Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on.
The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel.
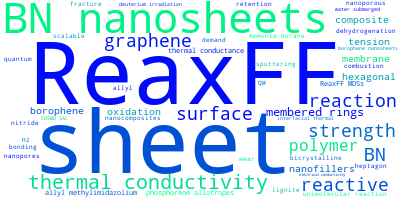
The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied.
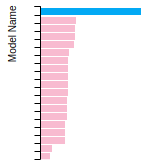
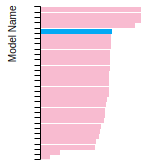
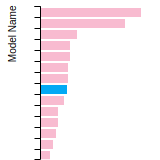
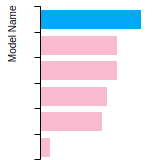
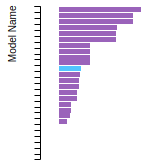
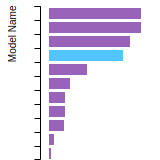
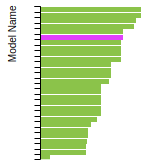
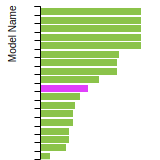
The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP).
Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis.
OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm.
|
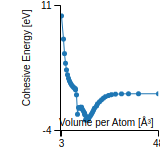
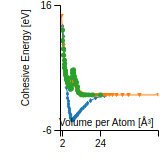
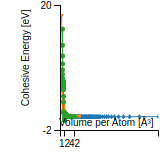
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information.
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (definite) F. Bedoya, J. Allain, F. J. Domínguez-Gutiérrez, and P. Krstic, “Effect of deuterium irradiation on graphite boronized in the NSTX-U tokamak,” Scientific Reports. 2019. link Times cited: 3 USED (definite) C. Gautam et al., “Synthesis and 3D Interconnected Nanostructured h-BN-Based Biocomposites by Low-Temperature Plasma Sintering: Bone Regeneration Applications,” ACS Omega. 2018. link Times cited: 21 Abstract: Recent advances and demands in biomedical applications drive… read moreAbstract: Recent advances and demands in biomedical applications drive a large amount of research to synthesize easily scalable low-density, high-strength, and wear-resistant biomaterials. The chemical inertness with low density combined with high strength makes h-BN one of the promising materials for such application. In this work, three-dimensional hexagonal boron nitride (h-BN) interconnected with boron trioxide (B2O3) was prepared by easily scalable and energy efficient spark plasma sintering (SPS) process. The composite structure shows significant densification (1.6–1.9 g/cm3) and high surface area (0.97–14.5 m2/g) at an extremely low SPS temperature of 250 °C. A high compressive strength of 291 MPa with a reasonably good wear resistance was obtained for the composite structure. The formation of strong covalent bonds between h-BN and B2O3 was formulated and established by molecular dynamics simulation. The composite showed significant effect on cell viability/proliferation. It shows a high mineralized nodule formation over the control, which suggests its use as a possible osteogenic agent in bone formation. read less USED (definite) B. Mortazavi, M.-Q. Le, T. Rabczuk, and L. Pereira, “Anomalous strain effect on the thermal conductivity of borophene: a reactive molecular dynamics study,” Physica E-low-dimensional Systems & Nanostructures. 2017. link Times cited: 52 USED (high confidence) J. Gao, X. Luo, W. Xie, Y. Qin, R. M. M. Hasan, and P. Fan, “Atomistic Insights into Bias-Induced Oxidation on Passivated Silicon Surface Through Reaxff Md Simulation,” SSRN Electronic Journal. 2023. link Times cited: 1 USED (high confidence) B. Sharma and A. Parashar, “Mechanical strength of a nanoporous bicrystalline h-BN nanomembrane in a water submerged state.,” Physical chemistry chemical physics : PCCP. 2020. link Times cited: 18 Abstract: Due to superior water permeability, structural stability, an… read moreAbstract: Due to superior water permeability, structural stability, and adsorption capability, h-BN nanosheets are emerging as an efficient membrane for water desalination. In order to cater to the demand for potable water, large size membranes are required to maintain a high desalination rate from water purification systems. These large size membranes usually contain polycrystals with an offset in their mechanical properties from pristine h-BN nanosheets. In this article, molecular dynamics based simulations were performed in conjunction with a hybrid interatomic potential (reactive force field, TIP3P, and Lennard Jones) to simulate the mechanical strength of nanoporous single and bicrystalline h-BN nanosheets under water submerged conditions. The interaction between the atomic configuration of grain boundary atoms and nanopores in the presence of water molecules helps in investigating the viability of defective h-BN nanomembranes for underwater applications. Higher dislocation density enhances the mechanical strength of nanoporous bicrystalline h-BN nanosheets containing twin nanopores, which makes them a better substitute for water submerged applications as compared to the pristine nanosheets. The mechanical strength of nanoporous single crystalline h-BN nanosheets deteriorates with an increase in the number of nanopores, whereas a contrasting trend was observed with bicrystalline h-BN nanosheets. read less USED (high confidence) D. Mandelli, B. Hirshberg, and M. Parrinello, “Metadynamics of Paths.,” Physical review letters. 2020. link Times cited: 11 Abstract: We present a method to sample reactive pathways via biased m… read moreAbstract: We present a method to sample reactive pathways via biased molecular dynamics simulations in trajectory space. We show that the use of enhanced sampling techniques enables unconstrained exploration of multiple reaction routes. Time correlation functions are conveniently computed via reweighted averages along a single trajectory and kinetic rates are accessed at no additional cost. These abilities are illustrated analyzing a model potential and the umbrella inversion of NH_{3} in water. The algorithm allows a parallel implementation and promises to be a powerful tool for the study of rare events. read less USED (high confidence) F. J. Domínguez-Gutiérrez et al., “Deuterium uptake and sputtering of simultaneous lithiated, boronized, and oxidized carbon surfaces irradiated by low-energy deuterium,” Journal of Applied Physics. 2018. link Times cited: 5 Abstract: We study the effects of deuterium irradiation on D-uptake by… read moreAbstract: We study the effects of deuterium irradiation on D-uptake by simultaneously boronized, lithiated, oxidized, and deuterated carbon surfaces. We present analysis of the bonding chemistry of D for various concentrations of boron, lithium, oxygen, and deuterium on carbon surfaces using molecular dynamics with reactive force field potentials, which are here adapted to include the interaction of boron and lithium. We calculate D retention and sputtering yields of each constituent of the Li-C-B-O mixture and discuss the role of oxygen in these processes. The extent of the qualitative agreement between new experimental data for B-C-O-D obtained in this paper and computational data is provided. As in the case of the Li-C-O system, comparative studies where experimental and computational data complement each other (in this case on the B-Li-C-O system) provide deeper insights into the mechanisms behind the role that O plays in the retention of D, a relevant issue in fusion machines. read less USED (high confidence) M.-Q. Le, “Reactive molecular dynamics simulations of the mechanical properties of various phosphorene allotropes,” Nanotechnology. 2018. link Times cited: 19 Abstract: Although various phosphorene allotropes have been theoretica… read moreAbstract: Although various phosphorene allotropes have been theoretically predicted to be stable at 0 K, the mechanical properties and fracture mechanism at room temperature remain unclear for many of them. We investigate through reactive molecular dynamics simulations at room temperature the mechanical properties of phosphorene allotropes including: five sheets with hexagonal structures (β-, γ-, δ-, θ-, and α-phosphorene), one sheet with 4-8 membered rings (4-8-P), and two sheets with 5-7 membered rings. High, moderate and slight anisotropies in their mechanical properties are observed, depending on their crystal structures. Their Young’s moduli and tensile strength are approximately in the range from 7.3% through 25%, and from 8.6% through 22% of those of graphene, respectively. At the early stage of fracture, eye-shaped cracks are formed by local bond breaking and perpendicular to the tensile direction in hexagonal and 4-8-P sheets. Complete fractures take place with straight cracks in these hexagonal sheets under tension along the zigzag direction and under tension along the square edge direction in the 4-8-P sheet. Crack meandering and branching are observed during the tension of α-, β-, and γ-phosphorene along the armchair direction; and along the square diagonal direction in the 4-8-P sheet. Under uniaxial tension of two phosphorene sheets with 5-7 atom rings, 12 and 10 membered rings are formed by merging two neighbor heptagons, and a heptagon and its neighbor pentagon, respectively. These 12 and 10 membered rings coalesce subsequently, causing the failure of these two sheets. The results are of great importance in the design of these novel phosphorene allotropes. read less USED (high confidence) F. J. Domínguez-Gutiérrez and P. Krstic, “Chemical sputtering of boronized and oxidized carbon surfaces irradiated by low-energy deuterium atoms,” Journal of Applied Physics. 2017. link Times cited: 10 Abstract: We use molecular dynamics to study the chemical sputtering o… read moreAbstract: We use molecular dynamics to study the chemical sputtering of boronized and oxidized amorphous carbon surfaces by deuterium irradiation in the range of impact energies of 5–30 eV. We report the sputtering yield as well as mass, energy, and angular spectra of ejected atoms and molecules of both virgin and deuterium saturated BCO surfaces and compare them with our data for a deuterated BC surface and existing theoretical and experimental results for amorphous C:D surfaces. Boron significantly suppresses the erosion of carbon, while the presence of oxygen results in further suppression. read less USED (high confidence) B. Banerjee, G. Pugazhenthi, and T. Banerjee, “Experimental Insights into the Thermal Dehydrogenation of Ethylene Diamine Bisborane Using Allyl-Based Ionic Liquids,” Energy & Fuels. 2017. link Times cited: 21 Abstract: This work reports the use of allyl-based imidazolium cations… read moreAbstract: This work reports the use of allyl-based imidazolium cations for dehydrogenation of ethylene diaminebisborane (EDAB) at three different temperatures, namely, 95, 105, and 115 °C, under vacuum. The allyl-based ionic liquid (IL) was selected by using the infinite dilution activity coefficient (IDAC) as predicted from the COSMO-SAC (COnductor-like Screening MOdel–Segment Activity Coefficient) model. Based on the results of the COSMO-SAC model, the following allyl-based ILs were used for experimentation: 1-allyl-3-methylimidazolium dicyanamide ([AMIM][N(CN)2]), 1-allyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([AMIM][Tf2N]), and 1-allyl-3-methylimidazolium bromide ([AMIM][Br]). The highest amount of hydrogen (3.25 equiv) was measured from the EDAB/[AMIM][Br] system at 115 °C. Gas chromatography was conducted to confirm that the gas released was pure hydrogen. To better understand the reaction mechanism of EDAB dehydrogenation, the Reactive Force Field (ReaxFF) method was employed. Further analyse... read less USED (high confidence) I. Leven, I. Azuri, L. Kronik, and O. Hod, “Inter-layer potential for hexagonal boron nitride.,” The Journal of chemical physics. 2013. link Times cited: 63 Abstract: A new interlayer force-field for layered hexagonal boron nit… read moreAbstract: A new interlayer force-field for layered hexagonal boron nitride (h-BN) based structures is presented. The force-field contains three terms representing the interlayer attraction due to dispersive interactions, repulsion due to anisotropic overlaps of electron clouds, and monopolar electrostatic interactions. With appropriate parameterization, the potential is able to simultaneously capture well the binding and lateral sliding energies of planar h-BN based dimer systems as well as the interlayer telescoping and rotation of double walled boron-nitride nanotubes of different crystallographic orientations. The new potential thus allows for the accurate and efficient modeling and simulation of large-scale h-BN based layered structures. read less USED (high confidence) B. Chen et al., “ReaxFF Reactive Force Field for Molecular Dynamics Simulations of Lignite Depolymerization in Supercritical Methanol with Lignite-Related Model Compounds,” Energy & Fuels. 2012. link Times cited: 34 Abstract: To investigate the detailed mechanisms for lignite methanoly… read moreAbstract: To investigate the detailed mechanisms for lignite methanolysis, we used ReaxFF reactive force field to perform a series of molecular dynamics simulations (MDSs) on a unimolecular model compound. The α-O-4 and β-O-4 types of lignite-related model compounds were selected as representatives of linkages in lignites. The reaction products predicted by ReaxFF MDSs are consistent with those from experimental results reported. The initiation reaction observed in ReaxFF MDSs involving the ether linkage cleavage and methanol participation closely matches the results observed from previously reported experiments. The agreement of these results with available experimental observations demonstrates that ReaxFF MDSs can give an atomistic description of the initiation mechanism for methanolysis and provide useful insights into the complicated reaction processes. read less USED (high confidence) A. D. Pierro, B. Mortazavi, H. Noori, T. Rabczuk, and A. Fina, “A Multiscale Investigation on the Thermal Transport in Polydimethylsiloxane Nanocomposites: Graphene vs Borophene.” 2021. link Times cited: 5 Abstract: Graphene and borophene are highly attractive two-dimensional… read moreAbstract: Graphene and borophene are highly attractive two-dimensional materials with outstanding physical properties. In this study we employed a combined atomistic continuum multiscale modeling to explore the effective thermal conductivity of polymers nanocomposites made of PDMS polymer as the matrix and graphene and borophene as nanofillers. We first conduct classical molecular dynamics simulations to investigate the interfacial thermal conductance between graphene/PDMS and borophene/PDMS interfaces. Acquired results confirm that the interfacial thermal conductance between nanosheets and polymer increases from the single-layer to multilayered nanosheets and finally converges. The data provided by the atomistic simulations were then used in the finite element method simulations to evaluate the effective thermal conductivity of polymer nanocomposites at continuum level. We explore the effects of nanofillers type, their volume content, geometry aspect ratio and thickness on the nanocomposites effective thermal conductivity. As a very interesting finding, we show that borophene nanosheets, despite almost two orders of magnitude lower thermal conductivity than graphene, can yield very close enhancement in the effective thermal conductivity in comparison with graphene, particularly for low volume content and small aspect ratios and thicknesses. We conclude that for the polymer-based nanocomposites, significant improvement in the thermal conductivity can be reached by improving the bonding between the fillers and polymer or in another word enhancing the thermal conductance at the interface. By taking into account the high electrical conductivity of borophene, our results suggest borophene nanosheets as promising nanofillers to simultaneously enhance the polymers thermal and electrical conductivity. read less USED (low confidence) G. Liu, F. Shen, Y. Zhang, C. Liu, L. Yang, and H. Chang, “Reactive molecular dynamics study on carbon steel corrosion induced by chloride: Effects of applied potential and temperature,” Construction and Building Materials. 2024. link Times cited: 0 USED (low confidence) R. Chen et al., “A study on the pyrolysis of n-hexane initiated by 1-nitropropane: Molecular dynamics simulations and SVUV-PIMS experiments,” Journal of Analytical and Applied Pyrolysis. 2023. link Times cited: 0 USED (low confidence) M. Adouni, F. Alkhatib, A. Gouissem, and T. R. Faisal, “Knee joint biomechanics and cartilage damage prediction during landing: A hybrid MD-FE-musculoskeletal modeling,” PLOS ONE. 2023. link Times cited: 1 Abstract: Understanding the mechanics behind knee joint injuries and p… read moreAbstract: Understanding the mechanics behind knee joint injuries and providing appropriate treatment is crucial for improving physical function, quality of life, and employability. In this study, we used a hybrid molecular dynamics-finite element-musculoskeletal model to determine the level of loads the knee can withstand when landing from different heights (20, 40, 60 cm), including the height at which cartilage damage occurs. The model was driven by kinematics–kinetics data of asymptomatic subjects at the peak loading instance of drop landing. Our analysis revealed that as landing height increased, the forces on the knee joint also increased, particularly in the vastus muscles and medial gastrocnemius. The patellar tendon experienced more stress than other ligaments, and the medial plateau supported most of the tibial cartilage contact forces and stresses. The load was mostly transmitted through cartilage-cartilage interaction and increased with landing height. The critical height of 126 cm, at which cartilage damage was initiated, was determined by extrapolating the collected data using an iterative approach. Damage initiation and propagation were mainly located in the superficial layers of the tibiofemoral and patellofemoral cartilage. Finally, this study provides valuable insights into the mechanisms of landing-associated cartilage damage and could help limit joint injuries and improve training programs. read less USED (low confidence) F. Cao et al., “Effective parameters on the combustion performance of coated aluminum hydride nanoparticles: A molecular dynamics study,” Materials Today Communications. 2023. link Times cited: 1 USED (low confidence) J. Yang, T. Zhang, J. Cai, B. Niu, Y. Zhang, and D. Long, “Investigating the pyrolysis mechanisms of three archetypal ablative resins through pyrolysis experiments and ReaxFF MD simulations,” Materials Today Communications. 2023. link Times cited: 0 USED (low confidence) V. Mesilov, B. Zhuang, S. Xi, and S. Bernasek, “Poisoning of Copper Chabazite Catalyst by Biodiesel Metal Contaminants: Effect of Alkali and Alkaline Earth Metals,” The Journal of Physical Chemistry C. 2023. link Times cited: 0 USED (low confidence) Z. Yang, A. Xiao, D. Liu, Q. Shi, and Y. Li, “Damage of SARS‐CoV‐2 spike protein by atomic oxygen of cold atmospheric plasma: A molecular dynamics study,” Plasma Processes and Polymers. 2023. link Times cited: 2 Abstract: Recently, the cold atmospheric plasma (CAP) has demonstrated… read moreAbstract: Recently, the cold atmospheric plasma (CAP) has demonstrated a satisfactory ability to inactivate severe acute respiratory syndrome CoV‐2 (SARS‐CoV‐2), but the microscopic inactivation mechanism is still unclear. This paper takes the interaction process between O atoms generated by plasma and the spike protein of coronavirus as the research object. It uses the reaction molecular dynamics simulation method to study the reaction mechanism of different numbers of O atoms and the spike protein molecules. The results show that the O atom triggers a chain reaction by taking away hydrogen atoms in the spike protein molecule, destroying the molecular structure of the spike protein and making it inactive. The severity of the reaction and the destruction of the spike protein molecule also increases with increasing O atom numbers. [ FROM AUTHOR] Copyright of Plasma Processes & Polymers is the property of John Wiley & Sons, Inc. and its content may not be copied or emailed to multiple sites or posted to a listserv without the copyright holder's express written permission. However, users may print, download, or email articles for individual use. This may be abridged. No warranty is given about the accuracy of the copy. Users should refer to the original published version of the material for the full . (Copyright applies to all s.) read less USED (low confidence) Z. Liang, K. Li, F. Guo, H. Zhang, Y. Bu, and J. Zhang, “The Dynamic Nature of Graphene Active Sites in the H_2O Gasification process: A ReaxFF and DFT Study,” Journal of Molecular Modeling. 2023. link Times cited: 1 USED (low confidence) M. Wang et al., “Nano-deterioration of steel passivation film: chloride attack in material defects,” Materials and Structures. 2023. link Times cited: 9 USED (low confidence) Q. Xu, Y. Zhu, Y. Liu, Y. Shi, and Y.-B. Song, “Relationship between Organic Sulfur Occurrence and Coalification Stage:Insights from Thermodynamic Simulation and X-Ray Photoelectron Spectroscopy,” SSRN Electronic Journal. 2023. link Times cited: 1 USED (low confidence) M. Li et al., “Molecular progress of the corrosion of passivated Iron: The effects of structural strain,” Construction and Building Materials. 2022. link Times cited: 0 USED (low confidence) X. Dai et al., “Mineralization mechanism of carbon dioxide with illite interlayer cations using molecular dynamics simulation and experiments,” Journal of CO2 Utilization. 2022. link Times cited: 4 USED (low confidence) X. Huang and J. Wang, “Impacts of moisture on the thermal-oxygen pyrolysis of Kapton, a ReaxF MD simulation,” 2022 IEEE International Conference on High Voltage Engineering and Applications (ICHVE). 2022. link Times cited: 0 Abstract: Kapton-type polyimide films are always used as the solid ins… read moreAbstract: Kapton-type polyimide films are always used as the solid insulation medium in many power-electronic devices, which will decompose due to the high temperature caused by the partial discharge in the open air. In order to explore the influence of moisture on the thermal-oxygen decomposition characteristics of Kapton, simulation models containing 1polyimide, oxygen and water molecules are established in this paper. The ReaxFF molecular dynamics simulation is applied to indicate the pyrolysis process of polyimide, as well as the changes in production of small molecules. The results show that the moisture will encourage the degradation of main chains of polyimide by aggravating the broken of imide ring. And in the short effect of unbalanced PD, the benzene ring the Kapton could not be broken. read less USED (low confidence) S. Li, X. Yao, X. Wang, S.-Q. Tian, and Y. Zhang, “Reactive molecular dynamics simulation on degradation of aflatoxin B1 by cold atmospheric plasmas,” Innovative Food Science & Emerging Technologies. 2022. link Times cited: 8 USED (low confidence) L. An, R. Gu, B. Zhong, Y. Yu, and J. Zhang, “Water Icing Triggered Scalable and Controllable Exfoliation of Hexagonal Boron Nitride Nanosheets,” SSRN Electronic Journal. 2022. link Times cited: 6 Abstract: Scalable and controllable exfoliation for hexagonal boron ni… read moreAbstract: Scalable and controllable exfoliation for hexagonal boron nitride nanosheets ( h BNNSs) is still challenging. An et al. develop an environmentally friendly method to exfoliate h -BNNSs with controllable thickness and high yield by a rapid water freezing process. The as-obtained h -BNNSs can be used as polymer additives, thermal conductive fillers, and flame retardants. SUMMARY Hexagonal boron nitride nanosheets ( h -BNNSs) are a functional mate-rial with excellent performance; they have broad application prospects in the field of heat dissipation of electronicdevices. To achieve commercial application as soon as possible, there is an urgent need to develop a simple, controllable, and scalable method to produce high-quality h -BNNSs. Here, a scalable and controllable approach is proposed to exfoliate high-quality h -BNNSs from hexagonal boron nitride ( h -BN) flakes that relies on efficient reduction of h- BNNS interlayer interaction byrapidvolumeexpansion ofwaterinicing.Theeffectivenessandfeasi-bilityofthismethodare verifiedbymoleculardynamicssimulations.The thickness of h -BNNSs is determined by freezing and exfoliation cycles, and monolayer h -BNNSs can be obtained after five cycles of freezing and exfoliation. The as-obtained h -BNNSs have excellent dispersibility in water, enabling versatile application as polymer additives, thermal conductive fillers, flame retardants, and more. high-quality h -BNNSs from commercial h -BN flakes. This be achieved by freezing-induced volumetric expansion and subsequent ultrasonication-assisted exfoliation of commercial h -BN flakes. Hydroxyl groups local structure of demonstrated molecular dynamics (MD) simulations. photo-electron spectroscopy characterization. the XPS survey spectrum of the as-obtained h -BNNSs after five cycles of freezing and exfoliation, showing that the B1s peak located at 190.4 eV and N1s peak centered at 398.1 eV with high intensities can be easily detected. The atomic ratio of B:N is close to 1:1, indicating high purity of the as-obtained h -BNNSs. O1s and C1s peaks with very low intensities also appear in the XPS survey spectrum, which can be ascribed to the -OH groups attached to the edges/defects of the as-obtained h -BNNSs or ab-sorbed small amounts of contamination. Figure shows the Raman spectrum of theas-obtained h -BNNSsafterfivecyclesoffreezingandexfoliation;apeakcentered at cm (cid:3) 1 can clearly originated from an average of multiple specimens. The through-plane thermal diffusivity of the test samples were measured at 25 (cid:1) C using an LFA 467 analyzer (Netzsch). The specific heat capacity (Cp) of the samples was measured using a differential scanning calo-rimeter (DSC; TA Instruments) at a temperature range from room temperature to 200 (cid:1) C. The flame retardant test thermal infrared images were captured by a FLIR E5 camera. read less USED (low confidence) D. Hou et al., “The Corrosion Deterioration of Reinforced Passivation Film: The Impact of Defects,” Applied Surface Science. 2022. link Times cited: 14 USED (low confidence) K. Li et al., “Thermal behaviour, kinetics and mechanisms of CO2 interactions with graphene: An atomic scale reactive molecular dynamic study,” Chemical Engineering Journal. 2021. link Times cited: 11 USED (low confidence) J. Wang, B. Zhu, and Y. Sun, “Microscopic mechanism of α-rhombic crystal boron nanocluster oxidation in oxygen,” Fuel. 2021. link Times cited: 10 USED (low confidence) S. Liu, H. Jin, Y. Yang, and L. Yu, “Molecular dynamic investigation on sulfur migration during hydrogen production by benzothiophene gasification in supercritical water,” International Journal of Hydrogen Energy. 2021. link Times cited: 8 USED (low confidence) D. Zi-Zhao et al., “Boudouard reaction accompanied by graphitization of wrinkled carbon layers in coke gasification: A theoretical insight into the classical understanding,” Fuel. 2021. link Times cited: 12 USED (low confidence) P. Liu, P. Sui, and N. Song, “Adsorption behaviors of ethylenediamine on α-phase boron nanoparticle surfaces: first-principle calculation and MD simulation,” Journal of Nanoparticle Research. 2021. link Times cited: 1 USED (low confidence) A. Noroozi, N. Malih, and J. Davoodi, “The thermal transport characterization of borophene: A molecular dynamics study,” Computational Materials Science. 2021. link Times cited: 9 USED (low confidence) P. Gao and J. Zhang, “Understanding the Intra‐Molecular Proton Transfer of Octahydrotriborate and Exploring the Dehydrogenation Pathways of NH4B3H8 by DFT Calculations,” Advanced Theory and Simulations. 2021. link Times cited: 8 Abstract: The intra‐molecular proton transfer of octahydrotriborate, [… read moreAbstract: The intra‐molecular proton transfer of octahydrotriborate, [B3H8]−, is discovered by density functional theory (DFT) computational studies; such a transfer can largely impact its dehydrogenation pathways. The DFT calculation results further disclose the generation of another triborane intermediate product, B3H9, which is formed via inter‐molecular proton transfer from [NH4]+ to [B3H8]−. Additionally, this intermediate product is unstable and can release hydrogen easily, as the corresponding energy barrier via this pathway is only 12.0 kcal mol−1. Such a relative energy is much lower than that of the routinely raised pathway, which mainly depends on the dihydrogen interaction between the B−Hδ− and N−Hδ+ . This new mechanism is able to explain several experimental observations involving the dehydrogenation of NH4B3H8. Moreover, the detailed dehydrogenations of NH4B3H8 in different states are also studied, and the role of chemical environment during dehydrogenation is demonstrated. read less USED (low confidence) X. Sun, X. Luo, Y. Li, F. Yu, X. Zhao, and L. Yang, “Effects of H2 pre-etching on BN seed morphology and induced graphene synthesis on Cu substrate: A theoretical study,” Applied Surface Science. 2021. link Times cited: 0 USED (low confidence) Y. Q. Xing, J. K. Chi, and M. Xiao, “Reactive molecular dynamics simulation on the pyrolysis characteristics of epoxy resin under the effect of partial discharge active products,” High Performance Polymers. 2020. link Times cited: 5 Abstract: Aromatic amine cured Bisphenol F epoxy resin is used as a pi… read moreAbstract: Aromatic amine cured Bisphenol F epoxy resin is used as a pivotal solid insulation material in electrical equipment at low temperature and extreme environment. Partial discharge (PD) can lead to an increase in local temperature, which leads to pyrolysis, and the existence of PD active products will aggravate the pyrolysis process of insulating materials. In order to investigate the effect of PD active products on the pyrolysis characteristics of aromatic amine cured epoxy resin, the pyrolysis process of epoxy resin under partial discharge active products is simulated based on ReaxFF. The types and changing trend of small molecular products and the mechanism of PD active products acting on the pyrolysis process of epoxy resin are analyzed. The results show that the oxidation of hydroxyl and C-N bridge bonds by PD active products is the main reason for accelerating the pyrolysis of epoxy resin, which changes the main pyrolysis path of epoxy resin cross-linked structure. The thermal stability of cross-linked epoxy resin decreases under the action of active products, and the types and change trend of small molecular products generated in the pyrolysis process also changes greatly with the change of pyrolysis path. read less USED (low confidence) P. Liu et al., “Molecular-scale descriptions and experimental characterizations of nitrocellulose soaked in pure liquid ethanol or diethyl ether respectively at room temperature,” Materials Research Express. 2020. link Times cited: 3 Abstract: Studies on nitrocellulose (NC) mixtures with little solubili… read moreAbstract: Studies on nitrocellulose (NC) mixtures with little solubilities were neglected in many cases previously. This investigation was performed to provide supplemental characterizations of NC and its soaked state with pure liquid ethanol or diethyl ether by simulations and practical methods. Above all, a short-chained NC model (polymerisation degree: 8) and a dried NC specimen were characterized for their substitution of nitrate and microstructure. It was confirmed that both the numerical model and practical specimen belonged to low-nitrated NC. The bonding information of a glycosyl unit and nitrate ester were summarized via first-principle calculations. Then, ReaxFF potential based Molecular Dynamic (MD) simulations and soaking tests on binary organic mixtures demonstrated that both ethanol and diethyl ether had limited solubility for our specified NC. However, potential energies and diffusion coefficients of both computational models showed that the interactions from ethanol molecules were relatively stronger than diethyl ether molecules. The viscosities of saturated NC solutions also proved this consequence, as the difference between pure ether and its filtered NC solution was only 0.02 mm2 s−1. Finally, the strong volatility of diethyl ether itself could keep the wetness of NC upper surface shortly, because this was an upward volatilization effect. Due to this effect, the penetration of NC-diethyl ether mixture was higher in the early period of penetration tests. read less USED (low confidence) X. Sun et al., “Effect of BN seeds on locating and promoting the initial nucleation of graphene on Cu substrate and its mechanism: A theoretical study,” Applied Surface Science. 2020. link Times cited: 4 USED (low confidence) Y. Tian et al., “Molecular basis for coke strength: Stacking-fault structure of wrinkled carbon layers,” Carbon. 2020. link Times cited: 27 USED (low confidence) R. Sun, H. Qi, P. Liu, and F. Lv, “Molecular Dynamic Simulations of Diethyl Ether and its Mixture with Cellulose Dinitrate Tripolymer Molecules for their Thermal Diffusion Behaviors,” Journal of Molecular and Engineering Materials. 2020. link Times cited: 0 Abstract: In this paper, thermal diffusion states of pure diethyl ethe… read moreAbstract: In this paper, thermal diffusion states of pure diethyl ether and its mixture with cellulose dinitrate tripolymer were uncovered by LAMMPS-based Molecular Dynamic (MD) simulations. Those MD simulations were generally performed through specified ReaxFF reactive force field to obtain the properties of the chemical system such as molecular energy, density, mean square displacement (MSD) and molecular coordinate. The result of MD simulations presented the clear superheating phenomenon of pure liquid diethyl ether system in the studied environment. The obtained phase transition point was much higher than the reported one. The deviation between two temperatures was about 132.369[Formula: see text]K. It was also demonstrated that the transition process was associated with the sharp increment of potential energy, volume, diffusion coefficient and cohesive energy. However, the split of these diethyl ether molecules was not uniform. The cluster-like transition state was observed before the end of the vaporing process (460[Formula: see text]K). As for the addition of cellulose dinitrate tripolymer, these molecules were not agglomerated in the simulated organic mixture. However, the diffusion of cellulose dinitrate tripolymer was much weaker than those diethyl ether molecules. While the concentration of cellulose dinitrate tripolymer was higher, molecular interactions of this organic mixture were consequently improved, and this further limited the diffusion behavior of the entire chemical system. It could be concluded that the diffusion behavior of the entire organic system was decreased with more amount of cellulose dinitrate tripolymer molecules. read less USED (low confidence) C. Li, Y. Shi, Y. Hao, W.-sheng Li, S. Ren, and L. Wang, “Multilayer boron nitride nanofilm as an effective barrier for atomic oxygen irradiation,” Applied Surface Science. 2020. link Times cited: 8 USED (low confidence) F. Xu, H. Liu, Q. Wang, S. Pan, D. Zhao, and Y. Liu, “Study of non-isothermal pyrolysis mechanism of lignite using ReaxFF molecular dynamics simulations,” Fuel. 2019. link Times cited: 34 USED (low confidence) B. Mclean, G. Webber, and A. Page, “Boron Nitride Nanotube Nucleation during Ni-Catalyzed Boron Oxide Chemical Vapor Deposition,” The Journal of Physical Chemistry C. 2019. link Times cited: 6 Abstract: Boron oxide chemical vapor deposition (BOCVD) has proved to … read moreAbstract: Boron oxide chemical vapor deposition (BOCVD) has proved to be a valuable synthetic technique for the catalytic synthesis of boron nitride nanotubes (BNNTs) for almost two decades. However, the nucleation mechanism of BNNTs during BOCVD remains largely unknown. Here, we report a mechanism to explain BNNT nucleation on Ni catalyst nanoparticles during BOCVD with an ammonia precursor using nonequilibrium molecular dynamics simulations. The presence of oxygen is a significant impediment to the formation of BN hexagonal ring networks, due to the B–O bond strength and the rapid adsorption kinetics of BO monomers to the Ni catalyst. Despite H2O production being assumed to accompany the formation of BN during BOCVD, we do not observe Ni-catalyzed evolution of H2O, although significant amounts of H2 is evident. At low oxygen chemical potentials, defect-free BN ring networks are produced following the oligomerization of BN chain structures and the Ni-catalyzed cleavage of homoelemental B–B and N–N bonds. The BNNT ... read less USED (low confidence) B. Mclean, G. Webber, and A. Page, “Boron Nitride Nanotube Nucleation via Network Fusion during Catalytic Chemical Vapor Deposition.,” Journal of the American Chemical Society. 2019. link Times cited: 17 Abstract: Despite boron nitride nanotubes (BNNTs) first being synthesi… read moreAbstract: Despite boron nitride nanotubes (BNNTs) first being synthesized in the 1990s, their nucleation mechanism remains unknown. Here we report non-equilibrium molecular dynamics simulations showing how BNNT cap structures form during Ni-catalyzed chemical vapor deposition (CVD) of ammonia borane. BN hexagonal ring networks are produced following the catalytic evolution of H2 from the CVD feedstock, the formation and polymerization of B-N chain structures, and the repeated cleavage of homoelemental B-B / N-N bonds by the catalyst surface. Defect-free BNNT cap structures then form perpendicular to the catalyst surface via direct fusion of adjacent BN networks. This BNNT network fusion mechanism is a marked deviation from the established mechanism for carbon nanotube nucleation during CVD, and potentially explains why CVD-synthesized BNNTs are frequently observed having sharper tips and wider diameters, compared to CVD-synthesized carbon nanotubes. read less USED (low confidence) B. Mclean, G. Webber, and A. Page, “Boron Nitride Nucleation Mechanism during Chemical Vapor Deposition,” The Journal of Physical Chemistry C. 2018. link Times cited: 14 Abstract: We present nonequilibrium molecular dynamics simulations dem… read moreAbstract: We present nonequilibrium molecular dynamics simulations demonstrating how boron nitride (BN) nanomaterials nucleate during boron oxide chemical vapor deposition (CVD). Chemical reactions between g... read less USED (low confidence) V. Rizzi, D. Polino, E. Sicilia, N. Russo, and M. Parrinello, “The Onset of Dehydrogenation in Solid Ammonia Borane: An Ab Initio Metadynamics Study.,” Angewandte Chemie. 2018. link Times cited: 29 Abstract: The discovery of effective hydrogen storage materials is fun… read moreAbstract: The discovery of effective hydrogen storage materials is fundamental for the progress of a clean energy economy. Ammonia borane (H3 BNH3 , AB) has attracted great interest as a promising candidate but the reaction path that leads from its solid phase to hydrogen release is not yet fully understood. To address the need for insights in the atomistic details of such a complex solid state process, in this work we use ab-initio molecular dynamics and metadynamics to study the early stages of AB dehydrogenation. We show that the formation of ammonia diborane (H3 NBH2 (μ-H)BH3 ) leads to the release of NH4 + , which in turn triggers an autocatalytic H2 production cycle. Our calculations provide a model for how complex solid state reactions can be theoretically investigated and rely upon the presence of multiple ammonia borane molecules, as substantiated by standard quantum-mechanical simulations on a cluster. read less USED (low confidence) S. Sadeghzadeh, “Borophene sheets with in-plane chain-like boundaries; a reactive molecular dynamics study,” Computational Materials Science. 2018. link Times cited: 18 USED (low confidence) D. Hong and X. Guo, “Molecular dynamics simulations of Zhundong coal pyrolysis using reactive force field,” Fuel. 2017. link Times cited: 71 USED (low confidence) S. Sadeghzadeh, “The creation of racks and nanopores creation in various allotropes of boron due to the mechanical loads,” Superlattices and Microstructures. 2017. link Times cited: 16 USED (low confidence) U. Demirci, “Ammonia borane, a material with exceptional properties for chemical hydrogen storage,” International Journal of Hydrogen Energy. 2017. link Times cited: 202 USED (low confidence) B. T. Koo, R. Heden, and P. Clancy, “Nucleation and growth of 2D covalent organic frameworks: polymerization and crystallization of COF monomers.,” Physical chemistry chemical physics : PCCP. 2017. link Times cited: 20 Abstract: We establish a theoretical foundation for understanding the … read moreAbstract: We establish a theoretical foundation for understanding the nucleation and growth of 2D covalent organic frameworks (COFs) from solution. This foundation should make it easier to realize some of the unique properties of COFs in targeted applications by allowing us to understand how processing variables such as solvent choice and linkage chemistry lead to larger crystalline domains. We use free energy techniques to map out the reaction mechanisms and activation energies of three fundamental reactions that are responsible for the early stages of 2D COF nucleation for a prototypical and commonly used 2D boronate ester material, COF-5, in water and methanol solvents. We show that the presence of water and methanol greatly catalyzes the boronate ester formation reactions, lowering the activation energy barrier by about 10 kcal mol-1 relative to an uncatalyzed reaction pathway. This is in good agreement with experimental observations by Smith and Dichtel (JACS 2014). Our crystallization studies also conclusively eliminate certain proposed mechanisms of growth, such as polymerization of large sheets followed by stacking, while strengthening the case for templated polymerization as a likely growth mechanism for COF crystals. read less USED (low confidence) S. Shen, X. Lu, L. Liu, and C. Zhang, “Investigation of the influence of crack width on healing properties of asphalt binders at multi-scale levels,” Construction and Building Materials. 2016. link Times cited: 44 USED (low confidence) M.-Q. Le, B. Mortazavi, and T. Rabczuk, “Mechanical properties of borophene films: a reactive molecular dynamics investigation,” Nanotechnology. 2016. link Times cited: 54 Abstract: The most recent experimental advances could provide ways for… read moreAbstract: The most recent experimental advances could provide ways for the fabrication of several atomic thick and planar forms of boron atoms. For the first time, we explore the mechanical properties of five types of boron films with various vacancy ratios ranging from 0.1–0.15, using molecular dynamics simulations with ReaxFF force field. It is found that the Young’s modulus and tensile strength decrease with increasing the temperature. We found that boron sheets exhibit an anisotropic mechanical response due to the different arrangement of atoms along the armchair and zigzag directions. At room temperature, 2D Young’s modulus and fracture stress of these five sheets appear in the range 63–136 N m−1 and 12–19 N m−1, respectively. In addition, the strains at tensile strength are in the ranges of 9%–14%, 11%–19%, and 10%–16% at 1, 300, and 600 K, respectively. This investigation not only reveals the remarkable stiffness of 2D boron, but establishes relations between the mechanical properties of the boron sheets to the loading direction, temperature and atomic structures. read less USED (low confidence) J. Zhao, Z. Yang, N. Wei, and L. Kou, “Superhigh moduli and tension-induced phase transition of monolayer gamma-boron at finite temperatures,” Scientific Reports. 2016. link Times cited: 4 USED (low confidence) H. Wang, Y. Feng, X. Zhang, W. Lin, and Y. Zhao, “Study of coal hydropyrolysis and desulfurization by ReaxFF molecular dynamics simulation,” Fuel. 2015. link Times cited: 60 USED (low confidence) J. N. Sarma, R. Chowdhury, R. Jayaganthan, and F. Scarpa, “Atomistic Studies on Tensile Mechanics of BN Nanotubes in the Presence of Defects,” International Journal of Nanoscience. 2014. link Times cited: 8 Abstract: Boron nitride nanotubes (BNNTs) are of immense importance du… read moreAbstract: Boron nitride nanotubes (BNNTs) are of immense importance due to their many interesting functional features, notably biocompatibility and piezoelectricity and dominant mechanical strength as compared to carbon nanotubes (CNTs). The reliable implementation of these structures in an application is inherently related to its mechanical characteristics under external loads. The presence of defects in these structures severely affects the tensile properties. The effect of presence of point, line and Stone–Wales (SW) defects on the tensile behavior of BNNTs is systematically investigated by applying reactive force fields in molecular dynamics (MD) framework. Reactive force fields effectively describe the bond breaking and bond forming mechanism for BNNTs that are important for a practical situation. The Young's modulus of single-walled (10,0) BNNTs of length 100 nm has been found to be nearly 1.098 TPa, in good agreement with the available reports. The presence of defects has been shown to significantly reduce the tensile strength of the tube, while the number and separation of the defects effectively contribute to the percentage reduction. In addition, the effect of tube diameter and also the initial temperature are observed to strongly influence the tensile characteristics of BNNTs, indicating increased auxetic behavior than CNTs. read less USED (low confidence) K. Farah, M. Langeloth, M. Böhm, and F. Müller-Plathe, “Surface-Induced Interphases During Curing Processes Between Bi- and Pentafunctional Components: Reactive Coarse-Grained Molecular Dynamics Simulations,” The Journal of Adhesion. 2012. link Times cited: 7 Abstract: The present reactive molecular dynamics (RMD) simulations di… read moreAbstract: The present reactive molecular dynamics (RMD) simulations discuss the formation of interphase regions in cured polymer adhesives. The latter are obtained from the curing of reactive liquid mixtures composed of pentafunctional linkers and bifunctional monomers in contact with idealized surfaces. The present reactive scheme mimics the one of epoxies with amine linkers, i.e., processes investigated experimentally by Possart and co-workers. Generic RMD simulations are performed in a coarse-grained (CG) resolution to evaluate basic principles in curing characterized by preferential interactions. The creation of linker-rich domains is promoted by preferential surface-linker as well as linker-linker interactions in the reactive mixtures. The dimension of the interphase both in the starting mixture and the cured network depends on these preferential interactions which lead to a retardation of the curing velocity. This retardation behavior is mapped by conversion curves as a function of the number of reactive steps and by the spatially resolved profiles of the connected linkers. Although derived by generic potentials, the simulated reduction of the curing velocity is in agreement with experimental results in epoxies. The chosen interactions also imply a smaller number of linker bonds in the interphase than in the bulk region. The present RMD approach offers insight into key parameters of curing processes under the influence of preferential surface interactions coupled to selective attractions in the liquid starting mixture. read less USED (low confidence) B. Cui and H. Wang, “Oxidative aging mechanism of asphalt binder using experiment-derived average molecular model and ReaxFF molecular dynamics simulation,” Fuel. 2023. link Times cited: 5 USED (low confidence) B. Sharma and A. Parashar, “Effect of defects and functionalization on mechanical and fracture properties of two-dimensional nanomaterials,” Fundamentals and Properties of Multifunctional Nanomaterials. 2021. link Times cited: 0 NOT USED (low confidence) M. Yu, R. Lou, H. Li, F. Wang, J. Wang, and K. Wang, “Reactive force field molecular dynamics (ReaxFF-MD) simulation of lignite combustion under an external electric field,” Fuel. 2024. link Times cited: 0 NOT USED (low confidence) Y. Guo, H. Shi, H. Liu, Y. Xie, and Y. Guan, “Reactive molecular dynamics simulation and chemical kinetic modeling of ammonia/methane co-combustion,” Fuel. 2023. link Times cited: 0 NOT USED (low confidence) H. Jabraoui, A. Alpuche, C. Rossi, and A. Estève, “New insights into the mechanisms of TiB2(001) thermal oxidation combining molecular dynamics and density functional theory calculations,” Acta Materialia. 2023. link Times cited: 0 NOT USED (low confidence) P. Krstic et al., “Detailed studies of the processes in low energy H irradiation of Li and Li-compound surfaces,” Journal of Applied Physics. 2023. link Times cited: 0 Abstract: We have used a combination of pico-to-nano temporal/spatial … read moreAbstract: We have used a combination of pico-to-nano temporal/spatial scale computational physics and chemistry modeling of plasma–material interfaces in the tokamak fusion plasma edges to unravel the evolving characteristics, not readily accessible by empirical means, of lithium-, oxygen-, and hydrogen-containing materials of plasma-facing components under irradiation by hydrogen and its isotopes. In the present calculation, amorphous lithium compound surfaces containing oxygen, Li2O, and LiOH were irradiated by 1–100 eV particles at incident angles on the surface ranging from perpendicular to almost grazing angles. Consequential surface processes, reflection, retention, and sputtering were studied at “the same footing” and compared to earlier results from amorphous Li and LiH surfaces. The critical role of charging dynamics of lithium, oxygen, and hydrogen atoms in the surface chemistry during hydrogen-fuel irradiation was found to drive the kinetics and dynamics of these surfaces in unexpected ways that ultimately could have profound effects on fusion plasma confinement behavior and surface erosion. read less NOT USED (low confidence) Q. Mao, M. Feng, X. Jiang, Y. Ren, K. Luo, and A. V. van Duin, “Classical and reactive molecular dynamics: Principles and applications in combustion and energy systems,” Progress in Energy and Combustion Science. 2023. link Times cited: 10 NOT USED (low confidence) R. Cappabianca, P. D. Angelis, M. Fasano, E. Chiavazzo, and P. Asinari, “An Overview on Transport Phenomena within Solid Electrolyte Interphase and Their Impact on the Performance and Durability of Lithium-Ion Batteries,” Energies. 2023. link Times cited: 0 Abstract: The nature of the electrode–electrolyte interface has an imp… read moreAbstract: The nature of the electrode–electrolyte interface has an impact on the performance and durability of lithium-ion batteries (LIBs). The initial electrolyte’s thermodynamic instability at the anode–electrolyte interface in LIBs results in the formation of a passivation layer, called solid electrolyte interphase (SEI). The initial dense and intact layer allows Li+ transport and restricts electron tunneling, thus preventing electrolyte decomposition and ensuring the electrochemical stability of a battery. However, the growth of this layer can reduce the availability of active lithium and electrolyte, and ultimately lead to an irreversible battery capacity fade. Investigating the transport phenomena of lithium ions within SEI is crucial for understanding its formation and growth. Nonetheless, accurately describing all relevant mechanisms is challenging due to its complex and multiscale nature. An overview of current computational efforts to study Li+ transport within SEI is given in this article, ranging from electronic/atomistic scale simulations to macroscopic models. The drawbacks and advantages of the proposed numerical approaches are summarized along with the obstacles that need to be overcome to obtain accurate experimental data, identified on the basis of the most recent literature evidence. We highlight collaboration gaps between modeling and experimental approaches, as well as the urgent need for new multiscale models, to gain a better understanding of such a crucial transport phenomenon. read less NOT USED (low confidence) N. Nayir et al., “Modeling and simulations for 2D materials: a ReaxFF perspective,” 2D Materials. 2023. link Times cited: 5 Abstract: Recent advancements in the field of two-dimensional (2D) mat… read moreAbstract: Recent advancements in the field of two-dimensional (2D) materials have led to the discovery of a wide range of 2D materials with intriguing properties. Atomistic-scale simulation methods have played a key role in these discoveries. In this review, we provide an overview of the recent progress in ReaxFF force field developments and applications in modeling the following layered and nonlayered 2D materials: graphene, transition metal dichalcogenides, MXenes, hexagonal boron nitrides, groups III-, IV- and V-elemental materials, as well as the mixed dimensional van der Waals heterostructures. We further discuss knowledge gaps and challenges associated with synthesis and characterization of 2D materials. We close this review with an outlook addressing the challenges as well as plans regarding ReaxFF development and possible large-scale simulations, which should be helpful to guide experimental studies in a discovery of new materials and devices. read less NOT USED (low confidence) Q. Luo et al., “Insights into the Oxygen-Containing Groups Transformation During Coal Char Gasification in H2o/Co2 Atmosphere by Using Reaxff Reactive Force Field,” SSRN Electronic Journal. 2023. link Times cited: 2 NOT USED (low confidence) L. Liang et al., “Theoretical Insight into the Competitive Effect of Co2 and Additive H2o in Coke Gasification,” SSRN Electronic Journal. 2023. link Times cited: 1 NOT USED (low confidence) R. Huang, Y. Sun, Z. Yang, Y. Liu, and S. Yue, “A novel ReaxFF multi-scale method for analyzing the fracture behavior of the CeO2,” Computational Materials Science. 2023. link Times cited: 2 NOT USED (low confidence) P. Biswas, Y. Wang, S. A. Herrera, P. Ghildiyal, and M. Zachariah, “Catalytic Cleavage of the Dative Bond of Ammonia Borane by Polymeric Carbonyl Groups for Enhanced Energy Generation,” Chemistry of Materials. 2023. link Times cited: 1 NOT USED (low confidence) G. Li et al., “Molecular Insight into Pyrolysis Processes via Reactive Force Field Molecular Dynamics: A State-of-the-art Review,” Journal of Analytical and Applied Pyrolysis. 2022. link Times cited: 12 NOT USED (low confidence) Y. Yamada, H. Tanaka, Y. Tanaka, S. Kubo, T. Taguchi, and S. Sato, “Toward strategical bottom-up synthesis of carbon materials with exceptionally high pyridinic-nitrogen content: Development of screening techniques,” Carbon. 2022. link Times cited: 8 NOT USED (low confidence) X. Huang, J. Wang, J. Wang, H. Xie, and Q. Li, “A Reaxff Md Based Effect Investigation of Diamino Curing Agents in the Initial Thermo-Oxidative Pyrolysis of Epoxy Resins,” SSRN Electronic Journal. 2022. link Times cited: 3 NOT USED (low confidence) W. Zhang, F. Starr, K. Beers, and J. Douglas, “Reactive Molecular Dynamics Simulations of the Depolymerization of Polyethylene Using Graphene-Oxide-Supported Platinum Nanoparticles.,” The journal of physical chemistry. A. 2022. link Times cited: 3 Abstract: While plastic materials offer many benefits to society, the … read moreAbstract: While plastic materials offer many benefits to society, the slow degradation and difficulty in recycling plastics raise important environmental and sustainability concerns. Traditional recycling efforts often lead to materials with inferior properties and correspondingly lower value, making them uneconomical to recycle. Recent efforts have shown promising chemical pathways for converting plastic materials into a wide range of value-added products, feedstocks or monomers. This is commonly referred to as "chemical recycling". Here, we use reactive molecular dynamics (MD) simulations to study the catalytic process of depolymerization of polyethylene (PE) using platinum (Pt) nanoparticles (NPs) in comparison to PE pyrolysis (thermal degradation). We apply a simple kinetic model to our MD results for the catalytic reaction rate as a function of temperature, from which we obtain the activation energy of the reaction, which shows the that the Pt NPs reduce the barrier for depolymerization. We further evaluate the molecular mass distribution of the reaction products to gain insight into the influence of the Pt NPs on reaction selectivity. Our results demonstrate the potential for the reactive MD method to help the design of recycling approaches for polymer materials. read less NOT USED (low confidence) X. Wang, T. Zhao, Y. Wang, L. Zhang, and L. Zou, “Microscopic Pyrolytic and Electric Decomposition Mechanism of Insulating Polyimide/Boron Nitride Nanosheet Composites based on ReaxFF,” Polymers. 2022. link Times cited: 4 Abstract: High thermal conductivity insulating materials with excellen… read moreAbstract: High thermal conductivity insulating materials with excellent comprehensive properties can be obtained by doping boron nitride nanosheets (BNNSs) into polyimide (PI). To study the microscopic mechanism of composite material decomposition in an actual working environment and the inhibitory effect of BNNS doping on the decomposition process, molecular dynamics simulations were carried out at high temperatures, in intense electric fields, and with various reactive species in plasma based on the reactive force field (ReaxFF). The results showed that the decomposition was mainly caused by hydrogen capture and adsorption, which broke the benzene ring and C-N bond on the PI chains and led to serious damage to the PI structure. The BNNS filling was shown to inhibit the decomposition of the PI matrix at high temperatures and in intense electric fields. Moreover, the BNNS filling also inhibited the material decomposition caused by ·OH and ·NO. The erosive effect of the positive corona on the PI composites was more obvious than that of the negative corona. In this paper, the microscopic dynamic reaction paths of material pyrolysis in various environments were revealed at the atomic level, and it was concluded that BNNS doping could effectively inhibit the decomposition of PI in various environments. read less NOT USED (low confidence) J. Zhang et al., “Insights into the Molecular Structure of Yangchangwan Subbituminous Coal Based on the Combination of Experimental and Multi-Scale Computational Descriptions,” Solid Fuel Chemistry. 2022. link Times cited: 4 NOT USED (low confidence) A. Lele, P. Krstic, and A. V. van Duin, “ReaxFF Force Field Development for Gas-Phase hBN Nanostructure Synthesis.,” The journal of physical chemistry. A. 2022. link Times cited: 1 Abstract: Two-dimensional (2D) hexagonal boron nitride materials are i… read moreAbstract: Two-dimensional (2D) hexagonal boron nitride materials are isomorphs of carbon nanomaterials and hold promise for electronics applications owing to their unique properties. Despite the recent advances in synthesis, the current production capacity for boron nitride (BN) nanostructures is far behind that for carbon-based nanostructures. Understanding the growth mechanism of BN nanostructures through modeling and experiments is key to improving this situation. In the current work, we present the development of a ReaxFF-based force field capable of modeling the gas-phase chemistry important for the chemical vapor deposition (CVD) synthesis process. This force field is parameterized to model the boron nitride nanostructure (BNNS) formation in the gas phase using BN and HBNH as precursors. Our ReaxFF simulations show that BN is the best of these two precursors in terms of quality and the size of BNNSs. The BN precursors lead to the formation of closed BNNSs. However, BNNSs are replaced with complex polymeric structures at temperatures of 2500 K and higher due to entropic effects. Compared to the BN precursors, the HBNH precursors form relatively small, flat, and low-quality BNNSs, but this structure is less affected by temperature. Additives like H2 significantly affect the BNNS formation by preventing closed BNNS formation. Our results show the ReaxFF capability in predicting the BN gas-phase chemistry and BNNS formation, thus providing key insights for experimental synthesis. read less NOT USED (low confidence) H. Wang, T. Zhao, S. Yang, L. Zou, X. Wang, and Y. Zhang, “Reactive force field-based molecular dynamics simulation of the interaction between plasma reactive oxygen species and the receptor-binding domain of the spike protein in the capsid protein of SARS-CoV-2,” Journal of Physics D: Applied Physics. 2021. link Times cited: 2 Abstract: Under the pressures of the current global pandemic, research… read moreAbstract: Under the pressures of the current global pandemic, researchers have been working hard to find a reliable way to suppress infection by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and prevent the spread of COVID-19. Studies have shown that the recognition and binding of human angiotensin-converting enzyme 2 by the receptor-binding domain (RBD) of the spike protein on the surface of SARS-CoV-2 is a crucial step in viral invasion of human receptor cells, and blocking this process could inhibit the virus from invading normal human cells. Plasma treatment can disrupt the structure of the RBD and effectively block the binding process. However, the mechanism by which plasma blocks recognition and binding is not clear. In this study, the reaction between reactive oxygen species (ROS) in plasma and a molecular model of the RBD was simulated using a reactive molecular dynamics method. The results showed that the destruction of the RBD by ROS was triggered by hydrogen abstraction reactions: O and OH abstracted H atoms from the RBD, while the H atoms of H2O2 and HO2 were abstracted by the RBD. This hydrogen abstraction resulted in the breakage of C–H, N–H, O–H and C=O bonds and the formation of C=C and C=N bonds. The addition reaction of OH increased the number of O–H bonds and caused the formation of C–O, N–O and O–H bonds. The dissociation of N–H bonds led to the destruction of the original peptide bond structure and amino acid residues, changed the type of amino acid residues and caused the conversion of N–C and N=C and C=O and C–O. The simulation partially elucidated the microscopic mechanism of the interaction between ROS in plasma and the capsid protein of SARS-CoV-2, providing theoretical support for the control of SARS-CoV-2 infection by plasma, a contribution to overcoming the global pandemic. read less NOT USED (low confidence) D. Yilmaz, W. Woodward, and A. V. van Duin, “Machine Learning-Assisted Hybrid ReaxFF Simulations.,” Journal of chemical theory and computation. 2021. link Times cited: 5 Abstract: We have developed a machine learning (ML)-assisted Hybrid Re… read moreAbstract: We have developed a machine learning (ML)-assisted Hybrid ReaxFF simulation method ("Hybrid/Reax"), which alternates reactive and non-reactive molecular dynamics simulations with the assistance of ML models to simulate phenomena that require longer time scales and/or larger systems than are typically accessible to ReaxFF. Hybrid/Reax uses a specialized tracking tool during the reactive simulations to further accelerate chemical reactions. Non-reactive simulations are used to equilibrate the system after the reactive simulation stage. ML models are used between reactive and non-reactive stages to predict non-reactive force field parameters of the system based on the updated bond topology. Hybrid/Reax simulation cycles can be continued until the desired chemical reactions are observed. As a case study, this method was used to study the cross-linking of a polyethylene (PE) matrix analogue (decane) with the cross-linking agent dicumyl peroxide (DCP). We were able to run relatively long simulations [>20 million molecular dynamics (MD) steps] on a small test system (4660 atoms) to simulate cross-linking reactions of PE in the presence of DCP. Starting with 80 PE molecules, more than half of them cross-linked by the end of the Hybrid/Reax cycles on a single Xeon processor in under 48 h. This simulation would take approximately 1 month if run with pure ReaxFF MD on the same machine. read less NOT USED (low confidence) S. Attarian and S. Xiao, “Development of a 2NN-MEAM potential for boron.” 2021. link Times cited: 2 Abstract: In this paper, we present the first work in developing a sec… read moreAbstract: In this paper, we present the first work in developing a second nearest-neighbor modified embedded atom method (2NN-MEAM) potential function that can be used to model interatomic interactions in bo... read less NOT USED (low confidence) S. Arabha, A. Akbarzadeh, and A. Rajabpour, “Engineered porous borophene with tunable anisotropic properties,” Composites Part B-engineering. 2020. link Times cited: 17 NOT USED (low confidence) P. Gao and J. Zhang, “Understanding the Dehydrogenation Pathways of Ammonium Octahydrotriborate (NH4B3H8) by Molecular Dynamics Simulations with the Reactive Force Field (ReaxFF),” Advanced Theory and Simulations. 2020. link Times cited: 11 Abstract: Ammonium octahydrotriborate (NH4B3H8) is a potential candida… read moreAbstract: Ammonium octahydrotriborate (NH4B3H8) is a potential candidate for hydrogen storage, due to its chemical stability and high hydrogen content. To systematically investigate its initial decomposition and dehydrogenation pathways, molecular dynamics simulations with a reactive force field are conducted. Both temperature ramping and the canonical ensemble simulations are carried out. A compositional analysis for the simulated systems is also applied to monitor the possible intermediate products during its decomposition and dehydrogenation processes. At around 1000 K, the molecular hydrogen is released via inter‐molecular interaction between B‐H δ− and N‐H δ+ . It is also noticed that the formed NH3 and BH3 can react to generate BN bonds; as the temperature further increases, the isomers with a skeleton of BNB are observed, and the corresponding dehydrogenation is also increased. The present work provides detailed pictures of the decomposition and dehydrogenation pathways for NH4B3H8. read less NOT USED (low confidence) H. Sim, R. Yetter, S. Hong, A. V. van Duin, D. Dabbs, and I. Aksay, “Enhanced Fuel Decomposition in the Presence of Colloidal Functionalized Graphene Sheet-Supported Platinum Nanoparticles.” 2020. link Times cited: 9 Abstract: Experiments and simulations
were used to demonstrate that de… read moreAbstract: Experiments and simulations
were used to demonstrate that decorating
functionalized graphene sheets (FGSs) with platinum nanoparticles
(Pt@FGS) stabilized these particles. Addition of these particles to
liquid hydrocarbon fuels was observed to significantly affect decomposition
under supercritical conditions at a pressure of 4.75 MPa and temperatures
from 753 to 803 K. The suspension of only 50 ppmw Pt@FGS in the fuel
(equivalent to adding 10 ppmw Pt) enhanced fuel conversion rates (by
up to 24%) with a major effect on specific product yields. The production
of low-molecular-weight species increased in the pyrolysis products
(with the hydrogen yield increasing by a factor of 12.5). ReaxFF molecular
dynamics (MD) simulations supported a mechanism in which synergy between
Pt and FGS catalyzed dehydrogenation during n-C12H26 pyrolysis. The highest conversion rates and
greatest yields of hydrogen and low-molecular-weight species were
observed for fuels containing Pt@FGS particles rather than those containing
either FGSs or Pt-clusters alone. Analysis of the platinum decorated
FGSs post reaction indicated no deterioration of the composite particles. read less NOT USED (low confidence) W. Feng, Z. Li, H. Gao, Q. Wang, H.-cun Bai, and P. Li, “Understanding the molecular structure of HSW coal at atomic level: A comprehensive characterization from combined experimental and computational study,” Green Energy & Environment. 2020. link Times cited: 30 NOT USED (low confidence) G. Zhang, J. Li, and Z. Liu, “Multiple Objective NSGA-II-Based Optimization Program and Its Application in Reactive Force Field for 2,4,6-Trinitrotoluene Diffusion in the Aqueous Phase,” The Journal of Physical Chemistry C. 2019. link Times cited: 4 Abstract: The reactive force field (ReaxFF) molecular dynamics simulat… read moreAbstract: The reactive force field (ReaxFF) molecular dynamics simulation is an effective method to study the behavior of energetic materials in the aqueous phase. In order to optimize the ReaxFF of energeti... read less NOT USED (low confidence) N. Nayir, A. V. van Duin, and S. Erkoç, “Development of the ReaxFF Reactive Force Field for Inherent Point Defects in the Si/Silica System.,” The journal of physical chemistry. A. 2019. link Times cited: 16 Abstract: We redeveloped the ReaxFF force field parameters for Si/O/H … read moreAbstract: We redeveloped the ReaxFF force field parameters for Si/O/H interactions that enable molecular dynamics (MD) simulations of Si/SiO2 interfaces and O diffusion in bulk Si at high temperatures, in particular with respect to point defect stability and migration. Our calculations show that the new force field framework (ReaxFFpresent), which was guided by the extensive quantum mechanical-based training set, describes correctly the underlying mechanism of the O-migration in Si network, namely, the diffusion of O in bulk Si occurs by jumping between the neighboring bond-centered sites along a path in the (110) plane, and during the jumping, O goes through the asymmetric transition state at a saddle point. Additionally, the ReaxFFpresent predicts the diffusion barrier of O-interstitial in the bulk Si of 64.8 kcal/mol, showing a good agreement with the experimental and density functional theory values in the literature. The new force field description was further applied to MD simulations addressing O diffusion in bulk Si at different target temperatures ranging between 800 and 2400 K. According to our results, O diffusion initiates at the temperatures over 1400 K, and the atom diffuses only between the bond-centered sites even at high temperatures. In addition, the diffusion coefficient of O in Si matrix as a function of temperature is in overall good agreement with experimental results. As a further step of the force field validation, we also prepared amorphous SiO2 (a-SiO2) with a mass density of 2.21 gr/cm3, which excellently agrees with the experimental value of 2.20 gr/cm3, to model a-SiO2/Si system. After annealing the a-SiO2/Si system at high temperatures until below the computed melting point of bulk Si, the results show that ReaxFFpresent successfully reproduces the experimentally and theoretically defined diffusion mechanism in the system and succeeded in overcoming the diffusion problem observed with ReaxFFSiOH(2010), which results in O diffusion in the Si substrate even at the low temperature such as 300 K. read less NOT USED (low confidence) T. Liang, P. Zhang, P. Yuan, S. Zhai, and D. Yang, “A molecular dynamics study on the thermal conductivities of single- and multi-layer two-dimensional borophene,” Nano Futures. 2019. link Times cited: 23 Abstract: Borophene, a new two-dimensional (2D) structure of boron ato… read moreAbstract: Borophene, a new two-dimensional (2D) structure of boron atoms, has aroused a great deal of attention and research recently. However, research on the thermal conductivity of borophene is still scarce, although this is critical for the potential application of borophene. Accordingly, we investigate the in-plane and cross-plane thermal conductivities of single- and multi-layer borophene using the non-equilibrium molecular dynamics simulations. The effect on the thermal conductivity with respect to sample length, temperature, layer number and mechanical strain is systematically examined. It is found that the in-plane thermal conductivity of infinite-size single-layer borophene exhibits strong anisotropy, which is calculated to be 102.5 ± 1.9 (along the zigzag direction) and 233.3 ± 2.1 W m−1K−1 (along the armchair direction). Notably, we found that both the in-plane and cross-plane thermal conductivities of borophene are affected by temperature variations, which is the same as other 2D materials. Surprisingly, the in-plane thermal conductivity of multi-layer borophene is insensitive to the layer number. This is attributed to the out-of-plane flexural phonons mode vibration being maintained by the intrinsic bi-layer structure (buckled structure), resulting in a negligible effect of interlayer vdW interactions of the multi-layer structure on the out-of-plane flexural phonons mode. In particular, the cross-plane strain was found to be effective in modulating the cross-plane thermal conductivity of multi-layer borophene in our research. Our findings here are of significance for understanding the thermal transport behavior of single- and multi-layer borophene and promoting their future applications in thermal management and nanodevices. read less NOT USED (low confidence) N. Nayir, A. V. van Duin, and S. Erkoç, “Development of a ReaxFF Reactive Force Field for Interstitial Oxygen in Germanium and Its Application to GeO2/Ge Interfaces,” The Journal of Physical Chemistry C. 2019. link Times cited: 10 Abstract: We developed the ReaxFF force field parameters for Ge/O/H in… read moreAbstract: We developed the ReaxFF force field parameters for Ge/O/H interactions, specifically targeted for the applications of Ge/GeO2 interfaces and O-diffusion in bulk Ge. The original training set, taken... read less NOT USED (low confidence) F. Yin, C. Tang, Y. Tang, Y. Gui, and Z. Zhao, “Reactive Molecular Dynamics Study of Effects of Small-Molecule Organic Acids on PMIA Thermal Decomposition.,” The journal of physical chemistry. B. 2018. link Times cited: 8 Abstract: Reactive molecular dynamics was used to investigate the atom… read moreAbstract: Reactive molecular dynamics was used to investigate the atomic-level mechanism of formic acid-accelerated deterioration of meta-aramid (PMIA) fibers. The simulation results showed that formic acid promoted PMIA decomposition. The activation energy of a composite system (PF) consisting of formic acid and PMIA was 106.94 kJ/mol at 2000-3000 K, which is 11.95% lower than that of pure PMIA. The main small-molecule products of the PF system were H/C/O-containing molecules (H2O, CO, and CO2), hydrocarbon molecules (e.g., CH4, •C2H, C2H4, and C3H4), N-containing molecules (N2, NH3, and HCN), H2, and various free radicals. Formic acid can promote the production of small molecules such as CO, CO2, and H2O. The N-H bonds, C-N bonds and the amide C═O double bond of PMIA were vulnerable to CO, H ions, and free radicals produced by formic acid decomposition, and this decreased the PMIA stability. Temperature is an important factor in the thermal decomposition of PMIA and can accelerate reactions in the PF system. The initial reaction rate of PMIA at 3000 K was 8.1 times that at 2000 K, and the intermediate reaction rate was 6.2 times that at 2200 K; temperature also affects the types of pyrolysis products, for example, hydrocarbons are high-temperature products. read less NOT USED (low confidence) P. Krstic, J. Allain, F. J. Domínguez-Gutiérrez, and F. Bedoya, “Unraveling the surface chemistry processes in lithiated and boronized plasma material interfaces under extreme conditions,” Matter and Radiation at Extremes. 2018. link Times cited: 17 NOT USED (low confidence) S. Sadeghzadeh and M. M. Khatibi, “Vibrational modes and frequencies of borophene in comparison with graphene nanosheets,” Superlattices and Microstructures. 2018. link Times cited: 16 NOT USED (low confidence) Z. Sha, Q. Pei, K. Zhou, Z. Dong, and Y.-W. Zhang, “Temperature and strain-rate dependent mechanical properties of single-layer borophene,” Extreme Mechanics Letters. 2018. link Times cited: 22 NOT USED (low confidence) B. Liu and S. Yan, “DFT investigation on the decomposition of dihydrogen-bonded methylamine-borane octamer,” Computational and Theoretical Chemistry. 2018. link Times cited: 5 NOT USED (low confidence) Y. Yamada et al., “Carbon materials with controlled edge structures,” Carbon. 2017. link Times cited: 54 NOT USED (low confidence) G. Siegel, C. Ciobanu, B. Narayanan, M. Snure, and S. Badescu, “Heterogeneous Pyrolysis: A Route for Epitaxial Growth of hBN Atomic Layers on Copper Using Separate Boron and Nitrogen Precursors.,” Nano letters. 2017. link Times cited: 20 Abstract: Growth of hBN on metal substrates is often performed via che… read moreAbstract: Growth of hBN on metal substrates is often performed via chemical vapor deposition from a single precursor (e.g., borazine) and results in hBN monolayers limited by the substrates catalyzing effect. Departing from this paradigm, we demonstrate close control over the growth of mono-, bi-, and trilayers of hBN on copper using triethylborane and ammonia as independent sources of boron and nitrogen. Using density functional theory (DFT) calculations and reactive force field molecular dynamics, we show that the key factor enabling the growth beyond the first layer is the activation of ammonia through heterogeneous pyrolysis with boron-based radicals at the surface. The hBN layers grown are in registry with each other and assume a perfect or near perfect epitaxial relation with the substrate. From atomic force microscopy (AFM) characterization, we observe a moiré superstructure in the first hBN layer with an apparent height modulation and lateral periodicity of ∼10 nm. While this is unexpected given that the moiré pattern of hBN/Cu(111) does not have a significant morphological corrugation, our DFT calculations reveal a spatially modulated interface dipole layer which determines the unusual AFM response. These findings have improved our understanding of the mechanisms involved in growth of hBN and may help generate new growth methods for applications in which control over the number of layers and their alignment is crucial (such as tunneling barriers, ultrathin capacitors, and graphene-based devices). read less NOT USED (low confidence) C. M. Ashraf and A. V. van Duin, “Extension of the ReaxFF Combustion Force Field toward Syngas Combustion and Initial Oxidation Kinetics.,” The journal of physical chemistry. A. 2017. link Times cited: 170 Abstract: A detailed insight of key reactive events related to oxidati… read moreAbstract: A detailed insight of key reactive events related to oxidation and pyrolysis of hydrocarbon fuels further enhances our understanding of combustion chemistry. Though comprehensive kinetic models are available for smaller hydrocarbons (typically C3 or lower), developing and validating reaction mechanisms for larger hydrocarbons is a daunting task, due to the complexity of their reaction networks. The ReaxFF method provides an attractive computational method to obtain reaction kinetics for complex fuel and fuel mixtures, providing an accuracy approaching ab-initio-based methods but with a significantly lower computational expense. The development of the first ReaxFF combustion force field by Chenoweth et al. (CHO-2008 parameter set) in 2008 has opened new avenues for researchers to investigate combustion chemistry from the atomistic level. In this article, we seek to address two issues with the CHO-2008 ReaxFF description. While the CHO-2008 description has achieved significant popularity for studying large hydrocarbon combustion, it fails to accurately describe the chemistry of small hydrocarbon oxidation, especially conversion of CO2 from CO, which is highly relevant to syngas combustion. Additionally, the CHO-2008 description was obtained faster than expected H abstraction by O2 from hydrocarbons, thus underestimating the oxidation initiation temperature. In this study, we seek to systemically improve the CHO-2008 description and validate it for these cases. Additionally, our aim was to retain the accuracy of the 2008 description for larger hydrocarbons and provide similar quality results. Thus, we expanded the ReaxFF CHO-2008 DFT-based training set by including reactions and transition state structures relevant to the syngas and oxidation initiation pathways and retrained the parameters. To validate the quality of our force field, we performed high-temperature NVT-MD simulations to study oxidation and pyrolysis of four different hydrocarbon fuels, namely, syngas, methane, JP-10, and n-butylbenzene. Results obtained from syngas and methane oxidation simulation indicated that our redeveloped parameters (named as the CHO-2016 parameter set) has significantly improved the C1 chemistry predicted by ReaxFF and has solved the low-temperature oxidation initiation problem. Moreover, Arrhenius parameters of JP-10 decomposition and initiation mechanism pathways of n-butylbenzene pyrolysis obtained using the CHO-2016 parameter set are also in good agreement with both experimental and CHO-2008 simulation results. This demonstrated the transferability of the CHO-2016 description for a wide range of hydrocarbon chemistry. read less NOT USED (low confidence) W. Liu et al., “A two-dimensional conjugated aromatic polymer via C-C coupling reaction.,” Nature chemistry. 2017. link Times cited: 249 NOT USED (low confidence) F. Shayeganfar and R. Shahsavari, “Oxygen- and Lithium-Doped Hybrid Boron-Nitride/Carbon Networks for Hydrogen Storage.,” Langmuir : the ACS journal of surfaces and colloids. 2016. link Times cited: 56 Abstract: Hydrogen storage capacities have been studied on newly desig… read moreAbstract: Hydrogen storage capacities have been studied on newly designed three-dimensional pillared boron nitride (PBN) and pillared graphene boron nitride (PGBN). We propose these novel materials based on the covalent connection of BNNTs and graphene sheets, which enhance the surface and free volume for storage within the nanomaterial and increase the gravimetric and volumetric hydrogen uptake capacities. Density functional theory and molecular dynamics simulations show that these lithium- and oxygen-doped pillared structures have improved gravimetric and volumetric hydrogen capacities at room temperature, with values on the order of 9.1-11.6 wt % and 40-60 g/L. Our findings demonstrate that the gravimetric uptake of oxygen- and lithium-doped PBN and PGBN has significantly enhanced the hydrogen sorption and desorption. Calculations for O-doped PGBN yield gravimetric hydrogen uptake capacities greater than 11.6 wt % at room temperature. This increased value is attributed to the pillared morphology, which improves the mechanical properties and increases porosity, as well as the high binding energy between oxygen and GBN. Our results suggest that hybrid carbon/BNNT nanostructures are an excellent candidate for hydrogen storage, owing to the combination of the electron mobility of graphene and the polarized nature of BN at heterojunctions, which enhances the uptake capacity, providing ample opportunities to further tune this hybrid material for efficient hydrogen storage. read less NOT USED (low confidence) S. Bhunya, T. Malakar, G. Ganguly, and A. Paul, “Combining Protons and Hydrides by Homogeneous Catalysis for Controlling the Release of Hydrogen from Ammonia–Borane: Present Status and Challenges,” ACS Catalysis. 2016. link Times cited: 82 Abstract: Ammonia–borane (AB) has been in the spotlight for its much t… read moreAbstract: Ammonia–borane (AB) has been in the spotlight for its much touted potential as an onboard vehicular hydrogen delivery material. Over the past decade, catalyzed dehydrogenation/dehydrocoupling reactions for releasing H2 from the maximum available 3 equiv in AB have gained significant momentum. In this Perspective, we focus on the homogeneous AB dehydrogenation catalysis, by both transition metal (TM)-based and metal-free systems. Several questions pertaining to underlying mechanisms, nature of intermediates, and catalyst efficacy have surfaced as the multitude of discoveries in the field has built up at a fast pace. The varied fate of the dehydrogenation reactions of AB with different catalysts yielding different end products ranging from polyaminoborane (PAB) to polyborazylene (PBZ) and the ability/inability of catalysts to release more than 1 equiv of H2 from AB have fuelled the genesis of several mechanistic hypotheses. However, the copious investigations on the experimental and theoretical fronts have ... read less NOT USED (low confidence) Y. Han, D. D. Jiang, J. Zhang, W. Li, Z. Gan, and J. Gu, “Development, applications and challenges of ReaxFF reactive force field in molecular simulations,” Frontiers of Chemical Science and Engineering. 2016. link Times cited: 87 NOT USED (low confidence) S. J. Pai, B. C. Yeo, and S. Han, “Development of the ReaxFFCBN reactive force field for the improved design of liquid CBN hydrogen storage materials.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 15 Abstract: Liquid CBN (carbon-boron-nitrogen) hydrogen-storage material… read moreAbstract: Liquid CBN (carbon-boron-nitrogen) hydrogen-storage materials such as 3-methyl-1,2-BN-cyclopentane have the advantage of being easily accessible for use in current liquid-fuel infrastructure. To develop practical liquid CBN hydrogen-storage materials, it is of great importance to understand the reaction pathways of hydrogenation/dehydrogenation in the liquid phase, which are difficult to discover by experimental methods. Herein, we developed a reactive force field (ReaxFFCBN) from quantum mechanical (QM) calculations based on density functional theory for the storage of hydrogen in BN-substituted cyclic hydrocarbon materials. The developed ReaxFFCBN provides similar dehydrogenation pathways and energetics to those predicted by QM calculations. Moreover, molecular dynamics (MD) simulations with the developed ReaxFFCBN can predict the stability and dehydrogenation behavior of various liquid CBN hydrogen-storage materials. Our simulations reveal that a unimolecular dehydrogenation mechanism is preferred in liquid CBN hydrogen-storage materials. However, as the temperature in the simulation increases, the contribution of a bimolecular dehydrogenation mechanism also increases. Moreover, our ReaxFF MD simulations show that in terms of thermal stability and dehydrogenation kinetics, liquid CBN materials with a hexagonal structure are more suitable materials than those with a pentagonal structure. We expect that the developed ReaxFFCBN could be a useful protocol in developing novel liquid CBN hydrogen-storage materials. read less NOT USED (low confidence) R. Kumar and A. Parashar, “Atomistic modeling of BN nanofillers for mechanical and thermal properties: a review.,” Nanoscale. 2016. link Times cited: 69 Abstract: Due to their exceptional mechanical properties, thermal cond… read moreAbstract: Due to their exceptional mechanical properties, thermal conductivity and a wide band gap (5-6 eV), boron nitride nanotubes and nanosheets have promising applications in the field of engineering and biomedical science. Accurate modeling of failure or fracture in a nanomaterial inherently involves coupling of atomic domains of cracks and voids as well as a deformation mechanism originating from grain boundaries. This review highlights the recent progress made in the atomistic modeling of boron nitride nanofillers. Continuous improvements in computational power have made it possible to study the structural properties of these nanofillers at the atomistic scale. read less NOT USED (low confidence) S. Bhoi, T. Banerjee, and K. Mohanty, “Molecular dynamic simulation of spontaneous combustion and pyrolysis of brown coal using ReaxFF,” Fuel. 2014. link Times cited: 131 NOT USED (low confidence) B. Chen, Z.-J. Diao, and H. Lu, “Using the ReaxFF reactive force field for molecular dynamics simulations of the spontaneous combustion of lignite with the Hatcher lignite model,” Fuel. 2014. link Times cited: 61 NOT USED (low confidence) N. Kovtyukhova, Y. Wang, R. Lv, M. Terrones, V. Crespi, and T. Mallouk, “Reversible intercalation of hexagonal boron nitride with Brønsted acids.,” Journal of the American Chemical Society. 2013. link Times cited: 82 Abstract: Hexagonal boron nitride (h-BN) is an insulating compound tha… read moreAbstract: Hexagonal boron nitride (h-BN) is an insulating compound that is structurally similar to graphite. Like graphene, single sheets of BN are atomically flat, and they are of current interest in few-layer hybrid devices, such as transistors and capacitors, that contain insulating components. While graphite and other layered compounds can be intercalated by redox reactions and then converted chemically to suspensions of single sheets, insulating BN is not susceptible to oxidative intercalation except by extremely strong oxidizing agents. We report that stage-1 intercalation compounds can be formed by simple thermal drying of h-BN in Brønsted acids H2SO4, H3PO4, and HClO4. X-ray photoelectron and vibrational spectra, as well as electronic structure and molecular dynamics calculations, demonstrate that noncovalent interactions of these oxyacids with the basic N atoms of the sheets drive the intercalation process. read less NOT USED (low confidence) M. R. Weismiller, S. Wang, A. Chowdhury, S. Thynell, and R. Yetter, “Confined rapid thermolysis studies of ammonia borane,” Thermochimica Acta. 2013. link Times cited: 19 NOT USED (low confidence) J. Graetz, D. Wolstenholme, G. Pez, L. Klebanoff, S. Mcgrady, and A. Cooper, “Development of Off-Board Reversible Hydrogen Storage Materials.” 2012. link Times cited: 9 NOT USED (low confidence) X.-min Cheng, Q.-D. Wang, J.-Q. Li, J. Wang, and X. Li, “ReaxFF molecular dynamics simulations of oxidation of toluene at high temperatures.,” The journal of physical chemistry. A. 2012. link Times cited: 107 Abstract: Aromatic hydrocarbon fuels, such as toluene, are important c… read moreAbstract: Aromatic hydrocarbon fuels, such as toluene, are important components in real jet fuels. In this work, reactive molecular dynamics (MD) simulations employing the ReaxFF reactive force field have been performed to study the high-temperature oxidation mechanisms of toluene at different temperatures and densities with equivalence ratios ranging from 0.5 to 2.0. From the ReaxFF MD simulations, we have found that the initiation consumption of toluene is mainly through three ways, (1) the hydrogen abstraction reactions by oxygen molecules or other small radicals to form the benzyl radical, (2) the cleavage of the C-H bond to form benzyl and hydrogen radicals, and (3) the cleavage of the C-C bond to form phenyl and methyl radicals. These basic reaction mechanisms are in good agreement with available chemical kinetic models. The temperatures and densities have composite effects on toluene oxidation; concerning the effect of the equivalence ratio, the oxidation reaction rate is found to decrease with the increasing of equivalence ratio. The analysis of the initiation reaction of toluene shows that the hydrogen abstraction reaction dominates the initial reaction stage at low equivalence ratio (0.5-1.0), while the contribution from the pyrolysis reaction increases significantly as the equivalence ratio increases to 2.0. The apparent activation energies, E(a), for combustion of toluene extracted from ReaxFF MD simulations are consistent with experimental results. read less NOT USED (low confidence) B. D. Jensen, A. Bandyopadhyay, K. Wise, and G. Odegard, “Parametric Study of ReaxFF Simulation Parameters for Molecular Dynamics Modeling of Reactive Carbon Gases.,” Journal of chemical theory and computation. 2012. link Times cited: 35 Abstract: The development of innovative carbon-based materials can be … read moreAbstract: The development of innovative carbon-based materials can be greatly facilitated by molecular modeling techniques. Although the Reax Force Field (ReaxFF) can be used to simulate the chemical behavior of carbon-based systems, the simulation settings required for accurate predictions have not been fully explored. Using the ReaxFF, molecular dynamics (MD) simulations are used to simulate the chemical behavior of pure carbon and hydrocarbon reactive gases that are involved in the formation of carbon structures such as graphite, buckyballs, amorphous carbon, and carbon nanotubes. It is determined that the maximum simulation time step that can be used in MD simulations with the ReaxFF is dependent on the simulated temperature and selected parameter set, as are the predicted reaction rates. It is also determined that different carbon-based reactive gases react at different rates, and that the predicted equilibrium structures are generally the same for the different ReaxFF parameter sets, except in the case of the predicted formation of large graphitic structures with the Chenoweth parameter set under specific conditions. read less NOT USED (low confidence) L. Huang, K. L. Joshi, A. V. van Duin, T. Bandosz, and K. Gubbins, “ReaxFF molecular dynamics simulation of thermal stability of a Cu3(BTC)2 metal-organic framework.,” Physical chemistry chemical physics : PCCP. 2012. link Times cited: 44 Abstract: The thermal stability of a dehydrated Cu(3)(BTC)(2) (copper(… read moreAbstract: The thermal stability of a dehydrated Cu(3)(BTC)(2) (copper(II) benzene 1,3,5-tricarboxylate) metal-organic framework was studied by molecular dynamics simulation with a ReaxFF reactive force field. The results show that Cu(3)(BTC)(2) is thermally stable up to 565 K. When the temperature increases between 600 K and 700 K, the framework starts to partially collapse. The RDF analysis shows that the long range correlations between Cu dimers disappear, indicating the loss of the main channels of Cu(3)(BTC)(2). When the temperature is above 800 K, we find the decomposition of the Cu(3)(BTC)(2) framework. CO is the major product, and we also observe the release of CO(2), O(2), 1,3,5-benzenetricarboxylate (C(6)H(3)(CO(2))(3), BTC) and glassy carbon. The Cu dimer is stable up to 1100 K, but we find the formation of new copper oxide clusters at 1100 K. These results are consistent with experimental findings, and provide valuable information for future theoretical investigations of Cu(3)(BTC)(2) and its application in adsorption, separation and catalytic processes. read less NOT USED (low confidence) Y. Shin et al., “Variable charge many-body interatomic potentials,” MRS Bulletin. 2012. link Times cited: 56 Abstract: Recent developments in reactive potentials for the simulatio… read moreAbstract: Recent developments in reactive potentials for the simulation of complex bonding and complex chemistry are reviewed. In particular, the reactive force field and charged optimized many-body methods are two paradigms that enable atoms to autonomously determine their charge state and the nature of their local bonding environments. The capabilities of these methods are illustrated by examples involving ionic-covalent systems, a metal-covalent system, a high- k dielectric gate stack, and the interaction of water with an oxide. Prospects for future development and applications are also discussed. read less NOT USED (low confidence) S. Swinnen, V. S. Nguyen, and M. Nguyen, “Theoretical Study of the Hydrogen Release Mechanism from a Lithium Derivative of Ammonia: Borane LiNH2BH3-NH3BH3,” Chemical Physics Letters. 2011. link Times cited: 8 NOT USED (low confidence) F. Zhao, B. Li, D. Che, and S. Liu, “The mechanism of H2O in the superheated steam affecting the quality of in-situ pyrolysates of oil shale kerogen: Part B-favorable conversion of residues,” Fuel. 2023. link Times cited: 4 NOT USED (low confidence) G.-H. Wei et al., “Exploring the continuous cleavage-oxidation mechanism of the catalytic oxidation of cellulose to formic acid: A combined experimental and theoretical study,” Fuel. 2023. link Times cited: 4 NOT USED (low confidence) F. Zhao, B. Li, L. Zhang, D. Che, and S. Liu, “The mechanism of superheated steam affecting the quality of in-situ pyrolysates of oil shale kerogen: Part A-saturation of pyrolytic organics,” Fuel. 2022. link Times cited: 7 NOT USED (low confidence) Z. Liang et al., “Impact of oxidants O2, H2O, and CO2 on graphene oxidation: A critical comparison of reaction kinetics and gasification behavior,” Chemical Engineering Journal. 2022. link Times cited: 7 NOT USED (low confidence) K. Li et al., “Thermal behaviour during initial stages of graphene oxidation: Implications for reaction kinetics and mechanisms,” Chemical Engineering Journal. 2021. link Times cited: 13 NOT USED (low confidence) O. Hod, “Interlayer Interactions in Low-Dimensional Layered Hetero-structures: Modeling and Applications,” Handbook of Materials Modeling. 2020. link Times cited: 0 NOT USED (low confidence) M. R. Weismiller, M. F. Russo, A. Duin, and R. Yetter, “Using molecular dynamics simulations with a ReaxFF reactive force field to develop a kinetic mechanism for ammonia borane oxidation.” 2013. link Times cited: 12 NOT USED (high confidence) S. Yu et al., “A Review on the Application of Molecular Dynamics to the Study of Coalbed Methane Geology,” Frontiers in Earth Science. 2021. link Times cited: 7 Abstract: Over the last three decades, molecular dynamics (MD) has bee… read moreAbstract: Over the last three decades, molecular dynamics (MD) has been extensively utilized in the field of coalbed methane geology. These uses include but are not limited to 1) adsorption of gaseous molecules onto coal, 2) diffusion of gaseous molecules into coal, 3) gas adsorption-induced coal matrix swelling and shrinkage, and 4) coal pyrolysis and combustion. With the development of computation power, we are entering a period where MD can be widely used for the above higher level applications. Here, the application of MD for coalbed methane study was reviewed. Combining GCMC (grand canonical Monte Carlo) and MD simulation can provide microscopic understanding of the adsorption of gaseous molecules onto coal. The experimental observations face significant challenges when encountering the nanoscale diffusion process due to coal structure heterogeneity. Today, all types of diffusion coefficients, such as self-, corrected-, and transport-diffusion coefficients can be calculated based on MD and the Peng-Robinson equation. To date, the MD simulation for both pure and multi-components has reached a situation of unprecedented success. Meanwhile, the swelling deformation of coal has been attracting an increasing amount of attention both via experimental and mimetic angles, which can be successfully clarified using MD and a poromechanical model incorporating the geothermal gradient law. With the development of computational power and physical examination level, simulation sophistication and improvements in MD, GCMC, and other numerical models will provide more opportunities to go beyond the current informed approach, gaining researcher confidence in the engagement in the estimation of coal-swelling deformation behaviors. These reactive MD works have clarified the feasibility and capability of the reactive force field ReaxFF to describe initial reactive events for coal pyrolysis and combustion. In future, advancing MD simulation (primarily characterized by the ReaxFF force field) will allow the exploration of the more complex reaction process. The reaction mechanism of pyrolysis and spontaneous combustion should also be a positive trend, as well as the potential of MD for both visualization and microscopic mechanisms for more clean utilization processes of coal. Thus, it is expected that the availability of MD will continue to increase and be added to the extensive list of advanced analytical approaches to explore the multi-scaled behaviors in coalbed methane geology. read less NOT USED (high confidence) S. D. Silva-Santos et al., “High Pressure in Boron Nitride Nanotubes for Kirigami Nanoribbon Elaboration,” Journal of Physical Chemistry C. 2021. link Times cited: 3 NOT USED (high confidence) J. Hu, Z. Wilde, P. Peralta, C. Muhich, and J. Oswald, “Predicting Hugoniot equation of state in erythritol with ab initio and reactive molecular dynamics,” Journal of Applied Physics. 2021. link Times cited: 0 Abstract: Erythritol has been proposed as an inert surrogate for devel… read moreAbstract: Erythritol has been proposed as an inert surrogate for developing theoretical and computational models to study aging in energetic materials. In this work, we present a comparison of mechanical and shock properties of erythritol computed using the ReaxFF reactive force field and from ab initio calculations employing density functional theory (DFT). We screened eight different ReaxFF parameterizations, of which the CHO parameters developed for hydrocarbon oxidation provide the most accurate predictions of mechanical properties and the crystal structure of erythritol. Further validation of the applicability of this ReaxFF parameterization for modeling erythritol is demonstrated by comparing predictions of the elastic constants, crystal structure, vibrational density of states, and Hugoniot curves against DFT calculations. The ReaxFF predictions are in close agreement with the DFT simulations for the elastic constants and shock Hugoniot when the crystal is loaded along its c axis but show as much as 30% disagreement in the elastic constants in the a b plane and 12% difference in shock pressures when shocked along the a or b crystal axes. Last, we compare thermomechanical properties predicted from classical molecular dynamics with those calculated using the quasi-harmonic approximation and show that quantum mechanical effects produce large discrepancies in the computed values of heat capacity and thermal expansion coefficients compared with classical assumptions. Combining classical molecular dynamics predictions of mechanical behavior with phonon-based calculations of thermal behaviors, we show that predicted shock-induced temperatures for pressures up to 6.5 GPa do not exceed the pressure-dependent melting point of erythritol. read less NOT USED (high confidence) A. D. Pierro, B. Mortazavi, H. Noori, T. Rabczuk, and A. Fina, “A Multiscale Investigation on the Thermal Transport in Polydimethylsiloxane Nanocomposites: Graphene vs. Borophene,” Nanomaterials. 2021. link Times cited: 3 Abstract: Graphene and borophene are highly attractive two-dimensional… read moreAbstract: Graphene and borophene are highly attractive two-dimensional materials with outstanding physical properties. In this study we employed combined atomistic continuum multi-scale modeling to explore the effective thermal conductivity of polymer nanocomposites made of polydimethylsiloxane (PDMS) polymer as the matrix and graphene and borophene as nanofillers. PDMS is a versatile polymer due to its chemical inertia, flexibility and a wide range of properties that can be tuned during synthesis. We first conducted classical Molecular Dynamics (MD) simulations to calculate the thermal conductance at the interfaces between graphene and PDMS and between borophene and PDMS. Acquired results confirm that the interfacial thermal conductance between nanosheets and polymer increases from the single-layer to multilayered nanosheets and finally converges, in the case of graphene, to about 30 MWm−2 K−1 and, for borophene, up to 33 MWm−2 K−1. The data provided by the atomistic simulations were then used in the Finite Element Method (FEM) simulations to evaluate the effective thermal conductivity of polymer nanocomposites at the continuum level. We explored the effects of nanofiller type, volume content, geometry aspect ratio and thickness on the nanocomposite effective thermal conductivity. As a very interesting finding, we found that borophene nanosheets, despite having almost two orders of magnitude lower thermal conductivity than graphene, can yield very close enhancement in the effective thermal conductivity in comparison with graphene, particularly for low volume content and small aspect ratios and thicknesses. We conclude that, for the polymer-based nanocomposites, significant improvement in the thermal conductivity can be reached by improving the bonding between the fillers and polymer, or in other words, by enhancing the thermal conductance at the interface. By taking into account the high electrical conductivity of borophene, our results suggest borophene nanosheets as promising nanofillers to simultaneously enhance the polymers’ thermal and electrical conductivity. read less NOT USED (high confidence) F. Zhang, Y. Cao, X. Liu, H. Xu, D. Lu, and R. Yang, “How Small Molecules Affect the Thermo-Oxidative Aging Mechanism of Polypropylene: A Reactive Molecular Dynamics Study,” Polymers. 2021. link Times cited: 6 Abstract: Understanding the aging mechanism of polypropylene (PP) is f… read moreAbstract: Understanding the aging mechanism of polypropylene (PP) is fundamental for the fabrication and application of PP-based materials. In this paper, we present our study in which we first used reactive molecular dynamics (RMD) simulations to explore the thermo-oxidative aging of PP in the presence of acetic acid or acetone. We studied the effects of temperature and oxygen on the aging process and discussed the formation pathways of typical small molecule products (H2, CO, CO2, CH4, C2H4, and C2H6). The effect of two infection agents, acetic acid and acetone, on the aging reaction was analyzed emphatically. The simulation results showed that acetone has a weak impact on accelerating the aging process, while acetic acid has a significant effect, consistent with previous experimental studies. By tracking the simulation trajectories, both acetic acid and acetone produced small active free radicals to further react with other fragment products, thus accelerating the aging process. The first reaction step of acetic acid is often the shedding of the H atom on the hydroxyl group, while the reaction of acetone is often the shedding of the H atom or the methyl. The latter requires higher energy at lower temperatures. This is why the acceleration effect of acetone for the thermo-oxidative aging of PP was not so significant compared to acetic acid in the experimental temperature (383.15 K). read less NOT USED (high confidence) B. Sharma and A. Parashar, “Fracture behaviour of pristine and defective form of water submerged h-BN nanosheets,” Journal of Physics D: Applied Physics. 2020. link Times cited: 5 Abstract: Hexagonal boron nitride (h-BN) nanosheets are emerging as po… read moreAbstract: Hexagonal boron nitride (h-BN) nanosheets are emerging as potential candidates to replace polymeric membrane for water purification. Synthesizing the membrane size of immaculate h-BN is a fictitious concept. So far, no articles have reported on the fracture behaviour of pristine and defective h-BN nanosheets in a water-submerged state. In this article, classical mechanics-based simulations were employed to study the effect of the hydrogen functionalization of sp-hybridized crack edge atoms and spatial distribution of Stone–Thrower–Wales (STW) and dislocation defects on the fracture properties of water-submerged h-BN nanosheets. Classical mechanics-based simulations were carried out with the help of hybrid-type interatomic potential in which reactive force field and transferable intermolecular potentials were used for arresting the atomistic interaction in h-BN and water, respectively, while for interfacial interactions, Lennard–Jones potential was employed. Enhancement in the values of fracture toughness of h-BN was investigated in the prescence of water molecules compared to in a dry state. It was deduced from the results that passivation, as well as the spatial distribution of STW defects, have a deteriorating effect on the values of fracture toughness of h-BN in the dry state, but water molecules help in nullifying a negative affect. Compared to the spatial distribution of dislocations, the STW type of defects have a more pronounced effect on the values of fracture toughness of h-BN water-submerged state. It can be predicted from the simulations that these improved fracture toughness values in a water-submerged state will help in developing these nanosheets as an effective desalination membrane in water purification. read less NOT USED (high confidence) Shuhui 姝惠 YANG 杨 et al., “Molecular dynamics simulations of the interaction between OH radicals in plasma with poly-β-1–6-N-acetylglucosamine,” Plasma Science and Technology. 2020. link Times cited: 7 Abstract: Cold atmospheric plasma shows a satisfactory ability to inac… read moreAbstract: Cold atmospheric plasma shows a satisfactory ability to inactivate bacterial biofilms that are difficult to remove using conventional methods in some cases. However, the researches on the inactivation mechanism are not quite sufficient. Poly-β-1–6-N-acetylglucosamine (PNAG), which is one of the important components in some biofilms, was used as the research subject, and the related mechanism of action triggered by different concentrations of the OH in plasma was studied using reactive molecular dynamics simulations. The results showed that OH radicals could not only trigger the hydrogen abstraction reaction leading to cleavage of the PNAG molecular structure, but undergo an OH addition reaction with PNAG molecules. New reaction pathways appeared in the simulations as the OH concentration increased, but the reaction efficiency first increased and then decreased. The simulation study in this paper could, to some extent, help elucidate the microscopic mechanism of the interaction between OH radicals in plasma and bacterial biofilms at the atomic level. read less NOT USED (high confidence) F. Thiemann, P. Rowe, E. A. Müller, and A. Michaelides, “Machine Learning Potential for Hexagonal Boron Nitride Applied to Thermally and Mechanically Induced Rippling,” The Journal of Physical Chemistry C. 2020. link Times cited: 18 Abstract: We introduce an interatomic potential for hexagonal boron ni… read moreAbstract: We introduce an interatomic potential for hexagonal boron nitride (hBN) based on the Gaussian approximation potential (GAP) machine learning methodology. The potential is based on a training set of... read less NOT USED (high confidence) L. W. Bertels, L. B. Newcomb, M. Alaghemandi, J. R. Green, and M. Head‐Gordon, “Benchmarking the Performance of the ReaxFF Reactive Force Field on Hydrogen Combustion Systems.,” The journal of physical chemistry. A. 2020. link Times cited: 24 Abstract: A thorough understanding of the kinetics and dynamics of com… read moreAbstract: A thorough understanding of the kinetics and dynamics of combusting mixtures is of considerable interest, especially in regimes beyond the reach of current experimental validation. The ReaxFF reactive force field method has provided a way to simulate large-scale systems of hydrogen combustion via a parameterized potential that can simulate bond breaking. This modeling approach has been applied to hydrogen combustion, as well as myriad other reactive chemical systems. In this work, we benchmark the performance of several common parameterizations of this potential against higher-level quantum mechanical (QM) approaches. We demonstrate instances where these parameterizations of the ReaxFF potential fail both quantitatively and qualitatively to describe reactive events relevant for hydrogen combustion systems. read less NOT USED (high confidence) E. Armani and P. A. Autreto, “High-velocity impact of a hybrid CBN nanotubes,” arXiv: Materials Science. 2020. link Times cited: 3 Abstract: Nanomaterials under extreme conditions can behave in a compl… read moreAbstract: Nanomaterials under extreme conditions can behave in a completely different manner. High-velocity impact, for example, can produce nanoribbons without any chemical approach via carbon or boron nitride nanotubes unzipping. Although hybrid nanostructures have been used to create stronger structures, few studies on these materials under extreme conditions have been employed. In this work, we study an experimentally synthesized hybrid doubled walled CBN nanotube (boron nitride and carbon nanotubes concentrically assembled) under high-velocity impact. Our results show that the combination of elastic and brittle materials can produce different structures, such as nanoribbons and boron nitride atomic chains. These results can have a significant impact on the production of new nanostructures. read less NOT USED (high confidence) B. Sharma and A. Parashar, “A review on thermo-mechanical properties of bi-crystalline and polycrystalline 2D nanomaterials,” Critical Reviews in Solid State and Materials Sciences. 2020. link Times cited: 29 Abstract: Due to outstanding properties, graphene and h-BN nanosheets … read moreAbstract: Due to outstanding properties, graphene and h-BN nanosheets are emerging as a potential candidate for wide spectrum of applications in the field of engineering and bio-medical science. Graphene and h-BN nanosheets have comparable mechanical and thermal properties, whereas due to high band gap h-BN (∼5eV) have contrasting electrical conductivities. Large size graphene and h-BN nanosheets are synthesized by chemical vapor deposition technique, which results in polycrystalline atomic structure. These polycrystalline nanosheets are characterized either by experimental means or numerical simulations. Experimental techniques are considered as most accurate and practical, but cost and time involved in these techniques limits it application at the nanoscale level. On the other hand, atomistic modeling techniques are emerging as viable alternatives to the experimentations, and are accurate enough to predict the mechanical properties, fracture toughness, and thermal conductivities of polycrystalline graphene and h-BN nanosheets. This comprehensive review article encompasses different characterizing techniques used by the researchers for polycrystalline nanosheets. This review will help in elaborating the properties of polycrystalline graphene and h-BN, and also establishing a perspective on how the microstructure impacts its large-scale physical properties. read less NOT USED (high confidence) P. Gao, Z. Huang, and H. Yu, “Exploration of the Dehydrogenation Pathways of Ammonia Diborane and Diammoniate of Diborane by Molecular Dynamics Simulations Using Reactive Force Fields.,” The journal of physical chemistry. A. 2020. link Times cited: 16 Abstract: Ammonium aminodiboranate (AADB) and diammoniate of diborane … read moreAbstract: Ammonium aminodiboranate (AADB) and diammoniate of diborane (DADB) are two isomers of ammonia borane (AB), which have been intensively studied for hydrogen storage. Their high hydrogen contents give them the high potential to serve as hydrogen storage materials. To explore their dehydrogenation pathways, molecular dynamics (MD) simulations with a reactive force field (ReaxFF) were applied. Temperature ramping simulations of their thermolysis were carried out. For AADB, at low temperatures, its hydrogen release can be realized mainly via inter-molecular dehydrogenations. As the temperature of the simulated system increases, the formations of B-N bonds begin to occur. In the case of DADB, we found that this molecule could release hydrogen at a lower temperature with the cleavage of B-N bond. The compositional analysis of the simulated systems was also conducted to monitor the potential intermediates along their dehydrogenation pathways. Our current work provides a detailed picture of the initial dehydrogenation steps of AADB and DADB, and highlights the difference in their respective dehydrogenation processes. read less NOT USED (high confidence) S. J. Pai, H. W. Lee, and S. Han, “Improved Description of a Coordinate Bond in the ReaxFF Reactive Force Field.,” The journal of physical chemistry letters. 2019. link Times cited: 4 Abstract: To improve the description of a coordinate bond of the react… read moreAbstract: To improve the description of a coordinate bond of the reactive force field (ReaxFF), we have developed ReaxFFcoord by explicitly incorporating the coordinate bond contribution, Ecoord, into the original ReaxFF ( Chenoweth et al. J. Phys. Chem. A 2008 , 112 , 1040 - 1053 ), in which the auxiliary functions are newly suggested to describe the plug-in behavior of lone-pair electrons from a donor atom to a vacant orbital of an acceptor atom. To validate the developed ReaxFFcoord, we tested it in various systems, including a representative coordinate bond-containing molecule, namely, carbon monoxide or ammonia borane. Although the fitting abilities of the ReaxFFcoord and original ReaxFF are similar, their molecular dynamics (MD) simulations are significantly different, where MD simulations employing ReaxFFcoord provide more realistic dynamic behaviors of atoms. It is expected that the ReaxFFcoord will significantly help ReaxFF to successfully extend its applicability to the material and biological systems, including coordinate bonds in organometallic systems. read less NOT USED (high confidence) S. Y. Kim, J. Kwak, C. Ciobanu, and S. Y. Kwon, “Recent Developments in Controlled Vapor‐Phase Growth of 2D Group 6 Transition Metal Dichalcogenides,” Advanced Materials. 2019. link Times cited: 93 Abstract: An overview of recent developments in controlled vapor‐phase… read moreAbstract: An overview of recent developments in controlled vapor‐phase growth of 2D transition metal dichalcogenide (2D TMD) films is presented. Investigations of thin‐film formation mechanisms and strategies for realizing 2D TMD films with less‐defective large domains are of central importance because single‐crystal‐like 2D TMDs exhibit the most beneficial electronic and optoelectronic properties. The focus is on the role of the various growth parameters, including strategies for efficiently delivering the precursors, the selection and preparation of the substrate surface as a growth assistant, and the introduction of growth promoters (e.g., organic molecules and alkali metal halides) to facilitate the layered growth of (Mo, W)(S, Se, Te)2 atomic crystals on inert substrates. Critical factors governing the thermodynamic and kinetic factors related to chemical reaction pathways and the growth mechanism are reviewed. With modification of classical nucleation theory, strategies for designing and growing various vertical/lateral TMD‐based heterostructures are discussed. Then, several pioneering techniques for facile observation of structural defects in TMDs, which substantially degrade the properties of macroscale TMDs, are introduced. Technical challenges to be overcome and future research directions in the vapor‐phase growth of 2D TMDs for heterojunction devices are discussed in light of recent advances in the field. read less NOT USED (high confidence) T. Chatterjee and S. Thynell, “Quantum mechanics investigation on initial decomposition of ammonia borane in glyme,” International Journal of Chemical Kinetics. 2018. link Times cited: 4 NOT USED (high confidence) T. Maaravi, I. Leven, I. Azuri, L. Kronik, and O. Hod, “Interlayer Potential for Homogeneous Graphene and Hexagonal Boron Nitride Systems: Reparametrization for Many-Body Dispersion Effects,” Journal of Physical Chemistry C. 2017. link Times cited: 55 Abstract: A new parametrization of the anisotropic interlayer potentia… read moreAbstract: A new parametrization of the anisotropic interlayer potential for hexagonal boron nitride (h-BN ILP) is presented. The force-field is benchmarked against density functional theory calculations of several dimer systems within the Heyd-Scuseria-Ernzerhof hybrid density functional approximation, corrected for many-body dispersion effects. The latter, more advanced method for treating dispersion, is known to produce binding energies nearly twice as small as those obtained with pairwise correction schemes, used for an earlier ILP parametrization. The new parametrization yields good agreement with the reference calculations to within ∼1 and ∼0.5 meV/atom for binding and sliding energies, respectively. For completeness, we present a complementary parameter set for homogeneous graphitic systems. Together with our previously suggested ILP parametrization for the heterogeneous graphene/h-BN junction, this provides a powerful tool for consistent simulation of the structural, mechanical, tribological, and heat transp... read less NOT USED (high confidence) F. J. Dominguez-Gutierrez et al., “Unraveling the plasma-material interface with real time diagnosis of dynamic boron conditioning in extreme tokamak plasmas,” Nuclear Fusion. 2017. link Times cited: 9 Abstract: We present a study of the role of boron and oxygen in the ch… read moreAbstract: We present a study of the role of boron and oxygen in the chemistry of deuterium retention in boronized ATJ graphite irradiated by the extreme environment of a tokamak deuterium plasma. The experimental results were obtained by the first XPS measurements inside the plasma chamber of the National Spherical Torus Experiment Upgrade, between the plasma exposures. The subtle interplay of boron, carbon, oxygen and deuterium chemistry is explained by reactive molecular dynamics simulations, verified by quantum–classical molecular dynamics and successfully compared to the measured data. The calculations deciphered the roles of oxygen and boron for the deuterium retention and predict deuterium uptake into a boronized carbon surface close in value to that previously predicted for a lithiated and oxidized carbon surface. read less NOT USED (high confidence) S. Liu, A. V. van Duin, D. M. van Duin, B. Liu, and J. Edgar, “Atomistic Insights into Nucleation and Formation of Hexagonal Boron Nitride on Nickel from First-Principles-Based Reactive Molecular Dynamics Simulations.,” ACS nano. 2017. link Times cited: 38 Abstract: Atomistic-scale insights into the growth of a continuous, at… read moreAbstract: Atomistic-scale insights into the growth of a continuous, atomically thin hexagonal boron nitride (hBN) lattice from elemental boron and nitrogen on Ni substrates were obtained from multiscale modeling combining density functional theory (DFT) and reactive molecular dynamics. The quantum mechanical calculations focused on the adsorption and reaction energetics for the hBN building-block species, i.e., atomic B, N, BxNy (x, y = 1, 2), on Ni(111) and Ni(211), and the diffusion pathways of elemental B and N on these slab model surfaces and in the sublayer. B can diffuse competitively on both the surface and in the sublayer, while N diffuses strictly on the substrate surface. The DFT data were then used to generate a classical description of the Ni-B and Ni-N pair interactions within the formulation of the reactive force field, ReaxFF. Using the potential developed from this work, the elementary nucleation and growth process of an hBN monolayer structure from elemental B and N is shown at the atomistic scale. The nucleation initiates from the growth of linear BN chains, which evolve into branched and then hexagonal lattices. Subsequent DFT calculations confirmed the structure evolution energetically and validate the self-consistency of this multiscale modeling framework. On the basis of this framework, the fundamental aspects regarding crystal quality and the role of temperature and substrates used during hBN growth can also be understood. read less NOT USED (high confidence) R. Kumar and A. Parashar, “Fracture toughness enhancement of h-BN monolayers via hydrogen passivation of a crack edge,” Nanotechnology. 2017. link Times cited: 26 Abstract: Molecular dynamics-based simulations were performed in conju… read moreAbstract: Molecular dynamics-based simulations were performed in conjunction with reactive force-field potential parameters to investigate the effect of crack-edge passivation via hydrogenation on the fracture properties of h-BN nanosheets. In semi-hydrogenated (H is attached to either B or N) and fully hydrogenated (H is attached to both B and N) crack-edge atoms, three hybridisation states—sp2, sp3 and sp2 + sp3—were considered in the simulations. Significant improvement in the fracture toughness of h-BN nanosheets was predicted with semi- and fully hydrogenated crack-edge atoms. An overall improvement in fracture toughness of h-BN in the range of 16%–23% was estimated with the sp3 or sp2 + sp3 hybridisation state of crack-edge atoms. This significant shift in the fracture toughness of h-BN nanosheets was attributed to lowered crack-edge energy, a stress-relieving mechanism and blunting of the crack tip. Semi-hydrogenated crack-edge atoms with hydrogen attached only to N atoms have shown a negative response in terms of fracture toughness. read less NOT USED (high confidence) Z. Chen, W. Sun, and L. Zhao, “High-Temperature and High-Pressure Pyrolysis of Hexadecane: Molecular Dynamic Simulation Based on Reactive Force Field (ReaxFF).,” The journal of physical chemistry. A. 2017. link Times cited: 36 Abstract: As important products of heavy oil pyrolysis, heavier compon… read moreAbstract: As important products of heavy oil pyrolysis, heavier components such as gasoline and diesel supply the vast majority of energy demand through combustion, and lighter components such as ethylene and propylene are the main sources of industrial chemicals and plastic products. In this work, pyrolysis of hexadecane, as the model compound, was studied by reactive force field (ReaxFF) molecular simulation at high temperatures and high pressures. It was confirmed by unimolecular simulations that there exist eight different initial mechanisms all starting with C-C bond dissociation. The biradical mechanism was verified, through which the pyrolysis process can be accomplished within a shorter time. The enthalpy of reaction was calculated by the QM method, which was well consistent with ReaxFF calculation results. Multimolecular simulations showed that there is a strong dependency relationship between products distribution and temperature, as well as that between reaction rates and temperature. The optimal condition for ethylene formation in our work is 11.6 MPa and 2000 K, whereas it is best for hydrogen formation at conditions of 11.6 MPa and 3500 K. Kinetic analysis was performed with the activation energy of 113.03 kJ/mol and pre-exponential factor of 4.55 × 1012, and it is in good agreement with previous work. read less NOT USED (high confidence) L. Han and P. Krstic, “A path for synthesis of boron-nitride nanostructures in volume of arc plasma,” Nanotechnology. 2017. link Times cited: 18 Abstract: We find a possible channel for direct nanosynthesis of boron… read moreAbstract: We find a possible channel for direct nanosynthesis of boron-nitride (BN) nanostructures, including growth of BN nanotubes from a mixture of BN diatomic molecules by quantum–classical molecular dynamics simulations. No catalyst or boron nanoparticle is needed for this synthesis, however the conditions for the synthesis of each of the nanostructures, such as temperature and flux of the BN feedstock are identified and are compatible with the conditions in an electric arc at high pressure. We also find that BN nanostructures can be synthetized by feeding a boron nanoparticle by BN diatomic molecules, however if hydrogen rich molecules like NH3 or HBNH are used as a feedstock, two-dimensional nanoflake stable structures are formed. read less NOT USED (high confidence) V. Chaban, S. Pal, and O. Prezhdo, “Laser-Induced Explosion of Nitrated Carbon Nanotubes: Nonadiabatic and Reactive Molecular Dynamics Simulations.,” Journal of the American Chemical Society. 2016. link Times cited: 31 Abstract: Laser-initiated decomposition of carbon nanotubes (CNTs) can… read moreAbstract: Laser-initiated decomposition of carbon nanotubes (CNTs) can lead to medical, military, and other applications. In medicine, CNTs give rise to efficient remedies against diseases and malignant cells, since they encapsulate drug molecules, can be delivered inside living organisms, and absorb light that penetrates through biological tissues. As explosives, pyrotechnics, and propellants, CNTs can be activated remotely by a visible or infrared laser, avoiding the need for a detonating cord. The reported non-equilibrium investigation demonstrates the possibility of photoinduced polynitro-CNT explosion and provides a detailed chemical mechanism of the decomposition process, explicitly in the time domain. Nonadiabatic molecular dynamics (MD) performed with real-time time-dependent tight-binding density functional theory demonstrates that the photogenerated exciton deposits its energy into a broad range of phonon modes within less than a picosecond, resulting in a rapid polynitro-CNT heating. Following the heating, reactive MD demonstrates an explosion, during which the local temperature of polynitro-CNTs and its fragments rises as high as 4000 K. Photoexcitation of nitro groups by a high-energy laser is not required; the energy can be delivered to polynitro-CNTs using near-infrared light within the biological window. Furthermore, the explosion is possible both with and without an external oxygen source. Anaerobic explosion could be particularly beneficial in confined biological and nanoscale environments. The products of the polynitro-CNT decomposition are nontoxic: carbon dioxide and molecular nitrogen. The in silico demonstration of the laser-induced polynitro-CNT explosion, its chemical mechanism, and the time scales of physical and chemical transformations can be tested experimentally using time-resolved laser techniques. read less NOT USED (high confidence) R. Kumar, P. Mertiny, and A. Parashar, “Effects of Different Hydrogenation Regimes on Mechanical Properties of h-BN: A Reactive Force Field Study,” Journal of Physical Chemistry C. 2016. link Times cited: 34 Abstract: This article describes molecular dynamics based simulations,… read moreAbstract: This article describes molecular dynamics based simulations, which were performed to investigate the effects of different hydrogenation regimes on the mechanical properties of boron nitride nanosheets (h-BN). The reaction force field (ReaxFF) was used as the interatomic potential to capture atomistic interactions. Separate atomistic models were developed for pristine, semihydrogenated (hydrogen is attached either to boron or nitrogen) and fully hydrogenated h-BN (hydrogen is attached to both boron and nitrogen). The radial distribution function was used to study the structural integrity and stability of both pristine and hydrogenated structures. The simulations predicted an improvement in stability and integrity of the atomistic structures under the influence of hydrogenation compared to pristine h-BN. The semihydrogenated structure in which hydrogen was attached only to nitrogen was found to be the least stable configuration, while the fully hydrogenated structure was the most stable. Furthermore, the se... read less NOT USED (high confidence) V. Chaban and O. Prezhdo, “Haber Process Made Efficient by Hydroxylated Graphene: Ab Initio Thermochemistry and Reactive Molecular Dynamics.,” The journal of physical chemistry letters. 2016. link Times cited: 16 Abstract: The Haber-Bosch process is the main industrial method for pr… read moreAbstract: The Haber-Bosch process is the main industrial method for producing ammonia from diatomic nitrogen and hydrogen. We use a combination of ab initio thermochemical analysis and reactive molecular dynamics to demonstrate that a significant increase in the ammonia production yield can be achieved using hydroxylated graphene and related species. Exploiting the polarity difference between N2/H2 and NH3, as well as the universal proton acceptor behavior of NH3, we demonstrate a strong shift of the equilibrium of the Haber-Bosch process toward ammonia (ca. 50 kJ mol(-1) enthalpy gain and ca. 60-70 kJ mol(-1) free energy gain). The modified process is of significant importance to the chemical industry. read less NOT USED (high confidence) S. Bhoi, T. Banerjee, and K. Mohanty, “Insights on the combustion and pyrolysis behavior of three different ranks of coals using reactive molecular dynamics simulation,” RSC Advances. 2016. link Times cited: 39 Abstract: The process of combustion and pyrolysis of coal can be consi… read moreAbstract: The process of combustion and pyrolysis of coal can be considered to be convoluted where numerous intermediates are expected to form during the course of the reaction. In this work, we have investigated the reactive products using the ReaxFF force field for three different ranked (low to high) coals, namely lignite, bituminous, and anthracite. It was observed that during the pyrolysis and combustion processes, the gases CO and CO2 were predominant. The formation rate of CO and CO2 was found to be higher for lignite coal which agreed with the experimental trend reported in the literature. In a similar manner, the fraction of CO and CO2 was found to be higher in the pyrolysis process. Further, a large number of principal intermediates such as methane, ethane and ethylene are also generated for low to high ranking (lignite, bituminous, and anthracite) coal. The pyrolysis and combustion processes were affected by temperature (2000–4000 K) with respect to the formation of various intermediates (methane, ethane and ethylene). They were found to be high throughout irrespective of the rank of coal. A higher temperature (2000–4000 K) was adopted in the reactive molecular dynamics (MD) simulation so as to visualize the chemical reactions within a computationally affordable time. read less NOT USED (high confidence) N. Subramanian, A. Rai, and A. Chattopadhyay, “Atomistically informed stochastic multiscale model to predict the behavior of carbon nanotube-enhanced nanocomposites,” Carbon. 2015. link Times cited: 47 NOT USED (high confidence) R. Paupitz, C. Junkermeier, A. V. van Duin, and P. S. Branicio, “Fullerenes generated from porous structures.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 34 Abstract: A class of macromolecules based on the architecture of the w… read moreAbstract: A class of macromolecules based on the architecture of the well-known fullerenes is theoretically investigated. The building blocks used to geometrically construct these molecules are the two dimensional structures: porous graphene and biphenylene-carbon. Density functional-based tight binding methods as well as reactive molecular dynamics methods are applied to study the electronic and structural properties of these molecules. Our calculations predict that these structures can be stable up to temperatures of 2500 K. The atomization energies of carbon structures are predicted to be in the range of 0.45 eV per atom to 12.11 eV per atom (values relative to the C60 fullerene), while the hexagonal boron nitride analogues have atomization energies between -0.17 eV per atom and 12.01 eV per atom (compared to the B12N12 fullerene). Due to their high porosity, these structures may be good candidates for gas storage and/or molecular encapsulation. read less NOT USED (high confidence) L. Huang, T. Bandosz, K. L. Joshi, A. V. van Duin, and K. Gubbins, “Reactive adsorption of ammonia and ammonia/water on CuBTC metal-organic framework: a ReaxFF molecular dynamics simulation.,” The Journal of chemical physics. 2013. link Times cited: 41 Abstract: We report ReaxFF molecular dynamics simulations for reactive… read moreAbstract: We report ReaxFF molecular dynamics simulations for reactive adsorption of NH(3) on dehydrated CuBTC metal-organic framework. If the temperature is moderate (up to 125 °C), the dehydrated CuBTC demonstrates a good hydrostatic stability for water concentrations up to 4.0 molecules per copper site. However, if the temperature increases to 550 K, the dehydrated CuBTC will collapse even at a small water concentration, 1.0 H(2)O molecule per copper site. When NH(3) molecules are adsorbed in the channel and micropores of CuBTC, they prefer to chemisorb to the copper sites rather than forming a dimer with another NH(3) molecule. The formation of equimolar Cu(2)(NH(2))(4) and (NH(4))(3)BTC structures is observed at 348 K, which is in good agreement with previous experimental findings. The dehydrated CuBTC framework is partially collapsed upon NH(3) adsorption, while the Cu-Cu dimer structure remains stable under the investigated conditions. Further calculations reveal that the stability of CuBTC is related to the ammonia concentration. The critical NH(3) concentration after which the dehydrated CuBTC starts to collapse is determined to be 1.0 NH(3) molecule per copper site. Depending on whether NH(3) concentration is below or above the critical value, the dehydrated CuBTC can be stable to a higher temperature, 378 K, or can collapse at a lower temperature, 250 K. H(2)O∕NH(3) mixtures have also been studied, and we find that although water molecules do not demonstrate a strong interaction with the copper sites of CuBTC, the existence of water molecules can substantially prevent ammonia from interacting with CuBTC, and thus reduce the amount of chemisorbed NH(3) molecules on CuBTC and stabilize the CuBTC framework to some extent. read less NOT USED (high confidence) S. Xu et al., “A molecular understanding of the gas-phase reduction and doping of graphene oxide,” Nano Research. 2012. link Times cited: 15 NOT USED (high confidence) K. Farah, F. Müller-Plathe, and M. Böhm, “Classical reactive molecular dynamics implementations: state of the art.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2012. link Times cited: 71 Abstract: Reactive molecular dynamics (RMD) implementations equipped w… read moreAbstract: Reactive molecular dynamics (RMD) implementations equipped with force field approaches to simulate both the time evolution as well as chemical reactions of a broad class of materials are reviewed herein. We subdivide the RMD approaches developed during the last decade as well as older ones already reviewed in 1995 by Srivastava and Garrison and in 2000 by Brenner into two classes. The methods in the first RMD class rely on the use of a reaction cutoff distance and employ a sudden transition from the educts to the products. Due to their simplicity these methods are well suited to generate equilibrated atomistic or material-specific coarse-grained polymer structures. In connection with generic models they offer useful qualitative insight into polymerization reactions. The methods in the second RMD class are based on empirical reactive force fields and implement a smooth and continuous transition from the educts to the products. In this RMD class, the reactive potentials are based on many-body or bond-order force fields as well as on empirical standard force fields, such as CHARMM, AMBER or MM3 that are modified to become reactive. The aim with the more sophisticated implementations of the second RMD class is the investigation of the reaction kinetics and mechanisms as well as the evaluation of transition state geometries. Pure or hybrid ab initio, density functional, semi-empirical, molecular mechanics, and Monte Carlo methods for which no time evolution of the chemical systems is achieved are excluded from the present review. So are molecular dynamics techniques coupled with quantum chemical methods for the treatment of the reactive regions, such as Car-Parinello molecular dynamics. read less NOT USED (high confidence) L. Sun and W. Deng, “Recent developments of first‐principles force fields,” Wiley Interdisciplinary Reviews: Computational Molecular Science. 2017. link Times cited: 18 Abstract: Molecular mechanics force fields derived from first‐principl… read moreAbstract: Molecular mechanics force fields derived from first‐principles calculations represent the next generation of force fields for molecular dynamics simulations. In recent years, a large amount of first‐principles force fields have been developed in the fields of physical and biological fields. Here, we review the first‐principles force fields especially for simulating the adsorption of small molecules in nanoporous materials and on surfaces. We describe the latest developments in force field parameterization and application, primarily in the last 10 years. Emphasis is placed on the procedure in developing these first‐principles force fields. We discuss the selections of first‐principles methods and fragment models during the parameterization. As the first‐principles force fields are available in a wide range of simulation packages, it is anticipated that use of these force fields will lead to new discoveries of the adsorption phenomena in nanoporous materials and on surfaces. WIREs Comput Mol Sci 2017, 7:e1282. doi: 10.1002/wcms.1282 read less NOT USED (definite) N. Subramanian, B. Koo, A. Rai, and A. Chattopadhyay, “Molecular dynamics-based multiscale damage initiation model for CNT/epoxy nanopolymers,” Journal of Materials Science. 2018. link Times cited: 15 NOT USED (definite) R. A. Bizao, T. Botari, E. Perim, N. Pugno, and D. Galvão, “Mechanical properties and fracture patterns of graphene (graphitic) nanowiggles,” Carbon. 2017. link Times cited: 21 NOT USED (definite) B. Koo, N. Subramanian, and A. Chattopadhyay, “Molecular dynamics study of brittle fracture in epoxy-based thermoset polymer,” Composites Part B-engineering. 2016. link Times cited: 52 NOT USED (definite) T. Senftle et al., “The ReaxFF reactive force-field: development, applications and future directions.” 2016. link Times cited: 1212 |