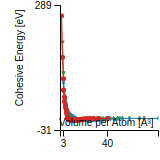
Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
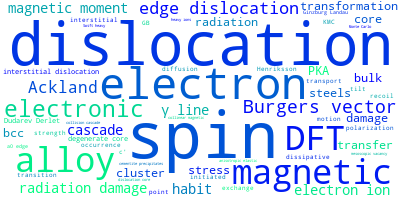
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
211 Citations (137 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) J. J. Möller and E. Bitzek, “BDA: A novel method for identifying defects in body-centered cubic crystals,” MethodsX. 2016. link Times cited: 17 USED (definite) D. Nguyen-Manh, P. Ma, M. Lavrentiev, and S. Dudarev, “Constrained non-collinear magnetism in disordered Fe and Fe-Cr alloys,” International Conference on Supercomputing. 2013. link Times cited: 8 Abstract: The development of quantitative models for radiation damage … read moreAbstract: The development of quantitative models for radiation damage effects in iron, iron alloys and steels, particularly for the high temperature properties of the alloys, requires understanding of magnetic interactions, which control the phase stability of ferritic-martensitic, ferritic, and austenitic steels. In this work, disordered magnetic configurations of pure iron and Fe-Cr alloys are investigated using Density Functional Theory (DFT) formalism, in the form of constrained non-collinear magnetic calculations, with the objective of creating a database of atomic magnetic moments and forces acting between the atoms. From a given disordered atomic configuration of either pure Fe or Fe-Cr alloy, a penalty contribution to the usual spin-polarized DFT total energy has been calculated by constraining the magnitude and direction of magnetic moments. An extensive database of non-collinear magnetic moment and force components for various atomic configurations has been generated and used for interpolating the spatially-dependent magnetic interaction parameters, for applications in large-scale spin-lattice dynamics and magnetic Monte-Carlo simulations. read less USED (high confidence) M. B. Salman, M. Kilic, and M. Banisalman, “Formation of Interstitial Dislocation Loops by Irradiation in Alpha-Iron under Strain: A Molecular Dynamics Study,” Crystals. 2021. link Times cited: 4 Abstract: The present work reports the formation of an interstitial di… read moreAbstract: The present work reports the formation of an interstitial dislocation loop with a lower primary knock-on atom (PKA) energy in alpha-iron under strain conditions by the use of molecular dynamics simulation. The study was conducted using a PKA energy of 1~10 keV and hydro-static strain from −1.4 to 1.6%. The application of 1.6% hydrostatic strain results in the formation of ½<111> dislocation loop with a low PKA of 3 keV. This result was associated with a threshold displacement energy decrement when moving from compression to tension strain, which resulted in more Frenkel pairs initiated at peak time. Furthermore, many of the initiated defects were energetically favorable by 2 eV in the form of the interstitial dislocation loop rather than a mono defect. read less USED (high confidence) N. Gao, Z. Yao, G. Lu, H. Deng, and F. Gao, “Mechanisms for <100> interstitial dislocation loops to diffuse in BCC iron,” Nature Communications. 2021. link Times cited: 23 USED (high confidence) N. Gao, Z. Yao, G. Lu, H. Deng, and F. Gao, “Mechanisms for <100> interstitial dislocation loops to diffuse in BCC iron,” Nature Communications. 2021. link Times cited: 0 USED (high confidence) J. Hellsvik et al., “General method for atomistic spin-lattice dynamics with first-principles accuracy,” Physical Review B. 2018. link Times cited: 23 Abstract: We present a computationally efficient and general first-pri… read moreAbstract: We present a computationally efficient and general first-principles based method for spin-lattice simulations for solids and clusters. The method is based on a coupling of atomistic spin dynamics a ... read less USED (high confidence) A. Bakaev, D. Terentyev, Z. Chang, M. Posselt, P. Olsson, and E. Zhurkin, “Effect of isotropic stress on dislocation bias factor in bcc iron: an atomistic study,” Philosophical Magazine. 2018. link Times cited: 5 Abstract: The effect of externally applied stress on the dislocation b… read moreAbstract: The effect of externally applied stress on the dislocation bias factor (BF) in bcc iron has been studied using a combination of atomistic static calculations and finite element integration. Three kinds of dislocations were considered, namely, a0/2〈1 1 1〉{1 1 0} screw, a0/2〈1 1 1〉{1 1 0} edge and a0〈1 0 0〉{0 0 1} edge dislocations. The computations reveal that the isotropic crystal expansion leads to an increasing or constant dislocation bias, depending on the Burgers vector and type of dislocation. On the other hand, compressive stress reduces the dislocation bias for all the dislocations studied. Variation of the dislocation BF depending on dislocation type and Burgers vector is discussed by analysing the modification of the interaction energy landscape and the capture efficiency values for the vacancy and self-interstitial atom. read less USED (high confidence) F. Granberg, J. Byggmästar, A. Sand, and K. Nordlund, “Cascade debris overlap mechanism of 〈100〉 dislocation loop formation in Fe and FeCr,” Europhysics Letters. 2017. link Times cited: 38 Abstract: Two types of dislocation loops are observed in irradiated α-… read moreAbstract: Two types of dislocation loops are observed in irradiated α-Fe, the 1/2〈111〉 loop and the 〈100〉 loop. Atomistic simulations consistently predict that only the energetically more favourable 1/2〈111〉 loops are formed directly in cascades, leaving the formation mechanism of 〈100〉 loops an unsolved question. We show how 〈100〉 loops can be formed when cascades overlap with random pre-existing primary radiation damage in Fe and FeCr. This indicates that there are no specific constraints involved in the formation of 〈100〉 loops, and can explain their common occurrence. read less USED (high confidence) N. Gao, J. Chen, R. Kurtz, Z. Wang, R. F. Zhang, and F. Gao, “New understanding of nano-scale interstitial dislocation loops in BCC iron,” Journal of Physics: Condensed Matter. 2017. link Times cited: 21 Abstract: Complex states of nanoscale interstitial dislocation loop ca… read moreAbstract: Complex states of nanoscale interstitial dislocation loop can be described by its habit plane and Burgers vector. Using atomistic simulations, we provide direct evidences on the change of the habit plane of a 1/2〈1 1 1〉 loop from {1 1 1} to {1 1 0} and {2 1 1}, in agreement with TEM observations. A new {1 0 0} habit plane of this loop is also predicted by simulations. The non-conservation of the Burgers vector is approved theoretically for: (1) dislocation reactions between loops with different Burgers vectors and (2) the transition between 〈1 0 0〉 loops and 1/2〈1 1 1〉 loops. The rotation from a 1/2〈1 1 1〉 to a 〈1 0 0〉 loop has also been explored, which occurs at 570 K for time on the order of 10 s. The dislocation-precipitate phase duality and change of habit plane are then proposed as new features for nano-scale dislocation loops. read less USED (high confidence) A. Mutter, B. Wang, J. Meiser, P. Umstätter, and H. Urbassek, “Magnetic structure of [0 0 1] tilt grain boundaries in bcc Fe studied via magnetic potentials,” Philosophical Magazine. 2017. link Times cited: 4 Abstract: Using magnetic potentials and a molecular statics approach, … read moreAbstract: Using magnetic potentials and a molecular statics approach, we study the changes in the magnetic structure of bcc Fe in the vicinity of grain boundaries (GBs). We focus on symmetric tilt GBs around the [0 0 1] axis with a tilt angle between 7 and 53. We find that immediately in the GB plane, the deviations in the magnetic moments from the bulk value are most pronounced. The distribution of moments in the GB plane is modulated according to the periodicity of the coincidence site lattice. In the direction perpendicular to the GB plane, the moments decay exponentially towards the bulk value; the decay length increases with decreasing tilt angle. This dependence can be explained by the well-known stress field around GBs. read less USED (high confidence) H. Zong, Z. Ni, X. Ding, T. Lookman, and J. Sun, “Origin of low thermal hysteresis in shape memory alloy ultrathin films,” Acta Materialia. 2016. link Times cited: 12 USED (high confidence) M. J. Aliaga, R. Schäublin, J. F. Löffler, and M. Caturla, “Surface-induced vacancy loops and damage dispersion in irradiated Fe thin films,” Acta Materialia. 2015. link Times cited: 40 USED (high confidence) Z. Chang, P. Olsson, D. Terentyev, and N. Sandberg, “Multiscale calculations of dislocation bias in fcc Ni and bcc Fe model lattices,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2015. link Times cited: 7 USED (high confidence) M. J. Aliaga, A. Prokhodtseva, R. Schaeublin, and M. Caturla, “Molecular dynamics simulations of irradiation of α-Fe thin films with energetic Fe ions under channeling conditions,” Journal of Nuclear Materials. 2014. link Times cited: 2 USED (high confidence) F. Granberg, D. Terentyev, K. Henriksson, F. Djurabekova, and K. Nordlund, “Interaction of Dislocations with Carbides in BCC Fe Studied by Molecular Dynamics,” Fusion Science and Technology. 2014. link Times cited: 21 Abstract: Iron carbide (Fe3C), also known as cementite, is present in … read moreAbstract: Iron carbide (Fe3C), also known as cementite, is present in many steels and has also been seen as nanosized precipitates in steels. We examine the interaction of edge dislocations with nanosized cementite precipitates in Fe by molecular dynamics. The simulations are carried out with a Tersoff-like bond order interatomic potential by Henriksson et al. for Fe-C-Cr systems. Comparing the results obtained with this potential for a defect free Fe system with results from previously used potentials, we find that the potential by Henriksson et al. gives significantly higher values for the critical stress, at least at low temperatures. The explanation was found to be the difference in the core structure of the edge dislocation. The results show that edge dislocations can unpin from cementite precipitates of sizes 1 nm and 2 nm even at a temperature of 1 K, although the stresses needed for this are high. On the other hand, a 4 nm precipitate is not sheared by edge dislocations at low temperatures (≤100 K) on our simulation timescale. read less USED (high confidence) P. Ma, S. Dudarev, and C. Woo, “Spin-lattice-electron dynamics simulations of magnetic materials,” Physical Review B. 2012. link Times cited: 48 Abstract: We develop a dynamic spin-lattice-electron model for simulat… read moreAbstract: We develop a dynamic spin-lattice-electron model for simulating the time-dependent evolution of coupled spin, atomic, and electronic degrees of freedom in a magnetic material. Using the model, we relate the dissipative parameters entering the Langevin equations for the lattice and spin degrees of freedom to the heat transfer coefficients of a phenomenological spin-lattice-electron three-temperature model. We apply spin-lattice-electron dynamics simulations to the interpretation of experiments on laser-induced demagnetization of iron thin films, and estimate the rates of heat transfer between the spins and electrons, and between atoms and electrons. To model the dynamics of energy dissipation in a magnetic material undergoing plastic deformation, we develop an algorithm that separates the local collective modes of motion of atoms from their random thermal motion. Using this approach, we simulate the propagation of compressive shock waves through magnetic iron. We also explore the microscopic dynamics of dissipative coupling between the spin and lattice subsystems, and show that the rate of spin-lattice heat transfer is proportional to the integral of the four-spin time-dependent correlation function. read less USED (high confidence) C. Björkas, K. Nordlund, and M. Caturla, “Influence of the picosecond defect distribution on damage accumulation in irradiated α-Fe,” Physical Review B. 2012. link Times cited: 49 Abstract: The importance of the defect distribution produced in the fi… read moreAbstract: The importance of the defect distribution produced in the first few picoseconds of a collision cascade on long-term damage evolution is studied with molecular dynamics and kinetic Monte Carlo (KMC) methods. Three different interatomic potentials are used to obtain the primary damage produced by energetic recoils in α-Fe. Contrary to previous results, a dependence of cluster-size distribution with recoil energy is obtained. Moreover, large variations in this distribution are observed depending on the interatomic potential. Using the results for 50 keV collision cascades, damage accumulation is modeled with KMC. The accumulation rate of damage visible under transmission electron microscopy predicted by KMC depends significantly on the database used for cascade damage and, therefore, on the interatomic potential. Based on these results, we show that the comparison of cluster-size distributions with experiments can be used to test the reliability of interatomic potentials. read less USED (high confidence) L. Ventelon and F. Willaime, “Generalized stacking-faults and screw-dislocation core-structure in bcc iron: A comparison between ab initio calculations and empirical potentials,” Philosophical Magazine. 2010. link Times cited: 65 Abstract: Generalized stacking fault energies and screw dislocation co… read moreAbstract: Generalized stacking fault energies and screw dislocation core structures are reported for two sets of models for iron: density functional theory (DFT) calculations and empirical potentials. A thorough comparison between various DFT approaches has been performed on {110} and {211} γ-lines, which give a first indication on dislocation properties: (i) the effect of the exchange-correlation functional, LDA versus GGA, is significant in the pseudopotential approximation but not in the PAW approximation or in paramagnetic calculations; and (ii) the discrepancy due to the basis set between SIESTA and plane-wave results is rather small. Three empirical potentials for iron have been benchmarked on these DFT results. They all yield similar energies, but different shapes for the γ-lines. Using the criterion suggested by Duesbery and Vitek, the γ-line results point to non-degenerate core structures for the DFT calculations and for the Ackland and Ackland–Mendelev potentials but not for the Dudarev–Derlet potential. The direct calculations of the dislocation core structures show that the Ackland potential is an exception to the Duesbery–Vitek rule. More insight into the stability of the core structure can be gained by looking at the response to the polarization of the core. The Dudarev–Derlet and Ackland potentials have similar polarizations, but the energy difference between degenerate and non-degenerate cores is much larger with the Dudarev–Derlet potential, as expected from the γ-lines. The polarizability of the non-degenerate core is smaller with the Ackland–Mendelev potential than in DFT, indicating that the energy landscape is flatter in this direction. read less USED (high confidence) C. Björkas and K. Nordlund, “Assessment of the relation between ion beam mixing, electron–phonon coupling and damage production in Fe,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2009. link Times cited: 36 USED (high confidence) M. R. Gilbert, S. Dudarev, P. Derlet, and D. Pettifor, “Structure and metastability of mesoscopic vacancy and interstitial loop defects in iron and tungsten,” Journal of Physics: Condensed Matter. 2008. link Times cited: 89 Abstract: The most recent observations of dynamical time-dependent flu… read moreAbstract: The most recent observations of dynamical time-dependent fluctuating behaviour of mesoscopic radiation defects in body-centred cubic metals (Arakawa et al 2006 Phys. Rev. Lett. 96 125506; 2007 Science 318 956–9; Yao et al 2008 Phil. Mag. at press) have highlighted the need to develop adequate quantitative models for the structural stability of defects in the mesoscopic limit where defects are accessible to direct in situ electron microscope imaging. In pursuit of this objective, we investigate and compare several types of mesoscopic vacancy and interstitial defects in iron and tungsten by simulating them using recently developed many-body interatomic potentials. We show that the mesoscopic vacancy dislocation loops observed in ion-irradiated materials are, without exception, metastable with respect to the transformation into spherical voids, but that the rate of this transformation and even the specific type of the transformation mechanism depend on the defect size and the properties of the material. read less USED (high confidence) S. Dudarev, R. Bullough, and P. Derlet, “Effect of the alpha-gamma phase transition on the stability of dislocation loops in bcc iron.,” Physical review letters. 2008. link Times cited: 162 Abstract: Body-centered-cubic iron develops an elastic instability, dr… read moreAbstract: Body-centered-cubic iron develops an elastic instability, driven by spin fluctuations, near the alpha-gamma phase transition temperature T(c) = 912 degrees C that is associated with the dramatic reduction of the shear stiffness constant c' (c(11)-c(12))/2 near T(c). This reduction of c' has a profound effect on the temperature dependence of the anisotropic elastic self-energies of dislocations in iron. It also affects the relative stability of the a[100] and a/2[111] prismatic edge dislocation loops formed during irradiation. The difference between the anisotropic elastic free energies provides the fundamental explanation for the observed dominant occurrence of the a[100], as opposed to the a/2[111], Burgers vector configurations of prismatic dislocation loops in iron and iron-based alloys at high temperatures. read less USED (high confidence) D. Duffy, N. Itoh, A. Rutherford, and A. Stoneham, “Making tracks in metals,” Journal of Physics: Condensed Matter. 2008. link Times cited: 46 Abstract: Swift heavy ions lose energy primarily by inelastic electron… read moreAbstract: Swift heavy ions lose energy primarily by inelastic electronic scattering and, above an energy threshold, electronic losses result in damage to the lattice. Such high energy radiation is beyond the range of validity of traditional cascade simulations, and predictive damage calculations are challenging. We use a novel methodology, which combines molecular dynamics with a consistent treatment of electronic energy transport and redistribution to the lattice, to model how swift heavy ions form damage tracks. We consider a range of material parameters (electron–phonon coupling strength, thermal conductivity and electronic specific heat) and show how these affect the maximum lattice temperature reached and the extent of residual damage. Our analysis also suggests that fission tracks may form in alloys of archaeological interest. read less USED (high confidence) A. Rutherford and D. Duffy, “The effect of electron–ion interactions on radiation damage simulations,” Journal of Physics: Condensed Matter. 2007. link Times cited: 169 Abstract: Classical cascade simulations of radiation damage generally … read moreAbstract: Classical cascade simulations of radiation damage generally neglect the effect of energy exchange between the lattice and the electrons; however electronic effects increase with increasing radiation energy. Indeed, even for low energy radiation events the electrons contribute to heat transport and increase the cooling rate, particularly in materials with strong electron–ion interactions. We use a method described in an earlier publication to include these effects in a series of 10 keV cascades in Fe, for a range of electron–ion interaction strengths. We find a non-monotonic relationship between the number of residual defects and the strength of the electron–ion interactions and we discuss the mechanisms involved. read less USED (high confidence) C. Ortiz and M. Caturla, “Cascade damage evolution: rate theory versus kinetic Monte Carlo simulations,” Journal of Computer-Aided Materials Design. 2007. link Times cited: 20 USED (high confidence) D. M. Duffy and A. Rutherford, “Including the effects of electronic stopping and electron–ion interactions in radiation damage simulations,” Journal of Physics: Condensed Matter. 2007. link Times cited: 240 Abstract: Radiation damage is traditionally modelled using cascade sim… read moreAbstract: Radiation damage is traditionally modelled using cascade simulations, and the effect of inelastic scattering by electrons, if included, is introduced via a friction term in the equation of motion. We have developed a model in which the molecular dynamics simulation is coupled to a model for the electronic energy, which evolves via the heat diffusion equation. Energy lost by the atoms, due electronic stopping or electron–ion interactions, is input to the electronic system via a source term in the diffusion equation. Energy is fed back to the atomic system from the hot electrons by means of a Langevin thermostat, which depends on the local electronic temperature. Results of the model are presented for 10 keV cascades in Fe. read less USED (low confidence) Y. Lei et al., “An Embedded-Atom Method Potential for studying the properties of Fe-Pb solid-liquid interface,” Journal of Nuclear Materials. 2022. link Times cited: 1 USED (low confidence) I. Toda-Caraballo, J. Wróbel, and D. Nguyen-Manh, “Generalized universal equation of states for magnetic materials: A novel formulation for an interatomic potential in Fe,” Physical Review Materials. 2022. link Times cited: 0 USED (low confidence) J. Gao, E. Gaganidze, and J. Aktaa, “Relative population of 1/2<111> and <100> interstitial loops in alpha-Fe under irradiation: Effects of C15 cluster stability and loop one-dimensional movement,” Acta Materialia. 2022. link Times cited: 5 USED (low confidence) Y. Wang et al., “Machine-learning interatomic potential for radiation damage effects in bcc-iron,” Computational Materials Science. 2022. link Times cited: 7 USED (low confidence) H. Min et al., “Development of an interatomic potential for Fe-He by neural network,” Computational Materials Science. 2021. link Times cited: 2 USED (low confidence) P. Maya et al., “Studies on the near-surface trapping of deuterium in implantation experiments,” Nuclear Fusion. 2020. link Times cited: 1 Abstract: Surface-shifted deuterium profiles are re-examined in deuter… read moreAbstract: Surface-shifted deuterium profiles are re-examined in deuterium-ion irradiation experiments by using a combined experimental and modelling approach. Recrystallized tungsten foil samples were irradiated with energetic deuterium ions and the defect and deuterium depth profiles were studied using positron annihilation spectroscopy and secondary ion mass spectroscopy. We report direct experimental evidence of trapping of deuterium at the vacancies created by the deuterium ions themselves during the implantation by using positron annihilation studies. The deuterium profile is simulated using a Monte-Carlo diffusion model by taking into account the defect-aided diffusion of deuterium due to the local strain field created by the vacancies. The simulations also elucidate the role of the anisotropy in the diffusion and trapping of deuterium in ion-implantation experiments in metals. read less USED (low confidence) R. Gröger and V. Vítek, “Single crystal yield criterion for chromium based on atomistic studies of isolated 1/2[111] screw dislocations,” International Journal of Plasticity. 2020. link Times cited: 12 USED (low confidence) G. D. Santos et al., “Size- and temperature-dependent magnetization of iron nanoclusters,” Physical Review B. 2020. link Times cited: 12 Abstract: The magnetic behavior of bcc iron nanoclusters, with diamete… read moreAbstract: The magnetic behavior of bcc iron nanoclusters, with diameters between 2 and 8 nm, is investigated by means of spin dynamics (SD) simulations coupled to molecular dynamics (MD-SD), using a distance-dependent exchange interaction. Finite-size effects in the total magnetization as well as the influence of the free surface and the surface/core proportion of the nanoclusters are analyzed in detail for a wide temperature range, going beyond the cluster and bulk Curie temperatures. Comparison is made with experimental data and with theoretical models based on the mean-field Ising model adapted to small clusters, and taking into account the influence of low coordinated spins at free surfaces. Our results for the temperature dependence of the average magnetization per atom M(T), including the thermalization of the transnational lattice degrees of freedom, are in very good agreement with available experimental measurements on small Fe nanoclusters. In contrast, significant discrepancies with experiment are observed if the translational degrees of freedom are artificially frozen. The finite-size effects on M(T) are found to be particularly important near the cluster Curie temperature. Simulated magnetization above the Curie temperature scales with cluster size as predicted by models assuming short-range magnetic ordering (SRMO). Analytical approximations to the magnetization as a function of temperature and size are proposed. read less USED (low confidence) R. Alexander et al., “Interatomic potentials for irradiation-induced defects in iron,” Journal of Nuclear Materials. 2020. link Times cited: 13 USED (low confidence) D. Karfaridis et al., “Influence of the Pt thickness on the structural and magnetic properties of epitaxial Fe/Pt bilayers,” Thin Solid Films. 2020. link Times cited: 4 USED (low confidence) S. Park, M. Banisalman, and T. Oda, “Characterization and quantification of numerical errors in threshold displacement energy calculated by molecular dynamics in bcc-Fe,” Computational Materials Science. 2019. link Times cited: 4 USED (low confidence) K. Li et al., “Determination of the accuracy and reliability of molecular dynamics simulations in estimating the melting point of iron: Roles of interaction potentials and initial system configurations,” Journal of Molecular Liquids. 2019. link Times cited: 8 USED (low confidence) P. Maya et al., “Evaluation of tungsten as divertor plasma-facing material: results from ion irradiation experiments and computer simulations,” Nuclear Fusion. 2019. link Times cited: 11 Abstract: The effect of the primary knock-on atom (PKA) spectrum in ra… read moreAbstract: The effect of the primary knock-on atom (PKA) spectrum in radiation damage and the subsequent defect structure formation and their impact in deuterium (D) trapping has been investigated using computer simulations and surrogate ion irradiation experiments. The neutron spectrum for an ‘ITER-like’ divertor shape and parameters has been generated using ATTILA and SPECTER codes to identify the relevant PKA energies. It has been observed that 10 MeV boron (B) produces a PKA spectrum similar to that obtained from a reactor-like neutron spectrum. Experiments have been carried out with ions of gold (Au), B, helium (He) and D with energies ranging from 0.1 MeV–80 MeV for a fluence range of ions m−2– ions m−2, and distinctly different PKA spectra have been produced. While 80 MeV Au ions produced dense and small clusters of interstitial defects (<10 nm), B produced large dislocation loops up to 60 nm in size. At room temperature, the imprint of the cascade is well captured by the vacancies due to their low mobility, and the vacancy defects observed in Au and B irradiation showed significant differences. Molecular dynamics simulations show that at PKA energies exceeding 150 keV, the fragmentation of the cascades takes place, which tends to limit the size of individual defects in the case of 80 MeV Au irradiation. A mechanism based on the competitive capture of mobile interstitials has been proposed to explain the observed large dislocation loops as well as dislocation lines in different irradiation experiments. read less USED (low confidence) T. Suzudo, T. Onitsuka, and K. Fukumoto, “Analyzing the cross slip motion of screw dislocations at finite temperatures in body-centered-cubic metals: molecular statics and dynamics studies,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 13 Abstract: The plasticity of body-centered-cubic metals at low temperat… read moreAbstract: The plasticity of body-centered-cubic metals at low temperatures is substantially determined by the screw-dislocation kinetics. Because the core of screw dislocations in these metals has a non-planar structure, its motion is complex. For example, although density functional theory predicts slip on a {110} plane, the actual slip plane at elevated temperatures differs from the prediction. In this work, we explored state-of-the-art atomistic modeling methods and successfully reproduced the transition of the slip plane through a temperature increase. We then devised an algorithm to analyze the activation of dislocation jump over the Peierls barrier and discovered a possible origin of this unexpected phenomenon: thermal fluctuation leads to the kink-pair nucleation for cross slip jumps with no transition of the dislocation core structure. read less USED (low confidence) M. Mock and K. Albe, “Modelling of dislocation-solute interaction in ODS steels: Analytic bond-order potential for the iron-yttrium system,” Journal of Nuclear Materials. 2018. link Times cited: 6 USED (low confidence) T. Yokoyama, A. Koide, and Y. Uemura, “Local thermal expansions and lattice strains in Elinvar and stainless steel alloys,” Physical Review Materials. 2018. link Times cited: 6 USED (low confidence) M. Mudrick, M. Eisenbach, D. Perera, G. M. Stocks, and D. Landau, “Combined molecular and spin dynamics simulation of bcc iron with lattice vacancies,” Journal of Physics: Conference Series. 2017. link Times cited: 5 Abstract: Using an atomistic model that treats both translational and … read moreAbstract: Using an atomistic model that treats both translational and spin degrees of freedom, we have performed combined molecular and spin dynamics simulations to study dynamic properties of BCC iron with varying vacancy concentrations. Atomic interactions are described by an empirical many-body potential while spin interactions are handled with a Heisenberg-like Hamiltonian with a coordinate dependent exchange interaction. By calculating the Fourier transform of spatial and temporal correlation functions, vibrational and magnetic excitations have been studied. The creation of vacancies in the material has shown splitting of the characteristic transverse spin-wave excitations, indicating the production of additional excitation modes. By merging two vacancies to form a nearest neighbor pair, we find that these modes become more distinct. Investigation of longitudinal spin-wave excitations reveals interactions between constituent components of the split transverse excitations. read less USED (low confidence) N. Gao, W. Setyawan, R. Kurtz, and Z. Wang, “Effects of applied strain on nanoscale self-interstitial cluster formation in BCC iron,” Journal of Nuclear Materials. 2017. link Times cited: 6 USED (low confidence) C. Gao, D. Tian, M. Li, and D.-zhi Qian, “Comparative study of displacement cascades simulated with ‘magnetic’ potentials and Mendelev-type potential in α-Fe,” Journal of Nuclear Materials. 2017. link Times cited: 7 USED (low confidence) W. Choi, Y. Kim, D. Seol, and B.-J. Lee, “Modified embedded-atom method interatomic potentials for the Co-Cr, Co-Fe, Co-Mn, Cr-Mn and Mn-Ni binary systems,” Computational Materials Science. 2017. link Times cited: 62 USED (low confidence) J. Bach, J. J. Möller, M. Göken, E. Bitzek, and H. Höppel, “On the transition from plastic deformation to crack initiation in the high- and very high-cycle fatigue regimes in plain carbon steels,” International Journal of Fatigue. 2016. link Times cited: 23 USED (low confidence) X.-Y. Liu et al., “Molecular dynamics simulation of thermal transport in UO2 containing uranium, oxygen, and fission-product defects,” Physical review applied. 2016. link Times cited: 36 Abstract: Uranium dioxide (UO2) is the most commonly used fuel in ligh… read moreAbstract: Uranium dioxide (UO2) is the most commonly used fuel in light water nuclear reactors and thermal conductivity controls the removal of heat produced by fission, therefore, governing fuel temperature during normal and accident conditions. The use of fuel performance codes by the industry to predict operational behavior is widespread. A primary source of uncertainty in these codes is thermal conductivity, and optimized fuel utilization may be possible if existing empirical models were replaced with models that incorporate explicit thermal conductivity degradation mechanisms during fuel burn-up. This approach is able to represent the degradation of thermal conductivity due to each individual defect type, rather than the overall burn-up measure typically used which is not an accurate representation of the chemical or microstructure state of the fuel that actually governs thermal conductivity and other properties. To generate a mechanistic thermal conductivity model, molecular dynamics (MD) simulations of UO2 thermal conductivity including representative intrinsic defects and fission products are carried out. These calculations employ a standard Buckingham type interatomic potential and a potential that combines the many-body embedded atom potential with Morse-Buckingham pair potentials. Potential parameters for UO2+x and ZrO2 are developed for the latter potential. Physical insights from the resonant phonon-spin scattering mechanism due to spins on the magnetic uranium ions have been introduced into the treatment of the MD results, with the corresponding relaxation time derived from existing experimental data. High defect scattering is predicted for Xe atoms compared to that of La and Zr ions. Intrinsic uranium defects reduce the thermal conductivity more than oxygen defects. For each defect and fission product, scattering parameters are derived for application in both a Callaway model and the corresponding high-temperature model typically used in fuel performance codes. The model is validated by comparison to low-temperature experimental measurements on single crystal hyper-stoichiometric UO2+x samples and high-temperature literature data. Ultimately, this work will enable more accurate fuel performance simulations as well as extension to new fuel types and operating conditions, all of which improve the fuel economics of nuclear energy and maintain high fuel reliability and safety. read less USED (low confidence) T. Swinburne, “Stochastic Dynamics of Crystal Defects.” 2015. link Times cited: 5 USED (low confidence) T. Shimada, K. Ouchi, I. Ikeda, Y. Ishii, and T. Kitamura, “Magnetic instability criterion for spin–lattice systems,” Computational Materials Science. 2015. link Times cited: 12 USED (low confidence) H. Wen and C. Woo, “Temperature dependence of enthalpies and entropies of formation and migration of mono-vacancy in BCC iron,” Journal of Nuclear Materials. 2014. link Times cited: 12 USED (low confidence) S. M. H. Haghighat et al., “Influence of the dislocation core on the glide of the ½ 110 edge dislocation in bcc-iron: An embedded atom method study,” Computational Materials Science. 2014. link Times cited: 14 USED (low confidence) R. Schäublin and S. M. H. Haghighat, “Molecular dynamics study of strengthening by nanometric void and Cr alloying in Fe,” Journal of Nuclear Materials. 2013. link Times cited: 18 USED (low confidence) J. Chen, N. Gao, P. Jung, and T. Sauvage, “A new mechanism of loop formation and transformation in bcc iron without dislocation reaction,” Journal of Nuclear Materials. 2013. link Times cited: 43 USED (low confidence) H. Wen, P. Ma, and C. Woo, “Spin-lattice dynamics study of vacancy formation and migration in ferromagnetic BCC iron,” Journal of Nuclear Materials. 2013. link Times cited: 20 USED (low confidence) J. Liu, R. Davidchack, and H. Dong, “Molecular dynamics calculation of solid–liquid interfacial free energy and its anisotropy during iron solidification,” Computational Materials Science. 2013. link Times cited: 41 USED (low confidence) K. Odbadrakh et al., “Coarse Grained Approach to First Principles Modeling of Radiation Cascade in Large Fe Supercells,” Journal of Physics: Conference Series. 2012. link Times cited: 2 Abstract: Classical Molecular Dynamics (MD) simulations characterizing… read moreAbstract: Classical Molecular Dynamics (MD) simulations characterizing dislocations and radiation damage typically treat 105-107 atoms. First principles techniques employed to understand systems at an atomistic level are not practical for such large systems consisting of millions of atoms. We present an efficient coarse grained (CG) approach to calculate local electronic and magnetic properties of large MD-generated structures from the first principles. Local atomic magnetic moments in crystalline Fe are perturbed by the presence of radiation generated vacancies and interstitials. The effects are most pronounced near the defect cores and decay slowly as the strain field of the defects decrease with distance. We develop the CG technique based on the Locally Self-consistent Multiple Scattering (LSMS) method that exploits the near-sightedness of the electron Green function. The atomic positions were determined by MD with an embedded atom force field. The local moments in the neighborhood of the defect cores are calculated with first-principles based on full local structure information. Atoms in the rest of the system are modeled by representative atoms with approximated properties. The calculations result in local moments near the defect centers with first-principles accuracy, while capturing coarse-grained details of local moments at greater length scales. This CG approach makes these large scale structures amenable to first principles study. read less USED (low confidence) M. Ojha, D. Nicholson, K. Odbadrakh, B. Radhakrishnan, R. Stoller, and T. Egami, “The use of atomic level stress to characterize the structure of irradiated iron,” Journal of Physics: Conference Series. 2012. link Times cited: 0 Abstract: The behaviour of irradiated material near a primary knock on… read moreAbstract: The behaviour of irradiated material near a primary knock on atom immediately after impact is of great importance for designing reactor materials. Currently, molecular dynamics simulations with classical force fields provide the foundation for understanding the resulting cascade. However, modern density functional calculations can now treat large enough numbers of atoms that they can provide additional details of the magnetic and electronic nature of irradiated samples. In this paper we calculate from first principles the atomic level stresses for an instantaneous configuration following the initiation of a low energy cascade in iron. read less USED (low confidence) D. Beaujouan, P. Thibaudeau, and C. Barreteau, “Anisotropic magnetic molecular dynamics of cobalt nanowires,” Physical Review B. 2012. link Times cited: 24 Abstract: An investigation of thermally induced spin and lattice dynam… read moreAbstract: An investigation of thermally induced spin and lattice dynamics of a cobalt nanowire on a (111)Pt substrate
is presented via magnetic molecular dynamics. This dynamical simulation model treats each atom as a particle
supporting a classical spin. A coordinate dependent on both exchange and anisotropic functions ensures a minimal
coupling between the spin and the lattice degrees of freedom to translate the magnetostrictive behavior of most
magnetic materials. A spin-pair model of anisotropy is proposed to connect to the lattice thermodynamics.
In order to solve linked spin-coordinate equations of motion, the efficiencies of algorithms based on SuzukiTrotter
decompositions are compared. The temperature dependence of the magnetic behavior of Co nanowires is
investigated through thermal stochastic connections with mechanical and spin Langevin noises. From a magnetic
Hamiltonian parametrized on ab initio calculations, the size dependence of the energy barriers and characteristic
time scales of the magnetization relaxation are computed. In the superparamagnetic limit, it is shown that all
spins in a nanowire evolve in a coherent rotation. When the size of the single nanowire increases, nucleations of
domain walls let the activation energy be independent of the length of the wire read less USED (low confidence) T. Swinburne, S. Dudarev, S. Fitzgerald, M. Gilbert, and A. Sutton, “Theory and simulation of the diffusion of kinks on dislocations in bcc metals,” Physical Review B. 2012. link Times cited: 58 Abstract: Isolated kinks on thermally fluctuating (1/2) screw, edge an… read moreAbstract: Isolated kinks on thermally fluctuating (1/2) screw, edge and (1/2) edge dislocations in bcc iron are simulated under zero stress conditions using molecular dynamics (MD). Kinks are seen to perform stochastic motion in a potential landscape that depends on the dislocation character and geometry, and their motion provides fresh insight into the coupling of dislocations to a heat bath. The kink formation energy, migration barrier and friction parameter are deduced from the simulations. A discrete Frenkel-Kontorova-Langevin (FKL) model is able to reproduce the coarse grained data from MD at a fraction of the computational cost, without assuming an a priori temperature dependence beyond the fluctuation-dissipation theorem. Analytic results reveal that discreteness effects play an essential r\^ole in thermally activated dislocation glide, revealing the existence of a crucial intermediate length scale between molecular and dislocation dynamics. The model is used to investigate dislocation motion under the vanishingly small stress levels found in the evolution of dislocation microstructures in irradiated materials. read less USED (low confidence) N. Gunkelmann, H. Ledbetter, and H. Urbassek, “Experimental and atomistic study of the elastic properties of α′ Fe–C martensite,” Acta Materialia. 2012. link Times cited: 38 USED (low confidence) H. Xu, Y. Osetsky, and R. Stoller, “Cascade annealing simulations of bcc iron using object kinetic Monte Carlo,” Journal of Nuclear Materials. 2012. link Times cited: 34 USED (low confidence) P. Ma, S. Dudarev, and C. Woo, “Spin-lattice dynamics model for magnon-phonon-electron heat transfer on a million atom scale,” Journal of Applied Physics. 2012. link Times cited: 4 Abstract: We develop an atomistic spin-lattice dynamics model for simu… read moreAbstract: We develop an atomistic spin-lattice dynamics model for simulating energy relaxation in magnetic materials. The model explicitly solves equations of motion for atoms and spins, and includes interaction with electron excitations. We apply the model to simulate the dynamics of propagation and attenuation of a compressive elastic wave in iron. We find that interaction between the lattice, spin and electron degrees of freedom does not have an appreciable effect on the velocity of the wave. At the same time, dissipative spin-lattice-electron interactions dominate the dynamics of attenuation of the wave in the material. read less USED (low confidence) N. N. Kumar, P. Durgaprasad, B. Dutta, and G. K. Dey, “Modeling of radiation hardening in ferritic/martensitic steel using multi-scale approach,” Computational Materials Science. 2012. link Times cited: 30 USED (low confidence) D. E. Smirnova, S. Starikov, S. Starikov, V. Stegailov, and V. Stegailov, “Interatomic potential for uranium in a wide range of pressures and temperatures,” Journal of Physics: Condensed Matter. 2012. link Times cited: 3 Abstract: Using the force-matching method we develop an interatomic po… read moreAbstract: Using the force-matching method we develop an interatomic potential that allows us to study the structure and properties of α-U, γ-U and liquid uranium. The potential is fitted to the forces, energies and stresses obtained from ab initio calculations. The model gives a good comparison with the experimental and ab initio data for the lattice constants of α-U and γ-U, the elastic constants, the room-temperature isotherm, the normal density isochore, the bond-angle distribution functions and the vacancy formation energies. The calculated melting line of uranium at pressures up to 80 GPa and the temperature of the α–γ transition at 3 GPa agree well with the experimental phase diagram of uranium. read less USED (low confidence) R. Drautz and D. Pettifor, “Valence-dependent analytic bond-order potential for magnetic transition metals,” Physical Review B. 2011. link Times cited: 39 Abstract: We extend the analytic bond-order potentials for transition … read moreAbstract: We extend the analytic bond-order potentials for transition metals [Phys. Rev. B 74, 174117 (2006)] to include ferro, antiferro, and noncollinear magnetism and charge transfer. This is achieved by first deriving a suitable tight-binding model through the expansion of the spin-density energy functional to second order with respect to magnetic and charge fluctuations. The tight-binding model is then approximated locally by the bond-order potential expansion, where the variational property of the bond-order potential expansion allows us to derive analytic expressions for the forces and torques on the atoms. From the bond-order potentials we then extract a hierarchy of multispin interactions beyond the conventional Heisenberg model. The explicit valence dependence of the bond-order potentials enables us to characterize the magnetic properties of the 3$d$ transition metals and to reproduce the trend from antiferromagnetic spin ordering close to the center of the $d$ band through noncollinear spin configurations to ferromagnetic ordering toward the edges of the $d$ band. The analytic representation of the energy within the bond-order potentials is then further expanded in the form of a Ginzburg-Landau expansion, deriving the prefactors explicitly from tight-binding and bond-order potentials. Thus, in this paper we present a coherent simplification from fundamental to empirical models of magnetism through coarse graining the electronic structure from spin-density functional theory to tight binding to bond-order potentials to the Ginzburg-Landau expansion. read less USED (low confidence) N. Gao, N. Gao, H. Swygenhoven, H. Swygenhoven, M. Victoria, and J. Chen, “Formation of dislocation loops during He clustering in bcc Fe,” Journal of Physics: Condensed Matter. 2011. link Times cited: 35 Abstract: The clustering of helium in bcc (body centered cubic) iron a… read moreAbstract: The clustering of helium in bcc (body centered cubic) iron and the growth of a helium bubble are simulated at the atomistic level for the helium-rich vacancy-poor condition. It is shown that a dislocation loop is formed as a sequential collection of 〈111〉 crowdions, the latter being the most stable self-interstitial atom configuration in the presence of a He cluster. read less USED (low confidence) F. Gao, H. Deng, H. Heinisch, and R. Kurtz, “A new Fe–He interatomic potential based on ab initio calculations in α-Fe,” Journal of Nuclear Materials. 2011. link Times cited: 82 USED (low confidence) J. Boutard, S. Dudarev, and M. Rieth, “Modelling structural and plasma facing materials for fusion power plants: Recent advances and outstanding issues in the EURATOM fusion materials programme,” Journal of Nuclear Materials. 2011. link Times cited: 11 USED (low confidence) A. A. Mazouzi, A. Álamo, D. Lidbury, D. Moinereau, and S. Dyck, “PERFORM 60: Prediction of the effects of radiation for reactor pressure vessel and in-core materials using multi-scale modelling – 60 years foreseen plant lifetime,” Nuclear Engineering and Design. 2011. link Times cited: 17 USED (low confidence) M. Všianská and M. Šob, “The effect of segregated sp-impurities on grain-boundary and surface structure, magnetism and embrittlement in nickel,” Progress in Materials Science. 2011. link Times cited: 119 USED (low confidence) B. Jelinek et al., “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Physical Review B. 2011. link Times cited: 218 Abstract: A set of modified embedded-atom method (MEAM) potentials for… read moreAbstract: A set of modified embedded-atom method (MEAM) potentials for the interactions between Al, Si, Mg, Cu, and Fe was developed from a combination of each element's MEAM potential in order to study metal alloying. Previously published MEAM parameters of single elements have been improved for better agreement to the generalized stacking fault energy (GSFE) curves when compared with ab initio generated GSFE curves. The MEAM parameters for element pairs were constructed based on the structural and elastic properties of element pairs in the NaCl reference structure garnered from ab initio calculations, with adjustment to reproduce the ab initio heat of formation of the most stable binary compounds. The new MEAM potentials were validated by comparing the formation energies of defects, equilibrium volumes, elastic moduli, and heat of formation for several binary compounds with ab initio simulations and experiments. Single elements in their ground-state crystal structure were subjected to heating to test the potentials at elevated temperatures. An Al potential was modified to avoid formation of an unphysical solid structure at high temperatures. The thermal expansion coefficient of a compound with the composition of AA 6061 alloy was evaluated and compared with experimental values. MEAM potential tests performed in this work, utilizing the universal atomistic simulation environment (ASE), are distributed to facilitate reproducibility of the results. read less USED (low confidence) B.-J. Lee, W. Ko, H.-K. Kim, and E.-H. Kim, “The modified embedded-atom method interatomic potentials and recent progress in atomistic simulations,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2010. link Times cited: 137 USED (low confidence) R. Anders and F. Haider, “Calculation of phase diagrams and simulation of segregation using Monte Carlo with lattice relaxation,” Journal of Nuclear Materials. 2010. link Times cited: 0 USED (low confidence) L. Malerba et al., “Comparison of empirical interatomic potentials for iron applied to radiation damage studies,” Journal of Nuclear Materials. 2010. link Times cited: 210 USED (low confidence) M. Lavrentiev, D. Nguyen-Manh, and S. Dudarev, “Cluster expansion models for Fe–Cr alloys, the prototype materials for a fusion power plant,” Computational Materials Science. 2010. link Times cited: 23 USED (low confidence) D. Terentyev, X. He, A. Serra, and J. Kuriplach, “Structure and strength of 〈1 1 0〉 tilt grain boundaries in bcc Fe: An atomistic study,” Computational Materials Science. 2010. link Times cited: 52 USED (low confidence) S. Dudarev et al., “Langevin model for real-time Brownian dynamics of interacting nanodefects in irradiated metals,” Physical Review B. 2010. link Times cited: 61 Abstract: In situ real-time electron microscope observations of metals… read moreAbstract: In situ real-time electron microscope observations of metals irradiated with ultrahigh-energy electrons or energetic ions show that the dynamics of microstructural evolution in these materials is strongly influenced by long-range elastic interactions between mobile nanoscale radiation defects. Treating long-range interactions is also necessary for modeling microstructures formed in ex situ high-dose-rate ion-beam irradiation experiments, and for interpolating the ion-beam irradiation data to the low-dose-rate limit characterizing the neutron irradiation environments of fission or fusion power plants. We show that simulations, performed using an algorithm where nanoscale radiation defects are treated as interacting Langevin particles, are able to match and explain the real-time dynamics of nanodefects observed in in situ electron microscope experiments. read less USED (low confidence) D. Terentyev, Y. Osetsky, and D. Bacon, “Effects of temperature on structure and mobility of the 〈1 0 0〉 edge dislocation in body-centred cubic iron,” Acta Materialia. 2010. link Times cited: 44 USED (low confidence) M. Gilbert and S. Dudarev, “Ab initio multi-string Frenkel–Kontorova model for a b = a/2[111] screw dislocation in bcc iron,” Philosophical Magazine. 2010. link Times cited: 23 Abstract: We formulate a multi-string Frenkel–Kontorova (MSFK) model f… read moreAbstract: We formulate a multi-string Frenkel–Kontorova (MSFK) model for a a/2[111] screw dislocation in bcc iron, and investigate the occurrence of degenerate and non-degenerate dislocation core structures as functionals of the law of interaction between the [111] strings of atoms forming the crystal. By comparing the effective inter-string interaction laws derived from ab initio density functional calculations and from semi-empirical interatomic potentials for α-iron, we show that it is the form of the function determining how the atomic strings interact with each other as a function of their relative one-dimensional displacement in the [111] direction that determines whether a degenerate or a non-degenerate screw dislocation core configuration has lower energy. We show that by constructing a one-dimensional inter-string interaction law, and by solving the MSFK equations, it is possible to easily predict the nature of the screw dislocation core, hence providing a simple yet effective check to aid the development of short-range semi-empirical interatomic potentials for bcc transition metals. Finally, we analyse the relation between the inter-string interaction law, and the shape and the height of the Peierls energy barriers separating the adjacent equilibrium configurations for a migrating screw dislocation. read less USED (low confidence) L. Sandoval, H. Urbassek, and P. Entel, “Solid-solid phase transitions and phonon softening in an embedded-atom method model for iron,” Physical Review B. 2009. link Times cited: 36 USED (low confidence) M. Samaras, “Multiscale Modelling: the role of helium in iron,” Materials Today. 2009. link Times cited: 73 USED (low confidence) D. Duffy, S. Khakshouri, and A. Rutherford, “Electronic effects in radiation damage simulations,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2009. link Times cited: 23 USED (low confidence) C. Fazio et al., “European cross-cutting research on structural materials for Generation IV and transmutation systems,” Journal of Nuclear Materials. 2009. link Times cited: 67 USED (low confidence) S. M. H. Haghighat, G. Lucas, and R. Schaeublin, “Atomistic simulation of He bubble in Fe as obstacle to dislocation.” 2009. link Times cited: 14 Abstract: Degradation of mechanical properties due to nanometric irrad… read moreAbstract: Degradation of mechanical properties due to nanometric irradiation induced defects is one of the challenging issues in designing materials for future fusion reactors. Various types of defects such as voids and He bubbles may be produced due to high dose of neutron irradiation due to fusion reaction. We study the influence of He bubble on the mobility of an edge dislocation in pure bcc-Fe using molecular dynamics simulation as a function of bubble size, He density and temperature. It appears that low contents He bubbles are penetrable defects, which size and temperature rise make them harder and softer, respectively. At high He contents a size dependent loop punching is observed, which at larger bubble sizes leads to a multistep dislocation-defect interaction. It also appears that the bubble surface curvature and temperature are the main parameters in the screw segments annihilation needed for the release of the dislocation from the bubble. read less USED (low confidence) G. Lucas and R. Schäublin, “Stability of helium bubbles in alpha-iron: A molecular dynamics study,” Journal of Nuclear Materials. 2009. link Times cited: 44 USED (low confidence) S. Chiesa, P. Derlet, S. Dudarev, and H. Swygenhoven, “Atomistic calculation of elastic constants of alpha-iron containing point defects by means of magnetic interatomic potentials,” Journal of Nuclear Materials. 2009. link Times cited: 5 USED (low confidence) S. Dudarev, P. Derlet, and R. Bullough, “The magnetic origin of anomalous high-temperature stability of dislocation loops in iron and iron-based alloys,” Journal of Nuclear Materials. 2009. link Times cited: 16 USED (low confidence) M. Gilbert, Z. Yao, M. Kirk, M. Jenkins, and S. Dudarev, “Vacancy defects in Fe: Comparison between simulation and experiment,” Journal of Nuclear Materials. 2009. link Times cited: 35 USED (low confidence) D. Duffy and A. Rutherford, “Including electronic effects in damage cascade simulations,” Journal of Nuclear Materials. 2009. link Times cited: 16 USED (low confidence) N. Gao, C. Fu, M. Samaras, R. Schäublin, M. Victoria, and W. Hoffelner, “Multiscale modelling of bi-crystal grain boundaries in bcc iron,” Journal of Nuclear Materials. 2009. link Times cited: 38 USED (low confidence) P. Korzhavyi, A. Ruban, J. Odqvist, J. Nilsson, and B. Johansson, “Electronic structure and effective chemical and magnetic exchange interactions in bcc Fe-Cr alloys,” Physical Review B. 2009. link Times cited: 90 Abstract: Electronic structure calculations are employed in order to i… read moreAbstract: Electronic structure calculations are employed in order to investigate the cohesive properties (lattice parameter, enthalpy of formation, and bulk modulus) of random Fe-Cr alloys as a function of composition and magnetic state, as well as to derive the chemical and magnetic exchange interactions of the constituent atoms. The calculations predict certain anomalies in the cohesive properties of ferromagnetic alloys at a concentration of about $7\text{ }\text{at}\text{ }%$ Cr; these anomalies may be related to the changes in Fermi-surface topology that occur with composition in this alloy system. The obtained interatomic interactions are used as parameters in the configurational (Ising) and magnetic (Heisenberg) Hamiltonians for modeling finite-temperature thermodynamic properties of the alloys. We discuss the approximations and limitations of similar modeling approaches, investigate the origin of existing difficulties, and analyze possible ways of extending the theoretical models in order to capture the essential physics of interatomic interactions in the Fe-Cr or similar alloys where magnetism plays a crucial role in the phase stability. read less USED (low confidence) D. Terentyev, L. Malerba, P. Klaver, and P. Olsson, “Formation of stable sessile interstitial complexes in reactions between glissile dislocation loops in bcc Fe,” Journal of Nuclear Materials. 2008. link Times cited: 34 USED (low confidence) S. M. H. Haghighat, J. Fikar, and R. Schäublin, “Effect of interatomic potential on the behavior of dislocation-defect interaction simulation in α-Fe,” Journal of Nuclear Materials. 2008. link Times cited: 35 USED (low confidence) M. Samaras and M. Victoria, “Modelling in nuclear energy environments,” Materials Today. 2008. link Times cited: 27 USED (low confidence) D. Nguyen-Manh, M. Lavrentiev, and S. Dudarev, “Ab initio and Monte Carlo modeling in Fe–Cr system: Magnetic origin of anomalous thermodynamic and kinetic properties,” Computational Materials Science. 2008. link Times cited: 15 USED (low confidence) J. Kuriplach, O. Melikhova, M. Hou, S. Petegem, E. Zhurkin, and M. Šob, “Positron annihilation in vacancies at grain boundaries in metals,” Applied Surface Science. 2008. link Times cited: 9 USED (low confidence) D. Hepburn and G. Ackland, “Metallic-covalent interatomic potential for carbon in iron,” Physical Review B. 2008. link Times cited: 118 Abstract: Existing interatomic potentials for the iron-carbon system s… read moreAbstract: Existing interatomic potentials for the iron-carbon system suffer from qualitative flaws in describing even the simplest of defects. In contrast to more accurate first-principles calculations, all previous potentials show strong bonding of carbon to overcoordinated defects (e.g., self-interstitials, dislocation cores) and a failure to accurately reproduce the energetics of carbon-vacancy complexes. Thus any results from their application in molecular dynamics to more complex environments are unreliable. The problem arises from a fundamental error in potential design--the failure to describe short-ranged covalent bonding of the carbon p electrons. We describe a resolution to the problem and present an empirical potential based on insights from density-functional theory, showing covalent-type bonding for carbon. The potential correctly describes the interaction of carbon and iron across a wide range of defect environments. It has the embedded atom method form and hence appropriate for billion atom molecular-dynamics simulations. read less USED (low confidence) D. Bacon, “Simulation of the interaction between an edge dislocation and a ’100’ interstitial dislocation loop in alpha-iron.” 2008. link Times cited: 113 USED (low confidence) D. Terentyev and L. Malerba, “Interaction of 〈1 0 0〉 and ½〈1 1 1〉 dislocation loops with point defects in ferritic alloys,” Journal of Nuclear Materials. 2008. link Times cited: 13 USED (low confidence) G. Ackland, A. Jones, and R. Noble-Eddy, “Molecular dynamics simulations of the martensitic phase transition process,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2008. link Times cited: 31 USED (low confidence) D. Nguyen-Manh, M. Lavrentiev, and S. Dudarev, “The Fe–Cr system: atomistic modelling of thermodynamics and kinetics of phase transformations.” 2008. link Times cited: 50 USED (low confidence) K. Nordlund and S. Dudarev, “Interatomic potentials for simulating radiation damage effects in metals,” Comptes Rendus Physique. 2008. link Times cited: 29 USED (low confidence) N. Negulyaev, V. Stepanyuk, W. Hergert, P. Bruno, and J. Kirschner, “Atomic-scale self-organization of Fe nanostripes on stepped Cu(111) surfaces: Molecular dynamics and kinetic Monte Carlo simulations,” Physical Review B. 2008. link Times cited: 24 Abstract: Growth of Fe nanostripes on a vicinal Cu(111) surface is inv… read moreAbstract: Growth of Fe nanostripes on a vicinal Cu(111) surface is investigated on the atomic scale by performing molecular dynamics and kinetic Monte Carlo simulations. We involve in our study the kinetic mechanisms of atomic incorporation recently reported by Mo et al. [Phys. Rev. Lett. 94, 155503 (2005)]. The atomistic processes responsible for the interlayer mass transport and the formation of Fe stripes of 1 ML height are identified. We demonstrate that strain relaxations at steps have a strong impact on the self-assembly of one-dimensional Fe atomic structures on vicinal Cu(111). read less USED (low confidence) M. Marinica and F. Willaime, “Orientation of Interstitials in Clusters in α-Fe: A Comparison between Empirical Potentials,” Solid State Phenomena. 2007. link Times cited: 22 Abstract: We have addressed two issues concerning the relative stabili… read moreAbstract: We have addressed two issues concerning the relative stabilities of various orienta- tions of interstitial clusters in iron by making a comprehensive comparison between four recent empirical potentials. First, we have investigated the effect of finite temperature on the com- petition between clusters made of a few dumbbells oriented along h111i or h110i. We show by quasi-harmonic calculations that h111i clusters have much larger vibrational formation en- tropies and that they are therefore stabilized with respect to h110i clusters at high temperature. Second, we have compared the formation energies of loops with several hundred atoms with Burgers vector 1 2 h111i or h100i. The 1 2 h111i loops are found to be always more stable, but the energy differences with h100i loops depend strongly on the potential. read less USED (low confidence) R. Lässer et al., “Structural materials for DEMO: The EU development, strategy, testing and modelling.” 2007. link Times cited: 66 USED (low confidence) N. Baluc et al., “Status of R&D activities on materials for fusion power reactors,” Nuclear Fusion. 2007. link Times cited: 136 Abstract: Current R&D activities on materials for fusion power reactor… read moreAbstract: Current R&D activities on materials for fusion power reactors are mainly focused on plasma facing, structural and tritium breeding materials for plasma facing (first wall, divertor) and breeding blanket components. Most of these activities are being performed in Europe, Japan, the People's Republic of China, Russia and the USA. They relate to the development of new high temperature, radiation resistant materials, the development of coatings that will act as erosion, corrosion, permeation and/or electrical/MHD barriers, characterization of candidate materials in terms of mechanical and physical properties, assessment of irradiation effects, compatibility experiments, development of reliable joints, and development and/or validation of design rules. Priorities defined worldwide in the field of materials for fusion power reactors are summarized, as well as the main achievements obtained during the last few years and the near-term perspectives in the different investigation areas. read less USED (low confidence) M. Samaras, W. Hoffelner, and M. Victoria, “Modelling of advanced structural materials for GEN IV reactors,” Journal of Nuclear Materials. 2007. link Times cited: 28 USED (low confidence) J. Kuriplach, O. Melikhova, M. Hou, S. Petegem, E. Zhurkin, and M. Šob, “Positron annihilation at grain boundaries in metals,” Physica Status Solidi (c). 2007. link Times cited: 5 Abstract: Positron annihilation at selected tilt and twist grain
bound… read moreAbstract: Positron annihilation at selected tilt and twist grain
boundaries in iron and nickel is examined theoretically. First
the atomic structure of studied perfect and imperfect grain
boundaries is obtained using molecular dynamics simulations.
Characteristics of positrons trapped at such GBs are then
calculated employing the atomic superposition method and are
related to free volumes found at GBs. It is observed that in
some cases vacancies introduced into ideal grain boundaries do
not result in an increase of the positron lifetime. read less USED (low confidence) P. V. Zwol, P. Derlet, H. Swygenhoven, and S. Dudarev, “BCC Fe surface and cluster magnetism using a magnetic potential,” Surface Science. 2007. link Times cited: 13 USED (low confidence) S. Dudarev and P. Derlet, “Molecular dynamics modelling of radiation defects in ferromagnetic α-iron,” Journal of Nuclear Materials. 2007. link Times cited: 4 USED (low confidence) L. Malerba et al., “Modelling of Radiation Damage in Fe-Cr Alloys,” Journal of Astm International. 2007. link Times cited: 29 Abstract: High-Cr ferritic/martensitic steels are being considered as … read moreAbstract: High-Cr ferritic/martensitic steels are being considered as structural materials for a large number of future nuclear applications, from fusion to accelerator-driven systems and GenIV reactors. Fe-Cr alloys can be used as model materials to investigate some of the mechanisms governing their microstructure evolution under irradiation and its correlation to changes in their macroscopic properties. Focusing on these alloys, we show an example of how the integration of computer simulation and theoretical models can provide keys for the interpretation of a host of relevant experimental observations. In particular we show that proper accounting for two basic features of these alloys, namely, the existence of a fairly strong attractive interaction between self-interstitials and Cr atoms and of a mixing enthalpy that changes sign from negative to positive around 8 to 10 % Cr, is a necessary and, to a certain extent, sufficient condition to rationalize and understand their behavior under irradiation. These features have been revealed by ab initio calculations, are supported by experimental evidence, and have been adequately transferred into advanced empirical interatomic potentials, which have been and are being used for the simulation of damage production, defect behavior, and phase transformation in these alloys. The results of the simulations have been and are being used to parameterize models capable of extending the description of radiation effects to scales beyond the reach of molecular dynamics. The present paper intends to highlight the most important achievements and results of this research activity. read less USED (low confidence) C. Ortiz and M. Caturla, “Simulation of defect evolution in irradiated materials: Role of intracascade clustering and correlated recombination,” Physical Review B. 2007. link Times cited: 73 Abstract: The evolution of damage produced by collision cascades in Fe… read moreAbstract: The evolution of damage produced by collision cascades in Fe is studied using both kinetic Monte Carlo (kMC) and rate theory (RT) approaches. The initial damage distribution is obtained from molecular-dynamics simulations of 30 keV recoils in Fe. An isochronal annealing is simulated to identify the different thermally activated mechanisms that govern defect evolution. When clusters form during collision cascades, kMC simulations show that additional recovery peaks should be expected, in comparison to recovery curves obtained under electron irradiation conditions. Detailed kMC and RT simulations reveal that some of these recovery peaks are due to correlated recombinations at low temperature between defects. In particular, we show that under cascade-damage conditions it is possible to observe correlated recombinations between vacancies and self-interstitial clusters. These correlated recombinations cannot be reproduced with a RT model, and therefore kMC and RT differ at low temperature. However, for the conditions presented here, the contribution of correlated recombination is very small and therefore no significant differences are observed at high temperatures between these two models. read less USED (low confidence) D. Nguyen-Manh, V. Vítek, and A. Horsfield, “Environmental dependence of bonding: A challenge for modelling of intermetallics and fusion materials,” Progress in Materials Science. 2007. link Times cited: 63 USED (low confidence) F. Djurabekova, L. Malerba, C. Domain, and C. Becquart, “Stability and mobility of small vacancy and copper-vacancy clusters in bcc-Fe: An atomistic kinetic Monte Carlo study,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 34 USED (low confidence) J. Wallenius, P. Olsson, L. Malerba, and D. Terentyev, “Simulation of thermal ageing and radiation damage in Fe–Cr,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 18 USED (low confidence) R. Drautz and D. Pettifor, “Valence-dependent analytic bond-order potential for transition metals,” Physical Review B. 2006. link Times cited: 79 Abstract: An analytic interatomic bond-order potential is derived that… read moreAbstract: An analytic interatomic bond-order potential is derived that depends explicitly on the valence of the transition-metal element. It generalizes the second-moment Finnis-Sinclair and fourth-moment Carlsson potentials to include higher moments. We find that the sixth-moment approximation predicts not only the structural trend from $\mathrm{hcp}\ensuremath{\rightarrow}\mathrm{bcc}\ensuremath{\rightarrow}\mathrm{hcp}\ensuremath{\rightarrow}\mathrm{fcc}$ that is observed across the nonmagnetic $4d$ and $5d$ transition-metal series, but also the different ferromagnetic moments of the bcc, fcc, and hcp phases of the $3d$ transition-metal iron. An analytic expression for the force is obtained and proved to converge to the Hellmann-Feynman force as higher moments are included. read less USED (low confidence) G. Ackland, “Magnetically induced immiscibility in the Ising model of FeCr stainless steel.,” Physical review letters. 2006. link Times cited: 39 Abstract: The iron-chromium alloy system has an unexplained anomaly: a… read moreAbstract: The iron-chromium alloy system has an unexplained anomaly: although there is a broad miscibility gap it appears to be favorable for chromium to dissolve in iron. This is consistent with ab initio calculation, but no simpler physically intuitive picture has been presented. Here it is shown that the Ising model, based on the bcc lattice with antiferromagnetic and ferromagnetic species, has the potential to exhibit similar behavior, with a skew miscibility gap arising from the solubility of antiferromagnetic species on nonadjacent sites. Essential characteristics of stainless steel (high Cr solubility and surface segregation) are correctly reproduced. Under some conditions, magnetization increases with temperature. The equilibrium miscibility gap due to mixed magnetism and segregation-driven positive dM/dT are fundamental features of the bcc Ising model itself, not just FeCr. read less USED (low confidence) I. Cook, “Materials research for fusion energy,” Nature Materials. 2006. link Times cited: 91 USED (low confidence) R. Devanathan, “Interatomic Potentials for Nuclear Materials,” Handbook of Materials Modeling. 2020. link Times cited: 1 USED (low confidence) L. Malerba, “Large Scale Integrated Materials Modeling Programs.” 2020. link Times cited: 2 USED (low confidence) M. Caturla, “Object kinetic Monte Carlo methods applied to modeling radiation effects in materials,” Computational Materials Science. 2019. link Times cited: 10 USED (low confidence) J. J. Möller and E. Bitzek, “Atomic-scale modeling of elementary processes during the fatigue of metallic materials: from crack initiation to crack-microstructure interactions.” 2018. link Times cited: 1 USED (low confidence) Y. Umeno, T. Shimada, Y. Kinoshita, and T. Kitamura, “Methodology of Quantum Mechanics/Atomic Simulations.” 2017. link Times cited: 0 USED (low confidence) M. Eisenbach, D. Perera, D. Landau, D. Nicholson, J. Yin, and G. K. Brown, “Magnetic Materials at finite Temperatures: thermodynamics and combined spin and molecular dynamics derived from first principles calculations,” Journal of Physics: Conference Series. 2015. link Times cited: 3 Abstract: We present a unified approach to describe the combined behav… read moreAbstract: We present a unified approach to describe the combined behavior of the atomic and magnetic degrees of freedom in magnetic materials. Using Monte Carlo simulations directly combined with first principles the Curie temperature can be obtained ab initio in good agreement with experimental values. The large scale constrained first principles calculations have been used to construct effective potentials for both the atomic and magnetic degrees of freedom that allow the unified study of influence of phonon-magnon coupling on the thermodynamics and dynamics of magnetic systems. The MC calculations predict the specific heat of iron in near perfect agreement with experimental results from 300K to above Tc and allow the identification of the importance of the magnon-phonon interaction at the phase-transition. Further Molecular Dynamics and Spin Dynamics calculations elucidate the dynamics of this coupling and open the potential for quantitative and predictive descriptions of dynamic structure factors in magnetic materials using first principles derived simulations. read less USED (low confidence) T. Swinburne, “Atomistic Simulations in bcc Metals.” 2015. link Times cited: 0 USED (low confidence) R. Stoller and E. Zarkadoula, “Primary Radiation Damage Formation in Solids.” 2014. link Times cited: 11 USED (low confidence) S. Lu, D. Li, and D. Brenner, “Molecular Dynamics Simulations of Plastic Damage in Metals.” 2014. link Times cited: 6 USED (low confidence) W. Hoffelner, “Advanced Mechanical Testing and Analysis Methods.” 2013. link Times cited: 0 USED (low confidence) G. Ackland, “1.10 – Interatomic Potential Development.” 2012. link Times cited: 10 USED (low confidence) H. Urbassek and L. Sandoval, “Molecular dynamics modeling of martensitic transformations in steels.” 2012. link Times cited: 17 Abstract: Abstract: Molecular dynamics simulation constitutes an appea… read moreAbstract: Abstract: Molecular dynamics simulation constitutes an appealing method to study, on an atomistic basis, the processes and mechanisms of martensitic phase transformations. Its use requires the existence of reliable interatomic potentials which adequately describe the properties of the phases. In this review we present a few recent examples demonstrating the application of this method to the study of the martensitic phase transition in iron. Besides phase changes in bulk materials, transformations in small systems (nanowires) are also considered. read less USED (low confidence) P. K. Soin, A. Horsfield, and D. Nguyen-Manh, “Magnetic Tight-Binding Simulations of Defects in Iron,” MRS Proceedings. 2011. link Times cited: 0 USED (low confidence) E. Hristova, R. Janisch, R. Drautz, and A. Hartmaier, “Solubility of carbon in α-iron under volumetric strain and close to the Σ5(3 1 0)[0 0 1] grain boundary: Comparison of DFT and empirical potential methods,” Computational Materials Science. 2011. link Times cited: 46 USED (low confidence) D. Bacon, Y. Osetsky, and D. Rodney, “Chapter 88 Dislocation–Obstacle Interactions at the Atomic Level.” 2009. link Times cited: 114 USED (low confidence) J. Boutard, S. Dudarev, and E. Diegele, “Radiation Effects in Structural Materials for Fusion Power Plants: the Outcomes of the EU Fusion Program.” 2008. link Times cited: 9 USED (low confidence) H. Swygenhoven and P. Derlet, “Chapter 81 – Atomistic Simulations of Dislocations in FCC Metallic Nanocrystalline Materials.” 2008. link Times cited: 27 USED (low confidence) M. Samaras, M. Victoria, and W. Hoffelner, “The Structure, Role and Flexibility of Grain Boundaries,” MRS Proceedings. 2008. link Times cited: 0 USED (low confidence) D. Duffy and A. Rutherford, “Including the Effects of Electronic Excitations and Electron-Phonon Coupling in Cascade Simulations.” 2006. link Times cited: 0 Abstract: Radiation damage has traditionally been modeled using cascad… read moreAbstract: Radiation damage has traditionally been modeled using cascade simulations however such simulations generally neglect the effects of electron-ion interactions, which may be significant in high energy cascades. A model has been developed which includes the effects of electronic stopping and electron-phonon coupling in Molecular Dynamics simulations by means of an inhomogeneous Langevin thermostat. The energy lost by the atoms to electronic excitations is gained by the electronic system and the energy evolution of the electronic system is modeled by the heat diffusion equation. Energy is exchanged between the electronic system and the atoms in the Molecular Dynamics simulation by means of a Langevin thermostat, the temperature of which is the local electronic temperature. The model is applied to a 10 keV cascade simulation for Fe. (authors) read less NOT USED (low confidence) M. Samaras, M. Victoria, and W. Hoffelner, “Advanced materials modelling – E.U. perspectives,” Journal of Nuclear Materials. 2009. link Times cited: 11 NOT USED (high confidence) L. Patra and B. Liao, “Indirect Exchange Interaction Leads to Large Lattice Contribution to Magnetocaloric Entropy Change.,” Physical review letters. 2023. link Times cited: 0 Abstract: Materials with a large magnetocaloric response are highly de… read moreAbstract: Materials with a large magnetocaloric response are highly desirable for magnetic cooling applications. It is suggested that a strong spin-lattice coupling tends to generate a large magnetocaloric effect, but no microscopic mechanism has been proposed. In this Letter, we use spin-lattice dynamics simulation to examine the lattice contribution to the magnetocaloric entropy change in bcc iron (Fe) and hcp gadolinium (Gd) with exchange interaction parameters determined from ab initio calculations. We find that indirect Ruderman-Kittel-Kasuya-Yosida (RKKY) exchange interaction in hcp Gd leads to longer-range spin-lattice coupling and more strongly influences the low-frequency long-wavelength phonons. This results in a higher lattice contribution toward the total magnetocaloric entropy change as compared to bcc Fe with short-range direct exchange interactions. Our analysis provides a framework for understanding the magnetocaloric effect in magnetic materials with strong spin-lattice couplings. Our finding suggests that long-range indirect RKKY-type exchange gives rise to a larger lattice contribution to the magnetocaloric entropy change and is, thus, beneficial for magnetocaloric materials. read less NOT USED (high confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 NOT USED (high confidence) A. Front et al., “Simulation of thermodynamic properties of magnetic transition metals from an efficient tight-binding model: The case of cobalt and beyond,” Physical Review B. 2021. link Times cited: 0 Abstract: Alexis Front, ∗ Georg Daniel Förster, 2, † Van-Truong Tran, … read moreAbstract: Alexis Front, ∗ Georg Daniel Förster, 2, † Van-Truong Tran, Chu-Chun Fu, Cyrille Barreteau, François Ducastelle, and Hakim Amara 5, ‡ Laboratoire d’Etude des Microstructures, ONERA-CNRS, UMR104, Université Paris-Saclay, BP 72, Châtillon Cedex, 92322, France Interfaces, Confinement, Matériaux et Nanostructures (ICMN), CNRS, Université d’Orléans, Orléans, France Université Paris-Saclay, CEA, Service de Recherches de Métallurgie Physique, 91191 Gif-sur-Yvette, France DRF-Service de Physique de l’Etat Condensé, CEA-CNRS, Université Paris-Saclay, F-91191 Gif-sur-Yvette, France Université de Paris, Laboratoire Matériaux et Phénomènes Quantiques (MPQ), CNRS-UMR7162, 75013 Paris, France read less NOT USED (high confidence) A. Goryaeva et al., “Efficient and transferable machine learning potentials for the simulation of crystal defects in bcc Fe and W,” Physical Review Materials. 2021. link Times cited: 20 NOT USED (high confidence) H. Zapolsky, A. Vaugeois, R. Patte, and G. Demange, “Size-Dependent Solute Segregation at Symmetric Tilt Grain Boundaries in α-Fe: A Quasiparticle Approach Study,” Materials. 2021. link Times cited: 2 Abstract: In the present work, atomistic modeling based on the quasipa… read moreAbstract: In the present work, atomistic modeling based on the quasiparticle approach (QA) was performed to establish general trends in the segregation of solutes with different atomic size at symmetric 〈100〉 tilt grain boundaries (GBs) in α-Fe. Three types of solute atoms X1, X2 and X3 were considered, with atomic radii smaller (X1), similar (X2) and larger (X3) than iron atoms, respectively, corresponding to phosphorus (P), antimony (Sb) and tin (Sn). With this, we were able to evidence that segregation is dominated by atomic size and local hydrostatic stress. For low angle GBs, where the elastic field is produced by dislocation walls, X1 atoms segregate preferentially at the limit between compressed and dilated areas. Contrariwise, the positions of X2 atoms at GBs reflect the presence of tensile and compressive areal regions, corresponding to extremum values of the σXX and σYY components of the strain tensor. Regarding high angle GBs Σ5 (310) (θ = 36.95°) and Σ29 (730), it was found that all three types of solute atoms form Fe9X clusters within B structural units (SUs), albeit being deformed in the case of larger atoms (X2 and X3). In the specific case of Σ29 (730) where the GB structure can be described by a sequence of |BC.BC| SUs, it was also envisioned that the C SU can absorb up to four X1 atoms vs. one X2 or X3 atom only. Moreover, a depleted zone was observed in the vicinity of high angle GBs for X2 or X3 atoms. The significance of this research is the development of a QA methodology capable of ascertaining the atomic position of solute atoms for a wide range of GBs, as a mean to highlight the impact of the solute atoms’ size on their locations at and near GBs. read less NOT USED (high confidence) M. Eckhoff and J. Behler, “High-dimensional neural network potentials for magnetic systems using spin-dependent atom-centered symmetry functions,” npj Computational Materials. 2021. link Times cited: 29 NOT USED (high confidence) M. Strungaru, M. O. A. Ellis, S. Ruta, O. Chubykalo-Fesenko, R. Evans, and R. Chantrell, “Spin-lattice dynamics model with angular momentum transfer for canonical and microcanonical ensembles,” Physical Review B. 2021. link Times cited: 10 Abstract: A unified model of molecular and atomistic spin dynamics is … read moreAbstract: A unified model of molecular and atomistic spin dynamics is presented enabling simulations both in microcanonical and canonical ensembles without the necessity of additional phenomenological spin damping. Transfer of energy and angular momentum between the lattice and the spin systems is achieved by a phenomenological coupling term representing the spin-orbit interaction. The characteristic spectra of the spin and phonon systems are analyzed for different coupling strength and temperatures. The spin spectral density shows magnon modes together with the uncorrelated noise induced by the coupling to the lattice. The effective damping parameter is investigated showing an increase with both coupling strength and temperature. The model paves the way to understanding magnetic relaxation processes beyond the phenomenological approach of the Gilbert damping and the dynamics of the energy transfer between lattice and spins. read less NOT USED (high confidence) I. Novikov, B. Grabowski, F. Körmann, and A. Shapeev, “Magnetic Moment Tensor Potentials for collinear spin-polarized materials reproduce different magnetic states of bcc Fe,” npj Computational Materials. 2020. link Times cited: 50 NOT USED (high confidence) R. Meyer et al., “Vibrational and magnetic signatures of extended defects in Fe,” The European Physical Journal B. 2020. link Times cited: 5 NOT USED (high confidence) R. Drautz, “Atomic cluster expansion of scalar, vectorial, and tensorial properties including magnetism and charge transfer,” Physical Review B. 2020. link Times cited: 55 Abstract: The atomic cluster expansion (Drautz, Phys. Rev. B 99, 01410… read moreAbstract: The atomic cluster expansion (Drautz, Phys. Rev. B 99, 014104 (2019)) is extended in two ways, the modelling of vectorial and tensorial atomic properties and the inclusion of atomic degrees of freedom in addition to the positions of the atoms. In particular, atomic species, magnetic moments and charges are attached to the atomic positions and an atomic cluster expansion that includes the different degrees of freedom on equal footing is derived. Expressions for the efficient evaluation of forces and torques are given. Relations to other methods are discussed. read less NOT USED (high confidence) S. Mi, H. Gong, and J. Fan, “Structural stability and mechanical property of Fe-W solid solutions from a constructed Fe-W potential,” Journal of Applied Physics. 2019. link Times cited: 15 Abstract: An Fe-W potential has been constructed by means of the embed… read moreAbstract: An Fe-W potential has been constructed by means of the embedded-atom method and has proven to be more realistic than the three published Fe-W potentials in the literature. Based on the constructed Fe-W potential, molecular dynamic simulation has been used to reveal structural stability, thermodynamic properties, and mechanical properties of BCC Fe-W solid solutions within the entire composition range. It is found that the Fe-W interaction in BCC Fe-W solid solutions should be weak and attractive with small and negative heats of formation, which agree well with those from the thermodynamic Miedema model and could clarify the controversy regarding heats of formation of Fe-W solid solutions in the literature. In addition, the derived coefficient of thermal expansion, heat capacity, elastic constants, and elastic moduli of Fe-W solid solutions from the present Fe-W potential are in good agreement with the corresponding data from ab initio calculation or experiments in the literature.An Fe-W potential has been constructed by means of the embedded-atom method and has proven to be more realistic than the three published Fe-W potentials in the literature. Based on the constructed Fe-W potential, molecular dynamic simulation has been used to reveal structural stability, thermodynamic properties, and mechanical properties of BCC Fe-W solid solutions within the entire composition range. It is found that the Fe-W interaction in BCC Fe-W solid solutions should be weak and attractive with small and negative heats of formation, which agree well with those from the thermodynamic Miedema model and could clarify the controversy regarding heats of formation of Fe-W solid solutions in the literature. In addition, the derived coefficient of thermal expansion, heat capacity, elastic constants, and elastic moduli of Fe-W solid solutions from the present Fe-W potential are in good agreement with the corresponding data from ab initio calculation or experiments in the literature. read less NOT USED (high confidence) E. Higgins, P. Hasnip, and M. Probert, “Simultaneous Prediction of the Magnetic and Crystal Structure of Materials Using a Genetic Algorithm,” Crystals. 2019. link Times cited: 6 Abstract: We introduce a number of extensions and enhancements to a ge… read moreAbstract: We introduce a number of extensions and enhancements to a genetic algorithm for crystal structure prediction, to make it suitable to study magnetic systems. The coupling between magnetic properties and crystal structure means that it is essential to take a holistic approach, and we present for the first time, a genetic algorithm that performs a simultaneous global optimisation of both magnetic structure and crystal structure. We first illustrate the power of this approach on a novel test system—the magnetic Lennard–Jones potential—which we define. Then we study the complex interface structures found at the junction of a Heusler alloy and a semiconductor substrate as found in a proposed spintronic device and show the impact of the magnetic interface structure on the device performance. read less NOT USED (high confidence) J. Tranchida, S. Plimpton, P. Thibaudeau, and A. Thompson, “Massively parallel symplectic algorithm for coupled magnetic spin dynamics and molecular dynamics,” J. Comput. Phys. 2018. link Times cited: 76 NOT USED (high confidence) I. Toda-Caraballo, J. Wróbel, D. Nguyen-Manh, P. Pérez, and P. Rivera-Díaz-del-Castillo, “Simulation and Modeling in High Entropy Alloys,” JOM. 2017. link Times cited: 27 NOT USED (high confidence) H. Lambert, Á. Fekete, J. Kermode, and A. Vita, “Imeall: A computational framework for the calculation of the atomistic properties of grain boundaries,” Comput. Phys. Commun. 2017. link Times cited: 10 NOT USED (high confidence) Z. Zheng et al., “Evolution of dislocation loops in austenitic stainless steels implanted with high concentration of hydrogen,” Philosophical Magazine. 2017. link Times cited: 5 Abstract: It has been found that under certain conditions, hydrogen re… read moreAbstract: It has been found that under certain conditions, hydrogen retention would be strongly enhanced in irradiated austenitic stainless steels. To investigate the effect of the retained hydrogen on the defect microstructure, AL-6XN stainless steel specimens were irradiated with low energy (100 keV) H2+ so that high concentration of hydrogen was injected into the specimens while considerable displacement damage dose (up to 7 dpa) was also achieved. Irradiation induced dislocation loops and voids were characterised by transmission electron microscopy. For specimens irradiated to 7 dpa at 290 °C, dislocation loops with high number density were found and the void swelling was observed. At 380 °C, most of dislocation loops were unfaulted and tangled at 7 dpa, and the void swellings were observed at 5 dpa and above. Combining the data from low dose in previous work to high dose, four stages of dislocation loops evolution with hydrogen retention were suggested. Finally, molecular dynamics simulation was made to elucidate the division of large dislocation loops under irradiation. read less NOT USED (high confidence) C. Angelie and J. Soudan, “Nanothermodynamics of iron clusters: Small clusters, icosahedral and fcc-cuboctahedral structures.,” The Journal of chemical physics. 2017. link Times cited: 3 Abstract: The study of the thermodynamics and structures of iron clust… read moreAbstract: The study of the thermodynamics and structures of iron clusters has been carried on, focusing on small clusters and initial icosahedral and fcc-cuboctahedral structures. Two combined tools are used. First, energy intervals are explored by the Monte Carlo algorithm, called σ-mapping, detailed in the work of Soudan et al. [J. Chem. Phys. 135, 144109 (2011), Paper I]. In its flat histogram version, it provides the classical density of states, gp(Ep), in terms of the potential energy of the system. Second, the iron system is described by a potential which is called "corrected EAM" (cEAM), explained in the work of Basire et al. [J. Chem. Phys. 141, 104304 (2014), Paper II]. Small clusters from 3 to 12 atoms in their ground state have been compared first with published Density Functional Theory (DFT) calculations, giving a complete agreement of geometries. The series of 13, 55, 147, and 309 atom icosahedrons is shown to be the most stable form for the cEAM potential. However, the 147 atom cluster has a special behaviour, since decreasing the energy from the liquid zone leads to the irreversible trapping of the cluster in a reproducible amorphous state, 7.38 eV higher in energy than the icosahedron. This behaviour is not observed at the higher size of 309 atoms. The heat capacity of the 55, 147, and 309 atom clusters revealed a pronounced peak in the solid zone, related to a solid-solid transition, prior to the melting peak. The corresponding series of 13, 55, and 147 atom cuboctahedrons has been compared, underscoring the unstability towards the icosahedral structure. This unstability occurs clearly in several steps for the 147 atom cluster, with a sudden transformation at a transition state. This illustrates the concerted icosahedron-cuboctahedron transformation of Buckminster Fuller-Mackay, which is calculated for the cEAM potential. Two other clusters of initial fcc structures with 24 and 38 atoms have been studied, as well as a 302 atom cluster. Each one relaxes towards a more stable structure without regularity. The 38 atom cluster exhibits a nearly glassy relaxation, through a cascade of six metastable states of long life. This behaviour, as that of the 147 atom cluster towards the amorphous state, shows that difficulties to reach ergodicity in the lower half of the solid zone are related to particular features of the potential energy landscape, and not necessarily to a too large size of the system. Comparisons of the cEAM iron system with published results about Lennard-Jones systems and DFT calculations are made. The results of the previous clusters have been combined with that of Paper II to plot the cohesive energy Ec and the melting temperature Tm in terms of the cluster atom number Nat. The Nat-1/3 linear dependence of the melting temperature (Pawlow law) is observed again for Nat > 150. In contrast, for Nat < 150, the curve diverges strongly from the Pawlow law, giving it an overall V-shape, with a linear increase of Tm when Nat goes from 55 to 13 atoms. Surprisingly, the 38 atom cluster is anomalously below the overall curve. read less NOT USED (high confidence) F. Calvo, N. Combe, J. Morillo, and M. Benoit, “Modeling Iron–Gold Nanoparticles Using a Dedicated Semi-Empirical Potential: Application to the Stability of Core–Shell Structures,” Journal of Physical Chemistry C. 2017. link Times cited: 16 Abstract: Core–shell nanoparticles made from iron embedded in gold hav… read moreAbstract: Core–shell nanoparticles made from iron embedded in gold have a strong potential interest in nanomedicine, the Au shell providing an efficient biocompatible coating for the magnetic Fe core. With the aim of determining theoretically the equilibrium morphologies of Fe–Au nanoparticles in a broad size range and with different compositions, a semiempirical many-body Fe–Au potential was designed combining well-established models for the pure metals and introducing dedicated contributions for the interactions involving mixed elements. The potential was parametrized against various energetic properties involving impurities, intermetallics, and finite clusters obtained using density functional calculations in the generalized gradient approximation. The potential was tested to investigate Fe–Au nanoparticles near equiconcentration containing about 1000–2000 atoms at finite temperature using parallel tempering Monte Carlo simulations initiated from the core–shell structure. The core–shell nanoparticles are found t... read less NOT USED (high confidence) M. G. D. V. Cuppari, R. Veiga, H. Goldenstein, J. E. G. Silva, and C. Becquart, “Lattice Instabilities and Phase Transformations in Fe from Atomistic Simulations,” Journal of Phase Equilibria and Diffusion. 2017. link Times cited: 2 NOT USED (high confidence) M. Cuppari, R. Veiga, H. Goldenstein, J. E. G. Silva, and C. Becquart, “Lattice Instabilities and Phase Transformations in Fe from Atomistic Simulations,” Journal of Phase Equilibria and Diffusion. 2017. link Times cited: 0 NOT USED (high confidence) P. Ma, S. Dudarev, and C. Woo, “SPILADY: A parallel CPU and GPU code for spin-lattice magnetic molecular dynamics simulations,” Comput. Phys. Commun. 2016. link Times cited: 28 NOT USED (high confidence) J. Janssen, N. Gunkelmann, and H. Urbassek, “Influence of C concentration on elastic moduli of α′-Fe1-xCx alloys,” Philosophical Magazine. 2016. link Times cited: 9 Abstract: The elastic constants of tetragonally distorted - crystallit… read moreAbstract: The elastic constants of tetragonally distorted - crystallites are calculated for several available interatomic interaction potentials. Besides embedded-atom-method-type potentials also a simple pair potential, modified embedded-atom-method and bond-order potentials are investigated. Care is taken to minimise the crystal structure properly in the presence of the C interstitials; we verify that the influence of statistics, i.e. the randomness of the C positions in the lattice, affects the elastic properties only little, as long as C is not allowed to cluster. We find that both sign and order of magnitude of the tetragonal elastic constants vary strongly between the predictions of the available potentials. Recent experimental data are available for the orientation-averaged elastic moduli; in contrast to the tetragonal constants, they feature only a mild dependence on C content. The experimental data are well reproduced by several of the potentials studied here. Existing deviations between experiment and predictions are discussed. read less NOT USED (high confidence) Y. Yang, S. Li, X. Ding, J. Sun, and E. Salje, “Interface Driven Pseudo‐Elasticity in a‐Fe Nanowires,” Advanced Functional Materials. 2016. link Times cited: 20 Abstract: Molecular dynamics simulations of bent [100] α‐Fe nanowires … read moreAbstract: Molecular dynamics simulations of bent [100] α‐Fe nanowires show the nucleation of twins and nanoscale interfaces that lead to pseudo‐elasticity during loading/unloading cycles. The new type of interfaces along {110} stems from the accumulation of individual <111>/{112} twin boundaries and stores high interfacial energies. These nonconventional interfaces provide a large part of the driving force for shape recovery upon unloading, while the minimization of surface energy is no longer the dominant driving force. This new pseudo‐elastic effect is not much affected by surface roughness, and can be extended over a wide range of wire diameters, if the sample is seeded with conventional twin boundaries, which will transform to the desired {110} interfaces under bending. read less NOT USED (high confidence) D. Terentyev, A. Bakaev, D. V. Neck, and E. Zhurkin, “Glide of dislocations in <1 1 1>3 2 1 slip system: an atomistic study,” Philosophical Magazine. 2016. link Times cited: 5 Abstract: Atomistic calculations are performed to investigate plastic … read moreAbstract: Atomistic calculations are performed to investigate plastic slip in the <1 1 1>{3 2 1} system in body-centred cubic iron. Several modern interatomic potentials, developed over the last decade, are applied to compute the stacking fault γ-line energy in the {3 2 1} plane and the results are compared with the ab initio prediction. The applied potentials have shown strong deviations, but several potentials acquired good qualitative agreement with the ab initio data. Depending on the applied potential, the lowest value of the Peierls stress for the edge dislocation (ED) is 50 MPa (Ackland and Bacon from 1997) and the highest is 550 MPa (Dudarev and Derlet from 2005), while for the screw dislocation it is much higher, in the range 1–2 GPa. At finite temperature, however, the flow stress of the ED is found to decrease exponentially reaching a negligible value at about 200 K, irrespective of the applied potential. On the basis of the data obtained using Ackland–Mendelev potential from 2004, we conclude that the slip resistance of the <1 1 1>{3 2 1} system is in between the resistance of the <1 1 1>{1 1 0} and <1 1 1>{1 1 2} slip systems. read less NOT USED (high confidence) Y. Osetsky and R. Stoller, “Atomic-scale mechanisms of helium bubble hardening in iron☆,” Journal of Nuclear Materials. 2015. link Times cited: 46 NOT USED (high confidence) D. Terentyev, A. Dubinko, V. Dubinko, S. Dmitriev, E. Zhurkin, and M. Sorokin, “Interaction of discrete breathers with primary lattice defects in bcc Fe,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 43 Abstract: The interaction of discrete breathers with the primary latti… read moreAbstract: The interaction of discrete breathers with the primary lattice defects in transition metals such as vacancy, dislocation, and surface is analyzed on the example of bcc iron employing atomistic simulations. Scattering of discrete breathers on the lattice defects induces localized atomic excitations, with intensity and relaxation time depending on the defect structure and breather kinetic energy. The dissipation of the intrinsic breather energy due to the scattering is computed and analyzed. It is concluded that the breather-to-defect energy transfer may stipulate the activation of the lattice defects causing unexpected athermal effects such as enhanced mass transfer or electroplasticity, already experimentally reported but so far not fully understood at the atomic-scale level. read less NOT USED (high confidence) Z. Chang, D. Terentyev, N. Sandberg, K. Samuelsson, and P. Olsson, “Anomalous bias factors of dislocations in bcc iron,” Journal of Nuclear Materials. 2015. link Times cited: 38 NOT USED (high confidence) M. E. Ford, D. Pettifor, and R. Drautz, “Non-collinear magnetism with analytic Bond-Order Potentials,” Journal of Physics: Condensed Matter. 2015. link Times cited: 8 Abstract: The theory of analytic Bond-Order Potentials as applied to n… read moreAbstract: The theory of analytic Bond-Order Potentials as applied to non-collinear magnetic structures of transition metals is extended to take into account explicit rotations of Hamiltonian and local moment matrix elements between locally and globally defined spin-coordinate systems. Expressions for the gradients of the energy with respect to the Hamiltonian matrix elements, the interatomic forces and the magnetic torques are derived. The method is applied to simulations of the rotation of magnetic moments in α iron, as well as α and β manganese, based on d-valent orthogonal tight-binding parametrizations of the electronic structure. A new weighted-average terminator is introduced to improve the convergence of the Bond-Order Potential energies and torques with respect to tight-binding reference values, although the general behavior is qualitatively correct for low-moment expansions. read less NOT USED (high confidence) M. Basire, J. Soudan, and C. Angelie, “Nanothermodynamics of large iron clusters by means of a flat histogram Monte Carlo method.,” The Journal of chemical physics. 2014. link Times cited: 2 Abstract: The thermodynamics of iron clusters of various sizes, from 7… read moreAbstract: The thermodynamics of iron clusters of various sizes, from 76 to 2452 atoms, typical of the catalyst particles used for carbon nanotubes growth, has been explored by a flat histogram Monte Carlo (MC) algorithm (called the σ-mapping), developed by Soudan et al. [J. Chem. Phys. 135, 144109 (2011), Paper I]. This method provides the classical density of states, gp(Ep) in the configurational space, in terms of the potential energy of the system, with good and well controlled convergence properties, particularly in the melting phase transition zone which is of interest in this work. To describe the system, an iron potential has been implemented, called "corrected EAM" (cEAM), which approximates the MEAM potential of Lee et al. [Phys. Rev. B 64, 184102 (2001)] with an accuracy better than 3 meV/at, and a five times larger computational speed. The main simplification concerns the angular dependence of the potential, with a small impact on accuracy, while the screening coefficients S(ij) are exactly computed with a fast algorithm. With this potential, ergodic explorations of the clusters can be performed efficiently in a reasonable computing time, at least in the upper half of the solid zone and above. Problems of ergodicity exist in the lower half of the solid zone but routes to overcome them are discussed. The solid-liquid (melting) phase transition temperature T(m) is plotted in terms of the cluster atom number N(at). The standard N(at)(-1/3) linear dependence (Pawlow law) is observed for N(at) >300, allowing an extrapolation up to the bulk metal at 1940 ±50 K. For N(at) <150, a strong divergence is observed compared to the Pawlow law. The melting transition, which begins at the surface, is stated by a Lindemann-Berry index and an atomic density analysis. Several new features are obtained for the thermodynamics of cEAM clusters, compared to the Rydberg pair potential clusters studied in Paper I. read less NOT USED (high confidence) K. Nordlund, C. Björkas, T. Ahlgren, A. Lasa, and A. Sand, “Multiscale modelling of plasma–wall interactions in fusion reactor conditions,” Journal of Physics D: Applied Physics. 2014. link Times cited: 58 Abstract: The interaction of fusion reactor plasma with the material o… read moreAbstract: The interaction of fusion reactor plasma with the material of the first wall involves a complex multitude of interlinked physical and chemical effects. Hence, modern theoretical treatment of it relies to a large extent on multiscale modelling, i.e. using different kinds of simulation approaches suitable for different length and time scales in connection with each other. In this review article, we overview briefly the physics and chemistry of plasma–wall interactions in tokamak-like fusion reactors, and present some of the most commonly used material simulation approaches relevant for the topic. We also give summaries of recent multiscale modelling studies of the effects of fusion plasma on the modification of the materials of the first wall, especially on swift chemical sputtering, mixed material formation and hydrogen isotope retention in tungsten. read less NOT USED (high confidence) J. J. Möller and E. Bitzek, “Comparative study of embedded atom potentials for atomistic simulations of fracture in α-iron,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 50 Abstract: Atomistic simulations play a crucial role in advancing our u… read moreAbstract: Atomistic simulations play a crucial role in advancing our understanding of the crack-tip processes that take place during fracture of semi-brittle materials like α-iron. As with all atomistic simulations, the results of such simulations however depend critically on the underlying atomic interaction model. Here, we present a systematic study of eight α-iron embedded atom method potentials used to model cracks subjected to plane strain mode-I loading conditions in six different crystal orientations. Molecular statics simulations are used to determine the fracture behavior (cleavage, dislocation emission, twinning) and the critical stress intensity factor KIc. The structural transformations in front of the crack tips, and in particular the occurrence of {1 1 0} planar faults, are analyzed in detail and related to the strain-dependent generalized stacking fault energy curve. The simulation results are discussed in terms of theoretical fracture criteria and compared to recent experimental data. The different potentials are ranked according to their capability to model the experimentally observed fracture behavior. read less NOT USED (high confidence) M. E. Ford, R. Drautz, T. Hammerschmidt, and D. Pettifor, “Convergence of an analytic bond-order potential for collinear magnetism in Fe,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 20 Abstract: Analytic bond-order potentials (BOPs) for magnetic transitio… read moreAbstract: Analytic bond-order potentials (BOPs) for magnetic transition metals are applied for pure iron as described by an orthogonal d-valent tight-binding (TB) model. Explicit analytic equations for the gradients of the binding energy with respect to the Hamiltonian on-site levels are presented, and are then used to minimize the energy with respect to the magnetic moments, which is equivalent to a TB self-consistency scheme. These gradients are also used to calculate the exact forces, consistent with the energy, necessary for efficient relaxations and molecular dynamics. The Jackson kernel is used to remove unphysical negative densities of states, and approximations for the asymptotic recursion coefficients are examined. BOP, TB and density functional theory results are compared for a range of bulk and defect magnetic structures. The BOP energies and magnetic moments for bulk structures are shown to converge with increasing numbers of moments, with nine moments sufficient for a quantitative comparison of structural energy differences. The formation energies of simple defects such as the monovacancies and divacancies also converge rapidly. Other physical quantities, such as the position of the high-spin to low-spin transition in ferromagnetic fcc (face centred cubic) iron, surface peaks in the local density of states, the elastic constants and the formation energies of the self-interstitial atom defects, require higher moments for convergence. read less NOT USED (high confidence) K. Nordlund and F. Djurabekova, “Multiscale modelling of irradiation in nanostructures,” Journal of Computational Electronics. 2014. link Times cited: 42 NOT USED (high confidence) D. Perera et al., “Phonon-magnon interactions in body centered cubic iron: A combined molecular and spin dynamics study,” Journal of Applied Physics. 2014. link Times cited: 12 Abstract: Combining an atomistic many-body potential with a classical … read moreAbstract: Combining an atomistic many-body potential with a classical spin Hamiltonian parameterized by first principles calculations, molecular-spin dynamics computer simulations were performed to investigate phonon-magnon interactions in body centered cubic iron. Results obtained for spin-spin and density-density dynamic structure factors show that noticeable softening and damping of magnon modes occur due to the presence of lattice vibrations. Furthermore, as a result of the phonon-magnon coupling, additional longitudinal spin wave excitations are observed, with the same frequencies as the longitudinal phonon modes. read less NOT USED (high confidence) M. Marinica et al., “Interatomic potentials for modelling radiation defects and dislocations in tungsten,” Journal of Physics: Condensed Matter. 2013. link Times cited: 258 Abstract: We have developed empirical interatomic potentials for study… read moreAbstract: We have developed empirical interatomic potentials for studying radiation defects and dislocations in tungsten. The potentials use the embedded atom method formalism and are fitted to a mixed database, containing various experimentally measured properties of tungsten and ab initio formation energies of defects, as well as ab initio interatomic forces computed for random liquid configurations. The availability of data on atomic force fields proves critical for the development of the new potentials. Several point and extended defect configurations were used to test the transferability of the potentials. The trends predicted for the Peierls barrier of the 1 2 ⟨ 111 ⟩ ?> screw dislocation are in qualitative agreement with ab initio calculations, enabling quantitative comparison of the predicted kink-pair formation energies with experimental data. read less NOT USED (high confidence) T. Lee, M. Baskes, S. Valone, and J. Doll, “Atomistic modeling of thermodynamic equilibrium and polymorphism of iron,” Journal of Physics: Condensed Matter. 2012. link Times cited: 52 Abstract: We develop two new modified embedded-atom method (MEAM) pote… read moreAbstract: We develop two new modified embedded-atom method (MEAM) potentials for elemental iron, intended to reproduce the experimental phase stability with respect to both temperature and pressure. These simple interatomic potentials are fitted to a wide variety of material properties of bcc iron in close agreement with experiments. Numerous defect properties of bcc iron and bulk properties of the two close-packed structures calculated with these models are in reasonable agreement with the available first-principles calculations and experiments. Performance at finite temperatures of these models has also been examined using Monte Carlo simulations. We attempt to reproduce the experimental iron polymorphism at finite temperature by means of free energy computations, similar to the procedure previously pursued by Müller et al (2007 J. Phys.: Condens. Matter 19 326220), and re-examine the adequacy of the conclusion drawn in the study by addressing two critical aspects missing in their analysis: (i) the stability of the hcp structure relative to the bcc and fcc structures and (ii) the compatibility between the temperature and pressure dependences of the phase stability. Using two MEAM potentials, we are able to represent all of the observed structural phase transitions in iron. We discuss that the correct reproductions of the phase stability among three crystal structures of iron with respect to both temperature and pressure are incompatible with each other due to the lack of magnetic effects in this class of empirical interatomic potential models. The MEAM potentials developed in this study correctly predict, in the bcc structure, the self-interstitial in the 〈110〉 orientation to be the most stable configuration, and the screw dislocation to have a non-degenerate core structure, in contrast to many embedded-atom method potentials for bcc iron in the literature. read less NOT USED (high confidence) H. Hou, R. Wang, J. Wang, X. Liu, G. Chen, and P. Huang, “An analytic bond-order potential for the Fe–Cu system,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 5 Abstract: An angular-dependent analytic bond-order potential (ABOP) fo… read moreAbstract: An angular-dependent analytic bond-order potential (ABOP) for copper and Fe–Cu system was developed, based on the ABOP of pure iron introduced by Müller et al (2007 J. Phys.: Condens. Matter 19 326220). The potential parameters for the present ABOP model of copper were determined by fitting to the experimental data of the basic properties of fcc Cu and ab initio calculated properties of bcc Cu. The model predicts the vacancy formation energy in good agreement with the experimental result, although no vacancy formation information was used in the fitting of the model parameters. The melting point of Cu is also properly reproduced. The Fe–Cu binary system was described by adding two independent cross parameters in the potential model. The cross parameters were fitted using the ab initio data of the formation energies and lattice parameters of fictitious Fe–Cu alloys. The potential was applied to investigate the point defects and small defect clusters in dilute Fe–Cu alloys. The results were compared with the ab initio data and the values obtained with other potentials. read less NOT USED (high confidence) D. Duffy, “Modelling materials for fusion power,” International Materials Reviews. 2011. link Times cited: 12 Abstract: Fusion has the potential for delivering safe, clean, low car… read moreAbstract: Fusion has the potential for delivering safe, clean, low carbon power; however, significant scientific and engineering hurdles must first be overcome. One such hurdle is the design of materials that will withstand the harsh conditions. The materials which line the vessel walls will experience exceptionally high heat and particle fluxes, which will gradually erode the materials and contaminate the plasma. The deuterium–tritium fusion reaction will produce high energy neutrons, which will create defects and transmutation reactions in the vessel walls. These defects, along with the transmutation gasses, evolve over time and change the microstructure and properties of the material. In order to design suitable materials for fusion, the radiation damage, and its evolution over time, must be understood and evaluated for a broad class of materials. Modelling has a vital role to play because it can provide details about processes that occur on length and timescales that are inaccessible to experiment. In this review, the challenges that face designers of fusion power plants are discussed. The modelling techniques that are used to model radiation effects are described and the links between modelling and experiment are discussed. The review concludes with a discussion about the future direction for fusion materials research. read less NOT USED (high confidence) N. Baluc et al., “From materials development to their test in IFMIF: an overview,” Nuclear Fusion. 2011. link Times cited: 6 Abstract: R&D activities on fusion reactor materials in Switzerland fo… read moreAbstract: R&D activities on fusion reactor materials in Switzerland focus on (1) the development of advanced metallic materials for structural applications in plasma-facing (first wall, divertor) and breeding blanket components of the future fusion power reactors, in particular oxide dispersion strengthened reduced activation ferritic steels and tungsten-base materials, (2) the modelling of radiation damage and radiation effects and (3) small specimen test technology for the future International Fusion Materials Irradiation Facility. The main objectives, examples of recent results and future activities are described in the case of these three R&D areas. read less NOT USED (high confidence) G. J. Ackland, K. D’Mellow, S. L. Daraszewicz, D. J. Hepburn, M. Uhrin, and K.Stratford, “The MOLDY short-range molecular dynamics package,” Comput. Phys. Commun. 2011. link Times cited: 37 NOT USED (high confidence) P. K. Soin, P. K. Soin, A. Horsfield, and D. Nguyen-Manh, “Efficient self-consistency for magnetic tight binding,” Comput. Phys. Commun. 2011. link Times cited: 12 NOT USED (high confidence) S. Chiesa, P. Derlet, S. Dudarev, and H. Swygenhoven, “Optimization of the magnetic potential for α-Fe,” Journal of Physics: Condensed Matter. 2011. link Times cited: 46 Abstract: A second generation of empirical potentials is produced for … read moreAbstract: A second generation of empirical potentials is produced for α-Fe within the framework of the magnetic potential formalism (Dudarev and Derlet 2005 J. Phys.: Condens. Matter 17 7097). A materials database that, in addition to ab initio-derived point defect formation energies, now includes third-order elastic constant and ab initio-derived string potential data controlling, respectively, the thermal expansion properties and the core structure of the 1/2⟨111⟩ screw dislocation. Three parameterizations are presented in detail, all of which exhibit positive thermal expansion and produce a non-degenerate configuration for the relaxed 1/2⟨111⟩ screw dislocation easy core structure. These potentials, along with two other published potentials, are investigated in terms of defect formation volume, early stage dislocation loop clustering energetics, ⟨110⟩ dumbbell interstitial diffusion, and the zero-stress 1/2⟨111⟩ screw dislocation Peierls barrier and its corresponding kink formation energies. read less NOT USED (high confidence) K. Odbadrakh et al., “Calculated electronic and magnetic structure of screw dislocations in alpha iron,” Journal of Applied Physics. 2011. link Times cited: 7 Abstract: Local atomic magnetic moments in crystalline Fe are perturbe… read moreAbstract: Local atomic magnetic moments in crystalline Fe are perturbed by the presence of dislocations. The effects are most pronounced near the dislocation core and decay slowly as the strain field of the dislocation decreases with distance. We have calculated local moments using the locally self-consistent multiple scattering (LSMS) method for a supercell containing a screw-dislocation quadrupole. Finite size effects are found to be significant indicating that dislocation cores affect the electronic structure and magnetic moments of neighboring dislocations. The influence of neighboring dislocations points to a need to study individual dislocations from first principles just as they appear amid surrounding atoms in large-scale classical force field simulations. An approach for the use of the LSMS to calculate local moments in subvolumes of large atomic configurations generated in the course of classical molecular dynamics simulation of dislocationdynamics is discussed. read less NOT USED (high confidence) G. Bonny, R. Pasianot, D. Terentyev, and L. Malerba, “Iron chromium potential to model high-chromium ferritic alloys,” Philosophical Magazine. 2011. link Times cited: 73 Abstract: We present an Fe–Cr interatomic potential to model high-Cr f… read moreAbstract: We present an Fe–Cr interatomic potential to model high-Cr ferritic alloys. The potential is fitted to thermodynamic and point-defect properties obtained from density functional theory (DFT) calculations and experiments. The developed potential is also benchmarked against other potentials available in literature. It shows particularly good agreement with the DFT obtained mixing enthalpy of the random alloy, the formation energy of intermetallics and experimental excess vibrational entropy and phase diagram. In addition, DFT calculated point-defect properties, both interstitial and substitutional, are well reproduced, as is the screw dislocation core structure. As a first validation of the potential, we study the precipitation hardening of Fe–Cr alloys via static simulations of the interaction between Cr precipitates and screw dislocations. It is concluded that the description of the dislocation core modification near a precipitate might have a significant influence on the interaction mechanisms observed in dynamic simulations. read less NOT USED (high confidence) L. Malerba et al., “Ab initio calculations and interatomic potentials for iron and iron alloys : Achievements within the Perfect Project,” Journal of Nuclear Materials. 2010. link Times cited: 65 NOT USED (high confidence) C. Race, D. Mason, M. Finnis, W. Foulkes, A. Horsfield, and A. Sutton, “The treatment of electronic excitations in atomistic models of radiation damage in metals,” Reports on Progress in Physics. 2010. link Times cited: 97 Abstract: Atomistic simulations are a primary means of understanding t… read moreAbstract: Atomistic simulations are a primary means of understanding the damage done to metallic materials by high energy particulate radiation. In many situations the electrons in a target material are known to exert a strong influence on the rate and type of damage. The dynamic exchange of energy between electrons and ions can act to damp the ionic motion, to inhibit the production of defects or to quench in damage, depending on the situation. Finding ways to incorporate these electronic effects into atomistic simulations of radiation damage is a topic of current major interest, driven by materials science challenges in diverse areas such as energy production and device manufacture. In this review, we discuss the range of approaches that have been used to tackle these challenges. We compare augmented classical models of various kinds and consider recent work applying semi-classical techniques to allow the explicit incorporation of quantum mechanical electrons within atomistic simulations of radiation damage. We also outline the body of theoretical work on stopping power and electron–phonon coupling used to inform efforts to incorporate electronic effects in atomistic simulations and to evaluate their performance. read less NOT USED (high confidence) H. Wang, P. Ma, and C. Woo, “Exchange interaction function for spin-lattice coupling in bcc iron,” Physical Review B. 2010. link Times cited: 27 Abstract: Functional representations of the spin polarization and the … read moreAbstract: Functional representations of the spin polarization and the exchange interaction in terms of the lattice configuration is necessary to model the dynamics of the coupled spin and lattice subsystems in large-scale atomistic simulation of magnetic materials. Data needed for this purpose have only existed in the regime of small displacements from the equilibrium perfect lattice configurations. In this paper, we report and discuss the results of our first-principles calculations for bcc iron over a wide range of lattice constants using the magnetic force theorem and the one-electron Green's function. Despite the relatively complex functional form of the exchange interaction function for bcc iron our results show that it can be expressed as a superposition of Bethe-Slater-type curves representing interatomic exchange interaction of the $3d$ electrons. read less NOT USED (high confidence) M. Lavrentiev, D. Nguyen-Manh, and S. Dudarev, “Magnetic cluster expansion model for bcc-fcc transitions in Fe and Fe-Cr alloys,” Physical Review B. 2010. link Times cited: 91 Abstract: An ab initio-based magnetic-cluster-expansion treatment deve… read moreAbstract: An ab initio-based magnetic-cluster-expansion treatment developed for body- and face-centered cubic phases of iron and iron-chromium alloys is applied to modeling the $\ensuremath{\alpha}\text{\ensuremath{-}}\ensuremath{\gamma}$ and $\ensuremath{\gamma}\text{\ensuremath{-}}\ensuremath{\delta}$ phase transitions in these materials. The Curie, N\'eel, and the structural phase-transition temperatures predicted by the model are in good agreement with experimental observations, indicating that it is the thermal excitation of magnetic and phonon degrees of freedom that stabilizes the fcc $\ensuremath{\gamma}$ phase. The model also describes the occurrence of the $\ensuremath{\gamma}$ loop in the phase diagram of Fe-Cr alloys for a realistic interval of temperatures and Cr concentrations. read less NOT USED (high confidence) F. Djurabekova, L. Malerba, R. Pasianot, P. Olsson, and K. Nordlund, “Kinetics versus thermodynamics in materials modeling: The case of the di-vacancy in iron,” Philosophical Magazine. 2010. link Times cited: 22 Abstract: Monte Carlo models are widely used for the study of microstr… read moreAbstract: Monte Carlo models are widely used for the study of microstructural and microchemical evolution of materials under irradiation. However, they often link explicitly the relevant activation energies to the energy difference between local equilibrium states. We provide a simple example (di-vacancy migration in iron) in which a rigorous activation energy calculation, by means of both empirical interatomic potentials and density functional theory methods, clearly shows that such a link is not granted, revealing a migration mechanism that a thermodynamics-linked activation energy model cannot predict. Such a mechanism is, however, fully consistent with thermodynamics. This example emphasizes the importance of basing Monte Carlo methods on models where the activation energies are rigorously calculated, rather than deduced from widespread heuristic equations. read less NOT USED (high confidence) G. Bonny and R. Pasianot, “Gauge transformations to combine multi-component many-body interatomic potentials,” Philosophical Magazine Letters. 2010. link Times cited: 19 Abstract: Many-body interatomic potentials play an important role in a… read moreAbstract: Many-body interatomic potentials play an important role in atomistic modelling of materials. For pure elements it is known that there exist gauge transformations that can change the form of the potential functions without modifying its properties. These same transformations, however, fail when applied to alloys. Even though different research groups may use the same potentials to describe pure elements, the gauges employed for fitting alloys will generally be different. In this scenario, it is a priori impossible to merge them into one potential describing the combined system, and thus no advantage is taken from state-of-the-art developments in the literature. Here, we generalise the gauge transformations applied to pure species in order to leave the properties of alloys invariant. Based on these transformations, a strategy to merge potentials developed within different gauges is presented, aiming at the description of the combined system. Advantage of existing state-of-the-art potentials is so taken, thus focusing the efforts on fitting only the missing interactions. Such a procedure constitutes a helpful tool for the development of potentials targeted to alloys of increased complexity, while maintaining the description quality of their constituents. read less NOT USED (high confidence) D. Terentyev, S. M. H. Haghighat, and R. Schäublin, “Strengthening due to Cr-rich precipitates in Fe–Cr alloys: Effect of temperature and precipitate composition,” Journal of Applied Physics. 2010. link Times cited: 39 Abstract: Molecular dynamics (MD) simulations were carried out to stud… read moreAbstract: Molecular dynamics (MD) simulations were carried out to study the interaction between nanometric Cr precipitates and a 1/2 >{110} edge dislocation (ED) in pure Fe and Fe-9 at. % Cr (Fe-9Cr) random alloy. The aim of this work is to estimate the variation in the pinning strength of the Cr precipitate as a function of temperature, its chemical composition and the matrix composition in which the precipitate is embedded. The dislocation was observed to shear Cr precipitates rather than by-pass via the formation of the Orowan loop, even though a pronounced screw dipole was emerged in the reactions with the precipitates of size larger than 4.5 nm. The screw arms of the formed dipole were not observed to climb thus no point defects were left inside the sheared precipitates, irrespective of simulation temperature. Both Cr solution and Cr precipitates, embedded in the Fe-9Cr matrix, were seen to contribute to the flow stress. The decrease in the flow stress with temperature in the alloy containing Cr precipitates is, therefore, related to the simultaneous change in the matrix friction stress, precipitate resistance, and dislocation flexibility. Critical stress estimated from MD simulations was seen to have a strong dependence on the precipitate composition. If the latter decreases from 95% down to 80%, the corresponding critical stress decreases almost as twice. The results presented here suggest a significant contribution to the flow stress due to the alpha-alpha(') separation, at least for EDs. The obtained data can be used to validate and to parameterize dislocation dynamics models, where the temperature dependence of the obstacle strength is an essential input data. read less NOT USED (high confidence) Y. Osetsky and D. Bacon, “Atomic-scale mechanisms of void hardening in bcc and fcc metals,” Philosophical Magazine. 2010. link Times cited: 98 Abstract: Strengthening due to voids can be a significant effect of rad… read moreAbstract: Strengthening due to voids can be a significant effect of radiation damage in metals, but treatment of this by elasticity theory of dislocations is difficult when the mechanisms controlling the obstacle strength are atomic in nature. Results are reported of atomic-scale modelling to compare edge dislocation–void interaction in fcc copper and bcc iron. Voids of up to 6 nm diameter in iron and 8 nm diameter in copper were studied over the temperature range 0 to 600 K at different applied strain rates. Voids in iron are strong obstacles, for the dislocation has to adopt a dipole-like configuration at the void before breaking away. The dipole unzips at the critical stress when the dislocation is able to climb by absorbing vacancies and leave the void surface. Dislocation dissociation into Shockley partials in copper prevents dislocation climb and affects the strength of small and large voids differently. Small voids are much weaker obstacles than those in iron because the partials break from a void individually. Large voids are at least as strong as those in iron, but the controlling mechanism depends on temperature. read less NOT USED (high confidence) S. H. Haghighat and R. Schäublin, “Influence of the stress field due to pressurized nanometric He bubbles on the mobility of an edge dislocation in iron,” Philosophical Magazine. 2010. link Times cited: 45 Abstract: Voids and He bubbles are strong obstacles to dislocation, wh… read moreAbstract: Voids and He bubbles are strong obstacles to dislocation, which induce hardening and loss of ductility. In Fe, molecular dynamics simulation is used to investigate the basic mechanisms of the interaction between a moving edge dislocation and a void or He bubble, as a function of its He content, temperature, interatomic potentials and interaction geometry. Different interatomic potentials for Fe–Fe and Fe–He interactions are used. It appears that temperature eases the dislocation release, due to the increased mobility of the screw segments appearing on the dislocation line upon bowing from the void or He bubble. The mobility includes the cross-slipping of these segments, which leads to the formation of a jog. It appears that the He bubble induces an inhomogeneous stress field in its surroundings, which strongly influences the dislocation passage depending on the geometry of the interaction. read less NOT USED (high confidence) Y. Mishin, M. Asta, and J. Li, “Atomistic modeling of interfaces and their impact on microstructure and properties,” Acta Materialia. 2010. link Times cited: 418 NOT USED (high confidence) S. Chiesa, M. R. Gilbert, S. Dudarev, P. Derlet, and H. Swygenhoven, “The non-degenerate core structure of a ½⟨111⟩ screw dislocation in bcc transition metals modelled using Finnis–Sinclair potentials: The necessary and sufficient conditions,” Philosophical Magazine. 2009. link Times cited: 30 Abstract: It is shown that semi-empirical potentials for bcc metals ba… read moreAbstract: It is shown that semi-empirical potentials for bcc metals based on the non-directional second-moment Finnis–Sinclair approximation are able to predict, as a matter of routine, the non-degenerate core structure for the perfect ½⟨111⟩ dislocation if they correctly describe the inter-string pair potential of a rigid multi-string Frenkel–Kontorova model for the corresponding ideal bcc lattice. We prove this by inspecting the previously published empirical potentials, and also by performing an extensive search in functional parameter space for an optimal parameterisation of the magnetic potential formalism for bcc ferromagnetic Fe. read less NOT USED (high confidence) M. Gruner and P. Entel, “Simulating functional magnetic materials on supercomputers,” Journal of Physics: Condensed Matter. 2009. link Times cited: 38 Abstract: The recent passing of the petaflop per second landmark by th… read moreAbstract: The recent passing of the petaflop per second landmark by the Roadrunner project at the Los Alamos National Laboratory marks a preliminary peak of an impressive world-wide development in the high-performance scientific computing sector. Also, purely academic state-of-the-art supercomputers such as the IBM Blue Gene/P at Forschungszentrum Jülich allow us nowadays to investigate large systems of the order of 103 spin polarized transition metal atoms by means of density functional theory. Three applications will be presented where large-scale ab initio calculations contribute to the understanding of key properties emerging from a close interrelation between structure and magnetism. The first two examples discuss the size dependent evolution of equilibrium structural motifs in elementary iron and binary Fe–Pt and Co–Pt transition metal nanoparticles, which are currently discussed as promising candidates for ultra-high-density magnetic data storage media. However, the preference for multiply twinned morphologies at smaller cluster sizes counteracts the formation of a single-crystalline L10 phase, which alone provides the required hard magnetic properties. The third application is concerned with the magnetic shape memory effect in the Ni–Mn–Ga Heusler alloy, which is a technologically relevant candidate for magnetomechanical actuators and sensors. In this material strains of up to 10% can be induced by external magnetic fields due to the field induced shifting of martensitic twin boundaries, requiring an extremely high mobility of the martensitic twin boundaries, but also the selection of the appropriate martensitic structure from the rich phase diagram. read less NOT USED (high confidence) S. Chiesa, P. Derlet, and S. Dudarev, “Free energy of a ⟨110⟩ dumbbell interstitial defect in bcc Fe: Harmonic and anharmonic contributions,” Physical Review B. 2009. link Times cited: 35 Abstract: S. Chiesa,1 P. M. Derlet,2 and S. L. Dudarev3,4 1NUM/ASQ-Mat… read moreAbstract: S. Chiesa,1 P. M. Derlet,2 and S. L. Dudarev3,4 1NUM/ASQ-Materials Science and Simulation, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland 2NUM-Condensed Matter Theory Group, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland 3Culham Science Centre, EURATOM/UKAEA Fusion Association, Oxfordshire OX14 3DB, United Kingdom 4Department of Physics, Imperial College, Exhibition Road, London SW7 2AZ, United Kingdom Received 7 April 2009; revised manuscript received 22 May 2009; published 17 June 2009 read less NOT USED (high confidence) L. Ventelon, F. Willaime, and P. Leyronnas, “Atomistic simulation of single kinks of screw dislocations in α-Fe,” Journal of Nuclear Materials. 2009. link Times cited: 42 NOT USED (high confidence) S. Dudarev et al., “The EU programme for modelling radiation effects in fusion reactor materials: An overview of recent advances and future goals,” Journal of Nuclear Materials. 2009. link Times cited: 80 NOT USED (high confidence) N. Juslin and K. Nordlund, “Pair potential for Fe-He,” Journal of Nuclear Materials. 2008. link Times cited: 88 NOT USED (high confidence) P. Ma, C. Woo, and S. Dudarev, “Large-scale simulation of the spin-lattice dynamics in ferromagnetic iron,” Physical Review B. 2008. link Times cited: 110 Abstract: combined application of the Langevin spin dynamics and the f… read moreAbstract: combined application of the Langevin spin dynamics and the fluctuation-dissipation theorem. We investigate several applications of the method, performing microcanonical ensemble simulations of adiabatic spin-lattice relaxation of periodic arrays of 180° domain walls, and isothermal-isobaric ensemble dynamical simulations of thermally equilibrated homogeneous systems at various temperatures. The predicted isothermal magnetization curve agrees well with the experimental data for a broad range of temperatures. The equilibrium as well as time-correlation functions of spin orientations exhibit the presence of short-range magnetic order above the Curie temperature. Furthermore, short-range order spin fluctuations are shown to contribute to the thermal expansion of the material. Our analysis illustrates the significant part played by the spin degrees of freedom in the dynamics of motion of atoms in magnetic iron and iron-based alloys. It also shows that the spin-lattice dynamics algorithm developed in this paper offers a viable way of performing large-scale dynamical atomistic simulations of magnetic materials. read less NOT USED (high confidence) C. Engin, L. Sandoval, and H. Urbassek, “Characterization of Fe potentials with respect to the stability of the bcc and fcc phase,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 64 Abstract: By calculating free energies, several published interatomic … read moreAbstract: By calculating free energies, several published interatomic interaction potentials for iron are investigated with respect to the stability of the low-temperature bcc phase and the high-temperature fcc phase. These are empirical many-body potentials for use in atomistic simulation. We find that in all of these potentials—except one—the bcc phase is the stable crystal structure for all temperatures up to the melting point. However, several potentials exhibit a metastable fcc phase in the sense that the fcc structure corresponds to a local minimum of the free energy. read less NOT USED (high confidence) L. Ventelon and F. Willaime, “Core structure and Peierls potential of screw dislocations in α-Fe from first principles: cluster versus dipole approaches,” Journal of Computer-Aided Materials Design. 2007. link Times cited: 126 NOT USED (high confidence) R. Schäublin and N. Baluc, “Radiation damage in ferritic/martensitic steels for fusion reactors: a simulation point of view,” Nuclear Fusion. 2007. link Times cited: 33 Abstract: Low activation ferritic/martensitic steels are good candidat… read moreAbstract: Low activation ferritic/martensitic steels are good candidates for the future fusion reactors, for, relative to austenitic steels, their lower damage accumulation and moderate swelling under irradiation by the 14 MeV neutrons produced by the fusion reaction. Irradiation of these steels, e.g. EUROFER97, is known to produce hardening, loss of ductility, shift in ductile to brittle transition temperature and a reduction of fracture toughness and creep resistance starting at the lowest doses. Helium, produced by transmutation by the 14 MeV neutrons, is known to impact mechanical properties, but its effect at the microstructure level is still unclear. The mechanisms underlying the degradation of mechanical properties are not well understood, despite numerous studies on the evolution of the microstructure under irradiation. This impedes our ability to predict materials' behaviour at higher doses for use in the future fusion reactors. Simulations of these effects are now essential. An overview is presented on molecular dynamics simulations of the primary state of damage in iron and of the mobility of a dislocation, vector of plasticity, in the presence of a defect. read less NOT USED (high confidence) A. Paxton and M. Finnis, “Magnetic tight binding and the iron-chromium enthalpy anomaly,” Physical Review B. 2007. link Times cited: 29 Abstract: We describe a self-consistent magnetic tight-binding theory … read moreAbstract: We describe a self-consistent magnetic tight-binding theory based in an expansion of the Hohenberg-Kohn density functional to second order, about a non-spin-polarized reference density. We show how a first order expansion about a density having a trial input magnetic moment leads to a fixed moment model. We employ a simple set of tight-binding parameters that accurately describes electronic structure and energetics, and show these to be transferable between first row transition metals and their alloys. We make a number of calculations of the electronic structure of dilute Cr impurities in Fe, which we compare with results using the local spin density approximation. The fixed moment model provides a powerful means for interpreting complex magnetic configurations in alloys; using this approach, we are able to advance a simple and readily understood explanation for the observed anomaly in the enthalpy of mixing. read less NOT USED (high confidence) P. Derlet, D. Nguyen-Manh, and S. Dudarev, “Multiscale modeling of crowdion and vacancy defects in body-centered-cubic transition metals,” Physical Review B. 2007. link Times cited: 391 Abstract: We investigate the structure and mobility of single self-int… read moreAbstract: We investigate the structure and mobility of single self-interstitial atom and vacancy defects in body-centered-cubic transition metals forming groups 5B (vanadium, niobium, and tantalum) and 6B (chromium, molybdenum, and tungsten) of the Periodic Table. Density-functional calculations show that in all these metals the axially symmetric self-interstitial atom configuration has the lowest formation energy. In chromium, the difference between the energies of the and the self-interstitial configurations is very small, making the two structures almost degenerate. Local densities of states for the atoms forming the core of crowdion configurations exhibit systematic widening of the "local" d band and an upward shift of the antibonding peak. Using the information provided by electronic structure calculations, we derive a family of Finnis-Sinclair-type interatomic potentials for vanadium, niobium, tantalum, molybdenum, and tungsten. Using these potentials, we investigate the thermally activated migration of self-interstitial atom defects in tungsten. We rationalize the results of simulations using analytical solutions of the multistring Frenkel-Kontorova model describing nonlinear elastic interactions between a defect and phonon excitations. We find that the discreteness of the crystal lattice plays a dominant part in the picture of mobility of defects. We are also able to explain the origin of the non-Arrhenius diffusion of crowdions and to show that at elevated temperatures the diffusion coefficient varies linearly as a function of absolute temperature. read less NOT USED (high confidence) M. Müller, P. Erhart, and K. Albe, “Analytic bond-order potential for bcc and fcc iron—comparison with established embedded-atom method potentials,” Journal of Physics: Condensed Matter. 2007. link Times cited: 177 Abstract: A new analytic bond-order potential for iron is presented th… read moreAbstract: A new analytic bond-order potential for iron is presented that has been fitted to experimental data and results from first-principles calculations. The angular-dependent functional form allows a proper description of a large variety of bulk, surface and defect properties, including the Bain path, phonon dispersions, defect diffusivities and defect formation energies. By calculating Gibbs free energies of body-centred cubic (bcc) and face-centred cubic (fcc) iron as a function of temperature, we show that this potential is able to reproduce the transitions from α-iron to γ-iron and δ-iron before the melting point. The results are compared to four widely used embedded-atom-method potentials for iron. read less NOT USED (high confidence) P. Ma, W. Liu, C. Woo, and S. Dudarev, “Large-scale molecular dynamics simulation of magnetic properties of amorphous iron under pressure,” Journal of Applied Physics. 2007. link Times cited: 19 Abstract: We perform large-scale molecular dynamics simulations to stu… read moreAbstract: We perform large-scale molecular dynamics simulations to study the magnetic properties of amorphous iron under pressure. Simulations, exceeding by at least two orders of magnitude those accessible to density functional calculations, use the recently developed magnetic interatomic potential for iron. The distributions of the size of atomic magnetic moments and parameters characterizing the structure of amorphous iron, such as radial distribution functions, are calculated as a function of the applied hydrostatic stress. As the density increases, there is a reduction in the magnitude of the mean magnetic moment of individual atoms, accompanied by the transformation of an increasing proportion of atoms from a magnetic to a nonmagnetic configuration. Beyond a critical density the proportion of nonmagnetic atoms increases sharply, yet homogeneously. The local magnetic moment of an atom correlates with the local Voronoi volume via a logarithmic relation. In addition, we observe a complex dependence of the local magnetic moment on the topological arrangement of neighboring atoms. © 2007 American Institute of Physics. DOI: 10.1063/1.2715753 read less NOT USED (high confidence) M. Lavrentiev, R. Drautz, D. Nguyen-Manh, T. Klaver, and S. Dudarev, “Monte Carlo study of thermodynamic properties and clustering in the bcc Fe-Cr system,” Physical Review B. 2007. link Times cited: 111 Abstract: Iron-chromium alloys are characterized by a complex phase di… read moreAbstract: Iron-chromium alloys are characterized by a complex phase diagram, by the small negative enthalpy of mixing at low Cr concentrations in the bcc -phase of Fe, and by the inversion of the short-range order parameter. We present Monte Carlo simulations of the binary Fe-Cr alloy based on the cluster expansion approximation for the enthalpy of the system. The set of cluster expansion coefficients is validated against density functional calculations of energies of small clusters of chromium in bcc structure. We show that in the limit of small Cr concentration the enthalpy of mixing remains negative up to fairly high temperatures, and individual Cr atoms remain well separated from each other. Clustering of Cr atoms begins at concentrations exceeding approximately 10% at 800 K and 20% at 1400 K, with Cr-Fe interfaces being parallel to the 110 planes. Calculations show that the first and the second short-range order parameters change sign at approximately 10.5% Cr, in agreement with experimental observations. Semi-grand-canonical ensemble simulations used together with experimental data on vibrational entropy of mixing give an estimate for the temperature of the top of the - miscibility gap. We find that the complex ordering reactions occurring in Fe-Cr, as well as the thermodynamic properties of the alloy, can be reasonably well described using a few concentrationindependent cluster expansion coefficients. read less NOT USED (high confidence) S. Koch, “Development of RF-MEAM interaction potentials for Fe-Y.” 2019. link Times cited: 0 Abstract: Der Fokus dieser Arbeit lag zunachst auf einer simulationsge… read moreAbstract: Der Fokus dieser Arbeit lag zunachst auf einer simulationsgestutzen Untersuchung uber die Entsteh- ungsmechanismen von Oxidteilchen in ODS-Stahlen. Hierbei bilden empirische Wechselwirkungs- potenziale von Eisen-Yttrium-Sauerstoff (Fe-Y-O) die Grundlage fur eine Beschreibung dieser Oxid- teilchen-Bildungs-Prozesse in Molekulardynamik (MD) Simulationen, die auch Eigenschaften von Versetzungen und anderen Bestrahlungs-Panomenen detailiert zur weiteren Aufklarung behandeln konnen.
Zu diesem Zweck ist das speziell auf die Simulation zugeschnittene Anfitten der o.g. MD Potenziale (hier fur Fe-Y-O) notwendig. Hierzu dienen die zuvor durchgefuhrten ab-initio (DFT) Rechnungen als Daten- referenzgrundlage (z.B. von Phasen oder Defekten) zur Optimierung der Potenzialparameter wahrend des Anfittens, um ein moglichst exaktes MD Potenzial zu erzeugen, dass die ab-initio Daten auf groseren MD Skalen detailgetreu abbildet. Im ersten Drittel dieses Projektes wurden mehrere Potenziale fur die einzelnen Metall-Komponenten, Fe-Fe und Y-Y, erzeugt. Dabei stellte sich heraus, dass etablierte Standardmethoden nicht in der Lage sind genaue Fe-Y Potenziale als Teillosung fur das Fe-Y-O Problem zu erzeugen. Dabei wurde eine Kombination aus dem (M)EAM Modell und zur Optimierung eine LSM gestutzte Software (POTFIT) genutzt. Die Komplexitat des Problems liegt in den richtungsabhangigen Atombindungen, die die hier entwickelten fortgeschrittenen Simulations- und Fitmethoden benotigen.
Im ersten Schritt von drei Schritten (chapter 3) wurden zunachst einmal die Defizite der Standard-Fittechniken evaluiert, indem die wahrend des Fitting-Prozesses gefundenen Parametersets im EAM Formalismus mit der flexiblen Software POTFIT auf ihre Eignung hin grundlich untersucht worden sind. Die hierfur genutzten Fitfunktionen wurden ursprunglich Anfang 2000 von Zhou und Wadley entwickelt. Hierbei liegt die Ursache fur die dann entdeckte Parameterset-Problematik darin, dass zur Beschreibung des Fe-Y Systems das Model aus drei Potentialkomponenten besteht: Fe-Fe, Y-Y und Fe-Y. Fur diese einzelnen Komponenten sind die Potentialparameter erfolgreich angefittet worden mit Bezug zur Gitterkonstante und Bindungs- bzw. Kohasionsenergie (beides mit 1% Genauigkeit bezgl. DFT Rechnungen) sowie zu allen elastischen Konstanten (5% Genauigkeit bezgl. Experimente). All dies unter Zuhilfenahme von Parametersuchraum-beschrankenden Techniken, die zur Einhaltung der oben genannten Eigenschaften dienen und urspurnglich von Johnson & Oh sind. Selbst kompliziertere Defekteigenschaften, wie Zwischengitter- und Leerstellenbildungsenergien wurden erfolgreich angefittet. Das hier entwickelte EAM Potenzial fur Y-Y ist z.B. in der Lage bei Eigenzwischengitteratomen die basal oktaedrische Position von Zwischengitteratomen (ZA) im Yttrium hcp-Gitter als Grundzustand und die Transition eines jeden ZAs aus einer anderen Position, wie zuvor in DFT berechnet, zu reproduzieren.
Zur Bildung des angestrebten Fe-Y Potenzials wurden diese beiden Komponenten, Fe-Fe und Y-Y, zum weiteren Fitten in dem weitgefacherten und komplexen Fe-Y Potzenzialsuchraum genutzt. Die Parametersets wurden mit sogenannten hier entwickelten Hauptparameter (Key Driver) systematisch untersucht. Ein flexibleres Konzept statt der starreren Universal Binding Relations in Abhangigkeit von der Rose Gleichung. Dieser Hauptparameter zeigte eindeutig, dass die Nutzung der Rose Gleichung zur Parametersuchraum-Minimierung den Suchraum dahingehend einschrankt, sodass ein akkurates Anfitten der hier genutzten 900 DFT Datensets nicht mehr moglich ist. Allerdings ist die Orientierung im Parametersuchraum mit dieser Rose Gleichung bei standardmasigen Optimierungsmethoden (wie LSM) unabdingbar, da ohne diese die benotigten globalen Optima fur die Parameter nicht auffindbar sind.
Als aufklarendes Testverfahren zur weiteren Ergrundung dieser Problematik und Prufung zur Eignung fur Fe-Y Potenziale und den anschliesenden Simulationen diente der Versuch, 9 verschiedene Bindungs-energien von Yttrium-Leerstellenclustern mit ansteigender Leerstellenzahl zu reproduzieren. Dieser Test konnte von diesen Potenzialen nur teilweise erfullt werden und wurde auf die fehlende Beschreibung der Bindungswinkelabhangigkeit im Modell zuruckgefuhrt. Die Erweiterung von EAM durch MEAM mit Winkelabhangigkeit ist jedoch keineswegs eine zufriedenstellende Losung, da MEAM alternativlos auf der irrefuhrenden Rose Gleichung beruht. Daher war die Benutzung des ubersichtlicheren EAM Typs aus zwei Grunden nutzlich: 1. MEAM braucht die Rose Gleichung um diesen komplexen Formalismus zu beherrschen mit denselben Problemen wie in EAM, aber dieses grundlegende Problem ist in MEAM deutlich schwerer zu identifizieren als in EAM. 2. Die mit EAM gefundenen, angefitteten Parameter sind eine hervorragende Startparameter-Grundlage fur den verbesserten darauffolgenden RF-MEAM Typ.
Im zweiten Schritt wurde das Problem aus dem ersten Schritt gelost, indem ein modifizierter MEAM Spezialtyp im referenzlosen Format (RF-MEAM) angewandt worden ist. Im Gegensatz zum herkommlichen MEAM wird hier die Rose Gleichung durch mehr DFT Daten und insbesondere einer intelligenteren Machine Learning ahnlichen Genetic Algorithmus (GA) Optimiertechnik ersetzt, die allerdings eine bedachte Startparameterwahl vorraussetzt, womit Schritt 1 wieder ins Spiel kommt. Die genutzte fortgeschrittene MEAMfit Software, die per GA funktioniert, wurde zwischen 2016 und 2017 funktionierend eigens dafur implementiert. Mit den in Schritt 1 gefitteten Parametern und Set-Auswahltechniken konnten die weiterfuhrenden Fits mit optimalen Startparametern durchgefuhrt werden.
Auf dieser Stufe waren diese Fits mit der speziell verbesserten Technik in der Lage ein detailgetreues Fe-Y Potenzial zu generieren, das sowohl alle Phasen (Fe2Y, Fe3Y, Fe5Y, Fe23Y6 und Fe17Y2 sowohl als auch reines Fe und Y) als auch die gesamte Defektdatenbasis mit einer durchschnittlichen Abweichung von ≈11% erfolgreich abbildet. Zusatzlich bestatigend zu dieser allgemeinen Ubereinstimmung wurde konsequenterweise der in Schritt 1 entwickelte Test hervorragend mit einmaliger Genauigkeit bestanden, mit max. 5% Abweichung von den komlexen o.g. Y-Leerstellen Bildungsenergien. Allerdings konnte ein systematischer Fehlertrend aufgespurt werden, der Schwachen in der Fe-Fe Komponente offenbarte. Als Folge dessen wurde umgehend diese Komponente durch ein anderes etabliertes Fe-Fe Potenzial von G. Ackland mit einer extrem genauen Schmelztemperatur (nur 3% Abweichung vom Exp.) ausgetauscht. Mit diesem genauen Potenzial konnte zum ersten Mal die Clusterbildung von gelosten Yttrium Atomen in einer Eisenschmelze erfolgreich per MD Simulation auf atomarer Ebene nachgestellt werden oberhalb von 1750 K. Temperaturen darunter hatten eine Ausscheidungsbildung von Y mit sehr geringer Y-Loslichkeit (<0.1%) in Ubereinstimmung mit den Experimenten zur Folge. Dies wurde durch den Pot. Typ A ermoglicht, der aber die energetische Reihenfolge bei den Fe-Y Phasen nicht ganz genau einhalt. Typ B hingegen halt diese ein, dort fehlt aber die Y-Clusterbildungsneigung. Durch den gebotenen Praxisbezug zur Metallurgie mussen die Loslichkeit und Clusterbildung gleichzeitig in der Simulation genau reproduzierbar sein, was aber weder Typ A noch B kann, was zum Typ A/B Dilemma fuhrt.
Dieses Typ A/B Dilemma (Phasen oder Defekt Genauigkeit) fuhrt zum letzten dritten Schritt (chapter 5). Darin ist zusatzlich die Strukturaufklarung von der Fe17Y2 Phase mit Vergleichen zu exp. EXAFS Spektren unserer Kollaborationspartner vom ISSP (Riga) enthalten. Diese Aufklarung dient auch dazu die fehlenden magnetischen Abhangigkeiten im Potenzial zu kompensieren, da die Phasenreihenfolge mit sehr feinen Energieunterschieden wohl stark von magnetischen Wechselwirkungen gepragt ist. Obwohl Potenzial Typ B diesen (Magnetismus) nicht direkt beachtet, ist es in der Lage das tatsachlich gemessene EXAFS Spektrum grostenteils genau wiederzugeben. Allerdings offenbart eine einzige ausgepragte Phasenverschiebung, dass die angenommene hcp Struktur durch eine unterschwellige rhombohedrale Komponente, die sporadisch in der c-Gitterrichtung auftritt, korrigiert werden muss. AIMD (DFT) Berechnungen in Kooperation mit der University of Edinburgh bestatigen dies und zeigen sogar, dass magnetische Wechselwirkungen diese Strukturmischung stabilisieren. Endgultig bestatigt werden konnte dies mit der genauen EXAFS Spektren Reproduktion mit dem durch AIMD verbesserten nochmals gefitteten Potenzialtyp B, der als neuer Typ C durch AIMD indirekt den Einfluss der magnetischen Wechselwirkungen mit einschliest. Diese erstmalige nahezu deckungsgleiche MD Simulation eines EXAFS Spektrums von einem komplexen metallischen Alloy, hier Fe-Y, stellt eine bisher unerreichte Verbesserung dar. Schlieslich lost Typ C das Typ A/B Dilemma und ernoglicht eine genaue gleichzeitige MD Modellierung von Phasen- und Defekten in Fe-Y – ein Durchbruch in der MD-Potenzialentwicklung. read less NOT USED (high confidence) R. Stoller, “1.11 – Primary Radiation Damage Formation.” 2012. link Times cited: 143 NOT USED (definite) J. J. Moller et al., “110
planar faults in strained bcc metals: Origins and implications of a commonly observed artifact of classical potentials,” Physical Review Materials. 2018. link Times cited: 18 Abstract: Large-scale atomistic simulations with classical potentials … read moreAbstract: Large-scale atomistic simulations with classical potentials can provide valuable insights into microscopic deformation mechanisms and defect-defect interactions in materials. Unfortunately, these assets often come with the uncertainty of whether the observed mechanisms are based on realistic physical phenomena or whether they are artifacts of the employed material models. One such example is the often reported occurrence of stable planar faults (PFs) in body-centered cubic (bcc) metals subjected to high strains, e.g., at crack tips or in strained nano-objects. In this paper, we study the strain dependence of the generalized stacking fault energy (GSFE) of {110} planes in various bcc metals with material models of increasing sophistication, i.e., (modified) embedded atom method, angular-dependent, Tersoff, and bond-order potentials as well as density functional theory. We show that under applied tensile strains the GSFE curves of many classical potentials exhibit a local minimum which gives rise to the formation of stable PFs. These PFs do not appear when more sophisticated material models are used and have thus to be regarded as artifacts of the potentials. We demonstrate that the local GSFE minimum is not formed for reasons of symmetry and we recommend including the determination of the strain-dependent (110) GSFE as a benchmark for newly developed potentials. read less NOT USED (definite) E. Güler and M. Güler, “A benchmark for some bulk properties of bcc iron,” The International Journal of Multiphysics. 2013. link Times cited: 7 Abstract: Some bulk properties of bcc iron were calculated. Structural… read moreAbstract: Some bulk properties of bcc iron were calculated. Structural and elastic properties such as cohesive energy, bulk modulus, typical elastic constants and vacancy formation energy were calculated for zero Kelvin temperature. All obtained results during the study were compared with the both previous experimental and theoretical results. Obtained results for the present study show well agreement with literature. read less |
 EAM_MagneticCubic_DudarevDerlet_2005_Fe__MO_135034229282_002
EAM_MagneticCubic_DudarevDerlet_2005_Fe__MO_135034229282_002