Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
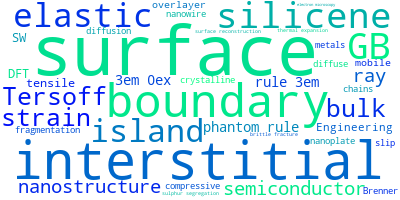
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
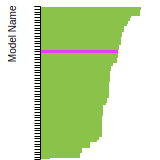
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
354 Citations (41 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) T. Zhu, J. Li, and S. Yip, “Atomistic characterization of three-dimensional lattice trapping barriers to brittle fracture,” Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2006. link Times cited: 39 Abstract: We present a detailed account of an atomistic study of three… read moreAbstract: We present a detailed account of an atomistic study of three-dimensional lattice trapping barriers to brittle fracture in Si. By means of a prototypical interatomic potential model, we map out the molecular details of the evolution of atomically sharp cracks in the (111) cleavage plane with straight crack fronts along the and directions, respectively. The thermally activated processes of bond rupturing along the crack front are quantitatively characterized using a reaction pathway sampling scheme. The calculated minimum energy paths reveal a mechanism of kink-pair formation and migration in facilitating the crack front advancement. We show that the physical origin of directional anisotropy in cleavage crack propagation can be attributed to a difference in the kink-pair formation energy for different crack orientations. The effects of interatomic potentials are delineated by comparing the Stillinger–Weber model with an environment-dependent model. read less USED (high confidence) H. Chen, V. Levitas, D. Popov, and N. Velisavljevic, “Nontrivial nanostructure, stress relaxation mechanisms, and crystallography for pressure-induced Si-I → Si-II phase transformation,” Nature Communications. 2022. link Times cited: 0 USED (high confidence) S. Yoo, B. Lee, and K. Kang, “Density functional theory study of the mechanical behavior of silicene and development of a Tersoff interatomic potential model tailored for elastic behavior,” Nanotechnology. 2021. link Times cited: 8 Abstract: Silicene, a graphene-like 2D material made from Si atoms, ha… read moreAbstract: Silicene, a graphene-like 2D material made from Si atoms, has been fabricated and studied for its promising applications in micro/nanoelectronics. For the reliable function of silicene devices, it is important to investigate silicene’s mechanical properties. In this study, the authors conducted density functional theory (DFT) simulations of mechanical tests of silicene and investigated the elastic modulus and mechanical response such as structural transformation. In addition, the authors optimized the Tersoff potential parameters using a gradient-based minimization with a grid search method in hyperdimensional parameter space, to match the DFT calculation results in the elastic regime. With the new parameter set, the elastic moduli of silicene in the zigzag (ZZ) and armchair (AC) directions were computed with molecular statics (MS) simulations and compared with those of other Si interatomic potential models and DFT results. In addition, uniaxial tensile tests along the ZZ and AC directions were performed to examine how far the Tersoff model is transferable with our new parameter set to describe the nonlinear mechanical behavior of silicene. The results of uniaxial tensile tests suggest that the angle penalty function in the Tersoff model needs to be modified and that the stress–strain curve predicted with this modification shows improvement compared to the original function. read less USED (high confidence) V. Levitas, H. Chen, and L. Xiong, “Lattice instability during phase transformations under multiaxial stress: Modified transformation work criterion,” Physical Review B. 2017. link Times cited: 41 Abstract: Valery I. Levitas,1,2 Hao Chen,3 and Liming Xiong3 1Departme… read moreAbstract: Valery I. Levitas,1,2 Hao Chen,3 and Liming Xiong3 1Departments of Aerospace Engineering, Mechanical Engineering, and Material Science and Engineering, Iowa State University, Ames, Iowa 50011, USA 2Ames Laboratory, Division of Materials Science and Engineering, Ames, Iowa 50011, USA 3Department of Aerospace Engineering, Iowa State University, Ames, Iowa 50011, USA (Received 15 December 2016; revised manuscript received 22 June 2017; published 29 August 2017) read less USED (high confidence) V. I. Tokar and H. Dreyss’e, “Size calibration of strained epitaxial islands due to dipole–monopole interaction,” Journal of Statistical Mechanics: Theory and Experiment. 2014. link Times cited: 2 Abstract: Irreversible growth of strained epitaxial nanoislands has be… read moreAbstract: Irreversible growth of strained epitaxial nanoislands has been studied with the use of the kinetic Monte Carlo (KMC) technique. It has been shown that the strain-inducing size misfit between the substrate and the overlayer produces long range dipole–monopole (d–m) interaction between the mobile adatoms and the islands. To simplify the account of the long range interactions in the KMC simulations, use has been made of a modified square island model. An analytic formula for the interaction between the point surface monopole and the dipole forces has been derived and used to obtain a simple expression for the interaction between the mobile adatom and the rectangular island. The d–m interaction was found to be longer ranged than the conventional dipole–dipole potential. The narrowing of the island size distributions (ISDs) observed in the simulations was shown to be a consequence of a weaker repulsion of adatoms from small islands than from large ones which led to the preferential growth of the former. Furthermore, similar to the unstrained case, the power-law behavior of the average island size and of the island density on the coverage has been found. In contrast to the unstrained case, the value of the scaling exponent was not universal but strongly dependent on the strength of the long range interactions. Qualitative agreement of the simulation results with some previously unexplained behaviors of experimental ISDs in the growth of semiconductor quantum dots was observed. read less USED (high confidence) L. Hale et al., “Dislocation morphology and nucleation within compressed Si nanospheres: A molecular dynamics study,” Computational Materials Science. 2012. link Times cited: 24 USED (high confidence) A. Ince and S. Erkoç, “Molecular‐dynamics simulations of silicene nanoribbons under strain,” physica status solidi (b). 2012. link Times cited: 7 Abstract: Structural properties of silicene nanoribbons (SiNRs) of var… read moreAbstract: Structural properties of silicene nanoribbons (SiNRs) of varying width have been investigated under 5% and 10% uniaxial strain via classical Molecular‐Dynamics simulations at 1 and 300 K temperatures by the aid of atomistic many‐body potential energy functions (PEFs). It has been found that under strain, SiNRs show such material properties: they are very ductile, with considerable toughness and a very long plastic range before fragmentation. read less USED (high confidence) L. Hale, X. W. Zhou, J. Zimmerman, N. Moody, R. Ballarini, and W. Gerberich, “Phase transformations, dislocations and hardening behavior in uniaxially compressed silicon nanospheres,” Computational Materials Science. 2010. link Times cited: 26 USED (high confidence) Z. Yang, Z.-X. Lu, and Y.-pu Zhao, “Shape effects on the yield stress and deformation of silicon nanowires: A molecular dynamics simulation,” Journal of Applied Physics. 2009. link Times cited: 49 Abstract: The tension and compression of single-crystalline silicon na… read moreAbstract: The tension and compression of single-crystalline silicon nanowires (SiNWs) with different cross-sectional shapes are studied systematically using molecular dynamics simulation. The shape effects on the yield stresses are characterized. For the same surface to volume ratio, the circular cross-sectional SiNWs are stronger than the square cross-sectional ones under tensile loading, but reverse happens in compressive loading. With the atoms colored by least-squares atomic local shear strain, the deformation processes reveal that the failure modes of incipient yielding are dependent on the loading directions. The SiNWs under tensile loading slip in {111} surfaces, while the compressive loading leads the SiNWs to slip in the {110} surfaces. The present results are expected to contribute to the design of the silicon devices in nanosystems. read less USED (high confidence) H. Zhao and N. Aluru, “Size and surface orientation effects on thermal expansion coefficient of one-dimensional silicon nanostructures,” Journal of Applied Physics. 2009. link Times cited: 13 Abstract: We perform classical molecular dynamics simulations based on… read moreAbstract: We perform classical molecular dynamics simulations based on the Tersoff interatomic potential to investigate the size and surface orientation dependence of lattice constant and thermal expansion coefficient of one-dimensional silicon nanostructures. Three different surface orientations of silicon are considered, i.e., Si(110), Si(111), and Si(100) with 2×1 reconstruction. For each surface orientation, we investigate nanostructures with thicknesses ranging from 0.3 to 5.0 nm. We compute the vibrational amplitude of surface atoms, lattice constant, and thermal expansion coefficient as a function of size and temperature, and compare them for different surface orientations. An analytical expression is developed to compute the variation of the thermal expansion coefficient with size of the nanostructure. read less USED (high confidence) M. Ghazisaeidi, J. Freund, and H. Johnson, “Statistical characterization of surface defects created by Ar ion bombardment of crystalline silicon,” Journal of Applied Physics. 2008. link Times cited: 1 Abstract: Ion bombardment of crystalline silicon targets induces patte… read moreAbstract: Ion bombardment of crystalline silicon targets induces pattern formation by the creation of mobile surface species that participate in forming nanometer-scale structures. The formation of these mobile species on a Si(001) surface, caused by sub-keV argon ion bombardment, is investigated through molecular dynamics simulation of Stillinger-Weber [Phys. Rev. B 31, 5262 (1985)] silicon. Specific criteria for identifying and classifying these mobile atoms based on their energy and coordination number are developed. The mobile species are categorized based on these criteria and their average concentrations are calculated. read less USED (high confidence) J. Schall, G. Gao, and J. Harrison, “Elastic constants of silicon materials calculated as a function of temperature using a parametrization of the second-generation reactive empirical bond-order potential,” Physical Review B. 2008. link Times cited: 48 Abstract: A parametrization for silicon is presented that is based on … read moreAbstract: A parametrization for silicon is presented that is based on the second-generation reactive empirical bondorder REBO formalism Brenner, Shenderova, Harrison, Stuart, Ni, and Sinnott J. Phys.: Condens. Matter 14, 783 2002 . Because it shares the same analytic form as Brenner’s second-generation REBO, this new potential is a step toward a single potential that can model many atom systems that contain C, Si, and H, where bond breaking and bond making are important. The widespread use of Brenner’s REBO potential, its ability to model both zero-Kelvin elastic constants of diamond and the temperature dependence of the elastic constants, and the existence of parameters for many atom types were the motivating factors for obtaining this parametrization for Si. While Si-C-H classical bond-order potentials do exist, they are based on Brenner’s original formalism. This new parametrization is validated by examining the structure and stability of a large number of crystalline silicon structures, by examining the relaxation energies of point defects, the energies of silicon surfaces, the effects of adatoms on surface energies, and the structures of both liquid silicon and amorphous silicon. Finally, the elastic constants of diamond-cubic and amorphous silicon between 0 and 1100 K are calculated with this new parametrization and compared to values calculated using a previously published potential. read less USED (high confidence) K. Kang and W. Cai, “Brittle and ductile fracture of semiconductor nanowires – molecular dynamics simulations,” Philosophical Magazine. 2007. link Times cited: 135 Abstract: Fracture of silicon and germanium nanowires in tension at ro… read moreAbstract: Fracture of silicon and germanium nanowires in tension at room temperature is studied by molecular dynamics simulations using several interatomic potential models. While some potentials predict brittle fracture initiated by crack nucleation from the surface, most potentials predict ductile fracture initiated by dislocation nucleation and slip. A simple parameter based on the ratio between the ideal tensile strength and the ideal shear strength is found to correlate very well with the observed brittle versus ductile behaviours for all the potentials used in this study. This parameter is then computed by ab initio methods, which predict brittle fracture at room temperature. A brittle-to-ductile transition (BDT) is observed in MD simulations at higher temperature. The BDT mechanism in semiconductor nanowires is different from that in the bulk, due to the lack of a pre-existing macrocrack that is always assumed in bulk BDT models. read less USED (high confidence) K. Nishio, T. Morishita, W. Shinoda, and M. Mikami, “Molecular dynamics simulations of self-organized polyicosahedral Si nanowire.,” The Journal of chemical physics. 2006. link Times cited: 24 Abstract: A novel polyicosahedral nanowire is spontaneously formed in … read moreAbstract: A novel polyicosahedral nanowire is spontaneously formed in a series of annealing molecular dynamics simulations of liquid Si inside a nanopore of 1.36 nm in diameter. The polyicosahedral Si nanowire is stable even in a vacuum up to about 77% of the melting temperature of bulk Si. Our structural energy calculations reveal that the polyicosahedral nanowire is energetically advantageous over the pentagonal one for a wire whose diameter is less than 6.02 nm, though the latter has been recently proposed as the lowest energy wire. These results suggest the possibility of the formation of a new stable polyicosahedral Si nanowire. read less USED (high confidence) K. Garikipati, M. Falk, M. Bouville, B. Puchala, and H. Narayanan, “The continuum elastic and atomistic viewpoints on the formation volume and strain energy of a point defect,” Journal of The Mechanics and Physics of Solids. 2005. link Times cited: 27 USED (high confidence) H. Shim, L. Zhou, H.-C. Huang, and T. Cale, “Nanoplate elasticity under surface reconstruction,” Applied Physics Letters. 2005. link Times cited: 71 Abstract: Using classical molecular statics simulations, we show that … read moreAbstract: Using classical molecular statics simulations, we show that nanoplate elasticity strongly depends on surface reconstruction and alignment of bond chains. Because of its well-established surface reconstructions and the readily available interatomic potential, diamond-cubic silicon is the prototype of this study. We focus on silicon nanoplates of high-symmetry surfaces, {111} and {100}; with 7×7 and 2×1 reconstructions. Nanoplates with unreconstructed {111} surfaces are elastically stiffer than bulk. In contrast, the same nanoplates with 7×7 reconstructed {111} surfaces are elastically softer than bulk. On {100} surfaces, the 2×1 surface reconstruction has little impact. The bond chains are along one of the two ⟨110⟩ directions, making the two ⟨110⟩ directions nonequivalent. The alignment of the bond chains on the opposite surfaces of a nanoplate dictates its elastic anisotropy. The sensitivity of nanoplate elasticity on details of surface atomic arrangements may impact the application of nanoplates (or nan... read less USED (high confidence) L. Marqués, L. Pelaz, P. Castrillo, and J. Barbolla, “Molecular dynamics study of the configurational and energetic properties of the silicon self-interstitial,” Physical Review B. 2005. link Times cited: 51 Abstract: We have carried out classical molecular dynamics simulations… read moreAbstract: We have carried out classical molecular dynamics simulations to study the configurational and energetic properties of the Si self-interstitial. We have shown that the Si self-interstitial can appear in four different configurations, characterized by different energetics. Along with the already known tetrahedral, dumbbell, and extended configurations, we have found a highly asymmetric configuration not previously reported in the literature. Using a data analysis technique based on time averages, we have extracted the formation enthalpies and the probability of finding the interstitial in a given configuration, both depending on temperature. By the use of thermodynamic integration techniques we have determined the Gibbs free energy and entropy of formation, and the relative concentration of each interstitial configuration as a function of temperature. We have demonstrated that the change of interstitial configuration is correlated with the diffusion process, and we have identified two different mechanisms for interstitial-mediated self-diffusion. In spite of the microscopic complexity of the interstitial-mediated diffusion process, our results predict a pure Arrhenius behavior with an activation energy of $4.60\phantom{\rule{0.3em}{0ex}}\mathrm{eV}$ in the temperature interval $900\char21{}1685\phantom{\rule{0.3em}{0ex}}\mathrm{K}$, in good agreement with experiments. This energy is decomposed in an effective interstitial formation enthalpy of $3.83\phantom{\rule{0.3em}{0ex}}\mathrm{eV}$ and a migration barrier of $0.77\phantom{\rule{0.3em}{0ex}}\mathrm{eV}$, which macroscopically represent the averaged behavior of the different interstitial configurations. read less USED (high confidence) M. Makeev, W. Yu, and A. Madhukar, “Atomic scale stresses and strains in Ge/Si(001) nanopixels: An atomistic simulation study,” Journal of Applied Physics. 2004. link Times cited: 6 Abstract: Recent progress in the growth of nanostructures on nonplanar… read moreAbstract: Recent progress in the growth of nanostructures on nonplanar (patterned) substrates has brought to the forefront issues related to atomic-level surface and subsurface stress and strain field variations, as these govern the process of formation of such nanostructures and strongly affect their physical properties. In this work, we use atomistic simulations to study the atomically resolved displacements, stresses, strains, and the strain energy in laterally finite nanoscale Si(001) mesas, uncovered and covered with the lattice-mismatched Ge overlayers. The spatial variations of the stress are examined both across the surface profile of the mesas and in the direction down to the substrate. We find that the hydrostatic stress and strain at the Ge∕Si interface undergo rapid changes from tensile in the interior of the Si mesa to compressive in the Ge overlayer, with the transition taking place over distances of the order of Si lattice constant. Substantial relaxation of the hydrostatic stress and strain, in both... read less USED (high confidence) P. Lorazo, D. Perez, L. J. Lewis, and M. Meunier, “Thermodynamics of absorbing solids during short-pulse laser ablation,” SPIE High-Power Laser Ablation. 2004. link Times cited: 4 Abstract: The fundamental mechanisms of matter removal involved in the… read moreAbstract: The fundamental mechanisms of matter removal involved in the interaction of short laser pulses with absorbing solids have been investigated using molecular-dynamics/Monte~Carlo simulations. This is accomplished under the two following assumptions: (i) the elementary thermodynamic properties of targets (metals and semiconductors) are adequately described by empirical potentials; (ii) in the regime where ablation is thermal, the complete time evolution of the system can be followed in p-T-P space and the result mapped onto the equilibrium phase diagram of the material. We find remarkable similarities in the physical pathways to ablation in metals and semiconductors for pulse durations ranging from 200 fs to 400 ps: (i) under conditions of isochoric heating and rapid adiabatic cooling with femtosecond pulses, several mechanisms can simultaneously account for matter removal in the target: spallation, phase explosion, vaporization, and fragmentation; the latter is identified for the first time in the context of laser ablation. (ii) Under nonadiabatic cooling with picosecond pulses, ablation is driven by a "trivial" fragmentation process in the metallic, supercritical fluid; this suggests a pulse duration upper limit for phase explosion of ~ 10-11 s. read less USED (high confidence) A. Barnard and P. Zapol, “A model for the phase stability of arbitrary nanoparticles as a function of size and shape.,” The Journal of chemical physics. 2004. link Times cited: 234 Abstract: A thermodynamic model describing relative stability of diffe… read moreAbstract: A thermodynamic model describing relative stability of different shapes for nanoparticles as a function of their size was developed for arbitrary crystalline solids and applied to group IV semiconductors. The model makes use of various surface, edge and corner energies, and takes into account surface tension. Approximations and importance of each term of the model were analyzed. The predictions for clean and hydrogenated diamond nanoparticles are compared to explicitly calculated density functional results. It is shown that diamond nanocrystal morphology is markedly different from silicon and germanium. read less USED (high confidence) A. Charaï et al., “Structural change induced on an atomie scale by equilibrium sulphur segregation in tilt germanium grain boundaries,” Philosophical Magazine B. 2001. link Times cited: 1 Abstract: In the present study, structural modifications induced by ea… read moreAbstract: In the present study, structural modifications induced by eauilibrium sulphur segregation in pure tilt germanium {710}<001>, ∑=25 (θ=16.26°) and {551}<011>, ∑=51 (θ=16.10°) grain boundaries (GBs) were investigated using high-resolution electron microscopy coupled to electron-energy-loss spectroscopy and supported by structural modelling and image simulations. Our results showed that the as-grown ∑=25 GB is composed of two parts: a stable structural region and a variable perturbed core. On the basis of our simulations, it is shown that this boundary can only be formed by a multiplicity of configurations which are energetically close to each other but differently configured along the boundary plane. When sulphurized, drastic changes in the structure of the GB were observed. Energy-filtered electron microscopy imaging revealed a sulphur enrichment at the perturbed part of the boundary. Although sulphur segregation at the boundary is detected, no information can at the present stage be extracted on segregation sites and bonding configurations because of the complexity of the boundary structure. To simplify this aspect, a simpler GB, that is germanium ∑=51, was studied. The structure of such a GB is a well-known configuration, that is a Lomer dislocation, which is basically a fivefold ring adjacent to a sevenfold ring. After sulphur treatment, high-resolution electron microscopy imaging also shows significant contrast modifications apparently concentrated on the dislocation core. Chemical imaging indicates again the presence of sulphur enrichment along the boundary plane strongly sustaining that eauilibrium sulphur segregation in the Ge(S) system oceurs into the GB and therefore confirms our previous results on the ∑= 25 GB. One can therefore argue that it is the presence of those odd-membered rings at the boundary, which should possess a specific crystallographic and electronic nature, coupled to the electronic properties of sulphur, that are responsible for the preferential segregation into the boundary. read less USED (high confidence) K. Nordlund, P. Partyka, R. Averback, I. Robinson, and P. Ehrhart, “Atomistic simulation of diffuse x-ray scattering from defects in solids,” Journal of Applied Physics. 2000. link Times cited: 21 Abstract: Diffuse x-ray scattering is a powerful means to study the st… read moreAbstract: Diffuse x-ray scattering is a powerful means to study the structure of defects in crystalline solids. The traditional analysis of diffuse x-ray scattering experiments relies on analytical and numerical methods which are not well suited for studying complicated defect configurations. We present here an atomistic simulation method by which the diffuse x-ray scattering can be calculated for an arbitrary finite-sized defect in any material where reliable interatomic force models exist. We present results of the method for point defects, defect clusters and dislocations in semiconductors and metals, and show that surface effects on diffuse scattering, which might be important for the investigation of shallow implantation damage, will be negligible in most practical cases. We also compare the results with x-ray experiments on defects in semiconductors to demonstrate how the method can be used to understand complex damage configurations. read less USED (low confidence) V. Gusakov, V. Belko, and N. Dorozhkin, “Formation and diffusion of self-interstitial atoms in silicon crystals under hydrostatic pressure: Quantum-chemical simulation,” Journal of Surface Investigation. X-ray, Synchrotron and Neutron Techniques. 2009. link Times cited: 2 USED (low confidence) V. Gusakov, V. I. Belko, and N. N. Dorozhkin, “Effect of Hydrostatic Pressure on Self-Interstitial Diffusion in Si, Ge, Si Crystals: Quantum-Chemical Simulations,” Solid State Phenomena. 2007. link Times cited: 0 Abstract: A theoretical modeling of the diffusion of self-interstitial… read moreAbstract: A theoretical modeling of the diffusion of self-interstitials in silicon and germanium crystals both at normal and high hydrostatic pressure has been carried out using molecular mechanics, semiempirical (PM3, PM5) and ab-initio (SIESTA) methods. According to the simulation for the Si and Ge neutral interstitials (I0) both in silicon and germanium crystals more stable configuration is <110> split interstitial. T is the stable configuration for the double positive interstitial I++, but the interstitial is displaced from the high-symmetry site. Stability of <110> splitinterstitial is not changed under hydrostatic pressure. The activation barriers for the diffusion of interstitials were determined and equal to ΔEa(Si)(<110> -> T1)=0.69 eV; ΔEa (Ge)(<110> -> T1)=1.1 eV. For mixed interstitials the calculated activation barriers equal Si Emix = 1.06 eV, Ge Emix = 0.86 eV. Hydrostatic pressure decreases the activation barriers ΔEa(Si), ΔEa (Ge). read less USED (low confidence) M. Bachlechner et al., “Mechanisms of pit formation at strained crystallineSi(111)∕Si3N4(0001)interfaces: Molecular-dynamics simulations,” Physical Review B. 2006. link Times cited: 8 USED (low confidence) P. Qian, N. Chen, and J. Shen, “Atomistic simulation for the phase stability, site preference and thermal expansion of YFe12−xTx (T=Ti, V, Cr, Mn, Zr, Nb, Mo, W),” Solid State Communications. 2005. link Times cited: 26 USED (low confidence) P. Erhart and K. Albe, “Analytical potential for atomistic simulations of silicon, carbon, and silicon carbide,” Physical Review B. 2005. link Times cited: 462 Abstract: We present an analytical bond-order potential for silicon, c… read moreAbstract: We present an analytical bond-order potential for silicon, carbon, and silicon carbide that has been optimized by a systematic fitting scheme. The functional form is adopted from a preceding work {\}Phys. Rev. B 65, 195124 (2002) and is built on three independently fitted potentials for Si-Si, C-C, and Si-C interaction. For elemental silicon and carbon, the potential perfectly reproduces elastic properties and agrees very well with first-principles results for high-pressure phases. The formation enthalpies of point defects are reasonably reproduced. In the case of silicon stuctural features of the melt agree nicely with data taken from literature. For silicon carbide the dimer as well as the solid phases B1, B2, and B3 were considered. Again, elastic properties are very well reproduced including internal relaxations under shear. Comparison with first-principles data on point defect formation enthalpies shows fair agreement. The successful validation of the potentials for configurations ranging from the molecular to the bulk regime indicates the transferability of the potential model and makes it a good choice for atomistic simulations that sample a large configuration space. read less USED (low confidence) S.-P. Huang and W.-C. Wang, “Structural and Dynamic Properties of Amorphous Silicon: Tight-Binding Molecular Dynamics Simulation,” Chinese Physics Letters. 2004. link Times cited: 3 Abstract: The tight-binding molecular dynamics simulation has been per… read moreAbstract: The tight-binding molecular dynamics simulation has been performed to study structural and dynamical properties of amorphous silicon. It is found that the radial distribution function and static structure factor are in good agreement with the experimental measurements. The bond order parameters Ql are sensitive to the structure change at different quenching rates. For the dynamical properties, we have calculated the vibration and electronic density of states. The simulation results show that the transverse acoustic is in good agreement with the experimental data, and the high frequency transverse optical (TO) peak shifts to the right of the experimental TO peak. read less USED (low confidence) M. Makeev, W. Yu, and A. Madhukar, “Stress distributions and energetics in the laterally ordered systems of buried pyramidal Ge/Si(001) islands: An atomistic simulation study,” Physical Review B. 2003. link Times cited: 11 Abstract: Stress distributions in laterally ordered arrays of coherent… read moreAbstract: Stress distributions in laterally ordered arrays of coherent Ge islands of shallow pyramidal shape buried in a Si(001) matrix are studied via large-scale atomistic simulations, using Stillinger-Weber Ge/Si systems as a vehicle. The existence of tensile hydrostatic stress regions is observed on the spacer surface, above the buried islands. Our previously reported finding [M. A. Makeev and A. Madhukar, Phys. Rev. Lett. 86, 5542 (2001)] that the hydrostatic stress at the spacer layer surface above the island apex is nearly inversely proportional to the spacer layer thickness is validated by a comparison with experimental data. The lateral variations of the hydrostatic stress on the spacer layer surface show "bell-shape" profiles, with the effective size of the tensile regions above the island apex varying as a power law with the spacer layer thickness, with the power exponent being greater than 1. Studies of the energetics of twofold stacks of island systems show that the elastic interaction energy between the islands is minimized for the vertically aligned geometry. The spacer layer thickness dependence of the hydrostatic and biaxial stress field distributions in the interior of the Si(001) matrix are presented as these define the behavior of the electron and hole three-dimensional confinement potentials that determine the electronic properties of the pyramidal island quantum dots. read less USED (low confidence) E. Haddeman and B. Thijsse, “Transient sputtering of silicon by argon studied by molecular dynamics simulations,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 21 USED (low confidence) M. Makeev and A. Madhukar, “Large-scale atomistic simulations of atomic displacements, stresses, and strains in nanoscale mesas: Effect of mesa edges, corners, and interfaces,” Applied Physics Letters. 2002. link Times cited: 8 Abstract: Large-scale atomistic simulations are performed to study the… read moreAbstract: Large-scale atomistic simulations are performed to study the atomic displacements, stresses, and strains in the Stillinger–Weber model of nanoscale Si(001) bare and Ge overlayer covered mesas. Considerable inhomogeneity in the atomic displacement fields in the vicinity of the mesa edges, corners, and at the lattice-mismatched Ge/Si interface is observed, maximum displacements being as large as 0.6 A even for an uncovered mesa. For Ge overlayer covered mesas, relaxation in the mesa interior and deep into the substrate is observed. The relationship between the off-diagonal components of the stress and strain tensors is found to become non-linear in the vicinity of the mesa edges for both bare and covered mesas. read less USED (low confidence) J. Oh and C. Grein, “Epitaxial growth simulations of CdTe(1 1 1)B on Si(0 0 1),” Journal of Crystal Growth. 1998. link Times cited: 15 USED (low confidence) I. Štich, “First-principles finite-temperature simulation of surface dynamics: Si(111)-(7 × 7),” Surface Science. 1996. link Times cited: 5 USED (low confidence) A. Natori, T. Suzuki, and H. Yasunaga, “Atomic structures and atomic dynamics on ‘1 × 1’ Si(111) at high temperatures,” Surface Science. 1996. link Times cited: 11 USED (low confidence) A. Dyson and P. V. Smith, “Empirical molecular dynamics calculations for the (001) and (111) 2×1 reconstructed surfaces of diamond,” Surface Science. 1994. link Times cited: 12 USED (low confidence) Q. Yu and P. Clancy, “Molecular dynamics simulation of the surface reconstruction and strain relief in Si1-xGex/Si(100) heterostructures,” Modelling and Simulation in Materials Science and Engineering. 1994. link Times cited: 5 Abstract: The structure of a variety of Si1-xGex/Si heterostructures, … read moreAbstract: The structure of a variety of Si1-xGex/Si heterostructures, as well as bulk Si(100) and Ge(100) modelled by the Stillinger-Weber potential, have been simulated by molecular dynamics to investigate the surface reconstruction and strain relief in the SiGe thin films. It was found that the strain in SiGe/Si(100) thin films was relaxed by the segregation of Ge to the surface. Rebonding of sub-surface atoms into dimers in the presence of a vacancy or cluster of vacancies above them was observed in the ensuing surface reconstruction. For SiGe/Si, the amount of 'rebonded missing dimers' in the surface increased with increasing Ge composition. However, for Ge/Si(100), a V-shaped defect was observed in the Ge thin film. For bulk Si, several rebonded missing dimers were found at the surface, while for bulk Ge(100) the surface showed a typical 2*1 reconstruction. All these findings corroborate recent related experimental studies and theoretical predictions. read less USED (low confidence) S. Hara, T. Kumagai, S. Izumi, and S. Sakai, “Evaluation of Structural and Mechanical Properties of Amorphous Silicon Surabaces by a Combination Approach of Ab-initio and Classical Molecular Dynamics,” Journal of The Society of Materials Science, Japan. 2005. link Times cited: 0 Abstract: In this study, we use a combiend method of a classical molec… read moreAbstract: In this study, we use a combiend method of a classical molecular dynamics with the Tersoff potential and an ab-initio calculation based on density functional theory. This combination method can provide quantitative evaluation of the surface energy and surface stress of well-relaxed amorphous silicon in addition to its structure. Using this method, a surface energy of 1.05 ± 0.14 J/m2 and a surface stress of 1.5 ± 1.2N/m was obtained. This calculation also led to a new discovery of the microscopic characteristic of a-Si surfaces, which was not revealed through the use of an empirical potential. It was shown that there are two types of threefold coordinated atoms at the surface region; one with p3-like bonding and the other with sp2-like bonding. In addition, the investigation indicated that the microstructures of these coordination defects were different from those of threefold coordinated atoms observed in the bulk region. read less USED (low confidence) L. Nurminen, F. Tavazza, D. Landau, A. Kuronen, and K. Kaski, “Monte Carlo Simulation of the Surface Structure of Ge on Si(00l).” 2003. link Times cited: 1 USED (low confidence) J. Chen, A. Hairie, B. Lebouvffir, G. Nouet, and E. Paumter, “Potential Energy Calculation of two Structures of the Σ=11 Tilt Grain Boundary in Silicon and Germanium,” MRS Proceedings. 1997. link Times cited: 0 USED (low confidence) W. Neumann, H. Hofmeister, D. Conrad, K. Scheerschmidt, and S. Ruvimov, “Characterization of interface structures in nanocrystalline germanium by means of high-resolution electron microscopy and molecular dynamics simulations,” Zeitschrift Fur Kristallographie. 1996. link Times cited: 5 Abstract: The atomic structure of nanocrystalline particles formed by … read moreAbstract: The atomic structure of nanocrystalline particles formed by vapor deposition and subsequent annealing of amorphous thin films of germanium was studied by high resolution electron microscopy (HREM). The HREM images revealed a strongly varied multiply twinned structure. In some regions of adjacent twins contrast features were detected which were caused by an overlapping of twin lamellae. It will be shown by HREM contrast simulations that these interface types can be described by Σ = 3 n boundaries. The influence of lattice relaxations is taken into consideration by molecular dynamics simulations of the structure models. read less USED (low confidence) Q. Yu and P. Clancy, “Surface Reconstruction and Strain Relief in Si 1− x - Ge x Films on Si(100),” MRS Proceedings. 1992. link Times cited: 2 NOT USED (low confidence) L. Martín‐Encinar, L. Marqués, M. Aboy, P. López, I. Santos, and L. Pelaz, “Molecular Dynamics Study of Stress Relaxation During Ge Deposition on Si(100) 2\times 1 Substrates,” 2023 14th Spanish Conference on Electron Devices (CDE). 2023. link Times cited: 0 Abstract: We studied epitaxial growth of Ge films on Si(001) $2\times … read moreAbstract: We studied epitaxial growth of Ge films on Si(001) $2\times 1$ at different temperatures using classical molecular dynamics simulations. Ge-Si intermixing contributes to strain accommodation mostly in the original Si substrate surface and first grown Ge layer. Stress accumulation is further released by the generation of dislocations whose amount and type depend on temperature. At high temperatures, a larger amount and more variety of dislocations are formed, thus affecting the surface morphology and consequently the size of 3D islands. read less NOT USED (low confidence) M. Maździarz, “Transferability of interatomic potentials for silicene,” Beilstein Journal of Nanotechnology. 2023. link Times cited: 1 Abstract: The ability of various interatomic potentials to reproduce t… read moreAbstract: The ability of various interatomic potentials to reproduce the properties of silicene, that is, 2D single-layer silicon, polymorphs was examined. Structural and mechanical properties of flat, low-buckled, trigonal dumbbell, honeycomb dumbbell, and large honeycomb dumbbell silicene phases, were obtained using density functional theory and molecular statics calculations with Tersoff, MEAM, Stillinger–Weber, EDIP, ReaxFF, COMB, and machine-learning-based interatomic potentials. A quantitative systematic comparison and a discussion of the results obtained are reported. read less NOT USED (low confidence) M. Posselt, H. Bracht, M. Ghorbani-Asl, and D. Radić, “Atomic mechanisms of self-diffusion in amorphous silicon,” AIP Advances. 2022. link Times cited: 1 Abstract: Based on recent calculations of the self-diffusion (SD) coef… read moreAbstract: Based on recent calculations of the self-diffusion (SD) coefficient in amorphous silicon (a-Si) by classical Molecular Dynamics simulation [Posselt et al., J. Appl. Phys. 131, 035102 (2022)], detailed investigations on atomic mechanisms are performed. For this purpose, two Stillinger–Weber-type potentials are used, one strongly overestimates the SD coefficient, while the other leads to values much closer to the experimental data. By taking into account the individual squared displacements (or diffusion lengths) of atoms, the diffusional and vibrational contributions to the total mean squared displacement can be determined separately. It is shown that the diffusional part is not directly correlated with the concentration of coordination defects. The time-dependent distribution of squared displacements of atoms indicates that in a-Si, a well-defined elemental diffusion length does not exist, in contrast to SD in the crystalline Si. The analysis of atoms with large squared displacements reveals that the mechanisms of SD in a-Si are characterized by complex rearrangements of bonds or exchanges of neighbors. These are mono- and bi-directional exchanges of neighbors and neighbor replacements. Exchanges or replacements may concern up to three neighbors and may occur in relatively short periods of some ps. Bi- or mono-directional exchange or replacement of one neighbor atom happens more frequently than processes including more neighbors. A comparison of results for the two interatomic potentials shows that an increased three-body parameter only slows down the migration but does not change the migration mechanisms fundamentally. read less NOT USED (low confidence) R. Abram, D. Chrobak, J. Byggmästar, K. Nordlund, and R. Nowak, “Comprehensive structural changes in nanoscale-deformed silicon modelled with an integrated atomic potential,” Materialia. 2022. link Times cited: 2 NOT USED (low confidence) W. Wan, C. Tang, and W. Zou, “Exploring Silicon [001] Small Angle Symmetric Tilt Grain Boundaries: Structures, Energies and Stress fields,” Applied Surface Science. 2022. link Times cited: 4 NOT USED (low confidence) Y. Liu, W. Wan, Q. Li, Z. Xiong, C. Tang, and L. Zhou, “Revisiting the Rate-Dependent Mechanical Response of Typical Silicon Structures via Molecular Dynamics,” Nanomaterials. 2022. link Times cited: 1 Abstract: Strain rate is a critical parameter in the mechanical applic… read moreAbstract: Strain rate is a critical parameter in the mechanical application of nano-devices. A comparative atomistic study on both perfect monocrystalline silicon crystal and silicon nanowire was performed to investigate how the strain rate affects the mechanical response of these silicon structures. Using a rate response model, the strain rate sensitivity and the critical strain rate of two structures were given. The rate-dependent dislocation activities in the fracture process were also discussed, from which the dislocation nucleation and motion were found to play an important role in the low strain rate deformations. Finally, through the comparison of five equivalent stresses, the von Mises stress was verified as a robust yield criterion of the two silicon structures under the strain rate effects. read less NOT USED (low confidence) Q. Zhang et al., “Probing the displacement damage mechanism in Si, Ge, GaAs by defects evolution analysis,” Computational Materials Science. 2022. link Times cited: 1 NOT USED (low confidence) L. Zhang and J. Yan, “Evolution of high-pressure metastable phase Si-XIII during silicon nanoindentation: A molecular dynamics study,” Computational Materials Science. 2021. link Times cited: 5 NOT USED (low confidence) B. Zhang, Y. Wang, J. Chen, J. Li, and W. Lai, “Development of an angular-dependent potential for radiation damage study in Fe-Si solutions,” Journal of Nuclear Materials. 2020. link Times cited: 3 NOT USED (low confidence) K. Ye, J. Wang, and Y. Li, “Temperature effect on Young’s modulus of surface oxidized silicon nano-films,” Modern Physics Letters B. 2020. link Times cited: 0 Abstract: Based on the semi-continuum model, the effect of temperature… read moreAbstract: Based on the semi-continuum model, the effect of temperature on Young’s modulus in the presence of oxide layer in silicon nano-films was studied theoretically by using the anharmonic Keating deform... read less NOT USED (low confidence) M. Shi, N. Admal, and E. Tadmor, “Noise filtering in atomistic stress calculations for crystalline materials,” Journal of The Mechanics and Physics of Solids. 2020. link Times cited: 2 NOT USED (low confidence) Z. Liu, T. Yunqing, N. Liao, and P. Yang, “Study on interfacial interaction between Si and ZnO,” Ceramics International. 2019. link Times cited: 15 NOT USED (low confidence) M. A. Wilson, S. Grutzik, and M. Chandross, “Continuum stress intensity factors from atomistic fracture simulations,” Computer Methods in Applied Mechanics and Engineering. 2019. link Times cited: 14 NOT USED (low confidence) B. Helfrecht, R. Semino, G. Pireddu, S. Auerbach, and M. Ceriotti, “A new kind of atlas of zeolite building blocks.,” The Journal of chemical physics. 2019. link Times cited: 30 Abstract: We have analyzed structural motifs in the Deem database of h… read moreAbstract: We have analyzed structural motifs in the Deem database of hypothetical zeolites to investigate whether the structural diversity found in this database can be well-represented by classical descriptors, such as distances, angles, and ring sizes, or whether a more general representation of the atomic structure, furnished by the smooth overlap of atomic position (SOAP) method, is required to capture accurately structure-property relations. We assessed the quality of each descriptor by machine-learning the molar energy and volume for each hypothetical framework in the dataset. We have found that a SOAP representation with a cutoff length of 6 Å, which goes beyond near-neighbor tetrahedra, best describes the structural diversity in the Deem database by capturing relevant interatomic correlations. Kernel principal component analysis shows that SOAP maintains its superior performance even when reducing its dimensionality to those of the classical descriptors and that the first three kernel principal components capture the main variability in the dataset, allowing a 3D point cloud visualization of local environments in the Deem database. This "cloud atlas" of local environments was found to show good correlations with the contribution of a given motif to the density and stability of its parent framework. Local volume and energy maps constructed from the SOAP/machine learning analyses provide new images of zeolites that reveal smooth variations of local volumes and energies across a given framework and correlations between the contributions to volume and energy associated with each atom-centered environment. read less NOT USED (low confidence) J. V. Michelin, L. G. Gonçalves, and J. Rino, “On the transferability of interaction potentials for condensed phases of silicon,” Journal of Molecular Liquids. 2019. link Times cited: 6 NOT USED (low confidence) Y. Zuo et al., “A Performance and Cost Assessment of Machine Learning Interatomic Potentials.,” The journal of physical chemistry. A. 2019. link Times cited: 413 Abstract: Machine learning of the quantitative relationship between lo… read moreAbstract: Machine learning of the quantitative relationship between local environment descriptors and the potential energy surface of a system of atoms has emerged as a new frontier in the development of interatomic potentials (IAPs). Here, we present a comprehensive evaluation of ML-IAPs based on four local environment descriptors --- atom-centered symmetry functions (ACSF), smooth overlap of atomic positions (SOAP), the Spectral Neighbor Analysis Potential (SNAP) bispectrum components, and moment tensors --- using a diverse data set generated using high-throughput density functional theory (DFT) calculations. The data set comprising bcc (Li, Mo) and fcc (Cu, Ni) metals and diamond group IV semiconductors (Si, Ge) is chosen to span a range of crystal structures and bonding. All descriptors studied show excellent performance in predicting energies and forces far surpassing that of classical IAPs, as well as predicting properties such as elastic constants and phonon dispersion curves. We observe a general trade-off between accuracy and the degrees of freedom of each model, and consequently computational cost. We will discuss these trade-offs in the context of model selection for molecular dynamics and other applications. read less NOT USED (low confidence) D. V. Canle et al., “Practical realization of a sub-λ/2 acoustic jet,” Scientific Reports. 2019. link Times cited: 17 NOT USED (low confidence) S. Safaei, R. Tavakoli, and M. Jafary-Zadeh, “Molecular dynamics study of two dimensional silicon dioxides with in-plane negative Poisson’s ratio,” Computational Materials Science. 2018. link Times cited: 9 NOT USED (low confidence) J. Harrison, J. Schall, S. Maskey, P. Mikulski, M. T. Knippenberg, and B. Morrow, “Review of force fields and intermolecular potentials used in atomistic computational materials research,” Applied Physics Reviews. 2018. link Times cited: 99 Abstract: Molecular simulation is a powerful computational tool for a … read moreAbstract: Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferability. When selecting a potential energy function for use in molecular simulations, the accuracy, transferability, and computational speed must all be considered. In this focused review, some of the more commonly used potential energy functions for molecular simulations are reviewed with an eye toward presenting their general forms, strengths, and weaknesses.Molecular simulation is a powerful computational tool for a broad range of applications including the examination of materials properties and accelerating drug discovery. At the heart of molecular simulation is the analytic potential energy function. These functions span the range of complexity from very simple functions used to model generic phenomena to complex functions designed to model chemical reactions. The complexity of the mathematical function impacts the computational speed and is typically linked to the accuracy of the results obtained from simulations that utilize the function. One approach to improving accuracy is to simply add more parameters and additional complexity to the analytic function. This approach is typically used in non-reactive force fields where the functional form is not derived from quantum mechanical principles. The form of other types of potentials, such as the bond-order potentials, is based on quantum mechanics and has led to varying levels of accuracy and transferabilit... read less NOT USED (low confidence) N. Zarkevich, H. Chen, V. Levitas, and D. D. Johnson, “Lattice Instability during Solid-Solid Structural Transformations under a General Applied Stress Tensor: Example of Si I→Si II with Metallization.,” Physical review letters. 2018. link Times cited: 18 Abstract: The density functional theory was employed to study the stre… read moreAbstract: The density functional theory was employed to study the stress-strain behavior and elastic instabilities during the solid-solid phase transformation (PT) when subjected to a general stress tensor, as exemplified for semiconducting Si I and metallic Si II, where metallization precedes the PT, so stressed Si I can be a metal. The hydrostatic PT occurs at 76 GPa, while under uniaxial loading it is 11 GPa (3.7 GPa mean pressure), 21 times lower. The Si I→Si II PT is described by a critical value of the phase-field's modified transformation work, and the PT criterion has only two parameters given six independent stress elements. Our findings reveal novel, more practical synthesis routes for new or known high-pressure phases under predictable nonhydrostatic loading, where competition of instabilities can serve for phase selection rather than free energy minima used for equilibrium processing. read less NOT USED (low confidence) Y. Hong, N. Zhang, and M. A. Zaeem, “Metastable phase transformation and deformation twinning induced hardening-stiffening mechanism in compression of silicon nanoparticles,” Acta Materialia. 2018. link Times cited: 20 NOT USED (low confidence) W. Zhou, K. Gong, J. Wan, L. Quan, Y. Chu, and Y. Cao, “Molecular dynamics simulation study on ablation of silicon by water-jet-guided laser,” Proceedings of the Institution of Mechanical Engineers, Part E: Journal of Process Mechanical Engineering. 2017. link Times cited: 5 Abstract: Stillinger–Weber potential and Z-layer energy model were ado… read moreAbstract: Stillinger–Weber potential and Z-layer energy model were adopted in molecular dynamics simulation to study the ablation of silicon by water-jet-guided femtosecond laser, and comparison was made by ablating silicon with or without water-jet cooling in our simulations. Simulation results indicated that with water-jet cooling, the thermal-affected zone could be reduced in area, and the peak of density could disappear more quickly. It was therefore concluded that water-jet-guided laser could be used to considerably improve the ablation quality of silicon. read less NOT USED (low confidence) N. Zographos, C. Zechner, I. Martín-Bragado, K. Lee, and Y. Oh, “Multiscale modeling of doping processes in advanced semiconductor devices,” Materials Science in Semiconductor Processing. 2017. link Times cited: 12 NOT USED (low confidence) S. Xiao and B. Liu, “An Exploration Toward a Unified Failure Criterion,” Journal of Applied Mechanics. 2017. link Times cited: 0 NOT USED (low confidence) V. Levitas, H. Chen, and L. Xiong, “Triaxial-Stress-Induced Homogeneous Hysteresis-Free First-Order Phase Transformations with Stable Intermediate Phases.,” Physical review letters. 2017. link Times cited: 43 Abstract: Starting with thermodynamic predictions and following with m… read moreAbstract: Starting with thermodynamic predictions and following with molecular dynamics simulations, special triaxial compression-tension states were found for which the stresses for the instability of the crystal lattice of silicon (Si) are the same for direct and reverse phase transformations (PTs) between semiconducting Si I and metallic Si II phases. This leads to unique homogeneous and hysteresis-free first-order PTs, for which each intermediate crystal lattice along the transformation path is in indifferent thermodynamic equilibrium and can be arrested and studied by fixing the strain in one direction. By approaching these stress states, a traditional two-phase system continuously transforms to homogenous intermediate phases. Zero hysteresis and homogeneous transformations are the optimal property for various PT applications, which drastically reduce damage and energy dissipation. read less NOT USED (low confidence) H. N. Pishkenari, E. Mohagheghian, and A. Rasouli, “Molecular dynamics study of the thermal expansion coefficient of silicon,” Physics Letters A. 2016. link Times cited: 23 NOT USED (low confidence) S. Takamoto et al., “Charge-transfer interatomic potential for investigation of the thermal-oxidation growth process of silicon,” Journal of Applied Physics. 2016. link Times cited: 11 Abstract: A charge-transfer interatomic potential, based on the hybrid… read moreAbstract: A charge-transfer interatomic potential, based on the hybrid-Tersoff potential that incorporates a covalent-ionic mixed-bond nature, was developed to reproduce the growth process of the thermal oxidation of silicon. A fitting process was employed with various reference structures sampled by MD. Actively exploring and learning the wide-range of phase space enabled us to develop a robust interatomic potential. Our interatomic potential reproduced the bulk properties of Si and SiO2 polymorphs well, in addition to the radial distribution function and bond angle distribution of amorphous SiO2. The covalent-ionic mixed-bond nature of the interatomic potential well reproduced the dissociation process of an oxygen molecule on the Si/SiO2 interface. The initial oxidation simulation was performed on the silicon surface. We grew the amorphous SiO2 layer by incorporating the oxygen molecules into the silicon network at the interface. The density of the SiO2 layer and the charge distribution at the interface showed go... read less NOT USED (low confidence) A. Portavoce, J. P. Toinin, K. Hoummada, L. Raymond, and G. Tréglia, “Stress influence on substitutional impurity segregation on dislocation loops in IV–IV semiconductors,” Computational Materials Science. 2016. link Times cited: 3 NOT USED (low confidence) N. S. Mikhaleva, M. Visotin, Z. Popov, A. Kuzubov, and A. Fedorov, “Ab initio and empirical modeling of lithium atoms penetration into silicon,” Computational Materials Science. 2015. link Times cited: 4 NOT USED (low confidence) B. Liu, H. Zhang, J. Tao, X. Chen, and Y.-A. Zhang, “Comparative investigation of a newly optimized modified embedded atom method potential with other potentials for silicon,” Computational Materials Science. 2015. link Times cited: 7 NOT USED (low confidence) P. Hecquet, “Interaction energy between dipole lines applied on symmetric (2 × 1) reconstructed Si(001),” Surface Science. 2014. link Times cited: 1 NOT USED (low confidence) M. Kiran, B. Haberl, J. S. Williams, and J. Bradby, “Temperature dependent deformation mechanisms in pure amorphous silicon,” Journal of Applied Physics. 2014. link Times cited: 9 Abstract: High temperature nanoindentation has been performed on pure … read moreAbstract: High temperature nanoindentation has been performed on pure ion-implanted amorphous silicon (unrelaxed a-Si) and structurally relaxed a-Si to investigate the temperature dependence of mechanical deformation, including pressure-induced phase transformations. Along with the indentation load-depth curves, ex situ measurements such as Raman micro-spectroscopy and cross-sectional transmission electron microscopy analysis on the residual indents reveal the mode of deformation under the indenter. While unrelaxed a-Si deforms entirely via plastic flow up to 200 °C, a clear transition in the mode of deformation is observed in relaxed a-Si with increasing temperature. Up to 100 °C, pressure-induced phase transformation and the observation of either crystalline (r8/bc8) end phases or pressure-induced a-Si occurs in relaxed a-Si. However, with further increase of temperature, plastic flow rather than phase transformation is the dominant mode of deformation. It is believed that the elevated temperature and pressure together induce bond softening and “defect” formation in structurally relaxed a-Si, leading to the inhibition of phase transformation due to pressure-releasing plastic flow under the indenter. read less NOT USED (low confidence) A. Portavoce and G. Tréglia, “Theoretical investigation of Cottrell atmosphere in silicon,” Acta Materialia. 2014. link Times cited: 11 NOT USED (low confidence) J. Sun, L. Fang, J. Han, Y. Han, H. Chen, and K. Sun, “Phase transformations of mono-crystal silicon induced by two-body and three-body abrasion in nanoscale,” Computational Materials Science. 2014. link Times cited: 38 NOT USED (low confidence) U. Monteverde, M. Migliorato, J. Pal, and D. Powell, “Elastic and vibrational properties of group IV semiconductors in empirical potential modelling,” Journal of Physics: Condensed Matter. 2013. link Times cited: 8 Abstract: We have developed an interatomic potential that with a singl… read moreAbstract: We have developed an interatomic potential that with a single set of parameters is able to accurately describe at the same time the elastic, vibrational and thermodynamics properties of semiconductors. The simultaneous inclusion of radial and angular forces of the interacting atom pairs (short range) together with the influence of the broken crystal symmetry when the atomic arrangement is out of equilibrium (long range) results in correct predictions of all of the phonon dispersion spectrum and mode-Grüneisen parameters of silicon and germanium. The long range interactions are taken into account up to the second nearest neighbours, to correctly influence the elastic and vibrational properties, and therefore represent only a marginal computational cost compared to the full treatment of other proposed potentials. Results of molecular dynamics simulations are compared with those of ab initio calculations, showing that when our proposed potential is used to perform the initial stages of the structural relaxation, a significant reduction of the computational time needed during the geometry optimization of density functional theory simulations is observed. read less NOT USED (low confidence) M. L. Nietiadi, Y. Rosandi, Y. Rosandi, J. Lorincik, and H. Urbassek, “Sputtering of a silicon surface: Preferential sputtering of surface impurities,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2013. link Times cited: 4 NOT USED (low confidence) T. Vazhappilly and D. Micha, “Atomic modeling of structural and optical properties of amorphous silicon,” Chemical Physics Letters. 2013. link Times cited: 7 NOT USED (low confidence) C. Hou, J. Xu, P. Wang, W. Huang, and X. Wang, “Efficient GPU-accelerated molecular dynamics simulation of solid covalent crystals,” Comput. Phys. Commun. 2013. link Times cited: 28 NOT USED (low confidence) V. Tomar, “Timescaling in Multiscale Mechanics of Nanowires and Nanocrystalline Materials.” 2013. link Times cited: 0 NOT USED (low confidence) V. Brázdová and D. Bowler, “Calculating Energies and Forces.” 2013. link Times cited: 0 NOT USED (low confidence) C. Ciobanu, C. Wang, and K. Ho, “Other Methodologies for Investigating Atomic Structure.” 2013. link Times cited: 0 NOT USED (low confidence) M. L. Nietiadi, Y. Rosandi, M. Kopnarski, and H. Urbassek, “Sputtering of dimers off a silicon surface,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2012. link Times cited: 5 NOT USED (low confidence) J. Adhikari, “Design of Compound Semiconductor Alloys Using Molecular Simulations.” 2012. link Times cited: 0 NOT USED (low confidence) E. Tadmor and R. E. Miller, “Modeling Materials: Continuum, Atomistic and Multiscale Techniques.” 2011. link Times cited: 395 Abstract: 1. Introduction Part I. Continuum Mechanics and Thermodynami… read moreAbstract: 1. Introduction Part I. Continuum Mechanics and Thermodynamics: 2. Essential continuum mechanics and thermodynamics Part II. Atomistics: 3. Lattices and crystal structures 4. Quantum mechanics of materials 5. Empirical atomistic models of materials 6. Molecular statics Part III. Atomistic Foundations of Continuum Concepts: 7. Classical equilibrium statistical mechanics 8. Microscopic expressions for continuum fields 9. Molecular dynamics Part IV. Multiscale Methods: 10. What is multiscale modeling? 11. Atomistic constitutive relations for multilattice crystals 12. Atomistic/continuum coupling: static methods 13. Atomistic/continuum coupling: finite temperature and dynamics Appendix References Index. read less NOT USED (low confidence) H. Kim and V. Tomar, “Nanometer to Micron Scale Atomistic Mechanics of Silicon Using Atomistic Simulations at Accelerated Time Steps,” Journal of Nanomechanics and Micromechanics. 2011. link Times cited: 2 Abstract: Atomistic simulations have a unique capability to reveal the… read moreAbstract: Atomistic simulations have a unique capability to reveal the material deformation mechanisms and the corresponding deformation-based constitutive behavior. However, atomistic simulations are limited by the accessible length and time scales. In the present work, an equivalent crystal lattice method is used to analyze atomistic mechanical deformation of nanometer- to micrometer-sized polycrystalline silicon (Si) samples at accelerated time steps. The equivalent crystal lattice method’s validity is verified by the results of classical molecular dynamics (MD) simulations at MD strain rates. The method is then used to predict material behavior at subcontinuum length scales. An extrapolation of the thin film polycrystalline silicon stress-strain relationships to lower strain-rate values indicates that the thin film peak stress values at the experimental strain rates are in agreement with experimental values. Analyses reveal that the peak stress values in the case of polycrystalline Si follow inverse Hall-Petch ... read less NOT USED (low confidence) W. Ge et al., “Meso-scale oriented simulation towards virtual process engineering (VPE)-The EMMS Paradigm,” Chemical Engineering Science. 2011. link Times cited: 126 NOT USED (low confidence) C.-ying Wang, Z. Wang, and Q. Meng, “Comparative study of the empirical interatomic potentials and density-functional simulations of divacancy and hexavacancy in silicon,” Physica B-condensed Matter. 2011. link Times cited: 3 NOT USED (low confidence) M. Timonova and B. Thijsse, “Thermodynamic properties and phase transitions of silicon using a new MEAM potential,” Computational Materials Science. 2010. link Times cited: 13 NOT USED (low confidence) A. V. der Ven, J. Thomas, Q. Xu, and J. Bhattacharya, “Linking the electronic structure of solids to their thermodynamic and kinetic properties,” Math. Comput. Simul. 2010. link Times cited: 154 NOT USED (low confidence) C. Wang, Y. Zhang, and Y. Jia, “New tetramer structures in the initial process of Si homoepitaxial growth on Si (0 0 1),” Applied Surface Science. 2009. link Times cited: 0 NOT USED (low confidence) L. Pelaz, L. Marqués, M. Aboy, P. López, and I. Santos, “Front-end process modeling in silicon,” The European Physical Journal B. 2009. link Times cited: 32 NOT USED (low confidence) S. Ghasemi et al., “The energy landscape of silicon systems and its description by force fields, tight binding schemes, density functional methods and Quantum Monte Carlo methods,” arXiv: Computational Physics. 2009. link Times cited: 27 Abstract: The accuracy of the energy landscape of silicon systems obta… read moreAbstract: The accuracy of the energy landscape of silicon systems obtained from various density functional methods, a tight binding scheme and force fields is studied. Quantum Monte Carlo results serve as quasi exact reference values. In addition to the well known accuracy of DFT methods for geometric ground states and metastable configurations we find that DFT methods give a similar accuracy for transition states and thus a good overall description of the energy landscape. On the other hand, force fields give a very poor description of the landscape that are in most cases too rugged and contain many fake local minima and saddle points or ones that have the wrong height. read less NOT USED (low confidence) C. Wang, Y. Zhang, and Y. Jia, “A new Si tetramer structure on Si (001),” Solid State Sciences. 2009. link Times cited: 4 NOT USED (low confidence) M. Posselt, F. Gao, and H. Bracht, “Correlation between self-diffusion in Si and the migration mechanisms of vacancies and self-interstitials: An atomistic study,” Physical Review B. 2008. link Times cited: 28 Abstract: The migration of point defects in silicon and the correspond… read moreAbstract: The migration of point defects in silicon and the corresponding atomic mobility are investigated by classical molecular dynamics simulations using the Stillinger-Weber potential and the Tersoff potential. In contrast to most of the previous studies both the point defect diffusivity and the self-diffusion coefficient per defect are calculated separately so that the diffusion-correlation factor can be determined. Simulations with both the Stillinger-Weber and the Tersoff potential show that vacancy migration is characterized by the transformation of the tetrahedral vacancy to the split vacancy and vice versa and the diffusion-correlation factor is about 0.5. This value was also derived by the statistical diffusion theory under the assumption of the same migration mechanism. The mechanisms of self-interstitial migration are more complex. The detailed study, including a visual analysis and investigations with the nudged elastic band method, reveals a variety of transformations between different self-interstitial configurations. Molecular dynamics simulations using the Stillinger-Weber potential show, that the self-interstitial migration is dominated by a dumbbell mechanism, whereas the interstitialcy mechanism prevails with the Tersoff potental. The corresponding values of the correlation factor are different, namely 0.59 and 0.69 for the dumbbell and the interstitialcy mechanism, respectively. The latter value is nearly equal to that obtainedmore » by the statistical theory which assumes the interstitialcy mechanism. Recent analysis of experimental results demonstrated, that in the framework of state-of-the-art diffusion and reaction models the best interpretation of point defect data can be given by assuming . The comparison with the present atomistic study leads to the conclusion that a dumbbell mechanism governs the self-interstitial migration in Si. Simulations using the Stillinger-Weber potential reveal two dominating migration paths which are characterized by transformation between the extended dumbbell and the dumbbell and vice versa. This process occurs either in a single {110} plane or includes a change into an equivalent {110} plane.« less read less NOT USED (low confidence) T. Kouno and S. Ogata, “Activation Energy for Oxygen Diffusion in Strained Silicon : A Hybrid Quantum-Classical Simulation Study with the Nudged Elastic Band Method(Condensed matter : electronic structure and electrical, magnetic, and optical properties),” Journal of the Physical Society of Japan. 2008. link Times cited: 7 Abstract: The activation energy for oxygen diffusion in strained silic… read moreAbstract: The activation energy for oxygen diffusion in strained silicon crystal is investigated using the hybrid quantum-classical simulation scheme in combination with the nudged elastic band method. The electronic density-functional theory is applied to a local region containing the oxygen atom, while the classical inter-atomic potential, to the rest of the system. The system is stretched to three mutually perpendicular directions at a wide range of degree between -2 and 9%. We thereby find that the activation energy changes by between -0.4 and 0.2 eV depending sensitively on both direction and degree of the stretch, and that the peripheral atoms located far from the oxygen atom in the system contribute little to the change. Microscopic mechanisms of the strain-dependence of the activation energy are elucidated through combined analyses about the atomic and electronic structures. read less NOT USED (low confidence) K. Yan and A. Soh, “Simulation of surface effects in energy dissipation of ultra-high-frequency (UHF) nanocantilevers,” SPIE Smart Structures and Materials + Nondestructive Evaluation and Health Monitoring. 2008. link Times cited: 0 Abstract: Devices composed of nanoelectromechanical systems (NEMS) pos… read moreAbstract: Devices composed of nanoelectromechanical systems (NEMS) possess distinguished properties which make them quite suitable for a variety of applications including ultra-high-frequency (UHF) resonators. However, most GHz resonators have low quality factor even though it has been well above 103 ~ 105 for very-high-frequency (VHF) microresonators. The motivation for our investigation of single crystal silicon nanoresonator arises from both its technological importance and its extraordinary surface effects. Our simulation results show that the quality factor decreased in a nearly linear manner as the surface area to volume ratio (SVR) was increased, which suggests that surface losses play a significant role in determining the quality factor of nanoresonators. read less NOT USED (low confidence) E.-H. Kim, Y.-H. Shin, and B.-J. Lee, “A modified embedded-atom method interatomic potential for Germanium,” Calphad-computer Coupling of Phase Diagrams and Thermochemistry. 2008. link Times cited: 86 NOT USED (low confidence) P. Śpiewak et al., “Molecular dynamics simulation of intrinsic point defects in germanium,” Journal of Crystal Growth. 2007. link Times cited: 14 NOT USED (low confidence) T. Kumagai, S. Izumi, S. Hara, and S. Sakai, “Development of bond-order potentials that can reproduce the elastic constants and melting point of silicon for classical molecular dynamics simulation,” Computational Materials Science. 2007. link Times cited: 148 NOT USED (low confidence) S. Munetoh, T. Motooka, K. Moriguchi, and A. Shintani, “Interatomic potential for Si–O systems using Tersoff parameterization,” Computational Materials Science. 2007. link Times cited: 382 NOT USED (low confidence) V. Ivashchenko, P. Turchi, and V. Shevchenko, “Simulations of the mechanical properties of crystalline, nanocrystalline, and amorphous SiC and Si,” Physical Review B. 2007. link Times cited: 84 Abstract: Molecular-dynamics simulations of crystalline (c), nanocryst… read moreAbstract: Molecular-dynamics simulations of crystalline (c), nanocrystalline (nc), and amorphous (a) silicon carbides and silicon were carried out to investigate their vibrational and mechanical properties. The atomic configurations, vibrational spectra, and stress-strain curves were calculated at room temperature. In the case of the nanocrystalline structures, these characteristics were analyzed as functions of grain size. Young's and bulk modul and yield and flow stresses were determined from uniaxial deformation of samples under periodic boundary constraints and from experiments on rod extension. For silicon carbides, Young's modulus and flow stress decrease in the sequence ``c-nc-a,'' and with decreasing grain size, which is attributed to a weakening of the Si--C bonds in the amorphous matrix. The enhancement of the strength properties of the homopolar nc--Si structures is attributed to their deformation anisotropy. The calculated vibrational spectra and Young's moduli are in rather good agreement with the corresponding experimental characteristics. read less NOT USED (low confidence) M. Timonova, B.-J. Lee, and B. Thijsse, “Sputter erosion of Si(001) using a new silicon MEAM potential and different thermostats,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 32 NOT USED (low confidence) S. Bukkapatnam, M. Malshe, P. Agrawal, L. Raff, and R. Komanduri, “Parametrization of interatomic potential functions using a genetic algorithm accelerated with a neural network,” Physical Review B. 2006. link Times cited: 17 NOT USED (low confidence) S. Wang et al., “THE CALCULATION OF THE SURFACE ENERGY OF HIGH-INDEX SURFACES OF SILICON AT ZERO TEMPERATURE,” Surface Review and Letters. 2006. link Times cited: 1 Abstract: We used the molecular dynamics simulation based on the Still… read moreAbstract: We used the molecular dynamics simulation based on the Stillinger–Weber (SW) interatomic potential to calculate the high-index surface energies of surfaces containing any of the stereographic surfaces of silicon at zero temperature. An empirical formula based on the structural unit model was generalized for high-index surfaces. Our simulated results show that the generalized formula can give a good estimation of the surface energy and structural feature of the high-index surfaces not only on the edge of stereographic but also within it. Our simulation and empirical formula results reveal that the closest surface has the lowest energy, so the closest (101) surface has the lowest surface energy and the (101), (111) and (001) surfaces are the extremum on the curve of surface energy versus orientation angle. Both the theoretical simulation results and the empirical formula calculation results are consistent with the available first-principles theoretical data. read less NOT USED (low confidence) P. Śpiewak et al., “Simulation of intrinsic point defect properties and vacancy clustering during Czochralski germanium crystal growth,” Materials Science in Semiconductor Processing. 2006. link Times cited: 8 NOT USED (low confidence) M. Demkowicz and A. Argon, “Liquidlike atomic environments act as plasticity carriers in amorphous silicon,” Physical Review B. 2005. link Times cited: 88 NOT USED (low confidence) M. Demkowicz and A. Argon, “Autocatalytic avalanches of unit inelastic shearing events are the mechanism of plastic deformation in amorphous silicon,” Physical Review B. 2005. link Times cited: 54 NOT USED (low confidence) D. Murdick, X. W. Zhou, and H. Wadley, “Assessment of interatomic potentials for molecular dynamics simulations of GaAs deposition,” Physical Review B. 2005. link Times cited: 22 Abstract: Computational studies of atomic assembly processes during Ga… read moreAbstract: Computational studies of atomic assembly processes during GaAs vapor deposition require interatomic potentials that are able to reasonably predict the structures and energies of a molecular arsenic vapor, a variety of elemental gallium and arsenic lattices, binary GaAs lattices, GaAs lattice defects, and 001 GaAs surfaces. These properties were systematically evaluated and compared to ab initio and experimental data for one Tersoff and two Stillinger-Weber SW GaAs interatomic potentials. It was observed that bulk and arsenic molecular properties calculated by the Tersoff parametrization matched density functional predictions and experimental observations significantly better than either of the SW parametrizations. These trends can be related to the bonding physics included in each potential format. Surface free energy calculations indicate that none of these potentials correctly predict the low-energy surface reconstructions of the GaAs 001 surface. Simulated As2 molecular bonding with gallium-rich GaAs 001 surfaces indicate a high sticking probability for SW potentials, which is in good agreement with experimental observations at low growth temperatures. However, the Tersoff parametrization resulted in an unphysically high desorption probability for As2 over a wide range of surface temperatures. read less NOT USED (low confidence) R. Wagner and E. Gulari, “Thermodynamic control of germanium quantum dot growth on silicon,” Surface Science. 2005. link Times cited: 5 NOT USED (low confidence) L. Zhang and J. Feng, “Molecular-dynamics simulation of germanium film growth by cluster deposition,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 4 NOT USED (low confidence) M. Posselt, F. Gao, and D. Zwicker, “Atomistic Study of the Migration of Di- and Tri-Interstitials in Silicon,” Physical Review B. 2005. link Times cited: 40 Abstract: A comprehensive study on the migration of di- and tri-inters… read moreAbstract: A comprehensive study on the migration of di- and tri-interstitials in silicon is performed using classical molecular dynamics simulations with a Stillinger-Weber potential. At first the structures and energetics of the di- and the tri-interstitial are investigated, and the accuracy of the interatomic potential is tested by comparing the results with literature data obtained by tight-binding and density-functional-theory calculations. Then the migration is investigated for temperatures between 800 and 1600 K. Very long simulation times, large computational cells and different initial conditions are considered. The defect diffusivity, the self-diffusion coefficient per defect and the corresponding effective migration barriers are calculated. Compared to a mono-interstitial, the di-interstitial migrates faster, whereas the tri-interstitial diffuses slower. The mobility of the di- and the mono-interstitial is higher than the mobility of the lattice atoms during the diffusion of these defects. On the other hand, the tri-interstitial mobility is lower than the corresponding atomic mobility. The migration mechanism of the di-interstitial shows a pronounced dependence on the temperature. At low temperature a high mobility on zigzag-like lines along a axis within a {l_brace}110{r_brace} plane is found, whereas the change between equivalent directions or equivalent {l_brace}110{r_brace} planes occurs seldomly and requires a long simulationmore » time, but the rate of directional change increases with increasing temperature. During the diffusion within {l_brace}110{r_brace} planes the di-interstitial moves like a wave packet so that the atomic mobility is lower than that of the defect. On the other hand, the change between equivalent {l_brace}110{r_brace} migration planes is characterized by frequent atomic rearrangements. The visual analysis of the tri-interstitial diffusion reveals complex migration mechanisms and a high atomic mobility. The diffusivities and effective migration barriers obtained are compared with the few data from the literature. The implications of the present results for the explanation of experimental data on defect evolution and migration are discussed.« less read less NOT USED (low confidence) S. G. Mayr and R. Averback, “Ion-irradiation-induced stresses and swelling in amorphous Ge thin films,” Physical Review B. 2005. link Times cited: 33 Abstract: Mechanical stresses and morphology during growth and ion bom… read moreAbstract: Mechanical stresses and morphology during growth and ion bombardment of amorphous Ge thin films are investigated by a combination of in situ stress measurements and molecular dynamics computer simulations. Strong compressive stresses are generated during irradiation that subsequently lead to severe swelling. The simulations indicate that interstitial-mediated viscous flow in combination with well-localized vacancy defects are the main ingredients responsible for the observed phenomena. read less NOT USED (low confidence) S. Zhang and N. Chen, “Lattice inversion for interatomic potentials in AlN, GaN and InN,” Chemical Physics. 2005. link Times cited: 23 NOT USED (low confidence) Y. Mo, M. Bazant, and E. Kaxiras, “Sulfur point defects in crystalline and amorphous silicon,” Physical Review B. 2004. link Times cited: 44 Abstract: We present first-principles calculations for the behavior of… read moreAbstract: We present first-principles calculations for the behavior of sulfur point defects in crystalline and amorphous silicon structures. By introducing the sulfur point defects at various representative positions in the samples, including substitutional and interstitial sites in the crystal and fourfold coordinated or miscoordinated sites (dangling bond and floating bond sites ) in the amorphous, we analyze the energetics in detail and determine the most stable structures. Two important conclusions we draw are: (a) in crystalline Si, the S defects form pairs in which the two S atoms are energetically bound but not covalently bonded; (b) in amorphous Si, they preferentially occupy threefold coordinated sites, even when the starting configuration has higher coordination (four- or fivefold). The implications of these results for the electronic structure of sulfur-doped Si samples are also analyzed in the context of the present calculations. read less NOT USED (low confidence) J. Tarus and K. Nordlund, “Molecular dynamics simulation of Ge surface segregation,” Thin Solid Films. 2004. link Times cited: 4 NOT USED (low confidence) S. Izumi, Y. Sato, S. Hara, and S. Sakai, “Development of a molecular dynamics potential for Si–H systems and its application to CVD reaction processes,” Surface Science. 2004. link Times cited: 6 NOT USED (low confidence) B. Thijsse, T. Klaver, and E. Haddeman, “Molecular Dynamics simulation of silicon sputtering: sensitivity to the choice of potential,” Applied Surface Science. 2004. link Times cited: 24 NOT USED (low confidence) J. Tarus and G. Zollo, “Ion-irradiation-induced effects inSimGensuperlattices,” Physical Review B. 2004. link Times cited: 2 NOT USED (low confidence) P. Gunes, Şi̇mşek S., and S. Erkoç, “a Comparative Study of Empirical Potential Energy Functions,” International Journal of Modern Physics C. 2004. link Times cited: 2 Abstract: A comparative study has been performed for silicon microclus… read moreAbstract: A comparative study has been performed for silicon microclusters, Si3 and Si4, considering fifteen different empirical potential energy functions. It has been found that only two of the empirical potential energy functions give linear structure more stable for Si3, the remaining potential functions give triangular structure as more stable. In the case of Si4 microclusters eight potential functions give open tetrahedral structure as more stable, two functions give perfect tetrahedral as more stable, three functions give square structure as more stable, and two functions give linear structure as more stable. read less NOT USED (low confidence) T. Hawa and M. Zachariah, “Molecular dynamics study of particle-particle collisions between hydrogen-passivated silicon nanoparticles,” Physical Review B. 2004. link Times cited: 39 Abstract: One of the significant challenges in the use of nanoparticle… read moreAbstract: One of the significant challenges in the use of nanoparticles is the control of primary particle size and extent of agglomeration when grown from the gas phase. In this paper we evaluate a possible strategy of surface passivation. Here the particle--particle interaction of hydrogen-surface-terminated silicon nanoparticles has been evaluated using molecular dynamics simulation. Nanoparticles of the size between 200 and 6400 silicon atoms at 300--1800 K were studied with a reparametrized Kohen-Tully-Stillinger empirical interatomic potential. A hydrogen monolayer is shown to prevent coalescence between particles under thermal collision conditions. The critical approach energy for coalescence was found to increase with increasing particle size but decreases with increasing temperature. Both solid and liquid droplets were seen to bounce at thermal energies, and in some cases, ``superelastic'' collisions are observed, where the rebound kinetic energy of the droplet is higher than the approach energy. These results suggest that surface coatings can significantly retard nanoaerosol growth. read less NOT USED (low confidence) J. Tarus and K. Nordlund, “Molecular dynamics study on Si20 cluster deposition on Si(0 0 1),” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2003. link Times cited: 8 NOT USED (low confidence) K. Garikipati, “Couple stresses in crystalline solids: origins from plastic slip gradients, dislocation core distortions, and three-body interatomic potentials,” Journal of The Mechanics and Physics of Solids. 2003. link Times cited: 17 NOT USED (low confidence) M. Makeev and A. Madhukar, “Stress and strain fields from an array of spherical inclusions in semi-infinite elastic media: Ge nanoinclusions in Si,” Physical Review B. 2003. link Times cited: 13 Abstract: Atomically resolved stress and strain fields from arrays of … read moreAbstract: Atomically resolved stress and strain fields from arrays of laterally ordered spherical Ge nanoinclusions in a semi-infinite Si(001) matrix are studied via atomistic simulations. We find that the hydrostatic stress and strain on the Si(001) matrix surface, induced by the inclusion buried at depth d, are tensile and follow the inverse cubic dependence for both small and intermediate d. The magnitudes of the stress and strain fields from inclusions of different volumes are found to be nearly proportional to the volume of the inclusion for large radii of inclusions, while for small radii the volume dependence overestimates the effect. Furthermore, we find that the magnitude of the stress and strain fields on the matrix surface is nearly proportional to the lattice mismatch between the inclusion and host material. The obtained simulation results are compared with the predictions of continuum-elasticity-based models and an overall good agreement is found. read less NOT USED (low confidence) S. Erkoç, K. Leblebicioğlu, and U. Halici, “Application of Genetic Algorithms to Geometry Optimization of Microclusters: A Comparative Study of Empirical Potential Energy Functions for Silicon,” Materials and Manufacturing Processes. 2003. link Times cited: 14 Abstract: Evolutionary computation techniques (in particular, genetic … read moreAbstract: Evolutionary computation techniques (in particular, genetic algorithms) have been applied to optimize the structure of microclusters. Various empirical potential energy functions have been used to describe the interactions among the atoms in the clusters. A comparative study of silicon microclusters has been performed. read less NOT USED (low confidence) Y. Kadiri, N. Jakse, J. Wax, and J. Bretonnet, “Structure factor and atomic dynamics of stable and supercooled liquid silicon by molecular dynamics,” Journal of Non-crystalline Solids. 2002. link Times cited: 4 NOT USED (low confidence) P. Keblinski, M. Bazant, R. Dash, and M. Treacy, “Thermodynamic behavior of a model covalent material described by the environment-dependent interatomic potential,” Physical Review B. 2002. link Times cited: 38 Abstract: Using molecular-dynamics simulations we study the thermodyna… read moreAbstract: Using molecular-dynamics simulations we study the thermodynamic behavior of a single-component covalent material described by the recently proposed environment-dependent interatomic potential (EDIP). The parametrization of EDIP for silicon exhibits a range of unusual properties typically found in more complex materials, such as the existence of two structurally distinct disordered phases, a density increase upon melting of the low-temperature amorphous phase, and negative thermal-expansion coefficients for both the crystal (at high temperatures) and the amorphous phase (at all temperatures). Structural differences between the two disordered phases also lead to a first-order transition between them, which suggests the existence of a second critical point, as is believed to exist for amorphous forms of frozen water. For EDIP-Si, however, the unusual behavior is associated not only with the open nature of tetrahedral bonding but also with a competition between fourfold (covalent) and fivefold (metallic) coordination. The unusual behavior of the model and its unique ability to simulate the liquid/amorphous transition on molecular-dynamics time scales make it a suitable prototype for fundamental studies of anomalous thermodynamics in disordered systems. read less NOT USED (low confidence) A. Barnard and S. Russo, “Development of an improved Stillinger-Weber potential for tetrahedral carbon using ab initio (Hartree-Fock and MP2) methods,” Molecular Physics. 2002. link Times cited: 28 Abstract: An improved interatomic potential for tetrahedral carbon is … read moreAbstract: An improved interatomic potential for tetrahedral carbon is presented. This potential is of the Stillinger-Weber (SW) type and has been determined from calculations performed on a select group of small hydrocarbon molecules, chosen for their similarities to the tetrahedral lattice of bulk diamond. Counterpoise corrected Hartree-Fock (HF) and second-order Møller-Plesset perturbation theory (MP2) calculations were performed on ethane, 2,2-dimethylpropane (neo-pentane, (C5H12), 2-dimethyl-3-dimethylbutane (neobutane, C8H18) and cyclohexane (C6H12) in order to determine the two-body (stretching) and three-body (bond bending) energies. The suitability of these molecules to model the properties of diamond was determined by comparison of CC bond length, well depth, CCC bond angle, simultaneous stretch and bend energy and force constants to those of bulk diamond. It was found that neopentane provided the best overall description of tetrahedral bonded carbon. The ab initio derived stretch and bend energies were fitted to the SW potential energy terms and the SW parameters calculated. The newly parametrized SW potential was then evaluated by calculating the stretch force constants, elastic constants and the X-point phonon modes of bulk diamond. read less NOT USED (low confidence) K. Scheerschmidt, D. Conrad, and A. Belov, “Atomic processes at bonded Si-interfaces studied by molecular dynamics: tayloring densities and bandgaps?,” Computational Materials Science. 2002. link Times cited: 4 NOT USED (low confidence) N. Chaâbane, H. Vach, and G. H. Peslherbe, “Complex dynamics during the reactive scattering of Si+ (2P) and H2,” Journal of Non-crystalline Solids. 2002. link Times cited: 2 NOT USED (low confidence) K. Nordlund, “Computational materials science of ion irradiation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2002. link Times cited: 18 NOT USED (low confidence) M. Koster and H. Urbassek, “Atomistic simulation of stress effects in a-Si due to low-energy Si impact,” Surface Science. 2002. link Times cited: 12 NOT USED (low confidence) G. Smith, E. Tadmor, N. Bernstein, and E. Kaxiras, “Multiscale simulations of silicon nanoindentation,” Acta Materialia. 2001. link Times cited: 106 NOT USED (low confidence) X. P. Xie, M. Liang, Z. M. Choo, and S. Li, “A COMPARATIVE SIMULATION STUDY OF SILICON (001) SURFACE RECONSTRUCTION USING DIFFERENT INTERATOMIC POTENTIALS,” Surface Review and Letters. 2001. link Times cited: 3 Abstract: We have performed a comparative study of Si(001) surface rec… read moreAbstract: We have performed a comparative study of Si(001) surface reconstruction employing molecular dynamics simulation using the interatomic potentials of Stillinger–Weber, Tersoff and Bazant–Kaxiras. Simulations were carried out for temperatures at 300 K and 1000 K using each of these three potentials. At 300 K, the three potentials were found to generate surface features comprising mainly the simple (2 × 1) reconstruction. At 1000 K, more complex reconstruction similar to the p(2 × 2) and c(2 × 2) patterns was observed on the surfaces of Stillinger–Weber and Tersoff crystals while the surface generated on Bazant–Kaxiras crystal is characterized by disorderliness with no identifiable pattern of reconstruction. read less NOT USED (low confidence) M. Mazzarolo, L. Colombo, G. Lulli, and E. Albertazzi, “Low-energy recoils in crystalline silicon: Quantum simulations,” Physical Review B. 2001. link Times cited: 19 NOT USED (low confidence) G. Ackland, “High-pressure phases of group IV and III-V semiconductors,” Reports on Progress in Physics. 2001. link Times cited: 212 Abstract: The currently known structures and properties of group?IV el… read moreAbstract: The currently known structures and properties of group?IV elements and III-V compounds at high pressure are reviewed. Structural properties of various phases, as determined by experimental techniques, predominantly x-ray crystallography using diamond anvil cells, are covered first. The relative equilibrium stability of these phases, as determined by theoretical methods, is also discussed. Metastable phases and the processing techniques by which they can be made are examined, introducing the importance of phase transition kinetics in determining what is actually seen. Elastic and vibrational properties are then considered, looking at how elastic constants and phonon frequencies are affected by increasing pressure and how this can help us to understand the phase diagram and transition kinetics. Finally, using these ideas, it is shown how one can formulate equilibrium pressure-temperature equations of state for these materials. Throughout, the review draws on both experimental and theoretical work, and emphasizes features which seem to be generic to these tetrahedral semiconductors and their high-pressure phases. read less NOT USED (low confidence) R. Vink, G. Barkema, W. F. Weg, and N. Mousseau, “Fitting the Stillinger–Weber potential to amorphous silicon,” Journal of Non-crystalline Solids. 2001. link Times cited: 137 NOT USED (low confidence) J. Cai and J.-S. Wang, “Reconstruction of Si(001) : A comparison study of many body potential calculations,” Physica Status Solidi B-basic Solid State Physics. 2001. link Times cited: 2 Abstract: The Tersoff potential and a recently developed potential fun… read moreAbstract: The Tersoff potential and a recently developed potential function for covalent materials (phys. stat. sol. (b) 212, 9 (1999)) are used to simulate the reconstruction of Si(001) surface. We obtain a dimered (2 x 1) reconstruction with an asymmetric rearrangement of atoms in deeper layers in Z-direction using Tersoffs potential and an asymmetric buckled dimered (2 × 1) reconstruction using the recently developed potential. The latter is in agreement with results from the first principles calculations or experiments. read less NOT USED (low confidence) R. E. Miller and V. Shenoy, “SIZE-DEPENDENT ELASTIC PROPERTIES OF NANOSIZED STRUCTURAL ELEMENTS,” Nanotechnology. 2000. link Times cited: 1762 Abstract: Effective stiffness properties (D) of nanosized structural e… read moreAbstract: Effective stiffness properties (D) of nanosized structural elements such as plates and beams differ from those predicted by standard continuum mechanics (Dc). These differences (D-Dc)/Dc depend on the size of the structural element. A simple model is constructed to predict this size dependence of the effective properties. The important length scale in the problem is identified to be the ratio of the surface elastic modulus to the elastic modulus of the bulk. In general, the non-dimensional difference in the elastic properties from continuum predictions (D-Dc)/Dc is found to scale as αS/Eh, where α is a constant which depends on the geometry of the structural element considered, S is a surface elastic constant, E is a bulk elastic modulus and h a length defining the size of the structural element. Thus, the quantity S/E is identified as a material length scale for elasticity of nanosized structures. The model is compared with direct atomistic simulations of nanoscale structures using the embedded atom method for FCC Al and the Stillinger-Weber model of Si. Excellent agreement between the simulations and the model is found. read less NOT USED (low confidence) K. Nordlund, J. Nord, J. Frantz, and J. Keinonen, “Strain-induced Kirkendall mixing at semiconductor interfaces,” Computational Materials Science. 2000. link Times cited: 48 NOT USED (low confidence) T. Sinno, E. Dornberger, W. Ammon, R. A. Brown, and F. Dupret, “Defect engineering of Czochralski single-crystal silicon,” Materials Science & Engineering R-reports. 2000. link Times cited: 112 NOT USED (low confidence) M. Mäki-Jaskari, K. Kaski, and A. Kuronen, “Simulations of crack initiation in silicon,” Computational Materials Science. 2000. link Times cited: 6 NOT USED (low confidence) Pérez-Martı́n A., J. Domínguez-Vázquez, and Jiménez-Rodrı́guez J. J., “A MD study of low energy boron bombardment on silicon.,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2000. link Times cited: 11 NOT USED (low confidence) T. Halicioǧlu and D. Barnett, “Formation and migration energies of interstitials in silicon under strain conditions,” Surface Science. 1999. link Times cited: 5 NOT USED (low confidence) J. F. Justo, V. Bulatov, and S. Yip, “DISLOCATION CORE RECONSTRUCTION AND ITS EFFECT ON DISLOCATION MOBILITY IN SILICON,” Journal of Applied Physics. 1999. link Times cited: 31 Abstract: Through atomistic calculations of kink nucleation and migrat… read moreAbstract: Through atomistic calculations of kink nucleation and migration in the core of partial dislocations in silicon we demonstrate that symmetry-breaking structural reconstructions will strongly affect dislocation mobility. Core reconstructions give rise to multiple kink species, and, relative to kinks in an unreconstructed dislocation, an increase in kink formation and migration energies. These factors provide additional resistance to dislocation motion which scales with the energy reconstruction. Our results indicate that the observed variations of dislocation mobility in going from elemental to IV–IV, and further to III–V and II–VI zinc-blende semiconductors, can be attributed in part to the weakening of core reconstruction across the series. read less NOT USED (low confidence) A. Natori and H. Harada, “Surface melting of vicinal Si(111) surfaces,” Surface Science. 1999. link Times cited: 5 NOT USED (low confidence) A. Dyson and P. Smith, “Improved empirical interatomic potential for C—Si—H systems,” Molecular Physics. 1999. link Times cited: 30 Abstract: The Brenner hydrocarbon potential was extended recently to i… read moreAbstract: The Brenner hydrocarbon potential was extended recently to include interactions with silicon. This extended Brenner potential has now been improved by the fitting of bond order correction terms, and the introduction of an adjustable parameter into the angular function. The new potential gives an excellent description of small Si m H n molecules and radicals. Its treatment of the low index surfaces of silicon and β-SiC is also significantly improved, although the recently proposed non-dimerized structure for the silicon terminated (001) surface of β-SiC is not described properly. Calculations of the chemisorption of C2H2 and CH3 onto the (001) surfaces of silicon and β-SiC using this improved potential are reported. Also presented are some initial results of molecular dynamics simulations of the Si(111) 7 × 7:CH3 and hydrogenated Si(001) 2 × 1:C2H2 chemisorption systems. read less NOT USED (low confidence) P. Klein and H. Gao, “Crack nucleation and growth as strain localization in a virtual-bond continuum,” Engineering Fracture Mechanics. 1998. link Times cited: 185 NOT USED (low confidence) K. Moriguchi and A. Shintani, “VERIFICATION OF TERSOFF’S POTENTIAL FOR STATIC STRUCTURAL ANALYSIS OF SOLIDS OF GROUP-IV ELEMENTS,” Japanese Journal of Applied Physics. 1998. link Times cited: 24 Abstract: We have carried out several stringent tests of Tersoff'… read moreAbstract: We have carried out several stringent tests of Tersoff's potential through static structural analysis of solids of group-IV elements and examined the chemical properties of the Tersoff's potential for C, Si and Ge. It is clear that Tersoff's potential has a limited ability to describe the differences in chemical reactivity between elemental Ge and Si, but well describes the chemical differences of valence s and p electron properties between elemental C and Si. Tersoff's potential has higher force constants for angular distortions than those seen in actual systems. We assume that the well-known higher melting points of Tersoff's potential are due to the larger force constants for angular distortions. The calculated elastic constants and Poisson ratios indicate congruity with the experimental values in every element. Tersoff's potential has good transferability and will be an effective tool for evaluating new materials composed of group-IV elements. read less NOT USED (low confidence) A. Dyson and P. V. Smith, “Empirical potential study of the chemisorption of C2H2 and CH3 on the β-SiC(001) surface,” Surface Science. 1998. link Times cited: 26 NOT USED (low confidence) R. Averback and T. D. Rubia, “Displacement damage in irradiated metals and semiconductors,” Journal of Physics C: Solid State Physics. 1997. link Times cited: 310 NOT USED (low confidence) L. Marqués, M. Jaraíz, J. Rubio, J. Vicente, L. Bailón, and J. Barbolla, “Molecular dynamics simulations of ion bombardment processes,” Materials Science and Technology. 1997. link Times cited: 3 Abstract: AbstractAn improved molecular dynamics technique that allows… read moreAbstract: AbstractAn improved molecular dynamics technique that allows reduction of the computation time required in ion bombardment simulations is presented. This technique has been used to study the influence of the target temperature and structure on the argon sputtering of silicon. Molecular dynamics simulations of l keV Ar+ ion bombardment of silicon were carried out for several types of sample: (100) crystalline at 0 K, (100) crystalline at 300 K, and amorphous at 300 K. The yield of the sputtering process and the energy distribution of the sputtered atoms have been obtained. These results show that the sputtering process depends on the target surface binding energy which, in turn, is very sensitive to the structure of the sample surface. read less NOT USED (low confidence) S. A. Fedotov, A. A. Efimchik, and A. Byeli, “DLC growth by ion beam assisted deposition : a molecular simulation,” Diamond and Related Materials. 1997. link Times cited: 8 NOT USED (low confidence) H. Hensel and H. Urbassek, “Disordering and annealing of a Si surface under low-energy Si bombardment,” Radiation Effects and Defects in Solids. 1997. link Times cited: 4 Abstract: We simulated the bombardment of Si(100)(2 × 1) by Si atoms u… read moreAbstract: We simulated the bombardment of Si(100)(2 × 1) by Si atoms using molecular dynamics. The kinetic energies of the projectiles were 100 and 50 eV. To model the Si–Si-interactions the empirical potential of Stillinger and Weber with the two body part of the potential splined to the universal potential was used. A geometric criterion based on the Lindemann radius was defined to study damage in the Si target. We observed clusters of disordered Si atoms in the target induced by the bombardment. Large clusters of about 50 atoms are formed in the beginning of the bombardment; they shrink and decay into smaller clusters until and equilibrium cluster size of about 10 atoms is reached. Upon annealing at elevated temperature the disordered zones dissolve into point defects. read less NOT USED (low confidence) M. Tang, L. Colombo, J. Zhu, and T. D. Rubia, “Intrinsic point defects in crystalline silicon: Tight-binding molecular dynamics studiesof self-diffusion, interstitial-vacancy recombination, and formation volumes,” Physical Review B. 1997. link Times cited: 236 Abstract: Tight-binding molecular dynamics simulations are performed t… read moreAbstract: Tight-binding molecular dynamics simulations are performed to study self-diffusion, interstitial-vacancy recombination, and formation volumes of point defects in crystalline silicon. The results show that (i) self-diffusion is dominated by vacancies (V) at low temperature and by interstitials (I) at high temperature; (ii) interstitial-vacancy recombination at room temperature leads to formation of a metastable I-V complex, which has an annihilation energy barrier of 1.1 eV; (iii) interstitial and vacancy relaxation volumes in silicon are approximately equal in magnitude and opposite in sign. {copyright} {ital 1997} {ital The American Physical Society} read less NOT USED (low confidence) R. Chatterjee and B. Garrison, “Pushing the limits of classical modeling of bombardment events in solids,” Radiation Effects and Defects in Solids. 1997. link Times cited: 5 Abstract: Bombardment of solids with keV atoms leads to violent collis… read moreAbstract: Bombardment of solids with keV atoms leads to violent collisions with subsequent ejection of target particles. This review discusses how classical molecular dynamics simulations designed to describe the bombardment events can give insight into microscopic processes where not only classical but also quantum effects such as electronic excitation and organic reactions play an important role. By incorporating a simple excitation/de-excitation model into the simulation, we have shown that collisional events are important to describe the distribution of excited state atoms measured experimentally. Molecular dynamics simulations employing a reactive many-body potential of small hydrocarbon molecules adsorbed on a metal surface predict the occurrence of various collision induced organic reactions prior to ejection. Lateral motion of particles in the region right above the surface plays an important role in signal enhancement. The calculations predict several processes such as direct molecular ejection, d... read less NOT USED (low confidence) R. Winkler and S. Pantelides, “Charge transfer and dipole moments of polyatomic systems,” Journal of Chemical Physics. 1997. link Times cited: 22 Abstract: Heteronuclear molecules have electric dipole moments because… read moreAbstract: Heteronuclear molecules have electric dipole moments because of electronic charge transfer among the constituent atoms. Quantum mechanical calculations reproduce the values of observed dipoles quite well, but easy-to-use model theories have so far failed to produce dipole moments in agreement with experiment. By combining density-functional theory and classical concepts, we obtain a simple predictive model for charge transfer which overcomes the shortcomings of earlier models based on the concept of electronegativity equalization. It yields dipole moments for many diatomic molecules and for the water molecule that are in satisfactory agreement with experiment. The model has promise as a supplement of classical molecular dynamics simulations for multicomponent polyatomic systems. read less NOT USED (low confidence) S. A. Fedotov, A. A. Efimchik, and A. Byeli, “Ion Beam Assisted Deposition: A Molecular Dynamics Simulation,” Materials and Manufacturing Processes. 1997. link Times cited: 2 Abstract: Molecular dynamics simulation of C film growth by ion assist… read moreAbstract: Molecular dynamics simulation of C film growth by ion assisted deposition is reported. The developed kinetic model of ion assistance offers a possibility to consider relatively low energies of assisting ions (lower than 1 keV) compared to traditional models. C-C atomic interactions are calculated using the Tersoff potential (1). Effects of deposition and assistance modes on the film properties are discussed. A conclusion is made that the Tersoff potential cannot provide accurate simulation of the low-density C phase formation, because it does not describe explicitly the passage between sp3-sp sp3 hybridizations. read less NOT USED (low confidence) R. Herrmann, J. Gerlach, and E. Campbell, “Molecular dynamics simulation of laser ablation of silicon,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1997. link Times cited: 27 NOT USED (low confidence) V. Bulatov, M. Nastar, J. F. Justo, and S. Yip, “Atomistic modeling of crystal-defect mobility and interactions,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1997. link Times cited: 3 NOT USED (low confidence) Yu, Wang, and Stroud, “Empirical molecular-dynamics study of diffusion in liquid semiconductors.,” Physical review. B, Condensed matter. 1996. link Times cited: 46 Abstract: We report the results of an extensive molecular-dynamics stu… read moreAbstract: We report the results of an extensive molecular-dynamics study of diffusion in liquid Si and Ge (l-Si and l-Ge) and of impurities in l-Ge, using empirical Stillinger-Weber (SW) potentials with several choices of parameters. We use a numerical algorithm in which the three-body part of the SW potential is decomposed into products of two-body potentials, thereby permitting the study of large systems. One choice of SW parameters agrees very well with the observed l-Ge structure factors. The diffusion coefficients D(T) at melting are found to be approximately 6.4\ifmmode\times\else\texttimes\fi{}${10}^{\mathrm{\ensuremath{-}}5}$ ${\mathrm{cm}}^{2}$/s for l-Si, in good agreement with previous calculations, and about 4.2\ifmmode\times\else\texttimes\fi{}${10}^{\mathrm{\ensuremath{-}}5}$ and 4.6\ifmmode\times\else\texttimes\fi{}${10}^{\mathrm{\ensuremath{-}}5}$ ${\mathrm{cm}}^{2}$/s for two models of l-Ge. In all cases, D(T) can be fitted to an activated temperature dependence, with activation energies ${\mathit{E}}_{\mathit{d}}$ of about 0.42 eV for l-Si, and 0.32 or 0.26 eV for two models of l-Ge, as calculated from either the Einstein relation or from a Green-Kubo-type integration of the velocity autocorrelation function. D(T) for Si impurities in l-Ge is found to be very similar to the self-diffusion coefficient of l-Ge. We briefly discuss possible reasons why the SW potentials give D(T)'s substantially lower than ab initio predictions. \textcopyright{} 1996 The American Physical Society. read less NOT USED (low confidence) P. Stephenson, M. Radny, and P. V. Smith, “A modified Stillinger-Weber potential for modelling silicon surfaces,” Surface Science. 1996. link Times cited: 19 NOT USED (low confidence) E. Kaxiras, “Review of atomistic simulations of surface diffusion and growth on semiconductors,” Computational Materials Science. 1996. link Times cited: 17 NOT USED (low confidence) A. Dyson and P. V. Smith, “Extension of the Brenner empirical interatomic potential to CSiH systems,” Surface Science. 1996. link Times cited: 70 NOT USED (low confidence) H.-C. Huang, N. Ghoniem, J. Wong, and M. Baskes, “Molecular dynamics determination of defect energetics in beta -SiC using three representative empirical potentials,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 102 Abstract: The determination of formation and migration energies of poi… read moreAbstract: The determination of formation and migration energies of point and clustered defects in SiC is of critical importance to a proper understanding of atomic phenomena in a wide range of applications. We present here calculations of formation and migration energies of a number of point and clustered defect configurations. A newly developed set of parameters for the modified embedded-atom method (MEAM) is presented. Detailed molecular dynamics calculations of defect energetics using three representative potentials, namely the Pearson potential, the Tersoff potential and the MEAM, have been performed. Results of the calculations are compared to first-principles calculations and to available experimental data. The results are analysed in terms of developing a consistent empirical interatomic potential and are used to discuss various atomic migration processes. read less NOT USED (low confidence) V. Bulatov, S. Yip, and A. Argon, “Atomic modes of dislocation mobility in silicon,” Philosophical Magazine. 1995. link Times cited: 128 Abstract: Mechanisms of partial dislocation mobility in the {111} glid… read moreAbstract: Mechanisms of partial dislocation mobility in the {111} glide system of silicon have been studied in full atomistic detail by applying novel effective relaxation and sampling algorithms in conjunction with the Stillinger-Weber empirical interatomic potential and simulation models of up to 90000 atoms. Low-energy pathways are determined for the generation, annihilation and motion of in-core defects of the 30°-partial dislocation, specifically, the individual left and right components of a double-kink, an antiphase defect (APD), and various kink-APD complexes. It is shown that the underlying mechanisms in these defect reactions fall into three distinct categories, characterized by the processes of bond-breaking, bond switching, and bond exchange, respectively. The quantitative results reveal a strong left-right asymmetry in the kinetics of kink propagation and a strong APD-kink binding; these have not been recognized previously and therefore hold implications for further experiments. The present wo... read less NOT USED (low confidence) A. A. Valuev, A. S. Kaklyugin, and H. E. Norman, “Molecular modelling of the chemical interaction of atoms and molecules with a surface,” Russian Chemical Reviews. 1995. link Times cited: 3 Abstract: The modelling of a surface as an assembly of moving atoms in… read moreAbstract: The modelling of a surface as an assembly of moving atoms interacting with one another and with an incident particle is examined. Both dynamic methods for the modelling of a surface (for short times) and probability methods (for long times) are analysed. The Massey adiabaticity criterion has been used to determine the regions of applicability of the methods of molecular dynamics. Within the framework of probability methods, the chemical bond is described with the aid of Harrison's generalised periodic system of the elements. Together with the general modeling problems, the reconstruction of the surface, physical and chemical sorption, as well as the modification of the surface and of its morphology as a result of the multiple repetition of elementary processes (precipitation, etching, corrosion) are discussed. The bibliography includes 169 references. read less NOT USED (low confidence) J. Crain, G. Ackland, and S. Clark, “Exotic structures of tetrahedral semiconductors,” Reports on Progress in Physics. 1995. link Times cited: 29 Abstract: Recent experimental and theoretical studies of exotic forms … read moreAbstract: Recent experimental and theoretical studies of exotic forms of tetrahedrally coordinated semiconductors are reviewed. These unusual phases are synthesized as long-lived metastable forms of the elemental semiconductors silicon and germanium by the application and subsequent removal of high pressure. Rather than being simply crystallographic oddities, the bonding arrangements in these phases show many similarities to those found in amorphous semiconductors. As a result, these dense structures have been used as so-called 'complex crystal' models for the amorphous state. Advances in experimental and computational techniques have recently allowed for detailed study of the structural, electronic and vibrational properties of these phases to be made under variable temperature and pressure conditions. In view of the considerable difficulties associated with performing theoretical studies of non-crystalline solids, the BC8 and ST12 structures are useful in that an understanding of their properties provides insight into the essential physics of amorphous tetrahedral semiconductors. read less NOT USED (low confidence) T. Ito, “RECENT PROGRESS IN COMPUTER-AIDED MATERIALS DESIGN FOR COMPOUND SEMICONDUCTORS,” Journal of Applied Physics. 1995. link Times cited: 50 Abstract: Recent progress in computational materials science in the ar… read moreAbstract: Recent progress in computational materials science in the area of semiconductor materials is reviewed. Reliable predictions can now be made for a wide range of problems, such as band structure and structural and thermodynamic properties of various compound semiconductors, using electronic theories such as the pseudopotential method. Further applications are examined by investigating the behavior of various atomic species in semiconductors, including the stability and band structure of heterostructures, superlattices, lattice defects, alloy systems, and surface‐related properties such as surface reconstruction, surface passivation, and adatom migration during thin film growth. The empirical interatomic potentials, pseudopotential, and stochastic Monte Carlo methods are used. An overview of these issues is provided and the latest achievements are presented to illustrate the capability of the theoretical‐computational approach by comparing experimental results. The constituents of the semiconductors that are... read less NOT USED (low confidence) A. Skinner and J. Broughton, “Neural networks in computational materials science: training algorithms,” Modelling and Simulation in Materials Science and Engineering. 1995. link Times cited: 70 Abstract: Neural networks can be used in principle in an unbiased way … read moreAbstract: Neural networks can be used in principle in an unbiased way for a multitude of pattern recognition and interpolation problems within computational material science. Reliably finding the weights of large feed-forward neural networks with both accuracy and speed is crucial to their use. In this paper, the rate of convergence of numerous optimization techniques that can be used to determine the weights is compared for two problems related to the construction of atomistic potentials. Techniques considered were back propagation (steepest descent), conjugate gradient methods, real-string genetic algorithms, simulated annealing and a new swarm search algorithm. For small networks, where only a few optimal solutions exist, we find that conjugate-gradient methods are most successful. However, for larger networks where the parameter space to be searched is more complex, a hybrid scheme is most effective; genetic algorithm or simulated annealing to find a good initial starting set of weights, followed by a conjugate-gradient approach to home in on a final solution. These hybrid approaches are now our method of choice for training large networks. read less NOT USED (low confidence) J. B. Adams et al., “Atomic-level computer simulation,” Journal of Nuclear Materials. 1994. link Times cited: 17 NOT USED (low confidence) H. Yan, X. Hu, and H. Jónsson, “Atomic structure of β-SiC( 100) surfaces: a study using the Tersoff potential,” Surface Science. 1994. link Times cited: 24 NOT USED (low confidence) M. Tang and S. Yip, “Lattice instability in β‐SiC and simulation of brittle fracture,” Journal of Applied Physics. 1994. link Times cited: 47 Abstract: Brittle fracture of β‐SiC (polytype 3C) under hydrostatic te… read moreAbstract: Brittle fracture of β‐SiC (polytype 3C) under hydrostatic tension has been modeled by molecular dynamics simulation using an interatomic potential function that treats the solid as fully covalent. The critical stress at which the lattice becomes structurally unstable is shown to agree quantitatively with that predicted by stability analysis based on elastic stiffness coefficients. The instability mode is the spinodal (vanishing of bulk modulus), and decohesion occurs as spontaneous nucleation of cracking on {111} shuffle planes. Atomic relaxation on the newly generated cracked surfaces appears to take place immediately following crack opening. read less NOT USED (low confidence) T. Halicioǧlu, “Binding energies for carbon atoms and clusters deposited on the Si(100) surface,” Thin Solid Films. 1994. link Times cited: 2 NOT USED (low confidence) K. M. Nelson, C. Cornwell, and L. Wille, “Massively parallel computer simulations of fullerenes and Si-clusters,” Computational Materials Science. 1994. link Times cited: 7 NOT USED (low confidence) J. Weinstein, R. Fisher, S. Vasanawala, M. Shapiro, and T. Tombrello, “Molecular dynamics simulation of cluster-ion fragmentation,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1994. link Times cited: 6 NOT USED (low confidence) D. Vvedensky, N. Haider, T. Shitara, S̆milauer P., and J. L. Beeby, “Evolution of surface morphology during epitaxial growth,” Philosophical Transactions of the Royal Society of London. Series A: Physical and Engineering Sciences. 1993. link Times cited: 11 Abstract: We examine the type of information that can be obtained from… read moreAbstract: We examine the type of information that can be obtained from Monte Carlo simulations of epitaxial growth. A basic model will be first introduced and some of the features that make it suitable for describing both atomic-scale processes and large-scale morphologies will be pointed out. The ability of this model to reproduce experimental data will then be addressed. The first example discussed will be growth on GaAs(OO1) vicinal surfaces, where the density of surface steps on the simulated surfaces reproduces quantitatively the evolution of the reflection high-energy electron diffraction (RHEED) intensity oscillations for appropriately chosen growth and diffraction conditions. This work will then be used as a basis for examining the predictions of the simulated surface morphologies on patterned substrates, based on comparisons with micro-RHEED measurements. Extensions of the basic model to more complex growth scenarios where the atomic constituents are delivered in the form of heteroatomic molecules will also be discussed. read less NOT USED (low confidence) X. Gong, Q. Zheng, and Y.-zhen He, “Structural properties of silicon clusters: an empirical potential study,” Journal of Physics: Condensed Matter. 1993. link Times cited: 14 Abstract: By using our newly proposed empirical interatomic potential … read moreAbstract: By using our newly proposed empirical interatomic potential for silicon, the structure and some dynamical properties of the silicon cluster Sin (10 read less NOT USED (low confidence) C. S. Carmer, B. Weiner, and M. Frenklach, “Molecular dynamics with combined quantum and empirical potentials: C2H2 adsorption on Si(100),” Journal of Chemical Physics. 1993. link Times cited: 71 Abstract: Classical trajectory calculations were employed to study the… read moreAbstract: Classical trajectory calculations were employed to study the reaction of acetylene with dimer sites on the Si(100) surface at 105 K. Two types of potential energy functions were combined to describe interactions for different regions of the model surface. A quantum mechanical potential based on the semiempirical AM1 Hamiltonian was used to describe interactions between C2H2 and a portion of the silicon surface, while an empirically parametrized potential was developed to extend the size of the surface and simulate the dynamics of the surrounding silicon atoms. Reactions of acetylene approaching different sites were investigated, directly above a surface dimer, and between atoms from separate dimers. In all cases, the outcome of C2H2 surface collisions was controlled by the amount of translational energy possessed by the incoming molecule. Acetylene molecules with high translational energy reacted with silicon dimers to form surface species with either one or two Si–C bonds. Those molecules with low transl... read less NOT USED (low confidence) T. Halicioǧlu, “Carbon atoms on the (2 × 1) reconstructed Si(100) surface,” Surface Science. 1993. link Times cited: 4 NOT USED (low confidence) W. Tiller, “Fundamental aspects of film nucleation and growth in chemical vapor deposition,” Surface & Coatings Technology. 1992. link Times cited: 0 NOT USED (low confidence) M. Marder, “Molecular dynamics of cracks,” Comput. Sci. Eng. 1989. link Times cited: 38 Abstract: Brittle objects fail because of cracks. But how and why do t… read moreAbstract: Brittle objects fail because of cracks. But how and why do they move? The answers to these questions hide down at the atomic scale. Simple analytical models point to numerical simulations of brittle fracture that can be compared directly with laboratory experiments. These simulations do not yet agree with experimental results because the atomic force laws on which the computations rest are not yet known well enough. The author discusses how atomic discreteness affects crack motion, and explains three qualitative phenomena: lattice trapping, a velocity gap, and crack-tip instabilities. The mathematics that originally predicted lattice trapping and the velocity gap is elaborate, but the author has found explanations that rely on simple intuition about atomic motions. He then shows how scaling ideas make it possible to extrapolate with confidence from nanometers to centimeters, and picoseconds to microseconds, so as to compare theory and experiment for brittle fracture. read less NOT USED (low confidence) K. Nordlund and F. Djurabekova, “Molecular Dynamics Simulations of Non-equilibrium Systems,” Handbook of Materials Modeling. 2020. link Times cited: 3 NOT USED (low confidence) “References,” Basic Physics of Nanoscience. 2019. link Times cited: 0 NOT USED (low confidence) S. Goel, X. Luo, A. Agrawal, and R. Reuben, “Diamond machining of silicon: A review of advances in molecular dynamics simulation,” International Journal of Machine Tools & Manufacture. 2015. link Times cited: 314 NOT USED (low confidence) T. Sinno, “Atomistic Calculation of Defect Thermodynamics in Crystalline Silicon.” 2015. link Times cited: 0 NOT USED (low confidence) R. Khanna and V. Sahajwalla, “Atomistic Simulations of Properties and Phenomena at High Temperatures.” 2014. link Times cited: 3 NOT USED (low confidence) N. Zuckerman and J. Lukes, “Combined Kinetic Monte Carlo—Molecular Dynamics Approach for Modeling Phonon Transport in Quantum Dot Superlattices,” Journal of Heat Transfer-transactions of The Asme. 2014. link Times cited: 1 Abstract: A new kinetic Monte Carlo method for modeling phonon transpo… read moreAbstract: A new kinetic Monte Carlo method for modeling phonon transport in quantum dot superlattices is presented. The method uses phonon scattering phase functions and cross sections to describe collisions between phonons and quantum dots. The phase functions and cross sections are generated using molecular dynamics simulation, which is capable of including atomistic effects otherwise unavailable in Monte Carlo approaches. The method is demonstrated for a test case featuring a Si-Ge quantum dot superlattice, and the model is compared against published experiments. It is found that molecular dynamics-derived cross sections must be weighted by diffuse mismatch model-type weighting factors in order to satisfy detailed balance considerations. Additionally, it is found that thin alloy “base layer” films strongly reduce thermal conductivity in these systems and must be included in the modeling to obtain agreement with published experimental data. read less NOT USED (low confidence) H. S. Park, “Computational Modeling of Surface Effects: Distinctions from Classical Surface Elasticity Theory.” 2013. link Times cited: 0 NOT USED (low confidence) J. Li et al., “Complete Realization of the EMMS Paradigm.” 2013. link Times cited: 1 NOT USED (low confidence) J. Li et al., “Many-Core Programming.” 2013. link Times cited: 0 NOT USED (low confidence) A. Ince and S. Erkoç, “Silicene nanoribbons: Molecular-dynamics simulations,” Computational Materials Science. 2011. link Times cited: 18 NOT USED (low confidence) R. W. Nunes and J. F. Justo, “Silicon Nanowires: From Empirical to First Principles Modeling.” 2010. link Times cited: 0 NOT USED (low confidence) H. S. Park and P. Klein, “Multiscale Modeling of Surface Effects on the Mechanical Behavior and Properties of Nanowires.” 2010. link Times cited: 0 NOT USED (low confidence) T. Kumagai, S. Hara, S. Izumi, and S. Sakai, “Development of a Bond-Order Potential that can Reproduce the Elastic Constants and Melting Point of Silicon,” Journal of The Society of Materials Science, Japan. 2006. link Times cited: 1 Abstract: The Tersoff potential is one of the most widely used interat… read moreAbstract: The Tersoff potential is one of the most widely used interatomic potentials for silicon. However, its poor descrip-tion of the elastic constants and melting point of diamond silicon is well known. In this research, a new bond-order type interatomic potential has been developed that can reproduce the elastic constants and melting point of diamond silicon as well as the cohesive energies and equilibrium bond lengths of polytypes of silicon. We improved the original Tersoff potential function through the introduction of a flexible angular dependent term. In order to increase the robustness of the potential, systems that include a wide range of local atomic environments are employed for fitting. Optimized potential parameters were found using a genetic algorithm. The elastic constants and melting point of diamond silicon calculated using the developed potential turned out to be C 11 = 166.4GPa, C 12 = 65.3GPa, C 44 = 77.1GPa and T m = 1681K. It was also found that only elastic constants can be reproduced using the original Tersoff potential function, and that our proposed angular dependent term is a key to reproducing the melting point. read less NOT USED (low confidence) B. Puchala, M. Falk, and K. Garikipati, “Using Elasticity to Correct for Boundary Effects in Calculations of Stress-Diffusion Coupling Parameters.” 2006. link Times cited: 0 NOT USED (low confidence) M. Demkowicz and A. Argon, “Unit Shearing Events in Plasticity of Amorphous Silicon,” MRS Proceedings. 2005. link Times cited: 0 NOT USED (low confidence) J. Tarus, M. Tantarimäki, and K. Nordlund, “Segregation in SiGe clusters,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 12 NOT USED (low confidence) J. F. Justo, “Modeling Covalent Bond with Interatomic Potentials.” 2005. link Times cited: 1 NOT USED (low confidence) R. Wagner and E. Gulari, “Germanium Island Size Distribution by Atomistic Simulation,” MRS Proceedings. 2003. link Times cited: 0 Abstract: Strained epitaxial growth of Ge on Si(001) produces self-ass… read moreAbstract: Strained epitaxial growth of Ge on Si(001) produces self-assembled, nanometer scale islands, or quantum dots. We study this growth by atomistic simulation, computing the energy of island structures to determine when and how islanding occurs. The distribution of island sizes on a surface is determined by the relation of island energy to size. Applying the calculated chemical potential to the Boltzmann-Gibbs distribution, we predict size distributions as functions of coverage and temperature. The peak populations around 80 000 atoms (35 nm wide) compare favorably with experiment. read less NOT USED (low confidence) J. Adams, “Bonding Energy Models.” 2001. link Times cited: 7 NOT USED (low confidence) T. Ito, “Atomistic simulation of epitaxial growth processes.” 2001. link Times cited: 0 NOT USED (low confidence) J. Que, M. Radny, P. V. Smith, and A. Dyson, “Application of the extended Brenner potential to the Si(111)7 × 7:H system I : cluster calculations,” Surface Science. 2000. link Times cited: 13 NOT USED (low confidence) K. Nordlund and R. Averback, “Collision cascades in metals and semiconductors: defect creation and interface behavior,” Journal of Nuclear Materials. 2000. link Times cited: 30 NOT USED (low confidence) R. E. Miller and V. B. Shenoy, “Size-dependent elastic properties of nanosized structural elements,” Nanotechnology. 2000. link Times cited: 16 Abstract: Effective stiffness properties (D) of nanosized structural e… read moreAbstract: Effective stiffness properties (D) of nanosized structural elements such as plates and beams differ from those predicted by standard continuum mechanics (Dc). These differences (D-Dc)/Dc depend on the size of the structural element. A simple model is constructed to predict this size dependence of the effective properties. The important length scale in the problem is identified to be the ratio of the surface elastic modulus to the elastic modulus of the bulk. In general, the non-dimensional difference in the elastic properties from continuum predictions (D-Dc)/Dc is found to scale as αS/Eh, where α is a constant which depends on the geometry of the structural element considered, S is a surface elastic constant, E is a bulk elastic modulus and h a length defining the size of the structural element. Thus, the quantity S/E is identified as a material length scale for elasticity of nanosized structures. The model is compared with direct atomistic simulations of nanoscale structures using the embedded atom method for FCC Al and the Stillinger-Weber model of Si. Excellent agreement between the simulations and the model is found. read less NOT USED (low confidence) M. Frenklach, “Numerical Modeling of Surface Reactions.” 1999. link Times cited: 2 NOT USED (low confidence) K. Nordlund, P. Partyka, Y. Zhong, I. Robinson, R. Averback, and P. Ehrhart, “Glancing incidence diffuse X-ray scattering studies of implantation damage in Si,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 1999. link Times cited: 6 NOT USED (low confidence) F. B. Mota, J. F. Justo, and A. Fazzio, “Structural and electronic properties of silicon nitride materials,” International Journal of Quantum Chemistry. 1998. link Times cited: 20 Abstract: The authors developed an empirical potential for interaction… read moreAbstract: The authors developed an empirical potential for interactions between Si and N to describe silicon nitride systems using the Tersoff functional form. With this model, they explored the structural properties of amorphous silicon nitride through the Monte Carlo simulations and compared them to available experimental data. The empirical model provided a very good description of such properties for a-SiN{sub x} (0 < x < 1.5). Electronic structure of amorphous and point defects in crystalline silicon nitride were then studied using first-principles calculations. For such calculations, the configurations were created by the empirical model, with the relaxed structures used as input for the first-principles calculations. Atomic relaxation was later allowed in the first-principles calculations. read less NOT USED (low confidence) M. Bazant, E. Kaxiras, and J. F. Justo, “The Environment-Dependent Interatomic Potential Applied To Silicon Disordered Structures And Phase Transitions,” MRS Proceedings. 1997. link Times cited: 7 NOT USED (low confidence) J. F. Justo, M. Bazant, E. Kaxiras, V. Bulatov, and S. Yip, “Interatomic Potential for Condensed Phases and Bulk Defects in Silicon,” MRS Proceedings. 1997. link Times cited: 5 NOT USED (low confidence) T. Frauenheim, D. Porezag, T. Köhler, and F. Weich, “Molecular-Dynamic Simulations of Structure Formation in Complex Materials.” 1996. link Times cited: 1 NOT USED (low confidence) D. Conrad, K. Scheerschmidt, and U. Gösele, “Molecular dynamics simulations of silicon wafer bonding,” Applied Physics A. 1996. link Times cited: 34 NOT USED (low confidence) M. Bazant and E. Kaxiras, “Derivation of Interatomic Potentials by Inversion of Ab Initio Cohesive Energy Curves,” MRS Proceedings. 1995. link Times cited: 3 NOT USED (low confidence) N. Bernstein and E. Kaxiras, “O( N ) Scaling Simulations of Silicon Bulk and Surface Properties Based on a Non-Orthogonal Tight-Binding Hamiltonian,” MRS Proceedings. 1995. link Times cited: 0 Abstract: We have implemented a molecular-dynamics algorithm for silic… read moreAbstract: We have implemented a molecular-dynamics algorithm for silicon using a non-orthogonal tight-binding Hamiltonian with the functional form of Menon and Subbaswamy. Parameters for this Hamiltonian were determined by fitting to a database of first-principles total energy calculations of bulk phases and point defect formation energies. These geometries were chosen to reproduce the configurations seen in defective crystalline and amorphous silicon. We have also implemented the non-orthogonal density-matrix method, paying particular attention to data motion locality to facilitate efficient parallelization of the algorithm. The necessary sparse matrix operations (trace, transpose, matrix multiplication) have also been implemented on a single processor workstation with an algorithm which takes O ( N ) time. Tests of the method's accuracy involved calculations of surface energies and structural reconstructions and activation energies for bulk diffusion through concerted exchange. We present results of a simulation of the melting and rapid quenching of a silicon sample using molecular-dynamics, and examine the resulting structures. read less NOT USED (low confidence) B. R. Eggen, R. Johnston, and J. Murrell, “Carbon cluster structures and stabilities predicted from solid-state potentials,” Journal of the Chemical Society, Faraday Transactions. 1994. link Times cited: 23 Abstract: An empirical potential-energy function comprising two- and t… read moreAbstract: An empirical potential-energy function comprising two- and three-body terms, whose parameters have been determined from the properties of diamond and graphite, is used to study the structures and energies of carbon microclusters.The binding energy per atom of smaller linear clusters increases monotonically with the number of atoms, whereas cyclic clusters display an optimal energy per atom for six-membered rings. The energies of fullerenes are sensitive to nuclearity and shape, with icosahedral C60 and D5h C70 being the most stable clusters. The potential predicts the binding energy of C60 to be 7.25 eV per atom, in good agreement with experimental measurements.For larger clusters, spherical fragments of cubic bulk structures have been investigated; diamond fragments become relatively more stable than other cubic fragments for more than approximately 100 atoms. Open nanotubes are found to be most stable for circumferences containing five hexagons. Vibrational frequencies were calculated and correlated with experimental results for some clusters. read less NOT USED (low confidence) D. Vvedensky, “Theory of Atomic-Scale Processes during Epitaxial Growth: Current Status.” 1993. link Times cited: 3 NOT USED (low confidence) I. Štich, M. Payne, A. Vita, M. Gillan, and L. Clarke, “First-Principles Studies of Semiconductor Surfaces: Reconstruction and Dissociative Chemisorption.” 1993. link Times cited: 0 NOT USED (high confidence) J.-C. Griesser, L. Frérot, J. A. Oldenstaedt, M. Müser, and L. Pastewka, “Analytic elastic coefficients in molecular calculations: Finite strain, nonaffine displacements, and many-body interatomic potentials,” Physical Review Materials. 2023. link Times cited: 1 Abstract: Elastic constants are among the most fundamental and importa… read moreAbstract: Elastic constants are among the most fundamental and important properties of solid materials, which is why they are routinely characterized in both experiments and simulations. While conceptually simple, the treatment of elastic constants is complicated by two factors not yet having been concurrently discussed: finite-strain and non-affine, internal displacements. Here, we revisit the theory behind zero-temperature, finite-strain elastic constants and extend it to explicitly consider non-affine displacements. We further present analytical expressions for second-order derivatives of the potential energy for two-body and generic many-body interatomic potentials, such as cluster and empirical bond-order potentials. Specifically, we revisit the elastic constants of silicon, silicon carbide and silicon dioxide under hydrostatic compression and dilatation. Based on existing and new results, we outline the effect of multiaxial stress states as opposed to volumetric deformation on the limits of stability of their crystalline lattices. read less NOT USED (high confidence) Y. Kurniawan et al., “Bayesian, frequentist, and information geometric approaches to parametric uncertainty quantification of classical empirical interatomic potentials.,” The Journal of chemical physics. 2021. link Times cited: 6 Abstract: In this paper, we consider the problem of quantifying parame… read moreAbstract: In this paper, we consider the problem of quantifying parametric uncertainty in classical empirical interatomic potentials (IPs) using both Bayesian (Markov Chain Monte Carlo) and frequentist (profile likelihood) methods. We interface these tools with the Open Knowledgebase of Interatomic Models and study three models based on the Lennard-Jones, Morse, and Stillinger-Weber potentials. We confirm that IPs are typically sloppy, i.e., insensitive to coordinated changes in some parameter combinations. Because the inverse problem in such models is ill-conditioned, parameters are unidentifiable. This presents challenges for traditional statistical methods, as we demonstrate and interpret within both Bayesian and frequentist frameworks. We use information geometry to illuminate the underlying cause of this phenomenon and show that IPs have global properties similar to those of sloppy models from fields, such as systems biology, power systems, and critical phenomena. IPs correspond to bounded manifolds with a hierarchy of widths, leading to low effective dimensionality in the model. We show how information geometry can motivate new, natural parameterizations that improve the stability and interpretation of uncertainty quantification analysis and further suggest simplified, less-sloppy models. read less NOT USED (high confidence) W. Wan, C. Tang, J. Zhang, and L. Zhou, “General Molecular Dynamics Approach to Understand the Mechanical Anisotropy of Monocrystalline Silicon under the Nanoscale Effects of Point Defect,” Nanomaterials. 2021. link Times cited: 7 Abstract: Mechanical anisotropy and point defects would greatly affect… read moreAbstract: Mechanical anisotropy and point defects would greatly affect the product quality while producing silicon wafers via diamond-wire cutting. For three major orientations concerned in wafer production, their mechanical performances under the nanoscale effects of a point defect were systematically investigated through molecular dynamics methods. The results indicated anisotropic mechanical performance with fracture phenomena in the uniaxial deformation process of monocrystalline silicon. Exponential reduction caused by the point defect has been demonstrated for some properties like yield strength and elastic strain energy release. Dislocation analysis suggested that the slip of dislocations appeared and created hexagonal diamond structures with stacking faults in the [100] orientation. Meanwhile, no dislocation was observed in [110] and [111] orientations. Visualization of atomic stress proved that the extreme stress regions of the simulation models exhibited different geometric and numerical characteristics due to the mechanical anisotropy. Moreover, the regional evolution of stress concentration and crystal fracture were interrelated and mutually promoted. This article contributes to the research towards the mechanical and fracture anisotropy of monocrystalline silicon. read less NOT USED (high confidence) W. Wan, C. Tang, A. Qiu, and Y. Xiang, “The Size Effects of Point Defect on the Mechanical Properties of Monocrystalline Silicon: A Molecular Dynamics Study,” Materials. 2021. link Times cited: 7 Abstract: The molecular dynamics method was used to simulate the fract… read moreAbstract: The molecular dynamics method was used to simulate the fracture process of monocrystalline silicon with different sizes of point defect under a constant strain rate. The mechanism of the defect size on the mechanical properties of monocrystalline silicon was also investigated. The results suggested that the point defect significantly reduces the yield strength of monocrystalline silicon. The relationships between the yield strength variation and the size of point defect fitted an exponential function. By statistically analyzing the internal stress in monocrystalline silicon, it was found that the stress concentration induced by the point defect led to the decrease in the yield strength. A comparison between the theoretical strength given by the four theories of strength and actual strength proved that the Mises theory was the best theory of strength to describe the yield strength of monocrystalline silicon. The dynamic evolution process of Mises stress and dislocation showed that the fracture was caused by the concentration effect of Mises stress and dislocation slip. Finally, the fractured microstructures were similar to a kind of two-dimensional grid which distributed along the cleavage planes while visualizing the specimens. The results of this article provide a reference for evaluating the size effects of point defects on the mechanical properties of monocrystalline silicon. read less NOT USED (high confidence) F. Dai, D. Zhao, and L. Zhang, “Atomic Simulations of Packing Structures, Local Stress and Mechanical Properties for One Silicon Lattice with Single Vacancy on Heating,” Materials. 2021. link Times cited: 2 Abstract: The effect of vacancy defects on the structure and mechanica… read moreAbstract: The effect of vacancy defects on the structure and mechanical properties of semiconductor silicon materials is of great significance to the development of novel microelectronic materials and the processes of semiconductor sensors. In this paper, molecular dynamics is used to simulate the atomic packing structure, local stress evolution and mechanical properties of a perfect lattice and silicon crystal with a single vacancy defect on heating. In addition, their influences on the change in Young’s modulus are also analyzed. The atomic simulations show that in the lower temperature range, the existence of vacancy defects reduces the Young’s modulus of the silicon lattice. With the increase in temperature, the local stress distribution of the atoms in the lattice changes due to the migration of the vacancy. At high temperatures, the Young’s modulus of the silicon lattice changes in anisotropic patterns. For the lattice with the vacancy, when the temperature is higher than 1500 K, the number and degree of distortion in the lattice increase significantly, the obvious single vacancy and its adjacent atoms contracting inward structure disappears and the defects in the lattice present complex patterns. By applying uniaxial tensile force, it can be found that the temperature has a significant effect on the elasticity–plasticity behaviors of the Si lattice with the vacancy. read less NOT USED (high confidence) V. Danesh, H. N. Pishkenari, and H. Zohoor, “Twisted-shape selection of self-assembled Si ⟨100⟩ nanobelts and nanowires,” Journal of Physics D: Applied Physics. 2021. link Times cited: 0 Abstract: This letter discusses the surface-reconstruction-induced sel… read moreAbstract: This letter discusses the surface-reconstruction-induced self-twisting behavior of Si〈100〉 nanobelts and nanowires (NWs) with rectangular cross section. Giving a thorough physical interpretation, we explain the reason behind this phenomenon and present a continuum-based model. It is revealed that these structures can self-assemble into both right- and left-handed helicoids depending on their crystal arrangements. More specifically, for NWs with the same number of layers in each of their cross sections directions, two distinct values of torsion angle are possible for each of right- and left-handed twisted morphologies. In conclusion, four modes of torsion can be observed in Si〈100〉 NWs. Furthermore, some atomistic simulations are conducted to substantiate analytical results by utilizing Tersoff’s potential. These results confirm the precision of the analytical discussions and are in good agreement with the continuum model predictions. Finally, the results of Tersoff’s potential are validated by a density functional based tight-binding model. read less NOT USED (high confidence) M. Farzinpour, D. Toghraie, B. Mehmandoust, F. Aghadavoudi, and A. Karimipour, “Molecular dynamics simulation of ferronanofluid behavior in a nanochannel in the presence of constant and time-dependent magnetic fields,” Journal of Thermal Analysis and Calorimetry. 2020. link Times cited: 22 NOT USED (high confidence) Y. Lysogorskiy, T. Hammerschmidt, J. Janssen, J. Neugebauer, and R. Drautz, “Transferability of interatomic potentials for molybdenum and silicon,” Modelling and Simulation in Materials Science and Engineering. 2019. link Times cited: 14 Abstract: Interatomic potentials are widely used in computational mate… read moreAbstract: Interatomic potentials are widely used in computational materials science, in particular for simulations that are too computationally expensive for density functional theory (DFT). Most interatomic potentials have a limited application range and often there is very limited information available regarding their performance for specific simulations. We carried out high-throughput calculations for molybdenum and silicon with DFT and a number of interatomic potentials. We compare the DFT reference calculations and experimental data to the predictions of the interatomic potentials. We focus on a large number of basic materials properties, including the cohesive energy, atomic volume, elastic coefficients, vibrational properties, thermodynamic properties, surface energies and vacancy formation energies, which enables a detailed discussion of the performance of the different potentials. We further analyze correlations between properties as obtained from DFT calculations and how interatomic potentials reproduce these correlations, and suggest a general measure for quantifying the accuracy and transferability of an interatomic potential. From our analysis we do not establish a clearcut ranking of the potentials as each potential has its strengths and weaknesses. It is therefore essential to assess the properties of a potential carefully before application of the potential in a specific simulation. The data presented here will be useful for selecting a potential for simulations of Mo or Si. read less NOT USED (high confidence) L. Wang, W. Yu, and S. Shen, “Revisiting the structures and energies of silicon 〈110〉 symmetric tilt grain boundaries,” Journal of Materials Research. 2019. link Times cited: 19 Abstract: Atomistic simulations of 18 silicon 〈110〉 symmetric tilt gra… read moreAbstract: Atomistic simulations of 18 silicon 〈110〉 symmetric tilt grain boundaries are performed using Stillinger Weber, Tersoff, and the optimized Modified Embedded Atom Method potentials. We define a novel structural unit classification through dislocation core analysis to characterize the relaxed GB structures. GBs with the misorientation angle θ ranging from 13.44° to 70.53° are solely composed of Lomer dislocation cores. For GBs with θ less than but close to 70.53°, GB ‘step’ appears and the equilibrated states with lowest GB energies can be attained only when such GB ‘step’ is located in the middle of each single periodic GB structure. For the misorientation angles in the range of 93.37° ≤ θ ≤ 148.41°, GB structures become complicated since they contain multiple types of dislocation cores. This work not only facilitates the structural characterization of silicon 〈110〉 STGBs, but also may provide new insights into mirco-structure design in multicrystalline silicon. read less NOT USED (high confidence) D. Prasad and N. Mitra, “An atomistic study of phase transition in cubic diamond Si single crystal subjected to static compression,” Computational Materials Science. 2019. link Times cited: 7 NOT USED (high confidence) L. Martín, I. Santos, P. López, L. Marqués, M. Aboy, and L. Pelaz, “Modeling SiGe Through Classical Molecular Dynamics Simulations: Chasing an Appropriate Empirical Potential,” 2018 Spanish Conference on Electron Devices (CDE). 2018. link Times cited: 2 Abstract: We used classical molecular dynamics simulations to reproduc… read moreAbstract: We used classical molecular dynamics simulations to reproduce basic properties of Si, Ge and SiGe using different empirical potentials available in the literature. The empirical potential that offered the better compromise with experimental data was used to study the surface stability of these materials. We considered the (100), $(100)2\times 1$ and (111) surfaces, and we found the processing temperature range to avoid the structural degradation of studied surfaces. read less NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 38 NOT USED (high confidence) L. N. Abdulkadir, K. Abou-El-Hossein, A. I. Jumare, M. Liman, T. A. Olaniyan, and P. B. Odedeyi, “Review of molecular dynamics/experimental study of diamond-silicon behavior in nanoscale machining,” The International Journal of Advanced Manufacturing Technology. 2018. link Times cited: 0 NOT USED (high confidence) A. Bartók, J. Kermode, N. Bernstein, and G. Csányi, “Machine Learning a General-Purpose Interatomic Potential for Silicon,” Physical Review X. 2018. link Times cited: 291 Abstract: The success of first principles electronic structure calcula… read moreAbstract: The success of first principles electronic structure calculation for predictive modeling in chemistry, solid state physics, and materials science is constrained by the limitations on simulated length and time scales due to computational cost and its scaling. Techniques based on machine learning ideas for interpolating the Born-Oppenheimer potential energy surface without explicitly describing electrons have recently shown great promise, but accurately and efficiently fitting the physically relevant space of configurations has remained a challenging goal. Here we present a Gaussian Approximation Potential for silicon that achieves this milestone, accurately reproducing density functional theory reference results for a wide range of observable properties, including crystal, liquid, and amorphous bulk phases, as well as point, line, and plane defects. We demonstrate that this new potential enables calculations that would be extremely expensive with a first principles electronic structure method, such as finite temperature phase boundary lines, self-diffusivity in the liquid, formation of the amorphous by slow quench, and dynamic brittle fracture. We show that the uncertainty quantification inherent to the Gaussian process regression framework gives a qualitative estimate of the potential's accuracy for a given atomic configuration. The success of this model shows that it is indeed possible to create a useful machine-learning-based interatomic potential that comprehensively describes a material, and serves as a template for the development of such models in the future. read less NOT USED (high confidence) T. Zhu, K. Swaminathan-Gopalan, K. Cruse, K. Stephani, and E. Ertekin, “Vibrational Energy Transport in Hybrid Ordered/Disordered Nanocomposites: Hybridization and Avoided Crossings of Localized and Delocalized Modes,” Advanced Functional Materials. 2018. link Times cited: 22 Abstract: Vibrational energy transport in disordered media is of funda… read moreAbstract: Vibrational energy transport in disordered media is of fundamental importance to several fields spanning from sustainable energy to biomedicine to thermal management. This work investigates hybrid ordered/disordered nanocomposites that consist of crystalline membranes decorated by regularly patterned disordered regions formed by ion beam irradiation. The presence of the disordered regions results in reduced thermal conductivity, rendering these systems of interest for use as nanostructured thermoelectrics and thermal device components, yet their vibrational properties are not well understood. Here, the mechanism of vibrational transport and the reason underlying the observed reduction is established in detail. The hybrid systems are found to exhibit glass‐crystal duality in vibrational transport. Lattice dynamics reveals substantial hybridization between the localized and delocalized modes, which induces avoided crossings and harmonic broadening in the dispersion. Allen/Feldman theory shows that the hybridization and avoided crossings are the dominant drivers of the reduction. Anharmonic scattering is also enhanced in the patterned nanocomposites, further contributing to the reduction. The systems exhibit features reminiscent of both nanophononic materials and locally resonant nanophononic metamaterials, but operate in a manner distinct to both. These findings indicate that such “patterned disorder” can be a promising strategy to tailor vibrational transport through hybrid nanostructures. read less NOT USED (high confidence) H. Li, R. Xu, Z. Bi, X. Shen, and K. Han, “Melting Properties of Medium-Sized Silicon Nanoclusters:
A Molecular Dynamics Study,” Journal of Electronic Materials. 2017. link Times cited: 7 NOT USED (high confidence) E. Dontsova and R. Ballarini, “Atomistic modeling of the fracture toughness of silicon and silicon-silicon interfaces,” International Journal of Fracture. 2017. link Times cited: 6 NOT USED (high confidence) G. P. P. Pun and Y. Mishin, “Optimized interatomic potential for silicon and its application to thermal stability of silicene,” Physical Review B. 2017. link Times cited: 35 Abstract: An optimized interatomic potential has been constructed for … read moreAbstract: An optimized interatomic potential has been constructed for silicon using a modified Tersoff model. The potential reproduces a wide range of properties of Si and improves over existing potentials with respect to point defect structures and energies, surface energies and reconstructions, thermal expansion, melting temperature, and other properties. The proposed potential is compared with three other potentials from the literature. The potentials demonstrate reasonable agreement with first-principles binding energies of small Si clusters as well as single-layer and bilayer silicenes. The four potentials are used to evaluate the thermal stability of free-standing silicenes in the form of nanoribbons, nanoflakes, and nanotubes. While single-layer silicene is found to be mechanically stable at zero Kelvin, it is predicted to become unstable and collapse at room temperature. By contrast, the bilayer silicene demonstrates a larger bending rigidity and remains stable at and even above room temperature. The results suggest that bilayer silicene might exist in a free-standing form at ambient conditions. read less NOT USED (high confidence) L. Wang, J. D. Lee, and C. Kan, “Work conjugate pair of stress and strain in molecular dynamics,” International Journal of Smart and Nano Materials. 2016. link Times cited: 1 Abstract: ABSTRACT Certain stress and strain form a thermodynamic conj… read moreAbstract: ABSTRACT Certain stress and strain form a thermodynamic conjugate pair such that their strain energy equals to a scalar-valued potential energy. Different atomistic stresses and strains are analytically derived based on the work conjugate relation. It is numerically verified with both two-body and three-body potentials that the atomistic Kirchhoff stress, first-order Piola–Kirchhoff stress and second-order Piola–Kirchhoff stress are conjugates to atomistic logarithmic strain, deformation gradient and Lagrangian strain, respectively. Virial stress at 0 K based on original volume is the special form of atomistic Kirchhoff stress for pair potential. It is numerically verified that Hencky strain is not conjugate to any stress. read less NOT USED (high confidence) S. Mukherjee, J. Song, and S. Vengallatore, “Atomistic simulations of material damping in amorphous silicon nanoresonators,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 1 Abstract: Atomistic simulations using molecular dynamics (MD) are emer… read moreAbstract: Atomistic simulations using molecular dynamics (MD) are emerging as a valuable tool for exploring dissipation and material damping in nanomechanical resonators. In this study, we used isothermal MD to simulate the dynamics of the longitudinal-mode oscillations of an amorphous silicon nanoresonator as a function of frequency (2 GHz–50 GHz) and temperature (15 K–300 K). Damping was characterized by computing the loss tangent with an estimated uncertainty of 7%. The dissipation spectrum displays a sharp peak at 50 K and a broad peak at around 160 K. Damping is a weak function of frequency at room temperature, and the loss tangent has a remarkably high value of ~0.01. In contrast, at low temperatures (15 K), the loss tangent increases monotonically from 4×10−4 to 4×10−3 as the frequency increases from 2 GHz to 50 GHz. The mechanisms of dissipation are discussed. read less NOT USED (high confidence) C. L. D. Prinzio and R. G. Pereyra, “Molecular dynamics simulations of 〈1 0 1¯ 0〉/ψ tilt grain boundaries in ice,” Modelling and Simulation in Materials Science and Engineering. 2016. link Times cited: 6 Abstract: In this paper, molecular dynamics simulations (MDS) of 〈1 0 … read moreAbstract: In this paper, molecular dynamics simulations (MDS) of 〈1 0 1¯ 0〉/ψ symmetric tilt ice grain boundaries are presented. The MDS were carried out using the GROMACS v4.5.5 program, and the water molecules were described using the TIP5P-Ew model. The grain boundary energies, γgb, relative to those of the surface free energies, γs, were obtained as a function of the misorientation angle Ψ, and compared with the γgb/γs values experimentally obtained. The results show a good correspondence between the experimental and simulated values. The planar density of coincidence sites at the grain boundary planes, Γ, was obtained as a function of ψ. The Γ values were compared with the simulated γgb/γs values and a relation between the minimum of the simulated γgb/γs values and the maximum of the Γ values was observed, suggesting that the CSL theory is a good starting point to detect low energy ice GBs. read less NOT USED (high confidence) L. Sang, V. V. Hoang, and D. T. N. Tranh, “Melting of crystalline Si nanoparticle investigated by simulation,” The European Physical Journal D. 2015. link Times cited: 6 NOT USED (high confidence) T.-H. Chen, R. Xu, and Q. Li, “Effect of Strain Rate on Tensile Strength of Defective Silicon Nanorods,” Acta Mechanica Solida Sinica. 2015. link Times cited: 4 NOT USED (high confidence) V. Lipp, B. Rethfeld, M. E. Garcia, and D. Ivanov, “Atomistic-continuum modeling of short laser pulse melting of Si targets,” Physical Review B. 2014. link Times cited: 41 Abstract: We present an atomistic-continuum model to simulate ultrasho… read moreAbstract: We present an atomistic-continuum model to simulate ultrashort laser-induced melting processes in semiconductor solids on the example of silicon. The kinetics of transient non-equilibrium phase transition mechanisms is addressed with a Molecular Dynamics method at atomic level, whereas the laser light absorption, strong generated electron-phonon non-equilibrium, fast diffusion and heat conduction due to photo-excited free carriers are accounted for in the continuum. We give a detailed description of the model, which is then applied to study the mechanism of short laser pulse melting of free standing Si films. The effect of laser-induced pressure and temperature of the lattice on the melting kinetics is investigated. Two competing melting mechanisms, heterogeneous and homogeneous, were identified. Apart of classical heterogeneous melting mechanism, the nucleation of the liquid phase homogeneously inside the material significantly contributes to the melting process. The simulations showed, that due to the open diamond structure of the crystal, the laser-generated internal compressive stresses reduce the crystal stability against the homogeneous melting. Consequently, the latter can take a massive character within several picoseconds upon the laser heating. Due to negative volume of melting of modeled Si material, -7.5%, the material contracts upon the phase transition, relaxes the compressive stresses and the subsequent melting proceeds heterogeneously until the excess of thermal energy is consumed. The threshold fluence value, at which homogeneous nucleation of liquid starts contributing to the classical heterogeneous propagation of the solid-liquid interface, is found from the series of simulations at different laser input fluences. On the example of Si, the laser melting kinetics of semiconductors was found to be noticeably different from that of metals with fcc crystal structure. read less NOT USED (high confidence) R. Skorpa, J. Simon, D. Bedeaux, and S. Kjelstrup, “Equilibrium properties of the reaction H2 ⇌ 2H by classical molecular dynamics simulations.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 9 Abstract: We have developed a classical molecular dynamics model for t… read moreAbstract: We have developed a classical molecular dynamics model for the hydrogen dissociation reaction, containing two- and three-particle potentials derived by Kohen, Tully and Stillinger. Two fluid densities were investigated for a wide range of temperatures, and 11 fluid densities were considered for one temperature. We report the temperature range where the degree of reaction is significant, and also where a stable molecule dominates the population in the energy landscape. The three-particle potential, which is essential for the reaction model and seldom studied, together with the two-particle interaction lead to a large effective excluded volume diameter of the molecules in the molecular fluid. The three-particle interaction was also found to give a large positive contribution to the pressure of the reacting mixture at high density and/or low temperatures. From knowledge of the dissociation constant of the reaction and the fluid pressure, we estimated the standard enthalpy of the dissociation reaction to be 430 kJ mol(-1) (ρ = 0.0695 g cm(-3)) and 380 kJ mol(-1) (ρ = 0.0191 g cm(-3)). These values are in good agreement with the experimental vaule of 436 kJ mol(-1) under ambient pressure. The model is consistent with a Lennard-Jones model of the molecular fluid, and may facilitate studies of the impact of chemical reactions on transport systems. read less NOT USED (high confidence) L. Xie, P. Brault, A. Thomann, and J. Bauchire, “AlCoCrCuFeNi high entropy alloy cluster growth and annealing on silicon: A classical molecular dynamics simulation study,” Applied Surface Science. 2013. link Times cited: 102 NOT USED (high confidence) P. Zhang and D. Trinkle, “Database optimization for empirical interatomic potential models,” Modelling and Simulation in Materials Science and Engineering. 2013. link Times cited: 8 Abstract: Weighted least squares fitting to a database of quantum mech… read moreAbstract: Weighted least squares fitting to a database of quantum mechanical calculations can determine the optimal parameters of empirical potential models. While algorithms exist to provide optimal potential parameters for a given fitting database of structures with corresponding energy-related predictions and to estimate prediction errors using Bayesian sampling, defining an optimal fitting database based on potential predictions remains elusive. A testing set of structures and energy-related predictions provides an empirical measure of potential transferability. Here, we propose an objective function for fitting databases based on testing set errors. The objective function allows the optimization of the weights in a fitting database, the assessment of the adding or removing of structures in the fitting database, or the comparison of two different fitting databases. To showcase this technique, we consider an example Lennard-Jones potential for Ti, where modeling multiple complicated crystal structures is difficult for a radial pair potential. The algorithm finds different optimal fitting databases, depending on the objective function of potential prediction error for a testing set. read less NOT USED (high confidence) Z. Li and R. C. Picu, “Shuffle-glide dislocation transformation in Si,” Journal of Applied Physics. 2013. link Times cited: 23 Abstract: The transformation of dislocation cores from the shuffle to … read moreAbstract: The transformation of dislocation cores from the shuffle to the glide set of {111} glide planes in Si is examined in this work. The transformation is thermally activated and is favored by a resolved shear stress which applies no force on the original perfect shuffle dislocation. A resolved shear stress driving dislocation motion in the glide plane is not observed to promote the transition. The stress-dependent activation energy for the described shuffle-glide transformation mechanism is evaluated using a statistical analysis. It is observed that the transformation is not associated with an intermediate metastable state, as has been previously suggested in the literature. read less NOT USED (high confidence) A. Masolin, P. Bouchard, R. Martini, and M. Bernacki, “Thermo-mechanical and fracture properties in single-crystal silicon,” Journal of Materials Science. 2013. link Times cited: 124 NOT USED (high confidence) L. Pizzagalli et al., “A new parametrization of the Stillinger–Weber potential for an improved description of defects and plasticity of silicon,” Journal of Physics: Condensed Matter. 2013. link Times cited: 66 Abstract: A new parametrization of the widely used Stillinger–Weber po… read moreAbstract: A new parametrization of the widely used Stillinger–Weber potential is proposed for silicon, allowing for an improved modelling of defects and plasticity-related properties. The performance of the new potential is compared to the original version, as well as to another parametrization (Vink et al 2001 J. Non-Cryst. Solids, 282 248), in the case of several situations: point defects and dislocation core stability, threshold displacement energies, bulk shear, generalized stacking fault energy surfaces, fracture, melting temperature, amorphous structure, and crystalline phase stability. A significant improvement is obtained in the case of dislocation cores, bulk behaviour under high shear stress, the amorphous structure, and computation of threshold displacement energies, while most of the features of the original version (elastic constants, point defects) are retained. However, despite a slight improvement, a complex process like fracture remains difficult to model. read less NOT USED (high confidence) P. Howell, “Comparison of molecular dynamics methods and interatomic potentials for calculating the thermal conductivity of silicon.,” The Journal of chemical physics. 2012. link Times cited: 74 Abstract: We compare the molecular dynamics Green-Kubo and direct meth… read moreAbstract: We compare the molecular dynamics Green-Kubo and direct methods for calculating thermal conductivity κ, using as a test case crystalline silicon at temperatures T in the range 500-1000 K (classical regime). We pay careful attention to the convergence with respect to simulation size and duration and to the procedures used to fit the simulation data. We show that in the Green-Kubo method the heat current autocorrelation function is characterized by three decay processes, of which the slowest lasts several tens of picoseconds so that convergence requires several tens of nanoseconds of data. Using the Stillinger-Weber potential we find excellent agreement between the two methods. We also use the direct method to calculate κ(T) for the Tersoff potential and find that the magnitude and the temperature-dependence are different for the two potentials and that neither potential agrees with experimental data. We argue that this implies that using the Stillinger-Weber or Tersoff potentials to predict trends in kappa as some system parameter is varied may yield results which are specific to the potential but not intrinsic to Si. read less NOT USED (high confidence) T. Yamaguchi and K. Saitoh, “Molecular Dynamics Study on Mechanical Properties in the Structure of Self-Assembled Quantum Dot,” World Journal of Nano Science and Engineering. 2012. link Times cited: 1 Abstract: Stress and strain in the structure of self-assembled quantum… read moreAbstract: Stress and strain in the structure of self-assembled quantum dots constructed in the Ge/Si(001) system is calculated by using molecular dynamics simulation. Pyramidal hut cluster composed of Ge crystal with {105} facets surfaces observed in the early growth stage are computationally modeled. We calculate atomic stress and strain in relaxed pyramidal structure. Atomic stress for triplet of atoms is approximately defined as an average value of pairwise (virial) quantity inside triplet, which is the product of vectors between each two atoms. Atomic strain by means of atomic strain measure (ASM) which is formulated on the Green’s definition of continuum strain. We find the stress (strain) relaxation in pyramidal structure and stress (strain) concentration in the edge of pyramidal structure. We discuss size dependency of stress and strain distribution in pyramidal structure. The relationship between hydrostatic stress and atomic volumetric strain is basically linear for all models, but for the surface of pyramidal structure and Ge-Si interface. This means that there is a reasonable correlation between atomic stress proposed in the present study and atomic strain measure, ASM. read less NOT USED (high confidence) A. Furmanchuk, O. Isayev, T. Dinadayalane, D. Leszczyńska, and J. Leszczynski, “Mechanical properties of silicon nanowires,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2012. link Times cited: 12 Abstract: Silicon nanowires (SiNWs) are at the top of the list of mate… read moreAbstract: Silicon nanowires (SiNWs) are at the top of the list of materials used in conventional electromechanical devices as well as in strained nanotechnology. Both experimental and theoretical studies showed the size‐dependent character of mechanical properties of SiNWs. However, the surface contaminations, local surface strains, ‘boundary conditions’, native oxide, equipment‐induced errors, and the errors caused by postprocessing of results lead to softening of Young's modulus and extension of the region where the size dependency is seen by experimentalists. Application of improved potentials or advanced theoretical modeling such as inclusion of explicit treatment of temperature and quantum‐mechanical effects allows to show specificity of Young's modulus to the size and shape in case of small (width <4 nm) nanowires. The ductile‐brittle transitions of SiNWs at different temperatures are revealed. Some suggestions on postprocessing techniques are discussed. © 2012 John Wiley & Sons, Ltd. read less NOT USED (high confidence) C. Yang, Y. Wang, and X. Xu, “Molecular dynamics studies of ultrafast laser-induced phase and structural change in crystalline silicon,” International Journal of Heat and Mass Transfer. 2012. link Times cited: 17 NOT USED (high confidence) M. T. Knippenberg, P. Mikulski, K. E. Ryan, S. Stuart, G. Gao, and J. Harrison, “Bond-order potentials with split-charge equilibration: application to C-, H-, and O-containing systems.,” The Journal of chemical physics. 2012. link Times cited: 25 Abstract: A method for extending charge transfer to bond-order potenti… read moreAbstract: A method for extending charge transfer to bond-order potentials, known as the bond-order potential/split-charge equilibration (BOP/SQE) method [P. T. Mikulski, M. T. Knippenberg, and J. A. Harrison, J. Chem. Phys. 131, 241105 (2009)], is integrated into a new bond-order potential for interactions between oxygen, carbon, and hydrogen. This reactive potential utilizes the formalism of the adaptive intermolecular reactive empirical bond-order potential [S. J. Stuart, A. B. Tutein, and J. A. Harrison, J. Chem. Phys. 112, 6472 (2000)] with additional terms for oxygen and charge interactions. This implementation of the reactive potential is able to model chemical reactions where partial charges change in gas- and condensed-phase systems containing oxygen, carbon, and hydrogen. The BOP/SQE method prevents the unrestricted growth of charges, often observed in charge equilibration methods, without adding significant computational time, because it makes use of a quantity which is calculated as part of the underlying covalent portion of the potential, namely, the bond order. The implementation of this method with the qAIREBO potential is designed to provide a tool that can be used to model dynamics in a wide range of systems without significant computational cost. To demonstrate the usefulness and flexibility of this potential, heats of formation for isolated molecules, radial distribution functions of liquids, and energies of oxygenated diamond surfaces are calculated. read less NOT USED (high confidence) A. Dongare, B. Lamattina, D. Irving, A. Rajendran, M. Zikry, and D. Brenner, “An angular-dependent embedded atom method (A-EAM) interatomic potential to model thermodynamic and mechanical behavior of Al/Si composite materials,” Modelling and Simulation in Materials Science and Engineering. 2012. link Times cited: 23 Abstract: A new interatomic potential is developed for the Al/Si syste… read moreAbstract: A new interatomic potential is developed for the Al/Si system in the formulation of the recently developed angular-dependent embedded atom method (A-EAM). The A-EAM is formulated by combining the embedded atom method potential for Al with the Stillinger–Weber potential for Si. The parameters of the Al/Si cross-interactions are fitted to reproduce the structural energetics of Al/Si bulk alloys determined based on the results of density functional theory calculations and the experimentally observed mixing behavior of the AlSi liquid alloy at high temperatures. The ability to investigate the thermodynamic properties of the Al/Si system is demonstrated by computing the binary phase diagram of the Al–Si system as predicted by the A-EAM potential and comparing with that obtained using experiments. The ability to study the mechanical behavior of the Al/Si composite systems is demonstrated by investigating the micromechanisms related to dynamic failure of the Al/Si nanocomposites using MD simulations. read less NOT USED (high confidence) G. Yun and H. S. Park, “Bridging the gap between experimental measurements and atomistic predictions of the elastic properties of silicon nanowires using multiscale modeling,” Finite Elements in Analysis and Design. 2012. link Times cited: 9 NOT USED (high confidence) C. Hou and W. Ge, “GPU-accelerated molecular dynamics simulation of solid covalent crystals,” Molecular Simulation. 2012. link Times cited: 17 Abstract: Graphics processing unit (GPU) is becoming a powerful comput… read moreAbstract: Graphics processing unit (GPU) is becoming a powerful computational tool in science and engineering. In this paper, different from previous molecular dynamics (MD) simulation with pair potentials and many-body potentials, two MD simulation algorithms implemented on a single GPU are presented to describe a special category of many-body potentials – bond order potentials used frequently in solid covalent materials, such as the Tersoff potentials for silicon crystals. The simulation results reveal that the performance of GPU implementations is apparently superior to their CPU counterpart. Furthermore, the proposed algorithms are generalised, transferable and scalable, and can be extended to the simulations with general many-body interactions such as Stillinger–Weber potential and so on. read less NOT USED (high confidence) N. Jakse, T. L. Nguyen, and A. Pasturel, “Ordering effects in disordered systems: the Au–Si system,” Journal of Physics: Condensed Matter. 2011. link Times cited: 15 Abstract: We have characterized the short-range order in the liquid an… read moreAbstract: We have characterized the short-range order in the liquid and undercooled states of Au–Si alloy at the eutectic composition using molecular dynamics simulations. The interactions are described via a modified embedded-atom model refined to take into account the liquid properties. For the eutectic liquid, the local structure is characterized by a strong Au–Si affinity, namely a well-pronounced chemical short-range order which leads to the slowing down of the formation of icosahedral local motifs in the undercooled regime. Moreover we discuss the influence of this peculiar local structure on the dynamic and thermodynamic properties of the liquid phase and compare our results with available experimental data. read less NOT USED (high confidence) H. Kim and V. Tomar, “Nanometer to micron scale mechanics of [100] silicon nanowires using atomistic simulations at accelerated time steps,” physica status solidi (a). 2011. link Times cited: 10 Abstract: Atomistic simulations have a unique capability to reveal the… read moreAbstract: Atomistic simulations have a unique capability to reveal the material deformation mechanisms and the corresponding deformation‐based constitutive behavior. However, atomistic simulations are limited by the accessible length and time scales. In the present work an equivalent crystal lattice method is used to perform mechanical deformation atomistic simulations of nanometer to micrometer sized silicon (Si) nanowires at accelerated time steps. The equivalent crystal lattice method's validity is verified by comparing the method's results with the results of classical molecular dynamics (MD) simulations at MD strain rates. The simulations predict that when the nanowire cross‐sectional size exceeds 50 nm, the dependence of the nanowire Young's moduli values on the changes in nanowire cross‐sectional size is considerably reduced. Analyses show a transition in nanowire failure mechanism from being ductile to being brittle with increase in the nanowire cross‐sectional size. Examinations of the surface effect reveal that below a critical surface to volume ratio value of 0.05 nm−1, the peak nanowire strength is independent of further reduction in the surface to volume ratio value. This finding places a size limit on the surface effect observed in Si nanowires. read less NOT USED (high confidence) F. Zirkelbach, B. Stritzker, K. Nordlund, J. Lindner, W. Schmidt, and E. Rauls, “Combined ab initio and classical potential simulation study on silicon carbide precipitation in silicon,” Physical Review B. 2011. link Times cited: 22 Abstract: Atomistic simulations on the silicon carbide precipitation i… read moreAbstract: Atomistic simulations on the silicon carbide precipitation in bulk silicon employing both, classical potential and first-principles methods are presented. The calculations aim at a comprehensive, microscopic understanding of the precipitation mechanism in the context of controversial discussions in the literature. For the quantum-mechanical treatment, basic processes assumed in the precipitation process are calculated in feasible systems of small size. The migration mechanism of a carbon 〈1 0 0〉 interstitial and silicon 〈11 0〉 self-interstitial in otherwise defect-free silicon are investigated using density functional theory calculations. The influence of a nearby vacancy, another carbon interstitial and a substitutional defect as well as a silicon self-interstitial has been investigated systematically. Interactions of various combinations of defects have been characterized including a couple of selected migration pathways within these configurations. Most of the investigated pairs of defects tend to agglomerate allowing for a reduction in strain. The formation of structures involving strong carbon–carbon bonds turns out to be very unlikely. In contrast, substitutional carbon occurs in all probability. A long range capture radius has been observed for pairs of interstitial carbon as well as interstitial carbon and vacancies. A rather small capture radius is predicted for substitutional carbon and silicon self-interstitials. Initial assumptions regarding the precipitation mechanism of silicon carbide in bulk silicon are established and conformability to experimental findings is discussed. Furthermore, results of the accurate first-principles calculations on defects and carbon diffusion in silicon are compared to results of classical potential simulations revealing significant limitations of the latter method. An approach to work around this problem is proposed. Finally, results of the classical potential molecular dynamics simulations of large systems are examined, which reinforce previous assumptions and give further insight into basic processes involved in the silicon carbide transition. read less NOT USED (high confidence) B. Jelinek et al., “Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys,” Physical Review B. 2011. link Times cited: 218 Abstract: A set of modified embedded-atom method (MEAM) potentials for… read moreAbstract: A set of modified embedded-atom method (MEAM) potentials for the interactions between Al, Si, Mg, Cu, and Fe was developed from a combination of each element's MEAM potential in order to study metal alloying. Previously published MEAM parameters of single elements have been improved for better agreement to the generalized stacking fault energy (GSFE) curves when compared with ab initio generated GSFE curves. The MEAM parameters for element pairs were constructed based on the structural and elastic properties of element pairs in the NaCl reference structure garnered from ab initio calculations, with adjustment to reproduce the ab initio heat of formation of the most stable binary compounds. The new MEAM potentials were validated by comparing the formation energies of defects, equilibrium volumes, elastic moduli, and heat of formation for several binary compounds with ab initio simulations and experiments. Single elements in their ground-state crystal structure were subjected to heating to test the potentials at elevated temperatures. An Al potential was modified to avoid formation of an unphysical solid structure at high temperatures. The thermal expansion coefficient of a compound with the composition of AA 6061 alloy was evaluated and compared with experimental values. MEAM potential tests performed in this work, utilizing the universal atomistic simulation environment (ASE), are distributed to facilitate reproducibility of the results. read less NOT USED (high confidence) A. Furmanchuk, O. Isayev, T. Dinadayalane, and J. Leszczynski, “Car-parrinello molecular dynamics simulations of tensile tests on Si〈001〉 nanowires,” Journal of Physical Chemistry C. 2011. link Times cited: 5 Abstract: Theoretical simulations of tensile tests on Si⟨001⟩ nanowire… read moreAbstract: Theoretical simulations of tensile tests on Si⟨001⟩ nanowires have been carried out using Car–Parrinello molecular dynamics. H-passivation was used to model experimentally occurring passivation in Si nanowires. First-principle molecular dynamics simulations at ambient temperature reveal the governing role of size, overall shape, and composition of the surface layer for the mechanical properties. Our results indicate that SiH2 groups in the outer layer and the octahedral shape of the wire soften Young’s modulus and allow wire to handle larger transverse strains than SiH groups in wires with the tetrahedral shape. The importance of the overall shape of the wire has been discussed by comparing the behavior of surface layers of {100} and {110} facets. The presence of the {100} facets helps to relax the transverse strain during tension. On the basis of changes in structural parameters, we have presented the schematic motion of Si atoms in core and surface layers before the fracture appeared. read less NOT USED (high confidence) F. Zirkelbach, B. Stritzker, K. Nordlund, J. Lindner, W. Schmidt, and E. Rauls, “Defects in carbon implanted silicon calculated by classical potentials and first-principles methods,” Physical Review B. 2010. link Times cited: 7 Abstract: A comparative theoretical investigation of carbon interstiti… read moreAbstract: A comparative theoretical investigation of carbon interstitials in silicon is presented. Calculations using classical potentials are compared to first-principles density-functional theory calculations of the geometries, formation, and activation energies of the carbon dumbbell interstitial, showing the importance of a quantummechanical description of this system. In contrast to previous studies, the present first-principles calculations of the interstitial carbon migration path yield an activation energy that excellently matches the experiment. The bond-centered interstitial configuration shows a net magnetization of two electrons, illustrating the need for spin-polarized calculations. read less NOT USED (high confidence) J. Guénolé, J. Godet, and L. Pizzagalli, “Determination of activation parameters for the core transformation of the screw dislocation in silicon,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 13 Abstract: The non-dissociated screw dislocation in a model covalent ma… read moreAbstract: The non-dissociated screw dislocation in a model covalent material like silicon is known to exist in three possible stable core configurations. We performed calculations combining the nudged elastic band technique and a semi-empirical description in order to determine mechanisms and activation parameters for transforming one core into another. Our results showed that a glide core is necessarily reconstructed, since the energy barrier for reconstruction is easily overcome by thermal activation. Conversely, a transformation between a shuffle and a glide core appears unlikely at low temperature, which raises questions about the existence of the double-period glide configuration. read less NOT USED (high confidence) A. Kerrache, N. Mousseau, and L. J. Lewis, “Amorphous silicon under mechanical shear deformations: Shear velocity and temperature effects,” Physical Review B. 2010. link Times cited: 8 Abstract: Mechanical shear deformations lead, in some cases, to effect… read moreAbstract: Mechanical shear deformations lead, in some cases, to effects similar to those resulting from ion irradiation. Here we characterize the effects of shear velocity and temperature on amorphous silicon (\aSi) modelled using classical molecular dynamics simulations based on the empirical Environment Dependent Inter-atomic Potential (EDIP). With increasing shear velocity at low temperature, we find a systematic increase in the internal strain leading to the rapid appearance of structural defects (5-fold coordinated atoms). The impacts of externally applied strain can be almost fully compensated by increasing the temperature, allowing the system to respond more rapidly to the deformation. In particular, we find opposite power-law relations between the temperature and the shear velocity and the deformation energy. The spatial distribution of defects is also found to strongly depend on temperature and strain velocity. For low temperature or high shear velocity, defects are concentrated in a few atomic layers near the center of the cell while, with increasing temperature or decreasing shear velocity, they spread slowly throughout the full simulation cell. This complex behavior can be related to the structure of the energy landscape and the existence of a continuous energy-barrier distribution. read less NOT USED (high confidence) H. S. Park, “A multiscale finite element method for the dynamic analysis of surface‐dominated nanomaterials,” International Journal for Numerical Methods in Engineering. 2010. link Times cited: 5 Abstract: The purpose of this article is to present a multiscale finit… read moreAbstract: The purpose of this article is to present a multiscale finite element method that captures nanoscale surface stress effects on the dynamic mechanical behavior of nanomaterials. The method is based upon arguments from crystal elasticity, i.e. the Cauchy–Born rule, but significantly extends the capability of the standard Cauchy–Born rule by accounting for critical nanoscale surface stress effects, which are well known to have a significant effect on the mechanics of crystalline nanostructures. We present the governing equations of motion including surface stress effects, and demonstrate that the methodology is general and thus enables simulations of both metallic and semiconducting nanostructures. The numerical examples on elastic wave propagation and dynamic tensile and compressive loading show the ability of the proposed approach to capture surface stress effects on the dynamic behavior of both metallic and semiconducting nanowires, and demonstrate the advantages of the proposed approach in studying the deformation of nanostructures at strain rates and time scales that are inaccessible to classical molecular dynamics simulations. Copyright © 2010 John Wiley & Sons, Ltd. read less NOT USED (high confidence) X. W. Zhou, R. Jones, and S. Aubry, “Molecular Dynamics Prediction of Thermal Conductivity of GaN Films and Wires at Realistic Length Scales,” Physical Review B. 2010. link Times cited: 23 Abstract: Recent molecular dynamics simulation methods have enabled th… read moreAbstract: Recent molecular dynamics simulation methods have enabled thermal conductivity of bulk materials to be estimated. In these simulations, periodic boundary conditions are used to extend the system dimensions to the thermodynamic limit. Such a strategy cannot be used for nanostructures with finite dimensions which are typically much larger than it is possible to simulate directly. To bridge the length scales between the simulated and the actual nanostructures, we perform large-scale molecular dynamics calculations of thermal conductivities at different system dimensions to examine a recently developed conductivity vs dimension scaling theory for both film and wire configurations. We demonstrate that by an appropriate application of the scaling law, reliable interpolations can be used to accurately predict thermal conductivity of films and wires as a function of film thickness or wire radius at realistic length scales from molecular dynamics simulations. We apply this method to predict thermal conductivities for GaN wurtzite nanostructures. read less NOT USED (high confidence) G. Lucas, M. Bertolus, and L. Pizzagalli, “An environment-dependent interatomic potential for silicon carbide: calculation of bulk properties, high-pressure phases, point and extended defects, and amorphous structures,” Journal of Physics: Condensed Matter. 2010. link Times cited: 41 Abstract: An interatomic potential has been developed to describe inte… read moreAbstract: An interatomic potential has been developed to describe interactions in silicon, carbon and silicon carbide, based on the environment-dependent interatomic potential (EDIP) (Bazant et al 1997 Phys. Rev. B 56 8542). The functional form of the original EDIP has been generalized and two sets of parameters have been proposed. Tests with these two potentials have been performed for many properties of SiC, including bulk properties, high-pressure phases, point and extended defects, and amorphous structures. One parameter set allows us to keep the original EDIP formulation for silicon, and is shown to be well suited for modelling irradiation-induced effects in silicon carbide, with a very good description of point defects and of the disordered phase. The other set, including a new parametrization for silicon, has been shown to be efficient for modelling point and extended defects, as well as high-pressure phases. read less NOT USED (high confidence) L. Hale, X. W. Zhou, J. Zimmerman, N. Moody, R. Ballarini, and W. Gerberich, “Molecular dynamics simulation of delamination of a stiff, body-centered-cubic crystalline film from a compliant Si substrate,” Journal of Applied Physics. 2009. link Times cited: 7 Abstract: Compliant substrate technology offers an effective approach … read moreAbstract: Compliant substrate technology offers an effective approach to grow high-quality multilayered films, of importance to microelectronics and microelectromechanical systems devices. By using a thin, soft substrate to relieve the mismatch strain of an epitaxial film, the critical thickness of misfit dislocation formation in the overlayer is effectively increased. Experiments have indicated that stiff films deposited onto Si substrates can delaminate at the interface. However, the atomic mechanisms of the deformation and the fracture of the films have not been well studied. Here, we have applied molecular dynamics simulations to study the delamination of a stiff body-centered-cubic crystalline film from a compliant Si substrate due to tensile loading. The observed mechanical behavior is shown to be relatively independent of small changes in temperature, loading rate, and system size. Fracture occurs at the interface between the two materials resulting in nearly atomically clean surfaces. Dislocations are seen ... read less NOT USED (high confidence) A. Dongare, L. Zhigilei, A. Rajendran, and B. Lamattina, “Interatomic potentials for atomic scale modeling of metal–matrix ceramic particle reinforced nanocomposites,” Composites Part B-engineering. 2009. link Times cited: 15 NOT USED (high confidence) S. Huang, S. Zhang, T. Belytschko, S. S. Terdalkar, and T. Zhu, “Mechanics of nanocrack: Fracture, dislocation emission, and amorphization,” Journal of The Mechanics and Physics of Solids. 2009. link Times cited: 77 NOT USED (high confidence) H. S. Park, “Quantifying the size-dependent effect of the residual surface stress on the resonant frequencies of silicon nanowires if finite deformation kinematics are considered,” Nanotechnology. 2009. link Times cited: 68 Abstract: There are two major objectives to the present work. The firs… read moreAbstract: There are two major objectives to the present work. The first objective is to demonstrate that, in contrast to predictions from linear surface elastic theory, when nonlinear, finite deformation kinematics are considered, the residual surface stress does impact the resonant frequencies of silicon nanowires. The second objective of this work is to delineate, as a function of nanowire size, the relative contributions of both the residual (strain-independent) and the surface elastic (strain-dependent) parts of the surface stress to the nanowire resonant frequencies. Both goals are accomplished by using the recently developed surface Cauchy–Born model, which accounts for nanoscale surface stresses through a nonlinear, finite deformation continuum mechanics model that leads to the solution of a standard finite element eigenvalue problem for the nanowire resonant frequencies. In addition to demonstrating that the residual surface stress does impact the resonant frequencies of silicon nanowires, we further show that there is a strong size dependence to its effect; in particular, we find that consideration of the residual surface stress alone leads to significant errors in predictions of the nanowire resonant frequency, with an increase in error with decreasing nanowire size. Correspondingly, the strain-dependent part of the surface stress is found to have an increasingly important effect on the resonant frequencies of the nanowires with decreasing nanowire size. read less NOT USED (high confidence) K. Yan and A. Soh, “Simulation of surface effects on the intrinsic dissipation of nanooscillators,” Philosophical Magazine Letters. 2009. link Times cited: 2 Abstract: The rapid increase of surface area-volume ratio (SVR) with s… read moreAbstract: The rapid increase of surface area-volume ratio (SVR) with shrinking structure size has a great impact on surface-related intrinsic dissipation, which usually leads to low quality factors for the devices composed of nanoelectromechanical systems. In the present study, the flexural oscillations of nanocantilevers with varying thicknesses and lengths were simulated using the molecular dynamics method, in which the surface effects on the energy dissipation was evaluated when SVR was increased to extremely large values (between 0.4 and 2.0 nm−1). And, it is also interesting to note that the prediction of the size-dependent Young's modulus by means of resonant frequency of the underdamped oscillation showed good agreement with previous findings. read less NOT USED (high confidence) V. Kharlamov, Y. Trushin, E. E. Zhurkin, M. Lubov, and J. Pezoldt, “Study of Si and C adatoms and SiC clusters on the silicon surface by the molecular dynamics method,” Technical Physics. 2008. link Times cited: 7 NOT USED (high confidence) H. S. Park and P. Klein, “Surface stress effects on the resonant properties of metal nanowires: The importance of finite deformation kinematics and the impact of the residual surface stress,” Journal of The Mechanics and Physics of Solids. 2008. link Times cited: 150 NOT USED (high confidence) K. Yan and A. Soh, “Effects of grain boundary cavities on the thermal resistance under ultrashort laser pulse,” Journal of Physics D: Applied Physics. 2008. link Times cited: 2 Abstract: Molecular dynamics simulations were carried out for nonequil… read moreAbstract: Molecular dynamics simulations were carried out for nonequilibrium phonon heat transport across the Si Σ = 5, (3 1 0) symmetric tilt grain boundaries subjected to an ultrashort laser pulse. It has been found that the existence of cavities at the grain boundary will increase the grain boundary potential energy and decrease the heat dissipation, which is evident from the approximately 20% longer relaxation time required for the system to reach thermodynamic equilibrium. Thus, the existence of grain boundary cavities is one of the main hindrances in the thermal management of nanostructures. read less NOT USED (high confidence) H. S. Park and P. Klein, “A Surface Cauchy-Born model for silicon nanostructures,” Computer Methods in Applied Mechanics and Engineering. 2008. link Times cited: 81 NOT USED (high confidence) H. S. Park, “Surface stress effects on the resonant properties of silicon nanowires,” Journal of Applied Physics. 2008. link Times cited: 78 Abstract: The purpose of the present work is to quantify the coupled e… read moreAbstract: The purpose of the present work is to quantify the coupled effects of surface stresses and boundary conditions on the resonant properties of silicon nanowires. We accomplish this by using the surface Cauchy–Born model, which is a nonlinear, finite deformation continuum mechanics model that enables the determination of the nanowire resonant frequencies including surface stress effects through solution of a standard finite element eigenvalue problem. By calculating the resonant frequencies of both fixed/fixed and fixed/free ⟨100⟩ silicon nanowires with unreconstructed {100} surfaces using two formulations, one that accounts for surface stresses and one that does not, it is quantified how surface stresses cause variations in nanowire resonant frequencies from those expected from continuum beam theory. We find that surface stresses significantly reduce the resonant frequencies of fixed/fixed nanowires as compared to continuum beam theory predictions, while small increases in resonant frequency with respect to... read less NOT USED (high confidence) P. A. Apte and X. Zeng, “Anisotropy of crystal-melt interfacial free energy of silicon by simulation,” Applied Physics Letters. 2008. link Times cited: 58 Abstract: We extend the cleaving wall method to a nonpairwise additive… read moreAbstract: We extend the cleaving wall method to a nonpairwise additive potential. Using this method, we compute the anisotropy of crystal-melt interfacial free energy γ for Stillinger–Weber potential of silicon [F. H. Stillinger and T. A. Weber, Phys. Rev. B 31, 5262 (1985)]. The calculated γ for (100), (111), and (110) orientations are 0.42±0.02, 0.34±0.02, and 0.35±0.03J∕m2, respectively. The anisotropy in γ we found is consistent with the experimental observation that Si(100)-melt interface develops (111) facets and also helps in explaining a higher undercooling observed for Si(111)-melt interface in Czochralski method. read less NOT USED (high confidence) M. McDowell, A. Leach, and K. Gall, “Bending and tensile deformation of metallic nanowires,” Modelling and Simulation in Materials Science and Engineering. 2008. link Times cited: 112 Abstract: Using molecular statics simulations and the embedded atom me… read moreAbstract: Using molecular statics simulations and the embedded atom method, a technique for bending silver nanowires and calculating Young's modulus via continuum mechanics has been developed. The measured Young's modulus values extracted from bending simulations were compared with modulus values calculated from uniaxial tension simulations for a range of nanowire sizes, orientations and geometries. Depending on axial orientation, the nanowires exhibit stiffening or softening under tension and bending as size decreases. Bending simulations typically result in a greater variation of Young's modulus values with nanowire size compared with tensile deformation, which indicates a loading-method-dependent size effect on elastic properties at sub-5 nm wire diameters. Since the axial stress is maximized at the lateral surfaces in bending, the loading-method-dependent size effect is postulated to be primarily a result of differences in nanowire surface and core elastic modulus. The divergence of Young's modulus from the bulk modulus in these simulations occurs at sizes below the range in which experiments have demonstrated a size scale effect on elastic properties of metallic nanowires. This difference indicates that other factors beyond native metallic surface properties play a role in experimentally observed nanowire elastic modulus size effects. read less NOT USED (high confidence) D.-B. Zhang, M. Hua, and T. Dumitricǎ, “Stability of polycrystalline and wurtzite Si nanowires via symmetry-adapted tight-binding objective molecular dynamics.,” The Journal of chemical physics. 2008. link Times cited: 45 Abstract: The stability of the most promising ground state candidate S… read moreAbstract: The stability of the most promising ground state candidate Si nanowires with less than 10 nm in diameter is comparatively studied with objective molecular dynamics coupled with nonorthogonal tight-binding and classical potential models. The computationally expensive tight-binding treatment becomes tractable due to the substantial simplifications introduced by the presented symmetry-adapted scheme. It indicates that the achiral polycrystalline of fivefold symmetry and the wurtzite wires of threefold symmetry are the most favorable quasi-one-dimensional Si arrangements. Quantitative differences with the classical model description are noted over the whole diameter range. Using a Wulff energy decomposition approach it is revealed that these differences are caused by the inability of the classical potential to accurately describe the interaction of Si atoms on surfaces and strained morphologies. read less NOT USED (high confidence) B. Puchala, M. Falk, and K. Garikipati, “Elastic effects on relaxation volume tensor calculations,” Physical Review B. 2008. link Times cited: 16 Abstract: Relaxation volume tensors quantify the effect of stress on d… read moreAbstract: Relaxation volume tensors quantify the effect of stress on diffusion of crystal defects. Continuum linear elasticity predicts that calculations of these parameters using periodic boundary conditions do not suffer from systematic deviations due to elastic image effects and should be independent of the supercell size or symmetry. In practice, however, calculations of formation volume tensors of the $⟨110⟩$ interstitial in Stillinger--Weber silicon demonstrate that changes in bonding at the defect affect the elastic moduli and result in system-size dependent relaxation volumes. These vary with the inverse of the system size. Knowing the rate of convergence permits accurate estimates of these quantities from modestly sized calculations. Furthermore, within the continuum linear elasticity assumptions, the average stress can be used to estimate the relaxation volume tensor from constant volume calculations. read less NOT USED (high confidence) R. E. Rudd and B. L. Lawrence, “Mechanics of silicon nanowires: size-dependent elasticity from first principles,” Molecular Simulation. 2008. link Times cited: 38 Abstract: We discuss size-dependent elastic properties in the context … read moreAbstract: We discuss size-dependent elastic properties in the context of our recent work on the mechanics of silicon nanowires. The results are based on first-principles density functional theory calculations. We focus especially on the size dependence of the Young's modulus, but also comment on the size dependence of the residual stress and the equilibrium length of the hydrogen-passivated Si nanowires. We compare these results to prior results from classical molecular dynamics based on empirical potentials. read less NOT USED (high confidence) W. Zhang, N. Mingo, and T. Fisher, “Simulation of phonon transport across a non-polar nanowire junction using an atomistic Green’s function method,” Physical Review B. 2007. link Times cited: 57 Abstract: Phonon transport across a non-polar nanowire situated betwee… read moreAbstract: Phonon transport across a non-polar nanowire situated between two semi-infinite contacts is simulated in this paper using the atomistic Green's function method. Abrupt geometric changes between the nanowire and bulk contacts are handled by self-energy matrices obtained from bare surface Green's functions. Transport properties such as phonon transmission functions and thermal conductances are calculated, and their dependencies on the interatomic potential, length, diameter, shape, and lattice orientation are investigated. The results reveal that the overall thermal conductance of the nanowire\char21{}bulk-contact structure increases with nanowire diameter while the normalized thermal conductance approaches an asymptotic value. Thermal conductance decreases significantly with increasing nanowire length and converges to that of the single-contact case. This method can be generalized to study phonon transport through a variety of nanostructures between bulk contacts. read less NOT USED (high confidence) P. Valentini and T. Dumitricǎ, “Molecular Dynamics Simulations of Nanoparticle-Surface Collisions in Crystalline Silicon,” Journal of Nano Research. 2007. link Times cited: 7 Abstract: We present a microscopic description for the impacting proce… read moreAbstract: We present a microscopic description for the impacting process of silicon nanospheres onto a silicon substrate. In spite of the relatively low energy regime considered (up to 1 eV/atom), the impacting process exhibits a rich behavior: A rigid Hertzian model is valid for speeds below 500 m/s, while a quasi-ellipsoidal deformation regime emerges at larger speeds. Furthermore, for speeds up to 1000 m/s the particle undergoes a soft landing and creates a long-lived coherent surface phonon. Higher speeds lead to a rapid attenuation of the coherent phonon due to a partial diamond cubic to-tin phase transformation occurring in the particle. read less NOT USED (high confidence) M. Demkowicz, A. Argon, D. Farkas, and M. Frary, “Simulation of plasticity in nanocrystalline silicon,” Philosophical Magazine. 2007. link Times cited: 46 Abstract: Molecular dynamics investigation of plasticity in a model na… read moreAbstract: Molecular dynamics investigation of plasticity in a model nanocrystalline silicon system demonstrates that inelastic deformation localizes in intergranular regions. The carriers of plasticity in these regions are atomic environments, which can be described as high-density liquid-like amorphous silicon. During fully developed flow, plasticity is confined to system-spanning intergranular zones of easy flow. As an active flow zone rotates out of the plane of maximum resolved shear stress during deformation to large strain, new zones of easy flow are formed. Compatibility of the microstructure is accommodated by processes such as grain rotation and formation of new grains. Nano-scale voids or cracks may form if stress concentrations emerge which cannot be relaxed by a mechanism that simultaneously preserves microstructural compatibility. read less NOT USED (high confidence) K. Sastry, D. Goldberg, and D. D. Johnson, “Scalability of a Hybrid Extended Compact Genetic Algorithm for Ground State Optimization of Clusters,” Materials and Manufacturing Processes. 2007. link Times cited: 28 Abstract: We analyze the utility and scalability of extended compact g… read moreAbstract: We analyze the utility and scalability of extended compact genetic algorithm (eCGA)—a genetic algorithm (GA) that automatically and adaptively mines the regularities of the fitness landscape using machine learning methods and information theoretic measures—for ground state optimization of clusters. In order to reduce the computational time requirements while retaining the high reliability of predicting near-optimal structures, we employ two efficiency-enhancement techniques: (1) hybridizing eCGA with a local search method, and (2) seeding the initial population with lowest energy structures of a smaller cluster. The proposed method is exemplified by optimizing silicon clusters with 4–20 atoms. The results indicate that the population size required to obtain near-optimal solutions with 98% probability scales sub linearly (as Θ(n 0.83)) with the cluster size. The total number of function evaluations (cluster energy calculations) scales sub-cubically (as Θ(n 2.45)), which is a significant improvement over exponential scaling of poorly designed evolutionary algorithms. read less NOT USED (high confidence) B. Yang and V. Tewary, “Multiscale modeling of point defects in Si-Ge(001) quantum wells,” Physical Review B. 2007. link Times cited: 8 Abstract: A computationally efficient hybrid Green's function (GF… read moreAbstract: A computationally efficient hybrid Green's function (GF) technique is developed for multiscale modeling of point defects in a trilayer lattice system that links seamlessly the length scales from lattice (subnanometers) to continuum (bulk). The model accounts for the discrete structure of the lattice including nonlinear effects at the atomistic level and full elastic anisotropy at the continuum level. The model is applied to calculate the discrete core structure of point defects (vacancies and substitutional impurities) in Si-Ge(001) quantum wells (QWs) that are of contemporary technological interest. Numerical results are presented for the short range and long range lattice distortions and strains in the lattice caused by the defects and their formation energy and Kanzaki forces that are basic characteristics of the defects. The continuum and the lattice GFs of the material system are used to link the different length scales, which enables us to model the point defects and extended defects such as the quantum well in a unified formalism. Nonlinear effects in the core of the point defects are taken into account by using an iterative scheme. The Tersoff potential is used to set up the lattice structure, compute the unrelaxed forces and force constants in the lattice, andmore » derive the elastic constants required for the continuum GF. It is found that the overall elastic properties of the material and the properties of defects vary considerably when the material is strained from the bulk to the QW state. This change in the defect properties is very significant and can provide a characteristic signature of the defect. For example, in the case of a single vacancy in Ge, the strain reverses the sign of the relaxation volume. It is also found that the defect properties, such as the defect core structures, change abruptly across a Ge/Si interface. The transition occurs over a region extending from two to four lattice constants, depending upon the defect species.« less read less NOT USED (high confidence) R. D. Menezes, J. F. Justo, and L. Assali, “Energetics of silicon nanowires: a molecular dynamics investigation,” physica status solidi (a). 2007. link Times cited: 4 Abstract: Silicon nanowires, with the 〈100〉 and 〈110〉 growth direction… read moreAbstract: Silicon nanowires, with the 〈100〉 and 〈110〉 growth directions and at several surface facet configurations, were investigated by molecular dynamics simulations. We considered three commonly used interatomic potentials for silicon, and tested the reliability of each model to describe silicon nanowires. We find that, for each growth direction, the facet family plays a central role on the nanowire energy, which follows a universal scaling law as a function of the nanowire perimeter. Those results were discussed in the context of recent experimental and ab initio data. (© 2007 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim) read less NOT USED (high confidence) B. Lee and R. Rudd, “First-principles calculation of mechanical properties of Si nanowires and comparison to nanomechanical theory,” Physical Review B. 2007. link Times cited: 109 Abstract: We report the results of first-principles density functional… read moreAbstract: We report the results of first-principles density functional theory calculations of the Young's modulus and other mechanical properties of hydrogen-passivated Si {l_angle}001{r_angle} nanowires. The nanowires are taken to have predominantly {l_brace}100{r_brace}surfaces, with small {l_brace}110{r_brace} facets according to the Wulff shape. The Young's modulus, the equilibrium length and the constrained residual stress of a series of prismatic beams of differing sizes are found to have size dependences that scale like the surface area to volume ratio for all but the smallest beam. The results are compared with a continuum model and the results of classical atomistic calculations based on an empirical potential. We attribute the size dependence to specific physical structures and interactions. In particular, the hydrogen interactions on the surface and the charge density variations within the beam are quantified and used both to parameterize the continuum model and to account for the discrepancies between the two models and the first-principles results. read less NOT USED (high confidence) R. Drautz, X. W. Zhou, D. Murdick, B. Gillespie, H. Wadley, and D. Pettifor, “Analytic bond-order potentials for modelling the growth of semiconductor thin films,” Progress in Materials Science. 2007. link Times cited: 28 NOT USED (high confidence) J. Samela, K. Nordlund, J. Keinonen, and V. Popok, “Comparison of silicon potentials for cluster bombardment simulations,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2007. link Times cited: 26 NOT USED (high confidence) B. Yang and V. Tewary, “Efficient Green’s Function Modeling of Line and Surface Defects in Multilayered Anisotropic Elastic and Piezoelectric Materials,” Cmes-computer Modeling in Engineering & Sciences. 2006. link Times cited: 17 Abstract: Green’s function (GF) modeling of defects may take effect on… read moreAbstract: Green’s function (GF) modeling of defects may take effect only if the GF as well as its various integrals over a line, a surface and/or a volume can be efficiently evaluated. The GF is needed in modeling a point defect, while integrals are needed in modeling line, surface and volumetric defects. In a matrix of multilayered, generally anisotropic and linearly elastic and piezoelectric materials, the GF has been derived by applying 2D Fourier transforms and the Stroh formalism. Its use involves another two dimensions of integration in the Fourier inverse transform. A semi-analytical scheme has been developed previously for efficient evaluation of the GF. In this paper, an efficient scheme for evaluation of the line and surface integrals of the GF is presented. These integrals are obtained by carrying out the integration over the physical domain analytically and then over the transform domain numerically. The efficiency is thus comparable to that in the evaluation of the GF. The high efficiency in the evaluation of the surface integral is of particular value to the modeling of dislocations due to the lack of a line-defect treatment of this group of defects (originally, of uniform planar distribution of force dipoles) in a multilayered heterogeneous matrix. Numerical examples of nitride semiconductors with strong piezoelectric effect are presented to demonstrate the efficiency and accuracy of the present scheme. keyword: Anisotropy, Defects, Dislocations, Elasticity, Fourier transforms, Green’s function, Multilayers, Piezoelectricity, Semiconductors, Steps, Stroh formalism. 1 Publication of the National Institute of Standards and Technology, an agency of the US Government; not subject to copyright. 2 Correspondence author. Department of Mechanical and Aerospace Engineering, Florida Institute of Technology, Melbourne, FL 32901. Email: boyang@fit.edu. 3 Materials Reliability Division, National Institute of Standards and Technology, Boulder, CO 80305 read less NOT USED (high confidence) A. Argon and M. Demkowicz, “Atomistic simulation and analysis of plasticity in amorphous silicon,” Philosophical Magazine. 2006. link Times cited: 33 Abstract: The principal findings of a comprehensive computational simu… read moreAbstract: The principal findings of a comprehensive computational simulation of plastic flow in amorphous Si – presented elsewhere in detail – are summarized. The unit plastic events have been identified to consist of discrete shear transformations triggered at characteristic thresholds of stress that result in transformation shear strains of about 0.015. Based on these findings, a kinetic model of plastic flow is proposed that provides for the temperature dependence of the plastic flow resistance and explains the evolution of a unique flow state starting from different amorphous structures. It is proposed that these findings should be broadly applicable to other strongly bonded glassy covalent compounds. §Dedicated to F. R. N. Nabarro on the occasion of his 90th birthday, in recognition of six and a half decades of insightful contributions to materials science. read less NOT USED (high confidence) Z. Tang, H. Zhao, G. Li, and N. Aluru, “Finite-temperature quasicontinuum method for multiscale analysis of silicon nanostructures,” Physical Review B. 2006. link Times cited: 99 Abstract: In this paper, we extend the quasicontinuum approach for a m… read moreAbstract: In this paper, we extend the quasicontinuum approach for a multiscale analysis of silicon nanostructures at finite temperature. The quasicontinuum method uses the classical continuum mechanics framework, but the constitutive response of the system is determined by employing an atomistic description. For finite-temperature solid systems under isothermal conditions, the constitutive response is determined by using the Helmholtz free energy density. The static part of the Helmholtz free energy density is obtained directly from the interatomic potential while the vibrational part is calculated by using the theory of quantum-mechanical lattice dynamics. Specifically, we investigate three quasiharmonic models, namely the real space quasiharmonic model, the local quasiharmonic model, and the reciprocal space quasiharmonic model, to compute the vibrational free energy. Using the finite-temperature quasicontinuum method, we compute the effect of the temperature and strain on the phonon density of states, phonon Gruneisen parameters, and the elastic properties of the Tersoff silicon. We also compute the mechanical response of silicon nanostructures for various external loads and the results are compared to molecular dynamics simulations. read less NOT USED (high confidence) R. Zhu, E. Pan, P. W. Chung, X. Cai, K. M. Liew, and A. Buldum, “Atomistic calculation of elastic moduli in strained silicon,” Semiconductor Science and Technology. 2006. link Times cited: 113 Abstract: Strained silicon is becoming a new technology in silicon ind… read moreAbstract: Strained silicon is becoming a new technology in silicon industry where the novel strain-induced features are utilized. In this paper we present a molecular dynamic prediction for the elastic stiffnesses C11, C12 and C44 in strained silicon as functions of the volumetric strain level. Our approach combines basic continuum mechanics with the classical molecular dynamic approach, supplemented with the Stillinger–Weber potential. Using our approach, the bulk modulus, effective elastic stiffnesses C11, C12 and C44 of the strained silicon, including also the effective Young's modulus and Poisson's ratio, are all calculated and presented in terms of figures and formulae. In general, our simulation indicates that the bulk moduli, C11 and C12, increase with increasing volumetric strain whilst C44 is almost independent of the volumetric strain. The difference between strained moduli and those at zero strain can be very large, and therefore use of standard free-strained moduli should be cautious. read less NOT USED (high confidence) P. Lorazo, L. J. Lewis, and M. Meunier, “Thermodynamic pathways to melting, ablation, and solidification in absorbing solids under pulsed laser irradiation,” Physical Review B. 2006. link Times cited: 293 Abstract: The thermodynamic pathways involved in laser irradiation of … read moreAbstract: The thermodynamic pathways involved in laser irradiation of absorbing solids are investigated in silicon for pulse durations of $500\phantom{\rule{0.3em}{0ex}}\mathrm{fs}$ and $100\phantom{\rule{0.3em}{0ex}}\mathrm{ps}$. This is achieved by accounting for carrier and atom dynamics within a combined Monte Carlo and molecular-dynamics scheme and simultaneously tracking the time evolution of the irradiated material in $\ensuremath{\rho}\text{\ensuremath{-}}T\text{\ensuremath{-}}P$ space. Our simulations reveal thermal changes in long-range order and state of aggregation driven, in most cases, by nonequilibrium states of rapidly heated or promptly cooled matter. Under femtosecond irradiation near the ablation threshold, the system is originally pulled to a near-critical state following rapid $(\ensuremath{\lesssim}{10}^{\ensuremath{-}12}\phantom{\rule{0.3em}{0ex}}\mathrm{s})$ disordering of the mechanically unstable crystal and isochoric heating of the resulting metallic liquid. The latter is then adiabatically cooled to the liquid-vapor regime where phase explosion of the subcritical, superheated melt is initiated by a direct conversion of translational, mechanical energy into surface energy on a $\ensuremath{\sim}{10}^{\ensuremath{-}12}--{10}^{\ensuremath{-}11}\phantom{\rule{0.3em}{0ex}}\mathrm{s}$ time scale. At higher fluences, matter removal involves, instead, the fragmentation of an initially homogeneous fluid subjected to large strain rates upon rapid, supercritical expansion in vacuum. Under picosecond irradiation, homogeneous and, at later times, heterogeneous melting of the superheated solid are followed by nonisochoric heating of the molten metal. In this case, the subcritical liquid material is subsequently cooled onto the binodal by thermal conduction and explosive boiling does not take place; as a result, ablation is associated with a ``trivial'' fragmentation process, i.e., the relatively slow expansion and dissociation into liquid droplets of supercritical matter near thermodynamic equilibrium. This implies a liquid-vapor equilibration time of $\ensuremath{\sim}{10}^{\ensuremath{-}11}--{10}^{\ensuremath{-}10}\phantom{\rule{0.3em}{0ex}}\mathrm{s}$ and heating along the binodal under nanosecond irradiation. Solidification of the nonablated, supercooled molten material is eventually observed on a $\ensuremath{\sim}{10}^{\ensuremath{-}11}--{10}^{\ensuremath{-}9}\phantom{\rule{0.3em}{0ex}}\mathrm{s}$ time scale, irrespective of the pulse duration. read less NOT USED (high confidence) D. E. Kim and S. I. Oh, “Atomistic simulation of structural phase transformations in monocrystalline silicon induced by nanoindentation,” Nanotechnology. 2006. link Times cited: 111 Abstract: Structural phase transformations of silicon during nanoinden… read moreAbstract: Structural phase transformations of silicon during nanoindentation were investigated in detail at the atomic level. The molecular dynamics simulations of nanoindentation on the (100) and (111) surface of single crystalline silicon were simulated, and this supported the theoretical prediction of the anisotropic behaviour of structural phase transformations. Simulations showed that microscopic aspects of phase transformation varied according to the crystallographic orientation of the contact surface and were directly linked to the slip system of silicon. In the transformed region along the centreline, the crystalline structure of Si-II and the amorphous structure were observed when silicon was loaded in the [100] and [111] directions, respectively. Simultaneously, metastable phases with fourfold coordination, such as Si-III and Si-XII, were formed by the inhomogeneous distortion in the slip direction of silicon and observed along the direction. Additionally, our results indicated that the deviatoric stress added to the hydrostatic pressure induced by loading was an indispensable factor for the structural phase transformation to Si-II during nanoindentation on the (100) surface. read less NOT USED (high confidence) P. Erhart and K. Albe, “Molecular Dynamics Simulations of Gas Phase Condensation of Silicon Carbide Nanoparticles,” Advanced Engineering Materials. 2005. link Times cited: 12 Abstract: Gas phase condensation of silicon and silicon carbide nanopa… read moreAbstract: Gas phase condensation of silicon and silicon carbide nanoparticles is studied by molecular-dynamics simulations. By using a recently developed bond-order potential for Si, C and SiC we investigate the fundamental processes governing nucleation and growth of SiC nanoparticles. For the case of elemental silicon particles we show that variations in the binding energy of dimers, which represent stable nuclei for the condensation process, significantly affect the long time evolution of the cluster formation process. A detailed analysis of the molecular reactions during the early stages of SiC particle growth is presented. Reactions, in which silicon monomers are formed, are dominant in case of stoichiometric composition of the precursor gas. Moreover, we find the formation of carbon-dominated species to be preferred and a sensitive dependence of the particle composition and morphology on the processing conditions, especially the cooling and precursor gas composition. read less NOT USED (high confidence) S. Hara, S. Izumi, T. Kumagai, and S. Sakai, “Surface energy, stress and structure of well-relaxed amorphous silicon: A combination approach of ab initio and classical molecular dynamics,” Surface Science. 2005. link Times cited: 58 NOT USED (high confidence) R. Holenstein, S. Kirkwood, R. Fedosejevs, and Y. Tsui, “Simulation of femtosecond laser ablation of silicon,” Photonics North. 2004. link Times cited: 16 Abstract: Femtosecond laser ablation is an important process in microm… read moreAbstract: Femtosecond laser ablation is an important process in micromachining and nanomachining of microelectronic, optoelectronic, biophotonic and MEMS components. The process of laser ablation of silicon is being studied on an atomic level using molecular dynamics simulations. We investigate ablation thresholds for Gaussian laser pulses of 800 nm wavelength, in the range of a few hundred femtoseconds in duration. Absorption is modelled via linear and 2-photon absorption processes into a hot electron bath which then transfers energy into the crystal lattice. The simulation box is a narrow column approximately 5.4 nm x 5.4 nm x 81 nm with periodic boundaries in the x and y transverse directions and a 1-D heat flow model at the bottom coupled to a heat bath to simulate an infinite bulk medium corresponding to the solid bulk material. A modified Stillinger-Weber potential is used to model the silicon atoms. The calculated thresholds are compared to various reported experimental values for the ablation threshold of silicon. We provide an overview of the code and discuss the simulation techniques used. read less NOT USED (high confidence) C. L. Allred, X. Yuan, M. Bazant, and L. Hobbs, “Elastic constants of defected and amorphous silicon with the environment-dependent interatomic potential,” Physical Review B. 2004. link Times cited: 31 Abstract: The elastic constants of a wide range of models of defected … read moreAbstract: The elastic constants of a wide range of models of defected crystalline and amorphous silicon are calculated, using the environment-dependent interatomic potential (EDIP). The defected crystalline simulation cells contain randomly generated defect distributions. An extensive characterization of point defects is performed, including structure, energy and influence on elastic constants. Three important conclusions are drawn. (1) Defects have independent effects on the elastic constants of silicon up to (at least) a defect concentration of 0.3%. (2) The linear effect of Frenkel pairs on the Young's modulus of silicon is -1653 GPa per defect fraction. (3) 17 different point defect types cause a very similar decrease in the Young's modulus: -(0.28{+-}0.05)% when calculated in isolation using a 1728-atom cell. These principles will be very useful for predicting the effect of radiation damage on the elastic modulus of silicon in the typical case in which point-defect concentrations can be estimated, but the exact distribution and species of defects is unknown. We also study amorphous samples generated in quenching the liquid with EDIP, including an ideal structure of perfect fourfold coordination, samples with threefold and fivefold coordinated defects, one with a nanovoid, and one with an amorphous inclusion in a crystalline matrix.more » In the last case, a useful finding is that the change in the Young's modulus is simply related to the volume fraction of amorphous material, as has also been observed by experiment.« less read less NOT USED (high confidence) T. Lim, “Connection between the 2-body energy of the Kaxiras-Pandey and the Biswas-Hamann potentials,” Czechoslovak Journal of Physics. 2004. link Times cited: 10 NOT USED (high confidence) P. Beaucage and N. Mousseau, “Liquid–liquid phase transition in Stillinger–Weber silicon,” Journal of Physics: Condensed Matter. 2004. link Times cited: 39 Abstract: It was recently demonstrated that Stillinger–Weber silicon u… read moreAbstract: It was recently demonstrated that Stillinger–Weber silicon undergoes a liquid–liquid first-order phase transition deep into the supercooled region (Sastry and Angell 2003 Nat. Mater. 2 739). Here we study the effects of perturbations on this phase transition. We show that the order of the liquid–liquid transition changes with negative pressure. We also find that the liquid–liquid transition disappears when the three-body term of the potential is strengthened by as little as 5%. This implies that the details of the potential could affect strongly the nature and even the existence of the liquid–liquid phase. read less NOT USED (high confidence) R. Wagner and E. Gulari, “Simulation of Ge’Si intermixing during heteroepitaxy,” Physical Review B. 2004. link Times cited: 26 Abstract: During epitaxial growth of Ge on Si~001!, intermixing can oc… read moreAbstract: During epitaxial growth of Ge on Si~001!, intermixing can occur between the deposited Ge and the Si substrate. We show that although Ge prefers to wet the surface, entropy drives some fraction into the underlying layers. We present a simple model of intermixing by equilibration of the top crystal layers in the absence of bulk diffusion. The equilibration is performed with a flexible lattice Monte Carlo simulation. Ultimately, intermixing leads to a temperature-dependent graded Ge concentration. The resulting evolution of chemical potential is consistent with the onset of islanding after 3‐ 4 monolayers of deposition. read less NOT USED (high confidence) T. Lim, “Relationship between the 2-body Parameters of the Biswas-Hamann and the Bauer-Maysenholder-Seeger Potential Functions,” Czechoslovak Journal of Physics. 2004. link Times cited: 9 NOT USED (high confidence) K. Shintani, T. Nakajima, and S. Kameoka, “Atomistic model of limited-thickness Si(001) epitaxy at low temperatures,” Journal of Applied Physics. 2004. link Times cited: 7 Abstract: Limited-thickness homoepitaxial growth on a Si(001) surface … read moreAbstract: Limited-thickness homoepitaxial growth on a Si(001) surface at low temperatures is investigated by using the classical molecular-dynamics method with the Stillinger-Weber potential. The simulation begins with preliminary equilibration of the substrate at a specified temperature. 256 silicon atoms with the energy of 0.2 eV are then deposited one by one on the substrate. The simulations are performed at the temperatures 300, 500, 700, and 1000 K. At 300 and 500 K, the initial three or four monolayers grow epitaxially, and the subsequent layers form amorphouslike structures. At 700 and 1000 K, the deposited atoms form epitaxial structures throughout the simulation. In the epitaxial growth mode, 2×1 dimer rows are observed to align along alternately perpendicular 〈110〉 directions in successive atomic layers. Tracking a few atoms on the substrate surface reveals that these transient anisotropic surface structures are created by the breaking and reconstruction of dimers due to the impingement of deposited atoms... read less NOT USED (high confidence) R. Rudd, “Coarse-Grained Molecular Dynamics for Computer Modeling of Nanomechanical Systems,” International Journal for Multiscale Computational Engineering. 2003. link Times cited: 28 Abstract: Unique challenges for computer modeling and simulation arise… read moreAbstract: Unique challenges for computer modeling and simulation arise in the course of the development and design of nanoscale mechanical systems. Materials often exhibit unconventional behavior at the nanoscale that can affect device operation and failure. This uncertainty poses a problem because of the limited experimental characterization at these ultra-small length scales. In this Article we give an overview of how we have used concurrent multiscale modeling techniques to address some of these issues. Of particular interest are the dynamic and temperature-dependent processes found in nanomechanical systems. We focus on the behavior of sub-micron mechanical components of Micro-Electro-Mechanical Systems (MEMS) and Nano-Electro-Mechanical Systems (NEMS), especially flexural-mode resonators. The concurrent multiscale methodology we have developed for NEMS employs an atomistic description of millions of atoms in relatively small but key regions of the system, coupled to, and run concurrently with, a generalized finite element model of the periphery. We describe two such techniques. The more precise model, Coarse-Grained Molecular Dynamics (CGMD), describes the dynamics on a mesh of elements, but the equations of motion are built up from the underlying atomistic physics to ensure a smooth coupling between regions governed by different length scales. In many cases the degrees of smoothness ofmore » the coupling provided by CGMD is not necessary. The hybrid Coupling of Length Scales (CLS) methodology, combining molecular dynamics with conventional finite element modeling, provides a suitable technique for these cases at a greatly reduced computation expense. We review these models and some of the results we have obtained regarding size effects in the elasticity and dissipation of nanomechanical systems.« less read less NOT USED (high confidence) J. Godet, L. Pizzagalli, S. Brochard, and P. Beauchamp, “Comparison between classical potentials and ab initio methods for silicon under large shear,” Journal of Physics: Condensed Matter. 2003. link Times cited: 47 Abstract: The homogeneous shear of the {111} planes along the directio… read moreAbstract: The homogeneous shear of the {111} planes along the direction of bulk silicon has been investigated using ab initio techniques, to better understand the strain properties of both shuffle and glide set planes. Similar calculations have been done with three empirical potentials, Stillinger–Weber, Tersoff and EDIP, in order to find the one giving the best results under large shear strains. The generalized stacking fault energies have also been calculated with these potentials to complement this study. It turns out that the Stillinger–Weber potential better reproduces the ab initio results, for the smoothness and the amplitude of the energy variation as well as the localization of shear in the shuffle set. read less NOT USED (high confidence) S. Sastry and C. A. Angell, “Liquid–liquid phase transition in supercooled silicon,” Nature Materials. 2003. link Times cited: 451 NOT USED (high confidence) M. Prasad and T. Sinno, “Internally Consistent Approach for Modeling Solid-State Aggregation: I. Atomistic Calculations of Vacancy Clustering in Silicon,” Physical Review B. 2003. link Times cited: 33 Abstract: A computational framework is presented for describing the nu… read moreAbstract: A computational framework is presented for describing the nucleation and growth of vacancy clusters in crystalline silicon. The overall approach is based on a parametrically consistent comparison between two representations of the process in order to provide a systematic method for probing the details of atomic mechanisms responsible for aggregation. In this paper, the atomistic component of the overall framework is presented. First, a detailed set of targeted atomistic simulations are described that characterize fully the thermodynamic and transport properties of vacancy clusters over a wide range of sizes. It is shown that cluster diffusion is surprisingly favorable because of the availability of multiple, almost degenerate configurations. A single large-scale parallel molecular dynamics simulation is then used to compute directly the evolution of the vacancy cluster size distribution in a supersaturated system initially containing 1000 uniformly distributed vacancies in a host lattice of 216 000 Si atoms at 1600 K. The results of this simulation are interpreted in the context of mean-field scaling theory based on the observed power-law evolution of the size distribution moments. It is shown that the molecular dynamics results for aggregation of vacancy clusters, particularly the evolution of the average cluster size, can be very well represented by a highly simplified mean-field model. A direct comparison to a detailed continuum model is made in a subsequent article. read less NOT USED (high confidence) C. R. Miranda, R. W. Nunes, and A. Antonelli, “Temperature effects on dislocation core energies in silicon and germanium,” Physical Review B. 2003. link Times cited: 14 Abstract: Temperature effects on the energetics of the 90° partial dis… read moreAbstract: Temperature effects on the energetics of the 90° partial dislocation in silicon and germanium are investigated, using nonequilibrium methods to estimate free energies, coupled with Monte Carlo simulations. Atomic interactions are described by Tersoff and environment-dependent interatomic potentials. Our results indicate that the vibrational entropy has the effect of increasing the difference in free energy between the two possible reconstructions of the 90° partial, namely, the single-period and the double-period geometries. This effect further increases the energetic stability of the double-period reconstruction at high temperatures. The results also indicate that anharmonic effects may play an important role in determining the structural properties of these defects in the high-temperature regime. read less NOT USED (high confidence) C. Köhler, “Atomistic simulations of strain distributions in quantum dot nanostructures,” Journal of Physics: Condensed Matter. 2003. link Times cited: 6 Abstract: Strain distributions around a Ge quantum dot (QD) buried in … read moreAbstract: Strain distributions around a Ge quantum dot (QD) buried in a Si spacer layer are investigated theoretically by means of classical molecular dynamics simulations using the Tersoff potential. Applying periodic boundary conditions laterally, two-dimensional superlattices of QDs are obtained. Strain distributions in systems of different sizes and lattice misorientations are computed in order to explain possible vertical correlations in self-organized three-dimensional QD superstructures. Generally, the strain of relaxed systems displays an oscillatory behaviour as a function of the distance from the QD. For QD systems with growth direction [001], a simple fitting function is used to describe the strain along a vertical path above the QD by an oscillation and a decay according to a power law. For QDs with the shape of a truncated pyramid, the planar strain decays by a power of approximately −3. The period of the oscillation is nearly proportional to the QD superlattice constant and decreases with increasing coordination number of the QD superlattice. In misoriented systems with a small tilt angle about the [110] axis, the region of tensile planar strain above the QD is bent in the direction opposite to the misorientation causing a vertical correlation with lateral shift. For a tilt angle ≈55°, no strain oscillation is found which implies a perfect vertical correlation. read less NOT USED (high confidence) W. Cai, V. V. Bulatob, J. Chang, J. Li, and S. Yip, “Periodic image effects in dislocation modelling,” Philosophical Magazine. 2003. link Times cited: 181 Abstract: The use of periodic boundary conditions for modelling crysta… read moreAbstract: The use of periodic boundary conditions for modelling crystal dislocations is predicated on one's ability to handle the inevitable image effects. This communication deals with an often overlooked mathematical subtlety involved in dealing with the periodic dislocation arrays, that is conditional convergence of the lattice sums of image fields. By analysing the origin of conditional convergence and the numerical artefacts associated with it, we establish a mathematically consistent and numerically efficient procedure for regularization of the lattice sums and the corresponding image fields. The regularized solutions are free from the artefacts caused by conditional convergence and regain periodicity and translational invariance of the periodic supercells. Unlike the other existing methods, our approach is applicable to general anisotropic elasticity and arbitrary dislocation arrangements. The capabilities of this general methodology are demonstrated by application to a variety of situations encountered in atomistic and continuum modelling of crystal dislocations. The applications include introduction of dislocations in the periodic supercell for subsequent atomistic simulations, atomistic calculations of the core energies and the Peierls stress and continuum dislocation dynamics simulations in three dimensions. read less NOT USED (high confidence) L. Pizzagalli, P. Beauchamp, and J. Rabier, “Stability and core structure of undissociated screw dislocations in group IV materials investigated by means of atomistic calculations,” Journal of Physics: Condensed Matter. 2002. link Times cited: 11 Abstract: We have examined the various possible configurations for an … read moreAbstract: We have examined the various possible configurations for an undissociated screw dislocation in group IV materials (Ge, Si, 3C-SiC, diamond) by means of semi-empirical atomistic calculations. A complete structural characterization and a determination of the relative stability are performed. We found that, in contrast to the case for Ge and Si, a geometry with the presence of sp 2 atoms in th ec ore is the most stable structure for 3C-SiC and diamond. This yields a stable screw dislocation configuration i nt he ‘shuffle’ set for Si and Ge, and in th e‘ glide’ set for 3C-SiC and diamond. read less NOT USED (high confidence) J. Godet, L. Pizzagalli, S. Brochard, and P. Beauchamp, “Surface step effects on Si (100) under uniaxial tensile stress, by atomistic calculations,” Scripta Materialia. 2002. link Times cited: 13 NOT USED (high confidence) A. S. Barnard, S. Russo, and G. Leach, “Nearest neighbour considerations in Stillinger-Weber type potentials for diamond,” Molecular Simulation. 2002. link Times cited: 5 Abstract: Results of a preliminary investigation into the effect of va… read moreAbstract: Results of a preliminary investigation into the effect of varying the interaction cutoff on the bulk properties of diamond using a Stillinger-Weber (SW) type potential for C (Diamond) are presented. The interaction cutoff is varied over a range that includes and excludes the second-nearest neighbours. Whilst the original SW potential for silicon only included first-nearest neighbours inside the interaction cut-off, subsequent parameterizations for carbon (diamond) have also included second-nearest neighbours. Elastic and vibration properties of diamond were calculated over a range of cutoff distances used and the results show that certain lattice properties exhibit an approximately linear dependence on the interaction cut-off. read less NOT USED (high confidence) M. Zeifman, B. Garrison, and L. Zhigilei, “Combined molecular dynamics–direct simulation Monte Carlo computational study of laser ablation plume evolution,” Journal of Applied Physics. 2002. link Times cited: 54 Abstract: A two-stage computational model of evolution of a plume gene… read moreAbstract: A two-stage computational model of evolution of a plume generated by laser ablation of an organic solid is proposed and developed. The first stage of the laser ablation, which involves laser coupling to the target and ejection of molecules and clusters, is described by the molecular dynamics (MD) method. The second stage of a long-term expansion of the ejected plume is modeled by the direct simulation Monte Carlo (DSMC) method. The presence of clusters, which comprise a major part of the overall plume at laser fluences above the ablation threshold, presents the main computational challenge in the development of the combined model. An extremely low proportion of large-sized clusters hinders both the statistical estimation of their characteristics from the results of the MD model and the following representation of each cluster size as a separate species, as required in the conventional DSMC. A number of analytical models are proposed and verified for the statistical distributions of translational and inter... read less NOT USED (high confidence) M. Ishimaru, “Atomistic simulations of structural relaxation processes in amorphous silicon,” Journal of Applied Physics. 2002. link Times cited: 36 Abstract: Structural relaxation processes in amorphous silicon (a-Si) … read moreAbstract: Structural relaxation processes in amorphous silicon (a-Si) have been examined by molecular-dynamics (MD) simulations using the Tersoff interatomic potential. The a-Si networks generated by rapid quenching from liquid Si were annealed. Structural changes due to the relaxation of a-Si networks were observed. The present MD simulations reproduce well experimental measurements of changes in radial distribution functions, static structure factors, bond angle distributions, and phonon densities of states due to structural relaxation. read less NOT USED (high confidence) S. Ogata, E. Lidorikis, F. Shimojo, A. Nakano, P. Vashishta, and R. Kalia, “Hybrid finite-element/molecular-dynamics/electronic-density-functional approach to materials simulations on parallel computers,” Computer Physics Communications. 2001. link Times cited: 142 NOT USED (high confidence) N.-xian Chen, S. Jiang, and X. Su, “Theoretical study on the phase stability, site preference, and lattice parameters for Gd(Fe, T)12,” Journal of Physics: Condensed Matter. 2001. link Times cited: 69 Abstract: The stability of the intermetallics Gd(Fe, T)12 and the site… read moreAbstract: The stability of the intermetallics Gd(Fe, T)12 and the site preferences of the ternary 3d or 4d transition element T are investigated by using a series of interatomic pair potentials, ΦFe-Fe(r), ΦFe-Gd(r), ΦFe-T(r), ΦT-T(r), ΦT-Gd(r), and ΦGd-Gd(r), for the first time. The calculated results show that adding either Cr, Mo, Ti, or V atoms makes the crystal cohesive energy of Gd(Fe, T)12 decrease markedly, proving that these atoms can stabilize Gd(Fe, T)12 with ThMn12 structure even though the GdFe12 crystal structure is itself metastable. The calculated lattice parameters are in good agreement with experiment. The amount of cohesive energy decrease is correlated with the species and occupation site of the ternary atoms. The order of site preference of these stabilizing elements T is 8i, 8j, and 8f, with 8i corresponding to the greatest energy decrease. The calculated results further show that the addition of Co, Cu, Ni, Sc, and Zn does not stabilize the GdFe12 phase in the ThMn12 structure. The calculated results reported correspond well to available experimental data indicating that the ab initio interatomic potentials can be used to describe rare-earth materials. read less NOT USED (high confidence) T. Halicioǧlu, “Calculated energetics for adsorption and desorption steps during etching of Si(110) surface by Cl,” Journal of Vacuum Science and Technology. 2001. link Times cited: 3 Abstract: Energetics and the configurational aspects related to the ad… read moreAbstract: Energetics and the configurational aspects related to the adsorption of Cl on the (110) index plane of Si and the subsequent desorption of SiClx species from the surface (leading to etching) were investigated. Calculations were conducted for varying surface Cl concentrations. First and second chlorination steps for surface Si atoms were analyzed and the role played by surface vacancies was investigated. On the Si(110) surface, steric effects coming from repulsive forces among the adsorbed Cl atoms, were found to be quite significant. Results indicate that the second chlorination step which leads to the formation of attached SiCl2 species, is very important in the overall eching process. read less NOT USED (high confidence) T. Lenosky et al., “Highly optimized empirical potential model of silicon,” Modelling and Simulation in Materials Science and Engineering. 2000. link Times cited: 145 Abstract: We fit an empirical potential for silicon using the modified… read moreAbstract: We fit an empirical potential for silicon using the modified embedded atom (MEAM) functional form, which contains a nonlinear function of a sum of pairwise and three-body terms. The three-body term is similar to the Stillinger-Weber form. We parametrized our model using five cubic splines, each with 10 fitting parameters, and fitted the parameters to a large database using the force-matching method. Our model provides a reasonable description of energetics for all atomic coordinations, Z, from the dimer (Z = 1) to fcc and hcp (Z = 12). It accurately reproduces phonons and elastic constants, as well as point defect energetics. It also provides a good description of reconstruction energetics for both the 30° and 90° partial dislocations. Unlike previous models, our model accurately predicts formation energies and geometries of interstitial complexes - small clusters, interstitial-chain and planar {311} defects. read less NOT USED (high confidence) Scheerschmidt, Conrad, Kirmse, Schneider, and Neumann, “Electron microscope characterization of CdSe/ZnSe quantum dots based on molecular dynamics structure relaxations,” Ultramicroscopy. 2000. link Times cited: 20 NOT USED (high confidence) K. Scheerschmidt, D. Conrad, A. Belov, and D. Timpel, “Enhanced semi-empirical potentials in molecular dynamics simulations of wafer bonding,” Materials Science in Semiconductor Processing. 2000. link Times cited: 4 NOT USED (high confidence) M. Schaible, “Empirical Molecular Dynamics Modeling of Silicon and Silicon Dioxide: A Review,” Critical Reviews in Solid State and Materials Sciences. 1999. link Times cited: 28 Abstract: A number of computational methods have been developed over t… read moreAbstract: A number of computational methods have been developed over the last 40 years to simulate the behavior of solid materials with small dimensions. On the macro-scale, Finite Element analysis calculates mechanical stress on micron-sized cantilevers and motors. On the atomic level, newer ab initio methods compute nuclear and electronic behavior of hundred atom models with unprecedented rigor. By implementing the laws of classic mechanics, empirical Molecular Dynamics (MD) programs help bridge these two computational extremes. MD identifies nonelectronic, particle motion for large 100,000 atom cells with good success. MD derives both equilibrium and nonequilibrium properties for many complex condensed regimes; quantitatively (and qualitatively) reaffirms empirical data; aids discovery of new materials processing techniques, and helps predict unknown physical phenomena often only observable under extreme environmental settings. One material of great technical importance to the semiconductor industry is silicon (... read less NOT USED (high confidence) C. T. Reeves, B. A. Ferguson, C. Mullins, G. Sitz, B. A. Helmer, and D. Graves, “Trapping dynamics of ethane on Si(100)-(2×1): Molecular beam experiments and molecular dynamics simulations,” Journal of Chemical Physics. 1999. link Times cited: 14 Abstract: The trapping probability, or physical adsorption probability… read moreAbstract: The trapping probability, or physical adsorption probability, of ethane on a clean Si(100)-(2×1) surface has been measured as a function of the incident translational energy and incident polar angle of the molecule at a surface temperature of 65 K. At all incident angles the trapping probability decreases as the translational energy of the incoming ethane molecule is increased from 0.05 to 1.3 eV. As the incident polar angle, with respect to the surface normal, is increased, the trapping probability decreases. This decrease in trapping probability with increasing polar angle contradicts the idea of normal energy scaling and has been seen in very few cases. Classical molecular dynamics calculations have been employed to study the cause of this unusual angular dependence. This simulation predicts trapping probabilities in good agreement with the experimental data. Analysis of the computed trajectories indicates that the initial site of impact within the unit cell, as well as energy exchange on initial impact with the surface, is important in determining the fate of an incident molecule. Normal momentum of the incident molecule is dissipated during the first impact much more efficiently than is parallel momentum. The simulations also indicate that the observed angular dependence can be explained in terms of parallel momentum accommodation. Large amounts of parallel momentum remaining after initial impact may be converted to normal momentum on subsequent impacts, causing molecules to scatter from the surface. Therefore, molecules that impact the surface at glancing angles and high translational kinetic energies are more likely to scatter from the surface than those at normal incidence or with lower translational kinetic energy. read less NOT USED (high confidence) R. Sahara, H. Mizuseki, K. Ohno, S. Uda, T. Fukuda, and Y. Kawazoe, “Body-centered-cubic lattice model with many-body interactions representing the melting transition in Si,” Journal of Chemical Physics. 1999. link Times cited: 11 Abstract: A body-centered-cubic (BCC) lattice model with realistic man… read moreAbstract: A body-centered-cubic (BCC) lattice model with realistic many-body interactions is introduced and investigated by means of the Metropolis’ Monte Carlo method to describe both crystalline and molten states of Si. Under the simplest assumption that atoms surrounded by tetrahedral first-neighbors only have an energy lower than the other atoms, a clear first-order phase transition including hysteresis is observed between a solid with diamond structure and a melt. Nucleation and domain growth are dynamically observed in certain range of the supercooling. In order to introduce more realistic and accurate lattice-gas models, the Tersoff potential is renormalized and the interactions are mapped onto a BCC lattice. Then, it is found that the phase transition temperature and other thermodynamic properties are dramatically improved compared with the case using the Tersoff potential directly in the lattice model without renormalization. read less NOT USED (high confidence) G. Krasko, B. Rice, and S. Yip, “A bond-order potential for atomistic simulations in iron,” Journal of Computer-Aided Materials Design. 1999. link Times cited: 8 NOT USED (high confidence) D. Hanson, J. Kress, and A. Voter, “AN INTERATOMIC POTENTIAL FOR REACTIVE ION ETCHING OF SI BY CL IONS,” Journal of Chemical Physics. 1999. link Times cited: 26 Abstract: An interatomic potential has been developed to describe the … read moreAbstract: An interatomic potential has been developed to describe the dynamics of Si/Cl systems, with particular relevance to reactive ion etching of Si by energetic Cl ions. We have modified the Stillinger–Weber (SW) potential of Feil et al. by adding two new terms: (1) an embedding term that corrects for the variation in Si–Cl bond strength as a function of the number of neighbors, and (2) a four-body term to describe the variation of the Si–Si bond strength as a function of the number of neighbors of each Si atom and the atom types (a bond order correction). Calculated Si etch rates obtained from molecular dynamics simulations using the new potential are in better agreement with recent experimental results than those obtained with the unmodified potential. Predictions of the stoichiometry of the etch products are also markedly different between the two potentials. read less NOT USED (high confidence) P. Keblinski, D. Wolf, S. Phillpot, and H. Gleiter, “Role of bonding and coordination in the atomic structure and energy of diamond and silicon grain boundaries,” Journal of Materials Research. 1998. link Times cited: 59 Abstract: The high-temperature equilibrated atomic structures and ener… read moreAbstract: The high-temperature equilibrated atomic structures and energies of large-unit-cell grain boundaries (GB’s) in diamond and silicon are determined by means of Monte-Carlo simulations using Tersoff’s potentials for the two materials. Silicon provides a relatively simple basis for understanding GB structural disorder in a purely sp ^3 bonded material against which the greater bond stiffness in diamond combined with its ability to change hybridization in a defected environment from sp ^3 to sp ^2 can be elucidated. We find that due to the purely sp ^3-type bonding in Si, even in highly disordered, high-energy GB’s at least 80% of the atoms are fourfold coordinated in a rather dense confined amorphous structure. By contrast, in diamond even relatively small bond distortions exact a considerable price in energy that favors a change to sp ^2-type local bonding; these competing effects translate into considerably more ordered diamond GB’s; however, at the price of as many as 80% of the atoms being only threefold coordinated. Structural disorder in the Si GB’s is therefore partially replaced by coordination disorder in the diamond GB’s. In spite of these large fractions of three-coordinated GB carbon atoms, however, the three-coordinated atoms are rather poorly connected amongst themselves, thus likely preventing any type of graphite-like electrical conduction through the GB’s. read less NOT USED (high confidence) K. Nordlund, M. Ghaly, and R. Averback, “Mechanisms of ion beam mixing in metals and semiconductors,” Journal of Applied Physics. 1998. link Times cited: 69 Abstract: Ion beam mixing was investigated in crystalline and amorphou… read moreAbstract: Ion beam mixing was investigated in crystalline and amorphous semiconductors and metals using molecular dynamics simulations. The magnitude of mixing in an amorphous element compared to its crystalline counterpart was found to be larger by a factor of 2 or more. Mixing in semiconductors was found to be significantly larger than in a face-centered-cubic (fcc) metal of corresponding mass and atomic density. The difference in mixing between amorphous and crystalline materials is attributed to local relaxation mechanisms occurring during the cooling down phase of the cascade. Comparison of mixing in semiconductors and metals shows that short range structural order also has a significant influence on mixing. The mixing results in fcc metals indicate that the role of the electron–phonon coupling in the evolution of collision cascades may be less significant than previously thought. read less NOT USED (high confidence) T. Sinno, F. von Gottberg, and R. A. Brown, “Investigation of point defect clusters in silicon using parallel molecular dynamics,” Journal of Computer-Aided Materials Design. 1997. link Times cited: 2 NOT USED (high confidence) P. Keblinski, S. Phillpot, D. Wolf, and H. Gleiter, “On the Thermodynamic Stability of Amorphous Intergranular Films in Covalent Materials,” Journal of the American Ceramic Society. 1997. link Times cited: 68 Abstract: The thermodynamic origin, structure, and stability of the th… read moreAbstract: The thermodynamic origin, structure, and stability of the thin amorphous films commonly found in grain boundaries in covalent ceramics are investigated by molecular-dynamics simulation. to focus on the purely thermodynamic aspects, any kinetic effects associated with impurity-controlled interface chemistry are excluded by investigating pure silicon (described by the Stillinger-Weber three-body potential). For this single-component covalent model material, the authors demonstrate that all high-energy boundaries exhibit a universal amorphous structure, with a width of {approximately}0.25 nm, whereas low-energy boundaries are crystalline and much sharper. They also demonstrate that introduction of an amorphous film into a crystalline interface lowers the excess energy to a level similar to the energy of two bulk crystal-amorphous interfaces. The competition between a narrow crystalline boundary structure and a wider amorphous boundary structure is shown to be governed by the relative excess energies of the atoms in the grain boundaries and in the bulk amorphous phase. These observations suggest that, in principle, amorphous grain-boundary films do not require impurities for their stabilization and that, as first proposed by Clarke, an equilibrium grain-boundary phase of uniform thickness can be the result of purely thermodynamic rather than kinetic factors. read less NOT USED (high confidence) L. Marqués, M. Caturla, T. D. Rubia, and G. Gilmer, “Ion beam induced recrystallization of amorphous silicon: A molecular dynamics study,” Journal of Applied Physics. 1996. link Times cited: 43 Abstract: We use molecular dynamics techniques to study the ion beam i… read moreAbstract: We use molecular dynamics techniques to study the ion beam induced enhancement in the growth rate of microcrystals embedded in an amorphous silicon matrix. The influence of the ion beam on the amorphous‐to‐crystal transformation was separated into thermal annealing effects and defect production effects. Thermal effects were simulated by heating the sample above the amorphous melting point, and damage induced effects by introducing several low energy recoils in the amorphous matrix directed at the crystalline grain. In both cases, the growth rate of the microcrystals is enhanced several orders of magnitude with respect to the pure thermal process, in agreement with experimental results. The dynamics of the crystallization process and the defect structures generated during the growth were analyzed and will be discussed. read less NOT USED (high confidence) R. Venkatesh, W. Marlow, R. Lucchese, and J. Schulte, “THE EFFECT OF THE NATURE OF THE INTERACTION POTENTIAL ON CLUSTER REACTION RATES,” Journal of Chemical Physics. 1996. link Times cited: 5 Abstract: The effect of two different interaction potentials, a two‐bo… read moreAbstract: The effect of two different interaction potentials, a two‐body and a many‐body potential, on thermal cluster reaction rates was studied for 2–13 atom nickel clusters using the classical trajectory method. The reaction rates were computed for cluster–monomer and cluster–cluster collisions at T=1200 K, using the bulk and dimer parametrized Lennard‐Jones (LJ) potentials and were compared with the rates previously obtained for these collisional events by using a more realistic many‐body tight‐binding second moment approximation (TB‐SMA) potential. For cluster–monomer collisions, close agreement exists between the reaction cross section results for dimer fitted LJ (LJD) potential and TB‐SMA potential suggesting that the cluster–monomer collisions may be dominated by pairwise interactions. The bulk fitted LJ potential (LJB) underestimates the sticking cross section results of the other two potentials for most cluster sizes. This discrepancy however appears to be due to the relatively smaller cluster binding ene... read less NOT USED (high confidence) A.-Q. Chen and L. Corrales, “Semiempirical methodology for simulating covalently bonded materials: Application to silicon,” Journal of Chemical Physics. 1996. link Times cited: 4 Abstract: A recently introduced semiempirical methodology is used to m… read moreAbstract: A recently introduced semiempirical methodology is used to model and simulate silicon via molecular dynamics. This approach is capable of grasping essential qualitative and quantitative features of the coupling between the electronic coordinates and the geometric structure. Properties of the bulk diamond crystal, the melt and amorphous solid states are obtained using optimization techniques and molecular dynamics simulations. The pair distribution function of the amorphous state is in excellent agreement with experimental and other molecular dynamics simulation results. read less NOT USED (high confidence) A. P. Smith et al., “Si adatom binding and diffusion on the Si(100) surface: Comparison of ab initio, semiempirical and empirical potential results,” Journal of Chemical Physics. 1995. link Times cited: 51 Abstract: The binding energies and configurations for single Si adatom… read moreAbstract: The binding energies and configurations for single Si adatoms on the Si(100) surface are investigated theoretically. Detailed comparisons between previously published and new calculations using classical potentials, semiempirical formulations, and density functional theory (DFT) are made. The DFT calculations used both the plane‐wave‐pseudopotential approach in a periodic slab geometry and the Gaussian‐orbital based all‐electron approach employing cluster geometries. In the local‐density approximation excellent agreement between the cluster and slab results was obtained. Inclusion of gradient corrections to the exchange‐correlation energy significantly improves absolute binding energies and changes relative energies by as much as 0.3–0.5 eV depending on the particular exchange‐correlation functional used. Binding energies and relative energies obtained using the classical potentials disagree with the gradient corrected DFT energies at about the 0.6–0.9 eV level, and most find qualitatively different local... read less NOT USED (high confidence) X. Luo and Z. Tong, “Nano-grooving by Using Multi-tip Diamond Tools.” 2018. link Times cited: 2 NOT USED (high confidence) V. Tewary, “Multiscale Green’s functions for modeling of nanomaterials.” 2015. link Times cited: 3 NOT USED (high confidence) H. S. Park, “Surface stress effects on the critical buckling strains of silicon nanowires,” Computational Materials Science. 2012. link Times cited: 62 NOT USED (high confidence) T. Dumitricǎ, “Computational Nanomechanics of Quasi-one-dimensional Structures in a Symmetry-Adapted Tight Binding Framework.” 2010. link Times cited: 2 NOT USED (high confidence) M. Timonova and B. Thijsse, “Optimizing the MEAM potential for silicon,” Modelling and Simulation in Materials Science and Engineering. 2010. link Times cited: 25 Abstract: By applying simulated annealing techniques we fit the modifi… read moreAbstract: By applying simulated annealing techniques we fit the modified embedded atom method (MEAM) potential to a database of ab initio energies for silicon and construct an improved parametrization of this potential. In addition, we introduce a new, reference-free version of the MEAM potential. This MEAM version is also fitted to the silicon data and shows an even better agreement, although the improvement is modest. Finally, we investigate whether increasing the number of different angular terms in the MEAM potential from 3 to 4 will lead to a better potential. The aim of this work is to determine a broad-ranged potential, one that is reliable in many different low- and high-energy atomic geometries in silicon crystals, molecules, near defects and under strain. To verify this, the performance of the new potentials is tested in different circumstances that were not explicitly included in the fit: relaxed defect energies, thermal expansion, melting temperature and liquid silicon. The new MEAM parametrizations found in this work, called MEAM-M and RF-MEAM, are shown to be overall more accurate than previous potentials—although a few defect energies are exceptions—and we recommend them for future work. The melting temperatures are closer to the experiment than those of other MEAM potentials, but they are still too high. read less NOT USED (high confidence) K. Scheerschmidt and M. Planck, “Empirical Molecular Dynamics: Possibilities, Requirements, and Limitations.” 2007. link Times cited: 9 NOT USED (high confidence) S. Sinha and K. Goodson, “Review: Multiscale Thermal Modeling in Nanoelectronics,” International Journal for Multiscale Computational Engineering. 2005. link Times cited: 48 Abstract: Subcontinuum phonon conduction phenomena impede the cooling … read moreAbstract: Subcontinuum phonon conduction phenomena impede the cooling of field-effect transistors with gate lengths less than 100 nm, which degrades their performance and reliability. Thermal modeling of these nanodevices requires attention to a broad range of length scales and physical phenomena, ranging from continuum heat diffusion to atomic-scale interactions and phonon confinement. This review describes the state of the art in subcontinuum thermal modeling. Although the focus is on the silicon field-effect transistor, the models are general enough to apply to other semiconductor devices as well. Special attention is given to the recent advances in applying statistical and atomistic simulation methods to thermal transport. read less NOT USED (high confidence) M. Posselt, F. Gao, and D. Zwicker, “Migration of Di- and Tri-Interstitials in Silicon,” Nuclear Instruments & Methods in Physics Research Section B-beam Interactions With Materials and Atoms. 2005. link Times cited: 5 NOT USED (high confidence) T. Lim, “A Relationship Between the 2-body Energy of Kaxiras–Pandey and Pearson–Takai–Halicioglu–Tiller Potential Functions,” Physica Scripta. 2004. link Times cited: 22 Abstract: A parametric relationship between the Pearson–Takai–Haliciog… read moreAbstract: A parametric relationship between the Pearson–Takai–Halicioglu–Tiller (PTHT) and the Kaxiras–Pandey (KP) empirical potential energy functions is developed for the case of 2-body interaction. The need for such relationship arises when preferred parametric data and adopted software correspond to different potential functions. The analytical relationship was obtained by equating the potential functions' derivatives at zeroth, first and second order with respect to the interatomic distance at the equilibrium bond length, followed by comparison of coefficients in the repulsive and attractive terms. Plots of non-dimensional 2-body energy versus the nondimensional interatomic distance verified the analytical relationships developed herein. The discrepancy revealed in theoretical plots suggests that the 2-body PTHT and KP potentials are more suitable for curve-fitting "softer" and "harder" bonds respectively. read less NOT USED (high confidence) A. Dudka, “Structure analysis by reduced data. III. Method of interexperimental minimization,” Crystallography Reports. 2002. link Times cited: 8 NOT USED (high confidence) K. Scheerschmidt and P. Werner, “Characterization of Structure and Composition of Quantum Dots by Transmission Electron Microscopy.” 2002. link Times cited: 16 NOT USED (high confidence) K. Scheerschmidt, “Molecular dynamics simulations of wafer bonding,” MRS Proceedings. 2001. link Times cited: 0 Abstract: Molecular dynamics simulations using empirical potentials ha… read moreAbstract: Molecular dynamics simulations using empirical potentials have been employed to describe atomic inte ractions at interfaces created by the macroscopic wafer bonding process. Investigating perfect o r distorted s urfaces of different s emiconductor materials as well as of silica enables one to study the elementary processes and the resulting defects at the interfaces, and to characterize the ability of the potentials used. T wist r otation due to misalignment and bonding over steps influence strongly the bondability of larger areas. Empirical potentials developed by the bond o rder tight-binding approximation incl ude -bonds and yield enhanced interface structures, energies, and transferability to new materials systems. read less NOT USED (high confidence) S. W. Yim, N. Sonwalkar, and N. Saka, “Molecular dynamics simulation of boundary lubricated interfaces,” Journal of Computer-Aided Materials Design. 1999. link Times cited: 8 NOT USED (high confidence) A. Belov**, K. Scheerschmidt, and U. Gösele, “Extended Point Defect Structures at Intersections of Screw Dislocations in Si: A Molecular Dynamics Study,” Physica Status Solidi (a). 1999. link Times cited: 12 Abstract: Molecular dynamics computer simulations have been employed w… read moreAbstract: Molecular dynamics computer simulations have been employed with the Tersoff interatomic potential to examine the atomic structure of (a/2) 〈110〉 screw dislocations forming regular two-dimensional arrays in silicon. The main attention is focused on the atomic configurations of dislocation intersections. The dislocations are assumed to be undissociated, following HREM observations on the low-angle (001) twist boundaries produced by silicon wafer bonding in ultrahigh vacuum. It is shown that cores of the dislocation intersections are formed by closed characteristic groups of atoms (extended point defects). The symmetry of these defects strongly depends on the fact whether the screw dislocation arrays generate a twist or a shear boundary. read less NOT USED (high confidence) K. Nordlund, P. Partyka, and R. Averback, “Fully Atomistic Analysis of Diffuse X-Ray Scattering Spectra of Silicon Defects,” MRS Proceedings. 1997. link Times cited: 6 Abstract: Diffuse X-ray scattering is a useful method for studying def… read moreAbstract: Diffuse X-ray scattering is a useful method for studying defects in silicon and metals. Although the traditional approaches of analyzing experimental diffuse X-ray scattering data have given much information about the size of defects and defect clusters, they are not very well suited for determining the atomic configuration. We present a fully atomistic computational method to calculate the diffuse X-ray scattering line profile of an arbitrary atomic configuration, and compare line profiles of point defects and Frenkel pair configurations with experiment. read less NOT USED (definite) Y. He et al., “Multilayer hexagonal silicon forming in slit nanopore,” Scientific Reports. 2015. link Times cited: 5 NOT USED (definite) K. Eriguchi, “Application of Molecular Dynamics Simulations to Plasma Etch Damage in Advanced Metal-Oxide-Semiconductor Field-Effect Transistors.” 2012. link Times cited: 0 Abstract: According to "the international technology roadmap for … read moreAbstract: According to "the international technology roadmap for semiconductors (ITRS)" (SIA, 2009), the shrinkage of silicon-based metal–oxide–semiconductor field-effect transistor (MOSFET) – an elemental device (unit) in ultra-large-scale integrated (ULSI) circuits – has been accelerating due to expanding demands for the higher performance and the lower power operation. The characteristic dimensions of current MOSFETs in mass productions are around 30 – 50 nm. Figure 1 shows the scaling trend of the key feature sizes in ULSI circuits predicted by Semiconductor Industry Association, USA. Various types of MOSFETs are designed for the specific purposes, i.e., low standby power (LSP), low operation power (LOP), and high performance (HP) operations, and built in ULSI circuits such as dynamic random access memory (DRAM) and micro-processing unit (MPU). New structured MOSFETs such as fully-depleted (FD) and metal-gate (MG) devices have been recently proposed. Since physical gate length (Lg) and source / drain extension depth (Ext) are the key feature sizes determining MOSFET performance (Sze & Ng, 2007), the shrinkage of Lg and Ext is a primal focus in the development of MOSFETs. These sizes have become a few nanometers, comparable to the scale of atomistic simulation domain. read less NOT USED (definite) “The Tersoff potential for extreme environment,” arXiv: Materials Science. 2018. link Times cited: 0 Abstract: A novel modification of the Tersoff potential for Si is pres… read moreAbstract: A novel modification of the Tersoff potential for Si is presented. The modification improves the transferability of the Tersoff potential for liquid states without the change of original parameters and with no alteration of bulk properties. Also, the modification introduces a correction term for high-pressure states. The modification is meaningful considering that by high energy irradiations local liquid structures and unstable high-pressure manifolds may occur, therefore an interatomic potential must have an acceptable reliability on high thermal/pressure situations to simulate such phenomenon. Particularly, in the modification, a new screening function replaces the radial cutoff function and the bond order function is slightly changed. Also, a repulsive energy function is replaced by a correction function within a specific pair distance. read less |
 SW_BalamaneHaliciogluTiller_1992_Si__MO_113686039439_005
SW_BalamaneHaliciogluTiller_1992_Si__MO_113686039439_005