Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
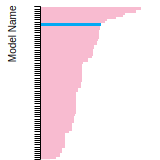
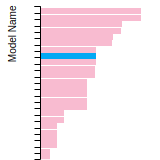
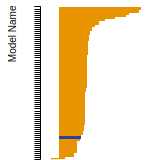
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
162 Citations (52 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (high confidence) C. Larsen, S. Kaappa, A. L. Vishart, T. Bligaard, and K. Jacobsen, “Machine-learning-enabled optimization of atomic structures using atoms with fractional existence,” Physical Review B. 2022. link Times cited: 0 Abstract: We introduce a method for global optimization of the structu… read moreAbstract: We introduce a method for global optimization of the structure of atomic systems that uses additional atoms with fractional existence. The method allows for movement of atoms over long distances bypassing energy barriers encountered in the conventional position space. The method is based on Gaussian processes, where the extrapolation to fractional existence is performed with a vectorial fingerprint. The method is applied to clusters and two-dimensional systems, where the fractional existence variables are optimized while keeping the atomic positions fixed on a lattice. Simultaneous optimization of atomic coordinates and existence variables is demonstrated on copper clusters of varying size. The existence variables are shown to speed up the global optimization of large and particularly difficult-to-optimize clusters. read less USED (high confidence) D. G. Sentürk, A. D. Backer, T. Friedrich, and S. V. Aert, “Optimal experiment design for element specific atom counting using multiple annular dark field scanning transmission electron microscopy detectors.,” Ultramicroscopy. 2022. link Times cited: 3 USED (high confidence) E. Pervolarakis, G. Tritsaris, P. Rosakis, and I. Remediakis, “Machine Learning for the edge energies of high symmetry Au nanoparticles,” Surface Science. 2022. link Times cited: 1 USED (high confidence) I. Bell, J. Dyre, and T. Ingebrigtsen, “Excess-entropy scaling in supercooled binary mixtures,” Nature Communications. 2020. link Times cited: 34 USED (high confidence) S. Subedi, S. M. Handrigan, L. Morrissey, and S. Nakhla, “Mechanical properties of nanocrystalline aluminium: a molecular dynamics investigation,” Molecular Simulation. 2020. link Times cited: 3 Abstract: ABSTRACT Uniaxial deformation was performed using molecular … read moreAbstract: ABSTRACT Uniaxial deformation was performed using molecular dynamics to estimate the mechanical properties of nanocrystalline aluminium. It was observed that the stacking faults and sliding of the grain boundaries affected the mechanical properties. In addition, accumulation of atoms near grain boundaries during deformation hardened the nanocrystalline material as the grain diameter increased (reverse Hall-Petch relation). Further, the effects of strain rate and temperature were investigated with various mean grain diameters. Investigation showed that mechanical properties were independent of tested strain rates (109–1010 s−1) and that the nanocrystalline material softened with increasing temperature. The elastic modulus was then compared to experimental results from literature at room temperature. The change in crystalline structure was observed with respect to percent strain and various mean grain diameters of nanocrystalline aluminium. It was observed that stacking faults increased with decreased mean grain diameter, which led to reduced mechanical properties. read less USED (high confidence) V. Pryadchenko et al., “The effect of thermal treatment on the atomic structure of core–shell PtCu nanoparticles in PtCu/C electrocatalysts,” Physics of the Solid State. 2017. link Times cited: 6 USED (high confidence) I. Pobelov et al., “Dynamic breaking of a single gold bond,” Nature Communications. 2017. link Times cited: 25 USED (high confidence) J. R. Boes and J. Kitchin, “Modeling Segregation on AuPd(111) Surfaces with Density Functional Theory and Monte Carlo Simulations,” Journal of Physical Chemistry C. 2017. link Times cited: 44 Abstract: The simulation of segregation in multicomponent alloy surfac… read moreAbstract: The simulation of segregation in multicomponent alloy surfaces is challenging with atomistic approaches because of the need to model a very large number of possible configurations with a high degree of accuracy. Density functional theory (DFT) is too expensive to use directly, and atomistic potentials are often a compromise between accuracy and computational speed. In this work we develop a neural network (NN) atomistic potential capable of predicting accurate energies for any configuration of a AuPd(111) slab. The fully trained neural network spanning all configurations and lattice constants of a AuPd binary alloy is trained from only 3914 DFT calculations. Using this NN, segregation profiles are created spanning bulk compositions between 10 and 90% Au, and at temperatures ranging from 700 to 1000 K using Monte Carlo simulations. These profiles are then fit to the Langmuir–McLean formulation of the Gibbs-isotherm with a model for the enthalpy of segregation. The simulation results are in excellent agreem... read less USED (high confidence) J. R. Boes and J. Kitchin, “Neural network predictions of oxygen interactions on a dynamic Pd surface,” Molecular Simulation. 2017. link Times cited: 57 Abstract: Artificial neural networks (NNs) are increasingly common in … read moreAbstract: Artificial neural networks (NNs) are increasingly common in quantum chemistry applications. These models can be trained to higher-level ab-initio calculations and are capable of achieving arbitrary levels of accuracy. The most common applications thus far have been specialised for either bulk or surface structures of up to two chemical components. However, very few of these studies utilise NNs trained to high-dimensional potential energy surfaces, and there are even fewer studies which examine adsorbate–adsorbate and adsorbate–surface interactions with those NNs. The goal of this work is to determine the feasibility of and develop methodologies for producing a high-dimensional NN capable of reproducing coverage-dependent oxygen interactions with a dynamic Pd fcc(1 1 1) surface. We utilise the atomistic machine-learning potential software package to generate a Behler–Parrinello local symmetry function NN trained on a large database of density functional theory (DFT) calculations. These training methods are flexible, and thus easily expanded upon as demonstrated in previous work. This allows the database of high quality PdO DFT calculations to be used as a basis for future work, such as the inclusion of a third chemical species, for example a binary Pd alloy, or another adsorbate atom such as hydrogen. read less USED (high confidence) A. Magyarkuti et al., “Temporal correlations and structural memory effects in break junction measurements,” arXiv: Mesoscale and Nanoscale Physics. 2016. link Times cited: 8 Abstract: We review data analysis techniques that can be used to study… read moreAbstract: We review data analysis techniques that can be used to study temporal correlations among conductance traces in break junction measurements. We show that temporal histograms are a simple but efficient tool to check the temporal homogeneity of the conductance traces, or to follow spontaneous or triggered temporal variations, like structural modifications in trained contacts, or the emergence of single-molecule signatures after molecule dosing. To statistically analyze the presence and the decay time of temporal correlations, we introduce shifted correlation plots. Finally, we demonstrate that correlations between opening and subsequent closing traces may indicate structural memory effects in atomic-sized metallic and molecular junctions. Applying these methods on measured and simulated gold metallic contacts as a test system, we show that the surface diffusion induced flattening of the broken junctions helps to produce statistically independent conductance traces at room temperature, whereas at low temperature repeating tendencies are observed as long as the contacts are not closed to sufficiently high conductance setpoints. Applying opening-closing correlation analysis on Pt-CO-Pt single-molecule junctions, we demonstrate pronounced contact memory effects and recovery of the molecule for junctions breaking before atomic chains are formed. However, if chains are pulled the random relaxation of the chain and molecule after rupture prevents opening-closing correlations. read less USED (high confidence) P. M. Larsen, S. Schmidt, and J. Schiøtz, “Robust structural identification via polyhedral template matching,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 532 Abstract: Successful scientific applications of large-scale molecular … read moreAbstract: Successful scientific applications of large-scale molecular dynamics often rely on automated methods for identifying the local crystalline structure of condensed phases. Many existing methods for structural identification, such as common neighbour analysis, rely on interatomic distances (or thresholds thereof) to classify atomic structure. As a consequence they are sensitive to strain and thermal displacements, and preprocessing such as quenching or temporal averaging of the atomic positions is necessary to provide reliable identifications. We propose a new method, polyhedral template matching (PTM), which classifies structures according to the topology of the local atomic environment, without any ambiguity in the classification, and with greater reliability than e.g. common neighbour analysis in the presence of thermal fluctuations. We demonstrate that the method can reliably be used to identify structures even in simulations near the melting point, and that it can identify the most common ordered alloy structures as well. In addition, the method makes it easy to identify the local lattice orientation in polycrystalline samples, and to calculate the local strain tensor. An implementation is made available under a Free and Open Source Software license. read less USED (high confidence) V. Pryadchenko et al., “Atomic Structure of Bimetallic Nanoparticles in PtAg/C Catalysts: Determination of Components Distribution in the Range from Disordered Alloys to ‘Core–Shell’ Structures,” Journal of Physical Chemistry C. 2015. link Times cited: 30 Abstract: Nanocatalysts PtAg/C with different distributions of compone… read moreAbstract: Nanocatalysts PtAg/C with different distributions of components in bimetallic PtAg nanoparticles were synthesized by the methods of simultaneous and sequential reduction of Ag+ and Pt(IV) in carbon suspension, prepared on the basis of ethylene glycol–water solvent. The characterization of atomic structure of as prepared PtAg nanoparticles and PtAg nanoparticles obtained after acid treatment at 90 °C was performed by Pt L3-edge extended X-ray absorption fine structure (EXAFS) using the technique for determining local structure parameters of the absorbing Pt atoms, which have the nearest surrounding consisting of Pt and Ag atoms. This technique enabled stable and unambiguous values of parameters for the local structure of Pt atoms in PtAg nanoparticles to be obtained under severe correlations between some of these parameters. The effects of averaging over the sizes and compositions of PtAg nanoparticles on the obtained values of structural parameters were studied by the method of cluster simulations. Analys... read less USED (high confidence) N. Galanis, I. Remediakis, and G. Kopidakis, “Structure and mechanical properties of ultra-nanocrystalline diamond and nanocrystalline Cu from atomistic simulations,” Mechanics of Materials. 2013. link Times cited: 12 USED (high confidence) F. Pauly, J. Viljas, M. Bürkle, M. Dreher, P. Nielaba, and J. Cuevas, “Molecular dynamics study of the thermopower of Ag, Au, and Pt nanocontacts,” Bulletin of the American Physical Society. 2011. link Times cited: 33 Abstract: Using molecular dynamics simulations of many junction stretc… read moreAbstract: Using molecular dynamics simulations of many junction stretching processes combined with tight-bindingbased electronic structure and transport calculations, we analyze the thermopower of silver (Ag), gold (Au), and platinum (Pt) atomic contacts. In all cases we observe that the thermopower vanishes on average within the standard deviation and that its fluctuations increase for a decreasing minimum cross section of the junctions. However, we find a suppression of the fluctuations of the thermopower for the s-valent metals Ag and Au, when the conductance originates from a single, perfectly transmitting channel. Essential features of the experimental results for Au, Ag, and copper (Cu) of Ludoph and van Ruitenbeek [Phys. Rev. B 59, 12290 (1999)], as yet unaddressed by atomistic studies, can hence be explained by considering the atomic and electronic structure at the disordered narrowest constriction of the contacts. For the multivalent metal Pt our calculations predict the fluctuations of the thermopower to be larger by one order of magnitude as compared to Ag and Au, and suppressions of the fluctuations as a function of the conductance are absent. Main features of our results are explained in terms of an extended single-level model. read less USED (high confidence) A. Pedersen, J. Berthet, and H. Jónsson, “Simulated Annealing with Coarse Graining and Distributed Computing,” Workshop on Applied Parallel Computin. 2010. link Times cited: 20 USED (high confidence) T. Anniyev et al., “Complementarity between high-energy photoelectron and L-edge spectroscopy for probing the electronic structure of 5d transition metal catalysts.,” Physical chemistry chemical physics : PCCP. 2010. link Times cited: 23 Abstract: We demonstrate the successful use of hard X-ray photoelectro… read moreAbstract: We demonstrate the successful use of hard X-ray photoelectron spectroscopy (HAXPES) for selectively probing the platinum partial d-density of states (DOS) in a Pt-Cu nanoparticle catalyst which shows activity superior to pure Pt towards the oxygen-reduction reaction (ORR). The information about occupied Pt d-band states was complemented by Pt L(2)-edge X-ray absorption near-edge spectroscopy (XANES), which probes unoccupied valence states. We found a significant electronic perturbation of the Pt projected d-DOS which was narrowed and shifted to higher binding energy compared to pure platinum. The effect of this electronic structure perturbation on the chemical properties of the nanoparticle surface is discussed in terms of the d-band model. We have thereby demonstrated that the combination of L-edge spectroscopy and HAXPES allows for an experimental derivation of the valence electronic structure in an element-specific way for 5d metal catalysts. read less USED (high confidence) A. Pedersen, G. Henkelman, J. Schiøtz, and H. Jónsson, “Long time scale simulation of a grain boundary in copper,” New Journal of Physics. 2009. link Times cited: 20 Abstract: A general, twisted and tilted, grain boundary in copper has … read moreAbstract: A general, twisted and tilted, grain boundary in copper has been simulated using the adaptive kinetic Monte Carlo method to study the atomistic structure of the non-crystalline region and the mechanism of annealing events that occur at low temperature. The simulated time interval spanned 67 μs at 135 K. Similar final configurations were obtained starting from different initial structures: (i) by bringing the two grains into contact without any intermediate layer, and (ii) by inserting an amorphous region between the grains. The results obtained were analyzed with a radial distribution function and a common neighbor analysis. Annealing events leading to lowering of the energy typically involved concerted displacement of several atoms—even as many as 10 atoms displaced by more than half an Ångström. Increased local icosahedral ordering is observed in the boundary layer, but local HCP coordination was also observed. In the final low-energy configurations, the thickness of the region separating the crystalline grains corresponds to just one atomic layer, in good agreement with reported experimental observations. The simulated system consists of 1307 atoms and atomic interactions were described using effective medium theory. read less USED (high confidence) N. Bailey, J. Schiøtz, A. Lemaître, and K. Jacobsen, “Avalanche size scaling in sheared three-dimensional amorphous solid.,” Physical review letters. 2007. link Times cited: 74 Abstract: We study the statistics of plastic rearrangement events in a… read moreAbstract: We study the statistics of plastic rearrangement events in a simulated amorphous solid at T=0. Events are characterized by the energy release and the "slip volume", the product of plastic strain and system volume. Their distributions for a given system size L appear to be exponential, but a characteristic event size cannot be inferred, because the mean values of these quantities increase as Lalpha with alpha approximately 3/2. In contrast with results obtained in 2D models, we do not see simply connected avalanches. The exponent suggests a fractal shape of the avalanches, which is also evidenced by the mean fractal dimension and participation ratio. read less USED (high confidence) J. S. Engbæk, J. Schiøtz, B. Dahl-Madsen, and S. Horch, “Atomic structure of screw dislocations intersecting the Au(111) surface : A combined scanning tunneling microscopy and molecular dynamics study,” Physical Review B. 2006. link Times cited: 19 Abstract: The atomic-scale structure of naturally occurring screw disl… read moreAbstract: The atomic-scale structure of naturally occurring screw dislocations intersecting a Au (cid:1) 111 (cid:2) surface has been investigated both experimentally by scanning tunneling microscopy (cid:1) STM (cid:2) and theoretically using molecular dynamics (cid:1) MD (cid:2) simulations. The step profiles of 166 dislocations were measured using STM. Many of them exhibit noninteger step-height plateaus with different widths. Clear evidence was found for the existence of two different populations at the surface with distinct (cid:1) narrowed or widened (cid:2) partial-splitting widths. All findings are fully confirmed by the MD simulations. The MD simulations extend the STM-, i.e., surface, investigation to the subsurface region. Due to this additional insight, we can explain the different partial-splitting widths as the result of the interaction between the partial dislocations and the surface. read less USED (high confidence) N. Bailey, J. Schiøtz, and K. Jacobsen, “Atomistic simulation study of the shear-band deformation mechanism in Mg-Cu metallic glasses,” Physical Review B. 2006. link Times cited: 81 Abstract: We have simulated plastic deformation of a model Mg-Cu metal… read moreAbstract: We have simulated plastic deformation of a model Mg-Cu metallic glass in order to study shear banding. In uniaxial tension, we find a necking instability occurs rather than shear banding. We can force the latter to occur by deforming in plane strain, forbidding the change of length in one of the transverse directions. Furthermore, in most of the simulations a notch is used to initiate shear bands, which lie at a 45 deg. angle to the tensile loading direction. The shear bands are characterized by the Falk and Langer local measure of plastic deformation D{sub min}{sup 2}, averaged here over volumes containing many atoms. The D{sub min}{sup 2} profile has a peak whose width is around 10 nm; this width is largely independent of the strain rate. Most of the simulations were, at least nominally, at 100 K, about T{sub g}/3 for this system. The development of the shear bands takes a few tens of ps, once plastic flow has started, more or less independent of strain rate. The shear bands can also be characterized using a correlation function defined in terms of D{sub min}{sup 2}, which, moreover, can detect incipient shear bands in cases where they do not fullymore » form. By averaging the kinetic energy over small regions, the local temperature can be calculated, and this is seen to be higher in the shear bands by about 50-100 K. Increases in temperature appear to initiate from interactions of the shear bands with the free surfaces and with each other, and are delayed somewhat with respect to the localization of plastic flow itself. We observe a slight decrease in density, up to 1%, within the shear band, which is consistent with notions of increased free volume or disorder within a plastically deforming amorphous material.« less read less USED (high confidence) J. Schiøtz and K. Jacobsen, “A Maximum in the Strength of Nanocrystalline Copper,” Science. 2003. link Times cited: 1262 Abstract: We used molecular dynamics simulations with system sizes up … read moreAbstract: We used molecular dynamics simulations with system sizes up to 100 million atoms to simulate plastic deformation of nanocrystalline copper. By varying the grain size between 5 and 50 nanometers, we show that the flow stress and thus the strength exhibit a maximum at a grain size of 10 to 15 nanometers. This maximum is because of a shift in the microscopic deformation mechanism from dislocation-mediated plasticity in the coarse-grained material to grain boundary sliding in the nanocrystalline region. The simulations allow us to observe the mechanisms behind the grain-size dependence of the strength of polycrystalline metals. read less USED (high confidence) S. Xu, J. Li, C. Li, and F. Chan, “IMMERSIVE VISUALISATION OF NANO-INDENTATION SIMULATION OF CU.” 2002. link Times cited: 2 Abstract: This paper introduces the immersive visualisation of nano-in… read moreAbstract: This paper introduces the immersive visualisation of nano-indentation simulation of Cu. The molecular dynamics simulation is performed in a system consisting of 326,592 atoms of size 18.6 × 17.0 × 18.4 nm, under periodic boundary conditions. Based on the coordination number calculation, a practical method for the extraction of “extraordinary” atoms is developed. The simulation has been visualised in the CAVE – an advanced full-immersive virtual environment. read less USED (high confidence) J. Schiøtz and A. E. Carlsson, “The influence of surface stress on dislocation emission from sharp and blunt cracks in fcc metals,” Philosophical Magazine A. 1997. link Times cited: 10 Abstract: We use computer simulations to study the behaviour of atomic… read moreAbstract: We use computer simulations to study the behaviour of atomically sharp and blunted cracks in various fcc metals. The simulations use effective medium potentials which contain many-body interactions. We find that when using potentials representing platinum and gold a sharp crack is stable with respect to the emission of a dislocation from the crack tip, whereas for all other metals studied the sharp crack is unstable. This result cannot be explained by existing criteria for the intrinsic ductile/brittle behaviour of crack tips, but is probably caused by surface stresses. When the crack is no longer atomically sharp dislocation emission becomes easier in all the studied metals. The effect is relatively strong; the critical stress intensity factor for emission to occur is reduced by up to 20%. This behaviour appears to be caused by the surface stress near the crack tip. The surface stress is a consequence of the many-body nature of the interatomic interactions. The enhanced dislocation emission can cause an order-of-magnitude increase in the fracture toughness of certain materials, in which a sharp crack would propagate by cleavage. Collisions with already existing dislocations will blunt the crack, if this prevents further propagation of the crack the toughness of the material is dramatically increased. read less USED (low confidence) S. Canepa et al., “Initiation and Progression of Anisotropic Galvanic Replacement Reactions in a Single Ag Nanowire: Implications for Nanostructure Synthesis,” ACS Applied Nano Materials. 2021. link Times cited: 5 USED (low confidence) G. Jang et al., “Microstructural evolution and mechanical properties of nanocrystalline Fe–Mn–Al–C steel processed by high-pressure torsion,” Materials Science and Engineering: A. 2021. link Times cited: 9 USED (low confidence) M. Kappeler, A. Marusczyk, and B. Ziebarth, “Simulation of nickel surfaces using ab-initio and empirical methods,” Materialia. 2020. link Times cited: 2 USED (low confidence) K. Sato, Q. Xu, and T. Yoshiie, “Simulation of the effect of volume size factor of solute atoms and their clusters on one dimensional motion of interstitial clusters in Ni binary alloys,” Computational Materials Science. 2017. link Times cited: 1 USED (low confidence) S. H. Brodersen, U. Grønbjerg, B. Hvolbæk, and J. Schiøtz, “Understanding the catalytic activity of gold nanoparticles through multi-scale simulations,” Journal of Catalysis. 2011. link Times cited: 63 USED (low confidence) A. Paduraru, U. G. Andersen, A. Thyssen, N. Bailey, K. Jacobsen, and J. Schiøtz, “Computer simulations of nanoindentation in Mg–Cu and Cu–Zr metallic glasses,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 17 Abstract: The formation of shear bands during plastic deformation of C… read moreAbstract: The formation of shear bands during plastic deformation of Cu0.50Zr0.50 and Mg0.85Cu0.15 metallic glasses is studied using atomic-scale computer simulations. The atomic interactions are described using realistic many-body potentials within the effective medium theory, and are compared with similar simulations using a Lennard-Jones description of the material. The metallic glasses are deformed both in simple shear and in a simulated nanoindentation experiment. Plastic shear localizes into shear bands with a width of approximately 5 nm in CuZr and 8 nm in MgCu. In simple shear, the shear band formation is very clear, whereas only incipient shear bands are seen in nanoindentation. The shear band formation during nanoindentation is sensitive to the indentation velocity, indenter radius and the cooling rate during the formation of the metallic glass. For comparison, a similar nanoindentation simulation was made with a nanocrystalline sample, showing how the presence of a polycrystalline structure leads to a different and more spatially distributed deformation pattern, where dislocation avalanches play an important role. read less USED (low confidence) M. Liao, M. Weng, S. Ju, and H.-J. Chiang, “Molecular Dynamics Simulation on the Nanoindentation Behavior of a Copper Bilayered Thin Film,” Chinese Journal of Catalysis. 2008. link Times cited: 7 USED (low confidence) K. Sato, T. Yoshiie, and Q. Xu, “One dimensional motion of interstitial clusters in Ni–Au alloy,” Journal of Nuclear Materials. 2007. link Times cited: 14 USED (low confidence) T. Fujii and Y. Akiniwa, “Molecular Dynamics Analysis for Deformation and Fracture Behavior of Copper Thin Films,” Key Engineering Materials. 2007. link Times cited: 3 Abstract: Molecular dynamics simulation is conducted to investigate th… read moreAbstract: Molecular dynamics simulation is conducted to investigate the effect of notch depth on the deformation and fracture behavior of a single crystal copper which is expected to a conductive material of micro-devices. In the stress – strain relationship, a normal stress increases with increasing applied strain. Then, the normal stress decreases rapidly. When the stress decreases, the dislocation emits from a notch root and the stacking fault is formed on the {111} plane, which is slip plane of the fcc crystalline structure. The maximum stress decreases with notch depth. The non-damaging defect size is quite small. The shear stress in the slip direction at dislocation emission is constant irrespective of the notch depth. The criterion of the dislocation emission is given by the critical value of the resolved shear stress in the sliding direction. read less USED (low confidence) K. Sato, T. Yoshiie, T. Ishizaki, and Q. Xu, “Behavior of vacancies near edge dislocations in Ni and α-Fe : Positron annihilation experiments and rate theory calculations,” Physical Review B. 2007. link Times cited: 44 USED (low confidence) S. Baud et al., “Comparative study of ab initio and tight-binding electronic structure calculations applied to platinum surfaces,” Physical Review B. 2004. link Times cited: 34 Abstract: We have applied the full-potential linearized augmented plan… read moreAbstract: We have applied the full-potential linearized augmented plane-wave (FLAPW) ab initio method and the $spd$ tight-binding (TB) model to the calculations of the surface energies ${E}_{S}(hkl)$ and relaxations of the three low-index [(111), (100), (110)] surfaces of platinum. The two methods give similar results, and in particular the anisotropy ratios ${E}_{S}(110)∕{E}_{S}(111)$ and ${E}_{S}(100)∕{E}_{S}(111)$ are very close. The calculation of surface energy of reconstructed $(1\ifmmode\times\else\texttimes\fi{}2)$ Pt(110) confirms that this face undergoes a missing-row reconstruction and the corresponding structural parameters agree well with experiment. The local densities of states (LDOS) calculated by each of the methods on the flat surfaces are almost the same. We have also investigated the $6(111)\ifmmode\times\else\texttimes\fi{}(\overline{1}11)$ vicinal surface and found a similar agreement for the LDOS. read less USED (low confidence) S. L. Frederiksen, K. Jacobsen, and J. Schiøtz, “Simulations of intergranular fracture in nanocrystalline molybdenum,” Acta Materialia. 2004. link Times cited: 72 USED (low confidence) J. Schiøtz, “Atomic-scale modeling of plastic deformation of nanocrystalline copper,” Scripta Materialia. 2004. link Times cited: 147 USED (low confidence) J. Schiøtz, “Strain-induced coarsening in nanocrystalline metals under cyclic deformation,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2004. link Times cited: 84 USED (low confidence) M. Dreher, F. Pauly, J. Heurich, J. Cuevas, E. Scheer, and P. Nielaba, “Structure and conductance histogram of atomic-sized Au contacts,” Physical Review B. 2004. link Times cited: 109 Abstract: Many experiments have shown that the conductance histograms … read moreAbstract: Many experiments have shown that the conductance histograms of metallic atomic-sized contacts exhibit a peak structure, which is characteristic of the corresponding material. The origin of these peaks still remains as an open problem. In order to shed some light on this issue, we present a theoretical analysis of the conductance histograms of Au atomic contacts. We have combined classical molecular dynamics simulations of the breaking of nanocontacts with conductance calculations based on a tight-binding model. This combination gives us access to crucial information such as contact geometries, forces, minimum cross section, total conductance, and transmission coefficients of the individual conduction channels. The ensemble of our results suggests that the low-temperature Au conductance histograms are a consequence of a subtle interplay between mechanical and electrical properties of these nanocontacts. At variance with other suggestions in the literature, our results indicate that the peaks in the Au conductance histograms are not a simple consequence of conductance quantization or the existence of exceptionally stable radii. We show that the main peak in the histogram close to one quantum of conductance is due to the formation of single-atom contacts and chains of gold atoms. Moreover, we present a detailed comparison with experimental results on Au atomic contacts where the individual channel transmissions have been determined. read less USED (low confidence) H. Swygenhoven, P. Derlet, and A. Frøseth, “Stacking fault energies and slip in nanocrystalline metals,” Nature Materials. 2004. link Times cited: 853 USED (low confidence) R. Huang, X. Ma, and Y. Wang, “Computational investigations of the monolayer heteroepitaxial growth of Au on Ni(110),” Surface Science. 2004. link Times cited: 5 USED (low confidence) N. Bailey, J. Schiøtz, and K. Jacobsen, “Simulation of Cu-Mg metallic glass: Thermodynamics and Structure,” Physical Review B. 2003. link Times cited: 61 Abstract: We have obtained effective medium theory interatomic potenti… read moreAbstract: We have obtained effective medium theory interatomic potential parameters suitable for studying Cu-Mg metallic glasses. We present thermodynamic and structural results from simulations of such glasses over a range of compositions. We have produced low-temperature configurations by cooling from the melt at as slow a rate as practical, using constant temperature and pressure molecular dynamics. During the cooling process we have carried out thermodynamic analyses based on the temperature dependence of the enthalpy and its derivative, the specific heat, from which the glass transition temperature may be determined. We have also carried out structural analyses using the radial distribution function (RDF) and common neighbor analysis (CNA). Our analysis suggests that the splitting of the second peak, commonly associated with metallic glasses, in fact. has little to do with the glass transition itself, but is simply a consequence of the narrowing of peaks associated with structural features present in the liquid state. In fact, the splitting temperature for the Cu-Cu RDF is well above T g . The CNA also highlights a strong similarity between the structure of the intermetallic alloys and the amorphous alloys of similar composition. We have also investigated the diffusivity in the supercooled regime. Its temperature dependence indicates fragile-liquid behavior, typical of binary metallic glasses. On the other hand, the relatively low specific-heat jump of around 1.5k B /atom indicates apparent strung-liquid behavior, but this can be explained by the width of the transition due to the high cooling rates. read less USED (low confidence) T. Vegge, T. Leffers, O. B. Pedersen, and K. Jacobsen, “Atomistic simulations of jog migration on extended screw dislocations,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2001. link Times cited: 12 USED (low confidence) D. Spišák, “Structure and energetics of Cu(100) vicinal surfaces,” Surface Science. 2001. link Times cited: 19 USED (low confidence) T. Vegge, “Atomistic simulations of screw dislocation cross slip in copper and nickel,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 2001. link Times cited: 18 USED (low confidence) A. Bilić, B. King, and D. J. O’connor, “EMBEDDED ATOM METHOD STUDY OF SURFACE ALLOYING OF Al ON Pd(001),” Surface Review and Letters. 1999. link Times cited: 0 Abstract: We have simulated the structure and energetics of thin films… read moreAbstract: We have simulated the structure and energetics of thin films created by the deposition of Al onto Pd(001). The study has been carried out within the semiempirical embedded atom method (EAM), utilizing a Pd–Al potential from the literature and two other alloy potentials generated from elemental potentials. Only one of the potentials reproduces the experimentally observed reconstruction. Problems with the construction and validity of the alloy potentials are highlighted. read less USED (low confidence) J. Strömquist, L. Bengtsson, M. Persson, and B. Hammer, “The dynamics of H absorption in and adsorption on Cu(111),” Surface Science. 1998. link Times cited: 101 USED (low confidence) J. Merikoski, I. Vattulainen, J. Heinonen, and T. Ala‐Nissila, “Effect of kinks and concerted diffusion mechanisms on mass transport and growth on stepped metal surfaces,” Surface Science. 1997. link Times cited: 58 USED (low confidence) T. Rasmussen, K. Jacobsen, T. Leffers, and O. B. Pedersen, “Atomic structure and energetics of constricted screw dislocations in copper,” Materials Science and Engineering A-structural Materials Properties Microstructure and Processing. 1997. link Times cited: 5 USED (low confidence) A. Marusczyk, S. Ramakers, M. Kappeler, P. Haremski, M. Wieler, and P. Lupetin, “Atomistic Simulation of Nickel Surface and Interface Properties.” 2021. link Times cited: 0 USED (low confidence) T. Shimokawa, T. Kinari, and S. Shintaku, “Relationship between Grain Boundary Structures and Mechanical Properties of Nanocrystalline Metals with Different Stacking Fault Energy Using Atomic Scale Computational Experiments,” Journal of The Society of Materials Science, Japan. 2008. link Times cited: 0 Abstract: Mechanical properties of nanocrystalline metals of which the… read moreAbstract: Mechanical properties of nanocrystalline metals of which the grainsize is below about 100nm are affected by the relationship betweenthe intergranular and intragranular deformation. Intragranulardeformation in nanocrystalline metals could be caused by the movementof perfect dislocations or Shockley's partial dislocations, hencestacking fault energy is one of the important value to control theinternal structure. Moreover, the proportion of the grain boundaryregion dramatically increases with grain size decreasing, hence it isalso important to investigate the effects of individual grain boundarystructures and the distribution of grain boundary characteristics onthe macroscopic mechanical properties of polycrystalline materials. Inthis study, we investigate deformation mechanism and mechanicalproperties of nanocrystalline Al and Cu that show the differentstacking fault energy using molecular dynamics simulations, and wealso consider the effects of grain boundary misorientationdistributions on the mechanical properties of such materials. read less USED (low confidence) T. Rasmussen, “Atomic Scale Simulation of Cross Slip and Screw Dislocation Dipole Annihilation,” MRS Proceedings. 1998. link Times cited: 1 Abstract: Atomistic simulations are used to study cross slip of a sing… read moreAbstract: Atomistic simulations are used to study cross slip of a single screw dislocation as well as screw dislocation dipole annihilation in Cu. A configuration space path techniquex is applied to determine, without presumptions about the saddle point, the minimum energy path of transition for cross slip. The cross slip process is that proposed by Friedel and Escaig, and the energy of the in-plane constriction initiating cross slip is determined. A minimum stable dipole height much smaller than previously inferred from experimental studies is found. Relaxed screw dislocation dipoles adopt a skew configuration due to the anisotropy of Cu. The path technique is applied to investigate annihilation of stable screw dislocation dipoles, and the energy barrier for annihilation as a function of dipole height is determined for both homogeneous and heterogeneous cross slip leading to the annihilation. The results might be used as quantitative input into meso-/macro-scopical modelling approaches which rely on parameters deduced from either simulation or experiment. read less NOT USED (low confidence) S. Han, S. Lysgaard, T. Vegge, and H. Hansen, “Rapid mapping of alloy surface phase diagrams via Bayesian evolutionary multitasking,” npj Computational Materials. 2023. link Times cited: 0 NOT USED (low confidence) D. Loevlie, B. Ferreira, and G. Mpourmpakis, “Demystifying the Chemical Ordering of Multimetallic Nanoparticles,” Accounts of Chemical Research. 2023. link Times cited: 1 Abstract: Conspectus Multimetallic nanoparticles (NPs) have highly tun… read moreAbstract: Conspectus Multimetallic nanoparticles (NPs) have highly tunable properties due to the synergy between the different metals and the wide variety of NP structural parameters such as size, shape, composition, and chemical ordering. The major problem with studying multimetallic NPs is that as the number of different metals increases, the number of possible chemical orderings (placements of different metals) for a NP of fixed size explodes. Thus, it becomes infeasible to explore NP energetic differences with highly accurate computational methods, such as density functional theory (DFT), which has a high computational cost and is typically applied to up to a couple of hundred metal atoms. Here, we demonstrate a methodology advancing NP simulations by effectively exploring the vast materials space of multimetallic NPs and accurately identifying the ones with the most thermodynamically preferred chemical orderings. With accuracies reaching that of DFT, our methodology is applicable to practically any NP size, shape, and metal composition. We achieve this by significantly advancing the bond-centric (BC) model, a physics-based model that has been previously shown to rapidly predict bimetallic NP cohesive energies (CEs). Specifically, the BC model is trained in a way to understand how the bimetallic bond strength changes under different coordination environments present on a NP and how the metal composition of every site affects the detailed coordination environment using fractional coordination numbers. This newly modified BC model leads to an improvement from 0.331 (original model) to 0.089 eV/atom in CE predictions when compared to DFT values on a robust data set of 90 different NPs consisting of PtPd, AuPt, and AuPd NPs with varying compositions and chemical orderings. By incorporating the modified BC model into an in-house-developed genetic algorithm (GA) we can effectively and accurately predict the most stable chemical orderings of large, realistic bimetallic NPs consisting of thousands of metal atoms. This is demonstrated on AuPd bimetallic NPs, a challenging system due to the similarity in the cohesion of the two metals. By training our BC model using a unique DFT calculation on a bimetallic NP (one calculation for two metals combining together), we expand to explore the chemical ordering of multimetallic NPs. We first demonstrate the application of our methodology on a AuPdPt NP and validate our stability predictions with literature data. Then, we effectively explore the vast materials space of multimetallic NPs consisting of combinations of Au, Pt, and Pd as a function of metal composition. Our thermodynamic stability trends are presented in a ternary diagram revealing detailed, and yet, unexpected chemical ordering trends. Our computational framework can aid both experimental and computational researchers toward effectively screening multimetallic NP stability. Moreover, we provide an outlook of how this framework can be applied to catalyst discovery, high-entropy alloys, and single-atom alloys. read less NOT USED (low confidence) A. Farahvash, M. Agrawal, A. Peterson, and A. Willard, “Modeling Surface Vibrations and Their Role in Molecular Adsorption: A Generalized Langevin Approach.,” Journal of chemical theory and computation. 2023. link Times cited: 0 Abstract: The atomic vibrations of a solid surface can play a signific… read moreAbstract: The atomic vibrations of a solid surface can play a significant role in the reactions of surface-bound molecules, as well as their adsorption and desorption. Relevant phonon modes can involve the collective motion of atoms over a wide array of length scales. In this paper, we demonstrate how the generalized Langevin equation can be utilized to describe these collective motions weighted by their coupling to individual sites. Our approach builds upon the generalized Langevin oscillator (GLO) model originally developed by Tully. We extend the GLO by deriving parameters from atomistic simulation data. We apply this approach to study the memory kernel of a model platinum surface and demonstrate that the memory kernel has a bimodal form due to coupling to both low-energy acoustic modes and high-energy modes near the Debye frequency. The same bimodal form was observed across a wide variety of solids of different elemental compositions, surface structures, and solvation states. By studying how these dominant modes depend on the simulation size, we argue that the acoustic modes are frozen in the limit of macroscopic lattices. By simulating periodically replicated slabs of various sizes, we quantify the influence of phonon confinement effects in the memory kernel and their concomitant effect on simulated sticking coefficients. read less NOT USED (low confidence) H. Deng, J. Comer, and B. Liu, “A high-dimensional neural network potential for molecular dynamics simulations of condensed phase nickel and phase transitions,” Molecular Simulation. 2022. link Times cited: 0 Abstract: ABSTRACT A high-dimensional neural network interatomic poten… read moreAbstract: ABSTRACT A high-dimensional neural network interatomic potential was developed and used in molecular dynamics simulations of condensed phase Ni and Ni systems with liquid–solid phase coexistence. The reference data set was generated by sampling the potential energy surface over a broad temperature-pressure domain using ab initio MD simulations to train a unified potential. Excellent agreement was achieved between bulk face-centred cubic nickel thermal expansion simulations and relevant experimental data. The same potential also yields accurate structures and diffusivities in the liquid state. The phase transition between liquid and solid phases was simulated using the two-phase interface method. The predicted melting point temperature is within a few kelvins of the literature value. The general methodology could be applied to describe crystals with much more complex phase behaviours. read less NOT USED (low confidence) J. I. Garcia-Peiro, J. Bonet-Aleta, J. Santamaría, and J. L. Hueso, “Platinum nanoplatforms: classic catalysts claiming a prominent role in cancer therapy.,” Chemical Society reviews. 2022. link Times cited: 9 Abstract: Platinum nanoparticles (Pt NPs) have a well-established role… read moreAbstract: Platinum nanoparticles (Pt NPs) have a well-established role as a classic heterogeneous catalyst. Also, Pt has traditionally been employed as a component of organometallic drug formulations for chemotherapy. However, a new role in cancer therapy is emerging thanks to its outstanding catalytic properties, enabling novel approaches that are surveyed in this review. Herein, we critically discuss results already obtained and attempt to ascertain future perspectives for Pt NPs as catalysts able to modify key processes taking place in the tumour microenvironment (TME). In addition, we explore relevant parameters affecting the cytotoxicity, biodistribution and clearance of Pt nanosystems. We also analyze pros and cons in terms of biocompatibility and potential synergies that emerge from combining the catalytic capabilities of Pt with other agents such as co-catalysts, external energy sources (near-infrared light, X-ray, electric currents) and conventional therapies. read less NOT USED (low confidence) A. Medford, P. G. Moses, K. Jacobsen, and A. Peterson, “A Career in Catalysis: Jens Kehlet Nørskov,” ACS Catalysis. 2022. link Times cited: 14 NOT USED (low confidence) Y. Wang, Y. Liang, T. Bo, S. Meng, and M. Liu, “Orbital Dependence in Single-Atom Electrocatalytic Reactions.,” The journal of physical chemistry letters. 2022. link Times cited: 14 Abstract: Transition metal single-atom catalysts supported on N-doped … read moreAbstract: Transition metal single-atom catalysts supported on N-doped graphene (TM-N-C) could serve as an ideal model for studying orbital dependence in electrocatalytic reactions because the atom on the catalytic active site has discrete single-atom-like orbitals. In this work, the catalytic efficiency of Fe-N-C for the oxygen evolution reaction (OER) under a small structural perturbation has been comprehensively investigated with density functional theory calculations. The results suggest that the subtle local environment of a single atom can significantly modulate the catalytic reactivity. Further analysis demonstrates that the energy level of the TM dz2 orbital center, rather than the d-band center, is responsible for the OER catalytic efficiency as the dz2 orbital participates mainly in the reactions. Essentially, the d-band theory can be extended to the sub-d orbital level, and a small perturbation of the crystal field, induced by lattice strain or z-direction displacement of the TM atom, can prominently change the sub-d orbital associated with the reaction and in turn affect the catalytic activity. read less NOT USED (low confidence) L. Avakyan et al., “Ultimate sensitivity of radial distribution functions to architecture of PtCu bimetallic nanoparticles,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) A. Mirzoev, B. Gelchinski, and A. A. Rempel, “Neural Network Prediction of Interatomic Interaction in Multielement Substances and High-Entropy Alloys: A Review,” Doklady Physical Chemistry. 2022. link Times cited: 2 NOT USED (low confidence) J. Yoon et al., “Deep reinforcement learning for predicting kinetic pathways to surface reconstruction in a ternary alloy,” Machine Learning: Science and Technology. 2021. link Times cited: 9 Abstract: The majority of computational catalyst design focuses on the… read moreAbstract: The majority of computational catalyst design focuses on the screening of material components and alloy composition to optimize selectivity and activity for a given reaction. However, predicting the metastability of the alloy catalyst surface at realistic operating conditions requires an extensive sampling of possible surface reconstructions and their associated kinetic pathways. We present CatGym, a deep reinforcement learning (DRL) environment for predicting the thermal surface reconstruction pathways and their associated kinetic barriers in crystalline solids under reaction conditions. The DRL agent iteratively changes the positions of atoms in the near-surface region to generate kinetic pathways to accessible local minima involving changes in the surface compositions. We showcase our agent by predicting the surface reconstruction pathways of a ternary Ni3Pd3Au2(111) alloy catalyst. Our results show that the DRL agent can not only explore more diverse surface compositions than the conventional minima hopping method, but also generate the kinetic surface reconstruction pathways. We further demonstrate that the kinetic pathway to a global minimum energy surface composition and its associated transition state predicted by our agent is in good agreement with the minimum energy path predicted by nudged elastic band calculations. read less NOT USED (low confidence) J. A. Gauthier, J. Stenlid, F. Abild-Pedersen, M. Head‐Gordon, and A. Bell, “The Role of Roughening to Enhance Selectivity to C2+ Products during CO2 Electroreduction on Copper,” ACS Energy Letters. 2021. link Times cited: 24 NOT USED (low confidence) I. M. P. Espinosa, T. Jacobs, and A. Martini, “Evaluation of Force Fields for Molecular Dynamics Simulations of Platinum in Bulk and Nanoparticle Forms.,” Journal of chemical theory and computation. 2021. link Times cited: 7 Abstract: Understanding the size- and shape-dependent properties of pl… read moreAbstract: Understanding the size- and shape-dependent properties of platinum nanoparticles is critical for enabling the design of nanoparticle-based applications with optimal and potentially tunable functionality. Toward this goal, we evaluated nine different empirical potentials with the purpose of accurately modeling faceted platinum nanoparticles using molecular dynamics simulation. First, the potentials were evaluated by computing bulk and surface properties-surface energy, lattice constant, stiffness constants, and the equation of state-and comparing these to prior experimental measurements and quantum mechanics calculations. Then, the potentials were assessed in terms of the stability of cubic and icosahedral nanoparticles with faces in the {100} and {111} planes, respectively. Although none of the force fields predicts all the evaluated properties with perfect accuracy, one potential-the embedded atom method formalism with a specific parameter set-was identified as best able to model platinum in both bulk and nanoparticle forms. read less NOT USED (low confidence) M. Liu, Y. Yang, and J. Kitchin, “Semi-grand canonical Monte Carlo simulation of the acrolein induced surface segregation and aggregation of AgPd with machine learning surrogate models.,” The Journal of chemical physics. 2021. link Times cited: 7 Abstract: The single atom alloy of AgPd has been found to be a promisi… read moreAbstract: The single atom alloy of AgPd has been found to be a promising catalyst for the selective hydrogenation of acrolein. It is also known that the formation of Pd islands on the surface will greatly reduce the selectivity of the reaction. As a result, the surface segregation and aggregation of Pd on the AgPd surface under reaction conditions of selective hydrogenation of acrolein are of great interest. In this work, we lay out a workflow that can predict the surface segregation and aggregation of Pd on a FCC(111) AgPd surface with and without the presence of acrolein. We use machine learning surrogate models to predict the AgPd bulk energy, AgPd slab energy, and acrolein adsorption energy on AgPd slabs. Then, we use the semi-grand canonical Monte Carlo simulation to predict the surface segregation and aggregation under different bulk Pd concentrations. Under vacuum conditions, our method predicts that only trace amount of Pd will exist on the surface at Pd bulk concentrations less than 20%. However, with the presence of acrolein, Pd will start to aggregate as dimers on the surface at Pd bulk concentrations as low as 6.5%. read less NOT USED (low confidence) G. H. Gu et al., “Autobifunctional Mechanism of Jagged Pt Nanowires for Hydrogen Evolution Kinetics via End-to-End Simulation.,” Journal of the American Chemical Society. 2021. link Times cited: 21 Abstract: The extraordinary mass activity of jagged Pt nanowires can s… read moreAbstract: The extraordinary mass activity of jagged Pt nanowires can substantially improve the economics of the hydrogen evolution reaction (HER). However, it is a great challenge to fully unveil the HER kinetics driven by the jagged Pt nanowires with their multiscale morphology. Herein we present an end-to-end framework that combines experiment, machine learning, and multiscale advances of the past decade to elucidate the HER kinetics catalyzed by jagged Pt nanowires under alkaline conditions. The bifunctional catalysis conventionally refers to the synergistic increase in activity by the combination of two different catalysts. We report that monometals, such as jagged Pt nanowires, can exhibit bifunctional characteristics owing to its complex surface morphology, where one site prefers electrochemical proton adsorption and another is responsible for activation, resulting in a 4-fold increase in the activity. We find that the conventional design guideline that the sites with a 0 eV Gibbs free energy of adsorption are optimal for HER does not hold under alkaline conditions, and rather, an energy between -0.2 and 0.0 eV is shown to be optimal. At the reaction temperatures, the high activity arises from low-coordination-number (≤7) Pt atoms exposed by the jagged surface. Our current demonstration raises an emerging prospect to understand highly complex kinetic phenomena on the nanoscale in full by implementing end-to-end multiscale strategies. read less NOT USED (low confidence) P. Acar, “Recent progress of uncertainty quantification in small-scale materials science,” Progress in Materials Science. 2020. link Times cited: 16 NOT USED (low confidence) M. Liu and J. Kitchin, “SingleNN: Modified Behler–Parrinello Neural Network with Shared Weights for Atomistic Simulations with Transferability,” Journal of Physical Chemistry C. 2020. link Times cited: 18 Abstract: In this article, we introduce the SingleNN, which is a modif… read moreAbstract: In this article, we introduce the SingleNN, which is a modified version of the Behler–Parrinello Neural Network (BPNN) where the neural networks for the prediction of atomic energy for different el... read less NOT USED (low confidence) C. B. Saltonstall et al., “Complexion dictated thermal resistance with interface density in reactive metal multilayers,” Physical Review B. 2020. link Times cited: 5 NOT USED (low confidence) J. Dean, M. J. Cowan, J. Estes, M. Ramadan, and G. Mpourmpakis, “Rapid Prediction of Bimetallic Mixing Behavior at the Nanoscale.,” ACS nano. 2020. link Times cited: 12 Abstract: The nanoparticle (NP) design space allows for variations in … read moreAbstract: The nanoparticle (NP) design space allows for variations in size, shape, composition, and chemical ordering. In the search for low-energy structures, this results in an extremely large search space which cannot be screened by brute force methods. In this work, we develop a genetic algorithm to predict stable bimetallic NPs of any size, shape, and metal composition. Our method predicts nanostructures in agreement with experimental trends and it captures the detailed chemical ordering of an experimental 23,196-atom FePt NP with nearly atom-by-atom accuracy. Our developed screening process is extremely fast, allowing us to generate and analyze a database of 5,454 low-energy bimetallic NPs. By identifying thermodynamically stable NPs, we rationalize bimetallic mixing at the nanoscale and reveal metal-, size-, and temperature-dependent mixing behavior. Importantly, our method is applicable to any bimetallic NP size, bridging the materials gap in nanoscale simulations, and guides experimentation in the lab by elucidating stability, mixing, and detailed chemical ordering behavior of bimetallic NPs. read less NOT USED (low confidence) J. Nevalaita and P. Koskinen, “Free-standing 2D metals from binary metal alloys,” AIP Advances. 2020. link Times cited: 10 Abstract: Recent experiment demonstrated the formation of free-standin… read moreAbstract: Recent experiment demonstrated the formation of free-standing Au monolayers by exposing Au-Ag alloy to electron beam irradiation. Inspired by this discovery, we used semi-empirical effective medium theory simulations to investigate monolayer formation in 30 different binary metal alloys composed of late d-series metals Ni, Cu, Pd, Ag, Pt, and Au. In qualitative agreement with the experiment, we find that the beam energy required to dealloy Ag atoms from Au-Ag alloy is smaller than the energy required to break the dealloyed Au monolayer. Our simulations suggest that similar method could also be used to form Au monolayers from Au-Cu alloy and Pt monolayers from Pt-Cu, Pt-Ni, and Pt-Pd alloys. read less NOT USED (low confidence) M. Rueck, B. Garlyyev, F. Mayr, A. Bandarenka, and A. Gagliardi, “Oxygen Reduction Activities of Strained Platinum Core-Shell Electrocatalysts Predicted by Machine Learning.,” The journal of physical chemistry letters. 2020. link Times cited: 19 Abstract: Core-shell nanocatalyst activities are chiefly controlled by… read moreAbstract: Core-shell nanocatalyst activities are chiefly controlled by bimetallic material composition, shell thickness, and nanoparticle size. We present a machine learning framework predicting strain with site-specific precision to rationalize how strain on Pt core-shell nanocatalysts can enhance oxygen reduction activities. Large compressive strain on Pt@Cu and Pt@Ni induces optimal mass activities at 1.9 nm nanoparticle size. It is predicted that bimetallic Pt@Au and Pt@Ag have best mass activities at 2.8 nm, where active sites are exposed to weak compressive strain. We demonstrate that optimal strain depends on the nanoparticle size: for instance, strengthening compressive strain on 1.92 nm sized Pt@Cu and Pt@Ni, or weakening compressive strain on 2.83 nm sized Pt@Ag and Pt@Au, can lead to further enhanced mass activities. read less NOT USED (low confidence) P. Zhang, H. Xue, and N. Suen, “Intermetallic compounds with high hydrogen evolution reaction performance: a case study of a MCo2 (M = Ti, Zr, Hf and Sc) series.,” Chemical communications. 2019. link Times cited: 11 Abstract: Noble metals (e.g., Ru, Ir and Pt) or their derivatives exhi… read moreAbstract: Noble metals (e.g., Ru, Ir and Pt) or their derivatives exhibit very appealing activity toward the hydrogen evolution reaction (HER), but their high price and low reserves impede their wide use. Herein, we propose a strategy in which, through the manipulation of crystal and electronic structure, one can convert a common metal to have a Pt-like performance for HER. To achieve this goal, a series of MCo2 (M = Ti, Zr, Hf and Sc) has been synthesized by using a rapid arc-melting method. TiCo2 exhibits comparable HER activity to that of Pt/C, for which it requires only -70 mV (V vs. RHE) to reach 10 mA cm-2 with a Tafel slope of 33 mV decade-1 in 1.0 M KOH. Moreover, X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) indicate that the lower adsorption energy (ΔGH*) of H on the Co atom in TiCo2, due to the change in Co electronic state, is another key factor to account for its high HER activity. This case study offers a good illustration of how to transform a non-noble metal so it behaves like a noble metal toward HER and can potentially be applied under other conditions. read less NOT USED (low confidence) P. G. Lindgren, G. Kastlunger, and A. Peterson, “Scaled and Dynamic Optimizations of Nudged Elastic Bands.,” Journal of chemical theory and computation. 2019. link Times cited: 17 Abstract: We present a modified nudged elastic band routine that can r… read moreAbstract: We present a modified nudged elastic band routine that can reduce the number of force calls by more than 50% for bands with nonuniform convergence. The method, which we call "dyNEB", dynamically and selectively optimizes images on the basis of the perpendicular PES-derived forces and parallel spring forces acting on that region of the band. The convergence criteria are scaled to focus on the region of interest, i.e., the saddle point, while maintaining continuity of the band and avoiding truncation. We show that this method works well for solid state reaction barriers-nonelectrochemical in general and electrochemical in particular-and that the number of force calls can be significantly reduced without loss of resolution at the saddle point. read less NOT USED (low confidence) L. T. Roling, T. S. Choksi, and F. Abild-Pedersen, “A coordination-based model for transition metal alloy nanoparticles.,” Nanoscale. 2019. link Times cited: 25 Abstract: We present a simple approach for predicting the relative ene… read moreAbstract: We present a simple approach for predicting the relative energies of bimetallic nanoparticles spanning a wide-ranging combinatorial space, using only the identity and nearest-neighbor coordination number of individual metal atoms as independent parameters. By performing straightforward metal atom adsorption calculations on surface slab models, we parameterize expressions for the energy of metal atoms as a function of their coordination number in 21 bimetallic pairings of fcc metals. We rigorously establish the transferability of our model by predicting relative energies of a series of nanoparticles across a large number of morphologies, sizes, atomic compositions, and arrangements. The model is particularly accurate in predicting atomic rearrangements at or near the metal surfaces, which is essential for its potential applications when studying segregation phenomena or dynamic processes in heterogeneous catalysis. By rapidly forecasting site stabilities with atomic specificity across generic structural and compositional features, our model is able to reverse engineer thermodynamically feasible motifs of active sites in bimetallic nanoparticles through robust property ⇔ structure relations. read less NOT USED (low confidence) P. Schlexer, A. Andersen, B. Sebők, I. Chorkendorff, J. Schiøtz, and T. Hansen, “Size‐Dependence of the Melting Temperature of Individual Au Nanoparticles,” Particle & Particle Systems Characterization. 2019. link Times cited: 30 Abstract: Nanoparticles have an immense importance in various fields, … read moreAbstract: Nanoparticles have an immense importance in various fields, such as medicine, catalysis, and various technological applications. Nanoparticles exhibit a significant depression in melting point as their size goes below ≈10 nm. However, nanoparticles are frequently used in high temperature applications such as catalysis where temperatures often exceed several 100 degrees which makes it interesting to study not only the melting temperature depression, but also how the melting progresses through the particle. Using high‐resolution transmission electron microscopy, the melting process of gold nanoparticles in the size range of 2–20 nm Au nanoparticles combined with molecular dynamics studies is investigated. A linear dependence of the melting temperature on the inverse particle size is confirmed; electron microscopy imaging reveals that the particles start melting at the surface and the liquid shell formed then rapidly expands to the particle core. read less NOT USED (low confidence) L. Friedeheim, J. Dyre, and N. Bailey, “Hidden scale invariance at high pressures in gold and five other face-centered-cubic metal crystals.,” Physical review. E. 2018. link Times cited: 9 Abstract: Recent density functional theory simulations showed that met… read moreAbstract: Recent density functional theory simulations showed that metals have a hitherto overlooked symmetry termed "hidden scale invariance" [Hummel et al., Phys. Rev. B 92, 174116 (2015)PRBMDO1098-012110.1103/PhysRevB.92.174116]. This scaling property implies the existence of lines in the thermodynamic phase diagram, so-called isomorphs, along which structure and dynamics are invariant to a good approximation when given in properly reduced units. This means that the phase diagram becomes effectively one-dimensional with regard to several physical properties. This paper investigates consequences and implications of the isomorph theory in six metallic crystals: Au, Ni, Cu, Pd, Ag, and Pt. The data are obtained from molecular dynamics simulations employing many-body effective medium theory (EMT) to model the atomic interactions realistically. We test the predictions from isomorph theory for structure and dynamics by means of the radial distribution and the velocity autocorrelation functions, as well as the prediction of instantaneous equilibration after a jump between two isomorphic state points. Many properties of crystals tend to be dominated by defects, and many of the properties associated with these defects are expected to be isomorph invariant as well. This is investigated in this paper for the case of vacancy diffusion. In regard to the perfect crystal properties, we find the predicted invariance of structure and also, though less perfectly, of dynamics. We show results on the variation of the density-scaling exponent γ, which can be related to the Grüneisen parameter, for all six metals. We consider large density changes up to a factor of two, corresponding to very high pressures. Unlike systems modeled using the Lennard-Jones potential where the density-scaling exponent γ is almost constant, this quantity varies substantially when using the EMT potential and is also strongly material dependent. read less NOT USED (low confidence) G.-U. Jeong, C. S. Park, H.-S. Do, S.-M. Park, and B.-J. Lee, “Second nearest-neighbor modified embedded-atom method interatomic potentials for the Pd-M (M = Al, Co, Cu, Fe, Mo, Ni, Ti) binary systems,” Calphad. 2018. link Times cited: 12 NOT USED (low confidence) V. Pryadchenko et al., “Effect of Thermal Treatment on the Atomic Structure and Electrochemical Characteristics of Bimetallic PtCu Core–Shell Nanoparticles in PtCu/C Electrocatalysts,” The Journal of Physical Chemistry C. 2018. link Times cited: 16 Abstract: Supported PtCu/C electrocatalysts containing core–shell bime… read moreAbstract: Supported PtCu/C electrocatalysts containing core–shell bimetallic PtCu nanoparticles were synthesized by sequential chemical reduction of Cu2+ and Pt(IV) in a carbon suspension, prepared on the basis of ethylene glycol–water solvent, and then treated at different temperatures in the range from 250 to 350 °C. The structural characterization of “as-prepared” PtCu nanoparticles and of those obtained after the thermal treatments was performed by transmission electron microscopy, X-ray diffraction, and Pt L3- and Cu K-edge extended X-ray absorption fine structure spectroscopy. The atomic cluster models of PtCu nanoparticles before and after the thermal treatment, reflecting the character of the components’ distribution, were generated. The electrochemical performance of the obtained PtCu/C electrocatalysts in oxygen reduction reaction was studied by cycling and linear voltammetry. read less NOT USED (low confidence) E. L. Kolsbjerg, A. Peterson, and B. Hammer, “Neural-network-enhanced evolutionary algorithm applied to supported metal nanoparticles,” Physical Review B. 2018. link Times cited: 65 NOT USED (low confidence) A. Garden, A. Pedersen, and H. Jónsson, “Reassignment of ’magic numbers’ for Au clusters of decahedral and FCC structural motifs.,” Nanoscale. 2018. link Times cited: 18 Abstract: Calculations of low free energy structures of gold clusters … read moreAbstract: Calculations of low free energy structures of gold clusters with a few hundred to a few thousand atoms have been performed. For the study to be computationally feasible, a systematic scheme for generating low energy atomic structures with a given structural motif and a chosen size is used. Comparison of the relative stability of clusters with the decahedral and FCC motifs reveals periodic windows of stability for asymmetric, open-shell FCC clusters while the stability of the decahedral structural motif shows a smoother variation in stability with cluster size. As a result, the FCC structural motif is most stable for clusters with around 440, 610, 800, 1050… atoms, while the decahedral motif is more stable for atomic numbers of around 525, 705, 925, 1175… These new 'magic numbers' represent regions around which a number of asymmetric, open shell clusters of a given motif are most stable, in contrast to the discrete set of highly symmetric magic size structures that are commonly cited. As temperature is raised, the decahedral motif gains stability over the FCC motif. These results help explain reported experimental observations and can guide future laboratory preparations of shape selected clusters. read less NOT USED (low confidence) K. Lauritzen, A. Magyarkuti, Z. Balogh, A. Halbritter, and G. Solomon, “Classification of conductance traces with recurrent neural networks.,” The Journal of chemical physics. 2018. link Times cited: 19 Abstract: We present a new automated method for structural classificat… read moreAbstract: We present a new automated method for structural classification of the traces obtained in break junction experiments. Using recurrent neural networks trained on the traces of minimal cross-sectional area in molecular dynamics simulations, we successfully separate the traces into two classes: point contact or nanowire. This is done without any assumptions about the expected features of each class. The trained neural network is applied to experimental break junction conductance traces, and it separates the classes as well as the previously used experimental methods. The effect of using partial conductance traces is explored, and we show that the method performs equally well using full or partial traces (as long as the trace just prior to breaking is included). When only the initial part of the trace is included, the results are still better than random chance. Finally, we show that the neural network classification method can be used to classify experimental conductance traces without using simulated results for training, but instead training the network on a few representative experimental traces. This offers a tool to recognize some characteristic motifs of the traces, which can be hard to find by simple data selection algorithms. read less NOT USED (low confidence) J.-S. Kim, D. Seol, J. Ji, H.-S. Jang, Y. Kim, and B.-J. Lee, “Second nearest-neighbor modified embedded-atom method interatomic potentials for the Pt-M (M = Al, Co, Cu, Mo, Ni, Ti, V) binary systems,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2017. link Times cited: 31 NOT USED (low confidence) X. Li, T. Gao, C. Jiang, and R. Huang, “Surface Alloying of Au Atoms on a Ni (110) Surface: a Theoretical Study,” IOP Conference Series: Materials Science and Engineering. 2017. link Times cited: 1 Abstract: We investigated the surface alloying of Au atoms on a Ni (11… read moreAbstract: We investigated the surface alloying of Au atoms on a Ni (110) surface using a molecular dynamics (MD) method. It is found that the concentration of deposited Au atoms and the depositing temperature determine the topological surface structures, which can be ascribed to the variation in Gibbs free energy of the system. Dimer formation on the over layer is used to explain the surface alloying of the miscible system and in good agreement with the experimental results. read less NOT USED (low confidence) M. Kammler, S. Janke, A. Kandratsenka, and A. Wodtke, “Genetic algorithm approach to global optimization of the full-dimensional potential energy surface for hydrogen atom at fcc-metal surfaces,” Chemical Physics Letters. 2017. link Times cited: 13 NOT USED (low confidence) A. H. Larsen et al., “The atomic simulation environment-a Python library for working with atoms.,” Journal of physics. Condensed matter : an Institute of Physics journal. 2017. link Times cited: 1824 Abstract: The atomic simulation environment (ASE) is a software packag… read moreAbstract: The atomic simulation environment (ASE) is a software package written in the Python programming language with the aim of setting up, steering, and analyzing atomistic simulations. In ASE, tasks are fully scripted in Python. The powerful syntax of Python combined with the NumPy array library make it possible to perform very complex simulation tasks. For example, a sequence of calculations may be performed with the use of a simple 'for-loop' construction. Calculations of energy, forces, stresses and other quantities are performed through interfaces to many external electronic structure codes or force fields using a uniform interface. On top of this calculator interface, ASE provides modules for performing many standard simulation tasks such as structure optimization, molecular dynamics, handling of constraints and performing nudged elastic band calculations. read less NOT USED (low confidence) A. Peterson, R. Christensen, and A. Khorshidi, “Addressing uncertainty in atomistic machine learning.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 99 Abstract: Machine-learning regression has been demonstrated to precise… read moreAbstract: Machine-learning regression has been demonstrated to precisely emulate the potential energy and forces that are output from more expensive electronic-structure calculations. However, to predict new regions of the potential energy surface, an assessment must be made of the credibility of the predictions. In this perspective, we address the types of errors that might arise in atomistic machine learning, the unique aspects of atomistic simulations that make machine-learning challenging, and highlight how uncertainty analysis can be used to assess the validity of machine-learning predictions. We suggest this will allow researchers to more fully use machine learning for the routine acceleration of large, high-accuracy, or extended-time simulations. In our demonstrations, we use a bootstrap ensemble of neural network-based calculators, and show that the width of the ensemble can provide an estimate of the uncertainty when the width is comparable to that in the training data. Intriguingly, we also show that the uncertainty can be localized to specific atoms in the simulation, which may offer hints for the generation of training data to strategically improve the machine-learned representation. read less NOT USED (low confidence) J. Wang, L. Niu, and Y. Zhang, “Ab initio study of He-He interactions in homogeneous electron gas,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2017. link Times cited: 2 NOT USED (low confidence) A. Khorshidi and A. Peterson, “Amp: A modular approach to machine learning in atomistic simulations,” Comput. Phys. Commun. 2016. link Times cited: 270 NOT USED (low confidence) S. Malekie, F. Ziaie, and A. Esmaeli, “Study on dosimetry characteristics of polymer–CNT nanocomposites: Effect of polymer matrix,” Nuclear Instruments & Methods in Physics Research Section A-accelerators Spectrometers Detectors and Associated Equipment. 2016. link Times cited: 24 NOT USED (low confidence) S. Lysgaard, J. S. G. Mýrdal, H. Hansen, and T. Vegge, “A DFT-based genetic algorithm search for AuCu nanoalloy electrocatalysts for CO₂ reduction.,” Physical chemistry chemical physics : PCCP. 2015. link Times cited: 58 Abstract: Using a DFT-based genetic algorithm (GA) approach, we have d… read moreAbstract: Using a DFT-based genetic algorithm (GA) approach, we have determined the most stable structure and stoichiometry of a 309-atom icosahedral AuCu nanoalloy, for potential use as an electrocatalyst for CO2 reduction. The identified core-shell nano-particle consists of a copper core interspersed with gold atoms having only copper neighbors and a gold surface with a few copper atoms in the terraces. We also present an adsorbate-dependent correction scheme, which enables an accurate determination of adsorption energies using a computationally fast, localized LCAO-basis set. These show that it is possible to use the LCAO mode to obtain a realistic estimate of the molecular chemisorption energy for systems where the computation in normal grid mode is not computationally feasible. These corrections are employed when calculating adsorption energies on the Cu, Au and most stable mixed particles. This shows that the mixed Cu135@Au174 core-shell nanoalloy has a similar adsorption energy, for the most favorable site, as a pure gold nano-particle. Cu, however, has the effect of stabilizing the icosahedral structure because Au particles are easily distorted when adding adsorbates. read less NOT USED (low confidence) N. Bailey et al., “RUMD: A general purpose molecular dynamics package optimized to utilize GPU hardware down to a few thousand particles,” arXiv: Computational Physics. 2015. link Times cited: 83 Abstract: RUMD is a general purpose, high-performance molecular dynami… read moreAbstract: RUMD is a general purpose, high-performance molecular dynamics (MD) simulation package running on graphical processing units (GPU's). RUMD addresses the challenge of utilizing the many-core nature of modern GPU hardware when simulating small to medium system sizes (roughly from a few thousand up to hundred thousand particles). It has a performance that is comparable to other GPU-MD codes at large system sizes and substantially better at smaller sizes.RUMD is open-source and consists of a library written in C++ and the CUDA extension to C, an easy-to-use Python interface, and a set of tools for set-up and post-simulation data analysis. The paper describes RUMD's main features, optimizations and performance benchmarks. read less NOT USED (low confidence) M. M. Melander, K. Laasonen, and H. Jónsson, “Removing External Degrees of Freedom from Transition-State Search Methods using Quaternions.,” Journal of chemical theory and computation. 2015. link Times cited: 23 Abstract: In finite systems, such as nanoparticles and gas-phase molec… read moreAbstract: In finite systems, such as nanoparticles and gas-phase molecules, calculations of minimum energy paths (MEPs) connecting initial and final states of transitions as well as searches for saddle points are complicated by the presence of external degrees of freedom, such as overall translation and rotation. A method based on quaternion algebra for removing the external degrees of freedom is described here and applied in calculations using two commonly used methods: the nudged elastic band (NEB) method for MEPs and the DIMER method for finding the minimum mode in minimum mode following searches of first-order saddle points. With the quaternion approach, fewer images in the NEB are needed to represent MEPs accurately. In both NEB and DIMER calculations of finite systems, the number of iterations required to reach convergence is significantly reduced. The algorithms have been implemented in the Atomic Simulation Environment (ASE) open source software. read less NOT USED (low confidence) A. Pedersen and M. Luiser, “Bowl breakout: escaping the positive region when searching for saddle points.,” The Journal of chemical physics. 2014. link Times cited: 7 Abstract: We present a scheme improving the minimum-mode following met… read moreAbstract: We present a scheme improving the minimum-mode following method for finding first order saddle points by confining the displacements of atoms to the subset of those subject to the largest force. By doing so it is ensured that the displacement remains of a local character within regions where all eigenvalues of the Hessian matrix are positive. However, as soon as a region is entered where an eigenvalue turns negative all atoms are released to maintain the ability of determining concerted moves. Applying the proposed scheme reduces the required number of force calls for the determination of connected saddle points by a factor two or more compared to a free search. Furthermore, a wider distribution of the relevant low barrier saddle points is obtained. Finally, the dependency on the initial distortion and the applied maximal step size is reduced making minimum-mode guided searches both more robust and applicable. read less NOT USED (low confidence) U. Nerle, R. M. Hodlur, and M. Rabinal, “A sharp and visible range plasmonic in heavily doped metal oxide films,” Materials Research Express. 2014. link Times cited: 9 Abstract: As alternatives to conventional metals, metal oxide semicond… read moreAbstract: As alternatives to conventional metals, metal oxide semiconductors offer many advantages in the field of plasmonic applications. Herein, the influence of antimony doping on the plasmon resonance properties of tin oxide films prepared by spray pyrolysis has been investigated. The colour of heavily doped antimony tin oxide films was tuned from colourless (transparent) to dark bluish. The crystallinity and morphology of these films were studied by x-ray diffraction and scanning electron microscopy. X-ray diffraction reveals that these films have purely cassetirite structure and no secondary phases of tin and antimony oxides are present. The surface morphology shows films have grainy structure which decreases with increase in antimony content. The Hall effect and electrical conductivity measurements show that the majority carriers are electrons and conductivity decreases by three orders of magnitude with increase in antimony content up to 50 wt%. The optical absorption shows that with increase in doping the plasmon peak becomes sharp and shifts to the visible region. Such plasmonic features are rarely reported on such materials. These results are analysed in terms of classical theory of surface plasmon resonance. The better plasmonic features, low cost, ease of synthesis and good chemical stability make these materials potential candidates for plasmonic applications. read less NOT USED (low confidence) S. Lysgaard, D. D. Landis, T. Bligaard, and T. Vegge, “Genetic Algorithm Procreation Operators for Alloy Nanoparticle Catalysts,” Topics in Catalysis. 2014. link Times cited: 28 NOT USED (low confidence) A. Chernatynskiy, S. Phillpot, and R. LeSar, “Uncertainty Quantification in Multiscale Simulation of Materials: A Prospective,” Annual Review of Materials Research. 2013. link Times cited: 102 Abstract: Simulation has long since joined experiment and theory as a … read moreAbstract: Simulation has long since joined experiment and theory as a valuable tool to address materials problems. Analysis of errors and uncertainties in experiment and theory is well developed; such analysis for simulations, particularly for simulations linked across length scales and timescales, is much less advanced. In this prospective, we discuss salient issues concerning uncertainty quantification (UQ) from a variety of fields and review the sparse literature on UQ in materials simulations. As specific examples, we examine the development of atomistic potentials and multiscale simulations of crystal plasticity. We identify needs for conceptual advances, needs for the development of best practices, and needs for specific implementations. read less NOT USED (low confidence) T. Yoshiie et al., “Multi-scale modeling of irradiation effects in spallation neutron source materials,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2011. link Times cited: 3 NOT USED (low confidence) J. C. Silva, R. F. B. Souza, L. Parreira, É. T. Neto, M. L. Calegaro, and M. C. Santos, “Ethanol oxidation reactions using SnO2@Pt/C as an electrocatalyst,” Applied Catalysis B-environmental. 2010. link Times cited: 84 NOT USED (low confidence) F. Delogu, “A numerical investigation of the stability of nanometer-sized amorphous structures,” Intermetallics. 2010. link Times cited: 2 NOT USED (low confidence) G. Somorjai and Y. Li, “Major Successes of Theory-and-Experiment-Combined Studies in Surface Chemistry and Heterogeneous Catalysis,” Topics in Catalysis. 2009. link Times cited: 46 NOT USED (low confidence) R. G. Freitas, E. P. Antunes, and E. Pereira, “CO and methanol electrooxidation on Pt/Ir/Pt multilayers electrodes,” Electrochimica Acta. 2009. link Times cited: 48 NOT USED (low confidence) M. Dreher et al., “Numerical investigations of complex nano-systems,” Phase Transitions. 2005. link Times cited: 1 Abstract: The nature of the melting transition for a system of hard di… read moreAbstract: The nature of the melting transition for a system of hard disks with translational degrees of freedom in two spatial dimensions has been analysed by a combination of computer simulation methods and a finite size scaling technique. The behaviour of the system is consistent with the predictions of the Kosterlitz–Thouless–Halperin–Nelson–Young (KTHNY) theory. The structural and elastic properties of binary colloidal mixtures in two and three spatial dimensions are discussed as well as those of colloidal systems with quenched point impurities. Hard and soft disks in external periodic (light) fields show rich phase diagrams, including freezing and melting transitions when the density of the system is varied. Using Monte Carlo simulation methods we have investigated the phase diagrams of such systems for various values of the density and the amplitude of the external potential. The conductance quantization of atomic Au wires is discussed as well as the properties of Si clusters. read less NOT USED (low confidence) R. Oliveira, M. C. Santos, B. G. Marcussi, P. Nascente, L. Bulhões, and E. Pereira, “The use of a metallic bilayer for the oxidation of small organic molecules,” Journal of Electroanalytical Chemistry. 2005. link Times cited: 37 NOT USED (low confidence) A. Christensen, “ACCELERATING CONVERGENCE OF MOLECULAR DYNAMICS-BASED STRUCTURAL RELAXATION,” International Journal of Modern Physics C. 2005. link Times cited: 0 Abstract: We describe strategies to accelerate the terminal stage of m… read moreAbstract: We describe strategies to accelerate the terminal stage of molecular dynamics (MD)-based relaxation algorithms, where a large fraction of the computational resources are used. First, we analyze the qualitative and quantitative behavior of the QuickMin family of MD relaxation algorithms and explore the influence of spectral properties and dimensionality of the molecular system on the algorithm efficiency. We test two algorithms, the MinMax and Lanczos, for spectral estimation from an MD trajectory, and use this to derive a practical scheme of time step adaptation in MD relaxation algorithms to improve efficiency. We also discuss the implementation aspects. Secondly, we explore the final state refinement acceleration by a combination with the conjugate gradient technique, where the key ingredient is an implicit corrector step. Finally, we test the feasibility of passive Hessian matrix accumulation from an MD trajectory, as another route for final phase acceleration. Our suggestions may be implemented within most MD quench implementations with a few, straightforward lines of code, thus maintaining the appealing simplicity of the MD quench algorithms. In this paper, we also bridge the conceptual gap between the MD quench algorithms inspired from physics and the mathematically rooted line search algorithms. read less NOT USED (low confidence) J. Meier, J. Schiøtz, P. Liu, J. Nørskov, and U. Stimming, “Nano-scale effects in electrochemistry,” Chemical Physics Letters. 2004. link Times cited: 114 NOT USED (low confidence) C. Barreteau, F. Raouafi, M. Desjonquéres, and D. Spanjaard, “Modelling of transition and noble metal vicinal surfaces: energetics, vibrations and stability,” Journal of Physics: Condensed Matter. 2003. link Times cited: 13 Abstract: The energetics of transition and noble metal (Rh, Pd, Cu) vi… read moreAbstract: The energetics of transition and noble metal (Rh, Pd, Cu) vicinal surfaces, i.e., the surface energy, step energy, kink energy and electronic interactions between steps, is studied at 0 K from electronic structure calculations in the tight binding approximation using an s, p and d valence orbital basis set. Then, the surface phonon spectra of copper are investigated in the harmonic approximation with the help of a semi-empirical inter-atomic potential. This allows one to derive the contribution of phonons at finite temperatures to the step free energy and to the interactions between steps. The last part is devoted to the stability of vicinal surfaces relative to faceting with special attention paid to the domain of orientations (100)–(111). Semi-empirical potentials are shown to be not realistic enough to give a reliable answer for this problem. The results derived from electronic structure calculations predict a variety of behaviours and, in particular, a possible faceting into two other vicinal orientations. Finally, temperature effects are discussed. Comparisons are made with other theoretical works and available experiments. read less NOT USED (low confidence) T. Vegge, J. Sethna, S.-A. Cheong, K. W. Jacobsen, C. R. Myers, and D. C. Ralph, “Calculation of quantum tunneling for a spatially extended defect: the dislocation kink in copper has a low effective mass.,” Physical review letters. 2000. link Times cited: 31 Abstract: Several experiments indicate that there are atomic tunneling… read moreAbstract: Several experiments indicate that there are atomic tunneling defects in plastically deformed metals. How this is possible has not been clear, given the large mass of the metal atoms. Using a classical molecular-dynamics calculation, we determine the structures, energy barriers, effective masses, and quantum tunneling rates for dislocation kinks and jogs in copper screw dislocations. We find that jogs are unlikely to tunnel, but the kinks should have large quantum fluctuations. The kink motion involves hundreds of atoms each shifting a tiny amount, leading to a small effective mass and tunneling barrier. read less NOT USED (low confidence) J. Schiøtz and T. Vegge, “Computer Simulations of the Mechanical Properties of Metals,” Science Progress. 1999. link Times cited: 1 Abstract: Atomic-scale computer simulations can be used to gain a bett… read moreAbstract: Atomic-scale computer simulations can be used to gain a better understanding of the mechanical properties of materials. In this paper we demonstrate how this can be done in the case of nanocrystalline copper, and give a brief overview of how simulations may be extended to larger length scales. Nanocrystalline metals are metals with grain sizes in the nanometre range, they have a number of technologically interesting properties such as much increased hardness and yield strength. Our simulations show that the deformation mechanisms are different in these materials than in coarsegrained materials. The main deformation is occurring in the grain boundaries, and only little dislocation activity is seen inside the grains. This leads to a hardening of the material as the grain size is increased, and the volume fraction of grain boundaries is decreased. read less NOT USED (low confidence) J. Andzelm et al., “Heterogeneous catalysis: looking forward with molecular simulation,” Catalysis Today. 1999. link Times cited: 20 NOT USED (low confidence) J. Schiøtz, F. D. Tolla, and K. Jacobsen, “Softening of nanocrystalline metals at very small grain sizes,” Nature. 1998. link Times cited: 1510 NOT USED (low confidence) A. Christensen, A. Ruban, and H. Skriver, “Surface segregation profile for Ni50Pd50(100),” Surface Science. 1997. link Times cited: 17 NOT USED (low confidence) C. Mottet, “Structure and Chemical Ordering in Nanoalloys: Toward Nanoalloy Phase Diagrams.” 2013. link Times cited: 3 NOT USED (low confidence) J. Li, X. Dai, S. Liang, K. Tai, Y. Kong, and B. Liu, “Interatomic potentials of the binary transition metal systems and some applications in materials physics,” Physics Reports. 2008. link Times cited: 110 NOT USED (low confidence) P. Henseler et al., “Nano-Systems in External Fields and Reduced Geometry: Numerical Investigations.” 2007. link Times cited: 0 NOT USED (low confidence) J. Schiøtz and S. L. Frederiksen, “Competing Deformation Mechanisms in Nanocrystalline Metals.” 2004. link Times cited: 0 NOT USED (low confidence) M. Dreher et al., “Numerical Studies of Collective Effects in Nano-Systems.” 2003. link Times cited: 0 NOT USED (low confidence) W. Püschl, “Models for dislocation cross-slip in close-packed crystal structures: a critical review,” Progress in Materials Science. 2002. link Times cited: 294 NOT USED (low confidence) E. Iesulauro, T. Cretegny, C.-S. Chen, K. Dodhia, C. Myers, and A. Ingraffea, “Multiscale Modeling of Crack Growth in Polycrystals.” 2002. link Times cited: 0 NOT USED (low confidence) J. Newsam, “Design of Catalysts and Catalyst Libraries.” 2000. link Times cited: 4 NOT USED (low confidence) T. Vegge, O. B. Pedersen, T. Leffers, and K. Jacobsen, “Atomic-scale modeling of the annihilation of jogged screw dislocation dipoles,” MRS Proceedings. 1999. link Times cited: 7 Abstract: Using atomistic simulations we investigate the annihilation … read moreAbstract: Using atomistic simulations we investigate the annihilation of screw dislocation dipoles in Cu. In particular we determine the influence of jogs on the annihilation barrier for screw dislocation dipoles. The simulations involve energy minimizations, molecular dynamics, and the Nudged Elastic Band method. We find that jogs on screw dislocations substantially reduce the annihilation barrier, hence leading to an increase in the minimum stable dipole height. read less NOT USED (low confidence) F. Besenbacher, L. P. Nielsen, and P. Sprunger, “Chapter 6 Surface alloying in heteroepitaxial metal-on-metal growth,” The Chemical Physics of Solid Surfaces. 1997. link Times cited: 26 NOT USED (high confidence) R. Martin-Barrios, N. Hertl, O. Galparsoro, A. Kandratsenka, A. Wodtke, and P. Larrégaray, “H atom scattering from W(110): A benchmark for molecular dynamics with electronic friction.,” Physical Chemistry Chemical Physics. 2022. link Times cited: 2 Abstract: Molecular dynamics with electronic friction (MDEF) at the le… read moreAbstract: Molecular dynamics with electronic friction (MDEF) at the level of the local density friction approximation (LDFA) has been applied to describe electronically non-adiabatic energy transfer accompanying H atom collisions with many solid metal surfaces. When implemented with full dimensional potential energy and electron density functions, excellent agreement with experiment is found. Here, we compare the performance of a reduced dimensional MDEF approach involving a simplified description of H atom coupling to phonons to that of full dimensional MDEF calculations known to yield accurate results. Both approaches give remarkably similar results for H atom energy loss distributions with a 300 K W(110) surface. At low surface temperature differences are seen; but, quantities like average energy loss are still accurately reproduced. Both models predict similar conditions under which H atoms that have penetrated into the subsurface regions could be observed in scattering experiments. read less NOT USED (high confidence) S. Han, G. Barcaro, A. Fortunelli, S. Lysgaard, T. Vegge, and H. Hansen, “Unfolding the structural stability of nanoalloys via symmetry-constrained genetic algorithm and neural network potential,” npj Computational Materials. 2022. link Times cited: 5 NOT USED (high confidence) N. Hertl, A. Kandratsenka, and A. Wodtke, “Effective medium theory for bcc metals: electronically non-adiabatic H atom scattering in full dimensions,” Physical Chemistry Chemical Physics. 2022. link Times cited: 1 Abstract: We report a newly derived Effective Medium Theory (EMT) form… read moreAbstract: We report a newly derived Effective Medium Theory (EMT) formalism for bcc metals and apply it for the construction of a full-dimensional PES for H atoms interacting with molybdenum (Mo) and tungsten (W). We construct PESs for the (111) and (110) facets of both metals. The EMT-PESs have the advantage that they automatically provide the background electron density on the fly which makes incorporation of ehp excitation within the framework of electronic friction straightforward. Using molecular dynamics with electronic friction (MDEF) with these new PESs, we simulated 2.76 eV H atoms scattering and adsorption. The large energy losses at a surface temperature of 300 K is very similar those seen for H atom scattering from the late fcc metals and is dominated by ehp excitation. We see significant differences in the scattering from different surface facets of the same metal. For the (110) facet, we see strong evidence of sub-surface scattering, which should be observable in experiment and we predict the best conditions for observing this novel type of scattering process. At low temperatures the MD simulations predict that H atom scattering is surface specific due to the reduced influence of the random force. read less NOT USED (high confidence) L. Chen, X. Zhang, A. Chen, S. Yao, X. Hu, and Z. Zhou, “Targeted design of advanced electrocatalysts by machine learning,” Chinese Journal of Catalysis. 2022. link Times cited: 20 NOT USED (high confidence) C. Martínez-Alonso, J. M. Guevara‐Vela, and J. Llorca, “The effect of elastic strains on the adsorption energy of H, O, and OH in transition metals.,” Physical chemistry chemical physics : PCCP. 2021. link Times cited: 3 Abstract: The influence of elastic strains on the adsorption of H, O, … read moreAbstract: The influence of elastic strains on the adsorption of H, O, and OH on the (111) surfaces of 8 fcc (Ni, Cu, Pd, Ag, Pt, Au, Rh, Ir) and on the (0001) surfaces of 3 hcp (Co, Zn, Cd) transition metals was analyzed by means of density functional theory calculations. To this end, surface slabs were subjected to different strain states (uniaxial, biaxial, shear, and a combination of them) up to strains dictated by the mechanical stability limits indicated by phonon calculations. It was found that the adsorption energy followed the predictions of the d-band theory but - surprisingly - the variations in the adsorption energy only depended on the area of the adsorption hole and not on the particular elastic strain tensor applied to achieve this area. The analysis of the electronic structure showed that the applied strains did not modify the shape of the Projected Density of States (PDOS) of the d-orbitals of the transition metals but only led to a shift in the energy levels. Moreover, the presence of the adsorbates on the surfaces led to negligible changes in the PDOS. Thus, the adsorption energies were a function of the Fermi energy which in turn was associated with the change of the area of the adsorption through a general linear law that was valid for all metals. The information in this paper allows the immediate and accurate estimation of the effect of any elastic strain on the adsorption energies of H, O, and OH in 11 transition metals with more than half-filled d-orbitals. read less NOT USED (high confidence) L. Friedeheim, N. Bailey, and J. Dyre, “Effectively one-dimensional phase diagram of CuZr liquids and glasses,” Physical Review B. 2021. link Times cited: 2 Abstract: This paper presents computer simulations of CuxZr100−x (x = … read moreAbstract: This paper presents computer simulations of CuxZr100−x (x = 36, 50, 64) in the liquid and glass phases. The simulations are based on the effective-medium theory (EMT) potentials. We find good invariance of both structure and dynamics in reduced units along the isomorphs of the systems. The state points studied involve a density variation of almost a factor of two and temperatures going from 1500 K to above 4000 K for the liquids and from 500 K to above 1500 K for the glasses. For comparison, results are presented also for similar temperature variations along isochores, showing little invariance. In general for a binary system the phase diagram has three axes: composition, temperature and pressure (or density). When isomorphs are present, there are effectively only two axes, and for a fixed composition just one. We conclude that the thermodynamic phase diagram of this metallic glass former for a fixed composition is effectively one-dimensional in the sense that many physical properties are invariant along the same curves, implying that in order to investigate the phase diagram, it is only necessary to go across these curves. read less NOT USED (high confidence) N. Gerrits, J. I. Juaristi, and J. Meyer, “Electronic friction coefficients from the atom-in-jellium model for
Z=1–92,” Physical Review B. 2020. link Times cited: 4 Abstract: The break-down of the Born-Oppenheimer approximation is an i… read moreAbstract: The break-down of the Born-Oppenheimer approximation is an important topic in chemical dynamics on metal surfaces. In this context, the most frequently used "work-horse" is electronic friction theory, commonly relying on friction coefficients obtained from density functional theory (DFT) calculations from the early 80s based on the atom-in-jellium model. However, results are only available for a limited set of jellium densities and elements ($Z=1-18$). In this work, these calculations are revisited by investigating the corresponding friction coefficients for the entire periodic table ($Z=1-92$). Furthermore, friction coefficients obtained by including the electron density gradient on the Generalized Gradient Approximation (GGA) level are presented. Finally, we show that spin polarization and relativistic effects can have sizeable effects on these friction coefficients for some elements. read less NOT USED (high confidence) A. Bacher, U. R. Pedersen, T. Schrøder, and J. Dyre, “The EXP pair-potential system. IV. Isotherms, isochores, and isomorphs in the two crystalline phases.,” The Journal of chemical physics. 2020. link Times cited: 5 Abstract: This paper studies numerically the solid phase of a system o… read moreAbstract: This paper studies numerically the solid phase of a system of particles interacting by the exponentially repulsive pair potential, which is a face-centered cubic (fcc) crystal at low densities and a body-centered cubic (bcc) crystal at higher densities [U. R. Pedersen et al., J. Chem. Phys. 150, 174501 (2019)]. Structure is studied via the pair-distribution function and dynamics via the velocity autocorrelation function and the phonon density of states. These quantities are evaluated along isotherms, isochores, and three isomorphs in both crystal phases. Isomorphs are traced out by integrating the density-temperature relation characterizing configurational adiabats, starting from state points in the middle of the fcc-bcc coexistence region. Good isomorph invariance of structure and dynamics is seen in both crystal phases, which is notable in view of the large density variations studied. This is consistent with the fact that the virial potential-energy correlation coefficient is close to unity in the entire fcc phase and in most of the bcc phase (basically below the re-entrant density). Our findings confirm that the isomorph theory, developed and primarily studied for liquids, applies equally well for solids. read less NOT USED (high confidence) T. S. Choksi, V. Streibel, and F. Abild-Pedersen, “Predicting metal-metal interactions. II. Accelerating generalized schemes through physical insights.,” The Journal of chemical physics. 2020. link Times cited: 7 Abstract: Operando-computational frameworks that integrate descriptors… read moreAbstract: Operando-computational frameworks that integrate descriptors for catalyst stability within catalyst screening paradigms enable predictions of rates and selectivity on chemically faithful representations of nanoparticles under reaction conditions. These catalyst stability descriptors can be efficiently predicted by density functional theory (DFT)-based models. The alloy stability model, for example, predicts the stability of metal atoms in nanoparticles with site-by-site resolution. Herein, we use physical insights to present accelerated approaches of parameterizing this recently introduced alloy-stability model. These accelerated approaches meld quadratic functions for the energy of metal atoms in terms of the coordination number with linear correlations between model parameters and the cohesive energies of bulk metals. By interpolating across both the coordination number and chemical space, these accelerated approaches shrink the training set size for 12 fcc p- and d-block metals from 204 to as few as 24 DFT calculated total energies without sacrificing the accuracy of our model. We validate the accelerated approaches by predicting adsorption energies of metal atoms on extended surfaces and 147 atom cuboctahedral nanoparticles with mean absolute errors of 0.10 eV and 0.24 eV, respectively. This efficiency boost will enable a rapid and exhaustive exploration of the vast material space of transition metal alloys for catalytic applications. read less NOT USED (high confidence) U. R. Pedersen, A. Bacher, T. Schrøder, and J. Dyre, “The EXP pair-potential system. III. Thermodynamic phase diagram.,” The Journal of chemical physics. 2019. link Times cited: 7 Abstract: This paper determines the thermodynamic phase diagram of the… read moreAbstract: This paper determines the thermodynamic phase diagram of the EXP system of particles interacting by the purely repulsive exponential pair potential. The solid phase is face-centered cubic (fcc) at low densities and pressures. At higher densities and pressures, the solid phase is body-centered cubic (bcc) with a re-entrant liquid phase at the highest pressures simulated. The investigation first identifies the phase diagram at zero temperature at which the following four crystal structures are considered: fcc, bcc, hexagonal close packed, and cubic diamond. There is a T = 0 phase transition at pressure 2.651 × 10-3 with the thermodynamically stable structure being fcc below and bcc above this pressure. The densities of the two crystal structures at the phase transition are 1.7469 × 10-2 (fcc) and 1.7471 × 10-2 (bcc). At finite temperatures, the fcc-bcc, fcc-liquid, and bcc-liquid coexistence lines are determined by numerical integration of the Clausius-Clapeyron equation and validated by interface-pinning simulations at selected state points. The bcc-fcc phase transition is a weak first-order transition. The liquid-fcc-bcc triple point, which is determined by the interface-pinning method, has temperature 5.9 × 10-5 and pressure 2.5 × 10-6; the triple-point densities are 1.556 × 10-3 (liquid), 1.583 × 10-3 (bcc), and 1.587 × 10-3 (fcc). read less NOT USED (high confidence) E. G. del Río, J. Mortensen, and K. Jacobsen, “Local Bayesian optimizer for atomic structures,” Physical Review B. 2018. link Times cited: 53 Abstract: A local optimization method based on Bayesian Gaussian Proce… read moreAbstract: A local optimization method based on Bayesian Gaussian Processes is developed and applied to atomic structures. The method is applied to a variety of systems including molecules, clusters, bulk materials, and molecules at surfaces. The approach is seen to compare favorably to standard optimization algorithms like conjugate gradient or BFGS in all cases. The method relies on prediction of surrogate potential energy surfaces, which are fast to optimize, and which are gradually improved as the calculation proceeds. The method includes a few hyperparameters, the optimization of which may lead to further improvements of the computational speed. read less NOT USED (high confidence) Y. Dorenkamp et al., “Hydrogen collisions with transition metal surfaces: Universal electronically nonadiabatic adsorption.,” The Journal of chemical physics. 2018. link Times cited: 28 Abstract: Inelastic scattering of H and D atoms from the (111) surface… read moreAbstract: Inelastic scattering of H and D atoms from the (111) surfaces of six fcc transition metals (Au, Pt, Ag, Pd, Cu, and Ni) was investigated, and in each case, excitation of electron-hole pairs dominates the inelasticity. The results are very similar for all six metals. Differences in the average kinetic energy losses between metals can mainly be attributed to different efficiencies in the coupling to phonons due to the different masses of the metal atoms. The experimental observations can be reproduced by molecular dynamics simulations based on full-dimensional potential energy surfaces and including electronic excitations by using electronic friction in the local density friction approximation. The determining factors for the energy loss are the electron density at the surface, which is similar for all six metals, and the mass ratio between the impinging atoms and the surface atoms. Details of the electronic structure of the metal do not play a significant role. The experimentally validated simulations are used to explore sticking over a wide range of incidence conditions. We find that the sticking probability increases for H and D collisions near normal incidence-consistent with a previously reported penetration-resurfacing mechanism. The sticking probability for H or D on any of these metals may be represented as a simple function of the incidence energy, Ein, metal atom mass, M, and incidence angle, 𝜗in. S=(S0+a⋅Ein+b⋅M)*(1-h(𝜗in-c)(1-cos(𝜗in-c)d⋅h(Ein-e)(Ein-e))), where h is the Heaviside step function and for H, S0 = 1.081, a = -0.125 eV-1, b=-8.40⋅10-4 u-1, c = 28.88°, d = 1.166 eV-1, and e = 0.442 eV; whereas for D, S0 = 1.120, a = -0.124 eV-1, b=-1.20⋅10-3 u-1, c = 28.62°, d = 1.196 eV-1, and e = 0.474 eV. read less NOT USED (high confidence) P. M. Larsen, K. Jacobsen, and J. Schiøtz, “Rich Ground-State Chemical Ordering in Nanoparticles: Exact Solution of a Model for Ag-Au Clusters.,” Physical review letters. 2017. link Times cited: 13 Abstract: We show that nanoparticles can have very rich ground-state c… read moreAbstract: We show that nanoparticles can have very rich ground-state chemical order. This is illustrated by determining the chemical ordering of Ag-Au 309-atom Mackay icosahedral nanoparticles. The energy of the nanoparticles is described using a cluster expansion model, and a mixed integer programming approach is used to find the exact ground-state configurations for all stoichiometries. The chemical ordering varies widely between the different stoichiometries and displays a rich zoo of structures with nontrivial ordering. read less NOT USED (high confidence) S. Bhattacharjee, U. Waghmare, and S.-C. Lee, “An improved d-band model of the catalytic activity of magnetic transition metal surfaces,” Scientific Reports. 2016. link Times cited: 146 NOT USED (high confidence) E. L. Kolsbjerg, M. Groves, and B. Hammer, “An automated nudged elastic band method.,” The Journal of chemical physics. 2016. link Times cited: 74 Abstract: A robust, efficient, dynamic, and automated nudged elastic b… read moreAbstract: A robust, efficient, dynamic, and automated nudged elastic band (AutoNEB) algorithm to effectively locate transition states is presented. The strength of the algorithm is its ability to use fewer resources than the nudged elastic band (NEB) method by focusing first on converging a rough path before improving upon the resolution around the transition state. To demonstrate its efficiency, it has been benchmarked using a simple diffusion problem and a dehydrogenation reaction. In both cases, the total number of force evaluations used by the AutoNEB method is significantly less than the NEB method. Furthermore, it is shown that for a fast and robust relaxation to the transition state, a climbing image elastic band method where the full spring force, rather than only the component parallel to the local tangent to the path, is preferred especially for pathways through energy landscapes with multiple local minima. The resulting corner cutting does not affect the accuracy of the transition state as long as this is located with the climbing image method. Finally, a number of pitfalls often encountered while locating the true transition state of a reaction are discussed in terms of systematically exploring the multidimensional energy landscape of a given process. read less NOT USED (high confidence) N. Admal, J. Marian, and G. Po, “The atomistic representation of first strain-gradient elastic tensors,” Journal of The Mechanics and Physics of Solids. 2016. link Times cited: 36 NOT USED (high confidence) L. Zosiak, C. Goyhenex, R. Kozubski, and G. Tréglia, “Electronic structure of CoPt based systems: from bulk to nanoalloys,” Journal of Physics: Condensed Matter. 2015. link Times cited: 4 Abstract: An accurate description of the local electronic structure is… read moreAbstract: An accurate description of the local electronic structure is necessary for guiding the design of materials with targeted properties in a controlled way. For complex materials like nanoalloys, self-consistent tight-binding calculations should be a good alternative to ab initio methods, for handling the most complex and large systems (hundreds to thousands of atoms), provided that these parameterized method is well founded from ab initio ones that they intend to replace. Ab initio calculations (density functional theory) enabled us to derive rules for charge distribution as a function of structural change and alloying effects in Co and Pt based systems, from bulk to nanoalloys. A general local neutrality rule per site, orbital and species was found. Based on it, self-consistent tight-binding calculations could be implemented and applied to CoPt nanoalloys. A very good agreement is obtained between tight-binding and DFT calculations in terms of local electronic structure. read less NOT USED (high confidence) S. Janke, D. Auerbach, A. Wodtke, and A. Kandratsenka, “An accurate full-dimensional potential energy surface for H-Au(111): Importance of nonadiabatic electronic excitation in energy transfer and adsorption.,” The Journal of chemical physics. 2015. link Times cited: 57 Abstract: We have constructed a potential energy surface (PES) for H-a… read moreAbstract: We have constructed a potential energy surface (PES) for H-atoms interacting with fcc Au(111) based on fitting the analytic form of the energy from Effective Medium Theory (EMT) to ab initio energy values calculated with density functional theory. The fit used input from configurations of the H-Au system with Au atoms at their lattice positions as well as configurations with the Au atoms displaced from their lattice positions. It reproduces the energy, in full dimension, not only for the configurations used as input but also for a large number of additional configurations derived from ab initio molecular dynamics (AIMD) trajectories at finite temperature. Adiabatic molecular dynamics simulations on this PES reproduce the energy loss behavior of AIMD. EMT also provides expressions for the embedding electron density, which enabled us to develop a self-consistent approach to simulate nonadiabatic electron-hole pair excitation and their effect on the motion of the incident H-atoms. For H atoms with an energy of 2.7 eV colliding with Au, electron-hole pair excitation is by far the most important energy loss pathway, giving an average energy loss ≈3 times that of the adiabatic case. This increased energy loss enhances the probability of the H-atom remaining on or in the Au slab by a factor of 2. The most likely outcome for H-atoms that are not scattered also depends prodigiously on the energy transfer mechanism; for the nonadiabatic case, more than 50% of the H-atoms which do not scatter are adsorbed on the surface, while for the adiabatic case more than 50% pass entirely through the 4 layer simulation slab. read less NOT USED (high confidence) J. Jalkanen and M. Müser, “Systematic analysis and modification of embedded-atom potentials: case study of copper,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 10 Abstract: In this study, we evaluate the functionals of different embe… read moreAbstract: In this study, we evaluate the functionals of different embedded-atom methods (EAM) by fitting their free parameters to ab-initio results for copper. Our emphasis lies on testing the transferability of the potentials between systems which vary in their spatial dimension and geometry. The model structures encompass zero-dimensional clusters, one-dimensional chains, two-dimensional tilings, and three-dimensional bulk systems. To avoid having to mimic charge transfer, which is outside the scope of conventional EAM potentials, we focus on structures, in which all atoms are symmetrically equivalent. We find that the simple, four-parameter Gupta EAM potential is overall satisfactory. Adding complexity to it decreases the errors on our set of structures only by marginal amounts, unless EAM is modified to depend also on density gradients, higher-order derivatives, or related terms. All tested conventional EAM functions reveal similar problems: the binding energy of closed-packed systems is overestimated in comparison to open or planar geometries, and structures with small coordination tend to be too rigid. These deficiencies can be fixed in terms of a systematically modified embedded-atom method (SMEAM), which reproduces DFT results on bond lengths, binding energies, and stiffnesses or bulk moduli by typically O(1%), O(5%), and O(15%) accuracy, respectively. SMEAM also predicts the radial distribution function of liquid copper quite accurately. Yet, it does not overcome the difficulty to reproduce the elastic tensor elements of a hypothetical diamond lattice. read less NOT USED (high confidence) G. Kroes, M. Pavanello, M. Blanco-Rey, M. Alducin, and D. Auerbach, “Ab initio molecular dynamics calculations on scattering of hyperthermal H atoms from Cu(111) and Au(111).,” The Journal of chemical physics. 2014. link Times cited: 43 Abstract: Energy loss from the translational motion of an atom or mole… read moreAbstract: Energy loss from the translational motion of an atom or molecule impinging on a metal surface to the surface may determine whether the incident particle can trap on the surface, and whether it has enough energy left to react with another molecule present at the surface. Although this is relevant to heterogeneous catalysis, the relative extent to which energy loss of hot atoms takes place to phonons or electron-hole pair (ehp) excitation, and its dependence on the system's parameters, remain largely unknown. We address these questions for two systems that present an extreme case of the mass ratio of the incident atom to the surface atom, i.e., H + Cu(111) and H + Au(111), by presenting adiabatic ab initio molecular dynamics (AIMD) predictions of the energy loss and angular distributions for an incidence energy of 5 eV. The results are compared to the results of AIMDEFp calculations modeling energy loss to ehp excitation using an electronic friction ("EF") model applied to the AIMD trajectories, so that the energy loss to the electrons is calculated "post" ("p") the computation of the AIMD trajectory. The AIMD calculations predict average energy losses of 0.38 eV for Cu(111) and 0.13-0.14 eV for Au(111) for H-atoms that scatter from these surfaces without penetrating the surface. These energies closely correspond with energy losses predicted with Baule models, which is suggestive of structure scattering. The predicted adiabatic integral energy loss spectra (integrated over all final scattering angles) all display a lowest energy peak at an energy corresponding to approximately 80% of the average adiabatic energy loss for non-penetrative scattering. In the adiabatic limit, this suggests a way of determining the approximate average energy loss of non-penetratively scattered H-atoms from the integral energy loss spectrum of all scattered H-atoms. The AIMDEFp calculations predict that in each case the lowest energy loss peak should show additional energy loss in the range 0.2-0.3 eV due to ehp excitation, which should be possible to observe. The average non-adiabatic energy losses for non-penetrative scattering exceed the adiabatic losses to phonons by 0.9-1.0 eV. This suggests that for scattering of hyperthermal H-atoms from coinage metals the dominant energy dissipation channel should be to ehp excitation. These predictions can be tested by experiments that combine techniques for generating H-atom beams that are well resolved in translational energy and for detecting the scattered atoms with high energy-resolution. read less NOT USED (high confidence) S. Chill et al., “EON: software for long time simulations of atomic scale systems,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 56 Abstract: The EON software is designed for simulations of the state-to… read moreAbstract: The EON software is designed for simulations of the state-to-state evolution of atomic scale systems over timescales greatly exceeding that of direct classical dynamics. States are defined as collections of atomic configurations from which a minimization of the potential energy gives the same inherent structure. The time evolution is assumed to be governed by rare events, where transitions between states are uncorrelated and infrequent compared with the timescale of atomic vibrations. Several methods for calculating the state-to-state evolution have been implemented in EON, including parallel replica dynamics, hyperdynamics and adaptive kinetic Monte Carlo. Global optimization methods, including simulated annealing, basin hopping and minima hopping are also implemented. The software has a client/server architecture where the computationally intensive evaluations of the interatomic interactions are calculated on the client-side and the state-to-state evolution is managed by the server. The client supports optimization for different computer architectures to maximize computational efficiency. The server is written in Python so that developers have access to the high-level functionality without delving into the computationally intensive components. Communication between the server and clients is abstracted so that calculations can be deployed on a single machine, clusters using a queuing system, large parallel computers using a message passing interface, or within a distributed computing environment. A generic interface to the evaluation of the interatomic interactions is defined so that empirical potentials, such as in LAMMPS, and density functional theory as implemented in VASP and GPAW can be used interchangeably. Examples are given to demonstrate the range of systems that can be modeled, including surface diffusion and island ripening of adsorbed atoms on metal surfaces, molecular diffusion on the surface of ice and global structural optimization of nanoparticles. read less NOT USED (high confidence) A. Sengupta, D. Saha, T. Niehaus, and S. Mahapatra, “Effect of Line Defects on the Electrical Transport Properties of Monolayer MoS _\bf 2 Sheet,” IEEE Transactions on Nanotechnology. 2014. link Times cited: 6 Abstract: We present a computational study on the impact of line defec… read moreAbstract: We present a computational study on the impact of line defects on the electronic properties of monolayer MoS2. Four different kinds of line defects with Mo and S as the bridging atoms, consistent with recent theoretical and experimental observations, are considered herein. We employ the density functional tightbinding (DFTB) method with a Slater-Koster-type DFTB-CP2K basis set for evaluating the material properties of perfect and the various defective MoS2 sheets. The transmission spectra are computed with a DFTB-non-equilibrium Green's function formalism. We also perform a detailed analysis of the carrier transmission pathways under a small bias and investigate the phase of the transmission eigenstates of the defective MoS2 sheets. Our simulations show a two to four fold decrease in carrier conductance of MoS2 sheets in the presence of line defects as compared to that for the perfect sheet. read less NOT USED (high confidence) A. Pedersen, S. Hafstein, and H. Jónsson, “Efficient Sampling of Saddle Points with the Minimum-Mode Following Method,” SIAM J. Sci. Comput. 2011. link Times cited: 31 Abstract: The problem of sampling low lying, first-order saddle points… read moreAbstract: The problem of sampling low lying, first-order saddle points on a high dimensional surface is discussed and a method presented for improving the sampling efficiency. The discussion is in the context of an energy surface for a system of atoms and thermally activated transitions in solids treated within the harmonic approximation to transition state theory. Given a local minimum as an initial state and a small, initial displacement, the minimum-mode following method is used to climb up to a saddle point. The goal is to sample as many of the low lying saddle points as possible when such climbs are repeated from different initial displacements. Various choices for the distribution of initial displacements are discussed and a comparison made between (1)displacements along eigenmodes at the minimum, (2)purely random displacements with a maximum cutoff, and (3) Gaussian distribution of displacements. The last choice is found to give best overall results in two test problems studied, a heptamer island on a surface and a grain boundary in a metal. A method referred to as “skipping-path method” is presented to reduce redundant calculations when a climb heads towards a saddle point that has already been identified. The method is found to reduce the computational effort of finding new saddle points to as little as a third, especially when a thorough sampling is performed. read less NOT USED (high confidence) A. Pedersen and H. Jónsson, “Simulations of hydrogen diffusion at grain boundaries in aluminum,” Acta Materialia. 2009. link Times cited: 73 NOT USED (high confidence) H. Jarmakani et al., “Molecular dynamics simulations of shock compression of nickel: From monocrystals to nanocrystals,” Acta Materialia. 2008. link Times cited: 109 NOT USED (high confidence) N. Bailey, U. R. Pedersen, N. Gnan, T. Schrøder, and J. Dyre, “Pressure-energy correlations in liquids. I. Results from computer simulations.,” The Journal of chemical physics. 2008. link Times cited: 187 Abstract: We show that a number of model liquids at fixed volume exhib… read moreAbstract: We show that a number of model liquids at fixed volume exhibit strong correlations between equilibrium fluctuations of the configurational parts of (instantaneous) pressure and energy. We present detailed results for 13 systems, showing in which systems these correlations are significant. These include Lennard-Jones liquids (both single- and two-component) and several other simple liquids, neither hydrogen-bonding liquids such as methanol and water, nor the Dzugutov liquid, which has significant contributions to pressure at the second nearest neighbor distance. The pressure-energy correlations, which for the Lennard-Jones case are shown to also be present in the crystal and glass phases, reflect an effective inverse power-law potential dominating fluctuations, even at zero and slightly negative pressure. An exception to the inverse power-law explanation is a liquid with hard-sphere repulsion and a square-well attractive part, where a strong correlation is observed, but only after time averaging. The companion paper [N. P. Bailey et al., J. Chem. Phys. 129, 184508 (2008)] gives a thorough analysis of the correlations, with a focus on the Lennard-Jones liquid, and a discussion of some experimental and theoretical consequences. read less NOT USED (high confidence) A. Paduraru, A. Kenoufi, N. Bailey, and J. Schiøtz, “An Interatomic Potential for Studying CuZr Bulk Metallic Glasses,” Advanced Engineering Materials. 2007. link Times cited: 22 Abstract: Binary alloys capable of forming metallic glasses have been … read moreAbstract: Binary alloys capable of forming metallic glasses have been discovered recently. The mechanical properties of BMGs are remarkably different from the ones of ordinary metallic alloys due to the atomic level disorder in the glassy state. Unlike crystalline materials plastic deformation in metallic glasses cannot be caused by lattice defects but takes place through atomic-scale deformation events and may furthermore involve localization through formation of shear bands. For understanding the origin of their mechanical properties it is important to get the basic understanding of fundamental theoretical problems through atomistic simulations. Molecular dynamics (MD) treats atomic systems according to Newtonian mechanic laws. Atoms are point particles, interacting through an interatomic potential, describing the energy of an atom as a function of the positions of all atoms in a neighboring region of space. The time evolution of the system is obtained by numerically integrating Newton’s second law. Often the interatomic potential is a classical potential, no quantum mechanical description of the material is attempted, but the functional form of the potential may be derived from quantum mechanical arguments. MD is able to treat systems with millions of atoms, and permits the average calculations of transport (diffusion, thermal conductivities, viscosity), or mechanical quantities (elastic constant, plastic yield), and also the modeling of complex phenomena (shear band localization, fracture appearance, neutronic cascades). The quality of the results will depend on the quality of the interatomic potential, simple potentials giving a less accurate description of the interatomic interactions while allowing very large simulations, more complicated potentials may give a better description of the interaction, but limit the simulation size. Many-body potentials such as the Embedded Atom Method and the Effective Medium Theory (EMT) have been shown to give a good description of the late transition metals crystallizing in close-packed structures, and their alloys, while still allowing simulations with millions of atoms. In this paper, we create an EMT potential optimized for modeling the mechanical and thermodynamic properties of CuZr bulk metallic glass. CuZr was recently discovered to be a binary bulk metallic glass. Since binary alloys are easier to model than alloys with more elements, this makes CuZr an attractive bulk metallic glass to study theoretically. Previously, an interatomic force field has been fitted to CuZr by Duan et al., but we find that the EMT force field described here provides a better description of the structure of the metallic glass. The potential developed here will be used to model the mechanical properties of CuZr, to be published elsewhere. read less NOT USED (high confidence) T. Shimokawa, T. Kinari, S. Shintaku, A. Nakatani, and H. Kitagawa, “Defect-induced anisotropy in mechanical properties of nanocrystalline metals by molecular dynamics simulations,” Modelling and Simulation in Materials Science and Engineering. 2005. link Times cited: 13 Abstract: The influence of defects in nanograins, e.g. stacking faults… read moreAbstract: The influence of defects in nanograins, e.g. stacking faults and twinnings, on mechanical properties of nanocrystalline materials is studied by molecular dynamics simulations. Two types of many-body interatomic potential based on aluminium are adopted to investigate the influence of stacking fault energy on the deformation mechanism of nanocrystalline metals: one accurately reproduces the energy value of stacking faults for aluminium; the other underestimates the energy value for aluminium. Three different deformation processes are performed to nanocrystalline models with high or low stacking fault energy, and crystal slips occur in both models. In the case of the high stacking fault energy, crystal deformation occurs by perfect dislocations and no defects exist in the grains. Therefore, the strength almost recovers after relaxation. On the other hand, for low stacking fault energy, stacking faults remain through the grains after the crystal slips by partial dislocations. Consequently, these defects cause anisotropy in the mechanical properties of the simulated nanocrystalline metals. read less NOT USED (high confidence) K. Franzrahe et al., “Two-dimensional model colloids and nano wires: phase transitions, effects of external potentials and quantum effects,” Comput. Phys. Commun. 2005. link Times cited: 7 NOT USED (high confidence) S. L. Frederiksen, K. Jacobsen, K. S. Brown, and J. Sethna, “Bayesian ensemble approach to error estimation of interatomic potentials.,” Physical review letters. 2004. link Times cited: 106 Abstract: Using a Bayesian approach a general method is developed to a… read moreAbstract: Using a Bayesian approach a general method is developed to assess error bars on predictions made by models fitted to data. The error bars are estimated from fluctuations in ensembles of models sampling the model-parameter space with a probability density set by the minimum cost. The method is applied to the development of interatomic potentials for molybdenum using various potential forms and databases based on atomic forces. The calculated error bars on elastic constants, gamma-surface energies, structural energies, and dislocation properties are shown to provide realistic estimates of the actual errors for the potentials. read less NOT USED (high confidence) K. Sato, T. Yoshiie, Y. Satoh, and Q. Xu, “Computer simulation of formation energy and migration energy of vacancies under high strain in Cu,” Materials Transactions. 2004. link Times cited: 9 Abstract: Recently it has been reported that many vacancy clusters are… read moreAbstract: Recently it has been reported that many vacancy clusters are generated at sawtooth-like fractured tips of thin foils. The elastic strain of the torn portion is more than 10% before the fracture, which is expected to cause the generation of many vacancy clusters. In this paper, the formation and migration behaviors of vacancies in Cu under high elastic strain from 10% compression to 20% elongation were studied by computer simulation using the effective medium theory (EMT) potential. The model lattice was elastically deformed along the ‹110› and ‹100› directions. Poisson's ratio was determined to minimize the total energy. After full relaxation of the lattice by the static method (Newton-Raphson method) under fixed boundary conditions, a vacancy was introduced and the change of the total energy (formation energy) was calculated. The migration energy of vacancies was obtained as the total energy difference between the model lattice with an atom at the lattice point and the atom at the saddle point. High strain dependence of these energies is obtained. For example, the formation and the migration energies of vacancies are 1.21 eV and 0.79 eV in the absence of deformation, respectively. The formation energy in the ‹100› deformation is 1.09 eV and 1.13 eV by 10% compression and 10% elongation, respectively. The migration energies and the migration distances vary with the migration direction. For example, the migration energy for the shortest migration distance in the ‹100› deformation is reduced to 0.45 eV and 0.26 eV by 10% compression and 10% elongation, respectively. While that for the longest migration distance increases to 1.29 eV by 10% compression. These results are explained by the configuration of neighboring atoms nearest to the vacancy. read less NOT USED (high confidence) T. Vegge and K. Jacobsen, “Atomistic simulations of dislocation processes in copper,” Journal of Physics: Condensed Matter. 2002. link Times cited: 38 Abstract: We discuss atomistic simulations of dislocation processes in… read moreAbstract: We discuss atomistic simulations of dislocation processes in copper based on effective medium theory interatomic potentials. Results on screw dislocation structures and processes are reviewed with particular focus on point defect mobilities and processes involving cross slip. For example, the stability of screw dislocation dipoles is discussed. We show that the presence of jogs will strongly influence cross slip barriers and dipole stability. We furthermore present some new results on jogged edge dislocations and edge dislocation dipoles. The jogs are found to be extended, and simulations of vacancy controlled climb show the jogs to climb easily in their extended form. The stability of small vacancy dipoles is discussed and it is seen that the introduction of jogs may lead to the formation of Z-type faulted vacancy dipoles. read less NOT USED (high confidence) J. Schiøtz, T. Leffers, and B. Singh, “Modeling of Dislocation Generation and Interaction During High-Speed Deformation of Metals,” Radiation Effects and Defects in Solids. 2000. link Times cited: 11 Abstract: Recent experiments by Kiritani et al. [1] have revealed a su… read moreAbstract: Recent experiments by Kiritani et al. [1] have revealed a surprisingly high rate of vacancy production during high-speed deformation of thin foils of fcc metals. Virtually no dislocations are seen after the deformation. This is interpreted as evidence for a dislocation-free deformation mechanism at very high strain rates. We have used molecular-dynamics simulations to investigate high-speed deformation of copper crystals. Even though no pre-existing dislocation sources are present in the initial system, dislocations are quickly nucleated and a very high dislocation density is reached during the deformation. Due to the high density of dislocations, many inelastic interactions occur between dislocations, resulting in the generation of vacancies. After the deformation, a very high density of vacancies is observed, in agreement with the experimental observations. The processes responsible for the generation of vacancies are investigated. The main process is found to be incomplete annihilation of segments of edge dislocations on adjacent slip planes. The dislocations are also seen to be participating in complicated dislocation reactions, where sessile dislocation segments are constantly formed and destroyed. read less NOT USED (high confidence) T. Klamroth and P. Saalfrank, “Effect of substrate vibrations on the sticking of atoms at surfaces: A critical comparison of different propagation methods for the H/Cu(100) system,” Journal of Chemical Physics. 2000. link Times cited: 11 Abstract: Several effects due to the coupling of the translational mot… read moreAbstract: Several effects due to the coupling of the translational motion of a gas phase atom (H), to the vibrations of a substrate [Cu(100)], are investigated here with time-dependent wave packet methods. Three different propagation techniques, namely, reduced-dimensionality but “exact” wave packet propagation, the time-dependent-self-consistent-field (TDSCF) method, and the “mean-field” (Ehrenfest) mixed quantum-classical-molecular-dynamics scheme (QCMD), are tested against each other and compared with classical trajectory results, and with rigid-surface calculations. Our key findings are: (1) The Cu(100) substrate is very “open” for impinging H atoms, leading to large subsurface and bulk absorption yields; (2) the H atoms can be “hot” for several picoseconds after hitting the surface before they finally settle down in adsorption or absorption sites; (3) while classical mechanics agrees reasonably well with the exact quantum calculation, the mixed quantum-classical and TDSCF approaches which both rely on a single... read less NOT USED (high confidence) T. Rasmussen, Vegge, T. Leffers, O. B. Pedersen, and K. Jacobsen, “Simulation of structure and annihilation of screw dislocation dipoles,” Philosophical Magazine A. 2000. link Times cited: 42 Abstract: Large scale atomistic simulations are used to investigate th… read moreAbstract: Large scale atomistic simulations are used to investigate the properties of screw dislocation dipoles in copper. Spontaneous annihilation is observed for dipole heights less than 1 nm. Equilibrated dipoles of heights larger than 1 nm adopt a skew configuration due to the elastic anisotropy of Cu. The equilibrium splitting width of the screw dislocations decreases with decreasing dipole height, as expected from elasticity theory. The energy barriers, and corresponding transition states for annihilation of stable dipoles are determined for straight and for flexible dislocations for dipole heights up to 5.2 nm. In both cases the annihilation is initiated by cross-slip of one of the dislocations. For straight dislocations the activation energy shows a linear dependence on the inverse dipole height, and for flexible dislocations the dependence is roughly linear for the dipoles investigated. read less NOT USED (high confidence) A. Christensen et al., “Phase diagrams for surface alloys,” Physical Review B. 1997. link Times cited: 312 Abstract: We discuss surface alloy phases and their stability based on… read moreAbstract: We discuss surface alloy phases and their stability based on surface phase diagrams constructed from the surface energy as a function of the surface composition. We show that in the simplest cases of pseudomorphic overlayers there are four generic classes of systems, characterized by the sign of the heat of segregation from the bulk and the sign of the excess interactions between the atoms in the surface (the surface mixing energy). We also consider the more complicated cases with ordered surface phases, nonpseudomorphic overlayers, second layer segregation, and multilayers. The discussion is based on density-functional calculations using the coherent-potential approximation and on effective-medium theory. We give self-consistent density-functional results for the segregation energy and surface mixing energy for all combinations of the transition and noble metals. Finally we discuss in detail the cases Ag/Cu(100), Pt/Cu(111), Ag/Pt(111), Co/Cu(111), Fe/Cu(111), and Pd/Cu(110) in connection with available experimental results. read less NOT USED (high confidence) B. Fruëhberger, J. Eng, and J. G. Chen, “Observation of anomalous reactivities of Ni/Pt(111) bimetallic surfaces,” Catalysis Letters. 1997. link Times cited: 35 NOT USED (high confidence) S. Lemos, R. T. S. Oliveira, M. C. Santos, P. Nascente, L. Bulhões, and E. Pereira, “Electrocatalysis of methanol, ethanol and formic acid using a Ru/Pt metallic bilayer,” Journal of Power Sources. 2007. link Times cited: 40 NOT USED (high confidence) M. Duesbery, “Dislocation motion, constriction and cross-slip in fcc metals,” Modelling and Simulation in Materials Science and Engineering. 1998. link Times cited: 74 Abstract: A Finnis/Sinclair-type interatomic potential for copper is u… read moreAbstract: A Finnis/Sinclair-type interatomic potential for copper is used to examine the properties of dissociated screw dislocations in the face-centred cubic lattice. The critical stress for dislocation motion is found to be a sensitive function of the partial dislocation separation, with a lower limit at least 85% smaller than the Peierls stress. The constriction of Heidenreich-Shockley partials is modelled using an applied stress which interacts only with the edge Burgers vectors; recombination is not observed, but there is a critical separation below which the potential energy of the dislocation rises very rapidly. The classical model of cross-slip, in which the dislocation cannot leave its slip plane unless it is fully constricted, is found to be incorrect. Instead, cross-slip is possible at all partial separations, provided the driving stress is large enough. Finally, a new mechanism for cross-slip nucleation is described. read less NOT USED (high confidence) J. Schiøtz, T. Vegge, and K. Jacobsen, “Atomic-scale modeling of the deformation of nanocrystalline metals,” MRS Proceedings. 1998. link Times cited: 6 Abstract: Nanocrystalline metals, i.e. metals with grain sizes from 5 … read moreAbstract: Nanocrystalline metals, i.e. metals with grain sizes from 5 to 50 nm, display technologically interesting properties, such as dramatically increased hardness, increasing with decreasing grain size. Due to the small grain size, direct atomic-scale simulations of plastic deformation of these materials are possible, as such a polycrystalline system can be modeled with the computational resources available today. The authors present molecular dynamics simulations of nanocrystalline copper with grain sizes up to 13 nm. Two different deformation mechanisms are active, one is deformation through the motion of dislocations, the other is sliding in the grain boundaries. At the grain sizes studied here the latter dominates, leading to a softening as the grain size is reduced. This implies that there is an optimal grain size, where the hardness is maximal. Since the grain boundaries participate actively in the deformation, it is interesting to study the effects of introducing impurity atoms in the grain boundaries. The authors study how silver atoms in the grain boundaries influence the mechanical properties of nanocrystalline copper. read less |
 EMT_Asap_Standard_JacobsenStoltzeNorskov_1996_AlAgAuCuNiPdPt__MO_115316750986_001
EMT_Asap_Standard_JacobsenStoltzeNorskov_1996_AlAgAuCuNiPdPt__MO_115316750986_001