 ThreeBodyCluster_BH_BiswasHamann_1987_Si__MO_019616213550_000
ThreeBodyCluster_BH_BiswasHamann_1987_Si__MO_019616213550_000
| Title
A single sentence description.
|
Three-body cluster potential for Si by Biswas and Hamann (1987) v000 |
|---|---|
| Description
A short description of the Model describing its key features including for example: type of model (pair potential, 3-body potential, EAM, etc.), modeled species (Ac, Ag, ..., Zr), intended purpose, origin, and so on.
|
A theory of classical two- and three-body interatomic potentials is developed. The ability of the classical potentials to model quantum-mechanical local-density-functional calculations for a wide range of silicon structures is explored. In developing classical models it was found to be necessary to perform new local-density-functional calculations for self-interstitial and layered silicon structures. The potential was derived from fits and tests to energies of bulk, surface, layered, and self-interstitial structures and is designed for tetrahedral silicon silicon structures (diamond and amorphous phases). |
| Species
The supported atomic species.
| Si |
| Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
|
None |
| Contributor |
Dipanjan Ghosh |
| Maintainer |
Dipanjan Ghosh |
| Developer |
Rajesh Biswas Donald. R. Hamann |
| Published on KIM | 2019 |
| How to Cite |
This Model originally published in [1] is archived in OpenKIM [2-5]. [1] Biswas R, Hamann DR. New classical models for silicon structural energies. Phys Rev B. 1987;36(12):6434–45. doi:10.1103/PhysRevB.36.6434 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information. [2] Biswas R, Hamann DR. Three-body cluster potential for Si by Biswas and Hamann (1987) v000. OpenKIM; 2019. doi:10.25950/68983f48 [3] Ghosh D, Karls DS, Biswas R, Hamann DR. Three-body cluster potential by Biswas and Hamann (1987) v000. OpenKIM; 2019. doi:10.25950/972369ee [4] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6 [5] Elliott RS, Tadmor EB. Knowledgebase of Interatomic Models (KIM) Application Programming Interface (API). OpenKIM; 2011. doi:10.25950/ff8f563a Click here to download the above citation in BibTeX format. |
| Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on. The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel. The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied. The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP). Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis. OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm. |
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information. 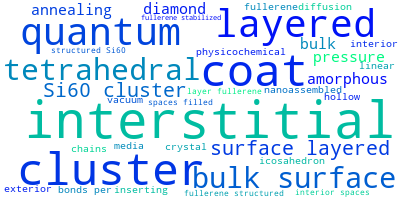
124 Citations (13 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) A. Galashev, “Molecular dynamics study of hydrogenated silicon clusters at high temperatures,” Molecular Physics. 2009. link Times cited: 5 Abstract: This paper reports on a study of the stability of silicon cl… read more USED (high confidence) A. Galashev, “Thermal instability of silicon fullerenes stabilized with hydrogen: Computer simulation,” Semiconductors. 2008. link Times cited: 4 USED (high confidence) A. Shalabi, K. Kamel, and M. M. Assem, “Theoretical characterization and many-body expansion analysis of BF3, BCl3, AlF3 and AlCl3 interactions,” Theoretica chimica acta. 1995. link Times cited: 4 USED (high confidence) W. K. Liu and C. McVeigh, “Predictive multiscale theory for design of heterogeneous materials,” Computational Mechanics. 2008. link Times cited: 57 USED (low confidence) P. Käshammer, N. Borgardt, M. Seibt, and T. Sinno, “Quantitative assessment of molecular dynamics-grown amorphous silicon and germanium films on silicon (111),” Surface Science. 2016. link Times cited: 3 USED (low confidence) P. Ghosh and M. Ranganathan, “Submonolayer growth study using a solid-on-solid model for 2 × 1 reconstructed surfaces of diamond-like lattices,” Surface Science. 2014. link Times cited: 7 USED (low confidence) H. Lu, J. Q. Xie, and J. Feng, “Simulation study on Si and Ge film growth by cluster deposition,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2000. link Times cited: 10 USED (low confidence) V. Konoplev, A. Gras-marti, E. P. Andribet, Pérez-Martı́n A., and Jiménez-Rodrı́guez J. J., “Effect of temperature on the bulk atomic relocation in low-energy collision cascades in silicon: a molecular dynamics study,” Radiation Effects and Defects in Solids. 1995. link Times cited: 4 Abstract: The production of damage in a Si lattice by internally start… read more USED (low confidence) E. Beam, “Computer simulation of the surface topology of (001) silicon resulting from the termination of 12〈110〉 edge dislocations with Burgers vectors parallel to the surface,” Materials Science and Engineering B-advanced Functional Solid-state Materials. 1993. link Times cited: 0 USED (low confidence) T. Weber and F. Stillinger, “Dynamical branching during fluorination of the dimerized Si(100) surface: A molecular dynamics study,” Journal of Chemical Physics. 1990. link Times cited: 70 Abstract: Collections of classical trajectories have been numerically … read more USED (low confidence) Stansfield, Broomfield, and Clary, “Classical-trajectory calculations on Ar+ sputtering of a Si(001) surface using an ab initio potential.,” Physical review. B, Condensed matter. 1989. link Times cited: 37 Abstract: We describe classical-trajectory calculations of sputtering … read more USED (low confidence) J. Lampinen, R. Nieminen, and K. Kaski, “Molecular dynamics simulation of epitaxial growth of the Si(001) surface,” Surface Science. 1988. link Times cited: 16 USED (low confidence) P. Fedders, “Defects, tight binding, and ab initio molecular dynamics simulations on a-Si,” Journal of Non-crystalline Solids. 1991. link Times cited: 0 NOT USED (low confidence) S. Surulere, M. Shatalov, and J. Ehigie, “Analysis of oscillations in 1D nanostructures influenced by different concrete potential functions,” International Journal of Non-Linear Mechanics. 2022. link Times cited: 0 NOT USED (low confidence) L. J. Lewis, “Fifty years of amorphous silicon models : the end of the story?,” Journal of Non-Crystalline Solids. 2022. link Times cited: 8 NOT USED (low confidence) R. Drautz, “Atomic cluster expansion for accurate and transferable interatomic potentials,” Physical Review B. 2019. link Times cited: 260 NOT USED (low confidence) H. Li et al., “Cost-effective synthesis of carbazole/triphenylsilyl host materials with multiple σ-π conjugation for blue phosphorescent organic light-emitting diodes,” Dyes and Pigments. 2018. link Times cited: 7 NOT USED (low confidence) T. Gao, W. Yan, X. Guo, Y. Qin, and Q. Xie, “Structural properties in liquid Si during rapid cooling processes,” Physica B-condensed Matter. 2013. link Times cited: 5 NOT USED (low confidence) A. P. Bart’ok, R. Kondor, and G. Csányi, “On representing chemical environments,” Physical Review B. 2012. link Times cited: 1246 Abstract: We review some recently published methods to represent atomi… read more NOT USED (low confidence) A. Gufan, O. V. Kukin, Y. M. Gufan, and A. Smolin, “Models of three-particle interactions and theory of nonlinear deformations of crystals,” Physics of the Solid State. 2012. link Times cited: 6 NOT USED (low confidence) J. Yu, S. Sinnott, and S. Phillpot, “Charge optimized many-body potential for the Si/SiO2 system,” Physical Review B. 2007. link Times cited: 151 Abstract: A dynamic-charge, many-body potential for the Si/SiO{sub 2} … read more NOT USED (low confidence) S. Billeter, A. Curioni, D. Fischer, and W. Andreoni, “Ab initio derived augmented Tersoff potential for silicon oxynitride compounds and their interfaces with silicon,” Physical Review B. 2006. link Times cited: 42 Abstract: Coordination-dependent interatomic potentials are proposed f… read more NOT USED (low confidence) A. Tekin and B. Hartke, “GLOBAL GEOMETRY OPTIMIZATION OF SILICON CLUSTERS EMPLOYING EMPIRICAL POTENTIALS, DENSITY FUNCTIONALS, AND AB INITIO CALCULATIONS,” Journal of Theoretical and Computational Chemistry. 2005. link Times cited: 13 Abstract: Sin clusters in the size range n = 4–30 have been investigat… read more NOT USED (low confidence) P. Gunes, Şi̇mşek S., and S. Erkoç, “a Comparative Study of Empirical Potential Energy Functions,” International Journal of Modern Physics C. 2004. link Times cited: 2 Abstract: A comparative study has been performed for silicon microclus… read more NOT USED (low confidence) Y. Umeno, T. Kitamura, K. Date, M. Hayashi, and T. Iwasaki, “Optimization of interatomic potential for Si/SiO2 system based on force matching,” Computational Materials Science. 2002. link Times cited: 25 NOT USED (low confidence) A. Barnard and S. Russo, “Development of an improved Stillinger-Weber potential for tetrahedral carbon using ab initio (Hartree-Fock and MP2) methods,” Molecular Physics. 2002. link Times cited: 28 Abstract: An improved interatomic potential for tetrahedral carbon is … read more NOT USED (low confidence) B. Marsen, M. Lonfat, P. Scheier, and K. Sattler, “The energy gap of pristine silicon clusters,” Journal of Electron Spectroscopy and Related Phenomena. 2000. link Times cited: 10 NOT USED (low confidence) C. Herrero, “Path-integral Monte Carlo study of amorphous silicon,” Journal of Non-crystalline Solids. 2000. link Times cited: 8 NOT USED (low confidence) H. Cox, R. Johnston, and J. Murrell, “Empirical Potentials for Modeling Solids, Surfaces, and Clusters,” IEEE Journal of Solid-state Circuits. 1999. link Times cited: 48 Abstract: A review of studies that have been made using the Murrell–Mo… read more NOT USED (low confidence) M. M. J. Treacy, J. Gibson, and P. J. Keblinski, “Paracrystallites found in evaporated amorphous tetrahedral semiconductors,” Journal of Non-crystalline Solids. 1998. link Times cited: 142 NOT USED (low confidence) J. Q. Xie and J. Feng, “Molecular-dynamics simulation of silicon film growth from cluster beams,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1998. link Times cited: 8 NOT USED (low confidence) X. Liu, Z. Zhen, and J.-cheng Zhang, “New Potential Energy Functions for Diamond and α-Tin Crystals,” Chinese Physics Letters. 1998. link Times cited: 1 Abstract: A new model of potential energy functions for atomic solids … read more NOT USED (low confidence) L. Marqués, M. Jaraíz, J. Rubio, J. Vicente, L. Bailón, and J. Barbolla, “Molecular dynamics simulations of ion bombardment processes,” Materials Science and Technology. 1997. link Times cited: 3 Abstract: AbstractAn improved molecular dynamics technique that allows… read more NOT USED (low confidence) E. Kaxiras, “Review of atomistic simulations of surface diffusion and growth on semiconductors,” Computational Materials Science. 1996. link Times cited: 17 NOT USED (low confidence) X. Liu, “New model of potential energy functions for atomic solids. Part 2. New potentials of silicon and germanium crystals,” Journal of Molecular Structure-theochem. 1995. link Times cited: 1 NOT USED (low confidence) L. Xin-hou, “New Model of Potential Energy Functions for Atomic Solids and Application to Silicon Crystal,” Chinese Physics Letters. 1995. link Times cited: 1 Abstract: A new theoretical model of potential energy functions for at… read more NOT USED (low confidence) A. A. Valuev, A. S. Kaklyugin, and H. E. Norman, “Molecular modelling of the chemical interaction of atoms and molecules with a surface,” Russian Chemical Reviews. 1995. link Times cited: 3 Abstract: The modelling of a surface as an assembly of moving atoms in… read more NOT USED (low confidence) V. Konoplev and A. Gras-marti, “Molecular dynamics simulation of low-energy collision cascades and atomic mixing in silicon,” Philosophical Magazine. 1995. link Times cited: 4 Abstract: We investigate atomic relocation processes in silicon at OK,… read more NOT USED (low confidence) T. Ito, “RECENT PROGRESS IN COMPUTER-AIDED MATERIALS DESIGN FOR COMPOUND SEMICONDUCTORS,” Journal of Applied Physics. 1995. link Times cited: 50 Abstract: Recent progress in computational materials science in the ar… read more NOT USED (low confidence) K. C. Low, H. Lim, and C. Ong, “Vacancies on the Si(001) c(4*2) surface,” Journal of Physics: Condensed Matter. 1994. link Times cited: 5 Abstract: We have employed a parametrized tight-binding molecular-dyna… read more NOT USED (low confidence) M. Schreiber and B. Lamberts, “Determination of parameter-free model potentials for the molecular dynamics simulation of amorphous semiconductors — application to silicon,” Journal of Molecular Structure-theochem. 1994. link Times cited: 0 NOT USED (low confidence) Z. Jiang and R. A. Brown, “Modelling oxygen defects in silicon crystals using an empirical interatomic potential,” Chemical Engineering Science. 1994. link Times cited: 14 NOT USED (low confidence) K. Roos and U. C. Tringides, “Low-temperature, flux-independent epitaxy in Ag/Si(111),” Surface Science. 1994. link Times cited: 7 NOT USED (low confidence) A. Shalabi and A. M. E. Misiery, “Routes for LiF unit cell growth,” Chemical Physics Letters. 1993. link Times cited: 3 NOT USED (low confidence) A. Mistriotis, A. Zdetsis, G. Froudakis, and M. Menon, “Reproduction of quantum tight-binding effects in silicon clusters by a four-body classical model,” Journal of Physics: Condensed Matter. 1993. link Times cited: 0 Abstract: The results obtained by a recently proposed empirical potent… read more NOT USED (low confidence) C. S. Carmer, B. Weiner, and M. Frenklach, “Molecular dynamics with combined quantum and empirical potentials: C2H2 adsorption on Si(100),” Journal of Chemical Physics. 1993. link Times cited: 71 Abstract: Classical trajectory calculations were employed to study the… read more NOT USED (low confidence) Tsumuraya, Ishibashi, and Kusunoki, “Statistics of Voronoi polyhedra in a model silicon glass.,” Physical review. B, Condensed matter. 1993. link Times cited: 14 Abstract: We clarify the local structure in a model silicon glass by u… read more NOT USED (low confidence) V. M. Bedanov and D. Mukhin, “The mechanism of anisotropic island growth in molecular-beam epitaxy of Si on Si(001),” Surface Science. 1992. link Times cited: 7 NOT USED (low confidence) R. Fournier, S. Sinnott, and A. Depristo, “Density functional study of the bonding in small silicon clusters,” Journal of Chemical Physics. 1992. link Times cited: 90 Abstract: We report the ground electronic state, equilibrium geometry,… read more NOT USED (low confidence) W. Niessen and V. G. Zakrzewski, “Complex Electron Affinity Processes in Clusters of S and Si.” 1992. link Times cited: 2 Abstract: Vertical and in some cases adiabatic electron affinities are… read more NOT USED (low confidence) C. L. Cleveland and U. Landman, “Dynamics of Cluster-Surface Collisions,” Science. 1992. link Times cited: 208 Abstract: The structure, energetics, and dynamics of shock conditions … read more NOT USED (low confidence) W. Tiller, “The role of ledges in stress tensor-mediated surface processes for Si and GaAs,” Metallurgical Transactions A. 1991. link Times cited: 0 NOT USED (low confidence) M. Heggie, “Semiclassical interatomic potential for carbon and its application to the self-interstitial in graphite,” Journal of Physics: Condensed Matter. 1991. link Times cited: 33 Abstract: A semiclassical interatomic potential for carbon is discusse… read more NOT USED (low confidence) M. Kohyama, “On the transferable SETB method for Si,” Journal of Physics: Condensed Matter. 1991. link Times cited: 21 Abstract: The two types of transferable semi-empirical tight-binding (… read more NOT USED (low confidence) A. Silverman, J. Adler, and R. Weil, “Computer modelling of the diffusion mechanisms of fluorine in amorphous silicon,” Thin Solid Films. 1990. link Times cited: 9 NOT USED (low confidence) J. Hafner and W. Jank, “The electronic structure of liquid metals,” Journal of Physics: Condensed Matter. 1990. link Times cited: 1 Abstract: The authors present first-principles calculations of the ele… read more NOT USED (low confidence) U. Ray, M. Jarrold, K. Creegan, and J. Bower, “Studies of the chemistry of large semiconductor cluster ions,” International Journal of Mass Spectrometry and Ion Processes. 1990. link Times cited: 5 NOT USED (low confidence) M. Kohyama, S. Kose, M. Kinoshita, and R. Yamamoto, “The self-consistent tight-binding method: application to silicon and silicon carbide,” Journal of Physics: Condensed Matter. 1990. link Times cited: 27 Abstract: The self-consistent tight-binding (SCTB) model proposed by M… read more NOT USED (low confidence) K. Raghavachari, “Theoretical studies on silicon clusters,” Phase Transitions. 1990. link Times cited: 48 Abstract: This is a brief review of the theoretical studies which have… read more NOT USED (low confidence) M. Jarrold, U. Ray, and K. Creegan, “Chemistry of semiconductor clusters: Large silicon clusters are much less reactive towards oxygen than the bulk,” Journal of Chemical Physics. 1990. link Times cited: 70 Abstract: The chemical reactions of Si+n (n=10–65) with O2 have been i… read more NOT USED (low confidence) G. Schoeck and W. Pichl, “Bond trapping of cracks,” Physica Status Solidi (a). 1990. link Times cited: 17 Abstract: A model is presented for stabilization of cracks in an atomi… read more NOT USED (low confidence) H. Iyetomi and P. Vashishta, “Generalisation of the density-functional theory and three-body interactions in classical fluids,” Journal of Physics: Condensed Matter. 1989. link Times cited: 9 Abstract: An external field coupled with the two-body density distribu… read more NOT USED (low confidence) A. Arnold, N. Mauser, and J. Hafner, “A molecular dynamics study of the structure of liquid germanium,” Journal of Physics: Condensed Matter. 1989. link Times cited: 54 Abstract: The structure of liquid Ge as a function of temperature and … read more NOT USED (low confidence) K. Ohno, K. Esfarjani, and Y. Kawazoe, “Tight-Binding Methods.” 2018. link Times cited: 0 NOT USED (low confidence) R. Khanna and V. Sahajwalla, “Atomistic Simulations of Properties and Phenomena at High Temperatures.” 2014. link Times cited: 3 NOT USED (low confidence) K. Sattler, “The energy gap of clusters, nanoparticles, and quantum dots.” 2002. link Times cited: 48 NOT USED (low confidence) K. Sattler, “Nanodots and Nanowires of Silicon.” 2001. link Times cited: 0 NOT USED (low confidence) T. Ito, “Atomistic simulation of epitaxial growth processes.” 2001. link Times cited: 0 NOT USED (low confidence) L. Xin-hou, “New Potentials of Silicon and Germanium Crystals,” Chinese Physics Letters. 1996. link Times cited: 2 Abstract: In this paper, a new model of potential energy functions for… read more NOT USED (low confidence) T. Frauenheim, D. Porezag, T. Köhler, and F. Weich, “Molecular-Dynamic Simulations of Structure Formation in Complex Materials.” 1996. link Times cited: 1 NOT USED (low confidence) X. Liu, “NEW MODEL OF POTENTIAL-ENERGY FUNCTIONS FOR ATOMIC SOLIDS,” Journal of the Chemical Society, Faraday Transactions. 1995. link Times cited: 2 Abstract: A new theoretical model of potential-energy functions for at… read more NOT USED (low confidence) M. M. Souza and G. Amaratunga, “Self Diffusion in Silicon Using the Ackland Potential.” 1993. link Times cited: 2 NOT USED (low confidence) P. Fedders, “First Principles Molecular Dynamics Studies of a-Si and a-Si:H,” MRS Proceedings. 1993. link Times cited: 1 NOT USED (low confidence) R. Biswas, K. Roos, and M. Tringides, “Low Temperature Growth Mechanisms for Rheed Oscillations,” MRS Proceedings. 1993. link Times cited: 1 NOT USED (low confidence) G. Gadiyak, Y. Morokov, and D. Mukhin, “Simulation of fluorine interaction with a silicon surface,” Applied Surface Science. 1992. link Times cited: 3 NOT USED (low confidence) D. Wales and M. C. Waterworth, “Structures and rearrangements of model silicon clusters,” Journal of the Chemical Society, Faraday Transactions. 1992. link Times cited: 12 Abstract: We present a study of small silicon clusters bound by the em… read more NOT USED (low confidence) S. Li, R. Johnston, and J. Murrell, “Cluster structures and stabilities from solid-state potentials. Application to silicon clusters,” Journal of the Chemical Society, Faraday Transactions. 1992. link Times cited: 40 Abstract: An empirical potential-energy function comprising two- and t… read more NOT USED (low confidence) S. Sarma and K. E. Khor, “Empirical potential approach to the stability and energetics of thin films and surfaces,” Applied Surface Science. 1992. link Times cited: 1 NOT USED (low confidence) F. Ercolessi and J. B. Adams, “Interatomic Potentials From First-Principles Calculations,” MRS Proceedings. 1992. link Times cited: 22 Abstract: We propose a new scheme to extract “optimal” interatomic pot… read more NOT USED (low confidence) M. Robinson, “Computer Simulation of Atomic Collision Processes in Solids,” MRS Proceedings. 1992. link Times cited: 2 Abstract: Computer simulation is a major tool for studying the interac… read more NOT USED (low confidence) P. Fedders and D. D. Drabold, “Results from First Principles Molecular Dynamics Simulations on a-Si,” MRS Proceedings. 1991. link Times cited: 2 NOT USED (low confidence) R. Biswas, I. Kwon, and C. Soukoulis, “Molecular Dynamics Simulations of the Structural, Vibrational and Electronic Properties of Amorphous Silicon,” MRS Proceedings. 1990. link Times cited: 1 NOT USED (low confidence) A. Carlsson, “Beyond Pair Potentials in Elemental Transition Metals and Semiconductors,” Journal of Physics C: Solid State Physics. 1990. link Times cited: 169 NOT USED (low confidence) M. Heggie, “A New Interatomic Potential for Non-Metals.” 1989. link Times cited: 0 NOT USED (low confidence) W. Kamitakahara, R. Biswas, A. M. Bouchard, F. Gompf, and J. Suck, “Vibrational Spectra for Hydrogenated Amorphous Semiconductors,” MRS Proceedings. 1989. link Times cited: 0 Abstract: Hydrogen vibration spectra have been measured by neutron sca… read more NOT USED (low confidence) A. Carlsson, “Angular Forces in Transition Metals and Diamond Structure Semiconductors.” 1989. link Times cited: 1 NOT USED (low confidence) J. Hafner, “Quantum Theory of Structure: sp-Bonded Systems.” 1989. link Times cited: 11 NOT USED (low confidence) W. Andreoni, “On the Electronic and Structural Properties of Small Clusters.” 1989. link Times cited: 0 NOT USED (low confidence) V. Vítek, D. Srolovitz, and W. Morgan, “MOLECULAR DYNAMICS SIMULATION OF THE PHYSICS OF THIN FILM GROWTH ON SILICON: EFFECTS OF THE PROPERTIES OF INTERATOMIC POTENTIAL MODELS.” 1989. link Times cited: 0 NOT USED (low confidence) D. J. Oh and R. Johnson, “A Semi-Empirical Potential for Graphite,” MRS Proceedings. 1988. link Times cited: 4 NOT USED (low confidence) R. Biswas, A. M. Bouchard, W. Kamitakahara, G. Grest, and C. Soukoulis, “Vibrational Localization and Vibrational Spectra in Amorphous Silicon,” MRS Proceedings. 1988. link Times cited: 0 Abstract: Amorphous silicon structures have been generated by quenchin… read more NOT USED (high confidence) J. Utterson and R. Erban, “Symmetries of many-body systems imply distance-dependent potentials.,” Physical review. E. 2023. link Times cited: 0 Abstract: Considering an interatomic potential U(q), where q=[q_{1},q_… read more NOT USED (high confidence) S. Surulere, M. Shatalov, and E. Olayiwola, “Optimal interatomic potentials using modified method of least squares: Optimal form of interatomic potentials,” Open Physics. 2023. link Times cited: 0 Abstract: The problem of optimization of interatomic potentials is for… read more NOT USED (high confidence) J. Thomas, H. Chen, and C. Ortner, “Body-Ordered Approximations of Atomic Properties,” Archive for Rational Mechanics and Analysis. 2022. link Times cited: 1 NOT USED (high confidence) F. Berthier, Q. Lullien, and B. Legrand, “Effective site energy and cluster expansion approaches for the study of phase diagrams,” Physical Review B. 2021. link Times cited: 0 Abstract: We apply the cluster expansion (CE) method to determine the … read more NOT USED (high confidence) J. Thomas, H. Chen, and C. Ortner, “Rigorous body-order approximations of an electronic structure potential energy landscape.” 2021. link Times cited: 4 NOT USED (high confidence) D. Prasad and N. Mitra, “An atomistic study of phase transition in cubic diamond Si single crystal subjected to static compression,” Computational Materials Science. 2019. link Times cited: 7 NOT USED (high confidence) T. Gao et al., “Microstructural properties and evolution of nanoclusters in liquid Si during a rapid cooling process,” JETP Letters. 2017. link Times cited: 2 NOT USED (high confidence) J. C. Castro-Palacio, L. Velazquez-Abad, M. Fernández, and J. Q. Cuador-Gil, “Molecular dynamics study of one dimensional nanoscale Si/SiO2 interfaces,” The European Physical Journal D. 2013. link Times cited: 1 NOT USED (high confidence) A. Gufan, O. V. Kukin, and I. A. Osipenko, “An invariant form of the potential energy function used to simulate properties of condensed matter,” Bulletin of the Russian Academy of Sciences: Physics. 2012. link Times cited: 3 NOT USED (high confidence) S. Mahajan, G. Subbarayan, and B. Sammakia, “Estimating Kapitza Resistance Between \rm Si\hbox-\rm SiO_2 Interface Using Molecular Dynamics Simulations,” IEEE Transactions on Components, Packaging and Manufacturing Technology. 2011. link Times cited: 20 Abstract: The interface between nano-scale films is of relevance in ma… read more NOT USED (high confidence) A. Galashev, “Simulation of silicon nanoparticles stabilized by hydrogen at high temperatures,” Journal of Nanoparticle Research. 2010. link Times cited: 4 NOT USED (high confidence) A. M. Ukpong, “Studies of the electronic and vibrational signatures of the unusual bonding geometries in melt-quenched amorphous silicon,” Molecular Physics. 2009. link Times cited: 2 Abstract: Tight-binding molecular dynamics simulations have been perfo… read more NOT USED (high confidence) S. Mahajan, G. Subbarayan, and B. Sammakia, “Estimating Kapitza resistance between Si-SiO2 interface using molecular dynamics simulations,” 2008 11th Intersociety Conference on Thermal and Thermomechanical Phenomena in Electronic Systems. 2008. link Times cited: 13 Abstract: The interface between nano-scale films is of relevance in ma… read more NOT USED (high confidence) A. Galashev and I. A. Izmodenov, “Computer investigation of the structure of Si73 clusters surrounded by hydrogen,” Glass Physics and Chemistry. 2008. link Times cited: 5 NOT USED (high confidence) J. Schall, G. Gao, and J. Harrison, “Elastic constants of silicon materials calculated as a function of temperature using a parametrization of the second-generation reactive empirical bond-order potential,” Physical Review B. 2008. link Times cited: 48 Abstract: A parametrization for silicon is presented that is based on … read more NOT USED (high confidence) M. M. J. Treacy, J. Gibson, L. Fan, D. Paterson, and I. McNulty, “Fluctuation microscopy: a probe of medium range order,” Reports on Progress in Physics. 2005. link Times cited: 168 Abstract: Fluctuation microscopy is a hybrid diffraction-imaging techn… read more NOT USED (high confidence) M. Rao and S. Sengupta, “A mesoscopic model of a two-dimensional solid state structural transformation: statics and dynamics,” Journal of Physics: Condensed Matter. 2004. link Times cited: 6 Abstract: We study the equilibrium properties of a system of particles… read more NOT USED (high confidence) W. K. Liu, E. Karpov, S. Zhang, and H. S. Park, “An introduction to computational nanomechanics and materials,” Computer Methods in Applied Mechanics and Engineering. 2004. link Times cited: 405 NOT USED (high confidence) A. S. Barnard, S. Russo, and G. Leach, “Nearest neighbour considerations in Stillinger-Weber type potentials for diamond,” Molecular Simulation. 2002. link Times cited: 5 Abstract: Results of a preliminary investigation into the effect of va… read more NOT USED (high confidence) T. Lenosky et al., “Highly optimized empirical potential model of silicon,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 145 Abstract: We fit an empirical potential for silicon using the modified… read more NOT USED (high confidence) C. Herrero, “Quantum atomic dynamics in amorphous silicon; a path-integral Monte Carlo simulation,” Journal of Physics: Condensed Matter. 2000. link Times cited: 10 Abstract: The quantum dynamics of atoms in amorphous silicon has been … read more NOT USED (high confidence) M. Schaible, “Empirical Molecular Dynamics Modeling of Silicon and Silicon Dioxide: A Review,” Critical Reviews in Solid State and Materials Sciences. 1999. link Times cited: 28 Abstract: A number of computational methods have been developed over t… read more NOT USED (high confidence) C. Bittencourt, “Formation of a SiC buffer layer by reaction of Si (100) with methane and hydrogen plasma,” Journal of Physics D. 1999. link Times cited: 5 Abstract: The reaction of Si (100) surfaces at T = 950 °C with radical… read more NOT USED (high confidence) C. Bittencourt, “Reaction of Si (100) with silane–methane low-power plasma: SiC buffer-layer formation,” Journal of Applied Physics. 1999. link Times cited: 14 Abstract: The formation of a SiC buffer layer on Si (100) at substrate… read more NOT USED (high confidence) J. Q. Xie, J. Feng, and H. Lu, “Molecular-dynamics simulation of low-temperature growth of silicon films by cluster deposition,” Modelling and Simulation in Materials Science and Engineering. 1999. link Times cited: 8 Abstract: Silicon thin-film growth from cluster beams at a substrate t… read more NOT USED (high confidence) L. J. Lewis and N. Mousseau, “Tight-binding molecular-dynamics studies of defects and disorder in covalently bonded materials,” Computational Materials Science. 1998. link Times cited: 13 NOT USED (high confidence) J. Fang, R. Johnston, and J. Murrell, “Potential energy functions for atomic solids: V. Application to alkali metal solids,” Molecular Physics. 1993. link Times cited: 18 Abstract: Empirical potential functions comprising two-body and three-… read more NOT USED (high confidence) J. Holender and G. J. Morgan, “Generation of a large structure (105 atoms) of amorphous Si using molecular dynamics,” Journal of Physics: Condensed Matter. 1991. link Times cited: 29 Abstract: A method for generating amorphous tetrahedral structures hav… read more NOT USED (high confidence) A. Al-Derzi, R. Johnston, J. Murrell, and J. Rodriguez-Ruiz, “Potential energy functions for atomic solids: III. Fitting phonon frequencies and elastic constants of diamond structures,” Molecular Physics. 1991. link Times cited: 39 NOT USED (high confidence) S. Wong, “Controlling Indentation-induced Phases of Silicon.” 2017. link Times cited: 0 Abstract: Silicon (Si) is the backbone of the semiconductor industry. … read more NOT USED (high confidence) K. Scheerschmidt and M. Planck, “Empirical Molecular Dynamics: Possibilities, Requirements, and Limitations.” 2007. link Times cited: 9 |
| Funding | Not available |
| Short KIM ID
The unique KIM identifier code.
| MO_019616213550_000 |
| Extended KIM ID
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
| ThreeBodyCluster_BH_BiswasHamann_1987_Si__MO_019616213550_000 |
| DOI |
10.25950/68983f48 https://doi.org/10.25950/68983f48 https://commons.datacite.org/doi.org/10.25950/68983f48 |
| KIM Item Type
Specifies whether this is a Portable Model (software implementation of an interatomic model); Portable Model with parameter file (parameter file to be read in by a Model Driver); Model Driver (software implementation of an interatomic model that reads in parameters).
| Portable Model using Model Driver ThreeBodyCluster_BH__MD_043141570610_000 |
| Driver | ThreeBodyCluster_BH__MD_043141570610_000 |
| KIM API Version | 2.0 |
| Potential Type | bh |
| Grade | Name | Category | Brief Description | Full Results | Aux File(s) |
|---|---|---|---|---|---|
| P | vc-species-supported-as-stated | mandatory | The model supports all species it claims to support; see full description. |
Results | Files |
| F | vc-periodicity-support | mandatory | Periodic boundary conditions are handled correctly; see full description. |
Results | Files |
| P | vc-permutation-symmetry | mandatory | Total energy and forces are unchanged when swapping atoms of the same species; see full description. |
Results | Files |
| A | vc-forces-numerical-derivative | consistency | Forces computed by the model agree with numerical derivatives of the energy; see full description. |
Results | Files |
| P | vc-dimer-continuity-c1 | informational | The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description. |
Results | Files |
| P | vc-objectivity | informational | Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description. |
Results | Files |
| P | vc-inversion-symmetry | informational | Total energy is unchanged and forces change sign when inverting a configuration through the origin; see full description. |
Results | Files |
| P | vc-memory-leak | informational | The model code does not have memory leaks (i.e. it releases all allocated memory at the end); see full description. |
Results | Files |
| P | vc-thread-safe | mandatory | The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description. |
Results | Files |
| P | vc-unit-conversion | mandatory | The model is able to correctly convert its energy and/or forces to different unit sets; see full description. |
Results | Files |
BCC Lattice Constant
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
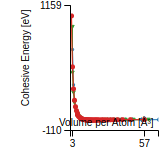
Cohesive Energy Graph
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
Diamond Lattice Constant
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Dislocation Core Energies
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
(No matching species)FCC Elastic Constants
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
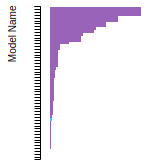
FCC Lattice Constant
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
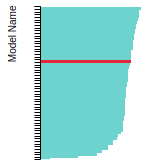
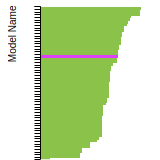
FCC Stacking Fault Energies
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
(No matching species)FCC Surface Energies
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
(No matching species)SC Lattice Constant
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Cubic Crystal Basic Properties Table
Species: SiCreators: Daniel S. Karls
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/b47dd4c4
Given an xyz file corresponding to a finite cluster of atoms, this Test Driver computes the total potential energy and atomic forces on the configuration. The positions are then relaxed using conjugate gradient minimization and the final positions and forces are recorded. These results are primarily of interest for training machine-learning algorithms.
Creators:
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/64cb38c5
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Cohesive energy versus lattice constant curve for bcc Si v004 | view | 2735 | |
| Cohesive energy versus lattice constant curve for diamond Si v004 | view | 3620 | |
| Cohesive energy versus lattice constant curve for fcc Si v004 | view | 3481 | |
| Cohesive energy versus lattice constant curve for sc Si v004 | view | 2198 |
Creators: Junhao Li and Ellad Tadmor
Contributor: tadmor
Publication Year: 2019
DOI: https://doi.org/10.25950/5853fb8f
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for bcc Si at zero temperature v006 | view | 2847 | |
| Elastic constants for diamond Si at zero temperature v001 | view | 5950 | |
| Elastic constants for fcc Si at zero temperature v006 | view | 2527 | |
| Elastic constants for sc Si at zero temperature v006 | view | 6142 |
Creators: Junhao Li
Contributor: jl2922
Publication Year: 2019
DOI: https://doi.org/10.25950/d794c746
Computes the elastic constants for hcp crystals by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for hcp Si at zero temperature v004 | view | 1783 |
Creators:
Contributor: ilia
Publication Year: 2024
DOI: https://doi.org/10.25950/2f2c4ad3
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/2765e3bf
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium zero-temperature lattice constant for bcc Si v007 | view | 2271 | |
| Equilibrium zero-temperature lattice constant for diamond Si v007 | view | 4574 | |
| Equilibrium zero-temperature lattice constant for fcc Si v007 | view | 2367 | |
| Equilibrium zero-temperature lattice constant for sc Si v007 | view | 1983 |
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/c339ca32
Calculates lattice constant of hexagonal bulk structures at zero temperature and pressure by using simplex minimization to minimize the potential energy.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium lattice constants for hcp Si v005 | view | 72077 |
Creators:
Contributor: mjwen
Publication Year: 2024
DOI: https://doi.org/10.25950/9d9822ec
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Linear thermal expansion coefficient of diamond Si at 293.15 K under a pressure of 0 MPa v002 | view | 42234215 |
Creators: Daniel S. Karls
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/c3dca28e
Given an extended xyz file corresponding to a non-orthogonal periodic box of atoms, use LAMMPS to compute the total potential energy and atomic forces.
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/fca89cea
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Monovacancy formation energy and relaxation volume for diamond Si | view | 3667700 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/c27ba3cd
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Vacancy formation and migration energy for diamond Si | view | 3333683 |
EquilibriumCrystalStructure__TD_457028483760_002
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium crystal structure and energy for Si in AFLOW crystal prototype A_hR8_148_cf v002 | other | view |
| Equilibrium crystal structure and energy for Si in AFLOW crystal prototype A_oF16_69_gh v002 | other | view |
No Driver
| Verification Check | Error Categories | Link to Error page |
|---|---|---|
| MemoryLeak__VC_561022993723_004 | other | view |
| PeriodicitySupport__VC_895061507745_004 | other | view |
| ThreeBodyCluster_BH_BiswasHamann_1987_Si__MO_019616213550_000.txz | Tar+XZ | Linux and OS X archive |
| ThreeBodyCluster_BH_BiswasHamann_1987_Si__MO_019616213550_000.zip | Zip | Windows archive |
This Model requires a Model Driver. Archives for the Model Driver ThreeBodyCluster_BH__MD_043141570610_000 appear below.
| ThreeBodyCluster_BH__MD_043141570610_000.txz | Tar+XZ | Linux and OS X archive |
| ThreeBodyCluster_BH__MD_043141570610_000.zip | Zip | Windows archive |
Parameters for the ‘new’ potential
This parameterization is optimized for tetrahedral structures of silicon (referred to in the paper as the ‘new’ potential in contrast to an ‘old’ version published two years earlier more suitable for high-pressure phases of silicon. The parameters for the ‘old’ potential were by fitting the model to structural energies calculated using density functional theory within the local-density approximation (LDA). The method used for determining the parameters for the ‘new’ potential is not mentioned explicitly, so we presume that the authors followed the same framework for both of their models. The influence distance parameter
Table of parameters
| Parameter | Value | Units |
|---|---|---|
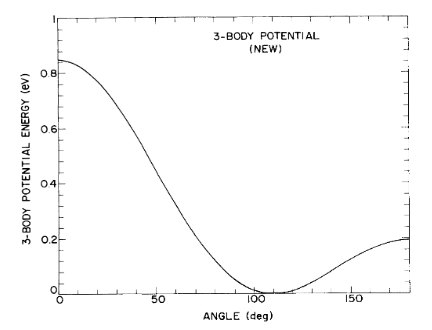
Representative plots
A plot from the original article depicting the angular variation of the three-body potential for the new classical Si model is shown. The bond lengths were set equal to 2.35 angstroms, which is the equilibrium bond length of Si.
Login to edit Wiki content
Wiki Contributors
| 2019-05-10T08:56:10.900217 | tadmor |
| 2019-05-09T19:26:36.079304 | dipghosh |
| 2019-05-09T19:22:51.679195 | dipghosh |