Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
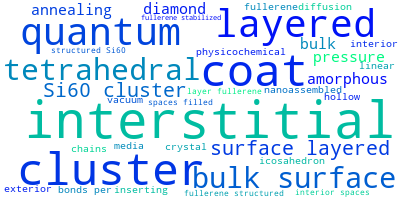
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) A. Galashev, “Molecular dynamics study of hydrogenated silicon clusters at high temperatures,” Molecular Physics. 2009. link Times cited: 5 Abstract: This paper reports on a study of the stability of silicon cl… read moreAbstract: This paper reports on a study of the stability of silicon clusters of intermediate size at a high temperature. The temperature dependence of the physicochemical properties of 60- and 73-atom silicon nanoparticles are investigated using the molecular dynamics method. The 73-atom particles have a crystal structure, a random atomic packing, and a packing formed by inserting a 13-atom icosahedron into a 60-atom fullerene. They are surrounded by a ‘coat’ from 60 atoms of hydrogen. The nanoassembled particle at the presence of a hydrogen ‘coat’ has the most stable number (close to four) of Si–Si bonds per atom. The structure and kinetic properties of a hollow single-layer fullerene-structured Si60 cluster are considered in the temperature range 10 K ≤ T ≤ 1760 K. Five series of calculations are conducted, with a simulation of several media inside and outside the Si60 cluster, specifically, the vacuum and interior spaces filled with 30 and 60 hydrogen atoms with and without the exterior hydrogen environment of 60 atoms. Fullerene surrounded by a hydrogen ‘coat’ and containing 60 hydrogen atoms in the interior space has a higher stability. Such clusters have smaller self-diffusion coefficients at high temperatures. The fullerene stabilized with hydrogen is stable to the formation of linear atomic chains up to the temperatures 270–280 K. read less USED (high confidence) A. Galashev, “Thermal instability of silicon fullerenes stabilized with hydrogen: Computer simulation,” Semiconductors. 2008. link Times cited: 4 USED (high confidence) A. Shalabi, K. Kamel, and M. M. Assem, “Theoretical characterization and many-body expansion analysis of BF3, BCl3, AlF3 and AlCl3 interactions,” Theoretica chimica acta. 1995. link Times cited: 4 USED (high confidence) W. K. Liu and C. McVeigh, “Predictive multiscale theory for design of heterogeneous materials,” Computational Mechanics. 2008. link Times cited: 57 USED (low confidence) P. Käshammer, N. Borgardt, M. Seibt, and T. Sinno, “Quantitative assessment of molecular dynamics-grown amorphous silicon and germanium films on silicon (111),” Surface Science. 2016. link Times cited: 3 USED (low confidence) P. Ghosh and M. Ranganathan, “Submonolayer growth study using a solid-on-solid model for 2 × 1 reconstructed surfaces of diamond-like lattices,” Surface Science. 2014. link Times cited: 7 USED (low confidence) H. Lu, J. Q. Xie, and J. Feng, “Simulation study on Si and Ge film growth by cluster deposition,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2000. link Times cited: 10 USED (low confidence) V. Konoplev, A. Gras-marti, E. P. Andribet, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “Effect of temperature on the bulk atomic relocation in low-energy collision cascades in silicon: a molecular dynamics study,” Radiation Effects and Defects in Solids. 1995. link Times cited: 4 Abstract: The production of damage in a Si lattice by internally start… read moreAbstract: The production of damage in a Si lattice by internally starting 100 eV self-recoils has been studied using a MD simulation. Different initial lattice temperatures below the Debye temperature for Si have been considered. The number of stable atomic displacements and the amount of atomic mixing increase with the initial target temperature. The increase with temperature of atomic mixing is nonlinear -appreciable changes take place between 300 and 500 K, while the difference between the amount of mixing corresponding to 0 and 300 K is negligibly small. The size of the cascade zone in which stable atomic displacements occur doubles itself for temperature changes between 0 and 300 K, with a value for 500 K lying in between. This nonmonotonic variation with the initial target temperature of the size of the cascade zone may have its origin in the correlation between the initial direction of motion of the starting recoil and the directions of thermal velocities of the neighbouring atoms around this recoil. read less USED (low confidence) E. Beam, “Computer simulation of the surface topology of (001) silicon resulting from the termination of 12〈110〉 edge dislocations with Burgers vectors parallel to the surface,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 1993. link Times cited: 0 USED (low confidence) T. Weber and F. Stillinger, “Dynamical branching during fluorination of the dimerized Si(100) surface: A molecular dynamics study,” Journal of Chemical Physics. 1990. link Times cited: 70 Abstract: Collections of classical trajectories have been numerically … read moreAbstract: Collections of classical trajectories have been numerically generated for individual F2 molecules impinging at normal incidence on a Si(100) surface at 0 K dimerized in a p(2×1) pattern. A linear combination of two‐atom and three‐atom interaction functions represents the potential energy. Trajectories fall into four categories: (a) non‐reactive F2 rebound, (b) monofluorination at a surface dangling bond with energetic expulsion into the vacuum of the remaining F atom, (c) difluorination of a pair of dangling bonds, and (d) monofluorination with retention of the second F in a weakly bound Si–F⋅⋅⋅F surface complex. Surface patterns for difluorination, (c), indicate absence of surface diffusion during this mode of chemisorption. Increasing either the translational kinetic energy or the vibrational excitation of the incident F2 appears to enhance its surface reactivity. read less USED (low confidence) Stansfield, Broomfield, and Clary, “Classical-trajectory calculations on Ar+ sputtering of a Si(001) surface using an ab initio potential.,” Physical review. B, Condensed matter. 1989. link Times cited: 37 Abstract: We describe classical-trajectory calculations of sputtering … read moreAbstract: We describe classical-trajectory calculations of sputtering yields for Ar/sup +/-ion collisions with a Si(001) surface. The Ar/sup +/-Si and short-ranged Si-Si interaction potentials were calculated using the ab initio Hartree-Fock and configuration-interaction methods. The low-energy potential describing the silicon solid is the two- and three-body form due to Stillinger and Weber. We compare the calculated sputtering yields with experiment. The potential-energy surface strongly influences the calculated sputtering yields, and it is found that the most reasonable agreement is obtained from our potentials using the (2 x 1) Si(001) reconstructed surface rather than the bulk-terminated surface. Analysis of the kinetic energy and angular distributions of the sputtered silicon atoms and of cluster yields has provided a mechanism of ejection. read less USED (low confidence) J. Lampinen, R. Nieminen, and K. Kaski, “Molecular dynamics simulation of epitaxial growth of the Si(001) surface,” Surface Science. 1988. link Times cited: 16 USED (low confidence) P. Fedders, “Defects, tight binding, and ab initio molecular dynamics simulations on a-Si,” Journal of Non-crystalline Solids. 1991. link Times cited: 0 NOT USED (low confidence) S. Surulere, M. Shatalov, and J. Ehigie, “Analysis of oscillations in 1D nanostructures influenced by different concrete potential functions,” International Journal of Non-Linear Mechanics. 2022. link Times cited: 0 NOT USED (low confidence) L. J. Lewis, “Fifty years of amorphous silicon models : the end of the story?,” Journal of Non-Crystalline Solids. 2022. link Times cited: 8 NOT USED (low confidence) R. Drautz, “Atomic cluster expansion for accurate and transferable interatomic potentials,” Physical Review B. 2019. link Times cited: 260 NOT USED (low confidence) H. Li et al., “Cost-effective synthesis of carbazole/triphenylsilyl host materials with multiple σ-π conjugation for blue phosphorescent organic light-emitting diodes,” Dyes and Pigments. 2018. link Times cited: 7 NOT USED (low confidence) T. Gao, W. Yan, X. Guo, Y. Qin, and Q. Xie, “Structural properties in liquid Si during rapid cooling processes,” Physica B-condensed Matter. 2013. link Times cited: 5 NOT USED (low confidence) A. P. Bart’ok, R. Kondor, and G. Csányi, “On representing chemical environments,” Physical Review B. 2012. link Times cited: 1246 Abstract: We review some recently published methods to represent atomi… read moreAbstract: We review some recently published methods to represent atomic neighborhood environments, and analyze their relative merits in terms of their faithfulness and suitability for fitting potential energy surfaces. The crucial properties that such representations (sometimes called descriptors) must have are differentiability with respect to moving the atoms and invariance to the basic symmetries of physics: rotation, reflection, translation, and permutation of atoms of the same species. We demonstrate that certain widely used descriptors that initially look quite different are specific cases of a general approach, in which a finite set of basis functions with increasing angular wave numbers are used to expand the atomic neighborhood density function. Using the example system of small clusters, we quantitatively show that this expansion needs to be carried to higher and higher wave numbers as the number of neighbors increases in order to obtain a faithful representation, and that variants of the descriptors converge at very different rates. We also propose an altogether different approach, called Smooth Overlap of Atomic Positions, that sidesteps these difficulties by directly defining the similarity between any two neighborhood environments, and show that it is still closely connected to the invariant descriptors. We test the performance of the various representations by fitting models to the potential energy surface of small silicon clusters and the bulk crystal. read less NOT USED (low confidence) A. Gufan, O. V. Kukin, Y. M. Gufan, and A. Smolin, “Models of three-particle interactions and theory of nonlinear deformations of crystals,” Physics of the Solid State. 2012. link Times cited: 6 NOT USED (low confidence) J. Yu, S. Sinnott, and S. Phillpot, “Charge optimized many-body potential for the Si/SiO2 system,” Physical Review B. 2007. link Times cited: 151 Abstract: A dynamic-charge, many-body potential for the Si/SiO{sub 2} … read moreAbstract: A dynamic-charge, many-body potential for the Si/SiO{sub 2} system, based on an extended Tersoff potential for semiconductors, is proposed and implemented. The validity of the potential function is tested for both pure silicon and for five polymorphs of silica, for which good agreement is found between the calculated and experimental structural parameters and energies. The dynamic charge transfer intrinsic to the potential function allows the interface properties to be captured automatically, as demonstrated for the silicon/{beta}-cristobalite interface. read less NOT USED (low confidence) S. Billeter, A. Curioni, D. Fischer, and W. Andreoni, “Ab initio derived augmented Tersoff potential for silicon oxynitride compounds and their interfaces with silicon,” Physical Review B. 2006. link Times cited: 42 Abstract: Coordination-dependent interatomic potentials are proposed f… read moreAbstract: Coordination-dependent interatomic potentials are proposed for silicon oxides and oxynitrides\char22{}also hydrogenated ones\char22{}with a functional form based on the widely used Tersoff silicon potential. They are intended for an accurate sampling of the configurational space of realistic silicon oxynitride systems and their interfaces with silicon, including defects and changes of oxidation states. The parameters, which are given in the text, are obtained by simultaneously mapping forces and energies onto the results of density-functional-theory calculations performed for a set of diverse systems and configurations and a wide composition range. Application to a larger set of systems and configurations shows the transferability of these augmented Tersoff potentials and their validity in predicting bulk lattice parameters, energetics of defect relaxation, and vibrational spectra. read less NOT USED (low confidence) A. Tekin and B. Hartke, “GLOBAL GEOMETRY OPTIMIZATION OF SILICON CLUSTERS EMPLOYING EMPIRICAL POTENTIALS, DENSITY FUNCTIONALS, AND AB INITIO CALCULATIONS,” Journal of Theoretical and Computational Chemistry. 2005. link Times cited: 13 Abstract: Sin clusters in the size range n = 4–30 have been investigat… read moreAbstract: Sin clusters in the size range n = 4–30 have been investigated using a combination of global structure optimization methods with DFT and ab initio calculations. One of the central aims is to provide explanations for the structural transition from prolate to spherical outer shapes at about n = 25, as observed in ion mobility measurements. Firstly, several existing empirical potentials for silicon and a newly generated variant of one of them were better adapted to small silicon clusters, by global optimization of their parameters. The best resulting empirical potentials were then employed in global cluster structure optimizations. The most promising structures from this stage were relaxed further at the DFT level with the hybrid B3LYP functional. For the resulting structures, single point energies have been calculated at the LMP2 level with a reasonable medium-sized basis set, cc-pVTZ. These DFT and LMP2 calculations were also carried out for the best structures proposed in the literature, including the most recent ones, to obtain the currently best and most complete overall picture of the structural preferences of silicon clusters. In agreement with recent findings, results obtained at the DFT level do support the shape transition from prolate to spherical structures, beginning with Si26 (albeit not completely without problems). In stark contrast, at the LMP2 level, the dominance of spherical structures after the transition region could not be confirmed. Instead, just as below the transition region, prolate isomers are obtained as the lowest-energy structures for n ≤ 29. We conclude that higher (probably multireference) levels of theoretical treatments are needed before the puzzle of the silicon cluster shape transition at n = 25 can safely be considered as explained. read less NOT USED (low confidence) P. Gunes, Şi̇mşek S., and S. Erkoç, “a Comparative Study of Empirical Potential Energy Functions,” International Journal of Modern Physics C. 2004. link Times cited: 2 Abstract: A comparative study has been performed for silicon microclus… read moreAbstract: A comparative study has been performed for silicon microclusters, Si3 and Si4, considering fifteen different empirical potential energy functions. It has been found that only two of the empirical potential energy functions give linear structure more stable for Si3, the remaining potential functions give triangular structure as more stable. In the case of Si4 microclusters eight potential functions give open tetrahedral structure as more stable, two functions give perfect tetrahedral as more stable, three functions give square structure as more stable, and two functions give linear structure as more stable. read less NOT USED (low confidence) Y. Umeno, T. Kitamura, K. Date, M. Hayashi, and T. Iwasaki, “Optimization of interatomic potential for Si/SiO2 system based on force matching,” Computational Materials Science. 2002. link Times cited: 25 NOT USED (low confidence) A. Barnard and S. Russo, “Development of an improved Stillinger-Weber potential for tetrahedral carbon using ab initio (Hartree-Fock and MP2) methods,” Molecular Physics. 2002. link Times cited: 28 Abstract: An improved interatomic potential for tetrahedral carbon is … read moreAbstract: An improved interatomic potential for tetrahedral carbon is presented. This potential is of the Stillinger-Weber (SW) type and has been determined from calculations performed on a select group of small hydrocarbon molecules, chosen for their similarities to the tetrahedral lattice of bulk diamond. Counterpoise corrected Hartree-Fock (HF) and second-order Møller-Plesset perturbation theory (MP2) calculations were performed on ethane, 2,2-dimethylpropane (neo-pentane, (C5H12), 2-dimethyl-3-dimethylbutane (neobutane, C8H18) and cyclohexane (C6H12) in order to determine the two-body (stretching) and three-body (bond bending) energies. The suitability of these molecules to model the properties of diamond was determined by comparison of CC bond length, well depth, CCC bond angle, simultaneous stretch and bend energy and force constants to those of bulk diamond. It was found that neopentane provided the best overall description of tetrahedral bonded carbon. The ab initio derived stretch and bend energies were fitted to the SW potential energy terms and the SW parameters calculated. The newly parametrized SW potential was then evaluated by calculating the stretch force constants, elastic constants and the X-point phonon modes of bulk diamond. read less NOT USED (low confidence) B. Marsen, M. Lonfat, P. Scheier, and K. Sattler, “The energy gap of pristine silicon clusters,” Journal of Electron Spectroscopy and Related Phenomena. 2000. link Times cited: 10 NOT USED (low confidence) C. Herrero, “Path-integral Monte Carlo study of amorphous silicon,” Journal of Non-crystalline Solids. 2000. link Times cited: 8 NOT USED (low confidence) H. Cox, R. Johnston, and J. Murrell, “Empirical Potentials for Modeling Solids, Surfaces, and Clusters,” IEEE Journal of Solid-state Circuits. 1999. link Times cited: 48 Abstract: A review of studies that have been made using the Murrell–Mo… read moreAbstract: A review of studies that have been made using the Murrell–Mottram two-plus-three-body empirical potential is presented. The explicit many-body nature of the potential is described and the fitting of these potentials to experimental data on one or more solid phases is detailed. Comparisons are made between potentials for various nonmetallic and metallic elements, from which trends in the parameters defining the potentials can clearly be seen. Examples of the many applications of these potentials to the study of solids (relative stabilities and phase transitions), surfaces (energies, relaxation and reconstructions), melting (both of the bulk and of the surfaces), and clusters (structures, growth, and dynamics) are given. read less NOT USED (low confidence) M. M. J. Treacy, J. Gibson, and P. J. Keblinski, “Paracrystallites found in evaporated amorphous tetrahedral semiconductors,” Journal of Non-crystalline Solids. 1998. link Times cited: 142 NOT USED (low confidence) J. Q. Xie and J. Feng, “Molecular-dynamics simulation of silicon film growth from cluster beams,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1998. link Times cited: 8 NOT USED (low confidence) X. Liu, Z. Zhen, and J.-cheng Zhang, “New Potential Energy Functions for Diamond and α-Tin Crystals,” Chinese Physics Letters. 1998. link Times cited: 1 Abstract: A new model of potential energy functions for atomic solids … read moreAbstract: A new model of potential energy functions for atomic solids is given and applied to diamond and α-tin crystals. In the new model, a factor expressing the characters of covalent bonds has been included. Therefore it is suitable for covalent crystals. New potentials of C and α-Sn crystals accurately reproduce experimental elastic constants and phonon dispersion curves and so on. The set of new potentials is accurate enough for computer simulations. read less NOT USED (low confidence) L. Marqués, M. Jaraíz, J. Rubio, J. Vicente, L. Bailón, and J. Barbolla, “Molecular dynamics simulations of ion bombardment processes,” Materials Science and Technology. 1997. link Times cited: 3 Abstract: AbstractAn improved molecular dynamics technique that allows… read moreAbstract: AbstractAn improved molecular dynamics technique that allows reduction of the computation time required in ion bombardment simulations is presented. This technique has been used to study the influence of the target temperature and structure on the argon sputtering of silicon. Molecular dynamics simulations of l keV Ar+ ion bombardment of silicon were carried out for several types of sample: (100) crystalline at 0 K, (100) crystalline at 300 K, and amorphous at 300 K. The yield of the sputtering process and the energy distribution of the sputtered atoms have been obtained. These results show that the sputtering process depends on the target surface binding energy which, in turn, is very sensitive to the structure of the sample surface. read less NOT USED (low confidence) E. Kaxiras, “Review of atomistic simulations of surface diffusion and growth on semiconductors,” Computational Materials Science. 1996. link Times cited: 17 NOT USED (low confidence) X. Liu, “New model of potential energy functions for atomic solids. Part 2. New potentials of silicon and germanium crystals,” Journal of Molecular Structure-theochem. 1995. link Times cited: 1 NOT USED (low confidence) L. Xin-hou, “New Model of Potential Energy Functions for Atomic Solids and Application to Silicon Crystal,” Chinese Physics Letters. 1995. link Times cited: 1 Abstract: A new theoretical model of potential energy functions for at… read moreAbstract: A new theoretical model of potential energy functions for atomic solids is developed. An angular factor is included in this model and its effect is discussed. Using this new model, a new preliminary potential for silicon crystal is derived. Calculated phonon dispersion curves, using this new potential, is given. A good agreement has been found. read less NOT USED (low confidence) A. A. Valuev, A. S. Kaklyugin, and H. E. Norman, “Molecular modelling of the chemical interaction of atoms and molecules with a surface,” Russian Chemical Reviews. 1995. link Times cited: 3 Abstract: The modelling of a surface as an assembly of moving atoms in… read moreAbstract: The modelling of a surface as an assembly of moving atoms interacting with one another and with an incident particle is examined. Both dynamic methods for the modelling of a surface (for short times) and probability methods (for long times) are analysed. The Massey adiabaticity criterion has been used to determine the regions of applicability of the methods of molecular dynamics. Within the framework of probability methods, the chemical bond is described with the aid of Harrison's generalised periodic system of the elements. Together with the general modeling problems, the reconstruction of the surface, physical and chemical sorption, as well as the modification of the surface and of its morphology as a result of the multiple repetition of elementary processes (precipitation, etching, corrosion) are discussed. The bibliography includes 169 references. read less NOT USED (low confidence) V. Konoplev and A. Gras-marti, “Molecular dynamics simulation of low-energy collision cascades and atomic mixing in silicon,” Philosophical Magazine. 1995. link Times cited: 4 Abstract: We investigate atomic relocation processes in silicon at OK,… read moreAbstract: We investigate atomic relocation processes in silicon at OK, initiated by an internal 100eV silicon recoil. The molecular dynamics code MODYSEM is used, based on a Tersoff potential for silicon. A fitting procedure was used for the generation of 8 potential valid over the whole energy range of interest. The contribution of the collisional, spontaneous relaxation and thermalization stages to the atomic relocation process are discussed. A threshold distance for the definition of relocated atoms is determined, which separates atomic displacements into stable and unstable (or transient) groups. The atomic mixing process is quantified in terms of the first and second spatial moments over the relocation cross-section. These moments depend on the criterion used to define a relocated Si atom, with short-distance thermal-like atomic displacements, which appear during the thermalization stage, dominating the values of the spatial moments. However, the moments of the relocation cross-section calculated by c... read less NOT USED (low confidence) T. Ito, “RECENT PROGRESS IN COMPUTER-AIDED MATERIALS DESIGN FOR COMPOUND SEMICONDUCTORS,” Journal of Applied Physics. 1995. link Times cited: 50 Abstract: Recent progress in computational materials science in the ar… read moreAbstract: Recent progress in computational materials science in the area of semiconductor materials is reviewed. Reliable predictions can now be made for a wide range of problems, such as band structure and structural and thermodynamic properties of various compound semiconductors, using electronic theories such as the pseudopotential method. Further applications are examined by investigating the behavior of various atomic species in semiconductors, including the stability and band structure of heterostructures, superlattices, lattice defects, alloy systems, and surface‐related properties such as surface reconstruction, surface passivation, and adatom migration during thin film growth. The empirical interatomic potentials, pseudopotential, and stochastic Monte Carlo methods are used. An overview of these issues is provided and the latest achievements are presented to illustrate the capability of the theoretical‐computational approach by comparing experimental results. The constituents of the semiconductors that are... read less NOT USED (low confidence) K. C. Low, H. Lim, and C. Ong, “Vacancies on the Si(001) c(4*2) surface,” Journal of Physics: Condensed Matter. 1994. link Times cited: 5 Abstract: We have employed a parametrized tight-binding molecular-dyna… read moreAbstract: We have employed a parametrized tight-binding molecular-dynamics scheme in the study of the phenomenon of vacancies on the Si(001) c(4*2) surface. Simulated annealing is performed with a 'fictitious-Lagrangian' procedure to determine the optimal structures of a single and a dimer vacancy on this surface. A monovacancy is found to be less stable than a dimer vacancy, which agrees with experimental observations. We also show that there is a possible anisotropy in the surface migration of a dimer vacancy on the surface. The calculated activation energy for dimer-vacancy diffusion is 0.6 eV higher than that estimated experimentally at high temperatures. read less NOT USED (low confidence) M. Schreiber and B. Lamberts, “Determination of parameter-free model potentials for the molecular dynamics simulation of amorphous semiconductors — application to silicon,” Journal of Molecular Structure-theochem. 1994. link Times cited: 0 NOT USED (low confidence) Z. Jiang and R. A. Brown, “Modelling oxygen defects in silicon crystals using an empirical interatomic potential,” Chemical Engineering Science. 1994. link Times cited: 14 NOT USED (low confidence) K. Roos and U. C. Tringides, “Low-temperature, flux-independent epitaxy in Ag/Si(111),” Surface Science. 1994. link Times cited: 7 NOT USED (low confidence) A. Shalabi and A. M. E. Misiery, “Routes for LiF unit cell growth,” Chemical Physics Letters. 1993. link Times cited: 3 NOT USED (low confidence) A. Mistriotis, A. Zdetsis, G. Froudakis, and M. Menon, “Reproduction of quantum tight-binding effects in silicon clusters by a four-body classical model,” Journal of Physics: Condensed Matter. 1993. link Times cited: 0 Abstract: The results obtained by a recently proposed empirical potent… read moreAbstract: The results obtained by a recently proposed empirical potential for silicon which includes four-body terms are compared with the results of quantum-mechanical tight-binding calculations. In particular, the ground-state energy and structure of the Si33 cluster were computed by both methods. By performing an equivalent calculation using only up to three-body interactions the authors demonstrate that the four-body term is absolutely necessary in order to achieve good agreement with the quantum method. read less NOT USED (low confidence) C. S. Carmer, B. Weiner, and M. Frenklach, “Molecular dynamics with combined quantum and empirical potentials: C2H2 adsorption on Si(100),” Journal of Chemical Physics. 1993. link Times cited: 71 Abstract: Classical trajectory calculations were employed to study the… read moreAbstract: Classical trajectory calculations were employed to study the reaction of acetylene with dimer sites on the Si(100) surface at 105 K. Two types of potential energy functions were combined to describe interactions for different regions of the model surface. A quantum mechanical potential based on the semiempirical AM1 Hamiltonian was used to describe interactions between C2H2 and a portion of the silicon surface, while an empirically parametrized potential was developed to extend the size of the surface and simulate the dynamics of the surrounding silicon atoms. Reactions of acetylene approaching different sites were investigated, directly above a surface dimer, and between atoms from separate dimers. In all cases, the outcome of C2H2 surface collisions was controlled by the amount of translational energy possessed by the incoming molecule. Acetylene molecules with high translational energy reacted with silicon dimers to form surface species with either one or two Si–C bonds. Those molecules with low transl... read less NOT USED (low confidence) Tsumuraya, Ishibashi, and Kusunoki, “Statistics of Voronoi polyhedra in a model silicon glass.,” Physical review. B, Condensed matter. 1993. link Times cited: 14 Abstract: We clarify the local structure in a model silicon glass by u… read moreAbstract: We clarify the local structure in a model silicon glass by use of Voronoi-polyhedron analysis. The glass is produced by molecular dynamics with a Stillinger-Weber potential. The atoms in the glass are nearly distinguishable: there are about 200 types in the system with 216 atoms. The analysis clarifies that the polyhedra are formed by a small number of large-area polygons or by a large number of small-area polygons. This feature is different from those in Lennard-Jones glasses or metallic glasses and is attributed to the loose-packed structure even in the glass state, in which the atoms still have directional bonding read less NOT USED (low confidence) V. M. Bedanov and D. Mukhin, “The mechanism of anisotropic island growth in molecular-beam epitaxy of Si on Si(001),” Surface Science. 1992. link Times cited: 7 NOT USED (low confidence) R. Fournier, S. Sinnott, and A. Depristo, “Density functional study of the bonding in small silicon clusters,” Journal of Chemical Physics. 1992. link Times cited: 90 Abstract: We report the ground electronic state, equilibrium geometry,… read moreAbstract: We report the ground electronic state, equilibrium geometry, vibrational frequencies, and binding energy for various isomers of Sin(n = 2–8) obtained with the linear combination of atomic orbitals‐density functional method. We used both a local density approximation approach and one with gradient corrections. Our local density approximation results concerning the relative stability of electronic states and isomers are in agreement with Hartree–Fock and Mo/ller–Plesset (MP2) calculations [K. Raghavachari and C. M. Rohlfing, J. Chem. Phys. 89, 2219 (1988)]. The binding energies calculated with the gradient corrected functional are in good agreement with experiment (Si2 and Si3) and with the best theoretical estimates. Our analysis of the bonding reveals two limiting modes of bonding and classes of silicon clusters. One class of clusters is characterized by relatively large s atomic populations and a large number of weak bonds, while the other class of clusters is characterized by relatively small s atomic p... read less NOT USED (low confidence) W. Niessen and V. G. Zakrzewski, “Complex Electron Affinity Processes in Clusters of S and Si.” 1992. link Times cited: 2 Abstract: Vertical and in some cases adiabatic electron affinities are… read moreAbstract: Vertical and in some cases adiabatic electron affinities are calculated for the clusters S4 and Sin, n = 3 – 7 with large basis sets. The effects of electron correlation are taken into account by CI and Green function techniques. The clusters show a complex behaviour upon electron attachment. The isomers of 84 show normal electron capture processes as well as electron attachment with shake-up. The Si clusters show multiple affinity states resulting from capture of an electron into different orbitals: Si3 C2v has at least three, Si4 D2h four, Si5 D3h two, Si6 D4v one, Si6 C2v three and Si7 D5h two affinity states (vertical processes: Sin + e− ± Sin + hν). For the Sin clusters in some cases shake-up affinities are calculated which are positive. The effects of electron correlation on the electron affinities are extremely large for the Si clusters in particular. In several cases the differences between the adiabatic and vertical electron affinities are very large amounting up to 1.5 eV. read less NOT USED (low confidence) C. L. Cleveland and U. Landman, “Dynamics of Cluster-Surface Collisions,” Science. 1992. link Times cited: 208 Abstract: The structure, energetics, and dynamics of shock conditions … read moreAbstract: The structure, energetics, and dynamics of shock conditions generated in a nano-cluster upon impact on a crystalline surface are investigated with molecular-dynamics simulations for a 561-atom argon cluster incident with a velocity of 3 kilometers per second onto a sodium chloride surface. The "piling-up" shock phenomenon occurring upon impact, coupled with cascades of energy and momentum transfer processes and inertial confinement of material in the interior of the cluster, creates a transient medium lasting for about a picosecond and characterized by extreme local density, pressure, and kinetic temperature. The nano-shock conditions and impulsive nature of interactions in the newly formed compressed nonequilibrium environment open avenues for studying chemical reactivity and dynamics catalysed via cluster impact. read less NOT USED (low confidence) W. Tiller, “The role of ledges in stress tensor-mediated surface processes for Si and GaAs,” Metallurgical Transactions A. 1991. link Times cited: 0 NOT USED (low confidence) M. Heggie, “Semiclassical interatomic potential for carbon and its application to the self-interstitial in graphite,” Journal of Physics: Condensed Matter. 1991. link Times cited: 33 Abstract: A semiclassical interatomic potential for carbon is discusse… read moreAbstract: A semiclassical interatomic potential for carbon is discussed which is based on the proximity cell (the Wigner-Seitz cell) around each atom. It introduces three internal degrees of freedom per atom, representing the magnitude and direction of the p orbital that is not involved in sp hybridization. Its direct interpolation between sp2 and sp3 configurations combined with good elastic properties allows its use on problematic defects, such as the interplanar interstitial in graphite, which is given as an example. read less NOT USED (low confidence) M. Kohyama, “On the transferable SETB method for Si,” Journal of Physics: Condensed Matter. 1991. link Times cited: 21 Abstract: The two types of transferable semi-empirical tight-binding (… read moreAbstract: The two types of transferable semi-empirical tight-binding (SETB) method for Si recently proposed by Goodwin et al. (1989) and by Sawada, which are intended to reproduce the binding energies and equilibrium volumes of variously coordinated structures of Si, have been examined and compared with each other. It has been found that there are some drawbacks in the method proposed by Goodwin et al, and that the method proposed by Sawada is much superior. The parameters in the Sawada method have been readjusted in order to apply this method to lattice defects or disordered systems of Si. The present results indicate the importance of incorporating the dependence on the local environment into the repulsive energy in the transferable SETB method. This can be explained by the origin of the repulsive energy. read less NOT USED (low confidence) A. Silverman, J. Adler, and R. Weil, “Computer modelling of the diffusion mechanisms of fluorine in amorphous silicon,” Thin Solid Films. 1990. link Times cited: 9 NOT USED (low confidence) J. Hafner and W. Jank, “The electronic structure of liquid metals,” Journal of Physics: Condensed Matter. 1990. link Times cited: 1 Abstract: The authors present first-principles calculations of the ele… read moreAbstract: The authors present first-principles calculations of the electronic structure of molten simple and transition metals. read less NOT USED (low confidence) U. Ray, M. Jarrold, K. Creegan, and J. Bower, “Studies of the chemistry of large semiconductor cluster ions,” International Journal of Mass Spectrometry and Ion Processes. 1990. link Times cited: 5 NOT USED (low confidence) M. Kohyama, S. Kose, M. Kinoshita, and R. Yamamoto, “The self-consistent tight-binding method: application to silicon and silicon carbide,” Journal of Physics: Condensed Matter. 1990. link Times cited: 27 Abstract: The self-consistent tight-binding (SCTB) model proposed by M… read moreAbstract: The self-consistent tight-binding (SCTB) model proposed by Majewski and Vogl (1987) has been extended to be applicable for calculations of lattice defects in solids or disordered systems with both ionic and covalent characters that cannot be treated using other types of tight-binding theories. The precise formulation of electronic structure, total energy and atomic forces in the supercell technique has been presented. In order to apply this method to lattice defects in SiC, the parameters and functional forms have been examined so as to reproduce the basic properties of Si, SiC and C. The nature of the bonding and the phase stability in Si and SiC have been analysed by the present SCTB method. read less NOT USED (low confidence) K. Raghavachari, “Theoretical studies on silicon clusters,” Phase Transitions. 1990. link Times cited: 48 Abstract: This is a brief review of the theoretical studies which have… read moreAbstract: This is a brief review of the theoretical studies which have been carried out to understand the nature of the structures, stabilities and fragmentation behavior of silicon clusters. For the small clusters Si2-Si10, accurate quantum chemical or local density functional calculations have been carried out. These studies have shown that the small clusters are more compact and considerably different from any structures which may be inferred from microcrystalline models based on the diamond lattice for silicon. Clusters containing 4, 6, 7 and 10 atoms have been identified as “magic numbers” for small silicon clusters and the ground state energetics of these clusters are consistent with the observations from recent photofragmentation and photoelectron experiments. Recent attempts to study larger silicon clusters including the efforts to derive accurate silicon-silicon interaction potentials are also described. read less NOT USED (low confidence) M. Jarrold, U. Ray, and K. Creegan, “Chemistry of semiconductor clusters: Large silicon clusters are much less reactive towards oxygen than the bulk,” Journal of Chemical Physics. 1990. link Times cited: 70 Abstract: The chemical reactions of Si+n (n=10–65) with O2 have been i… read moreAbstract: The chemical reactions of Si+n (n=10–65) with O2 have been investigated using selected ion drift tube techniques. The smaller clusters are etched by O2 to give Si+n−2 (and two SiO molecules) and the larger clusters chemisorb oxygen forming an SinO+2 adduct. The transition occurs between n=29 and 36 under the conditions employed. There are large variations in the reactivity of the smaller clusters: Si+13, Si+14, and Si+23 are particularly inert. The variations in reactivity are rapidly damped with increasing cluster size and for clusters with 40–65 atoms the reactivity is nearly independent of size. However, these large clusters are ∼102 times less reactive towards O2 than most bulk silicon surfaces. Studies of the temperature dependence of the reactions reveal that they proceed through a metastable precursor state which is probably molecular O2 physisorbed to the cluster surface. Variations in the size of the activation barrier for dissociative chemisorption account for the changes in reactivity with clus... read less NOT USED (low confidence) G. Schoeck and W. Pichl, “Bond trapping of cracks,” Physica Status Solidi (a). 1990. link Times cited: 17 Abstract: A model is presented for stabilization of cracks in an atomi… read moreAbstract: A model is presented for stabilization of cracks in an atomistic solid by “bond trapping”. In contrast to the normal “lattice trapping” which results from the periodicity of the atomic lattice, “bond trapping” depends on special features of the interatomic potentials and will lead to energy dissipation by phonons. It is expected to occur mainly in structures with open lattices with covalent binding, in glass, or in amorphous materials where lattice trapping is not to be expected.
Ein Modell wird vorgestellt fur die Stabilisierung von Rissen in atomaren Festkorpem durch “bond trapping”. Im Gegensatz zum normalen “lattice trapping” das auf der Periodizitat des Atomgitters beruht, hangt “bond trapping” von den speziellen Eigenschaften des interatomaren Potentials ab und gibt Anlas zu Energiedissipation durch Phononen. Es tritt hauptsachlich in offenen Gittern mit homoopolarer Bindung, in Glas oder in amorphen Materialien auf, in denen “lattice trapping” nicht erwartet wird. read less NOT USED (low confidence) H. Iyetomi and P. Vashishta, “Generalisation of the density-functional theory and three-body interactions in classical fluids,” Journal of Physics: Condensed Matter. 1989. link Times cited: 9 Abstract: An external field coupled with the two-body density distribu… read moreAbstract: An external field coupled with the two-body density distribution function leads to a generalisation of the density-functional theory in which the free energy is regarded as a functional not only of the density but also of the two-body distribution function. The generalised formalism is applied to the liquid structure theory in the presence of a weak three-body potential. The first-order calculation shows that the three-body effects are fully incorporated into the theory through modification of the pair potential by virtue of the variational property of the free-energy functional. read less NOT USED (low confidence) A. Arnold, N. Mauser, and J. Hafner, “A molecular dynamics study of the structure of liquid germanium,” Journal of Physics: Condensed Matter. 1989. link Times cited: 54 Abstract: The structure of liquid Ge as a function of temperature and … read moreAbstract: The structure of liquid Ge as a function of temperature and density was investigated using molecular dynamics and interatomic forces derived from the pseudopotential theory. The authors present results for the pair correlation function, the static structure factor and the bond-angle distribution function as a function of temperature and density. Our results are in good agreement with diffraction experiments. The density derivative of the structure factor deviates from the prediction of the uniform-fluid model due to the density dependence of the interatomic potential. The bond-angle distribution shows that the local order in liquid Ge is different from that of both the semiconducting and the metallic crystalline phases. With progressive undercooling the structure of the liquid does not tend towards the structure of amorphous Ge, because solidification is associated with a metal-semiconductor transition resulting in state-dependent interatomic potentials. read less NOT USED (low confidence) “References,” Basic Physics of Nanoscience. 2019. link Times cited: 0 NOT USED (low confidence) K. Ohno, K. Esfarjani, and Y. Kawazoe, “Tight-Binding Methods.” 2018. link Times cited: 0 NOT USED (low confidence) R. Khanna and V. Sahajwalla, “Atomistic Simulations of Properties and Phenomena at High Temperatures.” 2014. link Times cited: 3 NOT USED (low confidence) K. Sattler, “The energy gap of clusters, nanoparticles, and quantum dots.” 2002. link Times cited: 48 NOT USED (low confidence) K. Sattler, “Nanodots and Nanowires of Silicon.” 2001. link Times cited: 0 NOT USED (low confidence) T. Ito, “Atomistic simulation of epitaxial growth processes.” 2001. link Times cited: 0 NOT USED (low confidence) L. Xin-hou, “New Potentials of Silicon and Germanium Crystals,” Chinese Physics Letters. 1996. link Times cited: 2 Abstract: In this paper, a new model of potential energy functions for… read moreAbstract: In this paper, a new model of potential energy functions for atomic solids is given and applied to silicon and germanium crystals. Obtained potentials of Si and Ge crystals accurately reproduce experimental elastic constants and phonon dispersion curves. read less NOT USED (low confidence) T. Frauenheim, D. Porezag, T. Köhler, and F. Weich, “Molecular-Dynamic Simulations of Structure Formation in Complex Materials.” 1996. link Times cited: 1 NOT USED (low confidence) X. Liu, “NEW MODEL OF POTENTIAL-ENERGY FUNCTIONS FOR ATOMIC SOLIDS,” Journal of the Chemical Society, Faraday Transactions. 1995. link Times cited: 2 Abstract: A new theoretical model of potential-energy functions for at… read moreAbstract: A new theoretical model of potential-energy functions for atomic solids has been developed. An angular factor has been included in this model and its effect has been discussed. Using this new model a new preliminary potential for silicon crystal has been derived. The calculated phonon dispersion curve along the [q00] direction, using this new potential, has been given. A good agreement has been found with experiment. read less NOT USED (low confidence) M. M. Souza and G. Amaratunga, “Self Diffusion in Silicon Using the Ackland Potential.” 1993. link Times cited: 2 NOT USED (low confidence) P. Fedders, “First Principles Molecular Dynamics Studies of a-Si and a-Si:H,” MRS Proceedings. 1993. link Times cited: 1 NOT USED (low confidence) R. Biswas, K. Roos, and M. Tringides, “Low Temperature Growth Mechanisms for Rheed Oscillations,” MRS Proceedings. 1993. link Times cited: 1 NOT USED (low confidence) G. Gadiyak, Y. Morokov, and D. Mukhin, “Simulation of fluorine interaction with a silicon surface,” Applied Surface Science. 1992. link Times cited: 3 NOT USED (low confidence) D. Wales and M. C. Waterworth, “Structures and rearrangements of model silicon clusters,” Journal of the Chemical Society, Faraday Transactions. 1992. link Times cited: 12 Abstract: We present a study of small silicon clusters bound by the em… read moreAbstract: We present a study of small silicon clusters bound by the empirical potential-energy function of Li, Johnston and Murrell (LJM). Analytic first and second derivatives of the potential are employed in molecular dynamics simulations and geometry optimisations of both minima and transition states. Frequency analyses of all the resulting stationary points enable us to define the topology of the potential-energy surfaces, and rearrangement mechanisms are characterised for various clusters containing up to 50 atoms. read less NOT USED (low confidence) S. Li, R. Johnston, and J. Murrell, “Cluster structures and stabilities from solid-state potentials. Application to silicon clusters,” Journal of the Chemical Society, Faraday Transactions. 1992. link Times cited: 40 Abstract: An empirical potential-energy function comprising two- and t… read moreAbstract: An empirical potential-energy function comprising two- and three-body terms, whose parameters have been determined from the properties of solid silicon, is used to study the structures and energies of silicon microclusters. For small clusters, densely packed (non-diamond) structures are found which are in broad agreement with ab initio calculations. For larger clusters, optimisations starting from fragments of the cubic bulk solids indicate that close-packed structures are favoured initially and that diamond structures become relatively more stable only for clusters of well above 100 atoms. read less NOT USED (low confidence) S. Sarma and K. E. Khor, “Empirical potential approach to the stability and energetics of thin films and surfaces,” Applied Surface Science. 1992. link Times cited: 1 NOT USED (low confidence) F. Ercolessi and J. B. Adams, “Interatomic Potentials From First-Principles Calculations,” MRS Proceedings. 1992. link Times cited: 22 Abstract: We propose a new scheme to extract “optimal” interatomic pot… read moreAbstract: We propose a new scheme to extract “optimal” interatomic potentials starting from a large number of atomic configurations (and their forces) obtained from first-principles calculations. The method appears to be able to overcome the difficulties encountered by traditional fitting approaches when using rich and complex analytical forms, and constitute a step forward towards large-scale simulations of condensed matter systems with a degree of accuracy comparable to that obtained by ab initio methods. A first exploratory application to aluminum is presented. read less NOT USED (low confidence) M. Robinson, “Computer Simulation of Atomic Collision Processes in Solids,” MRS Proceedings. 1992. link Times cited: 2 Abstract: Computer simulation is a major tool for studying the interac… read moreAbstract: Computer simulation is a major tool for studying the interactions of swift ions with solids which underlie processes such as particle backscattering, ion implantation, radiation damage, and sputtering. Numerical models are classed as molecular dynamics or binary collision models, along with some intermediate types. Binary collision models are divided into those for crystalline targets and those for structureless ones. The foundations of such models are reviewed, including interatomic potentials, electron excitations, and relationships among the various types of codes. Some topics of current interest are summarized. read less NOT USED (low confidence) P. Fedders and D. D. Drabold, “Results from First Principles Molecular Dynamics Simulations on a-Si,” MRS Proceedings. 1991. link Times cited: 2 NOT USED (low confidence) R. Biswas, I. Kwon, and C. Soukoulis, “Molecular Dynamics Simulations of the Structural, Vibrational and Electronic Properties of Amorphous Silicon,” MRS Proceedings. 1990. link Times cited: 1 NOT USED (low confidence) A. Carlsson, “Beyond Pair Potentials in Elemental Transition Metals and Semiconductors,” Journal of Physics C: Solid State Physics. 1990. link Times cited: 169 NOT USED (low confidence) M. Heggie, “A New Interatomic Potential for Non-Metals.” 1989. link Times cited: 0 NOT USED (low confidence) W. Kamitakahara, R. Biswas, A. M. Bouchard, F. Gompf, and J. Suck, “Vibrational Spectra for Hydrogenated Amorphous Semiconductors,” MRS Proceedings. 1989. link Times cited: 0 Abstract: Hydrogen vibration spectra have been measured by neutron sca… read moreAbstract: Hydrogen vibration spectra have been measured by neutron scattering for several amorphous semiconductor materials, including a-Ge:H and a-SiC:H samples containing about 10 at. % H. The data for a-Ge:H are compared in detail with the results of realistic computer simulations. read less NOT USED (low confidence) A. Carlsson, “Angular Forces in Transition Metals and Diamond Structure Semiconductors.” 1989. link Times cited: 1 NOT USED (low confidence) J. Hafner, “Quantum Theory of Structure: sp-Bonded Systems.” 1989. link Times cited: 11 NOT USED (low confidence) W. Andreoni, “On the Electronic and Structural Properties of Small Clusters.” 1989. link Times cited: 0 NOT USED (low confidence) V. Vítek, D. Srolovitz, and W. Morgan, “MOLECULAR DYNAMICS SIMULATION OF THE PHYSICS OF THIN FILM GROWTH ON SILICON: EFFECTS OF THE PROPERTIES OF INTERATOMIC POTENTIAL MODELS.” 1989. link Times cited: 0 NOT USED (low confidence) D. J. Oh and R. Johnson, “A Semi-Empirical Potential for Graphite,” MRS Proceedings. 1988. link Times cited: 4 NOT USED (low confidence) R. Biswas, A. M. Bouchard, W. Kamitakahara, G. Grest, and C. Soukoulis, “Vibrational Localization and Vibrational Spectra in Amorphous Silicon,” MRS Proceedings. 1988. link Times cited: 0 Abstract: Amorphous silicon structures have been generated by quenchin… read moreAbstract: Amorphous silicon structures have been generated by quenching liquid silicon configurations using molecular-dynamics simulations. Localized vibrational modes have been identified in these models. The presence of under-coordinated atoms in these a-Si models leads to extra resonant modes at low frequencies. The vibrational densities of states, and dynamic structure factors for localized, resonant and extended modes, are discussed and compared with neutron scattering data. The amorphous networks have also been adapted to model amorphous silicon-germanium systems. Densities of states and localization characteristics have been calculated for a-Si x Ge 1-x alloys and a-Si/a-Ge superlattices, and are compared to Raman measurements. read less NOT USED (high confidence) J. Utterson and R. Erban, “Symmetries of many-body systems imply distance-dependent potentials.,” Physical review. E. 2023. link Times cited: 0 Abstract: Considering an interatomic potential U(q), where q=[q_{1},q_… read moreAbstract: Considering an interatomic potential U(q), where q=[q_{1},q_{2},⋯,q_{N}]∈R^{3N} is a vector describing positions q_{i}∈R^{3}, it is shown that U can be defined as a function of the interatomic distance variables r_{ij}=|q_{i}-q_{j}| provided the potential U satisfies some symmetry assumptions. Moreover, the potential U can be defined as a function of a proper subset of the distance variables r_{ij}, provided N>5, with the number of distance variables used scaling linearly with the number of atoms N. read less NOT USED (high confidence) S. Surulere, M. Shatalov, and E. Olayiwola, “Optimal interatomic potentials using modified method of least squares: Optimal form of interatomic potentials,” Open Physics. 2023. link Times cited: 0 Abstract: The problem of optimization of interatomic potentials is for… read moreAbstract: The problem of optimization of interatomic potentials is formulated and solved by means of generalization of the Morse, Kaxiras–Pandey, and Rydberg potentials. The interatomic potentials are treated as solutions of some second-order ordinary differential equations which will be classified and analyzed. The most appropriate analytic form of the understudied potentials will be proposed based on a one-dimensional search for the parameter, γ \gamma , which is the power of the interatomic distance, r r . The optimal analytic form will also be proposed for metals such as gold, copper, aluminium, titanium, and the silver–copper alloy. The method of least squares will be used to estimate the potential parameters. Phenomenological potentials such as the classical Rydberg, classical Morse, generalized Morse, Kaxiras–Pandey, and classical Lennard–Jones will be studied, and new potentials based on the combination of some of the aforementioned potentials will also be proposed. Metrics such as the goal function values, will be used to identify which optimal value of the parameter, γ \gamma , is most appropriate to introduce into the preferred interatomic potential for interaction between atoms. read less NOT USED (high confidence) J. Thomas, H. Chen, and C. Ortner, “Body-Ordered Approximations of Atomic Properties,” Archive for Rational Mechanics and Analysis. 2022. link Times cited: 1 NOT USED (high confidence) F. Berthier, Q. Lullien, and B. Legrand, “Effective site energy and cluster expansion approaches for the study of phase diagrams,” Physical Review B. 2021. link Times cited: 0 Abstract: We apply the cluster expansion (CE) method to determine the … read moreAbstract: We apply the cluster expansion (CE) method to determine the effective cluster interactions (ECIs) from a simple energetic model that depends on both local and global composition. This model is defined by the site energies of random solid solutions of a one-dimensional alloy Co-Pt. We explore how these local and global dependencies are reflected on the cluster interactions. The energies of the structures are not well reproduced with concentration-independent interactions. Moreover, the interactions have a larger range than the energetic model which is limited to the nearest neighbors. By fitting the ECIs on the site energies, we suggest a mean-field-type weighting of the excess variables present in clusters of large size. We show that the site energy formalism controls the size of the clusters required for CE convergence and their concentration dependence. Finally, we take advantage of the site energy formalism to describe the elastic and chemical effects that control the thermodynamics of the alloy as a function of the ECIs. read less NOT USED (high confidence) J. Thomas, H. Chen, and C. Ortner, “Rigorous body-order approximations of an electronic structure potential energy landscape.” 2021. link Times cited: 4 NOT USED (high confidence) D. Prasad and N. Mitra, “An atomistic study of phase transition in cubic diamond Si single crystal subjected to static compression,” Computational Materials Science. 2019. link Times cited: 7 NOT USED (high confidence) T. Gao et al., “Microstructural properties and evolution of nanoclusters in liquid Si during a rapid cooling process,” JETP Letters. 2017. link Times cited: 2 NOT USED (high confidence) J. C. Castro-Palacio, L. Velazquez-Abad, M. Fernández, and J. Q. Cuador-Gil, “Molecular dynamics study of one dimensional nanoscale Si/SiO2 interfaces,” The European Physical Journal D. 2013. link Times cited: 1 NOT USED (high confidence) A. Gufan, O. V. Kukin, and I. A. Osipenko, “An invariant form of the potential energy function used to simulate properties of condensed matter,” Bulletin of the Russian Academy of Sciences: Physics. 2012. link Times cited: 3 NOT USED (high confidence) S. Mahajan, G. Subbarayan, and B. Sammakia, “Estimating Kapitza Resistance Between \rm Si\hbox-\rm SiO_2 Interface Using Molecular Dynamics Simulations,” IEEE Transactions on Components, Packaging and Manufacturing Technology. 2011. link Times cited: 20 Abstract: The interface between nano-scale films is of relevance in ma… read moreAbstract: The interface between nano-scale films is of relevance in many critical applications. Specifically, recent technological advances in semiconductor industry that utilize silicon-on-insulator devices have given importance to the understanding of thermal transport across ${\rm Si}{\hbox{-}}{\rm SiO}_{2}$ interface. Estimates of interfacial (Kapitza) resistance to the thermal transport across ${\rm Si}{\hbox{-}}{\rm SiO}_{2}$ films do not appear to exist at the present time. In this paper, we develop and carryout reverse non-equilibrium molecular dynamics simulations by imposing known heat flux to determine the Kapitza resistance between ${\rm Si}{\hbox{-}}{\rm SiO}_{2}$ thin films. For the ${\rm Si}{\hbox{-}}{\rm SiO}_{2}$ interface, the average Kapitza resistance for a ${\sim}{8}~{\rm\AA}$ thick oxide layer system was 0.503 ${\times}10^{-9}~{\rm m}^{2}{\rm K}/{\rm W}$ and for a ${\sim}{\rm 11.5}~{\rm\AA}$ thick oxide layer system was 0.518 $\,\times 10^{-9}~{\rm m}^{2}{\rm K}/{\rm W}$. These values were of the same order of magnitude as the Kapitza resistance values determined from the acoustic mismatch model and the diffuse mismatch model for the ${\rm Si}\hbox{-}{\rm SiO}_{2}$ interface. read less NOT USED (high confidence) A. Galashev, “Simulation of silicon nanoparticles stabilized by hydrogen at high temperatures,” Journal of Nanoparticle Research. 2010. link Times cited: 4 NOT USED (high confidence) A. M. Ukpong, “Studies of the electronic and vibrational signatures of the unusual bonding geometries in melt-quenched amorphous silicon,” Molecular Physics. 2009. link Times cited: 2 Abstract: Tight-binding molecular dynamics simulations have been perfo… read moreAbstract: Tight-binding molecular dynamics simulations have been performed to investigate the effect of quenching rate of the Si melt on the resulting local structure of amorphous silicon. Different quenching rates were used to cool liquid silicon in the simulations to demonstrate that the choice of quenching rates significantly influences the resulting local structure. The calculated pair correlation functions show that the local structure is sensitive to the thermal processing of the liquid silicon melt. The use of cooling rates higher than 10−13 K s−1 appears to prevent the activation of the required structural re-arrangements necessary to stabilise the networks, causing unexpected bonding geometries to develop. The electronic signatures of the defects show that only the triangular defect structure contributes resonance states to the conduction band tail. Also, the vibrational signature of the triangular structure shows a high energy transverse optical mode at 95 meV, indicating that the defect is likely to be unstable at 300 K, although both defects contribute minimal states to the mid-gap level. read less NOT USED (high confidence) S. Mahajan, G. Subbarayan, and B. Sammakia, “Estimating Kapitza resistance between Si-SiO2 interface using molecular dynamics simulations,” 2008 11th Intersociety Conference on Thermal and Thermomechanical Phenomena in Electronic Systems. 2008. link Times cited: 13 Abstract: The interface between nano-scale films is of relevance in ma… read moreAbstract: The interface between nano-scale films is of relevance in many critical applications. Specifically, recent technological advances in semiconductor industry that utilize Silicon-on-Insulator (SOI) devices have given urgency to understanding thermal transport across Si-SiO2 interface. Estimates of interfacial (Kapitza) resistance to thermal transport across Si-SiO2 films do not appear to exist at the present time. In this paper, we develop and carryout reverse non-equilibrium molecular dynamics (NEMD) simulations by imposing known heat flux to determine the Kapitza resistance between Si-SiO2 thin films. For the Si-SiO2 interface, the average Kapitza resistance for a ~8 Aring thick oxide layer system was 0.503 times 10-9 m K/W and for a ~11.5 Aring thick oxide layer system was 0.518 times 10-9 m K/W. These values were of the same order of magnitude as the Kapitza resistance values determined from the acoustic mismatch model (AMM) and the diffuse mismatch model (DMM) for the Si-SiO2 interface. read less NOT USED (high confidence) A. Galashev and I. A. Izmodenov, “Computer investigation of the structure of Si73 clusters surrounded by hydrogen,” Glass Physics and Chemistry. 2008. link Times cited: 5 NOT USED (high confidence) J. Schall, G. Gao, and J. Harrison, “Elastic constants of silicon materials calculated as a function of temperature using a parametrization of the second-generation reactive empirical bond-order potential,” Physical Review B. 2008. link Times cited: 48 Abstract: A parametrization for silicon is presented that is based on … read moreAbstract: A parametrization for silicon is presented that is based on the second-generation reactive empirical bondorder REBO formalism Brenner, Shenderova, Harrison, Stuart, Ni, and Sinnott J. Phys.: Condens. Matter 14, 783 2002 . Because it shares the same analytic form as Brenner’s second-generation REBO, this new potential is a step toward a single potential that can model many atom systems that contain C, Si, and H, where bond breaking and bond making are important. The widespread use of Brenner’s REBO potential, its ability to model both zero-Kelvin elastic constants of diamond and the temperature dependence of the elastic constants, and the existence of parameters for many atom types were the motivating factors for obtaining this parametrization for Si. While Si-C-H classical bond-order potentials do exist, they are based on Brenner’s original formalism. This new parametrization is validated by examining the structure and stability of a large number of crystalline silicon structures, by examining the relaxation energies of point defects, the energies of silicon surfaces, the effects of adatoms on surface energies, and the structures of both liquid silicon and amorphous silicon. Finally, the elastic constants of diamond-cubic and amorphous silicon between 0 and 1100 K are calculated with this new parametrization and compared to values calculated using a previously published potential. read less NOT USED (high confidence) M. M. J. Treacy, J. Gibson, L. Fan, D. Paterson, and I. McNulty, “Fluctuation microscopy: a probe of medium range order,” Reports on Progress in Physics. 2005. link Times cited: 168 Abstract: Fluctuation microscopy is a hybrid diffraction-imaging techn… read moreAbstract: Fluctuation microscopy is a hybrid diffraction-imaging technique that detects medium range order in amorphous materials by examining spatial fluctuations in coherent scattering. These fluctuations appear as speckle in images and diffraction patterns. The volume of material contributing to the speckle is determined by the point-spread function (the resolution) of the imaging optics and the sample thickness. The spatial periodicities being probed are related to the diffraction vector. Statistical analysis of the speckle allows the random and non-random (ordered) contributions to be discriminated. The image resolution that gives the maximum speckle contrast, as determined by the normalized variance of the image intensity, is determined by the characteristic length scale of the ordering. Because medium range ordering length scales can extend out to about the tenth coordination shell, fluctuation microscopy tends to be a low image resolution technique. This review presents the kinematical scattering theory underpinning fluctuation microscopy and a description of fluctuation electron microscopy as it has been employed in the transmission electron microscope for studying amorphous materials. Recent results using soft x-rays for studying nanoscale materials are also presented. We summarize outstanding issues and point to possible future directions for fluctuation microscopy as a technique. read less NOT USED (high confidence) M. Rao and S. Sengupta, “A mesoscopic model of a two-dimensional solid state structural transformation: statics and dynamics,” Journal of Physics: Condensed Matter. 2004. link Times cited: 6 Abstract: We study the equilibrium properties of a system of particles… read moreAbstract: We study the equilibrium properties of a system of particles in two dimensions, interacting via pair and three-body potentials. This system undergoes a structural transition from a square to a rhombic lattice and thus constitutes a simple model for a generic tetragonal to orthorhombic transition. We aim at an intermediate level of description lying in between that of coarse grained elastic strain Hamiltonians and microscopic ab initio approaches. We obtain macroscopic thermodynamic properties and the phase diagram at zero and finite temperatures as a function of the density and the relative strengths of the pair and three-body energies using lattice sums, an approximate 'cell model' theory and molecular dynamics simulations in the NV T ensemble. In addition, we study the dynamics of nucleation following a quench from the square to the triangular phase (Rao and Sengupta 2003 Phys. Rev. Lett. 91 045502). As in real solids, the final microstructure depends sensitively on the depth of the quench—a shallow quench results in an equilibrium ferrite while a deep quench gives rise to a metastable twinned martensite. We find, in accordance with experiments, that the twinned martensite is associated with a diffusionless transformation. We propose that this model solid may be used as a test bed for studies of the statics and dynamics of structural transitions. read less NOT USED (high confidence) W. K. Liu, E. Karpov, S. Zhang, and H. S. Park, “An introduction to computational nanomechanics and materials,” Computer Methods in Applied Mechanics and Engineering. 2004. link Times cited: 405 NOT USED (high confidence) A. S. Barnard, S. Russo, and G. Leach, “Nearest neighbour considerations in Stillinger-Weber type potentials for diamond,” Molecular Simulation. 2002. link Times cited: 5 Abstract: Results of a preliminary investigation into the effect of va… read moreAbstract: Results of a preliminary investigation into the effect of varying the interaction cutoff on the bulk properties of diamond using a Stillinger-Weber (SW) type potential for C (Diamond) are presented. The interaction cutoff is varied over a range that includes and excludes the second-nearest neighbours. Whilst the original SW potential for silicon only included first-nearest neighbours inside the interaction cut-off, subsequent parameterizations for carbon (diamond) have also included second-nearest neighbours. Elastic and vibration properties of diamond were calculated over a range of cutoff distances used and the results show that certain lattice properties exhibit an approximately linear dependence on the interaction cut-off. read less NOT USED (high confidence) T. Lenosky et al., “Highly optimized empirical potential model of silicon,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 145 Abstract: We fit an empirical potential for silicon using the modified… read moreAbstract: We fit an empirical potential for silicon using the modified embedded atom (MEAM) functional form, which contains a nonlinear function of a sum of pairwise and three-body terms. The three-body term is similar to the Stillinger-Weber form. We parametrized our model using five cubic splines, each with 10 fitting parameters, and fitted the parameters to a large database using the force-matching method. Our model provides a reasonable description of energetics for all atomic coordinations, Z, from the dimer (Z = 1) to fcc and hcp (Z = 12). It accurately reproduces phonons and elastic constants, as well as point defect energetics. It also provides a good description of reconstruction energetics for both the 30° and 90° partial dislocations. Unlike previous models, our model accurately predicts formation energies and geometries of interstitial complexes - small clusters, interstitial-chain and planar {311} defects. read less NOT USED (high confidence) C. Herrero, “Quantum atomic dynamics in amorphous silicon; a path-integral Monte Carlo simulation,” Journal of Physics: Condensed Matter. 2000. link Times cited: 10 Abstract: The quantum dynamics of atoms in amorphous silicon has been … read moreAbstract: The quantum dynamics of atoms in amorphous silicon has been addressed by using path-integral Monte Carlo simulations. Structural results (radial distribution functions) found from these simulations agree well with experimental data. We study the quantum delocalization of the silicon atoms around their equilibrium positions. This delocalization is larger for coordination defects (fivefold-coordinated Si atoms). Correlations in the atomic displacements are analysed as a function of the interatomic distance and compared with those derived from classical Monte Carlo simulations. At high temperatures, the classical limit is recovered. Our results are also compared with those derived from similar quantum simulations for crystalline silicon. Structural disorder favours a larger vibrational amplitude for the atoms in amorphous silicon. read less NOT USED (high confidence) M. Schaible, “Empirical Molecular Dynamics Modeling of Silicon and Silicon Dioxide: A Review,” Critical Reviews in Solid State and Materials Sciences. 1999. link Times cited: 28 Abstract: A number of computational methods have been developed over t… read moreAbstract: A number of computational methods have been developed over the last 40 years to simulate the behavior of solid materials with small dimensions. On the macro-scale, Finite Element analysis calculates mechanical stress on micron-sized cantilevers and motors. On the atomic level, newer ab initio methods compute nuclear and electronic behavior of hundred atom models with unprecedented rigor. By implementing the laws of classic mechanics, empirical Molecular Dynamics (MD) programs help bridge these two computational extremes. MD identifies nonelectronic, particle motion for large 100,000 atom cells with good success. MD derives both equilibrium and nonequilibrium properties for many complex condensed regimes; quantitatively (and qualitatively) reaffirms empirical data; aids discovery of new materials processing techniques, and helps predict unknown physical phenomena often only observable under extreme environmental settings. One material of great technical importance to the semiconductor industry is silicon (... read less NOT USED (high confidence) C. Bittencourt, “Formation of a SiC buffer layer by reaction of Si (100) with methane and hydrogen plasma,” Journal of Physics D. 1999. link Times cited: 5 Abstract: The reaction of Si (100) surfaces at T = 950 °C with radical… read moreAbstract: The reaction of Si (100) surfaces at T = 950 °C with radicals of methane obtained in a low-power-density glow discharge plasma, has been studied by combining in situ surface science techniques (x-ray photoemission spectroscopy and high electron energy diffraction) and ex situ analytical techniques (atomic force microscopy and infrared absorption). An analysis of C 1s and Si 2p core-level shifts combined with the examination of the valence-band curves showed that the obtained buffer layers were stoichiometric. For long carbonization times (>30 min) the formation of a carbon rich surface was observed. To understand the mechanism of hetero-epitaxial silicon carbide (SiC) buffer layer growth, the early stage of SiC nucleation was observed by atomic force microscopy and reflection high-energy electron diffraction. The results suggest that three-dimensional epitaxial islands nucleate at the earliest growth stage followed by a further Volmer-Weber growth until the formation of a carbon rich surface. The growth mechanism of the SiC buffer layer is discussed on the basis of a reported model. read less NOT USED (high confidence) C. Bittencourt, “Reaction of Si (100) with silane–methane low-power plasma: SiC buffer-layer formation,” Journal of Applied Physics. 1999. link Times cited: 14 Abstract: The formation of a SiC buffer layer on Si (100) at substrate… read moreAbstract: The formation of a SiC buffer layer on Si (100) at substrate temperature as low as 950 °C using radicals of methane molecules obtained in a low-power-density glow-discharge plasma, is presented. The x-ray photoemission spectroscopy and low-energy-yield spectroscopy performed in the constant final-state mode suggest that the layers obtained were stoichiometric. To understand the mechanism of heteroepitaxial silicon carbide growth, the early stage of SiC nucleation was observed by atomic force microscopy and reflection high-energy electron diffraction. The results reveal that three-dimensional epitaxial crystallites nucleate at the earliest growth stage followed by a further Volmer–Weber growth. read less NOT USED (high confidence) J. Q. Xie, J. Feng, and H. Lu, “Molecular-dynamics simulation of low-temperature growth of silicon films by cluster deposition,” Modelling and Simulation in Materials Science and Engineering. 1999. link Times cited: 8 Abstract: Silicon thin-film growth from cluster beams at a substrate t… read moreAbstract: Silicon thin-film growth from cluster beams at a substrate temperature of 300 K has been investigated with molecular-dynamics simulations utilizing the Stillinger-Weber two- and three-body interaction potential. The spreading of Si-atom clusters and the structure of grown films have been studied as a function of the incident cluster velocity. Our simulation results show that the films grown at a low substrate temperature of 300 K are amorphous and the substrates suffer heavier damage with an increase in the cluster velocity. As compared with our previous results on Si thin-film growth at a substrate temperature of 1000 K, we found that substrate temperature and cluster velocity had a significant impact in determining the structure of the grown films and the cluster spreading on the substrate. read less NOT USED (high confidence) L. J. Lewis and N. Mousseau, “Tight-binding molecular-dynamics studies of defects and disorder in covalently bonded materials,” Computational Materials Science. 1998. link Times cited: 13 NOT USED (high confidence) J. Fang, R. Johnston, and J. Murrell, “Potential energy functions for atomic solids: V. Application to alkali metal solids,” Molecular Physics. 1993. link Times cited: 18 Abstract: Empirical potential functions comprising two-body and three-… read moreAbstract: Empirical potential functions comprising two-body and three-body terms have been derived for the alkali metals by fitting parameters to the phonon frequencies, elastic constants, lattice energies and lattice distances of the body-centred-cubic (b.c.c.) solids. These potentials give, in all cases, very similar energies for the b.c.c., face-centred-cubic (f.c.c.) and hexagonal closest packing (h.c.p.) structures, with simple cubic (s.c.), diamond and various two-dimensional structures being much less stable. The lithium potential has been used to predict structures and stabilities of neutral Li n microclusters. No ‘magic number’ stabilities have been found. For n ≥ 6 the structures can, in the main, be described as face-fused tetrahedra, and ab initio calculations support these structures in some important cases (e.g. n = 6, 7). read less NOT USED (high confidence) J. Holender and G. J. Morgan, “Generation of a large structure (105 atoms) of amorphous Si using molecular dynamics,” Journal of Physics: Condensed Matter. 1991. link Times cited: 29 Abstract: A method for generating amorphous tetrahedral structures hav… read moreAbstract: A method for generating amorphous tetrahedral structures having 13824 and 110592 atoms is presented. The authors took the Wooten, Winer and Weaire amorphous model (1985) of 216 atoms and put together a number of these blocks. This larger structure was annealed using molecular dynamics and then cooled. Comparison with experiment was carried out using the structure factors calculated directly. Very good agreement has been attained. The generated structures contrary to the original www model, contain coordination defects. read less NOT USED (high confidence) A. Al-Derzi, R. Johnston, J. Murrell, and J. Rodriguez-Ruiz, “Potential energy functions for atomic solids: III. Fitting phonon frequencies and elastic constants of diamond structures,” Molecular Physics. 1991. link Times cited: 39 NOT USED (high confidence) S. Wong, “Controlling Indentation-induced Phases of Silicon.” 2017. link Times cited: 0 Abstract: Silicon (Si) is the backbone of the semiconductor industry. … read moreAbstract: Silicon (Si) is the backbone of the semiconductor industry. The widespread use of Si is largely due to the useful electrical and optical properties of the material in its standard, diamond cubic (dc) crystal structure. However, in recent years, there has been an increasing interest in the properties of Si with different crystal structures (phases). In particular, the metastable phases formed through pressure application have been the topic of much study due to their promising properties. For example, the body-centred cubic phase (bc8-Si) has been reported to have an ultra-narrow band-gap whereas the rhombohedral phase (r8-Si) has been predicted to have an improved absorption coefficient across the solar spectrum. A mixture of these two exotic phases can be formed directly from a standard dc-Si semiconductor wafer using the application of pressure through point loading via indentation. This thesis addresses several challenges regarding the formation, stability, and properties of this bc8/r8 structure. The process involving the nucleation of the bc8/r8 phase via indentation was investigated in detail. Specifically, the interplay between the nucleation of the bc8/r8 phase with other plastic deformation processes within the surrounding crystalline lattice (labelled collectively as “crystalline defects” within this work) that occur is studied. It was shown that both phase transformation and the formation of crystalline defects are nucleation limited. Thus, holding a volume of Si at high pressure for a duration increases the likelyhood that th material will plastically deform. It is also shown that these two forms of plastic deformation act as competing mechanisms, with the mode of incipient plasticity playing a dominant role in the shape and volume of the final phase transformed region. Indentations in which phase trans- read less NOT USED (high confidence) K. Scheerschmidt and M. Planck, “Empirical Molecular Dynamics: Possibilities, Requirements, and Limitations.” 2007. link Times cited: 9 NOT USED (definite) K. Eriguchi, “Application of Molecular Dynamics Simulations to Plasma Etch Damage in Advanced Metal-Oxide-Semiconductor Field-Effect Transistors.” 2012. link Times cited: 0 Abstract: According to "the international technology roadmap for … read moreAbstract: According to "the international technology roadmap for semiconductors (ITRS)" (SIA, 2009), the shrinkage of silicon-based metal–oxide–semiconductor field-effect transistor (MOSFET) – an elemental device (unit) in ultra-large-scale integrated (ULSI) circuits – has been accelerating due to expanding demands for the higher performance and the lower power operation. The characteristic dimensions of current MOSFETs in mass productions are around 30 – 50 nm. Figure 1 shows the scaling trend of the key feature sizes in ULSI circuits predicted by Semiconductor Industry Association, USA. Various types of MOSFETs are designed for the specific purposes, i.e., low standby power (LSP), low operation power (LOP), and high performance (HP) operations, and built in ULSI circuits such as dynamic random access memory (DRAM) and micro-processing unit (MPU). New structured MOSFETs such as fully-depleted (FD) and metal-gate (MG) devices have been recently proposed. Since physical gate length (Lg) and source / drain extension depth (Ext) are the key feature sizes determining MOSFET performance (Sze & Ng, 2007), the shrinkage of Lg and Ext is a primal focus in the development of MOSFETs. These sizes have become a few nanometers, comparable to the scale of atomistic simulation domain. read less |
 ThreeBodyCluster_BH_BiswasHamann_1987_Si__MO_019616213550_000
ThreeBodyCluster_BH_BiswasHamann_1987_Si__MO_019616213550_000