Current potential: ThreeBodyCluster_KP_KaxirasPandey_1988_Si__MO_072486242437_000
Deep Citation determination:
Does the citing paper use the current potential to generate results displayed in the paper?
Provide us with identifying information so that we know you are not a bot (you will not be added to a mailing list):
Title
A single sentence description.
Three-body cluster potential for Si by Kaxiras and Pandey (1988) v000
Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
In a critical evaluation, we show that existing classical potentials are not suitable for calculating the energy of realistic atomic processes in Si. We present a new potential which is especially suited to simulate processes in the diamond lattice rather than in high-energy bulk structures of Si. Our potential is based on a very large quantum-mechanical data base. It consists of two- and three-body terms with short-range separable forms, and reproduces accurately the energy surface for atomic exchange in Si. Thus, it is ideally suited for molecular dynamics simulations of atomic processes in Si.
Species
The supported atomic species.
Si
Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
This Model originally published in [1] is archived in OpenKIM [2-5].
[1] Kaxiras E, Pandey KC. New classical potential for accurate simulation of atomic processes in Si. Phys Rev B. 1988;38(17):12736–9. doi:10.1103/PhysRevB.38.12736 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information.
[2] Pandey KC, Kaxiras E. Three-body cluster potential for Si by Kaxiras and Pandey (1988) v000. OpenKIM; 2019. doi:10.25950/b5b6ae37
[3] Vievering J, Karls DS, Kaxiras E, Pandey KC. Three-body cluster potential by Kaxiras and Pandey (1988) v000. OpenKIM; 2019. doi:10.25950/477dbd2b
[4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
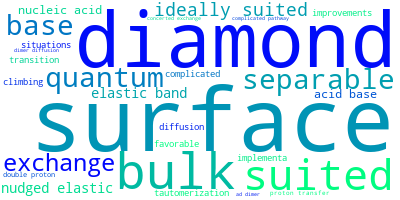
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
36 Citations (4 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) P. Maragakis, S. Andreev, Y. Brumer, D. Reichman, and E. Kaxiras, “Adaptive nudged elastic band approach for transition state calculation,” Journal of Chemical Physics. 2002. link Times cited: 150
Abstract: We present a method for the location of transition states in… read more
Abstract: We present a method for the location of transition states in complicated physical systems. Our algorithm is a variation of the well-established nudged elastic band method and leads to significant improvements in efficiency and accuracy. We assess the applicability of our method by testing it on several systems of practical interest representing a variety of physical situations. At the molecular level, we apply the method to tautomerization processes in nucleic acid bases and the double proton transfer in nucleic acid base pairs. For bulk systems, we considered the concerted exchange mechanism in Si, which is a complicated pathway for defect-free diffusion in the diamond lattice. For surface systems, we considered ad-dimer diffusion mechanisms on Si(100). We incorporated the climbing image extension of the nudged elastic band method and compared it against the original approach on two-dimensional model potential energy surfaces. Based on favorable comparisons with related methods and the general implementa... read less
USED (low confidence) T. Weber and F. Stillinger, “Dynamical branching during fluorination of the dimerized Si(100) surface: A molecular dynamics study,” Journal of Chemical Physics. 1990. link Times cited: 70
Abstract: Collections of classical trajectories have been numerically … read more
Abstract: Collections of classical trajectories have been numerically generated for individual F2 molecules impinging at normal incidence on a Si(100) surface at 0 K dimerized in a p(2×1) pattern. A linear combination of two‐atom and three‐atom interaction functions represents the potential energy. Trajectories fall into four categories: (a) non‐reactive F2 rebound, (b) monofluorination at a surface dangling bond with energetic expulsion into the vacuum of the remaining F atom, (c) difluorination of a pair of dangling bonds, and (d) monofluorination with retention of the second F in a weakly bound Si–F⋅⋅⋅F surface complex. Surface patterns for difluorination, (c), indicate absence of surface diffusion during this mode of chemisorption. Increasing either the translational kinetic energy or the vibrational excitation of the incident F2 appears to enhance its surface reactivity. read less
USED (low confidence) R. Smith, D. E. Harrison, and B. Garrison, “Simulation of keV particle bombardment of covalent materials: An investigation of the yield dependence on incidence angle,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1990. link Times cited: 18
USED (low confidence) J. Spence, H. Kolar, Y. M. Huang, and H. Alexandera, “Dislocation Kink Motion - Ab-Initio Calculations and Atomic Resolution Movies,” MRS Proceedings. 1995. link Times cited: 1
NOT USED (low confidence) L. Pelaz, L. Marqués, M. Aboy, P. López, and I. Santos, “Front-end process modeling in silicon,” The European Physical Journal B. 2009. link Times cited: 32
NOT USED (low confidence) S. Billeter, A. Curioni, D. Fischer, and W. Andreoni, “Ab initio derived augmented Tersoff potential for silicon oxynitride compounds and their interfaces with silicon,” Physical Review B. 2006. link Times cited: 42
Abstract: Coordination-dependent interatomic potentials are proposed f… read more
Abstract: Coordination-dependent interatomic potentials are proposed for silicon oxides and oxynitrides\char22{}also hydrogenated ones\char22{}with a functional form based on the widely used Tersoff silicon potential. They are intended for an accurate sampling of the configurational space of realistic silicon oxynitride systems and their interfaces with silicon, including defects and changes of oxidation states. The parameters, which are given in the text, are obtained by simultaneously mapping forces and energies onto the results of density-functional-theory calculations performed for a set of diverse systems and configurations and a wide composition range. Application to a larger set of systems and configurations shows the transferability of these augmented Tersoff potentials and their validity in predicting bulk lattice parameters, energetics of defect relaxation, and vibrational spectra. read less
NOT USED (low confidence) L. Teik-Cheng, “Relationship and Discrepancies Among Typical Interatomic Potential Functions,” Chinese Physics Letters. 2004. link Times cited: 13
Abstract: We develop a simultaneous relationship among parameters of t… read more
Abstract: We develop a simultaneous relationship among parameters of the generalized version of the Lennard-Jones, Morse, Rydberg and Buckingham pair potentials, and the two-body portion of the Kaxiras–Pandey potential function by introducing a set of scaling factors. These potential functions are selected according to their frequent adoption in condensed matter and molecular computation. In addition to verifying the parametric relations, theoretical plots of these potential curves show that each of these potential functions is unique in terms of their characteristic shape. However, gaps between these potential functions are narrowed for interatomic interactions possessing lower separation energy and longer interatomic equilibrium distance. Finally, comparison with the ab initio results shows that the extended-Rydberg potential energy curve gives the best agreement among the five empirical potential functions, for the specific case of hydrogen molecule. read less
NOT USED (low confidence) P. Gunes, Şi̇mşek S., and S. Erkoç, “a Comparative Study of Empirical Potential Energy Functions,” International Journal of Modern Physics C. 2004. link Times cited: 2
Abstract: A comparative study has been performed for silicon microclus… read more
Abstract: A comparative study has been performed for silicon microclusters, Si3 and Si4, considering fifteen different empirical potential energy functions. It has been found that only two of the empirical potential energy functions give linear structure more stable for Si3, the remaining potential functions give triangular structure as more stable. In the case of Si4 microclusters eight potential functions give open tetrahedral structure as more stable, two functions give perfect tetrahedral as more stable, three functions give square structure as more stable, and two functions give linear structure as more stable. read less
NOT USED (low confidence) C. R. Zacharias, M. R. Lemes, and A. D. Pino, “Combining genetic algorithm and simulated annealing: A molecular geometry optimization study,” Journal of Molecular Structure-theochem. 1998. link Times cited: 42
NOT USED (low confidence) E. Kaxiras, “Review of atomistic simulations of surface diffusion and growth on semiconductors,” Computational Materials Science. 1996. link Times cited: 17
NOT USED (low confidence) A. A. Valuev, A. S. Kaklyugin, and H. E. Norman, “Molecular modelling of the chemical interaction of atoms and molecules with a surface,” Russian Chemical Reviews. 1995. link Times cited: 3
Abstract: The modelling of a surface as an assembly of moving atoms in… read more
Abstract: The modelling of a surface as an assembly of moving atoms interacting with one another and with an incident particle is examined. Both dynamic methods for the modelling of a surface (for short times) and probability methods (for long times) are analysed. The Massey adiabaticity criterion has been used to determine the regions of applicability of the methods of molecular dynamics. Within the framework of probability methods, the chemical bond is described with the aid of Harrison's generalised periodic system of the elements. Together with the general modeling problems, the reconstruction of the surface, physical and chemical sorption, as well as the modification of the surface and of its morphology as a result of the multiple repetition of elementary processes (precipitation, etching, corrosion) are discussed. The bibliography includes 169 references. read less
NOT USED (low confidence) A. Tomasulo and M. V. Ramakrishna, “Quantum confinement effects in semiconductor clusters. II,” Journal of Chemical Physics. 1995. link Times cited: 64
Abstract: The band gaps and spectral shifts of CdS, CdSe, CdTe, AlP, G… read more
Abstract: The band gaps and spectral shifts of CdS, CdSe, CdTe, AlP, GaP, GaAs, and InP semiconductor clusters are calculated from band structure calculations using accurate local and nonlocal empirical pseudopotentials. The effect of spin‐orbit coupling on the band structures is included in the calculations when they are important. The complete set of pseudopotential parameters and full computational details are reported for all these semiconductors. The calculated spectral shifts of zinc‐blende and wurtzite CdS, wurtzite CdSe, zinc‐blende CdTe, and zinc‐blende InP clusters are in good agreement with experiments over a range of cluster sizes. The effect of crystal structure on the band gaps is small in large clusters but becomes important in small clusters. Spin‐orbit coupling splits the valence band into A, B, and C sub‐bands and we identify transitions arising from these sub‐bands in the spectra of both CdSe and CdTe clusters. These results demonstrate that the empirical pseudopotential method yields unique insi... read less
NOT USED (low confidence) M. V. Ramakrishna and J.-W. Pan, “Chemical reactions of silicon clusters,” Journal of Chemical Physics. 1994. link Times cited: 27
Abstract: Smalley and co‐workers discovered that chemisorption reactiv… read more
Abstract: Smalley and co‐workers discovered that chemisorption reactivities of silicon clusters vary over three orders of magnitude as a function of cluster size. In particular, they found that Si33, Si39, and Si45 clusters are least reactive towards various reagents compared to their immediate neighbors in size. We explain these observations based on our stuffed fullerene model. This structural model consists of bulk‐like core of five atoms surrounded by fullerene‐like surface. Reconstruction of the ideal fullerene geometry gives rise to fourfold coordinated crown atoms and π‐bonded dimer pairs. This model yields unique structures for Si33, Si39, and Si45 clusters without any dangling bonds and thus explains their lowest reactivity towards chemisorption of closed shell reagents. This model is also consistent with the experimental finding of Jarrold and Constant that silicon clusters undergo a transition from prolate to spherical shapes at Si27. We justify our model based on an in depth analysis of the differences ... read less
NOT USED (low confidence) Tsumuraya, Ishibashi, and Kusunoki, “Statistics of Voronoi polyhedra in a model silicon glass.,” Physical review. B, Condensed matter. 1993. link Times cited: 14
Abstract: We clarify the local structure in a model silicon glass by u… read more
Abstract: We clarify the local structure in a model silicon glass by use of Voronoi-polyhedron analysis. The glass is produced by molecular dynamics with a Stillinger-Weber potential. The atoms in the glass are nearly distinguishable: there are about 200 types in the system with 216 atoms. The analysis clarifies that the polyhedra are formed by a small number of large-area polygons or by a large number of small-area polygons. This feature is different from those in Lennard-Jones glasses or metallic glasses and is attributed to the loose-packed structure even in the glass state, in which the atoms still have directional bonding read less
NOT USED (low confidence) K. Raghavachari and C. Rohlfing, “Structures of Si10. Are there conventionally bonded low-energy isomers?,” Chemical Physics Letters. 1992. link Times cited: 53
NOT USED (low confidence) W. Niessen and V. G. Zakrzewski, “Complex Electron Affinity Processes in Clusters of S and Si.” 1992. link Times cited: 2
Abstract: Vertical and in some cases adiabatic electron affinities are… read more
Abstract: Vertical and in some cases adiabatic electron affinities are calculated for the clusters S4 and Sin, n = 3 – 7 with large basis sets. The effects of electron correlation are taken into account by CI and Green function techniques. The clusters show a complex behaviour upon electron attachment. The isomers of 84 show normal electron capture processes as well as electron attachment with shake-up. The Si clusters show multiple affinity states resulting from capture of an electron into different orbitals: Si3 C2v has at least three, Si4 D2h four, Si5 D3h two, Si6 D4v one, Si6 C2v three and Si7 D5h two affinity states (vertical processes: Sin + e− ± Sin + hν). For the Sin clusters in some cases shake-up affinities are calculated which are positive. The effects of electron correlation on the electron affinities are extremely large for the Si clusters in particular. In several cases the differences between the adiabatic and vertical electron affinities are very large amounting up to 1.5 eV. read less
NOT USED (low confidence) R. Smith, “A semi-empirical many-body interatomic potential for modelling dynamical processes in gallium arsenide,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1992. link Times cited: 55
NOT USED (low confidence) D. A. Jelski, B. Swift, T. Rantala, X. Xia, and T. George, “Structure of the Si45 cluster,” Journal of Chemical Physics. 1991. link Times cited: 46
Abstract: Six structures for the Si45 cluster are compared using a tig… read more
Abstract: Six structures for the Si45 cluster are compared using a tight‐binding model. Two new structures are proposed which appear to be the low‐energy isomers and to explain much of the existing experimental data. Cluster reactivity is distinguished from cluster stability, and several reasons are discussed which may lead to a reactive or unreactive species. These criteria are applied to the Si45 isomers, and the results are also correlated with experimental data. read less
NOT USED (low confidence) E. Kaxiras, “Structural model for a covalently bonded Si45 cluster,” Chemical Physics Letters. 1989. link Times cited: 45
NOT USED (low confidence) Smith, Harrison, and Garrison, “keV particle bombardment of semiconductors: A molecular-dynamics simulation.,” Physical review. B, Condensed matter. 1989. link Times cited: 126
Abstract: Molecular-dynamics simulations have been performed for the k… read more
Abstract: Molecular-dynamics simulations have been performed for the keV particlebombardment of Si/l brace/110/r brace/ and Si/l brace/100/r brace/ using a many-body potential developed byTersoff. Energy and angle distributions are presented along with an analysis ofthe important ejection mechanisms. We have developed a computer logic that onlyintegrates the equations of motion of the atoms that are struck, thusdecreasing the computer time by a factor of 3 from a completemolecular-dynamics simulation. read less
NOT USED (low confidence) S. Phillpot and D. Wolf, “Grain boundaries in silicon from zero temperature through melting,” Journal of the American Ceramic Society. 1988. link Times cited: 11
Abstract: The results of atomistic simulations of twist grain boundari… read more
Abstract: The results of atomistic simulations of twist grain boundaries in covalent silicon are presented and compared with similar studies in metals. Three aspects are discussed in detail: (i) the zero-temperature structure-energy correlation, (ii) the elastic anomalies near a twist boundary at zero temperature, and (iii) the high-temperature stability of a boundary and its role in thermodynamic melting. In each case striking similarities with studies on metals are found, which are attributed to the important role played by atoms in close proximity. By contrast the covalent nature of bonding in silicon appears to play only a minor role. read less
NOT USED (low confidence) T. Lim, “Obtaining the Varshni potential function using the 2-body Kaxiras-Pandey parameters,” Journal of The Serbian Chemical Society. 2009. link Times cited: 6
Abstract: A generalized version of the Varshni potential function was … read more
Abstract: A generalized version of the Varshni potential function was adopted by Kaxiras and Pandey for describing the 2-body energy portion of multi-body condensed matter. The former's simplicity and resemblance to a Morse potential allows faster computation while the latter's greater number of parameters allows better curve-fitting of spectroscopic data. This paper shows one set of parameter conversion from the Varshni function to the 2-body portion of the Kaxiras-Pandey function, and vice versa two sets of parameter conversion. The latter two sets reveal good correlation between plotted curves, and were verified by the imposition of equal energy curvatures at equilibrium and equal energy integral from equilibrium to dissociation. These parameter conversions can also be attained more easily by equating the product of indices (for short range) and the summation of index reciprocals (for long range). read less
NOT USED (low confidence) J. F. Justo, M. Bazant, E. Kaxiras, V. Bulatov, and S. Yip, “Interatomic Potential for Condensed Phases and Bulk Defects in Silicon,” MRS Proceedings. 1997. link Times cited: 5
NOT USED (low confidence) G. S. Dubey and G. Gumbs, “Molecular Dynamic Simulations of Semiconductor Clusters,” MRS Proceedings. 1996. link Times cited: 0
Abstract: Molecular dynamics simulations are carried out for silicon c… read more
Abstract: Molecular dynamics simulations are carried out for silicon clusters to analyze the effect of heating and cooling on their structural properties. The calculations are based on the Kaxiras and Pandey potential which has been derived from a microscopic calculation with the density functional method. Results for the cluster formation are presented for different rates of cooling and heating. Our simulations clearly show the dependence of the patterns on the cooling and heating rates as well as the way in which these clusters evolve. Our results indicate that the structures are most stable when the rate of loss of thermal energy is slow and the potential energy is lowest. read less
NOT USED (low confidence) Maris and Tamura, “Anharmonic decay and the propagation of phonons in an isotopically pure crystal at low temperatures: Application to dark-matter detection.,” Physical review. B, Condensed matter. 1993. link Times cited: 75
Abstract: We consider the propagation and anharmonic decay of high-ene… read more
Abstract: We consider the propagation and anharmonic decay of high-energy phonons introduced into a perfect dielectric crystal at low temperatures. The phonon-decay rate is calculated for an fcc model with central forces between nearest neighbors. We give an approximate relation between the parameters entering into this model and the experimentally known properties of real crystals. A discussion is given of the range of wave vectors over which slow transverse phonons are stable against anharmonic decay. These results are relevant to the design of experiments to detect dark matter via the study of the phonons excited in a crystal when a dark-matter particle scatters off a nucleus. We discuss the primary phonon production mechanism and the possibility that there is an anisotropy of the phonon flux that is related to the direction in which the nucleus recoils. read less
NOT USED (low confidence) G. Dereli, “Stillinger-Weber Type Potentials in Monte Carlo Simulation of Amorphous Silicon,” Molecular Simulation. 1992. link Times cited: 16
Abstract: The growth of amorphous silicon on a substrate of a two-laye… read more
Abstract: The growth of amorphous silicon on a substrate of a two-layer slab of crystalline silicon with various surface indices is simulated with Stillinger-Weber type interatomic potentials. The growth is realized by means of a continuum Monte Carlo method and the radial distribution functions are evaluated for various cases. read less
NOT USED (low confidence) S. Sarma and K. E. Khor, “Empirical potential approach to the stability and energetics of thin films and surfaces,” Applied Surface Science. 1992. link Times cited: 1
NOT USED (low confidence) J. Chelikowsky and K. M. Glassford, “Classical Potentials for Covalent Solids and Clusters: Application to Silicon and Silicon Dioxide,” MRS Proceedings. 1990. link Times cited: 0
NOT USED (high confidence) S. Surulere, M. Shatalov, and E. Olayiwola, “Optimal interatomic potentials using modified method of least squares: Optimal form of interatomic potentials,” Open Physics. 2023. link Times cited: 0
Abstract: The problem of optimization of interatomic potentials is for… read more
Abstract: The problem of optimization of interatomic potentials is formulated and solved by means of generalization of the Morse, Kaxiras–Pandey, and Rydberg potentials. The interatomic potentials are treated as solutions of some second-order ordinary differential equations which will be classified and analyzed. The most appropriate analytic form of the understudied potentials will be proposed based on a one-dimensional search for the parameter, γ \gamma , which is the power of the interatomic distance, r r . The optimal analytic form will also be proposed for metals such as gold, copper, aluminium, titanium, and the silver–copper alloy. The method of least squares will be used to estimate the potential parameters. Phenomenological potentials such as the classical Rydberg, classical Morse, generalized Morse, Kaxiras–Pandey, and classical Lennard–Jones will be studied, and new potentials based on the combination of some of the aforementioned potentials will also be proposed. Metrics such as the goal function values, will be used to identify which optimal value of the parameter, γ \gamma , is most appropriate to introduce into the preferred interatomic potential for interaction between atoms. read less
NOT USED (high confidence) T. Lim and R. A. Udyavara, “Relations between Varshni and Morse potential energy parameters,” Central European Journal of Physics. 2009. link Times cited: 7
Abstract: A set of relationships between the Morse and Varshni potenti… read more
Abstract: A set of relationships between the Morse and Varshni potential functions for describing covalent bondstretching energy has been developed by imposing equal force constant and equal energy integral. In view of the extensive adoption of Morse function in molecular force fields, this paper suggests two sets of parameter conversions from Varshni to Morse. The parameter conversion based on equal force constant is applicable for small change in bond length, while the parameter conversion based on equal energy integral is more applicable for significant bond-stretching. Plotted results reveal that the Varshni potential function is more suitable for describing hard bonds rather than soft bonds. read less
NOT USED (high confidence) T. Lim, “Application of Extended-Rydberg Parameters for Extracting the 2-Body Portion of Kaxiras–Pandey Function,” Journal of Mathematical Chemistry. 2007. link Times cited: 4
NOT USED (high confidence) T. Lim, “Connection between the 2-body energy of the Kaxiras-Pandey and the Biswas-Hamann potentials,” Czechoslovak Journal of Physics. 2004. link Times cited: 10
NOT USED (high confidence) D. Brenner, O. Shenderova, J. Harrison, S. Stuart, B. Ni, and S. Sinnott, “A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons,” Journal of Physics: Condensed Matter. 2002. link Times cited: 3204
Abstract: A second-generation potential energy function for solid carb… read more
Abstract: A second-generation potential energy function for solid carbon and hydrocarbon molecules that is based on an empirical bond order formalism is presented. This potential allows for covalent bond breaking and forming with associated changes in atomic hybridization within a classical potential, producing a powerful method for modelling complex chemistry in large many-atom systems. This revised potential contains improved analytic functions and an extended database relative to an earlier version (Brenner D W 1990 Phys. Rev. B 42 9458). These lead to a significantly better description of bond energies, lengths, and force constants for hydrocarbon molecules, as well as elastic properties, interstitial defect energies, and surface energies for diamond. read less
NOT USED (high confidence) C. Herrero, “Quantum atomistic simulations of silicon and germanium,” Journal of Materials Research. 2001. link Times cited: 4
Abstract: Quantum atomistic simulations of crystalline silicon and ger… read more
Abstract: Quantum atomistic simulations of crystalline silicon and germanium have been carried out by the path-integral Monte Carlo method. The interatomic interactions were modeled by Stillinger–Weber-type potentials, with parameters adequate to quantum simulations. Quantum zero-point motion together with anharmonicity of the interatomic potential led to a lattice expansion of 7 × 10^−3 Å for both Si and Ge. Results for the equation-of-state (volume versus pressure) and for the thermal expansion coefficient agreed well with experimental results for both materials at T > 100 K and for hydrostatic pressures up to 100 kbar. read less
NOT USED (high confidence) M. V. Ramakrishna and A. Bahel, “Combined tight‐binding and density functional molecular dynamics investigation of Si12 cluster structure,” Journal of Chemical Physics. 1996. link Times cited: 38
Abstract: An extensive search for the lowest energy structure of Si12 … read more
Abstract: An extensive search for the lowest energy structure of Si12 has been carried out using a combination of simulated annealing studies based on tight‐binding molecular dynamics and density functional based Car–Parrinello calculations. This investigation revealed three families of cluster structures that are low in energy. The potential energy surface in the vicinity of these structures has corrugated landscape, similar to that associated with the conformations of long chain polymers and proteins. The lowest energy structure is a hexacapped trigonal prism, which is a continuation of the growth pattern started at Si6, whereby the faces of a trigonal prism or anti prism seed are terminated by adatoms. This finding reveals emergence of a nucleation pattern in the growth of silicon clusters in the 6–13 atom size range. read less
NOT USED (high confidence) T. Lim, “A Relationship Between the 2-body Energy of Kaxiras–Pandey and Pearson–Takai–Halicioglu–Tiller Potential Functions,” Physica Scripta. 2004. link Times cited: 22
Abstract: A parametric relationship between the Pearson–Takai–Haliciog… read more
Abstract: A parametric relationship between the Pearson–Takai–Halicioglu–Tiller (PTHT) and the Kaxiras–Pandey (KP) empirical potential energy functions is developed for the case of 2-body interaction. The need for such relationship arises when preferred parametric data and adopted software correspond to different potential functions. The analytical relationship was obtained by equating the potential functions' derivatives at zeroth, first and second order with respect to the interatomic distance at the equilibrium bond length, followed by comparison of coefficients in the repulsive and attractive terms. Plots of non-dimensional 2-body energy versus the nondimensional interatomic distance verified the analytical relationships developed herein. The discrepancy revealed in theoretical plots suggests that the 2-body PTHT and KP potentials are more suitable for curve-fitting "softer" and "harder" bonds respectively. read less
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
The letter grade A was assigned because the normalized error in the computation was 1.25037e-09 compared with a machine precision of 2.22045e-16. The letter grade was based on 'score=log10(error/eps)', with ranges A=[0, 7.5], B=(7.5, 10.0], C=(10.0, 12.5], D=(12.5, 15.0), F>15.0. 'A' is the best grade, and 'F' indicates failure.
vc-forces-numerical-derivative
consistency
Forces computed by the model agree with numerical derivatives of the energy; see full description.
The model is C^0 continuous. This means that the model has continuous energy, but a discontinuous first derivative.
vc-dimer-continuity-c1
informational
The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description.
Model energy and forces are invariant with respect to rigid-body motion (translation and rotation) for all configurations the model was able to compute.
vc-objectivity
informational
Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description.
All threads give identical results for tested case. Model appears to be thread-safe.
vc-thread-safe
mandatory
The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description.
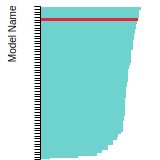
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
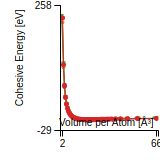
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
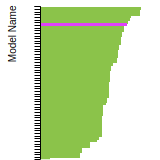
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Given an xyz file corresponding to a finite cluster of atoms, this Test Driver computes the total potential energy and atomic forces on the configuration. The positions are then relaxed using conjugate gradient minimization and the final positions and forces are recorded. These results are primarily of interest for training machine-learning algorithms.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the elastic constants for an arbitrary crystal. A robust computational protocol is used, attempting multiple methods and step sizes to achieve an acceptably low error in numerical differentiation and deviation from material symmetry. The crystal structure is specified using the AFLOW prototype designation as part of the Crystal Genome testing framework. In addition, the distance from the obtained elasticity tensor to the nearest isotropic tensor is computed.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the elastic constants for hcp crystals by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Calculates lattice constant of hexagonal bulk structures at zero temperature and pressure by using simplex minimization to minimize the potential energy.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Given an extended xyz file corresponding to a non-orthogonal periodic box of atoms, use LAMMPS to compute the total potential energy and atomic forces.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
Test
Test Results
Link to Test Results page
Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI)
 ThreeBodyCluster_KP_KaxirasPandey_1988_Si__MO_072486242437_000
ThreeBodyCluster_KP_KaxirasPandey_1988_Si__MO_072486242437_000