Current potential: ThreeBodyBondOrder_WR_WangRockett_1991_Si__MO_081872846741_000
Deep Citation determination:
Does the citing paper use the current potential to generate results displayed in the paper?
Provide us with identifying information so that we know you are not a bot (you will not be added to a mailing list):
Title
A single sentence description.
Three-body bond-order potential for Si by Wang and Rockett (1991) v000
Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
A modified form of the Tersoff empirical interatomic potential for Si is proposed to improve simulation of adatom behaviors on Si surfaces. The modified form of the potential is consistent with local-density-approximation calculations of the surface electronic band structure of Si(001) 2x1. It is demonstrated that the addition of a screened-Morse-potential tail to the bulk Tersoff interaction behavior when tetrahedral coordination is disrupted improves the results significantly. The surface structure is calculated and shown to yield substantial differences with respect to the original potential form. In particular, anomalous abrupt variations in adatom bonding energy are eliminated and the probability of a successful deposition of the adatom on a lattice site is increased.
Species
The supported atomic species.
Si
Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
This Model originally published in [1] is archived in OpenKIM [2-5].
[1] Wang J, Rockett A. Simulating diffusion on Si(001) 2x1 surfaces using a modified interatomic potential. Phys Rev B. 1991;43(15):12571–9. doi:10.1103/PhysRevB.43.12571 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information.
[2] Wang J, Rockett A. Three-body bond-order potential for Si by Wang and Rockett (1991) v000. OpenKIM; 2019. doi:10.25950/6b836b28
[3] Singh R, Karls DS, Wang J, Rockett A. Three-body bond-order (Tersoff-style) potential by Wang and Rockett (1991) v000. OpenKIM; 2019. doi:10.25950/29dfb23c
[4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
34 Citations (18 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (low confidence) U. Wad, A. V. Limaye, and S. Ogale, “Monte Carlo simulated annealing study of mixed Si–Ge and C–C dimer adsorption on a Si (001) 2×1 surface,” Solid State Communications. 2002. link Times cited: 0
USED (low confidence) K. Mae, “Single adatom diffusion in homo- and heteroepitaxies of Si and Ge on (100)-2 × 1 surfaces modeled by MEAM,” Thin Solid Films. 2001. link Times cited: 5
USED (low confidence) U. Wad, A. V. Limaye, M. Gokhale, and S. Ogale, “Comparative study of adatom induced relaxations and energetics for Si, Ge and carbon adsorption on a (2×1) Si(001) surface,” Bulletin of Materials Science. 1999. link Times cited: 1
USED (low confidence) J. Oh and C. Grein, “Epitaxial growth simulations of CdTe(1 1 1)B on Si(0 0 1),” Journal of Crystal Growth. 1998. link Times cited: 15
USED (low confidence) Z. Zhang, F. Wu, and M. Lagally, “AN ATOMISTIC VIEW OF Si(001) HOMOEPITAXY1,” Annual Review of Materials Science. 1997. link Times cited: 25
Abstract: ▪ Abstract Growth of thin films from atoms deposited from th… read more
Abstract: ▪ Abstract Growth of thin films from atoms deposited from the gas phase is intrinsically a non-equilibrium phenomenon dictated by a competition between kinetics and thermodynamics. Precise control of the growth becomes possible only after achieving an understanding of this competition. In this review, we present an atomistic view of the various kinetic aspects in a model system, the epitaxy of Si on Si(001), as revealed by scanning tunneling microscopy and total-energy calculations. Fundamentally important issues investigated include adsorption dynamics and energetics, adatom diffusion, nucleation, sticking, and detachment. We also briefly discuss the inverse process of growth, removal by sputtering or etching. We aim our discussions to an understanding at a quantitative level whenever possible. read less
USED (low confidence) E. Kaxiras, “Review of atomistic simulations of surface diffusion and growth on semiconductors,” Computational Materials Science. 1996. link Times cited: 17
USED (low confidence) V. Milman, S. Pennycook, and D. Jesson, “Ab initio total energy study of adsorption and diffusion on the Si(100) surface,” Thin Solid Films. 1996. link Times cited: 17
USED (low confidence) R. Bogusławski, Q. Zhang, Z. Zhang, C. Roland, and J. Bernholc, “Ab initio studies of single-height Si(001) steps,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 1995. link Times cited: 3
USED (low confidence) S. Liu, Z. Zhang, J. Nørskov, and H. Metiu, “The mobility of Pt atoms and small Pt clusters on Pt(111) and its implications for the early stages of epitaxial growth,” Surface Science. 1994. link Times cited: 35
USED (low confidence) A. Rockett, “A Monte Carlo simulation of the growth of Si(001)2×1: datom/SA step interactions and growth mechanisms,” Surface Science. 1994. link Times cited: 9
USED (low confidence) C. Toh and C. K. Ong, “Adatom-dimer interaction on the Si(001)-2 × 1 surface,” Surface Science. 1994. link Times cited: 3
USED (low confidence) V. M. Bedanov and D. Mukhin, “Anisotropic diffusion and island formation in the MBE growth of Si(001),” Surface Science. 1993. link Times cited: 5
USED (low confidence) C. Toh and C. K. Ong, “Diffusion and interconversion of ’defect’ ad-dimers on the Si(001) 2*1 surface: a molecular statics study,” Journal of Physics: Condensed Matter. 1993. link Times cited: 7
Abstract: The authors use a modified form of the Stillinger-Weber pote… read more
Abstract: The authors use a modified form of the Stillinger-Weber potential to obtain the binding energy and geometry of a number of Si ad-dimer structures on the fully relaxed Si(001) 2*1 surface by canonical Monte Carlo simulation. At low temperatures they show the possible existence of two kinds of 'defect' ad-dimers which may hinder normal 1*2 growth. The mechanisms for both diffusion and interconversion of such dimers are then elucidated by examining their associated minimum-energy paths. read less
USED (low confidence) H. Metiu, Y. Lu, and Z. Zhang, “Epitaxial Growth and the Art of Computer Simulations,” Science. 1992. link Times cited: 102
Abstract: The results of kinetic simulations of the aggregates formed … read more
Abstract: The results of kinetic simulations of the aggregates formed during the deposition of atoms on a semiconductor surface are reviewed. Because the kinetic parameters are poorly known and the accuracy of the existing interatomic potentials has not been sufficiently tested, the goal has been to reach a qualitative understanding of the formation of unusual patterns during growth, such as the segregation of aluminum during the growth of aluminum-gallium-arsenide (AlGaAs) coherent tilted superlattices and the formation of thin, long, and parallel islands during the deposition of Si on an Si(100) surface. Kinetic mechanisms for these phenomena are proposed. read less
USED (low confidence) Z. Zhang, Y. Lu, and H. Metiu, “Pathways for dimer string growth during Si deposition on Si(100)−2 × 1☆,” Surface Science. 1991. link Times cited: 19
USED (low confidence) G. Dereli, “Stillinger-Weber Type Potentials in Monte Carlo Simulation of Amorphous Silicon,” Molecular Simulation. 1992. link Times cited: 16
Abstract: The growth of amorphous silicon on a substrate of a two-laye… read more
Abstract: The growth of amorphous silicon on a substrate of a two-layer slab of crystalline silicon with various surface indices is simulated with Stillinger-Weber type interatomic potentials. The growth is realized by means of a continuum Monte Carlo method and the radial distribution functions are evaluated for various cases. read less
USED (low confidence) M. Grabow, P. Feibelman, G. Gilmer, B. Cooper, and Y. Mo, “Surface Diffusion: Atomistics and Surface Morphology (Summary of MRS Symposium B Panel Discussion),” MRS Proceedings. 1992. link Times cited: 1
Abstract: The paper gives some of the highlights of a panel discussion… read more
Abstract: The paper gives some of the highlights of a panel discussion on surface diffusion held Monday, November 30, 1992 at the Fall MRS Meeting in Boston, Massachusetts. Four invited speakers discussed computer modeling techniques and scanning tunneling microscopy experiments that have been used to provide new understanding of the atomistic processes that occur at surfaces. We present a summary of each of the invited talks, indicate other presentations on surface diffusion in this proceedings, and provide a transcript of the two discussion sessions. read less
USED (low confidence) C. Roland and G. Gilmer, “Growth Properties of the Si(100) Steps: A Molecular Dynamics Study,” MRS Proceedings. 1991. link Times cited: 0
Abstract: We have mapped out the energy surfaces seen by a single sili… read more
Abstract: We have mapped out the energy surfaces seen by a single silicon adatom over the Si(100) surface and Si(100) steps, using Molecular Dynamics methods. This identifies the most likely binding sites as well as the activation energies for diffusion over the terraces and steps. We find that only the 5e step with no rebonded atoms is a good sink for adatoms - the S A , rebonded S b and D b steps are weak sinks. Because of a higher density of binding sites and lower activation energies for surface diffusion along die step edge, we expect mat growth at the S b and D b steps take place much more readily man at the S A step. read less
NOT USED (low confidence) D. Ward, X. W. Zhou, B. M. Wong, F. Doty, and J. Zimmerman, “Accuracy of existing atomic potentials for the CdTe semiconductor compound.,” The Journal of chemical physics. 2011. link Times cited: 35
Abstract: CdTe and CdTe-based Cd(1-x)Zn(x)Te (CZT) alloys are importan… read more
Abstract: CdTe and CdTe-based Cd(1-x)Zn(x)Te (CZT) alloys are important semiconductor compounds that are used in a variety of technologies including solar cells, radiation detectors, and medical imaging devices. Performance of such systems, however, is limited due to the propensity of nano- and micro-scale defects that form during crystal growth and manufacturing processes. Molecular dynamics simulations offer an effective approach to study the formation and interaction of atomic scale defects in these crystals, and provide insight on how to minimize their concentrations. The success of such a modeling effort relies on the accuracy and transferability of the underlying interatomic potential used in simulations. Such a potential must not only predict a correct trend of structures and energies of a variety of elemental and compound lattices, defects, and surfaces but also capture correct melting behavior and should be capable of simulating crystalline growth during vapor deposition as these processes sample a variety of local configurations. In this paper, we perform a detailed evaluation of the performance of two literature potentials for CdTe, one having the Stillinger-Weber form and the other possessing the Tersoff form. We examine simulations of structures and the corresponding energies of a variety of elemental and compound lattices, defects, and surfaces compared to those obtained from ab initio calculations and experiments. We also perform melting temperature calculations and vapor deposition simulations. Our calculations show that the Stillinger-Weber parameterization produces the correct lowest energy structure. This potential, however, is not sufficiently transferrable for defect studies. Origins of the problems of these potentials are discussed and insights leading to the development of a more transferrable potential suitable for molecular dynamics simulations of defects in CdTe crystals are provided. read less
NOT USED (low confidence) L. Pelaz, L. Marqués, M. Aboy, P. López, and I. Santos, “Front-end process modeling in silicon,” The European Physical Journal B. 2009. link Times cited: 32
NOT USED (low confidence) D. Powell, M. Migliorato, and A. Cullis, “Optimized Tersoff potential parameters for tetrahedrally bonded III-V semiconductors,” Physical Review B. 2007. link Times cited: 64
Abstract: We address the issue of accurate parametrization for the Abe… read more
Abstract: We address the issue of accurate parametrization for the Abell-Tersoff empirical potential applied to tetrahedrally bonded semiconductor materials. Empirical potential methods for structural relaxation are widely used for group IV semiconductors while, with few notable exceptions, work on III-V materials has not been extensive. In the case of the Abell-Tersoff potential parametrizations exist only for III-As and III-N, and are designed to correctly predict only a limited number of cohesive and elastic properties. In this work we show how by fitting to a larger set of cohesive and elastic properties calculated from density functional theory, we are able to obtain parameters for III-As, III-N, III-P, and III-Sb zinc blende semiconductors, which can also correctly predict important nonlinear effects in the strain. read less
NOT USED (low confidence) C. Toh and C. K. Ong, “Effects of an anomalous strain accommodating behavior of Si ad-dimers on the nucleation of regular and defect islands,” Surface Science. 1994. link Times cited: 0
NOT USED (low confidence) D. Savage, F. Liu, V. Zielasek, and M. Lagally, “Chapter 2 Fundamental Mechanisms of Film Growth,” Semiconductors and Semimetals. 1998. link Times cited: 9
NOT USED (low confidence) F. Liu and M. Lagally, “Chapter 7 Epitaxial growth of Si on Si(001),” The Chemical Physics of Solid Surfaces. 1997. link Times cited: 2
NOT USED (high confidence) X. W. Zhou et al., “A prediction of dislocation‐free CdTe/CdS photovoltaic multilayers via nano‐patterning and composition grading,” Progress in Photovoltaics: Research and Applications. 2015. link Times cited: 12
Abstract: Defects in multilayered films have long been a performance‐l… read more
NOT USED (high confidence) D. Ward, X. W. Zhou, B. M. Wong, and F. Doty, “A refined parameterization of the analytical Cd–Zn–Te bond-order potential,” Journal of Molecular Modeling. 2013. link Times cited: 13
NOT USED (high confidence) U. Monteverde, M. Migliorato, J. Pal, and D. Powell, “Elastic and vibrational properties of group IV semiconductors in empirical potential modelling,” Journal of Physics: Condensed Matter. 2013. link Times cited: 8
Abstract: We have developed an interatomic potential that with a singl… read more
Abstract: We have developed an interatomic potential that with a single set of parameters is able to accurately describe at the same time the elastic, vibrational and thermodynamics properties of semiconductors. The simultaneous inclusion of radial and angular forces of the interacting atom pairs (short range) together with the influence of the broken crystal symmetry when the atomic arrangement is out of equilibrium (long range) results in correct predictions of all of the phonon dispersion spectrum and mode-Grüneisen parameters of silicon and germanium. The long range interactions are taken into account up to the second nearest neighbours, to correctly influence the elastic and vibrational properties, and therefore represent only a marginal computational cost compared to the full treatment of other proposed potentials. Results of molecular dynamics simulations are compared with those of ab initio calculations, showing that when our proposed potential is used to perform the initial stages of the structural relaxation, a significant reduction of the computational time needed during the geometry optimization of density functional theory simulations is observed. read less
NOT USED (high confidence) C. Henager, F. Gao, S. Hu, G. Lin, E. Bylaska, and N. Zabaras, “Simulating Interface Growth and Defect Generation in CZT – Simulation State of the Art and Known Gaps.” 2012. link Times cited: 1
Abstract: This one-year, study topic project will survey and investiga… read more
Abstract: This one-year, study topic project will survey and investigate the known state-of-the-art of modeling and simulation methods suitable for performing fine-scale, fully 3-D modeling, of the growth of CZT crystals at the melt-solid interface, and correlating physical growth and post-growth conditions with generation and incorporation of defects into the solid CZT crystal. In the course of this study, this project will also identify the critical gaps in our knowledge of modeling and simulation techniques in terms of what would be needed to be developed in order to perform accurate physical simulations of defect generation in melt-grown CZT. The transformational nature of this study will be, for the first time, an investigation of modeling and simulation methods for describing microstructural evolution during crystal growth and the identification of the critical gaps in our knowledge of such methods, which is recognized as having tremendous scientific impacts for future model developments in a wide variety of materials science areas. read less
NOT USED (high confidence) D. Ward, X. W. Zhou, B. M. Wong, J. Zimmerman, and F. Doty, “Analytical bond-order potential for the cadmium telluride binary system.” 2012. link Times cited: 69
Abstract: CdTe and Cd${}_{1\ensuremath{-}x}$Zn${}_{x}$Te are the leadi… read more
Abstract: CdTe and Cd${}_{1\ensuremath{-}x}$Zn${}_{x}$Te are the leading semiconductor compounds for both photovoltaic and radiation detection applications. The performance of these materials is sensitive to the presence of atomic-scale defects in the structures. To enable accurate studies of these defects using modern atomistic simulation technologies, we have developed a high-fidelity analytical bond-order potential for the CdTe system. This potential incorporates primary ($\ensuremath{\sigma}$) and secondary ($\ensuremath{\pi}$) bonding and the valence dependence of the heteroatom interactions. The functional forms of the potential are directly derived from quantum-mechanical tight-binding theory under the condition that the first two and first four levels of the expanded Green's function for the $\ensuremath{\sigma}$- and $\ensuremath{\pi}$-bond orders, respectively, are retained. The potential parameters are optimized using iteration cycles that include first-fitting properties of a variety of elemental and compound configurations (with coordination varying from 1 to 12) including small clusters, bulk lattices, defects, and surfaces, and then checking crystalline growth through vapor deposition simulations. It is demonstrated that this CdTe bond-order potential gives structural and property trends close to those seen in experiments and quantum-mechanical calculations and provides a good description of melting temperature, defect characteristics, and surface reconstructions of the CdTe compound. Most importantly, this potential captures the crystalline growth of the ground-state structures for Cd, Te, and CdTe phases in vapor deposition simulations. read less
NOT USED (high confidence) D. Alfonso and S. Ulloa, “Simulation of hyperthermal deposition of Si and C on SiC surfaces,” Applied Physics Letters. 1999. link Times cited: 4
Abstract: We describe the adsorption dynamics of Si and C atoms at sup… read more
Abstract: We describe the adsorption dynamics of Si and C atoms at supersonic velocities on Si- and C-terminated 6H–SiC(0001) substrates using molecular dynamics simulations. The sticking probabilities of adatoms are found to be very high and not to change substantially with increasing incident kinetic energy. We identify two mechanisms responsible for the high sticking probabilities of the adatoms: (a) efficient transfer of adatom energy to the substrate and (b) strong attractive forces experienced by the impinging adatom over the entire surface. The calculated potential energy surfaces reveal possible binding sites of the adatoms on the substrates. read less
NOT USED (high confidence) E. Kaxiras, “Atomistic aspects of diffusion and growth on the Si and Ge (111) surfaces,” Thin Solid Films. 1995. link Times cited: 18
NOT USED (high confidence) A. P. Smith et al., “Si adatom binding and diffusion on the Si(100) surface: Comparison of ab initio, semiempirical and empirical potential results,” Journal of Chemical Physics. 1995. link Times cited: 51
Abstract: The binding energies and configurations for single Si adatom… read more
Abstract: The binding energies and configurations for single Si adatoms on the Si(100) surface are investigated theoretically. Detailed comparisons between previously published and new calculations using classical potentials, semiempirical formulations, and density functional theory (DFT) are made. The DFT calculations used both the plane‐wave‐pseudopotential approach in a periodic slab geometry and the Gaussian‐orbital based all‐electron approach employing cluster geometries. In the local‐density approximation excellent agreement between the cluster and slab results was obtained. Inclusion of gradient corrections to the exchange‐correlation energy significantly improves absolute binding energies and changes relative energies by as much as 0.3–0.5 eV depending on the particular exchange‐correlation functional used. Binding energies and relative energies obtained using the classical potentials disagree with the gradient corrected DFT energies at about the 0.6–0.9 eV level, and most find qualitatively different local... read less
NOT USED (high confidence) D. Srivastava and B. Garrison, “The dynamics of surface rearrangements in Si adatom diffusion on the Si100(2 × 1) surface,” Journal of Chemical Physics. 1991. link Times cited: 33
Abstract: The Si adatom adsorption and diffusion on the fully relaxed … read more
Abstract: The Si adatom adsorption and diffusion on the fully relaxed Si{100}(2×1) surface is studied by a combination of molecular dynamics simulations with Tersoff’s potential for the Si interactions, a simplified transition state theory of Voter and lattice gas simulations. Six local minima for adsorption are found on the surface and the activation energies between each are determined. The Arrhenius behavior for the macroscopic diffusion is found to be D=5.67×10−3 exp(−0.75 eV/kT) cm2/s. In addition, it is found that the adatom diffusion is strongly anisotropic in nature and the direction of easy diffusion is perpendicular to the dimers (i.e., parallel to the dimer rows) of the original surface. The minimum energy path for the diffusion is found to be activated by the local unreconstruction (dimer opening) of the otherwise fully reconstructed surface. read less
NOT USED (definite) X. W. Zhou, M. E. Foster, R. Jones, P. Yang, H. Fan, and F. Doty, “A modified Stillinger-Weber potential for TlBr and its polymorphic extension,” Journal of Materials Science Research. 2015. link Times cited: 6
Abstract: TlBr is promising for g- and x- radiation detection, but suf… read more
Abstract: TlBr is promising for g- and x- radiation detection, but suffers from rapid performance degradation under the operating external electric fields. To enable molecular dynamics (MD) studies of this degradation, we have developed a Stillinger-Weber type of TlBr interatomic potential. During this process, we have also addressed two problems of wider interests. First, the conventional Stillinger-Weber potential format is only applicable for tetrahedral structures (e.g., diamond-cubic, zinc-blende, or wurtzite). Here we have modified the analytical functions of the Stillinger-Weber potential so that it can now be used for other crystal structures. Second, past modifications of interatomic potentials cannot always be applied by a broad community because any new analytical functions of the potential would require corresponding changes in the molecular dynamics codes. Here we have developed a polymorphic potential model that simultaneously incorporates Stillinger-Weber, Tersoff, embedded-atom method, and any variations (i.e., modified functions) of these potentials. We have implemented this polymorphic model in MD code LAMMPS, and demonstrated that our TlBr potential enables stable MD simulations under external electric fields. read less
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
The letter grade A was assigned because the normalized error in the computation was 8.89250e-10 compared with a machine precision of 2.22045e-16. The letter grade was based on 'score=log10(error/eps)', with ranges A=[0, 7.5], B=(7.5, 10.0], C=(10.0, 12.5], D=(12.5, 15.0), F>15.0. 'A' is the best grade, and 'F' indicates failure.
vc-forces-numerical-derivative
consistency
Forces computed by the model agree with numerical derivatives of the energy; see full description.
The model is C^1 continuous. This means that the model has continuous energy and continuous first derivative.
vc-dimer-continuity-c1
informational
The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description.
Model energy and forces are invariant with respect to rigid-body motion (translation and rotation) for all configurations the model was able to compute.
vc-objectivity
informational
Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description.
All threads give identical results for tested case. Model appears to be thread-safe.
vc-thread-safe
mandatory
The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description.
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
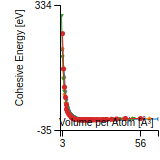
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Given an xyz file corresponding to a finite cluster of atoms, this Test Driver computes the total potential energy and atomic forces on the configuration. The positions are then relaxed using conjugate gradient minimization and the final positions and forces are recorded. These results are primarily of interest for training machine-learning algorithms.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the elastic constants for hcp crystals by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Calculates lattice constant of hexagonal bulk structures at zero temperature and pressure by using simplex minimization to minimize the potential energy.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Given an extended xyz file corresponding to a non-orthogonal periodic box of atoms, use LAMMPS to compute the total potential energy and atomic forces.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
 ThreeBodyBondOrder_WR_WangRockett_1991_Si__MO_081872846741_000
ThreeBodyBondOrder_WR_WangRockett_1991_Si__MO_081872846741_000