Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
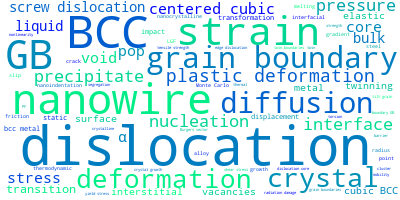
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
1016 Citations (492 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) S. Stephan, S. Schmitt, H. Hasse, and H. Urbassek, “Molecular dynamics simulation of the Stribeck curve: Boundary lubrication, mixed lubrication, and hydrodynamic lubrication on the atomistic level,” Friction. 2023. link Times cited: 3 USED (high confidence) W. Lou, L. Cheng, R. Wang, C.-yang Hu, and K. Wu, “Atomistic Investigation of the Influence of Hydrogen on Mechanical Response during Nanoindentation in Pure Iron,” Acta Metallurgica Sinica (English Letters). 2023. link Times cited: 1 USED (high confidence) I. A. Alhafez, M. Kopnarski, and H. Urbassek, “Multiple Scratching: An Atomistic Study,” Tribology Letters. 2023. link Times cited: 1 USED (high confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (high confidence) C. Kura, M. Wakeda, K. Hayashi, and T. Ohmura, “Energetic and atomic structural analyses of the screw dislocation absorption at tilt grain boundaries in BCC-Fe,” Scientific Reports. 2022. link Times cited: 1 USED (high confidence) L. Reali, M. Gilbert, M. Boleininger, and S. Dudarev, “Intense
γ
-Photon and High-Energy Electron Production by Neutron Irradiation: Effects of Nuclear Excitations on Reactor Materials,” PRX Energy. 2022. link Times cited: 1 Abstract: The effects of neutron irradiation on materials are often in… read moreAbstract: The effects of neutron irradiation on materials are often interpreted in terms of atomic recoils, initiated by neutron impacts and producing crystal lattice defects. In addition, there is a remarkable two-step process, strongly pronounced in the medium-weight and heavy elements. This process involves the generation of energetic {\gamma} photons in nonelastic collisions of neutrons with atomic nuclei, achieved via capture and inelastic reactions. Subsequently, high-energy electrons are excited through the scattering of {\gamma} photons by the atomic electrons. We derive and validate equations enabling a fast and robust evaluation of photon and electron fluxes produced by the neutrons in the bulk of materials. The two-step n-{\gamma}-e scattering creates a nonequilibrium dynamically fluctuating steady-state population of high-energy electrons, with the spectra of photon and electron energies extending well into the mega-electron-volt range. This stimulates vacancy diffusion through electron-triggered atomic recoils, primarily involving vacancy-impurity dissociation, even if thermal activation is ineffective. Tungsten converts the energy of fusion or fission neutrons into a flux of {\gamma} radiation at the conversion efficiency approaching 99%, with implications for structural materials, superconductors, and insulators, as well as phenomena like corrosion, and helium and hydrogen isotope retention. read less USED (high confidence) I. Chesser and Y. Mishin, “Point-defect avalanches mediate grain boundary diffusion,” Communications Materials. 2022. link Times cited: 7 USED (high confidence) G. Sainath and A. Nagesha, “Twin interaction with \Sigma11 tilt grain boundaries in BCC Fe : Formation of new grain boundaries.” 2022. link Times cited: 0 USED (high confidence) N. Bertin, L. Zepeda-Ruiz, and V. Bulatov, “Sweep-tracing algorithm: in silico slip crystallography and tension-compression asymmetry in BCC metals,” Materials Theory. 2022. link Times cited: 10 USED (high confidence) G. Sainath and A. Nagesha, “Atomistic Simulations of Twin Boundary Effect on the Crack Growth Behaviour in BCC Fe,” Transactions of the Indian National Academy of Engineering. 2021. link Times cited: 4 USED (high confidence) J. Veerababu, G. Sainath, and A. Nagesh, “Twin boundary reversibility characteristics in α –Fe,” Materials Today Communications. 2021. link Times cited: 1 USED (high confidence) S. Eder et al., “Experimentally validated atomistic simulation of the effect of relevant grinding parameters on work piece topography, internal stresses, and microstructure,” Friction. 2021. link Times cited: 11 USED (high confidence) S. Eder et al., “Experimentally validated atomistic simulation of the effect of relevant grinding parameters on work piece topography, internal stresses, and microstructure,” Friction. 2021. link Times cited: 1 USED (high confidence) Y. Wang, X.-guo Zeng, H. Chen, X. Yang, F. Wang, and J. Ding, “Hugoniot States and Mie–Grüneisen Equation of State of Iron Estimated Using Molecular Dynamics,” Crystals. 2021. link Times cited: 8 Abstract: The objective of this study was to develop a micromechanical… read moreAbstract: The objective of this study was to develop a micromechanical approach for determining the Mie–Grüneisen EOS parameters of iron under the Hugoniot states. The multiscale shock technique (MSST) coupled with molecular dynamics (MD) simulations was employed to describe the shocked Hugoniot relation of single-crystal (SC) and nanocrystalline (NC) iron under high pressures. The Mie–Grüneisen equation of state (EOS) parameters, the cold pressure (Pc), the cold energy (Ec), the Grüneisen coefficient (γ), and the melting temperature (Tm) are discussed. The error between SC and NC iron results was found to be less than 1.5%. Interestingly, the differences in Hugoniot state (PH) and the internal energy between SC and NC iron were insignificant, which shows that the effect of grain size (GS) under high pressures was not significant. The Pc and Ec of SC and NC iron calculated based on the Morse potential were almost the same with those calculated based on the Born–Mayer potential; however, those calculated based on the Born–Mayer potential were a little larger at high pressures. In addition, several empirical and theoretical models were compared for the calculation of γ and Tm. The Mie–Grüneisen EOSs were shown on the 3D contour space; the pressure obtained with the Hugoniot curves as the reference was larger than that obtained with the cold curves as the reference. read less USED (high confidence) Y. Yang, X. Ding, J. Sun, and E. Salje, “Twisting of a Pristine α-Fe Nanowire: From Wild Dislocation Avalanches to Mild Local Amorphization,” Nanomaterials. 2021. link Times cited: 3 Abstract: The torsion of pristine α-Fe nanowires was studied by molecu… read moreAbstract: The torsion of pristine α-Fe nanowires was studied by molecular dynamics simulations. Torsion-induced plastic deformation in pristine nanowires is divided into two regimes. Under weak torsion, plastic deformation leads to dislocation nucleation and propagation. Twisting-induced dislocations are mainly 12<111> screw dislocations in a <112>-oriented nanowire. The nucleation and propagation of these dislocations were found to form avalanches which generate the emission of energy jerks. Their probability distribution function (PDF) showed power laws with mixing between different energy exponents. The mixing stemmed from simultaneous axial and radial dislocation movements. The power-law distribution indicated strongly correlated ‘wild’ dislocation dynamics. At the end of this regime, the dislocation pattern was frozen, and further twisting of the nanowire did not change the dislocation pattern. Instead, it induced local amorphization at the grip points at the ends of the sample. This “melting” generated highly dampened, mild avalanches. We compared the deformation mechanisms of twinned and pristine α-Fe nanowires under torsion. read less USED (high confidence) C. Xu and D. Yang, “Helium Effects on the Mechanical Properties of Nanocrystalline Fe: Based on Molecular Dynamics,” Crystals. 2021. link Times cited: 1 Abstract: A molecular dynamics (MD) simulation study was performed to … read moreAbstract: A molecular dynamics (MD) simulation study was performed to investigate the effects of helium (He) on the mechanical properties of nanocrystalline body-centered cubic iron (BCC Fe). Simulated X-ray diffraction (XRD) was used to explore the relationship between the generation of cracks and the change of the crystal structure in nanocrystalline BCC Fe during tensile deformation. It is observed that the peak stress and the elastic modulus decrease with increasing concentration of He atoms, which are introduced into the grain boundary (GB) region of nanocrystalline Fe. The generation and connection of intergranular cracks are enhanced by He atoms. Significant peak separation, which is associated with the generation of cracks, is found in the simulated XRD patterns of nanocrystalline Fe during the tensile process. The lower diffraction angle of the {211}′ peak suggests a more serious lattice distortion during loading. For all nanocrystalline Fe deformed to 6% strain, the degree and fraction of the lattice distortion increases with the increasing loading stress. read less USED (high confidence) K. Lyon et al., “Theory of magnon diffuse scattering in scanning transmission electron microscopy,” Physical Review B. 2021. link Times cited: 2 Abstract: Keenan Lyon, ∗ Anders Bergman, Paul Zeiger, Demie Kepaptsogl… read moreAbstract: Keenan Lyon, ∗ Anders Bergman, Paul Zeiger, Demie Kepaptsoglou, 3 Quentin M. Ramasse, 4, 5 Juan Carlos Idrobo, and Ján Rusz † Department of Physics and Astronomy, Uppsala University, Lägerhyddsvägen 1, Uppsala, Sweden SuperSTEM Laboratory, SciTech Daresbury Campus, Daresbury WA4 4AD, United Kingdom Department of Physics, University of York, York YO10 5DD, United Kingdom School of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, United Kingdom School of Physics and Astronomy, University of Leeds, Leeds LS2 9JT, United Kingdom Center for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN, USA (Dated: May 11, 2021) read less USED (high confidence) L. Wu, Y. Zhu, H. Wang, and M. Li, “Crystal–melt interface kinetic behaviors of iron,” AIP Advances. 2021. link Times cited: 5 Abstract: While the crystal–melt interface kinetic equation predicts v… read moreAbstract: While the crystal–melt interface kinetic equation predicts various kinetic behaviors, the realization of these scenarios and the corresponding thermodynamic conditions remain unclear. In this work, six representative interface kinetic behaviors of Fe were modeled and examined by molecular dynamics simulations. For the flat interface, several models were designed to study the migration, fluctuation, and recovery of the interface. For the cylindrical or curved interface, different models were also designed to test the equilibrium, migration, and instability of the interface. By comparing the kinetic behaviors of the two types of interfaces, we can observe the effect of interface curvature. During the simulations, two crucial material-specific parameters, the crystal–melt interface free energy and kinetic coefficient, were determined and compared among different models. read less USED (high confidence) N. Zhou, K. Elkhodary, L. Zhang, and S. Tang, “Understanding the linear relation between pop-in excursion length and critical force for spherical nanoindentation,” Philosophical Magazine. 2021. link Times cited: 1 Abstract: ABSTRACT Pop-in is a widely observed phenomenon in nanoinden… read moreAbstract: ABSTRACT Pop-in is a widely observed phenomenon in nanoindentation. In this paper, dislocation evolution in pop-in processes is analysed in detail through molecular dynamics (MD) simulations. We found that a large number of dislocations nucleate homogeneously at the initiation of pop-in, followed by extensive dislocation propagation, which is the dominant mode of plastic deformation during pop-in. Moreover, we noted that establishing the correct dislocation evolution mechanisms of pop-in can serve to explain the overshoot phenomenon observed in nanoindentation experiments. Through our MD analysis on the obtained dislocation structures, therefore, we were able to propose a model that can predict the total length of dislocations associated with the plastic processes underneath a spherical indenter. In addition, the Taylor model was used to verify that our proposed dislocation length model sits well with the MD simulated force-displacement curves of nanoindentation. In fact, the MD simulated linear relation between critical force and indentation depth during pop-in is consistent with the Hertzian and Taylor models. Our MD simulations, therefore, can provide significant insight into the experimentally observed pop-in phenomena. read less USED (high confidence) P. Tripathi, S. Karewar, Y. Lo, and S. Bhowmick, “Role of interface morphology on the martensitic transformation in pure Fe,” Materialia. 2021. link Times cited: 7 USED (high confidence) T. D. Ta, H. Ta, K. Tieu, and B. Tran, “Impact of chosen force fields and applied load on thin film lubrication,” Friction. 2021. link Times cited: 7 USED (high confidence) T. D. Ta, H. Ta, K. Tieu, and B. Tran, “Impact of chosen force fields and applied load on thin film lubrication,” Friction. 2021. link Times cited: 0 USED (high confidence) Z. Wang, X. Shi, X. Yang, Z. H. Liu, S. Shi, and X. Ma, “The Effects of Hydrogen Distribution on the Elastic Properties and Hydrogen-Induced Hardening and Softening of α-Fe,” Applied Sciences. 2020. link Times cited: 2 Abstract: In this work, we conducted a high-throughput atomistic simul… read moreAbstract: In this work, we conducted a high-throughput atomistic simulation of the interstitial solid solutions of hydrogen in α-Fe. The elastic constants and moduli were calculated. Through statistical analysis of structures and results, the influences of the microscopic distribution of hydrogen on the elastic moduli, as well as hydrogen-induced hardening and softening, are discussed. We found that even though the uniformly distributed hydrogen caused slight softening in α-Fe, the distribution of hydrogen at different adjacent positions significantly affected the elastic moduli. For example, hydrogen increased the Young’s modulus and shear modulus at the 5th and 10th nearest neighbors, resulting in hardening, but decreased the bulk modulus at the 7th nearest neighbor, making the material easier to compress. These phenomena are related to the distribution densities of the positions that hydrogen atoms can occupy on the two major slip families, {110} and {112}, at different nearest neighbors distinguished by distances. read less USED (high confidence) J. G. Sevillano, I. Aldazabal, and J. Aldazabal, “Plastically-Induced Volume Deformation of Nanocrystalline α-Fe with a <110> Columnar Structure.” 2020. link Times cited: 0 Abstract: Volume changes accompanying the plastic deformation at 300 K… read moreAbstract: Volume changes accompanying the plastic deformation at 300 K of nanocrystalline samples of α-Fe with a columnar grain structure possessing a ⟨11¯0⟩ random fiber texture has been obtained from molecular dynamics (MD) simulations. The samples were strained in tension along the common axial direction of the columnar grains. After removal of the elastic volume change, the evolution of plastic volume strain was obtained. Small but non-negligible volume dilations or contractions are observed depending on size (samples of very small grain size show volume contraction). The rate of volume change is high during the first 10% plastic deformation and continues at a low pace thereafter; the first 10% deformation represents a transient in the stress–strain behavior too. The complex behavior observed is reasonably explained by the superposition of contributions from different plastically-induced structural changes to the mass density change: Mainly from changes of grain size, grain boundary structure, dislocation density and density of point-defects. The results are of interest for the development of crystal plasticity theories not restricted by the volume conserving assumption. read less USED (high confidence) G. Nikoulis, P. Grammatikopoulos, S. Steinhauer, and J. Kioseoglou, “NanoMaterialsCAD: Flexible Software for the Design of Nanostructures,” Advanced Theory and Simulations. 2020. link Times cited: 1 Abstract: NanoMaterialsCAD is a new open source tool dedicated to the … read moreAbstract: NanoMaterialsCAD is a new open source tool dedicated to the creation, manipulation, and 3D visualization of crystalline structures at the nanoscale. It is designed for preprocessing atomistic configurations to be used as input for atomistic (e.g., molecular dynamics or Monte Carlo) or ab initio (e.g., density functional theory) computer simulations. It offers several tools for designing complex nanostructures (including nanoparticles, nanowires, nanotubes, nanoscrolls, etc., and combinations/permutations of them) which are either lacking or cumbersome in other existing packages. Through its intuitive graphical user interface (GUI) it enables facile ways to design and modify the size/shape and relative positions of nanoobjects while observing the changes in real time. NanoMaterialsCAD is written in C++, and exploits Open Graphics Library (OpenGL) (for the GUI), Win32API (for interaction with Windows), and Assembly (for fast data management). The source code and executable file are available for download from GitHub (https://github.com/cossphy/NanoMaterialsCAD). It is aspired that NanoMaterialsCAD will be adopted by the nanomaterials modeling community as a valuable resource; to this end it will be kept improving, incorporating more nanostructures, and adding extra functionalities to its toolbox. read less USED (high confidence) K. Zolnikov, D. Kryzhevich, and A. Korchuganov, “Regularities of Structural Rearrangements in Single- and Bicrystals Near the Contact Zone,” Springer Tracts in Mechanical Engineering. 2020. link Times cited: 0 USED (high confidence) D. Louzguine-Luzgin and A. Bazlov, “Crystallization of FCC and BCC Liquid Metals Studied by Molecular Dynamics Simulation,” Metals. 2020. link Times cited: 17 Abstract: The atomic structure variations on cooling, vitrification an… read moreAbstract: The atomic structure variations on cooling, vitrification and crystallization processes in liquid metals face centered cubic (FCC) Cu are simulated in the present work in comparison with body centered cubic (BCC) Fe. The process is done on continuous cooling and isothermal annealing using a classical molecular-dynamics computer simulation procedure with an embedded-atom method potential at constant pressure. The structural changes are monitored with direct structure observation in the simulation cells containing from about 100 k to 1 M atoms. The crystallization process is analyzed under isothermal conditions by monitoring density and energy variation as a function of time. A common-neighbor cluster analysis is performed. The results of thermodynamic calculations on estimating the energy barrier for crystal nucleation and a critical nucleus size are compared with those obtained from simulation. The differences in crystallization of an FCC and a BCC metal are discussed. read less USED (high confidence) X. Xing et al., “Molecular Dynamics Studies of Hydrogen Effect on Intergranular Fracture in α-Iron,” Materials. 2020. link Times cited: 5 Abstract: In the current study, the effect of hydrogen atoms on the in… read moreAbstract: In the current study, the effect of hydrogen atoms on the intergranular failure of α-iron is examined by a molecular dynamics (MD) simulation. The effect of hydrogen embrittlement on the grain boundary (GB) is investigated by diffusing hydrogen atoms into the grain boundaries using a bicrystal body-centered cubic (BCC) model and then deforming the model with a uniaxial tension. The Debye Waller factors are applied to illustrate the volume change of GBs, and the simulation results suggest that the trapped hydrogen atoms in GBs can therefore increase the excess volume of GBs, thus enhancing intergranular failure. When a constant displacement loading is applied to the bicrystal model, the increased strain energy can barely be released via dislocation emission when H is present. The hydrogen pinning effect occurs in the current dislocation slip system, <111>{112}. The hydrogen atoms facilitate cracking via a decrease of the free surface energy and enhance the phase transition via an increase in the local pressure. Hence, the failure mechanism is prone to intergranular failure so as to release excessive pressure and energy near GBs. This study provides a mechanistic framework of intergranular failure, and a theoretical model is then developed to predict the intergranular cracking rate. read less USED (high confidence) Z. Wang, X. Shi, X. Yang, W. He, S. Shi, and X. Ma, “Atomistic simulation of martensitic transformations induced by deformation of α-Fe single crystal during the mode-I fracture,” Journal of Materials Science. 2020. link Times cited: 4 USED (high confidence) R. Ishraaq, S. Chhetri, O. Gautam, S. Nahid, and A. Afsar, “Molecular dynamics simulation for the analysis of mechanical properties and effect of Stone-Wales and bi-vacancy defect on carbon nanotube reinforced iron composites,” arXiv: Materials Science. 2020. link Times cited: 0 Abstract: Carbon nanotube (CNT) reinforced metal matrix composites (MM… read moreAbstract: Carbon nanotube (CNT) reinforced metal matrix composites (MMCs) are gaining the attention of the researchers because of their demand in space and automobile industries for having low weight and high mechanical properties. Iron is the most used metal in all engineering fields. Therefore, reinforcing iron with CNT can reduce its required amount, which might have a positive economic impact due to the reduced cost of production. However, before the industrial application of any material the mechanical properties under different conditions must be known. In this study, the mechanical properties of iron reinforced separately with single, double and triple wall CNTs are investigated by Molecular Dynamics (MD) simulation. The study revealed that the strength and stiffness of pure iron could be enhanced up to 80.4 % and 57.4 %, respectively, by adding CNTs into iron. We also investigated the effect of fiber volume percentage and temperature on the mechanical properties of the composite having single, double and triplewalled carbon nanotubes individually. As the stone-wales and bi-vacancy defects are inherently introduced in CNTs during manufacturing, their effect on mechanical properties are also investigated in the present study read less USED (high confidence) J. Fang, L. Liu, N. Gao, W. Hu, F. Gao, and H. Deng, “Molecular dynamics simulation of the diffusion of self-interstitial atoms and interstitial loops under temperature gradient field in tungsten,” Journal of Applied Physics. 2020. link Times cited: 5 Abstract: Tungsten (W) and W-based alloys are potential candidates for… read moreAbstract: Tungsten (W) and W-based alloys are potential candidates for next-generation fusion reactors, which would withstand both irradiation damages and heavy heat load. In this work, we employed the molecular dynamic method to simulate the behaviors of different radiation defects under the effect of the temperature gradient field, which is induced by heat load. The rotation of the ⟨ 111 ⟩ dumbbell and habit plane of 1/2 ⟨ 111 ⟩ interstitial loops is analyzed in detail. The results show that these two behaviors are not significantly affected by the temperature gradient. Contrary to the thermal equilibrium state, temperature gradient results in the directional diffusion of ⟨ 111 ⟩ dumbbell and 1/2 ⟨ 111 ⟩ interstitial loops in tungsten from the cold to the hot region. The energy barrier is also reduced in the temperature gradient field, which accelerates the defect diffusion. These results indicate that the accumulation of radiation defects in the high-temperature region is expected in temperature gradient fields, which would lead to more severe radiation damages and degradation of materials. read less USED (high confidence) J. Herman, M. Govednik, S. P. Patil, and B. Markert, “Molecular Dynamics Simulation Study of the Mechanical Properties of Nanocrystalline Body-Centered Cubic Iron,” Surfaces. 2020. link Times cited: 4 Abstract: In the present work, the mechanical properties of nanocrysta… read moreAbstract: In the present work, the mechanical properties of nanocrystalline body-centered cubic (BCC) iron with an average grain size of 10 Å were investigated using molecular dynamics (MD) simulations. The structure has one layer of crystal grains, which means such a model could represent a structure with directional crystallization. A series of uniaxial tensile tests with different strain rates and temperatures was performed until the full rupture of the model. Moreover, tensile tests of the models with a void at the center and shear tests were carried out. In the tensile test simulations, peak stress and average values of flow stress increase with strain rate. However, the strain rate does not affect the elasticity modulus. Due to the presence of void, stress concentrations in structure have been observed, which leads to dislocation pile-up and grain boundary slips at lower strains. Furthermore, the model with the void reaches lower values of peak stresses as well as stress overshoot compared to the no void model. The study results provide a better understanding of the mechanical response of nanocrystalline BCC iron under various loadings. read less USED (high confidence) W. Xie, C. Liu, and D. Jiang, “Normal behavior of single-asperity contact of bcc iron: a molecular dynamics simulation study,” 2020 Asia-Pacific International Symposium on Advanced Reliability and Maintenance Modeling (APARM). 2020. link Times cited: 0 Abstract: Characteristics of contact surface are important for the ope… read moreAbstract: Characteristics of contact surface are important for the operation of precision machines, which need to transmit force and torque, such as the interface contact between rotating disks of gas turbine rotor system. Meanwhile, the contact behavior of microscopic contact interface is proven to be different from that of macroscopic contact. This work presents molecular dynamic simulation of microscopic contact behaviors with single-asperity, including single-asperity contact with same asperity radius, single-asperity contact with different asperity radius and effect of overlapping ratio of the adjacent asperity. Through molecular dynamic simulation results, comparisons are conducted between microscopic contact and macroscopic contact behavior as well as evolution process of the dislocation. The simulation results show that dislocation has important effects on single-asperity contact, resulting in the reduction of contact force comparing to Hertz contact theory. read less USED (high confidence) Y. Yang, S. Li, X. Ding, J. Sun, J. Weiss, and E. Salje, “Twisting of pre-twinned α-Fe nanowires: from mild to wild avalanche dynamics,” Acta Materialia. 2020. link Times cited: 16 USED (high confidence) S. S. Pezeshki, M. Silani, M. S. Talaei, and S. Ziaei-Rad, “An atomistic perspective into the fracture behaviour of Fe-bicrystal,” Molecular Simulation. 2020. link Times cited: 0 Abstract: ABSTRACT The interaction between the crack and the grain bou… read moreAbstract: ABSTRACT The interaction between the crack and the grain boundary has been investigated by molecular dynamics simulation. The focus of this research is to study fracture resistance of grain boundary in a three-dimensional pre-cracked Fe-bicrystal. The fracture resistance of Σ5 < 100> {013} symmetric tilt grain boundary (STGB) has been compared with Σ5 < 100> {012} STGB in terms of crack length-time curve, temperature per time diagram, and the stress–strain curve to understand the detailed mechanism of fracture in Fe-bicrystal. Crack delay time at the grain boundary is proposed as a parameter for comparing the fracture resistance of grain boundaries. The results show that the crack delay time at the grain boundary is inversely related to the grain boundary energy. Hence, crack delay time for Σ5{013} STGB with 986 energy is more than Σ5{012} STGB with 1098 energy. The findings show that Σ5{013} STGB resists more than Σ5{012} STGB against crack propagation. The required stress, which is needed to overcome the grain boundary resistance and cause the crack penetration to the adjacent grain, has been calculated by using stress–strain curve. Modified BCC defect analysis algorithm and centrosymmetry parameter are also employed to analyze propagated defects and their interaction with the grain boundary. read less USED (high confidence) J. Byggmastar, K. Nordlund, and F. Djurabekova, “Gaussian approximation potentials for body-centered-cubic transition metals,” Physical Review Materials. 2020. link Times cited: 22 Abstract: We develop a set of machine-learning interatomic potentials … read moreAbstract: We develop a set of machine-learning interatomic potentials for elemental V, Nb, Mo, Ta, and W using the Gaussian approximation potential framework. The potentials show good accuracy and transferability for elastic, thermal, liquid, defect, and surface properties. All potentials are augmented with accurate repulsive potentials, making them applicable to radiation damage simulations involving high-energy collisions. We study melting and liquid properties in detail and use the potentials to provide melting curves up to 400 GPa for all five elements. read less USED (high confidence) I. A. Alhafez and H. Urbassek, “Influence of the Rake Angle on Nanocutting of Fe Single Crystals: A Molecular-Dynamics Study,” Crystals. 2020. link Times cited: 9 Abstract: Using molecular dynamics simulation, we study the cutting of… read moreAbstract: Using molecular dynamics simulation, we study the cutting of an Fe single crystal using tools with various rake angles α . We focus on the (110)[001] cut system, since here, the crystal plasticity is governed by a simple mechanism for not too strongly negative rake angles. In this case, the evolution of the chip is driven by the generation of edge dislocations with the Burgers vector b = 1 2 [ 111 ] , such that a fixed shear angle of ϕ = 54.7 ∘ is established. It is independent of the rake angle of the tool. The chip form is rectangular, and the chip thickness agrees with the theoretical result calculated for this shear angle from the law of mass conservation. We find that the force angle χ between the direction of the force and the cutting direction is independent of the rake angle; however, it does not obey the predictions of macroscopic cutting theories, nor the correlations observed in experiments of (polycrystalline) cutting of mild steel. Only for (strongly) negative rake angles, the mechanism of plasticity changes, leading to a complex chip shape or even suppressing the formation of a chip. In these cases, the force angle strongly increases while the friction angle tends to zero. read less USED (high confidence) J. Meiser and H. Urbassek, “α ↔ γ phase transformation in iron: comparative study of the influence of the interatomic interaction potential,” Modelling and Simulation in Materials Science and Engineering. 2020. link Times cited: 6 Abstract: Only few available interatomic interaction potentials implem… read moreAbstract: Only few available interatomic interaction potentials implement the α ↔ γ phase transformation in iron by featuring a stable low-temperature bcc and high-temperature fcc lattice structure. Among these are the potentials by Meyer and Entel (1998 Phys. Rev. B 57 5140), by Müller et al (2007 J. Phys.: Condens. Matter 19 326220) and by Lee et al (2012 J. Phys.: Condens. Matter 24 225404). We study how these potentials model the phase transformation during heating and cooling; in order to help initiating the transformation, the simulation volume contains a grain boundary. For the martensitic transformation occurring on cooling an fcc structure, we additionally study two potentials that only implement a stable bcc structure of iron, by Zhou et al (2004 Phys. Rev. B 69 144113) and by Mendelev et al (2003 Philos. Mag. 83 3977). We find that not only the transition temperature depends on the potential, but that also the height of the energy barrier between fcc and bcc phase governs whether the transformation takes place at all. In addition, details of the emerging microstructure depend on the potential, such as the fcc/hcp fraction formed in the α → γ transformation, or the twinning induced in and the lattice orientation of the bcc phase in the γ → α transformation. read less USED (high confidence) Y. Wu, K. Zhang, J. Xiao, Y. Jiang, and L. Lv, “Conjugated bilayer structure of the homogeneous solid-liquid interface of metals.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 1 Abstract: The concept of "interfacial region" has long preva… read moreAbstract: The concept of "interfacial region" has long prevailed for over half century for describing the homogeneous solid-liquid (SL) interface of metals, but its intrinsic structure is still unclear due to the homogeneity. In this study, we reveal, for the first time, the intrinsic structure of these homogeneous SL interfaces consisting of two conjugated monoatomic layers of interfacial solid (IS) and interfacial liquid (IL) with a certain degree of corrugation via molecular dynamics simulations. We named it as the conjugated bilayer structure (CBS). In the framework of CBS, only the IS + IL bilayer plays stepwise transition roles from the solid to the liquid, which defines the four-terrace nature of the interface and act simultaneously as the boundaries of the bilateral bulk phases. The inherent diffuse nature of the "interfacial region" is proven originating from the corrugation of the IS + IL bilayer and its four-terrace nature. More importantly, the CBS also explains that the interfacial free energy originates mainly from the increase in the potential energy of the IS layer relative to its counterpart bulk solid instead of the previously argued entropy loss of the liquid phase. After all these verifications and interpretations, the CBS was verified as the intrinsic structure of the homogeneous SL interface of metals. Accordingly, we argue that the concept of CBS also resolves the volume-bearing flaw of the "interfacial region" concept and can definitively locate the intrinsic surface according to the capillary wave theory. read less USED (high confidence) X. Ou, J. Sietsma, and M. Santofimia, “Coalescence of martensite under uniaxial tension of iron crystallites by atomistic simulations,” Materials Science and Technology. 2020. link Times cited: 6 Abstract: Molecular dynamics simulations are used to study the effects… read moreAbstract: Molecular dynamics simulations are used to study the effects of tensile loading on nucleation and subsequent growth of bcc phase in pure fcc iron. The results show that orientation variant selection occurs during the stress-induced fcc-to-bcc transformation, which leads to the coalescence of neighbouring bcc platelets with identical orientation. The bcc phase nucleates mainly following Nishiyama–Wassermann and Kurdjumov–Sachs orientation relationships with the parent fcc phase. The present simulations contribute to a better understanding of mechanisms controlling mechanically induced martensitic transformation as well as coalescence of bcc platelets in steels. read less USED (high confidence) “Impulsive generation of 〈100〉 dislocation loops in BCC iron,” Modelling and Simulation in Materials Science and Engineering. 2020. link Times cited: 2 Abstract: The conditions for the formation of 〈100〉 dislocation loops … read moreAbstract: The conditions for the formation of 〈100〉 dislocation loops in body-centered cubic (BCC) iron were investigated via molecular dynamics simulations using a simplified model intended to mimic conditions in high energy collision cascades, focusing on the possible coherent displacement of atoms at the boundary of a subcascade. We report on the formation of 〈100〉 dislocation loops due to the fast displacement of a few hundred atoms with a coherent acceleration, in agreement with previous results for much larger cascade simulations. We analyze in detail the resulting atomic velocities and pressures, and find that they cannot be described within the usual formalism for a shock regime, since the pressure pulse only lasts less than 1 ps and does not match expected values from a Hugoniot shock. Our simulations include two interatomic potentials: Mendelev, which is extensively used for radiation damage simulations, and Ackland, which has been used for shock simulations because it can reproduce the experimentally observed transition from BCC to hexagonal close-packed structure at around 25 GPa, at high deformation rates. They both show similar evolution of defects, also indicating departure from a shock regime which is extremely different for these potentials. read less USED (high confidence) G. Sainath, S. Goyal, and A. Nagesha, “Plasticity through De-Twinning in Twinned BCC Nanowires,” Crystals. 2020. link Times cited: 10 Abstract: The deformation behaviour of twinned FCC nanowires has been … read moreAbstract: The deformation behaviour of twinned FCC nanowires has been extensively investigated in recent years. However, the same is not true for their BCC counterparts. Very few studies exist concerning the deformation behaviour of twinned BCC nanowires. In view of this, molecular dynamics (MD) simulations have been performed to understand the deformation mechanisms in twinned BCC Fe nanowires. The twin boundaries (TBs) were oriented parallel to the loading direction [110] and the number of TBs is varied from one to three. MD simulation results indicate that deformation under the compressive loading of twinned BCC Fe nanowires is dominated by a unique de-twinning mechanism involving the migration of a special twin–twin junction. This de-twinning mechanism results in the complete annihilation of pre-existing TBs along with reorientation of the nanowire. Further, it has been observed that the annihilation of pre-existing TBs has occurred through two different mechanisms, one without any resolved shear stress and other with finite and small resolved shear stress. The present study enhances our understanding of de-twinning in BCC nanowires. read less USED (high confidence) W. Zhou, X. Ren, Y. Yang, Z. Tong, and L. Chen, “Dislocation behavior in nickel and iron during laser shock-induced plastic deformation,” The International Journal of Advanced Manufacturing Technology. 2020. link Times cited: 14 USED (high confidence) H. Zong, H. Wiebe, and G. Ackland, “Understanding high pressure molecular hydrogen with a hierarchical machine-learned potential,” Nature Communications. 2020. link Times cited: 7 USED (high confidence) M. Zonana, C. Ruestes, E. Bringa, and H. Urbassek, “Effect of Tip Roundness on the Nanoindentation of Fe Crystals,” Tribology Letters. 2020. link Times cited: 7 USED (high confidence) N. Zhou, K. Elkhodary, X. Huang, S. Tang, and Y. Li, “Dislocation structure and dynamics govern pop-in modes of nanoindentation on single-crystal metals,” Philosophical Magazine. 2020. link Times cited: 14 Abstract: ABSTRACT There are two types of pop-in mode that have been w… read moreAbstract: ABSTRACT There are two types of pop-in mode that have been widely observed in nanoindentation experiments: the single pop-in, and the successive pop-in modes. Here we employ the molecular dynamics (MD) modelling to simulate nanoindentation for three face-centred cubic (FCC) metals, including Al, Cu and Ni, and two body-centred cubic (BCC) metals, such as Fe and Ta. We aim to examine the deformation mechanisms underlying these pop-in modes. Our simulation results indicate that the dislocation structures formed in single crystals during nanoindentation are mainly composed of half prismatic dislocation loops. These half prismatic dislocation loops in FCC metals are primarily constituted of extended dislocations. Lomer–Cottrell locks that result from the interactions between these extended dislocations can resist the slipping of half dislocation loops. These locks can build up the elastic energy that is needed to activate the nucleation of new half dislocation loops. A repetition of this sequence results in successive pop-in events in Al and other FCC metals. Conversely, the half prismatic dislocation loops that form in BCC metals after first pop-in are prone to slip into the bulk, which sustains plastic indentation process after first pop-in and prevents subsequent pop-ins. We thus conclude that pop-in modes are correlated with lattice structures during nanoindentation, regardless of their crystal orientations. read less USED (high confidence) P. Derlet and S. Dudarev, “Microscopic structure of a heavily irradiated material,” Physical Review Materials. 2020. link Times cited: 48 Abstract: New generation nuclear fission and future fusion reactors pr… read moreAbstract: New generation nuclear fission and future fusion reactors provide one approach to address the world's increasing energy requirements. The irradiation of fission/fusion components can lead to fundamental changes in material properties that affect the stability and performance of not only the material component but that of the entire reactor. How does the material state evolve with respect to the irradiation dose, and can there exist a microstructure resistant to further irradiation? The present work develops a new computationally efficient approach to answer these questions. read less USED (high confidence) J. A. Hofer, C. Ruestes, E. Bringa, and H. Urbassek, “Effect of subsurface voids on the nanoindentation of Fe crystals,” Modelling and Simulation in Materials Science and Engineering. 2020. link Times cited: 3 Abstract: Subsurface voids may strongly affect the response of materia… read moreAbstract: Subsurface voids may strongly affect the response of materials to nanoindentation. We explore these effects for a bcc single-crystalline Fe sample using molecular dynamics simulation. Deformation occurs mainly by nucleation and propagation of dislocations. As dislocations impinge into the voids, these suffer a reduction in volume, consistent with mass transfer mechanisms. Our results show that voids act as highly efficient absorbers of dislocations, effectively limiting the extension of the plastic zone. Surprisingly, mechanical properties are marginally affected by the presence of voids in the range of sizes and spatial distributions tested, except for voids a few nanometers below the surface. Deformation twinning is observed as a transient effect in some cases; however, for voids close enough to the indentation area, no twinning was found. read less USED (high confidence) G. Sun, A. Hawken, and P. Harrowell, “The displacement field associated with the freezing of a melt and its role in determining crystal growth kinetics,” Proceedings of the National Academy of Sciences. 2020. link Times cited: 11 Abstract: Significance We demonstrate that an accurate estimation of t… read moreAbstract: Significance We demonstrate that an accurate estimation of the displacements associated with the transformation of liquid into crystal is necessary to explain the striking variations in the temperature dependence of the addition rate of liquid atoms to the growing crystal interface during freezing. An assignment algorithm, adapted from operations theory, is shown to provide a good estimate of these atomic displacements. As the assignment algorithm requires only initial and target locations, it is applicable to all forms of structural transformations. In resolving a fundamental feature of the kinetics of freezing, a phenomenon of central importance to material fabrication, this paper also provides the tools to open lines of research into the kinetics of structural transformation. The atomic displacements associated with the freezing of metals and salts are calculated by treating crystal growth as an assignment problem through the use of an optimal transport algorithm. Converting these displacements into timescales based on the dynamics of the bulk liquid, we show that we can predict the activation energy for crystal growth rates, including activation energies significantly smaller than those for atomic diffusion in the liquid. The exception to this success, pure metals that freeze into face-centered cubic crystals with little to no activation energy, are discussed. The atomic displacements generated by the assignment algorithm allows us to quantify the key roles of crystal structure and liquid caging length in determining the temperature dependence of crystal growth kinetics. read less USED (high confidence) W. Zhou, X. Ren, Y. Yang, Z. Tong, and L. Chen, “Dislocation behavior in nickel and iron during laser shock-induced plastic deformation,” The International Journal of Advanced Manufacturing Technology. 2019. link Times cited: 0 USED (high confidence) S. Kim, K. Kang, and S. Y. Kim, “Dynamic drags acting on moving defects in discrete dispersive media: From dislocation to low-angle grain boundary,” arXiv: Materials Science. 2019. link Times cited: 6 USED (high confidence) A. Rajput and S. Paul, “Cyclic Plastic Deformation Response of Nanocrystalline BCC Iron,” Metals and Materials International. 2019. link Times cited: 11 USED (high confidence) R. Essajai, Y. Benhouria, A. Rachadi, M. Qjani, A. Mzerd, and N. Hassanain, “Shape-dependent structural and magnetic properties of Fe nanoparticles studied through simulation methods,” RSC Advances. 2019. link Times cited: 14 Abstract: Studying the shape-dependent structural and magnetic propert… read moreAbstract: Studying the shape-dependent structural and magnetic properties of nanoparticles is one of the most necessary scientific challenges in order to match these nano-objects for adequate applications. In this research paper, the shape effect of iron nanoparticles (FeNPs) on structural and magnetic properties was investigated on the basis of a combination of Molecular Statics (MS) and Monte Carlo (MC) simulations. To this end, three kinds of FeNP shapes (such as spherical, planar and rod) in an equal volume have been considered. The coordination number distribution of FeNPs obtained from the data extracted by MS simulations was exploited for performing MC simulations on the familiar Ising model. The numerical findings obtained showed that the structural stability, the Curie temperature as well as the shape of the hysteresis loop are correlated with the FeNP shape. read less USED (high confidence) I. A. Alhafez and H. Urbassek, “Influence of tip adhesion on nanoindentation and scratching,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 9 Abstract: Using molecular dynamics simulation, we study the influence … read moreAbstract: Using molecular dynamics simulation, we study the influence of tip adhesion on nanoindentation and scratching. By using a model pair potential between tip atoms and substrate atoms, we can arbitrarily change the adhesion strength. For the prototypical case of a diamond tip and a bcc Fe substrate, we find that with increasing adhesion strength, the indentation hardness and also the normal hardness during scratching decreases. Even more pronounced is a strong increase of the transverse force and hence of the friction coefficient during scratching. The indent pit becomes atomically rough, and the pileup produced during scratch increases with increasing adhesion strength. On the other hand, the length of the dislocations produced and the spatial extent of the plastic zone shrinks. read less USED (high confidence) S. Deldar, I. A. Alhafez, M. Smaga, T. Beck, and H. Urbassek, “Cyclic Indentation of Iron: A Comparison of Experimental and Atomistic Simulations,” Metals. 2019. link Times cited: 7 Abstract: Cyclic indentation is a technique used to characterize mater… read moreAbstract: Cyclic indentation is a technique used to characterize materials by indenting repeatedly on the same location. This technique allows information to be obtained on how the plastic material response changes under repeated loading. We explore the processes underlying this technique using a combined experimental and simulative approach. We focus on the loading–unloading hysteresis and the dependence of the hysteresis width ha,p on the cycle number. In both approaches, we obtain a power-law demonstrating ha,p with respect to the hardening exponent e. A detailed analysis of the atomistic simulation results shows that changes in the dislocation network under repeated indentation are responsible for this behavior. read less USED (high confidence) R. Mohammadzadeh, “Analysis of plastic strain-enhanced diffusivity in nanocrystalline iron by atomistic simulation,” Journal of Applied Physics. 2019. link Times cited: 8 Abstract: Plastic deformation may affect bulk diffusion in nanocrystal… read moreAbstract: Plastic deformation may affect bulk diffusion in nanocrystalline materials by altering the rates of point defect production and annihilation. In the present work, a detailed analysis of this phenomenon is given by a series of molecular dynamics (MD) simulations to clarify the effect of preplastic strain on the diffusivity of iron atoms. The embedded atom method interatomic potential was used to perform MD simulations. The self-diffusion coefficient of iron atoms in unstrained and prestrained samples was measured over temperatures ranging from 600 to 1000 K and at total strains of 5%–20%. The results reveal that the diffusivity is indeed enhanced as a result of plastic straining, especially at low temperatures. The calculated diffusion coefficient of iron atoms in the prestrained samples is 10–80 times higher than that in the unstrained samples. The atomic structure analysis results indicated that the generation of excess vacancies and unstructured region by preplastic deformation contributes to the enhancement of self-diffusion under plastic straining conditions. At low temperatures, preplastic straining has a considerable effect in the peak shifting and broadening of the radial distribution function, which probably lowers the activation barrier height for diffusion.Plastic deformation may affect bulk diffusion in nanocrystalline materials by altering the rates of point defect production and annihilation. In the present work, a detailed analysis of this phenomenon is given by a series of molecular dynamics (MD) simulations to clarify the effect of preplastic strain on the diffusivity of iron atoms. The embedded atom method interatomic potential was used to perform MD simulations. The self-diffusion coefficient of iron atoms in unstrained and prestrained samples was measured over temperatures ranging from 600 to 1000 K and at total strains of 5%–20%. The results reveal that the diffusivity is indeed enhanced as a result of plastic straining, especially at low temperatures. The calculated diffusion coefficient of iron atoms in the prestrained samples is 10–80 times higher than that in the unstrained samples. The atomic structure analysis results indicated that the generation of excess vacancies and unstructured region by preplastic deformation contributes to the enhanc... read less USED (high confidence) A. Hawken, G. Sun, and P. Harrowell, “Role of interfacial inherent structures in the fast crystal growth from molten salts and metals,” Physical Review Materials. 2019. link Times cited: 12 Abstract: Molecular dynamics simulations of the temperature dependent … read moreAbstract: Molecular dynamics simulations of the temperature dependent crystal growth rates of the salts, NaCl and ZnS, from their melts are reported, along with those of a number of pure metals. The growth rate of NaCl and the FCC-forming metals show little evidence of activated control, while that of ZnS and Fe, a BCC forming metal, exhibit activation barriers similar to those observed for diffusion in the melt. Unlike ZnS and Fe, the interfacial inherent structures of NaCl and Cu and Ag are found to be crystalline. We calculate the median displacement between the interfacial liquid and crystalline states and show that this distance is smaller than the cage length, demonstrating that crystal growth in the fast crystallizers can occur via local vibrations and so largely avoid the activated kinetics associated with the larger displacements associated with particle transport. read less USED (high confidence) J.-Y. Zhang and W.-Z. Zhang, “A general method to construct dislocations in atomistic simulations,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 10 Abstract: An important aspect of atomistic simulations of dislocations… read moreAbstract: An important aspect of atomistic simulations of dislocations is the construction of the initial dislocation configurations. However, limited configurations can be constructed by previous methods, impeding the simulations of a general dislocation configuration in real materials. In this paper, we develop a simple and general method for constructing dislocations with arbitrary shapes specified by the users, realising the Volterra process at the atomic level. Examples of its applications to a dislocation helix, the partial dislocations, the multi-dislocation configurations, and the dislocations in the imperfect crystal are presented, showing the capacity and robustness of the present method. read less USED (high confidence) A. Hasanzadeh, A. Hamedani, G. Alahyarizadeh, A. Minuchehr, and M. Aghaei, “The role of chromium and nickel on the thermal and mechanical properties of FeNiCr austenitic stainless steels under high pressure and temperature: a molecular dynamics study,” Molecular Simulation. 2019. link Times cited: 8 Abstract: ABSTRACT The effect of Cr and Ni content on thermo-mechanica… read moreAbstract: ABSTRACT The effect of Cr and Ni content on thermo-mechanical properties of FeNiCr austenitic stainless steel under ambient and high pressure and temperature were investigated by MD simulations. The FCC structure was selected as optimum structure for FeNiCr system based on obtained MD results from Bonny EAM potential and valid experimental results. The structural and mechanical properties of pure Fe, Ni, and Cr were also estimated based on this potential, indicating good agreement with experimental results. These properties were computed for four experimental case studies which showed less than 10% error. Moreover, the elastic constants of the Fe–(8–18)Ni–(18–25)Cr systems were estimated. Results showed that bulk modulus increases by increasing the Ni and Cr contents, which can be connected to the changes in bonding electrons. The thermal properties of FeNiCr were calculated in ambient and high pressure. Although thermo-mechanical properties confirm good agreement with experimental results at the ambient condition, however, they indicate that FeNiCr Bonny potential is not applicable at high pressure. In order to tackle this issue, a hybrid potential was used at high Pressure/Temperature. The results illustrate enhanced mechanical properties, increase of melting point and reduction of LTE in high pressure and deteriorated mechanical properties at high temperature. read less USED (high confidence) A. Tsukanov, A. Lozhkomoev, M. Lerner, I. Gotman, E. Gutmanas, and S. Psakhie, “Molecular dynamics study of bimetallic Fe–Cu Janus nanoparticles formation by electrical explosion of wires,” Philosophical Magazine. 2019. link Times cited: 3 Abstract: ABSTRACT Bimetallic nanoparticles comprised of two elements … read moreAbstract: ABSTRACT Bimetallic nanoparticles comprised of two elements which are immiscible in the bulk present a unique combination of physical–chemical properties that strongly depend on the atomic arrangement within the particle. In this study, molecular dynamics (MD) simulations of bimetallic Fe–Cu nanoparticles formation by high-velocity collision of individual metal nanoparticles (IMNPs) were performed. Physically these conditions model fast electrical explosion of two metal wires (Fe and Cu). By varying the size, temperature and velocity of colliding IMNPs, the conditions under which phase-segregated Janus nanoparticles are formed were determined. The model predictions showed good agreement with the experimental results. The present work is a step forward to understanding the formation mechanisms of bimetallic nanoparticles with different chemical configurations. read less USED (high confidence) W. Tucker, A. Dove, and P. Schelling, “Dissipation and plastic deformation in collisions between metallic nanoparticles,” Computational Materials Science. 2019. link Times cited: 3 USED (high confidence) C. Xu and W. Wang, “Simulation Study of Helium Effect on the Microstructure of Nanocrystalline Body-Centered Cubic Iron,” Materials. 2018. link Times cited: 8 Abstract: Helium (He) effect on the microstructure of nanocrystalline … read moreAbstract: Helium (He) effect on the microstructure of nanocrystalline body-centered cubic iron (BCC-Fe) was studied through Molecular Dynamics (MD) simulation and simulated X-ray Diffraction (XRD). The crack generation and the change of lattice constant were investigated under a uniaxial tensile strain at room temperature to explore the roles of He concentration and distribution played in the degradation of mechanical properties. The simulation results show that the expansion of the lattice constant decreases and the swelling rate increases while the He in the BCC region diffuses into the grain boundary (GB) region. The mechanical property of nanocrystalline BCC-Fe shows He concentration and distribution dependence, and the existence of He in GB is found to benefit the generation and growth of cracks and to affect the strength of GB during loading. It is observed that the reduction of tensile stress contributed by GB He is more obvious than that contributed by grain interior He. read less USED (high confidence) S. Paul, S. Kumar, and S. Tarafder, “Orientation-dependent crack-tip blunting and crack propagation in a single crystal BCC iron,” Bulletin of Materials Science. 2018. link Times cited: 3 USED (high confidence) A. Porras-Vazquez, L. Martinie, P. Vergne, and N. Fillot, “Independence between friction and velocity distribution in fluids subjected to severe shearing and confinement.,” Physical chemistry chemical physics : PCCP. 2018. link Times cited: 22 Abstract: Friction reduction is more than ever a key point in saving n… read moreAbstract: Friction reduction is more than ever a key point in saving natural resources and energy, and the question of how to achieve this concerns first and foremost every lubricated system. Among the observed phenomena related to lubricated friction, limiting shear stress (LSS) appears to be one of the most challenging to explain, since its origin is still uncertain. Various scenarios have been proposed involving a transition to a glassy state under high pressure, shear banding, shear localization, or even the occurrence of slip at the solid-liquid interface, none of which have proven conclusive. This work provides new insights into the mechanisms leading to LSS and the underlying flow organization. It bridges a gap between the scenarios previously discussed in the literature to explain the mechanisms behind LSS and friction experiments, which provide macroscopic results only. We first present some general molecular dynamics (MD) simulations developed to characterize molecular fluids in their bulk state and then proceed to study their response under severe shearing and confinement. Results from simulations are compared to experimental data derived from friction tests. A further analysis of the pressure and temperature involved hints that LSS and the physical state of the lubricant are strongly interconnected concepts. Additionally, the friction results were uncorrelated to the choice of surfaces, contrary to velocity distribution. read less USED (high confidence) J. J. Moller et al., “110
planar faults in strained bcc metals: Origins and implications of a commonly observed artifact of classical potentials,” Physical Review Materials. 2018. link Times cited: 18 Abstract: Large-scale atomistic simulations with classical potentials … read moreAbstract: Large-scale atomistic simulations with classical potentials can provide valuable insights into microscopic deformation mechanisms and defect-defect interactions in materials. Unfortunately, these assets often come with the uncertainty of whether the observed mechanisms are based on realistic physical phenomena or whether they are artifacts of the employed material models. One such example is the often reported occurrence of stable planar faults (PFs) in body-centered cubic (bcc) metals subjected to high strains, e.g., at crack tips or in strained nano-objects. In this paper, we study the strain dependence of the generalized stacking fault energy (GSFE) of {110} planes in various bcc metals with material models of increasing sophistication, i.e., (modified) embedded atom method, angular-dependent, Tersoff, and bond-order potentials as well as density functional theory. We show that under applied tensile strains the GSFE curves of many classical potentials exhibit a local minimum which gives rise to the formation of stable PFs. These PFs do not appear when more sophisticated material models are used and have thus to be regarded as artifacts of the potentials. We demonstrate that the local GSFE minimum is not formed for reasons of symmetry and we recommend including the determination of the strain-dependent (110) GSFE as a benchmark for newly developed potentials. read less USED (high confidence) A. Herron, S. Coleman, K. Dang, D. Spearot, and E. Homer, “Simulation of kinematic Kikuchi diffraction patterns from atomistic structures,” MethodsX. 2018. link Times cited: 3 USED (high confidence) S. Stephan, M. P. Lautenschlaeger, I. A. Alhafez, M. Horsch, H. Urbassek, and H. Hasse, “Molecular Dynamics Simulation Study of Mechanical Effects of Lubrication on a Nanoscale Contact Process,” Tribology Letters. 2018. link Times cited: 22 USED (high confidence) M. H. Hamza, M. A. Hendy, T. Hatem, and J. El-Awady, “Impact of angular deviation from coincidence site lattice grain boundaries on hydrogen segregation and diffusion in α-iron,” MRS Communications. 2018. link Times cited: 5 Abstract: Coincidence site lattice (CSL) grain boundaries (GBs) are be… read moreAbstract: Coincidence site lattice (CSL) grain boundaries (GBs) are believed to be low-energy, resistant to intergranular fracture, as well as to hydrogen embrittlement. Nevertheless, the behavior of CSL-GBs are generally confused with their angular deviations. In the current study, the effect of angular deviation from the perfect ∑3(111)[110] GBs in α-iron on the hydrogen diffusion and the susceptibility of the GB to hydrogen embrittlement is investigated through molecular static and dynamics simulations. By utilizing Rice-Wang model, it is shown that the ideal GB shows the highest resistance to decohesion below the hydrogen saturation limit. Finally, the hydrogen diff usivity along the ideal GB is observed to be the highest. read less USED (high confidence) M. Fellinger, A. M. Tan, L. Hector, and D. Trinkle, “Geometries of edge and mixed dislocations in bcc Fe from first-principles calculations,” Physical Review Materials. 2018. link Times cited: 21 Abstract: We use DFT to compute core structures of $a_0[100](010)$ edg… read moreAbstract: We use DFT to compute core structures of $a_0[100](010)$ edge, $a_0[100](011)$ edge, $a_0/2[\bar{1}\bar{1}1](1\bar{1}0)$ edge, and $a_0/2[111](1\bar{1}0)$ $71^{\circ}$ mixed dislocations in bcc Fe. The calculations use flexible boundary conditions (FBC), which allow dislocations to relax as isolated defects by coupling the core to an infinite harmonic lattice through the lattice Green function (LGF). We use LGFs of dislocated geometries in contrast to previous FBC-based dislocation calculations that use the bulk crystal LGF. Dislocation LGFs account for changes in topology in the core as well as strain throughout the lattice. A bulk-like approximation for the force constants in a dislocated geometry leads to LGFs that optimize the cores of the $a_0[100](010)$ edge, $a_0[100](011)$ edge, and $a_0/2[111](1\bar{1}0)$ $71^{\circ}$ mixed dislocations. This approximation fails for the $a_0/2[\bar{1}\bar{1}1](1\bar{1}0)$ dislocation, so here we derive the LGF using accurate force constants from a Gaussian approximation potential. The standard deviations of dislocation Nye tensor distributions quantify the widths of the cores. The relaxed cores are compact, and the magnetic moments on the Fe atoms closely follow the volumetric strain distributions in the cores. We also compute the core structures of these dislocations using eight different classical interatomic potentials, and quantify symmetry differences between the cores using the Fourier coefficients of their Nye tensor distributions. Most of the core structures computed using the classical potentials agree well with DFT results. The DFT geometries provide benchmarking for classical potential studies of work-hardening, as well as substitutional and interstitial sites for computing solute-dislocation interactions that serve as inputs for mesoscale models of solute strengthening and solute diffusion near dislocations. read less USED (high confidence) W. Setyawan, C. Henager, and S. Hu, “Nonlinear ultrasonic response of voids and Cu precipitates in body-centered cubic Fe,” Journal of Applied Physics. 2018. link Times cited: 9 Abstract: Interpreting nonlinear ultrasonic signals detected in a nond… read moreAbstract: Interpreting nonlinear ultrasonic signals detected in a nondestructive evaluation of radiation damage requires the knowledge of the correlation between defects and nonlinearity. In this work, molecular dynamics simulations are performed to study the effect of distributed vacancies, voids, Cu atoms, and Cu precipitates on the nonlinear ultrasonic response in body-centered cubic (bcc) Fe. The nonlinearity parameter calculated from the second harmonic amplitude in the perfect lattice is 2.73. Vacancies are found to increase the nonlinearity. However, clusters of vacancies in the form of spherical voids show an opposite effect. This finding can be used to conveniently distinguish vacancies from voids in the material. Unlike vacancies, individual Cu atoms decrease the nonlinearity. Clustering of Cu atoms into Cu precipitates further decreases the nonlinearity. Interestingly, precipitates with a diameter of 2 nm and larger exhibit a similar effect despite their different structure and coherency with the Fe matrix.Interpreting nonlinear ultrasonic signals detected in a nondestructive evaluation of radiation damage requires the knowledge of the correlation between defects and nonlinearity. In this work, molecular dynamics simulations are performed to study the effect of distributed vacancies, voids, Cu atoms, and Cu precipitates on the nonlinear ultrasonic response in body-centered cubic (bcc) Fe. The nonlinearity parameter calculated from the second harmonic amplitude in the perfect lattice is 2.73. Vacancies are found to increase the nonlinearity. However, clusters of vacancies in the form of spherical voids show an opposite effect. This finding can be used to conveniently distinguish vacancies from voids in the material. Unlike vacancies, individual Cu atoms decrease the nonlinearity. Clustering of Cu atoms into Cu precipitates further decreases the nonlinearity. Interestingly, precipitates with a diameter of 2 nm and larger exhibit a similar effect despite their different structure and coherency with the Fe matrix. read less USED (high confidence) S. Hayakawa, T. Okita, M. Itakura, M. Aichi, and K. Suzuki, “Interactions between clusters of self-interstitial atoms via a conservative climb in BCC–Fe,” Philosophical Magazine. 2018. link Times cited: 8 Abstract: ABSTRACT We conduct kinetic Monte Carlo simulations for the … read moreAbstract: ABSTRACT We conduct kinetic Monte Carlo simulations for the conservative climb motion of a cluster of self-interstitial atoms (SIAs) towards another SIA cluster in BCC–Fe; the conservative climb velocity is inversely proportional to the fourth power of the distance between them, as per the prediction based on Einstein’s equation. The size of the climbing cluster significantly affects its conservative climb velocity, while the size of the cluster that originates the stress field does not. The activation energy for the conservative climb is considerably greater than that derived in previous studies and strongly dependent on the climbing cluster size. The results presented in this study are the atomistic evaluation of the behaviour of SIA clusters through three-dimensional motion, which cannot be achieved using molecular dynamics techniques alone. read less USED (high confidence) M. H. Nazir, Z. Khan, A. Saeed, A. Siddaiah, and P. Menezes, “Synergistic wear-corrosion analysis and modelling of nanocomposite coatings,” Tribology International. 2018. link Times cited: 31 USED (high confidence) M. Guerdane and M. Berghoff, “Crystal-melt interface mobility in bcc Fe: Linking molecular dynamics to phase-field and phase-field crystal modeling,” Physical Review B. 2018. link Times cited: 13 Abstract: By combining molecular dynamics (MD) simulations with phase-… read moreAbstract: By combining molecular dynamics (MD) simulations with phase-field (PF) and phase-field crystal (PFC) modeling we study collision-controlled growth kinetics from the melt for pure Fe. The MD/PF comparison shows, on the one hand, that the PF model can be properly designed to reproduce quantitatively different aspects of the growth kinetics and anisotropy of planar and curved solid-liquid interfaces. On the other hand, this comparison demonstrates the ability of classical MD simulations to predict morphology and dynamics of moving curved interfaces up to a length scale of about 0.15 μm. After mapping the MD model to the PF one, the latter permits to analyze the separate contribution of different anisotropies to the interface morphology. The MD/PFC agreement regarding the growth anisotropy and morphology extends the trend already observed for the here used PFC model in describing structural and elastic properties of bcc Fe. read less USED (high confidence) M. Banisalman and T. Oda, “Iron Interstitial Defects Stability: Under the Uniaxial Stress Effect,” Cumhuriyet Science Journal. 2018. link Times cited: 0 Abstract: Stres alanlarindaki kusur kinetiginin anlasilmasi, nukleer m… read moreAbstract: Stres alanlarindaki kusur kinetiginin anlasilmasi, nukleer maddelerin bozulmasinin cok boyutlu modellenmesi icin onemlidir. Molekuler dinamik (MD) simulasyonu ile, formasyon ve goc enerjisi enerjileri, alfa Fe'de kendiliginden olusan atom (SIA) ve SIA kumeleri (1 ~ 3 gecis reklamlari) icin degerlendirilmistir. % 0 ~ 3 tek eksenli surdurulebilir [100] gerilme etkileri SIAs ve halter konfigurasyonlari icin test edilmistir. Kararlilik ile ilgili olarak, halter konfigurasyonlari daha buyuk suslarda ve daha buyuk kumelerde daha kararli hale gelir. Hareketlilik icin, surdurulebilir gerilmeler altinda tek SIA kusurlarinin difuzyonu izlenmistir. Serbest gerilme kosullarinda, SIA kumelerinin difuzivitesi, doymus gerilmede uc boyutlu (3D) ile bir boyutlu (1D) asamali bir gecise sahiptir. 3D gecis kucuk kumeler ve alt gerilmeler icin iken ve buyuk olcude SIA hizalama konfigurasyonu icin sunulurken, 1D gecisi buyuk kumeler ve buyuk gerginlik icin gozlenmistir. Cekme gerilmesi altinda ve kucuk kumeler icin, difuzyon artirimi daha yuksek bir sicaklikta daha buyuktur. Bununla birlikte, sicaklik etkisi daha buyuk kumeler icin kucuktur. Gerinim alanlarinin bu etkileri, kusurlar ve uygulanan stres alanlari arasindaki elastik etkilesim ile aciklanabilir. read less USED (high confidence) X. Liu, X. Wen, and R. Hoffmann, “Surface Activation of Transition Metal Nanoparticles for Heterogeneous Catalysis: What We Can Learn from Molecular Dynamics,” ACS Catalysis. 2018. link Times cited: 43 Abstract: Many heterogeneous reactions catalyzed by nanoparticles occu… read moreAbstract: Many heterogeneous reactions catalyzed by nanoparticles occur at relatively high temperatures, which may modulate the surface morphology of nanoparticles during reaction. Inspired by the discovery of dynamic formation of active sites on gold nanoparticles, we explore theoretically the nature of the highly mobile atoms on the surface of nanoparticles of various sizes for 11 transition metals. Using molecular dynamics simulations, on a 3 nm Fe nanoparticle as an example, the effect of surface premelting and overall melting on the structure and physical properties of the nanoparticles is analyzed. When the nanoparticle is heated up, the atoms in the outer shell appear amorphous already at 900 K. Surface premelting is reached at 1050 K, with more than three liquid atoms, based on the Lindemann criterion. The activated atoms may transfer their extra kinetic energy to the rest of the nanoparticle and activate other atoms. The dynamic studies indicate that the number of highly mobile atoms on the surface increas... read less USED (high confidence) S. Liu et al., “Refinement effect of TiC on ferrite by molecular statics/dynamics simulations and first-principles calculations,” Journal of Alloys and Compounds. 2018. link Times cited: 3 USED (high confidence) Y. Zhang, D. Schwen, and X. Bai, “Molecular dynamics simulations of concentration-dependent defect production in Fe-Cr and Fe-Cu alloys,” Journal of Applied Physics. 2017. link Times cited: 13 Abstract: Molecular dynamics simulations are conducted to study the ef… read moreAbstract: Molecular dynamics simulations are conducted to study the effects of alloying elements on the primary damage behaviors in three Fe-based ferritic alloy systems: (1) a Fe-Cr system in which the heat of mixing changes its sign with the Cr concentration; (2) a Fe-Cu system that has a positive heat of mixing; and (3) an ideal but artificial Fe-Cr system that has a zero heat of mixing, which is used as a reference system to investigate solute interstitial formation based on probability. It is found that in these alloys, the solute type and concentration do not have a significant effect on the total number of surviving Frenkel pairs. However, the fraction of solute interstitials has distinct behaviors. In Fe-Cr, the Cr interstitial fraction is much higher than the Cr solute concentration and the Cr interstitial production efficiency decreases with the increasing Cr concentration. By contrast, in Fe-Cu, Cu interstitials are barely produced. In the ideal alloy, the solute interstitial fraction is close to the sol... read less USED (high confidence) L. Scherthan et al., “Effects of mechanical loading on the magnetic and dynamic properties of engineering materials,” Hyperfine Interactions. 2017. link Times cited: 2 USED (high confidence) F. Granberg, J. Byggmästar, A. Sand, and K. Nordlund, “Cascade debris overlap mechanism of 〈100〉 dislocation loop formation in Fe and FeCr,” Europhysics Letters. 2017. link Times cited: 38 Abstract: Two types of dislocation loops are observed in irradiated α-… read moreAbstract: Two types of dislocation loops are observed in irradiated α-Fe, the 1/2〈111〉 loop and the 〈100〉 loop. Atomistic simulations consistently predict that only the energetically more favourable 1/2〈111〉 loops are formed directly in cascades, leaving the formation mechanism of 〈100〉 loops an unsolved question. We show how 〈100〉 loops can be formed when cascades overlap with random pre-existing primary radiation damage in Fe and FeCr. This indicates that there are no specific constraints involved in the formation of 〈100〉 loops, and can explain their common occurrence. read less USED (high confidence) L. Scherthan et al., “Effects of mechanical loading on the magnetic and dynamic properties of engineering materials,” Hyperfine Interactions. 2017. link Times cited: 0 USED (high confidence) N. Gao, J. Chen, R. Kurtz, Z. Wang, R. F. Zhang, and F. Gao, “New understanding of nano-scale interstitial dislocation loops in BCC iron,” Journal of Physics: Condensed Matter. 2017. link Times cited: 21 Abstract: Complex states of nanoscale interstitial dislocation loop ca… read moreAbstract: Complex states of nanoscale interstitial dislocation loop can be described by its habit plane and Burgers vector. Using atomistic simulations, we provide direct evidences on the change of the habit plane of a 1/2〈1 1 1〉 loop from {1 1 1} to {1 1 0} and {2 1 1}, in agreement with TEM observations. A new {1 0 0} habit plane of this loop is also predicted by simulations. The non-conservation of the Burgers vector is approved theoretically for: (1) dislocation reactions between loops with different Burgers vectors and (2) the transition between 〈1 0 0〉 loops and 1/2〈1 1 1〉 loops. The rotation from a 1/2〈1 1 1〉 to a 〈1 0 0〉 loop has also been explored, which occurs at 570 K for time on the order of 10 s. The dislocation-precipitate phase duality and change of habit plane are then proposed as new features for nano-scale dislocation loops. read less USED (high confidence) B. Cheng and M. Ceriotti, “Computing the absolute Gibbs free energy in atomistic simulations: Applications to defects in solids,” Physical Review B. 2017. link Times cited: 40 Abstract: The Gibbs free energy is the fundamental thermodynamic poten… read moreAbstract: The Gibbs free energy is the fundamental thermodynamic potential underlying the relative stability of different states of matter under constant-pressure conditions. However, computing this quantity from atomic-scale simulations is far from trivial. As a consequence, all too often the potential energy of the system is used as a proxy, overlooking entropic and anharmonic effects. Here we discuss a combination of different thermodynamic integration routes to obtain the absolute Gibbs free energy of a solid system starting from a harmonic reference state. This approach enables the direct comparison between the free energy of different structures, circumventing the need to sample the transition paths between them. We showcase this thermodynamic integration scheme by computing the Gibbs free energy associated with a vacancy in BCC iron, and the intrinsic stacking fault free energy of nickel. These examples highlight the pitfalls of estimating the free energy of crystallographic defects only using the minimum potential energy, which overestimates the vacancy free energy by 60% and the stacking-fault energy by almost 300% at temperatures close to the melting point. read less USED (high confidence) Z. Zheng, E. Li, N. Ding, and X. Xu, “Beat phenomenon in metal nanowires: A molecular dynamics study,” Computational Materials Science. 2017. link Times cited: 6 USED (high confidence) H. Winkelmann, H. Rojacz, S. Eder, M. Varga, and S. Nugent, “Influence of Momentum and Energy on Materials: An Experimental and Molecular Dynamics Approach for Impact Phenomena,” steel research international. 2017. link Times cited: 1 Abstract: Single‐impact tests and molecular dynamics (MD) simulations … read moreAbstract: Single‐impact tests and molecular dynamics (MD) simulations are performed to evaluate effects at energy‐ and momentum‐variable impact phenomena at two distinct scales and velocity ranges. Therefore, a carbon steel in various heat treatment conditions is examined using the single impact test to evaluate the influence of varying energies and momenta on the deformation behavior. Experimentally found material parameters “momentum‐sensitivity” ps(E) and “minimum deformation momentum” p0(E) are introduced for a better mathematical description of impact phenomena or deformation processes using energy and momentum. Empirical laws are found, where the minimum deformation momentum is a linear function of the impact energy (E) and the deformation at low momenta is an inverse function of E, which has significant influence on the deformation. The proposed empirical law are recalculated via down‐scaled molecular dynamics simulation (rigid indenter impacting on an iron block) and is found applicable for macro and nano scale impact phenomena. read less USED (high confidence) X.-yan Li et al., “Annihilating vacancies via dynamic reflection and emission of interstitials in nano-crystal tungsten,” Nuclear Fusion. 2017. link Times cited: 15 Abstract: Radiation damage not only seriously degrades the mechanical … read moreAbstract: Radiation damage not only seriously degrades the mechanical properties of tungsten (W) but also enhances hydrogen retention in the material. Introducing a large amount of defect sinks, e.g. grain boundaries (GBs) is an effective method for improving radiation-resistance of W. However, the mechanism by which the vacancies are dynamically annihilated at long timescale in nano-crystal W is still not clear. The dynamic picture for eliminating vacancies with single interstitials and small interstitial-clusters has been investigated by combining molecular dynamics, molecular statics and object Kinetic Monte Carlo methods. On one hand, the annihilation of bulk vacancies was enhanced due to the reflection of an interstitial-cluster of parallel 〈111〉 crowdions by the GB. The interstitial-cluster was observed to be reflected back into the grain interior when approaching a locally dense GB region. Near this region, the energy landscape for the interstitial was featured by a shoulder, different to the decreasing energy landscape of the interstitial near a locally loose region as indicative of the sink role of the GB. The bulk vacancy on the reflection path was annihilated. On the other hand, the dynamic interstitial emission efficiently anneals bulk vacancies. The single interstitial trapped at the GB firstly moved along the GB quickly and clustered to be the di-interstitial therein, reducing its mobility to a value comparable to that that for bulk vacancy diffusion. Then, the bulk vacancy was recombined via the coupled motion of the di-interstitial along the GB, the diffusion of the vacancy towards the GB and the accompanying interstitial emission. These results suggest that GBs play an efficient role in improving radiation-tolerance of nano-crystal W via reflecting highly-mobile interstitials and interstitial-clusters into the bulk and annihilating bulk vacancies, and via complex coupling of in-boundary interstitial diffusion, clustering of the interstitial and vacancy diffusion in the bulk. read less USED (high confidence) V. Mazhukin, A. V. Shapranov, V. E. Perezhigin, O. Koroleva, and A. Mazhukin, “Kinetic melting and crystallization stages of strongly superheated and supercooled metals,” Mathematical Models and Computer Simulations. 2017. link Times cited: 17 USED (high confidence) Z. Hu and S. Mahadevan, “Uncertainty quantification and management in additive manufacturing: current status, needs, and opportunities,” The International Journal of Advanced Manufacturing Technology. 2017. link Times cited: 103 USED (high confidence) A. Fedorov, A. V. Shul’gin, and S. Lavruk, “Investigation of the physical properties of iron nanoparticles in the course of the melting and solidification,” Physics of Metals and Metallography. 2017. link Times cited: 6 USED (high confidence) V. Mazhukin, A. V. Shapranov, V. E. Perezhigin, O. Koroleva, and A. Mazhukin, “Kinetic melting and crystallization stages of strongly superheated and supercooled metals,” Mathematical Models and Computer Simulations. 2017. link Times cited: 0 USED (high confidence) A. Al-Motasem, A. Al-Motasem, J. Bergström, A. Gåård, P. Krakhmalev, and L. J. Holleboom, “Atomistic Insights on the Wear/Friction Behavior of Nanocrystalline Ferrite During Nanoscratching as Revealed by Molecular Dynamics,” Tribology Letters. 2017. link Times cited: 48 USED (high confidence) Z. Zheng, E. Li, N. Ding, and X. Xu, “A Molecular Dynamic Study on Nonlinear Vibration Behaviors of Fe Nanowires,” International Journal of Computational Methods. 2017. link Times cited: 1 Abstract: In this paper, vibration behaviors of Fe nanowires are inves… read moreAbstract: In this paper, vibration behaviors of Fe nanowires are investigated by using the large-scale molecular dynamics (MD) simulations. It is observed that the vibration frequency of nanowires rises slightly and nonlinearly with the increase of initial actuation amplitude. Based on the atomic arrangement, a discrete spring-mass model is developed. Its nonlinear elastic relation is used to explain this phenomenon. In addition, Fe nanowires with different lengths and heights show different vibration properties in this work. The ratio between the length ([Formula: see text]) and the height ([Formula: see text]) of nanowires has a significant influence on vibration behaviors. The vibration properties of nanowires can be explained by the Euler–Bernoulli model when the ratio is relatively large, while they can be illustrated by the Timoshenko model when the ratio is relatively small. read less USED (high confidence) N. Anento, A. Serra, and Y. Osetsky, “Effect of nickel on point defects diffusion in Fe – Ni alloys,” Acta Materialia. 2017. link Times cited: 30 USED (high confidence) L. Li and M. Han, “Molecular dynamics simulations on tensile behaviors of single-crystal bcc Fe nanowire: effects of strain rates and thermal environment,” Applied Physics A. 2017. link Times cited: 21 USED (high confidence) L. Li and M. Han, “Molecular dynamics simulations on tensile behaviors of single-crystal bcc Fe nanowire: effects of strain rates and thermal environment,” Applied Physics A. 2017. link Times cited: 2 USED (high confidence) A. Korchuganov, K. Zolnikov, D. Kryzhevich, and S. Psakhie, “Primary Ion-Irradiation Damage of BCC-Iron Surfaces,” Russian Physics Journal. 2017. link Times cited: 31 USED (high confidence) S. Eder, U. Cihak-Bayr, D. Bianchi, G. Feldbauer, and G. Betz, “Thermostat Influence on the Structural Development and Material Removal during Abrasion of Nanocrystalline Ferrite.,” ACS applied materials & interfaces. 2017. link Times cited: 30 Abstract: We consider a nanomachining process of hard, abrasive partic… read moreAbstract: We consider a nanomachining process of hard, abrasive particles grinding on the rough surface of a polycrystalline ferritic work piece. Using extensive large-scale molecular dynamics (MD) simulations, we show that the mode of thermostating, i.e., the way that the heat generated through deformation and friction is removed from the system, has crucial impact on tribological and materials related phenomena. By adopting an electron-phonon coupling approach to parametrize the thermostat of the system, thus including the electronic contribution to the thermal conductivity of iron, we can reproduce the experimentally measured values that yield realistic temperature gradients in the work piece. We compare these results to those obtained by assuming the two extreme cases of only phononic heat conduction and instantaneous removal of the heat generated in the machining interface. Our discussion of the differences between these three cases reveals that although the average shear stress is virtually temperature independent up to a normal pressure of approximately 1 GPa, the grain and chip morphology as well as most relevant quantities depend heavily on the mode of thermostating beyond a normal pressure of 0.4 GPa. These pronounced differences can be explained by the thermally activated processes that guide the reaction of the Fe lattice to the external mechanical and thermal loads caused by nanomachining. read less USED (high confidence) C. Kang, Q. Wang, and L. Shao, “Kinetics of interstitial defects in α-Fe: The effect from uniaxial stress,” Journal of Nuclear Materials. 2017. link Times cited: 15 USED (high confidence) R. Chinnappan and B. Panigrahi, “Theoretical study of high-pressure phase stability of NaZr2(PO4)3 via elastic constants and equation of state,” Indian Journal of Physics. 2017. link Times cited: 1 USED (high confidence) P. Scardi, L. Rebuffi, M. Abdellatief, A. Flor, and A. Leonardi, “Debye–Waller coefficient of heavily deformed nanocrystalline iron1,” Journal of Applied Crystallography. 2017. link Times cited: 17 Abstract: Extensive deformation of an iron alloy powder increases the … read moreAbstract: Extensive deformation of an iron alloy powder increases the static disorder contribution to the thermal factor, with an increase of ∼20% in the Debye–Waller coefficient observed by both X-ray diffraction and extended X-ray absorption fine structure. Molecular dynamics simulations shed light on the underlying mechanisms, confirming the major role played by the grain boundary. read less USED (high confidence) J. Varillas, J. Očenášek, J. Torner, and J. Alcalá, “Unraveling deformation mechanisms around FCC and BCC nanocontacts through slip trace and pileup topography analyses,” Acta Materialia. 2017. link Times cited: 24 USED (high confidence) N. Gunkelmann, I. A. Alhafez, D. Steinberger, H. Urbassek, and S. Sandfeld, “Nanoscratching of iron: A novel approach to characterize dislocation microstructures,” Computational Materials Science. 2017. link Times cited: 16 USED (high confidence) J. Wang, J. Bian, X. Niu, and G. Wang, “A universal method to calculate the surface energy density of spherical surfaces in crystals,” Acta Mechanica Sinica. 2017. link Times cited: 6 USED (high confidence) J. Skogsrud, M. Jørum, and C. Thaulow, “Nanomechanical modeling of a (100)[001] crack in a single crystal bcc iron cantilever beam,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 0 Abstract: An atomistic model of a fully 3D, nano-sized, pre-cracked ca… read moreAbstract: An atomistic model of a fully 3D, nano-sized, pre-cracked cantilever beam has been made and MD simulations have been performed to deflect the beam and initiate crack growth. The crucial process zone in front of the crack has been investigated with respect to linear elastic and elastic-plastic fracture mechanics and plastic deformation mechanisms such as dislocations and twinning. The effect of crack geometry and loading rate has been studied. Two crack geometries were compared, one atomically sharp and one blunted. The sharper crack was shown to lead to a cleaner crack extension on (110)-planes, while the rounded crack showed extension along the initial (100)-plane in accordance with experiments on micro-sized 3 wt% Si α-Fe cantilevers. The effect of strain rate was also investigated, and it was found that lower strain rate correlated better with experimental observations. However, the strain rate used is still several magnitudes higher than for experiments, limiting the usefulness of strain rate observations for predicting behavior in experiments. A brief post-deformation comparison between simulations and SEM-images of focused ion beam-fabricated micro-cantilevers was also done, showing possible signs of similar deformation mechanisms and dislocation systems between them. read less USED (high confidence) G. Sainath and B. Choudhary, “Formation of pentagonal atomic chains in BCC Fe nanowires,” Materials Research Express. 2016. link Times cited: 4 Abstract: For the first time, we report the formation of pentagonal at… read moreAbstract: For the first time, we report the formation of pentagonal atomic chains during tensile deformation of ultra thin BCC Fe nanowires. Extensive molecular dynamics simulations have been performed on 〈100〉/{110} BCC Fe nanowires with different cross section width varying from 0.404 to 3.634 nm at temperatures ranging from 10 to 900 K. The results indicate that above certain temperature, long and stable pentagonal atomic chains form in BCC Fe nanowires with cross section width less than 2.83 nm. The temperature, above which the pentagonal chains form, increases with increase in nanowire size. The pentagonal chains have been observed to be highly stable over large plastic strains and contribute to high ductility in Fe nanowires. read less USED (high confidence) G. Sainath and B. Choudhary, “Influence of twist boundary on deformation behaviour of 〈1 0 0〉 BCC Fe nanowires,” Philosophical Magazine Letters. 2016. link Times cited: 10 Abstract: Molecular dynamics simulations revealed significant differen… read moreAbstract: Molecular dynamics simulations revealed significant difference in deformation behaviour of 〈1 0 0〉 BCC Fe nanowires with and without twist boundary. The plastic deformation in perfect 〈1 0 0〉 BCC Fe nanowire was dominated by twinning and reorientation to 〈1 1 0〉 followed by further deformation by slip mode. On the contrary, 〈1 0 0〉 BCC Fe nanowire with a twist boundary deformed by slip at low plastic strains followed by twinning at high strains and absence of full reorientation. The results suggest that the deformation in 〈1 0 0〉 BCC Fe nanowire by dislocation slip is preferred over twinning in the presence of initial dislocations or dislocation networks. The results also explain the absence of extensive twinning in bulk materials, which inherently contains large number of dislocations. read less USED (high confidence) J. Wang, J. Bian, X. Niu, and G. Wang, “A universal method to calculate the surface energy density of spherical surfaces in crystals,” Acta Mechanica Sinica. 2016. link Times cited: 0 USED (high confidence) S. Xu and Y.-Q. Su, “Nanovoid growth in BCC α-Fe: influences of initial void geometry,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 20 Abstract: The growth of voids has a great impact on the mechanical pro… read moreAbstract: The growth of voids has a great impact on the mechanical properties of ductile materials by altering their microstructures. Exploring the process of void growth at the nanoscale helps in understanding the dynamic fracture of metals. While some very recent studies looked into the effects of the initial geometry of an elliptic void on the plastic deformation of face-centered cubic metals, a systematic study of the initial void ellipticity and orientation angle in body-centered cubic (BCC) metals is still lacking. In this paper, large scale molecular dynamics simulations with millions of atoms are conducted, investigating the void growth process during tensile loading of metallic thin films in BCC α-Fe. Our simulations elucidate the intertwined influences on void growth of the initial ellipticity and initial orientation angle of the void. It is shown that these two geometric parameters play an important role in the stress–strain response, the nucleation and evolution of defects, as well as the void size/outline evolution in α-Fe thin films. Results suggest that, together with void size, different initial void geometries should be taken into account if a continuum model is to be applied to nanoscale damage progression. read less USED (high confidence) H. Xie 谢, T. Yu 于, W. Fang 方, F. Yin 殷, and D. Khan, “Strain-rate-induced bcc-to-hcp phase transformation of Fe nanowires,” Chinese Physics B. 2016. link Times cited: 3 Abstract: Using molecular dynamics simulation method, the plastic defo… read moreAbstract: Using molecular dynamics simulation method, the plastic deformation mechanism of Fe nanowires is studied by applying uniaxial tension along the [110] direction. The simulation result shows that the bcc-to-hcp martensitic phase transformation mechanism controls the plastic deformation of the nanowires at high strain rate or low temperature; however, the plastic deformation mechanism will transform into a dislocation nucleation mechanism at low strain rate and higher temperature. Furthermore, the underlying cause of why the bcc-to-hcp martensitic phase transition mechanism is related to high strain rate and low temperature is also carefully studied. Based on the present study, a strain rate-temperature plastic deformation map for Fe nanowires has been proposed. read less USED (high confidence) G. Sainath and B. Choudhary, “Deformation behaviour of body centered cubic iron nanopillars containing coherent twin boundaries,” Philosophical Magazine. 2016. link Times cited: 33 Abstract: Molecular dynamics simulations were performed to understand … read moreAbstract: Molecular dynamics simulations were performed to understand the role of twin boundaries on deformation behaviour of body-centred cubic (BCC) iron (Fe) nanopillars. The twin boundaries varying from 1 to 5 providing twin boundary spacing in the range 8.5–2.8 nm were introduced perpendicular to the loading direction. The simulation results indicated that the twin boundaries in BCC Fe play a contrasting role during deformation under tensile and compressive loadings. During tensile deformation, a large reduction in yield stress was observed in twinned nanopillars compared to perfect nanopillar. However, the yield stress exhibited only marginal variation with respect to twin boundary spacing. On the contrary, a decrease in yield stress with increase in twin boundary spacing was obtained during compressive deformation. This contrasting behaviour originates from difference in operating mechanisms during yielding and subsequent plastic deformation. It has been observed that the deformation under tensile loading was dominated mainly by twin growth mechanism. On the other hand, the deformation was dominated by nucleation and slip of full dislocations under compressive loading. The twin boundaries offer a strong repulsive force on full dislocations resulting in the yield stress dependence on twin boundary spacing. The occurrence of twin–twin interaction during tensile deformation and dislocation–twin interaction during compressive deformation has been discussed. read less USED (high confidence) A. M. Tan and D. Trinkle, “Computation of the lattice Green function for a dislocation.,” Physical review. E. 2016. link Times cited: 13 Abstract: Modeling isolated dislocations is challenging due to their l… read moreAbstract: Modeling isolated dislocations is challenging due to their long-ranged strain fields. Flexible boundary condition methods capture the correct long-range strain field of a defect by coupling the defect core to an infinite harmonic bulk through the lattice Green function (LGF). To improve the accuracy and efficiency of flexible boundary condition methods, we develop a numerical method to compute the LGF specifically for a dislocation geometry; in contrast to previous methods, where the LGF was computed for the perfect bulk as an approximation for the dislocation. Our approach directly accounts for the topology of a dislocation, and the errors in the LGF computation converge rapidly for edge dislocations in a simple cubic model system as well as in BCC Fe with an empirical potential. When used within the flexible boundary condition approach, the dislocation LGF relaxes dislocation core geometries in fewer iterations than when the perfect bulk LGF is used as an approximation for the dislocation, making a flexible boundary condition approach more efficient. read less USED (high confidence) J. Ewen, C. Gattinoni, F. Thakkar, N. Morgan, H. Spikes, and D. Dini, “Nonequilibrium Molecular Dynamics Investigation of the Reduction in Friction and Wear by Carbon Nanoparticles Between Iron Surfaces,” Tribology Letters. 2016. link Times cited: 46 USED (high confidence) X. Ou, J. Sietsma, and M. Santofimia, “Molecular dynamics simulations of the mechanisms controlling the propagation of bcc/fcc semi-coherent interfaces in iron,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 24 Abstract: Molecular dynamics simulations have been used to study the e… read moreAbstract: Molecular dynamics simulations have been used to study the effects of different orientation relationships between fcc and bcc phases on the bcc/fcc interfacial propagation in pure iron systems at 300 K. Three semi-coherent bcc/fcc interfaces have been investigated. In all the cases, results show that growth of the bcc phase starts in the areas of low potential energy and progresses into the areas of high potential energy at the original bcc/fcc interfaces. The phase transformation in areas of low potential energy is of a martensitic nature while that in the high potential energy areas involves occasional diffusional jumps of atoms. read less USED (high confidence) L. Dezerald, D. Rodney, E. Clouet, L. Ventelon, and F. Willaime, “Plastic anisotropy and dislocation trajectory in BCC metals,” Nature Communications. 2016. link Times cited: 122 USED (high confidence) J. Bean and K. McKenna, “Origin of differences in the excess volume of copper and nickel grain boundaries,” Acta Materialia. 2016. link Times cited: 61 USED (high confidence) D. Belashchenko, “Universal Algorithm for Reconstruction of Atomic Models of Noncrystalline Systems,” Russian Journal of Physical Chemistry A. 2016. link Times cited: 1 USED (high confidence) B. Beeler, M. Asta, P. Hosemann, and N. Grønbech-Jensen, “Effect of strain and temperature on the threshold displacement energy in body-centered cubic iron,” Journal of Nuclear Materials. 2016. link Times cited: 29 USED (high confidence) J. J. Möller and E. Bitzek, “BDA: A novel method for identifying defects in body-centered cubic crystals,” MethodsX. 2016. link Times cited: 17 USED (high confidence) C. Qiao et al., “Molecular dynamics simulation studies on the plastic behaviors of an iron nanowire under torsion,” RSC Advances. 2016. link Times cited: 9 Abstract: The plastic deformation mechanism of iron (Fe) nanowires und… read moreAbstract: The plastic deformation mechanism of iron (Fe) nanowires under torsion is studied using the molecular dynamics (MD) method by applying an external driving force at a constant torsion speed. We find that the deformation behavior depends on the orientation of the wire. The dislocations in 〈100〉 and 〈111〉 oriented nanowires propagate through the nanowires under torsion, whereas those in 〈110〉 oriented nanowires divide the wire into two parts. The situation that there is a low angle twist grain boundary (GB) in the nanowires is also under consideration. The results reveal that the dislocations are concentrated on the GB in the initial state, presenting different patterns of dislocation network. The networks change depending on the twist direction. They shrink with increase in twist angle but expand with the decreasing twist angle, presenting an asymmetric phenomenon. Our findings can help us more thoroughly understand the plastic deformation mechanism of Fe nanowires under torsion. read less USED (high confidence) Z. Shi and C. V. Singh, “Competing twinning mechanisms in body-centered cubic metallic nanowires,” Scripta Materialia. 2016. link Times cited: 39 USED (high confidence) G. Sainath and B. Choudhary, “Directional Anisotropy of Crack Propagation Along Σ3 Grain Boundary in BCC Fe,” Transactions of the Indian Institute of Metals. 2016. link Times cited: 3 USED (high confidence) S. Parviainen, F. Djurabekova, S. Fitzgerald, A. Ruzibaev, and K. Nordlund, “Atomistic simulations of field assisted evaporation in atom probe tomography,” Journal of Physics D: Applied Physics. 2016. link Times cited: 19 Abstract: Atom probe tomography (APT) is an extremely powerful techniq… read moreAbstract: Atom probe tomography (APT) is an extremely powerful technique for determining the three-dimensional structure and chemical composition of a given sample. Although it is designed to provide images of material structure with atomic scale resolution, reconstruction artifacts, well-known to be present in reconstructed images, reduce their accuracy. No existing simulation technique has been able to describe the origin of these artifacts. Here we develop a simulation technique which allows for atomistic simulations of the atom emission process in the presence of high electric fields in APT experiments. Our code combines hybrid concurrent electrodynamics—molecular dynamics and a Monte Carlo approach. We use this technique to demonstrate the atom-level origin of artifacts in APT image reconstructions on examples of inclusions and voids in investigated samples. The results show that even small variations in the surface topology give rise to distortions in the local electric field, limiting the accuracy of conventional APT reconstruction algorithms. read less USED (high confidence) J. G. Sevillano, I. Aldazabal, A. Luque, and J. Aldazabal, “Atomistic simulation of the elongation response of a <011> oriented columnar nano-grain bcc Fe polycrystalline sample,” Meccanica. 2016. link Times cited: 6 USED (high confidence) Y. Yang, S. Li, X. Ding, J. Sun, and E. Salje, “Interface Driven Pseudo‐Elasticity in a‐Fe Nanowires,” Advanced Functional Materials. 2016. link Times cited: 20 Abstract: Molecular dynamics simulations of bent [100] α‐Fe nanowires … read moreAbstract: Molecular dynamics simulations of bent [100] α‐Fe nanowires show the nucleation of twins and nanoscale interfaces that lead to pseudo‐elasticity during loading/unloading cycles. The new type of interfaces along {110} stems from the accumulation of individual <111>/{112} twin boundaries and stores high interfacial energies. These nonconventional interfaces provide a large part of the driving force for shape recovery upon unloading, while the minimization of surface energy is no longer the dominant driving force. This new pseudo‐elastic effect is not much affected by surface roughness, and can be extended over a wide range of wire diameters, if the sample is seeded with conventional twin boundaries, which will transform to the desired {110} interfaces under bending. read less USED (high confidence) S. Pan, S. Feng, J. Qiao, B. Dong, and J. Qin, “The shells of atomic structure in metallic glasses,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 3 Abstract: We proposed a scheme to describe the spatial correlation bet… read moreAbstract: We proposed a scheme to describe the spatial correlation between two atoms in metallic glasses. Pair distribution function in a model iron was fully decomposed into several shells and can be presented as the spread of nearest neighbor correlation via distance. Moreover, angle distribution function can also be decomposed into groups. We demonstrate that there is close correlation between pair distribution function and angle distribution function for metallic glasses. We think that our results are very helpful understanding the atomic structure of metallic glasses. read less USED (high confidence) J. Han, C. Wang, X. Liu, Y. Wang, Z.-kui Liu, and J.-Z. Jiang, “Atomic-Level Mechanisms of Nucleation of Pure Liquid Metals during Rapid Cooling.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2015. link Times cited: 13 Abstract: To obtain a material with the desired performance, the atomi… read moreAbstract: To obtain a material with the desired performance, the atomic-level mechanisms of nucleation from the liquid to solid phase must be understood. Although this transition has been investigated experimentally and theoretically, its atomic-level mechanisms remain debatable. In this work, the nucleation mechanisms of pure Fe under rapid cooling conditions are investigated. The local atomic packing stability and liquid-to-solid transition-energy pathways of Fe are studied using molecular dynamics simulations and first-principle calculations. The results are expressed as functions of cluster size in units of amorphous clusters (ACs) and body-centered cubic crystalline clusters (BCC-CCs). We found the prototypes of ACs in supercooled liquids and successfully divided these ACs to three categories according to their transition-energy pathways. The information obtained in this study could contribute to our current understanding of the crystallization of metallic melts during rapid cooling. read less USED (high confidence) J. G. Sevillano, I. Aldazabal, A. Luque, and J. Aldazabal, “Atomistic simulation of the elongation response of a <011> oriented columnar nano-grain bcc Fe polycrystalline sample,” Meccanica. 2015. link Times cited: 0 USED (high confidence) D. Olson, H. Gao, C. Tang, W. Tysoe, and A. Martini, “Pressure dependence of the interfacial structure of potassium chloride films on iron,” Thin Solid Films. 2015. link Times cited: 2 USED (high confidence) S. Eder, U. Cihak-Bayr, A. Vernes, and G. Betz, “Evolution of topography and material removal during nanoscale grinding,” Journal of Physics D: Applied Physics. 2015. link Times cited: 19 Abstract: In this work we perform molecular dynamics simulations to qu… read moreAbstract: In this work we perform molecular dynamics simulations to quantify and parametrize the evolution of a bcc Fe work piece topography during nanometric grinding with multiple hard abrasive particles. The final surface quality depends on both the normal pressure and the abrasive geometry. We fit the time development of the substrate’s root mean squared roughness to an exponential function, allowing the definition of a run-in regime, during which the surface ‘forgets’ about its initial state, and a steady-state regime where the roughness no longer changes. The time constants associated with smoothing and material removal are almost inversely proportional to each other, highlighting the distinctiveness of these two simultaneously occurring processes. We also describe an attempt to reduce the time required to achieve the smoothest possible surface finish by periodically re-adjusting the normal pressure during the grinding process. read less USED (high confidence) W. Li, F. Yuan, and X. Wu, “Atomistic Tensile Deformation Mechanisms of Fe with Gradient Nano-Grained Structure,” Heterostructured Materials. 2015. link Times cited: 13 Abstract: Large-scale molecular dynamics (MD) simulations have been pe… read moreAbstract: Large-scale molecular dynamics (MD) simulations have been performed to investigate the tensile properties and the related atomistic deformation mechanisms of the gradient nano-grained (GNG) structure of bcc Fe (gradient grains with d from 25 nm to 105 nm), and comparisons were made with the uniform nano-grained (NG) structure of bcc Fe (grains with d = 25 nm). The grain size gradient in the nano-scale converts the applied uniaxial stress to multi-axial stresses and promotes the dislocation behaviors in the GNG structure, which results in extra hardening and flow strength. Thus, the GNG structure shows slightly higher flow stress at the early plastic deformation stage when compared to the uniform NG structure (even with smaller grain size). In the GNG structure, the dominant deformation mechanisms are closely related to the grain sizes. For grains with d = 25 nm, the deformation mechanisms are dominated by GB migration, grain rotation and grain coalescence although a few dislocations are observed. For grains with d = 54 nm, dislocation nucleation, propagation and formation of dislocation wall near GBs are observed. Moreover, formation of dislocation wall and dislocation pile-up near GBs are observed for grains with d = 105 nm, which is the first observation by MD simulations to our best knowledge. The strain compatibility among different layers with various grain sizes in the GNG structure should promote the dislocation behaviors and the flow stress of the whole structure, and the present results should provide insights to design the microstructures for developing strong-and-ductile metals. (C) 2015 Author(s). All article content, except where otherwise noted, is licensed under a Creative Commons Attribution 3.0 Unported License. read less USED (high confidence) K. Nakashima, R. Stoller, and H. Xu, “Recombination radius of a Frenkel pair and capture radius of a self-interstitial atom by vacancy clusters in bcc Fe,” Journal of Physics: Condensed Matter. 2015. link Times cited: 23 Abstract: The recombination radius of a Frenkel pair is a fundamental … read moreAbstract: The recombination radius of a Frenkel pair is a fundamental parameter for the object kinetic Monte Carlo (OKMC) and mean field rate theory (RT) methods that are used to investigate irradiation damage accumulation in irradiated materials. The recombination radius in bcc Fe has been studied both experimentally and numerically, however there is no general consensus about its value. The detailed atomistic processes of recombination also remain uncertain. Values from 1.0a0 to 3.3a0 have been employed as a recombination radius in previous studies using OKMC and RT. The recombination process of a Frenkel pair is investigated at the atomic level using the self-evolved atomistic kinetic Monte Carlo (SEAKMC) method in this paper. SEAKMC calculations reveal that a self-interstitial atom recombines with a vacancy in a spontaneous reaction from several nearby sites following characteristic pathways. The recombination radius of a Frenkel pair is estimated to be 2.26a0 by taking the average of the recombination distances from 80 simulation cases. In addition, we apply these procedures to the capture radius of a self-interstitial atom by a vacancy cluster. The capture radius is found to gradually increase with the size of the vacancy cluster. The fitting curve for the capture radius is obtained as a function of the number of vacancies in the cluster. read less USED (high confidence) D. Medlin, K. Hattar, J. Zimmerman, F. Abdeljawad, and S. Foiles, “Defect Character at Grain Boundary Facet Junctions: A Combined HRSTEM and Atomistic Modeling Study of a Σ=5 Grain Boundary in Fe,” Microscopy and Microanalysis. 2015. link Times cited: 0 Abstract: Electron microscopy plays an important role in motivating an… read moreAbstract: Electron microscopy plays an important role in motivating and testing our theoretical understanding of grain boundary structure and behavior across the atomistic and continuum length scales. One of the key foundational challenges in grain boundary science is to establish meaningful links between atomic-scale interfacial configurations, which can often be described in terms of sets of characteristic structural units [1], and more macro-scale descriptors of the interfacial geometry, such as grain misorientation and boundary inclination. A useful approach for establishing such linkages is to identify and to characterize the sets of interfacial line defects that accommodate departures from low-energy singular reference configurations [2]. read less USED (high confidence) S. Ratanaphan, D. Olmsted, V. Bulatov, E. Holm, A. Rollett, and G. Rohrer, “Grain boundary energies in body-centered cubic metals,” Acta Materialia. 2015. link Times cited: 176 USED (high confidence) G. Lv, H. Zhang, X. He, W. Yang, and Y. Su, “Atomistic simulation of Cu–Ni precipitates hardening in α-iron,” Journal of Physics D: Applied Physics. 2015. link Times cited: 7 Abstract: In this paper, we investigated the interaction of an edge di… read moreAbstract: In this paper, we investigated the interaction of an edge dislocation with Cu precipitates with a spherical geometry and with Cu–Ni precipitates that possess a Cu core with an outer Ni shell, commonly observed in reactor pressure vessel (RPV) steels. We applied molecular dynamics techniques to explore the critical stress required to unpin the dislocation (CSRUD), the breakaway dislocation line shape when the dislocation leaves the precipitates and the transition of Cu atoms within precipitates. The results indicate that the CSRUD of the Cu–Ni precipitates with a diameter less than 2.38 nm is larger than that of Cu precipitates that contain the same number of Cu atoms, while for a diameter larger than 2.38 nm, the CSRUD of Cu–Ni precipitates is weaker, which is related to the bcc to fcc-like or hcp-like atoms transformation in precipitates. The dislocations interact with Cu and Cu–Ni precipitates via the cut mechanism. read less USED (high confidence) L. Dezerald, L. Proville, L. Ventelon, F. Willaime, and D. Rodney, “First-principles prediction of kink-pair activation enthalpy on screw dislocations in bcc transition metals: V, Nb, Ta, Mo, W, and Fe,” Physical Review B. 2015. link Times cited: 74 Abstract: extension. Interestingly, we find that the atomistic line te… read moreAbstract: extension. Interestingly, we find that the atomistic line tension is more than twice the usual elastic estimates. The calculations also show interesting group tendencies with the line tension and kink-pair width larger in group V than in group VI elements. Finally, the present kink-pair activation energies are shown to compare qualitatively with experimental data and potential origins of quantitative discrepancies are discussed. read less USED (high confidence) M. Janish, P. Kotula, B. Boyce, and C. B. Carter, “Observations of fcc and hcp tantalum,” Journal of Materials Science. 2015. link Times cited: 17 USED (high confidence) N. Gunkelmann, D. Tramontina, E. Bringa, and H. Urbassek, “Morphological changes in polycrystalline Fe after compression and release,” Journal of Applied Physics. 2015. link Times cited: 21 Abstract: Despite a number of large-scale molecular dynamics simulatio… read moreAbstract: Despite a number of large-scale molecular dynamics simulations of shock compressed iron, the morphological properties of simulated recovered samples are still unexplored. Key questions remain open in this area, including the role of dislocation motion and deformation twinning in shear stress release. In this study, we present simulations of homogeneous uniaxial compression and recovery of large polycrystalline iron samples. Our results reveal significant recovery of the body-centered cubic grains with some deformation twinning driven by shear stress, in agreement with experimental results by Wang et al. [Sci. Rep. 3, 1086 (2013)]. The twin fraction agrees reasonably well with a semi-analytical model which assumes a critical shear stress for twinning. On reloading, twins disappear and the material reaches a very low strength value. read less USED (high confidence) D. Belashchenko and O. Kuskov, “Molecular-dynamic modeling of thermodynamic properties of the lunar Fe-S core,” Doklady Earth Sciences. 2015. link Times cited: 5 USED (high confidence) X. Tong, H. Zhang, and D. Li, “Effect of Annealing Treatment on Mechanical Properties of Nanocrystalline α-iron: an Atomistic Study,” Scientific Reports. 2015. link Times cited: 33 USED (high confidence) D. Belashchenko, “On the geometry and thermodynamics of nanoclusters,” Russian Journal of Physical Chemistry A. 2015. link Times cited: 2 USED (high confidence) R. Meyer and C. M. Mangiardi, “Parallelization of Molecular-Dynamics Simulations Using Tasks,” MRS Proceedings. 2015. link Times cited: 5 Abstract: This article discusses novel algorithms for molecular-dynami… read moreAbstract: This article discusses novel algorithms for molecular-dynamics (MD) simulations with short-ranged forces on modern multi- and many-core processors like the Intel Xeon Phi. A task-based approach to the parallelization of MD on shared-memory computers and a tiling scheme to facilitate the SIMD vectorization of the force calculations is described. The algorithms have been tested with three different potentials and the resulting speed-ups on Intel Xeon Phi coprocessors are shown. read less USED (high confidence) A. Mayer, “Dynamic shear and tensile strength of iron: Continual and atomistic simulation,” Mechanics of Solids. 2014. link Times cited: 19 USED (high confidence) A. Mayer, “Dynamic shear and tensile strength of iron: Continual and atomistic simulation,” Mechanics of Solids. 2014. link Times cited: 0 USED (high confidence) M. Y. Romashka and A. Yanilkin, “Simulation of the low-temperature stage of annealing of radiation defects in BCC iron using the molecular dynamics method,” The Physics of Metals and Metallography. 2014. link Times cited: 5 USED (high confidence) K. Chockalingam, R. Janisch, and A. Hartmaier, “Coupled atomistic-continuum study of the effects of C atoms at α-Fe dislocation cores,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 9 Abstract: The influence of carbon at dislocation cores in α-Fe is stud… read moreAbstract: The influence of carbon at dislocation cores in α-Fe is studied to determine the Peierls stress, i.e. the critical stress required to move the dislocation at 0 K. The effect of carbon on both edge and screw dislocations is investigated. A coupled molecular statics (MS) and extended finite element method (XFEM) is employed for this study, where the dislocation core is modeled atomistically. The results on pure Fe are found to be in good agreement with a fully atomistic study. The coupled approach captures the right core behavior and significantly reduces the size of the atomistic region, while describing the behavior of a single dislocation in an infinite anisotropic elastic medium. Furthermore, mechanical boundary conditions can be applied consistently. It was found that the influence of carbon on edge dislocations is much stronger than that on screw dislocations, and that carbon causes a directionally dependent Peierls stress in the case of a screw dislocation. Even though the increase of the Peierls stress is much more pronounced for edge dislocations, the total value does not reach the level of the Peierls stress for screw dislocations, either with or without carbon at the core. Hence, we conclude that the motion of screw dislocations remains the rate limiting factor for plastic deformation of α-Fe. read less USED (high confidence) B. Beeler, M. Asta, P. Hosemann, and N. Grønbech-Jensen, “Effects of applied strain on radiation damage generation in body-centered cubic iron,” Journal of Nuclear Materials. 2014. link Times cited: 41 USED (high confidence) V. Munizaga, G. García, E. Bringa, M. Weissmann, R. Ramírez, and M. Kiwi, “Atomistic simulation of soldering iron filled carbon nanotubes,” Computational Materials Science. 2014. link Times cited: 4 USED (high confidence) D. Wang, N. Gao, F. Gao, and Z. Wang, “Cu Segregation at Σ5 Symmetrical Grain Boundary in α-Fe: Atomic-Level Simulations,” Chinese Physics Letters. 2014. link Times cited: 3 Abstract: Cu-rich precipitation is regarded as one of the main issues … read moreAbstract: Cu-rich precipitation is regarded as one of the main issues causing embrittlement of ferritic steels. In the present work, the Cu segregation at Σ5 {012} symmetrical grain boundary (GB) in BCC iron is investigated by combining Metropolis Monte Carlo and molecular statics approaches. The segregation driven energies of Cu clusters decrease with increasing the distance from GB and also depend on the cluster size. The length scales associated with Cu segregation at GB are determined. All these results indicate that Cu atoms prefer to segregate at S5 GB, which may account for the embrittlement of ferritic steels. The present results provide important knowledge to understand the detailed mechanisms of Cu segregation at GB and also the possible effects on mechanical properties of α-Fe. read less USED (high confidence) X. Yang, S. Sun, X.-L. Wu, E. Ma, and T.-Y. Zhang, “Dissecting the Mechanism of Martensitic Transformation via Atomic-Scale Observations,” Scientific Reports. 2014. link Times cited: 85 USED (high confidence) X. Tong, H. Zhang, and D. Li, “Effects of misorientation and inclination on mechanical response of 〈1 1 0〉 tilt grain boundaries in α-Fe to external stresses,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 21 Abstract: Due to the industrial importance of α-iron-based polycrystal… read moreAbstract: Due to the industrial importance of α-iron-based polycrystalline materials, their grain boundary (GB) structures and properties need to be well characterized and understood in order to optimize the materials through effective GB engineering. In this study, a molecular dynamics (MD) simulation study was performed to investigate a series of 〈1 1 0〉 symmetric tilt grain boundaries (STGBs) and asymmetric tilt grain boundaries (ATGBs) in α-iron. It is shown that the GB energy is proportional to the GB volumetric expansion. During uniaxial deformation, 〈1 1 1〉{1 1 2} twinning appears to be more competitive or easier than 〈1 1 1〉{1 1 2} dislocation emission from the GB at yielding. For bicrystal systems containing STGBs the yield strength obeys the Schmid law, while for ATGB bicrystal systems the yield strengths are mainly determined by the local stress rather than overall stress and average GB energy. The higher degree of atomic disordering in the ATGB regions generates larger local stress fluctuation and thus facilitates local defect emission when subjected to external stresses. read less USED (high confidence) T. Cheng, W. Li, and D. Fang, “Modeling of the temperature-dependent ideal tensile strength of solids,” Physica Scripta. 2014. link Times cited: 11 Abstract: To reveal the fracture failure mechanisms of single crystals… read moreAbstract: To reveal the fracture failure mechanisms of single crystals at elevated temperatures, a new temperature-dependent ideal tensile strength model for solids has been developed, based on the critical strain principle. At the same time, the uniaxial tensile strength model, based on the critical failure energy density principle for isotropic materials that was presented in the previous study, is generalized to multi-axial loading and to cubic single crystals. The relationship between the two models is discussed, and how to obtain the material properties needed in the calculations is summarized. The two well-established models are used to predict the temperature-dependent ideal tensile strength of W, Fe and Al single crystals. The predictions from the critical strain principle agree well with the predictions from the critical failure energy density principle. The theoretical values from the critical strain principle at 0 K is in reasonable agreement with the ab initio results. The study shows that the temperature dependence of the ideal tensile strength is similar to that of Young’s modulus; that is, the ideal tensile strength firstly remains approximately constant and then decreases linearly with the temperature. The fracture failure for single crystals at elevated temperatures has been identified, for the first time, as a strain-controlled criterion. read less USED (high confidence) D. Belashchenko, “Estimation of the thermodynamic characteristics of the earth’s core using the embedded atom model,” Geochemistry International. 2014. link Times cited: 7 USED (high confidence) C. Healy and G. Ackland, “Molecular dynamics simulations of compression–tension asymmetry in plasticity of Fe nanopillars,” Acta Materialia. 2014. link Times cited: 79 USED (high confidence) A. Ojha, H. Sehitoglu, L. Patriarca, and H. Maier, “Twin migration in Fe-based bcc crystals: theory and experiments,” Philosophical Magazine. 2014. link Times cited: 33 Abstract: We establish an overall energy expression to determine the t… read moreAbstract: We establish an overall energy expression to determine the twin migration stress in bcc metals. Twin migration succeeds twin nucleation often after a load drop, and a model to establish twin migration stress is of paramount importance. We compute the planar fault energy barriers and determine the elastic energies of twinning dislocations including the role of residual dislocations (br) and twin intersection types such as 1 1 0, 1 1 3 and 2 1 0. The energy expression derived provides the twin migration stress in relation to the twin nucleation stress with a ratio of 0.5–0.8 depending on the resultant residual burgers vector and the intersection types. Utilizing digital image correlation, it was possible to differentiate the twin nucleation and twin advancement events experimentally, and transmission electron microscopy observations provided further support to the modelling efforts. Overall, the methodology developed provides an enhanced understanding of twin progression in bcc metals, and most importantly the proposed model does not rely on empirical constants. We utilize Fe-50at.%Cr in our experiments, and subsequently predict the twin migration stress for pure Fe, and Fe-3at.%V from the literature showing excellent agreement with experiments. read less USED (high confidence) H. Jin, I. Elfimov, and M. Militzer, “Study of the interaction of solutes with Σ5 (013) tilt grain boundaries in iron using density-functional theory,” Journal of Applied Physics. 2014. link Times cited: 70 Abstract: Substitutional alloying elements significantly affect the re… read moreAbstract: Substitutional alloying elements significantly affect the recrystallization and austenite-ferrite phase transformation rates in steels. The atomistic mechanisms of their interaction with the interfaces are still largely unexplored. Using density functional theory, we determine the segregation energies between commonly used alloying elements and the Σ5 (013) tilt grain boundary in bcc iron. We find a strong solute-grain boundary interaction for Nb, Mo, and Ti that is consistent with experimental observations of the effects of these alloying elements on delaying recrystallization and the austenite-to-ferrite transformation in low-carbon steels. In addition, we compute the solute-solute interactions as a function of solute pair distance in the grain boundary, which suggest co-segregation for these large solutes at intermediate distances in striking contrast to the bulk. read less USED (high confidence) W. J. Zhou, H. Luan, Y. L. He, J. Sun, and W. Tao, “A study on boundary force model used in multiscale simulations with non-periodic boundary condition,” Microfluidics and Nanofluidics. 2014. link Times cited: 25 USED (high confidence) S. Muto et al., “Quantitative characterization of nanoscale polycrystalline magnets with electron magnetic circular dichroism,” Nature Communications. 2014. link Times cited: 41 USED (high confidence) S. Eder, A. Vernes, and G. Betz, “On the Derjaguin offset in boundary-lubricated nanotribological systems.,” Langmuir : the ACS journal of surfaces and colloids. 2013. link Times cited: 41 Abstract: We performed molecular dynamics simulations of boundary-lubr… read moreAbstract: We performed molecular dynamics simulations of boundary-lubricated sliding, varying the boundary lubricant type, its molecular surface coverage, the substrate roughness, and the load. The resulting load versus friction behavior was then analyzed to study how changes in lubricant type, coverage, and roughness affect the extrapolated friction force at zero load, the so-called Derjaguin offset. A smooth-particle-based evaluation method by the authors, applied here for the first time to visualize the sliding interface between the two layers of boundary lubricant, allowed the definition and calculation of a dimensionless normalized sliding resistance area, which was then related to the Derjaguin offset. This relationship excellently reflects the molecular surface coverage, which determines the physical condition of the lubricant, and can differentiate between some lubricant-specific frictional properties. read less USED (high confidence) Z. Chen and J. Qu, “Dislocation-induced acoustic nonlinearity parameter in crystalline solids,” Journal of Applied Physics. 2013. link Times cited: 27 Abstract: Based on an orientation-dependent dislocation line energy, a… read moreAbstract: Based on an orientation-dependent dislocation line energy, a solution to the acoustic nonlinearity parameter is obtained for pure and mixed dislocations in anisotropic crystals. The solution is validated by comparison with molecular dynamic simulations. Parametric studies using this new solution show that (i) elastic anisotropy can significantly change the nonlinear behavior of dislocations including “corners” in the bowed dislocation line, much reduced critical stress for instability, sharp peaks in the β versus applied shear relationship, etc., (ii) mixed dislocations may have distinct behavior that is not bounded by the pure edge and screw dislocations, (iii) asymptotic solutions of the acoustic nonlinearity parameter in terms of power series (as high as 5th order) may not be valid even for pure dislocations in isotropic solids. read less USED (high confidence) M. Rajagopalan, M. Tschopp, and K. Solanki, “Grain Boundary Segregation of Interstitial and Substitutional Impurity Atoms in Alpha-Iron,” JOM. 2013. link Times cited: 81 USED (high confidence) S. Wang, N. Hashimoto, and S. Ohnuki, “Hydrogen-induced change in core structures of 110[111] edge and 110[111] screw dislocations in iron,” Scientific Reports. 2013. link Times cited: 27 USED (high confidence) J. J. Möller, A. Prakash, and E. Bitzek, “FE2AT—finite element informed atomistic simulations,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 17 Abstract: Atomistic simulations play an important role in advancing ou… read moreAbstract: Atomistic simulations play an important role in advancing our understanding of the mechanical properties of materials. Currently, most atomistic simulations are performed using relatively simple geometries under homogeneous loading conditions, and a significant part of the computer time is spent calculating the elastic response of the material, while the focus of the studies lies usually on the mechanisms of plastic deformation and failure. Here we present a simple but versatile approach called FE2AT to use finite element calculations to provide appropriate initial and boundary conditions for atomistic simulations. FE2AT allows us to forgo the simulation of large parts of the elastic loading process, even in the case of complex sample geometries and loading conditions. FE2AT is open source and can be used in combination with different atomistic simulation codes and methods. Its application is demonstrated using the bending of a nano-beam and the determination of the displacement field around a crack tip as examples. read less USED (high confidence) P. Erhart, J. Marian, and B. Sadigh, “Thermodynamic and mechanical properties of copper precipitates in alpha-iron from atomistic simulations,” Physical Review B. 2013. link Times cited: 28 Abstract: Precipitate hardening is commonly used in materials science … read moreAbstract: Precipitate hardening is commonly used in materials science to control strength by acting on the number density, size distribution, and shape of solute precipitates in the hardened matrix. The Fe-Cu system has attracted much attention over the last several decades due to its technological importance as a model alloy for Cu steels. In spite of these efforts several aspects of its phase diagram remain unexplained. Here we use atomistic simulations to characterize the polymorphic phase diagram of Cu precipitates in body-centered cubic (BCC) Fe and establish a consistent link between their thermodynamic and mechanical properties in terms of thermal stability, shape, and strength. The size at which Cu precipitates transform from BCC to a close-packed 9R structure is found to be strongly temperature dependent, ranging from approximately 4 nm in diameter (similar to 2700 atoms) at 200 K to about 8 nm (similar to 22 800 atoms) at 700 K. These numbers are in very good agreement with the interpretation of experimental data given Monzen et al. [Philos. Mag. A 80, 711 (2000)]. The strong temperature dependence originates from the entropic stabilization of BCC Cu, which is mechanically unstable as a bulk phase. While at high temperatures the transition exhibits first-order characteristics, the hysteresis, and thus the nucleation barrier, vanish at temperatures below approximately 300 K. This behavior is explained in terms of the mutual cancellation of the energy differences between core and shell (wetting layer) regions of BCC and 9R nanoprecipitates, respectively. The proposed mechanism is not specific for the Fe-Cu system but could generally be observed in immiscible systems, whenever the minority component is unstable in the lattice structure of the host matrix. Finally, we also study the interaction of precipitates with screw dislocations as a function of both structure and orientation. The results provide a coherent picture of precipitate strength that unifies previous calculations and experimental observations. read less USED (high confidence) D. Chen, J. Wang, T. Chen, and L. Shao, “Defect annihilation at grain boundaries in alpha-Fe,” Scientific Reports. 2013. link Times cited: 98 USED (high confidence) K. Solanki, M. Tschopp, M. A. Bhatia, and N. Rhodes, “Atomistic Investigation of the Role of Grain Boundary Structure on Hydrogen Segregation and Embrittlement in α-Fe,” Metallurgical and Materials Transactions A. 2013. link Times cited: 84 USED (high confidence) E. Hayward, C. Deo, B. Uberuaga, and C. Tomé, “The interaction of a screw dislocation with point defects in bcc iron,” Philosophical Magazine. 2012. link Times cited: 30 Abstract: In this study, we calculate the interaction energy of intrin… read moreAbstract: In this study, we calculate the interaction energy of intrinsic point defects vacancies and interstitials) with screw dislocations in body-centered cubic iron. First (we calculate the dipole tensor of a defect in the bulk crystal using molecular statics. Using a formulation based on linear elasticity theory, we calculate the interaction energy of the defect and the dislocation using both isotropic and anisotropic strain fields. Second, we perform atomistic calculations using molecular statics methods to directly calculate the interaction energy. Results from these two methods are compared. We verify that continuum methods alone are unable to correctly predict the interactions of defects and dislocations near the core. Although anisotropic theory agrees qualitatively with atomistics far from the core, it cannot predict which dumbbell orientations are stable and any continuum calculations must be used with caution. Spontaneous absorption by the core of both vacancies and dumbbells is seen. This paper demonstrates and discusses the differences between continuum and atomistic calculations of interaction energy between a dislocation core and a point defect. read less USED (high confidence) K. Kang, J. Wang, and I. Beyerlein, “Atomic structure variations of mechanically stable fcc-bcc interfaces,” Journal of Applied Physics. 2012. link Times cited: 77 Abstract: It has recently been shown that under severe plastic deforma… read moreAbstract: It has recently been shown that under severe plastic deformation processing bi-metal fcc/bcc composites develop a mechanically stable heterophase interface that joins the {112}fcc//{112}bcc planes in the Kurdjumov-Sachs orientation relationship. In this article, we study variations in the relaxed equilibrium atomic structure of this interface with changes in fcc stacking fault energy (SFE) and lattice mismatch between the two crystals. Using molecular statics/dynamics simulations for three fcc/bcc systems, Cu-Nb, Al-Fe, and Al-Nb, we find that the number of distinct sets of intrinsic interfacial dislocations and their core structures vary significantly among these three systems. The impact of these atomic-scale structural differences on interfacial properties is demonstrated through their interactions with point defects. The interfaces studied here are shown to exhibit a wide variation in ability, ranging from being a poor to an excellent sink for vacancies. read less USED (high confidence) D. Molnár, P. Binkele, S. Hocker, and S. Schmauder, “Atomistic multiscale simulations on the anisotropic tensile behaviour of copper-alloyed alpha-iron at different states of thermal ageing,” Philosophical Magazine. 2012. link Times cited: 18 Abstract: The mechanical behaviour of steels is strongly related to th… read moreAbstract: The mechanical behaviour of steels is strongly related to their underlying atomistic structures which evolve during thermal treatment. Cu-alloyed α-Fe undergoes a change in material behaviour during the ageing process, especially at temperatures of above 300°C, where precipitates form on a large time-scale within the α-Fe matrix, yielding first a precipitation strengthening of the material. As the precipitates grow further in time, the material strength decreases again. This complex process is modelled with a multiscale approach, combining Kinetic Monte Carlo (KMC) with Molecular Dynamics (MD) simulations in a sequential way and exploiting the advantages of both methods while simultaneously circumventing their particular disadvantages. The formation of precipitates is modelled on a single-crystal lattice with a diffusion based KMC approach. Transferring selected precipitation states at different ageing times to MD simulations allows the performance of nano tensile tests and the analysis of failure initiation. The anisotropic tensile behaviour is investigated in the [100], [110] and [111] directions, showing monotonically decreasing tensile strengths and deformation strains. Hence precipitation strengthening is mainly due to dislocation–precipitate interactions which are non-existent at small tensile loadings in this scenario. At the point of ductile failure, dislocations are generated at the interfaces between precipitates and the Fe matrix. Straining in the [100] direction, they lie on {110} and {112} glide planes, as expected. With the method presented here, the changes of the anisotropic tensile moduli are related to different states of thermal ageing, i.e., to nucleation, growth and Ostwald ripening of Cu precipitates. read less USED (high confidence) M. Gilbert, S. Queyreau, and J. Marian, “Stress and temperature dependence of screw dislocation mobility in α -Fe by molecular dynamics,” Physical Review B. 2011. link Times cited: 155 Abstract: The low-temperature plastic yield of α-Fe single crystals is… read moreAbstract: The low-temperature plastic yield of α-Fe single crystals is known to display a strong temperature dependence and to be controlled by the thermally activated motion of screw dislocations. In this paper, we present molecular dynamics simulations of 12 〈111〉{112} screw dislocation motion as a function of temperature and stress in order to extract mobility relations that describe the general dynamic behavior of screw dislocations in pure α-Fe. We find two dynamic regimes in the stress-velocity space governed by different mechanisms of motion. Consistent with experimental evidence, at low stresses and temperatures, the dislocations move by thermally activated nucleation and propagation of kink pairs. Then, at a critical stress, a temperature-dependent transition to a viscous linear regime is observed. Critical output from the simulations, such as threshold stresses and the stress dependence of the kink activation energy, are compared to experimental data and other atomistic works with generally very good agreement. Contrary to some experimental interpretations, we find that glide on {112} planes is only apparent, as slip always occurs by elementary kink-pair nucleation/propagation events on {110} planes. Additionally, a dislocation core transformation from compact to dissociated has been identified above room temperature, although its impact on the general mobility is seen to be limited. This and other observations expose the limitations of inferring or presuming dynamic behavior on the basis of only static calculations. We discuss the relevance and applicability of our results and provide a closed-form functional mobility law suitable for mesoscale computational techniques. read less USED (high confidence) X. Liu, W. Xie, W. Chen, and H. Zhang, “Effects of grain boundary and boundary inclination on hydrogen diffusion in a-iron,” Journal of Materials Research. 2011. link Times cited: 28 Abstract: Diffusion of interstitial hydrogen atoms in a-iron was inves… read moreAbstract: Diffusion of interstitial hydrogen atoms in a-iron was investigated using molecular dynamic simulation. In particular, hydrogen diffusivities in bulk, on (001) surface and within a Σ5 [100]/(013) symmetric tilt grain boundary (STGB) were estimated in a temperature range of 400 and 700 K. Furthermore, hydrogen diffusivities in a series of Σ5 [100] tilt grain boundaries with different inclinations were also determined as a function of temperature. The inclination dependence of activation energy for diffusion exhibits two local maxima, which correspond to two STGBs. Additional calculation of inclination dependence of boundary energy and boundary specific excess volume shows two local minima at the same STGBs. This suggests hydrogen diffusion into and within a grain boundary might be assisted by grain boundary excess volume and stress. Simulation of effects of hydrostatic pressure on diffusion shows tensile stress can promote hydrogen diffusion in lattice into grain boundary or surface traps, while compressive stress leads to a decrease in diffusivity, and a slower rate of filling these traps. read less USED (high confidence) M. Itakura, H. Kaburaki, and M. Yamaguchi, “First-principles study on the mobility of screw dislocations in bcc iron,” Acta Materialia. 2011. link Times cited: 87 USED (high confidence) P. Gordon, T. Neeraj, and M. Mendelev, “Screw dislocation mobility in BCC Metals: a refined potential description for α-Fe,” Philosophical Magazine. 2011. link Times cited: 56 Abstract: In this work, we seek to develop a new interatomic potential… read moreAbstract: In this work, we seek to develop a new interatomic potential for α-Fe that is able to rationalize experimental flow stress data. We generate a series of potentials with similar bulk and point defect properties, but exhibit different energetic landscapes for the Peierls potential. The family of potentials all possess a compact core structure, which we find necessitates a camel-hump shaped Peierls potential. Within this constraint, we analyze the relationships between the Peierls potential, the 3-D kink nucleation energetics, and the resulting shape of the kink structures for the screw dislocation. We find that one of our models, labeled MPG20, gives very good agreement with experimental flow stress data over the entire stress range considered. read less USED (high confidence) K. Odbadrakh et al., “Calculated electronic and magnetic structure of screw dislocations in alpha iron,” Journal of Applied Physics. 2011. link Times cited: 7 Abstract: Local atomic magnetic moments in crystalline Fe are perturbe… read moreAbstract: Local atomic magnetic moments in crystalline Fe are perturbed by the presence of dislocations. The effects are most pronounced near the dislocation core and decay slowly as the strain field of the dislocation decreases with distance. We have calculated local moments using the locally self-consistent multiple scattering (LSMS) method for a supercell containing a screw-dislocation quadrupole. Finite size effects are found to be significant indicating that dislocation cores affect the electronic structure and magnetic moments of neighboring dislocations. The influence of neighboring dislocations points to a need to study individual dislocations from first principles just as they appear amid surrounding atoms in large-scale classical force field simulations. An approach for the use of the LSMS to calculate local moments in subvolumes of large atomic configurations generated in the course of classical molecular dynamics simulation of dislocationdynamics is discussed. read less USED (high confidence) Y. Gao, Y. Yang, and D. Sun, “Wetting of Liquid Iron in Carbon Nanotubes and on Graphene Sheets: A Molecular Dynamics Study,” Chinese Physics Letters. 2011. link Times cited: 10 Abstract: Using molecular dynamics simulations, we study the wetting o… read moreAbstract: Using molecular dynamics simulations, we study the wetting of liquid iron in a carbon nanotube and on a graphene sheet. It is found that the contact angle of a droplet in a carbon nanotube increases linearly with the increase of wall curvature but is independent of the length of the filled liquid. The contact angle for a droplet on a graphene sheet decreases with the increasing droplet size. The line tension of a droplet on a graphene sheet is also obtained. Detailed studies show that liquid iron near the carbon walls exhibits the ordering tendencies in both the normal and tangential directions. read less USED (high confidence) Y. Zhang, F. Zhang, L. Qian, and T. Wang, “Atomic-scale simulation of α/γ-iron phase boundary affecting crack propagation using molecular dynamics method,” Computational Materials Science. 2011. link Times cited: 15 USED (high confidence) M. Tschopp, M. Horstemeyer, F. Gao, X. Sun, and M. Khaleel, “Energetic driving force for preferential binding of self-interstitial atoms to Fe grain boundaries over vacancies,” Scripta Materialia. 2010. link Times cited: 62 USED (high confidence) P. Gordon, T. Neeraj, Y. Li, and J. Li, “Screw dislocation mobility in BCC metals: the role of the compact core on double-kink nucleation,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 78 Abstract: In this work, we examine the kink-nucleation process in BCC … read moreAbstract: In this work, we examine the kink-nucleation process in BCC screw dislocations using atomistic simulation and transition pathway analysis, with a particular focus on the compact core structure. We observe the existence of a threshold stress, which results in an abrupt change in the minimum energy path of the kink-nucleation process, and hence, a discontinuity in the activation energy versus stress for the process. The magnitude of the discontinuity is found to be related to the degree of metastability of an intermediate split-core structure. This feature appears to be a direct consequence of the so-called ‘camel-hump’ nature of the Peierls potential, which manifests itself in the existence of a metastable, intermediate split-core structure. The effect is observed in a number of empirical EAM potentials, including Fe, Ta, V, Nb and Mo, suggesting a generality to the observations. read less USED (high confidence) P. Erhart and J. Marian, “Calculation of the substitutional fraction of ion-implanted He in an α-Fe target,” Journal of Nuclear Materials. 2010. link Times cited: 14 USED (high confidence) J. Yun-fei, M. Chen, Y. Xiangxi, W. Wei-min, and N. Xi-Jing, “A simple theoretical model for evaluating the ability to form a single crystal,” Chinese Physics B. 2010. link Times cited: 0 Abstract: A simple theoretical model proposed recently to evaluate the… read moreAbstract: A simple theoretical model proposed recently to evaluate the ability of bulk materials to form single crystals is further tested via vast molecular dynamics simulations of growth for fcc (Ni, Cu, Al, Ar) and hcp (Mg) crystals, especially applied to the growth of bcc (Fe) crystal, showing that the validity of the model is independent of crystal types and the interaction potentials of the constitute atoms. read less USED (high confidence) E. Dolgusheva and V. Trubitsin, “Influence of the size and shape of free nanoparticles on the local changes in the lattice parameter and on the structural stability of body-centered cubic zirconium and iron,” Physics of the Solid State. 2010. link Times cited: 2 USED (high confidence) S. Kotrechko and A. Ovsjannikov, “Temperature dependence of the yield stress of metallic nano-sized crystals,” Philosophical Magazine. 2009. link Times cited: 22 Abstract: It is shown that the temperature effect on the variance of l… read moreAbstract: It is shown that the temperature effect on the variance of local shear stresses is the main factor pre-determining the temperature law of the yield stress of nano-sized crystals. The results of molecular dynamics simulations of uniaxial tension of Mo, α-Fe and W nanowires in three crystallographic directions ([100], [110] and [111]) over the temperature range 100–1000 K are presented. It is found that within this temperature range, the yield stress of nano-sized crystals varies not exponentially, as for bulk single crystals, but is a parabolic function of temperature. read less USED (high confidence) M. Mendelev, M. J. Kramer, R. T. Ott, and D. Sordelet, “Molecular dynamics simulation of diffusion in supercooled Cu–Zr alloys,” Philosophical Magazine. 2009. link Times cited: 60 Abstract: Molecular dynamics (MD) simulations of diffusion in Cu–Zr al… read moreAbstract: Molecular dynamics (MD) simulations of diffusion in Cu–Zr alloys in their liquid and supercooled liquid states were performed using a recently developed Finnis–Sinclair many-body interatomic potential. To help assess how well the interatomic potential describes the energetics of the Cu–Zr system, the liquid structure determined by MD simulations was compared with wide-angle X-ray scattering measurements of the liquid structure for a Cu64.5Zr35.5 alloy. Diffusion was examined as a function of composition, pressure and temperature. The simulations reveal that the diffusion exhibits strong compositional dependence, with both species exhibiting minimum diffusivities at ∼70% Cu. Moreover, the MD simulations show that the activation volumes for Zr and Cu atoms exhibit a maximum near 70% Cu. Evidence is obtained that the glass transition temperature also changes strongly with composition, thereby contributing to the diffusion behaviour. The relationship between this minimum in diffusion and the apparent best glass-forming composition in the Cu–Zr system is discussed. read less USED (high confidence) G. Balasubramanian, S. Banerjee, and I. Puri, “Unsteady nanoscale thermal transport across a solid-fluid interface,” Journal of Applied Physics. 2008. link Times cited: 40 Abstract: We simulate unsteady nanoscale thermal transport at a solid-… read moreAbstract: We simulate unsteady nanoscale thermal transport at a solid-fluid interface by placing cooler liquid-vapor Ar mixtures adjacent to warmer Fe walls. The equilibration of the system towards a uniform overall temperature is investigated using nonequilibrium molecular dynamics simulations from which the heat flux is also determined explicitly. The Ar–Fe intermolecular interactions induce the migration of fluid atoms into quasicrystalline interfacial layers adjacent to the walls, creating vacancies at the migration sites. This induces temperature discontinuities between the solidlike interfaces and their neighboring fluid molecules. The interfacial temperature difference and thus the heat flux decrease as the system equilibrates over time. The averaged interfacial thermal resistance Rk,av decreases as the imposed wall temperature Tw is increased, as Rk,av∝Tw−4.8. The simulated temperature evolution deviates from an analytical continuum solution due to the overall system heterogeneity. read less USED (high confidence) E. Clouet, S. Garruchet, H. Nguyen, M. Perez, and C. Becquart, “Dislocation interaction with C in α-Fe: A comparison between atomic simulations and elasticity theory,” Acta Materialia. 2008. link Times cited: 169 USED (high confidence) S. Dudarev, R. Bullough, and P. Derlet, “Effect of the alpha-gamma phase transition on the stability of dislocation loops in bcc iron.,” Physical review letters. 2008. link Times cited: 162 Abstract: Body-centered-cubic iron develops an elastic instability, dr… read moreAbstract: Body-centered-cubic iron develops an elastic instability, driven by spin fluctuations, near the alpha-gamma phase transition temperature T(c) = 912 degrees C that is associated with the dramatic reduction of the shear stiffness constant c' (c(11)-c(12))/2 near T(c). This reduction of c' has a profound effect on the temperature dependence of the anisotropic elastic self-energies of dislocations in iron. It also affects the relative stability of the a[100] and a/2[111] prismatic edge dislocation loops formed during irradiation. The difference between the anisotropic elastic free energies provides the fundamental explanation for the observed dominant occurrence of the a[100], as opposed to the a/2[111], Burgers vector configurations of prismatic dislocation loops in iron and iron-based alloys at high temperatures. read less USED (high confidence) L. Ventelon and F. Willaime, “Core structure and Peierls potential of screw dislocations in α-Fe from first principles: cluster versus dipole approaches,” Journal of Computer-Aided Materials Design. 2007. link Times cited: 126 USED (high confidence) C. Ortiz and M. Caturla, “Cascade damage evolution: rate theory versus kinetic Monte Carlo simulations,” Journal of Computer-Aided Materials Design. 2007. link Times cited: 20 USED (high confidence) A. Kuksin, G. Norman, V. Stegailov, and A. Yanilkin, “Surface melting of superheated crystals. Atomistic simulation study,” Comput. Phys. Commun. 2007. link Times cited: 20 USED (high confidence) T. Bazhirov, A. Kuksin, G. Norman, and V. Stegailov, “On similarity relations for the stability limits of metastable metals,” Doklady Physics. 2007. link Times cited: 0 USED (high confidence) J. Hoyt, M. Asta, and D. Sun, “Molecular dynamics simulations of the crystal–melt interfacial free energy and mobility in Mo and V,” Philosophical Magazine. 2006. link Times cited: 40 Abstract: Molecular dynamics simulations, based on embedded-atom metho… read moreAbstract: Molecular dynamics simulations, based on embedded-atom method potentials, have been used to compute thermodynamic and kinetic properties of crystal–melt interfaces in the bcc metals Mo and V. The interfacial free energy and its associated crystalline anisotropy have been obtained with the capillary fluctuation method and for both metals the anisotropy and the value of the Turnbull coefficient are found to be significantly lower than for the case of fcc materials. The interface mobility, or kinetic coefficient, which relates the isothermal crystallization rate to interface undercooling, was computed by non-equilibrium molecular dynamics simulations. Mobilities in the range 9-16 cm s−1K−1 are obtained. For Mo the mobility in the (110) crystallographic growth direction is larger than in the (100) and (111) directions, whereas for V the growth is found to be isotropic within numerical uncertainty. The kinetic-coefficient results are discussed within the framework of a density-functional-based theory of crystal growth. read less USED (high confidence) J. Chaussidon, M. Fivel, and D. Rodney, “The glide of screw dislocations in bcc Fe: Atomistic static and dynamic simulations ☆,” Acta Materialia. 2006. link Times cited: 208 USED (high confidence) G. Norman, V. Stegailov, and A. Yanilkin, “Fracture of crystalline iron subjected to high-rate tension. Molecular dynamics simulation,” Doklady Physics. 2005. link Times cited: 2 USED (high confidence) R. A. Pérez and M. Weissmann, “Ab initio study of magnetic effects on diffusion in α-Fe,” Journal of Physics: Condensed Matter. 2004. link Times cited: 14 Abstract: A deviation from the Arrhenius law in ?-Fe self-diffusion an… read moreAbstract: A deviation from the Arrhenius law in ?-Fe self-diffusion and also in the diffusion of substitutional impurities is found experimentally. Below the Curie temperature the diffusion coefficients have lower values than those extrapolated from the paramagnetic region and the Arrhenius plot shows an upward curvature. In this work we attempt to understand this behaviour from first-principles calculations. Formation and migration energies for self-diffusion and also for the diffusion of some substitutional impurities are calculated. Spin-polarized and non-spin-polarized calculations are assumed to approximately represent ferromagnetic and paramagnetic ?-Fe respectively. The calculations were performed using the WIEN97 code, with a supercell of 36?atoms which allows us to include both vacancies and impurities and therefore to study the migration along the direction. The increment in the diffusion barrier due to the total magnetic alignment at 0?K, with respect to the paramagnetic case, is almost constant for non-magnetic impurities, as it is in the experiments, whereas for magnetic impurities it depends on the diffusing atom. read less USED (high confidence) L. Wu, Y. Zhu, H. Wang, and M. Li, “Crystal–melt coexistence in fcc and bcc metals: a molecular-dynamics study of kinetic coefficients,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 5 Abstract: As a sequel to the previous paper on the calculation of the … read moreAbstract: As a sequel to the previous paper on the calculation of the crystal–melt interface free energy (2021 Materialia 15 100962), here we report the results on the kinetic coefficients using molecular dynamics simulations performed on six fcc metals and four bcc metals with the intention to compare the crystal structural influence. We found that the calculated kinetic coefficients are well described by the model by Broughton, Gilmer and Jackson (1982 Phys. Rev. Lett. 49 1496), and in particular, they exhibit varying degrees of anisotropy. We reveal that the anisotropies are related to the fluctuation of the crystal–melt interfaces, which causes the increase of the actual interface area in melting or solidification. The kinetic coefficients always display asymmetry between the solidification and melting process, and the difference is much more pronounced for the (111) interfaces in fcc metals which have the highest anisotropy. We found that the atomic mechanisms of the kinetic behaviors of these interfaces are closely related to the formation of twin-crystal domains during solidification, which delays the solidification process and consequently causes a decrease in the calculated kinetic coefficients. read less USED (high confidence) V. Mazhukin, A. V. Shapranov, and O. Koroleva, “Atomistic modeling of crystal-melt interface mobility of fcc (Al, Cu) and bcc (Fe) metals in STRONG superheating/undercooling states.” 2020. link Times cited: 5 Abstract: A detailed study of the mobility and kinetic properties of s… read moreAbstract: A detailed study of the mobility and kinetic properties of solid liquid interfaces (SLI) with different types of crystal lattices (fcc Al, Cu) and (bcc Fe) metals in a wide range of temperatures and pressures was carried out using atomistic modeling. The ranges of maximum permissible values of superheated/undercooled states for each metal have been determined. The ultimate goal of the study was to determine the temperature dependences of the stationary front velocity υsl(ΔT) describing the SLI mobility in each of the metals in an analytical form. The analytical dependence υsl(ΔT) was constructed by comparing the results of atomistic modeling in the area of maximum permissible superheating/undercooling values with the data of the main kinetic models of Wilson Frenkel (WF) and Broughton, Gilmer and Jackson (BGJ). An acceptable agreement was achieved by introducing appropriate correction parameters into the kinetic models using the least squares method. The influence of the crystallographic orientation of metals and external pressure on the SLI mobility is investigated. read less USED (high confidence) B. Cheng, “Predicting homogeneous nucleation rate from atomistic simulations.” 2019. link Times cited: 0 Abstract: Nucleation is ubiquitous, from the formation of clouds to th… read moreAbstract: Nucleation is ubiquitous, from the formation of clouds to the preparation of pharmaceutical compounds, from metal casting to the tempering of chocolates, and from the growth of beautiful nautilus shells to the assembly of microtubules in cells. The first experiment for observing a nucleation event was performed by Fahrenheit in 1724, and it can be easily replicated in a kitchen: simply place a bottle of purified water in the freezer. The liquid water can be cooled to far below zero degrees Celsius without freezing due to the lack of microscopic nuclei, which are the embryos from which the freezing phase transition can occur. After that, shaking the bottle will induce nucleation which in turn prompts a rapid ice crystallization. Despite its pivotal importance and long history, we only have a rough idea about the underlying mechanism of nucleation. The classical nucleation theory says that, during the growth of a nucleus inside a bulk phase, an interface that surrounds the nucleus has to be created. This interface is associated with an energy penalty, which the system has to overcome for the nucleus to grow into a critical size which precedes an avalanche of structural transitions. It is analogous to being stuck in a valley on the Alps, so that a great deal of time and energy have to be spent to climb over a peak in order to reach another valley. And yet, we have not reached a quantitative understanding of how high the energy barrier is and how long is the waiting time of nucleation for specific systems. Taking again the example of ice nucleation from bulk liquid water, there has been a long-standing discrepancy by more than 10 orders of magnitude between the measured and the predicted expectation time of nucleation. While the classical nucleation theory is able to paint a physical picture of nucleation, for many systems it is insufficient and thus needs extension. Despite substantial improvements in recent years, experimental characterization of the dynamical nucleation processes is extremely difficult, which motivates atomistic modelling efforts that use numerical simulation techniques. However, atomistic simulations also faces a number of challenges: firstly the typical time scales accessible to atomistic simulations are confined to below microseconds, while nucleation can take hours or days to occur. We have mitigated the challenge by employing state-of-the-art enhanced sampling methods in the simulation studies of nucleation [1–5]. In a nutshell, instead of naively waiting for a rare event to happen, we place a bias to help the system overcome the energy penalty of nucleation. To return to the earlier analogy with Alpine hiking, we can flatten out the Alps by depositing (a lot of) sand into all the valleys, making the landscape level and easy to explore. Secondly, only microscopic quantities such as the coordinates and the velocities of each atom can be directly obtained from simulations. On the contrary, macroscopic observables read less USED (high confidence) D. Sun, “Proliferation of Twinning in Metals: Application to Magnesium Alloys.” 2018. link Times cited: 2 Abstract: In the search for new alloys with a great strength-to-weight… read moreAbstract: In the search for new alloys with a great strength-to-weight ratio, magnesium has emerged at the forefront. With a strength rivaling that of steel and aluminum alloys --- materials which are deployed widely in real world applications today --- but only a fraction of the density, magnesium holds great promise in a variety of next-generation applications. Unfortunately, the widespread adoption of magnesium is hindered by the fact that it fails in a brittle fashion, which is undesirable when it comes to plastic deformation mechanisms. Consequently, one must design magnesium alloys to navigate around this shortcoming and fail in a more ductile fashion. However, such designs are not possible without a thorough understanding of the underlying mechanisms of deformation in magnesium, which is somewhat contested at the moment. In addition to slip, which is one of the dominant mechanisms in metallic alloys, a mechanism known as twinning is also present, especially in hexagonal close-packed (HCP) materials such as magnesium. Twinning involves the reorientation of the material lattice about a planar discontinuity and has been shown as one of the preferred mechanisms by which magnesium accommodates out-of-plane deformation. Unfortunately, twinning is not particularly well-understood in magnesium, and needs to be addressed before progress can be made in materials design. In particular, though two specific modes of twinning have been acknowledged, various works in the literature have identified a host of additional modes, many of which have been cast aside as "anomalous" observations. To this end, we introduce a new framework for predicting the modes by which a material can twin, for any given material. Focusing on magnesium, we begin our investigation by introducing a kinematic framework that predicts novel twin configurations, cataloging these twins modes by their planar normal and twinning shear. We then subject the predicted twin modes to a series of atomistic simulations, primarily in molecular statics but with supplementary calculations using density functional theory, giving us insight on both the energy of the twin interface and barriers to formation. We then perform a stress analysis and identify the twin modes which are most likely to be activated, thus finding the ones most likely to affect the yield surface of magnesium. Over the course of our investigation, we show that many different modes actually participate on the yield surface of magnesium; the two classical modes which are accepted by the community are confirmed, but many additional modes --- some of which are close to modes which have been previously regarded as anomalies --- are also observed. We also perform some extensional work, showing the flexibility of our framework in predicting twins in other materials and in other environments and highlighting the complicated nature of twinning, especially in HCP materials. read less USED (high confidence) A. Sobolev, V. Starukhin, I. Buldashev, and A. Mirzoev, “New potentials for multiscale simulations of liquid metals.” 2017. link Times cited: 1 Abstract: We present the technique and results for finding norm-conser… read moreAbstract: We present the technique and results for finding norm-conserving pseudopotentials and EAM potentials that can be used to recover atomic and electronic structure of liquid iron at and above the melting point. Pseudopotentials were found by minimizing the energy differences of our results with all-electron reference methods; EAM potentials — by the modified hybridization method proposed earlier by Belashchenko. We show that these potentials are at least as accurate in describing liquid iron as the established potentials in the field. read less USED (high confidence) S. Eder, A. Vernes, and G. Betz, “Methods and numerical aspects of nanoscopic contact area estimation in atomistic tribological simulations,” Comput. Phys. Commun. 2014. link Times cited: 13 USED (low confidence) S. S. Mishra and S. Bhattacharjee, “Collisional cooling in confined gases at cryogenic temperatures: Time dependent near-wall accumulation and gas dynamics,” International Journal of Heat and Mass Transfer. 2024. link Times cited: 0 USED (low confidence) S. Kumar, A. Rajput, S. K. Paul, M. Tiwari, and D. K. Prajapati, “Friction and wear study of metallic contacts under dry sliding conditions: A molecular dynamics simulation-based approach,” Proceedings of the Institution of Mechanical Engineers, Part J: Journal of Engineering Tribology. 2023. link Times cited: 0 Abstract: Sliding friction originating due to ploughing and adhesive w… read moreAbstract: Sliding friction originating due to ploughing and adhesive wear significantly affects the performance of small-scale components, that is, nano-electromechanical systems/micro-electromechanical. To get a comprehensive understanding of the friction mechanisms, a comprehensive study of surface interactions at the nanoscale is crucial, particularly when dealing with nano-electromechanical and micro-electromechanical components. This study performed molecular dynamics simulation to explore the interactions between asperities (made of similar/dissimilar materials) at the nanoscale under dry sliding conditions. The research framework focuses on modelling the contact between two hemispherical asperities during dry sliding by considering three material combinations: soft-to-soft (Cu–Cu), hard-to-hard (Fe–Fe), and hard-to-soft (Fe–Cu). The study assesses plastic deformation and atomic wear at specific sliding speeds. Notably, the results indicate that the frictional force on the lower asperity increases as interference increases. Additionally, atomic wear rises with increased interference in the case of the Fe–Cu tribopair. Particularly high atomic wear is observed in the Cu–Cu tribopair due to the ease of slip within the face-centred cubic crystal structure of copper. read less USED (low confidence) P. Lin, S. Cui, J. Nie, L. He, and W. Cui, “Molecular Dynamics Simulations of Displacement Cascades in BCC-Fe: Effects of Dislocation, Dislocation Loop and Grain Boundary,” Materials. 2023. link Times cited: 0 Abstract: The interactions between displacement cascades and three typ… read moreAbstract: The interactions between displacement cascades and three types of structures, dislocations, dislocation loops and grain boundaries, in BCC-Fe are investigated through molecular dynamics simulations. Wigner–Seitz analysis is used to calculate the number of point defects induced in order to illustrate the effects of three special structures on the displacement cascade. The displacement cascades in systems interacting with all three types of structure tend to generate more total defects compared to bulk Fe. The surviving number of point defects in the grain boundary case is the largest of the three types of structures. The changes in the atomic structures of dislocations, dislocation loops and grain boundaries after displacement cascades are analyzed to understand how irradiation damage affects them. These results could reveal irradiation damage at the microscale. Varied defect production numbers and efficiencies are investigated, which could be used as the input parameters for higher scale simulation. read less USED (low confidence) A. V. Markidonov, M. Starostenkov, A. Gostevskaya, D. A. Lubyanoy, and P. V. Zakharov, “Molecular Dynamics Simulation of Reduction of the Surface Layer Porosity in a BCC Crystal Induced by Laser Pulses,” Physics of the Solid State. 2023. link Times cited: 0 USED (low confidence) J. S. Lee et al., “Atomistic investigation into the formation of axial weak twins during the compression of single-crystal Mg nanopillars,” Acta Materialia. 2023. link Times cited: 0 USED (low confidence) T. Ye et al., “Simulation of threshold displacement energy in Fe-Cr-Al alloys using molecular dynamics,” Journal of Nuclear Materials. 2023. link Times cited: 0 USED (low confidence) M. M. Rahman, F. El-Mellouhi, and N. Mousseau, “Structural evolution of vacancy clusters in
α
-iron: A kinetic activation-relaxation technique study,” Physical Review Materials. 2023. link Times cited: 0 USED (low confidence) A. Gostevskaya, A. Markidonov, M. Starostenkov, I. A. Panchenko, and V. K. Drobyshev, “Simulation of High Temperatures Influence on Metal Structure during Laser Ablation,” High Energy Chemistry. 2023. link Times cited: 0 USED (low confidence) M. Wakeda and T. Ohmura, “Atomistic evaluation of the dislocation transmission across tilt and twist low-angle grain boundaries in body-centered cubic iron,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) W. Huang, J. Tang, W. Zhou, J. Wen, and M. Yi, “Molecular dynamics simulations of ultrasonic vibration-assisted grinding of polycrystalline iron: Nanoscale plastic deformation mechanism and microstructural evolution,” Applied Surface Science. 2023. link Times cited: 0 USED (low confidence) J. Veerababu and A. Nagesha, “Competition between full slip and twinning in BCC-Fe: Effect of preloaded stress and temperature,” Journal of Applied Physics. 2023. link Times cited: 0 Abstract: Slip or twinning is one of the fundamental questions in the … read moreAbstract: Slip or twinning is one of the fundamental questions in the deformation studies of metals and alloys. Internal parameters such as generalized stacking fault energy and size and external parameters such as pressure, strain rate, and temperature influence the competition between the full slip and twinning, thus dictating the predominance of one mechanism over the other. In the present investigation, we studied the influence of preloaded stress and temperature on the deformation behavior of BCC-Fe nanowires using molecular dynamics simulations and theoretical analysis. Based on detailed investigations into the energetics associated with slip and twinning, we observed that twinning is the preferred deformation mechanism in BCC-Fe. However, this has been modified by preloaded stresses applied in normal, transverse, and both directions on the nanowire. We observed a slip on {110}, on {112}, and even on {123} planes. The temperature did not alter the inherent twinning nature but linearly decreased the various fault energies. read less USED (low confidence) Y. Yang, J. Zhao, J. Cui, and B. Jiang, “Enhanced mechanical and tribological properties of polymer nanocomposites by improving interfacial properties by hexagonal boron nitride nanosheets: Molecular dynamics simulations,” Journal of Applied Polymer Science. 2023. link Times cited: 0 USED (low confidence) T. Li et al., “Molecular Dynamics Investigation on Micro-Friction Behavior of Cylinder Liner-Piston Ring Assembly,” Tribology Letters. 2023. link Times cited: 0 USED (low confidence) D. Rapp, S. Hocker, H. Lipp, and S. Schmauder, “Strengthening and failure of iron-graphene composites: A molecular dynamics study,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) M. P. Hazarika, A. Tripathi, and S. N. Chakraborty, “Two-temperature molecular dynamics simulation study of copper thin film irradiation with femtosecond and picosecond laser pulses,” Journal of Laser Applications. 2023. link Times cited: 0 Abstract: Metal targets irradiated with laser pulses have a wide range… read moreAbstract: Metal targets irradiated with laser pulses have a wide range of applications in thin film preparation, nanomaterial synthesis, bio-medical imaging, and metal ablation. Here, using two-temperature model based molecular dynamics simulation, we investigate laser mediated ablation in copper. Ablation of the film starts with the formation of voids within it. This void forming mechanism at low laser fluences ([Formula: see text] mJ/cm[Formula: see text]) is studied using both picosecond and femtosecond pulses. At the same fluence, shorter laser pulse transfers more energy to the atoms generating temperatures greater than the melting temperature of the crystal. This increases the kinetic energy of the atoms and they start vibrating with different velocities. If these vibrations cross a threshold of 5 Å per picosecond (500 m/s), voids and faults start appearing in the system. At the same fluence, higher concentration of voids are also created at a faster rate with the femtosecond pulse. read less USED (low confidence) W. Xie, C. Liu, G. Huang, Z.-ye Qin, K. Zong, and D. Jiang, “Trans-scale rough surface contact model based on molecular dynamics method: Simulation, modeling and experimental verification,” European Journal of Mechanics - A/Solids. 2023. link Times cited: 2 USED (low confidence) N. Daghbouj et al., “Microstructure evolution of iron precipitates in (Fe, He)-irradiated 6H-SiC: A combined TEM and multiscale modeling,” Journal of Nuclear Materials. 2023. link Times cited: 3 USED (low confidence) S. Kazanç and C. Canbay, “Investigation of microstructural development of liquid Nb in dependence of cooling rate: Molecular dynamics simulation study,” Vacuum. 2023. link Times cited: 1 USED (low confidence) J. Gou et al., “Hydrogen-induced cracking of welded X80 steel studies by experimental testing and molecular dynamics modeling,” Corrosion Science. 2023. link Times cited: 11 USED (low confidence) M. Wang, F. Wang, J. Zhang, H. Wang, Y. Wang, and H. Wu, “Effects of h-BN additives on tensile mechanical behavior of Fe matrix: A molecular dynamics study,” Computational Materials Science. 2023. link Times cited: 1 USED (low confidence) “The structure and energy of symmetric tilt grain boundaries in tungsten,” Journal of Nuclear Materials. 2023. link Times cited: 1 USED (low confidence) X. Li et al., “Vacancy accumulation mechanism at iron grain boundaries: The influence of grain boundary character and its coupling with grain size,” Journal of Nuclear Materials. 2023. link Times cited: 1 USED (low confidence) J. C. Stimac, C. Serrao, and J. Mason, “Dependence of simulated radiation damage on crystal structure and atomic misfit in metals,” Journal of Nuclear Materials. 2023. link Times cited: 1 USED (low confidence) H. Ghaffarian and D. Jang, “Anisotropic deformation mechanism of 110 hexagonal dislocation networks in BCC Iron,” Scripta Materialia. 2023. link Times cited: 0 USED (low confidence) T. Ye et al., “Molecular dynamics simulation of tensile deformation behavior of single-crystal Fe–Cr–Al before and after irradiation,” Journal of Materials Research. 2022. link Times cited: 1 Abstract: Fe–Cr–Al alloy is one of the candidate materials for reactor… read moreAbstract: Fe–Cr–Al alloy is one of the candidate materials for reactor fuel cladding due to excellent high-temperature oxidation resistance; however, it has significant irradiation embrittlement and hardening. To understand the effect of Cr and Al and the defects (point defects, clusters, and nanocracks) produced from radiation damage on the mechanical properties, the uniaxial tensile property of single-crystal Fe–Cr–Al is investigated. The results show that, due to the presence of Cr and Al, the phase transformation from body-centered-cubic to face-centered-cubic is impeded and the formation of defects and amorphous structures is promoted, leading to the reduction of Young’s modulus and the ultimate tensile stress. Interstitials are the main factor in Frenkel pairs contributing to the reduction of mechanical properties due to the high shear stress and lattice distortion. The collapse of the nanocrack causes the increase of Young’s modulus and the decrease of the ultimate tensile strength. Graphical abstract read less USED (low confidence) S. Nandi and S. Kumar, “Atomistic structural transformation of iron single crystal under bi-axial stretching using classical molecular dynamics simulation,” Bulletin of Materials Science. 2022. link Times cited: 0 USED (low confidence) L. Zhao et al., “Strong size effect on deformation twin-mediated plasticity in body-centered-cubic iron,” Journal of Materials Science & Technology. 2022. link Times cited: 2 USED (low confidence) T. Xu, W. Wang, H. Jiang, and G. He, “Study on micro crack propagation mechanism of ferrite–pearlite gas transmission pipeline steel with lamellar structure,” Scientific Reports. 2022. link Times cited: 1 USED (low confidence) Z. Wei, X. Ma, C. Ke, and X. P. Zhang, “Distinct migration mechanisms of stepped FCC/BCC martensitic interfaces associated with typical orientation relationships: a molecular dynamics study,” Journal of Materials Science. 2022. link Times cited: 0 USED (low confidence) Z.-Q. Shen et al., “OKMC simulation of vacancy-enhanced Cu solute segregation affected by temperature/irradiation in the Fe–Cu system,” Nuclear Science and Techniques. 2022. link Times cited: 0 USED (low confidence) W. Xie, D. Jiang, J. Jin, and C. Liu, “Single-asperity failure mechanism driven by morphology and multiaxial loading using molecular dynamics simulation,” Computational Materials Science. 2022. link Times cited: 0 USED (low confidence) Z. Zhao, B. Safaei, Y. Wang, Y. Liu, F.-H. Chu, and Y. Wei, “Atomistic scale behaviors of intergranular crack propagation along twist grain boundary in iron under dynamic loading,” Engineering Fracture Mechanics. 2022. link Times cited: 3 USED (low confidence) K. Nakamura, T. Kumagai, and T. Ohnuma, “Atomistic Simulation of Shear Deformation at Bcc-Fe Grain Boundary and Precipitation Strengthening by Cr23c6,” SSRN Electronic Journal. 2022. link Times cited: 0 USED (low confidence) X.-yan Li et al., “Radiation damage accumulation mechanisms at iron grain boundaries revealed by coupled atomic and coarse-grained simulations via the parameter-passing and structural feedback,” Journal of Nuclear Materials. 2022. link Times cited: 1 USED (low confidence) Y. Zhu et al., “Molecular dynamic simulation of Cs corrosion in Cs oven for negative ion source applications,” AIP Advances. 2022. link Times cited: 0 Abstract: Molecular dynamic simulation is used to simulate the corrosi… read moreAbstract: Molecular dynamic simulation is used to simulate the corrosion process of Fe or Ni in liquid Cs by Large-scale Atomic/Molecular Massively Parallel Simulator. The embedded-atom method potential is used to describe the interaction of Fe–Fe, Ni–Ni, and Cs–Cs, and Morse two-body potential is used to describe the Fe–Cs and Ni–Cs atomic interaction. Temperature is considered as a critical condition in this work. Results indicate that corrosion is easy to occur in the systems. The increase in temperature can help the process of Cs corrosion. Compared to the Ni–Cs system, the Fe–Cs system has a higher atomic concentration function. The radial distribution function shows that Cs atoms are dissolved into the substrates, but the Fe and Ni substrates are still crystalline structures. Moreover, Cs in Fe or Ni is still a liquid phase. read less USED (low confidence) A. Markidonov, M. Starostenkov, A. Gostevskaya, D. A. Lubyanoy, and P. Zakharov, “Modeling by a Molecular Dynamics Method of Structural Changes of a BCC Metal Surface Layer with Short-Term High-Energy External Action,” Metal Science and Heat Treatment. 2022. link Times cited: 0 USED (low confidence) W. Ma et al., “Evolution of Symmetrical Grain Boundaries under External Strain in Iron Investigated by Molecular Dynamics Method,” Metals. 2022. link Times cited: 2 Abstract: In the present work, the evolution of atomic structures and … read moreAbstract: In the present work, the evolution of atomic structures and related changes in energy state, atomic displacement and free volume of symmetrical grain boundaries (GB) under the effects of external strain in body-centered cubic (bcc) iron are investigated by the molecular dynamics (MD) method. The results indicate that without external strain, full MD relaxations at high temperatures are necessary to obtain the lower energy states of GBs, especially for GBs that have lost the symmetrical feature near GB planes following MD relaxations. Under external strain, two mechanisms are explored for the failure of these GBs, including slip system activation, dislocation nucleation and dislocation network formation induced directly by either the external strain field or by phase transformation from the initial bcc to fcc structure under the effects of external strain. Detailed analysis shows that the change in free volume is related to local structure changes in these two mechanisms, and can also lead to increases in local stress concentration. These findings provide a new explanation for the failure of GBs in BCC iron systems. read less USED (low confidence) W. Xie, C. Liu, G. Huang, D. Jiang, and J. Jin, “Nano-sized single-asperity friction behavior: Insight from molecular dynamics simulations,” European Journal of Mechanics - A/Solids. 2022. link Times cited: 3 USED (low confidence) Y. Han, L. Pan, H. Zhang, Y. Zeng, and Z. Yin, “Effect of lubricant additives of Cu, Fe and bimetallic CuFe nanoparticles on tribological properties,” Wear. 2022. link Times cited: 7 USED (low confidence) S. M. N. Souq, F. A. Ghasemi, and M. M. S. Fakhrabadi, “Effects of Various Cross Sections on Elastoplastic Behavior of Fe Nanowires under Tension/Compression,” Journal of Materials Engineering and Performance. 2022. link Times cited: 3 USED (low confidence) Z. Zhao, B. Safaei, Y. Wang, F.-H. Chu, and Y. Wei, “Grain boundary elimination by twinning and dislocation nucleation in front of intergranular crack tips in BCC iron,” Materials & Design. 2022. link Times cited: 4 USED (low confidence) H. Sun and L. Béland, “Statistical distribution of spontaneous recombination radii of Frenkel pairs in FCC and BCC metals,” Acta Materialia. 2022. link Times cited: 6 USED (low confidence) D. H. Ta, “Adhesive Strength of Hexadecane on Different Iron Compounds: an MD Approach,” Journal of Technical Education Science. 2022. link Times cited: 0 Abstract: The lubricity of alkane is a research target for numerous tr… read moreAbstract: The lubricity of alkane is a research target for numerous tribological applications in either industrial area or fundamental scientific studies. In the current work, a comparative investigation using a classical molecular dynamics (MD) method is carried out to investigate the effect of pure iron and its oxide surfaces on structural properties, adsorption ability of hexadecane (C16H34). A reliable force field (FF) of condensed-phase optimized molecular potentials for atomistic simulation studies (COMPASS) is employed to describe the intra- and intermolecular interactions for hexadecane and its interaction with iron oxide surfaces, while the interaction between hexadecane and pure iron is derived from an ab initio result. Regarding the surfaces, the pure iron surfaces are considered using embedded-atom method/Finnis-Sinclair potential (EAM/FS), while the iron oxide surfaces are constructed using the traditional Buckingham force field. The results reveal that hexadecane shows preferential adsorption on iron oxide surfaces compared to pure iron. read less USED (low confidence) Y. Chen and K. Morishita, “Molecular Dynamics Simulation of Defect Production in Fe due to Irradiation,” Nuclear Materials and Energy. 2022. link Times cited: 2 USED (low confidence) L. Sang, N. Sugimura, and H. Washizu, “Graphene as solid lubricant vertically buried into iron contact surface by annealing for superlubricity,” Tribology International. 2022. link Times cited: 15 USED (low confidence) Z. Wang et al., “New mechanisms of dislocation line-loop interactions in BCC-Fe explored by molecular dynamics method,” Results in Physics. 2022. link Times cited: 4 USED (low confidence) G. Poletaev and V. Kovalenko, “Development of Potentials for Description of Interatomic Interactions in Hadfield Steel for Molecular Dynamic Simulation,” Himičeskaâ fizika i mezoskopiâ. 2021. link Times cited: 0 Abstract: Summary. Hadfield steel, due to its excellent work hardening… read moreAbstract: Summary. Hadfield steel, due to its excellent work hardening ability read less USED (low confidence) J. Yin, H. Hou, J. Wang, X. Liu, and F. Xue, “Atomistic simulation of [100](001) crack propagation with Cu precipitates in α-iron,” International Journal of Pressure Vessels and Piping. 2021. link Times cited: 4 USED (low confidence) W. Lu et al., “Atomistic Simulation Study of the FCC and BCC Crystal-Melt Interface Stresses,” Surfaces and Interfaces. 2021. link Times cited: 4 USED (low confidence) G. F. B. Moladje, L. Thuinet, C. Becquart, and A. Legris, “Radiation induced segregation near dislocations and symmetric tilt grain boundaries in Fe-Cr alloys: a phase-field study,” Acta Materialia. 2021. link Times cited: 8 USED (low confidence) M. Kroll, T. Schmalofski, H. Dette, and R. Janisch, “Efficient Prediction of Grain Boundary Energies from Atomistic Simulations via Sequential Design,” Advanced Theory and Simulations. 2021. link Times cited: 1 Abstract: With the goal of improving data based materials design, it i… read moreAbstract: With the goal of improving data based materials design, it is shown that by a sequential design of experiment scheme the process of generating and learning from the data can be combined to discover the relevant sections of the parameter space. The application is the energy of grain boundaries as a function of their geometric degrees of freedom, calculated from a simple model, or via atomistic simulations. The challenge is to predict the deep cusps of the energy, which are located at irregular intervals of the geometric parameters. Existing sampling approaches either use large sets of datapoints or a priori knowledge of the cusps' positions. By contrast, the authors' technique can find unknown cusps automatically with a minimal amount of datapoints. Key point is a Kriging interpolator with Matérn kernel to estimate the energy function. Using the jackknife variance, the next point in the sequential design is a compromise between sampling the region of largest fluctuations and avoiding a clustering of datapoints. In this way, the cusps of the energy can be found within only a few iterations, and refined as desired. This approach will be advantageous for any application with strong, localized fluctuations in the values of the unknown function. read less USED (low confidence) Z. Zhao, Y. Wang, B. Safaei, H. Long, F.-H. Chu, and Y. Wei, “Orientation effect on intergranular fracture behaviors along symmetrical tilt grain boundaries in bcc iron,” Materials Today Communications. 2021. link Times cited: 4 USED (low confidence) Y. Wang et al., “Prediction of vacancy formation energies at tungsten grain boundaries from local structure via machine learning method,” Journal of Nuclear Materials. 2021. link Times cited: 6 USED (low confidence) H. Ghaffarian, Y.-E. Na, and D. Jang, “Interfacial Plasticity Mediated by Lath Boundaries in Reduced-Activation Ferritic/Martensitic Steels,” Journal of Nuclear Materials. 2021. link Times cited: 5 USED (low confidence) S. Stephan, M. Dyga, I. A. Alhafez, J. Lenhard, H. Urbassek, and H. Hasse, “Reproducibility of atomistic friction computer experiments: a molecular dynamics simulation study,” Molecular Simulation. 2021. link Times cited: 2 Abstract: ABSTRACT The elementary processes of friction in contact pro… read moreAbstract: ABSTRACT The elementary processes of friction in contact processes of two solid bodies occur on the nanoscale and are difficult to study experimentally. Therefore, molecular dynamics simulations are often used for their elucidation. In these studies, usually, only a single simulation is carried out for each scenario and the resulting observables are evaluated. In the present work, the reliability and reproducibility of measured observables from such nanoscopic contact process simulations are assessed by means of their statistical uncertainties. Therefore, the computer experiment is carried out not only once, but it is repeated eight times, with individual runs that only differ in the initial thermal motion. This set of replicas enables an assessment of observations for distinguishing reproducible physical phenomena from stochastic coincidence. In this way, a dry and a lubricated nanoscale scratching process were studied, in which a cylindrical indenter carried out two sequential movements: it first penetrates a substrate vertically and then scratches laterally, which causes elastic and plastic deformation of the substrate. In the lubricated case, the indenter was fully immersed in the fluid. Substrate, indenter, and fluid were described by suitably parametrised Lennard–Jones potentials. Various mechanical and thermodynamic process properties were monitored in all simulation runs. The results are compared and evaluated statistically. read less USED (low confidence) A. Kedharnath, R. Kapoor, and A. Sarkar, “Classical molecular dynamics simulations of the deformation of metals under uniaxial monotonic loading: A review,” Computers & Structures. 2021. link Times cited: 16 USED (low confidence) N. Zotov and B. Grabowski, “Molecular dynamics simulations of screw dislocation mobility in bcc Nb,” Modelling and Simulation in Materials Science and Engineering. 2021. link Times cited: 8 Abstract: The screw dislocation mobility in bcc Nb has been studied by… read moreAbstract: The screw dislocation mobility in bcc Nb has been studied by molecular dynamics (MD) simulations at different strain rates and temperatures using an embedded-atom method (EAM) potential. Static properties of the screw dislocation, as determined with the EAM potential, are in agreement with previous density-functional-theory calculations. The elementary slip plane of the screw dislocation remains (110) for all studied strain rates (in the range 6.3 × 107–6.3 × 109 s−1) and temperatures (5 to 550 K). However, the consecutive cross-slip on different symmetry-equivalent (110) planes leads to an effective glide on (112) planes. It is demonstrated that the screw dislocation trajectories, velocities and waviness of the screw dislocation depend on the crystallographic indices, (110) or (112), of the maximum resolved shear stress plane. The waiting time for the start of the screw dislocation motion increases exponentially with decreasing strain rate, substantiating the necessity to apply in future accelerated MD techniques in order to compare with macroscopic stress-strain experiments. read less USED (low confidence) Z. Yuan, J. Zhao, and S. Huang, “Molecular Dynamics Simulations on the Thermal Effect of Interfacial Friction During Asperity Shearing,” International Journal of Modern Physics C. 2021. link Times cited: 0 Abstract:
To explore the thermal effect of interfacial friction at t… read moreAbstract:
To explore the thermal effect of interfacial friction at the nano/microscale, a solid-solid contact model of a rough surface with a single peak was established to research single-crystal Fe. The friction characteristics, stress distributions, temperature changes, and energy changes under different indentation depths and lattice orientations during the shearing process were analyzed. From the perspective of temperature and energy, the mechanism of the thermal effect was revealed. The relationship between the friction force, temperature, and energy at the atomic scale was clarified. The results showed that the temperature of the asperities gradually increased during the shearing process, and a stress concentration formed in the shearing zone. After contact, the asperities had undergone unrecoverable plastic deformation, and there was wear at the contact interface accompanied by the loss of atoms from the asperity. At each indentation depth, as the rotation angle of the crystal increased, the friction force, average temperature, and the sum of the changes in thermal kinetic and thermal potential energy all first increased and then decreased; the trends of the three parameters changing with the rotation angle of the crystal were consistent. The average decreases in the friction force, average temperature, and the sum of the changes in thermal kinetic and thermal potential energy were 52.47%, 30.91%, and 56.75%, respectively, for a crystal structure with a rotation angle of 45° compared to a crystal structure with a rotation angle of 0°. The methods used in this study provide a reference for the design of frictional pairs and the reduction of the thermal effect of interfacial friction. read less USED (low confidence) J. Varillas, J. Očenášek, J. Torner, and J. Alcalá, “Understanding imprint formation, plastic instabilities and hardness evolutions in FCC, BCC and HCP metal surfaces,” Acta Materialia. 2021. link Times cited: 25 USED (low confidence) X. Zheng, L. Su, G. Deng, J. Zhang, H. Zhu, and A. K. Tieu, “Study on Lubrication Characteristics of C4-Alkane and Nanoparticle during Boundary Friction by Molecular Dynamics Simulation,” Metals. 2021. link Times cited: 5 Abstract: Lubricant has been widely applied to reduce wear and frictio… read moreAbstract: Lubricant has been widely applied to reduce wear and friction between the contact surfaces when they are in relative motion. In the current study, a nonequilibrium molecular dynamics (NEMD) simulation was specifically established to conduct a comprehensive investigation on the dynamic contact between two iron surfaces in a boundary friction system considering the mixed C4-alkane and nanoparticles as lubricant. The main research objective was to explore the effects of fluid and nanoparticles addition on the surface contact and friction force. It was found that nanoparticles acted like ball bearings between the contact surfaces, leading to a change of sliding friction mode to rolling friction mode. Under normal loads, plastic deformation occurred at the top surface because nanoparticles were mainly supporting the normal load. By increasing the number of C4-alkane molecules between two contact surfaces, the contact condition has been changed from partial to full lubrication. In addition, an attractive force from the solid–liquid LJ interaction between C4-alkane and surfaces was observed at the early stage of sliding, due to the large space formed by wall surfaces and nanoparticles. The findings in this paper would be beneficial for understanding the frictional behavior of a simple lubricant with or without nanoparticles addition in a small confinement. read less USED (low confidence) S. Jiang et al., “Effects of vacancies on plasticity and phase transformation in single-crystal iron under shock loading,” Journal of Applied Physics. 2021. link Times cited: 5 Abstract: A characteristic region with vacancy concentration ranging f… read moreAbstract: A characteristic region with vacancy concentration ranging from 0% to 2% was introduced into the single-crystal iron to investigate the effects of vacancies on plasticity and phase transformation of single-crystal iron under shock loading. The simulations were implemented by applying non-equilibrium molecular dynamics simulations with an excellent modified analytic embedded-atom method (MAEAM) potential. A fixed piston velocity of vp = 0.5 km/s was applied in our simulations, under which no plasticity or phase transformation occurred in the perfect single-crystal iron based on the description of the used MAEAM potential. The plasticity and phase transformation in iron were observably influenced by the vacancies as shown in this work. Significant anisotropy of shock response was distinctly exhibited. The nucleation and growth of dislocation loops emitting from the vacancy region were clearly observed in the sample that was shocked along the [110] direction, and the activated slip systems were determined as ( 11 2 ¯ )[111] and (112) [ 11 1 ¯ ]. The vacancies and the vacancies-induced dislocation loops provided preferential nucleation positions for the subsequent phase transformation, which resulted in the phenomenon that the phase transformation product (HCP phase) always preferentially appeared in the vacancy region. The influences of different vacancy concentrations on plasticity and phase transformation were also discussed. read less USED (low confidence) Q. Wu, Y. Wang, T. Han, H. Wang, L. Han, and L. Bao, “Molecular Dynamics Simulations of the Effect of Temperature and Strain Rate on the Plastic Deformation of Body-Centered Cubic Iron Nanowires,” Journal of Engineering Materials and Technology-transactions of The Asme. 2021. link Times cited: 4 Abstract:
The tensile tests of body-centered cubic (BCC) Fe nanowire… read moreAbstract:
The tensile tests of body-centered cubic (BCC) Fe nanowires were simulated through molecular dynamics methods. The temperature and strain rate effects on the mechanical properties as well as the orientation-dependent plastic deformation mechanism were analyzed. For [001]-oriented BCC Fe nanowires, as the temperature increased, the yield stress and Young’s modulus decreased. While the yield stress and Young’s modulus increased as the strain rate increased. With the increase in temperature, when the temperature was less than 400 K, the twin propagation stress decreased dramatically, and then tended to reach a saturation value at higher temperatures. Under different temperatures and strain rates, the [001]-oriented Fe nanowires all deformed by twinning. The oscillation stage in the stress–strain curve corresponds to the process from the nucleation of the twin to the reorientation of the nanowire. For [110]-oriented Fe nanowires, the plastic deformation is dominated by dislocation slip. The independent events such as the nucleation, slip, and annihilation of dislocations are the causes of the unsteady fluctuations in the stress–strain curve. The Fe nanowires eventually undergo shear damage along the dominant slip surface. read less USED (low confidence) S. Starikov et al., “Angular-dependent interatomic potential for large-scale atomistic simulation of iron: Development and comprehensive comparison with existing interatomic models,” Physical Review Materials. 2021. link Times cited: 16 Abstract: The development of classical interatomic potential for iron … read moreAbstract: The development of classical interatomic potential for iron is a quite demanding task with a long history background. A new interatomic potential for simulation of iron was created with a focus on description of crystal defects properties. In contrast with previous studies, here the potential development was based on force-matching method that requires only ab initio data as reference values. To verify our model, we studied various features of body-centered-cubic iron including the properties of point defects (vacancy and self-interstitial atom), the Peierls energy barrier for dislocations (screw and mix types), and the formation energies of planar defects (surfaces, grain boundaries, and stacking fault). The verification also implies thorough comparison of a potential with 11 other interatomic potentials reported in literature. This potential correctly reproduces the largest number of iron characteristics which ensures its advantage and wider applicability range compared to the other considered classical potentials. Here application of the model is illustrated by estimation of self-diffusion coefficients and the calculation of fcc lattice properties at high temperature. read less USED (low confidence) P. Lin, J. Nie, and M. Liu, “Investigation of the effects of point defects on the tensile strength of BCC-Fe using molecular dynamics,” Applied Physics A. 2021. link Times cited: 2 USED (low confidence) I. A. Alhafez, C. Ruestes, E. Bringa, and H. Urbassek, “Indentation and scratching of iron by a rotating tool – a molecular dynamics study,” Computational Materials Science. 2021. link Times cited: 7 USED (low confidence) M. Wakeda, Y. Chang, Seiichiro, and T. Ohmura, “Multiscale analyses of the interaction between dislocation and Σ9 symmetric tilt grain boundaries in Fe–Si bicrystals by nanoindentation technique,” International Journal of Plasticity. 2021. link Times cited: 6 USED (low confidence) W. Xie, C. Liu, D. Jiang, and J. Jin, “Inelastic contact behaviors of nanosized single-asperity and multi-asperity on α-Fe surface: Molecular dynamic simulations,” International Journal of Mechanical Sciences. 2021. link Times cited: 10 USED (low confidence) H. Luu, S. Dang, T. Hoang, and N. Gunkelmann, “Molecular dynamics simulation of nanoindentation in Al and Fe: On the influence of system characteristics,” Applied Surface Science. 2021. link Times cited: 33 USED (low confidence) J. Zhu, X. He, D.-S. Yang, Z. Bie, H. Mei, and X. Tian, “A peridynamic model for fracture analysis of polycrystalline BCC-Fe associated with molecular dynamics simulation,” Theoretical and Applied Fracture Mechanics. 2021. link Times cited: 2 USED (low confidence) J. Alizadeh, A. Salati, M. R. E. Fordoei, and M. Panjepour, “Investigation of Grain Boundary Influence on the Thermodynamic Phase Stability of Nanocrystalline Iron by Using the Molecular Dynamics Simulation Method,” Journal of Materials Engineering and Performance. 2021. link Times cited: 2 USED (low confidence) Z. Zhao, F. Chu, and Y. Wei, “Atomistic scale behaviors of crack propagation in nanocrystalline bcc iron,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2021. link Times cited: 7 USED (low confidence) L. Wu, H. Wang, Y. Zhu, and M. Li, “Crystal-melt coexistence in FCC and BCC metals: A molecular-dynamics study of crystal-melt interface free energies,” Materialia. 2021. link Times cited: 6 USED (low confidence) T. Yu, I. Chesser, S. Ratanaphan, E. Holm, S. Yang, and C. Deng, “Survey of shear coupling behavior in FCC Ni and BCC Fe grain boundaries,” Materialia. 2021. link Times cited: 7 USED (low confidence) M. M. Rahman, A. Ashikuzzaman, and R. Saha, “Atomistic investigation of the grain boundary effect on fracture mechanisms in BCC Fe bamboolike polycrystal nanowire.” 2021. link Times cited: 0 Abstract: Nanowires (NWs) have many enhanced properties such as high y… read moreAbstract: Nanowires (NWs) have many enhanced properties such as high yield strength, high ductility and large elongation under tensile loading. Though many molecular dynamics simulations have been performed exhibiting the benefit of having grain boundaries in nanowires, this study focuses on the mechanical properties of a specific grain formation similar to a structure of bamboo that indicates the opposite phenomena. In this experiment, BCC Fe nanowires were strained under tensile loading and with the assistance of molecular dynamics simulation it was demonstrated that the behavior of polycrystalline Fe NW exhibits dramatic differences from their single crystalline NW counterparts. In single crystalline Fe NW, dislocations were generated from the surface in the directions of specific planes for BCC materials; whereas in the case of nanowire with bamboolike polycrystalline shape, dislocation were generated from grain boundary due to the localized high stress and the heterogeneous character of atoms in the grain boundary. Ultimate strengths for 〈111〉, 〈1-1-2〉, 〈102〉 and 〈110〉 oriented single crystal NWs were found 34.57 GPa, 19.15 GPa, 14.94 GPa and 25.48 GPa respectively. A substantial low strength (8.89 GPa) was observed for the polycrystalline Fe structure, which was mainly caused due to the deformation in the interface of the two subsequent grains. Necking was observed followed by fracture, resulting in an overall drop in toughness. These findings can be a matter of concern in certain applications where materials with high strength and stability are required. read less USED (low confidence) Y. Yang, S. Li, X. Ding, and J. Sun, “Pseudoelasticity in twinned α-Fe nanowires under bending,” Computational Materials Science. 2020. link Times cited: 3 USED (low confidence) G. F. B. Moladje, L. Thuinet, C. Becquart, and A. Legris, “A phase field model for dislocation climb under irradiation: Formalism and applications to pure bcc iron and ferritic alloys,” International Journal of Plasticity. 2020. link Times cited: 14 USED (low confidence) Z. Wang, X. Shi, X. Yang, W. He, S. Shi, and X. Ma, “Atomistic simulation of the effect of the dissolution and adsorption of hydrogen atoms on the fracture of α-Fe single crystal under tensile load,” International Journal of Hydrogen Energy. 2020. link Times cited: 7 USED (low confidence) P. Grützmacher, C. Gachot, and S. Eder, “Visualization of microstructural mechanisms in nanocrystalline ferrite during grinding,” Materials & Design. 2020. link Times cited: 23 USED (low confidence) G. F. B. Moladje, L. Thuinet, C. Domain, C. Becquart, and A. Legris, “Phase-field calculations of sink strength in Al, Ni, and Fe: A detailed study of elastic effects,” Computational Materials Science. 2020. link Times cited: 8 USED (low confidence) S. Zahabi, N. Nouri, S. Ziaei-Rad, and M. S. Talaei, “Effect of iron bicrystal orientation on mechanical properties and dislocation density using molecular dynamics simulations of nanoindentation,” Mechanics of Advanced Materials and Structures. 2020. link Times cited: 1 Abstract: This study concerns the effect of bicrystal spatial orientat… read moreAbstract: This study concerns the effect of bicrystal spatial orientation on the iron Σ5 boundary under nanoindentation tests. The orientation of the bicrystal is changed relative to the indenter by the parameter “nanoindentation angle” to create dislocation collisions with grain boundary in different directions and to determine the possibility of transmission of these dislocations through the grain boundary. Simulations were carried out for nanoindentation angles between −90° and 90° with a resolution of 18°. Molecular dynamics with EAM atomic potential functions are employed for the simulations. The effect of nanoindentation angle on the crystals Young’s modulus, shear stress, hardness, and Poisson’s ratio were determined. Due to the periodicity which has been observed in all of the aforementioned quantities of the rotated crystal, analytical equations were extracted by adapting the Fourier series expansion on the calculated data. Some explanations were provided for the dislocation behaviors based on the physics of the problem and also by the results reported in the literature. Simultaneous effects of Poisson’s ratio and pile-ups on each other were also investigated and interpreted. read less USED (low confidence) F. Shuang and K. Aifantis, “Using molecular dynamics to determine mechanical grain boundary energies and capture their dependence on residual Burgers vector, segregation and grain size,” Acta Materialia. 2020. link Times cited: 14 USED (low confidence) M. Z. H. Khan, L. Khanal, and Y. Qiang, “Radiation Effect on BCC Fe Nanoparticle with Varying Radiation Energy by Molecular Dynamics Simulation,” Chemical Engineering (Engineering) eJournal. 2020. link Times cited: 3 Abstract: Molecular dynamics simulations were conducted to explore pri… read moreAbstract: Molecular dynamics simulations were conducted to explore primary radiation damages on body-centered cubic (BCC) Fe nanoparticle. A series of 6 cascades for each primary knock-on atom (PKA) energy (5 keV, 10 keV, 20 keV, 30 keV and 40 keV) was simulated to assure statistical precision. It has been observed that defects created due to the interactions have stayed as a single to several size clusters. Among them, most of the clusters are either single interstitials (SIAs) or vacancies (Vs). In each of the energy cases, it produces one block of a big cluster. The block cluster of SIAs stays at the near surface of the nanoparticle, however, Vs stay inside the nanoparticle. The study has shown that the total number of vacancy defects is larger than the total number of interstitial defects because more PKA energy gives more mobility that some SIAs can get enough energy to leave the Fe nanoparticle. read less USED (low confidence) Y. Wei, N. Gao, Z. Shen, C. Chen, Z. Xie, and L. Guo, “Interactions between hydrogen bubbles and prismatic interstitial dislocation loops in BCC iron,” Computational Materials Science. 2020. link Times cited: 1 USED (low confidence) A. Bondarev, A. Fraile, T. Polcar, and D. Shtansky, “Mechanisms of friction and wear reduction by h-BN nanosheet and spherical W nanoparticle additives to base oil: Experimental study and molecular dynamics simulation,” Tribology International. 2020. link Times cited: 35 USED (low confidence) X. Xing et al., “Atomistic simulation of hydrogen-induced plastic zone compression during cyclic loading,” International Journal of Hydrogen Energy. 2020. link Times cited: 8 USED (low confidence) S. Peeters, P. Restuccia, S. Loehlé, B. Thiébaut, and M. Righi, “Tribochemical Reactions of MoDTC Lubricant Additives with Iron by Quantum Mechanics/Molecular Mechanics Simulations,” The Journal of Physical Chemistry C. 2020. link Times cited: 19 Abstract: The remarkable lubricant properties of molybdenum dithiocarb… read moreAbstract: The remarkable lubricant properties of molybdenum dithiocarbamates (MoDTCs) make this class of oil additives well-known in the automotive industry. However, the mechanism of function of these compo... read less USED (low confidence) M. Zhang, J. Chen, K. Sun, and L. Fang, “The effect of point defects caused by particle bombardment on the deformation behaviours of alpha-iron: A molecular dynamics simulation,” Materials Chemistry and Physics. 2020. link Times cited: 6 USED (low confidence) I. A. Alhafez, C. Ruestes, E. Bringa, and H. Urbassek, “Influence of pre-existing plasticity on nanoindentation – an atomistic analysis of the dislocation fields produced,” Journal of the Mechanics and Physics of Solids. 2019. link Times cited: 14 USED (low confidence) S. Shinzato, M. Wakeda, and S. Ogata, “An atomistically informed kinetic Monte Carlo model for predicting solid solution strengthening of body-centered cubic alloys,” International Journal of Plasticity. 2019. link Times cited: 26 USED (low confidence) S. Starikov, M. Mrovec, and R. Drautz, “Study of Grain Boundary Self-Diffusion in Iron with Different Atomistic Models,” MatSciRN: Computational Studies of Inorganic & Organic Materials (Topic). 2019. link Times cited: 22 USED (low confidence) Y. Sato, S. Shinzato, T. Ohmura, and S. Ogata, “Atomistic prediction of the temperature- and loading-rate-dependent first pop-in load in nanoindentation,” International Journal of Plasticity. 2019. link Times cited: 32 USED (low confidence) A. Kedharnath, R. Kapoor, and A. Sarkar, “Atomistic simulation of interaction of collision cascade with different types of grain boundaries in α-Fe,” Journal of Nuclear Materials. 2019. link Times cited: 14 USED (low confidence) R. Essajai et al., “Structural and magnetic properties of iron nanoparticles: insights from Monte-Carlo and molecular-statics simulations,” Materials Research Express. 2019. link Times cited: 9 Abstract: In this Letter, the structural stability, local structure ch… read moreAbstract: In this Letter, the structural stability, local structure characterization and magnetic properties of iron nanoparticles (FeNPs) were investigated by using a combination of Molecular Statics (MS) and Monte Carlo (MC) simulations. To do so, six kinds of spherical-shaped FeNPs with diameters in the range of 3.14 to 5.42 nm have been considered. The coordination number distribution of FeNPs obtained from the data extracted by MS simulations was exploited for performing MC simulations on the familiar Ising model. The numerical findings obtained in the current study show that the structural and magnetic properties correlate with the size of the FeNP. read less USED (low confidence) Y. Qi, Y. Che, S. Pan, and H. Zhang, “Study on micro thermodynamic process of gas flow in pulse tube by unequilibrium molecular dynamics simulations,” International Journal of Heat and Mass Transfer. 2019. link Times cited: 5 USED (low confidence) T. Sharma, N. Kumar, R. Mondal, K. Krishna, I. Samajdar, and V. Kain, “Ductile-to-Brittle Transition in Low-Alloy Steel: A Combined Experimental and Numerical Investigation,” Journal of Materials Engineering and Performance. 2019. link Times cited: 5 USED (low confidence) J. Delgado, “A molecular dynamics study of nanocontact plasticity and dislocation avalanches in FCC and BCC crystals.” 2019. link Times cited: 3 Abstract: This study aims to investigate the underlying mechanisms whi… read moreAbstract: This study aims to investigate the underlying mechanisms which govern the development of dense defect networks in nanoscale crystal plasticity, either under contact and uniaxial loading conditions, with emphasis on the onset of intermittent avalanche phenomena. The investigation is based on a comprehensive set of massive molecular dynamics (MD) simulations performed with embedded-atom method potentials in face-centered cubic (FCC) and body-centered cubic (BCC) crystals.
The first part of the thesis concerns the combined role of elasticity and plasticity in nanocontact loadings, where attention is given to the mechanisms leading to the formation of a permanent nanoimprint as well as to the onset of material pile-up at the contact vicinity. It is found that the topographical arrangement of the slip traces emitted at the surface into specific deformation patterns is a distinctive feature of the underlying dislocation glide and twinning processes occurring in FCC and BCC crystals as a function of temperature and surface orientation. A mechanistic analysis is made on the influence of the defect nucleation events in conjunction with the development of entangled defect networks upon the material hardness and its evolution towards a plateau level with increasing indenter-tip penetration. Complementary MD simulations of the uniaxial stress-strain curve of the plastically deformed region are carried out with the purpose of establishing a direct correlation between nanoscale material responses attaining under uniaxial and contact loading conditions. The results of this comparison illustrate on the key role played by defect nucleation processes on the formation of permanent nanoimprints, which differs from the conventional view in that in micro and macroscopic scales imprint formation is essentially governed by the evolutionary character of a preexisting (entangled) defect network: the greater the dislocation density, the larger the measured hardness. In overall, this work provides a fundamental insight into the understanding of why BCC surfaces are harder than FCC surfaces at the nanoscale.
A statistical physics background is devised to investigate the influence of the dislocation mechanisms on the onset of avalanche events that are inherent to crystal plasticity. The analysis is predicated upon the notion in that the size distribution of such avalanches follows power-law scaling. To investigate the avalanche size distributions in cubic crystals, a group of novel MD simulations are performed where the computational cells containing a periodic arrangement of a preexisting dislocation network are subjected to uniaxial straining under displacement control at different strain rates and temperatures. Under sufficiently slow driving, the dislocation networks evolve through the emission of dislocation avalanches which do not overlap in time. This illustrates that the mobilized entangled dislocation arrangements exhibit quiescent periods during each plastic (dissipative) event, enabling comparison with experimental results which are also performed under strict displacement controlled conditions. The results illustrate on the attainment of a transitional slip size separating two power-law avalanche regimes as a function of the fundamental dislocation glide processes at the crossroads of self-organized and tuned criticality models. Detailed analyses of the MD simulations furnish specific mechanisms characterizing dislocation avalanche emission and propagation in FCC and BCC metals throughout a wide temperature range, which is central in supporting the onset of the aforementioned two power-law regimes.
En este estudio se investigan los mecanismos fundamentales para el desarrollo de las densas redes de defectos que se producen durante la deformación plástica de metales mediante ensayos uniaxiales y de indentación en escalas nanométricas. Estos procesos de deformación plástica se caracterizan por la producción de eventos intermitentes o avalanchas de dislocaciones. La investigación se basa en un extenso grupo de simulaciones de dinámica molecular en las que se emplean potenciales interatómicos del tipo embedded-atom method en cristales cúbicos centrados en las caras (CCC) y cúbicos centrados en el cuerpo (CC). La primera parte de esta tesis discute el papel combinado de la elasticidad y plasticidad en los nanocontactos. Se presta una especial atención a los mecanismos que llevan a la formación de nanohuellas plásticas así como al desarrollo de apilamiento de material alrededor del nanocontacto. Se encuentra que los arreglos topográficos de trazas de deslizamiento (emitidas a la superficie) muestran patrones específicos de deformación, los cuales son a su vez un rasgo distintivo de los mecanismos de deslizamiento de dislocaciones y procesos de nanomaclado que ocurren en los materiales CCC y CC en función de la temperatura y la orientación de la superficie. Se presenta un estudio mecanístico sobre la influencia de los eventos de nucleación de defectos, que llevan al desarrollo de una compleja red de defectos, sobre la nanodureza y su convergencia hacia un valor relativamente constante a medida que el indentador penetra en la superficie La modelización del comportamiento uniaxial de la zona deformada debajo de las nanoindentaciones permite la correlación entre ambos tipos de ensayos. Los resultados de esta comparación ilustran el importante papel que juegan los procesos de nucleación de dislocaciones sobre la formación de nanohuellas plásticas, lo que difiere (en términos mecanísticos) del comportamiento plástico convencional encontrado en escales micro y macroscópicas, donde el carácter evolutivo de una red de defectos preexistente gobierna la formación de huella, cumpliéndose así que cuanto mayor es la densidad de defectos, mayores son también las macro y microdurezas. En general, este trabajo aporta un trasfondo fundamental para comprender la razón por la que las superficies CC son más duras que las CCC en la nano escala. En la última parte de esta investigación se utilizan modelos de física estadística para investigar la influencia de los mecanismos de propagación de dislocaciones sobre la emisión de avalanchas plásticas. El análisis se basa en la noción de que la distribución del tamaño de las avalanchas sigue una ley potencial universal. Para investigar esta distribución en cristales cúbicos, se realizan un grupo de simulaciones novedosas donde las celdas computaciones, que contienen arreglos periódicos de las redes de dislocaciones, son sometidas a cargas uniaxiales a diferentes temperaturas y velocidades de deformación. A velocidades de deformación suficientemente lentas, las redes de dislocaciones evolucionan a través de la emisión de avalanchas que no se sobreponen en el tiempo, lo que ilustra que la movilización de las redes ocurre de tal manera que se garantiza una alternancia entre periodos de inactividad y cada evento plástico. La comparación entre resultados experimentales y computacionales lleva a encontrar la existencia de una magnitud de deslizamiento crítico que separa a dos regímenes de avalanchas cuya distribución de tamaños obedece leyes potenciales. Este resultado demuestra que los procesos de avalanchas son claramente dependientes de los mecanismos de deslizamiento e interacción de dislocaciones presentes en el material; aspecto que describe la transición entre el modelo de criticalidad gobernada por la tensión y el de criticalidad auto-organizada. Las simulaciones muestran los mecanismos específicos que caracterizan la emisión y propagación de avalanchas en metales CC y CCC en un amplio rango de temperatura, lo que es de gran importancia para justificar la utilización de estos modelos de criticalidad. read less USED (low confidence) T. Suzudo, T. Onitsuka, and K. Fukumoto, “Analyzing the cross slip motion of screw dislocations at finite temperatures in body-centered-cubic metals: molecular statics and dynamics studies,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 13 Abstract: The plasticity of body-centered-cubic metals at low temperat… read moreAbstract: The plasticity of body-centered-cubic metals at low temperatures is substantially determined by the screw-dislocation kinetics. Because the core of screw dislocations in these metals has a non-planar structure, its motion is complex. For example, although density functional theory predicts slip on a {110} plane, the actual slip plane at elevated temperatures differs from the prediction. In this work, we explored state-of-the-art atomistic modeling methods and successfully reproduced the transition of the slip plane through a temperature increase. We then devised an algorithm to analyze the activation of dislocation jump over the Peierls barrier and discovered a possible origin of this unexpected phenomenon: thermal fluctuation leads to the kink-pair nucleation for cross slip jumps with no transition of the dislocation core structure. read less USED (low confidence) X. Wang et al., “Molecular dynamics study on the Burgers vector transition of nanometric dislocation loops induced by cascade in bcc-iron,” Journal of Nuclear Materials. 2019. link Times cited: 7 USED (low confidence) H. Luu and N. Gunkelmann, “Pressure-induced phase transformations in Fe-C: Molecular dynamics approach,” Computational Materials Science. 2019. link Times cited: 24 USED (low confidence) S. M. Zamzamian, M. Samadfam, S. Feghhi, and A. Arjhangmehr, “Atomistic simulation of the effect of carbon content and carbon-rich region on irradiation response of α-Fe on picosecond timescale,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2019. link Times cited: 7 USED (low confidence) Z. Zhao and F. Chu, “Dynamic meshing force and blunt effects on atomistic scale ductile-brittle behavior of the crack in bcc iron,” Engineering Fracture Mechanics. 2019. link Times cited: 9 USED (low confidence) L. Fu and H. Fang, “Atomistic investigation of hydrogen embrittlement effect for symmetric and asymmetric grain boundary structures of bcc Fe,” Computational Materials Science. 2019. link Times cited: 5 USED (low confidence) Z. Zhao, Z.-ye Qin, and F. Chu, “Atomistic scale fracture behavior of the bcc iron with 1 1 0 crack under dynamic rectangular loading rate,” Computational Materials Science. 2019. link Times cited: 12 USED (low confidence) C. Zheng, R. Schoell, P. Hosemann, and D. Kaoumi, “Ion irradiation effects on commercial PH 13-8 Mo maraging steel Corrax,” Journal of Nuclear Materials. 2019. link Times cited: 3 USED (low confidence) A. Hosseini, M. N. Nasrabadi, and A. Esfandiarpour, “Investigation of primary radiation damage near free surfaces in iron nanofoam with a model cylindrical nanovoids structure,” Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2019. link Times cited: 5 USED (low confidence) K. Ueno and Y. Shibuta, “Solute partition at solid-liquid interface of binary alloy from molecular dynamics simulation,” Materialia. 2018. link Times cited: 6 USED (low confidence) Y. Fan and P. Cao, “Long Time-Scale Atomistic Modeling and Simulation of Deformation and Flow in Solids,” Handbook of Materials Modeling. 2018. link Times cited: 5 USED (low confidence) S. Rawat and P. M. Raole, “Molecular dynamics investigation of void evolution dynamics in single crystal iron at extreme strain rates,” Computational Materials Science. 2018. link Times cited: 23 USED (low confidence) T. Lu et al., “Atomistic study of hydrogen behavior around dislocations in α iron,” Journal of Nuclear Materials. 2018. link Times cited: 18 USED (low confidence) J. Aldazabal, I. Aldazabal, and J. G. Sevillano, “Elasto-plastic behaviour of a columnar structure of nanocrystalline iron with sharp 〈011〉 fibre texture,” Materialia. 2018. link Times cited: 4 USED (low confidence) M. Mock and K. Albe, “Modelling of dislocation-solute interaction in ODS steels: Analytic bond-order potential for the iron-yttrium system,” Journal of Nuclear Materials. 2018. link Times cited: 6 USED (low confidence) K. Lu et al., “Grain Boundary Plays a Key Role in Carbon Diffusion in Carbon Irons Revealed by a ReaxFF Study,” The Journal of Physical Chemistry C. 2018. link Times cited: 25 Abstract: Carbon diffusion is a critical process to the manufacture of… read moreAbstract: Carbon diffusion is a critical process to the manufacture of many industry products, such as iron carbides, stainless steels, and carbon materials. Here we investigate carbon diffusion induced by tensile, screw dislocation, edge dislocation, and polycrystal boundary through reactive molecular dynamics simulations with ReaxFF potentials. The temperature enhances the dynamics and therefore the carbon diffusion. The pre-existing defects promote the carbon diffusion with a linear relationship between carbon diffusion barrier and strain as well as line defect concentrations. Furthermore, we also observed a linear relationship between the carbon diffusion barrier and the volume fractions of the polycrystalline boundary, indicating that the grain boundary mechanism is prominent in carbon diffusion in the carbon iron. read less USED (low confidence) S. Jung, Y. Kwon, C. Lee, and B.-J. Lee, “Influence of hydrogen on the grain boundary crack propagation in bcc iron: A molecular dynamics simulation,” Computational Materials Science. 2018. link Times cited: 26 USED (low confidence) J. Yin, Y. Wang, X. Yan, H. Hou, and J. Wang, “Atomistic simulation of shear-coupled motion of [1 1 0] symmetric tilt grain boundary in α-iron,” Computational Materials Science. 2018. link Times cited: 9 USED (low confidence) X. Zhao, C. Lu, A. K. Tieu, L. Zhan, L. Pei, and M. Huang, “Deformation mechanisms and slip-twin interactions in nanotwinned body-centered cubic iron by molecular dynamics simulations,” Computational Materials Science. 2018. link Times cited: 11 USED (low confidence) E. Baibuz et al., “Data sets of migration barriers for atomistic Kinetic Monte Carlo simulations of Fe self-diffusion,” Data in Brief. 2018. link Times cited: 1 USED (low confidence) Q.-ul-ain Sahi and Y.-S. Kim, “Primary radiation damage characterization of α-iron under irradiation temperature for various PKA energies,” Materials Research Express. 2018. link Times cited: 17 Abstract: The understanding of radiation-induced microstructural defec… read moreAbstract: The understanding of radiation-induced microstructural defects in body-centered cubic (BCC) iron is of major interest to those using advanced steel under extreme conditions in nuclear reactors. In this study, molecular dynamics (MD) simulations were implemented to examine the primary radiation damage in BCC iron with displacement cascades of energy 1, 5, 10, 20, and 30 keV at temperatures ranging from 100 to 1000 K. Statistical analysis of eight MD simulations of collision cascades were carried out along each [110], [112], [111] and a high index [135] direction and the temperature dependence of the surviving number of point defects and the in-cascade clustering of vacancies and interstitials were studied. The peak time and the corresponding number of defects increase with increasing irradiation temperature and primary knock-on atom (PKA) energy. However, the final number of surviving point defects decreases with increasing lattice temperature. This is associated with the increase of thermal spike at high PKA energy and its long timespan at higher temperatures. Defect production efficiency (i.e., surviving MD defects, per Norgett-Robinson-Torrens displacements) also showed a continuous decrease with the increasing irradiation temperature and PKA energy. The number of interstitial clusters increases with both irradiation temperature and PKA energy. However, the increase in the number of vacancy clusters with PKA energy is minimal-to-constant and decreases as the irradiation temperature increases. Similarly, the probability and cluster size distribution for larger interstitials increase with temperature, whereas only smaller size vacancy clusters were observed at higher temperatures. read less USED (low confidence) H. Wang, J. Tian, W. Zhou, X. Chen, B. Bai, and J. Xue, “Collision cascades interact with an edge dislocation in bcc Fe: a molecular dynamics study,” RSC Advances. 2018. link Times cited: 16 Abstract: The interactions of an edge dislocation (ED) with collision … read moreAbstract: The interactions of an edge dislocation (ED) with collision cascades induced by 5 keV primary knocked-on atoms (PKAs) towards the ED in bcc Fe are studied using classical molecular dynamics (MD) simulations. It is found that the number and distribution of the residual point defects are related to the distance between the initial PKAs and the ED. Based on this result, we provide a comprehensive summary of four characteristic phenomena for cascade–ED interactions, including few interactions, the formation of a vacancy cluster, the sink effect for point defects, and the sub-cascade area affection, depending on the overlap of the peak cascades' area with the ED line. Then a qualitative model is proposed to clearly elucidate the underlying mechanisms of the four situations. Considering that dislocations constitute an essential part of the micro-structure of crystalline solids, our work demonstrates that: the pre-existing dislocations in crystalline materials could induce diverse effects under irradiation environments, which should be taken into account for designing and improving the radiation resistant materials. read less USED (low confidence) V. P. Ramunni, “Hydrogen migration modeling in a symmetric tilt boundary of the Iron-Chromium system,” Journal of Nuclear Materials. 2018. link Times cited: 4 USED (low confidence) I. A. Alhafez and H. Urbassek, “Orientation dependence in nanocutting of Fe single crystals: A molecular-dynamics study,” Computational Materials Science. 2018. link Times cited: 10 USED (low confidence) I. Svistunov and A. S. Kolokol, “An analysis of interatomic potentials for vacancy diffusion simulation in concentrated Fe-Cr alloys.” 2018. link Times cited: 1 Abstract: В данном исследовании проверялась корректность работы трех м… read moreAbstract: В данном исследовании проверялась корректность работы трех межатомных потенциалов взаимодействия, доступных в научной литературе, в молекулярно-динамическом моделировании вакансионной диффузии в концентрированных сплавах Fe–Cr. Проведенная работа была необходима для дальнейшего детального исследования механизма вакансионной диффузии в данных сплавах с содержанием хрома 5–25 ат.% в температурном диапазоне 600–1000 К. Анализ был выполнен на моделях сплава с содержанием хрома 10, 20, 50 ат.%. Рассмотрение модели сплава с 50 ат.% хрома было необходимо для дальнейшего исследования диффузионных процессов в обогащенных хромом преципитатах данных сплавов. Для всех потенциалов были рассчитаны и проанализированы энергии формирования вакансии в сплавах и диффузионные подвижности атомов железа и хрома через искусственно созданную одиночную вакансию. В качестве основной характеристики для анализа подвижностей атомов была выбрана временная зависимость их среднеквадратичного смещения. Моделирование энергий формирования вакансий не выявило качественных различий между исследуемыми моделями потенциалов. Проведенное исследование атомных подвижностей показало плохое воспроизведение диффузии вакансии в исследуемых сплавах концентрационно-зависимой моделью (CDM), которая сильно занижала подвижность атомов хрома через вакансию во всем исследуемом диапазоне температур и концентраций хрома. Установлено, что двусвязная модель потенциала (2BM) в своей оригинальной и модифицированной версии подобных недостатков не имеет. Это позволяет использовать эти потенциалы в моделированиях вакансионного механизма диффузии в исследуемых сплавах. Для обоих 2BM-потенциалов была зафиксирована существенная зависимость соотношения подвижностей хрома и железа от температуры и содержания хрома в сплавах. Количественные данные коэффициентов диффузии атомов, полученные этими потенциалами, также существенно различаются. read less USED (low confidence) X. Meng, C. Fang, and K. Niu, “Tribological behavior anisotropy in sliding interaction of asperities on single-crystal α-iron: A quasi-continuum study,” Tribology International. 2018. link Times cited: 10 USED (low confidence) A. Kohnert, M. Cusentino, and B. Wirth, “Molecular statics calculations of the biases and point defect capture volumes of small cavities,” Journal of Nuclear Materials. 2018. link Times cited: 16 USED (low confidence) M. Ahmadi, B. Sonderegger, S. D. Yadav, and M. Poletti, “Modelling and simulation of diffusion driven pore formation in martensitic steels during creep,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2018. link Times cited: 9 USED (low confidence) J. Fikar, R. Gröger, and R. Schäublin, “Effect of orientation of prismatic dislocation loops on interaction with free surfaces in BCC iron,” Journal of Nuclear Materials. 2017. link Times cited: 8 USED (low confidence) Z. Zhao and F. Chu, “Atomic behaviors of crack propagation in bcc iron under dynamic loading rate with rectangular fluctuation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2017. link Times cited: 17 USED (low confidence) Y. Yang, X. Wang, G. Zhang, Y. Zhang, and Z. Yang, “Molecular dynamics simulations of single crystal copper nanocubes under triaxial tensile loading,” Computational Materials Science. 2017. link Times cited: 13 USED (low confidence) G. Sainath and B. Choudhary, “Atomistic simulations on ductile-brittle transition in ⟨111⟩ BCC Fe nanowires,” Journal of Applied Physics. 2017. link Times cited: 23 Abstract: Molecular dynamics simulations have been performed to unders… read moreAbstract: Molecular dynamics simulations have been performed to understand the influence of temperature on the tensile deformation and fracture behavior of ⟨111⟩ BCC Fe nanowires. The simulations have been carried out at different temperatures in the range 10–1000 K employing a constant strain rate of 1 × 108 s−1. The results indicate that at low temperatures (10–375 K), the nanowires yield through the nucleation of a sharp crack and fails in brittle manner. On the other hand, nucleation of multiple 1/2⟨111⟩ dislocations at yielding followed by significant plastic deformation leading to ductile failure has been observed at high temperatures in the range 450–1000 K. At 400 K, the nanowire yields through nucleation of crack associated with many mobile 1/2⟨111⟩ and immobile ⟨100⟩ dislocations at the crack tip and fails in ductile manner. The ductile-brittle transition observed in ⟨111⟩ BCC Fe nanowires is appropriately reflected in the stress-strain behavior and plastic strain at failure. The ductile-brittle transitio... read less USED (low confidence) R. Kositski and D. Mordehai, “Role of dislocation pile-ups in nucleation-controlled size-dependent strength of Fe nanowires,” Acta Materialia. 2017. link Times cited: 22 USED (low confidence) A. Mutter, B. Wang, J. Meiser, P. Umstätter, and H. Urbassek, “Magnetic structure of [0 0 1] tilt grain boundaries in bcc Fe studied via magnetic potentials,” Philosophical Magazine. 2017. link Times cited: 4 Abstract: Using magnetic potentials and a molecular statics approach, … read moreAbstract: Using magnetic potentials and a molecular statics approach, we study the changes in the magnetic structure of bcc Fe in the vicinity of grain boundaries (GBs). We focus on symmetric tilt GBs around the [0 0 1] axis with a tilt angle between 7 and 53. We find that immediately in the GB plane, the deviations in the magnetic moments from the bulk value are most pronounced. The distribution of moments in the GB plane is modulated according to the periodicity of the coincidence site lattice. In the direction perpendicular to the GB plane, the moments decay exponentially towards the bulk value; the decay length increases with decreasing tilt angle. This dependence can be explained by the well-known stress field around GBs. read less USED (low confidence) M. A. Ghaffari, Y. Zhang, and S. Xiao, “Molecular dynamics modeling and simulation of lubricant between sliding solids.” 2017. link Times cited: 11 Abstract: This paper presents molecular dynamic modeling and simulatio… read moreAbstract: This paper presents molecular dynamic modeling and simulation of lubricant between sliding solids. Linear n-alkanes with united atoms were used to model lubricant while iron sliding solids were modeled with body-centered cubic crystal lattices. We employed various potential functions, including the embedded atom method, the multibody force field and the Lennard–Jones potential, to approximate the interatomic interactions in the molecular model. Hydrodynamic lubrication was considered in this paper. We found that the temperature and the chain length of alkanes had effects on the friction between lubricated sliding solids. In addition, one debris, modeled as a nanoparticle, was added in the lubricant to study its effect on the friction. It was observed that nanoparticles would increase the friction in hydrodynamic lubrication. read less USED (low confidence) R. Khanna, Sahajwalla, and S. Seetharaman, “Atomistic Monte Carlo Simulations on the Melting Transition of Iron at Ambient Pressure.” 2017. link Times cited: 2 Abstract: Atomistic computer simulations are playing an increasingly i… read moreAbstract: Atomistic computer simulations are playing an increasingly important role in high temperature processes, bridging the gap between theory and experiment, and taking cutting edge research into unchartered territories. A fundamental understanding of the melting behaviour of BCC iron is of great significance in steelmaking operations. We report Monte Carlo simulations on the melting transition of iron using three well known interaction potentials, namely the Rosato potential, the Mendelev potential and the Sutton-Chen potential. Depending on the potential used, the interactions between iron atoms varied from 1/r2, as well as 1/r8.14 at short distances, and the range of applicability was found to vary from 3.5 A to 9.5 A. These atomic level computer simulations were carried out using a range of simulation variables such as the size of the simulation cell, step size, boundary conditions and statistical ensemble. The melting transition was identified clearly in all cases through discontinuities in energy and increases in local disorder. The simulation data however showed a significant scatter. Interaction potentials of iron based on system characteristics at 0 K were found somewhat limited for quantitative high temperature simulations (~1800K). Iron potentials developed for earth science applications were not found to be suitable for these investigations. These computer simulations therefore point towards a significant gap in the knowledge base in the atomistic modelling of molten iron, especially for steelmaking applications. read less USED (low confidence) H.-jie Yan, J.-cai Zhuang, P. Zhou, Q. Li, C. Q. Zhou, and P. Fu, “Molecular dynamics simulation of thermal physical properties of molten iron,” International Journal of Heat and Mass Transfer. 2017. link Times cited: 18 USED (low confidence) A. Korchuganov, K. Zolnikov, and D. S. Kryzhevich, “Atomistic simulation of structural damage during ion irradiation of iron single crystals,” Journal of Physics: Conference Series. 2017. link Times cited: 1 Abstract: The evolution of atomic displacement cascades initiated near… read moreAbstract: The evolution of atomic displacement cascades initiated near free surfaces with different crystallographic orientations in bcc iron specimens was studied on the base of the molecular dynamics approach. The craters surrounded by adatom mounds were formed in the case of the (111) surface irradiation. The dislocation loops consisted of vacancies were generated after the (110) surface irradiation. The dislocation Burgers vector was a/2 <111> or a <100>. It was shown that the type of structural damage is determined by the anisotropy of propagation of shock waves generated by atomic displacement cascades. For energies of the atomic displacement cascade lower than 20 keV the number of adatoms and survived point defects was higher for specimen with the (110) free surface due to the different character of surface damage. The increase in the cascade energy up to 20 keV results in formation of almost the equal number of survived point defects for the (110) and (111) free surfaces as displacement cascades were developed on larger distance from the irradiated surfaces. read less USED (low confidence) B. D. Snartland, A. B. Hagen, and C. Thaulow, “Fracture mechanical testing of single crystal notched α-iron micro-cantilevers,” Engineering Fracture Mechanics. 2017. link Times cited: 11 USED (low confidence) J. Ewen, S. Restrepo, N. Morgan, and D. Dini, “Nonequilibrium molecular dynamics simulations of stearic acid adsorbed on iron surfaces with nanoscale roughness,” Tribology International. 2017. link Times cited: 52 USED (low confidence) S. Eder, D. Bianchi, U. Cihak-Bayr, and K. Gkagkas, “Methods for atomistic abrasion simulations of laterally periodic polycrystalline substrates with fractal surfaces,” Comput. Phys. Commun. 2017. link Times cited: 19 USED (low confidence) X. Xing, W. Chen, and H. Zhang, “Atomistic study of hydrogen embrittlement during cyclic loading: Quantitative model of hydrogen accumulation effects,” International Journal of Hydrogen Energy. 2017. link Times cited: 27 USED (low confidence) S. Alkan and H. Sehitoglu, “Non-Schmid response of Fe3Al: The twin-antitwin slip asymmetry and non-glide shear stress effects,” Acta Materialia. 2017. link Times cited: 23 USED (low confidence) A. Dutta, “Compressive deformation of Fe nanopillar at high strain rate: Modalities of dislocation dynamics,” Acta Materialia. 2017. link Times cited: 33 USED (low confidence) J. Skogsrud and C. Thaulow, “Effect of crystallographic orientation on nanomechanical modelling of an iron single crystal cracked cantilever beam,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2017. link Times cited: 13 USED (low confidence) X. Xing, M. Yu, W. Chen, and H. Zhang, “Atomistic simulation of hydrogen-assisted ductile-to-brittle transition in α-iron,” Computational Materials Science. 2017. link Times cited: 29 USED (low confidence) G. Lv and Y. Su, “Molecular dynamics simulation and first principles calculations of radiation-induced Cu clusters in Fe-3 at.% Cu alloy,” Comput. Phys. Commun. 2017. link Times cited: 5 USED (low confidence) D. Medlin, K. Hattar, J. Zimmerman, F. Abdeljawad, and S. Foiles, “Defect character at grain boundary facet junctions: Analysis of an asymmetric Σ = 5 grain boundary in Fe,” Acta Materialia. 2017. link Times cited: 47 USED (low confidence) A. Silva et al., “Confined chemical and structural states at dislocations in Fe–9wt%Mn steels: A correlative TEM-atom probe study combined with multiscale modelling,” Acta Materialia. 2017. link Times cited: 71 USED (low confidence) M. Abu-Shams, W. Haider, and I. Shabib, “Evolution of displacement cascades in Fe–Cr structures with different [001] tilt grain boundaries,” Radiation Effects and Defects in Solids. 2017. link Times cited: 3 Abstract: ABSTRACT Reduced-activation ferritic/martensitic steels of C… read moreAbstract: ABSTRACT Reduced-activation ferritic/martensitic steels of Cr concentration between 2.25 and 12 wt% are candidate structural materials for next-generation nuclear reactors. In this study, molecular dynamics (MD) simulation is used to generate the displacement cascades in Fe–Cr structures with different Cr concentrations by using different primary knock-on atom (PKA) energies between 2 and 10 keV. A concentration-dependent model potential has been used to describe the interactions between Fe and Cr. Single crystals (SCs) of three different coordinate bases (e.g. [310], [510], and [530]) and bi-crystal (BC) structures with three different [001] tilt grain boundaries (GBs) (e.g. Σ5, Σ13, and Σ17) have been simulated. The Wigner–Seitz cell criterion has been used to identify the produced Frenkel pairs. The results show a marked difference between collisions observed in SCs and those in BC structures. The numbers of vacancies and interstitials are found to be significantly higher in BC structures than those found in SCs. The number of point defects exhibits a power relationship with the PKA energies; however, the Cr concentration does not seem to have any influence on the number of survived point defects. In BC models, a large fraction of the total survived point defects (between 59% and 93%) tends accumulate at the GBs, which seem to trap the generated point defects. The BC structure with Σ17 GB is found to trap more defects than Σ5 and Σ13 GBs. The defect trapping is found to be dictated by the crystallographic parameters of the GBs. For all studied GBs, self-interstitial atoms (SIAs) are easily trapped within the GB region than vacancies. An analysis of defect composition reveals an enrichment of Cr in SIAs, and in BC cases, more than half of the Cr-SIAs are found to be located within the GB region. read less USED (low confidence) A. B. Hagen and C. Thaulow, “Low temperature in-situ micro-compression testing of iron pillars,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2016. link Times cited: 25 USED (low confidence) Y. Gao and H. Urbassek, “Scratching of nanocrystalline metals: A molecular dynamics study of Fe,” Applied Surface Science. 2016. link Times cited: 34 USED (low confidence) S. Y. Korostelev, S. Psakhie, E. E. Slyadnikov, and I. Y. Turchanovskii, “Simulation of a nonequilibrium phase transition initiated by the volumetric heat source using a molecular dynamics method.” 2016. link Times cited: 4 Abstract: A nonequilibrium phase transition (melting) initiated by the… read moreAbstract: A nonequilibrium phase transition (melting) initiated by the volumetric heat source is investigated using a molecular dynamics method (MMD). Physical parameters characterizing the study process are computed. It is shown that at the critical level of the heat source power density the phase transition is realized under nonequilibrium conditions of considerable local overheating and may be accompanied by the formation of the locally unstable state of a microcrystal and complex dynamics: the origin of phase transformation fronts both on the surface and deep within the microcrystal. read less USED (low confidence) M. S. Talaei, N. Nouri, and S. Ziaei-Rad, “Grain boundary effects on nanoindentation of Fe bicrystal using molecular dynamic,” Mechanics of Materials. 2016. link Times cited: 19 USED (low confidence) O. Kuskov and D. Belashchenko, “Thermodynamic properties of Fe-S alloys from molecular dynamics modeling: Implications for the lunar fluid core,” Physics of the Earth and Planetary Interiors. 2016. link Times cited: 15 USED (low confidence) S. Kim, D. T. Ho, K. Kang, and S. Y. Kim, “Phonon scattering during dislocation motion inducing stress-drop in cubic metals,” Acta Materialia. 2016. link Times cited: 9 USED (low confidence) D. Terentyev, A. Zinovev, and G. Bonny, “Displacement cascades in FeNiMnCu alloys: RVP model alloys,” Journal of Nuclear Materials. 2016. link Times cited: 7 USED (low confidence) R. Hoffman, A. P. Moore, and C. Deo, “Examination of the Effect of Vacancy Detachment Rates on Kinetic Monte Carlo Simulations of bcc Metals,” MRS Advances. 2016. link Times cited: 1 Abstract: A Kinetic Monte Carlo simulation, using a modified version o… read moreAbstract: A Kinetic Monte Carlo simulation, using a modified version of the SPPARKS code, of simple defects and complex vacancy clusters was run on a bcc lattice. In this simulation the complexity of void formation was varied by introducing a detachment rate for individual vacancies leaving the void and either treating this value as constant for all size voids or having this value be dependent on the size of the void. Molecular Dynamics simulations were used to determine the binding energies of vacancies for voids of varying size. The simulation was then run over long time periods to determine the number of defects in the simulation under irradiation conditions. It was found that the additional complexity of size dependent void detachment rates had little effect on the defect concentrations and thus a constant barrier should be sufficient for simulations of voids in bcc metals. read less USED (low confidence) L. You, L. Hu, Y. Xie, and S. Zhao, “Influence of Cu precipitation on tensile properties of Fe–Cu–Ni ternary alloy at different temperatures by molecular dynamics simulation,” Computational Materials Science. 2016. link Times cited: 14 USED (low confidence) G. A. Nematollahi, B. Grabowski, D. Raabe, and J. Neugebauer, “Multiscale description of carbon-supersaturated ferrite in severely drawn pearlitic wires,” Acta Materialia. 2016. link Times cited: 37 USED (low confidence) O. Kuskov and D. Belashchenko, “Molecular dynamics estimates for the thermodynamic properties of the Fe–S liquid cores of the Moon, Io, Europa, and Ganymede,” Solar System Research. 2016. link Times cited: 4 USED (low confidence) M. Dholakia, S. Chandra, and S. Jaya, “Molecular dynamics studies of displacement cascades in Fe-Y2TiO5 system.” 2016. link Times cited: 1 Abstract: The effect of displacement cascade on Fe-Y2TiO5 bilayer is s… read moreAbstract: The effect of displacement cascade on Fe-Y2TiO5 bilayer is studied using classical molecular dynamics simulations. Different PKA species – Fe, Y, Ti and O – with the same PKA energy of 8 keV are used to produce displacement cascades that encompass the interface. It is shown that Ti atom has the highest movement in the ballistic regime of cascades which can lead to Ti atoms moving out of the oxide clusters into the Fe matrix in ODS alloys. read less USED (low confidence) G. Bonny, D. Terentyev, J. Elena, A. Zinovev, B. Minov, and E. Zhurkin, “Assessment of hardening due to dislocation loops in bcc iron: Overview and analysis of atomistic simulations for edge dislocations,” Journal of Nuclear Materials. 2016. link Times cited: 24 USED (low confidence) A. Löfgren, P. Zeiger, V. Kocevski, and J. Rusz, “Influence of nuclear quantum effects on frozen phonon simulations of electron vortex beam HAADF-STEM images.,” Ultramicroscopy. 2016. link Times cited: 9 USED (low confidence) H. Song and J. Hoyt, “A molecular dynamics study of heterogeneous nucleation at grain boundaries during solid-state phase transformations,” Computational Materials Science. 2016. link Times cited: 29 USED (low confidence) O. Kuskov and D. Belashchenko, “Molecular dynamics estimates for the thermodynamic properties of the Fe–S liquid cores of the Moon, Io, Europa, and Ganymede,” Solar System Research. 2016. link Times cited: 0 USED (low confidence) A. Al-Motasem, N. Mai, S. Choi, and M. Posselt, “Atomistic study on mixed-mode fracture mechanisms of ferrite iron interacting with coherent copper and nickel nanoclusters,” Journal of Nuclear Materials. 2016. link Times cited: 10 USED (low confidence) T. Okita, S. Hayakawa, M. Itakura, M. Aichi, S. Fujita, and K. Suzuki, “Conservative climb motion of a cluster of self-interstitial atoms toward an edge dislocation in BCC-Fe,” Acta Materialia. 2016. link Times cited: 15 USED (low confidence) S. Pan, S. Feng, J. Qiao, W.-M. Wang, and J. Qin, “Crystallization pathways of liquid-bcc transition for a model iron by fast quenching,” Scientific Reports. 2015. link Times cited: 29 USED (low confidence) F. T. Latypov and A. Mayer, “Shear strength of metals under uniaxial deformation and pure shear,” Journal of Physics: Conference Series. 2015. link Times cited: 5 Abstract: In this paper, we investigate the dynamic shear strength of … read moreAbstract: In this paper, we investigate the dynamic shear strength of perfect monocrystalline metals using the molecular dynamics simulation. Three types of deformation (single shear, uniaxial compression and tension) are investigated for five metals of different crystallographic systems (fcc, bcc and hcp). A strong dependence of the calculated shear strength on the deformation type is observed. In the case of bcc (iron) and hcp (titanium) metals, the maximal shear strength is achieved at the uniaxial compression, while the minimal shear strength is observed at the uniaxial tension. In the case of fcc metals (aluminum, copper, nickel) the largest strength is achieved at the pure shear, the lowest strength is obtained at the uniaxial compression. read less USED (low confidence) A. Esfandiarpour, S. Feghhi, and A. Shokri, “Effects of atomic grain boundary structures on primary radiation damage in α-Fe,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2015. link Times cited: 21 USED (low confidence) D. Kryzhevich, A. Korchuganov, K. Zolnikov, and S. Psakhie, “Plastic deformation nucleation in BCC crystallites under nanoindentation.” 2015. link Times cited: 0 Abstract: Molecular dynamics investigation of metal crystallite with b… read moreAbstract: Molecular dynamics investigation of metal crystallite with bcc lattice under nanoindentation was carried out. Potentials of interatomic interactions were calculated on the base of the approximation of the Finnis-Sinclair method. For clarity and simpler indentation data interpretation, an extended cylindrical indenter was used in the investigation. The features of the bcc iron structural response at nanoindentation of surfaces with different crystallographic orientations were revealed. Generation of structural defects in the contact zone always resulted in the decrease in the rate of growth of the reaction force. read less USED (low confidence) J. Fikar and R. Gröger, “Interactions of prismatic dislocation loops with free surfaces in thin foils of body-centered cubic iron,” Acta Materialia. 2015. link Times cited: 12 USED (low confidence) S. Eder, U. Cihak-Bayr, and A. Pauschitz, “Nanotribological simulations of multi-grit polishing and grinding,” Wear. 2015. link Times cited: 34 USED (low confidence) S. Huang, M. Feng, B. Wang, X. Xu, X. Cao, and Y. Wang, “Molecular dynamics simulation on the fabrication of graphene nanoscrolls with ferromagnetic nanowire templates,” Applied Surface Science. 2015. link Times cited: 2 USED (low confidence) G. Sainath and B. Choudhary, “Molecular dynamics simulations on size dependent tensile deformation behaviour of [110] oriented body centred cubic iron nanowires,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2015. link Times cited: 37 USED (low confidence) V. P. Ramunni and A. Rivas, “Diffusion behavior of Cr diluted in bcc and fcc Fe: Classical and quantum simulation methods,” Materials Chemistry and Physics. 2015. link Times cited: 12 USED (low confidence) M. Chiapetto, L. Malerba, and C. Becquart, “Nanostructure evolution under irradiation in FeMnNi alloys: A ‘grey alloy’ object kinetic Monte Carlo model,” Journal of Nuclear Materials. 2015. link Times cited: 21 USED (low confidence) E. Asadi and M. A. Zaeem, “Quantifying a Two-Mode Phase-Field Crystal Model for BCC Metals at Melting Point,” Computational Materials Science. 2015. link Times cited: 15 USED (low confidence) G. Sainath, B. Choudhary, and T. Jayakumar, “Molecular dynamics simulation studies on the size dependent tensile deformation and fracture behaviour of body centred cubic iron nanowires,” Computational Materials Science. 2015. link Times cited: 73 USED (low confidence) Y. Gao, A. Brodyanski, M. Kopnarski, and H. Urbassek, “Nanoscratching of iron: A molecular dynamics study of the influence of surface orientation and scratching direction,” Computational Materials Science. 2015. link Times cited: 66 USED (low confidence) T. D. Ta, A. K. Tieu, H. Zhu, and B. Kosasih, “Adsorption of Normal-Alkanes on Fe(110), FeO(110), and Fe2O3(0001): Influence of Iron Oxide Surfaces,” Journal of Physical Chemistry C. 2015. link Times cited: 45 Abstract: A comparative analysis of adsorption of six normal-alkanes (… read moreAbstract: A comparative analysis of adsorption of six normal-alkanes (CNH2N+2, N = 4, 6, 8, 10, 12, 16) on Fe(110), FeO(110), and Fe2O3(0001) was carried out using classical molecular dynamics (MD) simulation. A realistic model system for adsorbed alkanes was employed using the COMPASS force field (FF), while the appropriate relaxed surfaces and an effective interfacial potential were obtained from ab initio calculations. The results show that butane molecules orient randomly on Fe(110) and Fe2O3(0001) surfaces, but they preferentially orient in the (010) direction on FeO(110) at low temperature. Additionally, alkanes adsorb physically on Fe(110), FeO(110), and Fe2O3(0001), in the following decreasing order Fe(110) > FeO(110) > Fe2O3(0001). The adsorption energies per saturated carbon site decrease with an increase of molecular chain length, and this propensity is similar for different surface potentials. In contrast, the saturated carbon density is insensitive to the surface potentials and shows an increasing tren... read less USED (low confidence) R. Kositski and D. Mordehai, “Depinning-controlled plastic deformation during nanoindentation of BCC iron thin films and nanoparticles,” Acta Materialia. 2015. link Times cited: 23 USED (low confidence) Y. Zhang, P. Millett, M. Tonks, X. Bai, and S. B. Biner, “Preferential Cu precipitation at extended defects in bcc Fe: An atomistic study,” Computational Materials Science. 2015. link Times cited: 22 USED (low confidence) R. Murzaev, A. Kistanov, V. Dubinko, D. Terentyev, and S. Dmitriev, “Moving discrete breathers in bcc metals V, Fe and W,” Computational Materials Science. 2015. link Times cited: 43 USED (low confidence) P. Franciosi, L. T. Le, G. Monnet, C. Kahloun, and M.-H. Chavanne, “Investigation of slip system activity in iron at room temperature by SEM and AFM in-situ tensile and compression tests of iron single crystals,” International Journal of Plasticity. 2015. link Times cited: 85 USED (low confidence) D. Terentyev, X. He, G. Bonny, A. Bakaev, E. Zhurkin, and L. Malerba, “Hardening due to dislocation loop damage in RPV model alloys: Role of Mn segregation,” Journal of Nuclear Materials. 2015. link Times cited: 36 USED (low confidence) W. Gao, L. Kong, and P. Hodgson, “Molecular Dynamics Simulation of Heat Transfer during Quenching in CNT Nanofluids,” Materials Performance and Characterization. 2015. link Times cited: 0 Abstract: Nanofluids exhibit superior thermal properties to convention… read moreAbstract: Nanofluids exhibit superior thermal properties to conventional fluid and particle-fluid suspensions and show a great potential as quenching media for quench hardening of steel components. The heat transfer mechanism in nanofluid is very complex and unclear. In this paper, molecular dynamics (MD) simulation method is used to theoretically study the heat transfer from a metal surface at different temperatures to a water-based nanofluid with functionalized carbon nanotubes (FCNTs). To model the quenching process, an initial temperature jump between the nanofluid and an iron slab is employed, and non-equilibrium molecular dynamics (NEMD) simulations are performed. The MD results reveal the heat transfer process in the initial stage of quenching and at the first moment of contact of a liquid nanofluid with a hot metal surface. The thermodynamics and transport properties of the nanofluid and the heat transfer characteristics are discussed with the atomistic details of the interactions of the FCNT with the iron atoms and the water molecules. read less USED (low confidence) E. Asadi, M. A. Zaeem, S. Nouranian, and M. Baskes, “Quantitative Modeling of the Equilibration of Two-Phase Solid-Liquid Fe by Atomistic Simulations on Diffusive Time Scales,” Physical Review B. 2015. link Times cited: 61 Abstract: (Received 10 July 2014; revised manuscript received 10 Decem… read moreAbstract: (Received 10 July 2014; revised manuscript received 10 December 2014; published 12 January 2015) In this paper, molecular dynamics (MD) simulations based on the modified-embedded atom method (MEAM) and a phase-field crystal (PFC) model are utilized to quantitatively investigate the solid-liquid properties of Fe. A set of second nearest-neighbor MEAM parameters for high-temperature applications are developed for Fe, and the solid-liquid coexisting approach is utilized in MD simulations to accurately calculate the melting point, expansion in melting, latent heat, and solid-liquid interface free energy, and surface anisotropy. The required input properties to determine the PFC model parameters, such as liquid structure factor and fluctuations of atoms in the solid, are also calculated from MD simulations. The PFC parameters are calculated utilizing an iterative procedure from the inputs of MD simulations. The solid-liquid interface free energy and surface anisotropy are calculated using the PFC simulations. Very good agreement is observed between the results of our calculations from MEAM-MD and PFC simulations and the available modeling and experimental results in the literature. As an application of the developed model, the grain boundary free energy of Fe is calculated using the PFC model and the results are compared against experiments. read less USED (low confidence) Y. Qi, L. Wang, S. Wang, X. Li, and W. Cui, “Structural and dynamical heterogeneity of undercooled Fe75Cu25 melts with miscibility gap,” Journal of Alloys and Compounds. 2014. link Times cited: 11 USED (low confidence) H. Khater, G. Monnet, D. Terentyev, and A. Serra, “Dislocation glide in Fe–carbon solid solution: From atomistic to continuum level description,” International Journal of Plasticity. 2014. link Times cited: 31 USED (low confidence) Y. Gao and H. Urbassek, “Evolution of plasticity in nanometric cutting of Fe single crystals,” Applied Surface Science. 2014. link Times cited: 31 USED (low confidence) S. Eder, D. Bianchi, U. Cihak-Bayr, A. Vernes, and G. Betz, “An analysis method for atomistic abrasion simulations featuring rough surfaces and multiple abrasive particles,” Comput. Phys. Commun. 2014. link Times cited: 41 USED (low confidence) R. Matsumoto, S. Seki, S. Taketomi, and N. Miyazaki, “Hydrogen-related phenomena due to decreases in lattice defect energies—Molecular dynamics simulations using the embedded atom method potential with pseudo-hydrogen effects,” Computational Materials Science. 2014. link Times cited: 18 USED (low confidence) G. Bonny, D. Terentyev, E. Zhurkin, and L. Malerba, “Monte Carlo study of decorated dislocation loops in FeNiMnCu model alloys,” Journal of Nuclear Materials. 2014. link Times cited: 45 USED (low confidence) Y. Gao, C. Ruestes, and H. Urbassek, “Nanoindentation and nanoscratching of iron: Atomistic simulation of dislocation generation and reactions,” Computational Materials Science. 2014. link Times cited: 114 USED (low confidence) S. M. H. Haghighat et al., “Influence of the dislocation core on the glide of the ½ 110 edge dislocation in bcc-iron: An embedded atom method study,” Computational Materials Science. 2014. link Times cited: 14 USED (low confidence) X. Zheng, H. Zhu, A. K. Tieu, and K. Chen, “Molecular dynamics simulation of confined n-alkanes: ordered structure and crystalline bridges,” International Journal of Surface Science and Engineering. 2014. link Times cited: 6 Abstract: A molecular dynamics simulation of confined n-alkanes with d… read moreAbstract: A molecular dynamics simulation of confined n-alkanes with different chain length and thickness has been performed. It discusses the film structure and shear stress of thin film at the atomic scale. Specifically, layered structure in thin film is observed due to the strong liquid-solid interaction and small confinement. An explicit two-layer lubricant film that has ‘tetratic’ order is observed. Without crystalline bridges, the shear stress drops dramatically. For a thin film with up to three layers, the shear stress varies with thickness. This is due to that the number of crystalline bridges varies between ordered layers. When the film thickness increases further, the effect of the interfacial interaction towards the inner film becomes blunt, and the shear stress becomes stable and is dominated by the lubricant chain length. read less USED (low confidence) E. Sak-Saracino and H. Urbassek, “Free energies of austenite and martensite Fe–C alloys: an atomistic study,” Philosophical Magazine. 2014. link Times cited: 12 Abstract: We investigate the influence of C interstitials on the phase… read moreAbstract: We investigate the influence of C interstitials on the phase stability of Fe–C crystals. We employ the Meyer–Entel interatomic interaction potential which is able to reproduce the austenite-martensite phase transition for pure Fe, and supplement it by a simple pairwise Fe–C interaction potential. Using two different thermodynamic methods, we calculate the free energies of the martensite and austenite phases. We find that C destabilizes the ground-state bcc phase. The decrease in the equilibrium transformation temperature with increasing C content parallels the one found in the experiment. This destabilization is found even if C is added for a potential in which only the bcc phase is stable until the melting point; here, for sufficiently high C addition, a stable fcc phase is established in the phase diagram. read less USED (low confidence) S. M. H. Haghighat, R. Schäublin, and D. Raabe, “Atomistic simulation of the a0 binary junction formation and its unzipping in body-centered cubic iron,” Acta Materialia. 2014. link Times cited: 22 USED (low confidence) M. Tikhonchev, V. Svetukhin, and E. Gaganidze, “MD simulation of atomic displacement cascades near chromium-rich clusters in FeCr alloy,” Journal of Nuclear Materials. 2013. link Times cited: 23 USED (low confidence) D. Terentyev, G. Bonny, C. Domain, G. Monnet, and L. Malerba, “Mechanisms of radiation strengthening in Fe–Cr alloys as revealed by atomistic studies,” Journal of Nuclear Materials. 2013. link Times cited: 52 USED (low confidence) G. Bonny et al., “On the thermal stability of late blooming phases in reactor pressure vessel steels: An atomistic study,” Journal of Nuclear Materials. 2013. link Times cited: 77 USED (low confidence) R. Schäublin and S. M. H. Haghighat, “Molecular dynamics study of strengthening by nanometric void and Cr alloying in Fe,” Journal of Nuclear Materials. 2013. link Times cited: 18 USED (low confidence) D. Kryzhevich, A. V. Korhuganov, K. Zolnikov, and S. G. Psakhye, “Structural Response of Metal Crystallite with BCC Lattice on Atomic Level under Nanoindentation,” Key Engineering Materials. 2013. link Times cited: 1 Abstract: Molecular dynamics investigation of metal crystallite with b… read moreAbstract: Molecular dynamics investigation of metal crystallite with bcc lattice under nanoindentation was carried out. Potentials of interatomic interactions were calculated on the base of the approximation of the embedded atom method. The potentials chosen make it possible to describe with a high accuracy the elastic and surface properties of the simulated metal and energy parameters of defects, which is important for solution of the task posed in the work. For clarity and simpler indentation data interpretation, an extended cylindrical indenter was used in the investigation and loading was realized by its lateral surface. The simulated crystallite had a parallelepiped shape. The loaded plane of crystallite was modeled as a free surface while the positions of atoms in the opposite plane of crystallite were fixed along the indentation direction. Other planes of crystallite were simulated as free surfaces. The indenter velocity varied from 5 to 25 m/s in different calculations. The loading of the model crystallite was realized at 300 K. Influence of interfaces (free surfaces and grain boundaries) on peculiarities of plastic deformation nucleation and interactions of generated structural defects with interfaces in simulated crystallite under nanoindentation were investigated. read less USED (low confidence) N. Lazarev and A. Bakai, “Atomistic simulation of primary damages in Fe, Ni and Zr,” Journal of Supercritical Fluids. 2013. link Times cited: 9 USED (low confidence) Y. Qi, L. Wang, and T. Fang, “Demixing behaviour in binary Cu-Co melt,” Physics and Chemistry of Liquids. 2013. link Times cited: 8 Abstract: Molecular dynamics simulation has been performed to explore … read moreAbstract: Molecular dynamics simulation has been performed to explore the structure, thermodynamics and dynamics properties of Cu-Co melt based upon embedded atom method (EAM). The pair correlation function of liquid Cu50Co50 show stronger interaction of homogeneous atom pairs. The coordination number (CN) of Cu-Cu and Co-Co in Cu50Co50 melt is a little higher than that of Cu-Co at temperature of 1800 K. The calculated enthalpy of mixing is positive in the whole concentration range and S CC(q) increases sharply at lower q, which are the typical features of dense fluid that exhibits phase segregation tendency. The interdiffusion coefficient shows same concentration dependence as that of demixing alloy. Our work indicates that Cu-Co melt exhibits weak demixing behaviour even at temperatures greater than those of bimodal curve. read less USED (low confidence) X.-yan Li et al., “Energetic and kinetic behaviors of small vacancy clusters near a symmetric Σ5(3 1 0)/[0 0 1] tilt grain boundary in bcc Fe,” Journal of Nuclear Materials. 2013. link Times cited: 24 USED (low confidence) X. Shu et al., “Fe self-diffusion and Cu and Ni diffusion in bulk and grain boundary of Fe: A molecular dynamics study,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2013. link Times cited: 10 USED (low confidence) J. Liu, R. Davidchack, and H. Dong, “Molecular dynamics calculation of solid–liquid interfacial free energy and its anisotropy during iron solidification,” Computational Materials Science. 2013. link Times cited: 41 USED (low confidence) X. Zheng, H. Zhu, B. Kosasih, and A. K. Tieu, “A molecular dynamics simulation of boundary lubrication: The effect of n-alkanes chain length and normal load,” Wear. 2013. link Times cited: 58 USED (low confidence) P. Staikov and N. Djourelov, “Simulations of 〈1 0 0〉 edge and 1/2〈1 1 1〉 screw dislocations in α-iron and tungsten and positron lifetime calculations,” Physica B-condensed Matter. 2013. link Times cited: 23 USED (low confidence) I. Vatne, A. Stukowski, C. Thaulow, E. Østby, and J. Marian, “Three-dimensional crack initiation mechanisms in bcc-Fe under loading modes I, II and III,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2013. link Times cited: 54 USED (low confidence) B. Radhakrishnan, S. Gorti, D. Nicholson, and J. Dantzig, “Comparison of Phase Field Crystal and Molecular Dynamics Simulations for a Shrinking Grain,” Journal of Physics: Conference Series. 2012. link Times cited: 8 Abstract: The Phase-Field Crystal (PFC) model represents the atomic de… read moreAbstract: The Phase-Field Crystal (PFC) model represents the atomic density as a continuous function, whose spatial distribution evolves at diffusional, rather than vibrational time scales. PFC provides a tool to study defect interactions at the atomistic level but over longer time scales than in molecular dynamics (MD). We examine the behavior of the PFC model with the goal of relating the PFC parameters to physical parameters of real systems, derived from MD simulations. For this purpose we model the phenomenon of the shrinking of a spherical grain situated in a matrix. By comparing the rate of shrinking of the central grain using MD and PFC we obtain a relationship between PFC and MD time scales for processes driven by grain boundary diffusion. The morphological changes in the central grain including grain shape and grain rotation are also examined in order to assess the accuracy of the PFC in capturing the evolution path predicted by MD. read less USED (low confidence) D. Molnár et al., “Multiscale simulations on the coarsening of Cu-rich precipitates in α-Fe using kinetic Monte Carlo, molecular dynamics and phase-field simulations,” Acta Materialia. 2012. link Times cited: 59 USED (low confidence) T. Swinburne, S. Dudarev, S. Fitzgerald, M. Gilbert, and A. Sutton, “Theory and simulation of the diffusion of kinks on dislocations in bcc metals,” Physical Review B. 2012. link Times cited: 58 Abstract: Isolated kinks on thermally fluctuating (1/2) screw, edge an… read moreAbstract: Isolated kinks on thermally fluctuating (1/2) screw, edge and (1/2) edge dislocations in bcc iron are simulated under zero stress conditions using molecular dynamics (MD). Kinks are seen to perform stochastic motion in a potential landscape that depends on the dislocation character and geometry, and their motion provides fresh insight into the coupling of dislocations to a heat bath. The kink formation energy, migration barrier and friction parameter are deduced from the simulations. A discrete Frenkel-Kontorova-Langevin (FKL) model is able to reproduce the coarse grained data from MD at a fraction of the computational cost, without assuming an a priori temperature dependence beyond the fluctuation-dissipation theorem. Analytic results reveal that discreteness effects play an essential r\^ole in thermally activated dislocation glide, revealing the existence of a crucial intermediate length scale between molecular and dislocation dynamics. The model is used to investigate dislocation motion under the vanishingly small stress levels found in the evolution of dislocation microstructures in irradiated materials. read less USED (low confidence) D. Terentyev, E. Zhurkin, and G. Bonny, “Emission of full and partial dislocations from a crack in BCC and FCC metals: An atomistic study,” Computational Materials Science. 2012. link Times cited: 18 USED (low confidence) H. Xu, Y. Osetsky, and R. Stoller, “Cascade annealing simulations of bcc iron using object kinetic Monte Carlo,” Journal of Nuclear Materials. 2012. link Times cited: 34 USED (low confidence) Y. Zhang, Y. Zhao, and F. Zhang, “Effect of Orientation Relationships on the Stiffness and the Strength of the Dual-Phase Metals: Molecular Dynamics Simulation,” Advanced Materials Research. 2012. link Times cited: 0 Abstract: Phase boundary is an important kind of interfaces for the du… read moreAbstract: Phase boundary is an important kind of interfaces for the dual-phase metals. Orientation relationship (OR) is a crucial factor affecting the performance of the phase boundaries and the dual-phase metals. Molecular dynamics simulation is performed to examine the structures of the bcc/fcc iron phase boundaries in Nishiyama–Wassermann (N–W) and Kurdjumov–Sachs (K–S) orientation relationships, and the performances of the dual-phase model with these ORs. The structural relaxation shows that the phase boundary with N-W relation has the lower energy than that in K-S relation. Stress-strain curves show that dual-phase model in N-W relation has the higher stiffness and strength than that in K-S relation. Simulation results show that phase boundary in N-W relation has a more exellent performance, and is preferred to be processed in heat treatment. read less USED (low confidence) E. Hayward and C. Deo, “Atomic Scale Modeling of Hydrogen and Helium in BCC Iron,” Fusion Science and Technology. 2012. link Times cited: 2 Abstract: Understanding the interactions of hydrogen and helium within… read moreAbstract: Understanding the interactions of hydrogen and helium within ferritic steels will aid in the development of materials appropriate for next generation nuclear reactors. We discuss interatomic potentials appropriate for simulating these elements, presenting a potential for the H-He interactions. Preliminary results for small H-He bubbles in bcc iron are given and discussed. read less USED (low confidence) W. Xie, X. Liu, W. Chen, and H. Zhang, “Hydrogen hardening effect in heavily deformed single crystal α-Fe,” Computational Materials Science. 2011. link Times cited: 27 USED (low confidence) D. Terentyev, L. Malerba, G. Bonny, A. Al-Motasem, and M. Posselt, “Interaction of an edge dislocation with Cu–Ni-vacancy clusters in bcc iron,” Journal of Nuclear Materials. 2011. link Times cited: 24 USED (low confidence) J. Maisonneuve, T. Oda, and S. Tanaka, “Molecular Statics Study of Hydrogen Isotope Trapping in BCC-Iron Vacancy Clusters,” Fusion Science and Technology. 2011. link Times cited: 10 Abstract: The stability of hydrogen atoms trapped in vacancy clusters … read moreAbstract: The stability of hydrogen atoms trapped in vacancy clusters of a bcc iron structure is investigated by molecular statics calculations of the hydrogen binding energy to these clusters. The configurations having a minimum potential energy are obtained from the relaxation of a large number of different initial atomic configurations. Calculations of hydrogen binding energy to a mono-vacancy illustrate a relatively large gain of energy in trapping up to two hydrogen atoms in a monovacancy and the increasing difficulty to trap additional atoms due to hydrogen mutual repulsion. Comparison with ab-initio reference calculations of the hydrogen binding energy shows good agreement for up to three trapped hydrogen atoms. Based on the calculations conducted on the most stable vacancy-hydrogen complexes containing two to six vacancies, the maximum capacity of hydrogen atoms per vacancy was found to decrease with the size of vacancy cluster. The calculations of hydrogen binding energies to these clusters show that trapping two hydrogen atoms per vacancy is still a particularly favorable process for vacancy clusters. read less USED (low confidence) W. Xie, “Hydrogen induced hardening effects on alpha iron: a molecular dynamics study.” 2011. link Times cited: 0 USED (low confidence) S. M. H. Haghighat, D. Terentyev, and R. Schäublin, “Atomistic simulation of the influence of Cr on the mobility of the edge dislocation in Fe(Cr) alloys,” Journal of Nuclear Materials. 2011. link Times cited: 16 USED (low confidence) D. Stewart, Y. Osetskiy, and R. Stoller, “Atomistic studies of formation and diffusion of helium clusters and bubbles in BCC iron,” Journal of Nuclear Materials. 2011. link Times cited: 82 USED (low confidence) G. Bonny, D. Terentyev, and L. Malerba, “Interaction of screw and edge dislocations with chromium precipitates in ferritic iron: An atomistic study,” Journal of Nuclear Materials. 2011. link Times cited: 21 USED (low confidence) S. Cuesta-López and J. Perlado, “Nanoscale View of Shock Wave Propagation in Single Crystal Fe, W, and Ta for Nuclear Fusion Technology,” Fusion Science and Technology. 2011. link Times cited: 2 Abstract: We report non-equilibrium Molecular Dynamics simulations pro… read moreAbstract: We report non-equilibrium Molecular Dynamics simulations providing a nanoscale view for the modeling of shock wave generation, propagation and melting in single crystalline materials Fe, Ta, W, of clear interest for Nuclear Fusion Technology. Our methodology successfully uses massive parallel molecular dynamics in an attempt to cover similar times and length scales as laser-shock experiments. Response of the materials are analyzed in terms of modern atomistic visualization and evolution of their structural properties. Preliminary results point that Wand Ta behave more efficiently in terms of uniformity under shock propagation than lighter materials like Fe. This kind of materials must attract our attention in the short term as possible designs in inertial confinement fusion (ICF) targets. read less USED (low confidence) H. Kimizuka and S. Ogata, “Slow diffusion of hydrogen at a screw dislocation core in α-iron,” Physical Review B. 2011. link Times cited: 44 Abstract: Here we demonstrate and characterize the H-diffusion behavio… read moreAbstract: Here we demonstrate and characterize the H-diffusion behavior around a screw dislocation in body-centered cubic (bcc) $\ensuremath{\alpha}$-Fe by performing path-integral molecular dynamics modeling and adopting an ab initio\char21{}based potential. Counterintuitively, our results indicate that the H diffusivity along the dislocation line is significantly lower than lattice diffusion. Thus, the ``fast'' pipe diffusion does not occur for H in $\ensuremath{\alpha}$-Fe. read less USED (low confidence) I. Vatne, E. Østby, C. Thaulow, and D. Farkas, “Quasicontinuum simulation of crack propagation in bcc-Fe,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2011. link Times cited: 43 USED (low confidence) H. Kimizuka, H. Mori, and S. Ogata, “Effect of temperature on fast hydrogen diffusion in iron: A path-integral quantum dynamics approach,” Physical Review B. 2011. link Times cited: 45 USED (low confidence) J. Jeon, B.-J. Lee, and Y. Chang, “Molecular dynamics simulation study of the effect of grain size on the deformation behavior of nanocrystalline body-centered cubic iron,” Scripta Materialia. 2011. link Times cited: 87 USED (low confidence) W. Luo, W. Hu, S. Xiao, H. Deng, and F. Gao, “Phase transition in nanocrystalline iron: Atomistic-level simulations,” International Journal of Materials Research. 2010. link Times cited: 5 Abstract: Molecular dynamics simulations, along with the modified anal… read moreAbstract: Molecular dynamics simulations, along with the modified analytic embedded atom method, have been employed to study the bcc → fcc phase transition of nanocrystalline iron. The Gibbs free energies of bulk fcc and bcc iron phases are calculated as a function of temperature, and used to determine the bulk phase-transition temperature. Furthermore, the transformation temperature in the nanocrystalline iron, with a mean grain size of 3 nm, is determined to be 975 ± 25 K using the bond-order parameter method. The radial-distribution function and common neighbor analysis are used to understand the phase structure of the nanocrystalline iron and the evolution of local atomic structure. The snapshots of a two atomic layer thick slice provide a visible scenario of structural evolution during phase transition. read less USED (low confidence) Y. Gao, Y. Yang, D. Sun, M. Asta, and J. Hoyt, “Molecular dynamics simulations of the crystal–melt interface mobility in HCP Mg and BCC Fe,” Journal of Crystal Growth. 2010. link Times cited: 28 USED (low confidence) D. Terentyev and X. He, “Dimensionality of interstitial He migration in 〈1 1 0〉 tilt grain boundaries in α-Fe,” Computational Materials Science. 2010. link Times cited: 24 USED (low confidence) D. Terentyev, X. He, A. Serra, and J. Kuriplach, “Structure and strength of 〈1 1 0〉 tilt grain boundaries in bcc Fe: An atomistic study,” Computational Materials Science. 2010. link Times cited: 52 USED (low confidence) E. K. Njeim and D. Bahr, “Atomistic simulations of nanoindentation in the presence of vacancies,” Scripta Materialia. 2010. link Times cited: 50 USED (low confidence) D. Terentyev, S. M. H. Haghighat, and R. Schäublin, “Strengthening due to Cr-rich precipitates in Fe–Cr alloys: Effect of temperature and precipitate composition,” Journal of Applied Physics. 2010. link Times cited: 39 Abstract: Molecular dynamics (MD) simulations were carried out to stud… read moreAbstract: Molecular dynamics (MD) simulations were carried out to study the interaction between nanometric Cr precipitates and a 1/2 >{110} edge dislocation (ED) in pure Fe and Fe-9 at. % Cr (Fe-9Cr) random alloy. The aim of this work is to estimate the variation in the pinning strength of the Cr precipitate as a function of temperature, its chemical composition and the matrix composition in which the precipitate is embedded. The dislocation was observed to shear Cr precipitates rather than by-pass via the formation of the Orowan loop, even though a pronounced screw dipole was emerged in the reactions with the precipitates of size larger than 4.5 nm. The screw arms of the formed dipole were not observed to climb thus no point defects were left inside the sheared precipitates, irrespective of simulation temperature. Both Cr solution and Cr precipitates, embedded in the Fe-9Cr matrix, were seen to contribute to the flow stress. The decrease in the flow stress with temperature in the alloy containing Cr precipitates is, therefore, related to the simultaneous change in the matrix friction stress, precipitate resistance, and dislocation flexibility. Critical stress estimated from MD simulations was seen to have a strong dependence on the precipitate composition. If the latter decreases from 95% down to 80%, the corresponding critical stress decreases almost as twice. The results presented here suggest a significant contribution to the flow stress due to the alpha-alpha(') separation, at least for EDs. The obtained data can be used to validate and to parameterize dislocation dynamics models, where the temperature dependence of the obstacle strength is an essential input data. read less USED (low confidence) M. Gilbert and S. Dudarev, “Ab initio multi-string Frenkel–Kontorova model for a b = a/2[111] screw dislocation in bcc iron,” Philosophical Magazine. 2010. link Times cited: 23 Abstract: We formulate a multi-string Frenkel–Kontorova (MSFK) model f… read moreAbstract: We formulate a multi-string Frenkel–Kontorova (MSFK) model for a a/2[111] screw dislocation in bcc iron, and investigate the occurrence of degenerate and non-degenerate dislocation core structures as functionals of the law of interaction between the [111] strings of atoms forming the crystal. By comparing the effective inter-string interaction laws derived from ab initio density functional calculations and from semi-empirical interatomic potentials for α-iron, we show that it is the form of the function determining how the atomic strings interact with each other as a function of their relative one-dimensional displacement in the [111] direction that determines whether a degenerate or a non-degenerate screw dislocation core configuration has lower energy. We show that by constructing a one-dimensional inter-string interaction law, and by solving the MSFK equations, it is possible to easily predict the nature of the screw dislocation core, hence providing a simple yet effective check to aid the development of short-range semi-empirical interatomic potentials for bcc transition metals. Finally, we analyse the relation between the inter-string interaction law, and the shape and the height of the Peierls energy barriers separating the adjacent equilibrium configurations for a migrating screw dislocation. read less USED (low confidence) E. Hayward and C. Deo, “A Molecular Dynamics Study of Irradiation Induced Cascades in Iron Containing Hydrogen,” Cmc-computers Materials & Continua. 2010. link Times cited: 5 Abstract: Damage cascades representative of those that would be induce… read moreAbstract: Damage cascades representative of those that would be induced by neutron irradiation have been simulated in systems of pure iron and iron containing 0.01 at.% hydrogen. Results from molecular dynamics simulations using three different embedded-atom method (EAM) type potentials are compared for primary knock-on atom energies of 5, 10, and 20 keV to assess the effect of hydrogen on the primary damage state. We examine the influence of hydrogen on the primary damage state due to a single radiation cascade. These results can serve as an atomistic database for methods and simulations for long time scale evolution of radiation damage. read less USED (low confidence) D. Belashchenko, N. Kravchunovskaya, and O. Ostrovski, “Molecular dynamics calculation of surface tension of liquid metals using the embedded atom model,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2010. link Times cited: 9 USED (low confidence) G. Bonny, R. Pasianot, N. Castin, and L. Malerba, “Ternary Fe–Cu–Ni many-body potential to model reactor pressure vessel steels: First validation by simulated thermal annealing,” Philosophical Magazine. 2009. link Times cited: 206 Abstract: In recent years, the development of atomistic models dealing… read moreAbstract: In recent years, the development of atomistic models dealing with microstructure evolution and subsequent mechanical property change in reactor pressure vessel steels has been recognised as an important complement to experiments. In this framework, a literature study has shown the necessity of many-body interatomic potentials for multi-component alloys. In this paper, we develop a ternary many-body Fe–Cu–Ni potential for this purpose. As a first validation, we used it to perform a simulated thermal annealing study of the Fe–Cu and Fe–Cu–Ni alloys. Good qualitative agreement with experiments is found, although fully quantitative comparison proved impossible, due to limitations in the used simulation techniques. These limitations are also briefly discussed. read less USED (low confidence) Z. Chen, N. Kioussis, and N. Ghoniem, “Influence of nanoscale Cu precipitates inα-Feon dislocation core structure and strengthening,” Physical Review B. 2009. link Times cited: 39 Abstract: Atomistic simulations of the interaction of a screw dislocat… read moreAbstract: Atomistic simulations of the interaction of a screw dislocation in (cid:1) -Fe with different size bcc Cu precipitates suggest two plausible strengthening mechanisms. For precipitate diameters in the range 1.5 nm (cid:2) d (cid:2) 3.3 nm, the dislocation core structure within the Cu precipitate undergoes a polarized to nonpolarized transformation, leading to the dislocation pinning at the precipitate-matrix interface and the bowing out of the dislocation line. The calculated bow-out angle and resolved shear stress required to detach the dislocation from the precipitate are in agreement with recent experiments. The structural transition of larger (cid:1) d (cid:3) 3.3 nm (cid:2) Cu precipitates under high shear stress is responsible for the loss of slip systems and hence for dislocation pinning. read less USED (low confidence) G. Monnet, C. Domain, S. Queyreau, S. Naamane, and B. Devincre, “Atomic and dislocation dynamics simulations of plastic deformation in reactor pressure vessel steel,” Journal of Nuclear Materials. 2009. link Times cited: 15 USED (low confidence) D. Terentyev and N. Castin, “Mobility of small clusters of self-interstitial atoms in dilute Fe-Cr alloy studied by means of atomistic calculations,” Computational Materials Science. 2009. link Times cited: 9 USED (low confidence) M. Ruda, D. Farkas, and G. García, “Atomistic simulations in the Fe–C system,” Computational Materials Science. 2009. link Times cited: 79 USED (low confidence) N. Gao, C. Fu, M. Samaras, R. Schäublin, M. Victoria, and W. Hoffelner, “Multiscale modelling of bi-crystal grain boundaries in bcc iron,” Journal of Nuclear Materials. 2009. link Times cited: 38 USED (low confidence) D. Terentyev, P. Olsson, T. Klaver, and L. Malerba, “On the migration and trapping of single self-interstitial atoms in dilute and concentrated Fe–Cr alloys: Atomistic study and comparison with resistivity recovery experiments,” Computational Materials Science. 2008. link Times cited: 60 USED (low confidence) R. Gröger, R. Gröger, A. G. Bailey, A. G. Bailey, and V. Vítek, “Multiscale modeling of plastic deformation of molybdenum and tungsten: I. Atomistic studies of the core structure and glide of 1/2〈1 1 1〉 screw dislocations at 0 K,” Acta Materialia. 2008. link Times cited: 231 USED (low confidence) X. Liu and S. Biner, “Molecular dynamics simulations of the interactions between screw dislocations and self-interstitial clusters in body-centered cubic Fe,” Scripta Materialia. 2008. link Times cited: 53 USED (low confidence) C. Fu and F. Willaime, “First principles calculations in iron: structure and mobility of defect clusters and defect complexes for kinetic modelling.” 2008. link Times cited: 41 USED (low confidence) H. Heinisch, F. Gao, and R. Kurtz, “Atomistic Modeling of Helium Interacting with Screw Dislocations in α-Fe,” Journal of Nuclear Materials. 2007. link Times cited: 41 USED (low confidence) H. Heinisch, F. Gao, and R. Kurtz, “Modeling the Interaction of Helium with Dislocations and Grain Boundaries in Alpha-Iron,” Journal of Astm International. 2007. link Times cited: 5 Abstract: Helium is a ubiquitous impurity in nuclear materials that ca… read moreAbstract: Helium is a ubiquitous impurity in nuclear materials that can have significant deleterious effects on mechanical properties, including deformation and fracture. To determine ways to mitigate the effects of helium it is necessary to understand the behavior of helium with respect to its interaction with various microstructural features. Toward that end, we have employed molecular statics, molecular dynamics, and the dimer method of potential surface mapping to study the fate of helium in the vicinity of dislocations and grain boundaries in alpha-iron. Even at very low temperatures interstitial helium atoms can migrate to dislocations and grain boundaries, where they are strongly bound. The binding energies of helium to these microstructural features relative to the perfect crystal and the migration energies of helium diffusing within them have a strong correlation to the excess atomic volume that exists in these extended defects. Helium atom migration energies within the dislocations and grain boundaries studied are in the range of 0.4–0.5 eV. Helium “kick out” mechanisms have been identified within dislocations and grain boundaries by which interstitial helium atoms replace an Fe lattice atom, creating a stable He-vacancy complex that may be a nucleation site for an He bubble. read less USED (low confidence) Y. Ashkenazy and R. Averback, “Atomic mechanisms controlling crystallization behaviour in metals at deep undercoolings,” EPL (Europhysics Letters). 2007. link Times cited: 45 Abstract: Understanding the liquid-solid phase transition has long bee… read moreAbstract: Understanding the liquid-solid phase transition has long been of scientific interest, owing to its singular importance in the processing of materials with desired microstructures. Presently, however, little is known about the atomic mechanisms controlling this process. Using molecular dynamics simulations, we find a surprising connection between the crystallization behavior of metals at extreme undercoolings and the properties of interstitial atoms in the crystalline phase. We show first that the activation energy of crystallization in a number of metals at the kinetically controlled regime is precisely the same as the migration energy of self-interstitials atom in the crystalline state. We then show, contrary to the present thought, that the advance of a planar solid-liquid interface in Fe at low temperatures is controlled by thermally activated jumps of a small fraction of the atoms on the liquid side of the interface, and remarkably these atoms have the same ⟨110⟩ dumbbell interstitialcy structure as observed for interstitials in crystalline Fe. read less USED (low confidence) A. Kuksin, G. Norman, V. Stegailov, and A. Yanilkin, “Atomistic simulations of structure transitions and fracture in Fe and Al single crystals,” Comput. Phys. Commun. 2007. link Times cited: 1 USED (low confidence) C. Ortiz and M. Caturla, “Simulation of defect evolution in irradiated materials: Role of intracascade clustering and correlated recombination,” Physical Review B. 2007. link Times cited: 73 Abstract: The evolution of damage produced by collision cascades in Fe… read moreAbstract: The evolution of damage produced by collision cascades in Fe is studied using both kinetic Monte Carlo (kMC) and rate theory (RT) approaches. The initial damage distribution is obtained from molecular-dynamics simulations of 30 keV recoils in Fe. An isochronal annealing is simulated to identify the different thermally activated mechanisms that govern defect evolution. When clusters form during collision cascades, kMC simulations show that additional recovery peaks should be expected, in comparison to recovery curves obtained under electron irradiation conditions. Detailed kMC and RT simulations reveal that some of these recovery peaks are due to correlated recombinations at low temperature between defects. In particular, we show that under cascade-damage conditions it is possible to observe correlated recombinations between vacancies and self-interstitial clusters. These correlated recombinations cannot be reproduced with a RT model, and therefore kMC and RT differ at low temperature. However, for the conditions presented here, the contribution of correlated recombination is very small and therefore no significant differences are observed at high temperatures between these two models. read less USED (low confidence) P. Derlet and S. Dudarev, “Million-atom molecular dynamics simulations of magnetic iron,” Progress in Materials Science. 2007. link Times cited: 61 USED (low confidence) F. Djurabekova, L. Malerba, C. Domain, and C. Becquart, “Stability and mobility of small vacancy and copper-vacancy clusters in bcc-Fe: An atomistic kinetic Monte Carlo study,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 34 USED (low confidence) P. Olsson, C. Domain, and J. Wallenius, “Ab initio study of Cr interactions with point defects in bcc Fe,” Physical Review B. 2007. link Times cited: 244 Abstract: The properties of Cr in alpha Fe have been investigated by a… read moreAbstract: The properties of Cr in alpha Fe have been investigated by ab initio calculations based on density functional theory. The intrinsic point defect formation energies were found to be larger in model ... read less USED (low confidence) J. Shim, H.-J. Lee, and B. Wirth, “Molecular dynamics simulation of primary irradiation defect formation in Fe–10%Cr alloy,” Journal of Nuclear Materials. 2006. link Times cited: 69 USED (low confidence) M. Samaras, P. Derlet, H. Swygenhoven, and M. Victoria, “Atomic scale modelling of the primary damage state of irradiated fcc and bcc nanocrystalline metals,” Journal of Nuclear Materials. 2006. link Times cited: 82 USED (low confidence) C. Domain, “Ab initio modelling of defect properties with substitutional and interstitials elements in steels and Zr alloys,” Journal of Nuclear Materials. 2006. link Times cited: 96 USED (low confidence) D. Terentyev et al., “Displacement cascades in Fe–Cr: A molecular dynamics study,” Journal of Nuclear Materials. 2006. link Times cited: 107 USED (low confidence) D. Sun et al., “Crystal-melt interfacial free energies in hcp metals: A molecular dynamics study of Mg,” Physical Review B. 2006. link Times cited: 327 Abstract: Crystal-melt interfacial free energies $(\ensuremath{\gamma}… read moreAbstract: Crystal-melt interfacial free energies $(\ensuremath{\gamma})$ are computed for hcp Mg by employing equilibrium molecular-dynamics (MD) simulations and the capillary-fluctuation method (CFM). This work makes use of a newly developed embedded-atom-method (EAM) interatomic potential for Mg fit to crystal, liquid, and melting properties. We describe how the CFM, which has previously been applied to cubic systems only, can be generalized for studies of hcp metals by employing a parametrization for the orientation dependence of $\ensuremath{\gamma}$ in terms of hexagonal harmonics. The method is applied in the calculation of the Turnbull coefficient $(\ensuremath{\alpha})$ and crystalline anisotropies of $\ensuremath{\gamma}$. We obtain a value of $\ensuremath{\alpha}=0.48$, with interfacial free energies for different high-symmetry orientations differing by approximately 1%. These results are compared to those obtained in previous MD-CFM studies for cubic EAM metals as well as experimental studies of solid-liquid interfaces in hcp alloys. In addition, the implications of our results for the prediction of dendrite growth directions in hcp metals are discussed. read less USED (low confidence) K. Kadau, T. Germann, P. Lomdahl, and B. Holian, “Atomistic simulations of shock-induced transformations and their orientation dependence in bcc Fe single crystals,” Physical Review B. 2005. link Times cited: 155 USED (low confidence) C. Domain and C. Becquart, “Diffusion of phosphorus in α-Fe : An ab initio study,” Physical Review B. 2005. link Times cited: 83 USED (low confidence) B.-J. Lee, B. Wirth, J. Shim, J. Kwon, S. Kwon, and J.-H. Hong, “Modified embedded-atom method interatomic potential for the Fe-Cu alloy system and cascade simulations on pure Fe and Fe-Cu alloys,” Physical Review B. 2005. link Times cited: 55 Abstract: A modified embedded-atom method (MEAM) interatomic potential… read moreAbstract: A modified embedded-atom method (MEAM) interatomic potential for the Fe-Cu binary system has been developed using previously developed MEAM potentials of Fe and Cu. The Fe-Cu potential was determined by fitting to data on the mixing enthalpy and the composition dependencies of the lattice parameters in terminal solid solutions. The potential gives a value of 0.65 eV for the dilute heat of solution and reproduces the increase of lattice parameter of Fe with addition of Cu in good agreement with experiments. The potential was used to investigate the primary irradiation defect formation in pure Fe and Fe-0.5 at. % Cu alloy by a molecular dynamics cascade simulation study with a PKA energy of 2 keV at 573 K. A tendency for self-interstitial atom-Cu binding, the formation of mixed (Fe-Cu) dumbbells and even Cu-Cu dumbbells was observed. Given a positive binding energy between Cu atoms and self-interstitials, Cu transport by an interstitial diffusion mechanism could be proposed to contribute to the formation of Cu-rich precipitates and irradiation-induced embrittlement in nuclear structural steels. read less USED (low confidence) M. Mendelev, D. Srolovitz, G. Ackland, and S. Han, “Effect of Fe segregation on the migration of a non-symmetric ∑5 tilt grain boundary in Al,” Journal of Materials Research. 2005. link Times cited: 106 Abstract: We present an analysis, based upon atomistic simulation data… read moreAbstract: We present an analysis, based upon atomistic simulation data, of the effect of Fe impurities on grain boundary migration in Al. The first step is the development of a new interatomic potential for Fe in Al. This potential provides an accurate description of Al–Fe liquid diffraction data and the bulk diffusivity of Fe in Al. We use this potential to determine the physical parameters in the Cahn–Lücke–Stüwe (CLS) model for the effect of impurities on grain boundary mobility. These include the heat of segregation of Fe to grain boundaries in Al and the diffusivity of Fe in Al. Using the simulation-parameterized CLS model, we predict the grain boundary mobility in Al in the presence of Fe as a function of temperature and Fe concentration. The order of magnitude and the trends in the mobility from the simulations are in agreement with existing experimental results. read less USED (low confidence) C. Domain, C. Becquart, and L. Malerba, “Simulation of radiation damage in Fe alloys: an object kinetic Monte Carlo approach,” Journal of Nuclear Materials. 2004. link Times cited: 288 USED (low confidence) D. Sun, M. Asta, J. Hoyt, M. Mendelev, and D. Srolovitz, “Crystal-melt interfacial free energies in metals: fcc versus bcc,” Physical Review B. 2004. link Times cited: 86 Abstract: The structural dependence of crystal-melt interfacial free e… read moreAbstract: The structural dependence of crystal-melt interfacial free energies ( y) is investigated for fcc and bcc solids through molecular-dynamics calculations employing interatomic potentials for Fe. We compute 30-35 % lower values of y for the bcc structure, and find that our results cannot be explained simply in terms of differences in latent heats (L) or densities (p) for bulk bcc and fcc phases. We observe a strong structural dependence of the Turnbull coefficient a= γ/Lρ 2 / 3 , and find a trend towards lower crystalline anisotropies of y for the bcc structure relative to fcc. read less USED (low confidence) S.-B. Fei and K. Aifantis, “Modelling dislocation-graphene interactions in a BCC Fe matrix by molecular dynamics simulations and gradient plasticity theory,” Applied Surface Science. 2021. link Times cited: 31 USED (low confidence) Y. Zhang, Z. Xiao, and X. Bai, “Effect of Cr Concentration on ½<111> to <100> Dislocation Loop Transformation in Fe-Cr alloys,” Journal of Nuclear Materials. 2021. link Times cited: 10 USED (low confidence) Y. Yang, S. Li, X. Ding, J. Sun, J. Weiss, and E. Salje, “Twisting of Pre-Twinned Alpha-Fe Nanowires: From Mild to Wild Avalanche Dynamics,” MatSciRN: Other Nanomaterials (Topic). 2020. link Times cited: 2 Abstract: Alpha-Fe nanowires are seeded with twin walls (TBs) with ori… read moreAbstract: Alpha-Fe nanowires are seeded with twin walls (TBs) with orientations perpendicular to the wire direction. Twisting the wire generates topological defects in the twin walls, namely new twin boundaries (kinks) inside the twin walls for small twist angles, and junctions between kinks for large twist angles. During twisting the kink motion is jerky and uncorrelated at small twisting angles. The probability density function (PDF) of jerk energies follows approximately a Gaussian distribution, indicating a mild deformation mode. The kink dynamics transforms from mild to wild at larger twist angles when complex twin patterns with a high density of junctions are generated. The collective motion of kinks now shows avalanche behavior with the energy being power-law distributed. The wildness, which measures the proportion of strain energy relaxed through such avalanches, is correlated with the junction density, and controlled by the external length scale (wire diameter) as well as an internal length scale (twin boundary spacing). Good strain-stress recoverability is achieved when unloading the wire before the formation of complex twin patterns. We correlate the evolution of twin patterns with a statistical analysis of jerk dynamics, which identifies the unique mechanical properties governed by twin boundary motion in nanowires. read less USED (low confidence) N. Kumar, K. Krishna, S. Chandra, and R. Tewari, “Influence of dislocations and grain boundaries on diffraction line profiles of nano-crystalline materials: A numerical study,” Computational Materials Science. 2020. link Times cited: 4 USED (low confidence) A. Nikonov and A. Nikonova, “Study of polycrystalline metal surface treatment. molecular dynamics simulation.” 2020. link Times cited: 0 USED (low confidence) M. Ecke, O. Michael, M. Wilke, S. Hütter, M. Krüger, and T. Halle, “Deformation Twinning in bcc Iron - Experimental Investigation of Twin Formation Assisted by Molecular Dynamics Simulation.” 2019. link Times cited: 2 USED (low confidence) Z. Zhao, Z.-ye Qin, X. Xu, and F. Chu, “Molecular Dynamics Simulation on Intergranular Crack Propagation Along ∑3 Tilt Grain Boundary in Bcc Iron,” Lecture Notes in Mechanical Engineering. 2019. link Times cited: 0 USED (low confidence) S. Taketomi and R. Matsumoto, “Atomistic Simulations of Hydrogen Effects on Lattice Defects in Alpha Iron,” Handbook of Mechanics of Materials. 2019. link Times cited: 5 USED (low confidence) S. A. Etesami and E. Asadi, “Molecular dynamics for near melting temperatures simulations of metals using modified embedded-atom method,” Journal of Physics and Chemistry of Solids. 2018. link Times cited: 71 USED (low confidence) A. Nikonov, “Molecular dynamics study of the behavior of single- and polycrystals of BCC Fe under shear loading conditions.” 2018. link Times cited: 1 USED (low confidence) P. Dungriyal, S. Singh, and R. Prasad, “Grain size Dependency, Plasticity and Dynamic Property Evaluation for Nano-crystalline BCC-Fe using Molecular Dynamic Simulations ☆,” Procedia Engineering. 2017. link Times cited: 6 USED (low confidence) G. Sainath and B. Choudhary, “Orientation dependent deformation behaviour of BCC iron nanowires,” Computational Materials Science. 2016. link Times cited: 108 USED (low confidence) M. Eisenbach, D. Perera, D. Landau, D. Nicholson, J. Yin, and G. K. Brown, “Magnetic Materials at finite Temperatures: thermodynamics and combined spin and molecular dynamics derived from first principles calculations,” Journal of Physics: Conference Series. 2015. link Times cited: 3 Abstract: We present a unified approach to describe the combined behav… read moreAbstract: We present a unified approach to describe the combined behavior of the atomic and magnetic degrees of freedom in magnetic materials. Using Monte Carlo simulations directly combined with first principles the Curie temperature can be obtained ab initio in good agreement with experimental values. The large scale constrained first principles calculations have been used to construct effective potentials for both the atomic and magnetic degrees of freedom that allow the unified study of influence of phonon-magnon coupling on the thermodynamics and dynamics of magnetic systems. The MC calculations predict the specific heat of iron in near perfect agreement with experimental results from 300K to above Tc and allow the identification of the importance of the magnon-phonon interaction at the phase-transition. Further Molecular Dynamics and Spin Dynamics calculations elucidate the dynamics of this coupling and open the potential for quantitative and predictive descriptions of dynamic structure factors in magnetic materials using first principles derived simulations. read less USED (low confidence) T. Swinburne, “Atomistic Simulations in bcc Metals.” 2015. link Times cited: 0 USED (low confidence) A. Rajabpour, L. Seidabadi, and M. Soltanpour, “Calculating the Bulk Modulus of Iron and Steel Using Equilibrium Molecular Dynamics Simulation,” Procedia Materials Science. 2015. link Times cited: 18 USED (low confidence) D. Molnár, P. Binkele, A. Mora, R. Mukherjee, B. Nestler, and S. Schmauder, “Molecular Dynamics virtual testing of thermally aged Fe–Cu microstructures obtained from multiscale simulations,” Computational Materials Science. 2014. link Times cited: 9 USED (low confidence) R. Khanna and V. Sahajwalla, “Atomistic Simulations of Properties and Phenomena at High Temperatures.” 2014. link Times cited: 3 USED (low confidence) D. Li, H. Zbib, X. Sun, and M. Khaleel, “Predicting plastic flow and irradiation hardening of iron single crystal with mechanism-based continuum dislocation dynamics,” International Journal of Plasticity. 2014. link Times cited: 107 USED (low confidence) N. Gao, A. Ghoniem, X. Gao, P. Luo, K. Wei, and Z. G. Wang, “Molecular dynamics simulation of Cu atoms interaction with symmetrical grain boundaries of BCC Fe,” Journal of Nuclear Materials. 2014. link Times cited: 11 USED (low confidence) D. Ilnitski, V. Krasnikov, A. Kuksin, A. Mayer, and A. Yanilkin, “Dynamics and Kinetics of Dislocations in Metals and Alloys Under Dynamic Loading,” MRS Proceedings. 2013. link Times cited: 2 USED (low confidence) V. Srinivasan, G. Sainath, B. Choudhary, M. D. Mathew, and T. Jayakumar, “Effect of Temperature and Strain Amplitude on Fatigue Behaviour of BCC Iron Single Crystal using Molecular Dynamics Simulation,” Procedia Engineering. 2013. link Times cited: 2 USED (low confidence) S. Seki, R. Matsumoto, Y. Inoue, S. Taketomi, and N. Miyazaki, “Development of EAM Potential for Fe with Pseudo-Hydrogen Effects and Molecular Dynamics Simulation of Hydrogen Embrittlement,” Journal of The Society of Materials Science, Japan. 2012. link Times cited: 6 Abstract: Numerous studies have reported that solute hydrogen atoms an… read moreAbstract: Numerous studies have reported that solute hydrogen atoms and lattice defects have strong interactions, and that hydrogen atoms significantly change the stability and/or mobility of lattice defects. Although molecular dynamics (MD) simulations can treat complicated interactions of various lattice defects, the time scale is insufficient to treat hydrogen diffusion so as to influence the lattice-defect generation and cooperative motion of hydrogen atoms and lattice defects. Here we developed an interatomic potential for Fe with pseudo-hydrogen effects on lattice-defect energies and performed MD simulations of tensile loading. First, we estimated the lattice-defect energies of Fe and hydrogen-trap energies of lattice defects by using first-principle calculations and evaluated the lattice-defect energies under a practical gaseous hydrogen environment. Second, we refitted the existing embedded-atom-method potential for Fe to represent the lattice-defect energies amended by hydrogen effects. Finally, we confirmed that our potential is applicable for various phenomena by estimating the reproducibility of grain-boundary energies that are not employed for potential fitting. Our tensile-loading simulations of a nano specimen show that hydrogen reduces elongation at rupture. read less USED (low confidence) E. Hristova, R. Janisch, R. Drautz, and A. Hartmaier, “Solubility of carbon in α-iron under volumetric strain and close to the Σ5(3 1 0)[0 0 1] grain boundary: Comparison of DFT and empirical potential methods,” Computational Materials Science. 2011. link Times cited: 46 USED (low confidence) I. Mastorakos, N. Le, M. Zeine, H. Zbib, and M. Khaleel, “Multiscale Modeling of Irradiation Induced Hardening in a-Fe, Fe-Cr and Fe-Ni Systems,” MRS Proceedings. 2010. link Times cited: 6 Abstract: Structural materials in the new Generation IV reactors will … read moreAbstract: Structural materials in the new Generation IV reactors will operate in harsh radiation conditions coupled with high levels of hydrogen and helium production, thus experiencing severe degradation of mechanical properties. The development of structural materials for use in such a hostile environment is predicated on understanding the underlying physical mechanisms responsible for microstructural evolution along with corresponding dimensional instabilities and mechanical property changes. As the phenomena involved are very complex and span in several length scales, a multiscale approach is necessary in order to fully understand the degradation of materials in irradiated environments. The purpose of this work is to study the behavior of Fe systems (namely a-Fe, Fe-Cr and Fe-Ni) under irradiation using both Molecular Dynamics (MD) and Dislocation Dynamics (DD) simulations. Critical information is passed from the atomistic (MD) to the microscopic scale (DD) in order to study the degradation of the material under examination. In particular, information pertaining to the dislocation-defects (such as voids, helium bubbles and prismatic loops) interactions is obtained from MD simulations. Then this information is used by DD to simulate large systems with high dislocation and defect densities. read less USED (low confidence) C. Engin and H. Urbassek, “Molecular-dynamics investigation of the fcc → bcc phase transformation in Fe,” Computational Materials Science. 2008. link Times cited: 49 USED (low confidence) F. Willaime, C. Fu, M. Marinica, and J. D. Torre, “Stability and mobility of self-interstitials and small interstitial clusters in α-iron: ab initio and empirical potential calculations,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 125 USED (low confidence) J. Wallenius, P. Olsson, and C. Lagerstedt, “Relation between thermal expansion and interstitial formation energy in pure Fe and Cr,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 12 USED (low confidence) S. Imamova, P. Atanasov, N. Nedialkov, F. Dausinger, and P. Berger, “Molecular dynamics simulation using pair and many body interatomic potentials: ultrashort laser ablation of Fe,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 26 NOT USED (low confidence) S. Liu, X. Yu, and N. Zhang, “An embedded-atom method interatomic potential for lithium-iron binary system and its applications in the liquid first wall system,” Journal of Nuclear Materials. 2024. link Times cited: 0 NOT USED (low confidence) T. Kumagai, K. Suzuki, A. Nomoto, S. Hara, and A. Takahashi, “Prediction of the Binding Energy of Self Interstitial Atoms in Alpha Iron by a Graph Neural Network,” Materialia. 2023. link Times cited: 0 NOT USED (low confidence) Y. Deng, Y. Wang, K. Xu, and Y. Wang, “Lightweight Extendable Stacking Framework for Structure Classification in Atomistic Simulations.,” Journal of chemical theory and computation. 2023. link Times cited: 0 Abstract: Identifying an atom's local crystal structure is one cr… read moreAbstract: Identifying an atom's local crystal structure is one crucial step in many atomistic simulation analyses. However, many traditional methods are available to only a few limited types of structures, and their performance often relies on manually determined parameters, which may lead to poor classification results in complex material systems. Machine learning models can enhance accuracy and generalizability, but they typically require large amounts of data and computation. This issue could be more severe for deep-learning-based frameworks, especially when confronted with unfamiliar crystal structures. To address this challenge, we propose a lightweight and extendable stacked structure (LESS) classifier, which adopts bond orientational order parameters as features and assembles several efficient machine learning methods as based models. The LESS classifier can recognize a variety of crystal structures, e.g., amorphous, mono, and binary structures, with over 98.8% accuracy on our validation data set, outperforming many current methods even including some deep-learning methods. Our model can also conduct probabilistic classification that aids in the interpretation of atomic structures in complicated environments such as heterogeneous interfaces. Furthermore, when exposed to a completely unknown crystal structure, the LESS framework can efficiently incorporate this new knowledge with generative sampled data from the current model. Overall, our model exhibits great potential as an accurate and flexible atomic structure identification tool featuring high efficiency in both learning and retraining. read less NOT USED (low confidence) F. Wang, Z. Yang, F. Li, J.-L. Shao, and L.-C. Xu, “Strategic sampling with stochastic surface walking for machine learning force fields in iron’s bcc–hcp phase transitions,” RSC Advances. 2023. link Times cited: 0 Abstract: This study developed a machine learning-based force field fo… read moreAbstract: This study developed a machine learning-based force field for simulating the bcc–hcp phase transitions of iron. By employing traditional molecular dynamics sampling methods and stochastic surface walking sampling methods, combined with Bayesian inference, we construct an efficient machine learning potential for iron. By using SOAP descriptors to map structural data, we find that the machine learning force field exhibits good coverage in the phase transition space. Accuracy evaluation shows that the machine learning force field has small errors compared to DFT calculations in terms of energy, force, and stress evaluations, indicating excellent reproducibility. Additionally, the machine learning force field accurately predicts the stable crystal structure parameters, elastic constants, and bulk modulus of bcc and hcp phases of iron, and demonstrates good performance in predicting higher-order derivatives and phase transition processes, as evidenced by comparisons with DFT calculations and existing experimental data. Therefore, our study provides an effective tool for investigating the phase transitions of iron using machine learning methods, offering new insights and approaches for materials science and solid-state physics research. read less NOT USED (low confidence) T. D. Cuong and A. D. Phan, “Reconstructing the phase diagram of iron in the terapascal region via the statistical moment method,” Physical Review B. 2023. link Times cited: 0 NOT USED (low confidence) D. V. Shastri and K. Arunachalam, “‘A concise study of radiation-induced lattice strain in fast reactor first-wall materials through coupled computational modeling and experiments,’” Nuclear and Particle Physics Proceedings. 2023. link Times cited: 0 NOT USED (low confidence) M. Kajihara, K. Nagaami, T. Miyagawa, T. Kondo, and A. Yonezu, “Development of a Velocity Measurement Method for a Microparticle Projectile and High-Speed Impact Testing of Metallic Materials for Grain Refinement,” Acta Materialia. 2023. link Times cited: 0 NOT USED (low confidence) A. Gostevskaya, A. Markidonov, M. Starostenkov, and V. K. Drobyshev, “A Molecular Dynamics Model for Studying the Influence of High Temperatures under Laser Irradiation on Changes in a BCC Crystal Structure,” Izvestiya of Altai State University. 2023. link Times cited: 0 Abstract: The paper discusses the changes in the structures of BCC cry… read moreAbstract: The paper discusses the changes in the structures of BCC crystals subjected to high-temperature exposure. The interest in the study is explained by the processes occurring in the liquid surface layer and their subsequent impact on layer crystallization. They will further affect various physical and geometric characteristics of the material surface as a whole. The presented model helps observe the imperfections of the structure caused by the appearance of pores on the surface layers of the metal. The computational cell temperature in the designed model is distributed according to the solution of the linear problem of heat conduction. The model allows for revealing a surface layer continuity violation when the excess free volume localizes in the form of a group of spherical pores. The dimensions of such imperfections, as well as the duration of their existence, differ when modeling different laser radiation energy densities. Further research reveals the conditions for the pores to remain stable throughout the entire simulation time, as well as the relationship between the crystallographic orientation of the “solid-liquid” interphase boundary and the sizes of the formed pores” interphase boundary and the sizes of the pores formed. Keywords: bcc crystal, molecular dynamics model, interface boundary, porosity. read less NOT USED (low confidence) M. Wang, F. Wang, H. Wang, J. Zhang, X. Zhao, and H. Wu, “Effects of bismuth nanoparticles on the nano-cutting properties of single-crystal iron materials: a molecular dynamics study,” Applied Physics A. 2023. link Times cited: 1 NOT USED (low confidence) X. Zhu, X. Wang, Y. Liu, Y. Luo, and H. Zhang, “Probing the Effect of Cuttings Particle Size on the Friction and Wear Mechanism at the Casing Friction Interface: A Molecular Dynamics Study.,” Langmuir : the ACS journal of surfaces and colloids. 2023. link Times cited: 0 Abstract: Cuttings particles of different sizes in the drilling fluid … read moreAbstract: Cuttings particles of different sizes in the drilling fluid are the leading cause of wear at the casing and drill pipe joints, and diamond-like carbon (DLC) films have excellent research potential in reducing tool wear due to their ultra-low friction coefficient and high wear resistance. In this paper, a corresponding molecular dynamics model was developed using LAMMPS to investigate the effect of silica particles of different particle sizes on the friction and wear mechanisms of Fe/DLC friction pairs at the microscale. The results show that small cuttings particles in a dry environment are more likely to cause interface wear between the casing and drill pipe joint, while in a water environment, the opposite is true. The main reason is that small particles in a dry environment have smaller contact areas and greater indentation depth, leading to greater wear at the friction interface. The movement of water molecules in the water environment will promote the composite movement of large particles, thereby exacerbating the wear of the interface. Moreover, the relevant research results at the micro-scale indicate that DLC films can effectively reduce wear, which provides theoretical support for its application in drill pipe joints. read less NOT USED (low confidence) L. Chalamet, D. Rodney, and Y. Shibuta, “Coarse-grained molecular dynamic model for metallic materials,” Computational Materials Science. 2023. link Times cited: 3 NOT USED (low confidence) F. Aquistapace, N. Amigo, J. F. Troncoso, O. Deluigi, and E. Bringa, “MultiSOM: Multi-layer Self Organizing Maps for local structure identification in crystalline structures,” Computational Materials Science. 2023. link Times cited: 2 NOT USED (low confidence) H. Sun and L. Béland, “Calculation of the recombination radii between point defects and defect clusters in nickel via kinetic Activation Relaxation Technique,” Journal of Nuclear Materials. 2023. link Times cited: 0 NOT USED (low confidence) C. Yang, C. Yin, Y. Wu, Q. Zhou, and X. Liu, “Atomic insights into the deformation mechanism of an amorphous wrapped nanolamellar heterostructure and its effect on self-lubrication,” Journal of Materials Research and Technology. 2023. link Times cited: 10 NOT USED (low confidence) R. Barik, S. Biswal, K. K. Bhandari, A. Ghosh, and D. Chakrabarti, “Micromechanics of cleavage fracture and the associated tongue formation in ferritic steel,” Materials Science and Engineering: A. 2023. link Times cited: 0 NOT USED (low confidence) P. Lafourcade et al., “Robust crystal structure identification at extreme conditions using a density-independent spectral descriptor and supervised learning,” Computational Materials Science. 2023. link Times cited: 0 NOT USED (low confidence) H. Mori, T. Tsuru, M. Okumura, D. Matsunaka, Y. Shiihara, and M. Itakura, “Dynamic interaction between dislocations and obstacles in bcc iron based on atomic potentials derived using neural networks,” Physical Review Materials. 2023. link Times cited: 0 NOT USED (low confidence) R. Xu, L. Martinie, P. Vergne, L. Joly, and N. Fillot, “An Approach for Quantitative EHD Friction Prediction Based on Rheological Experiments and Molecular Dynamics Simulations,” Tribology Letters. 2023. link Times cited: 1 NOT USED (low confidence) R. Wang, L. Cheng, C. Yin, W. Lou, and K. Wu, “The effects of hydrogen and vacancy on the tensile deformation behavior of Σ3 symmetric tilt grain boundaries in pure fe,” International Journal of Hydrogen Energy. 2023. link Times cited: 1 NOT USED (low confidence) S. Paul, D. Schwen, M. Short, and K. Momeni, “A Modified Embedded-Atom Method Potential for a Quaternary Fe-Cr-Si-Mo Solid Solution Alloy,” Materials. 2023. link Times cited: 0 Abstract: Ferritic-martensitic steels, such as T91, are candidate mate… read moreAbstract: Ferritic-martensitic steels, such as T91, are candidate materials for high-temperature applications, including superheaters, heat exchangers, and advanced nuclear reactors. Considering these alloys’ wide applications, an atomistic understanding of the underlying mechanisms responsible for their excellent mechano-chemical properties is crucial. Here, we developed a modified embedded-atom method (MEAM) potential for the Fe-Cr-Si-Mo quaternary alloy system—i.e., four major elements of T91—using a multi-objective optimization approach to fit thermomechanical properties reported using density functional theory (DFT) calculations and experimental measurements. Elastic constants calculated using the proposed potential for binary interactions agreed well with ab initio calculations. Furthermore, the computed thermal expansion and self-diffusion coefficients employing this potential are in good agreement with other studies. This potential will offer insightful atomistic knowledge to design alloys for use in harsh environments. read less NOT USED (low confidence) Y. Yang, J. Sun, and X. Ding, “Superelasticity Induced by a Strain Gradient,” Shape Memory and Superelasticity. 2023. link Times cited: 0 NOT USED (low confidence) M. H. Nazir, Z. Khan, M. M. Hussain, A. Rahil, and S. J. Zaidi, “A comprehensive experimental study and numerical analysis of coefficient of friction of nanocomposite coatings,” Materials Chemistry and Physics. 2023. link Times cited: 1 NOT USED (low confidence) R. Jana and M. A. Caro, “Searching for iron nanoparticles with a general-purpose Gaussian approximation potential,” Physical Review B. 2023. link Times cited: 2 Abstract: We present a general-purpose machine learning Gaussian appro… read moreAbstract: We present a general-purpose machine learning Gaussian approximation potential (GAP) for iron that is applicable to all bulk crystal structures found experimentally under diverse thermodynamic conditions, as well as surfaces and nanoparticles (NPs). By studying its phase diagram, we show that our GAP remains stable at extreme conditions, including those found in the Earth's core. The new GAP is particularly accurate for the description of NPs. We use it to identify new low-energy NPs, whose stability is verified by performing density functional theory calculations on the GAP structures. Many of these NPs are lower in energy than those previously available in the literature up to $N_\text{atoms}=100$. We further extend the convex hull of available stable structures to $N_\text{atoms}=200$. For these NPs, we study characteristic surface atomic motifs using data clustering and low-dimensional embedding techniques. With a few exceptions, e.g., at magic numbers $N_\text{atoms}=59$, $65$, $76$ and $78$, we find that iron tends to form irregularly shaped NPs without a dominant surface character or characteristic atomic motif, and no reminiscence of crystalline features. We hypothesize that the observed disorder stems from an intricate balance and competition between the stable bulk motif formation, with bcc structure, and the stable surface motif formation, with fcc structure. We expect these results to improve our understanding of the fundamental properties and structure of low-dimensional forms of iron, and to facilitate future work in the field of iron-based catalysis. read less NOT USED (low confidence) M. Mi’nkowski and L. Laurson, “Predicting elastic and plastic properties of small iron polycrystals by machine learning,” Scientific Reports. 2023. link Times cited: 1 NOT USED (low confidence) J. ’. Castellanos-Reyes et al., “Unveiling the impact of temperature on magnon diffuse scattering detection in the transmission electron microscope,” Physical Review B. 2023. link Times cited: 0 Abstract: Magnon diffuse scattering (MDS) signals could be studied wit… read moreAbstract: Magnon diffuse scattering (MDS) signals could be studied with high spatial resolution in scanning transmission electron microscopy (STEM), thanks to recent technological progress in electron energy loss spectroscopy. However, detecting MDS signals in STEM is challenging due to their overlap with stronger thermal diffuse scattering (TDS) signals. In bcc Fe at 300 K, MDS signals greater than or comparable to TDS signals occur under the central Bragg disk, into a currently inaccesible energy-loss region. Therefore, to detect MDS in STEM, it is necessary to find conditions in which TDS and MDS signals can be separated. Temperature may be a key factor due to the distinct thermal signatures of magnon and phonon signals. In this work, we present a study on the effects of temperature on MDS and TDS in bcc Fe -- considering a detector outside the central Bragg disk and a fixed convergent electron probe -- using the frozen phonon and frozen magnon multislice methods. Our study reveals that neglecting the effects of atomic vibrations causes the MDS signal to grow approximately linearly up to the Curie temperature of Fe, after which it exhibits less variation. The MDS signal displays an alternating behavior due to dynamical diffraction, instead of increasing monotonically as a function of thickness. Including the effects of atomic vibrations through a complex atomic electrostatic potential causes the linear growth of the MDS signal to change to a non-linear behavior that exhibits a predominant peak for a sample of thickness 16.072 nm at 1100 K. In contrast, the TDS signal grows more linearly than the MDS signal but still exhibits appreciable dynamical diffraction effects. An analysis of the signal-to-noise ratio (SNR) shows that the MDS signal can be a statistically significant contribution to the total scattering intensity under realizable measurement conditions and acquisition times. read less NOT USED (low confidence) Y. Yang, J. Zhao, J. Cui, and B. Jiang, “Molecular dynamics study on enhancement of mechanical and tribological properties of polytetrafluoroethylene composites by incorporating hexagonal boron nitride nanosheets,” Journal of Applied Polymer Science. 2023. link Times cited: 2 NOT USED (low confidence) T. Schmalofski, M. Kroll, H. Dette, and R. Janisch, “Towards active learning: A stopping criterion for the sequential sampling of grain boundary degrees of freedom,” Materialia. 2023. link Times cited: 0 NOT USED (low confidence) Y. Xu, G. Wang, J. Shen, P. Qian, and Y. Su, “Structural features, thermal stability and catalytic implication of Fe–Ni nanoparticles,” Journal of Solid State Chemistry. 2023. link Times cited: 0 NOT USED (low confidence) I. Bedarev and S. Lavruk, “Study of the Dependence of the Melting Temperature of Aluminum Nanoparticles on the Particle Size,” Journal of Engineering Physics and Thermophysics. 2022. link Times cited: 0 NOT USED (low confidence) R. Barik, A. Ghosh, and D. Chakrabarti, “Fundamental insights on ductile to brittle transition phenomenon in ferritic steel,” Materialia. 2022. link Times cited: 3 NOT USED (low confidence) D. Louzguine-Luzgin, “Structural Changes in Metallic Glass-Forming Liquids on Cooling and Subsequent Vitrification in Relationship with Their Properties,” Materials. 2022. link Times cited: 13 Abstract: The present review is related to the studies of structural c… read moreAbstract: The present review is related to the studies of structural changes observed in metallic glass-forming liquids on cooling and subsequent vitrification in terms of radial distribution function and its analogues. These structural changes are discussed in relationship with liquid’s properties, especially the relaxation time and viscosity. These changes are found to be directly responsible for liquid fragility: deviation of the temperature dependence of viscosity of a supercooled liquid from the Arrhenius equation through modification of the activation energy for viscous flow. Further studies of this phenomenon are necessary to provide direct mathematical correlation between the atomic structure and properties. read less NOT USED (low confidence) A. Markidonov, A. Gostevskaya, V. Gromov, M. Starostenkov, and P. A. Zykov, “Simulation of the Structural Changes in the Surface Layer of a Deformed BCC Crystal during a Short-Term External High-Intense Action,” Russian Metallurgy (Metally). 2022. link Times cited: 0 NOT USED (low confidence) Y. Lei et al., “An Embedded-Atom Method Potential for studying the properties of Fe-Pb solid-liquid interface,” Journal of Nuclear Materials. 2022. link Times cited: 1 NOT USED (low confidence) B. Yao, Z. R. Liu, and R. F. Zhang, “EAPOTc: An integrated empirical interatomic potential optimization platform for compound solids,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) Y. Chen et al., “Ion-beam radiation-induced Eshelby transformations: The mean and variance in hydrostatic and shear residual stresses,” Extreme Mechanics Letters. 2022. link Times cited: 1 NOT USED (low confidence) S. Mishra and S. Bhattacharjee, “Temperature guided behavioral transitions in confined helium: gas-wall interaction effects on dynamics and transport in the cryogenic limit,” Chemical Thermodynamics and Thermal Analysis. 2022. link Times cited: 2 NOT USED (low confidence) Y. Zhao, Q. Song, H. Ji, W. Cai, Z. Liu, and Y. Cai, “Multi-scale modeling method for polycrystalline materials considering grain boundary misorientation angle,” Materials & Design. 2022. link Times cited: 3 NOT USED (low confidence) C. Yang, G. Deng, X. Xing, Q. Han, and H. Liu, “Molecular dynamics study on the effect of Ni atoms on the crack arrest performance of Fe–Ni alloy,” Materials and Corrosion. 2022. link Times cited: 1 Abstract: Molecular dynamic simulations are applied to test the nickel… read moreAbstract: Molecular dynamic simulations are applied to test the nickel's modification mechanism of Fe–Ni alloy. Mono displacement loading is applied to a perfect single crystal model, a single crystal model with vacancies, and a model with transgranular crack. Moreover, constant strain load is applied to the polycrystal model to test the Ni effect on intergranular crack initiation. The results elucidate that Ni atoms could decrease the free surface energy and the stacking fault energy simultaneously. However, Ni atoms have a more significant effect on the reduction of stacking fault energy. If the Ni concentration is above 0.03, the transgranular crack constantly emits dislocations under loading, thus, postponing the cleavage cracking. Particularly, as the Ni concentration is above 0.05, the recrystallization process could be a favorable energy‐releasing behavior compared with the intergranular cracking. The findings suggest that a low concentration of Ni might degrade the physical property of Fe–Ni alloy. Increasing the Ni atomic concentration above specific critical values, for example, 0.03 or 0.05, could enhance the fracture toughness. read less NOT USED (low confidence) B. Lv, C. Chen, F. Zhang, G. Poletaev, and R. Rakitin, “Potentials for Describing Interatomic Interactions in γFe-Mn-C-N System,” Metals. 2022. link Times cited: 1 Abstract: Potentials for describing interatomic interactions in a γFe-… read moreAbstract: Potentials for describing interatomic interactions in a γFe-Mn-C-N multicomponent system, modified Hadfield steel, where face-centered cubic (f.c.c.) iron is the main component, are proposed. To describe the Fe-Fe interactions in austenite, it is proposed to use Lau EAM potential. For all other interactions, Morse potentials are proposed, the parameters of which were found from various experimental characteristics: in particular, the energy of dissolution and migration of an impurity in an f.c.c. iron crystal, the radius of atoms, their electronegativity, mutual binding energy, etc. The found potentials are intended for modeling the atomic structures and processes occurring at the atomic level in Hadfield steel using relatively large computational cells by the molecular dynamics method. read less NOT USED (low confidence) A. Kohnert and L. Capolungo, “The kinetics of static recovery by dislocation climb,” npj Computational Materials. 2022. link Times cited: 15 NOT USED (low confidence) W. Qian, Y. Anfeng, K. Zetian, K.-H. Chen, and L. Huan, “Molecular Dynamics Study on the Microscopic Mechanism of In-service Welding Damage and Failure,” Engineering Failure Analysis. 2022. link Times cited: 3 NOT USED (low confidence) W. Huang et al., “Revealing nanoscale material deformation mechanism and surface/subsurface characteristics in vibration-assisted nano-grinding of single-crystal iron,” Applied Surface Science. 2022. link Times cited: 13 NOT USED (low confidence) I. Toda-Caraballo, J. Wróbel, and D. Nguyen-Manh, “Generalized universal equation of states for magnetic materials: A novel formulation for an interatomic potential in Fe,” Physical Review Materials. 2022. link Times cited: 0 NOT USED (low confidence) J. Byggmästar, K. Nordlund, and F. Djurabekova, “Simple machine-learned interatomic potentials for complex alloys,” Physical Review Materials. 2022. link Times cited: 5 Abstract: Developing data-driven machine-learning interatomic potentia… read moreAbstract: Developing data-driven machine-learning interatomic potentials for materials containing many elements becomes increasingly challenging due to the vast configuration space that must be sampled by the training data. We study the learning rates and achievable accuracy of machine-learning interatomic potentials for many-element alloys with different combinations of descriptors for the local atomic environments. We show that for a five-element alloy system, potentials using simple low-dimensional descriptors can reach meV/atom-accuracy with modestly sized training datasets, significantly outperforming the high-dimensional SOAP descriptor in data efficiency, accuracy, and speed. In particular, we develop a computationally fast machine-learned and tabulated Gaussian approximation potential (tabGAP) for Mo–Nb–Ta–V–W alloys with a combination of two-body, three-body, and a new simple scalar many-body density descriptor based on the embedded atom method. read less NOT USED (low confidence) A. Bakaev et al., “Effect of radiation defects on the early stages of nanoindentation tests in bcc Fe and Fe-Cr alloys,” Computational Materials Science. 2022. link Times cited: 3 NOT USED (low confidence) A. Mahata and M. Kivy, “Computational study of nanoscale mechanical properties of Fe–Cr–Ni alloy,” Molecular Simulation. 2022. link Times cited: 1 Abstract: ABSTRACT Mechanical properties of Fe–Cr–Ni alloy nanowires h… read moreAbstract: ABSTRACT Mechanical properties of Fe–Cr–Ni alloy nanowires have been investigated using molecular dynamics simulation with embedded atom method and first principles approach. Various cases of uniaxial tension, compression and shear deformations have been performed and studied in this work. From the first principles calculations, the higher magnitudes of uniaxial and shear deformations resulted in higher probability of martensitic transformations. Before the first yielding, nanowires preserved the elastic stage and then the mechanical deformation proceeded in alternating quasi-elastic and yielding stages. The plastic behaviour was not observed in compression while both tensile and shear deformations showed apparent plastic behaviour. In shear deformation, due to the martensitic phase transformation, the plastic behaviour persisted for total strain of 0.6 which was much larger than that during tensile and compression. This validated the previous experimental observations. In the studied Fe–Cr–Ni nanowires, deformations were controlled by dislocations. Dislocation-mediated twinnings were captured by common neighbour analysis. Twin quantification showed that the twin activity increased with increasing strain rate. Twinnings originated from stacking faults led by 1/6 <112> Shockley partial dislocations. At elevated temperature (beyond 500 K), the materials softening happened, and 316L nanowire became more plastic under a lower stress. read less NOT USED (low confidence) Y. Wang et al., “Machine-learning interatomic potential for radiation damage effects in bcc-iron,” Computational Materials Science. 2022. link Times cited: 7 NOT USED (low confidence) L. V. Sang, N. Sugimura, K. Khajeh, and H. Washizu, “Solid Lubricants of Combined Graphene and Iron Nanoparticles for Study of Friction and Stability.,” Langmuir : the ACS journal of surfaces and colloids. 2022. link Times cited: 1 Abstract: This study focuses on designing solid lubricant particles by… read moreAbstract: This study focuses on designing solid lubricant particles by combining graphene and iron nanoparticles (namely, graphene-iron (GI) particles) and carrying out studies for behaviors of their lubrication for the iron contact by molecular dynamics simulations. By the annealing process of melting and cooling iron, we can create the lubricant particle, where the iron nanoparticle tightly holds the graphene sheet. In the sliding friction investigations, it is found that the influences of orientation of the graphene sheets inside the contact, size and configuration of the GI particles, and lubrication with the bare iron nanoparticles on friction are strong at low pressure and very slight at high pressure. The GI particles provide stability of the friction coefficient over a wide range of pressure; however, it strongly increases with pressure in the lubrication behaviors by the bare iron particles due to the deformation of the particles. The iron contact in the presence of the GI particles can achieve the ultralow values of the friction coefficient from 0.009 to 0.042. The contact surfaces are not nearly damaged (slightly elastic deformation) with the pressure up to 2.0 GPa. From the comparisons between the results in this study and previous reports, the GI particles have better lubrication than graphene coated on a surface and well stabilize under pressure compared to the different lubricant nanoparticles. The main reason for this is due to the contributions of graphene, besides reduction of the contact area resulted from the configuration of the nanoparticle, which promotes sliding and sharing of the pressure, preventing collision between the lubricant particles. read less NOT USED (low confidence) A. Allera, F. Ribeiro, M. Perez, and D. Rodney, “Carbon-induced strengthening of bcc iron at the atomic scale,” Physical Review Materials. 2022. link Times cited: 6 NOT USED (low confidence) I. Camacho et al., “On the anticorrosion mechanism of molten salts based nanofluids,” Solar Energy Materials and Solar Cells. 2022. link Times cited: 5 NOT USED (low confidence) X. Ou, J. Sietsma, and M. Santofimia, “Fundamental study of nonclassical nucleation mechanisms in iron,” Acta Materialia. 2022. link Times cited: 6 NOT USED (low confidence) A. Mishra, J. Lind, M. Kumar, and A. Dongare, “Understanding the phase transformation mechanisms that affect the dynamic response of Fe-based microstructures at the atomic scales,” Journal of Applied Physics. 2021. link Times cited: 8 NOT USED (low confidence) K. Hamad, Y. Choi, and U. M. Chaudry, “Atomic scale insights into the plasticity of iron-phosphorus alloy,” Materials Letters. 2021. link Times cited: 1 NOT USED (low confidence) Y. Sun, X. Huang, F. Liu, and H. Chu, “Equivalent surface energy of nanovoids in metallic crystals,” Computational Materials Science. 2021. link Times cited: 3 NOT USED (low confidence) L. Malerba et al., “Physical mechanisms and parameters for models of microstructure evolution under irradiation in Fe alloys – Part I: Pure Fe,” Nuclear Materials and Energy. 2021. link Times cited: 8 NOT USED (low confidence) X. Xing et al., “Hydrogen effect on the intergranular failure in polycrystal ɑ-iron with different crystal sizes,” International Journal of Hydrogen Energy. 2021. link Times cited: 7 NOT USED (low confidence) E. Antillon, C. Woodward, S. Rao, and B. Akdim, “Chemical short range order strengthening in BCC complex concentrated alloys,” Acta Materialia. 2021. link Times cited: 35 NOT USED (low confidence) H. Min et al., “Development of an interatomic potential for Fe-He by neural network,” Computational Materials Science. 2021. link Times cited: 2 NOT USED (low confidence) S. Peeters, C. Charrin, I. Duron, S. Loehl’e, B. Thiébaut, and M. Righi, “Importance of the catalytic effect of the substrate in the functionality of lubricant additives: the case of molybdenum dithiocarbamates,” Materials Today Chemistry. 2021. link Times cited: 7 NOT USED (low confidence) J. Çamkıran, F. Parsch, and G. Hibbard, “A local orientational order parameter for systems of interacting particles.,” The Journal of chemical physics. 2021. link Times cited: 3 Abstract: Many physical systems are well modeled as collections of int… read moreAbstract: Many physical systems are well modeled as collections of interacting particles. Nevertheless, a general approach to quantifying the absolute degree of order immediately surrounding a particle has yet to be described. Motivated thus, we introduce a quantity E that captures the amount of pairwise informational redundancy among the bonds formed by a particle. Particles with larger E have less diversity in bond angles and thus simpler neighborhoods. We show that E possesses a number of intuitive mathematical properties, such as increasing monotonicity in the coordination number of Platonic polyhedral geometries. We demonstrate analytically that E is, in principle, able to distinguish a wide range of structures and conjecture that it is maximized by the icosahedral geometry under the constraint of equal sphere packing. An algorithm for computing E is described and is applied to the structural characterization of crystals and glasses. The findings of this study are generally consistent with existing knowledge on the structure of such systems. We compare E to the Steinhardt order parameter Q6 and polyhedral template matching (PTM). We observe that E has resolution comparable to Q6 and robustness similar to PTM despite being much simpler than the former and far more informative than the latter. read less NOT USED (low confidence) C. Canbay and S. Kazanç, “Fe Elementinin Kristal ve Camsı Faza Dönüşümünün Hidrostatik Basınç Altında İncelenmesi: Moleküler Dinamik Benzetim Çalışması.” 2021. link Times cited: 0 NOT USED (low confidence) T. Ye et al., “Primary radiation damage characteristics in displacement cascades of FeCrAl alloys,” Journal of Nuclear Materials. 2021. link Times cited: 14 NOT USED (low confidence) J. Johny et al., “Multidimensional thermally-induced transformation of nest-structured complex Au-Fe nanoalloys towards equilibrium,” Nano Research. 2021. link Times cited: 12 NOT USED (low confidence) I. Srivastava, A. Kotia, S. Ghosh, and M. Ali, “Recent advances of molecular dynamics simulations in nanotribology,” Journal of Molecular Liquids. 2021. link Times cited: 26 NOT USED (low confidence) S. Eder, S. Leroch, P. Grützmacher, T. Spenger, and H. Heckes, “A multiscale simulation approach to grinding ferrous surfaces for process optimization,” International Journal of Mechanical Sciences. 2021. link Times cited: 26 NOT USED (low confidence) H. Xie, T. Ma, T. Yu, and F. Yin, “Body-centered-cubic to face-centered-cubic phase transformation of iron under compressive loading along [100] direction,” Materials today communications. 2021. link Times cited: 1 NOT USED (low confidence) N. Bertin, W. Cai, S. Aubry, and V. Bulatov, “Core energies of dislocations in bcc metals,” Physical Review Materials. 2021. link Times cited: 5 Abstract: Accurate methods and an efficient workflow for computing and… read moreAbstract: Accurate methods and an efficient workflow for computing and documenting dislocation core energies are developed and applied to $\frac{1}{2}\ensuremath{\langle}111\ensuremath{\rangle}$ and $\ensuremath{\langle}100\ensuremath{\rangle}$ dislocations in five body-centered cubic (bcc) metals W, Ta, V, Mo, and $\ensuremath{\alpha}$-Fe represented by 13 model interatomic potentials. For each dislocation type, dislocation core energies are extracted for a large number of dislocation characters thoroughly sampling the entire 2-space of crystallographic line orientations of the bcc lattice. Of particular interest, core energies of the $\frac{1}{2}\ensuremath{\langle}111\ensuremath{\rangle}{110}$ dislocations are found to be distinctly asymmetric with respect to the sign of the character angle, whereas core energies of $\ensuremath{\langle}100\ensuremath{\rangle}{110}$ junction dislocations exhibit marked cusps for line orientations vicinal to the closed-packed $\ensuremath{\langle}111\ensuremath{\rangle}$ directions. Our findings furnish substantial insights for developing accurate models of dislocation core energies employed in mesoscale dislocation dynamics simulations of crystal plasticity. read less NOT USED (low confidence) N. Gao, Z. Yao, G. Lu, H. Deng, and F. Gao, “Mechanisms for <100> interstitial dislocation loops to diffuse in BCC iron,” Nature Communications. 2021. link Times cited: 23 NOT USED (low confidence) N. Gao, Z. Yao, G. Lu, H. Deng, and F. Gao, “Mechanisms for <100> interstitial dislocation loops to diffuse in BCC iron,” Nature Communications. 2021. link Times cited: 0 NOT USED (low confidence) K. Lai, K. Li, H. Wen, Q. Guo, B. Wang, and Y. Zheng, “Synergistic effects of applied strain and cascade overlap on irradiation damage in BCC iron,” Journal of Nuclear Materials. 2020. link Times cited: 9 NOT USED (low confidence) L. A. Mistryukova, N. P. Kryuchkov, I. Aliev, and S. Yurchenko, “Efficient approach to calculating radial distribution function in bcc Fe lattice,” Journal of Physics: Conference Series. 2020. link Times cited: 1 Abstract: Many properties of condensed matter systems can be described… read moreAbstract: Many properties of condensed matter systems can be described by means of pair correlation functions that makes them an important structural characteristic. The shortest-graph interpolation method allows us to calculate pair correlation functions of classical crystals with pairwise interactions between particles. However, there is still no just so simple and practical approach to predict correlation functions in crystals with many-body interactions that are ubiquitous in nature. In this work, a simple modification of the interpolation method is suggested allowing to describe pair correlations bcc Fe lattice, considered as a classical crystal with many-body interactions of embedded atom model type. It is shown that the radial distribution function of the crystal can be calculated with high accuracy if mean square displacements are known. The obtained results would be useful in various fields of condensed matter physics, materials science, and crystallography. read less NOT USED (low confidence) X. He, Q. Bai, R. Shen, F.-hu Zhang, and Y.-bo Guo, “The evolution of configuration and final state of graphene on rough iron surface,” Applied Surface Science. 2020. link Times cited: 10 NOT USED (low confidence) B. Zhang, Y. Wang, J. Chen, J. Li, and W. Lai, “Development of an angular-dependent potential for radiation damage study in Fe-Si solutions,” Journal of Nuclear Materials. 2020. link Times cited: 3 NOT USED (low confidence) S. E. Restrepo, H. Lambert, and A. Paxton, “Effect of hydrogen on vacancy diffusion,” Physical Review Materials. 2020. link Times cited: 4 NOT USED (low confidence) D. Yavas, T. Phan, L. Xiong, K. Hebert, and A. Bastawros, “Mechanical degradation due to vacancies produced by grain boundary corrosion of steel,” Acta Materialia. 2020. link Times cited: 7 NOT USED (low confidence) K. Zhao and R. Aghababaei, “Interfacial plasticity controls material removal rate during adhesive sliding contact,” Physical Review Materials. 2020. link Times cited: 12 NOT USED (low confidence) K. Zhao and R. Aghababaei, “Adhesive wear law at the single asperity level,” Journal of The Mechanics and Physics of Solids. 2020. link Times cited: 26 NOT USED (low confidence) X.-D. Pan et al., “Effect of H on the formation of vacancy dislocation loops in α-Fe,” Journal of Nuclear Materials. 2020. link Times cited: 5 NOT USED (low confidence) W. Wei and X. Yu, “Molecular dynamics study of the effect of lithium on the tensile behaviors of bcc iron,” Materials Today Communications. 2020. link Times cited: 5 NOT USED (low confidence) Y. Sato, S. Shinzato, T. Ohmura, T. Hatano, and S. Ogata, “Unique universal scaling in nanoindentation pop-ins,” Nature Communications. 2020. link Times cited: 31 NOT USED (low confidence) R. Alexander et al., “Interatomic potentials for irradiation-induced defects in iron,” Journal of Nuclear Materials. 2020. link Times cited: 13 NOT USED (low confidence) B. Chen, S. Li, H. Zong, X. Ding, J. Sun, and E. Ma, “Unusual activated processes controlling dislocation motion in body-centered-cubic high-entropy alloys,” Proceedings of the National Academy of Sciences of the United States of America. 2020. link Times cited: 90 Abstract: Significance This work demonstrates dislocation dynamics in … read moreAbstract: Significance This work demonstrates dislocation dynamics in body-centered-cubic (BCC) high-entropy alloys (HEAs). The local composition inhomogeneity in these concentrated solutions leads to an unconventional rate-controlling mechanism that governs dislocation mobility. The activated process becomes nanoscale detrapping, of kinks on screw dislocations and of trapped segments of edge dislocations, presenting an activation barrier of similar magnitude for both dislocation types. Their sluggish mobility explains the elevated strength and strain hardening in BCC HEAs. Atomistic simulations of dislocation mobility reveal that body-centered cubic (BCC) high-entropy alloys (HEAs) are distinctly different from traditional BCC metals. HEAs are concentrated solutions in which composition fluctuation is almost inevitable. The resultant inhomogeneities, while locally promoting kink nucleation on screw dislocations, trap them against propagation with an appreciable energy barrier, replacing kink nucleation as the rate-limiting mechanism. Edge dislocations encounter a similar activated process of nanoscale segment detrapping, with comparable activation barrier. As a result, the mobility of edge dislocations, and hence their contribution to strength, becomes comparable to screw dislocations. read less NOT USED (low confidence) F. Shuang and K. Aifantis, “Relating the strength of graphene/metal composites to the graphene orientation and position,” Scripta Materialia. 2020. link Times cited: 38 NOT USED (low confidence) H. Mori and T. Ozaki, “Neural network atomic potential to investigate the dislocation dynamics in bcc iron,” Physical Review Materials. 2020. link Times cited: 23 Abstract: To design the mechanical strength of body-centered-cubic (bc… read moreAbstract: To design the mechanical strength of body-centered-cubic (bcc) iron, clarifying the dislocation dynamics is very important. Using systematically constructed reference data based on density functional theory (DFT) calculations, we construct an atomic artificial neural network (ANN) potential to investigate the dislocation dynamics in bcc iron with the accuracy of DFT calculations. The bulk properties and defect formation energies predicted by the constructed ANN potential are in good agreement with the reference DFT calculations. The ${a}_{0}/2\ensuremath{\langle}111\ensuremath{\rangle}{110}$ screw dislocation core structure predicted by the ANN potential is compact and nondegenerate. The Peierls barrier predicted by the ANN potential is 35.3 meV per length of the Burgers vector. These results are consistent with the DFT results. Furthermore, not only the Peierls barrier, but also the two-dimensional energy profile of the screw dislocation core position predicted by the ANN potential are in excellent agreement with the DFT results. These results clearly demonstrate the reproducibility and transferability of the constructed ANN potential for investigating dislocation dynamics with the accuracy of the DFT. Combined with advanced atomistic techniques, the ANN potential will be highly useful for investigating the dislocation dynamics in bcc iron at finite temperatures. read less NOT USED (low confidence) B. Faria, C. Guarda, N. Silvestre, and J. Lopes, “CNT-reinforced iron and titanium nanocomposites: Strength and deformation mechanisms,” Composites Part B-engineering. 2020. link Times cited: 23 NOT USED (low confidence) A. F. Galvis, P. A. Santos-Flórez, P. Sollero, M. de Koning, and L. Wrobel, “Multiscale model of the role of grain boundary structures in the dynamic intergranular failure of polycrystal aggregates,” Computer Methods in Applied Mechanics and Engineering. 2020. link Times cited: 6 NOT USED (low confidence) A. Dahlström, F. Danoix, P. Hedström, J. Odqvist, and H. Zapolsky, “Nanostructure in Fe0.65Cr0.35 close to the upper limit of the miscibility gap,” Scripta Materialia. 2020. link Times cited: 2 NOT USED (low confidence) R. Ishraaq, S. Nahid, S. Chhetri, O. Gautam, and A. Afsar, “A molecular dynamics investigation for predicting the optimum fiber radius and the effect of various parameters on the mechanical properties of carbon nanotube reinforced iron composite,” Computational Materials Science. 2020. link Times cited: 6 NOT USED (low confidence) S. Kim, H. Kim, K. Kang, and S. Y. Kim, “Relativistic effect inducing drag on fast-moving dislocation in discrete system,” International Journal of Plasticity. 2020. link Times cited: 19 NOT USED (low confidence) Q. Ye et al., “Theoretical development and experimental validation on the measurement of temperature by extended X-ray absorption fine structure.,” Journal of synchrotron radiation. 2020. link Times cited: 1 Abstract: A systematic investigation on the theoretical framework of t… read moreAbstract: A systematic investigation on the theoretical framework of the ultra-fast measurement of temperature by extended X-ray absorption fine structure (EXAFS) applied in laser-driven-compression experiments has been carried out and a new temperature measurement scheme based on the EXAFS cumulant expansion analysis and anharmonic correlated Debye model has been advanced. By considering the anharmonic effect of thermal vibration and avoiding the employment of the empirical model as well as parameters which have large inherent uncertainties in the temperature determination, this new scheme is theoretically more accurate than traditional ones. Then the performance of the new measurement scheme and traditional methods were validated on a synchrotron radiation platform by temperature-dependent EXAFS (TDEXAFS) experiments on Au, Fe, V and Ti; the results showed that the new scheme could provide the most accurate measured temperatures with much lower uncertainties. This accurate scheme gives a firmer physical ground to the EXAFS temperature measurement technique and can expect to be applied in laser-driven compression experiments and promote the development of matter state research at extreme conditions. read less NOT USED (low confidence) B. Yao and R. F. Zhang, “AADIS: An atomistic analyzer for dislocation character and distribution,” Comput. Phys. Commun. 2020. link Times cited: 19 NOT USED (low confidence) Z. Zhao, Z.-ye Qin, and F. Chu, “Asymmetrical propagation mechanism of the crack in bcc iron,” Computational Materials Science. 2020. link Times cited: 11 NOT USED (low confidence) H. Ma, P. Gao, P. Qian, and Y. Su, “Size-Dependent Electrochemical Properties of Pure Metallic Nanoparticles,” The Journal of Physical Chemistry C. 2020. link Times cited: 8 Abstract: A generalized size-dependent thermodynamic model was derived… read moreAbstract: A generalized size-dependent thermodynamic model was derived to describe the electrochemical properties of nanoparticles, which takes into account the effects of size-dependent stress distributions... read less NOT USED (low confidence) H. Gao and M. Müser, “Why liquids can appear to solidify during squeeze-out - Even when they don’t.,” Journal of colloid and interface science. 2019. link Times cited: 7 NOT USED (low confidence) Z. Sun, F. Z. Dai, B. Xu, and W.-Z. Zhang, “Dislocation-mediated migration of interphase boundaries,” Journal of Materials Science & Technology. 2019. link Times cited: 7 NOT USED (low confidence) A. Nikonov and A. M. Zharmukhambetova, “Molecular dynamic investigation of acoustic emission during mechanical treatment,” PROCEEDINGS OF THE INTERNATIONAL CONFERENCE ON ADVANCED MATERIALS WITH HIERARCHICAL STRUCTURE FOR NEW TECHNOLOGIES AND RELIABLE STRUCTURES 2019. 2019. link Times cited: 0 NOT USED (low confidence) M. A. A. Hasan, J. Wang, Y. Lim, A. Hu, and S. Shin, “Concentration dependence of hydrogen diffusion in α-iron from atomistic perspectives,” International Journal of Hydrogen Energy. 2019. link Times cited: 9 NOT USED (low confidence) K. Li et al., “Determination of the accuracy and reliability of molecular dynamics simulations in estimating the melting point of iron: Roles of interaction potentials and initial system configurations,” Journal of Molecular Liquids. 2019. link Times cited: 8 NOT USED (low confidence) S. M. Zamzamian, S. Feghhi, M. Samadfam, and M. Darvishzadeh, “Atomistic investigation of the effects of symmetric tilt grain boundary structures on irradiation response of the α-Fe containing carbon in solution,” Computational Materials Science. 2019. link Times cited: 4 NOT USED (low confidence) Y. Zhang, D. Schwen, Y. Zhang, and X. Bai, “Effects of oversized tungsten on the primary damage behavior in Fe-W alloys,” Journal of Alloys and Compounds. 2019. link Times cited: 8 NOT USED (low confidence) S. Teus and V. Gavriljuk, “Hydrogen Effect on the Electron Structure and Grain Boundary Mobility in the Alpha-Iron,” Mechanical Engineering eJournal. 2019. link Times cited: 0 Abstract: The hydrogen-caused change in the electron structure of the … read moreAbstract: The hydrogen-caused change in the electron structure of the alpha-iron along with hydrogen effect on the mobility of grain boundaries has been studied using the first-principles atomic calculations and molecular dynamics. It is shown that hydrogen entry into the alpha-iron crystal lattice increases the density of electron states at the Fermi level, which suggests the increase in the concentration of free electrons. The studied spatial distribution of electron density gives the evidence for corresponding enhancement of the metallic character of interatomic bonds. The obtained result is consistent with the electron approach to the hypothesis of hydrogen-enhanced localized plasticity (HELP) for a mechanism of hydrogen embrittlement of the iron-based alloys. Using the molecular dynamics simulation, it is obtained that hydrogen segregation at the grain boundaries increases their mobility. This effect is attributed to the hydrogen-increased concentration of free electrons, which weakens rigidity of interatomic bonds along with the decrease of the grain boundary surface tension. read less NOT USED (low confidence) K. H. Lee, V. Vuong, V. Fung, D. Jiang, and S. Irle, “Density-Functional Tight-Binding for Platinum Clusters and Bulk: Electronic vs Repulsive Parameters,” MRS Advances. 2019. link Times cited: 2 Abstract: We present a general purpose Pt-Pt density-functional tight-… read moreAbstract: We present a general purpose Pt-Pt density-functional tight-binding (DFTB) parameter for Pt clusters as well as bulk, using a genetic algorithm (GA) to automatize the parameterization effort. First we quantify the improvement possible by only optimizing the repulsive potential alone, and second we investigate the effect of improving the electronic parameter as well. During both parameterization efforts we employed our own training set and test sets, with one set containing ∼20,000 spin-polarized DFT structures. We analyze the performance of our two DFTB Pt-Pt parameter sets against density functional theory (DFT) as well as an earlier DFTB Pt-Pt parameters. Our study sheds light on the role of both repulsive and electronic parameters with regards to DFTB performance. read less NOT USED (low confidence) X. Xing, H. Zhang, G. Cui, J. Liu, and Z. Li, “Hydrogen inhibited phase transition near crack tip – An atomistic mechanism of hydrogen embrittlement,” International Journal of Hydrogen Energy. 2019. link Times cited: 11 NOT USED (low confidence) M. Zhang, W. Peng, H. Zhang, B.-jie Wu, K. Sun, and L. Fang, “The effect of PKA directions on the primary radiation damage in the alpha iron nanowires,” Materials Chemistry and Physics. 2019. link Times cited: 2 NOT USED (low confidence) M. S. Talaei, N. Nouri, and S. Ziaei-Rad, “An optimized approach for computing coincidence-site-lattice grain boundary energy,” Computational Condensed Matter. 2019. link Times cited: 3 NOT USED (low confidence) X. Ou and M. Song, “Deformation mechanisms of mechanically induced phase transformations in iron,” Computational Materials Science. 2019. link Times cited: 13 NOT USED (low confidence) Z. Liu et al., “Development of interatomic potentials for Fe-Cr-Al alloy with the particle swarm optimization method,” Journal of Alloys and Compounds. 2019. link Times cited: 20 NOT USED (low confidence) Y. Pachaury and Y. Shin, “Assessment of sub-surface damage during machining of additively manufactured Fe-TiC metal matrix composites,” Journal of Materials Processing Technology. 2019. link Times cited: 21 NOT USED (low confidence) X. Wang et al., “Formation of ⟨100⟩ dislocation loop in bcc-Fe via the ternary loop reaction,” Scripta Materialia. 2019. link Times cited: 10 NOT USED (low confidence) H. S. Kim, J. H. Kim, S.-H. Cha, and S. Cho, “Optimal determination of force field parameters for reduced molecular dynamics model,” Comput. Phys. Commun. 2019. link Times cited: 2 NOT USED (low confidence) J.-Y. Zhang, Y. Gao, Y. Wang, and W.-Z. Zhang, “A generalized O-element approach for analyzing interface structures,” Acta Materialia. 2019. link Times cited: 5 NOT USED (low confidence) M. H. Nazir, Z. Khan, A. Saeed, V. Bakolas, W. Braun, and R. Bajwa, “Experimental analysis and modelling for reciprocating wear behaviour of nanocomposite coatings,” Wear. 2018. link Times cited: 19 NOT USED (low confidence) K. Zhao, A. Mayer, J. He, and Z. Zhang, “Dislocation based plasticity in the case of nanoindentation,” International Journal of Mechanical Sciences. 2018. link Times cited: 21 NOT USED (low confidence) B. D. Snartland, A. Alvaro, V. Osen, and C. Thaulow, “Crack arrest testing at the micro-scale,” Engineering Fracture Mechanics. 2018. link Times cited: 4 NOT USED (low confidence) J. Byggmästar, F. Granberg, and K. Nordlund, “Effects of the short-range repulsive potential on cascade damage in iron,” Journal of Nuclear Materials. 2018. link Times cited: 52 NOT USED (low confidence) W. Jeong, K. Lee, D. Yoo, D. Lee, and S. Han, “Toward Reliable and Transferable Machine Learning Potentials: Uniform Training by Overcoming Sampling Bias,” The Journal of Physical Chemistry C. 2018. link Times cited: 29 Abstract: The neural network interatomic potential (NNP) is anticipate… read moreAbstract: The neural network interatomic potential (NNP) is anticipated to be a promising next-generation atomic potential for its self-learning capability and universal mathematical structure. While various examples demonstrate the usefulness of NNPs, we find that the NNP suffers from highly inhomogeneous feature-space sampling in the training set. As a result, underrepresented atomic configurations, often critical for simulations, cause large errors even though they are included in the training set. Using the Gaussian density function (GDF) that quantifies the sparsity of training points, we propose a weighting scheme that can effectively rectify the sampling bias. Various examples confirm that GDF weighting significantly improves the reliability and transferability of NNPs compared to the conventional training method, which is attributed to accurate mapping of atomic energies. By addressing a detrimental problem that is inherent in every machine learning potential, the present work will extend the application ra... read less NOT USED (low confidence) T. Sipkens and K. Daun, “Effect of Surface Interatomic Potential on Thermal Accommodation Coefficients Derived from Molecular Dynamics,” The Journal of Physical Chemistry C. 2018. link Times cited: 14 Abstract: This work investigates how the interatomic surface potential… read moreAbstract: This work investigates how the interatomic surface potential influences molecular dynamics (MD)-derived thermal accommodation coefficients (TACs). Iron, copper, and silicon surfaces are considered over a range of temperatures that include their melting points. Several classes of potentials are reviewed, including two-body, three-body, and bond-order force fields. MD-derived densities and visualization of the surfaces are used to explain the differences in the parameterizations of these potentials within the context of gas–surface scattering. Finally, TACs are predicted for a range of gas–surface combinations, and recommended values of the TAC are selected that take into account the robustness and uncertainties of each of the considered parameterizations. Further, it is observed that there is a significant change in the TAC about phase changes that must be taken into account for applications with a large range of surface temperatures. read less NOT USED (low confidence) P. Tripathi, S. K. Maurya, and S. Bhowmick, “Role of disconnections in mobility of the austenite-ferrite interphase boundary in Fe,” Physical Review Materials. 2018. link Times cited: 6 Abstract: Austenite ({\gamma}-Fe, face centered cubic (FCC)) to ferrit… read moreAbstract: Austenite ({\gamma}-Fe, face centered cubic (FCC)) to ferrite ({\alpha}-Fe, body centered cubic (BCC)) phase transformation in steel is of great significance from the point of view of industrial applications. In this work, using classical molecular dynamics simulations, we study the atomistic mechanisms involved during the nucleation and growth of the ferrite phase embedded in an austenite phase. We find that the disconnections present at the inter-phase boundary can act as the nucleation centers for the ferrite phase. Relatively small interface velocities (1.19 - 4.67 m/s) confirm a phase change via massive transformation mechanism. Boundary mobilities obtained in a temperature range of 1000 to 1400 K show an Arrhenius behavior, with activation energies ranging from 30 - 40 kJ/mol. read less NOT USED (low confidence) G. Lv, M. Zhang, H. Zhang, and Y. Su, “Hydrogen diffusion and vacancy clusterization in iron,” International Journal of Hydrogen Energy. 2018. link Times cited: 13 NOT USED (low confidence) M. Widom, “Modeling the structure and thermodynamics of high-entropy alloys,” Journal of Materials Research. 2018. link Times cited: 72 Abstract: High-entropy and multiprincipal element alloys present excit… read moreAbstract: High-entropy and multiprincipal element alloys present exciting opportunities and challenges for computational modeling of their structure and phase stability. Recent interest has catalyzed rapid development of techniques and equally rapid growth of new results. This review surveys the essential concepts of thermodynamics and total energy calculation, and the bridge between them provided by statistical mechanics. Specifically, we review the electronic density functional theory of alloy total energy as applied to supercells and special quasirandom structures. We contrast these with the coherent potential approximation and semi-empirical approximations. Statistical mechanical approaches include cluster expansions, hybrid Monte Carlo/molecular dynamics simulations, and extraction of entropy from correlation functions. We also compare first-principles approaches with Calculation of Phase Diagrams (CALPHAD) and highlight the need to augment experimental databases with first-principles derived data. Numerous example applications are given highlighting recent progress utilizing the concepts and methods that are introduced. read less NOT USED (low confidence) S. Eder, U. Cihak-Bayr, C. Gachot, and M. R. Ripoll, “Interfacial Microstructure Evolution Due to Strain Path Changes in Sliding Contacts.,” ACS applied materials & interfaces. 2018. link Times cited: 16 Abstract: We performed large-scale molecular dynamics (MD) simulations… read moreAbstract: We performed large-scale molecular dynamics (MD) simulations to study the transient softening stage that has been observed experimentally in sliding interfaces subject to strain path changes. The occurrence of this effect can be of crucial importance for the energy efficiency and wear resistance of systems that experience changes in the sliding direction, such as bearings or gears in wind parks, piston rings in combustion engines, or wheel-rail contacts for portal cranes. We therefore modeled the sliding of a rough counterbody against two polycrystalline substrates of face-centered cubic (fcc) copper and body-centered cubic (bcc) iron with initial near-surface grain sizes of 40 nm. The microstructural development of these substrates was monitored and quantified as a function of time, depth, and applied pressure during unidirectional sliding for 7 ns. The results were then compared to the case of sliding in one direction for 5 ns and reversing the sliding direction for an additional 2 ns. We observed the generation of partial dislocations, grain refinement, and rotation as well as twinning (for fcc) in the near-surface region. All microstructures were increasingly affected by these processes when maintaining the sliding direction but recovered to a great extent upon sliding reversal up to applied pressures of 0.4 GPa in the case of fcc Cu and 1.5 GPa for bcc Fe. We discuss the applicability and limits of our polycrystalline MD model for reproducing well-known bulk phenomena such as the Bauschinger effect in interfacial processes. read less NOT USED (low confidence) M. Mendelev et al., “Molecular dynamics simulation of the solid-liquid interface migration in terbium.,” The Journal of chemical physics. 2018. link Times cited: 16 Abstract: We developed a Tb embedded atom method potential which prope… read moreAbstract: We developed a Tb embedded atom method potential which properly reproduces the liquid structure obtained from the ab initio molecular dynamics simulation, the hexagonal close packed (hcp)-body-centered cubic (bcc) phase transformation, and melting temperatures. At least three crystal phases [hcp, face-centered cubic (fcc), and bcc] described by this potential can coexist with the liquid phase. Thus, the developed potential provides an excellent test bed for studies of the completive phase nucleation and growth in a single component system. The molecular dynamics simulation showed that all crystal phases can grow from the liquid phase close to their melting temperatures. However, in the cases of the hcp and fcc growth from the liquid phase at very large supercoolings, the bcc phase forms at the solid-liquid interface in the close packed orientations in spite of the fact that both hcp and fcc phases are more stable than the bcc phase at these temperatures. This bcc phase closes the hcp and fcc phase from the liquid such that the remaining liquid solidifies into the bcc phase. The initial hcp phase then slowly continues growing in expense of the bcc phase. read less NOT USED (low confidence) F. Ye, K. Tong, Y. K. Wang, Z. Li, and F. Zhou, “First-principles study of interaction between vacancies and nitrogen atoms in fcc iron,” Computational Materials Science. 2018. link Times cited: 8 NOT USED (low confidence) N. Castin et al., “Advanced atomistic models for radiation damage in Fe-based alloys: Contributions and future perspectives from artificial neural networks,” Computational Materials Science. 2018. link Times cited: 21 NOT USED (low confidence) D. Lin et al., “Shock engineering the additive manufactured graphene-metal nanocomposite with high density nanotwins and dislocations for ultra-stable mechanical properties,” Acta Materialia. 2018. link Times cited: 69 NOT USED (low confidence) Y. Dou, X. He, D. Wang, W. Shi, L. Jia, and W. Yang, “The Study of Nanosized Cu–Mn Precipitates Contribution to Hardening in Body Centered Cubic Fe Matrix,” Journal of Nuclear Engineering and Radiation Science. 2018. link Times cited: 1 Abstract: In order to study the contribution of manganese (Mn) atoms i… read moreAbstract: In order to study the contribution of manganese (Mn) atoms in copper (Cu) precipitates to hardening in body centered cubic (BCC) structure iron (Fe) matrix, the interactions of a 1/2 〈111〉 {110} edge dislocations with nanosized Cu and Cu–Mn precipitates in BCC Fe have been investigated by using molecular dynamics method (MD). The results indicate that the critical resolved shear stresses (τc) of the Cu–Mn precipitates are larger than that of Cu precipitates. Meanwhile, τc of the Cu–Mn precipitates show a much more significant dependence on temperature and size compared to Cu precipitates. Mn atoms exhibit strong attraction to dislocation segment in Cu precipitate and improve the fraction of transformed atoms from BCC phase to nine rhombohedron (R) phase for big size precipitates. Those all lead to the higher resistance to the dislocation glide. Eventually, these features confirmed that the appearance of Mn atoms in Cu precipitates greatly facilitates the hardening in BCC Fe matrix. read less NOT USED (low confidence) X. He, Q. Bai, and R. Shen, “Atomistic perspective of how graphene protects metal substrate from surface damage in rough contacts,” Carbon. 2018. link Times cited: 40 NOT USED (low confidence) M. A. Zaeem and E. Asadi, “Phase‐Field Crystal Modeling: Integrating Density Functional Theory, Molecular Dynamics, and Phase‐Field Modeling.” 2018. link Times cited: 2 NOT USED (low confidence) X.-yan Li et al., “On the possibility of universal interstitial emission induced annihilation in metallic nanostructures,” Journal of Nuclear Materials. 2018. link Times cited: 9 NOT USED (low confidence) N. Ren, B. Shang, P. Guan, and L. Hu, “General structural and dynamic characteristics beneficial to glass-forming ability of Fe-based glass-forming liquids,” Journal of Non-crystalline Solids. 2018. link Times cited: 7 NOT USED (low confidence) A. Nikonov, “Influence of vibration on acoustic emission during mechanical treatment. Molecular dynamics study.” 2017. link Times cited: 0 NOT USED (low confidence) G. Vetterick et al., “Achieving Radiation Tolerance through Non-Equilibrium Grain Boundary Structures,” Scientific Reports. 2017. link Times cited: 36 NOT USED (low confidence) X. Yang, S. Sun, H. Ruan, S. Shi, and T.-Y. Zhang, “Shear and shuffling accomplishing polymorphic fcc γ → hcp ε → bct α martensitic phase transformation,” Acta Materialia. 2017. link Times cited: 71 NOT USED (low confidence) T. D. Ta, A. K. Tieu, H. Zhu, B. Kosasih, Q. Zhu, and H. Phan, “The structural, tribological, and rheological dependency of thin hexadecane film confined between iron and iron oxide surfaces under sliding conditions,” Tribology International. 2017. link Times cited: 20 NOT USED (low confidence) C. Ruestes, E. Bringa, Y. Gao, and H. Urbassek, “Molecular Dynamics Modeling of Nanoindentation.” 2017. link Times cited: 35 NOT USED (low confidence) A. Filippov, A. Nikonov, V. Rubtsov, A. Dmitriev, and S. Tarasov, “Vibration and acoustic emission monitoring the stability of peakless tool turning: Experiment and modeling,” Journal of Materials Processing Technology. 2017. link Times cited: 56 NOT USED (low confidence) A. Elzas and B. Thijsse, “Cohesive law describing crack growth at iron/precipitate interfaces,” Computational Materials Science. 2017. link Times cited: 8 NOT USED (low confidence) P. Saidi, C. Dai, T. Power, Z. Yao, and M. Daymond, “An embedded atom method interatomic potential for the zirconium-iron system,” Computational Materials Science. 2017. link Times cited: 5 NOT USED (low confidence) J. Mo et al., “Effects of pressure on structure and mechanical property in monatomic metallic glass,” Journal of Non-crystalline Solids. 2017. link Times cited: 16 NOT USED (low confidence) A. B. Hagen, B. D. Snartland, and C. Thaulow, “Temperature and orientation effects on the deformation mechanisms of α-Fe micropillars,” Acta Materialia. 2017. link Times cited: 41 NOT USED (low confidence) W. Ko and J. Jeon, “Interatomic potential that describes martensitic phase transformations in pure lithium,” Computational Materials Science. 2017. link Times cited: 10 NOT USED (low confidence) S. Li, E. Salje, S. Jun, and X. Ding, “Large recovery of six-fold twinned nanowires of α-Fe,” Acta Materialia. 2017. link Times cited: 11 NOT USED (low confidence) G. Bonny, A. Bakaev, P. Olsson, C. Domain, E. Zhurkin, and M. Posselt, “Interatomic potential to study the formation of NiCr clusters in high Cr ferritic steels,” Journal of Nuclear Materials. 2017. link Times cited: 17 NOT USED (low confidence) J. Fan, R. Stewart, and T. Xu, “Simulation accuracy of crack-tip parameters with extended GP methods,” Engineering Fracture Mechanics. 2017. link Times cited: 4 NOT USED (low confidence) R. Ocaya and J. J. Terblans, “C-language package for standalone embedded atom method molecular dynamics simulations of fcc structures,” SoftwareX. 2016. link Times cited: 4 NOT USED (low confidence) N. Karkalos and A. Markopoulos, “Modeling Nano-Metric Manufacturing Processes with Molecular Dynamics Method: A Review,” Current Nanoscience. 2016. link Times cited: 9 NOT USED (low confidence) S. Hayakawa, T. Okita, M. Itakura, M. Aichi, S. Fujita, and K. Suzuki, “Behavior of a self-interstitial-atom type dislocation loop in the periphery of an edge dislocation in BCC-Fe,” Nuclear materials and energy. 2016. link Times cited: 8 NOT USED (low confidence) T. Lu et al., “Molecular dynamics study of the diffusion properties of H in Fe with point defects,” Fusion Engineering and Design. 2016. link Times cited: 17 NOT USED (low confidence) K. Zhao, I. G. Ringdalen, J. Wu, J. Wu, J. He, and Z. Zhang, “Ductile mechanisms of metals containing pre-existing nanovoids,” Computational Materials Science. 2016. link Times cited: 12 NOT USED (low confidence) A. Korchuganov, “Free surface damage induced by irradiation of BCC iron.” 2016. link Times cited: 1 Abstract: The influence of the crystallographic orientation of bcc iro… read moreAbstract: The influence of the crystallographic orientation of bcc iron samples on the character of structural changes near the free surface irradiated with ions was studied in the framework of a molecular dynamics method. Irradiation of the (111) surface leads to the formation of craters surrounded by atoms escaped on the surface (adatoms). In the case of the (110) surface irradiation, a vacancy-type dislocation loop with the Burgers vector a 〈100〉 or a/2 〈111〉 was formed. The number of adatoms and survived point defects was greater in the sample with the (110) surface than in the sample with the (111) surface for the atomic displacement cascade energies lower than 20 keV. The influence of the irradiated surface orientation on the number of generated point defects decreased with the increasing atomic displacement cascade energy. read less NOT USED (low confidence) C. M. Mangiardi and R. Meyer, “A hybrid algorithm for parallel molecular dynamics simulations,” Comput. Phys. Commun. 2016. link Times cited: 9 NOT USED (low confidence) A. Fedorov, A. Shulgin, and S. Lavruk, “Study of iron nanoparticle melting.” 2016. link Times cited: 5 Abstract: In paper melting process of iron nanoparticles was investiga… read moreAbstract: In paper melting process of iron nanoparticles was investigated with molecular dynamics method. Melting temperatures was found for particles with radius from 1.5 to 4 nm. Results match with data of other authors. Heat capacity was calculated based on investigation of caloric curves. Dependence between heat capacity and temperature for different size of nanoparticles was approximated. Heat conductivity of iron nanoparticles was calculated. read less NOT USED (low confidence) R. Aghababaei, D. Warner, and J. Molinari, “Critical length scale controls adhesive wear mechanisms,” Nature Communications. 2016. link Times cited: 201 NOT USED (low confidence) H. Ma et al., “Eigenstress model for electrochemistry of solid surfaces,” Scientific Reports. 2016. link Times cited: 18 NOT USED (low confidence) C. M. Mangiardi and R. Meyer, “Molecular-Dynamics Simulations Using Spatial Decomposition and Task-Based Parallelism.” 2016. link Times cited: 2 NOT USED (low confidence) R. Kositski, O. Kovalenko, S.-W. Lee, J. Greer, E. Rabkin, and D. Mordehai, “Cross-Split of Dislocations: An Athermal and Rapid Plasticity Mechanism,” Scientific Reports. 2016. link Times cited: 20 NOT USED (low confidence) X.-yan Li et al., “Radiation resistance of nano-crystalline iron: Coupling of the fundamental segregation process and the annihilation of interstitials and vacancies near the grain boundaries,” Acta Materialia. 2016. link Times cited: 60 NOT USED (low confidence) T. Xu, R. Stewart, J. Fan, X.-guo Zeng, and A. Yao, “Bridging crack propagation at the atomistic and mesoscopic scale for BCC-Fe with hybrid multiscale methods,” Engineering Fracture Mechanics. 2016. link Times cited: 19 NOT USED (low confidence) X.-yan Li et al., “Energetic and kinetic dataset on interaction of the vacancy and self-interstitial atom with the grain boundary in α-iron,” Data in Brief. 2016. link Times cited: 5 NOT USED (low confidence) J. Zhao et al., “Formation Mechanism of Fe Nanocubes by Magnetron Sputtering Inert Gas Condensation.,” ACS nano. 2016. link Times cited: 88 Abstract: In this work, we study the formation mechanisms of iron nano… read moreAbstract: In this work, we study the formation mechanisms of iron nanoparticles (Fe NPs) grown by magnetron sputtering inert gas condensation and emphasize the decisive kinetics effects that give rise specifically to cubic morphologies. Our experimental results, as well as computer simulations carried out by two different methods, indicate that the cubic shape of Fe NPs is explained by basic differences in the kinetic growth modes of {100} and {110} surfaces rather than surface formation energetics. Both our experimental and theoretical investigations show that the final shape is defined by the combination of the condensation temperature and the rate of atomic deposition onto the growing nanocluster. We, thus, construct a comprehensive deposition rate-temperature diagram of Fe NP shapes and develop an analytical model that predicts the temporal evolution of these properties. Combining the shape diagram and the analytical model, morphological control of Fe NPs during formation is feasible; as such, our method proposes a roadmap for experimentalists to engineer NPs of desired shapes for targeted applications. read less NOT USED (low confidence) S.-C. Lin, M.-W. Liu, M. Gururajan, and K.-A. Wu, “Modified Young’s equation for equilibrium dihedral angles of grain boundary grooves in thin films at the nanoscale,” Acta Materialia. 2016. link Times cited: 7 NOT USED (low confidence) T. D. Ta et al., “Tribological Behavior of Aqueous Copolymer Lubricant in Mixed Lubrication Regime.,” ACS applied materials & interfaces. 2016. link Times cited: 26 Abstract: Although a number of experiments have been attempted to inve… read moreAbstract: Although a number of experiments have been attempted to investigate the lubrication of aqueous copolymer lubricant, which is applied widely in metalworking operations, a comprehensive theoretical investigation at atomistic level is still lacking. This study addresses the influence of loading pressure and copolymer concentration on the structural properties and tribological performance of aqueous copolymer solution of poly(propylene oxide)-poly(ethylene oxide)-poly(propylene oxide) (PPO-PEO-PPO) at mixed lubrication using a molecular dynamic (MD) simulation. An effective interfacial potential, which has been derived from density functional theory (DFT) calculations, was employed for the interactions between the fluid's molecules and iron surface. The simulation results have indicated that the triblock copolymer is physisorption on iron surface. Under confinement by iron surfaces, the copolymer molecules form lamellar structure in aqueous solution and behave differently from its bulk state. The lubrication performance of aqueous copolymer lubricant increases with concentration, but the friction reduction is insignificant at high loading pressure. Additionally, the plastic deformation of asperity is dependent on both copolymer concentration and loading pressure, and the wear behavior shows a linear dependence of friction force on the number of transferred atoms between contacting asperities. read less NOT USED (low confidence) J. J. Möller and E. Bitzek, “On the influence of crack front curvature on the fracture behavior of nanoscale cracks,” Engineering Fracture Mechanics. 2015. link Times cited: 26 NOT USED (low confidence) S. Kotrechko et al., “Atomic mechanisms governing upper limit on the strength of nanosized crystals,” Engineering Fracture Mechanics. 2015. link Times cited: 4 NOT USED (low confidence) J. Skogsrud and C. Thaulow, “Application of CTOD in atomistic modeling of fracture,” Engineering Fracture Mechanics. 2015. link Times cited: 10 NOT USED (low confidence) E. S. Wise, M. Liu, and T. Miller, “Sputtering of cubic metal crystals by low-energy xenon-ions,” Computational Materials Science. 2015. link Times cited: 5 NOT USED (low confidence) X. Yang, S. Sun, and T.-Y. Zhang, “The mechanism of bcc α′ nucleation in single hcp ε laths in the fcc γ → hcp ε → bcc α′ martensitic phase transformation,” Acta Materialia. 2015. link Times cited: 104 NOT USED (low confidence) T. Swinburne, “Stochastic Dynamics of Crystal Defects.” 2015. link Times cited: 5 NOT USED (low confidence) S. Eich, D. Beinke, and G. Schmitz, “Embedded-atom potential for an accurate thermodynamic description of the iron–chromium system,” Computational Materials Science. 2015. link Times cited: 24 NOT USED (low confidence) A. B. Sivak, P. A. Sivak, V. A. Romanov, and V. M. Chernov, “Energetic, crystallographic and diffusion characteristics of hydrogen isotopes in iron,” Journal of Nuclear Materials. 2015. link Times cited: 7 NOT USED (low confidence) F. Z. Dai and W.-Z. Zhang, “An automatic and simple method for specifying dislocation features in atomistic simulations,” Comput. Phys. Commun. 2015. link Times cited: 26 NOT USED (low confidence) Y. Gao, C. Ruestes, D. Tramontina, and H. Urbassek, “Comparative simulation study of the structure of the plastic zone produced by nanoindentation,” Journal of The Mechanics and Physics of Solids. 2015. link Times cited: 111 NOT USED (low confidence) Z. W. Wu, M. Li, W. Wang, and K. Liu, “Hidden topological order and its correlation with glass-forming ability in metallic glasses,” Nature Communications. 2015. link Times cited: 111 NOT USED (low confidence) K. Gaminchev and H. Chamati, “Dynamic stability of Fe under high pressure,” Journal of Physics: Conference Series. 2014. link Times cited: 1 Abstract: We study the dynamic stability of bcc iron at both high pres… read moreAbstract: We study the dynamic stability of bcc iron at both high pressure and temperature via Molecular Dynamics in conjunction with three different interatomic potentials constructed within the embedded-atom method. We computed the phonon dispersions, the phonon density of states, as well as the radial distribution functions. It is found that these quantities exhibit different behaviours depending on the potential used. Furthermore it is revealed that the simulated sample remains dynamically stable over a wide range of temperature and pressure for all potentials. read less NOT USED (low confidence) N. Dubinin, N. Vatolin, and V. Filippov, “Thermodynamic perturbation theory in studies of metal melts,” Russian Chemical Reviews. 2014. link Times cited: 30 Abstract: The review concerns methods of the thermodynamic perturbatio… read moreAbstract: The review concerns methods of the thermodynamic perturbation theory (TPT) used to study liquid metals and alloys. Basic relations of the TPT are presented. Various reference systems are analyzed, their advantages and drawbacks are described. The results of calculations of the structure and thermodynamic properties of metal melts by various methods are discussed. Promising avenues of research in the title field are outlined. The bibliography includes 272 references. read less NOT USED (low confidence) T. Liu and S. Groh, “Atomistic modeling of the crack–void interaction in α-Fe,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2014. link Times cited: 32 NOT USED (low confidence) C. Wu, N. Karayiannis, M. Laso, D. Qu, Q. Luo, and J. Shen, “A metric to gauge local distortion in metallic glasses and supercooled liquids,” Acta Materialia. 2014. link Times cited: 9 NOT USED (low confidence) I. Mastorakos and H. Zbib, “A multiscale approach to study the effect of chromium and nickel concentration in the hardening of iron alloys,” Journal of Nuclear Materials. 2014. link Times cited: 7 NOT USED (low confidence) A. Dunn, L. Agudo-Mérida, I. Martín-Bragado, M. Mcphie, M. Cherkaoui, and L. Capolungo, “A novel method for computing effective diffusivity: Application to helium implanted α-Fe thin films,” Journal of Nuclear Materials. 2014. link Times cited: 11 NOT USED (low confidence) S. Teus, V. Mazanko, J. Olive, and V. Gavriljuk, “Grain boundary migration of substitutional and interstitial atoms in α-iron,” Acta Materialia. 2014. link Times cited: 33 NOT USED (low confidence) E. Asadi, M. A. Zaeem, A. Moitra, and M. Tschopp, “Effect of vacancy defects on generalized stacking fault energy of fcc metals,” Journal of Physics: Condensed Matter. 2014. link Times cited: 28 Abstract: Molecular dynamics (MD) and density functional theory (DFT) … read moreAbstract: Molecular dynamics (MD) and density functional theory (DFT) studies were performed to investigate the influence of vacancy defects on generalized stacking fault (GSF) energy of fcc metals. MEAM and EAM potentials were used for MD simulations, and DFT calculations were performed to test the accuracy of different common parameter sets for MEAM and EAM potentials in predicting GSF with different fractions of vacancy defects. Vacancy defects were placed at the stacking fault plane or at nearby atomic layers. The effect of vacancy defects at the stacking fault plane and the plane directly underneath of it was dominant compared to the effect of vacancies at other adjacent planes. The effects of vacancy fraction, the distance between vacancies, and lateral relaxation of atoms on the GSF curves with vacancy defects were investigated. A very similar variation of normalized SFEs with respect to vacancy fractions were observed for Ni and Cu. MEAM potentials qualitatively captured the effect of vacancies on GSF. read less NOT USED (low confidence) C.-B. Wang, W. Zhang, C. Ren, P. Huai, and Z. Zhu, “The effect of temperature on primary defect formation in Ni–Fe alloy,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2014. link Times cited: 16 NOT USED (low confidence) R. Veiga, C. Becquart, and M. Perez, “Comments on ‘Atomistic modeling of an Fe system with a small concentration of C,’” Computational Materials Science. 2014. link Times cited: 35 NOT USED (low confidence) J. Chen, N. Gao, P. Jung, and T. Sauvage, “A new mechanism of loop formation and transformation in bcc iron without dislocation reaction,” Journal of Nuclear Materials. 2013. link Times cited: 43 NOT USED (low confidence) W. J. Zhou, H. Luan, Y. L. He, J. Sun, and W. Tao, “A study on boundary force model used in multiscale simulations with non-periodic boundary condition,” Microfluidics and Nanofluidics. 2013. link Times cited: 0 NOT USED (low confidence) E. Zarkadoula et al., “Electronic effects in high-energy radiation damage in iron,” Journal of Physics: Condensed Matter. 2013. link Times cited: 52 Abstract: Electronic effects have been shown to be important in high-e… read moreAbstract: Electronic effects have been shown to be important in high-energy radiation damage processes where a high electronic temperature is expected, yet their effects are not currently understood. Here, we perform molecular dynamics simulations of high-energy collision cascades in α-iron using a coupled two-temperature molecular dynamics (2T-MD) model that incorporates both the effects of electronic stopping and electron–phonon interaction. We subsequently compare it with the model employing electronic stopping only, and find several interesting novel insights. The 2T-MD results in both decreased damage production in the thermal spike and faster relaxation of the damage at short times. Notably, the 2T-MD model gives a similar amount of final damage at longer times, which we interpret to be the result of two competing effects: a smaller amount of short-time damage and a shorter time available for damage recovery. read less NOT USED (low confidence) S. Dudarev, “Density Functional Theory Models for Radiation Damage,” Annual Review of Materials Research. 2013. link Times cited: 79 Abstract: Density functional theory models developed over the past dec… read moreAbstract: Density functional theory models developed over the past decade provide unique information about the structure of nanoscale defects produced by irradiation and about the nature of short-range interaction between radia- tion defects, clustering of defects, and their migration pathways. These ab initio models, involving no experimental input parameters, appear to be as quantitatively accurate and informative as the most advanced experimental techniques developed for the observation of radiation damage phenomena. Density functional theory models have effectively created a new paradigm for the scientific investigation and assessment of radiation damage effects, offering new insight into the origin of temperature- and dose-dependent response of materials to irradiation, a problem of pivotal significance for applications. read less NOT USED (low confidence) G. Norman and V. Stegailov, “Stochastic theory of the classical molecular dynamics method,” Mathematical Models and Computer Simulations. 2013. link Times cited: 0 NOT USED (low confidence) C. R. Weinberger, B. L. Boyce, and C. Battaile, “Slip planes in bcc transition metals,” International Materials Reviews. 2013. link Times cited: 214 Abstract: Slip in face centred cubic (fcc) metals is well documented t… read moreAbstract: Slip in face centred cubic (fcc) metals is well documented to occur on {111} planes in 〈110〉 directions. In body centred cubic (bcc) metals, the slip direction is also well established to be 〈111〉, but it is much less clear as to the slip planes on which dislocations move. Since plasticity in metals is governed by the collective motion and interaction of dislocations, the nature of the relevant slip planes is of critical importance in understanding and modelling plasticity in bcc metals. This review attempts to address two fundamental questions regarding the slip planes in bcc metals. First, on what planes can slip, and thus crystallographic rotation, be observed to occur, i.e. what are the effective slip planes? Second, on what planes do kinks form along the dislocation lines, i.e. what are the fundamental slip planes? We review the available literature on direct and indirect characterisation of slip planes from experiments, and simulations using atomistic models. Given the technological importance of bcc transition metals, this review focuses specifically on those materials. read less NOT USED (low confidence) Y. Abe, “Application of hyper-molecular dynamics to self-interstitial diffusion in α-iron,” Computational Materials Science. 2013. link Times cited: 7 NOT USED (low confidence) H. Askari, H. Zbib, and X. Sun, “Multiscale Modeling of Inclusions and Precipitation Hardening in Metal Matrix Composites: Application to Advanced High-Strength Steels,” Journal of Nanomechanics and Micromechanics. 2013. link Times cited: 18 Abstract: AbstractThe strengthening effect of precipitates in metals i… read moreAbstract: AbstractThe strengthening effect of precipitates in metals is investigated within a multiscale approach that utilizes models of various length scales; namely, molecular mechanics (MM), discrete dislocation dynamics (DD), and an equivalent inclusion method (EIM). In particular, precipitates are modeled as particles whose stress fields interact with dislocations. The stress field resulting from the elastic mismatch between the particles and the matrix is accounted for by using the EIM, whereas the MM method is employed to develop rules for the DD method for short range interactions between a single dislocation and an inclusion. The DD method is used to predict the strength of the composite structure resulting from the interaction between ensembles of dislocations and particles. As an application to this method, the mechanical behavior of advanced high strength steel is investigated and the results are compared to the experimental data published in previous studies. The results show that the finely dispersiv... read less NOT USED (low confidence) P. Armstrong and W. Peukert, “Size effects in the elastic deformation behavior of metallic nanoparticles,” Journal of Nanoparticle Research. 2012. link Times cited: 26 NOT USED (low confidence) N. Gunkelmann, E. Bringa, K. Kang, G. Ackland, C. Ruestes, and H. Urbassek, “Polycrystalline iron under compression: Plasticity and phase transitions,” Physical Review B. 2012. link Times cited: 85 NOT USED (low confidence) L. Proville, D. Rodney, and M. Marinica, “Quantum effect on thermally activated glide of dislocations.,” Nature materials. 2012. link Times cited: 192 NOT USED (low confidence) J. Jeon, B. J. Lee, and Y. Chang, “Multi-scale Modeling of Plasticity for Single Crystal Iron,” Transactions of materials processing. 2012. link Times cited: 0 Abstract: Atomistic simulations have become useful tools for exploring… read moreAbstract: Atomistic simulations have become useful tools for exploring new insights in materials science, but the length and time scale that can be handled with atomistic simulations are seriously limiting their practical applications. In order to make meaningful quantitative predictions, atomistic simulations are necessarily combined with higher-scale modeling. The present research is thus concerned with the development of a multi-scale model and its application to the prediction of the mechanical properties of body-centered cubic(BCC) iron with an emphasis on the coupling of atomistic molecular dynamics with meso-scale discrete dislocation dynamics modeling. In order to achieve predictive multi-scale simulations, it is necessary to properly incorporate atomistic details into the meso-scale approach. This challenge is handled with the proposed hierarchical information passing strategy from atomistic to meso-scale by obtaining material properties and dislocation mobility. Finally, this fundamental and physics-based meso-scale approach is employed for quantitative predictions of the mechanical response of single crystal iron. read less NOT USED (low confidence) Q. Shu, Y. Yang, Y.-teng Zhai, D. Sun, H. Xiang, and X. Gong, “Size-dependent melting behavior of iron nanoparticles by replica exchange molecular dynamics.,” Nanoscale. 2012. link Times cited: 40 Abstract: Using the replica-exchange molecular dynamics method (REMD),… read moreAbstract: Using the replica-exchange molecular dynamics method (REMD), we have investigated the size dependence of the melting behavior of iron nanoparticles. Comparing to conventional molecular dynamics (MD), the REMD method is found to be very efficient in determining the melting point by avoiding superheating and undercooling phenomena. With accurate determination of the melting point, we find that the melting temperature does not follow linearly with the inverse of size. By incorporating the size dependent thickness of surface liquid layer which is observed in our simulation, we propose a revised liquid skin melting model to describe the size dependent melting temperature. read less NOT USED (low confidence) K. Issa, “Molecular dynamics study of nanoscale heat transfer at liquid-solid interfaces (LSIs).” 2012. link Times cited: 1 NOT USED (low confidence) W. Gao, L. Kong, and P. Hodgson, “Atomic interaction of functionalized carbon nanotube-based nanofluids with a heating surface and its effect on heat transfer,” International Journal of Heat and Mass Transfer. 2012. link Times cited: 14 NOT USED (low confidence) N. Gunkelmann, H. Ledbetter, and H. Urbassek, “Experimental and atomistic study of the elastic properties of α′ Fe–C martensite,” Acta Materialia. 2012. link Times cited: 38 NOT USED (low confidence) M. Tschopp, K. Solanki, M. Baskes, F. Gao, X. Sun, and M. Horstemeyer, “Generalized framework for interatomic potential design: Application to Fe–He system,” Journal of Nuclear Materials. 2012. link Times cited: 20 NOT USED (low confidence) A. Koester, A. Ma, and A. Hartmaier, “Atomistically informed crystal plasticity model for body-centered cubic iron,” Acta Materialia. 2012. link Times cited: 77 NOT USED (low confidence) K. Trachenko, E. Zarkadoula, I. Todorov, M. Dove, D. Dunstan, and K. Nordlund, “Modeling high-energy radiation damage in nuclear and fusion applications,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2012. link Times cited: 25 NOT USED (low confidence) A. Ishii, H. Kimizuka, and S. Ogata, “Multi-replica molecular dynamics modeling,” Computational Materials Science. 2012. link Times cited: 3 NOT USED (low confidence) M. Tschopp, K. Solanki, F. Gao, X. Sun, M. Khaleel, and M. Horstemeyer, “Probing grain boundary sink strength at the nanoscale: Energetics and length scales of vacancy and interstitial absorption by grain boundaries in α -Fe,” Physical Review B. 2012. link Times cited: 271 Abstract: The energetics and length scales associated with the interac… read moreAbstract: The energetics and length scales associated with the interaction between point defects (vacancies and self-interstitial atoms) and grain boundaries in bcc Fe was explored. Molecular statics simulations were used to generate a grain boundary structure database that contained {approx}170 grain boundaries with varying tilt and twist character. Then, vacancy and self-interstitial atom formation energies were calculated at all potential grain boundary sites within 15 {angstrom} of the boundary. The present results provide detailed information about the interaction energies of vacancies and self-interstitial atoms with symmetric tilt grain boundaries in iron and the length scales involved with absorption of these point defects by grain boundaries. Both low- and high-angle grain boundaries were effective sinks for point defects, with a few low-{Sigma} grain boundaries (e.g., the {Sigma}3{l_brace}112{r_brace} twin boundary) that have properties different from the rest. The formation energies depend on both the local atomic structure and the distance from the boundary center. Additionally, the effect of grain boundary energy, disorientation angle, and {Sigma} designation on the boundary sink strength was explored; the strongest correlation occurred between the grain boundary energy and the mean point defect formation energies. Based on point defect binding energies, interstitials have {approx}80% more grain boundary sites permore » area and {approx}300% greater site strength than vacancies. Last, the absorption length scale of point defects by grain boundaries is over a full lattice unit larger for interstitials than for vacancies (mean of 6-7 {angstrom} versus 10-11 {angstrom} for vacancies and interstitials, respectively).« less read less NOT USED (low confidence) N. N. Kumar, P. Durgaprasad, B. Dutta, and G. K. Dey, “Modeling of radiation hardening in ferritic/martensitic steel using multi-scale approach,” Computational Materials Science. 2012. link Times cited: 30 NOT USED (low confidence) D. E. Smirnova, S. Starikov, S. Starikov, V. Stegailov, and V. Stegailov, “Interatomic potential for uranium in a wide range of pressures and temperatures,” Journal of Physics: Condensed Matter. 2012. link Times cited: 3 Abstract: Using the force-matching method we develop an interatomic po… read moreAbstract: Using the force-matching method we develop an interatomic potential that allows us to study the structure and properties of α-U, γ-U and liquid uranium. The potential is fitted to the forces, energies and stresses obtained from ab initio calculations. The model gives a good comparison with the experimental and ab initio data for the lattice constants of α-U and γ-U, the elastic constants, the room-temperature isotherm, the normal density isochore, the bond-angle distribution functions and the vacancy formation energies. The calculated melting line of uranium at pressures up to 80 GPa and the temperature of the α–γ transition at 3 GPa agree well with the experimental phase diagram of uranium. read less NOT USED (low confidence) S. Cuesta-López and J. Perlado, “Progress in Advanced Materials under Extreme Conditions for Nuclear Fusion Technology,” Fusion Science and Technology. 2012. link Times cited: 0 Abstract: We report non-equilibrium Molecular Dynamics simulations tha… read moreAbstract: We report non-equilibrium Molecular Dynamics simulations that provide a nanoscale view for the modeling of shock wave generation in any kind of material. Our methodology reported here is able to cover similar times and length scales as experiments. We are studying the propagation of shock waves, and their consequences: structural transformations and induced melting. We apply our methodology not only to single crystalline materials like Ta, W, but also in double layer conformations of bcc/fcc/bcc and bcc/bcc/bcc materials, with clear interest for Nuclear Fusion Technology. Preliminary results point that W and Ta behave more efficiently in terms of uniformity under shock propagation than lighter materials. Moreover, we show that shocks in double layer structures propagate and generate pressure more efficiently than common structures. read less NOT USED (low confidence) R. Drautz and D. Pettifor, “Valence-dependent analytic bond-order potential for magnetic transition metals,” Physical Review B. 2011. link Times cited: 39 Abstract: We extend the analytic bond-order potentials for transition … read moreAbstract: We extend the analytic bond-order potentials for transition metals [Phys. Rev. B 74, 174117 (2006)] to include ferro, antiferro, and noncollinear magnetism and charge transfer. This is achieved by first deriving a suitable tight-binding model through the expansion of the spin-density energy functional to second order with respect to magnetic and charge fluctuations. The tight-binding model is then approximated locally by the bond-order potential expansion, where the variational property of the bond-order potential expansion allows us to derive analytic expressions for the forces and torques on the atoms. From the bond-order potentials we then extract a hierarchy of multispin interactions beyond the conventional Heisenberg model. The explicit valence dependence of the bond-order potentials enables us to characterize the magnetic properties of the 3$d$ transition metals and to reproduce the trend from antiferromagnetic spin ordering close to the center of the $d$ band through noncollinear spin configurations to ferromagnetic ordering toward the edges of the $d$ band. The analytic representation of the energy within the bond-order potentials is then further expanded in the form of a Ginzburg-Landau expansion, deriving the prefactors explicitly from tight-binding and bond-order potentials. Thus, in this paper we present a coherent simplification from fundamental to empirical models of magnetism through coarse graining the electronic structure from spin-density functional theory to tight binding to bond-order potentials to the Ginzburg-Landau expansion. read less NOT USED (low confidence) E. Clouet, L. Ventelon, and F. Willaime, “Dislocation core field.II. Screw dislocation in iron,” Physical Review B. 2011. link Times cited: 44 Abstract: The dislocation core field, which comes in addition to the V… read moreAbstract: The dislocation core field, which comes in addition to the Volterra elastic field, is studied for the screw dislocation in alpha-iron. This core field, evidenced and characterized using ab initio calculations, corresponds to a biaxial dilatation, which we modeled within the anisotropic linear elasticity. We show that this core field needs to be considered when extracting quantitative information from atomistic simulations, such as dislocation core energies. Finally, we look at how dislocation properties are modified by this core field, by studying the interaction between two dislocations composing a dipole, as well as the interaction of a screw dislocation with a carbon atom. read less NOT USED (low confidence) A. Caro et al., “Properties of Helium bubbles in Fe and FeCr alloys,” Journal of Nuclear Materials. 2011. link Times cited: 73 NOT USED (low confidence) M. Talati, M. Posselt, G. Bonny, A. Al-Motasem, and F. Bergner, “Vibrational contribution to the thermodynamics of nanosized precipitates: vacancy–copper clusters in bcc-Fe,” Journal of Physics: Condensed Matter. 2011. link Times cited: 9 Abstract: The effects of lattice vibration on the thermodynamics of na… read moreAbstract: The effects of lattice vibration on the thermodynamics of nanosized coherent clusters in bcc-Fe consisting of vacancies and/or copper are investigated within the harmonic approximation. A combination of on-lattice simulated annealing based on Metropolis Monte Carlo simulations and off-lattice relaxation by molecular dynamics is applied to obtain the most stable cluster configurations at T = 0 K. The most recent interatomic potential built within the framework of the embedded-atom method for the Fe–Cu system is used. The total free energy of pure bcc-Fe and fcc-Cu as well as the total formation free energy and the total binding free energy of the vacancy–copper clusters are determined for finite temperatures. Our results are compared with the available data from previous investigations performed using many-body interatomic potentials and first-principles methods. For further applications in rate theory and object kinetic Monte Carlo simulations, the vibrational effects evaluated in the present study are included in the previously developed analytical fitting formulae. read less NOT USED (low confidence) J. Sampedro, E. Río, M. Caturla, M. Victoria, and J. Perlado, “Defect energetics in Fe–Cr alloys from empirical interatomic potentials,” Journal of Nuclear Materials. 2011. link Times cited: 4 NOT USED (low confidence) J. Boutard, S. Dudarev, and M. Rieth, “Modelling structural and plasma facing materials for fusion power plants: Recent advances and outstanding issues in the EURATOM fusion materials programme,” Journal of Nuclear Materials. 2011. link Times cited: 11 NOT USED (low confidence) K. Vörtler, N. Juslin, N. Juslin, G. Bonny, L. Malerba, and K. Nordlund, “The effect of prolonged irradiation on defect production and ordering in Fe–Cr and Fe–Ni alloys,” Journal of Physics: Condensed Matter. 2011. link Times cited: 54 Abstract: The understanding of the primary radiation damage in Fe-base… read moreAbstract: The understanding of the primary radiation damage in Fe-based alloys is of interest for the use of advanced steels in future fusion and fission reactors. In this work Fe–Cr alloys (with 5, 6.25, 10 and 15% Cr content) and Fe–Ni alloys (with 10, 40, 50 and 75% Ni content) were used as model materials for studying the features of steels from a radiation damage perspective. The effect of prolonged irradiation (neglecting diffusion), i.e. the overlapping of single 5 keV displacement cascade events, was studied by molecular dynamics simulation. Up to 200 single cascades were simulated, randomly induced in sequence in one simulation cell, to study the difference between fcc and bcc lattices, as well as initially ordered and random crystals. With increasing numbers of cascades we observed a saturation of Frenkel pairs in the bcc alloys. In fcc Fe–Ni, in contrast, we saw a continuous accumulation of defects: the growth of stacking-fault tetrahedra and a larger number of self-interstitial atom clusters were seen in contrast to bcc alloys. For all simulations the defect clusters and the short range order parameter were analysed in detail depending on the number of cascades in the crystal. We also report the modification of the repulsive part of the Fe–Ni interaction potential, which was needed to study the non-equilibrium processes. read less NOT USED (low confidence) X. Liu et al., “Atomic packing symmetry in the metallic liquid and glass states,” Acta Materialia. 2011. link Times cited: 43 NOT USED (low confidence) B. Jelinek et al., “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Physical Review B. 2011. link Times cited: 218 Abstract: A set of modified embedded-atom method (MEAM) potentials for… read moreAbstract: A set of modified embedded-atom method (MEAM) potentials for the interactions between Al, Si, Mg, Cu, and Fe was developed from a combination of each element's MEAM potential in order to study metal alloying. Previously published MEAM parameters of single elements have been improved for better agreement to the generalized stacking fault energy (GSFE) curves when compared with ab initio generated GSFE curves. The MEAM parameters for element pairs were constructed based on the structural and elastic properties of element pairs in the NaCl reference structure garnered from ab initio calculations, with adjustment to reproduce the ab initio heat of formation of the most stable binary compounds. The new MEAM potentials were validated by comparing the formation energies of defects, equilibrium volumes, elastic moduli, and heat of formation for several binary compounds with ab initio simulations and experiments. Single elements in their ground-state crystal structure were subjected to heating to test the potentials at elevated temperatures. An Al potential was modified to avoid formation of an unphysical solid structure at high temperatures. The thermal expansion coefficient of a compound with the composition of AA 6061 alloy was evaluated and compared with experimental values. MEAM potential tests performed in this work, utilizing the universal atomistic simulation environment (ASE), are distributed to facilitate reproducibility of the results. read less NOT USED (low confidence) G. Bonny, R. Pasianot, E. Zhurkin, and M. Hou, “Determination of the phase diagram from interatomic potentials: The iron-chromium case,” Computational Materials Science. 2011. link Times cited: 17 NOT USED (low confidence) D. Terentyev et al., “Further development of large-scale atomistic modelling techniques for Fe–Cr alloys,” Journal of Nuclear Materials. 2011. link Times cited: 15 NOT USED (low confidence) L. Malerba et al., “Comparison of empirical interatomic potentials for iron applied to radiation damage studies,” Journal of Nuclear Materials. 2010. link Times cited: 210 NOT USED (low confidence) S. Huang, D. L. Worthington, M. Asta, V. Ozoliņš, G. Ghosh, and P. Liaw, “Calculation of impurity diffusivities in α-Fe using first-principles methods,” Acta Materialia. 2010. link Times cited: 110 NOT USED (low confidence) C. Becquart and C. Domain, “Molecular dynamics simulations of damage and plasticity: The role of ab initio calculations in the development of interatomic potentials,” Philosophical Magazine. 2009. link Times cited: 7 Abstract: Predicting the behaviour of a component under irradiation or… read moreAbstract: Predicting the behaviour of a component under irradiation or submitted to an external load often requires understanding the evolution of its microstructure. This usually requires knowledge of the mechanisms taking place at the atomic level, which are introduced in multiscale-type modelling suites. In this context, interatomic potentials are necessary ingredients for most simulation techniques at the atomic level. They have been used for more than 40 years in various areas of materials science and, in particular, in the fields of radiation damage and plasticity. These simulations have shed particular light on the role of solute atoms in the formation of the primary damage or the motion of dislocations. However, ab initio calculations, as well as comparison of the results obtained with different interatomic potentials have indicated some failures in these potentials, which led to the building of new potentials. This article highlights how ab initio calculations, which nowadays constitute the state of the art method to predict atomic properties can (and will) increasingly contribute to the assessment, validation and building of interatomic potentials. read less NOT USED (low confidence) M. Tikhonchev, V. Svetukhin, A. Kadochkin, and E. Gaganidze, “MD simulation of atomic displacement cascades in Fe-10 at.%Cr binary alloy,” Journal of Nuclear Materials. 2009. link Times cited: 30 NOT USED (low confidence) P. Olsson, “Semi-empirical atomistic study of point defect properties in BCC transition metals,” Computational Materials Science. 2009. link Times cited: 61 NOT USED (low confidence) M. Samaras, “Multiscale Modelling: the role of helium in iron,” Materials Today. 2009. link Times cited: 73 NOT USED (low confidence) F. Gao, H. Heinisch, and R. Kurtz, “Migration of vacancies, He interstitials and He-vacancy clusters at grain boundaries in α-Fe,” Journal of Nuclear Materials. 2009. link Times cited: 41 NOT USED (low confidence) G. Bonny, R. Pasianot, L. Malerba, A. Caro, P. Olsson, and M. Lavrentiev, “Numerical prediction of thermodynamic properties of iron–chromium alloys using semi-empirical cohesive models: The state of the art,” Journal of Nuclear Materials. 2009. link Times cited: 72 NOT USED (low confidence) X. He, W. Yang, Z. Qu, and S.-W. Fan, “Effects of irradiation on chromium’s behavior in ferritic/martensitic FeCr alloy,” Frontiers of Energy and Power Engineering in China. 2009. link Times cited: 2 NOT USED (low confidence) D. Rodney and L. Proville, “Stress-dependent Peierls potential: Influence on kink-pair activation,” Physical Review B. 2009. link Times cited: 67 Abstract: Atomistic calculations based on the nudged elastic band meth… read moreAbstract: Atomistic calculations based on the nudged elastic band method for a Lomer dislocation in aluminum evidence a dependence of the Peierls potential on the applied shear stress in such a way that the Peierls stress predicted from the zero-stress potential is half its true value for the case considered here. Stress-dependent Peierls potentials that are extracted are then introduced as substrate potentials in a string model with a line tension (LT) adjusted to match the dislocation kink width obtained from atomistic simulations. The LT model is found to predict accurately dislocation saddle configurations and corresponding kink-pair activation enthalpies for a wide range of stresses. In particular, it is shown that the stress dependence of the Peierls potential is required to model with accuracy the nonlinearity of the enthalpy-stress curve. read less NOT USED (low confidence) P. Korzhavyi, A. Ruban, J. Odqvist, J. Nilsson, and B. Johansson, “Electronic structure and effective chemical and magnetic exchange interactions in bcc Fe-Cr alloys,” Physical Review B. 2009. link Times cited: 90 Abstract: Electronic structure calculations are employed in order to i… read moreAbstract: Electronic structure calculations are employed in order to investigate the cohesive properties (lattice parameter, enthalpy of formation, and bulk modulus) of random Fe-Cr alloys as a function of composition and magnetic state, as well as to derive the chemical and magnetic exchange interactions of the constituent atoms. The calculations predict certain anomalies in the cohesive properties of ferromagnetic alloys at a concentration of about $7\text{ }\text{at}\text{ }%$ Cr; these anomalies may be related to the changes in Fermi-surface topology that occur with composition in this alloy system. The obtained interatomic interactions are used as parameters in the configurational (Ising) and magnetic (Heisenberg) Hamiltonians for modeling finite-temperature thermodynamic properties of the alloys. We discuss the approximations and limitations of similar modeling approaches, investigate the origin of existing difficulties, and analyze possible ways of extending the theoretical models in order to capture the essential physics of interatomic interactions in the Fe-Cr or similar alloys where magnetism plays a crucial role in the phase stability. read less NOT USED (low confidence) L. Malerba, A. Caro, and J. Wallenius, “Multiscale modelling of radiation damage and phase transformations: The challenge of FeCr alloys,” Journal of Nuclear Materials. 2008. link Times cited: 123 NOT USED (low confidence) S. M. H. Haghighat, J. Fikar, and R. Schäublin, “Effect of interatomic potential on the behavior of dislocation-defect interaction simulation in α-Fe,” Journal of Nuclear Materials. 2008. link Times cited: 35 NOT USED (low confidence) R. Kurtz, H. Heinisch, and F. Gao, “Modeling of He–defect interactions in ferritic alloys for fusion,” Journal of Nuclear Materials. 2008. link Times cited: 32 NOT USED (low confidence) M. Samaras and M. Victoria, “Modelling in nuclear energy environments,” Materials Today. 2008. link Times cited: 27 NOT USED (low confidence) D. Hepburn and G. Ackland, “Metallic-covalent interatomic potential for carbon in iron,” Physical Review B. 2008. link Times cited: 118 Abstract: Existing interatomic potentials for the iron-carbon system s… read moreAbstract: Existing interatomic potentials for the iron-carbon system suffer from qualitative flaws in describing even the simplest of defects. In contrast to more accurate first-principles calculations, all previous potentials show strong bonding of carbon to overcoordinated defects (e.g., self-interstitials, dislocation cores) and a failure to accurately reproduce the energetics of carbon-vacancy complexes. Thus any results from their application in molecular dynamics to more complex environments are unreliable. The problem arises from a fundamental error in potential design--the failure to describe short-ranged covalent bonding of the carbon p electrons. We describe a resolution to the problem and present an empirical potential based on insights from density-functional theory, showing covalent-type bonding for carbon. The potential correctly describes the interaction of carbon and iron across a wide range of defect environments. It has the embedded atom method form and hence appropriate for billion atom molecular-dynamics simulations. read less NOT USED (low confidence) D. Terentyev and L. Malerba, “Effect of Cr atoms on the formation of double kinks in screw dislocations in Fe and its correlation with solute hardening and softening in Fe–Cr alloys,” Computational Materials Science. 2008. link Times cited: 13 NOT USED (low confidence) J. Sienicki et al., “Status report on the Small Secure Transportable Autonomous Reactor (SSTAR) /Lead-cooled Fast Reactor (LFR) and supporting research and development.” 2008. link Times cited: 39 Abstract: This report provides an update on development of a pre-conce… read moreAbstract: This report provides an update on development of a pre-conceptual design for the Small Secure Transportable Autonomous Reactor (SSTAR) Lead-Cooled Fast Reactor (LFR) plant concept and supporting research and development activities. SSTAR is a small, 20 MWe (45 MWt), natural circulation, fast reactor plant for international deployment concept incorporating proliferation resistance for deployment in non-fuel cycle states and developing nations, fissile self-sufficiency for efficient utilization of uranium resources, autonomous load following making it suitable for small or immature grid applications, and a high degree of passive safety further supporting deployment in developing nations. In FY 2006, improvements have been made at ANL to the pre-conceptual design of both the reactor system and the energy converter which incorporates a supercritical carbon dioxide Brayton cycle providing higher plant efficiency (44 %) and improved economic competitiveness. The supercritical CO2 Brayton cycle technology is also applicable to Sodium-Cooled Fast Reactors providing the same benefits. One key accomplishment has been the development of a control strategy for automatic control of the supercritical CO2 Brayton cycle in principle enabling autonomous load following over the full power range between nominal and essentially zero power. Under autonomous load following operation, the reactor core power adjusts itself to equal the heat removal from the reactor system to the power converter through the large reactivity feedback of the fast spectrum core without the need for motion of control rods, while the automatic control of the power converter matches the heat removal from the reactor to the grid load. The report includes early calculations for an international benchmarking problem for a LBE-cooled, nitride-fueled fast reactor core organized by the IAEA as part of a Coordinated Research Project on Small Reactors without Onsite Refueling; the calculations use the same neutronics computer codes and methodologies applied to SSTAR. Another section of the report details the SSTAR safety design approach which is based upon defense-in-depth providing multiple levels of protection against the release of radioactive materials and how the inherent safety features of the lead coolant, nitride fuel, fast neutron spectrum core, pool vessel configuration, natural circulation, and containment meet or exceed the requirements for each level of protection. The report also includes recent results of a systematic analysis by LANL of data on corrosion of candidate cladding and structural material alloys of interest to SSTAR by LBE and Pb coolants; the data were taken from a new database on corrosion by liquid metal coolants created at LANL. The analysis methodology that considers penetration of an oxidation front into the alloy and dissolution of the trailing edge of the oxide into the coolant enables the long-term corrosion rate to be extracted from shorter-term corrosion data thereby enabling an evaluation of alloy performance over long core lifetimes (e.g., 30 years) that has heretofore not been possible. A number of candidate alloy specimens with special treatments or coatings which might enhance corrosion resistance at the temperatures at which SSTAR would operate were analyzed following testing in the DELTA loop at LANL including steels that were treated by laser peening at LLNL; laser peening is an approach that alters the oxide-metal bonds which could potentially improve corrosion resistance. LLNL is also carrying out Multi-Scale Modeling of the Fe-Cr system with the goal of assisting in the development of cladding and structural materials having greater resistance to irradiation. read less NOT USED (low confidence) D. Terentyev et al., “Self-trapped interstitial-type defects in iron.,” Physical review letters. 2008. link Times cited: 80 Abstract: Small interstitial-type defects in iron with complex structu… read moreAbstract: Small interstitial-type defects in iron with complex structures and very low mobilities are revealed by molecular dynamics simulations. The stability of these defect clusters formed by nonparallel {110} dumbbells is confirmed by density functional theory calculations, and it is shown to increase with increasing temperature due to large vibrational formation entropies. This new family of defects provides an explanation for the low mobility of clusters needed to account for experimental observations of microstructure evolution under irradiation at variance with the fast migration obtained from previous atomistic simulations for conventional self-interstitial clusters. read less NOT USED (low confidence) M. Fivel, “Discrete dislocation dynamics: an important recent break-through in the modelling of dislocation collective behaviour,” Comptes Rendus Physique. 2008. link Times cited: 30 NOT USED (low confidence) D. Rodney, “Atomic modeling of irradiation-induced hardening,” Comptes Rendus Physique. 2008. link Times cited: 18 NOT USED (low confidence) K. Nordlund and S. Dudarev, “Interatomic potentials for simulating radiation damage effects in metals,” Comptes Rendus Physique. 2008. link Times cited: 29 NOT USED (low confidence) D. Belashchenko and B. Gelchinski, “Application of embedded atom model to the liquid metals.” 2008. link Times cited: 0 Abstract: The productive method of creation the inter-particle potenti… read moreAbstract: The productive method of creation the inter-particle potential in Embedded Atom Model (EAM) is presented that uses directly the structure data of liquid metal near the melting point. The embedding potential is written as the power series of the difference between the effective EAM electronic density ρ and its standard value ρ0 (ρ0 is taken as 1). The mean value = ρ0 in the standard state of a liquid (usually near the melting point). At this definition of EAM potential, the pair term in potential coincides with the effective pair potential that can be reconstructed using the pair correlation function (PCF) of liquid metal (for example with Schommers algorithm). The parameters of embedding part of potential are determined using the potential energy of liquid in the standard state, module of compression and parameters of a liquid in any extreme state (at high temperature or pressure). The method presented was applied to the creation of EAM potentials and simulations of liquid metals bismuth, gallium, mercury, rubidium, cesium, iron and iron - sulfur solutions in wide intervals of parameters, including states near the critical point (bismuth, gallium, mercury, rubidium and cesium) and at the conditions of Earth core. The accuracy of predictions of structure properties, thermodynamic properties and diffusivity is rather good. EAM potentials constructed especially for liquid metals describe their properties in wide temperature and pressure limits much better than the EAM potentials created for crystal metals. In the states typical for the shock waves tests (at very strong compression) crystal metal EAM potentials may give very erroneous pressure data. read less NOT USED (low confidence) S. Kotrechko, O. Filatov, and O. Ovsjannikov, “Peculiarities of Plastic Deformation and Failure of Nanoparticles of B.C.C. Transition Metals,” Materials Science Forum. 2007. link Times cited: 9 Abstract: Atomic mechanisms of the beginning of plastic deformation an… read moreAbstract: Atomic mechanisms of the beginning of plastic deformation and failure initiation in nanoparticles of b.c.c. transition metals are presented in this report. It is shown that strength level of nanoparticles of b.c.c. transition metals is pre-determined by the lattice instability within the local region of the crystal. At uniaxial tension even at low temperatures perfect crystal becomes unstable to shear („orthorhombic“ path), i.e. local shear instability is the main mechanism of stress relaxation in nanoparticles of b.c.c. metals. Specific features of local instability of nanoparticle under hydrostatic tension are considered. A model of the temperature dependence of strength is offered. It is shown that nanoparticle strength decreases as square root function of temperature with temperature growth. Just this is essential difference of the temperature dependence of nanoparticle strength from the same for “ordinary” single- and polycrystals. read less NOT USED (low confidence) M. Marinica and F. Willaime, “Orientation of Interstitials in Clusters in α-Fe: A Comparison between Empirical Potentials,” Solid State Phenomena. 2007. link Times cited: 22 Abstract: We have addressed two issues concerning the relative stabili… read moreAbstract: We have addressed two issues concerning the relative stabilities of various orienta- tions of interstitial clusters in iron by making a comprehensive comparison between four recent empirical potentials. First, we have investigated the effect of finite temperature on the com- petition between clusters made of a few dumbbells oriented along h111i or h110i. We show by quasi-harmonic calculations that h111i clusters have much larger vibrational formation en- tropies and that they are therefore stabilized with respect to h110i clusters at high temperature. Second, we have compared the formation energies of loops with several hundred atoms with Burgers vector 1 2 h111i or h100i. The 1 2 h111i loops are found to be always more stable, but the energy differences with h100i loops depend strongly on the potential. read less NOT USED (low confidence) P. V. Zwol, P. Derlet, H. Swygenhoven, and S. Dudarev, “BCC Fe surface and cluster magnetism using a magnetic potential,” Surface Science. 2007. link Times cited: 13 NOT USED (low confidence) T. Seletskaia, Y. Osetskiy, R. Stoller, and G. M. Stocks, “Development of a Fe-He interatomic potential based on electronic structure calculations,” Journal of Nuclear Materials. 2007. link Times cited: 51 NOT USED (low confidence) L. Malerba et al., “Modelling of Radiation Damage in Fe-Cr Alloys,” Journal of Astm International. 2007. link Times cited: 29 Abstract: High-Cr ferritic/martensitic steels are being considered as … read moreAbstract: High-Cr ferritic/martensitic steels are being considered as structural materials for a large number of future nuclear applications, from fusion to accelerator-driven systems and GenIV reactors. Fe-Cr alloys can be used as model materials to investigate some of the mechanisms governing their microstructure evolution under irradiation and its correlation to changes in their macroscopic properties. Focusing on these alloys, we show an example of how the integration of computer simulation and theoretical models can provide keys for the interpretation of a host of relevant experimental observations. In particular we show that proper accounting for two basic features of these alloys, namely, the existence of a fairly strong attractive interaction between self-interstitials and Cr atoms and of a mixing enthalpy that changes sign from negative to positive around 8 to 10 % Cr, is a necessary and, to a certain extent, sufficient condition to rationalize and understand their behavior under irradiation. These features have been revealed by ab initio calculations, are supported by experimental evidence, and have been adequately transferred into advanced empirical interatomic potentials, which have been and are being used for the simulation of damage production, defect behavior, and phase transformation in these alloys. The results of the simulations have been and are being used to parameterize models capable of extending the description of radiation effects to scales beyond the reach of molecular dynamics. The present paper intends to highlight the most important achievements and results of this research activity. read less NOT USED (low confidence) Y. Kubota, R. Matsumoto, and M. Nakagaki, “Molecular Dynamics Analysis on Crack Growth Behavior in Single and Nano-Crystalline Fe by the Use of FS-2NNMEAM Hybrid Potential,” Key Engineering Materials. 2007. link Times cited: 2 Abstract: In recent years, nano-crystalline materials have attracted m… read moreAbstract: In recent years, nano-crystalline materials have attracted many researchers’ attention, but the fracture mechanism has not been fully clarified. In a molecular dynamics (MD) simulation, grain size and crystal orientation can be chosen, and their effects on the mechanical properties of nano-crystalline materials can be evaluated clearly. This research first compares the results of crack growth behavior in single crystalline Fe for three typical interatomic potentials (Embedded Atom Method (EAM), Finnis Sinclair (FS), and Second Nearest Neighbor Modified EAM (2NNMEAM) potentials) and a Hybrid potential method, which uses FS potential for bcc structure atoms and 2NNMEAM potential for non-bcc structure atoms. The 2NNMEAM potential is accurate, but the computation time is dozens of times that of FS potential, which is the simplest of the three interatomic potentials. Therefore, the 2NNMEAM potential requires too much calculation for the purpose of this research that analyzes the crack growth behavior in nano-crystalline metals. However, Hybrid potential is able to give results similar to those of the 2NNMEAM potential, and the calculation time is close to that of the FS potential. From these results, the crack extension behavior in relatively large nano-crystalline models is analyzed using the Hybrid potential, and we demonstrate the grain-size dependency of the fracture behavior. read less NOT USED (low confidence) D. Terentyev, L. Malerba, and M. Hou, “Dimensionality of interstitial cluster motion in bcc-Fe,” Physical Review B. 2007. link Times cited: 110 NOT USED (low confidence) R. Pasianot and L. Malerba, “Interatomic potentials consistent with thermodynamics: The Fe–Cu system,” Journal of Nuclear Materials. 2007. link Times cited: 68 NOT USED (low confidence) V. Pontikis, V. Russier, and J. Wallenius, “An analytic n-body potential for bcc Iron,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 1 NOT USED (low confidence) J. Ferrante and F. Zypman, “Generalization of equivalent crystal theory to include angular dependence,” Computational Materials Science. 2006. link Times cited: 3 NOT USED (low confidence) M. Samaras, W. Hoffelner, and M. Victoria, “Irradiation of pre-existing voids in nanocrystalline iron,” Journal of Nuclear Materials. 2006. link Times cited: 31 NOT USED (low confidence) T. Seletskaia, Y. Osetsky, R. Stoller, and G. M. Stocks, “Calculation of helium defect clustering properties in iron using a multi-scale approach,” Journal of Nuclear Materials. 2006. link Times cited: 87 NOT USED (low confidence) V. Tomar and M. Zhou, “Classical molecular-dynamics potential for the mechanical strength of nanocrystalline composite fccAl+α−Fe2O3,” Physical Review B. 2006. link Times cited: 26 Abstract: A classical molecular-dynamics potential for analyzing mecha… read moreAbstract: A classical molecular-dynamics potential for analyzing mechanical deformation in the -Fe2O3+fcc-Al material system is developed. The potential includes an embedded atom method cluster functional, a Morsetype pair function, and a second-order electrostatic interaction function. It is fitted to the lattice constants, elastic constants, and cohesive energies of fcc Al, bcc Fe, -Fe2O3, -Al2O3, and B2-FeAl, accounting for the fact that mixtures of Al and Fe2O3 are chemically reactive and deformation may cause the formation of these components as reaction products or intermediates. To obtain close approximations of the behavior of mixtures with any combination of the atomic elements, the potential is formulated and fitted such that the Al-Al, Fe-Fe, Al-Fe, O-O, Fe-O, and Al-O interactions are accounted for in an explicit and interdependent manner. In addition to being fitted to the lattice constants, elastic constants, and cohesive energies, the potential gives predictions of the surface and stacking fault energies for the crystalline components that compare well with the predictions of established potentials in the literature for the corresponding crystalline components. The potential is applied to analyze quasistatic tensile deformation in nanocrystalline Al, in nanocrystalline Fe2O3, and in nanocrystalline Al+Fe2O3 composites. Application of the potential to nanocrystalline Al reveals the features of mechanical deformation, such as the formation of unit dislocations, flow strength approaching ideal shear strength, and the Hall-Petch relationships, that are in close agreement with experiments and with the predictions of established potentials for Al in the literature. Analyses of deformation in nanocrystalline Fe2O3 and in nanocrystalline Al+Fe2O3 composites point to the possibility that the strength of the nanocomposites can only be calculated using the mixture theory if the average grain size is above a critical value. Below the critical grain size, an accurate account of interfacial stresses is important to the prediction of the strength. For composites with grain sizes above the critical value, the observed dependence of strength on volume fraction is in agreement with experimental observations. read less NOT USED (low confidence) H. Chamati, N. Papanicolaou, Y. Mishin, and D. Papaconstantopoulos, “Embedded-atom potential for Fe and its application to self-diffusion on Fe(1 0 0),” Surface Science. 2006. link Times cited: 178 NOT USED (low confidence) C. Fu, J. D. Torre, F. Willaime, J. Bocquet, and A. Barbu, “Multiscale modelling of defect kinetics in irradiated iron,” Nature Materials. 2004. link Times cited: 415 NOT USED (low confidence) J. Hoyt et al., “Crystal–Melt Interfaces and Solidification Morphologies in Metals and Alloys,” MRS Bulletin. 2004. link Times cited: 100 Abstract: When liquids solidify, the interface between a crystal and i… read moreAbstract: When liquids solidify, the interface between a crystal and its melt often forms branching structures (dendrites), just as frost spreads across a window.The development of a quantitative understanding of dendritic evolution continues to present a major theoretical and experimental challenge within the metallurgical community. This article looks at key parameters that describe the interface—excess free energy and mobility—and discusses how these important properties relate to our understanding of crystal growth and other interfacial phenomena such as wetting and spreading of droplets and nucleation of the solid phase from the melt. In particular, two new simulation methods have emerged for computing the interfacial free energy and its anisotropy: the cleaving technique and the capillary fluctuation method. These are presented, along with methods for extracting the kinetic coefficient and a comparison of the results to several theories of crystal growth rates. read less NOT USED (low confidence) C. Fu, F. Willaime, and P. Ordejón, “Stability and mobility of mono- and di-interstitials in alpha-Fe.,” Physical review letters. 2004. link Times cited: 370 Abstract: We report a detailed ab initio study of the stability and mi… read moreAbstract: We report a detailed ab initio study of the stability and migration of self-interstitial atoms (SIAs) and di-interstitials (di-SIAs) in alpha-Fe. The <110> dumbbell is confirmed to be the most stable SIA configuration, 0.7 eV below the <111> dumbbell. The lowest-energy migration path corresponds to a nearest-neighbor translation-rotation jump with a barrier of 0.34 eV. The most stable configuration for di-SIAs consists of <110> parallel dumbbells. Their migration mechanism is similar to that for SIAs, with an activation energy of 0.42 eV. These results are at variance with predictions from existing empirical potentials and allow one to reconcile theory with experiments. read less NOT USED (low confidence) O. Chirayutthanasak et al., “Universal function for grain boundary energies in bcc metals,” Scripta Materialia. 2024. link Times cited: 0 NOT USED (low confidence) T. Ohmura and M. Wakeda, “Nano Mechanical Characterization and Physical Modeling of Plastic Deformation Chapter 1: Dislocation-Grain Boundary Interaction as a Strengthening Factor,” Journal of the Japan Institute of Metals and Materials. 2023. link Times cited: 0 NOT USED (low confidence) X. Liu et al., “A statistics-based study and machine-learning of stacking fault energies in HEAs,” Journal of Alloys and Compounds. 2023. link Times cited: 0 NOT USED (low confidence) S. Y. Korostelev, E. E. Slyadnikov, and I. Turchanovsky, “The resistance of amorphous metals to thermal effects. Molecular dynamics modeling,” PHYSICAL MESOMECHANICS OF CONDENSED MATTER: Physical Principles of Multiscale Structure Formation and the Mechanisms of Nonlinear Behavior: MESO2022. 2023. link Times cited: 0 NOT USED (low confidence) C. McElfresh, Y. Cui, Y. Cui, S. Dudarev, G. Po, and J. Marian, “Discrete stochastic model of point defect-dislocation interaction for simulating dislocation climb,” International Journal of Plasticity. 2021. link Times cited: 21 NOT USED (low confidence) D. Mejía-Burgos, S. Berrios, J. Mazo‐Zuluaga, and J. Mejía‐López, “Structural stability, shape memory and mechanical properties of Fe/Ni core/shell nanorods,” Journal of Alloys and Compounds. 2021. link Times cited: 0 NOT USED (low confidence) V. Mazhukin, A. V. Shapranov, O. Koroleva, and A. Mazhukin, “Atomistic modeling of the propagation of the melting/crystallization front for metals based on the generalization of the modified transition state theory,” Keldysh Institute Preprints. 2021. link Times cited: 0 Abstract: Based on the modification of the well-known kinetic model wi… read moreAbstract: Based on the modification of the well-known kinetic model with the Wilson-Frenkel diffusion constraint, a new kinetic model of the propagation velocity of the solid/liquid interface in various metal crystals (fcc - Al, Cu) and (bcc - Fe) has been developed in a wide temperature range, including the range of maximum permissible overheating/subcooling values. Molecular dynamics modeling of melting/crystallization processes of Al, Cu and Fe under deep overheating/overcooling conditions has been performed using 3 interaction potentials of the EAM family. By comparing the simulation results with the data of the modified kinetic model, the interface speed response function in the region of the maximum permissible values of overheating/overcooling in metals is constructed. The temperature dependence of the velocity of the interface is diffusion-limited and is described by the same equation for each metal in the entire temperature range. read less NOT USED (low confidence) D. McDowell, “Uncertainty Quantification in Multiscale Materials Modeling.” 2020. link Times cited: 12 NOT USED (low confidence) R. Devanathan, “Interatomic Potentials for Nuclear Materials,” Handbook of Materials Modeling. 2020. link Times cited: 1 NOT USED (low confidence) J. Crocombette and F. Willaime, “Ab Initio Electronic Structure Calculations for Nuclear Materials,” Comprehensive Nuclear Materials. 2020. link Times cited: 11 NOT USED (low confidence) Y. Wang and D. McDowell, “Uncertainty quantification in materials modeling,” Uncertainty Quantification in Multiscale Materials Modeling. 2020. link Times cited: 3 NOT USED (low confidence) A. Nikonov, “Construction of response functions of Cu, Fe and C mesoparticles in MCA method based on molecular dynamics calculations,” PROCEEDINGS OF THE INTERNATIONAL CONFERENCE ON ADVANCED MATERIALS WITH HIERARCHICAL STRUCTURE FOR NEW TECHNOLOGIES AND RELIABLE STRUCTURES 2019. 2019. link Times cited: 1 NOT USED (low confidence) J. J. Möller and E. Bitzek, “Atomic-scale modeling of elementary processes during the fatigue of metallic materials: from crack initiation to crack-microstructure interactions.” 2018. link Times cited: 1 NOT USED (low confidence) A. Nikonov, “Molecular dynamics study of acoustic emission from individual lattice defects.” 2017. link Times cited: 0 NOT USED (low confidence) A. Samin, M. Kurth, and L. Cao, “An analysis of radiation effects on NdFeB permanent magnets,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2015. link Times cited: 12 NOT USED (low confidence) K. Kinoshita, T. Shimokawa, T. Kinari, H. Sawada, K. Kawakami, and K. Ushioda, “Influence of non-glide stresses on the peierls energy of screw dislocations.” 2014. link Times cited: 2 Abstract: We investigate the influence of non-glide stresses on the Pe… read moreAbstract: We investigate the influence of non-glide stresses on the Peierls energy of screw dislocation by using Nudged-Elastic-Band method. The influence of the applied non-glide stress fields on the Peierls energy of a screw dislocation is clearly observed. Moreover, we find that the stress field dependence of the Peierls energy is changed by the moving direction of the screw dislocation under a specific applied stress field. Geometrical parameters, which can measure the atomic elastic deformation around the screw dislocation core, are introduced to explain the stress field dependence of the Peierls energy. Finally, the cross slip of a screw dislocation around a precipitate with a misfit strain is discussed by combining the analytical solution of stress fields around the precipitate with the geometrical parameters obtained by our atomic simulations. * Dr.Eng., Fundamental Metallurgy Research Lab., Advanced Technology Research Laboratories 1-8 Fuso-cho, Amagasaki, Hyogo Pref. 660-0891 NIPPON STEEL & SUMITOMO METAL TECHNICAL REPORT No. 114 MARCH 2017 62 ic simulation, open-sourced LAMMPS 5) is used and the stress field dependency of the Peierls energy is obtained by using the Nudged Elastic Band (NEB) method 6). In this paper, Chapter 2 describes the analysis model and the inter-atomic potential energy. Chapter 3 describes the influence of the non-glide stress on the Peierls energy of the screw dislocation, and the validity of the results obtained is investigated under different periodic boundary conditions and the inter-atomic potential energy. In Chapter 4, why the Peierls energy is changed by the non-glide stress is considered by focusing on the change of the atomic structure near the dislocation core. In addition, how the stress field around the coherent precipitate exerts influence on the cross-slip of the screw dislocation is considered. Lastly, the conclusion of this paper is given in Chapter 5. 2. Analysis Model and Analysis Conditions 2.1 Analysis model In this study, the analysis target is α-Fe. The crystal orientations in the directions of x, y and z are [112 _ ], [111] and [11 _ 0], respectively. Here, the lattice constant of α-Fe is a0. Three vectors are defined as v[112] = a0 [112 _ ] / 3, v[111] = a0 [111] / 2 and v[110] = a0 [11 _ 0]. Using these vectors, analysis zone ei , which has two different periodic boundary conditions is indicated as follows. First model ei s is: e1 s = 14 v[112], e2 s = 16 v[111], e3 s = 24 v[110] + v[111] (1) In this study, this is called the square model. Second model ei p is: e1 p = 14 v[112], e2 p = 16 v[111], e3 p = 24 v[110] + 7 v[112] + v[111] (2) This is called the parallelogram model. Figure 1 (a)(b) shows the analysis zone of each model, indicating that the difference between these two analysis models is the periodic boundary condition in the z direction. For each model, a screw dislocation pair with a distance of 5 nm in between in the x direction is placed at the center. Figure 1 (a)(b) shows the τyz stress field of each model including the screw dislocation pair. In this study, the screw dislocation on the left is referred to as S1 and the screw dislocation on the right is referred to as S2. Since the screw dislocation of S1 has the Burgers vector of bS1 = 1/2 [111], the screw dislocation of S2 has the Burgers vector of bS2 = −bS1. As described later, since this study focuses on the motion of easy-core screw dislocation, in this analysis model to which the periodic boundary conditions are applied, it is necessary to note that the distance in the x direction (distance between S1 and S2 and distance between S2 and S1') between adjacent screw dislocations is not strictly equal. (The difference of the distances is smaller than a0.) Each model uses different periodic boundary conditions in the z direction. As shown in Fig. 1 (a), in the square model, the dislocations that have the same Burgers vector are periodically aligned in the z direction. In contrast, as shown in Fig. 1 (b), in the parallelogram model, the screw dislocations that have different Burgers vector in the z direction are periodically aligned. The interaction between adjacent dislocations is different, and the different stress field in the analysis zone can be confirmed from Fig. 1 (a)(b). Here, e3 is inclined in the y direction by 1/2 v[111] for both models. This is equivalent to the plastic strain generated by putting the screw dislocation pair in the calculation cell. Considering this 1/2 v[111], average stress τyz in the system can be made zero. Using the analysis model above, the influence of the non-glide stress on the Peierls energy of the screw dislocation is considered. In addition, by comparing the results obtained from two analysis models, the influence of the difference of the periodic structure of the screw dislocation is considered. The screw dislocation core of bcc metal has energetically stable easy-cores and unstable hard-cores depending on its atomic geometry. Figure 1 (c) shows the {111} plane of the bcc structure. Here, the circles indicate atoms and the color difference indicates the depth difference in the [111] direction. From this figure, it is confirmed that the {111} plane has the three-layer periodic structure. In an easy-core, if displacement of the screw dislocation is superposed onto a bcc structure, each atomic configuration of the dislocation core maintains the three-layer structure that is the same as a perfect crystal. (The positions indicated by plotting squares in Fig. 1 (c) correspond to easy-cores.) However, in a hard-core, atomic configurations of the dislocation core exist on the same {111} plane. (In other words, atomic configuration of the dislocation core has the same color.) Therefore, the distance between adjacent atoms of a hardcore is shorter than that of an easy-core and the energy of dislocation becomes higher.7) In this study, a transfer phenomenon of the screw dislocation that exists in an easy-core to another adjacent easy-core is considered. 2.2 Inter-atomic potential Two inter-atomic potentials to indicate α-Fe as the inter-atomic interaction are used. One is the embedded atomic method (EAM) 8) by Chamati, et al. and the other is the EAM potential by Mendelev, et al.9) Mendelev, et al. have studied five types of potentials, from which a potential that best describes a defect structure in bcc iron is used as the other one. By comparing the influence of non-glide stress on the Peierls energy of the screw dislocation obtained from these two inter-atomic potentials, the validity of the result obtained 1 2 read less NOT USED (low confidence) X.-yan Li et al., “Principal physical parameters characterizing the interactions between irradiation-induced point defects and several tilt symmetric grain boundaries in Fe, Mo and W,” Journal of Nuclear Materials. 2014. link Times cited: 29 NOT USED (low confidence) Z. Chen, R. Chen, and B. Shan, “Nanomaterial Design and Computational Modeling.” 2014. link Times cited: 0 NOT USED (low confidence) I. Mastorakos, H. Zbib, D. Li, M. Khaleel, and X. Sun, “Multiscale Modeling of Irradiation Induced Hardening in Iron Alloys,” MRS Proceedings. 2012. link Times cited: 0 NOT USED (low confidence) H. Urbassek and L. Sandoval, “Molecular dynamics modeling of martensitic transformations in steels.” 2012. link Times cited: 17 Abstract: Abstract: Molecular dynamics simulation constitutes an appea… read moreAbstract: Abstract: Molecular dynamics simulation constitutes an appealing method to study, on an atomistic basis, the processes and mechanisms of martensitic phase transformations. Its use requires the existence of reliable interatomic potentials which adequately describe the properties of the phases. In this review we present a few recent examples demonstrating the application of this method to the study of the martensitic phase transition in iron. Besides phase changes in bulk materials, transformations in small systems (nanowires) are also considered. read less NOT USED (low confidence) A. Koester, A. Ma, and A. Hartmaier, “Atomistically informed continuum model for body centered cubic iron,” MRS Proceedings. 2011. link Times cited: 0 NOT USED (low confidence) L. Malerba, “Multi-scale modelling of irradiation effects in nuclear power plant materials.” 2010. link Times cited: 3 Abstract: Abstract: This chapter surveys the computer-based multi-scal… read moreAbstract: Abstract: This chapter surveys the computer-based multi-scale modelling approaches currently being used to develop physical models of the effects of radiation on nuclear materials. The focus is on the problem of radiation-induced hardening (and embrittlement) in steels, limited to the scales ranging from the nucleus to the single crystal. First, the multi-scale nature of radiation effects is illustrated, including examples of microstructural and mechanical property changes observed in steels used in nuclear reactors. Then the chapter discusses the fundamental ideas upon which the multi-scale modelling approach is based. Next, an overview of the techniques of use in a multi-scale modelling framework is given, with an example of how these can be integrated. A discussion of the state-of-the-art and other general remarks conclude the chapter. read less NOT USED (low confidence) 英喜 森, 肇 君塚, and 成信 尾方, “第一原理計算による BCC 鉄の一般化積層欠陥エネルギー表面に基づいた転位構造とパイエルス応力の解析,” Journal of The Japan Institute of Metals. 2009. link Times cited: 5 NOT USED (low confidence) 肇 君塚, 英喜 森, 裕己 牛田, and 成信 尾方, “経路積分セントロイド分子動力学法による BCC 金属中の水素拡散性とその温度依存性の評価,” Journal of The Japan Institute of Metals. 2009. link Times cited: 5 Abstract: We have analyzed the diffusion behavior of interstitial hydr… read moreAbstract: We have analyzed the diffusion behavior of interstitial hydrogen in bcc iron and niobium using path-integral centroid molecular dynamics (CMD) method, which can describe the real-time evolution of particles based on quantum statistical mechanics. In this study, the embedded-atom-method (EAM) potential model for the iron-hydrogen interaction is developed to reproduce the ab initio minimum energy path of hydrogen migration based on the density functional theory (DFT) data in the literature, while the description of niobium-hydrogen interaction is based on an empirical potential model. Time evolutions of mean-square displacements of hydrogen atoms in the two bulk metals are calculated at various temperatures, and then diffusion coefficients and activation energies of hydrogen migration are evaluated. Especially in the case of iron, the results are in good agreement with experimental measurements over a wide temperature range. In order to characterize the quantum effects on the hydrogen diffusion process, the CMD results are compared with those obtained from classical molecular dynamics method. The obtained results indicate that the quantum effects can play a significant role in hydrogen diffusivity over a wide temperature range in these bcc metals. read less NOT USED (low confidence) J. Boutard, S. Dudarev, and E. Diegele, “Radiation Effects in Structural Materials for Fusion Power Plants: the Outcomes of the EU Fusion Program.” 2008. link Times cited: 9 NOT USED (low confidence) M. Samaras, M. Victoria, and W. Hoffelner, “The Structure, Role and Flexibility of Grain Boundaries,” MRS Proceedings. 2008. link Times cited: 0 NOT USED (low confidence) V. Vítek and V. Paidar, “Chapter 87 - Non-planar Dislocation Cores: A Ubiquitous Phenomenon Affecting Mechanical Properties of Crystalline Materials.” 2008. link Times cited: 104 NOT USED (low confidence) P. Geysermans, “Numerical Modeling of Radiation Effects in Solids: Principal Features, Limitations and Perspectives.” 2008. link Times cited: 0 NOT USED (low confidence) D. Farkas and J. Rickman, “Multiscale modeling of deformation and fracture in metallic materials.” 2007. link Times cited: 0 NOT USED (low confidence) C. Becquart, “RPV steel microstructure evolution under irradiation: a multiscale approach,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 17 NOT USED (high confidence) S. Signetti and A. Heine, “Quantification of the Kinetic Energy Conversion to Temperature Increase in Metal-on-Metal Impacts up to Hypervelocity Conditions by Molecular Dynamics Simulation,” Journal of Dynamic Behavior of Materials. 2023. link Times cited: 2 NOT USED (high confidence) T. Suzudo, K. Ebihara, T. Tsuru, and H. Mori, “Cleavages along 110 in bcc iron emit dislocations from the curved crack fronts,” Scientific Reports. 2022. link Times cited: 5 NOT USED (high confidence) L.-J. Huang, M. Nastar, L. Messina, and T. Schuler, “Elastodiffusion calculation of asymptotic absorption efficiencies and bias factors of dislocations in Al, Ni and Fe,” Journal of Nuclear Materials. 2022. link Times cited: 2 NOT USED (high confidence) I. A. Alhafez and H. Urbassek, “Indentation and Scratching with a Rotating Adhesive Tool: A Molecular Dynamics Simulation Study,” Tribology Letters. 2022. link Times cited: 0 NOT USED (high confidence) T. Vasina, J. Bernard, M. Benoit, and F. Calvo, “Debye temperature of iron nanoparticles: Finite-size effects in the scaling regime,” Physical Review B. 2022. link Times cited: 0 NOT USED (high confidence) J. Chapman and P. Ma, “A machine-learned spin-lattice potential for dynamic simulations of defective magnetic iron,” Scientific Reports. 2022. link Times cited: 5 NOT USED (high confidence) T. D. Pham, T. Nguyen, T. Terai, Y. Shibutani, M. Sugiyama, and K. Sato, “Interaction of Carbon and Extended Defects in α-Fe Studied by First-Principles Based Interatomic Potential,” MATERIALS TRANSACTIONS. 2022. link Times cited: 1 NOT USED (high confidence) F. Meng et al., “General-purpose neural network interatomic potential for the
α
-iron and hydrogen binary system: Toward atomic-scale understanding of hydrogen embrittlement,” Physical Review Materials. 2021. link Times cited: 13 Abstract: To understand the physics of hydrogen embrittlement at the a… read moreAbstract: To understand the physics of hydrogen embrittlement at the atomic scale, a general-purpose neural network interatomic potential (NNIP) for the α -iron and hydrogen binary system is presented. It is trained using an extensive reference database produced by density functional theory (DFT) calculations. The NNIP can properly describe the interactions of hydrogen with various defects in α -iron, such as vacancies, surfaces, grain boundaries, and dislocations; in addition to the basic properties of α -iron itself, the NNIP also handles the defect properties in α -iron, hydrogen behavior in α -iron, and hydrogen-hydrogen interactions in α -iron and in vacuum, including the hydrogen molecule formation and dissociation at the α -iron surface. These are superb challenges for the existing empirical interatomic potentials, like the embedded-atom method based potentials, for the α -iron and hydrogen binary system. In this study, the NNIP was applied to several key phenomena necessary for understanding hydrogen embrittlement, such as hydrogen charging and discharging to α -iron, hydrogen transportation in defective α -iron, hydrogen trapping and desorption at the defects, and hydrogen-assisted cracking at the grain boundary. Unlike the existing interatomic potentials, the NNIP simulations quantitatively described the atomistic details of hydrogen behavior in the defective α -iron system with DFT accuracy. read less NOT USED (high confidence) A. Dahlström, F. Danoix, P. Hedström, J. Odqvist, and H. Zapolsky, “Effect of Stress on Spinodal Decomposition in Binary Alloys: Atomistic Modeling and Atom Probe Tomography,” Metallurgical and Materials Transactions A. 2021. link Times cited: 2 NOT USED (high confidence) W. Ko, J. S. Lee, and D.-H. Kim, “Atomistic simulations of Ag–Cu–Sn alloys based on a new modified embedded-atom method interatomic potential,” Journal of Materials Research. 2021. link Times cited: 3 Abstract: An interatomic potential for the ternary Ag–Cu–Sn system, an… read moreAbstract: An interatomic potential for the ternary Ag–Cu–Sn system, an important material system related to the applications of lead-free solders, is developed on the basis of the second nearest-neighbor modified embedded-atom-method formalism. Potential parameters for the ternary and related binary systems are determined based on the recently improved unary description of pure Sn and the present improvements to the unary descriptions of pure Ag and Cu. To ensure the sufficient performance of atomistic simulations in various applications, the optimization of potential parameters is conducted based on the force-matching method that utilizes density functional theory predictions of energies and forces on various atomic configurations. We validate that the developed interatomic potential exhibits sufficient accuracy and transferability to various physical properties of pure metals, intermetallic compounds, solid solutions, and liquid solutions. The proposed interatomic potential can be straightforwardly used in future studies to investigate atomic-scale phenomena in soldering applications. read less NOT USED (high confidence) I. Chesser, B. Runnels, and E. A. Holm, “A taxonomy of grain boundary migration mechanisms via displacement texture characterization,” Acta Materialia. 2021. link Times cited: 6 NOT USED (high confidence) H. Zapolsky, A. Vaugeois, R. Patte, and G. Demange, “Size-Dependent Solute Segregation at Symmetric Tilt Grain Boundaries in α-Fe: A Quasiparticle Approach Study,” Materials. 2021. link Times cited: 2 Abstract: In the present work, atomistic modeling based on the quasipa… read moreAbstract: In the present work, atomistic modeling based on the quasiparticle approach (QA) was performed to establish general trends in the segregation of solutes with different atomic size at symmetric 〈100〉 tilt grain boundaries (GBs) in α-Fe. Three types of solute atoms X1, X2 and X3 were considered, with atomic radii smaller (X1), similar (X2) and larger (X3) than iron atoms, respectively, corresponding to phosphorus (P), antimony (Sb) and tin (Sn). With this, we were able to evidence that segregation is dominated by atomic size and local hydrostatic stress. For low angle GBs, where the elastic field is produced by dislocation walls, X1 atoms segregate preferentially at the limit between compressed and dilated areas. Contrariwise, the positions of X2 atoms at GBs reflect the presence of tensile and compressive areal regions, corresponding to extremum values of the σXX and σYY components of the strain tensor. Regarding high angle GBs Σ5 (310) (θ = 36.95°) and Σ29 (730), it was found that all three types of solute atoms form Fe9X clusters within B structural units (SUs), albeit being deformed in the case of larger atoms (X2 and X3). In the specific case of Σ29 (730) where the GB structure can be described by a sequence of |BC.BC| SUs, it was also envisioned that the C SU can absorb up to four X1 atoms vs. one X2 or X3 atom only. Moreover, a depleted zone was observed in the vicinity of high angle GBs for X2 or X3 atoms. The significance of this research is the development of a QA methodology capable of ascertaining the atomic position of solute atoms for a wide range of GBs, as a mean to highlight the impact of the solute atoms’ size on their locations at and near GBs. read less NOT USED (high confidence) P. Yu, L. Zhang, and L. Du, “Atomic Simulations for Packing Changes of Nano-Sized Cu Clusters Embedded in the Febulk on Heating,” Metals. 2021. link Times cited: 3 Abstract: Understanding of the defect evolution mechanism under irradi… read moreAbstract: Understanding of the defect evolution mechanism under irradiation is very important for the research of pressure vessel steel embrittlement. In this paper, the embedded atom method (EAM) based canonical ensemble molecular dynamics (MD) method was used to study the evolution of the stacking structure of different nano-sized Cun (n = 13, 43 and 87) clusters in an Febulk embedded with BCC lattice structure during continuous heating. The mean square displacement, pair distribution functions and atomic structures of Cu atom clusters at the nanometer scale were calculated at different temperatures. The structural changes present apparent differences, for the Febulks contain nano-sized Cu clusters with different atom numbers during heating. For the Febulk–Cu13 system, since the ability to accommodate the atomic Cu in the Fe substrate is lesser, a small number of Cu atoms in BCC lattice positions cannot influence the whole structure of the Fe-Cu system. For the Febulk–Cu43 system, with an increase in temperature, a Cu atomic pile structural change happened, and the strain areas decreased significantly in the Febulk, but a single strain area grew large. For the Febulk–Cu87 system, when the Cu atoms are constrained by the Fe atoms in bulk, only a few of the Cu atoms adjust their positions. With the increase in temperature, strain in the Fe eased. read less NOT USED (high confidence) S. Kumar, M.-W. Liu, K.-A. Wu, and M. Gururajan, “Anisotropy in interface stress at the bcc-iron solid–melt interface: Molecular dynamics and phase field crystal modelling,” Computational Materials Science. 2021. link Times cited: 2 NOT USED (high confidence) E. Clouet, B. Bienvenu, L. Dezerald, and D. Rodney, “Screw dislocations in BCC transition metals: from ab initio modeling to yield criterion,” Comptes Rendus. Physique. 2021. link Times cited: 19 Abstract: We show here how density functional theory calculations can … read moreAbstract: We show here how density functional theory calculations can be used to predict the temperatureand orientation-dependence of the yield stress of body-centered cubic (BCC) metals in the thermallyactivated regime where plasticity is governed by the glide of screw dislocations with a 1/2 Burgers vector. Our numerical model incorporates non-Schmid effects, both the twinning/antitwinning asymmetry and non-glide effects, characterized through ab initio calculations on straight dislocations. The model uses the stress-dependence of the kink-pair nucleation enthalpy predicted by a line tension model also fully parameterized on ab initio calculations. The methodology is illustrated here on BCC tungsten but is applicable to all BCC metals. Comparison with experimental data allows to highlight both the successes and remaining limitations of our modeling approach. read less NOT USED (high confidence) S. Kazanç and C. Canbay, “Cu’nun Mekanik Özelliklerine Tek Eksenli Germe Zorlanmasının Etkisi: Moleküler Dinamik Yöntemi,” Fırat Üniversitesi Mühendislik Bilimleri Dergisi. 2021. link Times cited: 0 NOT USED (high confidence) M. Rahman, F. El-Mellouhi, O. Bouhali, C. Becquart, and N. Mousseau, “Pressure effect on diffusion of carbon at the
85.91∘〈100〉
symmetric tilt grain boundary of
α
-iron,” Physical Review Materials. 2021. link Times cited: 1 NOT USED (high confidence) R. Ocaya, “Lattice elasticity, waves and temperature from an interaction potential,” European Journal of Physics. 2021. link Times cited: 0 Abstract: Kinetic theory is taught to first-year students of physics a… read moreAbstract: Kinetic theory is taught to first-year students of physics as a plausible account of thermal equilibrium on the microscopic scale. However, it does not adequately clarify important properties like elasticity, lattice waves, phonons, optical oscillations, bandgap and so on, that are postulated through atomic vibrations. In this article, we apply simple computational tools in four computational experiments to study the effects of the vibrations of 373 atoms of a face-centered cubic crystal such as copper. We also discuss the perturbation modelling approach, the Sutton–Chen potential, and discrete integration. The experiments have different aims e.g. determining the steady-state atom arrangement starting from an arbitrarily specified cluster, the elasticity, the ensuing wave motion, lattice resonance, acoustic properties, and temperature rise. The calculated results agree well with their literature values. read less NOT USED (high confidence) S. Kazanç and C. Canbay, “Fe elementindeki αγδ Katı-Katı Faz Geçişlerinin Moleküler Dinamik Benzetimi ile İncelenmesi,” Fırat Üniversitesi Mühendislik Bilimleri Dergisi. 2021. link Times cited: 0 Abstract: When the phase diagram of the element Fe is examined, it is … read moreAbstract: When the phase diagram of the element Fe is examined, it is seen that it has different crystal structures at different temperatures below its melting temperature. In this study, the solid-solid phase transformations occurring at different temperatures in the Fe model system consisting of 4000 atoms were investigated using molecular dynamic simulation method. The Embedded Atom Method(EAM), which includes many body interactions, was used to calculate interactions between atoms. For the element Fe, the α, γ and δ phases formed below the melting temperature and the transformation temperatures for these phases were determined and the results were compared with the experimental values. Radial distribution function, changes in thermodynamic quantities and Ackland-Jones analysis method were used in the structural analysis of the model system. read less NOT USED (high confidence) S. Sun and P. Guan, “The critical model size for simulating the structure-dynamics correlation in bulk metallic glasses,” Science China Materials. 2021. link Times cited: 5 NOT USED (high confidence) L. Morrissey and S. Nakhla, “Considerations when calculating the mechanical properties of single crystals and bulk polycrystals from molecular dynamics simulations,” Molecular Simulation. 2020. link Times cited: 4 Abstract: ABSTRACT The choice of a proper interatomic potential is cri… read moreAbstract: ABSTRACT The choice of a proper interatomic potential is critical to obtaining accurate and realistic molecular dynamics results. However, previous studies that have tested the suitability of a potential to predict mechanical properties often do so using elastic constants from a triaxial stress state that ignores Poisson’s effect. While this method is suitable it is not consistent with macroscale experimental methods and cannot provide the complete loading behaviour. Further, there is a lack of knowledge as to whether accuracy in predicting elastic constants from a fixed volume condition indicates accuracy for elastic moduli from uniaxial tensile simulations. Moreover, those studies that did account for Poisson’s effect studied only one crystal orientation and thus assumed potential accuracy is independent of crystal orientation. Results from the current study demonstrated that accuracy of a potential is dependent on the crystal direction. Further, the most accurate potentials for elastic constants calculated using a fixed volume condition were not necessarily the most accurate at predicting elastic moduli from a physically realisable tension test. Finally, the Voigt Reuss Hill (VRH) method was shown to accurately predict polycrystalline mechanical properties from single crystal data as a function of temperature. read less NOT USED (high confidence) T. Suzudo, K. Ebihara, and T. Tsuru, “Brittle-fracture simulations of curved cleavage cracks in α-iron: A molecular dynamics study,” AIP Advances. 2020. link Times cited: 9 Abstract: Although body-centered-cubic (bcc) metals and alloys are ubi… read moreAbstract: Although body-centered-cubic (bcc) metals and alloys are ubiquitous as structural materials, they are brittle, particularly at low temperatures; however, the mechanism of their brittle fracture is not fully understood. In this study, we conduct a series of three-dimensional molecular dynamics simulations of the cleavage fracture of α-iron. In particular, we focus on mode-I loading starting from curved crack fronts or the so-called penny-shaped cracks. In the simulations, brittle fractures are observed at cleavages on the {100} plane, while the initial cracks become blunted on other planes as a result of dislocation emissions. Our modeling results agreed with a common experimental observation, that is, {100} is the preferential cleavage plane in bcc transition metals. In addition, dislocation emissions from the crack front were analyzed; the result supported the notion that plasticity in the vicinity of the crack front determines the preferential cleavage plane. read less NOT USED (high confidence) R. Ishraaq, M. Rashid, and A. Afsar, “A molecular dynamics investigation of mechanical properties of graphene reinforced iron composite and the effect of vacancy defect distance from the matrix-fiber interface,” arXiv: Materials Science. 2020. link Times cited: 1 Abstract: Graphene is a material of excellent mechanical properties, w… read moreAbstract: Graphene is a material of excellent mechanical properties, which make it an ideal fiber for reinforcing metal. Since iron is the most used metal in the world, reinforcing iron with graphene can reduce the overall requirement of material in any application where strength is demanded. However, the effect of graphene reinforcement on the mechanical properties of iron needs to be known before the industrial application of the composite. In this paper, we have investigated the mechanical properties of graphene-reinforced iron composite by Molecular Dynamics (MD) method for various conditions. The properties were investigated by applying uniaxial tension on a modeled representative volume element (RVE). The effect of temperature on the mechanical property of the composite was also studied because the knowledge is required for manufacturing products with the composite operating at a wide temperature range. MD analysis also revealed that the initiation of fracture is from the matrix-fiber interface. We also investigated how the distance of vacancy defects from the matrix-fiber interface affects the mechanical properties of the composite, which can be used to select a suitable manufacturing process. The results obtained from this study show that vacancy defects lower the strength at a greater extent as it gets closer to the interface. read less NOT USED (high confidence) S. Combettes et al., “How interface properties control the equilibrium shape of core-shell Fe-Au and Fe-Ag nanoparticles.,” Nanoscale. 2020. link Times cited: 6 Abstract: While combining two metals in the same nanoparticle can lead… read moreAbstract: While combining two metals in the same nanoparticle can lead to remarkable novel applications, the resulting structure in terms of crystallinity and shape remains difficult to predict. It is thus essential to provide a detailed atomistic picture of the underlying growth processes. In the present work we address the case of core-shell Fe-Au and Fe-Ag nanoparticles. Interface properties between Fe and the noble metals Au and Ag, computed using DFT, were used to parameterize Fe-Au and Fe-Ag pairwise interactions in combination with available many-body potentials for the pure elements. The growth of Au or Ag shells on nanometric Fe cores with prescribed shapes was then modelled by means of Monte Carlo simulations. The shape of the obtained Fe-Au nanoparticles is found to strongly evolve with the amount of metal deposited on the Fe core, a transition from the polyhedral Wulff shape of bare iron to a cubic shape taking place as the amount of deposited gold exceeds two monolayers. In striking contrast, the growth of silver proceeds in a much more anisotropic, Janus-like way and with a lesser dependence on the iron core shape. In both cases, the predicted morphologies are found to be in good agreement with experimental observations in which the nanoparticles are grown by physical deposition methods. Understanding the origin of these differences, which can be traced back to subtle variations in the electronic structure of the Au/Fe and Ag/Fe interfaces, should further contribute to the better design of core-shell bimetallic nanoparticles. read less NOT USED (high confidence) Y. Jiang, B. Wang, C. Xu, and J. Zhang, “Atomistic Simulation of the Strain Driven Phase Transition in Pure Iron Thin Films Containing Twin Boundaries,” Metals. 2020. link Times cited: 0 Abstract: Using molecular dynamics (MD) simulation, the strain-induced… read moreAbstract: Using molecular dynamics (MD) simulation, the strain-induced phase transitions in pure body-centered-cubic (bcc) iron (Fe) thin films containing twin boundaries (TBs) with different TB fractions and orientations are studied. Two groups of bcc thin films with different TB-surface orientation relationships are designed. In film group 1, the (112) [ 11 1 ¯ ] TBs are perpendicular to the ( 11 1 ¯ ) free surfaces, while the (112) [ 11 1 ¯ ] TBs are parallel to the free surfaces in film group 2. We vary the TB numbers inserted into the films to study the effect of TB fraction on the phase transition. Biaxial strains are applied to the films to induce the bcc to close packed (cp) phase transition. The critical strain, at which the first phase transition takes place, decreases with the TB fraction increase in film group 1 with a perpendicular TB-surface orientation, while such a relationship is not observed in film group 2 with parallel TB-surface orientation. We focus on the free surface and TB as the nucleation positions of the new phase and the afterward growth. In addition, the dynamics of the phase transition is discussed. This work may help to understand the mechanism of phase transition in nanoscale or surface-dominant systems with pre-existing defects. read less NOT USED (high confidence) P. Wang, Z. Song, Q. Li, and H. Wang, “Atomistic simulation for the interaction between dislocation and solute atoms, clusters, and associated physical insights,” Journal of Applied Physics. 2020. link Times cited: 1 Abstract: Solid-solution hardening (SSH), originated mainly from the p… read moreAbstract: Solid-solution hardening (SSH), originated mainly from the point obstacles with prescribed resistance (short-range) or spherical inclusions with purely dilatational eigenstrain (long-range), is critical to materials science and technological applications. Dislocation gliding in solid-solution hardening alloys generally undergoes both short-range and long-range interactions. However, the respective contribution of each aspect remains unclear. Here, we successfully decouple the short-range lattice distortion and long-range size misfit of the solid-solution hardening effect by introducing two scaling factors (s1 and s3) and analyzing the contributions of each aspect on the solute/dislocation interaction, respectively. For scaling factor s1, the interaction energy is localized, resembling the short-range interactions without volume change. The scaling factor s3 is equivalent to a dilatation/constriction center with volume change. The interaction energy is a long-range parameter and well predicted from the pure continuum elasticity perspective. Large-scale molecular dynamics (MD) simulations reveal the unique impacts of two strengthening mechanisms on dislocations with different scaling factors. It is found that the energy landscape and size misfit effect of the solute atoms play important roles in the SSH effect. With deeply understanding the SSH effects and the rapidly increasing computational power, it may pave a practical way to apply MD simulations on complex strengthening mechanism studies. read less NOT USED (high confidence) R. Meyer et al., “Vibrational and magnetic signatures of extended defects in Fe,” The European Physical Journal B. 2020. link Times cited: 5 NOT USED (high confidence) P. Yu, G. Zhu, and M. Wen, “Application of grand-canonical ensemble Monte Carlo simulation in metals using cavity-biased method,” Molecular Simulation. 2020. link Times cited: 1 Abstract: ABSTRACT A critic issue of the application of the convention… read moreAbstract: ABSTRACT A critic issue of the application of the conventional grand-canonical Monte Carlo (GCMC) method in high-density systems is the low acceptance ratio of insertion. Previous studies have revealed that this can be overcome by the cavity-biased (CB) insertion method in simulations of vapours, fluids and liquids. Here, we demonstrate that the method is also highly efficient in metals. Using the Fe–H system as an example, we find that the acceptance ratio of inserting H into Fe lattice is increased by several times using the CB GCMC method. The method is more valid than the conventional one at bulk H concentration over 5‰, implying that the CB GCMC method is highly efficient when there are deep traps for H in simulation systems, i.e. dislocations and interfaces. Application of the method in nanocrystalline Fe shows that the CPU time required for obtaining an equilibrium distribution of H is reduced by 60%. read less NOT USED (high confidence) X. Yang, Y. Zheng, and J. Li, “Evaluation of Cr Concentration Effect on Displacement Cascades in Fe–Cr Alloys with Piecewise Potential,” Acta Mechanica Solida Sinica. 2020. link Times cited: 0 NOT USED (high confidence) X. W. Zhou, M. E. Foster, J. Ronevich, and C. S. Marchi, “Review and construction of interatomic potentials for molecular dynamics studies of hydrogen embrittlement in Fe─C based steels,” Journal of Computational Chemistry. 2020. link Times cited: 7 Abstract: Reducing hydrogen embrittlement in the low‐cost Fe─C based s… read moreAbstract: Reducing hydrogen embrittlement in the low‐cost Fe─C based steels have the potential to significantly impact the development of hydrogen energy technologies. Molecular dynamics studies of hydrogen interactions with Fe─C steels provide fundamental information about the behavior of hydrogen at microstructural length scales, although such studies have not been performed due to the lack of an Fe─C─H ternary interatomic potential. In this work, the literature on interatomic potentials related to the Fe─C─H systems are reviewed with the aim of constructing an Fe─C─H potential from the published binary potentials. We found that Fe─C, Fe─H, and C─H bond order potentials exist and can be combined to construct an Fe─C─H ternary potential. Therefore, we constructed two such Fe─C─H potentials and demonstrate that these ternary potentials can reasonably capture hydrogen effects on deformation characteristics and deformation mechanisms for a variety of microstructural variations of the Fe─C steels, including martensite that results from γ to α phase transformation, and pearlite that results from the eutectic formation of the Fe3C cementite compound. read less NOT USED (high confidence) A. Tsukanov et al., “Effect of Cold-Sintering Parameters on Structure, Density, and Topology of Fe–Cu Nanocomposites,” Materials. 2020. link Times cited: 8 Abstract: The design of advanced nanostructured materials with predete… read moreAbstract: The design of advanced nanostructured materials with predetermined physical properties requires knowledge of the relationship between these properties and the internal structure of the material at the nanoscale, as well as the dependence of the internal structure on the production (synthesis) parameters. This work is the first report of computer-aided analysis of high pressure consolidation (cold sintering) of bimetallic nanoparticles of two immiscible (Fe and Cu) metals using the embedded atom method (EAM). A detailed study of the effect of cold sintering parameters on the internal structure and properties of bulk Fe–Cu nanocomposites was conducted within the limitations of the numerical model. The variation of estimated density and bulk porosity as a function of Fe-to-Cu ratio and consolidation pressure was found in good agreement with the experimental data. For the first time, topological analysis using Minkowski functionals was applied to characterize the internal structure of a bimetallic nanocomposite. The dependence of topological invariants on input processing parameters was described for various components and structural phases. The model presented allows formalizing the relationship between the internal structure and properties of the studied nanocomposites. Based on the obtained topological invariants and Hadwiger’s theorem we propose a new tool for computer-aided design of bimetallic Fe–Cu nanocomposites. read less NOT USED (high confidence) W. Jian, D. Hui, and D. Lau, “Nanoengineering in biomedicine: Current development and future perspectives,” Nanotechnology Reviews. 2020. link Times cited: 35 Abstract: Recent advances in biomedicine largely rely on the developme… read moreAbstract: Recent advances in biomedicine largely rely on the development in nanoengineering. As the access to unique properties in biomaterials is not readily available from traditional techniques, the nanoengineering becomes an effective approach for research and development, by which the performance as well as the functionalities of biomaterials has been greatly improved and enriched. This review focuses on the main materials used in biomedicine, including metallic materials, polymers, and nanocomposites, as well as the major applications of nanoengineering in developing biomedical treatments and techniques. Research that provides an in-depth understanding of material properties and efficient enhancement of material performance using molecular dynamics simulations from the nanoengineering perspective are discussed. The advanced techniques which facilitate nanoengineering in biomedical applications are also presented to inspire further improvement in the future. Furthermore, the potential challenges of nanoengineering in biomedicine are evaluated by summarizing concerned issues and possible solutions. Graphical abstract read less NOT USED (high confidence) S. Yin, J. Ding, M. Asta, and R. Ritchie, “Ab initio modeling of the energy landscape for screw dislocations in body-centered cubic high-entropy alloys,” npj Computational Materials. 2019. link Times cited: 55 NOT USED (high confidence) S. Stephan, M. Dyga, H. Urbassek, and H. Hasse, “The Influence of Lubrication and the Solid-Fluid Interaction on Thermodynamic Properties in a Nanoscopic Scratching Process.,” Langmuir : the ACS journal of surfaces and colloids. 2019. link Times cited: 23 Abstract: Liquid lubricants play an important role in contact processe… read moreAbstract: Liquid lubricants play an important role in contact processes; for example, they reduce friction and cool the contact zone. To gain better understanding of the influence of lubrication on the nanoscale, both dry and lubricated scratching processes in a model system are compared in the present work using molecular dynamics simulations. The entire range between total dewetting and total wetting is investigated by tuning the solid-fluid interaction energy. The investigated scratching process consists of three sequential movements: A cylindrical indenter penetrates an initially flat substrate, then scratches in the lateral direction, and is finally retracted out of the contact with the substrate. The indenter is fully submersed in the fluid in the lubricated cases. The substrate, the indenter, and the fluid are described by suitably parametrized Lennard-Jones model potentials. The presence of the lubricant is found to have a significant influence on the friction and on the energy balance of the process. The thermodynamic properties of the lubricant are evaluated in detail. A correlation of the simulation results for the profiles of the temperature, density, and pressure of the fluid in the vicinity of the chip is developed. The work done by the indenter is found to mainly dissipate and thereby heat up the substrate and eventually the fluid. Only a minor part of the work causes plastic deformation of the substrate. read less NOT USED (high confidence) L. Wang, J. Jin, P. Yang, Y. Zong, and Q. Peng, “Graphene Adhesion Mechanics on Iron Substrates: Insight from Molecular Dynamic Simulations,” Crystals. 2019. link Times cited: 8 Abstract: The adhesion feature of graphene on metal substrates is impo… read moreAbstract: The adhesion feature of graphene on metal substrates is important in graphene synthesis, transfer and applications, as well as for graphene-reinforced metal matrix composites. We investigate the adhesion energy of graphene nanosheets (GNs) on iron substrate using molecular dynamic (MD) simulations. Two Fe–C potentials are examined as Lennard–Jones (LJ) pair potential and embedded-atom method (EAM) potential. For LJ potential, the adhesion energies of monolayer GN are 0.47, 0.62, 0.70 and 0.74 J/m2 on the iron {110}, {111}, {112} and {100} surfaces, respectively, compared to the values of 26.83, 24.87, 25.13 and 25.01 J/m2 from EAM potential. When the number of GN layers increases from one to three, the adhesion energy from EAM potential increases. Such a trend is not captured by LJ potential. The iron {110} surface is the most adhesive surface for monolayer, bilayer and trilayer GNs from EAM potential. The results suggest that the LJ potential describes a weak bond of Fe–C, opposed to a hybrid chemical and strong bond from EAM potential. The average vertical distances between monolayer GN and four iron surfaces are 2.0–2.2 Å from LJ potential and 1.3–1.4 Å from EAM potential. These separations are nearly unchanged with an increasing number of layers. The ABA-stacked GN is likely to form on lower-index {110} and {100} surfaces, while the ABC-stacked GN is preferred on higher-index {111} surface. Our insights of the graphene adhesion mechanics might be beneficial in graphene growing, surface engineering and enhancement of iron using graphene sheets. read less NOT USED (high confidence) S. Teus and V. Gavriljuk, “Molecular Dynamics Study of the Hydrogen and Carbon Effect on Mobility of Grain Boundaries in α-Iron,” METALLOFIZIKA I NOVEISHIE TEKHNOLOGII. 2019. link Times cited: 0 NOT USED (high confidence) X. Yang, Y. Zheng, and J. Li, “Evaluation of Cr Concentration Effect on Displacement Cascades in Fe–Cr Alloys with Piecewise Potential,” Acta Mechanica Solida Sinica. 2019. link Times cited: 0 NOT USED (high confidence) D. Lenev and G. Norman, “Molecular Modeling of the Thermal Accommodation of Argon Atoms on Clusters of Iron Atoms,” High Temperature. 2019. link Times cited: 7 NOT USED (high confidence) S. M. Handrigan, L. Morrissey, and S. Nakhla, “Investigating various many-body force fields for their ability to predict reduction in elastic modulus due to vacancies using molecular dynamics simulations,” Molecular Simulation. 2019. link Times cited: 6 Abstract: ABSTRACT Molecular dynamics simulations are more frequently … read moreAbstract: ABSTRACT Molecular dynamics simulations are more frequently being utilised to predict macroscale mechanical properties as a result of atomistic defects. However, the interatomic force field can significantly affect the resulting mechanical properties. While several studies exist which demonstrate the ability of various force fields to predict mechanical properties, the investigation into which is most accurate for the investigation of vacancies is limited. To obtain meaningful predictions of mechanical properties, a clear understanding of force field parameterisation is required. As such, the current study evaluates various many-body force fields to demonstrate the reduction in mechanical properties of iron and iron–chromium due to the presence of vacancies while undergoing room temperature atomistic uniaxial tension. Reduction was normalised in each case with the zero-vacancy elastic modulus, removing the need to predict an accurate nominal elastic modulus. Comparisons were made to experimental data and an empirical model from literature. It was demonstrated that accurate fitting to vacancy formation and migration energy allowed for accurate predictions. In addition, bond-order based force fields showed enhanced predictions regardless of fitting procedure. Overall, these findings highlight the need to understand capabilities and limitations of available force fields, as well as the need for enhanced parameterisation of force fields. read less NOT USED (high confidence) Z.-G. Yu, Q. Luo, J.-yu Zhang, and K. Chou, “An insight into the viscosity prediction of ternary alloys with limited solubility,” Philosophical Magazine. 2019. link Times cited: 9 Abstract: ABSTRACT Geometrical models have been successfully applied i… read moreAbstract: ABSTRACT Geometrical models have been successfully applied into the calculation of viscosity of alloys. However, traditional geometrical (TG) models are feasible only in systems with a complete solubility area. Otherwise, they will not work. In this paper, a new method has been introduced for the calculation of alloy systems with limited solubility. How the new method overcomes the drawbacks of the TG models is discussed and analysed. The viscosity of two alloy systems with limited solubility is calculated by the present model. Comparisons between the experimental viscosity and the calculated values by different models show that our model gives the best results, especially for the data nearby the limited solubility area. The introduction of this model provides a way to solve the calculation problems of ternary alloys with limited solubility, which will extend geometrical models to more practical systems. read less NOT USED (high confidence) S. Zhi-peng, D. Fuzhi, X. Ben, and Z. Wen-zheng, “Three-Dimensional Growth of Coherent Ferrite in Austenite: A Molecular Dynamics Study,” Acta Metallurgica Sinica (english Letters). 2019. link Times cited: 1 NOT USED (high confidence) M. Ladinek and T. Hofer, “On the Influence of Loading Order in Nanostructural Fatigue Crack Propagation in BCC Iron—A Molecular Dynamics Study,” Metals. 2019. link Times cited: 4 Abstract: Most investigations dealing with fatigue crack propagation o… read moreAbstract: Most investigations dealing with fatigue crack propagation on the nanoscale, limit their studies on a loading scenario of constant stress or strain amplitudes. Since such a load scenario is rather rare, this paper aims to examine the influence of the load sequence on the crack growth using bcc iron. For this purpose, a specimen containing a central crack was loaded repeatedly by varying the load amplitude. All computations were carried out using molecular dynamics methods (MD), and the material behaviour was represented by utilising an embedded atom method (EAM) potential. Significant deviation in the crack growth behaviour was observed when loading the specimens with variable amplitudes rather than with constant amplitudes. Cracks did not only extend during the loading phase but also in the initial phase of the unloading process where cracks expanded from voids that had been formed in the last phase of loading. These voids coalesced with the main crack as the specimen was subjected to further loading. read less NOT USED (high confidence) X. Wu et al., “Softening effects due to reorientations of Cu precipitates in α-iron: Atomistic simulations of dislocations-obstacles interactions,” Journal of Applied Physics. 2019. link Times cited: 3 Abstract: Radiation-induced hardening by precipitates, which essential… read moreAbstract: Radiation-induced hardening by precipitates, which essentially has a dislocation pinning effect, is a major issue in nuclear reactor pressure vessels research. In this study, simulations of interactions between edge dislocations and copper precipitates are conducted as an example to investigate size-dependent pinning effects. Using molecular dynamics simulations, we discover a new two-stage mechanism that includes the processes of reorientations and atomistic collective migrations during the interactions. Both of these result in a weakening of the pinning effect on dislocations when the phase transition occurs in copper precipitates, which can be reflected in the decrease of the critical shear stress in stress and strain curves. Our studies considered the atomistic arrangement of the obstacle during the interaction, which constructively provide a new perspective for research studies of dislocation–obstacle pinning interaction and offer a more comprehensive estimation on the pinning strength of dislocations.Radiation-induced hardening by precipitates, which essentially has a dislocation pinning effect, is a major issue in nuclear reactor pressure vessels research. In this study, simulations of interactions between edge dislocations and copper precipitates are conducted as an example to investigate size-dependent pinning effects. Using molecular dynamics simulations, we discover a new two-stage mechanism that includes the processes of reorientations and atomistic collective migrations during the interactions. Both of these result in a weakening of the pinning effect on dislocations when the phase transition occurs in copper precipitates, which can be reflected in the decrease of the critical shear stress in stress and strain curves. Our studies considered the atomistic arrangement of the obstacle during the interaction, which constructively provide a new perspective for research studies of dislocation–obstacle pinning interaction and offer a more comprehensive estimation on the pinning strength of dislocations. read less NOT USED (high confidence) F. Bianchini, A. Glielmo, J. Kermode, and A. Vita, “Enabling QM-accurate simulation of dislocation motion in
γ−Ni
and
α−Fe
using a hybrid multiscale approach,” Physical Review Materials. 2019. link Times cited: 11 Abstract: We present an extension of the ‘learn on the fly’ method to … read moreAbstract: We present an extension of the ‘learn on the fly’ method to the study of the motion of dislocations in metallic systems, developed with the aim of producing information-efficient force models that can be systematically validated against reference QM calculations. Nye tensor analysis is used to dynamically track the quantum region centered at the core of a dislocation, thus enabling quantum mechanics/molecular mechanics simulations. The technique is used to study the motion of screw dislocations in Ni-Al systems, relevant to plastic deformation in Ni-based alloys, at a variety of temperature/strain conditions. These simulations reveal only a moderate spacing ( ∼ 5 A ) between Shockley partial dislocations, at variance with the predictions of traditional molecular dynamics (MD) simulation using interatomic potentials, which yields a much larger spacing in the high stress regime. The discrepancy can be rationalized in terms of the elastic properties of an hcp crystal, which influence the behavior of the stacking fault region between Shockley partial dislocations. The transferability of this technique to more challenging systems is addressed, focusing on the expected accuracy of such calculations. The bcc α − Fe phase is a prime example, as its magnetic properties at the open surfaces make it particularly challenging for embedding-based QM/MM techniques. Our tests reveal that high accuracy can still be obtained at the core of a dislocation, albeit at a significant computational cost for fully converged results. However, we find this cost can be reduced by using a machine learning approach to progressively reduce the rate of expensive QM calculations required during the dynamical simulations, as the size of the QM database increases. read less NOT USED (high confidence) L. Morrissey, S. M. Handrigan, S. Subedi, and S. Nakhla, “Atomistic uniaxial tension tests: investigating various many-body potentials for their ability to produce accurate stress strain curves using molecular dynamics simulations,” Molecular Simulation. 2019. link Times cited: 13 Abstract: ABSTRACT Molecular dynamics simulations, which take place on… read moreAbstract: ABSTRACT Molecular dynamics simulations, which take place on the atomistic scale, are now being used to predict the influence of atomistic processes on macro-scale mechanical properties. However, there is a lack of clear understanding on which potential should be used when attempting to obtain these properties. Moreover, many MD studies that do test mechanical properties do not actually simulate the macro-scale laboratory tension tests used to obtain them. As such, the purpose of the current study was to evaluate the various types of potentials for their accuracy in predicting the mechanical properties of iron from an atomistic uniaxial tension test at room temperature. Results demonstrated that while EAM and MEAM potentials all under predicted the elastic modulus at room temperature, the Tersoff and ReaxFF potentials were significantly more accurate. Unlike EAM and MEAM, both the Tersoff and ReaxFF potentials are bond order based. Therefore, these results demonstrate the importance of considering bonding between atoms when modelling tensile tests. In addition, the ReaxFF potential also accurately predicted the Poisson's ratio, allowing for complete characterisation of the material's behaviour. Overall, these findings highlight the need to understand the capabilities and limitations of each potential before application to a problem outside of the initial intended use. read less NOT USED (high confidence) Z. Sun, F. Z. Dai, B. Xu, and W.-Z. Zhang, “Three-Dimensional Growth of Coherent Ferrite in Austenite: A Molecular Dynamics Study,” Acta Metallurgica Sinica (English Letters). 2019. link Times cited: 5 NOT USED (high confidence) J. Ewen, H. Gao, M. Müser, and D. Dini, “Shear heating, flow, and friction of confined molecular fluids at high pressure.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 21 Abstract: Understanding the molecular-scale behavior of fluids confine… read moreAbstract: Understanding the molecular-scale behavior of fluids confined and sheared between solid surfaces is important for many applications, particularly tribology where this often governs the macroscopic frictional response. In this study, nonequilibrium molecular dynamics simulations are performed to investigate the effects of fluid and surface properties on the spatially resolved temperature and flow profiles, as well as friction. The severe pressure and shear rate conditions studied are representative of the elastohydrodynamic lubrication regime. In agreement with tribology experiments, flexible lubricant molecules give low friction, which increases linearly with logarithmic shear rate, while bulky traction fluids show higher friction, but a weaker shear rate dependence. Compared to lubricants, traction fluids show more significant shear heating and stronger shear localization. Models developed for macroscopic systems can be used to describe both the spatially resolved temperature profile shape and the mean film temperature rise. The thermal conductivity of the fluids increases with pressure and is significantly higher for lubricants compared to traction fluids, in agreement with experimental results. In a subset of simulations, the efficiency of the thermostat in one of the surfaces is reduced to represent surfaces with lower thermal conductivity. For these unsymmetrical systems, the flow and the temperature profiles become strongly asymmetric and some thermal slip can occur at the solid-fluid interface, despite the absence of velocity slip. The larger temperature rises and steeper velocity gradients in these cases lead to large reductions in friction, particularly at high pressure and shear rate. read less NOT USED (high confidence) Q. Zhang et al., “Molecular dynamics investigation of the local structure in iron melts and its role in crystal nucleation during rapid solidification.,” Physical chemistry chemical physics : PCCP. 2019. link Times cited: 19 Abstract: A comprehensive investigation on local structures in iron me… read moreAbstract: A comprehensive investigation on local structures in iron melts and their role in nucleation under various cooling rates was performed by means of large-scale molecular dynamics simulations. The embedded atoms method (EAM) was adopted to describe the interactions between iron atoms. Connections between short-range order (SRO), medium-range order (MRO), and crystalline nucleation from iron melts were constructed using several structural analysis techniques, including the radial distribution function, common neighbor analysis method, the Voronoi tessellation, and bond order analysis. The simulation results showed that abundant types of atomic clusters with SRO, mainly including the icosahedral-like (ICO-like) and fcc-like clusters, were predominant in undercooled iron melts. The obtained microstructures were determined by the competition between the ICO-like and crystal-like configurations. There existed a critical cooling rate, below which the fcc-like configurations gain the advantage upon cooling and where crystallization could take place; otherwise, the ICO-like configurations are favored and the glass phases could be obtained. Furthermore, it was proved that the crystal nucleation could be divided into three stages: first, a fluctuation and competition between crystal-like and ICO-like clusters in undercooled melts; second, the formation and growth of MRO clusters via the transformation of atomic configurations from ICO-like to crystal-like; finally, the nucleation of bcc nuclei from the core of steady MRO clusters. This process agrees with the Ostwald's step rule and the findings from other investigations. Based on the analysis of the compositional origin of MRO clusters, we further found that the MRO clusters were mainly composed of fcc-like instead of ICO-like configurations, indicating a negative role of ICO-like configurations in crystal nucleation. read less NOT USED (high confidence) W. Ko, D.-H. Kim, Y. Kwon, and M. Lee, “Atomistic Simulations of Pure Tin Based on a New Modified Embedded-Atom Method Interatomic Potential,” Metals. 2018. link Times cited: 22 Abstract: A new interatomic potential for the pure tin (Sn) system is … read moreAbstract: A new interatomic potential for the pure tin (Sn) system is developed on the basis of the second-nearest-neighbor modified embedded-atom-method formalism. The potential parameters were optimized based on the force-matching method utilizing the density functional theory (DFT) database of energies and forces of atomic configurations under various conditions. The developed potential significantly improves the reproducibility of many fundamental physical properties compared to previously reported modified embedded-atom method (MEAM) potentials, especially properties of the β phase that is stable at the ambient condition. Subsequent free energy calculations based on the quasiharmonic approximation and molecular-dynamics simulations verify that the developed potential can be successfully applied to study the allotropic phase transformation between α and β phases and diffusion phenomena of pure tin. read less NOT USED (high confidence) A. Nikonov, “Simulation of Atomic Mechanisms of Nucleation and Development of Plastic Deformation under Conditions of Shear Loading,” Metal Working and Material Science. 2018. link Times cited: 1 Abstract: Introduction Methods of surface treatment of materials are o… read moreAbstract: Introduction Methods of surface treatment of materials are one of the effective ways to improve its per- formance characteristics. One of the methods for fi nishing the surfaces of parts is the method of surface plastic deformation (nanostructuring burnishing), at which a layer with a nanocrystalline structure is formed. The study of the structural changes that occur directly in such burnishing process is extremely diffi cult. In this regard, numerical simulation methods can be an important addition to experimental studies. The purpose of the work is a numerical study of the mechanical response of a metal sample in a mono and nanoscale polycrystalline state to shear loading. In this paper, atomic mechanisms of nucleation and development of plastic deformation in a crystalline material under conditions of loading identical to local stresses arising during the processing of a material by surface plastic deformation are investigated . The methods of investigation. The research is carried out within the framework of high- performance parallel computations using the molecular dynamics method. The monocrystalline and polycrystalline body-centered cubic iron were chosen as the subject of research. Results and Discussion . The results of the studies show that under conditions of local shear loading in an initially defect-free α-iron crystal, it is possible to form a system of mutually intersecting dislocations, which subsequently leads to misorientation of individual parts of the crystallite and the formation of a nanofragmented structure of the surface layer. On the other hand, simulation data show that a shear in a nanoscale polycrystal is realized by the means of two competing mechanisms: grain boundary slip on one side and the process of recrystallization of individual grains on the other. With the growth of grains to di-mensions comparable with the size of the crystallite under study, the deformation in it begins to develop mainly due to the formation of structure defects, just as it occurs in a single crystal. Thus, the grain sizes and the orientation of its crystal lattice in relation to the direction of the external load determine the type of plastic deformation mechanisms of the surface material being realized. The obtained results can be used for a better understanding of the laws of processes and mechanisms realized in the surface layer of the material under surface plastic deformation conditions. read less NOT USED (high confidence) F. Baras, V. Turlo, O. Politano, S. Vadchenko, A. Rogachev, and A. Mukasyan, “SHS in Ni/Al Nanofoils: A Review of Experiments and Molecular Dynamics Simulations,” Advanced Engineering Materials. 2018. link Times cited: 38 Abstract: Non‐isothermal processes in nanometric metallic multilayers … read moreAbstract: Non‐isothermal processes in nanometric metallic multilayers are reviewed, both experimentally and theoretically. The Ni/Al nanofoil is considered as a model system. On the one hand, the experimental methods of elaboration and analysis are presented and, on the other hand, the modeling approach at the macroscopic and atomic scale. The basic experimental features are reported together with recent achievements. Molecular dynamics investigation of the reactivity of Ni/Al systems is reported for bulk systems and nanosystems including nanoparticles, nanowires, nanofilms, and multilayers. The focus is on atomic‐scale modeling versus experiments. Molecular dynamics approaches allow us to elucidate the mechanisms of non‐isothermal processes occurring in nanoscale systems, such as phase transformations and self‐propagation reactions. read less NOT USED (high confidence) D. King, S. Middleburgh, P. Burr, T. M. Whiting, P. Fossati, and M. Wenman, “Density functional theory study of the magnetic moment of solute Mn in bcc Fe,” Physical Review B. 2018. link Times cited: 12 Abstract: . An unexplained discrepancy exists between the experimental… read moreAbstract: . An unexplained discrepancy exists between the experimentally measured and theoretically calculated magnetic moments of Mn in -Fe. In this study, we use density functional theory to suggest that this discrepancy is likely due to the local strain environment of a Mn atom in the Fe structure. The ferromagnetic coupling, found by experiment, was shown to be metastable and could be stabilised by a 2% hydrostatic compressive strain. The effects of Mn concentration, vacancies and interstitial defects on the magnetic moment of Mn are also discussed. It was found that the ground state, anti-ferromagnetic (AFM) coupling of Mn to Fe requires long range tensile relaxations of the neighbouring atoms along <111> which is hindered in the presence of other Mn atoms. Vacancies and Fe interstitial defects stabilise the AFM coupling but are not expected to have a large effect on the average measured magnetic a stochastic manner with an average moment of ~0.06 B and median of ~0.70 B . By providing a statistically significant dataset, we show that the previous theory read less NOT USED (high confidence) N. Chandramoorthy and N. Hadjiconstantinou, “Solving lubrication problems at the nanometer scale,” Microfluidics and Nanofluidics. 2018. link Times cited: 4 NOT USED (high confidence) N. Chandramoorthy and N. Hadjiconstantinou, “Solving lubrication problems at the nanometer scale,” Microfluidics and Nanofluidics. 2018. link Times cited: 0 NOT USED (high confidence) L. Pártay, “On the performance of interatomic potential models of iron: Comparison of the phase diagrams,” Computational Materials Science. 2018. link Times cited: 19 NOT USED (high confidence) G. Sainath and B. Choudhary, “Twinning to slip transition in ultrathin BCC Fe nanowires,” Physics Letters A. 2018. link Times cited: 7 NOT USED (high confidence) L. Wan et al., “Hydrogen embrittlement controlled by reaction of dislocation with grain boundary in alpha-iron,” International Journal of Plasticity. 2018. link Times cited: 75 NOT USED (high confidence) M. Melnykov and R. Davidchack, “Characterization of melting properties of several Fe-C model potentials,” Computational Materials Science. 2018. link Times cited: 8 NOT USED (high confidence) I. A. Alhafez, C. Ruestes, and H. Urbassek, “Size of the Plastic Zone Produced by Nanoscratching,” Tribology Letters. 2018. link Times cited: 21 NOT USED (high confidence) Ó. Restrepo, N. Mousseau, M. Trochet, F. El-Mellouhi, O. Bouhali, and C. Becquart, “Carbon diffusion paths and segregation at high-angle tilt grain boundaries in α -Fe studied by using a kinetic activation-relation technique,” Physical Review B. 2018. link Times cited: 15 Abstract: Oscar A. Restrepo,1,2 Normand Mousseau,1 Mickaël Trochet,1 F… read moreAbstract: Oscar A. Restrepo,1,2 Normand Mousseau,1 Mickaël Trochet,1 Fedwa El-Mellouhi,3 Othmane Bouhali,2 and Charlotte S. Becquart4 1Département de physique and Regroupement québécois sur les matériaux de pointe, Université de Montréal, Case postale 6128, succursale centre-ville, Montréal (QC) Canada H3C 3J7 2Texas A&M University at Qatar, Doha, Qatar 3Qatar Environment and Energy Research Institute, Hamad Bin Khalifa University, Doha, Qatar 4Université de Lille, CNRS, INRA, ENSCL, UMR 8207, UMET, Unité Matériaux et Transformations, F 59 000 Lille, France read less NOT USED (high confidence) Z. Zhao and F. Chu, “Molecular dynamics simulation of crack initiation and propagation in bcc iron under load within spur gear tooth root,” Fatigue & Fracture of Engineering Materials & Structures. 2018. link Times cited: 12 Abstract: Spur gears are widely used in practice, and one of their typ… read moreAbstract: Spur gears are widely used in practice, and one of their typical failures is tooth breakage. In general, the tooth breakage occurs at tooth root, and the amount of crack growth during a meshing cycle is in atomistic scale. This work aims at identifying the mechanisms of crack initiation and propagation at tooth root by using molecular dynamics simulation. The results prove that there are phase transition regions and edge dislocations at crack tips. According to the distribution characteristic of the atomic potential, its concentration can be observed obviously by visualization software. In these concentration regions, microvoids come into being and expand gradually, which results in the subcrack initiation. Additionally, the microvoids and subcracks propagate along the high potential direction and then come together to accelerate the crack growth. Through carrying out a comparative simulation, the effects of heavy load at single meshing area on crack initiation and propagation are addressed. read less NOT USED (high confidence) A. Glielmo, C. Zeni, and A. Vita, “Efficient nonparametric n -body force fields from machine learning,” Physical Review B. 2018. link Times cited: 92 Abstract: The authors present a scheme to construct classical $n$-body… read moreAbstract: The authors present a scheme to construct classical $n$-body force fields using Gaussian Process (GP) Regression, appropriately mapped over explicit n-body functions (M-FFs). The procedure is possible, and will yield accurate forces, whenever prior knowledge allows to restrict the interactions to a finite order $n$, so that the ``universal approximator'' resolving power of standard GPs or Neural Networks is not needed. Under these conditions, the proposed construction preserves flexibility of training, systematically improvable accuracy, and a clear framework for validation of the underlying machine learning technique. Moreover, the M-FFs are as fast as classical parametrized potentials, since they avoid lengthy summations over database entries or weight parameters. read less NOT USED (high confidence) Z.-L. Wang, Z.-F. Huang, and Z.-rong Liu, “Elastic constants of stressed and unstressed materials in the phase-field crystal model,” Physical Review B. 2017. link Times cited: 15 Abstract: A general procedure to investigate the elastic response and … read moreAbstract: A general procedure to investigate the elastic response and calculate the elastic constants of stressed and unstressed materials through continuum field modeling, particularly the phase field crystal (PFC) models, is presented. It is found that for a complete description of system response to elastic deformation, the variations of all the quantities of lattice wave vectors, their density amplitudes (including the corresponding anisotropic variation and degeneracy breaking), the average atomic density, and system volume should be incorporated. The quantitative and qualitative results of elastic constant calculations highly depend on the physical interpretation of the density field used in the model, and also importantly, on the intrinsic pressure that usually pre-exists in the model system. A formulation based on thermodynamics is constructed to account for the effects caused by constant pre-existing stress during the homogeneous elastic deformation, through the introducing of a generalized Gibbs free energy and an effective finite strain tensor used for determining the elastic constants. The elastic properties of both solid and liquid states can be well produced by this unified approach, as demonstrated by an analysis for the liquid state and numerical evaluations for the bcc solid phase. The numerical calculations of bcc elastic constants and Poisson's ratio through this method generate results that are consistent with experimental conditions, and better match the data of bcc Fe given by molecular dynamics simulations as compared to previous work. The general theory developed here is applicable to the study of different types of stressed or unstressed material systems under elastic deformation. read less NOT USED (high confidence) J. Wang, G. Madsen, and R. Drautz, “Grain boundaries in bcc-Fe: a density-functional theory and tight-binding study,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 30 Abstract: Grain boundaries (GBs) have a significant influence on mater… read moreAbstract: Grain boundaries (GBs) have a significant influence on material properties. In the present paper, we calculate the energies of eleven low-Σ ( Σ ≤ 13 ) symmetrical tilt GBs and two twist GBs in ferromagnetic bcc iron using first-principles density functional theory (DFT) calculations. The results demonstrate the importance of a sufficient sampling of initial rigid body translations in all three directions. We show that the relative GB energies can be explained by the miscoordination of atoms at the GB region. While the main features of the studied GB structures were captured by previous empirical interatomic potential calculations, it is shown that the absolute values of GB energies calculated were substantially underestimated. Based on DFT-calculated GB structures and energies, we construct a new d-band orthogonal tight-binding (TB) model for bcc iron. The TB model is validated by its predictive power on all the studied GBs. We apply the TB model to block boundaries in lath martensite and demonstrate that the experimentally observed GB character distribution can be explained from the viewpoint of interface energy. read less NOT USED (high confidence) H. Lambert, Á. Fekete, J. Kermode, and A. Vita, “Imeall: A computational framework for the calculation of the atomistic properties of grain boundaries,” Comput. Phys. Commun. 2017. link Times cited: 10 NOT USED (high confidence) Ó. Restrepo, C. Becquart, F. El-Mellouhi, O. Bouhali, and N. Mousseau, “Diffusion mechanisms of C in 100, 110 and 111 Fe surfaces studied using kinetic activation-relaxation technique,” Acta Materialia. 2017. link Times cited: 11 NOT USED (high confidence) V. Turlo, F. Baras, and O. Politano, “Comparative study of embedded-atom methods applied to the reactivity in the Ni–Al system,” Modelling and Simulation in Materials Science and Engineering. 2017. link Times cited: 23 Abstract: Structural, thermodynamic, atomic and thermal transport prop… read moreAbstract: Structural, thermodynamic, atomic and thermal transport properties of solid and liquid phases of the Ni–Al system were studied by means of MD simulations using three embedded-atom method (EAM) potentials developed by Mishin and colleagues (Mishin et al 2002 Phys. Rev. B 65 224114; Mishin 2004 Acta Mater. 52 145167; Purja Pun and Mishin 2009 Phil. Mag. 89 32453267). The extracted properties (lattice parameter, enthalpy, heat capacity, mass diffusivity and thermal conductivity) were compared with experimental data. The limitations of EAM potentials for studying different aspects of reactivity were assessed for each potential separately. read less NOT USED (high confidence) R. Ocaya and J. J. Terblans, “Addressing the challenges of standalone multi-core simulations in molecular dynamics,” Physical Sciences Reviews. 2017. link Times cited: 0 Abstract: Computational modelling in material science involves mathema… read moreAbstract: Computational modelling in material science involves mathematical abstractions of force fields between particles with the aim to postulate, develop and understand materials by simulation. The aggregated pairwise interactions of the material’s particles lead to a deduction of its macroscopic behaviours. For practically meaningful macroscopic scales, a large amount of data are generated, leading to vast execution times. Simulation times of hours, days or weeks for moderately sized problems are not uncommon. The reduction of simulation times, improved result accuracy and the associated software and hardware engineering challenges are the main motivations for many of the ongoing researches in the computational sciences. This contribution is concerned mainly with simulations that can be done on a “standalone” computer based on Message Passing Interfaces (MPI), parallel code running on hardware platforms with wide specifications, such as single/multi- processor, multi-core machines with minimal reconfiguration for upward scaling of computational power. The widely available, documented and standardized MPI library provides this functionality through the MPI_Comm_size (), MPI_Comm_rank () and MPI_Reduce () functions. A survey of the literature shows that relatively little is written with respect to the efficient extraction of the inherent computational power in a cluster. In this work, we discuss the main avenues available to tap into this extra power without compromising computational accuracy. We also present methods to overcome the high inertia encountered in single-node-based computational molecular dynamics. We begin by surveying the current state of the art and discuss what it takes to achieve parallelism, efficiency and enhanced computational accuracy through program threads and message passing interfaces. Several code illustrations are given. The pros and cons of writing raw code as opposed to using heuristic, third-party code are also discussed. The growing trend towards graphical processor units and virtual computing clouds for high-performance computing is also discussed. Finally, we present the comparative results of vacancy formation energy calculations using our own parallelized standalone code called Verlet–Stormer velocity (VSV) operating on 30,000 copper atoms. The code is based on the Sutton–Chen implementation of the Finnis–Sinclair pairwise embedded atom potential. A link to the code is also given. read less NOT USED (high confidence) E. Baibuz et al., “Migration barriers for surface diffusion on a rigid lattice: challenges and solutions,” Computational Materials Science. 2017. link Times cited: 22 NOT USED (high confidence) L. Hale and C. Becker, “Vacancy dissociation in body-centered cubic screw dislocation cores,” Computational Materials Science. 2017. link Times cited: 9 NOT USED (high confidence) N. Gao, W. Setyawan, S. H. Zhang, and Z. Wang, “Defect-induced change of temperature-dependent elastic constants in BCC iron,” Journal of Nuclear Materials. 2017. link Times cited: 3 NOT USED (high confidence) D. Dragoni, T. Daff, G. Csányi, and N. Marzari, “Achieving DFT accuracy with a machine-learning interatomic potential: thermomechanics and defects in bcc ferromagnetic iron,” arXiv: Materials Science. 2017. link Times cited: 167 Abstract: We show that the Gaussian Approximation Potential machine le… read moreAbstract: We show that the Gaussian Approximation Potential machine learning framework can describe complex magnetic potential energy surfaces, taking ferromagnetic iron as a paradigmatic challenging case. The training database includes total energies, forces, and stresses obtained from density-functional theory in the generalized-gradient approximation, and comprises approximately 150,000 local atomic environments, ranging from pristine and defected bulk configurations to surfaces and generalized stacking faults with different crystallographic orientations. We find the structural, vibrational and thermodynamic properties of the GAP model to be in excellent agreement with those obtained directly from first-principles electronic-structure calculations. There is good transferability to quantities, such as Peierls energy barriers, which are determined to a large extent by atomic configurations that were not part of the training set. We observe the benefit and the need of using highly converged electronic-structure calculations to sample a target potential energy surface. The end result is a systematically improvable potential that can achieve the same accuracy of density-functional theory calculations, but at a fraction of the computational cost. read less NOT USED (high confidence) Y. Sun and W. Lai, “Molecular dynamics simulations of cascade damage near the Y 2 Ti 2 O 7 nanocluster/ferrite interface in nanostructured ferritic alloys,” Chinese Physics B. 2017. link Times cited: 2 NOT USED (high confidence) X. Ou, “Molecular dynamics simulations of fcc-to-bcc transformation in pure iron: a review,” Materials Science and Technology. 2017. link Times cited: 51 Abstract: Molecular dynamics (MD) simulation has been used to study th… read moreAbstract: Molecular dynamics (MD) simulation has been used to study the martensitic transformation in iron at the atomic scale. The paper reviews the available interatomic interaction potentials for iron, which describe the properties of different phases present in that system. Cases on the fcc-to-bcc transformation in iron by MD simulations were included in the present paper. Factors affecting the fcc-to-bcc transformation in iron were analysed: (a) structural factors, such as grain/phase boundaries, grain sizes and stacking faults; (b) simulation conditions, such as the presence of free surfaces, external stress/strain and studied temperatures; (c) the interatomic interaction potential. The main emphasis of the present paper is on results giving insight on the mechanisms of the nucleation and growth of bcc phase in iron. This review was submitted as part of the 2016 Materials Literature Review Prize of the Institute of Materials, Minerals and Mining run by the Editorial Board of MST. Sponsorship of the prize by TWI Ltd is gratefully acknowledged. read less NOT USED (high confidence) F. Calvo, N. Combe, J. Morillo, and M. Benoit, “Modeling Iron–Gold Nanoparticles Using a Dedicated Semi-Empirical Potential: Application to the Stability of Core–Shell Structures,” Journal of Physical Chemistry C. 2017. link Times cited: 16 Abstract: Core–shell nanoparticles made from iron embedded in gold hav… read moreAbstract: Core–shell nanoparticles made from iron embedded in gold have a strong potential interest in nanomedicine, the Au shell providing an efficient biocompatible coating for the magnetic Fe core. With the aim of determining theoretically the equilibrium morphologies of Fe–Au nanoparticles in a broad size range and with different compositions, a semiempirical many-body Fe–Au potential was designed combining well-established models for the pure metals and introducing dedicated contributions for the interactions involving mixed elements. The potential was parametrized against various energetic properties involving impurities, intermetallics, and finite clusters obtained using density functional calculations in the generalized gradient approximation. The potential was tested to investigate Fe–Au nanoparticles near equiconcentration containing about 1000–2000 atoms at finite temperature using parallel tempering Monte Carlo simulations initiated from the core–shell structure. The core–shell nanoparticles are found t... read less NOT USED (high confidence) M. G. D. V. Cuppari, R. Veiga, H. Goldenstein, J. E. G. Silva, and C. Becquart, “Lattice Instabilities and Phase Transformations in Fe from Atomistic Simulations,” Journal of Phase Equilibria and Diffusion. 2017. link Times cited: 2 NOT USED (high confidence) M. Cuppari, R. Veiga, H. Goldenstein, J. E. G. Silva, and C. Becquart, “Lattice Instabilities and Phase Transformations in Fe from Atomistic Simulations,” Journal of Phase Equilibria and Diffusion. 2017. link Times cited: 0 NOT USED (high confidence) D. Rodney, L. Ventelon, E. Clouet, L. Pizzagalli, and F. Willaime, “Ab initio modeling of dislocation core properties in metals and semiconductors,” Acta Materialia. 2017. link Times cited: 209 NOT USED (high confidence) V. Zhakhovsky, K. Migdal, N. Inogamov, and S. Anisimov, “MD simulation of steady shock-wave fronts with phase transition in single-crystal iron.” 2017. link Times cited: 18 Abstract: Overdriven shock waves propagating in main crystallographic … read moreAbstract: Overdriven shock waves propagating in main crystallographic directions of single-crystal bcc iron were studied with moving-window molecular dynamics (MD) technique. To simulate correctly the shock-induced bcc-to-hcp phase transition in iron a new EAM potential fitted to the cold pressure curves and pressure transition at 13 GPa was developed with the stress matching method. We demonstrate that structure of shock fronts depends on orientation of crystal. A peculiar structure of steady shock-wave front in [100] direction is observed. While the ultra-fast α → e transition initiated in uniaxially compressed crystal along [100] in elastic zone transforms bcc completely to hcp phase, transformation in other directions is performed only partially with production of metastable composition of nanometer-sized bcc-hcp-fcc grains. read less NOT USED (high confidence) I. A. Alhafez, A. Brodyanski, M. Kopnarski, and H. Urbassek, “Influence of Tip Geometry on Nanoscratching,” Tribology Letters. 2017. link Times cited: 36 NOT USED (high confidence) S. Rao et al., “Atomistic simulations of dislocations in a model BCC multicomponent concentrated solid solution alloy,” Acta Materialia. 2016. link Times cited: 160 NOT USED (high confidence) A. Tsukanov and S. Psakhie, “ADHESION EFFECTS WITHIN THE HARD MATTER – SOFT MATTER INTERFACE: MOLECULAR DYNAMICS.” 2016. link Times cited: 17 Abstract: In the present study three soft matter – hard matter systems… read moreAbstract: In the present study three soft matter – hard matter systems consisting of different nanomaterials and organic molecules were studied using the steered molecular dynamics approach in order to reveal regularities in the formation of organic-inorganic hybrids and the stability of multimolecular complexes, as well as to analyze the energy aspects of adhesion between bio-molecules and layered ceramics. The combined process free energy estimation (COPFEE) procedure was used for quantitative and qualitative assessment of the considered heterogeneous systems. Interaction of anionic and cationic amino acids with the surface of a [Mg4Al2(OH)122+ 2Cl–] layered double hydroxide (LDH) nanosheet was considered. In both cases, strong adhesion was observed despite the opposite signs of electric charge. The free energy of the aspartic amino acid anion, which has two deprotonated carboxylic groups, was determined to be –45 kJ/mol for adsorption on the LDH surface. For the cationic arginine, with only one carboxylic group and a positive net charge, the energy of adsorption was –26 kJ/mol, which is twice higher than that of chloride anion adsorption on the same cationic nanosheet. This fact clearly demonstrates the capability of “soft matter” species to adjust themselves and fit into the surface, minimizing energy of the system. The adsorption of protonated histamine, having no carboxylic groups, on a boehmite nanosheet is also energetically favorable, but the depth of free energy well is quite small at 3.6 kJ/mol. In the adsorbed state the protonated amino-group of histamine plays the role of proton donor, while the hydroxyl oxygens of the layered hydroxide have the role of proton acceptor, which is unusual. The obtained results represent a small step towards further understanding of the adhesion effects within the hard matter – soft matter contact zone. read less NOT USED (high confidence) E. Vitkovská and P. Ballo, “Reconstruction of α–Iron 〈100〉 Symmetric Tilt Grain Boundaries Σ17(410) and Σ13(510),” Journal of Electrical Engineering. 2016. link Times cited: 0 Abstract: A detailed numerical study on structure of symmetric tilt gr… read moreAbstract: A detailed numerical study on structure of symmetric tilt grain boundaries in α-iron is presented. The study is focused on structural and energetic optimization of 〈100〉 grain boundaries Σ5(210), Σ5(310), Σ17(410) and Σ13(510). Particular attention is given to grain boundary reconstruction, which is characterized by increased atomic density in grain boundary plane compared to bulk. The results of our numerical experiments significantly improved our knowledge about the migration of atoms between planes perpendicular as well as parallel to GB plane as an essential part of grain boundary reconstruction. read less NOT USED (high confidence) A. Glielmo, P. Sollich, and A. Vita, “Accurate interatomic force fields via machine learning with covariant kernels,” Physical Review B. 2016. link Times cited: 147 Abstract: We present a novel scheme to accurately predict atomic force… read moreAbstract: We present a novel scheme to accurately predict atomic forces as vector quantities, rather than sets of scalar components, by Gaussian process (GP) regression. This is based on matrix-valued kernel functions, on which we impose the requirements that the predicted force rotates with the target configuration and is independent of any rotations applied to the configuration database entries. We show that such covariant GP kernels can be obtained by integration over the elements of the rotation group $\mathit{SO}(d)$ for the relevant dimensionality $d$. Remarkably, in specific cases the integration can be carried out analytically and yields a conservative force field that can be recast into a pair interaction form. Finally, we show that restricting the integration to a summation over the elements of a finite point group relevant to the target system is sufficient to recover an accurate GP. The accuracy of our kernels in predicting quantum-mechanical forces in real materials is investigated by tests on pure and defective Ni, Fe, and Si crystalline systems. read less NOT USED (high confidence) A. Elzas and B. Thijsse, “Dislocation impacts on iron/precipitate interfaces under shear loading,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 12 Abstract: Molecular dynamics simulations are performed to obtain a bet… read moreAbstract: Molecular dynamics simulations are performed to obtain a better understanding of the interactions of single dislocations and dislocation pile-ups with interfaces between iron and a precipitate. The material properties of the precipitate material and the iron-precipitate interaction are varied to understand the influence of interface structure, interface strength and precipitate stiffness on these interactions under shear loading. Our main findings are: (1) the interface adhesion is determined by a combination of the atomic interactions across the interface and the interface structure, (2) the interface structure is the key factor determining the dislocation accommodation capability of the interface: very strong semi-coherent interfaces do accommodate dislocations, while only very weak coherent interfaces are capable of doing this, and (3) a strong precipitate prevents slip transfer into the precipitate. Results of this study combined with those of a forthcoming study under tensile loading can be used to improve the description of interface decohesion in existing larger-scale models, such as discrete dislocation plasticity. read less NOT USED (high confidence) I. Adlakha and K. Solanki, “Atomic-scale investigation of triple junction role on defects binding energetics and structural stability in α-Fe,” Acta Materialia. 2016. link Times cited: 17 NOT USED (high confidence) S. Teus and V. Gavriljuk, “Grain-Boundary Diffusion of Hydrogen Atoms in the α-Iron,” Metallofizika I Noveishie Tekhnologii. 2016. link Times cited: 6 NOT USED (high confidence) D. Dragoni, D. Ceresoli, and N. Marzari, “Vibrational and thermoelastic properties of bcc iron from selected EAM potentials,” Computational Materials Science. 2016. link Times cited: 7 NOT USED (high confidence) R. Babicheva et al., “Elastic moduli of nanocrystalline binary Al alloys with Fe, Co, Ti, Mg and Pb alloying elements,” Philosophical Magazine. 2016. link Times cited: 13 Abstract: The paper studies the elastic moduli of nanocrystalline (NC)… read moreAbstract: The paper studies the elastic moduli of nanocrystalline (NC) Al and NC binary Al–X alloys (X is Fe, Co, Ti, Mg or Pb) by using molecular dynamics simulations. X atoms in the alloys are either segregated to grain boundaries (GBs) or distributed randomly as in disordered solid solution. At 0 K, the rigidity of the alloys increases with decrease in atomic radii of the alloying elements. An addition of Fe, Co or Ti to the NC Al leads to increase in the Young’s E and shear μ moduli, while an alloying with Pb decreases them. The elastic moduli of the alloys depend on a distribution of the alloying elements. The alloys with the random distribution of Fe or Ti demonstrate larger E and μ than those for the corresponding alloys with GB segregations, while the rigidity of the Al–Co alloy is higher for the case of the GB segregations. The moduli E and μ for polycrystalline aggregates of Al and Al–X alloys with randomly distributed X atoms are estimated based on the elastic constants of corresponding single-crystals according to the Voigt-Reuss-Hill approximation, which neglects the contribution of GBs to the rigidity. The results show that GBs in NC materials noticeably reduce their rigidity. Furthermore, the temperature dependence of μ for the NC Al–X alloys is analyzed. Only the Al–Co alloy with GB segregations shows the decrease in μ to the lowest extent in the temperature range of 0–600 K in comparison with the NC pure Al. read less NOT USED (high confidence) G. Lv, H. Zhang, X. He, W. Yang, and Y. Su, “Vacancy enhanced formation and phase transition of Cu-rich precipitates in α - iron under neutron irradiation,” AIP Advances. 2016. link Times cited: 2 Abstract: In this paper, we employed both molecular statics and molecu… read moreAbstract: In this paper, we employed both molecular statics and molecular dynamics simulation methods to investigate the role of vacancies in the formation and phase transition of Cu-rich precipitates in α-iron. The results indicated that vacancies promoted the diffusion of Cu atoms to form Cu-rich precipitates. After Cu-rich precipitates formed, they further trapped vacancies. The supersaturated vacancy concentration in the Cu-rich precipitate induced a shear strain, which triggered the phase transition from bcc to fcc structure by transforming the initial bcc (110) plane into fcc (111) plane. In addition, the formation of the fcc-twin structure and the stacking fault structure in the Cu-rich precipitates was observed in dynamics simulations. read less NOT USED (high confidence) J. Janssen, N. Gunkelmann, and H. Urbassek, “Influence of C concentration on elastic moduli of α′-Fe1-xCx alloys,” Philosophical Magazine. 2016. link Times cited: 9 Abstract: The elastic constants of tetragonally distorted - crystallit… read moreAbstract: The elastic constants of tetragonally distorted - crystallites are calculated for several available interatomic interaction potentials. Besides embedded-atom-method-type potentials also a simple pair potential, modified embedded-atom-method and bond-order potentials are investigated. Care is taken to minimise the crystal structure properly in the presence of the C interstitials; we verify that the influence of statistics, i.e. the randomness of the C positions in the lattice, affects the elastic properties only little, as long as C is not allowed to cluster. We find that both sign and order of magnitude of the tetragonal elastic constants vary strongly between the predictions of the available potentials. Recent experimental data are available for the orientation-averaged elastic moduli; in contrast to the tetragonal constants, they feature only a mild dependence on C content. The experimental data are well reproduced by several of the potentials studied here. Existing deviations between experiment and predictions are discussed. read less NOT USED (high confidence) K. Wang, W. Zhu, S. Xiao, J. Chen, and W. Hu, “A new embedded-atom method approach based on the pth moment approximation,” Journal of Physics: Condensed Matter. 2016. link Times cited: 5 Abstract: Large scale atomistic simulations with suitable interatomic … read moreAbstract: Large scale atomistic simulations with suitable interatomic potentials are widely employed by scientists or engineers of different areas. The quick generation of high-quality interatomic potentials is urgently needed. This largely relies on the developments of potential construction methods and algorithms in this area. Quantities of interatomic potential models have been proposed and parameterized with various methods, such as the analytic method, the force-matching approach and multi-object optimization method, in order to make the potentials more transferable. Without apparently lowering the precision for describing the target system, potentials of fewer fitting parameters (FPs) are somewhat more physically reasonable. Thus, studying methods to reduce the FP number is helpful in understanding the underlying physics of simulated systems and improving the precision of potential models. In this work, we propose an embedded-atom method (EAM) potential model consisting of a new manybody term based on the pth moment approximation to the tight binding theory and the general transformation invariance of EAM potentials, and an energy modification term represented by pairwise interactions. The pairwise interactions are evaluated by an analytic-numerical scheme without the need to know their functional forms a priori. By constructing three potentials of aluminum and comparing them with a commonly used EAM potential model, several wonderful results are obtained. First, without losing the precision of potentials, our potential of aluminum has fewer potential parameters and a smaller cutoff distance when compared with some constantly-used potentials of aluminum. This is because several physical quantities, usually serving as target quantities to match in other potentials, seem to be uniquely dependent on quantities contained in our basic reference database within the new potential model. Second, a key empirical parameter in the embedding term of the commonly used EAM model is found to be related to the effective order of moments of local density of states. This may provide a way to improve the precision of EAM potentials further through more precise approximations to tight binding theory. In addition, some critical details about construction procedures are discussed. read less NOT USED (high confidence) E. Hintsala, A. J. Wagner, W. Gerberich, and K. Mkhoyan, “The role of back stress in sub-50 nm Si nanocubes,” Scripta Materialia. 2016. link Times cited: 10 NOT USED (high confidence) F. Abdeljawad, D. Medlin, J. Zimmerman, K. Hattar, and S. Foiles, “A diffuse interface model of grain boundary faceting,” Journal of Applied Physics. 2016. link Times cited: 17 Abstract: Interfaces, free or internal, greatly influence the physical… read moreAbstract: Interfaces, free or internal, greatly influence the physical properties and stability of materials microstructures. Of particular interest are the processes that occur due to anisotropic interfacial properties. In the case of grain boundaries (GBs) in metals, several experimental observations revealed that an initially flat GB may facet into hill-and-valley structures with well defined planes and corners/edges connecting them. Herein, we present a diffuse interface model that is capable of accounting for strongly anisotropic GB properties and capturing the formation of hill-and-valley morphologies. The hallmark of our approach is the ability to independently examine the various factors affecting GB faceting and subsequent facet coarsening. More specifically, our formulation incorporates higher order expansions to account for the excess energy due to facet junctions and their non-local interactions. As a demonstration of the modeling capability, we consider the Σ5 〈001〉 tilt GB in body-centered-cubic iron,... read less NOT USED (high confidence) H. Gao, W. Tysoe, and A. Martini, “Identification of the Shear Plane During Sliding of Solid Boundary Films: Potassium Chloride Films on Iron,” Tribology Letters. 2016. link Times cited: 2 NOT USED (high confidence) L. Rebuffi et al., “On the reliability of powder diffraction Line Profile Analysis of plastically deformed nanocrystalline systems,” Scientific Reports. 2016. link Times cited: 28 NOT USED (high confidence) R. Ocaya and J. J. Terblans, “Coding considerations for standalone molecular dynamics simulations of atomistic structures,” Journal of Physics: Conference Series. 2016. link Times cited: 1 Abstract: The laws of Newtonian mechanics allow ab-initio molecular dy… read moreAbstract: The laws of Newtonian mechanics allow ab-initio molecular dynamics to model and simulate particle trajectories in material science by defining a differentiable potential function. This paper discusses some considerations for the coding of ab-initio programs for simulation on a standalone computer and illustrates the approach by C language codes in the context of embedded metallic atoms in the face-centred cubic structure. The algorithms use velocity-time integration to determine particle parameter evolution for up to several thousands of particles in a thermodynamical ensemble. Such functions are reusable and can be placed in a redistributable header library file. While there are both commercial and free packages available, their heuristic nature prevents dissection. In addition, developing own codes has the obvious advantage of teaching techniques applicable to new problems. read less NOT USED (high confidence) Ó. Restrepo, N. Mousseau, F. El-Mellouhi, O. Bouhali, M. Trochet, and C. Becquart, “Diffusion properties of Fe-C systems studied by using kinetic activation-relaxation technique,” Computational Materials Science. 2016. link Times cited: 20 NOT USED (high confidence) H. Song and J. Hoyt, “An atomistic simulation study of the crystallographic orientation relationships during the austenite to ferrite transformation in pure Fe,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 21 Abstract: Molecular dynamics (MD) simulations on a model of pure Fe ha… read moreAbstract: Molecular dynamics (MD) simulations on a model of pure Fe have been used in the investigation of solid-state nucleation of a body-centered-cubic (BCC) phase from a polycrystalline face-centered-cubic (FCC) matrix. A neighbor vector analysis (NVA) method has been introduced and it is shown how the NVA can be used to determine the misorientation of grain or interphase boundaries. In particular, the NVA was utilized to identify the orientation relationships (ORs) of several BCC nuclei and three special ORs were tested, namely the Kurdjumov-Sachs (KS), Nishiyama–Wassermann (NW) and Pitsch (P). From several quasi-2D simulations, it was found that all stable nuclei at grain boundaries formed at least one orientation relationship with the parent grains that was consistent with either the KS or NW relationship. Several initial MD simulation cells, which prohibited the formation of special ORs, were also examined and in these simulations no nucleation was observed after long run times. In addition, the {1 1 1}γ//{1 1 0}α ?> orientation was detected in all mobile phase boundaries. Consistent with experimental findings, these observations demonstrate the importance of this high coherency atomic plane during both the nucleation and growth process. The nucleation and phase boundary characteristics identified here may provide important insights into the nucleation rate and grain orientation of more general solid state nucleation processes. read less NOT USED (high confidence) D. Ta, A. K. Tieu, H. Zhu, and B. Kosasih, “Thin film lubrication of hexadecane confined by iron and iron oxide surfaces: A crucial role of surface structure.,” The Journal of chemical physics. 2015. link Times cited: 29 Abstract: A comparative analysis of thin film lubrication of hexadecan… read moreAbstract: A comparative analysis of thin film lubrication of hexadecane between different iron and its oxide surfaces has been carried out using classical molecular dynamic simulation. An ab initio force-field, COMPASS, was applied for n-hexadecane using explicit atom model. An effective potential derived from density functional theory calculation was utilized for the interfacial interaction between hexadecane and the tribo-surfaces. A quantitative surface parameterization was introduced to investigate the influence of surface properties on the structure, rheological properties, and tribological performance of the lubricant. The results show that although the wall-fluid attraction of hexadecane on pure iron surfaces is significantly stronger than its oxides, there is a considerable reduction of shear stress of confined n-hexadecane film between Fe(100) and Fe(110) surfaces compared with FeO(110), FeO(111), Fe2O3(001), and Fe2O3(012). It was found that, in thin film lubrication of hexadecane between smooth iron and iron oxide surfaces, the surface corrugation plays a role more important than the wall-fluid adhesion strength. read less NOT USED (high confidence) Y. Osetsky and R. Stoller, “Atomic-scale mechanisms of helium bubble hardening in iron☆,” Journal of Nuclear Materials. 2015. link Times cited: 46 NOT USED (high confidence) V. Dremov et al., “MD modeling of screw dislocation influence upon initiation and mechanism of BCC-HCP polymorphous transition in iron.” 2015. link Times cited: 5 Abstract: The present work is devoted to classical molecular dynamics … read moreAbstract: The present work is devoted to classical molecular dynamics investigation into microscopic mechanisms of the bcc-hcp transition in iron. The interatomic potential of EAM type used in the calculations was tested for the capability to reproduce ab initio data on energy evolution along the bcc-hcp transformation path (Burgers deformation + shuffe) and then used in the large-scale MD simulations. The large-scale simulations included constant volume deformation along the Burgers path to study the origin and nature of the plasticity, hydrostatic volume compression of defect free samples above the bcc to hcp transition threshold to observe the formation of new phase embryos, and the volume compression of samples containing screw dislocations to study the effect of the dislocations on the probability of the new phase critical embryo formation. The volume compression demonstrated high level of metastability. The transition starts at pressure much higher than the equilibrium one. Dislocations strongly affect the probability of the critical embryo formation and significantly reduce the onset pressure of transition. The dislocations affect also the resulting structure of the samples upon the transition. The formation of layered structure is typical for the samples containing the dislocations. The results of the simulations were compared with the in-situ experimental data on the mechanism of the bcc-hcp transition in iron. read less NOT USED (high confidence) Z. Chang, D. Terentyev, N. Sandberg, K. Samuelsson, and P. Olsson, “Anomalous bias factors of dislocations in bcc iron,” Journal of Nuclear Materials. 2015. link Times cited: 38 NOT USED (high confidence) Z. Chen and J. Qu, “A fluctuation method to calculate the third order elastic constants in crystalline solids,” Journal of Applied Physics. 2015. link Times cited: 1 Abstract: This paper derives exact expressions of the isothermal third… read moreAbstract: This paper derives exact expressions of the isothermal third order elastic constants (TOE) in crystalline solids in terms of the kinetic and potential energies of the system. These expressions reveal that the TOE constants consist of a Born component and a relaxation component. The Born component is simply the third derivative of the system's potential energy with respect to the deformation, while the relaxation component is related to the non-uniform rearrangements of the atoms when the system is subject to a macroscopic deformation. Further, based on the general expressions derived here, a direct (fluctuation) method of computing the isothermal TOE constants is developed. Numerical examples of using this fluctuation method are given to compute the TOE constants of single crystal iron. read less NOT USED (high confidence) M. Mendelev et al., “Development of interatomic potentials appropriate for simulation of devitrification of Al90Sm10 alloy,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 59 Abstract: A semi-empirical potential for the Al90Sm10 alloy is present… read moreAbstract: A semi-empirical potential for the Al90Sm10 alloy is presented. The potential provides satisfactory reproduction of pure Al properties, the formation energies of a set of Al–Sm crystal phases with Sm content about 10%, and the structure of the liquid Al90Sm10 alloy. During molecular dynamics simulation in which the liquid alloy is cooled at a rate of 1010 K s−1, the developed potential produces a glass structure with lower ab initio energy than that produced by ab initio molecular dynamics (AIMD) itself using a typical AIMD cooling rate of 8·1013 K s−1. Based on these facts the developed potential should be suitable for simulations of phase transformations in the Al90Sm10 alloy. read less NOT USED (high confidence) S. Wilson, K. Gunawardana, and M. Mendelev, “Solid-liquid interface free energies of pure bcc metals and B2 phases.,” The Journal of chemical physics. 2015. link Times cited: 32 Abstract: The solid-liquid interface (SLI) free energy was determined … read moreAbstract: The solid-liquid interface (SLI) free energy was determined from molecular dynamics (MD) simulation for several body centered cubic (bcc) metals and B2 metallic compounds (space group: Pm3̄m; prototype: CsCl). In order to include a bcc metal with a low melting temperature in our study, a semi-empirical potential was developed for Na. Two additional synthetic "Na" potentials were also developed to explore the effect of liquid structure and latent heat on the SLI free energy. The obtained MD data were compared with the empirical Turnbull, Laird, and Ewing relations. All three relations are found to predict the general trend observed in the MD data for bcc metals obtained within the present study. However, only the Laird and Ewing relations are able to predict the trend obtained within the sequence of "Na" potentials. The Laird relation provides the best prediction for our MD data and other MD data for bcc metals taken from the literature. Overall, the Laird relation also agrees well with our B2 data but requires a proportionality constant that is substantially different from the bcc case. It also fails to explain a considerable difference between the SLI free energies of some B2 phases which have nearly the same melting temperature. In contrast, this difference is satisfactorily described by the Ewing relation. Moreover, the Ewing relation obtained from the bcc dataset also provides a reasonable description of the B2 data. read less NOT USED (high confidence) Y. Zhang, X. Bai, M. Tonks, and S. B. Biner, “Formation of prismatic loops from C15 Laves phase interstitial clusters in body-centered cubic iron,” Scripta Materialia. 2015. link Times cited: 48 NOT USED (high confidence) A. Kuksin and A. Yanilkin, “Dislocation nucleation and motion in metals and alloys at high-rate deformation: Molecular dynamic simulation,” Mechanics of Solids. 2015. link Times cited: 14 NOT USED (high confidence) Y. Osetsky, N. Anento, A. Serra, and D. Terentyev, “The role of nickel in radiation damage of ferritic alloys,” Acta Materialia. 2015. link Times cited: 18 NOT USED (high confidence) Y. Sun, Y. Wu, X.-ming Lu, R.-B. Li, and J. Xiao, “Anisotropy and roughness of the solid-liquid interface of BCC Fe,” Journal of Molecular Modeling. 2015. link Times cited: 5 NOT USED (high confidence) A. Kuksin and A. Yanilkin, “Dislocation nucleation and motion in metals and alloys at high-rate deformation: Molecular dynamic simulation,” Mechanics of Solids. 2015. link Times cited: 0 NOT USED (high confidence) A. Ojha, H. Sehitoglu, L. Patriarca, and H. Maier, “Twin nucleation in Fe-based bcc alloys—modeling and experiments,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 43 Abstract: We develop an analytical expression for twin nucleation stre… read moreAbstract: We develop an analytical expression for twin nucleation stress in bcc metal and alloys considering generalized planar fault energy and the dislocations bounding the twin nucleus. We minimize the total energy to predict the twinning stress relying only on parameters that are obtained through atomistic calculations, thus excluding the need for any empirical constants. We validate the present approach by means of precise measurements of the onset of twinning in bcc Fe–50at% Cr single crystals showing excellent agreement. The experimental observations of the three activated slip systems of symmetric configuration in relation to the twinning mechanism are demonstrated via transmission electron microscopy techniques along with digital image correlation. We then confirm the validity of the model for Fe, Fe–25at% Ni and Fe–3at% V alloys compared with experiments from the literature to show general applicability. read less NOT USED (high confidence) T. Lazauskas, S. Kenny, and R. Smith, “Influence of the prefactor to defect motion in α-Iron during long time scale simulations,” Journal of Physics: Condensed Matter. 2014. link Times cited: 12 Abstract: We present a study of the influence of the prefactor in the … read moreAbstract: We present a study of the influence of the prefactor in the Arrhenius equation for the long time scale motion of defects in α-Fe. It is shown that calculated prefactors vary widely between different defect types and it is thus important to determine these accurately when implementing on-the-fly kinetic Monte Carlo (otf-KMC) simulations. The results were verified by reproducing many events using Molecular Dynamics (MD) and Temperature-Accelerated Dynamics (TAD). The calculated prefactor was shown to increase the relative interstitial-vacancy diffusion rates by an order of magnitude compared to the assumption of a constant prefactor value and the ordering of the rate table for the interstitial defect migration mechanisms was also changed. In addition, low prefactor values were observed for the 4 interstitial dumbbells configuration with low barrier transitions. read less NOT USED (high confidence) N. Gunkelmann, D. Tramontina, E. Bringa, and H. Urbassek, “Interplay of plasticity and phase transformation in shock wave propagation in nanocrystalline iron,” New Journal of Physics. 2014. link Times cited: 30 Abstract: Strong shock waves create not only plasticity in Fe, but als… read moreAbstract: Strong shock waves create not only plasticity in Fe, but also phase transform the material from its bcc phase to the high-pressure hcp phase. We perform molecular-dynamics simulations of large, 8-million atom nanocrystalline Fe samples to study the interplay between these two mechanisms. We compare results for a potential that describes dislocation generation realistically but excludes phase change with another which in addition faithfully features the bcc → hcp transformation. With increasing shock strength, we find a transition from a two-wave structure (elastic and plastic wave) to a three-wave structure (an additional phase-transformation wave), in agreement with experiment. Our results demonstrate that the phase transformation is preceded by dislocation generation at the grain boundaries (GBs). Plasticity is mostly given by the formation of dislocation loops, which cross the grains and leave behind screw dislocations. We find that the phase transition occurs for a particle velocity between 0.6 and 0.7 km s−1. The phase transition takes only about 10 ps, and the transition time decreases with increasing shock pressure. read less NOT USED (high confidence) S. Kiselev, “Method of molecular dynamics in mechanics of deformable solids,” Journal of Applied Mechanics and Technical Physics. 2014. link Times cited: 8 NOT USED (high confidence) G. Bonny, D. Terentyev, A. Bakaev, P. Grigorev, and D. V. Neck, “Many-body central force potentials for tungsten,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 79 Abstract: Tungsten and tungsten-based alloys are the primary candidate… read moreAbstract: Tungsten and tungsten-based alloys are the primary candidate materials for plasma facing components in fusion reactors. The exposure to high-energy radiation, however, severely degrades the performance and lifetime limits of the in-vessel components. In an effort to better understand the mechanisms driving the materials' degradation at the atomic level, large-scale atomistic simulations are performed to complement experimental investigations. At the core of such simulations lies the interatomic potential, on which all subsequent results hinge. In this work we review 19 central force many-body potentials and benchmark their performance against experiments and density functional theory (DFT) calculations. As basic features we consider the relative lattice stability, elastic constants and point-defect properties. In addition, we also investigate extended lattice defects, namely: free surfaces, symmetric tilt grain boundaries, the 1/2〈1 1 1〉{1 1 0} and 1/2〈1 1 1〉 {1 1 2} stacking fault energy profiles and the 1/2〈1 1 1〉 screw dislocation core. We also provide the Peierls stress for the 1/2〈1 1 1〉 edge and screw dislocations as well as the glide path of the latter at zero Kelvin. The presented results serve as an initial guide and reference list for both the modelling of atomically-driven phenomena in bcc tungsten, and the further development of its potentials. read less NOT USED (high confidence) H.-L. Jang, J. H. Kim, Y. Park, and S. Cho, “Adjoint design sensitivity analysis of molecular dynamics in parallel computing environment,” International Journal of Mechanics and Materials in Design. 2014. link Times cited: 10 NOT USED (high confidence) H.-L. Jang, J.-H. Kim, Y. Park, and S. Cho, “Adjoint design sensitivity analysis of molecular dynamics in parallel computing environment,” International Journal of Mechanics and Materials in Design. 2014. link Times cited: 0 NOT USED (high confidence) S. Kiselev, “Method of molecular dynamics in mechanics of deformable solids,” Journal of Applied Mechanics and Technical Physics. 2014. link Times cited: 1 NOT USED (high confidence) J. J. Möller and E. Bitzek, “Comparative study of embedded atom potentials for atomistic simulations of fracture in α-iron,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 50 Abstract: Atomistic simulations play a crucial role in advancing our u… read moreAbstract: Atomistic simulations play a crucial role in advancing our understanding of the crack-tip processes that take place during fracture of semi-brittle materials like α-iron. As with all atomistic simulations, the results of such simulations however depend critically on the underlying atomic interaction model. Here, we present a systematic study of eight α-iron embedded atom method potentials used to model cracks subjected to plane strain mode-I loading conditions in six different crystal orientations. Molecular statics simulations are used to determine the fracture behavior (cleavage, dislocation emission, twinning) and the critical stress intensity factor KIc. The structural transformations in front of the crack tips, and in particular the occurrence of {1 1 0} planar faults, are analyzed in detail and related to the strain-dependent generalized stacking fault energy curve. The simulation results are discussed in terms of theoretical fracture criteria and compared to recent experimental data. The different potentials are ranked according to their capability to model the experimentally observed fracture behavior. read less NOT USED (high confidence) S. Narayanan, D. McDowell, and T. Zhu, “Crystal plasticity model for BCC iron atomistically informed by kinetics of correlated kinkpair nucleation on screw dislocation,” Journal of The Mechanics and Physics of Solids. 2014. link Times cited: 72 NOT USED (high confidence) Y.-S. Lin, M. Mrovec, and V. Vitek, “A new method for development of bond-order potentials for transition bcc metals,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 22 Abstract: A new development of numerical bond-order potentials (BOPs) … read moreAbstract: A new development of numerical bond-order potentials (BOPs) for the non-magnetic transition metals V, Nb, Ta, Cr, Mo and W is presented. The principles on which the BOPs have been set up are the same as in earlier developments (Aoki et al 2007 Prog. Mater. Sci. 52 154). However, the bond integrals are based on the recently advanced method of parametrization of tight-binding from DFT calculations (Madsen et al 2011 Phys. Rev. B 83 4119, Urban et al 2011 Phys. Rev. B 84 155119) and do not require any screening. At the same time, the functional form of the environmentally dependent repulsion is identified with the functional form of the repulsion arising from the overlap of s and p electrons in argon as proposed in Aoki and Kurokawa (2007 J. Phys.: Condens. Matter 19 136228). This is justified by the same physical origin of the environment dependent repulsion, which in transition metals arises from the overlap of s electrons that are being squeezed into the ion core regions under the influence of the strong covalent d bonds. The testing of the developed BOPs involves investigation of alternative higher energy structures, transformation paths connecting the bcc structure with other structures via continuously distorted configurations, evaluation of the vacancy formation energy and calculation of phonon spectra. In all cases, the BOP calculations are in more than satisfactory agreement with either DFT calculations and/or available experimental data. The calculated γ-surfaces for {1 0 1} planes all suggest that the core of 1/2〈1 1 1〉 screw dislocations is non-degenerate in the transition metals. This is also in full agreement with available calculations that account fully for the quantum-mechanical nature of the d electrons that provide the bulk of the bonding in transition metals. The testing of developed BOPs clearly demonstrates that they are transferable to structures well outside the regime of the ideal bcc lattice and are suitable for investigating the atomic structure and behaviour of extended crystal defects. read less NOT USED (high confidence) D. Belashchenko, “Computer simulation of liquid metals,” Physics—Uspekhi. 2013. link Times cited: 84 Abstract: Methods for and the results of the computer simulation of li… read moreAbstract: Methods for and the results of the computer simulation of liquid metals are reviewed. Two basic methods, classical molecular dynamics with known interparticle potentials and the ab initio method, are considered. Most attention is given to the simulated results obtained using the embedded atom model (EAM). The thermodynamic, structural, and diffusion properties of liquid metal models under normal and extreme (shock) pressure conditions are considered. Liquid-metal simulated results for the Groups I–IV elements, a number of transition metals, and some binary systems (Fe–C, Fe–S) are examined. Possibilities for the simulation to account for the thermal contribution of delocalized electrons to energy and pressure are considered. Solidification features of supercooled metals are also discussed. read less NOT USED (high confidence) K. Henriksson, C. Björkas, and K. Nordlund, “Atomistic simulations of stainless steels: a many-body potential for the Fe–Cr–C system,” Journal of Physics: Condensed Matter. 2013. link Times cited: 65 Abstract: Stainless steels found in real-world applications usually ha… read moreAbstract: Stainless steels found in real-world applications usually have some C content in the base Fe–Cr alloy, resulting in hard and dislocation-pinning carbides—Fe3C (cementite) and Cr23C6—being present in the finished steel product. The higher complexity of the steel microstructure has implications, for example, for the elastic properties and the evolution of defects such as Frenkel pairs and dislocations. This makes it necessary to re-evaluate the effects of basic radiation phenomena and not simply to rely on results obtained from purely metallic Fe–Cr alloys. In this report, an analytical interatomic potential parameterization in the Abell–Brenner–Tersoff form for the entire Fe–Cr–C system is presented to enable such calculations. The potential reproduces, for example, the lattice parameter(s), formation energies and elastic properties of the principal Fe and Cr carbides (Fe3C, Fe5C2, Fe7C3, Cr3C2, Cr7C3, Cr23C6), the Fe–Cr mixing energy curve, formation energies of simple C point defects in Fe and Cr, and the martensite lattice anisotropy, with fair to excellent agreement with empirical results. Tests of the predictive power of the potential show, for example, that Fe–Cr nanowires and bulk samples become elastically stiffer with increasing Cr and C concentrations. High-concentration nanowires also fracture at shorter relative elongations than wires made of pure Fe. Also, tests with Fe3C inclusions show that these act as obstacles for edge dislocations moving through otherwise pure Fe. read less NOT USED (high confidence) M. Marinica et al., “Interatomic potentials for modelling radiation defects and dislocations in tungsten,” Journal of Physics: Condensed Matter. 2013. link Times cited: 258 Abstract: We have developed empirical interatomic potentials for study… read moreAbstract: We have developed empirical interatomic potentials for studying radiation defects and dislocations in tungsten. The potentials use the embedded atom method formalism and are fitted to a mixed database, containing various experimentally measured properties of tungsten and ab initio formation energies of defects, as well as ab initio interatomic forces computed for random liquid configurations. The availability of data on atomic force fields proves critical for the development of the new potentials. Several point and extended defect configurations were used to test the transferability of the potentials. The trends predicted for the Peierls barrier of the 1 2 ⟨ 111 ⟩ ?> screw dislocation are in qualitative agreement with ab initio calculations, enabling quantitative comparison of the predicted kink-pair formation energies with experimental data. read less NOT USED (high confidence) B. Barvinschi, L. Proville, and D. Rodney, “Quantum Peierls stress of straight and kinked dislocations and effect of non-glide stresses,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 17 Abstract: It was recently shown (Proville et al 2012 Nature Mater. 11 … read moreAbstract: It was recently shown (Proville et al 2012 Nature Mater. 11 845) that to predict reliable Peierls stresses from atomistic simulations, one has to correct the Peierls barrier by the zero-point energy difference between the initial and activated states of the dislocation. The corresponding quantum Peierls stresses are studied here in α-Fe modeled with two embedded atom method potentials. First, we show that the quantum correction arises from modes localized near the dislocation core, such that partial Hessian matrices built on small cylinders centered on the dislocation core can be used to compute the zero-point energy difference. Second, we compute quantum Peierls stresses for straight and kinked dislocations and show that the former is smaller than the latter with both α-Fe models. Finally, we compare quantum Peierls stresses obtained in simple shear and in traction along two orientations considered experimentally by Kuramoto et al (1979 Phil. Mag. 39 717), evidencing a strong effect of non-glide stresses on the quantum Peierls stress. read less NOT USED (high confidence) V. Jansson and L. Malerba, “Simulation of the nanostructure evolution under irradiation in Fe-C alloys,” Journal of Nuclear Materials. 2013. link Times cited: 51 NOT USED (high confidence) E. Barker, D. Li, H. Zbib, and X. Sun, “Gradient Plasticity Model and its Implementation into MARMOT.” 2013. link Times cited: 0 Abstract: The influence of strain gradient on deformation behavior of … read moreAbstract: The influence of strain gradient on deformation behavior of nuclear structural materials, such as boby centered cubic (bcc) iron alloys has been investigated. We have developed and implemented a dislocation based strain gradient crystal plasticity material model. A mesoscale crystal plasticity model for inelastic deformation of metallic material, bcc steel, has been developed and implemented numerically. Continuum Dislocation Dynamics (CDD) with a novel constitutive law based on dislocation density evolution mechanisms was developed to investigate the deformation behaviors of single crystals, as well as polycrystalline materials by coupling CDD and crystal plasticity (CP). The dislocation density evolution law in this model is mechanism-based, with parameters measured from experiments or simulated with lower-length scale models, not an empirical law with parameters back-fitted from the flow curves. read less NOT USED (high confidence) G. Norman and V. Stegailov, “Stochastic theory of the classical molecular dynamics method,” Mathematical Models and Computer Simulations. 2013. link Times cited: 137 NOT USED (high confidence) G. Bonny, N. Castin, J. Bullens, A. Bakaev, T. Klaver, and D. Terentyev, “On the mobility of vacancy clusters in reduced activation steels: an atomistic study in the Fe–Cr–W model alloy,” Journal of Physics: Condensed Matter. 2013. link Times cited: 29 Abstract: Reduced activation steels are considered as structural mater… read moreAbstract: Reduced activation steels are considered as structural materials for future fusion reactors. Besides iron and the main alloying element chromium, these steels contain other minor alloying elements, typically tungsten, vanadium and tantalum. In this work we study the impact of chromium and tungsten, being major alloying elements of ferritic Fe–Cr–W-based steels, on the stability and mobility of vacancy defects, typically formed under irradiation in collision cascades. For this purpose, we perform ab initio calculations, develop a many-body interatomic potential (EAM formalism) for large-scale calculations, validate the potential and apply it using an atomistic kinetic Monte Carlo method to characterize the lifetime and diffusivity of vacancy clusters. To distinguish the role of Cr and W we perform atomistic kinetic Monte Carlo simulations in Fe–Cr, Fe–W and Fe–Cr–W alloys. Within the limitation of transferability of the potentials it is found that both Cr and W enhance the diffusivity of vacancy clusters, while only W strongly reduces their lifetime. The cluster lifetime reduction increases with W concentration and saturates at about 1–2 at.%. The obtained results imply that W acts as an efficient ‘breaker’ of small migrating vacancy clusters and therefore the short-term annealing process of cascade debris is modified by the presence of W, even in small concentrations. read less NOT USED (high confidence) L. Ventelon, F. Willaime, E. Clouet, and D. Rodney, “Ab initio investigation of the Peierls potential of screw dislocations in bcc Fe and W,” Acta Materialia. 2013. link Times cited: 103 NOT USED (high confidence) A. Sirenko and D. Belashchenko, “Molecular dynamics study of Ag, Ar, Cu, Ni, Al, Fe, Ta, K, and Cs nanoclusters within the embedded atom model,” Nanotechnologies in Russia. 2013. link Times cited: 4 NOT USED (high confidence) A. Kuksin and A. Yanilkin, “Atomistic simulation of the motion of dislocations in metals under phonon drag conditions,” Physics of the Solid State. 2013. link Times cited: 51 NOT USED (high confidence) M.-G. Kim, H.-L. Jang, and S. Cho, “Adjoint design sensitivity analysis of reduced atomic systems using generalized Langevin equation for lattice structures,” J. Comput. Phys. 2013. link Times cited: 12 NOT USED (high confidence) D. Belashchenko, “Account for electron contributions in embedded atom model for iron and nickel in molecular dynamics simulation,” Russian Journal of Physical Chemistry A. 2013. link Times cited: 6 NOT USED (high confidence) K. Hammond, H.-J. L. Voigt, L. A. Marus, N. Juslin, and B. Wirth, “Simple pair-wise interactions for hybrid Monte Carlo–molecular dynamics simulations of titania/yttria-doped iron,” Journal of Physics: Condensed Matter. 2013. link Times cited: 13 Abstract: We present pair-wise, charge-neutral model potentials for an… read moreAbstract: We present pair-wise, charge-neutral model potentials for an iron–titanium–yttrium–oxygen system. These simple models are designed to provide a tractable method of simulating nanostructured ferritic alloys (NFAs) using off-lattice Monte Carlo and molecular dynamics techniques without deviating significantly from the formalism employed in existing Monte Carlo simulations. The model is fitted to diamagnetic density functional theory (DFT) calculations of the various species over a range of densities and concentrations. The resulting model potentials provide reasonable and in some cases even excellent mechanical and thermodynamic properties for the pure metals. The model replicates the qualitative trends in formation energy predicted by DFT, though the energies of formation do not agree as well for dilute systems as they do for more concentrated systems. We find that on-lattice models will consistently favor tetrahedral oxygen interstitial sites over octahedral interstitial sites, while relaxed systems typically favor octahedral sites. This emphasizes the need for the off-lattice simulations for which this potential was designed. read less NOT USED (high confidence) N. Pisutha-Arnond, V. W. Chan, K. Elder, and K. Thornton, “Calculations of isothermal elastic constants in the phase-field crystal model,” Physical Review B. 2013. link Times cited: 26 Abstract: The phase-field crystal (PFC) method is an emerging coarse-g… read moreAbstract: The phase-field crystal (PFC) method is an emerging coarse-grained atomistic model that can be used to predict material properties. In this work, we describe procedures for calculating isothermal elastic constants using the PFC method. We find that the conventional procedures used in the PFC method for calculating the elastic constants are inconsistent with those defined from a theory of thermoelasticity of stressed materials. Therefore we present an alternative procedure for calculating the elastic constants that are consistent with the definitions from the thermoelasticity theory, and show that the two procedures result in different predictions. Furthermore, we employ a thermodynamic formulation of stressed solids to quantify the differences between the elastic constants obtained from the two procedures in terms of thermodynamic quantities such as the pressure evaluated at the undeformed state. read less NOT USED (high confidence) V. Starukhin, D. Belashchenko, A. Mirzoev, and A. Vorontsov, “Application of the Schommers method for constructing a hybrid pair potential,” Russian Metallurgy (Metally). 2012. link Times cited: 0 NOT USED (high confidence) M. Mendelev, M. Kramer, S. Hao, K. Ho, and C. Z. Wang, “Development of interatomic potentials appropriate for simulation of liquid and glass properties of NiZr2 alloy,” Philosophical Magazine. 2012. link Times cited: 116 Abstract: A new interatomic potential for the Ni–Zr system is presente… read moreAbstract: A new interatomic potential for the Ni–Zr system is presented. This potential was developed specifically to match experimental scattering data from Ni, Zr and NiZr2 liquids. Both ab initio and published thermodynamic data were used to optimise the potential to study the liquid and amorphous structure of the NiZr2 alloy. This potential has the C 16 phase, being more stable than C 11b phase in the NiZr2 alloy, consistent with experiments. The potential leads to the correct glass structure in the molecular dynamics simulation and, therefore, can be used to study the liquid–glass transformation in the NiZr2 alloy. read less NOT USED (high confidence) E. Zarkadoula et al., “The nature of high-energy radiation damage in iron,” Journal of Physics: Condensed Matter. 2012. link Times cited: 95 Abstract: Understanding and predicting a material’s performance in res… read moreAbstract: Understanding and predicting a material’s performance in response to high-energy radiation damage, as well as designing future materials to be used in intense radiation environments, requires knowledge of the structure, morphology and amount of radiation-induced structural changes. We report the results of molecular dynamics simulations of high-energy radiation damage in iron in the range 0.2–0.5 MeV. We analyze and quantify the nature of collision cascades both at the global and the local scale. We observe three distinct types of damage production and relaxation, including reversible deformation around the cascade due to elastic expansion, irreversible structural damage due to ballistic displacements and smaller reversible deformation due to the shock wave. We find that the structure of high-energy collision cascades becomes increasingly continuous as opposed to showing sub-cascade branching as reported previously. At the local length scale, we find large defect clusters and novel small vacancy and interstitial clusters. These features form the basis for physical models aimed at understanding the effects of high-energy radiation damage in structural materials. read less NOT USED (high confidence) D. Terentyev, N. Anento, and A. Serra, “Interaction of dislocations with carbon-decorated dislocation loops in bcc Fe: an atomistic study,” Journal of Physics: Condensed Matter. 2012. link Times cited: 16 Abstract: Properties of ferritic Fe-based alloys are highly sensitive … read moreAbstract: Properties of ferritic Fe-based alloys are highly sensitive to the carbon content dissolved in the matrix because interstitial carbon is known to strongly interact with lattice point defects and dislocations. As a result, the accumulation of radiation defects and its impact on the change of mechanical properties is also affected by the presence of dissolved interstitial carbon. This work contributes to an understanding of how interstitial carbon atoms influence the properties of small dislocation loops, which form directly in collision cascades upon neutron or ion irradiation and are ‘invisible’ to (i.e. undetectable by) standard experimental techniques applied to reveal nano-structural damage in metals. We have carried out MD simulations to investigate how the trapping of 1/2〈111〉 dislocation loops at thermally stable carbon–vacancy complexes, known to form under irradiation, affects the interaction of these dislocation loops with dislocations in bcc Fe. We have considered loops of size 1 and 3.5 nm, which represent experimentally invisible and visible defects, respectively. The obtained results point at the strong suppression of the drag of carbon-decorated loops by dislocations. In the case of direct interaction between dislocation and carbon-decorated loops, invisible loops are found to act as obstacles whose strength is at least twice as high compared to that of undecorated ones. Additional strengthening due to the carbon decoration on the visible loops was also regularly registered. The reasons for the additional strengthening have been rationalized and discussed. It is demonstrated that carbon decoration/segregation at dislocation loops affects not only accumulation of radiation damage under prolonged irradiation but also alters the post-irradiation plastic deformation mechanisms. For the first time, we provide evidence that undetectable dislocation loops decorated by carbon do contribute to the radiation hardening. read less NOT USED (high confidence) T. Yoshikawa, T. Takayanagi, H. Kimizuka, and M. Shiga, “Quantum–Thermal Crossover of Hydrogen and Tritium Diffusion in α-Iron,” Journal of Physical Chemistry C. 2012. link Times cited: 15 Abstract: The diffusion coefficients of hydrogen (H) and tritium (T) i… read moreAbstract: The diffusion coefficients of hydrogen (H) and tritium (T) in α-Fe have been computed using two approximate quantum dynamical techniques, that is, centroid molecular dynamics (CMD) and ring polymer molecular dynamics (RPMD), in the temperature range of T = 100–1000 K using the embedded atom method (EAM) potential. It has been found that the RPMD and CMD methods give very similar results. From a further analysis based on quantum transition-state theory (centroid density QTST) combined with path integral molecular dynamics (PIMD), it has been clear that there is a crossover between thermal and quantum mechanisms at about T = 500 and 300 K for H and T diffusions, respectively. The importance of nuclear quantum effects at low temperatures has been illustrated in terms of the effective free-energy surface map. read less NOT USED (high confidence) C. H. Ersland, I. Vatne, and C. Thaulow, “Atomistic modeling of penny-shaped and through-thickness cracks in bcc iron,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 39 Abstract: Atomistic simulations of penny-shaped embedded cracks in bod… read moreAbstract: Atomistic simulations of penny-shaped embedded cracks in body-centered cubic (bcc) iron are performed using molecular dynamics. The results reveal that the original circular crack geometry can change shape gradually upon loading, depending on the crystallographic orientation. This new geometry generally favors emission of dislocation loops instead of unstable fracture. A comparison is made between through-thickness cracks in six different orientations and penny-shaped cracks on the same crack planes. We find that changes in crack shape and the interaction of events in different directions play an important role in how fracture mechanisms evolve when cracks in full 3D simulations extend, and that dislocation emission and mechanical twins ‘win’ over unstable crack growth by bond breaking. read less NOT USED (high confidence) S. G. Mayr and A. Arabi-Hashemi, “Structural defects in Fe–Pd-based ferromagnetic shape memory alloys: tuning transformation properties by ion irradiation and severe plastic deformation,” New Journal of Physics. 2012. link Times cited: 8 Abstract: Fe–Pd-based ferromagnetic shape memory alloys constitute an … read moreAbstract: Fe–Pd-based ferromagnetic shape memory alloys constitute an exciting class of magnetically switchable smart materials that reveal excellent mechanical properties and biocompatibility. However, their application is severely hampered by a lack of understanding of the physics at the atomic scale. A many-body potential is presented that matched ab inito calculations and can account for the energetics of martensite austenite transition along the Bain path and relative phase stabilities in the ordered and disordered phases of Fe–Pd. Employed in massively parallel classical molecular dynamics simulations, the impact of order/disorder, point defects and severe plastic deformation in the presence of single- and polycrystalline microstructures are explored as a function of temperature. The model predictions are in agreement with experiments on phase changes induced by ion irradiation, cold rolling and hammering, which are also presented. read less NOT USED (high confidence) F. Yuan, “Atomistic simulation study of tensile deformation in bulk nanocrystalline bcc iron,” Science China Physics, Mechanics and Astronomy. 2012. link Times cited: 24 NOT USED (high confidence) E. Hayward and C. Deo, “Synergistic effects in hydrogen–helium bubbles,” Journal of Physics: Condensed Matter. 2012. link Times cited: 67 Abstract: The detrimental effects of hydrogen and helium on structural… read moreAbstract: The detrimental effects of hydrogen and helium on structural materials undergoing irradiation are well documented, if not well understood. There is experimental evidence to suggest that a synergistic effect between the two elements exists, which results in increased damage when both are present. This situation is expected in the next generation of fusion and fission reactors, so a fundamental understanding of these synergistic interactions is needed to predict materials performance. We perform atomistic simulations of hydrogen and helium bubbles in body-centered cubic iron to determine the mechanism behind this effect. We first develop an interatomic potential suitable for describing the interactions between hydrogen and helium. Through analysis of the energetics and structure of these bubbles, we explain the observed synergy as a consequence of bubble growth through helium induced loop punching, aided by the presence of hydrogen, instead of as a direct interaction between hydrogen and helium. The hydrogen benefits from an increased area of free surface on which to bind. read less NOT USED (high confidence) J. Liu and H. Dong, “Molecular dynamics calculation of thermodynamic properties of iron solidification,” IOP Conference Series: Materials Science and Engineering. 2012. link Times cited: 3 Abstract: The aim of this study is to identify the best available inte… read moreAbstract: The aim of this study is to identify the best available inter-atomic potentials for molecular dynamics (MD) calculation of solidification of iron and then to use the best potential to calculate thermodynamic properties such as equilibrium melting temperature, enthalpy, heat capacity and solid-liquid interfacial free energy. Our study reveals that embedded atom method (EAM) potential developed by Ackland et al. [2004 J. Phys.: Condens Matter. 16 S2629] appears to be the most accurate model for MD simulation of iron solidification. Simulations with the above EAM model predict the equilibrium melting temperature of iron is 1790K, the solid-liquid interfacial energy 214 mJ/m2. The difference with the experimental data is 1.2%, and 4.9% respectively. read less NOT USED (high confidence) T. Lee, M. Baskes, S. Valone, and J. Doll, “Atomistic modeling of thermodynamic equilibrium and polymorphism of iron,” Journal of Physics: Condensed Matter. 2012. link Times cited: 52 Abstract: We develop two new modified embedded-atom method (MEAM) pote… read moreAbstract: We develop two new modified embedded-atom method (MEAM) potentials for elemental iron, intended to reproduce the experimental phase stability with respect to both temperature and pressure. These simple interatomic potentials are fitted to a wide variety of material properties of bcc iron in close agreement with experiments. Numerous defect properties of bcc iron and bulk properties of the two close-packed structures calculated with these models are in reasonable agreement with the available first-principles calculations and experiments. Performance at finite temperatures of these models has also been examined using Monte Carlo simulations. We attempt to reproduce the experimental iron polymorphism at finite temperature by means of free energy computations, similar to the procedure previously pursued by Müller et al (2007 J. Phys.: Condens. Matter 19 326220), and re-examine the adequacy of the conclusion drawn in the study by addressing two critical aspects missing in their analysis: (i) the stability of the hcp structure relative to the bcc and fcc structures and (ii) the compatibility between the temperature and pressure dependences of the phase stability. Using two MEAM potentials, we are able to represent all of the observed structural phase transitions in iron. We discuss that the correct reproductions of the phase stability among three crystal structures of iron with respect to both temperature and pressure are incompatible with each other due to the lack of magnetic effects in this class of empirical interatomic potential models. The MEAM potentials developed in this study correctly predict, in the bcc structure, the self-interstitial in the 〈110〉 orientation to be the most stable configuration, and the screw dislocation to have a non-degenerate core structure, in contrast to many embedded-atom method potentials for bcc iron in the literature. read less NOT USED (high confidence) L. Ventelon, F. Willaime, C. Fu, M. Héran, and I. Ginoux, “Ab initio investigation of radiation defects in tungsten: Structure of self-interstitials and specificity of di-vacancies compared to other bcc transition metals,” Journal of Nuclear Materials. 2012. link Times cited: 94 NOT USED (high confidence) H. Hou, R. Wang, J. Wang, X. Liu, G. Chen, and P. Huang, “An analytic bond-order potential for the Fe–Cu system,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 5 Abstract: An angular-dependent analytic bond-order potential (ABOP) fo… read moreAbstract: An angular-dependent analytic bond-order potential (ABOP) for copper and Fe–Cu system was developed, based on the ABOP of pure iron introduced by Müller et al (2007 J. Phys.: Condens. Matter 19 326220). The potential parameters for the present ABOP model of copper were determined by fitting to the experimental data of the basic properties of fcc Cu and ab initio calculated properties of bcc Cu. The model predicts the vacancy formation energy in good agreement with the experimental result, although no vacancy formation information was used in the fitting of the model parameters. The melting point of Cu is also properly reproduced. The Fe–Cu binary system was described by adding two independent cross parameters in the potential model. The cross parameters were fitted using the ab initio data of the formation energies and lattice parameters of fictitious Fe–Cu alloys. The potential was applied to investigate the point defects and small defect clusters in dilute Fe–Cu alloys. The results were compared with the ab initio data and the values obtained with other potentials. read less NOT USED (high confidence) H. Zbib, D. Li, X. Sun, and M. Khaleel, “Large Scale DD Simulation Results for Crystal Plasticity Parameters in Fe-Cr And Fe-Ni Systems.” 2012. link Times cited: 1 Abstract: The development of viable nuclear energy source depends on e… read moreAbstract: The development of viable nuclear energy source depends on ensuring structural materials integrity. Structural materials in nuclear reactors will operate in harsh radiation conditions coupled with high level hydrogen and helium production, as well as formation of high density of point defects and defect clusters, and thus will experience severe degradation of mechanical properties. Therefore, the main objective of this work is to develop a capability that predicts aging behavior and in-service lifetime of nuclear reactor components and, thus provide an instrumental tool for tailoring materials design and development for application in future nuclear reactor technologies. Towards this end goal, the long term effort is to develop a physically based multiscale modeling hierarchy, validated and verified, to address outstanding questions regarding the effects of irradiation on materials microstructure and mechanical properties during extended service in the fission and fusion environments. The focus of the current investigation is on modern steels for use in nuclear reactors including high strength ferritic-martensitic steels (Fe-Cr-Ni alloys). The effort is to develop a predicative capability for the influence of irradiation on mechanical behavior. Irradiation hardening is related to structural information crossing different length scales, such as composition, dislocation, and crystal orientation distribution. To predict effectivemore » hardening, the influence factors along different length scales should be considered. Therefore, a hierarchical upscaling methodology is implemented in this work in which relevant information is passed between models at three scales, namely, from molecular dynamics to dislocation dynamics to dislocation-based crystal plasticity. The molecular dynamics (MD) was used to predict the dislocation mobility in body centered cubic (bcc) Fe and its Ni and Cr alloys. The results are then passed on to dislocation dynamics to predict the critical resolved shear stress (CRSS) from the evolution of local dislocation and defects. In this report the focus is on the results obtained from large scale dislocation dynamics simulations. The effect of defect density, materials structure was investigated, and evolution laws are obtained. These results will form the bases for the development of evolution and hardening laws for a dislocation-based crystal plasticity framework. The hierarchical upscaling method being developed in this project can provide a guidance tool to evaluate performance of structural materials for next-generation nuclear reactors. Combined with other tools developed in the Nuclear Energy Advanced Modeling and Simulation (NEAMS) program, the models developed will have more impact in improving the reliability of current reactors and affordability of new reactors.« less read less NOT USED (high confidence) M. Byshkin and M. Hou, “Phase transformations and segregation in Fe–Ni alloys and nanoalloys,” Journal of Materials Science. 2012. link Times cited: 16 NOT USED (high confidence) I. Vatne, E. Østby, and C. Thaulow, “Multiscale simulations of mixed-mode fracture in bcc-Fe,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 18 Abstract: Mixed-mode fracture of bcc-Fe has been investigated using th… read moreAbstract: Mixed-mode fracture of bcc-Fe has been investigated using the quasicontinuum method with an embedded atom method potential. A modified boundary layer approach has been employed to simulate a semi-infinite crack and to investigate the influence of the degree of mode II and mode III loading on the crack tip mechanisms. The analyses have been carried out with four crystallographic orientations and different degrees of mixed-mode loading. The results show that the critical stress intensity factor and the mechanisms at the crack tip are sensitive to both the crystallographic orientation and the mode of loading. Phenomena such as crack propagation, twinning and emission of edge and screw dislocations are observed. read less NOT USED (high confidence) N. Baluc et al., “From materials development to their test in IFMIF: an overview,” Nuclear Fusion. 2011. link Times cited: 6 Abstract: R&D activities on fusion reactor materials in Switzerland fo… read moreAbstract: R&D activities on fusion reactor materials in Switzerland focus on (1) the development of advanced metallic materials for structural applications in plasma-facing (first wall, divertor) and breeding blanket components of the future fusion power reactors, in particular oxide dispersion strengthened reduced activation ferritic steels and tungsten-base materials, (2) the modelling of radiation damage and radiation effects and (3) small specimen test technology for the future International Fusion Materials Irradiation Facility. The main objectives, examples of recent results and future activities are described in the case of these three R&D areas. read less NOT USED (high confidence) M. Tikhonchev, V. Svetukhin, A. Kadochkin, and E. Gaganidze, “Molecular dynamics simulation of atomic displacement cascades in Fe-9 at % Cr and Fe-9 at % Cr-0.1 at % C alloys,” Russian Metallurgy (Metally). 2011. link Times cited: 5 NOT USED (high confidence) Y. Zhao and G. Lu, “QM/MM study of dislocation—hydrogen/helium interactions in α-Fe,” Modelling and Simulation in Materials Science and Engineering. 2011. link Times cited: 63 Abstract: Impurities such as hydrogen (H) and helium (He) interact str… read moreAbstract: Impurities such as hydrogen (H) and helium (He) interact strongly with dislocations in metals. Using a multiscale quantum-mechanics/molecular-mechanics (QM/MM) approach, we have examined the interactions between the impurities (H and He) with dislocations (edge and screw) in α-Fe. The impurity trapping at the dislocation core is examined by calculating the impurity-dislocation binding energy and the impurity solution energy. We find that in general both H and He prefer the tetrahedral sites at the dislocation core, as well as in the bulk; the exceptions are due to deformed structures at the dislocation cores. Both H and He have a greater solution energy and binding energy to the edge dislocation than to the screw dislocation. The impurity pipe diffusion along the dislocation core is investigated using the QM/MM nudged-elastic-band method. We find that the diffusion barrier along the screw dislocation is lower than the bulk value for both H and He impurities. For the edge dislocation, although H has similar diffusion barriers as in the bulk, He has much higher diffusion energy barriers compared with the bulk. Finally we have examined the impurity effect on the dislocation mobility. We find that both H and He can lower the Peierls energy barrier for the screw dislocation significantly. The H enhanced dislocation mobility is consistent with experimental observations. read less NOT USED (high confidence) S. Chiesa, P. Derlet, S. Dudarev, and H. Swygenhoven, “Optimization of the magnetic potential for α-Fe,” Journal of Physics: Condensed Matter. 2011. link Times cited: 46 Abstract: A second generation of empirical potentials is produced for … read moreAbstract: A second generation of empirical potentials is produced for α-Fe within the framework of the magnetic potential formalism (Dudarev and Derlet 2005 J. Phys.: Condens. Matter 17 7097). A materials database that, in addition to ab initio-derived point defect formation energies, now includes third-order elastic constant and ab initio-derived string potential data controlling, respectively, the thermal expansion properties and the core structure of the 1/2⟨111⟩ screw dislocation. Three parameterizations are presented in detail, all of which exhibit positive thermal expansion and produce a non-degenerate configuration for the relaxed 1/2⟨111⟩ screw dislocation easy core structure. These potentials, along with two other published potentials, are investigated in terms of defect formation volume, early stage dislocation loop clustering energetics, ⟨110⟩ dumbbell interstitial diffusion, and the zero-stress 1/2⟨111⟩ screw dislocation Peierls barrier and its corresponding kink formation energies. read less NOT USED (high confidence) D. Belashchenko and O. I. Ostrovskii, “Molecular dynamics simulation of shock compression of metals: Iron and iron-sulfur solutions,” Russian Journal of Physical Chemistry A. 2011. link Times cited: 12 NOT USED (high confidence) S. J. Eder, A. Vernes, G. Vorlaufer, and G. Betz, “Molecular dynamics simulations of mixed lubrication with smooth particle post-processing,” Journal of Physics: Condensed Matter. 2011. link Times cited: 33 Abstract: A post-processing method for molecular dynamics (MD) simulat… read moreAbstract: A post-processing method for molecular dynamics (MD) simulations of friction based on the smooth particle approach is proposed, allowing—among other features—the introduction and evaluation of a solid–solid contact area arising due to direct asperity interaction. In order to illustrate the feasibility of this scheme, a large number of MD calculations of lubricated nanotribological systems with various asperity geometries and carefully selected numbers of lubricant molecules were carried out and analysed. In this manner, it is shown that the friction force as a function of load agrees very well with a three-parameter friction law which, in addition to the adhesion- and the load-controlled terms, contains a load-independent offset. read less NOT USED (high confidence) C. Becquart, C. Becquart, and C. Domain, “Modeling Microstructure and Irradiation Effects,” Metallurgical and Materials Transactions A. 2011. link Times cited: 53 NOT USED (high confidence) A. C. Tsoumanis and C. I. Siettos, “Detection of coarse-grained unstable states of microscopic/stochastic systems: a timestepper-based iterative protocol,” Nonlinear Dynamics. 2011. link Times cited: 0 NOT USED (high confidence) G. Bonny, R. Pasianot, D. Terentyev, and L. Malerba, “Iron chromium potential to model high-chromium ferritic alloys,” Philosophical Magazine. 2011. link Times cited: 73 Abstract: We present an Fe–Cr interatomic potential to model high-Cr f… read moreAbstract: We present an Fe–Cr interatomic potential to model high-Cr ferritic alloys. The potential is fitted to thermodynamic and point-defect properties obtained from density functional theory (DFT) calculations and experiments. The developed potential is also benchmarked against other potentials available in literature. It shows particularly good agreement with the DFT obtained mixing enthalpy of the random alloy, the formation energy of intermetallics and experimental excess vibrational entropy and phase diagram. In addition, DFT calculated point-defect properties, both interstitial and substitutional, are well reproduced, as is the screw dislocation core structure. As a first validation of the potential, we study the precipitation hardening of Fe–Cr alloys via static simulations of the interaction between Cr precipitates and screw dislocations. It is concluded that the description of the dislocation core modification near a precipitate might have a significant influence on the interaction mechanisms observed in dynamic simulations. read less NOT USED (high confidence) A. Cao, “Shape memory effects and pseudoelasticity in bcc metallic nanowires,” Journal of Applied Physics. 2010. link Times cited: 45 Abstract: In this paper, using molecular dynamic simulation and ab ini… read moreAbstract: In this paper, using molecular dynamic simulation and ab initio calculations, a novel pseudoelasticity is uncovered in a variety of bcc single crystalline nanowires. Specifically, an initial wire with a ⟨100⟩ axis and {100} surfaces has been transformed to a new configuration with a ⟨110⟩ axis and {111} lateral surfaces under uniaxial tensile loading. The loaded ⟨110⟩ wire spontaneously reorients back to the original one upon unloading, giving rise to about 41% recoverable strains. The primary deformation mechanisms associated with the reversible lattice reorientation are twinning and detwinning, i.e., forward and backward twin boundary migration on adjacent {112} slip planes. We reveal that the physics underlying the novel behavior in these bcc nanowires is the high propensity for twinning and detwinning, which is characterized as the small ratio of twin boundary migration energy to twin boundary formation energy. Furthermore, the relatively weaker temperature dependence of shape memory effects and large... read less NOT USED (high confidence) L. Malerba et al., “Ab initio calculations and interatomic potentials for iron and iron alloys : Achievements within the Perfect Project,” Journal of Nuclear Materials. 2010. link Times cited: 65 NOT USED (high confidence) A. C. Tsoumanis and C. Siettos, “Detection of coarse-grained unstable states of microscopic/stochastic systems: a timestepper-based iterative protocol,” Nonlinear Dynamics. 2010. link Times cited: 3 NOT USED (high confidence) E. Beamish, C. Campañá, and T. Woo, “Grain boundary sliding in irradiated stressed Fe–Ni bicrystals: a molecular dynamics study,” Journal of Physics: Condensed Matter. 2010. link Times cited: 5 Abstract: Molecular dynamics simulations were used to model grain boun… read moreAbstract: Molecular dynamics simulations were used to model grain boundary sliding in stressed Fe–Ni bicrystals exposed to low energy neutron irradiation. We studied how sliding stress thresholds and sliding mechanisms changed with variations in the Ni boundary morphology and boundary geometry. Simulations corresponding to ordered boundary Ni distributions and coincident-site lattice (CSL) geometries relaxed stress through a dislocation-mediated sliding mechanism. Such a mechanism was found to follow Orowan’s law, in which the stress relaxation rate is proportional to the dislocation velocity. Alternatively, simulations of disordered Ni distributions and non-CSL boundary geometries were described by a random shuffling process with time-dependent stress relaxation rate. Nevertheless, irrespective of the stress relaxation process followed by the bicrystals, after reaching equilibrium, the amounts of boundary displacement and stress relaxation were always found to be proportionally related. These observations might prove useful to groups working on building continuum (macroscopic) models of deformations in irradiated materials. read less NOT USED (high confidence) F. Djurabekova, L. Malerba, R. Pasianot, P. Olsson, and K. Nordlund, “Kinetics versus thermodynamics in materials modeling: The case of the di-vacancy in iron,” Philosophical Magazine. 2010. link Times cited: 22 Abstract: Monte Carlo models are widely used for the study of microstr… read moreAbstract: Monte Carlo models are widely used for the study of microstructural and microchemical evolution of materials under irradiation. However, they often link explicitly the relevant activation energies to the energy difference between local equilibrium states. We provide a simple example (di-vacancy migration in iron) in which a rigorous activation energy calculation, by means of both empirical interatomic potentials and density functional theory methods, clearly shows that such a link is not granted, revealing a migration mechanism that a thermodynamics-linked activation energy model cannot predict. Such a mechanism is, however, fully consistent with thermodynamics. This example emphasizes the importance of basing Monte Carlo methods on models where the activation energies are rigorously calculated, rather than deduced from widespread heuristic equations. read less NOT USED (high confidence) G. Bonny and R. Pasianot, “Gauge transformations to combine multi-component many-body interatomic potentials,” Philosophical Magazine Letters. 2010. link Times cited: 19 Abstract: Many-body interatomic potentials play an important role in a… read moreAbstract: Many-body interatomic potentials play an important role in atomistic modelling of materials. For pure elements it is known that there exist gauge transformations that can change the form of the potential functions without modifying its properties. These same transformations, however, fail when applied to alloys. Even though different research groups may use the same potentials to describe pure elements, the gauges employed for fitting alloys will generally be different. In this scenario, it is a priori impossible to merge them into one potential describing the combined system, and thus no advantage is taken from state-of-the-art developments in the literature. Here, we generalise the gauge transformations applied to pure species in order to leave the properties of alloys invariant. Based on these transformations, a strategy to merge potentials developed within different gauges is presented, aiming at the description of the combined system. Advantage of existing state-of-the-art potentials is so taken, thus focusing the efforts on fitting only the missing interactions. Such a procedure constitutes a helpful tool for the development of potentials targeted to alloys of increased complexity, while maintaining the description quality of their constituents. read less NOT USED (high confidence) A. Duff and M. Sluiter, “Diagnostic Structures for Interatomic Potentials,” Materials Transactions. 2010. link Times cited: 3 Abstract: unrelaxed structures are easily reproducible, enabling them … read moreAbstract: unrelaxed structures are easily reproducible, enabling them to be used by other authors to readily test potential formalisms other than those considered in this work. Specifically, we will perform our calculations on unrelaxed bcc � -iron (ferrite) with interstitial carbon. This will enable us to make a contribution to the ongoing debate as to how the ferritic phase of steel might be best represented using a semi-empirical potential. 3–6) This is an important topic because many properties of steel originate from the interaction of carbon (and nitrogen) with point and extended defects. These interactions must be treated using a semiempirical or empirical approach, since the numbers of atoms involved in such interactions place them well out of reach of current ab-initio methods such as density functional theory (DFT). 7,8) read less NOT USED (high confidence) L. Ventelon and F. Willaime, “Generalized stacking-faults and screw-dislocation core-structure in bcc iron: A comparison between ab initio calculations and empirical potentials,” Philosophical Magazine. 2010. link Times cited: 65 Abstract: Generalized stacking fault energies and screw dislocation co… read moreAbstract: Generalized stacking fault energies and screw dislocation core structures are reported for two sets of models for iron: density functional theory (DFT) calculations and empirical potentials. A thorough comparison between various DFT approaches has been performed on {110} and {211} γ-lines, which give a first indication on dislocation properties: (i) the effect of the exchange-correlation functional, LDA versus GGA, is significant in the pseudopotential approximation but not in the PAW approximation or in paramagnetic calculations; and (ii) the discrepancy due to the basis set between SIESTA and plane-wave results is rather small. Three empirical potentials for iron have been benchmarked on these DFT results. They all yield similar energies, but different shapes for the γ-lines. Using the criterion suggested by Duesbery and Vitek, the γ-line results point to non-degenerate core structures for the DFT calculations and for the Ackland and Ackland–Mendelev potentials but not for the Dudarev–Derlet potential. The direct calculations of the dislocation core structures show that the Ackland potential is an exception to the Duesbery–Vitek rule. More insight into the stability of the core structure can be gained by looking at the response to the polarization of the core. The Dudarev–Derlet and Ackland potentials have similar polarizations, but the energy difference between degenerate and non-degenerate cores is much larger with the Dudarev–Derlet potential, as expected from the γ-lines. The polarizability of the non-degenerate core is smaller with the Ackland–Mendelev potential than in DFT, indicating that the energy landscape is flatter in this direction. read less NOT USED (high confidence) S. H. Haghighat and R. Schäublin, “Influence of the stress field due to pressurized nanometric He bubbles on the mobility of an edge dislocation in iron,” Philosophical Magazine. 2010. link Times cited: 45 Abstract: Voids and He bubbles are strong obstacles to dislocation, wh… read moreAbstract: Voids and He bubbles are strong obstacles to dislocation, which induce hardening and loss of ductility. In Fe, molecular dynamics simulation is used to investigate the basic mechanisms of the interaction between a moving edge dislocation and a void or He bubble, as a function of its He content, temperature, interatomic potentials and interaction geometry. Different interatomic potentials for Fe–Fe and Fe–He interactions are used. It appears that temperature eases the dislocation release, due to the increased mobility of the screw segments appearing on the dislocation line upon bowing from the void or He bubble. The mobility includes the cross-slipping of these segments, which leads to the formation of a jog. It appears that the He bubble induces an inhomogeneous stress field in its surroundings, which strongly influences the dislocation passage depending on the geometry of the interaction. read less NOT USED (high confidence) H. Heinisch, F. Gao, and R. Kurtz, “Atomic-scale modeling of interactions of helium, vacancies and helium–vacancy clusters with screw dislocations in alpha-iron,” Philosophical Magazine. 2010. link Times cited: 31 Abstract: The interactions of He and vacancy defects with ⟨111⟩ screw … read moreAbstract: The interactions of He and vacancy defects with ⟨111⟩ screw dislocations in alpha-Fe were modeled using molecular statics, molecular dynamics and transition state energy determinations. The formation energies and binding energies of interstitial He atoms, vacancies and He–vacancy clusters near and within dislocations in alpha-Fe were determined at various locations relative to the dislocation core. Using the dimer transition state method, the migration energies and trajectories of the He and vacancy defects near and within the screw dislocation were also determined. Both interstitial He atoms and single vacancies are attracted to and trapped in the dislocation core region, and they both migrate along the dislocation line with a migration energy of about 0.4 eV, which is about half the migration energy of vacancies in the perfect crystal and about five times the migration energy for interstitial He in the perfect crystal. Divacancies and He–divacancy complexes have migration properties within the dislocation core that are similar to those in the perfect crystal, although the stability of these defects within the dislocation may be somewhat less than in the perfect crystal. read less NOT USED (high confidence) Y. Mishin, M. Asta, and J. Li, “Atomistic modeling of interfaces and their impact on microstructure and properties,” Acta Materialia. 2010. link Times cited: 418 NOT USED (high confidence) S. Chiesa, M. R. Gilbert, S. Dudarev, P. Derlet, and H. Swygenhoven, “The non-degenerate core structure of a ½⟨111⟩ screw dislocation in bcc transition metals modelled using Finnis–Sinclair potentials: The necessary and sufficient conditions,” Philosophical Magazine. 2009. link Times cited: 30 Abstract: It is shown that semi-empirical potentials for bcc metals ba… read moreAbstract: It is shown that semi-empirical potentials for bcc metals based on the non-directional second-moment Finnis–Sinclair approximation are able to predict, as a matter of routine, the non-degenerate core structure for the perfect ½⟨111⟩ dislocation if they correctly describe the inter-string pair potential of a rigid multi-string Frenkel–Kontorova model for the corresponding ideal bcc lattice. We prove this by inspecting the previously published empirical potentials, and also by performing an extensive search in functional parameter space for an optimal parameterisation of the magnetic potential formalism for bcc ferromagnetic Fe. read less NOT USED (high confidence) G. Bonny, R. Pasianot, and L. Malerba, “Fitting interatomic potentials consistent with thermodynamics: Fe, Cu, Ni and their alloys,” Philosophical Magazine. 2009. link Times cited: 24 Abstract: In computational materials science, many atomistic methods h… read moreAbstract: In computational materials science, many atomistic methods hinge on an interatomic potential to describe material properties. In alloys, besides a proper description of problem-specific properties, a reasonable reproduction of the experimental phase diagram by the potential is essential. In this framework, two complementary methods were developed to fit interatomic potentials to the thermodynamic properties of an alloy. The first method involves the zero Kelvin phase diagram and makes use of the concept of the configuration polyhedron. The second method involves phase boundaries at finite temperature and is based on the cluster variation method. As an example for both techniques, they are applied to the Fe–Cu, Fe–Ni and Cu–Ni systems. The resulting potentials are compared to those found in the literature and are found to reproduce the experimental phase diagram more consistently than the latter. read less NOT USED (high confidence) R. Gröger and V. Vitek, “Directional versus central-force bonding in studies of the structure and glide of 1/2⟨111⟩ screw dislocations in bcc transition metals,” Philosophical Magazine. 2009. link Times cited: 33 Abstract: In this paper, we address the differences between Finnis–Sin… read moreAbstract: In this paper, we address the differences between Finnis–Sinclair potentials and bond-order potentials (BOPs) when studying 1/2⟨111⟩ screw dislocations in bcc transition metals, specifically Mo and W. These two types of potentials differ in that the former is central-force, whereas the latter include angular bonding. The cores of 1/2⟨111⟩ screw dislocations have two variants, one invariant with respect to the ⟨101⟩ diad and the other not. Hence, the latter core is degenerate. The BOPs always lead to the invariant type, whereas for Finnis–Sinclair potentials both variants occur. However, the symmetry of the core does not play a decisive role in the glide of dislocations. It is the description of interatomic forces that governs both the core structure and the glide behaviour of dislocations. The general characteristics of dislocation glide, the twinning–antitwinning asymmetry and a lower Peierls stress for tension than compression are the same for both types of potentials. Whereas the results obtained with BOPs are similar for the two cases studied, Finnis–Sinclair potentials lead to a broader variety. Particularly, the slip plane at 0 K is always {110} for BOPs but it is either {110} or {112} for Finnis–Sinclair potentials. The reason is that, in the latter case, the core configuration and core transformations are less constrained than in the former case. Hence, in bcc transition metals the BOPs are a more reliable description of atomic interactions than Finnis–Sinclair potentials, but when the d band does not play any important role, the Finnis–Sinclair potentials are fully applicable. read less NOT USED (high confidence) M. Mendelev, M. Asta, M. J. Rahman, and J. Hoyt, “Development of interatomic potentials appropriate for simulation of solid–liquid interface properties in Al–Mg alloys,” Philosophical Magazine. 2009. link Times cited: 126 Abstract: Different approaches are analyzed for construction of semi-e… read moreAbstract: Different approaches are analyzed for construction of semi-empirical potentials for binary alloys, focusing specifically on the capability of these potentials to describe solid–liquid phase equilibria, as a pre-requisite to studies of solidification phenomena. Fitting ab initio compound data does not ensure correct reproduction of the dilute solid-solution formation energy, and explicit inclusion of this quantity in the potential development procedure does not guarantee that the potential will predict the correct solid–liquid phase diagram. Therefore, we conclude that fitting only to solid phase properties, as is done in most potential development procedures, generally is not sufficient to develop a semi-empirical potential suitable for the simulation of solidification. A method is proposed for the incorporation of data for liquid solution energies in the potential development procedure, and a new semi-empirical potential developed suitable for simulations of dilute alloys of Mg in Al. The potential correctly reproduces both zero-temperature solid properties and solidus and liquid lines on the Al-rich part of the Al–Mg phase diagram. read less NOT USED (high confidence) A. Stukowski, B. Sadigh, P. Erhart, and A. Caro, “Efficient implementation of the concentration-dependent embedded atom method for molecular-dynamics and Monte-Carlo simulations,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 83 Abstract: The concentration-dependent embedded atom method (CD-EAM) is… read moreAbstract: The concentration-dependent embedded atom method (CD-EAM) is a powerful model for atomistic simulation of concentrated alloys with arbitrarily complex mixing enthalpy curves. In this paper, we show that in spite of explicit three-body forces, this model can be implemented quite simply with a computational efficiency comparable to the standard EAM for molecular-dynamics (MD) simulations. Ready-to-use subroutines for the parallel MD code LAMMPS can be provided by the authors upon request. We further propose an improved version of this potential that allows for very efficient calculations of single-particle displacement/transmutation energies, while retaining the complexity implicit in the three-body interactions. This enables large-scale Monte-Carlo simulations of alloys with the interatomic interactions described by the CD-EAM model. read less NOT USED (high confidence) E. Clouet, “Elastic energy of a straight dislocation and contribution from core tractions,” Philosophical Magazine. 2009. link Times cited: 39 Abstract: We derive an expression of the core traction contribution to… read moreAbstract: We derive an expression of the core traction contribution to the dislocation elastic energy within linear anisotropic elasticity theory using the sextic formalism. With this contribution, the elastic energy is a state variable consistent with the work of the Peach–Koehler forces. This contribution needs also to be considered when extracting from atomic simulations core energies. The core energies thus obtained are real intrinsic dislocation properties: they do not depend on the presence and position of other defects. This is illustrated by calculating core energies of edge dislocation in bcc iron, where we show that dislocations gliding in {110} planes are more stable than those gliding in {112} planes. read less NOT USED (high confidence) S. Chiesa, P. Derlet, and S. Dudarev, “Free energy of a ⟨110⟩ dumbbell interstitial defect in bcc Fe: Harmonic and anharmonic contributions,” Physical Review B. 2009. link Times cited: 35 Abstract: S. Chiesa,1 P. M. Derlet,2 and S. L. Dudarev3,4 1NUM/ASQ-Mat… read moreAbstract: S. Chiesa,1 P. M. Derlet,2 and S. L. Dudarev3,4 1NUM/ASQ-Materials Science and Simulation, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland 2NUM-Condensed Matter Theory Group, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland 3Culham Science Centre, EURATOM/UKAEA Fusion Association, Oxfordshire OX14 3DB, United Kingdom 4Department of Physics, Imperial College, Exhibition Road, London SW7 2AZ, United Kingdom Received 7 April 2009; revised manuscript received 22 May 2009; published 17 June 2009 read less NOT USED (high confidence) D. Terentyev, N. Juslin, K. Nordlund, and N. Sandberg, “Fast three dimensional migration of He clusters in bcc Fe and Fe–Cr alloys,” Journal of Applied Physics. 2009. link Times cited: 81 Abstract: In this work, we perform atomistic molecular dynamics simula… read moreAbstract: In this work, we perform atomistic molecular dynamics simulations to assess the properties of small helium vacancy (He-V) and pure He clusters in body-centered cubic Fe and in Fe90–Cr10 (Fe–10Cr) random alloy. The following two goals are pursued: determining diffusion mechanisms of He-V clusters occurring in dynamic simulations and revealing a possible influence of Cr on the mobility/stability of He-V clusters in the Fe–10Cr alloy. We also present a newly developed set of interatomic potentials for the Fe–Cr–He system, fitted to a set of specially performed density functional theory calculations. The obtained results show that the dissociation energies of the studied He-V clusters, as well as the migration energy of He interstitial, are not significantly affected in the alloy compared to pure Fe. It was found that small pure He clusters with sizes up to four atoms, that were assumed to be immobile in many previous studies devoted to He-release/accumulation kinetics, in fact, exhibit fast three dimensional... read less NOT USED (high confidence) A. Ramasubramaniam, M. Itakura, and E. Carter, “Interatomic potentials for hydrogen in α –iron based on density functional theory,” Physical Review B. 2009. link Times cited: 158 Abstract: We present two interatomic potentials for hydrogen in –iron … read moreAbstract: We present two interatomic potentials for hydrogen in –iron based on the embedded atom method potentials for iron developed by Mendelev et al. Philos. Mag. 83, 3977 2003 and Ackland et al. J. Phys.: Condens. Matter 16, S2629 2004 . Since these latter potentials are unique among existing iron potentials in their ability to produce the same core structure for screw dislocations as density functional theory DFT calculations, our interatomic potentials for hydrogen in iron also inherit this important feature. We use an extensive database of energies and atomic configurations from DFT calculations to fit the cross interaction of hydrogen with iron. Detailed tests on the dissolution and diffusion of hydrogen in bulk –iron, as well as the binding of H to vacancies, free surfaces, and dislocations, indicate that our potentials are in excellent overall agreement with DFT calculations. read less NOT USED (high confidence) D. Belashchenko, “Application of the embedded atom model to liquid metals: Liquid sodium,” High Temperature. 2009. link Times cited: 33 NOT USED (high confidence) M. Mendelev, M. Kramer, R. Ott, D. Sordelet, D. Yagodin, and P. Popel,’ “Development of suitable interatomic potentials for simulation of liquid and amorphous Cu–Zr alloys,” Philosophical Magazine. 2009. link Times cited: 334 Abstract: We present a new semi-empirical potential suitable for molec… read moreAbstract: We present a new semi-empirical potential suitable for molecular dynamics simulations of liquid and amorphous Cu–Zr alloys. To provide input data for developing the potential, new experimental measurements of the structure factors for amorphous Cu64.5Zr35.5 alloy were performed. In this work, we propose a new method to include diffraction data in the potential development procedure, which also includes fitting to first-principles and liquid density and enthalpy of mixing data. To refine the new potential, we used first-principles and liquid enthalpy of mixing data published earlier combined with the densities of liquid Cu64.5Zr35.5 measured over a range of temperatures. We show that the potential predicts a liquid-to-glass transition temperature that agrees reasonably well with experimental data. Finally, we compare the new potential with two previously developed semi-empirical potentials for Cu–Zr alloys and examine their comparative and contrasting descriptions of structure and properties for Cu64.5Zr35.5 liquids and glasses. read less NOT USED (high confidence) Y. Abe and S. Jitsukawa, “Lowest energy structures of self-interstitial atom clusters in α-iron from a combination of Langevin molecular dynamics and the basin-hopping technique,” Philosophical Magazine. 2009. link Times cited: 9 Abstract: A combination of simulated annealing with Langevin molecular… read moreAbstract: A combination of simulated annealing with Langevin molecular dynamics and the basin-hopping with occasional jumping (BHOJ) technique was used to systematically determine the most stable configurations of self-interstitial atom (SIA) clusters I n (n = 1–38) in α-iron. In addition to the original BHOJ technique, we introduced an additional long jumping process in which a randomly selected less-bounded atom is moved to a neighbouring site of another SIA in the cluster to enhance the probability of locating the global minimum structure. With the obtained putative lowest energy structures, the binding energies as a function of cluster size were estimated. We also determined the sizes of particular stable clusters based on their geometrical symmetry. Furthermore, the values were extrapolated based on accurately determined formation energies, and are available for immediate use in kinetic Monte Carlo or rate theory models. read less NOT USED (high confidence) G. Bonny, R. Pasianot, and L. Malerba, “Fe–Ni many-body potential for metallurgical applications,” Modelling and Simulation in Materials Science and Engineering. 2009. link Times cited: 119 Abstract: A many-body interatomic potential for the Fe–Ni system is fi… read moreAbstract: A many-body interatomic potential for the Fe–Ni system is fitted, capable of describing both the ferritic and austenitic phase. The Fe–Ni system exhibits two stable ordered intermetallic phases, namely, L10 FeNi and L12 FeNi3, that are key issues to be tackled when creating a Fe–Ni potential consistent with thermodynamics. A procedure, based on a rigid lattice Ising model and the theory of correlation functions space, is developed to address all the intermetallics that are possible ground states of the system. While controlling the ground states of the system, the mixing enthalpy and defect properties were fitted. Both bcc and fcc defect properties are compared with density functional theory calculations and other potentials found in the literature. Finally, the potential is thermodynamically validated by constructing the alloy phase diagram. It is shown that the experimental phase diagram is reproduced reasonably well and that our potential gives a globally improved description of the Fe–Ni system in the whole concentration range with respect to the potentials found in the literature. read less NOT USED (high confidence) M. R. Gilbert, S. Dudarev, P. Derlet, and D. Pettifor, “Structure and metastability of mesoscopic vacancy and interstitial loop defects in iron and tungsten,” Journal of Physics: Condensed Matter. 2008. link Times cited: 89 Abstract: The most recent observations of dynamical time-dependent flu… read moreAbstract: The most recent observations of dynamical time-dependent fluctuating behaviour of mesoscopic radiation defects in body-centred cubic metals (Arakawa et al 2006 Phys. Rev. Lett. 96 125506; 2007 Science 318 956–9; Yao et al 2008 Phil. Mag. at press) have highlighted the need to develop adequate quantitative models for the structural stability of defects in the mesoscopic limit where defects are accessible to direct in situ electron microscope imaging. In pursuit of this objective, we investigate and compare several types of mesoscopic vacancy and interstitial defects in iron and tungsten by simulating them using recently developed many-body interatomic potentials. We show that the mesoscopic vacancy dislocation loops observed in ion-irradiated materials are, without exception, metastable with respect to the transformation into spherical voids, but that the rate of this transformation and even the specific type of the transformation mechanism depend on the defect size and the properties of the material. read less NOT USED (high confidence) D. Belashchenko and N. Nikitin, “The embedded atom model of liquid cesium,” Russian Journal of Physical Chemistry A, Focus on Chemistry. 2008. link Times cited: 12 NOT USED (high confidence) A. Fortini, M. Mendelev, S. Buldyrev, and D. Srolovitz, “Asperity contacts at the nanoscale: Comparison of Ru and Au,” Journal of Applied Physics. 2008. link Times cited: 52 Abstract: We develop and validate an interatomic potential for rutheni… read moreAbstract: We develop and validate an interatomic potential for ruthenium based on the embedded atom method framework with the Finnis/Sinclair representation. We confirm that the potential yields a stable hcp lattice with reasonable lattice and elastic constants and surface and stacking fault energies. We employ molecular dynamics simulations to bring two surfaces together, one flat and the other with a single asperity. We compare the process of asperity contact formation and breaking in Au and Ru, two materials currently in use in microelectromechanical system switches. While Au is very ductile at 150 and 300 K, Ru shows considerably less plasticity at 300 and 600 K (approximately the same homologous temperature). In Au, the asperity necks down to a single atom thick bridge at separation. While similar necking occurs in Ru at 600 K, it is much more limited than in Au. On the other hand, at 300 K, Ru breaks by a much more brittle process of fracture/decohesion with limited plastic deformation. read less NOT USED (high confidence) I. Valikova and A. Nazarov, “Simulation of characteristics determining pressure effects on the concentration and diffusivity of vacancies in BCC metals: A new approach,” The Physics of Metals and Metallography. 2008. link Times cited: 10 NOT USED (high confidence) É. Cancès, F. Legoll, M. Marinica, K. Minoukadeh, and F. Willaime, “Some improvements of the activation-relaxation technique method for finding transition pathways on potential energy surfaces.,” The Journal of chemical physics. 2008. link Times cited: 96 Abstract: The activation-relaxation technique nouveau is an eigenvecto… read moreAbstract: The activation-relaxation technique nouveau is an eigenvector following method for systematic search of saddle points and transition pathways on a given potential energy surface. We propose a variation in this method aiming at improving the efficiency of the local convergence close to the saddle point. The efficiency of the method is demonstrated in the case of point defects in body centered cubic iron. We also prove the convergence and robustness of a simplified version of this new algorithm. read less NOT USED (high confidence) P. Gordon, T. Neeraj, and M. J. Luton, “Atomistic simulation of dislocation nucleation barriers from cracktips in α-Fe,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 16 Abstract: In this work, we demonstrate that activation pathways for di… read moreAbstract: In this work, we demonstrate that activation pathways for dislocation loop nucleation from cracktips can be explored with full atomistic detail using an efficient form of the nudged elastic band method. The approach is demonstrated in detail with an example of edge emission from an Fe crack under mode II loading, wherein activation energy barriers are obtained as a function of sub-critical stress intensity and the energy barriers for loop formation are compared with 2D calculations. Activation energy barriers are also computed for an intrinsically ductile cracktip orientation under mode I loading, from which we can infer the frequency of nucleation from the cracktip. read less NOT USED (high confidence) M. Mendelev, M. Kramer, C. Becker, and M. Asta, “Analysis of semi-empirical interatomic potentials appropriate for simulation of crystalline and liquid Al and Cu,” Philosophical Magazine. 2008. link Times cited: 365 Abstract: We investigate the application of embedded atom method (EAM)… read moreAbstract: We investigate the application of embedded atom method (EAM) interatomic potentials in the study of crystallization kinetics from deeply undercooled melts, focusing on the fcc metals Al and Cu. For this application, it is important that the EAM potential accurately reproduces melting properties and liquid structure, in addition to the crystalline properties most commonly fit in its development. To test the accuracy of previously published EAM potentials and to guide the development of new potential in this work, first-principles calculations have been performed and new experimental measurements of the Al and Cu liquid structure factors have been undertaken by X-ray diffraction. We demonstrate that the previously published EAM potentials predict a liquid structure that is too strongly ordered relative to measured diffraction data. We develop new EAM potentials for Al and Cu to improve the agreement with the first-principles and measured liquid diffraction data. Furthermore, we calculate liquid-phase diffusivities and find that this quantity correlates well with the liquid structure. Finally, we perform molecular dynamics simulations of crystal nucleation from the melt during quenching at constant cooling rate. We find that EAM potentials, which predict the same zero-temperature crystal properties but different liquid structures, can lead to quite different crystallization kinetics. More interestingly, we find that two potentials predicting very similar equilibrium solid and liquid properties can still produce very different crystallization kinetics under far-from-equilibrium conditions characteristic of the rapid quenching simulations employed here. read less NOT USED (high confidence) P. Erhart, A. Caro, M. Caro, and B. Sadigh, “Short-range order and precipitation in Fe-rich Fe-Cr alloys: Atomistic off-lattice Monte Carlo simulations,” Physical Review B. 2008. link Times cited: 67 Abstract: Short-range order (SRO) in Fe-rich Fe-Cr alloys is investiga… read moreAbstract: Short-range order (SRO) in Fe-rich Fe-Cr alloys is investigated by means of atomistic off-lattice Monte Carlo simulations in the semi-grand-canonical ensemble using classical interatomic potentials. The SRO parameter defined by Cowley [Phys. Rev. 77, 669 (1950)] is used to quantify the degree of ordering. In agreement with experiments a strong ordering tendency in the Cr distribution at low Cr concentrations $(\ensuremath{\lesssim}5%)$ is observed, as manifested in negative values of the SRO parameters. For intermediate Cr concentrations $(5%\ensuremath{\lesssim}{c}_{\mathrm{Cr}}\ensuremath{\lesssim}15%)$, the SRO parameter for the $\ensuremath{\alpha}$ phase goes through a minimum, but at the solubility limit, the $\ensuremath{\alpha}$-phase still displays a rather strong SRO. In thermodynamic equilibrium for concentrations within the two-phase region the SRO parameter measured over the entire sample therefore comprises the contributions from both the $\ensuremath{\alpha}$ and ${\ensuremath{\alpha}}^{\ensuremath{'}}$ phases. If both of these contributions are taken into account, it is possible to quantitatively reproduce the experimental results and interpret their physical implications. It is thereby shown that the inversion of the SRO observed experimentally is due to the formation of stable (supercritical) ${\ensuremath{\alpha}}^{\ensuremath{'}}$ precipitates. It is not related to the loss of SRO in the $\ensuremath{\alpha}$ phase or to the presence of unstable (subcritical) Cr precipitates in the $\ensuremath{\alpha}$ phase. read less NOT USED (high confidence) R. Asaro, D. Farkas, and Y. Kulkarni, “The Soret effect in diffusion in crystals,” Acta Materialia. 2008. link Times cited: 17 NOT USED (high confidence) C. Engin, L. Sandoval, and H. Urbassek, “Characterization of Fe potentials with respect to the stability of the bcc and fcc phase,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 64 Abstract: By calculating free energies, several published interatomic … read moreAbstract: By calculating free energies, several published interatomic interaction potentials for iron are investigated with respect to the stability of the low-temperature bcc phase and the high-temperature fcc phase. These are empirical many-body potentials for use in atomistic simulation. We find that in all of these potentials—except one—the bcc phase is the stable crystal structure for all temperatures up to the melting point. However, several potentials exhibit a metastable fcc phase in the sense that the fcc structure corresponds to a local minimum of the free energy. read less NOT USED (high confidence) D. Belashchenko, N. Kravchunovskaya, and O. Ostrovski, “Properties of iron under Earth’s core conditions: Molecular dynamics simulation with an embedded-atom method potential,” Inorganic Materials. 2008. link Times cited: 2 NOT USED (high confidence) M. Mendelev, S. Han, W. Son, G. Ackland, and D. Srolovitz, “Simulation of the interaction between Fe impurities and point defects in V,” Physical Review B. 2007. link Times cited: 56 Abstract: We report improved results of atomistic modeling of V-Fe all… read moreAbstract: We report improved results of atomistic modeling of V-Fe alloys. We introduced an electronic structure embedding approach to improve the description of the point defects in first-principles calculations, by including the semicore electrons in some V atoms those near the interstitial where the semicore levels are broadened but not those further from the point defect. This enables us to combine good accuracy for the defect within large supercells and to expand the data set of first-principles point defect calculations in vanadium with and without small amounts of iron. Based on these data, previous first-principles work, and new calculations on the alloy liquid, we fitted an interatomic potential for the V-Fe system which describes the important configurations likely to arise when such alloys are exposed to radiation. This potential is in a form suitable for molecular dynamics MD simulations of large systems. Using the potential, we have calculated the migration barriers of vacancies in the presence of iron, showing that these are broadly similar. On the other hand, MD simulations show that V self-diffusion at high temperatures and Fe diffusion are greatly enhanced by the presence of interstitials. read less NOT USED (high confidence) R. Schäublin and N. Baluc, “Radiation damage in ferritic/martensitic steels for fusion reactors: a simulation point of view,” Nuclear Fusion. 2007. link Times cited: 33 Abstract: Low activation ferritic/martensitic steels are good candidat… read moreAbstract: Low activation ferritic/martensitic steels are good candidates for the future fusion reactors, for, relative to austenitic steels, their lower damage accumulation and moderate swelling under irradiation by the 14 MeV neutrons produced by the fusion reaction. Irradiation of these steels, e.g. EUROFER97, is known to produce hardening, loss of ductility, shift in ductile to brittle transition temperature and a reduction of fracture toughness and creep resistance starting at the lowest doses. Helium, produced by transmutation by the 14 MeV neutrons, is known to impact mechanical properties, but its effect at the microstructure level is still unclear. The mechanisms underlying the degradation of mechanical properties are not well understood, despite numerous studies on the evolution of the microstructure under irradiation. This impedes our ability to predict materials' behaviour at higher doses for use in the future fusion reactors. Simulations of these effects are now essential. An overview is presented on molecular dynamics simulations of the primary state of damage in iron and of the mobility of a dislocation, vector of plasticity, in the presence of a defect. read less NOT USED (high confidence) M. Müller, P. Erhart, and K. Albe, “Analytic bond-order potential for bcc and fcc iron—comparison with established embedded-atom method potentials,” Journal of Physics: Condensed Matter. 2007. link Times cited: 177 Abstract: A new analytic bond-order potential for iron is presented th… read moreAbstract: A new analytic bond-order potential for iron is presented that has been fitted to experimental data and results from first-principles calculations. The angular-dependent functional form allows a proper description of a large variety of bulk, surface and defect properties, including the Bain path, phonon dispersions, defect diffusivities and defect formation energies. By calculating Gibbs free energies of body-centred cubic (bcc) and face-centred cubic (fcc) iron as a function of temperature, we show that this potential is able to reproduce the transitions from α-iron to γ-iron and δ-iron before the melting point. The results are compared to four widely used embedded-atom-method potentials for iron. read less NOT USED (high confidence) C. Becquart et al., “Atomistic modeling of an Fe system with a small concentration of C,” Computational Materials Science. 2007. link Times cited: 156 NOT USED (high confidence) T. Bazhirov, A. Kuksin, G. Norman, and V. Stegailov, “On thermodynamic similarity of the stability boundaries of metastable metal states,” Russian Journal of Physical Chemistry A. 2007. link Times cited: 1 NOT USED (high confidence) K.-A. Wu and A. Karma, “Phase-field crystal modeling of equilibrium bcc-liquid interfaces,” Physical Review B. 2007. link Times cited: 122 Abstract: We investigate the equilibrium properties of bcc-liquid inte… read moreAbstract: We investigate the equilibrium properties of bcc-liquid interfaces modeled with a continuum phase-field crystal (PFC) approach [K. R. Elder and M. Grant, Phys. Rev. E 70, 051605 (2004)]. A multiscale analysis of the PFC model is carried out which exploits the fact that the amplitudes of crystal density waves decay slowly into the liquid in the physically relevant limit where the freezing transition is weakly first order. This analysis yields a set of coupled equations for these amplitudes that is similar to the set of equations derived from Ginzburg-Landau (GL) theory [K.-A. Wu et al., Phys. Rev. B 73, 094101 (2006)]. The two sets only differ in the details of higher order nonlinear couplings between different density waves, which is determined by the form of the nonlinearity assumed in the PFC model and by the ansatz that all polygons with the same number of sides have equal weight in GL theory. Despite these differences, for parameters (liquid structure factor and solid density wave amplitude) of Fe determined from molecular dynamics (MD) simulations, the PFC and GL amplitude equations yield very similar predictions for the overall magnitude and anisotropy of the interfacial free-energy and density wave profiles. These predictions are compared with MD simulations as well as numerical solutions of the PFC model. read less NOT USED (high confidence) C. Björkas and K. Nordlund, “Comparative study of cascade damage in Fe simulated with recent potentials,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 102 NOT USED (high confidence) V. Stegailov and A. Yanilkin, “Structural transformations in single-crystal iron during shock-wave compression and tension: Molecular dynamics simulation,” Journal of Experimental and Theoretical Physics. 2007. link Times cited: 18 NOT USED (high confidence) A. Caro, M. Caro, P. Klaver, B. Sadigh, E. M. Lopasso, and S. G. Srinivasan, “The computational modeling of alloys at the atomic scale: From ab initio and thermodynamics to radiation-induced heterogeneous precipitation,” JOM. 2007. link Times cited: 19 NOT USED (high confidence) P. Gordon, T. Neeraj, M. J. Luton, and D. Farkas, “Crack-Tip Deformation Mechanisms in α-Fe and Binary Fe Alloys: An Atomistic Study on Single Crystals,” Metallurgical and Materials Transactions A. 2007. link Times cited: 41 NOT USED (high confidence) M. Mendelev and G. Ackland, “Development of an interatomic potential for the simulation of phase transformations in zirconium,” Philosophical Magazine Letters. 2007. link Times cited: 266 Abstract: In recent years, some 30 studies have been published on the … read moreAbstract: In recent years, some 30 studies have been published on the molecular dynamics (MD) of zirconium, primarily of its twinning deformation and response to radiation damage. Its low thermal neutron absorption makes it uniquely suited for the latter application. Surprisingly, currently used interatomic potentials do not encapsulate the unique properties of Zr, namely its high stacking-fault energy, anomolous self-diffusion, melting and phase transformation under temperature and pressure (or alloying). Ab initio calculations have shown deficiencies in the description of point defects, both vacancies and interstitials, using existing interatomic potentials, deficiencies that can now be rectified by refitting. Here, we show the calculation of phase transitions self-consistently and present a potential for Zr that correctly reproduces the energetics of our extended database of ab initio configurations and high-temperature phase transitions. The potential has an analytic many-body form, making it suitable for existing large-scale MD codes. We also present a best-fit potential for the hcp structure and its defects. read less NOT USED (high confidence) G. Norman, V. Stegailov, and A. Yanilkin, “The modeling of high-rate tension of crystalline iron by the method of molecular dynamics,” High Temperature. 2007. link Times cited: 9 NOT USED (high confidence) P. Ma, W. Liu, C. Woo, and S. Dudarev, “Large-scale molecular dynamics simulation of magnetic properties of amorphous iron under pressure,” Journal of Applied Physics. 2007. link Times cited: 19 Abstract: We perform large-scale molecular dynamics simulations to stu… read moreAbstract: We perform large-scale molecular dynamics simulations to study the magnetic properties of amorphous iron under pressure. Simulations, exceeding by at least two orders of magnitude those accessible to density functional calculations, use the recently developed magnetic interatomic potential for iron. The distributions of the size of atomic magnetic moments and parameters characterizing the structure of amorphous iron, such as radial distribution functions, are calculated as a function of the applied hydrostatic stress. As the density increases, there is a reduction in the magnitude of the mean magnetic moment of individual atoms, accompanied by the transformation of an increasing proportion of atoms from a magnetic to a nonmagnetic configuration. Beyond a critical density the proportion of nonmagnetic atoms increases sharply, yet homogeneously. The local magnetic moment of an atom correlates with the local Voronoi volume via a logarithmic relation. In addition, we observe a complex dependence of the local magnetic moment on the topological arrangement of neighboring atoms. © 2007 American Institute of Physics. DOI: 10.1063/1.2715753 read less NOT USED (high confidence) D. Belashchenko, “Embedded atom model application to liquid metals: Liquid rubidium,” Russian Journal of Physical Chemistry. 2006. link Times cited: 13 NOT USED (high confidence) A. Caro, M. Caro, E. M. Lopasso, and D. Crowson, “Implications of ab initio energetics on the thermodynamics of Fe–Cr alloys,” Applied Physics Letters. 2006. link Times cited: 30 Abstract: The authors analyze the implications of the recently reporte… read moreAbstract: The authors analyze the implications of the recently reported results of ab initio calculations of formation energies of the Fe–Cr alloy. The formation energies show a change in sign from negative to positive as Cr composition increases above ∼10%. By developing a classic potential to evaluate the thermodynamic properties, they determine the location of the solubility limit and compare it with earlier results. A significant difference appears in a region of temperature and composition that is relevant for the nuclear applications of this alloy. Experimental results seem to confirm the validity of the location of the new solvus line. read less NOT USED (high confidence) F. Zypman and J. Ferrante, “Gradient equivalent crystal theory,” Journal of Physics: Condensed Matter. 2006. link Times cited: 0 Abstract: This paper presents an extension of the formalism of equival… read moreAbstract: This paper presents an extension of the formalism of equivalent crystal theory (ECT) by introducing an electron density gradient term so that the total model density becomes a more accurate representation of the real local density. Specifically, we allow for the electron density around a lattice site to have directionality, in addition to an average value, as assumed in ECT. We propose that an atom senses its neighbouring density as a weighted sum—the weights given by the its own electronic probability. As a benchmark, the method is used to compute vacancy migration energy curves of iron. These energies are in good agreement with previously published results. read less NOT USED (high confidence) L. Ventelon, B. Wirth, and C. Domain, “Helium–self-interstitial atom interaction in α-iron,” Journal of Nuclear Materials. 2006. link Times cited: 66 NOT USED (high confidence) D. Belashchenko, “Embedded atom model for liquid metals: Liquid iron,” Russian Journal of Physical Chemistry. 2006. link Times cited: 21 NOT USED (high confidence) K. Nordlund, J. Wallenius, and L. Malerba, “Molecular dynamics simulations of threshold displacement energies in Fe,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2006. link Times cited: 164 NOT USED (high confidence) S. Dudarev, “Angular-dependent matrix potentials for fast molecular-dynamics simulations of transition metals,” Journal of Physics: Condensed Matter. 2006. link Times cited: 5 Abstract: The significance of the part played by the angular-dependent… read moreAbstract: The significance of the part played by the angular-dependent components of forces associated with d–d bonding between atoms in a transition metal has long remained a subject of debate. While almost all the large-scale molecular dynamics simulations of collision cascades and radiation damage in transition metals and alloys are currently performed using spherically symmetric many-body potentials, density functional calculations exhibit a highly anisotropic pattern of charge density deformation in and around the core of interstitial atom defects. This paper describes a fast second-order matrix recursion-based algorithm for including effects of angular anisotropy of d–d bonds in a large-scale molecular dynamics simulation. read less NOT USED (high confidence) D. Belashchenko and O. I. Ostrovskii, “The embedded atom model for liquid metals: Liquid gallium and bismuth,” Russian Journal of Physical Chemistry. 2006. link Times cited: 23 NOT USED (high confidence) D. Kulikov, L. Malerba, and M. Hou, “On the binding energies and configurations of vacancy and copper–vacancy clusters in bcc Fe–Cu:a computational study,” Philosophical Magazine. 2006. link Times cited: 21 Abstract: Vacancy and copper–vacancy clusters in bcc Fe–Cu alloys have… read moreAbstract: Vacancy and copper–vacancy clusters in bcc Fe–Cu alloys have been studied using a combination of metropolis Monte Carlo (MMC) and molecular dynamics (MD) techniques, to investigate their lowest energy configurations and corresponding binding energies, for sizes up to a few hundreds of elements (∼2 nm). Two different many-body interatomic potentials were used to perform the calculations, in order to assess the robustness of the results 1, 2. Empirical expressions for the binding energies, of immediate use in kinetic Monte Carlo (KMC) or rate theory (RT) models, have been obtained. It is observed that vacancy clusters are three-dimensional cavities whose shape is primarily determined by a criterion of maximisation of the number of first and second nearest neighbour pairs. Copper atoms, when present, tend to coat an inner vacancy cluster, while remaining first nearest neighbours to each other. The inner vacancy cluster, when completely coated, tends to be as close as possible to the surface of the hollow precipitate. These findings are consistent with previous experimental and computational work. The binding energy of these complexes is a monotonously growing function of the ratio number of vacancies to number of copper atoms. Pure copper precipitates appear to follow a loose criterion of maximisation of first nearest neighbour pairs. While the two interatomic potentials used in this work provide largely similar values for the binding energies and comparable configurations, some differences are found and discussed. Subtle differences observed in comparison with ab initio calculations are also discussed. read less NOT USED (high confidence) Y. Mishin, M. Mehl, D. Papaconstantopoulos, and D. Papaconstantopoulos, “Phase stability in the Fe–Ni system: Investigation by first-principles calculations and atomistic simulations,” Acta Materialia. 2005. link Times cited: 261 NOT USED (high confidence) A. Barashev, “Monte Carlo simulation of phosphorus diffusion in α-iron via the vacancy mechanism,” Philosophical Magazine. 2005. link Times cited: 24 Abstract: Monte Carlo simulations of the vacancy and phosphorus (P) at… read moreAbstract: Monte Carlo simulations of the vacancy and phosphorus (P) atom diffusion in body centred cubic (bcc) iron are presented. The input parameters for the calculations, namely the activation energies of atomic jumps, have been obtained using a potential set developed recently for a dilute Fe–P alloy using ab initio data. The diffusion coefficients entering equations for the fluxes of vacancies and solute atoms are evaluated. The results show that, in the temperature range of practical importance for P segregation, P atoms move down the vacancy gradient; hence, under irradiation conditions, vacancies should drag P atoms towards sinks of point defects. This is because of the high binding energy between a P atom and a vacancy in the first and second nearest neighbour sites from each other, which allows a vacancy to move around a P atom without loss of bonding and, hence, co-migrate with it. read less NOT USED (high confidence) G. Ackland, “Two-band second moment model for transition metals and alloys,” Journal of Nuclear Materials. 2005. link Times cited: 35 NOT USED (high confidence) G. Ackland, M. Mendelev, D. Srolovitz, S. Han, and A. Barashev, “Development of an interatomic potential for phosphorus impurities in α-iron,” Journal of Physics: Condensed Matter. 2004. link Times cited: 542 Abstract: We present the derivation of an interatomic potential for th… read moreAbstract: We present the derivation of an interatomic potential for the iron–phosphorus system based primarily on ab initio data. Transferability in this system is extremely problematic, and the potential is intended specifically to address the problem of radiation damage and point defects in iron containing low concentrations of phosphorus atoms. Some preliminary molecular dynamics calculations show that P strongly affects point defect migration. read less NOT USED (high confidence) S. Ogata, “Free-energy-based Atomistic Study of Nucleation Kinetics and Thermodynamics of Defects in Metals; Plastic Strain Carrier ‘Plaston,’” The Plaston Concept. 2022. link Times cited: 0 NOT USED (high confidence) Z. SHEN, D. AI, S. LV, J. Gao, W. LAI, and Z. LI, “The evolution of matrix damage under irradiation in Fe-C alloy by OKMC simulation,” Acta Physica Sinica. 2022. link Times cited: 0 Abstract: The effect of carbon traps in Fe-C alloys on matrix defects … read moreAbstract: The effect of carbon traps in Fe-C alloys on matrix defects and the evolution of matrix defects in Fe-C alloys under irradiation were investigated in this paper. The Object kinetic Monte Carlo (OKMC) modeling is carried out and to establish a bridge between the micro-computational simulation data and the macro-experimental data. The simulation results verify the evolution of the carbon (C)-vacancy (Vac) complex under ideal conditions, and at relatively low temperatures, the complex is mainly C-Vac2. Based on the assumption of complex traps, the paper simulates the evolution of matrix defects in Fe-C systems under irradiation conditions. It is verified that the carbon vacancy complex has an obvious trapping effect on matrix defects, and the evolution simulation of matrix defects in the Fe-C system under irradiation conditions can obtain results consistent with the experiments. Furthermore, the effective approximate parameters used in the simulation are compared and discussed, which would provide basic support for the research on the evolution of iron-based alloy irradiation defects. read less NOT USED (high confidence) S. Koch, “Development of RF-MEAM interaction potentials for Fe-Y.” 2019. link Times cited: 0 Abstract: Der Fokus dieser Arbeit lag zunachst auf einer simulationsge… read moreAbstract: Der Fokus dieser Arbeit lag zunachst auf einer simulationsgestutzen Untersuchung uber die Entsteh- ungsmechanismen von Oxidteilchen in ODS-Stahlen. Hierbei bilden empirische Wechselwirkungs- potenziale von Eisen-Yttrium-Sauerstoff (Fe-Y-O) die Grundlage fur eine Beschreibung dieser Oxid- teilchen-Bildungs-Prozesse in Molekulardynamik (MD) Simulationen, die auch Eigenschaften von Versetzungen und anderen Bestrahlungs-Panomenen detailiert zur weiteren Aufklarung behandeln konnen.
Zu diesem Zweck ist das speziell auf die Simulation zugeschnittene Anfitten der o.g. MD Potenziale (hier fur Fe-Y-O) notwendig. Hierzu dienen die zuvor durchgefuhrten ab-initio (DFT) Rechnungen als Daten- referenzgrundlage (z.B. von Phasen oder Defekten) zur Optimierung der Potenzialparameter wahrend des Anfittens, um ein moglichst exaktes MD Potenzial zu erzeugen, dass die ab-initio Daten auf groseren MD Skalen detailgetreu abbildet. Im ersten Drittel dieses Projektes wurden mehrere Potenziale fur die einzelnen Metall-Komponenten, Fe-Fe und Y-Y, erzeugt. Dabei stellte sich heraus, dass etablierte Standardmethoden nicht in der Lage sind genaue Fe-Y Potenziale als Teillosung fur das Fe-Y-O Problem zu erzeugen. Dabei wurde eine Kombination aus dem (M)EAM Modell und zur Optimierung eine LSM gestutzte Software (POTFIT) genutzt. Die Komplexitat des Problems liegt in den richtungsabhangigen Atombindungen, die die hier entwickelten fortgeschrittenen Simulations- und Fitmethoden benotigen.
Im ersten Schritt von drei Schritten (chapter 3) wurden zunachst einmal die Defizite der Standard-Fittechniken evaluiert, indem die wahrend des Fitting-Prozesses gefundenen Parametersets im EAM Formalismus mit der flexiblen Software POTFIT auf ihre Eignung hin grundlich untersucht worden sind. Die hierfur genutzten Fitfunktionen wurden ursprunglich Anfang 2000 von Zhou und Wadley entwickelt. Hierbei liegt die Ursache fur die dann entdeckte Parameterset-Problematik darin, dass zur Beschreibung des Fe-Y Systems das Model aus drei Potentialkomponenten besteht: Fe-Fe, Y-Y und Fe-Y. Fur diese einzelnen Komponenten sind die Potentialparameter erfolgreich angefittet worden mit Bezug zur Gitterkonstante und Bindungs- bzw. Kohasionsenergie (beides mit 1% Genauigkeit bezgl. DFT Rechnungen) sowie zu allen elastischen Konstanten (5% Genauigkeit bezgl. Experimente). All dies unter Zuhilfenahme von Parametersuchraum-beschrankenden Techniken, die zur Einhaltung der oben genannten Eigenschaften dienen und urspurnglich von Johnson & Oh sind. Selbst kompliziertere Defekteigenschaften, wie Zwischengitter- und Leerstellenbildungsenergien wurden erfolgreich angefittet. Das hier entwickelte EAM Potenzial fur Y-Y ist z.B. in der Lage bei Eigenzwischengitteratomen die basal oktaedrische Position von Zwischengitteratomen (ZA) im Yttrium hcp-Gitter als Grundzustand und die Transition eines jeden ZAs aus einer anderen Position, wie zuvor in DFT berechnet, zu reproduzieren.
Zur Bildung des angestrebten Fe-Y Potenzials wurden diese beiden Komponenten, Fe-Fe und Y-Y, zum weiteren Fitten in dem weitgefacherten und komplexen Fe-Y Potzenzialsuchraum genutzt. Die Parametersets wurden mit sogenannten hier entwickelten Hauptparameter (Key Driver) systematisch untersucht. Ein flexibleres Konzept statt der starreren Universal Binding Relations in Abhangigkeit von der Rose Gleichung. Dieser Hauptparameter zeigte eindeutig, dass die Nutzung der Rose Gleichung zur Parametersuchraum-Minimierung den Suchraum dahingehend einschrankt, sodass ein akkurates Anfitten der hier genutzten 900 DFT Datensets nicht mehr moglich ist. Allerdings ist die Orientierung im Parametersuchraum mit dieser Rose Gleichung bei standardmasigen Optimierungsmethoden (wie LSM) unabdingbar, da ohne diese die benotigten globalen Optima fur die Parameter nicht auffindbar sind.
Als aufklarendes Testverfahren zur weiteren Ergrundung dieser Problematik und Prufung zur Eignung fur Fe-Y Potenziale und den anschliesenden Simulationen diente der Versuch, 9 verschiedene Bindungs-energien von Yttrium-Leerstellenclustern mit ansteigender Leerstellenzahl zu reproduzieren. Dieser Test konnte von diesen Potenzialen nur teilweise erfullt werden und wurde auf die fehlende Beschreibung der Bindungswinkelabhangigkeit im Modell zuruckgefuhrt. Die Erweiterung von EAM durch MEAM mit Winkelabhangigkeit ist jedoch keineswegs eine zufriedenstellende Losung, da MEAM alternativlos auf der irrefuhrenden Rose Gleichung beruht. Daher war die Benutzung des ubersichtlicheren EAM Typs aus zwei Grunden nutzlich: 1. MEAM braucht die Rose Gleichung um diesen komplexen Formalismus zu beherrschen mit denselben Problemen wie in EAM, aber dieses grundlegende Problem ist in MEAM deutlich schwerer zu identifizieren als in EAM. 2. Die mit EAM gefundenen, angefitteten Parameter sind eine hervorragende Startparameter-Grundlage fur den verbesserten darauffolgenden RF-MEAM Typ.
Im zweiten Schritt wurde das Problem aus dem ersten Schritt gelost, indem ein modifizierter MEAM Spezialtyp im referenzlosen Format (RF-MEAM) angewandt worden ist. Im Gegensatz zum herkommlichen MEAM wird hier die Rose Gleichung durch mehr DFT Daten und insbesondere einer intelligenteren Machine Learning ahnlichen Genetic Algorithmus (GA) Optimiertechnik ersetzt, die allerdings eine bedachte Startparameterwahl vorraussetzt, womit Schritt 1 wieder ins Spiel kommt. Die genutzte fortgeschrittene MEAMfit Software, die per GA funktioniert, wurde zwischen 2016 und 2017 funktionierend eigens dafur implementiert. Mit den in Schritt 1 gefitteten Parametern und Set-Auswahltechniken konnten die weiterfuhrenden Fits mit optimalen Startparametern durchgefuhrt werden.
Auf dieser Stufe waren diese Fits mit der speziell verbesserten Technik in der Lage ein detailgetreues Fe-Y Potenzial zu generieren, das sowohl alle Phasen (Fe2Y, Fe3Y, Fe5Y, Fe23Y6 und Fe17Y2 sowohl als auch reines Fe und Y) als auch die gesamte Defektdatenbasis mit einer durchschnittlichen Abweichung von ≈11% erfolgreich abbildet. Zusatzlich bestatigend zu dieser allgemeinen Ubereinstimmung wurde konsequenterweise der in Schritt 1 entwickelte Test hervorragend mit einmaliger Genauigkeit bestanden, mit max. 5% Abweichung von den komlexen o.g. Y-Leerstellen Bildungsenergien. Allerdings konnte ein systematischer Fehlertrend aufgespurt werden, der Schwachen in der Fe-Fe Komponente offenbarte. Als Folge dessen wurde umgehend diese Komponente durch ein anderes etabliertes Fe-Fe Potenzial von G. Ackland mit einer extrem genauen Schmelztemperatur (nur 3% Abweichung vom Exp.) ausgetauscht. Mit diesem genauen Potenzial konnte zum ersten Mal die Clusterbildung von gelosten Yttrium Atomen in einer Eisenschmelze erfolgreich per MD Simulation auf atomarer Ebene nachgestellt werden oberhalb von 1750 K. Temperaturen darunter hatten eine Ausscheidungsbildung von Y mit sehr geringer Y-Loslichkeit (<0.1%) in Ubereinstimmung mit den Experimenten zur Folge. Dies wurde durch den Pot. Typ A ermoglicht, der aber die energetische Reihenfolge bei den Fe-Y Phasen nicht ganz genau einhalt. Typ B hingegen halt diese ein, dort fehlt aber die Y-Clusterbildungsneigung. Durch den gebotenen Praxisbezug zur Metallurgie mussen die Loslichkeit und Clusterbildung gleichzeitig in der Simulation genau reproduzierbar sein, was aber weder Typ A noch B kann, was zum Typ A/B Dilemma fuhrt.
Dieses Typ A/B Dilemma (Phasen oder Defekt Genauigkeit) fuhrt zum letzten dritten Schritt (chapter 5). Darin ist zusatzlich die Strukturaufklarung von der Fe17Y2 Phase mit Vergleichen zu exp. EXAFS Spektren unserer Kollaborationspartner vom ISSP (Riga) enthalten. Diese Aufklarung dient auch dazu die fehlenden magnetischen Abhangigkeiten im Potenzial zu kompensieren, da die Phasenreihenfolge mit sehr feinen Energieunterschieden wohl stark von magnetischen Wechselwirkungen gepragt ist. Obwohl Potenzial Typ B diesen (Magnetismus) nicht direkt beachtet, ist es in der Lage das tatsachlich gemessene EXAFS Spektrum grostenteils genau wiederzugeben. Allerdings offenbart eine einzige ausgepragte Phasenverschiebung, dass die angenommene hcp Struktur durch eine unterschwellige rhombohedrale Komponente, die sporadisch in der c-Gitterrichtung auftritt, korrigiert werden muss. AIMD (DFT) Berechnungen in Kooperation mit der University of Edinburgh bestatigen dies und zeigen sogar, dass magnetische Wechselwirkungen diese Strukturmischung stabilisieren. Endgultig bestatigt werden konnte dies mit der genauen EXAFS Spektren Reproduktion mit dem durch AIMD verbesserten nochmals gefitteten Potenzialtyp B, der als neuer Typ C durch AIMD indirekt den Einfluss der magnetischen Wechselwirkungen mit einschliest. Diese erstmalige nahezu deckungsgleiche MD Simulation eines EXAFS Spektrums von einem komplexen metallischen Alloy, hier Fe-Y, stellt eine bisher unerreichte Verbesserung dar. Schlieslich lost Typ C das Typ A/B Dilemma und ernoglicht eine genaue gleichzeitige MD Modellierung von Phasen- und Defekten in Fe-Y – ein Durchbruch in der MD-Potenzialentwicklung. read less NOT USED (high confidence) N. Anento, L. Malerba, and A. Serra, “Edge dislocations as sinks for sub-nanometric radiation induced defects in α-iron,” Journal of Nuclear Materials. 2018. link Times cited: 4 NOT USED (high confidence) P. Wang et al., “Atomistic simulation for deforming complex alloys with application toward TWIP steel and associated physical insights,” Journal of The Mechanics and Physics of Solids. 2017. link Times cited: 42 NOT USED (high confidence) H. D. Aristizabal, P. Parra, P. López, and E. Restrepo‐Parra, “Atomic-scale simulations of material behaviors and tribology properties for BCC metal film,” Chinese Physics B. 2015. link Times cited: 6 Abstract: This work has two main purposes: (i) introducing the basic c… read moreAbstract: This work has two main purposes: (i) introducing the basic concepts of molecular dynamics analysis to material scientists and engineers, and (ii) providing a better understanding of instrumented indentation measurements, presenting an example of nanoindentation and scratch test simulations. To reach these purposes, three-dimensional molecular dynamics (MD) simulations of nanoindentation and scratch test technique were carried out for generic thin films that present BCC crystalline structures. Structures were oriented in the plane (100) and placed on FCC diamond substrates. A pair wise potential was employed to simulate the interaction between atoms of each layer and a repulsive radial potential was used to represent a spherical tip indenting the sample. Mechanical properties of this generic material were obtained by varying the indentation depth and dissociation energy. The load-unload curves and coefficient of friction were found for each test; on the other hand, dissociation energy was varied showing a better mechanical response for films that present grater dissociation energy. Structural change evolution was observed presenting vacancies and slips as the depth was varied. read less NOT USED (high confidence) H. Jin, “Atomistic simulations of solute-interface interactions in iron.” 2014. link Times cited: 6 Abstract: The kinetics of the recrystallization and austenite-ferrite … read moreAbstract: The kinetics of the recrystallization and austenite-ferrite (fcc-bcc) phase transformation in steels are markedly affected by substitutional alloying elements. Nevertheless, the detailed mechanisms of their interaction with the grain boundaries and interfaces are not fully understood. Using density functional theory, we determine the segregation energies of commonly used alloying elements (e.g. Nb, Mo, Mn, Si, Cr, Ni) in the Σ5 (013) tilt grain boundary in bcc and fcc Fe, and the bcc-fcc interfaces. We find a strong interaction between large solutes (e.g. Nb, Mo and Ti) and grain boundaries or interfaces that is consistent with experimental observations of the effects of these alloying elements on delaying recrystallization and the austenite-to-ferrite transformation in low-carbon steels. In addition, we compute the solute-solute interactions as a function of solute pair distance in the grain boundaries and interfaces, which suggest co-segregation for these large solutes at intermediate distances in striking contrast to the bulk. Besides the prediction of solute segregation, the selfand solute-diffusion in Febased system are also investigated within a framework combining density functional theory calculations and kinetic Monte Carlo simulations. Good agreement between our calculations and the measurements for selfand solute diffusion in bulk Fe is achieved. For the first time, the effective activation energies and diffusion coefficients for various solutes in the α-Fe Σ5 (013) grain boundary are determined. The results demonstrate that grain boundary diffusion is significantly faster than for lattice diffusion, confirming grain boundaries are fast diffusion paths. By contrast, the effective activation energy of self-diffusion in a bcc-fcc Fe interface is close to the value of fcc bulk self-diffusion, and the investigated bcc-fcc interface provides a moderate “fast diffusion” path. read less NOT USED (high confidence) E. Asadi, M. A. Zaeem, and M. Baskes, “Phase-Field Crystal Model for Fe Connected to MEAM Molecular Dynamics Simulations,” JOM. 2014. link Times cited: 31 NOT USED (high confidence) C. Tackes, “Thermal analysis of undercooled metallic liquids by electromagnetic levitation drop calorimetry.” 2013. link Times cited: 2 Abstract: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . … read moreAbstract: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xvi CHAPTER read less NOT USED (high confidence) X. Zheng, H. Zhu, A. K. Tieu, and B. Kosasih, “Roughness and Lubricant Effect on 3D Atomic Asperity Contact,” Tribology Letters. 2013. link Times cited: 32 NOT USED (high confidence) H. Xie, B. Liu, F. Yin, and T. Yu, “Effect of grain boundary sliding on the toughness of ultrafine grain structure steel: A molecular dynamics simulation study,” Chinese Physics B. 2013. link Times cited: 5 Abstract: Molecular dynamics simulations are carried out to investigat… read moreAbstract: Molecular dynamics simulations are carried out to investigate the mechanisms of low-temperature impact toughness of the ultrafine grain structure steel. The simulation results suggest that the sliding of the {001}/{110} type and {110}/{111} type grain boundary can improve the impact toughness. Then, the mechanism of grain boundary sliding is studied and it is found that the motion of dislocations along the grain boundary is the underlying cause of the grain boundary sliding. Finally, the sliding of the grain boundary is analyzed from the standpoint of the energy. We conclude that the measures which can increase the quantity of the {001}/{110} type and {110}/{111} type grain boundary and elongate the free gliding distance of dislocations along these grain boundaries will improve the low-temperature impact toughness of the ultrafine grain structure steel. read less NOT USED (high confidence) M. Byshkin, B. Zhu, and M. Hou, “The effect of encapsulation in carbon nanotubes on properties of Fe–Ni nanoalloys with cubic and helical structures,” Journal of Materials Science. 2012. link Times cited: 1 NOT USED (high confidence) S. Naamane, G. Monnet, and B. Devincre, “Low temperature deformation in iron studied with dislocation dynamics simulations,” International Journal of Plasticity. 2010. link Times cited: 73 |
 EAM_MagneticCubic_MendelevHanSrolovitz_2003_Fe__MO_856295952425_002
EAM_MagneticCubic_MendelevHanSrolovitz_2003_Fe__MO_856295952425_002