Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
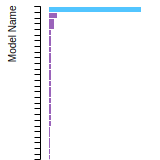
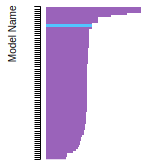
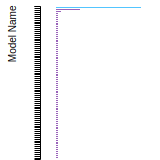
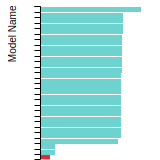
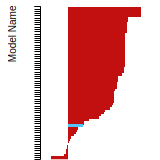
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
90 Citations (44 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) F. Zheng et al., “Structure and motifs of iron oxides from 1 to 3 TPa.” 2021. link Times cited: 1 Abstract: Iron oxides are fundamental components of planet-forming mat… read moreAbstract: Iron oxides are fundamental components of planet-forming materials. Understanding the FeO system ' s behavior and properties under high pressure can help us identify many new phases and states possible in exoplanetary interiors, especially terrestrial ones. Using the adaptive genetic algorithm (AGA), we investigate the structure of iron oxides for a wide range of stoichiometries (0.25 ≤ x O ≤ 0.8) at 1, 2, and 3 TPa. Five unreported ground-state structures with Fe 2 O, FeO, Fe 3 O 5 , FeO 2 , and FeO 4 compositions are identified. Phonon calculations confirm their dynamical stability. The ab initio molecular dynamics simulations confirm the thermal stability of Fe-rich phases at high temperatures. The calculated density of states (DOS) suggests that, except for FeO 4 , all phases are metallic, but their carrier densities decrease with increasing pressure and oxygen content. The cluster alignment analysis of stable and metastable phases shows that several motifs may co-exist in a structure of iron oxides with low O content. In contrast, most iron oxides with high O content adopt a simple BCC motif at TPa pressures. Our results provide a crystal structure database of iron oxides for modeling and understanding the interiors of exoplanets. read less USED (high confidence) Y. Fang et al., “Unconventional iron-magnesium compounds at terapascal pressures,” Physical Review B. 2021. link Times cited: 2 Abstract: Yimei Fang,1 Yang Sun,2, ∗ Renhai Wang,3, 4 Feng Zheng,1 Shu… read moreAbstract: Yimei Fang,1 Yang Sun,2, ∗ Renhai Wang,3, 4 Feng Zheng,1 Shunqing Wu,1, † Cai-Zhuang Wang,4 Renata M. Wentzcovitch,2, 5, 6, ‡ and Kai-Ming Ho4 Department of Physics, OSED, Key Laboratory of Low Dimensional Condensed Matter Physics (Department of Education of Fujian Province) Jiujiang Research institute, Xiamen University, Xiamen 361005, China. Department of Applied Physics and Applied Mathematics, Columbia University, New York, NY, 10027, USA Department of Physics, University of Science and Technology of China, Hefei 230026, China Department of Physics, Iowa State University, Ames, Iowa 50011, United States Department of Earth and Environmental Sciences, Columbia University, New York, NY, 10027, USA Lamont-Doherty Earth Observatory, Columbia University, Palisades, NY, 10964, USA (Dated: April 19, 2021) read less USED (high confidence) R. Wang et al., “Theoretical search for possible Li–Ni–B crystal structures using an adaptive genetic algorithm,” Journal of Applied Physics. 2020. link Times cited: 8 Abstract: The structural diversity of rare-earth and transition metal … read moreAbstract: The structural diversity of rare-earth and transition metal borides indicates that alkali-transition metal borides (A-T-B) show tremendous promise in exhibiting a variety of crystal structures with different dimensionalities of T-B frameworks. On the other hand, the A-T-B ternary systems are severely underexplored because of the synthetic challenges associated with their preparation. Accurate and efficient computational predictions of low-energy stable and metastable phases can identify the optimal compositions of the hypothetical compounds in the A-T-B systems to guide the synthesis. In this work, we have computationally discovered several new phases in the Li–Ni–B ternary system. The newly discovered LiNiB, Li2Ni3B, and Li2NiB phases expand the existing theoretical database, and the convex-hull surface of Li–Ni–B has been re-constructed. The lowest energy structure of the LiNiB compound has been found by an adaptive genetic algorithm with layered motif, which matches with the experimentally determined structure. According to our electrochemical calculations, LiNiB and another predicted layered Li2NiB compounds have great potential as anode materials for lithium batteries. The Li2Ni3B compound with the space group P4332 was predicted to crystallize in a cubic structure composed of distorted octahedral units of BNi6, which is isostructural to two noncentrosymmetric superconductors Li2Pd3B and Li2Pt3B. While we were unable to experimentally confirm the Li2Ni3B compound utilizing the hydride synthetic route, attempts to synthesize this compound by alternate methods remain highly desirable, considering its potential superconducting properties. read less USED (high confidence) L. Bulat, A. Ivanov, V. Osvenskiĭ, D. Pshenay-Severin, and A. Sorokin, “Thermal conductivity of Cu2Se taking into account the influence of mobile copper ions,” Physics of the Solid State. 2017. link Times cited: 3 USED (high confidence) Y. Shi et al., “Structures of defects on anatase TiO2(001) surfaces.,” Nanoscale. 2017. link Times cited: 19 Abstract: Defects on oxide surfaces play a crucial role in surface rea… read moreAbstract: Defects on oxide surfaces play a crucial role in surface reactivity and thus it is crucial to understand their atomic and electronic structures. The defects on anatase TiO2(001)-(1 × 4) surfaces are found to be highly reactive; however, due to the surface reconstruction, the defects exhibit a complicated character in different experiments that make it very challenging to determine their atomic structures. Here we present a systematic first-principles investigation of the defects on anatase TiO2(001)-(1 × 4) surfaces based on a global-search adaptive genetic algorithm (AGA) and density functional theory (DFT). For different Ti-O ratios, we identify the low energy defect structures, investigate their electronic structure using a hybrid functional, and map their regions of stability under realistic conditions. We successfully find novel oxygen vacancy (OV) and Ti interstitial (Tiini) structures that are different from the conventional ones in terms of their charge localization, magnetic state, and their scanning-tunneling-microscopy bright-dark image signature. This provides an insight into the complex geometric and electronic structure of the surface defects, and resolves several experimental discrepancies. read less USED (high confidence) S. Wu et al., “Exploring new phases of Fe3−xCoxC for rare-earth-free magnets,” Journal of Physics D: Applied Physics. 2017. link Times cited: 10 Abstract: Structures, magnetic moments, and magnetocrystalline anisotr… read moreAbstract: Structures, magnetic moments, and magnetocrystalline anisotropy energies of the Fe3−xCoxC intermetallic compounds are systematically investigated using adaptive genetic algorithm (AGA) crystal-structure predictions and first-principles calculations. Besides reproducing the known cementite (Pnma) structure of Fe3C, i.e. x = 0, the AGA searches also capture several new metastable phases within the room-temperature range. In particular, a bainite (P6322) structure exhibits the largest magnetic moment among all low-energy structures, and its energy is only 4 meV/atom higher than the cementite (Pnma) phase. The atomic structure of the Pnma Fe2CoC phase, i.e. x = 1, is also identified, and the calculated x-ray diffraction spectrum, magnetocrystalline anisotropy energy, and saturation magnetization based on the structure from our theoretical study are in good agreement with experiment. read less USED (high confidence) H. Mu, B. Xu, C. Ouyang, and X. Lei, “Highly optimized embedding atom method potential for Pt-Cu alloys,” Journal of Alloys and Compounds. 2017. link Times cited: 6 USED (high confidence) A. Iskandarov, Y. Ding, and Y. Umeno, “Effect of cation dopants in zirconia on interfacial properties in nickel/zirconia systems: an atomistic modeling study,” Journal of Physics: Condensed Matter. 2017. link Times cited: 6 Abstract: Cation doping is often used to stabilize the cubic or tetrag… read moreAbstract: Cation doping is often used to stabilize the cubic or tetragonal phase of zirconia for enhanced thermomechanical and electrochemical properties. In the present paper we report a combined density functional theory (DFT) and molecular dynamics study of the effect of Sc, Y, and Ce dopants on properties of Ni/ZrO2 interfaces and nickel sintering. First, we develop an MD model that is based on DFT data for various nickel/zirconia interfaces. Then, we employ the model to simulate Ni nanoparticles coalescing on a zirconia surface. The results show the possibility of particle migration by means of fast sliding over the surface when the work of separation is small (<1.0J m−2). The sliding observed for the O-terminated Ni(1 1 1)/ZrO2(1 1 1) interface is not affected by dopants in zirconia because the work of separation of the doped interface stays small. The most pronounced effect of the dopants is observed for the Zr-terminated Ni(1 1 1)/ZrO2(1 1 1) interface, which possesses a large work of separation (4.4J m−2) and thus restricts the sliding mechanism of Ni nanoparticle migration. DFT calculations for the interface revealed that dopants with a smaller covalent radius result in a larger energy barriers for Ni diffusion. We analyze this effect and discuss how it can be used to suppress nickel sintering by using the dopant selection. read less USED (high confidence) X. Zhao, L. Ke, C. Wang, and K. Ho, “Metastable cobalt nitride structures with high magnetic anisotropy for rare-earth free magnets.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 23 Abstract: Metastable structures of cobalt nitrides and Fe-substituted … read moreAbstract: Metastable structures of cobalt nitrides and Fe-substituted cobalt nitrides are explored as possible candidates for rare-earth free permanent magnets. Through crystal structure searches using an adaptive genetic algorithm, new structures of ConN (n = 3…8) are found to have lower energies than those previously discovered by experiments. Some structures exhibit large magnetic anisotropy energy, reaching as high as 200 μeV per Co atom (or 2.45 MJ m-3) based on first-principles density functional calculation. Substituting a fraction of Co with Fe helps in stabilizing new structures and at the same time further improves the magnetic properties. Our theoretical predictions provide useful insights into a promising system for the discovery of new rare-earth free magnets by experiment. read less USED (high confidence) N. Wang et al., “Embedded atom model for the liquid U–10Zr alloy based on density functional theory calculations,” RSC Advances. 2015. link Times cited: 5 Abstract: Understanding the structural characteristics of U–Zr alloys … read moreAbstract: Understanding the structural characteristics of U–Zr alloys is significant because they are very promising candidates for core materials in Gen IV nuclear reactors. In this work, we developed an embedded atom model for the liquid U–10Zr alloy based on density functional theory calculations and studied the structural, thermodynamic, and atomic-transport characteristics of liquid U–Zr alloys by molecular dynamics simulations based on this embedded atom model. The results showed that the mixed volume of U and Zr increased and the formation enthalpy of mixing was positive. The self-diffusion in liquid U–10Zr alloy obeyed the Arrhenius activation process; linear correlation coefficients were about 0.98 for the Arrhenius plots. The activation energies were evaluated as 33.9 and 32.6 kJ mol−1 and the preexponential factors at 23.73 and 21.88 × 10−5 cm2 s−1 for U and Zr, respectively. read less USED (high confidence) X. Zhao, L. Ke, M. C. Nguyen, C. Wang, and K. Ho, “Structures and magnetic properties of Co-Zr-B magnets studied by first-principles calculations,” arXiv: Materials Science. 2015. link Times cited: 14 Abstract: The structures and magnetic properties of the Co-Zr-B alloys… read moreAbstract: The structures and magnetic properties of the Co-Zr-B alloys near the Co5Zr composition were studied using adaptive genetic algorithm and first-principles calculations to guide further experimental effort on optimizing their magnetic performances. Through extensive structural searches, we constructed the contour maps of the energetics and magnetic moments of the Co-Zr-B magnet alloys as a function of composition. We found that the Co-Zr-B system exhibits the same structural motif as the "Co11Zr2" polymorphs, which plays a key role in achieving high coercivity. Boron atoms can either substitute selective cobalt atoms or occupy the interstitial sites. First-principles calculation shows that the magnetocrystalline anisotropy energies can be significantly improved through proper boron doping. read less USED (high confidence) F. Zhang et al., “Composition-dependent stability of the medium-range order responsible for metallic glass formation,” Acta Materialia. 2014. link Times cited: 24 USED (high confidence) S. Starikov et al., “Soft picosecond X-ray laser nanomodification of gold and aluminum surfaces,” Applied Physics B. 2014. link Times cited: 25 USED (high confidence) M. C. Nguyen, L. Ke, X. Zhao, V. Antropov, C. Wang, and K. Ho, “Atomic Structure and Magnetic Properties of HfCo_7 Alloy,” IEEE Transactions on Magnetics. 2013. link Times cited: 6 Abstract: Co rich Hf-Co alloys demonstrate promising properties as pot… read moreAbstract: Co rich Hf-Co alloys demonstrate promising properties as potential permanent magnet systems. However their structure has not been well established by experiment. We performed a search for low-energy structures of the HfCo7 system with recently developed adaptive genetic algorithm. Among the low-energy structures with 16 and 32 atoms per unit cell, we found structural motifs similar to the permanent magnet SmCo5. For the unit cell size larger than 40 atoms, we found a phase separation into pure hcp Co and Hf2Co7 in agreement with the experiment. Calculated magnetization and temperature of magnetic phase transition are similar to those for hcp Co. Overall considered HfCo7 system represents a mix of in- and out-of-plane magnetic anisotropies systems with possible site dependence of magnetic anisotropy. read less USED (high confidence) G. Norman, S. Starikov, V. Stegailov, I. Saitov, and P. Zhilyaev, “Atomistic Modeling of Warm Dense Matter in the Two‐Temperature State,” Contributions to Plasma Physics. 2013. link Times cited: 45 Abstract: Warm dense matter is the state between the heated condensed … read moreAbstract: Warm dense matter is the state between the heated condensed matter and plasma. The importance of the development of warm dense matter theoretical description is determined by the fact that such conditions may arise in the variety of different scientific and industrial applications. For instance, warm dense matter is formed: in the matter impacted by femto‐ and picoseconds laser pulses; in nuclear materials at the formation of radiation track, etc. In these phenomena, the initial state of the system is a two‐temperature state and the electron temperature may be several orders higher than the ion one. In this work, the attempt of development of the united atomistic model of a warm dense matter is carried out. The special consideration is given to the twotemperature effects and the influence of the electron pressure on the behavior of ions. (© 2013 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less USED (high confidence) P. Brommer, F. Gähler, and M. Mihalkovi[cbreve], “Ordering and correlation of cluster orientations in CaCd6,” Philosophical Magazine. 2007. link Times cited: 10 Abstract: In order to study the low-temperature phase transition in Ca… read moreAbstract: In order to study the low-temperature phase transition in CaCd6, which is attributed to a reordering of the innermost tetrahedral cluster shells, accurate Embedded-Atom-Method potentials are developed for this system. With these potentials, the ideal cluster structure and the couplings between neighbouring clusters are determined. From these data, an effective Hamiltonian for the cluster orientations is derived. The Hamiltonian is used in Monte Carlo simulations, which exhibit a sharp jump in the internal energy near the expected transition temperature. read less USED (high confidence) F. Rösch, H. Trebin, and P. Gumbsch, “Interatomic potentials and the simulation of fracture: C15
NbCr2,” International Journal of Fracture. 2006. link Times cited: 12 USED (high confidence) S. Hocker, F. Gähler, and P. Brommer, “Molecular dynamics simulation of aluminium diffusion in decagonal quasicrystals,” Philosophical Magazine. 2006. link Times cited: 10 Abstract: Al diffusion in decagonal Al–Ni–Co and Al–Cu–Co quasicrystal… read moreAbstract: Al diffusion in decagonal Al–Ni–Co and Al–Cu–Co quasicrystals is investigated by molecular dynamics simulations. Results obtained with newly developed embedded-atom method potentials are compared with our previous work with effective pair potentials. With both types of potential, strong aluminium diffusion is observed above two-thirds of the melting temperature, and the general behaviours of the system are quite similar. The diffusion constant is measured as a function of temperature and pressure, and the activation enthalpies and activation volumes are determined from the resulting Arrhenius plot. For a number of important diffusion processes, the energy barriers are determined with molecular statics simulations. The qualitative behaviour of the dynamics is also confirmed by ab-initio simulations. read less USED (high confidence) F. Rösch, “Atomistic dynamics of crack propagation in complex metallic alloys.” 2008. link Times cited: 0 Abstract: The failure of solid matter is familiar to us from everyday … read moreAbstract: The failure of solid matter is familiar to us from everyday life. A broken dish or a shattered glass is annoying. The breakage of a ship or an airplane hull, however, can cost human lives. Nevertheless, insight into the fundamental mechanisms leading to fracture has been gained only within the last couple of years. One reason for this is that fracture is a multi-scale phenomenon. A macroscopic external strain is directed to the crack tip, where it breaks atomic bonds. Thus, to understand fracture mechanisms in macroscopic devices, one also has to know the processes on the atomic scale. These are hardly accessible by experiments, in particular when dynamic aspects are the center of interest. They are also not included in the classical elastic theory of fracture. Computer simulations have proven to be a useful tool to examine fracture processes on an atomic level. In molecular dynamics simulations the trajectories of the atoms are calculated by integration of Newtons equations of motion. Numerical experiments with simple crystal structures and model interactions reveal phenomena that are related to the discrete nature of matter and therefore cannot be explained by continuum theories. In more complex systems the mechanisms are not yet clear.
In the current work two extreme cases of complex metallic alloys are investigated. The model quasicrystal is built-up mainly from atomic clusters. Due to the aperiodic long-range order, no unit cell exists. A major building block of the quasicrystal can also be used to obtain a periodic Friauf-Laves compound. The Friauf-Laves phases are topologically close-packed binary structures and form a huge class of intermetallic compounds. Among them are many candidates for high-temperature use. However, their brittleness at low and ambient temperature limits applications. Molecular dynamics simulations are performed to examine this brittle behavior at low temperature. First, model potentials are applied to qualitatively probe the influence of the underlying structure on crack propagation. In a second step, a specific Friauf-Laves compound is chosen. Interatomic potentials are constructed, which reproduce quantities obtained by quantum-mechanical calculations. Systematic fracture simulations then are performed.
Der Bruch fester Materie geschieht durch die Ausbreitung von Rissen und ist uns aus dem Alltag vertraut. Ein zerbrochener Teller oder ein zerschlagenes Glas ist lastig. Versagt dagegen der Rumpf eines Schiffes oder der eines Flugzeuges, kann dies Menschen das Leben kosten. Gleichwohl wurde erst in den letzten Jahren Einblick in die grundlegenden Mechanismen gewonnen, die zum Versagen von Festkorpern durch Rissbildung fuhren. Ein Grund hierfur ist, dass Bruch ein Multi-Skalen-Phanomen darstellt. Eine makroskopisch von ausen angelegte Spannung wird an der Rissspitze verstarkt. Daraufhin werden atomare Bindungen gebrochen. Um Bruchmechanismen makroskopischer Bauteile zu verstehen, wird somit die Kenntnis von Prozessen auf atomarer Ebene benotigt. Diese sind durch heutige experimentelle Techniken kaum zuganglich, insbesondere dann nicht, wenn dynamische Aspekte im Mittelpunkt des Interesses stehen. Solche Prozesse sind auch in der klassischen Elastizitatstheorie des Bruchs nicht berucksichtigt. Computersimulationen haben sich als nutzliches Hilfsmittel zur Untersuchung atomarer Vorgange erwiesen. In Molekulardynamik-Simulationen werden die Bahnen der Atome durch Integration der Newton'schen Bewegungsgleichungen berechnet. Numerische Experimente mit einfachen Kristallstrukturen und Modellwechselwirkungen lassen Effekte erkennen, fur die der diskrete Aufbau der Materie verantwortlich ist. Kontinuumstheorien sind daher nicht geeignet, die erwahnten Phanomene zu erklaren. Die Vorgange in komplexeren Festkorpern sind bis heute noch nicht vollstandig verstanden. Um einen Einblick in die grundlegenden Mechanismen zu erhalten, wird in der vorliegende Arbeit das Bruchverhalten komplexer metallischer Verbindungen bei tiefen Temperaturen mit Hilfe der Molekulardynamik untersucht. read less USED (low confidence) S. Hayakawa and H. Xu, “Development of an interatomic potential for L12 precipitates in Fe–Ni–Al alloys,” Computational Materials Science. 2024. link Times cited: 0 USED (low confidence) T. Yang, X. Han, W. Li, X. Chen, and P. Liu, “Angular dependent potential for Al-Zr binary system to study the initial heterogeneous nucleation behavior of liquid Al on L12-Al3Zr,” Computational Materials Science. 2023. link Times cited: 0 USED (low confidence) Y. Fang et al., “Structural prediction of Fe-Mg-O compounds at super-Earth’s pressures,” Physical Review Materials. 2023. link Times cited: 0 Abstract: Terrestrial exoplanets are of great interest for being simul… read moreAbstract: Terrestrial exoplanets are of great interest for being simultaneously similar to and different from Earth. Their compositions are likely comparable to those of solar-terrestrial objects, but their internal pressures and temperatures can vary significantly with their masses/sizes. The most abundant non-volatile elements are O, Mg, Si, Fe, Al, and Ca, and there has been much recent progress in understanding the nature of magnesium silicates up to and beyond ~3 TPa. However, a critical element, Fe, has yet to be systematically included in materials discovery studies of potential terrestrial planet-forming phases at ultra-high pressures. Here, using the adaptive genetic algorithm (AGA) crystal structure prediction method, we predict several unreported stable crystalline phases in the binary Fe-Mg and ternary Fe-Mg-O systems up to pressures of 3 TPa. The analysis of the local packing motifs of the low-enthalpy Fe-Mg-O phases reveals that the Fe-Mg-O system favors a BCC motif under ultra-high pressures regardless of chemical composition. Besides, oxygen enrichment is conducive to lowering the enthalpies of the Fe-Mg-O phases. Our results extend the current knowledge of structural information of the Fe-Mg-O system to exoplanet pressures. read less USED (low confidence) W. Angermeier, B. Scheiner, N. Shaffer, and T. White, “Disentangling the effects of non-adiabatic interactions upon ion self-diffusion within warm dense hydrogen,” Philosophical Transactions of the Royal Society A. 2023. link Times cited: 1 Abstract: Warm dense matter is a material state in the region of param… read moreAbstract: Warm dense matter is a material state in the region of parameter space connecting condensed matter to classical plasma physics. In this intermediate regime, we investigate the significance of non-adiabatic electron-ion interactions upon ion dynamics. To disentangle non-adiabatic from adiabatic electron-ion interactions, we compare the ion self-diffusion coefficient from the non-adiabatic electron force field computational model with an adiabatic, classical molecular dynamics simulation. A classical pair potential developed through a force-matching algorithm ensures the only difference between the models is due to the electronic inertia. We implement this new method to characterize non-adiabatic effects on the self-diffusion of warm dense hydrogen over a wide range of temperatures and densities. Ultimately we show that the impact of non-adiabatic effects is negligible for equilibrium ion dynamics in warm dense hydrogen. This article is part of the theme issue ‘Dynamic and transient processes in warm dense matter’. read less USED (low confidence) J. Sun et al., “Layered Li-Co-B as a Low-Potential Anode for Lithium-Ion Batteries.,” Inorganic chemistry. 2023. link Times cited: 0 Abstract: An anode material is one of the key factors affecting the ca… read moreAbstract: An anode material is one of the key factors affecting the capacity, cycle, and rate (fast charge) performance of lithium-ion batteries. Using the adaptive genetic algorithm, we found a new ground-state Li2CoB and two metastable states LiCoB and LiCo2B2 in the Li-Co-B system. The Li2CoB phase is a lithium-rich layered structure, and it has an equivalent lithium-ion migration barrier (0.32 eV) in addition to the lower voltage platform (0.05 V) than graphite, which is the most important commercial anode material at present. Moreover, we analyzed the mechanism of delithiation for Li2CoB and found that it maintained metallicity in the process of delithiation, indicating its good conductivity as an electrode material. Therefore, it is an excellent potential anode material for lithium-ion batteries. Our work provides a promising theoretical basis for the experimental synthesis of Li-Co-B and similar new materials. read less USED (low confidence) S. Zhang et al., “Wide-Temperature Tunable Phonon Thermal Switch Based on Ferroelectric Domain Walls of Tetragonal KTN Single Crystal,” Nanomaterials. 2023. link Times cited: 0 Abstract: Ferroelectric domain walls (DWs) of perovskite oxide materia… read moreAbstract: Ferroelectric domain walls (DWs) of perovskite oxide materials, which can be written and erased by an external electric field, offer the possibility to dynamically manipulate phonon scattering and thermal flux behavior. Different from previous ferroelectric materials, such as BaTiO3, PbTiO3, etc., with an immutable and low Curie temperature. The Curie temperature of perovskite oxide KTa1−xNbxO3 (KTN) crystal can be tuned by altering the Ta/Nb ratio. In this work, the ferroelectric KTa0.6Nb0.4O3 (KTN) single crystal is obtained by the Czochralski method. To understand the role of ferroelectric domains in thermal transport behavior, we perform a nonequilibrium molecular dynamics (NEMD) calculation on monodomain and 90° DWs of KTN at room temperature. The calculated thermal conductivity of monodomain KTN is 9.84 W/(m·k), consistent with experimental results of 8.96 W/(m·k), and distinctly decreased with the number of DWs indicating the outstanding performance of the thermal switch. We further evaluate the thermal boundary resistance (TBR) of KTN DWs. An interfacial thermal resistance value of 2.29 × 10−9 K·m2/W and a large thermal switch ratio of 4.76 was obtained for a single DW of KTN. Our study shows that the ferroelectric KTN can provide great potential for the application of thermal switch at room temperature and over a broad temperature range. read less USED (low confidence) F. Zheng et al., “Superconductivity in the Li-B-C system at 100 GPa,” Physical Review B. 2023. link Times cited: 4 USED (low confidence) H. Wang, X. Gao, S. Chen, L. Yiming, Z.-wang Wu, and H. Ren, “Effects of Al on the precipitation of B2 Cu-rich particles in Fe–Cu ferritic alloy: Experimental and theoretical study,” Journal of Alloys and Compounds. 2020. link Times cited: 5 USED (low confidence) L. Stanek, R. Clay, M. Dharma-wardana, M. Wood, K. Beckwith, and M. Murillo, “Efficacy of the radial pair potential approximation for molecular dynamics simulations of dense plasmas,” arXiv: Plasma Physics. 2020. link Times cited: 12 Abstract: Macroscopic simulations of dense plasmas rely on detailed mi… read moreAbstract: Macroscopic simulations of dense plasmas rely on detailed microscopic information that can be computationally expensive and is difficult to verify experimentally. In this work, we delineate the accuracy boundary between microscale simulation methods by comparing Kohn-Sham density functional theory molecular dynamics (KS-MD) and radial pair potential molecular dynamics (RPP- MD) for a range of elements, temperature, and density. By extracting the optimal RPP from KS-MD data using force-matching, we constrain its functional form and dismiss classes of potentials that assume a constant power law for small interparticle distances. Our results show excellent agreement between RPP-MD and KS-MD for multiple metrics of accuracy at temperatures of only a few electron volts. The use of RPPs offers orders of magnitude decrease in computational cost and indicates that three-body potentials are not required beyond temperatures of a few eV. Due to its efficiency, the validated RPP-MD provides an avenue for reducing errors due to finite-size effects that can be on the order of $\sim20\%$. read less USED (low confidence) B. Lin, J. Li, Z. Wang, and J. Wang, “Dislocation nucleation from Zr–Nb bimetal interfaces cooperating with the dynamic evolution of interfacial dislocations,” International Journal of Plasticity. 2020. link Times cited: 13 USED (low confidence) Y. Wang, Y. Shi, C. Zhao, Q. Zheng, and J. Zhao, “Photogenerated carrier dynamics at the anatase/rutile
TiO2
interface,” Physical Review B. 2019. link Times cited: 10 USED (low confidence) S. Starikov, L. Kolotova, A. Kuksin, D. Smirnova, and V. Tseplyaev, “Atomistic simulation of cubic and tetragonal phases of U-Mo alloy: Structure and thermodynamic properties,” Journal of Nuclear Materials. 2018. link Times cited: 46 USED (low confidence) S. Starikov, A. Kuksin, D. Smirnova, A. Dolgodvorov, and V. Ozrin, “Multiscale Modeling of Uranium Mononitride: Point Defects Diffusion, Self-Diffusion, Phase Composition,” Defect and Diffusion Forum. 2017. link Times cited: 5 Abstract: Multiscale computational approach is used to evaluate micros… read moreAbstract: Multiscale computational approach is used to evaluate microscopic parameters for description of nitride nuclear fuel. The results of atomistic simulation and thermodynamic modeling allow to estimate diffusivity and concentrations of point defects at various stoichiometric ratios of UN1+x. The diffusivities of Xe atom were calculated in various equilibrium states. In addition, we obtained the dependence of partial nitrogen pressure on x and temperature. The results of atomistic simulation were used for modeling of nuclear fuel behavior with use of mechanistic fuel codes. read less USED (low confidence) D. Smirnova and S. Starikov, “An interatomic potential for simulation of Zr-Nb system,” Computational Materials Science. 2017. link Times cited: 37 USED (low confidence) K. Migdal, P. Pokatashkin, and A. Yanilkin, “Thermodynamic properties and phase transitions of γ and liquid uranium: QMD and classical MD modeling.” 2017. link Times cited: 1 Abstract: The application of molecular dynamics allows us to take into… read moreAbstract: The application of molecular dynamics allows us to take into account the influence of temperature on thermodynamic properties and phase transitions. In this work different uranium phases are investigated at finite temperatures by means of quantum and classical molecular dynamics. The stability of high temperature γ phase is discussed. The boundaries of phase stability are estimated based on quantum molecular results. In order to investigate phase transitions new interatomic potential is developed by force-matching method. The melting curve up to 750 GPa is obtained by Z-modified method. The results are in a good agreement with experimental data and classical molecular dynamics simulation by two phase methods up to 100 GPa.The application of molecular dynamics allows us to take into account the influence of temperature on thermodynamic properties and phase transitions. In this work different uranium phases are investigated at finite temperatures by means of quantum and classical molecular dynamics. The stability of high temperature γ phase is discussed. The boundaries of phase stability are estimated based on quantum molecular results. In order to investigate phase transitions new interatomic potential is developed by force-matching method. The melting curve up to 750 GPa is obtained by Z-modified method. The results are in a good agreement with experimental data and classical molecular dynamics simulation by two phase methods up to 100 GPa. read less USED (low confidence) P. Korotaev, A. Kuksin, P. Pokatashkin, and A. Yanilkin, “Quantum and classical molecular dynamics simulation of boron carbide behavior under pressure.” 2017. link Times cited: 3 Abstract: We present the study of boron carbide behavior under pressur… read moreAbstract: We present the study of boron carbide behavior under pressure using a multiscale approach. Both quantum and classical molecular dynamics simulations are implemented at this work. Specific phase transitions of boron carbide: chain bending and disordering are discussed and stress-phase diagram is constructed. Interatomic angular dependent potential is obtained. We present a study of grain slipping along amorphous zones, as this phenomenon is to be investigated for the construction of the microscopic model of deformation under shock wave loading. read less USED (low confidence) N. Wang et al., “Energetics of gaseous and volatile fission products in molten U–10Zr alloy: A density functional theory study,” Journal of Nuclear Materials. 2015. link Times cited: 1 USED (low confidence) D. Smirnova, A. Kuksin, and S. Starikov, “Investigation of point defects diffusion in bcc uranium and U–Mo alloys,” Journal of Nuclear Materials. 2015. link Times cited: 47 USED (low confidence) D. Schopf, H. Euchner, and H. Trebin, “Effective potentials for simulations of the thermal conductivity of type-I semiconductor clathrate systems,” Physical Review B. 2014. link Times cited: 13 USED (low confidence) X. Zhao, M. C. Nguyen, C. Wang, and K. Ho, “Structures and stabilities of alkaline earth metal peroxides XO2 (X = Ca, Be, Mg) studied by a genetic algorithm,” RSC Advances. 2013. link Times cited: 19 Abstract: The structures and stabilities of alkaline earth metal perox… read moreAbstract: The structures and stabilities of alkaline earth metal peroxides XO2 (X = Ca, Be, Mg) were studied using an adaptive genetic algorithm (GA) for global structure optimization in combination with first-principles calculations. From the adaptive GA search, we obtained an orthorhombic structure for CaO2 with 12 atoms in the unit cell, which is energetically more favorable than the previously proposed structures. Reaction energy of the decomposition CaO2 → CaO + 1/2O2 determined by density functional theory (DFT) calculation shows that this orthorhombic calcium peroxide structure is thermodynamically stable. The simulated X-ray diffraction (XRD) pattern using our predicted structure is in excellent agreement with experimental data. We also show that crystal phase BeO2 is unlikely to exist under normal conditions. MgO2 has a cubic pyrite structure, but it is not stable against decomposition: MgO2 → MgO + 1/2O2. read less USED (low confidence) M. C. Nguyen, J.-H. Choi, X. Zhao, C. Wang, Z. Zhang, and K. Ho, “New layered structures of cuprous chalcogenides as thin film solar cell materials: Cu2Te and Cu2Se.,” Physical review letters. 2013. link Times cited: 79 Abstract: The stable crystal structures of two cuprous chalcogenides o… read moreAbstract: The stable crystal structures of two cuprous chalcogenides of Cu2X (X=Te or Se) are predicted using an adaptive genetic algorithm in combination with first-principles density functional theory calculations. Both systems are found to prefer a unique and previously unrecognized layered structure, with the total energies much lower than all structures proposed in the literature so far. The newly discovered structures are further shown to be dynamically and mechanically stable, and possess electronic properties consistent with existing experimental observations. In particular, their layered nature is expected to prevail over other structural forms at the interfaces of thin-film solar cells, and knowledge about the precise atomic structures of the interfaces is a prerequisite for achieving long-term stability and high efficiency of CdTe and Cu(In,Ga)Se2 solar cells. read less USED (low confidence) C. Ciobanu, C. Wang, and K. Ho, “Crystal Structure Prediction.” 2013. link Times cited: 3 USED (low confidence) D. E. Smirnova, S. Starikov, S. Starikov, V. Stegailov, and V. Stegailov, “Interatomic potential for uranium in a wide range of pressures and temperatures,” Journal of Physics: Condensed Matter. 2012. link Times cited: 3 Abstract: Using the force-matching method we develop an interatomic po… read moreAbstract: Using the force-matching method we develop an interatomic potential that allows us to study the structure and properties of α-U, γ-U and liquid uranium. The potential is fitted to the forces, energies and stresses obtained from ab initio calculations. The model gives a good comparison with the experimental and ab initio data for the lattice constants of α-U and γ-U, the elastic constants, the room-temperature isotherm, the normal density isochore, the bond-angle distribution functions and the vacancy formation energies. The calculated melting line of uranium at pressures up to 80 GPa and the temperature of the α–γ transition at 3 GPa agree well with the experimental phase diagram of uranium. read less USED (low confidence) B. Bauerhenne, “Empirical MD Simulations of Laser-Excited Matter,” Materials Interaction with Femtosecond Lasers. 2021. link Times cited: 0 USED (low confidence) A. Kiselev, J. Roth, and H. Trebin, “Molecular Dynamics Simulations of Silicon: The Influence of Electron-Temperature Dependent Interactions.” 2016. link Times cited: 2 NOT USED (low confidence) W. Xia et al., “Accelerating the discovery of novel magnetic materials using machine learning–guided adaptive feedback,” Proceedings of the National Academy of Sciences of the United States of America. 2022. link Times cited: 6 Abstract: Significance Discovering rare earth (RE)–free magnets that c… read moreAbstract: Significance Discovering rare earth (RE)–free magnets that can meet the performance and cost goals for advanced electromagnetic devices has been the dream of many scientists over several decades. We present the efficient discovery and synthesis of an RE-free magnetic Fe3CoB2 compound in this paper. Our machine learning (ML)–guided framework greatly reduces the complexity of high-throughput screening and makes ab initio calculations and further structure searches using an adaptive genetic algorithm much more effective than previous approaches. We demonstrate that our ML-guided framework enables the computational discovery and experimental synthesis of an Fe3CoB2 compound to be accomplished in days. Our ML-guided framework presents a paradigm for efficient materials design and discovery in the digital era. read less NOT USED (low confidence) A. Krishnamoorthy et al., “EZFF: Python library for multi-objective parameterization and uncertainty quantification of interatomic forcefields for molecular dynamics,” arXiv: Computational Physics. 2020. link Times cited: 6 NOT USED (low confidence) X. Zhao, S. Wu, M. C. Nguyen, K. Ho, and C. Wang, “Adaptive Genetic Algorithm for Structure Prediction and Application to Magnetic Materials,” Handbook of Materials Modeling. 2018. link Times cited: 4 NOT USED (low confidence) X. Zhao, C. Wang, M. Kim, and K. Ho, “Fe-Cluster Compounds of Chalcogenides: Candidates for Rare-Earth-Free Permanent Magnet and Magnetic Nodal-Line Topological Material.,” Inorganic chemistry. 2017. link Times cited: 3 Abstract: Fe-cluster-based crystal structures are predicted for chalco… read moreAbstract: Fe-cluster-based crystal structures are predicted for chalcogenides Fe3X4 (X = S, Se, Te) using an adaptive genetic algorithm. Topologically different from the well-studied layered structures of iron chalcogenides, the newly predicted structures consist of Fe clusters that are either separated by the chalcogen atoms or connected via sharing of the vertex Fe atoms. Using first-principles calculations, we demonstrate that these structures have competitive or even lower formation energies than the experimentally synthesized Fe3X4 compounds and exhibit interesting magnetic and electronic properties. In particular, we show that Fe3Te4 can be a good candidate as a rare-earth-free permanent magnet and Fe3S4 can be a magnetic nodal-line topological material. read less NOT USED (low confidence) P. Brommer, A. Kiselev, D. Schopf, P. Beck, J. Roth, and H. Trebin, “Classical interaction potentials for diverse materials from ab initio data: a review of potfit,” Modelling and Simulation in Materials Science and Engineering. 2014. link Times cited: 76 Abstract: Force matching is an established technique to generate effec… read moreAbstract: Force matching is an established technique to generate effective potentials for molecular dynamics simulations from first-principles data. This method has been implemented in the open source code potfit. Here, we present a review of the method and describe the main features of the code. Particular emphasis is placed on the features added since the initial release: interactions represented by analytical functions, differential evolution as optimization method, and a greatly extended set of interaction models. Beyond the initially present pair and embedded-atom method potentials, potfit can now also optimize angular dependent potentials, charge and dipolar interactions, and electron-temperature-dependent potentials. We demonstrate the functionality of these interaction models using three example systems: phonons in type I clathrates, fracture of α-alumina, and laser-irradiated silicon. read less NOT USED (low confidence) X. Zhao, “Accelerating materials discovery and design: computational study of the structure and properties of materials.” 2015. link Times cited: 0 Abstract: This thesis summarizes our efforts to study the structure an… read moreAbstract: This thesis summarizes our efforts to study the structure and properties of materials computationally. The adaptive genetic algorithm (AGA) developed by us to predict crystal/surface/interface structures is presented. Applications of AGA to a variety of systems, such as non-rare earth magnetic materials, ultra-hard transition metal borides and SrTiO3 grain boundaries, are discussed. We demonstrated by AGA the capability of solving crystal structures with more than 100 atoms per unit cell and rapidly accessing the structures and phase stabilities of different compositions in multicomponent systems. We also introduced a motif-network scheme to study the complex crystal structures in silicate cathodes. In addition, we explored different computational methods for atomistic simulations of materials behavior, such as Monte Carlo modeling of the alnico magnets. read less NOT USED (low confidence) P. Brommer, “Development and test of interaction potentials for complex metallic alloys.” 2008. link Times cited: 8 Abstract: Complex metallic alloys and quasicrystals show extraordinary… read moreAbstract: Complex metallic alloys and quasicrystals show extraordinary physical properties relevant for technological applications, for example hardness at low density. In the study of these systems, atomistic simulation with classical interaction potentials is a very promising tool. Such simulations require classical effective potentials describing the cohesive energy as a function of the atomic coordinates. The quality of the simulation depends crucially on the accuracy with which this potential describes the real interactions. One way to generate physically relevant potentials is the force matching method, where the parameters of a potential are adjusted to optimally reproduce the forces on individual atoms determined from quantum-mechanical calculation. The programme package potfit developed as part of this thesis implements the force matching method efficiently. Potentials are generated for a number of complex metallic alloy systems. A potential for the decagonal basic Ni-rich Al-Co-Ni quasicrystal is used to simulate diffusion processes and melting. In the CaCd6 system built from multishelled clusters, the shape and orientation of the innermost cluster shell is studied. Finally, phonon dispersion in the Mg-Zn system is determined and compared to experiment. The programme potfit is shown to be an effective tool for generating physically justified effective potentials. Potentials created with potfit can greatly improve the understanding of complex metallic alloys through atomistic simulations.
Komplexe intermetallische Verbindungen und Quasikristalle zeigen ausergewohnliche physikalische Eigenschaften, wie z.B. Harte bei geringer Dichte. Bei der Untersuchung dieser Systeme sind atomistische Simulationen mit klassischen Wechselwirkungspotenzialen ein wichtiges Werkzeug. Fur solche Simulationen benotigt man klassische effektive Potenziale, die die Bindungsenergie als eine Funktion der Atomkoordinaten beschreiben. Die Qualitat der Simulation hangt entscheidend von der Genauigkeit ab, mit der diese Potenziale die echten Wechselwirkungen wiedergeben. Eine Moglichkeit, physikalisch relevante Potenziale zu erzeugen, ist die Force-Matching-Methode. Dabei werden die Parameter eines Potenzials so angepasst, dass die mit quantenmechanischen Methoden bestimmten Krafte auf die einzelnen Atome bestmoglich reproduziert werden. Das als Teil dieser Arbeit entwickelte Programmpaket potfit implementiert die Force-Matching-Methode effizient. Fur einige komplexe intermetallische Verbindungen werden Potenziale bestimmt. In dekagonalen Al-Co-Ni-Quasikristallen werden mit Hilfe eines Potenzials Diffusionsprozesse und Schmelzvorgange simuliert. In der aus mehrschaligen Clustern bestehenden CaCd6-Verbindung wird die Struktur der innersten Clusterschale untersucht. Schlieslich wird die Phononendispersion im Mg-Zn-System bestimmt und mit experimentellen Ergebnissen verglichen. Es wird gezeigt, dass das Programm potfit ein effektives Werkzeug zur Erzeugung physikalisch gerechtfertigter Wechselwirkungen ist. Potenziale, die mit potfit erzeugt werden, konnen zum Verstandnis komplexer metallischer Verbindungen durch atomistische Simulationen viel beitragen. read less NOT USED (high confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 NOT USED (high confidence) M. Dharma-wardana, L. Stanek, and M. Murillo, “Yukawa-Friedel-tail pair potentials for warm dense matter applications.,” Physical review. E. 2022. link Times cited: 3 Abstract: Accurate equations of state (EOS) and plasma transport prope… read moreAbstract: Accurate equations of state (EOS) and plasma transport properties are essential for numerical simulations of warm dense matter encountered in many high-energy-density situations. Molecular dynamics (MD) is a simulation method that generates EOS and transport data using an externally provided potential to dynamically evolve the particles without further reference to the electrons. To minimize computational cost, pair potentials needed in MD may be obtained from the neutral-pseudoatom (NPA) approach, a form of single-ion density functional theory (DFT), where many-ion effects are included via ion-ion correlation functionals. Standard N-ion DFT-MD provides pair potentials via the force matching technique but at much greater computational cost. Here we propose a simple analytic model for pair potentials with physically meaningful parameters based on a Yukawa form with a thermally damped Friedel tail (YFT) applicable to systems containing free electrons. The YFT model accurately fits NPA pair potentials or the nonparametric force-matched potentials from N-ion DFT-MD, showing excellent agreement for a wide range of conditions. The YFT form provides accurate extrapolations of the NPA or force-matched potentials for small and large particle separations within a physical model. Our method can be adopted to treat plasma mixtures, allowing for large-scale simulations of multispecies warm dense matter. read less NOT USED (high confidence) R. Wang et al., “Prediction of crystal structures and motifs in the Fe–Mg–O system at Earth’s core pressures,” New Journal of Physics. 2021. link Times cited: 2 Abstract: Fe, Mg, and O are among the most abundant elements in terres… read moreAbstract: Fe, Mg, and O are among the most abundant elements in terrestrial planets. While the behavior of the Fe–O, Mg–O, and Fe–Mg binary systems under pressure have been investigated, there are still very few studies of the Fe–Mg–O ternary system at relevant Earth’s core and super-Earth’s mantle pressures. Here, we use the adaptive genetic algorithm (AGA) to study ternary Fe x Mg y O z phases in a wide range of stoichiometries at 200 GPa and 350 GPa. We discovered three dynamically stable phases with stoichiometries FeMg2O4, Fe2MgO4, and FeMg3O4 with lower enthalpy than any known combination of Fe–Mg–O high-pressure compounds at 350 GPa. With the discovery of these phases, we construct the Fe–Mg–O ternary convex hull. We further clarify the composition- and pressure-dependence of structural motifs with the analysis of the AGA-found stable and metastable structures. Analysis of binary and ternary stable phases suggest that O, Mg, or both could stabilize a BCC iron alloy at inner core pressures. read less NOT USED (high confidence) B. Lin, J. Wang, J. Li, and Z. Wang, “A neural-network based framework of developing cross interaction in alloy embedded-atom method potentials: application to Zr–Nb alloy,” Journal of Physics: Condensed Matter. 2020. link Times cited: 2 Abstract: Interaction potentials are critical to molecular dynamics si… read moreAbstract: Interaction potentials are critical to molecular dynamics simulations on fundamental mechanisms at atomic scales. Combination of well-developed single-element empirical potentials via cross interaction (CI) is an important and effective way to develop alloy embedded-atom method (EAM) potentials. In this work, based on neural-network (NN) models, firstly we proposed a framework to construct CI potential functions via utilizing single-element potentials. The framework contained four steps: (1) extracting characteristic points from single-element potential functions, (2) constructing CI functions by cubic spline interpolation, (3) evaluating the accuracy of CI functions by referring to first-principle (FP) data, and (4) searching for reasonable CI functions via NN models. Then with this framework, we developed a Zr–Nb alloy CI potential utilizing the MA-III (pure Zr potential developed by Mendelev and Ackland in 2007) and the Fellinger, Park and Wilkins (FPW) (pure Nb potential developed by FPW in 2010) potentials as single-element parts. The calculated results with this Zr–Nb alloy potential showed that: (1) the newly developed CI potential functions could simultaneously present the potential-function features of Zr and Nb; (2) the normalized energy–volume curves of L12 Zr3Nb, B2 ZrNb and L12 ZrNb3 calculated by this CI potential reasonably agreed with FP results; (3) the referred MA-III Zr and FPW Nb potentials can satisfactorily reproduce the priority of prismatic slip in Zr and the tension–compression asymmetry of 〈111〉{112} slip in Nb, while other ab initio developed Zr–Nb alloy potentials cannot. Our study indicates that, this NN based framework can take full advantage of single-element potentials, and is very convenient to develop EAM potentials of alloys; moreover, the new-developed Zr–Nb alloy EAM potential can reasonably describe the complicated deformation behaviors in Zr–Nb systems. read less NOT USED (high confidence) B. Balasubramanian et al., “Synergistic computational and experimental discovery of novel magnetic materials,” Molecular Systems Design & Engineering. 2020. link Times cited: 9 Abstract: We review recent results on discoveries of new magnetic comp… read moreAbstract: We review recent results on discoveries of new magnetic compounds by combining experiments, adaptive genetic algorithm searches, and advanced electronic-structure computational methods. read less NOT USED (high confidence) D. S. Karls, M. Bierbaum, A. A. Alemi, R. Elliott, J. Sethna, and E. Tadmor, “The OpenKIM processing pipeline: A cloud-based automatic material property computation engine.,” The Journal of chemical physics. 2020. link Times cited: 7 Abstract: The Open Knowledgebase of Interatomic Models (OpenKIM) is a … read moreAbstract: The Open Knowledgebase of Interatomic Models (OpenKIM) is a framework intended to facilitate access to standardized implementations of interatomic models for molecular simulations along with computational protocols to evaluate them. These protocols include tests to compute material properties predicted by models and verification checks to assess their coding integrity. While housing this content in a unified, publicly available environment constitutes a major step forward for the molecular modeling community, it further presents the opportunity to understand the range of validity of interatomic models and their suitability for specific target applications. To this end, OpenKIM includes a computational pipeline that runs tests and verification checks using all available interatomic models contained within the OpenKIM Repository at https://openkim.org. The OpenKIM Processing Pipeline is built on a set of Docker images hosted on distributed, heterogeneous hardware and utilizes open-source software to automatically run test-model and verification check-model pairs and resolve dependencies between them. The design philosophy and implementation choices made in the development of the pipeline are discussed as well as an example of its application to interatomic model selection. read less NOT USED (high confidence) S. Longbottom and P. Brommer, “Uncertainty quantification for classical effective potentials: an extension to potfit,” Modelling and Simulation in Materials Science and Engineering. 2018. link Times cited: 12 Abstract: Effective potentials are an essential ingredient of classica… read moreAbstract: Effective potentials are an essential ingredient of classical molecular dynamics (MD) simulations. Little is understood of the consequences of representing the complex energy landscape of an atomic configuration by an effective potential or force field containing considerably fewer parameters. The probabilistic potential ensemble method has been implemented in the potfit force matching code. This introduces uncertainty quantification into the interatomic potential generation process. Uncertainties in the effective potential are propagated through MD to obtain uncertainties in quantities of interest (QoI), which are a measure of the confidence in the model predictions. We demonstrate the technique using three potentials for nickel: two simple pair potentials, Lennard-Jones and Morse, and a local density dependent embedded atom method potential. A potential ensemble fit to density functional theory (DFT) reference data is constructed for each potential to calculate the uncertainties in lattice constants, elastic constants and thermal expansion. We quantitatively illustrate the cases of poor model selection and fit, highlighted by the uncertainties in the quantities calculated. This shows that our method can capture the effects of the error incurred in QoI resulting from the potential generation process without resorting to comparison with experiment or DFT, which is an essential part to assess the predictive power of MD simulations. read less NOT USED (high confidence) Z. J. Yang et al., “Structures and magnetic properties of iron silicide from adaptive genetic algorithm and first-principles calculations,” Journal of Applied Physics. 2018. link Times cited: 9 Abstract: We performed a systematic search for low-energy structures o… read moreAbstract: We performed a systematic search for low-energy structures of binary iron silicide over a wide range of compositions using the crystal structure prediction method based on adaptive genetic algorithm. 36 structures with formation energies within 50 meV/atom (11 of them are within 20 meV) above the convex hull formed by experimentally known stable structures are predicted. Magnetic properties of these low-energy structures are investigated. Some of these structures can be promising candidates for rare-earth-free permanent magnet. read less NOT USED (high confidence) M. Wen, S. Shirodkar, P. Plecháč, E. Kaxiras, R. Elliott, and E. Tadmor, “A force-matching Stillinger-Weber potential for MoS2: Parameterization and Fisher information theory based sensitivity analysis,” Journal of Applied Physics. 2017. link Times cited: 25 Abstract: Two-dimensional molybdenum disulfide (MoS2) is a promising m… read moreAbstract: Two-dimensional molybdenum disulfide (MoS2) is a promising material for the next generation of switchable transistors and photodetectors. In order to perform large-scale molecular simulations of the mechanical and thermal behavior of MoS2-based devices, an accurate interatomic potential is required. To this end, we have developed a Stillinger-Weber potential for monolayer MoS2. The potential parameters are optimized to reproduce the geometry (bond lengths and bond angles) of MoS2 in its equilibrium state and to match as closely as possible the forces acting on the atoms along a dynamical trajectory obtained from ab initio molecular dynamics. Verification calculations indicate that the new potential accurately predicts important material properties including the strain dependence of the cohesive energy, the elastic constants, and the linear thermal expansion coefficient. The uncertainty in the potential parameters is determined using a Fisher information theory analysis. It is found that the parameters are... read less NOT USED (high confidence) K. Migdal, P. Pokatashkin, and A. Yanilkin, “Investigation of melting at the uranium γ phase by quantum and classical molecular dynamics methods,” High Temperature. 2017. link Times cited: 3 NOT USED (high confidence) A. Rohskopf, H. Seyf, K. Gordiz, T. Tadano, and A. Henry, “Empirical interatomic potentials optimized for phonon properties,” npj Computational Materials. 2017. link Times cited: 35 NOT USED (high confidence) A. Rokhmanenkov, A. Kuksin, and A. Yanilkin, “Simulation of hydrogen diffusion in TiHx structures,” Physics of Metals and Metallography. 2017. link Times cited: 9 NOT USED (high confidence) M. Wen, J. Li, P. Brommer, R. Elliott, J. Sethna, and E. Tadmor, “A KIM-compliant potfit for fitting sloppy interatomic potentials: application to the EDIP model for silicon,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 16 Abstract: Fitted interatomic potentials are widely used in atomistic s… read moreAbstract: Fitted interatomic potentials are widely used in atomistic simulations thanks to their ability to compute the energy and forces on atoms quickly. However, the simulation results crucially depend on the quality of the potential being used. Force matching is a method aimed at constructing reliable and transferable interatomic potentials by matching the forces computed by the potential as closely as possible, with those obtained from first principles calculations. The potfit program is an implementation of the force-matching method that optimizes the potential parameters using a global minimization algorithm followed by a local minimization polish. We extended potfit in two ways. First, we adapted the code to be compliant with the KIM Application Programming Interface (API) standard (part of the Knowledgebase of Interatomic Models project). This makes it possible to use potfit to fit many KIM potential models, not just those prebuilt into the potfit code. Second, we incorporated the geodesic Levenberg–Marquardt (LM) minimization algorithm into potfit as a new local minimization algorithm. The extended potfit was tested by generating a training set using the KIM environment-dependent interatomic potential (EDIP) model for silicon and using potfit to recover the potential parameters from different initial guesses. The results show that EDIP is a ‘sloppy model’ in the sense that its predictions are insensitive to some of its parameters, which makes fitting more difficult. We find that the geodesic LM algorithm is particularly efficient for this case. The extended potfit code is the first step in developing a KIM-based fitting framework for interatomic potentials for bulk and two-dimensional materials. The code is available for download via https://www.potfit.net. read less NOT USED (high confidence) P. Wu et al., “Fe-Si networks in Na2FeSiO4 cathode materials.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 24 Abstract: Using a combination of adaptive genetic algorithm search, mo… read moreAbstract: Using a combination of adaptive genetic algorithm search, motif-network search scheme and first-principles calculations, we have systematically studied the low-energy crystal structures of Na2FeSiO4. We show that the low-energy crystal structures with different space group symmetries can be classified into several families based on the topologies of their Fe-Si networks. In addition to the diamond-like network which is shared by most of the low-energy structures, another three robust Fe-Si networks are also found to be stable during the charge/discharge process. The electrochemical properties of representative structures from these four different Fe-Si networks in Na2FeSiO4 and Li2FeSiO4 are investigated and found to be strongly correlated with the Fe-Si network topologies. Our studies provide a new route to characterize the crystal structures of Na2FeSiO4 and Li2FeSiO4 and offer useful guidance for the design of promising cathodes for Na/Li ion batteries. read less NOT USED (high confidence) J. Roth, A. Kiselev, and H. Trebin, “Studying laser ablation with molecular dynamics simulations: From metals to covalent materials,” 2016 Progress in Electromagnetic Research Symposium (PIERS). 2016. link Times cited: 2 Abstract: Simulating laser ablation on an atomistic scale is a demandi… read moreAbstract: Simulating laser ablation on an atomistic scale is a demanding task which requires millions to billions of atoms for sample sizes in the nano- to micrometer range. read less NOT USED (high confidence) P. Pokatashkin, A. Kuksin, and A. Yanilkin, “Angular dependent potential for α-boron and large-scale molecular dynamics simulations,” Modelling and Simulation in Materials Science and Engineering. 2015. link Times cited: 9 Abstract: Both quantum mechanical and molecular-dynamics (MD) simulati… read moreAbstract: Both quantum mechanical and molecular-dynamics (MD) simulations of α-boron are done at this work. Angular dependent interatomic potential (ADP) for boron is obtained using force-matching technique. Fitting data are based on ab initio results within −20..100 GPa pressure range and temperatures up to 2000 K. Characteristics of α-boron, obtained using ADP potential such as bond lengths at equilibrium condition, bulk modulus, pressure-volume relations, Gruneisen coefficient, thermal expansion coefficient are in good agreement with both ab initio data, obtained in this work and known experimental data. As an example of application, the propagation of shock waves through a single crystal of α-boron is also explored by large-scale MD simulations. read less NOT USED (high confidence) H. Geng, “Accelerating ab initio path integral molecular dynamics with multilevel sampling of potential surface,” J. Comput. Phys. 2014. link Times cited: 13 NOT USED (high confidence) X. Zhao et al., “Interface Structure Prediction from First-Principles,” Journal of Physical Chemistry C. 2014. link Times cited: 32 Abstract: Information about the atomic structures at solid–solid inter… read moreAbstract: Information about the atomic structures at solid–solid interfaces is crucial for understanding and predicting the performance of materials. Due to the complexity of the interfaces, it is very challenging to resolve their atomic structures using either experimental techniques or computer simulations. In this paper, we present an efficient first-principles computational method for interface structure prediction based on an adaptive genetic algorithm. This approach significantly reduces the computational cost, while retaining the accuracy of first-principles prediction. The method is applied to the investigation of both stoichiometric and nonstoichiometric SrTiO3 Σ3(112)[110] grain boundaries with unit cell containing up to 200 atoms. Several novel low-energy structures are discovered, which provide fresh insights into the structure and stability of the grain boundaries. read less NOT USED (high confidence) S. Wu et al., “An adaptive genetic algorithm for crystal structure prediction,” Journal of Physics: Condensed Matter. 2013. link Times cited: 140 Abstract: We present a genetic algorithm (GA) for structural search th… read moreAbstract: We present a genetic algorithm (GA) for structural search that combines the speed of structure exploration by classical potentials with the accuracy of density functional theory (DFT) calculations in an adaptive and iterative way. This strategy increases the efficiency of the DFT-based GA by several orders of magnitude. This gain allows a considerable increase in the size and complexity of systems that can be studied by first principles. The performance of the method is illustrated by successful structure identifications of complex binary and ternary intermetallic compounds with 36 and 54 atoms per cell, respectively. The discovery of a multi-TPa Mg-silicate phase with unit cell containing up to 56 atoms is also reported. Such a phase is likely to be an essential component of terrestrial exoplanetary mantles. read less NOT USED (high confidence) D. Smirnova et al., “A ternary EAM interatomic potential for U–Mo alloys with xenon,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 71 Abstract: A new interatomic potential for a uranium–molybdenum system … read moreAbstract: A new interatomic potential for a uranium–molybdenum system with xenon is developed in the framework of an embedded atom model using a force-matching technique and a dataset of ab initio atomic forces. The verification of the potential proves that it is suitable for the investigation of various compounds existing in the system as well as for simulation of pure elements: U, Mo and Xe. Computed lattice constants, thermal expansion coefficients, elastic properties and melting temperatures of U, Mo and Xe are consistent with the experimentally measured values. The energies of the point defect formation in pure U and Mo are proved to be comparable to the density-functional theory calculations. We compare this new U–Mo–Xe potential with the previously developed U and Mo–Xe potentials. A comparative study between the different potential functions is provided. The key purpose of the new model is to study the atomistic processes of defect evolution taking place in the U–Mo nuclear fuel. Here we use the potential to simulate bcc alloys containing 10 wt% of intermetallic Mo and U2Mo. read less NOT USED (high confidence) P. Beck, P. Brommer, J. Roth, and H. Trebin, “Influence of polarizability on metal oxide properties studied by molecular dynamics simulations,” Journal of Physics: Condensed Matter. 2012. link Times cited: 10 Abstract: We have studied the dependence of metal oxide properties in … read moreAbstract: We have studied the dependence of metal oxide properties in molecular dynamics (MD) simulations on the polarizability of oxygen ions. We present studies of both liquid and crystalline structures of silica (SiO2), magnesia (MgO) and alumina (Al2O3). For each of the three oxides, two separately optimized sets of force fields were used: (i) long-range Coulomb interactions between oxide and metal ions combined with a short-range pair potential; (ii) extension of force field (i) by adding polarizability to the oxygen ions. We show that while an effective potential of type (i) without polarizable oxygen ions can describe radial distributions and lattice constants reasonably well, potentials of type (ii) are required to obtain correct values for bond angles and the equation of state. The importance of polarizability for metal oxide properties decreases with increasing temperature. read less NOT USED (high confidence) D. Smirnova, S. Starikov, and V. Stegailov, “New interatomic potential for computation of mechanical and thermodynamic properties of uranium in a wide range of pressures and temperatures,” The Physics of Metals and Metallography. 2012. link Times cited: 12 NOT USED (high confidence) D. Schopf, P. Brommer, B. Frigan, and H. Trebin, “Embedded atom method potentials for Al-Pd-Mn phases,” Physical Review B. 2011. link Times cited: 23 Abstract: A novel embedded atom method (EAM) potential for the Ξ phase… read moreAbstract: A novel embedded atom method (EAM) potential for the Ξ phases of Al-Pd-Mn has been determined with the force-matching method. Different combinations of analytic functions were tested for the pair and transfer part. The best results are obtained if one allows for oscillations on two different length scales. These potentials stabilize structure models of the Ξ phases and describe their energy with high accuracy. Simulations at temperatures up to 1200 K show very good agreement with ab initio results with respect to stability and dynamics of the system. read less NOT USED (high confidence) P. Beck, P. Brommer, J. Roth, and H. Trebin, “Ab initio based polarizable force field generation and application to liquid silica and magnesia.,” The Journal of chemical physics. 2011. link Times cited: 20 Abstract: We extend the program potfit, which generates effective atom… read moreAbstract: We extend the program potfit, which generates effective atomic interaction potentials from ab initio data, to electrostatic interactions and induced dipoles. The potential parametrization algorithm uses the Wolf direct, pairwise summation method with spherical truncation. The polarizability of oxygen atoms is modeled with the Tangney-Scandolo interatomic force field approach. Due to the Wolf summation, the computational effort in simulation scales linearly in the number of particles, despite the presence of electrostatic interactions. Thus, this model allows to perform large-scale molecular dynamics simulations of metal oxides with realistic potentials. Details of the implementation are given, and the generation of potentials for SiO(2) and MgO is demonstrated. The approach is validated by simulations of microstructural, thermodynamic, and vibrational properties of liquid silica and magnesia. read less NOT USED (high confidence) M. Ji, K. Umemoto, C. Wang, K. Ho, and R. Wentzcovitch, “Ultrahigh-pressure phases of H2O ice predicted using an adaptive genetic algorithm,” Physical Review B. 2011. link Times cited: 55 Abstract: We propose three new phases of H2O under ultrahigh pressure.… read moreAbstract: We propose three new phases of H2O under ultrahigh pressure. Our structural search was performed using an adaptive genetic algorithm which allows an extensive exploration of crystal structure. The new sequence of pressure-induced transitions beyond ice X at 0 K should be ice X! Pbcm ! Pbca ! Pmc21 ! P21 ! P21=c phases. Across the Pmc21-P21 transition, the coordination number of oxygen increases from 4 to 5 with a significant increase of density. All stable crystalline phases have nonmetallic band structures up to 7 TPa. read less NOT USED (high confidence) M. Mihalkovič and C. Henley, “Energetics and dynamics of caged Zn4 in i-ScZn,” Philosophical Magazine. 2011. link Times cited: 9 Abstract: In i-ScZn, like other quasicrystals of the i-CaCd class, the… read moreAbstract: In i-ScZn, like other quasicrystals of the i-CaCd class, the innermost shell of the icosahedral cluster is a Zn4 tetrahedron, which thus breaks the symmetry of the outer cluster shells. We investigate theoretically the dynamics of individual tetrahedra, using interatomic pair potentials, fitted from an ab initio database, and molecular dynamics (MD). This includes the formulation of an effective Hamiltonian written in terms of a rigid-body rotation representing the state of each tetrahedron. We characterize the minimum-energy orientations of a tetrahedron, as well as the paths of the transitions between minima (reorientations). The velocity autocorrelations were evaluated for the tetrahedral atoms in the MD dynamics; the corresponding spectral density S(ω) is fairly well fitted by a simplified model in which each atom hops in a double well. read less NOT USED (high confidence) L. P. Wang and T. V. Voorhis, “Communication: Hybrid ensembles for improved force matching.,” The Journal of chemical physics. 2010. link Times cited: 21 Abstract: Force matching is a method for parameterizing empirical pote… read moreAbstract: Force matching is a method for parameterizing empirical potentials in which the empirical parameters are fitted to a reference potential energy surface (PES). Typically, training data are sampled from a canonical ensemble generated with either the empirical potential or the reference PES. In this Communication, we show that sampling from either ensemble risks excluding critical regions of configuration space, leading to fitted potentials that deviate significantly from the reference PES. We present a hybrid ensemble which combines the Boltzmann probabilities of both potential surfaces into the fitting procedure, and we demonstrate that this technique improves the quality and stability of empirical potentials. read less NOT USED (high confidence) S. Sonntag, J. Roth, and H. Trebin, “Molecular dynamics simulations of laser induced surface melting in orthorhombic Al13Co4,” Applied Physics A. 2010. link Times cited: 6 NOT USED (high confidence) M. Fellinger, H. Park, and J. Wilkins, “Force-matched embedded-atom method potential for niobium,” Physical Review B. 2010. link Times cited: 115 Abstract: Large-scale simulations of plastic deformation and phase tra… read moreAbstract: Large-scale simulations of plastic deformation and phase transformations in alloys require reliable classical interatomic potentials. We construct an embedded-atom method potential for niobium as the first step in alloy potential development. Optimization of the potential parameters to a well-converged set of density-functional theory (DFT) forces, energies, and stresses produces a reliable and transferable potential for molecular dynamics simulations. The potential accurately describes properties related to the fitting data, and also produces excellent results for quantities outside the fitting range. Structural and elastic properties, defect energetics, and thermal behavior compare well with DFT results and experimental data, e.g., DFT surface energies are reproduced with less than 4% error, generalized stacking-fault energies differ from DFT values by less than 15%, and the melting temperature is within 2% of the experimental value. read less NOT USED (high confidence) P. Brommer and F. Gähler, “Potfit: effective potentials from ab initio data,” Modelling and Simulation in Materials Science and Engineering. 2007. link Times cited: 180 Abstract: We present a program called potfit which generates an effect… read moreAbstract: We present a program called potfit which generates an effective atomic interaction potential by matching it to a set of reference data computed in first-principles calculations. It thus allows one to perform large-scale atomistic simulations of materials with physically justified potentials. We describe the fundamental principles behind the program, emphasizing its flexibility in adapting to different systems and potential models, while also discussing its limitations. The program has been used successfully in creating effective potentials for a number of complex intermetallic alloys, notably quasicrystals. read less NOT USED (high confidence) E. Bitzek, P. Koskinen, F. Gähler, M. Moseler, and P. Gumbsch, “Structural relaxation made simple.,” Physical review letters. 2006. link Times cited: 1093 Abstract: We introduce a simple local atomic structure optimization al… read moreAbstract: We introduce a simple local atomic structure optimization algorithm which is significantly faster than standard implementations of the conjugate gradient method and often competitive with more sophisticated quasi-Newton schemes typically used in ab initio calculations. It is based on conventional molecular dynamics with additional velocity modifications and adaptive time steps. The surprising efficiency and especially the robustness and versatility of the method is illustrated using a variety of test cases from nanoscience, solid state physics, materials research, and biochemistry. read less NOT USED (high confidence) F. Rösch, H. Trebin, and P. Gumbsch, “Fracture of complex metallic alloys: an atomistic study of model systems,” Philosophical Magazine. 2006. link Times cited: 10 Abstract: Molecular dynamics simulations of crack propagation are perf… read moreAbstract: Molecular dynamics simulations of crack propagation are performed for two extreme cases of complex metallic alloys. In a model quasicrystal the structure is determined by clusters of atoms, whereas the model C15 Laves phase is a simple periodic stacking of a unit cell. The simulations reveal that the basic building units of the structures also govern their fracture behaviour. Atoms in the Laves phase play a comparable role with the clusters in the quasicrystal. Although the latter are not rigid units, they have to be regarded as significant physical entities. read less NOT USED (high confidence) M. Mihalkovič and M. Widom, “Canonical cell model of cadmium-based icosahedral alloys,” Philosophical Magazine. 2006. link Times cited: 26 Abstract: Cadmium-based quasicrystals (Cd--Ca and Cd--Yb) were the fir… read moreAbstract: Cadmium-based quasicrystals (Cd--Ca and Cd--Yb) were the first binary alloys discovered to form thermodynamically stable quasicrystals. As binary alloys, and with a strong size difference between atomic species, they are ideal systems for structural and thermodynamic analysis. Observed quasicrystal approximants with crystallographically determined structures can be interpreted as decorations of Henley's canonical cells. We use first-principles total energy calculations to resolve details of the most favourable decorations. read less NOT USED (high confidence) A. Kiselev, “Molecular dynamics simulations of laser ablation in covalent materials.” 2017. link Times cited: 1 Abstract: 15 Deutsche Zusammenfassung 18 I. Theoretical background 27… read moreAbstract: 15 Deutsche Zusammenfassung 18 I. Theoretical background 27 read less NOT USED (high confidence) P. Beck, “Molecular dynamics of metal oxides with induced electrostatic dipole moments.” 2012. link Times cited: 1 Abstract: Metal oxides belong to the most important material classes i… read moreAbstract: Metal oxides belong to the most important material classes in industrial technology. These high-tech ceramics are irreplacable in lots of modern microelectronic devices due to their excellent insulating properties, high melting points and high degrees of hardness. In the theoretical study of these systems, the atomistic modelling with molecular dynamics simulations and classical effective interaction force fields is a very powerful tool. There, fundamental properties can be uncovered and understood due to the atomic resolution. Both force field generation and simulation of oxide systems are computationally much more demanding than those of metals or covalent materials due to long-range electrostatic interactions. Furthermore, it is often not sufficient to only take Coulomb interactions into account, but to include electrostatic dipole moments. The latter can be integrated with the Tangney Scandolo polarizable force field model, where dipole moments are determined by a self-consistent iterative solution during each simulation time step. Applying the direct, pairwise Wolf summation to interactions between charges and its extension to dipole moments avoids high computational effort due to its linear scaling properties in the number of particles. Three relevant metal oxides have been selected to apply the new high-performance force field generation model. Therewith, a detailed investigation of crack propagation was possible. Both crack propagation insights and the influence of cracks on the dipole field are shown. Finally, the coupling of strain and – even more meaningful – strain gradient with the dipole moments is presented, which gives rise to flexoelectric effects in non-piezoeletric materials.
Die vorliegende Arbeit vertieft die breit angelegte Modellierungs-Studie dreier wichtiger Metalloxide. Detaillierte Untersuchungen durch Molekulardynamik- Simulationen mit Kraftfeldern fur Siliziumdioxid, Magnesiumoxid und Alpha-Aluminiumoxid werden dargestellt. Speziell angepasste Visualisierungs-Techniken erweitern die numerischen Einblicke und verhelfen zu neuen Erkenntnissen in den untersuchten ionischen Systemen. Im Wesentlichen leistet die Arbeit einen dreistufigen Beitrag zur numerischen Erforschung ionischer kondensierter Materie:
1. Zunachst wird die neue Art der Kraftfelderstellung dargestellt, welche
das Potenzial-Modell von Tangney und Scandolo mit der direkten Wolfsummation verknupft. Gezeigt wird die gewissenhafte Prufung, dass die Vereinigung der Vorteile des TS Modells, welches elektrostatische Dipolmomente mit einbezieht, mit den linearen Skalierungs-Eigenschaften der Wolfsumme gelungen ist. Die Implementierung der Kraftfelderstellung in potfit durch den Autor gibt anderen Simulations-Gruppen die Moglichkeit, diese prazise, effiziente und flexible
Methode fur ihre individuellen Studien ionischer Materie einzusetzen.
2. Weiterhin werden die mithilfe der neuen Methodik generierten Kraftfelder fur Siliziumdioxid, Magnesiumoxid und Alpha-Aluminiumoxid vorgestellt. Neben ihrer Erstellung wird in jedem Einzelfall die sorgfaltige Validierung gezeigt. Die neuen Kraftfelder werden nicht nur fur eigene Simulationen eingesetzt, sondern anderen Gruppen offentlich zuganglich gemacht. Dadurch konnen nun vielerorts Simulationen durchgefuhrt werden, die bisher aufgrund von Beschrankungen bezuglich Zeit-, Langenskalen oder Randbedingungen nicht annehmbar realisierbar waren.
3. Unter Einsatz der neuen Kraftfelder wurden verschiedene Simulationen durchgefuhrt, die – auch mithilfe der speziell angepassten Visualisierung der fraktionalen Anisotropie – zu neuen Entdeckungen und grundlegenden Erkenntnissen gefuhrt haben:
• Das fur Alpha-Aluminiumoxid erstellte Kraftfeld wurde zur MD Simulation von sich ausbreitenden Rissen verwendet, welche die vor zwei Jahren elektronenmikroskopisch untersuchten Verlaufs-Richtungen von in verschiedenen kristallinen Ebenen existierenden Rissen vollstandig reproduzieren konnte. Daruberhinaus konnte – erstmalig in atomistischen Simulationen von Metalloxiden – der Einfluss von sich ausbreitenden Rissen auf die Orientierung der elektrostatischen Dipolmomente beobachtet und analysiert werden.
• Derartige flexoelektrische Phanomene wurden schlieslich sowohl in MD Simulationen als auch mithilfe der Darstellungstheorie untersucht. Die Kombination liefert eine konsistente Vorhersage flexoelektrischen Verhaltens in Alpha-Aluminiumoxid und auch in Periklas, obwohl beide Materialien aufgrund ihrer inversionssymmetrischen Kristallstruktur keine Piezoelektrizitat aufweisen. Die analytisch angenommene und experimentell vorhergesagte lineare Kopplung zwischen Spannungsgradient und flexoelektrischer Polarisation wurde von MD Simulationen bestatigt. Erstmalig wurde flexoelektrische Domanenausbildung modelliert. In Periklas zeigt sich, dass flexoelektrische Domanen exakt von Neel-Wanden abgetrennt werden. read less NOT USED (high confidence) S. Sonntag, “Computer simulations of laser ablation from simple metals to complex metallic alloys.” 2011. link Times cited: 10 Abstract: In this work, a method for computer simulations of laser abl… read moreAbstract: In this work, a method for computer simulations of laser ablation in metals is presented. The ambitious task to model the physical processes, that occur on different time and length scales, is overcome to some extent by the combination of two techniques: Molecular dynamics and finite differences. The former is needed to achieve atomistic resolution of the processes involved. Material failure like melting, vaporization or spallation occur on the atomic scale. Light absorption and electronic heat conduction, which plays the major role in metals, is described by a generalized heat conduction equation solved by the finite differences method. From the so-called Two-Temperature Model temperature, density and pressure evolution - both in time and space - can be derived. With this, various studies on laser heated metals were done. For reasons discussed in more detail later, aluminum was chosen as a model system for most simulations on isotropic materials. As a more complex structure, the metallic alloy Al13Co4 was used because of its special material properties. As an approximant to the decagonal phase of Al-Ni-Co, the alloy shows an anisotropy in its transport properties, e.g. an anisotropic heat conduction.
It will be shown, that the model is able to describe the physics in laser heated solids on time scales from 100 fs up to the ns-scale properly. Great insight was gained about the processes occuring during and shortly after the laser pulse. Many of the quantities interesting for experimentalists can be predicted by the theory. From the simulations relevant parameters like the electron-cooling time or the important ablation threshold were calculated. All values match their experimental counterpart very well.
Die vorliegende Arbeit beschaftigt sich mit der Laserablation in Metallen. Ziel ist es, mit Hilfe von numerischen Simulationen das Verhalten von Metallen nach der Bestrahlung mit intensiven Laserpulsen vorherzusagen. Die Arbeit ist inhaltlich in zwei Teile gegliedert. In der ersten Halfte werden theoretische Grundlagen, eine qualitative Beschreibung der Ablation und die Implementierung des Modells gegeben. Im zweiten Teil folgen Ergebnisse sowie, falls vorhanden, Vergleiche mit Experimenten. Die Arbeit schliest mit einer Zusammenfassung und einem Ausblick. read less NOT USED (high confidence) H. Trebin et al., “Simulating structure and physical properties of complex metallic alloys.” 2008. link Times cited: 3 Abstract: An introduction is presented to numerical methods, by which … read moreAbstract: An introduction is presented to numerical methods, by which the behavior of complex metallic alloys can be simulated. We primarily consider the molecular dynamics (MD) technique as implemented in our software package IMD, where Newton’s equations of motion are solved for all atoms in a solid. After a short discourse on integration algorithms, some possible types of interactions are addressed. Already simple model potentials, as for example the Lennard-Jones-Gauss potential, can give rise to complex structures, where the characteristic length scales of the order by far exceed the range of the pair interaction. Realistic interactions are modelled by highly parametrized effective potentials, like the EAM (Embedded Atom Method) potential. Our program potfit allows to fit the parameters such that data from experiment or from ab-initio calculations are well reproduced. Several applications of the methods are outlined, notably the simulation of aluminium diffusion in quasicrystalline d-Al-Ni-Co, the computation of the phonon dispersion via the dynamical structure factor of MgZn2, the propagation of cracks in NbCr2, and an order-disorder phase transition in CaCd6. read less NOT USED (high confidence) S. Hocker, “Molekulardynamiksimulation der Diffusion in dekagonalen Quasikristallen mit optimierten Wechselwirkungspotentialen.” 2007. link Times cited: 1 Abstract: Die etwa 25 Jahre zuruckliegende Entdeckung der Quasikristal… read moreAbstract: Die etwa 25 Jahre zuruckliegende Entdeckung der Quasikristalle erweiterte das Gebiet der Festkorperphysik um einen neuen Strukturtyp. In der Erforschung dieser geordneten, jedoch nicht periodischen Strukturen wurden seither bedeutende Erfolge erzielt. Von verschiedenen Quasikristalltypen konnten zunehmend detaillierte Strukturmodelle erstellt werden. Zunachst wurden in der numerischen Simulation einfache binare Modellsysteme untersucht, inzwischen sind realistische ternare Strukturen Gegenstand der numerischen Forschung.
Fur die Molekulardynamiksimulation solcher Strukturen ist, neben einem geeigneten Strukturmodell, eine gute Modellierung der atomaren Wechselwirkungen erforderlich. Besonders fur Simulationen nahe der Schmelztemperatur besteht Bedarf nach einem realistischen Wechselwirkungspotential. Da herkommliche Potentiale ublicherweise an Parameter der Grundzustandsstruktur angepasst wurden, tendieren diese dazu, bei hohen Temperaturen zu versagen.
Die numerische Exploration in diesem Temperaturbereich kann einen wichtigen Beitrag zum Verstandnis der Quasikristalle liefern. Diffusionsprozesse, die bedeutend fur die Entstehung von quasikristallinen Hochtemperaturphasen sind, konnen im Fall von Aluminium nur in der Simulation erforscht werden, weil fur das ubliche Messverfahren kein geeignetes Radioisotop existiert. Da Aluminium ein wesentlicher Bestandteil vieler Quasikristalle ist, ist das Verstandnis der Aluminiumdiffusion von groser Bedeutung.
Ziel dieser Arbeit ist die Untersuchung der Aluminiumdiffusion in den dekagonalen Quasikristallen AlNiCo und AlCoCu mittels Molekulardynamiksimulation. Dazu werden zunachst, durch Anpassung an Ab-initio-Daten, Wechselwirkungspotentiale fur die Strukturen entwickelt. Mit diesen Potentialen werden Simulationen bei hohen Temperaturen durchgefuhrt. Die dabei auftretende Diffusion wird ausgewertet und mit Daten aus Ab-initio-Rechnungen verglichen.
Die in der vorliegenden Arbeit konstruierten EAM-Potentiale fur Hochtemperatursimulationen an dekagonalen Quasikristallen erwiesen sich als geeignet, um grundlegende Eigenschaften der Systeme richtig zu modellieren. Der Vergleich mit der Ab-initio-Rechnung lieferte fur alle Atomsorten eine sehr gute Ubereinstimmung in den aus der thermischen Bewegung resultierenden Atomaufenthaltswahrscheinlichkeiten der Strukturen. Auch einzelne mit den EAM-Potentialen untersuchten Al-Diffusionsprozesse konnten in der Ab-initio-MD-Simulation bestatigt werden.
In den quasikristallinen Strukturen wurde in der klassischen Molekulardynamiksimulation weit reichende Aluminiumdiffusion beobachtet. Die durch Ab-initio-Rechnungen bestatigten Al-Diffusionsprozesse wurden detailliert untersucht. Dabei wurde deutlich, dass die Diffusion in der dekagonalen Ebene uber quasikristallspezifische Mechanismen verlauft. Bedeutend ist hierbei die Besonderheit, dass Gebiete existieren, die zur Atomabgabe neigen, wahrend andere ein zusatzliches Atom aufnehmen konnen. Es finden Kettenprozesse statt, an deren Start- und Endpositionen sich diese Regionen befinden. Charakterisch fur diese Gebiete ist, dass die Al-Positionen nicht scharf lokalisiert sind, sondern innerhalb eines Bereichs eine kontinuierliche Al-Aufenthaltswahrscheinlichkeit besteht. In der periodischen Richtung existieren durchgangige Kanale kontinuierlicher Al-Aufenthaltswahrscheinlichkeit. Durch diese Kanale verlauft die Al-Diffusion in periodischer Richtung. Der Diffusionsmechanismus ist hierbei die direkte, synchron verlaufende, Bewegung innerhalb einer Reihe von Atomen. Dabei befinden sich immer drei Atome pro Schicht im Kanal. Bei mehr als drei Schichten findet meist eine Kopplung mit einem Prozess der dekagonalen Ebene statt, wobei ein Atom aus der dekagonalen Ebene in den Diffusionskanal springt, wahrend ein anderes diesen verlasst. In diesem Fall diffundieren nur die sich dazwischen befindenden Atome entlang des Kanals.
Zur Diffusion in der dekagonalen Ebene konnen deutlich mehr Al-Atome beitragen als zu jener in der periodischen Richtung, welche auf die Diffusionskanale beschrankt ist. Da jedoch die Mobilitat der Al-Atome in den Diffusionskanalen deutlich hoher ist, liegen die Diffusionskoeffizienten fur die periodische Richtung uber jenen der dekagonalen Ebene. Die geringe Energiebarriere innerhalb der Diffusionskanale wurde mit Ab-initio-Rechnungen bestatigt.
With the discovery of quasicrystals in 1982, condensed matter physics was extended by a new kind of structure. Since then, the exploration of these ordered, but nonperiodic structures has lead to significant new results. There was an increase of detailed structure models of several quasicrystals. For a long time, in numerical simulations only binary model systems were studied, but real quasicrystals are mostly ternary or even quaternary. Nowadays these structures can be explored in numerical simulations.
Molecular dynamics simulations of such realistic structures require, apart from an appropriate model of the structure, a well adjusted model of the atomic interactions. Especially for simulations at temperatures near the melting point, there is demand for realistic potentials. Since conventional potentials were usually constructed by adjustment to the ground state structure, these potentials tend to fail at high temperatures.
Simulations at high temperatures provide an important contribution to the understandig of quasicrystals. Diffusion processes are fundamental in the formation of high temperature phases and the motion of defects. In the case of aluminium the diffusion cannot be measured experimentally, due to the lack of suitable radiotracers. Therefore numerical simulation is the sole possibilty to study the Al diffusion processes. Since aluminium is a basic component of many quasicrystals, the understanding of aluminum diffusion is of great importance.
The goal of this thesis is the exploration of diffusion in the decagonal AlNiCo and AlCoCu quasicrystals via molecular dynamics simulation. For this purpose, atomic interaction potentials for these structures are developed by adjusting them to ab initio data.
The newly developed EAM potentials for molecular dynamics simulations at high temperatures in AlNiCo and AlCoCu quasicrystals turned out to be well adapted for modelling the basic properties of these decagonal quasicrystals. The comparison with ab initio calculations shows very good agreement in the atom density maps of both structures. Similarly some specific diffusion processes, which were found in the simulations with EAM potentials, were validated in ab initio calculations.
In the molecular dynamics simulation, long range Al diffusion was observed in both decagonal quasicrystal structures. Al diffusion processes, which were validated by ab initio calculations, were studied in detail. It was found that the diffusion in the decagonal plane proceeds via mechanisms which are specific to quasicrystals. Of great importance are sites which tend to emit atoms, whereas other sites can absorb atoms. Chain processes occur, where the initial and the final positions are at these sites. The important characteristic of these sites is that the Al positions are not localized, but there is a continuous Al density in these regions. In the periodic direction, channels of continuous Al density extend through the structure. The diffusion in this direction runs via such channels. The diffusion mechanism is a synchronous motion of atoms within a column. In each layer there are three atoms which are part of this column. With more than three layers there is usually a coupling with a jump process in the decagonal plane. An atom of the decagonal plane jumps into the diffusion channel, whereas another atom leaves the channel. In this case, only the atoms in-between diffuse along the channel.
There are clearly more Al atoms which contribute to the diffusion in the decagonal plane than in the periodic direction, in which the diffusion is limited to the channels. However, since the mobility of Al atoms in the diffusion channels is significantly higher, the diffusion coefficients in the periodic direction are larger than those in the decagonal plane. The small energy barriers in the diffusion channels were validated in ab inito calculations. read less NOT USED (high confidence) F. Gähler and K. Benkert, “Atomistic Simulations on Scalar and Vector Computers.” 2006. link Times cited: 6 |
 EAM_IMD_BrommerGaehler_2006A_AlNiCo__MO_122703700223_003
EAM_IMD_BrommerGaehler_2006A_AlNiCo__MO_122703700223_003