Sim_LAMMPS_SNAP_ChenDengTran_2017_Mo__SM_003882782678_000
| Title
A single sentence description.
|
LAMMPS SNAP potential for Mo developed by Chen et al. (2017) v000 |
|---|---|
| Description | In this work, we present a highly accurate spectral neighbor analysis potential (SNAP) model for molybdenum (Mo) developed through the rigorous application of machine learning techniques on large materials data sets. Despite Mo's importance as a structural metal, existing force fields for Mo based on the embedded atom and modified embedded atom methods do not provide satisfactory accuracy on many properties. We will show that by fitting to the energies, forces, and stress tensors of a large density functional theory (DFT)-computed dataset on a diverse set of Mo structures, a Mo SNAP model can be developed that achieves close to DFT accuracy in the prediction of a broad range of properties, including elastic constants, melting point, phonon spectra, surface energies, grain boundary energies, etc. We will outline a systematic model development process, which includes a rigorous approach to structural selection based on principal component analysis, as well as a differential evolution algorithm for optimizing the hyperparameters in the model fitting so that both the model error and the property prediction error can be simultaneously lowered. We expect that this newly developed Mo SNAP model will find broad applications in large and long-time scale simulations. |
| Species
The supported atomic species.
| Mo |
| Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
|
None |
| Content Origin | LAMMPS package 22-Sep-2017 |
| Contributor |
Daniel S. Karls |
| Maintainer |
Daniel S. Karls |
| Developer |
Richard Tran Hanmei Tang lek-Heng Chu Deng, Zhi Chi Chen Ong, Shyue Ping |
| Published on KIM | 2019 |
| How to Cite |
This Simulator Model originally published in [1] is archived in OpenKIM [2-4]. [1] Chen C, Deng Z, Tran R, Tang H, Chu I-H, Ong SP. Accurate force field for molybdenum by machine learning large materials data. Phys Rev Materials [Internet]. 2017Sep;1(4):043603. Available from: https://link.aps.org/doi/10.1103/PhysRevMaterials.1.043603 doi:10.1103/PhysRevMaterials.1.043603 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information. [2] Tran R, Tang H, Chu lek-Heng, Deng Z, Chen C, Ong SP. LAMMPS SNAP potential for Mo developed by Chen et al. (2017) v000. OpenKIM; 2019. doi:10.25950/968529b5 [3] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6 [4] Elliott RS, Tadmor EB. Knowledgebase of Interatomic Models (KIM) Application Programming Interface (API). OpenKIM; 2011. doi:10.25950/ff8f563a Click here to download the above citation in BibTeX format. |
| Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on. The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel. The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied. The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP). Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis. OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm. |
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information. 
85 Citations (5 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) B. Waters, D. S. Karls, I. Nikiforov, R. Elliott, E. Tadmor, and B. Runnels, “Automated determination of grain boundary energy and potential-dependence using the OpenKIM framework,” Computational Materials Science. 2022. link Times cited: 5 USED (low confidence) T. Miyagawa, Y. Sakai, K. Mori, N. Kato, A. Yonezu, and K. Ishibashi, “Distribution of the Mechanical Properties of Ti–Cu Combinatorial Thin Film Evaluated Using Nanoindentation Experiments and Molecular Dynamics with a Neural Network Potential,” SSRN Electronic Journal. 2022. link Times cited: 1 USED (low confidence) A. Shrestha, X. Gao, J. Hicks, and C. Paolucci, “Nanoparticle Size Effects on Phase Stability for Molybdenum and Tungsten Carbides,” Chemistry of Materials. 2021. link Times cited: 15 USED (low confidence) S. Fujii and A. Seko, “Structure and lattice thermal conductivity of grain boundaries in silicon by using machine learning potential and molecular dynamics,” Computational Materials Science. 2021. link Times cited: 8 USED (low confidence) X. Gu and C. Y. Zhao, “Thermal conductivity of single-layer MoS2(1−x)Se2x alloys from molecular dynamics simulations with a machine-learning-based interatomic potential,” Computational Materials Science. 2019. link Times cited: 47 NOT USED (low confidence) X. Qian, B.-J. Yoon, R. Arróyave, X. Qian, and E. R. Dougherty, “Knowledge-driven learning, optimization, and experimental design under uncertainty for materials discovery,” Patterns. 2023. link Times cited: 0 NOT USED (low confidence) R. Feng et al., “PolyGET: Accelerating Polymer Simulations by Accurate and Generalizable Forcefield with Equivariant Transformer,” ArXiv. 2023. link Times cited: 0 Abstract: Polymer simulation with both accuracy and efficiency is a ch… read more NOT USED (low confidence) J. P. Stoppelman, A. Wilkinson, and J. G. McDaniel, “Equation of state predictions for ScF3 and CaZrF6 with neural network-driven molecular dynamics.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: In silico property prediction based on density functional th… read more NOT USED (low confidence) D. F. T. du Toit and V. L. Deringer, “Cross-platform hyperparameter optimization for machine learning interatomic potentials.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: Machine-learning (ML)-based interatomic potentials are incre… read more NOT USED (low confidence) W. Du, X. Fan, H. Li, D. Zhai, and Y. Liu, “Development of a Ni-Al Reactive Force Field for Ni-Based Superalloy: Revealing Electrostatic Effects on Mechanical Deformation,” SSRN Electronic Journal. 2023. link Times cited: 0 NOT USED (low confidence) R. Guo, G. Li, J. Tang, Y. Wang, and X. Song, “Small-data-based Machine Learning Interatomic Potentials for Graphene Grain Boundaries Enabled by Structural Unit Model,” Carbon Trends. 2023. link Times cited: 2 NOT USED (low confidence) J. Jiang, L.-C. Xu, F. Li, and J. Shao, “Machine Learning Potential Model Based on Ensemble Bispectrum Feature Selection and Its Applicability Analysis,” Metals. 2023. link Times cited: 2 Abstract: With the continuous improvement of machine learning methods,… read more NOT USED (low confidence) S. Blücher, K.-R. Müller, and S. Chmiela, “Reconstructing Kernel-Based Machine Learning Force Fields with Superlinear Convergence,” Journal of Chemical Theory and Computation. 2022. link Times cited: 2 Abstract: Kernel machines have sustained continuous progress in the fi… read more NOT USED (low confidence) H. Deng, J. Comer, and B. Liu, “A high-dimensional neural network potential for molecular dynamics simulations of condensed phase nickel and phase transitions,” Molecular Simulation. 2022. link Times cited: 0 Abstract: ABSTRACT A high-dimensional neural network interatomic poten… read more NOT USED (low confidence) K. Pitike and W. Setyawan, “Accurate Fe–He machine learning potential for studying He effects in BCC-Fe,” Journal of Nuclear Materials. 2022. link Times cited: 1 NOT USED (low confidence) B. Bishnoi, “Lagrangian Density Space-Time Deep Neural Network Topology,” ArXiv. 2022. link Times cited: 1 Abstract: As a network-based functional approximator, we have proposed… read more NOT USED (low confidence) T. Miyagawa, K. Mori, N. Kato, and A. Yonezu, “Development of neural network potential for MD simulation and its application to TiN,” Computational Materials Science. 2022. link Times cited: 3 NOT USED (low confidence) T. Wen, L. Zhang, H. Wang, W. E, and D. Srolovitz, “Deep Potentials for Materials Science,” Materials Futures. 2022. link Times cited: 54 Abstract:
To fill the gap between accurate (and expensive) ab initio… read more NOT USED (low confidence) K. Xie et al., “Neural network potential for Zr–Rh system by machine learning,” Journal of Physics: Condensed Matter. 2021. link Times cited: 3 Abstract: Zr–Rh metallic glass has enabled its many applications in ve… read more NOT USED (low confidence) X. Chen et al., “Machine learning enhanced empirical potentials for metals and alloys,” Comput. Phys. Commun. 2021. link Times cited: 5 NOT USED (low confidence) M. Gilbert et al., “Perspectives on multiscale modelling and experiments to accelerate materials development for fusion,” Journal of Nuclear Materials. 2021. link Times cited: 33 NOT USED (low confidence) X. Wang, S. Xu, W. Jian, X.-G. Li, Y. Su, and I. Beyerlein, “Generalized stacking fault energies and Peierls stresses in refractory body-centered cubic metals from machine learning-based interatomic potentials,” Computational Materials Science. 2021. link Times cited: 30 NOT USED (low confidence) R. V. Babu, G.Ayyappan, and A.Kumaravel, “EXPLORATORY DATA ANALYSIS ON MACROSCOPIC MATERIAL BEHAVIOR USING MICROMECHANICAL SIMULATIONS BY APPLYING THE GAUSSIAN PROCESSES WITH VARIOUS KERNELS,” Indian Journal of Computer Science and Engineering. 2021. link Times cited: 1 Abstract: Pro Vice Chancellor, Galgotias University, Greater Noida, Ut… read more NOT USED (low confidence) H. Yanxon, D. Zagaceta, B. Tang, D. Matteson, and Q. Zhu, “PyXtal_FF: a python library for automated force field generation,” Machine Learning: Science and Technology. 2020. link Times cited: 15 Abstract: We present PyXtal_FF—a package based on Python programming l… read more NOT USED (low confidence) P. Pattnaik, S. Raghunathan, T. Kalluri, P. Bhimalapuram, C. V. Jawahar, and U. Priyakumar, “Machine Learning for Accurate Force Calculations in Molecular Dynamics Simulations.,” The journal of physical chemistry. A. 2020. link Times cited: 32 Abstract: The computationally expensive nature of ab initio molecular … read more NOT USED (low confidence) X. Chen, X. Gao, Y. Zhao, D. Lin, W. Chu, and H. Song, “TensorAlloy: An automatic atomistic neural network program for alloys,” Comput. Phys. Commun. 2020. link Times cited: 10 NOT USED (low confidence) G. Pilania, P. Balachandran, J. Gubernatis, and T. Lookman, “Data-Based Methods for Materials Design and Discovery: Basic Ideas and General Methods.” 2020. link Times cited: 11 Abstract: Machine learning methods are changing the way we design and … read more NOT USED (low confidence) J. Chapman, R. Batra, and R. Ramprasad, “Machine learning models for the prediction of energy, forces, and stresses for Platinum,” Computational Materials Science. 2020. link Times cited: 18 NOT USED (low confidence) A. V. der Ven, Z. Deng, S. Banerjee, and S. Ong, “Rechargeable Alkali-Ion Battery Materials: Theory and Computation.,” Chemical reviews. 2020. link Times cited: 116 Abstract: Since its development in the 1970s, the rechargeable alkali-… read more NOT USED (low confidence) T. J. Oweida, A.-U. Mahmood, M. D. Manning, S. Rigin, and Y. G. Yingling, “Merging Materials and Data Science: Opportunities, Challenges, and Education in Materials Informatics,” MRS Advances. 2020. link Times cited: 6 Abstract: Since the launch of the Materials Genome Initiative (MGI) th… read more NOT USED (low confidence) L. Tang, Z.-J. Yang, T. Wen, K. Ho, M. Kramer, and C. Wang, “Development of interatomic potential for Al-Tb alloys using a deep neural network learning method.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 17 Abstract: An interatomic potential for the Al-Tb alloy around the comp… read more NOT USED (low confidence) D. Smirnova et al., “Atomistic description of self-diffusion in molybdenum: A comparative theoretical study of non-Arrhenius behavior,” Physical Review Materials. 2020. link Times cited: 16 Abstract: According to experimental observations, the temperature depe… read more NOT USED (low confidence) W. Zhang, S. Byna, C. Niu, and Y. Chen, “Exploring Metadata Search Essentials for Scientific Data Management,” 2019 IEEE 26th International Conference on High Performance Computing, Data, and Analytics (HiPC). 2019. link Times cited: 10 Abstract: Scientific experiments and observations store massive amount… read more NOT USED (low confidence) W. Zhang, S. Byna, H. Tang, B. Williams, and Y. Chen, “MIQS: metadata indexing and querying service for self-describing file formats,” Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. 2019. link Times cited: 10 Abstract: Scientific applications often store datasets in self-describ… read more NOT USED (low confidence) T. D. Huan, R. Batra, J. Chapman, C. Kim, A. Chandrasekaran, and R. Ramprasad, “Iterative-Learning Strategy for the Development of Application-Specific Atomistic Force Fields,” The Journal of Physical Chemistry C. 2019. link Times cited: 18 Abstract: Emerging data-driven approaches in materials science have tr… read more NOT USED (low confidence) A. Goryaeva, J. Maillet, and M. Marinica, “Towards better efficiency of interatomic linear machine learning potentials,” Computational Materials Science. 2019. link Times cited: 34 NOT USED (low confidence) Y. Zuo et al., “A Performance and Cost Assessment of Machine Learning Interatomic Potentials.,” The journal of physical chemistry. A. 2019. link Times cited: 413 Abstract: Machine learning of the quantitative relationship between lo… read more NOT USED (low confidence) S. Ong, “Accelerating materials science with high-throughput computations and machine learning,” Computational Materials Science. 2019. link Times cited: 66 NOT USED (low confidence) A. Hernandez, A. Balasubramanian, F. Yuan, S. Mason, and T. Mueller, “Fast, accurate, and transferable many-body interatomic potentials by symbolic regression,” npj Computational Materials. 2019. link Times cited: 51 NOT USED (low confidence) C. Chen, W. Ye, Y. Zuo, C. Zheng, and S. Ong, “Graph Networks as a Universal Machine Learning Framework for Molecules and Crystals,” Chemistry of Materials. 2018. link Times cited: 602 Abstract: Graph networks are a new machine learning (ML) paradigm that… read more NOT USED (low confidence) W. Zhang, H. Tang, S. Byna, and Y. Chen, “DART: distributed adaptive radix tree for efficient affix-based keyword search on HPC systems,” Proceedings of the 27th International Conference on Parallel Architectures and Compilation Techniques. 2018. link Times cited: 10 Abstract: Affix-based search is a fundamental functionality for storag… read more NOT USED (low confidence) M. Wood and A. Thompson, “Extending the accuracy of the SNAP interatomic potential form.,” The Journal of chemical physics. 2017. link Times cited: 130 Abstract: The Spectral Neighbor Analysis Potential (SNAP) is a classic… read more NOT USED (low confidence) R. Hu, “Random neural networks for dimensionality reduction and regularized supervised learning.” 2019. link Times cited: 1 Abstract: This dissertation explores Random Neural Networks (RNNs) in … read more NOT USED (high confidence) A. Diggs et al., “Hydrogen-induced degradation dynamics in silicon heterojunction solar cells via machine learning,” Communications Materials. 2023. link Times cited: 1 NOT USED (high confidence) M. D. K. Jones, J. Dawson, S. Campbell, V. Barrioz, L. D. Whalley, and Y. Qu, “Modelling Interfaces in Thin-Film Photovoltaic Devices,” Frontiers in Chemistry. 2022. link Times cited: 2 Abstract: Developing effective device architectures for energy technol… read more NOT USED (high confidence) G. Baldinozzi and V. Pontikis, “Phenomenological potentials for the refractory metals Cr, Mo and W,” Journal of Physics: Condensed Matter. 2022. link Times cited: 1 Abstract: Cohesion in the refractory metals Cr, Mo, and W is phenomeno… read more NOT USED (high confidence) T. Lee et al., “Atomic-scale origin of the low grain-boundary resistance in perovskite solid electrolyte Li0.375Sr0.4375Ta0.75Zr0.25O3,” Nature Communications. 2022. link Times cited: 9 NOT USED (high confidence) H. Zheng et al., “Multi-scale investigation of short-range order and dislocation glide in MoNbTi and TaNbTi multi-principal element alloys,” npj Computational Materials. 2022. link Times cited: 8 NOT USED (high confidence) Y. Chen et al., “A Focused Review on Engineering Application of Multi-Principal Element Alloy,” Frontiers in Materials. 2022. link Times cited: 3 Abstract: Compared with traditional alloys with one principal componen… read more NOT USED (high confidence) W. Zhang, S. Byna, H. Sim, S. Lee, S. S. Vazhkudai, and Y. Chen, “Exploiting User Activeness for Data Retention in HPC Systems,” SC21: International Conference for High Performance Computing, Networking, Storage and Analysis. 2021. link Times cited: 2 Abstract: HPC systems typically rely on the fixed-lifetime (FLT) data … read more NOT USED (high confidence) R. Tamura, M. Matsuda, J. Lin, Y. Futamura, T. Sakurai, and T. Miyazaki, “Structural analysis based on unsupervised learning: Search for a characteristic low-dimensional space by local structures in atomistic simulations,” Physical Review B. 2021. link Times cited: 0 Abstract: Owing to the advances in computational techniques and the in… read more NOT USED (high confidence) S. Moayedpour, D. Dardzinski, S. Yang, A. Hwang, and N. Marom, “Structure prediction of epitaxial inorganic interfaces by lattice and surface matching with Ogre.,” The Journal of chemical physics. 2021. link Times cited: 8 Abstract: We present a new version of the Ogre open source Python pack… read more NOT USED (high confidence) J. Qi et al., “Bridging the gap between simulated and experimental ionic conductivities in lithium superionic conductors,” Materials Today Physics. 2021. link Times cited: 33 NOT USED (high confidence) Y. Mishin, “Machine-Learning Interatomic Potentials for Materials Science,” Electrical Engineering eJournal. 2021. link Times cited: 103 NOT USED (high confidence) Y.-S. Lin, G. P. P. Pun, and Y. Mishin, “Development of a physically-informed neural network interatomic potential for tantalum,” Computational Materials Science. 2021. link Times cited: 9 NOT USED (high confidence) G. P. P. Pun, V. Yamakov, J. Hickman, E. Glaessgen, and Y. Mishin, “Development of a general-purpose machine-learning interatomic potential for aluminum by the physically informed neural network method,” Physical Review Materials. 2020. link Times cited: 13 Abstract: Interatomic potentials constitute the key component of large… read more NOT USED (high confidence) J. Nigam, S. Pozdnyakov, and M. Ceriotti, “Recursive evaluation and iterative contraction of N-body equivariant features.,” The Journal of chemical physics. 2020. link Times cited: 45 Abstract: Mapping an atomistic configuration to a symmetrized N-point … read more NOT USED (high confidence) J. Byggmastar, K. Nordlund, and F. Djurabekova, “Gaussian approximation potentials for body-centered-cubic transition metals,” Physical Review Materials. 2020. link Times cited: 22 Abstract: We develop a set of machine-learning interatomic potentials … read more NOT USED (high confidence) C. Lapointe et al., “Machine learning surrogate models for prediction of point defect vibrational entropy,” Physical Review Materials. 2020. link Times cited: 5 Abstract: The temperature variation of the defect densities in a cryst… read more NOT USED (high confidence) A. Tran, J. Tranchida, T. Wildey, and A. Thompson, “Multi-fidelity machine-learning with uncertainty quantification and Bayesian optimization for materials design: Application to ternary random alloys,” The Journal of chemical physics. 2020. link Times cited: 42 Abstract: We present a scale-bridging approach based on a multi-fideli… read more NOT USED (high confidence) D. Zagaceta, H. Yanxon, and Q. Zhu, “Spectral neural network potentials for binary alloys,” Journal of Applied Physics. 2020. link Times cited: 4 Abstract: In this work, we present a numerical implementation to compu… read more NOT USED (high confidence) C. M. Andolina, P. Williamson, and W. Saidi, “Optimization and validation of a deep learning CuZr atomistic potential: Robust applications for crystalline and amorphous phases with near-DFT accuracy.,” The Journal of chemical physics. 2020. link Times cited: 32 Abstract: We show that a deep-learning neural network potential (DP) b… read more NOT USED (high confidence) S. Desai, S. Reeve, and J. Belak, “Implementing a neural network interatomic model with performance portability for emerging exascale architectures,” Comput. Phys. Commun. 2020. link Times cited: 9 NOT USED (high confidence) C. Chen, Y. Zuo, W. Ye, X.-G. Li, Z. Deng, and S. Ong, “A Critical Review of Machine Learning of Energy Materials,” Advanced Energy Materials. 2020. link Times cited: 268 Abstract: Machine learning (ML) is rapidly revolutionizing many fields… read more NOT USED (high confidence) X.-G. Li, C. Chen, H. Zheng, Y. Zuo, and S. Ong, “Complex strengthening mechanisms in the NbMoTaW multi-principal element alloy,” npj Computational Materials. 2019. link Times cited: 114 NOT USED (high confidence) T. Wen et al., “Development of a deep machine learning interatomic potential for metalloid-containing Pd-Si compounds,” Physical Review B. 2019. link Times cited: 30 Abstract: Interatomic potentials based on neural-network machine learn… read more NOT USED (high confidence) S. Xiao, R. Hu, Z. Li, S. Attarian, K.-M. Björk, and A. Lendasse, “A machine-learning-enhanced hierarchical multiscale method for bridging from molecular dynamics to continua,” Neural Computing and Applications. 2019. link Times cited: 17 NOT USED (high confidence) J. Schmidt, M. R. G. Marques, S. Botti, and M. A. L. Marques, “Recent advances and applications of machine learning in solid-state materials science,” npj Computational Materials. 2019. link Times cited: 1226 NOT USED (high confidence) H. Zheng et al., “Grain boundary properties of elemental metals,” Acta Materialia. 2019. link Times cited: 98 NOT USED (high confidence) S. Bishnoi et al., “Predicting Young’s modulus of oxide glasses with sparse datasets using machine learning,” Journal of Non-Crystalline Solids. 2019. link Times cited: 61 NOT USED (high confidence) M. Wood, M. Cusentino, B. Wirth, and A. Thompson, “Data-driven material models for atomistic simulation,” Physical Review B. 2019. link Times cited: 37 Abstract: The central approximation made in classical molecular dynami… read more NOT USED (high confidence) K. Butler, G. S. Gautam, and P. Canepa, “Designing interfaces in energy materials applications with first-principles calculations,” npj Computational Materials. 2019. link Times cited: 69 NOT USED (high confidence) Z. Deng, C. Chen, X.-G. Li, and S. Ong, “An electrostatic spectral neighbor analysis potential for lithium nitride,” npj Computational Materials. 2019. link Times cited: 62 NOT USED (high confidence) J. Wang, D. Shin, and S. Shin, “Comprehensive evaluation and parametric sensitivity of interatomic potential models for diffusion kinetics of Cr2O3 in molecular dynamics,” AIP Advances. 2019. link Times cited: 4 Abstract: While molecular dynamics (MD) has proven to be a promising a… read more NOT USED (high confidence) A. Seko, A. Togo, and I. Tanaka, “Group-theoretical high-order rotational invariants for structural representations: Application to linearized machine learning interatomic potential,” Physical Review B. 2019. link Times cited: 22 Abstract: Many rotational invariants for crystal structure representat… read more NOT USED (high confidence) S. A. Miller, M. Dylla, S. Anand, K. Gordiz, G. J. Snyder, and E. Toberer, “Empirical modeling of dopability in diamond-like semiconductors,” npj Computational Materials. 2018. link Times cited: 17 NOT USED (high confidence) S. Xiao, A. Lendasse, and R. Hu, “Data-Enabled Computational Multiscale Method in Materials Science and Engineering,” 2018 International Conference on Computational Science and Computational Intelligence (CSCI). 2018. link Times cited: 1 Abstract: In the community of computational materials science, one of … read more NOT USED (high confidence) W. Ye, C. Chen, S. Dwaraknath, A. Jain, S. Ong, and K. Persson, “Harnessing the Materials Project for machine-learning and accelerated discovery,” MRS Bulletin. 2018. link Times cited: 21 Abstract: Improvements in computational resources over the last decade… read more NOT USED (high confidence) X.-G. Li, C. Hu, C. Chen, Z. Deng, J. Luo, and S. Ong, “Quantum-accurate spectral neighbor analysis potential models for Ni-Mo binary alloys and fcc metals,” Physical Review B. 2018. link Times cited: 61 Abstract: In recent years, efficient interatomic potentials approachin… read more NOT USED (high confidence) Q. Bai, L. Yang, H. Chen, and Y. Mo, “Computational Studies of Electrode Materials in Sodium‐Ion Batteries,” Advanced Energy Materials. 2018. link Times cited: 120 Abstract: Sodium‐ion batteries have attracted extensive interest as a … read more NOT USED (high confidence) Y. Zhang and C. Ling, “A strategy to apply machine learning to small datasets in materials science,” npj Computational Materials. 2018. link Times cited: 406 NOT USED (high confidence) A. Bartók, J. Kermode, N. Bernstein, and G. Csányi, “Machine Learning a General-Purpose Interatomic Potential for Silicon,” Physical Review X. 2018. link Times cited: 291 Abstract: The success of first principles electronic structure calcula… read more NOT USED (high confidence) A. Takahashi, A. Seko, and I. Tanaka, “Linearized machine-learning interatomic potentials for non-magnetic elemental metals: Limitation of pairwise descriptors and trend of predictive power.,” The Journal of chemical physics. 2017. link Times cited: 20 Abstract: Machine-learning interatomic potential (MLIP) has been of gr… read more NOT USED (high confidence) D. Dragoni, T. Daff, G. Csányi, and N. Marzari, “Achieving DFT accuracy with a machine-learning interatomic potential: thermomechanics and defects in bcc ferromagnetic iron,” arXiv: Materials Science. 2017. link Times cited: 167 Abstract: We show that the Gaussian Approximation Potential machine le… read more |
| Funding | Not available |
| Short KIM ID
The unique KIM identifier code.
| SM_003882782678_000 |
| Extended KIM ID
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
| Sim_LAMMPS_SNAP_ChenDengTran_2017_Mo__SM_003882782678_000 |
| DOI |
10.25950/968529b5 https://doi.org/10.25950/968529b5 https://commons.datacite.org/doi.org/10.25950/968529b5 |
| KIM Item Type | Simulator Model |
| KIM API Version | 2.1 |
| Simulator Name
The name of the simulator as defined in kimspec.edn.
| LAMMPS |
| Potential Type | snap |
| Simulator Potential | snap |
| Run Compatibility | portable-models |
| Grade | Name | Category | Brief Description | Full Results | Aux File(s) |
|---|---|---|---|---|---|
| P | vc-species-supported-as-stated | mandatory | The model supports all species it claims to support; see full description. |
Results | Files |
| P | vc-periodicity-support | mandatory | Periodic boundary conditions are handled correctly; see full description. |
Results | Files |
| P | vc-permutation-symmetry | mandatory | Total energy and forces are unchanged when swapping atoms of the same species; see full description. |
Results | Files |
| A | vc-forces-numerical-derivative | consistency | Forces computed by the model agree with numerical derivatives of the energy; see full description. |
Results | Files |
| P | vc-dimer-continuity-c1 | informational | The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description. |
Results | Files |
| P | vc-objectivity | informational | Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description. |
Results | Files |
| P | vc-inversion-symmetry | informational | Total energy is unchanged and forces change sign when inverting a configuration through the origin; see full description. |
Results | Files |
| F | vc-memory-leak | informational | The model code does not have memory leaks (i.e. it releases all allocated memory at the end); see full description. |
Results | Files |
| N/A | vc-thread-safe | mandatory | The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description. |
Results | Files |
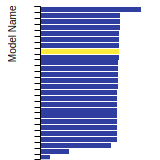
BCC Lattice Constant
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
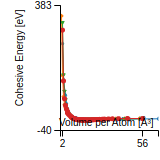
Cohesive Energy Graph
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
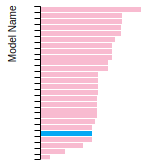
Diamond Lattice Constant
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
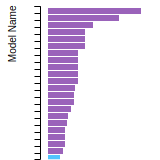
Dislocation Core Energies
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
(No matching species)FCC Elastic Constants
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
FCC Lattice Constant
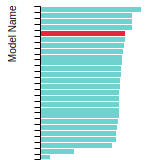
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
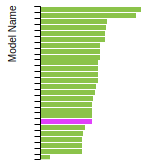
FCC Stacking Fault Energies
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
(No matching species)FCC Surface Energies
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
(No matching species)SC Lattice Constant
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Cubic Crystal Basic Properties Table
Species: MoCreators:
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/64cb38c5
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Cohesive energy versus lattice constant curve for bcc Mo v004 | view | 2061 | |
| Cohesive energy versus lattice constant curve for diamond Mo v004 | view | 2429 | |
| Cohesive energy versus lattice constant curve for fcc Mo v004 | view | 1930 | |
| Cohesive energy versus lattice constant curve for sc Mo v004 | view | 1820 |
Creators: Junhao Li and Ellad Tadmor
Contributor: tadmor
Publication Year: 2019
DOI: https://doi.org/10.25950/5853fb8f
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for bcc Mo at zero temperature v006 | view | 2047 | |
| Elastic constants for diamond Mo at zero temperature v001 | view | 40210 | |
| Elastic constants for fcc Mo at zero temperature v006 | view | 6302 | |
| Elastic constants for sc Mo at zero temperature v006 | view | 2399 |
Creators: Junhao Li
Contributor: jl2922
Publication Year: 2019
DOI: https://doi.org/10.25950/d794c746
Computes the elastic constants for hcp crystals by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for hcp Mo at zero temperature v004 | view | 2483 |
Creators:
Contributor: ilia
Publication Year: 2025
DOI: https://doi.org/10.25950/866c7cfa
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium crystal structure and energy for Mo in AFLOW crystal prototype A_cF4_225_a v003 | view | 202017 | |
| Equilibrium crystal structure and energy for Mo in AFLOW crystal prototype A_cI2_229_a v003 | view | 147951 | |
| Equilibrium crystal structure and energy for Mo in AFLOW crystal prototype A_hP1_191_a v003 | view | 177480 | |
| Equilibrium crystal structure and energy for Mo in AFLOW crystal prototype A_hP4_194_ac v003 | view | 170675 |
Creators: Brandon Runnels
Contributor: brunnels
Publication Year: 2019
DOI: https://doi.org/10.25950/4723cee7
Computes grain boundary energy for a range of tilt angles given a crystal structure, tilt axis, and material.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Relaxed energy as a function of tilt angle for a 100 symmetric tilt grain boundary in bcc Mo v000 | view | 30472091 | |
| Relaxed energy as a function of tilt angle for a 110 symmetric tilt grain boundary in bcc Mo v000 | view | 102861967 | |
| Relaxed energy as a function of tilt angle for a 111 symmetric tilt grain boundary in bcc Mo v000 | view | 53383187 | |
| Relaxed energy as a function of tilt angle for a 112 symmetric tilt grain boundary in bcc Mo v000 | view | 265816471 |
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/2765e3bf
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium zero-temperature lattice constant for bcc Mo v007 | view | 6686 | |
| Equilibrium zero-temperature lattice constant for diamond Mo v007 | view | 10524 | |
| Equilibrium zero-temperature lattice constant for fcc Mo v007 | view | 11644 | |
| Equilibrium zero-temperature lattice constant for sc Mo v007 | view | 7134 |
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/c339ca32
Calculates lattice constant of hexagonal bulk structures at zero temperature and pressure by using simplex minimization to minimize the potential energy.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium lattice constants for hcp Mo v005 | view | 46321 |
Creators:
Contributor: mjwen
Publication Year: 2024
DOI: https://doi.org/10.25950/9d9822ec
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Linear thermal expansion coefficient of bcc Mo at 293.15 K under a pressure of 0 MPa v002 | view | 14150396 |
Creators: Matt Bierbaum
Contributor: mattbierbaum
Publication Year: 2019
DOI: https://doi.org/10.25950/6c43a4e6
Calculates the surface energy of several high symmetry surfaces and produces a broken-bond model fit. In latex form, the fit equations are given by:
E_{FCC} (\vec{n}) = p_1 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (2 ( |x+ 2y + z| + |x+2y-z| + |x-2y + z| + |x-2y-z| + |2x+y+z| + |2x+y-z| +|2x-y+z| +|2x-y-z| +|x+y+2z| +|x+y-2z| +|x-y+2z| +|x-y-2z| ) + c
E_{BCC} (\vec{n}) = p_1 (6 \left( | x+y+z| + |x+y-z| + |-x+y-z| + |x-y+z| \right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) +c.
In Python, these two fits take the following form:
def BrokenBondFCC(params, index):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*(abs(x+2*y+z) + abs(x+2*y-z) +abs(x-2*y+z) +abs(x-2*y-z) + abs(2*x+y+z) +abs(2*x+y-z) +abs(2*x-y+z) +abs(2*x-y-z) + abs(x+y+2*z) +abs(x+y-2*z) +abs(x-y+2*z) +abs(x-y-2*z))+params[3]
def BrokenBondBCC(params, x, y, z):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*6*(abs(x+y+z) + abs(x-y-z) + abs(x-y+z) + abs(x+y-z)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[3]
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Broken-bond fit of high-symmetry surface energies in bcc Mo v004 | view | 376287 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/fca89cea
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Monovacancy formation energy and relaxation volume for bcc Mo | view | 29356690 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/c27ba3cd
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Vacancy formation and migration energy for bcc Mo | view | 9383522 |
| Test | Error Categories | Link to Error page |
|---|---|---|
| Cohesive energy versus lattice constant curve for bcc Mo v004 | other | view |
| Cohesive energy versus lattice constant curve for diamond Mo v004 | other | view |
| Cohesive energy versus lattice constant curve for fcc Mo v004 | other | view |
| Cohesive energy versus lattice constant curve for sc Mo v004 | other | view |
EquilibriumCrystalStructure__TD_457028483760_000
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium crystal structure and energy for Mo in AFLOW crystal prototype A_hP4_194_ac v000 | other | view |
No Driver
| Verification Check | Error Categories | Link to Error page |
|---|---|---|
| MemoryLeak__VC_561022993723_004 | other | view |
| PeriodicitySupport__VC_895061507745_004 | other | view |
| Sim_LAMMPS_SNAP_ChenDengTran_2017_Mo__SM_003882782678_000.txz | Tar+XZ | Linux and OS X archive |
| Sim_LAMMPS_SNAP_ChenDengTran_2017_Mo__SM_003882782678_000.zip | Zip | Windows archive |