 EAM_Dynamo_FarkasCaro_2018_FeNiCrCoCu__MO_803527979660_000
EAM_Dynamo_FarkasCaro_2018_FeNiCrCoCu__MO_803527979660_000
Use this Potential
| Title
A single sentence description.
|
EAM potential (LAMMPS cubic hermite tabulation) for the Fe-Ni-Cr-Co-Cu system developed by Farkas and Caro (2018) v000 |
|---|---|
| Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
|
This is a set of embedded atom method model interatomic potentials capable of representing a high-entropy alloy with five components. The set is developed to resemble but not model precisely face-centered cubic (fcc) near-equiatomic mixtures of Fe–Ni–Cr–Co–Cu. The individual components have atomic sizes deviating up to 3%. With the heats of mixing of all binary equiatomic random fcc mixtures being less than 0.7 kJ/mol and the corresponding value for the quinary being −0.0002 kJ/mol, the potentials predict the random equiatomic fcc quinary mixture to be stable with respect to phase separation or ordering and with respect to bcc and hcp random mixtures. |
| Species
The supported atomic species.
| Co, Cr, Cu, Fe, Ni |
| Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
|
None |
| Content Origin | https://www.ctcms.nist.gov/potentials/entry/2018--Farkas-D-Caro-A--Fe-Ni-Cr-Co-Cu/ |
| Contributor |
I Nikiforov |
| Maintainer |
I Nikiforov |
| Developer |
Diana Farkas A. Caro |
| Published on KIM | 2022 |
| How to Cite |
This Model originally published in [1] is archived in OpenKIM [2-5]. [1] Farkas D, Caro A. Model interatomic potentials and lattice strain in a high-entropy alloy. Journal of Materials Research [Internet]. 2018Oct1;33(19):3218–25. Available from: https://doi.org/10.1557/jmr.2018.245 doi:10.1557/jmr.2018.245 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information. [2] Farkas D, Caro A. EAM potential (LAMMPS cubic hermite tabulation) for the Fe-Ni-Cr-Co-Cu system developed by Farkas and Caro (2018) v000. OpenKIM; 2022. doi:10.25950/8c2ffcf6 [3] Foiles SM, Baskes MI, Daw MS, Plimpton SJ. EAM Model Driver for tabulated potentials with cubic Hermite spline interpolation as used in LAMMPS v005. OpenKIM; 2018. doi:10.25950/68defa36 [4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6 [5] Elliott RS, Tadmor EB. Knowledgebase of Interatomic Models (KIM) Application Programming Interface (API). OpenKIM; 2011. doi:10.25950/ff8f563a Click here to download the above citation in BibTeX format. |
| Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on. The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel. The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied. The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP). Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis. OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm. |
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information. 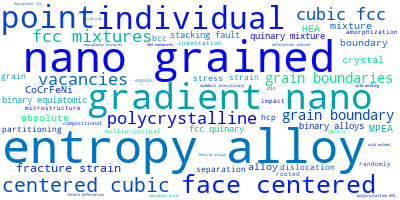
152 Citations (132 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) Y. Tao, W. Cheng, and W. Wang, “Atomistic tensile deformation mechanisms in a CrCoNi medium-entropy alloy with gradient nano-grained structure at cryogenic temperature,” Frontiers in Materials. 2023. link Times cited: 0 Abstract: Large-scale molecular dynamics (MD) simulations have been ut… read more USED (high confidence) O. K. Orhan, M. A. Hendy, and M. Ponga, “Electronic effects on the radiation damage in high-entropy alloys,” Acta Materialia. 2022. link Times cited: 6 USED (high confidence) M. McCarthy et al., “Emergence of near-boundary segregation zones in face-centered cubic multiprincipal element alloys,” Physical Review Materials. 2021. link Times cited: 8 Abstract: Grain boundaries have been shown to dramatically influence th… read more USED (high confidence) A. Manzoor, G. Arora, B. Jerome, N. Linton, B. Norman, and D. Aidhy, “Machine Learning Based Methodology to Predict Point Defect Energies in Multi-Principal Element Alloys,” Frontiers in Materials. 2021. link Times cited: 16 Abstract: Multi-principal element alloys (MPEAs) are a new class of al… read more USED (high confidence) Y. Qi, T. He, and M. Feng, “The effect of Cu and Mn elements on the mechanical properties of single-crystal CoCrFeNi-based high-entropy alloy under nanoindentation,” Journal of Applied Physics. 2021. link Times cited: 18 Abstract: Considering the impact of chemical compositions, the mechani… read more USED (high confidence) Y. Cui, Y. Toku, and Y. Ju, “Nanotwinning and tensile behavior in cold-welded high-entropy-alloy nanowires,” Nanotechnology. 2021. link Times cited: 8 Abstract: Since the fabrication technique for high-entropy alloy (HEA)… read more USED (high confidence) Y. Ma, M. Yang, F. Yuan, and X. Wu, “Deformation induced hcp nano-lamella and its size effect on the strengthening in a CoCrNi medium-entropy alloy,” Journal of Materials Science & Technology. 2021. link Times cited: 38 USED (high confidence) D. Farkas and A. Caro, “Model interatomic potentials for Fe–Ni–Cr–Co–Al high-entropy alloys,” Journal of Materials Research. 2020. link Times cited: 76 Abstract: A set of embedded atom model (EAM) interatomic potentials wa… read more USED (high confidence) M. Cusentino, M. Wood, and R. Dingreville, “Compositional and structural origins of radiation damage mitigation in high-entropy alloys,” Journal of Applied Physics. 2020. link Times cited: 20 Abstract: The ability of high-entropy alloys to resist radiation damag… read more USED (high confidence) A. Makarov, R. Konchakov, Y. P. Mitrofanov, M. Kretova, N. Kobelev, and V. Khonik, “A simple kinetic parameter indicating the origin of the relaxations induced by point(-like) defects in metallic crystals and glasses,” Journal of Physics: Condensed Matter. 2020. link Times cited: 2 Abstract: Computer simulation shows that an increase of the volume V d… read more USED (high confidence) D. Utt et al., “The origin of jerky dislocation motion in high-entropy alloys,” Nature Communications. 2020. link Times cited: 20 USED (high confidence) F. Yuan, W. Cheng, S.-Q. Zhang, X. Liu, and X. Wu, “Atomistic simulations of tensile deformation in a CrCoNi medium-entropy alloy with heterogeneous grain structures,” Materialia. 2020. link Times cited: 35 USED (low confidence) H. Kristoffersen, J. K. Pedersen, and J. Rossmeisl, “Role of vacancies in structural thermalization of binary and high-entropy alloys,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) D. T. H. Hue et al., “Unusual plastic deformation mechanism in copper-high entropy alloy nanocomposites: Insights from molecular dynamics simulations,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) H. Dong et al., “TRIP-TWIP effect in deformation mechanisms of Co–Cr–Ni medium entropy via molecular dynamics simulations,” Journal of Materials Research and Technology. 2023. link Times cited: 0 USED (low confidence) W. Huang, D. Farkas, and X.-M. Bai, “High-Throughput Machine Learning - Kinetic Monte Carlo Framework for Diffusion Studies in Equiatomic and Non-equiatomic FeNiCrCoCu High-Entropy Alloys,” Materialia. 2023. link Times cited: 0 USED (low confidence) J. Wang, N. Lu, and C.-H. Zhang, “Study of high temperature mechanical properties and irradiation behavior of Zr3Al alloy by Chen’s Lattice Inversion EAM,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) D.-K. Nguyen, T.-H. Fang, Y.-R. Cai, and C.-C. Huang, “Machining mechanism of polycrystalline nickel-based alloy under ultrasonic elliptical vibration-assisted cutting,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 0 Abstract: This work investigates the machining mechanism and deformati… read more USED (low confidence) Y. Wang, Y. Wang, F. Tan, J. Li, B. Liu, and Q. Fang, “Unrevealing Grain Boundary Mobility in the Precipitate Hardening High Entropy Alloys,” SSRN Electronic Journal. 2023. link Times cited: 0 USED (low confidence) S. Yan, Y. Nie, and A. Paradowska, “Effect of specimen size and crystallographic orientation on the nano/microscale mechanical properties and deformation behavior of CrCoNi medium-entropy alloy,” Materials & Design. 2023. link Times cited: 0 USED (low confidence) S. Yoon, Y. Kimura, S. Gu, Y. Toku, Y. Ju, and Y. Cui, “Thermal stress-assisted formation of submicron pillars from a thin film of CoCrCuFeNi high entropy alloy: experiments and simulations,” RSC Advances. 2023. link Times cited: 0 Abstract: In this work, for the first time, the thermal stress-assiste… read more USED (low confidence) Y.-H. Wu, R. Huang, and Y. Wen, “Crystallization kinetics, microstructure evolution, and mechanical responses of Cr-Co alloys,” Modelling and Simulation in Materials Science and Engineering. 2023. link Times cited: 0 Abstract: Understanding the crystallization kinetics of Cr-Co alloys a… read more USED (low confidence) Y. Wang, Y. Qi, T. He, and M. Feng, “Grain refinement induced by grain boundary segregation in FeNiCrCoCu high-entropy alloys using molecular dynamics simulation of nanoindentation,” Materials Chemistry and Physics. 2023. link Times cited: 0 USED (low confidence) S. Singh and A. Parashar, “Effect of Frenkel pairs on the tensile and shock compression strength of multi-elemental alloys,” Physica Scripta. 2023. link Times cited: 0 Abstract: In this article, molecular dynamics simulations were perform… read more USED (low confidence) X. Liu, L. Chang, T. Ma, and C. Zhou, “Molecular dynamics simulation of tension and compression deformation behavior in CoCrCuFeNi high-entropy alloy: Effects of temperature and orientation,” Materials Today Communications. 2023. link Times cited: 3 USED (low confidence) Z. Zhang, Q. Huang, and H. Zhou, “High-entropy alloy nanocrystals with low-angle grain boundary for superb plastic deformability and recoverability,” International Journal of Plasticity. 2023. link Times cited: 3 USED (low confidence) J. Li, Z. Zheng, X. Zang, Y. Sun, L. Dong, and J. Liu, “Study on micro-crack propagation mechanism in different positions of single crystal titanium at nanoscale,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) R. Li, L. Guo, Y. Liu, Q.-H. Xu, and Q. Peng, “Irradiation Resistance of CoCrCuFeNi High Entropy Alloy under Successive Bombardment,” Acta Metallurgica Sinica (English Letters). 2023. link Times cited: 0 USED (low confidence) O. Elgack, B. Almomani, J. Syarif, M. Elazab, M. Irshaid, and M. Al-Shabi, “Molecular dynamics simulation and machine learning-based analysis for predicting tensile properties of high-entropy FeNiCrCoCu alloys,” Journal of Materials Research and Technology. 2023. link Times cited: 3 USED (low confidence) B. Zhu, D. Zhao, Y.-hong Niu, Z. Zhang, and H. Zhao, “Atomic study on the deformation behavior of nanotwinned CoCrCuFeNi high entropy alloy during nanoscratching,” Journal of Materials Research and Technology. 2023. link Times cited: 1 USED (low confidence) A. Liang, D. C. Goodelman, A. Hodge, D. Farkas, and P. S. Branicio, “CoFeNiTi and CrFeNiTi high entropy alloy thin films microstructure formation,” Acta Materialia. 2023. link Times cited: 1 USED (low confidence) H. Xie, Z. Ma, W. Zhang, H. Zhao, and L. Ren, “Graphene enables equiatomic FeNiCrCoCu high-entropy alloy with improved TWIP and TRIP effects under shock compression,” Journal of Materials Science & Technology. 2023. link Times cited: 1 USED (low confidence) X. Wang, H. Liu, J. Mo, S. Wang, H. Li, and W. Yang, “Atomic-level understanding of enhanced mechanical properties in FeNiCrCoCu high-entropy alloy,” Materials Today Communications. 2023. link Times cited: 1 USED (low confidence) I. A. Alhafez, O. Deluigi, D. Tramontina, C. Ruestes, E. Bringa, and H. Urbassek, “Simulated nanoindentation into single-phase fcc Fe\documentclass[12pt]minimal \usepackageamsmath \usepackagewasysym \usepackageamsfonts \usepackageamssymb \usepackageamsbsy \usepackagemathrsfs \usepackageupgreek \setlength\oddsidemargin-69pt \begindocument$_x$\enddocument,” Scientific Reports. 2023. link Times cited: 1 USED (low confidence) Y. Sun, H. Xin, W. Song, D. Zhao, and S. Ma, “Molecular dynamics simulation of nanoindentation on nano-twinned FeCoCrNiCu high entropy alloy,” Molecular Simulation. 2023. link Times cited: 0 Abstract: ABSTRACT Twin boundary (TB) plays an important role in the d… read more USED (low confidence) R. Li, Y. Li, Y. Liu, and Q. Peng, “The effect of grain boundary on irradiation resistance of CoCrCuFeNi high entropy alloy,” Computational Materials Science. 2023. link Times cited: 3 USED (low confidence) S. Hayakawa, J. Li, J. Bommidi, and H. Xu, “Compositional effects on dislocation properties in NiCo and NiFe alloys using atomistic simulations,” Computational Materials Science. 2023. link Times cited: 3 USED (low confidence) O. Deluigi et al., “Plastic behavior of a nanoporous high-entropy alloy under compression,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) J. Qiu, Z. Xu, J. Song, C. Hu, L. Miao, and X. He, “Molecular dynamics simulation of a new inhomogeneous concentration distribution model based on frictional behavior of FeNiCrCoCu high-entropy alloy,” Materials Today Communications. 2023. link Times cited: 2 USED (low confidence) A. Makarov et al., “Defect-induced ordering and disordering in metallic glasses,” Intermetallics. 2023. link Times cited: 1 USED (low confidence) A. Seoane, D. Farkas, and X. Bai, “Molecular dynamics studies of sluggish grain boundary diffusion in equiatomic FeNiCrCoCu high-entropy alloy,” Journal of Materials Science. 2023. link Times cited: 2 USED (low confidence) T. Shu, N. Hu, F. Liu, and G. Cheng, “Nanoparticles induced intragranular and dislocation substructures in powder bed fusion for strengthening of high-entropy-alloy,” Materials Science and Engineering: A. 2023. link Times cited: 2 USED (low confidence) M. Duan, T. Fu, Y. Pan, X. Liu, and X. Peng, “Stacking fault strengthening in CoCrFeMnNi high-entropy alloy,” Applied Physics Letters. 2023. link Times cited: 1 Abstract: The effects of stacking faults (SFs) on the mechanical prope… read more USED (low confidence) G. Lei, Y. Zhang, H. Gao, X. Cui, and H. Yu, “Nano-tribological behavior of CuCoCrFeNi high-entropy alloys at cryogenic temperature: A molecular dynamics study,” Journal of Applied Physics. 2023. link Times cited: 5 Abstract: High-entropy alloys exhibit great potential for cryogenic ap… read more USED (low confidence) Q. Li, C. Jiang, and Y. Du, “Molecular dynamics study on dynamic mechanical behaviour of FeCoCrCuNi high entropy alloy,” Materials Technology. 2023. link Times cited: 1 Abstract: ABSTRACT In this paper, molecular dynamics simulation method… read more USED (low confidence) Y. Xiong, J. Zhang, S. Ma, S. Huang, B. Xu, and S. Zhao, “Multiscale modeling of irradiation-induced defect evolution in BCC multi principal element alloys,” Journal of Alloys and Compounds. 2023. link Times cited: 1 USED (low confidence) J. Ren et al., “Atomistic Simulation of Tribology Behaviors Of Ti-Based Feconiti High Entropy Alloy Coating During Nanoscratching,” SSRN Electronic Journal. 2023. link Times cited: 1 USED (low confidence) Y. Tong et al., “Real-time atomic deformation behavior of nano CoCrCuFeNi high-entropy alloy,” Transactions of Nonferrous Metals Society of China. 2023. link Times cited: 2 USED (low confidence) A. M. Kazakov, A. V. Yakhin, E. Z. Karimov, R. Babicheva, A. Kistanov, and E. Korznikova, “Effect of Segregation on Deformation Behaviour of Nanoscale CoCrCuFeNi High-Entropy Alloy,” Applied Sciences. 2023. link Times cited: 3 Abstract: A molecular dynamics (MD) simulation method is used to inves… read more USED (low confidence) H. Joress et al., “Why is EXAFS for complex concentrated alloys so hard? Challenges and opportunities for measuring ordering with X-ray absorption spectroscopy,” Matter. 2023. link Times cited: 2 USED (low confidence) J. Yu et al., “Combining Machine Learning and Molecular Dynamics to Predict Mechanical Properties and Microstructural Evolution of FeNiCrCoCu High-Entropy Alloys,” Nanomaterials. 2023. link Times cited: 0 Abstract: Compared with traditional alloys, high-entropy alloys have b… read more USED (low confidence) Z. Zhang et al., “Atomistic understanding towards twin boundary on the effect of crack propagation in FeNiCrCoCu high-entropy alloy and Ni,” Materials Today Communications. 2023. link Times cited: 1 USED (low confidence) L. Li et al., “Shock Compression and Spall Damage of Dendritic High-Entropy Alloy Cocrfenicu,” SSRN Electronic Journal. 2023. link Times cited: 2 USED (low confidence) S. Singh, S. S. Sharma, A. Chaurasia, and A. Parashar, “Atomistic insights to study the effects of nano-voids on shock compression behaviour of single crystal and bicrystal nickel,” Materials Today: Proceedings. 2023. link Times cited: 2 USED (low confidence) F. Tan, L. Li, J. Li, B. Liu, P. Liaw, and Q. Fang, “Multiscale modelling of irradiation damage behavior in high entropy alloys,” Advanced Powder Materials. 2023. link Times cited: 3 USED (low confidence) Z. Lou, Y. Yan, C. Li, and Y. Geng, “Deformation behavior of high-entropy alloys under dual-tip probe scratching,” Journal of Alloys and Compounds. 2023. link Times cited: 1 USED (low confidence) T. He, Y. Qi, Y. Ji, and M. Feng, “Grain boundary segregation-induced strengthening-weakening transition and its ideal maximum strength in nanopolycrystalline FeNiCrCoCu high-entropy alloys,” International Journal of Mechanical Sciences. 2023. link Times cited: 14 USED (low confidence) K. Kuptsov et al., “High-entropy Fe-Cr-Ni-Co-(Cu) coatings produced by vacuum electro-spark deposition for marine and coastal applications,” Surface and Coatings Technology. 2023. link Times cited: 5 USED (low confidence) N. Lopanitsyna, G. Fraux, M. A. Springer, S. De, and M. Ceriotti, “Modeling high-entropy transition metal alloys with alchemical compression,” Physical Review Materials. 2022. link Times cited: 7 Abstract: Alloys composed of several elements in roughly equimolar com… read more USED (low confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (low confidence) R. Babicheva et al., “Effect of short-range ordering and grain boundary segregation on shear deformation of CoCrFeNi high-entropy alloys with Al addition,” Computational Materials Science. 2022. link Times cited: 13 USED (low confidence) D. Yan, Z. Yun, and J. Li, “Twin boundary spacing and loading direction dependent tensile deformation of nano-twinned Al10(CrCoFeNi)90 high-entropy alloy: An atomic study,” International Journal of Mechanical Sciences. 2022. link Times cited: 7 USED (low confidence) S. Mishra, K. G. Vishnu, and A. Strachan, “Comparing the accuracy of melting temperature prediction methods for high entropy alloys,” Journal of Applied Physics. 2022. link Times cited: 1 Abstract: Refractory complex concentrated alloys (RCCAs) are a relativ… read more USED (low confidence) O. Deluigi, F. Valencia, N. Amigo, F. Aquistapace, R. González, and E. Bringa, “Atomistic simulations of tensile deformation of a nanoporous high-entropy alloy,” Journal of Materials Science. 2022. link Times cited: 4 USED (low confidence) C. Cheng, S. Ma, and S. Wang, “The Role of Phonon Anharmonicity on the Structural Stability and Phonon Heat Transport of Crfeconicux High-Entropy Alloys at Finite Temperatures,” SSRN Electronic Journal. 2022. link Times cited: 0 USED (low confidence) D.-Q. Doan, A.-S. Tran, and N.-C. Vu, “Grain and twin boundaries dependent mechanical behavior of FeCoCrNiCu high-entropy alloy,” Materials Today Communications. 2022. link Times cited: 4 USED (low confidence) H. Li, Y. Li, Y. Nie, and S. Yan, “Small-scale mechanical properties and deformation mechanisms of a laser welded CrCoNi medium-entropy alloy joint,” Materials Science and Engineering: A. 2022. link Times cited: 2 USED (low confidence) T. Gao et al., “Molecular dynamics simulations of tensile response for FeNiCrCoCu high-entropy alloy with voids,” International Journal of Mechanical Sciences. 2022. link Times cited: 22 USED (low confidence) S. Liu, G.-zhu Feng, L. Xiao, Y. Guan, and W. Song, “Shock-induced dynamic response in single and nanocrystalline high-entropy alloy FeNiCrCoCu,” International Journal of Mechanical Sciences. 2022. link Times cited: 14 USED (low confidence) H. Xie, Z. Ma, W. Zhang, H. Zhao, and L. Ren, “Phase transition in shock compressed high-entropy alloy FeNiCrCoCu,” International Journal of Mechanical Sciences. 2022. link Times cited: 15 USED (low confidence) D. Hua et al., “Formation mechanism of hierarchical twins in the CoCrNi medium entropy alloy,” Journal of Materials Science & Technology. 2022. link Times cited: 17 USED (low confidence) S. Singh and A. Parashar, “Shock resistance capability of multi-principal elemental alloys as a function of lattice distortion and grain size,” Journal of Applied Physics. 2022. link Times cited: 7 Abstract: This article aims to study the shock resistance capability o… read more USED (low confidence) S. Hayakawa and H. Xu, “Interaction between a dislocation and nanotwin–hcp lamella in Ni-based concentrated alloys from atomistic simulations,” Scripta Materialia. 2022. link Times cited: 2 USED (low confidence) D.-Q. Doan, “Effects of crystal orientation and twin boundary distance on mechanical properties of FeNiCrCoCu high-entropy alloy under nanoindentation,” Materials Chemistry and Physics. 2022. link Times cited: 9 USED (low confidence) G. Li et al., “Stabilizing defective coherent twin boundaries for strong and stable nanocrystalline nanotwinned Cu,” Acta Materialia. 2022. link Times cited: 5 USED (low confidence) S. K. Singh and A. Parashar, “Effect of lattice distortion and grain size on the crack tip behaviour in Co-Cr-Cu-Fe-Ni under mode-I and mode-II loading,” Engineering Fracture Mechanics. 2022. link Times cited: 9 USED (low confidence) H. Xie, Z. Ma, W. Zhang, H. Zhao, and L. Ren, “Probing the atomic-scale origins of anti-friction and wear-resisting in graphene-coated high-entropy alloys,” Materials & Design. 2022. link Times cited: 7 USED (low confidence) D. Trong, V. C. Long, and Ș. Ţălu, “Effects of Number of Atoms and Doping Concentration on the Structure, Phase Transition, and Crystallization Process of Fe1-x-yNixCoy Alloy: A Molecular Dynamic Study,” Applied Sciences. 2022. link Times cited: 6 Abstract: In this study, molecular dynamics simulations are employed t… read more USED (low confidence) R. Salloom, M. Baskes, and S. G. Srinivasan, “Atomic level simulations of the phase stability and stacking fault energy of FeCoCrMnSi high entropy alloy,” Modelling and Simulation in Materials Science and Engineering. 2022. link Times cited: 4 Abstract: High entropy alloys (HEAs) have many promising properties be… read more USED (low confidence) D.-Q. Doan and T. Fang, “Effect of vibration parameters on the material removal characteristics of high-entropy alloy in scratching,” International Journal of Mechanical Sciences. 2022. link Times cited: 8 USED (low confidence) H. Xie, Z. Ma, W. Zhang, H. Zhao, and L. Ren, “Strengthening effect of high-entropy alloys endowed by monolayer graphene,” Materials Today Physics. 2022. link Times cited: 7 USED (low confidence) J. Chen, Y. Ding, X. Zhang, Y. Gao, and Y. Ma, “Intrinsic stacking fault energy and mechanism for deformation twin formation of solid solution matrix in Ni-based superalloys,” Vacuum. 2022. link Times cited: 5 USED (low confidence) S. Chen, T. Wang, X. Li, Y. Cheng, G. Zhang, and H. Gao, “Short-Range Ordering and its Impact on Thermodynamic Property of High-Entropy Alloys,” Acta Materialia. 2022. link Times cited: 15 USED (low confidence) T.-X. Bui, T. Fang, and C.-I. Lee, “Deformation and machining mechanism of nanocrystalline NiCoCrFe high entropy alloys,” Journal of Alloys and Compounds. 2022. link Times cited: 8 USED (low confidence) Y. Luo, Z.-an Tian, Q. Zheng, L. Hu, and K. Dong, “Crystallization insights revealed by simulation solidification study of Fe63Ni33Co4 alloy melt at subcritical cooling rate,” Journal of Non-Crystalline Solids. 2022. link Times cited: 1 USED (low confidence) S. K. Singh and A. Parashar, “Effect of lattice distortion and nanovoids on the shock compression behavior of (Co-Cr-Cu-Fe-Ni) high entropy alloy,” Computational Materials Science. 2022. link Times cited: 22 USED (low confidence) X. Feng, K. Cao, X. Huang, G. Li, and Y. Lu, “Nanolayered CoCrFeNi/Graphene Composites with High Strength and Crack Resistance,” Nanomaterials. 2022. link Times cited: 4 Abstract: Emerging high-entropy alloy (HEA) films achieve high strengt… read more USED (low confidence) H.-Y. Xie, Z. Ma, H. Zhao, and L. Ren, “Atomic perspective of contact protection in graphene-coated high-entropy films,” Tribology International. 2022. link Times cited: 10 USED (low confidence) I. Mastorakos and J. Ma, “Composition influence on edge dislocation mobility in an FCC high-entropy alloy,” MRS Advances. 2022. link Times cited: 0 Abstract: Molecular dynamics simulations were conducted to investigate… read more USED (low confidence) M. Muralles, J. Oh, and Z. Chen, “Molecular Dynamics Study of Feco Phase Transitions and Thermal Properties Based on an Improved 2nn Meam Potential,” SSRN Electronic Journal. 2022. link Times cited: 3 USED (low confidence) M. Grant et al., “Integrating atomistic simulations and machine learning to design multi-principal element alloys with superior elastic modulus,” Journal of Materials Research. 2022. link Times cited: 6 Abstract: Multi-principal element, high entropy alloys (HEAs) are an e… read more USED (low confidence) A. Seoane, D. Farkas, and X. Bai, “Influence of compositional complexity on species diffusion behavior in high-entropy solid-solution alloys,” Journal of Materials Research. 2022. link Times cited: 5 Abstract: Detailed comparative molecular dynamics simulations of the d… read more USED (low confidence) C. Ruestes and D. Farkas, “Dislocation emission and propagation under a nano-indenter in a model high entropy alloy,” Computational Materials Science. 2022. link Times cited: 19 USED (low confidence) Q. Liang, S. Weng, T. Fu, S. Hu, and X. Peng, “Dislocation reaction-based formation mechanism of stacking fault tetrahedra in FCC high-entropy alloy,” Materials Chemistry and Physics. 2022. link Times cited: 11 USED (low confidence) S. K. Singh, A. Chaurasia, and A. Parashar, “Atomistic simulations to study shock and ultrashort pulse response of high entropy alloy,” Materials Today: Proceedings. 2022. link Times cited: 6 USED (low confidence) J. Li, X. Yang, P. Wang, and Q. An, “Healing stacking fault tetrahedron in NiFe solid solution alloys through grain boundary migration,” Journal of Nuclear Materials. 2022. link Times cited: 2 USED (low confidence) Y. Gao et al., “Growth Pattern of Homogeneous and Heterogeneous Nucleation in High-Entropy FeNiCrCoCu Alloys,” Crystal Growth & Design. 2022. link Times cited: 5 USED (low confidence) T. Shen, H. Y. Song, M. An, and Y. L. Li, “Uncovering strengthening and softening mechanisms of nano-twinned CoCrFeCuNi high entropy alloys by molecular dynamics simulation,” Journal of Applied Physics. 2022. link Times cited: 6 Abstract: High-entropy alloys (HEAs) break the design concept of tradi… read more USED (low confidence) J. Li, X. Yang, P. Wang, and Q. An, “Dynamic interactions between low-angle grain boundary and stacking fault tetrahedron in Ni-Fe solid solution alloys,” Journal of Alloys and Compounds. 2022. link Times cited: 7 USED (low confidence) R. Konchakov, A. Makarov, A. Aronin, N. Kobelev, and V. Khonik, “Elastic Dipoles in Crystal and Glassy Aluminum and High-Entropy Fe20Ni20Cr20Co20Cu20 Alloy,” JETP Letters. 2022. link Times cited: 1 USED (low confidence) M. Abere et al., “A predictive analytical model of thermal conductivity for aluminum/transition metal high-entropy alloys,” Scripta Materialia. 2022. link Times cited: 6 USED (low confidence) Z. Bai, T. Fu, S. Weng, Y. Zhao, and X. Peng, “Deformation characteristics of nanolayered dual-phase CrCoNi medium-entropy alloy nanowires,” Materials Today Communications. 2022. link Times cited: 1 USED (low confidence) S. Yan, H. Zhou, Z. Zhu, Y. Fu, and J. Tian, “High strength-ductility synergy in a laser welded dissimilar joint of CrCoNi medium-entropy alloy and stainless steel,” Materials Science and Engineering: A. 2022. link Times cited: 11 USED (low confidence) Y. Cui, Z. Chen, S. Gu, W. Yang, and Y. Ju, “Investigating size dependence in nanovoid-embedded high-entropy-alloy films under biaxial tension,” Archive of Applied Mechanics. 2022. link Times cited: 4 USED (low confidence) T. Fu et al., “Twin boundary migration and reactions with stacking fault tetrahedron in Cu and CoCrCuFeNi high-entropy alloy,” Journal of Materials Research and Technology. 2022. link Times cited: 10 USED (low confidence) J. Peng et al., “Vacancy dependent mechanical behaviors of high-entropy alloy,” International Journal of Mechanical Sciences. 2022. link Times cited: 19 USED (low confidence) J. Li et al., “Study on wear behavior of FeNiCrCoCu high entropy alloy coating on Cu substrate based on molecular dynamics,” Applied Surface Science. 2021. link Times cited: 48 USED (low confidence) Y. Liu, R. Li, and Q. Peng, “The preexisting edge dislocations as recombination center of point defects enhancing irradiation tolerance in CoCrCuFeNi high entropy alloy,” Materialia. 2021. link Times cited: 7 USED (low confidence) C. Zhang et al., “Laser powder bed fusion of high-entropy alloy particle-reinforced stainless steel with enhanced strength, ductility, and corrosion resistance,” Materials & Design. 2021. link Times cited: 17 USED (low confidence) H. Liu, C. Peng, X. Li, S. Wang, and L. Wang, “The Effect of Phase Separation on the Mechanical Behavior of the Co–Cr–Cu–Fe–Ni High-Entropy Alloy,” Materials. 2021. link Times cited: 4 Abstract: Phase separation phenomena in high-entropy alloys (HEAs) hav… read more USED (low confidence) Y. Shen and D. Spearot, “Mobility of dislocations in FeNiCrCoCu high entropy alloys,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 18 Abstract: Dislocations in high entropy alloys (HEAs) are wavy and have… read more USED (low confidence) D. E. Farache, J. C. Verduzco, Z. D. McClure, S. Desai, and A. Strachan, “Active learning and molecular dynamics simulations to find high melting temperature alloys,” Computational Materials Science. 2021. link Times cited: 9 USED (low confidence) S. Yuan et al., “Gradient nanotwinned CrCoNi medium-entropy alloy with strength-ductility synergy,” Scripta Materialia. 2021. link Times cited: 31 USED (low confidence) L. Zhang, K. Qian, B. Schuller, C. Lu, Y. Shibuta, and X. Huang, “Prediction on Mechanical Properties of Non-Equiatomic High-Entropy Alloy by Atomistic Simulation and Machine Learning,” Metals. 2021. link Times cited: 21 Abstract: High-entropy alloys (HEAs) with multiple constituent element… read more USED (low confidence) J. Li, X. Yang, P. Wang, and Q. An, “Concentration-temperature superposition principle for grain boundary migration in Ni(Cu) bicrystals,” Materials today communications. 2021. link Times cited: 6 USED (low confidence) O. Deluigi, R. Pasianot, F. J. Valencia, A. Caro, D. Farkas, and E. Bringa, “Simulations of primary damage in a High Entropy Alloy: Probing enhanced radiation resistance,” Acta Materialia. 2021. link Times cited: 81 USED (low confidence) S. Hayakawa and H. Xu, “Temperature-dependent mechanisms of dislocation–twin boundary interactions in Ni-based equiatomic alloys,” Acta Materialia. 2021. link Times cited: 18 USED (low confidence) D. Farkas, “Deformation behavior of a model high entropy alloy from atomistic simulations,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2021. link Times cited: 13 USED (low confidence) H. Wang et al., “Deformation-induced crystalline-to-amorphous phase transformation in a CrMnFeCoNi high-entropy alloy,” Science Advances. 2021. link Times cited: 77 Abstract: In situ TEM experiments reveal the crystalline-to-amorphous … read more USED (low confidence) M. Wang, S. Guo, X. Lin, and W. Huang, “Research on the nucleation and growth of high-entropy alloy,” Materials Letters. 2021. link Times cited: 6 USED (low confidence) X. Jinkun, H. Tan, J. Chen, A. Martini, and C. Zhang, “Effect of carbon content on microstructure, hardness and wear resistance of CoCrFeMnNiCx high-entropy alloys,” Journal of Alloys and Compounds. 2020. link Times cited: 74 USED (low confidence) H. Feng et al., “A molecular dynamics investigation into deformation mechanism of nanotwinned Cu/high entropy alloy FeCoCrNi nanolaminates,” Surface & Coatings Technology. 2020. link Times cited: 8 USED (low confidence) Z. Zhu, S. Yan, H. Chen, and G. Gou, “Unprecedented combination of strength and ductility in laser welded NiCoCr medium entropy alloy joints,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2020. link Times cited: 12 USED (low confidence) L. Zhang and Y. Shibuta, “Inverse Hall-Petch relationship of high-entropy alloy by atomistic simulation,” Materials Letters. 2020. link Times cited: 52 USED (low confidence) J. Liu, “Molecular dynamic study of temperature dependence of mechanical properties and plastic inception of CoCrCuFeNi high-entropy alloy,” Physics Letters A. 2020. link Times cited: 36 USED (low confidence) S. Yan, Q. H. Qin, and Z. Zhong, “On the real-time atomistic deformation of nano twinned CrCoFeNi high entropy alloy,” Nanotechnology. 2020. link Times cited: 18 Abstract: High entropy alloys (HEAs) holding several principal element… read more USED (low confidence) M. Kretova, R. Konchakov, N. Kobelev, and V. Khonik, “Point Defects and Their Properties in the Fe20Ni20Cr20Co20Cu20 High-Entropy Alloy,” JETP Letters. 2020. link Times cited: 5 USED (low confidence) W. Li, S. Rao, Q. Wang, H. Fan, J. Yang, and J. El-Awady, “Core structure and mobility of edge dislocations in face-centered-cubic chemically complex NiCoFe and NiCoFeCu equiatomic solid-solution alloys,” Materialia. 2020. link Times cited: 18 USED (low confidence) R. Pasianot and D. Farkas, “Atomistic modeling of dislocations in a random quinary high-entropy alloy,” Computational Materials Science. 2020. link Times cited: 34 USED (low confidence) K. Shiotani, T. Niiyama, and T. Shimokawa, “Dislocation Emission from Grain Boundaries in High-Entropy Alloys: Influence of Atomic Compositions at Grain Boundaries,” Journal of the Society of Materials Science, Japan. 2020. link Times cited: 0 Abstract: High-Entropy Alloys (HEAs) are alloys in solid solution with… read more USED (low confidence) G. Ackland and G. Bonny, “Interatomic Potential Development,” Comprehensive Nuclear Materials. 2020. link Times cited: 4 USED (low confidence) K. Yashiro, S. Honda, R. Terada, and K. Naito, “Molecular dynamics simulation of indentation-cutting on Ni and Cu by rigid Fe tool (Focus on combination of surface structure of tool and work),” Transactions of the JSME (in Japanese). 2020. link Times cited: 0 Abstract: 2D-like indentations are performed by molecular dynamics sim… read more NOT USED (low confidence) F. Aquistapace, N. Amigo, J. F. Troncoso, O. Deluigi, and E. Bringa, “MultiSOM: Multi-layer Self Organizing Maps for local structure identification in crystalline structures,” Computational Materials Science. 2023. link Times cited: 2 NOT USED (low confidence) B. Yao, Z. R. Liu, and R. F. Zhang, “EAPOTc: An integrated empirical interatomic potential optimization platform for compound solids,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) E. Huang et al., “Machine-learning and high-throughput studies for high-entropy materials,” Materials Science and Engineering: R: Reports. 2022. link Times cited: 39 NOT USED (low confidence) S. Risal, W. Zhu, P. Guillén, and L. Sun, “Improving phase prediction accuracy for high entropy alloys with Machine learning,” Computational Materials Science. 2021. link Times cited: 31 NOT USED (high confidence) A. Naghdi et al., “Dislocation plasticity in equiatomic NiCoCr alloys: Effect of short-range order,” Physical Review B. 2022. link Times cited: 3 Abstract: Equiatomic NiCoCr solid solutions have been recently shown t… read more NOT USED (high confidence) F. Aquistapace et al., “Atomistic Simulations of Ductile Failure in a b.c.c. High-Entropy Alloy,” High Entropy Alloys & Materials. 2022. link Times cited: 5 NOT USED (high confidence) J. Qi, C. Oberdorfer, W. Windl, and E. Marquis, “Ab initio
simulation of field evaporation,” Physical Review Materials. 2022. link Times cited: 5 Abstract: A new simulation approach of field evaporation is presented.… read more NOT USED (high confidence) R. Jagatramka, C. Wang, and M. Daly, “An analytical method to quantify the statistics of energy landscapes in random solid solutions,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (high confidence) J. Mayandi et al., “Controlling the Electrical Properties of Reactively Sputtered High Entropy Alloy CrFeNiCoCu Films,” Journal of Electronic Materials. 2021. link Times cited: 2 NOT USED (high confidence) L. Xu, L. Casillas-Trujillo, Y. Gao, and H. Xu, “Compositional effects on stacking fault energies in Ni-based alloys using first-principles and atomistic simulations,” Computational Materials Science. 2021. link Times cited: 5 NOT USED (high confidence) C. Ruestes and D. Farkas, “Deformation response of high entropy alloy nanowires,” Journal of Materials Science. 2021. link Times cited: 12 NOT USED (high confidence) J. S. Andrade, I. N. Bastos, and L. C. R. Aliaga, “Determinação das características estruturais e mecânicas da liga de alta entropia Hf-Nb-Ta-Zr,” VETOR - Revista de Ciências Exatas e Engenharias. 2021. link Times cited: 0 Abstract: Neste artigo, os comportamentos estrutural e mecânico da lig… read more NOT USED (high confidence) J. Zhao et al., “Liquid Structure and Thermophysical Properties of Ternary Ni-Fe-Co Alloys Explored by Molecular Dynamics Simulations and Electrostatic Levitation Experiments,” Metallurgical and Materials Transactions A. 2021. link Times cited: 6 NOT USED (high confidence) Z. Hao, X. Han, Y. Fan, and Z. Lou, “Microscopic Study on the Mechanism of Tool Bond Wear in Cutting Ni–Fe-Cr-Co–Cu Series Nickel-Base Superalloy,” International Journal of Precision Engineering and Manufacturing. 2021. link Times cited: 2 NOT USED (high confidence) D. Farkas, “Varying Diffusion Kinetics Along Random Grain Boundaries in a Model Austenitic Stainless Steel,” Metallurgical and Materials Transactions A. 2021. link Times cited: 4 NOT USED (high confidence) J. Li, X. Yang, and P. Wang, “Shear-coupled grain boundary migration in bicrystal Ni with metallic dopant segregation,” Journal of Materials Research. 2021. link Times cited: 5 Abstract: The shear-coupled grain boundary (GB) migration in bicrystal… read more NOT USED (high confidence) J. Li, Q. Fang, and P. Liaw, “Microstructures and Properties of High‐Entropy Materials: Modeling, Simulation, and Experiments,” Advanced Engineering Materials. 2020. link Times cited: 32 Abstract: The performances of high‐entropy materials (HEMs), including… read more NOT USED (high confidence) M. M. Aish and S. A. Aljasar, “Thermo-mechanical properties of V-5Cr-5Ti alloy: 3D molecular dynamics simulation,” International Journal of Physical Sciences. 2020. link Times cited: 0 Abstract: This paper presents the results of the study of 3D molecular… read more NOT USED (high confidence) H. Feng et al., “Indentation-induced plastic behaviour of nanotwinned Cu/high entropy alloy FeCoCrNi nanolaminate: an atomic simulation,” RSC Advances. 2020. link Times cited: 17 Abstract: Using large-scale molecular dynamics (MD) simulations, the e… read more NOT USED (high confidence) D. Farkas, “Grain boundary structure in high-entropy alloys,” Journal of Materials Science. 2020. link Times cited: 23 |
| Funding |
Award Number: 1507846 Funder: Division of Materials Research |
| Short KIM ID
The unique KIM identifier code.
| MO_803527979660_000 |
| Extended KIM ID
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
| EAM_Dynamo_FarkasCaro_2018_FeNiCrCoCu__MO_803527979660_000 |
| DOI |
10.25950/8c2ffcf6 https://doi.org/10.25950/8c2ffcf6 https://commons.datacite.org/doi.org/10.25950/8c2ffcf6 |
| KIM Item Type
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
| Portable Model using Model Driver EAM_Dynamo__MD_120291908751_005 |
| Driver | EAM_Dynamo__MD_120291908751_005 |
| KIM API Version | 2.2 |
| Potential Type | eam |
| Grade | Name | Category | Brief Description | Full Results | Aux File(s) |
|---|---|---|---|---|---|
| P | vc-species-supported-as-stated | mandatory | The model supports all species it claims to support; see full description. |
Results | Files |
| P | vc-periodicity-support | mandatory | Periodic boundary conditions are handled correctly; see full description. |
Results | Files |
| P | vc-permutation-symmetry | mandatory | Total energy and forces are unchanged when swapping atoms of the same species; see full description. |
Results | Files |
| B | vc-forces-numerical-derivative | consistency | Forces computed by the model agree with numerical derivatives of the energy; see full description. |
Results | Files |
| F | vc-dimer-continuity-c1 | informational | The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description. |
Results | Files |
| P | vc-objectivity | informational | Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description. |
Results | Files |
| P | vc-inversion-symmetry | informational | Total energy is unchanged and forces change sign when inverting a configuration through the origin; see full description. |
Results | Files |
| P | vc-memory-leak | informational | The model code does not have memory leaks (i.e. it releases all allocated memory at the end); see full description. |
Results | Files |
| P | vc-thread-safe | mandatory | The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description. |
Results | Files |
| P | vc-unit-conversion | mandatory | The model is able to correctly convert its energy and/or forces to different unit sets; see full description. |
Results | Files |
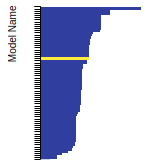
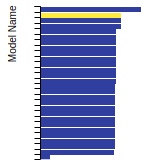
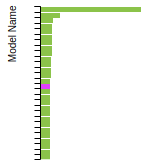
BCC Lattice Constant
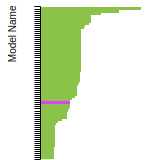
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
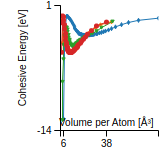
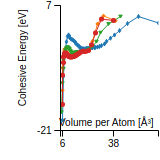
Cohesive Energy Graph
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
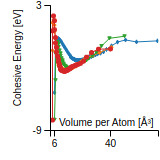
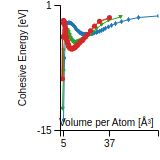
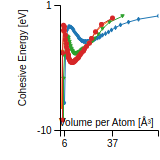
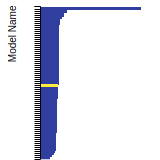
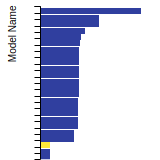
Diamond Lattice Constant
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
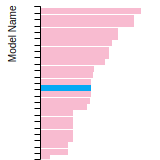
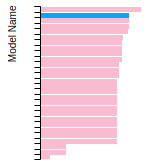

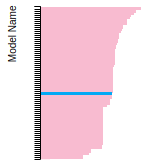
Dislocation Core Energies
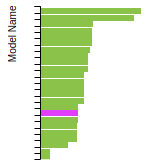
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
(No matching species)FCC Elastic Constants
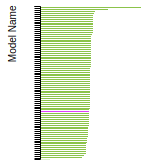
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
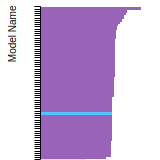
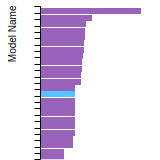

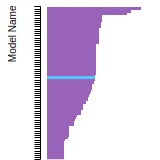
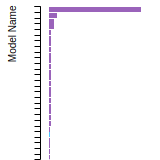
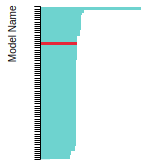
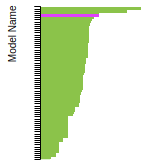
FCC Lattice Constant
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.

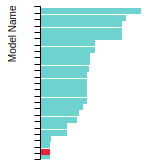

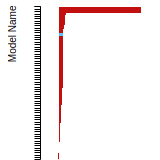
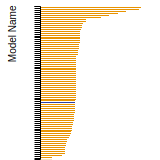
FCC Stacking Fault Energies
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
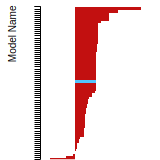
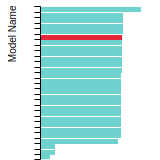
FCC Surface Energies
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
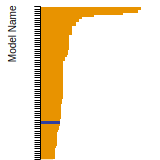
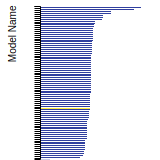
SC Lattice Constant
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Cubic Crystal Basic Properties Table
Species: CoSpecies: Cr
Species: Cu
Species: Fe
Species: Ni
Creators:
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/64cb38c5
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
Creators:
Contributor: ilia
Publication Year: 2024
DOI: https://doi.org/10.25950/888f9943
Computes the elastic constants for an arbitrary crystal. A robust computational protocol is used, attempting multiple methods and step sizes to achieve an acceptably low error in numerical differentiation and deviation from material symmetry. The crystal structure is specified using the AFLOW prototype designation as part of the Crystal Genome testing framework. In addition, the distance from the obtained elasticity tensor to the nearest isotropic tensor is computed.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for CrFe in AFLOW crystal prototype A2B_cF24_227_c_b at zero temperature and pressure v000 | view | 121459 | |
| Elastic constants for CrNi in AFLOW crystal prototype A2B_cF24_227_c_b at zero temperature and pressure v000 | view | 179929 | |
| Elastic constants for FeNi in AFLOW crystal prototype A2B_cF24_227_c_b at zero temperature and pressure v000 | view | 136934 |
Creators: Junhao Li and Ellad Tadmor
Contributor: tadmor
Publication Year: 2019
DOI: https://doi.org/10.25950/5853fb8f
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
Creators:
Contributor: ilia
Publication Year: 2023
DOI: https://doi.org/10.25950/e8a7ed84
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium crystal structure and energy for Cu in AFLOW crystal prototype A_cF4_225_a v001 | view | 60590 | |
| Equilibrium crystal structure and energy for Cu in AFLOW crystal prototype A_cI2_229_a v001 | view | 61988 |
Creators:
Contributor: ilia
Publication Year: 2024
DOI: https://doi.org/10.25950/2f2c4ad3
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Creators: Brandon Runnels
Contributor: brunnels
Publication Year: 2019
DOI: https://doi.org/10.25950/4723cee7
Computes grain boundary energy for a range of tilt angles given a crystal structure, tilt axis, and material.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Relaxed energy as a function of tilt angle for a 111 symmetric tilt grain boundary in bcc Fe v000 | view | 12196471 | |
| Relaxed energy as a function of tilt angle for a 112 symmetric tilt grain boundary in bcc Fe v000 | view | 114732758 | |
| Relaxed energy as a function of tilt angle for a 111 symmetric tilt grain boundary in fcc Fe v000 | view | 11074951 | |
| Relaxed energy as a function of tilt angle for a 112 symmetric tilt grain boundary in fcc Fe v000 | view | 66521053 |
Creators:
Contributor: brunnels
Publication Year: 2022
DOI: https://doi.org/10.25950/2c59c9d6
Computes grain boundary energy for a range of tilt angles given a crystal structure, tilt axis, and material.
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/2765e3bf
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/c339ca32
Calculates lattice constant of hexagonal bulk structures at zero temperature and pressure by using simplex minimization to minimize the potential energy.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium lattice constants for hcp Co v005 | view | 140945 | |
| Equilibrium lattice constants for hcp Cr v005 | view | 181999 | |
| Equilibrium lattice constants for hcp Cu v005 | view | 165014 | |
| Equilibrium lattice constants for hcp Fe v005 | view | 171946 | |
| Equilibrium lattice constants for hcp Ni v005 | view | 256282 |
Creators: Mingjian Wen
Contributor: mjwen
Publication Year: 2019
DOI: https://doi.org/10.25950/fc69d82d
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Linear thermal expansion coefficient of fcc Cu at 293.15 K under a pressure of 0 MPa v001 | view | 8071925 |
Creators:
Contributor: mjwen
Publication Year: 2024
DOI: https://doi.org/10.25950/9d9822ec
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Linear thermal expansion coefficient of bcc Cr at 293.15 K under a pressure of 0 MPa v002 | view | 501650 | |
| Linear thermal expansion coefficient of bcc Fe at 293.15 K under a pressure of 0 MPa v002 | view | 588301 | |
| Linear thermal expansion coefficient of fcc Ni at 293.15 K under a pressure of 0 MPa v002 | view | 701717 |
Creators: Matt Bierbaum
Contributor: mattbierbaum
Publication Year: 2019
DOI: https://doi.org/10.25950/64f4999b
Calculates the phonon dispersion relations for fcc lattices and records the results as curves.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Phonon dispersion relations for fcc Cu v004 | view | 99282 | |
| Phonon dispersion relations for fcc Ni v004 | view | 106148 |
Creators:
Contributor: SubrahmanyamPattamatta
Publication Year: 2019
DOI: https://doi.org/10.25950/b4cfaf9a
Intrinsic and extrinsic stacking fault energies, unstable stacking fault energy, unstable twinning energy, stacking fault energy as a function of fractional displacement, and gamma surface for a monoatomic FCC lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Stacking and twinning fault energies for fcc Cu v002 | view | 12515201 | |
| Stacking and twinning fault energies for fcc Ni v002 | view | 19596276 |
Creators: Matt Bierbaum
Contributor: mattbierbaum
Publication Year: 2019
DOI: https://doi.org/10.25950/6c43a4e6
Calculates the surface energy of several high symmetry surfaces and produces a broken-bond model fit. In latex form, the fit equations are given by:
E_{FCC} (\vec{n}) = p_1 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (2 ( |x+ 2y + z| + |x+2y-z| + |x-2y + z| + |x-2y-z| + |2x+y+z| + |2x+y-z| +|2x-y+z| +|2x-y-z| +|x+y+2z| +|x+y-2z| +|x-y+2z| +|x-y-2z| ) + c
E_{BCC} (\vec{n}) = p_1 (6 \left( | x+y+z| + |x+y-z| + |-x+y-z| + |x-y+z| \right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) +c.
In Python, these two fits take the following form:
def BrokenBondFCC(params, index):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*(abs(x+2*y+z) + abs(x+2*y-z) +abs(x-2*y+z) +abs(x-2*y-z) + abs(2*x+y+z) +abs(2*x+y-z) +abs(2*x-y+z) +abs(2*x-y-z) + abs(x+y+2*z) +abs(x+y-2*z) +abs(x-y+2*z) +abs(x-y-2*z))+params[3]
def BrokenBondBCC(params, x, y, z):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*6*(abs(x+y+z) + abs(x-y-z) + abs(x-y+z) + abs(x+y-z)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[3]
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Broken-bond fit of high-symmetry surface energies in bcc Cr v004 | view | 160200 | |
| Broken-bond fit of high-symmetry surface energies in bcc Fe v004 | view | 171399 | |
| Broken-bond fit of high-symmetry surface energies in fcc Cu v004 | view | 109128 | |
| Broken-bond fit of high-symmetry surface energies in fcc Ni v004 | view | 100912 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/fca89cea
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Monovacancy formation energy and relaxation volume for bcc Cr | view | 454533 | |
| Monovacancy formation energy and relaxation volume for bcc Fe | view | 1511723 | |
| Monovacancy formation energy and relaxation volume for fcc Cu | view | 404250 | |
| Monovacancy formation energy and relaxation volume for fcc Ni | view | 341452 | |
| Monovacancy formation energy and relaxation volume for hcp Co | view | 435833 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/c27ba3cd
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Vacancy formation and migration energy for bcc Cr | view | 3322420 | |
| Vacancy formation and migration energy for bcc Fe | view | 4699270 | |
| Vacancy formation and migration energy for fcc Cu | view | 1548165 | |
| Vacancy formation and migration energy for fcc Ni | view | 2059533 | |
| Vacancy formation and migration energy for hcp Co | view | 4681012 |
| Test | Error Categories | Link to Error page |
|---|---|---|
| Elastic constants for diamond Co at zero temperature v001 | other | view |
| Elastic constants for diamond Cr at zero temperature v001 | other | view |
| Elastic constants for diamond Cu at zero temperature v001 | other | view |
| Elastic constants for diamond Fe at zero temperature v001 | other | view |
| Elastic constants for diamond Ni at zero temperature v001 | other | view |
ElasticConstantsHexagonal__TD_612503193866_004
| Test | Error Categories | Link to Error page |
|---|---|---|
| Elastic constants for hcp Co at zero temperature v004 | other | view |
| Elastic constants for hcp Cr at zero temperature v004 | other | view |
| Elastic constants for hcp Cu at zero temperature v004 | other | view |
| Elastic constants for hcp Fe at zero temperature v004 | other | view |
| Elastic constants for hcp Ni at zero temperature v004 | other | view |
EquilibriumCrystalStructure__TD_457028483760_000
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium crystal structure and energy for CoFe in AFLOW crystal prototype A3B5_cI16_229_b_ac v000 | other | view |
| Equilibrium crystal structure and energy for FeNi in AFLOW crystal prototype AB2_cF24_227_a_d v000 | other | view |
EquilibriumCrystalStructure__TD_457028483760_002
GrainBoundaryCubicCrystalSymmetricTiltRelaxedEnergyVsAngle__TD_410381120771_003
PhononDispersionCurve__TD_530195868545_004
| Test | Error Categories | Link to Error page |
|---|---|---|
| Phonon dispersion relations for fcc Cu v004 | other | view |
| Phonon dispersion relations for fcc Ni v004 | other | view |
SurfaceEnergyCubicCrystalBrokenBondFit__TD_955413365818_004
| Test | Error Categories | Link to Error page |
|---|---|---|
| Broken-bond fit of high-symmetry surface energies in bcc Cr v004 | other | view |
| Broken-bond fit of high-symmetry surface energies in bcc Fe v004 | other | view |
| Broken-bond fit of high-symmetry surface energies in fcc Cu v004 | other | view |
| Broken-bond fit of high-symmetry surface energies in fcc Ni v004 | other | view |
No Driver
| Verification Check | Error Categories | Link to Error page |
|---|---|---|
| MemoryLeak__VC_561022993723_004 | other | view |
| EAM_Dynamo_FarkasCaro_2018_FeNiCrCoCu__MO_803527979660_000.txz | Tar+XZ | Linux and OS X archive |
| EAM_Dynamo_FarkasCaro_2018_FeNiCrCoCu__MO_803527979660_000.zip | Zip | Windows archive |
This Model requires a Model Driver. Archives for the Model Driver EAM_Dynamo__MD_120291908751_005 appear below.
| EAM_Dynamo__MD_120291908751_005.txz | Tar+XZ | Linux and OS X archive |
| EAM_Dynamo__MD_120291908751_005.zip | Zip | Windows archive |