Sim_LAMMPS_ReaxFF_KeithFantauzziJacob_2010_AuO__SM_974345878378_001
| Title
A single sentence description.
|
LAMMPS ReaxFF potential for Au-O systems developed by Keith et al. (2010) v001 |
|---|---|
| Description | LAMMPS ReaxFF potential for Au-O systems ('pair_style reaxff' with potential file ffield.reax.AuO and additional control and charge equilibration information). The force field optimization involved parameterization of the fcc, bcc, ideal-hcp, sc, diamond, and a15 bulk phases of Au. For these phases the ReaxFF force field gives good agreement for the binding energy, volume of minimum energy, and curvature of the binding well around the minimum compared to the QM calculations used for parameterization. |
| Species
The supported atomic species.
| Au, O |
| Disclaimer
A statement of applicability provided by the contributor, informing users of the intended use of this KIM Item.
|
None |
| Content Origin | LAMMPS package 29-Feb-2019 |
| Contributor |
Ellad B. Tadmor |
| Maintainer |
Ellad B. Tadmor |
| Developer |
John A. Keith Donato Fantauzzi Timo Jacob Adri C. T. van Duin |
| Published on KIM | 2020 |
| How to Cite |
This Simulator Model originally published in [1] is archived in OpenKIM [2-4]. [1] Keith JA, Fantauzzi D, Jacob T, Duin ACT van. Reactive forcefield for simulating gold surfaces and nanoparticles. Physical Review B. 2010Jun;81(23):235404. doi:10.1103/PhysRevB.81.235404 — (Primary Source) A primary source is a reference directly related to the item documenting its development, as opposed to other sources that are provided as background information. [2] Keith JA, Fantauzzi D, Jacob T, Duin ACT van. LAMMPS ReaxFF potential for Au-O systems developed by Keith et al. (2010) v001. OpenKIM; 2020. doi:10.25950/65389f7f [3] Tadmor EB, Elliott RS, Sethna JP, Miller RE, Becker CA. The potential of atomistic simulations and the Knowledgebase of Interatomic Models. JOM. 2011;63(7):17. doi:10.1007/s11837-011-0102-6 [4] Elliott RS, Tadmor EB. Knowledgebase of Interatomic Models (KIM) Application Programming Interface (API). OpenKIM; 2011. doi:10.25950/ff8f563a Click here to download the above citation in BibTeX format. |
| Citations
This panel presents information regarding the papers that have cited the interatomic potential (IP) whose page you are on. The OpenKIM machine learning based Deep Citation framework is used to determine whether the citing article actually used the IP in computations (denoted by "USED") or only provides it as a background citation (denoted by "NOT USED"). For more details on Deep Citation and how to work with this panel, click the documentation link at the top of the panel. The word cloud to the right is generated from the abstracts of IP principle source(s) (given below in "How to Cite") and the citing articles that were determined to have used the IP in order to provide users with a quick sense of the types of physical phenomena to which this IP is applied. The bar chart shows the number of articles that cited the IP per year. Each bar is divided into green (articles that USED the IP) and blue (articles that did NOT USE the IP). Users are encouraged to correct Deep Citation errors in determination by clicking the speech icon next to a citing article and providing updated information. This will be integrated into the next Deep Citation learning cycle, which occurs on a regular basis. OpenKIM acknowledges the support of the Allen Institute for AI through the Semantic Scholar project for providing citation information and full text of articles when available, which are used to train the Deep Citation ML algorithm. |
This panel provides information on past usage of this interatomic potential (IP) powered by the OpenKIM Deep Citation framework. The word cloud indicates typical applications of the potential. The bar chart shows citations per year of this IP (bars are divided into articles that used the IP (green) and those that did not (blue)). The complete list of articles that cited this IP is provided below along with the Deep Citation determination on usage. See the Deep Citation documentation for more information. 
56 Citations (18 used)
Help us to determine which of the papers that cite this potential actually used it to perform calculations. If you know, click the .
USED (high confidence) H. Min et al., “Modified Random Sequential Adsorption Model for Understanding Kinetics of Proteins Adsorption at a Liquid-Solid Interface.,” Langmuir : the ACS journal of surfaces and colloids. 2017. link Times cited: 8 Abstract: In this Article, we experimentally measure the adsorption ki… read more USED (high confidence) A. Ulvestad et al., “In Situ 3D Imaging of Catalysis Induced Strain in Gold Nanoparticles.,” The journal of physical chemistry letters. 2016. link Times cited: 30 Abstract: Multielectron transfer processes are crucially important in … read more USED (high confidence) L. Zhang, W. Wu, Y. Zhou, H. Ren, J. Dong, and H. Li, “Electronic transport properties of ultra-thin Ni and Ni-C nanowires.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 4 Abstract: The structures and electronic transport properties of ultra-… read more USED (high confidence) S. Ali, V. Myasnichenko, and E. Neyts, “Size-dependent strain and surface energies of gold nanoclusters.,” Physical chemistry chemical physics : PCCP. 2016. link Times cited: 60 Abstract: Gold nanocluster properties exhibit unique size-dependence. … read more USED (high confidence) M. Berritta, D. Manrique, and C. Lambert, “Interplay between quantum interference and conformational fluctuations in single-molecule break junctions.,” Nanoscale. 2015. link Times cited: 18 Abstract: We theoretically explored the combined role of conformationa… read more USED (high confidence) P. Kaghazchi, D. Fantauzzi, J. Anton, and T. Jacob, “Nanoscale-faceting of metal surfaces induced by adsorbates.,” Physical chemistry chemical physics : PCCP. 2010. link Times cited: 9 Abstract: Using density functional theory and thermodynamic considerat… read more USED (low confidence) S. Saha, D. Bhadyopadhyay, and N. Choudhury, “Solvation Structure and Dynamics of Aqueous Solutions of Au^+ Ions: A Molecular Dynamics Simulation Study,” Journal of Solution Chemistry. 2023. link Times cited: 0 USED (low confidence) H. Haouas, L. E. Atouani, K. Sbiaai, and A. Hasnaoui, “Size and temperature effects on surface energy of Au and Fe nanoparticles from atomistic simulations,” Computational Materials Science. 2022. link Times cited: 1 USED (low confidence) S. Li, X. Yao, X. Wang, S.-Q. Tian, and Y. Zhang, “Reactive molecular dynamics simulation on degradation of aflatoxin B1 by cold atmospheric plasmas,” Innovative Food Science & Emerging Technologies. 2022. link Times cited: 8 USED (low confidence) J. Yang, W. Zhang, D. Hou, G. Zhang, and Q. Ding, “Structure, dynamics and mechanical properties evolution of calcium silicate hydrate induced by dehydration and dehydroxylation,” Construction and Building Materials. 2021. link Times cited: 14 USED (low confidence) S. Starikov, I. Gordeev, Y. Lysogorskiy, L. Kolotova, and S. Makarov, “Optimized interatomic potential for study of structure and phase transitions in Si-Au and Si-Al systems,” Computational Materials Science. 2020. link Times cited: 19 USED (low confidence) J. Zhang, J. Yang, D. Hou, and Q. Ding, “Molecular dynamics study on calcium aluminosilicate hydrate at elevated temperatures: Structure, dynamics and mechanical properties,” Materials Chemistry and Physics. 2019. link Times cited: 14 USED (low confidence) S. Monti, G. Barcaro, L. Sementa, V. Carravetta, and H. Ågren, “Characterization of the adsorption dynamics of trisodium citrate on gold in water solution,” RSC Advances. 2017. link Times cited: 26 Abstract: Molecular dynamics simulations (MDs) based on a reactive for… read more USED (low confidence) D. Bhandary, V. Valechi, M. Cordeiro, and J. Singh, “Janus Gold Nanoparticles from Nanodroplets of Alkyl Thiols: A Molecular Dynamics Study.,” Langmuir : the ACS journal of surfaces and colloids. 2017. link Times cited: 10 Abstract: Janus particles provide an asymmetry in structure, which can… read more USED (low confidence) X. Zhang et al., “Site Stability on Cobalt Nanoparticles: A Molecular Dynamics ReaxFF Reactive Force Field Study,” Journal of Physical Chemistry C. 2014. link Times cited: 31 Abstract: The stability of step-edge-type surface sites on cobalt nano… read more USED (low confidence) X. Liu, F. Wang, H. S. Park, and H. Wu, “Defecting controllability of bombarding graphene with different energetic atoms via reactive force field model,” Journal of Applied Physics. 2013. link Times cited: 29 Abstract: We study the bombardment of a suspended monolayer graphene s… read more USED (low confidence) K. Kleiner et al., “Multiscale Modeling of Au-Island Ripening on Au(100),” Advances in Physical Chemistry. 2011. link Times cited: 11 Abstract: We describe a multiscale modeling hierarchy for the particul… read more USED (low confidence) X. Liu, “Defect-Induced Discontinuous Effects in Graphene Nanoribbon Under Torsion Loading,” Springer Theses. 2019. link Times cited: 0 NOT USED (low confidence) J. Tiihonen and H. Häkkinen, “Towards structural optimization of gold nanoclusters with quantum Monte Carlo.,” The Journal of chemical physics. 2023. link Times cited: 0 Abstract: We study the prospects of using quantum Monte Carlo techniqu… read more NOT USED (low confidence) S. Li et al., “Atomic-scale simulations of the deoxynivalenol degradation induced by reactive oxygen plasma species.,” Food research international. 2022. link Times cited: 2 NOT USED (low confidence) P. Niedziałkowski et al., “Deciphering the Molecular Mechanism of Substrate-Induced Assembly of Gold Nanocube Arrays toward an Accelerated Electrocatalytic Effect Employing Heterogeneous Diffusion Field Confinement,” Langmuir. 2022. link Times cited: 5 Abstract: The complex electrocatalytic performance of gold nanocubes (… read more NOT USED (low confidence) C. Dulong, B. Madebène, S. Monti, and J. Richardi, “Optimization of a new reactive force field for silver-based materials.,” Journal of chemical theory and computation. 2020. link Times cited: 3 Abstract: A new reactive force field based on the ReaxFF formalism is … read more NOT USED (low confidence) I. Shuttleworth, “Development of the ReaxFF Reactive Force-Field Description of Gold Oxides,” Journal of Physical Chemistry C. 2017. link Times cited: 2 Abstract: For the investigation of the interaction between oxygen and … read more NOT USED (low confidence) D. Depew and J. Wang, “Density Functional Theory Investigations on Bulk Iridium Structures for ReaxFF Catalysis Parameterization.” 2016. link Times cited: 0 NOT USED (low confidence) S. Monti, V. Carravetta, and H. Ågren, “Simulation of Gold Functionalization with Cysteine by Reactive Molecular Dynamics.,” The journal of physical chemistry letters. 2016. link Times cited: 41 Abstract: The anchoring mechanism of cysteine to gold in water solutio… read more NOT USED (low confidence) A. M. Raymunt, R. T. Bell, M. Thompson, and P. Clancy, “Effect of Laser Annealing on the Structure of Amorphous Porous SiCOH Materials,” Journal of Physical Chemistry C. 2015. link Times cited: 5 Abstract: The inherent trade-off of porosity and mechanical properties… read more NOT USED (low confidence) T. Liang et al., “Reactive Potentials for Advanced Atomistic Simulations,” Materials Research-ibero-american Journal of Materials. 2013. link Times cited: 180 Abstract: This article reviews recent advances in the development of r… read more NOT USED (low confidence) H. Heinz, T.-J. Lin, R. K. Mishra, and F. Emami, “Thermodynamically consistent force fields for the assembly of inorganic, organic, and biological nanostructures: the INTERFACE force field.,” Langmuir : the ACS journal of surfaces and colloids. 2013. link Times cited: 635 Abstract: The complexity of the molecular recognition and assembly of … read more NOT USED (low confidence) L. Morrissey, S. M. Handrigan, and S. Nakhla, “Discrepancies in the mechanical properties of gold nanowires: The importance of potential type and equilibration method,” Computational Materials Science. 2020. link Times cited: 6 NOT USED (high confidence) C. D. Griego et al., “Computationally Guided Searches for Efficient Catalysts through Chemical/Materials Space: Progress and Outlook,” Journal of Physical Chemistry C. 2021. link Times cited: 2 Abstract: Computational quantum chemistry promises to help guide the d… read more NOT USED (high confidence) R. K. Shayakhmetova and E. Khamitov, “ReaxFF Molecular Dynamics Simulation of the Cracking of Components of Vacuum Gasoil in the Presence of a Nickel Nanocluster,” Russian Journal of Physical Chemistry A. 2021. link Times cited: 0 NOT USED (high confidence) T. Loeffler, S. Manna, T. Patra, H. Chan, B. Narayanan, and S. Sankaranarayanan, “Active Learning A Neural Network Model For Gold Clusters & Bulk From Sparse First Principles Training Data,” ChemCatChem. 2020. link Times cited: 15 Abstract: Small metal clusters are of fundamental scientific interest … read more NOT USED (high confidence) A. C. D. López and E. Pehlke, “Initial steps toward Auad island nucleation on a c(2 × 2)-Cl Au(001) surface investigated by DFT.,” The Journal of chemical physics. 2020. link Times cited: 1 Abstract: Density functional theory calculations are reported that elu… read more NOT USED (high confidence) H. S. Huang, L. Ai, A. V. van Duin, M. Chen, and Y. Lü, “ReaxFF reactive force field for molecular dynamics simulations of liquid Cu and Zr metals.,” The Journal of chemical physics. 2019. link Times cited: 10 Abstract: We develop a ReaxFF reactive force field used for the molecu… read more NOT USED (high confidence) A. Antony et al., “Charge optimized many body (COMB) potentials for Pt and Au,” Journal of Physics: Condensed Matter. 2017. link Times cited: 12 Abstract: Interatomic potentials for Pt and Au are developed within th… read more NOT USED (high confidence) J. R. Boes, M. C. Groenenboom, J. Keith, and J. Kitchin, “Neural network and ReaxFF comparison for Au properties,” International Journal of Quantum Chemistry. 2016. link Times cited: 64 Abstract: We have studied how ReaxFF and Behler–Parrinello neural netw… read more NOT USED (high confidence) S. Monti, V. Carravetta, and H. Ågren, “Decoration of gold nanoparticles with cysteine in solution: reactive molecular dynamics simulations.,” Nanoscale. 2016. link Times cited: 19 Abstract: The dynamics of gold nanoparticle functionalization by means… read more NOT USED (high confidence) X. Li, V. Carravetta, C. Li, S. Monti, Ž. Rinkevičius, and H. Ågren, “Optical Properties of Gold Nanoclusters Functionalized with a Small Organic Compound: Modeling by an Integrated Quantum-Classical Approach.,” Journal of chemical theory and computation. 2016. link Times cited: 7 Abstract: Motivated by the growing importance of organometallic nanost… read more NOT USED (high confidence) Y. Han, D. D. Jiang, J. Zhang, W. Li, Z. Gan, and J. Gu, “Development, applications and challenges of ReaxFF reactive force field in molecular simulations,” Frontiers of Chemical Science and Engineering. 2016. link Times cited: 87 NOT USED (high confidence) E. Pohjolainen, X. Chen, S. Malola, G. Groenhof, and H. Häkkinen, “A Unified AMBER-Compatible Molecular Mechanics Force Field for Thiolate-Protected Gold Nanoclusters.,” Journal of chemical theory and computation. 2016. link Times cited: 62 Abstract: We present transferable AMBER-compatible force field paramet… read more NOT USED (high confidence) B. Narayanan et al., “Describing the Diverse Geometries of Gold from Nanoclusters to Bulk—A First-Principles-Based Hybrid Bond-Order Potential,” Journal of Physical Chemistry C. 2015. link Times cited: 27 Abstract: Molecular dynamics simulations using empirical force fields … read more NOT USED (high confidence) B. Narayanan, S. A. Deshmukh, S. Sankaranarayanan, and S. Ramanathan, “Strong correlations between structural order and passive state at water–copper oxide interfaces,” Electrochimica Acta. 2015. link Times cited: 9 NOT USED (high confidence) D. Thompson et al., “Formation Mechanism of Metal–Molecule–Metal Junctions: Molecule-Assisted Migration on Metal Defects,” Journal of Physical Chemistry C. 2015. link Times cited: 14 Abstract: Activation energies, Ea, measured from molecular exchange ex… read more NOT USED (high confidence) J. M. Cabrera-Trujillo, J. M. Montejano-Carrizales, F. Aguilera-Granja, and A. Posada-Amarillas, “Theoretical study of the thermally induced structural fluctuations in sub-nanometre size gold clusters,” The European Physical Journal D. 2015. link Times cited: 7 NOT USED (high confidence) D. Fantauzzi, J. Bandlow, L. Sabo, J. Mueller, A. V. van Duin, and T. Jacob, “Development of a ReaxFF potential for Pt-O systems describing the energetics and dynamics of Pt-oxide formation.,” Physical chemistry chemical physics : PCCP. 2014. link Times cited: 53 Abstract: ReaxFF force field parameters describing Pt-Pt and Pt-O inte… read more NOT USED (high confidence) H. Larsson, A. Duin, and B. Hartke, “Global optimization of parameters in the reactive force field ReaxFF for SiOH,” Journal of Computational Chemistry. 2013. link Times cited: 78 Abstract: We have used unbiased global optimization to fit a reactive … read more NOT USED (high confidence) O. Verners, Y. Shin, and A. Duin, “Molecular dynamics simulation of Al grain mixing in Fe/Ni matrices and its influence on oxidation,” Journal of Applied Physics. 2013. link Times cited: 1 Abstract: AlxNiyFe(1−x−y) alloys are structural materials with potenti… read more NOT USED (high confidence) E. Iype, M. Hütter, A. Jansen, S. S. Nedea, and C. Rindt, “Parameterization of a reactive force field using a Monte Carlo algorithm,” Journal of Computational Chemistry. 2013. link Times cited: 79 Abstract: Parameterization of a molecular dynamics force field is esse… read more NOT USED (high confidence) L. Wright, P. Rodger, S. Corni, and T. Walsh, “GolP-CHARMM: First-Principles Based Force Fields for the Interaction of Proteins with Au(111) and Au(100).,” Journal of chemical theory and computation. 2013. link Times cited: 202 Abstract: Computational simulation of peptide adsorption at the aqueou… read more NOT USED (high confidence) Y. Shin, H. Kwak, C. Zou, A. Vasenkov, and A. V. van Duin, “Development and validation of a ReaxFF reactive force field for Fe/Al/Ni alloys: molecular dynamics study of elastic constants, diffusion, and segregation.,” The journal of physical chemistry. A. 2012. link Times cited: 59 Abstract: We have developed a ReaxFF force field for Fe/Al/Ni binary a… read more NOT USED (high confidence) M. Backman, N. Juslin, and K. Nordlund, “Bond order potential for gold,” The European Physical Journal B. 2012. link Times cited: 11 NOT USED (high confidence) K. Farah, F. Müller-Plathe, and M. Böhm, “Classical reactive molecular dynamics implementations: state of the art.,” Chemphyschem : a European journal of chemical physics and physical chemistry. 2012. link Times cited: 71 Abstract: Reactive molecular dynamics (RMD) implementations equipped w… read more NOT USED (high confidence) J. Feng, J. Slocik, M. Sarikaya, R. Naik, B. Farmer, and H. Heinz, “Influence of the shape of nanostructured metal surfaces on adsorption of single peptide molecules in aqueous solution.,” Small. 2012. link Times cited: 90 Abstract: Self-assembly and function of biologically modified metal na… read more NOT USED (high confidence) C. Iacovella, W. R. French, B. Cook, P. Kent, and P. Cummings, “Role of polytetrahedral structures in the elongation and rupture of gold nanowires.,” ACS nano. 2011. link Times cited: 13 Abstract: We report comprehensive high-accuracy molecular dynamics sim… read more NOT USED (high confidence) E. Iype and A. A. Steenhoven, “In silico characterisation of magnesium salt hydrates as energy storage materials.” 2014. link Times cited: 3 Abstract: • A submitted manuscript is the author's version of the… read more NOT USED (definite) L. Wright, P. Rodger, T. Walsh, and S. Corni, “First-principles-based force field for the interaction of proteins with Au(100)(5 × 1) : an extension of GolP-CHARMM,” Journal of Physical Chemistry C. 2013. link Times cited: 54 Abstract: Noncovalent recognition between peptides and inorganic mater… read more |
| Funding | Not available |
| Short KIM ID
The unique KIM identifier code.
| SM_974345878378_001 |
| Extended KIM ID
The long form of the KIM ID including a human readable prefix (100 characters max), two underscores, and the Short KIM ID. Extended KIM IDs can only contain alpha-numeric characters (letters and digits) and underscores and must begin with a letter.
| Sim_LAMMPS_ReaxFF_KeithFantauzziJacob_2010_AuO__SM_974345878378_001 |
| DOI |
10.25950/65389f7f https://doi.org/10.25950/65389f7f https://commons.datacite.org/doi.org/10.25950/65389f7f |
| KIM Item Type | Simulator Model |
| KIM API Version | 2.1 |
| Simulator Name
The name of the simulator as defined in kimspec.edn.
| LAMMPS |
| Potential Type | reax |
| Simulator Potential | reaxff |
| Run Compatibility | portable-models |
| Previous Version | Sim_LAMMPS_ReaxFF_KeithFantauzziJacob_2010_AuO__SM_974345878378_000 |
| Grade | Name | Category | Brief Description | Full Results | Aux File(s) |
|---|---|---|---|---|---|
| P | vc-species-supported-as-stated | mandatory | The model supports all species it claims to support; see full description. |
Results | Files |
| N/A | vc-periodicity-support | mandatory | Periodic boundary conditions are handled correctly; see full description. |
Results | Files |
| F | vc-permutation-symmetry | mandatory | Total energy and forces are unchanged when swapping atoms of the same species; see full description. |
Results | Files |
| F | vc-dimer-continuity-c1 | informational | The energy versus separation relation of a pair of atoms is C1 continuous (i.e. the function and its first derivative are continuous); see full description. |
Results | Files |
| F | vc-objectivity | informational | Total energy is unchanged and forces transform correctly under rigid-body translation and rotation; see full description. |
Results | Files |
| F | vc-inversion-symmetry | informational | Total energy is unchanged and forces change sign when inverting a configuration through the origin; see full description. |
Results | Files |
| F | vc-memory-leak | informational | The model code does not have memory leaks (i.e. it releases all allocated memory at the end); see full description. |
Results | Files |
| N/A | vc-thread-safe | mandatory | The model returns the same energy and forces when computed in serial and when using parallel threads for a set of configurations. Note that this is not a guarantee of thread safety; see full description. |
Results | Files |
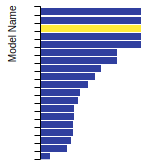
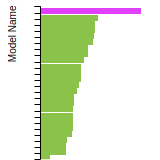
BCC Lattice Constant
This bar chart plot shows the mono-atomic body-centered cubic (bcc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
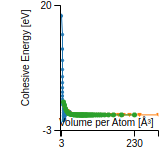
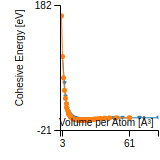
Cohesive Energy Graph
This graph shows the cohesive energy versus volume-per-atom for the current mode for four mono-atomic cubic phases (body-centered cubic (bcc), face-centered cubic (fcc), simple cubic (sc), and diamond). The curve with the lowest minimum is the ground state of the crystal if stable. (The crystal structure is enforced in these calculations, so the phase may not be stable.) Graphs are generated for each species supported by the model.
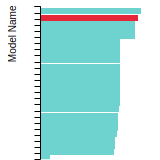
Diamond Lattice Constant
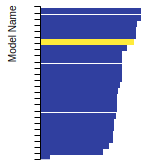
This bar chart plot shows the mono-atomic face-centered diamond lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
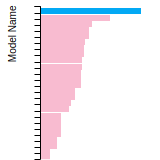
Dislocation Core Energies
This graph shows the dislocation core energy of a cubic crystal at zero temperature and pressure for a specific set of dislocation core cutoff radii. After obtaining the total energy of the system from conjugate gradient minimizations, non-singular, isotropic and anisotropic elasticity are applied to obtain the dislocation core energy for each of these supercells with different dipole distances. Graphs are generated for each species supported by the model.
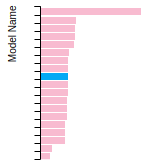
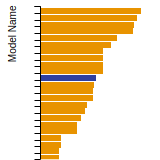
(No matching species)FCC Elastic Constants
This bar chart plot shows the mono-atomic face-centered cubic (fcc) elastic constants predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
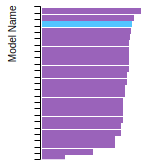
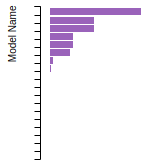
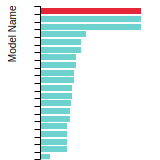
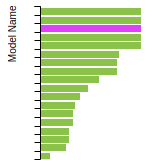
FCC Lattice Constant
This bar chart plot shows the mono-atomic face-centered cubic (fcc) lattice constant predicted by the current model (shown in red) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
FCC Stacking Fault Energies
This bar chart plot shows the intrinsic and extrinsic stacking fault energies as well as the unstable stacking and unstable twinning energies for face-centered cubic (fcc) predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
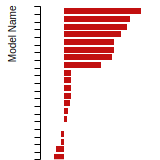
FCC Surface Energies
This bar chart plot shows the mono-atomic face-centered cubic (fcc) relaxed surface energies predicted by the current model (shown in blue) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
SC Lattice Constant
This bar chart plot shows the mono-atomic simple cubic (sc) lattice constant predicted by the current model (shown in the unique color) compared with the predictions for all other models in the OpenKIM Repository that support the species. The vertical bars show the average and standard deviation (one sigma) bounds for all model predictions. Graphs are generated for each species supported by the model.
Cubic Crystal Basic Properties Table
Species: AuSpecies: O
Creators:
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/64cb38c5
This Test Driver uses LAMMPS to compute the cohesive energy of a given monoatomic cubic lattice (fcc, bcc, sc, or diamond) at a variety of lattice spacings. The lattice spacings range from a_min (=a_min_frac*a_0) to a_max (=a_max_frac*a_0) where a_0, a_min_frac, and a_max_frac are read from stdin (a_0 is typically approximately equal to the equilibrium lattice constant). The precise scaling and number of lattice spacings sampled between a_min and a_0 (a_0 and a_max) is specified by two additional parameters passed from stdin: N_lower and samplespacing_lower (N_upper and samplespacing_upper). Please see README.txt for further details.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Cohesive energy versus lattice constant curve for bcc Au v003 | view | 3996 | |
| Cohesive energy versus lattice constant curve for bcc O v003 | view | 1886 | |
| Cohesive energy versus lattice constant curve for diamond Au v003 | view | 2174 | |
| Cohesive energy versus lattice constant curve for diamond O v004 | view | 38798 | |
| Cohesive energy versus lattice constant curve for fcc Au v004 | view | 19265 | |
| Cohesive energy versus lattice constant curve for fcc O v004 | view | 3849 | |
| Cohesive energy versus lattice constant curve for sc Au v004 | view | 10334 | |
| Cohesive energy versus lattice constant curve for sc O v004 | view | 3083 |
Creators: Junhao Li and Ellad Tadmor
Contributor: tadmor
Publication Year: 2019
DOI: https://doi.org/10.25950/5853fb8f
Computes the cubic elastic constants for some common crystal types (fcc, bcc, sc, diamond) by calculating the hessian of the energy density with respect to strain. An estimate of the error associated with the numerical differentiation performed is reported.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Elastic constants for bcc Au at zero temperature v006 | view | 14769 | |
| Elastic constants for bcc O at zero temperature v006 | view | 4348 | |
| Elastic constants for diamond Au at zero temperature v001 | view | 34684 | |
| Elastic constants for diamond O at zero temperature v001 | view | 140303 | |
| Elastic constants for fcc Au at zero temperature v006 | view | 17486 | |
| Elastic constants for fcc O at zero temperature v006 | view | 4699 | |
| Elastic constants for sc Au at zero temperature v006 | view | 14161 | |
| Elastic constants for sc O at zero temperature v006 | view | 4188 |
Creators:
Contributor: ilia
Publication Year: 2023
DOI: https://doi.org/10.25950/e8a7ed84
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium crystal structure and energy for Au in AFLOW crystal prototype A_cF4_225_a v001 | view | 99535 |
Creators:
Contributor: ilia
Publication Year: 2024
DOI: https://doi.org/10.25950/2f2c4ad3
Computes the equilibrium crystal structure and energy for an arbitrary crystal at zero temperature and applied stress by performing symmetry-constrained relaxation. The crystal structure is specified using the AFLOW prototype designation. Multiple sets of free parameters corresponding to the crystal prototype may be specified as initial guesses for structure optimization. No guarantee is made regarding the stability of computed equilibria, nor that any are the ground state.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium crystal structure and energy for AuO in AFLOW crystal prototype A2B3_oF40_43_b_ab v002 | view | 103960 | |
| Equilibrium crystal structure and energy for O in AFLOW crystal prototype A_hP4_194_f v002 | view | 45570 | |
| Equilibrium crystal structure and energy for O in AFLOW crystal prototype A_hR2_166_c v002 | view | 91731 | |
| Equilibrium crystal structure and energy for O in AFLOW crystal prototype A_oC12_63_cg v002 | view | 110210 | |
| Equilibrium crystal structure and energy for O in AFLOW crystal prototype A_oP24_61_3c v002 | view | 762782 |
Creators:
Contributor: brunnels
Publication Year: 2022
DOI: https://doi.org/10.25950/2c59c9d6
Computes grain boundary energy for a range of tilt angles given a crystal structure, tilt axis, and material.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Relaxed energy as a function of tilt angle for a 100 symmetric tilt grain boundary in fcc Au v000 | view | 267721905 | |
| Relaxed energy as a function of tilt angle for a 111 symmetric tilt grain boundary in fcc Au v000 | view | 383484841 |
Creators: Daniel S. Karls and Junhao Li
Contributor: karls
Publication Year: 2019
DOI: https://doi.org/10.25950/2765e3bf
Equilibrium lattice constant and cohesive energy of a cubic lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Equilibrium zero-temperature lattice constant for bcc Au v007 | view | 7800 | |
| Equilibrium zero-temperature lattice constant for bcc O v007 | view | 3932 | |
| Equilibrium zero-temperature lattice constant for diamond Au v007 | view | 50828 | |
| Equilibrium zero-temperature lattice constant for diamond O v007 | view | 28099 | |
| Equilibrium zero-temperature lattice constant for fcc Au v007 | view | 14385 | |
| Equilibrium zero-temperature lattice constant for fcc O v007 | view | 6489 | |
| Equilibrium zero-temperature lattice constant for sc Au v007 | view | 5434 | |
| Equilibrium zero-temperature lattice constant for sc O v007 | view | 3229 |
Creators: Mingjian Wen
Contributor: mjwen
Publication Year: 2019
DOI: https://doi.org/10.25950/fc69d82d
This Test Driver uses LAMMPS to compute the linear thermal expansion coefficient at a finite temperature under a given pressure for a cubic lattice (fcc, bcc, sc, diamond) of a single given species.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Linear thermal expansion coefficient of fcc Au at 293.15 K under a pressure of 0 MPa v001 | view | 240598347 |
Creators: Matt Bierbaum
Contributor: mattbierbaum
Publication Year: 2019
DOI: https://doi.org/10.25950/64f4999b
Calculates the phonon dispersion relations for fcc lattices and records the results as curves.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Phonon dispersion relations for fcc Au v004 | view | 78671 |
Creators:
Contributor: SubrahmanyamPattamatta
Publication Year: 2019
DOI: https://doi.org/10.25950/b4cfaf9a
Intrinsic and extrinsic stacking fault energies, unstable stacking fault energy, unstable twinning energy, stacking fault energy as a function of fractional displacement, and gamma surface for a monoatomic FCC lattice at zero temperature and pressure.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Stacking and twinning fault energies for fcc Au v002 | view | 754126944 |
Creators: Matt Bierbaum
Contributor: mattbierbaum
Publication Year: 2019
DOI: https://doi.org/10.25950/6c43a4e6
Calculates the surface energy of several high symmetry surfaces and produces a broken-bond model fit. In latex form, the fit equations are given by:
E_{FCC} (\vec{n}) = p_1 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (2 ( |x+ 2y + z| + |x+2y-z| + |x-2y + z| + |x-2y-z| + |2x+y+z| + |2x+y-z| +|2x-y+z| +|2x-y-z| +|x+y+2z| +|x+y-2z| +|x-y+2z| +|x-y-2z| ) + c
E_{BCC} (\vec{n}) = p_1 (6 \left( | x+y+z| + |x+y-z| + |-x+y-z| + |x-y+z| \right)) + p_2 (8 \left( |x| + |y| + |z|\right)) + p_3 (4 \left( |x+y| + |x-y| + |x+z| + |x-z| + |z+y| +|z-y|\right)) +c.
In Python, these two fits take the following form:
def BrokenBondFCC(params, index):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*(abs(x+2*y+z) + abs(x+2*y-z) +abs(x-2*y+z) +abs(x-2*y-z) + abs(2*x+y+z) +abs(2*x+y-z) +abs(2*x-y+z) +abs(2*x-y-z) + abs(x+y+2*z) +abs(x+y-2*z) +abs(x-y+2*z) +abs(x-y-2*z))+params[3]
def BrokenBondBCC(params, x, y, z):
import numpy
x, y, z = index
x = x / numpy.sqrt(x**2.+y**2.+z**2.)
y = y / numpy.sqrt(x**2.+y**2.+z**2.)
z = z / numpy.sqrt(x**2.+y**2.+z**2.)
return params[0]*6*(abs(x+y+z) + abs(x-y-z) + abs(x-y+z) + abs(x+y-z)) + params[1]*8*(abs(x) + abs(y) + abs(z)) + params[2]*4* (abs(x+y) + abs(x-y) + abs(x+z) + abs(x-z) + abs(z+y) + abs(z-y)) + params[3]
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Broken-bond fit of high-symmetry surface energies in fcc Au v004 | view | 944785 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/fca89cea
Computes the monovacancy formation energy and relaxation volume for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Monovacancy formation energy and relaxation volume for fcc Au | view | 15441826 |
Creators:
Contributor: efuem
Publication Year: 2023
DOI: https://doi.org/10.25950/c27ba3cd
Computes the monovacancy formation and migration energies for cubic and hcp monoatomic crystals.
| Test | Test Results | Link to Test Results page | Benchmark time
Usertime multiplied by the Whetstone Benchmark. This number can be used (approximately) to compare the performance of different models independently of the architecture on which the test was run.
Measured in Millions of Whetstone Instructions (MWI) |
|---|---|---|---|
| Vacancy formation and migration energy for fcc Au | view | 13692456 | |
| Vacancy formation and migration energy for sc O | view | 6463438 |
| Test | Error Categories | Link to Error page |
|---|---|---|
| Cohesive energy versus lattice constant curve for bcc Au v004 | other | view |
| Cohesive energy versus lattice constant curve for bcc O v004 | other | view |
| Cohesive energy versus lattice constant curve for diamond Au v004 | other | view |
EquilibriumCrystalStructure__TD_457028483760_002
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium crystal structure and energy for O in AFLOW crystal prototype A_mC16_12_2ij v002 | other | view |
| Equilibrium crystal structure and energy for O in AFLOW crystal prototype A_mC4_12_i v002 | other | view |
GrainBoundaryCubicCrystalSymmetricTiltRelaxedEnergyVsAngle__TD_410381120771_003
| Test | Error Categories | Link to Error page |
|---|---|---|
| Relaxed energy as a function of tilt angle for a 110 symmetric tilt grain boundary in fcc Au v000 | other | view |
| Relaxed energy as a function of tilt angle for a 112 symmetric tilt grain boundary in fcc Au v000 | other | view |
LatticeConstantHexagonalEnergy__TD_942334626465_005
| Test | Error Categories | Link to Error page |
|---|---|---|
| Equilibrium lattice constants for hcp Au v005 | other | view |
| Equilibrium lattice constants for hcp O v005 | other | view |
VacancyFormationEnergyRelaxationVolume__TD_647413317626_001
| Test | Error Categories | Link to Error page |
|---|---|---|
| Monovacancy formation energy and relaxation volume for sc O | other | view |
No Driver
| Verification Check | Error Categories | Link to Error page |
|---|---|---|
| ForcesNumerDeriv__VC_710586816390_003 | other | view |
| MemoryLeak__VC_561022993723_004 | other | view |
| PeriodicitySupport__VC_895061507745_003 | other | view |
| Sim_LAMMPS_ReaxFF_KeithFantauzziJacob_2010_AuO__SM_974345878378_001.txz | Tar+XZ | Linux and OS X archive |
| Sim_LAMMPS_ReaxFF_KeithFantauzziJacob_2010_AuO__SM_974345878378_001.zip | Zip | Windows archive |