EquilibriumCrystalStructure_A_oI2_71_a_I__TE_231324911199_000
| Title
A single sentence description.
|
Equilibrium crystal structure and energy for I in AFLOW crystal prototype A_oI2_71_a v000 |
|---|---|
| Description |
Computes the equilibrium crystal structure and energy for I in AFLOW crystal prototype A_oI2_71_a at zero temperature and applied stress by performing symmetry-constrained relaxation. The parameters (representing cell and internal degrees of freedom) allowed to vary during the relaxation are: a, b/a, c/a. The initial guess for these parameters is: 8.7854, 0.43612129, 0.33737792, obtained from http://aflowlib.duke.edu/AFLOWDATA/ICSD_WEB/ORCI/I1_ICSD_109039/CONTCAR.relax.vasp, the relaxed structure corresponding to Aflowlib Unique IDentifier aflow:70b381f79a677520 |
| Species
The supported atomic species.
| I |
| Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
|
Computer generated |
| Contributor |
I Nikiforov |
| Maintainer |
I Nikiforov |
| Developer |
I Nikiforov Ellad B. Tadmor Daniel S. Karls Moon-ki Choi |
| Published on KIM | 2023 |
| How to Cite | Click here to download this citation in BibTeX format. |
| Funding | Not available |
| Short KIM ID
The unique KIM identifier code.
| TE_231324911199_000 |
| Extended KIM ID
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
| EquilibriumCrystalStructure_A_oI2_71_a_I__TE_231324911199_000 |
| Citable Link | https://openkim.org/cite/TE_231324911199_000 |
| KIM Item Type | Test |
| Driver | EquilibriumCrystalStructure__TD_457028483760_000 |
| Properties
Properties as defined in kimspec.edn.
These properties are inhereted from the Test Driver.
| |
| KIM API Version | 2.3 |
| Simulator Name
The name of the simulator as defined in kimspec.edn.
This Simulator Name is inhereted from the Test Driver.
| ase |
| Model | Error Categories | Link to Error page |
|---|---|---|
| LJ_ElliottAkerson_2015_Universal__MO_959249795837_003 | other | view |
No Driver
| Model | Error Categories | Link to Error page |
|---|---|---|
| Sim_LAMMPS_EIM_Zhou_2010_BrClCsFIKLiNaRb__SM_259779394709_000 | other | view |
| EquilibriumCrystalStructure_A_oI2_71_a_I__TE_231324911199_000.txz | Tar+XZ | Linux and OS X archive |
| EquilibriumCrystalStructure_A_oI2_71_a_I__TE_231324911199_000.zip | Zip | Windows archive |
This Test requires a Test Driver. Archives for the Test Driver EquilibriumCrystalStructure__TD_457028483760_000 appear below.
| EquilibriumCrystalStructure__TD_457028483760_000.txz | Tar+XZ | Linux and OS X archive |
| EquilibriumCrystalStructure__TD_457028483760_000.zip | Zip | Windows archive |
Parameters for the ‘new’ potential
This parameterization is optimized for tetrahedral structures of silicon (referred to in the paper as the ‘new’ potential in contrast to an ‘old’ version published two years earlier more suitable for high-pressure phases of silicon. The parameters for the ‘old’ potential were by fitting the model to structural energies calculated using density functional theory within the local-density approximation (LDA). The method used for determining the parameters for the ‘new’ potential is not mentioned explicitly, so we presume that the authors followed the same framework for both of their models. The influence distance parameter \(D\) is not reported originally by the authors, here we use a value of 6 angstroms for reasons of speedup. Also the values of parameters \(B_1\) and \(B_2\) reported in the original article correspond to half of the values given below. This is because in our model, the summation of three-body potential term is implemented more efficiently in an asymmetric manner, while Biswas and Hamann assumed a symmetric form for the three-body potential in their original article.
Table of parameters
| Parameter | Value | Units |
|---|---|---|
| \(A_1\) | \(142.2922\) | \(eV\) |
| \(A_2\) | \(-107.0338\) | \(eV\) |
| \(B_1\) | \(26.0598\) | \(eV\) |
| \(B_2\) | \(1.3441478\) | \(eV\) |
| \(\lambda_1\) | \(0.5200836\) | \(\overset{\circ}{A}^{-2}\) |
| \(\lambda_2\) | \(0.4206931\) | \(\overset{\circ}{A}^{-2}\) |
| \(\alpha_1\) | \(0.3034373\) | \(\overset{\circ}{A}^{-2}\) |
| \(\alpha_2\) | \(0.3191903\) | \(\overset{\circ}{A}^{-2}\) |
| \(r_c\) | \(3.9527357\) | \(\overset{\circ}{A}\) |
| \(\mu\) | \(0.3120580\) | \(\overset{\circ}{A}\) |
| \(D\) | \(6.0\) | \(\overset{\circ}{A}\) |
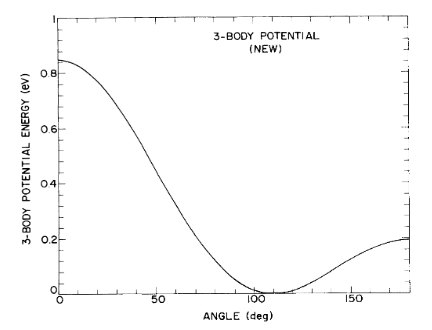
Representative plots
A plot from the original article depicting the angular variation of the three-body potential for the new classical Si model is shown. The bond lengths were set equal to 2.35 angstroms, which is the equilibrium bond length of Si.
Login to edit Wiki content