistent with the available experimental data. © 2011 Wiley Periodicals, Inc. J Comput Chem, 2011 read less NOT USED (high confidence) K. Fichthorn, Y. Tiwary, T. Hammerschmidt, P. Kratzer, and M. Scheffler, “Analytic many-body potential for GaAs(001) homoepitaxy: Bulk and surface properties,” Physical Review B. 2011. link Times cited: 14 Abstract: We employ atomic-scale simulation methods to investigate bul… read moreAbstract: We employ atomic-scale simulation methods to investigate bulk and surface properties of an analytic TersoffAbell type potential for describing interatomic interactions in GaAs. The potential is a modified form of that proposed by Albe and colleagues [Phys. Rev. B 66, 035205 (2002)] in which the cut-off parameters for the As-As interaction have been shortened. With this modification, many bulk properties predicted by the potential for solid GaAs are the same as those in the original potential, but properties of the GaAs(001) surface better match results from first-principles calculations with density-functional theory (DFT). We tested the ability of the potential to reproduce the phonon dispersion and heat capacity of bulk solid GaAs by comparing it to experiment and the overall agreement is good. In the modified potential, the GaAs(001) β2(2 × 4) reconstruction is favored under As-rich growth conditions in agreement with DFT calculations. Additionally, the binding energies and diffusion barriers for a Ga adatom on the β2(2 × 4) reconstruction generally match results from DFT calculations. These studies indicate that the potential is suitable for investigating aspects of GaAs(001) homoepitaxy. read less NOT USED (high confidence) J. Carrete, R. Longo, and L. J. Gallego, “Prediction of phonon thermal transport in thin GaAs, InAs and InP nanowires by molecular dynamics simulations: influence of the interatomic potential,” Nanotechnology. 2011. link Times cited: 12 Abstract: A number of different potentials are currently being used in… read moreAbstract: A number of different potentials are currently being used in molecular dynamics simulations of semiconductor nanostructures. Confusion can arise if an inappropriate potential is used. To illustrate this point, we performed direct molecular dynamics simulations to predict the room temperature lattice thermal conductivity λ of thin GaAs, InAs and InP nanowires. In each case, simulations performed using the classical Harrison potential afforded values of λ about an order of magnitude smaller than those obtained using more elaborate potentials (an Abell–Tersoff, as parameterized by Hammerschmidt et al for GaAs and InAs, and a potential of Vashishta type for InP). These results will be a warning to those wishing to use computer simulations to orient the development of quasi-one-dimensional systems as heat sinks or thermoelectric devices. read less NOT USED (high confidence) T. Zhao, G. Li, L. Liu, Y. Liu, and T.-hu Li, “Physical model for the growth of amorphous carbon nanotubes,” Applied Physics Letters. 2011. link Times cited: 3 Abstract: A physical model for the growth mechanism of amorphous carbo… read moreAbstract: A physical model for the growth mechanism of amorphous carbon nanotubes (ACNTs) (namely, “open tips growth and carbon clusters (Cn,n>6) adding”) has been proposed in this letter. Based on Tersoff–Brenner [Phys. Rev. B 37, 6991 (1988) and Phys. Rev. B 42, 9458 (1990)] and Lennard-Jones potential energy functions, a mathematic relationship for the growth of ACNTs was established. The predicted diameters of ACNTs from this physical model were in the range of 5–25 nm. The predicted values were in agreement with the experimental measurements in the range of 7–20 nm. read less NOT USED (high confidence) R. J. Rees, I. Snook, and E. R. Smith, “The use of analytic continuation to increase the accuracy in modelling fluid–surface interactions in cylindrical nanopores,” Molecular Simulation. 2011. link Times cited: 2 Abstract: In this paper, the mathematical formulation of a rigid/smoot… read moreAbstract: In this paper, the mathematical formulation of a rigid/smooth-walled semi-empirical fluid–surface interaction potential is discussed. Potentials of this type may be used to model the adsorption of simple fluids in cylindrical nanopores, for example, cavities in a solid material or free standing single- or multi-wall nanotubes. Analysis of the properties of the hypergeometric series functions used to evaluate these potentials indicates that modelling of the fluid–surface interactions for particles very near to the surface is improved with analytically continuous series. The derivation and implementation of a potential representing a cylindrical cavity within a continuous solid material are also presented. read less NOT USED (high confidence) B. Lee, S. Park, Y. Choi, and J.-S. Lee, “Molecular dynamics study on bulk melting induced by ultrashort pulse laser,” Journal of Mechanical Science and Technology. 2011. link Times cited: 2 NOT USED (high confidence) M. Yasuda, R. Mimura, H. Kawata, and Y. Hirai, “Computational study on structural modification of single-walled carbon nanotubes by electron irradiation,” Journal of Applied Physics. 2011. link Times cited: 12 Abstract: Molecular dynamics simulation is carried out to investigate … read moreAbstract: Molecular dynamics simulation is carried out to investigate structural modifications of single-walled carbon nanotubes by electron irradiation. Electron irradiation effects are introduced by the Monte Carlo method using an elastic collision cross section. We demonstrate the applicability of the method to the analysis of structural modifications with electron beam such as cutting, shrinking, and bending. The behavior of the carbon atoms in the nanotube during the structural modification is revealed. The simulation results also show the variation of the mechanical properties of carbon nanotubes by electron irradiation. read less NOT USED (high confidence) E. Oh, “Elastic properties of various boron-nitride structures,” Metals and Materials International. 2011. link Times cited: 31 NOT USED (high confidence) C. Ribeiro-Silva, J. Rino, L. G. Gonçalves, and A. Picinin, “An effective interaction potential for gallium phosphide,” Journal of Physics: Condensed Matter. 2011. link Times cited: 14 Abstract: An effective interatomic potential consisting of two- and th… read moreAbstract: An effective interatomic potential consisting of two- and three-body covalent interactions is used here to study the properties of gallium phosphide by molecular dynamics simulations. The many-body interatomic potential accounts for the energy scale, length scale and mechanical properties of GaP. At atmospheric pressure, the calculated melting temperature, linear thermal expansion, vibrational density of states and specific heat are in excellent agreement with experimental results. The structural phase transition induced by hydrostatic pressure at 27 GPa is also in quite good agreement with experimental findings. We also studied the energy of vacancy formation in the GaP lattice and the surface energy, which is in reasonable agreement with experimental data. read less NOT USED (high confidence) H. N. Pishkenari, S. H. Mahboobi, and A. Meghdari, “Simulation of imaging in tapping-mode atomic-force microscopy: a comparison amongst a variety of approaches,” Journal of Physics D: Applied Physics. 2011. link Times cited: 18 Abstract: Models capable of accurate simulation of microcantilever dyn… read moreAbstract: Models capable of accurate simulation of microcantilever dynamics coupled with complex tip–sample interactions are essential for interpretation and prediction of the imaging results in amplitude modulation or tapping-mode atomic-force microscopy (AM-AFM or TM-AFM). In this paper, four approaches based on combinations of lumped and finite element methods for modelling of cantilever dynamics, and van der Waals and molecular dynamics for modelling of tip–sample interactions, are used to simulate the precise imaging by AM-AFM. Based on the simulated imaging and force determination, the efficiency of different modelling schemes is evaluated. This comparison is performed considering their coincidence with the realistic behaviour of AM-AFM in imaging of nanoscale features. In the conducted simulations, a diamond tip is used to scan a C60 molecule absorbed on a graphite substrate. The effects of amplitude set-point, cantilever stiffness and quality factor on the accuracy of different modelling approaches are studied. read less NOT USED (high confidence) X. W. Zhou and R. Jones, “Effects of cutoff functions of Tersoff potentials on molecular dynamics simulations of thermal transport,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 19 Abstract: Past molecular dynamics studies of thermal transport have pr… read moreAbstract: Past molecular dynamics studies of thermal transport have predominantly used Stillinger–Weber potentials. As materials continuously shrink, their properties increasingly depend on defect and surface effects. Unfortunately, Stillinger–Weber potentials are best used for diamond-cubic-like bulk crystals. They cannot represent the energies of many metastable phases, nor can they accurately predict the energetics of defective and surface regions. To study nanostructured materials, where these regions can dominate thermal transport, the accuracy of Tersoff potentials in representing these structures is more desirable. Based upon an analysis of thermal transport in a GaN system, we demonstrate that the cutoff function of the existing Tersoff potentials may lead to problems in determining the thermal conductivity. To remedy this issue, improved cutoff schemes are proposed and evaluated. read less NOT USED (high confidence) E. Oh, “Application of the Continuum-Lattice Thermodynamics.” 2011. link Times cited: 0 NOT USED (high confidence) S. Lu, C. Cho, K. Choi, W. Choi, S.-kyo Lee, and N. Wang, “An inscribed surface model for the elastic properties of armchair carbon nanotube,” Journal of Mechanical Science and Technology. 2010. link Times cited: 9 NOT USED (high confidence) Y. Li, “Twist-enhanced stretchability of graphene nanoribbons: a molecular dynamics study,” Journal of Physics D: Applied Physics. 2010. link Times cited: 23 Abstract: Graphene nanoribbons (GNRs) have many applications in electr… read moreAbstract: Graphene nanoribbons (GNRs) have many applications in electronics due to their exceptional mechanical, electronic and thermal properties. In order to utilize GNRs for stretchable electronics, it is an important issue to enhance the stretchability of GNRs. In this work, we report that the stretchability of GNRs can be considerably strengthened by a small twist angle through molecular dynamics simulations. Compared with the tension simulation on untwisted GNRs, twist effect can help the C–C covalent bond go into large nonlinear deformation, when the twisted GNR is under tension. Therefore, the breaking strain of a twisted GNR can be 37.6% larger than that of an untwisted one at room temperature. At the same time, the stiffness of the twisted GNR could also be enhanced. Such results could be useful for further application of GNRs in stretchable electronics with multi-functionality. read less NOT USED (high confidence) I. Berinskii and A. Krivtsov, “On using many-particle interatomic potentials to compute elastic properties of graphene and diamond,” Mechanics of Solids. 2010. link Times cited: 27 NOT USED (high confidence) S. Hollerer, “Buckling Analysis of Carbon Nanotubes,” PAMM. 2010. link Times cited: 1 Abstract: In this work, an atomistic‐continuum model is applied to sin… read moreAbstract: In this work, an atomistic‐continuum model is applied to single‐walled carbon nanotubes. The constitutive behaviour is described by the interatomic Tersoff‐Brenner potential. The coupling between atomistic deformation and the deformation of the continuum is done by an expanded Cauchy‐Born rule. With the help of this model, the buckling behaviour of carbon nanotubes under different loading conditions is studied. Numerical simulation results are given for two different types of loading (axial compression, torsion). (© 2010 Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) V. Favre-Nicolin, J. Coraux, M. Richard, and H. Renevier, “Fast computation of scattering maps of nanostructures using graphical processing units,” Journal of Applied Crystallography. 2010. link Times cited: 31 Abstract: Scattering maps from strained or disordered nanostructures a… read moreAbstract: Scattering maps from strained or disordered nanostructures around a Bragg reflection can be either computed quickly using approximations and a (fast) Fourier transform or obtained using individual atomic positions. In this article, it is shown that it is possible to compute up to 4 × 1010 reflections atoms s−1 using a single graphics card, and the manner in which this speed depends on the number of atoms and points in reciprocal space is evaluated. An open-source software library (PyNX) allowing easy scattering computations (including grazing-incidence conditions) in the Python language is described, with examples of scattering from non-ideal nanostructures. read less NOT USED (high confidence) A. Kerrache, N. Mousseau, and L. J. Lewis, “Amorphous silicon under mechanical shear deformations: Shear velocity and temperature effects,” Physical Review B. 2010. link Times cited: 8 Abstract: Mechanical shear deformations lead, in some cases, to effect… read moreAbstract: Mechanical shear deformations lead, in some cases, to effects similar to those resulting from ion irradiation. Here we characterize the effects of shear velocity and temperature on amorphous silicon (\aSi) modelled using classical molecular dynamics simulations based on the empirical Environment Dependent Inter-atomic Potential (EDIP). With increasing shear velocity at low temperature, we find a systematic increase in the internal strain leading to the rapid appearance of structural defects (5-fold coordinated atoms). The impacts of externally applied strain can be almost fully compensated by increasing the temperature, allowing the system to respond more rapidly to the deformation. In particular, we find opposite power-law relations between the temperature and the shear velocity and the deformation energy. The spatial distribution of defects is also found to strongly depend on temperature and strain velocity. For low temperature or high shear velocity, defects are concentrated in a few atomic layers near the center of the cell while, with increasing temperature or decreasing shear velocity, they spread slowly throughout the full simulation cell. This complex behavior can be related to the structure of the energy landscape and the existence of a continuous energy-barrier distribution. read less NOT USED (high confidence) P. Valentini, T. Schwartzentruber, and I. Cozmuta, “Molecular dynamics simulation of O2 sticking on Pt(111) using the ab initio based ReaxFF reactive force field.,” The Journal of chemical physics. 2010. link Times cited: 45 Abstract: The molecular dynamics technique with the ab initio based cl… read moreAbstract: The molecular dynamics technique with the ab initio based classical reactive force field ReaxFF is used to study the adsorption dynamics of O(2) on Pt(111) for both normal and oblique impacts. Overall, good quantitative agreement with the experimental data is found at low incident energies. Specifically, our simulations reproduce the characteristic minimum of the trapping probability at kinetic incident energies around 0.1 eV. This feature is determined by the presence of a physisorption well in the ReaxFF potential energy surface (PES) and the progressive suppression of a steering mechanism when increasing the translational kinetic energy (or the molecule's rotational energy) because of steric hindrance. In the energy range between 0.1 and 0.4 eV, the sticking probability increases, similar to molecular beam sticking data. For very energetic impacts (above 0.4 eV), ReaxFF predicts sticking probabilities lower than experimental sticking data by almost a factor of 3 due to an overall less attractive ReaxFF PES compared to experiments and density functional theory. For oblique impacts, the trapping probability is reduced by the nonzero parallel momentum because of the PES corrugation and does not scale with the total incident kinetic energy. Furthermore, our simulations predict quasispecular (slightly supraspecular) distributions of angles of reflection, in accordance with molecular beam experiments. Increasing the beam energy (between 1.2 and 1.7 eV) causes the angular distributions to broaden and to exhibit a tail toward the surface normal because molecules have enough momentum to get very near the surface and thus probe more corrugated repulsive regions of the PES. read less NOT USED (high confidence) S. Dunham, “Multiscale modeling of nanoscale device fabrication,” 10th IEEE International Conference on Nanotechnology. 2010. link Times cited: 1 Abstract: Density function theory calculations provide the foundation … read moreAbstract: Density function theory calculations provide the foundation for hierarchical modeling of the processes controlling fabrication of nanoscale devices. The roles of atomistic methods in nanotechnology modeling are described and examples of their application given. The resulting physical models provide both deeper insight into the processes controlling device fabrication, as well as tools for technology development and optimization. read less NOT USED (high confidence) M. P. Ariza, M. Ortiz, and R. Serrano, “Long-term dynamic stability of discrete dislocations in graphene at finite temperature,” International Journal of Fracture. 2010. link Times cited: 21 NOT USED (high confidence) M. P. Ariza and M. Ortiz, “Discrete dislocations in graphene,” Journal of The Mechanics and Physics of Solids. 2010. link Times cited: 71 NOT USED (high confidence) I. V. Lysova, A. Sabirov, and A. Stepanov, “The effect of atomic dynamics on the energy loss of ions channeled in carbon nanotubes,” Journal of Surface Investigation. X-ray, Synchrotron and Neutron Techniques. 2010. link Times cited: 8 NOT USED (high confidence) A. Mavromaras et al., “Computational materials engineering: Capabilities of atomic-scale prediction of mechanical, thermal, and electrical properties of microelectronic materials,” 2010 11th International Thermal, Mechanical & Multi-Physics Simulation, and Experiments in Microelectronics and Microsystems (EuroSimE). 2010. link Times cited: 3 Abstract: Atomic-scale computational materials engineering offers an e… read moreAbstract: Atomic-scale computational materials engineering offers an exciting complement to experimental observations, revealing critical materials property data, and providing understanding which can form the basis for innovation. This contribution reviews the current state of atomic-scale simulations and their capabilities to predict mechanical, thermal, and electric properties of microelectronics materials. Specific examples are the elastic moduli of compounds such as aluminum oxide, the strength of an aluminum/silicon nitride interface, the first-principles prediction of coefficients of thermal expansion of bulk aluminum and silicon nitride, thermal conductivity of polyethylene, the prediction of the diffusion coefficient of hydrogen in metallic nickel, the calculation of dielectric properties of zinc oxide and optical properties of silicon carbide. The final example illustrates the control of the work function in the HfO2/TiN interface of a CMOS gate stack. For an increasing number of materials properties, computed values possess accuracies similar to measured data. Such accuracy has become possible due to advances in theoretical approaches and numerical algorithms combined with the astounding increase in compute power. read less NOT USED (high confidence) A. Krasheninnikov and K. Nordlund, “Ion and electron irradiation-induced effects in nanostructured materials,” Journal of Applied Physics. 2010. link Times cited: 877 Abstract: A common misconception is that the irradiation of solids wit… read moreAbstract: A common misconception is that the irradiation of solids with energetic electrons and ions has exclusively detrimental effects on the properties of target materials. In addition to the well-known cases of doping of bulk semiconductors and ion beam nitriding of steels, recent experiments show that irradiation can also have beneficial effects on nanostructured systems. Electron or ion beams may serve as tools to synthesize nanoclusters and nanowires, change their morphology in a controllable manner, and tailor their mechanical, electronic, and even magnetic properties. Harnessing irradiation as a tool for modifying material properties at the nanoscale requires having the full microscopic picture of defect production and annealing in nanotargets. In this article, we review recent progress in the understanding of effects of irradiation on various zero-dimensional and one-dimensional nanoscale systems, such as semiconductor and metal nanoclusters and nanowires, nanotubes, and fullerenes. We also consider the t... read less NOT USED (high confidence) P. Eyben et al., “Analysis and modeling of the high vacuum scanning spreading resistance microscopy nanocontact on silicon,” Journal of Vacuum Science & Technology B. 2010. link Times cited: 40 Abstract: Within this paper, the authors propose a refined high vacuum… read moreAbstract: Within this paper, the authors propose a refined high vacuum scanning spreading resistance microscopy (HV-SSRM) electromechanical nanocontact model based on experimental results as well as molecular dynamics (MD) simulation results. The formation under the tip of a nanometer-sized pocket of β-tin, a metastable metalliclike phase of silicon (also named Si-II), acting as a virtual probe is demonstrated. This gives a reasonable explanation for the superior SSRM spatial resolution as well as for the electrical properties at the Schottky-like SSRM contact. Moreover, the impact of the doping concentration on the plastic deformation of silicon for different species using micro-Raman combined with indentation experiments is studied. In order to elucidate the superior results of SSRM measurements when performed under high vacuum conditions, the impact of humidity on the mechanical deformation and Si-II formation is also analyzed using MD and SSRM experimental results. read less NOT USED (high confidence) A. Galashev, “Simulation of silicon nanoparticles stabilized by hydrogen at high temperatures,” Journal of Nanoparticle Research. 2010. link Times cited: 4 NOT USED (high confidence) S. Kim, I. Szlufarska, and D. Morgan, “Ab initio study of point defect structures and energetics in ZrC,” Journal of Applied Physics. 2010. link Times cited: 41 Abstract: The potential use of ZrC for nuclear applications in irradia… read moreAbstract: The potential use of ZrC for nuclear applications in irradiated environments makes it important to determine the structure and energetics of its point defects. In this paper the structures and energies of potential vacancy and interstitial point defects are examined by means of ab initio calculations. It is shown that C vacancies are easily formed and that their ab initio energetics are consistent with thermodynamic models of phase stability of the off-stoichiometric ZrCx (x<1) material. C interstitials are shown to be the most stable interstitial defect and form a C–C–C trimer along the ⟨101⟩ direction. C vacancies and interstitials are found to be dramatically more stable than antisite defects or Zr vacancies or interstitials. read less NOT USED (high confidence) L. Hale, X. W. Zhou, J. Zimmerman, N. Moody, R. Ballarini, and W. Gerberich, “Phase transformations, dislocations and hardening behavior in uniaxially compressed silicon nanospheres,” Computational Materials Science. 2010. link Times cited: 26 NOT USED (high confidence) K. Dayal and R. James, “Nonequilibrium molecular dynamics for bulk materials and nanostructures,” Journal of The Mechanics and Physics of Solids. 2010. link Times cited: 55 NOT USED (high confidence) Y. Mishin, M. Asta, and J. Li, “Atomistic modeling of interfaces and their impact on microstructure and properties,” Acta Materialia. 2010. link Times cited: 418 NOT USED (high confidence) C. Hwang, Y. Wang, Q. Kuo, and J.-M. Lu, “Molecular dynamics study of multi-walled carbon nanotubes under uniaxial loading,” Physica E-low-dimensional Systems & Nanostructures. 2010. link Times cited: 30 NOT USED (high confidence) R. Soulairol and F. Cleri, “Interface structure of silicon nanocrystals embedded in an amorphous silica matrix,” Solid State Sciences. 2010. link Times cited: 17 NOT USED (high confidence) Y. Xiao, W. Dong, and H. F. Busnengo, “Reactive force fields for surface chemical reactions: A case study with hydrogen dissociation on Pd surfaces.,” The Journal of chemical physics. 2010. link Times cited: 35 Abstract: An approach based on reactive force fields is applied to the… read moreAbstract: An approach based on reactive force fields is applied to the parametrization of potential energy surface (PES) for chemical reactions on surfaces with a benchmark system, H(2)/Pd(111). We show that a simple reactive force field based on the second moment approximation does not allow for obtaining reliable results of reaction dynamics for the considered system. With a more elaborate reactive force field, i.e., reactive bond order (REBO) force field, we succeeded in obtaining a reliable PES for H(2)/Pd(111). The accuracy of the constructed REBO force field is carefully checked through various tests including the comparison not only between energies calculated with density functional theory and those with REBO force field but also between the available results of ab initio molecular dynamics simulations and those with our force field. Moreover, our REBO force field is endowed with some transferability since the force field constructed with a database containing only information on H(2)/Pd(111) allows for obtaining also accurate results for H(2)/Pd(100) and qualitatively correct results for H(2)/Pd(110) without any refitting. With the help of our reactive force field, the molecular dynamics simulation for the dissociation of H(2) on the considered Pd surfaces is speeded up by five orders of magnitude compared to ab initio molecular dynamics method. The demonstrated reliability and the very high computational efficiency of reactive force fields open extremely attractive perspectives for studying large-scale complex reacting systems. read less NOT USED (high confidence) A. Galashev, “Molecular dynamics study of hydrogenated silicon clusters at high temperatures,” Molecular Physics. 2009. link Times cited: 5 Abstract: This paper reports on a study of the stability of silicon cl… read moreAbstract: This paper reports on a study of the stability of silicon clusters of intermediate size at a high temperature. The temperature dependence of the physicochemical properties of 60- and 73-atom silicon nanoparticles are investigated using the molecular dynamics method. The 73-atom particles have a crystal structure, a random atomic packing, and a packing formed by inserting a 13-atom icosahedron into a 60-atom fullerene. They are surrounded by a ‘coat’ from 60 atoms of hydrogen. The nanoassembled particle at the presence of a hydrogen ‘coat’ has the most stable number (close to four) of Si–Si bonds per atom. The structure and kinetic properties of a hollow single-layer fullerene-structured Si60 cluster are considered in the temperature range 10 K ≤ T ≤ 1760 K. Five series of calculations are conducted, with a simulation of several media inside and outside the Si60 cluster, specifically, the vacuum and interior spaces filled with 30 and 60 hydrogen atoms with and without the exterior hydrogen environment of 60 atoms. Fullerene surrounded by a hydrogen ‘coat’ and containing 60 hydrogen atoms in the interior space has a higher stability. Such clusters have smaller self-diffusion coefficients at high temperatures. The fullerene stabilized with hydrogen is stable to the formation of linear atomic chains up to the temperatures 270–280 K. read less NOT USED (high confidence) I. Chang and B.-C. Chiang, “Mechanical buckling of single-walled carbon nanotubes: Atomistic simulations,” Journal of Applied Physics. 2009. link Times cited: 14 Abstract: Various geometric sizes and helical types (i.e., armchair, z… read moreAbstract: Various geometric sizes and helical types (i.e., armchair, zigzag, and chiral) of single-walled carbon nanotubes (CNTs) are considered in molecular dynamics simulations in order to systematically examine the length-to-radius ratio and chirality effects on the buckling mechanism. The buckling strain is getting smaller as the CNT becomes slender for most nanotubes, which implies that the slender nanotubes have lower buckling resistance regardless of the radius of the CNTs. The applicability of the continuum buckling theory, which has been well developed for thin tubes, on predicting the buckling strain of the CNT is also examined. In general, the corresponding buckling strain and buckling type predicted by the continuum buckling theory could agree reasonably well with simulation results except at the transition region due to the competition of two buckling mechanisms. read less NOT USED (high confidence) E. Salmon, A. Duin, F. Lorant, P. Marquaire, and W. Goddard, “Early maturation processes in coal. Part 2: Reactive dynamics simulations using the ReaxFF reactive force field on Morwell Brown coal structures,” Organic Geochemistry. 2009. link Times cited: 176 NOT USED (high confidence) N. Sdobnyakov, A. N. Bazulev, V. Samsonov, D. A. Kul’pin, and D. N. Sokolov, “Study on the free surface energy per unit area of aluminium nanodrops using the schommers potential,” Journal of Structural Chemistry. 2009. link Times cited: 0 NOT USED (high confidence) D. K. Samarakoon and X.-Q. Wang, “Chair and twist-boat membranes in hydrogenated graphene.,” ACS nano. 2009. link Times cited: 81 Abstract: Graphane is a two-dimensional system consisting of a single … read moreAbstract: Graphane is a two-dimensional system consisting of a single planar layer of fully saturated carbon atoms, which has recently been realized experimentally through hydrogenation of graphene membranes. We have studied the stability of chair, boat, and twist-boat graphane structures using first-principles density functional calculations. Our results indicate that locally stable twist-boat membranes significantly contribute to the experimentally observed lattice contraction. The band gaps of graphane nanoribbons decrease monotonically with the increase of the ribbon width and are insensitive to the edge structure. The implications of these results for future hydrogenated graphene applications are discussed. read less NOT USED (high confidence) P. Süle and K. Heinig, “The molecular dynamics simulation of ion-induced ripple growth.,” The Journal of chemical physics. 2009. link Times cited: 20 Abstract: The wavelength-dependence of ion-sputtering induced growth o… read moreAbstract: The wavelength-dependence of ion-sputtering induced growth of repetitive nanostructures, such as ripples has been studied by molecular dynamics (MD) simulations in Si. The early stage of the ion erosion driven development of ripples has been simulated on prepatterned Si stripes with a wavy surface. The time evolution of the height function and amplitude of the sinusoidal surface profile has been followed by simulated ion-sputtering. According to Bradley-Harper (BH) theory, we expect correlation between the wavelength of ripples and the stability of them. However, we find that in the small ripple wavelength (lambda) regime BH theory fails to reproduce the results obtained by molecular dynamics. We find that at short wavelengths (lambda<35 nm) the adatom yield drops hence no surface diffusion takes place which is sufficient for ripple growth. The MD simulations predict that the growth of ripples with lambda>35 nm is stabilized in accordance with the available experimental results. According to the simulations, few hundreds of ion impacts in lambda long and few nanometers wide Si ripples are sufficient for reaching saturation in surface growth for for lambda>35 nm ripples. In another words, ripples in the long wavelength limit seems to be stable against ion-sputtering. A qualitative comparison of our simulation results with recent experimental data on nanopatterning under irradiation is attempted. read less NOT USED (high confidence) A. M. Ukpong, “Studies of the electronic and vibrational signatures of the unusual bonding geometries in melt-quenched amorphous silicon,” Molecular Physics. 2009. link Times cited: 2 Abstract: Tight-binding molecular dynamics simulations have been perfo… read moreAbstract: Tight-binding molecular dynamics simulations have been performed to investigate the effect of quenching rate of the Si melt on the resulting local structure of amorphous silicon. Different quenching rates were used to cool liquid silicon in the simulations to demonstrate that the choice of quenching rates significantly influences the resulting local structure. The calculated pair correlation functions show that the local structure is sensitive to the thermal processing of the liquid silicon melt. The use of cooling rates higher than 10−13 K s−1 appears to prevent the activation of the required structural re-arrangements necessary to stabilise the networks, causing unexpected bonding geometries to develop. The electronic signatures of the defects show that only the triangular defect structure contributes resonance states to the conduction band tail. Also, the vibrational signature of the triangular structure shows a high energy transverse optical mode at 95 meV, indicating that the defect is likely to be unstable at 300 K, although both defects contribute minimal states to the mid-gap level. read less NOT USED (high confidence) C. Zhou, J. Wu, L. Chen, Y. Wang, H. Cheng, and R. C. Forrey, “Force field for copper clusters and nanoparticles,” Journal of Computational Chemistry. 2009. link Times cited: 2 Abstract: An atomic force field for simulating copper clusters and nan… read moreAbstract: An atomic force field for simulating copper clusters and nanoparticles is developed. More than 2000 cluster configurations of varying size and shape are used to constrain the parametrization of the copper force field. Binding energies for these training clusters were computed using density functional theory. Extensive testing shows that the copper force field is fast and reliable for near‐equilibrium structures of clusters, ranging from only a few atoms to large nanoparticles that approach bulk structure. Nonequilibrium dissociation and compression structures that are included in the training set are also well described by the force field. Implications for molecular dynamics simulations and extensions to other metallic and covalent systems are discussed. © 2009 Wiley Periodicals, Inc. J Comput Chem, 2009 read less NOT USED (high confidence) J. Crocombette and L. Gélébart, “Multiscale modeling of the thermal conductivity of polycrystalline silicon carbide,” Journal of Applied Physics. 2009. link Times cited: 53 Abstract: A multiscale modeling, involving molecular dynamics and fini… read moreAbstract: A multiscale modeling, involving molecular dynamics and finite element calculations, of the degradation of the thermal conductivity of polycrystalline silicon carbide due to the thermal (Kapitza) resistances of grain boundaries is presented. Molecular dynamics simulations focus on the ⟨111⟩ family of tilt grain boundaries in cubic SiC. For large tilt angles a simple symmetry and shift procedure is used to generate the grain boundaries while for small angles the boundary structure is obtained by inserting arrays of edge dislocations. The energy and thermal resistances of the grain boundaries are presented. The latter are fed into a finite element homogenization procedure, which enables to calculate the effective thermal conductivity of the SiC polycrystal as a function of its average grain size. The decrease in the thermal conductivity of a polycrystal as a function of its grain size is qualitatively reproduced. However, available experimental values of the thermal conductivity of polycrystalline SiC tend ... read less NOT USED (high confidence) X. Shao, X. Wu, and W. Cai, “Dynamic lattice searching methods for optimization of clusters,” Frontiers of Chemistry in China. 2009. link Times cited: 3 NOT USED (high confidence) C. Björkas et al., “Interatomic potentials for the Be–C–H system,” Journal of Physics: Condensed Matter. 2009. link Times cited: 65 Abstract: Analytical bond-order potentials for beryllium, beryllium ca… read moreAbstract: Analytical bond-order potentials for beryllium, beryllium carbide and beryllium hydride are presented. The reactive nature of the formalism makes the potentials suitable for simulations of non-equilibrium processes such as plasma–wall interactions in fusion reactors. The Be and Be–C potentials were fitted to ab initio calculations as well as to experimental data of several different atomic configurations and Be–H molecule and defect data were used in determining the Be–H parameter set. Among other tests, sputtering, melting and quenching simulations were performed in order to check the transferability of the potentials. The antifluorite Be2C structure is well described by the Be–C potential and the hydrocarbon interactions are modelled by the established Brenner potentials. read less NOT USED (high confidence) S. W. Cranford, D. Sen, and M. Buehler, “Meso-origami: Folding multilayer graphene sheets,” Applied Physics Letters. 2009. link Times cited: 179 Abstract: Graphene features unique electronic, thermal, and mechanical… read moreAbstract: Graphene features unique electronic, thermal, and mechanical properties, and the flexibility and strong attraction between graphene layers promotes the formation of self-folded nanostructures. Here we study the self-folding of mono- and multilayer graphene sheets, utilizing a coarse-grained hierarchical multiscale model derived directly from atomistic simulation. Our model, developed by enforcing assertion of energy conservation, enables the simulation of graphene folding across a range of length scales from nanometers to micrometers. Through theoretical and simulation analysis we show that the critical self-folded length is πC/γ, where C and γ are the bending stiffness per unit length and the surface energy per unit length. read less NOT USED (high confidence) A. Dongare, L. Zhigilei, A. Rajendran, and B. Lamattina, “Interatomic potentials for atomic scale modeling of metal–matrix ceramic particle reinforced nanocomposites,” Composites Part B-engineering. 2009. link Times cited: 15 NOT USED (high confidence) B. Lee, “Comparative Study of the Nanomechanics of Si Nanowires,” Transactions of The Korean Society of Mechanical Engineers A. 2009. link Times cited: 0 Abstract: Mechanical properties of silicon nanowires are presented. In… read moreAbstract: Mechanical properties of silicon nanowires are presented. In particular, predictions from the calculations based on different length scales, first principles calculations, atomistic calculations, and continuum nanomechanical theory, are compared for silicon nanowires. There are several elements that determine the mechanics of silicon nanowires, and the complicated balance between these elements is studied. Specifically, the role of the increasing surface effects and reduced dimensionality predicted from theories of different length scales are compared. As a prototype, a Tersoff-based empirical potential has been used to study the mechanical properties of silicon nanowires including the Young’s modulus. The results significantly deviates from the first principles predictions as the size of wire is decreased. 기호설명 V[N] : N개의 원자를 가진 시스템의 총 결합에너지 n : 푸아송 비(Poisson’s ratio) B : 체적 탄성률(Bulk modulus) (GPa) E read less NOT USED (high confidence) J. Adhikari, “Miscibility of In x Ga1−x As alloys: a study using atomistic simulations,” Molecular Physics. 2009. link Times cited: 4 Abstract: Atomistic simulations are used in combination with the two p… read moreAbstract: Atomistic simulations are used in combination with the two potential energy functions, namely, the Valence Force Field (VFF) model and the Tersoff model, to study the solution thermodynamics of In x Ga1−x As alloy. The simulation data, in the form of a T − x diagram, is contrasted with the results obtained by using the Ho and Stringfellow approach. It is observed that for the VFF model, the upper critical solution temperature obtained from simulation data is approximately 850 K, which is higher than the 729 K predicted by the Ho and Stringfellow treatment. The composition range for which the two-phase heterogeneous region exists is wider than that predicted by the Ho and Stringfellow approach. The Tersoff model predicts a complex miscibility diagram, where the 850 K temperature corresponds to the approximate ‘eutectic’ temperature. Further improvement of model predictions may be made possible by investigation of temperature and composition dependent interaction parameter in a modified regular solution theory, and investigation of non-random, non-ideal solution models in the Ho and Stringfellow treatment, development of temperature dependent VFF model parameters and adjustment of Tersoff model parameters to account for longer range interactions which exist at temperatures above 850 K. The miscibility diagram constructed using the Tersoff model simulation data can be used to provide information on the phase stability and equilibrium Indium content at any given temperature for the crystalline solid solution. read less NOT USED (high confidence) S. Ahmad and M. Wahab, “Atomistic study of elastic constants and thermodynamic properties of zinc – blende CuBr,” Crystal Research and Technology. 2009. link Times cited: 1 Abstract: Elastic constants and thermodynamic properties of zinc blend… read moreAbstract: Elastic constants and thermodynamic properties of zinc blende CuBr are calculated using a molecular dynamics simulation based on Tersoff empirical interatomic potential. We find that the elastic modulus C11 is bigger than the other theoretical and experimental data, while C12 is somewhat small. The elastic modulus C44 is in good agreement with the theoretical calculations and experiment. Thermal expansion coefficient, specific heat capacity at constant volume and thermal conductivity are in very well agreement with experimental data. (© 2009 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) C. Qin, W. Hengan, W. Yu, and W. Xiu-xi, “Orientation and Rate Dependence of Wave Propagation in Shocked Beta-SiC from Atomistic Simulations,” Chinese Physics Letters. 2009. link Times cited: 1 Abstract: The orientation dependence of planar wave propagation in bet… read moreAbstract: The orientation dependence of planar wave propagation in beta-SiC is studied via the molecular dynamics (MD) method. Simulations are implemented under impact loadings in four main crystal directions, i.e., (100), (110), (111), and (112). The dispersion of stress states in different directions increases with rising impact velocity, which implies the anisotropic characteristic of shock wave propagation for beta-SiC materials. We also obtain the Hugoniot relations between the shock wave velocity and the impact velocity, and find that the shock velocity falls into a plateau above a threshold of impact velocity. The shock velocity of the plateaux is dependent on the shock directions, while (111) and (112) can be regarded as equivalent directions as they almost reach the same plateau. A comparison between the atomic stress from MD and the stress from Rankine–Hugoniot jump conditions is also made, and it is found that they agree with each other very well. read less NOT USED (high confidence) B. Gillespie and H. Wadley, “Atomistic examinations of the solid-phase epitaxial growth of silicon,” Journal of Crystal Growth. 2009. link Times cited: 14 NOT USED (high confidence) C. L. Bishop and M. Wilson, “The filling of flexible carbon nanotubes by molten salts,” Journal of Materials Chemistry. 2009. link Times cited: 10 Abstract: The direct filling of flexible single-walled carbon nanotube… read moreAbstract: The direct filling of flexible single-walled carbon nanotubes by model molten salts are described. The simulations advance on previous work, in which the carbon nanotubes were treated as fixed atomistic entities, by considering the carbon nanotubes as fully-flexible. The molten salts are described using two potential models, which vary in the relative energetic stability of two key bulk crystal structures. Small diameter carbon nanotubes are found to favour relatively low symmetry “ladder” and “hinged” structures, which can both be considered as formed from sections of square net sheet, in contrast to cylindrical geometries favoured when filling fixed carbon tubes. The small diameter tubes are also found to favour structures based on folding square, rather than hexagonal, nets. Additional energy minimisation calculations are performed in order to understand both the formation of the less symmetric structures and the favouring of square net-based structures. The role of the flexible tubes in controlling the filling mechanisms and kinetics is discussed. Larger diameter carbon tubes are found to favour relatively disordered internal structures. read less NOT USED (high confidence) M. C. Wu and J. Hsu, “Thermal conductivity of carbon nanotubes with quantum correction via heat capacity,” Nanotechnology. 2009. link Times cited: 27 Abstract: The molecular dynamics simulation with the use of the empiri… read moreAbstract: The molecular dynamics simulation with the use of the empirical Tersoff potential is applied to study the thermal characteristics of carbon nanotubes (CNTs). A thermal reservoir is devised to control the temperature and to exact the heat flux input. The quantum effect defining the precise temperature from the absolute zero Kelvin and up is included by applying phonon (boson) statistics to the specific heat. At low temperature, the CNT thermal conductivity increases with increasing temperature. After reaching its peak, which is limited by the length of the CNT, it decreases with temperature due to phonon–phonon interactions. The scaling law of thermal conductivity as a function of temperature and length is inferred from the simulation results, allowing prediction for CNTs of much longer length beyond what MD could simulate. read less NOT USED (high confidence) M. Maslov, A. Podlivaev, and L. A. Openov, “Nonorthogonal tight-binding model for hydrocarbons,” Physics Letters A. 2009. link Times cited: 40 NOT USED (high confidence) R. Ansari, S. Ajori, and B. Motevalli, “Mechanical properties of defective single-layered graphene sheets via molecular dynamics simulation,” Superlattices and Microstructures. 2009. link Times cited: 176 NOT USED (high confidence) H. Jeschke, M. Diakhate, and M. E. Garcia, “Molecular dynamics simulations of laser-induced damage of nanostructures and solids,” Applied Physics A. 2009. link Times cited: 25 NOT USED (high confidence) C. L. Bishop and M. Wilson, “The mechanisms for filling carbon nanotubes with molten salts: carbon nanotubes as energy landscape filters,” Journal of Physics: Condensed Matter. 2009. link Times cited: 13 Abstract: The mechanisms for filling carbon nanotubes with molten salt… read moreAbstract: The mechanisms for filling carbon nanotubes with molten salts are investigated using molecular dynamics computer simulation. Inorganic nanotubular structures, whose morphologies can be rationalized in terms of the folding, or the removal of sections from, planes of square nets are found to form. The formation mechanisms are found to follow a ‘chain-by-chain’ motif in which the structures build systematically from charge neutral M–X–M–X chains. The formation mechanisms are rationalized in terms of the ion–ion interactions (intra-chain and inter-chain terms). In addition, the mechanisms of filling are discussed in terms of a ‘hopping’ between basins on the underlying energy landscape. The role of the carbon nanotube as an energy landscape filter is discussed. read less NOT USED (high confidence) D. Graves and P. Brault, “Molecular dynamics for low temperature plasma–surface interaction studies,” Journal of Physics D: Applied Physics. 2009. link Times cited: 112 Abstract: The mechanisms of physical and chemical interactions of low … read moreAbstract: The mechanisms of physical and chemical interactions of low temperature plasmas with surfaces can be fruitfully explored using molecular dynamics (MD) simulations. MD simulations follow the detailed motion of sets of interacting atoms through integration of atomic equations of motion, using inter-atomic potentials that can account for bond breaking and formation that result when energetic species from the plasma impact surfaces. This paper summarizes the current status of the technique for various applications of low temperature plasmas to material processing technologies. The method is reviewed, and commonly used inter-atomic potentials are described. Special attention is paid to the use of MD in understanding various representative applications, including tetrahedral amorphous carbon film deposition from energetic carbon ions, the interactions of radical species with amorphous hydrogenated silicon films, silicon nanoparticles in plasmas, and plasma etching. read less NOT USED (high confidence) H. Ohta, T. Nagaoka, K. Eriguchi, and K. Ono, “An Improvement of Stillinger–Weber Interatomic Potential Model for Reactive Ion Etching Simulations,” Japanese Journal of Applied Physics. 2009. link Times cited: 13 Abstract: An approach to improve the interatomic potential model by St… read moreAbstract: An approach to improve the interatomic potential model by Stillinger and Weber (SW), which has been frequently utilized for molecular dynamics simulations of energetic-particle-induced surface reactions, was proposed. It was found that this well-known model for Si/halogen systems had a flaw in its three-body potential form if it was applied to reactive ion etching simulations. The repulsive interaction is overestimated owing to the simple summation form ∑i, j,khjik when a halogen atom is surrounded by more than three atoms. This situation always occurs when a high-energy halogen penetrates a Si lattice and, in this case, the penetration energy into the lattice is overestimated. The test simulations using our model showed that the surface structures predicted were markedly different from those using the original model. This improved model has a profound effect on the prediction of surface structures. read less NOT USED (high confidence) M. A. Locascio et al., “Tailoring the Load Carrying Capacity of MWCNTs Through Inter-shell Atomic Bridging,” Experimental Mechanics. 2009. link Times cited: 48 NOT USED (high confidence) S. Maruyama, “Molecular Dynamics Method for Micro/Nano Systems.” 2009. link Times cited: 13 Abstract: Molecular dynamics simulations are becoming more important a… read moreAbstract: Molecular dynamics simulations are becoming more important and more practical for microscale and nanoscale heat transfer problems. For example, studies of basic mechanisms of heat transfer such as phase change demand the understanding of microscopic liquid-solid contact phenomena. The efficient heat transfer at a three-phase interface (evaporation and condensation of liquid on a solid surface) becomes the singular problem in the macroscopic treatment. The nucleation theory of liquid droplets in vapor or of vapor bubbles in liquid sometimes needs to take account of nuclei of the size of molecular clusters. The effect of the surfactant on the heat and mass transfer through liquid-vapor interface is also an example of the direct effect of molecular scale phenomena on the macroscopic heat and mass transfer. Even though there has been much effort of extending our macroscopic analysis to extremely microscopic conditions in space (micrometer and nanometer scales), time (microseconds, nanoseconds and picoseconds), and rate (extremely high heat flux), there are certain limitations in the extrapolations. Hence, the bottom-up approach from molecular level is strongly anticipated. On the other hand, recent advances in microscale and nanoscale heat transfer and in nanotechnology require the detailed understandings of phase change and heat and mass transfer in nanometer and micrometer scale regimes. The chemical engineering processes to generate nanoscale structures such as carbon nanotubes or mesoporous silica structures are examples. The wetting of liquid or absorption is also important since the adhesive force is extremely important for micro/nano system and the creation of extremely large surface area is possible with nanoscale structures. The use of molecular dynamics simulations is straightforward for such a nanoscale system. Here, again, it is important to compare such nanoscale phenomena with macroscopic phenomena, because an analogy to the macroscopic system is often an important strategy in understanding a nanoscale phenomenon. Important physics intrinsic to a nanoscale system is usually found through the rational comparison 4 with a macroscopic system. In this chapter, one of the promising numerical techniques, the classical molecular dynamics method, is overviewed with a special emphasis on applications to inter-phase and heat transfer problems. The molecular dynamics methods have long been used and are well developed as a tool in statistical mechanics and physical chemistry [1, 2]. However, it is a new challenge to extend the method to the spatial and temporal scales of macroscopic heat transfer phenomena [3-6]. On the other hand, the thin film technology related … read less NOT USED (high confidence) C. Ciobanu, C. Wang, and K. Ho, “Global Optimization of 2-Dimensional Nanoscale Structures: A Brief Review,” Materials and Manufacturing Processes. 2009. link Times cited: 11 Abstract: In the cluster structure community, global optimization meth… read moreAbstract: In the cluster structure community, global optimization methods are common tools for arriving at the atomic structure of molecular and atomic clusters. The large number of local minima of the potential energy surface (PES) of these clusters, and the fact that these local minima proliferate exponentially with the number of atoms in the cluster simply demands the use of fast stochastic methods to find the optimum atomic configuration. Therefore, most of the development work has come from (and mostly stayed within) the cluster structure community. Partly due to wide availability and landmark successes of scanning tunneling microscopy (STM) and other high resolution microscopy techniques, finding the structure of periodically reconstructed semiconductor surfaces was not posed as a problem of stochastic optimization until recently, when it was shown that high-index semiconductor surfaces can posses a rather large number of local minima with such low surface energies that the identification of the global minimum becomes problematic. We have therefore set out to develop global optimization methods for systems other than clusters, focusing on periodic systems in two dimensions (2-D) as such systems currently occupy a central place in the field of nanoscience. In this article, we review some of our recent theoretical work on finding the atomic structure of surfaces, with emphasis the global optimization methods. While focused mainly on atomic structure, our account will show examples of how these development efforts contributed to elucidating several physical problems, and we will attempt to make a case for widespread use of these methods for structural problems in one and two dimensions. read less NOT USED (high confidence) J. Huh and H. Huh, “EFFECT OF HELICITY ON THE BUCKLING BEHAVIOR OF SINGLE-WALL CARBON NANOTUBES,” International Journal of Modern Physics B. 2008. link Times cited: 3 Abstract: Simulations of single-wall carbon nanotube(SWCNT)s having a … read moreAbstract: Simulations of single-wall carbon nanotube(SWCNT)s having a different chiral vector under axial compression were carried out based on molecular dynamics to investigate the effect of the helicity on the buckling behavior. Calculation was performed at room temperature for (8,8) armchair, (14,0) zigzag and (6,10) chiral single-wall carbon nanotubes. The Tersoff potential was used as the interatomic potential since it describes the C-C bonds in carbon nanotubes reliably. A conjugate gradient (CG) method was used to obtain the equilibrium configuration. Compressive force was applied at the top of a nanotube by moving the top-most atoms downward with the constant velocity of 10m/s. The buckling load, the critical strain, and the Young's modulus were calculated from the result of MD simulation. A zigzag carbon nanotube has the largest Young's modulus and buckling load, while a chiral carbon nonotube has the lowest values. read less NOT USED (high confidence) P. Krstic, C. Reinhold, and S. Stuart, “Energy and angle spectra of sputtered particles for low-energy deuterium impact of deuterated amorphous carbon,” Journal of Applied Physics. 2008. link Times cited: 16 Abstract: We study the translational, vibrational, and rotational ener… read moreAbstract: We study the translational, vibrational, and rotational energy spectra of atoms and molecules reflected or sputtered from deuterated amorphous carbon surfaces by impact of low-energy (1–30 eV) deuterium atoms. Both the rovibrational and translational energies of sputtered deuterium molecules are found to be close to 1 eV over the whole impact energy range, with approximate equipartition between rotational and vibrational modes, particularly at the higher impact energies. Sputtered carbon-containing molecules are vibrationally energetic, with rovibrational energies in the range of 1.5–2.5 eV; translational and rotational motions are less energetic, close to 0.5 eV, but hotter, with more energy per degree of freedom. The energy distributions of ejected molecules confirm the partial thermalization of the impact cascade. We also study the angular spectrum of the velocity of the outgoing particles as well as their angular momentum. While the velocity vectors are described well by a cosine distribution, a prefe... read less NOT USED (high confidence) V. Kharlamov, Y. Trushin, E. E. Zhurkin, M. Lubov, and J. Pezoldt, “Study of Si and C adatoms and SiC clusters on the silicon surface by the molecular dynamics method,” Technical Physics. 2008. link Times cited: 7 NOT USED (high confidence) D. Konatham and A. Striolo, “Molecular design of stable graphene nanosheets dispersions.,” Nano letters. 2008. link Times cited: 94 Abstract: Graphene sheets, one-atom-thick layers of carbon atoms, are … read moreAbstract: Graphene sheets, one-atom-thick layers of carbon atoms, are receiving enormous scientific attention because of extraordinary electronic and mechanical properties. These intrinsic properties will lead to innovative nanocomposite materials that could be used to produce novel transistors and thermally conductive polymeric materials. Such applications are currently hindered by the difficulty of producing large quantities of individual graphene sheets and by the propensity of these nanoparticles to agglomerate when dispersed in aqueous and/or organic matrixes. We report here molecular dynamics simulations for pristine and functionalized graphene nanosheets of 54 and 96 carbon atoms each dispersed in liquid organic linear alkanes (oils) at room conditions. For the first time, our results show that, although pristine graphene sheets agglomerate in the oils considered, graphene sheets functionalized at their edges with short branched alkanes yield stable dispersions. We characterized the simulated systems by computing radial distribution functions between the graphene sheets centers of mass, pair potentials of mean force between the graphene sheets in solution, and site-site radial distribution functions. The latter were used to determine the preferential orientation between approaching graphene sheets and the packing of the organic oils on the graphene sheets. Our results are useful not only for designing practical recipes for stabilizing graphene sheets in organic systems, but also for comparing the molecular mechanisms responsible for the graphene sheets aggregation to those that stabilize graphene sheets-containing dispersions, and for controlling the coupling between organic oils and graphene sheets used as fillers. In particular, we demonstrated that excluded-volume effects, generated by the branched architecture of the functional groups grafted on the graphene sheets, are responsible for the stabilization of small graphene sheets in the organic systems considered here. read less NOT USED (high confidence) X. Zhang, C.-Y. Wang, and C.-Y. Wang, “Application of a hybrid quantum mechanics and empirical moleculardynamics multiscale method to carbon nanotubes,” The European Physical Journal B. 2008. link Times cited: 3 NOT USED (high confidence) Y. Lin, P.-F. Yang, S. Jian, Y. Lai, and T.-C. Chen, “Experimental and Molecular Dynamics Investigations of Nanoindentation-induced Phase Transformations in Monocrystalline Silicon,” 2008 3rd International Microsystems, Packaging, Assembly & Circuits Technology Conference. 2008. link Times cited: 0 Abstract: This work presents experimental and molecular dynamics appro… read moreAbstract: This work presents experimental and molecular dynamics approaches towards deformation and phase transformation mechanisms of monocrystalline Si(100) subjected to nanoindentation. The nanoindentation experiment was conducted with a Berkovich indenter. Following the analysis using cross-sectional transmission electron microscopy with the samples prepared by focused ion beam milling, upon pressure release, metastable Si-III and Si-XII phases were identified inside the indentation-induced deformed region beneath the indent. We also demonstrated phase distributions during loading and unloading stages of spherical and Berkovich nanoindentations through molecular dynamics simulations. By searching the presence of the fifth neighboring atom within a non-bonding length, Si-III and Si-XII have been successfully distinguished from Si-I. Crystallinity of this mixed-phase was further identified by radial distribution functions. read less NOT USED (high confidence) T. Hawa and M. Zachariah, “Understanding the Effect of Hydrogen Surface Passivation and Etching on the Shape of Silicon Nanocrystals,” Journal of Physical Chemistry C. 2008. link Times cited: 15 Abstract: One of the significant challenges in the use of nanocrystals… read moreAbstract: One of the significant challenges in the use of nanocrystals, is the control of crystal shape when grown from the gas-phase. Recently, the Kortshagen group has succeeded in generating cubic Si nanocrystals in a nonequilibrium plasma. In this paper we consider the energetics of various shaped Si nanocrystals, and the role that hydrogen surface termination plays. We consider cube, truncated octahedron, icosahedron, and spherical shapes for both bare and hydrogen coated silicon nanocrystals for sizes between 2 and 10 nm. From our molecular dynamics (MD) simulations, show that for bare Si crystals, icosahedron crystals are the most energetically stable, and cubic the least. On the other hand, when hydrogenated, the cubic structure comes about because 1) the cubic structure is energetically favored when hydrogen terminated and 2) the plasma that operates with hydrogen also provides a steady source of hydrogen atoms for etching. read less NOT USED (high confidence) C. Roman, S. Roche, and Á. Rubio, “Modeling the properties of carbon nanotubes for sensor-based devices.” 2008. link Times cited: 5 Abstract: We acknowledge funding from the European Community through N… read moreAbstract: We acknowledge funding from the European Community through NoE Nanoquanta (NMP4-CT-2004-500198), SANES (NMP4-T-2006-017310), DNA-NANODEVICES (IST-2006-029192) and NANO-ERA Chemistry projects, UPV/EHU (SGIker Arina) and the Basque Governement. read less NOT USED (high confidence) T. Lu, E. Goldfield, and S. Gray, “Classical Trajectory Studies of the D + H2 → HD + H Reaction Confined in Carbon Nanotubes: Parallel Trajectories,” Journal of Physical Chemistry C. 2008. link Times cited: 7 Abstract: We use full-dimensional classical trajectories to study how … read moreAbstract: We use full-dimensional classical trajectories to study how reaction probabilities for the D + H2 → DH + H reaction are altered when the system is confined to move within various-sized carbon nanotubes (CNTs). This study focuses on D atoms initially moving parallel to the long axis of the tube. We compare our results with standard gas-phase reaction probabilities. Enhanced reaction probabilities are found for the smaller diameter CNTs, and slight quenching is found for the largest diameter CNT studied. These results are also consistent with those of a reduced-dimensional, quantum study. The origins of the confinement effects are discussed in terms of how the CNT modifies the H2 reactant state and of the modified forces experienced by the incoming D atom. read less NOT USED (high confidence) Y. Sun and K. Liew, “Application of the higher‐order Cauchy–Born rule in mesh‐free continuum and multiscale simulation of carbon nanotubes,” International Journal for Numerical Methods in Engineering. 2008. link Times cited: 53 Abstract: This paper investigates the application of a recently propos… read moreAbstract: This paper investigates the application of a recently proposed higher‐order Cauchy–Born rule in the continuum simulation and multiscale analysis of carbon nanotubes (CNTs). A mesh‐free computational framework is developed to implement the numerical computation of the hyper‐elastic constitutive model that is derived from the higher‐order Cauchy–Born rule. The numerical computation reveals that the buckling pattern of a single‐walled carbon nanotube (SWCNT) can be accurately displayed by taking into consideration the second‐order deformation gradient, and fewer mesh‐free nodes can provide a good simulation of homogeneous deformation. The bridging domain method is employed to couple the developed mesh‐free method and the atomistic simulation. The coupling method is used to simulate the bending buckling of an SWCNT and the tensile failure of an SWCNT with a single‐atom vacancy defect, and good computational results are obtained. Copyright © 2008 John Wiley & Sons, Ltd. read less NOT USED (high confidence) J. Harrison, J. Schall, M. T. Knippenberg, G. Gao, and P. Mikulski, “Elucidating atomic-scale friction using molecular dynamics and specialized analysis techniques,” Journal of Physics: Condensed Matter. 2008. link Times cited: 51 Abstract: Because all quantities associated with a given atom are know… read moreAbstract: Because all quantities associated with a given atom are known as a function of time, molecular dynamics simulations can provide unparalleled insight into dynamic processes. Many quantities calculated from simulations can be directly compared to experimental values, while others provide information not available from experiment. For example, the tilt and methyl angles of chains within a self-assembled monolayer and the amount of hydrogen in a diamond-like carbon (DLC) film are measurable in an experiment. In contrast, the atomic contact force on a single substrate atom, i.e., the force on that atom due to the tip atoms only, and the changes in hybridization of a carbon atom within a DLC film during sliding are not quantities that are currently obtainable from experiments. Herein, the computation of many quantities, including the ones discussed above, and the unique insights that they provided into compression, friction, and wear are discussed. read less NOT USED (high confidence) Z. Li, C.-yu Wang, X. Zhang, S. Ke, and W. Yang, “Transport properties of an armchair carbon nanotube with a double vacancy under stretching,” Journal of Physics: Condensed Matter. 2008. link Times cited: 3 Abstract: Structural properties of metallic single-walled carbon nanot… read moreAbstract: Structural properties of metallic single-walled carbon nanotubes with a double vacancy under stretching are studied by using a multiscale hybrid energy density method. Based on the optimized structure, the single-particle Green function method is then used to investigate the transport property. It is found that a reconstruction of the structure occurs with an increase of the imposed axial force, which alters the transmission function around the Fermi energy and will reduce the current. This reconstruction cannot be found by running a molecular dynamics simulation without a quantum description. read less NOT USED (high confidence) M. Malshe, R. Narulkar, L. Raff, M. Hagan, S. Bukkapatnam, and R. Komanduri, “Parametrization of analytic interatomic potential functions using neural networks.,” The Journal of chemical physics. 2008. link Times cited: 29 Abstract: A generalized method that permits the parameters of an arbit… read moreAbstract: A generalized method that permits the parameters of an arbitrary empirical potential to be efficiently and accurately fitted to a database is presented. The method permits the values of a subset of the potential parameters to be considered as general functions of the internal coordinates that define the instantaneous configuration of the system. The parameters in this subset are computed by a generalized neural network (NN) with one or more hidden layers and an input vector with at least 3n-6 elements, where n is the number of atoms in the system. The Levenberg-Marquardt algorithm is employed to efficiently affect the optimization of the weights and biases of the NN as well as all other potential parameters being treated as constants rather than as functions of the input coordinates. In order to effect this minimization, the usual Jacobian employed in NN operations is modified to include the Jacobian of the computed errors with respect to the parameters of the potential function. The total Jacobian employed in each epoch of minimization is the concatenation of two Jacobians, one containing derivatives of the errors with respect to the weights and biases of the network, and the other with respect to the constant parameters of the potential function. The method provides three principal advantages. First, it obviates the problem of selecting the form of the functional dependence of the parameters upon the system's coordinates by employing a NN. If this network contains a sufficient number of neurons, it will automatically find something close to the best functional form. This is the case since Hornik et al., [Neural Networks 2, 359 (1989)] have shown that two-layer NNs with sigmoid transfer functions in the first hidden layer and linear functions in the output layer are universal approximators for analytic functions. Second, the entire fitting procedure is automated so that excellent fits are obtained rapidly with little human effort. Third, the method provides a procedure to avoid local minima in the multidimensional parameter hyperspace. As an illustrative example, the general method has been applied to the specific case of fitting the ab initio energies of Si(5) clusters that are observed in a molecular dynamics (MD) simulation of the machining of a silicon workpiece. The energies of the Si(5) configurations obtained in the MD calculations are computed using the B3LYP procedure with a 6-31G(**) basis set. The final ab initio database, which comprises the density functional theory energies of 10 202 Si(5) clusters, is fitted to an empirical Tersoff potential containing nine adjustable parameters, two of which are allowed to be the functions of the Si(5) configuration. The fitting error averaged over all 10 202 points is 0.0148 eV (1.43 kJ mol(-1)). This result is comparable to the accuracy achieved by more general fitting methods that do not rely on an assumed functional form for the potential surface. read less NOT USED (high confidence) E. Sanville, A. Bholoa, R. Smith, and S. Kenny, “Silicon potentials investigated using density functional theory fitted neural networks,” Journal of Physics: Condensed Matter. 2008. link Times cited: 34 Abstract: We present a method for fitting neural networks to geometric… read moreAbstract: We present a method for fitting neural networks to geometric and energetic data sets. We then apply this method by fitting a neural network to a set of data generated using the local density approximation for systems composed entirely of silicon. In order to generate atomic potential energy data, we use the Bader analysis scheme to partition the total system energy among the constituent atoms. We then demonstrate the transferability of the neural network potential by fitting to various bulk, surface, and cluster systems. read less NOT USED (high confidence) H. S. Park and P. Klein, “A Surface Cauchy-Born model for silicon nanostructures,” Computer Methods in Applied Mechanics and Engineering. 2008. link Times cited: 81 NOT USED (high confidence) A. Kutana and K. Giapis, “Analytical carbon-oxygen reactive potential.,” The Journal of chemical physics. 2008. link Times cited: 6 Abstract: We present a reactive empirical potential with environment-d… read moreAbstract: We present a reactive empirical potential with environment-dependent bond strengths for the carbon-oxygen (CO) system. The distinct feature of the potential is the use of three adjustable parameters characterizing the bond: the strength, length, and force constant, rather than a single bond order parameter, as often employed in these types of potentials. The values of the parameters are calculated by fitting results obtained from density functional theory. The potential is tested in a simulation of oxidative unzipping of graphene sheets and carbon nanotubes. Previous higher-level theoretical predictions of graphene unzipping by adsorbed oxygen atoms are confirmed. Moreover, nanotubes with externally placed oxygen atoms are found to unzip much faster than flat graphene sheets. read less NOT USED (high confidence) T. Hammerschmidt, P. Kratzer, and M. Scheffler, “Analytic many-body potential for InAs/GaAs surfaces and nanostructures: Formation energy of InAs quantum dots,” Physical Review B. 2008. link Times cited: 46 Abstract: A parametrization of the Abell‐Tersoff potential for In, Ga,… read moreAbstract: A parametrization of the Abell‐Tersoff potential for In, Ga, As, InAs, and GaAs is presented by using both experimental data and results from density-functional calculations as input. This parametrization is optimized for the description of structural and elastic properties of bulk In, Ga, As, InAs, and GaAs, as well as for the structure and energy of several reconstructed low-index GaAs and InAs surfaces. We demonstrate the transferability to GaAs and InAs high-index surfaces and compare the results to those obtained with previously published parametrizations. Furthermore, we demonstrate the applicability to epitaxial InAs/GaAs films by comparing the Poisson ratio and elastic energy for biaxial strain, as obtained numerically with our potential and analytically from continuum-elasticity theory. Limitations for the description of point defects and surface diffusion are pointed out. This parametrization enables us to perform atomically detailed studies of InAs/GaAs heterostructures. The formation energy of InAs quantum dots on GaAs001 obtained from our atomistic approach is in good agreement with previous results from a hybrid approach. read less NOT USED (high confidence) Y. H. Lin and T.-C. Chen, “A molecular dynamics study of phase transformations in mono-crystalline Si under nanoindentation,” Applied Physics A. 2008. link Times cited: 37 NOT USED (high confidence) J. Goicochea, M. Madrid, and C. Amon, “Hierarchical modeling of heat transfer in silicon-based electronic devices,” 2008 11th Intersociety Conference on Thermal and Thermomechanical Phenomena in Electronic Systems. 2008. link Times cited: 15 Abstract: Heat transfer modeling in electronic devices has gained impo… read moreAbstract: Heat transfer modeling in electronic devices has gained importance over the last decade in the design of better performing devices. The trend towards miniaturization of these devices has led to components that operate in the micro and nano-meter and in the micro and pico-second ranges. When the characteristic dimensions of the electronic components are comparable to or smaller than the mean free path of the energy carriers (in this case phonons), the thermal conductivity, which affects their performance and reliability, reduces due to the scattering of the energy carriers with the boundaries. Several modeling approaches have been proposed in the literature to describe sub-continuum heat transport; however, the hierarchical modeling of heat transfer in electronic devices has been limited. This has precluded, at the industry level, the analysis of how changes at sub-continuum level impact the overall performance and reliability of these devices. There are numerous devices and applications whose design, performance and reliability are suitable for optimization if a hierarchical model was available. In this work, we present a hierarchical model capable of integrating the scales involved in the thermal analysis of electronic components. The integration of participating scales is achieved in three steps. First, we use molecular dynamics simulations to estimate the thermal properties (i.e. phonon relaxation times, dispersion relations and group velocities, among others), required to solve the Boltzmann transport equation (BTE). Then we apply quantum corrections (QCs) to the MD results to make them suitable for BTE, and lastly, we solve the BTE on various domains, subject to different boundary and initial conditions. Our hierarchical model is applied to silicon-based devices. read less NOT USED (high confidence) S. Mahajan, G. Subbarayan, and B. Sammakia, “Estimating Kapitza resistance between Si-SiO2 interface using molecular dynamics simulations,” 2008 11th Intersociety Conference on Thermal and Thermomechanical Phenomena in Electronic Systems. 2008. link Times cited: 13 Abstract: The interface between nano-scale films is of relevance in ma… read moreAbstract: The interface between nano-scale films is of relevance in many critical applications. Specifically, recent technological advances in semiconductor industry that utilize Silicon-on-Insulator (SOI) devices have given urgency to understanding thermal transport across Si-SiO2 interface. Estimates of interfacial (Kapitza) resistance to thermal transport across Si-SiO2 films do not appear to exist at the present time. In this paper, we develop and carryout reverse non-equilibrium molecular dynamics (NEMD) simulations by imposing known heat flux to determine the Kapitza resistance between Si-SiO2 thin films. For the Si-SiO2 interface, the average Kapitza resistance for a ~8 Aring thick oxide layer system was 0.503 times 10-9 m K/W and for a ~11.5 Aring thick oxide layer system was 0.518 times 10-9 m K/W. These values were of the same order of magnitude as the Kapitza resistance values determined from the acoustic mismatch model (AMM) and the diffuse mismatch model (DMM) for the Si-SiO2 interface. read less NOT USED (high confidence) T.-Y. Zhang, M. Luo, and W. Chan, “Size-dependent surface stress, surface stiffness, and Young’s modulus of hexagonal prism [111] β-SiC nanowires,” Journal of Applied Physics. 2008. link Times cited: 96 Abstract: The present work studies the size-dependent surface stress, … read moreAbstract: The present work studies the size-dependent surface stress, surface stiffness, and Young’s modulus of a prism crystalline nanowire, which is theoretically treated to be composed of a hypothetical nanowire phase, a true two-dimensional geometric surface phase, and a true one-dimensional geometric edge phase. The hypothetical nanowire phase could be elastically deformed due to relaxation of a free-standing nanowire, without any applied load, with respect to its bulk counterpart. The initially deformed nanowire phase is taken as reference in the present work in the determination of excess surface and edge energies. The theoretical results indicate that the edge phase causes the nominal specific surface energy, surface stress, and surface stiffness to be size dependent, and the surface phase and the edge phase make the nominal Young’s modulus size dependent. The edge and surface effects are more significant as the cross-sectional area of a nanowire becomes smaller. Molecular dynamics simulations on hexagonal ... read less NOT USED (high confidence) C. Y. Wang and L. Zhang, “An elastic shell model for characterizing single-walled carbon nanotubes,” Nanotechnology. 2008. link Times cited: 69 Abstract: This paper proposes a two-dimensional elastic shell model to… read moreAbstract: This paper proposes a two-dimensional elastic shell model to characterize the deformation of single-walled carbon nanotubes using the in-plane rigidity, Poisson ratio, bending rigidity and off-plane torsion rigidity as independent elastic constants. It was found that the off-plane torsion rigidity of a single-walled carbon nanotube is not zero due to the off-plane change in the π-orbital electron density on both sides of the nanotube. It was concluded that a three-dimensional elastic shell model of single-walled carbon nanotubes can be established with well-defined effective thickness. read less NOT USED (high confidence) B. Morrow and A. Striolo, “Platinum nanoparticles on carbonaceous materials: the effect of support geometry on nanoparticle mobility, morphology, and melting,” Nanotechnology. 2008. link Times cited: 35 Abstract: Molecular dynamics simulations have been used to investigate… read moreAbstract: Molecular dynamics simulations have been used to investigate the morphology and mobility of platinum nanoparticles of various sizes supported by carbon materials. The embedded-atom method was used to model Pt–Pt interactions, and the Lennard-Jones potential was used to model the Pt–C interactions. The C atoms in the supports were held fixed during the simulations. The supports considered were a single graphite sheet and three bundles of carbon nanotubes. Three sizes of Pt nanoparticles were considered: 130 atoms, 249 atoms, and 498 atoms (Pt130, Pt249, and Pt498 respectively). It was found that for all three sizes, diffusion coefficients were approximately one order of magnitude higher for graphite-supported nanoparticles than for carbon nanotube-supported nanoparticles. In addition, increasing the size of the nanoparticle decreased its diffusion coefficient, with Pt130 having the highest and Pt498 the lowest diffusion coefficients. More interestingly, we found that for the Pt nanoparticles of all three sizes the diffusion coefficient increases as temperature increases, reaches a maximum at the melting temperature of the nanoparticle, and then decreases. The melting temperature was found to be strongly dependent on the particle size, but only slightly dependent on the features of the supports. While the size of the nanoparticle was seen to affect the particles’ mobility, it did not significantly affect their structure. The nanoparticles supported by graphite have density profiles that indicate a highly ordered, fcc-like structure, while the particles supported by carbon nanotubes have a more disordered structure. An order parameter confirms that the nanoparticles’ structure depends on the support morphology. read less NOT USED (high confidence) A. Galashev, “Thermal instability of silicon fullerenes stabilized with hydrogen: Computer simulation,” Semiconductors. 2008. link Times cited: 4 NOT USED (high confidence) A. Galashev and I. A. Izmodenov, “Computer investigation of the structure of Si73 clusters surrounded by hydrogen,” Glass Physics and Chemistry. 2008. link Times cited: 5 NOT USED (high confidence) J. Zhou and R. Huang, “Internal lattice relaxation of single-layer graphene under in-plane deformation,” Journal of The Mechanics and Physics of Solids. 2008. link Times cited: 163 NOT USED (high confidence) F. Gou, A. Kleyn, and M. Gleeson, “The application of molecular dynamics to the study of plasma–surface interactions: CF x with silicon,” International Reviews in Physical Chemistry. 2008. link Times cited: 24 Abstract: In this paper, we provide an overview of the use of molecula… read moreAbstract: In this paper, we provide an overview of the use of molecular dynamics for simulations involving energetic particles (Ar, F, and CF x ) interacting with silicon surfaces. The groups (including our own) that have performed this work are seeking to advance the fundamental understanding of plasma interactions at surfaces. Although this paper restricts itself largely to the systems bracketed above, the approach and general mechanisms involved are applicable to a much wider range of systems. Proper description of plasma-related systems generally requires a large number of atoms in order to correctly characterize the interactions. Consequently, the bulk of the present work, and the main focus of the text, is based on classical molecular dynamics. In MD simulations, one of the most critical considerations is the selection of the interatomic potential. For simulations involving silicon etching, the choice is typically made between the Stillinger–Weber and the Tersoff–Brenner potentials. An outline of the two potentials is given, including efforts that have been made to improve and optimize the potentials and their parameters. Subsequently, we focus on some of the practical details involved in establishing the simulation process and outline how various parameters (e.g. heat bath, relaxation time and cell size) influence the simulation results. These sections deal with the influences of the heat bath (application time, rising time), the time-step and total integration time of molecular trajectories, the relaxation of the sample (during and post-etching) and the sample size. The approach is essentially pedagogical in nature, and may be of interest to those less familiar with the techniques. To illustrate the type of results that can be produced we present a case study for 100 eV interacting with a Si(100)-2 × 1 surface at different sample temperatures (100–800 K). The simulations reveal details of the change in etch rate, the F-turnover and the standing coverage of functional groups as a function of the temperature. Our primary interest is in studies with relevance for plasma–surface interactions. We discuss the general mechanisms that are most important in plasma–surface interactions and give an overview of some of the wide range of results that have been produced for various systems. The results presented illustrate that careful consideration must be given to the precise configuration of the plasma system. Numerous factors, including the chemical species, the energy and chemical mix of the incident particles and the surface composition and structure can play a crucial role in determining the net outcome of the interaction. read less NOT USED (high confidence) J. Schall, G. Gao, and J. Harrison, “Elastic constants of silicon materials calculated as a function of temperature using a parametrization of the second-generation reactive empirical bond-order potential,” Physical Review B. 2008. link Times cited: 48 Abstract: A parametrization for silicon is presented that is based on … read moreAbstract: A parametrization for silicon is presented that is based on the second-generation reactive empirical bondorder REBO formalism Brenner, Shenderova, Harrison, Stuart, Ni, and Sinnott J. Phys.: Condens. Matter 14, 783 2002 . Because it shares the same analytic form as Brenner’s second-generation REBO, this new potential is a step toward a single potential that can model many atom systems that contain C, Si, and H, where bond breaking and bond making are important. The widespread use of Brenner’s REBO potential, its ability to model both zero-Kelvin elastic constants of diamond and the temperature dependence of the elastic constants, and the existence of parameters for many atom types were the motivating factors for obtaining this parametrization for Si. While Si-C-H classical bond-order potentials do exist, they are based on Brenner’s original formalism. This new parametrization is validated by examining the structure and stability of a large number of crystalline silicon structures, by examining the relaxation energies of point defects, the energies of silicon surfaces, the effects of adatoms on surface energies, and the structures of both liquid silicon and amorphous silicon. Finally, the elastic constants of diamond-cubic and amorphous silicon between 0 and 1100 K are calculated with this new parametrization and compared to values calculated using a previously published potential. read less NOT USED (high confidence) J. Kermode, “Multiscale hybrid simulation of brittle fracture.” 2008. link Times cited: 5 NOT USED (high confidence) A. Liu and S. Stuart, “Empirical bond‐order potential for hydrocarbons: Adaptive treatment of van der Waals interactions,” Journal of Computational Chemistry. 2008. link Times cited: 31 Abstract: Bond‐order potentials provide a powerful class of models for… read moreAbstract: Bond‐order potentials provide a powerful class of models for simulating chemically reactive systems with classical potentials. In these models, the covalent bonding interactions adapt to the environment, allowing bond strength to change in response to local chemical changes. However, the non‐bonded interactions should also adapt in response to chemical changes, an effect which is neglected in current bond‐order potentials. Here the AIREBO potential is extended to include adaptive Lennard‐Jones terms, allowing the van der Waals interactions to vary adaptively with the chemical environment. The resulting potential energy surface and its gradient remain continuous, allowing it to be used for dynamics simulations. This new potential is parameterized for hydrocarbons, and is fit to the energetics and densities of a variety of condensed phase molecular hydrocarbons. The resulting model is more accurate for modeling aromatic and other unsaturated hydrocarbon species, for which the original AIREBO potential had some deficiencies. Testing on compounds not used in the fitting procedure shows that the new model performs substantially better in predicting heats of vaporization and pressures (or densities) of condensed‐phase molecular hydrocarbons. © 2007 Wiley Periodicals, Inc. J Comput Chem, 2008 read less NOT USED (high confidence) Y. Ma and S. Garofalini, “Molecular dynamics simulations of beta-SiC using both fixed charge and variable charge models.,” The Journal of chemical physics. 2008. link Times cited: 6 Abstract: In this paper, molecular dynamics simulations have been perf… read moreAbstract: In this paper, molecular dynamics simulations have been performed using both fixed charge and variable charge models. In the fixed charge model, partial charges are introduced to Si and C atoms to model the charge transfer observed in first principles studies. The calculated phonon dispersions, elastic constants, and lattice constants are in good accuracy. Variable charge model is also used to obtain geometry and connectivity dependent atomic charges. Our results show that although the variable charge model may not be advantageous in the study of ordered structures, it is important in describing structural disorders such as vacancies. read less NOT USED (high confidence) C. Wang and L. Zhang, “A critical assessment of the elastic properties and effective wall thickness of single-walled carbon nanotubes,” Nanotechnology. 2008. link Times cited: 145 Abstract: This paper discusses the fundamental issues of the elastic p… read moreAbstract: This paper discusses the fundamental issues of the elastic properties and effective wall thickness of single-walled carbon nanotubes (SWCNTs). It provides an in-depth analysis based on the rationale of the nanoscale-to-macroscale deformation relationship of SWCNTs and carries out a critical assessment of the diverse theoretical predictions in the literature. It was found that the in-plane stiffness of SWCNTs is a mechanics quantity that has been consistently reflected by the majority of the existing models. However, a further systematic study is necessary to clarify the dilemma of the wall thickness of SWCNTs. read less NOT USED (high confidence) J. Yim, M. Falk, and I. Boyd, “Modeling low energy sputtering of hexagonal boron nitride by xenon ions,” Journal of Applied Physics. 2008. link Times cited: 20 Abstract: The sputtering of hexagonal boron nitride due to low energy … read moreAbstract: The sputtering of hexagonal boron nitride due to low energy xenon ion bombardments occurs in various applications including fabrication of cubic boron nitride and erosion of Hall thruster channel walls. At low ion energies, accurate experimental characterization of sputtering increases in difficulty due to the low yields involved. A molecular dynamics model is employed to simulate the sputtering process and to calculate sputter yields for ion energies ranging from 10 to 350 eV. The results are compared to experimental data and a semiempirical expression developed by Bohdansky [Nucl. Instrum. Methods Phys. Res. B 2, 587 (1984)] is found to adequately describe the simulation data. Surface temperature effects are also investigated, and the sputter yield at 850 K is approximately twice that at 423 K. read less NOT USED (high confidence) F. Shimojo, R. Kalia, A. Nakano, and P. Vashishta, “Divide-and-conquer density functional theory on hierarchical real-space grids: Parallel implementation and applications,” Physical Review B. 2008. link Times cited: 60 Abstract: A linear-scaling algorithm based on a divide-and-conquer ! D… read moreAbstract: A linear-scaling algorithm based on a divide-and-conquer ! DC" scheme has been designed to perform large-scale molecular-dynamics ! MD" simulations, in which interatomic forces are computed quantum mechanically in the framework of the density functional theory ! DFT" . Electronic wave functions are represented on a real-space grid, which is augmented with a coarse multigrid to accelerate the convergence of iterative solutions and with adaptive fine grids around atoms to accurately calculate ionic pseudopotentials. Spatial decomposition is employed to implement the hierarchical-grid DC-DFT algorithm on massively parallel computers. The largest benchmark tests include 11.8! 10 6 -atom ! 1.04! 10 12 electronic degrees of freedom" calculation on 131 072 IBM BlueGene/L processors. The DC-DFT algorithm has well-defined parameters to control the data locality, with which the solutions converge rapidly. Also, the total energy is well conserved during the MD simulation. We perform first-principles MD simulations based on the DC-DFT algorithm, in which large system sizes bring in excellent agreement with x-ray scattering measurements for the pairdistribution function of liquid Rb and allow the description of low-frequency vibrational modes of graphene. The band gap of a CdSe nanorod calculated by the DC-DFT algorithm agrees well with the available conventional DFT results. With the DC-DFT algorithm, the band gap is calculated for larger system sizes until the result reaches the asymptotic value. DOI: 10.1103/PhysRevB.77.085103 read less NOT USED (high confidence) E. Oh and J. C. Slattery, “Nanoscale thermodynamics of multicomponent, elastic, crystalline solids: diamond, silicon, and silicon carbide,” Philosophical Magazine. 2008. link Times cited: 9 Abstract: This paper extends the thermodynamic behaviour of two-dimens… read moreAbstract: This paper extends the thermodynamic behaviour of two-dimensional and simple three-dimensional crystalline solids developed by Oh et al. and Slattery and Lagoudas to more complex, multicomponent, three-dimensional, elastic, crystalline solids. The analysis recognizes that the Helmholtz free energy is an explicit function of the lattice vectors defining the crystalline structure. From this theory, we obtain the stress-deformation behaviour and the elastic properties of diamond, silicon, and silicon carbide, which are face-centred, cubic, crystal structure. These are compared with available experimental values. read less NOT USED (high confidence) K. Nomura, R. Kalia, A. Nakano, and P. Vashishta, “A scalable parallel algorithm for large-scale reactive force-field molecular dynamics simulations,” Comput. Phys. Commun. 2008. link Times cited: 77 NOT USED (high confidence) X. J. Liu, J. P. Yang, and Y. Yang, “Heat conduction analysis of nano-tip and storage medium in thermal-assisted data storage using molecular dynamics simulation,” Molecular Simulation. 2008. link Times cited: 2 Abstract: In the thermal-assisted data storage technologies, the behav… read moreAbstract: In the thermal-assisted data storage technologies, the behavior of heat transfer between the nano-tips and the storage medium during thermo-mechanical data bit formation process is a critical factor affecting the areal storage density, data bit writing/reading speed and system reliability. In this paper, the thermal properties of a nano-tip are analyzed using the non-equilibrium molecular dynamics simulation. The simulated results show that the effects of the nano-structural configuration and boundary conditions on the thermal transport are remarkable, which can be attributed to the phonon boundary-scattering and possible phonon spectrum modification. Furthermore, the heat transfer between the nano-tip and the silicon medium film is simulated. The results show that the medium film can be efficiently heated locally with no pressure force. For a tip-medium contact area of 5.31 nm2, an area of about 95.5 nm2 on the medium surface can be heated with a temporal resolution of 0.11 ns. This time period is much smaller than the conduction timescale ( ≈ 2 μs) on the nano-tip in the heat-assisted scanning probe-based data storage technology during data bit writing process. read less NOT USED (high confidence) R. Sauer and S. Li, “An atomic interaction‐based continuum model for computational multiscale contact mechanics,” PAMM. 2007. link Times cited: 38 Abstract: A computational multiscale contact mechanics model is presen… read moreAbstract: A computational multiscale contact mechanics model is presented which describes the interaction between deformable solids based on the interaction of individual atoms or molecules. The contact model is formulated in the framework of large deformation continuum mechanics and combines the approaches of molecular modelling [1] and continuum contact mechanics [2]. In the following a brief overview of the contact model is given. Further details can be found in [3], [4] and [5]. (© 2008 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) I. Lebedeva, A. Knizhnik, and B. Potapkin, “The kinetics of carbon nanostructure 2D–3D transformation,” Russian Journal of Physical Chemistry B. 2007. link Times cited: 6 NOT USED (high confidence) V. Verma, V. Jindal, and K. Dharamvir, “Elastic moduli of a boron nitride nanotube,” Nanotechnology. 2007. link Times cited: 177 Abstract: The elastic properties of boron nitride nanotubes have been … read moreAbstract: The elastic properties of boron nitride nanotubes have been calculated using the Tersoff–Brenner potential which is a bond order potential used successfully previously for carbon nanotubes. In the present calculation, the same form of potential is used with adjusted parameters for hexagonal boron nitride. The Young’s modulus and shear modulus for single-walled armchair and zigzag tubes of different radii have been calculated. The effects of tube diameter are investigated. The computational results show the variation of Young’s modulus and shear modulus of boron nitride nanotubes with nanotube diameter. The results have been compared with available data, experimental as well as calculated. read less NOT USED (high confidence) Y. Kowaki, A. Harada, F. Shimojo, and K. Hoshino, “Radius dependence of the melting temperature of single-walled carbon nanotubes: molecular-dynamics simulations,” Journal of Physics: Condensed Matter. 2007. link Times cited: 20 Abstract: We have investigated the radius dependence of the melting te… read moreAbstract: We have investigated the radius dependence of the melting temperature of single-walled carbon nanotubes (SWCNTs) by classical molecular-dynamics (MD) simulations using the environment-dependent interatomic potential (EDIP) proposed by Marks. Here we define the ‘melting temperature’ as a temperature at which there occurs a thermal instability of SWCNTs. We have carried out molecular-dynamics simulations at several temperatures for carbon nanotubes with various radii and estimated the ‘melting temperature’ based on the temperature dependence of the radial distribution functions, mean-square displacements and atomic configurations. It is shown that the ‘melting temperature’ of SWCNTs decreases with decreasing radius. The origin of this radius dependence of the melting temperature of SWCNTs is discussed in relation to the stability of SWCNTs energetically based on the strain energy of carbon nanotubes. read less NOT USED (high confidence) T. Roussel, A. Didion, R. Pellenq, R. Gadiou, C. Bichara, and C. Vix‐Guterl, “Experimental and Atomistic Simulation Study of the Structural and Adsorption Properties of Faujasite Zeolite−Templated Nanostructured Carbon Materials†,” Journal of Physical Chemistry C. 2007. link Times cited: 51 Abstract: Nanostructured carbon materials were obtained by templating … read moreAbstract: Nanostructured carbon materials were obtained by templating faujasite zeolites. This was achieved by liquid infiltration of furfuryl alcool and chemical vapor deposition of propylene and acetonitrile. These carbon materials were characterized by adsorption of gaseous nitrogen and carbon dioxide, and the carbon structure was investigated by X-ray diffraction (XRD). They exhibit a very large pore volume in the micropore region (i.e., narrower than 2 nm), and the XRD spectra show the presence of a nanostructured carbon material with a well-defined unit cell whose size and symmetry are imposed by the zeolite template. We made use of Grand Canonical Monte Carlo simulation of carbon adsorption in order to obtain numerical models of such materials and study their texture and mechanical and adsorption properties on an atomistic scale. The carbon−carbon interactions were modeled within the frame of the tight binding and the reactive bond order (REBO) formalisms, while carbon−zeolite interactions were assumed to be... read less NOT USED (high confidence) Z. Zhong, X. Wang, and X. Feng, “Effects of pressure and temperature on sp^3 fraction in diamondlike carbon materials,” Journal of Materials Research. 2007. link Times cited: 4 Abstract: In this work, formation of diamond coating is studied using … read moreAbstract: In this work, formation of diamond coating is studied using large-scale molecular dynamics (MD) simulation. The diamond coating is studied to explore how and to what extent the temperature and pressure affects the deposition structure. To analyze the coating results, the radial distribution function and the fraction of diamond (sp^3 bonds) is calculated. It is found that the sp^3 fraction in the deposition structure increases with the temperature and pressure. When the pressure becomes large enough (10 GPa), the effect of the pressure on the coating structure is quite small and the sp^3 fraction tends to be constant. read less NOT USED (high confidence) I. Berinskii, E. Ivanova, A. Krivtsov, and N. Morozov, “Application of moment interaction to the construction of a stable model of graphite crystal lattice,” Mechanics of Solids. 2007. link Times cited: 17 NOT USED (high confidence) S.-M. Jeong and T. Kitamura, “Atomistic Simulation on the Phase Transformation of Silicon under Nonhydrostatic Stress,” Japanese Journal of Applied Physics. 2007. link Times cited: 7 Abstract: Silicon transforms from the diamond cubic structure (Si I) t… read moreAbstract: Silicon transforms from the diamond cubic structure (Si I) to the β-tin structure (Si II) under mechanical stress. The phase transition is affected by the stress state. In this study, we conduct an atomistic simulation to determine the ideal phase transition criterion of silicon under nonhydrostatic stress. We also verify the effects of nonequivalent transverse stress and non-<100>-oriented transverse stress. As a result, we find that the magnitude of transverse stress mainly affects the phase transition, but both transverse differential stress and non<100>-oriented transverse stress have a negligible little effect. read less NOT USED (high confidence) T. Hawa and M. Zachariah, “Molecular dynamics simulation and continuum modeling of straight-chain aggregate sintering : Development of a phenomenological scaling law,” Physical Review B. 2007. link Times cited: 27 Abstract: Atomistic molecular dynamics simulation and a simple continu… read moreAbstract: Atomistic molecular dynamics simulation and a simple continuum viscous flow model are employed to investigate the sintering of straight-chain nanoparticle aggregates. The results are used to develop a phenomenological sintering scaling law. The chain aggregates investigated consist of up to 80 primary particles of silicon, with primary particles of 2.5–7 nm in diameter. We found that sintering of chain aggregates consists of three steps. In step a , reaction between particles to minimize surface defects and development of a cylindrical like shape comprised an induction period. Step b consisted of contraction of the cylinder, which actually consisted of two contraction stages. The first stage was the local contraction stage where sintering occurs only at the ends of the particle chain, and the second stage involved the global contraction. The last step was the nominal sintering process from an oval to spherical shape. As expected, sintering time increases with increasing chain length, with the exception that very long chains fragmented. The sintering times normalized by the primary particle diameter showed a universal relationship which only depends on chain length. These results were found to be consistent with a mathematical model we develop based on continuum viscous flow. The model was able to predict the sintering time in excellent agreement with results obtained from molecular dynamics simulation for any chain length and any primary particle size for straight nanoparticle chain aggregates. The results for sintering times for aggregate chains could be summarized with a power law modification of the Frenkel viscous flow equation, to include a dependence on the number of particle connections in a chain aggregate: t= tFrenkel * N−1 0.68. read less NOT USED (high confidence) J. N. Ding, B. Kan, G. Cheng, and Q. Wang, “A new atomic-scale finite element simulation method for nanomechanics of single-walled carbon nanotubes,” 2007 7th IEEE Conference on Nanotechnology (IEEE NANO). 2007. link Times cited: 1 Abstract: In this paper, a new atomic-scale finite element method base… read moreAbstract: In this paper, a new atomic-scale finite element method based on the nonlinear spring model is developed for SWCNTs (single-walled carbon nanotubes). Atoms are chosen as nodes and supposed to be connected with each other in the finite element model by line-springs and torsion springs, whose mechanical parameters are determined by Tersoff-Brenner potential. The process of establishing global stiffness matrix is given in detail. Some of the application examples are shown, and the results are compared with those obtained by other methods. It is found that by using this method, the simulation can be accelerated without losing accuracy. read less NOT USED (high confidence) T. Hawa and M. Zachariah, “Development of a phenomenological scaling law for fractal aggregate sintering from molecular dynamics simulation,” Journal of Aerosol Science. 2007. link Times cited: 28 NOT USED (high confidence) W.-D. Hsu, I. Jang, and S. Sinnott, “Chemical modification of the poly(vinylidene fluoride-trifluoroethylene) copolymer surface through fluorocarbon ion beam deposition,” Journal of Vacuum Science and Technology. 2007. link Times cited: 3 Abstract: Classical molecular dynamics simulations are used to study t… read moreAbstract: Classical molecular dynamics simulations are used to study the effects of continuous fluorocarbon ion beam deposition on a poly(vinylidene fluoride-trifluoroethylene) [P(VDF-trFE)] surface, a polymer with electromechanical properties. Fluorocarbon plasma processing is widely used to chemically modify surfaces and deposit thin films. It is well accepted that polyatomic ions and neutrals within low-energy plasmas have a significant effect on the surface chemistry induced by the plasma. The deposition of mass selected fluorocarbon ions is useful to isolate the effects specific to polyatomic ions. Here, the differences in the chemical interactions of C3F5+ and CF3+ ions with the P(VDF-trFE) surface are examined. The incident energy of the ions in both beams is 50eV. The CF3+ ions are predicted to be more effective at fluorinating the P(VDF-trFE) surface than C3F5+ ions. At the same time, the C3F5+ ions are predicted to be more effective at growing fluorocarbon thin films. The simulations also reveal how the d... read less NOT USED (high confidence) P. Krstic, C. Reinhold, and S. Stuart, “Chemical sputtering from amorphous carbon under bombardment by deuterium atoms and molecules,” New Journal of Physics. 2007. link Times cited: 46 Abstract: We perform classical molecular dynamics simulations of the c… read moreAbstract: We perform classical molecular dynamics simulations of the chemical sputtering of deuterated amorphous carbon surfaces by D and D2, at energies of 7.5–30 eV D−1. Particular attention is paid to the preparation of the target surfaces for varying impact projectile fluence, energy and species, to the vibrational state of D2 projectiles, as well as to the variation in sputtering yields with target surface and impact projectile. The methane and acetylene sputtering yields per deuteron, obtained with atomic and molecular projectiles, agree quantitatively with recent experimental values. We study the distribution of sputtered species, as well as their kinetic energy and angular spectra. read less NOT USED (high confidence) C. Björkas and K. Nordlund, “Comparative study of cascade damage in Fe simulated with recent potentials,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 102 NOT USED (high confidence) K. Sastry, D. Goldberg, and D. D. Johnson, “Scalability of a Hybrid Extended Compact Genetic Algorithm for Ground State Optimization of Clusters,” Materials and Manufacturing Processes. 2007. link Times cited: 28 Abstract: We analyze the utility and scalability of extended compact g… read moreAbstract: We analyze the utility and scalability of extended compact genetic algorithm (eCGA)—a genetic algorithm (GA) that automatically and adaptively mines the regularities of the fitness landscape using machine learning methods and information theoretic measures—for ground state optimization of clusters. In order to reduce the computational time requirements while retaining the high reliability of predicting near-optimal structures, we employ two efficiency-enhancement techniques: (1) hybridizing eCGA with a local search method, and (2) seeding the initial population with lowest energy structures of a smaller cluster. The proposed method is exemplified by optimizing silicon clusters with 4–20 atoms. The results indicate that the population size required to obtain near-optimal solutions with 98% probability scales sub linearly (as Θ(n 0.83)) with the cluster size. The total number of function evaluations (cluster energy calculations) scales sub-cubically (as Θ(n 2.45)), which is a significant improvement over exponential scaling of poorly designed evolutionary algorithms. read less NOT USED (high confidence) F. Gou, M. Chuanliang, C. Lingzhouting, and Q. Qian, “Atomic simulation of SiC etching by energetic SiF3,” Journal of Vacuum Science and Technology. 2007. link Times cited: 3 Abstract: The authors present results from molecular-dynamics simulati… read moreAbstract: The authors present results from molecular-dynamics simulations of SiF3 impact on SiC (100) surfaces at normal incidence and over a range of energies of 10, 50, and 150eV. The surface temperatures are set to 300K for all energies and 600K for 150eV. The uptake of Si atoms is sensitive to the incident energy and temperature, while the uptake of F atoms is not very sensitive to the incident energy and temperature. The simulation results show that the etching yield of Si is higher than that of C. After 30 ML (monolayers) fluence, SiF3 does not etch SiC. The F-containing reaction layer is sensitive to the incident energy. The thickness of the reaction layer increases with the incident energy. In the reaction layer, SiF, SiF2, CF, and CF2 species are dominant. In etch products, atomic F etch products are dominant. Si atoms in SiC are mainly sputtered as SiFx (x=1–4). C atoms in SiC are sputtered as larger SixCyFZ species. read less NOT USED (high confidence) N. Chakraborti, R. Jayakanth, S. Das, E. D. Çalişir, and S. Erkoç, “Evolutionary and Genetic Algorithms Applied to Li+-C System: Calculations Using Differential Evolution and Particle Swarm Algorithm,” Journal of Phase Equilibria and Diffusion. 2007. link Times cited: 28 NOT USED (high confidence) X. W. Zhou and H. Wadley, “A potential for simulating the atomic assembly of cubic AB compounds,” Computational Materials Science. 2007. link Times cited: 7 NOT USED (high confidence) N. Kaur, K. Dharamvir, and V. Jindal, “Dimerization and fusion of two C60 molecules,” Chemical Physics. 2007. link Times cited: 8 NOT USED (high confidence) S. L. Mielke, T. Belytschko, and G. Schatz, “Nanoscale fracture mechanics.,” Annual review of physical chemistry. 2007. link Times cited: 42 Abstract: Theoretical calculations on undefected nanoscale materials p… read moreAbstract: Theoretical calculations on undefected nanoscale materials predict impressive mechanical properties. In this review we summarize the status of experimental efforts to directly measure the fracture strengths of inorganic and carbon nanotubes and discuss possible explanations for the deviations between the predicted and observed values. We also summarize the role of theory in understanding the molecular-level origin of these deviations. In particular, we consider the role of defects such as vacancies, Stone-Wales defects, adatoms and ad-dimers, chemical functionalization, and oxidative pitting. read less NOT USED (high confidence) X. W. Zhou and H. Wadley, “A potential for simulating the atomic assembly of cubic elements,” Computational Materials Science. 2007. link Times cited: 10 NOT USED (high confidence) T. Dumitricǎ and R. James, “Objective Molecular Dynamics,” Bulletin of the American Physical Society. 2007. link Times cited: 95 NOT USED (high confidence) L. Sun, C. Le, F. Saied, and J. Murthy, “Performance of a Parallel Molecular Dynamics Program for Computation of Thermal Properties,” Numerical Heat Transfer, Part B: Fundamentals. 2007. link Times cited: 5 Abstract: The parallel performance of classical molecular dynamics sim… read moreAbstract: The parallel performance of classical molecular dynamics simulations of the thermal properties of solid-state materials is evaluated. Computations are validated by predicting the bulk silicon thermal conductivity as a function of temperature. The performance of the computational algorithm and software are tested on three different architectures, including the IBM BlueGene, the IBM Power 4 +, and an Intel Xeon Linux cluster, corresponding to different combinations of processor speeds, communications bandwidth, and latency. Two popular three-body potentials used for silicon simulation are evaluated and compared. In addition, the popular Lennard-Jones potential is used to investigate to role of cutoff distance on parallel performance. read less NOT USED (high confidence) F. Meyer, P. Krstic, L. Vergara, H. Krause, C. Reinhold, and S. Stuart, “Low energy chemical sputtering of ATJ graphite by atomic and molecular deuterium ions,” Physica Scripta. 2007. link Times cited: 15 Abstract: We present experimental chemical sputtering results for D+, … read moreAbstract: We present experimental chemical sputtering results for D+, D2+ and D3+ ions incident on ATJ graphite in the energy range 5–60 eV D−1, and compare them with simulations for deuterated amorphous carbon impacted by neutral D, D2 and D3. The measured methane yields/D for the different species compared at the same energy/D diverge below about 60 eV D−1, the incident triatomic molecular ions leading to the largest yields/D, and the atomic ions to the smallest, reaching a factor of two difference at 10 eV/D. The measured yields/D are in reasonable agreement with molecular dynamics simulations over the entire calculated energy range. The model surfaces were prepared by D, D2 and D3 impacts in a way that mimics the experiment. For D2 incident at energies below 15 eV/D, the simulations show a strong dependence of the sputtering yields on the vibrational state of the incident projectile. read less NOT USED (high confidence) A. Galashev, V. Polukhin, I. A. Izmodenov, and O. Rakhmanova, “Molecular dynamics simulation of the physicochemical properties of silicon nanoparticles containing 73 atoms,” Glass Physics and Chemistry. 2007. link Times cited: 12 NOT USED (high confidence) R. Drautz, X. W. Zhou, D. Murdick, B. Gillespie, H. Wadley, and D. Pettifor, “Analytic bond-order potentials for modelling the growth of semiconductor thin films,” Progress in Materials Science. 2007. link Times cited: 28 NOT USED (high confidence) A. Bholoa, S. Kenny, and R. Smith, “A new approach to potential fitting using neural networks,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 30 NOT USED (high confidence) J. Samela, K. Nordlund, J. Keinonen, and V. Popok, “Comparison of silicon potentials for cluster bombardment simulations,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 26 NOT USED (high confidence) Z. Huang, Z. Guo, X. Chen, Z. Yu, T. M. Yu, and W. Lee, “Microscopic machining mechanism of polishing based on vibrations of liquid,” Nanotechnology. 2007. link Times cited: 8 Abstract: A molecular dynamics method has been applied to study the me… read moreAbstract: A molecular dynamics method has been applied to study the mechanism of polishing based on vibrations of liquid. Movements of polishing particles and formations of impact dents are simulated and discussed. The abrasive effect between particle and machined substrate is evaluated empirically. Polishing qualities, including roughness and fractal character under multiple impacts, are obtained by numerical methods. Results show that the particle will vibrate and roll viscously on the substrate. Press, tear and self-organization effects will be responsible for the formation of impact dents. Simulation results are compared with experimental data to verify the conclusions. read less NOT USED (high confidence) J. Crocombette, G. Dumazer, N. Q. Hoang, F. Gao, and W. J. Weber, “Molecular Dynamics Modeling of the Thermal Conductivity of Irradiated SiC as a Function of Cascade Overlap,” Journal of Applied Physics. 2007. link Times cited: 34 Abstract: SiC thermal conductivity is known to decrease under irradiat… read moreAbstract: SiC thermal conductivity is known to decrease under irradiation. To understand this effect, we study the variation of the thermal conductivity of cubic SiC with defect accumulation induced by displacement cascades. We use an empirical potential of the Tersoff type in the framework of nonequilibrium molecular dynamics. The conductivity of SiC is found to decrease with dose, in very good quantitative agreement with low temperature irradiation experiments. The results are analyzed in view of the amorphization states that are created by the cascade accumulation simulations. The calculated conductivity values at lower doses are close to the smallest measured values after high temperature irradiation, indicating that the decrease of the conductivity observed at lower doses is related to the creation of point defects. A subsequent decrease takes place upon further cascade accumulation. It is characteristic of the amorphization of the material and is experimentally observed for low temperature irradiation only. read less NOT USED (high confidence) H. Muramatsu, T. Hayashi, K. Y. Ahm, M. Terrones, and M. Endo, “Formation of off-centered double-walled carbon nanotubes exhibiting wide interlayer spacing from bi-cables,” Chemical Physics Letters. 2006. link Times cited: 5 NOT USED (high confidence) K. Sugio, H. Fukushima, and O. Yanagisawa, “Molecular Dynamics Simulation of Grain Boundary Formation and Migration in Silicon,” Materials Transactions. 2006. link Times cited: 5 Abstract: Molecular dynamics simulation using Tersoff potential was ca… read moreAbstract: Molecular dynamics simulation using Tersoff potential was carried out to investigate the formation and the migration of (010) E5 twist boundary in silicon. Effects of carbon atoms on the grain boundary formation and the grain boundary migration were also investigated. Amorphous thin layers remained at the twist boundary even after crystallization, and changes in the thickness of this layers caused grain boundary migration. When carbon atoms were segregated at the twist boundary, these atoms prevented shrinkage of an amorphous thin layer, and the grain boundary migration was retarded. Precipitated carbon atoms within the grain produces a strain field and this strain field possibly became driving force for the grain boundary migration. read less NOT USED (high confidence) L. Rosales, M. Pacheco, Z. Barticevic, C. Rocha, and A. Latgé, “Magnetic-field effects on transport in carbon nanotube junctions,” Physical Review B. 2006. link Times cited: 16 Abstract: Here we address a theoretical study on the behavior of elect… read moreAbstract: Here we address a theoretical study on the behavior of electronic states of heterojunctions and quantum dots based on carbon nanotubes under magnetic fields. Emphasis is put on the analysis of the local density of states, the conductance, and on the characteristic curves of current voltage. The heterostructures are modeled by joining zigzag tubes through single pentagon-heptagon pair defects, and described within a simple tight-binding calculation. The conductance is calculated using the Landauer formula in the Green-functions formalism. The theoretical approach used incorporates the atomic details of the topological defects by performing an energy relaxation via Monte Carlo calculation. The effect of a magnetic field on the conductance gap of the system is investigated and compared to those of isolated constituent tubes. It is found that the conductance gap of the studied carbon nanotube heterostructure exhibits oscillations as a function of the magnetic flux. However, unlike the pristine tubes case, they are not Aharonov-Bohm periodic oscillations. read less NOT USED (high confidence) J. Servantie and P. Gaspard, “DYNAMICS AND FRICTION IN DOUBLE WALLED CARBON NANOTUBES.” 2006. link Times cited: 20 Abstract: We report on a study of the translational sliding motion and… read moreAbstract: We report on a study of the translational sliding motion and dynamic friction in systems of double-walled carbon nanotubes using molecular dynamics simulations combined with theoretical analysis. The sliding motion is described by a one-dimensional analytical model which includes the van der Waals force between the nanotubes, a dynamic friction force, and a small Langevin-type fluctuating force. The dynamic friction force is shown to be linear in the velocity over a large domain of initial conditions in armchair-armchair, zigzag-armchair, and zigzag-zigzag double-walled nanotubes. Beyond this domain, evidence is obtained for nonlinear eects which increase friction. In armchair-armchair systems, the dynamic friction is observed to be nonlinearly enhanced by the excitation of internal modes. In the linear domain, the coecient of proportionality between the dynamic friction force and the velocity is shown to be given by Kirkwood’s formula in terms of the force autocorrelation function. PACS numbers: 68.35.Af;85.35.Kt I. INTRODUCTION Since the experiment of Cumings and Zettl 1 showed the possibility of making nanoscale mechanical devices with multiwalled carbon nanotubes, many papers appeared on the translational motion in these systems. Carbon nanotubes interact by van der Waals forces which keep the nanotubes nested together. Mechanical considerations as well as molecular dynamics simulations have been carried out in double-walled carbon nanotubes (DWNT), showing that oscillations are possible with a frequency larger than gigahertz. 2‐4 These oscillations concern the one-dimensional translational motion of the two nanotubes sliding one with respect to the other along their axes. The energy of this one-dimensional motion can be dissipated into the many other degrees of freedom of the nanotubes, resulting in a damping of the mechanical oscillations. This damping is caused by the dynamic friction between both nanotubes. The dynamic friction force against sliding motion is observed to be two orders of magnitude smaller than the van der Waals restoring force. Accordingly, the damping is achieved over long time intervals of the order of hundreds of picoseconds or more 5,6 . Several recent papers have been devoted to the properties of dynamic friction in nanotubes. The role of the commensuration in the corrugation of the force between both nanotubes has been investigated and friction has been expected to be larger in commensurate than incommensurate systems 6‐8 . In a recent Letter, 9 it was shown that the commensuration does not lead to significantly increased friction forces while their velocity dependence as well as edge eects read less NOT USED (high confidence) Z. Tang, H. Zhao, G. Li, and N. Aluru, “Finite-temperature quasicontinuum method for multiscale analysis of silicon nanostructures,” Physical Review B. 2006. link Times cited: 99 Abstract: In this paper, we extend the quasicontinuum approach for a m… read moreAbstract: In this paper, we extend the quasicontinuum approach for a multiscale analysis of silicon nanostructures at finite temperature. The quasicontinuum method uses the classical continuum mechanics framework, but the constitutive response of the system is determined by employing an atomistic description. For finite-temperature solid systems under isothermal conditions, the constitutive response is determined by using the Helmholtz free energy density. The static part of the Helmholtz free energy density is obtained directly from the interatomic potential while the vibrational part is calculated by using the theory of quantum-mechanical lattice dynamics. Specifically, we investigate three quasiharmonic models, namely the real space quasiharmonic model, the local quasiharmonic model, and the reciprocal space quasiharmonic model, to compute the vibrational free energy. Using the finite-temperature quasicontinuum method, we compute the effect of the temperature and strain on the phonon density of states, phonon Gruneisen parameters, and the elastic properties of the Tersoff silicon. We also compute the mechanical response of silicon nanostructures for various external loads and the results are compared to molecular dynamics simulations. read less NOT USED (high confidence) H. Lu, N. Daphalapurkar, B. Wang, S. Roy, and R. Komanduri, “Multiscale simulation from atomistic to continuum – coupling molecular dynamics (MD) with the material point method (MPM),” Philosophical Magazine. 2006. link Times cited: 46 Abstract: A new multiscale simulation approach is introduced that coup… read moreAbstract: A new multiscale simulation approach is introduced that couples atomistic-scale simulations using molecular dynamics (MD) with continuum-scale simulations using the recently developed material point method (MPM). In MPM, material continuum is represented by a finite collection of material points carrying all relevant physical characteristics, such as mass, acceleration, velocity, strain and stress. The use of material points at the continuum level provides a natural connection with the atoms in the lattice at the atomistic scale. A hierarchical mesh refinement technique in MPM is presented to scale down the continuum level to the atomistic level, so that material points at the fine level in MPM are allowed to directly couple with the atoms in MD. A one-to-one correspondence of MD atoms and MPM points is used in the transition region and non-local elastic theory is used to assure compatibility between MD and MPM regions, so that seamless coupling between MD and MPM can be accomplished. A silicon single crystal under uniaxial tension is used in demonstrating the viability of the technique. A Tersoff-type, three-body potential was used in the MD simulations. The coupled MD/MPM simulations show that silicon under nanometric tension experiences, with increasing elongation in elasticity, dislocation generation and plasticity by slip, void formation and propagation, formation of amorphous structure, necking, and final rupture. Results are presented in terms of stress–strain relationships at several strain rates, as well as the rate dependence of uniaxial material properties. This new multiscale computational method has potential for use in cases where a detailed atomistic-level analysis is necessary in localized spatially separated regions whereas continuum mechanics is adequate in the rest of the material. read less NOT USED (high confidence) T. Hammerschmidt, E. Schöll, and M. Scheffler, “Growth simulations of InAs/GaAs quantum dots.” 2006. link Times cited: 6 Abstract: Semiconductor nanostructures, and particularly quantum dots … read moreAbstract: Semiconductor nanostructures, and particularly quantum dots (QDs), have promising potential for technical applications such as light-emitting diodes, lasers, new devices, and quantum computers. But the big number of QDs needed, less than billions are hardly useful, is far beyond the means of normal manufacturing methods. For this nanotechnology to prevail, the QDs have to build themselves by self-assembly and self-organization. In this work, we study the growth of InAs QDs on GaAs substrates. For this purpose we developed a many-body potential of the Abell-Tersoff type that is able to account for the energetic balance of strain relief and QD side-facet formation during QD growth. It simultaneously captures many microscopic quantities of In, Ga, As, GaAs, and InAs bulk phases, as well as GaAs and InAs surface structures as obtained from experiment and density-functional theory (DFT) calculations with good overall accuracy. Its predictions for biaxial strained GaAs and InAs are in good agreement with DFT calculations and analytic results of continuum-elasticity theory. Based on recent STM results, we set up detailed atomic structures of InAs QDs with InAs wetting layers and homogenous InAs films on GaAs, relax them with our potential, and compare the resulting total energies. We show that the lateral elastic interaction of ‘hut’-like QDs dominated by {317} facets is significantly larger than that of ‘dome’-like QDs dominated by {101} facets. A strain-tensor analysis suggests that this effect is due to the relative orientations of the QD side facets to the elastic principal axes. Our calculated onset of the Stranski-Krastanov growth mode with respect to the InAs coverage is in good agreement with experimentally deduced values. The critical nucleus for QD formation is approximately 70 In atoms in size and poses an energy barrier of 5.3 eV. Furthermore, we can explain the experimentally observed shape sequence of ‘hut’-like QDs and ‘dome’-like QDs through the finding of distinct stability regimes. The regime separation depends strongly on the chemical potentials and the QD density. The experimental finding of vertical growth correlation in QD stacks can be explained by a distinct minimum in the potential-energy-surface (PES) of freestanding QDs in different lateral positions above overgrown QDs. This effect vanishes with increasing distance between the stacked QDs. The energy gain observed in our calculations can lower the energy barrier for QD formation to 3.5 eV and the size of the critical nucleus to only 25 In atoms. Additionally, we calculated the PES for In adsorption on surfaces that correspond to major side facets of ‘hut’and ‘dome’-like QDs by means of DFT to study possible kinetic effects. The dominating diffusion paths are perpendicular and parallel to the QD contour lines on {317} facets, but only perpendicular on {101} facets. The In incorporation on {317} facets could be kinetically limited due to the high barrier of approximately 1 eV for breaking As dimers. The diffusion barriers on {101} facets are lowered near the bottom of ‘dome’-like QDs, which supports the interpretation of the {317} facets on top as kinetic effect. read less NOT USED (high confidence) P. Erhart, N. Juslin, O. Goy, K. Nordlund, R. Müller, and K. Albe, “Analytic bond-order potential for atomistic simulations of zinc oxide,” Journal of Physics: Condensed Matter. 2006. link Times cited: 75 Abstract: An interatomic potential for zinc oxide and its elemental co… read moreAbstract: An interatomic potential for zinc oxide and its elemental constituents is derived based on an analytical bond-order formalism. The model potential provides a good description of the bulk properties of various solid structures of zinc oxide including cohesive energies, lattice parameters, and elastic constants. For the pure elements zinc and oxygen the energetics and structural parameters of a variety of bulk phases and in the case of oxygen also molecular structures are reproduced. The dependence of thermal and point defect properties on the cutoff parameters is discussed. As exemplary applications the irradiation of bulk zinc oxide and the elastic response of individual nanorods are studied. read less NOT USED (high confidence) B. Mouffok, H. Feraoun, and H. Aourag, “Two‐body potential of the Buckingham type for copper halides,” physica status solidi (b). 2006. link Times cited: 4 Abstract: The ground state, and elastic properties of copper halides i… read moreAbstract: The ground state, and elastic properties of copper halides in their zinc blende, NaCl and intermediate structures have been calculated using a two‐body potential of the Buckingham type coupled with a molecular dynamics simulation. (© 2006 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) S. Stuart, M. T. Knippenberg, O. Kum, and P. Krstic, “Simulation of amorphous carbon with a bond-order potential,” Physica Scripta. 2006. link Times cited: 19 Abstract: Simulations of amorphous carbon were performed at densities … read moreAbstract: Simulations of amorphous carbon were performed at densities ranging from 2.0 to 3.0 g cm−3 with a reactive bond-order potential. The fraction of sp3 bonding increases with increasing density, as is observed experimentally, but with generally too much sp2 content. Ring size distributions are calculated, with a number of large rings observed. It is suggested that structural quantities that are more directly related to physical properties—such as void volumes and coordination numbers—are more useful than ring size distributions in characterizing the structure of amorphous carbon. Void fractions and void volume distributions are calculated, indicating that a percolating void network exists at 2.0 g cm−3, large, non-percolating voids exist at intermediate density, and no voids are found larger than atomic volumes at 3.0 g cm−3. read less NOT USED (high confidence) T. Akabane, Y. Sasajima, and J. Onuki, “Computer simulation of silicon nanoscratch test,” Materials Transactions. 2006. link Times cited: 4 Abstract: By the molecular dynamics method, a computer simulation of a… read moreAbstract: By the molecular dynamics method, a computer simulation of a scratch test with a nanometer scale was performed. The specimen was composed of 1008 silicon atoms with a diamond single-crystal structure. The indentor was assumed to be a perfect rigid body, and the Morse potential was utilized as the interaction between the indentor and a silicon atom. Two types of potential, i.e., Stillinger-Weber and Tersoff potentials, were examined as the interaction between silicon atoms. The present simulation clarified that the standard deviation of the friction constant increased with decreasing scratch depth and became maximum when the indentor just began to scratch the specimen surface at critical load. The friction coefficient, indentation hardness and scratch hardness at critical load were estimated to be 1.2–1.6, 80–90 GPa and 8.5– 9.4 GPa, respectively. read less NOT USED (high confidence) G. Cao and X. Chen, “Mechanisms of nanoindentation on single-walled carbon nanotubes: The effect of nanotube length,” Journal of Materials Research. 2006. link Times cited: 32 Abstract: The mechanisms of nanoindentation on single-walled carbon na… read moreAbstract: The mechanisms of nanoindentation on single-walled carbon nanotubes (SWCNTs) have been studied by using molecular dynamics simulation and continuum analysis during which a flat layer of diamond atoms is pressed down incrementally on a vertically aligned SWCNT. SWCNTs are divided into three distinct categories based on their aspect ratios, such that the nanotube behavior transits from a shell (short tube) to a beam (long tube). Molecular dynamics simulations are used to explore the diverse indentation characteristics in each domain, where the relationships between the strain energy and indentation depth during loading, unloading, and reloading are continuously recorded. The nanoindentation mechanisms are characterized by the critical indentation depth, maximum strain energy and force associated with buckling, as well as with the evolution of carbon bond length and morphology of the SWCNTs. Bifurcation behaviors are explored by investigating the loading-unloading-reloading behaviors of the nanotubes. Parallel finite element simulations are also used to study the pre- and post-buckling behaviors of SWCNT by incorporating the van der Waals interaction into the continuum code. It is found that, for the most part, continuum analysis can effectively capture the overall indentation characteristics, yet some details related to the atomic characteristics of nanoindentation may only be revealed by molecular dynamics simulation. Finally, an indentation mechanism map is derived by comparing behaviors of SWCNTs with different aspect and section ratios. Focusing on the effects of nanotube length, this paper is the first of a series of numerical studies on the indentation mechanisms of carbon nanotubes, which may be used to determine the intrinsic mechanical properties of SWCNTs by means of nanoindentation. read less NOT USED (high confidence) D. Caillerie, A. Mourad, and A. Raoult, “Discrete Homogenization in Graphene Sheet Modeling,” Journal of Elasticity. 2006. link Times cited: 88 NOT USED (high confidence) X. Chen and G. Cao, “A structural mechanics study of single-walled carbon nanotubes generalized from atomistic simulation,” Nanotechnology. 2006. link Times cited: 84 Abstract: A new structural mechanics model is developed to closely dup… read moreAbstract: A new structural mechanics model is developed to closely duplicate the atomic configuration and behaviours of single-walled carbon nanotubes (SWCNTs). The SWCNTs are effectively represented by a space frame, where primary and secondary beams are used to bridge the nearest and next-nearest carbon atoms, to mimic energies associated with bond stretching and angle variation, respectively. The elastic properties of the frame components are generalized from molecular dynamics (MD) simulation based on an accurate ab initio force field, and numerical analyses of tension, bending, and torsion are carried out on nine different SWCNTs. The space-frame model also closely duplicates the buckling behaviours of SWCNTs in torsion and bending. In addition, by repeating the same process with continuum shell and beam models, new elastic and section parameters are fitted from the MD benchmark experiments. As an application, all three models are employed to study the thermal vibration behaviours of SWCNTs, and excellent agreements with MD analyses are found. The present analysis is a systematic structural mechanics attempt to fit SWCNT properties for several basic deformation modes and applicable to a variety of SWCNTs. The continuum models and fitted parameters may be used to effectively simulate the overall deformation behaviours of SWCNTs at much larger length- and timescales than pure MD analysis. read less NOT USED (high confidence) X. W. Zhou, D. Murdick, B. Gillespie, and H. Wadley, “Atomic assembly during GaN film growth : Molecular dynamics simulations,” Physical Review B. 2006. link Times cited: 44 Abstract: Molecular dynamics simulations using a recently developed Ga… read moreAbstract: Molecular dynamics simulations using a recently developed Ga-N Tersoff type bond order interatomic potential have been used to investigate the growth mechanisms of 0001 wurtzite GaN films from thermalized atomic gallium and nitrogen fluxes. The crystallinity and stoichiometry of the deposited wurtzite lattice structures were determined as a function of growth temperature and N:Ga flux ratio. The lattice perfection was found to improve as the growth temperature was increased to 500 K. At a fixed growth temperature, the lattice quality and stoichiometry both reached optimum as the N:Ga ratio approached a value between two and three. The optimum flux ratio increased with increasing growth temperature. These three observations are consistent with experimental studies of growth on wurtzite phase promoting substrates. The atomic assembly mechanisms responsible for these effects have been explored using time-resolved atom position images. The analysis revealed that high quality crystalline growth only occurred when off-lattice atoms which are usually associated with amorphous embryos or defect complexes formed during deposition were able to move to unoccupied lattice sites by thermally activated diffusion processes. The need for a high N:Ga flux ratio to synthesize stochiometric films arises because many of the nitrogen adatoms that impact N-rich 0001 GaN surfaces are re-evaporated. Reductions of the substrate temperature reduce this reevaporation and as a result, the optimum N:Ga ratio for the stoichiometric film formation and best lattice perfection was reduced as the growth temperature was decreased. read less NOT USED (high confidence) G. Lulli, E. Albertazzi, S. Balboni, and L. Colombo, “Defect-induced homogeneous amorphization of silicon: the role of defect structure and population,” Journal of Physics: Condensed Matter. 2006. link Times cited: 7 Abstract: Molecular dynamics based on the environment-dependent intera… read moreAbstract: Molecular dynamics based on the environment-dependent interatomic potential is used to investigate the influence of the nature and distribution of defects on solid state, homogeneous amorphization of Si. To this end, different kinds of defects, including single interstitials and vacancies (both uncorrelated and correlated distributions), bond defects, and small interstitial and vacancy clusters, have been considered. It is shown that the threshold defect concentration for amorphization depends on the defect type, and, in the case of single defects, on the degree of correlation between interstitial and vacancy distributions. The threshold varies within the interval [0.18–0.28] atomic fraction, the upper value corresponding to the case of bond defects, the lower to the uncorrelated distributions of single split interstitials plus compensating vacancies. read less NOT USED (high confidence) C. Reddy, S. Rajendran, and K. M. Liew, “Equilibrium configuration and continuum elastic properties of finite sized graphene,” Nanotechnology. 2006. link Times cited: 352 Abstract: This paper presents a continuum mechanics approach to modell… read moreAbstract: This paper presents a continuum mechanics approach to modelling the elastic deformation of finite graphene sheets based on Brenner’s potential. The potential energy of the graphene sheet is minimized for determining the equilibrium configuration. The four edges of the initially rectangular graphene sheet become curved at the equilibrium configuration. The curving of the sides is attributed to smaller coordination number for the atoms at the edges compared to that of the interior atoms. Considering two graphene models, with only two or all four edges constrained to be straight, the continuum Young’s moduli of graphene are computed applying the Cauchy–Born rule. The computed elastic constants of the graphene sheet are found to conform to orthotropic material behaviour. The computed constants differ considerably depending on whether a minimized or unminimized configuration is used for computation. read less NOT USED (high confidence) D. Fischer, A. Curioni, S. Billeter, and W. Andreoni, “The structure of the SiO2∕Si(100) interface from a restraint-free search using computer simulations,” Applied Physics Letters. 2006. link Times cited: 25 Abstract: The structure of the interface between SiO2 and Si(100) is i… read moreAbstract: The structure of the interface between SiO2 and Si(100) is investigated using the replica-exchange method driven by classical molecular dynamics simulations based on ab initio-derived interatomic potentials. Abrupt interfaces are shown to be unstable, whereas a substoichiometric oxide forms at the transition between the two materials that exhibits Si atoms in all three intermediate oxidation states, in agreement with experiment. A number of physical characteristics are found to be consistent with experimental data, including the distribution of Si atoms with different oxidation states, the increase in atom density and the stability of a pseudo-cristobalite pattern at the interface as well as the presence of Si–O–Si bridge bonds between the substrate and the suboxide. read less NOT USED (high confidence) A. Marzegalli, F. Montalenti, and L. Miglio, “Atomistic simulation of a 60° shuffle dislocation segment migrating in a Ge/SiGe(001) epitaxial film,” Journal of Physics: Condensed Matter. 2005. link Times cited: 8 Abstract: We show that the migration process of a 60° shuffle dislocat… read moreAbstract: We show that the migration process of a 60° shuffle dislocation in an heteroepitaxial Ge/Si0.5Ge0.5(001) system can be analysed by classical molecular dynamics simulations. By following the misfit segment during its motion, we build a sequence of strain maps giving detailed information about the elastic-energy relaxation in the film. The atomic-scale mechanisms underlying the dislocation motion towards the interface are also monitored, showing, for instance, that kinks are actually present along the dislocation line. read less NOT USED (high confidence) S. Kapur, M. Prasad, J. Crocker, and T. Sinno, “Role of configurational entropy in the thermodynamics of clusters of point defects in crystalline solids,” Physical Review B. 2005. link Times cited: 36 Abstract: Received 28 March 2005; revised manuscript received 24 May 2… read moreAbstract: Received 28 March 2005; revised manuscript received 24 May 2005; published 20 July 2005The internal configurational entropy of point defect clusters in crystalline silicon is studied in detail byanalyzing their potential energy landscapes. Both on-lattice and off-lattice calculation approaches are employedto demonstrate the importance of off-lattice configurational states that arise due to a large number of inherentstructures local minima in the energy landscape generated by the interatomic potential function. The resultingcluster configurational entropy of formation is shown to exhibit behavior that is qualitatively similar to thatobserved in supercooled liquids and amorphous solids and substantially alters the thermodynamic properties ofpoint defect clusters in crystals at high temperature. This behavior is shown to be independent of interatomicpotential and cluster type, and suggests that defects in crystals at high temperature should be generally de-scribed by a quasicontinuous collection of nondegenerate states rather than as a single ground state structure.The modified thermodynamic properties of vacancy clusters at high temperature are found to explain a long-standing discrepancy between simulation predictions and experimental measurements of vacancy aggregationdynamics in silicon.DOI: 10.1103/PhysRevB.72.014119 PACS number s : 61.72.Bb, 61.72.Qq read less NOT USED (high confidence) E. Oh, J. C. Slattery, and D. Lagoudas, “Thermodynamics of two-dimensional single-component elastic crystalline solids: single-wall carbon nanotubes,” Philosophical Magazine. 2005. link Times cited: 12 Abstract: The thermodynamic behaviour of two-dimensional single-compon… read moreAbstract: The thermodynamic behaviour of two-dimensional single-component elastic crystalline solids is developed: the surface Euler's equation, the surface Gibbs equation, the surface Gibbs–Duhem equation, and the conditions to be expected at equilibrium, including the stress-deformation behaviour of the crystal. The analysis recognizes that the surface Helmholtz free energy is an explicit function of the lattice vectors defining the crystalline structure. As an application, we obtain the stress-deformation behaviour of single-wall carbon nanotubes which are composed of a regular two-dimensional array of hexagonal lattices of carbon atoms. Using two potentials, Tersoff [1]–Brenner [2] and Brenner et al. [3] to describe interatomic potentials and hence the specific surface Helmholtz free energy, we compute the surface elastic properties for the single-wall carbon nanotubes. These are compared with the available experimental values. read less NOT USED (high confidence) T. Kakinaga, A. Hatai, O. Tabata, and Y. Isono, “Silicon anisotropic wet etching simulation using molecular dynamics,” The 13th International Conference on Solid-State Sensors, Actuators and Microsystems, 2005. Digest of Technical Papers. TRANSDUCERS ’05. 2005. link Times cited: 2 Abstract: The paper describes a new approach of atomic simulation for … read moreAbstract: The paper describes a new approach of atomic simulation for silicon (Si) anisotropic wet etching using molecular dynamics (MD). The extended Tersoff potential was adopted as a potential function for MD, which was able to handle Si etching reaction with hydrogen (H) and oxygen (O) atoms. The potential function also made it possible to represent a transition of electric charge under chemical etching MD simulations as a function of atomic distance. Binding energies between Si, H and O atoms were statically calculated by the function, and they showed good agreement with previously reported experimental data. Finally, etching MD simulation of single crystal Si in water was carried out using the extended Tersoff potential for revealing the effect of hydrogen termination at the top of Si on the interaction between Si, H and O atoms. read less NOT USED (high confidence) S. Park, H. Kim, K. Kang, J. Lee, Y. Choi, and O. Kwon, “Experimental and molecular dynamics study on crystallization of amorphous silicon under external fields,” Journal of Physics D: Applied Physics. 2005. link Times cited: 17 Abstract: Solid-phase crystallization (SPC) of amorphous silicon (a-Si… read moreAbstract: Solid-phase crystallization (SPC) of amorphous silicon (a-Si) under an external force field is investigated experimentally and numerically. Experimental results show that the kinetics of crystallization can be greatly enhanced by applying induction fields without the heating problems of a-Si film and its substrate, since temperature rises during the crystallization process are negligibly small. To explore the underlying acceleration mechanisms for the SPC process under the external fields, molecular dynamics simulations are carried out using the Tersoff potential. The numerical amorphous structure is obtained by the liquid quenching method and is utilized to simulate the crystallization processes at various process temperatures with and without external force fields. While homogeneous crystallization of a-Si could not be achieved readily, it is shown that the heterogeneous crystallization can be significantly accelerated by external force fields. This enhancement is due to increased molecular jumping frequencies associated with the molecular potential energies being increased by external excitations, rather than due to thermal mechanisms dominant in conventional SPC processes. read less NOT USED (high confidence) U. Tartaglino, T. Zykova-Timan, F. Ercolessi, and E. Tosatti, “Material surfaces and nanosystems close to the melting temperature,” Journal of Materials Science. 2005. link Times cited: 7 NOT USED (high confidence) F. Shimojo, R. Kalia, A. Nakano, and P. Vashishta, “Embedded divide-and-conquer algorithm on hierarchical real-space grids: parallel molecular dynamics simulation based on linear-scaling density functional theory,” Comput. Phys. Commun. 2005. link Times cited: 65 NOT USED (high confidence) T. Hawa and M. Zachariah, “Coalescence kinetics of bare and hydrogen-coated silicon nanoparticles : A molecular dynamics study,” Physical Review B. 2005. link Times cited: 40 Abstract: One of the significant challenges in the use of nanoparticle… read moreAbstract: One of the significant challenges in the use of nanoparticles is the control of primary particle size and extent of agglomeration when grown from the gas phase. In this paper we consider the role of surface passivation of the rate of nanoparticle coalescence. We have studied the coalescence of bare and H-coated silicon nanoparticles of sizes between 2\char21{}6 nm using molecular dynamics simulation at 1000 and 1500 K. We found that coalescence of coated particles consists of two steps, where reaction between particles and relocations of surface atoms near the reacting region, occur in the first step, which comprise an induction period. The second step consists of the nominal coalescence event, which depends on the surface tension and solid-state diffusion in the particle. The hydrogen passivation layer was found to remain on the surface of coalescing pair of the particles during the entire coalescence event. We also develop a mathematical model to describe the dynamics of coalescence of coated particles. The model is able to describe both the initial induction period and the coalescence period, and the role of the extent of surface coverage on the coalescence rate. In general, the entire coalescence time of coated particles is about 3\char21{}5 times that of bare particles, and the exothermicity from coalescence is about half that for the unpassivated particles. read less NOT USED (high confidence) U. Tartaglino, T. Zykova-Timan, F. Ercolessi, and E. Tosatti, “Melting and nonmelting of solid surfaces and nanosystems,” Physics Reports. 2005. link Times cited: 134 NOT USED (high confidence) D. Tománek, “Carbon-based nanotechnology on a supercomputer,” Journal of Physics: Condensed Matter. 2005. link Times cited: 19 Abstract: The quantum nature of phenomena dominating the behaviour of … read moreAbstract: The quantum nature of phenomena dominating the behaviour of nanostructures raises new challenges when trying to predict and understand the physical behaviour of these systems. Addressing this challenge is imperative in view of the continuous reduction of device sizes, which is rapidly approaching the atomic level. Since even the most advanced experimental observations are subject to being fundamentally influenced by the measurement itself, new approaches must be sought to design and test future building blocks of nanotechnology. In this respect, high-performance computing, allowing predictive large-scale computer simulations, has emerged as an indispensable tool to foresee and interpret the physical behaviour of nanostructures, thus guiding and complementing the experiment. This contribution will review some of the more intriguing phenomena associated with nanostructured carbon, including fullerenes, nanotubes and diamondoids. Due to the stability of the sp2 bond, carbon fullerenes and nanotubes are thermally and mechanically extremely stable and chemically inert. They contract rather than expand at high temperatures, and are unparalleled thermal conductors. Nanotubes may turn into ballistic electron conductors or semiconductors, and even acquire a permanent magnetic moment. In nanostructures that form during a hierarchical self-assembly process, even defects may play a different, often helpful role. sp2 bonded nanostructures may change their shape globally by a sequence of bond rotations, which turn out to be intriguing multi-step processes. At elevated temperatures, and following photo-excitations, efficient self-healing processes may repair defects, thus answering an important concern in molecular electronics. read less NOT USED (high confidence) L. Raff, M. Malshe, M. Hagan, D. I. Doughan, M. Rockley, and R. Komanduri, “Ab initio potential-energy surfaces for complex, multichannel systems using modified novelty sampling and feedforward neural networks.,” The Journal of chemical physics. 2005. link Times cited: 120 Abstract: A neural network/trajectory approach is presented for the de… read moreAbstract: A neural network/trajectory approach is presented for the development of accurate potential-energy hypersurfaces that can be utilized to conduct ab initio molecular dynamics (AIMD) and Monte Carlo studies of gas-phase chemical reactions, nanometric cutting, and nanotribology, and of a variety of mechanical properties of importance in potential microelectromechanical systems applications. The method is sufficiently robust that it can be applied to a wide range of polyatomic systems. The overall method integrates ab initio electronic structure calculations with importance sampling techniques that permit the critical regions of configuration space to be determined. The computed ab initio energies and gradients are then accurately interpolated using neural networks (NN) rather than arbitrary parametrized analytical functional forms, moving interpolation or least-squares methods. The sampling method involves a tight integration of molecular dynamics calculations with neural networks that employ early stopping and regularization procedures to improve network performance and test for convergence. The procedure can be initiated using an empirical potential surface or direct dynamics. The accuracy and interpolation power of the method has been tested for two cases, the global potential surface for vinyl bromide undergoing unimolecular decomposition via four different reaction channels and nanometric cutting of silicon. The results show that the sampling methods permit the important regions of configuration space to be easily and rapidly identified, that convergence of the NN fit to the ab initio electronic structure database can be easily monitored, and that the interpolation accuracy of the NN fits is excellent, even for systems involving five atoms or more. The method permits a substantial computational speed and accuracy advantage over existing methods, is robust, and relatively easy to implement. read less NOT USED (high confidence) K. Matsunaga and Y. Iwamoto, “Molecular Dynamics Study of Atomic Structure and Diffusion Behavior in Amorphous Silicon Nitride Containing Boron,” Journal of the American Ceramic Society. 2004. link Times cited: 81 Abstract: We have performed molecular dynamics simulations of amorphou… read moreAbstract: We have performed molecular dynamics simulations of amorphous Si3N4 containing boron (Si-B-N). We have examined short-range atomic arrangements and self-diffusion constants of amorphous Si-B-N systems with various boron contents. Our simulations show that boron atoms are threefold coordinated by nitrogen atoms and that nitrogen atoms are bonded to both silicon and boron atoms in the amorphous network of Si-B-N. Also, the self-diffusion constant of nitrogen in Si-B-N is much decreased compared with that in amorphous Si3N4. This suggests that boron is important in decreasing the mobility of atoms in amorphous Si-B-N, which may explain the improved thermal stability of amorphous Si-B-N relative to amorphous Si3N4 observed experimentally. read less NOT USED (high confidence) D. Vvedensky, “Multiscale modelling of nanostructures,” Journal of Physics: Condensed Matter. 2004. link Times cited: 98 Abstract: Most materials phenomena are manifestations of processes tha… read moreAbstract: Most materials phenomena are manifestations of processes that are operative over a vast range of length and time scales. A complete understanding of the behaviour of materials thereby requires theoretical and computational tools that span the atomic-scale detail of first-principles methods and the more coarse-grained description provided by continuum equations. Recent efforts have focused on combining traditional methodologies—density functional theory, molecular dynamics, Monte Carlo methods and continuum descriptions—within a unified multiscale framework. This review covers the techniques that have been developed to model various aspects of materials behaviour with the ultimate aim of systematically coupling the atomistic to the continuum descriptions. The approaches described typically have been motivated by particular applications but can often be applied in wider contexts. The self-assembly of quantum dot ensembles will be used as a case study for the issues that arise and the methods used for all nanostructures. Although quantum dots can be obtained with all the standard growth methods and for a variety of material systems, their appearance is a quite selective process, involving the competition between equilibrium and kinetic effects, and the interplay between atomistic and long-range interactions. Most theoretical models have addressed particular aspects of the ordering kinetics of quantum dot ensembles, with far fewer attempts at a comprehensive synthesis of this inherently multiscale phenomenon. We conclude with an assessment of the current status of multiscale modelling strategies and highlight the main outstanding issues. read less NOT USED (high confidence) F. Chuang, C. Ciobanu, C. Predescu, C. Wang, and K. Ho, “Structure of Si(1 1 4) determined by global optimization methods,” Surface Science. 2004. link Times cited: 36 NOT USED (high confidence) J. Hsieh, S. Ju, S.-H. Li, and C. Hwang, “Temperature dependence in nanoindentation of a metal substrate by a diamondlike tip,” Physical Review B. 2004. link Times cited: 33 Abstract: In this investigation, we simulated the nanoindentation of a… read moreAbstract: In this investigation, we simulated the nanoindentation of a copper substrate by a diamondlike tip, using molecular dynamics method. A series of simulations according to distinct system temperatures were performed to analyze the temperature dependences of some important physical quantities occurring in the indentation. We found that the maximal normal forces on the tip atoms, both the repulsive and the attractive, the elastic modulus of the indentation system and the network done by the tip during the indentation cycle all decrease with increasing system temperature. By these dependences, we then identified the critical temperature for the transition of plastic flow mechanism in the substrate. The evolution of the crystalline structure in the substrate was analyzed by examining the variation of the structure factor, which measures the perfection of the crystalline structure, during the indentation cycle. An important physical quantity is the difference between the equilibrium absolute values of structure factor before and after the indentation, which can be used to measure the permanent deformation in the substrate produced by the indentation. We found that the difference increases with increasing temperature if the system temperature is below the critical temperature. read less NOT USED (high confidence) R. Holenstein, S. Kirkwood, R. Fedosejevs, and Y. Tsui, “Simulation of femtosecond laser ablation of silicon,” Photonics North. 2004. link Times cited: 16 Abstract: Femtosecond laser ablation is an important process in microm… read moreAbstract: Femtosecond laser ablation is an important process in micromachining and nanomachining of microelectronic, optoelectronic, biophotonic and MEMS components. The process of laser ablation of silicon is being studied on an atomic level using molecular dynamics simulations. We investigate ablation thresholds for Gaussian laser pulses of 800 nm wavelength, in the range of a few hundred femtoseconds in duration. Absorption is modelled via linear and 2-photon absorption processes into a hot electron bath which then transfers energy into the crystal lattice. The simulation box is a narrow column approximately 5.4 nm x 5.4 nm x 81 nm with periodic boundaries in the x and y transverse directions and a 1-D heat flow model at the bottom coupled to a heat bath to simulate an infinite bulk medium corresponding to the solid bulk material. A modified Stillinger-Weber potential is used to model the silicon atoms. The calculated thresholds are compared to various reported experimental values for the ablation threshold of silicon. We provide an overview of the code and discuss the simulation techniques used. read less NOT USED (high confidence) D. Donadio, L. Colombo, and G. Benedek, “Elastic moduli of nanostructured carbon films,” Physical Review B. 2004. link Times cited: 7 Abstract: We have computed the elastic constants of nanostructured car… read moreAbstract: We have computed the elastic constants of nanostructured carbon films as obtained from classical molecular dynamics simulations of a cluster beam deposition process. The calculations show that the elastic constants of the deposited films are related to the average size of the clusters by a power law. This allows us to extrapolate the present theoretical data to the scale of the experimental results obtained by Brillouin scattering. read less NOT USED (high confidence) O. Kum, B. Dickson, S. Stuart, B. Uberuaga, and A. Voter, “Parallel replica dynamics with a heterogeneous distribution of barriers: application to n-hexadecane pyrolysis.,” The Journal of chemical physics. 2004. link Times cited: 24 Abstract: Parallel replica dynamics simulation methods appropriate for… read moreAbstract: Parallel replica dynamics simulation methods appropriate for the simulation of chemical reactions in molecular systems with many conformational degrees of freedom have been developed and applied to study the microsecond-scale pyrolysis of n-hexadecane in the temperature range of 2100-2500 K. The algorithm uses a transition detection scheme that is based on molecular topology, rather than energetic basins. This algorithm allows efficient parallelization of small systems even when using more processors than particles (in contrast to more traditional parallelization algorithms), and even when there are frequent conformational transitions (in contrast to previous implementations of the parallel replica algorithm). The parallel efficiency for pyrolysis initiation reactions was over 90% on 61 processors for this 50-atom system. The parallel replica dynamics technique results in reaction probabilities that are statistically indistinguishable from those obtained from direct molecular dynamics, under conditions where both are feasible, but allows simulations at temperatures as much as 1000 K lower than direct molecular dynamics simulations. The rate of initiation displayed Arrhenius behavior over the entire temperature range, with an activation energy and frequency factor of E(a) = 79.7 kcal/mol and log A/s(-1) = 14.8, respectively, in reasonable agreement with experiment and empirical kinetic models. Several interesting unimolecular reaction mechanisms were observed in simulations of the chain propagation reactions above 2000 K, which are not included in most coarse-grained kinetic models. More studies are needed in order to determine whether these mechanisms are experimentally relevant, or specific to the potential energy surface used. read less NOT USED (high confidence) T. Hawa and M. Zachariah, “Internal pressure and surface tension of bare and hydrogen coated silicon nanoparticles.,” The Journal of chemical physics. 2004. link Times cited: 62 Abstract: We present a study of internal pressure and surface tension … read moreAbstract: We present a study of internal pressure and surface tension of bare and hydrogen coated silicon nanoparticles of 2-10 nm diameter as a function of temperature, using molecular dynamics simulations employing a reparametrized Kohen-Tully-Stillinger interatomic potential. The internal pressure was found to increase with decreasing particle size but the density was found to be independent of the particle size. We showed that for covalent bond structures, changes in surface curvature and the associated surface forces were not sufficient to significantly change bond lengths and angles. Thus, the surface tension was also found to be independent of the particle size. Surface tension was found to decrease with increasing particle temperature while the internal pressure did not vary with temperature. The presence of hydrogen on the surface of a particle significantly reduces surface tension (e.g., drops from 0.83 J/m(2) to 0.42 J/m(2) at 1500 K). The computed pressure of bare and coated particles was found to follow the classical Laplace-Young equation. read less NOT USED (high confidence) C. L. Allred, X. Yuan, M. Bazant, and L. Hobbs, “Elastic constants of defected and amorphous silicon with the environment-dependent interatomic potential,” Physical Review B. 2004. link Times cited: 31 Abstract: The elastic constants of a wide range of models of defected … read moreAbstract: The elastic constants of a wide range of models of defected crystalline and amorphous silicon are calculated, using the environment-dependent interatomic potential (EDIP). The defected crystalline simulation cells contain randomly generated defect distributions. An extensive characterization of point defects is performed, including structure, energy and influence on elastic constants. Three important conclusions are drawn. (1) Defects have independent effects on the elastic constants of silicon up to (at least) a defect concentration of 0.3%. (2) The linear effect of Frenkel pairs on the Young's modulus of silicon is -1653 GPa per defect fraction. (3) 17 different point defect types cause a very similar decrease in the Young's modulus: -(0.28{+-}0.05)% when calculated in isolation using a 1728-atom cell. These principles will be very useful for predicting the effect of radiation damage on the elastic modulus of silicon in the typical case in which point-defect concentrations can be estimated, but the exact distribution and species of defects is unknown. We also study amorphous samples generated in quenching the liquid with EDIP, including an ideal structure of perfect fourfold coordination, samples with threefold and fivefold coordinated defects, one with a nanovoid, and one with an amorphous inclusion in a crystalline matrix.more » In the last case, a useful finding is that the change in the Young's modulus is simply related to the volume fraction of amorphous material, as has also been observed by experiment.« less read less NOT USED (high confidence) S. Stuart, Y. Li, O. Kum, J. Mintmire, and A. Voter, “Reactive Bond-Order Simulations Using Both Spatial and Temporal Approaches to Parallelism,” Structural Chemistry. 2004. link Times cited: 11 NOT USED (high confidence) B. Ni, K.-H. Lee, and S. Sinnott, “A reactive empirical bond order (REBO) potential for hydrocarbon oxygen interactions,” Journal of Physics: Condensed Matter. 2004. link Times cited: 108 Abstract: The expansion of the second-generation reactive empirical bo… read moreAbstract: The expansion of the second-generation reactive empirical bond order (REBO) potential for hydrocarbons, as parametrized by Brenner and co-workers, to include oxygen is presented. This involves the explicit inclusion of C–O, H–O, and O–O interactions to the existing C–C, C–H, and H–H interactions in the REBO potential. The details of the expansion, including all parameters, are given. The new, expanded potential is then applied to the study of the structure and chemical stability of several molecules and polymer chains, and to modelling chemical reactions among a series of molecules, within classical molecular dynamics simulations. read less NOT USED (high confidence) M. Haftel, N. Bernstein, M. Mehl, and D. Papaconstantopoulos, “Interlayer surface relaxations and energies of fcc metal surfaces by a tight-binding method,” Physical Review B. 2004. link Times cited: 21 Abstract: The authors examine the interlayer surface relaxations and s… read moreAbstract: The authors examine the interlayer surface relaxations and surface energies for the low-index faces of fcc $\mathrm{Ni}$, $\mathrm{Pd}$, $\mathrm{Rh}$, $\mathrm{Pt}$, $\mathrm{Au}$, and $\mathrm{Ir}$ using the Naval Research Laboratory (NRL) tight-binding (TB) method. We compare the TB calculations, utilizing self-consistent charge transfer, with experimental measurements, density functional theory (DFT) calculations, and semiempirical methods. We find that for these metals the NRL-TB method largely reproduces the trends with respect to the exposed face and periodic table position obtained in DFT calculations and experimental measurements. We find that the inclusion of self-consistency in the TB surface calculations is essential for obtaining this agreement, as the TB calculations without it predict large first interlayer expansions for many of these surfaces. We also examine the energetics and relaxations of the $2\ifmmode\times\else\texttimes\fi{}1$ (011) missing row reconstruction for these metals. The TB method predicts that, in agreement with experiment, $\mathrm{Au}$ and $\mathrm{Pt}$ undergo this reconstruction, while $\mathrm{Ni}$, $\mathrm{Pd}$, and $\mathrm{Rh}$ do not, but predicts the $\mathrm{Ir}$ ground state structure to be unreconstructed $1\ifmmode\times\else\texttimes\fi{}1$, opposite to experiment. The interatomic relaxations of the (011) missing row structure for $\mathrm{Pt}$, $\mathrm{Au}$, and $\mathrm{Ir}$ are in good agreement with DFT calculations and experiment. Finally, we analyze the bonding characteristics of these metals using a decomposition of the TB total energy over neighboring atoms and angular momentum character. read less NOT USED (high confidence) O. Louchev, H. Kanda, A. Rosén, and K. Bolton, “Thermal physics in carbon nanotube growth kinetics.,” The Journal of chemical physics. 2004. link Times cited: 25 Abstract: The growth of single wall carbon nanotubes (SWNTs) mediated … read moreAbstract: The growth of single wall carbon nanotubes (SWNTs) mediated by metal nanoparticles is considered within (i) the surface diffusion growth kinetics model coupled with (ii) a thermal model taking into account heat release of carbon adsorption-desorption on nanotube surface and carbon incorporation into the nanotube wall and (iii) carbon nanotube-inert gas collisional heat exchange. Numerical simulations performed together with analytical estimates reveal various temperature regimes occurring during SWNT growth. During the initial stage, which is characterized by SWNT lengths that are shorter than the surface diffusion length of carbon atoms adsorbed on the SWNT wall, the SWNT temperature remains constant and is significantly higher than that of the ambient gas. After this stage the SWNT temperature decreases towards that of gas and becomes nonuniformly distributed over the length of the SWNT. The rate of SWNT cooling depends on the SWNT-gas collisional energy transfer that, from molecular dynamics simulations, is seen to be efficient only in the SWNT radial direction. The decreasing SWNT temperature may lead to solidification of the catalytic metal nanoparticle terminating SWNT growth or triggering nucleation of a new carbon layer and growth of multiwall carbon nanotubes. read less NOT USED (high confidence) W. K. Liu, E. Karpov, S. Zhang, and H. S. Park, “An introduction to computational nanomechanics and materials,” Computer Methods in Applied Mechanics and Engineering. 2004. link Times cited: 405 NOT USED (high confidence) M. Arroyo and T. Belytschko, “Finite crystal elasticity of carbon nanotubes based on the exponential Cauchy-Born rule,” Physical Review B. 2004. link Times cited: 407 Abstract: A finite deformation continuum theory is derived from intera… read moreAbstract: A finite deformation continuum theory is derived from interatomic potentials for the analysis of the mechanics of carbon nanotubes. This nonlinear elastic theory is based on an extension of the Cauchy-Born rule called the exponential Cauchy-Born rule. The continuum object replacing the graphene sheet is a surface without thickness. The method systematically addresses both the characterization of the small strain elasticity of nanotubes and the simulation at large strains. Elastic moduli are explicitly expressed in terms of the functional form of the interatomic potential. The expression for the flexural stiffness of graphene sheets, which cannot be obtained from standard crystal elasticity, is derived. We also show that simulations with the continuum model combined with the finite element method agree very well with zero temperature atomistic calculations involving severe deformations. read less NOT USED (high confidence) T. Hawa and M. Zachariah, “Molecular dynamics study of particle-particle collisions between hydrogen-passivated silicon nanoparticles,” Physical Review B. 2004. link Times cited: 39 Abstract: One of the significant challenges in the use of nanoparticle… read moreAbstract: One of the significant challenges in the use of nanoparticles is the control of primary particle size and extent of agglomeration when grown from the gas phase. In this paper we evaluate a possible strategy of surface passivation. Here the particle--particle interaction of hydrogen-surface-terminated silicon nanoparticles has been evaluated using molecular dynamics simulation. Nanoparticles of the size between 200 and 6400 silicon atoms at 300--1800 K were studied with a reparametrized Kohen-Tully-Stillinger empirical interatomic potential. A hydrogen monolayer is shown to prevent coalescence between particles under thermal collision conditions. The critical approach energy for coalescence was found to increase with increasing particle size but decreases with increasing temperature. Both solid and liquid droplets were seen to bounce at thermal energies, and in some cases, ``superelastic'' collisions are observed, where the rebound kinetic energy of the droplet is higher than the approach energy. These results suggest that surface coatings can significantly retard nanoaerosol growth. read less NOT USED (high confidence) S. Sriraman, E. Aydil, and D. Maroudas, “Growth and characterization of hydrogenated amorphous silicon thin films from SiH2 radical precursor: Atomic-scale analysis,” Journal of Applied Physics. 2004. link Times cited: 21 Abstract: Molecular-dynamics (MD) simulations of hydrogenated amorphou… read moreAbstract: Molecular-dynamics (MD) simulations of hydrogenated amorphous silicon (a-Si:H) film growth on an initially H-terminated Si(001)-(2×1) substrate at T=500 K was studied through repeated impingement of SiH2 radicals to elucidate the effects of this species on the structural quality of the deposited films. A detailed analysis of the radical–surface interaction trajectories revealed the important reactions contributing to film growth. These reactions include (i) adsorption of SiH2 onto the deposition surface, (ii) insertion of SiH2 into surface Si–Si bonds, (iii) surface dimerization of adsorbed SiH2 groups, (iv) formation of polysilane chains and islands, (SiH2)n, n⩾2, on the surface, (v) formation of higher surface hydrides through the exchange of hydrogen, and (vi) dangling-bond-mediated dissociation of surface hydrides. The MD simulations of a-Si:H film growth predict an overall surface reaction probability of 39% for the SiH2 radical. Structural and chemical characterization of the deposited films was car... read less NOT USED (high confidence) A. Tekin and B. Hartke, “Global geometry optimization of small silicon clusters with empirical potentials and at the DFT level,” Physical Chemistry Chemical Physics. 2004. link Times cited: 40 Abstract: We have performed global parameter optimization of selected … read moreAbstract: We have performed global parameter optimization of selected empirical potentials for silicon, resulting in improved performance for small to medium-sized silicon clusters, as judged by a comparison of globally optimized cluster structures to the structures accepted in the literature for the size range up to n = 10. Using global cluster structure optimizations with the resulting optimized model potential and ensuing local optimizations at the DFT level, we could find improved proposals for global minimum structures in the size region n = 10–16. This study confirms the applicability of our general global cluster optimization strategy for still larger silicon clusters. read less NOT USED (high confidence) J. Moreland, “THE DISPARATE THERMAL CONDUCTIVITY OF CARBON NANOTUBES AND DIAMOND NANOWIRES STUDIED BY ATOMISTIC SIMULATION,” Microscale Thermophysical Engineering. 2004. link Times cited: 81 Abstract: Molecular dynamics simulations were used to calculate the th… read moreAbstract: Molecular dynamics simulations were used to calculate the thermal conductivity of carbon nanotubes and diamond nanowires with atomic interactions modeled by the Brenner potential. The dependence of thermal conductivity on length, temperature, and temperature “boundary” condition was investigated. Lengths from 50 nm to 1 μm were simulated at a temperature of 290 K, and additional simulations were performed at 100 K and 400 K, for the 100 nm length. Thermal conductivity was found to be significantly suppressed for the shorter lengths. Two different artificial thermostats were used to impose the temperature difference: one rescaled velocities (the Berendsen thermostat), the other assigned velocities sampled from the appropriate Boltzmann distribution to randomly selected atoms for each numerical time step (the Andersen thermostat). Thus, the Berendsen thermostat amplifies existing atomic motions, while the Andersen thermostat, in a sense, disrupts the atomic motions. Nevertheless, results were very similar. All simulations were run for at least 200,000 time steps of 1 fsec each. read less NOT USED (high confidence) Y. Zhao, Y. Lin, and B. Yakobson, “Fullerene shape transformations via Stone-Wales bond rotations,” Physical Review B. 2003. link Times cited: 38 Abstract: Coalescence of fullerene cages and nanotubes can lead to the… read moreAbstract: Coalescence of fullerene cages and nanotubes can lead to the formation of novel and useful structures. We analyze the Stone-Wales (SW) paths for such fusion processes, compute and compare their energy costs, and demonstrate how the paths are determined by the initial orientation of the merging fragments. We also emphasize the versatility of SW transformation by presenting the topological possibility of the gradual penetration of a buckyball through the wall of a nanotube. read less NOT USED (high confidence) R. Ruoff, D. Qian, and W. K. Liu, “Mechanical properties of carbon nanotubes: theoretical predictions and experimental measurements,” Comptes Rendus Physique. 2003. link Times cited: 647 NOT USED (high confidence) O. Kum, F. Ree, S. Stuart, and C. J. Wu, “Molecular dynamics investigation on liquid–liquid phase change in carbon with empirical bond-order potentials,” Journal of Chemical Physics. 2003. link Times cited: 16 Abstract: A liquid–liquid phase transition in carbon is investigated w… read moreAbstract: A liquid–liquid phase transition in carbon is investigated with two recent bond-order potentials. In contrary to a previous bond-order model, they show no phase change in liquid carbon, which agrees with simulations based on the nonempirical density-functional theory (DFT). Ab initio and DFT studies carried out in this work show that the observed discrepancy lies not in any inherent shortcoming in using empirical models for the bonding process, but rather in the quality of individual expressions used to represent a conjugated local environment in liquid carbon. The present work shows that the current bond-order models and a slightly modified potential proposed in this work agree with recent quantum mechanical simulations and will provide a viable tool for a large-scale study of carbon over a wide range of pressures and temperatures. read less NOT USED (high confidence) E. Ivanova, A. Krivtsov, N. Morozov, and A. Firsova, “Inclusion of the moment interaction in the calculation of the flexural rigidity of nanostructures,” Doklady Physics. 2003. link Times cited: 26 NOT USED (high confidence) J. Los and A. Fasolino, “Intrinsic long-range bond-order potential for carbon: Performance in Monte Carlo simulations of graphitization,” Physical Review B. 2003. link Times cited: 233 Abstract: We propose a bond order potential for carbon with built-in l… read moreAbstract: We propose a bond order potential for carbon with built-in long-range interactions. The potential is defined as the sum of an angular and coordination dependent short-range part accounting for the strong covalent interactions and a radial long-range part describing the weak interactions responsible, e.g., for the interplanar binding in graphite. The short-range part is a Brenner type of potential, with several modifications introduced to get an improved description of elastic properties and conjugation. Contrary to previous long-range extensions of existing bond order potentials, we prevent the loss of accuracy by compensating for the additional long-range interactions by an appropriate parametrization of the short-range part. We also provide a short-range bond order potential. In Monte Carlo simulations our potential gives a good description of the diamond to graphite transformation. For thin (111) slabs graphitization proceeds perpendicular to the surface as found in ab initio simulations, whereas for thick layers we find that graphitization occurs layer by layer. read less NOT USED (high confidence) O. Louchev and J. Hester, “Kinetic pathways of carbon nanotube nucleation from graphitic nanofragments,” Journal of Applied Physics. 2003. link Times cited: 24 Abstract: A detailed analysis of nanotube (NT) nucleation from graphit… read moreAbstract: A detailed analysis of nanotube (NT) nucleation from graphitic nanofragments by thermal vibration is given, outlining the role of activation energy barriers which may be significantly decreased by the interaction with amorphous carbon, fullerene-like, and carbonized metal catalyst nanoparticles. This analysis predicts the variety of carbon nanotube chiralities observed experimentally. Heat dissipation by the inert gas and the cooling rate of the metal–carbon nanoparticle are suggested to play an important role in kinetic selection between (i) a carbon nanosheet wrapping around the metal nanoparticle, (ii) NT nucleation and growth on the nanoparticle surface by the so called “root mechanism,” and (iii) the metal nanoparticle surface being covered by an amorphous carbon layer. read less NOT USED (high confidence) P. A. Marcos, J. Alonso, L. M. Molina, Á. Rubio, and M. J. López, “Structural and thermal properties of silicon-doped fullerenes,” Journal of Chemical Physics. 2003. link Times cited: 33 Abstract: Extensive Molecular Dynamics simulations have been performed… read moreAbstract: Extensive Molecular Dynamics simulations have been performed to investigate the structural and thermal properties of Si-doped fullerenes containing one and two silicon atoms. Both, a many-body potential and ab initio Density Functional Theory (DFT) have been used to investigate the structural features of the heterofullerenes. The competition between the exohedral and the substitutional types of doping, as a function of fullerene size (both even and odd heterofullerenes have been considered) and Si concentration, is analyzed. The DFT calculations confirm the main structural trends obtained with the many-body potential. The thermal stability and the structural transformations of the heterofullerenes have been also studied as a function of temperature (T=0–5000 K). The structural transformations include, local rearrangement of atoms, isomerization transitions, diffusion of atoms, eventual destruction of the cage, and sublimation of atoms. The isomerization transition between exohedral and substitutional isom... read less NOT USED (high confidence) J. Li, D. Liao, S. Yip, R. Najafabadi, and L. Ecker, “Force-based many-body interatomic potential for ZrC,” Journal of Applied Physics. 2003. link Times cited: 48 Abstract: A classical potential for ZrC is developed in the form of a … read moreAbstract: A classical potential for ZrC is developed in the form of a modified second-moment approximation with emphasis on the strong directional dependence of the C–Zr interactions. The model has a minimal set of parameters, 4 for the pure metal and 6 for the cross interactions, which are fitted to the database of cohesive energies of B1–, B2–, and B3–ZrC, the heat of formation, and most importantly, the atomic force constants of B1–ZrC from first-principles calculations. The potential is then extensively tested against various physical properties, none of which were considered in the fitting. Finite temperature properties such as thermal expansion and melting point are in excellent agreement with experiments. We believe our model should be a good template for metallic ceramics. read less NOT USED (high confidence) J. Gale and A. Rohl, “The General Utility Lattice Program (GULP),” Molecular Simulation. 2003. link Times cited: 1866 Abstract: The General Utility Lattice Program (GULP) has been extended… read moreAbstract: The General Utility Lattice Program (GULP) has been extended to include the ability to simulate polymers and surfaces, as well as adding many other new features, and the current status of the program is fully documented. Both the background theory is described, as well as providing a concise review of some of the previous applications in order to demonstrate the range of its use. Examples are presented of work performed using the new compatibilities of the software, including the calculation of Born effective charges, mechanical properties as a function of applied pressure, calculation of frequency-dependent dielectric data, surface reconstructions of calcite and the performance of a linear-scaling algorithm for bond-order potentials. read less NOT USED (high confidence) M. López, Á. Rubio, and J. A. Alonso, “Deformations and thermal stability of carbon nanotube ropes,” IEEE Transactions on Nanotechnology. 2003. link Times cited: 28 Abstract: Structural and thermal characteristics of crystalline ropes … read moreAbstract: Structural and thermal characteristics of crystalline ropes of single-wall carbon nanotubes (SWCNTs) are investigated. Novel crystalline ropes of polygonized SWCNTs produced by laser irradiation exhibit rounded-hexagonal cross sections in contrast to earlier observations of circular tubes. Extensive molecular dynamics (MD) simulations lead to several metastable structures of the lattice characterized by different tube cross sections, hexagonal, rounded-hexagonal and circular, and increasing cell volume. The competition between different tube shapes is analyzed and compared to experiments. On the other hand, bundles of SWCNTs coalesce, forming multiwall carbon nanotubes under thermal treatment at high temperatures. Extensive MD simulations confirm the single-wall-to-multiwall transformation and suggest the physical patching-and-tearing mechanism underlying the concerted coalescence of the tubes. read less NOT USED (high confidence) K. Satake and D. Graves, “Molecular dynamics simulation of ion bombardment on hydrogen terminated Si(001)2×1 surface,” Journal of Vacuum Science and Technology. 2003. link Times cited: 13 Abstract: Molecular dynamics simulations were performed to investigate… read moreAbstract: Molecular dynamics simulations were performed to investigate H2+ and SiH3+ ion bombardment of hydrogen terminated Si(001)2×1 surfaces. Normal incidence ion bombardment effects on dangling bond generation, adatom diffusion, and nucleation were studied as a function of incident energy between 10 and 40 eV. The dangling bond generation rate due to H2+ impacts at 20 and 40 eV was about twice that of SiH3+. However these effects appeared to be insignificant compared to probable neutral radical effects under typical plasma-enhanced chemical vapor deposition conditions. The enhanced diffusion of Si adatoms due to ion bombardment was observed to be minor in comparison with thermal diffusion and the disruption of ledge sites due to SiH3+ ion bombardment is not significant, with ion incident energies up to 40 eV. Ion bombardment in the incident energy range between 10 and 20 eV can contribute the modification of surface kinetics without bulk damage. read less NOT USED (high confidence) P. Träskelin, E. Salonen, K. Nordlund, A. Krasheninnikov, J. Keinonen, and C. Wu, “Molecular dynamics simulations of CH3 sticking on carbon surfaces,” Journal of Applied Physics. 2003. link Times cited: 15 Abstract: Employing both quantum mechanical and empirical force models… read moreAbstract: Employing both quantum mechanical and empirical force models, we use molecular dynamics simulations to obtain sticking cross sections for CH3 radical chemisorption on unsaturated sites of carbon surfaces. Effects of the local atomic neighborhood on the chemisorption are examined for the comparison of the results with experiments. Our results show that the chemisorption of a CH3 radical onto a dangling bond is highly affected by the neighborhood of the unsaturated carbon atom sites. Notably, sticking cross sections of totally bare dangling bond sites at the surface and sites partly shielded by neighboring methyl groups are observed to differ by two orders of magnitude, (15.3±1.7) A2 and (0.2±0.1) A2, respectively. We describe a steering effect which explains the recent experimental observation that the sticking cross section can be larger than the average area per surface site. read less NOT USED (high confidence) J. Kang and H. Hwang, “Hypothetical silicon nanotubes under axial compression,” Nanotechnology. 2003. link Times cited: 23 Abstract: This study shows the response of silicon nanotubes (SiNTs) u… read moreAbstract: This study shows the response of silicon nanotubes (SiNTs) under axial compression using an atomistic simulation based on the Tersoff potential. The application of pressure, proportional to the deformation within Hook’s law, eventually led to a collapse of the SiNT and an abrupt change in structure. Using the sum of the cross sections of the atoms on the SiNT cross section and the SiNT pressure, we determined Young’s modulus for the SiNTs that was constant irrespective of the SiNTs’ diameter. As the SiNTs’ diameter increased, the collapse pressure, that is the critical stress, linearly decreased. However, the net forces on the SiNTs at their collapse were almost constant irrespective of the SiNTs’ diameter. We calculated the variations in the unit cell volume as a function of pressure, which were not dealt with in previous works considering carbon nanotubes under compression. Using properly chosen parameters for the SiNTs (Young’s modulus, effective spring constant, diameter, lattice constant and cylindrical volume modulus), the critical strain, the collapse pressure, the elastic energy and the critical volume at which the SiNT buckling occurs can be estimated by equations given in this work. read less NOT USED (high confidence) F. Benkabou, M. Certier, and H. Aourag, “Elastic Properties of Zinc-blende G a N, A l N and I n N from Molecular Dynamics,” Molecular Simulation. 2003. link Times cited: 42 Abstract: Molecular dynamics calculations of the adiabatic elastic con… read moreAbstract: Molecular dynamics calculations of the adiabatic elastic constants of group III-Nitrides for temperatures ranging from 300 to 900 K have been performed. The results show good agreement with first-principles calculations. The moduli decreased with increasing temperature. The structural properties of zinc-blende GaN, AlN and InN are reported. Good agreement between the calculated and experimental values of the lattice constant, the cohesion energy, and the bulk modulus and its derivative are obtained. read less NOT USED (high confidence) V. Chizhikov, “Plane decagonal quasicrystals with 3-coordinated atoms,” Crystallography Reports. 2002. link Times cited: 0 NOT USED (high confidence) Z. Peng, H. Yonggang, P. Geubelle, and H. Kehchih, “On the continuum modeling of carbon nanotubes,” Acta Mechanica Sinica. 2002. link Times cited: 54 NOT USED (high confidence) I. Jang, R. Phillips, and S. Sinnott, “Study of C3H5+ ion deposition on polystyrene and polyethylene surfaces using molecular dynamics simulations,” Journal of Applied Physics. 2002. link Times cited: 14 Abstract: Molecular dynamics simulations of ion deposition processes a… read moreAbstract: Molecular dynamics simulations of ion deposition processes are used to study the deposition of C3H5+ ions on crystalline polystyrene (PS) and polyethylene (PE) surfaces at energies of 50 and 25 eV. For each system, 80 trajectories are carried out on pristine surfaces and the incident angle in every case is normal to the surface. The forces are determined using the reactive empirical bond order method developed by Tersoff and parametrized for hydrocarbons by Brenner, coupled to long-range Lennard–Jones potentials. The simulations predict that the ions deposited at 50 eV either dissociate and stick to the surface or remain on the surface intact in 98% of the trajectories on PS, and in 89% of the trajectories on PE. At 25 eV, the ions are deposited intact in 70% of the trajectories on PS and dissociate in only 3%. No dissociation of the incident ions is predicted to occur on PE at 25 eV. Rather, the ions scatter away in 90% of the trajectories. Consequently, ion deposition on PE at 25 eV is predicted to be v... read less NOT USED (high confidence) C. Rountree, R. Kalia, E. Lidorikis, A. Nakano, L. Brutzel, and P. Vashishta, “ATOMISTIC ASPECTS OF CRACK PROPAGATION IN BRITTLE MATERIALS: Multimillion Atom Molecular Dynamics Simulations,” Annual Review of Materials Research. 2002. link Times cited: 173 Abstract: ▪ Abstract Atomistic aspects of dynamic fracture in a variet… read moreAbstract: ▪ Abstract Atomistic aspects of dynamic fracture in a variety of brittle crystalline, amorphous, nanophase, and nanocomposite materials are reviewed. Molecular dynamics (MD) simulations, ranging from a million to 1.5 billion atoms, are performed on massively parallel computers using highly efficient multiresolution algorithms. These simulations shed new light on (a) branching, deflection, and arrest of cracks; (b) growth of nanoscale pores ahead of the crack and how pores coalesce with the crack to cause fracture; and (c) the influence of these mechanisms on the morphology of fracture surfaces. Recent advances in novel multiscale simulation schemes combining quantum mechanical, molecular dynamics, and finite-element approaches and the use of these hybrid approaches in the study of crack propagation are also discussed. read less NOT USED (high confidence) S. Sriraman, S. Agarwal, E. Aydil, and D. Maroudas, “Mechanism of hydrogen-induced crystallization of amorphous silicon,” Nature. 2002. link Times cited: 363 NOT USED (high confidence) K. Matsunaga, Y. Iwamoto, and Y. Ikuhara, “Atomic Structure and Diffusion in Amorphous Si-B-C-N by Molecular Dynamics Simulation,” Materials Transactions. 2002. link Times cited: 8 Abstract: We carried out molecular dynamics simulation of amorphous si… read moreAbstract: We carried out molecular dynamics simulation of amorphous silicon nitride containing boron and carbon, in order to investigate the short-range atomic arrangement and diffusion behavior. In amorphous Si–B–N, boron atoms are in a nearly threefold coordinated state with nitrogen atoms, while boron atoms in amorphous Si–B–C–N have bonding with both carbon and nitrogen atoms. Carbon atoms in Si–B–C–N are also bonded to silicon atoms. The self-diffusion constant of nitrogen in Si–B–N becomes much smaller than that in amorphous Si3N4. Also, amorphous Si–B–C–N exhibits smaller self-diffusion constants of constituent atoms, even compared to Si–B–N. Addition of boron and carbon is important in decreasing atomic mobility in amorphous Si–B–C–N. This may explain the increased thermal stability of the amorphous state observed experimentally. read less NOT USED (high confidence) S. Sriraman, E. Aydil, and D. Maroudas, “Atomic-scale analysis of deposition and characterization of a-Si: H thin films grown from SiH radical precursor,” Journal of Applied Physics. 2002. link Times cited: 14 Abstract: Growth of hydrogenated amorphous silicon films (a-Si:H) on a… read moreAbstract: Growth of hydrogenated amorphous silicon films (a-Si:H) on an initial H-terminated Si(001)(2×1) substrate at T=500 K was studied through molecular-dynamics (MD) simulations of repeated impingement of SiH radicals to elucidate the effects of reactive minority species on the structural quality of the deposited films. The important reactions contributing to film growth were identified through detailed visualization of radical–surface interaction trajectories. These reactions include (i) insertion of SiH into Si–Si bonds, (ii) adsorption onto surface dangling bonds, (iii) surface H abstraction by impinging SiH radicals through an Eley–Rideal mechanism, (iv) surface adsorption by penetration into subsurface layers or dissociation leading to interstitial atomic hydrogen, (v) desorption of interstitial hydrogen into the gas phase, (vi) formation of higher surface hydrides through the exchange of hydrogen, and (vii) dangling-bond-mediated dissociation of surface hydrides into monohydrides. The MD simulations of a... read less NOT USED (high confidence) P. Keblinski, M. Bazant, R. Dash, and M. Treacy, “Thermodynamic behavior of a model covalent material described by the environment-dependent interatomic potential,” Physical Review B. 2002. link Times cited: 38 Abstract: Using molecular-dynamics simulations we study the thermodyna… read moreAbstract: Using molecular-dynamics simulations we study the thermodynamic behavior of a single-component covalent material described by the recently proposed environment-dependent interatomic potential (EDIP). The parametrization of EDIP for silicon exhibits a range of unusual properties typically found in more complex materials, such as the existence of two structurally distinct disordered phases, a density increase upon melting of the low-temperature amorphous phase, and negative thermal-expansion coefficients for both the crystal (at high temperatures) and the amorphous phase (at all temperatures). Structural differences between the two disordered phases also lead to a first-order transition between them, which suggests the existence of a second critical point, as is believed to exist for amorphous forms of frozen water. For EDIP-Si, however, the unusual behavior is associated not only with the open nature of tetrahedral bonding but also with a competition between fourfold (covalent) and fivefold (metallic) coordination. The unusual behavior of the model and its unique ability to simulate the liquid/amorphous transition on molecular-dynamics time scales make it a suitable prototype for fundamental studies of anomalous thermodynamics in disordered systems. read less NOT USED (high confidence) M. Medvedeva, I. A. Wojciechowski, and B. Garrison, “Effect of mass and incidence angle of keV energy polyatomic projectiles in silicon sputtering,” Surface Science. 2002. link Times cited: 12 NOT USED (high confidence) M. Kohyama, “TOPICAL REVIEW: Computational studies of grain boundaries in covalent materials,” Modelling and Simulation in Materials Science and Engineering. 2002. link Times cited: 74 Abstract: Computational studies of energetics, atomic and electronic s… read moreAbstract: Computational studies of energetics, atomic and electronic structures and various properties of grain boundaries in covalent materials such as semiconductors and covalent ceramics are reviewed. For coincidence tilt boundaries, atomic and electronic structures were investigated intensively by using various computational schemes such as many-body interatomic potentials, tight-binding method and first-principles method. Computational results were compared with experimental results using recent novel techniques of electron microscopy such as high-resolution transmission electron microscopy, atomic-resolution Z-contrast imaging and electron energy-loss spectroscopy. Such collaboration clarified the detailed nature of coincidence tilt boundaries constructed by structural units. The behaviour of dopants at semiconductor grain boundaries was also investigated by such collaboration. Computations of twist boundaries provided insight into the nature of disordered configurations at general grain boundaries, which should strongly affect the properties of polycrystalline semiconductors and structural ceramics. Recent computational studies dealt with the basic mechanical properties of grain boundaries in covalent materials, where the behaviour of interfacial bonds plays an essential role. read less NOT USED (high confidence) M. Prasad and T. Sinno, “Atomistic-to-continuum description of vacancy cluster properties in crystalline silicon,” Applied Physics Letters. 2002. link Times cited: 28 Abstract: A synergistic combination of molecular dynamics and statics … read moreAbstract: A synergistic combination of molecular dynamics and statics calculations based on the empirical Environment-Dependent Interatomic Potential (EDIP) is used to compute the thermodynamic properties of vacancy clusters (voids) in silicon. All cluster formation properties are found to follow a simple size scaling law, leading to a compact expression for void free energies. An estimate for the free energy of the unreconstructed Si (111) surface is found to compare well with experimental measurements. The results should be useful for the development of accurate process simulators for void formation during crystal growth and wafer thermal annealing. read less NOT USED (high confidence) T. Frauenheim et al., “Atomistic simulations of complex materials: ground-state and excited-state properties,” Journal of Physics: Condensed Matter. 2002. link Times cited: 456 Abstract: The present status of development of the density-functional-… read moreAbstract: The present status of development of the density-functional-based tightbinding (DFTB) method is reviewed. As a two-centre approach to densityfunctional theory (DFT), it combines computational efficiency with reliability and transferability. Utilizing a minimal-basis representation of Kohn–Sham eigenstates and a superposition of optimized neutral-atom potentials and related charge densities for constructing the effective many-atom potential, all integrals are calculated within DFT. Self-consistency is included at the level of Mulliken charges rather than by self-consistently iterating electronic spin densities and effective potentials. Excited-state properties are accessible within the linear response approach to time-dependent (TD) DFT. The coupling of electronic and ionic degrees of freedom further allows us to follow the non-adiabatic structure evolution via coupled electron–ion molecular dynamics in energetic particle collisions and in the presence of ultrashort intense laser pulses. We either briefly outline or give references describing examples of applications to ground-state and excited-state properties. Addressing the scaling problems in size and time generally and for biomolecular systems in particular, we describe the implementation of the parallel ‘divide-and-conquer’ order-N method with DFTB and the coupling of the DFTB approach as a quantum method with molecular mechanics force fields. read less NOT USED (high confidence) W. Sekkal and A. Zaoui, “Predictive study of thermodynamic properties of GeC,” New Journal of Physics. 2002. link Times cited: 47 Abstract: We present in this paper a molecular dynamics simulation of … read moreAbstract: We present in this paper a molecular dynamics simulation of structural and thermodynamic properties of the hypothetical IV-IV compound GeC in the zinc-blende structure. This study is performed with the use of the well-tested Tersoff potential. Various physical quantities including elastic constants, Debye temperature, thermal expansion coefficient, heat capacity, and Grüneisen parameter are predicted. The comparison with the corresponding results for SiC is also discussed. read less NOT USED (high confidence) D. Brenner, O. Shenderova, J. Harrison, S. Stuart, B. Ni, and S. Sinnott, “A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons,” Journal of Physics: Condensed Matter. 2002. link Times cited: 3204 Abstract: A second-generation potential energy function for solid carb… read moreAbstract: A second-generation potential energy function for solid carbon and hydrocarbon molecules that is based on an empirical bond order formalism is presented. This potential allows for covalent bond breaking and forming with associated changes in atomic hybridization within a classical potential, producing a powerful method for modelling complex chemistry in large many-atom systems. This revised potential contains improved analytic functions and an extended database relative to an earlier version (Brenner D W 1990 Phys. Rev. B 42 9458). These lead to a significantly better description of bond energies, lengths, and force constants for hydrocarbon molecules, as well as elastic properties, interstitial defect energies, and surface energies for diamond. read less NOT USED (high confidence) N. Mousseau and G. Barkema, “Fast bond-transposition algorithms for generating covalent amorphous structures,” Current Opinion in Solid State & Materials Science. 2001. link Times cited: 8 NOT USED (high confidence) R. Komanduri, N. Chandrasekaran, and L. Raff, “Molecular dynamics simulation of the nanometric cutting of silicon,” Philosophical Magazine B. 2001. link Times cited: 139 Abstract: Molecular dynamics simulations of nanometric cutting of sing… read moreAbstract: Molecular dynamics simulations of nanometric cutting of single-crystal, defect-free, pure silicon were performed using the Tersoff potential over a wide range of rake angles (from -60° to +60°), widths of cut (1.1 to 4.34 ran), depths of cut (0.01 to 2.72 nm) and clearance angles (10° to 30°) to hwestigate the nature of material removal and surface generation process in ultraprecision machining and grinding. The observed material removal mechanisms can be divided into four components: (i) compression of the work material ahead of the tool; (ii) chip formation akin to an extrusion-like process; (iii) side flow; and (iv) subsurface deformation in the machined surface. Unlike in conventional machining of most ductile materials, where no volume or phase change is observed in the plastic deformation process, significant volume changes (from 18.38 to 14.19Å3), resulting in a densification of about 23% occur owing to phase transition from a diamond cubic (or α-silicon) to a bet (or β-tin structure) in the case of machining silicon. Such a structural change is typical of silicon undergoing a pressure-induced phase transformation. The extent of structural changes and their contributions to each of the four material removal mechanisms depend on the tool rake angle and the width of cut. The ratio of the width of cut to depth of cut w/d is the primary factor affecting the extent of side flow and subsurface compression. The tool rake angle and the w/d ratio are found to be dominant factors affecting the chip flow and shear zone compression ahead of the tool. Subsurface or near-surface deformation was observed with all rake angles and all cut depths down to 0.01 nm, indicating the need for an alternate final polishing process such as chemomechanical polishing to produce defect-free surfaces of silicon on an atomie scale. read less NOT USED (high confidence) A. Charaï et al., “Structural change induced on an atomie scale by equilibrium sulphur segregation in tilt germanium grain boundaries,” Philosophical Magazine B. 2001. link Times cited: 1 Abstract: In the present study, structural modifications induced by ea… read moreAbstract: In the present study, structural modifications induced by eauilibrium sulphur segregation in pure tilt germanium {710}<001>, ∑=25 (θ=16.26°) and {551}<011>, ∑=51 (θ=16.10°) grain boundaries (GBs) were investigated using high-resolution electron microscopy coupled to electron-energy-loss spectroscopy and supported by structural modelling and image simulations. Our results showed that the as-grown ∑=25 GB is composed of two parts: a stable structural region and a variable perturbed core. On the basis of our simulations, it is shown that this boundary can only be formed by a multiplicity of configurations which are energetically close to each other but differently configured along the boundary plane. When sulphurized, drastic changes in the structure of the GB were observed. Energy-filtered electron microscopy imaging revealed a sulphur enrichment at the perturbed part of the boundary. Although sulphur segregation at the boundary is detected, no information can at the present stage be extracted on segregation sites and bonding configurations because of the complexity of the boundary structure. To simplify this aspect, a simpler GB, that is germanium ∑=51, was studied. The structure of such a GB is a well-known configuration, that is a Lomer dislocation, which is basically a fivefold ring adjacent to a sevenfold ring. After sulphur treatment, high-resolution electron microscopy imaging also shows significant contrast modifications apparently concentrated on the dislocation core. Chemical imaging indicates again the presence of sulphur enrichment along the boundary plane strongly sustaining that eauilibrium sulphur segregation in the Ge(S) system oceurs into the GB and therefore confirms our previous results on the ∑= 25 GB. One can therefore argue that it is the presence of those odd-membered rings at the boundary, which should possess a specific crystallographic and electronic nature, coupled to the electronic properties of sulphur, that are responsible for the preferential segregation into the boundary. read less NOT USED (high confidence) L. Chernozatonskii, “Diboride bifullerenes and binanotubes,” Journal of Experimental and Theoretical Physics Letters. 2001. link Times cited: 20 NOT USED (high confidence) C. Herrero, “Quantum atomistic simulations of silicon and germanium,” Journal of Materials Research. 2001. link Times cited: 4 Abstract: Quantum atomistic simulations of crystalline silicon and ger… read moreAbstract: Quantum atomistic simulations of crystalline silicon and germanium have been carried out by the path-integral Monte Carlo method. The interatomic interactions were modeled by Stillinger–Weber-type potentials, with parameters adequate to quantum simulations. Quantum zero-point motion together with anharmonicity of the interatomic potential led to a lattice expansion of 7 × 10^−3 Å for both Si and Ge. Results for the equation-of-state (volume versus pressure) and for the thermal expansion coefficient agreed well with experimental results for both materials at T > 100 K and for hydrostatic pressures up to 100 kbar. read less NOT USED (high confidence) R. Ramírez and T. López-Ciudad, “Low lying vibrational excitation energies from equilibrium path integral simulations,” Journal of Chemical Physics. 2001. link Times cited: 18 Abstract: The centroid density is a function defined for quantum syste… read moreAbstract: The centroid density is a function defined for quantum systems in thermodynamic equilibrium that is readily obtained by path integral simulations. The physical information provided by the centroid density is a static response of the system under isothermal conditions, namely, the change in the expectation value of the position operator of the quantum particles upon application of constant external forces. An interesting application of this function is the study of vibrational properties of atomic nuclei in molecules and solids. In particular, the analysis of the tensor defined by the second cumulants of the centroid density (i.e., the static isothermal susceptibility tensor) leads to the definition of the linear response vibrational modes, which are characterized by a response of the quantum system parallel to the applied force. The eigenvalues of the susceptibility tensor provide the linear response of the system. This response function is the basis for the formulation of two approximations to evaluate v... read less NOT USED (high confidence) S. Ramalingam, E. Aydil, and D. Maroudas, “Molecular dynamics study of the interactions of small thermal and energetic silicon clusters with crystalline and amorphous silicon surfaces,” Journal of Vacuum Science & Technology B. 2001. link Times cited: 9 Abstract: An atomic-scale analysis based on molecular dynamics simulat… read moreAbstract: An atomic-scale analysis based on molecular dynamics simulations of the interactions of small thermal and energetic SinHm, n>1, clusters observed in various plasmas with crystalline and amorphous Si surfaces is presented. The experimental literature has assumed and employed a unit reaction probability for clusters of various sizes on all Si surfaces in phenomenological models for obtaining hydrogenated amorphous Si film growth rates, while the reaction mechanisms of clusters with the deposition surfaces have remained unexplored. In addition, it is widely speculated that clusters have a detrimental effect on the film quality. Our study shows that the clusters react with high (>85%) probability with crystalline surfaces and with surfaces of amorphous Si films. The structure and energetics of the corresponding adsorbed cluster configurations on these surfaces are analyzed and discussed. Furthermore, the simulations provide insight into possible mechanisms for the formation of defects, such as voids and dangl... read less NOT USED (high confidence) W. Sekkal and A. Zaoui, “Molecular dynamics simulation of superhard phases in RuO2,” Journal of Physics: Condensed Matter. 2001. link Times cited: 9 Abstract: We present a molecular dynamics simulation study of structur… read moreAbstract: We present a molecular dynamics simulation study of structural and thermodynamic properties of RuO2 in the fluorite and Pa structures. Based on a three-body potential, our results are in agreement with experimental measurements and other ab initio calculations. The transferability of this potential model is tested by simulating the superhard phases of RuO2 for varying temperature. Various thermodynamic properties including the Debye temperature, heat capacity, linear thermal coefficient, Gruneisen parameter, and melting point are predicted. Calculations are extended to simulate also the liquid phase of RuO2 in the Pa structure. read less NOT USED (high confidence) H. Koga, Y. Nakamura, M. Watanabe, and T. Yoshida, “Molecular dynamics study of deposition mechanism of cubic boron nitride,” Science and Technology of Advanced Materials. 2001. link Times cited: 13 NOT USED (high confidence) K. Moriguchi et al., “Nano-tube-like surface structure in graphite particles and its formation mechanism: A role in anodes of lithium-ion secondary batteries,” Journal of Applied Physics. 2000. link Times cited: 37 Abstract: Nano-structures on the surface of graphite based carbon part… read moreAbstract: Nano-structures on the surface of graphite based carbon particles have been investigated by means of high resolution transmission electron microscopy. The surfaces consist of “closed-edge” structures in a similar manner as carbon nano-tube. That is, they are composed of coaxial carbon tubes consisting of adequate coupling of graphite layer edges. These graphite particles are chemically stable and, therefore, applicable for lithium-ion secondary battery anodes. Molecular dynamics simulations based on the Tersoff potential reveal that the vibrations of the graphite layers at the free edges play an important role in the formation of the closed-edge structures. In lithium-ion secondary batteries, Li ions can intrude into bulk carbon anodes through these closed-edge structures. In order to clarify this intrusion mechanism, we have studied the barrier potentials of Li intrusion through these closed edges using the first-principles cluster calculations. From electrochemical measurements, the carbon anodes compos... read less NOT USED (high confidence) R. Neuendorf, R. Palmer, and R. Smith, “Implantation of size-selected Si clusters into graphite,” Applied Physics Letters. 2000. link Times cited: 3 Abstract: Molecular dynamics simulations have been performed to explor… read moreAbstract: Molecular dynamics simulations have been performed to explore the implantation of silicon clusters into a graphite substrate to well-defined depths. The cluster sizes range from N=20 up to N=200 atoms per cluster, deposited with kinetic energies from E=500 eV up to E=5000 eV per cluster. We find that the clusters remain intact as coherent, amorphous structures after implantation. The implantation depth is well defined and scales with the kinetic energy of the clusters and the inverse of the cross-sectional area. This indicates a constant decelerating force, associated with the lateral displacement of carbon atoms as the cluster “drills a hole” in the substrate. The main dissipation channels for the energetic silicon clusters are the creation of phonons in the graphite substrate and the breaking of C–C bonds. read less NOT USED (high confidence) S. Hobday and R. Smith, “Applications of Genetic Algorithms in Cluster Optimisation,” Molecular Simulation. 2000. link Times cited: 5 Abstract: Applications of Genetic Algorithms for optimisation of atomi… read moreAbstract: Applications of Genetic Algorithms for optimisation of atomic clusters are reported. It is shown that the genetic algorithms are very useful tools for determining the minimum energy structures of clusters of atoms described by interatomic potential functions containing up to a few hundred atoms. The algorithm generally outperforms other optimisation methods for this task. A number of applications are given including covalent carbon and silicon clusters, close-packed structures such as argon and silver and the two-component C—H system. read less NOT USED (high confidence) H. Jäger and K. Albe, “Molecular-dynamics simulations of steady-state growth of ion-deposited tetrahedral amorphous carbon films,” Journal of Applied Physics. 2000. link Times cited: 122 Abstract: Molecular-dynamics calculations were performed to simulate i… read moreAbstract: Molecular-dynamics calculations were performed to simulate ion beam deposition of diamond-like carbon films. Using the computationally efficient analytical potentials of Tersoff and Brenner we are able to simulate more than 103 carbon atom impacts on {111} diamond, so that steady-state film properties can be computed and analyzed. For the Tersoff potential, we achieve sp3 fractions approximately half of the experimentally observed values. For the more refined hydrocarbon potentials of Brenner the fraction of tetrahedrally coordinated atoms is much too low, even if structures with densities close to diamond are obtained. We show, that the sp3 contents calculated with Tersoff’s potential are an artifact related to the overbinding of specific bonding configurations between three- and fourfold coordinated sites. On the other hand, we can prove, that the range for the binding orbitals represented by the cutoff function is too short in Brenner’s parametrization. If an increased C–C interaction cutoff value is c... read less NOT USED (high confidence) S. Stuart, A. B. Tutein, and J. Harrison, “A reactive potential for hydrocarbons with intermolecular interactions,” Journal of Chemical Physics. 2000. link Times cited: 3524 Abstract: A potential function is presented that can be used to model … read moreAbstract: A potential function is presented that can be used to model both chemical reactions and intermolecular interactions in condensed-phase hydrocarbon systems such as liquids, graphite, and polymers. This potential is derived from a well-known dissociable hydrocarbon force field, the reactive empirical bond-order potential. The extensions include an adaptive treatment of the nonbonded and dihedral-angle interactions, which still allows for covalent bonding interactions. Torsional potentials are introduced via a novel interaction potential that does not require a fixed hybridization state. The resulting model is intended as a first step towards a transferable, empirical potential capable of simulating chemical reactions in a variety of environments. The current implementation has been validated against structural and energetic properties of both gaseous and liquid hydrocarbons, and is expected to prove useful in simulations of hydrocarbon liquids, thin films, and other saturated hydrocarbon systems. read less NOT USED (high confidence) S. Berber, Y.-K. Kwon, and D. Tománek, “Unusually high thermal conductivity of carbon nanotubes,” Physical review letters. 2000. link Times cited: 2836 Abstract: Combining equilibrium and nonequilibrium molecular dynamics … read moreAbstract: Combining equilibrium and nonequilibrium molecular dynamics simulations with accurate carbon potentials, we determine the thermal conductivity lambda of carbon nanotubes and its dependence on temperature. Our results suggest an unusually high value, lambda approximately 6600 W/m K, for an isolated (10,10) nanotube at room temperature, comparable to the thermal conductivity of a hypothetical isolated graphene monolayer or diamond. Our results suggest that these high values of lambda are associated with the large phonon mean free paths in these systems; substantially lower values are predicted and observed for the basal plane of bulk graphite. read less NOT USED (high confidence) C. Herrero, “Quantum atomic dynamics in amorphous silicon; a path-integral Monte Carlo simulation,” Journal of Physics: Condensed Matter. 2000. link Times cited: 10 Abstract: The quantum dynamics of atoms in amorphous silicon has been … read moreAbstract: The quantum dynamics of atoms in amorphous silicon has been addressed by using path-integral Monte Carlo simulations. Structural results (radial distribution functions) found from these simulations agree well with experimental data. We study the quantum delocalization of the silicon atoms around their equilibrium positions. This delocalization is larger for coordination defects (fivefold-coordinated Si atoms). Correlations in the atomic displacements are analysed as a function of the interatomic distance and compared with those derived from classical Monte Carlo simulations. At high temperatures, the classical limit is recovered. Our results are also compared with those derived from similar quantum simulations for crystalline silicon. Structural disorder favours a larger vibrational amplitude for the atoms in amorphous silicon. read less NOT USED (high confidence) M. Schaible, “Empirical Molecular Dynamics Modeling of Silicon and Silicon Dioxide: A Review,” Critical Reviews in Solid State and Materials Sciences. 1999. link Times cited: 28 Abstract: A number of computational methods have been developed over t… read moreAbstract: A number of computational methods have been developed over the last 40 years to simulate the behavior of solid materials with small dimensions. On the macro-scale, Finite Element analysis calculates mechanical stress on micron-sized cantilevers and motors. On the atomic level, newer ab initio methods compute nuclear and electronic behavior of hundred atom models with unprecedented rigor. By implementing the laws of classic mechanics, empirical Molecular Dynamics (MD) programs help bridge these two computational extremes. MD identifies nonelectronic, particle motion for large 100,000 atom cells with good success. MD derives both equilibrium and nonequilibrium properties for many complex condensed regimes; quantitatively (and qualitatively) reaffirms empirical data; aids discovery of new materials processing techniques, and helps predict unknown physical phenomena often only observable under extreme environmental settings. One material of great technical importance to the semiconductor industry is silicon (... read less NOT USED (high confidence) W. Sekkal, A. Laref, A. Zaoui, H. Aourag, and M. Certier, “The Miscibility of Copper Halides Using a Three-Body Potential. I. CuCl x Br1−x Crystal,” Molecular Simulation. 1999. link Times cited: 3 Abstract: Mixed CuCl x Br1−x crystals are studied using a Tersoff pote… read moreAbstract: Mixed CuCl x Br1−x crystals are studied using a Tersoff potential. Structural and elastic properties of the solid solution are calculated and are in good agreement with experiments. Various thermodynamic quantities including thermal expansion coefficient, heat capacity, and Gruneisen coefficient are also predicted. read less NOT USED (high confidence) S. Ramalingam, P. Mahalingam, E. Aydil, and D. Maroudas, “Theoretical study of the interactions of SiH2 radicals with silicon surfaces,” Journal of Applied Physics. 1999. link Times cited: 27 Abstract: Silylene (SiH2) radicals created by electron impact dissocia… read moreAbstract: Silylene (SiH2) radicals created by electron impact dissociation of silane in reactive gas discharges can play an important role in plasma deposition of amorphous and nanocrystalline silicon thin films. In this article, we present a systematic computational analysis of the interactions of SiH2 radicals with a variety of crystalline and amorphous silicon surfaces based on atomistic simulations. The hydrogen coverage of the surface and, hence, the availability of surface dangling bonds is shown to exert the strongest influence on the radical-surface reaction mechanisms and the corresponding reaction probabilities. The SiH2 radical reacts with unit probability on the pristine Si(001)-(2×1) surface which has one dangling bond per Si atom; upon reaction, the Si atom of the radical forms strong Si–Si bonds with either one or two surface Si atoms. On the H-terminated Si(001)-(2×1) surface, the radical is found to react with a probability of approximately 50%. The SiH2 radical attaches itself to the surface eithe... read less NOT USED (high confidence) M. M. Ali, “A short range many-body potential for modelling bcc metals,” Pramana. 1999. link Times cited: 4 NOT USED (high confidence) S. Ramalingam, D. Maroudas, and E. Aydil, “Atomistic simulation study of the interactions of SiH3 radicals with silicon surfaces,” Journal of Applied Physics. 1999. link Times cited: 54 Abstract: SiH3 radicals created by electron impact dissociation of SiH… read moreAbstract: SiH3 radicals created by electron impact dissociation of SiH4 in reactive gas discharges are widely believed to be the dominant precursor for plasma deposition of amorphous and nanocrystalline silicon thin films. In this article, we present a systematic computational analysis of the interactions of SiH3 radicals with a variety of crystalline and amorphous silicon surfaces through atomistic simulations. The hydrogen coverage of the surface and, hence, the availability of surface dangling bonds has the strongest influence on the radical–surface reaction mechanisms and the corresponding reaction probabilities. The SiH3 radical reacts with unit probability on the pristine Si(001)-(2×1) surface which has one dangling bond per Si atom; upon reaction, the Si atom of the radical forms strong Si–Si bonds with either one or two surface Si atoms. On the H-terminated Si(001)-(2×1) surface, the radical is much less reactive; the SiH3 radical was reflected back into the gas phase in all but two of the 16 simulations of... read less NOT USED (high confidence) W. Sekkal, A. Zaoui, A. Laref, H. Aourag, and M. Certier, “Structural and thermodynamic properties of Cx(BN)1-x alloy,” Journal of Physics: Condensed Matter. 1999. link Times cited: 7 Abstract: In this work, structural and thermodynamic properties of C-B… read moreAbstract: In this work, structural and thermodynamic properties of C-BN solid solutions are investigated using the well tested Tersoff potential. The bulk modulus is lower than those of diamond and cubic BN and the value predicted from considering ideal mixing between C and BN. Various thermodynamics quantities including the thermal expansion coefficient, heat capacity, Debye temperature and Grüneisen coefficient are also predicted. read less NOT USED (high confidence) R. Sahara, H. Mizuseki, K. Ohno, S. Uda, T. Fukuda, and Y. Kawazoe, “Body-centered-cubic lattice model with many-body interactions representing the melting transition in Si,” Journal of Chemical Physics. 1999. link Times cited: 11 Abstract: A body-centered-cubic (BCC) lattice model with realistic man… read moreAbstract: A body-centered-cubic (BCC) lattice model with realistic many-body interactions is introduced and investigated by means of the Metropolis’ Monte Carlo method to describe both crystalline and molten states of Si. Under the simplest assumption that atoms surrounded by tetrahedral first-neighbors only have an energy lower than the other atoms, a clear first-order phase transition including hysteresis is observed between a solid with diamond structure and a melt. Nucleation and domain growth are dynamically observed in certain range of the supercooling. In order to introduce more realistic and accurate lattice-gas models, the Tersoff potential is renormalized and the interactions are mapped onto a BCC lattice. Then, it is found that the phase transition temperature and other thermodynamic properties are dramatically improved compared with the case using the Tersoff potential directly in the lattice model without renormalization. read less NOT USED (high confidence) T. Çagin et al., “Computational Materials Chemistry at the Nanoscale,” Journal of Nanoparticle Research. 1999. link Times cited: 25 NOT USED (high confidence) S. Ramalingam, D. Maroudas, and E. Aydil, “Interactions of SiH radicals with silicon surfaces: An atomic-scale simulation study,” Journal of Applied Physics. 1998. link Times cited: 53 Abstract: A comprehensive study is presented of the interactions of Si… read moreAbstract: A comprehensive study is presented of the interactions of SiH radicals originating in silane containing plasmas with crystalline and amorphous silicon surfaces based on a detailed atomic-scale analysis. The hydrogen concentration on the surface is established to be the main factor that controls both the surface reaction mechanism and the reaction probability; other important factors include the location of impingement of the radical on the surface, as well as the molecular orientation of the radical with respect to the surface. On the ordered crystalline surfaces, the radical reacts in such a way as to maximize the number of Si–Si bonds it can form even if such bond formation requires dissociation of the radical and introduction of defects in the crystal structure. The radical is established to be fully reactive with the pristine Si(001)-(2×1) surface. This chemical reactivity is reduced significantly for the corresponding H-terminated surface with a hydrogen coverage of one monolayer. SiH is found to be ... read less NOT USED (high confidence) W. Sekkal, B. Bouhafs, H. Aourag, and M. Certier, “Molecular-dynamics simulation of structural and thermodynamic properties of boron nitride,” Journal of Physics: Condensed Matter. 1998. link Times cited: 112 Abstract: Structural and thermodynamic properties of cubic boron nitri… read moreAbstract: Structural and thermodynamic properties of cubic boron nitride (c-BN) under pressure and for varying temperature are studied by molecular-dynamics (MD) simulation with the use of a well-tested Tersoff potential. Various physical quantities including the thermal expansion coefficient and heat capacity are predicted. Our simulation is extended to study liquid boron nitride at various densities. read less NOT USED (high confidence) M. Ali, C. Storey, and A. Törn, “Application of Stochastic Global Optimization Algorithms to Practical Problems,” Journal of Optimization Theory and Applications. 1997. link Times cited: 108 NOT USED (high confidence) L. Marqués, J. Rubio, M. Jaraíz, L. Bailón, and J. Barbolla, “Dose effects on amorphous silicon sputtering by argon ions: A molecular dynamics simulation,” Journal of Applied Physics. 1997. link Times cited: 11 Abstract: We have investigated, using molecular dynamics techniques, t… read moreAbstract: We have investigated, using molecular dynamics techniques, the sputtering yield enhancement of amorphous silicon produced by argon ion accumulation within the target. Several amorphous silicon samples, with different argon contents, were bombarded with 1 keV argon ions at normal incidence. To study the influence of the target structure, we considered samples with different argon arrangements, either uniformly distributed or within solid bubbles. We have observed that silicon sputtering yield increases linearly with dose until steady state conditions are reached. This enhancement is produced by the shallow argon atoms through the weakening of Si–Si bonds. We have also observed that argon release takes place even long after the end of the collisional phase, and it is produced by ion-induced desorption and bubble destabilization. This enhanced argon yield determines the dose where target saturation and steady state conditions are reached. read less NOT USED (high confidence) A. Ihlal, R. Rizk, and O. H. Duparc, “Correlation between the gettering efficiencies and the energies of interfaces in silicon bicrystals,” Journal of Applied Physics. 1996. link Times cited: 22 Abstract: A comparative study of the gettering efficiency of the twin … read moreAbstract: A comparative study of the gettering efficiency of the twin grain boundaries Σ=25, Σ=13, and Σ=9 has been carried out by means of electron‐beam‐induced current measurements performed on quenched silicon bicrystals precontaminated by Cu or Ni. The extent of the denuded zone appearing on both sides of each interface type has been considered as the ‘‘rating’’ of its gettering efficiency. For both contaminants, the same scaling of the gettering efficiencies of the boundaries has been observed and was found to be in the order Σ=9≪Σ=13<Σ=25. To account for this ranking, we have correlated the gettering efficiency to the excess energy of the grain boundary with respect to the bulk energy, as theoretically calculated. The computational procedures have been performed by means of molecular‐dynamics simulations using several potentials. On the basis of the specific disorder affecting the Σ=25 structure upon heat treatment, our calculations provided the same progression for the interfacial energies as that observed e... read less NOT USED (high confidence) B. Yakobson, C. J. Brabec, and J. Bernholc, “Nanomechanics of carbon tubes: Instabilities beyond linear response.,” Physical review letters. 1996. link Times cited: 2420 Abstract: Carbon nanotubes subject to large deformations reversibly sw… read moreAbstract: Carbon nanotubes subject to large deformations reversibly switch into different morphological patterns. Each shape change corresponds to an abrupt release of energy and a singularity in the stress-strain curve. These transformations, simulated using a realistic many-body potential, are explained by a continuum shell model. With properly chosen parameters, the model provides a remarkably accurate ``roadmap'' of nanotube behavior beyond Hooke's law. read less NOT USED (high confidence) G. Jungnickel et al., “Graphitization Effects on Diamond Surfaces and the Diamond/Graphite Interface,” Physica Status Solidi (a). 1996. link Times cited: 39 Abstract: Graphitic layers have previously been conjectured to play an… read moreAbstract: Graphitic layers have previously been conjectured to play an active role in diamond nucleation by Lambrecht et al. and may also be involved in a mechanism for homoepitaxial diamond growth since the surfaces of diamond may partially graphitize under high-temperature conditions typical of growth processes. Recent molecular dynamics simulations of the diamond {111} surface, briefly reviewed and discussed here, indicate a progressive graphitization with increasing temperature which is strongly facilitated by any kind of surface perturbation or roughness such as step-like adsorbates. Here we show specifically that also twin boundaries promote graphitization. The process of debonding of the surface layer which is a simple displacive motion of the outer layer is also shown to be closely related to the delamination of the tetrahedrally bonded icosahedral C100 molecule into two concentric C20 and (fullerene-like) C80 fragments. In contrast, the tetrahedrally bonded icosahedral C300 molecule which contains one more concentric shell, does not spontaneously graphitize into a bucky onion (consisting of concentric C80 and C240 fullerenes) although the latter has lower energy. Progressive graphitization at a surface towards deeper layers before the top layer is delaminated can occur under certain conditions and then may lead to graphite/diamond prism plane interfaces similar to those previously investigated in connection with nucleation. The structural stability of the prism plane interface between graphite and diamond is re-investigated here. While the initial calculations with a classical potential underestimated the interface energy, the structural stability of the models previously presented is confirmed by the present quantum mechanical simulations. read less NOT USED (high confidence) H.-C. Huang, N. Ghoniem, J. Wong, and M. Baskes, “Molecular dynamics determination of defect energetics in beta -SiC using three representative empirical potentials,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 102 Abstract: The determination of formation and migration energies of poi… read moreAbstract: The determination of formation and migration energies of point and clustered defects in SiC is of critical importance to a proper understanding of atomic phenomena in a wide range of applications. We present here calculations of formation and migration energies of a number of point and clustered defect configurations. A newly developed set of parameters for the modified embedded-atom method (MEAM) is presented. Detailed molecular dynamics calculations of defect energetics using three representative potentials, namely the Pearson potential, the Tersoff potential and the MEAM, have been performed. Results of the calculations are compared to first-principles calculations and to available experimental data. The results are analysed in terms of developing a consistent empirical interatomic potential and are used to discuss various atomic migration processes. read less NOT USED (high confidence) A. A. Valuev, A. S. Kaklyugin, and H. E. Norman, “Molecular modelling of the chemical interaction of atoms and molecules with a surface,” Russian Chemical Reviews. 1995. link Times cited: 3 Abstract: The modelling of a surface as an assembly of moving atoms in… read moreAbstract: The modelling of a surface as an assembly of moving atoms interacting with one another and with an incident particle is examined. Both dynamic methods for the modelling of a surface (for short times) and probability methods (for long times) are analysed. The Massey adiabaticity criterion has been used to determine the regions of applicability of the methods of molecular dynamics. Within the framework of probability methods, the chemical bond is described with the aid of Harrison's generalised periodic system of the elements. Together with the general modeling problems, the reconstruction of the surface, physical and chemical sorption, as well as the modification of the surface and of its morphology as a result of the multiple repetition of elementary processes (precipitation, etching, corrosion) are discussed. The bibliography includes 169 references. read less NOT USED (high confidence) D. Timpel and K. Scheerschmidt, “HREM simulations of Ag particles in sodium silicate glasses refined by molecular dynamic relaxations,” Physica Status Solidi (a). 1995. link Times cited: 6 Abstract: Providing local information at an atomic level high resoluti… read moreAbstract: Providing local information at an atomic level high resolution electron microscopy (HREM) is applied to investigate Ag particles in sodium silicate glasses. Here, the structure of the embedded metallic particles, which influence the properties of glasses, is described by molecular dynamics relaxation calculations. The possibility of HREM to vizualize the structural modifications owing to relaxations is discussed on the basis of simulated HREM micrographs.
Hochauflosende Elektronenmikroskopie (HREM) wird wegen der direkten atomaren Strukturabbildung zur Untersuchung von Ag-Teilchen in Natriumsilikat-Glasern verwendet. Die Struktur der eingelagerten Metallteilchen, die wesentlichen Einflus auf die Glaseigenschaften hat, wird dabei durch molekulardynamische Relaxationsrechnungen beschrieben. Die Moglichkeiten der Sichtbarmachung struktureller Veranderungen im HREM infolge der Relaxationseffekte werden anhand von computersimulierten Abbildungen diskutiert. read less NOT USED (high confidence) C. Yoon and J. Megusar, “Molecular dynamic simulation of amorphous carbon and graphite interface,” Interface Science. 1995. link Times cited: 8 NOT USED (high confidence) J. Berger and J. Avron, “Classification scheme for toroidal molecules,” Journal of the Chemical Society, Faraday Transactions. 1995. link Times cited: 25 Abstract: A class of planar periodic tilings, which correspond to toro… read moreAbstract: A class of planar periodic tilings, which correspond to toroidal arrangements of trivalent atoms, with pentagonal, hexagonal and heptagonal rings, has been constructed. Each tiling is characterized by a set of four integers and defines a toroidal molecule. The tiling rules are motivated by geometric considerations and the tiling patterns are rich enough to describe a wide class of toroidal carbon molecules, with a broad range of shapes and numbers of atoms. The molecular dimensions are simply related to the integers that determine the tiling. The configurational energy and the delocalisation energy of several molecules obtained in this way were computed for Tersoff and Huckel models. The results indicate that many of these molecules are not strained, and may be expected to be stable. The influence of size on the Huckel spectrum bears both similarities and differences compared with the case of tubules. read less NOT USED (high confidence) S. Serra, S. Sanguinetti, and L. Colombo, “Solid‐to‐liquid phase change and fragmentation in C60,” Journal of Chemical Physics. 1995. link Times cited: 10 Abstract: We present a study of the thermodynamical properties of C60 … read moreAbstract: We present a study of the thermodynamical properties of C60 in the microcanonical ensemble. Solidlike and metastable liquidlike form can be identified in the low energy and in the high energy range, respectively. The transition between the two phases is characterized by a finite energy range, in agreement with general theories of cluster melting. In particular, we have observed that the melting is preceded by a highly isomerized transition region where a sizeable atomic mobility is achieved via hopping between different isomer structures. read less NOT USED (high confidence) V. Bakaev, “Rumpled graphite basal plane as a model heterogeneous carbon surface,” Journal of Chemical Physics. 1995. link Times cited: 26 Abstract: A new model is suggested for the heterogeneous surfaces of n… read moreAbstract: A new model is suggested for the heterogeneous surfaces of nongraphitized carbon adsorbents. It may be called the rumpled graphite basal plane (RGBP). The atomic structure of RGBP can be obtained by squeezing a graphite basal plane in a molecular dynamics computer simulation under a random distribution of initial atomic velocities. The empirical Tersoff potential describes the carbon–carbon interactions. The degree of squeezing is chosen to reproduce the main features of the x‐ray interference function of nongraphitized carbon blacks. Grand canonical ensemble Monte Carlo simulation of the isotherms of adsorption of N2 on RGBP reproduce experimental isotherms on these carbon blacks reasonably well, especially in the BET region of relative pressures. read less NOT USED (high confidence) A. P. Smith et al., “Si adatom binding and diffusion on the Si(100) surface: Comparison of ab initio, semiempirical and empirical potential results,” Journal of Chemical Physics. 1995. link Times cited: 51 Abstract: The binding energies and configurations for single Si adatom… read moreAbstract: The binding energies and configurations for single Si adatoms on the Si(100) surface are investigated theoretically. Detailed comparisons between previously published and new calculations using classical potentials, semiempirical formulations, and density functional theory (DFT) are made. The DFT calculations used both the plane‐wave‐pseudopotential approach in a periodic slab geometry and the Gaussian‐orbital based all‐electron approach employing cluster geometries. In the local‐density approximation excellent agreement between the cluster and slab results was obtained. Inclusion of gradient corrections to the exchange‐correlation energy significantly improves absolute binding energies and changes relative energies by as much as 0.3–0.5 eV depending on the particular exchange‐correlation functional used. Binding energies and relative energies obtained using the classical potentials disagree with the gradient corrected DFT energies at about the 0.6–0.9 eV level, and most find qualitatively different local... read less NOT USED (high confidence) H. Yan, X. Hu, and H. Jónsson, “Atomic structure of β-SiC( 100) surfaces: a study using the Tersoff potential,” Surface Science. 1994. link Times cited: 24 NOT USED (high confidence) O. H. H. Duparc and M. Torrent, “A new type of periodie boundary condition useful for high-temperature atomistic simulations of grain boundaries: Applications in semiconductors,” Interface Science. 1994. link Times cited: 21 NOT USED (high confidence) R. Ehlich, E. Campbell, O. Knospe, and R. Schmidt, “Collisional dynamics of C60 with noble-gas-atoms studied by molecular dynamics with empirical two- and three-body forces,” Zeitschrift für Physik D Atoms, Molecules and Clusters. 1993. link Times cited: 19 NOT USED (high confidence) J. Holender and G. J. Morgan, “Generation of a large structure (105 atoms) of amorphous Si using molecular dynamics,” Journal of Physics: Condensed Matter. 1991. link Times cited: 29 Abstract: A method for generating amorphous tetrahedral structures hav… read moreAbstract: A method for generating amorphous tetrahedral structures having 13824 and 110592 atoms is presented. The authors took the Wooten, Winer and Weaire amorphous model (1985) of 216 atoms and put together a number of these blocks. This larger structure was annealed using molecular dynamics and then cooled. Comparison with experiment was carried out using the structure factors calculated directly. Very good agreement has been attained. The generated structures contrary to the original www model, contain coordination defects. read less NOT USED (high confidence) P. Ballone and P. Milani, “Simulated annealing and collision properties of carbon clusters,” Zeitschrift für Physik D Atoms, Molecules and Clusters. 1991. link Times cited: 7 NOT USED (high confidence) K. Laasonen and R. Nieminen, “Molecular dynamics using the tight-binding approximation,” Journal of Physics: Condensed Matter. 1990. link Times cited: 46 Abstract: The authors present an extension of classical molecular dyna… read moreAbstract: The authors present an extension of classical molecular dynamics (MD) to include the forces calculated from electronic degrees of freedom using the tight-binding (TB) approximation. The combined MD-TB problem is solved using simulated annealing techniques. As an example they study the structures and energetics of small silicon clusters, containing up to 10 Si atoms. read less NOT USED (high confidence) A. Nakano, R. Kalia, and P. Vashishta, “Scalable molecular-dynamics, visualization, and data management algorithms for materials simulations,” Comput. Sci. Eng. 1989. link Times cited: 31 Abstract: Highly efficient algorithms for massively parallel computers… read moreAbstract: Highly efficient algorithms for massively parallel computers, interactive virtual environments for analyzing and steering simulations in real time, and data compression and mining schemes for input/output and knowledge discovery have led to rapid progress in large scale molecular dynamics simulations involving millions of atoms. Consequently, dynamic fracture of materials with realistic microstructures can now be modeled atom-by-atom. read less NOT USED (high confidence) Y. Yang et al., “Taking materials dynamics to new extremes using machine learning interatomic potentials,” Journal of Materials Informatics. 2021. link Times cited: 5 Abstract: Understanding materials dynamics under extreme conditions of… read moreAbstract: Understanding materials dynamics under extreme conditions of pressure, temperature, and strain rate is a scientific quest that spans nearly a century. Atomic simulations have had a considerable impact on this endeavor because of their ability to uncover materials’ microstructure evolution and properties at the scale of the relevant physical phenomena. However, this is still a challenge for most materials as it requires modeling large atomic systems (up to millions of particles) with improved accuracy. In many cases, the availability of sufficiently accurate but efficient interatomic potentials has become a serious bottleneck for performing these simulations as traditional potentials fail to represent the multitude of bonding. A new class of potentials has emerged recently, based on a different paradigm from the traditional approach. The new potentials are constructed by machinelearning with a high degree of fidelity from quantum-mechanical calculations. In this review, a brief introduction to the central ideas underlying machine learning interatomic potentials is given. In particular, the coupling of machine learning models with domain knowledge to improve accuracy, computational efficiency, and interpretability is highlighted. Subsequently, we demonstrate the effectiveness of the domain knowledge-based approach in certain select problems related to the kinetic response of warm dense materials. It is hoped that this review will inspire further advances in the understanding of matter under extreme conditions. read less NOT USED (high confidence) K. Töpfer, “Understanding the Interaction of CO and O2 with MgO(001) and Supported Metal Atoms: Towards Single-Atom Catalysis.” 2020. link Times cited: 0 Abstract: This thesis contributes to the fundamental understanding of … read moreAbstract: This thesis contributes to the fundamental understanding of the interactions of a single gold atom supported by a defective and defect-free MgO(001) surface in a mixed CO/O2 atmosphere. Using cluster models and point charge embedding within a density functional theory framework, the CO oxidation reaction for a single gold atom is simulated on differently charged oxygen vacancies of MgO(001) to rationalise its experimentally observed lack of catalytic activity. The results show, that only the F0 colour centre promotes the electron redistribution towards an adsorbed oxygen molecule and sufficiently weakens the oxygen bond, as required for a sustainable catalytic cycle. The moderate adsorption energy of the gold atom, however, cannot prevent the insertion of oxygen atoms into the vacancy, which remains after the formation of the first CO2 molecule. The surface becomes invariably repaired, which set the focus on the chemistry on a defect-free MgO(001) surface. To contribute towards the field of heterogeneous single-atom catalysis, various analysis tools are used to shed light on the binding situation of supported group 11 metal atoms to the defect-free substrate and both CO and O2 molecules. Cooperative effects are found to enhance the stability of CO upon co-adsorption with O2 for all three metal centres. The results gives further insights to the lack of catalytic activity with respect to the CO oxidation under thermal conditions as a competition between OC-O2 bond activation and surface diffusion leading to metal atom agglomeration. For the simulation of surface dynamics, an accurate description of the potential energy surface is achieved for CO on a defect-free MgO(001) surface by parametrizing a reactive bond order force field to a new set of ab initio data. Theoretical investigation of the non-reactive scattering of CO from the surface are done by performing quasi-classical scattering dynamic simulations. The scattering behaviour for several incidence energies and different initial ro-vibrational states of impinging CO is evaluated, which illustrates the role of surface atom motion on energy transfer processes. The analysis of time of flight spectra and scattering angle distributions reveals two different scattering channels, which become particularly noticeable at low incidence energies due to the weak interaction potential of CO with MgO(001). The scattering process is strongly influenced by the anisotropy of the potential energy surface for CO impinging in upwards and downwards alignment. Eventually, the observations are in agreement with the established Baule model especially for the distinct scattering features at low incident energies. read less NOT USED (high confidence) M. Rozhkov, N. Abramenko, A. Kolesnikova, and A. Romanov, “Zero misorientation interfaces in graphene,” Letters on Materials. 2020. link Times cited: 6 Abstract: This article presents the results on the modeling of straigh… read moreAbstract: This article presents the results on the modeling of straight-line interfaces that induce no misorientation of adjacent regions in graphene: zero misorientation interfaces (ZMIs). The interfaces in the hexagonal graphene lattice are represented as ensembles of disclinated carbon rings with broken rotational symmetry of the sixth order. The basic elements of such ensembles are structural units — complexes of disclinated rings with zero disclination charge. Using molecular dynamics simulation, the energies and atomic densities for ZMIs are found. Calculations demonstrate that atomic densities in ZMIs are lower than the atomic density in defect-free graphene. No direct correlation has been revealed between the atomic density and the interface energy. It is assumed, that the elastic field caused by ZMI defect structure contributes significantly to the energy of interface. Low-energy ZMIs possess linear energies not exceeding ~0.6 – 0.8 eV / Å, that is comparable to the energies of the grain boundaries, i. e. boundaries with misorientation, in graphene. Based on a mesoscopic approach operating with disclination schemes, in which defective carbon rings are replaced by disclinations, strain maps are plotted, and energies are found for two selected low-energy ZMIs. It is demonstrated that, at the distance of ZMI half-period from interface line, strains decrease to values of ~0.05. The energies of low-energy ZMIs calculated within the framework of two approaches: atomistic and mesoscopic, although differ numerically, coincide by the order of magnitude. read less NOT USED (high confidence) H. S. Jahromi and A. Setoodeh, “Longitudinal, Transverse, and Torsional Free Vibrational and Mechanical Behavior of Silicon Nanotubes Using an Atomistic Model,” Materials Research-ibero-american Journal of Materials. 2020. link Times cited: 13 NOT USED (high confidence) A. Sakti, Y. Nishimura, and H. Nakai, “Recent advances in quantum‐mechanical molecular dynamics simulations of proton transfer mechanism in various water‐based environments,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2019. link Times cited: 9 Abstract: Proton transfer in water‐based environments occurs because o… read moreAbstract: Proton transfer in water‐based environments occurs because of hydrogen‐bond interaction. There are many interesting physicochemical phenomena in this field, causing fast structural diffusion of hydronium and hydroxide ions. During the last few decades, to support experimental observations and measurements, quantum‐mechanical molecular dynamics (QMMD) simulations with reasonable accuracy and efficiency have significantly unraveled structural, energetic, and dynamical properties of excess proton in aqueous environments. This review summarizes the state‐of‐the‐art QMMD studies of proton transfer processes in aqueous solutions and complex systems including bulk liquid water, ice phases, and confined water in nanochannel/nanoporous materials as well as reports on CO2 scrubbing by amine‐based chemical absorption. read less NOT USED (high confidence) S. Sharma, P. Kumar, and R. Chandra, “Introduction to Molecular Dynamics,” Molecular Dynamics Simulation of Nanocomposites Using BIOVIA Materials Studio, Lammps and Gromacs. 2019. link Times cited: 22 NOT USED (high confidence) S. Meguid, A. R. Alian, and M. Dewapriya, “Atomistic Modelling of Nanoindentation of Multilayered Graphene-Reinforced Nanocomposites.” 2018. link Times cited: 10 NOT USED (high confidence) A. Kiselev, “Molecular dynamics simulations of laser ablation in covalent materials.” 2017. link Times cited: 1 Abstract: 15 Deutsche Zusammenfassung 18 I. Theoretical background 27… read moreAbstract: 15 Deutsche Zusammenfassung 18 I. Theoretical background 27 read less NOT USED (high confidence) L. Zhigilei, R. N. Salaway, B. K. Wittmaack, and A. Volkov, “Computational Studies of Thermal Transport Properties of Carbon Nanotube Materials.” 2017. link Times cited: 5 NOT USED (high confidence) V. A. Tupik, V. Margolin, and C. T. Su, “Computer simulation of obtaining thin films process technology using a new kind of pair interaction potential for atoms with covalent bond,” 2017 IEEE Conference of Russian Young Researchers in Electrical and Electronic Engineering (EIConRus). 2017. link Times cited: 1 Abstract: Today the computer simulation is one of powerful tool for sy… read moreAbstract: Today the computer simulation is one of powerful tool for synthetic and optimization of processes in the tasks of nanotechnology to get high quality of product. In spite of two big approaches of computer simulation the main part of their using in large range of tasks is a “top-down” approach nowadays. In this article we will show the description and apply of new one of pair interaction potential for atoms with covalent bond in simulation of obtaining thin films process technology. Based on the specific of atomic orbital for anisotropic bonding a new model of an atom and interatomic interaction of bonding were created. In this work the results of computer simulation were shown, as well as limitations of the new given algorithm and a discussion of it. read less NOT USED (high confidence) Y. Luo, H. Gong, N. Zhou, H. Huang, and L. Zhou, “Molecular dynamics study about the effect of substrate temperature on a-Si:H structure,” Applied Physics A. 2017. link Times cited: 1 NOT USED (high confidence) E. Neyts and P. Brault, “Molecular Dynamics Simulations for Plasma‐Surface Interactions,” Plasma Processes and Polymers. 2017. link Times cited: 51 Abstract: Plasma-surface interactions are in general highly complex du… read moreAbstract: Plasma-surface interactions are in general highly complex due to the interplay of many concurrent processes. Molecular dynamics simulations provide insight in some of these processes, subject to the accessible time and length scales, and the availability of suitable force fields. In this introductory tutorial-style review, we aim to describe the current capabilities and limitations of molecular dynamics simulations in this field, restricting ourselves to low-temperature non-thermal plasmas. Attention is paid to the simulation of the various fundamental processes occurring, including sputtering, etching, implantation, and deposition, as well as to what extent the basic plasma components can be accounted for, including ground state and excited species, electric fields, ions, photons, and electrons. A number of examples is provided, giving an bird's eye overview of the current state of the field. read less NOT USED (high confidence) T. Hynninen, L. Himanen, V. Parkkinen, T. Musso, J. Corander, and A. Foster, “An object oriented Python interface for atomistic simulations,” Comput. Phys. Commun. 2016. link Times cited: 2 NOT USED (high confidence) J. Moxnes, T. L. Jensen, and E. Unneberg, “Study of thermal instability of HMX crystalline polymorphs with and without molecular vacancies using reactive force field molecular dynamics,” Advanced Studies in Theoretical Physics. 2016. link Times cited: 3 Abstract: John F. Moxnes, Tomas L. Jensen, Erik Unneberg Study of ther… read moreAbstract: John F. Moxnes, Tomas L. Jensen, Erik Unneberg Study of thermal instability of HMX crystalline polymorphs with and without molecular vacancies using reactive force field molecular dynamics Advanced Studies in Theoretical Physics, Vol. 10, 2016, no. 7, 331-349 read less NOT USED (high confidence) A. Infuso, “Silicon photovoltaics: experimental testing and modelling of fracture across scales.” 2016. link Times cited: 0 Abstract: The study of the properties of materials can be addressed th… read moreAbstract: The study of the properties of materials can be addressed through a multi-scale approach, in order to have the possibility to grasp at each of the levels of analysis the peculiar aspects. Tracing a path inside the state-of-the-art in the available bibliography, historically in the field of mechanics s are found in which the material is studied through nonlocal theories based on continuous or discrete local approaches. More recently, with the advent of great computatio- nal power computers, analytical methodologies based on theories also very complex deriving from the field of chemistry and physics have been developed, capable to discretize at the ato- mic scale the material and study its behavior by applying energy approaches. Starting from the analysis of some of these theories at the nano- and micro-scales, it is possible to investi- gate the separation mechanisms at the molecular level, which may be considered as cracking processes within the material according to the adopted scale of analysis. The application of theories of this kind to large portions of material, in which there are up to some millions of particles involved is reasonably not an applicable solution, since it would require a huge effort in terms of computation time. To work around this problem and find a method suitable for the study of cracking mechanisms, a mixed method (MDFEM) was byconjugating pure molecu- lar dynamics (MD) and the finite element method (FEM), in which the material is discretized by means of one-dimensional elements whose mechanical characteristics are derived from MD. This approach is based on the application of a nonlocal theory in which the contribution of a portion of material placed within a certain distance from the point of fracture is taken into account by means of a parameter of non-locality. Moreover, the study of the evolution of cracking is addressed at the meso-scale by the application of a cohesive non-linear model. Towards the analysis of the macroscale, the theories put forward so far have been ap- plied to the study of phenomena of breakage inside Silicon cells embedded into rigid or semi-flexible photovoltaic modules. By performing various laboratory tests, useful for the characterization of the material and for understating the evolution of cracking process due to multiple causes, a study on the main issues that may compromise the durability and mainte- nance of the expected service levels of photovoltaic panels has been conducted. Experimen- tally results have been interpreted by using appropriate macro-scopic continuum models. The research carried out had the purpose to provide an introduction to a correct design of these systems of energy production in order to increase their durability and resistance to cracking read less NOT USED (high confidence) S. Lai, I. Setiyawati, T. Yen, and Y. H. Tang, “Studying lowest energy structures of carbon clusters by bond-order empirical potentials,” Theoretical Chemistry Accounts. 2016. link Times cited: 10 NOT USED (high confidence) T. Oda, W. J. Weber, and H. Tanigawa, “Two-body potential model based on cosine series expansion for ionic materials,” Computational Materials Science. 2016. link Times cited: 1 NOT USED (high confidence) T. Asche, M. Duderstaedt, P. Behrens, and A. Schneider, “Atomistic Simulation of Sol–Gel-Derived Hybrid Materials.” 2016. link Times cited: 1 NOT USED (high confidence) X. Zhao, “Accelerating materials discovery and design: computational study of the structure and properties of materials.” 2015. link Times cited: 0 Abstract: This thesis summarizes our efforts to study the structure an… read moreAbstract: This thesis summarizes our efforts to study the structure and properties of materials computationally. The adaptive genetic algorithm (AGA) developed by us to predict crystal/surface/interface structures is presented. Applications of AGA to a variety of systems, such as non-rare earth magnetic materials, ultra-hard transition metal borides and SrTiO3 grain boundaries, are discussed. We demonstrated by AGA the capability of solving crystal structures with more than 100 atoms per unit cell and rapidly accessing the structures and phase stabilities of different compositions in multicomponent systems. We also introduced a motif-network scheme to study the complex crystal structures in silicate cathodes. In addition, we explored different computational methods for atomistic simulations of materials behavior, such as Monte Carlo modeling of the alnico magnets. read less NOT USED (high confidence) R. Alsayegh, “Vision-augmented molecular dynamics simulation of nanoindentation,” Journal of Nanomaterials. 2015. link Times cited: 7 Abstract: We present a user-friendly vision-augmented technique to car… read moreAbstract: We present a user-friendly vision-augmented technique to carry out atomic simulation using hand gestures. The system is novel in its concept as it enables the user to directly manipulate the atomic structures on the screen, in 3D space using hand gestures, allowing the exploration and visualisation of molecular interactions at different relative conformations. The hand gestures are used to pick and place atoms on the screen allowing thereby the ease of carrying out molecular dynamics simulation in a more efficient way. The end result is that users with limited expertise in developing molecular structures can now do so easily and intuitively by the use of body gestures to interact with the simulator to study the system in question. The proposed system was tested by simulating the crystal anisotropy of crystalline silicon during nanoindentation. A long-range (Screened bond order) Tersoff potential energy function was used during the simulation which revealed the value of hardness and elastic modulus being similar to what has been found previously from the experiments. We anticipate that our proposed system will open up new horizons to the current methods on how an MD simulation is designed and executed. read less NOT USED (high confidence) A. McGaughey and J. Larkin, “PREDICTING PHONON PROPERTIES FROM EQUILIBRIUM MOLECULAR DYNAMICS SIMULATIONS,” Annual Review of Heat Transfer. 2014. link Times cited: 84 Abstract: The objective of this chapter is to describe how equilibrium… read moreAbstract: The objective of this chapter is to describe how equilibrium molecular dynamics simulations (with the help of harmonic lattice dynamics calculations) can be used to predict phonon properties and thermal conductivity using normal mode decomposition. The molecular dynamics and lattice dynamics methods are reviewed and the normal mode decomposition technique is described in detail. The application of normal mode decomposition is demonstrated through case studies on crystalline, alloy, and amorphous Lennard-Jones phases. Notable works that used normal mode decomposition are presented and the future of molecular dynamics simulations in phonon transport modeling is discussed. read less NOT USED (high confidence) P. Seleson, M. Parks, and M. Gunzburger, “Peridynamic State-Based Models and the Embedded-Atom Model,” Communications in Computational Physics. 2014. link Times cited: 27 Abstract: We investigate connections betweennonlocal continuum models … read moreAbstract: We investigate connections betweennonlocal continuum models andmolec- ular dynamics. A continuous upscaling of molecular dynamics models of the form of the embedded-atom model is presented, providing means for simulating molecular dynamics systems at greatly reduced cost. Results are presented for structured and structureless material models, supported by computational experiments. The nonlocal continuum models are shown to be instances of the state-based peridynamics theory. Connections relating multibody peridynamic models and upscaled nonlocal contin- uum models are derived. read less NOT USED (high confidence) J. Yeo, “Modeling and simulation of the structural evolution and thermal properties of ultralight aerogel and 2D materials.” 2014. link Times cited: 1 NOT USED (high confidence) C. Jiang, D. Morgan, and I. Szlufarska, “Structures and stabilities of small carbon interstitial clusters in cubic silicon carbide,” Acta Materialia. 2014. link Times cited: 19 NOT USED (high confidence) C. Davini, “Homogenization of a graphene sheet,” Continuum Mechanics and Thermodynamics. 2014. link Times cited: 23 NOT USED (high confidence) D. Schopf, “Effective potentials for numerical investigations of complex intermetallic phases.” 2013. link Times cited: 0 Abstract: The class of Complex Metallic Alloys (CMAs) is interesting f… read moreAbstract: The class of Complex Metallic Alloys (CMAs) is interesting for its wide range of physical properties. There are materials that exhibit high hardness at low density or good corrosion resistance, which is important for technological applications. Other compounds are superconductors, have strong anisotropic transport coefficients or exhibit a novel magnetic memory effect. The theoretical investigation of CMAs is often very challenging because of their inherent complexity and large unit cells with up to several thousand atoms. Molecular dynamics simulations with classical interaction potentials are well suited for this task – they can handle hundreds of thousands of atoms in reasonable time. Such simulations can provide insight into static and dynamic processes at finite temperatures on an atomistic level.
The accuracy of these simulations depends strongly on the quality of the employed interaction potentials. To generate physically relevant potentials the force-matching method can be applied. A computer code called potfit has been developed at the Institute for Theoretical and Applied Physics (ITAP) especially for this task. It uses a large database of quantum-mechanically calculated reference data, forces on individual atoms and cohesive energies, to generate effective potentials. The parameters of the potential are optimized in such a way that the reference data are reproduced as accurately as possible.
The potfit program has been greatly enhanced as part of this thesis. The optimization of analytic potentials, new interaction models as well as a new optimization algorithm were implemented. Potentials for two different complex metallic alloy systems have been generated and used to study their properties with molecular dynamics simulations.
The first system is an approximant to the decagonal Al-Pd-Mn quasicrystal. A potential which can reproduce the cohesive energy with high accuracy was generated. With the help of this potential a refinement of the experimentally poorly determined structure model could be performed.
The second class of potentials was fitted for intermetallic clathrate systems. They have interesting thermoelectric properties which originate from their special structure. Silicon- and germanium-based clathrate potentials were derived and the influence of the complex structure on the thermal conductivity has been studied.
Komplexe Intermetallische Verbindungen (CMAs) sind aufgrund ihrer vielfaltigen physikalischen Eigenschaften sehr interessant fur technologische Anwendungen. Dabei ist z.B. hohe Harte bei geringer Dichte und Korrosionsbestandigkeit wichtig. Neben Supraleitern gibt es Materialien mit anisotropen Transporteigenschaften oder einem neuartigen magnetischen Memory Effekt. Theoretische Untersuchungen von CMAs stellen durch ihre inharente Komplexitat und die riesigen Einheitszellen mit mehreren tausend Atomen oft eine grose Herausforderung dar. Molekulardynamiksimulationen mit effektiven Potenzialen konnen dazu eingesetzt werden; sie ermoglichen die Berechnung von hunderttausenden von Atome in annehmbarer Zeit. Damit kann ein Einblick in sowohl statische als auch dynamische Prozesse auf atomarer Ebene gewonnen werden.
Die Ergebenisse solcher Simulationen hangen jedoch sehr stark von der Qualitat der eingesetzten Wechselwirkung ab. Um physikalisch gerechtfertigte Potenziale zu erzeugen, kann die Force-Matching-Methode angewandt werden. Dazu wurde am Institut fur Theoretische und Angewandte Physik (ITAP) ein Programm mit dem Namen potfit entwickelt. Es verwendet eine grose Datenbank von quantenmechanisch berechneten Referenzgrosen wie z.B. Krafte auf die einzelnen Atome und die Kohasionsenergie, um effektive Potenziale zu generieren. Die freien Parameter des Potenzials werden optimiert, um die Referenzdaten so gut wie moglich zu reproduzieren.
Fur diese Arbeit wurde potfit deutlich erweitert. Es konnen nun analytisch definierte Potenziale optimiert werden, neue Wechselwirkungen wurden implementiert und ein neuer Optimierungsalgorithmus wurde hinzugefugt. Damit wurden effektive Potenziale fur zwei verschiedene CMA Systeme gefittet und deren Eigenschaften mit Molekulardynamik untersucht.
Fur die Approximanten eines decagonalen Al-Pd-Mn Quasikristalls, den Xi-Phasen, wurde ein Potenzial fur die Strukturbestimmung erzeugt. Es kann die Kohasionsenergien verschiedener Strukturen mit groser Genauigkeit wiedergeben. Ein aus experimentellen Daten ungenau bestimmtes Strukturmodell konnte damit erheblich verbessert werden.
Auserdem wurden Potenziale fur Intermetallische Klathrate erzeugt. Diese Systeme besitzen interessante thermoelektrische Eigenschaften aufgrund ihrer speziellen Kafigstruktur. Effektive Wechselwirkungen fur silizium- und germaniumbasierte Klathrate wurden erzeugt. Damit wurde der Einfluss der komplexen Struktur auf die thermische Leitfahigkeit des Gitters untersucht. read less NOT USED (high confidence) E. Sandoz-Rosado, “The tribological behavior of graphene and its role as a protective coating.” 2013. link Times cited: 5 Abstract: The tribological behavior of graphene and its role as a prot… read moreAbstract: The tribological behavior of graphene and its role as a protective coating Emil Jose Sandoz-Rosado The scope of this thesis is to explore the fundamental tribological behavior of graphene as a two-dimensional (2-D) nanomaterial and evaluate its performance as a protective coating. Graphene is the strongest material ever measured, gas-impermeable, chemically and thermally stable, and atomically-thin, making it an excellent candidate as a protective coating. The fundamental tribological behavior of graphene and other 2-D materials under sliding conditions has only just begun to be explored. In particular, the wear of graphene has hardly been explored. The objective of this work is to investigate the tribological behavior of graphene through atomistic simulation as well as experimental testing under various sliding regimes and length scales. Wear in a graphene monolayer, after scratch tests with a nanoindenter, was characterized for the first time using Raman spectroscopy, revealing new insights into the failure of graphene after sliding. These sliding tests revealed a new frictional phenomenon where friction increased linearly with sliding length over large distances. This was caused by delamination likely due to the coalescence of small bubbles of gas trapped between the graphene monolayer and substrate during sliding, confirmed with atomic force microscopy. Furthermore, atomistic simulations of an asperity sliding over a graphene bubble mimicked experimental results, further supporting this bubble coalescence hypothesis. Graphene’s potential as an anti-corrosive coating was demonstrated for macro-scale, commercially-available electrical connectors. It was demonstrated that even a monolayer of graphene can prevent oxide and reduce electrical contact resistance by orders of magnitude. read less NOT USED (high confidence) A. Shakouri, “Study of scale effects on the vibration of graphene sheets with applications to NEMS.” 2012. link Times cited: 0 NOT USED (high confidence) J. Elliott, S. Hida, T. Shiga, S. Maruyama, and J. Shiomi, “Influence of Thermal Boundary Resistance and Interfacial Phonon Scattering on Heat Conduction of Carbon Nanotube/Polymer Composites,” Transactions of the Japan Society of Mechanical Engineers. B. 2012. link Times cited: 1 Abstract: Polymer nanocomposites are one of the most promising applica… read moreAbstract: Polymer nanocomposites are one of the most promising applications of carbon nanotubes (CNTs). In order to improve their thermal properties, it is important to understand their heat conduction characteristics from a microscopic viewpoint. In this study, we have investigated the heat conduction of CNT/polyethylene composites by using molecular dynamics simulations. We particularly focused on the thermal boundary conductance across CNT/polyethylene interfaces and thermal conductivity of CNT in polyethylene matrix, which govern the overall thermal conductivity of CNT/polyethylene composites. We found relatively low thermal boundary conductance across CNT/polyethylene interfaces (»10 MWm i2 K i1 ) and a moderate but non-negligible thermal conductivity reduction of CNT in polyethylene matrix (22 %). By the mode-dependent phonon transport analysis, the thermal conductivity reduction was identified to be mainly due to scattering of low frequency CNT phonons by polyethylene. The results obtained through an effective medium approximation model give overall thermal conductivity of the CNT/polyethylene composite that is in agreement with experiments. read less NOT USED (high confidence) E. S. Flores, S. Adhikari, M. Friswell, and F. Scarpa, “Hyperelastic finite element model for single wall carbon nanotubes in tension,” Computational Materials Science. 2011. link Times cited: 19 NOT USED (high confidence) M. Hamdi and A. Ferreira, “Methodology of Design and Characterization of Bionano- and Nanorobotic Devices.” 2011. link Times cited: 1 NOT USED (high confidence) J. Ding, B. Kan, G. Cheng, Z. Fan, N. Yuan, and Z. Ling, “Numerical Approach to Torsion Deformation of Armchair Single Walled Carbon Nanotubes,” International Journal of Nonlinear Sciences and Numerical Simulation. 2008. link Times cited: 7 Abstract: In this paper, a new atomic-scale finite element method (AFE… read moreAbstract: In this paper, a new atomic-scale finite element method (AFEM) based on the nonlinear spring model is used to investigate the torsional properties of armchair single walled carbon nanotubes (SWNTs). A simple iteration algorithm is applied to get the solution of the equilibrium function. The torsion deformations of 12 armchair SWNTs with different diameters and lengths are simulated. The development of buckling deformation is observed and discussed. Critical torsion angle of each nanotube is obtained. It is found the shear moduli of the SWNTs range from 391 GPa to 592 GPa, and it is closely related with the diameter of nanotube. However, the nanotube length has little effect on the shear modulus. In addition, it is observed the buckling morphology varies as the parameter of nanotube changes. read less NOT USED (high confidence) A. Al-ostaz, G. Pal, P. Mantena, and A. Cheng, “Molecular dynamics simulation of SWCNT–polymer nanocomposite and its constituents,” Journal of Materials Science. 2008. link Times cited: 87 NOT USED (high confidence) P. Valavala and G. Odegard, “Multiscale Constitutive Modeling of Polymer Materials.” 2007. link Times cited: 8 Abstract: A nonlinear multiscale constitutive modeling framework is us… read moreAbstract: A nonlinear multiscale constitutive modeling framework is used to predict the elastic modulus of a glassy polymer. A set of seven molecular models with different molecular configurations is utilized to study the effect of the minimized potential energy level on the predicted elastic moduli. It is found that the average modulus from the seven models considered in the current study is seen to be in reasonable agreement with the experimentally observed value, given the statistical uncertainly in the data.Copyright © 2007 by ASME read less NOT USED (high confidence) U. Zimmerli, “Computational nanofluidics for the study of biomolecular flows.” 2006. link Times cited: 2 Abstract: Nanotechnology and biology are two areas of scientific resea… read moreAbstract: Nanotechnology and biology are two areas of scientific resea rch where recent progress has disclosed a variety of new perspectives. The ad vances in both fields prepared the grounds for interdisciplinary studies and rec ent findings promise novel applications that could lead to a technological revol uti n in medicine. In particular the advent of carbon nanotubes (CNTs) gave rise t o peculations on future applications. CNTs are tubular carbon molecules which can be imagined to function as nanometer size pipes and pores. In this respect, th y could mimic transmembrane pores and channels and they could be used to manipul ate the transport across the membranes of cells and cellular organelles. Spec ifically, CNT based nanopores are envisioned to transport biomolecules across ellular membranes for biomedical purposes. In this thesis we assess the viability of transmembrane RNA transport through CNT nanopores. Life on earth has evolved in an environment that is character ized by the presence of water. Consequently, biological systems such as cells an d their cytoplasm consist, to a large percentage, of water. The interaction of tra nsmembrane pores with water will be highly influential to their transport properti s. To assess the viability of transmembrane pores based on CNTs, it is therefore mandat ory to understand their interaction with aqueous solutions. Throughout the first part of this thesis we study the water car bon interaction in detail. In an initial study we assess the interaction betwee n water and benzene and between water and naphthalene using a quantum mechanica l pproach. We compare and evaluate different correction schemes for dens ity functionals and we recommend a correction scheme to be used in future studies. Due to the limitations of quantum mechanical methods to smal l systems we then focus on molecular dynamics simulations. After an introduc tion to molecular dynamics we present a methodological advancement to steered m ol cular dynamics. In molecular dynamics simulations the interactions betwee n particles are modeled using empirical interaction potentials. This reduces the c omputational cost significantly and allows to simulate systems with several ten-t housand atoms. Due to the empirical nature of the interaction potentials, the m odel assumptions and simplifications need to be validated carefully to ensure the ir legitimacy. read less NOT USED (high confidence) J. Murthy, S. Narumanchi, J. A. Pascual-Gutiérrez, T. Wang, C. Ni, and S. Mathur, “Review of Multiscale Simulation in Submicron Heat Transfer,” International Journal for Multiscale Computational Engineering. 2005. link Times cited: 76 Abstract: Over thelastdecade, interestin thesimulationof microandnano-… read moreAbstract: Over thelastdecade, interestin thesimulationof microandnano-scale heattransferhasleadto thedevelopmentof a varietyof modelsandnumerical methods for phonon transpor t in semiconductorsanddielectrics.These models spandirect simulationusingmoleculardynamics,a rangeof modelsof varying fidelity basedon theBoltzmanntranspor t equation, aswell as simplerhyperbolic extensionsto theclassicalFourier heatconduction equation. Thepaperreviews thebasicsof phonontransport in crystals,available models for phonon transpor t, aswell asnumerical methods for solving the equationsresultingfrom thesemodels. Recommend ationsaremadefor futurework. read less NOT USED (high confidence) M. Prasad and T. Sinno, “Feature Activated Molecular Dynamics: Parallelization and Application to Systems with Globally Varying Mechanical Fields,” Journal of Computer-Aided Materials Design. 2005. link Times cited: 1 NOT USED (high confidence) J. Chang, C. Hwang, S. Ju, and S. Huang, “A molecular dynamics simulation investigation into the structure of fullerene C60 grown on a diamond substrate,” Carbon. 2004. link Times cited: 11 NOT USED (high confidence) J. Kang, K. Byun, and H. Hwang, “Twist of hypothetical silicon nanotubes,” Modelling and Simulation in Materials Science and Engineering. 2004. link Times cited: 26 Abstract: The responses of hypothetical silicon nanotubes (SiNTs) unde… read moreAbstract: The responses of hypothetical silicon nanotubes (SiNTs) under torsion have been investigated using an atomistic simulation based on the Tersoff potential. A torque, proportional to the deformation within Hooke's law, resulted in ribbon-like flattened shapes and eventually led to breaking of the hypothetical SiNTs. Each shape change of the hypothetical SiNTs corresponded to an abrupt energy change and a singularity in the strain energy curve as a function of the external tangential force, torque, or twist angle. The dynamics of SiNTs under torsion can be modelled in continuum elasticity theory. read less NOT USED (high confidence) L. Porter, S. Yip, M. Yamaguchi, H. Kaburaki, and M. Tang, “EMPIRICAL BOND-ORDER POTENTIAL DESCRIPTION OF THERMODYNAMIC PROPERTIES OF CRYSTALLINE SILICON,” Journal of Applied Physics. 1997. link Times cited: 67 Abstract: Thermodynamic properties of silicon (diamond cubic phase) ar… read moreAbstract: Thermodynamic properties of silicon (diamond cubic phase) are calculated using an empirical many-body potential developed by Tersoff [Phys. Rev. Lett. 56, 632 (1986)] based on the concept of bond order. It is shown that this model gives predictions in good agreement with experiment for those properties governed by energetics (free energy, entropy, and heat capacity). The thermal expansion coefficient is less well described, which is traced to the fact that the model potential, in its present version, is overly stiff and therefore unable to account properly for the volume dependence of the transverse acoustic modes. Furthermore, sensitivity of the potential to whether each atom remains bonded to only four neighbors indicates that the short-range nature of the potential may necessitate model improvement before it is suitable for studies of thermomechanical properties at elevated temperatures or large deformations. read less NOT USED (high confidence) A. Omeltchenko, “Nanoscale Structures and Fracture Processes in Advanced Ceramics: Million-Atom MD Simulations on Parallel Architectures.” 1997. link Times cited: 0 Abstract: Properties and processes in silicon nitride and graphite are… read moreAbstract: Properties and processes in silicon nitride and graphite are investigated using molecular-dynamics (MD) simulations. Scalable and portable multiresolution algorithms are developed and implemented on parallel architectures to simulate systems containing 106 atoms interacting via realistic potentials. Structural correlations, mechanical properties, and thermal transport are studied in microporous silicon nitride as a function of density. The formation of pores is observed when the density is reduced to 2.6 g/cc, and the percolation occurs at a density of 2.0 g/cc. The density variation of the thermal conductivity and the Young’s modulus are well described by power laws with scaling exponents of 1.5 and 3.6, respectively. Dynamic fracture in a single graphite sheet is investigated. For certain crystalline orientations, the crack becomes unstable with respect to branching at a critical speed of -60% of the Rayleigh velocity. The origin of the branching instability is investigated by calculating local-stress distributions. The branched fracture profile is characterized by a roughness exponent, a 0.7, above a crossover length of 50A. For smaller length scales and within the same branch, a 0.4. Crack propagation is studied in nanophase silicon nitride prepared by sintering nanoclusters of size 60A. The system consists of crystalline cluster interiors, amorphous intercluster regions, and isolated pores. These microstructures cause crack branching and meandering, and the clusters undergo significant rearrangement due to plastic deformation of interfacial regions. As a result, the system can withstand enormous deformation (30%). In contrast, a crystalline sample in the same geometry cleaves under an applied strain of only 3%. read less NOT USED (high confidence) M. Gupta, E. A. Walters, and N. Blais, “Simulation of chemical reaction initiation through high velocity collisions of NO clusters with a surface,” Journal of Chemical Physics. 1996. link Times cited: 18 Abstract: Some computational results have been obtained for a system o… read moreAbstract: Some computational results have been obtained for a system of diatomic molecules clustered together and driven to impact on a surface at sufficient energy to induce an observable quantity of chemical reactions. The diatomic molecules were modeled to be energetically similar to nitric oxide, NO, which is a detonable material when in the condensed phase. The system was intended to simulate an experiment devised to examine the initiation phase of a detonation of liquid NO stimulated by impact with a high‐speed flyer plate. Classical trajectories were computed for six different cluster sizes, from 4 molecules to 50, and the clusters were directed into a wall at five different impact speeds ranging from 3.0 to 11.8 km s−1. The interatomic forces used for the computations were based on a modification of an empirical potential suggested by Tersoff. The characteristics of the products (O2, N2, NO, and N and O atoms) are examined, as well as the dynamic features of the collisions of the clusters with the wall. The... read less NOT USED (definite) S. Yesudasan, “Extended MARTINI water model for heat transfer studies,” Molecular Physics. 2019. link Times cited: 9 Abstract: The computationally efficient classical MARTINI model is ext… read moreAbstract: The computationally efficient classical MARTINI model is extended to simulate heat transfer simulations of water. The current MARTINI model, variations of it and other coarse grain water models focus on reproducing the thermodynamic properties below or at room temperature, hence making them unsuitable for studying high temperature simulations especially evaporation at . In this work, the MARTINI model is reparametrised using a combination of Genetic Algorithm, Artificial Neural Network and Nelder–Mead optimisation technique to match the phase equilibrium properties of water. The reparametrised model (MARTINI-E) accurately reproduces density, enthalpy of vaporisation and surface tension at and outperforms other leading coarse grain water models. The model is also validated using the energy conservation and enthalpy change due to latent heat in a lamellar system. This new water model can be used for simulating phase change phenomena, thin film evaporation and other energy transport mechanisms accurately. GRAPHICAL ABSTRACT read less NOT USED (definite) A. Hernandez, A. Balasubramanian, F. Yuan, S. Mason, and T. Mueller, “Fast, accurate, and transferable many-body interatomic potentials by symbolic regression,” npj Computational Materials. 2019. link Times cited: 51 NOT USED (definite) A. C. Hansen-Dorr, L. Wilkens, A. Croy, A. Dianat, G. Cuniberti, and M. Kastner, “Combined molecular dynamics and phase-field modelling of crack propagation in defective graphene,” Computational Materials Science. 2019. link Times cited: 14 NOT USED (definite) S. I. Vishkayi and M. B. Tagani, “Tuning the thermoelectric efficiency of a polyaniline sheet using strain engineering,” Journal of Physics D: Applied Physics. 2019. link Times cited: 7 Abstract: Two-dimensional polyaniline monolayers (C3N) have been recen… read moreAbstract: Two-dimensional polyaniline monolayers (C3N) have been recently synthesized as indirect semiconductors with high electron mobility. We investigate the electrical and thermal properties of a C3N sheet using a combination of density functional theory and the Green function formalism. Tensile strain along a zigzag direction can drive the transition from an indirect to a direct semiconductor, whereas the sheet transitions from a semiconductor to a metal under compressive strain. The thermoelectric efficiency of an unstrained C3N sheet is higher in p-doping, and its maximum value is obtained when the transport is along the zigzag direction. A reduction in the figure of merit is found upon applying strain, independent of its direction. To overcome this reduction, we show that when the electrical transport and strain are perpendicular to each other, the thermoelectric efficiency of the C3N sheet can be significantly increased, depending on the type of strain (tensile or compression). The results support the potential application of C3N sheets in the thermoelectrics and optoelectronics industries through using strain engineering. read less NOT USED (definite) H. Shabbir, C. Dellago, and M. Hartmann, “A High Coordination of Cross-Links Is Beneficial for the Strength of Cross-Linked Fibers,” Biomimetics. 2019. link Times cited: 13 Abstract: The influence of the coordination of (reversible) cross-link… read moreAbstract: The influence of the coordination of (reversible) cross-links on the mechanical properties of aligned fiber bundles is investigated. Two polymeric systems containing cross-links of different coordination (two- and three-fold coordination) but having the same binding energy are investigated. In particular, the response to loading of these systems is compared. Mechanical parameters (strength, stiffness and work-to-fracture) are obtained by computational loading tests. The influence of coordination is studied for simple test systems with pre-defined topologies that maximize strength as well as for more realistic fiber bundles containing nine chains. The results show that a higher coordination of cross-links has a beneficial effect on the strength and the stiffness of the systems, while the work-to-fracture was found larger for the system having a smaller coordination of cross-links. It can be concluded that controlling the coordination of cross-links is a versatile tool to specifically tailor the mechanical properties of polymeric structures. read less NOT USED (definite) G. P. P. Pun, R. Batra, R. Ramprasad, and Y. Mishin, “Physically informed artificial neural networks for atomistic modeling of materials,” Nature Communications. 2018. link Times cited: 188 NOT USED (definite) V. Vijayaraghavan and L. Zhang, “Effective Mechanical Properties and Thickness Determination of Boron Nitride Nanosheets Using Molecular Dynamics Simulation,” Nanomaterials. 2018. link Times cited: 37 Abstract: Research in boron nitride nanosheets (BNNS) has evoked signi… read moreAbstract: Research in boron nitride nanosheets (BNNS) has evoked significant interest in the field of nano-electronics, nanoelectromechanical (NEMS) devices, and nanocomposites due to its excellent physical and chemical properties. Despite this, there has been no reliable data on the effective mechanical properties of BNNS, with the literature reporting a wide scatter of strength data for the same material. To address this challenge, this article presents a comprehensive analysis on the effect of vital factors which can result in variations of the effective mechanical properties of BNNS. Additionally, the article also presents the computation of the correct wall thickness of BNNS from elastic theory equations, which is an important descriptor for any research to determine the mechanical properties of BNNS. It was predicted that the correct thickness of BNNS should be 0.106 nm and the effective Young’s modulus to be 2.75 TPa. It is anticipated that the findings from this study could provide valuable insights on the true mechanical properties of BNNS that could assist in the design and development of efficient BN-based NEMS devices, nanosensors, and nanocomposites. read less NOT USED (definite) M. Friedrich and U. Stefanelli, “Graphene ground states,” Zeitschrift für angewandte Mathematik und Physik. 2018. link Times cited: 0 NOT USED (definite) A. Dmitriev, A. Nikonov, and W. Österle, “Molecular Dynamics Modeling of the Sliding Performance of an Amorphous Silica Nano-Layer—The Impact of Chosen Interatomic Potentials,” Lubricants. 2018. link Times cited: 7 Abstract: The sliding behavior of an amorphous silica sample between t… read moreAbstract: The sliding behavior of an amorphous silica sample between two rigid surfaces is in the focus of the present paper. Molecular Dynamics using a classical Tersoff’s potential and a recently developed ReaxFF potential was applied for simulating sliding within a thin film corresponding to a tribofilm formed from silica nanoparticles. The simulations were performed at different temperatures corresponding to moderate and severe tribological stressing conditions. Simulations with both potentials revealed the need of considering different temperatures in order to obtain a sound interpretation of experimental findings. The results show the striking differences between the two potentials not only in terms of magnitude of the resistance stress (about one order of magnitude) but also in terms of friction mechanisms. The expected smooth sliding regime under high temperature conditions was predicted by both simulations, although with Tersoff’s potential smooth sliding was obtained only at the highest temperature. On the other hand, at room temperature Tersoff-style calculations demonstrate stick-slip behavior, which corresponds qualitatively with our experimental findings. Nevertheless, comparison with a macroscopic coefficient of friction is not possible because simulated resistance stresses do not depend on the applied normal pressure. read less NOT USED (definite) M. Stockett, M. Wolf, M. Gatchell, H. Schmidt, H. Zettergren, and H. Cederquist, “The threshold displacement energy of buckminsterfullerene C60 and formation of the endohedral defect fullerene He@C59,” Carbon. 2018. link Times cited: 4 NOT USED (definite) R. B. Hudson and A. Sinha, “Vibration of carbon nanotubes with defects: order reduction methods,” Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2018. link Times cited: 3 Abstract: Order reduction methods are widely used to reduce computatio… read moreAbstract: Order reduction methods are widely used to reduce computational effort when calculating the impact of defects on the vibrational properties of nearly periodic structures in engineering applications, such as a gas-turbine bladed disc. However, despite obvious similarities these techniques have not yet been adapted for use in analysing atomic structures with inevitable defects. Two order reduction techniques, modal domain analysis and modified modal domain analysis, are successfully used in this paper to examine the changes in vibrational frequencies, mode shapes and mode localization caused by defects in carbon nanotubes. The defects considered are isotope defects and Stone–Wales defects, though the methods described can be extended to other defects. read less NOT USED (definite) M. Friedrich and U. Stefanelli, “Ripples in Graphene: A Variational Approach,” Communications in Mathematical Physics. 2018. link Times cited: 5 NOT USED (definite) T. Jacobs and A. Martini, “Measuring and Understanding Contact Area at the Nanoscale: A Review,” Applied Mechanics Reviews. 2017. link Times cited: 80 Abstract: The size of the mechanical contact between nanoscale bodies … read moreAbstract: The size of the mechanical contact between nanoscale bodies that are pressed together under load has implications for adhesion, friction, and electrical and thermal transport at small scales. Yet, because the contact is buried between the two bodies, it is challenging to accurately measure the true contact area and to understand its dependence on load and material properties. Recent advancements in both experimental techniques and simulation methodologies have provided unprecedented insights into nanoscale contacts. This review provides a detailed look at the current understanding of nanocontacts. Experimental methods for determining contact area are discussed, including direct measurements using in situ electron microscopy, as well as indirect methods based on measurements of contact resistance, contact stiffness, lateral forces, and topography. Simulation techniques are also discussed, including the types of nanocontact modeling that has been performed and the various methods for extracting the magnitude of the contact area from a simulation. To describe and predict contact area, three different theories of nanoscale contact are reviewed: single-contact continuum mechanics; multi-contact continuum mechanics; and atomistic accounting. Representative results from nanoscale experimental and simulation investigations are presented in the context of these theories. Finally, the critical challenges are described, as well as the opportunities on the path to establishing a fundamental and actionable understanding of what it means to be “in contact” at the nanoscale. read less NOT USED (definite) C. Ruestes, I. A. Alhafez, and H. Urbassek, “Atomistic Studies of Nanoindentation—A Review of Recent Advances.” 2017. link Times cited: 45 Abstract: This review covers areas where our understanding of the mech… read moreAbstract: This review covers areas where our understanding of the mechanisms underlying nanoindentation has been increased by atomistic studies of the nanoindentation process. While such studies have been performed now for more than 20 years, recent investigations have demonstrated that the peculiar features of nanoplasticity generated during indentation can be analyzed in considerable detail by this technique. Topics covered include: nucleation of dislocations in ideal crystals, effect of surface orientation, effect of crystallography (fcc, bcc, hcp), effect of surface and bulk damage on plasticity, nanocrystalline samples, and multiple (sequential) indentation. In addition we discuss related features, such as the influence of tip geometry on the indentation and the role of adhesive forces, and how pre-existing plasticity affects nanoindentation. read less NOT USED (definite) R. J. Wang, C. Wang, and Y. Feng, “Effective geometric size and bond-loss effect in nanoelasticity of GaN nanowires,” International Journal of Mechanical Sciences. 2017. link Times cited: 7 NOT USED (definite) D. Damasceno, E. Mesquita, and R. Rajapakse, “Mechanical Behavior of Nano Structures Using Atomic-Scale Finite Element Method (AFEM),” Latin American Journal of Solids and Structures. 2017. link Times cited: 5 Abstract: THIS WORK PRESENTS A DETAILED DESCRIPTION OF THE FORMULATION… read moreAbstract: THIS WORK PRESENTS A DETAILED DESCRIPTION OF THE FORMULATION AND IM-PLEMENTATION OF THE ATOMISTIC FINITE ELEMENT METHOD AFEM, EXEMPLI-FIED IN THE ANALYSIS OF ONE- AND TWO-DIMENSIONAL ATOMIC DOMAINS GOV-ERNED BY THE LENNARD JONES INTERATOMIC POTENTIAL. THE METHODOLOGY TO SYNTHESIZE ELEMENT STIFFNESS MATRICES AND LOAD VECTORS, THE POTENTIAL ENERGY MODIFICATION OF THE ATOMISTIC FINITE ELEMENTS (AFE) TO ACCOUNT FOR BOUNDARY EDGE EFFECTS, THE INCLUSION OF BOUNDARY CONDITIONS IS CARE-FULLY DESCRIBED. THE CONCEPTUAL RELATION BETWEEN THE CUT-OFF RADIUS OF INTERATOMIC POTENTIALS AND THE NUMBER OF NODES IN THE AFE IS ADDRESSED AND EXEMPLIFIED FOR THE 1D CASE. FOR THE 1D CASE ELEMENTS WITH 3, 5 AND 7 NODES WERE ADDRESSED. THE AFEM HAS BEEN USED TO DESCRIBE THE ME-CHANICAL BEHAVIOR OF ONE-DIMENSIONAL ATOMIC ARRAYS AS WELL AS TWO-DIMENSIONAL LATTICES OF ATOMS. THE EXAMPLES ALSO INCLUDED THE ANALYSIS OF PRISTINE DOMAINS, AS WELL AS DOMAINS WITH MISSING ATOMS, DEFECTS, OR VACANCIES. RESULTS ARE COMPARED WITH CLASSICAL MOLECULAR DYNAMIC SIMULATIONS (MD) PERFORMED USING A COMMERCIAL PACKAGE. THE RESULTS HAVE BEEN VERY ENCOURAGING IN TERMS OF ACCURACY AND IN THE COMPUTA-TIONAL EFFORT NECESSARY TO EXECUTE BOTH METHODOLOGIES, AFEM AND MD. THE METHODOLOGY CAN BE EXPANDED TO MODEL ANY DOMAIN DESCRIBED BY AN INTERATOMIC ENERGY POTENTIAL. read less NOT USED (definite) S. Ono, “Nonequilibrium phonon dynamics beyond the quasiequilibrium approach,” Physical Review B. 2017. link Times cited: 13 Abstract: The description of nonequilibrium states of solids in a simp… read moreAbstract: The description of nonequilibrium states of solids in a simplified manner is a challenge in the field of ultrafast dynamics. Here, the phonon thermalization in solids through the three-phonon scatterings is investigated by solving the Boltzmann transport equation (BTE). The numerical solution of the BTE shows that the transverse acoustic and longitudinal acoustic (LA) phonon temperatures are not well-defined during the relaxation, indicating the breakdown of the quasiequilibrium approximation. The development of hot and cold phonons and the backward energy flow from low to high energy phonons are observed in the initial and final stage of the relaxation, respectively. A minimal model is presented to relate the latter with the power-law decay of the LA phonon energy. read less NOT USED (definite) T. Han, F. Scarpa, and N. Allan, “Super stretchable hexagonal boron nitride Kirigami,” Thin Solid Films. 2017. link Times cited: 20 NOT USED (definite) B. Mortazavi, M.-Q. Le, T. Rabczuk, and L. Pereira, “Anomalous strain effect on the thermal conductivity of borophene: a reactive molecular dynamics study,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 52 NOT USED (definite) G. P. P. Pun and Y. Mishin, “Optimized interatomic potential for silicon and its application to thermal stability of silicene,” Physical Review B. 2017. link Times cited: 35 Abstract: An optimized interatomic potential has been constructed for … read moreAbstract: An optimized interatomic potential has been constructed for silicon using a modified Tersoff model. The potential reproduces a wide range of properties of Si and improves over existing potentials with respect to point defect structures and energies, surface energies and reconstructions, thermal expansion, melting temperature, and other properties. The proposed potential is compared with three other potentials from the literature. The potentials demonstrate reasonable agreement with first-principles binding energies of small Si clusters as well as single-layer and bilayer silicenes. The four potentials are used to evaluate the thermal stability of free-standing silicenes in the form of nanoribbons, nanoflakes, and nanotubes. While single-layer silicene is found to be mechanically stable at zero Kelvin, it is predicted to become unstable and collapse at room temperature. By contrast, the bilayer silicene demonstrates a larger bending rigidity and remains stable at and even above room temperature. The results suggest that bilayer silicene might exist in a free-standing form at ambient conditions. read less NOT USED (definite) P. G. Stubley, A. Higginbotham, and J. Wark, “Simulations of the inelastic response of silicon to shock compression,” Computational Materials Science. 2017. link Times cited: 2 NOT USED (definite) Z. Tong et al., “Review on FIB-induced damage in diamond materials,” Current Nanoscience. 2016. link Times cited: 5 Abstract: Background: Although various advanced FIB processing methods… read moreAbstract: Background: Although various advanced FIB processing methods for the fabrication of 3D nanostructures have been successfully developed by many researchers, the FIB milling has an unavoidable result in terms of the implantation of ion source materials and the formation of damaged layer at the near surface. Understanding the ion-solid interactions physics provides a unique way to control the FIB produced defects in terms of their shape and location. Methods: We have carefully selected peer-reviewed papers which mainly focusing on the review questions of this paper. A deductive content analysis method was used to analyse the methods, findings and conclusions of these papers. Based on their research methods, we classify their works in different groups. The theory of ion-matter interaction and the previous investigation on ion-induced damage in diamond were reviewed and discussed. Results: The previous research work has provided a systematic analysis of ion-induced damage in diamond. Both experimental and simulation methods have been developed to understand the damage process. The damaged layers created in FIB processing process can significantly degrade/alter the device performance and limit the applications of FIB nanofabrication technique. There are still challenges involved in fabricating large, flat, and uniform TEM samples in undoped non-conductive diamond. Conclusions: The post-facto-observation leaves a gap in understanding the formation process of ioninduced damage, forcing the use of assumptions. In contrast, MD simulations of ion bombardment have shed much light on ion beam mixing for decades. These activities make it an interesting and important task to understand what the fundamental effects of energetic particles on matter are. read less NOT USED (definite) A. K. Cuentas-Gallegos et al., “Porosity and Surface Modifications on Carbon Materials for Capacitance Improvement,” Open Material Sciences. 2016. link Times cited: 5 Abstract: Supercapacitors (SC) are energy storage devices with higher … read moreAbstract: Supercapacitors (SC) are energy storage devices with higher power but lower energy density than Li batteries. SC store energy based on two mechanisms: double layer capacitance (non-Faradaic) and pseudocapacitance (faradaic). Porous carbon materials have been extensively used as electrodes in SC, where their great surface area and pore size distribution have been the main properties for capacitance improvement. Nevertheless, these properties have shown limitations since they cannot be highly increased without losing electric conductivity, which is detrimental for the power requirements of SC. An alternative approach to increase capacitance has been the surface modification of carbon materials by introducing faradaic contributions. In this mini review, the effect of surface area, porosity, surface modification by doping or functionalization, and introduction of electroactive oxides are discussed to show how these factors influences the intrinsic capacitance values of different carbon materials; and some examples from our work are provided. The manipulation of such properties, on carbon materials (porosity and /or surface chemistry) not only are useful for devices such as SC, but also are very useful for a wide variety of Bio-applications (Bio-sensors, labelling and drug delivery, impregnation with microorganisms for its use as biochar, or for bio-fuel cells, etc.) read less NOT USED (definite) M. Friedrich, P. Piovano, and U. Stefanelli, “The Geometry of C_60,” Siam Journal on Applied Mathematics. 2016. link Times cited: 8 Abstract: Molecular Mechanics describes molecules as particle configur… read moreAbstract: Molecular Mechanics describes molecules as particle configurations interacting via classical potentials. These configurational energies usually consist of the sum of different phenomenological terms which are tailored to the description of specific bonding geometries. This approach is followed here to model the fullerene $C_{60}$, an allotrope of carbon corresponding to a specific hollow spherical structure of sixty atoms. We rigorously address different modeling options and advance a set of minimal requirements on the configurational energy able to deliver an accurate prediction of the fine three-dimensional geometry of $C_{60}$ as well as of its remarkable stability. In particular, the experimentally observed truncated-icosahedron structure with two different bond lengths is shown to be a strict local minimizer. read less NOT USED (definite) B. Mortazavi, O. Rahaman, T. Rabczuk, and L. Pereira, “Thermal conductivity and mechanical properties of nitrogenated holey graphene,” Carbon. 2016. link Times cited: 111 NOT USED (definite) B. Mortazavi, Z. Fan, Z. Fan, L. Pereira, A. Harju, and T. Rabczuk, “Amorphized graphene: A stiff material with low thermal conductivity,” Carbon. 2016. link Times cited: 74 NOT USED (definite) A. Dmitriev, A. Nikonov, and W. Österle, “MD Sliding Simulations of Amorphous Tribofilms Consisting of either SiO2 or Carbon,” Lubricants. 2016. link Times cited: 35 Abstract: The sliding behaviors of two simplified tribofilms with amor… read moreAbstract: The sliding behaviors of two simplified tribofilms with amorphous structure consisting either of SiO2 molecules or C atoms were simulated by molecular dynamics modeling. The objective was to identify mechanisms explaining the experimentally observed lubricating properties of the two amorphous films. The impacts of layer thickness, normal pressure, temperature and different substrate materials were studied systematically, while the sliding velocity was kept constant at 30 m/s. While the layer thickness was not critical, all the other parameters showed special effects under certain conditions. Normal pressure impeded void formation and could even eliminate voids if applied at high temperature. Stick-slip sliding was changed to smooth sliding at high temperature due to void healing. Considering the carbon film, high friction forces and shearing of the entire film was observed with diamond substrates, whereas interface sliding at low friction forces and an amorphous layer of iron mixed with carbon was observed if the supporting substrates consisted of α-Fe. Both films show a decrease of friction forces and smooth sliding behavior at elevated temperature, corresponding well to the tribological behavior of an advanced nanocomposite sliding against a steel disc under severe stressing conditions when high flash temperatures can be expected. read less NOT USED (definite) A. Sgouros, A. Sgouros, G. Kalosakas, G. Kalosakas, C. Galiotis, and K. Papagelis, “Uniaxial compression of suspended single and multilayer graphenes,” 2D Materials. 2016. link Times cited: 19 Abstract: The mechanical response of single and multiple graphene shee… read moreAbstract: The mechanical response of single and multiple graphene sheets under uniaxial compressive loads was studied with molecular dynamics (MD) simulations, using different semi-empirical force fields at different boundary conditions or constrains. Compressive stress–strain curves were obtained and the critical stress/strain values were derived. The MD results are compared to the linear elasticity continuum theory for loaded slabs. Concerning the length dependence of critical values, qualitatively similar behavior is observed between the theory and numerical simulations for single layer graphenes, as the critical stress/strain for buckling was found to scale to the inverse squared length. However discrepancies were noted for multilayer graphenes, where the critical buckling stress also decreased with increasing length, though at a slower rate than expected from elastic buckling analysis. read less NOT USED (definite) D. Michels and M. Desbrun, “A semi-analytical approach to molecular dynamics,” J. Comput. Phys. 2015. link Times cited: 15 NOT USED (definite) M. Stockett et al., “Threshold energies for single-carbon knockout from polycyclic aromatic hydrocarbons.,” The journal of physical chemistry letters. 2015. link Times cited: 24 Abstract: We have measured absolute cross sections for ultrafast (femt… read moreAbstract: We have measured absolute cross sections for ultrafast (femtosecond) single-carbon knockout from polycyclic aromatic hydrocarbon (PAH) cations as functions of He–PAH center-of-mass collision energy in the 10–200 eV range. Classical molecular dynamics (MD) simulations cover this range and extend up to 105 eV. The shapes of the knockout cross sections are well-described by a simple analytical expression yielding experimental and MD threshold energies of EthExp = 32.5 ± 0.4 eV and EthMD = 41.0 ± 0.3 eV, respectively. These are the first measurements of knockout threshold energies for molecules isolated in vacuo. We further deduce semiempirical (SE) and MD displacement energies, i.e., the energy transfers to the PAH molecules at the threshold energies for knockout, of TdispSE = 23.3 ± 0.3 eV and TdispMD = 27.0 ± 0.3 eV. The semiempirical results compare favorably with measured displacement energies for graphene (Tdisp = 23.6 eV). read less NOT USED (definite) P. Brault and E. Neyts, “Molecular dynamics simulations of supported metal nanocatalyst formation by plasma sputtering,” Catalysis Today. 2015. link Times cited: 26 NOT USED (definite) I. Tejada, L. Brochard, T. Lelièvre, G. Stoltz, F. Legoll, and É. Cancès, “Coupling a reactive potential with a harmonic approximation for atomistic simulations of material failure,” Computer Methods in Applied Mechanics and Engineering. 2015. link Times cited: 1 NOT USED (definite) A. Sadeghirad, N. Su, and F. Liu, “Mechanical modeling of graphene using the three-layer-mesh bridging domain method,” Computer Methods in Applied Mechanics and Engineering. 2015. link Times cited: 12 NOT USED (definite) U. Monteverde et al., “Under pressure: control of strain, phonons and bandgap opening in rippled graphene,” Carbon. 2015. link Times cited: 59 NOT USED (definite) L. Aversa, S. Taioli, M. Nardi, R. Tatti, R. Verucchi, and S. Iannotta, “The Interaction of C60 on Si(111) 7 × 7 Studied by Supersonic Molecular Beams: Interplay between Precursor Kinetic Energy and Substrate Temperature in Surface Activated Processes,” Frontiers in Materials. 2015. link Times cited: 6 Abstract: Buckminsterfullerene (C60) is a molecule fully formed of car… read moreAbstract: Buckminsterfullerene (C60) is a molecule fully formed of carbon that can be used, owing to its electronic and mechanical properties, as “clean” precursor for the growth of carbon-based materials, ranging from -conjugated systems (graphenes) to synthesized species, e.g. carbides such as silicon carbide (SiC). To this goal, C60 cage rupture is the main physical process that triggers material growth. Cage breaking can be obtained either thermally by heating up the substrate to high temperatures (630°C), after C60 physisorption, or kinetically by using Supersonic Molecular Beam Epitaxy (SuMBE) techniques. In this work, aiming at demonstrating the growth of SiC thin films by C60 supersonic beams, we present the experimental investigation of C60 impacts on Si(111) 7x7 kept at 500°C for translational kinetic energies ranging from 18 to 30 eV. The attained kinetically activated synthesis of SiC submonolayer films is probed by in-situ surface electron spectroscopies (XPS and UPS). Furthermore, in these experimental conditions the C60-Si(111) 7×7 collision has been studied by computer simulations based on a tight-binding approximation to Density Functional Theory, DFT. Our theoretical and experimental findings point towards a kinetically driven growth of SiC on Si, where C60 precursor kinetic energy plays a crucial role, while temperature is relevant only after cage rupture to enhance Si and carbon reactivity. In particular, we observe a counterintuitive effect in which for low kinetic energy (below 22 eV), C60 bounces back without breaking more effectively at high temperature due to energy transfer from excited phonons. At higher kinetic energy (22 < K < 30 eV), for which cage rupture occurs, temperature enhances reactivity without playing a major role in the cage break. These results are in good agreement with ab-initio molecular dynamics simulations. SuMBE is thus a technique able to drive materials growth at low temperature regime. read less NOT USED (definite) S. K. Jain, G. Barkema, N. Mousseau, C. Fang, and M. Huis, “Strong Long-Range Relaxations of Structural Defects in Graphene Simulated Using a New Semiempirical Potential,” Journal of Physical Chemistry C. 2015. link Times cited: 22 Abstract: We present a new semiempirical potential for graphene, which… read moreAbstract: We present a new semiempirical potential for graphene, which includes also an out-of-plane energy term. This novel potential is developed from density functional theory (DFT) calculations for small numbers of atoms and can be used for configurations with millions of atoms. Our simulations show that buckling caused by typical defects such as the Stone–Wales (SW) defect extends to hundreds of nanometers. Surprisingly, this long-range relaxation lowers the defect formation energy dramatically—by a factor of 2 or 3—implying that previously published DFT-calculated defect formation energies suffer from large systematic errors. We also show the applicability of the novel potential to other long-range defects including line dislocations and grain boundaries, all of which exhibit pronounced out-of-plane relaxations. We show that the energy as a function of dislocation separation diverges logarithmically for flat graphene but converges to a constant for free-standing buckled graphene. A potential in which the atom... read less NOT USED (definite) Z. Tong, X. Luo, J. Sun, Y. Liang, and X. Jiang, “Investigation of a scale-up manufacturing approach for nanostructures by using a nanoscale multi-tip diamond tool,” The International Journal of Advanced Manufacturing Technology. 2015. link Times cited: 0 NOT USED (definite) B. Mortazavi and T. Rabczuk, “Multiscale modeling of heat conduction in graphene laminates,” Carbon. 2015. link Times cited: 103 NOT USED (definite) B. Mortazavi, B. Mortazavi, G. Cuniberti, and T. Rabczuk, “Mechanical properties and thermal conductivity of graphitic carbon nitride: A molecular dynamics study,” Computational Materials Science. 2015. link Times cited: 105 NOT USED (definite) M. U. Kucukkal and S. Stuart, “Simulation of carbon nanotube welding through Ar bombardment,” Journal of Molecular Modeling. 2014. link Times cited: 4 NOT USED (definite) P. Brommer, A. Kiselev, D. Schopf, P. Beck, J. Roth, and H. Trebin, “Classical interaction potentials for diverse materials from ab initio data: a review of potfit,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 76 Abstract: Force matching is an established technique to generate effec… read moreAbstract: Force matching is an established technique to generate effective potentials for molecular dynamics simulations from first-principles data. This method has been implemented in the open source code potfit. Here, we present a review of the method and describe the main features of the code. Particular emphasis is placed on the features added since the initial release: interactions represented by analytical functions, differential evolution as optimization method, and a greatly extended set of interaction models. Beyond the initially present pair and embedded-atom method potentials, potfit can now also optimize angular dependent potentials, charge and dipolar interactions, and electron-temperature-dependent potentials. We demonstrate the functionality of these interaction models using three example systems: phonons in type I clathrates, fracture of α-alumina, and laser-irradiated silicon. read less NOT USED (definite) T. Zhou, L. Liu, W. Goddard, S. Zybin, and F. Huang, “ReaxFF reactive molecular dynamics on silicon pentaerythritol tetranitrate crystal validates the mechanism for the colossal sensitivity.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 16 Abstract: Recently quantum mechanical (QM) calculations on a single Si… read moreAbstract: Recently quantum mechanical (QM) calculations on a single Si-PETN (silicon-pentaerythritol tetranitrate) molecule were used to explain its colossal sensitivity observed experimentally in terms of a unique Liu carbon-silyl nitro-ester rearrangement (R3Si-CH2-O-R2→ R3Si-O-CH2-R2). In this paper we expanded the study of Si-PETN from a single molecule to a bulk system by extending the ReaxFF reactive force field to describe similar Si-C-H-O-N systems with parameters optimized to reproduce QM results. The reaction mechanisms and kinetics of thermal decomposition of solid Si-PETN were investigated using ReaxFF reactive molecular dynamics (ReaxFF-RMD) simulations at various temperatures to explore the origin of the high sensitivity. We find that at lower temperatures, the decomposition of Si-PETN is initiated by the Liu carbon-silyl nitro-ester rearrangement forming Si-O bonds which is not observed in PETN. As the reaction proceeds, the exothermicity of Si-O bond formation promotes the onset of NO2 formation from N-OC bond cleavage which does not occur in PETN. At higher temperatures PETN starts to react by the usual mechanisms of NO2 dissociation and HONO elimination; however, Si-PETN remains far more reactive. These results validate the predictions from QM that the significantly increased sensitivity of Si-PETN arises from a unimolecular process involving the unusual Liu rearrangement but not from multi-molecular collisions. It is the very low energy barrier and the high exothermicity of the Si-O bond formation providing energy early in the decomposition process that is responsible. read less NOT USED (definite) H.-Y. Cheng et al., “Evolution of Carbon Nanofiber-Supported Pt Nanoparticles of Different Particle Sizes: A Molecular Dynamics Study,” Journal of Physical Chemistry C. 2014. link Times cited: 18 Abstract: Molecular dynamics simulations employing the ReaxFF reactive… read moreAbstract: Molecular dynamics simulations employing the ReaxFF reactive force field have been carried out to analyze the structural evolution of fishbone-type carbon nanofiber-supported Pt nanoparticles, with particle size ranging from 5.6 to 30.7 A. Simulated results indicate that upon adsorption the distribution of first-shell Pt–Pt coordination number and radial distribution function change significantly in Pt nanoparticles up to 2 nm in size and that the restructuring degree of the Pt nanoparticles decreases with particle size, which is attributed both to the reduced binding energy per Pt atom bonded to support and to the increased cohesive energy of the Pt nanoparticles. In the Pt10 particle, the majority of the Pt atoms are detached from the metal particle, leading to atomic adsorption of single Pt atoms on the support. As the Pt particle size is increased to ∼3 nm, however, the crystalline degree of Pt nanoparticles is even higher than that of the corresponding isolated nanoparticles because the strong metal–... read less NOT USED (definite) H. Tetlow, J. Boer, I. Ford, D. Vvedensky, J. Coraux, and L. Kantorovich, “Growth of Epitaxial Graphene: Theory and Experiment,” arXiv: Materials Science. 2014. link Times cited: 222 NOT USED (definite) X. Wang and B. Huang, “Computational Study of In-Plane Phonon Transport in Si Thin Films,” Scientific Reports. 2014. link Times cited: 82 NOT USED (definite) R. Ansari, A. Momen, S. Rouhi, and S. Ajori, “On the Vibration of Single-Walled Carbon Nanocones: Molecular Mechanics Approach versus Molecular Dynamics Simulations,” Shock and Vibration. 2014. link Times cited: 23 Abstract: The vibrational behavior of single-walled carbon nanocones i… read moreAbstract: The vibrational behavior of single-walled carbon nanocones is studied using molecular structural method and molecular dynamics simulations. In molecular structural approach, point mass and beam elements are employed to model the carbon atoms and the connecting covalent bonds, respectively. Single-walled carbon nanocones with different apex angles are considered. Besides, the vibrational behavior of nanocones under various types of boundary conditions is studied. Predicted natural frequencies are compared with the existing results in the literature and also with the ones obtained by molecular dynamics simulations. It is found that decreasing apex angle and the length of carbon nanocone results in an increase in the natural frequency. Comparing the vibrational behavior of single-walled carbon nanocones under different boundary conditions shows that the effect of end condition on the natural frequency is more prominent for nanocones with smaller apex angles. read less NOT USED (definite) Z. Budrikis, A. Sellerio, Z. Bertalan, and S. Zapperi, “Wrinkle motifs in thin films,” Scientific Reports. 2014. link Times cited: 15 NOT USED (definite) P. Ballone, “Modeling Potential Energy Surfaces: From First-Principle Approaches to Empirical Force Fields,” Entropy. 2013. link Times cited: 9 Abstract: Explicit or implicit expressions of potential energy surface… read moreAbstract: Explicit or implicit expressions of potential energy surfaces (PES) represent the basis of our ability to simulate condensed matter systems, possibly understanding and sometimes predicting their properties by purely computational methods. The paper provides an outline of the major approaches currently used to approximate and represent PESs and contains a brief discussion of what still needs to be achieved. The paper also analyses the relative role of empirical and ab initio methods, which represents a crucial issue affecting the future of modeling in chemical physics and materials science. read less NOT USED (definite) A. Guz’ and J. Rushchitsky, “Some Fundamental Aspects of Mechanics of Nanocomposite Materials and Structural Members,” Journal of Nanotechnology. 2013. link Times cited: 7 Abstract: This paper is devoted to formulation and analysis of fundame… read moreAbstract: This paper is devoted to formulation and analysis of fundamental aspects of mechanics of nanocomposite materials and structural members. These aspects most likely do not exhaust all of the possible fundamental characteristics of mechanics of nanocomposite materials and structural members, but, nevertheless, they permit to form the skeleton of direction of mechanics in hand. The proposed nine aspects are described and commented briefly. read less NOT USED (definite) Y. Cong, J. Yvonnet, and H. Zahrouni, “Simulation of instabilities in thin nanostructures by a perturbation approach,” Computational Mechanics. 2013. link Times cited: 0 NOT USED (definite) A. Favata, A. Micheletti, and P. Podio-Guidugli, “A nonlinear theory of prestressed elastic stick-and-spring structures,” International Journal of Engineering Science. 2013. link Times cited: 23 NOT USED (definite) A. Ilie, S. Crampin, L. Karlsson, and M. Wilson, “Repair and stabilization in confined nanoscale systems — inorganic nanowires within single-walled carbon nanotubes,” Nano Research. 2012. link Times cited: 12 NOT USED (definite) A. Oluwajobi, “Molecular Dynamics Simulation of Nanoscale Machining.” 2012. link Times cited: 6 Abstract: Product miniaturization is a major motivation for the develo… read moreAbstract: Product miniaturization is a major motivation for the development of ultra-precision technologies and processes which can achieve high form and excellent surface finish. Of all the available manufacturing approaches, mechanical nanometric machining is still a good option for machining complex 3D devices in a controllable way (Jackson, 2008). As the dimension goes down to the nanoscale, the machining phenomena take place in a limited region of tool-workpiece interface. At this length scale and interface, the material removal mechanisms are not fully understood, so more insight is needed, which on the long run will help to achieve high precision manufacturing with predictability, repeatability and productivity (Luo, 2004). At present, it is very difficult to observe the diverse microscopic physical phenomena occurring through experiments at the nanoscale (Rentsch, 2008). Subsequently, the other alternative is to explore available simulation techniques. Continuum mechanics approach is not adequate, as the point of interest/interface cannot be assumed to be homogeneous, but rather discrete, so, atomistic simulation methods are the suitable techniques for modelling at the nanoscale. read less NOT USED (definite) K. Eriguchi, “Application of Molecular Dynamics Simulations to Plasma Etch Damage in Advanced Metal-Oxide-Semiconductor Field-Effect Transistors.” 2012. link Times cited: 0 Abstract: According to "the international technology roadmap for … read moreAbstract: According to "the international technology roadmap for semiconductors (ITRS)" (SIA, 2009), the shrinkage of silicon-based metal–oxide–semiconductor field-effect transistor (MOSFET) – an elemental device (unit) in ultra-large-scale integrated (ULSI) circuits – has been accelerating due to expanding demands for the higher performance and the lower power operation. The characteristic dimensions of current MOSFETs in mass productions are around 30 – 50 nm. Figure 1 shows the scaling trend of the key feature sizes in ULSI circuits predicted by Semiconductor Industry Association, USA. Various types of MOSFETs are designed for the specific purposes, i.e., low standby power (LSP), low operation power (LOP), and high performance (HP) operations, and built in ULSI circuits such as dynamic random access memory (DRAM) and micro-processing unit (MPU). New structured MOSFETs such as fully-depleted (FD) and metal-gate (MG) devices have been recently proposed. Since physical gate length (Lg) and source / drain extension depth (Ext) are the key feature sizes determining MOSFET performance (Sze & Ng, 2007), the shrinkage of Lg and Ext is a primal focus in the development of MOSFETs. These sizes have become a few nanometers, comparable to the scale of atomistic simulation domain. read less NOT USED (definite) K. Termentzidis and S. Merabia, “Molecular Dynamics Simulations and Thermal Transport at the Nano-Scale.” 2012. link Times cited: 14 Abstract: This chapter presents an overview of the Molecular Dynamics … read moreAbstract: This chapter presents an overview of the Molecular Dynamics (MD) simulation technique to predict thermal transport properties of nanostructured materials. This covers systems having characteristic lengths of the order of a few nanometers like carbon nanotubes, nanowires and also superlattices, i.e. composite materials made of submicronic thickness of solid layers. The common features of these systems is the small ratio between their characteristic system size and the phonon mean free path, which leads to ballistic heat transport and deviations from the classical Fourier law. Also when the density of interfaces gets large, the energy transport properties of the materials can not longer be described solely by the thermal conductivities of the constituents of the material, but depend also on the thermal boundary resistance which measures the transmission of phonons across an interface. In this context, molecular dynamics was proven to be a very useful technique to study heat transport in nanostructured materials. The main reasons are; the length scale probed by the method is in the nanometer range, and it does not make any assumption on the phonons dynamics except their classical nature. read less NOT USED (definite) M. Jafary-Zadeh, C. Reddy, V. Sorkin, and Y.-W. Zhang, “Kinetic nanofriction: a mechanism transition from quasi-continuous to ballistic-like Brownian regime,” Nanoscale Research Letters. 2012. link Times cited: 29 NOT USED (definite) A. Kumar, S. Mukherjee, J. T. Paci, K. Chandraseker, and G. Schatz, “A rod model for three dimensional deformations of single-walled carbon nanotubes,” International Journal of Solids and Structures. 2011. link Times cited: 35 NOT USED (definite) A. Page, K. Chandrakumar, Y. Wang, S. Irle, and K. Morokuma, “Mechanisms of Single-Walled Carbon Nanotube Nucleation, Growth and Chirality-Control: Insights from QM/MD Simulations.” 2011. link Times cited: 4 Abstract: The experimental characterisations of carbon nanotubes (CNTs… read moreAbstract: The experimental characterisations of carbon nanotubes (CNTs) (Iijima, 1991) and in particular single-walled CNTs (SWNTs) (Iijima & Ichihashi, 1993) in the early 1990s were landmark moments in 20th century science. The potential uses of these remarkable nanostructures are now becoming realised, as their synthesis is now routinely performed on the industrial scale. The initial successes in this respect were generally experimental techniques that were previously well established in other fields. This is particularly true of the chemical vapor deposition (CVD) and arc-discharge processes. The original experimental characterisation of SWNTs was in fact accomplished using nanotubes synthesised with the former method (Iijima & Ichihashi, 1993). The understanding of the way in which CNTs nucleate and grow was therefore synergic with the evolution and refinement of these synthetic methods. Indeed, the original mechanisms of CNT nucleation and growth were conceived from experimental observations. The most prevalent of these today is the vapor-liquid-solid (VLS) mechanism (Saito, 1995). According to this mechanism, SWNT nucleation growth is postulated to consist of three distinct stages. The first of these features a mixed carbon/catalyst vapor phase, from which co-condensation yields liquid catalyst-carbide nanoparticles. Typical catalysts in the growth of SWNTs are traditionally transition metals such as Fe, Ni, Co, Mo, and alloys thereof (see (Journet et al., 1997; Moisala et al., 2003; Harris, 2007), and references therein). The precipitation of atomic carbon from this liquid carbide phase takes place once the carbide phase is saturated with carbon. This precipitation yields the formation of solid phase CNTs. Due to the inherent limits in spatial and temporal resolutions that are furnished by experimental techniques and instrumentation, there inevitably remain questions regarding the VLS mechanism and CNT growth that, for now, cannot be answered from an experimental standpoint. There are several infamous examples in this respect. For instance, the mechanism of so-called ‘catalyst-free’ SWNT nucleation growth remains unknown, following the recent read less NOT USED (definite) J. Wang, H. W. Zhang, X. Guo, and M. Tian, “Study of Carbon Nanotube Based on Higher Order Cauchy-Born Rule.” 2011. link Times cited: 5 Abstract: Since single-walled carbon nanotube (SWCNT) and multi-walled… read moreAbstract: Since single-walled carbon nanotube (SWCNT) and multi-walled carbon nanotube (MWCNT) are found by Iijima (1991, 1993), these nanomaterials have stimulated extensive interest in the material research communities in the past decades. It has been found that carbon nanotubes possess many interesting and exceptional mechanical and electronic properties (Ruoff et al., 2003; Popov, 2004). Therefore, it is expected that they can be used as promising materials for applications in nanoengineering. In order to make good use of these nanomaterials, it is important to have a good knowledge of their mechanical properties. Experimentally, Tracy et al. (1996) estimated that the Young’s modulus of 11 MWCNTs vary from 0.4TPa to 4.15TPa with an average of 1.8TPa by measuring the amplitude of their intrinsic thermal vibrations, and it is concluded that carbon nanotubes appear to be much stiffer than their graphite counterpart. Based on the similar experiment method, Krishnan et al. (1998) reported that the Young’s modulus is in the range of 0.9TPa to 1.70TPa with an average of 1.25TPa for 27 SWCNTs. Direct tensile loading tests of SWCNTs and MWCNTs have also been performed by Yu et al. (2000) and they reported that the Young’s modulus are 0.32-1.47TPa for SWCNTs and 0.27-0.95TPa for MWCNTS, respectively. In the experiment, however, it is very difficult to measure the mechanical properties of carbon nanotues directly due to their very small size. Based on molecular dynamics simulation and Tersoff-Brenner atomic potential, Yakobson et al. (1996) predicted that the axial modulus of SWCNTs are ranging from 1.4 to 5.5 TPa (Note here that in their study, the wall thickness of SWNT was taken as 0.066nm); Liang & Upmanyu (2006) investigated the axial-strain-induced torsion (ASIT) response of SWCNTs, and Zhang et al. (2008) studied ASIT in multi-walled carbon nanotubes. By employing a non-orthogonal tight binding theory, Goze et al. (1999) investigated the Young’s modulus of armchair and zigzag SWNTs with diameters of 0.5-2.0 nm. It was found that the Young’s modulus is dependent on the diameter of the tube noticeably as the tube diameter is small. Popov et al. (2000) predicted the mechanical properties of SWCNTs using Born’s perturbation technique with a lattice-dynamical model. The results they obtained showed that the Young’s modulus and the Poisson’s ratio of both armchair and zigzag SWCNTs depend on the tube radius as the tube radius are small. Other atomic modeling studies read less NOT USED (definite) I. Chang, “Structural Instability of Carbon Nanotube.” 2011. link Times cited: 0 Abstract: Since Iijima reported MWCNTs in 1991, CNTs have captured the… read moreAbstract: Since Iijima reported MWCNTs in 1991, CNTs have captured the intensive attention of researchers worldwide due to the combination of their expected structural perfection, small size, low density, high stiffness, high strength, and excellent electronic properties. CNTs have been widely adopted as microscopic probing tips (Dai et al., 1996; Hafner et al., 2001), nanocomposites reinforcements (Bower et al., 1998; Jin et al., 1998), nanotweezers (Kim & Lieber, 1999), and nanoactuators (Baughman et al., 1999; Fennimore et al., 2003) due to their slender and high aspect ratio structures. Meanwhile, nanotubes are also highly susceptible to buckling under compression, which is a structural instability. Once the buckling of CNTs occurs, the load-carrying capability would suddenly reduce and lead to possible catastrophic failure of the nanotubes, which significantly limit the loading strengths of the probing tips and compressive strengths of nanocomposite structures. Even the physical properties such as conductance of carbon nanotube can be influenced by the occurrence of buckling (Postma et al., 2001). Hence, it is crucial to understand the mechanism of nanotube buckling and even predict the onset of buckling in order to improve the nanotube applications. A review of the relevant literature shows that significant studies have employed both experimental (Falvo et al., 1997; Iijima et al., 1996; Thostenson & Chou, 2004; Waters et al., 2004) and theoretical (Ru, 2000; Yakobson & Avouris, 2001) approaches to investigate the bucking behaviors of CNTs. However due to the difficulties encountered at nanoscale, the experimental investigation of the buckling behaviors of CNTs remains a challenging problem and individual factors that affect buckling could not be easily identified. In theoretical study, the CNTs are commonly treated as beams or thin-shell tubes with certain wall thickness and elastic constants and, thus, it is difficult to consider the chirality and size effects on buckling behavior of CNTs because the continuum assumption disregards the discrete nature of atomic structures (Ru, 2000; Yakobson & Avouris, 2001). Some researchers attempted to introduce the atomic-continuum method combining the atomic detail in the continuum description and examine the various properties of CNTs (Chang, 2004; Guo et al., 2008; Li & Chou, 2003a, 2003b). The atomic-continuum method could shorten the computational time in larger atomic system. As the fast development and rapid advancement of computers, molecular approaches have become important tools and are widely applied to study the factors that would influence the buckling of CNTs (Buehler et al., 2004; Cao & Chen, 2006a, 2006b; Huh & Huh, 2008; Liew et al., 2004; Ozaki et al., 2000). Although some researchers already discussed various aspects of read less NOT USED (definite) M. Bernardi, M. Giulianini, and J. Grossman, “Self-assembly and its impact on interfacial charge transfer in carbon nanotube/P3HT solar cells.,” ACS nano. 2010. link Times cited: 92 Abstract: Charge transfer at the interface of conjugated polymer and n… read moreAbstract: Charge transfer at the interface of conjugated polymer and nanoscale inorganic acceptors is pivotal in determining the efficiency of excitonic solar cells. Despite intense efforts, carbon nanotube/polymer solar cells have resulted in disappointing efficiencies (<2%) due in large part to poor charge transfer at the interface. While the interfacial energy level alignment is clearly important, the self-assembly and the interface structure also play a major role in facilitating this charge transfer. To understand and control this effect to our advantage, we study the interface of commonly used conductive polymer poly-3-hexylthiophene (P3HT) and single-walled carbon nanotubes (SWNTs) with a combination of molecular dynamics simulations, absorption spectra experiments, and an analysis of charge transfer effects. Classical molecular dynamics simulations show that the P3HT wraps around the SWNTs in a number of different conformations, including helices, bundles, and more elongated conformations that maximize planar π-π stacking, in agreement with recent experimental observations. Snapshots from the MD simulations reveal that the carbon nanotubes play an important templating role of increasing the π-conjugation in the system, an effect deriving from the π-π stacking interaction at the interface and the 1-dimensional (1D) nature of the SWNTs, and independent of the SWNT chirality. We show how this increase in the system conjugation could largely improve the charge transfer in P3HT-SWNT type II heterojunctions and support our results with absorption spectra measurements of mixtures of carbon nanotubes and P3HT. These findings open possibilities for improved preparation of polymeric solar cells based on carbon nanotubes and on 1D nanomaterials in general. read less NOT USED (definite) P. Belík and M. Luskin, “Analysis of the Quasi-Nonlocal Approximation of Linear and Circular Chains in the Plane,” Multiscale Model. Simul. 2010. link Times cited: 2 Abstract: We give an analysis of the stability and displacement error … read moreAbstract: We give an analysis of the stability and displacement error for linear and circular atomistic chains in the plane when the atomistic energy is approximated by the Cauchy–Born continuum energy and by the quasi-nonlocal atomistic-to-continuum coupling energy. We consider atomistic energies that include Lennard-Jones-type nearest neighbor and next nearest neighbor pair-potential interactions. Previous analyses for linear chains have shown that the Cauchy–Born and quasi-nonlocal approximations reproduce (up to the order of the lattice spacing) the atomistic lattice stability for perturbations that are constrained to the line of the chain. However, we show that the Cauchy–Born gives a finite increase for the lattice stability of a linear or circular chain under compression when general perturbations in the plane are allowed. We also analyze the increase of the lattice stability under compression when pair-potential energies are augmented by bond-angle energies. Our estimates of the largest strain for lattice s... read less NOT USED (definite) S. Rubaiyat and S. Chowdhury, “STUDY OF PRISTINE CARBON NANOTUBE UNDER TENSILE AND COMPRESSIVE LOADS USING MOLECULAR DYNAMICS SIMULATION,” Journal of Mechanical Engineering. 2010. link Times cited: 3 Abstract: After the discovery, carbon nanotubes (CNTs) have received t… read moreAbstract: After the discovery, carbon nanotubes (CNTs) have received tremendous scientific and industrial interests. This is due to their exceptional mechanical, electrical, and thermal properties. CNTs having pristine structure (i.e., structure without any defect) hold very high mechanical properties. In this article, mechanical properties of CNTs are studied under both tensile and compressive loads using molecular dynamics (MD) simulations. Four armchair single-walled nanotubes (SWNTs) having indexes of (3,3), (4,4), (5,5) and (6,6) with pristine structure are simulated with MD. Molecular simulations are carried out using the classical MD method, in which the Newtonian equations of motion are solved numerically for a set of atoms. The velocity- Verlet algorithm is used for solving the Newtonian equations of motion. The Brenner potential is used for carbon-carbon interaction in the CNT and temperature of the system is controlled by velocity scaling. Simulation results show that modulus of elasticity of CNTs varies significantly with CNT diameter. The results obtained from the compressive test by MD simulations are in well agreement with the results obtained from theoretical Euler equation and parabolic equation for long and short column respectively. Keywords: Carbon nanotubes; Molecular dynamics; Young’s modulus; Failure strength; Failure strain. DOI: 10.3329/jme.v40i2.5346 Journal of Mechanical Engineering , Vol. ME 40, No. 2, December 2009 72-78 read less NOT USED (definite) C. Björkas, K. Vörtler, K. Nordlund, D. Nishijima, and R. Doerner, “Chemical sputtering of Be due to D bombardment,” New Journal of Physics. 2009. link Times cited: 66 Abstract: While covalently bonded materials such as carbon are well kn… read moreAbstract: While covalently bonded materials such as carbon are well known to be eroded by chemical sputtering when exposed to plasmas or low-energy ion irradiation, pure metals have been believed to sputter only physically. The erosion of Be when subject to D bombardment was in this work measured at the PISCES-B facility and modelled with molecular dynamics simulations. During the experiments, a chemical effect was observed, since a fraction of the eroded Be was in the form of BeD molecules. This fraction decreased with increasing ion energy. The same trend was seen in the simulations and was explained by the swift chemical sputtering mechanism, showing that pure metals can, indeed, be sputtered chemically. D ions of only 7 eV can erode Be through this mechanism. read less NOT USED (definite) B. Sadigh, P. Erhart, A. Stukowski, and A. Caro, “Composition-dependent interatomic potentials: A systematic approach to modelling multicomponent alloys,” Philosophical Magazine. 2009. link Times cited: 16 Abstract: We propose a simple scheme to construct composition-dependen… read moreAbstract: We propose a simple scheme to construct composition-dependent interatomic potentials for multicomponent systems that, when superposed onto the potentials for the pure elements, can reproduce not only the heat of mixing of the solid solution in the entire concentration range but also the energetics of a wider range of configurations including intermetallic phases. We show that an expansion in cluster interactions provides a way to systematically increase the accuracy of the model, and that it is straightforward to generalise this procedure to multicomponent systems. Concentration-dependent interatomic potentials can be built upon almost any type of potential for the pure elements including embedded atom method (EAM), modified EAM, bond-order, and Stillinger–Weber type potentials. In general, composition-dependent N-body terms in the total energy lead to explicit (N + 1)-body forces, which potentially render them computationally expensive. We present an algorithm that overcomes this problem and that can speed up the calculation of the forces for composition-dependent pair potentials in such a way as to make them computationally comparable in efficiency and scaling behaviour to standard EAM potentials. We also discuss the implementation in Monte Carlo simulations. Finally, we exemplarily review the composition-dependent EAM model for the Fe–Cr system. read less NOT USED (definite) S. Irle, Y. Ohta, Y. Okamoto, A. Page, Y. Wang, and K. Morokuma, “Milestones in molecular dynamics simulations of single-walled carbon nanotube formation: A brief critical review,” Nano Research. 2009. link Times cited: 47 NOT USED (definite) C. L. Dias, T. Ala‐Nissila, M. Grant, and M. Karttunen, “Three-dimensional ‘Mercedes-Benz’ model for water.,” The Journal of chemical physics. 2009. link Times cited: 54 Abstract: In this paper we introduce a three-dimensional version of th… read moreAbstract: In this paper we introduce a three-dimensional version of the Mercedes-Benz model to describe water molecules. In this model van der Waals interactions and hydrogen bonds are given explicitly through a Lennard-Jones potential and a Gaussian orientation-dependent terms, respectively. At low temperature the model freezes forming Ice-I and it reproduces the main peaks of the experimental radial distribution function of water. In addition to these structural properties, the model also captures the thermodynamical anomalies of water: The anomalous density profile, the negative thermal expansivity, the large heat capacity, and the minimum in the isothermal compressibility. read less NOT USED (definite) K. V. Bets and B. Yakobson, “Spontaneous twist and intrinsic instabilities of pristine graphene nanoribbons,” Nano Research. 2009. link Times cited: 148 NOT USED (definite) Q. Lu, M. Arroyo, and R. Huang, “Elastic bending modulus of monolayer graphene,” Journal of Physics D: Applied Physics. 2009. link Times cited: 382 Abstract: An analytic formula is derived for the elastic bending modul… read moreAbstract: An analytic formula is derived for the elastic bending modulus of monolayer graphene based on an empirical potential for solid-state carbon atoms. Two physical origins are identified for the non-vanishing bending stiffness of the atomically thin graphene sheet, one due to the bond-angle effect and the other resulting from the bond-order term associated with the dihedral angles. The analytical prediction compares closely with ab initio energy calculations. Pure bending of graphene monolayers into cylindrical tubes is simulated by a molecular mechanics approach, showing slight nonlinearity and anisotropy in the tangent bending modulus as the bending curvature increases. An intrinsic coupling between bending and in-plane strain is noted for graphene monolayers rolled into carbon nanotubes. read less NOT USED (definite) T. Edler and S. G. Mayr, “Mechanisms of stress generation during bombardment of Ge with keV ions: experiments and molecular dynamics simulations,” New Journal of Physics. 2007. link Times cited: 8 Abstract: The present contribution focuses on the phenomenology and me… read moreAbstract: The present contribution focuses on the phenomenology and mechanisms of stress generation in Ge thin films during keV ion bombardment. Experimentally, amorphous Ge (a-Ge) thin films were grown from vapor, and subsequently bombarded with Ar+ ions with energies of up to 3 keV. Stress generation is monitored by a laser beam deflection method. In order to identify the underlying nanoscale physics, molecular dynamics simulations were performed, in which crystalline and (a-Ge) films of different densities were irradiated. Experiments and simulations both show generation of compressive stresses, which saturate at ≈200 MPa and can be attributed to generation of voids with sizes of approximately 1 nm several nanometres below the surface. read less NOT USED (definite) J. Yim, M. Falk, M. Keidar, and I. Boyd, “Calculation of Boron Nitride Sputter Yields Under Low Energy Xenon Ion Bombardment.” 2007. link Times cited: 4 Abstract: An accurate description of the sputter yield of boron nitrid… read moreAbstract: An accurate description of the sputter yield of boron nitride (BN) from xenon ion bombardment at low energies is needed for improving the prediction capabilities of Hall thruster erosion models and in turn for lifetime prediction models. However, sputter yield data at low (< 300 eV) energies does not exist. The molecular dynamics method is employed to model the sputtering of BN at low energies. The results are compared to existing experimental data at higher energies. A qualitative comparison with a quantumstatistical model is also performed. read less NOT USED (definite) K. Volokh and K. Ramesh, “An approach to multi-body interactions in a continuum-atomistic context: Application to analysis of tension instability in carbon nanotubes,” International Journal of Solids and Structures. 2006. link Times cited: 11 NOT USED (definite) M. Ali, R. Smith, and S. Hobday, “The structure of atomic and molecular clusters, optimised using classical potentials,” Comput. Phys. Commun. 2006. link Times cited: 17 NOT USED (definite) M. Arroyo and T. Belytschko, “Finite element methods for the non‐linear mechanics of crystalline sheets and nanotubes,” International Journal for Numerical Methods in Engineering. 2004. link Times cited: 157 Abstract: The formulation and finite element implementation of a finit… read moreAbstract: The formulation and finite element implementation of a finite deformation continuum theory for the mechanics of crystalline sheets is described. This theory generalizes standard crystal elasticity to curved monolayer lattices by means of the exponential Cauchy–Born rule. The constitutive model for a two‐dimensional continuum deforming in three dimensions (a surface) is written explicitly in terms of the underlying atomistic model. The resulting hyper‐elastic potential depends on the stretch and the curvature of the surface, as well as on internal elastic variables describing the rearrangements of the crystal within the unit cell. Coarse grained calculations of carbon nanotubes (CNTs) are performed by discretizing this continuum mechanics theory by finite elements. A smooth discrete representation of the surface is required, and subdivision finite elements, proposed for thin‐shell analysis, are used. A detailed set of numerical experiments, in which the continuum/finite element solutions are compared to the corresponding full atomistic calculations of CNTs, involving very large deformations and geometric instabilities, demonstrates the accuracy of the proposed approach. Simulations for large multi‐million systems illustrate the computational savings which can be achieved. Copyright © 2003 John Wiley & Sons, Ltd. read less NOT USED (definite) K. VijayaSekhar, S. G. Acharyya, S. Debroy, V. P. K. Miriyala, and A. Acharyya, “Self-healing phenomena of graphene: potential and applications,” Open Physics. 2016. link Times cited: 17 Abstract: The present study investigates the self healing behavior of … read moreAbstract: The present study investigates the self healing behavior of both pristine and defected single layer graphene using a molecular dynamic simulation. Single layer graphene containing various defects such as preexisting vacancies and differently oriented pre-existing cracks were subjected to uniaxial tensile loading till fracture occurred. Once the load was relaxed, the graphene was found to undergo self healing. It was observed that this self healing behaviour of cracks holds irrespective of the nature of pre-existing defects in the graphene sheet. Cracks of any length were found to heal provided the critical crack opening distance lies within 0.3-0.5 nm for a pristine sheet and also for a sheet with pre-existing defects. Detailed bond length analysis of the graphene sheet was done to understand the mechanism of self healing of graphene. The paper also discusses the immense potential of the self healing phenomena of graphene in the field of graphene based sub-nano sensors for crack sensing. read less NOT USED (definite) “The Tersoff potential for extreme environment,” arXiv: Materials Science. 2018. link Times cited: 0 Abstract: A novel modification of the Tersoff potential for Si is pres… read moreAbstract: A novel modification of the Tersoff potential for Si is presented. The modification improves the transferability of the Tersoff potential for liquid states without the change of original parameters and with no alteration of bulk properties. Also, the modification introduces a correction term for high-pressure states. The modification is meaningful considering that by high energy irradiations local liquid structures and unstable high-pressure manifolds may occur, therefore an interatomic potential must have an acceptable reliability on high thermal/pressure situations to simulate such phenomenon. Particularly, in the modification, a new screening function replaces the radial cutoff function and the bond order function is slightly changed. Also, a repulsive energy function is replaced by a correction function within a specific pair distance. read less NOT USED (definite) “Fast, accurate, and transferable many-body interatomic potentials by genetic programming,” ArXiv. 2019. link Times cited: 0 Abstract: The length and time scales of atomistic simulations are limi… read moreAbstract: The length and time scales of atomistic simulations are limited by the computational cost of the methods used to predict material properties. In recent years there has been great progress in the use of machine learning algorithms to develop fast and accurate interatomic potential models, but it remains a challenge to develop models that generalize well and are fast enough to be used at extreme time and length scales. To address this challenge, we have developed a machine learning algorithm based on genetic programming that is capable of discovering accurate, computationally efficient many-body potential models. The key to our approach is to explore a hypothesis space of models based on fundamental physical principles and select models within this hypothesis space based on their accuracy, speed, and simplicity. The focus on simplicity reduces the risk of overfitting the training data and increases the chances of discovering a model that generalizes well. Our algorithm was validated by rediscovering an exact Lennard Jones potential and a Sutton Chen embedded atom method potential from training data generated using these models. By using training data generated from density functional theory calculations, we found potential models for elemental copper that are simple, as fast as embedded atom models, and capable of accurately predicting properties outside of their training set. Our approach requires relatively small sets of training data, making it possible to generate training data using highly accurate methods at a reasonable computational cost. We present our approach, the forms of the discovered models, and assessments of their transferability, accuracy and speed. read less NOT USED (definite) “Exploring molecular dynamics with forces from n-body potentials using Matlab,” arXiv: Chemical Physics. 2009. link Times cited: 0 Abstract: We present methods for exploratory studies of molecular dyna… read moreAbstract: We present methods for exploratory studies of molecular dynamics using MATLAB. Such methods are not suitable for large scale applications, but they can be used for developement and testing of new types of interactions and other aspects of the simulations, or simply for instruction and education purposes. We also present exploration of forces obtained from 3-body potentials in Molecular Dynamics in this framework. The methods are based on use of matrices and multidimensional arrays for which MATLAB has a set of both linear algebra based as well as element-wise operations. Applications to three-body interactions are the main aspect of this work, but extension to any general form of n-body interactions is also discussed. The methods discussed can be also applied without any change using the latest versions of the package GNU OCTAVE as a replacement for MATLAB. The code examples are listed in some detail, a full package of the MATLAB and OCTAVE codes is available for download. read less NOT USED (definite) “Geometry and Self-stress of Single-Wall Carbon Nanotubes and Graphene via a Discrete Model Based on a 2nd-Generation REBO Potential,” Journal of Elasticity. 2016. link Times cited: 0 NOT USED (definite) “Characterization of Optimal Carbon Nanotubes Under Stretching and Validation of the Cauchy–Born Rule,” Archive for Rational Mechanics and Analysis. 2018. link Times cited: 0 NOT USED (definite) “Ion implantation in nanodiamonds: size effect and energy dependence,” Scientific Reports. 2018. link Times cited: 0 |
 Tersoff_LAMMPS_Tersoff_1988T2_Si__MO_245095684871_004
Tersoff_LAMMPS_Tersoff_1988T2_Si__MO_245095684871_004